
ポリクローナル抗体およびモノクローナル抗体のハイブリドーマ不要のエクスビボ産生法ならびに不死化細胞集団の作製法
本発明は、ハイブリドーマの形成を必要とせずに抗体産生細胞を作製するための方法および組成物を含む。1つの態様において、本方法は、不死化細胞集団を有するトランスジェニックマウスから細胞を収集することによるモノクローナル抗体産生細胞の作製を含む。1つの態様において、本方法は、不死化ヒトモノクローナル抗体産生細胞株を作製するための、ヒト化抗体を産生し得るトランスジェニックマウスと交配させた不死化細胞集団を有するトランスジェニックマウスの使用を含む。
【発明の詳細な説明】
【技術分野】
【0001】
発明の背景
1. 発明の分野
本発明は、分子生物学、細胞生物学、および免疫学の分野に関する。より具体的には、本発明はハイブリドーマを使用しない抗体合成の方法および用途に関する。なお、本出願は2003年4月14日に出願された米国仮特許出願第60/462,631号および2003年11月24日に出願された米国仮特許出願第60/524,701号の優先権を主張し、これらは完全に参照により本明細書に組み入れられる。
【背景技術】
【0002】
2. 背景
モノクローナル抗体は、標的分子上の特定部位との反応において高い特異性および感度を有するタンパク質である。モノクローナル抗体は、年月を経て、ヒトの疾患の解析および治療といった近代の生物学的研究および医学における主要な試薬となった。しかし、モノクローナル抗体は、導入されてから四半世紀を超えても、いまだに多発性骨髄腫由来細胞に融合させた脾細胞の体細胞クローン(ハイブリドーマ)によってのみ産生される(Kohler and Milstein, 1975)。これらの「ハイブリドーマ」は何年もの間モノクローナル抗体を産生し得るが、その作製は、絶えず続く汚染のリスク、再三にわたるフィーダー細胞の必要性、および遺伝的不安定性の可能性によって制限される手間のかかる多段階の過程を含む(Harlow and Lane, 1988)。ハイブリドーマ作製の過程は2ヶ月以内に完了することは稀であり、たっぷり1年以上を要する場合が多い。
【0003】
モノクローナル抗体を作製するための従来のアプローチは、少なくも2つの制限を受ける;(i) 主として遺伝的不安定性によるハイブリドーマ細胞株の安定性の欠如および(ii) ハイブリドーマが培養において2〜3週間の存続期間しか有さず、この期間内に結合の特異性に関してスクリーニングしなければならないことによる、クローンの選択およびスクリーニングに関する時間の制限。モノクローナル抗体をインビトロで作製するためのファージディスプレイ技術(Winter et al., 1994;Barbas et al., 2000)、キメラ化またはヒト化戦略(Winte and Milstein, 1991)、およびハイブリドーマの形成に適したヒト骨髄腫細胞株(Karpas et al., 2001)等の最近の技術開発にもかかわらず、不死化モノクローナル抗体産生細胞を作製するためのさらなる戦略および方法はやはり必要であり、本分野において大きな進展を意味すると考えられる。
【発明の開示】
【0004】
発明の概要
本発明の態様は、抗体産生細胞を不死化細胞に融合させる(例えばハイブリドーマ作製)必要なしに、所望の抗原に対する抗体を産生する抗体産生細胞を作製するための方法を含む。本発明の局面は、ハイブリドーマを形成することなく不死化され得る抗体産生細胞を、抗原に対する抗体を産生するよう細胞を誘導するのに効率的な様式で所望の抗原に接触させる段階;および抗体産生細胞を不死化する段階を含む。抗体産生細胞は典型的に、条件的に機能するかまたは条件的に発現される形質転換発癌遺伝子を含み、抗体産生細胞の不死化は形質転換発癌遺伝子の発現または機能の誘導(形質転換発癌遺伝子の誘導)によってもたらされる。特定の態様において、条件的に機能する形質転換発癌遺伝子は温度感受性SV40大型腫瘍抗原(tsSV40Tag)であり、好ましくはtsSV40TagはA58S-SV40Tagである。本発明の特定の局面において、形質転換発癌遺伝子は、抗体産生細胞を25℃〜35℃、好ましくは30℃〜35℃、より好ましくは約33℃で培養することによって誘導する。典型的に、抗体産生細胞はハイブリドーマ培地で培養する。
【0005】
さらなる態様において、本方法は抗体産生細胞の抗体産生能を評価する段階を含む。抗体産生細胞の評価は、所望の抗原ならびに類似の抗原および異なる抗原に対する抗体結合をアッセイする段階を含み得る。モノクローナル抗体を産生するモノクローナル細胞集団を作製するには、典型的に単一細胞を選択して培養する。単一細胞は希釈クローニングにより選択し得る。本発明の他の局面では、ポリクローナル抗体を産生するポリクローナル細胞集団を作製するために、複数の細胞を選択して培養する。特定の態様において、抗体産生細胞は脾臓細胞(脾細胞)を含む。抗原は、ペプチド;タンパク質;糖タンパク質;リポタンパク質;炭水化物;ウイルス;細菌;病原微生物;組織;細胞全体;生検組織;患者由来の細胞;組織抽出物;新鮮なまたは培養した組織;アポトーシス細胞;細胞および組織に由来する膜、細胞質、および核画分等の細胞成分;精製タンパク質;部分精製タンパク質;レーザー捕獲した組織;またはパラフィン包埋し固定した組織であってよい。好ましい態様において、組織は対象由来の腫瘍組織を含む。
【0006】
本発明の他の局面において、抗体産生細胞は、ハイブリドーマを形成することなく不死化され得る抗体産生細胞を有するトランスジェニックマウスから取得することができる。トランスジェニックマウスは、ヒト抗体を産生するための遺伝子相補体(genetic complement)を含み得る。本発明の抗体産生細胞はマウスにおいて構成されてもよく、選択された抗原は、抗体を産生するよう抗体産生細胞を誘導するのに効率的な様式でマウスに投与される。特定の態様において、本方法は抗体産生細胞を抗原提示細胞と共培養することにより抗体産生細胞を所望の抗原と接触させる段階を含み、好ましくは抗原提示細胞は樹状細胞である。種々の態様において、抗体産生細胞はヒト抗体を産生するための遺伝子相補体を含み、ヒト抗体を産生する。本方法は、抗体産生細胞によって産生された抗体を精製する段階をさらに含み得る。特定の態様において、本方法は治療抗体を必要とする対象に抗体を投与する段階をさらに含み得る。
【0007】
本発明のさらなる態様は、所望の抗原に対するヒト抗体を産生する抗体産生細胞を作製するための方法を含む。本方法は、形質転換発癌遺伝子を条件的に発現するかまたは条件的に機能する形質転換発癌遺伝子を発現し、かつヒト抗体を産生するための遺伝子相補体を発現する抗体産生細胞を取得する段階を含み得る。抗体産生細胞の不死化は典型的に、形質転換発癌遺伝子の発現または機能を誘導することによってもたらされる。本発明はまた、ハイブリドーマを形成することなく不死化され得る抗体産生細胞を、抗原に対するヒト抗体を産生するよう細胞を誘導するのに効率的な様式で所望の抗原に接触させる段階、および抗体産生細胞を不死化する段階を含む。条件的に機能する形質転換発癌遺伝子は温度感受性SV40大型腫瘍抗原(tsSV40Tagであり)、好ましくはtsSV40TagはA58S-SV40Tagである。本発明の特定の局面は、25℃〜35℃、好ましくは30℃〜35℃、より好ましくは約33℃で抗体産生細胞を培養することによる、形質転換発癌遺伝子の発現または機能性の誘導を含む。
【0008】
本発明はまた、モノクローナル抗体を産生するモノクローナル集団を作製するために、単一細胞を選択および培養する段階を含み得る。他の局面において、本方法は、ポリクローナル抗体を産生するポリクローナル集団を作製するための、複数の細胞の選択および培養を含み得る。特定の局面において、抗体産生細胞は脾臓細胞を含む。抗原には、1つまたは複数のペプチド;タンパク質;糖タンパク質;リポタンパク質;炭水化物;ウイルス;細菌;病原微生物;組織;細胞全体;生検組織;患者由来の細胞;組織抽出物;新鮮なまたは培養した組織;アポトーシス細胞;細胞および組織に由来する膜、細胞質、および核画分等の細胞成分;精製タンパク質;部分精製タンパク質;レーザー捕獲した組織;またはパラフィン包埋し固定した組織が含まれ得る。組織は対象由来の腫瘍組織を含み得る。さらなる態様において、抗体産生細胞は、ハイブリドーマを形成することなく不死化され得る抗体産生細胞を有するトランスジェニックマウスから取得することができる。抗体産生細胞はマウスにおいて構成されてもよく、選択された抗原は、抗体を産生するよう抗体産生細胞を誘導するのに効率的な様式でマウスに投与される。他の局面において、本方法は抗体産生細胞によって産生された抗体を精製する段階をさらに含み得る。治療抗体を必要とする対象に抗体を投与することができる。
【0009】
本明細書に記載する任意の方法または組成物を、本明細書に記載する任意の他の方法または組成物に関して実行し得ることが意図される。
【0010】
特許請求の範囲および/または明細書における「含む」という用語と併せて用いらる場合の「1つの(a)」または「1つの(an)」という語の使用は「1つの(one)」を意味し得るが、「1つまたは複数の」、「少なくとも1つの」、および「1つまたは2つ以上の」の意味とも一致する。
【0011】
特許請求の範囲における「または」という用語の使用は、本開示では選択肢のみと「および/または」とを指す定義が支持されるものの、選択肢のみを指すことを明白に示すかまたは選択肢が相互に排他的である場合を除き、「および/または」を意味するために用いられる。
【0012】
本発明の他の目的、特徴、および利点は、以下の詳細な説明から明らかになると考えられる。しかしながら、この詳細な説明により本発明の精神および範囲内の種々の変更および修正が当業者に明らかになると考えられる上は、詳細な説明および特定の実施例は本発明の特定の態様を示すものの、説明のためのみに提供されることが理解されるべきである。
【0013】
例示的態様の説明
モノクローナル抗体の産生は典型的に、骨髄細胞株パートナーとの体細胞融合による脾細胞の不死化(ハイブリドーマ形成)を必要とする。ハイブリドーマは不死となり得るが、ハイブリドーマはフィーダー細胞層に依存し、遺伝的安定性を欠く可能性がある。ハイブリドーマ技術の開始以来、モノクローナル抗体産生細胞株の効率および安定性を改善する試みは実質的な進展をもたらしていない。さらに、ヒト抗体の産生に適したヒト多発性骨髄腫由来細胞株は、開発が非常に困難である。本発明者らは、抗体の産生を大いに単純化し、ハイブリドーマの必要性を排除する戦略について説明する。
【0014】
特定の態様において、抗体産生細胞、例えば脾細胞、または抗原提示細胞、例えば樹状細胞は、その発現が適切なプロモーターの制御下にあり、ポリヌクレオチドを含む細胞を許容温度において条件的に不死化させる変異体温度感受性発癌遺伝子をコードするポリヌクレオチドを有するトランスジェニックマウスに由来する。好ましい態様において、温度感受性発癌遺伝子は、マウス主要組織適合性プロモーターの制御下にあるシミアンウイルス40大型腫瘍抗原(tsSV40Tag)である。この脾細胞は許容温度(例えば33℃)において不死化され、ハイブリドーマを形成する必要なく抗体を産生する。このアプローチは、ポリクローナル抗体およびモノクローナル抗体の両方を作製し産生するために用いることができる。この無ハイブリドーマの細胞(hybridoma free cell)の増殖特性および安定性により、ハイスループット発見および抗体に基づく免疫療法のためのさらなる組成物および方法が提供される。
【0015】
本発明のさらなる態様は、無ハイブリドーマの抗体(hybridoma-free antibody)、すなわちハイブリドーマを形成することなく産生される抗体を作製および使用するための過程、組成物、および方法を含む。本発明の1つの態様は、無ハイブリドーマのマウスモノクローナル抗体またはポリクローナル抗体を作製するための組成物および方法を含む。
【0016】
さらなる態様において、本方法は、温度感受性発癌遺伝子、例えばシミアンウイルス40大型T抗原(tsSV40Tag)をインビトロ、エクスビボ、またはインビボで発現する適切な細胞種を、抗体を産生させるための抗原と接触させる段階を含み得る。特定の局面において、抗体産生細胞、好ましくは脾細胞は、tsSV40Tagを発現するマウスから単離され不死化される。そのようなマウス、ImmortoMouse(登録商標)、H-2Kb-tsA58トランスジェニックマウスでは、tsSV40Tagをコードする核酸の発現は主要組織適合性プロモーターの制御下にある(Jat et al., 1991、完全に参照として本明細書により組み入れられる)。ImmortoMouse(登録商標)に由来する細胞は、33℃で培養した場合に依然として不死のままである(Jat et al., 1991)。tsSV40Tagを発現するトランスジェニックマウスおよびtsSV40Tagを発現するトランスジェニックマウスに由来する細胞を含む種々の方法および組成物が特許文献に記載されており、例えばそれぞれ完全に本明細書に組み入れられる米国特許第6,399,384号;米国特許第5,866,759号;米国特許第5,688,692号;および米国特許第5,270,191号を参照されたい。他の局面において、抗体産生細胞または抗原提示細胞は、これらに限定されないが、骨髄、胸腺、脳、または生殖組織を含む、tsSV40Tag発現動物の種々の組織から単離および培養され得る。さらなる他の局面においては、そのような組織試料から幹細胞を単離し培養することができる。
【0017】
さらなる態様においては、(例えば、ImmortoMouse(登録商標)から)収集された細胞を1つまたは複数の抗原と接触させ、その後、抗原結合の特異性の評価を含む抗原に対する抗体産生の評価を行い得る。細胞の種々の亜集団を作製またはクローニングし、後に使用するために保存することができる。
【0018】
本発明のさらなる態様では、条件的に発現されるかまたは機能する形質転換発癌遺伝子を有するマウスを、ヒト抗体を産生するための遺伝子相補体を含むトランスジェニックマウスと交配させる段階を含む。1つの態様においては、条件的に発現されるかまたは機能する発癌遺伝子およびヒト抗体を産生するための遺伝子相補体の両方を有するマウスによる細胞を収集し(例えば、脾細胞、胸腺細胞、B細胞)、ヒト抗体を産生する不死化細胞集団を作製することができる。ヒトまたは異種間抗体の作製においてマウスを用いる組成物および方法について記載している種々の特許が存在し、例えばこの技術の特許については、完全に参照により本明細書に組み入れられる米国特許第6,673,986号;米国特許第6,657,103号;米国特許第6,162,963号;米国特許第6,235,883号;米国特許第6,150,584号;米国特許第6,114,598号;米国特許第6,075,181号;および米国特許第5,939,598号を参照されたい。
【0019】
I. 抗体産生
典型的に、本発明の抗体は、条件的に不死化が可能である細胞を有するマウスもしくは他の動物を免疫することによるか、または不死化細胞もしくは潜在的に不死化可能な細胞を関心対象の抗原と接触させることによって作製する。不死化細胞または潜在的不死化細胞は、条件的に機能する、例えば特定の温度以下でのみ機能するまたは特定の培養条件下でのみ発現される形質転換遺伝子を発現し得る。例えば、シミアンウイルス40初期領域変異体tsA58によってコードされる熱不安定性ラージT抗原(tsSV40Tag)を用いて、トランスジェニックマウスまたは不死化可能な細胞株を樹立することができる。これらの細胞株は、許容温度(例えば33℃)においてまたは特定の環境において(例えばテトラサイクリンの存在下において)持続的に増殖し得るが、非許容温度(37〜39.5℃)に上げるかまたは制御因子を除去するかもしくは添加した際に細胞増殖の停止を示す。増殖停止は細胞周期のG1期またはG2期において起こる。増殖が停止した後でも、一般的なタンパク質合成およびトリパンブルー排除能によってアッセイされるように、細胞は代謝的に活性を有したままである。これらの細胞株は、非許容温度または条件において分裂することはできない。
【0020】
トランスジェニック動物を免疫した後、モノクローナル抗体作製手順において使用するために、抗体産生能または抗原提示能を有する体細胞、それぞれ特にBリンパ球(B細胞)または樹状細胞(DC)を選択する。これらの細胞は、これらに限定されないが、マウス、ラット、ウサギ、イヌ、ネコ、ヤギ、ウシ、ウマ、ヒツジ、またはヒトを含む1匹(人)または複数匹(人)の対象から生検採取した脾臓、扁桃腺、リンパ節、または末梢血生検試料から取得し得る。脾臓細胞および末梢血細胞が好ましく、脾臓細胞は分裂形質芽球期にある抗体産生細胞のより豊富な供給源であるためであり、末梢血細胞は入手しやすいためである。一連の動物を免疫し、最も高い抗体価を有する動物の脾臓を摘出し、脾臓をシリンジでホモジナイズすることによって脾臓のリンパ球を得る場合が多い。典型的に、免疫したマウスの脾臓は、約5 x 107〜2 x 108個のリンパ球を含む。
【0021】
関心対象の抗原に結合する抗体の産生を誘導するために、形質転換タンパク質を発現する動物を免疫するか(例えば、H-2Kb-tsA58 ImmortoMouse(登録商標))、または形質転換タンパク質を発現する細胞を抗原(例えば、繊維状fd-tetファージ(Zacher et al., 1980))に曝露する。免疫または接触は様々な期間にわたって1回または複数回繰り返してよく、例えば、免疫または抗原曝露は1、2、3、4、5、6、7、8、9、10、11、12日、もしくはそれ以上の日数、または1、2、3、4、5、6、7、8、9、10、11、12週、もしくはそれ以上の週に1回であってよい。特定の局面において、免疫または抗原曝露は、1日おきもしくは1週間おき、または3日、4日、5日、もしくはそれ以上の日数に1回、または3週、4週、5週、もしくはそれ以上の週に1回であってよい。免疫または抗原曝露は、2、3、4、5、6、7、8、9、10、11、12、13、14、15週、もしくはそれ以上の週、さらには2、3、4、5、6、7、8、9、10、11、12、13、14、15ヶ月、もしくはそれ以上の月数にわたって行ってよく、好ましくは12週間行い得る。抗原調製物は、静脈内(i.v.)、腹腔内(i.p.)、皮内、皮下(s.c.)、またはそれらの様々な組み合わせを含む1つまたは複数の経路により投与する。各追加免疫後に動物から採血し、ELISAを用いて血清中の抗抗原抗体価をモニターし得る。
【0022】
免疫した動物の臓器を摘出もしくは生検採取するか(例えば脾臓)、または抗原曝露細胞を収集し、細胞培養液中に入れる。細胞は典型的に、2枚の冷却スライドグラスの間に挟んだ臓器の被膜に穏やかに圧力を加えることによって臓器から放出させる。次いで、抗体産生細胞(例えば脾細胞)を適切な増殖培地、好ましくはハイブリドーマ培地に再懸濁し、低密度で培養する。細胞を新たな容器に連続して移すことにより、組織残屑を重力により除去する。典型的に全部で約2 x 108個の細胞を6、24、および96ウェルプレートに分配し、33℃で培養する。2〜3週間の間に少なくとも2回、3回、4回、またはそれ以上の回数、培地を完全に交換する。クローンは典型的に、3週間後に90%を超えるウェルで観察される。プレートをモニターし、各ウェルに新鮮な培地をおよそ3週間ごとに添加する。限界希釈により、陽性ウェルをサブクローニングし得る(Harlow and Lane, 1988);いくつかのクローンはまた24ウェルおよび96ウェルプレートに拡大し、長期培養後に反応性をモニターする。各ウェルにおいて細胞を慎重に懸濁することにより、および無血清培地を使用することにより、脾細胞の「凝集」を回避することができる。典型的に、カウントし、ウェル当たり約0.1〜0.5個の細胞でプレーティングした後、各96ウェルプレートを顕微鏡下で系統的に観察する。
【0023】
抗体産生細胞の集団が単離された後、それらをスクリーニングし、関心対象の特徴、例えば特定の病原体またはタンパク質に対する選択的結合を有する抗体に関して選択する。細胞のサブセットが選択されたならば、同定された1つまたは複数の細胞をクローニングし増殖させる。典型的に、関心対象の抗原、例えば繊維状ファージおよび/または組み換えタンパク質に対するELISAを用いて、関心対象の抗体を産生する細胞をスクリーニングし選択するが、例示的な方法は以下およびHarlow and Lane(1988)に記載されている。陰性対照には、免疫した動物から単離された細胞または抗原に曝露した細胞と比較するための、BSA、ハイブリドーマ培地のみ、免疫前血清、および二次抗体が含まれ得る。免疫ポリクローナル血清および抗抗原抗体は陽性細胞となり得る。特定の局面においては、抗体は培養上清から直接プレーティングする。陽性ウェルの細胞を限界希釈によりサブクローニングし、モノクローナル株を取得する。これらの手順により現れるサブクローンを、ELISA法により種々の抗原に対して試験する。反応性をELISAリーダーによってモニターする。
【0024】
有望な抗体を産生する細胞が同定されサブクローニングされたならば、ウェスタンブロット解析および他の抗原結合アッセイを行って得られた抗体を確認し、さらに特徴づける。典型的には、抗原をSDS-PAGE電気泳動により分離し、ポリフッ化ビニリデン膜(Bio-Rad)に電気的に転写する。膜を条片に分割し、PBSに溶解した5%ノンファットミルクによりブロッキングし、その後適切な洗浄緩衝液、例えば0.1% Tween 20を含むPBSで洗浄する。条片を免疫前血清(1:1000)、免疫後血清(1:1000)、陽性対照抗体、不死化細胞クローンから分泌されたIgGを含む上清、または細胞培養液と共にインキュベートする。何回か洗浄した後、検出試薬結合二次抗体(ペルオキシダーゼ結合二次Ab)(Bio-Rad)を条片に添加し、室温でインキュベートする。条片を洗浄し、例えば高感度化学発光(ECL)(Amersham Biosciences、ニュージャージー州、ピスカタウェイ)により反応性を検出する。
【0025】
マウスには、典型的に2ヶ月間にわたり2週間の間隔で抗原を腹腔内投与する。3回目および4回目の免疫後に、免疫した各マウスの血清をELISAアッセイにより解析し得る。典型的には、最も高い抗抗原抗体価を有する1匹または複数匹のマウスの脾臓を摘出し、脾細胞を単離する。限界希釈により単一クローンを取得することができる。抗体産生は、最初の抗原を用いて細胞培養上清においてELISAによってモニターする(Harlow and Lane, 1988)。
【0026】
培養により、特定のサブクローンを選択する不死化細胞の集団が提供される。典型的に、不死化細胞の選択は、マイクロタイタープレートにおいて単一クローン希釈により細胞を培養し、その後(約2〜3週後に)個々のクローンの上清を所望の反応性に関して試験することによって行う。アッセイ法は、放射性免疫測定法、酵素免疫測定法、細胞障害アッセイ法、プラークアッセイ法、ドット免疫結合アッセイ法等のように、感度がよく、簡便、かつ迅速であるべきである。抗体を調製するおよび特徴づける一般的な方法は、当技術分野において周知である(例えば、Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory, 1988(参照により本明細書により組み入れられる)を参照されたい)。
【0027】
細胞からの治療タンパク質の作製の成功に関する基準の1つは、産生の安定性を維持する細胞株を取得することである。これが達成されない場合、工程収率、時間および費用の効果的利用、ならびに産物の規制認可に関する問題が生じ得る。ハイブリドーマ細胞株によるタンパク質産生の不安定性に関して報告しているいくつかの研究が存在する。ハイブリドーマにおけるタンパク質産生の不安定性の原因は多様であり、多くの場合、正確な分子機構は未だ不明である。
【0028】
ポリクローナル抗体の産生に関しては、抗体はエクスビボで作製することができ、マウスまたはウサギ等の標的抗体産生動物における複数回の抗原注射および採血の必要性を排除することができる。この技術により、標的抗原または所与の組織、細胞集団、もしくはタンパク質に関連した抗原群への曝露に基づいて初回刺激および拡大した抗体、T細胞、またはナチュラルキラー細胞のポリクローナル集団を作製することが可能になる。ハイブリドーマでは特定のクローンが他よりも優位になるため、そのような過程はハイブリドーマを用いた場合にはより困難である。
