
ポリリンゴ酸ベースの多機能性薬物送達システム
【課題】特定の組織または細胞型への薬物ミサイル弾頭を送達するのに有用な体系化薬物システムを提供すること。
【解決手段】上記システムは、精製ポリリンゴ酸を基礎にする。天然原料から単離されたこのポリマーは、生物適合性であり、生物分解性でありそして非常に毒性が低い。本ポリマーは、極端に水溶性であり、多くの異なる活性分子を連結するのに使用され得る多数の遊離カルボキシル基を含む。開示された実施例において、カルボキシル基のN−ヒドロキシスクシンイミドエステルは、そのような分子を連結するのに使用される。上記活性分子とは、特定の細胞性取り込みを促進するモノクローナル抗体および、標的細胞の細胞性代謝を修飾するよう設計されたアンチセンス核酸のような特定のプロドラッグが挙げられる。上記のプロドラッグは、有利なように特定の条件下で放出されるよう、いくらか弱い結合により結合される。
【解決手段】上記システムは、精製ポリリンゴ酸を基礎にする。天然原料から単離されたこのポリマーは、生物適合性であり、生物分解性でありそして非常に毒性が低い。本ポリマーは、極端に水溶性であり、多くの異なる活性分子を連結するのに使用され得る多数の遊離カルボキシル基を含む。開示された実施例において、カルボキシル基のN−ヒドロキシスクシンイミドエステルは、そのような分子を連結するのに使用される。上記活性分子とは、特定の細胞性取り込みを促進するモノクローナル抗体および、標的細胞の細胞性代謝を修飾するよう設計されたアンチセンス核酸のような特定のプロドラッグが挙げられる。上記のプロドラッグは、有利なように特定の条件下で放出されるよう、いくらか弱い結合により結合される。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、2003年12月5日に出願した米国仮特許出願第60/527,330号を基礎に出願し、その出願からの優先権を主張する。
【0002】
(本発明の背景および先行技術の説明)
本発明は、薬物の標的化送達の分野に関し、より具体的には多機能性標的化薬物送達ビヒクルに関する。
【背景技術】
【0003】
現在、いくつかの異なる分子骨格が薬物ビヒクルの合成に使用され、注目すべき例として、N−(2−ヒドロキシプロピル)メタクリルアミド(HPMA)コポリマー(20〜30kDa)(例えば、非特許文献26参照)および他のポリアクリル酸(polycarylic acid)の誘導体である。しかしながら、これらは、それらの炭素−炭素主鎖の故に生物分解性と考えられておらず(例えば、非特許文献27および本明細書の参考文献参照)、有害なアクリル酸による避けがたい汚染により問題である(例えば、非特許文献28参照)。他の分解性骨格(例えば、ポリ(L−グルタミン酸)(例えば、非特許文献29参照)は、ペプチド結合の周りの回転制限もしくは化学合成および生成物の精製に望ましい有機溶媒における限られた溶解度のような好ましくない性質を有し得、そしてさらにペプチド主鎖の高い水素結合形成能力に起因する他の潜在的免疫原性構造に構造的近似において免疫原性の基盤になっている(例えば、非特許文献29、30、31参照)。
【0004】
(アンチセンス技術)
特定のRNA配列と結合し、そして不活性化するアンチセンスオリゴヌクレオチド(オリゴ)は、遺伝子機能、遺伝子発現の制御、遺伝子産物間の相互作用および薬物開発の新しい治療標的の実証の研究をするための最良の手段の1つであり得る。アンチセンスオリゴは、ウイルス疾患、癌および他の重篤な疾患に対する安全で有効な治療の有望性を提供する。RNA産生のためのDNAテンプレートを模倣する特定のアンチセンスオリゴは、相補的RNAへ結合し、そしてタンパク質の翻訳(例えば腫瘍マーカーのタンパク質の翻訳)を抑制するために使用される(例えば、非特許文献1参照)。
【0005】
神経膠腫においてアンチセンス技術の使用について有望なデータがある。インビトロおよびヌードマウスにおいて神経膠腫の増殖は、テロメラーゼに対するアンチセンスにより阻害され得る(例えば、非特許文献2参照)。パイロット研究は、IGF−Iレセプターに対するアンチセンスが神経膠腫細胞アポトーシスを誘発し、患者の臨床的改善という結果が得られたことを示した(例えば、非特許文献3参照)。いくつかの臨床試験が、他の癌の処置にアンチセンスオリゴを現在使用している(例えば、非特許文献4参照)。これらの研究は不十分な特異性、安定性および非アンチセンス効果の無い新世代のアンチセンスオリゴを活用する(例えば、非特許文献5参照)。最も有望な改善されたオリゴの種類は、モルホリノ(Morpholino)オリゴおよびペプチド核酸(PNA)オリゴである。これらの種類は、全てのアンチセンスタイプの中で最高の配列特異性を有し、そしてこの特異性を非常に広い濃度範囲にわたって維持する(例えば、非特許文献6、7、8、9、10および11参照)。新しい、早く進化するアンチセンスアプローチの変種は、非常に強力な遺伝子発現サイレンサーであり、潜在的な抗癌薬でもある小干渉RNA(siRNA)に代表される。
【0006】
(インビトロおよびインビボにおける腫瘍の進行を抑制するためのいくつかの分子マーカーの組合わせによるブロッキング)
このアプローチは癌化学療法において長く成功裏に使用されてきたが、特定の腫瘍マーカーを標的化することにはまだ適用されていなかった。遺伝子/タンパク質アレイアプローチの進展の後にのみ、腫瘍の進行および再発の間の特定の遺伝子の協奏的な変化に関するデータを得て、そして関連付けることが可能となった。そのような協奏的な変化は、腫瘍の発症および進行を効率的にブロックすることを期待していくつかの遺伝子の同時変化を阻止する可能性を提供する。神経膠腫の増殖および拡散をブロックするいくつかの候補遺伝子があり、それには腫瘍の増殖をより効率的に抑制するために化学療法剤との組合わせで潜在的に使用され得るチロシンキナーゼレセプター(例えば、EGFR)、いくつかの成長因子および抗アポトーシス遺伝子が含まれる(例えば、非特許文献12、13、14、15および16参照)。本発明者らの以前の研究で、別の潜在的な候補タンパク質、ラミニン−8を同定したが、それは脳腫瘍および乳癌で過剰発現され、神経膠腫の予後の悪さと関連し、神経膠腫侵襲と関連する。
【0007】
(薬物送達)
腫瘍を処置するために癌細胞を直接標的化する目的で、薬物、例えば、モノクロ−ル抗体、アンチセンスオリゴまたは低分子(例えばタルセバ(Tarceva)(エルロチニブ)(erlotinib))は、細胞膜を透過し得る。細胞内薬物送達について3種類の基本的な方法があり、それらは、膜の水性チャネルまたは孔を通る受動的拡散、膜の脂質への溶解による脂質溶解性薬物の受動的拡散およびキャリア媒介性の能動輸送(ウイルスベクター、リポソーム媒介性の遺伝子移送システム、特殊化学品)である(例えば、非特許文献16、17参照)。脳組織は、水溶性でイオン化または極性の薬物の透過を抑制する脳毛細血管内皮細胞間のタイトジャンクションによる特別の血液脳関門を有するので、薬物による処置が特に困難である(例えば、非特許文献18、19参照)。
【0008】
高分子量分子は、高分子および脂質(分子量>45kDa)について癌組織で観察される高い透過性および保持性効果(enhanced permeability and
retention)(EPR)の故に、最近特に注目を集めている(例えば、非特許文献20、21および22参照)。腫瘍を正常組織と区別しない今日使用される低分子抗癌剤と異なり、高分子(またはポリマー性)薬物は、EPR効果により高度の選択性で癌を標的化し得る(例えば、非特許文献22、23参照)。そのような有望な薬物キャリアの1つとして、ポリ−L−リンゴ酸(PMLA)が、本発明者らの1人により開発されてきた(例えば、非特許文献24、25参照)。
【非特許文献1】Astriab−Fisher A,Sergueev DS,Fisher M,Shaw BR,Juliano RL.「Antisense inhibition of P−glycoprotein expression using peptide−oligonucleotide conjugates.」 Biochem Pharmacol、2000年、第60巻、p83−90
【非特許文献2】Komata T,Kondo Y,Koga S,Ko SC,Chung LW,Kondo S.「Combination therapy of malignant glioma cells with 2−5A−antisense telomerase RNA and recombinant adenovirus p53.」 Gene Ther,2000年、第7巻、p2071−2079
【非特許文献3】Andrews DW,Resnicoff M,Flanders AE,Kenyon L,Curtis M,Merli G,Baserga R,Iliakis G,Aiken RD.「Results of a pilot study involving the use of an antisense oligodeoxynucleotide directed against the insulin−like growth factor type I receptor in malignant astrocytomas.」J Clin Oncol、2001年、第19巻、p2189−2200
【非特許文献4】Jansen B,Wacheck V,Heere−Ress E,Schlagbauer−Wadl H,Hoeller C,Lucas T,Hoermann M,Hollenstein U,Wolff K、Pehamberger H.「Chemosensitisation of malignant melanoma by BCL2 antisense therapy.」Lancet、2000年、第356巻、p1728−1733
【非特許文献5】Nielsen PE.「Peptide nucleic acid targeting of double−stranded DNA.」Methods Enzymol、2001年、第340巻、p329−340
【非特許文献6】Summerton J,Weller D.「Morpholino antisense oligomers:Design,preparation and properties.」Antisense Nucleic Acid Drug Dev、1997年、第7巻、p187−195
【非特許文献7】Lacerra G,Sierakowska H,Carestia C,Fucharoen S,Summerton J,Weller D,Kole R.「Restoration of hemoglobin A synthesis in erythroid cells from peripheral blood of thalassemic patients」Proc Natl Acad Sci USA、2000年、第97巻、p9591−9596
【非特許文献8】Taylor MF,Paulauskis JD,Weller DD,Kobzic L.「Comparison of efficacy of antisense oligomers directed toward TNF−α in helper T and macrophage cell lines.」Cytokine、1997年、第9巻、p672−681
【非特許文献9】Arora V,Knapp DC,Smith BL,Statdfield ML,Stein DA,Reddy MT,Weller DD,Iversen PL.「c−Myc antisense limits rat liver regeneration and indicates role for c−myc in regulating cytochrome P−450 3A activity.」J Pharmacol Exp Ther、2000年、第292巻、p921−928
【非特許文献10】Shi N,Boado RJ,Pardridge WM.「Antisense imaging of gene expression in the brain in vivo.」Proc Natl Acad Sci USA、2000年、第97巻、p14709−14714
【非特許文献11】Boado RJ,Kazantsev A,Apostol BL,Thompson LM,Pardridge WM.「Antisense−mediated down−regulation of the human hutingtin gene.」J Pharmacol Exp Ther、2000年、第295巻、p239−243
【非特許文献12】Tanabe K,Kim R,Inoue H,Emi M,Uchida Y,Toge T.「Antisense Bcl−2 and HER−2 oligonucleotide treatment of breast cancer cells enhances their sensitivity to anticancer drugs.」Int J Oncol、2003年、第22巻、p875−81
【非特許文献13】Cho YS,Cho−Chung YS.「Antisense protein kinase A RIalpha acts synergistically with hydroxycamptothecin to inhibit growth and induce apoptosis in human cancer cells:molecular basis for combinational therapy.」Clin Cancer Res、2003年、第9巻、p1171−1178
【非特許文献14】Tortora G,Caputo R,Damiano V,Caputo R,Troiani T,Veneziani BM,De Placido S,Bianco AR,Zangemeister−Wittke U,Ciardiello F.「Combined targeted inhibition of bcl−2,bcl−XL,epidermal growth factor receptor,and protein kinase A type I causes potent antitumor,apoptotic,and antiangiogenic activity.」Clin Cancer Res、2003年、第9巻、p866−871
【非特許文献15】Jiang Z,Zheng X,Rich KM.「Down−regulation of bcl−2 and bcl−XL expression with bispecific antisense treatment in glioblastoma in vitro induce to enhance caspase−dependent cell death.」J Neurochem、2003年、第84巻、p273−281
【非特許文献16】Mycek MJ,Harvey RA,Champe,PC.「Pharmacology.Lippincott−Raven」第2版、Philadelphia,New York、1997年、p475
【非特許文献17】Park JW.「Liposome−based drug delivery in breast cancer treatment.」Breast Cancer Res.、2002年、第4巻、p95−99
【非特許文献18】Matsukado K,Sugita M,Black KL.「Intracarotid low dose bradykinin infusion selectively increases tumor permeability through activation of bradykinin B2 receptors in malignant gliomas.」Brain Res、1998年、第792巻、p10−15
【非特許文献19】Ningaraj NS,Rao MK,Black KL.「Adenosine 5’−triphosphate−sensitive potassium channel−mediated blood−brain tumor barrier permeability increase in a rat brain tumor model.」Cancer Res、2003年、第63巻、p8899−8911
【非特許文献20】Torchilin VP,Lukyanov AN.「Peptide and protein drug delivery to and into tumors:challenges and solutions.」Drug Discov Today、2003年、第8巻、p259−66
【非特許文献21】Peterson CM,Shiah J.,Sun Y,Kopeckova P,Minko T,Straight RC,Kopecek J「HPMA copolymer delivery of chemotherapy and photodynamic therapy in ovarian cancer」Adv Exp Med Biol、2003年、第519巻、p101−23
【非特許文献22】Maeda H,Fang J.,Inutsuka T,Kitamoto Y.「Vascular permeability enhancement in solid tumor:various factors,mechanisms involved and its implications.」Int Immunopharmacol、2003年、第3巻、p319−28
【非特許文献23】Satchi−Fainaro R,Puder M,Davies JW,Tran HT,Sampson DA,Greene AK,Corfas G,Folkman J.「Targeting angiogenesis with a conjugate of HPMA copolymer and TNP−470.」Nat Med、2004年、第10巻、p255−261
【非特許文献24】Fischer H,Erdmann S,Holler E.「An unsual polyanion from Physarum polycephalum that inhibits homologous DNA polymerase α in vitro.」Biochemistry、1989年、第28巻、p5219−5226
【非特許文献25】Lee BS,Holler E.「Effects of culture conditions on β−poly(I−malate)production by Physarum polycephalum.」Appl Microbiol Biotechnol、1999年、第51巻、p647−652
【非特許文献26】Kopecek,J.,Kopeckova,P.,Minko,T.,Lu,Z.「HPMA copolymer−anticancer drug conjugates:desgn,activity,and mechanism of action」、Eur.J.Biopharm.2000年、第50巻、p61−81
【非特許文献27】Vincenzi,V.,Ferruti,P.,Ford,J.,Duncan,R.「Synthesis and preliminary evaluation of novel functionalized poly(ethylene glycol)−block−poly(ester−carbonate)copolymers as biodegradable carriers.」Macromolecular Bioscience、2001年、第1巻、p164−169
【非特許文献28】「Environmental health criteria 191. Acrylic acid.」[online]United Nations Environmental Programme/International Labour Organisation/World Health Organisation.International Programme on Chemical Safety.インターネット<URL:http://www.inchem.org/documents/ehe/ehc/ehc191.htm>
【非特許文献29】Li,C.「Poly(L−glutamic acid)−anticancer drug conjugates.」 Advanced Drug Delivery、2002年、第54巻、p695−713
【非特許文献30】Gill,T.J.,Kunz,H.W. Papermarker,D.S.1967.「Studies on synthetic polypeptide analogues.J.Biol.Chem.1967年、第242号、p3306−3318
【非特許文献31】Chiang,C.H−Yeh,M,K.「Contribution of poly(amino acids)to advances in pharmaceutical biotechmologyy.」、Current Pharmaceut.Biotechnol.2003年、第4巻、p8−16
【発明の概要】
【発明が解決しようとする課題】
【0009】
(発明の要旨)
ポリリンゴ酸を使用することによる以前に悩まされた薬物キャリアシステムの問題を解決することが本発明の目的である。そのポリリンゴ酸は多くの機能性カルボキシル基を担持し、そのような基は、質量数50,000のポリリンゴ酸1分子当たり、少なくとも約50であり、そして約500を担持する。
【課題を解決するための手段】
【0010】
ポリリンゴ酸は分子量2,500から少なくとも100,000のサイズの範囲で容易に入手できる。これは、以下のことを達成するために多数の生物学的に活性な機能的分子の連結を可能にする:
薬物キャリア分子あたりの組織標的化分子の種類および数における高い調整可能性;
薬物キャリア分子あたりの結合薬物モジュール(プロドラッグ)の種類および数における高い調整可能性;
上記機能的モジュールに加えて疎水性残基または親水性残基のいずれかを担持する基を有することにより溶解度を調節する能力;
上述の機能的モジュール以外に、例えば,PEGのような分解に対してキャリアシステムを保護する(例えば、血液循環系において寿命を増加する)ことに活性である、さらなる基を結合する能力;
上記薬物送達システムの構築単位として使用される多くの他の残基と共に、生分解性であるキャリア骨格;
ウイルス成分を使用しないシステム;
機能的モジュール(mABまたは他の腫瘍細胞表面レセプター)、上記種類の薬物(悪性に発現した遺伝子に対するアンチセンスオリゴヌクレオチド)、および腫瘍組織に対して高い特異性(EPR−効果)を担う全体的に高分子量の薬物送達システムを有する送達システム;ならびに
ラットの体重1kg当たり5mg程度の高い用量を可能にする低毒性の薬物システム。
【0011】
本発明のさらなる目的は:
複雑な合成方法および制御不能な副反応を避けることにより薬物送達システムの合成を容易にすること;
容易な精製を提供する好ましい溶解度および他の性質で作製すること;
各個別の化学結合反応の規定された化学量論および再現性を可能にする最大収率を達成すること;
結合された機能的モジュールの種類と数に関する高い調整可能性の構造的および合成の基礎を用意すること;ならびに
薬物送達システムの簡単なスケールアップを可能にする技術を提供することである。
【0012】
技術発明は以下を包含する:
非常に数多くの反応性カルボキシル基(分子量50,000の骨格に対して約500)を担持する主鎖または骨格としてポリリンゴ酸を用いる薬物送達システムの選択;
それらのNHS−エステルを形成することによる、これらのカルボキシル基の殆ど(理想的には全部)の活性化;
NHS−活性化カルボキシル基での置換の求核剤として単一のアミノ基を担持する機能的モジュールの化学的に独立した調製;
上記活性化された骨格および反応性機能的モジュールの各々を別々に調製すること;
上記NHS−活性化骨格および各反応性機能的モジュールを、独立しているがよく規定された順に組み合わせて反応させ、新しく添加した機能的モジュールのそれぞれの添加後に所望の中間体/生成物の精製および実証を可能にすること;
これらの反応を化学量論量で組合わせること;
結合体の高くかつ再現性のよい収率を達成すること;
