
ポリブチレンテレフタレート及びポリブチレンテレフタレートの製造方法
【課題】色調良好なPBTを提供すること、及び原料BGのTHFへの転化率が低く、PBTを効率的に製造できる方法を提供すること。
【解決手段】テレフタル酸由来の構成単位と1,4ブタンジオール由来の構成単位を含み、窒素原子含有量が0.01〜50質量ppmであることを特徴とするポリブチレンテレフタレート。このうち、特に、上記1,4−ブタンジオールがバイオマス資源由来であるものが好ましい。また、上記1,4−ブタンジオール中の窒素原子含有量が0.01〜50質量ppmであることが好ましい。
【解決手段】テレフタル酸由来の構成単位と1,4ブタンジオール由来の構成単位を含み、窒素原子含有量が0.01〜50質量ppmであることを特徴とするポリブチレンテレフタレート。このうち、特に、上記1,4−ブタンジオールがバイオマス資源由来であるものが好ましい。また、上記1,4−ブタンジオール中の窒素原子含有量が0.01〜50質量ppmであることが好ましい。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、窒素原子含有量が特定範囲で色調良好なポリブチレンテレフタレートに関する。
【背景技術】
【0002】
熱可塑性ポリエステル樹脂の中で代表的なエンジニアリンブプラスチックであるポリブチレンテレフタレート(以下PBTと表す)は、成形加工の容易性、機械的物性、耐熱性
、耐薬品性、保香性、その他の物理的・化学的特性に優れていることから、自動車部品、電気・電子部品、精密機器部品などの射出成形品に広く使用されている。また、近年は、その優れた性質を活かし、フィルム、シート、モノフィラメント、繊維などの一般消費材分野でも広く使用されており、これに伴って、色調良好なPBTが求められるようになってきている。
【0003】
PBTは、通常、テレフタル酸又はそのアルキルエステルと1,4−ブタンジオール(以下BGと表すことがある)とを反応させて製造するが、BGは反応中にテトラヒドロフラン(以下THFと表すことがある)に転化しやすいため、THF転化率の低いPBTの製造方法が求められている。
一方、循環型(サステイナブル)社会の構築を求める声の高まりとともに、樹脂材料分野においてもエネルギーと同様に化石燃料原料からの脱却が望まれている。化石燃料を原料としない樹脂材料を開発する場合、植物などを原料とするバイオマス資源が有力な原料候補であり、バイオマスプラスチックの実用化が進んでいる。
【0004】
原料である1,4ブタンジオールがバイオマス資源から得られたものである場合、化石燃料から得られたものである場合に比べ、PBTの色調が悪くなる。
特許文献1には、バイオマス資源を原料としてポリエステルを得る技術について、ジカルボン酸中の窒素含有量を制御することにより、窒素含有量1000質量ppm以下のポリエステルを得ることについて記載されている。しかしながら、該文献には、ジオール中の窒素含有量及びその影響については記載も示唆もない。又、PBTに関する記載も全く無い。(尚、以下、本発明において、特に断らない限り濃度表示「ppm」は、「質量ppm」を表す。)
【特許文献1】特開2005―139287号公報
【発明の開示】
【発明が解決しようとする課題】
【0005】
本発明は、上記の現状に鑑みてなされたものであり、色調良好なPBTを提供すること、及び原料BGのTHFへの転化率が低く、PBTを効率的に製造できる方法を提供することを目的とする。
【課題を解決するための手段】
【0006】
本発明者は、上記目的を達成すべく鋭意検討した。この結果、特定量の窒素原子を含有するPBTの色調が良好であることを見出し本発明に到達した。
即ち、本発明の要旨は、テレフタル酸由来の構成単位と1,4ブタンジオール由来の構成単位を含み、窒素原子含有量が0.01〜50質量ppmであることを特徴とするポリブチレンテレフタレートに存する。また、本発明の別の要旨は、テレフタル酸又はテレフタル酸アルキレートと窒素原子含有量が0.01〜50質量ppmの1,4ブタンジオールとをエステル化反応又はエステル交換反応させた後、重縮合反応させるポリブチレンテレフタレートの製造方法に存する。
【発明の効果】
【0007】
本発明によれば、色調良好で高品質なPBTを得ることができる。特に、バイオマス資源由来のBGを原料として、色調良好なPBTを得ることができる。また、原料BGのTHFへの転化率が低く、効率的にPBT原料を製造することができる。
【発明を実施するための最良の形態】
【0008】
以下、本発明を詳細に説明するが、以下に記載する各構成要件の説明は、本発明の実施態様の代表例であり、本発明はこれらに限定されるものではない。
尚、本明細書において、「〜」を用いて表される数値範囲は、「〜」の前後に記載される数値を下限値及び上限値として含む範囲を意味する。また、本明細書における、下限値又は上限値は、その下限値又は上限値の数値を含む範囲を意味する。
<PBT製造原料>
本発明のPBTは、テレフタル酸又はテレフタル酸アルキレートと1,4ブタンジオールとをエステル化反応又はエステル交換反応させた後、重縮合反応させることにより得られる。本発明のPBTの製造に用いる原料は、バイオマス資源由来であるのが、環境保護の点から好ましい。
【0009】
テレフタル酸又はテレフタル酸アルキルエステルは、全ジカルボン酸成分の80モル%以上であるのが好ましく、90モル%以上であるのが更に好ましく、100モル%であるのが最も好ましい。また、BG成分は、全ジオール成分の80モル%以上であるのが好ましく、90モル%以上であるのが更に好ましく、100モル%であるのが最も好ましい。テレフタル酸又はテレフタル酸アルキルエステルの全ジカルボン酸成分に占める割合及びBG成分の全ジオール成分に占める割合が前記範囲以上であると、電気部品等に成形する際の結晶化の点やフィルム、繊維等に成形する際の延伸による分子鎖の配向結晶化の点から、成形体としての機械的強度、耐熱性、保香性等が良好になりやすい。
【0010】
テレフタル酸又はテレフタル酸アルキルエステル以外のジカルボン酸成分としては、具体的には、例えば、フタル酸、イソフタル酸、ジブロモイソフタル酸、スルホイソフタル酸ナトリウム、フェニレンジオキシジカルボン酸、4,4’−ジフェニルジカルボン酸、4,4’−ジフェニルエーテルジカルボン酸、4,4’−ジフェニルケトンジカルボン酸、4,4’−ジフェノキシエタンジカルボン酸、4,4’−ジフェニルスルホンジカルボン酸、2,6−ナフタレンジカルボン酸等の芳香族ジカルボン酸およびこれらのエステル形成性誘導体;ヘキサヒドロテレフタル酸、ヘキサヒドロイソフタル酸等の脂環式ジカルボン酸およびこれらのエステル形成性誘導体;コハク酸、グルタル酸、アジピン酸、ピメリン酸、スベリン酸、アゼライン酸、セバシン酸、ウンデカジカルボン酸、ドデカジカルボン酸等の脂肪族鎖式ジカルボン酸よびこれらのエステル形成性誘導体等が挙げられる。
【0011】
BG以外のジオール成分としては、具体的には、例えば、エチレングリコール、トリメチレングリコール、ペンタメチレングリコール、ヘキサメチレングリコール、オクタメチレングリコール、デカメチレングリコール、ネオペンチルグリコール、2−エチル−2−ブチル−1,3−プロパンジオール、ポリエチレングリコール、ポリテトラメチレンエーテルグリコール等の脂肪族鎖式ジオール;1,2−シクロヘキサンジオール、1,4−シクロヘキサンジオール、1,1−シクロヘキサンジメチロール、1,4−シクロヘキサンジメチロール、2,5−ノルボルナンジメチロール等の脂環式ジオール;キシリレングリコール、4,4’−ジヒドロキシビフェニル、2,2−ビス(4’−ヒドロキシフェニル)プロパン、2,2−ビス(4’−β−ヒドロキシエトキシフェニル)プロパン、ビス(4−ヒドロキシフェニル)スルホン、ビス(4−β−ヒドロキシエトキシフェニル)スルホン酸等の芳香族ジオール;2,2−ビス(4’−ヒドロキシフェニル)プロパンのエチレンオキサイド付加物又はプロピレンオキサイド付加物;バイオマス資源由来でないBG等が挙げられる。
【0012】
なお、共重合成分として、更に、以下の成分が用いられていてもよい。例えば、グリコール酸、p−ヒドロキシ安息香酸、p−β−ヒドロキシエトキシ安息香酸等のヒドロキシカルボン酸やアルコキシカルボン酸、及び、ステアリルアルコール、ヘネイコサノール、オクタコサノール、ベンジルアルコール、ステアリン酸、ベヘン酸、安息香酸、t−ブチル安息香酸、ベンゾイル安息香酸等の単官能成分、トリカルバリル酸、トリメリット酸、トリメシン酸、ピロメリット酸、ナフタレンテトラカルボン酸、没食子酸、トリメチロールエタン、トリメチロールプロパン、グリセロール、ペンタエリスリトール、シュガーエステル等の三官能以上の多官能成分、等が挙げられる。
【0013】
<バイオマス資源由来BG>
本発明のPBTの製造に用いるジカルボン酸成分は、バイオマス資源由来であるのが架橋保護の点から好ましい。
バイオマス資源とは、植物の光合成作用で太陽の光エネルギーがデンプンやセルロースなどの形に変換されて蓄えられたもの、植物体を食べて成育する動物の体や、植物体や動物体を加工してできる製品等が含まれる。具体的には、木材、稲わら、籾殻、米ぬか、古米、とうもろこし、サトウキビ、キャッサバ、サゴヤシ、おから、コーンコブ、タピオカカス、バガス、植物油カス、芋、そば、大豆、油脂、古紙、製紙残渣、水産物残渣、家畜排泄物、下水汚泥、食品廃棄物等が挙げられる。この中でも、木材、稲わら、籾殻、米ぬか、古米、とうもろこし、サトウキビ、キャッサバ、サゴヤシ、おから、コーンコブ、タピオカカス、バガス、植物油カス、芋、そば、大豆、油脂、古紙、製紙残渣等の植物資源が好ましく、より好ましくは、木材、稲わら、籾殻、古米、とうもろこし、サトウキビ、キャッサバ、サゴヤシ、芋、油脂、古紙、製紙残渣が挙げられ、最も好ましくはとうもろこし、さとうきび、キャッサバ、サゴヤシが挙げられる。
【0014】
バイオマス資源は、一般に、窒素元素やNa、K、Mg、Ca等の多くのアルカリ金属及びアルカリ土類金属を含有する。
これらのバイオマス資源は、その方法は特に限定はされないが、例えば、酸やアルカリ等の化学処理、微生物を用いた生物学的処理、物理的処理等の公知の前処理・糖化の工程等を経て炭素源へと誘導される。その工程には、バイオマス資源をチップ化する、削る、擦り潰す等の前処理による微細化工程が含まれることが多く、必要に応じて、更にグラインダーやミルによる粉砕工程も含まれる。こうして微細化されたバイオマス資源は、通常、更に前処理・糖化の工程を経て炭素源へと誘導される。その具体的な方法としては、硫酸、硝酸、塩酸、燐酸等の強酸による酸処理、アルカリ処理、アンモニア凍結蒸煮爆砕法、溶媒抽出、超臨界流体処理、酸化剤処理等の化学的方法;微粉砕、蒸煮爆砕法、マイクロ波処理、電子線照射等の物理的方法;微生物や酵素処理による加水分解等生物学的処理等が挙げられる。
【0015】
上記のバイオマス資源から誘導される炭素源としては、通常、グルコース、マンノース、ガラクトース、フルクトース、ソルボース、タガトース等のヘキソース;アラビノース、キシロース、リボース、キシルロース、リブロース等のペントース;ペントサン、サッカロース、澱粉、セルロース等の2糖・多糖類;酪酸、カプロン酸、カプリル酸、カプリン酸、ラウリン酸、ミリスチン酸、パルミチン酸、パルミトレイン酸、ステアリン酸、オレイン酸、リノール酸、リノレン酸、モノクチン酸、アラキジン酸、エイコセン酸、アラキドン酸、ベヘニン酸、エルカ酸、ドコサペンタエン酸、ドコサヘキサエン酸、リグノセリン酸、セラコレン酸等の油脂;グリセリン、マンニトール、キシリトール、リビトール等のポリアルコール類等の発酵性糖質が用いられる。このうち、グルコース、フルクトース、キシロース等のヘキソース又はペントースが好ましく、特にグルコースが好ましい。より広義の植物資源由来の炭素源としては、紙の主成分であるセルロースも好ましい。
【0016】
通常、これらの炭素源を用いて、微生物変換による発酵法や加水分解・脱水反応・水和反応・酸化反応等の反応工程を含む化学変換法ならびにこれらの発酵法と化学変換法の組み合わせによりジオールが合成される。これらの中でも、微生物変換による発酵法が好ましい。
本発明のPBTを製造するのに用いるジオールは、窒素原子含有量が0.01〜50質量ppmであるのが好ましい。バイオマス資源のジオールを用いる場合、具体的には、例えば、グルコース等の炭素源から発酵法により直接ジオールを製造してもよいし、発酵法により得られたコハク酸、コハク酸無水物、コハク酸エステル等を化学反応によりジオール化合物に変換しても良いし、発酵法により得られた1,3−ブタジエンから1,4−ブタンジオールを製造する等してもよい。この中でも、発酵法により得られたコハク酸(又はコハク酸をコハク酸ジメチルに変成後に)を、還元触媒により水添して1,4−ブタンジオールを得る方法が効率的で好ましい。
【0017】
コハク酸を水添する触媒の例としては、例えば、Pd、Ru、Re、Rh、Ni、Cu、Co及びその化合物等が挙げられる。具体的には、Pd/Ag/Re、Ru/Ni/Co/ZnO、Cu/Zn酸化物、Cu/Zn/Cr酸化物、Ru/Re、Re/C、Ru/Sn、Ru/Pt/Sn、Pt/Re/アルカリ、Pt/Re、Pd/Co/Re、Cu/Si、Cu/Cr/Mn、ReO/CuO/ZnO、CuO/CrO、Pd/Re、Ni/Co、Pd/CuO/CrO3、リン酸Ru、Ni/Co、Co/Ru/Mn、Cu/Pd/KOH、Cu/Cr/Zn等が挙げられる。この中でも、Ru/Sn又はRu/Pt/Snが触媒活性の点で好ましい。
また、更に、バイオマス資源から公知の有機化学触媒反応との組み合わせによりBGを製造する方法も用いられる。例えば、バイオマス資源としてペントースを利用する場合には公知の脱水反応、触媒反応の組み合わせで容易にブタンジオール等のジオールを製造できる。
【0018】
<BG中の窒素原子含有量>
バイオマス資源由来から誘導されたBGには、発酵処理ならびに酸による中和工程を含む精製処理に起因して不純物として窒素原子が含まれてくる。具体的には、アミノ酸、蛋白質、アンモニア、尿素、発酵菌由来等の窒素原子が含まれてくる。
本発明においてPBTの原料となる、バイオマス資源から得られたBGの窒素原子含有量は、該BGに対して質量比で、上限は、通常50ppm、好ましくは10ppm、より好ましくは2ppmであるのよい。また、下限は特に制限されないが、通常、0.01ppm、好ましくは0.05ppmがよく、特に、精製工程の経済性の観点からは、0.1ppmであるのが好ましい。BG中の窒素原子含有量が上記上限以下であると、PBT製造における重縮合反応速度、生成するPBTの色調などが好ましくなる傾向が強い。一方、上記下限以上であると、精製工程が簡便としやすく経済的に有利な上、PBT製造反応中でのBGのTHFへの転化も低く押させやすい。
【0019】
上記上限以下であると重縮合反応速度や色調の点で好ましくなりやすい理由は定かではないが、発酵処理ならびに酸による中和処理を含む精製処理において、窒素源となる化合物以外に重縮合反応を阻害し、PBTの色調を悪化させる着色誘引物質が生成しやすくなっているのではないかと推定される。また、発酵における窒素源となるアンモニア、アミノ酸なども反応を遅延させ、PBTの色調悪化を誘引するものと考えられる。
【0020】
BG中の窒素原子量は、例えば、バイオマス資源の発酵により得られるコハク酸を水添してBGを得る場合は、その発酵条件、アンモニアによる中和条件、コハク酸の晶析条件などによりコハク酸中の窒素原子量を調節することにより調節可能である。また、コハク酸を水添して得られるBGは、蒸留を含む精製条件により、その含有窒素原子含有量を調
節できる。また、BGがバイオマス資源の発酵により直接得られる場合にも、その発酵条件、アンモニアによる中和条件、得られたBGの蒸留を含む精製条件などにより調節できる。
【0021】
<BG中の硫黄原子含有量>
発酵法により製造したBG等を用いる場合には、酸による中和工程を含む精製処理等により硫黄原子が含まれてくる場合がある。この場合、具体的に、硫黄原子が含有される不純物としては、硫酸、亜硫酸、有機スルホン酸塩等が挙げられる。
BG中に含まれる硫黄原子量は、該BGに対して質量比で、上限は通常100ppm、好ましくは20ppm、より好ましくは10ppm、特に好ましくは5ppm、最も好ましくは0.5ppmである。一方、下限は特に制限されないが、通常0.001ppm、好ましくは0.01ppm、より好ましくは0.05ppm、特に好ましくは、0.1ppmである。上記上限以下であると、重縮合反応速度やPBTの安定性が好ましくなる傾向がある。一方、上記下限以上であると、精製工程を簡便にして経済的に有利にしやすくなる。硫黄原子含有量は、公知の元素分析法により測定される値である。
【0022】
<BG中の1−アセトキシ−4−ヒドロキシブタン及び2−(4−ヒドロキシブチルオキシ)テトラヒドロフラン)の含有量>
窒素原子含有量が0.01〜50質量ppmであるBGを用いると、PBTを製造する際の重縮合速度が大きくなる。具体的には、実施例で後述する通り、バイオマス資源から得られ、窒素原子含有量が0.01〜50質量ppmであるBGを用いると、化石燃料原料から得られたBGを使用した場合に比べて、重縮合速度が大きくなっている。これは、本発明の実施例で用いたBGにおける1−アセトキシ−4−ヒドロキシブタン(1,4−HABと表すことが有る)及び/又は2−(4−ヒドロキシブチルオキシ)テトラヒドロフラン(BGTFと表すことが有る)の含有量が少ないために、重縮合反応の阻害が少ないためであると考えられる。従って、BG中の1−アセトキシ−4−ヒドロキシブタンは、50質量ppm以下が好ましく、10質量ppm以下がより好ましい。また、下限は少ないほうが良いが通常0.1質量ppmである。
【0023】
<PBT製造方法>
本発明のPBTは、これを製造できれば特に制限されない。PBTの公知の製造方法は、主原料としてテレフタル酸を用いるいわゆる直接重合法と、主原料としてテレフタル酸ジアルキルエステルを用いるエステル交換法とに大別される。前者は、初期のエステル化反応で水が生成し、後者は初期のエステル交換反応でアルコールが生成するという違いがあるが、原料の入手安定性、留出物の処理の容易さ、原料原単位の高さ、また本発明による改良効果という観点からは直接重合法が好ましい。
【0024】
直接重合法の一例としては、テレフタル酸とBGとを、単数若しくは複数段のエステル化反応槽内で、エステル化反応触媒の存在下に、温度が、通常180以上、好ましくは200℃以上、特に好ましくは210℃以上、通常260℃以下、好ましくは250℃以下、特に好ましくは245℃以下で、圧力が、通常10kPa以上、好ましくは13kPa以上、特に好ましくは50kPa以上、通常133kPa以下、好ましくは101kPa以下、特に好ましくは90kPa以下で、反応時間が、通常0.5時間以上、好ましくは1時間以上、通常5時間以下、好ましくは3時間以下で、連続的にエステル化反応させ、得られたエステル化反応生成物としてのオリゴマーを重縮合反応槽に移送し、複数段の重縮合反応槽内で、重縮合反応触媒の存在下で連続的に、温度が、通常210℃以上、好ましくは220℃以上、通常260℃以下、好ましくは250℃以下、特に好ましくは245℃以下で、圧力が、通常27kPa以下、好ましくは20kPa以下、より好ましくは13kPa以下、中でも少なくとも1つの重縮合反応槽においては好ましくは2kPa以下の減圧下で、攪拌しながら、通常2〜12時間好ましくは2〜10時間で縮合反応させる方法等が挙げられる。
【0025】
