
ポーラスシリカ前駆体組成物及びその作製方法、ポーラスシリカ膜及びその作製方法、並びに半導体素子
【課題】 膜強度を高くし、薬液耐性を向上させたポーラスシリカ膜を得るためのポーラスシリカ前駆体組成物及びその作製方法、この前駆体組成物を用いて作製したポーラスシリカ膜及びその作製方法、並びにこのポーラスシリカ膜を用いて得られた半導体素子の提供。
【解決手段】 界面活性剤をテンプレートとして用いるポーラスシリカ前駆体組成物の作製方法において、有機アルカリ触媒の存在下トリアルコキシシランの重縮合反応を行った後、酸触媒の存在下、ポーラスシリカ前駆体の骨格形成材料を添加してさらに重縮合反応を行うか、又は酸触媒の存在下、ポーラスシリカ前駆体の骨格形成材料及びさらにトリアルコキシシランを添加してさらに重縮合反応を行うことからなる。得られた前駆体組成物を用いて作製されたポーラスシリカ膜及びこのポーラスシリカ膜を用いて得られた半導体素子。
【解決手段】 界面活性剤をテンプレートとして用いるポーラスシリカ前駆体組成物の作製方法において、有機アルカリ触媒の存在下トリアルコキシシランの重縮合反応を行った後、酸触媒の存在下、ポーラスシリカ前駆体の骨格形成材料を添加してさらに重縮合反応を行うか、又は酸触媒の存在下、ポーラスシリカ前駆体の骨格形成材料及びさらにトリアルコキシシランを添加してさらに重縮合反応を行うことからなる。得られた前駆体組成物を用いて作製されたポーラスシリカ膜及びこのポーラスシリカ膜を用いて得られた半導体素子。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ポーラスシリカ前駆体組成物及びその作製方法、ポーラスシリカ膜及びその作製方法、並びに半導体素子に関し、特にケージ(Cage)構造の成分割合の多いポーラスシリカ膜を得るためのポーラスシリカ前駆体組成物及びその作製方法、このポーラスシリカ前駆体組成物を用いて得られるポーラスシリカ膜及びその作製方法、並びにこのポーラスシリカ膜を利用した半導体素子に関する。
【背景技術】
【0002】
半導体素子の層間絶縁膜等として、ポーラスシリカ膜が使用されており、このポーラスシリカ膜の作製方法には、大きく分けてCVD法と塗布法とがある。CVD法は、CVDによるシリカ成膜時に有機物であるポロジェンを加えて成膜し、熱、電子線、UV等により、ポロジェンを除去し、ポーラスシリカ膜を得る方法である。一方、塗布法は、界面活性剤等の有機物であるテンプレートが加えられた前駆体組成物(塗布液)をスピンコートして成膜し、焼成し、電子線、UV等により、テンプレートを除去し、ポーラスシリカ膜を得る方法である(例えば、特許文献1参照)。これらの場合、得られるポーラスシリカ膜は、空孔量を増加させるに従い膜強度が低下するという問題があり、また、比表面が増加するため、膜の欠陥があらわになる確立が高まり、薬液耐性も低下するという問題がある。そのため、膜強度が高く、薬液耐性に優れたポーラスシリカ膜が求められている。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】特開平07−133350号公報
【発明の概要】
【発明が解決しようとする課題】
【0004】
本発明の課題は、上述の従来技術の問題点を解決することにあり、ポーラスシリカ膜中のSi−Oケージ構造の成分割合を多くすることで、膜強度を高くし、また、薬液耐性を向上させたポーラスシリカ膜を得るための、ポーラスシリカ前駆体組成物及びその作製方法、この前駆体組成物を用いて作製したポーラスシリカ膜及びその作製方法、並びにこのポーラスシリカ膜を用いて得られた半導体素子を提供することにある。
【課題を解決するための手段】
【0005】
本発明者らは、上記課題を解決するために鋭意検討した結果、触媒を用いてトリアルコキシシラン系原料の加水分解・重縮合反応を進めた後、ポーラスシリカ前駆体の骨格形成材料を添加する手法を用いて作製されたポーラスシリカ前駆体組成物を用いることで、ポーラスシリカ膜中のケージ構造の成分割合が多くなり、膜強度を高くできると共に、薬液耐性を向上できることを見出し、本発明を完成させるに至った。
【0006】
本発明のポーラスシリカ前駆体組成物は、界面活性剤をテンプレートとして含むポーラスシリカ前駆体組成物であって、有機アルカリ触媒の存在下で得られたトリアルコキシシランの重縮合生成物とポーラスシリカ前駆体の骨格形成材料との酸触媒の存在下での重縮合生成物、又は該有機アルカリ触媒の存在下で得られたトリアルコキシシランの重縮合生成物とポーラスシリカ前駆体の骨格形成材料とトリアルコキシシランとの酸触媒の存在下での重縮合生成物を含有することを特徴とする。
【0007】
前記ポーラスシリカ前駆体組成物において、有機アルカリ触媒が、アンモニア、コリン、テトラメチルアンモニウムハイドロキシド、及びモノエタノールアミンから選ばれた少なくとも1種であることを特徴とする。
【0008】
前記ポーラスシリカ前駆体組成物において、酸触媒が、硝酸、塩酸、硫酸、及びフッ酸から選ばれた無機酸であることを特徴とする。
【0009】
前記ポーラスシリカ前駆体組成物において、骨格形成材料が、アルコキシシランから選ばれた少なくとも1種であることを特徴とする。
【0010】
本発明のポーラスシリカ前駆体組成物の作製方法は、界面活性剤をテンプレートとして用いるポーラスシリカ前駆体組成物の作製方法において、有機アルカリ触媒の存在下トリアルコキシシランの重縮合反応を行った後、酸触媒の存在下、ポーラスシリカ前駆体の骨格形成材料を添加してさらに重縮合反応を行うか、又は該酸触媒の存在下、ポーラスシリカ前駆体の骨格形成材料及びさらにトリアルコキシシランを添加してさらに重縮合反応を行うことを特徴とする。
【0011】
前記ポーラスシリカ前駆体組成物の作製方法において、有機アルカリ触媒が、アンモニア、コリン、テトラメチルアンモニウムハイドロキシド、及びモノエタノールアミンから選ばれた少なくとも1種であることを特徴とする。
【0012】
前記ポーラスシリカ前駆体組成物の作製方法において、酸触媒が、硝酸、塩酸、硫酸、及びフッ酸から選ばれた無機酸であることを特徴とする。
【0013】
前記ポーラスシリカ前駆体組成物の作製方法において、骨格形成材料が、アルコキシシランから選ばれた少なくとも1種であることを特徴とする。
【0014】
本発明のポーラスシリカ膜は、上記ポーラスシリカ前駆体組成物又は上記ポーラスシリカ前駆体組成物の作製方法に従って作製された前駆体組成物を用いて作製されたものであることを特徴とする。
【0015】
本発明のポーラスシリカ膜において、その膜のFT−IRスペクトルのSi−Oケージ構造のピークである1150cm−1近傍とSi−Oラダー構造のピークである1050cm−1近傍とのピーク高さの比が、1.0以上であることを特徴とする。
【0016】
上記Si−Oケージ構造のピークである1150cm−1近傍とSi−Oラダー構造のピークである1050cm−1近傍とのピーク高さの比が、1.0未満であると、目的とする膜強度、薬液耐性の向上を達成することができない傾向がある。
【0017】
本発明のポーラスシリカ膜の作製方法は、上記ポーラスシリカ前駆体組成物又は上記ポーラスシリカ前駆体組成物の作製方法に従って作製されたポーラスシリカ前駆体組成物を基板表面に塗布し、真空雰囲気下焼成処理し、界面活性剤を除去してポーラスシリカ膜を作製することを特徴とする。
【0018】
本発明の半導体素子は、上記ポーラスシリカ膜又は上記ポーラスシリカ膜の作製方法に従って作製したポーラスシリカ膜を用いて得られたものであることを特徴とする。
【発明の効果】
【0019】