新しく添加した反応性機能的モジュールと既に結合されたモジュールとの副反応を、次に入って来る機能的モジュールの結合に対して順番に添加する良く組織化された階層を選択することにより避けること。
【0013】
キャリアまたは骨格のデザインおよび合成について説明するが、それは共有結合的に結合した薬物を標的化組織に輸送し、その組織の細胞表面レセプターに結合し、エンドソームに内在化し、エンドソームから細胞質へ抜け出し、そして細胞質において細胞質内容物のグルタチオンおよび他のスルフヒドリル基との化学反応により活性遊離薬物を放出する。高分子量薬物ビヒクルまたは粒子の特異性は、腫瘍特異的結合体標的化分子による腫瘍組織標的化ならびに単にそれらが高分子量(>20000)であることに起因する腫瘍における高い透過性および保持性(EPR−効果)の両方を基礎にしている(40,41)。
【0014】
本特許出願で使用される骨格ポリ(リンゴ酸)(PMLAH)は、主鎖エステル結合を含み、生分解性であり(27およびその中の参考文献)そして高分子可撓性(49)であり、水(イオン化する場合)および有機溶媒(その酸性形態において)に溶解性であり、非毒性でありならびに非免疫原性(27およびその中の参考文献)である。薬物担持PMLAHは、誘導化されたリンゴ酸ラクトンの開環重合により主に合成されてきている(27およびその中の参考文献)。ドキソルビシン−ポリ(リンゴ酸)の合成は、化学的に合成されたポリ(β−D、L−リンゴ酸)から報告されている(49)。天然に存在するPMLAHからの薬物ビヒクルの合成は、行われなかった。本特許出願に記載する高度に機能的な薬物送達システムの種類は、以前に開示されたことは無かった。
【0015】
上記キャリアは、分子主鎖または分子骨格に対応するポリ(β−L−リンゴ酸)(PMLA)から構成され、それはそのカルボキシル基のところで、規定された比率で、以下の仕事を遂行する種々の機能的モジュールと化学的に結合される:(1)細胞質で有効になる放出可能な機能的モジュールによるプロドラッグの放出、(2)細胞(例えば、モノクローナル抗体(mAB))の表面に結合することにより特定の組織の方へキャリアを向けること、(3)エンドソームを通っての標的細胞への内在化(通常標的表面レセプターの内在化による)、(4)エンドソーム膜へ集積し、最終的にはエンドソーム膜を崩壊させる疎水性機能的単位の力でエンドソームから細胞質へ脱出し、リソソームへの過程でのエンドソームの酸性化の間に有効になること、(5)分解性酵素活性(例えばペプチダーゼ、プロテアーゼなど)に対してポリエチレングリコール(PEG)による保護。
【0016】
本発明において「モジュール」とは、小さい薬物分子または発色団分子から、例えば、抗体またはレシチンのような完全なタンパク質分子までの範囲にわたる生物学的活性分子構造である。本明細書に示される実施例の場合に、(1)ラミニン−8(34、51)のα−4鎖およびβ−1鎖に対するモルホリノアンチセンスオリゴヌクレオチドが示されており、それはそれらの3’−末端に人工的に導入された−NH2(アミノ)基によるアミド結合により介在スペーサーに結合されている。そのスペーサーは、細胞質の還元環境中でグルタチオンとのスルフヒドリル−ジスルフィド交換反応において切断可能なジスルフィド部分により、キャリアと連結されている(51、52)。(2)組織標的化は、モノクローナル抗体(mAB)を用いてラットトランスフェリンレセプターを認識し、そして結合するよう設計される。このレセプターは、血液脳関門(BBB)として機能する内皮細胞表面上で発現し、および特定の腫瘍では高いレベルで発現することが分かった(53、54)。インビトロおよびインビボの研究は、トランスフェリンレセプターがトランスフェリンまたはmAB OX−26または他の適切なmABと化学的に結合した薬物送達システムのための固定具として使用され得ることを示し、その抗体はトランスフェリンレセプターと結合し、そしてそれにより抗体のアロタイプに依存してラットまたはマウスまたは他の哺乳動物の血液脳関門(BBB)を通ってトランスサイトーシスを達成することを示す(5、56、57;45、58、59、60、および61)。(3)トランスフェリンレセプターと結合する抗体およびエンドソームへの内在化は、実証されている(57、55、および57)。トランスフェリンレセプターの場合、任意の適切な抗体、mAB、ヒト化抗体またはキメラ抗体またはレクチンまたはトランスフェリンレセプターに特異的な他のリガンドが使用され得ることは理解される。多数の細胞表面レセプターまたは抗原に対する適切なリガンドは、本発明に使用され得、そしてトランスフェリンレセプターは単なる例であることもまた、理解される。(4)エンドソームからの脱出は、ポリアクリル酸誘導体の場合、エンドソーム小胞からリソソームへと成熟化の間の酸性化により機能することが示された(51、62)。本特許出願で提案されている設計されたキャリアは、アミド結合によりポリリンゴ酸骨格と結合されている多数のバリン残基を担持する。リソソームへの過程の間の上記エンドソームの酸性化の間、上記キャリア分子のこれらの伸縮は、電荷が中和されそして疎水性になり、膜を崩壊させることができる。リソソームpHで電荷が中和される限り、他の分子が、バリンに代わって使用され得る。(5)PEG化は結合タンパク質の半減期を著しく増加させる(63)。循環時間を延長し、そして標的固体腫瘍の中への滲出を増進する(64)。半減期を増加させることが知られている任意の他の分子は、本発明に使用される。
例えば、本発明は以下を提供する。
(項目1)
薬物送達分子であって:
複数の遊離カルボン酸基を有するポリマー化したカルボン酸分子骨格;
複数の生物学的に活性な分子モジュールであり、それぞれ同じポリマー化したカルボン酸分子骨格に共有結合している、活性分子モジュールを含み、ここで該活性モジュールは:
標的化細胞により細胞性取り込みを促進する、少なくとも1つの標的化モジュール;および
標的化細胞の細胞性代謝を変える、少なくとも1つのプロドラッグモジュールを含む、薬物送達分子。
(項目2)
前記プロドラッグが、腫瘍特異的タンパク質の発現を阻害するように選択される、項目1に記載の薬物送達分子。
(項目3)
前記ポリマー化カルボン酸分子骨格がポリ(β−L−リンゴ酸)である、項目1に記載の薬物送達分子。
(項目4)
前記ポリ(β−L−リンゴ酸)が、2,500と100,000との間の分子量を有する、項目3に記載の薬物送達分子。
(項目5)
前記ポリ(β−L−リンゴ酸)が、少なくとも約5,000の分子量を有する、項目4に記載の薬物送達分子。
(項目6)
前記ポリマー化カルボン酸分子骨格のそれぞれの分子が、少なくとも約50の遊離カルボン酸基を有する、項目1に記載の薬物送達分子。
(項目7)
前記複数の分子モジュールが、生物膜の崩壊を促進する分子モジュールをさらに含む、項目1に記載の薬物送達分子。
(項目8)
項目7に記載の薬物送達分子であって、生物膜の崩壊を促進する前記分子モジュールが、生理的pHでは荷電し、リソソームのpHで荷電せず、それにより該分子モジュールの親油性を増加する親油性特性および親油性基を有する分子を含む、薬物送達分子。
(項目9)
前記複数の活性分子モジュールが、前記薬物送達分子の循環を延長する分子モジュールをさらに含む、項目1に記載の薬物送達分子。
(項目10)
項目9に記載の薬物送達分子であって、該薬物送達分子の循環を延長する前記分子モジュールが、ポリエチレングリコールを含む、薬物送達分子。
(項目11)
項目1に記載の薬物送達分子であって、前記複数の活性分子モジュールが、該薬物送達分子の細胞性取り込みを決定するレポータモジュールをさらに含む、薬物送達分子。
(項目12)
前記レポータモジュールが蛍光分子を含む、項目11に記載の薬物送達分子。
(項目13)
前記標的化分子が、血液脳関門の透過を促進するように選択される、項目1に記載の薬物送達分子。
(項目14)
前記標的化分子モジュールが抗体を含む、項目1に記載の薬物送達分子。
(項目15)
前記抗体が、トランスフェリンレセプターに結合する、項目14に記載の薬物送達分子。
(項目16)
前記抗体が、モノクローナル抗体である、項目14に記載の薬物送達分子。
(項目17)
前記抗体がヒト化抗体またはキメラ抗体である、項目14に記載の薬物送達分子。
(項目18)
項目1に記載の薬物送達分子であって、ここで前記プロドラッグ分子モジュールが、該薬物送達分子が細胞に入るときに切断される切断可能な結合で、前記ポリマー化カルボン酸分子骨格と結合される、薬物送達分子。
(項目19)
前記切断可能な結合が、ジスルフィド結合である、項目18に記載の薬物送達分子。
(項目20)
前記プロドラッグ分子モジュールが、アンチセンス分子を含む、項目1に記載の薬物送達分子。
(項目21)
前記アンチセンス分子がモルホリノアンチセンス分子である、項目20に記載の薬物送達分子。
(項目22)
前記アンチセンス分子が、ラミニン−8の産生を干渉する、項目20に記載の薬物送達分子。
(項目23)
項目22に記載の薬物送達分子であって、ここで前記アンチセンス分子が、α4ラミニンおよびβ1ラミニンからなる群より選択されるラミニンサブユニットの産生を変化させることによりラミニン−8の産生を干渉する、薬物送達分子。
(項目24)
薬物送達分子の合成方法であって、以下の工程:
複数の遊離カルボン酸基を有するポリマー化カルボン酸分子骨格を提供する工程;
該カルボキシル基を活性化する工程;
該活性化カルボキシル基をスルフヒドリル基およびアミノ基を含む化合物と反応させて、薬物送達分子にスルフヒドリル基を付けてスルフヒドリル薬物送達分子にする工程;
スルフヒドリル結合基を含む標的化分子を、該スルフヒドリル薬物送達分子と反応させて標的細胞による取り込みを促進する工程;ならびに
プロドラッグ分子を該標的細胞の細胞性代謝を変化させるために反応する工程を包含する、合成方法。
(項目25)
前記プロドラッグ分子が、スルフヒドリル結合基を含むアンチセンス分子である、項目24に記載の薬物送達分子の合成方法。
(項目26)
項目24に記載の薬物送達分子の合成方法であって、前記活性化カルボキシル基を、親油性部分を有し、エンドドームの酸性化の間に荷電しなくなる電荷基を含む分子と反応させ、それにより膜の崩壊を引き起こす工程をさらに含む、合成方法。
(項目27)
項目24に記載の薬物送達分子の合成方法であって、ここで複数の異なるプロドラッグ分子が、同じ薬物送達分子に結合し、それにより1を超えるプロドラッグ分子で、前記標的細胞を同時に処理することが可能になる、合成方法。
(項目28)
前記標的化分子が前記血液脳関門の透過を促進するよう選択される、項目24に記載の薬物送達分子の合成方法。
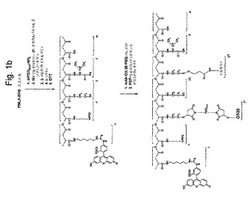
【図面の簡単な説明】
【0017】
【図1a】図1aは、本発明の薬物分子の全体的な構造を示す。
【図1b】図1bは、図1aの構造を構築するために使用される工程の全体的な順序を示す。
【図2】図2は、PMLA−NHSエステルの合成を示す略図である。
【図3】図3は、(PDP−モルホリノ)アンチセンスオリゴヌクレオチドの合成を示す略図である。
【図4】図4は、N−(フルオレセイン−5’−チオカルバモイル)ジアミノヘキサンの合成を示す略図である。
【図5】図5は、N,N’−ビス−(3−マレイミドプロピオニル)ポリ(エチレングリコール)の合成を示す略図である;
【図6】図6は、PMLA/L−バリン/2−メルカプトエチルアミン/mPEG5000−NH2結合体の合成を示す略図である。
【図7】図7は、PMLA/mAB OX−26/モルホリノアンチセンスオリゴヌクレオチド/L−バリン/2−メルカプトエチルアミン/mPEG5000−NH2結合体の合成を示す略図である。
【図8】図8は、FITCの、PMLA/mAB OX−26/モルホリノアンチセンスオリゴヌクレオチド/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体への結合を示す略図である。
【図9】図9は、ポリマーを試験する時に使用される溶血アッセイの結果の棒グラフを示す。
【図10a】図10aは、グルタチオンでの還元によるキャリアからのモルホリノオリゴの放出を示す。
【図10b】図10bは、グルタチオンでの還元に応答する時間の間のオリゴ放出の%を示す。
【図11】図11は、薬物2Aおよび/またはPBS(模倣)と比較して、有意差p<0.01でlog rank testにより分析された、薬物2による処置後のラットのKaplan−Meir生存曲線を示す;
【図12】図12は、培養神経膠腫U−87MG細胞における薬物2(30分)とマーカーエンドソームFM4−64との共分布である。共焦点顕微鏡により、FM4−64が細胞質エンドソーム(左上)に見られ、そして薬物2が同じ場所(右上)に見出される。両方の標識が共局在化する(左下、黄色)。
【図13】図13は、血管密度が正常脳と比較して腫瘍において増加し、そして薬物2が腫瘍血管密度を55%減少させることを示すグラフである。
【図14】図14は、U87/MG神経膠腫培養物のラミニン鎖に関する免疫蛍光染色を示す。PMLAビヒクル結合アンチセンスオリゴ(薬物2)は、ラミニン発現を抑制する(α4は赤色であり、β1は緑色である)。核は、DAPI(青色)で対比染色される。
【図15】図15は、ヒトラミニンβ1鎖に対するモノクロナル抗体を使用して異種移植された腫瘍の免疫蛍光分析を示す;ラミニンβ1鎖合成が、薬物2の投与後、GBMにおいて阻害された。
【図16】図16は、2つの多型膠芽腫(GBM)細胞培養物、U87MG、およびT98Gにおけるラミニン−8鎖発現の阻害のウェスタンブロッティング分析を示す:−、未処置、+、薬物2で処置。
【図17】図17は、薬物2が、上記BBBを通過することができることを示し、ここで赤色は、薬物の血管内投与の後の脳血管および移植腫瘍内で視覚化された薬物を示す。
【発明を実施するための形態】
【0018】
(発明の詳細な説明)
以下の説明は、当業者が発明物を作製し、そして利用することが可能なように提供され、発明を実施する発明者により企図された最良の様式を示す。しかしながら、種々の改変が、当業者には容易に明らかである。何故なら本発明の一般的な原理が、ポリ−L−リンゴ酸を基礎としたアンチセンス抗腫瘍薬物により実証された新規な薬物送達システムを提供するように本明細書に具体的に規定されているからである。
【0019】
薬剤および生物製剤のためのキャリアマトリックスまたは分子輸送ビヒクルとしてのPMLAの魅力的な性質は、以下の通りである:それは、非毒性でかつ非免疫原である;その疎水性は、疎水性側鎖またはスペーサーを導入することにより、調節され得る(30);それは、生分解性である(31);そしてそれは、血流で安定である。アンチセンスオリゴを特定の器官またはコンパートメントに標的化するために、例えば、レセプター媒介性エンドサイトーシスに有利な腫瘍特異的抗体のような標的化実体は、PMLAポリマーに結合され得る。理想的には、上記系は、分子輸送ビヒクルからその薬物を放出するための放出システムを含み;あり得る放出システムとしては:a)細胞内グルタチオンにより切断可能なジスルフィド結合、b)pH感受性ヒドラゾン結合、c)リソソームカテプシンB(または他のペプチダーゼ)(その活性は、種々の腫瘍において増加する)により切断されるテトラペプチド、;d)エンドソームからの本来の放出機能(32,33);およびe)エステル結合のような他の不安定な、または切断可能な結合、が挙げられる。最も重要なことに、多数の分子標的インヒビターが、1つのPMLA分子に容易に連結され得る。
【0020】
天然物、Physarum polycephalumの変形体(27およびその中の参考文献)由来のPMLAは、本特許出願に記載の薬物送達システムの合成のための出発物質であった。ここで用いられる化学合成の方法は、合成化学における一般的な公知の(fundus)方法からであり、ここで記載されているポリリンゴ酸ベースのシステムとは関係なく、他の系において記載されている。これらの方法の殆どは、本状況に適合されなければならず、特にキャリア系の化学的構築の進行の間の、出発物質の性質に適合されなければならず、そして生成物の精製方法に関しても適合されなければならなかった。上記ポリリンゴ酸骨格に結合される機能的部分の予測可能でかつ再現性のある化学量論的に誘導体化を成功裏に達成するために、上記骨格との反応の順序を、制御されていない反応は不可能であるような方法で確立および構築しなければならなかった。生成物の有効性およびその純度が達成され、段階的な合成の間に、定性化学アッセイおよび定量化学アッセイ、高速液体クロマトグラフィー(HPLC)、薄層クロマトグラフィー(TLC)、および紫外光/可視光/IR光スペクトル分析ならびにNMRスペクトル方法を含むインシチュ分析で制御された。完全に構築された薬物ビヒクルの膜崩壊的性質およびその合成の中間体の膜崩壊的性質もまた、慣用的で実験的な溶血膜アッセイ(51を参照のこと)により評価された。
【0021】
ここで使用された方法の開発を基礎とした標準的な合成方法参考は、特に以下である:
(69)の方法に類似する方法での、PMLAH−カルボキシル基のN−ヒドロキシスクシンイミド(NHS)エステルとしての活性化;
(51およびその中の参考文献)に記載の方法と類似の方法での、(2−ピリジルジチオ)プロピオニル基の、モルホリノ(PDP)−モルホリノ)アンチセンスオリゴヌクレオチドへの結合;
(71)のN−(フルオロセイン−5’−チオカルバミル)ジアミノヘキサンの形成の方法と類似の方法での、FITC(フルオロセインイソチオシアナート)−結合;
(70)に記載の方法と類似の方法での、N,N’−ビス−(3−マレイミドプロピオニル)ポリ(エチレングリコ−ル)の合成;
(72、51およびその中の参考文献)に記載の技術と類似の技術での、チオール基の抗体への導入;
(52および69)で実施された反応に類似して、mAB OX−26−スルフヒドリルとN,N’−ビス−(3−マレイミドプロピオニル)−PEGジアミド結合体との反応が行われた;
本明細書に記載されるようにNHS活性化されたカルボキシレートからのアミド形成により、PMLA/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体の合成が原理的に行われた;
スルフヒドリル基とマレイミドとの反応によるPMLA/mAB OX−26/モルホリノアンチセンスオリゴヌクレオチド/L−バリン/2−メルカプトエチルアミン/mPEG5000−NH2結合体の合成;
本明細書に記載されるようにNHS活性化されたカルボキシレートの求核攻撃により主にアミド形成を表す、FITC−スペーサーの、PMLA/mAB OX−26/モルホリノアンチセンスオリゴヌクレオチド/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体への結合。
【0022】
既存の薬物送達システムは以下の1つから数個の問題に悩まされる:
既存の薬物送達システムは多機能的ではなく、即ち、キャリア1分子あたりの組織標的化基の種類および量における可変性に関して限定される;
既存の薬物送達システムは、キャリア1分子あたりの結合薬物(プロドラッグ)の種類と量により限定される。
【0023】
既存の薬物送達システムは、生理学的流体中への溶解度により限定される。
【0024】
既存の薬物送達システムは、循環系における分解に対する不十分な安定性により限定される。
【0025】
既存の薬物送達システムは、生分解性ではない;
既存の薬物送達システムは、ウイルス核酸または他のウイルスフラグメントを含む;
既存の薬物送達システムは、腫瘍組織に特異的ではなく健全な宿主組織を損傷する;
既存の薬物送達システムは、毒性がある;
薬物送達システムの合成は、制御不可能な副反応に悩まされる;
薬物送達システムの合成は、反応生成物の精製を困難もしくは不可能にする溶解度または他の問題に悩まされる;
薬物送達システムの合成は、再現性のある結果が得られない;
新成分を含有し、従って薬物送達システムの特異性を増進し、薬物送達システムの抗腫瘍活性を強くする、構造的変更物/拡大物の合成は不可能である;
薬物送達システムの合成はスケールアップが容易にできない。
【0026】
各反応性機能的モジュールの、ポリリンゴ酸骨格のNHS(N−ヒドロキシスクシンイミド)−活性化カルボキシル基との制御された結合は、種々の異なる種類の反応性機能的モジュールを結合させることを可能にし、従って種々の異なる標的化分子、薬物(プロドラッグ)分子などを導入できる。種々のモジュールは、全く同一の骨格分子に結合され得るか、または2成分もしくは3成分などの薬物混成体を作製可能なように、または異なる骨格分子へ結合され得る。全く同一の骨格分子上の多機能性モジュールは、細胞に同時に導入される場合生物学的に相乗効果を示し得る。
【0027】
生分解性は、骨格および他の生分解性構築単位(アミノ酸、タンパク質)として生分解性リンゴ酸を用いることで達成され得る。
【実施例】
【0028】
(特定の実施例による本発明の説明)
(モルホリノアンチセンスオリゴヌクレオチドの腫瘍標的送達のためのポリリンゴ酸ベースの多機能性キャリアシステムの合成)
図1aは、本発明の代表的な薬物分子の全体的な構造を示す。ブロック(z+w)の合成のために、ポリリンゴ酸のカルボキシル基(ブロックw)を、NHS−エステルとして活性化し、そしてアミド結合を介してL−バリンに結合する。ブロックy3は、ジスルフィド薬物放出単位を含むプロドラッグモルホリノアンチセンスオリゴである。ブロックy1は、ポリエチレングリコール(PEG)スペーサーを介してブロックz+wに結合しているモノクローナル抗体(OX26)標的化分子である。ブロックxは、分解に対するPEG−保護基であって、アミド結合により連結されており、市販されているPEG−アミンから形成される。ブロックy2は、残っているスルフヒドリル固定基を表し、この時点でそれは合成に消費されず、置換N−エチルマレイミドの2重結合との反応により更なる機能性モジュールの結合に使用され得るか、または単に置換N−エチルマレイミドとの反応でブロックされ得る。ブロックn(蛍光レポータ基)をフルオロセインイソチオシアネート(FITC)およびN1−Boc−1,6−ジアミノヘキサンから調製する。上記薬物構造を、注意深い化学量論的制御下で、図1bに示されるように成長中の結合体への、上記ブロック(すなわち、モジュール)の段階的結合により構築する。工程の順序は、異なるシナリオに合うように容易に調整され得る。カルボジイミド試薬を用いて有機溶媒、好ましくはジメチルホルムアミド中で、結合反応を実施した。以下の標準的な方法で側鎖の適切な保護により副反能を抑制する。
【0029】
ブロックwについては、非常に精製度の高いポリリンゴ酸を、Physarum polycephalumの培養物から得る(24、25)。バイオポリマーを同時にそして酵素的にL−リンゴ酸((31、26)に分解し、それは炭酸ガスおよび水へと代謝され。そのナトリウム塩はマウスおよびウサギのそれぞれで毒性も無く、免疫原性もない(27およびその中の参考文献)。マウスに静脈内投与した後、ポリリンゴ酸は、腎臓経由の排泄により速く除かれた(73)。特定のポリリンゴ酸誘導体およびブロックポリマーは、実際にラットにおいて骨修復および筋再生に対して正の効果をを示した(30)。または特定のポリリンゴ酸誘導体およびブロックポリマーは、他の研究(32)において生体適合性であることがわかった。ポリリンゴ酸は、非常に多くの修飾可能なカルボキシル基の故に薬物キャリアデバイスの設計には優れた候補である。これらは、それらの化学量論および集積度に関して完全に制御可能な様式で種々の異なる生物学的に活性な分子に、容易に結合され得る。ブロックzは、L−バリンまたはL−ロイシンのような親油性基を含むポリマーを基礎にし、そしてエンドソームのリソソームへの成熟の間に周囲のpHが6以下に低下する場合、プロトン化され益々親油性になる。この増加する親油性で上記エンドソームの膜は漏れやすくなり、細胞質への高分子内容物の放出を生じる(33、74、75)。ブロックy3は、ジスルフィド結合を含有し、それは脳毛細血管を含む血液循環においては安定であるが、細胞の還元的環境においては切断される(38)。ブロックy1は、上記組織標的部分が標的細胞表面のレセプターに結合する事を可能にするポリエチレングリコールスペーサーを含む。それはまた、標的化ポリペプチドの分解に対しても保護する。(y3)例えばラミニンのα1鎖のような腫瘍必須遺伝子の発現を特異的にブロックするモルホリノオリゴヌクレオチドが、使用される。原則的に、任意の他の薬物またはプロドラッグがここでおよび単一キャリア分子上の異なる薬物のアレイで結合され得る。これらの結合体は、細胞質内で薬物放出単位でキャリアから切断されそして薬物(単数または複数)が有効になる。ブロックy1は、上記BBBの内皮細胞上のトランスフェリンレセプターに対するモノクローナル抗体により標的化されるBBBを突破し易くする(55)。ブラジキニン単独または他の分子と結合されたブラジキニンは、特異的レセプターに基づいて脳腫瘍に使用されるべき標的化分子になり得る(77、18および78)。脳腫瘍特異的標的化に関する更なる可能性は、ヒトEGFレセプターに対するモノクローナル抗体を使用することである(37,79)。ブラジキニンB2レセプターおよびEGFRは、腫瘍細胞上で過剰発現し、トランスフェリンレセプターとの組合わせで、脳腫瘍標的化部位として使用され得る。ブロックnは、任意の蛍光標識(ここではフルオロセイン)を付加し、受容体の腫瘍細胞のエンドソームにおいて上記キャリアのホーミング研究を容易にする薬物構造に結合する。