【0029】
本明細書で用いる「抗体」という用語は抗原結合領域を有する任意の抗体様分子を指し、これにはポリクローナル抗体およびモノクローナル抗体、ならびにFab'、Fab、F(ab)2'、単一ドメイン抗体(DAB)、Fv、scFv(一本鎖Fv)等のような抗体断片が含まれる。抗体に基づく様々な構築物および断片を調製するおよび用いる技法は、当技術分野において周知である。抗体を調製するおよび特徴づける手段も同様に、当技術分野において周知である(例えば、Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory, 1988(参照により本明細書に組み入れられる)を参照されたい)。
【0030】
本発明の特定の態様において、マウス主要組織適合性プロモーターの制御下に変異体温度感受性(ts)シミアンウイルス40(SV40)大型腫瘍抗原(Tag)を有するトランスジェニックマウス(H-2Kb-tsA58トランスジェニックマウス;ImmortoMouse(登録商標))と称される)に由来する不死化脾細胞は不死であり、ハイブリドーマを作製する必要性が回避される。
【0031】
記載した方法により抗体が同定されたならば、適切な1つまたは複数の遺伝子を増幅し、クローニングし、抗体を産生させるための別の細胞にトランスフェクトすることができる。例えば、それぞれ参照により本明細書に組み入れられる米国特許第5,658,570号、米国特許第6,165,745号、および6,602,503号、ならびにDaniell, 2001;Breitling and Dubel, 1999を参照されたい。
【0032】
II. トランスジェニックマウス
細胞株に関する研究により、多くの重要な生物学的疑問への我々の理解が大いに改善された。細胞株の作製は、関心対象の細胞種に不死化発癌遺伝子を導入することによって促進される(Jat et al., 1991)。多くの異なる細胞種をインビトロで不死化することが知られている1つの遺伝子は、SV40大型腫瘍抗原(SV40Tag)である。細胞株を作製するためにインビトロで遺伝子を挿入する必要性を回避するため、SV40TAg遺伝子を有するトランスジェニックマウスが作製された(Jat et al., 1991)。以前の研究により、トランスジェニックマウスにおけるSV40TAg発現が腫瘍形成および異常な発生に関連していることが示されたため、形質転換に関して熱不安定性であるSV40Tag、tsA58(tsSV40Tag)を用いて、生物体全体で見出される典型的な条件下においてインビボで存在する機能的なSV40TAgのレベルが低減された。
【0033】
広範な組織に発現を導くために、広範囲にわたって活性がありかつインターフェロンによって誘導され得るマウス主要組織適合遺伝子複合体H-2Kbプロモーターが用いられた。tsSV40TAg mRNAは、ハイブリッド構築物を有するすべての動物の組織において発現された。いずれの組織の発生も肉眼的に正常であった。H-2Kb-tsA58マウスの1つの系統はホモ接合性になるまで何世代かにわたって交配を繰り返され、導入遺伝子の機能的コピーを伝達している。これらのマウスは「ImmortoMice」と称される。ImmortoMouse(登録商標)はCharles River Labs、マサチューセッツ州、ウィルミントンから市販されている。多くの異なる種類の条件的不死化細胞株がImmortoMouse(登録商標)から導出されているが、この十分に樹立されたマウスモデルはモノクローナル抗体産生細胞を作製するために活用されていなかった。
【0034】
ImmortoMouse(登録商標)は、分化の細胞機構の研究において使用するため、および組織修復に関連した細胞移植実験のため、インビトロおよびインビボで正常な分化を起こし得る個々の細胞種の拡大集団を作製する能力に関して開発された。H-2Kb-tsA58マウスにより、単離された細胞を適切な条件下で培養することによって、種々の組織から条件的不死化細胞株を直接導出することが可能になる。これらのマウスにおいて、tsSV40Tagはインターフェロン誘導性クラスI抗原プロモーターによって制御される。インターフェロンの存在下または非存在下において33℃で培養することにより、インビボおよびインビトロにおいて正常な分化を起こす能力を依然として保持させつつ、細胞をインビトロで長期間培養することができる。
【0035】
III. ヒト抗体の産生
本発明の1つの態様においては、条件的に機能する形質転換発癌遺伝子を含むトランスジェニックマウス、例えばImmortoMouse(登録商標)を(以下に詳述するように)抗体産生のための種々の遺伝子成分をコードする他の種による遺伝子を有するトランスジェニックマウスと交配し、ヒト抗体等の他の種の抗体を産生する細胞株を作製することができる。
【0036】
完全なヒトモノクローナル抗体の多様なレパートリーを産生する能力は、ヒトの治療において用途を有する。治療用のヒトポリクローナル抗体またはモノクローナル抗体を作製するための最も有望なアプローチの1つは、マウスまたは他の非ヒト抗体の非存在下で豊富なレパートリーのヒト抗体を産生するように操作されたマウス系統の作製である(例えばXenoMouse(登録商標))。最近、遺伝子ターゲティングの結果としてマウス抗体産生を欠くマウスの生殖系列に、ヒト免疫グロブリン座位の部分を導入することによってマウスが作製された。これらのマウスは多様な成体様レパートリーを有する顕著なレベルの完全なヒト抗体を産生し、抗原による免疫化に際して抗原特異的ヒト抗体を作製する。XenoMouse(登録商標)には約80%のヒト重鎖抗体遺伝子およびかなりの量のヒト軽鎖遺伝子が備わっている。これらの遺伝子の複雑な構築およびそれらのセミランダムな対形成により、マウスが抗原構造の多様なレパートリーを認識することが可能になる。さらに、このマウスは非常に高い親和性の完全なヒト抗体をプロセシングし得る。入手可能なXenoMouse(登録商標)動物には複数系統が存在する。各系統は、種々の用途用に異なるクラスの抗体を産生し得る。そのような系統のマウスは、ヒト抗原を含む広範囲の抗原に対して高い親和性および特異性を有するヒト抗体を産生するための最適な供給源を提供し得る。
【0037】
XenoMouse(登録商標)は、マウス抗体遺伝子発現が抑制され、ヒト抗体遺伝子発現により機能的に置換されているが、マウス免疫系の残りの部分は原型を保ったままである遺伝子操作された系統のマウスを用いて、完全なヒトタンパク質配列を有する抗体を作製する。マウスゲノムにヒト抗体遺伝子を導入することにより、そのような形質を有するトランスジェニックマウスを永久に繁殖することができる。重要なことには、これらのトランスジェニックマウスは、マウスにおいて発現される(したがって、「自己」として認識される)ヒト産物のみが抗体自体であるために、ヒト抗原に対する抗体を産生し得る。他の機構はすべてマウスの機構であり、したがって任意の他のヒト組織またはタンパク質はマウスにより異物として認識され、免疫反応が開始されることになる。
【0038】
いくつかのヒトタンパク質、例えばサイトカイン、ホルモン、および増殖因子、またはそれらの受容体の異常な合成は、様々なヒト疾患の原因となる。ヒト抗体を用いた中和または完全な排除によりこれらのタンパク質の制御を行い、疾患を治療するまたは完全に排除することができる。これらのトランスジェニックマウスが、ヒト抗原に対するヒト抗体の産生において使用し得る細胞を生じる能力により、種々の病態の処置、診断、または治療における利点が提供されると考えられる。1つの課題は、安定した細胞株において所与の抗原に対するヒト抗体を十分に産生することである。この問題は本発明の態様によって解決し得る。1つの態様において、交配種のマウス集団(例えばImmortoMouse(登録商標)/XenoMouse(登録商標)交配)は、ハイブリドーマを作製する必要なしに、任意のヒト抗原に対する抗体を産生し得る不死化脾細胞を産生し得る。
【0039】
Xenomouseまたは同様の遺伝子修飾を有する動物は、キメラおよび他のヒト化技術とは異なる、100%ヒトタンパク質配列を有する抗体を産生する。これらのマウスを使用する他の利点は、XenoMouse(登録商標)技術を用いて産生される抗体がより優れた安全性プロファイルを提供し、またヒト体内からよりゆっくりと除去され、投与の頻度を減らすことが期待され得る点である。
【0040】
XenoMouse(登録商標)技術では、天然のインビボ親和性成熟過程を利用して、抗体産物候補を通常2〜4ヶ月のうちに作製する。これらの抗体産物候補は、ファージディスプレイで見られる親和性よりも100〜1000倍高い親和性を有し得る。ヒト化およびファージディスプレイ技術を用いて作製される抗体とは対照的に、時には困難であり時間を要することが判明している過程である、次の操作の必要性がない。したがって、抗体構造は最初の選択された抗体から最終的な市販抗体まで原型のまま残存し得る。
【0041】
従来、所望の特徴を有する抗体が同定された時点で、ハイブリドーマまたは組み換え細胞株から直接、前臨床物質を産生させることができる。ハイブリドーマ不要の産生(hybridoma-free production)により、時間節約の可能性に加えて、ハイブリドーマまたは組み換え細胞株において抗体を産生させる必要性が回避される。したがって、本発明の態様により、ハイブリドーマを必要とせずに長期培養においてヒト抗体を産生させる必要が満たされ得る。
【0042】
典型的に、マウスによって作製されるモノクローナル抗体は、それらがヒトタンパク質でないために、免疫系がそれらを異物として認識する患者によって拒絶される。患者は、ヒト抗マウス抗体またはHAMAを産生する場合が多い。この反応により、結合活性が中和されて抗体の有効性が減少する。マウス抗体の次の投与もまた毒性となり得る。本明細書に記載する方法を使用すると、ヒトまたはそれ以外の医学的に関連のあるほとんどすべての抗原に対する抗体を作製することができる。選択される複数の抗体を産生する能力は、最適な抗体産物を選択する上で重要である可能性がある。本発明の1つの態様において、ヒトモノクローナル抗体産生脾細胞の不死化集団は、開示の方法によって作製され得る。
【0043】
さらに、Medarex(ニュージャージー州、プリンストン)はUltiMAb Human Antibody Development SystemSMと称されるシステムを開発した。このシステムにより、様々な種類の完全なヒト(100%ヒトタンパク質配列)抗体が作製された。これらのマウスは、ヒト抗体をコードする遺伝子を含む。これらのモノクローナル抗体は、好ましい安全性プロファイルを有し、またヒト体内からよりゆっくりと除去され、疾患標的に影響を及ぼすために必要な投与の頻度および量が低減される可能性が高い。これらのマウスはまた、本明細書に記載するハイブリドーマ不要の抗体産生法と組み合わせて用いることもできる。
【0044】
要するに、本明細書に記載する方法により、治療または診断目的で、種々の病態(例えば炎症)において発現される腫瘍抗原またはタンパク質に対する抗体を作製することができる。1つの態様では、ヒトモノクローナル抗体を用いて、標的細胞集団、例えば腫瘍細胞に対する抗体の親和性および特異性により、標的細胞集団に対して送達薬剤(例えば細胞毒素)を標的するまたは強化することができる。一方、ヒトモノクローナル抗体を用いて、多くの病態および種々の種類の腫瘍において過剰発現される受容体(例えば、上皮増殖因子受容体(EGFR)等の増殖因子受容体)の機能を抑制することができる。EGFRシグナル伝達が阻止され、細胞死または増殖阻害がもたらされ得る。他の用途には、標的細胞集団、例えば腫瘍細胞に結合する蛍光抗体を用いた腫瘍の画像化が含まれる。
【0045】
特定の態様においては、本明細書に記載の方法により製造された抗体を用いて、以下に記載するファージディプレイ技術を用いて同定された血管郵便番号(vascular zip code)を同定するまたは標的することができる。血管郵便番号とは、薬剤がより効果的かつ効率的に標的され得る、ヒトの身体における特異的かつユニークなアドレスであり、例えば、それぞれ参照により本明細書に組み入れられる米国特許第5,622,699号、米国特許第6,174,687号、および米国特許第6,232,287号を参照されたい。血管標的化は、例えば身体の健常部分をそのままにしつつ腫瘍部位に照準を合わせることにより、治療の有効性を改善し得る。ファージと称される微視的粒子に提示された10億個を超える一群のペプチド配列を投与することにより、ペプチドは身体の特定領域に優先的にホーミングする。この大規模なスクリーニングにより、循環ペプチドの組織分布が無作為ではないこと、および特定のペプチドが異なる臓器に配向し結合することが示される。
【0046】
これらのペプチドは典型的に、臓器の組織および血管に存在する受容体に結合する。ペプチドは、体内を移動しつつ、リガンド(ペプチド結合タンパク質)の挙動を促進し、対象の細胞、組織、血管、または臓器における細胞受容体と相互作用し得る。ペプチドライブラリーをインビボでスクリーニングすることによってリガンドを同定することができ、次にこのリガンドを用いて受容体を同定することができる。次いで、本明細書に記載する方法により、受容体に対してヒトモノクローナル抗体を作製することができる。別の用途は、患者において循環する抗体レパートリーを同定することにより標的を同定し(Mintz et al., 2003)、次いで特定の症例における標的療法または受動免疫を開発するために、これらの標的に対するヒト抗体を作製することであってよい。
【0047】
従来、新規薬学的産物は、ヒト臨床試験が開始されるまでに前臨床開発におよそ6年を要する。完全なヒトモノクローナル抗体技術を用いれば、この期間を2年未満に短縮できる可能性がある。さらに、開発費用も、製薬産業により従来通りに開発される化合物に付随する費用のごく一部ですむ可能性が高い。
【0048】
IV. 抗体産生のための抗原
本発明の態様は、抗体産生細胞または抗原提示細胞の安定株を作製するために、条件的に機能する形質転換遺伝子を発現するトランスジェニックマウスを種々の抗原で免疫するか、または条件的に機能する形質転換遺伝子を発現する細胞を種々の抗原に曝露する段階を含む。本発明の範囲内の抗原には、これらに限定されないが、ペプチド、タンパク質、糖タンパク質、リポタンパク質、ウイルス、細菌、病原微生物、および罹患ヒト細胞を含む、対象において液性免疫応答および細胞免疫応答を誘発し得る任意の分子または巨大分子群が含まれ得る。特定の態様において、抗体作製のためのエクスビボ法(すなわち、処置する対象の外部における抗体作製)の使用により、ヒト等の哺乳動物内で作製され得ない抗原に対する抗体を作製することが可能になることに留意することが重要である。本発明の方法による抗体の作製により、対象内での寛容の効果が回避され得る。したがって、広範な種類の抗体を作製することできる。これらの抗体により、以前は標的することが困難であった抗原を標的することが可能になる。
【0049】
特定の態様において、同定された標的化ペプチド(例えば、特定の臓器を標的するペプチド)もしくはそれらの受容体またはさらには腫瘍細胞等の細胞全体に対する抗体(例えば、モノクローナルおよび/またはポリクローナル)を作製することが望ましい場合がある。適切な標的化ペプチドもしくは受容体またはそれらの部分を、リンカー、ポリリンカー、または誘導体化アミノ酸を介して、アジュバントを含む1つまたは複数の物質にカップリング、結合(bonded)、結合(bound)、共役、または化学的に連結させることができる。特定の局面において、アジュバントには、コロイド金の使用が含まれる(Dykman et al., 1996、完全に参照により本明細書に組み入れられる)。これは、二重特異性もしくは多価の組成物またはワクチンが産生されるように行ってもよい。これらの組成物の調製において用いられる方法は当業者に周知であり、ヒト対象に投与するために適していなければならない、すなわち薬学的に許容されなければならないことがさらに考えられる。好ましい物質には、キーホールリンペットヘモシアニン(KLH)またはウシ血清アルブミン(BSA)等の担体が含まれる。種々の態様において、対象は、これらに限定されないが、マウス、ウサギ、ニワトリ、ヤギ、ヒツジ、ウシ、イヌ、およびヒトを含む任意の高等脊椎動物であってよい。
【0050】
特定の態様においては、抗イディオタイプ抗体または標的化ペプチドの受容体に対する抗体を作製することができる。「標的化ペプチド」とは、対象の臓器または組織への選択的局在化を特徴とする、連続的なアミノ酸の配列を含むペプチドである。選択的局在化は、例えば、推定標的化ペプチド配列をファージの外表面上に提示されるタンパク質に組み込む、以下に開示する方法によって決定することができる。異なるアミノ酸配列のそのような標的化ペプチドを多数発現するように遺伝子操作したそのようなファージのライブラリーを対象に投与した後、対象から1つまたは複数の臓器または組織を採取し、その臓器または組織に見出されるファージを同定する。標的化ペプチド配列を発現するファージは、対照の組織または臓器と比較してある組織または臓器においてより多くの結合を示す場合に、その組織または臓器に選択的に局在すると見なされる。
【0051】
一般に、標的化ペプチドの選択的局在化は、対照の臓器または組織と比較して標的臓器または組織においてファージの少なくとも2倍の濃縮をもたらすべきである。対照の臓器または組織と比較して、標的臓器において少なくとも3倍、4倍、5倍、6倍、7倍、8倍、9倍、10倍、またはそれ以上の濃縮をもたらす選択的局在化が好ましい。または、選択的局在化を示す標的化ペプチド配列を発現するファージは、標的臓器から回収されたファージを次のラウンドのスクリーニングのために第二の宿主に再度注入した場合に、対照臓器と比較して標的臓器において濃縮の増加を示すべきである。選択的局在化を決定するためのもう一つの別の手段は、推定標的ペプチドを発現するファージが、非特異的ペプチドを発現するかまたは推定標的ペプチドを発現するように遺伝子操作されていない対照ファージと比較して、標的臓器において少なくとも2倍、より好ましくは3倍の濃縮を示すことである。選択的局在化を決定するための別の手段は、標的ペプチドを発現するファージの標的臓器への局在化が、標的ペプチド配列を含む合成ペプチドの共投与によって少なくとも部分的に阻止されることである。「標的化ペプチド」および「ホーミングペプチド」は、本明細書において同義に用いられる。
【0052】
抗体を産生させるための他の抗原には、生検試料、患者由来の細胞、患者由来の新鮮腫瘍組織、組織抽出物、新鮮なまたは培養した組織の試料が含まれ得る。関連のあるいくつかの抗原は細胞外成分中および/または間質内に存在する可能性があるため、単に細胞のみでない組織成分を含めることが重要である。抗体を作製するための他の抗原には、これらに限定されないが、アポトーシス細胞、細胞および組織に由来する膜成分、細胞質、核画分、精製タンパク質、部分精製タンパク質、レーザー捕獲した組織、またはパラフィン包埋するかもしくは固定した組織が含まれ得る。
【0053】
A. ファージディスプレイ
抗原または抗原候補は、ファージディスプレイを用いて同定することができる。本方法は、ファージディスプレイライブラリーのインビボ投与を含み得る。本発明の種々の態様では、リガンドを同定し、次いでこれらを用いてこれらリガンドに対する受容体をさらに同定することができ、次にこの受容体を用いてモノクローナル抗体産生不死化脾細胞を作製することができる。ファージディスプレイの様々な方法およびペプチドの多様な集団を作製する方法は、当技術分野において周知である。例えば、それぞれが参照により本明細書に組み入れられ、ファージライブラリーを調製する方法について記載している、米国特許第5,223,409号、第5,622,699号、および第6,068,829号を参照されたい。
【0054】
ファージディスプレイ技法は、小さいペプチドがその表面上に発現され得るようにバクテリオファージを遺伝子操作する段階を含む(Smith et al., 1985, 1993)。この技法の潜在的適用範囲はかなり広く、この10年の間に、ファージによって提示されるペプチドライブラリーの構築、およびライブラリーを用いてペプチドリガンドを単離するスクリーニング法の開発にはかなりの進展が見られた。例えば、ペプチドライブラリーを用いることにより、炎症反応に関与する抗体または細胞接着を媒介するインテグリンのような多くのタンパク質における相互作用部位および受容体-リガンド結合モチーフの特徴づけが可能となった。この方法はまた、ペプチド模倣剤または造影剤を開発するためのリードとなる新規ペプチドリガンドを同定するためにも用いられている(Arap et al., 1998a)。
【0055】
所与の臓器または組織を標的するための最も効率的なアミノ酸配列は、「バイオパニング」により単離することができる(Pasqualini and Ruoslahti, 1996;Pasqualini, 1999)。簡潔に説明すると、推定標的化ペプチドを含むファージのライブラリーを、細胞集団(例えば脾細胞)、動物またはヒト対象に投与するかまたはそれらと接触させ、ファージを含む臓器または組織の細胞抽出物または試料を回収し得る。繊維状ファージを利用する1つの態様においては、バイオパニングのラウンド間に、線毛陽性細菌においてファージをインビトロで増殖させ得る。この細菌はファージによって溶解せず、むしろ特定の挿入物を提示するファージを多コピー分泌する。標的分子に結合するファージを標的の臓器または組織から溶出させ、次いで宿主細菌中で培養することにより増幅することができる。必要に応じて、増幅されたファージをヒト宿主に投与し、臓器または組織の試料を再び回収してもよい。選択的結合剤の集団が得られるまで、複数ラウンドのバイオパニングを実施することができる。ペプチドのアミノ酸配列は、ファージゲノム中の標的化ペプチド挿入物に対応するDNAを配列決定することによって決定される。次に、標準的なタンパク質化学技法により、同定された標的化ペプチドを合成ペプチドとして産生することができる(Arap et al., 1998a, Smith et al., 1985)。このアプローチにより、その標的の性質に関していかなる概念も予め想定することなく、先入観にとらわれない機能的アッセイにおいて循環する標的化ペプチドを検出することができる。
【0056】
標的化ペプチドの受容体として候補標的が同定されたならば、標準的な生化学的方法を用いてこれを、単離、精製、およびクローニングすることができる(Pasqualini, 1999;Rajotte and Ruoslahti, 1999)。次いで、これらの精製タンパク質を、ImmortoMouse(登録商標)もしくはヒト化細胞集団を産生するImmortoMouse(登録商標)交雑種による脾細胞等の細胞集団、またはモノクローナル抗体産生脾細胞等の他の条件的不死化細胞を免疫または曝露するための抗原として使用することができる。次に、これらの抗体産生細胞を用いて、標的受容体または抗原に対して特異的な抗体集団を作製することができる。
【0057】
マウスにおいて行われたこれまでのインビボ選択研究では、fUSE5ベクター内の遺伝子III莢膜タンパク質との融合タンパク質として発現されるランダムペプチドのライブラリーが優先的に用いられた(Pasqualini and Ruoslahti, 1996)。所与のライブラリー中に存在する個々のクローンの数および多様性は、インビボ選択の成否にとって重要な要因である。欠損ファージクローンの過剰発現を有する可能性が低い一次ライブラリーを用いることが好ましい(Koivunen et al., 1999)。ライブラリーの調製は、108〜109形質導入単位(T.U.)/mlに最適化するべきである。特定の態様においては、各選択ラウンド間に大量増幅戦略を適用する。
【0058】
