
マイクロキャリア上での多能性幹細胞の培養
本発明は、多能性幹細胞をマイクロキャリア上で増殖、増量及び分化させる方法を目的とする。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、米国特許公開番号第61/116,447号(2008年11月20日出願)に対する優先権を請求する。
【0002】
(発明の概要)
本発明は、多能性幹細胞をマイクロキャリア上で増殖、増量(expansion)及び分化させるための方法を目的とする。
【背景技術】
【0003】
多能性幹細胞(例えば胚性幹細胞など)は、全ての成体型細胞へと分化する能力を有する。そのため、胚性幹細胞は、疾病、感染又は先天性異常の結果として損傷した器官のための、置換細胞源及び置換組織源になり得る。胚性幹細胞を置換細胞源として用いるのを可能にするには、試験管内で多能性を維持しつつ細胞を増殖することの困難さがネックになっている。
【0004】
現行の、未分化の胚性幹細胞を培養する方法では、例えばフィーダー細胞層の存在下で胚性幹細胞を培養するなどの、複雑な培養条件が必要とされる。あるいは、フィーダー細胞培養物に曝露することによって得られる培地を、胚性幹細胞の培養に使用する場合もある。これらの方法を用いる培養系では、多くの場合、培養される幹細胞の生物種とは異なる種から得られた細胞(異種細胞)が使用される。加えて、これらの培養系には動物の血清が添加される場合がある。
【0005】
胚性幹細胞は、研究及び薬剤スクリーニング用に将来性のある資源を提供する。現在のところ、ヒト胚性幹細胞株の大規模培養には問題が多く、大きな課題が提供される。現行の、インビトロでの多能性幹細胞増殖方法は、組織培養用フラスコにおいて、細胞外マトリックス(ECM)タンパク質又はフィーダー細胞でプレコートされた、平面状表面上で実施される。また、平面培養では、表面積が多能性幹細胞の長期増殖をサポートできないという限界性から、頻繁に継代培養する必要がある。マイクロキャリア系の多能性幹細胞培養法は、解決法を提供し得る。マイクロキャリアは表面積対容積比が大きく、したがって平面状表面上での、多能性幹細胞の増殖にまつわる表面積制限を排除する。
【0006】
例えば、Fokらは、未分化のESC−マイクロキャリアと凝集体の培養物を増殖させるための、撹拌懸濁培養系を開示する(Stem Cells 2005;23:1333〜1342.)。
【0007】
他の例では、Abranchesらは、表面にコラーゲン層を備えるデキストランマトリックスから構成された、ミクロ孔質のマイクロキャリアであるCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)が、スピナーフラスコ中でマウスS25 ES細胞株の増量を支持する能力について試験を開示する(Biotechnol.Bioeng.96(2007),pp.1211〜1221.)。
【0008】
他の例では、米国特許第20070264713号は、未分化の幹細胞を懸濁液中で培養するための方法、具体的には容器中のマイクロキャリア上で幹細胞を培養する方法について開示する。
【0009】
他の例では、国際公開第2006137787号は、細胞をマイクロキャリア上で培養するために使用される、マイクロタイタープレートなどの中実支持体に付着させたビーズなどの、微粒子状の物質すなわちマイクロキャリアを含む、スクリーニングツールを開示する。
【0010】
他の例では、国際公開第2008004990号は、培養時の幹細胞の付着、生存及び/又は増殖を促進する方法を開示し、この方法は、正電荷支持表面上で幹細胞を培養することを含む。
【0011】
他の例では、国際公開第2007012144号は、支持表面と、支持表面に結合させた合成付着性ポリペプチドと、を含むバイオリアクターを開示し、ここでこの合成付着性ポリペプチドは、胚性幹細胞又は多能性細胞への高い結合親和性により特徴付けられる。
【発明の概要】
【課題を解決するための手段】
【0012】
本発明は、多能性幹細胞をマイクロキャリア上で増殖、増量及び分化させる方法を提供する。
【0013】
一実施形態では、本発明は、多能性幹細胞の増殖方法を提供し、この方法は以下の
a.多能性幹細胞集団を、第1容量のマイクロキャリアに付着させる工程と、
b.多能性幹細胞を、第1容量のマイクロキャリア上で培養する工程と、
c.多能性幹細胞を、第1容量のマイクロキャリアから取り外す工程と、
d.多能性幹細胞集団を、第2容量のマイクロキャリアへと付着させる工程と、を含む。
【図面の簡単な説明】
【0014】
【図1】Rho−キナーゼ阻害剤は、マイクロキャリアへのヒト胚性幹細胞の付着と増殖を促進する。HILLEX(登録商標)IIマイクロキャリア(Solohill,MI)上で、2日間にわたって静置培養により増殖させた、H9細胞の画像。細胞は、10μMのRhoキナーゼ阻害剤、Y27632((Sigma−Aldrich,MO)を添加した、あるいは不添加の、マウス胚性線維芽細胞馴化培地(MEF−CM)で培養した(それぞれA及びB)。
【図2】マイクロキャリア上でのH9細胞の増殖。H9細胞を様々なマイクロキャリアに付着させ、37℃にて固定式プラットフォーム上に設置した。Plasticマイクロキャリアと、ProNectinFマイクロキャリアと、HILLEX(登録商標)IIマイクロキャリア(Solohill,MI)と、Plastic Plusマイクロキャリアを使用した(それぞれA、B、C、D)。3日間の増殖後に、細胞はHILLEX(登録商標)II(Solohill,MI)において、マイクロキャリアに対する最も良好な細胞付着性を示した。マイクロキャリアに付着せずに凝集塊を形成した細胞を矢印で示す。
【図3】マイクロキャリア上でのH9細胞の増殖。H9細胞を、37℃にて固定式プラットフォーム上の6ウェルディッシュ中で、10μMのY27632(Sigma−Aldrich,MO)及びMEF−CMの存在下で、HILLEX(登録商標)IIマイクロキャリア、ProNectinFマイクロキャリア、Plastic Plusマイクロキャリア及びPlasticマイクロキャリア(Solohill,MI)に付着させた。初期細胞播種密度は、0日での値である。3日目及び5日目の細胞数を示す。
【図4】マイクロキャリアに付着後のH1細胞の画像。ProNectinFマイクロキャリア、Plastic Plusマイクロキャリア、及びPlasticマイクロキャリアに付着させた細胞の3、5及び7日目の画像を示す。細胞は、37℃にて固定式プラットフォーム上の12ウェルディッシュ中で、10μMのY27632(Sigma−Aldrich,MO)を含有するMEF−CMで増殖させた。Plastic Plusマイクロキャリア及びPlasticマイクロキャリアに結合させた細胞は、独立して凝集塊を形成した(G、H中の矢印)。
【図5】マイクロキャリアに付着後のH1細胞の画像。Cytodex 1(登録商標)マイクロキャリア、Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)及びHILLEX(登録商標)IIマイクロキャリア(Solohill,MI)に付着させた細胞の3、5及び7日目の写真を示す。細胞は、37℃にて固定式プラットフォーム上の12ウェルディッシュ中で、10μMのY27632(Sigma−Aldrich,MO)を含有するMEF−CMで増殖させた。
【図6】マイクロキャリア上でのH1細胞の増殖。H1細胞を、HILLEX(登録商標)IIマイクロキャリア(Solohill,MI)、Cytodex 1(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)、Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)、ProNectinFマイクロキャリア(Solohill,MI)、Plastic Plusマイクロキャリア(Solohill,MI)、及びPlasticマイクロキャリア(Solohill,MI)に付着させ、37℃にて固定式プラットフォーム上の、10μMのY27632(Sigma−Aldrich,MO)及びMEF−CMが存在している12ウェルディッシュ中に設置した。初期細胞播種密度は、0日での値である。3、5及び7日目の細胞数を示す。最初の播種密度は、ラインにより示されるように13,333個細胞/cm2であった。
【図7】様々な濃度のRhoキナーゼ阻害剤存在下での、マイクロキャリア上でのH9細胞の増殖。固定式プラットフォーム上の、12ウェルプレート中で細胞を増殖させ、4日目及び7日目に計数して付着率と増殖率を決定した。A.H9細胞を1、2.5、5又は10μMのY27632(Sigma−Aldrich,MO)を含有するMEF−CM中で増殖させた。B.H9細胞を、0.5、1、2.5又は5μMのGlycyl−H 1152ジヒドロクロリド(Tocris,MO)含有MEF−CM中で増殖させた。
【図8】Rhoキナーゼ阻害剤濃度を減少させてH1細胞を増殖させた。2日間にわたって、濃度を減少させたY27632(Sigma−Aldrich,MO)又はGlycyl−H 1152ジヒドロクロリド(Tocris,MO)(10μM/5μM、2.5μM/0.5μM若しくは1.0μM/0.5μM)、又は0.25μMのGlycyl−H 1152ジヒドロクロリド(Tocris,MO)の存在下で、H1p38細胞を継続的に増殖させた。細胞はHILLEX(登録商標)II(Solohill,MI)、Cytodex 1(登録商標)、又はCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)に付着させた(それぞれA、B、C)。細胞を播種後3、5及び7日目に計数した。
【図9】スピナーフラスコ中での、異なった播種密度でのマイクロキャリアへの細胞付着の判定。H1細胞を、写真の左側に掲載した密度でCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)マイクロキャリア上に播種した;低密度(0.4×104個細胞/cm2)、中間密度(1.2×104個細胞/cm2)又は高密度(3×104個細胞/cm2)。3、5及び7日目に細胞を撮像し、細胞の付着したマイクロキャリアの百分率を判定した(画像に組み込んである)。
【図10】スピナーフラスコ中のマイクロキャリア上での細胞増殖は、初期播種密度により影響を受ける。H1細胞を、写真の左側に掲載した密度でCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)マイクロキャリア上に播種した;低密度(0.4×104個細胞/cm2)、中間密度(1.2×104個細胞/cm2)又は高密度(3×104個細胞/cm2)。3、5及び7日目で細胞をマイクロキャリアから解離させ計数した。
【図11】スピナーフラスコ中での、異なった播種密度でのマイクロキャリア上での細胞増殖率の判定。H1細胞を、異なる播種密度でCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)マイクロキャリア上に播種した;低密度(0.4×104個細胞/cm2)、中間密度(1.2×104個細胞/cm2)又は高密度(3×104個細胞/cm2)。3、5及び7日目で細胞をマイクロキャリアから解離させ計数した。細胞数の倍数増加を初期播種密度と比較して示す。
【図12】Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上で増殖させたH1細胞を培養の7日目以降に撮像した。細胞には3日目以降はRhoキナーゼ阻害剤不含MEF−CMを供給した。細胞はマイクロキャリアに付着したままであった。
【図13】H9細胞の増殖及びHILLEX(登録商標)IIマイクロキャリア(Solohill,MI)上のH9細胞の解離。A、B:HILLEX(登録商標)IIマイクロキャリア上(Solohill,MI)で6日間増殖させたH9細胞の10x及び20x画像。C:10分間にわたって0.05%トリプシン/EDTAでHILLEX(登録商標)IIマイクロキャリア(Solohill,MI)から解離させた細胞の20x画像。D:10分間にわたってTrypLE(商標)ExpressでHILLEX(登録商標)IIマイクロキャリア(Solohill,MI)から解離させた細胞の20x画像。
【図14】マイクロキャリアからのH9細胞の解離。固定式プラットフォーム上のHILLEX(登録商標)II(Solohill,MI)上で増殖させたH9細胞を、TrypLE(商標)Express又は0.05%トリプシン/EDTAで解離させた。細胞数及びそれらの生存率をそれぞれA及びBに示す。
【図15】マイクロキャリアからのH1細胞の解離。スピナーフラスコ中の、Cytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)上で増殖させたH1細胞を、TrypLE(商標)Express(Invitrogen,CA)、Accutase(商標)又はコラゲナーゼ(10mg/mL)で解離させた。細胞数及びそれらの生存率をそれぞれA及びBに示す。
【図16】HILLEX(登録商標)II(Solohill,MI)マイクロキャリア上で増殖させたH9細胞は、マイクロキャリア間を移動しなかった。
【図17】継代数43のH9を、スピナーフラスコ中の、HILLEX(登録商標)II(Solohill,MI)マイクロキャリア上で5継代にわたって増殖させた。細胞を2〜3日毎に計数し、細胞が1〜2×105個細胞/cm2に達した時点で継代した。
【図18】継代数43のH9細胞を、スピナーフラスコ中の、Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上で、5継代にわたって増殖させた。細胞を2〜3日毎に計数し、細胞が1〜2×105個細胞/cm2に達した時点で継代した。
【図19】蛍光標示式細胞分取(FACS)はスピナーフラスコ内で増殖させたH9細胞の多能性を示す。A:HILLEX(登録商標)II(Solohill,MI)マイクロキャリア上で増殖させた継代数43のH9細胞の大多数は多能性タンパク質を発現する。継代数1及び3の細胞は、TRA−1−81については評価しなかった。B:Cytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)マイクロキャリア上で増殖させた継代数43のH9細胞の大多数は多能性タンパク質を発現する。継代数1の細胞は、TRA−1−81については評価しなかった。
【図20】継代数49のH1細胞を、スピナーフラスコ中のCytodex 1(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上で、5継代にわたって増殖させた。細胞を2〜3日毎に計数し、細胞が4〜8×104個細胞/cm2に達した時点で継代した。
【図21】継代数49のH1細胞を、スピナーフラスコ中のCytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上で、5継代にわたって増殖させた。細胞を2〜3日毎に計数し、細胞が1〜2×105個細胞/cm2に達した時点で継代した。
【図22】蛍光標示式細胞分取(FACS)は、スピナーフラスコ内で増殖させたH1細胞の多能性を示す。
【図23】マイクロキャリア上のH1及びH9細胞の集団倍加。3日目から、以降経過させた日数についての集団倍加回数を算出した(5、6又は7日目)。
【図24】制限培地中のマイクロキャリア上でH9細胞を培養した。HILLEX(登録商標)II(HII,(Solohill,MI))又はCytodex 3(登録商標)(C3,(GE Healthcare Life Sciences,NJ))上で細胞を培養した。細胞を、以下の培地のいずれかで、マイクロキャリア上で培養した;mTESR(StemCell Technologies,Vancouver,Canada)、StemPro又はMEF−CM。10μMのY27632(Y,(Sigma−Aldrich,MO))又は2.5μMのGlycyl−H 1152ジヒドロクロリド(H,(Tocris,MO))を培地に加えた。播種後3、5及び7日目の増殖率を測定した。
【図25】制限培地中のマイクロキャリア上で、継代数38のH1細胞を培養した。HILLEX(登録商標)II(HII,(Solohill,MI))マイクロキャリア又はCytodex 3(登録商標)(C3,(GE Healthcare Life Sciences,NJ))マイクロキャリア上で細胞を培養した。細胞を、以下の培地のいずれかで、マイクロキャリア上で培養した;mTESR(StemCell Technologies,Vancouver,Canada)、StemPro及びMEF−CM。10μMのY27632(Y,(Sigma−Aldrich,MO))又は2.5μMのGlycyl−H 1152ジヒドロクロリド(H,(Tocris,MO))を培地に加えた。播種後3、5及び7日目の増殖率を測定した。
【図26】継代数50のH1細胞を、スピナーフラスコ中の、HILLEX(登録商標)II(Solohill,MI))マイクロキャリア上で、制限培地を用いて培養した。A:スピナーフラスコ中のMEF−CMで3、7又は9日間にわたって増殖させた後の、継代数50のH1細胞の画像。B:mTESR(StemCell Technologies,Vancouver,Canada)中で3、7又は9日間にわたって増殖させた継代数50のH1細胞の画像。矢印が指す細胞クラスターは、マイクロキャリアに付着していない。
【図27】スピナーフラスコ中で5回継代したヒト胚性幹細胞の分化。A:継代数43のH9細胞を、Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上で5回継代した。B:継代数49のH1細胞を、Cytodex 1(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上で5回継代した。両方の細胞型を共にマイクロキャリアから放出させ、MATRIGEL(BD Biosciences,CA)でコートしたプレートに播種した。80〜90%コンフルエントの細胞で、胚性幹細胞が胚体内胚葉へと分化し得るようなプロトコルを実施した。次いで胚体内胚葉マーカーのCXCR4を発現している細胞の百分率について、細胞をFACSで解析した。CXCR4陽性細胞のパーセントをプロットの右上の角に記載する。
【図28】マイクロキャリア上での、胚体内胚葉へのH1細胞の分化。このFACSプロットは、胚体内胚葉マーカーCXCR4を発現している細胞の百分率を示す。陽性パーセントを右上の角に記載する。処理前に全ての細胞をスピナーフラスコ中のマイクロキャリア上で増量させた。A:分化前に、継代数40のH1細胞を5回継代し、次いで6日間にわたってCytodex 1(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上で増殖させた。B:分化前に、継代数40のH1細胞を1回継代し、次いで8日間にわたってCytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上で増殖させた。C:分化前に、継代数50のH1細胞を1回継代し、次いで6日間にわたってHILLEX(登録商標)IIマイクロキャリア(Solohill,MI)上で増殖させた。
【図29】Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上での、胚体内胚葉へのH1細胞の分化。A:継代数40のH1細胞を、マイクロキャリア上で8日間にわたって増殖させた。B:継代数40のH1細胞を、マイクロキャリア上で11日間にわたって増殖させた。両方の細胞集団を共に、次いで37℃にて固定式プラットフォーム上で、胚体内胚葉へと分化させた。このFACSプロットは、胚体内胚葉マーカーCXCR4を発現している細胞の百分率を示す。陽性パーセントを右上の角に記載する。
【図30】胚体内胚葉への、マイクロキャリア上で培養したヒト胚性幹細胞株H1細胞の分化。CXCR4陽性細胞パーセントについてのFACS結果をY軸に示す。H1細胞は、分化前及び分化時にHILLEX(登録商標)II、Cytodex 1(登録商標)又はCytodex 3(登録商標)マイクロキャリア上で増殖させた。
【図31】膵臓内胚葉細胞への、マイクロキャリア上で培養したヒト胚性幹細胞株H1細胞の分化。CT値を、膵臓内胚葉マーカーのNgn3、Nkx6.1及びPdx1についてY軸に示す。H1細胞を、DMEM−高グルコース(HG)培地又はDMEM−F12(F12)培地のいずれかの培地中で、HILLEX(登録商標)II(HII)、Cytodex 1(登録商標)(C1)又はCytodex 3(登録商標)(C3)マイクロキャリア上で分化させた。分化プロトコルは13日間続いた。
【図32】膵臓ホルモン産生細胞への、マイクロキャリア上で培養したヒト胚性幹細胞株H1細胞の分化。FACSにより測定した陽性細胞パーセントを、膵臓ホルモンの細胞マーカーのシナプトフィジン、グルカゴン及びインスリンについてY軸で示す。H1細胞を2つの異なる濃度、10×105(10)又は20×105(20)で、Cytodex 3(登録商標)(C−3)マイクロキャリア上に播種した。DMEM−高グルコース(HG)中で、4〜9日間にわたって細胞を分化させ、更にHG又はDMEM−F12(F12)培地で10日〜24日間分化させた。
【図33】Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上での、内分泌細胞へのH1細胞の分化。H1細胞を、膵臓内胚葉(14日)を経て、膵内分泌細胞へと分化させ、膵内分泌細胞(21日)をインスリン発現細胞(28日)へと分化させた。Pdx1、グルカゴン及びインスリンの遺伝子発現レベルを測定した(それぞれA、B、C)。Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)(C3)上で増殖、分化させたH1細胞を、MATRIGEL(BD Biosciences,CA)コートした6ウェルディッシュ(平面)上で増殖、分化させた細胞と比較した。Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上で増殖させた細胞についての遺伝子発現の値は3つ組で採った。
【図34】Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上での、胚体内胚葉(DE)へのH9細胞の分化。CXCR4発現のFACSプロット。胚体内胚葉マーカーCXCR4についての陽性細胞パーセントを、右上の角に記載する。A:継代数39のH9細胞を、MATRIGEL(BD Biosciences,CA)コートした6ウェルディッシュ上で増殖させ、DEへと分化させた。B、C:スピナーからのCytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上のH9細胞の、2つ組サンプルを、12ウェルディッシュに設置し、固定式プラットフォーム上でインキュベートした。
【図35】Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上での、インスリン発現細胞へのH9細胞の分化。H9細胞を、膵臓内胚葉(14日)を経て、膵内分泌細胞へと分化させ、内分泌細胞(22日)をインスリン発現細胞(29日)へと分化させた。Pdx1、グルカゴン及びインスリンの遺伝子発現レベルを測定した(それぞれA、B、C)。Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)(C3)上で増殖、分化させたH9細胞を、MATRIGEL(BD Biosciences,CA)コートした6ウェルディッシュ(平面)上で増殖、分化させた細胞と比較した。
【図36】Cytodex 3(登録商標)マイクロキャリア上で5継代にわたって培養し、次いで記載の平面状基材上に移して培養し、Rhoキナーゼ阻害剤の存在下で培養したヒト胚性幹細胞での多能性の維持性。パネルAは、フローサイトメトリーにより検出されたものとして、多能性マーカーCD9、SSEA3、SSEA4、Tra−160及びTra−181の発現を記す。パネルBは、リアルタイムPCRにより検出されたものとして、多能性マーカーNanog、Pou5F1、SOX2及びZFP42、並びに分化マーカーFOXA2、FOXD3、GATA2、GATA4及びBrachyuryの発現を記す。
【図37】Cytodex 3(登録商標)マイクロキャリア上で5継代にわたって培養し、次いで記載の平面状基材上に移して培養し、Rhoキナーゼ阻害剤の存在下で培養したヒト胚性幹細胞による、胚体内胚葉形成。パネルAは、フローサイトメトリーにより検出されたものとして、CXCR4の発現を記す。パネルBは、リアルタイムPCRにより検出されたものとして、記載のマーカーの発現を記す。
【図38】ヒト胚性幹細胞をCytodex 3(登録商標)マイクロキャリア上で5継代にわたって培養し、次いでPRIMARIA(商標)平面状基材上に移し培養することによる胚体内胚葉形成。記載の遺伝子の発現はフローサイトメトリーにより測定した。
【図39】平面状基材上で培養したヒト胚性幹細胞は多能性を維持する。TrypLE(商標)、Accutase(商標)又はコラゲナーゼ処理したH1ヒトES細胞由来のmRNAサンプルを回収し、mRNAの多能性遺伝子発現について解析した。細胞を、1継代の4日間にわたって、MATRIGELにてMEF馴化培地で培養して増殖させ(A)、あるいは1継代にわたって、Primaria(商標)にて、Rock阻害剤を添加したMEF馴化培地で増殖させ(B)、あるいは2継代にわたって、Primaria(商標)にて、Rock阻害剤を添加したMEF馴化培地で増殖させた(C)。
【図40】Rhoキナーゼ阻害剤のGlycyl−H 1152ジヒドロクロリドの存在下で、PRIMARIAにてAccutase(商標)又はTrypLE(商標)を用いて分割比1:4、1:8又は1:16で継代して、7継代を超えてPRIMARIAにて増殖させたH1ヒト胚性幹細胞(継代数45を超える)を、多能性(A)及び胚体内胚葉への分化能(B)について試験した。対照は、1:30のMATRIGELにて増殖させ、コラゲナーゼで処理した、継代数48のH1ヒト胚性幹細胞である。10mA=Accutase(商標)に10分曝露して継代。10mT=TrypLE(商標)に10分曝露して継代。1:4、1:8又は1:16は継代比を意味する。P(X)は、MEFフィーダーからPrimaria(商標)プラスチックへと移動させて以降の継代数を意味する。
【図41】Rhoキナーゼ阻害剤のGlycyl−H 1152ジヒドロクロリドの存在下で、Accutase(商標)又はTrypLE(商標)を用いてPRIMARIAにて1:4比で継代して、7継代を超えてPRIMARIAにて増殖させたH1ヒト胚性幹細胞(継代数45を超える)を、多能性マーカー及び分化マーカーのmRNA発現について試験した。対照は、継代数37の細胞集団で開始する。10分Accutase(商標)=Accutase(商標)に10分曝露して継代。P(X)は、MEFフィーダーからPRIMARIA(商標)プラスチックへと移動させて以降の継代数を意味する。
【図42】Rhoキナーゼ阻害剤のGlycyl−H 1152ジヒドロクロリドの存在下で、Accutase(商標)又はTrypLE(商標)を用いてPRIMARIAにて分割比1:8で継代して、7継代を超えてPRIMARIA(商標)にて増殖させたH1ヒト胚性幹細胞(継代数45を超える)を、多能性マーカー及び分化マーカーのmRNA発現について試験した。対照は、継代数37の細胞集団で開始する。10分Accutase(商標)=Accutase(商標)に10分曝露して継代。P(X)は、MEFフィーダーからPRIMARIA(商標)プラスチックへと移動させて以降の継代数を意味する。
【図43】Rhoキナーゼ阻害剤のGlycyl−H 1152ジヒドロクロリドの存在下で、Accutase(商標)又はTrypLE(商標)を用いてPRIMARIAにて分割比1:16で継代して、7継代を超えてPRIMARIA(商標)にて増殖させたH1ヒト胚性幹細胞(継代数45を超える)を、多能性マーカー及び分化マーカーのmRNA発現について試験した。対照は、継代数37の細胞集団で開始する。10分Accutase(商標)=Accutase(商標)に10分曝露して継代。P(X)は、MEFフィーダーからPRIMARIA(商標)plasticへと移動させて以降の継代数を意味する。
【図44】Primaria(商標)平面状基材(カタログ番号353846,Becton Dickinson,Franklin Lakes,NJ)にて増殖させ、次いで播種の3日後にマイクロキャリアに移動させた、H1細胞の画像。A〜C:H1細胞をCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)マイクロキャリア上に播種した。D〜F:細胞をHILLEX(登録商標)II マイクロキャリア(Solohill,MI)上に播種した。A、D:H1細胞は、マイクロキャリアへと移す前に、10分間のTrypLE(商標)Express(Invitrogen,CA)処理により、Primaria(商標)平面状基材(カタログ番号353846,Becton Dickinson,Franklin Lakes,NJ)プレートにて継代した。A、E:H1細胞は、マイクロキャリア上に移す前に、10分間のAccutase(商標)処理を用いて、Primaria(商標)平面状基材(カタログ番号353846,Becton Dickinson,Franklin Lakes,NJ)プレートにて継代した。C、F:継代数46のH1細胞は、マイクロキャリアへと移す前に、コラゲナーゼ(1mg/mL)を用いて、MATIRGEL(BD Biosciences,CA)コートプレートにて継代した。
【図45】Primaria(商標)平面状基材(カタログ番号353846,Becton Dickinson,Franklin Lakes,NJ)にて増殖させ、Cytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)マイクロキャリア及びHILLEX(登録商標)IIマイクロキャリアへと移したH1細胞の多能性。FACS解析は、多能性細胞表面タンパク質の発現を示す。細胞を、Primaria(商標)(カタログ番号353846,Becton Dickinson,Franklin Lakes,NJ)での継代時に3〜10分間にわたってAccutase(商標)又はTrypLE(商標)Express(Invitrogen,CA)で処理した。
【図46】Primaria(商標)(カタログ番号353846,Becton Dickinson,Franklin Lakes,NJ)にて増殖させ、次いでCytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)へと移したH1細胞の分化。胚体内胚葉マーカーCXCR4の細胞表面発現についてのFACS解析。細胞を、Primaria(商標)(カタログ番号353846,Becton Dickinson,Franklin Lakes,NJ)での継代時に3〜10分間にわたってAccutase(商標)又はTrypLE(商標)Express(Invitrogen,CA)で処理した。
【図47】マイクロキャリア上での培養前にセルロース混合エステルからなる平面状基材にて培養したヒト胚性幹細胞のFACS解析。
【図48】マイクロキャリア上での培養及び分化の前に、セルロース混合エステルからなる平面状基材で培養したヒト胚性幹細胞の、胚体内胚葉系に特徴的なマーカーの発現に関するFACS解析。
【発明を実施するための形態】
【0015】
開示を明確にするために、本発明の「発明を実施するための形態」を、限定を目的とすることなく、本発明の特定の特徴、実施形態、又は応用を説明若しくは図示した以下の小項目に分ける。
【0016】
定義
幹細胞は、単一の細胞レベルで自己複製し、分化して後代細胞を生成する、それら両方の能力で定義される未分化細胞であり、後代細胞には、自己複製前駆細胞、非再生前駆細胞、及び最終分化細胞が含まれる。同様に、幹細胞は、インビトロで複数の胚葉(内胚葉、中胚葉及び外胚葉)から様々な細胞系の機能細胞へと分化する能力によって、並びに移植後に複数の胚葉の組織を生じ、胚盤胞への注入後、全てではないとしても殆どの組織を提供する能力によっても特徴付けられる。
【0017】
幹細胞は、それらの発達能力により:(1)全胚及び胚体外細胞型を生じる能力を意味する全能性、(2)全胚細胞型を生じる能力を意味する多能性、(3)細胞系統の小集合を生じるが、全て特定の組織、器官又は生理的システム内で生じる能力を有することを意味する多能性(例えば、造血幹細胞(HSC)は、HSC(自己複製)、血液細胞に限定された寡能性前駆細胞、並びに血液の通常の構成要素である全細胞型及び要素(例えば、血小板)を含む子孫を産生できる)、(4)多能性幹細胞と比較して限定された細胞系統の小集合を生じる能力を有することを意味する寡能性、並びに(5)1つの細胞系統を生じる能力を有することを意味する単能性(例えば、精子形成幹細胞)に分類される。
【0018】
分化は、特殊化されていない細胞(「中立の」)又は比較的特殊化されていない細胞が、例えば、神経細胞又は筋細胞などの特殊化した細胞の特徴を獲得するプロセスである。分化した、又は分化誘導された細胞は、細胞系内でより特殊化した(「コミットした」)状況を呈している細胞である。分化プロセスに適用した際の用語「コミットした」は、通常の環境下で特定の細胞型又は細胞型の小集合へと分化し続ける分化経路の地点に進行しており、通常の環境下で異なる細胞型に分化し、又はより分化されていない細胞型に戻ることができない細胞を指す。脱分化は、細胞が細胞系内で比較的特殊化されて(又は傾倒して)いない状況に戻るプロセスを指す。本明細書で使用するとき、細胞系は、細胞の遺伝、すなわちその細胞がどの細胞から来たか、またどの細胞を生じ得るかを規定する。細胞系は、細胞を発達及び分化の遺伝的スキーム内に設置する。系特異的なマーカーは、対象とする系の細胞の表現型に特異的に関連した特徴を指し、中立細胞の対象とする系への分化を評価する際に使用することができる。
【0019】
培養時の細胞を記載するために、様々な用語が使用される。「維持」は、一般に、細胞増殖及び/又は細胞分裂を促進する条件下で増殖培地に設置した細胞を指し、この細胞は細胞数のより大きな集団をもたらす場合ももたらさない場合もある。「継代」は、細胞増殖及び/又は細胞分裂を促進する条件下で、細胞を1番目の培養容器から取り出し、それらの細胞を2番目の培養容器へと設置するプロセスを指す。
【0020】
特定の細胞集団又は細胞株は、しばしば継代された回数によって呼称されるか特徴付けられる。例えば、10回継代された培養細胞集団はP10培養と呼ばれる場合がある。初代培養、すなわち組織から細胞を単離した後の最初の培養はP0と称される。最初の継代培養後、細胞は二次培養物(P1又は継代数1)と称される。2回目の継代培養の後では細胞は三次培養物(P2又は継代数2)となる、といった具合である。当業者であれば、継代期間中に集団は何度も倍加し得るものであり、したがってある培養の集団倍加の回数は継代数よりも大きいことが理解されるであろう。各継代間の期間中の細胞の増量(すなわち集団倍加数)は、限定するものではないが、播種密度、基質、培地、増殖条件、及び継代間の時間などを含む多くの因子に依存する。
【0021】