【0030】
(材料および方法)
ポリ(β−L−リンゴ酸)(PMLA)を、(25)から開発された方法を使用して培養Physarum polycephalum変形体のブロスから精製した。塩の形態の上記ポリマーをSephadex G25カラムでサイズ分画をした。数平均分子量50kDa(多分散度1.2)を有する画分をAmberlite IR−120(H+形態)を通して遊離酸ポリマー(PMLA−H)に変換しキャリア合成に使用するまで凍結乾燥で保存した。D2O中の1H−NMRは以下のδ値:3.3ppm(d、ポリエステル主鎖のメチレンのプロトン)、5.3ppm(t、ポリエステル主鎖のメチンのプロトン)、を示した。プロトン−ブロード−バンド−デカップル(Proton−broad−band−decoupled)13C−NMRは、以下のδ値:178.4ppm(−COOH)、74.5ppm(−CHOH−)、38.9ppm(−CH2−)および174.5ppm(−CO−)を示した。精製PMLA−Hは、220nm以下のみの波長にUV光吸収を示し、核酸およびタンパク質のそれぞれの代表的な260および280での吸収はない(更なる詳細は27に概説されている)。ラミニン−8のα−4鎖(AGC−TCA−AAG−GCA−TTT−CTC−CGC−TGA−C)およびラミニン−8のβ−1鎖(CTA−GCA−ACT−GGA−GAA−GCC−CCA−TGC−C)(50、34)に対するモルホリノTM−3’−NH2アンチセンスオリゴヌクレオチド((6)をGene Tools(USA)より購入した。10mMアジ化ナトリウムを含有する1mg/ml PBSの濃度におけるラットトランスフェリンレセプターCD71に対するマウスモノクローナル抗体(クローンOX−26、アイソタイプIgG2a)をChemicon Europe(UK)から得た。マウスIgG2a,κ(UPC10)をSigma(ドイツ)から購入した。クロマトグラフィーで純粋なmPEG−アミン(分子量 5000)およびアミン−PEG−アミン(分子量 3400)をNektar Therapeutics(USA)から得た。Merck(ドイツ)、Sigma(ドイツ)Pierce(USA)から得た試薬および溶媒は、入手可能な最高純度のものであった。ジクロロメタン(DCM)およびN,N−ジメチルホルムアミド(DMF)を、モレキュラーシーブ(0.4nm)で乾燥した。
【0031】
1H−NMRスペクトルをBruker Model DMX−500フーリエ変換スペクトル計に記録し、化学シフトを、TMSを内部標準として相対的にppm(δ)で示す。13C NMRスペクトルを125.8MHzで操作する同じスペクトル計で記録した。クロマトグラフ的な分離を、UVおよび蛍光の検出器を備え付けたMerck−Hitachi分析用LaChrom D−7000 HPLC系で実施した。1.5ml/分の流速で0.1%TFA(トリフルオロ酢酸)水溶液−0.07%TFAアセトニトリル溶液の2勾配のMacherey & Nagel C18−Nucleosil逆相(RP)カラム(250×4mm)、または流速0.75ml/分で50mMのリン酸ナトリウム緩衝液pH7.4を用いるサイズ排除カラムBio−Sil SEC250−5(5μm、300×7.8mm)のいずれかを使用した。ポリマーのNa塩またはK塩の分子量を、規定された分子量の標準ポリスチレンスルホン酸塩(Machery−Nagel)を用いてSEC−HPLCで決定した。薄層クロマトグラフィー(TLC)をMerckの予めコートされているシリカゲル60 F254アルミニウムシート上で実施した。溶出液は、n−ブタノール、水および酢酸(容量比ベースで4:2:1)を含有した。
【0032】
(合成)
(PMLA−NHSエステルの合成)
1.16gのPMLA−H(リンゴ酸モノマーに関して10mmol)を、30mlの無水ジメチルホルムアミド(DMF)に溶解した。N−ヒドロキシスクシンイミド(NHS)(15mmol)を無水ジメチルホルムアミド(DMF)(10m)に溶解し、これを、PMLA−H溶液に添加した。温度を氷浴で0℃まで下げて、その後10mlのDMFに溶解したジシクロヘキシルカルボジイミド(DCC)(15mmol)を添加した。その反応混合物を、気体の泡が発生しなくなるまで減圧下、室温で保持した。0℃で30分後に、その反応混合物を室温で48時間攪拌した。上記反応混合物を、上述のように減圧下で保持し、反応の最初の日の間は2時間ごとに、その後、反応の2日目の間は6時間ごとにインキュベーションをした。反応の2日後、ジシクロヘキシル尿素をろ過で除去し、そしてその反応容量を減圧下エバポレーションで減少させた。新鮮な無水DMF(10ml)を添加し、残りのジシクロヘキシル尿素を再び濾過により除去した。透明な反応混合物を、室温で12時間攪拌し、ジシクロヘキシル尿素の残量(もしあれば)を、ろ過で除去した。その容量を1〜3mlにまで、減圧下エバポレーションにより減少させ、そしてその生成物を酢酸エチルを加えて沈殿させた。その薄黄色の生成物(P1)を、ろ過で収集した。ジエチルエーテルを、最終比率1:1(酢酸エチル:ジエチルエーテル)を合わせるように、ろ液に加え、そしてさらなる明るい褐色の生成物(P2)を、ろ過で収集した。次いで、n−ヘキサンを、最終比率が1:1:1(酢酸エチル:ジエチルエーテル:n−ヘキサン)になるようにろ液に加えて、さらなる褐色の生成物(P3)をろ過で収集した。上記沈殿物を、それらの沈殿に使用したのと同じ溶媒に分散させ、低温(−20℃)で一晩放置した。その生成物を濾過して、同じ冷溶媒で繰り返し洗浄した。その生成物を、DMFを溶出液として重力で流してSephadex LH20を通すことによりさらに精製した。生成物含有画分を、収集しそして溶媒を減圧下、エバポレートさせた。最後にその生成物をジエチルエーテルに分散し、ろ過により収集し、真空で乾燥し、−20℃で保存した。
【0033】
PMLA−NHSエステルのこれらの調製物の純度/組成を、1H−NMRおよびUV−VIS分光法ににより分析した。NHS基の含量を、NHSエステル基のn−ブチルアミンでのアミノリシス後に決定した。10mgのPMLA−NHSエステルを、0.5mlのDMFに溶解した。0.5mlの10% n−ブチルアミンの1部分をこの溶液に加え、そしてその反応混合物を、室温で30分間インキュベートした。遠心分離後、20μLのサンプルを、80μLの水と混合し、水/0.1%(v/v)TFAを溶出液として用いてPR−HPLCにより分析した。NHS基を、260nmの吸光度でモニターした。それらの含量は、既知量のN−ヒドロキシスクシンイミド標準物質の吸光度と比較して計算した。PMLA−NHSエステルサンプル中のリンゴ酸残基とNHS基とのモル比を、これらの結果と、1H−NMRで測定したマリル残基の量と合わせて計算した。代表的には、その比はP1、P2、およびP3に対してそれぞれ35%、59%および85%であった。(CD3)2SO中での1H−NMRは、以下のδ値:2.8ppm(s、4H、N−CO−CH2−)、3.35ppm(d、上記ポリエステル主鎖のメチレンのプロトン)、5.85ppm(t、ポリエステル主鎖のメチンプロトン)を示した。上記反応は、図2に示される。
【0034】
((2−ピリジルジチオ)プロピオニル−モルホリノ(PDP−モルホリノ)アンチセンスオリゴヌクレオチドの合成)
モルホリノ−3(−NH2アンチセンスオリゴマー(1μmol)を、900μLのDMFおよび100μLの脱イオン水の混合物の中に溶解した。この混合物の中に、20μLの100mMのN−スクシンイミジル3−(2−ピリジルジチオ)プロピオネート(SPDP)のDMF溶液を加え室温で2時間放置した。その溶媒を、減圧下、室温でロータリーエバポレーターで除去した。その残渣を、10mM EDTAを含有する1mlの緩衝液A(0.1Mリン酸ナトリウム、0.15M NaCl、pH7.2)に溶解し、緩衝液Aで予め平衡化したSephadex G−25マイクロスピンカラムで精製した。PDP−モルホリノアンチセンスオリゴヌクレオチドの濃度を、1mMに調整して−20℃で保存した。
【0035】
その生成物の純度を、TLCおよびUV分光法によりNHSおよびSPDPの不存在を示して確認した。PDP基の含量を、以下のようなジスルフィド還元後の2−チオピリドンの濃度を測定して決定した:PDP−モルホリノアンチセンスオリゴヌクレオチドを、0.1M トリス緩衝液pH9.0中の0.2Mのジチオトレイトール(DTT)と室温で30分間インキュベートした。その反応混合物を、最初10分間蒸留水で洗浄し、次いで30分間、0〜60%のアセトニトリルの勾配で溶出することによりRP−HPLCにかけた。その反応生成物2−チオピリドンを、341nmのUV吸光度で検出した。2−チオピリドンの濃度を、還元された2−アルドリチオール(DPDS)の既知量の吸光度を標準として使用して測定した。PDP−モルホリノアンチセンスヌクレオチドの収率は、出発量のモルホリノ−3’−NH2オリゴヌクレオチドの80%より通常は高かった。この反応は、図3に示される。
【0036】
(N−(フルオレセイン−5’−チオカルバモイル)ジアミノヘキサンの合成)
フルオロセインイソチオシアネートアイソマーI(90mg)(FITC、最小98%、0.23mmol)を、3ml DMFに溶解し、そして76mgのN1−Boc−1,6−ジアミノヘキサン塩酸塩(0.3mmol)を添加した。結合反応を0.6mmolのトリエチルアミンを滴下することにより始めた。その反応混合物を、室温で2時間インキュベートし、その容量を、減圧下エバポレーションにより減少させた(最終容量は、約0.5ml)。冷水(5ml)を、残っている混合物に加え、そして1N HClで酸性化した。その沈殿を遠心分離で収集し、3回冷水で洗浄し、その後遠心分離し、上清にN1−Boc−1,6−ジアミノヘキサンがTLCおよびニンヒドリン試験により検出されなくなった。上記最終生成物をP2O5で乾燥した。この合成は図4に示される。
【0037】
そのBoc保護基を除去するために、その乾燥生成物を3mlのジクロロメタン(DCM)に溶解し、温度を氷浴で低下させた。2mlのTFAを、その溶液に加え、次いでこれを氷上で30分間、攪拌した。その反応を、TLCで追跡した。蛍光スポットは、UV光で見えた。上記溶媒を減圧下でエバポレートし、蝋状生成物をアセトンに溶解しそしてジエチルエーテルの添加により沈殿させた。精製のために上記生成物を、100mlの混合物の中に4mlの酢酸を含有する3mlのDCM/エタノール(3:2,v/v)に溶解し、同じ溶媒で平衡化した2cm×12cm SiO2カラムに通した。生成物はTLCによると純粋であった。そのRf値は、FITCに対しては、0.95であり、N1−(フルオレセイン−5’−チオカルバモイル)−N6−BOC−1,6−ジアミノヘキサンに対しては0.98であり、N−(フルオレセイン−5’−チオカルバモイル)ジアミノヘキサンに対しては0.64であった。
【0038】
(N,N’−ビス−(3−マレイミドプロピオニル)ポリ(エチレングリコール)の合成)
3mLの無水DMFに溶解した0.5gのNH2−PEG3400−NH2(0.147mmol)を、5mlの無水DMFに溶解した(3−マレイミドプロピオン酸NHSエステル)(106mg、0.4mmol)に室温で激しく攪拌しながら滴下した。上記反応の完結を、TLCおよびニンヒドリン試験陰性により確認した。室温で2時間インキュベーション後、その溶媒を減圧下、室温でロータリーエバポレータで除去した。生成物を、10mM EDTAを含有する2mlの緩衝液A(0.1Mリン酸ナトリウム、0.15M NaCl、pH7.2)に溶解した。不溶性の不純物を、遠心分離で除去した。透明な上清を、予め緩衝液Aで平衡化したSephadex G25カラムに通した。上記生生物はTLCおよびニンヒドリン試験では純粋であった。上記生成物の水溶液を、−20℃で保存した。
【0039】
(CD3)2SOに溶解した生成物の1H−NMRスペクトルは、以下のδ値:7.05ppm、s、4H、−CH=CH−;3.74ppm、t、2H N−CH2;3.5、s、PEGからの水素;3.03、t、2H CH2−CONHを示した。上記の値は、予測されるN,N’−ビス−(3−マレイミドプロピオニル)ポリ(エチレングリコール)ジアミドである生成物と一致した。マレイミド基の含量を、−HC=CH−と所定量の2−メルカプトエチルアミン(2−MEA)のスルフヒドリル基との反応に依存する方法および未反応スルフヒドリル基の、5,5’−ジチオビス−2−ニトロベンゾエート(DTNB,Ellman試薬)での以下のような滴定により、間接的に測定した。水中の適切な量の2−MEAを、N,N’−ビス−(3−マレイミドプロピオニル)ポリ(エチレングリコール)ジアミドの水溶液に加え、室温で30分間インキュベートした。DTNB(25μLの10mg/mLのエタノール溶液)を加え、そして室温で10分間インキュベートした後、412nmの吸光度を読んだ。その吸光度は、既知の量の2−MEAで標準化し、未反応スルフヒドリル基の量を計算した。N,N’−ビス−(3−マレイミドプロピオニル)ポリ(エチレングリコール)ジアミドのサンプル中のマレイミド基の含量を、2−MEAの量を最初の量から未反応2−MEAの引き算することにより計算した。この値からN,N’−ビス−(3−マレイミドプロピオニル)−ポリ(エチレングリコール)ジアミドの合成の収率は、65%と計算された。この合成は、図5に示される。
【0040】
(抗体へのチオール基の導入:固有のジスルフィド結合の、2−メルカプトエチルアミン(2−MEA)による還元)
ラットトランスフェリンレセプターCD71(クローンOX−26、アイソタイプIgG2a)に対するマウスモノクローナル抗体(mAB)を、10mMのアジ化ナトリウムを含有する1mg/mlのPBSの濃度の市販品を入手した。mAB OX−26の代わりにマウスモノクローナル抗体IgG2a,κ(UPC 10、Sigma)を使用し、コントロール結合体およびタンパク質測定用の標準曲線を作製そた。上記mAB OX26溶液をミクロ遠心膜フィルター(Sigma Ultrafree−CLミクロ遠心膜フィルター、再生セルロース、カットオフ30kDa)を使用して、4℃、5000×gで濃縮した。上記mAB貯蔵緩衝液を、10mM EDTAを含有する緩衝液A(0.1Mリン酸ナトリウム、0.15M NaCl、pH7.2)に変えた。mABの濃度を3〜5mg/mlに調整した。マウスmAB UPC 10を、緩衝液Aに溶解し、mAB
OX26と同じ濃度にした。固体の2−メルカプトエチルアミン塩酸塩(2−MEA)(6mg/ml)を、抗体溶液の中に攪拌し、その混合物を37℃で90分間インキュベートした。そのジスルフィドが還元された抗体を、ミクロ遠心膜フィルター(再生セルロース、カットオフ30kDa)を使用して、4℃、5000×gで、緩衝液A(N2下で脱気)を用いて膜分離精製した。その工程を、5,5’−ジチオビス(2−ニトロ安息香酸)(DTNB,Ellman試薬)とのインキュベーションの後に412nmで分光光学的に測定して、膜分離精製物に2−MEAが完全に無くなるまで実施した。ジスルフィド還元抗体溶液中のチオール基の数を、pH8.0で0.5mlの抗体溶液と25μlのDTNB溶液(エタノール中10mg/ml)とを10分間インキュベートした後で、412nmの吸光度(25℃で、e=14.15×103M−1cm−1)を読み取って計算した。その抗体タンパク質の濃度を、(65)の方法に従って測定した。結果は、上記ジスルフィド還元抗体が、Ig分子1モルあたり3.1〜3.5のチオール基を含むことを示した。SEC−HPLC分析は、分子量150kDaの1つのピークのみを示し、これは上記還元抗体がその分子の集積度を維持していることを示した。還元剤が不存在でさえも、EDTAの存在下では上記調製物はむしろ安定であって、そして遊離のチオール基の有意な損失は4℃で一晩では観察されなかった。
【0041】
(mAB OX−26/N,N’−ビス−(3−マレイミドプロピオニル)−PEGジアミド結合体の合成)
調製直後に、還元抗体の溶液を攪拌しているN,N’−ビス−(3−マレイミドプロピオニル)−PEGジアミド(抗体のチオール基に対して50倍のモル過剰なマレイミド基)の水溶液に、室温で滴下した。その反応混合物を、室温で2時間インキュベートし、未反応のN,N’−ビス−(3−マレイミドプロピオニル)−PEGジアミドを、緩衝液A(50kD膜、一晩で緩衝液交換3回)の中で透析により除去した。抗体−N,N’−ビス−(3−マレイミドプロピオニル)−PEGジアミド結合体の調製物を、さらなる合成反応のために直ちに使用した。
【0042】
上記結合反応の完結をSEC−HPLCの結果で示した。220nmで吸光度を測定している場合、溶出した物質のみが、結合体をあらわす7.20〜7.26分の位置にあり、一方、未反応N,N’−ビス−(3−マレイミドプロピオニル)−PEGジアミドに関する13.4〜13.7分の位置には、何も溶出しなかった。上記抗体結合体のマレイミド基の含量を、N,N’−ビス−(3−マレイミドプロピオニル)−PEGジアミドの合成に関する上述の2−MEAおよびEllman試薬を使用して間接的に決定した。抗体タンパク質の濃度を、(65)の方法に従って決定した。その結果は、抗体1分子あたり3.0のマレイミド基を示し、そしてジスルフィド還元抗体調製物の遊離のチオール基が、完全にN,N’−ビス−(3−マレイミドプロピオニル)−PEGジアミドと反応したとする仮定と一致した。さらに、SEC−HPLC分析は、分子量150kDaの1つのピークのみを示したので、架橋生成物は除外され得る。
【0043】
(PMLA/L−バリン/2−メルカプトエチルアミン/mPEG5000−NH2結合体の合成)
PMLA/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体の合成のために、1mmol(マリル単位に関して)のPMLA−NHSエステル(調製物P3:85%NHSエステル)を、10mlの無水DMFに溶解した。最初に、mPEG5000−NH2(2mL DMF中50μmol、NHS活性化マリル単位の5モル%に相当する)および200μmolのN−エチルモルホリンを、この順で加え、そして上記混合物を室温で30分、反応がTLCとニンヒドリン試験(原点でのニンヒドリン反応陽性に対して反応陰性)により完結するまで攪拌した。次にDMF中の200μmolの50mMの2−MEA(NHS−活性化マリル基の20モル%に相当する)のDMF溶液と200μmolのN−エチルモルホリンを、上記反応混合物に加えて、室温で30分間攪拌した。再び上記反応をTLC(2−MEAに対してRf=0.27、ポリマー結合体に対してRf=0)とニンヒドリン試験とで完結させた。この合成は、図6に示される。
【0044】
添加試薬の化学量論に従うと、PMLA−NHSエステル当量の4.5%および19.3%が、それぞれPEGおよび2−MEAに置き換えられた。残存する未反応NHSエステル当量を、L−バリン(5mlの水に1mmol)に結合させ、0.1gのNaHCO3(1.2mmol)の存在下で滴下した。その反応混合物を、室温で1時間攪拌し、冷却しながら0.1M HClで中和した。その溶媒を、減圧下30℃でエバポレートさせた。乾燥生成物を10mM EDTAおよび1mlの0.5MのDTTを含有する10mlの緩衝液A(0.1Mリン酸ナトリウム、0.15M NaCl、pH7.2)に溶解した。室温で10分後、その混合物を20000×gで、5分間遠心分離し、透明な上清を、予め緩衝液Aで平衡化したSephadex G25カラム(2.5cm×60cm)を通し、そして生成物含有画分を凍結乾燥した。
【0045】
PMLA/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体の組成を1H−NMRおよびUV−VIS分光光学分析により分析した。チオール基の含量を、1mlのリン酸ナトリウム緩衝液(pH8.0、100mM)に溶解した1mgの凍結乾燥結合体に25μlのDTNB溶液(エタノール中10mg/ml)を加え、室温で30分間インキュベーションした後412nmの波長での吸光度を読取ることにより測定した。PMLA/L−バリン/2−メルカプトエチルアミン/mPEG−NH2/FITC結合体の調製の場合(以下を参照のこと)、反応混合物はまた、ミクロ遠心分離膜フィルター(再生セルロース、5kDaカットオフ)を使用して、5000×gで膜分離精製し、412nmの吸光度を読んだ。チオールを分析するEllman方法は、チオールと発色性DTNB(5,5’−ジチオビス−2−ニトロ安息香酸、FW 396.4)との反応に基づいており、それにより黄色の5−チオ−2−ニトロ安息香酸(TNB)の形成が測定される。
【0046】
【化1】

FITC−結合体の場合のろ過の理由は、蛍光の存在がTNBの検出を不可能にするので、TNBをFITC結合体から分離するためである。そのろ過の間、TNBは膜を通過し、FITC−結合体が留まり、そして、ろ液の吸光度が測定される(上記ろ過は遊離の色素の除去のためではない。上記遊離の色素はこの反応の前に既に除去されている)。
【0047】
スルフヒドリル含量を、2−MEA標準に関して計算した。L−バリン部分の含量を、以下のようにトリニトロベンゼンスルホン酸(TNBS)アッセイを使用した加水分解およびRP−HPLCの後に、遊離のアミノ基を定量することにより決定した:1mgの上記結合体と30〜50μLの6N HClを、100μLキャピラリー試験管に入れた。封をしたきゃピラリーを100℃で12〜16時間、オーブンでインキュベートした。加水分解の後に、内容物をEppendorfチューブ(定量的にするためにそのキャピラリー試験管を水で濯ぐ)に移して、穏やかに加温することにより完全に乾固するまでエバポレートさせた。この物質を水に溶解して、そして遠心分離した。10〜30μlアリコートの上記上清を、300μLの重炭酸ナトリウム緩衝液(10mlの水に0.4gの重炭酸ナトリウム、pH8.5)に添加した。150μLの0.1%(w/v)TNBS水溶液の添加後に、上記混合物を37℃で30分間インキュベートした。遠心分離後、20μLのその反応混合物を30分の線型勾配(0〜10分、100%水、10〜40分、0〜60%アセトニトリル)を使用してRP−HPLCにより分離した。バリル部分の含量を既知量のL−バリンを標準として溶出物の340nmの吸光度を基礎にして計算した。mPEG:バリン:2−MEAのモル比を1H−NMRにより決定した。
【0048】
(PMLA/mAB OX−26/モルホリノアンチセンスオリゴヌクレオチド/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体の合成)
新しく調製されたmAB OX−26−PEG−マレイミドを、PMLA/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体に、4℃で、攪拌しながら滴下した。マレイミド基に対して100モル過剰の遊離チオール基(結合体)を用いて、全ての抗体が上記ポリマーと結合するようにした。30分のインキュベーション後、上記反応を完結した。上記反応の完結度をSEC−HPLCにより確認した。上記PMLA/mAB OX−26/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体を、ミクロ遠心分離膜フィルター(再生セルロース、分子量 100kDaカットオフ)を使用して、緩衝液Aの中で4℃、5000×gでの膜分離精製により精製した。mAB含有PMLA結合体は、フィルターに留められ、そしてタンパク質フリーのPMLA/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体のみがそのフィルターを通過した。上記膜分離精製を、SEC−HPLCで確認して膜分離精製物にタンパク質フリーののポリマー結合体の痕跡も無くなるまで繰り返した。