本発明によれば、有機アルカリ触媒を用いてトリアルコキシシラン系原料の重縮合を進めた後に、ポーラスシリカ前駆体の骨格形成材料を添加する手法を用いて作製されたポーラスシリカ前駆体組成物を用いることで、ポーラスシリカ膜中のケージ構造の成分割合がラダー構造の成分割合と同等又はより多くなり、膜強度を高くできると共に、薬液耐性を向上させることができるという効果を奏する。
【図面の簡単な説明】
【0020】
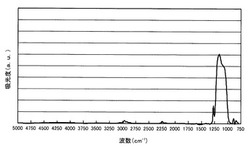
【図1】実施例1において得られたポーラスシリカ膜のFT−IRスペクトルを示すスペクトル図。
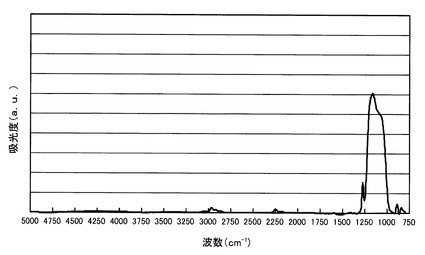
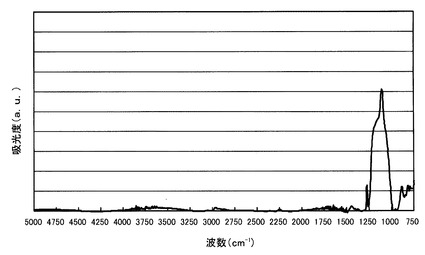
【図2】比較例1において得られたポーラスシリカ膜のFT−IRスペクトルを示すスペクトル図。
【発明を実施するための形態】
【0021】
本発明に係るポーラスシリカ前駆体組成物(塗布液)の実施の形態によれば、この前駆体組成物は、界面活性剤をテンプレートとして含む前駆体組成物であって、有機アルカリ触媒(例えば、アンモニア、コリン、テトラメチルアンモニウムハイドロキシド、及びモノエタノールアミン等から選ばれた少なくとも1種)の存在下で得られたトリアルコキシシラン(以下列挙するメチルトリメトキシシラン(MTMS)等から選ばれた少なくとも1種)の重縮合生成物と、酸触媒(例えば、硝酸、塩酸、硫酸、及びフッ酸等から選ばれた少なくとも1種の無機酸)の存在下、ポーラスシリカ前駆体の骨格形成材料(以下列挙するアルコキシシランから選ばれた少なくとも1種)との重縮合生成物、又は上記有機アルカリ触媒の存在下に得られたトリアルコキシシランの重縮合生成物と上記ポーラスシリカ前駆体の骨格形成材料とさらに上記トリアルコキシシランとの酸触媒の存在下での重縮合生成物を含有することからなる。
【0022】
本発明に係るポーラスシリカ前駆体組成物の作製方法の実施の形態によれば、この作製方法は、界面活性剤をテンプレートとして用いるポーラスシリカ前駆体組成物の作製方法において、有機アルカリ触媒(例えば、アンモニア、コリン、テトラメチルアンモニウムハイドロキシド、及びモノエタノールアミンから選ばれた少なくとも1種)の存在下、トリアルコキシシラン(以下列挙するメチルトリメトキシシラン(MTMS)等から選ばれた少なくとも1種)の重縮合反応を行った後、この重縮合生成物に、酸触媒(例えば、硝酸、塩酸、硫酸、及びフッ酸等から選ばれた少なくとも1種の無機酸)の存在下、ポーラスシリカ前駆体の骨格形成材料(以下列挙するアルコキシシランから選ばれた少なくとも1種)を添加してさらに重縮合反応を行うか、又は上記酸触媒の存在下、上記ポーラスシリカ前駆体の骨格形成材料及びさらに上記トリアルコキシシランを添加してさらに重縮合反応を行うことからなる。
【0023】
本発明に係るポーラスシリカ膜の実施の形態によれば、このポーラスシリカ膜は、上記ポーラスシリカ前駆体組成物又は上記ポーラスシリカ前駆体組成物の作製方法に従って作製された前駆体組成物を用いて作製されたものであり、そのポーラスシリカ膜のFT−IRスペクトルのSi−Oケージ構造のピークである1150cm−1近傍とSi−Oラダー構造のピークである1050cm−1近傍とのピーク高さの比が、1.0以上であることからなる。このピーク高さの比は、ポーラスシリカ膜中のケージ構造の成分割合をAとし、ラダー構造の成分割合をBとした場合、50%≦A/(A+B)≦100%を満足することと同義である。以下記載する実施例及び比較例によれば、ケージ構造のピーク高さとラダー構造のピーク高さとの比が1.0以上であると、すなわち、ケージ構造の成分割合が増加するに従って、膜強度及び薬液耐性が向上することが分かる。
【0024】
本発明では、有機アルカリ触媒を用いてトリアルコキシシラン系原料の加水分解・重縮合反応を進めた後、この重縮合生成物にポーラスシリカ前駆体の骨格形成材料を添加するか、又はこの重縮合生成物にポーラスシリカ前駆体の骨格形成材料及びトリアルコキシシラン系原料を添加する手法を用いて作製されたポーラスシリカ前駆体組成物を用いることで、ポーラスシリカ膜中のケージ構造の成分割合が多くなり、膜強度を高くできると共に、薬液耐性を向上させることができる。
【0025】
本発明で使用し得るトリアルコキシシランとしては、特に制限はないが、例えばメチルトリメトキシシラン(MTMS)、メチルトリエトキシシラン、エチルトリメトキシシラン、エチルトリエトキシシラン(ETES)、アリルトリメトキシシラン、アリルトリエトキシシラン、ベンジルトリメトキシシラン、ベンジルトリエトキシシラン、1,2−ビス(トリメトキシシリル)エタン、シクロヘキシルトリメトキシシラン、ドデシルトリメトキシシラン、ヘキシルトリエトキシシラン、ヘキシルトリメトキシシラン、オクタデシルトリエトキシシラン、オクタデシルトリメトキシシラン、n−オクチルトリエトキシシラン、ペンタトリエトキシシラン、トリエトキシエチルシラン、トリエトキシメチルシラン、トリエトキシフェニルシラン、トリエトキシシラン、3−(トリエトキシシリル)プロピルメタクリレート、トリエトキシビニルシラン、トリメトキシ(メチル)シラン、トリメトキシフェニルシラン、トリメトキシ(プロピル)シラン、トリメトキシ(p−トリル)シラン、トリメトキシシラン、ビニルトリメトキシシラン等が挙げられる。また、これらを2種類以上用いてもよい。
【0026】
本発明で使用しうる有機アルカリ触媒としては、トリアルコキシシランを重縮合(加水分解及び重縮合)することができる触媒であれば特に限定されず、例えば、上記したように、アンモニア、コリン、テトラメチルアンモニウムハイドロキシド、及びモノエタノールアミンから選ばれた少なくとも1種を用いることができる。
【0027】
本発明で使用しうるポーラスシリカ前駆体の骨格形成材料であるアルコキシシランとしては特に制限されず、例えば、テトラエトキシシラン(TEOS)、テトラメトキシシラン(TMOS)、テトライソプロポキシシラン、テトラブトキシシランなどの4級アルコキシシラン;トリメトキシフルオロシラン、トリエトキシフルオロシラン、トリイソプロポキシフルオロシラン、トリブトキシフルオロシランなどの3級アルコキシフルオロシラン;トリメトキシメチルシラン、トリエトキシメチルシラン、トリメトキシエチルシラン、トリエトキシエチルシラン、トリメトキシプロピルシラン、トリエトキシプロピルシランなどの3級アルコキシアルキルシラン;トリメトキシフェニルシラン、トリエトキシフェニルシラン、トリメトキシクロロフェニルシラン、トリエトキシクロロフェニルシランなどの3級アルコキシアリールシラン;トリメトキシフェネチルシラン、トリエトキシフェネチルシランなどの3級アルコキシフェネチルシラン;ジメトキシジメチルシラン、ジエトキシジメチルシランなどの2級アルコキシアルキルシラン;アルコキシシランの2量体等のオリゴマーなどが挙げられる。また、2種類以上用いてもよい。
【0028】
本発明で使用しうる酸触媒は、トリアルコキシシランの重縮合生成物とアルコキシシランとの重縮合反応を行うことができる触媒であれば特に限定されず、例えば、塩酸、硝酸、硫酸等の無機酸や、酢酸等の有機酸からなる酸触媒を挙げることができ、また、2種類以上を組み合わせて用いてもよい。但し、有機酸を用いると、有機酸の残渣が、得られるシリカ膜中に残存してしまうことや、無機酸に比べて酸素プラズマ耐性に優れたシリカ膜を形成することができるという効果が低くなるため、無機酸を用いることが好ましい。
【0029】
