
マイクロ流動化された水中油型乳剤及びワクチン組成物
【課題】抗原の免疫原性を増強するためのワクチンアジュバントとして有用なサブミクロン水中油型乳剤の提供。
【解決手段】該抗原がウイルス抗原、細菌抗原またはその組み合わせであり、アジュバントがQuilA、コレステロール、GPI−0100、臭化ジメチルジオクタデシルアンモニウム等であるサブミクロン水中油型乳剤。また、該乳剤と内在的または外来的に組み合わされた抗原を含有するワクチン組成物、さらに該乳剤及びワクチンの調製方法。
【解決手段】該抗原がウイルス抗原、細菌抗原またはその組み合わせであり、アジュバントがQuilA、コレステロール、GPI−0100、臭化ジメチルジオクタデシルアンモニウム等であるサブミクロン水中油型乳剤。また、該乳剤と内在的または外来的に組み合わされた抗原を含有するワクチン組成物、さらに該乳剤及びワクチンの調製方法。
【発明の詳細な説明】
【発明の詳細な説明】
【0001】
【発明の開示】
【0002】
発明の分野
本発明は、概して、ワクチン分野に関し、そして具体的には、家畜動物において免疫応答を増強するためのアジュバント処方物に関する。具体的には、本発明は、抗原の免疫原性を増強するための、サブミクロン水中油型乳剤のワクチンアジュバントとしての使用に関する。サブミクロン水中油型乳剤処方物、このような乳剤に取り込まれた抗原を含有するワクチン組成物、ならびに当該の乳剤及びワクチン調製方法が、本発明により提供される。本発明はまた、ワクチンにおける使用に適したサポニン配糖体及びステロールにより形成される複合体を含有する組成物も、提供する。
【0003】
発明の背景
細菌、ウイルス、寄生虫及びマイコプラズマ感染は、ウシ、ブタ及びペットなどの家畜動物において、幅広く広まっている。これらの感染性因子によって引き起こされる疾患は、多くの場合、抗微生物医薬療法に耐性であり、有効な治療の手段を残さない。結論として、ワクチン学的なアプローチが、家畜動物における感染性疾患を制御するために、ますます用いられている。感染性病原体全体を、化学的不活化または適切な遺伝子操作の後に、ワクチン処方物における使用に適するようにすることが可能である。あるいは、病原体のタンパク質サブユニットを、組換え発現系にて発現させ、そしてワクチン処方物における使用のために精製することが可能である。
【0004】
アジュバントは、概して、抗原に対する液性及び/または細胞性免疫応答を増加させる任意の材料を指す。従来のワクチンは、死滅した病原性微生物の粗調製物から構成され、そして、病原性微生物の培養に伴う不純物が、免疫応答を増強するためのアジュバントとして作用し得る。しかしながら、病原性微生物の均一な調製物または精製タンパク質サブユニットをワクチン接種用抗原として用いる場合、このような抗原によって引き起こされる免疫は不十分であり、それ故に、アジュバントとしての特定の外因性材料の添加が、必要となってくる。さらに、合成ワクチン及びサブユニットワクチンは、生産コストが高い。したがって、アジュバントを用いると、免疫応答を刺激するために必要とされる投与量がより少量で、それによってワクチンの生産コストが抑えられる可能性がある。
【0005】
アジュバントは、免疫応答を増強するための多くの異なる経路で作用することが知られている。多くのアジュバントは、免疫応答に関連するサイトカインネットワークを修飾する。これらの免疫調節アジュバントは、それらが抗原と一緒でない場合でさえも、その効果を発揮することが可能である。一般に、免疫調節アジュバントは、特定のサイトカインの一般的な亢進、及び同時に他のサイトカインの低下を引き起こし、細胞性Th1及び/または液性Th2応答へと導く。
【0006】
あるアジュバントは、抗原の立体配座の完全性を保存する能を有し、そのために抗原が適切な免疫エフェクター細胞に効率的に提示されることが可能である。アジュバント処方物による抗原の立体配座のこの保存の結果として、ワクチンは、免疫刺激複合体(ISCOM)に関して示されるように、有効期間が増すことになる。Ozel M.ら;Quarternary Structure of the
Immunestimmulating Complex (Iscom), J.of Ultrastruc. and Molec. Struc. Res.
102, 240-248(1989年)。
【0007】
あるアジュバントは、注射部位にデポー(depot)として抗原を保持する特性を有する。このデポー効果の結果として、抗原は、肝クリアランスによって急速には消失しない。アルミニウム塩及び油中水型乳剤は、このデポー効果を介して、より短期間に作用する。例えば、油中水型乳剤であるフロイント完全アジュバント(FCA)を用いることによって、長期のデポーを得ることが可能である。FCAは、典型的には、抗原提示細胞による抗原の除去が生分解により可能となるまで、注射部位に留まり続ける。
【0008】
それらの物理的な性質に基づき、アジュバントを、二つの非常に大まかなカテゴリー、すなわち、粒子状アジュバントと非粒子状アジュバントとに分類することが可能である。粒子状アジュバントは、ミクロ粒子として存在する。免疫原は、ミクロ粒子を取り込むか、あるいはそれと会合することが可能である。アルミニウム塩、油中水型乳剤、水中油型乳剤、免疫刺激複合体、リポソーム、ならびにナノ及びミクロ粒子は、粒子状アジュバントの例である。非粒子状アジュバントは、一般には、免疫調節物質であり、そしてこれらは、一般には、粒子状アジュバントと併せて用いられる。ムラミルジペプチド(マイコバクテリアから抽出されるペプチドグリカンのアジュバント活性成分)、非イオン性ブロック共重合体、サポニン(シャボンノキ(Quillaja saponaria)の樹皮から抽出されるトリテルペノイドの複合混合物)、リピドA(2個のリン酸基及び5または6個の概してC12〜C16長の脂肪酸鎖を有する、グルコサミンの二糖類)、サイトカイン、炭水化物重合体、誘導体化多糖類、ならびに、コレラ毒素及び大腸菌(E. coli)不安定毒素(LT)などの細菌毒素が、非粒子状アジュバントの例である。
【0009】
最も知られているアジュバントの一部は、非粒子状免疫調節物質と、デポー効果をアジュバント処方物に授け得る粒子状材料との組み合わせである。例えば、FCAは、結核菌(Mycobacterium tuberculosis)成分の免疫調節特性を、油型乳剤の短期デポー効果とともに組み合わせている。
【0010】
油型乳剤は、ワクチンアジュバントとして長い間使われている。Le Moignic及びPinoyは、1916年に、死滅ネズミチフス菌(Salmonella typhimurium)の鉱油懸濁液が免疫応答を増加させることを、見いだした。これに続き1925年には、Ramonが、スターチ油をジフテリアトキソイドへの抗毒素応答を増大させる物質の一つとして記載した。しかしながら、油型乳剤は、フロイントが現在フロイント完全アジュバント(FCA)として公知のアジュバント処方物を公表した1937年まで、一般的にはならなかった。FCAは、死滅マイコバクテリア及びアラセルAと混合された鉱(パラフィン)油から構成される、油中水型乳剤である。アラセルAは、主にマンニドモノオレエートであり、そして乳化剤として用いられる。FCAは、抗体応答の誘導に優れているにもかかわらず、重度の疼痛、膿瘍形成、発熱及び肉芽腫性炎症を引き起こす。これらの望ましくない副作用を避けるために、不完全フロイントアジュバント(IFA)が開発された。IFAは、マイコバクテリア成分がないことを除けば、FCAとその組成が類似している。IFAは、注射部位におけるデポー形成、及び抗体産生細胞の刺激による抗原の徐放を介して、作用する。
【0011】
FCAを改善するための別のアプローチは、鉱油を生体適合性の油により置き換えることが、注射部位におけるFCAに関連する反応を除去するのに役立つであろうとの概念に基づいた。乳剤は油中水型乳剤よりもむしろ水中油型乳剤であるべきとも、前者が長期間持続するデポーを注射部位に作り出すことから、考えられていた。Hillemanらは、ピーナツ油86%、乳化剤としてのアラセルA 10%、及び安定剤としてのモノステアリン酸アルミニウム4%からなる油性アジュバント「アジュバント65」について記載した。Hilleman, 1966年、Prog. Med. Virol. 8:131-182;Hilleman及びBeale,
1983年、New Approaches to Vaccine Development(Bell, R.及びTorrigiani, G.編集)中、Schwabe, Basel。ヒトにおいて、アジュバント65は安全かつ強力であったが、しかしIFAよりも低いアジュバント活性を示した。それにもかかわらず、アジュバント65の使用は、特定ロットのヒトに対するワクチンでの反応原性、及び精製もしくは合成乳化剤がアラセルAの代わりに用いられる場合のアジュバント活性の低減により、中止された。米国特許第5,718,904及び第5,690,942は、安全性プロファイルを改善する目的で、水中油型乳剤中の鉱油を代謝可能な油に置き換えることが可能であることを、教示している。
【0012】
アジュバント活性及び安全性の他に、乳剤の物理的外観もまた、市販のために考慮すべき重要なことである。物理的外観は、乳剤の安定性に依存する。クリーミング、沈降及び癒合は、乳剤の不安定さの指標である。クリーミングは、乳剤の油相と水相とが異なる比重である場合に生じる。クリーミングはまた、乳剤の初期の液滴サイズが大きく、かつ乳剤の液滴が全くブラウン運動をしていない場合にも、生じる。液滴サイズが大きい場合、界面が破裂する傾向があり、そして液滴が大きな粒子に癒合する。乳剤の安定性は、用いられる乳化剤の性質及び量、乳剤中での液滴サイズのサイズ、ならびに油相と水相との間の密度の差異などの多数の要因によって決まる。
【0013】
乳化剤は、界面の自由エネルギーを低減し、そして液滴の癒合に対する物理的もしくは静電気的なバリアを作り出すことによって、分散している液滴の安定性を促進する。非イオン性及びイオン性界面活性剤は、乳化剤として使用されている。非イオン性乳化剤は、界面にて配向し、そして比較的分厚い構造を作り出すが、このことにより分散している液滴が立体的に回避される。陰イオンもしくは陽イオン乳化剤は、対イオンをひきつけることによって電気二重層の形成を誘導し;二重層の反発力は、液滴を、それらが接近するときに互いに反発させる。
【0014】
乳化剤を用いる他に、乳剤の安定性は、機械的な手段によって乳剤の液滴サイズを低減することによっても、達成可能である。典型的には、プロペラミキサー、タービンローター、コロイドミル、ホモジナイザー及びソニケーターが、乳剤を製造するために用いられている。マイクロ流動化は、乳剤における液滴サイズの均一性を増加させる別の方法である。マイクロ流動化は、サブミクロン範囲の一定した粒子サイズの繊細で物理的に安定な乳剤を作り出すことが可能である。乳剤の安定性を増加させる他に、マイクロ流動化の過程は、最終産物の無菌性を確実にする好ましい方法である最終ろ過を可能とする。その上、サブミクロンの油粒子は、注射部位からリンパ管へ入り、次いでドレナージ連鎖のリンパ節、血液及び脾臓を通ることが可能である。このことは、局所的炎症及び有意な注射部位反応を作り出す可能性のある油のデポーを注射部位に確立する可能性を低減する。
【0015】
マイクロ流動化装置は、現在、市販されている。乳剤の形成は、マイクロ流動化装置において、相互作用チャンバー内で二つの流動化された流れが高速で相互作用するときに生じる。マイクロ流動化装置は、空気または窒素を駆動し、そして過剰の20,000psiの内部圧力にて動作することが可能である。米国特許第4,908,154は、いかなる乳化剤も本質的に含まない乳剤を得るためのマイクロ流動化装置の使用を、教示している。
【0016】
多数のサブミクロン水中油型アジュバント処方物が、文献に記載されている。米国特許第5,376,369は、Syntaxアジュバント処方物(SAF)として公知のサブミクロン水中油型乳剤アジュバント処方物を、教示している。SAFは、油成分としてのスクアレンまたはスクアラン、乳剤を形成する量のPluronic L121(ポリオキシ−プロピレン−ポリオキシエチレン)ブロック重合体、及び免疫を増強する量のムラミルジペプチドを、含有する。スクアレンは、多くの組織中(特に、サメ及び他の魚類の肝臓中)に見いだされる、コレステロールの直線状の炭化水素前駆体である。スクアランは、スクアレンの水素添加により調製され、かつ完全に飽和している。スクアレンとスクアランの双方は、代謝されることが可能であり、そして毒性学試験の成績は良好である。スクアレンもしくはスクアラン乳剤は、ヒト癌ワクチンにおいて使用され、軽度の副作用及び望ましい有効性を伴う。例えば、Anthony C. Allison, 1999年、Squalene and
Squalane emulsions as adjuvants, Methods 19:87-93を参照されたい。
【0017】
米国特許第6,299,884及び国際特許公報WO 90/14837は、ポリオキシ−プロピレン−ポリオキシエチレンブロック共重合体がサブミクロン水中油型乳剤の形成に必須ではないことを、教示している。その上、これらの参考文献は、非毒性の代謝可能な油の使用を教示し、そして鉱油及び毒性石油蒸留油のその乳剤処方物における使用を明確に除外している。
【0018】
米国特許第5,961,970は、ワクチンアジュバントとして用いられるさらに別のサブミクロン水中油型乳剤を教示している。本特許に記載される乳剤において、疎水性成分は、中鎖トリグリセリド油、植物油及びその混合物からなる群より選択される。この乳剤に含まれる界面活性剤は、リン脂質(例えば、レシチン)などの天然の生物学的に適合性の界面活性剤、あるいはTWEEN−80などの医薬的に許容可能な非天然の界面活性剤であることが可能である。この特許はまた、乳剤を形成するときに抗原を乳剤中へ取り込むことを教示しており、乳剤を独立的かつ外来的に形成した後に抗原を乳剤と混合するのとは対照的である。
【0019】
米国特許第5,084,269は、鉱油と組み合わせてレシチンを含有するアジュバント処方物が、宿主動物内での刺激を減少させ、そして同時に全身性免疫の増加を誘導することを、教示している。米国特許第5,084,269から得られたアジュバント処方物は、商品名AMPHIGEN(商標登録)で、動物用ワクチンにおいて商業的に用いられている。AMPHIGEN(商標登録)処方物は、ミセル−レシチンに囲まれた油滴で構成される。これらのミセルは、従来の油に基づくアジュバントよりも多くの全細胞抗原の付着を可能にする。その上、AMPHIGEN(商標登録)に基づくワクチン処方物は、典型的には10%〜20%の油を含有する油アジュバントを含有する他のワクチン処方物と比較して、2.5〜5%の鉱油と、低い油量を含有する。この低い油量が、このアジュバントに基づくワクチン処方物の注射部位組織に対する刺激を低下させて、屠殺時により少ない病変及びより少ないトリムを生じる。加えて、油滴を囲んでいるレシチン被覆は、注射部位反応をさらに低減し、安全かつ有効なワクチンをもたらす。
【0020】
AMPHIGEN(商標登録)処方物は、多数の動物用ワクチンにおいてアジュバントとして用いられ、そしてワクチン製品の物理的外観は、短期及び長期の保管期間中ならびに再構成時に維持される必要がある。加えて、凍結乾燥させた抗原を、予め調製しておいたアジュバント処方物と、注射直前に混合する。この実行は、水中油型乳剤内での抗原の均一な分布を必ずしも確実にするわけではなく、そして乳剤の外観は望ましくない可能性がある。その上、均質化された乳剤は、静置すると相分離を示し得る。したがって、長期の有効期間に相分離を示さない安定なアジュバント処方物の必要性が存在する。相分離を防ぐ一つの方法は、液滴サイズを小さくし、そして乳剤の粒子均一性を増加させることである。代謝可能な油性乳剤処方物のマイクロ流動化の過程が文書化されているものの、AMPHIGEN(商標登録)処方物などの水中油型乳剤のマイクロ流動化は、まだ実行されていない。
【0021】
本発明では、マイクロ流動化を、レシチンに囲まれた鉱油の液滴サイズをサブミクロンサイズにするために用いている。予想外にも、本発明者は、レシチンと油との混合物からなる水中油型乳剤をアジュバントとして添加したワクチン処方物のマイクロ流動化が、処方物の物理的外観を改善するだけでなく、処方物の免疫化効果も増強することを、発見した。マイクロ流動化された処方物はまた、改善された安全性プロファイルによっても特徴付けられる。
【0022】
発明の概要
本発明者は、代謝可能でない油に基づく水中油型乳剤のアジュバント活性及び安全性プロファイルが、マイクロ流動化を介して改善可能であることを、予想外にも発見した。マイクロ流動化された乳剤に取り込まれた抗原は、抗原をマイクロ流動化の前に乳剤中に内在的に取り込む場合でも、安定である。
【0023】
したがって、一実施態様において、本発明は、ワクチンアジュバントとして有用なサブミクロン水中油型乳剤処方物を提供する。本発明のサブミクロン水中油型乳剤は、代謝可能でない油、少なくとも一つの界面活性剤、及び水溶性成分から構成され、ここで油は、サブミクロン範囲の平均の油滴サイズで水溶性成分中に分散している。好ましい代謝可能でない油は、軽質鉱油である。好ましい界面活性剤としては、レシチン、TWEEN(商標登録)−80及びSPAN(商標登録)−80が挙げられる。
【0024】
本発明により提供される好ましい水中油型乳剤は、AMPHIGEN(商標登録)処方物から構成される。
本発明の水中油型乳剤は、保存剤、浸透圧剤、生体接着分子及び免疫刺激分子などの適切かつ望ましい成分をさらに含むことが可能である。好ましい免疫刺激分子としては、例えば、Quil
A、コレステロール、GPI−0100、臭化ジメチルジオクタデシルアンモニウム(DDA)が挙げられる。
【0025】
別の実施態様において、本発明は、サブミクロン水中油型乳剤の調製方法を提供する。本発明によると、油、1またはそれより多くの界面活性剤、水溶性成分、及び乳剤における使用に適した任意の他の成分を始めとする乳剤の種々の成分を、混ぜ合わせる。該混合物を最初の乳化過程にかけて、水中油型乳剤を形成し、次いでこれをマイクロ流動化装置に通して、液滴が直径1ミクロン未満、好ましくは平均液滴サイズ0.5ミクロン未満の水中油型乳剤を得る。
【0026】
さらに別の実施態様において、本発明は、抗原及び上述のサブミクロン水中油型乳剤を含有するワクチン組成物を提供する。抗原は、外来的あるいは内在的に、好ましくは内在的に、乳剤中に取り込まれている。
【0027】
本発明のワクチン組成物に含まれることが可能な抗原は、細菌、真菌またはウイルスの抗原、あるいはその組み合わせであることが可能である。該抗原は、不活化した細胞またはウイルスの全体または一部の調製物の形態を取るか、あるいは慣用のタンパク質精製、遺伝子操作技術または化学合成によって得られる抗原分子の形態を取ることが可能である。
【0028】
さらなる実施態様において、本発明は、抗原(単数または複数)をサブミクロン水中油型乳剤と組み合わせて含有するワクチン組成物の調製方法を、提供する。
本発明のワクチン組成物を調製する際に、抗原を、内在的(例えば、マイクロ流動化の前に)あるいは外来的(例えば、マイクロ流動化の後に)に水中油型乳剤の成分と組み合わせることが可能である。
【0029】
さらに別の実施態様において、本発明は、マイクロカプセル化された抗原及び上述のサブミクロン水中油型乳剤を含有し、ここでマイクロカプセル化された抗原が乳剤と外来的に組み合わされているワクチン組成物を、提供する。
【0030】
サポニンとステロールが、溶液中で組み合わされる場合に、互いに会合して、らせん状ミセルの形状の複合体を形成することもまた、驚くべきことに発見された。本発明によると、これらのらせん状ミセル複合体は、免疫刺激活性を有し、そしてワクチン組成物におけるアジュバントとして特に有用である。
【0031】
したがって、本発明は、サポニン及びステロールを含有し、ここでサポニンとステロールがらせん状ミセルの形状の複合体を形成しているワクチン組成物を、提供する。本発明はまた、サポニン、ステロール及び抗原を含有し、ここでサポニンとステロールがらせん状ミセルの形状の複合体を形成し、そして該抗原がらせん状ミセルと混合されているが、しかしその内部には取り込まれていない組成物も、提供する。
【0032】
発明の詳細な説明
