
ポリサッカライドを精製する方法
本発明は、細胞壁などの細胞成分からのポリサッカライドを精製するための改良された方法に関する。その方法は、ポリサッカライドを加水分解して分離し、それによって、ポリサッカライド単独又は担体分子に結合したポリサッカライドを含む抗原類、抗体類、及びワクチン類を産生するために有用な精製したポリサッカライドを生じさせることに関する。この方法は、単純、迅速、効率的、拡大可能、及び再現可能である。
【発明の詳細な説明】
【技術分野】
【0001】
本出願は、2006年1月13日に出願した米国仮出願第60/758894号に対する優先権の特典を主張するものであり、この仮出願全体を完全に参照により組み込む。
【0002】
本発明は、ポリサッカライドを精製する改良された方法に関する。その抽出されたポリサッカライドは、ポリサッカライド単独又は担体分子に結合したポリサッカライドを含む抗原類、抗体類、及びワクチン類を産生するために有用である。
【背景技術】
【0003】
多種多様の微生物が、ヒトの疾患を引き起こす。細菌感染は、グラム陽性及びグラム陰性の細菌、例えば、スピロヘータ、マイコバクテリア、リケッチア、クラミジア、マイコプラズマなどによって引き起こされる。酵母及び全身性病原真菌もまた免疫適格性及び免疫低下宿主の両方における重大な健康問題である。癌は、ヒトにおけるもう1つの疾患である。
【0004】
グラム陽性細菌、例えば、連鎖球菌(Streptococcus)、ブドウ球菌(Staphylococcus)、腸球菌(Enterococcus)、桿菌(Bacillus)、コリネバクテリウム(Corynebacterium)、リステリア(Listeria)、エリシペロスリクス(Erysipelothrix)及びクロストリジウム(Clostridium)など、並びにグラム陰性細菌、例えば、ヘモフィルス(Haemophilus)、赤痢菌(Shigella)、コレラ菌(Vibrio cholerae)、ナイセリア(Neisseria)及び特定の型の大腸菌(Escherichia coli)によって引き起こされる細菌感染は、世界中で深刻な罹患率を引き起こしている。このことは、現在使用されている抗生物質に対して細菌によって示される耐性の出現と相まって、これらの有害な結果を避ける細菌ワクチンの開発の必要性を示す。多くの現行の細菌ワクチンの戦略は、細菌細胞壁と関連するポリサッカライドを含む抗原の使用を伴う(非特許文献1、3、4、11、14、15、16、23、30、31、32、34、35、37、45、46、48)。
【0005】
すべての細菌、真菌及びいくつかの原虫に共通の特徴は、それらの細胞壁の中に及び/又はそこに付着してポリサッカライドが存在していることである。ポリサッカライドは、細胞壁の重要な構築的特徴であり、微生物を免疫システムによる攻撃から保護するために貢献する。微生物は、宿主の免疫システムの先天性の力及び適応力の両方による認識及び破壊を避けるための様々な異なる細胞壁及び細胞壁と関連する構造を進化させている。例えば、先天性免疫システムの貪食細胞は、細胞壁及び細胞壁と関連する抗原を認識する特別な細胞表面受容体を有する。CD14受容体及びトール様受容体(TLR)は、ペプチドグリカン、リポテイコ酸、及びポリサッカライド抗原を認識し、その際、マクロファージを活性化して微生物を貪食させることが立証されている(非特許文献24、38、49)。免疫システムの適応力は、また、細胞壁ポリサッカライド構造に対して特異的である抗体を発生することにより、これらの抗原に対応することができる(非特許文献29、41)。
【0006】
グラム陽性菌及びグラム陰性菌は、それらの外側の細胞表面構造によって区別される。グラム陽性細胞壁は、多くの場合脂質を欠いており、ほとんどが、ペプチドグリカン、ペプチドグリカンに共有結合しているタイコ酸及び/又はタイクロン酸、並びにタンパク質から構成されている。ムレイン又はムコペプチドとしても知られているペプチドグリカンは、N−アセチルグルコサミン(NAG,N-acetylglucosamine)及びN−アセチルムラミン酸(NAM,N-acetylmuramic acid)のD−アミノ酸及びL−アミノ酸を含有するペプチド側鎖による交互の繰返しからなるポリサッカライド主鎖から構成されている。グラム陰性菌は、外膜及びその外膜と細胞質の内膜との間に組み込まれたペプチドグリカンの層からなる細胞壁を所有する。その外膜は、リポポリサッカライド(LPS,lipopolysaccharide)並びに脂質及びタンパク質を含有する。そのLPS分子は、その外膜中に組み込まれるリピドAの「頭部」及びその外膜から外側に伸びるポリサッカライドの尾部から構成される。そのポリサッカライドの尾部は、通常は、中核のオリゴポリサッカライドとO−ポリサッカライドとからなる。そのO−ポリサッカライドは、N−アセチルグルコサミン(NAG)及びN−アセチルムラミン酸(NAM)の交互の組成物からなる塩基性繰返しモチーフを有する点でペプチドグリカンと似ている。また、莢膜ポリサッカライド(CPS,capsular polysaccharide)及び被膜下ポリサッカライド(sub-capsular polysaccharide)は、グラム陽性及びグラム陰性細胞壁の外面に付着しており、細菌性細胞を貪食細胞による認識及び貪食から保護するためにさらに貢献する。
【0007】
細菌外面のポリサッカライド含有成分は、免疫受容体によって特異的に認識されるために、細菌は、その外面組成を変異させる。例えば、細胞壁ポリサッカライドは、主として繰返し(オリゴ)ポリサッカライド単位から構成されているけれども、細菌の亜系は、様々な修飾及び/又はポリサッカライド単位の順序付けを持たせることによってこの基本構造を変化させる。例えば、B群連鎖球菌は、すべての血清型に共通の群抗原(即ち、ポリサッカライド群(GPS,group polysaccharides))を有するが、主要な血清型(Ia型、Ib型、II型、III型、IV型、V型、VI型、VII型、及びVIII型)を区別する型特異性の莢膜ポリサッカライド(CPS)を有する。これらの様々な型特異性莢膜ポリサッカライドのそれぞれの構造が特性化されている(非特許文献19、20、21、22、43、44、47)。同様に、肺炎連鎖球菌は、様々な型特異性莢膜ポリサッカライドを除く共通の群抗原(C物質)を有する。現在、それらの莢膜ポリサッカライド被膜によって区別される肺炎球菌(S. pneumoniae)の血清型が90個知られている(非特許文献7、49)。外面のばらつきのために、細菌型(又は亜系)は、型特異性の抗体によって識別し、分類することができる。従って、細胞壁抗原ポリサッカライドをそれらの自然状態で又は抗原サブ構造として、効率的に簡単に精製することができる方法は、ワクチンの開発を大いに助けるであろう。
【0008】
群及び型特異性のポリサッカライドは、感染防御抗体のための標的として首尾よく使用されてきており、これらの抗体の産生は、能動免疫を刺激するワクチンにより提供される。型特異性のポリサッカライド成分を有するワクチンのワクチン接種に応答して発生した抗体もまた、受動免疫を提供するために有用である。ワクチン製造で使用するためのポリサッカライドの大規模な生産は、精製したポリサッカライドの適切な供給を必要とする。特定の細菌性細胞から莢膜ポリサッカライドを単離するためのいくつかの方法(非特許文献43、45)は、酵素ムタノリシン(mutanolysin)による細胞の処理に依存している。ムタノリシンは、細菌細胞壁を切断し、それによって細胞成分を遊離させる。この方法はまた細胞溶解物をさらなる酵素により処理してタンパク質及び核酸を分解することも含む。微生物からポリサッカライドを単離するためのその他の報告されている方法(非特許文献50)は、高温のフェノールによる細胞の処理に依存している。フェノールは、ポリサッカライドを水層中に分離する。その水層を限外濾過法によって濃縮して未精製のポリサッカライド画分を生じさせ、それを次にクロマトグラフィーによって精製する。これらの方法は、細胞壁に付随する有毒な分子(例えば、リピドA内毒素、リポタイコ酸及びムラミルペプチド)を除去することが不可欠であって、高価な酵素及び毒素を分解して除去するための複雑なクロマトグラフィーの使用を必要とするために、困難で費用もかかる。精製したポリサッカライドを得るより効率的で、収量が高く且つ簡単な手段が望まれる。
【0009】
莢膜ポリサッカライドを単離するためのさらに別の効率的な方法(米国特許第6248570号に記載されている)は、細胞壁成分からCPSを抽出するための塩基加水分解反応に依存している。この方法は、困難で費用のかかる酵素は使用せずに、ポリサッカライドから、核酸、タンパク質、及び毒素を分解し、除去する効果がある。この塩基加水分解法は、他のそれまでに報告された方法に対して、より大きな単純性、効率、安全性、及び一般的適用性を提供するが、この方法のさらに大きな改良が本発明においては達成されている。
【特許文献1】米国特許第6248570号
【非特許文献1】Anderson, P., G. Peter, R.B. Johnson, L.H. Wetterlow and D.H. Smith. 1972. Immunization of humans with polyribophosphate, the capsular antigen of Haemophilus influenzae type b. J.Clin.Invest. 51:39-44.
【非特許文献2】Avery, O.T. and W.F. Goebel. 1931. Chemo-immunological studies on conjugated carbohydrate-proteins V. The immunological specificity of an antigen prepared by combining the capsular polysaccharide of type 3 pneumococcus with foreign protein. J.Exp.Med. 54:43 7-447.
【非特許文献3】Baker, C.J. and D.L. Kasper. 1985. Group B streptococcal vaccines. Rev.Inf.Dis. 7:458-467.
【非特許文献4】Baker, C.J., M.A. Rench, M.S. Edwards, R.J. Carpenter, B.M. Hays and D.L. Kasper. 1988. Immunization of pregnant women with a polysaccharide vaccine of group B Streptococcus. N.Eng1.J.Med. 319:1180-1185.
【非特許文献5】Baker, C.J., M.A. Rench and D.L. Kasper. 1990. Response to Type III polysaccharide in women whose infants have had invasive Group B streptococcal infection. New EngI.J.Med. 322:1857-1860.
【非特許文献6】Baltimore, R.S., D.L. Kasper and J. Vecchitto. 1979. Mouse protection test for group B Streptococcus type III. J.Infect.Dis. 140:81-86.
【非特許文献7】Bednar, B. and J.P. Hennessey. 1993. Molecular size analysis of capsular polysaccharide preparations from Streptococcus pneumoniae. Carbohyd.Res. 243:115-130.
【非特許文献8】Beri, R.G., J. Walker, E.T. Reese and J.E. Rollings. 1993. Characterization of chitosans via coupled size-exclusion chromatography and multiple-angle laser light-scattering technique. Carbohyd.Res. 238:11-26.
【非特許文献9】Bradford, M.M.. 1.976. A Rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analyt.Biochem. 72:248-254.
【非特許文献10】D'Ambra, A.J., J.E. Baugher, P.E. Concannon, R.A. Pon and F. Michon. 1997. Direct and indirect methods for molar-mass analysis of fragments of the capsular polysaccharide ofHaemophilus influenzae type b. Anal.Biochem. 250:228-236
【非特許文献11】Dick, W.E., Jr. and M. Beurret. 1989. Glycoconjugates of bacterial carbohydrate antigens. p. 48-114. In: J.M. Cruse and R.E. Lewis,Jr., Contributions to microbiology and immunology. S.Karger, Basel.
【非特許文献12】Dillon, H.C., Jr., S. Khare and B.M. Gray. 1987. Group B streptococcal carriage and disease: A 6-year prospective study. J.Pediat. 110:31-36.
【非特許文献13】Goebel, W.F. and O.T. Avery. 1931. Chemo-immunological studies on conjugated carbohydrate-proteins IV. The synthesis of the p-aminobenzyl ether of the soluble specific substance of type 3 pneumococcus and its coupling with protein. J.Exp.Med. 54:431.436.
【非特許文献14】Gold, R., M.L. Lepow, I. Goldschneider, T.L. Draper and E.C. Gotschlich. 1975. Clinical evaluation of group A and group C meningococcal polysaccharide vaccines in infants. J.Clin.Invest. 56:1536-1547.
【非特許文献15】Gold, R., M.L. Lepow, I. Goldschneider and E.C. Gotschlich. 1977. Immune response of human infants to polysaccharide vaccines of Groups A and C Neisseria meningitidis. J.Infect.Dis. 136S:S31-S35.
【非特許文献16】Gold, R.M., M.L. Lepow, I. Goldschneider, T.F. Draper and E.C. Gotschlich. 1978. Antibody responses of human infants to three doses of group A Neisseria meningitides vaccine administered at two, four and six months of age. J.Infect.Dis. 138:731-735.
【非特許文献17】Hennessey, J.P., B. Bednar and V. Manam. 1993. Molecular size analysis of Haemophilus influenzae type b capsular polysaccharide. J.Liq.Chromat. 16:1715-1729.
【非特許文献18】Howard, J.G., G.H. Christie, B.M. Courtenay, E. Leuchars and A.J.S. Davies. 1971. Studies on immunological paralysis. VI. Thymic-independence of tolerance and immunity to type III pneumococcal polysaccharide. Cell.Immunol. 2:614-626.
【非特許文献19】Jennings, H.J., E. Katzenellenbogen, C. Lugowski and D.L. Kasper. 1983. Structure of the native polysaccharide antigens of type la and type Ib Group B Streptococcus. Biochemistry 22:1258-1263.
【非特許文献20】Jennings, H.J., K.-.G. Rosell and D.L. Kasper. 1980. Structural determination and serology of the native polysaccharide antigen of type III group B Streptococcus. Can.J.Biochem. 58:112-120.
【非特許文献21】Jennings, H.J., K.-.G. Rosell and D.L. Kasper. 1980. Structure and serology of the native polysaccharide antigen of type Ia group B Streptococcus. Proc.Nat.Acad.Sci.USA. 77:2931-2935.
【非特許文献22】Jennings, H.J., K.-.G. Rosell, E. Katzenellenbogen and D.L. Kasper. 1983. Structural determination of the capsular polysaccharide antigen of type II Group B Streptococcus. J.Biol.Chem. 258:1793-1798.
【非特許文献23】Jennings, H.J. and R.K. Sood. 1994. Synthetic glycoconjugates as human vaccines. p. 325-371. In: Y.C. Lee and R.T. Lee, Neoglycoconjugates: Preparation and applications. Academic Press, New York.
【非特許文献24】Kang, D., Liu, G., Lundstrom, A. et al. 1998 A peptidoglycan recognition protein in innate immunity conserved from insects to humans. Proc. Natl. Acad. Sci. USA. 95:10078-10082.
【非特許文献25】Kasper, D.L., C.J. Baker, R.S. Baltimore, J.H. Crabb, G. Schiffman and H.J. Jennings. 1979. Immunodeterminant specificity of human immunity to type III group B Streptococcus. J.Exp.Med. 149:327-339.
【非特許文献26】Knobloch, J.E. and P.N. Shaklee. 1997. Absolute molecular weight of low-molecular-weight heparins by size-exclusion chromatography with multiangle laser light scattering detection. Anal.Biochem. 245:231-241.
【非特許文献27】Lancefield, R.C.. 1933. A serological differentiation of human and other groups of haemolytic streptococci. J.Exp.Med. 57:571-595.
【非特許文献28】Lancefield, R.C.. 1938. A micro-precipitin technique for classifying hemolytic streptococci and improved methods for producing antigen. Proc.Soc.Exp.Biol.and Med. 38:473-478.
【非特許文献29】Lancefield, R.C., M. McCarty and W.N. Everly. 1975. Multiple mouse-protective antibodies directed against group B streptococci: Special reference to antibodies effective against protein antigens. J.Exp.Med. 142:165-179.
【非特許文献30】Madoff, L.C., L.C. Paoletti, J.Y. Tai and D.L. Kasper. 1994. Maternal immunization of mice with Group B streptococcal type III polysaccharide-beta C protein conjugate elicits protective antibody to multiple serotypes. J.Clin.Invest. 94:286-292.
【非特許文献31】Marques, M.B., D.L. Kasper, A. Shroff, F. Michon, H.J. Jennings and M.R. Wessels. 1994. Functional activity of antibodies to the group B polysaccharide of group B streptococci elicited by a polysaccharide-protein conjugate vaccine. Infect.Immun. 62:1593-1599.
【非特許文献32】Makela, P.R.H., H. Peltola, H. Kayhty, et al. 1977. Polysaccharide vaccines of group A Neisseria meningitidis and Haemophilus influenzae type b: A field trial in Finland. J.Infect.Dis. 136:543-50.
【非特許文献33】Michon, F., J.R. Brisson, A. Dell, D.L. Kasper and H.J. Jennings. 1988. Multiantennary group-specific polysaccharide of group B Streptococcus. Biochem. 27:5341-5351.
【非特許文献34】Peltola, A., H. Kayhty, A. Sivonen and P.R.H. Makela. 1977. Haemophilus influenzae type b capsular polysaccharide vaccine in children: A double blind field study of 100,000 vaccines 3 months to 5 years of age in Finland. Pediatrics 60:730-737.
【非特許文献35】Peltola, H., P.R.H. Makela, H. Jousimies, et al. 1977. Clinical efficacy of meningococcal group A vaccine in children three months to five years of age. N.Engl.J.Med. 297:686-691.
【非特許文献36】Reuter, G. and R. Schauer. 1994. Determination of sialic acids. p. 168-199. In: W.J. Lennarz and G.W. Hart, Methods in Enzymology Vol. 230 Techniques in Glycobiology. Academic Press, New York.
【非特許文献37】Robbins, J.B. and R. Schneerson. 1990. Polysaccharide-protein conjugates: A new generation of vaccines. J.Infect.Dis. 161:821-832.
【非特許文献38】Scwander, R., Dziarski R., Wesche, H. et al. 1999. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J. Biol. Chem. 274:17406-17409.
【非特許文献39】Smith, A.L. and J. Haas. 1991. Neonatal Bacterial Meningitis. p. 313-333. In: W.M. Scheld, R.J. Whitley and D.T. Durack, Infections of the Central Nervous System. Raven Press, Ltd., New York,
【非特許文献40】Tsunashima, T., K: Moro, B. Chu and T.-Y. Liu. 1978. Characterization of group C meningococcal polysaccharide by light-scattering spectroscopy. III. Determination of molecular weight, radius of gyration, and translational diffusional coefficient.. Biopolymers 17:251-265.
【非特許文献41】von Hunolstein, C., L. Nicolini, S. D'Ascenzi, C. Volpe, G. Alfarone and G. Orefici. 1993. Sialic acid and biomass production by Streptococcus agalactiae under different growth conditions. Appl.Microbiol.Biotechnol. 38:458-462.
【非特許文献42】Wessels, M.R., W.J. Benedi, H.J. Jennings, F. Michon, J.L. DiFabio and D.L. Kasper. 1989. Isolation and characterization of type IV group B Streptococcus capsular polysaccharide. Infect.Immun. 57:1089-1094.
【非特許文献43】Wessels, M.R., J.L. DiFabio, V.J. Benedi, et al. 1991. Structural determination and immunochemical characterization of the type V group B Streptococcus capsular polysaccharide. J.Biol.Chem. 266:6714-6719.
【非特許文献44】Wessels, M.R., L.C. Paoletti, D.L. Kasper, et al. 1990. Immunogenicity in animals of a polysaccharide-protein conjugate vaccine against type III group B Streptococcus. J.Clin.Invest. 86:1428.1433.
【非特許文献45】Wessels, M.R., L.C. Paoletti, A.K. Rodewald, et al. 1993. Stimulation of protective antibodies against type la and Ib group B streptococci by a type Ia polysaccharide-tetanus toxoid conjugate vaccine. Infect.Immun. 61:4760-4766.
【非特許文献46】Wessels, M.R., V. Pozsgay, D.L. Kasper and H.J. Jennings. 1987. Structure and immunochemistry of an oligopolysaccharide repeating unit of the capsule polysaccharide of Type III Group B Streptococcus: A revised structure for the Type III Group B streptococcal polysaccharide antigen. J.Biol.Chem. 262:8262-8267.
【非特許文献47】Wyle, S.A., M.S. Artenstein, B.L. Brandt, et al. 1972. Immunologic response of man to group B meningococcal polysaccharide vaccines. J.Infect.Dis. 126:514-522.
【非特許文献48】Yang, R.B., Mark, M.R., Gray, A., et al. 1998. Toll-like receptor 2 mediates lipopolysaccharide-induced cellular signaling. Nature. 395:284-288.
【非特許文献49】Tipson, R.S. and Horton, D., Advances in Carbohydrate Chemistry and Biochemistry, Vol. 41, 1983, Academic Press, NY.
【非特許文献50】Westphal et al., Methods in Carbohydrate Chemistry, vol. V, 1965, Academic Press, NY.
【非特許文献51】Theodora W. Greene and Peter G. M. Wuts, Protective Groups in Organic Syntheses, 2nd Ed. (1991).
【非特許文献52】Kohler and Milstein (1975) Nature 256:495-497.
【非特許文献53】Takeda et al. (1985) Nature 314:452.
【非特許文献54】Campbell (1985) Laboratory Techniques in Biochemistry and Molecular Biology, Vol. 13, Burdon, et al. (eds.), Elsevier Science Publishers, Amsterdam.
【非特許文献55】Schneerson, R., et al. (1980) J. Exp. Med. 1952:361-476.
【非特許文献56】Marburg, S., et al. (1986) J. Am. Chem. Soc. 108:5282-5287.
【非特許文献57】Grossman, M. and Cohen, S. N., in "Basic and Clinical Immunology", 7th Ed., (Stites, D. P. and Terr, A. T. eds., Appleton & Lange 1991) Chapter 58 "Immunization".
【非特許文献58】Paoletti, et al. (1997) J. Infectious Diseases, 175:1237-9.
【非特許文献59】L. Warren, (1959) 1 Biol. Chem. 234, 1971.
【発明の開示】
【発明が解決しようとする課題】
【0010】
本発明の利点としては、(1)単純性、(2)分子収量の増加、(3)スケーラビリティー(大規模生産)、(4)精製したポリサッカライドをその天然の抗原型に保持する又は戻すことができること、及び(5)DNA、RNA、及び毒素が、加水分解ステップにおいて分解され、それ故、本発明に従って産生された最終産物中には感知できるほどの量が存在しないこと、の少なくとも1つが挙げられる。
【課題を解決するための手段】
【0011】
いくつかの実施形態において、ポリサッカライド及び細胞成分を含むストックからのポリサッカライドの精製は、以下の方法により達成することができる。その方法は、該ストックを第1の試薬と接触させて第1の混合物を形成するステップを必要とする。その第1の試薬は、酸又は塩基であり、第1の混合物のpHは、0〜6の範囲又は9〜15の範囲である。分離組成物は、細胞成分の少なくとも一部からポリサッカライドを分離して形成する。その分離組成物は、ポリサッカライドと細胞成分の残量とを含む。その分離された組成物は、第2の試薬と接触させて第2の混合物を形成する。その第2の試薬は塩基であり、第2の混合物のpHは、9〜15の範囲である。精製組成物は、細胞成分の残量の少なくとも一部からのポリサッカライドを精製することにより形成される。
【0012】
いくつかの実施形態において、ポリサッカライド及び細胞成分を含むストックからのポリサッカライドの精製は、以下の方法により達成することができる。その方法は、該ストックを第1の試薬と接触させて第1の混合物を形成するステップを必要とする。その第1の試薬は、塩基及び還元剤を含み、第1の混合物のpHは、9〜15の範囲である。分離された組成物を、細胞成分の少なくとも一部からポリサッカライドを分離することによって形成する。精製したポリサッカライドを、その分離ステップ後に回収する。
【0013】
そのストックを第1の試薬と接触させるステップを、本明細書では時に「第1ステップの加水分解」と称する。該分離組成物を第2の試薬と接触させるステップを、本明細書では時に「第2ステップの加水分解」と称する。これらの呼び方は、明確にするためになされ、どのようにも限定するものと解釈すべきではない。
【0014】
本発明の用途の非限定の例としては、精製したポリサッカライドの組成物、ポリサッカライド−ポリペプチド結合体、医薬品組成物、診断キット、及び抗体を製造することが挙げられる。抗体は、ポリサッカライドのオリジナルソースと反応することができ、それらは他の有機体と交差反応性であり得る。
【0015】
いくつかの実施形態において、ポリサッカライド結合ワクチンは、以下の方法を用いて製造することができる。精製ポリサッカライドは、上記の方法を用いて獲得する。そのポリサッカライドをポリペプチドに結合し、それによってワクチンを製造する。いくつかの実施形態において、そのワクチンにはアジュバントを添加する。いくつかの実施形態において、その結合は、還元的アミノ化によって達成する。
【発明を実施するための最良の形態】
【0016】
本発明の方法は、細胞成分から大きな収量でポリサッカライドを抽出するために使用することができる。収量の比較のためには実施例1を参照されたい。いくつかの実施形態において、該方法は、また、高分子量ポリサッカライドを分解するためにも有利に使用することができる。さらに、いくつかの実施形態において、本発明の方法は、混入物、例えばタンパク質及び核酸などからポリサッカライドを分離し、精製するための便利な膜濾過方法の任意的な使用を可能にする。これまでは、実質的に純粋なポリサッカライドは、一般的には低処理量で複雑な沈殿及びカラムクロマトグラフ技術を用いることによって得ていた。抽出したポリサッカライドを分離し、精製するために膜濾過方法を使用することができることは、膜濾過が、単純で、高い処理能力があり、規模拡大が可能な精製したポリサッカライドの高い収量を維持する技術であるために望ましい。
【0017】
ポリサッカライド及び細胞成分を含むストックからのポリサッカライドの精製は、本発明のいくつかの実施形態によれば2ステップの加水分解法によって達成することができる。その2ステップの加水分解法は、塩基又は酸を用い、その塩基性又は酸性試薬がポリサッカライドと細胞成分例えば微生物細胞壁の間の1つ又は複数の結合の加水分解をもたらすことができる第1ステップの加水分解を伴う。これらの塩基に不安定又は酸に不安定な結合は、ポリサッカライドをペプチドグリカンに連結するホスホジエステル結合及び/又はポリラムノースをペプチドグリカンに連結するホスホジエステル結合であり得る。ポリサッカライドと細胞壁成分の間の結合の加水分解により、ポリサッカライドを細胞壁成分から分離することができる。第1ステップの加水分解後に、例えばポリサッカライドを非付着細胞成分、例えば細胞壁成分、タンパク質、核酸及び脂質から分離して、分離組成物を生み出すことができる分離ステップが続く。その分離組成物は、ポリサッカライド及び細胞成分の残量を含む。第2ステップの加水分解は、そのときその分離組成物について行うことができ、ポリサッカライドと細胞壁成分の間の残りの結合を加水分解し且つ/又は望ましくないタンパク質及び核酸を少なくとも部分的に分解することができる。その第2ステップの加水分解の後には、分離、並びに任意的なN−アシル化及び精製が続く。
【0018】
いくつかの実施形態において、ポリサッカライドは、2回を超える加水分解ステップを用いて単離することができる。2回の加水分解ステップで実質的に純粋なポリサッカライドを単離するためのクロマトグラフィーの使用の必要性を未然に防ぐためには十分であり得るが、2回を超える加水分解ステップは、収量及び純度の向上をもたらすことができ、本発明に記載の方法を用いて決定することができる。いくつかの実施形態においては、3回の加水分解ステップが用いられ、それぞれ分離ステップが続く。いくつかの実施形態においては、4回の加水分解ステップが用いられ、それぞれ分離ステップが続く。いくつかの実施形態においては、4回を超える加水分解ステップを用いることができる。
【0019】
いくつかの実施形態において、ポリサッカライドの精製は、1回目、2回目、及び/又はすべての加水分解ステップに対して精製したヌクレアーゼ及び/又は精製したプロテアーゼを試薬中に添加しないで達成することができる。これらの実施形態において、その反応混合物は、ヌクレアーゼ及び/又はプロテアーゼを、不純物を介して及び/又はポリサッカライドを含むストック中のそれらの存在を介して依然として含有することができる。
【0020】
いくつかの実施形態においては、1回だけの加水分解ステップを必要とする。これらの実施形態においては、第1の試薬が、塩基及び還元剤を含む。その還元剤は、例えば、加水分解ステップの間、ポリサッカライドを保護する助けをすることができる。
【0021】
いくつかの実施形態において、他の細胞成分からポリサッカライドを引き離すためのpH範囲は、9〜14、例えば10〜13、11〜13、又は12である。加水分解は、2℃から100℃まで、例えば、4℃〜100℃、20℃〜100℃、30℃〜100℃、40℃〜100℃、25℃〜90℃、30℃〜80℃、34℃〜41℃、又は37℃の温度で達成することができる。下端が25℃以上である温度範囲は、収量の増加をもたらすことができる。第2ステップの加水分解は、第1ステップの加水分解の方式で用いられる加水分解条件より好ましくは強い条件で達成することができ、その結果、タンパク質、内毒素、及び核酸が、少なくとも部分的に分解される。いくつかの実施形態において、第2ステップの加水分解は、第1ステップより10〜90℃高い(例えば、少なくとも10、20、30、40、50、60、又は70℃は高いが90℃より高くはない)温度で起こり得る。いくつかの実施形態において、第2ステップの加水分解中の最高温度は、第1ステップの加水分解中の最高温度より10〜90℃高い(例えば、少なくとも10、20、30、40、50、60、又は70℃は高いが90℃より高くはない)。例えば、分離された組成物を第2の試薬と接触させる間の最高温度は、ストックを第1の試薬と接触させる間の最高温度より30℃〜90℃高い。いくつかの実施形態において、第2のステップは、第1のステップより1〜45時間長く持続させることができる(例えば、少なくとも、1、5、10、15、20、25、30、35、又は40時間は長いが45時間より長くはない)。いくつかの実施形態において、第2ステップの混合物のpHは、第1ステップの混合物よりpH7からの絶対値で0.1〜5単位離れていることができる(例えば、少なくとも0.1、0.2、0.3、0.4、0.5、1、1.3、1.5、2、2.3、2.5、3、3.3、3.5、4、又は4.5単位離れているが5単位を超えて離れてはいない)。いくつかの実施形態において、第2の混合物中の塩基の規定度は、第1の混合物中で使用される酸又は塩基の規定度より0.25〜4N大であり得る(例えば、少なくとも0.25、0.5、0.75、1、1.5、2、3.5、3、又は3.5Nは大きいが、4Nを超える大きさではない)。例えば、第2ステップの加水分解は、第2の混合物が、第1の混合物中の酸又は塩基の規定度より0.5〜4N大きい塩基の規定度を有する状態で、第1ステップの加水分解より40℃〜90℃高温で行うことができる。例えば、第2ステップの加水分解条件は、2NのNaOHの最終濃度により80℃で15〜48時間(精製されるポリサッカライドによる)にわたって行うものとすることができる。上記条件は、核酸とタンパク質の両方を著しく分解するのに一般的には十分であり、それらのフラグメントは、次に濾過によって容易に除去することができる。いくつかの実施形態において、この2ステップの加水分解法は、高分子量のポリサッカライドの分解をもたらすことができ、それは当該方法によって製造される産物において特にそして最終的に有用であり得る。
【0022】
(出発材料)
本発明により精製されるポリサッカライドのソースは、多様である。ソースとしては、細菌、古細菌、及び真核生物が挙げられる。多細胞生物においては、そのソースは全有機体の一部であり得る(例えば、癌細胞又は癌性細胞)。
【0023】
