
ポリプロピレン系押出発泡体、および、その製造方法
【課題】従来の押出発泡装置を用いて製造でき、窒素ガスを発泡剤として用い、高発泡倍率のポリプロピレン系押出発泡体およびその製造方法を提供する。
【解決手段】ポリプロピレン系樹脂と窒素ガスとを含有する混合物を押出機内で溶融混練した後、ダイから押出発泡させてポリプロピレン系押出発泡体を得る。ポリプロピレン系樹脂は、以下のAおよびBを満たし、低圧領域に押出発泡した際、発泡倍率が3倍以上である。(A)測定温度210℃、剪断速度1216s-1の条件でのキャピラリーフローテストにおいて、バーグレー補正における圧力補正値が4MPa以上である。(B)メルトフローレート(MFR)が0.5g/10分以上である。
【解決手段】ポリプロピレン系樹脂と窒素ガスとを含有する混合物を押出機内で溶融混練した後、ダイから押出発泡させてポリプロピレン系押出発泡体を得る。ポリプロピレン系樹脂は、以下のAおよびBを満たし、低圧領域に押出発泡した際、発泡倍率が3倍以上である。(A)測定温度210℃、剪断速度1216s-1の条件でのキャピラリーフローテストにおいて、バーグレー補正における圧力補正値が4MPa以上である。(B)メルトフローレート(MFR)が0.5g/10分以上である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ポリプロピレン系の押出発泡体、および、その製造方法に関する。
【背景技術】
【0002】
従来、自動車の内外装あるいは住宅等の建物の内外装には樹脂発泡体が多用されている。例えば、自動車では、室内空間に面する天井、ドア、フロア等の内装、あるいはカウルやボディ等の外装には、樹脂発泡体を用いたボードやパネルが利用されている。また、住宅設備においても、住宅用断熱ボードなどの建材として樹脂発泡体が利用されている。
このような樹脂発泡体の材料としてポリスチレン等が用いられていたが、耐熱性、耐薬品性、軽量性という観点からポリプロピレン系樹脂が好適とされている。
【0003】
しかしながら、ポリプロピレン系樹脂はポリスチレンと比較して発泡成形性に劣るため、押出発泡成形では、発泡倍率が3倍を超える押出発泡成形体を安定供給するのは困難であった。特に、直鎖状ポリプロピレンでは、伸長粘度の歪硬化性に乏しく、発泡成形過程において、隣接する気泡壁が破れやすかった。
そこで、ポリプロピレンの分子鎖に分岐構造を与えることで高い溶融張力を発現させ、発泡成形性を改善させた材料が開発された(例えば、特許文献1参照)。
【0004】
また、超高分子量成分の分子量と量比を最適化することにより、気泡形成過程の高速伸長流動場で大きな伸長粘度を生じさせることにより気泡壁を破れにくくしたポリプロピレン系樹脂組成物が発明された(例えば、特許文献2参照)。
そして、上記ポリプロピレン系樹脂組成物によれば、超臨界二酸化炭素を用いた押出発泡成形により発泡倍率が10倍を超える高発泡体が得られている(例えば、特許文献3)。
【0005】
さらに、ポリプロピレン系樹脂による発泡成形として、超臨界状態の窒素を発泡剤として発泡成形する方法が開示されている(例えば、特許文献4参照)。
【0006】
【特許文献1】特公平7−45551号公報
【特許文献2】特開2007−119760号公報
【特許文献3】WO2006/118160
【特許文献4】特開2005−97389号公報
【発明の開示】
【発明が解決しようとする課題】
【0007】
しかしながら、特許文献1において、分岐構造をもつ高分子はその構造が壊れやすいため、押出成形においては流動過程でその粘弾特性が変化するおそれがある。また、分岐構造をもつ高分子はリサイクルに不向きであるという問題がある。さらに、分岐構造があるため、低歪速度下での伸長粘度の立ち上がりを大きくし、シートの引取性ドローダウン性の向上に有効であるものの、気泡の破壊に寄与する高速度下での伸長変形においては必ずしも良好な特性が得られるものではなかった。
【0008】
また、特許文献2および3では、発泡剤として窒素の使用も示唆されてはいるが、窒素を使った発泡倍率3〜20倍の発泡体の製造例については開示されていない。
さらに、特許文献4の発泡体では、分岐構造を持つ高分子を用いて、射出発泡成形により発泡倍率が3倍を超える発泡体が得られた実施の形態が示されているが、押出発泡成形に関しては、実施の形態は示されていない。射出発泡成形においては、溶融した樹脂材料を金型内に射出し、その後、金型を所定量移動させて発泡させる、という工程を取るため、金型の温度や、金型を移動させるまでの時間、金型の移動速度などを任意に設定する事ができるため、発泡倍率の調整は比較的容易である。しかしながら、押出発泡成形においては、ダイの出口以降は自由表面であり、発泡倍率の制御は困難である。
【0009】
また、ダイ出口の流路面積を小さくするとダイ壁面の剪断速度が大きくなり、メルトフラクチャが発生しやすくなるとともに、コルゲートマークなどの不良現象が発生しやすくなる。すなわち、発泡体の外観不良が生じやすいという問題がある。
【0010】
本発明の目的は、従来の押出発泡装置を用いて製造でき、比較的に高発泡倍率かつ表面外観の良好なポリプロピレン系押出発泡体、および、その製造方法を提供することである。
【課題を解決するための手段】
【0011】
本発明のポリプロピレン系押出発泡体の製造方法は、ポリプロピレン系樹脂と発泡材料とを含有する混合物を押出機内で溶融混練する溶融混練工程と、この溶融混練工程で溶融混練された組成物をダイから押出発泡させる押出発泡工程と、を実施するポリプロピレン系押出発泡体の製造方法であって、前記ポリプロピレン系樹脂は、(A)測定温度210℃、剪断速度1216s-1の条件でのキャピラリーフローテストにおいて、バーグレー補正における圧力補正値が4MPa以上、および(B)メルトフローレート(MFR)が0.5g/10分以上であり、前記溶融混練工程では、前記ポリプロピレン系樹脂に前記発泡材料として窒素ガスを前記ポリプロピレン系樹脂に対して0.6質量%以上5質量%以下で含有させ、前記ポリプロピレン系押出発泡体の発泡倍率が3倍以上となる条件とすることを特徴とする。
ここで、キャピラリーフローテストとは、「キャピラリーレオメータによる流れ特性試験方法(JIS K 7199)」の規定に基づいて行われる。
剪断速度は、ここでは見かけの剪断速度を表し、キャピラリーダイの直径と流量から求めることができる。
バーグレー補正における圧力補正値とは、キャピラリーダイの入口と出口の圧力損失の和である。キャピラリーダイの直径Dとキャピラリーダイの長さLの比(L/D)を複数設定して、それぞれのテストを行うことによって求められる。この圧力補正値が大きいと、入口圧損が大きいことを示しており、入口部での縮小流に対する抵抗が大きい。すなわち、伸長粘度が大きいことを示している。
【0012】
具体的には、例えば、バレルの直径9.55mm、キャピラリーダイの直径1.0mm、キャピラリーダイの流入角90°、L/Dが30、40、50の3種を設定して圧力補正値を求めることができる。しかしながら、これらの値に限られず、流入角が90°以上かつバレルとキャピラリーダイの直径の比が8以上であればよい。
なお、剪断速度1216s-1における測定値がない場合は、この剪断速度の上下の隣接する2点の剪断速度における圧力補正値のデータを用いて補間することにより、剪断速度1216s-1での圧力補正値を算出してもよい。
【0013】
この発明によれば、ポリプロピレン系押出発泡体の材料であるポリプロピレン系樹脂は、剪断速度1216s-1という高速度下での圧力補正値が4MPa以上であることにより、気泡形成時のような高速度下において伸長粘度が高いということが言える。すなわち、気泡が破けにくい。したがって、ポリプロピレン系樹脂には溶解しにくい窒素ガスでも、発泡倍率が3倍以上のポリプロピレン系押出発泡体が得られる。
【0014】
また、ポリプロピレン系樹脂のメルトフローレート(MFR)を0.5g/10分以上とした。MFRが0.5g/10分未満であると、樹脂の流動性が劣り、生産性が悪い。すなわち、樹脂のMFRを0.5g/10分以上とすることにより、押出機での製造を容易に行うことができる。一方、MFRが100g/10分を越えると、ポリプロピレン系樹脂の溶融張力および粘度が低くなり、押出成形が困難となる場合がある点を留意する。従って、MFRのより好ましい範囲は、1.0g/10分以上10.0g/10分以下であり、2.0g/10分以上5.0g/10分以下であることが特に好ましい。
【0015】
以上のような性質を満たすポリプロピレン系樹脂としては、以下に述べるプロピレン系多段重合体、プロピレン単独重合体、プロピレンと他のオレフィンとの共重合体、またはこれらをブレンドしたものが用いられる。
【0016】
プロピレン系多段重合体としては、下記成分(P1)および成分(P2)で構成される。
(P1)135℃、テトラリン溶媒中で測定した極限粘度[η]が10dL/g超のプロピレン単独重合体成分またはプロピレンと炭素数が2〜8のα−オレフィンとの共重合体成分を、全重合体中に5〜20質量%含有する。
(P2)135℃、テトラリン溶媒中で測定した極限粘度[η]が0.5〜3.0dL/gのプロピレン単独重合体成分またはプロピレンと炭素数が2〜8のα−オレフィンとの共重合体成分を、全重合体中に80〜95質量%含有する。
このプロピレン系多段重合体は、成分(P1)すなわち超高分子量プロピレン系重合体の付与により高溶融張力化を達成し、分子量分布の調整により粘弾性特性が調整された直鎖状のプロピレン系重合体である。
【0017】
成分(P1)の極限粘度が10dL/g以下では、溶融張力が不十分となり、所望の発泡性能を得ることができない場合がある。また、成分(P1)の質量分率が5質量%より小さいと、溶融張力が不十分となり、所望の発泡性能を得ることができない場合がある。一方、質量分率が20質量%を越えると、いわゆるメルトフラクチャが激しくなる場合があり、発泡成形体の肌荒れ等の原因となり、製品品質が低下する。
成分(P1)の極限粘度は、前記したように10dL/g超であることが好ましいが、12〜30dL/gの範囲内であることがより好ましく、13〜18dL/gの範囲内であることが特に好ましい。
また、成分(P1)の質量分率は、8〜18質量%の範囲内であることが好ましく、10〜16質量%の範囲内であることが特に好ましい。
【0018】
成分(P2)の極限粘度が0.5dL/gより小さいと、溶融張力が不十分となり、所望の発泡性能を得ることができない場合がある。一方、3.0dL/gを越えると、粘度が高すぎ、好適な発泡成形体を成形することができない場合がある。
また、成分(P2)の質量分率が80質量%より小さいと、好適な発泡成形の実施が困難となる場合があり、質量分率が95質量%を越えると、溶融張力が低くなり、これも好適な発泡成形体の成形が困難となる場合がある。
成分(P2)の極限粘度は、前記したように0.5〜3.0dL/gの範囲内であることが好ましいが、0.8〜2.0dL/gの範囲内であることがより好ましく、1.0〜1.5dL/gの範囲内であることがさらに好ましい。
また、成分(P2)の質量分率は、82〜92質量%の範囲内であることが好ましく、84〜90質量%の範囲内であることが特に好ましい。
さらに、プロピレン系多段重合体の極限粘度は、好ましくは1.0dL/g以上6.0dL/g以下、より好ましくは2.0dL/g以上4.0dL/g以下、さらに好ましくは3.0dL/g以上3.5dL/g以下である。1.0dL/g未満では発泡性が悪くなるおそれがあり、6.0dL/gを越えると成形が困難となるおそれがあるためである。
【0019】
本実施形態で用いるプロピレン系多段重合体において、共重合体成分を構成する炭素数2〜8のα−オレフィンとしては、例えば、プロピレン以外のα−オレフィンであるエチレン、1−ブテン等が挙げられる。このうち、エチレンを使用することが好ましい。
プロピレン系多段重合体は、230℃におけるメルトフローレート(MFR)と、230℃における溶融張力(MT)との関係が、下記式(I)を満たすことが好ましい。
【0020】
log(MT)>−1.33log(MFR)+1.2 …(I)
【0021】
ここで、230℃におけるメルトフローレート(MFR)と、230℃における溶融張力(MT)との関係が、前記式(I)を満たさない場合にあっては、高倍率の発泡成形の実施が困難となる場合がある。前記した定数(1.2)は、1.3以上とすることが好ましく、1.4以上とすることが特に好ましい。
なお、プロピレン系多段重合体が前記した式(I)の関係を具備するようにするには、成分(P1)を5〜20質量%含有させるようにすればよい。
【0022】
また、プロピレン系多段重合体は、溶融状態の動的粘弾性(角周波数ωと貯蔵弾性率G’との関係)として、高周波数側での貯蔵弾性率の傾きが一定量以上の大きさであることが好ましく、具体的には、角周波数が10rad/s(ラジアン/秒)の場合の貯蔵弾性率G’(10)と、角周波数が1rad/sの場合の貯蔵弾性率G’(1)との比であるG’(10)/G’(1)が2.0以上であることが好ましく、2.5以上であることが特に好ましい。この比G’(10)/G’(1)が2.0より小さいと、発泡成形体に延伸等の外的変化を加えた際の安定性が低下する場合がある。
【0023】
同様に、プロピレン系多段重合体は、溶融状態の動的粘弾性として、低周波数側での貯蔵弾性率の傾きが、一定量以下の大きさであることが好ましく、具体的には、角周波数が0.1rad/sの場合の貯蔵弾性率G’(0.1)と、角周波数が0.01rad/sの場合の貯蔵弾性率G’(0.01)との比であるG’(0.1)/G’(0.01)が6.0以下であることが好ましく、4.0以下であることが特に好ましい。かかる比G’(0.1)/G’(0.01)が6.0を越えると、低剪断速度下で発泡成形体の発泡倍率を高くすることが困難となる場合がある。
【0024】
このようなプロピレン系多段重合体は、下記成分(a)及び(b)、または下記成分(a)、(b)及び(c)からなるオレフィン重合用触媒を用い、2段階以上の重合工程で、プロピレンを重合またはプロピレンと炭素数2〜8のα−オレフィンとを共重合させて製造することができる。
【0025】
(a)四塩化チタンを有機アルミニウム化合物で還元して得られる三塩化チタンを、エーテル化合物及び電子受容体で処理して得られる固体触媒成分
(b)有機アルミニウム化合物
(c)環状エステル化合物
【0026】
固体触媒成分(a)において、四塩化チタンを還元する有機アルミニウム化合物としては、例えば、(a1)アルキルアルミニウムジハライド、具体的には、メチルアルミニウムジクロライド、エチルアルミニウムジクロライド、及びn−プロピルアルミニウムジクロライド、(a2)アルキルアルミニウムセスキハライド、具体的には、エチルアルミニウムセスキクロライド、(a3)ジアルキルアルミニウムハライド、具体的には、ジメチルアルミニウムクロライド、ジエチルアルミニウムクロライド、ジ−n−プロピルアルミニウムクロライド、及びジエチルアルミニウムブロマイド、(a4)トリアルキルアルミニウム、具体的には、トリメチルアルミニウム、トリエチルアルミニウム、及びトリイソブチルアルミニウム、(a5)ジアルキルアルミニウムハイドライド、具体的には、ジエチルアルミニウムハイドライド等をあげることができる。ここで、「アルキル」とは、メチル、エチル、プロピル、ブチル等の低級アルキルである。また、「ハライド」とは、クロライドまたはブロマイドであり、特に前者が通常である。
【0027】
三塩化チタンを得るための、有機アルミニウム化合物による還元反応は、−60〜60℃、好ましくは−30〜30℃の温度範囲で実施することが通常である。還元反応における温度が−60℃より低いと、還元反応に長時間が必要となり、一方、還元反応における温度が60℃を超えると、部分的に過還元が生じる場合があり好ましくない。還元反応は、ペンタン、ヘプタン、オクタン及びデカン等の不活性炭化水素溶媒下において実施することが好ましい。
【0028】
四塩化チタンの有機アルミニウム化合物による還元反応によって得られた三塩化チタンに対して、更にエーテル処理及び電子受容体処理を施すことが好ましい。
前記三塩化チタンのエーテル処理で好ましく用いられるエーテル化合物としては、例えば、ジエチルエーテル、ジ−n−プロピルエーテル、ジ−n−ブチルエーテル、ジイソアミルエーテル、ジネオペンチルエーテル、ジ−n−ヘキシルエーテル、ジ−n−オクチルエーテル、ジ−2−エチルヘキシルエーテル、メチル−n−ブチルエーテル及びエチル−イソブチルエーテル等の各炭化水素残基が炭素数2〜8の鎖状炭化水素であるエーテル化合物が挙げられ、これらの中でも特に、ジ−n−ブチルエーテルを用いることが好適である。
【0029】
三塩化チタンの処理で用いられる電子受容体としては、周期律表第III族〜第IV族及び第VIII族の元素のハロゲン化合物を使用することが好ましく、具体的には、四塩化チタン、四塩化ケイ素、三フッ化ホウ素、三塩化ホウ素、五塩化アンチモン、三塩化ガリウム、三塩化鉄、二塩化テルル、四塩化スズ、三塩化リン、五塩化リン、四塩化バナジウム及び四塩化ジルコニウム等を挙げることができる。
【0030】
固体触媒成分(a)を調製する際に、三塩化チタンのエーテル化合物及び電子受容体による処理は、両処理剤の混合物を用いて行ってもよく、また、一方の処理剤による処理後に、他方の処理剤による処理を行うようにしてもよい。なお、これらのうちでは、後者が好ましく、エーテル処理後に電子受容体で処理を行うことが更に好ましい。
【0031】