上記反応系で使用できる溶媒としては、トリアルコキシシラン及びアルコキシシランを溶解又は分散させることができるものであれば特に限定されず、例えば、メタノール、エタノール、1−プロパノールなどの一級アルコール;2−プロパノール、2−ブタノールなどの二級アルコール;ターシャリーブチルアルコールなどの三級アルコール;アセトン、アセトニトリル等が挙げられる。また、2種類以上を組み合わせて用いてもよい。
【0030】
本発明で使用できる界面活性剤としては、ポリアルキレンオキサイド構造を有する非イオン界面活性剤を使用することが好ましい。ポリアルキレンオキサイド構造としては、ポリエチレンオキシド構造、ポリプロピレンオキシド構造、ポリメチレンキシド構造、ポリブチレンオキシド構造等が挙げられる。このようなポリアルキレンオキサイド構造を有する化合物としては、例えば、ポリオキシエチレンポリオキシプロピレンブロックコポリマー、ポリオキシエチレンポリオキシプロピレンアルキルエーテル、ポリエチレンアルキルエーテル、ポリオキシエチレンアルキルフェニルエーテル、ポリエチレンアルキルエーテル、ポリオキシエチレンアルキルフェニルエーテル等のエーテル型化合物、ポリオキシエチレングリセリン脂肪酸エステル、ポリオキシエチレンソルビタン脂肪酸エステル、ポリエチレンソルビトール脂肪酸エステル、ソルビタン脂肪酸エステル、プロピレングリコール脂肪酸エステル、ショ糖脂肪酸エステル等のエーテルエステル型化合物等を挙げることができる。また、2種類以上を組み合わせて用いてもよい。界面活性剤の状態は問われず、固体状態でも、溶媒に溶解した状態でもよい。
【0031】
上記界面活性剤は、溶液中でミセルを形成し、規則的に配列する。このミセルをテンプレートとして複合体をつくり、後段の工程でテンプレートを除去すると、均一で規則的な細孔を有するポーラスシリカ膜を製造することができる。そして、ポーラスの膜とすることにより、例えば、低誘電率(k)、低屈折率を有する膜となる。
【0032】
上記したようにトリアルコキシシラン及び有機アルカリ触媒、アルコキシシラン及び酸触媒、並びにアルコキシシランとトリアルコキシシラン及び酸触媒や、必要に応じて含有させる界面活性剤等の添加剤を溶媒に溶解または分散させて、トリアルコキシシランの加水分解反応及び重縮合反応を生じさせ、次いで得られた重縮合生成物とアルコキシシランとの加水分解反応及び重縮合反応、又は上記得られた重縮合生成物とアルコキシシランとトリアルコキシシランとの加水分解反応及び重縮合反応を生じさせて、ポーラスシリカ前駆体組成物を形成する。これらの加水分解反応や重縮合反応は、水を添加することにより生じるが、必要に応じて加熱や撹拌等してもよい。
【0033】
本発明によれば、上記したように、トリアルコキシシランの重縮合を行い、次いでこの重縮合生成物とアルコキシシランとの重縮合を行うか、又はこの重縮合生成物とアルコキシシランとトリアルコキシシランとの重縮合を行うことにより、目的とするポーラスシリカ前駆体組成物を作製している。このように、最初にトリアルコキシシランの重縮合を行い、次の重縮合の際にアルコキシシランとトリアルコキシシランとを用いて酸触媒の存在下に重縮合を行っても、目的とするポーラスシリカ前駆体組成物を作製することができる。しかし、最初にトリアルコキシシラン及びアルコキシシランを同時に使用して有機アルカリ触媒又は酸触媒の存在下に重縮合反応を行っても、目的とする所望のポーラスシリカ前駆体組成物を作製することはできない。
【0034】
以下、本発明の実施例及び比較例について説明する。本発明はこれらの実施例に限定されるわけではない。本発明では、既知の加水分解反応・重縮合反応を利用するため、用いるトリアルコキシシラン、有機アルカリ触媒、アルコキシシラン、及び酸触媒等は、以下の実施例で使用したもの以外に、上記したものであれば、適宜、使用可能であることは当業者の理解するところである。
【実施例1】
【0035】
メチルトリメトキシシラン(MTMS)0.1モル及び10wt%アンモニア水溶液0.01モル(アンモニア換算)を、エタノール中25℃で3時間撹拌し、透明で均一な溶液を得た。この溶液に、テトラエトキシシラン(TEOS)0.05モル、H2O4.25モル、非イオン性界面活性剤(第一工業製薬(株)製、商品名:P450、平均分子量:2300、OH(CH2CH2O)13(CH(CH3)CH2O)20(CH2CH2O)13H)0.01モル、硝酸0.01モル、及びエタノールを加え、再度3時間撹拌し、目的とするポーラスシリカ前駆体組成物(塗布液)を得た。
【0036】
この塗布液を用いて、半導体Si基板上に1200rpmの条件でスピンコートした後、基板を真空雰囲気下、350℃で1時間焼成処理した。350℃までの昇温時間は15分であった。
【0037】
上記のようにして得られたポーラスシリカ膜の膜厚は193nmで、比誘電率kは2.1で、屈折率は1.23で、弾性率は5.0GPaであった。
【0038】
得られたポーラスシリカ膜のFT−IRを測定し、このスペクトルを図1に示す。図1から明らかなように、Si−Oケージ構造のピークである1165cm−1とSi−Oラダー構造のピークである1060cm−1とのピーク高さの比は、Si−Oケージ構造/Si−Oラダー構造=1.21であり、Si−Oケージ構造のピークの方が大きかった。このピーク高さの比から、ケージ構造の成分割合は、1.21/(1.21+1.00)=54.8%である。
【0039】
得られたポーラスシリカ膜を30mm角にカットし、50℃加熱の1wt%KOH水溶液に3分間浸す処理を行った。処理前後の膜厚変動は8nmであり、薬液耐性に優れていることが分かる。
【実施例2】
【0040】
MTMS0.05モル及び10wt%アンモニア水溶液0.005モル(アンモニア換算)を、エタノール中25℃で3時間撹拌し、透明で均一な溶液を得た。この溶液に、TEOS0.05モル、MTMS0.05モル、H2O4.25モル、非イオン性界面活性剤(第一工業製薬製(株)製、商品名:P450、平均分子量:2300、OH(CH2CH2O)13(CH(CH3)CH2O)20(CH2CH2O)13H)0.01モル、硝酸0.01モル、及びエタノールを加え、再度3時間撹拌し、目的とするポーラスシリカ前駆体組成物(塗布液)を得た。
【0041】
この塗布液を用いて、半導体Si基板上に1200rpmの条件でスピンコートした後、基板を真空雰囲気下、350℃で1時間焼成処理した。350℃までの昇温時間は15分であった。
【0042】
上記のようにして得られたポーラスシリカ膜の膜厚は201nmで、比誘電率kは2.1で、屈折率は1.22で、弾性率は4.8GPaであった。
【0043】
得られたポーラスシリカ膜のFT−IRを測定した。このスペクトルから、Si−Oケージ構造のピークとSi−Oラダー構造のピークとのピーク高さの比は、Si−Oケージ構造/Si−Oラダー構造=1.17であり、Si−Oケージ構造のピークの方が大きかった。このピーク高さの比から、ケージ構造の成分割合は、1.17/(1.17+1.00)=53.9%である。
【0044】
得られたポーラスシリカ膜を30mm角にカットし、50℃加熱の1wt%KOH水溶液に3分浸す処理を行った。処理前後の膜厚変動は9nmであり、薬液耐性に優れていることが分かる。
【実施例3】
【0045】
エチルトリエトキシシラン(ETES)0.1モル及び15wt%コリン水溶液0.01モル(コリン換算)を、エタノール中25℃で3時間撹拌し、透明で均一な溶液を得た。この溶液に、TEOS0.05モル、H2O4.25モル、非イオン性界面活性剤(第一工業製薬(株)製、商品名:P450、平均分子量:2300、OH(CH2CH2O)13(CH(CH3)CH2O)20(CH2CH2O)13H)0.01モル、硝酸0.01モル、及びエタノールを加え、再度3時間撹拌し、目的とするポーラスシリカ前駆体組成物(塗布液)を得た。
【0046】
