
マイクロチャンバアレイを用いた蛍光偏光解消法による標的物質測定方法
【課題】蛍光偏光解消法による試料溶液中の微量標的物質の濃度を測定する方法を提供する。
【解決手段】本発明の標的物質測定方法は、試料溶液と、試料溶液中に存在が疑われる標的物質に結合可能な蛍光標識物質を混合する工程と、得られた混合液を、マイクロチャンバアレイの各マイクロチャンバに分注する工程と、蛍光偏光解消法により、蛍光標識物質が結合した標的物質が分注されたマイクロチャンバを検出することにより、生体試料中の標的物質の濃度を測定する工程とを備える。
【解決手段】本発明の標的物質測定方法は、試料溶液と、試料溶液中に存在が疑われる標的物質に結合可能な蛍光標識物質を混合する工程と、得られた混合液を、マイクロチャンバアレイの各マイクロチャンバに分注する工程と、蛍光偏光解消法により、蛍光標識物質が結合した標的物質が分注されたマイクロチャンバを検出することにより、生体試料中の標的物質の濃度を測定する工程とを備える。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、蛍光偏光解消法による試料溶液中の標的物質の濃度を測定する方法に関する。
【背景技術】
【0002】
多数の検体について測定を行なう生理活性物質の探索におけるハイスループットスクリーニングあるいは体外診断システムにおいて、効率よく試験を実施するためには、簡便性、感度、安定性に優れた検出系を用いる必要がある。これまでこれらの検出系としては大きく分けて紫外可視吸光法、化学発光・生物発光法、放射性同位元素(RI)を用いる検出系が用いられてきた。
【0003】
このうち蛍光法は高選択性・高感度が期待できる検出系であり、紫外可視吸光法とともによく用いられている。また、蛍光法はRIを用いる方法に比べ測定時間が短くてすむだけでなく、管理区域内での操作の必要がない、放射性廃棄物の問題がないという点においても優れており、RIを用いた方法からの転換も行なわれてきている。
【0004】
しかし、化合物ライブラリ−あるいは血液成分を含む臨床検体などの多数の検体について試験を行なう場合、蛍光を発する検体も含まれていることがあり、その夾雑蛍光により測定がしばしば妨害されるという問題点がある。
【0005】
蛍光偏光解消法は、1926年にPerrinらによって確立された大きな分子ほど回転ブラウン運動の回転速度が遅くなることを利用した方法である。蛍光性分子により標識された小さな分子は、回転速度が速いため蛍光の偏光が解消され、蛍光偏光度Pは小さい値を示す。一方、大きな分子の場合は回転速度が遅いため蛍光の偏光は解消せず、Pは大きな値を示す(非特許文献1)。つまり、標的物質に結合可能な蛍光標識物質と標的物質が結合するとPは増大することになる。このPの変化を測定することで、該標的物質の試料溶液中の濃度を測定することができる。
【0006】
尚、蛍光偏光度は、以下の式によって計算される。
P=(IH−IL)/(IH+IL)
(式中、IHは、励起光の偏光面に平行な平面で偏光された発光強度であり、ILは励起光の偏光面に垂直な平面で偏光された発光強度である。)
【0007】
また、蛍光偏光解消法の指標として蛍光異方性rも用いられる。この場合も蛍光偏光度Pと同様に、蛍光異方性rの値の変化を測定することで、該標的物質の試料溶液中の濃度を測定することができる。蛍光偏光度Pと蛍光異方性rとの関係は以下の式で表すことができる。
r=2P/(3−P)
【0008】
蛍光偏光解消法による標的物質の試料溶液中の濃度を測定する具体例としては、蛍光偏光免疫測定法(FPIA法)が挙げられる。この方法は、特に血中薬剤濃度のような比較的分子量の小さい物質の測定に用いられ、抗原-抗体反応と、蛍光偏光度との関係から検量線を用いて標的物質の濃度を求めるものである。蛍光偏光免疫測定法は既に確立された技術である(特許文献1)。
【0009】
蛍光偏光解消法では、標的物質が結合した結合型の蛍光標識物質と、標的物質が結合していない遊離型の蛍光標識物質を分離(B/F分離)することなく、ホモジニアスアッセイ系で該標的物質の試料溶液中の濃度を測定することができる。しかしながら、試料溶液中に標的物質が極めて少ない場合、多量に存在する遊離型の蛍光標識物質の影響が強く、結合型の蛍光標識物質の正確な検出が困難になる等の問題があった。従って、蛍光偏光解消法では、極めて微量な標的物質の検出は難しく、より高感度に微量標的物質を測定する蛍光偏光解消法の開発が望まれていた。
【0010】
また、昨今、ゲノミクスやプロテオミクスで数多くの遺伝子やタンパク質の機能を解析するにあたり、微量なサンプルを用い、高速な解析を行うことが必要になってきている。このようなニ−ズに対応するために、微量かつ高速な解析用のDNAチップやプロテオチップなどの開発が進められている。そして、微細加工技術の進歩から極微小なチャンバ−(マイクロチャンバ)を作製することが可能になった。更に、CCDカメラおよびコンピューター処理を利用して、定性的、半定量的に解析することも可能になっている。
【0011】
マイクロチャンバアレイは、プレ−ト上に複数のマイクロチャンバを備えたものであり、既に酵素活性を有するタンパク質、微生物のスクリ−ニング及び一分子酵素活性検出等の利用が報告されている(特許文献2、特許文献3)。
【非特許文献1】Perrin、 F. J. Phys. Rad. 1、 390−401、 1926
【特許文献1】特開昭57−058695号公報
【特許文献2】特開2006−061023号公報
【特許文献3】特許第3727026号公報
【発明の開示】
【発明が解決しようとする課題】
【0012】
本発明はこのような事情に鑑みてなされたものであり、蛍光偏光解消法による試料溶液中の微量標的物質の濃度を測定する方法を提供することを目的とする。
【課題を解決するための手段】
【0013】
すなわち、本発明は、
(1)試料溶液と、試料溶液中に存在が疑われる標的物質に結合可能な蛍光標識物質を混合する工程と、得られた混合液を、マイクロチャンバアレイの各マイクロチャンバに分注する工程と、蛍光偏光解消法により、蛍光標識物質が結合した標的物質が分注されたマイクロチャンバを検出することにより、生体試料中の標的物質の濃度を測定する工程と、を備えた標的物質測定方法;
(2)前記試料溶液が、生体試料である(1)に記載の標的物質測定方法;
(3)前記標的物質が、タンパク質又はDNAである、(1)又は(2)に記載の標的物質測定方法;
(4)前記蛍光標識物質が、蛍光標識抗体、蛍光標識抗原又は蛍光標識プロ−ブである、(1)〜(3)のいずれか1に記載の標的物質測定方法;
(5)前記蛍光標識物質の蛍光発色団が有機系蛍光発色団からなる、(1)〜(4)のいずれか1に記載の標的物質測定方法;
(6)前記蛍光標識物質の蛍光発色団が6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリンである(1)〜(5)のいずれか1に記載の標的物質測定方法;
(7)前記マイクロチャンバの開口の直径が、0.1〜40μmである、(1)〜(6)のいずれか1に記載の標的物質測定方法;
(8)前記マイクロチャンバの深さが、100〜2000nmである、(1)〜(7)のいずれか1に記載の標的物質測定方法;
(9)前記マイクロチャンバの容積が0.001〜25000fLである、(1)〜(8)のいずれか1に記載の標的物質測定方法;
(10)前記マイクロチャンバアレイが有するマイクロチャンバの数が、1cm2あたり10〜1012個である(1)〜(9)のいずれか1に記載の標的物質測定方法を提供するものである。
【発明の効果】
【0014】
本発明によれば、遊離型の蛍光標識物質等の影響を受けない、蛍光偏光解消法による高感度な試料溶液中の微量標的物質の濃度を測定する、標的物質測定方法を提供することができる。
【発明を実施するための最良の形態】
【0015】
本発明の実施形態における、試料溶液としては、後述するマイクロチャンバに分注可能なものであれば、特に制限されるものではないが、生体試料が好ましい。生体試料としては、特に体液が好ましく、例えば、血液、血清、血漿、尿、汗、組織液あるいは組織の可溶化液等が挙げられる。
【0016】
本実施形態において、標的物質としては、溶液中に存在し後述する蛍光標識物質と結合するものであれば、特に制限されるものではないが、例えば、タンパク質、DNA、RNA、糖類及び細胞等が挙げられ、特にタンパク質及びDNAが好ましい。
【0017】
本実施形態において、蛍光標識物質としては、標的物質との特異的な結合能を有し、蛍光偏光解消法による検出が可能なものであれば、特に制限されるものではないが、例えば、蛍光標識抗体、蛍光標識抗原、蛍光標識タンパク、蛍光標識ペプチド及び蛍光標識DNAプロ−ブ等が挙げられる。
【0018】
また、蛍光標識物質の蛍光発色団としては、蛍光偏光解消法を用いて標識蛍光標識物質が結合した標的物質と遊離の蛍光標識物質を検出できる化合物であれば、特に制限されるものではないが、有機系蛍光発色団が好ましく、例えば、ロ−ダミン、ピレン、ジアルキルアミノナフタレン及びシアニンの骨格を有する化合物が挙げられる。また、特にピレン骨格を有する化合物が好ましく、より具体的には6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリンが挙げられる。
【0019】
尚、6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリンは公知の蛍光発色団であり、その合成方法等は特に制限されるものではなく、公知の化学合成法を用いて合成することができる。
【0020】
ここで、使用する蛍光発色団の骨格を選択する際は、標識蛍光標識物質及び標的物質から分子量変化を考慮して、適宜、励起波長、蛍光波長、ストークスシフト、及び蛍光寿命等の最適化を行うことができる。この際、励起波長と蛍光波長との波長差である、ストークスシフトは、5nm以上であることが好ましく、特に20nm以上であることが好ましい。蛍光発色団の蛍光寿命(蛍光緩和時間)は、1ナノ秒〜1000ナノ秒の範囲が好ましく、特に50ナノ秒〜500ナノ秒の範囲が好ましい。
