
ポリ乳酸分解酵素及びそれを産生する微生物

【課題】PLA分解能を有する新規酵素及び新規微生物、それらを用いた効率的なPLA分解方法を提供する。
【解決手段】下記(a)〜(c)からなる群から選択されるポリペプチドを含有するタンパク質。(a)ミクロモノスポラ(Micromonospora)属由来の特定のアミノ酸配列からなるポリ乳酸分解活性を有するポリペプチド。(b)ミクロモノスポラ(Micromonospora)属由来の特定のアミノ酸配列に対して1個または数個のアミノ酸が欠失、置換、付加、または挿入されたアミノ酸配列を有し、かつ、ポリ乳酸分解活性を有するポリペプチド。(c)ミクロモノスポラ(Micromonospora)属由来の特定のアミノ酸配列と90%以上の同一性を有するアミノ酸配列を有し、かつ、ポリ乳酸分解活性を有するポリペプチド。
【解決手段】下記(a)〜(c)からなる群から選択されるポリペプチドを含有するタンパク質。(a)ミクロモノスポラ(Micromonospora)属由来の特定のアミノ酸配列からなるポリ乳酸分解活性を有するポリペプチド。(b)ミクロモノスポラ(Micromonospora)属由来の特定のアミノ酸配列に対して1個または数個のアミノ酸が欠失、置換、付加、または挿入されたアミノ酸配列を有し、かつ、ポリ乳酸分解活性を有するポリペプチド。(c)ミクロモノスポラ(Micromonospora)属由来の特定のアミノ酸配列と90%以上の同一性を有するアミノ酸配列を有し、かつ、ポリ乳酸分解活性を有するポリペプチド。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規のポリ乳酸分解酵素、前記酵素をコードする核酸、前記酵素を産生する新規微生物、及び、前記核酸が組み込まれた組換え微生物に関する。
【背景技術】
【0002】
ポリ乳酸(PLA)は、乳酸を化学合成によって重合させた物質である。PLAは融点やガラス転移点が高いため耐熱性に優れる。さらに透明度が高く従来のプラスチックと同等の物理的性質を備えているため、日用品から農業用品、医療機器や自動車部品などさまざまな分野で利用することが可能である。
【0003】
PLAは生分解性プラスチックであり、現在までに、PLAを分解する酵素や該酵素を産生する微生物についての研究がおこなわれてきた。例えば、特許文献1〜5は、PLA分解能を有する酵素や微生物を開示する。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特許第3762990号公報
【特許文献2】特許第4441639号公報
【特許文献3】特開2007−319077号公報
【特許文献4】特開2007−319078号公報
【特許文献5】特開2008−167701号公報
【発明の概要】
【発明が解決しようとする課題】
【0005】
PLA分解能は酵素や微生物の種類によって異なり、PLAの利用拡大のためには、より高いPLA分解能を有する酵素や微生物が必要である。したがって、本発明は、PLA分解能を有する新規酵素及び新規微生物を提供し、さらに、それらを用いた効率的なPLA分解方法を提供することを目的とする。
【課題を解決するための手段】
【0006】
本発明は、下記(a)〜(c)からなる群から選択されるポリペプチドを含有するタンパク質:
(a)配列表の配列番号1に記載のアミノ酸配列からなるポリペプチド;
(b)配列表の配列番号1に記載のアミノ酸配列に対して1個または数個のアミノ酸が欠失、置換、付加、または挿入されたアミノ酸配列を有し、かつ、PLA分解活性を有するポリペプチド;
(c)配列表の配列番号1に記載のアミノ酸配列と90%以上の同一性を有するアミノ酸配列を有し、かつ、PLA分解活性を有するポリペプチド、を提供する。
本発明のタンパク質は、新規PLA分解酵素である。配列表の配列番号1に記載のアミノ酸配列からなるポリペプチドは、高いPLA分解活性を有する。
【0007】
上記タンパク質が、配列番号1のアミノ酸配列の第34〜44番目のアミノ酸からなるペプチド、第65〜75番目のアミノ酸からなるペプチド及び第217〜227番目のアミノ酸からなるペプチドを含むことが好ましい。これらのペプチドは、PLA分解酵素の活性中心を含む。
【0008】
また、本発明は、上記タンパク質をコードするポリヌクレオチドを含有する核酸を提供する。
さらに、本発明は、下記(1)〜(4)からなる群から選択されるポリヌクレオチドを含有する核酸;
(1)配列表の配列番号2に記載の塩基配列からなるポリヌクレオチド;
(2)配列表の配列番号2に記載の塩基配列に対して1個〜数個の塩基が欠失、置換、付加、または挿入された塩基配列を有し、かつ、ポリ乳酸分解活性を有するポリペプチドをコードするポリヌクレオチド;
(3)配列表の配列番号2に記載の塩基配列と90%以上の同一性を有する塩基配列を有し、かつ、ポリ乳酸分解活性を有するポリペプチドをコードするポリヌクレオチド;
(4)(1)のポリヌクレオチドとストリンジェントな条件下でハイブリダイズし、かつ、ポリ乳酸分解活性を有するポリペプチドをコードするポリヌクレオチド、を提供する。
配列番号2に記載の塩基配列は、配列番号1に記載のアミノ酸配列からなるポリペプチドをコードする塩基配列である。すなわち、本発明の核酸は、PLA分解酵素をコードする。したがって、上記核酸を微生物に組み込んで、本発明のPLA分解酵素を微生物に産生させることができる。
【0009】
上記核酸が、配列番号1のアミノ酸配列の第34〜44番目のアミノ酸からなるペプチド、第65〜75番目のアミノ酸からなるペプチド及び第217〜227番目のアミノ酸からなるペプチドを含むヌクレオチドを含むことが好ましい。上記ヌクレオチドは酵素の活性中心を含むペプチドをコードする。
【0010】
また、本発明は、ミクロモノスポラ(Micromonospora)属に属し、配列表の配列番号2で示される塩基配列の核酸を有する、受領番号NITE AP−1105として寄託されている放線菌B12−1株を提供する。この放線菌B12−1株は、本発明のPLA分解酵素を産生する微生物である。
【0011】
また、本発明は、上記核酸を含有する組換えベクターを含む組換え微生物を提供する。組換え微生物により、本発明のPLA分解酵素が産生される。上記組換え微生物は、ストレプトマイセス(Streptomyces)属に属することが好ましく、受領番号NITE AP−1106として寄託されている微生物であることがさらに好ましい。この組換え微生物は、極めて高いPLA分解活性を有する。
【0012】
また、本発明は、放線菌B12−1株又は上記組換え微生物を培養し、得られる培養物からPLA分解酵素を回収する、PLA分解酵素の製造方法を提供する。
【0013】
さらに、本発明は、上記本発明のタンパク質、放線菌B12−1株及び上記組換え微生物からなる群から選択される少なくとも1種とPLAを含む物質とを接触させる、PLAの分解方法を提供する。本発明の分解方法は、PLA分解活性の高い酵素や微生物を用いているため、効率的にPLAが分解される。
【発明の効果】
【0014】
本発明によれば、PLA分解能を有する新規酵素及び新規微生物、さらに、それらを用いた効率的なPLA分解方法が提供される。
【図面の簡単な説明】
【0015】
【図1】図1は、B12−1株を寒天培地に37℃、5日間培養した際にB12−1株が形成したクリアゾーンを示す図である。
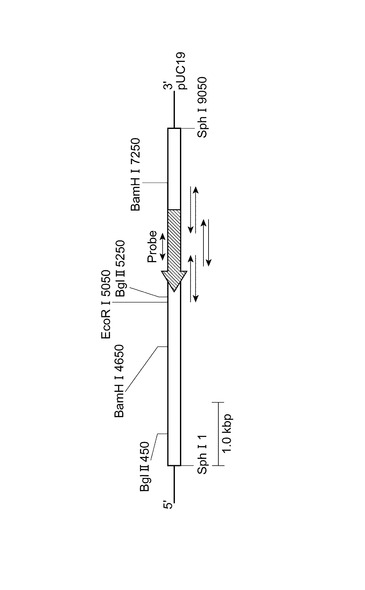
【図2】図2は、組換えプラスミドpG12を用いて作製した制限酵素地図を表す図である。
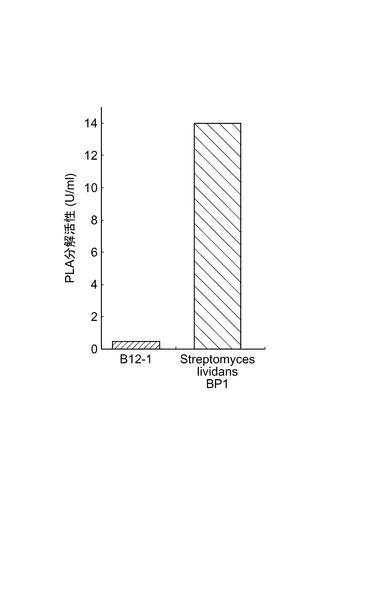
【図3】図3は、B12−1株の分解活性と組換え微生物Streptomyces lividans BP1の分解活性とを比較した図である。
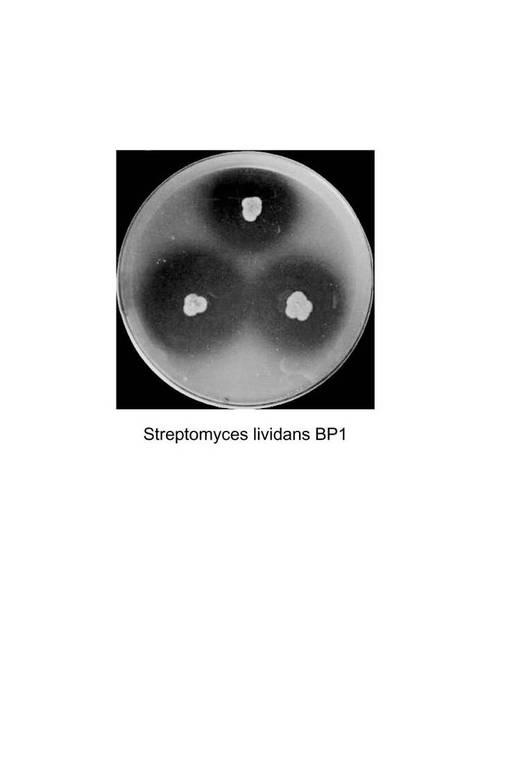
【図4】図4は、Streptomyces lividans BP1を寒天培地に30℃、5日間培養した際にStreptomyces lividans BP1が形成したクリアゾーンを示す図である。
【発明を実施するための形態】
【0016】
(タンパク質)
本発明が提供するタンパク質は、新規PLA分解酵素(以下、場合により単に「PLA分解酵素」と称する。)である。本発明のタンパク質は、PLAを分解することが知られている既知の酵素よりも高いPLA分解活性を有する。したがって、本発明のタンパク質を用いることで、従来よりも早く、またはより多くのPLAを分解することができる。本発明のタンパク質は、(a)配列表の配列番号1に記載のアミノ酸配列からなるポリペプチドを含有する。(a)配列表の配列番号1に記載のアミノ酸配列からなるポリペプチド(全長276アミノ酸残基)は、それ自身がPLA分解活性を有するポリペプチドである。
【0017】
本発明が提供するタンパク質が含有するポリペプチドは、(a)配列表の配列番号1に記載のアミノ酸配列からなるポリペプチドに限定されず、(b)配列表の配列番号1に記載のアミノ酸配列に対して1個または数個のアミノ酸が欠失、置換、付加、または挿入されたアミノ酸配列を有し、かつ、PLA分解活性を有するポリペプチドであってもよい。「1個または数個」とは1個から30個、好ましくは1個から10個、より好ましくは1個から5個、さらに好ましくは1個から3個である。さらに、本発明が提供するタンパク質が含有するポリペプチドは、(c)配列表の配列番号1に記載のアミノ酸配列と90%以上の同一性を有するアミノ酸配列を有し、かつ、PLA分解活性を有するポリペプチドであってもよい。該ポリペプチドのアミノ酸配列と配列番号1に記載のアミノ酸配列との同一性は、好ましくは93%以上、より好ましくは95%以上、さらに好ましくは98%以上である。
【0018】
本明細書における「PLA分解活性」は、例えば、実施例2に記載の酵素活性測定法により測定することができる。また、本明細書において「PLA分解活性を有する」とは、配列番号1に記載のアミノ酸配列からなるポリペプチドの、例えば50%以上、好ましくは70%以上、より好ましくは80%以上、さらに好ましくは95%以上のPLA分解活性を有することと定義することができる。
【0019】
本発明のタンパク質は、配列番号1に記載のアミノ酸配列から、セリンプロテアーゼの一種であると推定され、PLA分解活性の活性中心は、アスパラギン酸(配列番号1の第39番目)、ヒスチジン(配列番号1の第70番目)及びセリン(配列番号1の第222番目)であると推定される。したがって、本発明のタンパク質はこの活性中心を含むことが好ましい。具体的には、本発明のタンパク質は、配列番号1のアミノ酸配列の第34〜44番目のアミノ酸からなるペプチド、第65〜75番目のアミノ酸からなるペプチド及び第217〜227番目のアミノ酸からなるペプチドを含むことが好ましい。
【0020】
配列表の配列番号3に記載のアミノ酸配列からなるポリペプチドは、(a)配列番号1に記載のアミノ酸配列からなるポリペプチドに、シグナルペプチド(配列番号3の第1〜35番目、35アミノ酸残基)、及びプロペプチド(配列番号3の第36〜130番目、95アミノ酸残基)が付加されたポリペプチド(全長406アミノ酸残基)である。配列番号1に記載のポリペプチドは、配列番号3に記載のアミノ酸配列からなるPre−Pro−Enzymeとして生産され、シグナルペプチドにより細胞外に輸送された後、プロペプチドが切断され、活性化酵素として働くことができる。
【0021】
本発明のタンパク質は、(a)〜(c)から選択されるポリペプチドに、別種のタンパク質または物質が結合されたものでもよい。例えば、上記ポリペプチドの検出または精製を容易にするために、あるいは別の機能を付加するために、そのN末端側やC末端側に別種のタンパク質またはペプチド、例えばグルタチオンS−トランスフェラーゼ(GST)、ルシフェラーゼ、緑色蛍光タンパク質(GFP)、β−ガラクトシダーゼ、免疫グロブリン(IgG)、His−tag等が、直接的にまたはリンカーペプチド等を介して間接的に、遺伝子工学的手法等を用いて付加されたものであってもよい。
【0022】
(核酸)
本発明は、上記タンパク質をコードするポリヌクレオチドを含有する核酸、すなわちPLA分解酵素をコードするポリヌクレオチドを含有する核酸を提供する。具体的には、(1)配列表の配列番号2に記載の塩基配列からなるポリヌクレオチドを含有する核酸が挙げられる。(1)配列表の配列番号2に記載の塩基配列からなるポリヌクレオチド(全長831残基)は、(a)配列表の配列番号1に記載のアミノ酸配列からなるポリペプチドをコードするポリヌクレオチドである。
【0023】
本発明が提供する核酸が含有するポリヌクレオチドは、(1)配列表の配列番号2に記載の塩基配列からなるポリヌクレオチドに限定されず、(2)配列表の配列番号2に記載の塩基配列に対して1個〜数個の塩基が欠失、置換、付加、または挿入された塩基配列を有し、かつ、PLA分解活性を有するポリペプチドをコードするポリヌクレオチドであってもよい。「1個または数個」とは1個から30個、好ましくは1個から10個、より好ましくは1個から5個、さらに好ましくは1個から3個である。また、本発明が提供する核酸が含有するポリヌクレオチドは、(3)配列表の配列番号2に記載の塩基配列と90%以上の同一性を有する塩基配列を有し、かつ、PLA分解活性を有するポリペプチドをコードするポリヌクレオチドであってもよい。該ポリヌクレオチドの塩基配列と配列番号2に記載の塩基配列との同一性は、好ましくは93%以上、より好ましくは95%以上、さらに好ましくは98%以上である。さらに、本発明が提供する核酸が含有するポリヌクレオチドは、(4)配列表の配列番号2に記載の塩基配列からなるポリヌクレオチドとストリンジェントな条件下でハイブリダイズし、かつ、PLA分解活性を有するポリペプチドをコードするポリヌクレオチドであってもよい。