本発明の範囲内において、直鎖、環状、または二環式ペプチドを提示するファージライブラリーを用いることができる。しかし、単環式ペプチドは直鎖ペプチドよりも標的臓器に対して高い親和性を有する傾向があることから、環状挿入物において3〜10個のランダム残基(CX3〜10C)を提示するファージライブラリーが好ましい。二環式ペプチド(CX3C X3C X3C等;Rojotte et al., 1998)を提示するライブラリーも使用に成功している。しかし、同族合成ペプチドの産生は、可能ではあるが、異なるジスルフィド架橋配列を有する多数の配座異性体のために複雑となり得る。
【0059】
V. タンパク質およびペプチド
特定の態様において、抗原組成物は、抗体産生に使用し得る少なくとも1つのタンパク質、ペプチド、またはペプチド様化合物を含み得る。本明細書において使用するタンパク質またはペプチドは一般に、遺伝子から翻訳される全長配列までの約200アミノ酸を超えるタンパク質;約100アミノ酸を超えるポリペプチド;および/または約3〜約100アミノ酸のペプチドを指すが、これらに限定されるわけではない。便宜上、「タンパク質」、「ポリペプチド」、および「ペプチド」という用語は、本明細書において互換的に用いられる。特定の態様において、タンパク質は本明細書に記載の方法によって作製される抗体である。
【0060】
特定の態様において、少なくとも1つのタンパク質またはペプチドの大きさは、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100、約110、約120、約130、約140、約150、約160、約170、約180、約190、約200、約210、約220、約230、約240、約250、約275、約300、約325、約350、約375、約400、約425、約450、約475、約500、約525、約550、約575、約600、約625、約650、約675、約700、約725、約750、約775、約800、約825、約850、約875、約900、約925、約950、約975、約1000、約1100、約1200、約1300、約1400、約1500、約1750、約2000、約2250、約2500アミノ酸、またはそれ以上のアミノ酸残基を含み得るが、これらに限定されるわけではない。
【0061】
本明細書において用いる「アミノ酸残基」とは、当技術分野において周知である任意の天然アミノ酸、任意のアミノ酸誘導体、または任意のアミノ酸模倣体を指す。特定の態様において、タンパク質またはペプチドの残基は連続的であって、アミノ酸残基の配列を中断する任意の非アミノ酸は存在しない。他の態様においては、配列は1つまたは複数の非アミノ酸部分を含んでもよい。特定の態様において、タンパク質またはペプチドの残基の配列は、1つまたは複数の非アミノ酸部分によって中断されてもよい。
【0062】
したがって、「タンパク質またはペプチド」という用語は、天然タンパク質において認められる20個の一般的なアミノ酸の少なくとも1つ、または少なくとも1つの修飾アミノ酸もしくは稀なアミノ酸を含むアミノ酸配列を包含する。
【0063】
タンパク質またはペプチドは、標準的な分子生物学的技法によるタンパク質、ポリペプチド、もしくはペプチドの発現、天然源からのタンパク質もしくはペプチドの単離、またはタンパク質もしくはペプチドの化学合成を含む、当業者に周知の任意の技法によって作製し得る。様々な遺伝子に対応するヌクレオチド、ならびにタンパク質、ポリペプチド、およびペプチド配列がこれまでに開示されており、当業者に周知のコンピューターデータベースにおいて見出すことができる。1つのそのようなデータベースは、国立バイオテクノロジー情報センターのGenbankおよびGenPeptデータベースである(www.ncbi.nlm.nih.gov/)。既知遺伝子のコード領域は、本明細書に開示する技術を用いてまたは当業者に周知のように、増幅および/または発現させることができる。または、タンパク質、ポリペプチド、およびペプチドの様々な市販の調製物が当業者に周知である。
【0064】
A. ペプチド模倣体
本発明によるポリペプチドの調製に関する別の態様は、抗原として使用するためのペプチド模倣体の使用である。模倣体は、タンパク質二次構造の要素を模倣するペプチド含有分子である。例えば、完全に参照により本明細書に組み入れられる、Johnson et al., 1993を参照されたい。ペプチド模倣体の使用の背後にある基礎となる論理的根拠は、タンパク質のペプチド骨格が、抗体と抗原の相互作用のような分子相互作用を促進するようにアミノ酸側鎖を配向させるよう主として存在するというものである。ペプチド模倣体は、天然分子と類似した分子相互作用を可能にすると予想される。これらの原理を用いて、本明細書に開示する標的化ペプチドの天然特性の多くを有するが、改変およびさらには改善された特徴を有する抗原を操作してもよい。
【0065】
B. 融合タンパク質
本発明の他の態様は、抗原としての融合タンパク質の使用に関する。これらの分子は一般に、N末端またはC末端において第二のポリペプチドまたはタンパク質のすべてまたは一部に連結された、関心対象のペプチドのすべてまたは実質的部分を有する。例えば、融合物は、異種宿主においてタンパク質の組み換え発現を可能にするための他の種によるリーダー配列を用い得る。別の有用な融合には、融合タンパク質の精製を容易にするための、抗体エピトープ等の免疫学的に活性のあるドメインの付加が含まれる。融合接合部またはその近傍に切断部位を含めることにより、精製後の外来ポリペプチドの除去が促進されることになる。他の有用な融合には、酵素の活性部位、グリコシル化ドメイン、細胞標的化シグナル、または膜貫通領域等の機能的ドメインの連結が含まれる。好ましい態様において、本態様の融合タンパク質は、免疫応答を誘発するための抗原タンパク質またはペプチドに連結されたペプチドを含む。
【0066】
他の態様において、融合タンパク質は、治療ペプチドと融合され得る、本発明の方法によって作製される抗体を含む。融合タンパク質に組み入れ得るタンパク質またはペプチドの例には、細胞増殖抑制タンパク質、殺細胞タンパク質、アポトーシス促進剤、抗血管新生剤、ホルモン、サイトカイン、増殖因子、ペプチド薬、抗体、Fab断片抗体、抗原、受容体タンパク質、酵素、レクチン、MHCタンパク質、細胞接着タンパク質、および結合タンパク質が含まれる。
【0067】
これらの例は制限することを意図しておらず、本発明の範囲内において、実質的に任意のタンパク質またはペプチドを、本発明において使用するための融合タンパク質に組み入れ得ることを意図している。融合タンパク質を作製する方法は当業者に周知である。そのようなタンパク質は、例えば二官能性架橋試薬を用いる化学的結合によって、完全な融合タンパク質の新規合成によって、または第1ペプチドをコードするDNA配列を第2のペプチドまたはタンパク質をコードするDNA配列に結合させ、その後無傷の融合タンパク質を発現させることによって、作製することができる。
【0068】
C. タンパク質の精製
特定の態様において、タンパク質(例えば抗体)またはペプチドは単離または精製してもよい。タンパク質精製技法は当業者に周知である。これらの技法は、1つのレベルでの、細胞、組織、または臓器のホモジナイゼーションならびにポリペプチド画分および非ポリペプチド画分への粗分画を含む。関心対象のタンパク質またはポリペプチドは、部分的または完全な精製(または均一になるまでの精製)を達成するために、クロマトグラフィー法および電気泳法を用いてさらに精製してもよい。純粋なペプチドの調製に特に適した分析法は、イオン交換クロマトグラフィー、ゲル排除クロマトグラフィー、HPLC(高速(high)液体クロマトグラフィー)、FPLC(AP Biotech)、ポリアクリルアミドゲル電気泳動、アフィニティクロマトグラフィー、免疫アフィニティクロマトグラフィー、および等電点電気泳動である。アフィニティクロマトグラフィーによる受容体タンパク質精製の例は、その全文が参照により本明細書に組み入れられる米国特許第5,206,347号に開示されている。ペプチドを精製するさらに効率のよい方法の1つは、高速(fast)液体クロマトグラフィー(FPLC)またはHPLCである。
【0069】
精製タンパク質またはペプチドは、タンパク質またはペプチドがその天然に得られる状態と比較して任意の程度に精製されている、他の成分から単離可能な組成物を指すことが意図される。したがって、単離もしくは精製タンパク質またはペプチドはまた、それが天然に存在し得る環境を脱したタンパク質またはペプチドを指す。一般に、「精製された」とは、様々な他の成分を除去するために分画に供されたタンパク質またはペプチド組成物を指すことになり、その組成物は発現された生物活性を実質的に保持している。「実質的に精製された」という用語を用いる場合、この名称は、タンパク質またはペプチドが、組成物におけるタンパク質の約50%、60%、約70%、約80%、約90%、約95%、またはそれ以上を構成するような、組成物の主成分を形成する組成物を指すことになる。
【0070】
タンパク質またはペプチドの精製の程度を定量する様々な方法が、本開示に照らして当業者に周知である。これらには、例えば、活性画分の比活性の測定、またはSDS/PAGE分析による画分中のポリペプチド量の評価が含まれる。画分の純度を評価する好ましい方法は、画分の比活性の算出、それと最初の抽出物の比活性との比較、および「精製倍率〜倍」によって評価されるその純度の算出である。活性量を示すために用いられる実際の単位は、当然のことながら、精製後に選択される特定のアッセイ法、および発現されたタンパク質またはペプチドが検出可能な活性を示すか否かに依存することになる。
【0071】
タンパク質精製に用いるために適した様々な技法は、当業者に周知である。これらには、例えば、硫酸アンモニウム、PEG、抗体等を用いた、または熱変性による沈殿、その後の遠心分離;イオン交換、ゲル濾過、逆相、ヒドロキシアパタイト、およびアフィニティクロマトグラフィー等のクロマトグラフィー段階;等電点電気泳動;ゲル電気泳動;ならびにこれらおよび他の技法の組み合わせが含まれる。当技術分野において周知であるように、種々の精製段階を行う順序は変更してもよく、または特定の段階を省略してもよく、それでもなお実質的に精製されたタンパク質またはペプチドを調製するための適した方法が得られると考えられる。
【0072】
タンパク質またはペプチドは常にその最も精製された状態で提供すべきであるという一般要件はない。実際に、実質的にあまり精製されていない産物は特定の態様において有用であると考えられる。部分精製は、より少ない精製段階を組み合わせて用いて、または同じ一般的精製計画の異なる形態を用いて達成してもよい。例えば、HPLC装置を用いて行われる陽イオン交換カラムクロマトグラフィーは一般に、低圧クロマトグラフィーシステムを用いる同じ技法よりも高い精製「倍率」が得られることが認識される。相対的な精製のより低い程度を示す方法は、タンパク質産物の全体的な回収、または発現されたタンパク質の活性の維持において長所を有する可能性がある。
【0073】
アフィニティクロマトグラフィーは、単離すべき物質と、それが特異的に結合し得る分子との特異的親和性に依存するクロマトグラフィー手順である。これは、受容体-リガンド型の相互作用である。カラム材料は、結合パートナーの1つを不溶性の充填剤に共有結合させることによって合成される。次いで、カラム材料は溶液から物質を特異的に吸収し得る。条件を結合が起こらない条件に変更すると(例えば、pH、イオン強度、温度等の変化)、溶出が起こる。充填剤は、それ自体が有意な程度に分子を吸収せず、また広範囲の化学的、物理的、および熱安定性を有する物質であるべきである。リガンドは、その結合特性に影響を及ぼさないように結合すべきである。リガンドは、比較的堅固な結合を提供すべきである。さらに、試料またはリガンドを破壊することなく、物質を溶出することが可能であるべきである。
【0074】
D. 合成ペプチド
いくつかの抗原ペプチドは、その大きさが比較的小さいために、従来の技法に従って溶液中または固相支持体上で合成することができる。様々な自動合成機が市販されており、既知の手順に従ってこれらを用いることができる。例えば、それぞれ参照により本明細書に組み入れられる、Stewart and Young, (1984);Tam et al., (1983);Merrifield, (1986);およびBarany and Merrifield (1979)を参照されたい。通常、約6アミノ酸から約35〜50アミノ酸までの短いペプチド配列は、そのような方法によって容易に合成することができる。または、本発明のペプチドをコードするヌクレオチド配列を発現ベクターに挿入し、適当な宿主細胞に形質転換するかまたはトランスフェクトし、発現に適した条件下で培養する組み換え型DNA技術を用いてもよい。
【0075】
実施例
以下の実施例は、本発明の好ましい態様を実証するために含めるものである。以下の実施例に開示した技術は、本発明の実施に際して良好に機能するように本発明者らにより見出された技術を表し、従ってその実施のための好ましい様式を構成すると見なし得ることは、当業者に認識されるべきである。しかし当業者は、本開示に照らして、開示した特定の態様において多くの変更を行うことができ、それらも本発明の精神および範囲から逸脱することなく同様または類似の結果をなおも得ることができることを認識すべきである。
【0076】
実施例1:不死化脾臓細胞の作製
A. 方法
脾細胞の単離
H-2Kb-tsA58マウス(Charles River Laboratories、マサチューセッツ州、ウィルミントン)の脾臓をダルベッコ変法イーグル培地(DMEM)中に回収した。2枚の冷却スライドグラスの間に挟んだ臓器の被膜に穏やかに圧力を加え、細胞を放出させた。塩化アンモニウムを用いて赤血球を溶解し、10% CPSRおよびハイブリドーマ促進補充物を添加したハイブリドーマ培地15 mlに脾細胞を再懸濁した。ナイロンメッシュで濾過して組織残屑を除去した。細胞を24ウェルプレートに分配し(2 x 106個/ウェル)、33℃で培養した。1週間おきに培地を交換した。
【0077】
他のプレートサイズを用いてもよいが、24ウェル(脾臓55 mlによる脾臓懸濁液のうち1 ml)が好ましいことが判明した。培地は定期的に(例えば、1週間おきに)交換し得る。3週間後、90%を超えるウェルにおいてクローンが認められた。
【0078】
マウスの免疫化
H-2Kb-tsA58マウス(Charles River Laboratories、マサチューセッツ州、ウィルミントン)を、1週間おきに12週間、繊維状fd-tetファージで免疫した。簡潔に説明すると、107形質導入単位(TU)/μl(全量 = 1 ml)を含むファージ調製物を、4通りの経路(静脈内、腹腔内、皮内、および皮下)により投与した。追加免疫する度にマウスから採血し、ELISAにより血清中の抗ファージ抗体価をモニターした。動物実験は、テキサス大学M. D. Anderson癌センターの施設内動物管理および使用委員会によって概説され認可される標準的な定型手順を含んだ。
【0079】
クローン抗体産生脾細胞のスクリーニングおよび作製
以前に記載されている通りに、繊維状ファージおよび組み換えファージキャプシドpIIIタンパク質に対するELISAを行った(Harlow and Lane, 1998)。ウシ血清アルブミン(BSA)、ハイブリドーマ培地のみ、免疫前血清、および二次抗体を陰性対照とした。免疫ポリクローナル血清および抗ファージ抗体を陽性対照とした。抗体は、培養上清から直接プレーティングした。モノクローナル株を取得するため、限界希釈により(96ウェルプレートのウェル当たり0.1ないし0.5個の細胞)陽性ウェルの細胞をサブクローニングした。2ヵ月後に現れたサブクローンを、ELISA法により完全なファージ粒子およびpIIIファージキャプシドタンパク質に対して試験した。ELISAリーダーで反応性をモニターした。
【0080】
ウェスタンブロット解析
繊維状fd-tetファージ(109 TU/レーン)を煮沸し、4〜20%勾配SDS-PAGE(Invitrogen Corp.、カリフォルニア州、カールズバッド)により分離し、ポリフッ化ビニリデン膜(PVDF;Bio-Rad Laboratoried, Inc.、カリフォルニア州、ハーキュリーズ)に電気的に転写した。膜を条片に分割し、リン酸緩衝食塩水(PBS)に溶解した5%ノンファットミルクにより室温(RT)で1時間ブロッキングし、その後0.1% Tween 20を含むPBS(PBS-T)で1回洗浄した。条片を免疫前血清(1:1000)、免疫後血清(1:1000)、抗fd-tetファージ(Sigma-Aldrich、ミズーリ州、セントルイス)、不死化細胞クローンから分泌された抗ファージIgGを含む上清、または細胞培養液のみと共にRTで2時間インキュベートした。3回洗浄した後、ペルオキシダーゼ結合二次抗体(Bio-Rad Laboratoried, Inc.、カリフォルニア州、ハーキュリーズ)を条片に添加し、RTで1時間インキュベートした。条片を3回洗浄し、高感度化学発光(ECL;Amersham Biosciences Corp.、ニュージャージー州、ピスカタウェイ)により反応性を検出した。
【0081】
ELISA
以下は、誘導したモノクローナル抗体の産生の存在または非存在を評価するために使用するアッセイ法の一例である。選択した抗原を、PBSに溶解してHigh Binding Assay Plate(Costar、例えば24、48、または96ウェルプレート)に固定化し得る(109粒子または5μg/ウェル)。対照ウェルは、PBSに溶解した2 mgウシ血清アルブミン(BSA)を用いて、4℃で一晩コーティングする。次いで、一次抗体または対照ポリクローナル種IgG(Sigma)を一連の濃度で室温で1時間インキュベートする。二次抗体(抗種Fabアルカリホスファターゼ結合、Sigma、3% BSA中1:3000)を添加し、1時間インキュベートする。p-ニトロフェニルリン酸(Sigma)によりELISAを発色させ、1〜4時間後に405 nmで(Reader 520、Organon Teknika)測定値を得ることができる。
【0082】
免疫沈降およびウェスタンブロット解析
関心対象の抗原を、プロテアーゼインヒビターの存在下で、50 mM Tris-HCl pH 7.6、1% NP-40、150 mM NaCl、および0.1 mM ZnOAc中に希釈し得る。タンパク質濃度は、ローリー法(Bio-Rad)により決定し得る。プロテインG-セファロース(Pharmacia)の存在下で、約5μg/mlのモノクローナル抗体濃度での問題のクローンの存在下で、タンパク質を免疫沈降し得る。免疫沈降されたタンパク質をSDS-PAGEにより分離し、ニトロセルロース膜に転写し、抗モノクローナル抗体(例えば、マウスまたはヒト)IgG HRP(Jackson Laboratories)でブロッティングし、高感度化学発光(Renaissance、NEN)により可視化し得る。または、関心対象のタンパク質をまずSDS-PAGEゲルにより分離し、次いでタンパク質をニトロセルロース紙に転写し、問題のモノクローナル抗体集団でプロービングし、抗モノクローナル抗体(例えば、マウスまたはヒト)IgG HRP(Jackson Laboratories)を用いて結果を可視化し、高感度化学発光(Renaissance、NEN)により可視化してもよい。
【0083】
不死化脾臓培養物のエクスビボ免疫化
脾臓から回収した細胞をプレーティングしてから約5日後、例えば0.5 x 1010〜1 x 1012 TU/2 x 106 細胞の濃度のファージ(fd-Tet)の抗原用量をウェルに供し得る。この段階を「初回刺激段階」または「最初のエクスビボ免疫化」と見なすことができる。初回刺激から18日および25日後(脾臓I)、初回刺激から14日および21日後(脾臓II)に、さらなる追加免疫を行い得る(例えば、同量のファージを添加し得る)。または、平行実験において、予めファージに曝露した(ファージを負荷した)樹状細胞(DC)と共にインキュベートすることにより、脾細胞を初回刺激し得る。本実施例では、その後の追加免疫は行わなかった。エクスビボ免疫化は、図7の時系列に示される通りに行うことができる。細胞全体でエクスビボ免疫する場合には、脾細胞を、上記のように、アポトーシスB16-F10細胞を負荷したDCまたはアポトーシス細胞のみと共にインキュベートした。
【0084】
B.結果
Fd-Tetに対するインビトロ免疫化の結果を図1Aおよび1Bに示す。ELISAプレートをpIII精製タンパク質(5μg/ウェル)またはFd-Tet(1011 TU/ウェル)で4℃で一晩コーティングした。示したウェルの馴化培地を、「最初の免疫化」から7日後および2回目の免疫化から4日後に回収した。対照として、Fd-Tetを接種した(3回注射)1匹の動物の免疫前および免疫後血清を用いた。プレートは、抗マウス全Ig HRP結合(ZYMED)およびOPDにより発色させた。
【0085】
H-2Kb-tsA58マウスを規定の抗原(繊維状ファージ)で免疫し、血清中の抗ファージ抗体価をELISAによりモニターした。抗ファージIgG価は、最終追加免疫から7日後に高レベル(免疫前血清の<0.1と比較して、1:3,200希釈においてOD450>3)に達した(図8)。段階希釈によりさらに試験したところ、ファージに対するIgG価は平均約1:6,400であることが明らかになった。さらに、pIIIタンパク質(10μg/ウェルでコーティング)に対する血清価は、平均約1:1,600であった(データは示さず)。マウスの脾臓を摘出し、DMEM中で細胞懸濁液を調製した。細胞を96ウェルプレートに分配し、33℃で培養した。2〜3週間の間に、培地を3回完全に交換した。3週間後、>90%のウェルにおいてクローンが認められた。抗体の反応性を検出するため、ファージ粒子をコーティングしたマイクロタイターウェルプレートにおいて、上清を用いてELISAを行った。58%に到達するクローンが、ファージに対するIgG反応性に関して陽性であった。
【0086】
脾細胞は低細胞密度であっても健常であり、大半のウェルの上清において強力な反応性レベルをもたらしたことが認められた。モノクローナル株を取得するため、陽性ウェルの細胞を限界希釈によりクローニングしたが、ほとんどのクローンは陽性のままであった。モノクローナル株のサブクローニングを2回繰り返したところ、得られたクローンの実質的にすべてが陽性であり、作製された株が実際に単一クローンに由来する強力な証拠が提供された。
【0087】
4〜8週間後に出現したクローンを、ELISAにより、ファージ粒子に対しておよび微量なファージキャプシドタンパク質(pIII)に対して試験した。この場合も同様に、96ウェルプレートから24ウェルプレートに拡大した場合にも、または凍結および融解後にも、ほとんどの陽性クローンが反応を維持した(図9)。無傷のファージに対して強い反応性が認められ、いくつかのクローンは組み換えpIII融合タンパク質とも反応した(図10)。すべての段階の元のプレートおよびクローンも、最長で3ヶ月間培養において保存した。
【0088】
ELISAによりスクリーニングされた抗体がウェスタンブロットにおいて特定のタンパク質を認識し得るかどうかを判定するため、H-2Kb-tsA58トランスジェニックマウス由来不死化脾細胞による上清を、SDS-PAGEにより繊維状ファージ調製物を分離することにより、pIIIおよびpVIIIファージキャプシドタンパク質に対して評価した。ファージタンパク質を含むPVDF膜を、免疫前血清、免疫後血清、市販の抗ファージ、または不死化脾細胞クローンから分泌された抗ファージIgGを含む上清と共にインキュベートした。細胞培養液のみをさらなる陰性対象として用いた。pIIIおよびpVIIIファージキャプシドタンパク質に相当するバンドと特異的に反応する抗体が、H-2Kb-tsA58トランスジェニックマウス由来不死化脾細胞の上清中に検出された(図11)。この結果から、本明細書に記載の方法によって産生される抗体が、免疫ブロッティング(図11)または細胞表面抗原の蛍光活性化セルソーティング(FACS)(データは示さず)等の用途においても(データは示さず)使用できることが実証される。
【0089】
H-2Kb-tsA58トランスジェニックマウスの脾細胞は、規定の抗原に対する高力価のIgGを産生し得ると考えられる。この細胞培養系により、モノクローナル抗体の信頼性のあるかつ再現性のある供給源が確実になり、ハイブリドーマ作製の必要性が排除された。
【0090】