【0049】
PMLA/mAB OX−26/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体のタンパク質含量を、(66)の方法により測定した。非結合mAbの同じ量が、約10%高い吸光度を示し、従って、測定されたタンパク質含量を0.9のファクターで補正した(このファクターは、抗体が、膜分離精製で洩れないことが分かったが、留まった(結合)抗体は、原料抗体の90%しか達しなかった事実から実験的に引き出されたものであった)。この食い違いの理由は、不明である。タンパク質含有結合体の残存する遊離のチオール基の濃度を、上述の如く決定した。最初のチオール基の約70%が、まだ存在していたことを見出した。タンパク質含有結合体の濃度を、チオール基に関して3mMに調整した。
【0050】
次の工程で、ラミニン8のα4およびβ1鎖に対する5μmolのPDP−モルホリノアンチセンスオリゴヌクレオチド(5mlの1mM PDP−モルホリノアンチセンスオリゴヌクレオチド溶液)を、15μmol遊離のチオール基の濃度と等しくして、PMLA/mAB OX−26/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体(5mlの3mM タンパク質含有結合体溶液)の精製した溶液に、攪拌しながら滴下した。アンチセンスα4鎖:アンチセンスβ1鎖のモル比は、1:1であった。その反応混合物を4℃で一晩インキュベートした。上記反応の完結度を、260nmの吸収を有する溶出物の単一ピーク、および遊離のPDP−モルホリノアンチセンスオリゴヌクレオチドの位置における吸収の不存在を示すSEC−HPLCにより確認した。得られたPMLA/mAB OX−26/モルホリノアンチセンスオリゴヌクレオチド/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体を、ミクロ遠心分離膜フィルター(再生セルロース、分子量 100kDaカットオフ)を使用して、4℃、5000×gでの膜分離精製により精製した。この段階での遊離スルフヒドリル基の含量を、412nmで、Ellman試薬で決定した。アンチセンスモルホリノオリゴヌクレオチドの含量を、ジスルフィド基の50mM DTTによる37℃、2時間の還元およびSEC−HPLCによる分離後、260nmにおける吸光度により測定した。特に、その260nmの光吸収ピークを、還元されたPDP−モルホリノアンチセンスオリゴヌクレオチドを標準として得られるピーク比較した。
【0051】
PMLA/mAB OX−26/モルホリノアンチセンスオリゴヌクレオチド/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体のタンパク質含量を、(65)の方法で測定した。抗体:モルホリノアンチセンスオリゴヌクレオチドのモル比はは1:20〜1:26で変化したが、それでy1値が0.19%〜0.25%になる(全体の合成は、図7を参照のこと)。遊離のスルフヒドリル基を、N−エチルマレイミドでブロックし、未反応試薬を上のような膜分離精製により除去した。
【0052】
(FITCのPMLA/mAB OX−26/モルホリノアンチセンスオリゴヌクレオチド/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体への結合)
生物学的条件下での検出のために、PMLA/mAB OX−26/モルホリノアンチセンスオリゴヌクレオチド/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体を、フルオロセインイソチオシアネート(FITC)で、共有結合的に標識化した。この標識は、しかしながら、上で示されたPEG−NH2のPMLA−NHSエステルへの結合の後に導入された。N−(フルオレセイン−5’−チオカルバモイル)ジアミノ−ヘキサンの溶液を、DMFおよびPBS(1:1)の混合物中で最終濃度が25mMになるように調製した。mPEG−NH2/PMLA結合体の溶液(PMLA−NHSエステルのマリル単位に対して1mmol、調製P3:85% NHSエステル)に、25μmolのN−(フルオレセイン−5’−チオカルバモイル)ジアミノ−ヘキサン(図7のn=2.5%)および100μmolのN−エチレンモルホリンを加えて、それからその混合物を室温で30分間インキュベートした。その反応をTLCで追跡した。反応完結後に、蛍光を原点のみにおいて検出した。FITCのPMLA/mAB OX−26/モルホリノアンチセンスオリゴヌクレオチド/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体への構築については、色素の不存在下、上述と同じ経路に従った。上記結合体のFITC含量を、490nmでのFITC部分の吸収により測定した。FITC−担持結合体もまた、447nmにセットした励起波長および514nmにセットした発光波長でMerck−Hitach蛍光検出計により検出した。FITCの含量を、上述の如くポリマーを担持する変化する量のFITCの定量的な結合により形成した標準サンプルの吸光度または蛍光とサンプルの吸光度または蛍光を比較して計算した。上記結合体を、直接細胞培養実験に使用した。この合成は図8に示される。
【0053】
(溶血アッセイ)
図9は、赤血球(RBC)溶血アッセイを使用して測定したポリマーの膜崩壊活性を示す。新鮮なヒトRBCを、ヒト全血を2000gで5分間遠心分離することにより単離した。上記RBCを所望のpH(pH5.85またはpH5.5)の冷100mMリン酸ナトリウム緩衝液で3回洗浄した。最終ペレットを同じ緩衝液に再懸濁して、1mlあたり108 RBSを有する溶液にした。そのポリマーを、10mg/mlの濃度で、所望のpHにおける100mMの2塩基性のリン酸ナトリウム緩衝液に溶解した。上記ポリマー濃度は、2.5nmol/108 RBCであった。100%リーシスを達成するために、蒸留水中での溶血を使用した。ポリマーを有さない緩衝液中のRBCを、参照コントロールとして使用した。溶血アッセイを、1mlの適切な緩衝液に懸濁したRBCに上記ポリマーを添加することにより実施した。上記RBCを、数回試験管をひっくり返して混合し、37℃の水浴で1時間インキュベートした。インキュベーション後、上記RBCを13,500×gで10分間遠心分離をし、インタクトな細胞を沈降させ、リーシスを、RBCにより放出されたヘモグロビンの量を反映する541nmにおける上清の吸光度を測定することにより決定した。細胞のない上清の541nmの波長における吸光度の相対的増加100(A−A0)A合計を、膜崩壊の指標として測定した。その結果は、RBC膜を安定化するPMLA−PEGとは反対に、PMLA−PEG−バリン、PMLA−PEG−バリン−AS、およびPMLA−PEG−バリン−AS−mABは、膜不安定性を暗示することを示す。比較は、不安定性はPMLA−結合バリンの存在に起因することを示す。pHが下がっていく(エンドドームのリソソームへの成熟を模擬する)と、バリンのカルボキシル基がプロトン化され、電荷が中和されたバリン部分の増加した親油性に起因して、不安定性は、増加する。
【0054】
(モルホリノ−オリゴの放出)
図10aおよび図10bは、グルタチオン(グルタチオン−SH)によりジスルフィド結合の切断に起因する、薬物キャリアからのモルホリノアンチセンスオリゴヌクレオチドの放出を示す。ジスルフィド結合の切断は、2工程反応である。最初の工程は、1当量のグルタチオン−SHが、ジスルフィドと反応して混成ジスルフィドアンチセンスオリゴヌクレオチド−S−S−グルタチオン(縞の縦棒)(図10a)および1当量の遊離のアンチセンスオリゴヌクレオチド−SH(1色の縦棒)を形成する。時間が経つにつれて混成ジスルフィドが減少し、遊離のアンチセンスが増加する。この反応は、図10bに示されるように迅速である。2番目の工程は、混成ジスルフィドが第2当量のグルタチオンと反応してジスルフィドグルタチオン−S−S−グルタチオンおよび遊離のオリゴヌクレオチド−SHを生じる。この第2反応はゆっくりである。この反応の2工程のメカニズムおよび相対的速度は、いわゆるジスルフィド交換反応と呼ばれるこの反応に関して代表的である。その結果は、上記オリゴヌクレオチドが、細胞質で薬物キャリアから非常に効率的に切断され、それはこの所定の濃度のグルタチオンを含むことを示す。
【0055】
図に示されるように、薬物ビヒクルからアンチセンスモルホリノオリゴヌクレオチドの細胞質放出を模倣するために、5mM GSH(γ−L−グルタミル−L−システイニルグリシン、分子量307.33)を、室温で上記反応混合物に添加した。その混合物は、水中に0.25mMの薬物ビヒクルを含有した。いろいろな時に、全スルフヒドリル部分に対して過剰のN−エチルマレイミド(最終濃度、20mM)の添加により、上記反応を停止した。還元アンチセンスをN−エチルマレイミジルアンチセンスとして検出した。上記反応生成物を、HPLCにより分離し、放出されたアンチセンスモルホリノを260nmにおけるUV吸光度により検出した。その結果は、図10bに示される。完全な(100%)放出は、50mM DTTの存在下において37℃1時間の還元と関係付けられる。HPLC分析を、ゲルろ過カラムを使用してMerck−Hitach分析用HPLCで実施した。分離については、リン酸ナトリウム緩衝液(50mM、pH7.4)を使用して、流速0.75ml/分でMacherey & Nagel 125−5 GFC−HPLCカラム(300×7.7mm)で実施した。
【0056】
(ヌードラットの脳で増殖したヒト神経膠腫の、ポリリンゴ酸に結合したラミニン−8アンチセンスオリゴヌクレオチドでの処理)
腫瘍を処置し、そして正常細胞に対する副作用を減少させる特定の薬物送達は、すばらしいことである。タンパク質合成のレベルにおいて数種の分子標的を同時に阻害することは、腫瘍の増殖および進行を抑制することに非常に有効であり得る。ラミニン−8鎖の過剰発現は、神経膠腫の進行と関連し、ラミニン−8をブロッキングすることは、インビトロで神経膠腫侵襲を阻害する(34)。
【0057】
(方法)
(ポリリンゴ酸(PMLA))
多機能性薬物送達構築物は、ポリリンゴ酸(PMLA)のぶら下がっている(ペンダントな)カルボキシル基に結合するモジュールより構成される。そのポリマーは、Physarum polycephalumの天然産物である(27)。上記モジュールは、以下である:(1)ジスルフィド結合により上記骨格に結合するモルホリノアンチセンスオリゴヌクレオチド(その結合が、細胞質で切断され、遊離の薬物を放出する)、(2)癌細胞標的化およびレセプター媒介性エンドサイトーシスのためのトランスフェリンレセプターの抗体、(3)短鎖PEG結合L−ロイシンおよび直接結合L−バリン(両方がアミド結合を介して連結されているが、エンドソーム膜を崩壊させるpH依存性の親油性を提供する)、(4)循環時間を増加する長鎖PEG、および(5)組織/細胞内において構築分子を検出する蛍光レポータ分子(フルオレセイン、Cy5または同様な発色団)。
【0058】
(動物処置に使用された薬物の配合)
薬物1:ラミニンα4に対するアンチセンスオリゴ+ラミニンβ1に対するアンチセンスオリゴ(α4:β1=1:1);
薬物2:ラミニンα4に対するアンチセンスオリゴ+ラミニンβ1に対するアンチセンスオリゴ(α4:β1=1:1)+速く分裂している細胞に対する送達ビヒクルとしての抗トランスフェリンレセプターモノクローナル抗体(Chemicon InternationalからのラットCD71に対する抗体OX−26)(35、36、37および38);
薬物3:ラミニンα4に対するアンチセンスオリゴ+ラミニンβ1に対するアンチセンスオリゴ+EGFRに対するアンチセンスオリゴ(α4:β1:EGFR=1:1:1);
薬物4:ラミニンα4に対するアンチセンスオリゴ+ラミニンβ1に対するアンチセンスオリゴ+EGFRに対するアンチセンスオリゴ(α4:β1:EGFR=1:1:1)+抗トランスフェリンレセプター抗体;
薬物1A、2A、3Aおよび4Aは、対応する番号の薬物に対して同一である(即ち薬物1と薬物1Aは同じである)。対応するセンスオリゴを、アンチセンスオリゴの代わりに使用した。
【0059】
薬物1:PMLAに結合したラミニンα4鎖に対するアンチセンスオリゴヌクレオチドおよびラミニンβ1鎖に対するアンチセンスオリゴヌクレオチド;薬物2:PMLAに結合したラミニンα4鎖に対するアンチセンスオリゴヌクレオチド、ラミニンβ1鎖に対するアンチセンスオリゴヌクレオチド、抗トランスフェリンレセプターモノクローナル抗体(Chemicon Internationalからの抗体OX−26)。コントロール(薬物1Aおよび薬物2A)は、対応するセンスオリゴにより置き換えられたアンチセンスオリゴとの同じキャリア結合体であった。上記ヒトU−87MG神経芽膠腫細胞株を、インビトロ実験に使用し、異系勾配されたホモ接合体のNHRNU−M NIHヌードラット(Taconic Inc.)に、頭蓋内に注入した。
【0060】
異系勾配されたNIHヌードラット(Tac:N:NIH−Whn、Taconic)を、これらの試験に使用した。アンチセンス処置について、最も特異的で、安定でインビトロおよびインビボに有効であるとしてモルホリノオリゴ(Gene Tools、LLC)を使用した。モルホリノは、インビトロ研究で過去に成功裏に使用されたが、インビボでは、それらの送達はうまく行かなかった(6、7)。ポリ−L−リンゴ酸(PMLA)は、細胞にモルホリノを細胞に入れる送達キャリアとして使用された。PMLAの精製方法の原理が、(25)に記載されている。最近のPMLAのスケ−ルアップ生産方法(27)を使用した。PMLA官能基の化学が調査され(28、29)、そして有機溶媒および水性溶媒両方における生成物の誘導体化および精製は容易に達成し得る。我々の実験に関して、PMLAを、標的化腫瘍に特異的なラミニン−8鎖に加えて、PMLAはトランスフェリンレセプターに対するモノクローナル抗体に化学的に結合し、早く分裂する細胞に最も特異的な薬物を作製した(35、36、37および38)。
【0061】
予備的な毒性研究を、合成が完了した後に、モルホリノオリゴヌクレオチド、PMLAおよびそれらの結合体について実施した。それぞれの化学品を投与後30日に、肉眼での病理学的分析および顕微病理学的分析を行ったが、異常な変化は認められなかった。動物は、神経学的異常を発症しなかったし、それらの食欲もまた、正常であった。動物試験被験体は、定位的デバイスを使用してヒト経芽膠腫細胞U−87MGを頭蓋内に注入することにより作製した。薬物処置は、腫瘍細胞注入後3日目で開始した。
【0062】
頭蓋内処置について、以下に図示するように、ラットにアンチセンスオリゴを3日目、7日目、10日目および14日目(全部で4処置)に注入した。
【0063】
【化2】
各群12匹のラットにラミニン−8α4鎖に対するオリゴ+β1鎖に対するオリゴを、0.5mg/kgまたは2.5mg/kgの用量で注入した。各コントロール群11匹のラットに、α4鎖に対するセンスオリゴ+β1鎖に対するセンスオリゴを、0.5mg/kgまたは2.5mg/kgの用量で注入した。全ての外科的および非外科的操作を、2003年8月付けのIACUCプロトコル001118に従って行った。頚動脈内の処置について、腫瘍移植直後にカテーテルを頚動脈に移植した。カテーテルを、移植可能な皮下注入ポートに結合した。ラットに900μlのアンチセンスオリゴ溶液およびセンスオリゴ溶液(1分当たり0.06mlで15分間、蠕動ポンプで)を右頚動脈に皮下ポートチャンバ経由で潅注し、引き続きヘパリンロック(heparin flush)した。ラットを潅注終了後30分以内に安楽死させた。上記コントロールは:(a)正常なコントロール組織を得るために如何なる処置もせず、40日目に安楽死させた3匹のラット、および(b)腫瘍を有し、1日目、3日目、7日目、10日目および14日目にPBSで頭蓋内に擬似注入し、腫瘍の進行に起因する神経症状を発症させるとすぐに組織採取のために安楽死させた4匹のラットからなった。
【0064】
(結果)
(頭蓋内腫瘍処置)
薬物2の0.5および2.5mg/kgの用量は、生存率研究における処置と同等であった。頭蓋内投与による薬物2の4回の投薬後に、動物の生存時間が、PBS(擬似)またはセンスオリゴ(薬物2A)で処置されたラットと比較して、30%増加した、p<0.008(図11)。しかしながら、2つの処置は、僅かな効果しかなかった。トランスフェリンレセプター抗体の無い薬物1は、生存率には影響しなかった。従って、薬物細胞送達のメカニズムは、おそらくトランスフェリン媒介性エンドサイトーシスである。興味深いことに、アンチセンスをEGFR結合薬物2(薬物4)へ添加することにより、活性が無くなったが、おそらくEGFRが阻害された低酸素状態で、神経膠腫細胞の生存率の増加に起因するであろう(39)。
【0065】
薬物の内在化のメカニズムを、培養神経膠腫細胞において調査した。細胞がフルオレセイン標識化薬物2およびローダミン標識化エンドソームマーカーFM 4−64(Molecular Probes、Eugene、OR)で処理された場合、両方の化合物の染色は、共局在化を示した。10分以内に細胞膜の近くに共局在化し、30分以内に両方の標識がエンドソームに見出された(図12)。
【0066】
細胞がトランスフェリンレセプターで予め処理され、次いで10分以内に薬物2で処理された場合、上記薬物は、細胞質に見られなかった(データは、示されていない)。これらの結果は、トランスフェリンレセプター抗体が、活性薬物の一部として必要であることを示唆する。何故なら、それは薬物の細胞への透過を、レセプター媒介性のエンドサイトーシスにより可能にし、その後上記アンチセンスオリゴが標的細胞内に放出され得るからである。もし上記細胞がトランスフェリンに対する遊離抗体で処理される場合、上記レセプターは、ブロックされ、上記薬物上の抗体は結合できない。
【0067】
本発明者らは、薬物2がヌードラットに異種移植されたヒト神経膠腫における血管密度および特異的なラミニン鎖発現を減少させることを、発見した。本発明者らは、ラミニン−8発現を阻害するよう設計された薬物2が腫瘍における血管密度を減少させることを、示した。これらの血管を、von Willebrand因子を免疫染色することにより可視化した。血管の数を、薬物処置動物および未処置動物において、画像計測システム(Hamamatsu,Japan)に連結されたZeiss Axioscope顕微鏡を使用して、200倍の倍率で、3つの連続切片について5つの顕微鏡視野(1腫瘍ににつき15視野)において数えた。このデータをNIH ImageJソフトウエアに入力し血管密度を定量した。統計的有意差を、ANOVAにより決定した。
【0068】
(血管新生)
図13に示されるように、処置しないU87MGヒト腫瘍における毛細血管密度は、正常脳より有意に高かった。薬物2での4回の頭蓋内処置後、腫瘍血管密度は、55%減少した。データは、3匹の擬似手術した(正常)ラット(45顕微鏡視野)、未処置腫瘍を有する5匹のラット(75顕微鏡視野)および薬物2処置腫瘍を有する5匹のラット(75顕微鏡視野)について提示された。この結果は、ラミニン−8発現を阻害するよう設計された薬物2の作用の抗血管新生メカニズムを確証する。
【0069】
(ラミニン鎖免疫染色)
薬物2が事実上標的化ラミニン−8鎖の発現を阻害することを示すことは重要である。この目的に対して、インビボおよびインビトロで実験を実施した。腫瘍の免疫染色について、ラットラミニンを認識しないが腫瘍由来ラミニンとは反応するヒトラミニン−8β1鎖に対する抗体を、使用した。図14は、上記アンチセンスが免疫染色を、効果的に除去する細胞培養における効果を示す。図15に示されるように、薬物2がまた、異種移植したヒト腫瘍においてラミニンβ1鎖についての免疫染色を、効果的に減少させた。
【0070】
インビトロにおけるラミニン−8発現の阻害は、薬物2で3日間処置した2種の培養されたヒト神経膠腫、U87MGおよびT98G、の条件培地で評価した。図16のウエスタンブロット分析は、ラミニンα4鎖の著しい減少(密度計測により3倍の減少)およびラミニンβ1鎖の消失を示す。従って、薬物2は、インビボおよびインビトロの両方において、標的ラミニン鎖の発現を効果的に阻害する。
【0071】
(インビボにおける頸動脈内腫瘍処置および静脈内腫瘍処置)
頸動脈内処置のために、ラット脳にヒトU87−MG神経膠腫細胞を接種した14日後に、3匹のラットの群は、腫瘍移植後直ちに、頚動脈内にカテーテルを移植した。そのカテーテルを移植可能な注入ポートに連結した。そのラットに、900μlのアンチセンスおよびセンスオリゴ溶液(1分あたり0.06mlで、15分間蠕動ポンプで)を、皮下ポートチャンバー経由で右頚動脈に潅注し、ヘパリンロックした。薬物2を、頚動脈処置のために2.5μg/kgの濃度で注入したか、または5μg/kgの濃度で、尾静脈経由で注入した(同様に3匹のラット)。薬物分布を注入後1時間、3時間、12時間および24時間で調べた。蛍光単位を使用して、本発明者らは、移植された腫瘍細胞(濃い染色)および脳の血管細胞(より薄い染色)における薬物を検出した(図17)。薬物2を、薬物2により担持されるトランスフェリン抗体を(赤で)標識化するローダミン染色抗体を用いて可視化した。上記細胞核をDAPI(青)で対比染色する。移植されたU87MG腫瘍細胞(左パネル)は濃く標識化されるが、ラット脳(右パネル)は、限られた(主に血管の)染色を示す。これらの位置の最大濃度は、薬物注入後3時間および12時間の時点で達成された。これらの結果は、薬物2が血液脳関門(BBB)を、おそらくレセプター媒介性エンドサイトーシスで透過することを確証する。
【0072】
(結論)
ラミニン−8の発現およびヒト腫瘍におけるその阻害を研究するのに適切なインビボのモデルを開発した。ラミニン−8のα4鎖およびβ1鎖に対するアンチセンスオリゴ(ラミニン−8のブロッキング)と新規な薬物送達ビヒクル,PMLA、との組合わせは、ラットにおいて異種移植した頭蓋内ヒト神経膠腫におけるラミニン−8発現を効率的に阻害した。予備的な4回のアンチセンス処置後に、処置された神経膠腫を担持する動物の生存率が、有意に増加した、p<0.008。これらのデータは、治療標的としてラミニン−8を使用するPMLAベースのアンチセンス薬物が、ヒト脳腫瘍を阻害するのに有効であることを示す。
【0073】
従って、添付の特許請求の範囲は、上に具体的に説明されそして記載されたもの、概念的に等価のもの、自明に置換し得るものおよびまた、本発明の本質的な思想を本質的に取り込むものを含むことが理解されるべきである。当業者は、上述の好ましい実施形態の種々の改昨および改変は、本発明の範囲から逸脱しないで設定され得ることを理解する。説明された実施形態は、単なる例証の意図で示されたもので本発明を限定するとして考えられべきではない。従って、添付の特許請求の範囲の範囲内で、本発明は、本明細書に具体的に記載される以外でも実施され得ると理解されるべきである。
【0074】
(参考文献)
【0075】
【化3】
【0076】
【化4】
【0077】
【化5】
【0078】
【化6】
【技術分野】
【0001】
本発明は、2003年12月5日に出願した米国仮特許出願第60/527,330号を基礎に出願し、その出願からの優先権を主張する。
【0002】
(本発明の背景および先行技術の説明)
本発明は、薬物の標的化送達の分野に関し、より具体的には多機能性標的化薬物送達ビヒクルに関する。
【背景技術】
【0003】