エステル交換法の一例としては、テレフタル酸ジメチルとBGとを、単数若しくは複数段のエステル化反応槽内で、エステル交換反応触媒の存在下に、温度が110℃以上、好ましくは140℃以上、特に好ましくは180℃以上、260℃以下、好ましくは245℃以下、特に好ましくは220℃以下で、圧力が通常10kPa以上、好ましくは13kPa以上、特に好ましくは60kPa以上、通常133kPa以下、好ましくは101kPa以下、特に好ましくは90kPa以下で、反応時間が通常0.5時間以上、好ましくは1時間以上、通常5時間以下、好ましくは3時間以下で、連続的にエステル交換反応させ、得られたエステル交換反応生成物としてのオリゴマーを重縮合反応槽に移送し、複数段の重縮合反応槽内で、重縮合反応触媒の存在下で連続的に、温度が、通常210℃以上、好ましくは220℃以上、通常260℃以下、好ましくは250℃以下、特に好ましくは245℃以下で、圧力が、通常27kPa以下、好ましくは20kPa以下、特に好ましくは13kPa以下の減圧下、中でも少なくとも1つの重縮合反応槽においては好ましくは2kPa以下の減圧下で、攪拌しながら、通常2〜12時間好ましくは2〜10時間、重縮合反応させる方法等が挙げられる。
【0026】
エステル化反応またはエステル交換反応触媒としては、例えば、三酸化二アンチモン等のアンチモン化合物;二酸化ゲルマニウム、四酸化ゲルマニウム等のゲルマニウム化合物;テトラメチルチタネート、テトライソプロピルチタネート、テトラブチルチタネート等のチタンアルコラート、テトラフェニルチタネート等のチタンフェノラート等、のチタン化合物;ジブチルスズオキサイド、メチルフェニルスズオキサイド、テトラエチルスズ、ヘキサエチルジスズオキサイド、シクロヘキサヘキシルジスズオキサイド、ジドデシルスズオキサイド、トリエチルスズハイドロオキサイド、トリフェニルスズハイドロオキサイド、トリイソブチルスズアセテート、ジブチルスズジアセテート、ジフェニルスズジラウレート、モノブチルスズトリクロライド、トリブチルスズクロライド、ジブチルスズサルファイド、ブチルヒドロキシスズオキサイド、メチルスタンノン酸、エチルスタンノン酸、ブチルスタンノン酸等のスズ化合物;酢酸マグネシウム、水酸化マグネシウム、炭酸マグネシウム、酸化マグネシウム、マグネシウムアルコキサイド、燐酸水素マグネシウム等のマグネシウム化合物、酢酸カルシウム、水酸化カルシウム、炭酸カルシウム、酸化カルシウム、カルシウムアルコキサイド、燐酸水素カルシウム、等の等のカルシウム化合物等のアルカリ土類金属化合物の他、マンガン化合物、亜鉛化合物等を挙げることができる。中でも、チタン化合物、スズ化合物が好ましく、テトラブチルチタネートが特に好ましい。
【0027】
エステル化反応またはエステル交換反応触媒の使用量は特に限定されないが、PBT中の金属濃度(質量)として、通常1ppm以上、好ましくは5ppm以上、更に好ましくは10ppm以上、特に好ましくは20ppm以上、最も好ましくは30pmm以上、通常300ppm以下、好ましくは200ppm以下、より好ましくは150ppm以下、更に好ましくは100ppm以下、特に好ましくは90ppm以下、最も好ましくは60pmm以下が良い。上記上限以下であると、異物の原因となりにくい上、PBTの熱滞留時の劣化反応やガス発生が起こりにくい傾向があり、上記下限以上であると、主反応速度が速く副反応が起こりにくい。
【0028】
また、重縮合反応触媒としては、エステル化反応またはエステル交換反応の触媒をそのまま重縮合反応触媒として用いてもよいし、更に前記触媒を添加してもよい。重縮合反応触媒の使用量に特に制限はないが、上記のエステル化反応またはエステル交換反応の触媒と同様の理由から、PBT中の金属濃度(質量)として、通常300ppm以下、好ましくは200ppm以下、更に好ましくは100ppm以下、特に好ましくは50ppm以下、最も好ましくは30ppm以下がよい。
【0029】
また、触媒として有機チタン化合物を用いる場合には、異物抑制の観点から、最終的にはPBT中のチタン金属濃度(質量)は、250ppm以下であることが好ましく、100ppm以下であることが更に好ましく、60ppm以下であることが特に好ましく、50ppm以下であることが最も好ましい。
PBT中の金属濃度(質量)は、湿式灰化等の方法でPBT中の金属を回収した後、原子発光、Induced Coupled Plasma(ICP)法等を用いて測定することができる。
【0030】
また、前記のエステル化反応、エステル交換反応及び重縮合反応において、前記触媒の他に、正燐酸、亜燐酸、次亜燐酸、ポリ燐酸およびそれらのエステルや金属塩等の燐化合物;水酸化ナトリウム、安息香酸ナトリウム等のナトリウム化合物、酢酸リチウム、水酸化カリウム、酢酸カリウム等のカリウム化合物等のアルカリ金属化合物等の反応助剤;酢酸マグネシウム、酢酸カルシウム等のアルカリ土類金属化合物等の反応助剤;2,6−ジ−t−ブチル−4−オクチルフェノール、ペンタエリスリチル−テトラキス〔3−(3’,5’−t−ブチル−4’−ヒドロキシフェニル)プロピオネート〕等のフェノール化合物;ジラウリル−3,3’−チオジプロピオネート、ペンタエリスリチル−テトラキス(3−ラウリルチオジプロピオネート)等のチオエーテル化合物;トリフェニルホスファイト、トリス(ノニルフェニル)ホスファイト、トリス(2,4−ジ−t−ブチルフェニル)ホスファイト等の燐化合物等の抗酸化剤;パラフィンワックス、マイクロクリスタリンワックス、ポリエチレンワックス、モンタン酸やモンタン酸エステルに代表される長鎖脂肪酸およびそのエステル;シリコーンオイル等の離型剤等を使用しても良い。
【0031】
重縮合反応槽としては、縦型攪拌重合槽、横型攪拌重合槽、薄膜蒸発式重合槽等の公知のものを挙げることができる。反応液の粘度が上昇する重縮合の後期は、反応速度よりも物質移動が分子量増大の支配因子になる傾向があるため、副反応を抑制しつつ主反応を押し進めるには、可能な限り温度を下げ、表面更新性を上げた方が本発明の目的を達成するには有利であり、表面更新性とプラグフロー性、セルフクリーニング性に優れた薄膜蒸発機能を有した単数または複数の横型攪拌重合機を選定することが好ましい。
【0032】
また、本発明の製造法で得られたPBTは、引き続き公知の方法で固相重縮合させて分子量を上げることもできる。
重縮合反応により得られたPBTは、通常、重縮合反応槽の底部からポリマー抜出ダイに移送されてストランド状に抜き出され、水冷されながら若しくは水冷後、カッターで切断されてペレット状又はチップ状の粒状体とされる。粒状体は、引き続き公知の方法等で固相重縮合させて、その固有粘度を上ることもできる。
【0033】
<PBT>
本発明のPBTは、テレフタル酸由来の構成単位と1,4ブタンジオール由来の構成単位を含み、窒素原子含有量が0.01〜50質量ppmである。本発明のPBTにおける窒素原子含有量(質量比)で、上限は、好ましくは10ppm、より好ましくは2ppmであるのよく、下限は、好ましくは0.05ppm、より好ましくは0.1ppmであるのが良い。窒素原子含有量が上記範囲内のPBTは、上記の好ましいテレフタル酸又はテレフタル酸アルキレートと1,4ブタンジオールとを原料とすることにより得ることができる。
【0034】
本発明のPBTの固有粘度に特に制限はないが、機械的物性、ペレット化の安定性、成形性の観点からは、好ましくは0.50dL/g以上、更に好ましくは0.70dL/g以上、好ましくは1.50dL/gの以下、更に好ましくは1.35dL/g以下であるのが良い。PBTの固有粘度が上記下限以上であると成形品の機械物性の点で好ましく、
上記上限以下であると成形性の点好ましい傾向がある。
【0035】
本発明のPBTの末端カルボキシル基濃度に特に制限はないが、下限が、1当量/トンであることが好ましく、2当量/トンであることが更に好ましく、3当量/トンであることが特に好ましく、5当量/トンであることが最も好ましく、上限が、50当量/トンであることが好ましく、40当量/トンであることが更に好ましく、30当量/トンであることが特に好ましく、25当量/トンであることが最も好ましい。PBTの末端カルボキシル基濃度が上記上限以下であるとPBTの耐加水分解性を良好な傾向にあり、上記下限以上であると重縮合性が良好な傾向にある。PBTの末端カルボキシル基濃度は、樹脂を有機溶媒に溶解し、水酸化ナトリウム溶液等のアルカリ溶液を用いて滴定することにより求めることができる。
【0036】
本発明のPBTの末端ビニル基濃度に特に制限はないが、色調や重縮合性の点から、好ましくは15当量/トン以下、更に好ましくは10当量/トン以下、特には好ましくは7当量/トン以下がよい。PBTの末端ビニル基濃度は、PBTを溶媒に溶かしてから各磁気共鳴スペクトル(NMR)を測定することによって求められる。
<PBTの色調>
本発明のPBTは、色調が良好である。
【0037】
<PBT組成物>
本発明のPBTは、本発明の優れた効果を大幅に損なわない範囲で、PBT以外の成分を含んでいてもよい。具体例を挙げると、熱可塑性、熱硬化性などの各種樹脂、離型剤、充填剤、難燃剤、その他各種添加剤などが挙げられる。
熱可塑性樹脂としては、ポリエチレン、ポリプロピレン、ポリスチレン、ポリアクリロニトリル、ポリメタクリル酸エステル、ABS樹脂、ポリカーボネート、ポリアミド、ポリフェニレンサルファイド、ポリエチレンテレフタレート、液晶ポリエステル、ポリアセタール、ポリフェニレンオキサイド等が挙げられる。また、熱硬化性樹脂としては、フェノール樹脂、メラミン樹脂、シリコーン樹脂、エポキシ樹脂などが挙げられる。これらの樹脂は、1種のみ用いてもよいし、2種以上を組み合わせて使用することも出来る。このうち、熱可塑性樹脂をよく配合する。
【0038】
これらの樹脂の配合量(質量)は、本発明の優れた効果が発現されていればいくつでもよいが、樹脂全量に対して、PBTが、通常0.1質量%以上、好ましくは1質量%以上、更に好ましくは10質量%以上、通常99.9質量%以下、好ましくは99質量%以下、更に好ましくは90質量%以下がよい。
離型剤としては、特に制限されないが、例えば、2,6−ジ−t−ブチル−4−オクチルフェノール、ペンタエリスリチル−テトラキス〔3−(3',5'−t−ブチル−4'−
ヒドロキシフェニル)プロピオネート〕等のフェノール化合物、ジラウリル−3,3'−
チオジプロピオネート、ペンタエリスリチル-テトラキス(3-ラウリルチオジプロピオネート)等のチオエーテル化合物、トリフェニルホスファイト、トリス(ノニルフェニル)ホスファイト、トリス(2,4−ジ−t−ブチルフェニル)ホスファイト等の燐化合物などの抗酸化剤;パラフィンワックス、マイクロクリスタリンワックス、ポリエチレンワックス;モンタン酸やモンタン酸エステルに代表される長鎖脂肪酸およびそのエステル;シリコーンオイル等などが挙げられる。
【0039】
強化充填材としては、特に制限されないが、例えば、ガラス繊維、カーボン繊維、シリカ・アルミナ繊維、ジルコニア繊維、ホウ素繊維、窒化ホウ素繊維、窒化ケイ素チタン酸カリウム繊維、金属繊維などの無機繊維;芳香族ポリアミド繊維、フッ素樹脂繊維などの有機繊維などが挙げられる。このうち、無機充填材、特にガラス繊維が好適に使用される。強化充填材は、1種類のみ用いてもよいし、2種以上を組み合わせて使用してもよい。
【0040】
強化充填材が無機又は有機繊維である場合、その平均繊維径は、特に制限されないが、通常1〜100μm、好ましくは2〜50μm、更に好ましくは3〜30μm、特に好ましくは5〜20μmである。また、平均繊維長は、特に制限されないが、通常0.1〜20mm、好ましくは1〜10mmである。
強化充填材は、PBTとの界面密着性を向上させるため、収束剤または表面処理剤で表面処理されたものを用いるのが好ましい。収束剤または表面処理剤としては、例えば、エポキシ系化合物、アクリル系化合物、イソシアネート系化合物、シラン系化合物、チタネート系化合物などの官能性化合物が挙げられる。収束剤または表面処理剤による処理は、強化充填剤を予め表面処理しておいてもよいし、PBT組成物を調製する際に収束剤または表面処理剤と接触させてもよい。強化充填材の量は、PBT樹脂100質量部に対し、通常150質量部以下、好ましくは5〜100重量部である。
【0041】
本発明の製造方法で得られるPBTには、強化充填材以外の充填材を配合してもよい。具体的には、例えば、板状無機充填材、セラミックビーズ、アスベスト、ワラストナイト、タルク、クレー、マイカ、ゼオライト、カオリン、チタン酸カリウム、硫酸バリウム、酸化チタン、酸化ケイ素、酸化アルミニウム、水酸化マグネシウム等が挙げられる。板状無機充填材を配合することにより、成形品の異方性およびソリを低減することが出来る。板状無機充填材としては、例えば、ガラスフレーク、雲母、金属箔などを挙げることが出来る。これらの中ではガラスフレークが好適に使用される。
【0042】
難燃性を付与するために用いる難燃剤としては、特に制限されず、例えば、有機ハロゲン化合物、アンチモン化合物、リン化合物、その他の有機難燃剤、無機難燃剤などが挙げられる。有機ハロゲン化合物としては、例えば、臭素化ポリカーボネート、臭素化エポキシ樹脂、臭素化フェノキシ樹脂、臭素化ポリフェニレンエーテル樹脂、臭素化ポリスチレン樹脂、臭素化ビスフェノールA、ポリペンタブロモベンジルアクリレート等が挙げられる。アンチモン化合物としては、例えば、三酸化アンチモン、五酸化アンチモン、アンチモン酸ソーダ等が挙げられる。リン化合物としては、例えば、リン酸エステル、ポリリン酸、ポリリン酸アンモニウム、赤リン等が挙げられる。その他の有機難燃剤としては、例えば、メラミン、シアヌール酸などの窒素化合物などが挙げられる。その他の無機難燃剤としては、例えば、水酸化アルミニウム、水酸化マグネシウム、ケイ素化合物、ホウ素化合物などが挙げられる。
【0043】
その他の各種添加剤としては、特に制限されないが、例えば、酸化防止剤、耐熱安定剤などの安定剤の他、滑剤、離型剤、触媒失活剤、結晶核剤、結晶化促進剤などが挙げられる。これらの添加剤は、重縮合途中または重縮合後に添加してもよい。また、紫外線吸収剤、耐候安定剤などの安定剤、染顔料などの着色剤、帯電防止剤、発泡剤、可塑剤、耐衝撃性改良剤などを配合してもよい。
【0044】
上記のその他成分の配合方法は、特に制限されないが、例えば、ベント口から脱揮できる設備を有する1軸または2軸の押出機を混練機として使用する方法が好ましい。各成分は、付加的成分を含めて、混練機に一括して供給することが出来、あるいは、順次供給することも出来る。また、付加的成分を含めて、各成分から選ばれた2種以上の成分をあらかじめ混合しておくことも出来る。
【0045】
<PBTの成形加工>
PBTの成形加工方法は、特に制限されず、熱可塑性樹脂について一般に使用されている成形法等、具体的には、射出成形、中空成形、押し出し成形、プレス成形などを適用出来る。
本発明のPBT及びこれを含んだ組成物は、色調、熱安定性、透明性、品質安定性に優
れ、電気、電子部品、自動車用部品などの射出成形部品、フィルム、モノフィラメント、繊維などの押出し成形品用途において好適に使用できる。
【実施例】
【0046】
以下、実施例を用いて本発明の内容を更に具体的に説明するが、本発明はその要旨を越えない限り以下の実施例により限定されるものではない。
(分析方法)
<窒素原子含有量(質量ppm)の測定方法>
試料15mgを石英ボートへ採取して、微量全窒素分析装置(ダイヤインスツルメンツ
社製「TN−10型」)を用いて試料を燃焼し、燃焼・化学発光法により定量した。また
、その際に使用した標準試料は、トルエン中にアニリンを溶解し、窒素換算で0,0.5,1.0,2.0μg/mLを作製し使用した。
【0047】
<BG中の1,4−HAB、BGTF、THF量(質量ppm)及びTHF転化率>
島津製作所製ガスクロマト分析装置「島津GCC−14BPF型」にて、DB−WAXカラムを用い、修正面積百分率法により各ピークの成分の含有量を求めた。各ピークの係数は、1,4−ブタンジオールを1.000とした時に、有効炭素数に基づいて、1,4−HABは0.990、BGTFは0.922、THFは0.747として計算した。
エステル交換反応又はエステル化反応における留出液について、カールフィッシャー法(三菱化学社製「CA−03」で測定)にて水分量を求め、水分以外は有機成分とした。有機成分中のTHF量を上記ガスクロ法により求め、THF生成量とした。THF生成量をテレフタル酸又はジメチルテレフタレートに対するモル%で表し、転化率とした。
【0048】
<PBTの固有粘度、IV>
ウベローデ型粘度計を使用して以下の手順で求めた。すなわち、フェノール/テトラクロロエタン(質量比1/1)の混合溶媒を使用し、30℃において、濃度1.0g/dL
のポリマー溶液および溶媒のみの落下秒数を測定し、以下の式より求めた。
IV=((1+4KHηsp)0.5-1)/(2KHHC)
但し、ηsp=(η/η0)-1であり、ηはポリマー溶液落下秒数、η0は溶媒の落下秒
数、Cはポリマー溶液濃度(g/dL)、KHはハギンズの定数である。KHは0.33を採用した。
【0049】
<PBTの末端カルボキシル基濃度(当量/トンPBT)>
ベンジルアルコール25mLにPBT0.5gを溶解し、水酸化ナトリウムの0.01モル/Lベンジルアルコール溶液を使用して滴定した。
末端カルボキシル基濃度=(A−B)×0.1×f/W(当量/トン)
(ここで、Aは、滴定に要した0.01Nの水酸化ナトリウムのベンジルアルコール溶液の量(μL)、Bは、ブランクでの滴定に要した0.01モル/Lの水酸化ナトリウムのベンジルアルコール溶液の量(μL)、Wは、PBT試料の量(g)、fは、0.01モル/Lの水酸化ナトリウムのベンジルアルコール溶液の力価である。)
【0050】
<PBT 色調b値>
ペレット状のPBTを日本電色(株)製色差計「Z−300A型」を使用して、L、a、b表色系におけるb値で評価した。値が低いほど黄ばみが少なく色調が良好であることを示す。
【0051】
<引張試験片、曲げ試験片の成形方法>
ペレットを、120℃、2kPaで8時間減圧乾燥させた。次に乾燥したペレットを住友重機械工業社製「MINIMAT8/7A型射出成形機」にて、シリンダ温度250℃、金型温度80℃、射出保圧時間5秒、冷却20秒で成形し、引張試験片と曲げ試験片を
それぞれ得た。
引張試験片の形状は長さ55mm、厚み3mm、中央平行部長さ25mm、中央平行部幅3.2mm、両端幅6mmのダンベル状であり、曲げ試験片の形状は長さ53mm、厚み3mm、幅7mmの短冊状であった。引張試験片の形状を図1に示す。
【0052】
<成形品の引張強度 MPa>
上記で得られた引張試験片をJIS K7113に準拠して温度25℃、相対湿度50%の恒温恒湿下で、島津製作所社製「オートグラフAGS−5kNG型試験機」を用いて、チャック間距離32mm、引張試験速度10mm/分にて測定した。
【0053】
<成形品の曲げ弾性率 MPa>
上記で得られた曲げ試験片をJIS K7171に準拠して温度25℃、相対湿度50%の恒温恒湿下で、東洋精機社製「ストログラフR2型試験機」を用いて、スパン30mm、載荷速度2mm/分にて測定した。
(製造例)
参考例1から参考例5の<コハク酸醗酵液からのコハク酸精製>までは、特願2006−524976号と同様に行った。
【0054】
参考例1
<遺伝子破壊用ベクターの構築>
(A)枯草菌ゲノムDNAの抽出
LB培地[組成:トリプトン10g、イーストエキストラクト5g、NaCl5gを蒸留水1Lに溶解]10mLに、枯草菌(Bacillus subtilis ISW1214)を対数増殖期後期まで培養し、菌体を集めた。得られた菌体を10mg/mLの濃度にリゾチームを含む10mM NaCl/20mMトリス緩衝液(pH8.0)/1mM EDTA・2Na溶液0.15mLに懸濁した。
【0055】