エーテル化合物及び電子受容体による処理の前に、三塩化チタンを炭化水素で洗浄することが好ましい。前記した三塩化チタンによるエーテル処理は、三塩化チタンとエーテル化合物を接触させることによって行われ、また、エーテル化合物による三塩化チタンの処理は、希釈剤の存在下で両者を接触させることによって行うのが有利である。このような希釈剤には、ヘキサン、ヘプタン、オクタン、デカン、ベンゼン及びトルエン等の不活性炭化水素化合物を使用することが好適である。なお、エーテル処理における処理温度は、0〜100℃であることが好ましい。また、処理時間については特に制限されないが、通常20分〜5時間の範囲で行われる。
【0032】
エーテル化合物の使用量は、三塩化チタン1molあたり、一般に0.05〜3.0mol、好ましくは0.5〜1.5molの範囲とすればよい。エーテル化合物の使用量が0.05molより小さいと、生成される重合体の立体規則性を十分に向上させることができなくなるので好ましくない。一方、エーテル化合物の使用量が3.0molを越えると、生成される重合体の立体規則性は向上するものの、収率が低下することとなるので好ましくない。なお、有機アルミニウム化合物やエーテル化合物で処理した三塩化チタンは、厳密に言えば、三塩化チタンを主成分とする組成物である。
なお、このような固体触媒成分(a)としては、Solvay型三塩化チタンを好適に用いることができる。
【0033】
有機アルミニウム化合物(b)としては、前述した有機アルミニウム化合物と同様なものを使用すればよい。
環状エステル化合物(c)としては、例えば、γ−ラクトン、δ−ラクトン、ε−ラクトン等が挙げられるが、ε−ラクトンを使用することが好ましい。
以上の成分(a)〜(c)を混合することにより、本実施形態で用いるプロピレン系多段重合体を製造するためのオレフィン重合用触媒を得ることができる。
なお、この成分(a)〜(c)からなる触媒でプロピレン系樹脂を製造した場合、アイソタクティシティーが比較的高いポリプロピレン系樹脂が得られることが知られている。
【0034】
本実施形態で用いるプロピレン系多段重合体を得るには、2段階の重合方法のうち、水素不存在下でプロピレンを重合またはプロピレンと炭素数2〜8のα−オレフィンを共重合させることが好ましい。
ここで、「水素不存在下」とは、実質的に水素不存在下という意味であり、水素が全く存在しない場合だけでなく、水素が極微量存在する場合(例えば、10molppm程度)も含まれる。要は、135℃テトラリン溶媒中で測定した、1段階目のプロピレン系重合体またはプロピレン系共重合体の極限粘度[η]が10dL/g以下とならない程度に水素を含む場合でも、「水素不存在下」の意味には含まれる。
【0035】
このような水素不存在下でプロピレンの重合またはプロピレンとα−オレフィンとの共重合を行うことにより、超高分子量プロピレン系重合体、すなわち、プロピレン系多段重合体の成分(P1)および成分(P2)を製造することができる。
成分(P1)は、水素不存在下で、原料モノマーを重合温度として、好ましくは20〜80℃、より好ましくは40〜70℃、重合圧力として、一般に、常圧〜1.47MPa、好ましくは0.39〜1.18MPaの条件下でスラリー重合して製造することが好ましい。
【0036】
プロピレン系多段重合体の成分(P2)は、2段階目以降に製造することが好ましい。
成分(P2)の製造条件としては、前記したオレフィン重合用触媒を使用すること以外は特に制限はないが、原料モノマーを、重合温度として、好ましくは20〜80℃、より好ましくは60〜70℃、重合圧力として、一般に、常圧〜1.47MPa、好ましくは0.19〜1.18MPa、分子量調整剤としての水素が存在する条件下で重合して製造することが好ましい。
【0037】
なお、前述した製造方法では、本重合を実施する前に、予備重合を行うようにしてもよい。予備重合を実施すると、パウダーモルフォロジーを良好に維持することができる、予備重合は、一般的に、重合温度として、好ましくは0〜80℃、より好ましくは10〜60℃、重合量として、固体触媒成分1gあたり、好ましくは0.001〜100g、より好ましくは0.1〜10gのプロピレンを重合またはプロピレンと炭素数2〜8のα−オレフィンを共重合させることが好ましい。
【0038】
また、本発明で使用するポリプロピレン系樹脂として、プロピレン単独重合体とプロピレン−α−オレフィン共重合体をブレンドしたものを使用することができる。
プロピレン−α−オレフィン共重合体として、例えばプロピレン系ブロック共重合体が挙げられ、特開2003−286060に記載の製造方法によって製造することができる。
すなわち、下記成分[A],[B]及び[C]からなるオレフィン重合用触媒の存在下、プロピレンを重合又はエチレンとプロピレンとを共重合させて、エチレン含量が0〜5質量%のポリプロピレン成分又はプロピレン/エチレン共重合体成分を全重合量の1〜25質量%形成し、エチレンとプロピレンとを共重合させて、プロピレン/エチレン共重合体成分を全重合量の99〜75質量%形成し、エチレン含量を、全重合量の10〜80質量%とする。
【0039】
[A]下記化合物(a),(b),(c)又は下記化合物(a),(b),(c),(d)を反応させて得られる固体触媒成分
(a)マグネシウム化合物
(b)四塩化チタン
(c)フタル酸ジアルキル(アルキル基は、炭素数3〜20の直鎖状炭化水素基又は分岐状炭化水素基を表す)
(d)四塩化ケイ素
[B]有機アルミニウム化合物
[C]下記一般式(II)で表される有機ケイ素化合物
(R1)(R2CH2)Si(OR3)(OR4) …(II)
[式中、R1は、炭素数3〜12の脂環式炭化水素基、R2は、炭素数3〜20の分岐状炭化水素基、R3及びR4は、それぞれ独立であって、炭素数1〜20の炭化水素基を表す]
【0040】
固体触媒成分[A]は、チタン、マグネシウム、ハロゲン及び電子供与性化合物を含有する触媒成分であり、具体的には、上記化合物(a),(b),(c)又は上記化合物(a),(b),(c),(d)を反応させて得られる。
マグネシウム化合物(a)としては特に制限はないが、下記一般式(III)で表されるものを好ましく用いることができる。
MgR5R6 …(III)
上記の一般式(III)において、R5及びR6は、炭化水素基、OR7(R7は炭化水素基)またはハロゲン原子を示す。ここでR5、R6及びR7の炭化水素基としては、炭素数1〜12のアルキル基、炭素数3〜12のシクロアルキル基、炭素数6〜20のアリール基、炭素数7〜20のアラルキル基等を、R5及びR6のハロゲン原子としては、塩素、臭素、ヨウ素、フッ素を挙げることができる。また、R5、R6及びR7は同一でも異なってもよい。
【0041】
上記の一般式(III)で示されるマグネシウム化合物(a)の具体例としては、ジメチルマグネシウム、ジエチルマグネシウム、ジイソプロピルマグネシウム、ジブチルマグネシウム、ジヘキシルマグネシウム、ジオクチルマグネシウム、エチルブチルマグネシウム、ジフェニルマグネシウム、ジシクロへキシルマグネシウム、ブチルオクチルマグネシウム等のアルキルマグネシウムやアリールマグネシウム;ジメトキシマグネシウム、ジエトキシマグネシウム、ジプロポキシマグネシウム、ジブトキシマグネシウム、ジヘキシロキシマグネシウム、ジオクトキシマグネシウム、ジフェノキシマグネシウム、ジシクロヘキシロキシマグネシウム等のアルコキシマグネシウムやアリロキシマグネシウム;エチルマグネシウムクロリド、ブチルマグネシウムクロリド、ヘキシルマグネシウムクロリド、イソプロピルマグネシウムクロリド、イソブチルマグネシウムクロリド、t−ブチルマグネシウムクロリド、フェニルマグネシウムブロミド、ベンジルマグネシウムクロリド、エチルマグネシウムブロミド、ブチルマグネシウムブロミド、フェニルマグネシウムクロリド、ブチルマグネシウムイオダイド等のアルキルマグネシウムハライドやアリールマグネシウムハライド;ブトキシマグネシウムクロリド、シクロヘキシロキシマグネシウムクロリド、フェノキシマグネシウムクロリド、エトキシマグネシウムブロミド、ブトキシマグネシウムブロミド、エトキシマグネシウムイオダイド等のアルコキシマグネシウムハライドやアリロキシマグネシウムハライド;塩化マグネシウム、臭化マグネシウム、ヨウ化マグネシウム等のハロゲン化マグネシウム等が挙げられる。
これらのマグネシウム化合物(a)の中では、ハロゲン化マグネシウム、アルコキシマグネシウム、アルキルマグネシウムハライドが好適に使用できる。中でも、アルコキシマグネシウムが特に好ましい。
上記のマグネシウム化合物(a)は、金属マグネシウム又はマグネシウムを含有する化合物から調製することができる。
【0042】
フタル酸ジアルキル(c)のアルキル基は、炭素数3〜20の直鎖状炭化水素基又は分岐状炭化水素基であり、具体的には、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、t−ブチル基、n−ペンチル基、1−メチルブチル基、2−メチルブチル基、3−メチルブチル基、1,1−ジメチルプロピル基、1−メチルペンチル基、2−メチルペンチル基、3−メチルペンチル基、4−メチルペンチル基、1−エチルブチル基、2−エチルブチル基、n−ヘキシル基、シクロヘキシル基、n−ヘプチル基、n−オクチル基、n−ノニル基、2−メチルヘキシル基、3−メチルヘキシル基、4−メチルヘキシル基、2−エチルヘキシル基、3−エチルヘキシル基、4−エチルヘキシル基、2−メチルペンチル基、3−メチルペンチル基、2−エチルペンチル基、3−エチルペンチル基等が挙げられる。これらのアルキル基の中では、炭素数が4以上の直鎖状又は分岐状の脂肪族炭化水素基が好ましい。
これらの具体例としては、フタル酸ジ−n−ブチル、フタル酸ジイソブチル、フタル酸ジ−n−ヘプチル等が挙げられる。中でも、フタル酸ジ−n−ブチル、フタル酸ジイソブチルが特に好ましい。また、これらの化合物はそれぞれ単独で用いてもよいし、2種以上を組み合わせて用いてもよい。
【0043】
固体触媒成分[A]には、さらに、上記化合物(a),(b)及び(c)に、四塩化ケイ素(d)を加えることができる。四塩化ケイ素(d)は、マグネシウム化合物(a)に対するモル比が、通常0.01以上、好ましくは0.10以上となる割合で用いられる。このモル比が0.01未満では、触媒活性や立体規則性の向上効果が十分に発揮されない場合や、生成ポリマー中の微粉量が多くなる場合がある。
【0044】
上記の各化合物を接触させる方法としては、特に制限はなく、公知の方法で接触させればよい。例えば、特開昭53−43094号公報、同55−135102号公報、同55−135103号公報、同56−18606号公報等に記載の方法が挙げられる。具体的には、(1)マグネシウム化合物(a)又はマグネシウム化合物(a)とフタル酸ジアルキル(c)との錯化合物を、フタル酸ジアルキル(c)及び所望に応じて用いられる粉砕助剤等の存在下に粉砕して、四塩化チタン(b)と反応させる方法、(2)還元能を有しないマグネシウム化合物(a)の液状物と四塩化チタン(b)とを、フタル酸ジアルキル(c)の存在下において反応させて、固体状のチタン複合体を析出させる方法、(3)上記(1)又は(2)で得られたものに四塩化チタン(b)を反応させる方法、(4)上記(1)又は(2)で得られたものに、さらに、フタル酸ジアルキル(c)及び四塩化チタン(b)を反応させる方法、(5)マグネシウム化合物(a)又はマグネシウム化合物(a)とフタル酸ジアルキル(c)との錯化合物を、フタル酸ジアルキル(c)、四塩化チタン(b)及び所望に応じて用いられる粉砕助剤等の存在下で粉砕した後、必要に応じて四塩化ケイ素(d)で処理する方法等が挙げられる。
【0045】
さらには、これらの方法以外に、特開昭56−166205号公報、特開昭57−63309号公報、特開昭57−190004号公報、特開昭57−300407号公報、特開昭58−47003号公報等に記載の方法でも、固体触媒成分[A]を調製することができる。
四塩化チタン(b)の使用量は、マグネシウム化合物(a)のマグネシウム1モルに対して、通常、0.5〜100モル、好ましくは1〜50モルの範囲にするとよい。また、フタル酸ジアルキル(c)の使用量は、マグネシウム化合物(a)のマグネシウム1モルに対して、通常、0.01〜10モル、好ましくは0.05〜0.15モルの範囲にするとよい。さらに、四塩化ケイ素(d)を添加する場合には、その使用量を上記の割合にするとよい。
【0046】
固体触媒成分[A]の調製では、化合物(a)〜(d)の接触温度を、通常、−20〜200℃、好ましくは20〜150℃の範囲にするとよく、接触時間を、通常、1分〜24時間、好ましくは10分〜6時間の範囲にするとよい。
このとき、化合物(a)〜(d)の接触手順については特に問わない。例えば、各化合物を炭化水素等の不活性溶媒の存在下で接触させてもよいし、予め炭化水素等の不活性溶媒で各化合物を希釈して接触させてもよい。この不活性溶媒としては、例えば、n−ペンタン,イソペンタン,n−ヘキサン,n−ヘプタン,n−オクタン,イソオクタン等の脂肪族炭化水素;ベンゼン,トルエン,キシレン等の芳香族炭化水素又はこれらの混合物を挙げることができる。
また、固体触媒成分[A]の調製では、四塩化チタン(b)の接触を2回以上行い、触媒担体としての役割をするマグネシウム化合物(a)に十分担持させるとよい。
以上の接触により得られる固体触媒成分[A]は、炭化水素等の不活性溶媒で洗浄してもよい。この不活性溶媒としては、上記と同様のものが挙げられる。また、この固体触媒成分[A]は、乾燥状態で保存することもできるし、また炭化水素等の不活性溶媒中でも保存することができる。
【0047】
有機アルミニウム化合物[B]としては、アルキル基、ハロゲン原子、水素原子、アルコキシ基を含有するもの、アルミノキサン及びそれらの混合物を好ましく用いることができる。具体的には、トリメチルアルミニウム,トリエチルアルミニウム,トリイソプロピルアルミニウム,トリイソブチルアルミニウム,トリオクチルアルミニウム等のトリアルキルアルミニウム;ジエチルアルミニウムモノクロリド,ジイソプロピルアルミニウムモノクロリド,ジイソブチルアルミニウムモノクロリド,ジオクチルアルミニウムモノクロリド等のジアルキルアルミニウムモノクロリド;エチルアルミニウムセスキクロリド等のアルキルアルミニウムセスキハライド;メチルアルミノキサン等の鎖状アルミノキサン等を挙げることができる。これらの有機アルミニウム化合物[B]の中では、炭素数1〜5の低級アルキル基を有するトリアルキルアルミニウム、特にトリメチルアルミニウム,トリエチルアルミニウム,トリプロピルアルミニウム及びトリイソブチルアルミニウムが好ましい。また、これらの有機アルミニウム化合物[B]は、それぞれ単独で用いてもよいし、2種以上を組み合わせて用いてもよい。
【0048】
有機ケイ素化合物[C]は、上記一般式(II)で表される。
具体的には、R1としては、シクロプロピル基、シクロブチル基、シクロペンチル基、シクロヘキシル基、シクロへプチル基、シクロオクチル基、1−ノルボルニル基、2−ノルボルニル基等が挙げられ、特に、シクロペンチル基、シクロヘキシル基が好ましい。R2としては、イソプロピル基、イソブチル基、sec−ブチル基、t−ブチル基、ネオペンチル基等が挙げられ、特に、イソプロピル基が好ましい。R3及びR4としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、ヘキシル基、オクチル基、シクロヘキシル基等のアルキル基、アリル基、プロペニル基、ブテニル基等のアルケニル基、フェニル基、トリル基、キシリル基等のアリール基、フェネチル基、3−フェニルプロピル基等のアラルキル基等が挙げられる。これらの中では、特に炭素数1〜10のアルキル基が好ましい。
【0049】
有機ケイ素化合物[C]としては、具体的に、シクロプロピルイソブチルジメトキシシラン、シクロプロピルイソペンチルジメトキシシラン、シクロプロピル−2−メチルブチルジメトキシシラン、シクロプロピルネオペンチルジメトキシシラン、シクロプロピル−2−メチルへキシルジメトキシシラン、シクロブチルイソブチルジメトキシシラン、シクロブチルイソペンチルジメトキシシラン、シクロブチル−2−メチルブチルジメトキシシラン、シクロブチルネオペンチルジメトキシシラン、シクロブチル−2−メチルへキシルジメトキシシラン、シクロペンチルイソブチルジメトキシシラン、シクロペンチルイソペンチルジメトキシシラン、シクロペンチル−2−メチルブチルジメトキシシラン、シクロペンチルネオペンチルジメトキシシラン、シクロペンチル−2−メチルへキシルジメトキシシラン、シクロヘキシルイソブチルジメトキシシラン、シクロヘキシルイソペンチルジメトキシシラン、シクロヘキシル−2−メチルブチルジメトキシシラン、シクロヘキシルネオペンチルジメトキシシラン、シクロヘキシル−2−メチルへキシルジメトキシシラン、シクロへプチルイソブチルジメトキシシラン、シクロへプチルイソペンチルジメトキシシラン、シクロへプチル−2−メチルブチルジメトキシシラン、シクロへプチルネオペンチルジメトキシシラン、シクロへプチル−2−メチルへキシルジメトキシシラン、シクロオクチルイソブチルジメトキシシラン、シクロオクチルイソペンチルジメトキシシラン、シクロオクチル−2−メチルブチルジメトキシシラン、シクロオクチルネオペンチルジメトキシシラン、シクロオクチル−2−メチルへキシルジメトキシシラン、1−ノルボルニルイソブチルジメトキシシラン、1−ノルボルニルイソペンチルジメトキシシラン、1−ノルボルニル−2−メチルブチルジメトキシシラン、1−ノルボルニルネオペンチルジメトキシシラン、1−ノルボルニル−2−メチルへキシルジメトキシシラン、2−ノルボルニルイソブチルジメトキシシラン、2−ノルボルニルイソペンチルジメトキシシラン、2−ノルボルニル−2−メチルブチルジメトキシシラン、2−ノルボルニルネオペンチルジメトキシシラン、2−ノルボルニル−2−メチルへキシルジメトキシシラン等が挙げられる。このうち、好ましくはシクロペンチルイソブチルジメトキシシラン、シクロヘキシルイソブチルジメトキシシランである。