この塗布液を用いて、半導体Si基板上に1200rpmの条件でスピンコートした後、基板を真空雰囲気下、350℃で1時間焼成処理した。350℃までの昇温時間は15分であった。
【0047】
上記のようにして得られたポーラスシリカ膜の膜厚は197nmで、比誘電率kは2.1で、屈折率は1.23で、弾性率は5.0GPaであった。
【0048】
得られたポーラスシリカ膜のFT−IRを測定した。このスペクトルから、Si−Oケージ構造のピークとSi−Oラダー構造のピークとの高さの比は、Si−Oケージ構造/Si−Oラダー構造=1.20であり、Si−Oケージ構造のピークの方が大きかった。このピーク高さの比から、ケージ構造の成分割合は、1.20/(1.20+1.00)=54.5%である。
【0049】
得られたポーラスシリカ膜を30mm角にカットし、50℃加熱の1wt%KOH水溶液に3分浸す処理を行った。処理前後の膜厚変動は9nmであり、薬液耐性に優れていることが分かる。
【0050】
(比較例1)
TEOS0.05モル、MTMS0.1モル、H2O4.25モル、非イオン性界面活性剤(商品名:P450、平均分子量:2300、OH(CH2CH2O)13(CH(CH3)CH2O)20(CH2CH2O)13H)0.01モル、硝酸0.01モルをエタノール中25℃で3時間撹拌し、透明で均一なポーラスシリカ前駆体組成物(塗布液)を得た。
【0051】
この塗布液を用いて、半導体Si基板上に1200rpmの条件でスピンコートした後、基板を真空雰囲気下、350℃で1時間焼成処理した。350℃までの昇温時間は15分であった。
【0052】
上記のようにして得られたポーラスシリカ膜の膜厚は190nmで、比誘電率kは2.1で、屈折率は1.21で、弾性率は3.6GPaであった。
【0053】
得られたポーラスシリカ膜のFT−IRを測定し、このスペクトルを図2に示す。図2から明らかなように、Si−Oケージ構造のピークとSi−Oラダー構造のピークとの高さの比は、Si−Oケージ構造/Si−Oラダー構造=0.90であり、Si−Oラダー構造のピークの方が大きかった。このピーク高さの比から、ケージ構造の成分割合は、0.90/(0.90+1.00)=47.4%である。
【0054】
得られたポーラスシリカ膜を30mm角にカットし、50℃加熱の1wt%KOH水溶液に3分浸す処理を行ったところ、膜が無くなった。そのため、処理時間を1分としたところ、処理前後の膜厚変動は130nmであった。
【0055】
上記結果から、最初にMTMS及びTEOSを同時に使用し、触媒として酸触媒を使用した場合には、ケージ構造の成分割合がラダー構造の成分割合より少ないポーラスシリカ膜が得られ、この膜は弾性率が低く、薬液耐性に乏しかったことが分かる。
【0056】
(比較例2)
TEOS0.05モル、MTMS0.1モル、H2O4.25モル、非イオン性界面活性剤(第一工業製薬(株)製、商品名:P450、平均分子量:2300、OH(CH2CH2O)13(CH(CH3)CH2O)20(CH2CH2O)13H)0.01モル、及び15wt%コリン水溶液0.01モル(コリン換算)をエタノール中25℃で3時間撹拌し、透明で均一なポーラスシリカ前駆体組成物(塗布液)を得た。
【0057】
この塗布液を用いて、半導体Si基板上に1200rpmの条件でスピンコートした後、基板を真空雰囲気下、350℃で1時間焼成処理した。350℃までの昇温時間は15分であった。
【0058】
上記のようにして得られたポーラスシリカ膜の膜厚は182nmで、比誘電率kは2.1で、屈折率は1.21で、弾性率は3.5GPaであった。
【0059】
得られたポーラスシリカ膜のFT−IRを測定した。このスペクトルから、Si−Oケージ構造のピークとSi−Oラダー構造のピークとの高さの比は、Si−Oケージ/Si−Oラダー=0.85であり、Si−Oラダー構造のピークの方が大きかった。このピーク高さの比から、ケージ構造の成分割合は、0.85/(0.85+1.00)=45.9%である。
【0060】
得られたポーラスシリカ膜を30mm角にカットし、50℃加熱の1wt%KOH水溶液に3分浸す処理を行ったところ、膜が無くなった。そのため、処理時間を1分としたところ、処理前後の膜厚変動は145nmであった。
【0061】
上記結果から、最初にMTMS及びTEOSを同時に使用し、触媒として有機アルカリ触媒を使用した場合には、ケージ構造の成分割合がラダー構造の成分割合より少ないポーラスシリカ膜が得られ、この膜は弾性率が低く、薬液耐性に乏しかったことが分かる。
【産業上の利用可能性】
【0062】
本発明によれば、ポーラスシリカ膜は、膜中のケージ構造の方がラダー構造よりも高い割合を有することができるので、ポーラスシリカ膜の膜強度を高くできると共に、薬液耐性を向上させることができ、本発明は、半導体装置の産業分野で利用可能である。
【技術分野】
【0001】
本発明は、ポーラスシリカ前駆体組成物及びその作製方法、ポーラスシリカ膜及びその作製方法、並びに半導体素子に関し、特にケージ(Cage)構造の成分割合の多いポーラスシリカ膜を得るためのポーラスシリカ前駆体組成物及びその作製方法、このポーラスシリカ前駆体組成物を用いて得られるポーラスシリカ膜及びその作製方法、並びにこのポーラスシリカ膜を利用した半導体素子に関する。
【背景技術】
【0002】
半導体素子の層間絶縁膜等として、ポーラスシリカ膜が使用されており、このポーラスシリカ膜の作製方法には、大きく分けてCVD法と塗布法とがある。CVD法は、CVDによるシリカ成膜時に有機物であるポロジェンを加えて成膜し、熱、電子線、UV等により、ポロジェンを除去し、ポーラスシリカ膜を得る方法である。一方、塗布法は、界面活性剤等の有機物であるテンプレートが加えられた前駆体組成物(塗布液)をスピンコートして成膜し、焼成し、電子線、UV等により、テンプレートを除去し、ポーラスシリカ膜を得る方法である(例えば、特許文献1参照)。これらの場合、得られるポーラスシリカ膜は、空孔量を増加させるに従い膜強度が低下するという問題があり、また、比表面が増加するため、膜の欠陥があらわになる確立が高まり、薬液耐性も低下するという問題がある。そのため、膜強度が高く、薬液耐性に優れたポーラスシリカ膜が求められている。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】特開平07−133350号公報
【発明の概要】
【発明が解決しようとする課題】
【0004】
本発明の課題は、上述の従来技術の問題点を解決することにあり、ポーラスシリカ膜中のSi−Oケージ構造の成分割合を多くすることで、膜強度を高くし、また、薬液耐性を向上させたポーラスシリカ膜を得るための、ポーラスシリカ前駆体組成物及びその作製方法、この前駆体組成物を用いて作製したポーラスシリカ膜及びその作製方法、並びにこのポーラスシリカ膜を用いて得られた半導体素子を提供することにある。
【課題を解決するための手段】
【0005】
本発明者らは、上記課題を解決するために鋭意検討した結果、触媒を用いてトリアルコキシシラン系原料の加水分解・重縮合反応を進めた後、ポーラスシリカ前駆体の骨格形成材料を添加する手法を用いて作製されたポーラスシリカ前駆体組成物を用いることで、ポーラスシリカ膜中のケージ構造の成分割合が多くなり、膜強度を高くできると共に、薬液耐性を向上できることを見出し、本発明を完成させるに至った。
【0006】
本発明のポーラスシリカ前駆体組成物は、界面活性剤をテンプレートとして含むポーラスシリカ前駆体組成物であって、有機アルカリ触媒の存在下で得られたトリアルコキシシランの重縮合生成物とポーラスシリカ前駆体の骨格形成材料との酸触媒の存在下での重縮合生成物、又は該有機アルカリ触媒の存在下で得られたトリアルコキシシランの重縮合生成物とポーラスシリカ前駆体の骨格形成材料とトリアルコキシシランとの酸触媒の存在下での重縮合生成物を含有することを特徴とする。