【0021】
より具体的には、分子量変化が5000〜5万、即ち、蛍光標識物質が結合した標的物質の分子量が数千から数万の場合の場合、1ナノ秒〜15ナノ秒の蛍光寿命を有する蛍光発色団が好ましく、このような蛍光発色団の例としては、シアニン、ローダミン等が挙げられる。分子量変化が5万〜50万程度の場合、即ち、蛍光標識物質が結合した標的物質の分子量が数万から数十万の場合、50ナノ秒〜500ナノ秒の蛍光寿命を有する蛍光発色団が好ましい。このような蛍光発色団の例としては、ジアルキルアミノナフタレン、ピレン誘導体等が挙げられ、特に6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリンが好ましい。分子量変化が約50万から約500万程度の場合、即ち、蛍光標識物質が結合した標的物質の分子量が数十万から数百万の場合、100ナノ秒〜約1000ナノ秒の蛍光寿命を有する蛍光発色団が好ましい。このような蛍光発色団の例として、ピレン誘導体、金属錯体等が挙げられる。
【0022】
本実施形態における、マイクロチャンバアレイとしては、標識蛍光標識物質が結合した標的物質を一つずつ分注できるマイクロチャンバを複数有するものであれば、特に制限されるものではないが、1cm2のマイクロチャンバアレイにマイクロチャンバを10〜1012個、特に100〜108個有するものが好ましい。
【0023】
ここで、マイクロチャンバとしては、標識蛍光標識物質が結合した標的物質の大きさによって、所望の開口、深さ及び容積を設定すればよく、特に制限されるものではないが、例えば、標的物質がタンパク質やDNAの場合、マイクロチャンバの開口の直径は0.1〜40μm、深さは100〜2000nm、容積は0.001〜25000fLのものが好ましく、特に、容積が1000fL以下のものが好ましい。
【0024】
また、マイクロチャンバアレイの作製方法は、公知の作製方法を使用すれば良く、特に制限されるものではないが、例えば、通常のフォトリソグラフィ法を用いて作製することができる。
【0025】
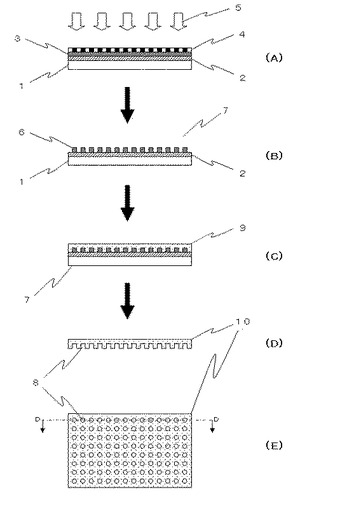
フォトリソグラフィ法によるマイクロチャンバアレイの作製方法の一実施形態を、図1に模式的に示した。ガラス基板1上に金膜2を蒸着し、金膜2上をフォトレジストにてコーティングしフォトレジストコート3を形成し、所定のパターンのクロム膜を有するパターンガラス4をフォトレジストコート3に置き、紫外線5を照射する(図1A)。次いで、紫外線5が照射されたフォトレジストコート3上のフォトレジストを除去し、フォトレジスト膜6が金膜2に残ったもの、即ち、フォトレジストをパターニングしたものを鑄型7とする(図1B)。フォトレジスト膜6が残留する部分は、マイクロチャンバ8に対応する鑄型部分である。従って、フォトレジスト膜6の寸法は、形成されるべきマイクロチャンバ8と実質的に同一の寸法を有するべきである。
【0026】
次に、PDMSと硬化剤を所定の比率で混合させた液状PDMS9を調製した後、その液状PDMS9を鑄型7上に適用し、その状態で液状PDMS9を硬化させる(図1C)。この過程において、硬化を促進させるために、液状PDMS9を担持した鑄型7は、約80℃で約60分間加熱することが好ましい。かくして、液状PDMS9の硬化が完了し、PMDSからなるマイクロチャンバアレイ10が鑄型7から剥がされる(図1D)。なお、この実施形態においては、マイクロチャンバアレイ10は、金膜2に載置された状態で硬化したPDMSであり、何等化学的な処理を施すことなく、容易に剥離することができる。
【0027】
図1Eは、マイクロチャンバアレイ10をマイクロチャンバ8の開口方向から見た図である。本実施形態のマイクロチャンバ8の形状は円柱であるが、その他の形状であってもよい。また、本実施形態では、紫外線により感光した部分が除去されるポジ型フォトレジストを用いて鋳型を作製したが、感光した部分が残るネガ型フォトレジストを用いても良い。
【0028】
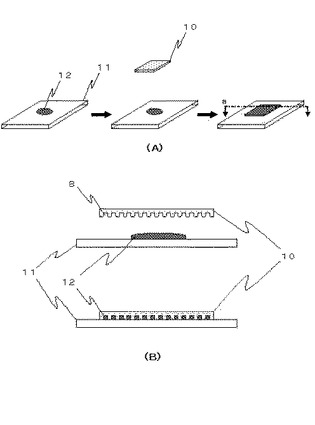
上記の方法でマイクロチャンバアレイ10を作製した場合、例えば、試料溶液12の液滴を保持したスライドグラス11上に、マイクロチャンバアレイ10を載置することにより、容易に、各マイクロチャンバ8に試料溶液12を分注することができる(図2)。このように、マイクロチャンバアレイ10をスライドグラス11に載置するだけで、試料溶液12を分注することができ、使用者が特にマイクロチャンバアレイ10の使用に熟練を要することはない(図2A)。また、該マイクロチャンバアレイを用いることで、分注される試料溶液12の体積は、マイクロチャンバ8により定められ、有機溶媒中に液滴を噴霧し懸架する場合に比べ、液滴のばらつきは、実質的に小さく若しくは無くなる(図2B)。
【0029】
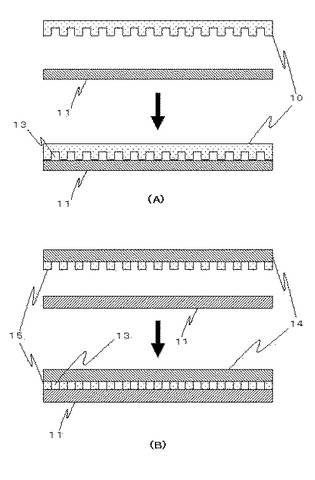
また、本実施形態では、マイクロチャンバアレイ10とスライドグラス11を載置することにより、容器部13を構成しているが(図3A)、例えば、スライドグラス14と貫通孔を有するPDMS15を貼り合わせることにより作製したマイクロチャンバアレイを、スライドグラス11に載置することにより、容器部13を構成するようになっていてよい(図3B)。
【0030】
ここで、容器部13の少なくとも一部は、水に対し実質的に不透過性を有する高分子樹脂から構成されているものが好ましく、また、高分子樹脂が空気に対して透過性を有するものが特に好ましい。これにより、封入された液滴の漏洩がなく、更に、空気透過性を有するため、空気が容器部13外へ透過して消散され、容器部13内に空気が残留することを確実に回避することができる。
【0031】
本実施形態において、マイクロチャンバアレイの材料としては、フォトリソグラフィ法により形成された鑄型を用いることで、マイクロチャンバアレイを形成できるものであれば、特に制限されるものではないが、水に対し実質的に不透過性を有する高分子樹脂から構成されているものが好ましく、また、高分子樹脂が空気に対して透過性を有するものが特に好ましい。例えば、ポリジメチルシロキサン 、シリコン、ポリスチレン、アクリル樹脂、ポリメチルメタクリレート及びポリカーボネートが挙げられ、ポリジメチルシロキサン(PDMS)が好ましい。また、特に容器部の少なくとも一部又は第二の部材は、PDMSにより形成されていることが好ましい。勿論、マイクロチャンバアレイ全体がPDMSにより形成されていてもよい。
【0032】
本実施形態において、蛍光偏光解消法により、蛍光標識物質が結合した標的物質が分注されたマイクロチャンバを検出する方法としては、マイクロチャンバアレイにおける蛍光偏光度を測定し、蛍光標識物質が結合した標的物質が分注されたマイクロチャンバを特定できるものであれば、特に制限されるものではないが、例えば、偏光フィルターを組み込んだ蛍光顕微鏡、偏光フィルターを組み込んだCCDカメラと励起光源を合わせたシステム、偏光フィルターを組み込んだ蛍光光度計などが用いられる。
【0033】
より具体的には、標的物質を含む試料と蛍光標識物質とを溶液中で混合した後、マイクロチャンバアレイに混合液を分注し、各マイクロチャンバにおける蛍光標識物質の蛍光偏光度を測定することで、蛍光標識物質が結合した標的物質が分注されたマイクロチャンバを検出する。必要であれば、標的物質の不在下での、蛍光標識物質の蛍光偏光度もまた測定する。測定は、穏和な温度(10℃〜40℃)で、一定温度で行うことが好ましい。
【0034】
蛍光偏光度の測定は、標的物質と蛍光標識物質との混合から所定の時間後に測定してもよく、あるいは、単位時間あたりの蛍光偏光度変化を測定してもよい。標的物質と蛍光標識物質との結合が完全に終了した時点で測定することにより、より再現性のある測定値が得られる。
【0035】
このように、蛍光標識物質が結合した標的物質が分注されたマイクロチャンバを検出し、検出されたマイクロチャンバの数とマイクロチャンバアレイが有するマイクロチャンバの数の比較等により、容易に試料溶液中に含まれる標的物質の濃度を測定することができる。
【0036】
例えば、1fLのマイクロチャンバを108個有するマイクロチャンバアレイを使用した場合、0.1μLの試料溶液がマイクロチャンバアレイに分注されることになる。ここで、蛍光標識物質が結合した標的物質が分注されたマイクロチャンバの数が6個であった場合、0.1μL中に標的物質が6個含まれていたことになるから、試料溶液中の標的物質の濃度は10−16Mとなる。
【0037】
以下、実施例を挙げて本発明を詳細に説明するが、本発明はこれらに限定されるものではない。
【実施例1】
【0038】
<マイクロチャンバアレイの作製>
ガラス基板上に金の薄膜を蒸着し、金の薄膜上をフォトレジストにてコ−ティングし、直径1μm、高さ1μmの円柱を1μm間隔で、縦横に10000個、総計108個有するようにクロム膜を形成するガラスをフォトレジストコ−ト上に載置し、紫外線を照射する。次いで、紫外線が照射されたフォトレジストを除去し、フォトレジスト膜が金膜に残ったもの、即ち、フォトレジストをパタ−ニングしたものを鑄型として使用した。
【0039】
PDMSと硬化剤を重量比10:1で混合させた液状PDMSを調製し、その液状PDMSを鑄型に適用し、その状態でPMDSを80℃で60分間硬化させる。PMDSの硬化が完了した後、PMDSを鑄型から剥がしマイクロチャンバアレイを作製した。
【実施例2】
【0040】
<6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリンの合成>
6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリン1は反応式Iに従って合成した。但し、反応式I中においてβ−シクロデキストリンに導入された置換基の位置は不定であり、理論上、位置混合物が生じる場合は位置異性体混合物として取り扱った。