【0024】
本明細書において、「ストリンジェントな条件下」とは、各塩基配列間の同一性の程度が、例えば、全体の平均で約80%以上、好ましくは約90%以上、より好ましくは約95%以上であるような、高い同一性を有する塩基配列間のみで、特異的にハイブリッドが形成されるような条件を意味する。具体的には、例えば、ナトリウム濃度が約19〜40mM、好ましくは約19〜20mMで、温度が約50〜70℃、好ましくは約60〜65℃の条件を示す。特に、ナトリウム濃度が約19mMで温度が約65℃の場合が好ましい。
【0025】
ハイブリダイゼーションは、当業界で公知の方法あるいはそれに準じる方法、例えば、モレキュラー・クローニング(Molecular Cloning)2nd(J. Sambrook et al.,Cold Spring Harbor Lab. Press, 1989)に記載の方法などに従って行うことができる。また、市販のライブラリーを使用する場合、添付の使用説明書に記載の方法に従って行なうことができる。
【0026】
(ミクロモノスポラ属に属する新規微生物B12−1株)
本発明のタンパク質が含有する(a)配列表の配列番号1に記載のアミノ酸配列からなるポリペプチドは、ミクロモノスポラ(Micromonospora)属に属する新規微生物B12−1株(千葉県木更津市かずさ鎌足2−5−8、独立行政法人製品評価技術基盤機構特許微生物寄託センター、受領番号:NITE AP−1105、2011年6月13日付受領にて寄託、以下、場合により「B12−1株」と称する。)から単離精製することができる。B12−1株は、PLA分解能を有する新菌種である。B12−1株は、PLAを分解可能な微生物として土壌から分離されたグラム陽性を示す放線菌であり、胞子を形成する。B12−1株は、PLAの分解について優れた能力を有する。
【0027】
B12−1株は、微生物の培養に用いられる通常の条件下で培養して、維持・増殖させることができる。例えば、pH6.0〜8.0、温度20〜50℃の培養条件で培養することができる。生育速度の点から、pH6.8〜7.3、温度37〜40℃で培養することが好ましい。
【0028】
B12−1株を維持可能な培地として、微生物培養に使用される炭素源、窒素源、無機塩類を適宜含む各種の培地を使用できる。例えばグルコース、フラクトース、マンノース、シュクロース、マンニトール、ソルビトール、糖蜜などの炭素源;例えば硫酸アンモニウム、リン酸アンモニウム、炭酸アンモニウム、ペプトン、酵母抽出物などの窒素源;例えばリン酸二カリウム、リン酸一ナトリウム、塩化マグネシウム、塩化カルシウム、硫酸第一鉄、モリブデン酸ナトリウム、塩化マンガンなどの無機塩類;を、それぞれ含むものであって、通常利用される固体又は液体培地であればいずれも好ましく使用できる。なお、培地には、上記成分の他、微生物の成育を促進させるための各種ビタミン、ミネラル、その他の栄養成分を含ませてもよい。
【0029】
(PLA分解酵素の製造方法)
本発明のPLA分解酵素は、B12−1株を培養し、得られる培養物からPLA分解酵素を回収することによって得ることができる。
【0030】
PLA分解酵素を得るための、B12−1株の培養に適した培地としては、具体的には、硫酸アンモニウム、リン酸水素二カリウム、リン酸二水素カリウム、硫酸マグネシウム、酵母エキス、オリーブ油等を含む液体培地を用いることができる。培地中の成分の濃度は限定されるものではなく、炭素源や窒素源などの種類や濃度によって適宜調整できる。
【0031】
PLA分解酵素は、B12−1株の菌体、菌体破砕物及び菌体の培養上清などの各形態から得ることができる。菌体の場合、その培養物から遠心分離等の集菌操作によって得られる生菌体、菌体を凍結乾燥した乾燥粉末、菌体を含む培養液のいずれであってもよい。破砕物としては、物理的手段又は化学的手段によって破砕されたものであればよく、物理的手段としては、超音波粉砕機、グラスビーズを用いた細胞破砕機などを挙げることができ、化学的手段としては、リゾチームなどの溶菌酵素を挙げることができる。物理的手段又は化学的手段を用いて菌体を破砕する場合には、それぞれ酵素が失活しない穏和な条件下で行うことが好ましい。培養上清としては、前述した液体培地中でB12−1株を培養したものであればよく、酵素産生量から、好ましくは培養2〜6日目、より好ましくは培養3〜4日目の培養上清を使用することができる。
【0032】
PLA分解酵素の回収法は、一般の酵素の回収の手段に準じて行うことができる。以下に示す方法に特に限定はされないが、例えば上記手段によって得られた菌体破砕物、あるいは培養上清を、粗酵素液として用いることができる。粗酵素液は、そのままで使用することもできるが、必要に応じて、例えば塩析法、沈澱法、限外濾過法等の分離手段、例えばイオン交換クロマトグラフィー、等電点クロマトグラフィー、疎水性クロマトグラフィー、ゲル濾過クロマトグラフィー、吸着クロマトグラフィー、アフィニティークロマトグラフィー、逆相クロマトグラフィー等の公知の方法を組み合わせて、更に分離精製することができる。
【0033】
PLA分解酵素を得るための別の方法としては、PLA分解酵素をコードするポリヌクレオチドを含有する核酸を含む組換え微生物を培養し、得られる培養物からPLA分解酵素を回収する方法が挙げられる。
【0034】
(組換え微生物)
本発明の組換え微生物は、PLA分解酵素のアミノ酸配列をコードするポリヌクレオチドを含有する核酸を含有する組換えベクターを含む。このような組換え微生物は、PLA分解酵素のアミノ酸配列をコードするポリヌクレオチドを含有する核酸をB12−1株より取得し、次いでこの核酸を適当なベクターに組込んで組換えベクターを作製し、この組換えベクターを用いて宿主を形質転換することにより作製することができる。
【0035】
本発明の組換え微生物を作製するために用いるベクターは、宿主の種類により適宜選択される。ベクターとして、宿主微生物体内で自律的に増殖しうるファージ又はプラスミドから構築されたベクターが適している。ファージベクターとしては、例えば、大腸菌(Escherichia coli)に属する微生物を宿主微生物とする場合にはλgt・λC、λgt・λBなどが使用できる。また、プラスミドベクターとしては、例えば、大腸菌を宿主微生物とする場合にはプラスミドpBR322、pBR325、pACYC184、pUC12、pUC13、pUC18、pUC19、pUC118、pINI、放線菌を宿主とする場合にはpIJ680、pIJ702、枯草菌を宿主とする場合にはpUB110、pKH300PLK、酵母を宿主とする場合にはYrp7、pYC1、Yep13などが使用でき、これらを組み合わせて使用してもよい。
【0036】
組換えベクターは、目的の遺伝子配列と、複製そして制御に関する情報を担持した遺伝子配列、例えばプロモーター、リボソーム結合部位、ターミネーター、シグナル配列、エンハンサー等とを構成要素とし、これらを公知の方法により組み合わせて作製される。ベクターに核酸を組み込む方法は、公知の方法を適用できる。例えば、適当な制限酵素を選択、処理して核酸を特定部位で切断し、次いで同様に特定部位で切断したベクターDNAと混合し、リガーゼによって再結合する方法が用いられる。あるいは、目的の核酸に適当なリンカーを結合し、これを目的に適したベクターのマルチクローニングサイトへ挿入することによっても、所望の組換えベクターが得られる。
【0037】
(宿主)
本発明の組換え微生物を作製するための宿主として、原核微生物又は真核微生物を用いることができる。原核微生物としては、例えば、ストレプトマイセス(Streptomyces)属、エシェリシア(Escherichia)属、バチルス(Bacillus)属等の菌を挙げることが出来、この中でもストレプトマイセス属が好ましい。具体的には、ストレプトマイセス・リビダンス(Streptomyces lividans)を挙げることができる。真核微生物の好適例としては、サッカロミセス(Saccharomyces)属及びピヒア(Pichia)属等の酵母、並びに、アスペルギルス(Aspergillus)属、ペニシリウム(Penicillium)属等の真菌を挙げることができる。
【0038】
(形質転換法)
核酸が組み込まれた組換えベクターを、宿主細胞に公知の方法で導入することにより組換え微生物が得られる。組換えベクターの宿主細胞への導入は、公知の方法により行うことができる。具体的には、リン酸カルシウムトランスフェクション、DEAE−デキストラン媒介トランスフェクション、マイクロインジェクション、陽イオン脂質媒介トランスフェクション、エレクトロポレーション、形質導入及び感染等が挙げられる。
【0039】
(組換え微生物Streptomyces lividans BP1)
上述のようにして、本発明の核酸を含有する組換えベクターを含む組換え微生物が得られる。本発明は、実施例5に記載の方法により得られた組換え微生物として、ストレプトマイセス・リビダンスを宿主とし、配列表の配列番号2に記載の塩基配列からなるポリヌクレオチドを含有する核酸を含有する組換えベクターを含む組換え微生物Streptomyces lividans BP1(千葉県木更津市かずさ鎌足2−5−8、独立行政法人製品評価技術基盤機構特許微生物寄託センター、受領番号:NITE AP−1106、2011年6月13日付受領にて寄託)を提供する。組換え微生物Streptomyces lividans BP1は、PLAの分解について優れた能力を有する。
【0040】
組換え微生物Streptomyces lividans BP1は、微生物の培養に用いられる通常の条件下で培養して、維持・増殖させることができる。例えば、pH6.0〜8.0、温度20〜37℃の培養条件で培養することができ、生育速度の観点から、pH6.8〜7.3、温度30〜35℃で培養することが好ましい。
【0041】
組換え微生物Streptomyces lividans BP1を維持可能な培地として、微生物培養に使用される炭素源、窒素源、無機塩類を適宜含む各種の培地を使用できる。例えばグルコース、フラクトース、マンノース、シュクロース、マンニトール、ソルビトール、糖蜜などの炭素源;例えば硫酸アンモニウム、リン酸アンモニウム、炭酸アンモニウムなどの窒素源;例えばリン酸二カリウム、リン酸一ナトリウム、塩化マグネシウム、塩化カルシウム、硫酸第一鉄、モリブデン酸ナトリウム、塩化マンガンなどの無機塩類;を、それぞれ含むものであって、通常利用される固体又は液体培地であればいずれも好ましく使用できる。なお、培地には、上記成分の他、微生物の成育を促進させるための各種ビタミン、ミネラル、その他の栄養成分を含ませてもよい。
【0042】
(組換え微生物によるPLA分解酵素の製造方法)
本発明の核酸を含有する組換えベクターを含む組換え微生物によっても、PLA分解酵素を得ることができる。本発明のPLA分解酵素は、該組換え微生物を培養し、得られる培養物からPLA分解酵素を回収することによって得ることができる。PLA分解酵素の産生のための培地は特に限定されないが、例えば、組換え微生物Streptomyces lividans BP1を培養する場合、PLA分解酵素の産生に適した培地として、具体的には、ポリペプトン、ソイペプトン、塩化ナトリウム、リン酸二水素カリウム、グルコース等を含む液体培地を用いることができる。PLA分解酵素は、組換え微生物の菌体、菌体破砕物及び培養上清などの各形態から得ることができ、それぞれB12−1株を用いた場合と同様の方法により調製できる。培養上清としては、酵素産生量から、好ましくは培養1〜3日目、より好ましくは2日目の培養上清を使用することができる。酵素の回収方法は、B12−1株を用いた場合と同様の方法を採用することができる。
【0043】
(PLAの分解方法)
本発明の、PLA分解酵素、B12−1株及び組換え微生物からなる群から選択される少なくとも1種とPLAを含む物質とを接触させることにより、PLAを分解することができる。本発明のPLA分解酵素は高いPLA分解活性を有するので、物質中に含まれるPLAを少ない時間で大量に分解することができ、効率的である。
【0044】
(PLA)
本発明の方法により分解されるPLAとしては、乳酸を主成分とする重合体であればよく、ポリL−乳酸及びポリD−乳酸のようなホモポリマー、ポリL/D−乳酸、これらと他の成分から構成される共重合体を挙げることができる。共重合体である場合には、乳酸成分は重量比率が10%以上のものであることが好ましい。また共重合体を構成する他の成分としては、ε−カプロラクトン、グリコリド、デプシペプチド等を挙げることができる。これらの他の成分は1種であっても、2種以上であってもよい。本発明によって分解可能なPLAの分子量に特に制限はないが、分解速度の点から、数平均分子量5000〜500000であることが好ましく、10000〜200000であることがより好ましい。また、PLAを含む物質の形態としては、例えば、フィルム(シート)、成型体、破砕物、粉末、懸濁液などを挙げることができる。また、分解時のPLAは、PLA単独であってもよいし、他のプラスチックとの混合物であってもよい。
【0045】