【0007】
前記ポーラスシリカ前駆体組成物において、有機アルカリ触媒が、アンモニア、コリン、テトラメチルアンモニウムハイドロキシド、及びモノエタノールアミンから選ばれた少なくとも1種であることを特徴とする。
【0008】
前記ポーラスシリカ前駆体組成物において、酸触媒が、硝酸、塩酸、硫酸、及びフッ酸から選ばれた無機酸であることを特徴とする。
【0009】
前記ポーラスシリカ前駆体組成物において、骨格形成材料が、アルコキシシランから選ばれた少なくとも1種であることを特徴とする。
【0010】
本発明のポーラスシリカ前駆体組成物の作製方法は、界面活性剤をテンプレートとして用いるポーラスシリカ前駆体組成物の作製方法において、有機アルカリ触媒の存在下トリアルコキシシランの重縮合反応を行った後、酸触媒の存在下、ポーラスシリカ前駆体の骨格形成材料を添加してさらに重縮合反応を行うか、又は該酸触媒の存在下、ポーラスシリカ前駆体の骨格形成材料及びさらにトリアルコキシシランを添加してさらに重縮合反応を行うことを特徴とする。
【0011】
前記ポーラスシリカ前駆体組成物の作製方法において、有機アルカリ触媒が、アンモニア、コリン、テトラメチルアンモニウムハイドロキシド、及びモノエタノールアミンから選ばれた少なくとも1種であることを特徴とする。
【0012】
前記ポーラスシリカ前駆体組成物の作製方法において、酸触媒が、硝酸、塩酸、硫酸、及びフッ酸から選ばれた無機酸であることを特徴とする。
【0013】
前記ポーラスシリカ前駆体組成物の作製方法において、骨格形成材料が、アルコキシシランから選ばれた少なくとも1種であることを特徴とする。
【0014】
本発明のポーラスシリカ膜は、上記ポーラスシリカ前駆体組成物又は上記ポーラスシリカ前駆体組成物の作製方法に従って作製された前駆体組成物を用いて作製されたものであることを特徴とする。
【0015】
本発明のポーラスシリカ膜において、その膜のFT−IRスペクトルのSi−Oケージ構造のピークである1150cm−1近傍とSi−Oラダー構造のピークである1050cm−1近傍とのピーク高さの比が、1.0以上であることを特徴とする。
【0016】
上記Si−Oケージ構造のピークである1150cm−1近傍とSi−Oラダー構造のピークである1050cm−1近傍とのピーク高さの比が、1.0未満であると、目的とする膜強度、薬液耐性の向上を達成することができない傾向がある。
【0017】
本発明のポーラスシリカ膜の作製方法は、上記ポーラスシリカ前駆体組成物又は上記ポーラスシリカ前駆体組成物の作製方法に従って作製されたポーラスシリカ前駆体組成物を基板表面に塗布し、真空雰囲気下焼成処理し、界面活性剤を除去してポーラスシリカ膜を作製することを特徴とする。
【0018】
本発明の半導体素子は、上記ポーラスシリカ膜又は上記ポーラスシリカ膜の作製方法に従って作製したポーラスシリカ膜を用いて得られたものであることを特徴とする。
【発明の効果】
【0019】
本発明によれば、有機アルカリ触媒を用いてトリアルコキシシラン系原料の重縮合を進めた後に、ポーラスシリカ前駆体の骨格形成材料を添加する手法を用いて作製されたポーラスシリカ前駆体組成物を用いることで、ポーラスシリカ膜中のケージ構造の成分割合がラダー構造の成分割合と同等又はより多くなり、膜強度を高くできると共に、薬液耐性を向上させることができるという効果を奏する。
【図面の簡単な説明】
【0020】
【図1】実施例1において得られたポーラスシリカ膜のFT−IRスペクトルを示すスペクトル図。
【図2】比較例1において得られたポーラスシリカ膜のFT−IRスペクトルを示すスペクトル図。
【発明を実施するための形態】
【0021】
本発明に係るポーラスシリカ前駆体組成物(塗布液)の実施の形態によれば、この前駆体組成物は、界面活性剤をテンプレートとして含む前駆体組成物であって、有機アルカリ触媒(例えば、アンモニア、コリン、テトラメチルアンモニウムハイドロキシド、及びモノエタノールアミン等から選ばれた少なくとも1種)の存在下で得られたトリアルコキシシラン(以下列挙するメチルトリメトキシシラン(MTMS)等から選ばれた少なくとも1種)の重縮合生成物と、酸触媒(例えば、硝酸、塩酸、硫酸、及びフッ酸等から選ばれた少なくとも1種の無機酸)の存在下、ポーラスシリカ前駆体の骨格形成材料(以下列挙するアルコキシシランから選ばれた少なくとも1種)との重縮合生成物、又は上記有機アルカリ触媒の存在下に得られたトリアルコキシシランの重縮合生成物と上記ポーラスシリカ前駆体の骨格形成材料とさらに上記トリアルコキシシランとの酸触媒の存在下での重縮合生成物を含有することからなる。
【0022】
本発明に係るポーラスシリカ前駆体組成物の作製方法の実施の形態によれば、この作製方法は、界面活性剤をテンプレートとして用いるポーラスシリカ前駆体組成物の作製方法において、有機アルカリ触媒(例えば、アンモニア、コリン、テトラメチルアンモニウムハイドロキシド、及びモノエタノールアミンから選ばれた少なくとも1種)の存在下、トリアルコキシシラン(以下列挙するメチルトリメトキシシラン(MTMS)等から選ばれた少なくとも1種)の重縮合反応を行った後、この重縮合生成物に、酸触媒(例えば、硝酸、塩酸、硫酸、及びフッ酸等から選ばれた少なくとも1種の無機酸)の存在下、ポーラスシリカ前駆体の骨格形成材料(以下列挙するアルコキシシランから選ばれた少なくとも1種)を添加してさらに重縮合反応を行うか、又は上記酸触媒の存在下、上記ポーラスシリカ前駆体の骨格形成材料及びさらに上記トリアルコキシシランを添加してさらに重縮合反応を行うことからなる。
【0023】
本発明に係るポーラスシリカ膜の実施の形態によれば、このポーラスシリカ膜は、上記ポーラスシリカ前駆体組成物又は上記ポーラスシリカ前駆体組成物の作製方法に従って作製された前駆体組成物を用いて作製されたものであり、そのポーラスシリカ膜のFT−IRスペクトルのSi−Oケージ構造のピークである1150cm−1近傍とSi−Oラダー構造のピークである1050cm−1近傍とのピーク高さの比が、1.0以上であることからなる。このピーク高さの比は、ポーラスシリカ膜中のケージ構造の成分割合をAとし、ラダー構造の成分割合をBとした場合、50%≦A/(A+B)≦100%を満足することと同義である。以下記載する実施例及び比較例によれば、ケージ構造のピーク高さとラダー構造のピーク高さとの比が1.0以上であると、すなわち、ケージ構造の成分割合が増加するに従って、膜強度及び薬液耐性が向上することが分かる。
【0024】
本発明では、有機アルカリ触媒を用いてトリアルコキシシラン系原料の加水分解・重縮合反応を進めた後、この重縮合生成物にポーラスシリカ前駆体の骨格形成材料を添加するか、又はこの重縮合生成物にポーラスシリカ前駆体の骨格形成材料及びトリアルコキシシラン系原料を添加する手法を用いて作製されたポーラスシリカ前駆体組成物を用いることで、ポーラスシリカ膜中のケージ構造の成分割合が多くなり、膜強度を高くできると共に、薬液耐性を向上させることができる。
【0025】