これらの有機ケイ素化合物[C]はそれぞれ単独で用いてもよいし、2種以上を組み合わせて用いてもよい。
【0050】
上記成分[A]〜[C]の使用量については、特に制限はないが、固体触媒成分[A]は、チタン原子に換算して、反応容積1リットル当たり、通常0.0005〜1ミリモルの範囲になるような量が用いられる。
有機アルミニウム化合物[B]は、アルミニウム/チタン(原子比)が、通常1〜1,000、好ましくは10〜500の範囲になるような量が用いられる。
この原子比が前記範囲を逸脱すると、触媒活性が不十分となることがある。
有機ケイ素化合物[C]は、有機ケイ素化合物[C]/有機金属化合物[B](モル比)が、通常0.02〜2.0、好ましくは0.05〜1.0の範囲になるような量が用いられる。このモル比が前記範囲を逸脱すると、十分な触媒活性が得られないことがある。
【0051】
本願で用いるプロピレン系ブロック共重合体は、上述したオレフィン重合用触媒を用いて、好ましくは、一段目に、プロピレンを重合またはエチレンとプロピレンとを共重合させて、エチレン含量が0〜5質量%のポリプロピレン成分またはプロピレン/エチレン共重合体成分、好ましくは、エチレン含量が0質量%のポリプロピレン成分、即ち、プロピレン単独重合部を、全重合量の1〜25質量%、好ましくは5〜20質量%形成し、次いで、二段目に、エチレンとプロピレンとを共重合させて、プロピレン/エチレン共重合体成分、即ち、共重合部を、全重合量の99〜75質量%、好ましくは95〜80質量%形成し、エチレン含量が、全重合量の10〜80質量%、好ましくは15〜45質量%のブロック共重合体を製造する。
【0052】
さらに、ポリプロピレン成分またはプロピレン/エチレン共重合体成分を形成する前に、オレフィン類、好ましくは、α−オレフィンの予備重合を行うことができる。
予備重合では、用いる触媒については特に制限されないが、好ましくは、上述したオレフィン重合用触媒を用いる。この場合、電子供与性成分として、上記有機化合物[C]に加え、さらに、ジシクロペンチルジメトキシシラン、シクロペンチルエチルジメトキシシラン、シクロペンチルイソプロピルジメトキシシラン、シクロペンチルターシャリブチルジメトキシシラン、テキシルシクロペンチルジメトキシシラン、テキシルシクロヘキシルジメトキシシラン、ジイソプロピルジメトキシシラン、ジイソブチルジメトキシシラン、ジターシャリブチルジメトキシシラン等を用いることができる。これらのうち、好ましくはジシクロペンチルジメトキシシランである。
α−オレフィンとしては、特に制限はないが、具体的には、エチレン、プロピレン、1−ブテン、1−ペンテン、1−ヘキセン、1−ヘプテン、1−オクテン、1−デセン、3−メチル−1−ペンテン、4−メチル−1−ペンテン、ビニルシクロヘキサン等が挙げられる。このうち、エチレン及びプロピレンが好ましい。これらのα−オレフィンは、1種単独で用いてもよいし、2種以上を組み合わせて用いてもよい。
【0053】
予備重合は、上記成分[A]、[B]及び[C]又はその他の有機ケイ素化合物の存在下、α−オレフィンを、通常1〜100℃の範囲の温度において、常圧〜5MPa(Gauge)の圧力で重合させればよい。重合時間は1分〜10時間、好ましくは10分〜5時間である。予備重合量は、固体触媒成分[A]に対して、通常0.1〜1,000質量%、好ましくは1.0〜500質量%重合させればよい。
さらに、予備重合で得られたオレフィン重合体を、予備重合触媒成分として用いることができる。即ち、この予備重合触媒成分をオレフィン重合用触媒成分として用い、本発明で用いるプロピレン系ブロック共重合体を製造することができる。
【0054】
プロピレン系ブロック共重合体の製造方法では、重合形式について特に制限はなく、溶液重合、スラリー重合、気相重合、バルク重合等のいずれにも適用可能である。また、重合方式としては回分式重合や連続式重合のどちらであってもよい。例えば、連続式で製造する場合は、前段の重合槽に原料プロピレンガス、分子量調整剤の水素ガス及び触媒を供給し、重合時間で重合量をコントロールしてプロピレン単独重合部を製造し、次いで後段の重合槽に移動してさらに原料プロピレンガスにエチレンガス、水素ガス、及び必要に応じて触媒を加えて共重合部を製造し、ブロック共重合体を得ることができる。
共重合部の製造に際しては、エチレンを単独で用いてもよいが、必要に応じて、エチレンとプロピレン以外の前記α−オレフィンを組み合わせてもよい。
プロピレン単独重合の条件としては、重合時に、上述した重合量が得られるものであれば特に制限されないが、その重合圧は、通常、大気圧〜8MPa(Gauge)、好ましくは0.2〜5MPa(Gauge)、重合温度は、通常、0〜200℃、好ましくは30〜100℃の範囲で適宜選ばれる。重合時間は、通常、5分〜20時間、好ましくは10分〜10時間程度である。
【0055】
エチレンとプロピレンとの共重合の条件としては、共重合時に、上述したエチレン含量及び重合量が得られるものであれば特に制限されないが、その重合圧は、通常、大気圧〜8MPa(Gauge)、好ましくは0.2〜5MPa(Gauge)、重合温度は、通常、0〜200℃、好ましくは20〜100℃の範囲で適宜選ばれる。重合時間は、通常、1分〜20時間、好ましくは1分〜10時間程度である。供給するエチレンとプロピレンとの比率は、エチレン/プロピレンのモル比で0.01〜9、好ましくは0.05〜2.3である。
【0056】
この発明では、ポリプロピレン系押出発泡体は押出発泡成形により成形される。特に、一般的に使用されている公知の押出発泡装置を用いることができる。
具体的には、前述のポリプロピレン系樹脂と各種添加物を押出発泡装置内に投入し、溶融混練後、例えば大気圧あるいは大気圧より減圧雰囲気や加圧雰囲気となる押出機内より低圧となる低圧領域に押し出して発泡させ、ポリプロピレン系押出発泡体を得る。
【0057】
ポリプロピレン系樹脂を発泡させる手段としては、発泡材料を含有させる。具体的には、成形時に溶融状態の樹脂原料に発泡材料としての窒素ガスを注入する物理発泡、樹脂原料に発泡材料として窒素ガスを発生させる化学発泡剤を混合させる化学発泡などを採用することができる。
物理発泡では、窒素ガスの注入量の制御によるポリプロピレン系押出発泡体の発泡倍率の設定が容易となり、製造性が容易である。窒素ガスとしては、特に、樹脂原料への溶解性がよい超臨界状態の窒素ガスが好ましい。
一方、化学発泡では、樹脂原料との均一混合が物理発泡より容易で、製造性が容易である。
【0058】
化学発泡で使用する発泡剤としては、例えば、アゾジカルボンアミドやバリウムアゾジカルボキシレートなどのアゾ化合物、N,N’−ジニトロソペンタメチレンテトラミンなどのニトロソ化合物、4,4’−オキシビス(ベンゼンスルホニルヒドラジド)などのヒドラジン誘導体などが例示できる。
なお、上述した発泡剤のように、窒素ガスのみならず、炭酸ガスやアンモニアなどを併せて生成するものを用いてもよい。
また、無機系化学発泡剤を使用する場合は、通常、取扱性、貯蔵安定性、ポリプロピレン系樹脂への分散性の点から、10質量%以上50質量%以下の濃度のポリオレフィン系樹脂のマスターバッチとして使用されることが好ましい。これら無機系化学発泡剤の添加量は種類、マスターバッチ中の濃度によって適宜選択すればよい。一般に、本発明のポリプロピレン系樹脂100質量部に対して、0.1質量部以上40質量部以下の範囲で含有されることが好ましく、より好ましくは0.5質量部以上30質量部以下の範囲で選択できる。
これらの発泡材料は、単独で用いてもよいし、2種以上を組み合わせて用いてもよい。
【0059】
発泡材料の使用量としては、ポリプロピレン系樹脂1gに対し、気泡を生成させるガスの発生量が2.2×10-4モル以上1.9×10-3モル以下となるように添加することが好ましい。より好ましくは、6.3×10-4モル以上1.9×10-3モル以下である。
具体的に、窒素ガス換算で、樹脂原料に対して0.6質量%以上5質量%以下で含有させることが好ましい。より好ましくは、1.7質量%以上5質量%以下で含有させる。0.6質量%より少ないと、断熱性などの発泡体として十分な機能が得られなくなるおそれがあり、5質量%より多く含有しても含有量に応じた発泡倍率の増大が期待できない上に、押出機内で樹脂原料と窒素ガスが分離し、押出された発泡体が不連続となる可能性がある。
化学発泡においても、押出過程で発生するガス量が上記モル数の範囲と同様となるように各種化学発泡剤の添加量を調整することが好ましい。
本発明では、発泡倍率が3倍以上となる条件で溶融混練を行う。このため、上記発泡剤の量とするとともに、シリンダ温度を170℃以上210℃以下、押出吐出量を1kg/hr以上150kg/hr以下で適宜選択すればよい。また、超臨界窒素ガスを使用する場合には、シリンダ内の圧力を10MPa以上20MPa以下で適宜選択すればよい。
【0060】
そして、押出発泡成形を行う際には、必要に応じて、酸化防止剤、中和剤、難燃剤、等の添加剤を使用することができる。添加剤の配合量は特に制限されず、適宜調節することができる。
また、粉末状または繊維状の多孔質フィラーとして、シリカ、活性炭、ゼオライト、シリカゲル、または繊維状活性炭を配合してもよい。
その他にも、結晶化核剤として、タルク、有機カルボン酸塩、有機リン酸塩、ソルビトール系核剤を配合してもよい。
【0061】
このようにして得られたポリプロピレン系押出発泡体は、発泡倍率が3倍以上であるため、断熱性能に優れている。
また、見かけの剪断速度が遅くても発泡倍率が3倍以上の高発泡体を製造可能であるので、ダイ出口のクリアランスを広くすることができる。したがって、厚みのある押出発泡体を形成することができるとともに、表面外観が良好となる。
さらに、本発明で使用するポリプロピレン系樹脂は、リサイクル性能にも優れ、また、耐薬品性や耐熱性も良好であることから、ポリプロピレン系押出発泡体もまた、リサイクル性能、耐薬品性および耐熱性に優れる。また、ポリプロピレン系樹脂は比較的に低コストであるので、材料費のコスト低減も図ることができる。
このように、厚みがあり、リサイクル性能、耐薬品性および耐熱性に優れた押出発泡体は、住設分野や自動車分野の内外装材として有用性が高い。
そして、公知の押出発泡装置をそのまま使用することができるので、新たな設備に投資する必要がない。したがって、製造工程においても大幅な変更がないので簡単に実施することができる。さらには、ポリプロピレン系押出発泡体をそのまま住設分野や自動車分野の内外装材などに利用できるとともに、さらにプレス加工などにより成形しても気泡がほとんど損なわれずに維持され、特性が損なわれることがないので、二次加工など、汎用性も向上できる。
【0062】
本発明のポリプロピレン系押出発泡体において、前記発泡倍率は、3倍以上20倍未満であることが好ましい。
この発明では、前述のポリプロピレン系樹脂材料として用いるからこそ、発泡倍率3倍以上のポリプロピレン系押出発泡体を形成することができる。なお、発泡倍率が20倍を超える発泡体を得るためには、発泡剤を高圧の状態で多量に供給する必要があり、特に二酸化炭素に比べて、ポリプロピレン系樹脂に対して溶解性の劣る窒素を発泡剤として用いた場合には、より高耐圧の設備が必要なため、設備投資のコストが膨大になり、また、成形の安定性にも問題が生じる可能性もある。
【0063】
本発明のポリプロピレン系押出発泡体において、前記ダイは、その出口の流路がスリット状、または円筒状であることが好ましい。
ポリプロピレン系押出発泡体の形状は、ダイに形成されたダイ出口の形状で決まる。このダイ出口の形状は、円形、菱形、スリット状、円筒状などの任意の形状とすることができるが、この中でも、厚みの調整が容易であり、成形性に優れるという点から、スリット状または円筒状であることが好ましい。
なお、発泡体の成形方法としては、複数のダイ出口から押し出された発泡体を集束して一つの発泡体とする、いわゆる細条集束体とすることも可能である。この場合、複数のダイ出口の形状を全て同じ形状としてもよいし、一つのダイに多種類の形状を複数形成するようにしてもよい。
【0064】
本発明のポリプロピレン系押出発泡体において、前記ダイの設定温度は、170℃以上210℃以下であることが好ましい。
ダイの設定温度が170℃未満であると、押出が困難となるおそれがあり、210℃を超えると、樹脂原料の発泡性能が低下するというおそれがある。
したがって、ダイの設定温度を170℃以上210℃以下とすることにより、成形性に優れ、高い発泡倍率のポリプロピレン系押出発泡体を得ることができる。
【0065】
本発明のポリプロピレン系押出発泡体は、厚みが2mm以上30mm以下に押出成形されたことが好ましい。
従来、ポリプロピレン系押出発泡体は、低剪断速度下で高発泡することが困難とされていたため、ダイのクリアランスを小さくする必要があり、厚みの小さいものとなっていた。
この発明によれば、ポリプロピレン系押出発泡体は2mm以上30mm以下という厚みがある。なお、ダイ出口が円形状のダイで押出発泡を行った場合は、直径が2mm以上30mm以下の太い発泡体を得ることができる。
したがって、ポリプロピレン系押出発泡体が厚い、または太いので、住設分野や自動車分野の内外装材として用いるのに有用性が大きい。
【発明を実施するための最良の形態】
【0066】
以下、本発明の実施形態を図面に基づいて説明する。
本発明で使用する押出発泡装置としては、樹脂原料を溶融状態に加熱し、適度の剪断応力を付与しながら混練し、押出発泡することができる公知の押出発泡装置を使用することができる。また、押出発泡装置を構成する押出機も、単軸押出機または二軸押出機のいずれのものも使用することができる。具体的には、例えば、特開2004−237729号公報に開示された、2台の押出機が接続されたタンデム型押出発泡成形装置などが挙げられる。
なお、本実施形態では、物理発泡により押出発泡する押出発泡装置を使用した。
図1は、本発明の一実施形態における押出発泡装置を模式的に示す概略図である。
【0067】
[押出発泡装置の構成]
押出発泡装置100は、図1に示すように、樹脂原料が投入されるホッパー110と、樹脂原料を溶融混練する押出機としてのシリンダ120と、シリンダ120に発泡剤ガスを導入するガス導入路130と、樹脂原料を押し出すギアポンプ140と、樹脂原料を成形するダイ150と、冷却用のマンドレル160と、を備えている。
【0068】
ホッパー110は、計量器としても機能し、樹脂原料が投入されると同時に計量することもできる。
シリンダ120は略円筒状に形成され、シリンダ120の内径よりも小さい径をもつ略円柱状のスクリュ121を有している。スクリュ121は、その外周面にらせん状の羽122を有しており、スクリュ121の軸を中心として回転可能に支持されている。シリンダ120の内部でスクリュ121が回転することにより、シリンダ120内の樹脂原料が溶融混練される。
なお、シリンダ120における押出温度は適宜設定すればよいが、例えば、170℃以上230℃以下に設定することが好ましい。押出温度が170℃未満であると押出が困難となるおそれがあり、230℃を超えると、発泡性能が低下するおそれがある。
【0069】
ガス導入路130は、シリンダ120の内部につながる流路であり、発泡ガス、すなわち窒素ガスを導入する。発泡ガスの導入により、シリンダ120内で樹脂原料と発泡ガスとが混合される。
ギアポンプ140は、シリンダ120で溶融混練された樹脂原料と発泡ガスとの混合物の流量を調整し、ダイ150へ安定的に押し出す。
【0070】
ダイ150は、樹脂原料を目的の形状に成形するものである。本実施形態では、図2に示すダイを用いた。
図2は、本実施形態にかかるダイの形状を示す模式図であり、(A)はダイの断面図、(B)はダイ出口側の側面図である。
ダイ150は、図2(A)および(B)に示すように、円環状の断面を有する円筒状の流路151を有している。この流路151のダイ出口側における円環の直径は、4mm以上1000mm以下であることが好ましい。直径が4mm未満であると、押出発泡した場合に一体化してしまい、円筒状に形成されないおそれがある。また、直径が1000mmを超えると、安定した連続成形が困難となる。
【0071】
なお、流路151は、ダイ出口151Aの円環の半径がダイ入口の円環の半径よりも大きく、ダイ150の内部で一連に形成されている。また、流路151はダイ出口151Aに近づくほど、そのクリアランスも小さくなる。クリアランスが最小となる部位を最小部位Sとすると、最小部位Sにおけるクリアランスは、適宜設定すればよいが、0.2mm以上とすることが好ましい。0.2mm未満であると、表面外観が悪化するとともに、十分な厚みの発泡体を得ることが難しくなる。クリアランスのより好ましい範囲は1mm以上5mm以下である。
【0072】
ダイ150の温度は、170℃以上210℃以下に設定することが好ましい。
ダイ150の温度が170℃未満であると、押出が困難となるおそれがあり、210℃を超えると、樹脂原料の発泡性能が低下するというおそれがある。
【0073】
マンドレル160は、水冷などによって、通常、10℃以上120℃以下に冷却されており、ダイ150から押し出された発泡体を冷却する。本実施形態では、マンドレル160は略円筒状に形成されており、その外周面161に、ダイ150から円筒状に押し出された発泡体が当接する。これにより、発泡体が成形および冷却される。
【0074】
[押出発泡装置の動作]
次に、押出発泡装置100で押出発泡を実施する際の動作について説明する。
まず、前述のポリプロピレン系樹脂をホッパー110に投入する。ホッパー110は計量器としても機能するため、所望の量の樹脂をホッパー110で計量することもできる。
ホッパー110に投入された樹脂は、シリンダ120に供給される。