「β−細胞系」は、転写因子PDX−1と、以下の転写因子:NGN−3、Nkx2.2、Nkx6.1、NeuroD、Isl−1、HNF−3 β、MAFA、Pax4、又はPax6のうちの少なくとも1つに関する遺伝子の発現が陽性である細胞を指す。β細胞系に特徴的なマーカーを発現する細胞としては、β細胞が挙げられる。
【0022】
本明細書で使用するとき、「胚体内胚葉系に特徴的なマーカーを発現している細胞」は、以下のマーカー:SOX−17、GATA−4、HNF−3 β、GSC、Cer1、Nodal、FGF8、Brachyury、Mix様ホメオボックスタンパク質、FGF4 CD48、eomesodermin(EOMES)、DKK4、FGF17、GATA−6、CXCR4、C−Kit、CD99又はOTX2のうちの少なくとも1つを発現している細胞を指す。胚体内胚葉系に特徴的なマーカーを発現している細胞には、原始線条前駆体細胞、原始線条細胞、中内胚葉細胞及び胚体内胚葉細胞を含む。
【0023】
本明細書で使用するとき、「膵臓内胚葉系に特徴的なマーカーを発現している細胞」は、以下のマーカーのうちの少なくとも1つを発現している細胞を指す:PDX−1、HNF−1β、PTF−1 α、HNF−6、又はHB9。膵臓内胚葉系に特徴的なマーカーを発現している細胞には、膵臓内胚葉細胞を含む。
【0024】
本明細書で使用するとき、「膵内分泌系に特徴的なマーカーを発現している細胞」は、以下のマーカーのうちの少なくとも1つを発現している細胞を指す:NGN−3、NeuroD、Islet−1、PDX−1、NKX6.1、Pax−4、Ngn−3、又はPTF−1 α。膵内分泌系に特徴的なマーカーを発現している細胞には、膵臓内分泌細胞、膵臓ホルモン発現細胞、及び膵臓ホルモン分泌細胞、並びにβ−細胞系の細胞を含む。
【0025】
本明細書で使用するとき、「胚体内胚葉」は、原腸形成中、胚盤葉上層から生じ、胃腸管及びその誘導体を形成する細胞の特徴を有する細胞を指す。胚体内胚葉細胞は、以下のマーカー、すなわちCXCR4、HNF−3 β、GATA−4、SOX−17、Cerberus、OTX2、goosecoid、c−Kit、CD99、及びMixl1を発現する。
【0026】
本明細書で使用するとき、「胚体外内胚葉」は、以下のマーカーのうちの少なくとも1つを発現している細胞の集団を指す:SOX−7、AFP又はSPARC。
【0027】
本明細書で使用するとき、「マーカー」は、対象とする細胞内で差異的に発現される核酸又はポリペプチド分子である。本文脈において、差異的な発現は、陽性マーカーのレベルの増大及び陰性マーカーのレベルの減少を意味する。検出可能なレベルの核酸マーカー又はポリペプチドマーカーは、他の細胞と比較して対象とする細胞内で十分高く又は低く、そのため当該技術分野において既知の多様な方法のいずれかを使用して、対象とする細胞を他の細胞から識別及び区別することができる。
【0028】
本明細書で使用するとき、「中内胚葉細胞」とは、以下のマーカーのうちの少なくとも1つを発現している細胞を指す:CD48、eomesodermin(EOMES)、SOX−17、DKK4、HNF−3 β、GSC、FGF17又はGATA−6。
【0029】
本明細書で使用するとき「膵内分泌細胞」又は「膵臓ホルモン発現細胞」とは、以下のホルモンのうちの少なくとも1つを発現することが可能な細胞を指す:インスリン、グルカゴン、ソマトスタチン、及び膵臓ポリペプチド。
【0030】
本明細書で使用するとき、「膵臓ホルモン分泌細胞」は、以下のホルモンのうちの少なくとも1つを分泌できる細胞を指す:インスリン、グルカゴン、ソマトスタチン、及び膵臓ポリペプチド。
【0031】
本明細書で使用するとき、「前原始線条細胞」は、以下のマーカーのうちの少なくとも1つを発現している細胞を指す:Nodal、又はFGF8。
【0032】
本明細書で使用するとき、「原始線条細胞」は、以下のマーカーのうちの少なくとも1つを発現している細胞を指す:Brachyury、Mix様ホメオボックスタンパク質、又はFGF4。
【0033】
マイクロキャリア
「マイクロキャリア」は、足場依存性の細胞を培養する際に、この細胞の付着及び増殖に有用な粒子、ビーズ又はペレットを指す。「マイクロキャリア」は以下の特性を有する:(a)(マイクロキャリア又は細胞に、有意なせん断ダメージを引き起こさないような撹拌速度での)懸濁培養で使用可能な程度に十分小さい;(b)中実である、あるいは表面に多孔性コーティングを備える中実コアを有している;及び(c)表面(多孔性キャリアの場合、外側表面と内側表面)が正又は負に帯電し得る。一態様では、マイクロキャリアは全般的に約150〜350μmの粒径を有し、かつ約0.8〜2.0meq/gの正電荷密度を有する。有用なマイクロキャリアとしては、限定するものではないが、Cytodex 1(登録商標)、Cytodex 2(登録商標)、又はCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)が挙げられる。
【0034】
他の態様では、マイクロキャリアは中実キャリアである。中実キャリアは、接着細胞(例えば、付着依存性の細胞)に特に好適である。キャリア粒子はまた、多孔性マイクロキャリアであってもよい。
【0035】
「多孔性マイクロキャリア」は、付着依存性の細胞を培養する際に、この細胞の付着及び増殖に有用な粒子を指す。多孔性マイクロキャリアは以下の特性を有する:(a)(マイクロキャリア又は細胞に、有意なせん断ダメージを引き起こさないような撹拌速度での)懸濁培養で使用可能な程度に、十分小さい;(b)細胞を粒子の内部空間へと遊走させるのに十分な大きさの孔と内部空間とを有する、及び(c)(外側及び内側)表面が正又は負に帯電し得る。一連の実施形態の1つでは、キャリアは(a)全般的に約150〜350μmの粒径を有し;(b)約15〜約40μmの平均孔開口径を有する孔を有し;及び(c)約0.8〜2.0meq/gの正電荷密度を有する。いくつかの実施形態では、正電荷はDEAE(N,N,−ジエチルアミノエチル)基により提供される。有用な多孔性マイクロキャリアとしては、限定するものではないが、Cytopore 1(登録商標)及びCytopore 2(登録商標)(GE Healthcare Life Sciences,Piscataway N.J.)が挙げられる。マイクロキャリアは任意の形状であってよいが、通常はほぼ球状の形状であり、かつマクロ若しくはマイクロ多孔性、又は中実性であり得る。
【0036】
多孔型及び中実型のマイクロ粒子キャリアのいずれもが供給元より市販されている。市販のマイクロキャリアの例としてはCytodex 1(登録商標)及びCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)が挙げられ、これらの両方がGE Healthcare Life Sciencesのデキストラン系マイクロキャリアである。市販の多孔性マイクロキャリアとしては、同様にGE Healthcare Life Sciencesの、Cytoline製品並びにCytopore製品が挙げられる。Biosilon(NUNC)及びCultispher(Percell Biolytica)もまた市販されている。更なる態様では、マイクロキャリアはポリカーボネート又はセルロース混合エステルから構成され、あるいはこれらでコートされ得る。
【0037】
本発明での使用に好適なマイクロキャリアは、天然由来の又は合成的に誘導された材料から構成され得る。例としては、コラーゲン系マイクロキャリア、デキストラン系マイクロキャリア、又はセルロース系マイクロキャリア、並びにガラス、セラミックス、ポリマー、又は金属が挙げられる。マイクロキャリアはタンパク質不含であっても、あるいはタンパク質(例えばコラーゲン)でコートされていてもよい。更なる態様では、マイクロキャリアは、マイクロキャリアへの細胞の結合性を高める、及びマイクロキャリアからの細胞の放出を高める化合物から構成されてもよく、あるいは化合物でコートされてもよく、限定するものではないが、このような化合物としてはポリ(モノステアロイルグリセリドココハク酸)、ポリ−D,L−ラクチド−コ−グリコリド、ヒアルロン酸ナトリウム、コラーゲン、フィブロネクチン、ラミニン、エラスチン、リジン、n−イソプロピルアクリルアミド、ビトロネクチンが挙げられる。
【0038】
細胞培養物用のマイクロキャリア
マイクロキャリア培養は、足場依存性の細胞(例えばヒト胚性幹細胞)の実用的な高収率培養を可能にする技術である。マイクロキャリアは、特に数ミリリットル〜1000Lを超える範囲の培養容量でヒト胚性幹細胞などの細胞を培養するために開発されてきた。マイクロキャリアは生物学的に不活性であり、撹拌式マイクロキャリア培養法に、丈夫であるが非剛性の基材を提供する。マイクロキャリアは透明であり得、付着した細胞の顕微鏡検査が可能である。Cytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)は、架橋デキストランマトリックスに化学結合させた、変性コラーゲンの薄い層から構成される。Cytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)の変性コラーゲン層は、トリプシン及びコラゲナーゼが挙げられる、様々なプロテアーゼにより消化されやすく、細胞の生存率、機能及び完全性を最大限維持しつつ、細胞をマイクロキャリアから取り外すという性能を提供する。
【0039】
タンパク質不含マイクロキャリアをヒト胚性幹細胞の培養に使用することもできる。例えば、商品名HILLEX(登録商標)(SoloHill Engineering,Inc.,MI.)で販売されている、製造及び研究又は調査用途に使用するためのマイクロキャリアは、表面に、マイクロキャリアに正電荷表面をもたらすカチオン性トリメチルアンモニウムが付着している、改質ポリスチレンビーズである。ビーズ直径範囲は、直径約90〜約200マイクロメートルである。
【0040】
マイクロキャリアに基づく細胞培養法は、多くの用途において、下流の加工のし易さを含む、多くの利点を提供した。マイクロキャリアは通常ほぼ球状の形状であり、かつ多孔性であるか中実であるかのいずれかであり得る。細胞付着用のマイクロキャリアの使用は、付着依存性の細胞の増殖を目的とする撹拌槽及び関連するリアクターの使用を容易にした。細胞は、容易に懸濁されたマイクロキャリアに付着した。懸濁可能である必要があることから、マイクロキャリアの物理的パラメーターは制限される。したがって、マイクロキャリアは一般には50〜2000マイクロメートルの範囲の中央粒径を有する。一部の用途では、中実型のマイクロキャリアが約100〜約250マイクロメートルの範囲である一方で、多孔型のマイクロキャリアは約250〜約2500マイクロメートルの範囲である。これらの寸法範囲は、多くの付着依存性の細胞を収容するのに十分な程度に大きく、かつ撹拌リアクター内での使用に好適な特性を有する懸濁液を形成するのに十分小さいマイクロキャリアの選別を可能にする。
【0041】
マイクロキャリア及び同様物などを使用するに当たり考慮される因子は:付着効率、免疫原性、生体適合性、生分解能、コンフルエントに到達するまでの時間、単位表面積あたりの最大付着可能密度が挙げられる付着細胞の増殖パラメーター、必要とされる箇所での脱着技術、及び脱着効率、培養条件の拡張性並びにスケールアップ条件下での培養均一性、スケールアップ脱着手順を成功裏に実施する能力、及びマイクロキャリアが移植に使用可能かどうか、である。これらの検討事項は、マイクロキャリアの表面特性、並びにマイクロキャリアの多孔性、直径、密度及び操作特性により影響を受け得る。
【0042】
例えば、マイクロキャリアの密度は検討事項である。密度が過剰だと、マイクロキャリアが懸濁液から沈殿する場合があり、あるいは培養容器の底に完全に沈殿したままである傾向があり、ひいてはリアクター内で混合する細胞、培養培地及びガス相の大容量性が乏しくなる場合がある。一方で、密度が低すぎる場合には、マイクロキャリアに過剰な流動性が生じ得る。1.02〜1.15g/cm3の密度が、多くのマイクロキャリアについて一般的である。
【0043】
リアクターに加えることのできる、マイクロキャリアの小さな直径と粒子容量は、マイクロキャリアに、ローラーボトル法又は付着依存性の細胞を増殖させるための他の方法(例えばプレート法)で見出されるものよりもはるかに大きい実質表面積を提供させる。多孔性マイクロキャリアは、単位容量又は単位重量当たりで更に大きい表面積を提供する。これらの多孔性マイクロキャリアは、付着依存性の細胞の増殖に利用可能な大きい空洞を保有する。これらの空洞は表面積を非常に増加させ、例えば混合時又はガス噴霧時のせん断ストレスなどのような、有害な機械的影響から細胞を保護し得る。
【0044】
マイクロキャリア表面は、細胞付着性と細胞増殖性を高めるためにテクスチャ加工処理することができる。マイクロキャリア表面のテクスチャ加工は、限定するものではないが、型成形、鋳造、リーチング及びエッチングが挙げられる技術によりなされる。テクスチャ加工表面の特徴の解像度(resolusion)は、ナノスケールであり得る。テクスチャ加工表面は、マイクロキャリア表面上に特異的な細胞設置を誘導するために使用することもできる。多孔性マイクロキャリア内の孔表面も同様に、細胞付着性と細胞増殖性を高めるためにテクスチャ加工することができる。孔表面のテクスチャ加工は、限定するものではないが、例えば型成形、鋳造、リーチング及びエッチングなどの技術によりなされる。
【0045】
マイクロキャリア表面は、マイクロキャリア表面に特異的な電荷を付与するためにプラズマコートされていてもよい。これらの電荷は、細胞付着性と細胞増殖性を高め得る。
【0046】
他の実施形態では、マイクロキャリアは、ポリ−N−イソプロピルアクリルアミドなどの熱応答性ポリマーから構成され、又はこのようなポリマーでコートされ、すなわち電気機械的特性を有する。
【0047】
多孔性でかつ中実型のマイクロ粒子キャリアのいずれもが供給元より市販されている。市販の中実マイクロキャリアの例としてはCytodex 1(登録商標)及びCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)が挙げられ、これらの両方がGE Healthcare Life Sciencesのデキストラン系マイクロキャリアである。市販の多孔性マイクロキャリアとしては、同様にGE Healthcare Life Sciencesの、Cytoline製品並びにCytopore製品が挙げられる。Biosilon(NUNC)及びCultispher(Percell Biolytica)もまた市販されている。
【0048】
マイクロキャリアには、生物活性剤を含有させることもできる。マイクロキャリアにはまた、細胞の増殖若しくは機能、又は組織環境を制御し得る生物活性剤を含有させることができ、限定するものではないがこれらの因子としては線維芽細胞増殖因子、エリスロポエチン、血管内皮細胞増殖因子、血小板由来増殖因子、骨形成タンパク質、トランスフォーミング増殖因子、腫瘍壊死因子、上皮増殖因子、インスリン様増殖因子が挙げられる。完全な因子、模倣体又はこれらの活性断片を使用することができる。
【0049】
マイクロキャリアに第2の細胞種を播種して、多能性幹細胞と共培養することもできる。一実施形態では、2種(又はそれ以上)の細胞型は、個々のマイクロキャリアに均一な割合で又は不均一な割合で付着し得る。2種以上の細胞種を、マイクロキャリア上に同時に播種することができ、あるいはそれらを異なる時点で播種することもできる。特異的な細胞型をマイクロキャリアの特異的な領域へと優先的に付着させるような様式で、マイクロキャリアを処理することができる。更なる実施形態では、単一の又は複数の細胞種が付着しているマイクロキャリアを、培養容器にて、懸濁液の状態で培養された第2の細胞種と共培養することができる。
【0050】
例えば第2の細胞種としては、上皮細胞(例えば口腔粘膜細胞、胃腸管細胞、鼻粘膜上皮細胞、呼吸器上皮細胞、膣上皮細胞、角膜上皮細胞)、骨髄細胞、脂肪細胞、幹細胞、ケラチノサイト、メラニン細胞、皮膚線維芽細胞、ケラチノサイト、血管内皮細胞(例えば大動脈内皮細胞、冠動脈内皮細胞、肺動脈内皮細胞、腸骨動脈内皮細胞、微小血管内皮細胞、臍動脈内皮細胞、臍静脈内皮細胞及び血管内皮前駆細胞(例えばCD34+、CD34+/CD117+細胞))、筋芽細胞、筋細胞、肝細胞、平滑筋細胞、横紋筋細胞、ストローマ細胞、及び他の軟組織細胞又は前駆細胞、軟骨細胞、骨芽細胞、島細胞、神経細胞(限定するものではないが、ニューロン、星状細胞、シュワン細胞、腸グリア細胞(enteric glial cells)、オリゴデンドロサイト)を挙げてもよい。
【0051】
多能性幹細胞
多能性幹細胞の特徴付け
多能性幹細胞は、ステージ特異的胚抗原(SSEA)3及び4の1種以上、並びにTra−1−60及びTra−1−81と呼ばれる抗体によって検出可能なマーカーを発現し得る(Thomsonら、Science 282:1145,1998)。インビトロで多能性幹細胞を分化させると、SSEA−4、Tra−1−60、及びTra−1−81の発現が消失し(存在する場合)、SSEA−1の発現が増大する。未分化の多能性幹細胞は、一般にアルカリホスファターゼ活性を有し、この活性は、製造業者(Vector Laboratories,Burlingame,Calif.)により説明されるように、細胞を4%パラホルムアルデヒドで固定した後、基質としてVector Redを使用して展開させることにより検出することができる。未分化の多能性幹細胞はまた、RT−PCRにより検出されるように、一般にOCT4及びTERTも発現する。
【0052】
増殖させた多能性幹細胞の別の望ましい表現型は、内胚葉、中胚葉、及び外胚葉組織の3胚葉の全ての細胞に分化し得るものである。幹細胞の多能性は、例えば、細胞を重症複合免疫不全症(SCID)マウスに注入し、形成される奇形腫を4%パラホルムアルデヒドで固定し、次いでこれを3胚葉からの細胞型のエビデンスについて組織学的に調べることによって確認することができる。代替的に、多能性は、胚様体を形成させ、この胚様体を3つの胚葉に関連したマーカーの存在について評価することにより決定することができる。
【0053】
増殖させた多能性幹細胞株は、標準的なGバンド法を使用して核型を決定し、次いで確立された、対応する霊長類種の核型と比較することができる。「正常な核型」を有する細胞を獲得することが望ましく、この「正常な核型」は、細胞が正倍数体であり、全てのヒト染色体が存在し、かつ著しく変更されてはいないことを意味する。
【0054】
多能性幹細胞源
使用が可能な多能性幹細胞の種類としては、妊娠期間中の任意の時期(必ずしもではないが、通常は妊娠約10〜12週よりも前)に採取した前胚性組織(例えば胚盤胞など)、胚性組織、胎児組織などの、妊娠後に形成される組織に由来する多能性細胞の株化細胞系が挙げられる。非限定的な例は、例えばヒト胚幹細胞株H1、H7、及びH9(WiCell)などのヒト胚幹細胞又はヒト胚生殖細胞の確立株である。それらの細胞の最初の樹立又は安定化中に本開示の組成物を使用することも想定され、その場合、源となる細胞は、源となる組織から直接採取した一次多能性細胞であろう。フィーダー細胞の非存在下で既に培養された多能性幹細胞集団から採取した細胞も好適である。例えば、BG01v(BresaGen,Athens,GA)などの変異ヒト胚性幹細胞株も好適である。同様に、例えば成体体細胞などの非多能性細胞由来の多能性幹細胞も好適である。
【0055】
本発明での使用に好適なマイクロキャリアへの多能性幹細胞の付着
多能性幹細胞は、マイクロキャリアに付着させる前に、当該技術分野の任意の方法により、平面状基材にて培養してもよい。例えば多能性幹細胞は、細胞外マトリックスタンパク質(例えばMATRIGEL)で処理した平面状基材にて培養することができる。別の方法としては、多能性幹細胞は、フィーダー細胞層を播種した平面状基材にて培養することもできる。
【0056】
一実施形態では、多能性幹細胞は胚性幹細胞である。別の実施形態では、胚性幹細胞はヒトのものである。
【0057】
本発明の一態様では、多能性幹細胞は、平面状基材から細胞を放出させるプロテアーゼで、多能性幹細胞を処理することにより、平面状基材から放出される。例えばこのようなプロテアーゼは、コラゲナーゼ、TrypLE(商標)Express、Accutase(商標)、トリプシン及び同様物などであり得る。
【0058】
一実施形態では、多能性幹細胞は、この細胞を約5〜約10分にわたってAccutase(商標)で処理することにより、マイクロキャリア基材から放出される。
【0059】
一実施形態では、多能性幹細胞は、この細胞を約10〜約20分にわたって0.05%トリプシン/EDTAで処理することにより、マイクロキャリア基材から放出される。
【0060】
一実施形態では、多能性幹細胞は、この細胞を約5〜約20分にわたってTrypLE(商標)Expressで処理することにより、マイクロキャリア基材から放出される。
【0061】
一実施形態では、多能性幹細胞は、この細胞を約5〜約10分にわたって10mg/mLコラゲナーゼで処理することにより、マイクロキャリア基材から放出される。
【0062】
放出された多能性細胞は、特定の密度でマイクロキャリアを含有している培地に加える。一実施形態では、多能性幹細胞は、約4,000〜約30,000個細胞/cm2マイクロキャリアで播種される。
【0063】
放出された多能性細胞は、マイクロキャリアを含有している培地に加える。一実施形態では、多能性幹細胞の付着性は、多能性幹細胞をRhoキナーゼ阻害剤で処理することにより増加する。Rhoキナーゼ阻害剤はY27632(Sigma−Aldrich,MO)であり得る。あるいは、Rhoキナーゼ阻害剤はGlycyl−H 1152ジヒドロクロリドである。
【0064】
一実施形態では、多能性幹細胞は約1μM〜約10μMの濃度のY27632で処理される。一実施形態では、多能性幹細胞は約10μMの濃度のY27632で処理される。
【0065】
一実施形態では、多能性幹細胞は約0.25μM〜約5μMの濃度のGlycyl−H 1152ジヒドロクロリドで処理される。一実施形態では、多能性幹細胞は約2.5μMの濃度のGlycyl−H 1152ジヒドロクロリドで処理される。
【0066】
マイクロキャリアを含有している培地は撹拌してもよい。本発明で使用するとき、「撹拌」は培養培地の運動であり得る。このような撹拌は、手動で、あるいは例えば固定式プラットフォーム、スピナーフラスコ、及び同様物などのような装置で実施することができる。一実施形態では、マイクロキャリアを含有している培地は、手による運動を用いることで撹拌される。マイクロキャリアと細胞とを含有しているディッシュは、30秒未満にわたって前後に動かされる。
【0067】
マイクロキャリアを含有している培地を撹拌してもよい。一実施形態では、マイクロキャリアを含有している培地は、スピナーフラスコの使用により撹拌される。スピナーフラスコ(Corning,Lowell,MA)は、ビーズ型に応じて30〜70RPMに設定した撹拌プレートに設置する。
【0068】
別の実施形態では、マイクロキャリアを含有している培地は、固定式プラットフォーム(Vari−mix,Barnstead,Dubuque,Iowa)の使用により撹拌される。固定式プラットフォームの速度は、2秒につき約1回転である。
【0069】
マイクロキャリア上の多能性幹細胞の分化
一実施形態では、多能性幹細胞は、胚体内胚葉系に特徴的なマーカーを発現している細胞へとマイクロキャリア上で分化し得る。あるいは、多能性幹細胞は、膵臓内胚葉系に特徴的なマーカーを発現している細胞へとマイクロキャリア上で分化し得る。あるいは、多能性幹細胞は、膵内分泌系に特徴的なマーカーを発現している細胞へとマイクロキャリア上で分化し得る。
【0070】
別の実施形態では、多能性幹細胞は、マイクロキャリア上で増殖させ、次いで胚体内胚葉系に特徴的なマーカーを発現している細胞へと平面状表面上で分化させることができる。あるいは、多能性幹細胞は、マイクロキャリア上で増殖させ、次いで胚体内胚葉系に特徴的なマーカーを発現している細胞へと平面状表面上で分化させることができる。あるいは、多能性幹細胞は、マイクロキャリア上で増殖させ、次いで膵内分泌系に特徴的なマーカーを発現している細胞へと平面状表面上で分化させることができる。
【0071】
本発明の方法に従って処理された多能性幹細胞は、当該術分野における任意の適当な方法によって他の様々な細胞型に分化させることができる。例えば、本発明の方法に従って処理した多能性幹細胞は、神経細胞、心臓細胞、肝細胞などに分化させることができる。
【0072】
例えば、本発明の方法に従って処理した多能性幹細胞は、国際公開第2007030870号に開示される方法に従って神経前駆細胞及び心筋細胞に分化させることができる。
【0073】
別の例では、本発明の方法に従って処理した多能性幹細胞は、米国特許第6,458,589号に開示される方法に従って肝細胞に分化させることができる。
【0074】
胚体内胚葉系に特徴的なマーカーを発現している細胞の形成
多能性幹細胞は、当該技術分野における任意の方法により、胚体内胚葉系に特徴的なマーカーを発現している細胞へと分化させることができる。
【0075】
例えば、多能性幹細胞は、D’Amourら、Nature Biotechnology 23、1534〜1541(2005年)に開示されている方法に従って、胚体内胚葉系統に特徴的なマーカーを発現している細胞へと分化させることができる。
【0076】
例えば、多能性幹細胞は、Shinozakiら、Development 131,1651〜1662(2004)により開示される方法に従って、胚体内胚葉系に特徴的なマーカーを発現している細胞へと分化させることができる。
【0077】
例えば、多能性幹細胞は、McLeanら、Stem Cells 25,29〜38(2007)により開示される方法に従って、胚体内胚葉系に特徴的なマーカーを発現している細胞へと分化させることができる。
【0078】
例えば、多能性幹細胞は、D’Amourら、Nature Biotechnology 24,1392〜1401(2006)に開示されている方法に従って、胚体内胚葉系に特徴的なマーカーを発現している細胞へと分化させることができる。
【0079】
胚体内胚葉系に特徴的なマーカーは、SOX17、GATA4、HNF−3 β、GSC、CER1、Nodal、FGF8、Brachyury、Mix−様ホメオボックスタンパク質、FGF4、CD48、eomesodermin(EOMES)、DKK4、FGF17、GATA6、CXCR4、C−Kit、CD99、及びOTX2からなる群より選択される。本発明での使用に好適なものは、胚体内胚葉系に特徴的なマーカーのうちの少なくとも1つを発現している細胞である。本発明の一態様において、胚体内胚葉系に特徴的なマーカーを発現している細胞は、原始線条前駆体細胞である。別の態様において、胚体内胚葉系に特徴的なマーカーを発現している細胞は、中内胚葉細胞である。別の態様において、胚体内胚葉系に特徴的なマーカーを発現している細胞は、胚体内胚葉細胞である。
【0080】
他の例では、本発明の方法に従って処理した多能性幹細胞は、血清は不含でアクチビンAを含有している培地中で多能性幹細胞を培養し、次いで細胞をアクチビンA及び血清と共に培養し、次いで細胞をアクチビンA及び様々な濃度の血清と共に培養することによって、胚体内胚葉系に特徴的なマーカーを発現している細胞へと分化させることができる。この方法の一例は、Nature Biotechnology 23,1534〜1541(2005)に開示されている。
【0081】
他の例では、本発明の方法に従って処理した多能性幹細胞は、血清は不含でアクチビンAを含有している培地中で多能性幹細胞を培養し、次いで細胞をアクチビンA及び異なる濃度の血清と共に培養することによって、胚体内胚葉系に特徴的なマーカーを発現している細胞へと分化させることができる。この方法の例は、D’Amourら、Nature Biotechnology(2005)に開示されている。
【0082】
他の例では、本発明の方法に従って処理した多能性幹細胞は、血清は不含でアクチビンA及びWntリガンドを含有している培地中で多能性幹細胞を培養し、次いでWntリガンドは除去し、細胞をアクチビンA及び血清と共に培養することによって、胚体内胚葉系に特徴的なマーカーを発現している細胞へと分化させることができる。この方法の例は、Nature Biotechnology 24,1392〜1401(2006年)に開示されている。
【0083】
他の例では、本発明の方法に従って処理した多能性幹細胞は、米国特許出願第11/736,908号(LifeScan,Inc.に譲渡)に開示される方法に従い、胚体内胚葉系に特徴的なマーカーを発現している細胞へと分化させることができる。
【0084】
他の例では、本発明の方法に従って処理した多能性幹細胞は、米国特許出願第11/779,311号に記載の方法に従って、胚体内胚葉系に特徴的なマーカーを発現している細胞へと分化させることができる。
【0085】
膵臓内胚葉系に特徴的なマーカーを発現している細胞の形成
多能性幹細胞は、当該技術分野における任意の方法により、膵臓内胚葉系に特徴的なマーカーを発現している細胞へと分化させることができる。
【0086】
例えば、多能性幹細胞は、D’Amourら、Nature Biotechnology 24,1392〜1401(2006)に開示されている方法に従って、膵臓内胚葉系に特徴的なマーカーを発現している細胞へと分化させることができる。
【0087】
例えば、本発明の方法に従って得られる、胚体内胚葉系に特徴的なマーカーを発現している細胞は、この胚体内胚葉系に特徴的なマーカーを発現している細胞を繊維芽細胞増殖因子及びヘッジホッグシグナル伝達経路阻害剤KAAD−シクロパミンで処理し、次いで繊維芽細胞増殖因子及びKAAD−シクロパミンを含有する培地を除去した後に、レチノイン酸、繊維芽細胞増殖因子及びKAAD−シクロパミンを含有する培地中で培養することにより、膵臓内胚葉系に特徴的なマーカーを発現している細胞へと更に分化する。この方法の例は、Nature Biotechnology 24,1392〜1401(2006)に開示されている。
【0088】
例えば、本発明の方法に従って得られる、胚体内胚葉系に特徴的なマーカーを発現している細胞は、この胚体内胚葉系に特徴的なマーカーを発現している細胞を米国特許出願第11/736,908号(LifeScan,Inc.に譲渡)に開示された方法に従いレチノイン酸及び少なくとも1種類の線維芽細胞増殖因子で所定の時間処理することによって、膵臓内胚葉系に特徴的なマーカーを発現している細胞へと更に分化する。
【0089】
例えば、本発明の方法に従って得られる、胚体内胚葉系に特徴的なマーカーを発現している細胞は、胚体内胚葉系に特徴的なマーカーを発現している細胞をレチノイン酸(Sigma−Aldrich,MO)及びエキセンディン4で処理し、次いでDAPT(Sigma−Aldrich,MO)及びエキセンディン4を含有している培地を除去し、続いてエキセンディン1、IGF−1及びHGFを含む培地で細胞を培養することで、膵臓内胚葉系に特徴的なマーカーを発現している細胞へと更に分化する。この方法の例は、Nature Biotechnology 24,1392〜1401(2006年)に開示されている。
【0090】
例えば、本発明の方法に従って得られる、膵臓内胚葉系に特徴的なマーカーを発現している細胞は、膵臓内胚葉系に特徴的なマーカーを発現している細胞をエキセンディン4を含有している培地で培養し、次にエキセンディン4を含有している培地を除去し、続いて細胞をエキセンディン1、IGF−1及びHGFを含有している培地で培養することにより、膵内分泌系に特徴的なマーカーを発現している細胞へと更に分化する。この方法の例は、D’Amourら、Nature Biotechnology,2006に開示されている。
【0091】
例えば、本発明の方法に従って得られる、膵臓内胚葉系に特徴的なマーカーを発現している細胞は、膵臓内胚葉系に特徴的なマーカーを発現している細胞を、DAPT(Sigma−Aldrich,MO)及びエキセンディン4を含有している培地で培養することにより、膵内分泌系に特徴的なマーカーを発現している細胞へと更に分化する。この方法の例は、D’Amourら、Nature Biotechnology,2006に開示されている。
【0092】
例えば、本発明の方法に従って得られる、膵臓内胚葉系に特徴的なマーカーを発現している細胞は、膵臓内胚葉系に特徴的なマーカーを発現している細胞をエキセンディン4を含有している培地で培養することにより、膵内分泌系に特徴的なマーカーを発現している細胞へと更に分化する。この方法の例は、D’Amourら、Nature Biotechnology,2006に開示されている。
【0093】
例えば、本発明の方法に従って得られる、膵臓内胚葉系に特徴的なマーカーを発現している細胞は、膵臓内胚葉系に特徴的なマーカーを発現している細胞を米国特許出願第11/736,908号(LifeScan,Inc.に譲渡)に開示された方法に従って、ノッチシグナル伝達経路を阻害する因子で処理することで、膵内分泌系に特徴的なマーカーを発現している細胞へと更に分化する。
【0094】
例えば、本発明の方法に従って得られる、膵臓内胚葉系に特徴的なマーカーを発現している細胞は、膵臓内胚葉系に特徴的なマーカーを発現している細胞を米国特許出願第11/779,311号(LifeScan,Inc.に譲渡)に開示された方法に従って、ノッチシグナル伝達経路を阻害する因子で処理することで、膵内分泌系に特徴的なマーカーを発現している細胞へと更に分化する。
【0095】
例えば、本発明の方法に従って得られる、膵臓内胚葉系に特徴的なマーカーを発現している細胞は、膵臓内胚葉系に特徴的なマーカーを発現している細胞を米国特許出願第11/736,908号(LifeScan,Inc.に譲渡)に開示された方法に従って、ノッチシグナル伝達経路を阻害する因子で処理することで、膵内分泌系に特徴的なマーカーを発現している細胞へと更に分化する。
【0096】
例えば、膵臓内胚葉系に特徴的なマーカーを発現している細胞は、膵臓内胚葉系に特徴的なマーカーを発現している細胞を米国特許出願第11/779,311号(LifeScan,Inc.に譲渡)に開示された方法に従って、ノッチシグナル伝達経路を阻害する因子で処理することで、膵内分泌系に特徴的なマーカーを発現している細胞へと更に分化する。
【0097】
膵内分泌系に特徴的なマーカーは、NGN3、NEUROD、ISL1、PDX1、NKX6.1、PAX4、NGN3及びPTF−1 αからなる群から選択される。一実施形態では、膵内分泌細胞は、以下のホルモンのうちの少なくとも1つを発現することができる:インスリン、グルカゴン、ソマトスタチン、及び膵臓ポリペプチド。本発明で使用するのに好適なものは、膵内分泌系の特徴を示すマーカーを少なくとも1つ発現している細胞である。本発明の一態様において、膵内分泌系に特徴的なマーカーを発現している細胞は、膵内分泌細胞である。膵臓内分泌細胞は、膵臓ホルモン発現細胞であってよい。また、膵臓内分泌細胞は膵臓ホルモン分泌細胞であってもよい。
【0098】
本発明の一態様では、膵臓内分泌細胞は、β細胞系に特徴的なマーカーを発現している細胞である。β細胞系に特徴的なマーカーを発現している細胞は、PDX1と、以下の転写因子、すなわち、NGN3、NKX2.2、NKX6.1、NEUROD、ISL1、HNF−3 β、MAFA、PAX4、又はPAX6のうちの少なくとも1つを発現する。本発明の一態様では、β細胞系に特徴的なマーカーを発現している細胞は、β細胞である。