分解に用いるPLA分解酵素、B12−1株及び組換え微生物は、乾燥形態又は適当な液体担体に混合された液体形態であってもよい。液体形態を構成するために用いられる液体担体としては、この用途に通常用いられるものであれば特に制限されず、水、緩衝液等を挙げることができる。
【0046】
PLA分解酵素をPLAの分解に用いる場合、PLA分解酵素として、上述の方法により精製した酵素又は粗酵素を用いることができる。PLAを含む物質と、PLA分解酵素とを接触させる接触方法としては、特に限定されないが、PLA分解酵素を含む酵素液にPLAを含む物質を投入する、PLAを含む物質が入った容器にPLA分解酵素を添加する、等の方法が挙げられる。分解時のpHは、分解速度の点から、pH6.0〜12.0とすることが好ましく、pH9.0〜10.0とすることがより好ましい。分解時の温度は、分解速度の点から、30℃〜60℃とすることが好ましく、45〜55℃とすることがより好ましい。分解時間は、処理効率の点から、1日間〜5日間とすることが好ましく、2日間〜3日間とすることがより好ましい。使用するPLA分解酵素の量は、基質となるPLAの種類及びPLAを含む物質の形態によって異なるが、例えば、分子量4×104のPLAを含む乳化液中のPLAを分解する場合、乳化液中に含まれるPLA1gに対して、0.01〜0.2gのPLA分解酵素を使用することが好ましく、0.05〜0.1gを使用することがより好ましい。
【0047】
B12−1株又は組換え微生物をPLAの分解に用いる場合、種々の形態で使用することができる。このような形態としては、微生物菌体、菌体破砕物及び微生物の培養上清などの各形態から適宜選択することができる。菌体の場合には、その培養物から遠心分離等の集菌操作によって得られる生菌体、菌体を凍結乾燥した乾燥粉末、微生物を含む培養液のいずれであってもよい。破砕物としては、物理的手段又は化学的手段によって破砕されたものであればよく、物理的手段としては、超音波粉砕機、グラスビーズを用いた細胞破砕機などを挙げることができ、化学的手段としては、リゾチームなどの溶菌酵素を挙げることができる。物理的手段又は化学的手段を用いて菌体を破砕する場合には、それぞれ破砕物が分解活性を損なわない穏和な条件下で行うことが好ましい。培養上清としては、前述した液体培地中で微生物を培養したものであればよく、培養上清における分解活性の強さから、B12−1株では好ましくは培養2〜6日目、より好ましくは3〜4日目の培養上清を使用することができ、組み換え微生物では好ましくは1〜3日目、より好ましくは2日目の培養上清を使用することができる。
【0048】
PLAを含む物質と、B12−1株又は組換え微生物とを接触させる接触方法としては、特に限定されないが、例えば、培養槽に、基本培地及び分解対象のPLAを添加し、さらに、種々の形態のB12−1株もしくは組換え微生物を添加することによって接触させることができる。分解時のpHは、分解速度の点から、pH6.0〜9.0とすることが好ましく、pH7.0〜8.0とすることがより好ましい。分解時の温度は、B12−1株では、分解速度の点から25〜50℃とすることが好ましく、30〜40℃とすることがより好ましい。また、組み換え微生物では、分解速度の点から20〜40℃とすることが好ましく、25〜35℃とすることがより好ましい。分解時間は、B12−1株では処理効率の点から2〜6日間とすることが好ましく、3〜4日間とすることがより好ましい。また組み換え微生物では処理効率の点から、1〜3日間とすることが好ましく、1〜2日間とすることがより好ましい。使用するB12−1株又は組換え微生物の量は、対象となるPLAの種類及びPLAを含む物質の形態によって異なるが、例えば、分子量4×104のPLAを含む乳化液中のPLAを分解する場合、乳化液中に含まれるPLA1gに対して、1.0×108〜2.0×109cfuの菌体を用いることが好ましく、5.0×108〜1.0×109cfuの菌体を用いることがより好ましい。
【実施例】
【0049】
以下、例を挙げて本発明を説明するが、本発明はこれらの実施例に限定されるものではない。
【0050】
<実施例1.ミクロモノスポラ属のB12−1株によるPLAの分解>
(菌株の培養とクリアゾーンの測定)
菌株としてミクロモノスポラ(Micromonospora)属のB12−1株(受領番号:NITE AP−1105)を用いた。試験管に分注したTryptic Soy Broth(TSB)培地(5mL)に一白金耳量の菌体を植菌後、振とう培養(40℃、120rpm)し、これを前培養液とした。本培養として、BM培地に前培養液を1%植菌し、振とう培養(40℃、120rpm)した。続いて固体培養をおこなった。固体培養にはPLA基本培地またはTSB培地にアガロース(BOL:清水食品)を1.5%添加した寒天培地を用いた。滅菌したつまようじでスラントから菌体を取りPLA基本培地の寒天培地に3点植菌した。37℃で5日間培養し、培養終了時にクリアゾーンの直径を測定した。その結果、B12−1株は直径19mmのクリアゾーンを形成した(図1)。
【0051】
(培地の組成)
各培地の組成を以下に示す。
【表1】

【表2】
【表3】
なお、乳化PLA(0.2%)は以下の方法で調製した。0.4gのポリ乳酸樹脂(Vyloecol BE−400、東洋紡)を20mLのジクロロメタンに溶解した。リン酸カリウム緩衝液(10mM、pH7.3)を200mL加え、超音波処理(Ultrtasonic generator US−300T:日本精機製作所、チップサイズ26、400W、5分間×3回)し、乳化した。
【0052】
<実施例2.PLA分解酵素の精製>
(酵素活性測定法)
本発明において、酵素活性を以下の方法により測定した。0.4gのポリ乳酸樹脂(Vyloecol BE−400、東洋紡)を20mLのジクロロメタンに溶解した。これにTris−HCl緩衝液(10mM、pH9.0)を200mL加え、超音波処理(Ultrtasonic generator US−300T:日本精機製作所、チップサイズ26、400W、5分間×3回)して乳化し、この乳化PLA(0.2%)を基質とした。乳化PLA650μLに酵素液50μLを添加し反応させた(50℃、10分間)。反応後、3分間煮沸し反応を停止させた。反応液の濁度(630nm)の減少量からPLA分解活性を求めた。なお、本明細書では、PLA分解活性1Uを、1分間に630nmにおける乳化PLAの濁度を1.0減少させる酵素量と定義した。
【0053】
(菌体の培養)
以下のように、前培養にはTSB培地を用い、本培養にはBMB12培地を用いた。試験管にTSB培地5mLを分注し、コロニーを一白金耳量植菌後、振とう培養し(37℃、180rpm、3日間)、これを前々培養液とした。500mL容三角フラスコにTSB培地100mLを分注し、前々培養液を1%植菌後、振とう培養し(37℃、180rpm、2日間)、これを前培養液とした。500mL容三角フラスコにBMB12培地を200mL分注し、前培養液を1%植菌後、振とう培養し(37℃、180rpm、4日間)、これを培養液とした。培養液を遠心分離(500mLボトル、5000rpm、4℃、10分間、himac SCR−20B:HITACHI)し、上清を回収した。
【0054】
培地の組成を以下に示す。
【表4】
【0055】
(酵素精製)
酵素精製は特に断らない限り低温室(7℃)で行った。
(1)硫安沈殿
培養液上清を限外濾過膜(microza SLP−0053:旭化成)で2〜3倍に濃縮後、その濃縮液を硫安沈殿に用いた。培養液上清に粉末状にした硫酸アンモニウムを60%飽和となるよう徐々に添加した。その後、1時間以上氷冷した。氷冷後、遠心分離(500mlボトル、4℃、12000rpm、30分、himac CR22GII:HITACHI)し、沈殿をリン酸カリウム緩衝液(20mM、pH8.0)に溶解した。同緩衝液で一晩透析し、活性画分を得た。活性画分を陰イオン交換クロマトグラフィーに供した。
【0056】
(2)陰イオン交換クロマトグラフィー
緩衝液はリン酸カリウム緩衝液(20mM、pH8.0)を用いた。充填剤としてTOYOPEARL DEAE−650M(東ソー株式会社)を用い、カラムに充填した。カラム(カラム容量:400mL)を5倍量の緩衝液で予め平衡化した。カラムにサンプルを通液しPLA分解酵素を吸着後、カラム容量の3倍量の同緩衝液で洗浄した。PLA分解酵素をNaClの濃度勾配(0〜0.5M)で溶出させた。活性画分を同緩衝液で透析後、TOYOPEARL DEAE−650M(東ソー株式会社)を充填したカラム容量100mLのカラムに換え、NaClの濃度勾配を0〜0.3Mとし、再度陰イオン交換クロマトグラフィーを行い、活性画分を得た。
【0057】
(3)疎水クロマトグラフィー
緩衝液はリン酸カリウム緩衝液(20mM、pH8.0)を用いた。充填剤としてTOYOPEARL Butyl−650M(東ソー株式会社)を用い、カラム(カラム容量:30mL)に充填した。カラムを、硫酸アンモニウム(30%飽和)を含むリン酸カリウム緩衝液で予め平衡化した。陰イオン交換クロマトグラフィーで得られた活性画分に硫酸アンモニウムを30%飽和となるように添加し、1時間氷冷後、遠心分離(500mLボトル、4℃、12000rpm、30分)した。これをカラムに通液し、PLA分解酵素を吸着させた。カラムを、硫酸アンモニウム(30%飽和)を含むリン酸カリウム緩衝液で洗浄した。PLA分解酵素を硫酸アンモニウムの濃度勾配(30〜0%飽和)を用いて溶出させ、活性画分を得た。
【0058】
(4)SDS−ポリアクリルアミドゲル電気泳動(SDS−PAGE)
疎水クロマトグラフィーで得られた活性画分をSDS−PAGEに供したところ、単一なバンドを示したため、これを精製酵素とした。また、SDS−PAGEからPLA分解酵素の分子量は31kDaと推定した。
【0059】
<実施例3.PLA分解酵素の性質>
(基質特異性)
PLA分解酵素の生分解性プラスチックに対する基質特異性を調べた。生分解性プラスチックの基質には、PLA(「Vyloecol BE−400」東洋紡、分子量4×104)、PLA(「LACEA #400」三井化学、分子量1.5×105)、PLA(「Lacty」島津、分子量2.2×105)、ポリカプロラクトン(PCL、「CELGREEN」ダイセル化学工業株式会社)、ポリブチレンサクシネートアジペート(PBSA「ビオノーレ3001」昭和高分子株式会社)、ポリブチレンサクシネート(PBS「ビオノーレ1001」昭和高分子株式会社)を用いた。基質特異性はPLA(Vyloecol BE−400)に対する分解活性を100とし、相対活性で示した。結果を表5に示す。表5に示す通り、本発明のPLA分解酵素は、PLAを分解したが、PCL、PBSAにはほとんど作用せず、PBSには全く作用しなかった。PLAでは分子量の小さい方がよく分解された。
【0060】
【表5】
【0061】
(至適温度および熱安定性)
PLA分解酵素の作用至適温度を以下のようにして求めた。酵素活性測定法における反応温度を30〜90℃まで10℃ずつ変化させて酵素反応を行い、各温度における活性を測定した。その結果、PLA分解酵素は70℃で最も活性が高く、70℃を至適温度とした。また、PLA分解酵素の熱安定性を調べるために、酵素溶液を所定の温度で30分間加熱処理した後、直ちに冷却し、残存活性を上述した酵素活性測定法に従って測定した。その結果、本発明のPLA分解酵素は、70℃の活性を100%として、50℃、30分間保温後も100%の活性を維持し、60℃、30分間保温後でも約50%の残存活性を有していた。
【0062】
(至適pHおよびpH安定性)
PLA分解酵素の作用至適pHを以下のようにして求めた。酵素活性測定における反応液中の緩衝液として、リン酸カリウム緩衝液(pH6.0〜8.0)、Tris−HCl緩衝液(pH7.5〜9.0)、グリシン−NaOH緩衝液(pH9.0〜11.0)、KCl−NaOH緩衝液(11.0〜12.0)を用いて反応を行い、活性を測定した。その結果、PLA分解酵素はpH9.0で最も活性が高く、pH9.0を至適pHとした。また、PLA分解酵素のpH安定性を調べるために、酵素溶液をpHの異なる緩衝液で透析し、4℃で20時間放置後、残存活性を酵素活性測定法に従って測定した。その結果、pH7.5の緩衝液を用いた場合の活性を100%として、本発明のPLA分解酵素はpH6.0〜pH9.0の範囲では90%以上の残存活性を示した。また、pH10.0〜pH12.0の範囲では約80%の残存活性を示した。
【0063】
<実施例4.PLA分解酵素遺伝子のクローニング>
(PLA分解酵素のN末端および内部アミノ酸配列の決定)
PLA分解酵素のアミノ酸配列を、プロテインシーケンサー(PPSQ−21A、島津製作所)を用いて分析した。N末端アミノ酸配列の分析には実施例2で得られた精製酵素をサンプルとした。その結果、N末端配列として、GTQTGATWGLDRIDQRNLPL(配列番号1の第1〜20番目)の配列を有していた。内部アミノ酸配列の分析には精製酵素を臭化シアンで断片化したものをサンプルとした。サンプルをSDS−PAGEに供したところ3種類の酵素断片(C1:14kDa、C2:11kDa、C3:0.6kDa)が確認できた。このうちC1、C2断片をPVDF膜に転写し、アミノ酸配列の決定に用いた。その結果、C1断片のアミノ酸配列は、N末端アミノ酸配列と等しかった。C2断片は、SLGGGASTTLDNAVANSIAS(配列番号1の第128〜147番目)の配列を有していた。
【0064】