シリンダ120の内部では、スクリュ121が軸を中心として回転しているので、このスクリュ121により樹脂が溶融混練される。このとき、ガス導入路130から発泡流体として窒素が導入されるので、樹脂と窒素が混合される。
溶融混練された樹脂と窒素の混合物はシリンダ120から押し出され、ギアポンプ140に導入される。ギアポンプ140で樹脂の流量が調節され、ダイ150へ押し出される。
【0075】
ダイ150では、内部の形状に沿って樹脂が押し出されることにより成形される。
そして、樹脂はダイ150から押し出されると同時に発泡して発泡体を形成し、マンドレル160に当接することによって賦形されるとともに冷却される。最後に、図示しないカッターにより切断され、シート状の発泡体が得られる。
【0076】
[実施形態の作用効果]
以上より、本実施形態では、次の作用効果を奏することができる。
本実施形態では、前述のポリプロピレン系樹脂を用いて公知の押出発泡装置により、発泡剤に窒素ガスを用いて押出発泡を行う。これにより、従来では困難であった窒素ガスでの発泡でも、発泡倍率3倍以上のポリプロピレン系押出発泡体として得ることができる。
このポリプロピレン系押出発泡体は、材料としてポリプロピレン系樹脂を用いるので、耐熱性、耐薬品性およびリサイクル性能に優れている。また、ポリプロピレン系樹脂は低コストであるので、材料のコスト削減を図ることができる。
【0077】
また、ポリプロピレン系押出発泡体は発泡倍率が3倍以上の高発泡体であるため、断熱性により優れており、住設分野や自動車分野の内外装材として有用性が高い。
そして、本実施形態では、公知の押出発泡装置で発泡倍率3倍以上のポリプロピレン系押出発泡体を製造することができる。すなわち、従来と同様の方法で製造することができる。したがって、新たな設備投資が必要なく、また、製造方法にも大きな変更がないので手間がかからず、簡単に製造することができる。
【0078】
[実施形態の変形例]
上記実施形態では、円筒形状のダイを使用したが、ダイの形状は目的に応じた形状を使用することができ、これに限られない。
例えば、図3に示すように、スリット状の断面を有する平行管状の流路を有するダイ170でもよい。
【0079】
また、図4に示すように、円形状の断面を有する円管状の流路を有するダイ180でもよい。
【0080】
さらに、ダイは、円形状の断面を有する円錐状の流路を有していてもよいし、任意形状の断面を有する平行管状の流路を有していても良い。このように、目的に応じた形状のダイを使用することができる。
【0081】
また、上記実施形態では、円筒状の出口が一つだけ形成されたダイを使用したが、複数の出口が形成されたダイを使用することにより、押し出された複数の発泡体を集束して一つの発泡体とする、いわゆる細条集束体とすることもできる。
この場合、複数の出口形状を全て同じ形状としてもよいし、各々の出口を異なる形状、例えば、円形、菱形、スリット状などとしてもよい。また、同じ円形であっても、それぞれの径の大きさを変えて複数の種類の円形とすることもできる。
【0082】
また、上記実施形態では、物理発泡により押出発泡を行ったが、化学発泡によるものでもよい。この場合は、前述の発泡剤等を添加することにより実施することができる。
【実施例1】
【0083】
以下、実施例及び比較例を挙げて本発明をより具体的に説明する。なお、本発明は実施例等の内容に何ら限定されるものではない。
<樹脂について>
本実施例では、以下に示す2種類のポリプロピレン系樹脂組成物を使用する。
【0084】
樹脂A:直鎖状ポリプロピレン
樹脂B:分岐状ポリプロピレン(商品名「PF814」、Basell社製)
【0085】
[樹脂A(プロピレン系多段重合体)の製造方法]
(1)予備重合触媒成分の調製:
内容積5リットルの攪拌機付き三つ口フラスコを十分に乾燥させ、窒素ガスで置換した後、脱水処理したヘプタンを4リットル、ジエチルアルミニウムクロライド140グラムを加え、市販品のSolvay型三塩化チタン触媒(東ソー・ファインケム(株)製)20gを加えた。これを攪拌しながら20℃に保持した状態で、プロピレンを連続的に導入した。80分後、攪拌を停止し、三塩化チタン触媒1gあたり0.8gのプロピレンが重合した予備重合触媒成分を得た。
【0086】
(2)プロピレン重合:
内容積10リットルの攪拌機付きステンレス製オートクレーブを十分乾燥し窒素ガスで置換した後、脱水処理したヘプタン6リットルを加え、系内の窒素をプロピレンで置換した。その後、攪拌しながらプロピレンを導入して内温50℃、全圧0.78MPaに系内が安定した後、上記予備重合触媒成分を固体触媒換算で0.75グラム含んだヘプタンスラリー200ミリリットルを加えて重合開始とした。プロピレンを1.0時間連続的に供給し、重合を行い、重合体(P1成分)を得た。その一部をサンプリングして分析した結果、極限粘度は15.4dL/gであった。その後、内温を40℃以下にまで降温し攪拌を弱め、脱圧を行った。
再び、内温を65℃として水素を0.10MPa加えて攪拌しながらプロピレンを導入した。全圧0.78MPaでプロピレンを連続的に供給しながら65℃で4時間重合を行い、重合体(P2成分)を得た。この時、重合体の一部をサンプリングして分析した結果、極限粘度は3.31dL/gであった。
重合終了後、50ミリリットルのメタノールを添加し降温、脱圧した。内容物を全量フィルター付きろ過槽へ移し、1−ブタノールを100ミリリットル加え、85℃で1時間撹拌した後に固液分離した。さらに、85℃のヘプタン6リットルで固体部を2回洗浄し、真空乾燥してプロピレン重合体3.3kgを得た。
以上の結果、重合体(P1)と重合体(P2)の重合成分の質量比は15.3:84.7であり、第二段目にて生成した重合体の極限粘度は1.13dL/gと求められた。
【0087】
[物性値などの測定方法]
(1)圧力補正値:
上記の4種類のポリプロピレン系樹脂組成物についてバーグレー補正における圧力補正値を測定した。バーグレー補正における圧力補正値は、「JIS K 7199」に規定されたキャピラリーレオメータによるプラスチックの流れ特性試験方法に基づいて測定することができる。
具体的には、上記の樹脂A、樹脂B、樹脂Cについて、キャピログラフ型式1C(商品名、(株)東洋精機製作所製)を用い、剪断速度を変えてそれぞれの圧力補正値を以下の条件で測定した。なお、キャピログラフ型式1Cは関連業界で広く用いられているものであり、これを用いて以下の条件で測定を実施すると、剪断速度は1216s-1となる。
【0088】
バレル直径(内径):9.55mm
キャピラリー直径D:1.0mm
ピストンの押出速度:100mm/min
測定温度 :210℃
キャピラリーの直径と長さの比L/D:30、40、50の3種
【0089】
(2)プロピレン重合体成分(P1成分)及びプロピレン重合体成分(P2成分)の質量分率:
重合時に連続的に供給するプロピレンの流量計積算値を用いた物質収支から求めた。
(3)極限粘度[η]:
135℃のテトラリン溶媒中で測定した。
プロピレン系多段重合体の第一段目(P1成分)の極限粘度[η1]とプロピレン重合体全体の極限粘度[ηtotal]は重合過程でサンプリングし評価し、プロピレン系多段重合体の第二段目(P2成分)の極限粘度[η2]は下記(IV)式により算出した。
【0090】
[η2]=([ηtotal]×100-[η1]×W1)/W2 …(IV)
[ηtotal] :プロピレン重合体全体の極限粘度(dL/g)
[η1] :P1成分の極限粘度(dL/g)
W1 :P1成分の質量分率(質量%)
W2 :P2成分の質量分率(質量%)
【0091】
(4)メルトフローレート(MFR):
JIS K7210に準拠し、温度を230℃、荷重を2.16kgf(21.2N)として測定した。
【0092】
(5)発泡倍率:
成形品の質量を、水没法により求めた体積で除することにより密度を求め、未発泡品の密度で除することにより、発泡倍率として算出した。
【0093】
(6)連続成形性:
樹脂中に混合する窒素ガス量が多すぎる場合は、成形機のシリンダ内で樹脂と窒素ガスが均一に分散せず、窒素ガスが分離した状態になる。
この状態で成形機から樹脂と窒素ガスの混合物を押出発泡させると、押出された成形品の途中で窒素ガスのみが押出される領域が発生し、発泡体が断絶される。
そこで、目視評価により発泡体の断絶の有無を確認した。
【0094】
[樹脂の物性評価]
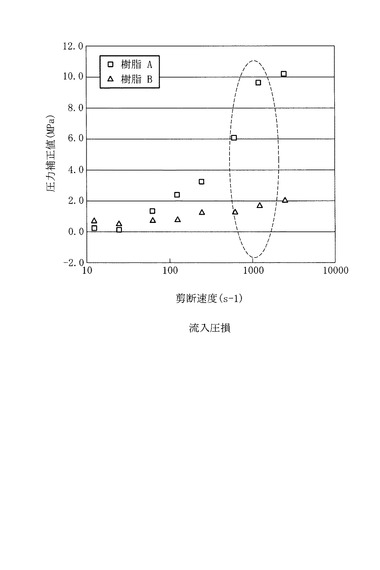
上記の樹脂Aおよび樹脂Bを上記の方法にて圧力補正値を測定した結果を図5に示す。図5は、剪断速度と圧力補正値との関係を表すグラフである。
【0095】
図5に示すように、従来のポリプロピレン系樹脂組成物である樹脂Bは、剪断速度が1000s-1以下で圧力補正値は大きく変化しない。一方、樹脂Aは、剪断速度が1000s-1以下で、圧力補正値が急激に大きくなっている。1000s-1付近で圧力補正値が大きいと、伸長粘度が高く気泡が破れにくいため、樹脂Aは、樹脂Bに比べて伸長粘度が高く、高発泡の発泡体が得られると言える。
【0096】
また、上記2種の樹脂について、剪断速度1216s-1における圧力補正値(バーグレー補正)とメルトフローレート(MFR)の測定結果を以下に示す。
【0097】
【表1】

【0098】
以下の表2に示すように、上記樹脂Aおよび樹脂Bを用いて、窒素ガスの物理発泡および化学発泡による発泡成形体の押出実験を実施した。
(1−1)物理発泡の製造装置および製造条件
成形機:日本製鋼所製 MuCell射出成形機、製品名「J180ELIII−MuCell」(金型をとりはずして使用)
押出時間:12秒
押出量:95g
シリンダ設定温度:180℃
(1−2)物理発泡の製造方法
MuCell射出成形機(金型をとりはずして使用)を用いて、前述した樹脂に超臨界状態の窒素ガスを15MPaで混合し、得られた混合物を大気中に押出すことによって発泡体を得た。
結果を以下の表2に示す。
【0099】
(2−1)化学発泡の製造装置および製造条件
押出機 :株式会社マックインターナショナルアソシエイツ製 押出機(スクリュー径45mm)
ダイ :円筒ダイ(直径45mm)
押出量 :10kg/hr
押出機 :シリンダ設定温度210℃
化学発泡剤A:アゾジカルボンアミドを主成分とし、試料0.500g、昇温速度2℃/min、流動パラフィン10mlの条件で昇温した際の発生ガス量が165℃で5ml、170℃で80mlとなるガス発生特性を有する発泡剤
化学発泡剤B:アゾジカルボンアミドを主成分とし、試料0.500g、昇温速度2℃/min、流動パラフィン10mlの条件で昇温した際の発生ガス量が175℃で5ml、180℃で80mlとなるガス発生特性を有する発泡剤
(2−2)化学発泡の製造方法
樹脂原料100重量部に対し、発泡剤5重量部(発生窒素0.7重量部相当)を加えて、ドライブレンドした混合物をホッパー110に投入する。そして、押出機の所定のスクリュ回転数に設定し、押出機内で樹脂原料を溶融化させるとともに発泡剤と混合し、一定の押出量で混合物をダイに供給する。
供給された混合物をダイから鉛直下方に押し出し、大気中で発泡させる。そして、筒状に押し出した溶融発泡体をダイ出口で切り取り、大気中で自然冷却させることにより、筒状の発泡体を得た。
結果を以下の表2に示す。
【0100】
【表2】
【0101】
表2に示すように、樹脂Aを用いた実施例1〜6では、物理発泡、化学発泡を問わず、十分な発泡が得られた。一方、樹脂Bを用いた比較例1の発泡体では、物理発泡でも発泡倍率が2倍に満たず、発泡倍率を上げるために注入する窒素ガスを増やしてもダイから窒素ガスが吹き出る状態となって押出発泡成形が出来なかった。
なお、図6に、実施例3と比較例1との成形後の外観を状況を示す。図中の左側が実施例3で右側が比較例1を示し、手前に比較として15cm定規を載置している。
また、窒素ガスが0.5重量%以下である比較例2においては、発泡倍率が3倍を下回る発泡体しか得られなかった。一方、比較例3、比較例4においては、発泡倍率は16倍を超えているものの、窒素ガスの分離による発泡体の断絶が発生しており、連続した発泡体を得る事は出来なかった。
なお、比較例1で得られた発泡体電子顕微鏡写真を、図7(倍率:50倍)および図8(倍率:180倍)にそれぞれ示す。また、実施例3で得られた発泡体電子顕微鏡写真を、図9(倍率:50倍)、図10(倍率:180倍)にそれぞれ示す。
これら電子顕微鏡写真でも分かるように、実施例では十分に発泡しているが、比較例では発泡が不十分であった。
【産業上の利用可能性】
【0102】
本発明のポリプロピレン系押出発泡体は、ポリプロピレン系樹脂としての特性を活かしつつ、軽量性、断熱性に優れるので、例えば、住設分野や自動車分野の内外装材に好適である。
【図面の簡単な説明】
【0103】
【図1】本発明の一実施形態の押出発泡装置を模式的に示す概略図。
【図2】本実施形態にかかるダイの形状を示す模式図であり、(A)はダイの断面図、(B)はダイ出口側の側面図。
【図3】上記実施形態にかかるダイの変形例を示す模式図であり、(A)はダイの断面図、(B)はダイ出口側の側面図。
【図4】上記実施形態にかかるダイの変形例を示す模式図であり、(A)はダイの断面図、(B)はダイ出口側の側面図。
【図5】剪断速度と圧力補正値との関係を示すグラフ。
【図6】実施例と比較例との発泡状況を示す外観図である。
【図7】比較例の発泡状況を示す電子顕微鏡写真(倍率:50倍)である。
【図8】比較例の発泡状況を示す電子顕微鏡写真(倍率:180倍)である。
【図9】実施例の発泡状況を示す電子顕微鏡写真(倍率:50倍)である。
【図10】実施例の発泡状況を示す電子顕微鏡写真(倍率:180倍)である。
【符号の説明】
【0104】
100…押出発泡装置
110…ホッパー
120…シリンダ
130…ガス導入路
140…ギアポンプ
150、170、180…ダイ
151…流路
160…マンドレル
S、T、U…クリアランスの最小部位
【技術分野】
【0001】
本発明は、ポリプロピレン系の押出発泡体、および、その製造方法に関する。
【背景技術】
【0002】
従来、自動車の内外装あるいは住宅等の建物の内外装には樹脂発泡体が多用されている。例えば、自動車では、室内空間に面する天井、ドア、フロア等の内装、あるいはカウルやボディ等の外装には、樹脂発泡体を用いたボードやパネルが利用されている。また、住宅設備においても、住宅用断熱ボードなどの建材として樹脂発泡体が利用されている。
このような樹脂発泡体の材料としてポリスチレン等が用いられていたが、耐熱性、耐薬品性、軽量性という観点からポリプロピレン系樹脂が好適とされている。
【0003】
しかしながら、ポリプロピレン系樹脂はポリスチレンと比較して発泡成形性に劣るため、押出発泡成形では、発泡倍率が3倍を超える押出発泡成形体を安定供給するのは困難であった。特に、直鎖状ポリプロピレンでは、伸長粘度の歪硬化性に乏しく、発泡成形過程において、隣接する気泡壁が破れやすかった。
そこで、ポリプロピレンの分子鎖に分岐構造を与えることで高い溶融張力を発現させ、発泡成形性を改善させた材料が開発された(例えば、特許文献1参照)。
【0004】
また、超高分子量成分の分子量と量比を最適化することにより、気泡形成過程の高速伸長流動場で大きな伸長粘度を生じさせることにより気泡壁を破れにくくしたポリプロピレン系樹脂組成物が発明された(例えば、特許文献2参照)。
そして、上記ポリプロピレン系樹脂組成物によれば、超臨界二酸化炭素を用いた押出発泡成形により発泡倍率が10倍を超える高発泡体が得られている(例えば、特許文献3)。
【0005】
さらに、ポリプロピレン系樹脂による発泡成形として、超臨界状態の窒素を発泡剤として発泡成形する方法が開示されている(例えば、特許文献4参照)。
【0006】
【特許文献1】特公平7−45551号公報
【特許文献2】特開2007−119760号公報
【特許文献3】WO2006/118160
【特許文献4】特開2005−97389号公報
【発明の開示】
【発明が解決しようとする課題】
【0007】
しかしながら、特許文献1において、分岐構造をもつ高分子はその構造が壊れやすいため、押出成形においては流動過程でその粘弾特性が変化するおそれがある。また、分岐構造をもつ高分子はリサイクルに不向きであるという問題がある。さらに、分岐構造があるため、低歪速度下での伸長粘度の立ち上がりを大きくし、シートの引取性ドローダウン性の向上に有効であるものの、気泡の破壊に寄与する高速度下での伸長変形においては必ずしも良好な特性が得られるものではなかった。
【0008】
また、特許文献2および3では、発泡剤として窒素の使用も示唆されてはいるが、窒素を使った発泡倍率3〜20倍の発泡体の製造例については開示されていない。
さらに、特許文献4の発泡体では、分岐構造を持つ高分子を用いて、射出発泡成形により発泡倍率が3倍を超える発泡体が得られた実施の形態が示されているが、押出発泡成形に関しては、実施の形態は示されていない。射出発泡成形においては、溶融した樹脂材料を金型内に射出し、その後、金型を所定量移動させて発泡させる、という工程を取るため、金型の温度や、金型を移動させるまでの時間、金型の移動速度などを任意に設定する事ができるため、発泡倍率の調整は比較的容易である。しかしながら、押出発泡成形においては、ダイの出口以降は自由表面であり、発泡倍率の制御は困難である。
【0009】
また、ダイ出口の流路面積を小さくするとダイ壁面の剪断速度が大きくなり、メルトフラクチャが発生しやすくなるとともに、コルゲートマークなどの不良現象が発生しやすくなる。すなわち、発泡体の外観不良が生じやすいという問題がある。
【0010】
本発明の目的は、従来の押出発泡装置を用いて製造でき、比較的に高発泡倍率かつ表面外観の良好なポリプロピレン系押出発泡体、および、その製造方法を提供することである。