【0099】
以下の実施例により本発明を更に例示するが、本発明はこれらの実施例により限定されるものではない。
【実施例】
【0100】
実施例1:マイクロキャリアに対するヒト胚性幹細胞の付着及び増殖
ヒト胚性幹細胞がマイクロキャリア上に付着しかつ増殖し得るか否かを判定するために、TrypLE(商標)Expressにより、継代数52のH9細胞をMATRIGEL(商標)(BD Biosciences,CA)コートプレートから放出させた。次いでそれらをマイクロキャリアとMEF−CMと共にインキュベートした。製造元からの取扱説明書に従い、ProNectinF(PN)、Plastic(P)、PlasticPlus(PP)、HILLEX(登録商標)II(H)、コラーゲン(Col)及びFACT III(SoloHill,MI)マイクロキャリアの懸濁液を調製した。毎日の観察結果に基づき、表1はH9細胞の、37℃にて2日経過後の、マイクロキャリアに対する付着と増殖を記載する。試験したほとんどのマイクロキャリアに対して、大部分の細胞は付着及び/又は増殖しなかった。H9細胞は、HILLEX(登録商標)IIマイクロキャリア(Solohill,MI)に対しては付着し増殖したが、画像で示される細胞−ビーズ凝集体は、2日間の静置培養後にはより少なくなった(図1B)。
【0101】
マイクロキャリアに対するヒト胚性幹細胞の付着性と増殖性を改善するために、Rho結合コイルドコイル形成タンパク質セリン/スレオニンキナーゼの低分子阻害剤である、Rhoキナーゼ阻害剤を培地に添加した。具体的には、Y27632,Y,(Sigma−Aldrich,MO)を使用した。10μMのY27632(Sigma−Aldrich,MO)添加MEF−CMは毎日交換した。10μMのY27632(Sigma−Aldrich,MO)の存在下で、H9細胞は試験した全てのマイクロキャリアに付着し、凝集体を形成した(表2)。画像解析によると、HILLEX(登録商標)IIマイクロキャリア(Solohill,MI)で増殖させたヒト胚性幹細胞は、他のマイクロキャリアについて試験したヒト胚性幹細胞よりも、付着しかつ増殖しているように見えた。加えてH9細胞は、Rhoキナーゼ阻害剤の存在下で、より良好にHILLEX(登録商標)II(Solohill,MI)に付着した(図1Aと図1Bとの比較による)。
【0102】
細胞治療用途のためのヒト胚性幹細胞の増量は、製品需要を満たす必要がある。現行の最も良好な増量技法としては、スピナーフラスコ法とバイオリアクター法が挙げられる。これらの技術のいずれもが、懸濁液中のマイクロキャリアの物理的な移動を必要とする。マイクロキャリア上でのヒト胚性幹細胞の増殖に対する移動効果を判定するために、6又は12ウェルディッシュを、37℃のインキュベーター内の固定式プラットフォームに設置した。3日間の増殖後、一部のマイクロキャリアから細胞凝集体が放出され始めた。図2のA、B、Dは、Plastic Plus、Plastic又はPronectinマイクロキャリアから脱着した細胞凝集体を記す。対照的に、図2Cの細胞は、HILLEX(登録商標)IIマイクロキャリア(Solohill,MI)に付着したまま留まりかつ増殖した。実施例4は、Viacount Flexを装備したGuava PCA−96(Guava Technologies,Hayward,CA)で細胞を計数するのに先立つ、脱着法を記載する。マイクロキャリア上の細胞の増殖速度を測定することで、播種時の開始細胞数と比較して、3日目の細胞数が減少していることが明らかになった。これは、5日目に実験が終了するまでの間増量が続けられる細胞の、マイクロキャリアへの初期付着が乏しいことに由来する可能性が高い。H9細胞は、HILLEX(登録商標)IIマイクロキャリア(Solohill,MI)に対し、他の種類のビーズと比較して最も速い増殖速度を有していたが、これはHILLEX(登録商標)IIマイクロキャリア(Solohill,MI)に対する、細胞のより良好な付着性に由来するものである可能性が高い(図2、3)。このことは、HILLEX(登録商標)IIマイクロキャリア(Solohill,MI)が、懸濁液中のH9細胞の増殖を支持し得ることを実証する。これは以降に繰り返される節の後に更に実証された。実施例5を参照されたい。
【0103】
ラージスケールでの増量のために、H1ヒト胚性細胞株をマイクロキャリア上での増殖性について同様に試験した。Rhoキナーゼ阻害剤のY27632(Sigma−Aldrich,MO)がH9細胞株の付着に必要とされたことから、この阻害剤はH1細胞についても同様に必要とされるであろうと推測された。Cytodex 1(登録商標)、Cytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)、HILLEX(登録商標)II、Plastic、ProNectinF、Plastic Plusマイクロキャリア(SoloHill Ann Arbor,MI)を、製造元からの取扱説明書に従って調製した。継代数47のH1ヒト胚性幹細胞を、約13,333個細胞/マイクロキャリアcm2(10μMのY27632(Sigma−Aldrich,MO)添加MEF−CM中)で播種した。細胞とマイクロキャリアを、37℃にて固定式プラットフォーム上の、12ウェルの組織培養用無処理ディッシュ(12ウェルあたり15cm2)内に設置して、マイクロキャリアと培地に運動させた。3、5及び7日目後に、1つのウェルを画像化し、採取し、計数した。細胞の付着能は、ビーズの種類に依存した。H9細胞株で観察されたものと同様の結果が、H1細胞株についても観察された。具体的には、Plastic、Plastic Plus又はProNectinFマイクロキャリア上に播種した細胞は十分に付着せず、及び/又は十分に増殖しなかった(図4)。HILLEX(登録商標)II(Solohill,MI)、Cytodex 1(登録商標)、又はCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)マイクロキャリア上に播種した細胞は十分に付着し、かつ十分に増殖した(図5)。実施例4に従って細胞を脱着し、収率について計数した。Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)で増殖させた細胞は、培養7日後に最も大きい細胞数を呈した(図6)。
【0104】
実施例2:細胞の付着及び増殖のためのY27632及び他のRhoキナーゼ阻害剤の、至適濃度
マイクロキャリアに対するヒト胚性幹細胞の付着と増殖を最も支持するRhoキナーゼ阻害剤濃度を決定するため、以下の実験を実施した。
【0105】
継代数44のH9細胞の、13,333個細胞/cm2の開始アリコートを、12ウェルの組織培養用無処理プレートの単一ウェル内で、15cm2マイクロキャリア上に播種した。37℃にて固定式プラットフォームに設置する前に、細胞を37℃にて少なくとも60分間静置した。細胞を加える前に、HILLEX(登録商標)II(Solohill,MI)マイクロキャリア及びCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)マイクロキャリアを、製造元から指示された通りに調製した。濃度範囲が10、5、2.5又は1μMのRhoキナーゼ阻害剤Y27632、あるいは濃度範囲が5、2.5、1又は0.5μMの(S)−(+)−4−グリシル−2−メチル−1−[(4−メチル−5−イソキノリニル)スルホニル]−ヘキサヒドロ−1H−1,4−ジアゼピンジヒドロクロリド(Glycyl−H 1152ジヒドロクロリド(H),Tocris,MO)を添加したMEF−CMで細胞を増殖させた。培地を毎日交換し、収率と生存率について、1ウェルの細胞を播種後4日目と7日目に計数した(図7A及びB)。全般的に、10又は5μMのY27632(Sigma−Aldrich,MO)が最も良好な細胞増殖を示し(7日目)、その一方で2.5及び1.0μMで最も良好な接着性を有しているように見えた(4日目)。濃度1及び0.5μMのGlycyl−H 1152ジヒドロクロリド(Tocris,MO)は最も良好な細胞増殖を示し(7日目)、その一方で5μMで最も良好な接着性を有しているように見えた(4日目)。
【0106】
Rhoキナーゼ阻害剤は、アポトーシスを促進すると特徴付けられてきたことから、次の用量設定法を試行した。継代数48のH1細胞を、TrypLE(商標)ExpressでMATRIGEL(商標)(BD Biosciences,CA)コートプレートから解離させた。次いで12ウェルの、組織培養用無処理プレートの単一ウェル内の15cm2マイクロキャリア上に、細胞を播種した。HILLEX(登録商標)II(Solohill,MI)、Cytodex 1(登録商標)、又はCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)マイクロキャリアを、Rhoキナーゼ阻害剤の量を減少させて試験した:10μMのY27632(Sigma−Aldrich,MO)を1日目に使用し、次いで0.5μMを2日目に使用(Y10/5μM);2.5μMのGlycyl−H 1152ジヒドロクロリド(Tocris,MO)を1日目に、続いて0.5μMを2日目に使用(H2.5/0.5μM);1μMのGlycyl−H 1152ジヒドロクロリド(Tocris,MO)を1日目に、0.5μMのGlycyl−H 1152ジヒドロクロリド(Tocris,MO)を2日目に使用(H1/0.5μM);あるいは0.25μMのGlycyl−H 1152ジヒドロクロリド(Tocris,MO)をMEF−CMに毎日添加(H0.25μM)。37℃にて固定式プラットフォームに設置する前に、37℃にてH1細胞及びマイクロキャリアを3時間にわたって45分毎に撹拌した。37℃にて固定式プラットフォームでの3、5及び7日目に細胞を計数した(図8)。全般的に、Glycyl−H 1152ジヒドロクロリド(Tocris,MO)の最良の濃度は、1日目では1〜2.5μMであり、2日目では0.5μMであり、以降では化合物は断った。細胞は、試験した全てのマイクロキャリアについて、これらの濃度のGlycyl−H 1152ジヒドロクロリド(Tocris,MO)で、10μMのY27632(Sigma−Aldrich,MO)と比較して同様の増殖速度を示した。0.25μMのGlycyl−H 1152ジヒドロクロリド(Tocris,MO)で細胞を維持した場合、細胞収率は乏しかった。Rhoキナーゼ阻害剤を最小量で用いることは、プロセス費用を低減することにもつながり、かつ細胞増殖についても有益なものであり得る。同様にこれらのデータは、ヒト胚性幹細胞がマイクロキャリアへの付着性及び増殖性を維持するためには、Rhoキナーゼ阻害剤は必要とされないことを示す。
【0107】
実施例3:マイクロキャリア上での接着及び増殖に対する細胞密度の効果
播種密度の改善は、必要とされる細胞の総数を減少させるための一手段である。適切な播種密度を決定するために、4x対物視野あたりのマイクロキャリア数を計数した。Cytodex 3マイクロキャリア(GE Healthcare Life Sciences,NJ)を含有している、10μMのY27632(Sigma−Aldrich,MO)添加MEF−CMの入った10cmプレートに、0.4×104個細胞/cm2(低密度)、1.2×104個細胞/cm2(中間密度)、又は3×104個細胞/cm2(高密度)の密度でH1細胞を播種した。次いでプレートを、37℃で6時間にわたり、45分毎に撹拌した。37℃にて30rpmのスピナーフラスコ(実施例5に記載)内の、10μMのY27632(Sigma−Aldrich,MO)添加MEF−CM 50mLに、細胞及びマイクロキャリアを移した。24時間後に、5μMのY27632(Sigma−Aldrich,MO)添加MEF−CM 25mLを添加した。24時間後、回転速度を40rpmに上昇させた。培養の3日目及び5日目に、75mLのうちの50mLを除去し、MEF−CMで交換した。6時間、3日、5日及び7日の時点で、スピナーフラスコからアリコートを取って画像を撮影した。細胞の付着したマイクロキャリアの百分率を、図9の画像の右下に記載する。播種後3日目では、細胞でコートされたマイクロキャリアの数は開始播種密度と対応したが、低密度で播種した細胞では、細胞でコートされたマイクロキャリアの数は5日目及び7日目でも増加しなかった。このことは、0.4×104個細胞/cm2が、細胞をマイクロキャリアへと組み込ませて凝集体にするのに十分な細胞数ではないことを意味する。3×104個細胞/cm2では、細胞が付着したマイクロキャリアの数は、1.2×104個細胞/cm2を播種した場合の5日目及び7日目と同様であった(図9)。細胞数を見た場合、高密度での細胞播種では、より多くの細胞がマイクロキャリアに付着していることは明らかである(図10)。開始播種細胞数と比較しての倍率変化(fold change)の解析では、高密度播種した培養物において、3日目及び5日目に、より多数の細胞が付着していたことを明らかになった(図11)。7日目には、対照と高密度播種培養物は、それらの開始播種密度から、細胞数について同様の倍率で変化した。これらのデータから、我々は、H1細胞をマイクロキャリア上に効果的に付着させ、増殖させるためには、1.2×104個細胞/cm2が最小の細胞数であると結論付けた。より高い播種密度へと移行させることは、細胞の増量に必要とされる日数を減少させる助けとなり得る。
【0108】
実施例4:マイクロキャリアからの細胞の解離
増殖速度を判定するためには、マイクロキャリアから細胞を解離させる必要がある。Rhoキナーゼ阻害剤Y27632(Sigma−Aldrich,MO)を除去しても、H1細胞はマイクロキャリアから解離しなかった(実施例2、図12)。マイクロキャリアから細胞を解離する前に、HILLEX(登録商標)IIマイクロキャリア(Solohill,MI)上のH9細胞を、倍率10x及び20xで画像解析した(それぞれ図13A,B)。HILLEX(登録商標)IIマイクロキャリア(Solohill,MI)上のH9細胞を酵素処理することで、生存細胞を脱着させた(図13C,D及び14)。37℃にて固定式プラットフォーム上で、HILLEX(登録商標)IIマイクロキャリア(Solohill,MI)を含有する6ウェルディッシュ中で、6日間にわたってH9細胞を増殖させた。マイクロキャリアに付着させた細胞を15mLコニカルチューブに設置し、マイクロキャリアを沈殿させた後に培地を吸引除去した。沈殿したマイクロキャリアを4mLのPBS(マグネシウムイオン及びカルシウムイオン不含)で3回洗浄し、重量沈降によりマイクロキャリアを沈殿させた。PBSを吸引除去し、1mLのPBSを加えた。細胞を有するマイクロキャリアを、12ウェルの組織培養用無処理プレートの単一ウェルに移した。プレートを、マイクロキャリアを沈殿させることができるような角度で静止させた。PBSを吸引除去し、1mLのTrypLE(商標)Express(Invitrogen,CA)又は0.05%トリプシン/EDTAをウェルに加えた。プレートを、37℃にて固定式プラットフォーム上に、10〜20分間にわたって設置した。プレートを取り外し、3mLのDMEM/F12又はMEF−CMをウェルに添加した。培地を激しくピペッティングし、細胞を放出させた(図13C,D)。顕微鏡下でのマイクロキャリアの観察は、細胞がマイクロキャリアから脱着したことを決定づけた。次いで細胞を、200×gで5分間にわたって遠心した。培地を吸引除去し、ペレットを1mLのDMEM/F12又はMEF−CM培地に再懸濁した。次いでViacount dyeを用いて、Guava PCA−96(Guava Technologies,Hayward,CA)で細胞を計数した。具体的には、培地で適切に希釈した200μL容量の細胞を、2μLのViacountと共に10分間にわたってインキュベートした。生存率と細胞数を決定した(図14)。TrypLE(商標)Express及びトリプシン/EDTAの両方が、細胞をマイクロキャリアから効率的に解離した。
【0109】
TrypLE(商標)Expressがマイクロキャリアから細胞を放出し、及びこれはGMP製品として利用可能なものであることから、他の可能性のある解離剤、具体的にはコラゲナーゼ及びAccutase(商標)(Sigma−Aldrich,MO)について試験を行った。継代数48のH1細胞を、スピナーフラスコ中(実施例5)で10日間にわたって増殖させた。次いでマイクロキャリアを回収し、50mLのコニカルチューブに移した。細胞を上記のようにPBSで洗浄し、12ウェルのプレートに移した。PBSを吸引除去し、1mLのTrypLE(商標)Express、Accutase(商標)又はコラゲナーゼ(10mg/mL)をウェルに添加して、37℃にて固定式プラットフォームに5〜10分にわたって設置した。細胞/マイクロキャリアをDMEM/F12に激しく再懸濁し、次いで解離した細胞とマイクロキャリアを50mLのコニカルチューブ上で40μmのセルストレイナーを通して濾過した。ウェルを追加の培地2mLで洗浄し、同様にセルストレイナーに加えて、200×gで5分にわたって遠心した。次いで細胞を1mLのDMEM/F12に再懸濁し、上記のように細胞を計数するために希釈した。細胞生存率は、試験した全ての酵素で類似していた。Accutase(商標)とTrypLE(商標)Expressは、5分及び10分のインキュベーション後に同様の数の細胞を放出した(図15)。これは、マイクロキャリア上のヒト胚性幹細胞のための細胞解離剤としての、Accutase(商標)とTrypLE(商標)Expressの適合性を実証する。
【0110】
実施例5:マイクロキャリア上の未分化の多能性幹細胞の増殖
マイクロキャリア上の細胞を増量させるためには、細胞を、マイクロキャリアから脱着又は酵素的に解離させ、かつ新しいマイクロキャリアへと再付着させることができなければならない。マイクロキャリア上での一般的な細胞増殖方法は、細胞の脱着及び再付着特性に依存する。以下の実験は、これがヒト胚性幹細胞の特徴ではなかったことを示す。具体的には、継代数43のH9細胞をHILLEX(登録商標)IIマイクロキャリア(Solohill,MI)上に播種し、125mLのスピナーフラスコ中でインキュベートした(以下を参照のこと)。フェノールレッドが培地中に存在し、かつHILLEX(登録商標)IIマイクロキャリアにより取り込まれた(Solohill,MI)。8日間の増殖後に、マイクロキャリア上の細胞のアリコート10mLを、フェノールレッド不含MEF−CMと、440mgのHILLEX(登録商標)IIマイクロキャリアと、5μMのY27632(Sigma−Aldrich,MO)とを含有している、新しいスピナーフラスコに設置した。37℃にて30rpm回転でのインキュベーションの5日目に、マイクロキャリアを取り出して画像を得た(図16)。黒っぽいマイクロキャリアは、フェノールレッドを含有している培地で増殖したH9細胞で覆われた、マイクロキャリアを示す。色味の明るいマイクロキャリアは、新しく加えたマイクロキャリアである。H9細胞が脱着し、新しいマイクロキャリアに再付着することが見込まれていたが、細胞は再付着する代わりに新しいマイクロキャリアと共に凝集体を形成した。色味の明るくないマイクロキャリアには細胞が付着しており、この細胞が黒っぽいマイクロキャリアとの凝集体を形成していないことは、細胞はマイクロキャリアから脱着しかつマイクロキャリアへと再付着することはできなかったことを示唆する。マイクロキャリア上の細胞の増殖を伝播させるためには、細胞をマイクロキャリアから酵素的に解離させる必要があった(実施例4を参照されたい)。
【0111】
現在では、ヒト胚性幹細胞をマイクロキャリア上で増殖させる方法が確立されているので、どのようにしてヒト胚性幹細胞をラージスケールのスピナーフラスコ中で増殖させるかを決定する必要がある。スピナーフラスコは、高密度系での細胞の増量を可能にする。これは空間の節約であり、バイオリアクター内で細胞を増量させるための第1工程であると考えられる。ヒト胚性幹細胞の、スピナーフラスコ中で増殖する能力について試験するために、継代数43のH9細胞を125mLのスピナーフラスコ中に播種した。細胞は、まず10cmプレート中でマイクロキャリアに付着させてからスピナーフラスコに移した。具体的には、TrypLE(商標)Expressと共に37℃で5分間インキュベートすることで、H9細胞を2枚の6ウェルディッシュから放出させた。細胞はコラゲナーゼ(1mg/mL)で処理し、1:30増殖因子低減MATRIGEL(商標)(BD Biosciences,CA)コートプレートに播種した後に、TrypLE(商標)Expressで処理した。細胞をDMEM/F12に再懸濁し、Viacountを用いてGuava instrumentで計数した。遠心後に、3×106個の細胞を、10μMのY27632(Sigma−Aldrich,MO)添加MEF−CMと、製造元からの取扱説明書に従って調製した250cm2のHILLEX(登録商標)IIマイクロキャリア(Solohill,MI)とを含有している、10cmのプレートに播種した。ディッシュを37℃に設置し、穏やかに回転させ、4.5時間にわたって45分毎に一度撹拌した。次いで細胞、マイクロキャリア及び培地を125mLのスピナーフラスコに移した。次いでスピナーフラスコには10μMのY27632(Sigma−Aldrich,MO)添加MEF−CM50mLを充填し、37℃にて撹拌プレートに40rpmで設置した。翌日、培地を交換し、5μMのY27632(Sigma−Aldrich,MO)添加MEF−CM75mLで充填した。撹拌速度は70rpmに上昇させた。培地にはY27632化合物(Sigma−Aldrich,MO)を加えずに1日おきに交換した。実施例4に記載の方法に従って細胞を継代し、3×106個の細胞を、250cm2の新しいマイクロキャリアに再播種した。培養物は、1〜2×105個細胞/cm2コンフルエントに達したときに継代した。この操作を5継代実施した(図17)。各継代での多能性マーカー発現では、細胞の80〜95%が多能性マーカーCD9、SSEA4、SSEA3、TRA−1−60及びTRA−1−81を発現していたと評価された(図19A)。同様の実験を、Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上で、継代数43のH9細胞についても実施した(図18,19B)。全般的に、細胞はHILLEX(登録商標)II(Solohill,MI)マイクロキャリア及びCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)マイクロキャリアのどちらでも良好に増殖し、かつ多能性を保持した。スピナーフラスコ中で5回継代した後に核型解析を実施したところ、細胞の1.5%で、12番染色体の異常なトリソミーが示された。これらの細胞は、実験の終了時には継代数50付近であることから、観察されるこのような異常は一般的に生じる恐れがある。細胞継代数が低い状態で開始した場合に、このような仮定が生じるか試験した。
【0112】
継代数48と継代数49のH1細胞株についても、同様の実験を実施した。回転速度と播種密度を除き、全てのパラメーターは同様のままであった。スピナーフラスコの回転速度は、初日はオーバーナイトで30rpmであり、以降の日では40rpmに上昇させた。Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)の場合の播種密度は約11,000個細胞/cm2であった一方で、Cytodex 1(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)の場合の播種密度は約7,000個細胞/cm2であった。播種した細胞数はスピナーフラスコあたり3×106個細胞で一定であった。マイクロキャリアの重量は、Cytodex 1(登録商標)及びCytodex 3(登録商標)の場合、100mgで一定であった。Cytodex 1(登録商標)及びCytodex 3(登録商標)がHILLEX(登録商標)IIマイクロキャリアを上回る1つの利点は、それらのより大きな表面積である。図20及び21は、Cytodex 1(登録商標)及びCytodex 3(登録商標)のそれぞれの上でのH1細胞の増量を示す。細胞は5回の継代にわたって多能性を維持していた(図22)。Cytodex 3(登録商標)マイクロキャリア上のH1細胞の核型解析は、試験した細胞の10%で、Y染色体に重複があることを明らかにした。これらのH1細胞はマイクロキャリア上に継代数48で継代され、その後5回継代された後に解析された。平面状表面上のMATRIGEL(商標)(BD Biosciences,CA)で増殖させた継代数55のH1細胞は、正常な核型を有していた。3日間と経過日数間(5、6又は7日間)との間での、これらの細胞の倍加速度解析では、全般的に倍加時間に変化は見られなかった(図23)。Cytodex 1(登録商標)マイクロキャリア上で増殖させたH1細胞及びHILLEX(登録商標)IIマイクロキャリア(Solohill,MI)上で増殖させたH9細胞は、ほとんど一定の倍加時間を示した(表3)。
【0113】
実施例6:制限培地中のマイクロキャリア上でのヒト胚性幹細胞の増殖
治療用製品を製造するためには、ヒト胚性幹細胞培養培地からいずれの動物成分も除外されていることが好ましい。現在、ヒト胚性幹細胞は、マウス胚性線維芽細胞を用いて馴化させた培地(MEF−CM)中の、MATRIGEL(商標)(BD Biosciences,CA)上で維持されている。MATRIGEL(商標)(BD Biosciences,CA)及びMEF−CMのいずれもがマウス細胞由来である。加えて、MEF−CMは高価であり、培地を生成するのに時間がかかる。ヒト胚性幹細胞が、制限培地を含むマイクロキャリア上で維持され得るかを判断するために、Stem Pro(Invitrogen,CA)、mTESR(StemCell Technologies,Vancouver,Canada)又はMEF−CM中の、Rhoキナーゼ阻害剤の10μMのY27632(Sigma−Aldrich,MO)又は2.5μMのGlycyl−H 1152ジヒドロクロリド(Tocris,MO)の存在下で、H9細胞をCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)及びHILLEX(登録商標)II(Solohill,MI)マイクロキャリア上に播種した。細胞を、37℃にて固定式プラットフォーム上の12ウェルディッシュ中に設置した。細胞を3、5及び7日目に計数した。継代数39のH9細胞はMEF−CM中で、どちらの種類のビーズ上でも増殖し、一般的な増量特性を示した(図24)。mTESR(StemCell Technologies,Vancouver,Canada)中で増殖させた同様の細胞は、10μMのY27632(Sigma−Aldrich,MO)の存在下で、Cytodex 3(登録商標)マイクロキャリア上で良好に増殖したが、HILLEX(登録商標)IIマイクロキャリア上では低い増殖率を呈した。20回にわたって継代することでStemPro培地に馴化された、継代数64のヒト胚性幹細胞株H9(継代数64のH9)細胞は、10μMのY27632(Sigma−Aldrich,MO)の存在下で、HILLEX(登録商標)II及びCytodex 3(登録商標)のいずれでも良好に増殖した。驚くべきことに、これらの細胞は、2.5μMのGlycyl−H 1152ジヒドロクロリド(Tocris,MO)の存在下では、Cytodex 3(登録商標)マイクロキャリア上では良好には増殖しなかった。したがって、マイクロキャリアの種類、Rhoキナーゼ阻害剤、及び培地の全てが、ヒト胚性幹細胞の増殖能を決定する役割を果たす。
【0114】
継代数38のH1ヒト胚性幹細胞を、12ウェルのディッシュに、mTESR(StemCell Technologies,Vancouver,Canada)又はMEF−CM中のRhoキナーゼ阻害剤の、10μMのY27632(Sigma−Aldrich,MO)又は2.5μMのGlycyl−H 1152ジヒドロクロリドの存在下で、Cytodex 3(登録商標)又はHILLEX(登録商標)IIマイクロキャリアのいずれかに播種した。細胞を37℃にて固定式プラットフォーム上に設置した。細胞を3、5及び7日目に計数した。MEF−CM中でY27632(Sigma−Aldrich,MO)の存在下で増殖させた細胞は、両方の種類のマイクロキャリアで一般的な増量特性を示したが、Glycyl−H 1152ジヒドロクロリド(Tocris,MO)の存在下では、Cytodex 3(登録商標)に対して乏しい増殖率を示した(図25)。mTESR培地(StemCell Technologies,Vancouver,Canada)は、どちらのRhoキナーゼ阻害剤の存在下でも、HILLEX(登録商標)II マイクロキャリア上でH1細胞を増殖させたが、Cytodex 3(登録商標)マイクロキャリアに対しては低い増殖率が示された。
【0115】
mTESR(StemCell Technologies,Vancouver,Canada)中で、HILLEX(登録商標)IIマイクロキャリアで良好に増殖した所与の継代数50のH1細胞のうちの3×106個を、250cm2のHILLEX(登録商標)IIマイクロキャリアに播種した。37℃で、5時間にわたって45分ごとに手で撹拌しながら、10cm2のディッシュ中でインキュベートした。10μMのY27632(Sigma−Aldrich,MO)を添加したmTESR(StemCell Technologies,Vancouver,Canada)を、1日おきに交換した。この操作を、MEF−CMで増殖させた細胞についても並行して実施した(実施例5)。MEF−CMで増殖させた細胞とは異なり、mTESR培地(StemCell Technologies,Vancouver,Canada)で増殖させた細胞は7日目にHILLEX(登録商標)IIマイクロキャリアから脱着し始めた(図26A対26B)。このことは、ヒト胚性幹細胞にHILLEX(登録商標)IIマイクロキャリア(Solohill,MI)に対する付着性及び増殖性を維持させるためには、mTESR(StemCell Technologies,Vancouver,Canada)に追加の成分を加える必要があることを示す。
【0116】
実施例7:マイクロキャリア上でのヒト胚性幹細胞の分化
ヒト胚性幹細胞がマイクロキャリア上で増量可能であることから、これらの細胞の分化の可能性が決定されるべきである。継代数43のヒト胚性幹細胞株H9の細胞を、Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上で5回継代した。5回目の継代で、細胞をマイクロキャリア上で6日間にわたって増殖させた後、TrypLE(商標)Expressを用いてマイクロキャリアから解離させた(実施例4を参照されたい)。次いで細胞を、1:30 MATRIGEL(商標):DMEM/F12コートしたプレートに設置した。細胞がプレート上で80〜90%コンフルエントに達した後、細胞を分化誘導剤に曝露させた。100ng/mLのアクチビンA(PeproTech,NJ)と、20ng/mLのWnt3a(R&D Biosciences,MN)と、8ng/mL bFGF(PeproTech,NJ)とを添加した、2%ウシ血清アルブミンフラクションV(脂肪酸不含)(FAF BSA,MP Biomedicals,Ohio)含有RPMIで細胞を2日間にわたって処理することで、胚体内胚葉へのヒト胚性幹細胞の分化を実施した。細胞は、100ng/mLのアクチビンA(PeproTech,NJ)と、8ng/mLのbFGF(PeproTech,NJ)とを添加した、2% FAF BSA含有RPMIで更に2日にわたって処理した。培地は毎日交換した。胚体内胚葉の細胞表面マーカーのCXCR4についてFACS解析を実施したところ、87%の細胞がマーカータンパク質を発現していたことが示された(図27A)。継代数49のヒト胚性幹細胞株H1の細胞を、Cytodex 1(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上で5回継代して増殖させて、同様の実験を実施したところ、マイクロキャリア上で分化した細胞の91%がCXCR4を発現していたことが明らかになった(図27B)。このことは、マイクロキャリア上で増殖させた細胞は、インスリン産生細胞に分化するための第1段階である、胚体内胚葉へと分化できるということを実証している。
【0117】
3種のマイクロキャリア、すなわちCytodex 1(登録商標)、Cytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)及びHILLEX(登録商標)II(Solohill,MI)は、H1細胞を付着させ、増殖させる。これらの3種のマイクロキャリア上で、H1細胞の分化を実施した。スピナーフラスコ中のこれらのマイクロキャリア上で、様々な継代数(1〜5回)で細胞を増殖させた(実施例5)。マイクロキャリアと細胞との懸濁液の、6〜8日間にわたる継代後のアリコートを、6又は12ウェルのプレートに移した。12ウェルプレートあたり合計15cm2のマイクロキャリアと細胞を、あるいは6ウェルプレートあたり30cm2マイクロキャリアと細胞を移した。次いで分化誘導培地をプレートのウェルに加え、このプレートを37℃にて固定式プラットフォーム上に設置した。100ng/mLのアクチビンA(PeproTech,NJ)と、20ng/mLのWnt3a(R&D Biosciences,MN)と、8ng/mLのbFGF(PeproTech,NJ)とを添加した、2%ウシ血清アルブミンフラクションV(脂肪酸不含)(MP Biomedicals,Ohio)含有RPMIで、2日間にわたって細胞を処理することで、胚体内胚葉へのヒト胚性幹細胞の分化を実施した。100ng/mLのアクチビンA(PeproTech,NJ)と、8ng/mLのbFGF(PeproTech,NJ)とを添加した2% FAF BSA含有RPMIで、更に2日間にわたって細胞を処理した。培地は毎日交換した。胚体内胚葉の細胞表面マーカーのCXCR4について、FACS解析を実施した(図28)。胚体内胚葉への分化を支持してCytodex 1(登録商標)マイクロキャリア、及びCytodex 3(登録商標)マイクロキャリアで細胞を増殖させたものではそれぞれ87%と92%であった一方で、HILLEX(登録商標)IIマイクロキャリアはこの実験で試験した他のマイクロキャリアと同程度には分化を支持しなかった(42%)。
【0118】
細胞密度がマイクロキャリア上の細胞の分化に影響を及ぼすか否かを判断するために、継代数40のヒト胚性幹細胞株H1の細胞を、スピナーフラスコ中のCytodex 3(登録商標)マイクロキャリア上で、8日間又は11日間のいずれかで増殖させた。次いで約15cm2当量のマイクロキャリアと細胞とを6ウェルディッシュに設置し、固定式プラットフォーム上に設置した。次いで細胞を、上記のような胚体内胚葉分化誘導培地中でインキュベートした。4日後に、CXCR4発現について細胞をFACSにより解析した。スピナーフラスコ中で6日間にわたって増殖させた細胞の87%がCXCR4を発現していた一方で、スピナーフラスコ中で11日間にわたって増殖させた細胞では、56%がCXCR4を発現していた(図29)。このことは、分化させる前の細胞の培養日数が重要であることを実証し、具体的には、細胞密度が高すぎる場合には、細胞が効率的に分化できなくなる場合があることを実証する。