本発明者は、レシチンと鉱油との混合物からなる水中油型乳剤をアジュバントとして添加したワクチン処方物のマイクロ流動化が、ワクチン処方物の物理的外観を改善するばかりでなく、ワクチン処方物の免疫化効果も増強することを、予想外にも発見した。マイクロ流動化されたワクチン処方物は、改善された安全性プロファイルによっても特徴付けられる。
【0033】
これらの発見に基づき、本発明は、ワクチン組成物におけるアジュバントとして有用なサブミクロン水中油型乳剤を提供する。これらのサブミクロン水中油型乳剤をマイクロ流動化装置を用いることによって作る方法もまた、提供される。さらに、本発明は、抗原がサブミクロン水中油型乳剤と組み合わされているサブミクロンワクチン組成物を、提供する。このようなワクチン組成物を作るための方法もまた、提供される。本発明は、サブミクロン水中油型乳剤と組み合わせてマイクロカプセル化された抗原を含有するワクチン組成物、ならびにこのようなワクチンを作るための方法を、さらに提供する。
【0034】
開示を明確にし、かつ限定しない目的で、発明の詳細な説明を、本発明の特定の特徴、実施態様または応用について記載または例示する以下のサブセクションに分割する。
サブミクロン水中油型乳剤
一実施態様において、本発明は、ワクチンアジュバントとして有用なサブミクロン水中油型乳剤処方物を提供する。本発明のサブミクロン水中油型乳剤は、ワクチン組成物における抗原の免疫原性を増強し、動物への投与に安全であり、かつ保管中は安定である。
【0035】
本発明のサブミクロン水中油型乳剤は、代謝可能でない油、少なくとも一つの界面活性剤、及び水溶性成分から構成され、ここで油は、サブミクロン範囲の平均的な油滴サイズで水溶性成分中に分散している。
【0036】
「サブミクロン」は、液滴が1μm(ミクロン)未満のサイズで、かつ平均の(average)または平均(mean)油滴サイズが1μm未満であることを意味する。好ましくは、乳剤の平均液滴サイズは、0.8μm未満;より好ましくは0.5μm未満;そしてよりいっそう好ましくは0.4μm未満、または約0.1〜0.3μm未満である。
【0037】
「平均液滴サイズ」は、粒子サイズの体積分布内での体積平均径(VMD)の粒子サイズとして定義される。VMDは、各粒子の直径をそのサイズの全粒子の体積とかけ合わせ、そして合計することにより、算出される。次いで、これを全粒子の総体積で割る。
【0038】
「代謝可能でない油」の語は、本明細書では、乳剤が投与される動物個体の体により代謝されることが可能でない油を指す。
「動物」及び「動物個体」の語は、本明細書では、例えばウシ、ヒツジ及びブタを始めとする、すべてのヒトでない動物を指す。
【0039】
本発明の乳剤における使用に適した代謝可能でない油としては、アルカン、アルケン、アルキン、ならびにそれに対応する酸及びアルコール、そのエーテル及びエステル、ならびにその混合物が挙げられる。好ましくは、油の個々の化合物は、軽質炭化水素化合物であって、すなわち、このような成分は6〜30個の炭素原子を有する。油は、合成的に調製されるか、または石油生成物から精製されることが可能である。本発明の乳剤における使用に好ましい代謝可能でない油としては、例えば、鉱油、パラフィン油及びシクロパラフィンが挙げられる。
【0040】
「鉱油」の語は、蒸留技術によってペトロラタムから得られる液体炭化水素の混合物を指す。該語は、「液化パラフィン」、「液体ペトロラタム」及び「白色鉱油」と同義である。該語はまた、「軽質鉱油」、すなわち、ペトロラタムの蒸留により同様に得られるが、ただし白色鉱油よりもわずかに低い比重である油を含むことも、意図する。例えば、Remington's Pharmaceutical Sciences、第18版(Easton、ペンシルバニア州:Mack Publishing Company,
1990年、788頁及び1323頁)を参照されたい。鉱油は、例えばJ.T. Baker(Phillipsburg、ペンシルバニア州)、USB Corporation(クリーブランド、オハイオ州)などの種々の市販元から得ることが可能である。好ましい鉱油は、DRAKEOL(商標登録)の名前で市販されている軽質鉱油である。
【0041】
典型的には、本発明のサブミクロン乳剤の油成分は、1%〜50%容量の量;好ましくは、10%〜45の量;より好ましくは、20%〜40%の量で、存在する。
本発明の水中油型乳剤は、典型的には、少なくとも一つ(すなわち、1またはそれより多く)の界面活性剤を含む。界面活性剤及び乳化剤(本明細書ではこれらの語を同義的に用いる)は、油滴の表面を安定化し、そして所望のサイズ内に油滴を維持する剤である。
【0042】
本発明の乳剤における使用に適した界面活性剤としては、天然の生物学的に適合性の界面活性剤、及び非天然の合成界面活性剤が挙げられる。生物学的に適合性の界面活性剤としては、リン脂質化合物またはリン脂質の混合物が挙げられる。好ましいリン脂質は、大豆もしくは卵レシチンなどのホスファチジルコリン(レシチン)である。レシチンは、粗植物油を水で洗浄し、そして得られた含水ゴムを分離及び乾燥することにより、ホスファチドとトリグリセリドの混合物として得られることが可能である。精製された生成物は、トリグリセリド及び植物油をアセトン洗浄により除去した後に残留する、アセトン不溶性リン脂質及び糖脂質の混合物を分画することにより、得られることが可能である。あるいは、レシチンを、種々の市販元から得ることが可能である。他の適切なリン脂質としては、ホスファチジルグリセロール、ホスファチジルイノシトール、ホスファチジルセリン、ホスファチジン酸、カルジオリピン及びホスファチジルエタノールアミンが挙げられる。リン脂質は、天然源から単離されても、または慣用的に合成されてもよい。
【0043】
本発明のサブミクロン乳剤における使用に適した非天然の合成界面活性剤としては、例えば脂肪酸置換ソルビタン界面活性剤(SPAN(商標登録)またはARLACEL(商標登録)の名前で市販されている)などのソルビタンに基づく非イオン性界面活性剤、ポリエトキシ化ソルビトールの脂肪酸エステル(TWEEN(商標登録))、ヒマシ油などの源から得られる脂肪酸のポリエチレングリコールエステル(EMULFOR);ポリエトキシ化脂肪酸(例えば、SIMULSOL
M−53の名前で入手可能なステアリン酸)、ポリエトキシ化イソオクチルフェノール/ホルムアルデヒド重合体(TYLOXAPOL)、ポリオキシエチレン脂肪酸アルコールエーテル(BRIJ(商標登録));ポリオキシエチレンノンフェニルエーテル(TRITON(商標登録)N)、ポリオキシエチレンイソオクチルフェニルエーテル(TRITON(商標登録)X))が挙げられる。好ましい合成界面活性剤は、SPAN(商標登録)及びTWEEN(商標登録)の名前で入手可能な界面活性剤である。
【0044】
本発明の水中油型乳剤における使用のための好ましい界面活性剤としては、レシチン、Tween−80及びSPAN−80が挙げられる。
一般的には、界面活性剤、または界面活性剤の組み合わせ(2またはそれより多くの界面活性剤が用いられる場合)は、0.01%〜10%容量、好ましくは0.1%〜6.0%、より好ましくは0.2〜5.0%の量にて、乳剤中に存在する。
【0045】
水溶性成分は、乳剤の連続相を構成し、そして水、緩衝生理食塩液または任意の他の適切な水溶液であることが可能である。
本発明の水中油型乳剤は、保存剤、浸透圧剤、生体接着分子及び免疫刺激分子を始めとする適切かつ望ましい成分を、さらに含むことが可能である。
【0046】
生体接着分子は、標的粘膜表面での、またはこれを介した抗原の送達及び付着を増強し、粘膜免疫を授与することが可能であると考えられている。適切な生体接着分子の例としては、ポリアクリル酸及びポリメタクリル酸(例えば、CARBOPOL(商標登録)、CARBOMER)などの、天然に生じるものではない酸性重合体;カルボキシメチルセルロースなどの合成的に修飾された酸性の天然重合体;(ヒドロキシプロピル)メチルセルロースなどの合成的に修飾された中性の天然重合体;キトサンなどの塩基性アミン保有重合体;アルギニン酸、ヒアルロン酸、ペクチン、トラガカントゴム及びカラヤゴムなどの天然源から得ることが可能な酸性重合体;ならびに、ポリビニルアルコールなどの天然に生じるものではない中性重合体;あるいはその組み合わせが挙げられる。
【0047】
「免疫刺激分子」の語句は、本明細書では、ワクチン組成物中の抗原性成分によって誘導される防御免疫応答を増強するこれらの分子を指す。適切な免疫刺激材料としては、細菌細胞壁成分(例えば、ムラブチド、トレオニル−MDP及びムラミルトリペプチドなどのN−アセチルムラミル−L−アラニル−D−イソグルタミン誘導体);サポニン配糖体及びその誘導体(例えば、Quil
A、QS 21及びGPI−0100);コレステロール;ならびに、第四級アンモニウム化合物(例えば、臭化ジメチルジオクタデシルアンモニウム(DDA)及びN,N−ジオクタデシル−N,N−ビス(2−ヒドロキシエチル)プロパンジアミン(「アブジリン」))が挙げられる。
【0048】
サポニンは、多様な植物種において二次代謝産物として産生される配糖体化合物である。サポニンの化学構造は、強力かつ効果的な免疫学的活性を始めとする、幅広い薬理学的及び生物学的活性を授ける。
【0049】
構造的には、サポニンは、1またはそれより多くの糖鎖に付着している任意のアグリコンからなる。サポニンは、そのアグリコン組成によって分類されることが可能である:トリテルペン配糖体、ステロイド配糖体及びステロイドアルカロイド配糖体。
【0050】
サポニンは、シャボンノキ(Quillaja saponaria)の樹皮から単離されることが可能である。サポニンは、免疫刺激剤として長く知られてきた。Dalsgaard, K.、「Evaluation of its adjuvant
activity with a special reference to the application in the vaccination of
cattle against foot-and-mouth disease」、Acta. Vet.
Scand. 69:1-40、1978年。サポニンを含有する植物の粗抽出物は、足口病ワクチンの効力を増強した。しかしながら、該粗抽出物は、ワクチンにおいて用いられた場合に副作用を伴った。これに続き、Dalsgaardは、サポニン由来のアジュバント活性成分を、透析、イオン交換及びゲルろ過クロマトグラフィによって部分精製した。Dalsgaard, K.ら、「Saponin adjuvants III.
Isolation of a substance from Quillaja saponaria Morina with adjuvant activity in
foot-and-mouth disease vaccines」、Arch. Gesamte.
Virusforsch. 44:243-254、1974年。この方法にて精製されたアジュバント活性成分は、「Quil
A」として公知である。重量に基づくと、粗サポニンと比較した場合に、Quil Aは効力の増加を示し、かつ局所反応を低減した。Quil Aは、動物用ワクチンにおいて広く用いられている。
【0051】
高速液体クロマトグラフィ(HPLC)によるQuil Aのさらなる分析によって、密接に関連するサポニンの不均一な混合物が示され、そして毒性が低減したかまたは最小限の強力なアジュバントであるQS21の発見が導びかれた。Kensil C.R.ら、「Separation and
characterization of saponins with adjuvant activity from Quillaja saponaria
Molina cortex」、J. Immunol. 146:431-437、1991年。ほとんどの他の免疫刺激剤とは異なり、QS21は、水溶性で、かつ乳剤型処方物の有無にかかわらずワクチンにおいて使用可能である。QS21は、マウスにおいてTh1型応答を誘発し、IgG2a及びIgG2b抗体の産生を刺激し、そしてサブユニット抗原に応答して抗原特異的CD8+
CTL(MHCクラスI)を誘導することが、示されている。ヒト臨床試験では、許容可能な毒性学的プロファイルを伴うそのアジュバント活性が、証明された。Kensil, C.R.ら、「Structural and imunological
charaterization of the vaccine adjuvant QS-21. In Vaccine Design: the subunit
and Adjvuant Approach」、Powell, M.F.及びNewman, M. J.編集、Plenum Publishing
Corporation、ニューヨーク州、1995年、525〜541頁。
【0052】
米国特許第6,080,725は、サポニン−リポフィル(lilpophile)結合体の作製方法及び使用方法を教示している。このサポニン−リポフィル結合体では、脂質、脂肪酸、ポリエチレングリコールまたはテルペンなどのリポフィル部分が、アシル化されていないかまたはデスアシル化されたトリテルペンサポニンと、トリテルペンサポニンの3−O−グルクロン酸上に存在するカルボキシ基を介して共有結合している。リポフィル部分と、Quillajaデスアシルサポニンなどのサポニン、lucyoside P、またはGypsophila属、saponaria属及びAcanthophyllum属由来サポニンの3−O−グルクロン酸との結合は、液性免疫及び細胞を介した免疫へのそのアジュバント効果を増強する。加えて、リポフィル部分と非アシルもしくはデスアシルサポニンの3−O−グルクロン酸残基との結合は、元のサポニンよりも精製し易く、低毒性で、化学的に安定で、かつ同等または優れたアジュバント特性を有するサポニン類似体を、もたらす。
【0053】
GPI−0100は、米国特許第6,080,725に記載されるサポニン−リポフィル結合体である。GPI−0100は、脂肪族アミンをグルクロン酸のカルボキシル基を介してデスアシルサポニンに付加することにより、生成される。
【0054】
第四級アンモニウム化合物−多数の脂肪族窒素含有塩基が、アミン、第四級アンモニウム化合物、グアニジン、ベンズアミジン及びチオウロニウムを始めとする免疫学的アジュバントとしての使用を提唱されている。具体的なこのような化合物としては、臭化ジメチルジオクタデシルアンモニウム(DDA)及びN,N−ジオクタデシル−N,N−ビス(2−ヒドロキシエチル)プロパンジアミン(「アブジリン」)が挙げられる。
【0055】
米国特許第5,951,988は、油成分と併せてDDAなどの第四級アンモニウム塩を含有するアジュバント処方物を、教示する。この処方物は、免疫原性応答を増強するために公知の免疫学的物質(例えばワクチン組成物中のウイルスもしくは細菌抗原)と併せて、有用である。該組成物は、非特異的な免疫刺激処方物として、取り込まれた抗原がなくても有用である。
【0056】
米国特許第4,310,550は、ワクチンアジュバントとして脂肪もしくは脂質乳剤とともに処方物化したN,N−高級アルキル−N,N’−ビス(2−ヒドロキシエチル)−プロパンジアミン及びN,N−高級アルキル−キシリレンジアミンの使用を、記載している。ヒトまたは動物においてアジュバント処方物の非経口投与を介して抗原の免疫原性応答を誘導または増強する方法は、米国特許第4,310,550に記載されている。
【0057】
好ましい実施態様において、本発明は、液滴サイズが1μm未満であり、かつ平均液滴サイズが約0.25μmのAMPHIGEN(商標登録)処方物から構成される、ワクチンアジュバントとして有用なサブミクロン水中油型乳剤を提供する。
【0058】
「AMPHIGEN(商標登録)処方物」の語は、本明細書では、DRAKEOL(商標登録)レシチン油溶液(Hydronics社、リンカーン、ネブラスカ州)と生理食塩溶液とをTWEEN(商標登録)80及びSPAN(商標登録)80の存在下で混合することによって形成される溶液を、指す。典型的なAMPHIGEN(商標登録)処方物は、軽質鉱油40%容量(v/v)、レシチン約25%w/v、TWEEN
80 約0.18%容量(v/v)及びSpan 80 約0.08%容量(v/v)を含有する。
【0059】
サブミクロン水中油型乳剤の調製方法
別の実施態様において、本発明は、上述のサブミクロン水中油型乳剤の調製方法を提供する。
【0060】
本発明によると、油、1またはそれより多くの界面活性剤、水溶性成分、及び乳剤における使用に適した任意の他の成分を始めとする乳剤の種々の成分を、組み合わせ、そして混ぜ合わせる。
【0061】
形成された混合物を、典型的には1もしくはそれより多くのホモジナイザーまたは乳化装置に1回以上通すことによる乳化過程にかけて、均一な外観及び約0.5μmの平均の液滴サイズの水中油型乳剤を形成する。例えばRoss乳化装置(Hauppauge、ニューヨーク州)、Gaulinホモジナイザー(Everett、マサチューセッツ州)などの市販されている任意のホモジナイザー及び乳化装置を、本目的のために用いることが可能である。
【0062】
次いで、このように形成された乳剤を、マイクロ流動化にかけて、サブミクロン範囲の液滴サイズをもたらす。マイクロ流動化は、Microfluidics社(ニュートン、マサチューセッツ州)から入手可能なモデル番号110Y;Gaulinモデル30CD(Gaulin, Inc.、Everett、マサチューセッツ州);及びRainnie Minilab 8.30H型(Miro Atomizer Food and Dairy, Inc.、ハドソン、ウィスコンシン州)などの市販のマイクロ流動化装置の使用によって、行うことが可能である。これらのマイクロ流動化装置は、二つの液流が相互作用チャンバー内で高速で相互作用して液滴がサブミクロンサイズの乳剤を形成するように、小さな開口部を介して高圧下で液体を強引に通すことによって、動作する。
【0063】
液滴サイズは、例えばレーザー回折、市販の定寸装置の使用によるなどの当該技術分野において公知の多様な方法によって、測定することが可能である。サイズは、用いる界面活性剤の種類、油に対する界面活性剤の割合、動作圧、温度等に応じて異なってもよい。当業者は、過度の実験を行うことなく所望の液滴サイズの乳剤を得るために、これらのパラメータの所望の組み合わせを決定することが可能である。本発明の乳剤の液滴サイズは、直径1μm未満、好ましくは平均液滴サイズ0.8μm未満、そしてより好ましくは平均液滴サイズ0.5μm未満、そしてよりいっそう好ましくは平均液滴サイズ0.3μm未満である。
【0064】
本発明の好ましい実施態様において、Hydronics社(リンカーン、ネブラスカ州)から市販され、そして軽質鉱油中に25%のレシチンを含有するDRAKEOLレシチン油溶液を、生理食塩液、ならびに界面活性剤であるTWEEN(商標登録)80及びSPAN(商標登録)80と組み合わせ、そして混合して、「AMPHIGEN(商標登録)溶液」または「AMPHIGEN(商標登録)処方物」を形成する。次いで、AMPHIGEN(商標登録)溶液をRoss(商標登録)(Hauppauge、ニューヨーク州、11788)乳化装置でおよそ3400rpmにて乳化して、水中油型乳剤を形成する。続いて、乳剤を、約4500±500psiで動作しているマイクロ流動化装置に、一度通す。マイクロ流動化された水中油型乳剤は、1μm未満のサイズの液滴を有し、平均液滴サイズ約0.25μmである。
【0065】
サブミクロン水中油型乳剤に取り込まれた抗原を含有する、ワクチン組成物
別の実施態様において、本発明は、抗原及び上述のサブミクロン水中油型乳剤を含有するワクチン組成物を、提供する。これらのワクチン組成物は、増強された免疫原性効果及び改善された物理的外観を有することによって、特徴付けられる(例えば、長期の保管後に相分離が認められない)。加えて、本発明のワクチン組成物は、動物への投与に関して安全である。
【0066】
本発明によると、抗原を、外来的に、また好ましくは内在的に乳剤と組み合わせることが可能である。「内在的に」の語は、マイクロ流動化工程の前に抗原を乳剤成分と組み合わせる過程を指す。「外来的に」の語は、乳剤をマイクロ流動化した後に抗原を乳剤に添加する過程を指す。外来的に添加された抗原は、遊離抗原であることが可能であり、あるいは以下にさらに記載するように、ミクロ粒子中にカプセル化されることが可能である。
【0067】