【課題を解決するための手段】
【0011】
本発明のポリプロピレン系押出発泡体の製造方法は、ポリプロピレン系樹脂と発泡材料とを含有する混合物を押出機内で溶融混練する溶融混練工程と、この溶融混練工程で溶融混練された組成物をダイから押出発泡させる押出発泡工程と、を実施するポリプロピレン系押出発泡体の製造方法であって、前記ポリプロピレン系樹脂は、(A)測定温度210℃、剪断速度1216s-1の条件でのキャピラリーフローテストにおいて、バーグレー補正における圧力補正値が4MPa以上、および(B)メルトフローレート(MFR)が0.5g/10分以上であり、前記溶融混練工程では、前記ポリプロピレン系樹脂に前記発泡材料として窒素ガスを前記ポリプロピレン系樹脂に対して0.6質量%以上5質量%以下で含有させ、前記ポリプロピレン系押出発泡体の発泡倍率が3倍以上となる条件とすることを特徴とする。
ここで、キャピラリーフローテストとは、「キャピラリーレオメータによる流れ特性試験方法(JIS K 7199)」の規定に基づいて行われる。
剪断速度は、ここでは見かけの剪断速度を表し、キャピラリーダイの直径と流量から求めることができる。
バーグレー補正における圧力補正値とは、キャピラリーダイの入口と出口の圧力損失の和である。キャピラリーダイの直径Dとキャピラリーダイの長さLの比(L/D)を複数設定して、それぞれのテストを行うことによって求められる。この圧力補正値が大きいと、入口圧損が大きいことを示しており、入口部での縮小流に対する抵抗が大きい。すなわち、伸長粘度が大きいことを示している。
【0012】
具体的には、例えば、バレルの直径9.55mm、キャピラリーダイの直径1.0mm、キャピラリーダイの流入角90°、L/Dが30、40、50の3種を設定して圧力補正値を求めることができる。しかしながら、これらの値に限られず、流入角が90°以上かつバレルとキャピラリーダイの直径の比が8以上であればよい。
なお、剪断速度1216s-1における測定値がない場合は、この剪断速度の上下の隣接する2点の剪断速度における圧力補正値のデータを用いて補間することにより、剪断速度1216s-1での圧力補正値を算出してもよい。
【0013】
この発明によれば、ポリプロピレン系押出発泡体の材料であるポリプロピレン系樹脂は、剪断速度1216s-1という高速度下での圧力補正値が4MPa以上であることにより、気泡形成時のような高速度下において伸長粘度が高いということが言える。すなわち、気泡が破けにくい。したがって、ポリプロピレン系樹脂には溶解しにくい窒素ガスでも、発泡倍率が3倍以上のポリプロピレン系押出発泡体が得られる。
【0014】
また、ポリプロピレン系樹脂のメルトフローレート(MFR)を0.5g/10分以上とした。MFRが0.5g/10分未満であると、樹脂の流動性が劣り、生産性が悪い。すなわち、樹脂のMFRを0.5g/10分以上とすることにより、押出機での製造を容易に行うことができる。一方、MFRが100g/10分を越えると、ポリプロピレン系樹脂の溶融張力および粘度が低くなり、押出成形が困難となる場合がある点を留意する。従って、MFRのより好ましい範囲は、1.0g/10分以上10.0g/10分以下であり、2.0g/10分以上5.0g/10分以下であることが特に好ましい。
【0015】
以上のような性質を満たすポリプロピレン系樹脂としては、以下に述べるプロピレン系多段重合体、プロピレン単独重合体、プロピレンと他のオレフィンとの共重合体、またはこれらをブレンドしたものが用いられる。
【0016】
プロピレン系多段重合体としては、下記成分(P1)および成分(P2)で構成される。
(P1)135℃、テトラリン溶媒中で測定した極限粘度[η]が10dL/g超のプロピレン単独重合体成分またはプロピレンと炭素数が2〜8のα−オレフィンとの共重合体成分を、全重合体中に5〜20質量%含有する。
(P2)135℃、テトラリン溶媒中で測定した極限粘度[η]が0.5〜3.0dL/gのプロピレン単独重合体成分またはプロピレンと炭素数が2〜8のα−オレフィンとの共重合体成分を、全重合体中に80〜95質量%含有する。
このプロピレン系多段重合体は、成分(P1)すなわち超高分子量プロピレン系重合体の付与により高溶融張力化を達成し、分子量分布の調整により粘弾性特性が調整された直鎖状のプロピレン系重合体である。
【0017】
成分(P1)の極限粘度が10dL/g以下では、溶融張力が不十分となり、所望の発泡性能を得ることができない場合がある。また、成分(P1)の質量分率が5質量%より小さいと、溶融張力が不十分となり、所望の発泡性能を得ることができない場合がある。一方、質量分率が20質量%を越えると、いわゆるメルトフラクチャが激しくなる場合があり、発泡成形体の肌荒れ等の原因となり、製品品質が低下する。
成分(P1)の極限粘度は、前記したように10dL/g超であることが好ましいが、12〜30dL/gの範囲内であることがより好ましく、13〜18dL/gの範囲内であることが特に好ましい。
また、成分(P1)の質量分率は、8〜18質量%の範囲内であることが好ましく、10〜16質量%の範囲内であることが特に好ましい。
【0018】
成分(P2)の極限粘度が0.5dL/gより小さいと、溶融張力が不十分となり、所望の発泡性能を得ることができない場合がある。一方、3.0dL/gを越えると、粘度が高すぎ、好適な発泡成形体を成形することができない場合がある。
また、成分(P2)の質量分率が80質量%より小さいと、好適な発泡成形の実施が困難となる場合があり、質量分率が95質量%を越えると、溶融張力が低くなり、これも好適な発泡成形体の成形が困難となる場合がある。
成分(P2)の極限粘度は、前記したように0.5〜3.0dL/gの範囲内であることが好ましいが、0.8〜2.0dL/gの範囲内であることがより好ましく、1.0〜1.5dL/gの範囲内であることがさらに好ましい。
また、成分(P2)の質量分率は、82〜92質量%の範囲内であることが好ましく、84〜90質量%の範囲内であることが特に好ましい。
さらに、プロピレン系多段重合体の極限粘度は、好ましくは1.0dL/g以上6.0dL/g以下、より好ましくは2.0dL/g以上4.0dL/g以下、さらに好ましくは3.0dL/g以上3.5dL/g以下である。1.0dL/g未満では発泡性が悪くなるおそれがあり、6.0dL/gを越えると成形が困難となるおそれがあるためである。
【0019】
本実施形態で用いるプロピレン系多段重合体において、共重合体成分を構成する炭素数2〜8のα−オレフィンとしては、例えば、プロピレン以外のα−オレフィンであるエチレン、1−ブテン等が挙げられる。このうち、エチレンを使用することが好ましい。
プロピレン系多段重合体は、230℃におけるメルトフローレート(MFR)と、230℃における溶融張力(MT)との関係が、下記式(I)を満たすことが好ましい。
【0020】
log(MT)>−1.33log(MFR)+1.2 …(I)
【0021】
ここで、230℃におけるメルトフローレート(MFR)と、230℃における溶融張力(MT)との関係が、前記式(I)を満たさない場合にあっては、高倍率の発泡成形の実施が困難となる場合がある。前記した定数(1.2)は、1.3以上とすることが好ましく、1.4以上とすることが特に好ましい。
なお、プロピレン系多段重合体が前記した式(I)の関係を具備するようにするには、成分(P1)を5〜20質量%含有させるようにすればよい。
【0022】
また、プロピレン系多段重合体は、溶融状態の動的粘弾性(角周波数ωと貯蔵弾性率G’との関係)として、高周波数側での貯蔵弾性率の傾きが一定量以上の大きさであることが好ましく、具体的には、角周波数が10rad/s(ラジアン/秒)の場合の貯蔵弾性率G’(10)と、角周波数が1rad/sの場合の貯蔵弾性率G’(1)との比であるG’(10)/G’(1)が2.0以上であることが好ましく、2.5以上であることが特に好ましい。この比G’(10)/G’(1)が2.0より小さいと、発泡成形体に延伸等の外的変化を加えた際の安定性が低下する場合がある。
【0023】
同様に、プロピレン系多段重合体は、溶融状態の動的粘弾性として、低周波数側での貯蔵弾性率の傾きが、一定量以下の大きさであることが好ましく、具体的には、角周波数が0.1rad/sの場合の貯蔵弾性率G’(0.1)と、角周波数が0.01rad/sの場合の貯蔵弾性率G’(0.01)との比であるG’(0.1)/G’(0.01)が6.0以下であることが好ましく、4.0以下であることが特に好ましい。かかる比G’(0.1)/G’(0.01)が6.0を越えると、低剪断速度下で発泡成形体の発泡倍率を高くすることが困難となる場合がある。
【0024】
このようなプロピレン系多段重合体は、下記成分(a)及び(b)、または下記成分(a)、(b)及び(c)からなるオレフィン重合用触媒を用い、2段階以上の重合工程で、プロピレンを重合またはプロピレンと炭素数2〜8のα−オレフィンとを共重合させて製造することができる。
【0025】
(a)四塩化チタンを有機アルミニウム化合物で還元して得られる三塩化チタンを、エーテル化合物及び電子受容体で処理して得られる固体触媒成分
(b)有機アルミニウム化合物
(c)環状エステル化合物
【0026】
固体触媒成分(a)において、四塩化チタンを還元する有機アルミニウム化合物としては、例えば、(a1)アルキルアルミニウムジハライド、具体的には、メチルアルミニウムジクロライド、エチルアルミニウムジクロライド、及びn−プロピルアルミニウムジクロライド、(a2)アルキルアルミニウムセスキハライド、具体的には、エチルアルミニウムセスキクロライド、(a3)ジアルキルアルミニウムハライド、具体的には、ジメチルアルミニウムクロライド、ジエチルアルミニウムクロライド、ジ−n−プロピルアルミニウムクロライド、及びジエチルアルミニウムブロマイド、(a4)トリアルキルアルミニウム、具体的には、トリメチルアルミニウム、トリエチルアルミニウム、及びトリイソブチルアルミニウム、(a5)ジアルキルアルミニウムハイドライド、具体的には、ジエチルアルミニウムハイドライド等をあげることができる。ここで、「アルキル」とは、メチル、エチル、プロピル、ブチル等の低級アルキルである。また、「ハライド」とは、クロライドまたはブロマイドであり、特に前者が通常である。
【0027】
三塩化チタンを得るための、有機アルミニウム化合物による還元反応は、−60〜60℃、好ましくは−30〜30℃の温度範囲で実施することが通常である。還元反応における温度が−60℃より低いと、還元反応に長時間が必要となり、一方、還元反応における温度が60℃を超えると、部分的に過還元が生じる場合があり好ましくない。還元反応は、ペンタン、ヘプタン、オクタン及びデカン等の不活性炭化水素溶媒下において実施することが好ましい。
【0028】
四塩化チタンの有機アルミニウム化合物による還元反応によって得られた三塩化チタンに対して、更にエーテル処理及び電子受容体処理を施すことが好ましい。
前記三塩化チタンのエーテル処理で好ましく用いられるエーテル化合物としては、例えば、ジエチルエーテル、ジ−n−プロピルエーテル、ジ−n−ブチルエーテル、ジイソアミルエーテル、ジネオペンチルエーテル、ジ−n−ヘキシルエーテル、ジ−n−オクチルエーテル、ジ−2−エチルヘキシルエーテル、メチル−n−ブチルエーテル及びエチル−イソブチルエーテル等の各炭化水素残基が炭素数2〜8の鎖状炭化水素であるエーテル化合物が挙げられ、これらの中でも特に、ジ−n−ブチルエーテルを用いることが好適である。
【0029】
三塩化チタンの処理で用いられる電子受容体としては、周期律表第III族〜第IV族及び第VIII族の元素のハロゲン化合物を使用することが好ましく、具体的には、四塩化チタン、四塩化ケイ素、三フッ化ホウ素、三塩化ホウ素、五塩化アンチモン、三塩化ガリウム、三塩化鉄、二塩化テルル、四塩化スズ、三塩化リン、五塩化リン、四塩化バナジウム及び四塩化ジルコニウム等を挙げることができる。
【0030】
固体触媒成分(a)を調製する際に、三塩化チタンのエーテル化合物及び電子受容体による処理は、両処理剤の混合物を用いて行ってもよく、また、一方の処理剤による処理後に、他方の処理剤による処理を行うようにしてもよい。なお、これらのうちでは、後者が好ましく、エーテル処理後に電子受容体で処理を行うことが更に好ましい。
【0031】
エーテル化合物及び電子受容体による処理の前に、三塩化チタンを炭化水素で洗浄することが好ましい。前記した三塩化チタンによるエーテル処理は、三塩化チタンとエーテル化合物を接触させることによって行われ、また、エーテル化合物による三塩化チタンの処理は、希釈剤の存在下で両者を接触させることによって行うのが有利である。このような希釈剤には、ヘキサン、ヘプタン、オクタン、デカン、ベンゼン及びトルエン等の不活性炭化水素化合物を使用することが好適である。なお、エーテル処理における処理温度は、0〜100℃であることが好ましい。また、処理時間については特に制限されないが、通常20分〜5時間の範囲で行われる。
【0032】
エーテル化合物の使用量は、三塩化チタン1molあたり、一般に0.05〜3.0mol、好ましくは0.5〜1.5molの範囲とすればよい。エーテル化合物の使用量が0.05molより小さいと、生成される重合体の立体規則性を十分に向上させることができなくなるので好ましくない。一方、エーテル化合物の使用量が3.0molを越えると、生成される重合体の立体規則性は向上するものの、収率が低下することとなるので好ましくない。なお、有機アルミニウム化合物やエーテル化合物で処理した三塩化チタンは、厳密に言えば、三塩化チタンを主成分とする組成物である。
なお、このような固体触媒成分(a)としては、Solvay型三塩化チタンを好適に用いることができる。
【0033】
有機アルミニウム化合物(b)としては、前述した有機アルミニウム化合物と同様なものを使用すればよい。
環状エステル化合物(c)としては、例えば、γ−ラクトン、δ−ラクトン、ε−ラクトン等が挙げられるが、ε−ラクトンを使用することが好ましい。
以上の成分(a)〜(c)を混合することにより、本実施形態で用いるプロピレン系多段重合体を製造するためのオレフィン重合用触媒を得ることができる。
なお、この成分(a)〜(c)からなる触媒でプロピレン系樹脂を製造した場合、アイソタクティシティーが比較的高いポリプロピレン系樹脂が得られることが知られている。
【0034】
本実施形態で用いるプロピレン系多段重合体を得るには、2段階の重合方法のうち、水素不存在下でプロピレンを重合またはプロピレンと炭素数2〜8のα−オレフィンを共重合させることが好ましい。
ここで、「水素不存在下」とは、実質的に水素不存在下という意味であり、水素が全く存在しない場合だけでなく、水素が極微量存在する場合(例えば、10molppm程度)も含まれる。要は、135℃テトラリン溶媒中で測定した、1段階目のプロピレン系重合体またはプロピレン系共重合体の極限粘度[η]が10dL/g以下とならない程度に水素を含む場合でも、「水素不存在下」の意味には含まれる。
【0035】
このような水素不存在下でプロピレンの重合またはプロピレンとα−オレフィンとの共重合を行うことにより、超高分子量プロピレン系重合体、すなわち、プロピレン系多段重合体の成分(P1)および成分(P2)を製造することができる。
成分(P1)は、水素不存在下で、原料モノマーを重合温度として、好ましくは20〜80℃、より好ましくは40〜70℃、重合圧力として、一般に、常圧〜1.47MPa、好ましくは0.39〜1.18MPaの条件下でスラリー重合して製造することが好ましい。
【0036】
プロピレン系多段重合体の成分(P2)は、2段階目以降に製造することが好ましい。
成分(P2)の製造条件としては、前記したオレフィン重合用触媒を使用すること以外は特に制限はないが、原料モノマーを、重合温度として、好ましくは20〜80℃、より好ましくは60〜70℃、重合圧力として、一般に、常圧〜1.47MPa、好ましくは0.19〜1.18MPa、分子量調整剤としての水素が存在する条件下で重合して製造することが好ましい。
【0037】
なお、前述した製造方法では、本重合を実施する前に、予備重合を行うようにしてもよい。予備重合を実施すると、パウダーモルフォロジーを良好に維持することができる、予備重合は、一般的に、重合温度として、好ましくは0〜80℃、より好ましくは10〜60℃、重合量として、固体触媒成分1gあたり、好ましくは0.001〜100g、より好ましくは0.1〜10gのプロピレンを重合またはプロピレンと炭素数2〜8のα−オレフィンを共重合させることが好ましい。
【0038】
また、本発明で使用するポリプロピレン系樹脂として、プロピレン単独重合体とプロピレン−α−オレフィン共重合体をブレンドしたものを使用することができる。
プロピレン−α−オレフィン共重合体として、例えばプロピレン系ブロック共重合体が挙げられ、特開2003−286060に記載の製造方法によって製造することができる。
すなわち、下記成分[A],[B]及び[C]からなるオレフィン重合用触媒の存在下、プロピレンを重合又はエチレンとプロピレンとを共重合させて、エチレン含量が0〜5質量%のポリプロピレン成分又はプロピレン/エチレン共重合体成分を全重合量の1〜25質量%形成し、エチレンとプロピレンとを共重合させて、プロピレン/エチレン共重合体成分を全重合量の99〜75質量%形成し、エチレン含量を、全重合量の10〜80質量%とする。
【0039】
[A]下記化合物(a),(b),(c)又は下記化合物(a),(b),(c),(d)を反応させて得られる固体触媒成分
(a)マグネシウム化合物
(b)四塩化チタン
(c)フタル酸ジアルキル(アルキル基は、炭素数3〜20の直鎖状炭化水素基又は分岐状炭化水素基を表す)
(d)四塩化ケイ素
[B]有機アルミニウム化合物