次に、上記懸濁液にプロテナーゼKを、最終濃度が100μg/mLになるように添加し、37℃で1時間保温した。更にドデシル硫酸ナトリウムを最終濃度が0.5質量%になるように添加し、50℃で6時間保温して溶菌した。この溶菌液に、等量のフェノール/クロロフォルム溶液を添加し、室温で10分間ゆるやかに振盪した後、全量を遠心分離(5,000×g、20分間、10〜12℃)し、上清画分を分取し、酢酸ナトリウムを0.3Mとなるように添加した後、2倍量のエタノールを加え混合した。遠心分離(15,000×g、2分)により回収した沈殿物を70質量%エタノールで洗浄した後、風乾した。得られたDNAに10mMトリス緩衝液(pH7.5)−1mM EDTA・2Na溶液5mLを加え、4℃で一晩静置し、以後のPCRの鋳型DNAに使用した。
【0056】
(B)PCRによるSacB遺伝子の増幅およびクローニング
枯草菌SacB遺伝子の取得は、上記(A)で調製したDNAを鋳型とし、既に報告されている該遺伝子の塩基配列(GenBank Database AccessionNo.X02730)を基に設計した合成DNA(配列番号1および配列番号2)を用いたPCRによって行った。
【0057】
反応液組成:鋳型DNA1μL、PfxDNAポリメラーゼ(インビトロジェン社製)0.2μL、1倍濃度添付バッファー、0.3μM各々プライマー、1mM MgSO4、0.25μMdNTPsを混合し、全量を20μLとした。
反応温度条件:DNAサーマルサイクラー PTC−200(MJResearch社製)を用い、94℃で20秒、68℃で2分からなるサイクルを35回繰り返した。但し、1サイクル目の94℃での保温は1分20秒、最終サイクルの68℃での保温は5分とした。
【0058】
増幅産物の確認は、0.75質量%アガロース(SeaKem GTG agarose:FMCBioProducts製)ゲル電気泳動により分離後、臭化エチジウム染色により可視化することにより行い、約2kbの断片を検出した。ゲルからの目的DNA断片の回収は、QIAQuickGel Extraction Kit(QIAGEN製)を用いて行った。
【0059】
回収したDNA断片は、T4ポリヌクレオチドキナーゼ(T4 Polynucleo
tide Kinase:宝酒造製)により5'末端をリン酸化した後、ライゲーションキットver.2(宝酒造製)を用いて大腸菌ベクター(pBluescriptII:STRATEGENE製)のEcoRV部位に結合し、得られたプラスミドDNAで大腸菌(DH5α株)を形質転換した。この様にして得られた組換え大腸菌を50μg/mLアンピシリンおよび50μg/mLX−Galを含むLB寒天培地[トリプトン10g、イーストエキストラクト5g、NaCl5g及び寒天15gを蒸留水1Lに溶解]に塗抹した。
【0060】
この培地上で白色のコロニーを形成したクローンを、次に50μg/mLアンピシリンおよび10質量%ショ糖を含むLB寒天培地に移し37℃で24時間培養した。これらのクローンのうち、ショ糖を含む培地で生育できなかったものについて、常法により液体培養した後、プラスミドDNAを精製した。SacB遺伝子が大腸菌内で機能的に発現する株は、ショ糖含有培地にて生育不能となるはずである。得られたプラスミドDNAを制限酵素SalIおよびPstIで切断することにより、約2kbの挿入断片が認められ、該プラスミドをpBS/SacBと命名した。
【0061】
(C)クロラムフェニコール耐性SacBベクターの構築
大腸菌プラスミドベクターpHSG396(宝酒造:クロラムフェニコール耐性マーカー)500ngに制限酵素PshBI10ユニットを37℃で1時間反応させた後、フェノール/クロロフォルム抽出およびエタノール沈殿により回収した。クレノウフラグメント(Klenow Fragment:宝酒造製)により両末端を平滑化した後、ライゲーションキットver.2(宝酒造製)を用いてMluIリンカー(宝酒造)を連結、環状化させ、大腸菌(DH5α株)を形質転換した。この様にして得られた組換え大腸菌を34μg/mLクロラムフェニコールを含むLB寒天培地に塗抹した。得られたクローンから常法によりプラスミドDNAを調製し、制限酵素MluIの切断部位を有するクローンを選抜し、pHSG396Mluと命名した。
【0062】
一方、上記(B)にて構築したpBS/SacBを制限酵素SalIおよびPstIで切断した後、クレノウフラグメントにて末端を平滑化した。これにライゲーションキットver.2(宝酒造製)を用いてMluIリンカーを連結したのち、0.75質量%アガロースゲル電気泳動によりSacB遺伝子を含む約2.0kbのDNA断片を分離、回収した。このSacB遺伝子断片を、制限酵素MluI切断後、アルカリフォスファターゼ(Alkaline Phosphatase Calf intestine:宝酒造)にて末端を脱リン酸化したpHSG396Mlu断片とライゲーションキットver.2(宝酒造製)を用いて連結させ、大腸菌(DH5α株)を形質転換した。この様にして得られた組換え大腸菌を34μg/mLクロラムフェニコールを含むLB寒天培地に塗抹した。こうして得られたコロニーを、次に34μg/mLクロラムフェニコールおよび10質量%ショ糖を含むLB寒天培地に移し37℃で24時間培養した。これらのクローンのうち、ショ糖を含む培地で生育できなかったものについて、常法によりプラスミドDNAを精製した。こうして得られたプラスミドDNAをMluI切断により解析した結果、約2.0kbの挿入断片を持つことが確認され、これをpCMB1と命名した。
【0063】
(D)カナマイシン耐性遺伝子の取得
カナマイシン耐性遺伝子の取得は、大腸菌プラスミドベクターpHSG299(宝酒造:カナマイシン耐性マーカー)のDNAを鋳型とし、配列番号3および配列番号4で示した合成DNAをプライマーとしたPCR法によって行った。
反応液組成:鋳型DNA1ng、PyrobestDNAポリメラーゼ(宝酒造)0.1μL、1倍濃度添付バッファー、0.5μM各々プライマー、0.25μMdNTPsを混合し、全量を20μLとした。
【0064】
反応温度条件:DNAサーマルサイクラー PTC−200(MJResearch社製)を用い、94℃で20秒、62℃で15秒、72℃で1分20秒からなるサイクルを20回繰り返した。但し、1サイクル目の94℃での保温は1分20秒、最終サイクルの72℃での保温は5分とした。
増幅産物の確認は、0.75質量%アガロース(SeaKem GTG agarose:FMCBioProducts製)ゲル電気泳動により分離後、臭化エチジウム染色により可視化することにより行い、約1.1kbの断片を検出した。ゲルからの目的DNA断片の回収は、QIAQuickGel Extraction Kit(QIAGEN製)を用いて行った。回収したDNA断片は、T4 ポリヌクレオチドキナーゼ(T4Polynucleotide Kinase:宝酒造製)により5'末端をリン酸化した。
【0065】
(E)カナマイシン耐性SacBベクターの構築
上記(C)で構築したpCMB1を制限酵素Van91IおよびScaIで切断して得られた約3.5kbのDNA断片を0.75質量%アガロースゲル電気泳動により分離、回収した。これを上記(D)で調製したカナマイシン耐性遺伝子と混合し、ライゲーションキットver.2(宝酒造製)を用いて連結し、得られたプラスミドDNAで大腸菌(DH5α株)を形質転換した。この様にして得られた組換え大腸菌を50μg/mLカナマイシンを含むLB寒天培地に塗抹した。
【0066】
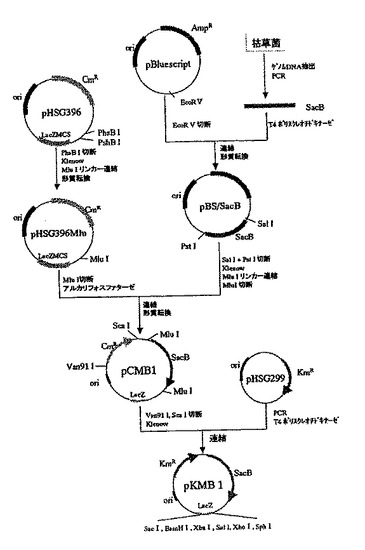
このカナマイシン含有培地上で生育した株は、ショ糖含有培地にて生育不能であることが確認された。また、同株から調製したプラスミドDNAは、制限酵素HindIII消化
により354、473、1807、1997bpの断片を生じたことから、図2に示した構造に間違いがないと判断し、該プラスミドをpKMB1と命名した。
【0067】
参考例2
<LDH遺伝子破壊株の作製>
(A)ブレビバクテリウム・フラバムMJ233−ES株ゲノムDNAの抽出
A培地[尿素 2g、(NH4)2SO4 7g、KH2PO4 0.5g、K2HPO40.5g、MgSO4・7H2O 0.5g、FeSO4・7H2O 6mg、MnSO4・4−5H2O6mg、ビオチン 200μg、チアミン 100μg、イーストエキストラクト 1g
、カザミノ酸 1g、グルコース20g、蒸留水1Lに溶解]10mLに、ブレビバクテ
リウム・フラバムMJ−233株を対数増殖期後期まで培養し、得られた菌体から上記参考例1の(A)に示す方法にてゲノムDNAを調製した。
【0068】
(B)ラクテートデヒドロゲナーゼ遺伝子のクローニング
MJ233株ラクテートデヒドロゲナーゼ遺伝子の取得は、上記(A)で調製したDNAを鋳型とし、特開平11−206385号公報に記載の該遺伝子の塩基配列を基に設計した合成DNA(配列番号5および配列番号6)を用いたPCRによって行った。
反応液組成:鋳型DNA1μL、TaqDNAポリメラーゼ(宝酒造)0.2μL、1倍濃度添付バッファー、0.2μM各々プライマー、0.25μMdNTPsを混合し、全量を20μLとした。
【0069】
反応温度条件:DNAサーマルサイクラー PTC−200(MJResearch社製)を用い、94℃で20秒、55℃で20秒、72℃で1分からなるサイクルを30回繰り返した。但し、1サイクル目の94℃での保温は1分20秒、最終サイクルの72℃での保温は5分とした。
増幅産物の確認は、0.75質量%アガロース(SeaKem GTG agarose:FMCBioProducts製)ゲル電気泳動により分離後、臭化エチジウム染色により可視化することにより行い、約0.95kbの断片を検出した。ゲルからの目的DNA断片の回収は、QIAQuickGel Extraction Kit(QIAGEN製)を用いて行った。
【0070】
回収したDNA断片を、PCR産物クローニングベクターpGEM−TEasy(Promega製)と混合し、ライゲーションキットver.2(宝酒造製)を用いて連結後、得られたプラスミドDNAで大腸菌(DH5α株)を形質転換した。この様にして得られた組換え大腸菌を50μg/mLアンピシリンおよび50μg/mLX−Galを含むLB寒天培地に塗抹した。
この培地上で白色のコロニーを形成したクローンを、常法により液体培養した後、プラスミドDNAを精製した。得られたプラスミドDNAを制限酵素SacIおよびSphIで切断することにより、約1.0kbの挿入断片が認められ、これをpGEMT/CgLDHと命名した。
【0071】
(C)ラクテートデヒドロゲナーゼ遺伝子破壊用プラスミドの構築
上記(B)で作製したpGEMT/CgLDHを制限酵素EcoRVおよびXbaIで切断することにより約0.25kbからなるラクテートデヒドロゲナーゼのコーディング領域を切り出した。残った約3.7kbのDNA断片の末端をクレノウフラグメントにて平滑化し、ライゲーションキットver.2(宝酒造製)を用いて環状化させ、大腸菌(DH5α株)を形質転換した。この様にして得られた組換え大腸菌を50μg/mLアンピシリンを含むLB寒天培地に塗抹した。この培地上で生育した株を、常法により液体培養した後、プラスミドDNAを精製した。得られたプラスミドDNAを制限酵素SacIおよびSphIで切断することにより、約0.75kbの挿入断片が認められたクローンを選抜し、これをpGEMT/ΔLDHと命名した。
【0072】
次に、上記pGEMT/ΔLDHを制限酵素SacIおよびSphIにて切断して生じる約0.75kbのDNA断片を、0.75質量%アガロースゲル電気泳動により分離、回収し、欠損領域を含むラクテートデヒドロゲナーゼ遺伝子断片を調製した。このDNA断片を、制限酵素SacIおよびSphIにて切断した参考例1にて構築したpKMB1と混合し、ライゲーションキットver.2(宝酒造製)を用いて連結後、得られたプラスミドDNAで大腸菌(DH5α株)を形質転換した。この様にして得られた組換え大腸菌を50μg/mLカナマイシンおよび50μg/mLX−Galを含むLB寒天培地に
塗抹した。
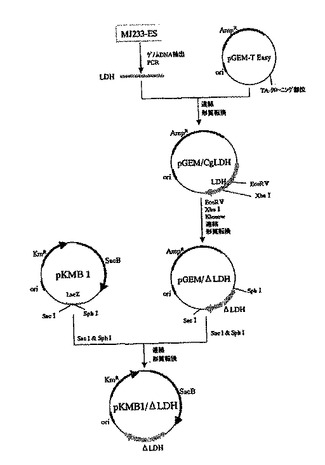
この培地上で白色のコロニーを形成したクローンを、常法により液体培養した後、プラスミドDNAを精製した。得られたプラスミドDNAを制限酵素SacIおよびSphIで切断することにより、約0.75kbの挿入断片が認められたものを選抜し、これをpKMB1/ΔLDHと命名した(図3)。
【0073】
(D)ブレビバクテリウム・フラバムMJ233−ES株由来ラクテートデヒドロゲナーゼ遺伝子破壊株の作製
ブレビバクテリウム・フラバムMJ−233株の形質転換に用いるプラスミドDNAは、pKMB1/ΔLDHを用いて塩化カルシウム法(Journal of Molecular Biology,53,159,1970)により形質転換した大腸菌JM110株から調製した。
【0074】
ブレビバクテリウム・フラバムMJ233−ES株の形質転換は、電気パルス法(Res.Microbiol., Vol.144, p.181−185, 1993)によ
って行い、得られた形質転換体をカナマイシン 50μg/mLを含むLBG寒天培地[
トリプトン10g、イーストエキストラクト5g、NaCl5g、グルコース 20g、
及び寒天15gを蒸留水1Lに溶解]に塗抹した。
【0075】
この培地上に生育した株は、pKMB1/ΔLDHがブレビバクテリウム・フラバムMJ233−ES株菌体内で複製不可能なプラスミドであるため、該プラスミドのラクテートデヒドロゲナーゼ遺伝子とブレビバクテリウム・フラバムMJ−233株ゲノム上の同遺伝子との間で相同組み換えを起こした結果、同ゲノム上に該プラスミドに由来するカナマイシン耐性遺伝子およびSacB遺伝子が挿入されているはずである。
【0076】
次に、上記相同組み換え株をカナマイシン50μg/mLを含むLBG培地にて液体培養した。この培養液の菌体数約100万相当分を10質量%ショ糖含有LBG培地に塗抹にした。結果、2回目の相同組み換えによりSacB遺伝子が脱落しショ糖非感受性となったと考えられる株約10個得た。
この様にして得られた株の中には、そのラクテートデヒドロゲナーゼ遺伝子がpKMB1/ΔLDHに由来する変異型に置き換わったものと野生型に戻ったものが含まれる。ラクテートデヒドロゲナーゼ遺伝子が変異型であるか野生型であるかの確認は、LBG培地にて液体培養して得られた菌体を直接PCR反応に供し、ラクテートデヒドロゲナーゼ遺伝子の検出を行うことによって容易に確認できる。ラクテートデヒドロゲナーゼ遺伝子をPCR増幅するためのプライマー(配列番号7および配列番号8)を用いて分析すると、野生型では720bp、欠失領域を持つ変異型では471bpのDNA断片を認めるはずである。
上記方法にてショ糖非感受性となった菌株を分析した結果、変異型遺伝子のみを有する株を選抜し、該株をブレビバクテリウム・フラバムMJ233/ΔLDHと命名した。
【0077】
(E)ラクテートデヒドロゲナーゼ活性の確認
上記(D)で作製したブレビバクテリウム・フラバムMJ233/ΔLDH株をA培地に植菌し、30℃で15時間好気的に振とう培養した。得られた培養物を遠心分離(3,000×g、4℃、20分間)して菌体を回収後、ナトリウム−リン酸緩衝液[組成:50mMリン酸ナトリウム緩衝液(pH7.3)]で洗浄した。
【0078】
次いで、洗浄菌体0.5g(湿重量)を上記ナトリウム−リン酸緩衝液2mLに懸濁し、氷冷下で超音波破砕器(ブランソン社製)にかけ菌体破砕物を得た。該破砕物を遠心分離(10,000×g,4℃,30分間)し、上清を粗酵素液として得た。対照として、ブレビバクテリウム・フラバムMJ233−ES株の粗酵素液を同様に調製し、以下の活性測定に供した。
【0079】
ラクテートデヒドロゲナーゼ酵素活性の確認は、両粗酵素液について、ピルビン酸を基質とした乳酸の生成に伴い、補酵素NADHがNAD+に酸化されるのを、340nmの
吸光度変化として測定した[L. Kanarek and R. L. Hill, J.Biol. Chem. 239, 4202(1964)]。反応は、50mM カリウム−リン酸緩衝液(pH7.2)、10mM ピルビン酸、0.4mMNADH存在下、37
℃にて行った。その結果、ブレビバクテリウム・フラバムMJ233−ES株から調製された粗酵素液におけるラクテートデヒドロゲナーゼ活性に対し、ブレビバクテリウム・フラバムMJ233/ΔLDH株から調製された粗酵素液におけるラクテートデヒドロゲナーゼ活性は、10分の1以下であった。
【0080】
参考例3
<コリネ型細菌発現ベクターの構築>
(A)コリネ型細菌用プロモーター断片の調製
コリネ型細菌で強力なプロモーター活性を有することが報告された特開平7−95891号公報の配列番号4に記載のDNA断片(以降TZ4プロモーターと称する)を利用することとした。本プロモーター断片の取得は、参考例2の(A)で調製したブレビバクテリウム・フラバムMJ233ゲノムDNAを鋳型とし、特開平7−95891号公報の配列番号4に記載の配列を基に設計した合成DNA(配列番号9および配列番号10)を用いたPCRによって行った。
【0081】
反応液組成:鋳型DNA1μL、PfxDNAポリメラーゼ(インビトロジェン社製) 0.2μL、1倍濃度添付バッファー、0.3μM各々プライマー、1mM MgSO4、0.25μMdNTPsを混合し、全量を20μLとした。
反応温度条件:DNAサーマルサイクラー PTC−200(MJResearch社製)を用い、94℃で20秒、60℃で20秒、72℃で30秒からなるサイクルを35回繰り返した。但し、1サイクル目の94℃での保温は1分20秒、最終サイクルの72℃での保温は2分とした。
【0082】
増幅産物の確認は、2.0質量%アガロース(SeaKem GTG agarose:FMCBioProducts製)ゲル電気泳動により分離後、臭化エチジウム染色により可視化することにより行い、約0.25kbの断片を検出した。ゲルからの目的DNA断片の回収は、QIAQuickGel Extraction Kit(QIAGEN製)を用いて行った。
【0083】
回収したDNA断片は、T4ポリヌクレオチドキナーゼ(T4 Polynucleo
tide Kinase:宝酒造製)により5'末端をリン酸化した後、ライゲーションキットver.2(宝酒造製)を用いて大腸菌ベクターpUC19(宝酒造)のSmaI部位に結合し、得られたプラスミドDNAで大腸菌(DH5α株)を形質転換した。この様にして得られた組換え大腸菌を50μg/mLアンピシリンおよび50μg/mLX−Galを含むLB寒天培地に塗抹した。
【0084】
この培地上で白色のコロニーを形成した6クローンについて、常法により液体培養した後、プラスミドDNAを精製し、塩基配列を決定した。これ中でTZ4プロモーターがpUC19のlacプロモーターと逆方向に転写活性を有するように挿入されたクローンを選抜し、これをpUC/TZ4と命名した。
次に、pUC/TZ4を制限酵素BamHIおよびPstIで切断して調製したDNA断片に、5’末端がリン酸化された合成DNA(配列番号11および配列番号12)から成り、両末端にそれぞれBamHIとPstIに対する粘着末端を有するDNAリンカーを混合し、ライゲーションキットver.2(宝酒造製)を用いて連結後、得られたプラスミドDNAで大腸菌(DH5α株)を形質転換した。本DNAリンカーには、リボソーム結合配列(AGGAGG)およびその下流に配したクローニングサイト(上流から順に、PacI、NotI、ApaI)が含まれている。
【0085】
この培地上で白色のコロニーを形成したクローンを、常法により液体培養した後、プラスミドDNAを精製した。得られたプラスミドDNAの中から制限酵素NotIによって切断されるものを選抜し、これをpUC/TZ4−SDと命名した。
この様にして構築したpUC/TZ4−SDを制限酵素PstIで切断後、クレノウフラグメントにて末端を平滑化し、次いで制限酵素KpnIで切断することにより生じた約0.3kbのプロモーター断片を、2.0質量%アガロースゲル電気泳動により分離、回収した。