本発明で使用し得るトリアルコキシシランとしては、特に制限はないが、例えばメチルトリメトキシシラン(MTMS)、メチルトリエトキシシラン、エチルトリメトキシシラン、エチルトリエトキシシラン(ETES)、アリルトリメトキシシラン、アリルトリエトキシシラン、ベンジルトリメトキシシラン、ベンジルトリエトキシシラン、1,2−ビス(トリメトキシシリル)エタン、シクロヘキシルトリメトキシシラン、ドデシルトリメトキシシラン、ヘキシルトリエトキシシラン、ヘキシルトリメトキシシラン、オクタデシルトリエトキシシラン、オクタデシルトリメトキシシラン、n−オクチルトリエトキシシラン、ペンタトリエトキシシラン、トリエトキシエチルシラン、トリエトキシメチルシラン、トリエトキシフェニルシラン、トリエトキシシラン、3−(トリエトキシシリル)プロピルメタクリレート、トリエトキシビニルシラン、トリメトキシ(メチル)シラン、トリメトキシフェニルシラン、トリメトキシ(プロピル)シラン、トリメトキシ(p−トリル)シラン、トリメトキシシラン、ビニルトリメトキシシラン等が挙げられる。また、これらを2種類以上用いてもよい。
【0026】
本発明で使用しうる有機アルカリ触媒としては、トリアルコキシシランを重縮合(加水分解及び重縮合)することができる触媒であれば特に限定されず、例えば、上記したように、アンモニア、コリン、テトラメチルアンモニウムハイドロキシド、及びモノエタノールアミンから選ばれた少なくとも1種を用いることができる。
【0027】
本発明で使用しうるポーラスシリカ前駆体の骨格形成材料であるアルコキシシランとしては特に制限されず、例えば、テトラエトキシシラン(TEOS)、テトラメトキシシラン(TMOS)、テトライソプロポキシシラン、テトラブトキシシランなどの4級アルコキシシラン;トリメトキシフルオロシラン、トリエトキシフルオロシラン、トリイソプロポキシフルオロシラン、トリブトキシフルオロシランなどの3級アルコキシフルオロシラン;トリメトキシメチルシラン、トリエトキシメチルシラン、トリメトキシエチルシラン、トリエトキシエチルシラン、トリメトキシプロピルシラン、トリエトキシプロピルシランなどの3級アルコキシアルキルシラン;トリメトキシフェニルシラン、トリエトキシフェニルシラン、トリメトキシクロロフェニルシラン、トリエトキシクロロフェニルシランなどの3級アルコキシアリールシラン;トリメトキシフェネチルシラン、トリエトキシフェネチルシランなどの3級アルコキシフェネチルシラン;ジメトキシジメチルシラン、ジエトキシジメチルシランなどの2級アルコキシアルキルシラン;アルコキシシランの2量体等のオリゴマーなどが挙げられる。また、2種類以上用いてもよい。
【0028】
本発明で使用しうる酸触媒は、トリアルコキシシランの重縮合生成物とアルコキシシランとの重縮合反応を行うことができる触媒であれば特に限定されず、例えば、塩酸、硝酸、硫酸等の無機酸や、酢酸等の有機酸からなる酸触媒を挙げることができ、また、2種類以上を組み合わせて用いてもよい。但し、有機酸を用いると、有機酸の残渣が、得られるシリカ膜中に残存してしまうことや、無機酸に比べて酸素プラズマ耐性に優れたシリカ膜を形成することができるという効果が低くなるため、無機酸を用いることが好ましい。
【0029】
上記反応系で使用できる溶媒としては、トリアルコキシシラン及びアルコキシシランを溶解又は分散させることができるものであれば特に限定されず、例えば、メタノール、エタノール、1−プロパノールなどの一級アルコール;2−プロパノール、2−ブタノールなどの二級アルコール;ターシャリーブチルアルコールなどの三級アルコール;アセトン、アセトニトリル等が挙げられる。また、2種類以上を組み合わせて用いてもよい。
【0030】
本発明で使用できる界面活性剤としては、ポリアルキレンオキサイド構造を有する非イオン界面活性剤を使用することが好ましい。ポリアルキレンオキサイド構造としては、ポリエチレンオキシド構造、ポリプロピレンオキシド構造、ポリメチレンキシド構造、ポリブチレンオキシド構造等が挙げられる。このようなポリアルキレンオキサイド構造を有する化合物としては、例えば、ポリオキシエチレンポリオキシプロピレンブロックコポリマー、ポリオキシエチレンポリオキシプロピレンアルキルエーテル、ポリエチレンアルキルエーテル、ポリオキシエチレンアルキルフェニルエーテル、ポリエチレンアルキルエーテル、ポリオキシエチレンアルキルフェニルエーテル等のエーテル型化合物、ポリオキシエチレングリセリン脂肪酸エステル、ポリオキシエチレンソルビタン脂肪酸エステル、ポリエチレンソルビトール脂肪酸エステル、ソルビタン脂肪酸エステル、プロピレングリコール脂肪酸エステル、ショ糖脂肪酸エステル等のエーテルエステル型化合物等を挙げることができる。また、2種類以上を組み合わせて用いてもよい。界面活性剤の状態は問われず、固体状態でも、溶媒に溶解した状態でもよい。
【0031】
上記界面活性剤は、溶液中でミセルを形成し、規則的に配列する。このミセルをテンプレートとして複合体をつくり、後段の工程でテンプレートを除去すると、均一で規則的な細孔を有するポーラスシリカ膜を製造することができる。そして、ポーラスの膜とすることにより、例えば、低誘電率(k)、低屈折率を有する膜となる。
【0032】
上記したようにトリアルコキシシラン及び有機アルカリ触媒、アルコキシシラン及び酸触媒、並びにアルコキシシランとトリアルコキシシラン及び酸触媒や、必要に応じて含有させる界面活性剤等の添加剤を溶媒に溶解または分散させて、トリアルコキシシランの加水分解反応及び重縮合反応を生じさせ、次いで得られた重縮合生成物とアルコキシシランとの加水分解反応及び重縮合反応、又は上記得られた重縮合生成物とアルコキシシランとトリアルコキシシランとの加水分解反応及び重縮合反応を生じさせて、ポーラスシリカ前駆体組成物を形成する。これらの加水分解反応や重縮合反応は、水を添加することにより生じるが、必要に応じて加熱や撹拌等してもよい。
【0033】
本発明によれば、上記したように、トリアルコキシシランの重縮合を行い、次いでこの重縮合生成物とアルコキシシランとの重縮合を行うか、又はこの重縮合生成物とアルコキシシランとトリアルコキシシランとの重縮合を行うことにより、目的とするポーラスシリカ前駆体組成物を作製している。このように、最初にトリアルコキシシランの重縮合を行い、次の重縮合の際にアルコキシシランとトリアルコキシシランとを用いて酸触媒の存在下に重縮合を行っても、目的とするポーラスシリカ前駆体組成物を作製することができる。しかし、最初にトリアルコキシシラン及びアルコキシシランを同時に使用して有機アルカリ触媒又は酸触媒の存在下に重縮合反応を行っても、目的とする所望のポーラスシリカ前駆体組成物を作製することはできない。
【0034】
以下、本発明の実施例及び比較例について説明する。本発明はこれらの実施例に限定されるわけではない。本発明では、既知の加水分解反応・重縮合反応を利用するため、用いるトリアルコキシシラン、有機アルカリ触媒、アルコキシシラン、及び酸触媒等は、以下の実施例で使用したもの以外に、上記したものであれば、適宜、使用可能であることは当業者の理解するところである。
【実施例1】
【0035】
メチルトリメトキシシラン(MTMS)0.1モル及び10wt%アンモニア水溶液0.01モル(アンモニア換算)を、エタノール中25℃で3時間撹拌し、透明で均一な溶液を得た。この溶液に、テトラエトキシシラン(TEOS)0.05モル、H2O4.25モル、非イオン性界面活性剤(第一工業製薬(株)製、商品名:P450、平均分子量:2300、OH(CH2CH2O)13(CH(CH3)CH2O)20(CH2CH2O)13H)0.01モル、硝酸0.01モル、及びエタノールを加え、再度3時間撹拌し、目的とするポーラスシリカ前駆体組成物(塗布液)を得た。
【0036】
この塗布液を用いて、半導体Si基板上に1200rpmの条件でスピンコートした後、基板を真空雰囲気下、350℃で1時間焼成処理した。350℃までの昇温時間は15分であった。
【0037】
上記のようにして得られたポーラスシリカ膜の膜厚は193nmで、比誘電率kは2.1で、屈折率は1.23で、弾性率は5.0GPaであった。