本発明のいくつかの利点は、さらに解説する価値がある。第一に、抗体合成細胞は、何ヶ月間も、場合によっては何年間も培養において安定であり、抗体産生の減少も不活性化も起こすことなく、限界希釈クローニング法および凍結融解法を許容する。ポリクローナル集団が凍結されており、所与のIgGを分泌する生存クローンが回収される(データは示さず)。
【0091】
第二に、不死化クローンは33℃でゆっくりと増殖し、遺伝的に安定であり、適時に多数の試料を処理することができる(および、論理的には「稀な」抗体を取得できる可能性がある)。H-2Kb-tsA58由来脾細胞により、長期培養したクローンを含むウェルからの特定ポリクローナルIgGの大量産生が可能になる。対照的に、ハイブリドーマは、クローンのランダムな混合物において、非分泌クローンが一般に分泌クローンを上回ることから問題がある。以前のデータから、H-2Kb-tsA58トランスジェニックマウスに由来するIgG分泌脾細胞と非分泌脾細胞の増殖速度は同様であることが示唆される(未発表知見)。
【0092】
第三に、インビトロ免疫化は致死脾細胞またはハイブリドーマでは効果がないのに対して、インビトロ免疫化は抗体産生を促進する他の脾臓由来不死化細胞種--マクロファージおよび線維芽細胞等--の存在により増強される。腹水産生における最近の制限を考えると、この新たな技術はモノクローナル抗体のエクスビボでの簡便な大量製造を支持する。
【0093】
第四に、H-2Kb-tsA58トランスジェニックマウスをヒト抗体産生のための遺伝子相補体を発現するマウスと交配することにより、ヒトモノクローナル抗体の作製もまた可能になる。本明細書に記載する戦略はハイブリドーマ作製に取って代わり、甚大かつ直接的な科学的および医学的利点によりマウスおよびヒトモノクローナル抗体作製を合理化し得る。
【0094】
fd-Tetを用いたエクスビボ免疫化の結果を図1Aおよび1Bに示す。ELISAプレートを、pIIIファージキャプシドタンパク質(5μg/ウェル)または無傷のファージ粒子(1011 TU/ウェル)で4℃で一晩コーティングした。異なる実験条件下で培養した細胞の馴化培地を、初回刺激してから11日目および22日目(脾臓I)、19日目および26日目(脾臓II)に回収した。ファージ反応性の陽性対照および陰性対照として、免疫前および免疫後抗ファージポリクローナル血清を使用した。Fd-Tetファージを12ヶ月間1週間おきに免疫したマウスに由来する血清を回収した。プレートは、二次抗マウスIg-ペルオキシダーゼ(ZYMED)で発現させ、TMB(Calbiochem)で発色させた。吸光度をELISAリーダーによりモニターした。
【0095】
不死化脾細胞の形態
免疫した動物による不死化脾細胞の一般的形態を、図3A、3B、および3Cに示す。培養してから2ヵ月後に写真を撮影した。濾胞性樹状細胞、形質細胞(抗体産生B細胞)のクローン、マクロファージ、および未同定の上皮様細胞(おそらく細網上皮細胞)を観察することができる。
【0096】
免疫したマウスの脾細胞の形態
免疫したマウスに由来する脾細胞を、培養してから2ヵ月に視覚的に分析した。例えば濾胞性樹状細胞、形質細胞(抗体産生B細胞)のクローン、マクロファージ、および未同定の上皮様細胞(おそらく細網上皮細胞)といったいくつかの異なる細胞が認められた。
【0097】
脾臓および骨髄細胞培養物は抗原提示に対して良好に機能することが実証されているが、データから、リンパ節もまた抗原提示に対して良好に機能することが示される(データは示さず)。
【0098】
実施例2:骨髄(BM)からの不死化樹状細胞(DC)の作製
平行した研究において、予めファージ(ファージを負荷した)または他の抗原に曝露した樹状細胞(DC)と共にインキュベートすることにより、脾細胞を初回刺激した。
【0099】
A. 方法
骨髄細胞の収集および単離
髄腔に27 G針を挿入することにより、H-2Kb-tsA58マウスの大腿骨、頸骨の長骨、および骨端から骨髄(BM)を収集した。塩化アンモニウムを用いて赤血球を溶解した。ペトリ皿に単一細胞懸濁液をプレーティングした。細胞を33℃で培養した。各プレートに、マウス(mu)GM-CSF(10 ng/ml)およびr-muIL-4(10 ng/ml)を補充した10% FBSを含む7 mlのRPMI 1640を添加した。3日後、完全培地およびサイトカイン3 mlをプレートに補充した。5日培養後、約50%の集団が未成熟樹状細胞によって表された。ゆるく接着した増殖DC凝集物を回収し、再度プレーティングした。未成熟DCを繊維状ファージ(fd-tet)(1.5 x 1012 TU/1 x 106未成熟DC)またはアポトーシスB16-F10細胞(2:1、DC/B16)と共にインキュベートした。インキュベートは、TNF-α(DCの成熟を誘導することが周知である因子)の存在下または非存在下において48時間継続した。
【0100】
アポトーシスB16-F10細胞の調製
約1 x 106細胞/mlのB16-F10細胞の懸濁液にUV照射することにより(UV Stratalinker、Stratagene 4ジュール/cm2で20分間)、B16-F10細胞においてアポトーシスを誘導した。24時間後、67%の細胞が、細胞がアポトーシスを起こしたことを示すアネキシン陽性であった。
【0101】
T細胞またはB細胞応答の誘導
T細胞および/またはB細胞が媒介する応答を誘導するため、DC細胞を脾臓由来細胞と混合し得る。次いで、T細胞および/またはB細胞媒介性応答を誘導するため、異なる実験条件下(抗原、例えばファージまたはアポトーシス細胞を負荷するか否か)のDC細胞を、単離した脾臓由来細胞(SDC)と共に5:1、SDC/DCの比率でインキュベートした。
【0102】
B. 結果
樹状細胞
インビトロでの樹状細胞の標準的な挙動は、それらの特性および生物学的因子に対する応答を踏まえると、条件的不死化DC細胞において変化する。同じウェル内の樹状細胞によって構成的に提示される抗原に応答するB細胞に対する選択圧が存在し得る。エクスビボ免疫化もまた、構成的な抗原提示を提供するために本発明の種々の態様に含まれ得る。
【0103】
「不死化」未成熟DCにおける特定の細胞表面抗原の存在は、FACS解析により評価することができる。プレーティングしてから5日後、細胞をいくつかの抗体、抗CD80、抗CD86、および抗H2k(BD)抗体を用いて表面抗原について評価した(Weigel et al., 2002)(図2)。骨髄からサイトカインを用いて分化させたDCの特徴的形態を図2に示す。
【0104】
幹細胞
不死化細胞の別の用途は、罹患患者の幹細胞集団の置換において考えられる。一例において、マウス等の動物の骨髄を照射し、それをImmortoMouse等の不死化動物の骨髄または幹細胞の任意の供給源と置換し得る。結果を解析するため、正常動物を屠殺し、immortomouseに由来する、不死である可能性のある任意の幹細胞を同定することができる。これにより、幹細胞がホーミングする特定臓器の同定が可能になる。また、ImmortoMouseを(すべての細胞においてLac Zを発現する)Rosaマウスと交配することは、増殖ばかりでなく、レシピエントにおける追跡が可能になるようImmortoMouseに由来する細胞をタグ化するのに役立つことになる。
【0105】
ImmortoMouseの脳幹細胞を単離し、特徴づけを行った。図5の右下パネル中の丸い細胞として増殖している最も小さい細胞は、脾臓幹細胞と考えられる。したがって、不死化幹細胞の単離およびこの集団の導入は、将来、癌患者等の障害を起こした患者の生存率を改善するために使用できる可能性がある。
【0106】
実施例3:無傷の細胞に対するモノクローナル抗体の作製
A. 方法
免疫化
H-2Kb-tsA58マウス(Charles River Laboratories、マサチューセッツ州、ウィルミントン)を、1週間おきに3週間、5 x 106個の間葉系幹細胞(MSC)で免疫した。ELISAおよびFACSにより、血清中の抗MSC抗体価を評価した。動物実験は、テキサス大学M. D. Anderson癌センターの施設内動物管理および使用委員会によって概説され認可される標準的な定型手順を含んだ
【0107】
不死化脾細胞の導出
マウスを屠殺し、それらの脾臓をダルベッコ変法イーグル培地(DMEM)中に回収した。2枚の冷却スライドグラスの間に挟んだ臓器の被膜に穏やかに圧力を加え、細胞を放出させた。次いで、10% CPSRおよびハイブリドーマ促進補充物を添加したハイブリドーマ培地15 mlに脾細胞を再懸濁した。ナイロンメッシュで濾過して組織残屑を除去した。細胞を培地55 mlに再懸濁し、異なる密度で6ウェル、24ウェル、および96ウェルプレートに分配した。プレートを33℃でインキュベートした。2〜3週間の間に、培地を3回完全に交換した。大部分のウェルにおいてクローンが認められた。
【0108】
プレーティングの図式は以下の通りであった:脾臓を55 mlに再懸濁した;6ウェルプレート1枚および24ウェルプレート1枚に播種し、残りの細胞を約280 mlに希釈し、20 x 96ウェル、5 x 24ウェル、および3 x 6ウェルに分配した。本発明者らは、研究を開始して3ヶ月経過後に、ELISAにより、96ウェルプレートによる59クローン(83%が+であった)、および24ウェルによる61クローン(82%が+であった)を評価した。55 mlから24ウェルプレートへの播種がこの系において最も有力な培養条件であることが明らかである。数ヵ月後にも細胞は健常であるように見え、すべてのウェルが陽性である。そのようなウェルからのサブクローニングは成功を収め得る。
【0109】
実施例4:カポジ肉腫(KS)細胞および間葉系幹細胞(MSC)に対する抗腫瘍反応性
A. 方法
血清の回収
免疫手順を開始する前、および2週間または3週間後に(免疫後血清1:2回注射後、および免疫後血清2:3回注射後)マウスから採血した。下記の通りに、マルチウェルプレートにプレーティングし、PAF 2%で固定したKS細胞およびMSC細胞に対して、血清の特異的反応性をアッセイした(図4A)。
【0110】
ELISAアッセイ
血清抗腫瘍反応性をELISAにより測定した。3 x 104個の対数増殖期KS(図4Aおよび4B)または1.5 x 104個のMSC細胞/ウェル(図4C)を96ウェルプレートにプレーティングした。37℃で一晩インキュベートした後、細胞をPBSで1回洗浄し、2% PFA中で室温で10分間固定し、PBSで1回リンスした。プレートは使用時まで-20℃で保存した。PBS-2% BSAを用いてRTで1時間ブロッキングした後、PBS-0.5% BSA中の血清希釈物1/500または1/1000(図4A)を二つ組で添加し、4℃で一晩インキュベートした。抗体は培養上清から直接プレーティングした(図4Bおよび4C)。翌日、プレートをPBS-0.5% BSA、0.01% Tween20で3回、およびPBSのみで1回洗浄した。PBS-0.5% BSAに溶解したウサギ抗マウスIg西洋ワサビペルオキシダーゼ(HRP)結合(ZYMED Laboratories Inc.、米国、カリフォルニア州)の1/2000希釈物100μlをプレートに添加した。振盪させながら室温で90分インキュベートした後、プレートを上記のように3回洗浄した。オルト-フェニレンジアミン(OPD)(Sigma-FAST、Sigma-Aldrich、米国、セントルイス Fast-tab)で反応を発色させ、50μl/ウェルの3 M硫酸で反応を停止した。マイクロプレートリーダーで450 nmの吸光度を測定した。100個を超えるクローンを評価した。
【0111】
陽性細胞のサブクローニング
モノクローナル株を取得するため、限界希釈により(96ウェルプレートのウェル当たり0.1ないし0.5個の細胞)陽性ウェルの細胞をサブクローニングした。
【0112】
実施例5:不死化胸腺細胞の作製
14日目にH-2Kb-tsA58細胞から胸腺を摘出し、ダルベッコ変法イーグル培地(DMEM)中に回収した。2枚の冷却スライドグラスの間に挟んだ臓器の被膜に穏やかに圧力を加え、細胞を放出させた。次いで、10% CPSRおよびハイブリドーマ促進補充物を添加したハイブリドーマ培地15 mlに胸腺細胞を再懸濁した。70μmナイロンメッシュで濾過して組織残屑を除去した。細胞を24ウェルプレートに分配し、33℃で培養した。
【0113】
残りの細胞を、抗マウス抗体: CD3、B220、H2k、CD86、CD80、CD11c、および(上皮細網細胞を認識する)MAdCAM-1を用いてFACSにより解析した。FACSの結果から、以下の割合の陽性細胞が示された(図6Aおよび6B)、それぞれCD3+:75%およびH2k+:20%。CD45RA(B220):3.5%(データは示さず)CD86+:4%(データは示さず)CD80+:0%(データは示さず)CD11c+:0%(データは示さず)MAdCAM-1+:0%(データは示さず)。
【0114】
本明細書において開示および主張する方法、組成物、および装置はすべて、本開示に照らして過度の実験を行うことなく作製し使用することができる。本主題が主張する概念、精神、および範囲から逸脱することなく、本明細書に記載した方法、組成物、および装置に変更が適用され得ることは、当業者には明らかであると考えられる。より具体的には、同一または類似の結果が得られれば、化学的および生理的に関連する特定の物質を本明細書に記載の物質と置換し得ることは明らかであると考えられる。当業者に明らかであるそのような類似の置換物および修飾物はすべて、主張する本主題の精神、範囲、および概念の範囲内であると見なされる。
【0115】
参考文献
以下の参考文献は、本明細書に記載したものに例示的な手法またはその他の詳細を提供する程度に、参照により特別に本明細書に組み込まれる。

【図面の簡単な説明】
【0116】
添付の図面は本明細書の一部を形成し、本発明の特定の局面をさらに示すために含める。本明細書に示す特定の態様の詳細な説明と組み合わせてこれらの図面の1つまたは複数を参照することにより、本発明をより良く理解することができる。
【図1】pIII精製タンパク質(5μg/ウェル)(図1A)またはファージ粒子Fd-Tet(1011 TU/ウェル)(図1B)に対するインビトロ抗体産生不死化脾細胞集団を作製する例示的な方法を示す。
【図2】樹状細胞を不死化する例示的な方法、および抗マウス抗体-CD80、-CD86、および-H2kを用いた不死化樹状細胞のFACS解析を示す。
【図3】未成熟不死化骨髄由来細胞(樹状細胞、DC)の形態の例示的な画像を示す。
【図4】抗腫瘍抗体を作製しアッセイする例示的な方法を示す。図4Aおよび図4Bは3 x 104個の対数増殖期KS細胞のELISAを示し、図4Cは1.5 x 104個のMSCのELISAを示す。抗体は培養上清から直接プレーティングした。図4Bおよび4Cでは、陽性対照としてポリクローナル血清を使用した。OPDにより反応を発色させ、450 nmで吸光度を測定した。
【図5】培養してから2ヵ月後の、免疫したマウスによる脾細胞培養物の例示的な形態を示す。濾胞性樹状細胞、形質細胞(抗体産生B細胞)のクローン、マクロファージ、および未同定の上皮様細胞(おそらく細網上皮細胞)が認められる。
【図6】胸腺細胞の不死化集団を作製し、胸腺細胞をスクリーニングする例示的な方法を示す。2つの細胞表面タンパク質集団のFACS解析を、CD3染色を示す図6AおよびH2K染色を示す図6Bにより示す。
【図7】培養において不死化細胞集団に抗原を導入するための例示的時間的経過を示す。
【図8】不死化脾臓細胞から産生された抗体、特に繊維状ファージ(fd-tet)810または組み換えファージキャプシドpIII 820に曝露したH-2Kb-tsA58トランスジェニックマウス由来不死化脾細胞から得られた抗体の例示的なスクリーニングおよび確認の結果を示す。
【図9】不死化脾臓細胞から産生された抗体、特に繊維状ファージ(fd-tet)910に曝露したH-2Kb-tsA58トランスジェニックマウス由来不死化脾細胞から得られた抗体の例示的なスクリーニングおよび確認の結果を示す。クローン1〜3は凍結/融解を行ったクローンに相当する。クローン4〜9は、96ウェルプレートから24ウェルプレートに拡大して6週間培養した異なるウェルに相当する;クローン10は、陰性対照としての培地のみを示す。含めた他の対照は免疫前および免疫後血清であった。
【図10】不死化脾臓細胞から産生された抗体、特に繊維状ファージ(fd-tet)1010、組み換えファージキャプシドpIII 1020、またはウシ血清アルブミン(BSA)1030に曝露したH-2Kb-tsA58トランスジェニックマウス由来不死化脾細胞から得られた抗体の例示的なスクリーニングおよび確認の結果を示す。クローン1、培地、陰性対照;クローン2、免疫前血清;クローン3〜7は、培養してから8週間後の異なるモノクローナル株の上清に相当する。バーは平均値に相当する。平均値の標準誤差は平均値の1%未満であった。
【図11】ファージタンパク質に対する抗体を産生するH-2Kb-tsA58トランスジェニックマウス由来不死化脾細胞による上清の反応性のウェスタンブロット解析を示す。反応性は、図のように、免疫前血清1110、免疫後血清1120、抗ファージ抗体1130、または不死化脾細胞クローンから分泌された抗ファージIgGを含む上清1140と共にインキュベートした後に評価した。pIIIファージキャプシドタンパク質1160およびpVIIIファージキャプシドタンパク質1170(矢印)に対して特異的に反応する抗体が、H-2Kb-tsA58トランスジェニックマウス由来不死化脾細胞の上清中に検出された。
【技術分野】
【0001】
発明の背景
1. 発明の分野
本発明は、分子生物学、細胞生物学、および免疫学の分野に関する。より具体的には、本発明はハイブリドーマを使用しない抗体合成の方法および用途に関する。なお、本出願は2003年4月14日に出願された米国仮特許出願第60/462,631号および2003年11月24日に出願された米国仮特許出願第60/524,701号の優先権を主張し、これらは完全に参照により本明細書に組み入れられる。
【背景技術】
【0002】
2. 背景
モノクローナル抗体は、標的分子上の特定部位との反応において高い特異性および感度を有するタンパク質である。モノクローナル抗体は、年月を経て、ヒトの疾患の解析および治療といった近代の生物学的研究および医学における主要な試薬となった。しかし、モノクローナル抗体は、導入されてから四半世紀を超えても、いまだに多発性骨髄腫由来細胞に融合させた脾細胞の体細胞クローン(ハイブリドーマ)によってのみ産生される(Kohler and Milstein, 1975)。これらの「ハイブリドーマ」は何年もの間モノクローナル抗体を産生し得るが、その作製は、絶えず続く汚染のリスク、再三にわたるフィーダー細胞の必要性、および遺伝的不安定性の可能性によって制限される手間のかかる多段階の過程を含む(Harlow and Lane, 1988)。ハイブリドーマ作製の過程は2ヶ月以内に完了することは稀であり、たっぷり1年以上を要する場合が多い。
【0003】
モノクローナル抗体を作製するための従来のアプローチは、少なくも2つの制限を受ける;(i) 主として遺伝的不安定性によるハイブリドーマ細胞株の安定性の欠如および(ii) ハイブリドーマが培養において2〜3週間の存続期間しか有さず、この期間内に結合の特異性に関してスクリーニングしなければならないことによる、クローンの選択およびスクリーニングに関する時間の制限。モノクローナル抗体をインビトロで作製するためのファージディスプレイ技術(Winter et al., 1994;Barbas et al., 2000)、キメラ化またはヒト化戦略(Winte and Milstein, 1991)、およびハイブリドーマの形成に適したヒト骨髄腫細胞株(Karpas et al., 2001)等の最近の技術開発にもかかわらず、不死化モノクローナル抗体産生細胞を作製するためのさらなる戦略および方法はやはり必要であり、本分野において大きな進展を意味すると考えられる。
【発明の開示】
【0004】
発明の概要
本発明の態様は、抗体産生細胞を不死化細胞に融合させる(例えばハイブリドーマ作製)必要なしに、所望の抗原に対する抗体を産生する抗体産生細胞を作製するための方法を含む。本発明の局面は、ハイブリドーマを形成することなく不死化され得る抗体産生細胞を、抗原に対する抗体を産生するよう細胞を誘導するのに効率的な様式で所望の抗原に接触させる段階;および抗体産生細胞を不死化する段階を含む。抗体産生細胞は典型的に、条件的に機能するかまたは条件的に発現される形質転換発癌遺伝子を含み、抗体産生細胞の不死化は形質転換発癌遺伝子の発現または機能の誘導(形質転換発癌遺伝子の誘導)によってもたらされる。特定の態様において、条件的に機能する形質転換発癌遺伝子は温度感受性SV40大型腫瘍抗原(tsSV40Tag)であり、好ましくはtsSV40TagはA58S-SV40Tagである。本発明の特定の局面において、形質転換発癌遺伝子は、抗体産生細胞を25℃〜35℃、好ましくは30℃〜35℃、より好ましくは約33℃で培養することによって誘導する。典型的に、抗体産生細胞はハイブリドーマ培地で培養する。
【0005】
さらなる態様において、本方法は抗体産生細胞の抗体産生能を評価する段階を含む。抗体産生細胞の評価は、所望の抗原ならびに類似の抗原および異なる抗原に対する抗体結合をアッセイする段階を含み得る。モノクローナル抗体を産生するモノクローナル細胞集団を作製するには、典型的に単一細胞を選択して培養する。単一細胞は希釈クローニングにより選択し得る。本発明の他の局面では、ポリクローナル抗体を産生するポリクローナル細胞集団を作製するために、複数の細胞を選択して培養する。特定の態様において、抗体産生細胞は脾臓細胞(脾細胞)を含む。抗原は、ペプチド;タンパク質;糖タンパク質;リポタンパク質;炭水化物;ウイルス;細菌;病原微生物;組織;細胞全体;生検組織;患者由来の細胞;組織抽出物;新鮮なまたは培養した組織;アポトーシス細胞;細胞および組織に由来する膜、細胞質、および核画分等の細胞成分;精製タンパク質;部分精製タンパク質;レーザー捕獲した組織;またはパラフィン包埋し固定した組織であってよい。好ましい態様において、組織は対象由来の腫瘍組織を含む。
【0006】
本発明の他の局面において、抗体産生細胞は、ハイブリドーマを形成することなく不死化され得る抗体産生細胞を有するトランスジェニックマウスから取得することができる。トランスジェニックマウスは、ヒト抗体を産生するための遺伝子相補体(genetic complement)を含み得る。本発明の抗体産生細胞はマウスにおいて構成されてもよく、選択された抗原は、抗体を産生するよう抗体産生細胞を誘導するのに効率的な様式でマウスに投与される。特定の態様において、本方法は抗体産生細胞を抗原提示細胞と共培養することにより抗体産生細胞を所望の抗原と接触させる段階を含み、好ましくは抗原提示細胞は樹状細胞である。種々の態様において、抗体産生細胞はヒト抗体を産生するための遺伝子相補体を含み、ヒト抗体を産生する。本方法は、抗体産生細胞によって産生された抗体を精製する段階をさらに含み得る。特定の態様において、本方法は治療抗体を必要とする対象に抗体を投与する段階をさらに含み得る。
【0007】
本発明のさらなる態様は、所望の抗原に対するヒト抗体を産生する抗体産生細胞を作製するための方法を含む。本方法は、形質転換発癌遺伝子を条件的に発現するかまたは条件的に機能する形質転換発癌遺伝子を発現し、かつヒト抗体を産生するための遺伝子相補体を発現する抗体産生細胞を取得する段階を含み得る。抗体産生細胞の不死化は典型的に、形質転換発癌遺伝子の発現または機能を誘導することによってもたらされる。本発明はまた、ハイブリドーマを形成することなく不死化され得る抗体産生細胞を、抗原に対するヒト抗体を産生するよう細胞を誘導するのに効率的な様式で所望の抗原に接触させる段階、および抗体産生細胞を不死化する段階を含む。条件的に機能する形質転換発癌遺伝子は温度感受性SV40大型腫瘍抗原(tsSV40Tagであり)、好ましくはtsSV40TagはA58S-SV40Tagである。本発明の特定の局面は、25℃〜35℃、好ましくは30℃〜35℃、より好ましくは約33℃で抗体産生細胞を培養することによる、形質転換発癌遺伝子の発現または機能性の誘導を含む。
【0008】