【0086】
(B)コリネ型細菌発現ベクターの構築
コリネ型細菌にて安定的に自立複製可能なプラスミドとして、特開平12−93183号公報記載のpHSG298par−repを利用する。本プラスミドは、ブレビバクテリウム・スタチオニスIFO12144株が保有する天然型プラスミドpBY503の複製領域および安定化機能を有する領域と大腸菌ベクターpHSG298(宝酒造)に由来するカナマイシン耐性遺伝子および大腸菌の複製領域を備える。pHSG298par−repを制限酵素SseIで切断後、クレノウフラグメントにて末端を平滑化し、次いで制限酵素KpnIで切断することによって調製したDNAを、上記(A)で調製したTZ4プロモーター断片と混合し、ライゲーションキットver.2(宝酒造製)を用いて連結後、得られたプラスミドDNAで大腸菌(DH5α株)を形質転換した。この様にして得られた組換え大腸菌を50μg/mLカナマイシンを含むLB寒天培地に塗抹した。
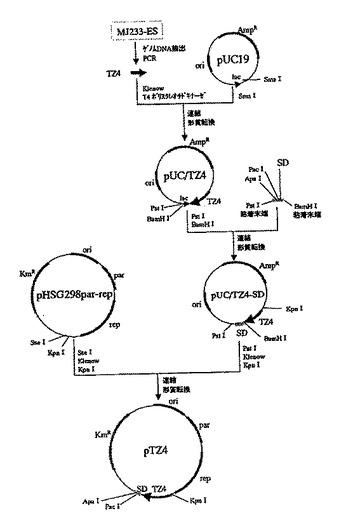
この培地上で生育した株を、常法により液体培養した後、プラスミドDNAを精製した。得られたプラスミドDNAの中から制限酵素NotIによって切断されるものを選抜し、該プラスミドをpTZ4と命名した(図3に構築手順を示した)。
【0087】
参考例4
<ピルベートカルボキシラーゼ活性増強株の作製>
(A)ピルベートカルボキシラーゼ遺伝子の取得
ブレビバクテリウム・フラバムMJ233株由来ピルベートカルボキシラーゼ遺伝子の取得は、参考例2の(A)で調製したDNAを鋳型とし、全ゲノム配列が報告されているコリネバクテリウム・グルタミカム ATCC13032株の該遺伝子の配列(GenBank Database Accession No.AP005276)を基に設計した合成DNA(配列番号13および配列番号14)を用いたPCRによって行った。
【0088】
反応液組成:鋳型DNA1μL、PfxDNAポリメラーゼ(インビトロジェン社製) 0.2μL、1倍濃度添付バッファー、0.3μM各々プライマー、1mM MgSO4、0.25μMdNTPsを混合し、全量を20μLとした。
反応温度条件:DNAサーマルサイクラー PTC−200(MJResearch社製)を用い、94℃で20秒、68℃で4分からなるサイクルを35回繰り返した。但し、1サイクル目の94℃での保温は1分20秒、最終サイクルの68℃での保温は10分とした。PCR反応終了後、Takara Ex Taq(宝酒造)を0.1μL加え、更に72℃で30分保温した。
【0089】
増幅産物の確認は、0.75質量%アガロース(SeaKem GTG agarose:FMCBioProducts製)ゲル電気泳動により分離後、臭化エチジウム染色により可視化することにより行い、約3.7kbの断片を検出した。ゲルからの目的DNA断片の回収は、QIAQuickGel Extraction Kit(QIAGEN製)を用いて行った。
【0090】
回収したDNA断片を、PCR産物クローニングベクターpGEM−TEasy(Promega製)と混合し、ライゲーションキットver.2(宝酒造製)を用いて連結後、得られたプラスミドDNAで大腸菌(DH5α株)を形質転換した。この様にして得られた組換え大腸菌を50μg/mLアンピシリンおよび50μg/mLX−Galを含むLB寒天培地に塗抹した。
【0091】
この培地上で白色のコロニーを形成したクローンを、常法により液体培養した後、プラスミドDNAを精製した。得られたプラスミドDNAを制限酵素PacIおよびApaIで切断することにより、約3.7kbの挿入断片が認められ、これをpGEM/MJPCと命名した。
pGEM/MJPCの挿入断片の塩基配列は、アプライドバイオシステム社製塩基配列解読装置(モデル377XL)およびビックダイターミネーターサイクルシークエンスキットver3を用いて決定した。その結果得られたDNA塩基配列および推測されるアミノ酸配列を配列番号15に記載する。また、アミノ酸配列のみを配列番号16に記載する。本アミノ酸配列はコリネバクテリウム・グルタミカムATCC13032株由来のそれと極めて高い相同性(99.4%)を示すことから、pGEM/MJPCの挿入断片がブレビバクテリウム・フラバムMJ233株由来のピルベートカルボキシラーゼ遺伝子であると断定した。
【0092】
(B)ピルベートカルボキシラーゼ活性増強用プラスミドの構築
上記(A)で作製したpGEM/MJPCを制限酵素PacIおよびApaIで切断することにより生じる約3.7kbからなるピルベートカルボキシラーゼ遺伝子断片を、0.75質量%アガロースゲル電気泳動により分離、回収した。
このDNA断片を、制限酵素PacIおよびApaIにて切断した、参考例3にて構築したpTZ4と混合し、ライゲーションキットver.2(宝酒造製)を用いて連結後、得られたプラスミドDNAで大腸菌(DH5α株)を形質転換した。この様にして得られた組換え大腸菌を50μg/mLカナマイシンを含むLB寒天培地に塗抹した。
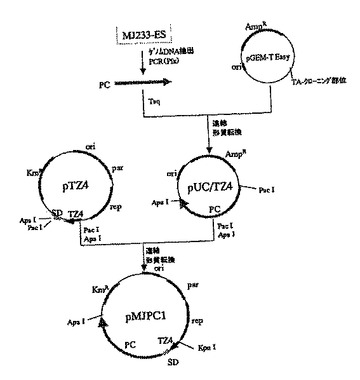
この培地上で生育した株を、常法により液体培養した後、プラスミドDNAを精製した。得られたプラスミドDNAを制限酵素PacIおよびApaIで切断することにより、約3.7kbの挿入断片が認められたものを選抜し、これをpMJPC1と命名した(図5)。
【0093】
(C)ブレビバクテリウム・フラバムMJ233/ΔLDH株への形質転換
ブレビバクテリウム・フラバムMJ233株内で複製可能なpMJPC1による形質転換用のプラスミドDNAは、上記(B)で形質転換した大腸菌(DH5α株)から調製した。
ブレビバクテリウム・フラバムMJ233/ΔLDH株への形質転換は、電気パルス法(Res. Microbiol., Vol.144,p.181−185, 1993)によって行い、得られた形質転換体をカナマイシン 50μg/mLを含むLBG寒天培
地[トリプトン10g、イーストエキストラクト5g、NaCl5g、グルコース 20
g、及び寒天15gを蒸留水1Lに溶解]に塗抹した。
この培地上に生育した株から、常法により液体培養した後、プラスミドDNAを抽出、制限酵素切断による解析を行った結果、同株がpMJPC1を保持していることを確認し、該株をブレビバクテリウム・フラバムMJ233/PC/ΔLDH株と命名した。
【0094】
(D)ピルベートカルボキシラーゼ酵素活性
上記(C)で得られた形質転換株ブレビバクテリウム・フラバムMJ233/PC/ΔLDH株をグルコース2質量%、カナマイシン25mg/Lを含むA培地100mlで終夜培養を行った。得られた菌体を集菌後、50mMリン酸カリウム緩衝液(pH7.5)50mlで洗浄し、同組成の緩衝液20mlに再度懸濁させた。懸濁液をSONIFIER350(BRANSON製)で破砕し、遠心分離した上清を無細胞抽出液とした。得られた無細胞抽出液を用いピルベートカルボキシラーゼ活性を測定した。酵素活性の測定は100mMTris/HCl緩衝液(pH7.5)、 0.1mg/10mlビオチン、
5mM塩化マグネシウム、50mM 炭酸水素ナトリウム、5mM ピルビン酸ナトリウム
、5mM アデノシン三リン酸ナトリウム、0.32mM NADH、20units/
1.5mlリンゴ酸デヒドロゲナーゼ(WAKO製、酵母由来)及び酵素を含む反応液中で25℃で反応させることにより行った。1Uは1分間に1μmolのNADHの減少を触媒する酵素量とした。ピルベートカルボキシラーゼを発現させた無細胞抽出液における比活性は0.2U/mg蛋白質であった。尚親株であるMJ233/△LDH株をA培地を用いて同様に培養した菌体では、本活性測定方法によりピルベートカルボキシラーゼ活性
は検出されなかった。
【0095】
参考例5
<発酵液の調製>
尿素:4g、硫酸アンモニウム:14g、リン酸1カリウム:0.5g、リン酸2カリウム0.5g、硫酸マグネシウム・7水和物:0.5g、硫酸第一鉄・7水和物:20mg、硫酸マンガン・水和物:20mg、D−ビオチン:200μg、塩酸チアミン:200μg、酵母エキス:1g、カザミノ酸:1g、及び蒸留水:1000mLの培地100mLを500mLの三角フラスコにいれ、120℃、20分加熱滅菌した。これを室温まで冷やし、あらかじめ滅菌した50質量%グルコース水溶液を4mL、無菌濾過した5質量%カナマイシン水溶液を50μL添加し、参考例4(C)で作製したブレビバクテリウム・フラバムMJ233/PC/ΔLDH株を接種して24時間30℃にて種培養した。
【0096】
尿素:12g、硫酸アンモニウム:42g、リン酸1カリウム:1.5g、リン酸2カリウム1.5g、硫酸マグネシウム・7水和物:1.5g、硫酸第一鉄・7水和物:60mg、硫酸マンガン・水和物:60mg、D−ビオチン:600μg、塩酸チアミン:600μg、酵母エキス3g、カザミノ酸3g、消泡剤(アデカノールLG294:旭電化製):1mL及び蒸留水:2500mLの培地を5Lの発酵糟に入れ、120℃、20分加熱滅菌した。これを室温まで冷やした後、あらかじめ滅菌した12質量%グルコース水溶液を500mL添加し、これに前述の種培養液を全量加えて、30℃に保温した。通気は毎分500mL、攪拌は毎分500回転で本培養を行った。12時間後にグルコースがほぼ消費されていた。
【0097】
硫酸マグネシウム・7水和物:1.5g、硫酸第一鉄・7水和物:60mg、硫酸マンガン・水和物:60mg、D−ビオチン:600μg、塩酸チアミン:600μg、消泡剤(アデカノールLG294:旭電化製):5ml及び蒸留水:1.5Lの培地を3Lの三角フラスコに入れ、120℃、20分加熱滅菌した。室温まで冷やした後、上記の本培養により得られた培養液を10000g、5分の遠心分離により集菌した菌体を添加して、O.D.(660nm)が60になるように再懸濁した。この懸濁液1.5Lとあらかじめ滅菌した20質量%グルコース溶液1.5Lを5Lのジャーファーメンターに入れて混合し、35℃に保温した。pHは2M炭酸アンモニウムを用いて7.6に保ち、毎分500mLで通気、毎分300回転で攪拌しながら反応を行った。反応開始後約50時間でグルコースがほぼ消費されていた。コハク酸が57g/L蓄積されていた。この発酵液を10000g、5分間の遠心分離、限外濾過(日東電工(株)製 NTU−3000−C1R)により菌体と上清に分離した。以上の操作を30回行うことにより、コハク酸発酵液上清を103L得ることが出来た。
【0098】
<コハク酸醗酵液からのコハク酸精製>
上記のようにして得られたコハク酸発酵液上清を103L(コハク酸含有量5.87kg)を、減圧しながらジャケット付き攪拌槽にて濃縮し、コハク酸の濃度が32.9質量
%、アンモニア11.9質量%の濃縮液:17.8kg(計算値)を得た。これに酢酸(ダイセル化学社製)を8.58kg加えて30℃まで冷却し、更にメタノール(キシダ化学社製)を4.0kg加えて15℃まで冷却し1時間攪拌した後、20℃にて4時間攪拌を継続した。
【0099】
結晶が析出しており、これを遠心ろ過器にてろ過を行い、コハク酸を74.6質量%、酢酸3.5質量%、アンモニア12.2質量%を含有する結晶4.95kgを得た。
酢酸11.3kgに得られた結晶4.9kgを入れ、85℃にて溶解し、直ちに20℃まで冷却した。既に結晶は析出していたが、そのまま更に3時間攪拌を続けた後、遠心ろ過器にてろ過を行い、コハク酸87.9質量%、酢酸8.4質量%、アンモニア0.6質
量%を含有する結晶2.44kgを得た。
【0100】
5℃に冷やした脱塩水3.5Lにて得られた結晶を懸洗し、これを遠心ろ過器にてろ過すると、コハク酸90質量%、酢酸1.7質量%、アンモニア0.05質量%(およそ500質量ppm)含有する2.08kgの結晶が得られた。
この粗コハク酸結晶2.0kgを28.5Lの脱塩水に溶解し、1Lのイオン交換樹脂(三菱化学社製SK1BH)をつめた塔にSV=2にて通液し、約33Lの処理液を得た。これを減圧したロータリーエバポレータに連続フィードしながら、およそ5.2Lまで濃縮した。この段階で既に結晶が析出していた。更に、5℃に冷却し、2時間攪拌を継続した後、これをろ過すると、コハク酸96.7質量%の結晶1.76kgを得た。 これを真空乾燥機にて乾燥すると1.68kgのコハク酸を得る事が出来た。
【0101】
<1,4−ブタンジオールの製造>
上記のような方法で得られたバイオマス資源由来コハク酸を用いて、以下の方法で1,4−ブタンジオールを得た。
バイオマス資源由来コハク酸100重量部、メタノール317重量部ならびに濃硫酸(97質量%)2重量部の混合液を、還流下で2時間攪拌させた。反応液を冷却後、炭酸水素ナトリウム3.6重量部を添加して60℃で30分間反応液を攪拌させた。常圧下での蒸留ならびにその蒸留残をろ過後、減圧蒸留することによりコハク酸ジメチル(収率93%)を得た。得られたコハク酸ジメチル100重量部をCuO−ZnO触媒(ズードケミー社製「T―8402」)15重量部存在下、仕込みコハク酸ジメチルに対して約4倍の体積容量を持つオートクレーブ(ハステロイC)を用いて水素5MPa加圧下で攪拌させながら1時間かけて230℃まで昇温させた。その後、230℃で15MPaの水素加圧下9時間反応液を攪拌させた。反応液を冷却後、脱ガスを行った。反応液からろ過により触媒を除去した。ろ液を減圧蒸留することにより精製1,4−ブタンジオールを得た(収率81%)。製造された精製1,4−ブタンジオール中には窒素原子が0.7質量ppm含まれたが、硫黄原子は含まれていなかった。また、1,4−ブタンジオール中には酸化生成物である2-(4-ヒドロキシブチルオキシ)テトラヒドロフランが584質量ppm含有されていた。
【0102】
[実施例1]
攪拌装置、窒素導入口、加熱装置、温度計、留出管、減圧用排気口を備えた反応容器に、ジメチルテレフタレート(帝人製)132質量部、窒素原子0.7質量ppmを含有する製造例1で得られたBG74質量部及び触媒としてテトラブチルチタネートをあらかじめ6質量%溶解させた1,4―ブタンジオール溶液1.7質量部を仕込み、窒素―減圧置換によって系内を窒素雰囲気下にした。
【0103】
次に、系内を撹拌しながら150℃まで加温後、215℃に昇温しながらエステル交換反応によって生成するメタノールを留出させつつ3時間反応した。次に、1.5時間かけて245℃まで昇温するとともに、1.5時間かけて0.07kPaになるように減圧し、同減圧度で1.5時間重縮合反応を行い、反応系を常圧に戻し重縮合を終了した。得られたPBTを反応槽の底部からストランドとして抜き出し、10℃の水中を潜らせた後、カッターでストランドをカットすることによりペレット状のPBTを得た。
【0104】
得られたPBT中の窒素原子含有量は、0.4質量ppm、PBTの固有粘度(IV)は1.06dL/g、末端カルボキシル基量は21当量/トンあった。減圧開始から重縮合終了までを重縮合時間として、固有粘度/重縮合時間を重縮合速度とした。
重縮合速度は0.35dL/g/hであった。
THF転化率は、エステル交換反応中の留出液をドライアイストラップで冷却採取したものについてTHF量を分析し、ジメチルテレフタレートあたりのモル%で表した。2モ
ル%であった。結果を表1に示す。
【0105】
[比較例1]
実施例1においてBGを化石燃料原料由来のBG(三菱化学製BG)に変え、重縮合時間を表1に示す時間に変えた以外は実施例1と同様に行い、PBTを得た。各種分析結果を表1に示す。
[実施例2]
攪拌装置、窒素導入口、加熱装置、温度計及び減圧用排気口を備えた反応容器に、テレフタル酸113質量部、窒素原子0.7質量ppmを含有する製造例1で得られたBG183質量部及び触媒としてテトラブチルチタネートをあらかじめ6質量%溶解させた1,4―ブタンジオール溶液0.7質量部を仕込み、窒素―減圧置換によって系内を窒素雰囲気下にした。
【0106】
次に、系内を撹拌しながら150℃まで加温後、圧力を78kPaになるよう減圧した後、220℃に1時間で昇温させて、さらに2時間生成する水を留出させつつエステル化反応した後窒素ガスにて常圧に戻した。
次に、酢酸マグネシウム4水塩を水に溶解し、さらにBGに溶解させた酢酸マグネシウム4水塩1質量%の1,4―ブタンジオール溶液(酢酸マグネシウム4水塩、水、1,4―ブタンジオールの質量比は1:2:97)1.3質量部を添加した。
【0107】
次に、1.5時間かけて245℃まで昇温するとともに、1.5時間かけて0.07kPaになるように減圧し、同減圧度で1.1時間重縮合反応を行い、反応系を常圧に戻し重縮合を終了した。得られたPBTを反応槽の底部からストランドとして抜き出し、10℃の水中を潜らせた後、カッターでストランドをカットすることによりペレット状のPBTを得た。
【0108】
酢酸マグネシウム添加後の減圧開始から重縮合終了までを重縮合時間として、固有粘度/重縮合時間を重縮合速度とした。重縮合速度は0.35dL/g/hであった。THF転化率は、エステル化反応中の留出液をドライアイストラップで冷却採取したものについてTHF量を分析し、仕込みテレフタル酸あたりのモル%で表した。54モル%であった。
得られたぺレット状のPBTを射出成形にて引張試験片、曲げ試験片に成形し引張強度と曲げ弾性率を測定した。
各種分析結果を表1に示す。
【0109】
[比較例2]
実施例2においてBGを化石燃料原料由来のBG(三菱化学製)に変え、重縮合時間を表1に示す時間に変えた以外は実施例2と同様に行い、PBTを得た。各種分析結果を表1に示す。
【0110】
[比較例3]
実施例2においてBGを化石燃料原料由来のBG(東燃化学製)に変え、重縮合時間を表1に示す時間に変えた以外は実施例2と同様に行い、PBTを得た。各種分析結果を表1に示す。
表1より、実施例は、重縮合速度が大きく、THF化への転化率が小さいこと、及び実施例のPBTは窒素含有量が少なく、色調が良好であること、等がわかる。
【産業上の利用可能性】
【0111】
本発明のPBTは、色調良好である。また、本発明のPBTの製法は、効率的に、THFへの転化が少ない。更に、バイオマス資源の有効利用にも寄与している。
【0112】
【表1】

【図面の簡単な説明】
【0113】
【図1】引張試験片の形状 単位mm。
【図2】pKMB1の構築の概略を示す。
【図3】pKMB1/ΔLDHの構築の概略を示す。
【図4】pTZベクターの構築の概略を示す。
【図5】pMJPC1の構築の概略を示す。
【図6】配列番号
【図7】配列番号
【図8】配列番号
【図9】配列番号
【図10】配列番号
【図11】配列番号
【図12】配列番号
【図13】配列番号
【図14】配列番号
【図15】配列番号
【図16】配列番号
【図17】配列番号
【技術分野】
【0001】
本発明は、窒素原子含有量が特定範囲で色調良好なポリブチレンテレフタレートに関する。
【背景技術】
【0002】
熱可塑性ポリエステル樹脂の中で代表的なエンジニアリンブプラスチックであるポリブチレンテレフタレート(以下PBTと表す)は、成形加工の容易性、機械的物性、耐熱性
、耐薬品性、保香性、その他の物理的・化学的特性に優れていることから、自動車部品、電気・電子部品、精密機器部品などの射出成形品に広く使用されている。また、近年は、その優れた性質を活かし、フィルム、シート、モノフィラメント、繊維などの一般消費材分野でも広く使用されており、これに伴って、色調良好なPBTが求められるようになってきている。
【0003】
PBTは、通常、テレフタル酸又はそのアルキルエステルと1,4−ブタンジオール(以下BGと表すことがある)とを反応させて製造するが、BGは反応中にテトラヒドロフラン(以下THFと表すことがある)に転化しやすいため、THF転化率の低いPBTの製造方法が求められている。