「抗原」の語は、本明細書では、動物において免疫原性であって、かつワクチン組成物が投与される動物において防御免疫応答を誘発するためにワクチン組成物中に含まれる、任意の分子、化合物または組成物を指す。
【0068】
抗原に関連して用いられる「免疫原性」の語は、動物において抗原に対する免疫応答を誘発する抗原の能を指す。該免疫応答は、細胞性T細胞によって主に媒介される細胞性免疫応答、あるいは次にB細胞を活性化して抗体産生へと導く、ヘルパーT細胞によって主に媒介される液性免疫応答であることが可能である。
【0069】
「防御免疫応答」は、抗原または抗原を含有する病原体により引き起こされる障害もしくは疾患の発生を防ぐかまたは検出可能に低下させるか、あるいはその重症度を除くかまたは検出可能に低減するか、あるいはその進行速度を検出可能に遅くする、動物において生じる、抗体または細胞のいずれかを介した免疫応答あるいはその双方である任意の免疫応答として、定義される。
【0070】
本発明のワクチン組成物に含まれることが可能な抗原としては、不活化した細胞の全体または一部の調製物の形態、あるいは慣用のタンパク質精製、遺伝子操作技術または化学合成によって得られる抗原分子の形態の、Mycoplasma hyopneumoniae、Haemophilus somnus、Haemophilus parasuis、Bordetella
bronchiseptica、Actinobacillus pleuropneumonie、Pasteurella multocida、Manheimia hemolytica、Mycoplasma bovis、Mycoplasma galanacieum、Mycobacterium bovis、Mycobacterium
paratuberculosis、クロストリジウム属、Streptococcus uberis、Streptococcus suis、Staphylococcus aureus、Erysipelothrix rhusopathiae、カンピロバクター属、Fusobacterium
necrophorum、大腸菌、Salmonella enterica serovars、レプトスピラ属などの病原性細菌;カンジダなどの病原性真菌;Cryptosporidium parvum、Neospora canium、Toxoplasma gondii、エイメリア属などの原虫; Ostertagia、Cooperia、Haemonchus、Fasciolaなどの蠕虫から調製される抗原が、挙げられる。抗原としては、さらに、不活化した細胞の全体または一部の調製物の形態、あるいは慣用のタンパク質精製、遺伝子操作技術または化学合成によって得られる抗原分子の形態の、ウシヘルペスウイルス−1、3、6、ウシウイルス性下痢ウイルス(BVDV)1型及び2型、ウシパラインフルエンザウイルス、ウシ呼吸器合胞体ウイルス、ウシ白血病ウイルス、牛疫ウイルス、足口病ウイルス、狂犬病、ブタコレラウイルス、アフリカブタコレラウイルス、ブタパルボウイルス、PRRSウイルス、ブタサーコウイルス、インフルエンザウイルス、ブタ水疱病ウイルス、Techen熱ウイルス、仮性狂犬病ウイルスなどの病原性ウイルスが、挙げられる。
【0071】
抗原の量は、水中油型乳剤と組み合わせた抗原が、動物において防御免疫応答を誘導するのに有効なようであるべきである。有効となる正確な抗原の量は、抗原の性質、活性及び純度に依存し、そして当業者により決定されることが可能である。
【0072】
ワクチン組成物中に存在する水中油型乳剤の量は、ワクチン組成物中の抗原の免疫原性を増強するのに十分であるべきである。望ましくかつ適切な場合には、界面活性剤の追加量または追加の界面活性剤を、水中油型乳剤により供される界面活性剤に加えて、ワクチン組成物中に加えることが可能である。一般的に言えば、油成分は、ワクチン組成物の最終容量において、1.0%〜20%容量の量;好ましくは、1.0%〜10%の量;より好ましくは、2.0%〜5.0%の量にて存在する。界面活性剤(単数)、あるいは2またはそれより多くの界面活性剤を用いる場合、界面活性剤の組み合わせは、ワクチン組成物の最終容量において、0.1%〜20%容量、好ましくは0.15%〜10%、より好ましくは0.2%〜6.0%の量にて存在する。
【0073】
抗原及び水中油型乳剤に加えて、ワクチン組成物は、水中油型乳剤に関連して上述のように、保存剤、浸透圧剤、生体接着分子及び免疫刺激分子(例えば、Quil
A、コレステロール、GPI−0100、臭化ジメチルジオクタデシルアンモニウム(DDA))などの適切かつ所望の他成分を含むことが可能である。
【0074】
本発明のワクチン組成物は、獣医学的に許容可能(veterinarily-acceptable)なキャリアーもまた含むことが可能である。「獣医学的に許容可能なキャリアー」の語には、任意及びすべての溶媒、分散媒体、被覆、アジュバント、安定剤、希釈剤、保存剤、抗細菌剤及び抗真菌剤、等張剤、吸収遅延剤等が含まれる。希釈剤としては、水、生理食塩液、ブドウ糖、エタノール、グリセロール等が挙げられてよい。等張剤としては、とりわけ、塩化ナトリウム、ブドウ糖、マンニトール、ソルビトール及びラクトースが挙げられてよい。安定剤としては、とりわけ、アルブミンが挙げられる。
【0075】
好ましい実施態様において、本発明は、1μm未満のサイズの液滴、好ましくは0.8μm未満、より好ましくは0.5μm未満の平均液滴サイズ、及びよりいっそう好ましくは約0.5μmの平均液滴サイズの水中油型乳剤中に内在的に取り込まれている、BVDV I型またはBVDV II型抗原のうち少なくとも一つを含むワクチン組成物を、提供する。BVDV
I型及び/またはII型抗原は、好ましくは、不活化ウイルス調製物の形態である。サブミクロン水中油型乳剤は、好ましくは、AMPHIGEN(商標登録)処方物(すなわち、軽質鉱油、レシチン、TWEEN(商標登録)80及びSPAN(商標登録)80を含有する処方物)から構成される。
【0076】
別の好ましい実施態様において、本発明は、レプトスピラ抗原、及びBVDV I型またはBVDV II型抗原のうち少なくとも一つを水中油型乳剤中に含むワクチン組成物を、提供する。好ましくは不活化した細胞もしくはウイルス調製物の形態である抗原は、
1μm未満のサイズの液滴、好ましくは0.8μm未満、より好ましくは0.5μm未満の平均液滴サイズ、及びよりいっそう好ましくは約0.5μmの平均液滴サイズの水中油型乳剤中に内在的に取り込まれている。サブミクロン水中油型乳剤は、好ましくは、AMPHIGEN処方物(すなわち、軽質鉱油、レシチン、TWEEN(商標登録)80及びSPAN(商標登録)80を含有する処方物)から構成される。該ワクチン組成物は、好ましくは、Quil−A、コレステロール、DDA、GPI−100及び水酸化アルミニウム(AlOH)から選択される1またはそれより多くの免疫刺激分子もまた含む。
【0077】
さらに別の好ましい実施態様において、本発明は、少なくとも一つの細菌抗原(例えば、組換えStreptococcus
uberis PauAタンパク質または大腸菌の細胞調製物、あるいはその双方の組み合わせ)を水中油型乳剤中に含むワクチン組成物を提供する。抗原は、1μm未満のサイズの液滴、好ましくは0.8μm未満、より好ましくは0.5μm未満の平均液滴サイズ、及びよりいっそう好ましくは約0.25μmの平均液滴サイズの水中油型乳剤と、内在的に組み合わされている。サブミクロン水中油型乳剤は、好ましくは、AMPHIGEN(商標登録)処方物(すなわち、軽質鉱油、レシチン、TWEEN(商標登録)80及びSPAN(商標登録)80を含有する処方物)から構成される。該ワクチン組成物は、好ましくは、Quil
A、DDA及びGPI−100から選択される1またはそれより多くの免疫刺激分子もまた含む。
【0078】
本発明のワクチン組成物を、経口、鼻腔内、粘膜、局所、経皮、及び非経口(例えば、静脈内、腹腔内、皮内、皮下または筋肉内)経路を始めとする公知の経路により動物に投与することが可能である。例えば、初回投与には非経口(parental)経路を用い、そしてそれに続く投与には粘膜経路を用いるなどの経路の組み合わせを用いて、投与を成し遂げることが可能である。
【0079】
ワクチン組成物の調製方法
さらなる実施態様において、本発明は、抗原(単数または複数)及びサブミクロン水中油型乳剤を含有するワクチン組成物の調製方法を提供する。
【0080】
本発明のワクチン組成物を調製する際に、抗原を、水中油型乳剤の成分と、内在的あるいは外来的に組み合わせることが可能である。「好ましくは」、抗原を、水中油型乳剤の成分と内在的に組み合わせる。
【0081】
抗原を、油、1またはそれより多くの界面活性剤、水溶性成分及び任意の他の適切な成分を含む乳剤の種々の成分と組み合わせて、混合物を形成することが可能である。該混合物を、典型的には1もしくはそれより多くのホモジナイザーまたは乳化装置に1回以上通すことによる第一混和過程にかけて、抗原を含有する水中油型乳剤を形成する。例えばRoss乳化装置(Hauppauge、ニューヨーク州)、Gaulinホモジナイザー(Everett、マサチューセッツ州)またはMicrofluidics(ニュートン、マサチューセッツ州)などの任意の市販されているホモジナイザーまたは乳化装置を、本目的のために用いることが可能である。あるいは、油、1またはそれより多くの界面活性剤及び水溶性成分を含む乳剤アジュバントの種々の成分を、まず組み合わせて、ホモジナイザーまたは乳化装置を用いることによって水中油型乳剤を形成し;次いで、抗原をこの乳剤に添加する。第一混和後の水中油型乳剤の平均液滴サイズは、およそ1.0〜1.2ミクロンである。
【0082】
次いで、抗原を含有する乳剤を、マイクロ流動化にかけて、サブミクロン範囲の液滴サイズを得る。マイクロ流動化は、Microfluidics社、ニュートン、マサチューセッツ州から入手可能なモデル番号110Y;Gaulinモデル30CD(Gaulin, Inc.、Everett、マサチューセッツ州);及びRainnie Minilab 8.30H型(Miro Atomizer Food and Dairy, Inc.、ハドソン、ウィスコンシン州)などの市販のマイクロ流動化装置の使用により、成し遂げることが可能である。
【0083】
液滴サイズは、例えばレーザー回折、市販の定寸装置の使用によるなどの、当該技術分野において公知の多様な方法によって、測定することが可能である。サイズは、用いる界面活性剤の種類、油に対する界面活性剤の割合、動作圧、温度等に応じて異なってもよい。所望の液滴サイズの乳剤を得るために、これらのパラメータの所望の組み合わせを決定することが可能である。本発明の乳剤の液滴サイズは、直径1μm未満である。好ましくは、平均液滴サイズは0.8μm未満である。より好ましくは、平均液滴サイズは0.5μm未満である。よりいっそう好ましくは、平均液滴サイズは約0.1〜0.3μmである。
【0084】
本発明の好ましい実施態様において、軽質鉱油中に25%のレシチンを含有するDRAKEOL(商標登録)レシチン油溶液を、TWEEN(商標登録)80及びSPAN(商標登録)80ならびに生理食塩液と組み合わせ、そして混合して、軽質鉱油40%、レシチン、TWEEN(商標登録)80
0.18%、及びSPAN(商標登録)80 0.08%を含有する混合物を形成する。次いで、該混合物を、Ross(商標登録)(Hauppauge、ニューヨーク州、11788)乳化装置でおよそ3400rpmにて乳化して乳剤生成物を形成し、これは「AMPHIGEN(商標登録)処方物」または「AMPHIGEN(商標登録)溶液」とも称される。続いて、所望の抗原を、乳化装置(たとえば、Rossホモジナイザー)を用いてAMPHIGEN(商標登録)溶液及び任意の他の適切な成分(例えば、免疫刺激分子)と組み合わせて、抗原を含有する水中油型乳剤を形成する。このような乳剤を、約10000±500psiで動作しているマイクロ流動化装置に一度通す。マイクロ流動化された水中油型乳剤は、平均液滴サイズ約0.25μmの、1μm未満のサイズの液滴を有する。
【0085】
別の好ましい実施態様において、水中油型乳剤(例えば、AMPHIGEN(商標登録)処方物)を所望の抗原と組み合わせる前に、抗原をサポニン配糖体(例えば、Quil
A)と組み合わせて、混合物を形成する。この抗原−サポニン混合物を、ホモジナイゼーション容器中でホモジナイゼーションにかける。次いで、例えばコレステロールなどのステロールを、ホモジナイズされた抗原−サポニン混合物に加える。次いで、抗原、サポニン及びステロールを含有する該混合物を、さらにホモジナイゼーションにかける。次いで、ホモジナイズされた抗原−サポニン−ステロール混合物を、例えばホモジナイザーを用いて、水中油型乳剤(例えば、AMPHIGEN(商標登録)処方物)と組み合わせる。次いで、抗原、サポニン及びステロールを含有するホモジナイズされた水中油型乳剤を、マイクロ流動化などの高圧ホモジナイゼーションにかける。
【0086】
マイクロカプセル化された抗原をサブミクロン水中油型乳剤中に含有するワクチン組成物、及び調製方法
さらに別の実施態様において、本発明は、ミクロ粒子中にカプセル化された抗原(または、「マイクロカプセル化された抗原」)を含有し、ここでマイクロカプセル化された抗原が前述のサブミクロン水中油型乳剤中に外来的に取り込まれているワクチン組成物を、提供する。
【0087】
抗原を粒子性キャリアー中に吸着または捕捉するための方法は、当該技術分野において公知である。例えば、Pharmaceutical Particulate Carriers: Therapeutic Applications(Justin Hanes、Masatoshi Chiba及びRobert Langer、「Vaccine design」中、Polymer microspheres for vaccine delivery、The
subunit and adjuvant approach. Michael F. Powell及びMark
J. Newman編集、1995年、Plenum Press、ニューヨーク州及びロンドン)を参照されたい。粒子性キャリアーは、動物個体において選択された抗原の複数のコピーを免疫系に提示し、そして局所リンパ節における抗原の捕捉及び保持を促進することが可能である。粒子は、マクロファージによって貪食されることが可能であり、かつサイトカイン放出を介して抗原提示を増強することが可能である。粒子性キャリアーは、当該技術分野においても記載されており、そして例えば、ポリメチルメタクリレート重合体由来のもの、ならびに、ポリ(ラクチド)及びPLGとして公知のポリ(ラクチド−共グリコリド)由来のものなどが挙げられる。ポリメチルメタクリレート重合体は非生分解性であるが、PLG粒子は、正常な代謝経路で排出される乳酸及びグリコール酸とのエステル結合のランダムな非酵素性加水分解によって生分解されることが可能である。
【0088】
生分解性ミクロスフェアもまた、 ワクチンの制御放出を成し遂げるのに用いられている。例えば、長期にわたり抗原の持続的な放出を成し遂げることが可能である。重合体の分子量、及び重合体におけるグリコール酸に対する乳酸の割合に応じて、PLGA重合体は、数日または数週間から数カ月または一年までの加水分解速度を有することが可能である。緩徐な制御放出により、複数回注射後に観察されるものと同様の高レベルの抗体の形成を生じる可能性がある。あるいは、ワクチン抗原のパルス放出を、異なる加水分解速度を伴う重合体を選択することによって成し遂げることが可能である。重合体の加水分解速度は、典型的には、重合体の分子量、及び重合体におけるグリコール酸に対する乳酸の割合に依存する。抗原放出速度の異なる2またはそれより多くの異なる重合体から作られたミクロ粒子は、抗原のパルス放出、及びワクチン接種の複数回投与レジメン(regime)の模倣を供する。
【0089】
本発明によると、前述のもののいずれかを始めとする抗原を、当該技術分野において公知の任意の手順(実施例17に例示されるものなど)を用いることにより粒子状重合体キャリアー(好ましくは、PLG重合体)に吸着させて、マイクロカプセル化された抗原調製物を形成することが可能である。次いで、マイクロカプセル化された抗原調製物を、前述したサブミクロン水中油型乳剤と混合し、そしてその中に分散させて、ワクチン組成物を形成する。
【0090】
好ましい実施態様において、本発明は、PLG重合体中にカプセル化された抗原を含有し、ここでマイクロカプセル化された抗原が軽質鉱油、レシチン、TWEEN80、SPAN80及び生理食塩液から構成されるマイクロ流動化された水中油型乳剤中に外来的に分散し、かつ1.0μm未満の平均液滴サイズを有するワクチン組成物を、提供する。
【0091】
サポニン及びステロールにより形成される複合体
一実施態様において、本発明は、サポニン及びステロールを含有し、ここでサポニン及びステロールがらせん状ミセルの形状の複合体を形成している組成物を、提供する。本発明によると、これらの複合体は免疫刺激活性を有する。
【0092】
「免疫刺激」とは、該複合体が抗原成分によって誘導される免疫応答を増強可能であること、あるいは該複合体が別の抗原成分とは独立に免疫応答を誘導可能であることを、意味する。
【0093】
本発明によると、本発明の組成物における使用に好ましいサポニンは、Quil Aである。
本発明のアジュバント組成物における使用に好ましいステロールとしては、β−シトステロール、スチグマステロール、エルゴステロール、エルゴカルシフェロール及びコレステロールが挙げられる。これらのステロールは当該技術分野において周知であって、例えばコレステロールは、メルクインデックス第11版、341頁に、動物脂肪中に見いだされる自然発生ステロールとして開示されている。最も好ましくは、ステロールはコレステロールである。
【0094】
組成物中のサポニン:ステロールの割合は、典型的には、およそ1:100〜5:1(重量対重量)である。好ましくは、割合は1:1である。
別の実施態様において、本発明は、サポニン、ステロール及び抗原を含有し、ここでサポニン及びステロールがらせん状ミセルの形状の複合体を形成しており、かつ抗原がらせん状ミセルと混合されているが、しかしその内部に取り込まれていないワクチン組成物を、提供する。
【0095】
以下は、本発明を実行するための具体的な実施態様の実施例である。当該実施例は、例示的な目的のみのために提供され、決して本発明の範囲を限定することを意図しない。
【実施例】
【0096】
(実施例1)
AMPHIGEN(商標登録)処方物の調製
AMPHIGEN(商標登録)処方物を、二工程の過程にて調製した。第一工程では、80リットルのDrakeolレシチン油溶液、116リットルの破傷風トキソイド生理食塩液、1.2リットルのSPAN
80、及び2.8リットルのTween 80を混ぜ合わせ、そしてRoss乳化装置を用いて乳化した。Drakeolレシチン油溶液は、大豆レシチン25%及び鉱油75%を含有した。乳化した生成物を、Ross乳化装置により最低5容量または最低10分間再循環させた。乳化した生成物を、さらなる処理のために、2〜7℃にて最大24時間保管した。Ross乳化装置のタンクから得られた乳剤を、Gaulinホモジナイザーに移し、そして4500psiの圧力下で20分間ホモジナイズした。次いで、得られた40%Drakeolレシチン油溶液(以下、「AMPHIGEN(商標登録)処方物」または「AMPHIGEN(商標登録)溶液」)を、無菌のポリプロピレンカルボキシ容器に分注した。分注は、クラス10,000の制御環境にあるクラス100の分注フードの中で行った。容器を、2〜7℃にて保管した。このAMPHIGEN(商標登録)処方物を、特に示さない限り、以下に記載の実験において用いた。
【0097】
(実施例2)
BVDワクチンの急速混和ホモジナイゼーションによる第一の混和
このホモジナイゼーション過程に用いられる装置を、図1に示す。無菌技術または蒸気用三方弁を用いて、BVD I型抗原(不活化したBVD
I型ウイルス調製物)を含有するボトルを、混和容器上の底部ポートに連結した。必要とされる容量のBVD I型抗原の移行が完了した後に、BVD I型のボトルを、不活化BVD
II型ウイルス調製物(不活化BVD II型ウイルス調製物)を含有するボトルと取り替えた。必要量のBVD II型抗原の移行が完了した後に、Rossホモジナイザーをポータブル容器に取り付け、そして再循環を最大のRPM(3300rpm)にて開始した。容器の撹拌を、中等度の速度にて維持した。