[C]下記一般式(II)で表される有機ケイ素化合物
(R1)(R2CH2)Si(OR3)(OR4) …(II)
[式中、R1は、炭素数3〜12の脂環式炭化水素基、R2は、炭素数3〜20の分岐状炭化水素基、R3及びR4は、それぞれ独立であって、炭素数1〜20の炭化水素基を表す]
【0040】
固体触媒成分[A]は、チタン、マグネシウム、ハロゲン及び電子供与性化合物を含有する触媒成分であり、具体的には、上記化合物(a),(b),(c)又は上記化合物(a),(b),(c),(d)を反応させて得られる。
マグネシウム化合物(a)としては特に制限はないが、下記一般式(III)で表されるものを好ましく用いることができる。
MgR5R6 …(III)
上記の一般式(III)において、R5及びR6は、炭化水素基、OR7(R7は炭化水素基)またはハロゲン原子を示す。ここでR5、R6及びR7の炭化水素基としては、炭素数1〜12のアルキル基、炭素数3〜12のシクロアルキル基、炭素数6〜20のアリール基、炭素数7〜20のアラルキル基等を、R5及びR6のハロゲン原子としては、塩素、臭素、ヨウ素、フッ素を挙げることができる。また、R5、R6及びR7は同一でも異なってもよい。
【0041】
上記の一般式(III)で示されるマグネシウム化合物(a)の具体例としては、ジメチルマグネシウム、ジエチルマグネシウム、ジイソプロピルマグネシウム、ジブチルマグネシウム、ジヘキシルマグネシウム、ジオクチルマグネシウム、エチルブチルマグネシウム、ジフェニルマグネシウム、ジシクロへキシルマグネシウム、ブチルオクチルマグネシウム等のアルキルマグネシウムやアリールマグネシウム;ジメトキシマグネシウム、ジエトキシマグネシウム、ジプロポキシマグネシウム、ジブトキシマグネシウム、ジヘキシロキシマグネシウム、ジオクトキシマグネシウム、ジフェノキシマグネシウム、ジシクロヘキシロキシマグネシウム等のアルコキシマグネシウムやアリロキシマグネシウム;エチルマグネシウムクロリド、ブチルマグネシウムクロリド、ヘキシルマグネシウムクロリド、イソプロピルマグネシウムクロリド、イソブチルマグネシウムクロリド、t−ブチルマグネシウムクロリド、フェニルマグネシウムブロミド、ベンジルマグネシウムクロリド、エチルマグネシウムブロミド、ブチルマグネシウムブロミド、フェニルマグネシウムクロリド、ブチルマグネシウムイオダイド等のアルキルマグネシウムハライドやアリールマグネシウムハライド;ブトキシマグネシウムクロリド、シクロヘキシロキシマグネシウムクロリド、フェノキシマグネシウムクロリド、エトキシマグネシウムブロミド、ブトキシマグネシウムブロミド、エトキシマグネシウムイオダイド等のアルコキシマグネシウムハライドやアリロキシマグネシウムハライド;塩化マグネシウム、臭化マグネシウム、ヨウ化マグネシウム等のハロゲン化マグネシウム等が挙げられる。
これらのマグネシウム化合物(a)の中では、ハロゲン化マグネシウム、アルコキシマグネシウム、アルキルマグネシウムハライドが好適に使用できる。中でも、アルコキシマグネシウムが特に好ましい。
上記のマグネシウム化合物(a)は、金属マグネシウム又はマグネシウムを含有する化合物から調製することができる。
【0042】
フタル酸ジアルキル(c)のアルキル基は、炭素数3〜20の直鎖状炭化水素基又は分岐状炭化水素基であり、具体的には、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、t−ブチル基、n−ペンチル基、1−メチルブチル基、2−メチルブチル基、3−メチルブチル基、1,1−ジメチルプロピル基、1−メチルペンチル基、2−メチルペンチル基、3−メチルペンチル基、4−メチルペンチル基、1−エチルブチル基、2−エチルブチル基、n−ヘキシル基、シクロヘキシル基、n−ヘプチル基、n−オクチル基、n−ノニル基、2−メチルヘキシル基、3−メチルヘキシル基、4−メチルヘキシル基、2−エチルヘキシル基、3−エチルヘキシル基、4−エチルヘキシル基、2−メチルペンチル基、3−メチルペンチル基、2−エチルペンチル基、3−エチルペンチル基等が挙げられる。これらのアルキル基の中では、炭素数が4以上の直鎖状又は分岐状の脂肪族炭化水素基が好ましい。
これらの具体例としては、フタル酸ジ−n−ブチル、フタル酸ジイソブチル、フタル酸ジ−n−ヘプチル等が挙げられる。中でも、フタル酸ジ−n−ブチル、フタル酸ジイソブチルが特に好ましい。また、これらの化合物はそれぞれ単独で用いてもよいし、2種以上を組み合わせて用いてもよい。
【0043】
固体触媒成分[A]には、さらに、上記化合物(a),(b)及び(c)に、四塩化ケイ素(d)を加えることができる。四塩化ケイ素(d)は、マグネシウム化合物(a)に対するモル比が、通常0.01以上、好ましくは0.10以上となる割合で用いられる。このモル比が0.01未満では、触媒活性や立体規則性の向上効果が十分に発揮されない場合や、生成ポリマー中の微粉量が多くなる場合がある。
【0044】
上記の各化合物を接触させる方法としては、特に制限はなく、公知の方法で接触させればよい。例えば、特開昭53−43094号公報、同55−135102号公報、同55−135103号公報、同56−18606号公報等に記載の方法が挙げられる。具体的には、(1)マグネシウム化合物(a)又はマグネシウム化合物(a)とフタル酸ジアルキル(c)との錯化合物を、フタル酸ジアルキル(c)及び所望に応じて用いられる粉砕助剤等の存在下に粉砕して、四塩化チタン(b)と反応させる方法、(2)還元能を有しないマグネシウム化合物(a)の液状物と四塩化チタン(b)とを、フタル酸ジアルキル(c)の存在下において反応させて、固体状のチタン複合体を析出させる方法、(3)上記(1)又は(2)で得られたものに四塩化チタン(b)を反応させる方法、(4)上記(1)又は(2)で得られたものに、さらに、フタル酸ジアルキル(c)及び四塩化チタン(b)を反応させる方法、(5)マグネシウム化合物(a)又はマグネシウム化合物(a)とフタル酸ジアルキル(c)との錯化合物を、フタル酸ジアルキル(c)、四塩化チタン(b)及び所望に応じて用いられる粉砕助剤等の存在下で粉砕した後、必要に応じて四塩化ケイ素(d)で処理する方法等が挙げられる。
【0045】
さらには、これらの方法以外に、特開昭56−166205号公報、特開昭57−63309号公報、特開昭57−190004号公報、特開昭57−300407号公報、特開昭58−47003号公報等に記載の方法でも、固体触媒成分[A]を調製することができる。
四塩化チタン(b)の使用量は、マグネシウム化合物(a)のマグネシウム1モルに対して、通常、0.5〜100モル、好ましくは1〜50モルの範囲にするとよい。また、フタル酸ジアルキル(c)の使用量は、マグネシウム化合物(a)のマグネシウム1モルに対して、通常、0.01〜10モル、好ましくは0.05〜0.15モルの範囲にするとよい。さらに、四塩化ケイ素(d)を添加する場合には、その使用量を上記の割合にするとよい。
【0046】
固体触媒成分[A]の調製では、化合物(a)〜(d)の接触温度を、通常、−20〜200℃、好ましくは20〜150℃の範囲にするとよく、接触時間を、通常、1分〜24時間、好ましくは10分〜6時間の範囲にするとよい。
このとき、化合物(a)〜(d)の接触手順については特に問わない。例えば、各化合物を炭化水素等の不活性溶媒の存在下で接触させてもよいし、予め炭化水素等の不活性溶媒で各化合物を希釈して接触させてもよい。この不活性溶媒としては、例えば、n−ペンタン,イソペンタン,n−ヘキサン,n−ヘプタン,n−オクタン,イソオクタン等の脂肪族炭化水素;ベンゼン,トルエン,キシレン等の芳香族炭化水素又はこれらの混合物を挙げることができる。
また、固体触媒成分[A]の調製では、四塩化チタン(b)の接触を2回以上行い、触媒担体としての役割をするマグネシウム化合物(a)に十分担持させるとよい。
以上の接触により得られる固体触媒成分[A]は、炭化水素等の不活性溶媒で洗浄してもよい。この不活性溶媒としては、上記と同様のものが挙げられる。また、この固体触媒成分[A]は、乾燥状態で保存することもできるし、また炭化水素等の不活性溶媒中でも保存することができる。
【0047】
有機アルミニウム化合物[B]としては、アルキル基、ハロゲン原子、水素原子、アルコキシ基を含有するもの、アルミノキサン及びそれらの混合物を好ましく用いることができる。具体的には、トリメチルアルミニウム,トリエチルアルミニウム,トリイソプロピルアルミニウム,トリイソブチルアルミニウム,トリオクチルアルミニウム等のトリアルキルアルミニウム;ジエチルアルミニウムモノクロリド,ジイソプロピルアルミニウムモノクロリド,ジイソブチルアルミニウムモノクロリド,ジオクチルアルミニウムモノクロリド等のジアルキルアルミニウムモノクロリド;エチルアルミニウムセスキクロリド等のアルキルアルミニウムセスキハライド;メチルアルミノキサン等の鎖状アルミノキサン等を挙げることができる。これらの有機アルミニウム化合物[B]の中では、炭素数1〜5の低級アルキル基を有するトリアルキルアルミニウム、特にトリメチルアルミニウム,トリエチルアルミニウム,トリプロピルアルミニウム及びトリイソブチルアルミニウムが好ましい。また、これらの有機アルミニウム化合物[B]は、それぞれ単独で用いてもよいし、2種以上を組み合わせて用いてもよい。
【0048】
有機ケイ素化合物[C]は、上記一般式(II)で表される。
具体的には、R1としては、シクロプロピル基、シクロブチル基、シクロペンチル基、シクロヘキシル基、シクロへプチル基、シクロオクチル基、1−ノルボルニル基、2−ノルボルニル基等が挙げられ、特に、シクロペンチル基、シクロヘキシル基が好ましい。R2としては、イソプロピル基、イソブチル基、sec−ブチル基、t−ブチル基、ネオペンチル基等が挙げられ、特に、イソプロピル基が好ましい。R3及びR4としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、ヘキシル基、オクチル基、シクロヘキシル基等のアルキル基、アリル基、プロペニル基、ブテニル基等のアルケニル基、フェニル基、トリル基、キシリル基等のアリール基、フェネチル基、3−フェニルプロピル基等のアラルキル基等が挙げられる。これらの中では、特に炭素数1〜10のアルキル基が好ましい。
【0049】
有機ケイ素化合物[C]としては、具体的に、シクロプロピルイソブチルジメトキシシラン、シクロプロピルイソペンチルジメトキシシラン、シクロプロピル−2−メチルブチルジメトキシシラン、シクロプロピルネオペンチルジメトキシシラン、シクロプロピル−2−メチルへキシルジメトキシシラン、シクロブチルイソブチルジメトキシシラン、シクロブチルイソペンチルジメトキシシラン、シクロブチル−2−メチルブチルジメトキシシラン、シクロブチルネオペンチルジメトキシシラン、シクロブチル−2−メチルへキシルジメトキシシラン、シクロペンチルイソブチルジメトキシシラン、シクロペンチルイソペンチルジメトキシシラン、シクロペンチル−2−メチルブチルジメトキシシラン、シクロペンチルネオペンチルジメトキシシラン、シクロペンチル−2−メチルへキシルジメトキシシラン、シクロヘキシルイソブチルジメトキシシラン、シクロヘキシルイソペンチルジメトキシシラン、シクロヘキシル−2−メチルブチルジメトキシシラン、シクロヘキシルネオペンチルジメトキシシラン、シクロヘキシル−2−メチルへキシルジメトキシシラン、シクロへプチルイソブチルジメトキシシラン、シクロへプチルイソペンチルジメトキシシラン、シクロへプチル−2−メチルブチルジメトキシシラン、シクロへプチルネオペンチルジメトキシシラン、シクロへプチル−2−メチルへキシルジメトキシシラン、シクロオクチルイソブチルジメトキシシラン、シクロオクチルイソペンチルジメトキシシラン、シクロオクチル−2−メチルブチルジメトキシシラン、シクロオクチルネオペンチルジメトキシシラン、シクロオクチル−2−メチルへキシルジメトキシシラン、1−ノルボルニルイソブチルジメトキシシラン、1−ノルボルニルイソペンチルジメトキシシラン、1−ノルボルニル−2−メチルブチルジメトキシシラン、1−ノルボルニルネオペンチルジメトキシシラン、1−ノルボルニル−2−メチルへキシルジメトキシシラン、2−ノルボルニルイソブチルジメトキシシラン、2−ノルボルニルイソペンチルジメトキシシラン、2−ノルボルニル−2−メチルブチルジメトキシシラン、2−ノルボルニルネオペンチルジメトキシシラン、2−ノルボルニル−2−メチルへキシルジメトキシシラン等が挙げられる。このうち、好ましくはシクロペンチルイソブチルジメトキシシラン、シクロヘキシルイソブチルジメトキシシランである。
これらの有機ケイ素化合物[C]はそれぞれ単独で用いてもよいし、2種以上を組み合わせて用いてもよい。
【0050】
上記成分[A]〜[C]の使用量については、特に制限はないが、固体触媒成分[A]は、チタン原子に換算して、反応容積1リットル当たり、通常0.0005〜1ミリモルの範囲になるような量が用いられる。
有機アルミニウム化合物[B]は、アルミニウム/チタン(原子比)が、通常1〜1,000、好ましくは10〜500の範囲になるような量が用いられる。
この原子比が前記範囲を逸脱すると、触媒活性が不十分となることがある。
有機ケイ素化合物[C]は、有機ケイ素化合物[C]/有機金属化合物[B](モル比)が、通常0.02〜2.0、好ましくは0.05〜1.0の範囲になるような量が用いられる。このモル比が前記範囲を逸脱すると、十分な触媒活性が得られないことがある。
【0051】
本願で用いるプロピレン系ブロック共重合体は、上述したオレフィン重合用触媒を用いて、好ましくは、一段目に、プロピレンを重合またはエチレンとプロピレンとを共重合させて、エチレン含量が0〜5質量%のポリプロピレン成分またはプロピレン/エチレン共重合体成分、好ましくは、エチレン含量が0質量%のポリプロピレン成分、即ち、プロピレン単独重合部を、全重合量の1〜25質量%、好ましくは5〜20質量%形成し、次いで、二段目に、エチレンとプロピレンとを共重合させて、プロピレン/エチレン共重合体成分、即ち、共重合部を、全重合量の99〜75質量%、好ましくは95〜80質量%形成し、エチレン含量が、全重合量の10〜80質量%、好ましくは15〜45質量%のブロック共重合体を製造する。
【0052】
さらに、ポリプロピレン成分またはプロピレン/エチレン共重合体成分を形成する前に、オレフィン類、好ましくは、α−オレフィンの予備重合を行うことができる。
予備重合では、用いる触媒については特に制限されないが、好ましくは、上述したオレフィン重合用触媒を用いる。この場合、電子供与性成分として、上記有機化合物[C]に加え、さらに、ジシクロペンチルジメトキシシラン、シクロペンチルエチルジメトキシシラン、シクロペンチルイソプロピルジメトキシシラン、シクロペンチルターシャリブチルジメトキシシラン、テキシルシクロペンチルジメトキシシラン、テキシルシクロヘキシルジメトキシシラン、ジイソプロピルジメトキシシラン、ジイソブチルジメトキシシラン、ジターシャリブチルジメトキシシラン等を用いることができる。これらのうち、好ましくはジシクロペンチルジメトキシシランである。
α−オレフィンとしては、特に制限はないが、具体的には、エチレン、プロピレン、1−ブテン、1−ペンテン、1−ヘキセン、1−ヘプテン、1−オクテン、1−デセン、3−メチル−1−ペンテン、4−メチル−1−ペンテン、ビニルシクロヘキサン等が挙げられる。このうち、エチレン及びプロピレンが好ましい。これらのα−オレフィンは、1種単独で用いてもよいし、2種以上を組み合わせて用いてもよい。
【0053】
予備重合は、上記成分[A]、[B]及び[C]又はその他の有機ケイ素化合物の存在下、α−オレフィンを、通常1〜100℃の範囲の温度において、常圧〜5MPa(Gauge)の圧力で重合させればよい。重合時間は1分〜10時間、好ましくは10分〜5時間である。予備重合量は、固体触媒成分[A]に対して、通常0.1〜1,000質量%、好ましくは1.0〜500質量%重合させればよい。
さらに、予備重合で得られたオレフィン重合体を、予備重合触媒成分として用いることができる。即ち、この予備重合触媒成分をオレフィン重合用触媒成分として用い、本発明で用いるプロピレン系ブロック共重合体を製造することができる。
【0054】
プロピレン系ブロック共重合体の製造方法では、重合形式について特に制限はなく、溶液重合、スラリー重合、気相重合、バルク重合等のいずれにも適用可能である。また、重合方式としては回分式重合や連続式重合のどちらであってもよい。例えば、連続式で製造する場合は、前段の重合槽に原料プロピレンガス、分子量調整剤の水素ガス及び触媒を供給し、重合時間で重合量をコントロールしてプロピレン単独重合部を製造し、次いで後段の重合槽に移動してさらに原料プロピレンガスにエチレンガス、水素ガス、及び必要に応じて触媒を加えて共重合部を製造し、ブロック共重合体を得ることができる。
共重合部の製造に際しては、エチレンを単独で用いてもよいが、必要に応じて、エチレンとプロピレン以外の前記α−オレフィンを組み合わせてもよい。
プロピレン単独重合の条件としては、重合時に、上述した重合量が得られるものであれば特に制限されないが、その重合圧は、通常、大気圧〜8MPa(Gauge)、好ましくは0.2〜5MPa(Gauge)、重合温度は、通常、0〜200℃、好ましくは30〜100℃の範囲で適宜選ばれる。重合時間は、通常、5分〜20時間、好ましくは10分〜10時間程度である。
【0055】
エチレンとプロピレンとの共重合の条件としては、共重合時に、上述したエチレン含量及び重合量が得られるものであれば特に制限されないが、その重合圧は、通常、大気圧〜8MPa(Gauge)、好ましくは0.2〜5MPa(Gauge)、重合温度は、通常、0〜200℃、好ましくは20〜100℃の範囲で適宜選ばれる。重合時間は、通常、1分〜20時間、好ましくは1分〜10時間程度である。供給するエチレンとプロピレンとの比率は、エチレン/プロピレンのモル比で0.01〜9、好ましくは0.05〜2.3である。