現在、いくつかの異なる分子骨格が薬物ビヒクルの合成に使用され、注目すべき例として、N−(2−ヒドロキシプロピル)メタクリルアミド(HPMA)コポリマー(20〜30kDa)(例えば、非特許文献26参照)および他のポリアクリル酸(polycarylic acid)の誘導体である。しかしながら、これらは、それらの炭素−炭素主鎖の故に生物分解性と考えられておらず(例えば、非特許文献27および本明細書の参考文献参照)、有害なアクリル酸による避けがたい汚染により問題である(例えば、非特許文献28参照)。他の分解性骨格(例えば、ポリ(L−グルタミン酸)(例えば、非特許文献29参照)は、ペプチド結合の周りの回転制限もしくは化学合成および生成物の精製に望ましい有機溶媒における限られた溶解度のような好ましくない性質を有し得、そしてさらにペプチド主鎖の高い水素結合形成能力に起因する他の潜在的免疫原性構造に構造的近似において免疫原性の基盤になっている(例えば、非特許文献29、30、31参照)。
【0004】
(アンチセンス技術)
特定のRNA配列と結合し、そして不活性化するアンチセンスオリゴヌクレオチド(オリゴ)は、遺伝子機能、遺伝子発現の制御、遺伝子産物間の相互作用および薬物開発の新しい治療標的の実証の研究をするための最良の手段の1つであり得る。アンチセンスオリゴは、ウイルス疾患、癌および他の重篤な疾患に対する安全で有効な治療の有望性を提供する。RNA産生のためのDNAテンプレートを模倣する特定のアンチセンスオリゴは、相補的RNAへ結合し、そしてタンパク質の翻訳(例えば腫瘍マーカーのタンパク質の翻訳)を抑制するために使用される(例えば、非特許文献1参照)。
【0005】
神経膠腫においてアンチセンス技術の使用について有望なデータがある。インビトロおよびヌードマウスにおいて神経膠腫の増殖は、テロメラーゼに対するアンチセンスにより阻害され得る(例えば、非特許文献2参照)。パイロット研究は、IGF−Iレセプターに対するアンチセンスが神経膠腫細胞アポトーシスを誘発し、患者の臨床的改善という結果が得られたことを示した(例えば、非特許文献3参照)。いくつかの臨床試験が、他の癌の処置にアンチセンスオリゴを現在使用している(例えば、非特許文献4参照)。これらの研究は不十分な特異性、安定性および非アンチセンス効果の無い新世代のアンチセンスオリゴを活用する(例えば、非特許文献5参照)。最も有望な改善されたオリゴの種類は、モルホリノ(Morpholino)オリゴおよびペプチド核酸(PNA)オリゴである。これらの種類は、全てのアンチセンスタイプの中で最高の配列特異性を有し、そしてこの特異性を非常に広い濃度範囲にわたって維持する(例えば、非特許文献6、7、8、9、10および11参照)。新しい、早く進化するアンチセンスアプローチの変種は、非常に強力な遺伝子発現サイレンサーであり、潜在的な抗癌薬でもある小干渉RNA(siRNA)に代表される。
【0006】
(インビトロおよびインビボにおける腫瘍の進行を抑制するためのいくつかの分子マーカーの組合わせによるブロッキング)
このアプローチは癌化学療法において長く成功裏に使用されてきたが、特定の腫瘍マーカーを標的化することにはまだ適用されていなかった。遺伝子/タンパク質アレイアプローチの進展の後にのみ、腫瘍の進行および再発の間の特定の遺伝子の協奏的な変化に関するデータを得て、そして関連付けることが可能となった。そのような協奏的な変化は、腫瘍の発症および進行を効率的にブロックすることを期待していくつかの遺伝子の同時変化を阻止する可能性を提供する。神経膠腫の増殖および拡散をブロックするいくつかの候補遺伝子があり、それには腫瘍の増殖をより効率的に抑制するために化学療法剤との組合わせで潜在的に使用され得るチロシンキナーゼレセプター(例えば、EGFR)、いくつかの成長因子および抗アポトーシス遺伝子が含まれる(例えば、非特許文献12、13、14、15および16参照)。本発明者らの以前の研究で、別の潜在的な候補タンパク質、ラミニン−8を同定したが、それは脳腫瘍および乳癌で過剰発現され、神経膠腫の予後の悪さと関連し、神経膠腫侵襲と関連する。
【0007】
(薬物送達)
腫瘍を処置するために癌細胞を直接標的化する目的で、薬物、例えば、モノクロ−ル抗体、アンチセンスオリゴまたは低分子(例えばタルセバ(Tarceva)(エルロチニブ)(erlotinib))は、細胞膜を透過し得る。細胞内薬物送達について3種類の基本的な方法があり、それらは、膜の水性チャネルまたは孔を通る受動的拡散、膜の脂質への溶解による脂質溶解性薬物の受動的拡散およびキャリア媒介性の能動輸送(ウイルスベクター、リポソーム媒介性の遺伝子移送システム、特殊化学品)である(例えば、非特許文献16、17参照)。脳組織は、水溶性でイオン化または極性の薬物の透過を抑制する脳毛細血管内皮細胞間のタイトジャンクションによる特別の血液脳関門を有するので、薬物による処置が特に困難である(例えば、非特許文献18、19参照)。
【0008】
高分子量分子は、高分子および脂質(分子量>45kDa)について癌組織で観察される高い透過性および保持性効果(enhanced permeability and
retention)(EPR)の故に、最近特に注目を集めている(例えば、非特許文献20、21および22参照)。腫瘍を正常組織と区別しない今日使用される低分子抗癌剤と異なり、高分子(またはポリマー性)薬物は、EPR効果により高度の選択性で癌を標的化し得る(例えば、非特許文献22、23参照)。そのような有望な薬物キャリアの1つとして、ポリ−L−リンゴ酸(PMLA)が、本発明者らの1人により開発されてきた(例えば、非特許文献24、25参照)。
【非特許文献1】Astriab−Fisher A,Sergueev DS,Fisher M,Shaw BR,Juliano RL.「Antisense inhibition of P−glycoprotein expression using peptide−oligonucleotide conjugates.」 Biochem Pharmacol、2000年、第60巻、p83−90
【非特許文献2】Komata T,Kondo Y,Koga S,Ko SC,Chung LW,Kondo S.「Combination therapy of malignant glioma cells with 2−5A−antisense telomerase RNA and recombinant adenovirus p53.」 Gene Ther,2000年、第7巻、p2071−2079
【非特許文献3】Andrews DW,Resnicoff M,Flanders AE,Kenyon L,Curtis M,Merli G,Baserga R,Iliakis G,Aiken RD.「Results of a pilot study involving the use of an antisense oligodeoxynucleotide directed against the insulin−like growth factor type I receptor in malignant astrocytomas.」J Clin Oncol、2001年、第19巻、p2189−2200
【非特許文献4】Jansen B,Wacheck V,Heere−Ress E,Schlagbauer−Wadl H,Hoeller C,Lucas T,Hoermann M,Hollenstein U,Wolff K、Pehamberger H.「Chemosensitisation of malignant melanoma by BCL2 antisense therapy.」Lancet、2000年、第356巻、p1728−1733
【非特許文献5】Nielsen PE.「Peptide nucleic acid targeting of double−stranded DNA.」Methods Enzymol、2001年、第340巻、p329−340
【非特許文献6】Summerton J,Weller D.「Morpholino antisense oligomers:Design,preparation and properties.」Antisense Nucleic Acid Drug Dev、1997年、第7巻、p187−195
【非特許文献7】Lacerra G,Sierakowska H,Carestia C,Fucharoen S,Summerton J,Weller D,Kole R.「Restoration of hemoglobin A synthesis in erythroid cells from peripheral blood of thalassemic patients」Proc Natl Acad Sci USA、2000年、第97巻、p9591−9596
【非特許文献8】Taylor MF,Paulauskis JD,Weller DD,Kobzic L.「Comparison of efficacy of antisense oligomers directed toward TNF−α in helper T and macrophage cell lines.」Cytokine、1997年、第9巻、p672−681
【非特許文献9】Arora V,Knapp DC,Smith BL,Statdfield ML,Stein DA,Reddy MT,Weller DD,Iversen PL.「c−Myc antisense limits rat liver regeneration and indicates role for c−myc in regulating cytochrome P−450 3A activity.」J Pharmacol Exp Ther、2000年、第292巻、p921−928
【非特許文献10】Shi N,Boado RJ,Pardridge WM.「Antisense imaging of gene expression in the brain in vivo.」Proc Natl Acad Sci USA、2000年、第97巻、p14709−14714
【非特許文献11】Boado RJ,Kazantsev A,Apostol BL,Thompson LM,Pardridge WM.「Antisense−mediated down−regulation of the human hutingtin gene.」J Pharmacol Exp Ther、2000年、第295巻、p239−243
【非特許文献12】Tanabe K,Kim R,Inoue H,Emi M,Uchida Y,Toge T.「Antisense Bcl−2 and HER−2 oligonucleotide treatment of breast cancer cells enhances their sensitivity to anticancer drugs.」Int J Oncol、2003年、第22巻、p875−81
【非特許文献13】Cho YS,Cho−Chung YS.「Antisense protein kinase A RIalpha acts synergistically with hydroxycamptothecin to inhibit growth and induce apoptosis in human cancer cells:molecular basis for combinational therapy.」Clin Cancer Res、2003年、第9巻、p1171−1178
【非特許文献14】Tortora G,Caputo R,Damiano V,Caputo R,Troiani T,Veneziani BM,De Placido S,Bianco AR,Zangemeister−Wittke U,Ciardiello F.「Combined targeted inhibition of bcl−2,bcl−XL,epidermal growth factor receptor,and protein kinase A type I causes potent antitumor,apoptotic,and antiangiogenic activity.」Clin Cancer Res、2003年、第9巻、p866−871
【非特許文献15】Jiang Z,Zheng X,Rich KM.「Down−regulation of bcl−2 and bcl−XL expression with bispecific antisense treatment in glioblastoma in vitro induce to enhance caspase−dependent cell death.」J Neurochem、2003年、第84巻、p273−281
【非特許文献16】Mycek MJ,Harvey RA,Champe,PC.「Pharmacology.Lippincott−Raven」第2版、Philadelphia,New York、1997年、p475
【非特許文献17】Park JW.「Liposome−based drug delivery in breast cancer treatment.」Breast Cancer Res.、2002年、第4巻、p95−99
【非特許文献18】Matsukado K,Sugita M,Black KL.「Intracarotid low dose bradykinin infusion selectively increases tumor permeability through activation of bradykinin B2 receptors in malignant gliomas.」Brain Res、1998年、第792巻、p10−15
【非特許文献19】Ningaraj NS,Rao MK,Black KL.「Adenosine 5’−triphosphate−sensitive potassium channel−mediated blood−brain tumor barrier permeability increase in a rat brain tumor model.」Cancer Res、2003年、第63巻、p8899−8911
【非特許文献20】Torchilin VP,Lukyanov AN.「Peptide and protein drug delivery to and into tumors:challenges and solutions.」Drug Discov Today、2003年、第8巻、p259−66
【非特許文献21】Peterson CM,Shiah J.,Sun Y,Kopeckova P,Minko T,Straight RC,Kopecek J「HPMA copolymer delivery of chemotherapy and photodynamic therapy in ovarian cancer」Adv Exp Med Biol、2003年、第519巻、p101−23
【非特許文献22】Maeda H,Fang J.,Inutsuka T,Kitamoto Y.「Vascular permeability enhancement in solid tumor:various factors,mechanisms involved and its implications.」Int Immunopharmacol、2003年、第3巻、p319−28
【非特許文献23】Satchi−Fainaro R,Puder M,Davies JW,Tran HT,Sampson DA,Greene AK,Corfas G,Folkman J.「Targeting angiogenesis with a conjugate of HPMA copolymer and TNP−470.」Nat Med、2004年、第10巻、p255−261
【非特許文献24】Fischer H,Erdmann S,Holler E.「An unsual polyanion from Physarum polycephalum that inhibits homologous DNA polymerase α in vitro.」Biochemistry、1989年、第28巻、p5219−5226
【非特許文献25】Lee BS,Holler E.「Effects of culture conditions on β−poly(I−malate)production by Physarum polycephalum.」Appl Microbiol Biotechnol、1999年、第51巻、p647−652
【非特許文献26】Kopecek,J.,Kopeckova,P.,Minko,T.,Lu,Z.「HPMA copolymer−anticancer drug conjugates:desgn,activity,and mechanism of action」、Eur.J.Biopharm.2000年、第50巻、p61−81
【非特許文献27】Vincenzi,V.,Ferruti,P.,Ford,J.,Duncan,R.「Synthesis and preliminary evaluation of novel functionalized poly(ethylene glycol)−block−poly(ester−carbonate)copolymers as biodegradable carriers.」Macromolecular Bioscience、2001年、第1巻、p164−169
【非特許文献28】「Environmental health criteria 191. Acrylic acid.」[online]United Nations Environmental Programme/International Labour Organisation/World Health Organisation.International Programme on Chemical Safety.インターネット<URL:http://www.inchem.org/documents/ehe/ehc/ehc191.htm>
【非特許文献29】Li,C.「Poly(L−glutamic acid)−anticancer drug conjugates.」 Advanced Drug Delivery、2002年、第54巻、p695−713
【非特許文献30】Gill,T.J.,Kunz,H.W. Papermarker,D.S.1967.「Studies on synthetic polypeptide analogues.J.Biol.Chem.1967年、第242号、p3306−3318
【非特許文献31】Chiang,C.H−Yeh,M,K.「Contribution of poly(amino acids)to advances in pharmaceutical biotechmologyy.」、Current Pharmaceut.Biotechnol.2003年、第4巻、p8−16
【発明の概要】
【発明が解決しようとする課題】
【0009】
(発明の要旨)
ポリリンゴ酸を使用することによる以前に悩まされた薬物キャリアシステムの問題を解決することが本発明の目的である。そのポリリンゴ酸は多くの機能性カルボキシル基を担持し、そのような基は、質量数50,000のポリリンゴ酸1分子当たり、少なくとも約50であり、そして約500を担持する。
【課題を解決するための手段】
【0010】
ポリリンゴ酸は分子量2,500から少なくとも100,000のサイズの範囲で容易に入手できる。これは、以下のことを達成するために多数の生物学的に活性な機能的分子の連結を可能にする:
薬物キャリア分子あたりの組織標的化分子の種類および数における高い調整可能性;
薬物キャリア分子あたりの結合薬物モジュール(プロドラッグ)の種類および数における高い調整可能性;
上記機能的モジュールに加えて疎水性残基または親水性残基のいずれかを担持する基を有することにより溶解度を調節する能力;
上述の機能的モジュール以外に、例えば,PEGのような分解に対してキャリアシステムを保護する(例えば、血液循環系において寿命を増加する)ことに活性である、さらなる基を結合する能力;
上記薬物送達システムの構築単位として使用される多くの他の残基と共に、生分解性であるキャリア骨格;
ウイルス成分を使用しないシステム;
機能的モジュール(mABまたは他の腫瘍細胞表面レセプター)、上記種類の薬物(悪性に発現した遺伝子に対するアンチセンスオリゴヌクレオチド)、および腫瘍組織に対して高い特異性(EPR−効果)を担う全体的に高分子量の薬物送達システムを有する送達システム;ならびに
ラットの体重1kg当たり5mg程度の高い用量を可能にする低毒性の薬物システム。
【0011】
本発明のさらなる目的は:
複雑な合成方法および制御不能な副反応を避けることにより薬物送達システムの合成を容易にすること;
容易な精製を提供する好ましい溶解度および他の性質で作製すること;
各個別の化学結合反応の規定された化学量論および再現性を可能にする最大収率を達成すること;
結合された機能的モジュールの種類と数に関する高い調整可能性の構造的および合成の基礎を用意すること;ならびに
薬物送達システムの簡単なスケールアップを可能にする技術を提供することである。
【0012】
技術発明は以下を包含する:
非常に数多くの反応性カルボキシル基(分子量50,000の骨格に対して約500)を担持する主鎖または骨格としてポリリンゴ酸を用いる薬物送達システムの選択;
それらのNHS−エステルを形成することによる、これらのカルボキシル基の殆ど(理想的には全部)の活性化;
NHS−活性化カルボキシル基での置換の求核剤として単一のアミノ基を担持する機能的モジュールの化学的に独立した調製;
上記活性化された骨格および反応性機能的モジュールの各々を別々に調製すること;
上記NHS−活性化骨格および各反応性機能的モジュールを、独立しているがよく規定された順に組み合わせて反応させ、新しく添加した機能的モジュールのそれぞれの添加後に所望の中間体/生成物の精製および実証を可能にすること;
これらの反応を化学量論量で組合わせること;
結合体の高くかつ再現性のよい収率を達成すること;
新しく添加した反応性機能的モジュールと既に結合されたモジュールとの副反応を、次に入って来る機能的モジュールの結合に対して順番に添加する良く組織化された階層を選択することにより避けること。
【0013】
キャリアまたは骨格のデザインおよび合成について説明するが、それは共有結合的に結合した薬物を標的化組織に輸送し、その組織の細胞表面レセプターに結合し、エンドソームに内在化し、エンドソームから細胞質へ抜け出し、そして細胞質において細胞質内容物のグルタチオンおよび他のスルフヒドリル基との化学反応により活性遊離薬物を放出する。高分子量薬物ビヒクルまたは粒子の特異性は、腫瘍特異的結合体標的化分子による腫瘍組織標的化ならびに単にそれらが高分子量(>20000)であることに起因する腫瘍における高い透過性および保持性(EPR−効果)の両方を基礎にしている(40,41)。
【0014】
本特許出願で使用される骨格ポリ(リンゴ酸)(PMLAH)は、主鎖エステル結合を含み、生分解性であり(27およびその中の参考文献)そして高分子可撓性(49)であり、水(イオン化する場合)および有機溶媒(その酸性形態において)に溶解性であり、非毒性でありならびに非免疫原性(27およびその中の参考文献)である。薬物担持PMLAHは、誘導化されたリンゴ酸ラクトンの開環重合により主に合成されてきている(27およびその中の参考文献)。ドキソルビシン−ポリ(リンゴ酸)の合成は、化学的に合成されたポリ(β−D、L−リンゴ酸)から報告されている(49)。天然に存在するPMLAHからの薬物ビヒクルの合成は、行われなかった。本特許出願に記載する高度に機能的な薬物送達システムの種類は、以前に開示されたことは無かった。
【0015】
上記キャリアは、分子主鎖または分子骨格に対応するポリ(β−L−リンゴ酸)(PMLA)から構成され、それはそのカルボキシル基のところで、規定された比率で、以下の仕事を遂行する種々の機能的モジュールと化学的に結合される:(1)細胞質で有効になる放出可能な機能的モジュールによるプロドラッグの放出、(2)細胞(例えば、モノクローナル抗体(mAB))の表面に結合することにより特定の組織の方へキャリアを向けること、(3)エンドソームを通っての標的細胞への内在化(通常標的表面レセプターの内在化による)、(4)エンドソーム膜へ集積し、最終的にはエンドソーム膜を崩壊させる疎水性機能的単位の力でエンドソームから細胞質へ脱出し、リソソームへの過程でのエンドソームの酸性化の間に有効になること、(5)分解性酵素活性(例えばペプチダーゼ、プロテアーゼなど)に対してポリエチレングリコール(PEG)による保護。