【0098】
無菌技術または蒸気用三方弁を用いて、Quil−Aを50mg/mlの濃度で含有するボトルを、混和容器上のホモジナイザーインラインポートに取り付けた。必要量のQuil−A溶液を、ライン吸引により容器へと通した。Quil−A溶液の移行が完了した後に、ボトルを取り外した。同じ方法で、必要量のコレステロール・エタノール溶液(18mg/ml)を、混和容器に移した。続いて、必要量のAMPHIGEN(商標登録)処方物、10%Thimerosol溶液、及び基本改変イーグル培地(「BME」)増量剤溶液を、混和容器に加えた。
【0099】
すべての添加が完了した後に、混合をさらに15分間続けた。得られた処方物を、2ml用量に分割し、そしてこれはマイクロ流動化されていないAMPHIGEN(商標登録)処方物に基づくBVDワクチンを表した。ワクチンの各用量は、Quil−A
500μg、コレステロール 500μg、AMPHIGEN(商標登録)処方物2.5%、及びThimerosol 0.009%を含有した。二つの異なるBVD株の抗原濃度を、gp53に対するELISA力価に関して測定した。
【0100】
(実施例3)
マイクロ流動化による第二の混和
図2は、マイクロ流動化を介した第二の混和に用いられる過程を示す。マイクロ流動化装置を、蒸気滅菌した。まず、補助の処理モジュールチャンバーをユニット内に設置し、そして空のチャンバーを二番目のチャンバー位置に設置した。実施例2に記載のように調製された、十分にアジュバント添加されているBVDワクチンを含有する容器を、供給容器のドレイン弁からマイクロ流動化装置の入口への移行ラインを取り付けることにより、マイクロ流動化装置に接続した。窒素ガスを、供給容器の空気フィルターの入口に接続し、そして容器の圧設定を20±5PSIに調整した。収集容器のドレイン弁を、マイクロ流動化装置の出口からの移行ラインに接続した。すべての必要な接続を行った後に、弁を開き、そしてマイクロ流動化を動作圧10,000±500PSIにて開始した。ワクチンの全量をマイクロ流動化装置に一度通し、そしてマイクロ流動化後チャンバー中に収集した。この調製物を、2mL用量に分割し、そしてこれはマイクロ流動化されたAMPHIGEN(商標登録)処方物に基づくBVDワクチンを表した。
【0101】
(実施例4)
ベンチ混和によるワクチン組成物の調製
実施例1に記載のように調製されたAMPHIGEN(商標登録)処方物を、BVD抗原及び増量剤を加えて2.5%に希釈した。得られた溶液を、ホモジナイザーを用いる代わりに撹拌子を用いて、ベンチにて混和した。最終調製物は、以下の組成物を含有した:BVD
1型及び2型抗原、2.5%AMPHIGEN(商標登録)処方物(実施例1に記載のように、油、レシチン、SPAN(商標登録)及びTWEEN(商標登録)を含有する)、ならびに生理食塩液。TWEEN
80及びSPAN 80は、最終ワクチン調製物中にそれぞれ0.18%及び0.08%容量にて存在する。
【0102】
(実施例5)
マイクロ流動化されていないAMPHIGEN(商標登録)処方物とマイクロ流動化されたAMPHIGEN(商標登録)処方物に基づくワクチン調製物との液滴サイズ分布の比較
実施例2に記載のように調製されたマイクロ流動化されていないAMPHIGEN(商標登録)処方物に基づくワクチン、実施例3に記載のように調製されたマイクロ流動化されたAMPHIGEN(商標登録)処方物に基づくワクチン、及び実施例4に記載のようにベンチで混和により作られた調製物を用いて、ワクチン調製物の液滴サイズを比較した。2ミリリットルの各調製物の試料を、Malvern 2000レーザー回折計測器に添加し、そして液滴サイズ分布を測定した。図3に示すように、結果は、マイクロ流動化されたAMPHIGEN(商標登録)処方物に基づくワクチン調製物の最大粒子体積がおよそ0.1ミクロンであり、一方、マイクロ流動化されていないAMPHIGEN(商標登録)処方物に基づくワクチン調製物の最大粒子分布体積がおよそ1ミクロンであったことを、示している。
【0103】
(実施例6)
ワクチンの相分離の低減
三つの異なるワクチン調製物:実施例2に記載のように調製されたマイクロ流動化されていないAMPHIGEN(商標登録)処方物に基づくワクチン、実施例3に記載のように調製されたマイクロ流動化されたAMPHIGEN(商標登録)処方物に基づくワクチン、及び実施例4に記載のようにベンチ混和により調製されたワクチン調製物を、並列に比較して、長期保管時のそれらの相分離の特性を判定した。すべてのこれらの調製物を4℃にて約1ヶ月間静置させ、そして相分離を、ワクチン調製物最上部のクリーム状の層の出現に関してモニターした。図4に示すように、他の二つの調製物と比較した場合、マイクロ流動化されたAMPHIGEN(商標登録)処方物に基づく調製物では相分離はなかった。
【0104】
(実施例7)
マイクロ流動化された、及びマイクロ流動化されていないウシウイルス下痢ウイルスに対するウシ用ワクチンの調製
ウシウイルス下痢ウイルス抗原を、マイクロ流動化を介してAMPHIGEN(商標登録)処方物中に内在的に取り込んだ。「内在的に取り込んだ」の語は、マイクロ流動化の前に抗原をAMPHIGEN(商標登録)処方物に添加することによる過程を指す。抗原をアジュバント処方物の成分とともに、マイクロ流動化過程の物理的な力にかけた。マイクロ流動化されていない対照群では、抗原調製物を、混和によりAMPHIGEN(商標登録)処方物中に分散させた。
【0105】
対照及びマイクロ流動化された調製物の双方の最終組成物は、以下のとおりであった:gp53に対する不活化後のELISA力価2535RU/用量のBVD
I型、1.25mg/用量の濃度のQuil−A、1.25mg/用量の濃度のコレステロール、2.5%の最終濃度のAMPHIGEN(商標登録)処方物、及び0.009%の最終濃度のThimerosol。ワクチン用量は、5mlであった。
【0106】
(実施例8)
マイクロ流動化されたAMPHIGEN(商標登録)処方物に基づくワクチン調製物における、内在的に取り込まれたBVDウイルス抗原の長期安定性
この実験は、内在的に取り込まれた抗原の長期保管中における安定性を判定するために行った。死滅BVD II型ウイルス抗原を、マイクロ流動化の過程中にAMPHIGEN(商標登録)処方物中に内在的に取り込んで、マイクロ流動化されたワクチン調製物(A907505)を得た。同抗原をマイクロ流動化されていないAMPHIGEN(商標登録)処方物(A904369、A904370及びA904371)中に含有する三つの他のワクチン調製物を、対照とした。マイクロ流動化されていない調製物では、抗原をAMPHIGEN(商標登録)処方物と混合し、そしてRossホモジナイザーを用いた混和により混合した。すべての四つのワクチン調製物を、4℃にて2年間保管した。保管中の異なる時点(0、6、12または24ヶ月)で、すべての四つの処方物を用いて、3ヶ月齢のウシにワクチン接種した。
【0107】
第0日及び第21日に、3ヶ月齢のウシを、皮下経路を介して、2mlのワクチン処方物でワクチン接種した。ワクチン接種された動物の血清を第35日に採取し、そしてワクチンに対する血清学的応答を、BVDV−E2
ELISAにより、抗体価に関して測定した。図5に示すように、マイクロ流動化されたワクチン調製物は、試験したすべての時点(0、6、12及び24ヶ月)で抗体価の上昇を示し、抗原調製物の安定性がマイクロ流動化の過程中における抗原の内在的な取り込みの間に失われないことが、示唆された。その上、マイクロ流動化されたワクチン調製物が、すべての時点で免疫応答の増強を誘導することもまた、驚くべきことに見いだされた。
【0108】
(実施例9)
マイクロ流動化後の、ワクチンにより誘導される直腸温増加の低減
実施例7に記載のように作られたマイクロ流動化されたワクチン調製物及びマイクロ流動化されていないワクチン調製物を用いて、第0日にウシにワクチン接種し、そして直腸温を、ワクチン接種1日前からワクチン接種後4日までの期間モニターした。ワクチン用量は、2mlであった。当該群を、1倍用量または2倍用量のいずれかのワクチンでワクチン接種した。直腸温を、第−1日から第4日まで毎日測定及び記録した。第0日における直腸温は、被験物質の投与前に測定した。
【0109】
図6に示すように、結果は、1倍用量または2倍用量いずれかのマイクロ流動化されていないワクチン処方物でワクチン接種されたこれらの動物において、ワクチン接種後約24時間に直腸温の急激な上昇があったことを、示している。しかしながら、マイクロ流動化された形状のワクチンでワクチン接種された動物においては、ワクチン接種後の直腸温の上昇は、ほんのわずかで、かつマイクロ流動化されていない処方物でワクチン接種された動物よりも有意に低かった(図6)。
【0110】
(実施例10)
マイクロ流動化されたワクチン処方物でワクチン接種した場合、注射部位反応体積はより速く回復した
実施例7に記載のように作られた、マイクロ流動化されたワクチン調製物及びマイクロ流動化されていないワクチン調製物を用いて、第0日にウシにワクチン接種した。この試験に含まれる動物は、交雑食用牛であった。プラセボ治療群(T01及びT02)のそれぞれには、3例の動物がいた。T03〜T06群のそれぞれには、6例の動物がいた。ワクチン用量は2mlであり、そして当該群を、第0日に1または2用量のいずれかのワクチンでワクチン接種した。第0日に、被験物質を、右首に投与した。2倍用量(4ml)の被験物質を投与される動物(T02、T04及びT06)には、片側に1回の注射として、全2倍用量を投与した。注射部位における反応サイズの評価を始めとする注射部位の観察を、第0日から第4日までの全日、及び第6、9及び14日に、首の右側で行った。第0日に、注射部位を、被験物質の投与前に観察した。1または2用量のプラセボでワクチン接種した群は、注射部位反応体積のいかなる有意な増加も示さず、そしてそれゆえにこれらのデータを図7に示していない。マイクロ流動化されていないワクチン処方物の場合、1用量と2用量のワクチン接種との間に注射部位反応体積の比例的な増加があった。他方で、マイクロ流動化されたワクチン処方物の場合、1倍用量がより大きな注射部位反応体積を誘導したが、二番目の用量での注射はいかなるさらなる増加も引き起こさなかった。その上、マイクロ流動化されたワクチン処方物で注射した動物の場合、注射部位反応部位体積は、マイクロ流動化されていないワクチン処方物で注射した動物と比較した場合に、より速い速度で回復した。これらの結果を、図7に示す。
【0111】
(実施例11)
内在的に取り込まれたBVDウイルス及びレプトスピラ抗原、ならびにQuil A及びDDAなどの免疫刺激分子を用いた、マイクロ流動化されたAMPHIGEN(商標登録)処方物に基づくワクチン調製物の調製
ホルマリンで不活化したLeptospira hardjo-bovis CLS株を、直接計数約1.4×109生物体/5ml用量にて、適切なアジュバント中に処方物化した。ホルマリンで不活化したLeptospira pomona T262株を、約2400比濁単位/5ml用量にて処方物化した。比濁単位は、処理前の発酵液の比濁測定に基づき算出した。BVDウイルス1型を、約3000相対単位/5ml用量のE2
Elisa力価にて処方物化した。BVDウイルス2型を、約3500相対単位/5ml用量のE2 Elisa力価にて処方物化した。相対単位は、合わせる前の不活化後のバルク液のE2
ELISA力価に基づき算出した。Quil−Aとコレステロールの双方を、0.5mg/用量の濃度にて用いた。Thimerosol及びAMPHIGEN(商標登録)処方物を、それぞれ0.009%及び2.5%の最終濃度にて用いた。水酸化アルミニウム(Rehydragel LV)を、2.0%の最終濃度にて用いた。DDAを免疫調節物質として用いた場合、DDAはAMPHIGEN(商標登録)処方物内に含まれた。AMPHIGEN(商標登録)処方物(すなわち、40%Drakeol−レシチンストック溶液)は1.6mg/mlのDDAを含有し、そして適切に希釈された場合、最終的なワクチン調製物は2.5%AMPHIGEN(商標登録)処方物及び0.1mg/mlのDDAを含有した。
【0112】
異なるワクチン処方物の調製において、BVD画分、レプト、Quil−A、コレステロール、Thimerosol、AMPHIGEN(商標登録)処方物、及び増量剤としての生理食塩液を、Silversonホモジナイザーに添加し、そして10,000±500RPMにて15分間混合した。次いで、成分を、200ミクロンのスクリーンを介して、10,000psiにてマイクロ流動化した。
【0113】
ワクチン処方物が水酸化アルミニウムを含有した場合、マイクロ流動化を水酸化アルミニウムなしで行った。マイクロ流動化が完了した後に、水酸化アルミニウムを添加し、そして撹拌子を用いて4℃にて一晩混合した。
【0114】
(実施例12)
曝露試験用BVDウイルスワクチンの調製
この実験に用いられるワクチン調製物は、BVDウイルス1型及びBVDウイルス2型の双方由来の抗原を含有した。BVD1−5960抗原を、gp53に対する不活化後のELISA力価2535RU/用量にて用いた。BVD2−890抗原を、gp53に対する不活化後のELISA力価3290RU/用量にて用いた。Quil
A及びコレステロールを、0.5mg/mlの濃度にて用いた。Thimersol及びAMPHIGEN(商標登録)処方物を、それぞれ0.009%及び2.5%の最終濃度にて用いた。DDAを免疫調節物質として用いた場合、DDAはAMPHIGEN(商標登録)処方物内に含まれた。AMPHIGEN(商標登録)ストック溶液(40%Drakeol−レシチン溶液)は様々な量のDDAを含有し、そして適切に希釈された場合、最終的なワクチン調製物は2.5%AMPHIGEN(商標登録)処方物及び0.5mg/用量〜2.0mg/用量のDDA濃度を含有した。アルミニウムゲル(Rehydragel-LV)を、2%の最終濃度にて用いた。GPI−0100を、2、3及び5mg/用量の範囲で用いた。
【0115】
すべての成分をSilversonホモジナイザーに添加し、そして10,500rpmにて15分間混和し、次いで10,000psiにて200ミクロンのチャンバーに通すことによりマイクロ流動化した。ワクチン調製物が水酸化アルミニウムを含有した場合、マイクロ流動化を水酸化アルミニウムなしで行った。マイクロ流動化が完了した後に、水酸化アルミニウムを添加し、そして撹拌子を用いて4℃にて一晩混合した。
【0116】
(実施例13)
レプトスピラ抗原を伴うマイクロ流動化されたAmphigenワクチン処方物でワクチン接種後の、レプトスピラ曝露に対する防御
【0117】
【表1】

【0118】
表1は、本試験において試験されたワクチン調製物におけるアジュバント処方物の組成を示す。ワクチン調製物を、実施例11に記載のように調製した。各群には、6例の動物がいた。約7ヶ月齢の交雑種未経産牛を、本試験において用いた。ワクチン接種を、第0日及び第21日に、皮下経路を介して5mlのワクチン容量にて行った。曝露を、国立動物疾病センター(NADC、National agricultural Disease Center)から得たL.
hardjo-bovis 203株を用いて行った。曝露は、第57〜59日に、1mlの接種により行った。曝露を、眼及び腟に同時投与した。曝露材料は、5.0X106レプトスピラ/mlを含有した。尿を、レプト培養、FA及びPCRのために一週間毎に採取した。腎臓の採取を、第112日及び第113日の間に行った。
【0119】
【表2】
【0120】
表2は、レプトスピラ曝露試験から得られたデータを示す。曝露された動物におけるレプトスピラ感染率を決定する際に、以下の基準を用いた。腎臓培養が一試料のみで陽性であったならば、動物はレプトスピラに陽性であると考えられる。動物がFAまたはPCRのいずれかに関して一試料のみで陽性であるならば、動物は陰性であると考えられる。試料がFA及びPCRの双方に関して一試料のみで陽性であるならば、レプトスピラに陽性であると考えられた。
【0121】
表2に示す結果は、すべての三つのアッセイに基づくすべてのワクチン群において、尿への排出期間が有意に短かったことを示している。尿及び腎臓のコロニー形成に関する限り、QAC及びDDAを含有するAlOHを含まない処方物の有効性は、同程度であった。AlOHは、本曝露試験においては、QACまたはDDAを含有するワクチンの有効性を改善せず、そしてむしろ低減した。
【0122】
【表3】
【0123】
ワクチン処方物中の双方のレプトスピラ抗原に対する血清学的応答を、ワクチン接種された動物において検出し、そしてピーク応答は第35日にみられた。血清学的応答と曝露に対する防御との間に、相関はなかった。血清学的応答がワクチン中のアルミニウムゲルの存在により増強されたにもかかわらず、ワクチン処方物中のアルミニウムゲルの存在は、防御レベルを低減させた。
【0124】
(実施例14)
AMPHIGEN(商標登録)処方物及びDDAを含有するマイクロ流動化されたワクチン調製物で免疫後の、BVDウイルス抗原に対する免疫応答の誘発、及びBVD
2型ウイルス曝露に対する防御
4〜7ヶ月齢の血清陰性の子ウシを、本実験において用いた。異なる6群があり、そして各群は10例の動物を有した(表4)。第0日及び第21日に、各動物は、2mlのワクチンまたはプラセボの皮下用量を、肩甲骨と頭蓋骨との中ほどにある側頸に1回受けた。
【0125】
【表4】
【0126】
5ml用量の曝露用ウイルス調製物(1鼻孔につき、およそ2.5ml)を、試験第44日に鼻腔内投与した。細胞変性していないBVD
2型ウイルスである単離番号24515(Ellis株)、ロット番号46325−70を、本試験において曝露用株として用いた。保持しておいた曝露用材料の試料を、曝露開始時及びその完了直後に力価測定した(1力価測定につき二つ組で)。5ml用量あたりの平均生存ウイルスは、曝露前は5.3log10FAID50/5ml、そして曝露後は5.4log10FAID50/5mlであった(FAIDは、TCID50に相当する)。
【0127】
動物を、第−3日から第58日まで、毎日モニターした。BVD 2感染に起因する臨床徴候に基づいた0、1、2または3の臨床疾患スコアを、第42日から第58日まで、各動物について行った。第44日におけるスコアは、曝露の前に記録した。BVD
1型及びBVD 2型ウイルス中和抗体の血清力価を測定するために、血液試料(二つの13ml血清分離用チューブ、SST)を、第0、21、35、44及び58日に各動物から採取した。
【0128】
血液試料を、第42日から第58日までの全日、各動物から採取し、そしてバフィーコート細胞におけるBVDウイルスの存在を判定した。第44日に、試料を曝露の前に得た。
【0129】
白血球細胞数を測定するために、血液試料(4ml EDTAチューブ1本)を、第42日から第58日までの全日、各動物から採取した。第44日に、試料を曝露の前に得た。
【0130】
白血球減少症を、40%またはそれを超えるベースライン(曝露2日前及び曝露当日の、曝露前の平均のWBC数)からのWBC数の減少として、定義した。
臨床疾患スコアを、以下のように疾患状態を定義するために用いた:スコアが≦1ならば、疾患=なし;スコアが>2ならば、疾患=あり。
【0131】
表5及び表6に示すように、AMPHIGEN(商標登録)処方物、Quil AまたはDDAとともにBVDウイルス抗原を含有し、かつマイクロ流動化されたワクチンでワクチン接種された群は、BVD
1型とBVD 2型ウイルスの双方に対する有意な血清ウイルス中和力価によって血清転換した。これらの群では、曝露後にウイルス血症を示す動物の率の有意な低減もあったが、対照群では、100%の動物がウイルス血症であった(表7)。加えて、これらのワクチン接種された群において、疾患の頻度もまた有意に低減した(表8)。同様に、白血球減少症を示す動物の率もワクチン群において低減し、そして白血球減少症の低減は、Quil
Aを含有する群よりもDDAを含有する群においてより有意であった(表9)。対照群では、ワクチン接種群と比較した場合に、体重増加の有意な降下があった(表10)。
【0132】
血清学
第0日におけるワクチン接種の前に、試験におけるすべての動物は、BVDウイルス 1型及び2型に対する抗体に血清陰性(SVN