【0041】
【化1】

反応式I
【0042】
(1)6A、6X−ジメシチル−β−シクロデキストリン2の合成
反応式Iに従い、β−シクロデキストリンから6A、6X−ジメシチル−β−シクロデキストリン2を合成した。すなわち、100mLのピリジンにβ−シクロデキストリン(5.675g、 5mmol)を撹拌しながら溶解し、これに室温にて塩化メシチレンスルホニル (1.094g、 5mmol)を撹拌しながら加え、2時間撹拌した。さらに塩化メシチレンスルホニル (1.094g、 5mmol)を追加し、2時間撹拌したのち塩化メシチレンスルホニル(1.094g、 5mmol)を追加し、3時間撹拌した。その後、さらに塩化メシチレンスルホニル(1.094g、 5mmol)を追加し、2時間撹拌した。20mmol(360μL)の水を加えてクエンチした後、ピリジンを留去、約50mLに濃縮し、アセトンにて結晶化した。粗結晶を温水(100mL)に溶解し、熱いままCHP−20Pカラムクロマト(60mL)にアプライした。温水400mLで洗浄後(β−シクロデキストリンが溶出)、温30% メタノ−ル(400mL)および常温の40%メタノ−ル(200mL)で6−O−メシチル−β−シクロデキストリン(2.77g、 2.1mmol、 42%)を溶出した後、6A、6X−O−ジメシチル−β−シクロデキストリン 2 (位置異性体混合物)を得た。
収量:1.15g(15%)
C60H90O39S2 (MW; 1499.5) LC−ESI/MS/MS:m/z 1521(M+NA)
【0043】
(2)6A−O−4−(1−ピレニル)ブタノイル−6X−メシチル−β−シクロデキストリン3の合成
続いて反応式Iに従い6A、6X−ジメシチル−β−シクロデキストリン2から6A−O−4−(1−ピレニル)ブタノイル−6X−メシチル−β−シクロデキストリン3を合成した。すなわち、15mLの乾燥させたジメチルスルホキシドに6A、6X−O−ジメシチル−β−シクロデキストリン 2 (1.25g、 0.83mmol)を撹拌しながら溶解した。室温にて撹拌しながらカリウムt−ブトキシド(93.6mg、 0.83mmol)及び1−ピレンブチリック酸(240mg、 0.83mmol)を溶解したジメチルスルホキシド溶液(5mL)を添加した。その後、80℃にて3時間加熱し反応後、1Lのアセトンを加え結晶化した。アセトンで洗浄後、粗結晶を50mLの水に溶解してCHP−20Pカラムクロマト(20mL)にアプライし、1Lの水、1Lの40% メタノ−ル、1Lの60% メタノ−ルで洗浄後、1Lの80%メタノ−ルで溶出し、6A−O−4−(1−ピレニル)ブタノイル−6X−O−メシチル−β−シクロデキストリン 3 (位置異性体混合物)を得た。
収量:410mg(31%)
C71H49O38S (MW; 1587.5) LC−ESI/MS/MS:m/z 1609(M+NA)
【0044】
(3)6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−スクシノイル−β−シクロデキストリン4の合成
続いて反応式Iに従い6A−O−4−(1−ピレニル)ブタノイル−6X−メシチル−β−シクロデキストリン3から6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−スクシノイル−β−シクロデキストリン4を合成した。すなわち、先に得た 6A−O−4−(1−ピレニル)ブタノイル−6X−メシチル−β−シクロデキストリン3 (80mg、 0.05mmol)を1mLの乾燥させたジメチルスルホキシドに撹拌しながら溶解し、これにコハク酸(118mg、1mmol)を続けて添加し、溶解した。室温にて撹拌しながらカリウムt−ブトキシド(112mg、 1mmol)のジメチルスルホキシド溶液(1mL)をゆっくり添加した。その後、80℃にて48時間撹拌した。反応後、反応液をろ過後、反応液に50mLのアセトンを加え、結晶化した。アセトンにて洗浄後、粗結晶を20mLの水に溶解してCHP−20Pカラムクロマトグラフィ−(20mL)にアプライした。500mLの水、500mLの40% メタノ−ルで洗浄後、500mLの60% メタノ−ル及び500mLの80% メタノ−ルにて溶出したが、原料の3と反応生成物が分離せず溶出したため、これらの溶出液を濃縮し、炭酸ナトリウムにより反応生成物である6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−スクシノイル−β−シクロデキストリン4をナトリウム塩とした後、HPLCにより分離・精製を行なった。HPLC条件は以下の通りである。
カラム:コスモシル 5C18−AR−300 4.6mm X 150mm、流速:1mL/min、検出波長:250−500nm、
溶離液:30−100% メタノ−ル
グラジエント:
【0045】
【表1】
【0046】
上記条件で11〜13minのピ−クを分取し、留去し6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−スクシノイル−β−シクロデキストリン4のナトリウム塩(16mg)を得た。これに1N塩酸を加え、4をカルボン酸体とし白濁した溶液を一旦、留去した。結晶をメタノ−ルに再溶解後、シリカゲル薄層クロマトグラフィ−(イソプロピルアルコ−ル:酢酸エチル:水=7:7:5)により展開し、Rf0.6のバンドから6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−スクシノイル−β−シクロデキストリン4 (位置異性体混合物)を得た。
収量:15mg(20%)
C66H88O39 (MW; 1505.4) LC−ESI/MS/MS:m/z 1549(M−H+2NA)
【0047】
(4)6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリン1の合成
続いて反応式Iに従い6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−スクシノイル−β−シクロデキストリン4から6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリン1を合成した。すなわち、反応容器に 6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−スクシノイル−β−シクロデキストリン4 (2.25mg、 1.5μmol)を量り取り、撹拌しながらN−ヒドロキシスクシンイミド(NHS、 17.3mg、 150μmol)のジメチルホルムアミド溶液(0.5mL)を添加した。これにジシクロヘキシルカルボジイミド(DCC、 31.0mg、 150μmol) のジメチルホルムアミド溶液(0.5mL)を撹拌しながら添加した。室温にて撹拌し反応開始1.5時間後からHPLCにて生成物ピ−クの分取・精製を行なった。位置異性体混合物であることに由来する分離不十分な3本の生成物のピ−クが認められたが、いずれもマススペクトルにより6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリン1(位置異性体)であることを確認したためこれら3本の画分(15から17min)を合わせて分取した( HPLC条件は6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−スクシノイル−β−シクロデキストリン4の精製時と同じ)。移動相を留去後、6.5mgの白色結晶を得た。マススペクトルにて確認したところ6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリン1以外にDCCの分解物であるジシクロヘキシルウレア(DCU)が含まれることが示されたが、これはUV領域において吸収を持たず、また、6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリン1が少量であること、DCUと6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリン1との分離が困難であること、DCUがこの後の6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリン1とタンパクとの反応を妨害しないことが予想されるため、この白色結晶をそのままタンパクの標識に用いた。
C70H91O41N (MW; 1602.45) LC−ESI/MS/MS:m/z 1624(M+NA)
【実施例3】
【0048】
<蛍光標識抗ウサギIgGヤギ抗体を用いた試料溶液中のウサギIgGの濃度測定>
1)蛍光標識抗ウサギIgGヤギ抗体の調製
6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリンを用いて抗ウサギIgGヤギ抗体の蛍光標識を行なった。すなわち、抗ウサギIgGヤギ抗体(0.8mg、5.3nmol)をセントリコン100にてバッファ交換し、500μLの50mM炭酸ナトリウムバッファpH9.76溶液とする。実施例2で得られた6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリンの粗結晶(1.3mg、理論最大含量は1μmol)を12.5μLのジメチルホルムアミドに溶解した後、ウサギIgGヤギ抗体の溶液に添加し、4℃にて15時間撹拌した。