(PLA分解酵素遺伝子の部分増幅)
ハイブリダイゼーションのプローブに用いるPLA分解酵素遺伝子断片を取得するために、PLA分解酵素遺伝子の部分増幅を、縮重プライマー(degenerated primer)を用いて、PCR装置(Thermal Cycler Dice TP600:Takara)で行った。DNAポリメラーゼとして、Hot star Taq(QIAGEN)を用いた。まず、PLA分解酵素のN末端およびC2断片の内部アミノ酸配列を基に、縮重プライマーを設計した。設計した縮重プライマーを以下に示す。
5’−ATCGACCAGCGCAACCTG−3’(F−aur427、配列番号5)
5’−GTTSGCSACSGCGTTGTC−3’(SはG又はC)(R−DNAVAN、配列番号6)
設計した縮重プライマーを用い、B12−1株の総DNAを鋳型としてPCRを行ったところ、約390bpの増幅断片を得た。この増幅断片をシークエンスしたところ、内部アミノ酸配列(SLGGGASTTL)(配列番号1の第128〜137番目)をコードする塩基配列が認められたため、この増幅断片をハイブリダイゼーションに用いるプローブとした。
【0065】
(サザンハイブリダイゼーション)
サザンハイブリダイゼーションと、後述するコロニーハイブリダイゼーションを、非放射性システムであるDIG−High Prime DNA Labeling and Detection Starter Kit II(ロシュダイアグノスティック社)を用いて行った。上記で得られた増幅断片をラベル化プローブ調製の鋳型とした。鋳型DNA(1μg)に滅菌蒸留水を加えて16μLにした。沸騰水中で10分間インキュベートして鋳型DNAを熱変性させた後、氷水で急速に冷却した。その後、DIG−high Primeを4μL加え、37℃で20時間反応させた。反応後、2μLのEDTA(0.2M、pH8.0)を加え、反応を停止させた。これをラベル化プローブとした。一方、B12−1株の総DNAを制限酵素であるKpnI、PstI、SacIおよびSphIで切断した(37℃、3時間)。切断後のDNA断片をアガロースゲル電気泳動に供し、サザンハイブリダイゼーションを行った。その結果、SphI断片の中で約9kbpのDNA断片がプローブとハイブリダイズした。そこで、この約9kbpのSphI断片をpUC19に連結し、コロニーハイブリダイゼーションを行った。
【0066】
(コロニーハイブリダイゼーション)
SphI断片(9kbp)をpUC19に連結した組換えプラスミドを大腸菌に形質転換した。コロニー500個に対しコロニーハイブリダイゼーションを行ったところ、陽性クローンが1個得られた。陽性クローンから得た組換えプラスミドのSphI断片に対しサザンハイブリダイゼーションを行ったところ、プローブがハイブリダイズした。よってこの9kbpのSphI断片にPLA分解酵素遺伝子が含まれると考えられた。このSphI断片(9kbp)を含む組換えプラスミドをpG12として以降の実験に用いた。
【0067】
(PLA分解酵素遺伝子の位置の推定)
組換えプラスミドpG12を各種の制限酵素で切断した後、サザンハイブリダイゼーションを行った。この結果より、9kbpのSphI断片中にEcoRI認識部位が1ヶ所、BamHI認識部位が2ヶ所、BglII認識部位が2ヶ所存在することが確認できた。次にSphI断片(9kbp)を、制限酵素EcoRI、BamHI、BglIIを組み合わせて切断し、断片長をアガロースゲル電気泳動から推定した。これらの断片に対しサザンハイブリダイゼーションを行い、制限酵素地図を作製し、遺伝子の位置を推定した(図2)。
【0068】
(PLA分解酵素遺伝子の塩基配列決定)
制限酵素地図から、PLA分解酵素遺伝子を含むと推定されるEcoRI−BamHI断片(2.2kbp)をpUC19ベクターにサブクローニングし、シークエンスを行った。シークエンスはベックス社に依頼した。シークエンス試薬は、BigDye Terminators v.1.1 Cycle Sequencing Kit(Applied Biosystems Japan)を用い、シーケンサーは、ABI Prism 3130 Genetic Analyzer (Applied Biosystems Japan)を使用した。決定した塩基配列は、Geneticx−Win Ver.8.0(Software development)で解析した。シークエンスの結果、開始コドン(ATG)、終止コドン(TGA)および開始コドンの上流にSD配列と推定される配列(AGGGAG)が認められた。配列は、配列番号4に示す1221bpからなり、配列番号3に示す406アミノ酸をコードしていた。シグナルペプチドをSignal iP 3.0を用いて推定した。また、シグナルペプチドの次のアミノ酸から、プロテインシーケンサーで明らかにしたPLA分解酵素のN末端アミノ酸の1つ前のアミノ酸までの配列をプロペプチドの配列とした。活性化酵素になる前の未成熟酵素は、シグナルペプチド(配列番号3の第1〜35番目、35アミノ酸残基)、プロペプチド(配列番号3の第36〜130番目、95アミノ酸残基)、成熟酵素(配列番号3の第131〜406番目、すなわち、配列番号1、276アミノ酸残基)から構成されると推定された。PLA分解酵素は、プロテアーゼで良く知られるPre−Pro−Enzymeとして生産され、シグナルペプチドにより細胞外に輸送された後、プロペプチドが切断され、活性化酵素として働くと推定された。
【0069】
本発明のPLA分解酵素のアミノ酸配列と類似性の高い酵素を検索したところ、Thermus aquaticu 由来Aqualysin1(類似性53%)、Thermus sp. Rt41A由来 Serine protease(53%)、Serratia sp. GF96由来Proteinase K(52%)が見出された。これらは、Pre−Pro−Enzymeとして生産され、かつ、セリンプロテアーゼに属する。よって遺伝子情報からは、本発明のPLA分解酵素はセリンプロテアーゼの一種であると推定された。本発明のPLA分解酵素では、活性中心と予測されるアスパラギン酸(配列番号2の第39番目)、ヒスチジン(配列番号2の第70番目)、セリン(配列番号2の第222番目)が保存されていた。
【0070】
<実施例5.PLA分解酵素遺伝子の組換え微生物への組み込みおよび遺伝子発現>
(プラスミドpBPa1の構築)
以下のようにして、組換えベクターとして、プラスミドpBPa1を構築した。遺伝子を組み込むプラスミドとして、放線菌大腸菌シャトルベクターである、pUC702を用いた。pUC702は、pUC19とpIJ702をKpnI、SacIサイトで連結したプラスミドである。PCRによりPLA分解酵素遺伝子増幅を行った。PCR用の鋳型として、PLA分解酵素遺伝子を含む組換えプラスミドpGBEを用い、プライマーとして、F−B12p702およびR−B12p702を用いた。これらのプライマーは、遺伝子断片がプロモーター領域またはターミネーター領域を含んで増幅されるように設計したものである。
F−B12p702およびR−B12p702プライマーの配列を以下に示す。
5’−GCGCGAATTCCCGCCCGGTAGTCGTTGC−3’(F−B12p702、配列番号7)
5’−AGAGGAATTCGCGACCATCCTGCCGGTC−3’(R−B12p702、配列番号8)
増幅された遺伝子断片を、pUC702のEcoRIサイトに挿入し、発現用プラスミドpBPa1を構築した。
【0071】
(放線菌プロトプラストの調製)
PLA分解酵素遺伝子を発現させる放線菌として、ストレプトマイセス・リビダンス(Streptomyces lividans)TK21を用いた。滅菌した試験管にTSB培地を5ml分注し、一白金耳量の菌体を植菌して振とう培養し(180rpm、30℃、48時間)、これを前培養液とした。次にYEME培地を100mL分注した500mL容三角フラスコに前培養液を2%植菌し、振とう培養し(180rpm、30℃、36時間)、これを培養液とした。培養液を集菌し、10.3%スクロース溶液で2回洗浄した。リゾチーム溶液(1mg/mLとなるようリン酸緩衝液に溶解)8mLを加え、よく懸濁し、30℃で15分間インキュベートした。さらに5mLのリン酸緩衝液を加え15分間インキュベートした。その後、反応液を滅菌したコットンでろ過し、ろ液を遠心分離(1500rpm、室温、7分間)した。上清を除き、沈殿したプロトプラストにリン酸緩衝液を3mL加え、穏やかに懸濁し、プロトプラスト溶液を調製した。トーマ血球計算版を用い、プロトプラストの濃度を計測した。マイクロチューブに分注し、−80℃で保存した。
【0072】
培地の組成を以下に示す。
【表6】
【0073】
(放線菌の形質転換)
プロトプラスト溶液を30℃のウォーターバスで素早く溶解した。プロトプラスト溶液150μLに適量のプラスミドpBPa1溶液を加え、穏やかに攪拌した。その後すぐにリン酸緩衝液(25%PEG含有)を加えゆっくりと3回ピペッティングした。この溶液をR5プレートに丁寧に塗布し、30℃で培養した。培養16〜20時間後にチオストレプトン含有軟寒天培地を重層し、さらに30℃で3日間培養し、プラスミドpBPa1を形質転換した組換え微生物Streptomyces lividans BP1を作製した。
【0074】
(組換え微生物の培養)
組換え微生物Streptomyces lividans BP1の培養は、終濃度5μg/mlとなるようチオストレプトンを加えたTSB培地で行った。まず、滅菌した試験管に上記TSB培地を5mL分注し、一白金耳量の菌体を植菌して振とう培養し(180rpm、30℃、2日間)、これを前培養液とした。次に100mLの上記TSB培地を分注した500mL容三角フラスコに前培養液を1%となるよう植菌し、振とう培養した(180rpm、30℃、2日間)。
【0075】
(組換え微生物のPLA分解能)
組換え微生物Streptomyces lividans BP1は14.0U/mlのPLA分解活性を示し、B12−1株の28倍の酵素生産性を示した(図3)。また、組換え微生物Streptomyces lividans BP1の、PLA基本培地の寒天培地でのクリアゾーンを測定した。B12−1株は37℃、5日間の培養で直径19mmのクリアゾーンを示したのに対し、組換え微生物Streptomyces lividans BP1は30℃、5日間の培養で直径45mmのクリアゾーンを示した(図4)。このことから組換え微生物Streptomyces lividans BP1は高いPLA分解活性を有することが示された。
【受託番号】
【0076】
ミクロモノスポラ(Micromonospora)属のB12−1株(千葉県木更津市かずさ鎌足2−5−8、独立行政法人製品評価技術基盤機構特許微生物寄託センター、受領番号:NITE AP−1105、2011年6月13日付受領にて寄託)
組換え微生物Streptomyces lividans BP1(千葉県木更津市かずさ鎌足2−5−8、独立行政法人製品評価技術基盤機構特許微生物寄託センター、受領番号:NITE AP−1106、2011年6月13日付受領にて寄託)
【技術分野】
【0001】
本発明は、新規のポリ乳酸分解酵素、前記酵素をコードする核酸、前記酵素を産生する新規微生物、及び、前記核酸が組み込まれた組換え微生物に関する。
【背景技術】
【0002】
ポリ乳酸(PLA)は、乳酸を化学合成によって重合させた物質である。PLAは融点やガラス転移点が高いため耐熱性に優れる。さらに透明度が高く従来のプラスチックと同等の物理的性質を備えているため、日用品から農業用品、医療機器や自動車部品などさまざまな分野で利用することが可能である。
【0003】
PLAは生分解性プラスチックであり、現在までに、PLAを分解する酵素や該酵素を産生する微生物についての研究がおこなわれてきた。例えば、特許文献1〜5は、PLA分解能を有する酵素や微生物を開示する。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特許第3762990号公報
【特許文献2】特許第4441639号公報
【特許文献3】特開2007−319077号公報
【特許文献4】特開2007−319078号公報
【特許文献5】特開2008−167701号公報
【発明の概要】
【発明が解決しようとする課題】
【0005】
PLA分解能は酵素や微生物の種類によって異なり、PLAの利用拡大のためには、より高いPLA分解能を有する酵素や微生物が必要である。したがって、本発明は、PLA分解能を有する新規酵素及び新規微生物を提供し、さらに、それらを用いた効率的なPLA分解方法を提供することを目的とする。
【課題を解決するための手段】
【0006】
本発明は、下記(a)〜(c)からなる群から選択されるポリペプチドを含有するタンパク質:
(a)配列表の配列番号1に記載のアミノ酸配列からなるポリペプチド;
(b)配列表の配列番号1に記載のアミノ酸配列に対して1個または数個のアミノ酸が欠失、置換、付加、または挿入されたアミノ酸配列を有し、かつ、PLA分解活性を有するポリペプチド;
(c)配列表の配列番号1に記載のアミノ酸配列と90%以上の同一性を有するアミノ酸配列を有し、かつ、PLA分解活性を有するポリペプチド、を提供する。
本発明のタンパク質は、新規PLA分解酵素である。配列表の配列番号1に記載のアミノ酸配列からなるポリペプチドは、高いPLA分解活性を有する。
【0007】