一方、循環型(サステイナブル)社会の構築を求める声の高まりとともに、樹脂材料分野においてもエネルギーと同様に化石燃料原料からの脱却が望まれている。化石燃料を原料としない樹脂材料を開発する場合、植物などを原料とするバイオマス資源が有力な原料候補であり、バイオマスプラスチックの実用化が進んでいる。
【0004】
原料である1,4ブタンジオールがバイオマス資源から得られたものである場合、化石燃料から得られたものである場合に比べ、PBTの色調が悪くなる。
特許文献1には、バイオマス資源を原料としてポリエステルを得る技術について、ジカルボン酸中の窒素含有量を制御することにより、窒素含有量1000質量ppm以下のポリエステルを得ることについて記載されている。しかしながら、該文献には、ジオール中の窒素含有量及びその影響については記載も示唆もない。又、PBTに関する記載も全く無い。(尚、以下、本発明において、特に断らない限り濃度表示「ppm」は、「質量ppm」を表す。)
【特許文献1】特開2005―139287号公報
【発明の開示】
【発明が解決しようとする課題】
【0005】
本発明は、上記の現状に鑑みてなされたものであり、色調良好なPBTを提供すること、及び原料BGのTHFへの転化率が低く、PBTを効率的に製造できる方法を提供することを目的とする。
【課題を解決するための手段】
【0006】
本発明者は、上記目的を達成すべく鋭意検討した。この結果、特定量の窒素原子を含有するPBTの色調が良好であることを見出し本発明に到達した。
即ち、本発明の要旨は、テレフタル酸由来の構成単位と1,4ブタンジオール由来の構成単位を含み、窒素原子含有量が0.01〜50質量ppmであることを特徴とするポリブチレンテレフタレートに存する。また、本発明の別の要旨は、テレフタル酸又はテレフタル酸アルキレートと窒素原子含有量が0.01〜50質量ppmの1,4ブタンジオールとをエステル化反応又はエステル交換反応させた後、重縮合反応させるポリブチレンテレフタレートの製造方法に存する。
【発明の効果】
【0007】
本発明によれば、色調良好で高品質なPBTを得ることができる。特に、バイオマス資源由来のBGを原料として、色調良好なPBTを得ることができる。また、原料BGのTHFへの転化率が低く、効率的にPBT原料を製造することができる。
【発明を実施するための最良の形態】
【0008】
以下、本発明を詳細に説明するが、以下に記載する各構成要件の説明は、本発明の実施態様の代表例であり、本発明はこれらに限定されるものではない。
尚、本明細書において、「〜」を用いて表される数値範囲は、「〜」の前後に記載される数値を下限値及び上限値として含む範囲を意味する。また、本明細書における、下限値又は上限値は、その下限値又は上限値の数値を含む範囲を意味する。
<PBT製造原料>
本発明のPBTは、テレフタル酸又はテレフタル酸アルキレートと1,4ブタンジオールとをエステル化反応又はエステル交換反応させた後、重縮合反応させることにより得られる。本発明のPBTの製造に用いる原料は、バイオマス資源由来であるのが、環境保護の点から好ましい。
【0009】
テレフタル酸又はテレフタル酸アルキルエステルは、全ジカルボン酸成分の80モル%以上であるのが好ましく、90モル%以上であるのが更に好ましく、100モル%であるのが最も好ましい。また、BG成分は、全ジオール成分の80モル%以上であるのが好ましく、90モル%以上であるのが更に好ましく、100モル%であるのが最も好ましい。テレフタル酸又はテレフタル酸アルキルエステルの全ジカルボン酸成分に占める割合及びBG成分の全ジオール成分に占める割合が前記範囲以上であると、電気部品等に成形する際の結晶化の点やフィルム、繊維等に成形する際の延伸による分子鎖の配向結晶化の点から、成形体としての機械的強度、耐熱性、保香性等が良好になりやすい。
【0010】
テレフタル酸又はテレフタル酸アルキルエステル以外のジカルボン酸成分としては、具体的には、例えば、フタル酸、イソフタル酸、ジブロモイソフタル酸、スルホイソフタル酸ナトリウム、フェニレンジオキシジカルボン酸、4,4’−ジフェニルジカルボン酸、4,4’−ジフェニルエーテルジカルボン酸、4,4’−ジフェニルケトンジカルボン酸、4,4’−ジフェノキシエタンジカルボン酸、4,4’−ジフェニルスルホンジカルボン酸、2,6−ナフタレンジカルボン酸等の芳香族ジカルボン酸およびこれらのエステル形成性誘導体;ヘキサヒドロテレフタル酸、ヘキサヒドロイソフタル酸等の脂環式ジカルボン酸およびこれらのエステル形成性誘導体;コハク酸、グルタル酸、アジピン酸、ピメリン酸、スベリン酸、アゼライン酸、セバシン酸、ウンデカジカルボン酸、ドデカジカルボン酸等の脂肪族鎖式ジカルボン酸よびこれらのエステル形成性誘導体等が挙げられる。
【0011】
BG以外のジオール成分としては、具体的には、例えば、エチレングリコール、トリメチレングリコール、ペンタメチレングリコール、ヘキサメチレングリコール、オクタメチレングリコール、デカメチレングリコール、ネオペンチルグリコール、2−エチル−2−ブチル−1,3−プロパンジオール、ポリエチレングリコール、ポリテトラメチレンエーテルグリコール等の脂肪族鎖式ジオール;1,2−シクロヘキサンジオール、1,4−シクロヘキサンジオール、1,1−シクロヘキサンジメチロール、1,4−シクロヘキサンジメチロール、2,5−ノルボルナンジメチロール等の脂環式ジオール;キシリレングリコール、4,4’−ジヒドロキシビフェニル、2,2−ビス(4’−ヒドロキシフェニル)プロパン、2,2−ビス(4’−β−ヒドロキシエトキシフェニル)プロパン、ビス(4−ヒドロキシフェニル)スルホン、ビス(4−β−ヒドロキシエトキシフェニル)スルホン酸等の芳香族ジオール;2,2−ビス(4’−ヒドロキシフェニル)プロパンのエチレンオキサイド付加物又はプロピレンオキサイド付加物;バイオマス資源由来でないBG等が挙げられる。
【0012】
なお、共重合成分として、更に、以下の成分が用いられていてもよい。例えば、グリコール酸、p−ヒドロキシ安息香酸、p−β−ヒドロキシエトキシ安息香酸等のヒドロキシカルボン酸やアルコキシカルボン酸、及び、ステアリルアルコール、ヘネイコサノール、オクタコサノール、ベンジルアルコール、ステアリン酸、ベヘン酸、安息香酸、t−ブチル安息香酸、ベンゾイル安息香酸等の単官能成分、トリカルバリル酸、トリメリット酸、トリメシン酸、ピロメリット酸、ナフタレンテトラカルボン酸、没食子酸、トリメチロールエタン、トリメチロールプロパン、グリセロール、ペンタエリスリトール、シュガーエステル等の三官能以上の多官能成分、等が挙げられる。
【0013】
<バイオマス資源由来BG>
本発明のPBTの製造に用いるジカルボン酸成分は、バイオマス資源由来であるのが架橋保護の点から好ましい。
バイオマス資源とは、植物の光合成作用で太陽の光エネルギーがデンプンやセルロースなどの形に変換されて蓄えられたもの、植物体を食べて成育する動物の体や、植物体や動物体を加工してできる製品等が含まれる。具体的には、木材、稲わら、籾殻、米ぬか、古米、とうもろこし、サトウキビ、キャッサバ、サゴヤシ、おから、コーンコブ、タピオカカス、バガス、植物油カス、芋、そば、大豆、油脂、古紙、製紙残渣、水産物残渣、家畜排泄物、下水汚泥、食品廃棄物等が挙げられる。この中でも、木材、稲わら、籾殻、米ぬか、古米、とうもろこし、サトウキビ、キャッサバ、サゴヤシ、おから、コーンコブ、タピオカカス、バガス、植物油カス、芋、そば、大豆、油脂、古紙、製紙残渣等の植物資源が好ましく、より好ましくは、木材、稲わら、籾殻、古米、とうもろこし、サトウキビ、キャッサバ、サゴヤシ、芋、油脂、古紙、製紙残渣が挙げられ、最も好ましくはとうもろこし、さとうきび、キャッサバ、サゴヤシが挙げられる。
【0014】
バイオマス資源は、一般に、窒素元素やNa、K、Mg、Ca等の多くのアルカリ金属及びアルカリ土類金属を含有する。
これらのバイオマス資源は、その方法は特に限定はされないが、例えば、酸やアルカリ等の化学処理、微生物を用いた生物学的処理、物理的処理等の公知の前処理・糖化の工程等を経て炭素源へと誘導される。その工程には、バイオマス資源をチップ化する、削る、擦り潰す等の前処理による微細化工程が含まれることが多く、必要に応じて、更にグラインダーやミルによる粉砕工程も含まれる。こうして微細化されたバイオマス資源は、通常、更に前処理・糖化の工程を経て炭素源へと誘導される。その具体的な方法としては、硫酸、硝酸、塩酸、燐酸等の強酸による酸処理、アルカリ処理、アンモニア凍結蒸煮爆砕法、溶媒抽出、超臨界流体処理、酸化剤処理等の化学的方法;微粉砕、蒸煮爆砕法、マイクロ波処理、電子線照射等の物理的方法;微生物や酵素処理による加水分解等生物学的処理等が挙げられる。
【0015】
上記のバイオマス資源から誘導される炭素源としては、通常、グルコース、マンノース、ガラクトース、フルクトース、ソルボース、タガトース等のヘキソース;アラビノース、キシロース、リボース、キシルロース、リブロース等のペントース;ペントサン、サッカロース、澱粉、セルロース等の2糖・多糖類;酪酸、カプロン酸、カプリル酸、カプリン酸、ラウリン酸、ミリスチン酸、パルミチン酸、パルミトレイン酸、ステアリン酸、オレイン酸、リノール酸、リノレン酸、モノクチン酸、アラキジン酸、エイコセン酸、アラキドン酸、ベヘニン酸、エルカ酸、ドコサペンタエン酸、ドコサヘキサエン酸、リグノセリン酸、セラコレン酸等の油脂;グリセリン、マンニトール、キシリトール、リビトール等のポリアルコール類等の発酵性糖質が用いられる。このうち、グルコース、フルクトース、キシロース等のヘキソース又はペントースが好ましく、特にグルコースが好ましい。より広義の植物資源由来の炭素源としては、紙の主成分であるセルロースも好ましい。
【0016】
通常、これらの炭素源を用いて、微生物変換による発酵法や加水分解・脱水反応・水和反応・酸化反応等の反応工程を含む化学変換法ならびにこれらの発酵法と化学変換法の組み合わせによりジオールが合成される。これらの中でも、微生物変換による発酵法が好ましい。
本発明のPBTを製造するのに用いるジオールは、窒素原子含有量が0.01〜50質量ppmであるのが好ましい。バイオマス資源のジオールを用いる場合、具体的には、例えば、グルコース等の炭素源から発酵法により直接ジオールを製造してもよいし、発酵法により得られたコハク酸、コハク酸無水物、コハク酸エステル等を化学反応によりジオール化合物に変換しても良いし、発酵法により得られた1,3−ブタジエンから1,4−ブタンジオールを製造する等してもよい。この中でも、発酵法により得られたコハク酸(又はコハク酸をコハク酸ジメチルに変成後に)を、還元触媒により水添して1,4−ブタンジオールを得る方法が効率的で好ましい。
【0017】
コハク酸を水添する触媒の例としては、例えば、Pd、Ru、Re、Rh、Ni、Cu、Co及びその化合物等が挙げられる。具体的には、Pd/Ag/Re、Ru/Ni/Co/ZnO、Cu/Zn酸化物、Cu/Zn/Cr酸化物、Ru/Re、Re/C、Ru/Sn、Ru/Pt/Sn、Pt/Re/アルカリ、Pt/Re、Pd/Co/Re、Cu/Si、Cu/Cr/Mn、ReO/CuO/ZnO、CuO/CrO、Pd/Re、Ni/Co、Pd/CuO/CrO3、リン酸Ru、Ni/Co、Co/Ru/Mn、Cu/Pd/KOH、Cu/Cr/Zn等が挙げられる。この中でも、Ru/Sn又はRu/Pt/Snが触媒活性の点で好ましい。
また、更に、バイオマス資源から公知の有機化学触媒反応との組み合わせによりBGを製造する方法も用いられる。例えば、バイオマス資源としてペントースを利用する場合には公知の脱水反応、触媒反応の組み合わせで容易にブタンジオール等のジオールを製造できる。
【0018】
<BG中の窒素原子含有量>
バイオマス資源由来から誘導されたBGには、発酵処理ならびに酸による中和工程を含む精製処理に起因して不純物として窒素原子が含まれてくる。具体的には、アミノ酸、蛋白質、アンモニア、尿素、発酵菌由来等の窒素原子が含まれてくる。
本発明においてPBTの原料となる、バイオマス資源から得られたBGの窒素原子含有量は、該BGに対して質量比で、上限は、通常50ppm、好ましくは10ppm、より好ましくは2ppmであるのよい。また、下限は特に制限されないが、通常、0.01ppm、好ましくは0.05ppmがよく、特に、精製工程の経済性の観点からは、0.1ppmであるのが好ましい。BG中の窒素原子含有量が上記上限以下であると、PBT製造における重縮合反応速度、生成するPBTの色調などが好ましくなる傾向が強い。一方、上記下限以上であると、精製工程が簡便としやすく経済的に有利な上、PBT製造反応中でのBGのTHFへの転化も低く押させやすい。
【0019】
上記上限以下であると重縮合反応速度や色調の点で好ましくなりやすい理由は定かではないが、発酵処理ならびに酸による中和処理を含む精製処理において、窒素源となる化合物以外に重縮合反応を阻害し、PBTの色調を悪化させる着色誘引物質が生成しやすくなっているのではないかと推定される。また、発酵における窒素源となるアンモニア、アミノ酸なども反応を遅延させ、PBTの色調悪化を誘引するものと考えられる。
【0020】
BG中の窒素原子量は、例えば、バイオマス資源の発酵により得られるコハク酸を水添してBGを得る場合は、その発酵条件、アンモニアによる中和条件、コハク酸の晶析条件などによりコハク酸中の窒素原子量を調節することにより調節可能である。また、コハク酸を水添して得られるBGは、蒸留を含む精製条件により、その含有窒素原子含有量を調
節できる。また、BGがバイオマス資源の発酵により直接得られる場合にも、その発酵条件、アンモニアによる中和条件、得られたBGの蒸留を含む精製条件などにより調節できる。
【0021】
<BG中の硫黄原子含有量>
発酵法により製造したBG等を用いる場合には、酸による中和工程を含む精製処理等により硫黄原子が含まれてくる場合がある。この場合、具体的に、硫黄原子が含有される不純物としては、硫酸、亜硫酸、有機スルホン酸塩等が挙げられる。
BG中に含まれる硫黄原子量は、該BGに対して質量比で、上限は通常100ppm、好ましくは20ppm、より好ましくは10ppm、特に好ましくは5ppm、最も好ましくは0.5ppmである。一方、下限は特に制限されないが、通常0.001ppm、好ましくは0.01ppm、より好ましくは0.05ppm、特に好ましくは、0.1ppmである。上記上限以下であると、重縮合反応速度やPBTの安定性が好ましくなる傾向がある。一方、上記下限以上であると、精製工程を簡便にして経済的に有利にしやすくなる。硫黄原子含有量は、公知の元素分析法により測定される値である。
【0022】
<BG中の1−アセトキシ−4−ヒドロキシブタン及び2−(4−ヒドロキシブチルオキシ)テトラヒドロフラン)の含有量>
窒素原子含有量が0.01〜50質量ppmであるBGを用いると、PBTを製造する際の重縮合速度が大きくなる。具体的には、実施例で後述する通り、バイオマス資源から得られ、窒素原子含有量が0.01〜50質量ppmであるBGを用いると、化石燃料原料から得られたBGを使用した場合に比べて、重縮合速度が大きくなっている。これは、本発明の実施例で用いたBGにおける1−アセトキシ−4−ヒドロキシブタン(1,4−HABと表すことが有る)及び/又は2−(4−ヒドロキシブチルオキシ)テトラヒドロフラン(BGTFと表すことが有る)の含有量が少ないために、重縮合反応の阻害が少ないためであると考えられる。従って、BG中の1−アセトキシ−4−ヒドロキシブタンは、50質量ppm以下が好ましく、10質量ppm以下がより好ましい。また、下限は少ないほうが良いが通常0.1質量ppmである。
【0023】
<PBT製造方法>
本発明のPBTは、これを製造できれば特に制限されない。PBTの公知の製造方法は、主原料としてテレフタル酸を用いるいわゆる直接重合法と、主原料としてテレフタル酸ジアルキルエステルを用いるエステル交換法とに大別される。前者は、初期のエステル化反応で水が生成し、後者は初期のエステル交換反応でアルコールが生成するという違いがあるが、原料の入手安定性、留出物の処理の容易さ、原料原単位の高さ、また本発明による改良効果という観点からは直接重合法が好ましい。
【0024】
直接重合法の一例としては、テレフタル酸とBGとを、単数若しくは複数段のエステル化反応槽内で、エステル化反応触媒の存在下に、温度が、通常180以上、好ましくは200℃以上、特に好ましくは210℃以上、通常260℃以下、好ましくは250℃以下、特に好ましくは245℃以下で、圧力が、通常10kPa以上、好ましくは13kPa以上、特に好ましくは50kPa以上、通常133kPa以下、好ましくは101kPa以下、特に好ましくは90kPa以下で、反応時間が、通常0.5時間以上、好ましくは1時間以上、通常5時間以下、好ましくは3時間以下で、連続的にエステル化反応させ、得られたエステル化反応生成物としてのオリゴマーを重縮合反応槽に移送し、複数段の重縮合反応槽内で、重縮合反応触媒の存在下で連続的に、温度が、通常210℃以上、好ましくは220℃以上、通常260℃以下、好ましくは250℃以下、特に好ましくは245℃以下で、圧力が、通常27kPa以下、好ましくは20kPa以下、より好ましくは13kPa以下、中でも少なくとも1つの重縮合反応槽においては好ましくは2kPa以下の減圧下で、攪拌しながら、通常2〜12時間好ましくは2〜10時間で縮合反応させる方法等が挙げられる。
【0025】
エステル交換法の一例としては、テレフタル酸ジメチルとBGとを、単数若しくは複数段のエステル化反応槽内で、エステル交換反応触媒の存在下に、温度が110℃以上、好ましくは140℃以上、特に好ましくは180℃以上、260℃以下、好ましくは245℃以下、特に好ましくは220℃以下で、圧力が通常10kPa以上、好ましくは13kPa以上、特に好ましくは60kPa以上、通常133kPa以下、好ましくは101kPa以下、特に好ましくは90kPa以下で、反応時間が通常0.5時間以上、好ましくは1時間以上、通常5時間以下、好ましくは3時間以下で、連続的にエステル交換反応させ、得られたエステル交換反応生成物としてのオリゴマーを重縮合反応槽に移送し、複数段の重縮合反応槽内で、重縮合反応触媒の存在下で連続的に、温度が、通常210℃以上、好ましくは220℃以上、通常260℃以下、好ましくは250℃以下、特に好ましくは245℃以下で、圧力が、通常27kPa以下、好ましくは20kPa以下、特に好ましくは13kPa以下の減圧下、中でも少なくとも1つの重縮合反応槽においては好ましくは2kPa以下の減圧下で、攪拌しながら、通常2〜12時間好ましくは2〜10時間、重縮合反応させる方法等が挙げられる。
【0026】
エステル化反応またはエステル交換反応触媒としては、例えば、三酸化二アンチモン等のアンチモン化合物;二酸化ゲルマニウム、四酸化ゲルマニウム等のゲルマニウム化合物;テトラメチルチタネート、テトライソプロピルチタネート、テトラブチルチタネート等のチタンアルコラート、テトラフェニルチタネート等のチタンフェノラート等、のチタン化合物;ジブチルスズオキサイド、メチルフェニルスズオキサイド、テトラエチルスズ、ヘキサエチルジスズオキサイド、シクロヘキサヘキシルジスズオキサイド、ジドデシルスズオキサイド、トリエチルスズハイドロオキサイド、トリフェニルスズハイドロオキサイド、トリイソブチルスズアセテート、ジブチルスズジアセテート、ジフェニルスズジラウレート、モノブチルスズトリクロライド、トリブチルスズクロライド、ジブチルスズサルファイド、ブチルヒドロキシスズオキサイド、メチルスタンノン酸、エチルスタンノン酸、ブチルスタンノン酸等のスズ化合物;酢酸マグネシウム、水酸化マグネシウム、炭酸マグネシウム、酸化マグネシウム、マグネシウムアルコキサイド、燐酸水素マグネシウム等のマグネシウム化合物、酢酸カルシウム、水酸化カルシウム、炭酸カルシウム、酸化カルシウム、カルシウムアルコキサイド、燐酸水素カルシウム、等の等のカルシウム化合物等のアルカリ土類金属化合物の他、マンガン化合物、亜鉛化合物等を挙げることができる。中でも、チタン化合物、スズ化合物が好ましく、テトラブチルチタネートが特に好ましい。