反応後、反応液に200μLの50mM トリス塩酸バッファ pH8.0を添加した。これを50mMトリス塩酸バッファpH8.0によりバッファ置換したHiTrap Desaltingカラム(5mL)にアプライした。50mM トリス塩酸バッファpH8.0により溶出。各フラクションは5滴ずつ分画し、UVスペクトルを測定してタンパク及びピレンの吸収の認められたフラクションを合わせた(600μL)。この溶液についてバイオラッド社製タンパク定量キットによりタンパク濃度を求めたところ1.1mg/mLとなった(抗ウサギIgGヤギ抗体のモル濃度は7.3μM)。また345nmの吸光度およびピレンのモル吸光係数(5x104)からピレンの濃度は20μMと求められた。このことから抗ウサギIgGヤギ抗体には1分子当たり平均2.7個の6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリンが結合したことが確かめられた。
【0049】
2)試料溶液中のウサギIgGと蛍光標識抗ウサギIgGヤギ抗体との混合
試料溶液は、ウサギIgGを20mM PBS (pH7.3)によって、10nM、1nM、100pM、10pM、1pM、100fM、10fM及び1fMにすることで調製した。それぞれの試料溶液5μLと、1)で調製した蛍光標識抗ウサギIgGヤギ抗体を20mM PBS (pH7.3)にて2nMに希釈した溶液5μLとを混合し反応液とした。尚、試料溶液及び蛍光標識抗ウサギIgGヤギ抗体溶液はいずれも37℃に設定し、また混合した時点を反応開始0分とした。
【0050】
3)蛍光偏光解消法による蛍光標識抗ウサギIgGヤギ抗体と結合したウサギIgGのマイクロチャンバの検出
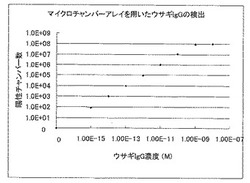
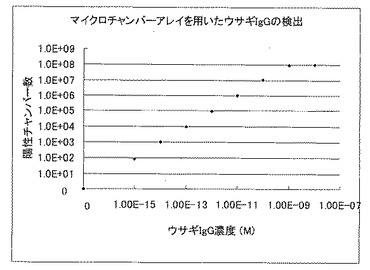
混合液5μLを速やかにスライドグラスに滴下し、実施例1で作製したマイクロチャンバアレイを、スライドグラス上の混合液に置くことで、マイクロチャンバアレイの各マイクロチャンバに混合液を分注した。スライドグラス上に残った混合液を、濾紙によって除去し、5分、8分、10分の蛍光偏光度を偏光フィルター励起光側と蛍光側ともに組み込んだ蛍光顕微鏡システム(オリンパス社)によって、蛍光標識抗ウサギIgGヤギ抗体と結合したウサギIgGの存在するマイクロチャンバを検出した。検出結果から導き出した試料溶液中のウサギIgGの測定結果を図4に示す。尚、測定温度は37℃に設定した。
<比較例1>
【0051】
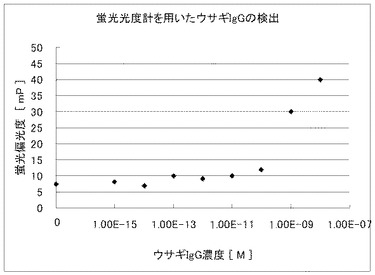
実施例3と同様の方法で調製した蛍光標識抗ウサギIgG抗体及び試料溶液を用いて、蛍光偏光度を蛍光偏光測定オプションつき蛍光分光光度計FLS920(浜松ホトニクス社)によって測定した。即ち、20mM PBS (pH7.3)にて2nMに希釈した蛍光標識抗ウサギIgG抗体溶液50μLを蛍光セルに入れた後、各濃度に調整した試料溶液50μLを該蛍光セルに添加して測定を開始し、5分、8分、10分の蛍光偏光度を測定した。尚、測定温度は37℃に設定した。測定結果を図5に示す。
【0052】
図4及び図5から明らかなように、マイクロチャンバアレイを用いた蛍光偏光解消法による試料溶液の濃度測定は、10fM以下の低濃度でも測定が可能であるのに対し、通常の蛍光偏光解消法による試料溶液の濃度測定では、10pM以下の濃度では、正確な濃度測定ができなかった。
【実施例4】
【0053】
<蛍光標識DNAプロ−ブを用いた試料溶液中のM13 mp18 ssDNAの濃度測定>
1)蛍光標識プロ−ブの調製
6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリンを用いてM13 mp18 ssDNAに対し相補的であり、5'末端にアミノ基を有する15塩基のDNAプローブ(以下M13プローブという)の蛍光標識を行なった。すなわち、M13プローブ(10nmol)を500μLの50mM炭酸ナトリウムバッファpH9.76溶液とする。実施例2で得られた6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリンの粗結晶(1.3mg、理論最大含量は1μmol)を12.5μLのジメチルホルムアミドに溶解した後、M13プローブの溶液に添加し、4℃にて15時間撹拌した。
【0054】
反応後、フォトダイオードアレイを検出器として持つHPLCにより分離・精製を行った。この際、目的とする蛍光標識M13プローブは、DNAとピレンの吸収を持つピークとして分取した。HPLCにより分離・精製を行なった。HPLC条件は以下の通りである。
【0055】
カラム:コスモシル 5C18−AR−300 4.6mm X 150mm
流速:1mL/min
検出波長:250−500nm
溶離液:30−100% アセトニトリル
グラジエント:
【0056】
【表2】
※TEAA トリエチルアミン-酢酸バッファ
【0057】
分取した溶液500μLについて260nmの吸光度によりDNA濃度を求めたところ5.0μMとなった。また345nmの吸光度およびピレンのモル吸光係数(5x104)からピレンの濃度は4.6μMと求められた。このことからM13プローブには1つ当たり平均0.92個の6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリンが結合したことが確かめられた。
2)試料溶液中のM13 ssDNAと蛍光標識プロ−ブとの混合
試料溶液は、(M13 mp18 ssDNA)を(0.1N NaClを含む10mM Tris-HCl (pH8.0))によって、10nM、(1)nM、100pM、10pM、1pM、100fM、10fM及び1fMにすることで調製した。それぞれの試料溶液(5)μLと、1)で調製した蛍光標識(M13)プローブを0.1N NaClを含む10mM Tris-HCl (pH8.0)で2nMに希釈した溶液(5)μLとを混合し混合液とした。尚、試料溶液及び蛍光標識(M13)プローブはいずれも37℃に設定し、また混合した時点を反応開始0分とした。
【0058】
3)蛍光偏光解消法による蛍光標識プロ−ブと結合したM13 ssDNAのマイクロチャンバの検出
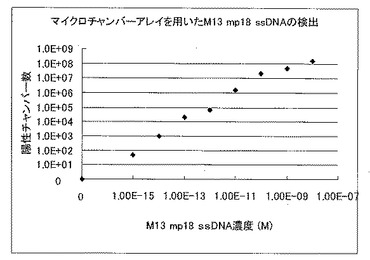
混合液5μLを速やかにスライドグラスに滴下し、実施例1で作製したマイクロチャンバアレイを、スライドグラス上の混合液に置くことで、マイクロチャンバアレイの各マイクロチャンバに混合液を分注した。スライドグラス上に残った混合液を、濾紙によって除去し、5分、8分、10分の蛍光偏光度を偏光フィルター励起光側と蛍光側ともに組み込んだ蛍光顕微鏡システム(オリンパス社)によって、蛍光標識M13プローブと結合したM13 mp18 ssDNAの存在するマイクロチャンバを検出した。検出結果から導き出した試料溶液中のM13 mp18 ssDNAの測定結果を図6に示す。尚、測定温度は37℃に設定した。
<比較例2>
【0059】
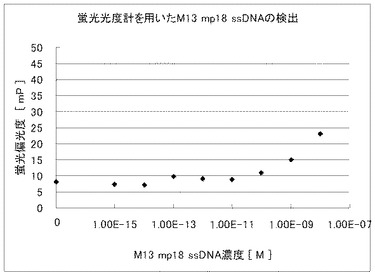
実施例4と同様の方法で調製した蛍光標識M13プローブ及び試料溶液を用いて、蛍光偏光度を蛍光偏光測定オプションつき蛍光分光光度計FLS920(浜松ホトニクス社)によって測定した。即ち、0.1N NaClを含む10mM Tris−HCl(pH8.0)により2nMに希釈した蛍光標識M13プローブ溶液 50μLを蛍光セルに入れた後、各濃度に調整した試料溶液50μLを該蛍光セルに添加して測定を開始し、5分、8分、10分の蛍光偏光度を測定した。尚、測定温度は37℃に設定した。測定結果を図7に示す。
【0060】
図6及び図7から明らかなように、マイクロチャンバアレイを用いた蛍光偏光解消法による試料溶液の濃度測定は、10fM以下の低濃度でも測定が可能であるのに対し、通常の蛍光偏光解消法による試料溶液の濃度測定では、10pM以下の濃度では、正確な濃度測定ができなかった。
【0061】
以上の結果から、本発明の標的物質測定方法は、従来の蛍光偏光解消法による試料溶液中の標的物質の濃度の測定方法に比し、極めて低濃度の標的物質の濃度が測定可能なことが明らかとなった。
【図面の簡単な説明】
【0062】
【図1】フォトリソグラフィ法によるマイクロチャンバアレイの作製方法の一実施形態を示した図である。
【図2】マイクロチャンバアレイ10の使用方法の一実施形態を示した図である。
【図3】容器部13の構成の実施形態を模式的に示した図である。
【図4】蛍光標識抗ウサギIgGヤギ抗体を用いた試料溶液中のウサギIgGのマイクロチャンバアレイを用いた蛍光偏光解消法による濃度測定結果を示した図である。
【図5】蛍光標識抗ウサギIgGヤギ抗体を用いた試料溶液中のウサギIgGの従来の蛍光偏光解消法による濃度測定結果を示した図である。
【図6】蛍光標識プロ−ブを用いた試料溶液中のM13 mp18 ssDNAのマイクロチャンバアレイを用いた蛍光偏光解消法による濃度測定結果を示した図である。
【図7】蛍光標識プロ−ブを用いた試料溶液中のM13 mp18 ssDNAの従来の蛍光偏光解消法による濃度測定結果を示した図である。
【技術分野】
【0001】
本発明は、蛍光偏光解消法による試料溶液中の標的物質の濃度を測定する方法に関する。
【背景技術】
【0002】
多数の検体について測定を行なう生理活性物質の探索におけるハイスループットスクリーニングあるいは体外診断システムにおいて、効率よく試験を実施するためには、簡便性、感度、安定性に優れた検出系を用いる必要がある。これまでこれらの検出系としては大きく分けて紫外可視吸光法、化学発光・生物発光法、放射性同位元素(RI)を用いる検出系が用いられてきた。
【0003】
このうち蛍光法は高選択性・高感度が期待できる検出系であり、紫外可視吸光法とともによく用いられている。また、蛍光法はRIを用いる方法に比べ測定時間が短くてすむだけでなく、管理区域内での操作の必要がない、放射性廃棄物の問題がないという点においても優れており、RIを用いた方法からの転換も行なわれてきている。
【0004】
しかし、化合物ライブラリ−あるいは血液成分を含む臨床検体などの多数の検体について試験を行なう場合、蛍光を発する検体も含まれていることがあり、その夾雑蛍光により測定がしばしば妨害されるという問題点がある。
【0005】