【0119】
付着性と増殖性が十分であると判断された3種のマイクロキャリアの全てで、ヒト胚性幹細胞が膵臓内胚葉細胞へと分化し得るか否かを判断するために、継代数41のヒト胚性幹細胞株H1の細胞(継代数41のH1)を、Cytodex 1(登録商標)、Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)及びHILLEX(登録商標)IIマイクロキャリア(Solohill,MI)上に播種した(実施例1を参照されたい)。製造元の取扱説明書に従ってマイクロキャリアを調製した。30cm2のマイクロキャリアを、低接着性の6ウェルプレートに移した。2枚の10cm2プレートから、製造元からの取扱説明書に従って、TrypLE(商標)ExpressによりH1細胞を解離させた。1ウェルあたり5×105個細胞で、細胞を播種した。実施例3に記載の方法に従って、ビーズへの細胞の付着を実施した。簡潔に述べると、細胞とマイクロキャリアを、37℃にて、10μMのY27632添加MEF馴化培地中で4時間にわたって、1時間ごとに簡単に撹拌しながらインキュベートした。HILLEX(登録商標)IIマイクロキャリア及びCytodex 1(登録商標)マイクロキャリア上の細胞を、固定式プラットフォーム上に設置した。Cytodex 3(登録商標)マイクロキャリア上の細胞は、一晩静置した。以降、培地はY27632不含の培地で毎日交換した。
【0120】
この実験では、乏しい付着性に起因して、Cytodex 1(登録商標)プレートの細胞の大多数が、もはやマイクロキャリアに付着していなかった。しかしながら、より長い付着時間及び/又はより低速に固定された速度では、細胞付着性は改善され得る。7日後に、2% BSA(脂肪酸不含(FAF))(Proliant,IA)含有RPMIと、以下の増殖因子:bFGF(8ng/mL,(PeproTech,NJ))、アクチビンA(100ng/mL,(PeproTech,NJ))、Wnt3a(20ng/mL,(R&D Biosciences,MN))とを用い、細胞を胚体内胚葉へと分化させた。4日間にわたる第2の分化を通して、Wnt3aは不含の同様の培地で細胞を処理した。4日後の、2つ組試料のFACS解析では、77〜83%の細胞でCXCR4レベルが陽性だったことが明らかになった。胚体内胚葉発現は、異なるマイクロキャリア上で増殖させた細胞間で、等価であった。図30を参照されたい。
【0121】
次いで、FGF7(50ng/mL,(R&D Systems,MN))、KAAD−シクロパミン(0.25μM,(Calbiochem,NJ))を添加した、2% FAF BSA(Proliant,IA)含有DMEM/F12又はDMEM−HGにより、細胞を更に2日間にわたって分化させた。これを、ノギン(100ng/mL,(R&D Biosciences,MN))、FGF7(50ng/mL,(R&D Systems,MN))、レチノイン酸(2μM,(Sigma−Aldrich,MO))、及びKAAD−シクロパミン(0.25μM,(Calbiochem,NJ))を添加した、1% B−27supplement(Invitrogen,CA)含有DMEM/F12又はDMEM−HGにより、4日間にわたって処理した。次いで、ノギン(100ng/mL,(R&D Biosciences,MN))、DAPT(1μM,(Sigma−Aldrich,MO))、Alk5阻害剤II(1μM,(Axxora,CA))を添加した、1% B−27 supplement(13日,膵臓内胚葉,(Invitrogen,CA))含有DMEM/F12又はDMEM−HGにより、細胞を3日間にわたって分化させた。図31は、Q−PCRにより、膵臓特異的な遺伝子のNKX6.1、PDX1及びNGN3の発現レベルを示す。CT値は、HILLEX(登録商標)IIマイクロキャリア上で分化させた細胞が、効率的に分化せず、必要なβ細胞前駆体細胞マーカーを発現しないことを明瞭に示す。細胞は、3種のマイクロキャリアの全てで効率的に胚体内胚葉へと分化したが、HILLEX(登録商標)IIマイクロキャリアでは、膵臓前駆体への更なる分化は効率的ではなかった。
【0122】
ヒト胚性幹細胞が、インスリン産生細胞へと更に分化し得るか否かを判断するために、継代数45のH1細胞をCytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上で増殖させ、上記と同様に分化させた。簡潔に述べると、製造元からの取扱説明書に従って、TrypLE(商標)Expressにより、10cm2プレートからH1細胞を解離させた。6ウェルプレートのウェルあたり1又は2×106個細胞で細胞を播種した。ビーズへの細胞の付着を実施例3に記載する。簡潔に述べると、細胞とマイクロキャリアとを、10μMのY27632を添加したMEF馴化培地中で、37℃にて4時間にわたって、1時間ごとに簡単に撹拌しながらインキュベートした。次いで細胞を、一晩静置してインキュベートした。2日目に、培地を5μMのY27632を添加したMEF馴化培地に交換し、プレートを固定式プラットフォーム上に設置した。以降毎日、培地をY27632は不含の培地と交換した。5日目に、培地を胚体内胚葉分化誘導培地(以下の増殖因子:bFGF(8ng/mL,(PeproTech,NJ))、アクチビンA(100ng/mL,(PeproTech,NJ))、Wnt3a(20ng/mL,(R&D Biosciences,MN))を添加した、2% BSA(脂肪酸不含(FAF))(Proliant,IA)含有RPMI)と交換した。分化誘導の2日目及び3日目に、Wnt3aは不含の同様の培地で細胞を処理した。3日後の、2つ組試料のFACS解析では、97〜98%の細胞でCXCR4レベルが陽性だったことが明らかになった。次いで、FGF7(50ng/mL,(R&D Systems,MN)),KAAD−シクロパミン(0.25μM,(Calbiochem,NJ))を添加した、2%FAF BSA(Proliant,IA)含有DMEM−高グルコース(HG)により、更に2日間にわたって細胞を分化させた。これを、ノギン(100ng/mL,(R&D Biosciences,MN))、FGF7(50ng/mL,(R&D Systems,MN))、レチノイン酸(2μM,(Sigma−Aldrich,MO))、及びKAAD−シクロパミン(0.25μM,(Calbiochem,NJ))を添加した、1% B−27supplement(Invitrogen,CA)含有DMEM−HGにより、4日間にわたって処理した。次いで、ノギン(100ng/mL,(R&D Biosciences,MN))、DAPT(1μM,(Sigma−Aldrich,MO))、Alk5阻害剤II(1μM,(Axxora,CA))を添加した、1% B−27 supplement(Invitrogen,CA)含有DMEM−HG又はDMEM−F12により、細胞を3日間にわたって分化させた。続いてこれを、Alk5阻害剤II(1μM,(Axxora,CA))を添加したDMEM−HG又はDMEM−F12中で、7日間にわたって分化させた。最終分化はそれぞれDMEM−HG又はDMEM−F12で5日間だった。合計24日間にわたる分化が、膵臓内分泌ホルモンの発現を誘導する。図32は、このエンドポイントの細胞の、FACS解析結果を示す。播種密度が最も高く、かつ6〜24日間にわたってDMEM−HGで分化させた細胞が、最も高レベルのインスリン発現を有していた(図32)。
【0123】
別の方法としては、ヒト胚性幹細胞がインスリン産生細胞へと分化し得るか否かを判断するために、7日間にわたって継代数44のH1細胞を、スピナーフラスコ中のCytodex 3(登録商標)マイクロキャリア上で増殖させた(実施例5を参照されたい)。15cm2/ウェルで、細胞とマイクロキャリアとを12ウェルプレートに移し、これを37℃にて固定式プラットフォーム上に設置した。RMPIの代わりにDMEM/F12を添加して、細胞を上記のように胚体内胚葉へと分化させた。4日後のFACS解析では、3つ組解析において、75〜77%の細胞でCXCR4レベルが陽性だったことが明らかになった。次いで、FGF7(50ng/mL,(R&D Systems,MN))、KAAD−シクロパミン(0.25μM,(Calbiochem,NJ))を添加した、2%ウシ血清アルブミンフラクションV(脂肪酸不含)含有DMEM/F12により、細胞を更に3日間にわたって分化させた。これを、ノギン(100ng/mL,(R&D Biosciences,MN))、FGF7(50ng/mL,(R&D Systems,MN))、レチノイン酸(2μM,(Sigma−Aldrich,MO))、及びKAAD−シクロパミン(0.25μM,(Calbiochem,NJ))を添加した、1% B−27supplement(Invitrogen,CA)含有DMEM/F12により、4日間にわたって処理した。次いで、ノギン(100ng/mL,(R&D Biosciences,MN))、ネトリン4(100ng/mL,(R&D Biosciences,MN))、DAPT(1μM,(Sigma−Aldrich,MO))、Alk5阻害剤II(1μM,(Axxora,CA))を添加した、1% B−27 supplement(15日,膵臓内胚葉,(Invitrogen,CA))含有DMEM/F12により、細胞を3日間にわたって分化させた。続いてこれを、Alk5阻害剤II(1μM,(Axxora,CA))を添加した、1% B−27 supplement(21日,膵内分泌細胞,(Invitrogen,CA))含有DMEM/F12で6日間処理した。7日間にわたる最終処理では、1% B−27 supplement(28日,インスリン発現細胞,(Invitrogen,CA))を添加したDMEM/F12を用いた。図33は、Q−PCRにより、膵臓特異的な遺伝子のインスリン、Pdx1及びグルカゴンの発現レベルを示す。マイクロキャリアについてのデータは、前出のMATRIGEL(商標)(BD Biosciences,CA)コートした平面状表面で分化させた継代数42のH1細胞と比較する。マイクロキャリアで分化させた細胞では、これらの膵臓特異的遺伝子の発現レベルは、平面状表面で分化させた細胞と比較して同様であるか、あるいはそれよりも勝っている。
【0124】
同様の実験を、Cytodex 3(登録商標)マイクロキャリア上で及び拡張型スピナーフラスコ中で継代した、継代数38のH9細胞で実施した。15cm2のマイクロキャリアと細胞とのアリコートを12ウェルプレートに設置し、分化誘導培地と共に、固定式プラットフォーム上に設置した。これを、MATRIGEL(商標)(BD Biosciences,CA)コートした6ウェルプレート上に設置した、細胞と比較した。RPMIとサプリメント(supplements)の中で胚体内胚葉への細胞の分化を実施したところ、平均して83%の細胞がCXCR4を発現していたのに対して(試料は2つ組)、平面状基材上では72%の細胞がCXCR4を発現していた(図34)。更に膵臓内胚葉(15日)、膵内分泌細胞(22日)及びインスリン発現細胞(29日)への分化では、マイクロキャリア上で増殖させた細胞と、平面状基材上で増殖させた細胞との間では、インスリン及びグルカゴンについて同様の発現レベルが示された(図35)。培地成分は、更に1日内分泌細胞分化誘導成分で処理した、上記のH1分化誘導実験について列挙したものと同一のものであった。インスリン発現ステージでは、細胞は22日のものと比較してインスリン発現の驚くべき減少を示した。この減少は、マイクロキャリア試料及び平面試料の双方で指摘されたことから、付着基材によるものである可能性は低い。このことは、H9細胞が、マイクロキャリア上でも少なくとも膵内分泌細胞へと良好に分化し得るということを示す。
【0125】
全般的に、2種の異なるヒト胚性幹細胞株のH1とH9は、Cytodex 3(登録商標)マイクロキャリア上で膵内分泌細胞へと分化でき、これらの細胞をラージスケールの培養系で増量及び分化させることの可能性を実証する(図17、21、33及び35)。ヒト胚性幹細胞は、少なくとも3種のマイクロキャリアビーズに付着及び増殖できたが、この細胞は少なくとも胚体内胚葉へと分化し得る(図28)。これらの結果は、治療用途のためにヒト胚性幹細胞を増殖させ分化させ得る方法を実証する。
【0126】
実施例8:3Dマイクロキャリア系培養法で単独の細胞として継代したヒト胚性幹細胞は、多能性を維持しつつ、ECM不含表面に移して培養することができる。
H1ヒト胚性幹細胞を、実施例5に記載の方法に従ってマイクロキャリア上で培養した。細胞をマイクロキャリアから取り外し、3μMのGlycyl−H 1152ジヒドロクロリドを添加したMEFCM16と共に、Nunc4、Nunc13、CELLBIND(商標)、又はPRIMARIA(商標)組織培養ポリスチレン(TCPS)平面状表面に蒔いた。細胞は6ウェルプレートに100,000個細胞/cm2の密度で播種し、次いで各表面上で更に1継代培養した。次いで細胞を、TrypLEで浮かせてフローサイトメトリーにより多能性マーカーについて試験するか、mRNA精製及びqRT−PCRのためにウェル中でRLTに溶解させるか、あるいは胚体内胚葉へと分化させるかした。2% BSA、100ng/mLのアクチビンA、20ng/mLのWnt3a、8ng/mLのbFGF及び3μMのGlycyl−H 1152ジヒドロクロリドを添加したRPMI培地で24時間にわたって細胞を処理することで、分化を誘導した。次いで培地を、2% BSA、100ng/mLのアクチビンA、8ng/mLのbFGF、及び3μMのGlycyl−H 1152ジヒドロクロリドを添加したRPMI培地に交換し、更に48時間にわたって培地を毎日交換した。
【0127】
フローサイトメトリー又はqRT−PCRのいずれかを用いて多能性マーカーにより測定したところ、マイクロキャリア上で培養した後にNunc4、Nunc13、CellBIND又はPrimaria組織培養ポリスチレン(TCPS)平面状表面へと移した細胞は、各平面状表面上で2回継代した後に多能性を維持していた(図36)。加えて細胞は、フローサイトメトリー又はqRT−PCRのいずれかにより測定されるものとして、胚体内胚葉運命への分化能を維持していた(図37)。Cytodex 3(登録商標)マイクロキャリア上で継代し、胚体内胚葉へと分化させたH1及びH9ヒト胚性幹細胞の比較試験でも、同様の結果が得られた(図38)。
【0128】
これらの結果は、ヒト胚性幹細胞を、マイクロキャリア上で継代した後に、次いで多能性を維持しつつ他の表面上で培養することができることを示す。細胞を他の表面へと移して効率的に分化誘導することもできる。
【0129】
実施例9:ヒト胚性幹細胞は、有糸分裂を不活性化した線維芽細胞フィーダー上でのクラスター/コロニー形式の培養から、少なくとも10回の継代にわたる、ECM不含表面上での単独細胞としての培養へと、多能性を損なうことなく、及び線維芽細胞フィーダーを手動で除去することなく直接移行させることができる。
ヒト胚性幹細胞株は、現在のところ、単独の細胞が増殖したコロニー、又は胚盤胞由来の少数の細胞のクラスターを発展させる方法に由来する。次いでこのコロニーを連続的に継代し、細胞株を構成するのに十分な細胞クラスター/コロニーが利用できるようになるまで増殖させる。ヒト胚性幹細胞の多能性と核型安定特性を維持することを目的として、一度細胞株が誘導されると、高度に再現性のあるヒト胚性幹細胞培養のための、当該技術分野の現行の標準方法では、有糸分裂不活性化線維芽細胞のフィーダー細胞層上の、ヒト胚性幹細胞クラスター/コロニーを維持し、及び手作業での撹乱(manual disruption)又はコラゲナーゼ若しくは中性プロテアーゼ若しくはこれらのブレンドでの穏やかな酵素的大容量継代を用いて、細胞を継代する。これらの継代法はヒト胚性幹細胞クラスターを維持し、かつヒト胚性幹細胞の、コロニー形式の増殖を促進する。安定したヒト胚性幹細胞株の確立後、この細胞をMATRIGEL(商標)などの細胞外マトリックス(ECM)基質へと移すことができる。しかしながら、細胞を線維芽細胞フィーダー上で増殖させるかあるいはECM基質上で増殖させるかのいずれにせよ、技術者に具体的に指示されるヒト胚性幹細胞に関して推奨される継代法は、ヒト胚性幹コロニーを十分に解離させはしない。
【0130】
現行の最も良好な、哺乳類細胞のラージスケール培養の実施法は、3次元培養容器を使用するものである。この容器は恒常性、つまり一定の状態を厳密に維持し、かつ足場依存性の細胞を支持するためにマイクロキャリアを組み込むことができる。しかしながら、ヒト胚性幹細胞培養(線維芽細胞フィーダー上又はECM基質上で増殖させる)、及びクラスター/コロニー形式での培養に使用される現行の標準法は、マイクロキャリア上でのヒト多能性胚性幹細胞培養物の良好な増殖及び維持に対して技術的な障害を提起することになるが、それは、これらの方法をラージスケールでのマイクロキャリア培養に転用するのは容易ではないからである。マイクロキャリアでヒト胚性幹細胞を効果的に増殖させるためには、ヒト胚性幹細胞培養物は、当該技術分野の現行標準法でのようなコロニー又はクラスターとしてではなく、単独細胞として継代可能であるべきである。加えて、ヒト胚性幹細胞は、フィーダー細胞層又はECM基質無しに増殖可能であるべきである。
【0131】
我々は、これらの技術的障害に取り組む方法を以下に記載する。我々は、有糸分裂不活性化線維芽細胞フィーダー層上のクラスター/コロニーに基づくヒト胚性幹細胞培養を、線維芽細胞フィーダー層、又はMATRIGEL若しくは他の細胞外マトリックス基質でコートした表面が下層として存在する必要のない単独細胞培養系へと、直接転換する方法を実証する。この方法は、何らかの方法により線維芽細胞フィーダー細胞を手動で除去する必要のない、あるいは総細胞集団から多能性細胞を選別する必要のない、ヒト胚性幹の大容量の継代を活用することで、培養を、コロニー形式の線維芽細胞フィーダー系培養から、Rhoキナーゼ(ROCK)阻害剤のGlycyl−H 1152ジヒドロクロリドの存在下で、PRIMARIAでの、フィーダー不含/マトリックス不含の培養へと直接転換する。この方法は、必要量を制御するように付着させるための密閉容器で達成でき、並びに多能性及び胚体内胚葉への分化能を保持し、かつ線維芽細胞の細胞集団は含有しない、非常に均質なヒト胚性幹培養物を生成する。
【0132】
方法:培地を吸引し、PBSで洗浄し、次いで細胞を解離酵素(コラゲナーゼ,Accutase(商標),又はTrypLE)で処理することにより、細胞をルーチン的に継代した。コラゲナーゼは濃度1mg/mLで使用した;Accutase(商標)又はTrypLEは、1X貯蔵濃度で使用した。全ての酵素は室温に戻してから使用した。2% BSAのDMEM/F12溶液を各ウェルに添加し、細胞を酵素で処理した後に、この細胞を溶液に均一に懸濁した。次いで細胞を200gで5分間にわたって遠心し、細胞ペレットと、追加の、2% BSA含有DMEM/F12溶液とを、再懸濁した細胞に加え、この細胞懸濁液を50mLの滅菌コニカルチューブに分注して200gで5分間にわたって遠心した。
【0133】
我々は連続的な方法を用い、MEF系培地をAccutase(商標)、TrypLE(商標)、又はコラゲナーゼのいずれかで処理し、クラスター/コロニー形式のヒト胚性幹細胞をPrimaria表面に高密度継代することで、線維芽細胞フィーダーを除去した。最初の継代で、細胞を、マウス胚性線維芽細胞(MEF)馴化培地(CM)に1:30希釈したMATRIGEL(商標)でコートしたT−25フラスコに蒔くか、あるいは細胞を、T−25 PRIMARIA(商標)培養フラスコ中の、3μMのGlycyl−H 1152ジヒドロクロリドを添加したMEF−CMに蒔くかした。全ての細胞を1:3.5の分割比で蒔き、かつ細胞を10分間にわたって酵素に曝露した。TrypLE(商標)又はAccutase(商標)で浮かせた細胞の細胞数を、トリパンブルー染色した細胞を血球計算板を用いて計数することで判定した。細胞を蒔いた後、培地は毎日交換し、MEF−CM+3μMのGlycyl−H 1152ジヒドロクロリドに蒔いたこの細胞に、毎日MEF−CM+1μMのGlycyl−H 1152ジヒドロクロリドを供給し、多能性及び分化に関するmRNAマーカーの発現について、試料を解析した。マトリックス不含条件下で、単独細胞として2回継代したhESCsは、多能性遺伝子の遺伝子発現を維持し、かつ分化遺伝子の発現は阻害した(図39)。
【0134】
2回目の継代:TrypLE(商標)又はAccutase(商標)に10分曝露することで、細胞を分割比1:4で継代した。我々はまた、細胞を処理し、脱着についてモニターすることにより経験的に決定した、より短い酵素曝露時間を導入した。我々は、細胞を浮き上がらせるのには、TrypLE(商標)に対しては3分間の曝露、及びAccutase(商標)に対しては5分間の曝露で十分であることを観察した。酵素で細胞を処理した後に、細胞を上記のように継代し、細胞mRNAのアリコートを、継代時にqRT−PCR用に採取した。
【0135】
3回目の継代:コンフルエンスに達したら細胞をPBSで洗浄し、3分若しくは10分(TrypLE(商標))又は5分若しくは10分(Accutase(商標))かけて酵素によりバラバラにし、2%BSA含有DMEM/F12に懸濁し、遠心し、2%BSA含有DMEM/F12で再度洗浄し、遠心し、次いで再懸濁して各培地に蒔いた。この継代時には、細胞を1:4比で、並びに同様に更に2種の分割比(1:8及び1:16)で蒔いた。各継代時に、細胞mRNAのアリコートをqRT−PCR用に採取した。
【0136】
4回目以降の継代:2回目及び3回目の継代時に採用された、酵素への曝露時間と継代比についての条件を、4回目以降の継代でも維持した。培養物がコンフルエンスまで増殖したら、細胞をPBSで洗浄し、特定の時間をかけて酵素によりバラバラにし、2%BSA含有DMEM/F12に懸濁し、遠心し、2% BSA含有DMEM/F12で再度洗浄し、遠心し、次いで再懸濁して各培地に特定の蒔き込み比で蒔いた。PRIMARIAに細胞を蒔くための培地には、蒔く際に3μMのGlycyl−H 1152ジヒドロクロリドを添加した。細胞を蒔いた後、培地は毎日交換し、MEF−CM+3μMのGlycyl−H 1152ジヒドロクロリドに蒔いたこの細胞に、毎日MEF−CM+1μMのGlycyl−H 1152ジヒドロクロリドを供給した。継代時に、qRT−PCR用に細胞mRNAのアリコートを採取した。
【0137】
8回を超える継代時には、継代終了時に、多能性表面マーカーに関するフローサイトメトリーによって(図40)、並びに多能性マーカー及び分化マーカーに関するqRT−PCRによって(図41、42及び43)、多能性について細胞を解析した。2% BSAと、100ng/mLのアクチビンAと、20ng/mLのWnt3aと、8ng/mLのbFGFと、3μMのGlycyl−H 1152ジヒドロクロリドとを添加したRPMI培地で24時間にわたって細胞を処理することで、細胞を同様に胚体内胚葉へと分化させた。次いで培地を、2% BSA、100ng/mLのアクチビンA、8ng/mLのbFGF、及び3μMのGlycyl−H1152ジヒドロクロリドを添加したRPMI培地に交換し、更に48時間にわたって培地を毎日交換した。次に、胚体内胚葉へと分化させた試料を、フローサイトメトリーによって、胚体内胚葉マーカーCXCR4の存在について試験した(図40)。
【0138】
これらの結果は、コロニー形式の、線維芽細胞フィーダー系培養物から、Rhoキナーゼ(ROCK)阻害剤のGlycyl−H 1152ジヒドロクロリドの存在下の、PRIMARIAでのフィーダー不含/マトリックス不含培養への大容量の継代は、多能性及び胚体内胚葉への分化能を保持し、かつ線維芽細胞の細胞集団は含有しない非常に均質なヒト胚性幹細胞培養物を生じることを示す。
【0139】
実施例10:組織培養用プラスチックからマイクロキャリアへのヒト胚性幹細胞の移動
H1細胞を、PRIMARIA(商標)(カタログ番号353846,Becton Dickinson,Franklin Lakes,NJ)組織培養プレート(実施例9の方法)で培養し、TrypLE(商標)Expressにより3〜5分処理することで放出させ、10μMのY27632(Sigma−Aldrich,MO)添加MEF−CM中にCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)又はHILLEX(登録商標)II(Solohill,MI)マイクロキャリアを含む6ウェルの組織培養用無処理プレートに播種した。対照として、MATRIGEL(BD Biosciences,CA)でコートしたプレートで増殖させ、かつコラゲナーゼ(1mg/mL)を用いて継代した、継代数46のH1細胞を放出させ、同様の方法でマイクロキャリアに播種した。プレートを、37℃で5時間にわたって、45分ごとに手で撹拌しながらインキュベートした。次いでプレートを37℃にて固定式プラットフォーム上に設置した。培地は、毎日10μMのY27632(Sigma−Aldrich,MO)添加MEF−CMで交換した。画像解析は、3日目の時点で、マイクロキャリアに対する細胞の良好な付着を示す(図44)。7日後に細胞を放出させ(以下の実施例4に記載)、多能性マーカーのCD9、SSEA−4、SSEA−3、TRA−1−60、TRA−1−81に関してFACSで解析した(図45)。多能性マーカーのほとんどが、90〜100%の細胞で発現していた。Cytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)マイクロキャリアで増殖させようが、HILLEX(登録商標)II(Solohill,MI)マイクロキャリアで増殖させようが、Accutase(商標)(Millipore,MA)で継代した細胞とTrypLE(商標)Express(Invitrogen,CA)で継代した細胞の間に明瞭な違いは存在しない。全般的に、細胞は、PRIMARIA(商標)(カタログ番号353846,Becton Dickinson,Franklin Lakes,NJ)細胞培養用プラスチックからマイクロキャリアに移した場合に多能性を保持した。
【0140】
次に、マイクロキャリア上のこれらのH1細胞は、胚体内胚葉へと分化した。この方法は実施例7に記載する。分化の4日後に、H1細胞をマイクロキャリアから放出させ、FACS解析を実施したところ、82%を超える細胞がCXCR4を発現していることが示された。図46を参照されたい。細胞は、マイクロキャリアの種類又はPRIMARIA(商標)(カタログ番号353846,Becton Dickinson,Franklin Lakes,NJ)から継代する際の酵素には関係なく、胚体内胚葉へと効率的に分化した。これは増量系の柔軟性を証明し、かつプラスチック及びマイクロキャリア上にマトリックスを必要とすることなく細胞を増殖させる。
【0141】
実施例11:セルロース混合エステルからなる平面状基材からマイクロキャリアへのヒト胚性幹細胞の移動
米国特許出願第61/116,452号に開示の方法に従って、セルロース混合エステルからなる平面状基材で、H1細胞を12継代にわたって培養した。TrypLE(商標)Expressで3〜5分処理することで平面状基材から細胞を放出させ、10mMのY27632(Sigma−Aldrich,MO)添加MEF−CM中にCYTODEX 3(登録商標)(GE Healthcare Life Sciences,NJ)マイクロキャリア又はHILLEX(登録商標)II(Solohill,MI)マイクロキャリアを含む6ウェルの組織培養用無処理プレートに播種した。対照として、MATRIGEL(商標)(BD Biosciences,CA)でコートしたプレートで増殖させ、かつコラゲナーゼ(1mg/mL)を用いて継代した、継代数44のH1細胞を放出させ、同様の方法でマイクロキャリアに播種した。プレートを、37℃で5時間にわたって、45分ごとに手で撹拌しながらインキュベートした。次いでプレートを37℃にて固定式プラットフォーム上に設置した。培地は毎日交換した。7日後に細胞を放出させ(実施例4に記載)、多能性マーカーのCD9、SSEA−4、SSEA−3、TRA−1−60、TRA−1−81に関してFACSで解析した(図47)。ほとんどの多能性マーカーが、90%を超える細胞で発現していた。CYTODEX 3(登録商標)マイクロキャリアで増殖させた細胞と、HILLEX(登録商標)IIマイクロキャリアで増殖させた細胞との間に明瞭な違いは存在しなかった。継代数44のH1対照細胞は、多能性が他の実験により確認されていたことから、HILLEX(登録商標)IIマイクロキャリアで増殖させた後の多能性について試験しなかった(実施例5を参照されたい)。全般的に、細胞は、セルロース混合エステルからなる平面状基材からマイクロキャリアへと移した場合にも多能性を維持していた。
【0142】
次に、実施例7に記載の方法に従って、マイクロキャリア上のH1細胞を胚体内胚葉へと分化させた。分化の4日後に、H1細胞をマイクロキャリアから放出させ、FACS解析を実施したところ、65%を超える細胞がCXCR4を発現していることが示された(図48)。細胞は、マイクロキャリアの種類とは無関係に効率的に胚体内胚葉へと分化した。HILLEX(登録商標)II(Solohill,MI)マイクロキャリア上で胚体内胚葉へと分化した細胞の数は少数であったように思われる。細胞の分化能は、増量系の柔軟性を証明する。加えて細胞を、動物成分マトリックスに対する任意の必要性を除去して、メンブレン及びマイクロキャリア上に直接増殖させかつ分化させることができる。
【0143】
【表1】
【0144】
【表2】
【0145】
【表3】
【0146】
本明細書を通して引用された刊行物は、その全体が参照により本明細書に組み込まれる。以上、本発明の様々な態様を実施例及び好ましい実施形態を参照して説明したが、本発明の範囲は、上記の説明文によってではなく、特許法の原則の下で適切に解釈される以下の「特許請求の範囲」によって定義されるものである点は認識されるであろう。
【技術分野】
【0001】
本発明は、米国特許公開番号第61/116,447号(2008年11月20日出願)に対する優先権を請求する。
【0002】
(発明の概要)
本発明は、多能性幹細胞をマイクロキャリア上で増殖、増量(expansion)及び分化させるための方法を目的とする。
【背景技術】
【0003】
多能性幹細胞(例えば胚性幹細胞など)は、全ての成体型細胞へと分化する能力を有する。そのため、胚性幹細胞は、疾病、感染又は先天性異常の結果として損傷した器官のための、置換細胞源及び置換組織源になり得る。胚性幹細胞を置換細胞源として用いるのを可能にするには、試験管内で多能性を維持しつつ細胞を増殖することの困難さがネックになっている。
【0004】
現行の、未分化の胚性幹細胞を培養する方法では、例えばフィーダー細胞層の存在下で胚性幹細胞を培養するなどの、複雑な培養条件が必要とされる。あるいは、フィーダー細胞培養物に曝露することによって得られる培地を、胚性幹細胞の培養に使用する場合もある。これらの方法を用いる培養系では、多くの場合、培養される幹細胞の生物種とは異なる種から得られた細胞(異種細胞)が使用される。加えて、これらの培養系には動物の血清が添加される場合がある。
【0005】
胚性幹細胞は、研究及び薬剤スクリーニング用に将来性のある資源を提供する。現在のところ、ヒト胚性幹細胞株の大規模培養には問題が多く、大きな課題が提供される。現行の、インビトロでの多能性幹細胞増殖方法は、組織培養用フラスコにおいて、細胞外マトリックス(ECM)タンパク質又はフィーダー細胞でプレコートされた、平面状表面上で実施される。また、平面培養では、表面積が多能性幹細胞の長期増殖をサポートできないという限界性から、頻繁に継代培養する必要がある。マイクロキャリア系の多能性幹細胞培養法は、解決法を提供し得る。マイクロキャリアは表面積対容積比が大きく、したがって平面状表面上での、多能性幹細胞の増殖にまつわる表面積制限を排除する。
【0006】
例えば、Fokらは、未分化のESC−マイクロキャリアと凝集体の培養物を増殖させるための、撹拌懸濁培養系を開示する(Stem Cells 2005;23:1333〜1342.)。
【0007】
他の例では、Abranchesらは、表面にコラーゲン層を備えるデキストランマトリックスから構成された、ミクロ孔質のマイクロキャリアであるCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)が、スピナーフラスコ中でマウスS25 ES細胞株の増量を支持する能力について試験を開示する(Biotechnol.Bioeng.96(2007),pp.1211〜1221.)。
【0008】
他の例では、米国特許第20070264713号は、未分化の幹細胞を懸濁液中で培養するための方法、具体的には容器中のマイクロキャリア上で幹細胞を培養する方法について開示する。
【0009】
他の例では、国際公開第2006137787号は、細胞をマイクロキャリア上で培養するために使用される、マイクロタイタープレートなどの中実支持体に付着させたビーズなどの、微粒子状の物質すなわちマイクロキャリアを含む、スクリーニングツールを開示する。
【0010】
他の例では、国際公開第2008004990号は、培養時の幹細胞の付着、生存及び/又は増殖を促進する方法を開示し、この方法は、正電荷支持表面上で幹細胞を培養することを含む。
【0011】
他の例では、国際公開第2007012144号は、支持表面と、支持表面に結合させた合成付着性ポリペプチドと、を含むバイオリアクターを開示し、ここでこの合成付着性ポリペプチドは、胚性幹細胞又は多能性細胞への高い結合親和性により特徴付けられる。
【発明の概要】
【課題を解決するための手段】
【0012】
本発明は、多能性幹細胞をマイクロキャリア上で増殖、増量及び分化させる方法を提供する。
【0013】
一実施形態では、本発明は、多能性幹細胞の増殖方法を提供し、この方法は以下の
a.多能性幹細胞集団を、第1容量のマイクロキャリアに付着させる工程と、
b.多能性幹細胞を、第1容量のマイクロキャリア上で培養する工程と、
c.多能性幹細胞を、第1容量のマイクロキャリアから取り外す工程と、
d.多能性幹細胞集団を、第2容量のマイクロキャリアへと付着させる工程と、を含む。
【図面の簡単な説明】
【0014】
【図1】Rho−キナーゼ阻害剤は、マイクロキャリアへのヒト胚性幹細胞の付着と増殖を促進する。HILLEX(登録商標)IIマイクロキャリア(Solohill,MI)上で、2日間にわたって静置培養により増殖させた、H9細胞の画像。細胞は、10μMのRhoキナーゼ阻害剤、Y27632((Sigma−Aldrich,MO)を添加した、あるいは不添加の、マウス胚性線維芽細胞馴化培地(MEF−CM)で培養した(それぞれA及びB)。
【図2】マイクロキャリア上でのH9細胞の増殖。H9細胞を様々なマイクロキャリアに付着させ、37℃にて固定式プラットフォーム上に設置した。Plasticマイクロキャリアと、ProNectinFマイクロキャリアと、HILLEX(登録商標)IIマイクロキャリア(Solohill,MI)と、Plastic Plusマイクロキャリアを使用した(それぞれA、B、C、D)。3日間の増殖後に、細胞はHILLEX(登録商標)II(Solohill,MI)において、マイクロキャリアに対する最も良好な細胞付着性を示した。マイクロキャリアに付着せずに凝集塊を形成した細胞を矢印で示す。