<1:2)であった(データは示さず)。二回目のワクチン接種(第35日)後14日に、プラセボを投与されたすべての動物(T01)は、BVDウイルス1型及び2型に対する抗体に血清陰性のままであり;そして、ITA(研究用被験抗原)でワクチン接種されたすべての動物(T02、T03、T04、T05及びT06)は、BVDウイルス1型及び2型に対する抗体に血清陽性(SVN
≧1:8)であった。AMPHIGEN(商標登録)処方物をアジュバントとして添加したDDA 2mg/用量のワクチンを投与された1例の動物は、第35日に、BVDウイルス2型に対する抗体に対してSVN力価3であった(表11及び表12)。
【0133】
第44日における曝露の前に、1例を除くすべての対照(T01)は、BVDウイルス1型及び2型に対する抗体に血清陰性(SVN
<1:2)であった(データは示さず)。1例の対照(#2497)は、BVDウイルス1型に対する抗体に血清陽性(SVN=10)、そしてBVDウイルス2型に対する抗体に血清陰性であった。曝露後14日に、試験におけるすべての動物は、BVDウイルス1型及び2型に対する抗体に血清陽性であった。
【0134】
【表5】
【0135】
【表6】
【0136】
【表7】
【0137】
【表8】
【0138】
【表9】
【0139】
【表10】
【0140】
ウイルスの単離
表13に示すデータのように、曝露期間(第44日〜第58日)中に、対照(T01)におけるすべての10例の動物は、ウイルス血症であった(BVDウイルスが、1日以上で単離された)。ITA投与群では、ウイルス血症の動物の頻度は、10例の各群(それぞれ、T02、T03、T04、T05及びT06)において1、0、3、2及び2例であった。対照とITA投与群との間の差は、統計学的に有意であった(P≦0.05)。ウイルス血症の最小二乗平均日数もまた、ITA投与群(0.0〜0.5日)と比較して、対照で有意に大きかった(10.4日)。
【0141】
臨床疾患
臨床徴候スコア2または3の動物は、BVD疾患の徴候を示していると考えられた。表14に示すように、BVDウイルス疾患の臨床徴候を伴う動物の頻度は、対照(T01)において10例中9例、そしてITA投与各群(それぞれ、T02、T03、T04、T05及びT06)において、10例中1、2、0、0及び0例であった。対照とITA投与群との間の差は、統計学的に有意であった(P≦0.05)。
【0142】
白血球減少症
表15に示すように、曝露期間(第44日〜第58日)中に、対照(T01)におけるすべての10例の動物は、白血球減少症であった(第42〜44日の曝露前ベースラインから、白血球数が40%低減)。白血球減少症を伴う動物の頻度は、ITA投与各群(それぞれ、T02、T03、T04、T05及びT06)において、10例中6、2、4、3及び2例であった。対照と、0.5mg/用量のAMPHIGEN(商標登録)処方物及び水酸化アルミニウムをアジュバントとして添加したワクチン投与群(T03)との間の差は、統計学的に有意であった(P≦0.05)。白血球減少症の最小二乗平均日数は、ITA投与群(0.2〜1.2日)と比較して、対照で有意に大きかった(7.8日)。
【0143】
(実施例15)
GPI−0100を含有するマイクロ流動化されたワクチン調製物で免疫後の、BVDウイルス抗原に対する免疫応答の誘発、及びBVD2型ウイルス曝露に対する防御
実施例14に記載の一連の実験条件にしたがい、そしてQuil AとGPI−0100との直接比較を行った。表11及び表12に示すように、マイクロ流動化されたAMPHIGEN(商標登録)処方物に基づいた、Quil
AまたはGPI−0100のいずれかを含有する調製物に含まれるBVD抗原でワクチン接種された動物は、BVD1型とBVD2型ウイルスの双方に対する有意な抗体価を有した。BVD1型ウイルスに対する抗体価は、BVD2型ウイルスに対するものよりもかなり高かった。しかしながら、それに続くBVD2型ウイルスによる曝露は、強力な防御を示し、そして疾患発生率は、マイクロ流動化されたAMPHIGEN(商標登録)処方物に基づくGPI−0100含有ワクチン調製物でワクチン接種された子ウシにおいて、低減した。
【0144】
【表11】
【0145】
【表12】
【0146】
【表13】
【0147】
【表14】
【0148】
【表15】
【0149】
結論として、各ワクチンの安全性は、ワクチン接種された動物において副作用または死亡がないことにより示された。各ワクチンの効力は、100%のワクチン接種された動物における血清転換(BVD−1及びBVD−2に対するSVN抗体価
>1:8)によって、示された。曝露に対する十分な耐性は、GPI−0100 2mgのみをアジュバントとして添加したワクチンにより示された。
【0150】
(実施例16)
マイクロ流動化された水中油型乳剤中にマイクロカプセル化された抗原を含有するワクチン調製物
3gのトレハロース(Fluka社)を水に加えて、333mg/mlのトレハロース溶液を得た。0.8%SDS溶液中に可溶化した組換えPauA抗原(SDS/rPauA)をトレハロース溶液に加えて、494μg rPauA/mlの最終濃度を得た。次の工程で、10gのポリ乳酸グリコール酸(PLG-Resomer
RE 503H、ベーリンガー・インゲルハイム社)を、200mlの塩化メチレン(MeCl2)に溶解した。得られたPLG/MeCl2溶液を、最初の工程で調製されたSDS−rPauA/トレハロース溶液と組み合わせた。組み合わせた溶液を、(MicrofluidicsモデルM110EHのマイクロ流動化装置)を用いてマイクロ流動化にかけ、そしてマイクロ流動化された調製物を、(テプコ噴霧乾燥機モデルSD−05)を用いて噴霧乾燥した。噴霧乾燥した材料を、500ミクロンのスクリーンを用いて収集した。
【0151】
この噴霧乾燥した材料中のrPauA濃度を、ウエスタンブロット解析を用いて定量した。1.04mgの噴霧乾燥した材料を、50μlのアセトン中に溶解し、そして室温にて10分間、13,200rpmで遠心分離した。上清を、除去した。上清画分及びペレット画分を、生物学的安全フード内で2.5時間乾燥させた。ペレットを、47.43μLの試料溶液(25μlの試料緩衝液+10μlの還元剤+65μlの水)中に再懸濁した。乾燥させた上清画分を、20μlの試料溶液で再懸濁した。ウエスタン解析において、精製PauAを、噴霧乾燥した材料のrPauA含量を定量するための標準品として用いた。
【0152】
20%マンニトールストック溶液を、100mgのマンニトール(シグマ社)を500mlの注射用水(WFI)に溶解することにより、調製した。溶液をホットプレート/撹拌機で40℃に加熱し、そして30℃に冷ました。溶液を、0.22ミクロンの無菌フィルター(ミリポア社)を介して無菌ろ過した。2.5%カルボキシメチルセルロース溶液を、12.5gのカルボキシメチルセルロース(シグマ社)を500mlのWFIに溶解することによって調製し、そして4℃にて一晩混合した。溶液を、121℃にて高圧滅菌した。
【0153】
噴霧乾燥により得られた粉末を、5%マンニトール、0.3%カルボキシメチルセルロース及び1:5000のThimerosolを含有する溶液中で再構成した。最終溶液を3mlバイアルに分割し、そして凍結乾燥機(USIFROID)を用いて凍結乾燥した。凍結乾燥粉末は、マイクロカプセル化されたrPauAを表した。マイクロカプセル化されたサブユニットタンパク質抗原を、2mlのAMPHIGEN(商標登録)処方物を含有するマイクロ流動化された水中油型乳剤(実施例20に記載されるマイクロ流動化された乳剤など)中に再懸濁する。
【0154】
(実施例17)
水中油型乳剤中に細菌全細胞抗原と組換えタンパク質抗原の双方を含有するマイクロ流動化されたワクチン処方物の調製
実施例2及び3に記載のように水中油型乳剤に内在的に添加された、組換えStreptococcus
uberis PauAタンパク質と大腸菌細菌細胞の双方を含有する二つのワクチン調製物を、作った。組換えPauA抗原は100μg/用量の濃度であり、そして大腸菌細胞は4X109/用量の最終数であった。二つのワクチン処方物の乳剤アジュバント組成物を、表16に示す。
【0155】
【表16】
【0156】
(実施例18)
水中油型乳剤中にrPauA及び全細胞細菌因子を含有するマイクロ流動化されたワクチンに対する免疫応答
成熟乳牛を、本実験において用いた。動物は、登録時に初回または二回目の授乳が終了したところであった。2mlの各ワクチン処方物を、乾乳時(D−0)に一度、28日後(D=28)、及び分娩後4〜10日(C+4〜C+10)に再び、3回皮下投与した。最初及び三回目の用量を首の左側に投与し、そして二回目の用量を首の右側に投与した。血液を、各ワクチン接種の前、ならびに三回目のワクチン接種後およそ14日及び32日に採取した。大腸菌及びrPauAに対する抗体価を、ELISAにより測定した。図8に示すように、結果は、rPauAに対する抗体価が、GPI−0100を免疫刺激剤として含有するワクチン処方物でワクチン接種された群においてより高く、そして最初のワクチン接種後第70日にピークとなったことを示している。大腸菌抗原に対する抗体価を、図9に示す。免疫刺激剤としてのGPI−0100の存在が、免疫刺激剤としてDDAを伴う処方物と比較した場合に相対的により高い抗体価を誘導したにもかかわらず、大腸菌抗原に対する抗体価は、双方のワクチン処方物で同程度であった。
【0157】
(実施例19)
マイクロ流動化されたAMPHIGEN(商標登録)処方物に基づくワクチン調製物の殺ウイルス活性の分析
マイクロ流動化がウイルスを不活化するかどうかを判定するために、三つのマイクロ流動化されたAMPHIGEN(商標登録)処方物に基づくウイルス調製物の殺ウイルス活性を測定した。この三つの調製物は、三つの異なるウシ感染性ウイルス、すなわちウシヘルペスウイルス(BHV)、パラインフルエンザウイルス3(PI3)及びウシ呼吸器合胞体ウイルス(BRSV)を含有した。
【0158】
三つのワクチン調製物における殺ウイルス活性の検出を、USDA 9CFR.113.35要件にしたがって行った。
表16に示した結果は、AMPHIGEN(商標登録)処方物に基づくワクチン調製物のマイクロ流動化がワクチン調製物のいかなる有意な不活化も引き起こさないことを、示している。
【0159】
【表17】
【0160】
(実施例20)
マイクロ流動化されたAMPHIGEN(商標登録)処方物の調製
AMPHIGEN(商標登録)処方物を、DRAKEOLレシチン油溶液(25%レシチンを伴う軽質鉱油)とTWEEN 80(0.18%の最終濃度で)とSpan
80(0.08%の最終濃度で)とを組み合わせて、36±1℃にて8〜22時間混合することにより、調製した。次いで、Ross(Hauppauge、ニューヨーク州、11788)乳化装置をおよそ3400rpmで用いて、油混合物を生理食塩液に加えた。続いて、該混合物を、200μmの相互作用チャンバーを伴うマイクロ流動化装置に、4500±500psiにて一度通した。図10A及び図10Bは、マイクロ流動化されたAMPHIGEN(商標登録)処方物の安定性を示す。レーザー回折により測定された、最初の開始時点における粒子サイズ分布(図10A)は、22ヶ月間の4℃保管後の粒子サイズ分布(図10B)とほとんど同一であった。
【0161】
(実施例21)
QuilA−コレステロール免疫原性複合体の電子顕微鏡による分析
Quil A及びコレステロールにより形成された免疫原性複合体の性質を判定するため、これらの二成分の混合物を、抗原の存在下及び非存在下のいずれかで作った。
【0162】
50mlのKR−Hals緩衝液を含有するビーカーに、溶液を磁気撹拌子で攪拌しながら、BVD I型抗原を加えた。その後、Quil
Aの濃縮ストック溶液(50mg/ml)を、溶液を攪拌しながら1滴ずつ加えて、50μg/mlの最終濃度に到達させた。Quil
Aの添加後に、コレステロール・エタノール溶液(18mg/ml)を50μg/mlの最終濃度となるように加えた。
【0163】
二番目のビーカーには、Quil A及びコレステロールを、BVD I型抗原を全く含まない50mlの緩衝液に同じ方法で加えた。
透過型電子顕微鏡用に、10μlの各試料を、formvar/炭素担体プラットホームを伴う400メッシュ銅グリッド(Electron
Microscopy Sciences, Inc.、フォートワシントン、ペンシルバニア州)上に吸着させた。該試料を、ろ過したpH5.2の2%リンタングステン酸10μlを造影剤として用いて、ネガティブ染色した。該試料を、80kVの加速電圧にてJEOL
1230透過型電子顕微鏡(JEOL Inc.、東京、日本)を用いて、検討した。デジタルイメージングを、Gatan BioScan 792カメラで行った。顕微鏡撮影を、4489 EMフィルムで行い、そしてコダブロムII RC F3紙(Eastman Kodak Company、ロチェスター、ニューヨーク州)に印刷した。
【0164】
BVD I型抗原を全く含まない、コレステロール及びQuil Aのみを含有する溶液では、らせん状ミセルが、Quil Aミセル及びコレステロール結晶とともに検出された(図11)。らせん状ミセルは、絡み合い、そしてメッシュのようにみえた。BVD
I型抗原を含有する試料では、らせん状ミセルは、ランダムに特定の高密度領域を取り囲むことがみられた(図12)。該高密度領域はBVD1型抗原を表し、そしてQuil
Aとコレステロールの会合により生じるらせん状免疫原性複合体は、BVD I型抗原の表面に吸着することが見いだされている。
【図面の簡単な説明】
【0165】
【図1】図1は、マイクロ流動化されていないワクチン組成物のバッチ調製のための過程を示す。この過程では、種々のワクチン成分を、左の添加容器に添加し、そして最終的には、成分を単純な機械的手段により混ぜ合わせる混合容器へと投入する。
【図2】図2は、内在的に取り込まれた抗原を含有するマイクロ流動化されたワクチン組成物を調製するための過程を示す。種々のワクチン成分を、添加容器に添加し、そして単純な機械的手段により混合するためのプレ乳剤混和ユニットに移す。
【図3】図3は、マイクロ流動化されていないAMPHIGEN(商標登録)処方物に基づくワクチン、マイクロ流動化されたAMPHIGEN(商標登録)処方物に基づくワクチン、及びベンチ混和ワクチン調製物の液滴サイズの分布を、示す。
【図4】図4は、マイクロ流動化されたワクチン調製物では相分離しないことを示す。
【図5】図5は、マイクロ流動化されたAMPHIGEN(商標登録)処方物に基づくワクチン調製物(A907505)、及び対照である三つのマイクロ流動化されていないAMPHIGEN(商標登録)処方物に基づくワクチン調製物(A904369、A904370及びA904371)中に内在的に取り込まれた抗原の安定性の比較を、示す。すべての四つのワクチン調製物を、4℃にて2年間保管した。保管中の異なる時点(0、6、12または24ヶ月)で、すべての四つの処方物を用いて、3ヶ月齢のウシをワクチン接種した。ワクチン接種を、2mlのワクチン用量で第0日及び第21日に行い、そして二回目のワクチン接種後2週間に、血清を採取した。BVDII型ウイルスに対する中和抗体価を、血清試料のそれぞれにおいて測定した。データを、5例の動物の幾何平均として示した。
【図6】図6は、マイクロ流動化されたワクチン及びマイクロ流動化されていないワクチンの投与前及び投与後のウシの直腸温の最小二乗平均を示す。T01:プラセボ群−1倍用量;T02:プラセボ群−2倍用量;T03:マイクロ流動化されていない処方物−1倍用量;T04:マイクロ流動化されていない処方物−2倍用量;T05:マイクロ流動化された処方物−1倍用量;T06:マイクロ流動化された処方物−2倍用量。
【図7】図7は、マイクロ流動化されていないワクチン処方物及びマイクロ流動化されたワクチン処方物の投与後にウシにおいて観察される注射部位反応体積の最小二乗平均を示す。T03:マイクロ流動化されていない処方物−1倍用量;T04:マイクロ流動化されていない処方物−2倍用量;T05:マイクロ流動化された処方物−1倍用量;T06:マイクロ流動化された処方物−2倍用量。
【図8】図8は、組換えPauA抗原と大腸菌全細胞抗原の双方を含有する種々のワクチン処方物でワクチン接種後の、Streptococcus uberis由来組換えPauA抗原のIgG力価の幾何平均を示す。
【図9】図9は、組換えPauA抗原と大腸菌全細胞抗原の双方を含有する種々のワクチン処方物でワクチン接種後の、Streptococcus uberis由来大腸菌全細胞抗原のIgG力価の幾何平均を示す。
【図10A】図10A及び図10Bは、最初の生成時(図10A)及び生成後22ヶ月(図10B)におけるマイクロ流動化されたAmphigen処方物の粒子サイズ分布を示す。
【図10B】図10A及び図10Bは、最初の生成時(図10A)及び生成後22ヶ月(図10B)におけるマイクロ流動化されたAmphigen処方物の粒子サイズ分布を示す。
【図11】図11は、Quil Aミセル及びコレステロール結晶とともに形成されたらせん状ミセルを示す電子顕微鏡写真である。
【図12】図12は、BVD I型抗原の表面にQuil A及びコレステロールにより形成されたらせん状免疫原性複合体を示す電子顕微鏡写真である。
【発明の詳細な説明】
【0001】
【発明の開示】
【0002】
発明の分野
本発明は、概して、ワクチン分野に関し、そして具体的には、家畜動物において免疫応答を増強するためのアジュバント処方物に関する。具体的には、本発明は、抗原の免疫原性を増強するための、サブミクロン水中油型乳剤のワクチンアジュバントとしての使用に関する。サブミクロン水中油型乳剤処方物、このような乳剤に取り込まれた抗原を含有するワクチン組成物、ならびに当該の乳剤及びワクチン調製方法が、本発明により提供される。本発明はまた、ワクチンにおける使用に適したサポニン配糖体及びステロールにより形成される複合体を含有する組成物も、提供する。
【0003】
発明の背景
細菌、ウイルス、寄生虫及びマイコプラズマ感染は、ウシ、ブタ及びペットなどの家畜動物において、幅広く広まっている。これらの感染性因子によって引き起こされる疾患は、多くの場合、抗微生物医薬療法に耐性であり、有効な治療の手段を残さない。結論として、ワクチン学的なアプローチが、家畜動物における感染性疾患を制御するために、ますます用いられている。感染性病原体全体を、化学的不活化または適切な遺伝子操作の後に、ワクチン処方物における使用に適するようにすることが可能である。あるいは、病原体のタンパク質サブユニットを、組換え発現系にて発現させ、そしてワクチン処方物における使用のために精製することが可能である。
【0004】
アジュバントは、概して、抗原に対する液性及び/または細胞性免疫応答を増加させる任意の材料を指す。従来のワクチンは、死滅した病原性微生物の粗調製物から構成され、そして、病原性微生物の培養に伴う不純物が、免疫応答を増強するためのアジュバントとして作用し得る。しかしながら、病原性微生物の均一な調製物または精製タンパク質サブユニットをワクチン接種用抗原として用いる場合、このような抗原によって引き起こされる免疫は不十分であり、それ故に、アジュバントとしての特定の外因性材料の添加が、必要となってくる。さらに、合成ワクチン及びサブユニットワクチンは、生産コストが高い。したがって、アジュバントを用いると、免疫応答を刺激するために必要とされる投与量がより少量で、それによってワクチンの生産コストが抑えられる可能性がある。
【0005】
アジュバントは、免疫応答を増強するための多くの異なる経路で作用することが知られている。多くのアジュバントは、免疫応答に関連するサイトカインネットワークを修飾する。これらの免疫調節アジュバントは、それらが抗原と一緒でない場合でさえも、その効果を発揮することが可能である。一般に、免疫調節アジュバントは、特定のサイトカインの一般的な亢進、及び同時に他のサイトカインの低下を引き起こし、細胞性Th1及び/または液性Th2応答へと導く。
【0006】
あるアジュバントは、抗原の立体配座の完全性を保存する能を有し、そのために抗原が適切な免疫エフェクター細胞に効率的に提示されることが可能である。アジュバント処方物による抗原の立体配座のこの保存の結果として、ワクチンは、免疫刺激複合体(ISCOM)に関して示されるように、有効期間が増すことになる。Ozel M.ら;Quarternary Structure of the
Immunestimmulating Complex (Iscom), J.of Ultrastruc. and Molec. Struc. Res.