蛍光偏光解消法は、1926年にPerrinらによって確立された大きな分子ほど回転ブラウン運動の回転速度が遅くなることを利用した方法である。蛍光性分子により標識された小さな分子は、回転速度が速いため蛍光の偏光が解消され、蛍光偏光度Pは小さい値を示す。一方、大きな分子の場合は回転速度が遅いため蛍光の偏光は解消せず、Pは大きな値を示す(非特許文献1)。つまり、標的物質に結合可能な蛍光標識物質と標的物質が結合するとPは増大することになる。このPの変化を測定することで、該標的物質の試料溶液中の濃度を測定することができる。
【0006】
尚、蛍光偏光度は、以下の式によって計算される。
P=(IH−IL)/(IH+IL)
(式中、IHは、励起光の偏光面に平行な平面で偏光された発光強度であり、ILは励起光の偏光面に垂直な平面で偏光された発光強度である。)
【0007】
また、蛍光偏光解消法の指標として蛍光異方性rも用いられる。この場合も蛍光偏光度Pと同様に、蛍光異方性rの値の変化を測定することで、該標的物質の試料溶液中の濃度を測定することができる。蛍光偏光度Pと蛍光異方性rとの関係は以下の式で表すことができる。
r=2P/(3−P)
【0008】
蛍光偏光解消法による標的物質の試料溶液中の濃度を測定する具体例としては、蛍光偏光免疫測定法(FPIA法)が挙げられる。この方法は、特に血中薬剤濃度のような比較的分子量の小さい物質の測定に用いられ、抗原-抗体反応と、蛍光偏光度との関係から検量線を用いて標的物質の濃度を求めるものである。蛍光偏光免疫測定法は既に確立された技術である(特許文献1)。
【0009】
蛍光偏光解消法では、標的物質が結合した結合型の蛍光標識物質と、標的物質が結合していない遊離型の蛍光標識物質を分離(B/F分離)することなく、ホモジニアスアッセイ系で該標的物質の試料溶液中の濃度を測定することができる。しかしながら、試料溶液中に標的物質が極めて少ない場合、多量に存在する遊離型の蛍光標識物質の影響が強く、結合型の蛍光標識物質の正確な検出が困難になる等の問題があった。従って、蛍光偏光解消法では、極めて微量な標的物質の検出は難しく、より高感度に微量標的物質を測定する蛍光偏光解消法の開発が望まれていた。
【0010】
また、昨今、ゲノミクスやプロテオミクスで数多くの遺伝子やタンパク質の機能を解析するにあたり、微量なサンプルを用い、高速な解析を行うことが必要になってきている。このようなニ−ズに対応するために、微量かつ高速な解析用のDNAチップやプロテオチップなどの開発が進められている。そして、微細加工技術の進歩から極微小なチャンバ−(マイクロチャンバ)を作製することが可能になった。更に、CCDカメラおよびコンピューター処理を利用して、定性的、半定量的に解析することも可能になっている。
【0011】
マイクロチャンバアレイは、プレ−ト上に複数のマイクロチャンバを備えたものであり、既に酵素活性を有するタンパク質、微生物のスクリ−ニング及び一分子酵素活性検出等の利用が報告されている(特許文献2、特許文献3)。
【非特許文献1】Perrin、 F. J. Phys. Rad. 1、 390−401、 1926
【特許文献1】特開昭57−058695号公報
【特許文献2】特開2006−061023号公報
【特許文献3】特許第3727026号公報
【発明の開示】
【発明が解決しようとする課題】
【0012】
本発明はこのような事情に鑑みてなされたものであり、蛍光偏光解消法による試料溶液中の微量標的物質の濃度を測定する方法を提供することを目的とする。
【課題を解決するための手段】
【0013】
すなわち、本発明は、
(1)試料溶液と、試料溶液中に存在が疑われる標的物質に結合可能な蛍光標識物質を混合する工程と、得られた混合液を、マイクロチャンバアレイの各マイクロチャンバに分注する工程と、蛍光偏光解消法により、蛍光標識物質が結合した標的物質が分注されたマイクロチャンバを検出することにより、生体試料中の標的物質の濃度を測定する工程と、を備えた標的物質測定方法;
(2)前記試料溶液が、生体試料である(1)に記載の標的物質測定方法;
(3)前記標的物質が、タンパク質又はDNAである、(1)又は(2)に記載の標的物質測定方法;
(4)前記蛍光標識物質が、蛍光標識抗体、蛍光標識抗原又は蛍光標識プロ−ブである、(1)〜(3)のいずれか1に記載の標的物質測定方法;
(5)前記蛍光標識物質の蛍光発色団が有機系蛍光発色団からなる、(1)〜(4)のいずれか1に記載の標的物質測定方法;
(6)前記蛍光標識物質の蛍光発色団が6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリンである(1)〜(5)のいずれか1に記載の標的物質測定方法;
(7)前記マイクロチャンバの開口の直径が、0.1〜40μmである、(1)〜(6)のいずれか1に記載の標的物質測定方法;
(8)前記マイクロチャンバの深さが、100〜2000nmである、(1)〜(7)のいずれか1に記載の標的物質測定方法;
(9)前記マイクロチャンバの容積が0.001〜25000fLである、(1)〜(8)のいずれか1に記載の標的物質測定方法;
(10)前記マイクロチャンバアレイが有するマイクロチャンバの数が、1cm2あたり10〜1012個である(1)〜(9)のいずれか1に記載の標的物質測定方法を提供するものである。
【発明の効果】
【0014】
本発明によれば、遊離型の蛍光標識物質等の影響を受けない、蛍光偏光解消法による高感度な試料溶液中の微量標的物質の濃度を測定する、標的物質測定方法を提供することができる。
【発明を実施するための最良の形態】
【0015】
本発明の実施形態における、試料溶液としては、後述するマイクロチャンバに分注可能なものであれば、特に制限されるものではないが、生体試料が好ましい。生体試料としては、特に体液が好ましく、例えば、血液、血清、血漿、尿、汗、組織液あるいは組織の可溶化液等が挙げられる。
【0016】
本実施形態において、標的物質としては、溶液中に存在し後述する蛍光標識物質と結合するものであれば、特に制限されるものではないが、例えば、タンパク質、DNA、RNA、糖類及び細胞等が挙げられ、特にタンパク質及びDNAが好ましい。
【0017】
本実施形態において、蛍光標識物質としては、標的物質との特異的な結合能を有し、蛍光偏光解消法による検出が可能なものであれば、特に制限されるものではないが、例えば、蛍光標識抗体、蛍光標識抗原、蛍光標識タンパク、蛍光標識ペプチド及び蛍光標識DNAプロ−ブ等が挙げられる。
【0018】
また、蛍光標識物質の蛍光発色団としては、蛍光偏光解消法を用いて標識蛍光標識物質が結合した標的物質と遊離の蛍光標識物質を検出できる化合物であれば、特に制限されるものではないが、有機系蛍光発色団が好ましく、例えば、ロ−ダミン、ピレン、ジアルキルアミノナフタレン及びシアニンの骨格を有する化合物が挙げられる。また、特にピレン骨格を有する化合物が好ましく、より具体的には6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリンが挙げられる。
【0019】
尚、6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリンは公知の蛍光発色団であり、その合成方法等は特に制限されるものではなく、公知の化学合成法を用いて合成することができる。
【0020】
ここで、使用する蛍光発色団の骨格を選択する際は、標識蛍光標識物質及び標的物質から分子量変化を考慮して、適宜、励起波長、蛍光波長、ストークスシフト、及び蛍光寿命等の最適化を行うことができる。この際、励起波長と蛍光波長との波長差である、ストークスシフトは、5nm以上であることが好ましく、特に20nm以上であることが好ましい。蛍光発色団の蛍光寿命(蛍光緩和時間)は、1ナノ秒〜1000ナノ秒の範囲が好ましく、特に50ナノ秒〜500ナノ秒の範囲が好ましい。
【0021】
より具体的には、分子量変化が5000〜5万、即ち、蛍光標識物質が結合した標的物質の分子量が数千から数万の場合の場合、1ナノ秒〜15ナノ秒の蛍光寿命を有する蛍光発色団が好ましく、このような蛍光発色団の例としては、シアニン、ローダミン等が挙げられる。分子量変化が5万〜50万程度の場合、即ち、蛍光標識物質が結合した標的物質の分子量が数万から数十万の場合、50ナノ秒〜500ナノ秒の蛍光寿命を有する蛍光発色団が好ましい。このような蛍光発色団の例としては、ジアルキルアミノナフタレン、ピレン誘導体等が挙げられ、特に6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリンが好ましい。分子量変化が約50万から約500万程度の場合、即ち、蛍光標識物質が結合した標的物質の分子量が数十万から数百万の場合、100ナノ秒〜約1000ナノ秒の蛍光寿命を有する蛍光発色団が好ましい。このような蛍光発色団の例として、ピレン誘導体、金属錯体等が挙げられる。
【0022】
本実施形態における、マイクロチャンバアレイとしては、標識蛍光標識物質が結合した標的物質を一つずつ分注できるマイクロチャンバを複数有するものであれば、特に制限されるものではないが、1cm2のマイクロチャンバアレイにマイクロチャンバを10〜1012個、特に100〜108個有するものが好ましい。
【0023】
ここで、マイクロチャンバとしては、標識蛍光標識物質が結合した標的物質の大きさによって、所望の開口、深さ及び容積を設定すればよく、特に制限されるものではないが、例えば、標的物質がタンパク質やDNAの場合、マイクロチャンバの開口の直径は0.1〜40μm、深さは100〜2000nm、容積は0.001〜25000fLのものが好ましく、特に、容積が1000fL以下のものが好ましい。
【0024】
また、マイクロチャンバアレイの作製方法は、公知の作製方法を使用すれば良く、特に制限されるものではないが、例えば、通常のフォトリソグラフィ法を用いて作製することができる。
【0025】