本発明はまた、モノクローナル抗体を産生するモノクローナル集団を作製するために、単一細胞を選択および培養する段階を含み得る。他の局面において、本方法は、ポリクローナル抗体を産生するポリクローナル集団を作製するための、複数の細胞の選択および培養を含み得る。特定の局面において、抗体産生細胞は脾臓細胞を含む。抗原には、1つまたは複数のペプチド;タンパク質;糖タンパク質;リポタンパク質;炭水化物;ウイルス;細菌;病原微生物;組織;細胞全体;生検組織;患者由来の細胞;組織抽出物;新鮮なまたは培養した組織;アポトーシス細胞;細胞および組織に由来する膜、細胞質、および核画分等の細胞成分;精製タンパク質;部分精製タンパク質;レーザー捕獲した組織;またはパラフィン包埋し固定した組織が含まれ得る。組織は対象由来の腫瘍組織を含み得る。さらなる態様において、抗体産生細胞は、ハイブリドーマを形成することなく不死化され得る抗体産生細胞を有するトランスジェニックマウスから取得することができる。抗体産生細胞はマウスにおいて構成されてもよく、選択された抗原は、抗体を産生するよう抗体産生細胞を誘導するのに効率的な様式でマウスに投与される。他の局面において、本方法は抗体産生細胞によって産生された抗体を精製する段階をさらに含み得る。治療抗体を必要とする対象に抗体を投与することができる。
【0009】
本明細書に記載する任意の方法または組成物を、本明細書に記載する任意の他の方法または組成物に関して実行し得ることが意図される。
【0010】
特許請求の範囲および/または明細書における「含む」という用語と併せて用いらる場合の「1つの(a)」または「1つの(an)」という語の使用は「1つの(one)」を意味し得るが、「1つまたは複数の」、「少なくとも1つの」、および「1つまたは2つ以上の」の意味とも一致する。
【0011】
特許請求の範囲における「または」という用語の使用は、本開示では選択肢のみと「および/または」とを指す定義が支持されるものの、選択肢のみを指すことを明白に示すかまたは選択肢が相互に排他的である場合を除き、「および/または」を意味するために用いられる。
【0012】
本発明の他の目的、特徴、および利点は、以下の詳細な説明から明らかになると考えられる。しかしながら、この詳細な説明により本発明の精神および範囲内の種々の変更および修正が当業者に明らかになると考えられる上は、詳細な説明および特定の実施例は本発明の特定の態様を示すものの、説明のためのみに提供されることが理解されるべきである。
【0013】
例示的態様の説明
モノクローナル抗体の産生は典型的に、骨髄細胞株パートナーとの体細胞融合による脾細胞の不死化(ハイブリドーマ形成)を必要とする。ハイブリドーマは不死となり得るが、ハイブリドーマはフィーダー細胞層に依存し、遺伝的安定性を欠く可能性がある。ハイブリドーマ技術の開始以来、モノクローナル抗体産生細胞株の効率および安定性を改善する試みは実質的な進展をもたらしていない。さらに、ヒト抗体の産生に適したヒト多発性骨髄腫由来細胞株は、開発が非常に困難である。本発明者らは、抗体の産生を大いに単純化し、ハイブリドーマの必要性を排除する戦略について説明する。
【0014】
特定の態様において、抗体産生細胞、例えば脾細胞、または抗原提示細胞、例えば樹状細胞は、その発現が適切なプロモーターの制御下にあり、ポリヌクレオチドを含む細胞を許容温度において条件的に不死化させる変異体温度感受性発癌遺伝子をコードするポリヌクレオチドを有するトランスジェニックマウスに由来する。好ましい態様において、温度感受性発癌遺伝子は、マウス主要組織適合性プロモーターの制御下にあるシミアンウイルス40大型腫瘍抗原(tsSV40Tag)である。この脾細胞は許容温度(例えば33℃)において不死化され、ハイブリドーマを形成する必要なく抗体を産生する。このアプローチは、ポリクローナル抗体およびモノクローナル抗体の両方を作製し産生するために用いることができる。この無ハイブリドーマの細胞(hybridoma free cell)の増殖特性および安定性により、ハイスループット発見および抗体に基づく免疫療法のためのさらなる組成物および方法が提供される。
【0015】
本発明のさらなる態様は、無ハイブリドーマの抗体(hybridoma-free antibody)、すなわちハイブリドーマを形成することなく産生される抗体を作製および使用するための過程、組成物、および方法を含む。本発明の1つの態様は、無ハイブリドーマのマウスモノクローナル抗体またはポリクローナル抗体を作製するための組成物および方法を含む。
【0016】
さらなる態様において、本方法は、温度感受性発癌遺伝子、例えばシミアンウイルス40大型T抗原(tsSV40Tag)をインビトロ、エクスビボ、またはインビボで発現する適切な細胞種を、抗体を産生させるための抗原と接触させる段階を含み得る。特定の局面において、抗体産生細胞、好ましくは脾細胞は、tsSV40Tagを発現するマウスから単離され不死化される。そのようなマウス、ImmortoMouse(登録商標)、H-2Kb-tsA58トランスジェニックマウスでは、tsSV40Tagをコードする核酸の発現は主要組織適合性プロモーターの制御下にある(Jat et al., 1991、完全に参照として本明細書により組み入れられる)。ImmortoMouse(登録商標)に由来する細胞は、33℃で培養した場合に依然として不死のままである(Jat et al., 1991)。tsSV40Tagを発現するトランスジェニックマウスおよびtsSV40Tagを発現するトランスジェニックマウスに由来する細胞を含む種々の方法および組成物が特許文献に記載されており、例えばそれぞれ完全に本明細書に組み入れられる米国特許第6,399,384号;米国特許第5,866,759号;米国特許第5,688,692号;および米国特許第5,270,191号を参照されたい。他の局面において、抗体産生細胞または抗原提示細胞は、これらに限定されないが、骨髄、胸腺、脳、または生殖組織を含む、tsSV40Tag発現動物の種々の組織から単離および培養され得る。さらなる他の局面においては、そのような組織試料から幹細胞を単離し培養することができる。
【0017】
さらなる態様においては、(例えば、ImmortoMouse(登録商標)から)収集された細胞を1つまたは複数の抗原と接触させ、その後、抗原結合の特異性の評価を含む抗原に対する抗体産生の評価を行い得る。細胞の種々の亜集団を作製またはクローニングし、後に使用するために保存することができる。
【0018】
本発明のさらなる態様では、条件的に発現されるかまたは機能する形質転換発癌遺伝子を有するマウスを、ヒト抗体を産生するための遺伝子相補体を含むトランスジェニックマウスと交配させる段階を含む。1つの態様においては、条件的に発現されるかまたは機能する発癌遺伝子およびヒト抗体を産生するための遺伝子相補体の両方を有するマウスによる細胞を収集し(例えば、脾細胞、胸腺細胞、B細胞)、ヒト抗体を産生する不死化細胞集団を作製することができる。ヒトまたは異種間抗体の作製においてマウスを用いる組成物および方法について記載している種々の特許が存在し、例えばこの技術の特許については、完全に参照により本明細書に組み入れられる米国特許第6,673,986号;米国特許第6,657,103号;米国特許第6,162,963号;米国特許第6,235,883号;米国特許第6,150,584号;米国特許第6,114,598号;米国特許第6,075,181号;および米国特許第5,939,598号を参照されたい。
【0019】
I. 抗体産生
典型的に、本発明の抗体は、条件的に不死化が可能である細胞を有するマウスもしくは他の動物を免疫することによるか、または不死化細胞もしくは潜在的に不死化可能な細胞を関心対象の抗原と接触させることによって作製する。不死化細胞または潜在的不死化細胞は、条件的に機能する、例えば特定の温度以下でのみ機能するまたは特定の培養条件下でのみ発現される形質転換遺伝子を発現し得る。例えば、シミアンウイルス40初期領域変異体tsA58によってコードされる熱不安定性ラージT抗原(tsSV40Tag)を用いて、トランスジェニックマウスまたは不死化可能な細胞株を樹立することができる。これらの細胞株は、許容温度(例えば33℃)においてまたは特定の環境において(例えばテトラサイクリンの存在下において)持続的に増殖し得るが、非許容温度(37〜39.5℃)に上げるかまたは制御因子を除去するかもしくは添加した際に細胞増殖の停止を示す。増殖停止は細胞周期のG1期またはG2期において起こる。増殖が停止した後でも、一般的なタンパク質合成およびトリパンブルー排除能によってアッセイされるように、細胞は代謝的に活性を有したままである。これらの細胞株は、非許容温度または条件において分裂することはできない。
【0020】
トランスジェニック動物を免疫した後、モノクローナル抗体作製手順において使用するために、抗体産生能または抗原提示能を有する体細胞、それぞれ特にBリンパ球(B細胞)または樹状細胞(DC)を選択する。これらの細胞は、これらに限定されないが、マウス、ラット、ウサギ、イヌ、ネコ、ヤギ、ウシ、ウマ、ヒツジ、またはヒトを含む1匹(人)または複数匹(人)の対象から生検採取した脾臓、扁桃腺、リンパ節、または末梢血生検試料から取得し得る。脾臓細胞および末梢血細胞が好ましく、脾臓細胞は分裂形質芽球期にある抗体産生細胞のより豊富な供給源であるためであり、末梢血細胞は入手しやすいためである。一連の動物を免疫し、最も高い抗体価を有する動物の脾臓を摘出し、脾臓をシリンジでホモジナイズすることによって脾臓のリンパ球を得る場合が多い。典型的に、免疫したマウスの脾臓は、約5 x 107〜2 x 108個のリンパ球を含む。
【0021】
関心対象の抗原に結合する抗体の産生を誘導するために、形質転換タンパク質を発現する動物を免疫するか(例えば、H-2Kb-tsA58 ImmortoMouse(登録商標))、または形質転換タンパク質を発現する細胞を抗原(例えば、繊維状fd-tetファージ(Zacher et al., 1980))に曝露する。免疫または接触は様々な期間にわたって1回または複数回繰り返してよく、例えば、免疫または抗原曝露は1、2、3、4、5、6、7、8、9、10、11、12日、もしくはそれ以上の日数、または1、2、3、4、5、6、7、8、9、10、11、12週、もしくはそれ以上の週に1回であってよい。特定の局面において、免疫または抗原曝露は、1日おきもしくは1週間おき、または3日、4日、5日、もしくはそれ以上の日数に1回、または3週、4週、5週、もしくはそれ以上の週に1回であってよい。免疫または抗原曝露は、2、3、4、5、6、7、8、9、10、11、12、13、14、15週、もしくはそれ以上の週、さらには2、3、4、5、6、7、8、9、10、11、12、13、14、15ヶ月、もしくはそれ以上の月数にわたって行ってよく、好ましくは12週間行い得る。抗原調製物は、静脈内(i.v.)、腹腔内(i.p.)、皮内、皮下(s.c.)、またはそれらの様々な組み合わせを含む1つまたは複数の経路により投与する。各追加免疫後に動物から採血し、ELISAを用いて血清中の抗抗原抗体価をモニターし得る。
【0022】
免疫した動物の臓器を摘出もしくは生検採取するか(例えば脾臓)、または抗原曝露細胞を収集し、細胞培養液中に入れる。細胞は典型的に、2枚の冷却スライドグラスの間に挟んだ臓器の被膜に穏やかに圧力を加えることによって臓器から放出させる。次いで、抗体産生細胞(例えば脾細胞)を適切な増殖培地、好ましくはハイブリドーマ培地に再懸濁し、低密度で培養する。細胞を新たな容器に連続して移すことにより、組織残屑を重力により除去する。典型的に全部で約2 x 108個の細胞を6、24、および96ウェルプレートに分配し、33℃で培養する。2〜3週間の間に少なくとも2回、3回、4回、またはそれ以上の回数、培地を完全に交換する。クローンは典型的に、3週間後に90%を超えるウェルで観察される。プレートをモニターし、各ウェルに新鮮な培地をおよそ3週間ごとに添加する。限界希釈により、陽性ウェルをサブクローニングし得る(Harlow and Lane, 1988);いくつかのクローンはまた24ウェルおよび96ウェルプレートに拡大し、長期培養後に反応性をモニターする。各ウェルにおいて細胞を慎重に懸濁することにより、および無血清培地を使用することにより、脾細胞の「凝集」を回避することができる。典型的に、カウントし、ウェル当たり約0.1〜0.5個の細胞でプレーティングした後、各96ウェルプレートを顕微鏡下で系統的に観察する。
【0023】
抗体産生細胞の集団が単離された後、それらをスクリーニングし、関心対象の特徴、例えば特定の病原体またはタンパク質に対する選択的結合を有する抗体に関して選択する。細胞のサブセットが選択されたならば、同定された1つまたは複数の細胞をクローニングし増殖させる。典型的に、関心対象の抗原、例えば繊維状ファージおよび/または組み換えタンパク質に対するELISAを用いて、関心対象の抗体を産生する細胞をスクリーニングし選択するが、例示的な方法は以下およびHarlow and Lane(1988)に記載されている。陰性対照には、免疫した動物から単離された細胞または抗原に曝露した細胞と比較するための、BSA、ハイブリドーマ培地のみ、免疫前血清、および二次抗体が含まれ得る。免疫ポリクローナル血清および抗抗原抗体は陽性細胞となり得る。特定の局面においては、抗体は培養上清から直接プレーティングする。陽性ウェルの細胞を限界希釈によりサブクローニングし、モノクローナル株を取得する。これらの手順により現れるサブクローンを、ELISA法により種々の抗原に対して試験する。反応性をELISAリーダーによってモニターする。
【0024】
有望な抗体を産生する細胞が同定されサブクローニングされたならば、ウェスタンブロット解析および他の抗原結合アッセイを行って得られた抗体を確認し、さらに特徴づける。典型的には、抗原をSDS-PAGE電気泳動により分離し、ポリフッ化ビニリデン膜(Bio-Rad)に電気的に転写する。膜を条片に分割し、PBSに溶解した5%ノンファットミルクによりブロッキングし、その後適切な洗浄緩衝液、例えば0.1% Tween 20を含むPBSで洗浄する。条片を免疫前血清(1:1000)、免疫後血清(1:1000)、陽性対照抗体、不死化細胞クローンから分泌されたIgGを含む上清、または細胞培養液と共にインキュベートする。何回か洗浄した後、検出試薬結合二次抗体(ペルオキシダーゼ結合二次Ab)(Bio-Rad)を条片に添加し、室温でインキュベートする。条片を洗浄し、例えば高感度化学発光(ECL)(Amersham Biosciences、ニュージャージー州、ピスカタウェイ)により反応性を検出する。
【0025】
マウスには、典型的に2ヶ月間にわたり2週間の間隔で抗原を腹腔内投与する。3回目および4回目の免疫後に、免疫した各マウスの血清をELISAアッセイにより解析し得る。典型的には、最も高い抗抗原抗体価を有する1匹または複数匹のマウスの脾臓を摘出し、脾細胞を単離する。限界希釈により単一クローンを取得することができる。抗体産生は、最初の抗原を用いて細胞培養上清においてELISAによってモニターする(Harlow and Lane, 1988)。
【0026】
培養により、特定のサブクローンを選択する不死化細胞の集団が提供される。典型的に、不死化細胞の選択は、マイクロタイタープレートにおいて単一クローン希釈により細胞を培養し、その後(約2〜3週後に)個々のクローンの上清を所望の反応性に関して試験することによって行う。アッセイ法は、放射性免疫測定法、酵素免疫測定法、細胞障害アッセイ法、プラークアッセイ法、ドット免疫結合アッセイ法等のように、感度がよく、簡便、かつ迅速であるべきである。抗体を調製するおよび特徴づける一般的な方法は、当技術分野において周知である(例えば、Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory, 1988(参照により本明細書により組み入れられる)を参照されたい)。
【0027】
細胞からの治療タンパク質の作製の成功に関する基準の1つは、産生の安定性を維持する細胞株を取得することである。これが達成されない場合、工程収率、時間および費用の効果的利用、ならびに産物の規制認可に関する問題が生じ得る。ハイブリドーマ細胞株によるタンパク質産生の不安定性に関して報告しているいくつかの研究が存在する。ハイブリドーマにおけるタンパク質産生の不安定性の原因は多様であり、多くの場合、正確な分子機構は未だ不明である。
【0028】
ポリクローナル抗体の産生に関しては、抗体はエクスビボで作製することができ、マウスまたはウサギ等の標的抗体産生動物における複数回の抗原注射および採血の必要性を排除することができる。この技術により、標的抗原または所与の組織、細胞集団、もしくはタンパク質に関連した抗原群への曝露に基づいて初回刺激および拡大した抗体、T細胞、またはナチュラルキラー細胞のポリクローナル集団を作製することが可能になる。ハイブリドーマでは特定のクローンが他よりも優位になるため、そのような過程はハイブリドーマを用いた場合にはより困難である。
【0029】
本明細書で用いる「抗体」という用語は抗原結合領域を有する任意の抗体様分子を指し、これにはポリクローナル抗体およびモノクローナル抗体、ならびにFab'、Fab、F(ab)2'、単一ドメイン抗体(DAB)、Fv、scFv(一本鎖Fv)等のような抗体断片が含まれる。抗体に基づく様々な構築物および断片を調製するおよび用いる技法は、当技術分野において周知である。抗体を調製するおよび特徴づける手段も同様に、当技術分野において周知である(例えば、Antibodies: A Laboratory Manual, Cold Spring Harbor Laboratory, 1988(参照により本明細書に組み入れられる)を参照されたい)。
【0030】
本発明の特定の態様において、マウス主要組織適合性プロモーターの制御下に変異体温度感受性(ts)シミアンウイルス40(SV40)大型腫瘍抗原(Tag)を有するトランスジェニックマウス(H-2Kb-tsA58トランスジェニックマウス;ImmortoMouse(登録商標))と称される)に由来する不死化脾細胞は不死であり、ハイブリドーマを作製する必要性が回避される。
【0031】
記載した方法により抗体が同定されたならば、適切な1つまたは複数の遺伝子を増幅し、クローニングし、抗体を産生させるための別の細胞にトランスフェクトすることができる。例えば、それぞれ参照により本明細書に組み入れられる米国特許第5,658,570号、米国特許第6,165,745号、および6,602,503号、ならびにDaniell, 2001;Breitling and Dubel, 1999を参照されたい。
【0032】
II. トランスジェニックマウス
細胞株に関する研究により、多くの重要な生物学的疑問への我々の理解が大いに改善された。細胞株の作製は、関心対象の細胞種に不死化発癌遺伝子を導入することによって促進される(Jat et al., 1991)。多くの異なる細胞種をインビトロで不死化することが知られている1つの遺伝子は、SV40大型腫瘍抗原(SV40Tag)である。細胞株を作製するためにインビトロで遺伝子を挿入する必要性を回避するため、SV40TAg遺伝子を有するトランスジェニックマウスが作製された(Jat et al., 1991)。以前の研究により、トランスジェニックマウスにおけるSV40TAg発現が腫瘍形成および異常な発生に関連していることが示されたため、形質転換に関して熱不安定性であるSV40Tag、tsA58(tsSV40Tag)を用いて、生物体全体で見出される典型的な条件下においてインビボで存在する機能的なSV40TAgのレベルが低減された。
【0033】
広範な組織に発現を導くために、広範囲にわたって活性がありかつインターフェロンによって誘導され得るマウス主要組織適合遺伝子複合体H-2Kbプロモーターが用いられた。tsSV40TAg mRNAは、ハイブリッド構築物を有するすべての動物の組織において発現された。いずれの組織の発生も肉眼的に正常であった。H-2Kb-tsA58マウスの1つの系統はホモ接合性になるまで何世代かにわたって交配を繰り返され、導入遺伝子の機能的コピーを伝達している。これらのマウスは「ImmortoMice」と称される。ImmortoMouse(登録商標)はCharles River Labs、マサチューセッツ州、ウィルミントンから市販されている。多くの異なる種類の条件的不死化細胞株がImmortoMouse(登録商標)から導出されているが、この十分に樹立されたマウスモデルはモノクローナル抗体産生細胞を作製するために活用されていなかった。
【0034】
ImmortoMouse(登録商標)は、分化の細胞機構の研究において使用するため、および組織修復に関連した細胞移植実験のため、インビトロおよびインビボで正常な分化を起こし得る個々の細胞種の拡大集団を作製する能力に関して開発された。H-2Kb-tsA58マウスにより、単離された細胞を適切な条件下で培養することによって、種々の組織から条件的不死化細胞株を直接導出することが可能になる。これらのマウスにおいて、tsSV40Tagはインターフェロン誘導性クラスI抗原プロモーターによって制御される。インターフェロンの存在下または非存在下において33℃で培養することにより、インビボおよびインビトロにおいて正常な分化を起こす能力を依然として保持させつつ、細胞をインビトロで長期間培養することができる。
【0035】
III. ヒト抗体の産生
本発明の1つの態様においては、条件的に機能する形質転換発癌遺伝子を含むトランスジェニックマウス、例えばImmortoMouse(登録商標)を(以下に詳述するように)抗体産生のための種々の遺伝子成分をコードする他の種による遺伝子を有するトランスジェニックマウスと交配し、ヒト抗体等の他の種の抗体を産生する細胞株を作製することができる。
【0036】
完全なヒトモノクローナル抗体の多様なレパートリーを産生する能力は、ヒトの治療において用途を有する。治療用のヒトポリクローナル抗体またはモノクローナル抗体を作製するための最も有望なアプローチの1つは、マウスまたは他の非ヒト抗体の非存在下で豊富なレパートリーのヒト抗体を産生するように操作されたマウス系統の作製である(例えばXenoMouse(登録商標))。最近、遺伝子ターゲティングの結果としてマウス抗体産生を欠くマウスの生殖系列に、ヒト免疫グロブリン座位の部分を導入することによってマウスが作製された。これらのマウスは多様な成体様レパートリーを有する顕著なレベルの完全なヒト抗体を産生し、抗原による免疫化に際して抗原特異的ヒト抗体を作製する。XenoMouse(登録商標)には約80%のヒト重鎖抗体遺伝子およびかなりの量のヒト軽鎖遺伝子が備わっている。これらの遺伝子の複雑な構築およびそれらのセミランダムな対形成により、マウスが抗原構造の多様なレパートリーを認識することが可能になる。さらに、このマウスは非常に高い親和性の完全なヒト抗体をプロセシングし得る。入手可能なXenoMouse(登録商標)動物には複数系統が存在する。各系統は、種々の用途用に異なるクラスの抗体を産生し得る。そのような系統のマウスは、ヒト抗原を含む広範囲の抗原に対して高い親和性および特異性を有するヒト抗体を産生するための最適な供給源を提供し得る。
【0037】
XenoMouse(登録商標)は、マウス抗体遺伝子発現が抑制され、ヒト抗体遺伝子発現により機能的に置換されているが、マウス免疫系の残りの部分は原型を保ったままである遺伝子操作された系統のマウスを用いて、完全なヒトタンパク質配列を有する抗体を作製する。マウスゲノムにヒト抗体遺伝子を導入することにより、そのような形質を有するトランスジェニックマウスを永久に繁殖することができる。重要なことには、これらのトランスジェニックマウスは、マウスにおいて発現される(したがって、「自己」として認識される)ヒト産物のみが抗体自体であるために、ヒト抗原に対する抗体を産生し得る。他の機構はすべてマウスの機構であり、したがって任意の他のヒト組織またはタンパク質はマウスにより異物として認識され、免疫反応が開始されることになる。
【0038】