【0038】
得られたポーラスシリカ膜のFT−IRを測定し、このスペクトルを図1に示す。図1から明らかなように、Si−Oケージ構造のピークである1165cm−1とSi−Oラダー構造のピークである1060cm−1とのピーク高さの比は、Si−Oケージ構造/Si−Oラダー構造=1.21であり、Si−Oケージ構造のピークの方が大きかった。このピーク高さの比から、ケージ構造の成分割合は、1.21/(1.21+1.00)=54.8%である。
【0039】
得られたポーラスシリカ膜を30mm角にカットし、50℃加熱の1wt%KOH水溶液に3分間浸す処理を行った。処理前後の膜厚変動は8nmであり、薬液耐性に優れていることが分かる。
【実施例2】
【0040】
MTMS0.05モル及び10wt%アンモニア水溶液0.005モル(アンモニア換算)を、エタノール中25℃で3時間撹拌し、透明で均一な溶液を得た。この溶液に、TEOS0.05モル、MTMS0.05モル、H2O4.25モル、非イオン性界面活性剤(第一工業製薬製(株)製、商品名:P450、平均分子量:2300、OH(CH2CH2O)13(CH(CH3)CH2O)20(CH2CH2O)13H)0.01モル、硝酸0.01モル、及びエタノールを加え、再度3時間撹拌し、目的とするポーラスシリカ前駆体組成物(塗布液)を得た。
【0041】
この塗布液を用いて、半導体Si基板上に1200rpmの条件でスピンコートした後、基板を真空雰囲気下、350℃で1時間焼成処理した。350℃までの昇温時間は15分であった。
【0042】
上記のようにして得られたポーラスシリカ膜の膜厚は201nmで、比誘電率kは2.1で、屈折率は1.22で、弾性率は4.8GPaであった。
【0043】
得られたポーラスシリカ膜のFT−IRを測定した。このスペクトルから、Si−Oケージ構造のピークとSi−Oラダー構造のピークとのピーク高さの比は、Si−Oケージ構造/Si−Oラダー構造=1.17であり、Si−Oケージ構造のピークの方が大きかった。このピーク高さの比から、ケージ構造の成分割合は、1.17/(1.17+1.00)=53.9%である。
【0044】
得られたポーラスシリカ膜を30mm角にカットし、50℃加熱の1wt%KOH水溶液に3分浸す処理を行った。処理前後の膜厚変動は9nmであり、薬液耐性に優れていることが分かる。
【実施例3】
【0045】
エチルトリエトキシシラン(ETES)0.1モル及び15wt%コリン水溶液0.01モル(コリン換算)を、エタノール中25℃で3時間撹拌し、透明で均一な溶液を得た。この溶液に、TEOS0.05モル、H2O4.25モル、非イオン性界面活性剤(第一工業製薬(株)製、商品名:P450、平均分子量:2300、OH(CH2CH2O)13(CH(CH3)CH2O)20(CH2CH2O)13H)0.01モル、硝酸0.01モル、及びエタノールを加え、再度3時間撹拌し、目的とするポーラスシリカ前駆体組成物(塗布液)を得た。
【0046】
この塗布液を用いて、半導体Si基板上に1200rpmの条件でスピンコートした後、基板を真空雰囲気下、350℃で1時間焼成処理した。350℃までの昇温時間は15分であった。
【0047】
上記のようにして得られたポーラスシリカ膜の膜厚は197nmで、比誘電率kは2.1で、屈折率は1.23で、弾性率は5.0GPaであった。
【0048】
得られたポーラスシリカ膜のFT−IRを測定した。このスペクトルから、Si−Oケージ構造のピークとSi−Oラダー構造のピークとの高さの比は、Si−Oケージ構造/Si−Oラダー構造=1.20であり、Si−Oケージ構造のピークの方が大きかった。このピーク高さの比から、ケージ構造の成分割合は、1.20/(1.20+1.00)=54.5%である。
【0049】
得られたポーラスシリカ膜を30mm角にカットし、50℃加熱の1wt%KOH水溶液に3分浸す処理を行った。処理前後の膜厚変動は9nmであり、薬液耐性に優れていることが分かる。
【0050】
(比較例1)
TEOS0.05モル、MTMS0.1モル、H2O4.25モル、非イオン性界面活性剤(商品名:P450、平均分子量:2300、OH(CH2CH2O)13(CH(CH3)CH2O)20(CH2CH2O)13H)0.01モル、硝酸0.01モルをエタノール中25℃で3時間撹拌し、透明で均一なポーラスシリカ前駆体組成物(塗布液)を得た。
【0051】
この塗布液を用いて、半導体Si基板上に1200rpmの条件でスピンコートした後、基板を真空雰囲気下、350℃で1時間焼成処理した。350℃までの昇温時間は15分であった。
【0052】
上記のようにして得られたポーラスシリカ膜の膜厚は190nmで、比誘電率kは2.1で、屈折率は1.21で、弾性率は3.6GPaであった。
【0053】
得られたポーラスシリカ膜のFT−IRを測定し、このスペクトルを図2に示す。図2から明らかなように、Si−Oケージ構造のピークとSi−Oラダー構造のピークとの高さの比は、Si−Oケージ構造/Si−Oラダー構造=0.90であり、Si−Oラダー構造のピークの方が大きかった。このピーク高さの比から、ケージ構造の成分割合は、0.90/(0.90+1.00)=47.4%である。
【0054】
得られたポーラスシリカ膜を30mm角にカットし、50℃加熱の1wt%KOH水溶液に3分浸す処理を行ったところ、膜が無くなった。そのため、処理時間を1分としたところ、処理前後の膜厚変動は130nmであった。
【0055】
上記結果から、最初にMTMS及びTEOSを同時に使用し、触媒として酸触媒を使用した場合には、ケージ構造の成分割合がラダー構造の成分割合より少ないポーラスシリカ膜が得られ、この膜は弾性率が低く、薬液耐性に乏しかったことが分かる。
【0056】
(比較例2)
TEOS0.05モル、MTMS0.1モル、H2O4.25モル、非イオン性界面活性剤(第一工業製薬(株)製、商品名:P450、平均分子量:2300、OH(CH2CH2O)13(CH(CH3)CH2O)20(CH2CH2O)13H)0.01モル、及び15wt%コリン水溶液0.01モル(コリン換算)をエタノール中25℃で3時間撹拌し、透明で均一なポーラスシリカ前駆体組成物(塗布液)を得た。
【0057】
この塗布液を用いて、半導体Si基板上に1200rpmの条件でスピンコートした後、基板を真空雰囲気下、350℃で1時間焼成処理した。350℃までの昇温時間は15分であった。
【0058】
上記のようにして得られたポーラスシリカ膜の膜厚は182nmで、比誘電率kは2.1で、屈折率は1.21で、弾性率は3.5GPaであった。
【0059】
得られたポーラスシリカ膜のFT−IRを測定した。このスペクトルから、Si−Oケージ構造のピークとSi−Oラダー構造のピークとの高さの比は、Si−Oケージ/Si−Oラダー=0.85であり、Si−Oラダー構造のピークの方が大きかった。このピーク高さの比から、ケージ構造の成分割合は、0.85/(0.85+1.00)=45.9%である。
【0060】
得られたポーラスシリカ膜を30mm角にカットし、50℃加熱の1wt%KOH水溶液に3分浸す処理を行ったところ、膜が無くなった。そのため、処理時間を1分としたところ、処理前後の膜厚変動は145nmであった。
【0061】
上記結果から、最初にMTMS及びTEOSを同時に使用し、触媒として有機アルカリ触媒を使用した場合には、ケージ構造の成分割合がラダー構造の成分割合より少ないポーラスシリカ膜が得られ、この膜は弾性率が低く、薬液耐性に乏しかったことが分かる。
【産業上の利用可能性】
【0062】