有機体のさらなる例としては、病原菌類、例えばアエロモナス細菌(Aeromonas hydrophila)及び他の菌種;炭疽菌(Bacillus anthracis);セレウス菌(Bacillus cereus);ボツリヌス神経毒素を産生する菌種のクロストリジウム属(Clostridium);ウシ流産菌(Brucella abortus);マルタ熱菌(Brucella melitensis);ブタ流産菌(Brucella suis);鼻疽菌(Burkholderia mallei)(旧学名はPseudomonas mallei);類鼻疽菌(Burkholderia pseudomallei)(旧学名はPseudomonas pseudomallei);カンピロバクター・ジェジュニ(Campylobacter jejuni);オウム病クラミジア(Chlamydia psittaci);ポツリヌス菌(Clostridium botulinum);ウェルシュ菌(Clostridium perfringens);コクシジオイデス・イミティス(Coccidioides immitis);コクシジオイデス・ポサダシ(Coccidioides posadasii);心糸状虫(Cowdria ruminantium)(心水病);Q熱コクシエラ(Coxiella burnetii);エンテロ毒性大腸菌群(EEC群,Enterovirulent Escherichia coli group)、例えば、毒素原生大腸菌(ETEC,Escherichia coli-enterotoxigenic)、腸管病原性大腸菌(EPEC,Escherichia coli-enteropathogenic)、O157:H7腸管出血性大腸菌(EHEC,Escherichia coli-O157: H7 enterohemorrhagic)、及び腸管組織侵入性大腸菌(EIEC,Escherichia coli-enteroinvasive)など;エーリキア(Ehrlichia)菌種、例えばエーリキア・シャフェンシス(Ehrlichia chaffeensis)など;野兎病菌(Francisella tularensis);在郷軍人病菌(Legionella pneumophilia);リベロバクター・アフリカヌス(Liberobacter africanus);リベロバクター・アジアティクス(Liberobacter asiaticus);リステリア菌(Listeria monocytogenes);多岐に渡る腸内細菌、例えば、クレブシエラ(Klebsiella)、エンテロバクター(Enterobacter)、プロテウス(Proteus)、シトロバクター(Citrobacter)、アエロバクター(Aerobacter)、プロビデンシア(Providencia)、及びセラチア(Serratia)など;マイコバクテリウム・ボビス(Mycobacterium bovis);結核菌(Mycobacterium tuberculosis);マイコプラズマ・カプリコルム(Mycoplasma capricolum);マイコプラズマ・ミコイデス亜種ミコイデス(Mycoplasma mycoides ssp mycoides);ペロノスクレロスポラ・フィリピネシス(Peronosclerospora philippinensis);ダイズさび病菌(Phakopsora pachyrhizi);プレシオモナス・シゲロイデス(Plesiomonas shigelloides);青枯病菌(Ralstonia solanacearum)品種3、次亜種2;発疹チフスリケッチイ(Rickettsia prowazekii);リケッチア・リケッチイ(Rickettsia rickettsii);サルモネラ菌種(Salmonella spp.);スクレロープソーラ・レイジアエ・バラエティー・ゼアエ(Schlerophthora rayssiae var zeae);赤痢菌菌種(Shigella spp.);黄色ブドウ球菌(Staphylococcus aureus);連鎖球菌(Streptococcus);ジャガイモがんしゅ病菌(Synchytrium endobioticum);非O1コレラ菌(Vibrio cholerae non-O1);O1コレラ菌(Vibrio cholerae O1);腸炎ビブリオ菌(Vibrio parahaemolyticus)及びその他のビブリオ菌;ビブリオ・バルニフィカス(Vibrio vulnificus);イメ白葉枯病菌(Xanthomonas oryzae);ピアス病菌(Xylella fastidiosa)(柑橘類ふ入り白化菌株);エンテロコリチカ菌(Yersinia enterocolitica)及び仮性結核菌(Yersinia pseudotuberculosis);並びにペスト菌(Yersinia pestis)が挙げられる。
【0024】
有機体のさらなる例としては、菌類、例えば、アスペルギルス菌種(Aspergillus spp.);ブラストミセス・デルマティティディス(Blastomyces dermatitidis);カンジダ(Candida);コクシジオイデス・イミチス(Coccidioides immitis);コクシジオイデス・ポサダシ(Coccidioides posadasii);クリプトコッカス・ネオフォルマンス(Cryptococcus neoformans);ヒストプラズマ・カプスラーツム(Histoplasma capsulatum);トウモロコシさび病菌(Maize rust);イネイモチ病菌(Rice blast);イネ褐斑病菌(Rice brown spot disease);ライムギイモチ病菌(Rye blast);スポロトリクス・シェンキイ(Sporothrix schenckii);及びコムギ菌(wheat fungus)などが挙げられる。
【0025】
有機体のさらなる例としては、寄生原虫及び寄生虫、例えば、アカントアメーバ(Acanthamoeba)及びその他の自由生活アメーバ;アニサキス種(Anisakis sp.)並びにその他の関連する寄生虫の回虫(Ascaris lumbricoides)及び鞭虫(Trichuris trichiura);クリプトスポリジウム・パルバム(Cryptosporidium parvum);シクロスポラ・カイエタネンシス(Cyclospora cayetanensis);裂頭条虫種(Diphyllobothrium spp.);赤痢アメーバ(Entamoeba histolytica);Eustrongylides sp.;ランブル鞭毛虫(Giardia lamblia);ナノフィエトゥス種(Nanophyetus spp.);住血吸虫種(Shistosoma spp.);トキソプラズマ原虫(Toxoplasma gondii);及び旋毛虫(Trichinella)が挙げられる。
【0026】
ソースのさらなる例としては、小細胞肺癌、神経芽細胞腫、乳癌、結腸癌などの癌細胞又は癌組織が挙げられる。これらの癌細胞又は癌組織は、例えば、ヒト、マウス、イヌ、ネコ、ヤギ、サル、又は雌ウシなどの哺乳類のものであり得る。
【0027】
本発明に従って使用するためのグラム陽性菌の非限定の例は、連鎖球菌(Streptococci)、ブドウ球菌(Staphylococci)、腸球菌(Enterococci)、桿菌(Bacillus)、コリネバクテリウム(Corynebacterium)、リステリア菌(Listeria)、エリシペロスリクス(Erysipelothrix)、及びクロストリジウム(Clostridium)である。本発明で使用するためのグラム陰性菌の非限定の例としては、ヘモフィルス(Haemophilus)(例えばインフルエンザ菌(Haemophilus influenzae))、ナイセリア(Neisseria)(例えば、髄膜炎菌(Neisseria meningitidis))及び大腸菌類(Escherichia)(例えば、大腸菌(Escherichia coli))が挙げられる。
【0028】
精製に対して望ましいポリサッカライドは、細胞壁などの細胞成分と関係するものであり得る。細胞壁と関係するとは、そのポリサッカライドが細胞壁それ自体の成分であり、且つ/又はその細胞壁に直接又は中間分子を介して間接的に付着しており、或いはその細胞壁を一時的に被覆するものである(例えば、一定の細菌の菌株は、技術的に「エキソポリサッカライド」としても知られる莢膜ポリサッカライドを滲出する)ことを意味する。
【0029】
いくつかの実施形態において、細菌から抽出されたポリサッカライドは、莢膜ポリサッカライド、被膜下ポリサッカライド、又はリポポリサッカライドであり得る。いくつかの実施形態において、該ポリサッカライドは、莢膜ポリサッカライドであり得る。
【0030】
いくつかの実施形態において、該ポリサッカライドは、A群連鎖球菌(GAS,group A streptococcus)の群特異的なポリサッカライドの被膜下ポリサッカライドであり得る。
【0031】
いくつかの実施形態において、該ポリサッカライドは、B群連鎖球菌(GBS,group B streptococcus)から抽出することができる。いくつかの実施形態において、該ポリサッカライドは、GBSタイプIa、Ib、II、III、IV、V、VI、VII及び/又はVIIIから抽出される型特異性のCPSであり得る。いくつかの実施形態において、該ポリサッカライドは、GBSタイプIa、Ib、II、III及びVから抽出されるCPSであり得る。
【0032】
いくつかの実施形態において、該ポリサッカライドは、C群連鎖球菌(GCS,group C streptococcus)からの群ポリサッカライドの被膜下ポリサッカライドであり得る。
【0033】
いくつかの実施形態において、該ポリサッカライドは、肺炎球菌(S. pneumoniae)から抽出することができる。いくつかの実施形態において、CPSは、肺炎球菌タイプ3及び14から抽出することができる。
【0034】
いくつかの実施形態において、該ポリサッカライドは、ナイセリア(Neisseria)又は大腸菌(Escherichia)から抽出することができる。いくつかの実施形態において、該ポリサッカライドは、髄膜炎菌(Neisseria meningitidis)B型、C型、Y型又はW135型或いは大腸菌K1(Escherichia coli K1)から抽出することができる。
【0035】
いくつかの実施形態においては、1種類のポリサッカライドを精製する。いくつかの実施形態においては、複数種のポリサッカライドを1つ又は複数のソースから共精製する。
【0036】
本明細書で使用する用語「ストック」とは、精製すべきポリサッカライドを含む出発材料を指す。典型的なストックは、上澄み、条件培地、均質化細胞、又は細胞ペレットである。エキソポリサッカライド(細菌から上澄み又は媒体中に滲みだされる莢膜ポリサッカライド)が所望のポリサッカライドである場合、典型的なストックは、遠心分離又は精密濾過によって細胞を分離し、その上澄みを一般的には10〜15倍濃縮することによって形成した濃厚な上澄みであり得る。いくつかの実施形態において、そのストックは、ポリサッカライドが5〜20mg/mLの濃度で存在するように濃縮した上澄み又は条件培地である。興味のあるポリサッカライドが細胞壁に付着している場合、そのときの典型的なストックは、ペレット化した又は均質化した細胞である。
【0037】
(第1ステップの加水分解)
本発明によれば、ポリサッカライドは、ストックを第1の試薬の酸試薬又は塩基試薬と接触させることによって抽出する。理論に縛られるものではないが、共有結合が、ポリサッカライドと細胞壁などの細胞成分(この細胞成分は、それ自体、例えばポリサッカライド、タンパク質、又は脂質であり得る)の間(又はポリサッカライドとポリサッカライドを細胞成分に連結している中間分子の間)で破壊されるものと考えられる。その他のタイプの結合としては、以下に限定はされないが、イオン結合及びファンデルワールス力によって形成される結合が挙げられる。ポリサッカライドと細胞壁成分を連結している結合は、ポリサッカライドと細胞壁成分の間の「直接」結合、又はポリサッカライドの細胞壁成分への付着をもたらす「介在する」又は「間接的」結合であり得る。その結合の破壊をもたらす化学反応は、塩基又は酸のいずれかによる加水分解反応である。
【0038】
一般に、シアル酸結合を有するB群連結球菌ポリサッカライドは、酸によって開裂されるが塩基にはならない。しかしながら、ホスホジエステル結合を含有する莢膜ポリサッカライドは、主として塩基によって開裂され、酸によってはそれほどではない。例えば、インフルエンザ菌(Haemophilus influenzae)b型及びいくつかの肺炎球菌莢膜ポリサッカライド類、例えば6A、6B、18、及び23などである。髄膜炎菌A(Meningococcal A)及び肺炎球菌(pneumococcal)19型は、酸及び塩基の両方によって開裂されるいくつかの莢膜ポリサッカライドの例である。
【0039】
(第1の試薬組成物)
いくつかの実施形態において、第1の試薬は、いろいろな塩基の1つ又は複数を含む。本発明に従って使用することができる塩基の非限定の例は、NaOH、KOH、LiOH、NaHCO3、Na2CO3、K2CO3、KCN、Et3N、NH3、H2N2H2、NaH、NaOMe、NaOEt及びKOtBuから選択される化合物を含む。いくつかの実施形態において、NaOH、KOH、LiOH、NaH、NaOMe又はKOtBuなどの塩基は、0.5N〜10Nの範囲(例えば、0.5N〜5N、0.5N〜2N、又は0.8〜1.5N)で使用することができる。いくつかの実施形態において、NaHCO3、Na2CO3、K2CO3及びKCNなどの塩基は、それらの溶解性が許容する高さの濃度で使用することができる。いくつかの実施形態において、トリエチルアミン(Et3N)などの有機塩基は、水又はアルコールなどの加水分解を生じる作用物質が存在する限り、中濃度から高濃度、例えば、1Nから7N(例えば2N〜4N)の範囲で使用することができる。アンモニア(NH3)又はヒドラジン(H2NNH2)などの塩基は、100%を含む殆んどどんな濃度でも使用することができる。溶媒、例えば、水、アルコール類(好ましくはC1−C4)、ジメチルスルホキシド、ジメチルホルムアミド、又はこれら及びその他の有機溶媒の混合物などを使用することができる。水を含む加水分解用の塩基溶液も使用することができる。
【0040】
いくつかの実施形態において、第1の試薬のpHは、9〜15の範囲内である。例えば、第1の試薬のpHは、10〜15、10〜14、11〜14、又は12〜14の任意の範囲内であり得る。いくつかの実施形態において、ストックと第1の試薬の混合物は、9〜14の範囲のpHを有し、例えば、その混合物は、9〜13、10〜13、又は11〜13のいずれかの範囲のpHを有することができる。いくつかの実施形態において、その混合物のpHは、12付近である。
【0041】
いくつかの実施形態において、第1の試薬は、いろいろな酸の1つ又は複数を含む。本発明に従って使用することができる酸の非限定の例は、HCl、H3PO4、クエン酸、酢酸、亜硝酸、及び硫酸から選択される化合物を含む。
【0042】
いくつかの実施形態において、第1の試薬のpHは、0〜6の範囲内であり得る。例えば、第1の試薬のpHは、0〜5、0〜4、0〜3、1〜2の任意の範囲内であり得る。いくつかの実施形態において、ストックと第1の試薬の混合物は、0〜6の範囲内のpHを有し、例えば、その混合物は、0〜5、1〜5、2〜5、3〜5、又は4〜5の任意の範囲内のpHを有することができる。いくつかの実施形態において、その混合物のpHは、4付近である。
【0043】
その第1の試薬は、さらなる物質、例えば、塩類(例えば、NaCl又はKCl)、酸化剤(例えば、H2O2、O3、又は次亜塩素酸塩)、ホウ素を含む還元剤(例えば、NaBH4、NaCNBH3、リチウムトリ−sec−ブチルボロヒドリド、又はNaBH(OCOCH3)3を含む)、その他の還元剤(例えば、水素化アルミニウムリチウム、ジチオスレイトール、又はβ−メルカプトエタノールが挙げられる)、酵素類(例えば、プロテアーゼ類又はヌクレアーゼ類が挙げられる)、キレート化剤(例えば、EDTA又はEGTAが挙げられる)、及び界面活性剤類(非イオン型、両性イオン型、アニオン型又はカチオン型)などを場合によって含むことができる。いくつかの実施形態において、界面活性剤は、その後の濾過ステップにおいて役立ち得る。いくつかの実施形態において、還元剤によって、例えばポリサッカライドの還元末端の3番目の炭素に連結している隣接する糖残基のβ−エリミネーションによって引き起こされるいわゆる「ピーリング」によるポリサッカライドの分解の可能性を避けることができる。
【0044】
いくつかの実施形態において、第1の試薬は、塩基及び還元剤を含む。いくつかの実施形態において、その塩基は、0.5N〜5Nの範囲内であり、その還元剤は、0.03mM〜300mMの範囲内である。例えば、その塩基は、0.5N〜2Nの範囲内の濃度を有することができ、その還元剤は、0.3mM〜30mMの範囲内の濃度を有することができる。典型的な第1の試薬は、塩基及び水素化ホウ素ナトリウムを含む。いくつかの実施形態において、その塩基の濃度は、0.5N〜5Nの範囲内であり、水素化ホウ素ナトリウムの濃度は、0.03mM〜300mMの範囲内である。例えば、その塩基の濃度は、0.5N〜2Nの範囲内であり得、水素化ホウ素ナトリウムの濃度は、0.3mM〜30mMの範囲内である。いくつかの実施形態において、その塩基は、NaOH、LiOH及びKOHから選択される。
【0045】
(使用)
いくつかの実施形態において、ストックは、第1の試薬1リットル当り0.1〜300gの細胞ペーストであることができる。例えば、そのストックは、第1の試薬1リットル当り0.5〜200gの細胞ペーストであることができる。例えば、そのストックは、第1の試薬1リットル当り1〜100gの細胞ペーストであることができる。例えば、そのストックは、第1の試薬1リットル当り5〜150gの細胞ペーストであることができる。例えば、そのストックは、第1の試薬1リットル当り20〜100gの細胞ペーストであることができる。いくつかの実施形態において、そのストックは、濃縮した上澄みであるか又は濃縮した第1の試薬(例えば、10NのNaOH)が、濃縮度のより低い混合物を得るために添加される(例えばその混合物のpHによって決定されるか又はその混合物中の第1の試薬の成分の濃度によって決定され、例えば、10NのNaOHが1NのNaOHに希釈される)条件培地である。
【0046】
加水分解は、2℃〜100℃の温度、例えば、4℃〜100℃、20℃〜100℃、30℃〜100℃、40℃〜100℃、25℃〜90℃、30℃〜80℃、34℃〜41℃、又は37℃で達成することができる。下端が25℃以上である温度範囲は、収量の向上をもたらすことができる。いくつかの実施形態において、混合物は、反応が起こることを可能にするために1〜48時間にわたってインキュベートする。例えば、そのインキュベーションは、1、2、3、4、5、6、7、8、9、10、12、14、15、16、18、20、24、36、若しくは48時間又はその間の任意の時間続けることができる。いくつかの実施形態において、その混合物はインキュベーションの間は、かき混ぜる。いくつかの実施形態において、その混合物はインキュベーションの間は、かき混ぜない。いくつかの実施形態において、第1ステップの加水分解は、15〜18時間のインキュベーションを用い、1NのNaOHの最終濃度で、37℃でかき混ぜて達成する。
【0047】
酸の第1試薬の典型的な使用としては、A群又はC群連鎖球菌の群特異的ポリサッカライド(例えば、GAS又はGCS被膜下ポリサッカライド)の加水分解を含む。かかるポリサッカライドについてのいくつかの実施形態において、第1の試薬は、0.1NのHCl、硝酸ナトリウム(NaNO2)、又はpH4〜5の緩衝酢酸溶液を含む。かかるポリサッカライドのいくつかの実施形態において、該混合物のpHは、4近くである。いくつかの実施形態において、第1ステップの酸加水分解は、混合物のpHが4近くである0.1NのHClの最終濃度で、室温で達成することができる。その他の酸濃度も、混合物中のストックの量、その混合物の望ましいpH、及び特定の酸の該ストックとの相互作用などの因子によって使用することができる。緩衝酢酸溶液pH4〜5、亜硝酸ナトリウム(NaNO2)を、25℃〜60℃の範囲の温度で同様に使用することができる。
【0048】
B群連鎖球菌(group B Streptococcus)、Y群髄膜炎菌(group Y meningococcus)、及びW−135群髄膜炎菌(group W-135 meningococcus)莢膜ポリサッカライドの精製についてのいくつかの実施形態において、1〜100グラム(例えば、1〜75グラム)の細胞ペーストを、750mLの1NのNaOH及び100mgの水素化ホウ素ナトリウムと接触させることができる。この反応混合物は、125rpmの速度で、例えば、37℃で16時間にわたって震盪させることができる。
【0049】
(第1ステップの分離)
別段の記述がない限り、この第1ステップの分離の項に以下で示されている本発明の態様は、本発明のすべてのその他の分離及び精製ステップに全面的に適用する。
【0050】
塩基又は酸の加水分解試薬中に存在する抽出されたポリサッカライドは、他の細胞成分に由来する不純物から、電荷、親水性、親和性、溶解性若しくは安定性、又は大きさに基づいて分離することができる。分離技術の非限定の例として、硫安塩析、クロマトグラフィー、及び膜濾過(接線流膜濾過を含む)が挙げられる。分離にクロマトグラフィーを利用する実施形態における典型的な方法としては、イオン交換(カチオン又はアニオン)、親和性クロマトグラフィー、親水性相互作用、疎水性相互作用、サイズ排除及びゲル浸透が挙げられる(米国特許第6248570号参照)。
【0051】
いくつかの実施形態において、その分離は、膜濾過によって行うことができ、それには、以下のものに限定はされないが、シングルパス、デッドエンド、ダイレクトフロー濾過(DFF,direct flow filtration)、及びクロスフロー又は接線流濾過(TFF,tangential flow filtration)が含まれる。本発明によれば、濾過は、細孔の大きさが規定範囲の半透膜を使用して大きさによって分子を分離する原理に基づいている。濾過方法及び膜のタイプの組合せを分離に使用することができることは当業者には知られている。
【0052】
本発明によれば、膜濾過は、ポリマー膜又は無機膜により生ずる細胞成分の分離である。当技術分野において、膜にはそれらが担体液から除去する材料の大きさによって定義される一般に是認されている4つのカテゴリーが存在する。細孔の大きさの最小の膜から最大の膜へと通す連続的濾過方法は、逆浸透(RO,Reverse Osmosis)、ナノ濾過(NF,Nanofiltration)、限外濾過(UF,Ultrafiltration)、及び精密濾過(MF,Microfiltration)である。
【0053】
上記の膜による濾過は、特定の細孔の大きさを有する膜を用いて、それらの分子量に応じて分子を分離する。例えば、0.001マイクロメートル未満の細孔の大きさを有するRO膜は、200ダルトン未満の分子量を有する分子を分離することを目的としている。0.001以上〜0.008マイクロメートル以下の細孔の大きさを有するNF膜による濾過は、200ダルトン以上〜15キロダルトン(kDa)以下の分子量を有する分子を分離することを目的としている。0.005以上〜0.1マイクロメートル以下の細孔の大きさを有するUF膜による濾過は、5kDa以上〜300kDa以下の分子量を有する分子を分離することを目的としている。0.05以上〜3.0マイクロメートル以下の細孔の大きさを有する精密濾過膜による濾過は、100kDa〜3000kDa以上の分子量を有する分子を分離することを目的としている。
【0054】
本発明によれば、膜濾過は、膜の細孔の大きさによって決まる特定の分画分子量(MWCO,Molecular Weight Cut-Off)を有する膜を利用することによるサイズ排除に基づいて、抽出したポリサッカライドを他の細胞成分から分離することができる。このMWCOは、公称分子量限界(NMWL,Nominal Molecular Weight Limit)又は公称分画分子量(NMWCO,Nominal Molecular Weight Cut-Off)とも呼ばれ、膜による濾過に対するキロダルトンの大きさの呼称である。このMWCOは、膜によって90%が保持される分子の分子量として定義される。例えば同じ分子量の分子は著しく異なる形状を有することがあり得るために、MWCOは、正確な測定基準ではないが、それにもかかわらず有用な測定基準であり、フィルターメーカーにより一般的に採用されている。疎水性並びに親水性の膜の両方を、本発明では使用することができる。そのような膜は、平らなシートとして、又はらせん状にねじれた形態で使用することができる。中空糸もまた使用することができる。UF膜の組成に関しては、可能性のあるいくつもの膜材料を使用することができ、以下に限定はされないが、再生セルロース、ポリエーテルスルホン(これはその固有の疎水性を改変するために修飾してもしなくてもよい)、ポリフッカビニリデン、並びにセラミックと金属酸化物との凝集体が挙げられる。多くのポリエーテルスルホンUF膜が、0.5〜13のpH範囲、及び85℃まで変動する温度に耐えることができる。MF膜用の材料は、UF膜用に使用されるすべて、並びに、ポリカーボネート、ポリプロピレン、ポリエチレン及びPTFE(テフロン(登録商標))さえも含む。
【0055】
いくつかの実施形態において、第1の加水分解ステップ後の反応混合物は、継続する膜又はクロマトグラフィーの使用を必要とする分離及び精製のステップを細胞残屑が妨げることを防止するために、大きな細胞残屑を小さい細胞成分から分離するために濾過することができる。これらの実施形態において、その透過液は、ポリサッカライドを含み、回収される。いくつかの実施形態において、第1の加水分解後の反応混合物は、0.05〜3.0マイクロメートルの間の細孔の大きさを有する精密濾過膜(例えば、500kDa〜3000kDa以上のMWCOを有するフィルター)により濾過することができる。いくつかの実施形態において、第1の加水分解後の反応混合物は、0.05〜0.5マイクロメートルの間(例えば、0.1〜0.2μm)の細孔の大きさを有する精密濾過膜により濾過することができる。いくつかの実施形態において、第1の加水分解ステップ後の反応混合物は、0.05〜3.0マイクロメートル(μm)のポリエーテルスルホン中空糸のフィルターモジュールにより濾過する。
【0056】
いくつかの実施形態において、膜は、5kDa〜300kDaの範囲内のMWCOを有する分離ステップにおいて使用することができる。例えば、そのMWCOは、5kDa〜200kDa、30kDa〜100kDa、10kDa〜60kDa又は50kDa〜200kDaであり得る。これらの実施形態において、その未透過液は、ポリサッカライドを含み、回収することができる。
【0057】
いくつかの実施形態において、500kDa〜3000kDa濾過ステップ及び5kDa〜300kDa濾過ステップの両方が実施される。例えば、500kDa〜3000kDa濾過による透過液を、次に5kDa〜300kDa膜により濾過し、その最後の未透過液を回収することができる。
【0058】
いくつかの実施形態において、接線流濾過は、両方のダイアフィルター(diafilter)(小さい分子を膜を通して洗浄し、興味のある大きい分子を未透過液中に残す分画プロセス)に作用し、反応混合物を濃縮することができる。本発明によれば、ダイアフィルトレーション(diafiltration)は、不連続式又は連続式ダイアフィルトレーションのいずれかである。不連続式ダイアフィルトレーションにおいては、溶液は濃縮され、失われた容積は新たな緩衝液により置換される。連続式ダイアフィルトレーションにおいては、溶液の容積は、新たな緩衝溶液の流入により維持され、その間古い緩衝溶液は除去される。
【0059】
いくつかの実施形態において、ポリサッカライドの分離及び精製は、限外濾過膜を用いる接線流濾過法によって実施することができる。
【0060】
いくつかの実施形態において、クロマトグラフィーは、ポリサッカライドの細胞成分からの分離に使用しない。
【0061】
いくつかの実施形態において、限外濾過膜を使用して、膜のMWCOが5kDa〜100kDaであり得る第1の分離ステップにおいて、GBS CPSを分離することができる。例えば、限外濾過膜のMWCOは、15kDa〜50kDaであり得る。例えば、GBS CPSを分離するために使用する限外濾過膜のMWCOは30kDaであり得る。
【0062】
いくつかの実施形態において、分離ステップは、5kDa〜200kDaのMWCOを有する限外濾過膜を使用する。そのUF膜は、第1の分離ステップにおいて、髄膜炎菌(Neisseria meningitidis)B型、C型、Y型又はW135型;大腸菌K1(E. coli K1);C群連鎖球菌サブCPS(group C Streptococcus sub-CPS);又はA群連鎖球菌サブCPS(group A Streptococcus sub-CPS)を分離するために使用することができる。いくつかの実施形態において、Y群髄膜炎菌莢膜ポリサッカライド(GYMP,group Y meningococcus capsular polysaccharide)及びW−135群髄膜炎菌莢膜ポリサッカライド(GWMP,group W-135 meningococcus capsular polysaccharide)を分離するために使用される限外濾過膜のMWCOは、100kDaであり得る。
【0063】
(第2ステップの加水分解)
第1ステップの分離に由来するポリサッカライドは、第2の試薬、塩基試薬と接触させて第2の混合物を形成する。理論に縛られるものではないが、この第2ステップの加水分解は、(1)ポリサッカライドの自然の構造又は天然の抗原性を維持しながら、ポリサッカライドに付着して残留している細胞成分、特に細胞壁成分を引き離すこと、及び(2)その他の細胞成分混入物質(例えば、タンパク質及び核酸など)を分解すること、の少なくとも1つに対して有用であり得る。いくつかの実施形態において、この第2ステップの加水分解は、実質的に純粋なポリサッカライドを得るための複雑なクロマトグラフ法に対する必要性を取り除くことができる。
【0064】
(第2の試薬組成物)
第2の試薬は、いろいろな塩基の1つ又は複数を含む。本発明に従って使用することができる塩基の非限定の例は、NaOH、KOH、LiOH、NaHCO3、Na2CO3、K2CO3、KCN、Et3N、NH3、H2N2H2、NaH、NaOMe、NaOEt及びKOtBuから選択される化合物を含む。いくつかの実施形態において、NaOH、KOH、LiOH、NaH、NaOMe又はKOtBuなどの塩基は、1N〜10Nの範囲(例えば、1N〜6N、又は2〜5N)で使用することができる。いくつかの実施形態において、NaHCO3、Na2CO3、K2CO3及びKCNなどの塩基は、それらの溶解性が許容する高さの濃度で使用することができる。いくつかの実施形態において、トリエチルアミン(Et3N)などの有機塩基は、水又はアルコールなどの加水分解を生じる作用物質が存在する限り、中濃度から高濃度、例えば、1Nから7N(例えば2N〜4N)の範囲で使用することができる。アンモニア(NH3)又はヒドラジン(H2NNH2)などの塩基は、100%を含む殆んどどんな濃度でも使用することができる。溶媒、例えば、水、アルコール類(好ましくはC1−C4)、ジメチルスルホキシド、ジメチルホルムアミド、又はこれら及びその他の有機溶媒の混合物などを使用することができる。水を含む加水分解用の塩基溶液も使用することができる。
【0065】
いくつかの実施形態において、第2の試薬のpHは、9〜15の範囲内である。例えば、第2の試薬のpHは、10〜15、10〜14、11〜14、又は12〜14の任意の範囲内であり得る。いくつかの実施形態において、第2の混合物は、9〜15の範囲のpHを有し、例えば、その第2の混合物は、10〜15、10〜14、10〜13、11〜14、又は11〜13のいずれかの範囲のpHを有することができる。
【0066】
その第2の試薬は、さらなる物質、例えば、塩類(例えば、NaCl又はKCl)、酸化剤(例えば、H2O2、O3、又は次亜塩素酸塩)、ホウ素を含む還元剤(例えば、NaBH4、NaCNBH3、リチウムトリ−sec−ブチルボロヒドリド、又はNaBH(OCOCH3)3を含む)、その他の還元剤(例えば、水素化アルミニウムリチウム、ジチオスレイトール、又はβ−メルカプトエタノールが挙げられる)、酵素類(例えば、プロテアーゼ類又はヌクレアーゼ類が挙げられる)、キレート化剤(例えば、EDTA又はEGTAが挙げられる)、及び界面活性剤類(非イオン型、両性イオン型、アニオン型又はカチオン型)などを場合によって含むことができる。いくつかの実施形態において、界面活性剤は、その後の濾過ステップにおいて有用であり得る。
【0067】
いくつかの実施形態において、第2の試薬は、塩基及び還元剤を含む。いくつかの実施形態において、その塩基は、1N〜10Nの範囲内であり、その還元剤は、0.03mM〜300mMの範囲内である。例えば、その塩基は、2N〜6Nの範囲内の濃度を有することができ、その還元剤は、0.3mM〜30mMの範囲内の濃度を有することができる。典型的な第1の試薬は、塩基及び水素化ホウ素ナトリウムを含む。いくつかの実施形態において、その塩基の濃度は、1N〜10Nの範囲内であり、水素化ホウ素ナトリウムは、0.03mM〜300mMの範囲内である。例えば、その塩基の濃度は、2N〜6Nの範囲内であり得、水素化ホウ素ナトリウムの濃度は、0.3mM〜30mMの範囲内である。いくつかの実施形態において、その塩基は、NaOH、LiOH及びKOHから選択される。
【0068】
(使用)
加水分解は、20℃〜100℃の温度、例えば、30℃〜100℃、37℃〜100℃、40℃〜100℃、50℃〜95℃、60℃〜90℃、70℃〜90℃、又は80℃で達成することができる。いくつかの実施形態において、混合物は、反応が起こることを可能にするために3〜72時間にわたってインキュベートする。例えば、そのインキュベーションは、4、5、6、7、8、9、10、12、14、15、16、18、20、24、36、48、60、若しくは72時間又はその間の任意の時間続けることができる。いくつかの実施形態において、その混合物はインキュベーションの間は、かき混ぜる。いくつかの実施形態において、その混合物はインキュベーションの間は、かき混ぜない。
【0069】
いくつかの実施形態において、第2ステップの加水分解は、第1ステップの加水分解と比較したとき、(1)より高温、(2)絶対値が7からずっと離れたpH、(3)モル濃度のより高い試薬、及び(4)より長いインキュベーション時間のいずれか1つを用いて達成される。いくつかの実施形態において、第2ステップの加水分解は、第1ステップより少なくとも10、20、30、40、50、60、又は70℃高い温度で起こり得る。いくつかの実施形態において、この第2ステップは、第1ステップより1、5、10、15、20、25、30、35、40、又は45時間長く、或いは1時間と45時間の間の任意の時間量だけ長く続けることができる。いくつかの実施形態において、第2ステップの混合物のpHは、第1ステップの混合物より、pH7からの絶対値で、0.1、0.2、0.3、0.4、0.5、1、1.3、1.5、2、2.3、2.5、3、3.3、3.5、4、4.5、又は5単位、或いは、0.1と5の間の任意の単位量だけ離れていることができる。第2ステップの加水分解でこれらのより強力な条件を用いる実施形態においては、他の細胞成分、例えば、タンパク質及び核酸などを、第1ステップの加水分解におけるよりもさらに効率的に分解することができる。
【0070】
1つの典型的な第2ステップの加水分解は、1〜2NのNaOHを、37℃で12〜48時間にわたって使用する。もう1つの典型的な第2ステップの加水分解は、2NのNaOHを、80℃で15〜48時間にわたって使用する。
【0071】
GBS CPSに対する第2の加水分解は、いくつかの実施形態において、2NのNaOHの最終濃度、80℃、16〜18時間の条件下で行うことができ、この条件は、GBS CPSに対する第1の加水分解に対する典型的な条件(1NのNaOHの最終濃度、37℃、16時間)より強い条件である。
【0072】
GYMPに対する第2の加水分解は、いくつかの実施形態において、2NのNaOHの最終濃度、80℃、48時間の条件下で行うことができ、この条件は、GYMPに対する第1の加水分解に対する典型的な条件(1NのNaOHの最終濃度、37℃、16時間)より強い条件である。
【0073】
GWMPに対する第2ステップの加水分解は、いくつかの実施形態において、2NのNaOHの最終濃度、80℃、一晩(本明細書で使用される「一晩」とは、15〜18時間を意味する)の条件下で行うことができ、この条件は、GWMPに対する第1ステップの加水分解に対する典型的な受験(1NのNaOHの最終濃度、37℃、16時間)より強い条件である。
【0074】
(第2ステップの分離)
第2ステップの加水分解に由来する抽出ポリサッカライドは、細胞成分に由来する不純物からの第1ステップの分離と同じように、以下に限定はされないが、クロマトグラフィー及び膜濾過などの分離技術によって分離することができる。クロマトグラフ分離法の非限定の例は、イオン交換(カチオン又はアニオン)、親水性相互作用、疎水性相互作用、又はゲル浸透クロマトグラフィーである。典型的な方法は、限外濾過膜を用いる接線流濾過である。
【0075】
いくつかの実施形態において、クロマトグラフィーは、ポリサッカライドの第2ステップの分離では使用しない。いくつかの実施形態において、クロマトグラフィーは、ポリサッカライドを精製するプロセスでは使用しない。
【0076】
いくつかの実施形態において、第2ステップの分離(例えば、GBS CPSに対して)は、20〜50kDa(例えば、30kDa)のMWCOを有する限外濾過膜により実施する。
【0077】
いくつかの実施形態において、ある程度大きなタンパク質(例えば、300kDaを超える)は、加水分解ステップによる分解に耐性を示す(例えば実施例2及び3参照)。これらのタンパク質を除去するために、第2の加水分解ステップからの混合物は、高いMWCO、例えば、200〜400kDa(例えば、300kDa)を有する限外濾過膜又は精密濾過膜に取り込む。その未透過液は、分解に耐性を示す大きなタンパク質を保持するので、その透過液を次に集める。その透過液は、濃縮し、別の膜、例えば、5〜100kDa、好ましくは5〜50kDa、最も好ましくは30kDaのMWCOを有する限外濾過膜に取り込む前にダイアフィルターにかけて、小さい分子量の不純物、例えば核酸及び分解したタンパク質を除去することができる。
【0078】
いくつかの実施形態において、第2ステップの分離(例えば、GYMP又はGWMPのための)は、塩基による加水分解の分解に耐性を示す大きなタンパク質を除去するために最初に300kDaのUF膜により行う。得たれた透過液を次に30kDaのUF膜に取り込んで小さい分子量の不純物を除去する。その30kDaUF膜濾過ステップからの未透過液を、後に続くステップのために用いる。
【0079】
(任意のN−アシル化)
ポリサッカライドを他の細胞成分から最終的に分離した後、加水分解手段によりポリサッカライドを脱アセチル化することができるので、生来のアセチル基が除去されている可能性のある遊離のアミノ基を再アシル化することができる。アシル化試薬及び反応条件を変化させることによって、その実行者は、アミノ基が再アシル化される度合いを制御することができる。そのアシル化試薬及びそれらの崩壊産物は、再アシル化されたポリサッカライドと比較して大きさが小さく、従って、サイズ排除法、例えば、ゲル浸透クロマトグラフィー又は膜濾過によってポリサッカライドから分離することができる。別法では、極性又は電荷の違いを、クロマトグラフィーに生かしてポリサッカライドを精製することができる。
【0080】
分離されたポリサッカライドは、様々なアシル化剤を用いることにより、任意で所望の程度まで再アシル化することができる。アシル化剤の非限定の例は、無水酢酸、塩化アセチル、酢酸ペンタフルオロフェニル、酢酸4−ニトロフェニル(非特許文献51)である。好ましいアシル化方法は、ポリサッカライドの遊離のアミノ基を再アシル化し、従って生来のポリサッカライド構造を再生するために、分離したポリサッカライドを、0.5M〜2M(例えば、0.7M〜1M)の濃度のアシル化剤と混合することに関連する。例えば、米国特許第5969130号、同第5576002号、同第4727136号、及び同第4356170号を参照されたい。
【0081】
(任意のN−アシル化後の精製)
いくつかの実施形態において、再アシル化したポリサッカライドは、クロマトグラフ精製又は膜濾過を用いて精製することができる。例えば、イオン交換(カチオン又はアニオン)、疎水性相互作用、親水性相互作用、ゲル浸透クロマトグラフィー、ダイレクトフロー濾過、又は接線流濾過は、すべて、再アシル化ポリサッカライドの反応成分からの分離を達成するために使用することができる。典型的な方法は、スーパーデックス(Superdex)(架橋したアガロース及びデキストラン)に基づくゲル浸透クロマトグラフィー又は残留混入物質を除去し、精製ポリサッカライドを提供する限外濾過膜による接線流濾過のいずれかを利用する。Superdexに基づくゲル浸透クロマトグラフィーを利用するいくつかの実施形態において、そのクロマトグラフィーは、溶離剤としてPBSを用いて、0.1mL/分〜10mL/分であり得る流速で、デキストランに対して1,000〜100,000分別範囲(MW)を有するSuperdex 200PGを使用することができる。接線流濾過を利用するいくつかの実施形態において、限外濾過膜は、例えば、5kDa〜100kDa(例えば、5kDa〜50kDa、又は5kDa〜30kDa)のMWCOを有するものを使用することができる。
【0082】
(純度)
精製したポリサッカライドの純度の程度は、修正マイクロスケールオルシノールアッセイ(非特許文献35)により評価された乾燥組成物中のシアル酸の重量百分率によって測定される。いくつかの実施形態において、精製ポリサッカライドの純度は、80%〜100%である。典型的な純度としては、85%〜100%、90%〜100%、94%〜100%、95%〜100%、及び99%以上が挙げられる。
【0083】
いくつかの実施形態において、純度は、質量百分率として表される260nmにおけるUV測光により検出される核酸の量によって特徴付けることができる。いくつかの実施形態において、精製したポリサッカライドの純度は、<5%の核酸、例えば、<4%、<3%、<2%、<1%、又は<0.5%の核酸であり得る。
【0084】
いくつかの実施形態において、純度は、Pierce社製(ロックフォード、イリノイ州)のCommie Plus試薬及び標準としてヒトIgGを用いるブラッドフォード法(非特許文献9)によって測定される精製ポリサッカライド中のタンパク質の質量百分率によって特徴付けることができる。いくつかの実施形態において、精製したポリサッカライドの純度は、<5%のタンパク質、例えば、<4%、<3%、<2%、<1%、<0.5%、<0.1%、又は<0.05%のタンパク質であり得る。
【0085】