102, 240-248(1989年)。
【0007】
あるアジュバントは、注射部位にデポー(depot)として抗原を保持する特性を有する。このデポー効果の結果として、抗原は、肝クリアランスによって急速には消失しない。アルミニウム塩及び油中水型乳剤は、このデポー効果を介して、より短期間に作用する。例えば、油中水型乳剤であるフロイント完全アジュバント(FCA)を用いることによって、長期のデポーを得ることが可能である。FCAは、典型的には、抗原提示細胞による抗原の除去が生分解により可能となるまで、注射部位に留まり続ける。
【0008】
それらの物理的な性質に基づき、アジュバントを、二つの非常に大まかなカテゴリー、すなわち、粒子状アジュバントと非粒子状アジュバントとに分類することが可能である。粒子状アジュバントは、ミクロ粒子として存在する。免疫原は、ミクロ粒子を取り込むか、あるいはそれと会合することが可能である。アルミニウム塩、油中水型乳剤、水中油型乳剤、免疫刺激複合体、リポソーム、ならびにナノ及びミクロ粒子は、粒子状アジュバントの例である。非粒子状アジュバントは、一般には、免疫調節物質であり、そしてこれらは、一般には、粒子状アジュバントと併せて用いられる。ムラミルジペプチド(マイコバクテリアから抽出されるペプチドグリカンのアジュバント活性成分)、非イオン性ブロック共重合体、サポニン(シャボンノキ(Quillaja saponaria)の樹皮から抽出されるトリテルペノイドの複合混合物)、リピドA(2個のリン酸基及び5または6個の概してC12〜C16長の脂肪酸鎖を有する、グルコサミンの二糖類)、サイトカイン、炭水化物重合体、誘導体化多糖類、ならびに、コレラ毒素及び大腸菌(E. coli)不安定毒素(LT)などの細菌毒素が、非粒子状アジュバントの例である。
【0009】
最も知られているアジュバントの一部は、非粒子状免疫調節物質と、デポー効果をアジュバント処方物に授け得る粒子状材料との組み合わせである。例えば、FCAは、結核菌(Mycobacterium tuberculosis)成分の免疫調節特性を、油型乳剤の短期デポー効果とともに組み合わせている。
【0010】
油型乳剤は、ワクチンアジュバントとして長い間使われている。Le Moignic及びPinoyは、1916年に、死滅ネズミチフス菌(Salmonella typhimurium)の鉱油懸濁液が免疫応答を増加させることを、見いだした。これに続き1925年には、Ramonが、スターチ油をジフテリアトキソイドへの抗毒素応答を増大させる物質の一つとして記載した。しかしながら、油型乳剤は、フロイントが現在フロイント完全アジュバント(FCA)として公知のアジュバント処方物を公表した1937年まで、一般的にはならなかった。FCAは、死滅マイコバクテリア及びアラセルAと混合された鉱(パラフィン)油から構成される、油中水型乳剤である。アラセルAは、主にマンニドモノオレエートであり、そして乳化剤として用いられる。FCAは、抗体応答の誘導に優れているにもかかわらず、重度の疼痛、膿瘍形成、発熱及び肉芽腫性炎症を引き起こす。これらの望ましくない副作用を避けるために、不完全フロイントアジュバント(IFA)が開発された。IFAは、マイコバクテリア成分がないことを除けば、FCAとその組成が類似している。IFAは、注射部位におけるデポー形成、及び抗体産生細胞の刺激による抗原の徐放を介して、作用する。
【0011】
FCAを改善するための別のアプローチは、鉱油を生体適合性の油により置き換えることが、注射部位におけるFCAに関連する反応を除去するのに役立つであろうとの概念に基づいた。乳剤は油中水型乳剤よりもむしろ水中油型乳剤であるべきとも、前者が長期間持続するデポーを注射部位に作り出すことから、考えられていた。Hillemanらは、ピーナツ油86%、乳化剤としてのアラセルA 10%、及び安定剤としてのモノステアリン酸アルミニウム4%からなる油性アジュバント「アジュバント65」について記載した。Hilleman, 1966年、Prog. Med. Virol. 8:131-182;Hilleman及びBeale,
1983年、New Approaches to Vaccine Development(Bell, R.及びTorrigiani, G.編集)中、Schwabe, Basel。ヒトにおいて、アジュバント65は安全かつ強力であったが、しかしIFAよりも低いアジュバント活性を示した。それにもかかわらず、アジュバント65の使用は、特定ロットのヒトに対するワクチンでの反応原性、及び精製もしくは合成乳化剤がアラセルAの代わりに用いられる場合のアジュバント活性の低減により、中止された。米国特許第5,718,904及び第5,690,942は、安全性プロファイルを改善する目的で、水中油型乳剤中の鉱油を代謝可能な油に置き換えることが可能であることを、教示している。
【0012】
アジュバント活性及び安全性の他に、乳剤の物理的外観もまた、市販のために考慮すべき重要なことである。物理的外観は、乳剤の安定性に依存する。クリーミング、沈降及び癒合は、乳剤の不安定さの指標である。クリーミングは、乳剤の油相と水相とが異なる比重である場合に生じる。クリーミングはまた、乳剤の初期の液滴サイズが大きく、かつ乳剤の液滴が全くブラウン運動をしていない場合にも、生じる。液滴サイズが大きい場合、界面が破裂する傾向があり、そして液滴が大きな粒子に癒合する。乳剤の安定性は、用いられる乳化剤の性質及び量、乳剤中での液滴サイズのサイズ、ならびに油相と水相との間の密度の差異などの多数の要因によって決まる。
【0013】
乳化剤は、界面の自由エネルギーを低減し、そして液滴の癒合に対する物理的もしくは静電気的なバリアを作り出すことによって、分散している液滴の安定性を促進する。非イオン性及びイオン性界面活性剤は、乳化剤として使用されている。非イオン性乳化剤は、界面にて配向し、そして比較的分厚い構造を作り出すが、このことにより分散している液滴が立体的に回避される。陰イオンもしくは陽イオン乳化剤は、対イオンをひきつけることによって電気二重層の形成を誘導し;二重層の反発力は、液滴を、それらが接近するときに互いに反発させる。
【0014】
乳化剤を用いる他に、乳剤の安定性は、機械的な手段によって乳剤の液滴サイズを低減することによっても、達成可能である。典型的には、プロペラミキサー、タービンローター、コロイドミル、ホモジナイザー及びソニケーターが、乳剤を製造するために用いられている。マイクロ流動化は、乳剤における液滴サイズの均一性を増加させる別の方法である。マイクロ流動化は、サブミクロン範囲の一定した粒子サイズの繊細で物理的に安定な乳剤を作り出すことが可能である。乳剤の安定性を増加させる他に、マイクロ流動化の過程は、最終産物の無菌性を確実にする好ましい方法である最終ろ過を可能とする。その上、サブミクロンの油粒子は、注射部位からリンパ管へ入り、次いでドレナージ連鎖のリンパ節、血液及び脾臓を通ることが可能である。このことは、局所的炎症及び有意な注射部位反応を作り出す可能性のある油のデポーを注射部位に確立する可能性を低減する。
【0015】
マイクロ流動化装置は、現在、市販されている。乳剤の形成は、マイクロ流動化装置において、相互作用チャンバー内で二つの流動化された流れが高速で相互作用するときに生じる。マイクロ流動化装置は、空気または窒素を駆動し、そして過剰の20,000psiの内部圧力にて動作することが可能である。米国特許第4,908,154は、いかなる乳化剤も本質的に含まない乳剤を得るためのマイクロ流動化装置の使用を、教示している。
【0016】
多数のサブミクロン水中油型アジュバント処方物が、文献に記載されている。米国特許第5,376,369は、Syntaxアジュバント処方物(SAF)として公知のサブミクロン水中油型乳剤アジュバント処方物を、教示している。SAFは、油成分としてのスクアレンまたはスクアラン、乳剤を形成する量のPluronic L121(ポリオキシ−プロピレン−ポリオキシエチレン)ブロック重合体、及び免疫を増強する量のムラミルジペプチドを、含有する。スクアレンは、多くの組織中(特に、サメ及び他の魚類の肝臓中)に見いだされる、コレステロールの直線状の炭化水素前駆体である。スクアランは、スクアレンの水素添加により調製され、かつ完全に飽和している。スクアレンとスクアランの双方は、代謝されることが可能であり、そして毒性学試験の成績は良好である。スクアレンもしくはスクアラン乳剤は、ヒト癌ワクチンにおいて使用され、軽度の副作用及び望ましい有効性を伴う。例えば、Anthony C. Allison, 1999年、Squalene and
Squalane emulsions as adjuvants, Methods 19:87-93を参照されたい。
【0017】
米国特許第6,299,884及び国際特許公報WO 90/14837は、ポリオキシ−プロピレン−ポリオキシエチレンブロック共重合体がサブミクロン水中油型乳剤の形成に必須ではないことを、教示している。その上、これらの参考文献は、非毒性の代謝可能な油の使用を教示し、そして鉱油及び毒性石油蒸留油のその乳剤処方物における使用を明確に除外している。
【0018】
米国特許第5,961,970は、ワクチンアジュバントとして用いられるさらに別のサブミクロン水中油型乳剤を教示している。本特許に記載される乳剤において、疎水性成分は、中鎖トリグリセリド油、植物油及びその混合物からなる群より選択される。この乳剤に含まれる界面活性剤は、リン脂質(例えば、レシチン)などの天然の生物学的に適合性の界面活性剤、あるいはTWEEN−80などの医薬的に許容可能な非天然の界面活性剤であることが可能である。この特許はまた、乳剤を形成するときに抗原を乳剤中へ取り込むことを教示しており、乳剤を独立的かつ外来的に形成した後に抗原を乳剤と混合するのとは対照的である。
【0019】
米国特許第5,084,269は、鉱油と組み合わせてレシチンを含有するアジュバント処方物が、宿主動物内での刺激を減少させ、そして同時に全身性免疫の増加を誘導することを、教示している。米国特許第5,084,269から得られたアジュバント処方物は、商品名AMPHIGEN(商標登録)で、動物用ワクチンにおいて商業的に用いられている。AMPHIGEN(商標登録)処方物は、ミセル−レシチンに囲まれた油滴で構成される。これらのミセルは、従来の油に基づくアジュバントよりも多くの全細胞抗原の付着を可能にする。その上、AMPHIGEN(商標登録)に基づくワクチン処方物は、典型的には10%〜20%の油を含有する油アジュバントを含有する他のワクチン処方物と比較して、2.5〜5%の鉱油と、低い油量を含有する。この低い油量が、このアジュバントに基づくワクチン処方物の注射部位組織に対する刺激を低下させて、屠殺時により少ない病変及びより少ないトリムを生じる。加えて、油滴を囲んでいるレシチン被覆は、注射部位反応をさらに低減し、安全かつ有効なワクチンをもたらす。
【0020】
AMPHIGEN(商標登録)処方物は、多数の動物用ワクチンにおいてアジュバントとして用いられ、そしてワクチン製品の物理的外観は、短期及び長期の保管期間中ならびに再構成時に維持される必要がある。加えて、凍結乾燥させた抗原を、予め調製しておいたアジュバント処方物と、注射直前に混合する。この実行は、水中油型乳剤内での抗原の均一な分布を必ずしも確実にするわけではなく、そして乳剤の外観は望ましくない可能性がある。その上、均質化された乳剤は、静置すると相分離を示し得る。したがって、長期の有効期間に相分離を示さない安定なアジュバント処方物の必要性が存在する。相分離を防ぐ一つの方法は、液滴サイズを小さくし、そして乳剤の粒子均一性を増加させることである。代謝可能な油性乳剤処方物のマイクロ流動化の過程が文書化されているものの、AMPHIGEN(商標登録)処方物などの水中油型乳剤のマイクロ流動化は、まだ実行されていない。
【0021】
本発明では、マイクロ流動化を、レシチンに囲まれた鉱油の液滴サイズをサブミクロンサイズにするために用いている。予想外にも、本発明者は、レシチンと油との混合物からなる水中油型乳剤をアジュバントとして添加したワクチン処方物のマイクロ流動化が、処方物の物理的外観を改善するだけでなく、処方物の免疫化効果も増強することを、発見した。マイクロ流動化された処方物はまた、改善された安全性プロファイルによっても特徴付けられる。
【0022】
発明の概要
本発明者は、代謝可能でない油に基づく水中油型乳剤のアジュバント活性及び安全性プロファイルが、マイクロ流動化を介して改善可能であることを、予想外にも発見した。マイクロ流動化された乳剤に取り込まれた抗原は、抗原をマイクロ流動化の前に乳剤中に内在的に取り込む場合でも、安定である。
【0023】
したがって、一実施態様において、本発明は、ワクチンアジュバントとして有用なサブミクロン水中油型乳剤処方物を提供する。本発明のサブミクロン水中油型乳剤は、代謝可能でない油、少なくとも一つの界面活性剤、及び水溶性成分から構成され、ここで油は、サブミクロン範囲の平均の油滴サイズで水溶性成分中に分散している。好ましい代謝可能でない油は、軽質鉱油である。好ましい界面活性剤としては、レシチン、TWEEN(商標登録)−80及びSPAN(商標登録)−80が挙げられる。
【0024】
本発明により提供される好ましい水中油型乳剤は、AMPHIGEN(商標登録)処方物から構成される。
本発明の水中油型乳剤は、保存剤、浸透圧剤、生体接着分子及び免疫刺激分子などの適切かつ望ましい成分をさらに含むことが可能である。好ましい免疫刺激分子としては、例えば、Quil
A、コレステロール、GPI−0100、臭化ジメチルジオクタデシルアンモニウム(DDA)が挙げられる。
【0025】
別の実施態様において、本発明は、サブミクロン水中油型乳剤の調製方法を提供する。本発明によると、油、1またはそれより多くの界面活性剤、水溶性成分、及び乳剤における使用に適した任意の他の成分を始めとする乳剤の種々の成分を、混ぜ合わせる。該混合物を最初の乳化過程にかけて、水中油型乳剤を形成し、次いでこれをマイクロ流動化装置に通して、液滴が直径1ミクロン未満、好ましくは平均液滴サイズ0.5ミクロン未満の水中油型乳剤を得る。
【0026】
さらに別の実施態様において、本発明は、抗原及び上述のサブミクロン水中油型乳剤を含有するワクチン組成物を提供する。抗原は、外来的あるいは内在的に、好ましくは内在的に、乳剤中に取り込まれている。
【0027】
本発明のワクチン組成物に含まれることが可能な抗原は、細菌、真菌またはウイルスの抗原、あるいはその組み合わせであることが可能である。該抗原は、不活化した細胞またはウイルスの全体または一部の調製物の形態を取るか、あるいは慣用のタンパク質精製、遺伝子操作技術または化学合成によって得られる抗原分子の形態を取ることが可能である。
【0028】
さらなる実施態様において、本発明は、抗原(単数または複数)をサブミクロン水中油型乳剤と組み合わせて含有するワクチン組成物の調製方法を、提供する。
本発明のワクチン組成物を調製する際に、抗原を、内在的(例えば、マイクロ流動化の前に)あるいは外来的(例えば、マイクロ流動化の後に)に水中油型乳剤の成分と組み合わせることが可能である。
【0029】
さらに別の実施態様において、本発明は、マイクロカプセル化された抗原及び上述のサブミクロン水中油型乳剤を含有し、ここでマイクロカプセル化された抗原が乳剤と外来的に組み合わされているワクチン組成物を、提供する。
【0030】
サポニンとステロールが、溶液中で組み合わされる場合に、互いに会合して、らせん状ミセルの形状の複合体を形成することもまた、驚くべきことに発見された。本発明によると、これらのらせん状ミセル複合体は、免疫刺激活性を有し、そしてワクチン組成物におけるアジュバントとして特に有用である。
【0031】
したがって、本発明は、サポニン及びステロールを含有し、ここでサポニンとステロールがらせん状ミセルの形状の複合体を形成しているワクチン組成物を、提供する。本発明はまた、サポニン、ステロール及び抗原を含有し、ここでサポニンとステロールがらせん状ミセルの形状の複合体を形成し、そして該抗原がらせん状ミセルと混合されているが、しかしその内部には取り込まれていない組成物も、提供する。
【0032】
発明の詳細な説明
本発明者は、レシチンと鉱油との混合物からなる水中油型乳剤をアジュバントとして添加したワクチン処方物のマイクロ流動化が、ワクチン処方物の物理的外観を改善するばかりでなく、ワクチン処方物の免疫化効果も増強することを、予想外にも発見した。マイクロ流動化されたワクチン処方物は、改善された安全性プロファイルによっても特徴付けられる。
【0033】
これらの発見に基づき、本発明は、ワクチン組成物におけるアジュバントとして有用なサブミクロン水中油型乳剤を提供する。これらのサブミクロン水中油型乳剤をマイクロ流動化装置を用いることによって作る方法もまた、提供される。さらに、本発明は、抗原がサブミクロン水中油型乳剤と組み合わされているサブミクロンワクチン組成物を、提供する。このようなワクチン組成物を作るための方法もまた、提供される。本発明は、サブミクロン水中油型乳剤と組み合わせてマイクロカプセル化された抗原を含有するワクチン組成物、ならびにこのようなワクチンを作るための方法を、さらに提供する。
【0034】
開示を明確にし、かつ限定しない目的で、発明の詳細な説明を、本発明の特定の特徴、実施態様または応用について記載または例示する以下のサブセクションに分割する。
サブミクロン水中油型乳剤
一実施態様において、本発明は、ワクチンアジュバントとして有用なサブミクロン水中油型乳剤処方物を提供する。本発明のサブミクロン水中油型乳剤は、ワクチン組成物における抗原の免疫原性を増強し、動物への投与に安全であり、かつ保管中は安定である。
【0035】
本発明のサブミクロン水中油型乳剤は、代謝可能でない油、少なくとも一つの界面活性剤、及び水溶性成分から構成され、ここで油は、サブミクロン範囲の平均的な油滴サイズで水溶性成分中に分散している。
【0036】
「サブミクロン」は、液滴が1μm(ミクロン)未満のサイズで、かつ平均の(average)または平均(mean)油滴サイズが1μm未満であることを意味する。好ましくは、乳剤の平均液滴サイズは、0.8μm未満;より好ましくは0.5μm未満;そしてよりいっそう好ましくは0.4μm未満、または約0.1〜0.3μm未満である。
【0037】
「平均液滴サイズ」は、粒子サイズの体積分布内での体積平均径(VMD)の粒子サイズとして定義される。VMDは、各粒子の直径をそのサイズの全粒子の体積とかけ合わせ、そして合計することにより、算出される。次いで、これを全粒子の総体積で割る。
【0038】
「代謝可能でない油」の語は、本明細書では、乳剤が投与される動物個体の体により代謝されることが可能でない油を指す。
「動物」及び「動物個体」の語は、本明細書では、例えばウシ、ヒツジ及びブタを始めとする、すべてのヒトでない動物を指す。
【0039】
本発明の乳剤における使用に適した代謝可能でない油としては、アルカン、アルケン、アルキン、ならびにそれに対応する酸及びアルコール、そのエーテル及びエステル、ならびにその混合物が挙げられる。好ましくは、油の個々の化合物は、軽質炭化水素化合物であって、すなわち、このような成分は6〜30個の炭素原子を有する。油は、合成的に調製されるか、または石油生成物から精製されることが可能である。本発明の乳剤における使用に好ましい代謝可能でない油としては、例えば、鉱油、パラフィン油及びシクロパラフィンが挙げられる。
【0040】
「鉱油」の語は、蒸留技術によってペトロラタムから得られる液体炭化水素の混合物を指す。該語は、「液化パラフィン」、「液体ペトロラタム」及び「白色鉱油」と同義である。該語はまた、「軽質鉱油」、すなわち、ペトロラタムの蒸留により同様に得られるが、ただし白色鉱油よりもわずかに低い比重である油を含むことも、意図する。例えば、Remington's Pharmaceutical Sciences、第18版(Easton、ペンシルバニア州:Mack Publishing Company,
1990年、788頁及び1323頁)を参照されたい。鉱油は、例えばJ.T. Baker(Phillipsburg、ペンシルバニア州)、USB Corporation(クリーブランド、オハイオ州)などの種々の市販元から得ることが可能である。好ましい鉱油は、DRAKEOL(商標登録)の名前で市販されている軽質鉱油である。
【0041】
典型的には、本発明のサブミクロン乳剤の油成分は、1%〜50%容量の量;好ましくは、10%〜45の量;より好ましくは、20%〜40%の量で、存在する。
本発明の水中油型乳剤は、典型的には、少なくとも一つ(すなわち、1またはそれより多く)の界面活性剤を含む。界面活性剤及び乳化剤(本明細書ではこれらの語を同義的に用いる)は、油滴の表面を安定化し、そして所望のサイズ内に油滴を維持する剤である。
【0042】
本発明の乳剤における使用に適した界面活性剤としては、天然の生物学的に適合性の界面活性剤、及び非天然の合成界面活性剤が挙げられる。生物学的に適合性の界面活性剤としては、リン脂質化合物またはリン脂質の混合物が挙げられる。好ましいリン脂質は、大豆もしくは卵レシチンなどのホスファチジルコリン(レシチン)である。レシチンは、粗植物油を水で洗浄し、そして得られた含水ゴムを分離及び乾燥することにより、ホスファチドとトリグリセリドの混合物として得られることが可能である。精製された生成物は、トリグリセリド及び植物油をアセトン洗浄により除去した後に残留する、アセトン不溶性リン脂質及び糖脂質の混合物を分画することにより、得られることが可能である。あるいは、レシチンを、種々の市販元から得ることが可能である。他の適切なリン脂質としては、ホスファチジルグリセロール、ホスファチジルイノシトール、ホスファチジルセリン、ホスファチジン酸、カルジオリピン及びホスファチジルエタノールアミンが挙げられる。リン脂質は、天然源から単離されても、または慣用的に合成されてもよい。
【0043】
本発明のサブミクロン乳剤における使用に適した非天然の合成界面活性剤としては、例えば脂肪酸置換ソルビタン界面活性剤(SPAN(商標登録)またはARLACEL(商標登録)の名前で市販されている)などのソルビタンに基づく非イオン性界面活性剤、ポリエトキシ化ソルビトールの脂肪酸エステル(TWEEN(商標登録))、ヒマシ油などの源から得られる脂肪酸のポリエチレングリコールエステル(EMULFOR);ポリエトキシ化脂肪酸(例えば、SIMULSOL
M−53の名前で入手可能なステアリン酸)、ポリエトキシ化イソオクチルフェノール/ホルムアルデヒド重合体(TYLOXAPOL)、ポリオキシエチレン脂肪酸アルコールエーテル(BRIJ(商標登録));ポリオキシエチレンノンフェニルエーテル(TRITON(商標登録)N)、ポリオキシエチレンイソオクチルフェニルエーテル(TRITON(商標登録)X))が挙げられる。好ましい合成界面活性剤は、SPAN(商標登録)及びTWEEN(商標登録)の名前で入手可能な界面活性剤である。
【0044】
本発明の水中油型乳剤における使用のための好ましい界面活性剤としては、レシチン、Tween−80及びSPAN−80が挙げられる。
一般的には、界面活性剤、または界面活性剤の組み合わせ(2またはそれより多くの界面活性剤が用いられる場合)は、0.01%〜10%容量、好ましくは0.1%〜6.0%、より好ましくは0.2〜5.0%の量にて、乳剤中に存在する。
【0045】
水溶性成分は、乳剤の連続相を構成し、そして水、緩衝生理食塩液または任意の他の適切な水溶液であることが可能である。
本発明の水中油型乳剤は、保存剤、浸透圧剤、生体接着分子及び免疫刺激分子を始めとする適切かつ望ましい成分を、さらに含むことが可能である。
【0046】
生体接着分子は、標的粘膜表面での、またはこれを介した抗原の送達及び付着を増強し、粘膜免疫を授与することが可能であると考えられている。適切な生体接着分子の例としては、ポリアクリル酸及びポリメタクリル酸(例えば、CARBOPOL(商標登録)、CARBOMER)などの、天然に生じるものではない酸性重合体;カルボキシメチルセルロースなどの合成的に修飾された酸性の天然重合体;(ヒドロキシプロピル)メチルセルロースなどの合成的に修飾された中性の天然重合体;キトサンなどの塩基性アミン保有重合体;アルギニン酸、ヒアルロン酸、ペクチン、トラガカントゴム及びカラヤゴムなどの天然源から得ることが可能な酸性重合体;ならびに、ポリビニルアルコールなどの天然に生じるものではない中性重合体;あるいはその組み合わせが挙げられる。
【0047】
「免疫刺激分子」の語句は、本明細書では、ワクチン組成物中の抗原性成分によって誘導される防御免疫応答を増強するこれらの分子を指す。適切な免疫刺激材料としては、細菌細胞壁成分(例えば、ムラブチド、トレオニル−MDP及びムラミルトリペプチドなどのN−アセチルムラミル−L−アラニル−D−イソグルタミン誘導体);サポニン配糖体及びその誘導体(例えば、Quil
A、QS 21及びGPI−0100);コレステロール;ならびに、第四級アンモニウム化合物(例えば、臭化ジメチルジオクタデシルアンモニウム(DDA)及びN,N−ジオクタデシル−N,N−ビス(2−ヒドロキシエチル)プロパンジアミン(「アブジリン」))が挙げられる。
【0048】
サポニンは、多様な植物種において二次代謝産物として産生される配糖体化合物である。サポニンの化学構造は、強力かつ効果的な免疫学的活性を始めとする、幅広い薬理学的及び生物学的活性を授ける。
【0049】
構造的には、サポニンは、1またはそれより多くの糖鎖に付着している任意のアグリコンからなる。サポニンは、そのアグリコン組成によって分類されることが可能である:トリテルペン配糖体、ステロイド配糖体及びステロイドアルカロイド配糖体。
【0050】
サポニンは、シャボンノキ(Quillaja saponaria)の樹皮から単離されることが可能である。サポニンは、免疫刺激剤として長く知られてきた。Dalsgaard, K.、「Evaluation of its adjuvant
activity with a special reference to the application in the vaccination of
cattle against foot-and-mouth disease」、Acta. Vet.