【0027】
エステル化反応またはエステル交換反応触媒の使用量は特に限定されないが、PBT中の金属濃度(質量)として、通常1ppm以上、好ましくは5ppm以上、更に好ましくは10ppm以上、特に好ましくは20ppm以上、最も好ましくは30pmm以上、通常300ppm以下、好ましくは200ppm以下、より好ましくは150ppm以下、更に好ましくは100ppm以下、特に好ましくは90ppm以下、最も好ましくは60pmm以下が良い。上記上限以下であると、異物の原因となりにくい上、PBTの熱滞留時の劣化反応やガス発生が起こりにくい傾向があり、上記下限以上であると、主反応速度が速く副反応が起こりにくい。
【0028】
また、重縮合反応触媒としては、エステル化反応またはエステル交換反応の触媒をそのまま重縮合反応触媒として用いてもよいし、更に前記触媒を添加してもよい。重縮合反応触媒の使用量に特に制限はないが、上記のエステル化反応またはエステル交換反応の触媒と同様の理由から、PBT中の金属濃度(質量)として、通常300ppm以下、好ましくは200ppm以下、更に好ましくは100ppm以下、特に好ましくは50ppm以下、最も好ましくは30ppm以下がよい。
【0029】
また、触媒として有機チタン化合物を用いる場合には、異物抑制の観点から、最終的にはPBT中のチタン金属濃度(質量)は、250ppm以下であることが好ましく、100ppm以下であることが更に好ましく、60ppm以下であることが特に好ましく、50ppm以下であることが最も好ましい。
PBT中の金属濃度(質量)は、湿式灰化等の方法でPBT中の金属を回収した後、原子発光、Induced Coupled Plasma(ICP)法等を用いて測定することができる。
【0030】
また、前記のエステル化反応、エステル交換反応及び重縮合反応において、前記触媒の他に、正燐酸、亜燐酸、次亜燐酸、ポリ燐酸およびそれらのエステルや金属塩等の燐化合物;水酸化ナトリウム、安息香酸ナトリウム等のナトリウム化合物、酢酸リチウム、水酸化カリウム、酢酸カリウム等のカリウム化合物等のアルカリ金属化合物等の反応助剤;酢酸マグネシウム、酢酸カルシウム等のアルカリ土類金属化合物等の反応助剤;2,6−ジ−t−ブチル−4−オクチルフェノール、ペンタエリスリチル−テトラキス〔3−(3’,5’−t−ブチル−4’−ヒドロキシフェニル)プロピオネート〕等のフェノール化合物;ジラウリル−3,3’−チオジプロピオネート、ペンタエリスリチル−テトラキス(3−ラウリルチオジプロピオネート)等のチオエーテル化合物;トリフェニルホスファイト、トリス(ノニルフェニル)ホスファイト、トリス(2,4−ジ−t−ブチルフェニル)ホスファイト等の燐化合物等の抗酸化剤;パラフィンワックス、マイクロクリスタリンワックス、ポリエチレンワックス、モンタン酸やモンタン酸エステルに代表される長鎖脂肪酸およびそのエステル;シリコーンオイル等の離型剤等を使用しても良い。
【0031】
重縮合反応槽としては、縦型攪拌重合槽、横型攪拌重合槽、薄膜蒸発式重合槽等の公知のものを挙げることができる。反応液の粘度が上昇する重縮合の後期は、反応速度よりも物質移動が分子量増大の支配因子になる傾向があるため、副反応を抑制しつつ主反応を押し進めるには、可能な限り温度を下げ、表面更新性を上げた方が本発明の目的を達成するには有利であり、表面更新性とプラグフロー性、セルフクリーニング性に優れた薄膜蒸発機能を有した単数または複数の横型攪拌重合機を選定することが好ましい。
【0032】
また、本発明の製造法で得られたPBTは、引き続き公知の方法で固相重縮合させて分子量を上げることもできる。
重縮合反応により得られたPBTは、通常、重縮合反応槽の底部からポリマー抜出ダイに移送されてストランド状に抜き出され、水冷されながら若しくは水冷後、カッターで切断されてペレット状又はチップ状の粒状体とされる。粒状体は、引き続き公知の方法等で固相重縮合させて、その固有粘度を上ることもできる。
【0033】
<PBT>
本発明のPBTは、テレフタル酸由来の構成単位と1,4ブタンジオール由来の構成単位を含み、窒素原子含有量が0.01〜50質量ppmである。本発明のPBTにおける窒素原子含有量(質量比)で、上限は、好ましくは10ppm、より好ましくは2ppmであるのよく、下限は、好ましくは0.05ppm、より好ましくは0.1ppmであるのが良い。窒素原子含有量が上記範囲内のPBTは、上記の好ましいテレフタル酸又はテレフタル酸アルキレートと1,4ブタンジオールとを原料とすることにより得ることができる。
【0034】
本発明のPBTの固有粘度に特に制限はないが、機械的物性、ペレット化の安定性、成形性の観点からは、好ましくは0.50dL/g以上、更に好ましくは0.70dL/g以上、好ましくは1.50dL/gの以下、更に好ましくは1.35dL/g以下であるのが良い。PBTの固有粘度が上記下限以上であると成形品の機械物性の点で好ましく、
上記上限以下であると成形性の点好ましい傾向がある。
【0035】
本発明のPBTの末端カルボキシル基濃度に特に制限はないが、下限が、1当量/トンであることが好ましく、2当量/トンであることが更に好ましく、3当量/トンであることが特に好ましく、5当量/トンであることが最も好ましく、上限が、50当量/トンであることが好ましく、40当量/トンであることが更に好ましく、30当量/トンであることが特に好ましく、25当量/トンであることが最も好ましい。PBTの末端カルボキシル基濃度が上記上限以下であるとPBTの耐加水分解性を良好な傾向にあり、上記下限以上であると重縮合性が良好な傾向にある。PBTの末端カルボキシル基濃度は、樹脂を有機溶媒に溶解し、水酸化ナトリウム溶液等のアルカリ溶液を用いて滴定することにより求めることができる。
【0036】
本発明のPBTの末端ビニル基濃度に特に制限はないが、色調や重縮合性の点から、好ましくは15当量/トン以下、更に好ましくは10当量/トン以下、特には好ましくは7当量/トン以下がよい。PBTの末端ビニル基濃度は、PBTを溶媒に溶かしてから各磁気共鳴スペクトル(NMR)を測定することによって求められる。
<PBTの色調>
本発明のPBTは、色調が良好である。
【0037】
<PBT組成物>
本発明のPBTは、本発明の優れた効果を大幅に損なわない範囲で、PBT以外の成分を含んでいてもよい。具体例を挙げると、熱可塑性、熱硬化性などの各種樹脂、離型剤、充填剤、難燃剤、その他各種添加剤などが挙げられる。
熱可塑性樹脂としては、ポリエチレン、ポリプロピレン、ポリスチレン、ポリアクリロニトリル、ポリメタクリル酸エステル、ABS樹脂、ポリカーボネート、ポリアミド、ポリフェニレンサルファイド、ポリエチレンテレフタレート、液晶ポリエステル、ポリアセタール、ポリフェニレンオキサイド等が挙げられる。また、熱硬化性樹脂としては、フェノール樹脂、メラミン樹脂、シリコーン樹脂、エポキシ樹脂などが挙げられる。これらの樹脂は、1種のみ用いてもよいし、2種以上を組み合わせて使用することも出来る。このうち、熱可塑性樹脂をよく配合する。
【0038】
これらの樹脂の配合量(質量)は、本発明の優れた効果が発現されていればいくつでもよいが、樹脂全量に対して、PBTが、通常0.1質量%以上、好ましくは1質量%以上、更に好ましくは10質量%以上、通常99.9質量%以下、好ましくは99質量%以下、更に好ましくは90質量%以下がよい。
離型剤としては、特に制限されないが、例えば、2,6−ジ−t−ブチル−4−オクチルフェノール、ペンタエリスリチル−テトラキス〔3−(3',5'−t−ブチル−4'−
ヒドロキシフェニル)プロピオネート〕等のフェノール化合物、ジラウリル−3,3'−
チオジプロピオネート、ペンタエリスリチル-テトラキス(3-ラウリルチオジプロピオネート)等のチオエーテル化合物、トリフェニルホスファイト、トリス(ノニルフェニル)ホスファイト、トリス(2,4−ジ−t−ブチルフェニル)ホスファイト等の燐化合物などの抗酸化剤;パラフィンワックス、マイクロクリスタリンワックス、ポリエチレンワックス;モンタン酸やモンタン酸エステルに代表される長鎖脂肪酸およびそのエステル;シリコーンオイル等などが挙げられる。
【0039】
強化充填材としては、特に制限されないが、例えば、ガラス繊維、カーボン繊維、シリカ・アルミナ繊維、ジルコニア繊維、ホウ素繊維、窒化ホウ素繊維、窒化ケイ素チタン酸カリウム繊維、金属繊維などの無機繊維;芳香族ポリアミド繊維、フッ素樹脂繊維などの有機繊維などが挙げられる。このうち、無機充填材、特にガラス繊維が好適に使用される。強化充填材は、1種類のみ用いてもよいし、2種以上を組み合わせて使用してもよい。
【0040】
強化充填材が無機又は有機繊維である場合、その平均繊維径は、特に制限されないが、通常1〜100μm、好ましくは2〜50μm、更に好ましくは3〜30μm、特に好ましくは5〜20μmである。また、平均繊維長は、特に制限されないが、通常0.1〜20mm、好ましくは1〜10mmである。
強化充填材は、PBTとの界面密着性を向上させるため、収束剤または表面処理剤で表面処理されたものを用いるのが好ましい。収束剤または表面処理剤としては、例えば、エポキシ系化合物、アクリル系化合物、イソシアネート系化合物、シラン系化合物、チタネート系化合物などの官能性化合物が挙げられる。収束剤または表面処理剤による処理は、強化充填剤を予め表面処理しておいてもよいし、PBT組成物を調製する際に収束剤または表面処理剤と接触させてもよい。強化充填材の量は、PBT樹脂100質量部に対し、通常150質量部以下、好ましくは5〜100重量部である。
【0041】
本発明の製造方法で得られるPBTには、強化充填材以外の充填材を配合してもよい。具体的には、例えば、板状無機充填材、セラミックビーズ、アスベスト、ワラストナイト、タルク、クレー、マイカ、ゼオライト、カオリン、チタン酸カリウム、硫酸バリウム、酸化チタン、酸化ケイ素、酸化アルミニウム、水酸化マグネシウム等が挙げられる。板状無機充填材を配合することにより、成形品の異方性およびソリを低減することが出来る。板状無機充填材としては、例えば、ガラスフレーク、雲母、金属箔などを挙げることが出来る。これらの中ではガラスフレークが好適に使用される。
【0042】
難燃性を付与するために用いる難燃剤としては、特に制限されず、例えば、有機ハロゲン化合物、アンチモン化合物、リン化合物、その他の有機難燃剤、無機難燃剤などが挙げられる。有機ハロゲン化合物としては、例えば、臭素化ポリカーボネート、臭素化エポキシ樹脂、臭素化フェノキシ樹脂、臭素化ポリフェニレンエーテル樹脂、臭素化ポリスチレン樹脂、臭素化ビスフェノールA、ポリペンタブロモベンジルアクリレート等が挙げられる。アンチモン化合物としては、例えば、三酸化アンチモン、五酸化アンチモン、アンチモン酸ソーダ等が挙げられる。リン化合物としては、例えば、リン酸エステル、ポリリン酸、ポリリン酸アンモニウム、赤リン等が挙げられる。その他の有機難燃剤としては、例えば、メラミン、シアヌール酸などの窒素化合物などが挙げられる。その他の無機難燃剤としては、例えば、水酸化アルミニウム、水酸化マグネシウム、ケイ素化合物、ホウ素化合物などが挙げられる。
【0043】
その他の各種添加剤としては、特に制限されないが、例えば、酸化防止剤、耐熱安定剤などの安定剤の他、滑剤、離型剤、触媒失活剤、結晶核剤、結晶化促進剤などが挙げられる。これらの添加剤は、重縮合途中または重縮合後に添加してもよい。また、紫外線吸収剤、耐候安定剤などの安定剤、染顔料などの着色剤、帯電防止剤、発泡剤、可塑剤、耐衝撃性改良剤などを配合してもよい。
【0044】
上記のその他成分の配合方法は、特に制限されないが、例えば、ベント口から脱揮できる設備を有する1軸または2軸の押出機を混練機として使用する方法が好ましい。各成分は、付加的成分を含めて、混練機に一括して供給することが出来、あるいは、順次供給することも出来る。また、付加的成分を含めて、各成分から選ばれた2種以上の成分をあらかじめ混合しておくことも出来る。
【0045】
<PBTの成形加工>
PBTの成形加工方法は、特に制限されず、熱可塑性樹脂について一般に使用されている成形法等、具体的には、射出成形、中空成形、押し出し成形、プレス成形などを適用出来る。
本発明のPBT及びこれを含んだ組成物は、色調、熱安定性、透明性、品質安定性に優
れ、電気、電子部品、自動車用部品などの射出成形部品、フィルム、モノフィラメント、繊維などの押出し成形品用途において好適に使用できる。
【実施例】
【0046】
以下、実施例を用いて本発明の内容を更に具体的に説明するが、本発明はその要旨を越えない限り以下の実施例により限定されるものではない。
(分析方法)
<窒素原子含有量(質量ppm)の測定方法>
試料15mgを石英ボートへ採取して、微量全窒素分析装置(ダイヤインスツルメンツ
社製「TN−10型」)を用いて試料を燃焼し、燃焼・化学発光法により定量した。また
、その際に使用した標準試料は、トルエン中にアニリンを溶解し、窒素換算で0,0.5,1.0,2.0μg/mLを作製し使用した。
【0047】
<BG中の1,4−HAB、BGTF、THF量(質量ppm)及びTHF転化率>
島津製作所製ガスクロマト分析装置「島津GCC−14BPF型」にて、DB−WAXカラムを用い、修正面積百分率法により各ピークの成分の含有量を求めた。各ピークの係数は、1,4−ブタンジオールを1.000とした時に、有効炭素数に基づいて、1,4−HABは0.990、BGTFは0.922、THFは0.747として計算した。
エステル交換反応又はエステル化反応における留出液について、カールフィッシャー法(三菱化学社製「CA−03」で測定)にて水分量を求め、水分以外は有機成分とした。有機成分中のTHF量を上記ガスクロ法により求め、THF生成量とした。THF生成量をテレフタル酸又はジメチルテレフタレートに対するモル%で表し、転化率とした。
【0048】
<PBTの固有粘度、IV>
ウベローデ型粘度計を使用して以下の手順で求めた。すなわち、フェノール/テトラクロロエタン(質量比1/1)の混合溶媒を使用し、30℃において、濃度1.0g/dL
のポリマー溶液および溶媒のみの落下秒数を測定し、以下の式より求めた。
IV=((1+4KHηsp)0.5-1)/(2KHHC)
但し、ηsp=(η/η0)-1であり、ηはポリマー溶液落下秒数、η0は溶媒の落下秒
数、Cはポリマー溶液濃度(g/dL)、KHはハギンズの定数である。KHは0.33を採用した。
【0049】
<PBTの末端カルボキシル基濃度(当量/トンPBT)>
ベンジルアルコール25mLにPBT0.5gを溶解し、水酸化ナトリウムの0.01モル/Lベンジルアルコール溶液を使用して滴定した。
末端カルボキシル基濃度=(A−B)×0.1×f/W(当量/トン)
(ここで、Aは、滴定に要した0.01Nの水酸化ナトリウムのベンジルアルコール溶液の量(μL)、Bは、ブランクでの滴定に要した0.01モル/Lの水酸化ナトリウムのベンジルアルコール溶液の量(μL)、Wは、PBT試料の量(g)、fは、0.01モル/Lの水酸化ナトリウムのベンジルアルコール溶液の力価である。)
【0050】
<PBT 色調b値>
ペレット状のPBTを日本電色(株)製色差計「Z−300A型」を使用して、L、a、b表色系におけるb値で評価した。値が低いほど黄ばみが少なく色調が良好であることを示す。
【0051】
<引張試験片、曲げ試験片の成形方法>
ペレットを、120℃、2kPaで8時間減圧乾燥させた。次に乾燥したペレットを住友重機械工業社製「MINIMAT8/7A型射出成形機」にて、シリンダ温度250℃、金型温度80℃、射出保圧時間5秒、冷却20秒で成形し、引張試験片と曲げ試験片を
それぞれ得た。
引張試験片の形状は長さ55mm、厚み3mm、中央平行部長さ25mm、中央平行部幅3.2mm、両端幅6mmのダンベル状であり、曲げ試験片の形状は長さ53mm、厚み3mm、幅7mmの短冊状であった。引張試験片の形状を図1に示す。
【0052】
<成形品の引張強度 MPa>
上記で得られた引張試験片をJIS K7113に準拠して温度25℃、相対湿度50%の恒温恒湿下で、島津製作所社製「オートグラフAGS−5kNG型試験機」を用いて、チャック間距離32mm、引張試験速度10mm/分にて測定した。
【0053】
<成形品の曲げ弾性率 MPa>
上記で得られた曲げ試験片をJIS K7171に準拠して温度25℃、相対湿度50%の恒温恒湿下で、東洋精機社製「ストログラフR2型試験機」を用いて、スパン30mm、載荷速度2mm/分にて測定した。
(製造例)
参考例1から参考例5の<コハク酸醗酵液からのコハク酸精製>までは、特願2006−524976号と同様に行った。
【0054】
参考例1
<遺伝子破壊用ベクターの構築>
(A)枯草菌ゲノムDNAの抽出
LB培地[組成:トリプトン10g、イーストエキストラクト5g、NaCl5gを蒸留水1Lに溶解]10mLに、枯草菌(Bacillus subtilis ISW1214)を対数増殖期後期まで培養し、菌体を集めた。得られた菌体を10mg/mLの濃度にリゾチームを含む10mM NaCl/20mMトリス緩衝液(pH8.0)/1mM EDTA・2Na溶液0.15mLに懸濁した。
【0055】
次に、上記懸濁液にプロテナーゼKを、最終濃度が100μg/mLになるように添加し、37℃で1時間保温した。更にドデシル硫酸ナトリウムを最終濃度が0.5質量%になるように添加し、50℃で6時間保温して溶菌した。この溶菌液に、等量のフェノール/クロロフォルム溶液を添加し、室温で10分間ゆるやかに振盪した後、全量を遠心分離(5,000×g、20分間、10〜12℃)し、上清画分を分取し、酢酸ナトリウムを0.3Mとなるように添加した後、2倍量のエタノールを加え混合した。遠心分離(15,000×g、2分)により回収した沈殿物を70質量%エタノールで洗浄した後、風乾した。得られたDNAに10mMトリス緩衝液(pH7.5)−1mM EDTA・2Na溶液5mLを加え、4℃で一晩静置し、以後のPCRの鋳型DNAに使用した。
【0056】
(B)PCRによるSacB遺伝子の増幅およびクローニング
枯草菌SacB遺伝子の取得は、上記(A)で調製したDNAを鋳型とし、既に報告されている該遺伝子の塩基配列(GenBank Database AccessionNo.X02730)を基に設計した合成DNA(配列番号1および配列番号2)を用いたPCRによって行った。
【0057】
反応液組成:鋳型DNA1μL、PfxDNAポリメラーゼ(インビトロジェン社製)0.2μL、1倍濃度添付バッファー、0.3μM各々プライマー、1mM MgSO4、0.25μMdNTPsを混合し、全量を20μLとした。
反応温度条件:DNAサーマルサイクラー PTC−200(MJResearch社製)を用い、94℃で20秒、68℃で2分からなるサイクルを35回繰り返した。但し、1サイクル目の94℃での保温は1分20秒、最終サイクルの68℃での保温は5分とした。
【0058】
増幅産物の確認は、0.75質量%アガロース(SeaKem GTG agarose:FMCBioProducts製)ゲル電気泳動により分離後、臭化エチジウム染色により可視化することにより行い、約2kbの断片を検出した。ゲルからの目的DNA断片の回収は、QIAQuickGel Extraction Kit(QIAGEN製)を用いて行った。
【0059】
回収したDNA断片は、T4ポリヌクレオチドキナーゼ(T4 Polynucleo
tide Kinase:宝酒造製)により5'末端をリン酸化した後、ライゲーションキットver.2(宝酒造製)を用いて大腸菌ベクター(pBluescriptII:STRATEGENE製)のEcoRV部位に結合し、得られたプラスミドDNAで大腸菌(DH5α株)を形質転換した。この様にして得られた組換え大腸菌を50μg/mLアンピシリンおよび50μg/mLX−Galを含むLB寒天培地[トリプトン10g、イーストエキストラクト5g、NaCl5g及び寒天15gを蒸留水1Lに溶解]に塗抹した。