【0056】
この発明では、ポリプロピレン系押出発泡体は押出発泡成形により成形される。特に、一般的に使用されている公知の押出発泡装置を用いることができる。
具体的には、前述のポリプロピレン系樹脂と各種添加物を押出発泡装置内に投入し、溶融混練後、例えば大気圧あるいは大気圧より減圧雰囲気や加圧雰囲気となる押出機内より低圧となる低圧領域に押し出して発泡させ、ポリプロピレン系押出発泡体を得る。
【0057】
ポリプロピレン系樹脂を発泡させる手段としては、発泡材料を含有させる。具体的には、成形時に溶融状態の樹脂原料に発泡材料としての窒素ガスを注入する物理発泡、樹脂原料に発泡材料として窒素ガスを発生させる化学発泡剤を混合させる化学発泡などを採用することができる。
物理発泡では、窒素ガスの注入量の制御によるポリプロピレン系押出発泡体の発泡倍率の設定が容易となり、製造性が容易である。窒素ガスとしては、特に、樹脂原料への溶解性がよい超臨界状態の窒素ガスが好ましい。
一方、化学発泡では、樹脂原料との均一混合が物理発泡より容易で、製造性が容易である。
【0058】
化学発泡で使用する発泡剤としては、例えば、アゾジカルボンアミドやバリウムアゾジカルボキシレートなどのアゾ化合物、N,N’−ジニトロソペンタメチレンテトラミンなどのニトロソ化合物、4,4’−オキシビス(ベンゼンスルホニルヒドラジド)などのヒドラジン誘導体などが例示できる。
なお、上述した発泡剤のように、窒素ガスのみならず、炭酸ガスやアンモニアなどを併せて生成するものを用いてもよい。
また、無機系化学発泡剤を使用する場合は、通常、取扱性、貯蔵安定性、ポリプロピレン系樹脂への分散性の点から、10質量%以上50質量%以下の濃度のポリオレフィン系樹脂のマスターバッチとして使用されることが好ましい。これら無機系化学発泡剤の添加量は種類、マスターバッチ中の濃度によって適宜選択すればよい。一般に、本発明のポリプロピレン系樹脂100質量部に対して、0.1質量部以上40質量部以下の範囲で含有されることが好ましく、より好ましくは0.5質量部以上30質量部以下の範囲で選択できる。
これらの発泡材料は、単独で用いてもよいし、2種以上を組み合わせて用いてもよい。
【0059】
発泡材料の使用量としては、ポリプロピレン系樹脂1gに対し、気泡を生成させるガスの発生量が2.2×10-4モル以上1.9×10-3モル以下となるように添加することが好ましい。より好ましくは、6.3×10-4モル以上1.9×10-3モル以下である。
具体的に、窒素ガス換算で、樹脂原料に対して0.6質量%以上5質量%以下で含有させることが好ましい。より好ましくは、1.7質量%以上5質量%以下で含有させる。0.6質量%より少ないと、断熱性などの発泡体として十分な機能が得られなくなるおそれがあり、5質量%より多く含有しても含有量に応じた発泡倍率の増大が期待できない上に、押出機内で樹脂原料と窒素ガスが分離し、押出された発泡体が不連続となる可能性がある。
化学発泡においても、押出過程で発生するガス量が上記モル数の範囲と同様となるように各種化学発泡剤の添加量を調整することが好ましい。
本発明では、発泡倍率が3倍以上となる条件で溶融混練を行う。このため、上記発泡剤の量とするとともに、シリンダ温度を170℃以上210℃以下、押出吐出量を1kg/hr以上150kg/hr以下で適宜選択すればよい。また、超臨界窒素ガスを使用する場合には、シリンダ内の圧力を10MPa以上20MPa以下で適宜選択すればよい。
【0060】
そして、押出発泡成形を行う際には、必要に応じて、酸化防止剤、中和剤、難燃剤、等の添加剤を使用することができる。添加剤の配合量は特に制限されず、適宜調節することができる。
また、粉末状または繊維状の多孔質フィラーとして、シリカ、活性炭、ゼオライト、シリカゲル、または繊維状活性炭を配合してもよい。
その他にも、結晶化核剤として、タルク、有機カルボン酸塩、有機リン酸塩、ソルビトール系核剤を配合してもよい。
【0061】
このようにして得られたポリプロピレン系押出発泡体は、発泡倍率が3倍以上であるため、断熱性能に優れている。
また、見かけの剪断速度が遅くても発泡倍率が3倍以上の高発泡体を製造可能であるので、ダイ出口のクリアランスを広くすることができる。したがって、厚みのある押出発泡体を形成することができるとともに、表面外観が良好となる。
さらに、本発明で使用するポリプロピレン系樹脂は、リサイクル性能にも優れ、また、耐薬品性や耐熱性も良好であることから、ポリプロピレン系押出発泡体もまた、リサイクル性能、耐薬品性および耐熱性に優れる。また、ポリプロピレン系樹脂は比較的に低コストであるので、材料費のコスト低減も図ることができる。
このように、厚みがあり、リサイクル性能、耐薬品性および耐熱性に優れた押出発泡体は、住設分野や自動車分野の内外装材として有用性が高い。
そして、公知の押出発泡装置をそのまま使用することができるので、新たな設備に投資する必要がない。したがって、製造工程においても大幅な変更がないので簡単に実施することができる。さらには、ポリプロピレン系押出発泡体をそのまま住設分野や自動車分野の内外装材などに利用できるとともに、さらにプレス加工などにより成形しても気泡がほとんど損なわれずに維持され、特性が損なわれることがないので、二次加工など、汎用性も向上できる。
【0062】
本発明のポリプロピレン系押出発泡体において、前記発泡倍率は、3倍以上20倍未満であることが好ましい。
この発明では、前述のポリプロピレン系樹脂材料として用いるからこそ、発泡倍率3倍以上のポリプロピレン系押出発泡体を形成することができる。なお、発泡倍率が20倍を超える発泡体を得るためには、発泡剤を高圧の状態で多量に供給する必要があり、特に二酸化炭素に比べて、ポリプロピレン系樹脂に対して溶解性の劣る窒素を発泡剤として用いた場合には、より高耐圧の設備が必要なため、設備投資のコストが膨大になり、また、成形の安定性にも問題が生じる可能性もある。
【0063】
本発明のポリプロピレン系押出発泡体において、前記ダイは、その出口の流路がスリット状、または円筒状であることが好ましい。
ポリプロピレン系押出発泡体の形状は、ダイに形成されたダイ出口の形状で決まる。このダイ出口の形状は、円形、菱形、スリット状、円筒状などの任意の形状とすることができるが、この中でも、厚みの調整が容易であり、成形性に優れるという点から、スリット状または円筒状であることが好ましい。
なお、発泡体の成形方法としては、複数のダイ出口から押し出された発泡体を集束して一つの発泡体とする、いわゆる細条集束体とすることも可能である。この場合、複数のダイ出口の形状を全て同じ形状としてもよいし、一つのダイに多種類の形状を複数形成するようにしてもよい。
【0064】
本発明のポリプロピレン系押出発泡体において、前記ダイの設定温度は、170℃以上210℃以下であることが好ましい。
ダイの設定温度が170℃未満であると、押出が困難となるおそれがあり、210℃を超えると、樹脂原料の発泡性能が低下するというおそれがある。
したがって、ダイの設定温度を170℃以上210℃以下とすることにより、成形性に優れ、高い発泡倍率のポリプロピレン系押出発泡体を得ることができる。
【0065】
本発明のポリプロピレン系押出発泡体は、厚みが2mm以上30mm以下に押出成形されたことが好ましい。
従来、ポリプロピレン系押出発泡体は、低剪断速度下で高発泡することが困難とされていたため、ダイのクリアランスを小さくする必要があり、厚みの小さいものとなっていた。
この発明によれば、ポリプロピレン系押出発泡体は2mm以上30mm以下という厚みがある。なお、ダイ出口が円形状のダイで押出発泡を行った場合は、直径が2mm以上30mm以下の太い発泡体を得ることができる。
したがって、ポリプロピレン系押出発泡体が厚い、または太いので、住設分野や自動車分野の内外装材として用いるのに有用性が大きい。
【発明を実施するための最良の形態】
【0066】
以下、本発明の実施形態を図面に基づいて説明する。
本発明で使用する押出発泡装置としては、樹脂原料を溶融状態に加熱し、適度の剪断応力を付与しながら混練し、押出発泡することができる公知の押出発泡装置を使用することができる。また、押出発泡装置を構成する押出機も、単軸押出機または二軸押出機のいずれのものも使用することができる。具体的には、例えば、特開2004−237729号公報に開示された、2台の押出機が接続されたタンデム型押出発泡成形装置などが挙げられる。
なお、本実施形態では、物理発泡により押出発泡する押出発泡装置を使用した。
図1は、本発明の一実施形態における押出発泡装置を模式的に示す概略図である。
【0067】
[押出発泡装置の構成]
押出発泡装置100は、図1に示すように、樹脂原料が投入されるホッパー110と、樹脂原料を溶融混練する押出機としてのシリンダ120と、シリンダ120に発泡剤ガスを導入するガス導入路130と、樹脂原料を押し出すギアポンプ140と、樹脂原料を成形するダイ150と、冷却用のマンドレル160と、を備えている。
【0068】
ホッパー110は、計量器としても機能し、樹脂原料が投入されると同時に計量することもできる。
シリンダ120は略円筒状に形成され、シリンダ120の内径よりも小さい径をもつ略円柱状のスクリュ121を有している。スクリュ121は、その外周面にらせん状の羽122を有しており、スクリュ121の軸を中心として回転可能に支持されている。シリンダ120の内部でスクリュ121が回転することにより、シリンダ120内の樹脂原料が溶融混練される。
なお、シリンダ120における押出温度は適宜設定すればよいが、例えば、170℃以上230℃以下に設定することが好ましい。押出温度が170℃未満であると押出が困難となるおそれがあり、230℃を超えると、発泡性能が低下するおそれがある。
【0069】
ガス導入路130は、シリンダ120の内部につながる流路であり、発泡ガス、すなわち窒素ガスを導入する。発泡ガスの導入により、シリンダ120内で樹脂原料と発泡ガスとが混合される。
ギアポンプ140は、シリンダ120で溶融混練された樹脂原料と発泡ガスとの混合物の流量を調整し、ダイ150へ安定的に押し出す。
【0070】
ダイ150は、樹脂原料を目的の形状に成形するものである。本実施形態では、図2に示すダイを用いた。
図2は、本実施形態にかかるダイの形状を示す模式図であり、(A)はダイの断面図、(B)はダイ出口側の側面図である。
ダイ150は、図2(A)および(B)に示すように、円環状の断面を有する円筒状の流路151を有している。この流路151のダイ出口側における円環の直径は、4mm以上1000mm以下であることが好ましい。直径が4mm未満であると、押出発泡した場合に一体化してしまい、円筒状に形成されないおそれがある。また、直径が1000mmを超えると、安定した連続成形が困難となる。
【0071】
なお、流路151は、ダイ出口151Aの円環の半径がダイ入口の円環の半径よりも大きく、ダイ150の内部で一連に形成されている。また、流路151はダイ出口151Aに近づくほど、そのクリアランスも小さくなる。クリアランスが最小となる部位を最小部位Sとすると、最小部位Sにおけるクリアランスは、適宜設定すればよいが、0.2mm以上とすることが好ましい。0.2mm未満であると、表面外観が悪化するとともに、十分な厚みの発泡体を得ることが難しくなる。クリアランスのより好ましい範囲は1mm以上5mm以下である。
【0072】
ダイ150の温度は、170℃以上210℃以下に設定することが好ましい。
ダイ150の温度が170℃未満であると、押出が困難となるおそれがあり、210℃を超えると、樹脂原料の発泡性能が低下するというおそれがある。
【0073】
マンドレル160は、水冷などによって、通常、10℃以上120℃以下に冷却されており、ダイ150から押し出された発泡体を冷却する。本実施形態では、マンドレル160は略円筒状に形成されており、その外周面161に、ダイ150から円筒状に押し出された発泡体が当接する。これにより、発泡体が成形および冷却される。
【0074】
[押出発泡装置の動作]
次に、押出発泡装置100で押出発泡を実施する際の動作について説明する。
まず、前述のポリプロピレン系樹脂をホッパー110に投入する。ホッパー110は計量器としても機能するため、所望の量の樹脂をホッパー110で計量することもできる。
ホッパー110に投入された樹脂は、シリンダ120に供給される。
シリンダ120の内部では、スクリュ121が軸を中心として回転しているので、このスクリュ121により樹脂が溶融混練される。このとき、ガス導入路130から発泡流体として窒素が導入されるので、樹脂と窒素が混合される。
溶融混練された樹脂と窒素の混合物はシリンダ120から押し出され、ギアポンプ140に導入される。ギアポンプ140で樹脂の流量が調節され、ダイ150へ押し出される。
【0075】
ダイ150では、内部の形状に沿って樹脂が押し出されることにより成形される。
そして、樹脂はダイ150から押し出されると同時に発泡して発泡体を形成し、マンドレル160に当接することによって賦形されるとともに冷却される。最後に、図示しないカッターにより切断され、シート状の発泡体が得られる。
【0076】
[実施形態の作用効果]
以上より、本実施形態では、次の作用効果を奏することができる。
本実施形態では、前述のポリプロピレン系樹脂を用いて公知の押出発泡装置により、発泡剤に窒素ガスを用いて押出発泡を行う。これにより、従来では困難であった窒素ガスでの発泡でも、発泡倍率3倍以上のポリプロピレン系押出発泡体として得ることができる。
このポリプロピレン系押出発泡体は、材料としてポリプロピレン系樹脂を用いるので、耐熱性、耐薬品性およびリサイクル性能に優れている。また、ポリプロピレン系樹脂は低コストであるので、材料のコスト削減を図ることができる。
【0077】
また、ポリプロピレン系押出発泡体は発泡倍率が3倍以上の高発泡体であるため、断熱性により優れており、住設分野や自動車分野の内外装材として有用性が高い。
そして、本実施形態では、公知の押出発泡装置で発泡倍率3倍以上のポリプロピレン系押出発泡体を製造することができる。すなわち、従来と同様の方法で製造することができる。したがって、新たな設備投資が必要なく、また、製造方法にも大きな変更がないので手間がかからず、簡単に製造することができる。
【0078】
[実施形態の変形例]
上記実施形態では、円筒形状のダイを使用したが、ダイの形状は目的に応じた形状を使用することができ、これに限られない。
例えば、図3に示すように、スリット状の断面を有する平行管状の流路を有するダイ170でもよい。
【0079】
また、図4に示すように、円形状の断面を有する円管状の流路を有するダイ180でもよい。
【0080】
さらに、ダイは、円形状の断面を有する円錐状の流路を有していてもよいし、任意形状の断面を有する平行管状の流路を有していても良い。このように、目的に応じた形状のダイを使用することができる。
【0081】
また、上記実施形態では、円筒状の出口が一つだけ形成されたダイを使用したが、複数の出口が形成されたダイを使用することにより、押し出された複数の発泡体を集束して一つの発泡体とする、いわゆる細条集束体とすることもできる。
この場合、複数の出口形状を全て同じ形状としてもよいし、各々の出口を異なる形状、例えば、円形、菱形、スリット状などとしてもよい。また、同じ円形であっても、それぞれの径の大きさを変えて複数の種類の円形とすることもできる。
【0082】
また、上記実施形態では、物理発泡により押出発泡を行ったが、化学発泡によるものでもよい。この場合は、前述の発泡剤等を添加することにより実施することができる。
【実施例1】
【0083】
以下、実施例及び比較例を挙げて本発明をより具体的に説明する。なお、本発明は実施例等の内容に何ら限定されるものではない。
<樹脂について>
本実施例では、以下に示す2種類のポリプロピレン系樹脂組成物を使用する。
【0084】
樹脂A:直鎖状ポリプロピレン
樹脂B:分岐状ポリプロピレン(商品名「PF814」、Basell社製)
【0085】
[樹脂A(プロピレン系多段重合体)の製造方法]
(1)予備重合触媒成分の調製:
内容積5リットルの攪拌機付き三つ口フラスコを十分に乾燥させ、窒素ガスで置換した後、脱水処理したヘプタンを4リットル、ジエチルアルミニウムクロライド140グラムを加え、市販品のSolvay型三塩化チタン触媒(東ソー・ファインケム(株)製)20gを加えた。これを攪拌しながら20℃に保持した状態で、プロピレンを連続的に導入した。80分後、攪拌を停止し、三塩化チタン触媒1gあたり0.8gのプロピレンが重合した予備重合触媒成分を得た。
【0086】
(2)プロピレン重合:
内容積10リットルの攪拌機付きステンレス製オートクレーブを十分乾燥し窒素ガスで置換した後、脱水処理したヘプタン6リットルを加え、系内の窒素をプロピレンで置換した。その後、攪拌しながらプロピレンを導入して内温50℃、全圧0.78MPaに系内が安定した後、上記予備重合触媒成分を固体触媒換算で0.75グラム含んだヘプタンスラリー200ミリリットルを加えて重合開始とした。プロピレンを1.0時間連続的に供給し、重合を行い、重合体(P1成分)を得た。その一部をサンプリングして分析した結果、極限粘度は15.4dL/gであった。その後、内温を40℃以下にまで降温し攪拌を弱め、脱圧を行った。
再び、内温を65℃として水素を0.10MPa加えて攪拌しながらプロピレンを導入した。全圧0.78MPaでプロピレンを連続的に供給しながら65℃で4時間重合を行い、重合体(P2成分)を得た。この時、重合体の一部をサンプリングして分析した結果、極限粘度は3.31dL/gであった。