フォトリソグラフィ法によるマイクロチャンバアレイの作製方法の一実施形態を、図1に模式的に示した。ガラス基板1上に金膜2を蒸着し、金膜2上をフォトレジストにてコーティングしフォトレジストコート3を形成し、所定のパターンのクロム膜を有するパターンガラス4をフォトレジストコート3に置き、紫外線5を照射する(図1A)。次いで、紫外線5が照射されたフォトレジストコート3上のフォトレジストを除去し、フォトレジスト膜6が金膜2に残ったもの、即ち、フォトレジストをパターニングしたものを鑄型7とする(図1B)。フォトレジスト膜6が残留する部分は、マイクロチャンバ8に対応する鑄型部分である。従って、フォトレジスト膜6の寸法は、形成されるべきマイクロチャンバ8と実質的に同一の寸法を有するべきである。
【0026】
次に、PDMSと硬化剤を所定の比率で混合させた液状PDMS9を調製した後、その液状PDMS9を鑄型7上に適用し、その状態で液状PDMS9を硬化させる(図1C)。この過程において、硬化を促進させるために、液状PDMS9を担持した鑄型7は、約80℃で約60分間加熱することが好ましい。かくして、液状PDMS9の硬化が完了し、PMDSからなるマイクロチャンバアレイ10が鑄型7から剥がされる(図1D)。なお、この実施形態においては、マイクロチャンバアレイ10は、金膜2に載置された状態で硬化したPDMSであり、何等化学的な処理を施すことなく、容易に剥離することができる。
【0027】
図1Eは、マイクロチャンバアレイ10をマイクロチャンバ8の開口方向から見た図である。本実施形態のマイクロチャンバ8の形状は円柱であるが、その他の形状であってもよい。また、本実施形態では、紫外線により感光した部分が除去されるポジ型フォトレジストを用いて鋳型を作製したが、感光した部分が残るネガ型フォトレジストを用いても良い。
【0028】
上記の方法でマイクロチャンバアレイ10を作製した場合、例えば、試料溶液12の液滴を保持したスライドグラス11上に、マイクロチャンバアレイ10を載置することにより、容易に、各マイクロチャンバ8に試料溶液12を分注することができる(図2)。このように、マイクロチャンバアレイ10をスライドグラス11に載置するだけで、試料溶液12を分注することができ、使用者が特にマイクロチャンバアレイ10の使用に熟練を要することはない(図2A)。また、該マイクロチャンバアレイを用いることで、分注される試料溶液12の体積は、マイクロチャンバ8により定められ、有機溶媒中に液滴を噴霧し懸架する場合に比べ、液滴のばらつきは、実質的に小さく若しくは無くなる(図2B)。
【0029】
また、本実施形態では、マイクロチャンバアレイ10とスライドグラス11を載置することにより、容器部13を構成しているが(図3A)、例えば、スライドグラス14と貫通孔を有するPDMS15を貼り合わせることにより作製したマイクロチャンバアレイを、スライドグラス11に載置することにより、容器部13を構成するようになっていてよい(図3B)。
【0030】
ここで、容器部13の少なくとも一部は、水に対し実質的に不透過性を有する高分子樹脂から構成されているものが好ましく、また、高分子樹脂が空気に対して透過性を有するものが特に好ましい。これにより、封入された液滴の漏洩がなく、更に、空気透過性を有するため、空気が容器部13外へ透過して消散され、容器部13内に空気が残留することを確実に回避することができる。
【0031】
本実施形態において、マイクロチャンバアレイの材料としては、フォトリソグラフィ法により形成された鑄型を用いることで、マイクロチャンバアレイを形成できるものであれば、特に制限されるものではないが、水に対し実質的に不透過性を有する高分子樹脂から構成されているものが好ましく、また、高分子樹脂が空気に対して透過性を有するものが特に好ましい。例えば、ポリジメチルシロキサン 、シリコン、ポリスチレン、アクリル樹脂、ポリメチルメタクリレート及びポリカーボネートが挙げられ、ポリジメチルシロキサン(PDMS)が好ましい。また、特に容器部の少なくとも一部又は第二の部材は、PDMSにより形成されていることが好ましい。勿論、マイクロチャンバアレイ全体がPDMSにより形成されていてもよい。
【0032】
本実施形態において、蛍光偏光解消法により、蛍光標識物質が結合した標的物質が分注されたマイクロチャンバを検出する方法としては、マイクロチャンバアレイにおける蛍光偏光度を測定し、蛍光標識物質が結合した標的物質が分注されたマイクロチャンバを特定できるものであれば、特に制限されるものではないが、例えば、偏光フィルターを組み込んだ蛍光顕微鏡、偏光フィルターを組み込んだCCDカメラと励起光源を合わせたシステム、偏光フィルターを組み込んだ蛍光光度計などが用いられる。
【0033】
より具体的には、標的物質を含む試料と蛍光標識物質とを溶液中で混合した後、マイクロチャンバアレイに混合液を分注し、各マイクロチャンバにおける蛍光標識物質の蛍光偏光度を測定することで、蛍光標識物質が結合した標的物質が分注されたマイクロチャンバを検出する。必要であれば、標的物質の不在下での、蛍光標識物質の蛍光偏光度もまた測定する。測定は、穏和な温度(10℃〜40℃)で、一定温度で行うことが好ましい。
【0034】
蛍光偏光度の測定は、標的物質と蛍光標識物質との混合から所定の時間後に測定してもよく、あるいは、単位時間あたりの蛍光偏光度変化を測定してもよい。標的物質と蛍光標識物質との結合が完全に終了した時点で測定することにより、より再現性のある測定値が得られる。
【0035】
このように、蛍光標識物質が結合した標的物質が分注されたマイクロチャンバを検出し、検出されたマイクロチャンバの数とマイクロチャンバアレイが有するマイクロチャンバの数の比較等により、容易に試料溶液中に含まれる標的物質の濃度を測定することができる。
【0036】
例えば、1fLのマイクロチャンバを108個有するマイクロチャンバアレイを使用した場合、0.1μLの試料溶液がマイクロチャンバアレイに分注されることになる。ここで、蛍光標識物質が結合した標的物質が分注されたマイクロチャンバの数が6個であった場合、0.1μL中に標的物質が6個含まれていたことになるから、試料溶液中の標的物質の濃度は10−16Mとなる。
【0037】
以下、実施例を挙げて本発明を詳細に説明するが、本発明はこれらに限定されるものではない。
【実施例1】
【0038】
<マイクロチャンバアレイの作製>
ガラス基板上に金の薄膜を蒸着し、金の薄膜上をフォトレジストにてコ−ティングし、直径1μm、高さ1μmの円柱を1μm間隔で、縦横に10000個、総計108個有するようにクロム膜を形成するガラスをフォトレジストコ−ト上に載置し、紫外線を照射する。次いで、紫外線が照射されたフォトレジストを除去し、フォトレジスト膜が金膜に残ったもの、即ち、フォトレジストをパタ−ニングしたものを鑄型として使用した。
【0039】
PDMSと硬化剤を重量比10:1で混合させた液状PDMSを調製し、その液状PDMSを鑄型に適用し、その状態でPMDSを80℃で60分間硬化させる。PMDSの硬化が完了した後、PMDSを鑄型から剥がしマイクロチャンバアレイを作製した。
【実施例2】
【0040】
<6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリンの合成>
6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリン1は反応式Iに従って合成した。但し、反応式I中においてβ−シクロデキストリンに導入された置換基の位置は不定であり、理論上、位置混合物が生じる場合は位置異性体混合物として取り扱った。
【0041】
【化1】
反応式I
【0042】
(1)6A、6X−ジメシチル−β−シクロデキストリン2の合成
反応式Iに従い、β−シクロデキストリンから6A、6X−ジメシチル−β−シクロデキストリン2を合成した。すなわち、100mLのピリジンにβ−シクロデキストリン(5.675g、 5mmol)を撹拌しながら溶解し、これに室温にて塩化メシチレンスルホニル (1.094g、 5mmol)を撹拌しながら加え、2時間撹拌した。さらに塩化メシチレンスルホニル (1.094g、 5mmol)を追加し、2時間撹拌したのち塩化メシチレンスルホニル(1.094g、 5mmol)を追加し、3時間撹拌した。その後、さらに塩化メシチレンスルホニル(1.094g、 5mmol)を追加し、2時間撹拌した。20mmol(360μL)の水を加えてクエンチした後、ピリジンを留去、約50mLに濃縮し、アセトンにて結晶化した。粗結晶を温水(100mL)に溶解し、熱いままCHP−20Pカラムクロマト(60mL)にアプライした。温水400mLで洗浄後(β−シクロデキストリンが溶出)、温30% メタノ−ル(400mL)および常温の40%メタノ−ル(200mL)で6−O−メシチル−β−シクロデキストリン(2.77g、 2.1mmol、 42%)を溶出した後、6A、6X−O−ジメシチル−β−シクロデキストリン 2 (位置異性体混合物)を得た。
収量:1.15g(15%)
C60H90O39S2 (MW; 1499.5) LC−ESI/MS/MS:m/z 1521(M+NA)
【0043】
(2)6A−O−4−(1−ピレニル)ブタノイル−6X−メシチル−β−シクロデキストリン3の合成
続いて反応式Iに従い6A、6X−ジメシチル−β−シクロデキストリン2から6A−O−4−(1−ピレニル)ブタノイル−6X−メシチル−β−シクロデキストリン3を合成した。すなわち、15mLの乾燥させたジメチルスルホキシドに6A、6X−O−ジメシチル−β−シクロデキストリン 2 (1.25g、 0.83mmol)を撹拌しながら溶解した。室温にて撹拌しながらカリウムt−ブトキシド(93.6mg、 0.83mmol)及び1−ピレンブチリック酸(240mg、 0.83mmol)を溶解したジメチルスルホキシド溶液(5mL)を添加した。その後、80℃にて3時間加熱し反応後、1Lのアセトンを加え結晶化した。アセトンで洗浄後、粗結晶を50mLの水に溶解してCHP−20Pカラムクロマト(20mL)にアプライし、1Lの水、1Lの40% メタノ−ル、1Lの60% メタノ−ルで洗浄後、1Lの80%メタノ−ルで溶出し、6A−O−4−(1−ピレニル)ブタノイル−6X−O−メシチル−β−シクロデキストリン 3 (位置異性体混合物)を得た。