いくつかのヒトタンパク質、例えばサイトカイン、ホルモン、および増殖因子、またはそれらの受容体の異常な合成は、様々なヒト疾患の原因となる。ヒト抗体を用いた中和または完全な排除によりこれらのタンパク質の制御を行い、疾患を治療するまたは完全に排除することができる。これらのトランスジェニックマウスが、ヒト抗原に対するヒト抗体の産生において使用し得る細胞を生じる能力により、種々の病態の処置、診断、または治療における利点が提供されると考えられる。1つの課題は、安定した細胞株において所与の抗原に対するヒト抗体を十分に産生することである。この問題は本発明の態様によって解決し得る。1つの態様において、交配種のマウス集団(例えばImmortoMouse(登録商標)/XenoMouse(登録商標)交配)は、ハイブリドーマを作製する必要なしに、任意のヒト抗原に対する抗体を産生し得る不死化脾細胞を産生し得る。
【0039】
Xenomouseまたは同様の遺伝子修飾を有する動物は、キメラおよび他のヒト化技術とは異なる、100%ヒトタンパク質配列を有する抗体を産生する。これらのマウスを使用する他の利点は、XenoMouse(登録商標)技術を用いて産生される抗体がより優れた安全性プロファイルを提供し、またヒト体内からよりゆっくりと除去され、投与の頻度を減らすことが期待され得る点である。
【0040】
XenoMouse(登録商標)技術では、天然のインビボ親和性成熟過程を利用して、抗体産物候補を通常2〜4ヶ月のうちに作製する。これらの抗体産物候補は、ファージディスプレイで見られる親和性よりも100〜1000倍高い親和性を有し得る。ヒト化およびファージディスプレイ技術を用いて作製される抗体とは対照的に、時には困難であり時間を要することが判明している過程である、次の操作の必要性がない。したがって、抗体構造は最初の選択された抗体から最終的な市販抗体まで原型のまま残存し得る。
【0041】
従来、所望の特徴を有する抗体が同定された時点で、ハイブリドーマまたは組み換え細胞株から直接、前臨床物質を産生させることができる。ハイブリドーマ不要の産生(hybridoma-free production)により、時間節約の可能性に加えて、ハイブリドーマまたは組み換え細胞株において抗体を産生させる必要性が回避される。したがって、本発明の態様により、ハイブリドーマを必要とせずに長期培養においてヒト抗体を産生させる必要が満たされ得る。
【0042】
典型的に、マウスによって作製されるモノクローナル抗体は、それらがヒトタンパク質でないために、免疫系がそれらを異物として認識する患者によって拒絶される。患者は、ヒト抗マウス抗体またはHAMAを産生する場合が多い。この反応により、結合活性が中和されて抗体の有効性が減少する。マウス抗体の次の投与もまた毒性となり得る。本明細書に記載する方法を使用すると、ヒトまたはそれ以外の医学的に関連のあるほとんどすべての抗原に対する抗体を作製することができる。選択される複数の抗体を産生する能力は、最適な抗体産物を選択する上で重要である可能性がある。本発明の1つの態様において、ヒトモノクローナル抗体産生脾細胞の不死化集団は、開示の方法によって作製され得る。
【0043】
さらに、Medarex(ニュージャージー州、プリンストン)はUltiMAb Human Antibody Development SystemSMと称されるシステムを開発した。このシステムにより、様々な種類の完全なヒト(100%ヒトタンパク質配列)抗体が作製された。これらのマウスは、ヒト抗体をコードする遺伝子を含む。これらのモノクローナル抗体は、好ましい安全性プロファイルを有し、またヒト体内からよりゆっくりと除去され、疾患標的に影響を及ぼすために必要な投与の頻度および量が低減される可能性が高い。これらのマウスはまた、本明細書に記載するハイブリドーマ不要の抗体産生法と組み合わせて用いることもできる。
【0044】
要するに、本明細書に記載する方法により、治療または診断目的で、種々の病態(例えば炎症)において発現される腫瘍抗原またはタンパク質に対する抗体を作製することができる。1つの態様では、ヒトモノクローナル抗体を用いて、標的細胞集団、例えば腫瘍細胞に対する抗体の親和性および特異性により、標的細胞集団に対して送達薬剤(例えば細胞毒素)を標的するまたは強化することができる。一方、ヒトモノクローナル抗体を用いて、多くの病態および種々の種類の腫瘍において過剰発現される受容体(例えば、上皮増殖因子受容体(EGFR)等の増殖因子受容体)の機能を抑制することができる。EGFRシグナル伝達が阻止され、細胞死または増殖阻害がもたらされ得る。他の用途には、標的細胞集団、例えば腫瘍細胞に結合する蛍光抗体を用いた腫瘍の画像化が含まれる。
【0045】
特定の態様においては、本明細書に記載の方法により製造された抗体を用いて、以下に記載するファージディプレイ技術を用いて同定された血管郵便番号(vascular zip code)を同定するまたは標的することができる。血管郵便番号とは、薬剤がより効果的かつ効率的に標的され得る、ヒトの身体における特異的かつユニークなアドレスであり、例えば、それぞれ参照により本明細書に組み入れられる米国特許第5,622,699号、米国特許第6,174,687号、および米国特許第6,232,287号を参照されたい。血管標的化は、例えば身体の健常部分をそのままにしつつ腫瘍部位に照準を合わせることにより、治療の有効性を改善し得る。ファージと称される微視的粒子に提示された10億個を超える一群のペプチド配列を投与することにより、ペプチドは身体の特定領域に優先的にホーミングする。この大規模なスクリーニングにより、循環ペプチドの組織分布が無作為ではないこと、および特定のペプチドが異なる臓器に配向し結合することが示される。
【0046】
これらのペプチドは典型的に、臓器の組織および血管に存在する受容体に結合する。ペプチドは、体内を移動しつつ、リガンド(ペプチド結合タンパク質)の挙動を促進し、対象の細胞、組織、血管、または臓器における細胞受容体と相互作用し得る。ペプチドライブラリーをインビボでスクリーニングすることによってリガンドを同定することができ、次にこのリガンドを用いて受容体を同定することができる。次いで、本明細書に記載する方法により、受容体に対してヒトモノクローナル抗体を作製することができる。別の用途は、患者において循環する抗体レパートリーを同定することにより標的を同定し(Mintz et al., 2003)、次いで特定の症例における標的療法または受動免疫を開発するために、これらの標的に対するヒト抗体を作製することであってよい。
【0047】
従来、新規薬学的産物は、ヒト臨床試験が開始されるまでに前臨床開発におよそ6年を要する。完全なヒトモノクローナル抗体技術を用いれば、この期間を2年未満に短縮できる可能性がある。さらに、開発費用も、製薬産業により従来通りに開発される化合物に付随する費用のごく一部ですむ可能性が高い。
【0048】
IV. 抗体産生のための抗原
本発明の態様は、抗体産生細胞または抗原提示細胞の安定株を作製するために、条件的に機能する形質転換遺伝子を発現するトランスジェニックマウスを種々の抗原で免疫するか、または条件的に機能する形質転換遺伝子を発現する細胞を種々の抗原に曝露する段階を含む。本発明の範囲内の抗原には、これらに限定されないが、ペプチド、タンパク質、糖タンパク質、リポタンパク質、ウイルス、細菌、病原微生物、および罹患ヒト細胞を含む、対象において液性免疫応答および細胞免疫応答を誘発し得る任意の分子または巨大分子群が含まれ得る。特定の態様において、抗体作製のためのエクスビボ法(すなわち、処置する対象の外部における抗体作製)の使用により、ヒト等の哺乳動物内で作製され得ない抗原に対する抗体を作製することが可能になることに留意することが重要である。本発明の方法による抗体の作製により、対象内での寛容の効果が回避され得る。したがって、広範な種類の抗体を作製することできる。これらの抗体により、以前は標的することが困難であった抗原を標的することが可能になる。
【0049】
特定の態様において、同定された標的化ペプチド(例えば、特定の臓器を標的するペプチド)もしくはそれらの受容体またはさらには腫瘍細胞等の細胞全体に対する抗体(例えば、モノクローナルおよび/またはポリクローナル)を作製することが望ましい場合がある。適切な標的化ペプチドもしくは受容体またはそれらの部分を、リンカー、ポリリンカー、または誘導体化アミノ酸を介して、アジュバントを含む1つまたは複数の物質にカップリング、結合(bonded)、結合(bound)、共役、または化学的に連結させることができる。特定の局面において、アジュバントには、コロイド金の使用が含まれる(Dykman et al., 1996、完全に参照により本明細書に組み入れられる)。これは、二重特異性もしくは多価の組成物またはワクチンが産生されるように行ってもよい。これらの組成物の調製において用いられる方法は当業者に周知であり、ヒト対象に投与するために適していなければならない、すなわち薬学的に許容されなければならないことがさらに考えられる。好ましい物質には、キーホールリンペットヘモシアニン(KLH)またはウシ血清アルブミン(BSA)等の担体が含まれる。種々の態様において、対象は、これらに限定されないが、マウス、ウサギ、ニワトリ、ヤギ、ヒツジ、ウシ、イヌ、およびヒトを含む任意の高等脊椎動物であってよい。
【0050】
特定の態様においては、抗イディオタイプ抗体または標的化ペプチドの受容体に対する抗体を作製することができる。「標的化ペプチド」とは、対象の臓器または組織への選択的局在化を特徴とする、連続的なアミノ酸の配列を含むペプチドである。選択的局在化は、例えば、推定標的化ペプチド配列をファージの外表面上に提示されるタンパク質に組み込む、以下に開示する方法によって決定することができる。異なるアミノ酸配列のそのような標的化ペプチドを多数発現するように遺伝子操作したそのようなファージのライブラリーを対象に投与した後、対象から1つまたは複数の臓器または組織を採取し、その臓器または組織に見出されるファージを同定する。標的化ペプチド配列を発現するファージは、対照の組織または臓器と比較してある組織または臓器においてより多くの結合を示す場合に、その組織または臓器に選択的に局在すると見なされる。
【0051】
一般に、標的化ペプチドの選択的局在化は、対照の臓器または組織と比較して標的臓器または組織においてファージの少なくとも2倍の濃縮をもたらすべきである。対照の臓器または組織と比較して、標的臓器において少なくとも3倍、4倍、5倍、6倍、7倍、8倍、9倍、10倍、またはそれ以上の濃縮をもたらす選択的局在化が好ましい。または、選択的局在化を示す標的化ペプチド配列を発現するファージは、標的臓器から回収されたファージを次のラウンドのスクリーニングのために第二の宿主に再度注入した場合に、対照臓器と比較して標的臓器において濃縮の増加を示すべきである。選択的局在化を決定するためのもう一つの別の手段は、推定標的ペプチドを発現するファージが、非特異的ペプチドを発現するかまたは推定標的ペプチドを発現するように遺伝子操作されていない対照ファージと比較して、標的臓器において少なくとも2倍、より好ましくは3倍の濃縮を示すことである。選択的局在化を決定するための別の手段は、標的ペプチドを発現するファージの標的臓器への局在化が、標的ペプチド配列を含む合成ペプチドの共投与によって少なくとも部分的に阻止されることである。「標的化ペプチド」および「ホーミングペプチド」は、本明細書において同義に用いられる。
【0052】
抗体を産生させるための他の抗原には、生検試料、患者由来の細胞、患者由来の新鮮腫瘍組織、組織抽出物、新鮮なまたは培養した組織の試料が含まれ得る。関連のあるいくつかの抗原は細胞外成分中および/または間質内に存在する可能性があるため、単に細胞のみでない組織成分を含めることが重要である。抗体を作製するための他の抗原には、これらに限定されないが、アポトーシス細胞、細胞および組織に由来する膜成分、細胞質、核画分、精製タンパク質、部分精製タンパク質、レーザー捕獲した組織、またはパラフィン包埋するかもしくは固定した組織が含まれ得る。
【0053】
A. ファージディスプレイ
抗原または抗原候補は、ファージディスプレイを用いて同定することができる。本方法は、ファージディスプレイライブラリーのインビボ投与を含み得る。本発明の種々の態様では、リガンドを同定し、次いでこれらを用いてこれらリガンドに対する受容体をさらに同定することができ、次にこの受容体を用いてモノクローナル抗体産生不死化脾細胞を作製することができる。ファージディスプレイの様々な方法およびペプチドの多様な集団を作製する方法は、当技術分野において周知である。例えば、それぞれが参照により本明細書に組み入れられ、ファージライブラリーを調製する方法について記載している、米国特許第5,223,409号、第5,622,699号、および第6,068,829号を参照されたい。
【0054】
ファージディスプレイ技法は、小さいペプチドがその表面上に発現され得るようにバクテリオファージを遺伝子操作する段階を含む(Smith et al., 1985, 1993)。この技法の潜在的適用範囲はかなり広く、この10年の間に、ファージによって提示されるペプチドライブラリーの構築、およびライブラリーを用いてペプチドリガンドを単離するスクリーニング法の開発にはかなりの進展が見られた。例えば、ペプチドライブラリーを用いることにより、炎症反応に関与する抗体または細胞接着を媒介するインテグリンのような多くのタンパク質における相互作用部位および受容体-リガンド結合モチーフの特徴づけが可能となった。この方法はまた、ペプチド模倣剤または造影剤を開発するためのリードとなる新規ペプチドリガンドを同定するためにも用いられている(Arap et al., 1998a)。
【0055】
所与の臓器または組織を標的するための最も効率的なアミノ酸配列は、「バイオパニング」により単離することができる(Pasqualini and Ruoslahti, 1996;Pasqualini, 1999)。簡潔に説明すると、推定標的化ペプチドを含むファージのライブラリーを、細胞集団(例えば脾細胞)、動物またはヒト対象に投与するかまたはそれらと接触させ、ファージを含む臓器または組織の細胞抽出物または試料を回収し得る。繊維状ファージを利用する1つの態様においては、バイオパニングのラウンド間に、線毛陽性細菌においてファージをインビトロで増殖させ得る。この細菌はファージによって溶解せず、むしろ特定の挿入物を提示するファージを多コピー分泌する。標的分子に結合するファージを標的の臓器または組織から溶出させ、次いで宿主細菌中で培養することにより増幅することができる。必要に応じて、増幅されたファージをヒト宿主に投与し、臓器または組織の試料を再び回収してもよい。選択的結合剤の集団が得られるまで、複数ラウンドのバイオパニングを実施することができる。ペプチドのアミノ酸配列は、ファージゲノム中の標的化ペプチド挿入物に対応するDNAを配列決定することによって決定される。次に、標準的なタンパク質化学技法により、同定された標的化ペプチドを合成ペプチドとして産生することができる(Arap et al., 1998a, Smith et al., 1985)。このアプローチにより、その標的の性質に関していかなる概念も予め想定することなく、先入観にとらわれない機能的アッセイにおいて循環する標的化ペプチドを検出することができる。
【0056】
標的化ペプチドの受容体として候補標的が同定されたならば、標準的な生化学的方法を用いてこれを、単離、精製、およびクローニングすることができる(Pasqualini, 1999;Rajotte and Ruoslahti, 1999)。次いで、これらの精製タンパク質を、ImmortoMouse(登録商標)もしくはヒト化細胞集団を産生するImmortoMouse(登録商標)交雑種による脾細胞等の細胞集団、またはモノクローナル抗体産生脾細胞等の他の条件的不死化細胞を免疫または曝露するための抗原として使用することができる。次に、これらの抗体産生細胞を用いて、標的受容体または抗原に対して特異的な抗体集団を作製することができる。
【0057】
マウスにおいて行われたこれまでのインビボ選択研究では、fUSE5ベクター内の遺伝子III莢膜タンパク質との融合タンパク質として発現されるランダムペプチドのライブラリーが優先的に用いられた(Pasqualini and Ruoslahti, 1996)。所与のライブラリー中に存在する個々のクローンの数および多様性は、インビボ選択の成否にとって重要な要因である。欠損ファージクローンの過剰発現を有する可能性が低い一次ライブラリーを用いることが好ましい(Koivunen et al., 1999)。ライブラリーの調製は、108〜109形質導入単位(T.U.)/mlに最適化するべきである。特定の態様においては、各選択ラウンド間に大量増幅戦略を適用する。
【0058】
本発明の範囲内において、直鎖、環状、または二環式ペプチドを提示するファージライブラリーを用いることができる。しかし、単環式ペプチドは直鎖ペプチドよりも標的臓器に対して高い親和性を有する傾向があることから、環状挿入物において3〜10個のランダム残基(CX3〜10C)を提示するファージライブラリーが好ましい。二環式ペプチド(CX3C X3C X3C等;Rojotte et al., 1998)を提示するライブラリーも使用に成功している。しかし、同族合成ペプチドの産生は、可能ではあるが、異なるジスルフィド架橋配列を有する多数の配座異性体のために複雑となり得る。
【0059】
V. タンパク質およびペプチド
特定の態様において、抗原組成物は、抗体産生に使用し得る少なくとも1つのタンパク質、ペプチド、またはペプチド様化合物を含み得る。本明細書において使用するタンパク質またはペプチドは一般に、遺伝子から翻訳される全長配列までの約200アミノ酸を超えるタンパク質;約100アミノ酸を超えるポリペプチド;および/または約3〜約100アミノ酸のペプチドを指すが、これらに限定されるわけではない。便宜上、「タンパク質」、「ポリペプチド」、および「ペプチド」という用語は、本明細書において互換的に用いられる。特定の態様において、タンパク質は本明細書に記載の方法によって作製される抗体である。
【0060】
特定の態様において、少なくとも1つのタンパク質またはペプチドの大きさは、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100、約110、約120、約130、約140、約150、約160、約170、約180、約190、約200、約210、約220、約230、約240、約250、約275、約300、約325、約350、約375、約400、約425、約450、約475、約500、約525、約550、約575、約600、約625、約650、約675、約700、約725、約750、約775、約800、約825、約850、約875、約900、約925、約950、約975、約1000、約1100、約1200、約1300、約1400、約1500、約1750、約2000、約2250、約2500アミノ酸、またはそれ以上のアミノ酸残基を含み得るが、これらに限定されるわけではない。
【0061】
本明細書において用いる「アミノ酸残基」とは、当技術分野において周知である任意の天然アミノ酸、任意のアミノ酸誘導体、または任意のアミノ酸模倣体を指す。特定の態様において、タンパク質またはペプチドの残基は連続的であって、アミノ酸残基の配列を中断する任意の非アミノ酸は存在しない。他の態様においては、配列は1つまたは複数の非アミノ酸部分を含んでもよい。特定の態様において、タンパク質またはペプチドの残基の配列は、1つまたは複数の非アミノ酸部分によって中断されてもよい。
【0062】
したがって、「タンパク質またはペプチド」という用語は、天然タンパク質において認められる20個の一般的なアミノ酸の少なくとも1つ、または少なくとも1つの修飾アミノ酸もしくは稀なアミノ酸を含むアミノ酸配列を包含する。
【0063】
タンパク質またはペプチドは、標準的な分子生物学的技法によるタンパク質、ポリペプチド、もしくはペプチドの発現、天然源からのタンパク質もしくはペプチドの単離、またはタンパク質もしくはペプチドの化学合成を含む、当業者に周知の任意の技法によって作製し得る。様々な遺伝子に対応するヌクレオチド、ならびにタンパク質、ポリペプチド、およびペプチド配列がこれまでに開示されており、当業者に周知のコンピューターデータベースにおいて見出すことができる。1つのそのようなデータベースは、国立バイオテクノロジー情報センターのGenbankおよびGenPeptデータベースである(www.ncbi.nlm.nih.gov/)。既知遺伝子のコード領域は、本明細書に開示する技術を用いてまたは当業者に周知のように、増幅および/または発現させることができる。または、タンパク質、ポリペプチド、およびペプチドの様々な市販の調製物が当業者に周知である。
【0064】
A. ペプチド模倣体
本発明によるポリペプチドの調製に関する別の態様は、抗原として使用するためのペプチド模倣体の使用である。模倣体は、タンパク質二次構造の要素を模倣するペプチド含有分子である。例えば、完全に参照により本明細書に組み入れられる、Johnson et al., 1993を参照されたい。ペプチド模倣体の使用の背後にある基礎となる論理的根拠は、タンパク質のペプチド骨格が、抗体と抗原の相互作用のような分子相互作用を促進するようにアミノ酸側鎖を配向させるよう主として存在するというものである。ペプチド模倣体は、天然分子と類似した分子相互作用を可能にすると予想される。これらの原理を用いて、本明細書に開示する標的化ペプチドの天然特性の多くを有するが、改変およびさらには改善された特徴を有する抗原を操作してもよい。
【0065】
B. 融合タンパク質
本発明の他の態様は、抗原としての融合タンパク質の使用に関する。これらの分子は一般に、N末端またはC末端において第二のポリペプチドまたはタンパク質のすべてまたは一部に連結された、関心対象のペプチドのすべてまたは実質的部分を有する。例えば、融合物は、異種宿主においてタンパク質の組み換え発現を可能にするための他の種によるリーダー配列を用い得る。別の有用な融合には、融合タンパク質の精製を容易にするための、抗体エピトープ等の免疫学的に活性のあるドメインの付加が含まれる。融合接合部またはその近傍に切断部位を含めることにより、精製後の外来ポリペプチドの除去が促進されることになる。他の有用な融合には、酵素の活性部位、グリコシル化ドメイン、細胞標的化シグナル、または膜貫通領域等の機能的ドメインの連結が含まれる。好ましい態様において、本態様の融合タンパク質は、免疫応答を誘発するための抗原タンパク質またはペプチドに連結されたペプチドを含む。
【0066】
他の態様において、融合タンパク質は、治療ペプチドと融合され得る、本発明の方法によって作製される抗体を含む。融合タンパク質に組み入れ得るタンパク質またはペプチドの例には、細胞増殖抑制タンパク質、殺細胞タンパク質、アポトーシス促進剤、抗血管新生剤、ホルモン、サイトカイン、増殖因子、ペプチド薬、抗体、Fab断片抗体、抗原、受容体タンパク質、酵素、レクチン、MHCタンパク質、細胞接着タンパク質、および結合タンパク質が含まれる。
【0067】
これらの例は制限することを意図しておらず、本発明の範囲内において、実質的に任意のタンパク質またはペプチドを、本発明において使用するための融合タンパク質に組み入れ得ることを意図している。融合タンパク質を作製する方法は当業者に周知である。そのようなタンパク質は、例えば二官能性架橋試薬を用いる化学的結合によって、完全な融合タンパク質の新規合成によって、または第1ペプチドをコードするDNA配列を第2のペプチドまたはタンパク質をコードするDNA配列に結合させ、その後無傷の融合タンパク質を発現させることによって、作製することができる。
【0068】
C. タンパク質の精製
特定の態様において、タンパク質(例えば抗体)またはペプチドは単離または精製してもよい。タンパク質精製技法は当業者に周知である。これらの技法は、1つのレベルでの、細胞、組織、または臓器のホモジナイゼーションならびにポリペプチド画分および非ポリペプチド画分への粗分画を含む。関心対象のタンパク質またはポリペプチドは、部分的または完全な精製(または均一になるまでの精製)を達成するために、クロマトグラフィー法および電気泳法を用いてさらに精製してもよい。純粋なペプチドの調製に特に適した分析法は、イオン交換クロマトグラフィー、ゲル排除クロマトグラフィー、HPLC(高速(high)液体クロマトグラフィー)、FPLC(AP Biotech)、ポリアクリルアミドゲル電気泳動、アフィニティクロマトグラフィー、免疫アフィニティクロマトグラフィー、および等電点電気泳動である。アフィニティクロマトグラフィーによる受容体タンパク質精製の例は、その全文が参照により本明細書に組み入れられる米国特許第5,206,347号に開示されている。ペプチドを精製するさらに効率のよい方法の1つは、高速(fast)液体クロマトグラフィー(FPLC)またはHPLCである。