【0060】
この培地上で白色のコロニーを形成したクローンを、次に50μg/mLアンピシリンおよび10質量%ショ糖を含むLB寒天培地に移し37℃で24時間培養した。これらのクローンのうち、ショ糖を含む培地で生育できなかったものについて、常法により液体培養した後、プラスミドDNAを精製した。SacB遺伝子が大腸菌内で機能的に発現する株は、ショ糖含有培地にて生育不能となるはずである。得られたプラスミドDNAを制限酵素SalIおよびPstIで切断することにより、約2kbの挿入断片が認められ、該プラスミドをpBS/SacBと命名した。
【0061】
(C)クロラムフェニコール耐性SacBベクターの構築
大腸菌プラスミドベクターpHSG396(宝酒造:クロラムフェニコール耐性マーカー)500ngに制限酵素PshBI10ユニットを37℃で1時間反応させた後、フェノール/クロロフォルム抽出およびエタノール沈殿により回収した。クレノウフラグメント(Klenow Fragment:宝酒造製)により両末端を平滑化した後、ライゲーションキットver.2(宝酒造製)を用いてMluIリンカー(宝酒造)を連結、環状化させ、大腸菌(DH5α株)を形質転換した。この様にして得られた組換え大腸菌を34μg/mLクロラムフェニコールを含むLB寒天培地に塗抹した。得られたクローンから常法によりプラスミドDNAを調製し、制限酵素MluIの切断部位を有するクローンを選抜し、pHSG396Mluと命名した。
【0062】
一方、上記(B)にて構築したpBS/SacBを制限酵素SalIおよびPstIで切断した後、クレノウフラグメントにて末端を平滑化した。これにライゲーションキットver.2(宝酒造製)を用いてMluIリンカーを連結したのち、0.75質量%アガロースゲル電気泳動によりSacB遺伝子を含む約2.0kbのDNA断片を分離、回収した。このSacB遺伝子断片を、制限酵素MluI切断後、アルカリフォスファターゼ(Alkaline Phosphatase Calf intestine:宝酒造)にて末端を脱リン酸化したpHSG396Mlu断片とライゲーションキットver.2(宝酒造製)を用いて連結させ、大腸菌(DH5α株)を形質転換した。この様にして得られた組換え大腸菌を34μg/mLクロラムフェニコールを含むLB寒天培地に塗抹した。こうして得られたコロニーを、次に34μg/mLクロラムフェニコールおよび10質量%ショ糖を含むLB寒天培地に移し37℃で24時間培養した。これらのクローンのうち、ショ糖を含む培地で生育できなかったものについて、常法によりプラスミドDNAを精製した。こうして得られたプラスミドDNAをMluI切断により解析した結果、約2.0kbの挿入断片を持つことが確認され、これをpCMB1と命名した。
【0063】
(D)カナマイシン耐性遺伝子の取得
カナマイシン耐性遺伝子の取得は、大腸菌プラスミドベクターpHSG299(宝酒造:カナマイシン耐性マーカー)のDNAを鋳型とし、配列番号3および配列番号4で示した合成DNAをプライマーとしたPCR法によって行った。
反応液組成:鋳型DNA1ng、PyrobestDNAポリメラーゼ(宝酒造)0.1μL、1倍濃度添付バッファー、0.5μM各々プライマー、0.25μMdNTPsを混合し、全量を20μLとした。
【0064】
反応温度条件:DNAサーマルサイクラー PTC−200(MJResearch社製)を用い、94℃で20秒、62℃で15秒、72℃で1分20秒からなるサイクルを20回繰り返した。但し、1サイクル目の94℃での保温は1分20秒、最終サイクルの72℃での保温は5分とした。
増幅産物の確認は、0.75質量%アガロース(SeaKem GTG agarose:FMCBioProducts製)ゲル電気泳動により分離後、臭化エチジウム染色により可視化することにより行い、約1.1kbの断片を検出した。ゲルからの目的DNA断片の回収は、QIAQuickGel Extraction Kit(QIAGEN製)を用いて行った。回収したDNA断片は、T4 ポリヌクレオチドキナーゼ(T4Polynucleotide Kinase:宝酒造製)により5'末端をリン酸化した。
【0065】
(E)カナマイシン耐性SacBベクターの構築
上記(C)で構築したpCMB1を制限酵素Van91IおよびScaIで切断して得られた約3.5kbのDNA断片を0.75質量%アガロースゲル電気泳動により分離、回収した。これを上記(D)で調製したカナマイシン耐性遺伝子と混合し、ライゲーションキットver.2(宝酒造製)を用いて連結し、得られたプラスミドDNAで大腸菌(DH5α株)を形質転換した。この様にして得られた組換え大腸菌を50μg/mLカナマイシンを含むLB寒天培地に塗抹した。
【0066】
このカナマイシン含有培地上で生育した株は、ショ糖含有培地にて生育不能であることが確認された。また、同株から調製したプラスミドDNAは、制限酵素HindIII消化
により354、473、1807、1997bpの断片を生じたことから、図2に示した構造に間違いがないと判断し、該プラスミドをpKMB1と命名した。
【0067】
参考例2
<LDH遺伝子破壊株の作製>
(A)ブレビバクテリウム・フラバムMJ233−ES株ゲノムDNAの抽出
A培地[尿素 2g、(NH4)2SO4 7g、KH2PO4 0.5g、K2HPO40.5g、MgSO4・7H2O 0.5g、FeSO4・7H2O 6mg、MnSO4・4−5H2O6mg、ビオチン 200μg、チアミン 100μg、イーストエキストラクト 1g
、カザミノ酸 1g、グルコース20g、蒸留水1Lに溶解]10mLに、ブレビバクテ
リウム・フラバムMJ−233株を対数増殖期後期まで培養し、得られた菌体から上記参考例1の(A)に示す方法にてゲノムDNAを調製した。
【0068】
(B)ラクテートデヒドロゲナーゼ遺伝子のクローニング
MJ233株ラクテートデヒドロゲナーゼ遺伝子の取得は、上記(A)で調製したDNAを鋳型とし、特開平11−206385号公報に記載の該遺伝子の塩基配列を基に設計した合成DNA(配列番号5および配列番号6)を用いたPCRによって行った。
反応液組成:鋳型DNA1μL、TaqDNAポリメラーゼ(宝酒造)0.2μL、1倍濃度添付バッファー、0.2μM各々プライマー、0.25μMdNTPsを混合し、全量を20μLとした。
【0069】
反応温度条件:DNAサーマルサイクラー PTC−200(MJResearch社製)を用い、94℃で20秒、55℃で20秒、72℃で1分からなるサイクルを30回繰り返した。但し、1サイクル目の94℃での保温は1分20秒、最終サイクルの72℃での保温は5分とした。
増幅産物の確認は、0.75質量%アガロース(SeaKem GTG agarose:FMCBioProducts製)ゲル電気泳動により分離後、臭化エチジウム染色により可視化することにより行い、約0.95kbの断片を検出した。ゲルからの目的DNA断片の回収は、QIAQuickGel Extraction Kit(QIAGEN製)を用いて行った。
【0070】
回収したDNA断片を、PCR産物クローニングベクターpGEM−TEasy(Promega製)と混合し、ライゲーションキットver.2(宝酒造製)を用いて連結後、得られたプラスミドDNAで大腸菌(DH5α株)を形質転換した。この様にして得られた組換え大腸菌を50μg/mLアンピシリンおよび50μg/mLX−Galを含むLB寒天培地に塗抹した。
この培地上で白色のコロニーを形成したクローンを、常法により液体培養した後、プラスミドDNAを精製した。得られたプラスミドDNAを制限酵素SacIおよびSphIで切断することにより、約1.0kbの挿入断片が認められ、これをpGEMT/CgLDHと命名した。
【0071】
(C)ラクテートデヒドロゲナーゼ遺伝子破壊用プラスミドの構築
上記(B)で作製したpGEMT/CgLDHを制限酵素EcoRVおよびXbaIで切断することにより約0.25kbからなるラクテートデヒドロゲナーゼのコーディング領域を切り出した。残った約3.7kbのDNA断片の末端をクレノウフラグメントにて平滑化し、ライゲーションキットver.2(宝酒造製)を用いて環状化させ、大腸菌(DH5α株)を形質転換した。この様にして得られた組換え大腸菌を50μg/mLアンピシリンを含むLB寒天培地に塗抹した。この培地上で生育した株を、常法により液体培養した後、プラスミドDNAを精製した。得られたプラスミドDNAを制限酵素SacIおよびSphIで切断することにより、約0.75kbの挿入断片が認められたクローンを選抜し、これをpGEMT/ΔLDHと命名した。
【0072】
次に、上記pGEMT/ΔLDHを制限酵素SacIおよびSphIにて切断して生じる約0.75kbのDNA断片を、0.75質量%アガロースゲル電気泳動により分離、回収し、欠損領域を含むラクテートデヒドロゲナーゼ遺伝子断片を調製した。このDNA断片を、制限酵素SacIおよびSphIにて切断した参考例1にて構築したpKMB1と混合し、ライゲーションキットver.2(宝酒造製)を用いて連結後、得られたプラスミドDNAで大腸菌(DH5α株)を形質転換した。この様にして得られた組換え大腸菌を50μg/mLカナマイシンおよび50μg/mLX−Galを含むLB寒天培地に
塗抹した。
この培地上で白色のコロニーを形成したクローンを、常法により液体培養した後、プラスミドDNAを精製した。得られたプラスミドDNAを制限酵素SacIおよびSphIで切断することにより、約0.75kbの挿入断片が認められたものを選抜し、これをpKMB1/ΔLDHと命名した(図3)。
【0073】
(D)ブレビバクテリウム・フラバムMJ233−ES株由来ラクテートデヒドロゲナーゼ遺伝子破壊株の作製
ブレビバクテリウム・フラバムMJ−233株の形質転換に用いるプラスミドDNAは、pKMB1/ΔLDHを用いて塩化カルシウム法(Journal of Molecular Biology,53,159,1970)により形質転換した大腸菌JM110株から調製した。
【0074】
ブレビバクテリウム・フラバムMJ233−ES株の形質転換は、電気パルス法(Res.Microbiol., Vol.144, p.181−185, 1993)によ
って行い、得られた形質転換体をカナマイシン 50μg/mLを含むLBG寒天培地[
トリプトン10g、イーストエキストラクト5g、NaCl5g、グルコース 20g、
及び寒天15gを蒸留水1Lに溶解]に塗抹した。
【0075】
この培地上に生育した株は、pKMB1/ΔLDHがブレビバクテリウム・フラバムMJ233−ES株菌体内で複製不可能なプラスミドであるため、該プラスミドのラクテートデヒドロゲナーゼ遺伝子とブレビバクテリウム・フラバムMJ−233株ゲノム上の同遺伝子との間で相同組み換えを起こした結果、同ゲノム上に該プラスミドに由来するカナマイシン耐性遺伝子およびSacB遺伝子が挿入されているはずである。
【0076】
次に、上記相同組み換え株をカナマイシン50μg/mLを含むLBG培地にて液体培養した。この培養液の菌体数約100万相当分を10質量%ショ糖含有LBG培地に塗抹にした。結果、2回目の相同組み換えによりSacB遺伝子が脱落しショ糖非感受性となったと考えられる株約10個得た。
この様にして得られた株の中には、そのラクテートデヒドロゲナーゼ遺伝子がpKMB1/ΔLDHに由来する変異型に置き換わったものと野生型に戻ったものが含まれる。ラクテートデヒドロゲナーゼ遺伝子が変異型であるか野生型であるかの確認は、LBG培地にて液体培養して得られた菌体を直接PCR反応に供し、ラクテートデヒドロゲナーゼ遺伝子の検出を行うことによって容易に確認できる。ラクテートデヒドロゲナーゼ遺伝子をPCR増幅するためのプライマー(配列番号7および配列番号8)を用いて分析すると、野生型では720bp、欠失領域を持つ変異型では471bpのDNA断片を認めるはずである。
上記方法にてショ糖非感受性となった菌株を分析した結果、変異型遺伝子のみを有する株を選抜し、該株をブレビバクテリウム・フラバムMJ233/ΔLDHと命名した。
【0077】
(E)ラクテートデヒドロゲナーゼ活性の確認
上記(D)で作製したブレビバクテリウム・フラバムMJ233/ΔLDH株をA培地に植菌し、30℃で15時間好気的に振とう培養した。得られた培養物を遠心分離(3,000×g、4℃、20分間)して菌体を回収後、ナトリウム−リン酸緩衝液[組成:50mMリン酸ナトリウム緩衝液(pH7.3)]で洗浄した。
【0078】
次いで、洗浄菌体0.5g(湿重量)を上記ナトリウム−リン酸緩衝液2mLに懸濁し、氷冷下で超音波破砕器(ブランソン社製)にかけ菌体破砕物を得た。該破砕物を遠心分離(10,000×g,4℃,30分間)し、上清を粗酵素液として得た。対照として、ブレビバクテリウム・フラバムMJ233−ES株の粗酵素液を同様に調製し、以下の活性測定に供した。
【0079】
ラクテートデヒドロゲナーゼ酵素活性の確認は、両粗酵素液について、ピルビン酸を基質とした乳酸の生成に伴い、補酵素NADHがNAD+に酸化されるのを、340nmの
吸光度変化として測定した[L. Kanarek and R. L. Hill, J.Biol. Chem. 239, 4202(1964)]。反応は、50mM カリウム−リン酸緩衝液(pH7.2)、10mM ピルビン酸、0.4mMNADH存在下、37
℃にて行った。その結果、ブレビバクテリウム・フラバムMJ233−ES株から調製された粗酵素液におけるラクテートデヒドロゲナーゼ活性に対し、ブレビバクテリウム・フラバムMJ233/ΔLDH株から調製された粗酵素液におけるラクテートデヒドロゲナーゼ活性は、10分の1以下であった。
【0080】
参考例3
<コリネ型細菌発現ベクターの構築>
(A)コリネ型細菌用プロモーター断片の調製
コリネ型細菌で強力なプロモーター活性を有することが報告された特開平7−95891号公報の配列番号4に記載のDNA断片(以降TZ4プロモーターと称する)を利用することとした。本プロモーター断片の取得は、参考例2の(A)で調製したブレビバクテリウム・フラバムMJ233ゲノムDNAを鋳型とし、特開平7−95891号公報の配列番号4に記載の配列を基に設計した合成DNA(配列番号9および配列番号10)を用いたPCRによって行った。
【0081】
反応液組成:鋳型DNA1μL、PfxDNAポリメラーゼ(インビトロジェン社製) 0.2μL、1倍濃度添付バッファー、0.3μM各々プライマー、1mM MgSO4、0.25μMdNTPsを混合し、全量を20μLとした。
反応温度条件:DNAサーマルサイクラー PTC−200(MJResearch社製)を用い、94℃で20秒、60℃で20秒、72℃で30秒からなるサイクルを35回繰り返した。但し、1サイクル目の94℃での保温は1分20秒、最終サイクルの72℃での保温は2分とした。
【0082】
増幅産物の確認は、2.0質量%アガロース(SeaKem GTG agarose:FMCBioProducts製)ゲル電気泳動により分離後、臭化エチジウム染色により可視化することにより行い、約0.25kbの断片を検出した。ゲルからの目的DNA断片の回収は、QIAQuickGel Extraction Kit(QIAGEN製)を用いて行った。
【0083】
回収したDNA断片は、T4ポリヌクレオチドキナーゼ(T4 Polynucleo
tide Kinase:宝酒造製)により5'末端をリン酸化した後、ライゲーションキットver.2(宝酒造製)を用いて大腸菌ベクターpUC19(宝酒造)のSmaI部位に結合し、得られたプラスミドDNAで大腸菌(DH5α株)を形質転換した。この様にして得られた組換え大腸菌を50μg/mLアンピシリンおよび50μg/mLX−Galを含むLB寒天培地に塗抹した。
【0084】
この培地上で白色のコロニーを形成した6クローンについて、常法により液体培養した後、プラスミドDNAを精製し、塩基配列を決定した。これ中でTZ4プロモーターがpUC19のlacプロモーターと逆方向に転写活性を有するように挿入されたクローンを選抜し、これをpUC/TZ4と命名した。
次に、pUC/TZ4を制限酵素BamHIおよびPstIで切断して調製したDNA断片に、5’末端がリン酸化された合成DNA(配列番号11および配列番号12)から成り、両末端にそれぞれBamHIとPstIに対する粘着末端を有するDNAリンカーを混合し、ライゲーションキットver.2(宝酒造製)を用いて連結後、得られたプラスミドDNAで大腸菌(DH5α株)を形質転換した。本DNAリンカーには、リボソーム結合配列(AGGAGG)およびその下流に配したクローニングサイト(上流から順に、PacI、NotI、ApaI)が含まれている。
【0085】
この培地上で白色のコロニーを形成したクローンを、常法により液体培養した後、プラスミドDNAを精製した。得られたプラスミドDNAの中から制限酵素NotIによって切断されるものを選抜し、これをpUC/TZ4−SDと命名した。
この様にして構築したpUC/TZ4−SDを制限酵素PstIで切断後、クレノウフラグメントにて末端を平滑化し、次いで制限酵素KpnIで切断することにより生じた約0.3kbのプロモーター断片を、2.0質量%アガロースゲル電気泳動により分離、回収した。
【0086】
(B)コリネ型細菌発現ベクターの構築
コリネ型細菌にて安定的に自立複製可能なプラスミドとして、特開平12−93183号公報記載のpHSG298par−repを利用する。本プラスミドは、ブレビバクテリウム・スタチオニスIFO12144株が保有する天然型プラスミドpBY503の複製領域および安定化機能を有する領域と大腸菌ベクターpHSG298(宝酒造)に由来するカナマイシン耐性遺伝子および大腸菌の複製領域を備える。pHSG298par−repを制限酵素SseIで切断後、クレノウフラグメントにて末端を平滑化し、次いで制限酵素KpnIで切断することによって調製したDNAを、上記(A)で調製したTZ4プロモーター断片と混合し、ライゲーションキットver.2(宝酒造製)を用いて連結後、得られたプラスミドDNAで大腸菌(DH5α株)を形質転換した。この様にして得られた組換え大腸菌を50μg/mLカナマイシンを含むLB寒天培地に塗抹した。
この培地上で生育した株を、常法により液体培養した後、プラスミドDNAを精製した。得られたプラスミドDNAの中から制限酵素NotIによって切断されるものを選抜し、該プラスミドをpTZ4と命名した(図3に構築手順を示した)。
【0087】
参考例4
<ピルベートカルボキシラーゼ活性増強株の作製>
(A)ピルベートカルボキシラーゼ遺伝子の取得
ブレビバクテリウム・フラバムMJ233株由来ピルベートカルボキシラーゼ遺伝子の取得は、参考例2の(A)で調製したDNAを鋳型とし、全ゲノム配列が報告されているコリネバクテリウム・グルタミカム ATCC13032株の該遺伝子の配列(GenBank Database Accession No.AP005276)を基に設計した合成DNA(配列番号13および配列番号14)を用いたPCRによって行った。
【0088】
反応液組成:鋳型DNA1μL、PfxDNAポリメラーゼ(インビトロジェン社製) 0.2μL、1倍濃度添付バッファー、0.3μM各々プライマー、1mM MgSO4、0.25μMdNTPsを混合し、全量を20μLとした。
反応温度条件:DNAサーマルサイクラー PTC−200(MJResearch社製)を用い、94℃で20秒、68℃で4分からなるサイクルを35回繰り返した。但し、1サイクル目の94℃での保温は1分20秒、最終サイクルの68℃での保温は10分とした。PCR反応終了後、Takara Ex Taq(宝酒造)を0.1μL加え、更に72℃で30分保温した。