収量:410mg(31%)
C71H49O38S (MW; 1587.5) LC−ESI/MS/MS:m/z 1609(M+NA)
【0044】
(3)6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−スクシノイル−β−シクロデキストリン4の合成
続いて反応式Iに従い6A−O−4−(1−ピレニル)ブタノイル−6X−メシチル−β−シクロデキストリン3から6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−スクシノイル−β−シクロデキストリン4を合成した。すなわち、先に得た 6A−O−4−(1−ピレニル)ブタノイル−6X−メシチル−β−シクロデキストリン3 (80mg、 0.05mmol)を1mLの乾燥させたジメチルスルホキシドに撹拌しながら溶解し、これにコハク酸(118mg、1mmol)を続けて添加し、溶解した。室温にて撹拌しながらカリウムt−ブトキシド(112mg、 1mmol)のジメチルスルホキシド溶液(1mL)をゆっくり添加した。その後、80℃にて48時間撹拌した。反応後、反応液をろ過後、反応液に50mLのアセトンを加え、結晶化した。アセトンにて洗浄後、粗結晶を20mLの水に溶解してCHP−20Pカラムクロマトグラフィ−(20mL)にアプライした。500mLの水、500mLの40% メタノ−ルで洗浄後、500mLの60% メタノ−ル及び500mLの80% メタノ−ルにて溶出したが、原料の3と反応生成物が分離せず溶出したため、これらの溶出液を濃縮し、炭酸ナトリウムにより反応生成物である6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−スクシノイル−β−シクロデキストリン4をナトリウム塩とした後、HPLCにより分離・精製を行なった。HPLC条件は以下の通りである。
カラム:コスモシル 5C18−AR−300 4.6mm X 150mm、流速:1mL/min、検出波長:250−500nm、
溶離液:30−100% メタノ−ル
グラジエント:
【0045】
【表1】
【0046】
上記条件で11〜13minのピ−クを分取し、留去し6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−スクシノイル−β−シクロデキストリン4のナトリウム塩(16mg)を得た。これに1N塩酸を加え、4をカルボン酸体とし白濁した溶液を一旦、留去した。結晶をメタノ−ルに再溶解後、シリカゲル薄層クロマトグラフィ−(イソプロピルアルコ−ル:酢酸エチル:水=7:7:5)により展開し、Rf0.6のバンドから6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−スクシノイル−β−シクロデキストリン4 (位置異性体混合物)を得た。
収量:15mg(20%)
C66H88O39 (MW; 1505.4) LC−ESI/MS/MS:m/z 1549(M−H+2NA)
【0047】
(4)6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリン1の合成
続いて反応式Iに従い6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−スクシノイル−β−シクロデキストリン4から6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリン1を合成した。すなわち、反応容器に 6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−スクシノイル−β−シクロデキストリン4 (2.25mg、 1.5μmol)を量り取り、撹拌しながらN−ヒドロキシスクシンイミド(NHS、 17.3mg、 150μmol)のジメチルホルムアミド溶液(0.5mL)を添加した。これにジシクロヘキシルカルボジイミド(DCC、 31.0mg、 150μmol) のジメチルホルムアミド溶液(0.5mL)を撹拌しながら添加した。室温にて撹拌し反応開始1.5時間後からHPLCにて生成物ピ−クの分取・精製を行なった。位置異性体混合物であることに由来する分離不十分な3本の生成物のピ−クが認められたが、いずれもマススペクトルにより6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリン1(位置異性体)であることを確認したためこれら3本の画分(15から17min)を合わせて分取した( HPLC条件は6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−スクシノイル−β−シクロデキストリン4の精製時と同じ)。移動相を留去後、6.5mgの白色結晶を得た。マススペクトルにて確認したところ6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリン1以外にDCCの分解物であるジシクロヘキシルウレア(DCU)が含まれることが示されたが、これはUV領域において吸収を持たず、また、6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリン1が少量であること、DCUと6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリン1との分離が困難であること、DCUがこの後の6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリン1とタンパクとの反応を妨害しないことが予想されるため、この白色結晶をそのままタンパクの標識に用いた。
C70H91O41N (MW; 1602.45) LC−ESI/MS/MS:m/z 1624(M+NA)
【実施例3】
【0048】
<蛍光標識抗ウサギIgGヤギ抗体を用いた試料溶液中のウサギIgGの濃度測定>
1)蛍光標識抗ウサギIgGヤギ抗体の調製
6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリンを用いて抗ウサギIgGヤギ抗体の蛍光標識を行なった。すなわち、抗ウサギIgGヤギ抗体(0.8mg、5.3nmol)をセントリコン100にてバッファ交換し、500μLの50mM炭酸ナトリウムバッファpH9.76溶液とする。実施例2で得られた6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリンの粗結晶(1.3mg、理論最大含量は1μmol)を12.5μLのジメチルホルムアミドに溶解した後、ウサギIgGヤギ抗体の溶液に添加し、4℃にて15時間撹拌した。
反応後、反応液に200μLの50mM トリス塩酸バッファ pH8.0を添加した。これを50mMトリス塩酸バッファpH8.0によりバッファ置換したHiTrap Desaltingカラム(5mL)にアプライした。50mM トリス塩酸バッファpH8.0により溶出。各フラクションは5滴ずつ分画し、UVスペクトルを測定してタンパク及びピレンの吸収の認められたフラクションを合わせた(600μL)。この溶液についてバイオラッド社製タンパク定量キットによりタンパク濃度を求めたところ1.1mg/mLとなった(抗ウサギIgGヤギ抗体のモル濃度は7.3μM)。また345nmの吸光度およびピレンのモル吸光係数(5x104)からピレンの濃度は20μMと求められた。このことから抗ウサギIgGヤギ抗体には1分子当たり平均2.7個の6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリンが結合したことが確かめられた。
【0049】
2)試料溶液中のウサギIgGと蛍光標識抗ウサギIgGヤギ抗体との混合
試料溶液は、ウサギIgGを20mM PBS (pH7.3)によって、10nM、1nM、100pM、10pM、1pM、100fM、10fM及び1fMにすることで調製した。それぞれの試料溶液5μLと、1)で調製した蛍光標識抗ウサギIgGヤギ抗体を20mM PBS (pH7.3)にて2nMに希釈した溶液5μLとを混合し反応液とした。尚、試料溶液及び蛍光標識抗ウサギIgGヤギ抗体溶液はいずれも37℃に設定し、また混合した時点を反応開始0分とした。
【0050】
3)蛍光偏光解消法による蛍光標識抗ウサギIgGヤギ抗体と結合したウサギIgGのマイクロチャンバの検出
混合液5μLを速やかにスライドグラスに滴下し、実施例1で作製したマイクロチャンバアレイを、スライドグラス上の混合液に置くことで、マイクロチャンバアレイの各マイクロチャンバに混合液を分注した。スライドグラス上に残った混合液を、濾紙によって除去し、5分、8分、10分の蛍光偏光度を偏光フィルター励起光側と蛍光側ともに組み込んだ蛍光顕微鏡システム(オリンパス社)によって、蛍光標識抗ウサギIgGヤギ抗体と結合したウサギIgGの存在するマイクロチャンバを検出した。検出結果から導き出した試料溶液中のウサギIgGの測定結果を図4に示す。尚、測定温度は37℃に設定した。
<比較例1>
【0051】
実施例3と同様の方法で調製した蛍光標識抗ウサギIgG抗体及び試料溶液を用いて、蛍光偏光度を蛍光偏光測定オプションつき蛍光分光光度計FLS920(浜松ホトニクス社)によって測定した。即ち、20mM PBS (pH7.3)にて2nMに希釈した蛍光標識抗ウサギIgG抗体溶液50μLを蛍光セルに入れた後、各濃度に調整した試料溶液50μLを該蛍光セルに添加して測定を開始し、5分、8分、10分の蛍光偏光度を測定した。尚、測定温度は37℃に設定した。測定結果を図5に示す。
【0052】
図4及び図5から明らかなように、マイクロチャンバアレイを用いた蛍光偏光解消法による試料溶液の濃度測定は、10fM以下の低濃度でも測定が可能であるのに対し、通常の蛍光偏光解消法による試料溶液の濃度測定では、10pM以下の濃度では、正確な濃度測定ができなかった。