【0069】
精製タンパク質またはペプチドは、タンパク質またはペプチドがその天然に得られる状態と比較して任意の程度に精製されている、他の成分から単離可能な組成物を指すことが意図される。したがって、単離もしくは精製タンパク質またはペプチドはまた、それが天然に存在し得る環境を脱したタンパク質またはペプチドを指す。一般に、「精製された」とは、様々な他の成分を除去するために分画に供されたタンパク質またはペプチド組成物を指すことになり、その組成物は発現された生物活性を実質的に保持している。「実質的に精製された」という用語を用いる場合、この名称は、タンパク質またはペプチドが、組成物におけるタンパク質の約50%、60%、約70%、約80%、約90%、約95%、またはそれ以上を構成するような、組成物の主成分を形成する組成物を指すことになる。
【0070】
タンパク質またはペプチドの精製の程度を定量する様々な方法が、本開示に照らして当業者に周知である。これらには、例えば、活性画分の比活性の測定、またはSDS/PAGE分析による画分中のポリペプチド量の評価が含まれる。画分の純度を評価する好ましい方法は、画分の比活性の算出、それと最初の抽出物の比活性との比較、および「精製倍率〜倍」によって評価されるその純度の算出である。活性量を示すために用いられる実際の単位は、当然のことながら、精製後に選択される特定のアッセイ法、および発現されたタンパク質またはペプチドが検出可能な活性を示すか否かに依存することになる。
【0071】
タンパク質精製に用いるために適した様々な技法は、当業者に周知である。これらには、例えば、硫酸アンモニウム、PEG、抗体等を用いた、または熱変性による沈殿、その後の遠心分離;イオン交換、ゲル濾過、逆相、ヒドロキシアパタイト、およびアフィニティクロマトグラフィー等のクロマトグラフィー段階;等電点電気泳動;ゲル電気泳動;ならびにこれらおよび他の技法の組み合わせが含まれる。当技術分野において周知であるように、種々の精製段階を行う順序は変更してもよく、または特定の段階を省略してもよく、それでもなお実質的に精製されたタンパク質またはペプチドを調製するための適した方法が得られると考えられる。
【0072】
タンパク質またはペプチドは常にその最も精製された状態で提供すべきであるという一般要件はない。実際に、実質的にあまり精製されていない産物は特定の態様において有用であると考えられる。部分精製は、より少ない精製段階を組み合わせて用いて、または同じ一般的精製計画の異なる形態を用いて達成してもよい。例えば、HPLC装置を用いて行われる陽イオン交換カラムクロマトグラフィーは一般に、低圧クロマトグラフィーシステムを用いる同じ技法よりも高い精製「倍率」が得られることが認識される。相対的な精製のより低い程度を示す方法は、タンパク質産物の全体的な回収、または発現されたタンパク質の活性の維持において長所を有する可能性がある。
【0073】
アフィニティクロマトグラフィーは、単離すべき物質と、それが特異的に結合し得る分子との特異的親和性に依存するクロマトグラフィー手順である。これは、受容体-リガンド型の相互作用である。カラム材料は、結合パートナーの1つを不溶性の充填剤に共有結合させることによって合成される。次いで、カラム材料は溶液から物質を特異的に吸収し得る。条件を結合が起こらない条件に変更すると(例えば、pH、イオン強度、温度等の変化)、溶出が起こる。充填剤は、それ自体が有意な程度に分子を吸収せず、また広範囲の化学的、物理的、および熱安定性を有する物質であるべきである。リガンドは、その結合特性に影響を及ぼさないように結合すべきである。リガンドは、比較的堅固な結合を提供すべきである。さらに、試料またはリガンドを破壊することなく、物質を溶出することが可能であるべきである。
【0074】
D. 合成ペプチド
いくつかの抗原ペプチドは、その大きさが比較的小さいために、従来の技法に従って溶液中または固相支持体上で合成することができる。様々な自動合成機が市販されており、既知の手順に従ってこれらを用いることができる。例えば、それぞれ参照により本明細書に組み入れられる、Stewart and Young, (1984);Tam et al., (1983);Merrifield, (1986);およびBarany and Merrifield (1979)を参照されたい。通常、約6アミノ酸から約35〜50アミノ酸までの短いペプチド配列は、そのような方法によって容易に合成することができる。または、本発明のペプチドをコードするヌクレオチド配列を発現ベクターに挿入し、適当な宿主細胞に形質転換するかまたはトランスフェクトし、発現に適した条件下で培養する組み換え型DNA技術を用いてもよい。
【0075】
実施例
以下の実施例は、本発明の好ましい態様を実証するために含めるものである。以下の実施例に開示した技術は、本発明の実施に際して良好に機能するように本発明者らにより見出された技術を表し、従ってその実施のための好ましい様式を構成すると見なし得ることは、当業者に認識されるべきである。しかし当業者は、本開示に照らして、開示した特定の態様において多くの変更を行うことができ、それらも本発明の精神および範囲から逸脱することなく同様または類似の結果をなおも得ることができることを認識すべきである。
【0076】
実施例1:不死化脾臓細胞の作製
A. 方法
脾細胞の単離
H-2Kb-tsA58マウス(Charles River Laboratories、マサチューセッツ州、ウィルミントン)の脾臓をダルベッコ変法イーグル培地(DMEM)中に回収した。2枚の冷却スライドグラスの間に挟んだ臓器の被膜に穏やかに圧力を加え、細胞を放出させた。塩化アンモニウムを用いて赤血球を溶解し、10% CPSRおよびハイブリドーマ促進補充物を添加したハイブリドーマ培地15 mlに脾細胞を再懸濁した。ナイロンメッシュで濾過して組織残屑を除去した。細胞を24ウェルプレートに分配し(2 x 106個/ウェル)、33℃で培養した。1週間おきに培地を交換した。
【0077】
他のプレートサイズを用いてもよいが、24ウェル(脾臓55 mlによる脾臓懸濁液のうち1 ml)が好ましいことが判明した。培地は定期的に(例えば、1週間おきに)交換し得る。3週間後、90%を超えるウェルにおいてクローンが認められた。
【0078】
マウスの免疫化
H-2Kb-tsA58マウス(Charles River Laboratories、マサチューセッツ州、ウィルミントン)を、1週間おきに12週間、繊維状fd-tetファージで免疫した。簡潔に説明すると、107形質導入単位(TU)/μl(全量 = 1 ml)を含むファージ調製物を、4通りの経路(静脈内、腹腔内、皮内、および皮下)により投与した。追加免疫する度にマウスから採血し、ELISAにより血清中の抗ファージ抗体価をモニターした。動物実験は、テキサス大学M. D. Anderson癌センターの施設内動物管理および使用委員会によって概説され認可される標準的な定型手順を含んだ。
【0079】
クローン抗体産生脾細胞のスクリーニングおよび作製
以前に記載されている通りに、繊維状ファージおよび組み換えファージキャプシドpIIIタンパク質に対するELISAを行った(Harlow and Lane, 1998)。ウシ血清アルブミン(BSA)、ハイブリドーマ培地のみ、免疫前血清、および二次抗体を陰性対照とした。免疫ポリクローナル血清および抗ファージ抗体を陽性対照とした。抗体は、培養上清から直接プレーティングした。モノクローナル株を取得するため、限界希釈により(96ウェルプレートのウェル当たり0.1ないし0.5個の細胞)陽性ウェルの細胞をサブクローニングした。2ヵ月後に現れたサブクローンを、ELISA法により完全なファージ粒子およびpIIIファージキャプシドタンパク質に対して試験した。ELISAリーダーで反応性をモニターした。
【0080】
ウェスタンブロット解析
繊維状fd-tetファージ(109 TU/レーン)を煮沸し、4〜20%勾配SDS-PAGE(Invitrogen Corp.、カリフォルニア州、カールズバッド)により分離し、ポリフッ化ビニリデン膜(PVDF;Bio-Rad Laboratoried, Inc.、カリフォルニア州、ハーキュリーズ)に電気的に転写した。膜を条片に分割し、リン酸緩衝食塩水(PBS)に溶解した5%ノンファットミルクにより室温(RT)で1時間ブロッキングし、その後0.1% Tween 20を含むPBS(PBS-T)で1回洗浄した。条片を免疫前血清(1:1000)、免疫後血清(1:1000)、抗fd-tetファージ(Sigma-Aldrich、ミズーリ州、セントルイス)、不死化細胞クローンから分泌された抗ファージIgGを含む上清、または細胞培養液のみと共にRTで2時間インキュベートした。3回洗浄した後、ペルオキシダーゼ結合二次抗体(Bio-Rad Laboratoried, Inc.、カリフォルニア州、ハーキュリーズ)を条片に添加し、RTで1時間インキュベートした。条片を3回洗浄し、高感度化学発光(ECL;Amersham Biosciences Corp.、ニュージャージー州、ピスカタウェイ)により反応性を検出した。
【0081】
ELISA
以下は、誘導したモノクローナル抗体の産生の存在または非存在を評価するために使用するアッセイ法の一例である。選択した抗原を、PBSに溶解してHigh Binding Assay Plate(Costar、例えば24、48、または96ウェルプレート)に固定化し得る(109粒子または5μg/ウェル)。対照ウェルは、PBSに溶解した2 mgウシ血清アルブミン(BSA)を用いて、4℃で一晩コーティングする。次いで、一次抗体または対照ポリクローナル種IgG(Sigma)を一連の濃度で室温で1時間インキュベートする。二次抗体(抗種Fabアルカリホスファターゼ結合、Sigma、3% BSA中1:3000)を添加し、1時間インキュベートする。p-ニトロフェニルリン酸(Sigma)によりELISAを発色させ、1〜4時間後に405 nmで(Reader 520、Organon Teknika)測定値を得ることができる。
【0082】
免疫沈降およびウェスタンブロット解析
関心対象の抗原を、プロテアーゼインヒビターの存在下で、50 mM Tris-HCl pH 7.6、1% NP-40、150 mM NaCl、および0.1 mM ZnOAc中に希釈し得る。タンパク質濃度は、ローリー法(Bio-Rad)により決定し得る。プロテインG-セファロース(Pharmacia)の存在下で、約5μg/mlのモノクローナル抗体濃度での問題のクローンの存在下で、タンパク質を免疫沈降し得る。免疫沈降されたタンパク質をSDS-PAGEにより分離し、ニトロセルロース膜に転写し、抗モノクローナル抗体(例えば、マウスまたはヒト)IgG HRP(Jackson Laboratories)でブロッティングし、高感度化学発光(Renaissance、NEN)により可視化し得る。または、関心対象のタンパク質をまずSDS-PAGEゲルにより分離し、次いでタンパク質をニトロセルロース紙に転写し、問題のモノクローナル抗体集団でプロービングし、抗モノクローナル抗体(例えば、マウスまたはヒト)IgG HRP(Jackson Laboratories)を用いて結果を可視化し、高感度化学発光(Renaissance、NEN)により可視化してもよい。
【0083】
不死化脾臓培養物のエクスビボ免疫化
脾臓から回収した細胞をプレーティングしてから約5日後、例えば0.5 x 1010〜1 x 1012 TU/2 x 106 細胞の濃度のファージ(fd-Tet)の抗原用量をウェルに供し得る。この段階を「初回刺激段階」または「最初のエクスビボ免疫化」と見なすことができる。初回刺激から18日および25日後(脾臓I)、初回刺激から14日および21日後(脾臓II)に、さらなる追加免疫を行い得る(例えば、同量のファージを添加し得る)。または、平行実験において、予めファージに曝露した(ファージを負荷した)樹状細胞(DC)と共にインキュベートすることにより、脾細胞を初回刺激し得る。本実施例では、その後の追加免疫は行わなかった。エクスビボ免疫化は、図7の時系列に示される通りに行うことができる。細胞全体でエクスビボ免疫する場合には、脾細胞を、上記のように、アポトーシスB16-F10細胞を負荷したDCまたはアポトーシス細胞のみと共にインキュベートした。
【0084】
B.結果
Fd-Tetに対するインビトロ免疫化の結果を図1Aおよび1Bに示す。ELISAプレートをpIII精製タンパク質(5μg/ウェル)またはFd-Tet(1011 TU/ウェル)で4℃で一晩コーティングした。示したウェルの馴化培地を、「最初の免疫化」から7日後および2回目の免疫化から4日後に回収した。対照として、Fd-Tetを接種した(3回注射)1匹の動物の免疫前および免疫後血清を用いた。プレートは、抗マウス全Ig HRP結合(ZYMED)およびOPDにより発色させた。
【0085】
H-2Kb-tsA58マウスを規定の抗原(繊維状ファージ)で免疫し、血清中の抗ファージ抗体価をELISAによりモニターした。抗ファージIgG価は、最終追加免疫から7日後に高レベル(免疫前血清の<0.1と比較して、1:3,200希釈においてOD450>3)に達した(図8)。段階希釈によりさらに試験したところ、ファージに対するIgG価は平均約1:6,400であることが明らかになった。さらに、pIIIタンパク質(10μg/ウェルでコーティング)に対する血清価は、平均約1:1,600であった(データは示さず)。マウスの脾臓を摘出し、DMEM中で細胞懸濁液を調製した。細胞を96ウェルプレートに分配し、33℃で培養した。2〜3週間の間に、培地を3回完全に交換した。3週間後、>90%のウェルにおいてクローンが認められた。抗体の反応性を検出するため、ファージ粒子をコーティングしたマイクロタイターウェルプレートにおいて、上清を用いてELISAを行った。58%に到達するクローンが、ファージに対するIgG反応性に関して陽性であった。
【0086】
脾細胞は低細胞密度であっても健常であり、大半のウェルの上清において強力な反応性レベルをもたらしたことが認められた。モノクローナル株を取得するため、陽性ウェルの細胞を限界希釈によりクローニングしたが、ほとんどのクローンは陽性のままであった。モノクローナル株のサブクローニングを2回繰り返したところ、得られたクローンの実質的にすべてが陽性であり、作製された株が実際に単一クローンに由来する強力な証拠が提供された。
【0087】
4〜8週間後に出現したクローンを、ELISAにより、ファージ粒子に対しておよび微量なファージキャプシドタンパク質(pIII)に対して試験した。この場合も同様に、96ウェルプレートから24ウェルプレートに拡大した場合にも、または凍結および融解後にも、ほとんどの陽性クローンが反応を維持した(図9)。無傷のファージに対して強い反応性が認められ、いくつかのクローンは組み換えpIII融合タンパク質とも反応した(図10)。すべての段階の元のプレートおよびクローンも、最長で3ヶ月間培養において保存した。
【0088】
ELISAによりスクリーニングされた抗体がウェスタンブロットにおいて特定のタンパク質を認識し得るかどうかを判定するため、H-2Kb-tsA58トランスジェニックマウス由来不死化脾細胞による上清を、SDS-PAGEにより繊維状ファージ調製物を分離することにより、pIIIおよびpVIIIファージキャプシドタンパク質に対して評価した。ファージタンパク質を含むPVDF膜を、免疫前血清、免疫後血清、市販の抗ファージ、または不死化脾細胞クローンから分泌された抗ファージIgGを含む上清と共にインキュベートした。細胞培養液のみをさらなる陰性対象として用いた。pIIIおよびpVIIIファージキャプシドタンパク質に相当するバンドと特異的に反応する抗体が、H-2Kb-tsA58トランスジェニックマウス由来不死化脾細胞の上清中に検出された(図11)。この結果から、本明細書に記載の方法によって産生される抗体が、免疫ブロッティング(図11)または細胞表面抗原の蛍光活性化セルソーティング(FACS)(データは示さず)等の用途においても(データは示さず)使用できることが実証される。
【0089】
H-2Kb-tsA58トランスジェニックマウスの脾細胞は、規定の抗原に対する高力価のIgGを産生し得ると考えられる。この細胞培養系により、モノクローナル抗体の信頼性のあるかつ再現性のある供給源が確実になり、ハイブリドーマ作製の必要性が排除された。
【0090】
本発明のいくつかの利点は、さらに解説する価値がある。第一に、抗体合成細胞は、何ヶ月間も、場合によっては何年間も培養において安定であり、抗体産生の減少も不活性化も起こすことなく、限界希釈クローニング法および凍結融解法を許容する。ポリクローナル集団が凍結されており、所与のIgGを分泌する生存クローンが回収される(データは示さず)。
【0091】
第二に、不死化クローンは33℃でゆっくりと増殖し、遺伝的に安定であり、適時に多数の試料を処理することができる(および、論理的には「稀な」抗体を取得できる可能性がある)。H-2Kb-tsA58由来脾細胞により、長期培養したクローンを含むウェルからの特定ポリクローナルIgGの大量産生が可能になる。対照的に、ハイブリドーマは、クローンのランダムな混合物において、非分泌クローンが一般に分泌クローンを上回ることから問題がある。以前のデータから、H-2Kb-tsA58トランスジェニックマウスに由来するIgG分泌脾細胞と非分泌脾細胞の増殖速度は同様であることが示唆される(未発表知見)。
【0092】
第三に、インビトロ免疫化は致死脾細胞またはハイブリドーマでは効果がないのに対して、インビトロ免疫化は抗体産生を促進する他の脾臓由来不死化細胞種--マクロファージおよび線維芽細胞等--の存在により増強される。腹水産生における最近の制限を考えると、この新たな技術はモノクローナル抗体のエクスビボでの簡便な大量製造を支持する。
【0093】
第四に、H-2Kb-tsA58トランスジェニックマウスをヒト抗体産生のための遺伝子相補体を発現するマウスと交配することにより、ヒトモノクローナル抗体の作製もまた可能になる。本明細書に記載する戦略はハイブリドーマ作製に取って代わり、甚大かつ直接的な科学的および医学的利点によりマウスおよびヒトモノクローナル抗体作製を合理化し得る。
【0094】
fd-Tetを用いたエクスビボ免疫化の結果を図1Aおよび1Bに示す。ELISAプレートを、pIIIファージキャプシドタンパク質(5μg/ウェル)または無傷のファージ粒子(1011 TU/ウェル)で4℃で一晩コーティングした。異なる実験条件下で培養した細胞の馴化培地を、初回刺激してから11日目および22日目(脾臓I)、19日目および26日目(脾臓II)に回収した。ファージ反応性の陽性対照および陰性対照として、免疫前および免疫後抗ファージポリクローナル血清を使用した。Fd-Tetファージを12ヶ月間1週間おきに免疫したマウスに由来する血清を回収した。プレートは、二次抗マウスIg-ペルオキシダーゼ(ZYMED)で発現させ、TMB(Calbiochem)で発色させた。吸光度をELISAリーダーによりモニターした。
【0095】
不死化脾細胞の形態
免疫した動物による不死化脾細胞の一般的形態を、図3A、3B、および3Cに示す。培養してから2ヵ月後に写真を撮影した。濾胞性樹状細胞、形質細胞(抗体産生B細胞)のクローン、マクロファージ、および未同定の上皮様細胞(おそらく細網上皮細胞)を観察することができる。
【0096】
免疫したマウスの脾細胞の形態
免疫したマウスに由来する脾細胞を、培養してから2ヵ月に視覚的に分析した。例えば濾胞性樹状細胞、形質細胞(抗体産生B細胞)のクローン、マクロファージ、および未同定の上皮様細胞(おそらく細網上皮細胞)といったいくつかの異なる細胞が認められた。
【0097】
脾臓および骨髄細胞培養物は抗原提示に対して良好に機能することが実証されているが、データから、リンパ節もまた抗原提示に対して良好に機能することが示される(データは示さず)。
【0098】
実施例2:骨髄(BM)からの不死化樹状細胞(DC)の作製
平行した研究において、予めファージ(ファージを負荷した)または他の抗原に曝露した樹状細胞(DC)と共にインキュベートすることにより、脾細胞を初回刺激した。
【0099】
A. 方法
骨髄細胞の収集および単離
髄腔に27 G針を挿入することにより、H-2Kb-tsA58マウスの大腿骨、頸骨の長骨、および骨端から骨髄(BM)を収集した。塩化アンモニウムを用いて赤血球を溶解した。ペトリ皿に単一細胞懸濁液をプレーティングした。細胞を33℃で培養した。各プレートに、マウス(mu)GM-CSF(10 ng/ml)およびr-muIL-4(10 ng/ml)を補充した10% FBSを含む7 mlのRPMI 1640を添加した。3日後、完全培地およびサイトカイン3 mlをプレートに補充した。5日培養後、約50%の集団が未成熟樹状細胞によって表された。ゆるく接着した増殖DC凝集物を回収し、再度プレーティングした。未成熟DCを繊維状ファージ(fd-tet)(1.5 x 1012 TU/1 x 106未成熟DC)またはアポトーシスB16-F10細胞(2:1、DC/B16)と共にインキュベートした。インキュベートは、TNF-α(DCの成熟を誘導することが周知である因子)の存在下または非存在下において48時間継続した。
【0100】
アポトーシスB16-F10細胞の調製
約1 x 106細胞/mlのB16-F10細胞の懸濁液にUV照射することにより(UV Stratalinker、Stratagene 4ジュール/cm2で20分間)、B16-F10細胞においてアポトーシスを誘導した。24時間後、67%の細胞が、細胞がアポトーシスを起こしたことを示すアネキシン陽性であった。
【0101】
T細胞またはB細胞応答の誘導
T細胞および/またはB細胞が媒介する応答を誘導するため、DC細胞を脾臓由来細胞と混合し得る。次いで、T細胞および/またはB細胞媒介性応答を誘導するため、異なる実験条件下(抗原、例えばファージまたはアポトーシス細胞を負荷するか否か)のDC細胞を、単離した脾臓由来細胞(SDC)と共に5:1、SDC/DCの比率でインキュベートした。
【0102】
B. 結果
樹状細胞
インビトロでの樹状細胞の標準的な挙動は、それらの特性および生物学的因子に対する応答を踏まえると、条件的不死化DC細胞において変化する。同じウェル内の樹状細胞によって構成的に提示される抗原に応答するB細胞に対する選択圧が存在し得る。エクスビボ免疫化もまた、構成的な抗原提示を提供するために本発明の種々の態様に含まれ得る。
【0103】
「不死化」未成熟DCにおける特定の細胞表面抗原の存在は、FACS解析により評価することができる。プレーティングしてから5日後、細胞をいくつかの抗体、抗CD80、抗CD86、および抗H2k(BD)抗体を用いて表面抗原について評価した(Weigel et al., 2002)(図2)。骨髄からサイトカインを用いて分化させたDCの特徴的形態を図2に示す。
【0104】
幹細胞
不死化細胞の別の用途は、罹患患者の幹細胞集団の置換において考えられる。一例において、マウス等の動物の骨髄を照射し、それをImmortoMouse等の不死化動物の骨髄または幹細胞の任意の供給源と置換し得る。結果を解析するため、正常動物を屠殺し、immortomouseに由来する、不死である可能性のある任意の幹細胞を同定することができる。これにより、幹細胞がホーミングする特定臓器の同定が可能になる。また、ImmortoMouseを(すべての細胞においてLac Zを発現する)Rosaマウスと交配することは、増殖ばかりでなく、レシピエントにおける追跡が可能になるようImmortoMouseに由来する細胞をタグ化するのに役立つことになる。
【0105】
ImmortoMouseの脳幹細胞を単離し、特徴づけを行った。図5の右下パネル中の丸い細胞として増殖している最も小さい細胞は、脾臓幹細胞と考えられる。したがって、不死化幹細胞の単離およびこの集団の導入は、将来、癌患者等の障害を起こした患者の生存率を改善するために使用できる可能性がある。