上記タンパク質が、配列番号1のアミノ酸配列の第34〜44番目のアミノ酸からなるペプチド、第65〜75番目のアミノ酸からなるペプチド及び第217〜227番目のアミノ酸からなるペプチドを含むことが好ましい。これらのペプチドは、PLA分解酵素の活性中心を含む。
【0008】
また、本発明は、上記タンパク質をコードするポリヌクレオチドを含有する核酸を提供する。
さらに、本発明は、下記(1)〜(4)からなる群から選択されるポリヌクレオチドを含有する核酸;
(1)配列表の配列番号2に記載の塩基配列からなるポリヌクレオチド;
(2)配列表の配列番号2に記載の塩基配列に対して1個〜数個の塩基が欠失、置換、付加、または挿入された塩基配列を有し、かつ、ポリ乳酸分解活性を有するポリペプチドをコードするポリヌクレオチド;
(3)配列表の配列番号2に記載の塩基配列と90%以上の同一性を有する塩基配列を有し、かつ、ポリ乳酸分解活性を有するポリペプチドをコードするポリヌクレオチド;
(4)(1)のポリヌクレオチドとストリンジェントな条件下でハイブリダイズし、かつ、ポリ乳酸分解活性を有するポリペプチドをコードするポリヌクレオチド、を提供する。
配列番号2に記載の塩基配列は、配列番号1に記載のアミノ酸配列からなるポリペプチドをコードする塩基配列である。すなわち、本発明の核酸は、PLA分解酵素をコードする。したがって、上記核酸を微生物に組み込んで、本発明のPLA分解酵素を微生物に産生させることができる。
【0009】
上記核酸が、配列番号1のアミノ酸配列の第34〜44番目のアミノ酸からなるペプチド、第65〜75番目のアミノ酸からなるペプチド及び第217〜227番目のアミノ酸からなるペプチドを含むヌクレオチドを含むことが好ましい。上記ヌクレオチドは酵素の活性中心を含むペプチドをコードする。
【0010】
また、本発明は、ミクロモノスポラ(Micromonospora)属に属し、配列表の配列番号2で示される塩基配列の核酸を有する、受領番号NITE AP−1105として寄託されている放線菌B12−1株を提供する。この放線菌B12−1株は、本発明のPLA分解酵素を産生する微生物である。
【0011】
また、本発明は、上記核酸を含有する組換えベクターを含む組換え微生物を提供する。組換え微生物により、本発明のPLA分解酵素が産生される。上記組換え微生物は、ストレプトマイセス(Streptomyces)属に属することが好ましく、受領番号NITE AP−1106として寄託されている微生物であることがさらに好ましい。この組換え微生物は、極めて高いPLA分解活性を有する。
【0012】
また、本発明は、放線菌B12−1株又は上記組換え微生物を培養し、得られる培養物からPLA分解酵素を回収する、PLA分解酵素の製造方法を提供する。
【0013】
さらに、本発明は、上記本発明のタンパク質、放線菌B12−1株及び上記組換え微生物からなる群から選択される少なくとも1種とPLAを含む物質とを接触させる、PLAの分解方法を提供する。本発明の分解方法は、PLA分解活性の高い酵素や微生物を用いているため、効率的にPLAが分解される。
【発明の効果】
【0014】
本発明によれば、PLA分解能を有する新規酵素及び新規微生物、さらに、それらを用いた効率的なPLA分解方法が提供される。
【図面の簡単な説明】
【0015】
【図1】図1は、B12−1株を寒天培地に37℃、5日間培養した際にB12−1株が形成したクリアゾーンを示す図である。
【図2】図2は、組換えプラスミドpG12を用いて作製した制限酵素地図を表す図である。
【図3】図3は、B12−1株の分解活性と組換え微生物Streptomyces lividans BP1の分解活性とを比較した図である。
【図4】図4は、Streptomyces lividans BP1を寒天培地に30℃、5日間培養した際にStreptomyces lividans BP1が形成したクリアゾーンを示す図である。
【発明を実施するための形態】
【0016】
(タンパク質)
本発明が提供するタンパク質は、新規PLA分解酵素(以下、場合により単に「PLA分解酵素」と称する。)である。本発明のタンパク質は、PLAを分解することが知られている既知の酵素よりも高いPLA分解活性を有する。したがって、本発明のタンパク質を用いることで、従来よりも早く、またはより多くのPLAを分解することができる。本発明のタンパク質は、(a)配列表の配列番号1に記載のアミノ酸配列からなるポリペプチドを含有する。(a)配列表の配列番号1に記載のアミノ酸配列からなるポリペプチド(全長276アミノ酸残基)は、それ自身がPLA分解活性を有するポリペプチドである。
【0017】
本発明が提供するタンパク質が含有するポリペプチドは、(a)配列表の配列番号1に記載のアミノ酸配列からなるポリペプチドに限定されず、(b)配列表の配列番号1に記載のアミノ酸配列に対して1個または数個のアミノ酸が欠失、置換、付加、または挿入されたアミノ酸配列を有し、かつ、PLA分解活性を有するポリペプチドであってもよい。「1個または数個」とは1個から30個、好ましくは1個から10個、より好ましくは1個から5個、さらに好ましくは1個から3個である。さらに、本発明が提供するタンパク質が含有するポリペプチドは、(c)配列表の配列番号1に記載のアミノ酸配列と90%以上の同一性を有するアミノ酸配列を有し、かつ、PLA分解活性を有するポリペプチドであってもよい。該ポリペプチドのアミノ酸配列と配列番号1に記載のアミノ酸配列との同一性は、好ましくは93%以上、より好ましくは95%以上、さらに好ましくは98%以上である。
【0018】
本明細書における「PLA分解活性」は、例えば、実施例2に記載の酵素活性測定法により測定することができる。また、本明細書において「PLA分解活性を有する」とは、配列番号1に記載のアミノ酸配列からなるポリペプチドの、例えば50%以上、好ましくは70%以上、より好ましくは80%以上、さらに好ましくは95%以上のPLA分解活性を有することと定義することができる。
【0019】
本発明のタンパク質は、配列番号1に記載のアミノ酸配列から、セリンプロテアーゼの一種であると推定され、PLA分解活性の活性中心は、アスパラギン酸(配列番号1の第39番目)、ヒスチジン(配列番号1の第70番目)及びセリン(配列番号1の第222番目)であると推定される。したがって、本発明のタンパク質はこの活性中心を含むことが好ましい。具体的には、本発明のタンパク質は、配列番号1のアミノ酸配列の第34〜44番目のアミノ酸からなるペプチド、第65〜75番目のアミノ酸からなるペプチド及び第217〜227番目のアミノ酸からなるペプチドを含むことが好ましい。
【0020】
配列表の配列番号3に記載のアミノ酸配列からなるポリペプチドは、(a)配列番号1に記載のアミノ酸配列からなるポリペプチドに、シグナルペプチド(配列番号3の第1〜35番目、35アミノ酸残基)、及びプロペプチド(配列番号3の第36〜130番目、95アミノ酸残基)が付加されたポリペプチド(全長406アミノ酸残基)である。配列番号1に記載のポリペプチドは、配列番号3に記載のアミノ酸配列からなるPre−Pro−Enzymeとして生産され、シグナルペプチドにより細胞外に輸送された後、プロペプチドが切断され、活性化酵素として働くことができる。
【0021】
本発明のタンパク質は、(a)〜(c)から選択されるポリペプチドに、別種のタンパク質または物質が結合されたものでもよい。例えば、上記ポリペプチドの検出または精製を容易にするために、あるいは別の機能を付加するために、そのN末端側やC末端側に別種のタンパク質またはペプチド、例えばグルタチオンS−トランスフェラーゼ(GST)、ルシフェラーゼ、緑色蛍光タンパク質(GFP)、β−ガラクトシダーゼ、免疫グロブリン(IgG)、His−tag等が、直接的にまたはリンカーペプチド等を介して間接的に、遺伝子工学的手法等を用いて付加されたものであってもよい。
【0022】
(核酸)
本発明は、上記タンパク質をコードするポリヌクレオチドを含有する核酸、すなわちPLA分解酵素をコードするポリヌクレオチドを含有する核酸を提供する。具体的には、(1)配列表の配列番号2に記載の塩基配列からなるポリヌクレオチドを含有する核酸が挙げられる。(1)配列表の配列番号2に記載の塩基配列からなるポリヌクレオチド(全長831残基)は、(a)配列表の配列番号1に記載のアミノ酸配列からなるポリペプチドをコードするポリヌクレオチドである。
【0023】
本発明が提供する核酸が含有するポリヌクレオチドは、(1)配列表の配列番号2に記載の塩基配列からなるポリヌクレオチドに限定されず、(2)配列表の配列番号2に記載の塩基配列に対して1個〜数個の塩基が欠失、置換、付加、または挿入された塩基配列を有し、かつ、PLA分解活性を有するポリペプチドをコードするポリヌクレオチドであってもよい。「1個または数個」とは1個から30個、好ましくは1個から10個、より好ましくは1個から5個、さらに好ましくは1個から3個である。また、本発明が提供する核酸が含有するポリヌクレオチドは、(3)配列表の配列番号2に記載の塩基配列と90%以上の同一性を有する塩基配列を有し、かつ、PLA分解活性を有するポリペプチドをコードするポリヌクレオチドであってもよい。該ポリヌクレオチドの塩基配列と配列番号2に記載の塩基配列との同一性は、好ましくは93%以上、より好ましくは95%以上、さらに好ましくは98%以上である。さらに、本発明が提供する核酸が含有するポリヌクレオチドは、(4)配列表の配列番号2に記載の塩基配列からなるポリヌクレオチドとストリンジェントな条件下でハイブリダイズし、かつ、PLA分解活性を有するポリペプチドをコードするポリヌクレオチドであってもよい。
【0024】
本明細書において、「ストリンジェントな条件下」とは、各塩基配列間の同一性の程度が、例えば、全体の平均で約80%以上、好ましくは約90%以上、より好ましくは約95%以上であるような、高い同一性を有する塩基配列間のみで、特異的にハイブリッドが形成されるような条件を意味する。具体的には、例えば、ナトリウム濃度が約19〜40mM、好ましくは約19〜20mMで、温度が約50〜70℃、好ましくは約60〜65℃の条件を示す。特に、ナトリウム濃度が約19mMで温度が約65℃の場合が好ましい。
【0025】
ハイブリダイゼーションは、当業界で公知の方法あるいはそれに準じる方法、例えば、モレキュラー・クローニング(Molecular Cloning)2nd(J. Sambrook et al.,Cold Spring Harbor Lab. Press, 1989)に記載の方法などに従って行うことができる。また、市販のライブラリーを使用する場合、添付の使用説明書に記載の方法に従って行なうことができる。
【0026】
(ミクロモノスポラ属に属する新規微生物B12−1株)
本発明のタンパク質が含有する(a)配列表の配列番号1に記載のアミノ酸配列からなるポリペプチドは、ミクロモノスポラ(Micromonospora)属に属する新規微生物B12−1株(千葉県木更津市かずさ鎌足2−5−8、独立行政法人製品評価技術基盤機構特許微生物寄託センター、受領番号:NITE AP−1105、2011年6月13日付受領にて寄託、以下、場合により「B12−1株」と称する。)から単離精製することができる。B12−1株は、PLA分解能を有する新菌種である。B12−1株は、PLAを分解可能な微生物として土壌から分離されたグラム陽性を示す放線菌であり、胞子を形成する。B12−1株は、PLAの分解について優れた能力を有する。
【0027】
B12−1株は、微生物の培養に用いられる通常の条件下で培養して、維持・増殖させることができる。例えば、pH6.0〜8.0、温度20〜50℃の培養条件で培養することができる。生育速度の点から、pH6.8〜7.3、温度37〜40℃で培養することが好ましい。
【0028】
B12−1株を維持可能な培地として、微生物培養に使用される炭素源、窒素源、無機塩類を適宜含む各種の培地を使用できる。例えばグルコース、フラクトース、マンノース、シュクロース、マンニトール、ソルビトール、糖蜜などの炭素源;例えば硫酸アンモニウム、リン酸アンモニウム、炭酸アンモニウム、ペプトン、酵母抽出物などの窒素源;例えばリン酸二カリウム、リン酸一ナトリウム、塩化マグネシウム、塩化カルシウム、硫酸第一鉄、モリブデン酸ナトリウム、塩化マンガンなどの無機塩類;を、それぞれ含むものであって、通常利用される固体又は液体培地であればいずれも好ましく使用できる。なお、培地には、上記成分の他、微生物の成育を促進させるための各種ビタミン、ミネラル、その他の栄養成分を含ませてもよい。
【0029】
(PLA分解酵素の製造方法)
本発明のPLA分解酵素は、B12−1株を培養し、得られる培養物からPLA分解酵素を回収することによって得ることができる。
【0030】
PLA分解酵素を得るための、B12−1株の培養に適した培地としては、具体的には、硫酸アンモニウム、リン酸水素二カリウム、リン酸二水素カリウム、硫酸マグネシウム、酵母エキス、オリーブ油等を含む液体培地を用いることができる。培地中の成分の濃度は限定されるものではなく、炭素源や窒素源などの種類や濃度によって適宜調整できる。
【0031】