重合終了後、50ミリリットルのメタノールを添加し降温、脱圧した。内容物を全量フィルター付きろ過槽へ移し、1−ブタノールを100ミリリットル加え、85℃で1時間撹拌した後に固液分離した。さらに、85℃のヘプタン6リットルで固体部を2回洗浄し、真空乾燥してプロピレン重合体3.3kgを得た。
以上の結果、重合体(P1)と重合体(P2)の重合成分の質量比は15.3:84.7であり、第二段目にて生成した重合体の極限粘度は1.13dL/gと求められた。
【0087】
[物性値などの測定方法]
(1)圧力補正値:
上記の4種類のポリプロピレン系樹脂組成物についてバーグレー補正における圧力補正値を測定した。バーグレー補正における圧力補正値は、「JIS K 7199」に規定されたキャピラリーレオメータによるプラスチックの流れ特性試験方法に基づいて測定することができる。
具体的には、上記の樹脂A、樹脂B、樹脂Cについて、キャピログラフ型式1C(商品名、(株)東洋精機製作所製)を用い、剪断速度を変えてそれぞれの圧力補正値を以下の条件で測定した。なお、キャピログラフ型式1Cは関連業界で広く用いられているものであり、これを用いて以下の条件で測定を実施すると、剪断速度は1216s-1となる。
【0088】
バレル直径(内径):9.55mm
キャピラリー直径D:1.0mm
ピストンの押出速度:100mm/min
測定温度 :210℃
キャピラリーの直径と長さの比L/D:30、40、50の3種
【0089】
(2)プロピレン重合体成分(P1成分)及びプロピレン重合体成分(P2成分)の質量分率:
重合時に連続的に供給するプロピレンの流量計積算値を用いた物質収支から求めた。
(3)極限粘度[η]:
135℃のテトラリン溶媒中で測定した。
プロピレン系多段重合体の第一段目(P1成分)の極限粘度[η1]とプロピレン重合体全体の極限粘度[ηtotal]は重合過程でサンプリングし評価し、プロピレン系多段重合体の第二段目(P2成分)の極限粘度[η2]は下記(IV)式により算出した。
【0090】
[η2]=([ηtotal]×100-[η1]×W1)/W2 …(IV)
[ηtotal] :プロピレン重合体全体の極限粘度(dL/g)
[η1] :P1成分の極限粘度(dL/g)
W1 :P1成分の質量分率(質量%)
W2 :P2成分の質量分率(質量%)
【0091】
(4)メルトフローレート(MFR):
JIS K7210に準拠し、温度を230℃、荷重を2.16kgf(21.2N)として測定した。
【0092】
(5)発泡倍率:
成形品の質量を、水没法により求めた体積で除することにより密度を求め、未発泡品の密度で除することにより、発泡倍率として算出した。
【0093】
(6)連続成形性:
樹脂中に混合する窒素ガス量が多すぎる場合は、成形機のシリンダ内で樹脂と窒素ガスが均一に分散せず、窒素ガスが分離した状態になる。
この状態で成形機から樹脂と窒素ガスの混合物を押出発泡させると、押出された成形品の途中で窒素ガスのみが押出される領域が発生し、発泡体が断絶される。
そこで、目視評価により発泡体の断絶の有無を確認した。
【0094】
[樹脂の物性評価]
上記の樹脂Aおよび樹脂Bを上記の方法にて圧力補正値を測定した結果を図5に示す。図5は、剪断速度と圧力補正値との関係を表すグラフである。
【0095】
図5に示すように、従来のポリプロピレン系樹脂組成物である樹脂Bは、剪断速度が1000s-1以下で圧力補正値は大きく変化しない。一方、樹脂Aは、剪断速度が1000s-1以下で、圧力補正値が急激に大きくなっている。1000s-1付近で圧力補正値が大きいと、伸長粘度が高く気泡が破れにくいため、樹脂Aは、樹脂Bに比べて伸長粘度が高く、高発泡の発泡体が得られると言える。
【0096】
また、上記2種の樹脂について、剪断速度1216s-1における圧力補正値(バーグレー補正)とメルトフローレート(MFR)の測定結果を以下に示す。
【0097】
【表1】
【0098】
以下の表2に示すように、上記樹脂Aおよび樹脂Bを用いて、窒素ガスの物理発泡および化学発泡による発泡成形体の押出実験を実施した。
(1−1)物理発泡の製造装置および製造条件
成形機:日本製鋼所製 MuCell射出成形機、製品名「J180ELIII−MuCell」(金型をとりはずして使用)
押出時間:12秒
押出量:95g
シリンダ設定温度:180℃
(1−2)物理発泡の製造方法
MuCell射出成形機(金型をとりはずして使用)を用いて、前述した樹脂に超臨界状態の窒素ガスを15MPaで混合し、得られた混合物を大気中に押出すことによって発泡体を得た。
結果を以下の表2に示す。
【0099】
(2−1)化学発泡の製造装置および製造条件
押出機 :株式会社マックインターナショナルアソシエイツ製 押出機(スクリュー径45mm)
ダイ :円筒ダイ(直径45mm)
押出量 :10kg/hr
押出機 :シリンダ設定温度210℃
化学発泡剤A:アゾジカルボンアミドを主成分とし、試料0.500g、昇温速度2℃/min、流動パラフィン10mlの条件で昇温した際の発生ガス量が165℃で5ml、170℃で80mlとなるガス発生特性を有する発泡剤
化学発泡剤B:アゾジカルボンアミドを主成分とし、試料0.500g、昇温速度2℃/min、流動パラフィン10mlの条件で昇温した際の発生ガス量が175℃で5ml、180℃で80mlとなるガス発生特性を有する発泡剤
(2−2)化学発泡の製造方法
樹脂原料100重量部に対し、発泡剤5重量部(発生窒素0.7重量部相当)を加えて、ドライブレンドした混合物をホッパー110に投入する。そして、押出機の所定のスクリュ回転数に設定し、押出機内で樹脂原料を溶融化させるとともに発泡剤と混合し、一定の押出量で混合物をダイに供給する。
供給された混合物をダイから鉛直下方に押し出し、大気中で発泡させる。そして、筒状に押し出した溶融発泡体をダイ出口で切り取り、大気中で自然冷却させることにより、筒状の発泡体を得た。
結果を以下の表2に示す。
【0100】
【表2】
【0101】
表2に示すように、樹脂Aを用いた実施例1〜6では、物理発泡、化学発泡を問わず、十分な発泡が得られた。一方、樹脂Bを用いた比較例1の発泡体では、物理発泡でも発泡倍率が2倍に満たず、発泡倍率を上げるために注入する窒素ガスを増やしてもダイから窒素ガスが吹き出る状態となって押出発泡成形が出来なかった。
なお、図6に、実施例3と比較例1との成形後の外観を状況を示す。図中の左側が実施例3で右側が比較例1を示し、手前に比較として15cm定規を載置している。
また、窒素ガスが0.5重量%以下である比較例2においては、発泡倍率が3倍を下回る発泡体しか得られなかった。一方、比較例3、比較例4においては、発泡倍率は16倍を超えているものの、窒素ガスの分離による発泡体の断絶が発生しており、連続した発泡体を得る事は出来なかった。
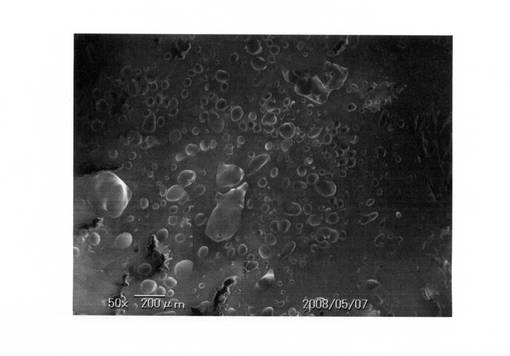
なお、比較例1で得られた発泡体電子顕微鏡写真を、図7(倍率:50倍)および図8(倍率:180倍)にそれぞれ示す。また、実施例3で得られた発泡体電子顕微鏡写真を、図9(倍率:50倍)、図10(倍率:180倍)にそれぞれ示す。
これら電子顕微鏡写真でも分かるように、実施例では十分に発泡しているが、比較例では発泡が不十分であった。
【産業上の利用可能性】
【0102】
本発明のポリプロピレン系押出発泡体は、ポリプロピレン系樹脂としての特性を活かしつつ、軽量性、断熱性に優れるので、例えば、住設分野や自動車分野の内外装材に好適である。
【図面の簡単な説明】
【0103】
【図1】本発明の一実施形態の押出発泡装置を模式的に示す概略図。
【図2】本実施形態にかかるダイの形状を示す模式図であり、(A)はダイの断面図、(B)はダイ出口側の側面図。
【図3】上記実施形態にかかるダイの変形例を示す模式図であり、(A)はダイの断面図、(B)はダイ出口側の側面図。
【図4】上記実施形態にかかるダイの変形例を示す模式図であり、(A)はダイの断面図、(B)はダイ出口側の側面図。
【図5】剪断速度と圧力補正値との関係を示すグラフ。
【図6】実施例と比較例との発泡状況を示す外観図である。
【図7】比較例の発泡状況を示す電子顕微鏡写真(倍率:50倍)である。
【図8】比較例の発泡状況を示す電子顕微鏡写真(倍率:180倍)である。
【図9】実施例の発泡状況を示す電子顕微鏡写真(倍率:50倍)である。
【図10】実施例の発泡状況を示す電子顕微鏡写真(倍率:180倍)である。
【符号の説明】
【0104】
100…押出発泡装置
110…ホッパー
120…シリンダ
130…ガス導入路
140…ギアポンプ
150、170、180…ダイ
151…流路
160…マンドレル
S、T、U…クリアランスの最小部位
【特許請求の範囲】
【請求項1】
ポリプロピレン系樹脂と発泡材料とを含有する混合物を押出機内で溶融混練する溶融混練工程と、この溶融混練工程で溶融混練された組成物をダイから押出発泡させる押出発泡工程と、を実施するポリプロピレン系押出発泡体の製造方法であって、
前記ポリプロピレン系樹脂は、(A)測定温度210℃、剪断速度1216s-1の条件でのキャピラリーフローテストにおいて、バーグレー補正における圧力補正値が4MPa以上、および(B)メルトフローレート(MFR)が0.5g/10分以上であり、
前記溶融混練工程では、前記ポリプロピレン系樹脂に前記発泡材料として窒素ガスを前記ポリプロピレン系樹脂に対して0.6質量%以上5質量%以下で含有させ、前記ポリプロピレン系押出発泡体の発泡倍率が3倍以上となる条件とする
ことを特徴とするポリプロピレン系押出発泡体の製造方法。
【請求項2】
請求項1に記載のポリプロピレン系押出発泡体の製造方法であって、
前記ポリプロピレン系樹脂が直鎖状であり、かつ、その極限粘度が1.0dL/g以上6.0dL/g以下である
ことを特徴とするポリプロピレン系押出発泡体の製造方法。
【請求項3】
請求項2に記載のポリプロピレン系押出発泡体の製造方法であって、
前記ポリプロピレン系樹脂の極限粘度が3.0dL/g以上6.0dL/g以下である
ことを特徴とするポリプロピレン系押出発泡体の製造方法。
【請求項4】
請求項1ないし請求項3のいずれか一項に記載のポリプロピレン系押出発泡体の製造方法であって、
前記ポリプロピレン系樹脂のメルトフローレート(MFR)が1.0g/10分以上10.0g/10分以下である
ことを特徴とするポリプロピレン系押出発泡体の製造方法。
【請求項5】
請求項1ないし請求項4のいずれか一項に記載のポリプロピレン系押出発泡体の製造方法であって、
前記発泡材料として、前記ポリプロピレン系樹脂との溶融混練された熱により分解して気体を発生する化学発泡剤を用いる
ことを特徴とするポリプロピレン系押出発泡体の製造方法。
【請求項6】
請求項1ないし請求項4のいずれか一項に記載のポリプロピレン系押出発泡体の製造方法であって、
前記窒素ガスを押出機内で溶融された前記ポリプロピレン系樹脂と混合させて溶解された
ことを特徴とするポリプロピレン系押出発泡体の製造方法。
【請求項7】
請求項1ないし請求項6のいずれか一項に記載のポリプロピレン系押出発泡体の製造方法であって、
前記発泡倍率は、3倍以上20倍未満である
ことを特徴とするポリプロピレン系押出発泡体の製造方法。
【請求項8】
請求項1ないし請求項7のいずれか一項に記載のポリプロピレン系押出発泡体の製造方法であって、
前記ダイとして、出口の流路がスリット状または円筒状のものを用いる
ことを特徴とするポリプロピレン系押出発泡体の製造方法。
【請求項9】
請求項1ないし請求項8のいずれか一項に記載のポリプロピレン系押出発泡体の製造方法であって、
前記ダイの設定温度は、170℃以上210℃以下である
ことを特徴とするポリプロピレン系押出発泡体の製造方法。
【請求項10】
請求項1ないし請求項9のいずれか一項に記載のポリプロピレン系押出発泡体の製造方法であって、
厚みが2mm以上30mm以下のポリプロピレン系押出発泡体に押出成形する
ことを特徴とするポリプロピレン系押出発泡体の製造方法。
【請求項11】
ポリプロピレン系樹脂と発泡材料とを含有する混合物が押出機内で溶融混練された後に、ダイから押出発泡されて得られるポリプロピレン系押出発泡体であって、
前記ポリプロピレン系樹脂は、(A)測定温度210℃、剪断速度1216s-1の条件でのキャピラリーフローテストにおいて、バーグレー補正における圧力補正値が4MPa以上、および(B)メルトフローレート(MFR)が0.5g/10分以上であり、
発泡倍率が3倍以上となる条件で、前記ポリプロピレン系樹脂に前記発泡材料として窒素ガスが前記ポリプロピレン系樹脂に対して0.6質量%以上5質量%以下で含有され、押出発泡された
ことを特徴としたポリプロピレン系押出発泡体。
【請求項1】
ポリプロピレン系樹脂と発泡材料とを含有する混合物を押出機内で溶融混練する溶融混練工程と、この溶融混練工程で溶融混練された組成物をダイから押出発泡させる押出発泡工程と、を実施するポリプロピレン系押出発泡体の製造方法であって、
前記ポリプロピレン系樹脂は、(A)測定温度210℃、剪断速度1216s-1の条件でのキャピラリーフローテストにおいて、バーグレー補正における圧力補正値が4MPa以上、および(B)メルトフローレート(MFR)が0.5g/10分以上であり、
前記溶融混練工程では、前記ポリプロピレン系樹脂に前記発泡材料として窒素ガスを前記ポリプロピレン系樹脂に対して0.6質量%以上5質量%以下で含有させ、前記ポリプロピレン系押出発泡体の発泡倍率が3倍以上となる条件とする
ことを特徴とするポリプロピレン系押出発泡体の製造方法。
【請求項2】
請求項1に記載のポリプロピレン系押出発泡体の製造方法であって、
前記ポリプロピレン系樹脂が直鎖状であり、かつ、その極限粘度が1.0dL/g以上6.0dL/g以下である
ことを特徴とするポリプロピレン系押出発泡体の製造方法。
【請求項3】
請求項2に記載のポリプロピレン系押出発泡体の製造方法であって、
前記ポリプロピレン系樹脂の極限粘度が3.0dL/g以上6.0dL/g以下である
ことを特徴とするポリプロピレン系押出発泡体の製造方法。
【請求項4】
請求項1ないし請求項3のいずれか一項に記載のポリプロピレン系押出発泡体の製造方法であって、
前記ポリプロピレン系樹脂のメルトフローレート(MFR)が1.0g/10分以上10.0g/10分以下である
ことを特徴とするポリプロピレン系押出発泡体の製造方法。
【請求項5】
請求項1ないし請求項4のいずれか一項に記載のポリプロピレン系押出発泡体の製造方法であって、
前記発泡材料として、前記ポリプロピレン系樹脂との溶融混練された熱により分解して気体を発生する化学発泡剤を用いる
ことを特徴とするポリプロピレン系押出発泡体の製造方法。
【請求項6】
請求項1ないし請求項4のいずれか一項に記載のポリプロピレン系押出発泡体の製造方法であって、
前記窒素ガスを押出機内で溶融された前記ポリプロピレン系樹脂と混合させて溶解された
ことを特徴とするポリプロピレン系押出発泡体の製造方法。
【請求項7】
請求項1ないし請求項6のいずれか一項に記載のポリプロピレン系押出発泡体の製造方法であって、
前記発泡倍率は、3倍以上20倍未満である
ことを特徴とするポリプロピレン系押出発泡体の製造方法。
【請求項8】
請求項1ないし請求項7のいずれか一項に記載のポリプロピレン系押出発泡体の製造方法であって、
前記ダイとして、出口の流路がスリット状または円筒状のものを用いる
ことを特徴とするポリプロピレン系押出発泡体の製造方法。
【請求項9】
請求項1ないし請求項8のいずれか一項に記載のポリプロピレン系押出発泡体の製造方法であって、
前記ダイの設定温度は、170℃以上210℃以下である
ことを特徴とするポリプロピレン系押出発泡体の製造方法。
【請求項10】
請求項1ないし請求項9のいずれか一項に記載のポリプロピレン系押出発泡体の製造方法であって、
厚みが2mm以上30mm以下のポリプロピレン系押出発泡体に押出成形する
ことを特徴とするポリプロピレン系押出発泡体の製造方法。
【請求項11】
ポリプロピレン系樹脂と発泡材料とを含有する混合物が押出機内で溶融混練された後に、ダイから押出発泡されて得られるポリプロピレン系押出発泡体であって、
前記ポリプロピレン系樹脂は、(A)測定温度210℃、剪断速度1216s-1の条件でのキャピラリーフローテストにおいて、バーグレー補正における圧力補正値が4MPa以上、および(B)メルトフローレート(MFR)が0.5g/10分以上であり、
発泡倍率が3倍以上となる条件で、前記ポリプロピレン系樹脂に前記発泡材料として窒素ガスが前記ポリプロピレン系樹脂に対して0.6質量%以上5質量%以下で含有され、押出発泡された
ことを特徴としたポリプロピレン系押出発泡体。
【図1】

【図2】

【図3】

【図4】
【図5】
【図6】

【図7】
【図8】
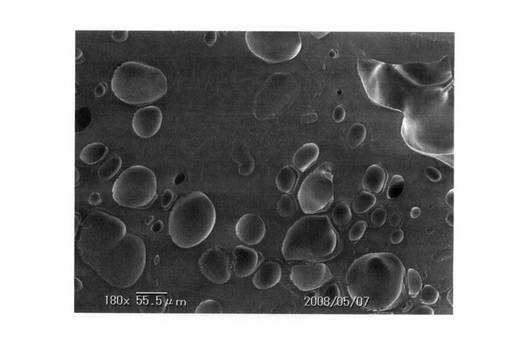
【図9】
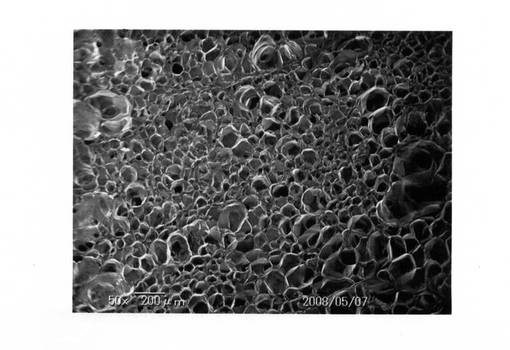
【図10】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【公開番号】特開2009−275150(P2009−275150A)
【公開日】平成21年11月26日(2009.11.26)
【国際特許分類】
【出願番号】特願2008−128852(P2008−128852)
【出願日】平成20年5月15日(2008.5.15)
【出願人】(505130112)株式会社プライムポリマー (180)
【Fターム(参考)】
【公開日】平成21年11月26日(2009.11.26)
【国際特許分類】
【出願日】平成20年5月15日(2008.5.15)
【出願人】(505130112)株式会社プライムポリマー (180)
【Fターム(参考)】
[ Back to top ]