いくつかの実施形態において、精製ポリサッカライドの純度は、<3%の核酸及び<1%のタンパク質であり得る。いくつかの実施形態において、精製ポリサッカライドの純度は、<1%の核酸及び<1%のタンパク質であり得る。いくつかの実施形態において、精製ポリサッカライドの純度は、<1%の核酸、<1%のタンパク質、及び90%〜100%のポリサッカライドであり得る。
【0086】
いくつかの実施形態において、精製ポリサッカライドの純度は、少なくとも、哺乳類(例えばヒト)を治療するワクチン、及びポリサッカライドとタンパク質との結合体を製造するタンパク質に連結するために使用するためには十分であり得る。
【0087】
(結合体分子)
いくつかの実施形態において、本発明のポリサッカライドは、グラム陰性及びグラム陽性細菌を含む様々な微生物に対する抗体応答を、個体中で、単独、又は別の免疫原性分子、例えばポリペプチド又は複数の担体タンパク質、単数の担体タンパク質に結合している場合のいずれかで引き出すために使用することができる。ポリサッカライドのポリペプチドへの結合は、一般的にはT細胞非依存性ポリサッカライドに対する免疫応答をT細胞依存性ポリサッカライドに転化する。従って、ポリペプチドの大きさは、T細胞非依存性からT細胞依存性への応答の転化を引き起こすのに十分なものが好ましい。第2の免疫原を提供するためには小さめのポリペプチドを使用することが役立つ。
【0088】
結合に役立つポリぺプチドの非限定の例としては、担体タンパク質、スカシガイヘモシアニン(KLH,keyhole limpet hemacyanin)、ウシ血清アルブミン(BSA,bovine serum albumin)、卵白アルブミン(OVA,ovalbumin)、ウサギ血清アルブミン(RSA,rabbit serum albumin)、破傷風トキソイド、ジフテリアトキソイド、ニューモリソイド(pneumolysoid)、外膜タンパク質、ウイルス糖タンパク質、並びにそれらの変異体、フラグメント、及び模倣体が挙げられる。破傷風毒素のCフラグメント(TTc,Fragment C of tetanus toxin)及び組換え型TTc(rTTc,recombinant TTc)は、ポリサッカライドのための担体として使用することができる。Cフラグメント(TTc)は、国際公開第2005/000346号によって報告されているように約865〜1315のアミノ酸を含む破傷風毒素のカルボキシル末端部分である。Cフラグメントの組換え発現は、米国特許第5443966号に報告されている。当業者であれば、ポリサッカライドに結合する免疫原性分子を選択することについて理解することができる。
【0089】
結合の任意の様式を採用して、ポリサッカライド成分をペプチドと結合することができる。好ましい方法は、米国特許第4356170号及び同第4902506号に記載されており、それらは、隣接ジオールの酸化的開裂を経てポリサッカライド中への末端アルデヒド基を導入し、そのアルデヒド基を還元的アミノ化によってペプチドのアミノ基に結合させることを記載している。
【0090】
しかしながら、本発明の結合分子は、還元的アミノ化を経て産生されるものに限定されないことを理解すべきである。従って、ワクチンは、当業者には既知の任意の連結方法、例えばSchneerson, R. et al.による(非特許文献55)及び米国特許第4644059号などに記載されているようなアジピン酸ジヒドラジドのスペーサー、又は、例えば、Marburg, S. et al.による(非特許文献56)に記載されているようなバイナリースペーサー技術を用いてポリサッカライドをペプチドと結合することによって産生することもできる。
【0091】
本発明に従って調製された精製ポリサッカライドは、ペプチドがポリサッカライド上の1つ又は複数の場所を介してポリサッカライドに連結している結合分子を産生するために使用することができる。従って、タンパク質成分に関して本発明に従って調製された結合分子は、ポリサッカライドが多様なタンパク質と一緒に架橋している、単量体、二量体、三量体、及びより高度に架橋した分子であり得る。
【0092】
(ワクチン類)
いくつかの実施形態において、ポリサッカライドワクチンを製造することができる。そのポリサッカライドは上記の方法によって精製することができる。これらのポリサッカライドは、ポリサッカライドに対して反応性であり従ってそのポリサッカライドが単離された有機体に対して反応性である抗体を発生する抗原として使用することができる。より具体的には、これらの実施形態は、例えば、ヒト又は動物を、一般的には当該ポリサッカライドが単離された、細菌、酵母菌、又は原虫の菌株による感染に対して保護することができる、単独での又はポリペプチドに結合したポリサッカライドワクチンの製品を提供する。ある場合には、本発明で使用されるポリサッカライドは、他の病原性微生物と交差反応性である抗体の産生を誘発することができ、それによってこれらの他の微生物による感染に対する保護を生み出す。ある場合には、本発明によるポリサッカライドの使用は、癌の治療又は予防に役立ち得る。
【0093】
さらに、本発明に従って精製したポリサッカライドにより調製されたワクチン類は、免疫原に対して宿主に免疫を予防的様式で与える免疫システムを活性化するのに役立つアジュバント類、例えば、ミョウバン、水酸化アルミニウム、ステアリルチロシン、及びフロイントアジュバントなども含むことができる(例えば、米国特許第5773007号参照)。いくつかの実施形態において、アジュバントは、CD14受容体アゴニスト、トール様受容体アゴニスト、QS21、MF59、モノホスホリル脂質A(MPL)から選択することができる。
【0094】
本発明のワクチン類は、能動免疫又は受動免疫を提供することができる。能動免疫を提供するためのワクチン類は、本発明の精製ポリサッカライドを含む。好ましくは、その精製ポリサッカライドは、莢膜ポリサッカライドである。
【0095】
いくつかの実施形態において、ポリサッカライド結合ワクチンを製造することができる。そのポリサッカライドは上記の方法によって精製することができる。その精製ポリサッカライドは、上の結合分子の項で記載したようにポリペプチドに結合することができる。
【0096】
本発明に従って精製したポリサッカライドから調製することができるワクチン類の非限定の例としては、A群連鎖球菌ワクチン(米国特許第5866135号参照)、B群連鎖球菌II型及びIII型ワクチン(米国特許第5302386号、同第6284884号参照)、及びB群髄膜炎菌ワクチン(米国特許第5969130号、同第5902586号、同第5969130号、同第5683699号、同第5576002号、同第5811102号を参照)が挙げられる。さらに、本発明に従って精製したポリサッカライドは、混合ワクチンに使用することもできる。
【0097】
(抗体類)
上に記載したポリサッカライドの加水分解及び分離の技術は、本発明の単離したポリサッカライドの大量生産を提供する。これは、ポリサッカライドに対して反応性の抗体の発生を促進する。本発明の1つの実施形態は、抗体の産生を引き出すことができる実質的に純粋な又は単離されたポリサッカライドを産生するための方法を提供する。使用するポリサッカライドに応じて、得られる抗体は、殺菌性、静菌性、殺真菌性、又は殺原虫性であり、抗体応答を引き出すために用いたポリサッカライドと交差反応性である抗体を含有する微生物による感染に対して動物を保護するものであり得る。
【0098】
単離されたポリサッカライド又はそこの免疫原性部分に対して特異的な抗体は、当技術分野では長期にわたって知られており、標準的に実施されている方法を用いて発生させることができる。かかる抗体としては、以下に限定はされないが、多クローン性の、単クローン性の、キメラの、単一鎖のFabフラグメント及びFab発現ライブラリーにより産生されたフラグメントを挙げることができる。中和抗体(即ち、二量体形成を阻害するもの)は、治療上の使用に対して特に好ましい。
【0099】
抗体の産生のために、ヤギ、ウサギ、ヒツジ、ネズミ、マウス、ウマ、雌ウシ、ヒトなどを含む様々な宿主を、免疫特性を有する単離したポリサッカライド、又はそれらの任意のフラグメント又は結合体を注射することによって免疫にすることができる。宿主の種類に応じて、様々なアジュバントを、免疫応答を増すために使用することができる。適当なアジュバントの非限定の例としては、フロイントアジュバント(完全又は不完全)、水酸化アルミニウム又はシリカ等のミネラルゲル類、及び界面活性物質、例えば、リゾレシチン、プルロニックポリオール類、多価アニオン類、ペプチド類、油エマルジョン類、KLH、及びジニトロフェノールなどが挙げられる。ヒトに一般的に使用されるアジュバントとしては、カルメット・ゲラン桿菌(BCG,bacilli Calmette Guerin)及びコリネバクテリウム・パルバム(Corynebacterium parvumn)が挙げられる。
【0100】
単離したポリサッカライドに対する単クローン抗体、又はそれの免疫原性フラグメントは、培養液中の連続継代細胞系による抗体分子の産生を提供する任意の技術を用いて調製することができる。これらには、以下に限定はされないが、ハイブリドーマ技術、位相表示(phase display)、ヒトB細胞ハイブリドーマ技術、及びEBV−ハイブリドーマ技術が含まれる(G. Kohler et al., 1975, Nature, 256:495-497;D. Kozbor et al., 1985, J. Immunol. Methods, 81:31-42;R.J. Cote et al., 1983, Proc. Natl. Acad. Sci. USA, 80:2026-2030;及び S.P. Cole et al., 1984, Mol. Cell Biol., 62:109-120)。この単クローン抗体の産生は、当技術分野では周知であり、ごく普通に使用されている。
【0101】
一方法によれば、ハイブリドーマ株化細胞の培養液が使用される(非特許文献52)。ポリサッカライドに対して向けた単クローン抗体類は、当技術分野では一般的に知られている技術により製造された、ヒト単クローン抗体、キメラ単クローン抗体、又はヒト化単クローン抗体であり得る。1つのアプローチによれば、キメラ単クローン抗体は、ヒトの定常部位と組み合わされたヒトではない(例えば、マウス)抗原結合領域を有するように発生させることができる(非特許文献53)。ヒト化抗体は、Queenらの米国特許第5585089号の手順に従って発生させることができる。単離されたポリサッカライド、ペプチド又はそれらのフラグメントは、単一のエピトープ又は抗原決定基或いは複数のエピトープを含むことができる。単離されたポリサッカライドの一部は、別のタンパク質のそれ、例えばKLHなどと融合することができ、抗体がキメラ分子に対して産生される。
【0102】
さらに、キメラ抗体の産生、適切な抗原特異性を備えた分子を得るためのヒト抗体遺伝子へのマウス抗体遺伝子のスプライシング及び生物活性のために開発された技術を使用することができる(S.L. Morrison et al., 1984, Proc. Natl. Acad. Sci. USA, 81:6851-6855;M.S. Neuberger et al., 1984, Nature, 312:604-608;及びS. Takeda et al., 1985, Nature, 314:452-454)。別法では、一本鎖抗体の産生について記載されている技術を、当技術分野で知られている方法を用いて、ポリサッカライド特異性の一本鎖抗体を産生するために適用することができる。関連するがはっきりとしたイディオタイプの組成物の特異性を備えた抗体を、無作為な免疫グロブリンの組合せライブラリーからのチェインシャフリング(chain shuffling)によって発生させることができる(D.R. Burton, 1991, Proc. Natl. Acad. Sci. USA, 88:11120-3)。抗体は、また、文献に開示されているように、リンパ球集団中のインビボ産生を引き起こすことによるか、非常に特異的な結合試薬の組換え型免疫グロブリンライブラリー又はパネルをスクリーニングすることによって産生することができる(R. Orlandi et al., 1989, Proc. Natl. Acad. Sci. USA, 86:3833-3837及びG. Winter et al., 1991, Nature, 349:293-299)。
【0103】
ポリサッカライドに対して向けられた抗体は、以下に限定はされないが、免疫吸収又は免疫親和性クロマトグラフィー或いはその他のクロマトグラフ法(例えば、HPLC)を含む当技術分野では周知の任意の技術によって精製することができる。抗体は、また、血清、血漿、又は細胞培養液からの免疫グロブリン画分として精製することもできる。
【0104】
単離されたポリサッカライドについての特定の結合サイトを含有する、Fab、Fab’、F(ab’)2、Facb、Fv、ScFv、Fd、VH、及びVLを含むがこれらには限定されない抗体フラグメントもまた発生させることができる。本発明の抗体分子は、インタクトな免疫グロブリン分子、実質的にインタクトな免疫グロブリン分子、又は抗原結合サイトを含有する免疫グロブリン分子のそのような部分、例えば、Fabフラグメントであり得る。ポリサッカライドに対して向けられた抗体のフラグメントは、当技術分野では一般的に知られている任意の技術(非特許文献54)によって発生させることができる。例えば、かかるフラグメントとしては、以下に限定されないが、抗体分子のペプシン消化によって産生することができるF(ab’)2フラグメント及びF(ab’)2フラグメントのジスルフィド架橋を還元することによって発生させることができるFabフラグメントが挙げられる。別法では、Fab発現ライブラリーは、所望の特異性を有する単クローンFabフラグメントの迅速で容易な同定を可能にするように構成することができる(W.D. Huse et al., 1989, Science, 254.1275-1281)。
【0105】
様々なイムノアッセイを、所望の特異性を有する抗体を同定するスクリーニングのために使用することができる。立証されている特異性を有する多クローン性又は単クローン性抗体を用いる競合結合アッセイ又は免疫放射線アッセイのための多数のプロトコールが当技術分野では周知である。かかるイムノアッセイは、単離されたポリサッカライドとその特定の抗体との間の錯体の形成を測定するステップを一般的には含む。2つの非干渉性ポリサッカライドエピトープと反応する単クローン性抗体を利用する2部位の単クローンに基づくイムノアッセイが好ましいが、競合結合アッセイを採用することもできる(Maddox、上記参照)。
【0106】
本発明の別の態様は、哺乳動物に、前記動物を、感染、例えば、細菌、真菌、原虫及びウイルス感染、特にHIV−1又はHIV−2によって引き起こされる感染から保護するための抗体及び/又はT細胞免疫応答を産生するのに適切な単離されたポリサッカライド又はそのフラグメントを接種することを含む哺乳動物における免疫応答を誘発するための方法に関する。本発明のさらに別の態様は、単離したポリサッカライドを、哺乳動物を疾患から保護する抗体を産生するような免疫応答を誘発するためにインビボでそのポリヌクレオチドの発現を方向付けるベクターを介して送達することを含む哺乳動物において免疫応答を誘発する方法に関する。
【0107】
(医薬品組成物)
本発明の医薬品組成物は、ポリサッカライド又はポリサッカライドと薬理学的に許容される担体、例えば、生理食塩水、ブドウ糖、グリセロール、エタノールなどを含む結合分子を含むことができる。別の実施形態において、前記医薬品組成物は、別の免疫原性部分、例えば、ペプチドなど、又は本発明のポリサッカライドの1つによって誘発される抗体を含む組成物を含む。前記医薬品組成物は、また、レシピエントの免疫応答を高めるアジュバントを含むこともできる。かかるアジュバントは、ミョウバン等のアルミニウム系又はステアリルチロシン等の長鎖アルキルアジュバントであり得る(1990年9月17日出願の米国特許第5773007号、欧州特許第0549617131号、Moloneyらの米国特許第4258029号参照)。Jenningsら(米国特許第5683699号)及びPaolettiらの(非特許文献58)も参照されたい。これらの医薬品組成物は、ワクチンとして特に有用である。
【0108】
受動免疫を誘発するために、医薬品組成物は、多クローン性抗体又は単クローン性抗体、或いはそれらの誘導体、或いはそれらのフラグメントを上記のように含むことができる。その抗体、フラグメント、又は誘導体の量は、標準的な臨床技術によって決定される治療的又は予防的に有効な量である。
【0109】
いくつかの実施形態において、本発明のポリサッカライドに対して向けられた抗体は、宿主の個体から別の個体に受動免疫を与えるための(即ち、グラム陰性又はグラム陽性細菌に対する個体の免疫応答を増大するため、又はエイズ患者を含む免疫無防備状態の又は免疫が枯渇した個体における応答を提供するための)治療的又は予防的適用における医薬品製剤として使用することができる。抗体の受動的伝達は、技術的に知られており、既知の任意の方法によって達成することができる。一方法によれば、本発明のポリサッカライド又はそれの結合体に対して向けられた抗体を、免疫適格性宿主(「ドナー」)動物において発生させ、その宿主動物から収集し、レシピエント個体に注入する。例えば、ヒトのドナーを、本発明のポリサッカライド又はポリサッカライド結合体に対して反応性の抗体を発生させるために使用することができる。その抗体を、次に、治療を必要としているヒトのレシピエントに治療的及び予防的に有効な量で投与し、それによってポリサッカライド成分によって誘発された抗体によって結合される細菌に対して、レシピエントにおける抵抗力を与えることができる(非特許文献57)。
【0110】
いくつかの実施形態において、本発明は、抗体の産生を誘発することができる実質的に純粋なポリサッカライドを産生するための方法を提供することができる。使用するポリサッカライドに応じて、得られる抗体は、殺菌性、殺真菌性、又は殺原虫性であり、抗体応答を引き出すために用いたポリサッカライドと交差反応性である抗体を含有する微生物による感染に対して動物を保護するものであり得る。
【0111】
本発明の医薬品製剤は、当技術分野で有効であることが知られている方法によって個体中に導入することができる。皮内、腹腔内、静脈内、皮下、経口、及び経鼻が導入のルートに入るがこれらだけではない。
【0112】
本発明の組成物は、以下に限定されないが、任意の適当な薬学的及び生理的に許容される担体、例えば、生理食塩水又はその他の注射可能な液体などを含むワクチンに適する当業者には一般的に知られている標準的な担体、緩衝剤、又は保存料を含むことができる。ワクチンには通例の添加剤、例えば、ラクトース又はソルビトール等の安定剤、及びリン酸アルミニウム、水酸化アルミニウム、又は硫酸アルミニウム及びステアリルチロシンなどの免疫原応答を高めるためのアジュバントも存在させることができる。本発明に従って産生されるワクチンは、また、複数の感染病原体に対する免疫応答を誘発する多価ワクチンの成分としても使用することができる。
【0113】
本発明のワクチンは、免疫原応答の一部としての抗体の産生を誘発するのに十分な量で投与する。投与量は、そのワクチンを受ける個体の大きさ、重量、又は年齢に基づいて調節することができる。個体における抗体応答は、抗体力価又は殺菌作用についてアッセイすることによって観測し、必要に応じて増量して応答を高めることができる。一般的に、乳幼児に対する1回の投与量は、1投与当り約10マイクログラム(μg)又は0.5μg〜20μg/kgの結合ワクチンである。成人は、0.5μg〜20μg/kgの結合ワクチンの投与量を受ける。CPSワクチンについては、一般的な投与量は、1投与量当り約25μgの各個体のCPSである。即ち、B群連鎖球菌に対するワクチンは、25μgの9個の血清型のそれぞれからのCPSのそれぞれを含む。各CPSの2μg〜15μgの範囲の投与量が、ポリサッカライド結合ワクチンについては一般に適切である。
【0114】
(診断キット)
別の実施形態において、本発明のポリサッカライド、それらの誘導体、又はフラグメントは、ニューモリシン毒素等の毒素は受け入れないが、グラム陰性又はグラム陽性細菌、或いはその他の微生物に対して向けられた抗体の存在を依然として示すことができるより安全な診断キットを産生するために使用することができる。かかる抗体の存在により、病原体にさらす前に示すことができ、感染に抵抗力があり得る個体を予測することができる。この診断キットは、少なくとも1つの本発明のポリサッカライド、それらの誘導体、又はフラグメント、及び変性したポリサッカライド、それらの誘導体、又はフラグメントが、グラム陰性又はグラム陽性細菌、又はその他の微生物に対して向けられた抗体を含有する試料と混合されるときの抗体反応の検知のための適当な試薬を含むことができる。抗体反応は、以下に限定されないが、ELISA、蛍光発光、比色分析、化学発光、又は電気化学発光アッセイを含む技術的に記載されている任意の方法によって同定することができる。かかる知見は重要であり、例えば、不必要なワクチン接種を避けることができる。
【0115】
別法では、該診断キットは、磁化可能な固体支持ビーズ、又はプラスチック母材、及び少なくとも1つの本発明のポリサッカライド、それらの誘導体又はフラグメントをさらに含むことができる。
【0116】
ある場合には、該ポリサッカライド、それらの誘導体又はフラグメントは標識化することが好ましいかもしれない。標識薬剤は技術的に周知である。例えば、標識薬剤としては、以下に限定されないが、放射能、化学発光、生体発光、発光、又は便利な分析のためのその他の同定及び/又は検出「タグ類」が挙げられる。体液又は組織の試料(例えば、血液、血清、唾液)を集めて精製し、診断キットに適用することができる。該ポリサッカライド、それらの誘導体又はフラグメントは、精製するか精製しなくてもよく、分子の混合物を構成することができる。
【0117】
固体マトリックスが技術的に知られており、利用可能であり、以下に限定されないが、例えば、試験管、ビーズ、マクロ粒子、ミクロ粒子、ディップスティック、プレート、チップなどの形状をした、ポリスチレン、ポリエチレン、ポリプロピレン、ポリカーボネート、又は任意の固体プラスチック材料であるポリマーが挙げられる。さらに、マトリックスとしては、以下に限定されないが、膜、96ウェルマイクロタイタープレート、試験管、及びエッペンドルフ管が挙げられる。一般に上記のマトリックス類は、リガンド結合剤が付着することができる任意の表面又はそれ自体がリガンド付着サイトを提供する表面を有する。
【0118】
[実施例]
以下の実施例は本発明を説明するために提供するが、本発明の範囲を限定するものとは決して解釈すべきでない。多くの変更及び置換えを、本発明の精神及び範囲から逸脱することなく行うことができることを、当業者であれば認識するであろう。
【0119】
(菌種、増殖培地、及び培養条件)
Ib型B群連鎖球菌菌株H36b(ATCC12401)を、アメリカン・タイプ・カルチャー・コレクション(American Type Culture Collection)(ロックビル、メリーランド州)から入手した。使用したその他の菌株、090(Ia型)、18RS21(II型)、M781(III型)、及びCJ11(V型)は、ハーバード・メディカル・スクール(Harvard Medical School)のD.L. Kasperにより、快く提供された。髄膜炎菌(Neisseria meningitidis)B、C、Y及びW135型は、FDA生物製剤評価センター(CBER, FDA)でCarl Fraschにより快く提供され、大腸菌(Escherichia coli)KIは、FDA生物製剤評価センターでWillie Vannにより快く提供された。
【0120】
その細菌菌株のそれぞれを、透析液(10,000MWCO)、6%のグルコースを補った3.5%のColumbia broth(Difco Laboratories, Inc.社製、デトロイト、ミシガン州)のPellicon cassette system(Millipore Corp.社製、ベッドフォード、マサチューセッツ州)中で個々に増殖した。振動三角フラスコ中、37℃で8時間にわたって増殖した150mLの種培養を用いて、14リットルのブロス(上記参照)を満たしたBioflo IV20リットル発酵層(New Brunswick Scientific Co.社製、エジソン、ニュージャージー州)に播種した。その発酵培養液を、37℃に維持し、10NのNaOHの添加によって持続的にpH7.1に調節し、1.5L/分で通気した。その細胞を、17時間後、MiniKros0.1μm孔の中空繊維カートリッジ(Microgon, Inc.社製、ラグナヒル、カリフォルニア州)を通す精密濾過法によって収集した。その培養上澄を、さらに処理が進むまで4℃で無菌状態で維持した。最終の細胞ペレットは、分離した細胞のSorvall GSAローター(DuPont Clinical & Instruments Div.製、ウィルミントン、デラウェア州)中、9000rpmで50分間の遠心分離によって得た。
【0121】
ポリサッカライドは、培養上澄から精製することもできる。細胞の除去後、ブロスを濃縮し、ポリサッカライドを産生するための一般的方法に記載されている手順に従ってダイアフィルターにかける。
【0122】
(モル質量測定)
ポリサッカライドの絶対モル質量分布は、インライン多角度レーザー光散乱光度計及び示差屈折による検出を備えた分析用ゲル浸透クロマトグラフィー(GPC−MALLS/RI)により測定した。この方法は、Jasco社製PU−980HPLCポンプ(イーストン、メリーランド州)、Rheodyne社製モデル7125噴射弁(Cotati、カリフォルニア州)、及びPBSと平衡しており、0.5mL/分の流速のSuperose 6HR 10/30カラムからなる液体クロマトグラフィーシステムに基づいて実施した。移動相は、超純水(Stephens Scientific社製、リバデール、ニュージャージー州)中で調製し、MilliporeタイプGVの0.22mm膜を装着した直径25mmのインラインフィルター(Millipore社製)を通して濾過した。ポリサッカライドの試料(1〜2mg)は、移動相中に10mg/mLの濃度で溶解し、得られた溶液を微小遠心分離機中14,000rpmで2〜3分間にわたり遠心分離して注入前に粒子状物質を除去した。カラム流出液は、Hewlett-Packard社製モデル1047A示差屈折計に連結しているインラインのDawn E eighteen角光散乱光度計(Wyatt Technology Corp.社製、サンタバーバラ、カリフォルニア州)により直接分析した。その屈折計のアナログ信号出力を補助入力チャネルを介してそのDOWN Eと接続した。光散乱データを獲得し、Wyatt's ASTRA 4.73.40ソフトウェアにより処理した。ピーク面積を、そのWyattソフトウェアにより、ピークの全範囲にわたって、200〜300の台形部分、又は「スライス」の面積の合計として計算した。従って得られたその面積から、所与のピークで溶離するポリサッカライドの重量平均及び数平均モル質量(それぞれ、Mw及びMn)を計算した。比屈折率増分(dn/dc)を0.141mL/gであるすべてのポリサッカライドについてオンラインHP 1047A屈折計を用いて測定した。この値は、他のポリサッカライドについて以前に得られた値(非特許文献7、8、40)と肩を並べた。
【0123】
(一次元1H NMRスペクトル)
D2O(Aldrich社製)中のポリサッカライド試料(4〜5mg/mL)の一次元1H NMRスペクトルを、Bruker Instruments社製AMX 500スペクトロメーター(ビルリカ、マサチューセッツ州)により500MHzで記録した。スペクトルデータは、50℃で取得し、化学シフトは、D2O中の外部の3−トリメチルシリルプロピオネートスルホン酸(Aldrich社製)を参照する。
【0124】
(精製ポリサッカライドの分析)
ステップの収量及び最終の精製試料の内容は、シアル酸定量分析に対する改良されたレゾルシノールアッセイ(非特許文献59)によって測定した。簡潔に記せば、5〜50μgのN−アセチルノイラミン酸(NeuAc)標準又は50μg/mLの莢膜ポリサッカライドを含有する1mLの試料又は対照に、1mLのレゾルシノール試薬(2%レゾルシノール溶液、0.25mLの0.1M CuSO4溶液、10mLの脱イオン水、濃HClで100mLの量にする)を加えた。試料をよく混合し、加熱ブロック(VWR)中110℃で25分間加熱した。試料を氷水中で5分間冷却した後、各試料に2mLの加水分解溶液(酢酸ブチルブタノール[85/15(v/v)])を加えた。各試料の有機相からの1mL部分を、石英のキュベットに移し、Shimadzu社製のUV160U、UV−可視記録分光光度計の580nmのところを読み取った。最終のポリサッカライド製剤の純度は、以下の式量:末端NeuAc残基に対しては309g/モル、GBSのIa、Ib型、又はIII型CPSの繰返し単位に対しては1004g/モル、GBSのII型又はV型CPSの繰り返し単位に対しては1328g/モルを用いてシアル酸含量から算出した。
【0125】
タンパク質含量は、PBS中に1ミリリットル当り20mgの莢膜ポリサッカライドを含有する試料についてPierce社(ロックフォード、イリノイ州)のCommie Plus試薬及び標準としてのヒトIgGを用いるBradford法(非特許文献9)により測定した。核酸含量は、260nmにおける直接的UV測光により測定した。これらのアッセイに対する光度測定は、Shimadzu社製のモデルUV 160U分光光度計(Shimadzu Scientific Inst.、コロンビア、メリーランド州)により行った。
【0126】
[実施例1]
(B群連鎖球菌(GBS)莢膜ポリサッカライドの加水分解及び単離)
図1は、この実施例で使用される本発明の実施形態の実験概略図を示す。
【0127】
(塩基を用いる第1ステップの加水分解)
750mLの1N NaOHを、前に述べた遠心分離ステップの直後の75gのGBS細胞ペーストに加え、細胞懸濁液を形成した。内容物を、2Lのビンに移し、100mgの水素化ホウ素ナトリウムをその細胞懸濁液に任意的に加えた。その反応混合物を37℃で16時間125rpmで震盪した。
【0128】
(膜濾過による分離)
一晩置いた細胞溶解物を、脱イオン(DI)水で10倍に希釈し、0.1Nの最終NaOH濃度とした。その希釈した細胞溶解物を、表面積が2000cm2の0.1gmのポリエーテルスルホン中空繊維フィルターモジュールを用いる精密濾過によって透明にした。その精密濾過モジュールをDI水を流して洗浄して残った貯蔵溶液を除去し、次いで、浸透ラインを閉じて希釈された細胞溶解物を10分間再循環させることにより平衡させた。その精密濾過は、一定の供給速度及び浸透速度で実施した。浸透液(出発容積の約90%)の収集に続いて、フィルターを2回1Lの生理食塩水で洗浄し、未透過液中のポリサッカライドを回収した。
【0129】
約13Lの精密濾過による細胞を含まない透過液を、Millipore(登録商標)Pellicon限外濾過装置(UF)の30kDa MWCOの膜により約1Lに濃縮した。その膜組成は、ポリエーテルスルホン(Biomax社製)である。一定容積を、最初に5Lの1M NaCl及び次に20LのDI水によりダイアフィルターにかけた。濃縮及びダイアフィルトレーションステップの後、その限外濾過未透過液を、30kDa MWCOのBiomax膜を装着した実験室規模の接線流濾過(TPF)装置(Millipore社製)により約70mLに濃縮した。
【0130】
(塩基を用いる第2ステップの加水分解)
30kDaの未透過液に5NのNaOHを加え、2N NaOHの最終濃度を達成した。ポリサッカライドを80℃で16〜18時間さらに抽出した後、その混合物を50℃未満まで冷却し、10LのDI水中に注いだ。
【0131】
(膜濾過による第2ステップの分離)
水酸化ナトリウム並びに小さい分子量の不純物を、30kDa MWCOのポリエーテルスルホン(Biomax社製)膜による一定容積のダイアフィルトレーションにより、5Lの1M NaCl及び20LのDI水によって除去し、次に、30kDa MWCOのBiomax膜を装着した実験室規模のTFF装置により約70mLの最小容積まで濃縮した。
【0132】
(任意的な再N−アシル化)
先に記した加水分解条件にポリサッカライドをさらすことにより、ポリサッカライドからN−アセチル基が放出されるために、無水酢酸(Aldrich Chemical Co.社製、ミルウォーキー、ウィスコンシン州)を、そのプールした部分に0.8Mの最終濃度まで滴下して加えることにより、そのポリサッカライドを再N−アセチル化した。この反応混合物を室温で1時間撹拌し、5NのNaOHを添加してpH9を維持した。その反応物のpHを次に13まで上げ、反応をさらに90分間続けた。その反応物のpHを、次に6Nの塩酸によりpH8に調整した。
【0133】
(膜濾過による精製)
再N−アセチル化した莢膜ポリサッカライドを含有するその溶液を、30kDa MWCOのBiomax限外濾過膜を装着した実験室規模のTFF装置により、5Lの0.9%NaClに対してほぼ70mLの最小容積までダイアフィルターにかけた。その精製した莢膜ポリサッカライドを、次に−70℃で冷凍保存するか、又は、下流部門の用途、例えば、抗体、医薬品組成物、診断キット、結合分子、及びワクチンの産生のために利用した。
【0134】
(結果)
1.精製莢膜ポリサッカライドの収量
表1は、実施例1の方法又はMichonらの米国特許第6248570号に記載の「1ステップの加水分解法」のいずれかを用いて得られた様々なBGS血清型の莢膜ポリサッカライド収量の比較を示す。すべての血清型について、莢膜ポリサッカライドは細胞ペレットから精製した。1ステップ法と比較して、本発明の方法は、試験した5個の血清型の4個の血清型に対してより大きい収量を生じた。
【0135】
2.精製莢膜ポリサッカライドの分析
検討したB群連鎖球菌血清型のそれぞれについて、260nmにおける直接のUV測光によって検出した核酸濃度は、0.5質量%を超えなかったが、Bradford法(非特許文献9)によってアッセイしたタンパク質は、<1質量%であった(表2)。すべてのポリサッカライドの純度は、改良レゾルシノールアッセイ(非特許文献36)により推定したそれらのシアル酸含量から計算して、約100%であった。本発明の方法及び1ステップの加水分解法の両方によって得られたすべてのポリサッカライド製剤に対して、測光データは、両方の方法ともタンパク質又は核酸による汚染が最小限の高純度の莢膜ポリサッカライドを産生することを示す。
【0136】
3.ポリサッカライドの分子の大きさ
別々の分析において、ポリサッカライドの絶対モル質量分布を、GPC−MALLS/RIにより測定した。この方法は、流速及び保持容量のようなクロマトグラフのパラメーターとは関係なく、且つ流体力学的性質が興味のある検体から大きく異なり得る二次標準物質を必要とすることなく、巨大分子のモル質量を直接推測することを可能にする。キャラクタリゼーション法としてのGPC−MALLS/RIの有用性は、医薬品として興味のあるポリサッカライドに対して十分に確立されている(非特許文献7、8、10、17、25)。モル質量分布は、重量平均モル質量(Mw)として通常は示される(表2)。
【0137】
【表1】

【0138】
【表2】
【0139】
[実施例2]
(Y群髄膜炎菌莢膜ポリサッカライドの加水分解及び単離)
図2は、この実施例で使用される本発明の実施形態の実験概略図を示す。
【0140】
(塩基を用いる第1ステップの加水分解)
750mLの1N NaOHを、細胞をペレット化した遠心分離ステップの直後の75gのGYM細胞ペーストに加えた。内容物を、2Lのビンに移し、100mgの水素化ホウ素ナトリウムをその細胞懸濁液に加えた。その反応混合物を37℃で16時間〜18時間125rpmで震盪した。
【0141】
(膜濾過による分離)
一晩置いた細胞溶解物を、脱イオン(DI)水で10倍に希釈し、約0.1Nの最終NaOH濃度とした。その希釈した細胞溶解物を、表面積が2000cm2の0.1μmのポリエーテルスルホン中空繊維フィルターモジュールを用いる精密濾過によって透明にした。その精密濾過モジュールをDI水を流して洗浄して残った貯蔵溶液を除去し、次いで、浸透ラインを閉じて希釈された細胞溶解物を10分間再循環させることにより平衡させた。その精密濾過は、一定の供給速度及び浸透速度で実施した。浸透液(出発容積の約90%)の収集に続いて、フィルターを2回1Lの生理食塩水で洗浄し、未透過液中のポリサッカライドを回収した。
【0142】
約7.5Lの精密濾過による細胞を含まない透過液を、Millipore Pellicon限外濾過装置(UF)の100kDa MWCOの膜により約1Lに濃縮した。その膜組成は、ポリエーテルスルホン(Biomax社製)である。一定容積を、最初に5Lの1M NaCl及び次に20LのDI水によりダイアフィルターにかけた。濃縮及びダイアフィルトレーションステップの後、その限外濾過未透過液を、100kDa MWCOのBiomax膜を装着した実験室規模の接線流濾過(TFF)装置(Millipore社製)により約70mLに濃縮した。
【0143】
(塩基を用いる第2ステップの加水分解)
100kDaの未透過液に5NのNaOHを加え、2N NaOHの最終濃度を達成した。ポリサッカライドを80℃で48時間さらに抽出した後、その混合物を50℃未満まで冷却し、10LのDI水中に注いだ。
【0144】
(膜濾過による第2ステップの分離)
GYMPの精製については、いくらかの大きいタンパク質(300kDaを超える大きさ)が、加水分解ステップによる分解に対して耐性を示すことが分かった。これらのタンパク質を除去するために、第2の加水分解ステップからの混合物を、Millipore Pellicon限外濾過装置(UF)の300kDa MWCOの膜にかけた。その透過液を、次にMillipore Pellicon限外濾過装置(UF)の5kDa MWCOの膜により約1Lに濃縮した。その膜組成は、ポリエーテルスルホン(Biomax社製)である。一定容積を、最初に5Lの1M NaCl及び次に20LのDI水によりダイアフィルターにかけた。濃縮及びダイアフィルトレーションステップの後、その限外濾過未透過液を、5kDa MWCOのBiomax膜を装着した実験室規模の接線流濾過(TFF)装置(Millipore社製)により約70mLに濃縮した。
【0145】
(任意的な再N−アシル化)
先に記した加水分解条件にポリサッカライドをさらすことにより、ポリサッカライドからN−アセチル基が放出されるために、無水酢酸(Aldrich Chemical Co.社製、ミルウォーキー、ウィスコンシン州)を、そのプールした部分に0.8Mの最終濃度まで滴下して加えることにより、そのポリサッカライドを再N−アセチル化した。この反応混合物を室温で1時間撹拌し、5NのNaOHを添加してpH9を維持した。その反応物のpHを次に13まで上げ、反応をさらに90分間続けた。その反応物のpHを、次に6Nの塩酸によりpH8に調整した。
【0146】
(膜濾過による精製)
再N−アセチル化した莢膜ポリサッカライドを含有するその溶液を、5kDa MWCOのBiomax限外濾過膜を装着した実験室規模のTFF装置により、5Lの0.9%NaClに対してほぼ70mLの最小容積までダイアフィルターにかけた。その精製した莢膜ポリサッカライドを、次に−70℃で冷凍保存したが、或いは、下流部門の用途、例えば、抗体、医薬品組成物、診断キット、結合分子、及びワクチンの産生のために利用することもできる。
【0147】
(結果)
1.Y群髄膜炎菌莢膜ポリサッカライドの収量
上記の方法を用いる精製Y群髄膜炎菌莢膜ポリサッカライドのおおよその収量は、1リットルの細菌培養液当り12mgであった。
【0148】
2.精製GYM莢膜ポリサッカライドの分析
精製GYM莢膜ポリサッカライドの一次元1H NMR分光分析は、非常に低い水準の汚染を示した。図4は、D2O中50℃で記録された精製GYM莢膜ポリサッカライドのNMRスペクトル(500MHz)を示す。直接の260nmでのUV測光により検出された核酸のレベルは、2.46質量%であり、一方Bradford法(非特許文献9)によりアッセイしたタンパク質は、このアッセイの検出の下限(1μg/mL)の上の検出できるものではなかった。最終精製物の純度は、改良型マイクロスケールオルシノールアッセイ(非特許文献35)により推測されるシアル酸含量により計算し、94%であった。従って、スペクトル及び測光データは、本発明の加水分解及び単離の方法が、タンパク質又は核酸による汚染が最低限である高純度のGYM莢膜ポリサッカライドを可能にすることを示す。
【0149】
[実施例3]
(W−135群髄膜炎菌莢膜ポリサッカライドの加水分解及び単離)
図3は、この実施例で使用される本発明の実施形態の実験概略図を示す。
【0150】
(塩基を用いる第1ステップの加水分解)
750mLの1N NaOHを、細胞をペレット化した遠心分離ステップの直後の75gのW−135群髄膜炎菌(GWM)細胞ペーストに加えた。内容物を、2本の2Lのビンに移し、100mgの水素化ホウ素ナトリウムをその細胞懸濁液に加えた。その反応混合物を37℃で16時間〜18時間150rpmで震盪した。
【0151】
(膜濾過による分離)
一晩置いた細胞溶解物を、脱イオン(DI)水で10倍に希釈し、約0.1Nの最終NaOH濃度とした。その希釈した細胞溶解物を、表面積が2000cm2の0.2μmのポリエーテルスルホン中空繊維フィルターモジュールを用いる精密濾過によって透明にした。その精密濾過モジュールをDI水を流して洗浄して残った貯蔵溶液を除去し、次いで、浸透ラインを閉じて希釈された細胞溶解物を10分間再循環させることにより平衡させた。その精密濾過は、一定の供給速度及び浸透速度で実施した。浸透液(出発容積の約90%)の収集に続いて、フィルターを2回1Lの生理食塩水で洗浄し、未透過液中のポリサッカライドを回収した。
【0152】
約12Lの精密濾過による細胞を含まない透過液を、Millipore Pellicon限外濾過装置(UF)の30kDa MWCOの膜により約1Lに濃縮した。その膜組成は、ポリエーテルスルホン(Biomax社製)である。一定容積を、最初に5Lの1M NaCl及び次に20LのDI水によりダイアフィルターにかけた。濃縮及びダイアフィルトレーションステップの後、その限外濾過未透過液を、100kDa MWCOのBiomax膜を装着した実験室規模の接線流濾過(TFF)装置(Millipore社製)により約70mLに濃縮した。
【0153】
(加水分解を用いる第2ステップの加水分解)
100kDaの未透過液に5NのNaOHを加え、2N NaOHの最終濃度を達成した。ポリサッカライドを80℃で一晩さらに抽出した後、その混合物を50℃未満まで冷却し、10LのDI水中に注いだ。
【0154】
(膜濾過による第2ステップの分離)
W−135群髄膜炎菌ポリサッカライド(GWMP)の精製については、いくらかの大きいタンパク質(300kDaを超える大きさ)が、加水分解ステップによる分解に対して耐性を示した。これらのタンパク質を除去するために、第2の加水分解ステップからの混合物を、Millipore Pellicon限外濾過装置(UF)の300kDa MWCOの膜にかけた。その透過液を、次にMillipore Pellicon限外濾過装置(UF)の30kDa MWCOのポリエーテルスルホン(Biomax社製)膜により約1Lに濃縮した。一定容積を、最初に5Lの1M NaCl及び次に20LのDI水によりダイアフィルターにかけた。濃縮及びダイアフィルトレーションステップの後、その限外濾過未透過液を、30kDa MWCOのBiomax膜を装着した実験室規模の接線流濾過(TFF)装置(Millipore社製)により約70mLに濃縮した。