【0016】
本発明において「モジュール」とは、小さい薬物分子または発色団分子から、例えば、抗体またはレシチンのような完全なタンパク質分子までの範囲にわたる生物学的活性分子構造である。本明細書に示される実施例の場合に、(1)ラミニン−8(34、51)のα−4鎖およびβ−1鎖に対するモルホリノアンチセンスオリゴヌクレオチドが示されており、それはそれらの3’−末端に人工的に導入された−NH2(アミノ)基によるアミド結合により介在スペーサーに結合されている。そのスペーサーは、細胞質の還元環境中でグルタチオンとのスルフヒドリル−ジスルフィド交換反応において切断可能なジスルフィド部分により、キャリアと連結されている(51、52)。(2)組織標的化は、モノクローナル抗体(mAB)を用いてラットトランスフェリンレセプターを認識し、そして結合するよう設計される。このレセプターは、血液脳関門(BBB)として機能する内皮細胞表面上で発現し、および特定の腫瘍では高いレベルで発現することが分かった(53、54)。インビトロおよびインビボの研究は、トランスフェリンレセプターがトランスフェリンまたはmAB OX−26または他の適切なmABと化学的に結合した薬物送達システムのための固定具として使用され得ることを示し、その抗体はトランスフェリンレセプターと結合し、そしてそれにより抗体のアロタイプに依存してラットまたはマウスまたは他の哺乳動物の血液脳関門(BBB)を通ってトランスサイトーシスを達成することを示す(5、56、57;45、58、59、60、および61)。(3)トランスフェリンレセプターと結合する抗体およびエンドソームへの内在化は、実証されている(57、55、および57)。トランスフェリンレセプターの場合、任意の適切な抗体、mAB、ヒト化抗体またはキメラ抗体またはレクチンまたはトランスフェリンレセプターに特異的な他のリガンドが使用され得ることは理解される。多数の細胞表面レセプターまたは抗原に対する適切なリガンドは、本発明に使用され得、そしてトランスフェリンレセプターは単なる例であることもまた、理解される。(4)エンドソームからの脱出は、ポリアクリル酸誘導体の場合、エンドソーム小胞からリソソームへと成熟化の間の酸性化により機能することが示された(51、62)。本特許出願で提案されている設計されたキャリアは、アミド結合によりポリリンゴ酸骨格と結合されている多数のバリン残基を担持する。リソソームへの過程の間の上記エンドソームの酸性化の間、上記キャリア分子のこれらの伸縮は、電荷が中和されそして疎水性になり、膜を崩壊させることができる。リソソームpHで電荷が中和される限り、他の分子が、バリンに代わって使用され得る。(5)PEG化は結合タンパク質の半減期を著しく増加させる(63)。循環時間を延長し、そして標的固体腫瘍の中への滲出を増進する(64)。半減期を増加させることが知られている任意の他の分子は、本発明に使用される。
例えば、本発明は以下を提供する。
(項目1)
薬物送達分子であって:
複数の遊離カルボン酸基を有するポリマー化したカルボン酸分子骨格;
複数の生物学的に活性な分子モジュールであり、それぞれ同じポリマー化したカルボン酸分子骨格に共有結合している、活性分子モジュールを含み、ここで該活性モジュールは:
標的化細胞により細胞性取り込みを促進する、少なくとも1つの標的化モジュール;および
標的化細胞の細胞性代謝を変える、少なくとも1つのプロドラッグモジュールを含む、薬物送達分子。
(項目2)
前記プロドラッグが、腫瘍特異的タンパク質の発現を阻害するように選択される、項目1に記載の薬物送達分子。
(項目3)
前記ポリマー化カルボン酸分子骨格がポリ(β−L−リンゴ酸)である、項目1に記載の薬物送達分子。
(項目4)
前記ポリ(β−L−リンゴ酸)が、2,500と100,000との間の分子量を有する、項目3に記載の薬物送達分子。
(項目5)
前記ポリ(β−L−リンゴ酸)が、少なくとも約5,000の分子量を有する、項目4に記載の薬物送達分子。
(項目6)
前記ポリマー化カルボン酸分子骨格のそれぞれの分子が、少なくとも約50の遊離カルボン酸基を有する、項目1に記載の薬物送達分子。
(項目7)
前記複数の分子モジュールが、生物膜の崩壊を促進する分子モジュールをさらに含む、項目1に記載の薬物送達分子。
(項目8)
項目7に記載の薬物送達分子であって、生物膜の崩壊を促進する前記分子モジュールが、生理的pHでは荷電し、リソソームのpHで荷電せず、それにより該分子モジュールの親油性を増加する親油性特性および親油性基を有する分子を含む、薬物送達分子。
(項目9)
前記複数の活性分子モジュールが、前記薬物送達分子の循環を延長する分子モジュールをさらに含む、項目1に記載の薬物送達分子。
(項目10)
項目9に記載の薬物送達分子であって、該薬物送達分子の循環を延長する前記分子モジュールが、ポリエチレングリコールを含む、薬物送達分子。
(項目11)
項目1に記載の薬物送達分子であって、前記複数の活性分子モジュールが、該薬物送達分子の細胞性取り込みを決定するレポータモジュールをさらに含む、薬物送達分子。
(項目12)
前記レポータモジュールが蛍光分子を含む、項目11に記載の薬物送達分子。
(項目13)
前記標的化分子が、血液脳関門の透過を促進するように選択される、項目1に記載の薬物送達分子。
(項目14)
前記標的化分子モジュールが抗体を含む、項目1に記載の薬物送達分子。
(項目15)
前記抗体が、トランスフェリンレセプターに結合する、項目14に記載の薬物送達分子。
(項目16)
前記抗体が、モノクローナル抗体である、項目14に記載の薬物送達分子。
(項目17)
前記抗体がヒト化抗体またはキメラ抗体である、項目14に記載の薬物送達分子。
(項目18)
項目1に記載の薬物送達分子であって、ここで前記プロドラッグ分子モジュールが、該薬物送達分子が細胞に入るときに切断される切断可能な結合で、前記ポリマー化カルボン酸分子骨格と結合される、薬物送達分子。
(項目19)
前記切断可能な結合が、ジスルフィド結合である、項目18に記載の薬物送達分子。
(項目20)
前記プロドラッグ分子モジュールが、アンチセンス分子を含む、項目1に記載の薬物送達分子。
(項目21)
前記アンチセンス分子がモルホリノアンチセンス分子である、項目20に記載の薬物送達分子。
(項目22)
前記アンチセンス分子が、ラミニン−8の産生を干渉する、項目20に記載の薬物送達分子。
(項目23)
項目22に記載の薬物送達分子であって、ここで前記アンチセンス分子が、α4ラミニンおよびβ1ラミニンからなる群より選択されるラミニンサブユニットの産生を変化させることによりラミニン−8の産生を干渉する、薬物送達分子。
(項目24)
薬物送達分子の合成方法であって、以下の工程:
複数の遊離カルボン酸基を有するポリマー化カルボン酸分子骨格を提供する工程;
該カルボキシル基を活性化する工程;
該活性化カルボキシル基をスルフヒドリル基およびアミノ基を含む化合物と反応させて、薬物送達分子にスルフヒドリル基を付けてスルフヒドリル薬物送達分子にする工程;
スルフヒドリル結合基を含む標的化分子を、該スルフヒドリル薬物送達分子と反応させて標的細胞による取り込みを促進する工程;ならびに
プロドラッグ分子を該標的細胞の細胞性代謝を変化させるために反応する工程を包含する、合成方法。
(項目25)
前記プロドラッグ分子が、スルフヒドリル結合基を含むアンチセンス分子である、項目24に記載の薬物送達分子の合成方法。
(項目26)
項目24に記載の薬物送達分子の合成方法であって、前記活性化カルボキシル基を、親油性部分を有し、エンドドームの酸性化の間に荷電しなくなる電荷基を含む分子と反応させ、それにより膜の崩壊を引き起こす工程をさらに含む、合成方法。
(項目27)
項目24に記載の薬物送達分子の合成方法であって、ここで複数の異なるプロドラッグ分子が、同じ薬物送達分子に結合し、それにより1を超えるプロドラッグ分子で、前記標的細胞を同時に処理することが可能になる、合成方法。
(項目28)
前記標的化分子が前記血液脳関門の透過を促進するよう選択される、項目24に記載の薬物送達分子の合成方法。
【図面の簡単な説明】
【0017】
【図1a】図1aは、本発明の薬物分子の全体的な構造を示す。
【図1b】図1bは、図1aの構造を構築するために使用される工程の全体的な順序を示す。
【図2】図2は、PMLA−NHSエステルの合成を示す略図である。
【図3】図3は、(PDP−モルホリノ)アンチセンスオリゴヌクレオチドの合成を示す略図である。
【図4】図4は、N−(フルオレセイン−5’−チオカルバモイル)ジアミノヘキサンの合成を示す略図である。
【図5】図5は、N,N’−ビス−(3−マレイミドプロピオニル)ポリ(エチレングリコール)の合成を示す略図である;
【図6】図6は、PMLA/L−バリン/2−メルカプトエチルアミン/mPEG5000−NH2結合体の合成を示す略図である。
【図7】図7は、PMLA/mAB OX−26/モルホリノアンチセンスオリゴヌクレオチド/L−バリン/2−メルカプトエチルアミン/mPEG5000−NH2結合体の合成を示す略図である。
【図8】図8は、FITCの、PMLA/mAB OX−26/モルホリノアンチセンスオリゴヌクレオチド/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体への結合を示す略図である。
【図9】図9は、ポリマーを試験する時に使用される溶血アッセイの結果の棒グラフを示す。
【図10a】図10aは、グルタチオンでの還元によるキャリアからのモルホリノオリゴの放出を示す。
【図10b】図10bは、グルタチオンでの還元に応答する時間の間のオリゴ放出の%を示す。
【図11】図11は、薬物2Aおよび/またはPBS(模倣)と比較して、有意差p<0.01でlog rank testにより分析された、薬物2による処置後のラットのKaplan−Meir生存曲線を示す;
【図12】図12は、培養神経膠腫U−87MG細胞における薬物2(30分)とマーカーエンドソームFM4−64との共分布である。共焦点顕微鏡により、FM4−64が細胞質エンドソーム(左上)に見られ、そして薬物2が同じ場所(右上)に見出される。両方の標識が共局在化する(左下、黄色)。
【図13】図13は、血管密度が正常脳と比較して腫瘍において増加し、そして薬物2が腫瘍血管密度を55%減少させることを示すグラフである。
【図14】図14は、U87/MG神経膠腫培養物のラミニン鎖に関する免疫蛍光染色を示す。PMLAビヒクル結合アンチセンスオリゴ(薬物2)は、ラミニン発現を抑制する(α4は赤色であり、β1は緑色である)。核は、DAPI(青色)で対比染色される。
【図15】図15は、ヒトラミニンβ1鎖に対するモノクロナル抗体を使用して異種移植された腫瘍の免疫蛍光分析を示す;ラミニンβ1鎖合成が、薬物2の投与後、GBMにおいて阻害された。
【図16】図16は、2つの多型膠芽腫(GBM)細胞培養物、U87MG、およびT98Gにおけるラミニン−8鎖発現の阻害のウェスタンブロッティング分析を示す:−、未処置、+、薬物2で処置。
【図17】図17は、薬物2が、上記BBBを通過することができることを示し、ここで赤色は、薬物の血管内投与の後の脳血管および移植腫瘍内で視覚化された薬物を示す。
【発明を実施するための形態】
【0018】
(発明の詳細な説明)
以下の説明は、当業者が発明物を作製し、そして利用することが可能なように提供され、発明を実施する発明者により企図された最良の様式を示す。しかしながら、種々の改変が、当業者には容易に明らかである。何故なら本発明の一般的な原理が、ポリ−L−リンゴ酸を基礎としたアンチセンス抗腫瘍薬物により実証された新規な薬物送達システムを提供するように本明細書に具体的に規定されているからである。
【0019】
薬剤および生物製剤のためのキャリアマトリックスまたは分子輸送ビヒクルとしてのPMLAの魅力的な性質は、以下の通りである:それは、非毒性でかつ非免疫原である;その疎水性は、疎水性側鎖またはスペーサーを導入することにより、調節され得る(30);それは、生分解性である(31);そしてそれは、血流で安定である。アンチセンスオリゴを特定の器官またはコンパートメントに標的化するために、例えば、レセプター媒介性エンドサイトーシスに有利な腫瘍特異的抗体のような標的化実体は、PMLAポリマーに結合され得る。理想的には、上記系は、分子輸送ビヒクルからその薬物を放出するための放出システムを含み;あり得る放出システムとしては:a)細胞内グルタチオンにより切断可能なジスルフィド結合、b)pH感受性ヒドラゾン結合、c)リソソームカテプシンB(または他のペプチダーゼ)(その活性は、種々の腫瘍において増加する)により切断されるテトラペプチド、;d)エンドソームからの本来の放出機能(32,33);およびe)エステル結合のような他の不安定な、または切断可能な結合、が挙げられる。最も重要なことに、多数の分子標的インヒビターが、1つのPMLA分子に容易に連結され得る。
【0020】
天然物、Physarum polycephalumの変形体(27およびその中の参考文献)由来のPMLAは、本特許出願に記載の薬物送達システムの合成のための出発物質であった。ここで用いられる化学合成の方法は、合成化学における一般的な公知の(fundus)方法からであり、ここで記載されているポリリンゴ酸ベースのシステムとは関係なく、他の系において記載されている。これらの方法の殆どは、本状況に適合されなければならず、特にキャリア系の化学的構築の進行の間の、出発物質の性質に適合されなければならず、そして生成物の精製方法に関しても適合されなければならなかった。上記ポリリンゴ酸骨格に結合される機能的部分の予測可能でかつ再現性のある化学量論的に誘導体化を成功裏に達成するために、上記骨格との反応の順序を、制御されていない反応は不可能であるような方法で確立および構築しなければならなかった。生成物の有効性およびその純度が達成され、段階的な合成の間に、定性化学アッセイおよび定量化学アッセイ、高速液体クロマトグラフィー(HPLC)、薄層クロマトグラフィー(TLC)、および紫外光/可視光/IR光スペクトル分析ならびにNMRスペクトル方法を含むインシチュ分析で制御された。完全に構築された薬物ビヒクルの膜崩壊的性質およびその合成の中間体の膜崩壊的性質もまた、慣用的で実験的な溶血膜アッセイ(51を参照のこと)により評価された。
【0021】
ここで使用された方法の開発を基礎とした標準的な合成方法参考は、特に以下である:
(69)の方法に類似する方法での、PMLAH−カルボキシル基のN−ヒドロキシスクシンイミド(NHS)エステルとしての活性化;
(51およびその中の参考文献)に記載の方法と類似の方法での、(2−ピリジルジチオ)プロピオニル基の、モルホリノ(PDP)−モルホリノ)アンチセンスオリゴヌクレオチドへの結合;
(71)のN−(フルオロセイン−5’−チオカルバミル)ジアミノヘキサンの形成の方法と類似の方法での、FITC(フルオロセインイソチオシアナート)−結合;
(70)に記載の方法と類似の方法での、N,N’−ビス−(3−マレイミドプロピオニル)ポリ(エチレングリコ−ル)の合成;
(72、51およびその中の参考文献)に記載の技術と類似の技術での、チオール基の抗体への導入;
(52および69)で実施された反応に類似して、mAB OX−26−スルフヒドリルとN,N’−ビス−(3−マレイミドプロピオニル)−PEGジアミド結合体との反応が行われた;
本明細書に記載されるようにNHS活性化されたカルボキシレートからのアミド形成により、PMLA/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体の合成が原理的に行われた;
スルフヒドリル基とマレイミドとの反応によるPMLA/mAB OX−26/モルホリノアンチセンスオリゴヌクレオチド/L−バリン/2−メルカプトエチルアミン/mPEG5000−NH2結合体の合成;
本明細書に記載されるようにNHS活性化されたカルボキシレートの求核攻撃により主にアミド形成を表す、FITC−スペーサーの、PMLA/mAB OX−26/モルホリノアンチセンスオリゴヌクレオチド/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体への結合。
【0022】
既存の薬物送達システムは以下の1つから数個の問題に悩まされる:
既存の薬物送達システムは多機能的ではなく、即ち、キャリア1分子あたりの組織標的化基の種類および量における可変性に関して限定される;
既存の薬物送達システムは、キャリア1分子あたりの結合薬物(プロドラッグ)の種類と量により限定される。
【0023】
既存の薬物送達システムは、生理学的流体中への溶解度により限定される。
【0024】
既存の薬物送達システムは、循環系における分解に対する不十分な安定性により限定される。
【0025】
既存の薬物送達システムは、生分解性ではない;
既存の薬物送達システムは、ウイルス核酸または他のウイルスフラグメントを含む;
既存の薬物送達システムは、腫瘍組織に特異的ではなく健全な宿主組織を損傷する;
既存の薬物送達システムは、毒性がある;
薬物送達システムの合成は、制御不可能な副反応に悩まされる;
薬物送達システムの合成は、反応生成物の精製を困難もしくは不可能にする溶解度または他の問題に悩まされる;
薬物送達システムの合成は、再現性のある結果が得られない;
新成分を含有し、従って薬物送達システムの特異性を増進し、薬物送達システムの抗腫瘍活性を強くする、構造的変更物/拡大物の合成は不可能である;
薬物送達システムの合成はスケールアップが容易にできない。
【0026】
各反応性機能的モジュールの、ポリリンゴ酸骨格のNHS(N−ヒドロキシスクシンイミド)−活性化カルボキシル基との制御された結合は、種々の異なる種類の反応性機能的モジュールを結合させることを可能にし、従って種々の異なる標的化分子、薬物(プロドラッグ)分子などを導入できる。種々のモジュールは、全く同一の骨格分子に結合され得るか、または2成分もしくは3成分などの薬物混成体を作製可能なように、または異なる骨格分子へ結合され得る。全く同一の骨格分子上の多機能性モジュールは、細胞に同時に導入される場合生物学的に相乗効果を示し得る。
【0027】
生分解性は、骨格および他の生分解性構築単位(アミノ酸、タンパク質)として生分解性リンゴ酸を用いることで達成され得る。
【実施例】
【0028】
(特定の実施例による本発明の説明)
(モルホリノアンチセンスオリゴヌクレオチドの腫瘍標的送達のためのポリリンゴ酸ベースの多機能性キャリアシステムの合成)
図1aは、本発明の代表的な薬物分子の全体的な構造を示す。ブロック(z+w)の合成のために、ポリリンゴ酸のカルボキシル基(ブロックw)を、NHS−エステルとして活性化し、そしてアミド結合を介してL−バリンに結合する。ブロックy3は、ジスルフィド薬物放出単位を含むプロドラッグモルホリノアンチセンスオリゴである。ブロックy1は、ポリエチレングリコール(PEG)スペーサーを介してブロックz+wに結合しているモノクローナル抗体(OX26)標的化分子である。ブロックxは、分解に対するPEG−保護基であって、アミド結合により連結されており、市販されているPEG−アミンから形成される。ブロックy2は、残っているスルフヒドリル固定基を表し、この時点でそれは合成に消費されず、置換N−エチルマレイミドの2重結合との反応により更なる機能性モジュールの結合に使用され得るか、または単に置換N−エチルマレイミドとの反応でブロックされ得る。ブロックn(蛍光レポータ基)をフルオロセインイソチオシアネート(FITC)およびN1−Boc−1,6−ジアミノヘキサンから調製する。上記薬物構造を、注意深い化学量論的制御下で、図1bに示されるように成長中の結合体への、上記ブロック(すなわち、モジュール)の段階的結合により構築する。工程の順序は、異なるシナリオに合うように容易に調整され得る。カルボジイミド試薬を用いて有機溶媒、好ましくはジメチルホルムアミド中で、結合反応を実施した。以下の標準的な方法で側鎖の適切な保護により副反能を抑制する。
【0029】
ブロックwについては、非常に精製度の高いポリリンゴ酸を、Physarum polycephalumの培養物から得る(24、25)。バイオポリマーを同時にそして酵素的にL−リンゴ酸((31、26)に分解し、それは炭酸ガスおよび水へと代謝され。そのナトリウム塩はマウスおよびウサギのそれぞれで毒性も無く、免疫原性もない(27およびその中の参考文献)。マウスに静脈内投与した後、ポリリンゴ酸は、腎臓経由の排泄により速く除かれた(73)。特定のポリリンゴ酸誘導体およびブロックポリマーは、実際にラットにおいて骨修復および筋再生に対して正の効果をを示した(30)。または特定のポリリンゴ酸誘導体およびブロックポリマーは、他の研究(32)において生体適合性であることがわかった。ポリリンゴ酸は、非常に多くの修飾可能なカルボキシル基の故に薬物キャリアデバイスの設計には優れた候補である。これらは、それらの化学量論および集積度に関して完全に制御可能な様式で種々の異なる生物学的に活性な分子に、容易に結合され得る。ブロックzは、L−バリンまたはL−ロイシンのような親油性基を含むポリマーを基礎にし、そしてエンドソームのリソソームへの成熟の間に周囲のpHが6以下に低下する場合、プロトン化され益々親油性になる。この増加する親油性で上記エンドソームの膜は漏れやすくなり、細胞質への高分子内容物の放出を生じる(33、74、75)。ブロックy3は、ジスルフィド結合を含有し、それは脳毛細血管を含む血液循環においては安定であるが、細胞の還元的環境においては切断される(38)。ブロックy1は、上記組織標的部分が標的細胞表面のレセプターに結合する事を可能にするポリエチレングリコールスペーサーを含む。それはまた、標的化ポリペプチドの分解に対しても保護する。(y3)例えばラミニンのα1鎖のような腫瘍必須遺伝子の発現を特異的にブロックするモルホリノオリゴヌクレオチドが、使用される。原則的に、任意の他の薬物またはプロドラッグがここでおよび単一キャリア分子上の異なる薬物のアレイで結合され得る。これらの結合体は、細胞質内で薬物放出単位でキャリアから切断されそして薬物(単数または複数)が有効になる。ブロックy1は、上記BBBの内皮細胞上のトランスフェリンレセプターに対するモノクローナル抗体により標的化されるBBBを突破し易くする(55)。ブラジキニン単独または他の分子と結合されたブラジキニンは、特異的レセプターに基づいて脳腫瘍に使用されるべき標的化分子になり得る(77、18および78)。脳腫瘍特異的標的化に関する更なる可能性は、ヒトEGFレセプターに対するモノクローナル抗体を使用することである(37,79)。ブラジキニンB2レセプターおよびEGFRは、腫瘍細胞上で過剰発現し、トランスフェリンレセプターとの組合わせで、脳腫瘍標的化部位として使用され得る。ブロックnは、任意の蛍光標識(ここではフルオロセイン)を付加し、受容体の腫瘍細胞のエンドソームにおいて上記キャリアのホーミング研究を容易にする薬物構造に結合する。
【0030】
(材料および方法)
ポリ(β−L−リンゴ酸)(PMLA)を、(25)から開発された方法を使用して培養Physarum polycephalum変形体のブロスから精製した。塩の形態の上記ポリマーをSephadex G25カラムでサイズ分画をした。数平均分子量50kDa(多分散度1.2)を有する画分をAmberlite IR−120(H+形態)を通して遊離酸ポリマー(PMLA−H)に変換しキャリア合成に使用するまで凍結乾燥で保存した。D2O中の1H−NMRは以下のδ値:3.3ppm(d、ポリエステル主鎖のメチレンのプロトン)、5.3ppm(t、ポリエステル主鎖のメチンのプロトン)、を示した。プロトン−ブロード−バンド−デカップル(Proton−broad−band−decoupled)13C−NMRは、以下のδ値:178.4ppm(−COOH)、74.5ppm(−CHOH−)、38.9ppm(−CH2−)および174.5ppm(−CO−)を示した。精製PMLA−Hは、220nm以下のみの波長にUV光吸収を示し、核酸およびタンパク質のそれぞれの代表的な260および280での吸収はない(更なる詳細は27に概説されている)。ラミニン−8のα−4鎖(AGC−TCA−AAG−GCA−TTT−CTC−CGC−TGA−C)およびラミニン−8のβ−1鎖(CTA−GCA−ACT−GGA−GAA−GCC−CCA−TGC−C)(50、34)に対するモルホリノTM−3’−NH2アンチセンスオリゴヌクレオチド((6)をGene Tools(USA)より購入した。10mMアジ化ナトリウムを含有する1mg/ml PBSの濃度におけるラットトランスフェリンレセプターCD71に対するマウスモノクローナル抗体(クローンOX−26、アイソタイプIgG2a)をChemicon Europe(UK)から得た。マウスIgG2a,κ(UPC10)をSigma(ドイツ)から購入した。クロマトグラフィーで純粋なmPEG−アミン(分子量 5000)およびアミン−PEG−アミン(分子量 3400)をNektar Therapeutics(USA)から得た。Merck(ドイツ)、Sigma(ドイツ)Pierce(USA)から得た試薬および溶媒は、入手可能な最高純度のものであった。ジクロロメタン(DCM)およびN,N−ジメチルホルムアミド(DMF)を、モレキュラーシーブ(0.4nm)で乾燥した。