PLA分解酵素は、B12−1株の菌体、菌体破砕物及び菌体の培養上清などの各形態から得ることができる。菌体の場合、その培養物から遠心分離等の集菌操作によって得られる生菌体、菌体を凍結乾燥した乾燥粉末、菌体を含む培養液のいずれであってもよい。破砕物としては、物理的手段又は化学的手段によって破砕されたものであればよく、物理的手段としては、超音波粉砕機、グラスビーズを用いた細胞破砕機などを挙げることができ、化学的手段としては、リゾチームなどの溶菌酵素を挙げることができる。物理的手段又は化学的手段を用いて菌体を破砕する場合には、それぞれ酵素が失活しない穏和な条件下で行うことが好ましい。培養上清としては、前述した液体培地中でB12−1株を培養したものであればよく、酵素産生量から、好ましくは培養2〜6日目、より好ましくは培養3〜4日目の培養上清を使用することができる。
【0032】
PLA分解酵素の回収法は、一般の酵素の回収の手段に準じて行うことができる。以下に示す方法に特に限定はされないが、例えば上記手段によって得られた菌体破砕物、あるいは培養上清を、粗酵素液として用いることができる。粗酵素液は、そのままで使用することもできるが、必要に応じて、例えば塩析法、沈澱法、限外濾過法等の分離手段、例えばイオン交換クロマトグラフィー、等電点クロマトグラフィー、疎水性クロマトグラフィー、ゲル濾過クロマトグラフィー、吸着クロマトグラフィー、アフィニティークロマトグラフィー、逆相クロマトグラフィー等の公知の方法を組み合わせて、更に分離精製することができる。
【0033】
PLA分解酵素を得るための別の方法としては、PLA分解酵素をコードするポリヌクレオチドを含有する核酸を含む組換え微生物を培養し、得られる培養物からPLA分解酵素を回収する方法が挙げられる。
【0034】
(組換え微生物)
本発明の組換え微生物は、PLA分解酵素のアミノ酸配列をコードするポリヌクレオチドを含有する核酸を含有する組換えベクターを含む。このような組換え微生物は、PLA分解酵素のアミノ酸配列をコードするポリヌクレオチドを含有する核酸をB12−1株より取得し、次いでこの核酸を適当なベクターに組込んで組換えベクターを作製し、この組換えベクターを用いて宿主を形質転換することにより作製することができる。
【0035】
本発明の組換え微生物を作製するために用いるベクターは、宿主の種類により適宜選択される。ベクターとして、宿主微生物体内で自律的に増殖しうるファージ又はプラスミドから構築されたベクターが適している。ファージベクターとしては、例えば、大腸菌(Escherichia coli)に属する微生物を宿主微生物とする場合にはλgt・λC、λgt・λBなどが使用できる。また、プラスミドベクターとしては、例えば、大腸菌を宿主微生物とする場合にはプラスミドpBR322、pBR325、pACYC184、pUC12、pUC13、pUC18、pUC19、pUC118、pINI、放線菌を宿主とする場合にはpIJ680、pIJ702、枯草菌を宿主とする場合にはpUB110、pKH300PLK、酵母を宿主とする場合にはYrp7、pYC1、Yep13などが使用でき、これらを組み合わせて使用してもよい。
【0036】
組換えベクターは、目的の遺伝子配列と、複製そして制御に関する情報を担持した遺伝子配列、例えばプロモーター、リボソーム結合部位、ターミネーター、シグナル配列、エンハンサー等とを構成要素とし、これらを公知の方法により組み合わせて作製される。ベクターに核酸を組み込む方法は、公知の方法を適用できる。例えば、適当な制限酵素を選択、処理して核酸を特定部位で切断し、次いで同様に特定部位で切断したベクターDNAと混合し、リガーゼによって再結合する方法が用いられる。あるいは、目的の核酸に適当なリンカーを結合し、これを目的に適したベクターのマルチクローニングサイトへ挿入することによっても、所望の組換えベクターが得られる。
【0037】
(宿主)
本発明の組換え微生物を作製するための宿主として、原核微生物又は真核微生物を用いることができる。原核微生物としては、例えば、ストレプトマイセス(Streptomyces)属、エシェリシア(Escherichia)属、バチルス(Bacillus)属等の菌を挙げることが出来、この中でもストレプトマイセス属が好ましい。具体的には、ストレプトマイセス・リビダンス(Streptomyces lividans)を挙げることができる。真核微生物の好適例としては、サッカロミセス(Saccharomyces)属及びピヒア(Pichia)属等の酵母、並びに、アスペルギルス(Aspergillus)属、ペニシリウム(Penicillium)属等の真菌を挙げることができる。
【0038】
(形質転換法)
核酸が組み込まれた組換えベクターを、宿主細胞に公知の方法で導入することにより組換え微生物が得られる。組換えベクターの宿主細胞への導入は、公知の方法により行うことができる。具体的には、リン酸カルシウムトランスフェクション、DEAE−デキストラン媒介トランスフェクション、マイクロインジェクション、陽イオン脂質媒介トランスフェクション、エレクトロポレーション、形質導入及び感染等が挙げられる。
【0039】
(組換え微生物Streptomyces lividans BP1)
上述のようにして、本発明の核酸を含有する組換えベクターを含む組換え微生物が得られる。本発明は、実施例5に記載の方法により得られた組換え微生物として、ストレプトマイセス・リビダンスを宿主とし、配列表の配列番号2に記載の塩基配列からなるポリヌクレオチドを含有する核酸を含有する組換えベクターを含む組換え微生物Streptomyces lividans BP1(千葉県木更津市かずさ鎌足2−5−8、独立行政法人製品評価技術基盤機構特許微生物寄託センター、受領番号:NITE AP−1106、2011年6月13日付受領にて寄託)を提供する。組換え微生物Streptomyces lividans BP1は、PLAの分解について優れた能力を有する。
【0040】
組換え微生物Streptomyces lividans BP1は、微生物の培養に用いられる通常の条件下で培養して、維持・増殖させることができる。例えば、pH6.0〜8.0、温度20〜37℃の培養条件で培養することができ、生育速度の観点から、pH6.8〜7.3、温度30〜35℃で培養することが好ましい。
【0041】
組換え微生物Streptomyces lividans BP1を維持可能な培地として、微生物培養に使用される炭素源、窒素源、無機塩類を適宜含む各種の培地を使用できる。例えばグルコース、フラクトース、マンノース、シュクロース、マンニトール、ソルビトール、糖蜜などの炭素源;例えば硫酸アンモニウム、リン酸アンモニウム、炭酸アンモニウムなどの窒素源;例えばリン酸二カリウム、リン酸一ナトリウム、塩化マグネシウム、塩化カルシウム、硫酸第一鉄、モリブデン酸ナトリウム、塩化マンガンなどの無機塩類;を、それぞれ含むものであって、通常利用される固体又は液体培地であればいずれも好ましく使用できる。なお、培地には、上記成分の他、微生物の成育を促進させるための各種ビタミン、ミネラル、その他の栄養成分を含ませてもよい。
【0042】
(組換え微生物によるPLA分解酵素の製造方法)
本発明の核酸を含有する組換えベクターを含む組換え微生物によっても、PLA分解酵素を得ることができる。本発明のPLA分解酵素は、該組換え微生物を培養し、得られる培養物からPLA分解酵素を回収することによって得ることができる。PLA分解酵素の産生のための培地は特に限定されないが、例えば、組換え微生物Streptomyces lividans BP1を培養する場合、PLA分解酵素の産生に適した培地として、具体的には、ポリペプトン、ソイペプトン、塩化ナトリウム、リン酸二水素カリウム、グルコース等を含む液体培地を用いることができる。PLA分解酵素は、組換え微生物の菌体、菌体破砕物及び培養上清などの各形態から得ることができ、それぞれB12−1株を用いた場合と同様の方法により調製できる。培養上清としては、酵素産生量から、好ましくは培養1〜3日目、より好ましくは2日目の培養上清を使用することができる。酵素の回収方法は、B12−1株を用いた場合と同様の方法を採用することができる。
【0043】
(PLAの分解方法)
本発明の、PLA分解酵素、B12−1株及び組換え微生物からなる群から選択される少なくとも1種とPLAを含む物質とを接触させることにより、PLAを分解することができる。本発明のPLA分解酵素は高いPLA分解活性を有するので、物質中に含まれるPLAを少ない時間で大量に分解することができ、効率的である。
【0044】
(PLA)
本発明の方法により分解されるPLAとしては、乳酸を主成分とする重合体であればよく、ポリL−乳酸及びポリD−乳酸のようなホモポリマー、ポリL/D−乳酸、これらと他の成分から構成される共重合体を挙げることができる。共重合体である場合には、乳酸成分は重量比率が10%以上のものであることが好ましい。また共重合体を構成する他の成分としては、ε−カプロラクトン、グリコリド、デプシペプチド等を挙げることができる。これらの他の成分は1種であっても、2種以上であってもよい。本発明によって分解可能なPLAの分子量に特に制限はないが、分解速度の点から、数平均分子量5000〜500000であることが好ましく、10000〜200000であることがより好ましい。また、PLAを含む物質の形態としては、例えば、フィルム(シート)、成型体、破砕物、粉末、懸濁液などを挙げることができる。また、分解時のPLAは、PLA単独であってもよいし、他のプラスチックとの混合物であってもよい。
【0045】
分解に用いるPLA分解酵素、B12−1株及び組換え微生物は、乾燥形態又は適当な液体担体に混合された液体形態であってもよい。液体形態を構成するために用いられる液体担体としては、この用途に通常用いられるものであれば特に制限されず、水、緩衝液等を挙げることができる。
【0046】
PLA分解酵素をPLAの分解に用いる場合、PLA分解酵素として、上述の方法により精製した酵素又は粗酵素を用いることができる。PLAを含む物質と、PLA分解酵素とを接触させる接触方法としては、特に限定されないが、PLA分解酵素を含む酵素液にPLAを含む物質を投入する、PLAを含む物質が入った容器にPLA分解酵素を添加する、等の方法が挙げられる。分解時のpHは、分解速度の点から、pH6.0〜12.0とすることが好ましく、pH9.0〜10.0とすることがより好ましい。分解時の温度は、分解速度の点から、30℃〜60℃とすることが好ましく、45〜55℃とすることがより好ましい。分解時間は、処理効率の点から、1日間〜5日間とすることが好ましく、2日間〜3日間とすることがより好ましい。使用するPLA分解酵素の量は、基質となるPLAの種類及びPLAを含む物質の形態によって異なるが、例えば、分子量4×104のPLAを含む乳化液中のPLAを分解する場合、乳化液中に含まれるPLA1gに対して、0.01〜0.2gのPLA分解酵素を使用することが好ましく、0.05〜0.1gを使用することがより好ましい。
【0047】
B12−1株又は組換え微生物をPLAの分解に用いる場合、種々の形態で使用することができる。このような形態としては、微生物菌体、菌体破砕物及び微生物の培養上清などの各形態から適宜選択することができる。菌体の場合には、その培養物から遠心分離等の集菌操作によって得られる生菌体、菌体を凍結乾燥した乾燥粉末、微生物を含む培養液のいずれであってもよい。破砕物としては、物理的手段又は化学的手段によって破砕されたものであればよく、物理的手段としては、超音波粉砕機、グラスビーズを用いた細胞破砕機などを挙げることができ、化学的手段としては、リゾチームなどの溶菌酵素を挙げることができる。物理的手段又は化学的手段を用いて菌体を破砕する場合には、それぞれ破砕物が分解活性を損なわない穏和な条件下で行うことが好ましい。培養上清としては、前述した液体培地中で微生物を培養したものであればよく、培養上清における分解活性の強さから、B12−1株では好ましくは培養2〜6日目、より好ましくは3〜4日目の培養上清を使用することができ、組み換え微生物では好ましくは1〜3日目、より好ましくは2日目の培養上清を使用することができる。
【0048】
PLAを含む物質と、B12−1株又は組換え微生物とを接触させる接触方法としては、特に限定されないが、例えば、培養槽に、基本培地及び分解対象のPLAを添加し、さらに、種々の形態のB12−1株もしくは組換え微生物を添加することによって接触させることができる。分解時のpHは、分解速度の点から、pH6.0〜9.0とすることが好ましく、pH7.0〜8.0とすることがより好ましい。分解時の温度は、B12−1株では、分解速度の点から25〜50℃とすることが好ましく、30〜40℃とすることがより好ましい。また、組み換え微生物では、分解速度の点から20〜40℃とすることが好ましく、25〜35℃とすることがより好ましい。分解時間は、B12−1株では処理効率の点から2〜6日間とすることが好ましく、3〜4日間とすることがより好ましい。また組み換え微生物では処理効率の点から、1〜3日間とすることが好ましく、1〜2日間とすることがより好ましい。使用するB12−1株又は組換え微生物の量は、対象となるPLAの種類及びPLAを含む物質の形態によって異なるが、例えば、分子量4×104のPLAを含む乳化液中のPLAを分解する場合、乳化液中に含まれるPLA1gに対して、1.0×108〜2.0×109cfuの菌体を用いることが好ましく、5.0×108〜1.0×109cfuの菌体を用いることがより好ましい。
【実施例】
【0049】
以下、例を挙げて本発明を説明するが、本発明はこれらの実施例に限定されるものではない。
【0050】
<実施例1.ミクロモノスポラ属のB12−1株によるPLAの分解>
(菌株の培養とクリアゾーンの測定)