【図3】マイクロキャリア上でのH9細胞の増殖。H9細胞を、37℃にて固定式プラットフォーム上の6ウェルディッシュ中で、10μMのY27632(Sigma−Aldrich,MO)及びMEF−CMの存在下で、HILLEX(登録商標)IIマイクロキャリア、ProNectinFマイクロキャリア、Plastic Plusマイクロキャリア及びPlasticマイクロキャリア(Solohill,MI)に付着させた。初期細胞播種密度は、0日での値である。3日目及び5日目の細胞数を示す。
【図4】マイクロキャリアに付着後のH1細胞の画像。ProNectinFマイクロキャリア、Plastic Plusマイクロキャリア、及びPlasticマイクロキャリアに付着させた細胞の3、5及び7日目の画像を示す。細胞は、37℃にて固定式プラットフォーム上の12ウェルディッシュ中で、10μMのY27632(Sigma−Aldrich,MO)を含有するMEF−CMで増殖させた。Plastic Plusマイクロキャリア及びPlasticマイクロキャリアに結合させた細胞は、独立して凝集塊を形成した(G、H中の矢印)。
【図5】マイクロキャリアに付着後のH1細胞の画像。Cytodex 1(登録商標)マイクロキャリア、Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)及びHILLEX(登録商標)IIマイクロキャリア(Solohill,MI)に付着させた細胞の3、5及び7日目の写真を示す。細胞は、37℃にて固定式プラットフォーム上の12ウェルディッシュ中で、10μMのY27632(Sigma−Aldrich,MO)を含有するMEF−CMで増殖させた。
【図6】マイクロキャリア上でのH1細胞の増殖。H1細胞を、HILLEX(登録商標)IIマイクロキャリア(Solohill,MI)、Cytodex 1(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)、Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)、ProNectinFマイクロキャリア(Solohill,MI)、Plastic Plusマイクロキャリア(Solohill,MI)、及びPlasticマイクロキャリア(Solohill,MI)に付着させ、37℃にて固定式プラットフォーム上の、10μMのY27632(Sigma−Aldrich,MO)及びMEF−CMが存在している12ウェルディッシュ中に設置した。初期細胞播種密度は、0日での値である。3、5及び7日目の細胞数を示す。最初の播種密度は、ラインにより示されるように13,333個細胞/cm2であった。
【図7】様々な濃度のRhoキナーゼ阻害剤存在下での、マイクロキャリア上でのH9細胞の増殖。固定式プラットフォーム上の、12ウェルプレート中で細胞を増殖させ、4日目及び7日目に計数して付着率と増殖率を決定した。A.H9細胞を1、2.5、5又は10μMのY27632(Sigma−Aldrich,MO)を含有するMEF−CM中で増殖させた。B.H9細胞を、0.5、1、2.5又は5μMのGlycyl−H 1152ジヒドロクロリド(Tocris,MO)含有MEF−CM中で増殖させた。
【図8】Rhoキナーゼ阻害剤濃度を減少させてH1細胞を増殖させた。2日間にわたって、濃度を減少させたY27632(Sigma−Aldrich,MO)又はGlycyl−H 1152ジヒドロクロリド(Tocris,MO)(10μM/5μM、2.5μM/0.5μM若しくは1.0μM/0.5μM)、又は0.25μMのGlycyl−H 1152ジヒドロクロリド(Tocris,MO)の存在下で、H1p38細胞を継続的に増殖させた。細胞はHILLEX(登録商標)II(Solohill,MI)、Cytodex 1(登録商標)、又はCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)に付着させた(それぞれA、B、C)。細胞を播種後3、5及び7日目に計数した。
【図9】スピナーフラスコ中での、異なった播種密度でのマイクロキャリアへの細胞付着の判定。H1細胞を、写真の左側に掲載した密度でCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)マイクロキャリア上に播種した;低密度(0.4×104個細胞/cm2)、中間密度(1.2×104個細胞/cm2)又は高密度(3×104個細胞/cm2)。3、5及び7日目に細胞を撮像し、細胞の付着したマイクロキャリアの百分率を判定した(画像に組み込んである)。
【図10】スピナーフラスコ中のマイクロキャリア上での細胞増殖は、初期播種密度により影響を受ける。H1細胞を、写真の左側に掲載した密度でCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)マイクロキャリア上に播種した;低密度(0.4×104個細胞/cm2)、中間密度(1.2×104個細胞/cm2)又は高密度(3×104個細胞/cm2)。3、5及び7日目で細胞をマイクロキャリアから解離させ計数した。
【図11】スピナーフラスコ中での、異なった播種密度でのマイクロキャリア上での細胞増殖率の判定。H1細胞を、異なる播種密度でCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)マイクロキャリア上に播種した;低密度(0.4×104個細胞/cm2)、中間密度(1.2×104個細胞/cm2)又は高密度(3×104個細胞/cm2)。3、5及び7日目で細胞をマイクロキャリアから解離させ計数した。細胞数の倍数増加を初期播種密度と比較して示す。
【図12】Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上で増殖させたH1細胞を培養の7日目以降に撮像した。細胞には3日目以降はRhoキナーゼ阻害剤不含MEF−CMを供給した。細胞はマイクロキャリアに付着したままであった。
【図13】H9細胞の増殖及びHILLEX(登録商標)IIマイクロキャリア(Solohill,MI)上のH9細胞の解離。A、B:HILLEX(登録商標)IIマイクロキャリア上(Solohill,MI)で6日間増殖させたH9細胞の10x及び20x画像。C:10分間にわたって0.05%トリプシン/EDTAでHILLEX(登録商標)IIマイクロキャリア(Solohill,MI)から解離させた細胞の20x画像。D:10分間にわたってTrypLE(商標)ExpressでHILLEX(登録商標)IIマイクロキャリア(Solohill,MI)から解離させた細胞の20x画像。
【図14】マイクロキャリアからのH9細胞の解離。固定式プラットフォーム上のHILLEX(登録商標)II(Solohill,MI)上で増殖させたH9細胞を、TrypLE(商標)Express又は0.05%トリプシン/EDTAで解離させた。細胞数及びそれらの生存率をそれぞれA及びBに示す。
【図15】マイクロキャリアからのH1細胞の解離。スピナーフラスコ中の、Cytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)上で増殖させたH1細胞を、TrypLE(商標)Express(Invitrogen,CA)、Accutase(商標)又はコラゲナーゼ(10mg/mL)で解離させた。細胞数及びそれらの生存率をそれぞれA及びBに示す。
【図16】HILLEX(登録商標)II(Solohill,MI)マイクロキャリア上で増殖させたH9細胞は、マイクロキャリア間を移動しなかった。
【図17】継代数43のH9を、スピナーフラスコ中の、HILLEX(登録商標)II(Solohill,MI)マイクロキャリア上で5継代にわたって増殖させた。細胞を2〜3日毎に計数し、細胞が1〜2×105個細胞/cm2に達した時点で継代した。
【図18】継代数43のH9細胞を、スピナーフラスコ中の、Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上で、5継代にわたって増殖させた。細胞を2〜3日毎に計数し、細胞が1〜2×105個細胞/cm2に達した時点で継代した。
【図19】蛍光標示式細胞分取(FACS)はスピナーフラスコ内で増殖させたH9細胞の多能性を示す。A:HILLEX(登録商標)II(Solohill,MI)マイクロキャリア上で増殖させた継代数43のH9細胞の大多数は多能性タンパク質を発現する。継代数1及び3の細胞は、TRA−1−81については評価しなかった。B:Cytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)マイクロキャリア上で増殖させた継代数43のH9細胞の大多数は多能性タンパク質を発現する。継代数1の細胞は、TRA−1−81については評価しなかった。
【図20】継代数49のH1細胞を、スピナーフラスコ中のCytodex 1(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上で、5継代にわたって増殖させた。細胞を2〜3日毎に計数し、細胞が4〜8×104個細胞/cm2に達した時点で継代した。
【図21】継代数49のH1細胞を、スピナーフラスコ中のCytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上で、5継代にわたって増殖させた。細胞を2〜3日毎に計数し、細胞が1〜2×105個細胞/cm2に達した時点で継代した。
【図22】蛍光標示式細胞分取(FACS)は、スピナーフラスコ内で増殖させたH1細胞の多能性を示す。
【図23】マイクロキャリア上のH1及びH9細胞の集団倍加。3日目から、以降経過させた日数についての集団倍加回数を算出した(5、6又は7日目)。
【図24】制限培地中のマイクロキャリア上でH9細胞を培養した。HILLEX(登録商標)II(HII,(Solohill,MI))又はCytodex 3(登録商標)(C3,(GE Healthcare Life Sciences,NJ))上で細胞を培養した。細胞を、以下の培地のいずれかで、マイクロキャリア上で培養した;mTESR(StemCell Technologies,Vancouver,Canada)、StemPro又はMEF−CM。10μMのY27632(Y,(Sigma−Aldrich,MO))又は2.5μMのGlycyl−H 1152ジヒドロクロリド(H,(Tocris,MO))を培地に加えた。播種後3、5及び7日目の増殖率を測定した。
【図25】制限培地中のマイクロキャリア上で、継代数38のH1細胞を培養した。HILLEX(登録商標)II(HII,(Solohill,MI))マイクロキャリア又はCytodex 3(登録商標)(C3,(GE Healthcare Life Sciences,NJ))マイクロキャリア上で細胞を培養した。細胞を、以下の培地のいずれかで、マイクロキャリア上で培養した;mTESR(StemCell Technologies,Vancouver,Canada)、StemPro及びMEF−CM。10μMのY27632(Y,(Sigma−Aldrich,MO))又は2.5μMのGlycyl−H 1152ジヒドロクロリド(H,(Tocris,MO))を培地に加えた。播種後3、5及び7日目の増殖率を測定した。
【図26】継代数50のH1細胞を、スピナーフラスコ中の、HILLEX(登録商標)II(Solohill,MI))マイクロキャリア上で、制限培地を用いて培養した。A:スピナーフラスコ中のMEF−CMで3、7又は9日間にわたって増殖させた後の、継代数50のH1細胞の画像。B:mTESR(StemCell Technologies,Vancouver,Canada)中で3、7又は9日間にわたって増殖させた継代数50のH1細胞の画像。矢印が指す細胞クラスターは、マイクロキャリアに付着していない。
【図27】スピナーフラスコ中で5回継代したヒト胚性幹細胞の分化。A:継代数43のH9細胞を、Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上で5回継代した。B:継代数49のH1細胞を、Cytodex 1(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上で5回継代した。両方の細胞型を共にマイクロキャリアから放出させ、MATRIGEL(BD Biosciences,CA)でコートしたプレートに播種した。80〜90%コンフルエントの細胞で、胚性幹細胞が胚体内胚葉へと分化し得るようなプロトコルを実施した。次いで胚体内胚葉マーカーのCXCR4を発現している細胞の百分率について、細胞をFACSで解析した。CXCR4陽性細胞のパーセントをプロットの右上の角に記載する。
【図28】マイクロキャリア上での、胚体内胚葉へのH1細胞の分化。このFACSプロットは、胚体内胚葉マーカーCXCR4を発現している細胞の百分率を示す。陽性パーセントを右上の角に記載する。処理前に全ての細胞をスピナーフラスコ中のマイクロキャリア上で増量させた。A:分化前に、継代数40のH1細胞を5回継代し、次いで6日間にわたってCytodex 1(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上で増殖させた。B:分化前に、継代数40のH1細胞を1回継代し、次いで8日間にわたってCytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上で増殖させた。C:分化前に、継代数50のH1細胞を1回継代し、次いで6日間にわたってHILLEX(登録商標)IIマイクロキャリア(Solohill,MI)上で増殖させた。
【図29】Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上での、胚体内胚葉へのH1細胞の分化。A:継代数40のH1細胞を、マイクロキャリア上で8日間にわたって増殖させた。B:継代数40のH1細胞を、マイクロキャリア上で11日間にわたって増殖させた。両方の細胞集団を共に、次いで37℃にて固定式プラットフォーム上で、胚体内胚葉へと分化させた。このFACSプロットは、胚体内胚葉マーカーCXCR4を発現している細胞の百分率を示す。陽性パーセントを右上の角に記載する。
【図30】胚体内胚葉への、マイクロキャリア上で培養したヒト胚性幹細胞株H1細胞の分化。CXCR4陽性細胞パーセントについてのFACS結果をY軸に示す。H1細胞は、分化前及び分化時にHILLEX(登録商標)II、Cytodex 1(登録商標)又はCytodex 3(登録商標)マイクロキャリア上で増殖させた。
【図31】膵臓内胚葉細胞への、マイクロキャリア上で培養したヒト胚性幹細胞株H1細胞の分化。CT値を、膵臓内胚葉マーカーのNgn3、Nkx6.1及びPdx1についてY軸に示す。H1細胞を、DMEM−高グルコース(HG)培地又はDMEM−F12(F12)培地のいずれかの培地中で、HILLEX(登録商標)II(HII)、Cytodex 1(登録商標)(C1)又はCytodex 3(登録商標)(C3)マイクロキャリア上で分化させた。分化プロトコルは13日間続いた。
【図32】膵臓ホルモン産生細胞への、マイクロキャリア上で培養したヒト胚性幹細胞株H1細胞の分化。FACSにより測定した陽性細胞パーセントを、膵臓ホルモンの細胞マーカーのシナプトフィジン、グルカゴン及びインスリンについてY軸で示す。H1細胞を2つの異なる濃度、10×105(10)又は20×105(20)で、Cytodex 3(登録商標)(C−3)マイクロキャリア上に播種した。DMEM−高グルコース(HG)中で、4〜9日間にわたって細胞を分化させ、更にHG又はDMEM−F12(F12)培地で10日〜24日間分化させた。
【図33】Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上での、内分泌細胞へのH1細胞の分化。H1細胞を、膵臓内胚葉(14日)を経て、膵内分泌細胞へと分化させ、膵内分泌細胞(21日)をインスリン発現細胞(28日)へと分化させた。Pdx1、グルカゴン及びインスリンの遺伝子発現レベルを測定した(それぞれA、B、C)。Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)(C3)上で増殖、分化させたH1細胞を、MATRIGEL(BD Biosciences,CA)コートした6ウェルディッシュ(平面)上で増殖、分化させた細胞と比較した。Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上で増殖させた細胞についての遺伝子発現の値は3つ組で採った。
【図34】Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上での、胚体内胚葉(DE)へのH9細胞の分化。CXCR4発現のFACSプロット。胚体内胚葉マーカーCXCR4についての陽性細胞パーセントを、右上の角に記載する。A:継代数39のH9細胞を、MATRIGEL(BD Biosciences,CA)コートした6ウェルディッシュ上で増殖させ、DEへと分化させた。B、C:スピナーからのCytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上のH9細胞の、2つ組サンプルを、12ウェルディッシュに設置し、固定式プラットフォーム上でインキュベートした。
【図35】Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上での、インスリン発現細胞へのH9細胞の分化。H9細胞を、膵臓内胚葉(14日)を経て、膵内分泌細胞へと分化させ、内分泌細胞(22日)をインスリン発現細胞(29日)へと分化させた。Pdx1、グルカゴン及びインスリンの遺伝子発現レベルを測定した(それぞれA、B、C)。Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)(C3)上で増殖、分化させたH9細胞を、MATRIGEL(BD Biosciences,CA)コートした6ウェルディッシュ(平面)上で増殖、分化させた細胞と比較した。
【図36】Cytodex 3(登録商標)マイクロキャリア上で5継代にわたって培養し、次いで記載の平面状基材上に移して培養し、Rhoキナーゼ阻害剤の存在下で培養したヒト胚性幹細胞での多能性の維持性。パネルAは、フローサイトメトリーにより検出されたものとして、多能性マーカーCD9、SSEA3、SSEA4、Tra−160及びTra−181の発現を記す。パネルBは、リアルタイムPCRにより検出されたものとして、多能性マーカーNanog、Pou5F1、SOX2及びZFP42、並びに分化マーカーFOXA2、FOXD3、GATA2、GATA4及びBrachyuryの発現を記す。
【図37】Cytodex 3(登録商標)マイクロキャリア上で5継代にわたって培養し、次いで記載の平面状基材上に移して培養し、Rhoキナーゼ阻害剤の存在下で培養したヒト胚性幹細胞による、胚体内胚葉形成。パネルAは、フローサイトメトリーにより検出されたものとして、CXCR4の発現を記す。パネルBは、リアルタイムPCRにより検出されたものとして、記載のマーカーの発現を記す。
【図38】ヒト胚性幹細胞をCytodex 3(登録商標)マイクロキャリア上で5継代にわたって培養し、次いでPRIMARIA(商標)平面状基材上に移し培養することによる胚体内胚葉形成。記載の遺伝子の発現はフローサイトメトリーにより測定した。
【図39】平面状基材上で培養したヒト胚性幹細胞は多能性を維持する。TrypLE(商標)、Accutase(商標)又はコラゲナーゼ処理したH1ヒトES細胞由来のmRNAサンプルを回収し、mRNAの多能性遺伝子発現について解析した。細胞を、1継代の4日間にわたって、MATRIGELにてMEF馴化培地で培養して増殖させ(A)、あるいは1継代にわたって、Primaria(商標)にて、Rock阻害剤を添加したMEF馴化培地で増殖させ(B)、あるいは2継代にわたって、Primaria(商標)にて、Rock阻害剤を添加したMEF馴化培地で増殖させた(C)。
【図40】Rhoキナーゼ阻害剤のGlycyl−H 1152ジヒドロクロリドの存在下で、PRIMARIAにてAccutase(商標)又はTrypLE(商標)を用いて分割比1:4、1:8又は1:16で継代して、7継代を超えてPRIMARIAにて増殖させたH1ヒト胚性幹細胞(継代数45を超える)を、多能性(A)及び胚体内胚葉への分化能(B)について試験した。対照は、1:30のMATRIGELにて増殖させ、コラゲナーゼで処理した、継代数48のH1ヒト胚性幹細胞である。10mA=Accutase(商標)に10分曝露して継代。10mT=TrypLE(商標)に10分曝露して継代。1:4、1:8又は1:16は継代比を意味する。P(X)は、MEFフィーダーからPrimaria(商標)プラスチックへと移動させて以降の継代数を意味する。
【図41】Rhoキナーゼ阻害剤のGlycyl−H 1152ジヒドロクロリドの存在下で、Accutase(商標)又はTrypLE(商標)を用いてPRIMARIAにて1:4比で継代して、7継代を超えてPRIMARIAにて増殖させたH1ヒト胚性幹細胞(継代数45を超える)を、多能性マーカー及び分化マーカーのmRNA発現について試験した。対照は、継代数37の細胞集団で開始する。10分Accutase(商標)=Accutase(商標)に10分曝露して継代。P(X)は、MEFフィーダーからPRIMARIA(商標)プラスチックへと移動させて以降の継代数を意味する。
【図42】Rhoキナーゼ阻害剤のGlycyl−H 1152ジヒドロクロリドの存在下で、Accutase(商標)又はTrypLE(商標)を用いてPRIMARIAにて分割比1:8で継代して、7継代を超えてPRIMARIA(商標)にて増殖させたH1ヒト胚性幹細胞(継代数45を超える)を、多能性マーカー及び分化マーカーのmRNA発現について試験した。対照は、継代数37の細胞集団で開始する。10分Accutase(商標)=Accutase(商標)に10分曝露して継代。P(X)は、MEFフィーダーからPRIMARIA(商標)プラスチックへと移動させて以降の継代数を意味する。
【図43】Rhoキナーゼ阻害剤のGlycyl−H 1152ジヒドロクロリドの存在下で、Accutase(商標)又はTrypLE(商標)を用いてPRIMARIAにて分割比1:16で継代して、7継代を超えてPRIMARIA(商標)にて増殖させたH1ヒト胚性幹細胞(継代数45を超える)を、多能性マーカー及び分化マーカーのmRNA発現について試験した。対照は、継代数37の細胞集団で開始する。10分Accutase(商標)=Accutase(商標)に10分曝露して継代。P(X)は、MEFフィーダーからPRIMARIA(商標)plasticへと移動させて以降の継代数を意味する。
【図44】Primaria(商標)平面状基材(カタログ番号353846,Becton Dickinson,Franklin Lakes,NJ)にて増殖させ、次いで播種の3日後にマイクロキャリアに移動させた、H1細胞の画像。A〜C:H1細胞をCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)マイクロキャリア上に播種した。D〜F:細胞をHILLEX(登録商標)II マイクロキャリア(Solohill,MI)上に播種した。A、D:H1細胞は、マイクロキャリアへと移す前に、10分間のTrypLE(商標)Express(Invitrogen,CA)処理により、Primaria(商標)平面状基材(カタログ番号353846,Becton Dickinson,Franklin Lakes,NJ)プレートにて継代した。A、E:H1細胞は、マイクロキャリア上に移す前に、10分間のAccutase(商標)処理を用いて、Primaria(商標)平面状基材(カタログ番号353846,Becton Dickinson,Franklin Lakes,NJ)プレートにて継代した。C、F:継代数46のH1細胞は、マイクロキャリアへと移す前に、コラゲナーゼ(1mg/mL)を用いて、MATIRGEL(BD Biosciences,CA)コートプレートにて継代した。
【図45】Primaria(商標)平面状基材(カタログ番号353846,Becton Dickinson,Franklin Lakes,NJ)にて増殖させ、Cytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)マイクロキャリア及びHILLEX(登録商標)IIマイクロキャリアへと移したH1細胞の多能性。FACS解析は、多能性細胞表面タンパク質の発現を示す。細胞を、Primaria(商標)(カタログ番号353846,Becton Dickinson,Franklin Lakes,NJ)での継代時に3〜10分間にわたってAccutase(商標)又はTrypLE(商標)Express(Invitrogen,CA)で処理した。
【図46】Primaria(商標)(カタログ番号353846,Becton Dickinson,Franklin Lakes,NJ)にて増殖させ、次いでCytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)へと移したH1細胞の分化。胚体内胚葉マーカーCXCR4の細胞表面発現についてのFACS解析。細胞を、Primaria(商標)(カタログ番号353846,Becton Dickinson,Franklin Lakes,NJ)での継代時に3〜10分間にわたってAccutase(商標)又はTrypLE(商標)Express(Invitrogen,CA)で処理した。
【図47】マイクロキャリア上での培養前にセルロース混合エステルからなる平面状基材にて培養したヒト胚性幹細胞のFACS解析。
【図48】マイクロキャリア上での培養及び分化の前に、セルロース混合エステルからなる平面状基材で培養したヒト胚性幹細胞の、胚体内胚葉系に特徴的なマーカーの発現に関するFACS解析。
【発明を実施するための形態】
【0015】
開示を明確にするために、本発明の「発明を実施するための形態」を、限定を目的とすることなく、本発明の特定の特徴、実施形態、又は応用を説明若しくは図示した以下の小項目に分ける。
【0016】
定義
幹細胞は、単一の細胞レベルで自己複製し、分化して後代細胞を生成する、それら両方の能力で定義される未分化細胞であり、後代細胞には、自己複製前駆細胞、非再生前駆細胞、及び最終分化細胞が含まれる。同様に、幹細胞は、インビトロで複数の胚葉(内胚葉、中胚葉及び外胚葉)から様々な細胞系の機能細胞へと分化する能力によって、並びに移植後に複数の胚葉の組織を生じ、胚盤胞への注入後、全てではないとしても殆どの組織を提供する能力によっても特徴付けられる。
【0017】
幹細胞は、それらの発達能力により:(1)全胚及び胚体外細胞型を生じる能力を意味する全能性、(2)全胚細胞型を生じる能力を意味する多能性、(3)細胞系統の小集合を生じるが、全て特定の組織、器官又は生理的システム内で生じる能力を有することを意味する多能性(例えば、造血幹細胞(HSC)は、HSC(自己複製)、血液細胞に限定された寡能性前駆細胞、並びに血液の通常の構成要素である全細胞型及び要素(例えば、血小板)を含む子孫を産生できる)、(4)多能性幹細胞と比較して限定された細胞系統の小集合を生じる能力を有することを意味する寡能性、並びに(5)1つの細胞系統を生じる能力を有することを意味する単能性(例えば、精子形成幹細胞)に分類される。
【0018】
分化は、特殊化されていない細胞(「中立の」)又は比較的特殊化されていない細胞が、例えば、神経細胞又は筋細胞などの特殊化した細胞の特徴を獲得するプロセスである。分化した、又は分化誘導された細胞は、細胞系内でより特殊化した(「コミットした」)状況を呈している細胞である。分化プロセスに適用した際の用語「コミットした」は、通常の環境下で特定の細胞型又は細胞型の小集合へと分化し続ける分化経路の地点に進行しており、通常の環境下で異なる細胞型に分化し、又はより分化されていない細胞型に戻ることができない細胞を指す。脱分化は、細胞が細胞系内で比較的特殊化されて(又は傾倒して)いない状況に戻るプロセスを指す。本明細書で使用するとき、細胞系は、細胞の遺伝、すなわちその細胞がどの細胞から来たか、またどの細胞を生じ得るかを規定する。細胞系は、細胞を発達及び分化の遺伝的スキーム内に設置する。系特異的なマーカーは、対象とする系の細胞の表現型に特異的に関連した特徴を指し、中立細胞の対象とする系への分化を評価する際に使用することができる。
【0019】
培養時の細胞を記載するために、様々な用語が使用される。「維持」は、一般に、細胞増殖及び/又は細胞分裂を促進する条件下で増殖培地に設置した細胞を指し、この細胞は細胞数のより大きな集団をもたらす場合ももたらさない場合もある。「継代」は、細胞増殖及び/又は細胞分裂を促進する条件下で、細胞を1番目の培養容器から取り出し、それらの細胞を2番目の培養容器へと設置するプロセスを指す。
【0020】
特定の細胞集団又は細胞株は、しばしば継代された回数によって呼称されるか特徴付けられる。例えば、10回継代された培養細胞集団はP10培養と呼ばれる場合がある。初代培養、すなわち組織から細胞を単離した後の最初の培養はP0と称される。最初の継代培養後、細胞は二次培養物(P1又は継代数1)と称される。2回目の継代培養の後では細胞は三次培養物(P2又は継代数2)となる、といった具合である。当業者であれば、継代期間中に集団は何度も倍加し得るものであり、したがってある培養の集団倍加の回数は継代数よりも大きいことが理解されるであろう。各継代間の期間中の細胞の増量(すなわち集団倍加数)は、限定するものではないが、播種密度、基質、培地、増殖条件、及び継代間の時間などを含む多くの因子に依存する。
【0021】
「β−細胞系」は、転写因子PDX−1と、以下の転写因子:NGN−3、Nkx2.2、Nkx6.1、NeuroD、Isl−1、HNF−3 β、MAFA、Pax4、又はPax6のうちの少なくとも1つに関する遺伝子の発現が陽性である細胞を指す。β細胞系に特徴的なマーカーを発現する細胞としては、β細胞が挙げられる。
【0022】
本明細書で使用するとき、「胚体内胚葉系に特徴的なマーカーを発現している細胞」は、以下のマーカー:SOX−17、GATA−4、HNF−3 β、GSC、Cer1、Nodal、FGF8、Brachyury、Mix様ホメオボックスタンパク質、FGF4 CD48、eomesodermin(EOMES)、DKK4、FGF17、GATA−6、CXCR4、C−Kit、CD99又はOTX2のうちの少なくとも1つを発現している細胞を指す。胚体内胚葉系に特徴的なマーカーを発現している細胞には、原始線条前駆体細胞、原始線条細胞、中内胚葉細胞及び胚体内胚葉細胞を含む。
【0023】
本明細書で使用するとき、「膵臓内胚葉系に特徴的なマーカーを発現している細胞」は、以下のマーカーのうちの少なくとも1つを発現している細胞を指す:PDX−1、HNF−1β、PTF−1 α、HNF−6、又はHB9。膵臓内胚葉系に特徴的なマーカーを発現している細胞には、膵臓内胚葉細胞を含む。
【0024】
本明細書で使用するとき、「膵内分泌系に特徴的なマーカーを発現している細胞」は、以下のマーカーのうちの少なくとも1つを発現している細胞を指す:NGN−3、NeuroD、Islet−1、PDX−1、NKX6.1、Pax−4、Ngn−3、又はPTF−1 α。膵内分泌系に特徴的なマーカーを発現している細胞には、膵臓内分泌細胞、膵臓ホルモン発現細胞、及び膵臓ホルモン分泌細胞、並びにβ−細胞系の細胞を含む。
【0025】
本明細書で使用するとき、「胚体内胚葉」は、原腸形成中、胚盤葉上層から生じ、胃腸管及びその誘導体を形成する細胞の特徴を有する細胞を指す。胚体内胚葉細胞は、以下のマーカー、すなわちCXCR4、HNF−3 β、GATA−4、SOX−17、Cerberus、OTX2、goosecoid、c−Kit、CD99、及びMixl1を発現する。
【0026】
本明細書で使用するとき、「胚体外内胚葉」は、以下のマーカーのうちの少なくとも1つを発現している細胞の集団を指す:SOX−7、AFP又はSPARC。
【0027】
本明細書で使用するとき、「マーカー」は、対象とする細胞内で差異的に発現される核酸又はポリペプチド分子である。本文脈において、差異的な発現は、陽性マーカーのレベルの増大及び陰性マーカーのレベルの減少を意味する。検出可能なレベルの核酸マーカー又はポリペプチドマーカーは、他の細胞と比較して対象とする細胞内で十分高く又は低く、そのため当該技術分野において既知の多様な方法のいずれかを使用して、対象とする細胞を他の細胞から識別及び区別することができる。
【0028】
本明細書で使用するとき、「中内胚葉細胞」とは、以下のマーカーのうちの少なくとも1つを発現している細胞を指す:CD48、eomesodermin(EOMES)、SOX−17、DKK4、HNF−3 β、GSC、FGF17又はGATA−6。
【0029】
本明細書で使用するとき「膵内分泌細胞」又は「膵臓ホルモン発現細胞」とは、以下のホルモンのうちの少なくとも1つを発現することが可能な細胞を指す:インスリン、グルカゴン、ソマトスタチン、及び膵臓ポリペプチド。
【0030】
本明細書で使用するとき、「膵臓ホルモン分泌細胞」は、以下のホルモンのうちの少なくとも1つを分泌できる細胞を指す:インスリン、グルカゴン、ソマトスタチン、及び膵臓ポリペプチド。
【0031】
本明細書で使用するとき、「前原始線条細胞」は、以下のマーカーのうちの少なくとも1つを発現している細胞を指す:Nodal、又はFGF8。
【0032】
本明細書で使用するとき、「原始線条細胞」は、以下のマーカーのうちの少なくとも1つを発現している細胞を指す:Brachyury、Mix様ホメオボックスタンパク質、又はFGF4。
【0033】
マイクロキャリア
「マイクロキャリア」は、足場依存性の細胞を培養する際に、この細胞の付着及び増殖に有用な粒子、ビーズ又はペレットを指す。「マイクロキャリア」は以下の特性を有する:(a)(マイクロキャリア又は細胞に、有意なせん断ダメージを引き起こさないような撹拌速度での)懸濁培養で使用可能な程度に十分小さい;(b)中実である、あるいは表面に多孔性コーティングを備える中実コアを有している;及び(c)表面(多孔性キャリアの場合、外側表面と内側表面)が正又は負に帯電し得る。一態様では、マイクロキャリアは全般的に約150〜350μmの粒径を有し、かつ約0.8〜2.0meq/gの正電荷密度を有する。有用なマイクロキャリアとしては、限定するものではないが、Cytodex 1(登録商標)、Cytodex 2(登録商標)、又はCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)が挙げられる。
【0034】
他の態様では、マイクロキャリアは中実キャリアである。中実キャリアは、接着細胞(例えば、付着依存性の細胞)に特に好適である。キャリア粒子はまた、多孔性マイクロキャリアであってもよい。
【0035】