【0155】
(任意的な再N−アシル化)
先に記した加水分解条件にポリサッカライドをさらすことにより、ポリサッカライドからN−アセチル基が放出されるために、無水酢酸(Aldrich Chemical Co.社製、ミルウォーキー、ウィスコンシン州)を、そのプールした部分に0.8Mの最終濃度まで滴下して加えることにより、そのポリサッカライドを再N−アセチル化した。この反応混合物を室温で1時間撹拌し、5NのNaOHを添加してpH9を維持した。その反応物のpHを次に13まで上げ、反応をさらに90分間続けた。その反応物のpHを、次に6Nの塩酸によりpH8に調整した。
【0156】
(膜濾過による精製)
再N−アセチル化した莢膜ポリサッカライドを含有するその溶液を、30kDa MWCOのBiomax限外濾過膜を装着した実験室規模のTFF装置により、5Lの0.9%NaClに対してほぼ70mLの最小容積までダイアフィルターにかけた。その精製した莢膜ポリサッカライドを、次に−70℃で冷凍保存したが、或いは、下流部門の用途、例えば、抗体、医薬品組成物、診断キット、結合分子、及びワクチンの産生のために利用することもできる。
【0157】
(結果)
1.W−135群髄膜炎菌莢膜ポリサッカライドの収量
上記の方法を用いる精製W−134群髄膜炎菌莢膜ポリサッカライドのおおよその収量は、1リットルの細菌培養液当り25.4mgであった。
【0158】
2.精製GWM莢膜ポリサッカライドの分析
直接の260nmでのUV測光により検出された核酸のレベルは、0.40質量%であり、一方Bradford法(非特許文献9)によりアッセイしたタンパク質は、このアッセイの検出の下限(1μg/mL)の上の検出できるものではなかった。最終精製物の純度は、改良型マイクロスケールオルシノールアッセイ(非特許文献35)により推測されるシアル酸含量により計算し、95%であった。従って、上記測光データは、本発明の加水分解及び単離の方法が、タンパク質又は核酸による汚染が最低限である高純度のGWM莢膜ポリサッカライドを可能にすることを示す。
【0159】
本明細書は、本明細書内に引用した参考文献の教示を踏まえて最も完全に理解される。本明細書中の実施形態は、本発明の実施形態の説明を提供するものであって、本発明を限定するものと解釈すべきではない。熟練技術者であれば、多くの他の実施形態が本発明によって包含されることを容易に認識する。本開示中に引用したすべての出版物及び特許は、参照によりそれら全体として組み込む。参照により組み込まれた材料が本明細書と矛盾するか一貫性がない限りにおいて、本明細書は、そのような材料のいずれにも優先する。本明細書におけるどの参考文献の引用も、かかる参考文献が本発明に対する先行技術であることを認めるものではない。
【0160】
別に示されていない限り、特許請求の範囲を含めて、本明細書で使用されている、成分の量、反応条件などを表すすべての数字は、すべての場合において、用語「約」によって修飾されるものと理解すべきである。従って、そうでないことが特に示されていない限り、数値パラメーターは、近似値であり、本発明によって得られることが求められる所望の特性によって変動し得る。少なくとも、そして特許請求の範囲に対する均等の原則の適用を限定しようとする試みとしてではなく、各数値パラメーターは、有効数字の数及び普通の丸め手法に照らして解釈すべきである。
【0161】
別に示されていない限り、範囲は境界点を含む。例えば、「5〜14」は、5以上から14以下の任意の値を指す。
【0162】
別に示されていない限り、一連の要素に先立つ用語「少なくとも」とは、その一連のあらゆる要素を指すものと理解すべきである。当業者であれば、ありふれたものにすぎない実験手段を用いて、本明細書に記載の本発明の特定の実施形態に対する多数の相当物を認識し、究明することができよう。かかる相当物は、添付の特許請求の範囲に含まれることが意図されている。
【0163】
(参考文献)
1. Anderson, P., G. Peter, R.B. Johnson, L.H. Wetterlow and D.H. Smith. 1972. Immunization of humans with polyribophosphate, the capsular antigen of Haemophilus influenzae type b. J.Clin.Invest. 51:39-44.
2. Avery, O.T. and W.F. Goebel. 1931. Chemo-immunological studies on conjugated carbohydrate-proteins V. The immunological specificity of an antigen prepared by combining the capsular polysaccharide of type 3 pneumococcus with foreign protein. J.Exp.Med. 54:43 7-447.
3. Baker, C.J. and D.L. Kasper. 1985. Group B streptococcal vaccines. Rev.Inf.Dis. 7:458-467.
4. Baker, C.J., M.A. Rench, M.S. Edwards, R.J. Carpenter, B.M. Hays and D.L. Kasper. 1988. Immunization of pregnant women with a polysaccharide vaccine of group B Streptococcus. N.Eng1.J.Med. 319:1180-1185.
5. Baker, C.J., M.A. Rench and D.L. Kasper. 1990. Response to Type III polysaccharide in women whose infants have had invasive Group B streptococcal infection. New EngI.J.Med. 322:1857-1860.
6. Baltimore, R.S., D.L. Kasper and J. Vecchitto. 1979. Mouse protection test for group B Streptococcus type III. J.Infect.Dis. 140:81-86.
7. Bednar, B. and J.P. Hennessey. 1993. Molecular size analysis of capsular polysaccharide preparations from Streptococcus pneumoniae. Carbohyd.Res. 243:115-130.
8. Beri, R.G., J. Walker, E.T. Reese and J.E. Rollings. 1993. Characterization of chitosans via coupled size-exclusion chromatography and multiple-angle laser light-scattering technique. Carbohyd.Res. 238:11-26.
9. Bradford, M.M.. 1.976. A Rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analyt.Biochem. 72:248-254.
10. D'Ambra, A.J., J.E. Baugher, P.E. Concannon, R.A. Pon and F. Michon. 1997. Direct and indirect methods for molar-mass analysis of fragments of the capsular polysaccharide ofHaemophilus influenzae type b. Anal.Biochem. 250:228-236
11. Dick, W.E., Jr. and M. Beurret. 1989. Glycoconjugates of bacterial carbohydrate antigens. p. 48-114. In: J.M. Cruse and R.E. Lewis,Jr., Contributions to microbiology and immunology. S.Karger, Basel.
12. Dillon, H.C., Jr., S. Khare and B.M. Gray. 1987. Group B streptococcal carriage and disease: A 6-year prospective study. J.Pediat. 110:31-36.
13. Goebel, W.F. and O.T. Avery. 1931. Chemo-immunological studies on conjugated carbohydrate-proteins IV. The synthesis of the p-aminobenzyl ether of the soluble specific substance of type 3 pneumococcus and its coupling with protein. J.Exp.Med. 54:431.436.
14. Gold, R., M.L. Lepow, I.Goldschneider, T.L. Draper and E.C. Gotschlich. 1975. Clinical evaluation of group A and group C meningococcal polysaccharide vaccines in infants. J.Clin.Invest. 56:1536-1547.
15. Gold, R., M.L. Lepow, I.Goldschneider and E.C. Gotschlich. 1977. Immune response of human infants to polysaccharide vaccines of Groups A and C Neisseria meningitidis. J.Infect.Dis. 136S:S31-S35.
16. Gold, R.M., M.L. Lepow, I. Goldschneider, T.F. Draper and E.C. Gotschlich. 1978. Antibody responses of human infants to three doses of group A Neisseria meningitides vaccine administered at two, four and six months of age. J.Infect.Dis. 138:731-735.
17. Hennessey, J.P., B. Bednar and V. Manam. 1993. Molecular size analysis of Haemophilus influenzae type b capsular polysaccharide. J.Liq.Chromat. 16:1715-1729.
18. Howard, J.G., G.H. Christie, B.M. Courtenay, E. Leuchars and A.J.S. Davies. 1971. Studies on immunological paralysis. VI. Thymic-independence of tolerance and immunity to type III pneumococcal polysaccharide. Cell.Immunol. 2:614-626.
19. Jennings, H.J., E. Katzenellenbogen, C. Lugowski and D.L. Kasper. 1983. Structure of the native polysaccharide antigens of type la and type Ib Group B Streptococcus. Biochemistry 22:1258-1263.
20. Jennings, H.J., K.-.G. Rosell and D.L. Kasper. 1980. Structural determination and serology of the native polysaccharide antigen of type III group B Streptococcus. Can.J.Biochem. 58:112-120.
21. Jennings, H.J., K.-.G. Rosell and D.L. Kasper. 1980. Structure and serology of the native polysaccharide antigen of type Ia group B Streptococcus. Proc.Nat.Acad.Sci.USA. 77:2931-2935.
22. Jennings, H.J., K.-.G. Rosell, E. Katzenellenbogen and D.L. Kasper. 1983. Structural determination of the capsular polysaccharide antigen of type II Group B Streptococcus. J.Biol.Chem. 258:1793-1798.
23. Jennings, H.J. and R.K. Sood. 1994. Synthetic glycoconjugates as human vaccines. p. 325-371. In: Y.C. Lee and R.T. Lee, Neoglycoconjugates: Preparation and applications. Academic Press, New York.
24. Kang, D., Liu, G., Lundstrom, A. et al. 1998 A peptidoglycan recognition protein in innate immunity conserved from insects to humans. Proc. Natl. Acad. Sci. USA. 95:10078-10082.
25. Kasper, D.L., C.J. Baker, R.S. Baltimore, J.H. Crabb, G. Schiffman and H.J. Jennings. 1979. Immunodeterminant specificity of human immunity to type III group B Streptococcus. J.Exp.Med. 149:327-339.
26. Knobloch, J.E. and P.N. Shaklee. 1997. Absolute molecular weight of low-molecular-weight heparins by size-exclusion chromatography with multiangle laser light scattering detection. Anal.Biochem. 245:231-241.
27. Lancefield, R.C.. 1933. A serological differentiation of human and other groups of haemolytic streptococci. J.Exp.Med. 57:571-595.
28. Lancefield, R.C.. 1938. A micro-precipitin technique for classifying hemolytic streptococci and improved methods for producing antigen. Proc.Soc.Exp.Biol.and Med. 38:473-478.
29. Lancefield, R.C., M. McCarty and W.N. Everly. 1975. Multiple mouse-protective antibodies directed against group B streptococci: Special reference to antibodies effective against protein antigens. J.Exp.Med. 142:165-179.
30. Madoff, L.C., L.C. Paoletti, J.Y. Tai and D.L. Kasper. 1994. Maternal immunization of mice with Group B streptococcal type III polysaccharide-beta C protein conjugate elicits protective antibody to multiple serotypes. J.Clin.Invest. 94:286-292.
31. Marques, M.B., D.L. Kasper, A. Shroff, F. Michon, H.J. Jennings and M.R. Wessels. 1994. Functional activity of antibodies to the group B polysaccharide of group B streptococci elicited by a polysaccharide-protein conjugate vaccine. Infect.Immun. 62:1593-1599.
32. Makela, P.R.H., H. Peltola, H. Kayhty, et al. 1977. Polysaccharide vaccines of group A Neisseria meningitidis and Haemophilus influenzae type b: A field trial in Finland. J.Infect.Dis. 136:543-50.
33. Michon, F., J.R. Brisson, A. Dell, D.L. Kasper and H.J. Jennings. 1988. Multiantennary group-specific polysaccharide of group B Streptococcus. Biochem. 27:5341-5351.
34. Peltola, A., H. Kayhty, A. Sivonen and P.R.H. Makela. 1977. Haemophilus influenzae type b capsular polysaccharide vaccine in children: A double blind field study of 100,000 vaccines 3 months to 5 years of age in Finland. Pediatrics 60:730-737.
35. Peltola, H., P.R.H. Makela, H. Jousimies, et al. 1977. Clinical efficacy of meningococcal group A vaccine in children three months to five years of age. N.Engl.J.Med. 297:686-691.
36. Reuter, G. and R. Schauer. 1994. Determination of sialic acids. p. 168-199. In: W.J. Lennarz and G.W. Hart, Methods in Enzymology Vol. 230 Techniques in Glycobiology. Academic Press, New York.
37. Robbins, J.B. and R. Schneerson. 1990. Polysaccharide-protein conjugates: A new generation of vaccines. J.Infect.Dis. 161:821-832.
38. Scwander, R., Dziarski R., Wesche, H. et al. 1999. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J. Biol. Chem. 274:17406-17409.
39. Smith, A.L. and J. Haas. 1991. Neonatal Bacterial Meningitis. p. 313-333. In: W.M. Scheld, R.J. Whitley and D.T. Durack, Infections of the Central Nervous System. Raven Press, Ltd., New York,
40. Tsunashima, T., K: Moro, B. Chu and T.-Y. Liu. 1978. Characterization of group C meningococcal polysaccharide by light-scattering spectroscopy. III. Determination of molecular weight, radius of gyration, and translational diffusional coefficient.. Biopolymers 17:251-265.
41. von Hunolstein, C., L. Nicolini, S. D'Ascenzi, C. Volpe, G. Alfarone and G. Orefici. 1993. Sialic acid and biomass production by Streptococcus agalactiae under different growth conditions. Appl.Microbiol.Biotechnol. 38:458-462.
42. Wessels, M.R., W.J. Benedi, H.J. Jennings, F. Michon, J.L. DiFabio and D.L. Kasper. 1989. Isolation and characterization of type IV group B Streptococcus capsular polysaccharide. Infect.Immun. 57:1089-1094.
43. Wessels, M.R., J.L. DiFabio, V.J. Benedi, et al. 1991. Structural determination and immunochemical characterization of the type V group B Streptococcus capsular polysaccharide. J.Biol.Chem. 266:6714-6719.
44. Wessels, M.R., L.C. Paoletti, D.L. Kasper, et al. 1990. Immunogenicity in animals of a polysaccharide-protein conjugate vaccine against type III group B Streptococcus. J.Clin.Invest. 86:1428.1433.
45. Wessels, M.R., L.C. Paoletti, A.K. Rodewald, et al. 1993. Stimulation of protective antibodies against type la and Ib group B streptococci by a type Ia polysaccharide-tetanus toxoid conjugate vaccine. Infect.Immun. 61:4760-4766.
46. Wessels, M.R., V. Pozsgay, D.L. Kasper and H.J. Jennings. 1987. Structure and immunochemistry of an oligopolysaccharide repeating unit of the capsule polysaccharide of Type III Group B Streptococcus: A revised structure for the Type III Group B streptococcal polysaccharide antigen. J.Biol.Chem. 262:8262-8267.
47. Wyle, S.A., M.S. Artenstein, B.L. Brandt, et al. 1972. Immunologic response of man to group B meningococcal polysaccharide vaccines. J.Infect.Dis. 126:514-522.
48. Yang, R.B., Mark, M.R., Gray, A., et al. 1998. Toll-like receptor 2 mediates lipopolysaccharide-induced cellular signaling. Nature. 395:284-288.
49. Tipson, R.S. and Horton, D., Advances in Carbohydrate Chemistry and Biochemistry, Vol. 41, 1983, Academic Press, NY.
50. Westphal et al., Methods in Carbohydrate Chemistry, vol. V, 1965, Academic Press, NY.
51. Theodora W. Greene and Peter G. M. Wuts, Protective Groups in Organic Syntheses, 2nd Ed. (1991).
52. Kohler and Milstein (1975) Nature 256:495-497.
53. Takeda et al. (1985) Nature 314:452.
54. Campbell (1985) Laboratory Techniques in Biochemistry and Molecular Biology, Vol. 13, Burdon, et al. (eds.), Elsevier Science Publishers, Amsterdam.
55. Schneerson, R., et al. (1980) J. Exp. Med. 1952:361-476.
56. Marburg, S., et al. (1986) J. Am. Chem. Soc. 108:5282-5287.
57. Grossman, M. and Cohen, S. N., in "Basic and Clinical Immunology", 7th Ed., (Stites, D. P. and Terr, A. T. eds., Appleton & Lange 1991) Chapter 58 "Immunization".
58. Paoletti, et al. (1997) J. Infectious Diseases, 175:1237-9.
59. L. Warren, (1959) 1 Biol. Chem. 234, 1971.
【図面の簡単な説明】
【0164】
【図1】B群連鎖球菌(GBS)莢膜ポリサッカライドを単離するために使用する本発明の実験概略図(実施例1)を示す図である。
【図2】Y群髄膜炎菌(GYM)莢膜ポリサッカライドを単離するために使用する本発明の実験概略図(実施例2)を示す図である。
【図3】W−135群髄膜炎菌(GWM)莢膜ポリサッカライドを単離するために使用する本発明の実験概略図(実施例3)を示す図である。
【図4】Y群髄膜炎菌莢膜ポリサッカライドからの莢膜ポリサッカライドのNMRスペクトル(実施例2)を示す図である。
【技術分野】
【0001】
本出願は、2006年1月13日に出願した米国仮出願第60/758894号に対する優先権の特典を主張するものであり、この仮出願全体を完全に参照により組み込む。
【0002】
本発明は、ポリサッカライドを精製する改良された方法に関する。その抽出されたポリサッカライドは、ポリサッカライド単独又は担体分子に結合したポリサッカライドを含む抗原類、抗体類、及びワクチン類を産生するために有用である。
【背景技術】
【0003】
多種多様の微生物が、ヒトの疾患を引き起こす。細菌感染は、グラム陽性及びグラム陰性の細菌、例えば、スピロヘータ、マイコバクテリア、リケッチア、クラミジア、マイコプラズマなどによって引き起こされる。酵母及び全身性病原真菌もまた免疫適格性及び免疫低下宿主の両方における重大な健康問題である。癌は、ヒトにおけるもう1つの疾患である。
【0004】
グラム陽性細菌、例えば、連鎖球菌(Streptococcus)、ブドウ球菌(Staphylococcus)、腸球菌(Enterococcus)、桿菌(Bacillus)、コリネバクテリウム(Corynebacterium)、リステリア(Listeria)、エリシペロスリクス(Erysipelothrix)及びクロストリジウム(Clostridium)など、並びにグラム陰性細菌、例えば、ヘモフィルス(Haemophilus)、赤痢菌(Shigella)、コレラ菌(Vibrio cholerae)、ナイセリア(Neisseria)及び特定の型の大腸菌(Escherichia coli)によって引き起こされる細菌感染は、世界中で深刻な罹患率を引き起こしている。このことは、現在使用されている抗生物質に対して細菌によって示される耐性の出現と相まって、これらの有害な結果を避ける細菌ワクチンの開発の必要性を示す。多くの現行の細菌ワクチンの戦略は、細菌細胞壁と関連するポリサッカライドを含む抗原の使用を伴う(非特許文献1、3、4、11、14、15、16、23、30、31、32、34、35、37、45、46、48)。
【0005】
すべての細菌、真菌及びいくつかの原虫に共通の特徴は、それらの細胞壁の中に及び/又はそこに付着してポリサッカライドが存在していることである。ポリサッカライドは、細胞壁の重要な構築的特徴であり、微生物を免疫システムによる攻撃から保護するために貢献する。微生物は、宿主の免疫システムの先天性の力及び適応力の両方による認識及び破壊を避けるための様々な異なる細胞壁及び細胞壁と関連する構造を進化させている。例えば、先天性免疫システムの貪食細胞は、細胞壁及び細胞壁と関連する抗原を認識する特別な細胞表面受容体を有する。CD14受容体及びトール様受容体(TLR)は、ペプチドグリカン、リポテイコ酸、及びポリサッカライド抗原を認識し、その際、マクロファージを活性化して微生物を貪食させることが立証されている(非特許文献24、38、49)。免疫システムの適応力は、また、細胞壁ポリサッカライド構造に対して特異的である抗体を発生することにより、これらの抗原に対応することができる(非特許文献29、41)。
【0006】
グラム陽性菌及びグラム陰性菌は、それらの外側の細胞表面構造によって区別される。グラム陽性細胞壁は、多くの場合脂質を欠いており、ほとんどが、ペプチドグリカン、ペプチドグリカンに共有結合しているタイコ酸及び/又はタイクロン酸、並びにタンパク質から構成されている。ムレイン又はムコペプチドとしても知られているペプチドグリカンは、N−アセチルグルコサミン(NAG,N-acetylglucosamine)及びN−アセチルムラミン酸(NAM,N-acetylmuramic acid)のD−アミノ酸及びL−アミノ酸を含有するペプチド側鎖による交互の繰返しからなるポリサッカライド主鎖から構成されている。グラム陰性菌は、外膜及びその外膜と細胞質の内膜との間に組み込まれたペプチドグリカンの層からなる細胞壁を所有する。その外膜は、リポポリサッカライド(LPS,lipopolysaccharide)並びに脂質及びタンパク質を含有する。そのLPS分子は、その外膜中に組み込まれるリピドAの「頭部」及びその外膜から外側に伸びるポリサッカライドの尾部から構成される。そのポリサッカライドの尾部は、通常は、中核のオリゴポリサッカライドとO−ポリサッカライドとからなる。そのO−ポリサッカライドは、N−アセチルグルコサミン(NAG)及びN−アセチルムラミン酸(NAM)の交互の組成物からなる塩基性繰返しモチーフを有する点でペプチドグリカンと似ている。また、莢膜ポリサッカライド(CPS,capsular polysaccharide)及び被膜下ポリサッカライド(sub-capsular polysaccharide)は、グラム陽性及びグラム陰性細胞壁の外面に付着しており、細菌性細胞を貪食細胞による認識及び貪食から保護するためにさらに貢献する。
【0007】
細菌外面のポリサッカライド含有成分は、免疫受容体によって特異的に認識されるために、細菌は、その外面組成を変異させる。例えば、細胞壁ポリサッカライドは、主として繰返し(オリゴ)ポリサッカライド単位から構成されているけれども、細菌の亜系は、様々な修飾及び/又はポリサッカライド単位の順序付けを持たせることによってこの基本構造を変化させる。例えば、B群連鎖球菌は、すべての血清型に共通の群抗原(即ち、ポリサッカライド群(GPS,group polysaccharides))を有するが、主要な血清型(Ia型、Ib型、II型、III型、IV型、V型、VI型、VII型、及びVIII型)を区別する型特異性の莢膜ポリサッカライド(CPS)を有する。これらの様々な型特異性莢膜ポリサッカライドのそれぞれの構造が特性化されている(非特許文献19、20、21、22、43、44、47)。同様に、肺炎連鎖球菌は、様々な型特異性莢膜ポリサッカライドを除く共通の群抗原(C物質)を有する。現在、それらの莢膜ポリサッカライド被膜によって区別される肺炎球菌(S. pneumoniae)の血清型が90個知られている(非特許文献7、49)。外面のばらつきのために、細菌型(又は亜系)は、型特異性の抗体によって識別し、分類することができる。従って、細胞壁抗原ポリサッカライドをそれらの自然状態で又は抗原サブ構造として、効率的に簡単に精製することができる方法は、ワクチンの開発を大いに助けるであろう。
【0008】
群及び型特異性のポリサッカライドは、感染防御抗体のための標的として首尾よく使用されてきており、これらの抗体の産生は、能動免疫を刺激するワクチンにより提供される。型特異性のポリサッカライド成分を有するワクチンのワクチン接種に応答して発生した抗体もまた、受動免疫を提供するために有用である。ワクチン製造で使用するためのポリサッカライドの大規模な生産は、精製したポリサッカライドの適切な供給を必要とする。特定の細菌性細胞から莢膜ポリサッカライドを単離するためのいくつかの方法(非特許文献43、45)は、酵素ムタノリシン(mutanolysin)による細胞の処理に依存している。ムタノリシンは、細菌細胞壁を切断し、それによって細胞成分を遊離させる。この方法はまた細胞溶解物をさらなる酵素により処理してタンパク質及び核酸を分解することも含む。微生物からポリサッカライドを単離するためのその他の報告されている方法(非特許文献50)は、高温のフェノールによる細胞の処理に依存している。フェノールは、ポリサッカライドを水層中に分離する。その水層を限外濾過法によって濃縮して未精製のポリサッカライド画分を生じさせ、それを次にクロマトグラフィーによって精製する。これらの方法は、細胞壁に付随する有毒な分子(例えば、リピドA内毒素、リポタイコ酸及びムラミルペプチド)を除去することが不可欠であって、高価な酵素及び毒素を分解して除去するための複雑なクロマトグラフィーの使用を必要とするために、困難で費用もかかる。精製したポリサッカライドを得るより効率的で、収量が高く且つ簡単な手段が望まれる。
【0009】
莢膜ポリサッカライドを単離するためのさらに別の効率的な方法(米国特許第6248570号に記載されている)は、細胞壁成分からCPSを抽出するための塩基加水分解反応に依存している。この方法は、困難で費用のかかる酵素は使用せずに、ポリサッカライドから、核酸、タンパク質、及び毒素を分解し、除去する効果がある。この塩基加水分解法は、他のそれまでに報告された方法に対して、より大きな単純性、効率、安全性、及び一般的適用性を提供するが、この方法のさらに大きな改良が本発明においては達成されている。
【特許文献1】米国特許第6248570号
【非特許文献1】Anderson, P., G. Peter, R.B. Johnson, L.H. Wetterlow and D.H. Smith. 1972. Immunization of humans with polyribophosphate, the capsular antigen of Haemophilus influenzae type b. J.Clin.Invest. 51:39-44.
【非特許文献2】Avery, O.T. and W.F. Goebel. 1931. Chemo-immunological studies on conjugated carbohydrate-proteins V. The immunological specificity of an antigen prepared by combining the capsular polysaccharide of type 3 pneumococcus with foreign protein. J.Exp.Med. 54:43 7-447.
【非特許文献3】Baker, C.J. and D.L. Kasper. 1985. Group B streptococcal vaccines. Rev.Inf.Dis. 7:458-467.
【非特許文献4】Baker, C.J., M.A. Rench, M.S. Edwards, R.J. Carpenter, B.M. Hays and D.L. Kasper. 1988. Immunization of pregnant women with a polysaccharide vaccine of group B Streptococcus. N.Eng1.J.Med. 319:1180-1185.
【非特許文献5】Baker, C.J., M.A. Rench and D.L. Kasper. 1990. Response to Type III polysaccharide in women whose infants have had invasive Group B streptococcal infection. New EngI.J.Med. 322:1857-1860.
【非特許文献6】Baltimore, R.S., D.L. Kasper and J. Vecchitto. 1979. Mouse protection test for group B Streptococcus type III. J.Infect.Dis. 140:81-86.
【非特許文献7】Bednar, B. and J.P. Hennessey. 1993. Molecular size analysis of capsular polysaccharide preparations from Streptococcus pneumoniae. Carbohyd.Res. 243:115-130.
【非特許文献8】Beri, R.G., J. Walker, E.T. Reese and J.E. Rollings. 1993. Characterization of chitosans via coupled size-exclusion chromatography and multiple-angle laser light-scattering technique. Carbohyd.Res. 238:11-26.
【非特許文献9】Bradford, M.M.. 1.976. A Rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analyt.Biochem. 72:248-254.
【非特許文献10】D'Ambra, A.J., J.E. Baugher, P.E. Concannon, R.A. Pon and F. Michon. 1997. Direct and indirect methods for molar-mass analysis of fragments of the capsular polysaccharide ofHaemophilus influenzae type b. Anal.Biochem. 250:228-236
【非特許文献11】Dick, W.E., Jr. and M. Beurret. 1989. Glycoconjugates of bacterial carbohydrate antigens. p. 48-114. In: J.M. Cruse and R.E. Lewis,Jr., Contributions to microbiology and immunology. S.Karger, Basel.
【非特許文献12】Dillon, H.C., Jr., S. Khare and B.M. Gray. 1987. Group B streptococcal carriage and disease: A 6-year prospective study. J.Pediat. 110:31-36.
【非特許文献13】Goebel, W.F. and O.T. Avery. 1931. Chemo-immunological studies on conjugated carbohydrate-proteins IV. The synthesis of the p-aminobenzyl ether of the soluble specific substance of type 3 pneumococcus and its coupling with protein. J.Exp.Med. 54:431.436.
【非特許文献14】Gold, R., M.L. Lepow, I. Goldschneider, T.L. Draper and E.C. Gotschlich. 1975. Clinical evaluation of group A and group C meningococcal polysaccharide vaccines in infants. J.Clin.Invest. 56:1536-1547.
【非特許文献15】Gold, R., M.L. Lepow, I. Goldschneider and E.C. Gotschlich. 1977. Immune response of human infants to polysaccharide vaccines of Groups A and C Neisseria meningitidis. J.Infect.Dis. 136S:S31-S35.