菌株としてミクロモノスポラ(Micromonospora)属のB12−1株(受領番号:NITE AP−1105)を用いた。試験管に分注したTryptic Soy Broth(TSB)培地(5mL)に一白金耳量の菌体を植菌後、振とう培養(40℃、120rpm)し、これを前培養液とした。本培養として、BM培地に前培養液を1%植菌し、振とう培養(40℃、120rpm)した。続いて固体培養をおこなった。固体培養にはPLA基本培地またはTSB培地にアガロース(BOL:清水食品)を1.5%添加した寒天培地を用いた。滅菌したつまようじでスラントから菌体を取りPLA基本培地の寒天培地に3点植菌した。37℃で5日間培養し、培養終了時にクリアゾーンの直径を測定した。その結果、B12−1株は直径19mmのクリアゾーンを形成した(図1)。
【0051】
(培地の組成)
各培地の組成を以下に示す。
【表1】
【表2】
【表3】
なお、乳化PLA(0.2%)は以下の方法で調製した。0.4gのポリ乳酸樹脂(Vyloecol BE−400、東洋紡)を20mLのジクロロメタンに溶解した。リン酸カリウム緩衝液(10mM、pH7.3)を200mL加え、超音波処理(Ultrtasonic generator US−300T:日本精機製作所、チップサイズ26、400W、5分間×3回)し、乳化した。
【0052】
<実施例2.PLA分解酵素の精製>
(酵素活性測定法)
本発明において、酵素活性を以下の方法により測定した。0.4gのポリ乳酸樹脂(Vyloecol BE−400、東洋紡)を20mLのジクロロメタンに溶解した。これにTris−HCl緩衝液(10mM、pH9.0)を200mL加え、超音波処理(Ultrtasonic generator US−300T:日本精機製作所、チップサイズ26、400W、5分間×3回)して乳化し、この乳化PLA(0.2%)を基質とした。乳化PLA650μLに酵素液50μLを添加し反応させた(50℃、10分間)。反応後、3分間煮沸し反応を停止させた。反応液の濁度(630nm)の減少量からPLA分解活性を求めた。なお、本明細書では、PLA分解活性1Uを、1分間に630nmにおける乳化PLAの濁度を1.0減少させる酵素量と定義した。
【0053】
(菌体の培養)
以下のように、前培養にはTSB培地を用い、本培養にはBMB12培地を用いた。試験管にTSB培地5mLを分注し、コロニーを一白金耳量植菌後、振とう培養し(37℃、180rpm、3日間)、これを前々培養液とした。500mL容三角フラスコにTSB培地100mLを分注し、前々培養液を1%植菌後、振とう培養し(37℃、180rpm、2日間)、これを前培養液とした。500mL容三角フラスコにBMB12培地を200mL分注し、前培養液を1%植菌後、振とう培養し(37℃、180rpm、4日間)、これを培養液とした。培養液を遠心分離(500mLボトル、5000rpm、4℃、10分間、himac SCR−20B:HITACHI)し、上清を回収した。
【0054】
培地の組成を以下に示す。
【表4】
【0055】
(酵素精製)
酵素精製は特に断らない限り低温室(7℃)で行った。
(1)硫安沈殿
培養液上清を限外濾過膜(microza SLP−0053:旭化成)で2〜3倍に濃縮後、その濃縮液を硫安沈殿に用いた。培養液上清に粉末状にした硫酸アンモニウムを60%飽和となるよう徐々に添加した。その後、1時間以上氷冷した。氷冷後、遠心分離(500mlボトル、4℃、12000rpm、30分、himac CR22GII:HITACHI)し、沈殿をリン酸カリウム緩衝液(20mM、pH8.0)に溶解した。同緩衝液で一晩透析し、活性画分を得た。活性画分を陰イオン交換クロマトグラフィーに供した。
【0056】
(2)陰イオン交換クロマトグラフィー
緩衝液はリン酸カリウム緩衝液(20mM、pH8.0)を用いた。充填剤としてTOYOPEARL DEAE−650M(東ソー株式会社)を用い、カラムに充填した。カラム(カラム容量:400mL)を5倍量の緩衝液で予め平衡化した。カラムにサンプルを通液しPLA分解酵素を吸着後、カラム容量の3倍量の同緩衝液で洗浄した。PLA分解酵素をNaClの濃度勾配(0〜0.5M)で溶出させた。活性画分を同緩衝液で透析後、TOYOPEARL DEAE−650M(東ソー株式会社)を充填したカラム容量100mLのカラムに換え、NaClの濃度勾配を0〜0.3Mとし、再度陰イオン交換クロマトグラフィーを行い、活性画分を得た。
【0057】
(3)疎水クロマトグラフィー
緩衝液はリン酸カリウム緩衝液(20mM、pH8.0)を用いた。充填剤としてTOYOPEARL Butyl−650M(東ソー株式会社)を用い、カラム(カラム容量:30mL)に充填した。カラムを、硫酸アンモニウム(30%飽和)を含むリン酸カリウム緩衝液で予め平衡化した。陰イオン交換クロマトグラフィーで得られた活性画分に硫酸アンモニウムを30%飽和となるように添加し、1時間氷冷後、遠心分離(500mLボトル、4℃、12000rpm、30分)した。これをカラムに通液し、PLA分解酵素を吸着させた。カラムを、硫酸アンモニウム(30%飽和)を含むリン酸カリウム緩衝液で洗浄した。PLA分解酵素を硫酸アンモニウムの濃度勾配(30〜0%飽和)を用いて溶出させ、活性画分を得た。
【0058】
(4)SDS−ポリアクリルアミドゲル電気泳動(SDS−PAGE)
疎水クロマトグラフィーで得られた活性画分をSDS−PAGEに供したところ、単一なバンドを示したため、これを精製酵素とした。また、SDS−PAGEからPLA分解酵素の分子量は31kDaと推定した。
【0059】
<実施例3.PLA分解酵素の性質>
(基質特異性)
PLA分解酵素の生分解性プラスチックに対する基質特異性を調べた。生分解性プラスチックの基質には、PLA(「Vyloecol BE−400」東洋紡、分子量4×104)、PLA(「LACEA #400」三井化学、分子量1.5×105)、PLA(「Lacty」島津、分子量2.2×105)、ポリカプロラクトン(PCL、「CELGREEN」ダイセル化学工業株式会社)、ポリブチレンサクシネートアジペート(PBSA「ビオノーレ3001」昭和高分子株式会社)、ポリブチレンサクシネート(PBS「ビオノーレ1001」昭和高分子株式会社)を用いた。基質特異性はPLA(Vyloecol BE−400)に対する分解活性を100とし、相対活性で示した。結果を表5に示す。表5に示す通り、本発明のPLA分解酵素は、PLAを分解したが、PCL、PBSAにはほとんど作用せず、PBSには全く作用しなかった。PLAでは分子量の小さい方がよく分解された。
【0060】
【表5】
【0061】
(至適温度および熱安定性)
PLA分解酵素の作用至適温度を以下のようにして求めた。酵素活性測定法における反応温度を30〜90℃まで10℃ずつ変化させて酵素反応を行い、各温度における活性を測定した。その結果、PLA分解酵素は70℃で最も活性が高く、70℃を至適温度とした。また、PLA分解酵素の熱安定性を調べるために、酵素溶液を所定の温度で30分間加熱処理した後、直ちに冷却し、残存活性を上述した酵素活性測定法に従って測定した。その結果、本発明のPLA分解酵素は、70℃の活性を100%として、50℃、30分間保温後も100%の活性を維持し、60℃、30分間保温後でも約50%の残存活性を有していた。
【0062】
(至適pHおよびpH安定性)
PLA分解酵素の作用至適pHを以下のようにして求めた。酵素活性測定における反応液中の緩衝液として、リン酸カリウム緩衝液(pH6.0〜8.0)、Tris−HCl緩衝液(pH7.5〜9.0)、グリシン−NaOH緩衝液(pH9.0〜11.0)、KCl−NaOH緩衝液(11.0〜12.0)を用いて反応を行い、活性を測定した。その結果、PLA分解酵素はpH9.0で最も活性が高く、pH9.0を至適pHとした。また、PLA分解酵素のpH安定性を調べるために、酵素溶液をpHの異なる緩衝液で透析し、4℃で20時間放置後、残存活性を酵素活性測定法に従って測定した。その結果、pH7.5の緩衝液を用いた場合の活性を100%として、本発明のPLA分解酵素はpH6.0〜pH9.0の範囲では90%以上の残存活性を示した。また、pH10.0〜pH12.0の範囲では約80%の残存活性を示した。
【0063】
<実施例4.PLA分解酵素遺伝子のクローニング>
(PLA分解酵素のN末端および内部アミノ酸配列の決定)
PLA分解酵素のアミノ酸配列を、プロテインシーケンサー(PPSQ−21A、島津製作所)を用いて分析した。N末端アミノ酸配列の分析には実施例2で得られた精製酵素をサンプルとした。その結果、N末端配列として、GTQTGATWGLDRIDQRNLPL(配列番号1の第1〜20番目)の配列を有していた。内部アミノ酸配列の分析には精製酵素を臭化シアンで断片化したものをサンプルとした。サンプルをSDS−PAGEに供したところ3種類の酵素断片(C1:14kDa、C2:11kDa、C3:0.6kDa)が確認できた。このうちC1、C2断片をPVDF膜に転写し、アミノ酸配列の決定に用いた。その結果、C1断片のアミノ酸配列は、N末端アミノ酸配列と等しかった。C2断片は、SLGGGASTTLDNAVANSIAS(配列番号1の第128〜147番目)の配列を有していた。
【0064】
(PLA分解酵素遺伝子の部分増幅)
ハイブリダイゼーションのプローブに用いるPLA分解酵素遺伝子断片を取得するために、PLA分解酵素遺伝子の部分増幅を、縮重プライマー(degenerated primer)を用いて、PCR装置(Thermal Cycler Dice TP600:Takara)で行った。DNAポリメラーゼとして、Hot star Taq(QIAGEN)を用いた。まず、PLA分解酵素のN末端およびC2断片の内部アミノ酸配列を基に、縮重プライマーを設計した。設計した縮重プライマーを以下に示す。
5’−ATCGACCAGCGCAACCTG−3’(F−aur427、配列番号5)
5’−GTTSGCSACSGCGTTGTC−3’(SはG又はC)(R−DNAVAN、配列番号6)
設計した縮重プライマーを用い、B12−1株の総DNAを鋳型としてPCRを行ったところ、約390bpの増幅断片を得た。この増幅断片をシークエンスしたところ、内部アミノ酸配列(SLGGGASTTL)(配列番号1の第128〜137番目)をコードする塩基配列が認められたため、この増幅断片をハイブリダイゼーションに用いるプローブとした。
【0065】
(サザンハイブリダイゼーション)
サザンハイブリダイゼーションと、後述するコロニーハイブリダイゼーションを、非放射性システムであるDIG−High Prime DNA Labeling and Detection Starter Kit II(ロシュダイアグノスティック社)を用いて行った。上記で得られた増幅断片をラベル化プローブ調製の鋳型とした。鋳型DNA(1μg)に滅菌蒸留水を加えて16μLにした。沸騰水中で10分間インキュベートして鋳型DNAを熱変性させた後、氷水で急速に冷却した。その後、DIG−high Primeを4μL加え、37℃で20時間反応させた。反応後、2μLのEDTA(0.2M、pH8.0)を加え、反応を停止させた。これをラベル化プローブとした。一方、B12−1株の総DNAを制限酵素であるKpnI、PstI、SacIおよびSphIで切断した(37℃、3時間)。切断後のDNA断片をアガロースゲル電気泳動に供し、サザンハイブリダイゼーションを行った。その結果、SphI断片の中で約9kbpのDNA断片がプローブとハイブリダイズした。そこで、この約9kbpのSphI断片をpUC19に連結し、コロニーハイブリダイゼーションを行った。
【0066】
(コロニーハイブリダイゼーション)
SphI断片(9kbp)をpUC19に連結した組換えプラスミドを大腸菌に形質転換した。コロニー500個に対しコロニーハイブリダイゼーションを行ったところ、陽性クローンが1個得られた。陽性クローンから得た組換えプラスミドのSphI断片に対しサザンハイブリダイゼーションを行ったところ、プローブがハイブリダイズした。よってこの9kbpのSphI断片にPLA分解酵素遺伝子が含まれると考えられた。このSphI断片(9kbp)を含む組換えプラスミドをpG12として以降の実験に用いた。
【0067】
(PLA分解酵素遺伝子の位置の推定)
組換えプラスミドpG12を各種の制限酵素で切断した後、サザンハイブリダイゼーションを行った。この結果より、9kbpのSphI断片中にEcoRI認識部位が1ヶ所、BamHI認識部位が2ヶ所、BglII認識部位が2ヶ所存在することが確認できた。次にSphI断片(9kbp)を、制限酵素EcoRI、BamHI、BglIIを組み合わせて切断し、断片長をアガロースゲル電気泳動から推定した。これらの断片に対しサザンハイブリダイゼーションを行い、制限酵素地図を作製し、遺伝子の位置を推定した(図2)。
【0068】
(PLA分解酵素遺伝子の塩基配列決定)