【実施例4】
【0053】
<蛍光標識DNAプロ−ブを用いた試料溶液中のM13 mp18 ssDNAの濃度測定>
1)蛍光標識プロ−ブの調製
6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリンを用いてM13 mp18 ssDNAに対し相補的であり、5'末端にアミノ基を有する15塩基のDNAプローブ(以下M13プローブという)の蛍光標識を行なった。すなわち、M13プローブ(10nmol)を500μLの50mM炭酸ナトリウムバッファpH9.76溶液とする。実施例2で得られた6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリンの粗結晶(1.3mg、理論最大含量は1μmol)を12.5μLのジメチルホルムアミドに溶解した後、M13プローブの溶液に添加し、4℃にて15時間撹拌した。
【0054】
反応後、フォトダイオードアレイを検出器として持つHPLCにより分離・精製を行った。この際、目的とする蛍光標識M13プローブは、DNAとピレンの吸収を持つピークとして分取した。HPLCにより分離・精製を行なった。HPLC条件は以下の通りである。
【0055】
カラム:コスモシル 5C18−AR−300 4.6mm X 150mm
流速:1mL/min
検出波長:250−500nm
溶離液:30−100% アセトニトリル
グラジエント:
【0056】
【表2】
※TEAA トリエチルアミン-酢酸バッファ
【0057】
分取した溶液500μLについて260nmの吸光度によりDNA濃度を求めたところ5.0μMとなった。また345nmの吸光度およびピレンのモル吸光係数(5x104)からピレンの濃度は4.6μMと求められた。このことからM13プローブには1つ当たり平均0.92個の6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリンが結合したことが確かめられた。
2)試料溶液中のM13 ssDNAと蛍光標識プロ−ブとの混合
試料溶液は、(M13 mp18 ssDNA)を(0.1N NaClを含む10mM Tris-HCl (pH8.0))によって、10nM、(1)nM、100pM、10pM、1pM、100fM、10fM及び1fMにすることで調製した。それぞれの試料溶液(5)μLと、1)で調製した蛍光標識(M13)プローブを0.1N NaClを含む10mM Tris-HCl (pH8.0)で2nMに希釈した溶液(5)μLとを混合し混合液とした。尚、試料溶液及び蛍光標識(M13)プローブはいずれも37℃に設定し、また混合した時点を反応開始0分とした。
【0058】
3)蛍光偏光解消法による蛍光標識プロ−ブと結合したM13 ssDNAのマイクロチャンバの検出
混合液5μLを速やかにスライドグラスに滴下し、実施例1で作製したマイクロチャンバアレイを、スライドグラス上の混合液に置くことで、マイクロチャンバアレイの各マイクロチャンバに混合液を分注した。スライドグラス上に残った混合液を、濾紙によって除去し、5分、8分、10分の蛍光偏光度を偏光フィルター励起光側と蛍光側ともに組み込んだ蛍光顕微鏡システム(オリンパス社)によって、蛍光標識M13プローブと結合したM13 mp18 ssDNAの存在するマイクロチャンバを検出した。検出結果から導き出した試料溶液中のM13 mp18 ssDNAの測定結果を図6に示す。尚、測定温度は37℃に設定した。
<比較例2>
【0059】
実施例4と同様の方法で調製した蛍光標識M13プローブ及び試料溶液を用いて、蛍光偏光度を蛍光偏光測定オプションつき蛍光分光光度計FLS920(浜松ホトニクス社)によって測定した。即ち、0.1N NaClを含む10mM Tris−HCl(pH8.0)により2nMに希釈した蛍光標識M13プローブ溶液 50μLを蛍光セルに入れた後、各濃度に調整した試料溶液50μLを該蛍光セルに添加して測定を開始し、5分、8分、10分の蛍光偏光度を測定した。尚、測定温度は37℃に設定した。測定結果を図7に示す。
【0060】
図6及び図7から明らかなように、マイクロチャンバアレイを用いた蛍光偏光解消法による試料溶液の濃度測定は、10fM以下の低濃度でも測定が可能であるのに対し、通常の蛍光偏光解消法による試料溶液の濃度測定では、10pM以下の濃度では、正確な濃度測定ができなかった。
【0061】
以上の結果から、本発明の標的物質測定方法は、従来の蛍光偏光解消法による試料溶液中の標的物質の濃度の測定方法に比し、極めて低濃度の標的物質の濃度が測定可能なことが明らかとなった。
【図面の簡単な説明】
【0062】
【図1】フォトリソグラフィ法によるマイクロチャンバアレイの作製方法の一実施形態を示した図である。
【図2】マイクロチャンバアレイ10の使用方法の一実施形態を示した図である。
【図3】容器部13の構成の実施形態を模式的に示した図である。
【図4】蛍光標識抗ウサギIgGヤギ抗体を用いた試料溶液中のウサギIgGのマイクロチャンバアレイを用いた蛍光偏光解消法による濃度測定結果を示した図である。
【図5】蛍光標識抗ウサギIgGヤギ抗体を用いた試料溶液中のウサギIgGの従来の蛍光偏光解消法による濃度測定結果を示した図である。
【図6】蛍光標識プロ−ブを用いた試料溶液中のM13 mp18 ssDNAのマイクロチャンバアレイを用いた蛍光偏光解消法による濃度測定結果を示した図である。
【図7】蛍光標識プロ−ブを用いた試料溶液中のM13 mp18 ssDNAの従来の蛍光偏光解消法による濃度測定結果を示した図である。
【特許請求の範囲】
【請求項1】
試料溶液と、試料溶液中に存在が疑われる標的物質に結合可能な蛍光標識物質を混合する工程と、
得られた混合液を、マイクロチャンバアレイの各マイクロチャンバに分注する工程と、
蛍光偏光解消法により、蛍光標識物質が結合した標的物質が分注されたマイクロチャンバを検出することにより、生体試料中の標的物質の濃度を測定する工程と、を備えた標的物質測定方法。
【請求項2】
前記試料溶液が、生体試料である請求項1に記載の標的物質測定方法。
【請求項3】
前記標的物質が、タンパク質又はDNAである、請求項1又は2に記載の標的物質測定方法。
【請求項4】
前記蛍光標識物質が、蛍光標識抗体、蛍光標識抗原又は蛍光標識プロ−ブである、請求項1〜3のいずれか1に記載の標的物質測定方法。
【請求項5】
前記蛍光標識物質の蛍光発色団が有機系蛍光発色団からなる、請求項1〜4のいずれか1に記載の標的物質測定方法。
【請求項6】
前記蛍光標識物質の蛍光発色団が6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリンである請求項1〜5のいずれか1に記載の標的物質測定方法。
【請求項7】
前記マイクロチャンバの開口の直径が、0.1〜40μmである、請求項1〜6のいずれか1に記載の標的物質測定方法。
【請求項8】
前記マイクロチャンバの深さが、100〜2000nmである、請求項1〜7のいずれか1に記載の標的物質測定方法。
【請求項9】
前記マイクロチャンバの容積が0.001〜25000fLである、請求項1〜8のいずれか1に記載の標的物質測定方法。
【請求項10】
前記マイクロチャンバアレイが有するマイクロチャンバの数が、1cm2あたり10〜1012個である請求項1〜9のいずれか1に記載の標的物質測定方法。
【請求項1】
試料溶液と、試料溶液中に存在が疑われる標的物質に結合可能な蛍光標識物質を混合する工程と、
得られた混合液を、マイクロチャンバアレイの各マイクロチャンバに分注する工程と、
蛍光偏光解消法により、蛍光標識物質が結合した標的物質が分注されたマイクロチャンバを検出することにより、生体試料中の標的物質の濃度を測定する工程と、を備えた標的物質測定方法。
【請求項2】
前記試料溶液が、生体試料である請求項1に記載の標的物質測定方法。
【請求項3】
前記標的物質が、タンパク質又はDNAである、請求項1又は2に記載の標的物質測定方法。
【請求項4】
前記蛍光標識物質が、蛍光標識抗体、蛍光標識抗原又は蛍光標識プロ−ブである、請求項1〜3のいずれか1に記載の標的物質測定方法。
【請求項5】
前記蛍光標識物質の蛍光発色団が有機系蛍光発色団からなる、請求項1〜4のいずれか1に記載の標的物質測定方法。
【請求項6】
前記蛍光標識物質の蛍光発色団が6A−O−4−(1−ピレニル)ブタノイル−6X−O−4−(O−スクシンイミジル)スクシノイル−β−シクロデキストリンである請求項1〜5のいずれか1に記載の標的物質測定方法。
【請求項7】
前記マイクロチャンバの開口の直径が、0.1〜40μmである、請求項1〜6のいずれか1に記載の標的物質測定方法。
【請求項8】
前記マイクロチャンバの深さが、100〜2000nmである、請求項1〜7のいずれか1に記載の標的物質測定方法。
【請求項9】
前記マイクロチャンバの容積が0.001〜25000fLである、請求項1〜8のいずれか1に記載の標的物質測定方法。
【請求項10】
前記マイクロチャンバアレイが有するマイクロチャンバの数が、1cm2あたり10〜1012個である請求項1〜9のいずれか1に記載の標的物質測定方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2008−233010(P2008−233010A)
【公開日】平成20年10月2日(2008.10.2)
【国際特許分類】
【出願番号】特願2007−76670(P2007−76670)
【出願日】平成19年3月23日(2007.3.23)
【出願人】(390014960)シスメックス株式会社 (810)
【Fターム(参考)】
【公開日】平成20年10月2日(2008.10.2)
【国際特許分類】
【出願日】平成19年3月23日(2007.3.23)
【出願人】(390014960)シスメックス株式会社 (810)
【Fターム(参考)】
[ Back to top ]