【0106】
実施例3:無傷の細胞に対するモノクローナル抗体の作製
A. 方法
免疫化
H-2Kb-tsA58マウス(Charles River Laboratories、マサチューセッツ州、ウィルミントン)を、1週間おきに3週間、5 x 106個の間葉系幹細胞(MSC)で免疫した。ELISAおよびFACSにより、血清中の抗MSC抗体価を評価した。動物実験は、テキサス大学M. D. Anderson癌センターの施設内動物管理および使用委員会によって概説され認可される標準的な定型手順を含んだ
【0107】
不死化脾細胞の導出
マウスを屠殺し、それらの脾臓をダルベッコ変法イーグル培地(DMEM)中に回収した。2枚の冷却スライドグラスの間に挟んだ臓器の被膜に穏やかに圧力を加え、細胞を放出させた。次いで、10% CPSRおよびハイブリドーマ促進補充物を添加したハイブリドーマ培地15 mlに脾細胞を再懸濁した。ナイロンメッシュで濾過して組織残屑を除去した。細胞を培地55 mlに再懸濁し、異なる密度で6ウェル、24ウェル、および96ウェルプレートに分配した。プレートを33℃でインキュベートした。2〜3週間の間に、培地を3回完全に交換した。大部分のウェルにおいてクローンが認められた。
【0108】
プレーティングの図式は以下の通りであった:脾臓を55 mlに再懸濁した;6ウェルプレート1枚および24ウェルプレート1枚に播種し、残りの細胞を約280 mlに希釈し、20 x 96ウェル、5 x 24ウェル、および3 x 6ウェルに分配した。本発明者らは、研究を開始して3ヶ月経過後に、ELISAにより、96ウェルプレートによる59クローン(83%が+であった)、および24ウェルによる61クローン(82%が+であった)を評価した。55 mlから24ウェルプレートへの播種がこの系において最も有力な培養条件であることが明らかである。数ヵ月後にも細胞は健常であるように見え、すべてのウェルが陽性である。そのようなウェルからのサブクローニングは成功を収め得る。
【0109】
実施例4:カポジ肉腫(KS)細胞および間葉系幹細胞(MSC)に対する抗腫瘍反応性
A. 方法
血清の回収
免疫手順を開始する前、および2週間または3週間後に(免疫後血清1:2回注射後、および免疫後血清2:3回注射後)マウスから採血した。下記の通りに、マルチウェルプレートにプレーティングし、PAF 2%で固定したKS細胞およびMSC細胞に対して、血清の特異的反応性をアッセイした(図4A)。
【0110】
ELISAアッセイ
血清抗腫瘍反応性をELISAにより測定した。3 x 104個の対数増殖期KS(図4Aおよび4B)または1.5 x 104個のMSC細胞/ウェル(図4C)を96ウェルプレートにプレーティングした。37℃で一晩インキュベートした後、細胞をPBSで1回洗浄し、2% PFA中で室温で10分間固定し、PBSで1回リンスした。プレートは使用時まで-20℃で保存した。PBS-2% BSAを用いてRTで1時間ブロッキングした後、PBS-0.5% BSA中の血清希釈物1/500または1/1000(図4A)を二つ組で添加し、4℃で一晩インキュベートした。抗体は培養上清から直接プレーティングした(図4Bおよび4C)。翌日、プレートをPBS-0.5% BSA、0.01% Tween20で3回、およびPBSのみで1回洗浄した。PBS-0.5% BSAに溶解したウサギ抗マウスIg西洋ワサビペルオキシダーゼ(HRP)結合(ZYMED Laboratories Inc.、米国、カリフォルニア州)の1/2000希釈物100μlをプレートに添加した。振盪させながら室温で90分インキュベートした後、プレートを上記のように3回洗浄した。オルト-フェニレンジアミン(OPD)(Sigma-FAST、Sigma-Aldrich、米国、セントルイス Fast-tab)で反応を発色させ、50μl/ウェルの3 M硫酸で反応を停止した。マイクロプレートリーダーで450 nmの吸光度を測定した。100個を超えるクローンを評価した。
【0111】
陽性細胞のサブクローニング
モノクローナル株を取得するため、限界希釈により(96ウェルプレートのウェル当たり0.1ないし0.5個の細胞)陽性ウェルの細胞をサブクローニングした。
【0112】
実施例5:不死化胸腺細胞の作製
14日目にH-2Kb-tsA58細胞から胸腺を摘出し、ダルベッコ変法イーグル培地(DMEM)中に回収した。2枚の冷却スライドグラスの間に挟んだ臓器の被膜に穏やかに圧力を加え、細胞を放出させた。次いで、10% CPSRおよびハイブリドーマ促進補充物を添加したハイブリドーマ培地15 mlに胸腺細胞を再懸濁した。70μmナイロンメッシュで濾過して組織残屑を除去した。細胞を24ウェルプレートに分配し、33℃で培養した。
【0113】
残りの細胞を、抗マウス抗体: CD3、B220、H2k、CD86、CD80、CD11c、および(上皮細網細胞を認識する)MAdCAM-1を用いてFACSにより解析した。FACSの結果から、以下の割合の陽性細胞が示された(図6Aおよび6B)、それぞれCD3+:75%およびH2k+:20%。CD45RA(B220):3.5%(データは示さず)CD86+:4%(データは示さず)CD80+:0%(データは示さず)CD11c+:0%(データは示さず)MAdCAM-1+:0%(データは示さず)。
【0114】
本明細書において開示および主張する方法、組成物、および装置はすべて、本開示に照らして過度の実験を行うことなく作製し使用することができる。本主題が主張する概念、精神、および範囲から逸脱することなく、本明細書に記載した方法、組成物、および装置に変更が適用され得ることは、当業者には明らかであると考えられる。より具体的には、同一または類似の結果が得られれば、化学的および生理的に関連する特定の物質を本明細書に記載の物質と置換し得ることは明らかであると考えられる。当業者に明らかであるそのような類似の置換物および修飾物はすべて、主張する本主題の精神、範囲、および概念の範囲内であると見なされる。
【0115】
参考文献
以下の参考文献は、本明細書に記載したものに例示的な手法またはその他の詳細を提供する程度に、参照により特別に本明細書に組み込まれる。
【図面の簡単な説明】
【0116】
添付の図面は本明細書の一部を形成し、本発明の特定の局面をさらに示すために含める。本明細書に示す特定の態様の詳細な説明と組み合わせてこれらの図面の1つまたは複数を参照することにより、本発明をより良く理解することができる。
【図1】pIII精製タンパク質(5μg/ウェル)(図1A)またはファージ粒子Fd-Tet(1011 TU/ウェル)(図1B)に対するインビトロ抗体産生不死化脾細胞集団を作製する例示的な方法を示す。
【図2】樹状細胞を不死化する例示的な方法、および抗マウス抗体-CD80、-CD86、および-H2kを用いた不死化樹状細胞のFACS解析を示す。
【図3】未成熟不死化骨髄由来細胞(樹状細胞、DC)の形態の例示的な画像を示す。
【図4】抗腫瘍抗体を作製しアッセイする例示的な方法を示す。図4Aおよび図4Bは3 x 104個の対数増殖期KS細胞のELISAを示し、図4Cは1.5 x 104個のMSCのELISAを示す。抗体は培養上清から直接プレーティングした。図4Bおよび4Cでは、陽性対照としてポリクローナル血清を使用した。OPDにより反応を発色させ、450 nmで吸光度を測定した。
【図5】培養してから2ヵ月後の、免疫したマウスによる脾細胞培養物の例示的な形態を示す。濾胞性樹状細胞、形質細胞(抗体産生B細胞)のクローン、マクロファージ、および未同定の上皮様細胞(おそらく細網上皮細胞)が認められる。
【図6】胸腺細胞の不死化集団を作製し、胸腺細胞をスクリーニングする例示的な方法を示す。2つの細胞表面タンパク質集団のFACS解析を、CD3染色を示す図6AおよびH2K染色を示す図6Bにより示す。
【図7】培養において不死化細胞集団に抗原を導入するための例示的時間的経過を示す。
【図8】不死化脾臓細胞から産生された抗体、特に繊維状ファージ(fd-tet)810または組み換えファージキャプシドpIII 820に曝露したH-2Kb-tsA58トランスジェニックマウス由来不死化脾細胞から得られた抗体の例示的なスクリーニングおよび確認の結果を示す。
【図9】不死化脾臓細胞から産生された抗体、特に繊維状ファージ(fd-tet)910に曝露したH-2Kb-tsA58トランスジェニックマウス由来不死化脾細胞から得られた抗体の例示的なスクリーニングおよび確認の結果を示す。クローン1〜3は凍結/融解を行ったクローンに相当する。クローン4〜9は、96ウェルプレートから24ウェルプレートに拡大して6週間培養した異なるウェルに相当する;クローン10は、陰性対照としての培地のみを示す。含めた他の対照は免疫前および免疫後血清であった。
【図10】不死化脾臓細胞から産生された抗体、特に繊維状ファージ(fd-tet)1010、組み換えファージキャプシドpIII 1020、またはウシ血清アルブミン(BSA)1030に曝露したH-2Kb-tsA58トランスジェニックマウス由来不死化脾細胞から得られた抗体の例示的なスクリーニングおよび確認の結果を示す。クローン1、培地、陰性対照;クローン2、免疫前血清;クローン3〜7は、培養してから8週間後の異なるモノクローナル株の上清に相当する。バーは平均値に相当する。平均値の標準誤差は平均値の1%未満であった。
【図11】ファージタンパク質に対する抗体を産生するH-2Kb-tsA58トランスジェニックマウス由来不死化脾細胞による上清の反応性のウェスタンブロット解析を示す。反応性は、図のように、免疫前血清1110、免疫後血清1120、抗ファージ抗体1130、または不死化脾細胞クローンから分泌された抗ファージIgGを含む上清1140と共にインキュベートした後に評価した。pIIIファージキャプシドタンパク質1160およびpVIIIファージキャプシドタンパク質1170(矢印)に対して特異的に反応する抗体が、H-2Kb-tsA58トランスジェニックマウス由来不死化脾細胞の上清中に検出された。
【特許請求の範囲】
【請求項1】
以下の段階を含む、所望の抗原に対する抗体を産生する抗体産生細胞を作製するための方法:
(a) ハイブリドーマを形成することなく不死化され得る抗体産生細胞を、抗原に対する抗体を産生するよう細胞を誘導するのに効率的な様式で所望の抗原に接触させる段階;および
(b) 抗体産生細胞を不死化する段階。
【請求項2】
抗体産生細胞が、条件的に機能するかまたは条件的に発現される形質転換発癌遺伝子を含み、該形質転換発癌遺伝子を誘導することにより不死化がもたらされる、請求項1記載の方法。
【請求項3】
条件的に機能する形質転換発癌遺伝子が温度感受性SV40大型腫瘍抗原(tsSV40Tag)である、請求項2記載の方法。
【請求項4】
tsSV40TagがA58S-SV40Tagである、請求項3記載の方法。
【請求項5】
抗体産生細胞を25℃〜35℃で培養することにより形質転換発癌遺伝子が誘導される、請求項4記載の方法。
【請求項6】
抗体産生細胞を30℃〜35℃で培養する、請求項5記載の方法。
【請求項7】
抗体産生細胞を33℃で培養する、請求項6記載の方法。
【請求項8】
抗体産生細胞をハイブリドーマ培地で培養する、請求項5記載の方法。
【請求項9】
抗体産生細胞の抗体産生能を評価する段階をさらに含む、請求項5記載の方法。
【請求項10】
評価する段階が、所望の抗原に対する抗体結合をアッセイする段階を含む、請求項9記載の方法。
【請求項11】
モノクローナル抗体を産生するモノクローナル集団を作製するために、単一細胞を選択して培養する、請求項1記載の方法。
【請求項12】
希釈クローニングにより単一細胞を選択する、請求項11記載の方法。
【請求項13】
ポリクローナル抗体を産生するポリクローナル集団を作製するために、複数の細胞を選択して培養する、請求項1記載の方法。
【請求項14】
抗体産生細胞が脾臓細胞を含む、請求項1記載の方法。
【請求項15】
抗原が、ペプチド、タンパク質、糖タンパク質、リポタンパク質、炭水化物、ウイルス、細菌、病原微生物、組織、細胞全体、生検組織、患者由来の細胞、組織抽出物、新鮮なまたは培養した組織、アポトーシス細胞、細胞成分、細胞および組織に由来する膜、細胞質、および核画分、精製タンパク質、部分精製タンパク質、レーザー捕獲した組織、パラフィン包埋し固定した組織からなる群より選択される、請求項1記載の方法。
【請求項16】
組織が、対象由来の腫瘍組織を含む、請求項15記載の方法。
【請求項17】
抗体産生細胞が、ハイブリドーマを形成することなく不死化され得る抗体産生細胞を有するトランスジェニックマウスから得られる、請求項1記載の方法。
【請求項18】
トランスジェニックマウスが、ヒト抗体を産生するための遺伝子相補体を含む、請求項17記載の方法。
【請求項19】
抗体産生細胞がマウスにおいて構成され、かつ選択された抗原が、抗体を産生するよう抗体産生細胞を誘導するのに効率的な様式でマウスに投与される、請求項1記載の方法。
【請求項20】
所望の抗原に接触させる段階が、抗体産生細胞を抗原提示細胞と共培養する段階を含む、請求項1記載の方法。
【請求項21】
抗原提示細胞が樹状細胞である、請求項20記載の方法。
【請求項22】
抗体産生細胞が、ヒト抗体を産生するための遺伝子相補体を含む、請求項1記載の方法。
【請求項23】
抗体産生細胞によって産生された抗体を精製する段階をさらに含む、請求項1記載の方法。
【請求項24】
治療抗体を必要とする対象に抗体を投与する段階をさらに含む、請求項23記載の方法。
【請求項25】
以下の段階を含む、所望の抗原に対するヒト抗体を産生する抗体産生細胞を作製するための方法:
(a) 形質転換発癌遺伝子を条件的に発現するかまたは条件的に機能する形質転換発癌遺伝子を発現し、かつヒト抗体を産生するための遺伝子相補体を発現する抗体産生細胞を取得する段階であって、該抗体産生細胞の不死化は、該形質転換発癌遺伝子の発現または機能を誘導することによってもたらされる、段階;および
(b) ハイブリドーマを形成することなく不死化され得る抗体産生細胞を、抗原に対するヒト抗体を産生するよう細胞を誘導するのに効率的な様式で所望の抗原に接触させる段階;および
(c) 抗体産生細胞を不死化する段階。
【請求項26】
条件的に機能する形質転換発癌遺伝子が温度感受性SV40大型腫瘍抗原(tsSV40Tag)である、請求項25記載の方法。
【請求項27】
tsSV40TagがA58S-SV40Tagである、請求項26記載の方法。
【請求項28】
抗体産生細胞を25℃〜35℃で培養することにより形質転換発癌遺伝子が誘導される、請求項27記載の方法。
【請求項29】
抗体産生細胞を30℃〜35℃で培養する、請求項28記載の方法。
【請求項30】
抗体産生細胞を33℃で培養する、請求項29記載の方法。
【請求項31】
モノクローナル抗体を産生するモノクローナル集団を作製するために、単一細胞を選択して培養する、請求項25記載の方法。
【請求項32】
ポリクローナル抗体を産生するポリクローナル集団を作製するために、複数の細胞を選択して培養する、請求項25記載の方法。
【請求項33】
抗体産生細胞が脾臓細胞を含む、請求項25記載の方法。
【請求項34】
抗原が、ペプチド、タンパク質、糖タンパク質、リポタンパク質、炭水化物、ウイルス、細菌、病原微生物、組織、細胞全体、生検組織、患者由来の細胞、組織抽出物、新鮮なまたは培養した組織、アポトーシス細胞、細胞成分、細胞および組織に由来する膜、細胞質、および核画分、精製タンパク質、部分精製タンパク質、レーザー捕獲した組織、パラフィン包埋し固定した組織からなる群より選択される、請求項25記載の方法。
【請求項35】
組織が、対象由来の腫瘍組織を含む、請求項34記載の方法。
【請求項36】
抗体産生細胞が、ハイブリドーマを形成することなく不死化され得る抗体産生細胞を有するトランスジェニックマウスから得られる、請求項25記載の方法。
【請求項37】
抗体産生細胞がマウスにおいて構成され、かつ選択された抗原が、抗体を産生するよう抗体産生細胞を誘導するのに効率的な様式でマウスに投与される、請求項25記載の方法。
【請求項38】
抗体産生細胞によって産生された抗体を精製する段階をさらに含む、請求項25記載の方法。
【請求項39】
治療抗体を必要とする対象に抗体を投与する段階をさらに含む、請求項38記載の方法。
【請求項1】
以下の段階を含む、所望の抗原に対する抗体を産生する抗体産生細胞を作製するための方法:
(a) ハイブリドーマを形成することなく不死化され得る抗体産生細胞を、抗原に対する抗体を産生するよう細胞を誘導するのに効率的な様式で所望の抗原に接触させる段階;および
(b) 抗体産生細胞を不死化する段階。
【請求項2】
抗体産生細胞が、条件的に機能するかまたは条件的に発現される形質転換発癌遺伝子を含み、該形質転換発癌遺伝子を誘導することにより不死化がもたらされる、請求項1記載の方法。
【請求項3】
条件的に機能する形質転換発癌遺伝子が温度感受性SV40大型腫瘍抗原(tsSV40Tag)である、請求項2記載の方法。
【請求項4】
tsSV40TagがA58S-SV40Tagである、請求項3記載の方法。
【請求項5】
抗体産生細胞を25℃〜35℃で培養することにより形質転換発癌遺伝子が誘導される、請求項4記載の方法。
【請求項6】
抗体産生細胞を30℃〜35℃で培養する、請求項5記載の方法。
【請求項7】
抗体産生細胞を33℃で培養する、請求項6記載の方法。
【請求項8】
抗体産生細胞をハイブリドーマ培地で培養する、請求項5記載の方法。
【請求項9】
抗体産生細胞の抗体産生能を評価する段階をさらに含む、請求項5記載の方法。
【請求項10】
評価する段階が、所望の抗原に対する抗体結合をアッセイする段階を含む、請求項9記載の方法。
【請求項11】
モノクローナル抗体を産生するモノクローナル集団を作製するために、単一細胞を選択して培養する、請求項1記載の方法。
【請求項12】
希釈クローニングにより単一細胞を選択する、請求項11記載の方法。
【請求項13】
ポリクローナル抗体を産生するポリクローナル集団を作製するために、複数の細胞を選択して培養する、請求項1記載の方法。
【請求項14】
抗体産生細胞が脾臓細胞を含む、請求項1記載の方法。
【請求項15】
抗原が、ペプチド、タンパク質、糖タンパク質、リポタンパク質、炭水化物、ウイルス、細菌、病原微生物、組織、細胞全体、生検組織、患者由来の細胞、組織抽出物、新鮮なまたは培養した組織、アポトーシス細胞、細胞成分、細胞および組織に由来する膜、細胞質、および核画分、精製タンパク質、部分精製タンパク質、レーザー捕獲した組織、パラフィン包埋し固定した組織からなる群より選択される、請求項1記載の方法。
【請求項16】
組織が、対象由来の腫瘍組織を含む、請求項15記載の方法。
【請求項17】
抗体産生細胞が、ハイブリドーマを形成することなく不死化され得る抗体産生細胞を有するトランスジェニックマウスから得られる、請求項1記載の方法。
【請求項18】
トランスジェニックマウスが、ヒト抗体を産生するための遺伝子相補体を含む、請求項17記載の方法。
【請求項19】
抗体産生細胞がマウスにおいて構成され、かつ選択された抗原が、抗体を産生するよう抗体産生細胞を誘導するのに効率的な様式でマウスに投与される、請求項1記載の方法。
【請求項20】
所望の抗原に接触させる段階が、抗体産生細胞を抗原提示細胞と共培養する段階を含む、請求項1記載の方法。
【請求項21】
抗原提示細胞が樹状細胞である、請求項20記載の方法。
【請求項22】
抗体産生細胞が、ヒト抗体を産生するための遺伝子相補体を含む、請求項1記載の方法。
【請求項23】
抗体産生細胞によって産生された抗体を精製する段階をさらに含む、請求項1記載の方法。
【請求項24】
治療抗体を必要とする対象に抗体を投与する段階をさらに含む、請求項23記載の方法。
【請求項25】
以下の段階を含む、所望の抗原に対するヒト抗体を産生する抗体産生細胞を作製するための方法:
(a) 形質転換発癌遺伝子を条件的に発現するかまたは条件的に機能する形質転換発癌遺伝子を発現し、かつヒト抗体を産生するための遺伝子相補体を発現する抗体産生細胞を取得する段階であって、該抗体産生細胞の不死化は、該形質転換発癌遺伝子の発現または機能を誘導することによってもたらされる、段階;および
(b) ハイブリドーマを形成することなく不死化され得る抗体産生細胞を、抗原に対するヒト抗体を産生するよう細胞を誘導するのに効率的な様式で所望の抗原に接触させる段階;および
(c) 抗体産生細胞を不死化する段階。
【請求項26】
条件的に機能する形質転換発癌遺伝子が温度感受性SV40大型腫瘍抗原(tsSV40Tag)である、請求項25記載の方法。
【請求項27】
tsSV40TagがA58S-SV40Tagである、請求項26記載の方法。
【請求項28】
抗体産生細胞を25℃〜35℃で培養することにより形質転換発癌遺伝子が誘導される、請求項27記載の方法。
【請求項29】
抗体産生細胞を30℃〜35℃で培養する、請求項28記載の方法。
【請求項30】
抗体産生細胞を33℃で培養する、請求項29記載の方法。
【請求項31】
モノクローナル抗体を産生するモノクローナル集団を作製するために、単一細胞を選択して培養する、請求項25記載の方法。
【請求項32】
ポリクローナル抗体を産生するポリクローナル集団を作製するために、複数の細胞を選択して培養する、請求項25記載の方法。
【請求項33】
抗体産生細胞が脾臓細胞を含む、請求項25記載の方法。
【請求項34】
抗原が、ペプチド、タンパク質、糖タンパク質、リポタンパク質、炭水化物、ウイルス、細菌、病原微生物、組織、細胞全体、生検組織、患者由来の細胞、組織抽出物、新鮮なまたは培養した組織、アポトーシス細胞、細胞成分、細胞および組織に由来する膜、細胞質、および核画分、精製タンパク質、部分精製タンパク質、レーザー捕獲した組織、パラフィン包埋し固定した組織からなる群より選択される、請求項25記載の方法。
【請求項35】
組織が、対象由来の腫瘍組織を含む、請求項34記載の方法。
【請求項36】
抗体産生細胞が、ハイブリドーマを形成することなく不死化され得る抗体産生細胞を有するトランスジェニックマウスから得られる、請求項25記載の方法。
【請求項37】
抗体産生細胞がマウスにおいて構成され、かつ選択された抗原が、抗体を産生するよう抗体産生細胞を誘導するのに効率的な様式でマウスに投与される、請求項25記載の方法。
【請求項38】
抗体産生細胞によって産生された抗体を精製する段階をさらに含む、請求項25記載の方法。
【請求項39】
治療抗体を必要とする対象に抗体を投与する段階をさらに含む、請求項38記載の方法。
【図2】

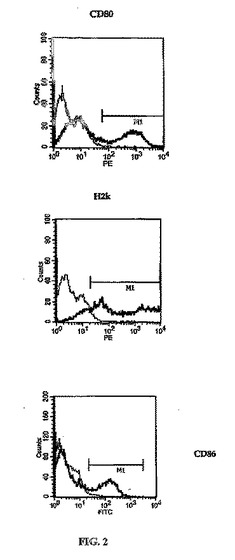

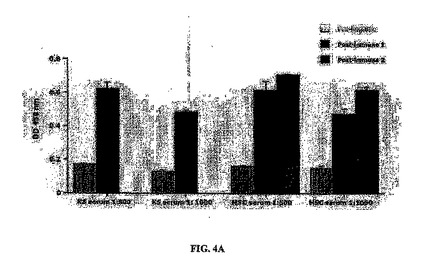
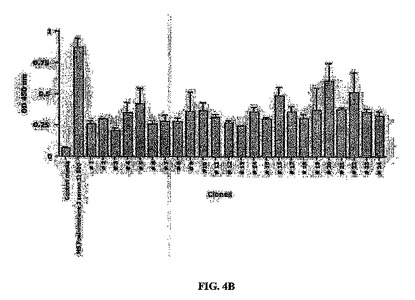

【図5】


【図7】
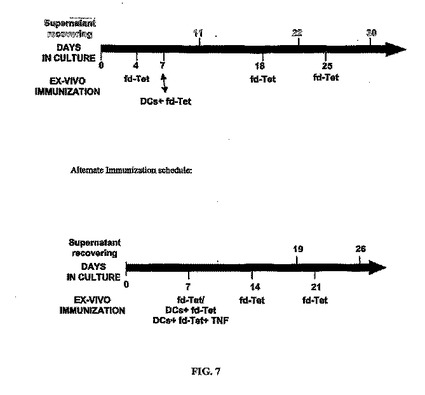
【図8】

【図9】

【図10】
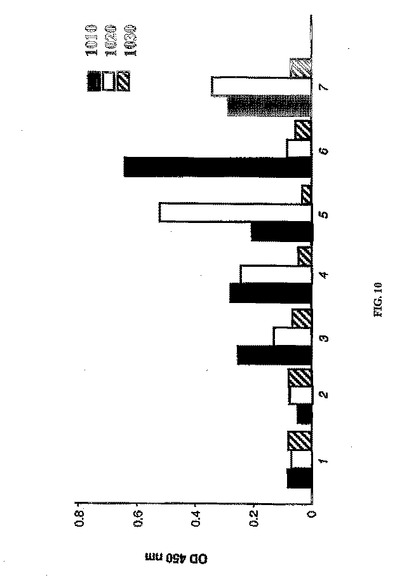
【図11】
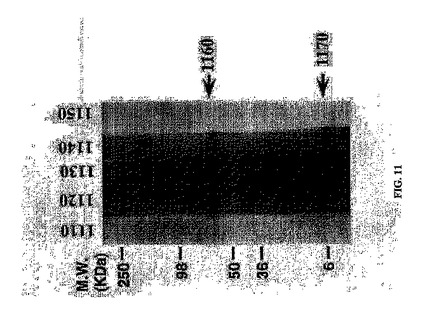
【図5】
【図7】
【図8】
【図9】
【図10】
【図11】
【公表番号】特表2007−525172(P2007−525172A)
【公表日】平成19年9月6日(2007.9.6)
【国際特許分類】
【出願番号】特願2006−510003(P2006−510003)
【出願日】平成16年4月14日(2004.4.14)
【国際出願番号】PCT/US2004/011427
【国際公開番号】WO2004/092220
【国際公開日】平成16年10月28日(2004.10.28)
【出願人】(505384335)
【Fターム(参考)】
【公表日】平成19年9月6日(2007.9.6)
【国際特許分類】
【出願日】平成16年4月14日(2004.4.14)
【国際出願番号】PCT/US2004/011427
【国際公開番号】WO2004/092220
【国際公開日】平成16年10月28日(2004.10.28)
【出願人】(505384335)
【Fターム(参考)】
[ Back to top ]