Scand. 69:1-40、1978年。サポニンを含有する植物の粗抽出物は、足口病ワクチンの効力を増強した。しかしながら、該粗抽出物は、ワクチンにおいて用いられた場合に副作用を伴った。これに続き、Dalsgaardは、サポニン由来のアジュバント活性成分を、透析、イオン交換及びゲルろ過クロマトグラフィによって部分精製した。Dalsgaard, K.ら、「Saponin adjuvants III.
Isolation of a substance from Quillaja saponaria Morina with adjuvant activity in
foot-and-mouth disease vaccines」、Arch. Gesamte.
Virusforsch. 44:243-254、1974年。この方法にて精製されたアジュバント活性成分は、「Quil
A」として公知である。重量に基づくと、粗サポニンと比較した場合に、Quil Aは効力の増加を示し、かつ局所反応を低減した。Quil Aは、動物用ワクチンにおいて広く用いられている。
【0051】
高速液体クロマトグラフィ(HPLC)によるQuil Aのさらなる分析によって、密接に関連するサポニンの不均一な混合物が示され、そして毒性が低減したかまたは最小限の強力なアジュバントであるQS21の発見が導びかれた。Kensil C.R.ら、「Separation and
characterization of saponins with adjuvant activity from Quillaja saponaria
Molina cortex」、J. Immunol. 146:431-437、1991年。ほとんどの他の免疫刺激剤とは異なり、QS21は、水溶性で、かつ乳剤型処方物の有無にかかわらずワクチンにおいて使用可能である。QS21は、マウスにおいてTh1型応答を誘発し、IgG2a及びIgG2b抗体の産生を刺激し、そしてサブユニット抗原に応答して抗原特異的CD8+
CTL(MHCクラスI)を誘導することが、示されている。ヒト臨床試験では、許容可能な毒性学的プロファイルを伴うそのアジュバント活性が、証明された。Kensil, C.R.ら、「Structural and imunological
charaterization of the vaccine adjuvant QS-21. In Vaccine Design: the subunit
and Adjvuant Approach」、Powell, M.F.及びNewman, M. J.編集、Plenum Publishing
Corporation、ニューヨーク州、1995年、525〜541頁。
【0052】
米国特許第6,080,725は、サポニン−リポフィル(lilpophile)結合体の作製方法及び使用方法を教示している。このサポニン−リポフィル結合体では、脂質、脂肪酸、ポリエチレングリコールまたはテルペンなどのリポフィル部分が、アシル化されていないかまたはデスアシル化されたトリテルペンサポニンと、トリテルペンサポニンの3−O−グルクロン酸上に存在するカルボキシ基を介して共有結合している。リポフィル部分と、Quillajaデスアシルサポニンなどのサポニン、lucyoside P、またはGypsophila属、saponaria属及びAcanthophyllum属由来サポニンの3−O−グルクロン酸との結合は、液性免疫及び細胞を介した免疫へのそのアジュバント効果を増強する。加えて、リポフィル部分と非アシルもしくはデスアシルサポニンの3−O−グルクロン酸残基との結合は、元のサポニンよりも精製し易く、低毒性で、化学的に安定で、かつ同等または優れたアジュバント特性を有するサポニン類似体を、もたらす。
【0053】
GPI−0100は、米国特許第6,080,725に記載されるサポニン−リポフィル結合体である。GPI−0100は、脂肪族アミンをグルクロン酸のカルボキシル基を介してデスアシルサポニンに付加することにより、生成される。
【0054】
第四級アンモニウム化合物−多数の脂肪族窒素含有塩基が、アミン、第四級アンモニウム化合物、グアニジン、ベンズアミジン及びチオウロニウムを始めとする免疫学的アジュバントとしての使用を提唱されている。具体的なこのような化合物としては、臭化ジメチルジオクタデシルアンモニウム(DDA)及びN,N−ジオクタデシル−N,N−ビス(2−ヒドロキシエチル)プロパンジアミン(「アブジリン」)が挙げられる。
【0055】
米国特許第5,951,988は、油成分と併せてDDAなどの第四級アンモニウム塩を含有するアジュバント処方物を、教示する。この処方物は、免疫原性応答を増強するために公知の免疫学的物質(例えばワクチン組成物中のウイルスもしくは細菌抗原)と併せて、有用である。該組成物は、非特異的な免疫刺激処方物として、取り込まれた抗原がなくても有用である。
【0056】
米国特許第4,310,550は、ワクチンアジュバントとして脂肪もしくは脂質乳剤とともに処方物化したN,N−高級アルキル−N,N’−ビス(2−ヒドロキシエチル)−プロパンジアミン及びN,N−高級アルキル−キシリレンジアミンの使用を、記載している。ヒトまたは動物においてアジュバント処方物の非経口投与を介して抗原の免疫原性応答を誘導または増強する方法は、米国特許第4,310,550に記載されている。
【0057】
好ましい実施態様において、本発明は、液滴サイズが1μm未満であり、かつ平均液滴サイズが約0.25μmのAMPHIGEN(商標登録)処方物から構成される、ワクチンアジュバントとして有用なサブミクロン水中油型乳剤を提供する。
【0058】
「AMPHIGEN(商標登録)処方物」の語は、本明細書では、DRAKEOL(商標登録)レシチン油溶液(Hydronics社、リンカーン、ネブラスカ州)と生理食塩溶液とをTWEEN(商標登録)80及びSPAN(商標登録)80の存在下で混合することによって形成される溶液を、指す。典型的なAMPHIGEN(商標登録)処方物は、軽質鉱油40%容量(v/v)、レシチン約25%w/v、TWEEN
80 約0.18%容量(v/v)及びSpan 80 約0.08%容量(v/v)を含有する。
【0059】
サブミクロン水中油型乳剤の調製方法
別の実施態様において、本発明は、上述のサブミクロン水中油型乳剤の調製方法を提供する。
【0060】
本発明によると、油、1またはそれより多くの界面活性剤、水溶性成分、及び乳剤における使用に適した任意の他の成分を始めとする乳剤の種々の成分を、組み合わせ、そして混ぜ合わせる。
【0061】
形成された混合物を、典型的には1もしくはそれより多くのホモジナイザーまたは乳化装置に1回以上通すことによる乳化過程にかけて、均一な外観及び約0.5μmの平均の液滴サイズの水中油型乳剤を形成する。例えばRoss乳化装置(Hauppauge、ニューヨーク州)、Gaulinホモジナイザー(Everett、マサチューセッツ州)などの市販されている任意のホモジナイザー及び乳化装置を、本目的のために用いることが可能である。
【0062】
次いで、このように形成された乳剤を、マイクロ流動化にかけて、サブミクロン範囲の液滴サイズをもたらす。マイクロ流動化は、Microfluidics社(ニュートン、マサチューセッツ州)から入手可能なモデル番号110Y;Gaulinモデル30CD(Gaulin, Inc.、Everett、マサチューセッツ州);及びRainnie Minilab 8.30H型(Miro Atomizer Food and Dairy, Inc.、ハドソン、ウィスコンシン州)などの市販のマイクロ流動化装置の使用によって、行うことが可能である。これらのマイクロ流動化装置は、二つの液流が相互作用チャンバー内で高速で相互作用して液滴がサブミクロンサイズの乳剤を形成するように、小さな開口部を介して高圧下で液体を強引に通すことによって、動作する。
【0063】
液滴サイズは、例えばレーザー回折、市販の定寸装置の使用によるなどの当該技術分野において公知の多様な方法によって、測定することが可能である。サイズは、用いる界面活性剤の種類、油に対する界面活性剤の割合、動作圧、温度等に応じて異なってもよい。当業者は、過度の実験を行うことなく所望の液滴サイズの乳剤を得るために、これらのパラメータの所望の組み合わせを決定することが可能である。本発明の乳剤の液滴サイズは、直径1μm未満、好ましくは平均液滴サイズ0.8μm未満、そしてより好ましくは平均液滴サイズ0.5μm未満、そしてよりいっそう好ましくは平均液滴サイズ0.3μm未満である。
【0064】
本発明の好ましい実施態様において、Hydronics社(リンカーン、ネブラスカ州)から市販され、そして軽質鉱油中に25%のレシチンを含有するDRAKEOLレシチン油溶液を、生理食塩液、ならびに界面活性剤であるTWEEN(商標登録)80及びSPAN(商標登録)80と組み合わせ、そして混合して、「AMPHIGEN(商標登録)溶液」または「AMPHIGEN(商標登録)処方物」を形成する。次いで、AMPHIGEN(商標登録)溶液をRoss(商標登録)(Hauppauge、ニューヨーク州、11788)乳化装置でおよそ3400rpmにて乳化して、水中油型乳剤を形成する。続いて、乳剤を、約4500±500psiで動作しているマイクロ流動化装置に、一度通す。マイクロ流動化された水中油型乳剤は、1μm未満のサイズの液滴を有し、平均液滴サイズ約0.25μmである。
【0065】
サブミクロン水中油型乳剤に取り込まれた抗原を含有する、ワクチン組成物
別の実施態様において、本発明は、抗原及び上述のサブミクロン水中油型乳剤を含有するワクチン組成物を、提供する。これらのワクチン組成物は、増強された免疫原性効果及び改善された物理的外観を有することによって、特徴付けられる(例えば、長期の保管後に相分離が認められない)。加えて、本発明のワクチン組成物は、動物への投与に関して安全である。
【0066】
本発明によると、抗原を、外来的に、また好ましくは内在的に乳剤と組み合わせることが可能である。「内在的に」の語は、マイクロ流動化工程の前に抗原を乳剤成分と組み合わせる過程を指す。「外来的に」の語は、乳剤をマイクロ流動化した後に抗原を乳剤に添加する過程を指す。外来的に添加された抗原は、遊離抗原であることが可能であり、あるいは以下にさらに記載するように、ミクロ粒子中にカプセル化されることが可能である。
【0067】
「抗原」の語は、本明細書では、動物において免疫原性であって、かつワクチン組成物が投与される動物において防御免疫応答を誘発するためにワクチン組成物中に含まれる、任意の分子、化合物または組成物を指す。
【0068】
抗原に関連して用いられる「免疫原性」の語は、動物において抗原に対する免疫応答を誘発する抗原の能を指す。該免疫応答は、細胞性T細胞によって主に媒介される細胞性免疫応答、あるいは次にB細胞を活性化して抗体産生へと導く、ヘルパーT細胞によって主に媒介される液性免疫応答であることが可能である。
【0069】
「防御免疫応答」は、抗原または抗原を含有する病原体により引き起こされる障害もしくは疾患の発生を防ぐかまたは検出可能に低下させるか、あるいはその重症度を除くかまたは検出可能に低減するか、あるいはその進行速度を検出可能に遅くする、動物において生じる、抗体または細胞のいずれかを介した免疫応答あるいはその双方である任意の免疫応答として、定義される。
【0070】
本発明のワクチン組成物に含まれることが可能な抗原としては、不活化した細胞の全体または一部の調製物の形態、あるいは慣用のタンパク質精製、遺伝子操作技術または化学合成によって得られる抗原分子の形態の、Mycoplasma hyopneumoniae、Haemophilus somnus、Haemophilus parasuis、Bordetella
bronchiseptica、Actinobacillus pleuropneumonie、Pasteurella multocida、Manheimia hemolytica、Mycoplasma bovis、Mycoplasma galanacieum、Mycobacterium bovis、Mycobacterium
paratuberculosis、クロストリジウム属、Streptococcus uberis、Streptococcus suis、Staphylococcus aureus、Erysipelothrix rhusopathiae、カンピロバクター属、Fusobacterium
necrophorum、大腸菌、Salmonella enterica serovars、レプトスピラ属などの病原性細菌;カンジダなどの病原性真菌;Cryptosporidium parvum、Neospora canium、Toxoplasma gondii、エイメリア属などの原虫; Ostertagia、Cooperia、Haemonchus、Fasciolaなどの蠕虫から調製される抗原が、挙げられる。抗原としては、さらに、不活化した細胞の全体または一部の調製物の形態、あるいは慣用のタンパク質精製、遺伝子操作技術または化学合成によって得られる抗原分子の形態の、ウシヘルペスウイルス−1、3、6、ウシウイルス性下痢ウイルス(BVDV)1型及び2型、ウシパラインフルエンザウイルス、ウシ呼吸器合胞体ウイルス、ウシ白血病ウイルス、牛疫ウイルス、足口病ウイルス、狂犬病、ブタコレラウイルス、アフリカブタコレラウイルス、ブタパルボウイルス、PRRSウイルス、ブタサーコウイルス、インフルエンザウイルス、ブタ水疱病ウイルス、Techen熱ウイルス、仮性狂犬病ウイルスなどの病原性ウイルスが、挙げられる。
【0071】
抗原の量は、水中油型乳剤と組み合わせた抗原が、動物において防御免疫応答を誘導するのに有効なようであるべきである。有効となる正確な抗原の量は、抗原の性質、活性及び純度に依存し、そして当業者により決定されることが可能である。
【0072】
ワクチン組成物中に存在する水中油型乳剤の量は、ワクチン組成物中の抗原の免疫原性を増強するのに十分であるべきである。望ましくかつ適切な場合には、界面活性剤の追加量または追加の界面活性剤を、水中油型乳剤により供される界面活性剤に加えて、ワクチン組成物中に加えることが可能である。一般的に言えば、油成分は、ワクチン組成物の最終容量において、1.0%〜20%容量の量;好ましくは、1.0%〜10%の量;より好ましくは、2.0%〜5.0%の量にて存在する。界面活性剤(単数)、あるいは2またはそれより多くの界面活性剤を用いる場合、界面活性剤の組み合わせは、ワクチン組成物の最終容量において、0.1%〜20%容量、好ましくは0.15%〜10%、より好ましくは0.2%〜6.0%の量にて存在する。
【0073】
抗原及び水中油型乳剤に加えて、ワクチン組成物は、水中油型乳剤に関連して上述のように、保存剤、浸透圧剤、生体接着分子及び免疫刺激分子(例えば、Quil
A、コレステロール、GPI−0100、臭化ジメチルジオクタデシルアンモニウム(DDA))などの適切かつ所望の他成分を含むことが可能である。
【0074】
本発明のワクチン組成物は、獣医学的に許容可能(veterinarily-acceptable)なキャリアーもまた含むことが可能である。「獣医学的に許容可能なキャリアー」の語には、任意及びすべての溶媒、分散媒体、被覆、アジュバント、安定剤、希釈剤、保存剤、抗細菌剤及び抗真菌剤、等張剤、吸収遅延剤等が含まれる。希釈剤としては、水、生理食塩液、ブドウ糖、エタノール、グリセロール等が挙げられてよい。等張剤としては、とりわけ、塩化ナトリウム、ブドウ糖、マンニトール、ソルビトール及びラクトースが挙げられてよい。安定剤としては、とりわけ、アルブミンが挙げられる。
【0075】
好ましい実施態様において、本発明は、1μm未満のサイズの液滴、好ましくは0.8μm未満、より好ましくは0.5μm未満の平均液滴サイズ、及びよりいっそう好ましくは約0.5μmの平均液滴サイズの水中油型乳剤中に内在的に取り込まれている、BVDV I型またはBVDV II型抗原のうち少なくとも一つを含むワクチン組成物を、提供する。BVDV
I型及び/またはII型抗原は、好ましくは、不活化ウイルス調製物の形態である。サブミクロン水中油型乳剤は、好ましくは、AMPHIGEN(商標登録)処方物(すなわち、軽質鉱油、レシチン、TWEEN(商標登録)80及びSPAN(商標登録)80を含有する処方物)から構成される。
【0076】
別の好ましい実施態様において、本発明は、レプトスピラ抗原、及びBVDV I型またはBVDV II型抗原のうち少なくとも一つを水中油型乳剤中に含むワクチン組成物を、提供する。好ましくは不活化した細胞もしくはウイルス調製物の形態である抗原は、
1μm未満のサイズの液滴、好ましくは0.8μm未満、より好ましくは0.5μm未満の平均液滴サイズ、及びよりいっそう好ましくは約0.5μmの平均液滴サイズの水中油型乳剤中に内在的に取り込まれている。サブミクロン水中油型乳剤は、好ましくは、AMPHIGEN処方物(すなわち、軽質鉱油、レシチン、TWEEN(商標登録)80及びSPAN(商標登録)80を含有する処方物)から構成される。該ワクチン組成物は、好ましくは、Quil−A、コレステロール、DDA、GPI−100及び水酸化アルミニウム(AlOH)から選択される1またはそれより多くの免疫刺激分子もまた含む。
【0077】
さらに別の好ましい実施態様において、本発明は、少なくとも一つの細菌抗原(例えば、組換えStreptococcus
uberis PauAタンパク質または大腸菌の細胞調製物、あるいはその双方の組み合わせ)を水中油型乳剤中に含むワクチン組成物を提供する。抗原は、1μm未満のサイズの液滴、好ましくは0.8μm未満、より好ましくは0.5μm未満の平均液滴サイズ、及びよりいっそう好ましくは約0.25μmの平均液滴サイズの水中油型乳剤と、内在的に組み合わされている。サブミクロン水中油型乳剤は、好ましくは、AMPHIGEN(商標登録)処方物(すなわち、軽質鉱油、レシチン、TWEEN(商標登録)80及びSPAN(商標登録)80を含有する処方物)から構成される。該ワクチン組成物は、好ましくは、Quil
A、DDA及びGPI−100から選択される1またはそれより多くの免疫刺激分子もまた含む。
【0078】
本発明のワクチン組成物を、経口、鼻腔内、粘膜、局所、経皮、及び非経口(例えば、静脈内、腹腔内、皮内、皮下または筋肉内)経路を始めとする公知の経路により動物に投与することが可能である。例えば、初回投与には非経口(parental)経路を用い、そしてそれに続く投与には粘膜経路を用いるなどの経路の組み合わせを用いて、投与を成し遂げることが可能である。
【0079】
ワクチン組成物の調製方法
さらなる実施態様において、本発明は、抗原(単数または複数)及びサブミクロン水中油型乳剤を含有するワクチン組成物の調製方法を提供する。
【0080】
本発明のワクチン組成物を調製する際に、抗原を、水中油型乳剤の成分と、内在的あるいは外来的に組み合わせることが可能である。「好ましくは」、抗原を、水中油型乳剤の成分と内在的に組み合わせる。
【0081】
抗原を、油、1またはそれより多くの界面活性剤、水溶性成分及び任意の他の適切な成分を含む乳剤の種々の成分と組み合わせて、混合物を形成することが可能である。該混合物を、典型的には1もしくはそれより多くのホモジナイザーまたは乳化装置に1回以上通すことによる第一混和過程にかけて、抗原を含有する水中油型乳剤を形成する。例えばRoss乳化装置(Hauppauge、ニューヨーク州)、Gaulinホモジナイザー(Everett、マサチューセッツ州)またはMicrofluidics(ニュートン、マサチューセッツ州)などの任意の市販されているホモジナイザーまたは乳化装置を、本目的のために用いることが可能である。あるいは、油、1またはそれより多くの界面活性剤及び水溶性成分を含む乳剤アジュバントの種々の成分を、まず組み合わせて、ホモジナイザーまたは乳化装置を用いることによって水中油型乳剤を形成し;次いで、抗原をこの乳剤に添加する。第一混和後の水中油型乳剤の平均液滴サイズは、およそ1.0〜1.2ミクロンである。
【0082】
次いで、抗原を含有する乳剤を、マイクロ流動化にかけて、サブミクロン範囲の液滴サイズを得る。マイクロ流動化は、Microfluidics社、ニュートン、マサチューセッツ州から入手可能なモデル番号110Y;Gaulinモデル30CD(Gaulin, Inc.、Everett、マサチューセッツ州);及びRainnie Minilab 8.30H型(Miro Atomizer Food and Dairy, Inc.、ハドソン、ウィスコンシン州)などの市販のマイクロ流動化装置の使用により、成し遂げることが可能である。
【0083】
液滴サイズは、例えばレーザー回折、市販の定寸装置の使用によるなどの、当該技術分野において公知の多様な方法によって、測定することが可能である。サイズは、用いる界面活性剤の種類、油に対する界面活性剤の割合、動作圧、温度等に応じて異なってもよい。所望の液滴サイズの乳剤を得るために、これらのパラメータの所望の組み合わせを決定することが可能である。本発明の乳剤の液滴サイズは、直径1μm未満である。好ましくは、平均液滴サイズは0.8μm未満である。より好ましくは、平均液滴サイズは0.5μm未満である。よりいっそう好ましくは、平均液滴サイズは約0.1〜0.3μmである。