【非特許文献16】Gold, R.M., M.L. Lepow, I. Goldschneider, T.F. Draper and E.C. Gotschlich. 1978. Antibody responses of human infants to three doses of group A Neisseria meningitides vaccine administered at two, four and six months of age. J.Infect.Dis. 138:731-735.
【非特許文献17】Hennessey, J.P., B. Bednar and V. Manam. 1993. Molecular size analysis of Haemophilus influenzae type b capsular polysaccharide. J.Liq.Chromat. 16:1715-1729.
【非特許文献18】Howard, J.G., G.H. Christie, B.M. Courtenay, E. Leuchars and A.J.S. Davies. 1971. Studies on immunological paralysis. VI. Thymic-independence of tolerance and immunity to type III pneumococcal polysaccharide. Cell.Immunol. 2:614-626.
【非特許文献19】Jennings, H.J., E. Katzenellenbogen, C. Lugowski and D.L. Kasper. 1983. Structure of the native polysaccharide antigens of type la and type Ib Group B Streptococcus. Biochemistry 22:1258-1263.
【非特許文献20】Jennings, H.J., K.-.G. Rosell and D.L. Kasper. 1980. Structural determination and serology of the native polysaccharide antigen of type III group B Streptococcus. Can.J.Biochem. 58:112-120.
【非特許文献21】Jennings, H.J., K.-.G. Rosell and D.L. Kasper. 1980. Structure and serology of the native polysaccharide antigen of type Ia group B Streptococcus. Proc.Nat.Acad.Sci.USA. 77:2931-2935.
【非特許文献22】Jennings, H.J., K.-.G. Rosell, E. Katzenellenbogen and D.L. Kasper. 1983. Structural determination of the capsular polysaccharide antigen of type II Group B Streptococcus. J.Biol.Chem. 258:1793-1798.
【非特許文献23】Jennings, H.J. and R.K. Sood. 1994. Synthetic glycoconjugates as human vaccines. p. 325-371. In: Y.C. Lee and R.T. Lee, Neoglycoconjugates: Preparation and applications. Academic Press, New York.
【非特許文献24】Kang, D., Liu, G., Lundstrom, A. et al. 1998 A peptidoglycan recognition protein in innate immunity conserved from insects to humans. Proc. Natl. Acad. Sci. USA. 95:10078-10082.
【非特許文献25】Kasper, D.L., C.J. Baker, R.S. Baltimore, J.H. Crabb, G. Schiffman and H.J. Jennings. 1979. Immunodeterminant specificity of human immunity to type III group B Streptococcus. J.Exp.Med. 149:327-339.
【非特許文献26】Knobloch, J.E. and P.N. Shaklee. 1997. Absolute molecular weight of low-molecular-weight heparins by size-exclusion chromatography with multiangle laser light scattering detection. Anal.Biochem. 245:231-241.
【非特許文献27】Lancefield, R.C.. 1933. A serological differentiation of human and other groups of haemolytic streptococci. J.Exp.Med. 57:571-595.
【非特許文献28】Lancefield, R.C.. 1938. A micro-precipitin technique for classifying hemolytic streptococci and improved methods for producing antigen. Proc.Soc.Exp.Biol.and Med. 38:473-478.
【非特許文献29】Lancefield, R.C., M. McCarty and W.N. Everly. 1975. Multiple mouse-protective antibodies directed against group B streptococci: Special reference to antibodies effective against protein antigens. J.Exp.Med. 142:165-179.
【非特許文献30】Madoff, L.C., L.C. Paoletti, J.Y. Tai and D.L. Kasper. 1994. Maternal immunization of mice with Group B streptococcal type III polysaccharide-beta C protein conjugate elicits protective antibody to multiple serotypes. J.Clin.Invest. 94:286-292.
【非特許文献31】Marques, M.B., D.L. Kasper, A. Shroff, F. Michon, H.J. Jennings and M.R. Wessels. 1994. Functional activity of antibodies to the group B polysaccharide of group B streptococci elicited by a polysaccharide-protein conjugate vaccine. Infect.Immun. 62:1593-1599.
【非特許文献32】Makela, P.R.H., H. Peltola, H. Kayhty, et al. 1977. Polysaccharide vaccines of group A Neisseria meningitidis and Haemophilus influenzae type b: A field trial in Finland. J.Infect.Dis. 136:543-50.
【非特許文献33】Michon, F., J.R. Brisson, A. Dell, D.L. Kasper and H.J. Jennings. 1988. Multiantennary group-specific polysaccharide of group B Streptococcus. Biochem. 27:5341-5351.
【非特許文献34】Peltola, A., H. Kayhty, A. Sivonen and P.R.H. Makela. 1977. Haemophilus influenzae type b capsular polysaccharide vaccine in children: A double blind field study of 100,000 vaccines 3 months to 5 years of age in Finland. Pediatrics 60:730-737.
【非特許文献35】Peltola, H., P.R.H. Makela, H. Jousimies, et al. 1977. Clinical efficacy of meningococcal group A vaccine in children three months to five years of age. N.Engl.J.Med. 297:686-691.
【非特許文献36】Reuter, G. and R. Schauer. 1994. Determination of sialic acids. p. 168-199. In: W.J. Lennarz and G.W. Hart, Methods in Enzymology Vol. 230 Techniques in Glycobiology. Academic Press, New York.
【非特許文献37】Robbins, J.B. and R. Schneerson. 1990. Polysaccharide-protein conjugates: A new generation of vaccines. J.Infect.Dis. 161:821-832.
【非特許文献38】Scwander, R., Dziarski R., Wesche, H. et al. 1999. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J. Biol. Chem. 274:17406-17409.
【非特許文献39】Smith, A.L. and J. Haas. 1991. Neonatal Bacterial Meningitis. p. 313-333. In: W.M. Scheld, R.J. Whitley and D.T. Durack, Infections of the Central Nervous System. Raven Press, Ltd., New York,
【非特許文献40】Tsunashima, T., K: Moro, B. Chu and T.-Y. Liu. 1978. Characterization of group C meningococcal polysaccharide by light-scattering spectroscopy. III. Determination of molecular weight, radius of gyration, and translational diffusional coefficient.. Biopolymers 17:251-265.
【非特許文献41】von Hunolstein, C., L. Nicolini, S. D'Ascenzi, C. Volpe, G. Alfarone and G. Orefici. 1993. Sialic acid and biomass production by Streptococcus agalactiae under different growth conditions. Appl.Microbiol.Biotechnol. 38:458-462.
【非特許文献42】Wessels, M.R., W.J. Benedi, H.J. Jennings, F. Michon, J.L. DiFabio and D.L. Kasper. 1989. Isolation and characterization of type IV group B Streptococcus capsular polysaccharide. Infect.Immun. 57:1089-1094.
【非特許文献43】Wessels, M.R., J.L. DiFabio, V.J. Benedi, et al. 1991. Structural determination and immunochemical characterization of the type V group B Streptococcus capsular polysaccharide. J.Biol.Chem. 266:6714-6719.
【非特許文献44】Wessels, M.R., L.C. Paoletti, D.L. Kasper, et al. 1990. Immunogenicity in animals of a polysaccharide-protein conjugate vaccine against type III group B Streptococcus. J.Clin.Invest. 86:1428.1433.
【非特許文献45】Wessels, M.R., L.C. Paoletti, A.K. Rodewald, et al. 1993. Stimulation of protective antibodies against type la and Ib group B streptococci by a type Ia polysaccharide-tetanus toxoid conjugate vaccine. Infect.Immun. 61:4760-4766.
【非特許文献46】Wessels, M.R., V. Pozsgay, D.L. Kasper and H.J. Jennings. 1987. Structure and immunochemistry of an oligopolysaccharide repeating unit of the capsule polysaccharide of Type III Group B Streptococcus: A revised structure for the Type III Group B streptococcal polysaccharide antigen. J.Biol.Chem. 262:8262-8267.
【非特許文献47】Wyle, S.A., M.S. Artenstein, B.L. Brandt, et al. 1972. Immunologic response of man to group B meningococcal polysaccharide vaccines. J.Infect.Dis. 126:514-522.
【非特許文献48】Yang, R.B., Mark, M.R., Gray, A., et al. 1998. Toll-like receptor 2 mediates lipopolysaccharide-induced cellular signaling. Nature. 395:284-288.
【非特許文献49】Tipson, R.S. and Horton, D., Advances in Carbohydrate Chemistry and Biochemistry, Vol. 41, 1983, Academic Press, NY.
【非特許文献50】Westphal et al., Methods in Carbohydrate Chemistry, vol. V, 1965, Academic Press, NY.
【非特許文献51】Theodora W. Greene and Peter G. M. Wuts, Protective Groups in Organic Syntheses, 2nd Ed. (1991).
【非特許文献52】Kohler and Milstein (1975) Nature 256:495-497.
【非特許文献53】Takeda et al. (1985) Nature 314:452.
【非特許文献54】Campbell (1985) Laboratory Techniques in Biochemistry and Molecular Biology, Vol. 13, Burdon, et al. (eds.), Elsevier Science Publishers, Amsterdam.
【非特許文献55】Schneerson, R., et al. (1980) J. Exp. Med. 1952:361-476.
【非特許文献56】Marburg, S., et al. (1986) J. Am. Chem. Soc. 108:5282-5287.
【非特許文献57】Grossman, M. and Cohen, S. N., in "Basic and Clinical Immunology", 7th Ed., (Stites, D. P. and Terr, A. T. eds., Appleton & Lange 1991) Chapter 58 "Immunization".
【非特許文献58】Paoletti, et al. (1997) J. Infectious Diseases, 175:1237-9.
【非特許文献59】L. Warren, (1959) 1 Biol. Chem. 234, 1971.
【発明の開示】
【発明が解決しようとする課題】
【0010】
本発明の利点としては、(1)単純性、(2)分子収量の増加、(3)スケーラビリティー(大規模生産)、(4)精製したポリサッカライドをその天然の抗原型に保持する又は戻すことができること、及び(5)DNA、RNA、及び毒素が、加水分解ステップにおいて分解され、それ故、本発明に従って産生された最終産物中には感知できるほどの量が存在しないこと、の少なくとも1つが挙げられる。
【課題を解決するための手段】
【0011】
いくつかの実施形態において、ポリサッカライド及び細胞成分を含むストックからのポリサッカライドの精製は、以下の方法により達成することができる。その方法は、該ストックを第1の試薬と接触させて第1の混合物を形成するステップを必要とする。その第1の試薬は、酸又は塩基であり、第1の混合物のpHは、0〜6の範囲又は9〜15の範囲である。分離組成物は、細胞成分の少なくとも一部からポリサッカライドを分離して形成する。その分離組成物は、ポリサッカライドと細胞成分の残量とを含む。その分離された組成物は、第2の試薬と接触させて第2の混合物を形成する。その第2の試薬は塩基であり、第2の混合物のpHは、9〜15の範囲である。精製組成物は、細胞成分の残量の少なくとも一部からのポリサッカライドを精製することにより形成される。
【0012】
いくつかの実施形態において、ポリサッカライド及び細胞成分を含むストックからのポリサッカライドの精製は、以下の方法により達成することができる。その方法は、該ストックを第1の試薬と接触させて第1の混合物を形成するステップを必要とする。その第1の試薬は、塩基及び還元剤を含み、第1の混合物のpHは、9〜15の範囲である。分離された組成物を、細胞成分の少なくとも一部からポリサッカライドを分離することによって形成する。精製したポリサッカライドを、その分離ステップ後に回収する。
【0013】
そのストックを第1の試薬と接触させるステップを、本明細書では時に「第1ステップの加水分解」と称する。該分離組成物を第2の試薬と接触させるステップを、本明細書では時に「第2ステップの加水分解」と称する。これらの呼び方は、明確にするためになされ、どのようにも限定するものと解釈すべきではない。
【0014】
本発明の用途の非限定の例としては、精製したポリサッカライドの組成物、ポリサッカライド−ポリペプチド結合体、医薬品組成物、診断キット、及び抗体を製造することが挙げられる。抗体は、ポリサッカライドのオリジナルソースと反応することができ、それらは他の有機体と交差反応性であり得る。
【0015】
いくつかの実施形態において、ポリサッカライド結合ワクチンは、以下の方法を用いて製造することができる。精製ポリサッカライドは、上記の方法を用いて獲得する。そのポリサッカライドをポリペプチドに結合し、それによってワクチンを製造する。いくつかの実施形態において、そのワクチンにはアジュバントを添加する。いくつかの実施形態において、その結合は、還元的アミノ化によって達成する。
【発明を実施するための最良の形態】
【0016】
本発明の方法は、細胞成分から大きな収量でポリサッカライドを抽出するために使用することができる。収量の比較のためには実施例1を参照されたい。いくつかの実施形態において、該方法は、また、高分子量ポリサッカライドを分解するためにも有利に使用することができる。さらに、いくつかの実施形態において、本発明の方法は、混入物、例えばタンパク質及び核酸などからポリサッカライドを分離し、精製するための便利な膜濾過方法の任意的な使用を可能にする。これまでは、実質的に純粋なポリサッカライドは、一般的には低処理量で複雑な沈殿及びカラムクロマトグラフ技術を用いることによって得ていた。抽出したポリサッカライドを分離し、精製するために膜濾過方法を使用することができることは、膜濾過が、単純で、高い処理能力があり、規模拡大が可能な精製したポリサッカライドの高い収量を維持する技術であるために望ましい。
【0017】
ポリサッカライド及び細胞成分を含むストックからのポリサッカライドの精製は、本発明のいくつかの実施形態によれば2ステップの加水分解法によって達成することができる。その2ステップの加水分解法は、塩基又は酸を用い、その塩基性又は酸性試薬がポリサッカライドと細胞成分例えば微生物細胞壁の間の1つ又は複数の結合の加水分解をもたらすことができる第1ステップの加水分解を伴う。これらの塩基に不安定又は酸に不安定な結合は、ポリサッカライドをペプチドグリカンに連結するホスホジエステル結合及び/又はポリラムノースをペプチドグリカンに連結するホスホジエステル結合であり得る。ポリサッカライドと細胞壁成分の間の結合の加水分解により、ポリサッカライドを細胞壁成分から分離することができる。第1ステップの加水分解後に、例えばポリサッカライドを非付着細胞成分、例えば細胞壁成分、タンパク質、核酸及び脂質から分離して、分離組成物を生み出すことができる分離ステップが続く。その分離組成物は、ポリサッカライド及び細胞成分の残量を含む。第2ステップの加水分解は、そのときその分離組成物について行うことができ、ポリサッカライドと細胞壁成分の間の残りの結合を加水分解し且つ/又は望ましくないタンパク質及び核酸を少なくとも部分的に分解することができる。その第2ステップの加水分解の後には、分離、並びに任意的なN−アシル化及び精製が続く。
【0018】
いくつかの実施形態において、ポリサッカライドは、2回を超える加水分解ステップを用いて単離することができる。2回の加水分解ステップで実質的に純粋なポリサッカライドを単離するためのクロマトグラフィーの使用の必要性を未然に防ぐためには十分であり得るが、2回を超える加水分解ステップは、収量及び純度の向上をもたらすことができ、本発明に記載の方法を用いて決定することができる。いくつかの実施形態においては、3回の加水分解ステップが用いられ、それぞれ分離ステップが続く。いくつかの実施形態においては、4回の加水分解ステップが用いられ、それぞれ分離ステップが続く。いくつかの実施形態においては、4回を超える加水分解ステップを用いることができる。
【0019】
いくつかの実施形態において、ポリサッカライドの精製は、1回目、2回目、及び/又はすべての加水分解ステップに対して精製したヌクレアーゼ及び/又は精製したプロテアーゼを試薬中に添加しないで達成することができる。これらの実施形態において、その反応混合物は、ヌクレアーゼ及び/又はプロテアーゼを、不純物を介して及び/又はポリサッカライドを含むストック中のそれらの存在を介して依然として含有することができる。
【0020】
いくつかの実施形態においては、1回だけの加水分解ステップを必要とする。これらの実施形態においては、第1の試薬が、塩基及び還元剤を含む。その還元剤は、例えば、加水分解ステップの間、ポリサッカライドを保護する助けをすることができる。
【0021】
いくつかの実施形態において、他の細胞成分からポリサッカライドを引き離すためのpH範囲は、9〜14、例えば10〜13、11〜13、又は12である。加水分解は、2℃から100℃まで、例えば、4℃〜100℃、20℃〜100℃、30℃〜100℃、40℃〜100℃、25℃〜90℃、30℃〜80℃、34℃〜41℃、又は37℃の温度で達成することができる。下端が25℃以上である温度範囲は、収量の増加をもたらすことができる。第2ステップの加水分解は、第1ステップの加水分解の方式で用いられる加水分解条件より好ましくは強い条件で達成することができ、その結果、タンパク質、内毒素、及び核酸が、少なくとも部分的に分解される。いくつかの実施形態において、第2ステップの加水分解は、第1ステップより10〜90℃高い(例えば、少なくとも10、20、30、40、50、60、又は70℃は高いが90℃より高くはない)温度で起こり得る。いくつかの実施形態において、第2ステップの加水分解中の最高温度は、第1ステップの加水分解中の最高温度より10〜90℃高い(例えば、少なくとも10、20、30、40、50、60、又は70℃は高いが90℃より高くはない)。例えば、分離された組成物を第2の試薬と接触させる間の最高温度は、ストックを第1の試薬と接触させる間の最高温度より30℃〜90℃高い。いくつかの実施形態において、第2のステップは、第1のステップより1〜45時間長く持続させることができる(例えば、少なくとも、1、5、10、15、20、25、30、35、又は40時間は長いが45時間より長くはない)。いくつかの実施形態において、第2ステップの混合物のpHは、第1ステップの混合物よりpH7からの絶対値で0.1〜5単位離れていることができる(例えば、少なくとも0.1、0.2、0.3、0.4、0.5、1、1.3、1.5、2、2.3、2.5、3、3.3、3.5、4、又は4.5単位離れているが5単位を超えて離れてはいない)。いくつかの実施形態において、第2の混合物中の塩基の規定度は、第1の混合物中で使用される酸又は塩基の規定度より0.25〜4N大であり得る(例えば、少なくとも0.25、0.5、0.75、1、1.5、2、3.5、3、又は3.5Nは大きいが、4Nを超える大きさではない)。例えば、第2ステップの加水分解は、第2の混合物が、第1の混合物中の酸又は塩基の規定度より0.5〜4N大きい塩基の規定度を有する状態で、第1ステップの加水分解より40℃〜90℃高温で行うことができる。例えば、第2ステップの加水分解条件は、2NのNaOHの最終濃度により80℃で15〜48時間(精製されるポリサッカライドによる)にわたって行うものとすることができる。上記条件は、核酸とタンパク質の両方を著しく分解するのに一般的には十分であり、それらのフラグメントは、次に濾過によって容易に除去することができる。いくつかの実施形態において、この2ステップの加水分解法は、高分子量のポリサッカライドの分解をもたらすことができ、それは当該方法によって製造される産物において特にそして最終的に有用であり得る。
【0022】
(出発材料)
本発明により精製されるポリサッカライドのソースは、多様である。ソースとしては、細菌、古細菌、及び真核生物が挙げられる。多細胞生物においては、そのソースは全有機体の一部であり得る(例えば、癌細胞又は癌性細胞)。
【0023】
有機体のさらなる例としては、病原菌類、例えばアエロモナス細菌(Aeromonas hydrophila)及び他の菌種;炭疽菌(Bacillus anthracis);セレウス菌(Bacillus cereus);ボツリヌス神経毒素を産生する菌種のクロストリジウム属(Clostridium);ウシ流産菌(Brucella abortus);マルタ熱菌(Brucella melitensis);ブタ流産菌(Brucella suis);鼻疽菌(Burkholderia mallei)(旧学名はPseudomonas mallei);類鼻疽菌(Burkholderia pseudomallei)(旧学名はPseudomonas pseudomallei);カンピロバクター・ジェジュニ(Campylobacter jejuni);オウム病クラミジア(Chlamydia psittaci);ポツリヌス菌(Clostridium botulinum);ウェルシュ菌(Clostridium perfringens);コクシジオイデス・イミティス(Coccidioides immitis);コクシジオイデス・ポサダシ(Coccidioides posadasii);心糸状虫(Cowdria ruminantium)(心水病);Q熱コクシエラ(Coxiella burnetii);エンテロ毒性大腸菌群(EEC群,Enterovirulent Escherichia coli group)、例えば、毒素原生大腸菌(ETEC,Escherichia coli-enterotoxigenic)、腸管病原性大腸菌(EPEC,Escherichia coli-enteropathogenic)、O157:H7腸管出血性大腸菌(EHEC,Escherichia coli-O157: H7 enterohemorrhagic)、及び腸管組織侵入性大腸菌(EIEC,Escherichia coli-enteroinvasive)など;エーリキア(Ehrlichia)菌種、例えばエーリキア・シャフェンシス(Ehrlichia chaffeensis)など;野兎病菌(Francisella tularensis);在郷軍人病菌(Legionella pneumophilia);リベロバクター・アフリカヌス(Liberobacter africanus);リベロバクター・アジアティクス(Liberobacter asiaticus);リステリア菌(Listeria monocytogenes);多岐に渡る腸内細菌、例えば、クレブシエラ(Klebsiella)、エンテロバクター(Enterobacter)、プロテウス(Proteus)、シトロバクター(Citrobacter)、アエロバクター(Aerobacter)、プロビデンシア(Providencia)、及びセラチア(Serratia)など;マイコバクテリウム・ボビス(Mycobacterium bovis);結核菌(Mycobacterium tuberculosis);マイコプラズマ・カプリコルム(Mycoplasma capricolum);マイコプラズマ・ミコイデス亜種ミコイデス(Mycoplasma mycoides ssp mycoides);ペロノスクレロスポラ・フィリピネシス(Peronosclerospora philippinensis);ダイズさび病菌(Phakopsora pachyrhizi);プレシオモナス・シゲロイデス(Plesiomonas shigelloides);青枯病菌(Ralstonia solanacearum)品種3、次亜種2;発疹チフスリケッチイ(Rickettsia prowazekii);リケッチア・リケッチイ(Rickettsia rickettsii);サルモネラ菌種(Salmonella spp.);スクレロープソーラ・レイジアエ・バラエティー・ゼアエ(Schlerophthora rayssiae var zeae);赤痢菌菌種(Shigella spp.);黄色ブドウ球菌(Staphylococcus aureus);連鎖球菌(Streptococcus);ジャガイモがんしゅ病菌(Synchytrium endobioticum);非O1コレラ菌(Vibrio cholerae non-O1);O1コレラ菌(Vibrio cholerae O1);腸炎ビブリオ菌(Vibrio parahaemolyticus)及びその他のビブリオ菌;ビブリオ・バルニフィカス(Vibrio vulnificus);イメ白葉枯病菌(Xanthomonas oryzae);ピアス病菌(Xylella fastidiosa)(柑橘類ふ入り白化菌株);エンテロコリチカ菌(Yersinia enterocolitica)及び仮性結核菌(Yersinia pseudotuberculosis);並びにペスト菌(Yersinia pestis)が挙げられる。
【0024】
有機体のさらなる例としては、菌類、例えば、アスペルギルス菌種(Aspergillus spp.);ブラストミセス・デルマティティディス(Blastomyces dermatitidis);カンジダ(Candida);コクシジオイデス・イミチス(Coccidioides immitis);コクシジオイデス・ポサダシ(Coccidioides posadasii);クリプトコッカス・ネオフォルマンス(Cryptococcus neoformans);ヒストプラズマ・カプスラーツム(Histoplasma capsulatum);トウモロコシさび病菌(Maize rust);イネイモチ病菌(Rice blast);イネ褐斑病菌(Rice brown spot disease);ライムギイモチ病菌(Rye blast);スポロトリクス・シェンキイ(Sporothrix schenckii);及びコムギ菌(wheat fungus)などが挙げられる。
【0025】
有機体のさらなる例としては、寄生原虫及び寄生虫、例えば、アカントアメーバ(Acanthamoeba)及びその他の自由生活アメーバ;アニサキス種(Anisakis sp.)並びにその他の関連する寄生虫の回虫(Ascaris lumbricoides)及び鞭虫(Trichuris trichiura);クリプトスポリジウム・パルバム(Cryptosporidium parvum);シクロスポラ・カイエタネンシス(Cyclospora cayetanensis);裂頭条虫種(Diphyllobothrium spp.);赤痢アメーバ(Entamoeba histolytica);Eustrongylides sp.;ランブル鞭毛虫(Giardia lamblia);ナノフィエトゥス種(Nanophyetus spp.);住血吸虫種(Shistosoma spp.);トキソプラズマ原虫(Toxoplasma gondii);及び旋毛虫(Trichinella)が挙げられる。
【0026】
ソースのさらなる例としては、小細胞肺癌、神経芽細胞腫、乳癌、結腸癌などの癌細胞又は癌組織が挙げられる。これらの癌細胞又は癌組織は、例えば、ヒト、マウス、イヌ、ネコ、ヤギ、サル、又は雌ウシなどの哺乳類のものであり得る。
【0027】
本発明に従って使用するためのグラム陽性菌の非限定の例は、連鎖球菌(Streptococci)、ブドウ球菌(Staphylococci)、腸球菌(Enterococci)、桿菌(Bacillus)、コリネバクテリウム(Corynebacterium)、リステリア菌(Listeria)、エリシペロスリクス(Erysipelothrix)、及びクロストリジウム(Clostridium)である。本発明で使用するためのグラム陰性菌の非限定の例としては、ヘモフィルス(Haemophilus)(例えばインフルエンザ菌(Haemophilus influenzae))、ナイセリア(Neisseria)(例えば、髄膜炎菌(Neisseria meningitidis))及び大腸菌類(Escherichia)(例えば、大腸菌(Escherichia coli))が挙げられる。
【0028】
精製に対して望ましいポリサッカライドは、細胞壁などの細胞成分と関係するものであり得る。細胞壁と関係するとは、そのポリサッカライドが細胞壁それ自体の成分であり、且つ/又はその細胞壁に直接又は中間分子を介して間接的に付着しており、或いはその細胞壁を一時的に被覆するものである(例えば、一定の細菌の菌株は、技術的に「エキソポリサッカライド」としても知られる莢膜ポリサッカライドを滲出する)ことを意味する。
【0029】
いくつかの実施形態において、細菌から抽出されたポリサッカライドは、莢膜ポリサッカライド、被膜下ポリサッカライド、又はリポポリサッカライドであり得る。いくつかの実施形態において、該ポリサッカライドは、莢膜ポリサッカライドであり得る。
【0030】
いくつかの実施形態において、該ポリサッカライドは、A群連鎖球菌(GAS,group A streptococcus)の群特異的なポリサッカライドの被膜下ポリサッカライドであり得る。
【0031】
いくつかの実施形態において、該ポリサッカライドは、B群連鎖球菌(GBS,group B streptococcus)から抽出することができる。いくつかの実施形態において、該ポリサッカライドは、GBSタイプIa、Ib、II、III、IV、V、VI、VII及び/又はVIIIから抽出される型特異性のCPSであり得る。いくつかの実施形態において、該ポリサッカライドは、GBSタイプIa、Ib、II、III及びVから抽出されるCPSであり得る。
【0032】
いくつかの実施形態において、該ポリサッカライドは、C群連鎖球菌(GCS,group C streptococcus)からの群ポリサッカライドの被膜下ポリサッカライドであり得る。
【0033】
いくつかの実施形態において、該ポリサッカライドは、肺炎球菌(S. pneumoniae)から抽出することができる。いくつかの実施形態において、CPSは、肺炎球菌タイプ3及び14から抽出することができる。
【0034】
いくつかの実施形態において、該ポリサッカライドは、ナイセリア(Neisseria)又は大腸菌(Escherichia)から抽出することができる。いくつかの実施形態において、該ポリサッカライドは、髄膜炎菌(Neisseria meningitidis)B型、C型、Y型又はW135型或いは大腸菌K1(Escherichia coli K1)から抽出することができる。
【0035】
いくつかの実施形態においては、1種類のポリサッカライドを精製する。いくつかの実施形態においては、複数種のポリサッカライドを1つ又は複数のソースから共精製する。
【0036】
本明細書で使用する用語「ストック」とは、精製すべきポリサッカライドを含む出発材料を指す。典型的なストックは、上澄み、条件培地、均質化細胞、又は細胞ペレットである。エキソポリサッカライド(細菌から上澄み又は媒体中に滲みだされる莢膜ポリサッカライド)が所望のポリサッカライドである場合、典型的なストックは、遠心分離又は精密濾過によって細胞を分離し、その上澄みを一般的には10〜15倍濃縮することによって形成した濃厚な上澄みであり得る。いくつかの実施形態において、そのストックは、ポリサッカライドが5〜20mg/mLの濃度で存在するように濃縮した上澄み又は条件培地である。興味のあるポリサッカライドが細胞壁に付着している場合、そのときの典型的なストックは、ペレット化した又は均質化した細胞である。
【0037】
(第1ステップの加水分解)
本発明によれば、ポリサッカライドは、ストックを第1の試薬の酸試薬又は塩基試薬と接触させることによって抽出する。理論に縛られるものではないが、共有結合が、ポリサッカライドと細胞壁などの細胞成分(この細胞成分は、それ自体、例えばポリサッカライド、タンパク質、又は脂質であり得る)の間(又はポリサッカライドとポリサッカライドを細胞成分に連結している中間分子の間)で破壊されるものと考えられる。その他のタイプの結合としては、以下に限定はされないが、イオン結合及びファンデルワールス力によって形成される結合が挙げられる。ポリサッカライドと細胞壁成分を連結している結合は、ポリサッカライドと細胞壁成分の間の「直接」結合、又はポリサッカライドの細胞壁成分への付着をもたらす「介在する」又は「間接的」結合であり得る。その結合の破壊をもたらす化学反応は、塩基又は酸のいずれかによる加水分解反応である。
【0038】
一般に、シアル酸結合を有するB群連結球菌ポリサッカライドは、酸によって開裂されるが塩基にはならない。しかしながら、ホスホジエステル結合を含有する莢膜ポリサッカライドは、主として塩基によって開裂され、酸によってはそれほどではない。例えば、インフルエンザ菌(Haemophilus influenzae)b型及びいくつかの肺炎球菌莢膜ポリサッカライド類、例えば6A、6B、18、及び23などである。髄膜炎菌A(Meningococcal A)及び肺炎球菌(pneumococcal)19型は、酸及び塩基の両方によって開裂されるいくつかの莢膜ポリサッカライドの例である。