【0031】
1H−NMRスペクトルをBruker Model DMX−500フーリエ変換スペクトル計に記録し、化学シフトを、TMSを内部標準として相対的にppm(δ)で示す。13C NMRスペクトルを125.8MHzで操作する同じスペクトル計で記録した。クロマトグラフ的な分離を、UVおよび蛍光の検出器を備え付けたMerck−Hitachi分析用LaChrom D−7000 HPLC系で実施した。1.5ml/分の流速で0.1%TFA(トリフルオロ酢酸)水溶液−0.07%TFAアセトニトリル溶液の2勾配のMacherey & Nagel C18−Nucleosil逆相(RP)カラム(250×4mm)、または流速0.75ml/分で50mMのリン酸ナトリウム緩衝液pH7.4を用いるサイズ排除カラムBio−Sil SEC250−5(5μm、300×7.8mm)のいずれかを使用した。ポリマーのNa塩またはK塩の分子量を、規定された分子量の標準ポリスチレンスルホン酸塩(Machery−Nagel)を用いてSEC−HPLCで決定した。薄層クロマトグラフィー(TLC)をMerckの予めコートされているシリカゲル60 F254アルミニウムシート上で実施した。溶出液は、n−ブタノール、水および酢酸(容量比ベースで4:2:1)を含有した。
【0032】
(合成)
(PMLA−NHSエステルの合成)
1.16gのPMLA−H(リンゴ酸モノマーに関して10mmol)を、30mlの無水ジメチルホルムアミド(DMF)に溶解した。N−ヒドロキシスクシンイミド(NHS)(15mmol)を無水ジメチルホルムアミド(DMF)(10m)に溶解し、これを、PMLA−H溶液に添加した。温度を氷浴で0℃まで下げて、その後10mlのDMFに溶解したジシクロヘキシルカルボジイミド(DCC)(15mmol)を添加した。その反応混合物を、気体の泡が発生しなくなるまで減圧下、室温で保持した。0℃で30分後に、その反応混合物を室温で48時間攪拌した。上記反応混合物を、上述のように減圧下で保持し、反応の最初の日の間は2時間ごとに、その後、反応の2日目の間は6時間ごとにインキュベーションをした。反応の2日後、ジシクロヘキシル尿素をろ過で除去し、そしてその反応容量を減圧下エバポレーションで減少させた。新鮮な無水DMF(10ml)を添加し、残りのジシクロヘキシル尿素を再び濾過により除去した。透明な反応混合物を、室温で12時間攪拌し、ジシクロヘキシル尿素の残量(もしあれば)を、ろ過で除去した。その容量を1〜3mlにまで、減圧下エバポレーションにより減少させ、そしてその生成物を酢酸エチルを加えて沈殿させた。その薄黄色の生成物(P1)を、ろ過で収集した。ジエチルエーテルを、最終比率1:1(酢酸エチル:ジエチルエーテル)を合わせるように、ろ液に加え、そしてさらなる明るい褐色の生成物(P2)を、ろ過で収集した。次いで、n−ヘキサンを、最終比率が1:1:1(酢酸エチル:ジエチルエーテル:n−ヘキサン)になるようにろ液に加えて、さらなる褐色の生成物(P3)をろ過で収集した。上記沈殿物を、それらの沈殿に使用したのと同じ溶媒に分散させ、低温(−20℃)で一晩放置した。その生成物を濾過して、同じ冷溶媒で繰り返し洗浄した。その生成物を、DMFを溶出液として重力で流してSephadex LH20を通すことによりさらに精製した。生成物含有画分を、収集しそして溶媒を減圧下、エバポレートさせた。最後にその生成物をジエチルエーテルに分散し、ろ過により収集し、真空で乾燥し、−20℃で保存した。
【0033】
PMLA−NHSエステルのこれらの調製物の純度/組成を、1H−NMRおよびUV−VIS分光法ににより分析した。NHS基の含量を、NHSエステル基のn−ブチルアミンでのアミノリシス後に決定した。10mgのPMLA−NHSエステルを、0.5mlのDMFに溶解した。0.5mlの10% n−ブチルアミンの1部分をこの溶液に加え、そしてその反応混合物を、室温で30分間インキュベートした。遠心分離後、20μLのサンプルを、80μLの水と混合し、水/0.1%(v/v)TFAを溶出液として用いてPR−HPLCにより分析した。NHS基を、260nmの吸光度でモニターした。それらの含量は、既知量のN−ヒドロキシスクシンイミド標準物質の吸光度と比較して計算した。PMLA−NHSエステルサンプル中のリンゴ酸残基とNHS基とのモル比を、これらの結果と、1H−NMRで測定したマリル残基の量と合わせて計算した。代表的には、その比はP1、P2、およびP3に対してそれぞれ35%、59%および85%であった。(CD3)2SO中での1H−NMRは、以下のδ値:2.8ppm(s、4H、N−CO−CH2−)、3.35ppm(d、上記ポリエステル主鎖のメチレンのプロトン)、5.85ppm(t、ポリエステル主鎖のメチンプロトン)を示した。上記反応は、図2に示される。
【0034】
((2−ピリジルジチオ)プロピオニル−モルホリノ(PDP−モルホリノ)アンチセンスオリゴヌクレオチドの合成)
モルホリノ−3(−NH2アンチセンスオリゴマー(1μmol)を、900μLのDMFおよび100μLの脱イオン水の混合物の中に溶解した。この混合物の中に、20μLの100mMのN−スクシンイミジル3−(2−ピリジルジチオ)プロピオネート(SPDP)のDMF溶液を加え室温で2時間放置した。その溶媒を、減圧下、室温でロータリーエバポレーターで除去した。その残渣を、10mM EDTAを含有する1mlの緩衝液A(0.1Mリン酸ナトリウム、0.15M NaCl、pH7.2)に溶解し、緩衝液Aで予め平衡化したSephadex G−25マイクロスピンカラムで精製した。PDP−モルホリノアンチセンスオリゴヌクレオチドの濃度を、1mMに調整して−20℃で保存した。
【0035】
その生成物の純度を、TLCおよびUV分光法によりNHSおよびSPDPの不存在を示して確認した。PDP基の含量を、以下のようなジスルフィド還元後の2−チオピリドンの濃度を測定して決定した:PDP−モルホリノアンチセンスオリゴヌクレオチドを、0.1M トリス緩衝液pH9.0中の0.2Mのジチオトレイトール(DTT)と室温で30分間インキュベートした。その反応混合物を、最初10分間蒸留水で洗浄し、次いで30分間、0〜60%のアセトニトリルの勾配で溶出することによりRP−HPLCにかけた。その反応生成物2−チオピリドンを、341nmのUV吸光度で検出した。2−チオピリドンの濃度を、還元された2−アルドリチオール(DPDS)の既知量の吸光度を標準として使用して測定した。PDP−モルホリノアンチセンスヌクレオチドの収率は、出発量のモルホリノ−3’−NH2オリゴヌクレオチドの80%より通常は高かった。この反応は、図3に示される。
【0036】
(N−(フルオレセイン−5’−チオカルバモイル)ジアミノヘキサンの合成)
フルオロセインイソチオシアネートアイソマーI(90mg)(FITC、最小98%、0.23mmol)を、3ml DMFに溶解し、そして76mgのN1−Boc−1,6−ジアミノヘキサン塩酸塩(0.3mmol)を添加した。結合反応を0.6mmolのトリエチルアミンを滴下することにより始めた。その反応混合物を、室温で2時間インキュベートし、その容量を、減圧下エバポレーションにより減少させた(最終容量は、約0.5ml)。冷水(5ml)を、残っている混合物に加え、そして1N HClで酸性化した。その沈殿を遠心分離で収集し、3回冷水で洗浄し、その後遠心分離し、上清にN1−Boc−1,6−ジアミノヘキサンがTLCおよびニンヒドリン試験により検出されなくなった。上記最終生成物をP2O5で乾燥した。この合成は図4に示される。
【0037】
そのBoc保護基を除去するために、その乾燥生成物を3mlのジクロロメタン(DCM)に溶解し、温度を氷浴で低下させた。2mlのTFAを、その溶液に加え、次いでこれを氷上で30分間、攪拌した。その反応を、TLCで追跡した。蛍光スポットは、UV光で見えた。上記溶媒を減圧下でエバポレートし、蝋状生成物をアセトンに溶解しそしてジエチルエーテルの添加により沈殿させた。精製のために上記生成物を、100mlの混合物の中に4mlの酢酸を含有する3mlのDCM/エタノール(3:2,v/v)に溶解し、同じ溶媒で平衡化した2cm×12cm SiO2カラムに通した。生成物はTLCによると純粋であった。そのRf値は、FITCに対しては、0.95であり、N1−(フルオレセイン−5’−チオカルバモイル)−N6−BOC−1,6−ジアミノヘキサンに対しては0.98であり、N−(フルオレセイン−5’−チオカルバモイル)ジアミノヘキサンに対しては0.64であった。
【0038】
(N,N’−ビス−(3−マレイミドプロピオニル)ポリ(エチレングリコール)の合成)
3mLの無水DMFに溶解した0.5gのNH2−PEG3400−NH2(0.147mmol)を、5mlの無水DMFに溶解した(3−マレイミドプロピオン酸NHSエステル)(106mg、0.4mmol)に室温で激しく攪拌しながら滴下した。上記反応の完結を、TLCおよびニンヒドリン試験陰性により確認した。室温で2時間インキュベーション後、その溶媒を減圧下、室温でロータリーエバポレータで除去した。生成物を、10mM EDTAを含有する2mlの緩衝液A(0.1Mリン酸ナトリウム、0.15M NaCl、pH7.2)に溶解した。不溶性の不純物を、遠心分離で除去した。透明な上清を、予め緩衝液Aで平衡化したSephadex G25カラムに通した。上記生生物はTLCおよびニンヒドリン試験では純粋であった。上記生成物の水溶液を、−20℃で保存した。
【0039】
(CD3)2SOに溶解した生成物の1H−NMRスペクトルは、以下のδ値:7.05ppm、s、4H、−CH=CH−;3.74ppm、t、2H N−CH2;3.5、s、PEGからの水素;3.03、t、2H CH2−CONHを示した。上記の値は、予測されるN,N’−ビス−(3−マレイミドプロピオニル)ポリ(エチレングリコール)ジアミドである生成物と一致した。マレイミド基の含量を、−HC=CH−と所定量の2−メルカプトエチルアミン(2−MEA)のスルフヒドリル基との反応に依存する方法および未反応スルフヒドリル基の、5,5’−ジチオビス−2−ニトロベンゾエート(DTNB,Ellman試薬)での以下のような滴定により、間接的に測定した。水中の適切な量の2−MEAを、N,N’−ビス−(3−マレイミドプロピオニル)ポリ(エチレングリコール)ジアミドの水溶液に加え、室温で30分間インキュベートした。DTNB(25μLの10mg/mLのエタノール溶液)を加え、そして室温で10分間インキュベートした後、412nmの吸光度を読んだ。その吸光度は、既知の量の2−MEAで標準化し、未反応スルフヒドリル基の量を計算した。N,N’−ビス−(3−マレイミドプロピオニル)ポリ(エチレングリコール)ジアミドのサンプル中のマレイミド基の含量を、2−MEAの量を最初の量から未反応2−MEAの引き算することにより計算した。この値からN,N’−ビス−(3−マレイミドプロピオニル)−ポリ(エチレングリコール)ジアミドの合成の収率は、65%と計算された。この合成は、図5に示される。
【0040】
(抗体へのチオール基の導入:固有のジスルフィド結合の、2−メルカプトエチルアミン(2−MEA)による還元)
ラットトランスフェリンレセプターCD71(クローンOX−26、アイソタイプIgG2a)に対するマウスモノクローナル抗体(mAB)を、10mMのアジ化ナトリウムを含有する1mg/mlのPBSの濃度の市販品を入手した。mAB OX−26の代わりにマウスモノクローナル抗体IgG2a,κ(UPC 10、Sigma)を使用し、コントロール結合体およびタンパク質測定用の標準曲線を作製そた。上記mAB OX26溶液をミクロ遠心膜フィルター(Sigma Ultrafree−CLミクロ遠心膜フィルター、再生セルロース、カットオフ30kDa)を使用して、4℃、5000×gで濃縮した。上記mAB貯蔵緩衝液を、10mM EDTAを含有する緩衝液A(0.1Mリン酸ナトリウム、0.15M NaCl、pH7.2)に変えた。mABの濃度を3〜5mg/mlに調整した。マウスmAB UPC 10を、緩衝液Aに溶解し、mAB
OX26と同じ濃度にした。固体の2−メルカプトエチルアミン塩酸塩(2−MEA)(6mg/ml)を、抗体溶液の中に攪拌し、その混合物を37℃で90分間インキュベートした。そのジスルフィドが還元された抗体を、ミクロ遠心膜フィルター(再生セルロース、カットオフ30kDa)を使用して、4℃、5000×gで、緩衝液A(N2下で脱気)を用いて膜分離精製した。その工程を、5,5’−ジチオビス(2−ニトロ安息香酸)(DTNB,Ellman試薬)とのインキュベーションの後に412nmで分光光学的に測定して、膜分離精製物に2−MEAが完全に無くなるまで実施した。ジスルフィド還元抗体溶液中のチオール基の数を、pH8.0で0.5mlの抗体溶液と25μlのDTNB溶液(エタノール中10mg/ml)とを10分間インキュベートした後で、412nmの吸光度(25℃で、e=14.15×103M−1cm−1)を読み取って計算した。その抗体タンパク質の濃度を、(65)の方法に従って測定した。結果は、上記ジスルフィド還元抗体が、Ig分子1モルあたり3.1〜3.5のチオール基を含むことを示した。SEC−HPLC分析は、分子量150kDaの1つのピークのみを示し、これは上記還元抗体がその分子の集積度を維持していることを示した。還元剤が不存在でさえも、EDTAの存在下では上記調製物はむしろ安定であって、そして遊離のチオール基の有意な損失は4℃で一晩では観察されなかった。
【0041】
(mAB OX−26/N,N’−ビス−(3−マレイミドプロピオニル)−PEGジアミド結合体の合成)
調製直後に、還元抗体の溶液を攪拌しているN,N’−ビス−(3−マレイミドプロピオニル)−PEGジアミド(抗体のチオール基に対して50倍のモル過剰なマレイミド基)の水溶液に、室温で滴下した。その反応混合物を、室温で2時間インキュベートし、未反応のN,N’−ビス−(3−マレイミドプロピオニル)−PEGジアミドを、緩衝液A(50kD膜、一晩で緩衝液交換3回)の中で透析により除去した。抗体−N,N’−ビス−(3−マレイミドプロピオニル)−PEGジアミド結合体の調製物を、さらなる合成反応のために直ちに使用した。
【0042】
上記結合反応の完結をSEC−HPLCの結果で示した。220nmで吸光度を測定している場合、溶出した物質のみが、結合体をあらわす7.20〜7.26分の位置にあり、一方、未反応N,N’−ビス−(3−マレイミドプロピオニル)−PEGジアミドに関する13.4〜13.7分の位置には、何も溶出しなかった。上記抗体結合体のマレイミド基の含量を、N,N’−ビス−(3−マレイミドプロピオニル)−PEGジアミドの合成に関する上述の2−MEAおよびEllman試薬を使用して間接的に決定した。抗体タンパク質の濃度を、(65)の方法に従って決定した。その結果は、抗体1分子あたり3.0のマレイミド基を示し、そしてジスルフィド還元抗体調製物の遊離のチオール基が、完全にN,N’−ビス−(3−マレイミドプロピオニル)−PEGジアミドと反応したとする仮定と一致した。さらに、SEC−HPLC分析は、分子量150kDaの1つのピークのみを示したので、架橋生成物は除外され得る。
【0043】
(PMLA/L−バリン/2−メルカプトエチルアミン/mPEG5000−NH2結合体の合成)
PMLA/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体の合成のために、1mmol(マリル単位に関して)のPMLA−NHSエステル(調製物P3:85%NHSエステル)を、10mlの無水DMFに溶解した。最初に、mPEG5000−NH2(2mL DMF中50μmol、NHS活性化マリル単位の5モル%に相当する)および200μmolのN−エチルモルホリンを、この順で加え、そして上記混合物を室温で30分、反応がTLCとニンヒドリン試験(原点でのニンヒドリン反応陽性に対して反応陰性)により完結するまで攪拌した。次にDMF中の200μmolの50mMの2−MEA(NHS−活性化マリル基の20モル%に相当する)のDMF溶液と200μmolのN−エチルモルホリンを、上記反応混合物に加えて、室温で30分間攪拌した。再び上記反応をTLC(2−MEAに対してRf=0.27、ポリマー結合体に対してRf=0)とニンヒドリン試験とで完結させた。この合成は、図6に示される。
【0044】
添加試薬の化学量論に従うと、PMLA−NHSエステル当量の4.5%および19.3%が、それぞれPEGおよび2−MEAに置き換えられた。残存する未反応NHSエステル当量を、L−バリン(5mlの水に1mmol)に結合させ、0.1gのNaHCO3(1.2mmol)の存在下で滴下した。その反応混合物を、室温で1時間攪拌し、冷却しながら0.1M HClで中和した。その溶媒を、減圧下30℃でエバポレートさせた。乾燥生成物を10mM EDTAおよび1mlの0.5MのDTTを含有する10mlの緩衝液A(0.1Mリン酸ナトリウム、0.15M NaCl、pH7.2)に溶解した。室温で10分後、その混合物を20000×gで、5分間遠心分離し、透明な上清を、予め緩衝液Aで平衡化したSephadex G25カラム(2.5cm×60cm)を通し、そして生成物含有画分を凍結乾燥した。
【0045】
PMLA/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体の組成を1H−NMRおよびUV−VIS分光光学分析により分析した。チオール基の含量を、1mlのリン酸ナトリウム緩衝液(pH8.0、100mM)に溶解した1mgの凍結乾燥結合体に25μlのDTNB溶液(エタノール中10mg/ml)を加え、室温で30分間インキュベーションした後412nmの波長での吸光度を読取ることにより測定した。PMLA/L−バリン/2−メルカプトエチルアミン/mPEG−NH2/FITC結合体の調製の場合(以下を参照のこと)、反応混合物はまた、ミクロ遠心分離膜フィルター(再生セルロース、5kDaカットオフ)を使用して、5000×gで膜分離精製し、412nmの吸光度を読んだ。チオールを分析するEllman方法は、チオールと発色性DTNB(5,5’−ジチオビス−2−ニトロ安息香酸、FW 396.4)との反応に基づいており、それにより黄色の5−チオ−2−ニトロ安息香酸(TNB)の形成が測定される。
【0046】
【化1】
FITC−結合体の場合のろ過の理由は、蛍光の存在がTNBの検出を不可能にするので、TNBをFITC結合体から分離するためである。そのろ過の間、TNBは膜を通過し、FITC−結合体が留まり、そして、ろ液の吸光度が測定される(上記ろ過は遊離の色素の除去のためではない。上記遊離の色素はこの反応の前に既に除去されている)。
【0047】
スルフヒドリル含量を、2−MEA標準に関して計算した。L−バリン部分の含量を、以下のようにトリニトロベンゼンスルホン酸(TNBS)アッセイを使用した加水分解およびRP−HPLCの後に、遊離のアミノ基を定量することにより決定した:1mgの上記結合体と30〜50μLの6N HClを、100μLキャピラリー試験管に入れた。封をしたきゃピラリーを100℃で12〜16時間、オーブンでインキュベートした。加水分解の後に、内容物をEppendorfチューブ(定量的にするためにそのキャピラリー試験管を水で濯ぐ)に移して、穏やかに加温することにより完全に乾固するまでエバポレートさせた。この物質を水に溶解して、そして遠心分離した。10〜30μlアリコートの上記上清を、300μLの重炭酸ナトリウム緩衝液(10mlの水に0.4gの重炭酸ナトリウム、pH8.5)に添加した。150μLの0.1%(w/v)TNBS水溶液の添加後に、上記混合物を37℃で30分間インキュベートした。遠心分離後、20μLのその反応混合物を30分の線型勾配(0〜10分、100%水、10〜40分、0〜60%アセトニトリル)を使用してRP−HPLCにより分離した。バリル部分の含量を既知量のL−バリンを標準として溶出物の340nmの吸光度を基礎にして計算した。mPEG:バリン:2−MEAのモル比を1H−NMRにより決定した。
【0048】
(PMLA/mAB OX−26/モルホリノアンチセンスオリゴヌクレオチド/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体の合成)
新しく調製されたmAB OX−26−PEG−マレイミドを、PMLA/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体に、4℃で、攪拌しながら滴下した。マレイミド基に対して100モル過剰の遊離チオール基(結合体)を用いて、全ての抗体が上記ポリマーと結合するようにした。30分のインキュベーション後、上記反応を完結した。上記反応の完結度をSEC−HPLCにより確認した。上記PMLA/mAB OX−26/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体を、ミクロ遠心分離膜フィルター(再生セルロース、分子量 100kDaカットオフ)を使用して、緩衝液Aの中で4℃、5000×gでの膜分離精製により精製した。mAB含有PMLA結合体は、フィルターに留められ、そしてタンパク質フリーのPMLA/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体のみがそのフィルターを通過した。上記膜分離精製を、SEC−HPLCで確認して膜分離精製物にタンパク質フリーののポリマー結合体の痕跡も無くなるまで繰り返した。
【0049】
PMLA/mAB OX−26/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体のタンパク質含量を、(66)の方法により測定した。非結合mAbの同じ量が、約10%高い吸光度を示し、従って、測定されたタンパク質含量を0.9のファクターで補正した(このファクターは、抗体が、膜分離精製で洩れないことが分かったが、留まった(結合)抗体は、原料抗体の90%しか達しなかった事実から実験的に引き出されたものであった)。この食い違いの理由は、不明である。タンパク質含有結合体の残存する遊離のチオール基の濃度を、上述の如く決定した。最初のチオール基の約70%が、まだ存在していたことを見出した。タンパク質含有結合体の濃度を、チオール基に関して3mMに調整した。
【0050】
次の工程で、ラミニン8のα4およびβ1鎖に対する5μmolのPDP−モルホリノアンチセンスオリゴヌクレオチド(5mlの1mM PDP−モルホリノアンチセンスオリゴヌクレオチド溶液)を、15μmol遊離のチオール基の濃度と等しくして、PMLA/mAB OX−26/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体(5mlの3mM タンパク質含有結合体溶液)の精製した溶液に、攪拌しながら滴下した。アンチセンスα4鎖:アンチセンスβ1鎖のモル比は、1:1であった。その反応混合物を4℃で一晩インキュベートした。上記反応の完結度を、260nmの吸収を有する溶出物の単一ピーク、および遊離のPDP−モルホリノアンチセンスオリゴヌクレオチドの位置における吸収の不存在を示すSEC−HPLCにより確認した。得られたPMLA/mAB OX−26/モルホリノアンチセンスオリゴヌクレオチド/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体を、ミクロ遠心分離膜フィルター(再生セルロース、分子量 100kDaカットオフ)を使用して、4℃、5000×gでの膜分離精製により精製した。この段階での遊離スルフヒドリル基の含量を、412nmで、Ellman試薬で決定した。アンチセンスモルホリノオリゴヌクレオチドの含量を、ジスルフィド基の50mM DTTによる37℃、2時間の還元およびSEC−HPLCによる分離後、260nmにおける吸光度により測定した。特に、その260nmの光吸収ピークを、還元されたPDP−モルホリノアンチセンスオリゴヌクレオチドを標準として得られるピーク比較した。