「多孔性マイクロキャリア」は、付着依存性の細胞を培養する際に、この細胞の付着及び増殖に有用な粒子を指す。多孔性マイクロキャリアは以下の特性を有する:(a)(マイクロキャリア又は細胞に、有意なせん断ダメージを引き起こさないような撹拌速度での)懸濁培養で使用可能な程度に、十分小さい;(b)細胞を粒子の内部空間へと遊走させるのに十分な大きさの孔と内部空間とを有する、及び(c)(外側及び内側)表面が正又は負に帯電し得る。一連の実施形態の1つでは、キャリアは(a)全般的に約150〜350μmの粒径を有し;(b)約15〜約40μmの平均孔開口径を有する孔を有し;及び(c)約0.8〜2.0meq/gの正電荷密度を有する。いくつかの実施形態では、正電荷はDEAE(N,N,−ジエチルアミノエチル)基により提供される。有用な多孔性マイクロキャリアとしては、限定するものではないが、Cytopore 1(登録商標)及びCytopore 2(登録商標)(GE Healthcare Life Sciences,Piscataway N.J.)が挙げられる。マイクロキャリアは任意の形状であってよいが、通常はほぼ球状の形状であり、かつマクロ若しくはマイクロ多孔性、又は中実性であり得る。
【0036】
多孔型及び中実型のマイクロ粒子キャリアのいずれもが供給元より市販されている。市販のマイクロキャリアの例としてはCytodex 1(登録商標)及びCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)が挙げられ、これらの両方がGE Healthcare Life Sciencesのデキストラン系マイクロキャリアである。市販の多孔性マイクロキャリアとしては、同様にGE Healthcare Life Sciencesの、Cytoline製品並びにCytopore製品が挙げられる。Biosilon(NUNC)及びCultispher(Percell Biolytica)もまた市販されている。更なる態様では、マイクロキャリアはポリカーボネート又はセルロース混合エステルから構成され、あるいはこれらでコートされ得る。
【0037】
本発明での使用に好適なマイクロキャリアは、天然由来の又は合成的に誘導された材料から構成され得る。例としては、コラーゲン系マイクロキャリア、デキストラン系マイクロキャリア、又はセルロース系マイクロキャリア、並びにガラス、セラミックス、ポリマー、又は金属が挙げられる。マイクロキャリアはタンパク質不含であっても、あるいはタンパク質(例えばコラーゲン)でコートされていてもよい。更なる態様では、マイクロキャリアは、マイクロキャリアへの細胞の結合性を高める、及びマイクロキャリアからの細胞の放出を高める化合物から構成されてもよく、あるいは化合物でコートされてもよく、限定するものではないが、このような化合物としてはポリ(モノステアロイルグリセリドココハク酸)、ポリ−D,L−ラクチド−コ−グリコリド、ヒアルロン酸ナトリウム、コラーゲン、フィブロネクチン、ラミニン、エラスチン、リジン、n−イソプロピルアクリルアミド、ビトロネクチンが挙げられる。
【0038】
細胞培養物用のマイクロキャリア
マイクロキャリア培養は、足場依存性の細胞(例えばヒト胚性幹細胞)の実用的な高収率培養を可能にする技術である。マイクロキャリアは、特に数ミリリットル〜1000Lを超える範囲の培養容量でヒト胚性幹細胞などの細胞を培養するために開発されてきた。マイクロキャリアは生物学的に不活性であり、撹拌式マイクロキャリア培養法に、丈夫であるが非剛性の基材を提供する。マイクロキャリアは透明であり得、付着した細胞の顕微鏡検査が可能である。Cytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)は、架橋デキストランマトリックスに化学結合させた、変性コラーゲンの薄い層から構成される。Cytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)の変性コラーゲン層は、トリプシン及びコラゲナーゼが挙げられる、様々なプロテアーゼにより消化されやすく、細胞の生存率、機能及び完全性を最大限維持しつつ、細胞をマイクロキャリアから取り外すという性能を提供する。
【0039】
タンパク質不含マイクロキャリアをヒト胚性幹細胞の培養に使用することもできる。例えば、商品名HILLEX(登録商標)(SoloHill Engineering,Inc.,MI.)で販売されている、製造及び研究又は調査用途に使用するためのマイクロキャリアは、表面に、マイクロキャリアに正電荷表面をもたらすカチオン性トリメチルアンモニウムが付着している、改質ポリスチレンビーズである。ビーズ直径範囲は、直径約90〜約200マイクロメートルである。
【0040】
マイクロキャリアに基づく細胞培養法は、多くの用途において、下流の加工のし易さを含む、多くの利点を提供した。マイクロキャリアは通常ほぼ球状の形状であり、かつ多孔性であるか中実であるかのいずれかであり得る。細胞付着用のマイクロキャリアの使用は、付着依存性の細胞の増殖を目的とする撹拌槽及び関連するリアクターの使用を容易にした。細胞は、容易に懸濁されたマイクロキャリアに付着した。懸濁可能である必要があることから、マイクロキャリアの物理的パラメーターは制限される。したがって、マイクロキャリアは一般には50〜2000マイクロメートルの範囲の中央粒径を有する。一部の用途では、中実型のマイクロキャリアが約100〜約250マイクロメートルの範囲である一方で、多孔型のマイクロキャリアは約250〜約2500マイクロメートルの範囲である。これらの寸法範囲は、多くの付着依存性の細胞を収容するのに十分な程度に大きく、かつ撹拌リアクター内での使用に好適な特性を有する懸濁液を形成するのに十分小さいマイクロキャリアの選別を可能にする。
【0041】
マイクロキャリア及び同様物などを使用するに当たり考慮される因子は:付着効率、免疫原性、生体適合性、生分解能、コンフルエントに到達するまでの時間、単位表面積あたりの最大付着可能密度が挙げられる付着細胞の増殖パラメーター、必要とされる箇所での脱着技術、及び脱着効率、培養条件の拡張性並びにスケールアップ条件下での培養均一性、スケールアップ脱着手順を成功裏に実施する能力、及びマイクロキャリアが移植に使用可能かどうか、である。これらの検討事項は、マイクロキャリアの表面特性、並びにマイクロキャリアの多孔性、直径、密度及び操作特性により影響を受け得る。
【0042】
例えば、マイクロキャリアの密度は検討事項である。密度が過剰だと、マイクロキャリアが懸濁液から沈殿する場合があり、あるいは培養容器の底に完全に沈殿したままである傾向があり、ひいてはリアクター内で混合する細胞、培養培地及びガス相の大容量性が乏しくなる場合がある。一方で、密度が低すぎる場合には、マイクロキャリアに過剰な流動性が生じ得る。1.02〜1.15g/cm3の密度が、多くのマイクロキャリアについて一般的である。
【0043】
リアクターに加えることのできる、マイクロキャリアの小さな直径と粒子容量は、マイクロキャリアに、ローラーボトル法又は付着依存性の細胞を増殖させるための他の方法(例えばプレート法)で見出されるものよりもはるかに大きい実質表面積を提供させる。多孔性マイクロキャリアは、単位容量又は単位重量当たりで更に大きい表面積を提供する。これらの多孔性マイクロキャリアは、付着依存性の細胞の増殖に利用可能な大きい空洞を保有する。これらの空洞は表面積を非常に増加させ、例えば混合時又はガス噴霧時のせん断ストレスなどのような、有害な機械的影響から細胞を保護し得る。
【0044】
マイクロキャリア表面は、細胞付着性と細胞増殖性を高めるためにテクスチャ加工処理することができる。マイクロキャリア表面のテクスチャ加工は、限定するものではないが、型成形、鋳造、リーチング及びエッチングが挙げられる技術によりなされる。テクスチャ加工表面の特徴の解像度(resolusion)は、ナノスケールであり得る。テクスチャ加工表面は、マイクロキャリア表面上に特異的な細胞設置を誘導するために使用することもできる。多孔性マイクロキャリア内の孔表面も同様に、細胞付着性と細胞増殖性を高めるためにテクスチャ加工することができる。孔表面のテクスチャ加工は、限定するものではないが、例えば型成形、鋳造、リーチング及びエッチングなどの技術によりなされる。
【0045】
マイクロキャリア表面は、マイクロキャリア表面に特異的な電荷を付与するためにプラズマコートされていてもよい。これらの電荷は、細胞付着性と細胞増殖性を高め得る。
【0046】
他の実施形態では、マイクロキャリアは、ポリ−N−イソプロピルアクリルアミドなどの熱応答性ポリマーから構成され、又はこのようなポリマーでコートされ、すなわち電気機械的特性を有する。
【0047】
多孔性でかつ中実型のマイクロ粒子キャリアのいずれもが供給元より市販されている。市販の中実マイクロキャリアの例としてはCytodex 1(登録商標)及びCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)が挙げられ、これらの両方がGE Healthcare Life Sciencesのデキストラン系マイクロキャリアである。市販の多孔性マイクロキャリアとしては、同様にGE Healthcare Life Sciencesの、Cytoline製品並びにCytopore製品が挙げられる。Biosilon(NUNC)及びCultispher(Percell Biolytica)もまた市販されている。
【0048】
マイクロキャリアには、生物活性剤を含有させることもできる。マイクロキャリアにはまた、細胞の増殖若しくは機能、又は組織環境を制御し得る生物活性剤を含有させることができ、限定するものではないがこれらの因子としては線維芽細胞増殖因子、エリスロポエチン、血管内皮細胞増殖因子、血小板由来増殖因子、骨形成タンパク質、トランスフォーミング増殖因子、腫瘍壊死因子、上皮増殖因子、インスリン様増殖因子が挙げられる。完全な因子、模倣体又はこれらの活性断片を使用することができる。
【0049】
マイクロキャリアに第2の細胞種を播種して、多能性幹細胞と共培養することもできる。一実施形態では、2種(又はそれ以上)の細胞型は、個々のマイクロキャリアに均一な割合で又は不均一な割合で付着し得る。2種以上の細胞種を、マイクロキャリア上に同時に播種することができ、あるいはそれらを異なる時点で播種することもできる。特異的な細胞型をマイクロキャリアの特異的な領域へと優先的に付着させるような様式で、マイクロキャリアを処理することができる。更なる実施形態では、単一の又は複数の細胞種が付着しているマイクロキャリアを、培養容器にて、懸濁液の状態で培養された第2の細胞種と共培養することができる。
【0050】
例えば第2の細胞種としては、上皮細胞(例えば口腔粘膜細胞、胃腸管細胞、鼻粘膜上皮細胞、呼吸器上皮細胞、膣上皮細胞、角膜上皮細胞)、骨髄細胞、脂肪細胞、幹細胞、ケラチノサイト、メラニン細胞、皮膚線維芽細胞、ケラチノサイト、血管内皮細胞(例えば大動脈内皮細胞、冠動脈内皮細胞、肺動脈内皮細胞、腸骨動脈内皮細胞、微小血管内皮細胞、臍動脈内皮細胞、臍静脈内皮細胞及び血管内皮前駆細胞(例えばCD34+、CD34+/CD117+細胞))、筋芽細胞、筋細胞、肝細胞、平滑筋細胞、横紋筋細胞、ストローマ細胞、及び他の軟組織細胞又は前駆細胞、軟骨細胞、骨芽細胞、島細胞、神経細胞(限定するものではないが、ニューロン、星状細胞、シュワン細胞、腸グリア細胞(enteric glial cells)、オリゴデンドロサイト)を挙げてもよい。
【0051】
多能性幹細胞
多能性幹細胞の特徴付け
多能性幹細胞は、ステージ特異的胚抗原(SSEA)3及び4の1種以上、並びにTra−1−60及びTra−1−81と呼ばれる抗体によって検出可能なマーカーを発現し得る(Thomsonら、Science 282:1145,1998)。インビトロで多能性幹細胞を分化させると、SSEA−4、Tra−1−60、及びTra−1−81の発現が消失し(存在する場合)、SSEA−1の発現が増大する。未分化の多能性幹細胞は、一般にアルカリホスファターゼ活性を有し、この活性は、製造業者(Vector Laboratories,Burlingame,Calif.)により説明されるように、細胞を4%パラホルムアルデヒドで固定した後、基質としてVector Redを使用して展開させることにより検出することができる。未分化の多能性幹細胞はまた、RT−PCRにより検出されるように、一般にOCT4及びTERTも発現する。
【0052】
増殖させた多能性幹細胞の別の望ましい表現型は、内胚葉、中胚葉、及び外胚葉組織の3胚葉の全ての細胞に分化し得るものである。幹細胞の多能性は、例えば、細胞を重症複合免疫不全症(SCID)マウスに注入し、形成される奇形腫を4%パラホルムアルデヒドで固定し、次いでこれを3胚葉からの細胞型のエビデンスについて組織学的に調べることによって確認することができる。代替的に、多能性は、胚様体を形成させ、この胚様体を3つの胚葉に関連したマーカーの存在について評価することにより決定することができる。
【0053】
増殖させた多能性幹細胞株は、標準的なGバンド法を使用して核型を決定し、次いで確立された、対応する霊長類種の核型と比較することができる。「正常な核型」を有する細胞を獲得することが望ましく、この「正常な核型」は、細胞が正倍数体であり、全てのヒト染色体が存在し、かつ著しく変更されてはいないことを意味する。
【0054】
多能性幹細胞源
使用が可能な多能性幹細胞の種類としては、妊娠期間中の任意の時期(必ずしもではないが、通常は妊娠約10〜12週よりも前)に採取した前胚性組織(例えば胚盤胞など)、胚性組織、胎児組織などの、妊娠後に形成される組織に由来する多能性細胞の株化細胞系が挙げられる。非限定的な例は、例えばヒト胚幹細胞株H1、H7、及びH9(WiCell)などのヒト胚幹細胞又はヒト胚生殖細胞の確立株である。それらの細胞の最初の樹立又は安定化中に本開示の組成物を使用することも想定され、その場合、源となる細胞は、源となる組織から直接採取した一次多能性細胞であろう。フィーダー細胞の非存在下で既に培養された多能性幹細胞集団から採取した細胞も好適である。例えば、BG01v(BresaGen,Athens,GA)などの変異ヒト胚性幹細胞株も好適である。同様に、例えば成体体細胞などの非多能性細胞由来の多能性幹細胞も好適である。
【0055】
本発明での使用に好適なマイクロキャリアへの多能性幹細胞の付着
多能性幹細胞は、マイクロキャリアに付着させる前に、当該技術分野の任意の方法により、平面状基材にて培養してもよい。例えば多能性幹細胞は、細胞外マトリックスタンパク質(例えばMATRIGEL)で処理した平面状基材にて培養することができる。別の方法としては、多能性幹細胞は、フィーダー細胞層を播種した平面状基材にて培養することもできる。
【0056】
一実施形態では、多能性幹細胞は胚性幹細胞である。別の実施形態では、胚性幹細胞はヒトのものである。
【0057】
本発明の一態様では、多能性幹細胞は、平面状基材から細胞を放出させるプロテアーゼで、多能性幹細胞を処理することにより、平面状基材から放出される。例えばこのようなプロテアーゼは、コラゲナーゼ、TrypLE(商標)Express、Accutase(商標)、トリプシン及び同様物などであり得る。
【0058】
一実施形態では、多能性幹細胞は、この細胞を約5〜約10分にわたってAccutase(商標)で処理することにより、マイクロキャリア基材から放出される。
【0059】
一実施形態では、多能性幹細胞は、この細胞を約10〜約20分にわたって0.05%トリプシン/EDTAで処理することにより、マイクロキャリア基材から放出される。
【0060】
一実施形態では、多能性幹細胞は、この細胞を約5〜約20分にわたってTrypLE(商標)Expressで処理することにより、マイクロキャリア基材から放出される。
【0061】
一実施形態では、多能性幹細胞は、この細胞を約5〜約10分にわたって10mg/mLコラゲナーゼで処理することにより、マイクロキャリア基材から放出される。
【0062】
放出された多能性細胞は、特定の密度でマイクロキャリアを含有している培地に加える。一実施形態では、多能性幹細胞は、約4,000〜約30,000個細胞/cm2マイクロキャリアで播種される。
【0063】
放出された多能性細胞は、マイクロキャリアを含有している培地に加える。一実施形態では、多能性幹細胞の付着性は、多能性幹細胞をRhoキナーゼ阻害剤で処理することにより増加する。Rhoキナーゼ阻害剤はY27632(Sigma−Aldrich,MO)であり得る。あるいは、Rhoキナーゼ阻害剤はGlycyl−H 1152ジヒドロクロリドである。
【0064】
一実施形態では、多能性幹細胞は約1μM〜約10μMの濃度のY27632で処理される。一実施形態では、多能性幹細胞は約10μMの濃度のY27632で処理される。
【0065】
一実施形態では、多能性幹細胞は約0.25μM〜約5μMの濃度のGlycyl−H 1152ジヒドロクロリドで処理される。一実施形態では、多能性幹細胞は約2.5μMの濃度のGlycyl−H 1152ジヒドロクロリドで処理される。
【0066】
マイクロキャリアを含有している培地は撹拌してもよい。本発明で使用するとき、「撹拌」は培養培地の運動であり得る。このような撹拌は、手動で、あるいは例えば固定式プラットフォーム、スピナーフラスコ、及び同様物などのような装置で実施することができる。一実施形態では、マイクロキャリアを含有している培地は、手による運動を用いることで撹拌される。マイクロキャリアと細胞とを含有しているディッシュは、30秒未満にわたって前後に動かされる。
【0067】
マイクロキャリアを含有している培地を撹拌してもよい。一実施形態では、マイクロキャリアを含有している培地は、スピナーフラスコの使用により撹拌される。スピナーフラスコ(Corning,Lowell,MA)は、ビーズ型に応じて30〜70RPMに設定した撹拌プレートに設置する。
【0068】
別の実施形態では、マイクロキャリアを含有している培地は、固定式プラットフォーム(Vari−mix,Barnstead,Dubuque,Iowa)の使用により撹拌される。固定式プラットフォームの速度は、2秒につき約1回転である。
【0069】
マイクロキャリア上の多能性幹細胞の分化
一実施形態では、多能性幹細胞は、胚体内胚葉系に特徴的なマーカーを発現している細胞へとマイクロキャリア上で分化し得る。あるいは、多能性幹細胞は、膵臓内胚葉系に特徴的なマーカーを発現している細胞へとマイクロキャリア上で分化し得る。あるいは、多能性幹細胞は、膵内分泌系に特徴的なマーカーを発現している細胞へとマイクロキャリア上で分化し得る。
【0070】
別の実施形態では、多能性幹細胞は、マイクロキャリア上で増殖させ、次いで胚体内胚葉系に特徴的なマーカーを発現している細胞へと平面状表面上で分化させることができる。あるいは、多能性幹細胞は、マイクロキャリア上で増殖させ、次いで胚体内胚葉系に特徴的なマーカーを発現している細胞へと平面状表面上で分化させることができる。あるいは、多能性幹細胞は、マイクロキャリア上で増殖させ、次いで膵内分泌系に特徴的なマーカーを発現している細胞へと平面状表面上で分化させることができる。
【0071】
本発明の方法に従って処理された多能性幹細胞は、当該術分野における任意の適当な方法によって他の様々な細胞型に分化させることができる。例えば、本発明の方法に従って処理した多能性幹細胞は、神経細胞、心臓細胞、肝細胞などに分化させることができる。
【0072】
例えば、本発明の方法に従って処理した多能性幹細胞は、国際公開第2007030870号に開示される方法に従って神経前駆細胞及び心筋細胞に分化させることができる。
【0073】
別の例では、本発明の方法に従って処理した多能性幹細胞は、米国特許第6,458,589号に開示される方法に従って肝細胞に分化させることができる。
【0074】
胚体内胚葉系に特徴的なマーカーを発現している細胞の形成
多能性幹細胞は、当該技術分野における任意の方法により、胚体内胚葉系に特徴的なマーカーを発現している細胞へと分化させることができる。
【0075】
例えば、多能性幹細胞は、D’Amourら、Nature Biotechnology 23、1534〜1541(2005年)に開示されている方法に従って、胚体内胚葉系統に特徴的なマーカーを発現している細胞へと分化させることができる。
【0076】
例えば、多能性幹細胞は、Shinozakiら、Development 131,1651〜1662(2004)により開示される方法に従って、胚体内胚葉系に特徴的なマーカーを発現している細胞へと分化させることができる。
【0077】
例えば、多能性幹細胞は、McLeanら、Stem Cells 25,29〜38(2007)により開示される方法に従って、胚体内胚葉系に特徴的なマーカーを発現している細胞へと分化させることができる。
【0078】
例えば、多能性幹細胞は、D’Amourら、Nature Biotechnology 24,1392〜1401(2006)に開示されている方法に従って、胚体内胚葉系に特徴的なマーカーを発現している細胞へと分化させることができる。
【0079】
胚体内胚葉系に特徴的なマーカーは、SOX17、GATA4、HNF−3 β、GSC、CER1、Nodal、FGF8、Brachyury、Mix−様ホメオボックスタンパク質、FGF4、CD48、eomesodermin(EOMES)、DKK4、FGF17、GATA6、CXCR4、C−Kit、CD99、及びOTX2からなる群より選択される。本発明での使用に好適なものは、胚体内胚葉系に特徴的なマーカーのうちの少なくとも1つを発現している細胞である。本発明の一態様において、胚体内胚葉系に特徴的なマーカーを発現している細胞は、原始線条前駆体細胞である。別の態様において、胚体内胚葉系に特徴的なマーカーを発現している細胞は、中内胚葉細胞である。別の態様において、胚体内胚葉系に特徴的なマーカーを発現している細胞は、胚体内胚葉細胞である。
【0080】
他の例では、本発明の方法に従って処理した多能性幹細胞は、血清は不含でアクチビンAを含有している培地中で多能性幹細胞を培養し、次いで細胞をアクチビンA及び血清と共に培養し、次いで細胞をアクチビンA及び様々な濃度の血清と共に培養することによって、胚体内胚葉系に特徴的なマーカーを発現している細胞へと分化させることができる。この方法の一例は、Nature Biotechnology 23,1534〜1541(2005)に開示されている。
【0081】
他の例では、本発明の方法に従って処理した多能性幹細胞は、血清は不含でアクチビンAを含有している培地中で多能性幹細胞を培養し、次いで細胞をアクチビンA及び異なる濃度の血清と共に培養することによって、胚体内胚葉系に特徴的なマーカーを発現している細胞へと分化させることができる。この方法の例は、D’Amourら、Nature Biotechnology(2005)に開示されている。
【0082】
他の例では、本発明の方法に従って処理した多能性幹細胞は、血清は不含でアクチビンA及びWntリガンドを含有している培地中で多能性幹細胞を培養し、次いでWntリガンドは除去し、細胞をアクチビンA及び血清と共に培養することによって、胚体内胚葉系に特徴的なマーカーを発現している細胞へと分化させることができる。この方法の例は、Nature Biotechnology 24,1392〜1401(2006年)に開示されている。
【0083】
他の例では、本発明の方法に従って処理した多能性幹細胞は、米国特許出願第11/736,908号(LifeScan,Inc.に譲渡)に開示される方法に従い、胚体内胚葉系に特徴的なマーカーを発現している細胞へと分化させることができる。
【0084】
他の例では、本発明の方法に従って処理した多能性幹細胞は、米国特許出願第11/779,311号に記載の方法に従って、胚体内胚葉系に特徴的なマーカーを発現している細胞へと分化させることができる。
【0085】
膵臓内胚葉系に特徴的なマーカーを発現している細胞の形成
多能性幹細胞は、当該技術分野における任意の方法により、膵臓内胚葉系に特徴的なマーカーを発現している細胞へと分化させることができる。
【0086】
例えば、多能性幹細胞は、D’Amourら、Nature Biotechnology 24,1392〜1401(2006)に開示されている方法に従って、膵臓内胚葉系に特徴的なマーカーを発現している細胞へと分化させることができる。
【0087】
例えば、本発明の方法に従って得られる、胚体内胚葉系に特徴的なマーカーを発現している細胞は、この胚体内胚葉系に特徴的なマーカーを発現している細胞を繊維芽細胞増殖因子及びヘッジホッグシグナル伝達経路阻害剤KAAD−シクロパミンで処理し、次いで繊維芽細胞増殖因子及びKAAD−シクロパミンを含有する培地を除去した後に、レチノイン酸、繊維芽細胞増殖因子及びKAAD−シクロパミンを含有する培地中で培養することにより、膵臓内胚葉系に特徴的なマーカーを発現している細胞へと更に分化する。この方法の例は、Nature Biotechnology 24,1392〜1401(2006)に開示されている。
【0088】
例えば、本発明の方法に従って得られる、胚体内胚葉系に特徴的なマーカーを発現している細胞は、この胚体内胚葉系に特徴的なマーカーを発現している細胞を米国特許出願第11/736,908号(LifeScan,Inc.に譲渡)に開示された方法に従いレチノイン酸及び少なくとも1種類の線維芽細胞増殖因子で所定の時間処理することによって、膵臓内胚葉系に特徴的なマーカーを発現している細胞へと更に分化する。
【0089】
例えば、本発明の方法に従って得られる、胚体内胚葉系に特徴的なマーカーを発現している細胞は、胚体内胚葉系に特徴的なマーカーを発現している細胞をレチノイン酸(Sigma−Aldrich,MO)及びエキセンディン4で処理し、次いでDAPT(Sigma−Aldrich,MO)及びエキセンディン4を含有している培地を除去し、続いてエキセンディン1、IGF−1及びHGFを含む培地で細胞を培養することで、膵臓内胚葉系に特徴的なマーカーを発現している細胞へと更に分化する。この方法の例は、Nature Biotechnology 24,1392〜1401(2006年)に開示されている。
【0090】
例えば、本発明の方法に従って得られる、膵臓内胚葉系に特徴的なマーカーを発現している細胞は、膵臓内胚葉系に特徴的なマーカーを発現している細胞をエキセンディン4を含有している培地で培養し、次にエキセンディン4を含有している培地を除去し、続いて細胞をエキセンディン1、IGF−1及びHGFを含有している培地で培養することにより、膵内分泌系に特徴的なマーカーを発現している細胞へと更に分化する。この方法の例は、D’Amourら、Nature Biotechnology,2006に開示されている。
【0091】
例えば、本発明の方法に従って得られる、膵臓内胚葉系に特徴的なマーカーを発現している細胞は、膵臓内胚葉系に特徴的なマーカーを発現している細胞を、DAPT(Sigma−Aldrich,MO)及びエキセンディン4を含有している培地で培養することにより、膵内分泌系に特徴的なマーカーを発現している細胞へと更に分化する。この方法の例は、D’Amourら、Nature Biotechnology,2006に開示されている。
【0092】
例えば、本発明の方法に従って得られる、膵臓内胚葉系に特徴的なマーカーを発現している細胞は、膵臓内胚葉系に特徴的なマーカーを発現している細胞をエキセンディン4を含有している培地で培養することにより、膵内分泌系に特徴的なマーカーを発現している細胞へと更に分化する。この方法の例は、D’Amourら、Nature Biotechnology,2006に開示されている。
【0093】
例えば、本発明の方法に従って得られる、膵臓内胚葉系に特徴的なマーカーを発現している細胞は、膵臓内胚葉系に特徴的なマーカーを発現している細胞を米国特許出願第11/736,908号(LifeScan,Inc.に譲渡)に開示された方法に従って、ノッチシグナル伝達経路を阻害する因子で処理することで、膵内分泌系に特徴的なマーカーを発現している細胞へと更に分化する。
【0094】
例えば、本発明の方法に従って得られる、膵臓内胚葉系に特徴的なマーカーを発現している細胞は、膵臓内胚葉系に特徴的なマーカーを発現している細胞を米国特許出願第11/779,311号(LifeScan,Inc.に譲渡)に開示された方法に従って、ノッチシグナル伝達経路を阻害する因子で処理することで、膵内分泌系に特徴的なマーカーを発現している細胞へと更に分化する。
【0095】
例えば、本発明の方法に従って得られる、膵臓内胚葉系に特徴的なマーカーを発現している細胞は、膵臓内胚葉系に特徴的なマーカーを発現している細胞を米国特許出願第11/736,908号(LifeScan,Inc.に譲渡)に開示された方法に従って、ノッチシグナル伝達経路を阻害する因子で処理することで、膵内分泌系に特徴的なマーカーを発現している細胞へと更に分化する。
【0096】
例えば、膵臓内胚葉系に特徴的なマーカーを発現している細胞は、膵臓内胚葉系に特徴的なマーカーを発現している細胞を米国特許出願第11/779,311号(LifeScan,Inc.に譲渡)に開示された方法に従って、ノッチシグナル伝達経路を阻害する因子で処理することで、膵内分泌系に特徴的なマーカーを発現している細胞へと更に分化する。
【0097】
膵内分泌系に特徴的なマーカーは、NGN3、NEUROD、ISL1、PDX1、NKX6.1、PAX4、NGN3及びPTF−1 αからなる群から選択される。一実施形態では、膵内分泌細胞は、以下のホルモンのうちの少なくとも1つを発現することができる:インスリン、グルカゴン、ソマトスタチン、及び膵臓ポリペプチド。本発明で使用するのに好適なものは、膵内分泌系の特徴を示すマーカーを少なくとも1つ発現している細胞である。本発明の一態様において、膵内分泌系に特徴的なマーカーを発現している細胞は、膵内分泌細胞である。膵臓内分泌細胞は、膵臓ホルモン発現細胞であってよい。また、膵臓内分泌細胞は膵臓ホルモン分泌細胞であってもよい。
【0098】
本発明の一態様では、膵臓内分泌細胞は、β細胞系に特徴的なマーカーを発現している細胞である。β細胞系に特徴的なマーカーを発現している細胞は、PDX1と、以下の転写因子、すなわち、NGN3、NKX2.2、NKX6.1、NEUROD、ISL1、HNF−3 β、MAFA、PAX4、又はPAX6のうちの少なくとも1つを発現する。本発明の一態様では、β細胞系に特徴的なマーカーを発現している細胞は、β細胞である。
【0099】
以下の実施例により本発明を更に例示するが、本発明はこれらの実施例により限定されるものではない。
【実施例】
【0100】
実施例1:マイクロキャリアに対するヒト胚性幹細胞の付着及び増殖
ヒト胚性幹細胞がマイクロキャリア上に付着しかつ増殖し得るか否かを判定するために、TrypLE(商標)Expressにより、継代数52のH9細胞をMATRIGEL(商標)(BD Biosciences,CA)コートプレートから放出させた。次いでそれらをマイクロキャリアとMEF−CMと共にインキュベートした。製造元からの取扱説明書に従い、ProNectinF(PN)、Plastic(P)、PlasticPlus(PP)、HILLEX(登録商標)II(H)、コラーゲン(Col)及びFACT III(SoloHill,MI)マイクロキャリアの懸濁液を調製した。毎日の観察結果に基づき、表1はH9細胞の、37℃にて2日経過後の、マイクロキャリアに対する付着と増殖を記載する。試験したほとんどのマイクロキャリアに対して、大部分の細胞は付着及び/又は増殖しなかった。H9細胞は、HILLEX(登録商標)IIマイクロキャリア(Solohill,MI)に対しては付着し増殖したが、画像で示される細胞−ビーズ凝集体は、2日間の静置培養後にはより少なくなった(図1B)。
【0101】
マイクロキャリアに対するヒト胚性幹細胞の付着性と増殖性を改善するために、Rho結合コイルドコイル形成タンパク質セリン/スレオニンキナーゼの低分子阻害剤である、Rhoキナーゼ阻害剤を培地に添加した。具体的には、Y27632,Y,(Sigma−Aldrich,MO)を使用した。10μMのY27632(Sigma−Aldrich,MO)添加MEF−CMは毎日交換した。10μMのY27632(Sigma−Aldrich,MO)の存在下で、H9細胞は試験した全てのマイクロキャリアに付着し、凝集体を形成した(表2)。画像解析によると、HILLEX(登録商標)IIマイクロキャリア(Solohill,MI)で増殖させたヒト胚性幹細胞は、他のマイクロキャリアについて試験したヒト胚性幹細胞よりも、付着しかつ増殖しているように見えた。加えてH9細胞は、Rhoキナーゼ阻害剤の存在下で、より良好にHILLEX(登録商標)II(Solohill,MI)に付着した(図1Aと図1Bとの比較による)。
【0102】
細胞治療用途のためのヒト胚性幹細胞の増量は、製品需要を満たす必要がある。現行の最も良好な増量技法としては、スピナーフラスコ法とバイオリアクター法が挙げられる。これらの技術のいずれもが、懸濁液中のマイクロキャリアの物理的な移動を必要とする。マイクロキャリア上でのヒト胚性幹細胞の増殖に対する移動効果を判定するために、6又は12ウェルディッシュを、37℃のインキュベーター内の固定式プラットフォームに設置した。3日間の増殖後、一部のマイクロキャリアから細胞凝集体が放出され始めた。図2のA、B、Dは、Plastic Plus、Plastic又はPronectinマイクロキャリアから脱着した細胞凝集体を記す。対照的に、図2Cの細胞は、HILLEX(登録商標)IIマイクロキャリア(Solohill,MI)に付着したまま留まりかつ増殖した。実施例4は、Viacount Flexを装備したGuava PCA−96(Guava Technologies,Hayward,CA)で細胞を計数するのに先立つ、脱着法を記載する。マイクロキャリア上の細胞の増殖速度を測定することで、播種時の開始細胞数と比較して、3日目の細胞数が減少していることが明らかになった。これは、5日目に実験が終了するまでの間増量が続けられる細胞の、マイクロキャリアへの初期付着が乏しいことに由来する可能性が高い。H9細胞は、HILLEX(登録商標)IIマイクロキャリア(Solohill,MI)に対し、他の種類のビーズと比較して最も速い増殖速度を有していたが、これはHILLEX(登録商標)IIマイクロキャリア(Solohill,MI)に対する、細胞のより良好な付着性に由来するものである可能性が高い(図2、3)。このことは、HILLEX(登録商標)IIマイクロキャリア(Solohill,MI)が、懸濁液中のH9細胞の増殖を支持し得ることを実証する。これは以降に繰り返される節の後に更に実証された。実施例5を参照されたい。
【0103】
ラージスケールでの増量のために、H1ヒト胚性細胞株をマイクロキャリア上での増殖性について同様に試験した。Rhoキナーゼ阻害剤のY27632(Sigma−Aldrich,MO)がH9細胞株の付着に必要とされたことから、この阻害剤はH1細胞についても同様に必要とされるであろうと推測された。Cytodex 1(登録商標)、Cytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)、HILLEX(登録商標)II、Plastic、ProNectinF、Plastic Plusマイクロキャリア(SoloHill Ann Arbor,MI)を、製造元からの取扱説明書に従って調製した。継代数47のH1ヒト胚性幹細胞を、約13,333個細胞/マイクロキャリアcm2(10μMのY27632(Sigma−Aldrich,MO)添加MEF−CM中)で播種した。細胞とマイクロキャリアを、37℃にて固定式プラットフォーム上の、12ウェルの組織培養用無処理ディッシュ(12ウェルあたり15cm2)内に設置して、マイクロキャリアと培地に運動させた。3、5及び7日目後に、1つのウェルを画像化し、採取し、計数した。細胞の付着能は、ビーズの種類に依存した。H9細胞株で観察されたものと同様の結果が、H1細胞株についても観察された。具体的には、Plastic、Plastic Plus又はProNectinFマイクロキャリア上に播種した細胞は十分に付着せず、及び/又は十分に増殖しなかった(図4)。HILLEX(登録商標)II(Solohill,MI)、Cytodex 1(登録商標)、又はCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)マイクロキャリア上に播種した細胞は十分に付着し、かつ十分に増殖した(図5)。実施例4に従って細胞を脱着し、収率について計数した。Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)で増殖させた細胞は、培養7日後に最も大きい細胞数を呈した(図6)。