【0039】
(第1の試薬組成物)
いくつかの実施形態において、第1の試薬は、いろいろな塩基の1つ又は複数を含む。本発明に従って使用することができる塩基の非限定の例は、NaOH、KOH、LiOH、NaHCO3、Na2CO3、K2CO3、KCN、Et3N、NH3、H2N2H2、NaH、NaOMe、NaOEt及びKOtBuから選択される化合物を含む。いくつかの実施形態において、NaOH、KOH、LiOH、NaH、NaOMe又はKOtBuなどの塩基は、0.5N〜10Nの範囲(例えば、0.5N〜5N、0.5N〜2N、又は0.8〜1.5N)で使用することができる。いくつかの実施形態において、NaHCO3、Na2CO3、K2CO3及びKCNなどの塩基は、それらの溶解性が許容する高さの濃度で使用することができる。いくつかの実施形態において、トリエチルアミン(Et3N)などの有機塩基は、水又はアルコールなどの加水分解を生じる作用物質が存在する限り、中濃度から高濃度、例えば、1Nから7N(例えば2N〜4N)の範囲で使用することができる。アンモニア(NH3)又はヒドラジン(H2NNH2)などの塩基は、100%を含む殆んどどんな濃度でも使用することができる。溶媒、例えば、水、アルコール類(好ましくはC1−C4)、ジメチルスルホキシド、ジメチルホルムアミド、又はこれら及びその他の有機溶媒の混合物などを使用することができる。水を含む加水分解用の塩基溶液も使用することができる。
【0040】
いくつかの実施形態において、第1の試薬のpHは、9〜15の範囲内である。例えば、第1の試薬のpHは、10〜15、10〜14、11〜14、又は12〜14の任意の範囲内であり得る。いくつかの実施形態において、ストックと第1の試薬の混合物は、9〜14の範囲のpHを有し、例えば、その混合物は、9〜13、10〜13、又は11〜13のいずれかの範囲のpHを有することができる。いくつかの実施形態において、その混合物のpHは、12付近である。
【0041】
いくつかの実施形態において、第1の試薬は、いろいろな酸の1つ又は複数を含む。本発明に従って使用することができる酸の非限定の例は、HCl、H3PO4、クエン酸、酢酸、亜硝酸、及び硫酸から選択される化合物を含む。
【0042】
いくつかの実施形態において、第1の試薬のpHは、0〜6の範囲内であり得る。例えば、第1の試薬のpHは、0〜5、0〜4、0〜3、1〜2の任意の範囲内であり得る。いくつかの実施形態において、ストックと第1の試薬の混合物は、0〜6の範囲内のpHを有し、例えば、その混合物は、0〜5、1〜5、2〜5、3〜5、又は4〜5の任意の範囲内のpHを有することができる。いくつかの実施形態において、その混合物のpHは、4付近である。
【0043】
その第1の試薬は、さらなる物質、例えば、塩類(例えば、NaCl又はKCl)、酸化剤(例えば、H2O2、O3、又は次亜塩素酸塩)、ホウ素を含む還元剤(例えば、NaBH4、NaCNBH3、リチウムトリ−sec−ブチルボロヒドリド、又はNaBH(OCOCH3)3を含む)、その他の還元剤(例えば、水素化アルミニウムリチウム、ジチオスレイトール、又はβ−メルカプトエタノールが挙げられる)、酵素類(例えば、プロテアーゼ類又はヌクレアーゼ類が挙げられる)、キレート化剤(例えば、EDTA又はEGTAが挙げられる)、及び界面活性剤類(非イオン型、両性イオン型、アニオン型又はカチオン型)などを場合によって含むことができる。いくつかの実施形態において、界面活性剤は、その後の濾過ステップにおいて役立ち得る。いくつかの実施形態において、還元剤によって、例えばポリサッカライドの還元末端の3番目の炭素に連結している隣接する糖残基のβ−エリミネーションによって引き起こされるいわゆる「ピーリング」によるポリサッカライドの分解の可能性を避けることができる。
【0044】
いくつかの実施形態において、第1の試薬は、塩基及び還元剤を含む。いくつかの実施形態において、その塩基は、0.5N〜5Nの範囲内であり、その還元剤は、0.03mM〜300mMの範囲内である。例えば、その塩基は、0.5N〜2Nの範囲内の濃度を有することができ、その還元剤は、0.3mM〜30mMの範囲内の濃度を有することができる。典型的な第1の試薬は、塩基及び水素化ホウ素ナトリウムを含む。いくつかの実施形態において、その塩基の濃度は、0.5N〜5Nの範囲内であり、水素化ホウ素ナトリウムの濃度は、0.03mM〜300mMの範囲内である。例えば、その塩基の濃度は、0.5N〜2Nの範囲内であり得、水素化ホウ素ナトリウムの濃度は、0.3mM〜30mMの範囲内である。いくつかの実施形態において、その塩基は、NaOH、LiOH及びKOHから選択される。
【0045】
(使用)
いくつかの実施形態において、ストックは、第1の試薬1リットル当り0.1〜300gの細胞ペーストであることができる。例えば、そのストックは、第1の試薬1リットル当り0.5〜200gの細胞ペーストであることができる。例えば、そのストックは、第1の試薬1リットル当り1〜100gの細胞ペーストであることができる。例えば、そのストックは、第1の試薬1リットル当り5〜150gの細胞ペーストであることができる。例えば、そのストックは、第1の試薬1リットル当り20〜100gの細胞ペーストであることができる。いくつかの実施形態において、そのストックは、濃縮した上澄みであるか又は濃縮した第1の試薬(例えば、10NのNaOH)が、濃縮度のより低い混合物を得るために添加される(例えばその混合物のpHによって決定されるか又はその混合物中の第1の試薬の成分の濃度によって決定され、例えば、10NのNaOHが1NのNaOHに希釈される)条件培地である。
【0046】
加水分解は、2℃〜100℃の温度、例えば、4℃〜100℃、20℃〜100℃、30℃〜100℃、40℃〜100℃、25℃〜90℃、30℃〜80℃、34℃〜41℃、又は37℃で達成することができる。下端が25℃以上である温度範囲は、収量の向上をもたらすことができる。いくつかの実施形態において、混合物は、反応が起こることを可能にするために1〜48時間にわたってインキュベートする。例えば、そのインキュベーションは、1、2、3、4、5、6、7、8、9、10、12、14、15、16、18、20、24、36、若しくは48時間又はその間の任意の時間続けることができる。いくつかの実施形態において、その混合物はインキュベーションの間は、かき混ぜる。いくつかの実施形態において、その混合物はインキュベーションの間は、かき混ぜない。いくつかの実施形態において、第1ステップの加水分解は、15〜18時間のインキュベーションを用い、1NのNaOHの最終濃度で、37℃でかき混ぜて達成する。
【0047】
酸の第1試薬の典型的な使用としては、A群又はC群連鎖球菌の群特異的ポリサッカライド(例えば、GAS又はGCS被膜下ポリサッカライド)の加水分解を含む。かかるポリサッカライドについてのいくつかの実施形態において、第1の試薬は、0.1NのHCl、硝酸ナトリウム(NaNO2)、又はpH4〜5の緩衝酢酸溶液を含む。かかるポリサッカライドのいくつかの実施形態において、該混合物のpHは、4近くである。いくつかの実施形態において、第1ステップの酸加水分解は、混合物のpHが4近くである0.1NのHClの最終濃度で、室温で達成することができる。その他の酸濃度も、混合物中のストックの量、その混合物の望ましいpH、及び特定の酸の該ストックとの相互作用などの因子によって使用することができる。緩衝酢酸溶液pH4〜5、亜硝酸ナトリウム(NaNO2)を、25℃〜60℃の範囲の温度で同様に使用することができる。
【0048】
B群連鎖球菌(group B Streptococcus)、Y群髄膜炎菌(group Y meningococcus)、及びW−135群髄膜炎菌(group W-135 meningococcus)莢膜ポリサッカライドの精製についてのいくつかの実施形態において、1〜100グラム(例えば、1〜75グラム)の細胞ペーストを、750mLの1NのNaOH及び100mgの水素化ホウ素ナトリウムと接触させることができる。この反応混合物は、125rpmの速度で、例えば、37℃で16時間にわたって震盪させることができる。
【0049】
(第1ステップの分離)
別段の記述がない限り、この第1ステップの分離の項に以下で示されている本発明の態様は、本発明のすべてのその他の分離及び精製ステップに全面的に適用する。
【0050】
塩基又は酸の加水分解試薬中に存在する抽出されたポリサッカライドは、他の細胞成分に由来する不純物から、電荷、親水性、親和性、溶解性若しくは安定性、又は大きさに基づいて分離することができる。分離技術の非限定の例として、硫安塩析、クロマトグラフィー、及び膜濾過(接線流膜濾過を含む)が挙げられる。分離にクロマトグラフィーを利用する実施形態における典型的な方法としては、イオン交換(カチオン又はアニオン)、親和性クロマトグラフィー、親水性相互作用、疎水性相互作用、サイズ排除及びゲル浸透が挙げられる(米国特許第6248570号参照)。
【0051】
いくつかの実施形態において、その分離は、膜濾過によって行うことができ、それには、以下のものに限定はされないが、シングルパス、デッドエンド、ダイレクトフロー濾過(DFF,direct flow filtration)、及びクロスフロー又は接線流濾過(TFF,tangential flow filtration)が含まれる。本発明によれば、濾過は、細孔の大きさが規定範囲の半透膜を使用して大きさによって分子を分離する原理に基づいている。濾過方法及び膜のタイプの組合せを分離に使用することができることは当業者には知られている。
【0052】
本発明によれば、膜濾過は、ポリマー膜又は無機膜により生ずる細胞成分の分離である。当技術分野において、膜にはそれらが担体液から除去する材料の大きさによって定義される一般に是認されている4つのカテゴリーが存在する。細孔の大きさの最小の膜から最大の膜へと通す連続的濾過方法は、逆浸透(RO,Reverse Osmosis)、ナノ濾過(NF,Nanofiltration)、限外濾過(UF,Ultrafiltration)、及び精密濾過(MF,Microfiltration)である。
【0053】
上記の膜による濾過は、特定の細孔の大きさを有する膜を用いて、それらの分子量に応じて分子を分離する。例えば、0.001マイクロメートル未満の細孔の大きさを有するRO膜は、200ダルトン未満の分子量を有する分子を分離することを目的としている。0.001以上〜0.008マイクロメートル以下の細孔の大きさを有するNF膜による濾過は、200ダルトン以上〜15キロダルトン(kDa)以下の分子量を有する分子を分離することを目的としている。0.005以上〜0.1マイクロメートル以下の細孔の大きさを有するUF膜による濾過は、5kDa以上〜300kDa以下の分子量を有する分子を分離することを目的としている。0.05以上〜3.0マイクロメートル以下の細孔の大きさを有する精密濾過膜による濾過は、100kDa〜3000kDa以上の分子量を有する分子を分離することを目的としている。
【0054】
本発明によれば、膜濾過は、膜の細孔の大きさによって決まる特定の分画分子量(MWCO,Molecular Weight Cut-Off)を有する膜を利用することによるサイズ排除に基づいて、抽出したポリサッカライドを他の細胞成分から分離することができる。このMWCOは、公称分子量限界(NMWL,Nominal Molecular Weight Limit)又は公称分画分子量(NMWCO,Nominal Molecular Weight Cut-Off)とも呼ばれ、膜による濾過に対するキロダルトンの大きさの呼称である。このMWCOは、膜によって90%が保持される分子の分子量として定義される。例えば同じ分子量の分子は著しく異なる形状を有することがあり得るために、MWCOは、正確な測定基準ではないが、それにもかかわらず有用な測定基準であり、フィルターメーカーにより一般的に採用されている。疎水性並びに親水性の膜の両方を、本発明では使用することができる。そのような膜は、平らなシートとして、又はらせん状にねじれた形態で使用することができる。中空糸もまた使用することができる。UF膜の組成に関しては、可能性のあるいくつもの膜材料を使用することができ、以下に限定はされないが、再生セルロース、ポリエーテルスルホン(これはその固有の疎水性を改変するために修飾してもしなくてもよい)、ポリフッカビニリデン、並びにセラミックと金属酸化物との凝集体が挙げられる。多くのポリエーテルスルホンUF膜が、0.5〜13のpH範囲、及び85℃まで変動する温度に耐えることができる。MF膜用の材料は、UF膜用に使用されるすべて、並びに、ポリカーボネート、ポリプロピレン、ポリエチレン及びPTFE(テフロン(登録商標))さえも含む。
【0055】
いくつかの実施形態において、第1の加水分解ステップ後の反応混合物は、継続する膜又はクロマトグラフィーの使用を必要とする分離及び精製のステップを細胞残屑が妨げることを防止するために、大きな細胞残屑を小さい細胞成分から分離するために濾過することができる。これらの実施形態において、その透過液は、ポリサッカライドを含み、回収される。いくつかの実施形態において、第1の加水分解後の反応混合物は、0.05〜3.0マイクロメートルの間の細孔の大きさを有する精密濾過膜(例えば、500kDa〜3000kDa以上のMWCOを有するフィルター)により濾過することができる。いくつかの実施形態において、第1の加水分解後の反応混合物は、0.05〜0.5マイクロメートルの間(例えば、0.1〜0.2μm)の細孔の大きさを有する精密濾過膜により濾過することができる。いくつかの実施形態において、第1の加水分解ステップ後の反応混合物は、0.05〜3.0マイクロメートル(μm)のポリエーテルスルホン中空糸のフィルターモジュールにより濾過する。
【0056】
いくつかの実施形態において、膜は、5kDa〜300kDaの範囲内のMWCOを有する分離ステップにおいて使用することができる。例えば、そのMWCOは、5kDa〜200kDa、30kDa〜100kDa、10kDa〜60kDa又は50kDa〜200kDaであり得る。これらの実施形態において、その未透過液は、ポリサッカライドを含み、回収することができる。
【0057】
いくつかの実施形態において、500kDa〜3000kDa濾過ステップ及び5kDa〜300kDa濾過ステップの両方が実施される。例えば、500kDa〜3000kDa濾過による透過液を、次に5kDa〜300kDa膜により濾過し、その最後の未透過液を回収することができる。
【0058】
いくつかの実施形態において、接線流濾過は、両方のダイアフィルター(diafilter)(小さい分子を膜を通して洗浄し、興味のある大きい分子を未透過液中に残す分画プロセス)に作用し、反応混合物を濃縮することができる。本発明によれば、ダイアフィルトレーション(diafiltration)は、不連続式又は連続式ダイアフィルトレーションのいずれかである。不連続式ダイアフィルトレーションにおいては、溶液は濃縮され、失われた容積は新たな緩衝液により置換される。連続式ダイアフィルトレーションにおいては、溶液の容積は、新たな緩衝溶液の流入により維持され、その間古い緩衝溶液は除去される。
【0059】
いくつかの実施形態において、ポリサッカライドの分離及び精製は、限外濾過膜を用いる接線流濾過法によって実施することができる。
【0060】
いくつかの実施形態において、クロマトグラフィーは、ポリサッカライドの細胞成分からの分離に使用しない。
【0061】
いくつかの実施形態において、限外濾過膜を使用して、膜のMWCOが5kDa〜100kDaであり得る第1の分離ステップにおいて、GBS CPSを分離することができる。例えば、限外濾過膜のMWCOは、15kDa〜50kDaであり得る。例えば、GBS CPSを分離するために使用する限外濾過膜のMWCOは30kDaであり得る。
【0062】
いくつかの実施形態において、分離ステップは、5kDa〜200kDaのMWCOを有する限外濾過膜を使用する。そのUF膜は、第1の分離ステップにおいて、髄膜炎菌(Neisseria meningitidis)B型、C型、Y型又はW135型;大腸菌K1(E. coli K1);C群連鎖球菌サブCPS(group C Streptococcus sub-CPS);又はA群連鎖球菌サブCPS(group A Streptococcus sub-CPS)を分離するために使用することができる。いくつかの実施形態において、Y群髄膜炎菌莢膜ポリサッカライド(GYMP,group Y meningococcus capsular polysaccharide)及びW−135群髄膜炎菌莢膜ポリサッカライド(GWMP,group W-135 meningococcus capsular polysaccharide)を分離するために使用される限外濾過膜のMWCOは、100kDaであり得る。
【0063】
(第2ステップの加水分解)
第1ステップの分離に由来するポリサッカライドは、第2の試薬、塩基試薬と接触させて第2の混合物を形成する。理論に縛られるものではないが、この第2ステップの加水分解は、(1)ポリサッカライドの自然の構造又は天然の抗原性を維持しながら、ポリサッカライドに付着して残留している細胞成分、特に細胞壁成分を引き離すこと、及び(2)その他の細胞成分混入物質(例えば、タンパク質及び核酸など)を分解すること、の少なくとも1つに対して有用であり得る。いくつかの実施形態において、この第2ステップの加水分解は、実質的に純粋なポリサッカライドを得るための複雑なクロマトグラフ法に対する必要性を取り除くことができる。
【0064】
(第2の試薬組成物)
第2の試薬は、いろいろな塩基の1つ又は複数を含む。本発明に従って使用することができる塩基の非限定の例は、NaOH、KOH、LiOH、NaHCO3、Na2CO3、K2CO3、KCN、Et3N、NH3、H2N2H2、NaH、NaOMe、NaOEt及びKOtBuから選択される化合物を含む。いくつかの実施形態において、NaOH、KOH、LiOH、NaH、NaOMe又はKOtBuなどの塩基は、1N〜10Nの範囲(例えば、1N〜6N、又は2〜5N)で使用することができる。いくつかの実施形態において、NaHCO3、Na2CO3、K2CO3及びKCNなどの塩基は、それらの溶解性が許容する高さの濃度で使用することができる。いくつかの実施形態において、トリエチルアミン(Et3N)などの有機塩基は、水又はアルコールなどの加水分解を生じる作用物質が存在する限り、中濃度から高濃度、例えば、1Nから7N(例えば2N〜4N)の範囲で使用することができる。アンモニア(NH3)又はヒドラジン(H2NNH2)などの塩基は、100%を含む殆んどどんな濃度でも使用することができる。溶媒、例えば、水、アルコール類(好ましくはC1−C4)、ジメチルスルホキシド、ジメチルホルムアミド、又はこれら及びその他の有機溶媒の混合物などを使用することができる。水を含む加水分解用の塩基溶液も使用することができる。
【0065】
いくつかの実施形態において、第2の試薬のpHは、9〜15の範囲内である。例えば、第2の試薬のpHは、10〜15、10〜14、11〜14、又は12〜14の任意の範囲内であり得る。いくつかの実施形態において、第2の混合物は、9〜15の範囲のpHを有し、例えば、その第2の混合物は、10〜15、10〜14、10〜13、11〜14、又は11〜13のいずれかの範囲のpHを有することができる。
【0066】
その第2の試薬は、さらなる物質、例えば、塩類(例えば、NaCl又はKCl)、酸化剤(例えば、H2O2、O3、又は次亜塩素酸塩)、ホウ素を含む還元剤(例えば、NaBH4、NaCNBH3、リチウムトリ−sec−ブチルボロヒドリド、又はNaBH(OCOCH3)3を含む)、その他の還元剤(例えば、水素化アルミニウムリチウム、ジチオスレイトール、又はβ−メルカプトエタノールが挙げられる)、酵素類(例えば、プロテアーゼ類又はヌクレアーゼ類が挙げられる)、キレート化剤(例えば、EDTA又はEGTAが挙げられる)、及び界面活性剤類(非イオン型、両性イオン型、アニオン型又はカチオン型)などを場合によって含むことができる。いくつかの実施形態において、界面活性剤は、その後の濾過ステップにおいて有用であり得る。
【0067】
いくつかの実施形態において、第2の試薬は、塩基及び還元剤を含む。いくつかの実施形態において、その塩基は、1N〜10Nの範囲内であり、その還元剤は、0.03mM〜300mMの範囲内である。例えば、その塩基は、2N〜6Nの範囲内の濃度を有することができ、その還元剤は、0.3mM〜30mMの範囲内の濃度を有することができる。典型的な第1の試薬は、塩基及び水素化ホウ素ナトリウムを含む。いくつかの実施形態において、その塩基の濃度は、1N〜10Nの範囲内であり、水素化ホウ素ナトリウムは、0.03mM〜300mMの範囲内である。例えば、その塩基の濃度は、2N〜6Nの範囲内であり得、水素化ホウ素ナトリウムの濃度は、0.3mM〜30mMの範囲内である。いくつかの実施形態において、その塩基は、NaOH、LiOH及びKOHから選択される。
【0068】
(使用)
加水分解は、20℃〜100℃の温度、例えば、30℃〜100℃、37℃〜100℃、40℃〜100℃、50℃〜95℃、60℃〜90℃、70℃〜90℃、又は80℃で達成することができる。いくつかの実施形態において、混合物は、反応が起こることを可能にするために3〜72時間にわたってインキュベートする。例えば、そのインキュベーションは、4、5、6、7、8、9、10、12、14、15、16、18、20、24、36、48、60、若しくは72時間又はその間の任意の時間続けることができる。いくつかの実施形態において、その混合物はインキュベーションの間は、かき混ぜる。いくつかの実施形態において、その混合物はインキュベーションの間は、かき混ぜない。
【0069】
いくつかの実施形態において、第2ステップの加水分解は、第1ステップの加水分解と比較したとき、(1)より高温、(2)絶対値が7からずっと離れたpH、(3)モル濃度のより高い試薬、及び(4)より長いインキュベーション時間のいずれか1つを用いて達成される。いくつかの実施形態において、第2ステップの加水分解は、第1ステップより少なくとも10、20、30、40、50、60、又は70℃高い温度で起こり得る。いくつかの実施形態において、この第2ステップは、第1ステップより1、5、10、15、20、25、30、35、40、又は45時間長く、或いは1時間と45時間の間の任意の時間量だけ長く続けることができる。いくつかの実施形態において、第2ステップの混合物のpHは、第1ステップの混合物より、pH7からの絶対値で、0.1、0.2、0.3、0.4、0.5、1、1.3、1.5、2、2.3、2.5、3、3.3、3.5、4、4.5、又は5単位、或いは、0.1と5の間の任意の単位量だけ離れていることができる。第2ステップの加水分解でこれらのより強力な条件を用いる実施形態においては、他の細胞成分、例えば、タンパク質及び核酸などを、第1ステップの加水分解におけるよりもさらに効率的に分解することができる。
【0070】
1つの典型的な第2ステップの加水分解は、1〜2NのNaOHを、37℃で12〜48時間にわたって使用する。もう1つの典型的な第2ステップの加水分解は、2NのNaOHを、80℃で15〜48時間にわたって使用する。
【0071】
GBS CPSに対する第2の加水分解は、いくつかの実施形態において、2NのNaOHの最終濃度、80℃、16〜18時間の条件下で行うことができ、この条件は、GBS CPSに対する第1の加水分解に対する典型的な条件(1NのNaOHの最終濃度、37℃、16時間)より強い条件である。
【0072】
GYMPに対する第2の加水分解は、いくつかの実施形態において、2NのNaOHの最終濃度、80℃、48時間の条件下で行うことができ、この条件は、GYMPに対する第1の加水分解に対する典型的な条件(1NのNaOHの最終濃度、37℃、16時間)より強い条件である。
【0073】
GWMPに対する第2ステップの加水分解は、いくつかの実施形態において、2NのNaOHの最終濃度、80℃、一晩(本明細書で使用される「一晩」とは、15〜18時間を意味する)の条件下で行うことができ、この条件は、GWMPに対する第1ステップの加水分解に対する典型的な受験(1NのNaOHの最終濃度、37℃、16時間)より強い条件である。
【0074】
(第2ステップの分離)
第2ステップの加水分解に由来する抽出ポリサッカライドは、細胞成分に由来する不純物からの第1ステップの分離と同じように、以下に限定はされないが、クロマトグラフィー及び膜濾過などの分離技術によって分離することができる。クロマトグラフ分離法の非限定の例は、イオン交換(カチオン又はアニオン)、親水性相互作用、疎水性相互作用、又はゲル浸透クロマトグラフィーである。典型的な方法は、限外濾過膜を用いる接線流濾過である。
【0075】
いくつかの実施形態において、クロマトグラフィーは、ポリサッカライドの第2ステップの分離では使用しない。いくつかの実施形態において、クロマトグラフィーは、ポリサッカライドを精製するプロセスでは使用しない。
【0076】
いくつかの実施形態において、第2ステップの分離(例えば、GBS CPSに対して)は、20〜50kDa(例えば、30kDa)のMWCOを有する限外濾過膜により実施する。
【0077】
いくつかの実施形態において、ある程度大きなタンパク質(例えば、300kDaを超える)は、加水分解ステップによる分解に耐性を示す(例えば実施例2及び3参照)。これらのタンパク質を除去するために、第2の加水分解ステップからの混合物は、高いMWCO、例えば、200〜400kDa(例えば、300kDa)を有する限外濾過膜又は精密濾過膜に取り込む。その未透過液は、分解に耐性を示す大きなタンパク質を保持するので、その透過液を次に集める。その透過液は、濃縮し、別の膜、例えば、5〜100kDa、好ましくは5〜50kDa、最も好ましくは30kDaのMWCOを有する限外濾過膜に取り込む前にダイアフィルターにかけて、小さい分子量の不純物、例えば核酸及び分解したタンパク質を除去することができる。
【0078】
いくつかの実施形態において、第2ステップの分離(例えば、GYMP又はGWMPのための)は、塩基による加水分解の分解に耐性を示す大きなタンパク質を除去するために最初に300kDaのUF膜により行う。得たれた透過液を次に30kDaのUF膜に取り込んで小さい分子量の不純物を除去する。その30kDaUF膜濾過ステップからの未透過液を、後に続くステップのために用いる。
【0079】
(任意のN−アシル化)
ポリサッカライドを他の細胞成分から最終的に分離した後、加水分解手段によりポリサッカライドを脱アセチル化することができるので、生来のアセチル基が除去されている可能性のある遊離のアミノ基を再アシル化することができる。アシル化試薬及び反応条件を変化させることによって、その実行者は、アミノ基が再アシル化される度合いを制御することができる。そのアシル化試薬及びそれらの崩壊産物は、再アシル化されたポリサッカライドと比較して大きさが小さく、従って、サイズ排除法、例えば、ゲル浸透クロマトグラフィー又は膜濾過によってポリサッカライドから分離することができる。別法では、極性又は電荷の違いを、クロマトグラフィーに生かしてポリサッカライドを精製することができる。
【0080】
分離されたポリサッカライドは、様々なアシル化剤を用いることにより、任意で所望の程度まで再アシル化することができる。アシル化剤の非限定の例は、無水酢酸、塩化アセチル、酢酸ペンタフルオロフェニル、酢酸4−ニトロフェニル(非特許文献51)である。好ましいアシル化方法は、ポリサッカライドの遊離のアミノ基を再アシル化し、従って生来のポリサッカライド構造を再生するために、分離したポリサッカライドを、0.5M〜2M(例えば、0.7M〜1M)の濃度のアシル化剤と混合することに関連する。例えば、米国特許第5969130号、同第5576002号、同第4727136号、及び同第4356170号を参照されたい。
【0081】
(任意のN−アシル化後の精製)
いくつかの実施形態において、再アシル化したポリサッカライドは、クロマトグラフ精製又は膜濾過を用いて精製することができる。例えば、イオン交換(カチオン又はアニオン)、疎水性相互作用、親水性相互作用、ゲル浸透クロマトグラフィー、ダイレクトフロー濾過、又は接線流濾過は、すべて、再アシル化ポリサッカライドの反応成分からの分離を達成するために使用することができる。典型的な方法は、スーパーデックス(Superdex)(架橋したアガロース及びデキストラン)に基づくゲル浸透クロマトグラフィー又は残留混入物質を除去し、精製ポリサッカライドを提供する限外濾過膜による接線流濾過のいずれかを利用する。Superdexに基づくゲル浸透クロマトグラフィーを利用するいくつかの実施形態において、そのクロマトグラフィーは、溶離剤としてPBSを用いて、0.1mL/分〜10mL/分であり得る流速で、デキストランに対して1,000〜100,000分別範囲(MW)を有するSuperdex 200PGを使用することができる。接線流濾過を利用するいくつかの実施形態において、限外濾過膜は、例えば、5kDa〜100kDa(例えば、5kDa〜50kDa、又は5kDa〜30kDa)のMWCOを有するものを使用することができる。
【0082】
(純度)
精製したポリサッカライドの純度の程度は、修正マイクロスケールオルシノールアッセイ(非特許文献35)により評価された乾燥組成物中のシアル酸の重量百分率によって測定される。いくつかの実施形態において、精製ポリサッカライドの純度は、80%〜100%である。典型的な純度としては、85%〜100%、90%〜100%、94%〜100%、95%〜100%、及び99%以上が挙げられる。
【0083】
いくつかの実施形態において、純度は、質量百分率として表される260nmにおけるUV測光により検出される核酸の量によって特徴付けることができる。いくつかの実施形態において、精製したポリサッカライドの純度は、<5%の核酸、例えば、<4%、<3%、<2%、<1%、又は<0.5%の核酸であり得る。
【0084】
いくつかの実施形態において、純度は、Pierce社製(ロックフォード、イリノイ州)のCommie Plus試薬及び標準としてヒトIgGを用いるブラッドフォード法(非特許文献9)によって測定される精製ポリサッカライド中のタンパク質の質量百分率によって特徴付けることができる。いくつかの実施形態において、精製したポリサッカライドの純度は、<5%のタンパク質、例えば、<4%、<3%、<2%、<1%、<0.5%、<0.1%、又は<0.05%のタンパク質であり得る。
【0085】
いくつかの実施形態において、精製ポリサッカライドの純度は、<3%の核酸及び<1%のタンパク質であり得る。いくつかの実施形態において、精製ポリサッカライドの純度は、<1%の核酸及び<1%のタンパク質であり得る。いくつかの実施形態において、精製ポリサッカライドの純度は、<1%の核酸、<1%のタンパク質、及び90%〜100%のポリサッカライドであり得る。
【0086】
いくつかの実施形態において、精製ポリサッカライドの純度は、少なくとも、哺乳類(例えばヒト)を治療するワクチン、及びポリサッカライドとタンパク質との結合体を製造するタンパク質に連結するために使用するためには十分であり得る。
【0087】
(結合体分子)
いくつかの実施形態において、本発明のポリサッカライドは、グラム陰性及びグラム陽性細菌を含む様々な微生物に対する抗体応答を、個体中で、単独、又は別の免疫原性分子、例えばポリペプチド又は複数の担体タンパク質、単数の担体タンパク質に結合している場合のいずれかで引き出すために使用することができる。ポリサッカライドのポリペプチドへの結合は、一般的にはT細胞非依存性ポリサッカライドに対する免疫応答をT細胞依存性ポリサッカライドに転化する。従って、ポリペプチドの大きさは、T細胞非依存性からT細胞依存性への応答の転化を引き起こすのに十分なものが好ましい。第2の免疫原を提供するためには小さめのポリペプチドを使用することが役立つ。
【0088】
結合に役立つポリぺプチドの非限定の例としては、担体タンパク質、スカシガイヘモシアニン(KLH,keyhole limpet hemacyanin)、ウシ血清アルブミン(BSA,bovine serum albumin)、卵白アルブミン(OVA,ovalbumin)、ウサギ血清アルブミン(RSA,rabbit serum albumin)、破傷風トキソイド、ジフテリアトキソイド、ニューモリソイド(pneumolysoid)、外膜タンパク質、ウイルス糖タンパク質、並びにそれらの変異体、フラグメント、及び模倣体が挙げられる。破傷風毒素のCフラグメント(TTc,Fragment C of tetanus toxin)及び組換え型TTc(rTTc,recombinant TTc)は、ポリサッカライドのための担体として使用することができる。Cフラグメント(TTc)は、国際公開第2005/000346号によって報告されているように約865〜1315のアミノ酸を含む破傷風毒素のカルボキシル末端部分である。Cフラグメントの組換え発現は、米国特許第5443966号に報告されている。当業者であれば、ポリサッカライドに結合する免疫原性分子を選択することについて理解することができる。
【0089】
結合の任意の様式を採用して、ポリサッカライド成分をペプチドと結合することができる。好ましい方法は、米国特許第4356170号及び同第4902506号に記載されており、それらは、隣接ジオールの酸化的開裂を経てポリサッカライド中への末端アルデヒド基を導入し、そのアルデヒド基を還元的アミノ化によってペプチドのアミノ基に結合させることを記載している。
【0090】
しかしながら、本発明の結合分子は、還元的アミノ化を経て産生されるものに限定されないことを理解すべきである。従って、ワクチンは、当業者には既知の任意の連結方法、例えばSchneerson, R. et al.による(非特許文献55)及び米国特許第4644059号などに記載されているようなアジピン酸ジヒドラジドのスペーサー、又は、例えば、Marburg, S. et al.による(非特許文献56)に記載されているようなバイナリースペーサー技術を用いてポリサッカライドをペプチドと結合することによって産生することもできる。
【0091】
本発明に従って調製された精製ポリサッカライドは、ペプチドがポリサッカライド上の1つ又は複数の場所を介してポリサッカライドに連結している結合分子を産生するために使用することができる。従って、タンパク質成分に関して本発明に従って調製された結合分子は、ポリサッカライドが多様なタンパク質と一緒に架橋している、単量体、二量体、三量体、及びより高度に架橋した分子であり得る。
【0092】
(ワクチン類)
いくつかの実施形態において、ポリサッカライドワクチンを製造することができる。そのポリサッカライドは上記の方法によって精製することができる。これらのポリサッカライドは、ポリサッカライドに対して反応性であり従ってそのポリサッカライドが単離された有機体に対して反応性である抗体を発生する抗原として使用することができる。より具体的には、これらの実施形態は、例えば、ヒト又は動物を、一般的には当該ポリサッカライドが単離された、細菌、酵母菌、又は原虫の菌株による感染に対して保護することができる、単独での又はポリペプチドに結合したポリサッカライドワクチンの製品を提供する。ある場合には、本発明で使用されるポリサッカライドは、他の病原性微生物と交差反応性である抗体の産生を誘発することができ、それによってこれらの他の微生物による感染に対する保護を生み出す。ある場合には、本発明によるポリサッカライドの使用は、癌の治療又は予防に役立ち得る。
【0093】
さらに、本発明に従って精製したポリサッカライドにより調製されたワクチン類は、免疫原に対して宿主に免疫を予防的様式で与える免疫システムを活性化するのに役立つアジュバント類、例えば、ミョウバン、水酸化アルミニウム、ステアリルチロシン、及びフロイントアジュバントなども含むことができる(例えば、米国特許第5773007号参照)。いくつかの実施形態において、アジュバントは、CD14受容体アゴニスト、トール様受容体アゴニスト、QS21、MF59、モノホスホリル脂質A(MPL)から選択することができる。
【0094】
本発明のワクチン類は、能動免疫又は受動免疫を提供することができる。能動免疫を提供するためのワクチン類は、本発明の精製ポリサッカライドを含む。好ましくは、その精製ポリサッカライドは、莢膜ポリサッカライドである。
【0095】
いくつかの実施形態において、ポリサッカライド結合ワクチンを製造することができる。そのポリサッカライドは上記の方法によって精製することができる。その精製ポリサッカライドは、上の結合分子の項で記載したようにポリペプチドに結合することができる。
【0096】
本発明に従って精製したポリサッカライドから調製することができるワクチン類の非限定の例としては、A群連鎖球菌ワクチン(米国特許第5866135号参照)、B群連鎖球菌II型及びIII型ワクチン(米国特許第5302386号、同第6284884号参照)、及びB群髄膜炎菌ワクチン(米国特許第5969130号、同第5902586号、同第5969130号、同第5683699号、同第5576002号、同第5811102号を参照)が挙げられる。さらに、本発明に従って精製したポリサッカライドは、混合ワクチンに使用することもできる。
【0097】
(抗体類)
上に記載したポリサッカライドの加水分解及び分離の技術は、本発明の単離したポリサッカライドの大量生産を提供する。これは、ポリサッカライドに対して反応性の抗体の発生を促進する。本発明の1つの実施形態は、抗体の産生を引き出すことができる実質的に純粋な又は単離されたポリサッカライドを産生するための方法を提供する。使用するポリサッカライドに応じて、得られる抗体は、殺菌性、静菌性、殺真菌性、又は殺原虫性であり、抗体応答を引き出すために用いたポリサッカライドと交差反応性である抗体を含有する微生物による感染に対して動物を保護するものであり得る。
【0098】
単離されたポリサッカライド又はそこの免疫原性部分に対して特異的な抗体は、当技術分野では長期にわたって知られており、標準的に実施されている方法を用いて発生させることができる。かかる抗体としては、以下に限定はされないが、多クローン性の、単クローン性の、キメラの、単一鎖のFabフラグメント及びFab発現ライブラリーにより産生されたフラグメントを挙げることができる。中和抗体(即ち、二量体形成を阻害するもの)は、治療上の使用に対して特に好ましい。
【0099】
抗体の産生のために、ヤギ、ウサギ、ヒツジ、ネズミ、マウス、ウマ、雌ウシ、ヒトなどを含む様々な宿主を、免疫特性を有する単離したポリサッカライド、又はそれらの任意のフラグメント又は結合体を注射することによって免疫にすることができる。宿主の種類に応じて、様々なアジュバントを、免疫応答を増すために使用することができる。適当なアジュバントの非限定の例としては、フロイントアジュバント(完全又は不完全)、水酸化アルミニウム又はシリカ等のミネラルゲル類、及び界面活性物質、例えば、リゾレシチン、プルロニックポリオール類、多価アニオン類、ペプチド類、油エマルジョン類、KLH、及びジニトロフェノールなどが挙げられる。ヒトに一般的に使用されるアジュバントとしては、カルメット・ゲラン桿菌(BCG,bacilli Calmette Guerin)及びコリネバクテリウム・パルバム(Corynebacterium parvumn)が挙げられる。
【0100】
単離したポリサッカライドに対する単クローン抗体、又はそれの免疫原性フラグメントは、培養液中の連続継代細胞系による抗体分子の産生を提供する任意の技術を用いて調製することができる。これらには、以下に限定はされないが、ハイブリドーマ技術、位相表示(phase display)、ヒトB細胞ハイブリドーマ技術、及びEBV−ハイブリドーマ技術が含まれる(G. Kohler et al., 1975, Nature, 256:495-497;D. Kozbor et al., 1985, J. Immunol. Methods, 81:31-42;R.J. Cote et al., 1983, Proc. Natl. Acad. Sci. USA, 80:2026-2030;及び S.P. Cole et al., 1984, Mol. Cell Biol., 62:109-120)。この単クローン抗体の産生は、当技術分野では周知であり、ごく普通に使用されている。