【0051】
PMLA/mAB OX−26/モルホリノアンチセンスオリゴヌクレオチド/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体のタンパク質含量を、(65)の方法で測定した。抗体:モルホリノアンチセンスオリゴヌクレオチドのモル比はは1:20〜1:26で変化したが、それでy1値が0.19%〜0.25%になる(全体の合成は、図7を参照のこと)。遊離のスルフヒドリル基を、N−エチルマレイミドでブロックし、未反応試薬を上のような膜分離精製により除去した。
【0052】
(FITCのPMLA/mAB OX−26/モルホリノアンチセンスオリゴヌクレオチド/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体への結合)
生物学的条件下での検出のために、PMLA/mAB OX−26/モルホリノアンチセンスオリゴヌクレオチド/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体を、フルオロセインイソチオシアネート(FITC)で、共有結合的に標識化した。この標識は、しかしながら、上で示されたPEG−NH2のPMLA−NHSエステルへの結合の後に導入された。N−(フルオレセイン−5’−チオカルバモイル)ジアミノ−ヘキサンの溶液を、DMFおよびPBS(1:1)の混合物中で最終濃度が25mMになるように調製した。mPEG−NH2/PMLA結合体の溶液(PMLA−NHSエステルのマリル単位に対して1mmol、調製P3:85% NHSエステル)に、25μmolのN−(フルオレセイン−5’−チオカルバモイル)ジアミノ−ヘキサン(図7のn=2.5%)および100μmolのN−エチレンモルホリンを加えて、それからその混合物を室温で30分間インキュベートした。その反応をTLCで追跡した。反応完結後に、蛍光を原点のみにおいて検出した。FITCのPMLA/mAB OX−26/モルホリノアンチセンスオリゴヌクレオチド/L−バリン/2−メルカプトエチルアミン/mPEG−NH2結合体への構築については、色素の不存在下、上述と同じ経路に従った。上記結合体のFITC含量を、490nmでのFITC部分の吸収により測定した。FITC−担持結合体もまた、447nmにセットした励起波長および514nmにセットした発光波長でMerck−Hitach蛍光検出計により検出した。FITCの含量を、上述の如くポリマーを担持する変化する量のFITCの定量的な結合により形成した標準サンプルの吸光度または蛍光とサンプルの吸光度または蛍光を比較して計算した。上記結合体を、直接細胞培養実験に使用した。この合成は図8に示される。
【0053】
(溶血アッセイ)
図9は、赤血球(RBC)溶血アッセイを使用して測定したポリマーの膜崩壊活性を示す。新鮮なヒトRBCを、ヒト全血を2000gで5分間遠心分離することにより単離した。上記RBCを所望のpH(pH5.85またはpH5.5)の冷100mMリン酸ナトリウム緩衝液で3回洗浄した。最終ペレットを同じ緩衝液に再懸濁して、1mlあたり108 RBSを有する溶液にした。そのポリマーを、10mg/mlの濃度で、所望のpHにおける100mMの2塩基性のリン酸ナトリウム緩衝液に溶解した。上記ポリマー濃度は、2.5nmol/108 RBCであった。100%リーシスを達成するために、蒸留水中での溶血を使用した。ポリマーを有さない緩衝液中のRBCを、参照コントロールとして使用した。溶血アッセイを、1mlの適切な緩衝液に懸濁したRBCに上記ポリマーを添加することにより実施した。上記RBCを、数回試験管をひっくり返して混合し、37℃の水浴で1時間インキュベートした。インキュベーション後、上記RBCを13,500×gで10分間遠心分離をし、インタクトな細胞を沈降させ、リーシスを、RBCにより放出されたヘモグロビンの量を反映する541nmにおける上清の吸光度を測定することにより決定した。細胞のない上清の541nmの波長における吸光度の相対的増加100(A−A0)A合計を、膜崩壊の指標として測定した。その結果は、RBC膜を安定化するPMLA−PEGとは反対に、PMLA−PEG−バリン、PMLA−PEG−バリン−AS、およびPMLA−PEG−バリン−AS−mABは、膜不安定性を暗示することを示す。比較は、不安定性はPMLA−結合バリンの存在に起因することを示す。pHが下がっていく(エンドドームのリソソームへの成熟を模擬する)と、バリンのカルボキシル基がプロトン化され、電荷が中和されたバリン部分の増加した親油性に起因して、不安定性は、増加する。
【0054】
(モルホリノ−オリゴの放出)
図10aおよび図10bは、グルタチオン(グルタチオン−SH)によりジスルフィド結合の切断に起因する、薬物キャリアからのモルホリノアンチセンスオリゴヌクレオチドの放出を示す。ジスルフィド結合の切断は、2工程反応である。最初の工程は、1当量のグルタチオン−SHが、ジスルフィドと反応して混成ジスルフィドアンチセンスオリゴヌクレオチド−S−S−グルタチオン(縞の縦棒)(図10a)および1当量の遊離のアンチセンスオリゴヌクレオチド−SH(1色の縦棒)を形成する。時間が経つにつれて混成ジスルフィドが減少し、遊離のアンチセンスが増加する。この反応は、図10bに示されるように迅速である。2番目の工程は、混成ジスルフィドが第2当量のグルタチオンと反応してジスルフィドグルタチオン−S−S−グルタチオンおよび遊離のオリゴヌクレオチド−SHを生じる。この第2反応はゆっくりである。この反応の2工程のメカニズムおよび相対的速度は、いわゆるジスルフィド交換反応と呼ばれるこの反応に関して代表的である。その結果は、上記オリゴヌクレオチドが、細胞質で薬物キャリアから非常に効率的に切断され、それはこの所定の濃度のグルタチオンを含むことを示す。
【0055】
図に示されるように、薬物ビヒクルからアンチセンスモルホリノオリゴヌクレオチドの細胞質放出を模倣するために、5mM GSH(γ−L−グルタミル−L−システイニルグリシン、分子量307.33)を、室温で上記反応混合物に添加した。その混合物は、水中に0.25mMの薬物ビヒクルを含有した。いろいろな時に、全スルフヒドリル部分に対して過剰のN−エチルマレイミド(最終濃度、20mM)の添加により、上記反応を停止した。還元アンチセンスをN−エチルマレイミジルアンチセンスとして検出した。上記反応生成物を、HPLCにより分離し、放出されたアンチセンスモルホリノを260nmにおけるUV吸光度により検出した。その結果は、図10bに示される。完全な(100%)放出は、50mM DTTの存在下において37℃1時間の還元と関係付けられる。HPLC分析を、ゲルろ過カラムを使用してMerck−Hitach分析用HPLCで実施した。分離については、リン酸ナトリウム緩衝液(50mM、pH7.4)を使用して、流速0.75ml/分でMacherey & Nagel 125−5 GFC−HPLCカラム(300×7.7mm)で実施した。
【0056】
(ヌードラットの脳で増殖したヒト神経膠腫の、ポリリンゴ酸に結合したラミニン−8アンチセンスオリゴヌクレオチドでの処理)
腫瘍を処置し、そして正常細胞に対する副作用を減少させる特定の薬物送達は、すばらしいことである。タンパク質合成のレベルにおいて数種の分子標的を同時に阻害することは、腫瘍の増殖および進行を抑制することに非常に有効であり得る。ラミニン−8鎖の過剰発現は、神経膠腫の進行と関連し、ラミニン−8をブロッキングすることは、インビトロで神経膠腫侵襲を阻害する(34)。
【0057】
(方法)
(ポリリンゴ酸(PMLA))
多機能性薬物送達構築物は、ポリリンゴ酸(PMLA)のぶら下がっている(ペンダントな)カルボキシル基に結合するモジュールより構成される。そのポリマーは、Physarum polycephalumの天然産物である(27)。上記モジュールは、以下である:(1)ジスルフィド結合により上記骨格に結合するモルホリノアンチセンスオリゴヌクレオチド(その結合が、細胞質で切断され、遊離の薬物を放出する)、(2)癌細胞標的化およびレセプター媒介性エンドサイトーシスのためのトランスフェリンレセプターの抗体、(3)短鎖PEG結合L−ロイシンおよび直接結合L−バリン(両方がアミド結合を介して連結されているが、エンドソーム膜を崩壊させるpH依存性の親油性を提供する)、(4)循環時間を増加する長鎖PEG、および(5)組織/細胞内において構築分子を検出する蛍光レポータ分子(フルオレセイン、Cy5または同様な発色団)。
【0058】
(動物処置に使用された薬物の配合)
薬物1:ラミニンα4に対するアンチセンスオリゴ+ラミニンβ1に対するアンチセンスオリゴ(α4:β1=1:1);
薬物2:ラミニンα4に対するアンチセンスオリゴ+ラミニンβ1に対するアンチセンスオリゴ(α4:β1=1:1)+速く分裂している細胞に対する送達ビヒクルとしての抗トランスフェリンレセプターモノクローナル抗体(Chemicon InternationalからのラットCD71に対する抗体OX−26)(35、36、37および38);
薬物3:ラミニンα4に対するアンチセンスオリゴ+ラミニンβ1に対するアンチセンスオリゴ+EGFRに対するアンチセンスオリゴ(α4:β1:EGFR=1:1:1);
薬物4:ラミニンα4に対するアンチセンスオリゴ+ラミニンβ1に対するアンチセンスオリゴ+EGFRに対するアンチセンスオリゴ(α4:β1:EGFR=1:1:1)+抗トランスフェリンレセプター抗体;
薬物1A、2A、3Aおよび4Aは、対応する番号の薬物に対して同一である(即ち薬物1と薬物1Aは同じである)。対応するセンスオリゴを、アンチセンスオリゴの代わりに使用した。
【0059】
薬物1:PMLAに結合したラミニンα4鎖に対するアンチセンスオリゴヌクレオチドおよびラミニンβ1鎖に対するアンチセンスオリゴヌクレオチド;薬物2:PMLAに結合したラミニンα4鎖に対するアンチセンスオリゴヌクレオチド、ラミニンβ1鎖に対するアンチセンスオリゴヌクレオチド、抗トランスフェリンレセプターモノクローナル抗体(Chemicon Internationalからの抗体OX−26)。コントロール(薬物1Aおよび薬物2A)は、対応するセンスオリゴにより置き換えられたアンチセンスオリゴとの同じキャリア結合体であった。上記ヒトU−87MG神経芽膠腫細胞株を、インビトロ実験に使用し、異系勾配されたホモ接合体のNHRNU−M NIHヌードラット(Taconic Inc.)に、頭蓋内に注入した。
【0060】
異系勾配されたNIHヌードラット(Tac:N:NIH−Whn、Taconic)を、これらの試験に使用した。アンチセンス処置について、最も特異的で、安定でインビトロおよびインビボに有効であるとしてモルホリノオリゴ(Gene Tools、LLC)を使用した。モルホリノは、インビトロ研究で過去に成功裏に使用されたが、インビボでは、それらの送達はうまく行かなかった(6、7)。ポリ−L−リンゴ酸(PMLA)は、細胞にモルホリノを細胞に入れる送達キャリアとして使用された。PMLAの精製方法の原理が、(25)に記載されている。最近のPMLAのスケ−ルアップ生産方法(27)を使用した。PMLA官能基の化学が調査され(28、29)、そして有機溶媒および水性溶媒両方における生成物の誘導体化および精製は容易に達成し得る。我々の実験に関して、PMLAを、標的化腫瘍に特異的なラミニン−8鎖に加えて、PMLAはトランスフェリンレセプターに対するモノクローナル抗体に化学的に結合し、早く分裂する細胞に最も特異的な薬物を作製した(35、36、37および38)。
【0061】
予備的な毒性研究を、合成が完了した後に、モルホリノオリゴヌクレオチド、PMLAおよびそれらの結合体について実施した。それぞれの化学品を投与後30日に、肉眼での病理学的分析および顕微病理学的分析を行ったが、異常な変化は認められなかった。動物は、神経学的異常を発症しなかったし、それらの食欲もまた、正常であった。動物試験被験体は、定位的デバイスを使用してヒト経芽膠腫細胞U−87MGを頭蓋内に注入することにより作製した。薬物処置は、腫瘍細胞注入後3日目で開始した。
【0062】
頭蓋内処置について、以下に図示するように、ラットにアンチセンスオリゴを3日目、7日目、10日目および14日目(全部で4処置)に注入した。
【0063】
【化2】
各群12匹のラットにラミニン−8α4鎖に対するオリゴ+β1鎖に対するオリゴを、0.5mg/kgまたは2.5mg/kgの用量で注入した。各コントロール群11匹のラットに、α4鎖に対するセンスオリゴ+β1鎖に対するセンスオリゴを、0.5mg/kgまたは2.5mg/kgの用量で注入した。全ての外科的および非外科的操作を、2003年8月付けのIACUCプロトコル001118に従って行った。頚動脈内の処置について、腫瘍移植直後にカテーテルを頚動脈に移植した。カテーテルを、移植可能な皮下注入ポートに結合した。ラットに900μlのアンチセンスオリゴ溶液およびセンスオリゴ溶液(1分当たり0.06mlで15分間、蠕動ポンプで)を右頚動脈に皮下ポートチャンバ経由で潅注し、引き続きヘパリンロック(heparin flush)した。ラットを潅注終了後30分以内に安楽死させた。上記コントロールは:(a)正常なコントロール組織を得るために如何なる処置もせず、40日目に安楽死させた3匹のラット、および(b)腫瘍を有し、1日目、3日目、7日目、10日目および14日目にPBSで頭蓋内に擬似注入し、腫瘍の進行に起因する神経症状を発症させるとすぐに組織採取のために安楽死させた4匹のラットからなった。
【0064】
(結果)
(頭蓋内腫瘍処置)
薬物2の0.5および2.5mg/kgの用量は、生存率研究における処置と同等であった。頭蓋内投与による薬物2の4回の投薬後に、動物の生存時間が、PBS(擬似)またはセンスオリゴ(薬物2A)で処置されたラットと比較して、30%増加した、p<0.008(図11)。しかしながら、2つの処置は、僅かな効果しかなかった。トランスフェリンレセプター抗体の無い薬物1は、生存率には影響しなかった。従って、薬物細胞送達のメカニズムは、おそらくトランスフェリン媒介性エンドサイトーシスである。興味深いことに、アンチセンスをEGFR結合薬物2(薬物4)へ添加することにより、活性が無くなったが、おそらくEGFRが阻害された低酸素状態で、神経膠腫細胞の生存率の増加に起因するであろう(39)。
【0065】
薬物の内在化のメカニズムを、培養神経膠腫細胞において調査した。細胞がフルオレセイン標識化薬物2およびローダミン標識化エンドソームマーカーFM 4−64(Molecular Probes、Eugene、OR)で処理された場合、両方の化合物の染色は、共局在化を示した。10分以内に細胞膜の近くに共局在化し、30分以内に両方の標識がエンドソームに見出された(図12)。
【0066】
細胞がトランスフェリンレセプターで予め処理され、次いで10分以内に薬物2で処理された場合、上記薬物は、細胞質に見られなかった(データは、示されていない)。これらの結果は、トランスフェリンレセプター抗体が、活性薬物の一部として必要であることを示唆する。何故なら、それは薬物の細胞への透過を、レセプター媒介性のエンドサイトーシスにより可能にし、その後上記アンチセンスオリゴが標的細胞内に放出され得るからである。もし上記細胞がトランスフェリンに対する遊離抗体で処理される場合、上記レセプターは、ブロックされ、上記薬物上の抗体は結合できない。
【0067】
本発明者らは、薬物2がヌードラットに異種移植されたヒト神経膠腫における血管密度および特異的なラミニン鎖発現を減少させることを、発見した。本発明者らは、ラミニン−8発現を阻害するよう設計された薬物2が腫瘍における血管密度を減少させることを、示した。これらの血管を、von Willebrand因子を免疫染色することにより可視化した。血管の数を、薬物処置動物および未処置動物において、画像計測システム(Hamamatsu,Japan)に連結されたZeiss Axioscope顕微鏡を使用して、200倍の倍率で、3つの連続切片について5つの顕微鏡視野(1腫瘍ににつき15視野)において数えた。このデータをNIH ImageJソフトウエアに入力し血管密度を定量した。統計的有意差を、ANOVAにより決定した。
【0068】
(血管新生)
図13に示されるように、処置しないU87MGヒト腫瘍における毛細血管密度は、正常脳より有意に高かった。薬物2での4回の頭蓋内処置後、腫瘍血管密度は、55%減少した。データは、3匹の擬似手術した(正常)ラット(45顕微鏡視野)、未処置腫瘍を有する5匹のラット(75顕微鏡視野)および薬物2処置腫瘍を有する5匹のラット(75顕微鏡視野)について提示された。この結果は、ラミニン−8発現を阻害するよう設計された薬物2の作用の抗血管新生メカニズムを確証する。
【0069】
(ラミニン鎖免疫染色)
薬物2が事実上標的化ラミニン−8鎖の発現を阻害することを示すことは重要である。この目的に対して、インビボおよびインビトロで実験を実施した。腫瘍の免疫染色について、ラットラミニンを認識しないが腫瘍由来ラミニンとは反応するヒトラミニン−8β1鎖に対する抗体を、使用した。図14は、上記アンチセンスが免疫染色を、効果的に除去する細胞培養における効果を示す。図15に示されるように、薬物2がまた、異種移植したヒト腫瘍においてラミニンβ1鎖についての免疫染色を、効果的に減少させた。
【0070】
インビトロにおけるラミニン−8発現の阻害は、薬物2で3日間処置した2種の培養されたヒト神経膠腫、U87MGおよびT98G、の条件培地で評価した。図16のウエスタンブロット分析は、ラミニンα4鎖の著しい減少(密度計測により3倍の減少)およびラミニンβ1鎖の消失を示す。従って、薬物2は、インビボおよびインビトロの両方において、標的ラミニン鎖の発現を効果的に阻害する。
【0071】
(インビボにおける頸動脈内腫瘍処置および静脈内腫瘍処置)
頸動脈内処置のために、ラット脳にヒトU87−MG神経膠腫細胞を接種した14日後に、3匹のラットの群は、腫瘍移植後直ちに、頚動脈内にカテーテルを移植した。そのカテーテルを移植可能な注入ポートに連結した。そのラットに、900μlのアンチセンスおよびセンスオリゴ溶液(1分あたり0.06mlで、15分間蠕動ポンプで)を、皮下ポートチャンバー経由で右頚動脈に潅注し、ヘパリンロックした。薬物2を、頚動脈処置のために2.5μg/kgの濃度で注入したか、または5μg/kgの濃度で、尾静脈経由で注入した(同様に3匹のラット)。薬物分布を注入後1時間、3時間、12時間および24時間で調べた。蛍光単位を使用して、本発明者らは、移植された腫瘍細胞(濃い染色)および脳の血管細胞(より薄い染色)における薬物を検出した(図17)。薬物2を、薬物2により担持されるトランスフェリン抗体を(赤で)標識化するローダミン染色抗体を用いて可視化した。上記細胞核をDAPI(青)で対比染色する。移植されたU87MG腫瘍細胞(左パネル)は濃く標識化されるが、ラット脳(右パネル)は、限られた(主に血管の)染色を示す。これらの位置の最大濃度は、薬物注入後3時間および12時間の時点で達成された。これらの結果は、薬物2が血液脳関門(BBB)を、おそらくレセプター媒介性エンドサイトーシスで透過することを確証する。
【0072】
(結論)
ラミニン−8の発現およびヒト腫瘍におけるその阻害を研究するのに適切なインビボのモデルを開発した。ラミニン−8のα4鎖およびβ1鎖に対するアンチセンスオリゴ(ラミニン−8のブロッキング)と新規な薬物送達ビヒクル,PMLA、との組合わせは、ラットにおいて異種移植した頭蓋内ヒト神経膠腫におけるラミニン−8発現を効率的に阻害した。予備的な4回のアンチセンス処置後に、処置された神経膠腫を担持する動物の生存率が、有意に増加した、p<0.008。これらのデータは、治療標的としてラミニン−8を使用するPMLAベースのアンチセンス薬物が、ヒト脳腫瘍を阻害するのに有効であることを示す。
【0073】
従って、添付の特許請求の範囲は、上に具体的に説明されそして記載されたもの、概念的に等価のもの、自明に置換し得るものおよびまた、本発明の本質的な思想を本質的に取り込むものを含むことが理解されるべきである。当業者は、上述の好ましい実施形態の種々の改昨および改変は、本発明の範囲から逸脱しないで設定され得ることを理解する。説明された実施形態は、単なる例証の意図で示されたもので本発明を限定するとして考えられべきではない。従って、添付の特許請求の範囲の範囲内で、本発明は、本明細書に具体的に記載される以外でも実施され得ると理解されるべきである。
【0074】
(参考文献)
【0075】
【化3】
【0076】
【化4】
【0077】
【化5】
【0078】
【化6】
【特許請求の範囲】
【請求項1】
本願明細書に記載の発明。
【請求項1】
本願明細書に記載の発明。
【図1a】

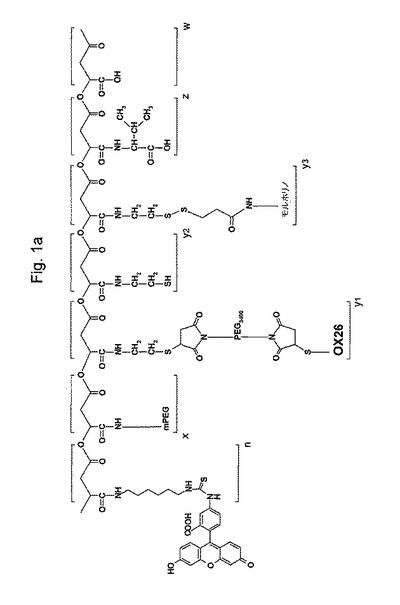
【図1b】
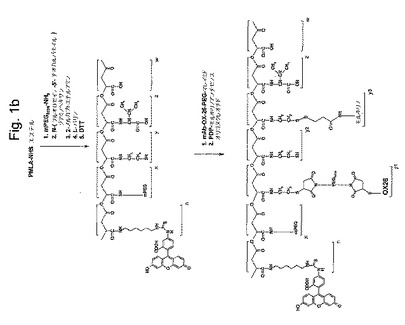
【図2】
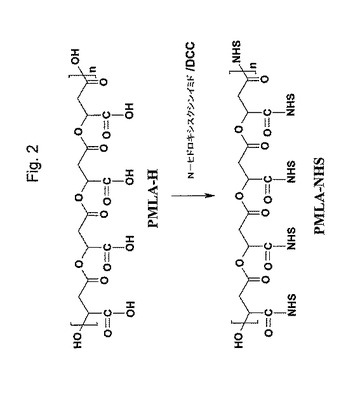
【図3】
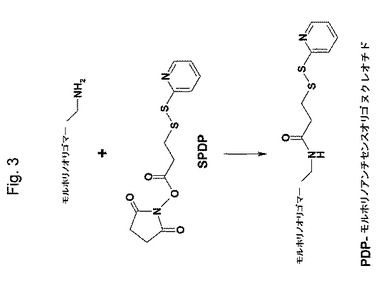
【図4】
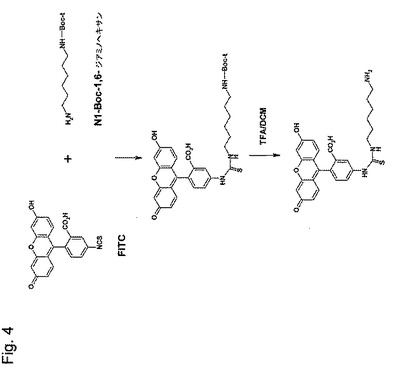
【図5】
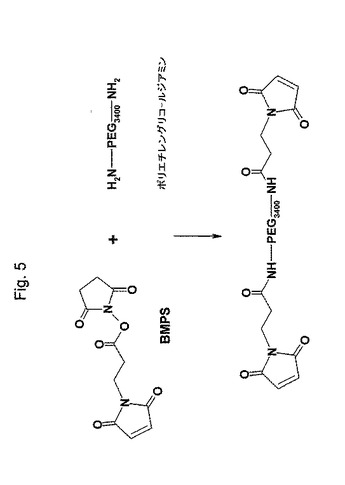
【図6】
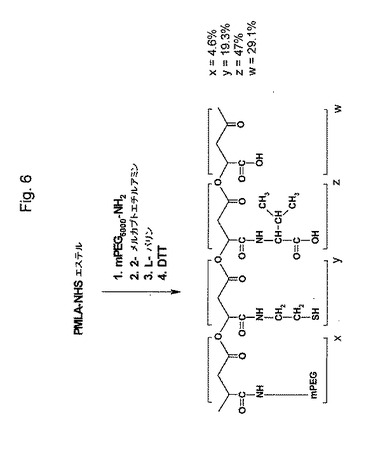
【図7】
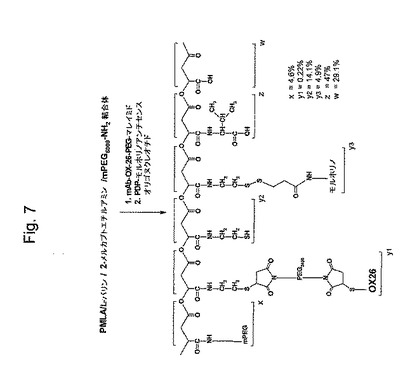
【図8】
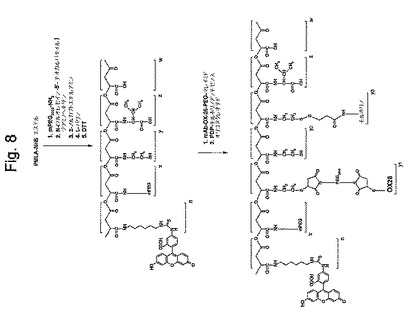
【図9】
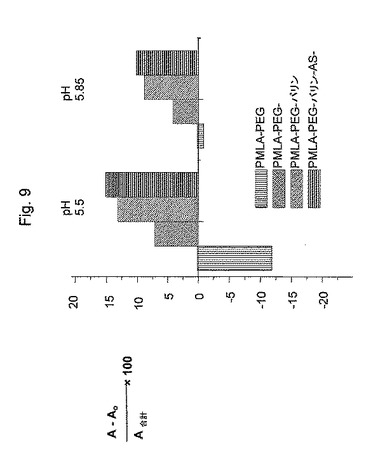
【図10a】
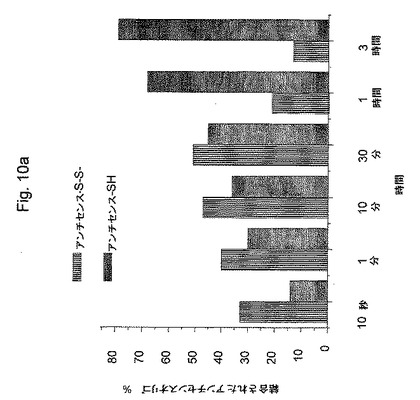
【図10b】
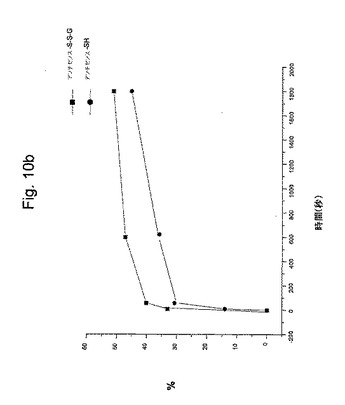
【図11】
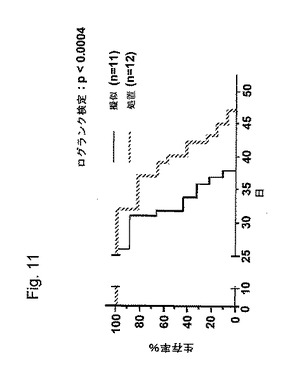
【図12】
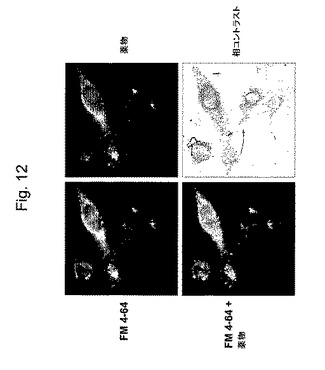
【図13】
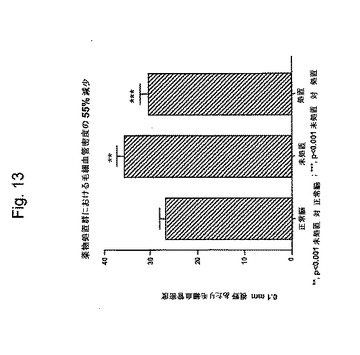
【図14】
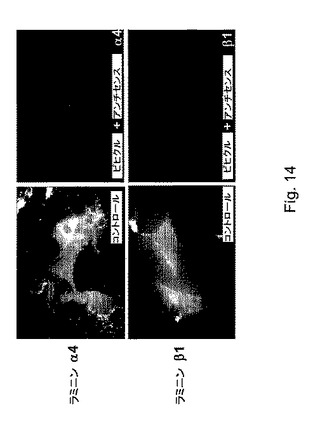
【図15】

【図16】
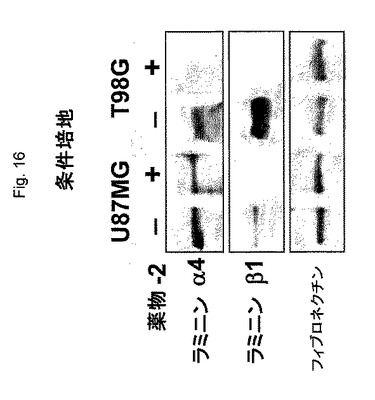
【図17】
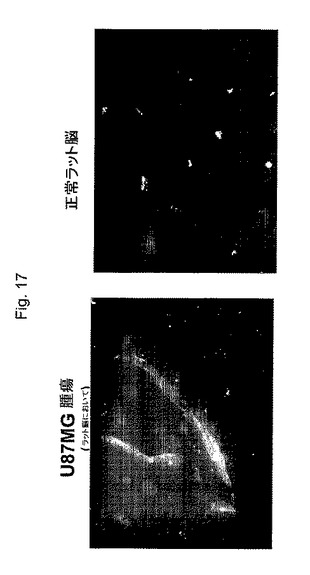
【図1b】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10a】
【図10b】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【公開番号】特開2010−163468(P2010−163468A)
【公開日】平成22年7月29日(2010.7.29)
【国際特許分類】
【出願番号】特願2010−107780(P2010−107780)
【出願日】平成22年5月7日(2010.5.7)
【分割の表示】特願2006−542822(P2006−542822)の分割
【原出願日】平成16年12月3日(2004.12.3)
【出願人】(506188312)アロジーン, インコーポレイテッド (2)
【出願人】(398062149)セダーズ−シナイ メディカル センター (34)
【Fターム(参考)】
【公開日】平成22年7月29日(2010.7.29)
【国際特許分類】
【出願日】平成22年5月7日(2010.5.7)
【分割の表示】特願2006−542822(P2006−542822)の分割
【原出願日】平成16年12月3日(2004.12.3)
【出願人】(506188312)アロジーン, インコーポレイテッド (2)
【出願人】(398062149)セダーズ−シナイ メディカル センター (34)
【Fターム(参考)】
[ Back to top ]