本発明によれば、ポーラスシリカ膜は、膜中のケージ構造の方がラダー構造よりも高い割合を有することができるので、ポーラスシリカ膜の膜強度を高くできると共に、薬液耐性を向上させることができ、本発明は、半導体装置の産業分野で利用可能である。
【特許請求の範囲】
【請求項1】
界面活性剤をテンプレートとして含むポーラスシリカ前駆体組成物であって、有機アルカリ触媒の存在下で得られたトリアルコキシシランの重縮合生成物とポーラスシリカ前駆体の骨格形成材料との酸触媒の存在下での重縮合生成物、又は該有機アルカリ触媒の存在下で得られたトリアルコキシシランの重縮合生成物とポーラスシリカ前駆体の骨格形成材料とトリアルコキシシランとの酸触媒の存在下での重縮合生成物を含有することを特徴とするポーラスシリカ前駆体組成物。
【請求項2】
前記有機アルカリ触媒が、アンモニア、コリン、テトラメチルアンモニウムハイドロキシド、及びモノエタノールアミンから選ばれた少なくとも1種であることを特徴とする請求項1記載のポーラスシリカ前駆体組成物。
【請求項3】
前記酸触媒が、硝酸、塩酸、硫酸、及びフッ酸から選ばれた無機酸であることを特徴とする請求項1又は2記載のポーラスシリカ前駆体組成物。
【請求項4】
前記骨格形成材料が、アルコキシシランから選ばれた少なくとも1種であることを特徴とする請求項1〜3のいずれか1項に記載のポーラスシリカ前駆体組成物。
【請求項5】
界面活性剤をテンプレートとして用いるポーラスシリカ前駆体組成物の作製方法において、有機アルカリ触媒の存在下トリアルコキシシランの重縮合反応を行った後、酸触媒の存在下、ポーラスシリカ前駆体の骨格形成材料を添加してさらに重縮合反応を行うか、又は該酸触媒の存在下、ポーラスシリカ前駆体の骨格形成材料及びさらにトリアルコキシシランを添加してさらに重縮合反応を行うことを特徴とするポーラスシリカ前駆体組成物の作製方法。
【請求項6】
前記有機アルカリ触媒が、アンモニア、コリン、テトラメチルアンモニウムハイドロキシド、及びモノエタノールアミンから選ばれた少なくとも1種であることを特徴とする請求項5記載のポーラスシリカ前駆体組成物の作製方法。
【請求項7】
前記酸触媒が、硝酸、塩酸、硫酸、及びフッ酸から選ばれた無機酸であることを特徴とする請求項5又は6記載のポーラスシリカ前駆体組成物の作製方法。
【請求項8】
前記骨格形成材料が、アルコキシシランから選ばれた少なくとも1種であることを特徴とする請求項5〜7のいずれか1項に記載のポーラスシリカ前駆体組成物の作製方法。
【請求項9】
請求項1〜4のいずれか1項に記載のポーラスシリカ前駆体組成物又は請求項5〜8のいずれか1項に記載のポーラスシリカ前駆体組成物の作製方法に従って作製されたポーラスシリカ前駆体組成物を用いて作製されたものであることを特徴とするポーラスシリカ膜。
【請求項10】
前記ポーラスシリカ膜のFT−IRスペクトルのSi−Oケージ構造のピークである1150cm−1近傍とSi−Oラダー構造のピークである1050cm−1近傍とのピーク高さの比が、1.0以上であることを特徴とする請求項9記載のポーラスシリカ膜。
【請求項11】
請求項1〜4のいずれか1項に記載のポーラスシリカ前駆体組成物又は請求項5〜8のいずれか1項に記載のポーラスシリカ前駆体組成物の作製方法に従って作製されたポーラスシリカ前駆体組成物を基板表面に塗布し、真空雰囲気下焼成処理し、界面活性剤を除去してポーラスシリカ膜を作製することを特徴とするポーラスシリカ膜の作製方法。
【請求項12】
請求項9若しくは10記載のポーラスシリカ膜又は請求項11記載のポーラスシリカ膜の作製方法に従って作製したポーラスシリカ膜を用いて得られたものであることを特徴とする半導体素子。
【請求項1】
界面活性剤をテンプレートとして含むポーラスシリカ前駆体組成物であって、有機アルカリ触媒の存在下で得られたトリアルコキシシランの重縮合生成物とポーラスシリカ前駆体の骨格形成材料との酸触媒の存在下での重縮合生成物、又は該有機アルカリ触媒の存在下で得られたトリアルコキシシランの重縮合生成物とポーラスシリカ前駆体の骨格形成材料とトリアルコキシシランとの酸触媒の存在下での重縮合生成物を含有することを特徴とするポーラスシリカ前駆体組成物。
【請求項2】
前記有機アルカリ触媒が、アンモニア、コリン、テトラメチルアンモニウムハイドロキシド、及びモノエタノールアミンから選ばれた少なくとも1種であることを特徴とする請求項1記載のポーラスシリカ前駆体組成物。
【請求項3】
前記酸触媒が、硝酸、塩酸、硫酸、及びフッ酸から選ばれた無機酸であることを特徴とする請求項1又は2記載のポーラスシリカ前駆体組成物。
【請求項4】
前記骨格形成材料が、アルコキシシランから選ばれた少なくとも1種であることを特徴とする請求項1〜3のいずれか1項に記載のポーラスシリカ前駆体組成物。
【請求項5】
界面活性剤をテンプレートとして用いるポーラスシリカ前駆体組成物の作製方法において、有機アルカリ触媒の存在下トリアルコキシシランの重縮合反応を行った後、酸触媒の存在下、ポーラスシリカ前駆体の骨格形成材料を添加してさらに重縮合反応を行うか、又は該酸触媒の存在下、ポーラスシリカ前駆体の骨格形成材料及びさらにトリアルコキシシランを添加してさらに重縮合反応を行うことを特徴とするポーラスシリカ前駆体組成物の作製方法。
【請求項6】
前記有機アルカリ触媒が、アンモニア、コリン、テトラメチルアンモニウムハイドロキシド、及びモノエタノールアミンから選ばれた少なくとも1種であることを特徴とする請求項5記載のポーラスシリカ前駆体組成物の作製方法。
【請求項7】
前記酸触媒が、硝酸、塩酸、硫酸、及びフッ酸から選ばれた無機酸であることを特徴とする請求項5又は6記載のポーラスシリカ前駆体組成物の作製方法。
【請求項8】
前記骨格形成材料が、アルコキシシランから選ばれた少なくとも1種であることを特徴とする請求項5〜7のいずれか1項に記載のポーラスシリカ前駆体組成物の作製方法。
【請求項9】
請求項1〜4のいずれか1項に記載のポーラスシリカ前駆体組成物又は請求項5〜8のいずれか1項に記載のポーラスシリカ前駆体組成物の作製方法に従って作製されたポーラスシリカ前駆体組成物を用いて作製されたものであることを特徴とするポーラスシリカ膜。
【請求項10】
前記ポーラスシリカ膜のFT−IRスペクトルのSi−Oケージ構造のピークである1150cm−1近傍とSi−Oラダー構造のピークである1050cm−1近傍とのピーク高さの比が、1.0以上であることを特徴とする請求項9記載のポーラスシリカ膜。
【請求項11】
請求項1〜4のいずれか1項に記載のポーラスシリカ前駆体組成物又は請求項5〜8のいずれか1項に記載のポーラスシリカ前駆体組成物の作製方法に従って作製されたポーラスシリカ前駆体組成物を基板表面に塗布し、真空雰囲気下焼成処理し、界面活性剤を除去してポーラスシリカ膜を作製することを特徴とするポーラスシリカ膜の作製方法。
【請求項12】
請求項9若しくは10記載のポーラスシリカ膜又は請求項11記載のポーラスシリカ膜の作製方法に従って作製したポーラスシリカ膜を用いて得られたものであることを特徴とする半導体素子。
【図1】


【図2】
【図2】
【公開番号】特開2012−251119(P2012−251119A)
【公開日】平成24年12月20日(2012.12.20)
【国際特許分類】
【出願番号】特願2011−127234(P2011−127234)
【出願日】平成23年6月7日(2011.6.7)
【出願人】(000231464)株式会社アルバック (1,740)
【Fターム(参考)】
【公開日】平成24年12月20日(2012.12.20)
【国際特許分類】
【出願日】平成23年6月7日(2011.6.7)
【出願人】(000231464)株式会社アルバック (1,740)
【Fターム(参考)】
[ Back to top ]