【0101】
一方法によれば、ハイブリドーマ株化細胞の培養液が使用される(非特許文献52)。ポリサッカライドに対して向けた単クローン抗体類は、当技術分野では一般的に知られている技術により製造された、ヒト単クローン抗体、キメラ単クローン抗体、又はヒト化単クローン抗体であり得る。1つのアプローチによれば、キメラ単クローン抗体は、ヒトの定常部位と組み合わされたヒトではない(例えば、マウス)抗原結合領域を有するように発生させることができる(非特許文献53)。ヒト化抗体は、Queenらの米国特許第5585089号の手順に従って発生させることができる。単離されたポリサッカライド、ペプチド又はそれらのフラグメントは、単一のエピトープ又は抗原決定基或いは複数のエピトープを含むことができる。単離されたポリサッカライドの一部は、別のタンパク質のそれ、例えばKLHなどと融合することができ、抗体がキメラ分子に対して産生される。
【0102】
さらに、キメラ抗体の産生、適切な抗原特異性を備えた分子を得るためのヒト抗体遺伝子へのマウス抗体遺伝子のスプライシング及び生物活性のために開発された技術を使用することができる(S.L. Morrison et al., 1984, Proc. Natl. Acad. Sci. USA, 81:6851-6855;M.S. Neuberger et al., 1984, Nature, 312:604-608;及びS. Takeda et al., 1985, Nature, 314:452-454)。別法では、一本鎖抗体の産生について記載されている技術を、当技術分野で知られている方法を用いて、ポリサッカライド特異性の一本鎖抗体を産生するために適用することができる。関連するがはっきりとしたイディオタイプの組成物の特異性を備えた抗体を、無作為な免疫グロブリンの組合せライブラリーからのチェインシャフリング(chain shuffling)によって発生させることができる(D.R. Burton, 1991, Proc. Natl. Acad. Sci. USA, 88:11120-3)。抗体は、また、文献に開示されているように、リンパ球集団中のインビボ産生を引き起こすことによるか、非常に特異的な結合試薬の組換え型免疫グロブリンライブラリー又はパネルをスクリーニングすることによって産生することができる(R. Orlandi et al., 1989, Proc. Natl. Acad. Sci. USA, 86:3833-3837及びG. Winter et al., 1991, Nature, 349:293-299)。
【0103】
ポリサッカライドに対して向けられた抗体は、以下に限定はされないが、免疫吸収又は免疫親和性クロマトグラフィー或いはその他のクロマトグラフ法(例えば、HPLC)を含む当技術分野では周知の任意の技術によって精製することができる。抗体は、また、血清、血漿、又は細胞培養液からの免疫グロブリン画分として精製することもできる。
【0104】
単離されたポリサッカライドについての特定の結合サイトを含有する、Fab、Fab’、F(ab’)2、Facb、Fv、ScFv、Fd、VH、及びVLを含むがこれらには限定されない抗体フラグメントもまた発生させることができる。本発明の抗体分子は、インタクトな免疫グロブリン分子、実質的にインタクトな免疫グロブリン分子、又は抗原結合サイトを含有する免疫グロブリン分子のそのような部分、例えば、Fabフラグメントであり得る。ポリサッカライドに対して向けられた抗体のフラグメントは、当技術分野では一般的に知られている任意の技術(非特許文献54)によって発生させることができる。例えば、かかるフラグメントとしては、以下に限定されないが、抗体分子のペプシン消化によって産生することができるF(ab’)2フラグメント及びF(ab’)2フラグメントのジスルフィド架橋を還元することによって発生させることができるFabフラグメントが挙げられる。別法では、Fab発現ライブラリーは、所望の特異性を有する単クローンFabフラグメントの迅速で容易な同定を可能にするように構成することができる(W.D. Huse et al., 1989, Science, 254.1275-1281)。
【0105】
様々なイムノアッセイを、所望の特異性を有する抗体を同定するスクリーニングのために使用することができる。立証されている特異性を有する多クローン性又は単クローン性抗体を用いる競合結合アッセイ又は免疫放射線アッセイのための多数のプロトコールが当技術分野では周知である。かかるイムノアッセイは、単離されたポリサッカライドとその特定の抗体との間の錯体の形成を測定するステップを一般的には含む。2つの非干渉性ポリサッカライドエピトープと反応する単クローン性抗体を利用する2部位の単クローンに基づくイムノアッセイが好ましいが、競合結合アッセイを採用することもできる(Maddox、上記参照)。
【0106】
本発明の別の態様は、哺乳動物に、前記動物を、感染、例えば、細菌、真菌、原虫及びウイルス感染、特にHIV−1又はHIV−2によって引き起こされる感染から保護するための抗体及び/又はT細胞免疫応答を産生するのに適切な単離されたポリサッカライド又はそのフラグメントを接種することを含む哺乳動物における免疫応答を誘発するための方法に関する。本発明のさらに別の態様は、単離したポリサッカライドを、哺乳動物を疾患から保護する抗体を産生するような免疫応答を誘発するためにインビボでそのポリヌクレオチドの発現を方向付けるベクターを介して送達することを含む哺乳動物において免疫応答を誘発する方法に関する。
【0107】
(医薬品組成物)
本発明の医薬品組成物は、ポリサッカライド又はポリサッカライドと薬理学的に許容される担体、例えば、生理食塩水、ブドウ糖、グリセロール、エタノールなどを含む結合分子を含むことができる。別の実施形態において、前記医薬品組成物は、別の免疫原性部分、例えば、ペプチドなど、又は本発明のポリサッカライドの1つによって誘発される抗体を含む組成物を含む。前記医薬品組成物は、また、レシピエントの免疫応答を高めるアジュバントを含むこともできる。かかるアジュバントは、ミョウバン等のアルミニウム系又はステアリルチロシン等の長鎖アルキルアジュバントであり得る(1990年9月17日出願の米国特許第5773007号、欧州特許第0549617131号、Moloneyらの米国特許第4258029号参照)。Jenningsら(米国特許第5683699号)及びPaolettiらの(非特許文献58)も参照されたい。これらの医薬品組成物は、ワクチンとして特に有用である。
【0108】
受動免疫を誘発するために、医薬品組成物は、多クローン性抗体又は単クローン性抗体、或いはそれらの誘導体、或いはそれらのフラグメントを上記のように含むことができる。その抗体、フラグメント、又は誘導体の量は、標準的な臨床技術によって決定される治療的又は予防的に有効な量である。
【0109】
いくつかの実施形態において、本発明のポリサッカライドに対して向けられた抗体は、宿主の個体から別の個体に受動免疫を与えるための(即ち、グラム陰性又はグラム陽性細菌に対する個体の免疫応答を増大するため、又はエイズ患者を含む免疫無防備状態の又は免疫が枯渇した個体における応答を提供するための)治療的又は予防的適用における医薬品製剤として使用することができる。抗体の受動的伝達は、技術的に知られており、既知の任意の方法によって達成することができる。一方法によれば、本発明のポリサッカライド又はそれの結合体に対して向けられた抗体を、免疫適格性宿主(「ドナー」)動物において発生させ、その宿主動物から収集し、レシピエント個体に注入する。例えば、ヒトのドナーを、本発明のポリサッカライド又はポリサッカライド結合体に対して反応性の抗体を発生させるために使用することができる。その抗体を、次に、治療を必要としているヒトのレシピエントに治療的及び予防的に有効な量で投与し、それによってポリサッカライド成分によって誘発された抗体によって結合される細菌に対して、レシピエントにおける抵抗力を与えることができる(非特許文献57)。
【0110】
いくつかの実施形態において、本発明は、抗体の産生を誘発することができる実質的に純粋なポリサッカライドを産生するための方法を提供することができる。使用するポリサッカライドに応じて、得られる抗体は、殺菌性、殺真菌性、又は殺原虫性であり、抗体応答を引き出すために用いたポリサッカライドと交差反応性である抗体を含有する微生物による感染に対して動物を保護するものであり得る。
【0111】
本発明の医薬品製剤は、当技術分野で有効であることが知られている方法によって個体中に導入することができる。皮内、腹腔内、静脈内、皮下、経口、及び経鼻が導入のルートに入るがこれらだけではない。
【0112】
本発明の組成物は、以下に限定されないが、任意の適当な薬学的及び生理的に許容される担体、例えば、生理食塩水又はその他の注射可能な液体などを含むワクチンに適する当業者には一般的に知られている標準的な担体、緩衝剤、又は保存料を含むことができる。ワクチンには通例の添加剤、例えば、ラクトース又はソルビトール等の安定剤、及びリン酸アルミニウム、水酸化アルミニウム、又は硫酸アルミニウム及びステアリルチロシンなどの免疫原応答を高めるためのアジュバントも存在させることができる。本発明に従って産生されるワクチンは、また、複数の感染病原体に対する免疫応答を誘発する多価ワクチンの成分としても使用することができる。
【0113】
本発明のワクチンは、免疫原応答の一部としての抗体の産生を誘発するのに十分な量で投与する。投与量は、そのワクチンを受ける個体の大きさ、重量、又は年齢に基づいて調節することができる。個体における抗体応答は、抗体力価又は殺菌作用についてアッセイすることによって観測し、必要に応じて増量して応答を高めることができる。一般的に、乳幼児に対する1回の投与量は、1投与当り約10マイクログラム(μg)又は0.5μg〜20μg/kgの結合ワクチンである。成人は、0.5μg〜20μg/kgの結合ワクチンの投与量を受ける。CPSワクチンについては、一般的な投与量は、1投与量当り約25μgの各個体のCPSである。即ち、B群連鎖球菌に対するワクチンは、25μgの9個の血清型のそれぞれからのCPSのそれぞれを含む。各CPSの2μg〜15μgの範囲の投与量が、ポリサッカライド結合ワクチンについては一般に適切である。
【0114】
(診断キット)
別の実施形態において、本発明のポリサッカライド、それらの誘導体、又はフラグメントは、ニューモリシン毒素等の毒素は受け入れないが、グラム陰性又はグラム陽性細菌、或いはその他の微生物に対して向けられた抗体の存在を依然として示すことができるより安全な診断キットを産生するために使用することができる。かかる抗体の存在により、病原体にさらす前に示すことができ、感染に抵抗力があり得る個体を予測することができる。この診断キットは、少なくとも1つの本発明のポリサッカライド、それらの誘導体、又はフラグメント、及び変性したポリサッカライド、それらの誘導体、又はフラグメントが、グラム陰性又はグラム陽性細菌、又はその他の微生物に対して向けられた抗体を含有する試料と混合されるときの抗体反応の検知のための適当な試薬を含むことができる。抗体反応は、以下に限定されないが、ELISA、蛍光発光、比色分析、化学発光、又は電気化学発光アッセイを含む技術的に記載されている任意の方法によって同定することができる。かかる知見は重要であり、例えば、不必要なワクチン接種を避けることができる。
【0115】
別法では、該診断キットは、磁化可能な固体支持ビーズ、又はプラスチック母材、及び少なくとも1つの本発明のポリサッカライド、それらの誘導体又はフラグメントをさらに含むことができる。
【0116】
ある場合には、該ポリサッカライド、それらの誘導体又はフラグメントは標識化することが好ましいかもしれない。標識薬剤は技術的に周知である。例えば、標識薬剤としては、以下に限定されないが、放射能、化学発光、生体発光、発光、又は便利な分析のためのその他の同定及び/又は検出「タグ類」が挙げられる。体液又は組織の試料(例えば、血液、血清、唾液)を集めて精製し、診断キットに適用することができる。該ポリサッカライド、それらの誘導体又はフラグメントは、精製するか精製しなくてもよく、分子の混合物を構成することができる。
【0117】
固体マトリックスが技術的に知られており、利用可能であり、以下に限定されないが、例えば、試験管、ビーズ、マクロ粒子、ミクロ粒子、ディップスティック、プレート、チップなどの形状をした、ポリスチレン、ポリエチレン、ポリプロピレン、ポリカーボネート、又は任意の固体プラスチック材料であるポリマーが挙げられる。さらに、マトリックスとしては、以下に限定されないが、膜、96ウェルマイクロタイタープレート、試験管、及びエッペンドルフ管が挙げられる。一般に上記のマトリックス類は、リガンド結合剤が付着することができる任意の表面又はそれ自体がリガンド付着サイトを提供する表面を有する。
【0118】
[実施例]
以下の実施例は本発明を説明するために提供するが、本発明の範囲を限定するものとは決して解釈すべきでない。多くの変更及び置換えを、本発明の精神及び範囲から逸脱することなく行うことができることを、当業者であれば認識するであろう。
【0119】
(菌種、増殖培地、及び培養条件)
Ib型B群連鎖球菌菌株H36b(ATCC12401)を、アメリカン・タイプ・カルチャー・コレクション(American Type Culture Collection)(ロックビル、メリーランド州)から入手した。使用したその他の菌株、090(Ia型)、18RS21(II型)、M781(III型)、及びCJ11(V型)は、ハーバード・メディカル・スクール(Harvard Medical School)のD.L. Kasperにより、快く提供された。髄膜炎菌(Neisseria meningitidis)B、C、Y及びW135型は、FDA生物製剤評価センター(CBER, FDA)でCarl Fraschにより快く提供され、大腸菌(Escherichia coli)KIは、FDA生物製剤評価センターでWillie Vannにより快く提供された。
【0120】
その細菌菌株のそれぞれを、透析液(10,000MWCO)、6%のグルコースを補った3.5%のColumbia broth(Difco Laboratories, Inc.社製、デトロイト、ミシガン州)のPellicon cassette system(Millipore Corp.社製、ベッドフォード、マサチューセッツ州)中で個々に増殖した。振動三角フラスコ中、37℃で8時間にわたって増殖した150mLの種培養を用いて、14リットルのブロス(上記参照)を満たしたBioflo IV20リットル発酵層(New Brunswick Scientific Co.社製、エジソン、ニュージャージー州)に播種した。その発酵培養液を、37℃に維持し、10NのNaOHの添加によって持続的にpH7.1に調節し、1.5L/分で通気した。その細胞を、17時間後、MiniKros0.1μm孔の中空繊維カートリッジ(Microgon, Inc.社製、ラグナヒル、カリフォルニア州)を通す精密濾過法によって収集した。その培養上澄を、さらに処理が進むまで4℃で無菌状態で維持した。最終の細胞ペレットは、分離した細胞のSorvall GSAローター(DuPont Clinical & Instruments Div.製、ウィルミントン、デラウェア州)中、9000rpmで50分間の遠心分離によって得た。
【0121】
ポリサッカライドは、培養上澄から精製することもできる。細胞の除去後、ブロスを濃縮し、ポリサッカライドを産生するための一般的方法に記載されている手順に従ってダイアフィルターにかける。
【0122】
(モル質量測定)
ポリサッカライドの絶対モル質量分布は、インライン多角度レーザー光散乱光度計及び示差屈折による検出を備えた分析用ゲル浸透クロマトグラフィー(GPC−MALLS/RI)により測定した。この方法は、Jasco社製PU−980HPLCポンプ(イーストン、メリーランド州)、Rheodyne社製モデル7125噴射弁(Cotati、カリフォルニア州)、及びPBSと平衡しており、0.5mL/分の流速のSuperose 6HR 10/30カラムからなる液体クロマトグラフィーシステムに基づいて実施した。移動相は、超純水(Stephens Scientific社製、リバデール、ニュージャージー州)中で調製し、MilliporeタイプGVの0.22mm膜を装着した直径25mmのインラインフィルター(Millipore社製)を通して濾過した。ポリサッカライドの試料(1〜2mg)は、移動相中に10mg/mLの濃度で溶解し、得られた溶液を微小遠心分離機中14,000rpmで2〜3分間にわたり遠心分離して注入前に粒子状物質を除去した。カラム流出液は、Hewlett-Packard社製モデル1047A示差屈折計に連結しているインラインのDawn E eighteen角光散乱光度計(Wyatt Technology Corp.社製、サンタバーバラ、カリフォルニア州)により直接分析した。その屈折計のアナログ信号出力を補助入力チャネルを介してそのDOWN Eと接続した。光散乱データを獲得し、Wyatt's ASTRA 4.73.40ソフトウェアにより処理した。ピーク面積を、そのWyattソフトウェアにより、ピークの全範囲にわたって、200〜300の台形部分、又は「スライス」の面積の合計として計算した。従って得られたその面積から、所与のピークで溶離するポリサッカライドの重量平均及び数平均モル質量(それぞれ、Mw及びMn)を計算した。比屈折率増分(dn/dc)を0.141mL/gであるすべてのポリサッカライドについてオンラインHP 1047A屈折計を用いて測定した。この値は、他のポリサッカライドについて以前に得られた値(非特許文献7、8、40)と肩を並べた。
【0123】
(一次元1H NMRスペクトル)
D2O(Aldrich社製)中のポリサッカライド試料(4〜5mg/mL)の一次元1H NMRスペクトルを、Bruker Instruments社製AMX 500スペクトロメーター(ビルリカ、マサチューセッツ州)により500MHzで記録した。スペクトルデータは、50℃で取得し、化学シフトは、D2O中の外部の3−トリメチルシリルプロピオネートスルホン酸(Aldrich社製)を参照する。
【0124】
(精製ポリサッカライドの分析)
ステップの収量及び最終の精製試料の内容は、シアル酸定量分析に対する改良されたレゾルシノールアッセイ(非特許文献59)によって測定した。簡潔に記せば、5〜50μgのN−アセチルノイラミン酸(NeuAc)標準又は50μg/mLの莢膜ポリサッカライドを含有する1mLの試料又は対照に、1mLのレゾルシノール試薬(2%レゾルシノール溶液、0.25mLの0.1M CuSO4溶液、10mLの脱イオン水、濃HClで100mLの量にする)を加えた。試料をよく混合し、加熱ブロック(VWR)中110℃で25分間加熱した。試料を氷水中で5分間冷却した後、各試料に2mLの加水分解溶液(酢酸ブチルブタノール[85/15(v/v)])を加えた。各試料の有機相からの1mL部分を、石英のキュベットに移し、Shimadzu社製のUV160U、UV−可視記録分光光度計の580nmのところを読み取った。最終のポリサッカライド製剤の純度は、以下の式量:末端NeuAc残基に対しては309g/モル、GBSのIa、Ib型、又はIII型CPSの繰返し単位に対しては1004g/モル、GBSのII型又はV型CPSの繰り返し単位に対しては1328g/モルを用いてシアル酸含量から算出した。
【0125】
タンパク質含量は、PBS中に1ミリリットル当り20mgの莢膜ポリサッカライドを含有する試料についてPierce社(ロックフォード、イリノイ州)のCommie Plus試薬及び標準としてのヒトIgGを用いるBradford法(非特許文献9)により測定した。核酸含量は、260nmにおける直接的UV測光により測定した。これらのアッセイに対する光度測定は、Shimadzu社製のモデルUV 160U分光光度計(Shimadzu Scientific Inst.、コロンビア、メリーランド州)により行った。
【0126】
[実施例1]
(B群連鎖球菌(GBS)莢膜ポリサッカライドの加水分解及び単離)
図1は、この実施例で使用される本発明の実施形態の実験概略図を示す。
【0127】
(塩基を用いる第1ステップの加水分解)
750mLの1N NaOHを、前に述べた遠心分離ステップの直後の75gのGBS細胞ペーストに加え、細胞懸濁液を形成した。内容物を、2Lのビンに移し、100mgの水素化ホウ素ナトリウムをその細胞懸濁液に任意的に加えた。その反応混合物を37℃で16時間125rpmで震盪した。
【0128】
(膜濾過による分離)
一晩置いた細胞溶解物を、脱イオン(DI)水で10倍に希釈し、0.1Nの最終NaOH濃度とした。その希釈した細胞溶解物を、表面積が2000cm2の0.1gmのポリエーテルスルホン中空繊維フィルターモジュールを用いる精密濾過によって透明にした。その精密濾過モジュールをDI水を流して洗浄して残った貯蔵溶液を除去し、次いで、浸透ラインを閉じて希釈された細胞溶解物を10分間再循環させることにより平衡させた。その精密濾過は、一定の供給速度及び浸透速度で実施した。浸透液(出発容積の約90%)の収集に続いて、フィルターを2回1Lの生理食塩水で洗浄し、未透過液中のポリサッカライドを回収した。
【0129】
約13Lの精密濾過による細胞を含まない透過液を、Millipore(登録商標)Pellicon限外濾過装置(UF)の30kDa MWCOの膜により約1Lに濃縮した。その膜組成は、ポリエーテルスルホン(Biomax社製)である。一定容積を、最初に5Lの1M NaCl及び次に20LのDI水によりダイアフィルターにかけた。濃縮及びダイアフィルトレーションステップの後、その限外濾過未透過液を、30kDa MWCOのBiomax膜を装着した実験室規模の接線流濾過(TPF)装置(Millipore社製)により約70mLに濃縮した。
【0130】
(塩基を用いる第2ステップの加水分解)
30kDaの未透過液に5NのNaOHを加え、2N NaOHの最終濃度を達成した。ポリサッカライドを80℃で16〜18時間さらに抽出した後、その混合物を50℃未満まで冷却し、10LのDI水中に注いだ。
【0131】
(膜濾過による第2ステップの分離)
水酸化ナトリウム並びに小さい分子量の不純物を、30kDa MWCOのポリエーテルスルホン(Biomax社製)膜による一定容積のダイアフィルトレーションにより、5Lの1M NaCl及び20LのDI水によって除去し、次に、30kDa MWCOのBiomax膜を装着した実験室規模のTFF装置により約70mLの最小容積まで濃縮した。
【0132】
(任意的な再N−アシル化)
先に記した加水分解条件にポリサッカライドをさらすことにより、ポリサッカライドからN−アセチル基が放出されるために、無水酢酸(Aldrich Chemical Co.社製、ミルウォーキー、ウィスコンシン州)を、そのプールした部分に0.8Mの最終濃度まで滴下して加えることにより、そのポリサッカライドを再N−アセチル化した。この反応混合物を室温で1時間撹拌し、5NのNaOHを添加してpH9を維持した。その反応物のpHを次に13まで上げ、反応をさらに90分間続けた。その反応物のpHを、次に6Nの塩酸によりpH8に調整した。
【0133】
(膜濾過による精製)
再N−アセチル化した莢膜ポリサッカライドを含有するその溶液を、30kDa MWCOのBiomax限外濾過膜を装着した実験室規模のTFF装置により、5Lの0.9%NaClに対してほぼ70mLの最小容積までダイアフィルターにかけた。その精製した莢膜ポリサッカライドを、次に−70℃で冷凍保存するか、又は、下流部門の用途、例えば、抗体、医薬品組成物、診断キット、結合分子、及びワクチンの産生のために利用した。
【0134】
(結果)
1.精製莢膜ポリサッカライドの収量
表1は、実施例1の方法又はMichonらの米国特許第6248570号に記載の「1ステップの加水分解法」のいずれかを用いて得られた様々なBGS血清型の莢膜ポリサッカライド収量の比較を示す。すべての血清型について、莢膜ポリサッカライドは細胞ペレットから精製した。1ステップ法と比較して、本発明の方法は、試験した5個の血清型の4個の血清型に対してより大きい収量を生じた。
【0135】
2.精製莢膜ポリサッカライドの分析
検討したB群連鎖球菌血清型のそれぞれについて、260nmにおける直接のUV測光によって検出した核酸濃度は、0.5質量%を超えなかったが、Bradford法(非特許文献9)によってアッセイしたタンパク質は、<1質量%であった(表2)。すべてのポリサッカライドの純度は、改良レゾルシノールアッセイ(非特許文献36)により推定したそれらのシアル酸含量から計算して、約100%であった。本発明の方法及び1ステップの加水分解法の両方によって得られたすべてのポリサッカライド製剤に対して、測光データは、両方の方法ともタンパク質又は核酸による汚染が最小限の高純度の莢膜ポリサッカライドを産生することを示す。
【0136】
3.ポリサッカライドの分子の大きさ
別々の分析において、ポリサッカライドの絶対モル質量分布を、GPC−MALLS/RIにより測定した。この方法は、流速及び保持容量のようなクロマトグラフのパラメーターとは関係なく、且つ流体力学的性質が興味のある検体から大きく異なり得る二次標準物質を必要とすることなく、巨大分子のモル質量を直接推測することを可能にする。キャラクタリゼーション法としてのGPC−MALLS/RIの有用性は、医薬品として興味のあるポリサッカライドに対して十分に確立されている(非特許文献7、8、10、17、25)。モル質量分布は、重量平均モル質量(Mw)として通常は示される(表2)。
【0137】
【表1】
【0138】
【表2】
【0139】
[実施例2]
(Y群髄膜炎菌莢膜ポリサッカライドの加水分解及び単離)
図2は、この実施例で使用される本発明の実施形態の実験概略図を示す。
【0140】
(塩基を用いる第1ステップの加水分解)
750mLの1N NaOHを、細胞をペレット化した遠心分離ステップの直後の75gのGYM細胞ペーストに加えた。内容物を、2Lのビンに移し、100mgの水素化ホウ素ナトリウムをその細胞懸濁液に加えた。その反応混合物を37℃で16時間〜18時間125rpmで震盪した。
【0141】
(膜濾過による分離)
一晩置いた細胞溶解物を、脱イオン(DI)水で10倍に希釈し、約0.1Nの最終NaOH濃度とした。その希釈した細胞溶解物を、表面積が2000cm2の0.1μmのポリエーテルスルホン中空繊維フィルターモジュールを用いる精密濾過によって透明にした。その精密濾過モジュールをDI水を流して洗浄して残った貯蔵溶液を除去し、次いで、浸透ラインを閉じて希釈された細胞溶解物を10分間再循環させることにより平衡させた。その精密濾過は、一定の供給速度及び浸透速度で実施した。浸透液(出発容積の約90%)の収集に続いて、フィルターを2回1Lの生理食塩水で洗浄し、未透過液中のポリサッカライドを回収した。
【0142】
約7.5Lの精密濾過による細胞を含まない透過液を、Millipore Pellicon限外濾過装置(UF)の100kDa MWCOの膜により約1Lに濃縮した。その膜組成は、ポリエーテルスルホン(Biomax社製)である。一定容積を、最初に5Lの1M NaCl及び次に20LのDI水によりダイアフィルターにかけた。濃縮及びダイアフィルトレーションステップの後、その限外濾過未透過液を、100kDa MWCOのBiomax膜を装着した実験室規模の接線流濾過(TFF)装置(Millipore社製)により約70mLに濃縮した。
【0143】
(塩基を用いる第2ステップの加水分解)
100kDaの未透過液に5NのNaOHを加え、2N NaOHの最終濃度を達成した。ポリサッカライドを80℃で48時間さらに抽出した後、その混合物を50℃未満まで冷却し、10LのDI水中に注いだ。
【0144】
(膜濾過による第2ステップの分離)
GYMPの精製については、いくらかの大きいタンパク質(300kDaを超える大きさ)が、加水分解ステップによる分解に対して耐性を示すことが分かった。これらのタンパク質を除去するために、第2の加水分解ステップからの混合物を、Millipore Pellicon限外濾過装置(UF)の300kDa MWCOの膜にかけた。その透過液を、次にMillipore Pellicon限外濾過装置(UF)の5kDa MWCOの膜により約1Lに濃縮した。その膜組成は、ポリエーテルスルホン(Biomax社製)である。一定容積を、最初に5Lの1M NaCl及び次に20LのDI水によりダイアフィルターにかけた。濃縮及びダイアフィルトレーションステップの後、その限外濾過未透過液を、5kDa MWCOのBiomax膜を装着した実験室規模の接線流濾過(TFF)装置(Millipore社製)により約70mLに濃縮した。
【0145】
(任意的な再N−アシル化)
先に記した加水分解条件にポリサッカライドをさらすことにより、ポリサッカライドからN−アセチル基が放出されるために、無水酢酸(Aldrich Chemical Co.社製、ミルウォーキー、ウィスコンシン州)を、そのプールした部分に0.8Mの最終濃度まで滴下して加えることにより、そのポリサッカライドを再N−アセチル化した。この反応混合物を室温で1時間撹拌し、5NのNaOHを添加してpH9を維持した。その反応物のpHを次に13まで上げ、反応をさらに90分間続けた。その反応物のpHを、次に6Nの塩酸によりpH8に調整した。
【0146】
(膜濾過による精製)
再N−アセチル化した莢膜ポリサッカライドを含有するその溶液を、5kDa MWCOのBiomax限外濾過膜を装着した実験室規模のTFF装置により、5Lの0.9%NaClに対してほぼ70mLの最小容積までダイアフィルターにかけた。その精製した莢膜ポリサッカライドを、次に−70℃で冷凍保存したが、或いは、下流部門の用途、例えば、抗体、医薬品組成物、診断キット、結合分子、及びワクチンの産生のために利用することもできる。
【0147】
(結果)
1.Y群髄膜炎菌莢膜ポリサッカライドの収量
上記の方法を用いる精製Y群髄膜炎菌莢膜ポリサッカライドのおおよその収量は、1リットルの細菌培養液当り12mgであった。
【0148】
2.精製GYM莢膜ポリサッカライドの分析
精製GYM莢膜ポリサッカライドの一次元1H NMR分光分析は、非常に低い水準の汚染を示した。図4は、D2O中50℃で記録された精製GYM莢膜ポリサッカライドのNMRスペクトル(500MHz)を示す。直接の260nmでのUV測光により検出された核酸のレベルは、2.46質量%であり、一方Bradford法(非特許文献9)によりアッセイしたタンパク質は、このアッセイの検出の下限(1μg/mL)の上の検出できるものではなかった。最終精製物の純度は、改良型マイクロスケールオルシノールアッセイ(非特許文献35)により推測されるシアル酸含量により計算し、94%であった。従って、スペクトル及び測光データは、本発明の加水分解及び単離の方法が、タンパク質又は核酸による汚染が最低限である高純度のGYM莢膜ポリサッカライドを可能にすることを示す。
【0149】
[実施例3]
(W−135群髄膜炎菌莢膜ポリサッカライドの加水分解及び単離)
図3は、この実施例で使用される本発明の実施形態の実験概略図を示す。
【0150】
(塩基を用いる第1ステップの加水分解)
750mLの1N NaOHを、細胞をペレット化した遠心分離ステップの直後の75gのW−135群髄膜炎菌(GWM)細胞ペーストに加えた。内容物を、2本の2Lのビンに移し、100mgの水素化ホウ素ナトリウムをその細胞懸濁液に加えた。その反応混合物を37℃で16時間〜18時間150rpmで震盪した。
【0151】
(膜濾過による分離)
一晩置いた細胞溶解物を、脱イオン(DI)水で10倍に希釈し、約0.1Nの最終NaOH濃度とした。その希釈した細胞溶解物を、表面積が2000cm2の0.2μmのポリエーテルスルホン中空繊維フィルターモジュールを用いる精密濾過によって透明にした。その精密濾過モジュールをDI水を流して洗浄して残った貯蔵溶液を除去し、次いで、浸透ラインを閉じて希釈された細胞溶解物を10分間再循環させることにより平衡させた。その精密濾過は、一定の供給速度及び浸透速度で実施した。浸透液(出発容積の約90%)の収集に続いて、フィルターを2回1Lの生理食塩水で洗浄し、未透過液中のポリサッカライドを回収した。
【0152】
約12Lの精密濾過による細胞を含まない透過液を、Millipore Pellicon限外濾過装置(UF)の30kDa MWCOの膜により約1Lに濃縮した。その膜組成は、ポリエーテルスルホン(Biomax社製)である。一定容積を、最初に5Lの1M NaCl及び次に20LのDI水によりダイアフィルターにかけた。濃縮及びダイアフィルトレーションステップの後、その限外濾過未透過液を、100kDa MWCOのBiomax膜を装着した実験室規模の接線流濾過(TFF)装置(Millipore社製)により約70mLに濃縮した。
【0153】
(加水分解を用いる第2ステップの加水分解)
100kDaの未透過液に5NのNaOHを加え、2N NaOHの最終濃度を達成した。ポリサッカライドを80℃で一晩さらに抽出した後、その混合物を50℃未満まで冷却し、10LのDI水中に注いだ。
【0154】
(膜濾過による第2ステップの分離)
W−135群髄膜炎菌ポリサッカライド(GWMP)の精製については、いくらかの大きいタンパク質(300kDaを超える大きさ)が、加水分解ステップによる分解に対して耐性を示した。これらのタンパク質を除去するために、第2の加水分解ステップからの混合物を、Millipore Pellicon限外濾過装置(UF)の300kDa MWCOの膜にかけた。その透過液を、次にMillipore Pellicon限外濾過装置(UF)の30kDa MWCOのポリエーテルスルホン(Biomax社製)膜により約1Lに濃縮した。一定容積を、最初に5Lの1M NaCl及び次に20LのDI水によりダイアフィルターにかけた。濃縮及びダイアフィルトレーションステップの後、その限外濾過未透過液を、30kDa MWCOのBiomax膜を装着した実験室規模の接線流濾過(TFF)装置(Millipore社製)により約70mLに濃縮した。
【0155】
(任意的な再N−アシル化)
先に記した加水分解条件にポリサッカライドをさらすことにより、ポリサッカライドからN−アセチル基が放出されるために、無水酢酸(Aldrich Chemical Co.社製、ミルウォーキー、ウィスコンシン州)を、そのプールした部分に0.8Mの最終濃度まで滴下して加えることにより、そのポリサッカライドを再N−アセチル化した。この反応混合物を室温で1時間撹拌し、5NのNaOHを添加してpH9を維持した。その反応物のpHを次に13まで上げ、反応をさらに90分間続けた。その反応物のpHを、次に6Nの塩酸によりpH8に調整した。
【0156】
(膜濾過による精製)
再N−アセチル化した莢膜ポリサッカライドを含有するその溶液を、30kDa MWCOのBiomax限外濾過膜を装着した実験室規模のTFF装置により、5Lの0.9%NaClに対してほぼ70mLの最小容積までダイアフィルターにかけた。その精製した莢膜ポリサッカライドを、次に−70℃で冷凍保存したが、或いは、下流部門の用途、例えば、抗体、医薬品組成物、診断キット、結合分子、及びワクチンの産生のために利用することもできる。
【0157】
(結果)
1.W−135群髄膜炎菌莢膜ポリサッカライドの収量
上記の方法を用いる精製W−134群髄膜炎菌莢膜ポリサッカライドのおおよその収量は、1リットルの細菌培養液当り25.4mgであった。
【0158】
2.精製GWM莢膜ポリサッカライドの分析
直接の260nmでのUV測光により検出された核酸のレベルは、0.40質量%であり、一方Bradford法(非特許文献9)によりアッセイしたタンパク質は、このアッセイの検出の下限(1μg/mL)の上の検出できるものではなかった。最終精製物の純度は、改良型マイクロスケールオルシノールアッセイ(非特許文献35)により推測されるシアル酸含量により計算し、95%であった。従って、上記測光データは、本発明の加水分解及び単離の方法が、タンパク質又は核酸による汚染が最低限である高純度のGWM莢膜ポリサッカライドを可能にすることを示す。
【0159】
本明細書は、本明細書内に引用した参考文献の教示を踏まえて最も完全に理解される。本明細書中の実施形態は、本発明の実施形態の説明を提供するものであって、本発明を限定するものと解釈すべきではない。熟練技術者であれば、多くの他の実施形態が本発明によって包含されることを容易に認識する。本開示中に引用したすべての出版物及び特許は、参照によりそれら全体として組み込む。参照により組み込まれた材料が本明細書と矛盾するか一貫性がない限りにおいて、本明細書は、そのような材料のいずれにも優先する。本明細書におけるどの参考文献の引用も、かかる参考文献が本発明に対する先行技術であることを認めるものではない。
【0160】
別に示されていない限り、特許請求の範囲を含めて、本明細書で使用されている、成分の量、反応条件などを表すすべての数字は、すべての場合において、用語「約」によって修飾されるものと理解すべきである。従って、そうでないことが特に示されていない限り、数値パラメーターは、近似値であり、本発明によって得られることが求められる所望の特性によって変動し得る。少なくとも、そして特許請求の範囲に対する均等の原則の適用を限定しようとする試みとしてではなく、各数値パラメーターは、有効数字の数及び普通の丸め手法に照らして解釈すべきである。
【0161】
別に示されていない限り、範囲は境界点を含む。例えば、「5〜14」は、5以上から14以下の任意の値を指す。
【0162】
別に示されていない限り、一連の要素に先立つ用語「少なくとも」とは、その一連のあらゆる要素を指すものと理解すべきである。当業者であれば、ありふれたものにすぎない実験手段を用いて、本明細書に記載の本発明の特定の実施形態に対する多数の相当物を認識し、究明することができよう。かかる相当物は、添付の特許請求の範囲に含まれることが意図されている。
【0163】
(参考文献)
1. Anderson, P., G. Peter, R.B. Johnson, L.H. Wetterlow and D.H. Smith. 1972. Immunization of humans with polyribophosphate, the capsular antigen of Haemophilus influenzae type b. J.Clin.Invest. 51:39-44.
2. Avery, O.T. and W.F. Goebel. 1931. Chemo-immunological studies on conjugated carbohydrate-proteins V. The immunological specificity of an antigen prepared by combining the capsular polysaccharide of type 3 pneumococcus with foreign protein. J.Exp.Med. 54:43 7-447.
3. Baker, C.J. and D.L. Kasper. 1985. Group B streptococcal vaccines. Rev.Inf.Dis. 7:458-467.
4. Baker, C.J., M.A. Rench, M.S. Edwards, R.J. Carpenter, B.M. Hays and D.L. Kasper. 1988. Immunization of pregnant women with a polysaccharide vaccine of group B Streptococcus. N.Eng1.J.Med. 319:1180-1185.
5. Baker, C.J., M.A. Rench and D.L. Kasper. 1990. Response to Type III polysaccharide in women whose infants have had invasive Group B streptococcal infection. New EngI.J.Med. 322:1857-1860.
6. Baltimore, R.S., D.L. Kasper and J. Vecchitto. 1979. Mouse protection test for group B Streptococcus type III. J.Infect.Dis. 140:81-86.