【0104】
実施例2:細胞の付着及び増殖のためのY27632及び他のRhoキナーゼ阻害剤の、至適濃度
マイクロキャリアに対するヒト胚性幹細胞の付着と増殖を最も支持するRhoキナーゼ阻害剤濃度を決定するため、以下の実験を実施した。
【0105】
継代数44のH9細胞の、13,333個細胞/cm2の開始アリコートを、12ウェルの組織培養用無処理プレートの単一ウェル内で、15cm2マイクロキャリア上に播種した。37℃にて固定式プラットフォームに設置する前に、細胞を37℃にて少なくとも60分間静置した。細胞を加える前に、HILLEX(登録商標)II(Solohill,MI)マイクロキャリア及びCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)マイクロキャリアを、製造元から指示された通りに調製した。濃度範囲が10、5、2.5又は1μMのRhoキナーゼ阻害剤Y27632、あるいは濃度範囲が5、2.5、1又は0.5μMの(S)−(+)−4−グリシル−2−メチル−1−[(4−メチル−5−イソキノリニル)スルホニル]−ヘキサヒドロ−1H−1,4−ジアゼピンジヒドロクロリド(Glycyl−H 1152ジヒドロクロリド(H),Tocris,MO)を添加したMEF−CMで細胞を増殖させた。培地を毎日交換し、収率と生存率について、1ウェルの細胞を播種後4日目と7日目に計数した(図7A及びB)。全般的に、10又は5μMのY27632(Sigma−Aldrich,MO)が最も良好な細胞増殖を示し(7日目)、その一方で2.5及び1.0μMで最も良好な接着性を有しているように見えた(4日目)。濃度1及び0.5μMのGlycyl−H 1152ジヒドロクロリド(Tocris,MO)は最も良好な細胞増殖を示し(7日目)、その一方で5μMで最も良好な接着性を有しているように見えた(4日目)。
【0106】
Rhoキナーゼ阻害剤は、アポトーシスを促進すると特徴付けられてきたことから、次の用量設定法を試行した。継代数48のH1細胞を、TrypLE(商標)ExpressでMATRIGEL(商標)(BD Biosciences,CA)コートプレートから解離させた。次いで12ウェルの、組織培養用無処理プレートの単一ウェル内の15cm2マイクロキャリア上に、細胞を播種した。HILLEX(登録商標)II(Solohill,MI)、Cytodex 1(登録商標)、又はCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)マイクロキャリアを、Rhoキナーゼ阻害剤の量を減少させて試験した:10μMのY27632(Sigma−Aldrich,MO)を1日目に使用し、次いで0.5μMを2日目に使用(Y10/5μM);2.5μMのGlycyl−H 1152ジヒドロクロリド(Tocris,MO)を1日目に、続いて0.5μMを2日目に使用(H2.5/0.5μM);1μMのGlycyl−H 1152ジヒドロクロリド(Tocris,MO)を1日目に、0.5μMのGlycyl−H 1152ジヒドロクロリド(Tocris,MO)を2日目に使用(H1/0.5μM);あるいは0.25μMのGlycyl−H 1152ジヒドロクロリド(Tocris,MO)をMEF−CMに毎日添加(H0.25μM)。37℃にて固定式プラットフォームに設置する前に、37℃にてH1細胞及びマイクロキャリアを3時間にわたって45分毎に撹拌した。37℃にて固定式プラットフォームでの3、5及び7日目に細胞を計数した(図8)。全般的に、Glycyl−H 1152ジヒドロクロリド(Tocris,MO)の最良の濃度は、1日目では1〜2.5μMであり、2日目では0.5μMであり、以降では化合物は断った。細胞は、試験した全てのマイクロキャリアについて、これらの濃度のGlycyl−H 1152ジヒドロクロリド(Tocris,MO)で、10μMのY27632(Sigma−Aldrich,MO)と比較して同様の増殖速度を示した。0.25μMのGlycyl−H 1152ジヒドロクロリド(Tocris,MO)で細胞を維持した場合、細胞収率は乏しかった。Rhoキナーゼ阻害剤を最小量で用いることは、プロセス費用を低減することにもつながり、かつ細胞増殖についても有益なものであり得る。同様にこれらのデータは、ヒト胚性幹細胞がマイクロキャリアへの付着性及び増殖性を維持するためには、Rhoキナーゼ阻害剤は必要とされないことを示す。
【0107】
実施例3:マイクロキャリア上での接着及び増殖に対する細胞密度の効果
播種密度の改善は、必要とされる細胞の総数を減少させるための一手段である。適切な播種密度を決定するために、4x対物視野あたりのマイクロキャリア数を計数した。Cytodex 3マイクロキャリア(GE Healthcare Life Sciences,NJ)を含有している、10μMのY27632(Sigma−Aldrich,MO)添加MEF−CMの入った10cmプレートに、0.4×104個細胞/cm2(低密度)、1.2×104個細胞/cm2(中間密度)、又は3×104個細胞/cm2(高密度)の密度でH1細胞を播種した。次いでプレートを、37℃で6時間にわたり、45分毎に撹拌した。37℃にて30rpmのスピナーフラスコ(実施例5に記載)内の、10μMのY27632(Sigma−Aldrich,MO)添加MEF−CM 50mLに、細胞及びマイクロキャリアを移した。24時間後に、5μMのY27632(Sigma−Aldrich,MO)添加MEF−CM 25mLを添加した。24時間後、回転速度を40rpmに上昇させた。培養の3日目及び5日目に、75mLのうちの50mLを除去し、MEF−CMで交換した。6時間、3日、5日及び7日の時点で、スピナーフラスコからアリコートを取って画像を撮影した。細胞の付着したマイクロキャリアの百分率を、図9の画像の右下に記載する。播種後3日目では、細胞でコートされたマイクロキャリアの数は開始播種密度と対応したが、低密度で播種した細胞では、細胞でコートされたマイクロキャリアの数は5日目及び7日目でも増加しなかった。このことは、0.4×104個細胞/cm2が、細胞をマイクロキャリアへと組み込ませて凝集体にするのに十分な細胞数ではないことを意味する。3×104個細胞/cm2では、細胞が付着したマイクロキャリアの数は、1.2×104個細胞/cm2を播種した場合の5日目及び7日目と同様であった(図9)。細胞数を見た場合、高密度での細胞播種では、より多くの細胞がマイクロキャリアに付着していることは明らかである(図10)。開始播種細胞数と比較しての倍率変化(fold change)の解析では、高密度播種した培養物において、3日目及び5日目に、より多数の細胞が付着していたことを明らかになった(図11)。7日目には、対照と高密度播種培養物は、それらの開始播種密度から、細胞数について同様の倍率で変化した。これらのデータから、我々は、H1細胞をマイクロキャリア上に効果的に付着させ、増殖させるためには、1.2×104個細胞/cm2が最小の細胞数であると結論付けた。より高い播種密度へと移行させることは、細胞の増量に必要とされる日数を減少させる助けとなり得る。
【0108】
実施例4:マイクロキャリアからの細胞の解離
増殖速度を判定するためには、マイクロキャリアから細胞を解離させる必要がある。Rhoキナーゼ阻害剤Y27632(Sigma−Aldrich,MO)を除去しても、H1細胞はマイクロキャリアから解離しなかった(実施例2、図12)。マイクロキャリアから細胞を解離する前に、HILLEX(登録商標)IIマイクロキャリア(Solohill,MI)上のH9細胞を、倍率10x及び20xで画像解析した(それぞれ図13A,B)。HILLEX(登録商標)IIマイクロキャリア(Solohill,MI)上のH9細胞を酵素処理することで、生存細胞を脱着させた(図13C,D及び14)。37℃にて固定式プラットフォーム上で、HILLEX(登録商標)IIマイクロキャリア(Solohill,MI)を含有する6ウェルディッシュ中で、6日間にわたってH9細胞を増殖させた。マイクロキャリアに付着させた細胞を15mLコニカルチューブに設置し、マイクロキャリアを沈殿させた後に培地を吸引除去した。沈殿したマイクロキャリアを4mLのPBS(マグネシウムイオン及びカルシウムイオン不含)で3回洗浄し、重量沈降によりマイクロキャリアを沈殿させた。PBSを吸引除去し、1mLのPBSを加えた。細胞を有するマイクロキャリアを、12ウェルの組織培養用無処理プレートの単一ウェルに移した。プレートを、マイクロキャリアを沈殿させることができるような角度で静止させた。PBSを吸引除去し、1mLのTrypLE(商標)Express(Invitrogen,CA)又は0.05%トリプシン/EDTAをウェルに加えた。プレートを、37℃にて固定式プラットフォーム上に、10〜20分間にわたって設置した。プレートを取り外し、3mLのDMEM/F12又はMEF−CMをウェルに添加した。培地を激しくピペッティングし、細胞を放出させた(図13C,D)。顕微鏡下でのマイクロキャリアの観察は、細胞がマイクロキャリアから脱着したことを決定づけた。次いで細胞を、200×gで5分間にわたって遠心した。培地を吸引除去し、ペレットを1mLのDMEM/F12又はMEF−CM培地に再懸濁した。次いでViacount dyeを用いて、Guava PCA−96(Guava Technologies,Hayward,CA)で細胞を計数した。具体的には、培地で適切に希釈した200μL容量の細胞を、2μLのViacountと共に10分間にわたってインキュベートした。生存率と細胞数を決定した(図14)。TrypLE(商標)Express及びトリプシン/EDTAの両方が、細胞をマイクロキャリアから効率的に解離した。
【0109】
TrypLE(商標)Expressがマイクロキャリアから細胞を放出し、及びこれはGMP製品として利用可能なものであることから、他の可能性のある解離剤、具体的にはコラゲナーゼ及びAccutase(商標)(Sigma−Aldrich,MO)について試験を行った。継代数48のH1細胞を、スピナーフラスコ中(実施例5)で10日間にわたって増殖させた。次いでマイクロキャリアを回収し、50mLのコニカルチューブに移した。細胞を上記のようにPBSで洗浄し、12ウェルのプレートに移した。PBSを吸引除去し、1mLのTrypLE(商標)Express、Accutase(商標)又はコラゲナーゼ(10mg/mL)をウェルに添加して、37℃にて固定式プラットフォームに5〜10分にわたって設置した。細胞/マイクロキャリアをDMEM/F12に激しく再懸濁し、次いで解離した細胞とマイクロキャリアを50mLのコニカルチューブ上で40μmのセルストレイナーを通して濾過した。ウェルを追加の培地2mLで洗浄し、同様にセルストレイナーに加えて、200×gで5分にわたって遠心した。次いで細胞を1mLのDMEM/F12に再懸濁し、上記のように細胞を計数するために希釈した。細胞生存率は、試験した全ての酵素で類似していた。Accutase(商標)とTrypLE(商標)Expressは、5分及び10分のインキュベーション後に同様の数の細胞を放出した(図15)。これは、マイクロキャリア上のヒト胚性幹細胞のための細胞解離剤としての、Accutase(商標)とTrypLE(商標)Expressの適合性を実証する。
【0110】
実施例5:マイクロキャリア上の未分化の多能性幹細胞の増殖
マイクロキャリア上の細胞を増量させるためには、細胞を、マイクロキャリアから脱着又は酵素的に解離させ、かつ新しいマイクロキャリアへと再付着させることができなければならない。マイクロキャリア上での一般的な細胞増殖方法は、細胞の脱着及び再付着特性に依存する。以下の実験は、これがヒト胚性幹細胞の特徴ではなかったことを示す。具体的には、継代数43のH9細胞をHILLEX(登録商標)IIマイクロキャリア(Solohill,MI)上に播種し、125mLのスピナーフラスコ中でインキュベートした(以下を参照のこと)。フェノールレッドが培地中に存在し、かつHILLEX(登録商標)IIマイクロキャリアにより取り込まれた(Solohill,MI)。8日間の増殖後に、マイクロキャリア上の細胞のアリコート10mLを、フェノールレッド不含MEF−CMと、440mgのHILLEX(登録商標)IIマイクロキャリアと、5μMのY27632(Sigma−Aldrich,MO)とを含有している、新しいスピナーフラスコに設置した。37℃にて30rpm回転でのインキュベーションの5日目に、マイクロキャリアを取り出して画像を得た(図16)。黒っぽいマイクロキャリアは、フェノールレッドを含有している培地で増殖したH9細胞で覆われた、マイクロキャリアを示す。色味の明るいマイクロキャリアは、新しく加えたマイクロキャリアである。H9細胞が脱着し、新しいマイクロキャリアに再付着することが見込まれていたが、細胞は再付着する代わりに新しいマイクロキャリアと共に凝集体を形成した。色味の明るくないマイクロキャリアには細胞が付着しており、この細胞が黒っぽいマイクロキャリアとの凝集体を形成していないことは、細胞はマイクロキャリアから脱着しかつマイクロキャリアへと再付着することはできなかったことを示唆する。マイクロキャリア上の細胞の増殖を伝播させるためには、細胞をマイクロキャリアから酵素的に解離させる必要があった(実施例4を参照されたい)。
【0111】
現在では、ヒト胚性幹細胞をマイクロキャリア上で増殖させる方法が確立されているので、どのようにしてヒト胚性幹細胞をラージスケールのスピナーフラスコ中で増殖させるかを決定する必要がある。スピナーフラスコは、高密度系での細胞の増量を可能にする。これは空間の節約であり、バイオリアクター内で細胞を増量させるための第1工程であると考えられる。ヒト胚性幹細胞の、スピナーフラスコ中で増殖する能力について試験するために、継代数43のH9細胞を125mLのスピナーフラスコ中に播種した。細胞は、まず10cmプレート中でマイクロキャリアに付着させてからスピナーフラスコに移した。具体的には、TrypLE(商標)Expressと共に37℃で5分間インキュベートすることで、H9細胞を2枚の6ウェルディッシュから放出させた。細胞はコラゲナーゼ(1mg/mL)で処理し、1:30増殖因子低減MATRIGEL(商標)(BD Biosciences,CA)コートプレートに播種した後に、TrypLE(商標)Expressで処理した。細胞をDMEM/F12に再懸濁し、Viacountを用いてGuava instrumentで計数した。遠心後に、3×106個の細胞を、10μMのY27632(Sigma−Aldrich,MO)添加MEF−CMと、製造元からの取扱説明書に従って調製した250cm2のHILLEX(登録商標)IIマイクロキャリア(Solohill,MI)とを含有している、10cmのプレートに播種した。ディッシュを37℃に設置し、穏やかに回転させ、4.5時間にわたって45分毎に一度撹拌した。次いで細胞、マイクロキャリア及び培地を125mLのスピナーフラスコに移した。次いでスピナーフラスコには10μMのY27632(Sigma−Aldrich,MO)添加MEF−CM50mLを充填し、37℃にて撹拌プレートに40rpmで設置した。翌日、培地を交換し、5μMのY27632(Sigma−Aldrich,MO)添加MEF−CM75mLで充填した。撹拌速度は70rpmに上昇させた。培地にはY27632化合物(Sigma−Aldrich,MO)を加えずに1日おきに交換した。実施例4に記載の方法に従って細胞を継代し、3×106個の細胞を、250cm2の新しいマイクロキャリアに再播種した。培養物は、1〜2×105個細胞/cm2コンフルエントに達したときに継代した。この操作を5継代実施した(図17)。各継代での多能性マーカー発現では、細胞の80〜95%が多能性マーカーCD9、SSEA4、SSEA3、TRA−1−60及びTRA−1−81を発現していたと評価された(図19A)。同様の実験を、Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上で、継代数43のH9細胞についても実施した(図18,19B)。全般的に、細胞はHILLEX(登録商標)II(Solohill,MI)マイクロキャリア及びCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)マイクロキャリアのどちらでも良好に増殖し、かつ多能性を保持した。スピナーフラスコ中で5回継代した後に核型解析を実施したところ、細胞の1.5%で、12番染色体の異常なトリソミーが示された。これらの細胞は、実験の終了時には継代数50付近であることから、観察されるこのような異常は一般的に生じる恐れがある。細胞継代数が低い状態で開始した場合に、このような仮定が生じるか試験した。
【0112】
継代数48と継代数49のH1細胞株についても、同様の実験を実施した。回転速度と播種密度を除き、全てのパラメーターは同様のままであった。スピナーフラスコの回転速度は、初日はオーバーナイトで30rpmであり、以降の日では40rpmに上昇させた。Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)の場合の播種密度は約11,000個細胞/cm2であった一方で、Cytodex 1(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)の場合の播種密度は約7,000個細胞/cm2であった。播種した細胞数はスピナーフラスコあたり3×106個細胞で一定であった。マイクロキャリアの重量は、Cytodex 1(登録商標)及びCytodex 3(登録商標)の場合、100mgで一定であった。Cytodex 1(登録商標)及びCytodex 3(登録商標)がHILLEX(登録商標)IIマイクロキャリアを上回る1つの利点は、それらのより大きな表面積である。図20及び21は、Cytodex 1(登録商標)及びCytodex 3(登録商標)のそれぞれの上でのH1細胞の増量を示す。細胞は5回の継代にわたって多能性を維持していた(図22)。Cytodex 3(登録商標)マイクロキャリア上のH1細胞の核型解析は、試験した細胞の10%で、Y染色体に重複があることを明らかにした。これらのH1細胞はマイクロキャリア上に継代数48で継代され、その後5回継代された後に解析された。平面状表面上のMATRIGEL(商標)(BD Biosciences,CA)で増殖させた継代数55のH1細胞は、正常な核型を有していた。3日間と経過日数間(5、6又は7日間)との間での、これらの細胞の倍加速度解析では、全般的に倍加時間に変化は見られなかった(図23)。Cytodex 1(登録商標)マイクロキャリア上で増殖させたH1細胞及びHILLEX(登録商標)IIマイクロキャリア(Solohill,MI)上で増殖させたH9細胞は、ほとんど一定の倍加時間を示した(表3)。
【0113】
実施例6:制限培地中のマイクロキャリア上でのヒト胚性幹細胞の増殖
治療用製品を製造するためには、ヒト胚性幹細胞培養培地からいずれの動物成分も除外されていることが好ましい。現在、ヒト胚性幹細胞は、マウス胚性線維芽細胞を用いて馴化させた培地(MEF−CM)中の、MATRIGEL(商標)(BD Biosciences,CA)上で維持されている。MATRIGEL(商標)(BD Biosciences,CA)及びMEF−CMのいずれもがマウス細胞由来である。加えて、MEF−CMは高価であり、培地を生成するのに時間がかかる。ヒト胚性幹細胞が、制限培地を含むマイクロキャリア上で維持され得るかを判断するために、Stem Pro(Invitrogen,CA)、mTESR(StemCell Technologies,Vancouver,Canada)又はMEF−CM中の、Rhoキナーゼ阻害剤の10μMのY27632(Sigma−Aldrich,MO)又は2.5μMのGlycyl−H 1152ジヒドロクロリド(Tocris,MO)の存在下で、H9細胞をCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)及びHILLEX(登録商標)II(Solohill,MI)マイクロキャリア上に播種した。細胞を、37℃にて固定式プラットフォーム上の12ウェルディッシュ中に設置した。細胞を3、5及び7日目に計数した。継代数39のH9細胞はMEF−CM中で、どちらの種類のビーズ上でも増殖し、一般的な増量特性を示した(図24)。mTESR(StemCell Technologies,Vancouver,Canada)中で増殖させた同様の細胞は、10μMのY27632(Sigma−Aldrich,MO)の存在下で、Cytodex 3(登録商標)マイクロキャリア上で良好に増殖したが、HILLEX(登録商標)IIマイクロキャリア上では低い増殖率を呈した。20回にわたって継代することでStemPro培地に馴化された、継代数64のヒト胚性幹細胞株H9(継代数64のH9)細胞は、10μMのY27632(Sigma−Aldrich,MO)の存在下で、HILLEX(登録商標)II及びCytodex 3(登録商標)のいずれでも良好に増殖した。驚くべきことに、これらの細胞は、2.5μMのGlycyl−H 1152ジヒドロクロリド(Tocris,MO)の存在下では、Cytodex 3(登録商標)マイクロキャリア上では良好には増殖しなかった。したがって、マイクロキャリアの種類、Rhoキナーゼ阻害剤、及び培地の全てが、ヒト胚性幹細胞の増殖能を決定する役割を果たす。
【0114】
継代数38のH1ヒト胚性幹細胞を、12ウェルのディッシュに、mTESR(StemCell Technologies,Vancouver,Canada)又はMEF−CM中のRhoキナーゼ阻害剤の、10μMのY27632(Sigma−Aldrich,MO)又は2.5μMのGlycyl−H 1152ジヒドロクロリドの存在下で、Cytodex 3(登録商標)又はHILLEX(登録商標)IIマイクロキャリアのいずれかに播種した。細胞を37℃にて固定式プラットフォーム上に設置した。細胞を3、5及び7日目に計数した。MEF−CM中でY27632(Sigma−Aldrich,MO)の存在下で増殖させた細胞は、両方の種類のマイクロキャリアで一般的な増量特性を示したが、Glycyl−H 1152ジヒドロクロリド(Tocris,MO)の存在下では、Cytodex 3(登録商標)に対して乏しい増殖率を示した(図25)。mTESR培地(StemCell Technologies,Vancouver,Canada)は、どちらのRhoキナーゼ阻害剤の存在下でも、HILLEX(登録商標)II マイクロキャリア上でH1細胞を増殖させたが、Cytodex 3(登録商標)マイクロキャリアに対しては低い増殖率が示された。
【0115】
mTESR(StemCell Technologies,Vancouver,Canada)中で、HILLEX(登録商標)IIマイクロキャリアで良好に増殖した所与の継代数50のH1細胞のうちの3×106個を、250cm2のHILLEX(登録商標)IIマイクロキャリアに播種した。37℃で、5時間にわたって45分ごとに手で撹拌しながら、10cm2のディッシュ中でインキュベートした。10μMのY27632(Sigma−Aldrich,MO)を添加したmTESR(StemCell Technologies,Vancouver,Canada)を、1日おきに交換した。この操作を、MEF−CMで増殖させた細胞についても並行して実施した(実施例5)。MEF−CMで増殖させた細胞とは異なり、mTESR培地(StemCell Technologies,Vancouver,Canada)で増殖させた細胞は7日目にHILLEX(登録商標)IIマイクロキャリアから脱着し始めた(図26A対26B)。このことは、ヒト胚性幹細胞にHILLEX(登録商標)IIマイクロキャリア(Solohill,MI)に対する付着性及び増殖性を維持させるためには、mTESR(StemCell Technologies,Vancouver,Canada)に追加の成分を加える必要があることを示す。
【0116】
実施例7:マイクロキャリア上でのヒト胚性幹細胞の分化
ヒト胚性幹細胞がマイクロキャリア上で増量可能であることから、これらの細胞の分化の可能性が決定されるべきである。継代数43のヒト胚性幹細胞株H9の細胞を、Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上で5回継代した。5回目の継代で、細胞をマイクロキャリア上で6日間にわたって増殖させた後、TrypLE(商標)Expressを用いてマイクロキャリアから解離させた(実施例4を参照されたい)。次いで細胞を、1:30 MATRIGEL(商標):DMEM/F12コートしたプレートに設置した。細胞がプレート上で80〜90%コンフルエントに達した後、細胞を分化誘導剤に曝露させた。100ng/mLのアクチビンA(PeproTech,NJ)と、20ng/mLのWnt3a(R&D Biosciences,MN)と、8ng/mL bFGF(PeproTech,NJ)とを添加した、2%ウシ血清アルブミンフラクションV(脂肪酸不含)(FAF BSA,MP Biomedicals,Ohio)含有RPMIで細胞を2日間にわたって処理することで、胚体内胚葉へのヒト胚性幹細胞の分化を実施した。細胞は、100ng/mLのアクチビンA(PeproTech,NJ)と、8ng/mLのbFGF(PeproTech,NJ)とを添加した、2% FAF BSA含有RPMIで更に2日にわたって処理した。培地は毎日交換した。胚体内胚葉の細胞表面マーカーのCXCR4についてFACS解析を実施したところ、87%の細胞がマーカータンパク質を発現していたことが示された(図27A)。継代数49のヒト胚性幹細胞株H1の細胞を、Cytodex 1(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上で5回継代して増殖させて、同様の実験を実施したところ、マイクロキャリア上で分化した細胞の91%がCXCR4を発現していたことが明らかになった(図27B)。このことは、マイクロキャリア上で増殖させた細胞は、インスリン産生細胞に分化するための第1段階である、胚体内胚葉へと分化できるということを実証している。
【0117】
3種のマイクロキャリア、すなわちCytodex 1(登録商標)、Cytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)及びHILLEX(登録商標)II(Solohill,MI)は、H1細胞を付着させ、増殖させる。これらの3種のマイクロキャリア上で、H1細胞の分化を実施した。スピナーフラスコ中のこれらのマイクロキャリア上で、様々な継代数(1〜5回)で細胞を増殖させた(実施例5)。マイクロキャリアと細胞との懸濁液の、6〜8日間にわたる継代後のアリコートを、6又は12ウェルのプレートに移した。12ウェルプレートあたり合計15cm2のマイクロキャリアと細胞を、あるいは6ウェルプレートあたり30cm2マイクロキャリアと細胞を移した。次いで分化誘導培地をプレートのウェルに加え、このプレートを37℃にて固定式プラットフォーム上に設置した。100ng/mLのアクチビンA(PeproTech,NJ)と、20ng/mLのWnt3a(R&D Biosciences,MN)と、8ng/mLのbFGF(PeproTech,NJ)とを添加した、2%ウシ血清アルブミンフラクションV(脂肪酸不含)(MP Biomedicals,Ohio)含有RPMIで、2日間にわたって細胞を処理することで、胚体内胚葉へのヒト胚性幹細胞の分化を実施した。100ng/mLのアクチビンA(PeproTech,NJ)と、8ng/mLのbFGF(PeproTech,NJ)とを添加した2% FAF BSA含有RPMIで、更に2日間にわたって細胞を処理した。培地は毎日交換した。胚体内胚葉の細胞表面マーカーのCXCR4について、FACS解析を実施した(図28)。胚体内胚葉への分化を支持してCytodex 1(登録商標)マイクロキャリア、及びCytodex 3(登録商標)マイクロキャリアで細胞を増殖させたものではそれぞれ87%と92%であった一方で、HILLEX(登録商標)IIマイクロキャリアはこの実験で試験した他のマイクロキャリアと同程度には分化を支持しなかった(42%)。
【0118】
細胞密度がマイクロキャリア上の細胞の分化に影響を及ぼすか否かを判断するために、継代数40のヒト胚性幹細胞株H1の細胞を、スピナーフラスコ中のCytodex 3(登録商標)マイクロキャリア上で、8日間又は11日間のいずれかで増殖させた。次いで約15cm2当量のマイクロキャリアと細胞とを6ウェルディッシュに設置し、固定式プラットフォーム上に設置した。次いで細胞を、上記のような胚体内胚葉分化誘導培地中でインキュベートした。4日後に、CXCR4発現について細胞をFACSにより解析した。スピナーフラスコ中で6日間にわたって増殖させた細胞の87%がCXCR4を発現していた一方で、スピナーフラスコ中で11日間にわたって増殖させた細胞では、56%がCXCR4を発現していた(図29)。このことは、分化させる前の細胞の培養日数が重要であることを実証し、具体的には、細胞密度が高すぎる場合には、細胞が効率的に分化できなくなる場合があることを実証する。
【0119】
付着性と増殖性が十分であると判断された3種のマイクロキャリアの全てで、ヒト胚性幹細胞が膵臓内胚葉細胞へと分化し得るか否かを判断するために、継代数41のヒト胚性幹細胞株H1の細胞(継代数41のH1)を、Cytodex 1(登録商標)、Cytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)及びHILLEX(登録商標)IIマイクロキャリア(Solohill,MI)上に播種した(実施例1を参照されたい)。製造元の取扱説明書に従ってマイクロキャリアを調製した。30cm2のマイクロキャリアを、低接着性の6ウェルプレートに移した。2枚の10cm2プレートから、製造元からの取扱説明書に従って、TrypLE(商標)ExpressによりH1細胞を解離させた。1ウェルあたり5×105個細胞で、細胞を播種した。実施例3に記載の方法に従って、ビーズへの細胞の付着を実施した。簡潔に述べると、細胞とマイクロキャリアを、37℃にて、10μMのY27632添加MEF馴化培地中で4時間にわたって、1時間ごとに簡単に撹拌しながらインキュベートした。HILLEX(登録商標)IIマイクロキャリア及びCytodex 1(登録商標)マイクロキャリア上の細胞を、固定式プラットフォーム上に設置した。Cytodex 3(登録商標)マイクロキャリア上の細胞は、一晩静置した。以降、培地はY27632不含の培地で毎日交換した。
【0120】
この実験では、乏しい付着性に起因して、Cytodex 1(登録商標)プレートの細胞の大多数が、もはやマイクロキャリアに付着していなかった。しかしながら、より長い付着時間及び/又はより低速に固定された速度では、細胞付着性は改善され得る。7日後に、2% BSA(脂肪酸不含(FAF))(Proliant,IA)含有RPMIと、以下の増殖因子:bFGF(8ng/mL,(PeproTech,NJ))、アクチビンA(100ng/mL,(PeproTech,NJ))、Wnt3a(20ng/mL,(R&D Biosciences,MN))とを用い、細胞を胚体内胚葉へと分化させた。4日間にわたる第2の分化を通して、Wnt3aは不含の同様の培地で細胞を処理した。4日後の、2つ組試料のFACS解析では、77〜83%の細胞でCXCR4レベルが陽性だったことが明らかになった。胚体内胚葉発現は、異なるマイクロキャリア上で増殖させた細胞間で、等価であった。図30を参照されたい。
【0121】
次いで、FGF7(50ng/mL,(R&D Systems,MN))、KAAD−シクロパミン(0.25μM,(Calbiochem,NJ))を添加した、2% FAF BSA(Proliant,IA)含有DMEM/F12又はDMEM−HGにより、細胞を更に2日間にわたって分化させた。これを、ノギン(100ng/mL,(R&D Biosciences,MN))、FGF7(50ng/mL,(R&D Systems,MN))、レチノイン酸(2μM,(Sigma−Aldrich,MO))、及びKAAD−シクロパミン(0.25μM,(Calbiochem,NJ))を添加した、1% B−27supplement(Invitrogen,CA)含有DMEM/F12又はDMEM−HGにより、4日間にわたって処理した。次いで、ノギン(100ng/mL,(R&D Biosciences,MN))、DAPT(1μM,(Sigma−Aldrich,MO))、Alk5阻害剤II(1μM,(Axxora,CA))を添加した、1% B−27 supplement(13日,膵臓内胚葉,(Invitrogen,CA))含有DMEM/F12又はDMEM−HGにより、細胞を3日間にわたって分化させた。図31は、Q−PCRにより、膵臓特異的な遺伝子のNKX6.1、PDX1及びNGN3の発現レベルを示す。CT値は、HILLEX(登録商標)IIマイクロキャリア上で分化させた細胞が、効率的に分化せず、必要なβ細胞前駆体細胞マーカーを発現しないことを明瞭に示す。細胞は、3種のマイクロキャリアの全てで効率的に胚体内胚葉へと分化したが、HILLEX(登録商標)IIマイクロキャリアでは、膵臓前駆体への更なる分化は効率的ではなかった。
【0122】