制限酵素地図から、PLA分解酵素遺伝子を含むと推定されるEcoRI−BamHI断片(2.2kbp)をpUC19ベクターにサブクローニングし、シークエンスを行った。シークエンスはベックス社に依頼した。シークエンス試薬は、BigDye Terminators v.1.1 Cycle Sequencing Kit(Applied Biosystems Japan)を用い、シーケンサーは、ABI Prism 3130 Genetic Analyzer (Applied Biosystems Japan)を使用した。決定した塩基配列は、Geneticx−Win Ver.8.0(Software development)で解析した。シークエンスの結果、開始コドン(ATG)、終止コドン(TGA)および開始コドンの上流にSD配列と推定される配列(AGGGAG)が認められた。配列は、配列番号4に示す1221bpからなり、配列番号3に示す406アミノ酸をコードしていた。シグナルペプチドをSignal iP 3.0を用いて推定した。また、シグナルペプチドの次のアミノ酸から、プロテインシーケンサーで明らかにしたPLA分解酵素のN末端アミノ酸の1つ前のアミノ酸までの配列をプロペプチドの配列とした。活性化酵素になる前の未成熟酵素は、シグナルペプチド(配列番号3の第1〜35番目、35アミノ酸残基)、プロペプチド(配列番号3の第36〜130番目、95アミノ酸残基)、成熟酵素(配列番号3の第131〜406番目、すなわち、配列番号1、276アミノ酸残基)から構成されると推定された。PLA分解酵素は、プロテアーゼで良く知られるPre−Pro−Enzymeとして生産され、シグナルペプチドにより細胞外に輸送された後、プロペプチドが切断され、活性化酵素として働くと推定された。
【0069】
本発明のPLA分解酵素のアミノ酸配列と類似性の高い酵素を検索したところ、Thermus aquaticu 由来Aqualysin1(類似性53%)、Thermus sp. Rt41A由来 Serine protease(53%)、Serratia sp. GF96由来Proteinase K(52%)が見出された。これらは、Pre−Pro−Enzymeとして生産され、かつ、セリンプロテアーゼに属する。よって遺伝子情報からは、本発明のPLA分解酵素はセリンプロテアーゼの一種であると推定された。本発明のPLA分解酵素では、活性中心と予測されるアスパラギン酸(配列番号2の第39番目)、ヒスチジン(配列番号2の第70番目)、セリン(配列番号2の第222番目)が保存されていた。
【0070】
<実施例5.PLA分解酵素遺伝子の組換え微生物への組み込みおよび遺伝子発現>
(プラスミドpBPa1の構築)
以下のようにして、組換えベクターとして、プラスミドpBPa1を構築した。遺伝子を組み込むプラスミドとして、放線菌大腸菌シャトルベクターである、pUC702を用いた。pUC702は、pUC19とpIJ702をKpnI、SacIサイトで連結したプラスミドである。PCRによりPLA分解酵素遺伝子増幅を行った。PCR用の鋳型として、PLA分解酵素遺伝子を含む組換えプラスミドpGBEを用い、プライマーとして、F−B12p702およびR−B12p702を用いた。これらのプライマーは、遺伝子断片がプロモーター領域またはターミネーター領域を含んで増幅されるように設計したものである。
F−B12p702およびR−B12p702プライマーの配列を以下に示す。
5’−GCGCGAATTCCCGCCCGGTAGTCGTTGC−3’(F−B12p702、配列番号7)
5’−AGAGGAATTCGCGACCATCCTGCCGGTC−3’(R−B12p702、配列番号8)
増幅された遺伝子断片を、pUC702のEcoRIサイトに挿入し、発現用プラスミドpBPa1を構築した。
【0071】
(放線菌プロトプラストの調製)
PLA分解酵素遺伝子を発現させる放線菌として、ストレプトマイセス・リビダンス(Streptomyces lividans)TK21を用いた。滅菌した試験管にTSB培地を5ml分注し、一白金耳量の菌体を植菌して振とう培養し(180rpm、30℃、48時間)、これを前培養液とした。次にYEME培地を100mL分注した500mL容三角フラスコに前培養液を2%植菌し、振とう培養し(180rpm、30℃、36時間)、これを培養液とした。培養液を集菌し、10.3%スクロース溶液で2回洗浄した。リゾチーム溶液(1mg/mLとなるようリン酸緩衝液に溶解)8mLを加え、よく懸濁し、30℃で15分間インキュベートした。さらに5mLのリン酸緩衝液を加え15分間インキュベートした。その後、反応液を滅菌したコットンでろ過し、ろ液を遠心分離(1500rpm、室温、7分間)した。上清を除き、沈殿したプロトプラストにリン酸緩衝液を3mL加え、穏やかに懸濁し、プロトプラスト溶液を調製した。トーマ血球計算版を用い、プロトプラストの濃度を計測した。マイクロチューブに分注し、−80℃で保存した。
【0072】
培地の組成を以下に示す。
【表6】
【0073】
(放線菌の形質転換)
プロトプラスト溶液を30℃のウォーターバスで素早く溶解した。プロトプラスト溶液150μLに適量のプラスミドpBPa1溶液を加え、穏やかに攪拌した。その後すぐにリン酸緩衝液(25%PEG含有)を加えゆっくりと3回ピペッティングした。この溶液をR5プレートに丁寧に塗布し、30℃で培養した。培養16〜20時間後にチオストレプトン含有軟寒天培地を重層し、さらに30℃で3日間培養し、プラスミドpBPa1を形質転換した組換え微生物Streptomyces lividans BP1を作製した。
【0074】
(組換え微生物の培養)
組換え微生物Streptomyces lividans BP1の培養は、終濃度5μg/mlとなるようチオストレプトンを加えたTSB培地で行った。まず、滅菌した試験管に上記TSB培地を5mL分注し、一白金耳量の菌体を植菌して振とう培養し(180rpm、30℃、2日間)、これを前培養液とした。次に100mLの上記TSB培地を分注した500mL容三角フラスコに前培養液を1%となるよう植菌し、振とう培養した(180rpm、30℃、2日間)。
【0075】
(組換え微生物のPLA分解能)
組換え微生物Streptomyces lividans BP1は14.0U/mlのPLA分解活性を示し、B12−1株の28倍の酵素生産性を示した(図3)。また、組換え微生物Streptomyces lividans BP1の、PLA基本培地の寒天培地でのクリアゾーンを測定した。B12−1株は37℃、5日間の培養で直径19mmのクリアゾーンを示したのに対し、組換え微生物Streptomyces lividans BP1は30℃、5日間の培養で直径45mmのクリアゾーンを示した(図4)。このことから組換え微生物Streptomyces lividans BP1は高いPLA分解活性を有することが示された。
【受託番号】
【0076】
ミクロモノスポラ(Micromonospora)属のB12−1株(千葉県木更津市かずさ鎌足2−5−8、独立行政法人製品評価技術基盤機構特許微生物寄託センター、受領番号:NITE AP−1105、2011年6月13日付受領にて寄託)
組換え微生物Streptomyces lividans BP1(千葉県木更津市かずさ鎌足2−5−8、独立行政法人製品評価技術基盤機構特許微生物寄託センター、受領番号:NITE AP−1106、2011年6月13日付受領にて寄託)
【特許請求の範囲】
【請求項1】
下記(a)〜(c)からなる群から選択されるポリペプチドを含有するタンパク質:
(a)配列表の配列番号1に記載のアミノ酸配列からなるポリペプチド;
(b)配列表の配列番号1に記載のアミノ酸配列に対して1個または数個のアミノ酸が欠失、置換、付加、または挿入されたアミノ酸配列を有し、かつ、ポリ乳酸分解活性を有するポリペプチド;
(c)配列表の配列番号1に記載のアミノ酸配列と90%以上の同一性を有するアミノ酸配列を有し、かつ、ポリ乳酸分解活性を有するポリペプチド。
【請求項2】
配列番号1のアミノ酸配列の第34〜44番目のアミノ酸からなるペプチド、第65〜75番目のアミノ酸からなるペプチド及び第217〜227番目のアミノ酸からなるペプチドを含む、請求項1に記載のタンパク質。
【請求項3】
請求項1または2に記載のタンパク質をコードするポリヌクレオチドを含有する核酸。
【請求項4】
下記(1)〜(4)からなる群から選択されるポリヌクレオチドを含有する核酸;
(1)配列表の配列番号2に記載の塩基配列からなるポリヌクレオチド;
(2)配列表の配列番号2に記載の塩基配列に対して1個〜数個の塩基が欠失、置換、付加、または挿入された塩基配列を有し、かつ、ポリ乳酸分解活性を有するポリペプチドをコードするポリヌクレオチド;
(3)配列表の配列番号2に記載の塩基配列と90%以上の同一性を有する塩基配列を有し、かつ、ポリ乳酸分解活性を有するポリペプチドをコードするポリヌクレオチド;
(4)(1)のポリヌクレオチドとストリンジェントな条件下でハイブリダイズし、かつ、ポリ乳酸分解活性を有するポリペプチドをコードするポリヌクレオチド。
【請求項5】
配列番号1のアミノ酸配列の第34〜44番目のアミノ酸からなるペプチド、第65〜75番目のアミノ酸からなるペプチド及び第217〜227番目のアミノ酸からなるペプチドをコードするヌクレオチドを含む、請求項4に記載の核酸。
【請求項6】
ミクロモノスポラ(Micromonospora)属に属し、配列表の配列番号2で示される塩基配列の核酸を有する、受領番号NITE AP−1105として寄託されている放線菌B12−1株。
【請求項7】
請求項3〜5のいずれか一項に記載の核酸を含有する組換えベクターを含む組換え微生物。
【請求項8】
ストレプトマイセス(Streptomyces)属に属する、請求項7に記載の組換え微生物。
【請求項9】
受領番号NITE AP−1106として寄託されている、請求項8に記載の組換え微生物。
【請求項10】
請求項6に記載の放線菌B12−1株又は請求項7〜9のいずれか一項に記載の組換え微生物を培養し、得られる培養物からポリ乳酸分解酵素を回収する、ポリ乳酸分解酵素の製造方法。
【請求項11】
請求項1若しくは2に記載のタンパク質、請求項6に記載の放線菌B12−1株及び請求項7〜9のいずれか一項に記載の組換え微生物からなる群から選択される少なくとも1種とポリ乳酸を含む物質とを接触させる、ポリ乳酸の分解方法。
【請求項1】
下記(a)〜(c)からなる群から選択されるポリペプチドを含有するタンパク質:
(a)配列表の配列番号1に記載のアミノ酸配列からなるポリペプチド;
(b)配列表の配列番号1に記載のアミノ酸配列に対して1個または数個のアミノ酸が欠失、置換、付加、または挿入されたアミノ酸配列を有し、かつ、ポリ乳酸分解活性を有するポリペプチド;
(c)配列表の配列番号1に記載のアミノ酸配列と90%以上の同一性を有するアミノ酸配列を有し、かつ、ポリ乳酸分解活性を有するポリペプチド。
【請求項2】
配列番号1のアミノ酸配列の第34〜44番目のアミノ酸からなるペプチド、第65〜75番目のアミノ酸からなるペプチド及び第217〜227番目のアミノ酸からなるペプチドを含む、請求項1に記載のタンパク質。
【請求項3】
請求項1または2に記載のタンパク質をコードするポリヌクレオチドを含有する核酸。
【請求項4】
下記(1)〜(4)からなる群から選択されるポリヌクレオチドを含有する核酸;
(1)配列表の配列番号2に記載の塩基配列からなるポリヌクレオチド;
(2)配列表の配列番号2に記載の塩基配列に対して1個〜数個の塩基が欠失、置換、付加、または挿入された塩基配列を有し、かつ、ポリ乳酸分解活性を有するポリペプチドをコードするポリヌクレオチド;
(3)配列表の配列番号2に記載の塩基配列と90%以上の同一性を有する塩基配列を有し、かつ、ポリ乳酸分解活性を有するポリペプチドをコードするポリヌクレオチド;
(4)(1)のポリヌクレオチドとストリンジェントな条件下でハイブリダイズし、かつ、ポリ乳酸分解活性を有するポリペプチドをコードするポリヌクレオチド。
【請求項5】
配列番号1のアミノ酸配列の第34〜44番目のアミノ酸からなるペプチド、第65〜75番目のアミノ酸からなるペプチド及び第217〜227番目のアミノ酸からなるペプチドをコードするヌクレオチドを含む、請求項4に記載の核酸。
【請求項6】
ミクロモノスポラ(Micromonospora)属に属し、配列表の配列番号2で示される塩基配列の核酸を有する、受領番号NITE AP−1105として寄託されている放線菌B12−1株。
【請求項7】
請求項3〜5のいずれか一項に記載の核酸を含有する組換えベクターを含む組換え微生物。
【請求項8】
ストレプトマイセス(Streptomyces)属に属する、請求項7に記載の組換え微生物。
【請求項9】
受領番号NITE AP−1106として寄託されている、請求項8に記載の組換え微生物。
【請求項10】
請求項6に記載の放線菌B12−1株又は請求項7〜9のいずれか一項に記載の組換え微生物を培養し、得られる培養物からポリ乳酸分解酵素を回収する、ポリ乳酸分解酵素の製造方法。
【請求項11】
請求項1若しくは2に記載のタンパク質、請求項6に記載の放線菌B12−1株及び請求項7〜9のいずれか一項に記載の組換え微生物からなる群から選択される少なくとも1種とポリ乳酸を含む物質とを接触させる、ポリ乳酸の分解方法。
【図2】

【図3】
【図1】

【図4】
【図3】
【図1】
【図4】
【公開番号】特開2013−99(P2013−99A)
【公開日】平成25年1月7日(2013.1.7)
【国際特許分類】
【出願番号】特願2011−137480(P2011−137480)
【出願日】平成23年6月21日(2011.6.21)
【出願人】(304023318)国立大学法人静岡大学 (416)
【Fターム(参考)】
【公開日】平成25年1月7日(2013.1.7)
【国際特許分類】
【出願日】平成23年6月21日(2011.6.21)
【出願人】(304023318)国立大学法人静岡大学 (416)
【Fターム(参考)】
[ Back to top ]