7. Bednar, B. and J.P. Hennessey. 1993. Molecular size analysis of capsular polysaccharide preparations from Streptococcus pneumoniae. Carbohyd.Res. 243:115-130.
8. Beri, R.G., J. Walker, E.T. Reese and J.E. Rollings. 1993. Characterization of chitosans via coupled size-exclusion chromatography and multiple-angle laser light-scattering technique. Carbohyd.Res. 238:11-26.
9. Bradford, M.M.. 1.976. A Rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analyt.Biochem. 72:248-254.
10. D'Ambra, A.J., J.E. Baugher, P.E. Concannon, R.A. Pon and F. Michon. 1997. Direct and indirect methods for molar-mass analysis of fragments of the capsular polysaccharide ofHaemophilus influenzae type b. Anal.Biochem. 250:228-236
11. Dick, W.E., Jr. and M. Beurret. 1989. Glycoconjugates of bacterial carbohydrate antigens. p. 48-114. In: J.M. Cruse and R.E. Lewis,Jr., Contributions to microbiology and immunology. S.Karger, Basel.
12. Dillon, H.C., Jr., S. Khare and B.M. Gray. 1987. Group B streptococcal carriage and disease: A 6-year prospective study. J.Pediat. 110:31-36.
13. Goebel, W.F. and O.T. Avery. 1931. Chemo-immunological studies on conjugated carbohydrate-proteins IV. The synthesis of the p-aminobenzyl ether of the soluble specific substance of type 3 pneumococcus and its coupling with protein. J.Exp.Med. 54:431.436.
14. Gold, R., M.L. Lepow, I.Goldschneider, T.L. Draper and E.C. Gotschlich. 1975. Clinical evaluation of group A and group C meningococcal polysaccharide vaccines in infants. J.Clin.Invest. 56:1536-1547.
15. Gold, R., M.L. Lepow, I.Goldschneider and E.C. Gotschlich. 1977. Immune response of human infants to polysaccharide vaccines of Groups A and C Neisseria meningitidis. J.Infect.Dis. 136S:S31-S35.
16. Gold, R.M., M.L. Lepow, I. Goldschneider, T.F. Draper and E.C. Gotschlich. 1978. Antibody responses of human infants to three doses of group A Neisseria meningitides vaccine administered at two, four and six months of age. J.Infect.Dis. 138:731-735.
17. Hennessey, J.P., B. Bednar and V. Manam. 1993. Molecular size analysis of Haemophilus influenzae type b capsular polysaccharide. J.Liq.Chromat. 16:1715-1729.
18. Howard, J.G., G.H. Christie, B.M. Courtenay, E. Leuchars and A.J.S. Davies. 1971. Studies on immunological paralysis. VI. Thymic-independence of tolerance and immunity to type III pneumococcal polysaccharide. Cell.Immunol. 2:614-626.
19. Jennings, H.J., E. Katzenellenbogen, C. Lugowski and D.L. Kasper. 1983. Structure of the native polysaccharide antigens of type la and type Ib Group B Streptococcus. Biochemistry 22:1258-1263.
20. Jennings, H.J., K.-.G. Rosell and D.L. Kasper. 1980. Structural determination and serology of the native polysaccharide antigen of type III group B Streptococcus. Can.J.Biochem. 58:112-120.
21. Jennings, H.J., K.-.G. Rosell and D.L. Kasper. 1980. Structure and serology of the native polysaccharide antigen of type Ia group B Streptococcus. Proc.Nat.Acad.Sci.USA. 77:2931-2935.
22. Jennings, H.J., K.-.G. Rosell, E. Katzenellenbogen and D.L. Kasper. 1983. Structural determination of the capsular polysaccharide antigen of type II Group B Streptococcus. J.Biol.Chem. 258:1793-1798.
23. Jennings, H.J. and R.K. Sood. 1994. Synthetic glycoconjugates as human vaccines. p. 325-371. In: Y.C. Lee and R.T. Lee, Neoglycoconjugates: Preparation and applications. Academic Press, New York.
24. Kang, D., Liu, G., Lundstrom, A. et al. 1998 A peptidoglycan recognition protein in innate immunity conserved from insects to humans. Proc. Natl. Acad. Sci. USA. 95:10078-10082.
25. Kasper, D.L., C.J. Baker, R.S. Baltimore, J.H. Crabb, G. Schiffman and H.J. Jennings. 1979. Immunodeterminant specificity of human immunity to type III group B Streptococcus. J.Exp.Med. 149:327-339.
26. Knobloch, J.E. and P.N. Shaklee. 1997. Absolute molecular weight of low-molecular-weight heparins by size-exclusion chromatography with multiangle laser light scattering detection. Anal.Biochem. 245:231-241.
27. Lancefield, R.C.. 1933. A serological differentiation of human and other groups of haemolytic streptococci. J.Exp.Med. 57:571-595.
28. Lancefield, R.C.. 1938. A micro-precipitin technique for classifying hemolytic streptococci and improved methods for producing antigen. Proc.Soc.Exp.Biol.and Med. 38:473-478.
29. Lancefield, R.C., M. McCarty and W.N. Everly. 1975. Multiple mouse-protective antibodies directed against group B streptococci: Special reference to antibodies effective against protein antigens. J.Exp.Med. 142:165-179.
30. Madoff, L.C., L.C. Paoletti, J.Y. Tai and D.L. Kasper. 1994. Maternal immunization of mice with Group B streptococcal type III polysaccharide-beta C protein conjugate elicits protective antibody to multiple serotypes. J.Clin.Invest. 94:286-292.
31. Marques, M.B., D.L. Kasper, A. Shroff, F. Michon, H.J. Jennings and M.R. Wessels. 1994. Functional activity of antibodies to the group B polysaccharide of group B streptococci elicited by a polysaccharide-protein conjugate vaccine. Infect.Immun. 62:1593-1599.
32. Makela, P.R.H., H. Peltola, H. Kayhty, et al. 1977. Polysaccharide vaccines of group A Neisseria meningitidis and Haemophilus influenzae type b: A field trial in Finland. J.Infect.Dis. 136:543-50.
33. Michon, F., J.R. Brisson, A. Dell, D.L. Kasper and H.J. Jennings. 1988. Multiantennary group-specific polysaccharide of group B Streptococcus. Biochem. 27:5341-5351.
34. Peltola, A., H. Kayhty, A. Sivonen and P.R.H. Makela. 1977. Haemophilus influenzae type b capsular polysaccharide vaccine in children: A double blind field study of 100,000 vaccines 3 months to 5 years of age in Finland. Pediatrics 60:730-737.
35. Peltola, H., P.R.H. Makela, H. Jousimies, et al. 1977. Clinical efficacy of meningococcal group A vaccine in children three months to five years of age. N.Engl.J.Med. 297:686-691.
36. Reuter, G. and R. Schauer. 1994. Determination of sialic acids. p. 168-199. In: W.J. Lennarz and G.W. Hart, Methods in Enzymology Vol. 230 Techniques in Glycobiology. Academic Press, New York.
37. Robbins, J.B. and R. Schneerson. 1990. Polysaccharide-protein conjugates: A new generation of vaccines. J.Infect.Dis. 161:821-832.
38. Scwander, R., Dziarski R., Wesche, H. et al. 1999. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J. Biol. Chem. 274:17406-17409.
39. Smith, A.L. and J. Haas. 1991. Neonatal Bacterial Meningitis. p. 313-333. In: W.M. Scheld, R.J. Whitley and D.T. Durack, Infections of the Central Nervous System. Raven Press, Ltd., New York,
40. Tsunashima, T., K: Moro, B. Chu and T.-Y. Liu. 1978. Characterization of group C meningococcal polysaccharide by light-scattering spectroscopy. III. Determination of molecular weight, radius of gyration, and translational diffusional coefficient.. Biopolymers 17:251-265.
41. von Hunolstein, C., L. Nicolini, S. D'Ascenzi, C. Volpe, G. Alfarone and G. Orefici. 1993. Sialic acid and biomass production by Streptococcus agalactiae under different growth conditions. Appl.Microbiol.Biotechnol. 38:458-462.
42. Wessels, M.R., W.J. Benedi, H.J. Jennings, F. Michon, J.L. DiFabio and D.L. Kasper. 1989. Isolation and characterization of type IV group B Streptococcus capsular polysaccharide. Infect.Immun. 57:1089-1094.
43. Wessels, M.R., J.L. DiFabio, V.J. Benedi, et al. 1991. Structural determination and immunochemical characterization of the type V group B Streptococcus capsular polysaccharide. J.Biol.Chem. 266:6714-6719.
44. Wessels, M.R., L.C. Paoletti, D.L. Kasper, et al. 1990. Immunogenicity in animals of a polysaccharide-protein conjugate vaccine against type III group B Streptococcus. J.Clin.Invest. 86:1428.1433.
45. Wessels, M.R., L.C. Paoletti, A.K. Rodewald, et al. 1993. Stimulation of protective antibodies against type la and Ib group B streptococci by a type Ia polysaccharide-tetanus toxoid conjugate vaccine. Infect.Immun. 61:4760-4766.
46. Wessels, M.R., V. Pozsgay, D.L. Kasper and H.J. Jennings. 1987. Structure and immunochemistry of an oligopolysaccharide repeating unit of the capsule polysaccharide of Type III Group B Streptococcus: A revised structure for the Type III Group B streptococcal polysaccharide antigen. J.Biol.Chem. 262:8262-8267.
47. Wyle, S.A., M.S. Artenstein, B.L. Brandt, et al. 1972. Immunologic response of man to group B meningococcal polysaccharide vaccines. J.Infect.Dis. 126:514-522.
48. Yang, R.B., Mark, M.R., Gray, A., et al. 1998. Toll-like receptor 2 mediates lipopolysaccharide-induced cellular signaling. Nature. 395:284-288.
49. Tipson, R.S. and Horton, D., Advances in Carbohydrate Chemistry and Biochemistry, Vol. 41, 1983, Academic Press, NY.
50. Westphal et al., Methods in Carbohydrate Chemistry, vol. V, 1965, Academic Press, NY.
51. Theodora W. Greene and Peter G. M. Wuts, Protective Groups in Organic Syntheses, 2nd Ed. (1991).
52. Kohler and Milstein (1975) Nature 256:495-497.
53. Takeda et al. (1985) Nature 314:452.
54. Campbell (1985) Laboratory Techniques in Biochemistry and Molecular Biology, Vol. 13, Burdon, et al. (eds.), Elsevier Science Publishers, Amsterdam.
55. Schneerson, R., et al. (1980) J. Exp. Med. 1952:361-476.
56. Marburg, S., et al. (1986) J. Am. Chem. Soc. 108:5282-5287.
57. Grossman, M. and Cohen, S. N., in "Basic and Clinical Immunology", 7th Ed., (Stites, D. P. and Terr, A. T. eds., Appleton & Lange 1991) Chapter 58 "Immunization".
58. Paoletti, et al. (1997) J. Infectious Diseases, 175:1237-9.
59. L. Warren, (1959) 1 Biol. Chem. 234, 1971.
【図面の簡単な説明】
【0164】
【図1】B群連鎖球菌(GBS)莢膜ポリサッカライドを単離するために使用する本発明の実験概略図(実施例1)を示す図である。
【図2】Y群髄膜炎菌(GYM)莢膜ポリサッカライドを単離するために使用する本発明の実験概略図(実施例2)を示す図である。
【図3】W−135群髄膜炎菌(GWM)莢膜ポリサッカライドを単離するために使用する本発明の実験概略図(実施例3)を示す図である。
【図4】Y群髄膜炎菌莢膜ポリサッカライドからの莢膜ポリサッカライドのNMRスペクトル(実施例2)を示す図である。
【特許請求の範囲】
【請求項1】
ポリサッカライド及び細胞成分を含むストックからの前記ポリサッカライドを精製する方法であって、
前記ストックを第1の試薬と接触させて第1の混合物を形成するステップ(前記第1の試薬は、酸又は塩基であり、前記第1の混合物のpHは、0〜6の範囲又は9〜15の範囲である)と、
前記ポリサッカライドを前記細胞成分の少なくとも一部から分離して前記ポリサッカライドと前記細胞成分の残量とを含む分離された組成物を形成するステップと、
前記分離された組成物を第2の試薬と接触させて第2の混合物を形成するステップ(前記第2の試薬は塩基であり、前記第2の混合物のpHは、9〜15の範囲である)と、
前記細胞成分の前記残量の少なくとも一部からのポリサッカライドを精製して、精製された組成物を形成するステップと
を含む方法。
【請求項2】
精製ステップの前にさらなる接触ステップ及び分離ステップが実施される、請求項1に記載の方法。
【請求項3】
ポリサッカライドが、N−アセチル基を含有し、これらのN−アセチル基の少なくとも一部が第2の試薬による処理によって加水分解される、請求項1又は2に記載の方法。
【請求項4】
加水分解されたポリサッカライド上に存在する一定割合のN−アセチル基が、未変性ポリサッカライドの免疫特性を維持するためにそのとき十分に再アシル化される、請求項3に記載の方法。
【請求項5】
精製したポリサッカライドをアシル化試薬と接触させるステップと、前記アシル化ポリサッカライドを精製するステップとをさらに含む、請求項3又は4に記載の方法。
【請求項6】
アシル化試薬が、無水酢酸、塩化アセチル、ペンタフルオロフェニル酢酸又は4−ニトロフェニル酢酸である、請求項5に記載の方法。
【請求項7】
第1の試薬が酸である、請求項1〜6のいずれかに記載の方法。
【請求項8】
第1の試薬が塩基である、請求項1〜6のいずれかに記載の方法。
【請求項9】
第1の試薬及び第2の試薬の少なくとも1つが有機塩基を含む、請求項8に記載の方法。
【請求項10】
第1の試薬及び第2の試薬の少なくとも1つが無機塩基を含む、請求項8に記載の方法。
【請求項11】
無機塩基が、NaOH、KOH又はLiOHを含む、請求項10に記載の方法。
【請求項12】
第1の試薬が、還元剤を含む、請求項1〜11のいずれかに記載の方法。
【請求項13】
還元剤が、NaBH4、NaCNBH3、リチウムトリ−sec−ブチルボロヒドリド、NaBH(OCOCH3)3、リチウムアルミニウムヒドリド、ジチオスレイトール、及びβ−メルカプトエタノールから選択される、請求項12に記載の方法。
【請求項14】
第1の混合物のpHが、約9から約13である、請求項1〜6、又は8〜13のいずれかに記載の方法。
【請求項15】
第2の混合物のpHが、約11から約14である、請求項1〜6、又は8〜14のいずれかに記載の方法。
【請求項16】
第1の試薬が、約1NのNaOH及びNaBH4を含み、第2の試薬が、約2NのNaOHを含む、請求項1〜6、又は8〜15のいずれかに記載の方法。
【請求項17】
細胞成分が、タンパク質及び核酸を含み、前記細胞成分の起源は微生物であり、第1の混合物が、ポリサッカライドの細胞成分との直接又は間接の付着を提供する1つ又は複数の結合を加水分解し、第2の混合物が、タンパク質及び核酸を分解する、請求項1〜16のいずれかに記載の方法。
【請求項18】
ポリサッカライドの分離が、クロマトグラフィーによるものである、請求項1〜17のいずれかに記載の方法。
【請求項19】
ポリサッカライドの分離が、膜濾過によるものである、請求項1〜17のいずれかに記載の方法。
【請求項20】
ポリサッカライドをクロマトグラフィーによって精製する、請求項1〜19のいずれかに記載の方法。
【請求項21】
ポリサッカライドを膜濾過によって精製する、請求項1〜19のいずれかに記載の方法。
【請求項22】
ポリサッカライドが、リポポリサッカライドである、請求項1〜21のいずれかに記載の方法。
【請求項23】
ポリサッカライドが、莢膜ポリサッカライド、被膜下ポリサッカライド、又はエキソポリサッカライドである、請求項1〜21のいずれかに記載の方法。
【請求項24】
ポリサッカライドが、連鎖球菌属のいずれかの細菌に由来する、請求項23に記載の方法。
【請求項25】
ポリサッカライドが、B群連鎖球菌莢膜ポリサッカライドである、請求項24に記載の方法。
【請求項26】
B群連鎖球菌莢膜ポリサッカライドが、B群連鎖球菌Ia型、Ib型、II型、III型、V型、VI型、又はVIII型に由来する、請求項25に記載の方法。
【請求項27】
ポリサッカライドが、A群連鎖球菌又はC群連鎖球菌被膜下ポリサッカライドである、請求項23に記載の方法。
【請求項28】
ポリサッカライドが、ナイセリア属のいずれかの細菌に由来する、請求項23に記載の方法。
【請求項29】
ポリサッカライドが、髄膜炎菌B型、C型、Y型又はW−135型に由来する莢膜ポリサッカライドである、請求項28に記載の方法。
【請求項30】
莢膜ポリサッカライドが、髄膜炎菌Y型に由来する、請求項29に記載の方法。
【請求項31】
莢膜ポリサッカライドが、髄膜炎菌W−135型に由来する、請求項29に記載の方法。
【請求項32】
ポリサッカライドが、肺炎球菌に由来する莢膜ポリサッカライドである、請求項23に記載の方法。
【請求項33】
莢膜ポリサッカライドが、肺炎球菌3型又は14型に由来する、請求項32に記載の方法。
【請求項34】
ポリサッカライドが、大腸菌属のいずれかの細菌に由来する莢膜ポリサッカライドである、請求項23に記載の方法。
【請求項35】
莢膜ポリサッカライドが、大腸菌K1に由来する、請求項34に記載の方法。
【請求項36】
精製したポリサッカライドをアシル化試薬と接触させるステップと、前記アシル化したポリサッカライドを精製するステップとをさらに含み、
第1の試薬が、還元剤及び塩基を含み、前記塩基が、NaOH、KOH、及びLiOHから選択され、
第2の試薬が、NaOH、KOH、及びLiOHから選択される塩基を含み、
ポリサッカライドを分離するステップが、限外濾過膜による接線流濾過の使用を含む膜濾過によるものであり、
ポリサッカライドを精製するステップが、限外濾過膜による接線流濾過の使用を含む膜濾過によるものであり、
前記アシル化試薬が、無水酢酸である、
請求項4に記載の方法。
【請求項37】
精製したポリサッカライドが、約5質量%未満の核酸及び約5質量%未満のタンパク質を含有する、請求項1〜36のいずれかに記載の方法。
【請求項38】
精製したポリサッカライドが、約1質量%未満の核酸及び約1質量%未満のタンパク質を含有し、精製したポリサッカライドの純度が、80%〜100%である、請求項37に記載の方法。
【請求項39】
精製したポリサッカライドの純度が、90%〜100%である、請求項38に記載の方法。
【請求項40】
分離された組成物を第2の試薬と接触させる間の最高温度が、ストックを第1の試薬と接触させる間の最高温度より30℃〜90℃高い、請求項1〜39のいずれかに記載の方法。
【請求項41】
ポリサッカライド及び細胞成分を含むストックからの前記ポリサッカライドを精製する方法であって、
前記ストックを第1の試薬と接触させて第1の混合物を形成するステップ(前記第1の試薬は、塩基及び還元剤を含み、前記第1の混合物のpHは、9〜15の範囲である)と、
前記ポリサッカライドを前記細胞成分の少なくとも一部から分離して前記ポリサッカライドと前記細胞成分の残量とを含む分離された組成物を形成するステップと、
前記精製したポリサッカライドを回収するステップと
を含む方法。
【請求項42】
精製したポリサッカライドを回収する前に、
前記分離された組成物を第2の試薬と接触させて第2の混合物を形成するステップ(前記第2の試薬は塩基であり、前記第2の混合物のpHは、9〜15の範囲である)と、
前記細胞成分の前記残量の少なくとも一部からのポリサッカライドを精製して、精製された組成物を形成するステップと
をさらに含む、請求項41に記載の方法。
【請求項43】
還元剤が、NaBH4、NaCNBH3、リチウムトリ−sec−ブチルボロヒドリド、NaBH(OCOCH3)3、リチウムアルミニウムヒドリド、ジチオスレイトール、及びβ−メルカプトエタノールから選択される、請求項41〜42のいずれかに記載の方法。
【請求項44】
ポリサッカライドの分離が、膜濾過によるものである、請求項41〜43のいずれかに記載の方法。
【請求項45】
膜濾過が、限外濾過膜による接線流濾過の使用を含む、請求項44に記載の方法。
【請求項46】
ポリサッカライド結合型ワクチンを製造する方法であって、
請求項1〜40のいずれかに記載の方法の使用によって精製したポリサッカライドを得るステップと、
前記精製したポリサッカライドをポリペプチドに結合し、それによってポリサッカライド結合型ワクチンを製造するステップと
を含む方法。
【請求項47】
ポリサッカライド結合型ワクチンを製造する方法であって、
請求項41〜45のいずれかに記載の方法の使用によって精製したポリサッカライドを得るステップと、
前記精製したポリサッカライドをポリペプチドに結合し、それによってポリサッカライド結合型ワクチンを製造するステップと
を含む方法。
【請求項48】
結合が、還元的アミノ化によって達成される、請求項46又は47に記載の方法。
【請求項49】
ポリペプチドが、担体タンパク質、スカシガイヘモシアニン(KLH)、ウシ血清アルブミン(BSA)、卵白アルブミン(OVA)、ウサギ血清アルブミン(RSA)、破傷風トキソイド、ジフテリアトキソイド、ニューモリソイド、外膜タンパク質、ウイルス糖タンパク質、並びにそれらのフラグメント、変異体、及び模倣体から選択される、請求項46〜48のいずれかに記載の方法。
【請求項50】
ポリペプチドが破傷風毒素フラグメントC(TTc)である、請求項49に記載の方法。
【請求項51】
ポリペプチドが組換え型破傷風毒素のフラグメントC(rTTc)である、請求項49に記載の方法。
【請求項52】
アジュバントを添加するステップをさらに含む、請求項46〜51のいずれかに記載の方法。
【請求項53】
アジュバントが、ミョウバン、水酸化アルミニウム、ステアリルチロシン、フロイント、OS21、MPL、MF59、トール様受容体アゴニスト、及びCD14受容体アゴニストから選択される、請求項52に記載の方法。
【請求項54】
精製したプロテアーゼを含む試薬を使用しないで行われる、請求項1〜53のいずれかに記載の方法。
【請求項55】
精製したヌクレアーゼを含む試薬を使用しないで行われる、請求項1〜54のいずれかに記載の方法。
【請求項1】
ポリサッカライド及び細胞成分を含むストックからの前記ポリサッカライドを精製する方法であって、
前記ストックを第1の試薬と接触させて第1の混合物を形成するステップ(前記第1の試薬は、酸又は塩基であり、前記第1の混合物のpHは、0〜6の範囲又は9〜15の範囲である)と、
前記ポリサッカライドを前記細胞成分の少なくとも一部から分離して前記ポリサッカライドと前記細胞成分の残量とを含む分離された組成物を形成するステップと、
前記分離された組成物を第2の試薬と接触させて第2の混合物を形成するステップ(前記第2の試薬は塩基であり、前記第2の混合物のpHは、9〜15の範囲である)と、
前記細胞成分の前記残量の少なくとも一部からのポリサッカライドを精製して、精製された組成物を形成するステップと
を含む方法。
【請求項2】
精製ステップの前にさらなる接触ステップ及び分離ステップが実施される、請求項1に記載の方法。
【請求項3】
ポリサッカライドが、N−アセチル基を含有し、これらのN−アセチル基の少なくとも一部が第2の試薬による処理によって加水分解される、請求項1又は2に記載の方法。
【請求項4】
加水分解されたポリサッカライド上に存在する一定割合のN−アセチル基が、未変性ポリサッカライドの免疫特性を維持するためにそのとき十分に再アシル化される、請求項3に記載の方法。
【請求項5】
精製したポリサッカライドをアシル化試薬と接触させるステップと、前記アシル化ポリサッカライドを精製するステップとをさらに含む、請求項3又は4に記載の方法。
【請求項6】
アシル化試薬が、無水酢酸、塩化アセチル、ペンタフルオロフェニル酢酸又は4−ニトロフェニル酢酸である、請求項5に記載の方法。
【請求項7】
第1の試薬が酸である、請求項1〜6のいずれかに記載の方法。
【請求項8】
第1の試薬が塩基である、請求項1〜6のいずれかに記載の方法。
【請求項9】
第1の試薬及び第2の試薬の少なくとも1つが有機塩基を含む、請求項8に記載の方法。
【請求項10】
第1の試薬及び第2の試薬の少なくとも1つが無機塩基を含む、請求項8に記載の方法。
【請求項11】
無機塩基が、NaOH、KOH又はLiOHを含む、請求項10に記載の方法。
【請求項12】
第1の試薬が、還元剤を含む、請求項1〜11のいずれかに記載の方法。
【請求項13】
還元剤が、NaBH4、NaCNBH3、リチウムトリ−sec−ブチルボロヒドリド、NaBH(OCOCH3)3、リチウムアルミニウムヒドリド、ジチオスレイトール、及びβ−メルカプトエタノールから選択される、請求項12に記載の方法。
【請求項14】
第1の混合物のpHが、約9から約13である、請求項1〜6、又は8〜13のいずれかに記載の方法。
【請求項15】
第2の混合物のpHが、約11から約14である、請求項1〜6、又は8〜14のいずれかに記載の方法。
【請求項16】
第1の試薬が、約1NのNaOH及びNaBH4を含み、第2の試薬が、約2NのNaOHを含む、請求項1〜6、又は8〜15のいずれかに記載の方法。
【請求項17】
細胞成分が、タンパク質及び核酸を含み、前記細胞成分の起源は微生物であり、第1の混合物が、ポリサッカライドの細胞成分との直接又は間接の付着を提供する1つ又は複数の結合を加水分解し、第2の混合物が、タンパク質及び核酸を分解する、請求項1〜16のいずれかに記載の方法。
【請求項18】
ポリサッカライドの分離が、クロマトグラフィーによるものである、請求項1〜17のいずれかに記載の方法。
【請求項19】
ポリサッカライドの分離が、膜濾過によるものである、請求項1〜17のいずれかに記載の方法。
【請求項20】
ポリサッカライドをクロマトグラフィーによって精製する、請求項1〜19のいずれかに記載の方法。
【請求項21】
ポリサッカライドを膜濾過によって精製する、請求項1〜19のいずれかに記載の方法。
【請求項22】
ポリサッカライドが、リポポリサッカライドである、請求項1〜21のいずれかに記載の方法。
【請求項23】
ポリサッカライドが、莢膜ポリサッカライド、被膜下ポリサッカライド、又はエキソポリサッカライドである、請求項1〜21のいずれかに記載の方法。
【請求項24】
ポリサッカライドが、連鎖球菌属のいずれかの細菌に由来する、請求項23に記載の方法。
【請求項25】
ポリサッカライドが、B群連鎖球菌莢膜ポリサッカライドである、請求項24に記載の方法。
【請求項26】
B群連鎖球菌莢膜ポリサッカライドが、B群連鎖球菌Ia型、Ib型、II型、III型、V型、VI型、又はVIII型に由来する、請求項25に記載の方法。
【請求項27】
ポリサッカライドが、A群連鎖球菌又はC群連鎖球菌被膜下ポリサッカライドである、請求項23に記載の方法。
【請求項28】
ポリサッカライドが、ナイセリア属のいずれかの細菌に由来する、請求項23に記載の方法。
【請求項29】
ポリサッカライドが、髄膜炎菌B型、C型、Y型又はW−135型に由来する莢膜ポリサッカライドである、請求項28に記載の方法。
【請求項30】
莢膜ポリサッカライドが、髄膜炎菌Y型に由来する、請求項29に記載の方法。
【請求項31】
莢膜ポリサッカライドが、髄膜炎菌W−135型に由来する、請求項29に記載の方法。
【請求項32】
ポリサッカライドが、肺炎球菌に由来する莢膜ポリサッカライドである、請求項23に記載の方法。
【請求項33】
莢膜ポリサッカライドが、肺炎球菌3型又は14型に由来する、請求項32に記載の方法。
【請求項34】
ポリサッカライドが、大腸菌属のいずれかの細菌に由来する莢膜ポリサッカライドである、請求項23に記載の方法。
【請求項35】
莢膜ポリサッカライドが、大腸菌K1に由来する、請求項34に記載の方法。
【請求項36】
精製したポリサッカライドをアシル化試薬と接触させるステップと、前記アシル化したポリサッカライドを精製するステップとをさらに含み、
第1の試薬が、還元剤及び塩基を含み、前記塩基が、NaOH、KOH、及びLiOHから選択され、
第2の試薬が、NaOH、KOH、及びLiOHから選択される塩基を含み、
ポリサッカライドを分離するステップが、限外濾過膜による接線流濾過の使用を含む膜濾過によるものであり、
ポリサッカライドを精製するステップが、限外濾過膜による接線流濾過の使用を含む膜濾過によるものであり、
前記アシル化試薬が、無水酢酸である、
請求項4に記載の方法。
【請求項37】
精製したポリサッカライドが、約5質量%未満の核酸及び約5質量%未満のタンパク質を含有する、請求項1〜36のいずれかに記載の方法。
【請求項38】
精製したポリサッカライドが、約1質量%未満の核酸及び約1質量%未満のタンパク質を含有し、精製したポリサッカライドの純度が、80%〜100%である、請求項37に記載の方法。
【請求項39】
精製したポリサッカライドの純度が、90%〜100%である、請求項38に記載の方法。
【請求項40】
分離された組成物を第2の試薬と接触させる間の最高温度が、ストックを第1の試薬と接触させる間の最高温度より30℃〜90℃高い、請求項1〜39のいずれかに記載の方法。
【請求項41】
ポリサッカライド及び細胞成分を含むストックからの前記ポリサッカライドを精製する方法であって、
前記ストックを第1の試薬と接触させて第1の混合物を形成するステップ(前記第1の試薬は、塩基及び還元剤を含み、前記第1の混合物のpHは、9〜15の範囲である)と、
前記ポリサッカライドを前記細胞成分の少なくとも一部から分離して前記ポリサッカライドと前記細胞成分の残量とを含む分離された組成物を形成するステップと、
前記精製したポリサッカライドを回収するステップと
を含む方法。
【請求項42】
精製したポリサッカライドを回収する前に、
前記分離された組成物を第2の試薬と接触させて第2の混合物を形成するステップ(前記第2の試薬は塩基であり、前記第2の混合物のpHは、9〜15の範囲である)と、
前記細胞成分の前記残量の少なくとも一部からのポリサッカライドを精製して、精製された組成物を形成するステップと
をさらに含む、請求項41に記載の方法。
【請求項43】
還元剤が、NaBH4、NaCNBH3、リチウムトリ−sec−ブチルボロヒドリド、NaBH(OCOCH3)3、リチウムアルミニウムヒドリド、ジチオスレイトール、及びβ−メルカプトエタノールから選択される、請求項41〜42のいずれかに記載の方法。
【請求項44】
ポリサッカライドの分離が、膜濾過によるものである、請求項41〜43のいずれかに記載の方法。
【請求項45】
膜濾過が、限外濾過膜による接線流濾過の使用を含む、請求項44に記載の方法。
【請求項46】
ポリサッカライド結合型ワクチンを製造する方法であって、
請求項1〜40のいずれかに記載の方法の使用によって精製したポリサッカライドを得るステップと、
前記精製したポリサッカライドをポリペプチドに結合し、それによってポリサッカライド結合型ワクチンを製造するステップと
を含む方法。
【請求項47】
ポリサッカライド結合型ワクチンを製造する方法であって、
請求項41〜45のいずれかに記載の方法の使用によって精製したポリサッカライドを得るステップと、
前記精製したポリサッカライドをポリペプチドに結合し、それによってポリサッカライド結合型ワクチンを製造するステップと
を含む方法。
【請求項48】
結合が、還元的アミノ化によって達成される、請求項46又は47に記載の方法。
【請求項49】
ポリペプチドが、担体タンパク質、スカシガイヘモシアニン(KLH)、ウシ血清アルブミン(BSA)、卵白アルブミン(OVA)、ウサギ血清アルブミン(RSA)、破傷風トキソイド、ジフテリアトキソイド、ニューモリソイド、外膜タンパク質、ウイルス糖タンパク質、並びにそれらのフラグメント、変異体、及び模倣体から選択される、請求項46〜48のいずれかに記載の方法。
【請求項50】
ポリペプチドが破傷風毒素フラグメントC(TTc)である、請求項49に記載の方法。
【請求項51】
ポリペプチドが組換え型破傷風毒素のフラグメントC(rTTc)である、請求項49に記載の方法。
【請求項52】
アジュバントを添加するステップをさらに含む、請求項46〜51のいずれかに記載の方法。
【請求項53】
アジュバントが、ミョウバン、水酸化アルミニウム、ステアリルチロシン、フロイント、OS21、MPL、MF59、トール様受容体アゴニスト、及びCD14受容体アゴニストから選択される、請求項52に記載の方法。
【請求項54】
精製したプロテアーゼを含む試薬を使用しないで行われる、請求項1〜53のいずれかに記載の方法。
【請求項55】
精製したヌクレアーゼを含む試薬を使用しないで行われる、請求項1〜54のいずれかに記載の方法。
【図1】

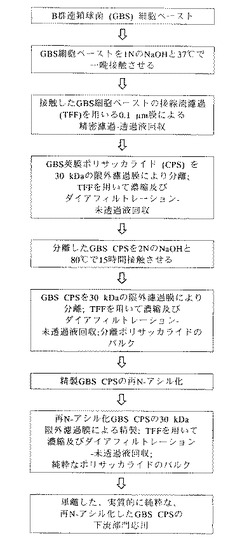
【図2】
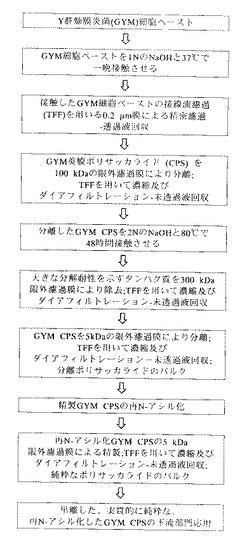
【図3】
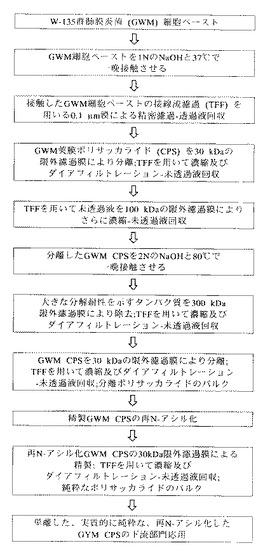
【図4】

【図2】
【図3】
【図4】
【公表番号】特表2009−523736(P2009−523736A)
【公表日】平成21年6月25日(2009.6.25)
【国際特許分類】
【出願番号】特願2008−550535(P2008−550535)
【出願日】平成19年1月12日(2007.1.12)
【国際出願番号】PCT/US2007/060497
【国際公開番号】WO2007/084856
【国際公開日】平成19年7月26日(2007.7.26)
【出願人】(591013229)バクスター・インターナショナル・インコーポレイテッド (448)
【氏名又は名称原語表記】BAXTER INTERNATIONAL INCORP0RATED
【出願人】(508211580)バクスター ヘルスケア エス.エイ. (2)
【Fターム(参考)】
【公表日】平成21年6月25日(2009.6.25)
【国際特許分類】
【出願日】平成19年1月12日(2007.1.12)
【国際出願番号】PCT/US2007/060497
【国際公開番号】WO2007/084856
【国際公開日】平成19年7月26日(2007.7.26)
【出願人】(591013229)バクスター・インターナショナル・インコーポレイテッド (448)
【氏名又は名称原語表記】BAXTER INTERNATIONAL INCORP0RATED
【出願人】(508211580)バクスター ヘルスケア エス.エイ. (2)
【Fターム(参考)】
[ Back to top ]