【0089】
増幅産物の確認は、0.75質量%アガロース(SeaKem GTG agarose:FMCBioProducts製)ゲル電気泳動により分離後、臭化エチジウム染色により可視化することにより行い、約3.7kbの断片を検出した。ゲルからの目的DNA断片の回収は、QIAQuickGel Extraction Kit(QIAGEN製)を用いて行った。
【0090】
回収したDNA断片を、PCR産物クローニングベクターpGEM−TEasy(Promega製)と混合し、ライゲーションキットver.2(宝酒造製)を用いて連結後、得られたプラスミドDNAで大腸菌(DH5α株)を形質転換した。この様にして得られた組換え大腸菌を50μg/mLアンピシリンおよび50μg/mLX−Galを含むLB寒天培地に塗抹した。
【0091】
この培地上で白色のコロニーを形成したクローンを、常法により液体培養した後、プラスミドDNAを精製した。得られたプラスミドDNAを制限酵素PacIおよびApaIで切断することにより、約3.7kbの挿入断片が認められ、これをpGEM/MJPCと命名した。
pGEM/MJPCの挿入断片の塩基配列は、アプライドバイオシステム社製塩基配列解読装置(モデル377XL)およびビックダイターミネーターサイクルシークエンスキットver3を用いて決定した。その結果得られたDNA塩基配列および推測されるアミノ酸配列を配列番号15に記載する。また、アミノ酸配列のみを配列番号16に記載する。本アミノ酸配列はコリネバクテリウム・グルタミカムATCC13032株由来のそれと極めて高い相同性(99.4%)を示すことから、pGEM/MJPCの挿入断片がブレビバクテリウム・フラバムMJ233株由来のピルベートカルボキシラーゼ遺伝子であると断定した。
【0092】
(B)ピルベートカルボキシラーゼ活性増強用プラスミドの構築
上記(A)で作製したpGEM/MJPCを制限酵素PacIおよびApaIで切断することにより生じる約3.7kbからなるピルベートカルボキシラーゼ遺伝子断片を、0.75質量%アガロースゲル電気泳動により分離、回収した。
このDNA断片を、制限酵素PacIおよびApaIにて切断した、参考例3にて構築したpTZ4と混合し、ライゲーションキットver.2(宝酒造製)を用いて連結後、得られたプラスミドDNAで大腸菌(DH5α株)を形質転換した。この様にして得られた組換え大腸菌を50μg/mLカナマイシンを含むLB寒天培地に塗抹した。
この培地上で生育した株を、常法により液体培養した後、プラスミドDNAを精製した。得られたプラスミドDNAを制限酵素PacIおよびApaIで切断することにより、約3.7kbの挿入断片が認められたものを選抜し、これをpMJPC1と命名した(図5)。
【0093】
(C)ブレビバクテリウム・フラバムMJ233/ΔLDH株への形質転換
ブレビバクテリウム・フラバムMJ233株内で複製可能なpMJPC1による形質転換用のプラスミドDNAは、上記(B)で形質転換した大腸菌(DH5α株)から調製した。
ブレビバクテリウム・フラバムMJ233/ΔLDH株への形質転換は、電気パルス法(Res. Microbiol., Vol.144,p.181−185, 1993)によって行い、得られた形質転換体をカナマイシン 50μg/mLを含むLBG寒天培
地[トリプトン10g、イーストエキストラクト5g、NaCl5g、グルコース 20
g、及び寒天15gを蒸留水1Lに溶解]に塗抹した。
この培地上に生育した株から、常法により液体培養した後、プラスミドDNAを抽出、制限酵素切断による解析を行った結果、同株がpMJPC1を保持していることを確認し、該株をブレビバクテリウム・フラバムMJ233/PC/ΔLDH株と命名した。
【0094】
(D)ピルベートカルボキシラーゼ酵素活性
上記(C)で得られた形質転換株ブレビバクテリウム・フラバムMJ233/PC/ΔLDH株をグルコース2質量%、カナマイシン25mg/Lを含むA培地100mlで終夜培養を行った。得られた菌体を集菌後、50mMリン酸カリウム緩衝液(pH7.5)50mlで洗浄し、同組成の緩衝液20mlに再度懸濁させた。懸濁液をSONIFIER350(BRANSON製)で破砕し、遠心分離した上清を無細胞抽出液とした。得られた無細胞抽出液を用いピルベートカルボキシラーゼ活性を測定した。酵素活性の測定は100mMTris/HCl緩衝液(pH7.5)、 0.1mg/10mlビオチン、
5mM塩化マグネシウム、50mM 炭酸水素ナトリウム、5mM ピルビン酸ナトリウム
、5mM アデノシン三リン酸ナトリウム、0.32mM NADH、20units/
1.5mlリンゴ酸デヒドロゲナーゼ(WAKO製、酵母由来)及び酵素を含む反応液中で25℃で反応させることにより行った。1Uは1分間に1μmolのNADHの減少を触媒する酵素量とした。ピルベートカルボキシラーゼを発現させた無細胞抽出液における比活性は0.2U/mg蛋白質であった。尚親株であるMJ233/△LDH株をA培地を用いて同様に培養した菌体では、本活性測定方法によりピルベートカルボキシラーゼ活性
は検出されなかった。
【0095】
参考例5
<発酵液の調製>
尿素:4g、硫酸アンモニウム:14g、リン酸1カリウム:0.5g、リン酸2カリウム0.5g、硫酸マグネシウム・7水和物:0.5g、硫酸第一鉄・7水和物:20mg、硫酸マンガン・水和物:20mg、D−ビオチン:200μg、塩酸チアミン:200μg、酵母エキス:1g、カザミノ酸:1g、及び蒸留水:1000mLの培地100mLを500mLの三角フラスコにいれ、120℃、20分加熱滅菌した。これを室温まで冷やし、あらかじめ滅菌した50質量%グルコース水溶液を4mL、無菌濾過した5質量%カナマイシン水溶液を50μL添加し、参考例4(C)で作製したブレビバクテリウム・フラバムMJ233/PC/ΔLDH株を接種して24時間30℃にて種培養した。
【0096】
尿素:12g、硫酸アンモニウム:42g、リン酸1カリウム:1.5g、リン酸2カリウム1.5g、硫酸マグネシウム・7水和物:1.5g、硫酸第一鉄・7水和物:60mg、硫酸マンガン・水和物:60mg、D−ビオチン:600μg、塩酸チアミン:600μg、酵母エキス3g、カザミノ酸3g、消泡剤(アデカノールLG294:旭電化製):1mL及び蒸留水:2500mLの培地を5Lの発酵糟に入れ、120℃、20分加熱滅菌した。これを室温まで冷やした後、あらかじめ滅菌した12質量%グルコース水溶液を500mL添加し、これに前述の種培養液を全量加えて、30℃に保温した。通気は毎分500mL、攪拌は毎分500回転で本培養を行った。12時間後にグルコースがほぼ消費されていた。
【0097】
硫酸マグネシウム・7水和物:1.5g、硫酸第一鉄・7水和物:60mg、硫酸マンガン・水和物:60mg、D−ビオチン:600μg、塩酸チアミン:600μg、消泡剤(アデカノールLG294:旭電化製):5ml及び蒸留水:1.5Lの培地を3Lの三角フラスコに入れ、120℃、20分加熱滅菌した。室温まで冷やした後、上記の本培養により得られた培養液を10000g、5分の遠心分離により集菌した菌体を添加して、O.D.(660nm)が60になるように再懸濁した。この懸濁液1.5Lとあらかじめ滅菌した20質量%グルコース溶液1.5Lを5Lのジャーファーメンターに入れて混合し、35℃に保温した。pHは2M炭酸アンモニウムを用いて7.6に保ち、毎分500mLで通気、毎分300回転で攪拌しながら反応を行った。反応開始後約50時間でグルコースがほぼ消費されていた。コハク酸が57g/L蓄積されていた。この発酵液を10000g、5分間の遠心分離、限外濾過(日東電工(株)製 NTU−3000−C1R)により菌体と上清に分離した。以上の操作を30回行うことにより、コハク酸発酵液上清を103L得ることが出来た。
【0098】
<コハク酸醗酵液からのコハク酸精製>
上記のようにして得られたコハク酸発酵液上清を103L(コハク酸含有量5.87kg)を、減圧しながらジャケット付き攪拌槽にて濃縮し、コハク酸の濃度が32.9質量
%、アンモニア11.9質量%の濃縮液:17.8kg(計算値)を得た。これに酢酸(ダイセル化学社製)を8.58kg加えて30℃まで冷却し、更にメタノール(キシダ化学社製)を4.0kg加えて15℃まで冷却し1時間攪拌した後、20℃にて4時間攪拌を継続した。
【0099】
結晶が析出しており、これを遠心ろ過器にてろ過を行い、コハク酸を74.6質量%、酢酸3.5質量%、アンモニア12.2質量%を含有する結晶4.95kgを得た。
酢酸11.3kgに得られた結晶4.9kgを入れ、85℃にて溶解し、直ちに20℃まで冷却した。既に結晶は析出していたが、そのまま更に3時間攪拌を続けた後、遠心ろ過器にてろ過を行い、コハク酸87.9質量%、酢酸8.4質量%、アンモニア0.6質
量%を含有する結晶2.44kgを得た。
【0100】
5℃に冷やした脱塩水3.5Lにて得られた結晶を懸洗し、これを遠心ろ過器にてろ過すると、コハク酸90質量%、酢酸1.7質量%、アンモニア0.05質量%(およそ500質量ppm)含有する2.08kgの結晶が得られた。
この粗コハク酸結晶2.0kgを28.5Lの脱塩水に溶解し、1Lのイオン交換樹脂(三菱化学社製SK1BH)をつめた塔にSV=2にて通液し、約33Lの処理液を得た。これを減圧したロータリーエバポレータに連続フィードしながら、およそ5.2Lまで濃縮した。この段階で既に結晶が析出していた。更に、5℃に冷却し、2時間攪拌を継続した後、これをろ過すると、コハク酸96.7質量%の結晶1.76kgを得た。 これを真空乾燥機にて乾燥すると1.68kgのコハク酸を得る事が出来た。
【0101】
<1,4−ブタンジオールの製造>
上記のような方法で得られたバイオマス資源由来コハク酸を用いて、以下の方法で1,4−ブタンジオールを得た。
バイオマス資源由来コハク酸100重量部、メタノール317重量部ならびに濃硫酸(97質量%)2重量部の混合液を、還流下で2時間攪拌させた。反応液を冷却後、炭酸水素ナトリウム3.6重量部を添加して60℃で30分間反応液を攪拌させた。常圧下での蒸留ならびにその蒸留残をろ過後、減圧蒸留することによりコハク酸ジメチル(収率93%)を得た。得られたコハク酸ジメチル100重量部をCuO−ZnO触媒(ズードケミー社製「T―8402」)15重量部存在下、仕込みコハク酸ジメチルに対して約4倍の体積容量を持つオートクレーブ(ハステロイC)を用いて水素5MPa加圧下で攪拌させながら1時間かけて230℃まで昇温させた。その後、230℃で15MPaの水素加圧下9時間反応液を攪拌させた。反応液を冷却後、脱ガスを行った。反応液からろ過により触媒を除去した。ろ液を減圧蒸留することにより精製1,4−ブタンジオールを得た(収率81%)。製造された精製1,4−ブタンジオール中には窒素原子が0.7質量ppm含まれたが、硫黄原子は含まれていなかった。また、1,4−ブタンジオール中には酸化生成物である2-(4-ヒドロキシブチルオキシ)テトラヒドロフランが584質量ppm含有されていた。
【0102】
[実施例1]
攪拌装置、窒素導入口、加熱装置、温度計、留出管、減圧用排気口を備えた反応容器に、ジメチルテレフタレート(帝人製)132質量部、窒素原子0.7質量ppmを含有する製造例1で得られたBG74質量部及び触媒としてテトラブチルチタネートをあらかじめ6質量%溶解させた1,4―ブタンジオール溶液1.7質量部を仕込み、窒素―減圧置換によって系内を窒素雰囲気下にした。
【0103】
次に、系内を撹拌しながら150℃まで加温後、215℃に昇温しながらエステル交換反応によって生成するメタノールを留出させつつ3時間反応した。次に、1.5時間かけて245℃まで昇温するとともに、1.5時間かけて0.07kPaになるように減圧し、同減圧度で1.5時間重縮合反応を行い、反応系を常圧に戻し重縮合を終了した。得られたPBTを反応槽の底部からストランドとして抜き出し、10℃の水中を潜らせた後、カッターでストランドをカットすることによりペレット状のPBTを得た。
【0104】
得られたPBT中の窒素原子含有量は、0.4質量ppm、PBTの固有粘度(IV)は1.06dL/g、末端カルボキシル基量は21当量/トンあった。減圧開始から重縮合終了までを重縮合時間として、固有粘度/重縮合時間を重縮合速度とした。
重縮合速度は0.35dL/g/hであった。
THF転化率は、エステル交換反応中の留出液をドライアイストラップで冷却採取したものについてTHF量を分析し、ジメチルテレフタレートあたりのモル%で表した。2モ
ル%であった。結果を表1に示す。
【0105】
[比較例1]
実施例1においてBGを化石燃料原料由来のBG(三菱化学製BG)に変え、重縮合時間を表1に示す時間に変えた以外は実施例1と同様に行い、PBTを得た。各種分析結果を表1に示す。
[実施例2]
攪拌装置、窒素導入口、加熱装置、温度計及び減圧用排気口を備えた反応容器に、テレフタル酸113質量部、窒素原子0.7質量ppmを含有する製造例1で得られたBG183質量部及び触媒としてテトラブチルチタネートをあらかじめ6質量%溶解させた1,4―ブタンジオール溶液0.7質量部を仕込み、窒素―減圧置換によって系内を窒素雰囲気下にした。
【0106】
次に、系内を撹拌しながら150℃まで加温後、圧力を78kPaになるよう減圧した後、220℃に1時間で昇温させて、さらに2時間生成する水を留出させつつエステル化反応した後窒素ガスにて常圧に戻した。
次に、酢酸マグネシウム4水塩を水に溶解し、さらにBGに溶解させた酢酸マグネシウム4水塩1質量%の1,4―ブタンジオール溶液(酢酸マグネシウム4水塩、水、1,4―ブタンジオールの質量比は1:2:97)1.3質量部を添加した。
【0107】
次に、1.5時間かけて245℃まで昇温するとともに、1.5時間かけて0.07kPaになるように減圧し、同減圧度で1.1時間重縮合反応を行い、反応系を常圧に戻し重縮合を終了した。得られたPBTを反応槽の底部からストランドとして抜き出し、10℃の水中を潜らせた後、カッターでストランドをカットすることによりペレット状のPBTを得た。
【0108】
酢酸マグネシウム添加後の減圧開始から重縮合終了までを重縮合時間として、固有粘度/重縮合時間を重縮合速度とした。重縮合速度は0.35dL/g/hであった。THF転化率は、エステル化反応中の留出液をドライアイストラップで冷却採取したものについてTHF量を分析し、仕込みテレフタル酸あたりのモル%で表した。54モル%であった。
得られたぺレット状のPBTを射出成形にて引張試験片、曲げ試験片に成形し引張強度と曲げ弾性率を測定した。
各種分析結果を表1に示す。
【0109】
[比較例2]
実施例2においてBGを化石燃料原料由来のBG(三菱化学製)に変え、重縮合時間を表1に示す時間に変えた以外は実施例2と同様に行い、PBTを得た。各種分析結果を表1に示す。
【0110】
[比較例3]
実施例2においてBGを化石燃料原料由来のBG(東燃化学製)に変え、重縮合時間を表1に示す時間に変えた以外は実施例2と同様に行い、PBTを得た。各種分析結果を表1に示す。
表1より、実施例は、重縮合速度が大きく、THF化への転化率が小さいこと、及び実施例のPBTは窒素含有量が少なく、色調が良好であること、等がわかる。
【産業上の利用可能性】
【0111】
本発明のPBTは、色調良好である。また、本発明のPBTの製法は、効率的に、THFへの転化が少ない。更に、バイオマス資源の有効利用にも寄与している。
【0112】
【表1】
【図面の簡単な説明】
【0113】
【図1】引張試験片の形状 単位mm。
【図2】pKMB1の構築の概略を示す。
【図3】pKMB1/ΔLDHの構築の概略を示す。
【図4】pTZベクターの構築の概略を示す。
【図5】pMJPC1の構築の概略を示す。
【図6】配列番号
【図7】配列番号
【図8】配列番号
【図9】配列番号
【図10】配列番号
【図11】配列番号
【図12】配列番号
【図13】配列番号
【図14】配列番号
【図15】配列番号
【図16】配列番号
【図17】配列番号
【特許請求の範囲】
【請求項1】
テレフタル酸由来の構成単位と1,4ブタンジオール由来の構成単位を含み、窒素原子含有量が0.01〜50質量ppmであることを特徴とするポリブチレンテレフタレート。
【請求項2】
請求項1記載のポリブチレンテレフタレートであって、前記1,4ブタンジオールがバイオマス資源由来であることを特徴とするポリブチレンテレフタレート。
【請求項3】
請求項1又は2記載のポリブチレンテレフタレートであって、前記1,4ブタンジオールの窒素原子含有量が0.01〜50質量ppmであることを特徴とするポリブチレンテレフタレート。
【請求項4】
請求項1〜3記載のポリブチレンテレフタレートであって、前記1,4ブタンジオール中の1−アセトキシ4−ヒドロキシブタンの含有量が0.1〜50質量ppmであることを特徴とするポリブチレンテレフタレート。
【請求項5】
請求項1〜4記載のポリブチレンテレフタレート99.9〜0.1質量%に対して、熱可塑性樹脂0.1〜99.9質量%を配合することにより得られる樹脂組成物。
【請求項6】
請求項1〜4記載のポリブチレンテレフタレート又は請求項5記載の樹脂組成物を成形してなる成形体。
【請求項7】
テレフタル酸又はテレフタル酸アルキレートと窒素原子含有量が0.01〜50質量ppmの1,4ブタンジオールとをエステル化反応又はエステル交換反応させた後、重縮合反応させるポリブチレンテレフタレートの製造方法。
【請求項1】
テレフタル酸由来の構成単位と1,4ブタンジオール由来の構成単位を含み、窒素原子含有量が0.01〜50質量ppmであることを特徴とするポリブチレンテレフタレート。
【請求項2】
請求項1記載のポリブチレンテレフタレートであって、前記1,4ブタンジオールがバイオマス資源由来であることを特徴とするポリブチレンテレフタレート。
【請求項3】
請求項1又は2記載のポリブチレンテレフタレートであって、前記1,4ブタンジオールの窒素原子含有量が0.01〜50質量ppmであることを特徴とするポリブチレンテレフタレート。
【請求項4】
請求項1〜3記載のポリブチレンテレフタレートであって、前記1,4ブタンジオール中の1−アセトキシ4−ヒドロキシブタンの含有量が0.1〜50質量ppmであることを特徴とするポリブチレンテレフタレート。
【請求項5】
請求項1〜4記載のポリブチレンテレフタレート99.9〜0.1質量%に対して、熱可塑性樹脂0.1〜99.9質量%を配合することにより得られる樹脂組成物。
【請求項6】
請求項1〜4記載のポリブチレンテレフタレート又は請求項5記載の樹脂組成物を成形してなる成形体。
【請求項7】
テレフタル酸又はテレフタル酸アルキレートと窒素原子含有量が0.01〜50質量ppmの1,4ブタンジオールとをエステル化反応又はエステル交換反応させた後、重縮合反応させるポリブチレンテレフタレートの製造方法。
【図1】


【図2】
【図3】
【図4】
【図5】
【図6】

【図7】

【図8】

【図9】

【図10】

【図11】

【図12】

【図13】

【図14】

【図15】

【図16】

【図17】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【公開番号】特開2008−101143(P2008−101143A)
【公開日】平成20年5月1日(2008.5.1)
【国際特許分類】
【出願番号】特願2006−285493(P2006−285493)
【出願日】平成18年10月19日(2006.10.19)
【出願人】(000005968)三菱化学株式会社 (4,356)
【Fターム(参考)】
【公開日】平成20年5月1日(2008.5.1)
【国際特許分類】
【出願日】平成18年10月19日(2006.10.19)
【出願人】(000005968)三菱化学株式会社 (4,356)
【Fターム(参考)】
[ Back to top ]