ヒト胚性幹細胞が、インスリン産生細胞へと更に分化し得るか否かを判断するために、継代数45のH1細胞をCytodex 3(登録商標)マイクロキャリア(GE Healthcare Life Sciences,NJ)上で増殖させ、上記と同様に分化させた。簡潔に述べると、製造元からの取扱説明書に従って、TrypLE(商標)Expressにより、10cm2プレートからH1細胞を解離させた。6ウェルプレートのウェルあたり1又は2×106個細胞で細胞を播種した。ビーズへの細胞の付着を実施例3に記載する。簡潔に述べると、細胞とマイクロキャリアとを、10μMのY27632を添加したMEF馴化培地中で、37℃にて4時間にわたって、1時間ごとに簡単に撹拌しながらインキュベートした。次いで細胞を、一晩静置してインキュベートした。2日目に、培地を5μMのY27632を添加したMEF馴化培地に交換し、プレートを固定式プラットフォーム上に設置した。以降毎日、培地をY27632は不含の培地と交換した。5日目に、培地を胚体内胚葉分化誘導培地(以下の増殖因子:bFGF(8ng/mL,(PeproTech,NJ))、アクチビンA(100ng/mL,(PeproTech,NJ))、Wnt3a(20ng/mL,(R&D Biosciences,MN))を添加した、2% BSA(脂肪酸不含(FAF))(Proliant,IA)含有RPMI)と交換した。分化誘導の2日目及び3日目に、Wnt3aは不含の同様の培地で細胞を処理した。3日後の、2つ組試料のFACS解析では、97〜98%の細胞でCXCR4レベルが陽性だったことが明らかになった。次いで、FGF7(50ng/mL,(R&D Systems,MN)),KAAD−シクロパミン(0.25μM,(Calbiochem,NJ))を添加した、2%FAF BSA(Proliant,IA)含有DMEM−高グルコース(HG)により、更に2日間にわたって細胞を分化させた。これを、ノギン(100ng/mL,(R&D Biosciences,MN))、FGF7(50ng/mL,(R&D Systems,MN))、レチノイン酸(2μM,(Sigma−Aldrich,MO))、及びKAAD−シクロパミン(0.25μM,(Calbiochem,NJ))を添加した、1% B−27supplement(Invitrogen,CA)含有DMEM−HGにより、4日間にわたって処理した。次いで、ノギン(100ng/mL,(R&D Biosciences,MN))、DAPT(1μM,(Sigma−Aldrich,MO))、Alk5阻害剤II(1μM,(Axxora,CA))を添加した、1% B−27 supplement(Invitrogen,CA)含有DMEM−HG又はDMEM−F12により、細胞を3日間にわたって分化させた。続いてこれを、Alk5阻害剤II(1μM,(Axxora,CA))を添加したDMEM−HG又はDMEM−F12中で、7日間にわたって分化させた。最終分化はそれぞれDMEM−HG又はDMEM−F12で5日間だった。合計24日間にわたる分化が、膵臓内分泌ホルモンの発現を誘導する。図32は、このエンドポイントの細胞の、FACS解析結果を示す。播種密度が最も高く、かつ6〜24日間にわたってDMEM−HGで分化させた細胞が、最も高レベルのインスリン発現を有していた(図32)。
【0123】
別の方法としては、ヒト胚性幹細胞がインスリン産生細胞へと分化し得るか否かを判断するために、7日間にわたって継代数44のH1細胞を、スピナーフラスコ中のCytodex 3(登録商標)マイクロキャリア上で増殖させた(実施例5を参照されたい)。15cm2/ウェルで、細胞とマイクロキャリアとを12ウェルプレートに移し、これを37℃にて固定式プラットフォーム上に設置した。RMPIの代わりにDMEM/F12を添加して、細胞を上記のように胚体内胚葉へと分化させた。4日後のFACS解析では、3つ組解析において、75〜77%の細胞でCXCR4レベルが陽性だったことが明らかになった。次いで、FGF7(50ng/mL,(R&D Systems,MN))、KAAD−シクロパミン(0.25μM,(Calbiochem,NJ))を添加した、2%ウシ血清アルブミンフラクションV(脂肪酸不含)含有DMEM/F12により、細胞を更に3日間にわたって分化させた。これを、ノギン(100ng/mL,(R&D Biosciences,MN))、FGF7(50ng/mL,(R&D Systems,MN))、レチノイン酸(2μM,(Sigma−Aldrich,MO))、及びKAAD−シクロパミン(0.25μM,(Calbiochem,NJ))を添加した、1% B−27supplement(Invitrogen,CA)含有DMEM/F12により、4日間にわたって処理した。次いで、ノギン(100ng/mL,(R&D Biosciences,MN))、ネトリン4(100ng/mL,(R&D Biosciences,MN))、DAPT(1μM,(Sigma−Aldrich,MO))、Alk5阻害剤II(1μM,(Axxora,CA))を添加した、1% B−27 supplement(15日,膵臓内胚葉,(Invitrogen,CA))含有DMEM/F12により、細胞を3日間にわたって分化させた。続いてこれを、Alk5阻害剤II(1μM,(Axxora,CA))を添加した、1% B−27 supplement(21日,膵内分泌細胞,(Invitrogen,CA))含有DMEM/F12で6日間処理した。7日間にわたる最終処理では、1% B−27 supplement(28日,インスリン発現細胞,(Invitrogen,CA))を添加したDMEM/F12を用いた。図33は、Q−PCRにより、膵臓特異的な遺伝子のインスリン、Pdx1及びグルカゴンの発現レベルを示す。マイクロキャリアについてのデータは、前出のMATRIGEL(商標)(BD Biosciences,CA)コートした平面状表面で分化させた継代数42のH1細胞と比較する。マイクロキャリアで分化させた細胞では、これらの膵臓特異的遺伝子の発現レベルは、平面状表面で分化させた細胞と比較して同様であるか、あるいはそれよりも勝っている。
【0124】
同様の実験を、Cytodex 3(登録商標)マイクロキャリア上で及び拡張型スピナーフラスコ中で継代した、継代数38のH9細胞で実施した。15cm2のマイクロキャリアと細胞とのアリコートを12ウェルプレートに設置し、分化誘導培地と共に、固定式プラットフォーム上に設置した。これを、MATRIGEL(商標)(BD Biosciences,CA)コートした6ウェルプレート上に設置した、細胞と比較した。RPMIとサプリメント(supplements)の中で胚体内胚葉への細胞の分化を実施したところ、平均して83%の細胞がCXCR4を発現していたのに対して(試料は2つ組)、平面状基材上では72%の細胞がCXCR4を発現していた(図34)。更に膵臓内胚葉(15日)、膵内分泌細胞(22日)及びインスリン発現細胞(29日)への分化では、マイクロキャリア上で増殖させた細胞と、平面状基材上で増殖させた細胞との間では、インスリン及びグルカゴンについて同様の発現レベルが示された(図35)。培地成分は、更に1日内分泌細胞分化誘導成分で処理した、上記のH1分化誘導実験について列挙したものと同一のものであった。インスリン発現ステージでは、細胞は22日のものと比較してインスリン発現の驚くべき減少を示した。この減少は、マイクロキャリア試料及び平面試料の双方で指摘されたことから、付着基材によるものである可能性は低い。このことは、H9細胞が、マイクロキャリア上でも少なくとも膵内分泌細胞へと良好に分化し得るということを示す。
【0125】
全般的に、2種の異なるヒト胚性幹細胞株のH1とH9は、Cytodex 3(登録商標)マイクロキャリア上で膵内分泌細胞へと分化でき、これらの細胞をラージスケールの培養系で増量及び分化させることの可能性を実証する(図17、21、33及び35)。ヒト胚性幹細胞は、少なくとも3種のマイクロキャリアビーズに付着及び増殖できたが、この細胞は少なくとも胚体内胚葉へと分化し得る(図28)。これらの結果は、治療用途のためにヒト胚性幹細胞を増殖させ分化させ得る方法を実証する。
【0126】
実施例8:3Dマイクロキャリア系培養法で単独の細胞として継代したヒト胚性幹細胞は、多能性を維持しつつ、ECM不含表面に移して培養することができる。
H1ヒト胚性幹細胞を、実施例5に記載の方法に従ってマイクロキャリア上で培養した。細胞をマイクロキャリアから取り外し、3μMのGlycyl−H 1152ジヒドロクロリドを添加したMEFCM16と共に、Nunc4、Nunc13、CELLBIND(商標)、又はPRIMARIA(商標)組織培養ポリスチレン(TCPS)平面状表面に蒔いた。細胞は6ウェルプレートに100,000個細胞/cm2の密度で播種し、次いで各表面上で更に1継代培養した。次いで細胞を、TrypLEで浮かせてフローサイトメトリーにより多能性マーカーについて試験するか、mRNA精製及びqRT−PCRのためにウェル中でRLTに溶解させるか、あるいは胚体内胚葉へと分化させるかした。2% BSA、100ng/mLのアクチビンA、20ng/mLのWnt3a、8ng/mLのbFGF及び3μMのGlycyl−H 1152ジヒドロクロリドを添加したRPMI培地で24時間にわたって細胞を処理することで、分化を誘導した。次いで培地を、2% BSA、100ng/mLのアクチビンA、8ng/mLのbFGF、及び3μMのGlycyl−H 1152ジヒドロクロリドを添加したRPMI培地に交換し、更に48時間にわたって培地を毎日交換した。
【0127】
フローサイトメトリー又はqRT−PCRのいずれかを用いて多能性マーカーにより測定したところ、マイクロキャリア上で培養した後にNunc4、Nunc13、CellBIND又はPrimaria組織培養ポリスチレン(TCPS)平面状表面へと移した細胞は、各平面状表面上で2回継代した後に多能性を維持していた(図36)。加えて細胞は、フローサイトメトリー又はqRT−PCRのいずれかにより測定されるものとして、胚体内胚葉運命への分化能を維持していた(図37)。Cytodex 3(登録商標)マイクロキャリア上で継代し、胚体内胚葉へと分化させたH1及びH9ヒト胚性幹細胞の比較試験でも、同様の結果が得られた(図38)。
【0128】
これらの結果は、ヒト胚性幹細胞を、マイクロキャリア上で継代した後に、次いで多能性を維持しつつ他の表面上で培養することができることを示す。細胞を他の表面へと移して効率的に分化誘導することもできる。
【0129】
実施例9:ヒト胚性幹細胞は、有糸分裂を不活性化した線維芽細胞フィーダー上でのクラスター/コロニー形式の培養から、少なくとも10回の継代にわたる、ECM不含表面上での単独細胞としての培養へと、多能性を損なうことなく、及び線維芽細胞フィーダーを手動で除去することなく直接移行させることができる。
ヒト胚性幹細胞株は、現在のところ、単独の細胞が増殖したコロニー、又は胚盤胞由来の少数の細胞のクラスターを発展させる方法に由来する。次いでこのコロニーを連続的に継代し、細胞株を構成するのに十分な細胞クラスター/コロニーが利用できるようになるまで増殖させる。ヒト胚性幹細胞の多能性と核型安定特性を維持することを目的として、一度細胞株が誘導されると、高度に再現性のあるヒト胚性幹細胞培養のための、当該技術分野の現行の標準方法では、有糸分裂不活性化線維芽細胞のフィーダー細胞層上の、ヒト胚性幹細胞クラスター/コロニーを維持し、及び手作業での撹乱(manual disruption)又はコラゲナーゼ若しくは中性プロテアーゼ若しくはこれらのブレンドでの穏やかな酵素的大容量継代を用いて、細胞を継代する。これらの継代法はヒト胚性幹細胞クラスターを維持し、かつヒト胚性幹細胞の、コロニー形式の増殖を促進する。安定したヒト胚性幹細胞株の確立後、この細胞をMATRIGEL(商標)などの細胞外マトリックス(ECM)基質へと移すことができる。しかしながら、細胞を線維芽細胞フィーダー上で増殖させるかあるいはECM基質上で増殖させるかのいずれにせよ、技術者に具体的に指示されるヒト胚性幹細胞に関して推奨される継代法は、ヒト胚性幹コロニーを十分に解離させはしない。
【0130】
現行の最も良好な、哺乳類細胞のラージスケール培養の実施法は、3次元培養容器を使用するものである。この容器は恒常性、つまり一定の状態を厳密に維持し、かつ足場依存性の細胞を支持するためにマイクロキャリアを組み込むことができる。しかしながら、ヒト胚性幹細胞培養(線維芽細胞フィーダー上又はECM基質上で増殖させる)、及びクラスター/コロニー形式での培養に使用される現行の標準法は、マイクロキャリア上でのヒト多能性胚性幹細胞培養物の良好な増殖及び維持に対して技術的な障害を提起することになるが、それは、これらの方法をラージスケールでのマイクロキャリア培養に転用するのは容易ではないからである。マイクロキャリアでヒト胚性幹細胞を効果的に増殖させるためには、ヒト胚性幹細胞培養物は、当該技術分野の現行標準法でのようなコロニー又はクラスターとしてではなく、単独細胞として継代可能であるべきである。加えて、ヒト胚性幹細胞は、フィーダー細胞層又はECM基質無しに増殖可能であるべきである。
【0131】
我々は、これらの技術的障害に取り組む方法を以下に記載する。我々は、有糸分裂不活性化線維芽細胞フィーダー層上のクラスター/コロニーに基づくヒト胚性幹細胞培養を、線維芽細胞フィーダー層、又はMATRIGEL若しくは他の細胞外マトリックス基質でコートした表面が下層として存在する必要のない単独細胞培養系へと、直接転換する方法を実証する。この方法は、何らかの方法により線維芽細胞フィーダー細胞を手動で除去する必要のない、あるいは総細胞集団から多能性細胞を選別する必要のない、ヒト胚性幹の大容量の継代を活用することで、培養を、コロニー形式の線維芽細胞フィーダー系培養から、Rhoキナーゼ(ROCK)阻害剤のGlycyl−H 1152ジヒドロクロリドの存在下で、PRIMARIAでの、フィーダー不含/マトリックス不含の培養へと直接転換する。この方法は、必要量を制御するように付着させるための密閉容器で達成でき、並びに多能性及び胚体内胚葉への分化能を保持し、かつ線維芽細胞の細胞集団は含有しない、非常に均質なヒト胚性幹培養物を生成する。
【0132】
方法:培地を吸引し、PBSで洗浄し、次いで細胞を解離酵素(コラゲナーゼ,Accutase(商標),又はTrypLE)で処理することにより、細胞をルーチン的に継代した。コラゲナーゼは濃度1mg/mLで使用した;Accutase(商標)又はTrypLEは、1X貯蔵濃度で使用した。全ての酵素は室温に戻してから使用した。2% BSAのDMEM/F12溶液を各ウェルに添加し、細胞を酵素で処理した後に、この細胞を溶液に均一に懸濁した。次いで細胞を200gで5分間にわたって遠心し、細胞ペレットと、追加の、2% BSA含有DMEM/F12溶液とを、再懸濁した細胞に加え、この細胞懸濁液を50mLの滅菌コニカルチューブに分注して200gで5分間にわたって遠心した。
【0133】
我々は連続的な方法を用い、MEF系培地をAccutase(商標)、TrypLE(商標)、又はコラゲナーゼのいずれかで処理し、クラスター/コロニー形式のヒト胚性幹細胞をPrimaria表面に高密度継代することで、線維芽細胞フィーダーを除去した。最初の継代で、細胞を、マウス胚性線維芽細胞(MEF)馴化培地(CM)に1:30希釈したMATRIGEL(商標)でコートしたT−25フラスコに蒔くか、あるいは細胞を、T−25 PRIMARIA(商標)培養フラスコ中の、3μMのGlycyl−H 1152ジヒドロクロリドを添加したMEF−CMに蒔くかした。全ての細胞を1:3.5の分割比で蒔き、かつ細胞を10分間にわたって酵素に曝露した。TrypLE(商標)又はAccutase(商標)で浮かせた細胞の細胞数を、トリパンブルー染色した細胞を血球計算板を用いて計数することで判定した。細胞を蒔いた後、培地は毎日交換し、MEF−CM+3μMのGlycyl−H 1152ジヒドロクロリドに蒔いたこの細胞に、毎日MEF−CM+1μMのGlycyl−H 1152ジヒドロクロリドを供給し、多能性及び分化に関するmRNAマーカーの発現について、試料を解析した。マトリックス不含条件下で、単独細胞として2回継代したhESCsは、多能性遺伝子の遺伝子発現を維持し、かつ分化遺伝子の発現は阻害した(図39)。
【0134】
2回目の継代:TrypLE(商標)又はAccutase(商標)に10分曝露することで、細胞を分割比1:4で継代した。我々はまた、細胞を処理し、脱着についてモニターすることにより経験的に決定した、より短い酵素曝露時間を導入した。我々は、細胞を浮き上がらせるのには、TrypLE(商標)に対しては3分間の曝露、及びAccutase(商標)に対しては5分間の曝露で十分であることを観察した。酵素で細胞を処理した後に、細胞を上記のように継代し、細胞mRNAのアリコートを、継代時にqRT−PCR用に採取した。
【0135】
3回目の継代:コンフルエンスに達したら細胞をPBSで洗浄し、3分若しくは10分(TrypLE(商標))又は5分若しくは10分(Accutase(商標))かけて酵素によりバラバラにし、2%BSA含有DMEM/F12に懸濁し、遠心し、2%BSA含有DMEM/F12で再度洗浄し、遠心し、次いで再懸濁して各培地に蒔いた。この継代時には、細胞を1:4比で、並びに同様に更に2種の分割比(1:8及び1:16)で蒔いた。各継代時に、細胞mRNAのアリコートをqRT−PCR用に採取した。
【0136】
4回目以降の継代:2回目及び3回目の継代時に採用された、酵素への曝露時間と継代比についての条件を、4回目以降の継代でも維持した。培養物がコンフルエンスまで増殖したら、細胞をPBSで洗浄し、特定の時間をかけて酵素によりバラバラにし、2%BSA含有DMEM/F12に懸濁し、遠心し、2% BSA含有DMEM/F12で再度洗浄し、遠心し、次いで再懸濁して各培地に特定の蒔き込み比で蒔いた。PRIMARIAに細胞を蒔くための培地には、蒔く際に3μMのGlycyl−H 1152ジヒドロクロリドを添加した。細胞を蒔いた後、培地は毎日交換し、MEF−CM+3μMのGlycyl−H 1152ジヒドロクロリドに蒔いたこの細胞に、毎日MEF−CM+1μMのGlycyl−H 1152ジヒドロクロリドを供給した。継代時に、qRT−PCR用に細胞mRNAのアリコートを採取した。
【0137】
8回を超える継代時には、継代終了時に、多能性表面マーカーに関するフローサイトメトリーによって(図40)、並びに多能性マーカー及び分化マーカーに関するqRT−PCRによって(図41、42及び43)、多能性について細胞を解析した。2% BSAと、100ng/mLのアクチビンAと、20ng/mLのWnt3aと、8ng/mLのbFGFと、3μMのGlycyl−H 1152ジヒドロクロリドとを添加したRPMI培地で24時間にわたって細胞を処理することで、細胞を同様に胚体内胚葉へと分化させた。次いで培地を、2% BSA、100ng/mLのアクチビンA、8ng/mLのbFGF、及び3μMのGlycyl−H1152ジヒドロクロリドを添加したRPMI培地に交換し、更に48時間にわたって培地を毎日交換した。次に、胚体内胚葉へと分化させた試料を、フローサイトメトリーによって、胚体内胚葉マーカーCXCR4の存在について試験した(図40)。
【0138】
これらの結果は、コロニー形式の、線維芽細胞フィーダー系培養物から、Rhoキナーゼ(ROCK)阻害剤のGlycyl−H 1152ジヒドロクロリドの存在下の、PRIMARIAでのフィーダー不含/マトリックス不含培養への大容量の継代は、多能性及び胚体内胚葉への分化能を保持し、かつ線維芽細胞の細胞集団は含有しない非常に均質なヒト胚性幹細胞培養物を生じることを示す。
【0139】
実施例10:組織培養用プラスチックからマイクロキャリアへのヒト胚性幹細胞の移動
H1細胞を、PRIMARIA(商標)(カタログ番号353846,Becton Dickinson,Franklin Lakes,NJ)組織培養プレート(実施例9の方法)で培養し、TrypLE(商標)Expressにより3〜5分処理することで放出させ、10μMのY27632(Sigma−Aldrich,MO)添加MEF−CM中にCytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)又はHILLEX(登録商標)II(Solohill,MI)マイクロキャリアを含む6ウェルの組織培養用無処理プレートに播種した。対照として、MATRIGEL(BD Biosciences,CA)でコートしたプレートで増殖させ、かつコラゲナーゼ(1mg/mL)を用いて継代した、継代数46のH1細胞を放出させ、同様の方法でマイクロキャリアに播種した。プレートを、37℃で5時間にわたって、45分ごとに手で撹拌しながらインキュベートした。次いでプレートを37℃にて固定式プラットフォーム上に設置した。培地は、毎日10μMのY27632(Sigma−Aldrich,MO)添加MEF−CMで交換した。画像解析は、3日目の時点で、マイクロキャリアに対する細胞の良好な付着を示す(図44)。7日後に細胞を放出させ(以下の実施例4に記載)、多能性マーカーのCD9、SSEA−4、SSEA−3、TRA−1−60、TRA−1−81に関してFACSで解析した(図45)。多能性マーカーのほとんどが、90〜100%の細胞で発現していた。Cytodex 3(登録商標)(GE Healthcare Life Sciences,NJ)マイクロキャリアで増殖させようが、HILLEX(登録商標)II(Solohill,MI)マイクロキャリアで増殖させようが、Accutase(商標)(Millipore,MA)で継代した細胞とTrypLE(商標)Express(Invitrogen,CA)で継代した細胞の間に明瞭な違いは存在しない。全般的に、細胞は、PRIMARIA(商標)(カタログ番号353846,Becton Dickinson,Franklin Lakes,NJ)細胞培養用プラスチックからマイクロキャリアに移した場合に多能性を保持した。
【0140】
次に、マイクロキャリア上のこれらのH1細胞は、胚体内胚葉へと分化した。この方法は実施例7に記載する。分化の4日後に、H1細胞をマイクロキャリアから放出させ、FACS解析を実施したところ、82%を超える細胞がCXCR4を発現していることが示された。図46を参照されたい。細胞は、マイクロキャリアの種類又はPRIMARIA(商標)(カタログ番号353846,Becton Dickinson,Franklin Lakes,NJ)から継代する際の酵素には関係なく、胚体内胚葉へと効率的に分化した。これは増量系の柔軟性を証明し、かつプラスチック及びマイクロキャリア上にマトリックスを必要とすることなく細胞を増殖させる。
【0141】
実施例11:セルロース混合エステルからなる平面状基材からマイクロキャリアへのヒト胚性幹細胞の移動
米国特許出願第61/116,452号に開示の方法に従って、セルロース混合エステルからなる平面状基材で、H1細胞を12継代にわたって培養した。TrypLE(商標)Expressで3〜5分処理することで平面状基材から細胞を放出させ、10mMのY27632(Sigma−Aldrich,MO)添加MEF−CM中にCYTODEX 3(登録商標)(GE Healthcare Life Sciences,NJ)マイクロキャリア又はHILLEX(登録商標)II(Solohill,MI)マイクロキャリアを含む6ウェルの組織培養用無処理プレートに播種した。対照として、MATRIGEL(商標)(BD Biosciences,CA)でコートしたプレートで増殖させ、かつコラゲナーゼ(1mg/mL)を用いて継代した、継代数44のH1細胞を放出させ、同様の方法でマイクロキャリアに播種した。プレートを、37℃で5時間にわたって、45分ごとに手で撹拌しながらインキュベートした。次いでプレートを37℃にて固定式プラットフォーム上に設置した。培地は毎日交換した。7日後に細胞を放出させ(実施例4に記載)、多能性マーカーのCD9、SSEA−4、SSEA−3、TRA−1−60、TRA−1−81に関してFACSで解析した(図47)。ほとんどの多能性マーカーが、90%を超える細胞で発現していた。CYTODEX 3(登録商標)マイクロキャリアで増殖させた細胞と、HILLEX(登録商標)IIマイクロキャリアで増殖させた細胞との間に明瞭な違いは存在しなかった。継代数44のH1対照細胞は、多能性が他の実験により確認されていたことから、HILLEX(登録商標)IIマイクロキャリアで増殖させた後の多能性について試験しなかった(実施例5を参照されたい)。全般的に、細胞は、セルロース混合エステルからなる平面状基材からマイクロキャリアへと移した場合にも多能性を維持していた。
【0142】
次に、実施例7に記載の方法に従って、マイクロキャリア上のH1細胞を胚体内胚葉へと分化させた。分化の4日後に、H1細胞をマイクロキャリアから放出させ、FACS解析を実施したところ、65%を超える細胞がCXCR4を発現していることが示された(図48)。細胞は、マイクロキャリアの種類とは無関係に効率的に胚体内胚葉へと分化した。HILLEX(登録商標)II(Solohill,MI)マイクロキャリア上で胚体内胚葉へと分化した細胞の数は少数であったように思われる。細胞の分化能は、増量系の柔軟性を証明する。加えて細胞を、動物成分マトリックスに対する任意の必要性を除去して、メンブレン及びマイクロキャリア上に直接増殖させかつ分化させることができる。
【0143】
【表1】
【0144】
【表2】
【0145】
【表3】
【0146】
本明細書を通して引用された刊行物は、その全体が参照により本明細書に組み込まれる。以上、本発明の様々な態様を実施例及び好ましい実施形態を参照して説明したが、本発明の範囲は、上記の説明文によってではなく、特許法の原則の下で適切に解釈される以下の「特許請求の範囲」によって定義されるものである点は認識されるであろう。
【特許請求の範囲】
【請求項1】
多能性幹細胞の増殖方法であって、
a.多能性幹細胞集団を、第1容量のマイクロキャリアに付着させる工程と、
b.前記多能性幹細胞を、前記第1容量のマイクロキャリア上で培養する工程と、
c.前記多能性幹細胞を、前記第1容量のマイクロキャリアから取り外す工程と、
d.前記多能性幹細胞集団を、第2容量のマイクロキャリアに付着させる工程と、を含む、方法。
【請求項2】
前記マイクロキャリア上の多能性幹細胞を培養する工程、取り外す工程及び付着させる工程が、連続容量のマイクロキャリアを用いて繰り返される、請求項1に記載の方法。
【請求項3】
前記第1容量のマイクロキャリアが、デキストランマイクロキャリア及びポリスチレンマイクロキャリアからなる群から選択される、請求項1に記載の方法。
【請求項4】
前記第2容量のマイクロキャリアが、デキストランマイクロキャリア及びポリスチレンマイクロキャリアからなる群から選択される、請求項1に記載の方法。
【請求項5】
前記多能性幹細胞を、Rhoキナーゼ阻害剤を含有している培地中で、前記第1容量のマイクロキャリアに付着させる、請求項1に記載の方法。
【請求項6】
前記多能性幹細胞を、Rhoキナーゼ阻害剤を含有している培地中で、前記第2容量のマイクロキャリアに付着させる、請求項1に記載の方法。
【請求項7】
前記多能性幹細胞を、酵素処理により前記第1容量のマイクロキャリアから取り外す、請求項1に記載の方法。
【請求項8】
前記多能性幹細胞を、酵素処理により前記第2容量のマイクロキャリアから取り外す、請求項1に記載の方法。
【請求項9】
前記多能性幹細胞を前記第2容量のマイクロキャリアに付着させる前に、前記細胞から前記第1容量のマイクロキャリアを取り外す、請求項1に記載の方法。
【請求項10】
多能性幹細胞を、インスリンを発現している細胞へとマイクロキャリア上で分化させる方法であって、
a.前記多能性幹細胞を、ある容量のマイクロキャリアに付着させる工程と、
b.前記多能性幹細胞を、胚体内胚葉系に特徴的なマーカーを発現している細胞へと分化させる工程と、
c.前記胚体内胚葉系に特徴的なマーカーを発現している細胞を、膵臓内胚葉系に特徴的なマーカーを発現している細胞へと分化させる工程と、
d.前記膵臓内胚葉系に特徴的なマーカーを発現している細胞を、膵内分泌系に特徴的なマーカーを発現している細胞へと分化させる工程と、
e.前記膵内分泌系に特徴的なマーカーを発現している細胞を、インスリンを発現している細胞へと分化させる工程と、を含む、方法。
【請求項1】
多能性幹細胞の増殖方法であって、
a.多能性幹細胞集団を、第1容量のマイクロキャリアに付着させる工程と、
b.前記多能性幹細胞を、前記第1容量のマイクロキャリア上で培養する工程と、
c.前記多能性幹細胞を、前記第1容量のマイクロキャリアから取り外す工程と、
d.前記多能性幹細胞集団を、第2容量のマイクロキャリアに付着させる工程と、を含む、方法。
【請求項2】
前記マイクロキャリア上の多能性幹細胞を培養する工程、取り外す工程及び付着させる工程が、連続容量のマイクロキャリアを用いて繰り返される、請求項1に記載の方法。
【請求項3】
前記第1容量のマイクロキャリアが、デキストランマイクロキャリア及びポリスチレンマイクロキャリアからなる群から選択される、請求項1に記載の方法。
【請求項4】
前記第2容量のマイクロキャリアが、デキストランマイクロキャリア及びポリスチレンマイクロキャリアからなる群から選択される、請求項1に記載の方法。
【請求項5】
前記多能性幹細胞を、Rhoキナーゼ阻害剤を含有している培地中で、前記第1容量のマイクロキャリアに付着させる、請求項1に記載の方法。
【請求項6】
前記多能性幹細胞を、Rhoキナーゼ阻害剤を含有している培地中で、前記第2容量のマイクロキャリアに付着させる、請求項1に記載の方法。
【請求項7】
前記多能性幹細胞を、酵素処理により前記第1容量のマイクロキャリアから取り外す、請求項1に記載の方法。
【請求項8】
前記多能性幹細胞を、酵素処理により前記第2容量のマイクロキャリアから取り外す、請求項1に記載の方法。
【請求項9】
前記多能性幹細胞を前記第2容量のマイクロキャリアに付着させる前に、前記細胞から前記第1容量のマイクロキャリアを取り外す、請求項1に記載の方法。
【請求項10】
多能性幹細胞を、インスリンを発現している細胞へとマイクロキャリア上で分化させる方法であって、
a.前記多能性幹細胞を、ある容量のマイクロキャリアに付着させる工程と、
b.前記多能性幹細胞を、胚体内胚葉系に特徴的なマーカーを発現している細胞へと分化させる工程と、
c.前記胚体内胚葉系に特徴的なマーカーを発現している細胞を、膵臓内胚葉系に特徴的なマーカーを発現している細胞へと分化させる工程と、
d.前記膵臓内胚葉系に特徴的なマーカーを発現している細胞を、膵内分泌系に特徴的なマーカーを発現している細胞へと分化させる工程と、
e.前記膵内分泌系に特徴的なマーカーを発現している細胞を、インスリンを発現している細胞へと分化させる工程と、を含む、方法。
【図1】

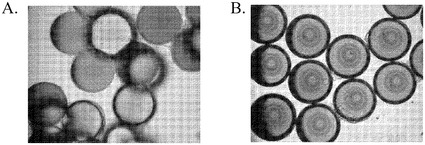
【図2】

【図3】

【図4】
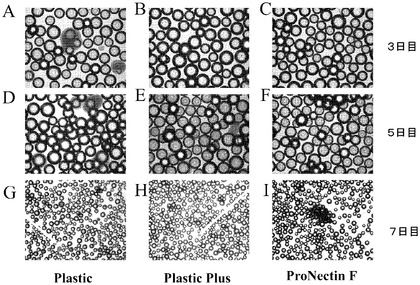
【図5】
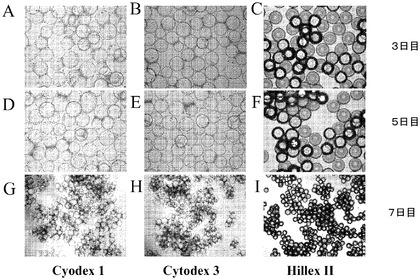
【図6】
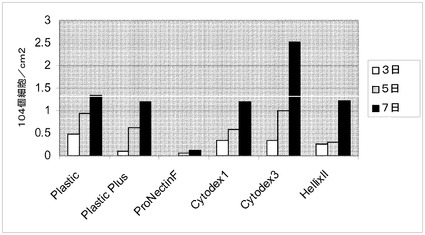
【図7】
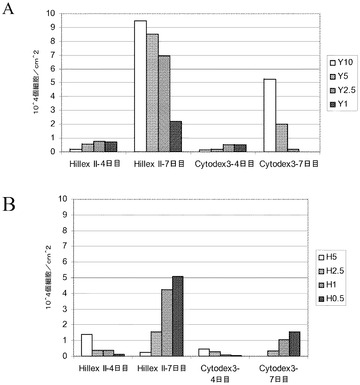
【図8】
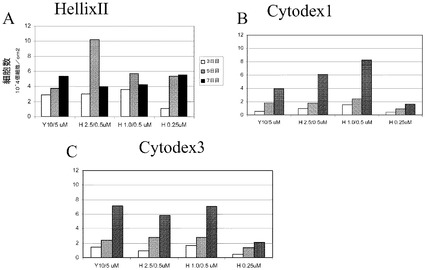
【図9】

【図10】
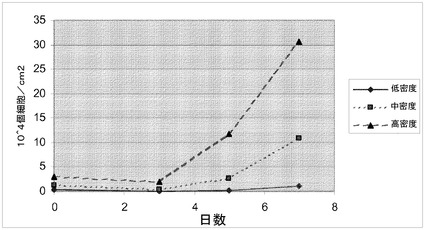
【図11】
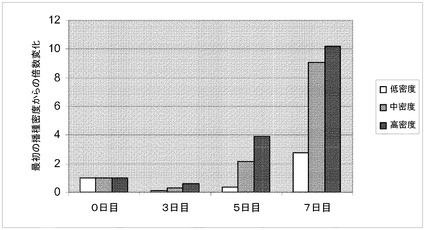
【図12】
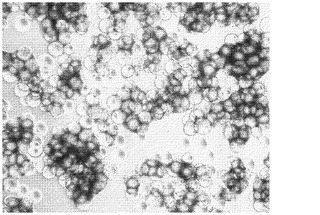
【図13】
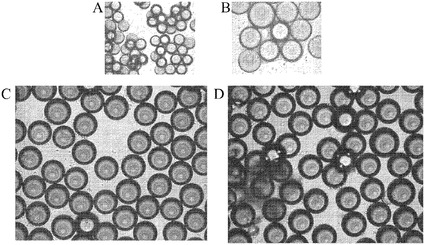
【図14】
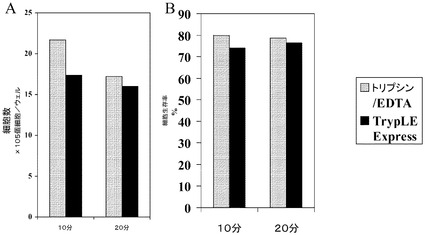
【図15】
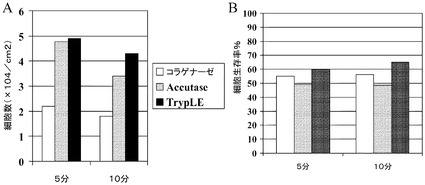
【図16】

【図17】
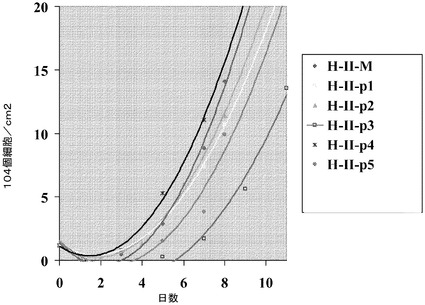
【図18】
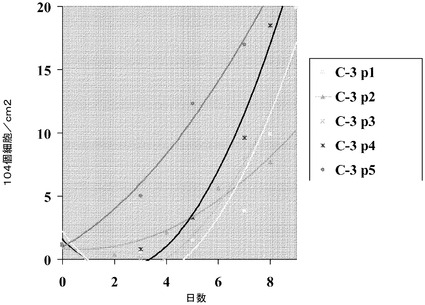
【図19】
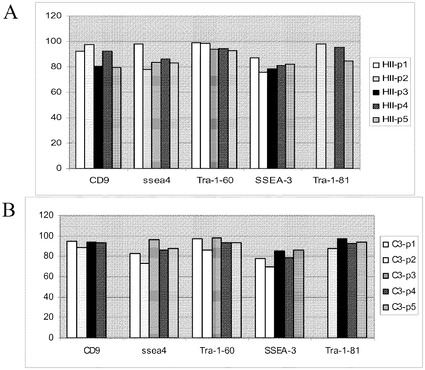
【図20】
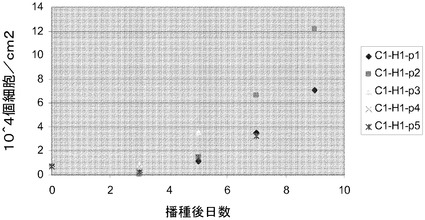
【図21】
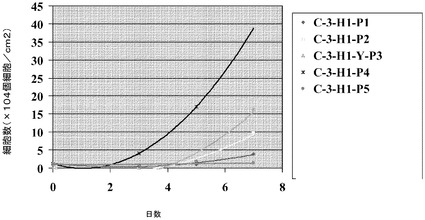
【図22】
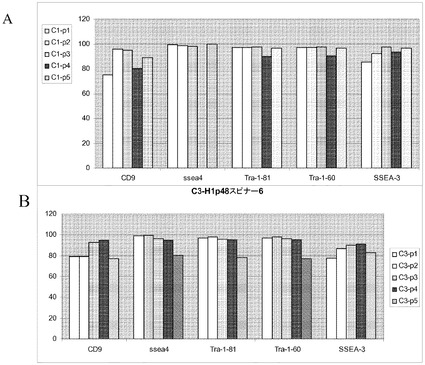
【図23】
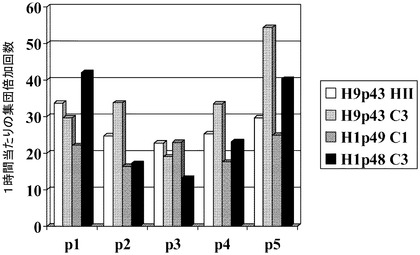
【図24】
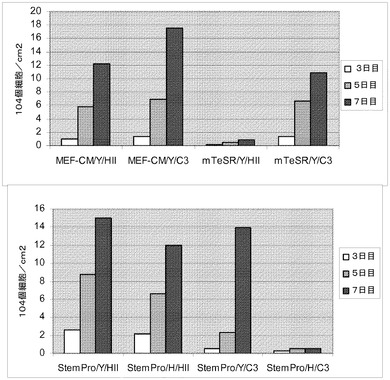
【図25】
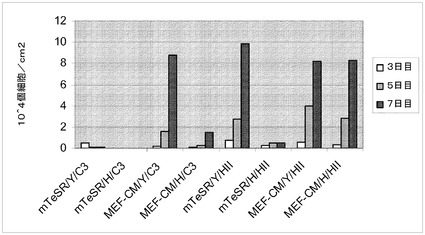
【図26】

【図27】
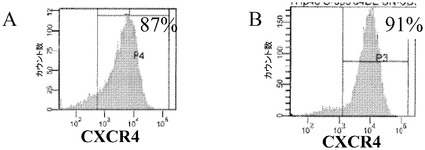
【図28】

【図29】
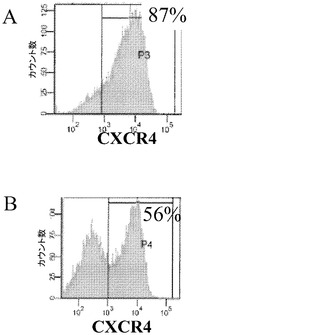
【図30】
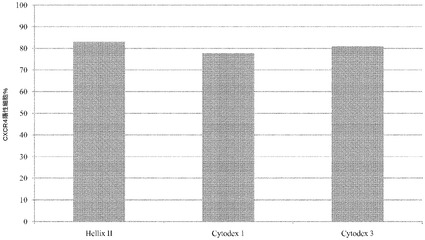
【図31】
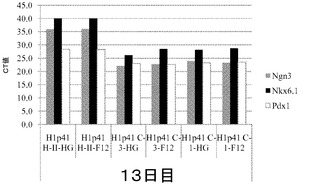
【図32】
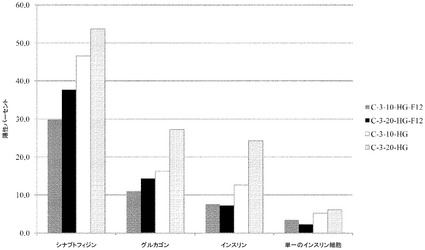
【図33】
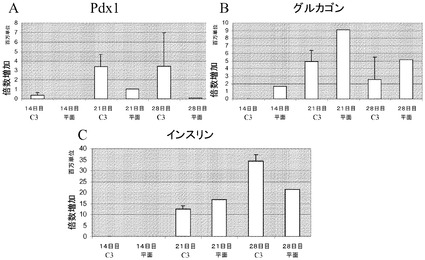
【図34】

【図35】
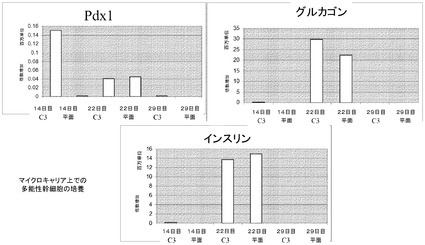
【図36】
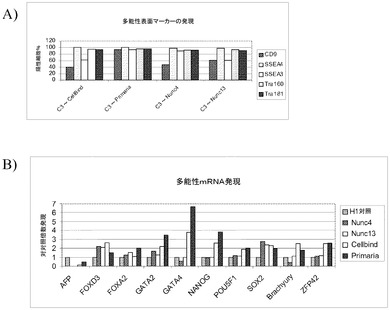
【図37】
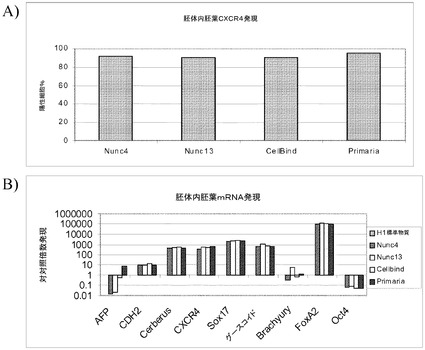
【図38】
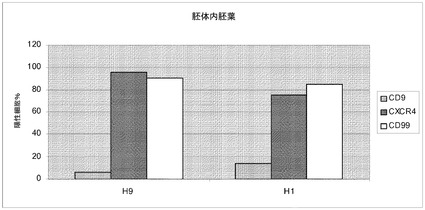
【図39】
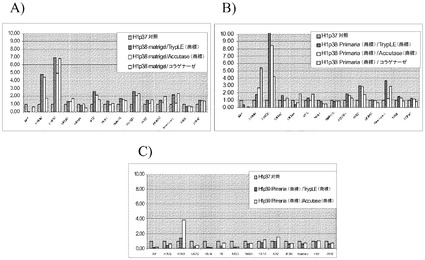
【図40】
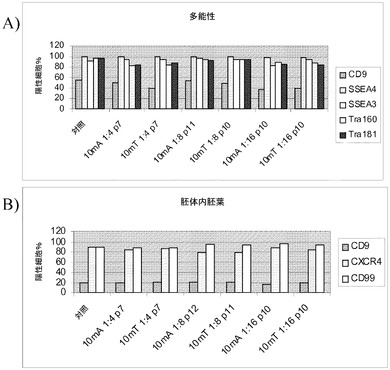
【図41】
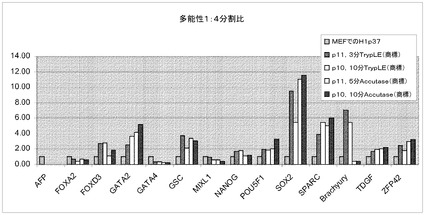
【図42】
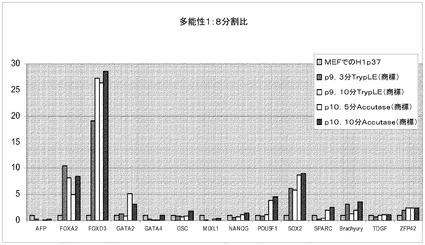
【図43】
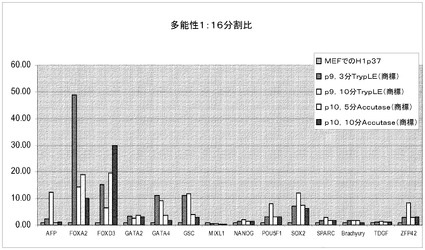
【図44】
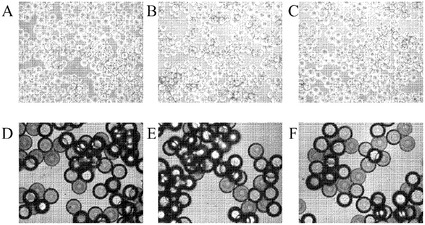
【図45】
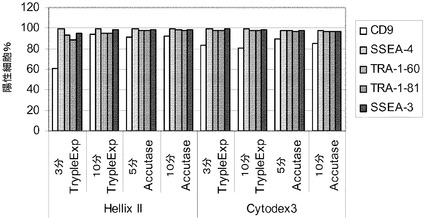
【図46】
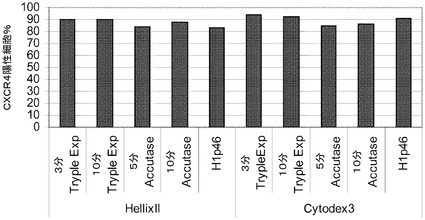
【図47】
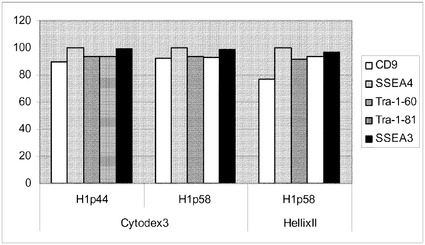
【図48】
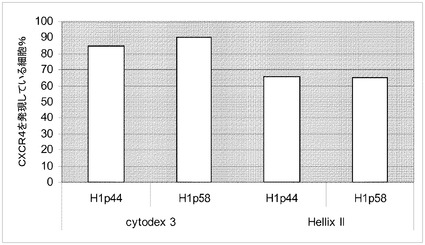
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【図31】
【図32】
【図33】
【図34】
【図35】
【図36】
【図37】
【図38】
【図39】
【図40】
【図41】
【図42】
【図43】
【図44】
【図45】
【図46】
【図47】
【図48】
【公表番号】特表2012−509085(P2012−509085A)
【公表日】平成24年4月19日(2012.4.19)
【国際特許分類】
【出願番号】特願2011−537603(P2011−537603)
【出願日】平成21年11月19日(2009.11.19)
【国際出願番号】PCT/US2009/065062
【国際公開番号】WO2010/059775
【国際公開日】平成22年5月27日(2010.5.27)
【出願人】(509087759)ヤンセン バイオテツク,インコーポレーテツド (77)
【Fターム(参考)】
【公表日】平成24年4月19日(2012.4.19)
【国際特許分類】
【出願日】平成21年11月19日(2009.11.19)
【国際出願番号】PCT/US2009/065062
【国際公開番号】WO2010/059775
【国際公開日】平成22年5月27日(2010.5.27)
【出願人】(509087759)ヤンセン バイオテツク,インコーポレーテツド (77)
【Fターム(参考)】
[ Back to top ]