【0084】
本発明の好ましい実施態様において、軽質鉱油中に25%のレシチンを含有するDRAKEOL(商標登録)レシチン油溶液を、TWEEN(商標登録)80及びSPAN(商標登録)80ならびに生理食塩液と組み合わせ、そして混合して、軽質鉱油40%、レシチン、TWEEN(商標登録)80
0.18%、及びSPAN(商標登録)80 0.08%を含有する混合物を形成する。次いで、該混合物を、Ross(商標登録)(Hauppauge、ニューヨーク州、11788)乳化装置でおよそ3400rpmにて乳化して乳剤生成物を形成し、これは「AMPHIGEN(商標登録)処方物」または「AMPHIGEN(商標登録)溶液」とも称される。続いて、所望の抗原を、乳化装置(たとえば、Rossホモジナイザー)を用いてAMPHIGEN(商標登録)溶液及び任意の他の適切な成分(例えば、免疫刺激分子)と組み合わせて、抗原を含有する水中油型乳剤を形成する。このような乳剤を、約10000±500psiで動作しているマイクロ流動化装置に一度通す。マイクロ流動化された水中油型乳剤は、平均液滴サイズ約0.25μmの、1μm未満のサイズの液滴を有する。
【0085】
別の好ましい実施態様において、水中油型乳剤(例えば、AMPHIGEN(商標登録)処方物)を所望の抗原と組み合わせる前に、抗原をサポニン配糖体(例えば、Quil
A)と組み合わせて、混合物を形成する。この抗原−サポニン混合物を、ホモジナイゼーション容器中でホモジナイゼーションにかける。次いで、例えばコレステロールなどのステロールを、ホモジナイズされた抗原−サポニン混合物に加える。次いで、抗原、サポニン及びステロールを含有する該混合物を、さらにホモジナイゼーションにかける。次いで、ホモジナイズされた抗原−サポニン−ステロール混合物を、例えばホモジナイザーを用いて、水中油型乳剤(例えば、AMPHIGEN(商標登録)処方物)と組み合わせる。次いで、抗原、サポニン及びステロールを含有するホモジナイズされた水中油型乳剤を、マイクロ流動化などの高圧ホモジナイゼーションにかける。
【0086】
マイクロカプセル化された抗原をサブミクロン水中油型乳剤中に含有するワクチン組成物、及び調製方法
さらに別の実施態様において、本発明は、ミクロ粒子中にカプセル化された抗原(または、「マイクロカプセル化された抗原」)を含有し、ここでマイクロカプセル化された抗原が前述のサブミクロン水中油型乳剤中に外来的に取り込まれているワクチン組成物を、提供する。
【0087】
抗原を粒子性キャリアー中に吸着または捕捉するための方法は、当該技術分野において公知である。例えば、Pharmaceutical Particulate Carriers: Therapeutic Applications(Justin Hanes、Masatoshi Chiba及びRobert Langer、「Vaccine design」中、Polymer microspheres for vaccine delivery、The
subunit and adjuvant approach. Michael F. Powell及びMark
J. Newman編集、1995年、Plenum Press、ニューヨーク州及びロンドン)を参照されたい。粒子性キャリアーは、動物個体において選択された抗原の複数のコピーを免疫系に提示し、そして局所リンパ節における抗原の捕捉及び保持を促進することが可能である。粒子は、マクロファージによって貪食されることが可能であり、かつサイトカイン放出を介して抗原提示を増強することが可能である。粒子性キャリアーは、当該技術分野においても記載されており、そして例えば、ポリメチルメタクリレート重合体由来のもの、ならびに、ポリ(ラクチド)及びPLGとして公知のポリ(ラクチド−共グリコリド)由来のものなどが挙げられる。ポリメチルメタクリレート重合体は非生分解性であるが、PLG粒子は、正常な代謝経路で排出される乳酸及びグリコール酸とのエステル結合のランダムな非酵素性加水分解によって生分解されることが可能である。
【0088】
生分解性ミクロスフェアもまた、 ワクチンの制御放出を成し遂げるのに用いられている。例えば、長期にわたり抗原の持続的な放出を成し遂げることが可能である。重合体の分子量、及び重合体におけるグリコール酸に対する乳酸の割合に応じて、PLGA重合体は、数日または数週間から数カ月または一年までの加水分解速度を有することが可能である。緩徐な制御放出により、複数回注射後に観察されるものと同様の高レベルの抗体の形成を生じる可能性がある。あるいは、ワクチン抗原のパルス放出を、異なる加水分解速度を伴う重合体を選択することによって成し遂げることが可能である。重合体の加水分解速度は、典型的には、重合体の分子量、及び重合体におけるグリコール酸に対する乳酸の割合に依存する。抗原放出速度の異なる2またはそれより多くの異なる重合体から作られたミクロ粒子は、抗原のパルス放出、及びワクチン接種の複数回投与レジメン(regime)の模倣を供する。
【0089】
本発明によると、前述のもののいずれかを始めとする抗原を、当該技術分野において公知の任意の手順(実施例17に例示されるものなど)を用いることにより粒子状重合体キャリアー(好ましくは、PLG重合体)に吸着させて、マイクロカプセル化された抗原調製物を形成することが可能である。次いで、マイクロカプセル化された抗原調製物を、前述したサブミクロン水中油型乳剤と混合し、そしてその中に分散させて、ワクチン組成物を形成する。
【0090】
好ましい実施態様において、本発明は、PLG重合体中にカプセル化された抗原を含有し、ここでマイクロカプセル化された抗原が軽質鉱油、レシチン、TWEEN80、SPAN80及び生理食塩液から構成されるマイクロ流動化された水中油型乳剤中に外来的に分散し、かつ1.0μm未満の平均液滴サイズを有するワクチン組成物を、提供する。
【0091】
サポニン及びステロールにより形成される複合体
一実施態様において、本発明は、サポニン及びステロールを含有し、ここでサポニン及びステロールがらせん状ミセルの形状の複合体を形成している組成物を、提供する。本発明によると、これらの複合体は免疫刺激活性を有する。
【0092】
「免疫刺激」とは、該複合体が抗原成分によって誘導される免疫応答を増強可能であること、あるいは該複合体が別の抗原成分とは独立に免疫応答を誘導可能であることを、意味する。
【0093】
本発明によると、本発明の組成物における使用に好ましいサポニンは、Quil Aである。
本発明のアジュバント組成物における使用に好ましいステロールとしては、β−シトステロール、スチグマステロール、エルゴステロール、エルゴカルシフェロール及びコレステロールが挙げられる。これらのステロールは当該技術分野において周知であって、例えばコレステロールは、メルクインデックス第11版、341頁に、動物脂肪中に見いだされる自然発生ステロールとして開示されている。最も好ましくは、ステロールはコレステロールである。
【0094】
組成物中のサポニン:ステロールの割合は、典型的には、およそ1:100〜5:1(重量対重量)である。好ましくは、割合は1:1である。
別の実施態様において、本発明は、サポニン、ステロール及び抗原を含有し、ここでサポニン及びステロールがらせん状ミセルの形状の複合体を形成しており、かつ抗原がらせん状ミセルと混合されているが、しかしその内部に取り込まれていないワクチン組成物を、提供する。
【0095】
以下は、本発明を実行するための具体的な実施態様の実施例である。当該実施例は、例示的な目的のみのために提供され、決して本発明の範囲を限定することを意図しない。
【実施例】
【0096】
(実施例1)
AMPHIGEN(商標登録)処方物の調製
AMPHIGEN(商標登録)処方物を、二工程の過程にて調製した。第一工程では、80リットルのDrakeolレシチン油溶液、116リットルの破傷風トキソイド生理食塩液、1.2リットルのSPAN
80、及び2.8リットルのTween 80を混ぜ合わせ、そしてRoss乳化装置を用いて乳化した。Drakeolレシチン油溶液は、大豆レシチン25%及び鉱油75%を含有した。乳化した生成物を、Ross乳化装置により最低5容量または最低10分間再循環させた。乳化した生成物を、さらなる処理のために、2〜7℃にて最大24時間保管した。Ross乳化装置のタンクから得られた乳剤を、Gaulinホモジナイザーに移し、そして4500psiの圧力下で20分間ホモジナイズした。次いで、得られた40%Drakeolレシチン油溶液(以下、「AMPHIGEN(商標登録)処方物」または「AMPHIGEN(商標登録)溶液」)を、無菌のポリプロピレンカルボキシ容器に分注した。分注は、クラス10,000の制御環境にあるクラス100の分注フードの中で行った。容器を、2〜7℃にて保管した。このAMPHIGEN(商標登録)処方物を、特に示さない限り、以下に記載の実験において用いた。
【0097】
(実施例2)
BVDワクチンの急速混和ホモジナイゼーションによる第一の混和
このホモジナイゼーション過程に用いられる装置を、図1に示す。無菌技術または蒸気用三方弁を用いて、BVD I型抗原(不活化したBVD
I型ウイルス調製物)を含有するボトルを、混和容器上の底部ポートに連結した。必要とされる容量のBVD I型抗原の移行が完了した後に、BVD I型のボトルを、不活化BVD
II型ウイルス調製物(不活化BVD II型ウイルス調製物)を含有するボトルと取り替えた。必要量のBVD II型抗原の移行が完了した後に、Rossホモジナイザーをポータブル容器に取り付け、そして再循環を最大のRPM(3300rpm)にて開始した。容器の撹拌を、中等度の速度にて維持した。
【0098】
無菌技術または蒸気用三方弁を用いて、Quil−Aを50mg/mlの濃度で含有するボトルを、混和容器上のホモジナイザーインラインポートに取り付けた。必要量のQuil−A溶液を、ライン吸引により容器へと通した。Quil−A溶液の移行が完了した後に、ボトルを取り外した。同じ方法で、必要量のコレステロール・エタノール溶液(18mg/ml)を、混和容器に移した。続いて、必要量のAMPHIGEN(商標登録)処方物、10%Thimerosol溶液、及び基本改変イーグル培地(「BME」)増量剤溶液を、混和容器に加えた。
【0099】
すべての添加が完了した後に、混合をさらに15分間続けた。得られた処方物を、2ml用量に分割し、そしてこれはマイクロ流動化されていないAMPHIGEN(商標登録)処方物に基づくBVDワクチンを表した。ワクチンの各用量は、Quil−A
500μg、コレステロール 500μg、AMPHIGEN(商標登録)処方物2.5%、及びThimerosol 0.009%を含有した。二つの異なるBVD株の抗原濃度を、gp53に対するELISA力価に関して測定した。
【0100】
(実施例3)
マイクロ流動化による第二の混和
図2は、マイクロ流動化を介した第二の混和に用いられる過程を示す。マイクロ流動化装置を、蒸気滅菌した。まず、補助の処理モジュールチャンバーをユニット内に設置し、そして空のチャンバーを二番目のチャンバー位置に設置した。実施例2に記載のように調製された、十分にアジュバント添加されているBVDワクチンを含有する容器を、供給容器のドレイン弁からマイクロ流動化装置の入口への移行ラインを取り付けることにより、マイクロ流動化装置に接続した。窒素ガスを、供給容器の空気フィルターの入口に接続し、そして容器の圧設定を20±5PSIに調整した。収集容器のドレイン弁を、マイクロ流動化装置の出口からの移行ラインに接続した。すべての必要な接続を行った後に、弁を開き、そしてマイクロ流動化を動作圧10,000±500PSIにて開始した。ワクチンの全量をマイクロ流動化装置に一度通し、そしてマイクロ流動化後チャンバー中に収集した。この調製物を、2mL用量に分割し、そしてこれはマイクロ流動化されたAMPHIGEN(商標登録)処方物に基づくBVDワクチンを表した。
【0101】
(実施例4)
ベンチ混和によるワクチン組成物の調製
実施例1に記載のように調製されたAMPHIGEN(商標登録)処方物を、BVD抗原及び増量剤を加えて2.5%に希釈した。得られた溶液を、ホモジナイザーを用いる代わりに撹拌子を用いて、ベンチにて混和した。最終調製物は、以下の組成物を含有した:BVD
1型及び2型抗原、2.5%AMPHIGEN(商標登録)処方物(実施例1に記載のように、油、レシチン、SPAN(商標登録)及びTWEEN(商標登録)を含有する)、ならびに生理食塩液。TWEEN
80及びSPAN 80は、最終ワクチン調製物中にそれぞれ0.18%及び0.08%容量にて存在する。
【0102】
(実施例5)
マイクロ流動化されていないAMPHIGEN(商標登録)処方物とマイクロ流動化されたAMPHIGEN(商標登録)処方物に基づくワクチン調製物との液滴サイズ分布の比較
実施例2に記載のように調製されたマイクロ流動化されていないAMPHIGEN(商標登録)処方物に基づくワクチン、実施例3に記載のように調製されたマイクロ流動化されたAMPHIGEN(商標登録)処方物に基づくワクチン、及び実施例4に記載のようにベンチで混和により作られた調製物を用いて、ワクチン調製物の液滴サイズを比較した。2ミリリットルの各調製物の試料を、Malvern 2000レーザー回折計測器に添加し、そして液滴サイズ分布を測定した。図3に示すように、結果は、マイクロ流動化されたAMPHIGEN(商標登録)処方物に基づくワクチン調製物の最大粒子体積がおよそ0.1ミクロンであり、一方、マイクロ流動化されていないAMPHIGEN(商標登録)処方物に基づくワクチン調製物の最大粒子分布体積がおよそ1ミクロンであったことを、示している。
【0103】
(実施例6)
ワクチンの相分離の低減
三つの異なるワクチン調製物:実施例2に記載のように調製されたマイクロ流動化されていないAMPHIGEN(商標登録)処方物に基づくワクチン、実施例3に記載のように調製されたマイクロ流動化されたAMPHIGEN(商標登録)処方物に基づくワクチン、及び実施例4に記載のようにベンチ混和により調製されたワクチン調製物を、並列に比較して、長期保管時のそれらの相分離の特性を判定した。すべてのこれらの調製物を4℃にて約1ヶ月間静置させ、そして相分離を、ワクチン調製物最上部のクリーム状の層の出現に関してモニターした。図4に示すように、他の二つの調製物と比較した場合、マイクロ流動化されたAMPHIGEN(商標登録)処方物に基づく調製物では相分離はなかった。
【0104】
(実施例7)
マイクロ流動化された、及びマイクロ流動化されていないウシウイルス下痢ウイルスに対するウシ用ワクチンの調製
ウシウイルス下痢ウイルス抗原を、マイクロ流動化を介してAMPHIGEN(商標登録)処方物中に内在的に取り込んだ。「内在的に取り込んだ」の語は、マイクロ流動化の前に抗原をAMPHIGEN(商標登録)処方物に添加することによる過程を指す。抗原をアジュバント処方物の成分とともに、マイクロ流動化過程の物理的な力にかけた。マイクロ流動化されていない対照群では、抗原調製物を、混和によりAMPHIGEN(商標登録)処方物中に分散させた。
【0105】
対照及びマイクロ流動化された調製物の双方の最終組成物は、以下のとおりであった:gp53に対する不活化後のELISA力価2535RU/用量のBVD
I型、1.25mg/用量の濃度のQuil−A、1.25mg/用量の濃度のコレステロール、2.5%の最終濃度のAMPHIGEN(商標登録)処方物、及び0.009%の最終濃度のThimerosol。ワクチン用量は、5mlであった。
【0106】
(実施例8)
マイクロ流動化されたAMPHIGEN(商標登録)処方物に基づくワクチン調製物における、内在的に取り込まれたBVDウイルス抗原の長期安定性
この実験は、内在的に取り込まれた抗原の長期保管中における安定性を判定するために行った。死滅BVD II型ウイルス抗原を、マイクロ流動化の過程中にAMPHIGEN(商標登録)処方物中に内在的に取り込んで、マイクロ流動化されたワクチン調製物(A907505)を得た。同抗原をマイクロ流動化されていないAMPHIGEN(商標登録)処方物(A904369、A904370及びA904371)中に含有する三つの他のワクチン調製物を、対照とした。マイクロ流動化されていない調製物では、抗原をAMPHIGEN(商標登録)処方物と混合し、そしてRossホモジナイザーを用いた混和により混合した。すべての四つのワクチン調製物を、4℃にて2年間保管した。保管中の異なる時点(0、6、12または24ヶ月)で、すべての四つの処方物を用いて、3ヶ月齢のウシにワクチン接種した。
【0107】
第0日及び第21日に、3ヶ月齢のウシを、皮下経路を介して、2mlのワクチン処方物でワクチン接種した。ワクチン接種された動物の血清を第35日に採取し、そしてワクチンに対する血清学的応答を、BVDV−E2
ELISAにより、抗体価に関して測定した。図5に示すように、マイクロ流動化されたワクチン調製物は、試験したすべての時点(0、6、12及び24ヶ月)で抗体価の上昇を示し、抗原調製物の安定性がマイクロ流動化の過程中における抗原の内在的な取り込みの間に失われないことが、示唆された。その上、マイクロ流動化されたワクチン調製物が、すべての時点で免疫応答の増強を誘導することもまた、驚くべきことに見いだされた。
【0108】
(実施例9)
マイクロ流動化後の、ワクチンにより誘導される直腸温増加の低減
実施例7に記載のように作られたマイクロ流動化されたワクチン調製物及びマイクロ流動化されていないワクチン調製物を用いて、第0日にウシにワクチン接種し、そして直腸温を、ワクチン接種1日前からワクチン接種後4日までの期間モニターした。ワクチン用量は、2mlであった。当該群を、1倍用量または2倍用量のいずれかのワクチンでワクチン接種した。直腸温を、第−1日から第4日まで毎日測定及び記録した。第0日における直腸温は、被験物質の投与前に測定した。
【0109】
図6に示すように、結果は、1倍用量または2倍用量いずれかのマイクロ流動化されていないワクチン処方物でワクチン接種されたこれらの動物において、ワクチン接種後約24時間に直腸温の急激な上昇があったことを、示している。しかしながら、マイクロ流動化された形状のワクチンでワクチン接種された動物においては、ワクチン接種後の直腸温の上昇は、ほんのわずかで、かつマイクロ流動化されていない処方物でワクチン接種された動物よりも有意に低かった(図6)。
【0110】
(実施例10)
マイクロ流動化されたワクチン処方物でワクチン接種した場合、注射部位反応体積はより速く回復した
実施例7に記載のように作られた、マイクロ流動化されたワクチン調製物及びマイクロ流動化されていないワクチン調製物を用いて、第0日にウシにワクチン接種した。この試験に含まれる動物は、交雑食用牛であった。プラセボ治療群(T01及びT02)のそれぞれには、3例の動物がいた。T03〜T06群のそれぞれには、6例の動物がいた。ワクチン用量は2mlであり、そして当該群を、第0日に1または2用量のいずれかのワクチンでワクチン接種した。第0日に、被験物質を、右首に投与した。2倍用量(4ml)の被験物質を投与される動物(T02、T04及びT06)には、片側に1回の注射として、全2倍用量を投与した。注射部位における反応サイズの評価を始めとする注射部位の観察を、第0日から第4日までの全日、及び第6、9及び14日に、首の右側で行った。第0日に、注射部位を、被験物質の投与前に観察した。1または2用量のプラセボでワクチン接種した群は、注射部位反応体積のいかなる有意な増加も示さず、そしてそれゆえにこれらのデータを図7に示していない。マイクロ流動化されていないワクチン処方物の場合、1用量と2用量のワクチン接種との間に注射部位反応体積の比例的な増加があった。他方で、マイクロ流動化されたワクチン処方物の場合、1倍用量がより大きな注射部位反応体積を誘導したが、二番目の用量での注射はいかなるさらなる増加も引き起こさなかった。その上、マイクロ流動化されたワクチン処方物で注射した動物の場合、注射部位反応部位体積は、マイクロ流動化されていないワクチン処方物で注射した動物と比較した場合に、より速い速度で回復した。これらの結果を、図7に示す。
【0111】
(実施例11)
内在的に取り込まれたBVDウイルス及びレプトスピラ抗原、ならびにQuil A及びDDAなどの免疫刺激分子を用いた、マイクロ流動化されたAMPHIGEN(商標登録)処方物に基づくワクチン調製物の調製
ホルマリンで不活化したLeptospira hardjo-bovis CLS株を、直接計数約1.4×109生物体/5ml用量にて、適切なアジュバント中に処方物化した。ホルマリンで不活化したLeptospira pomona T262株を、約2400比濁単位/5ml用量にて処方物化した。比濁単位は、処理前の発酵液の比濁測定に基づき算出した。BVDウイルス1型を、約3000相対単位/5ml用量のE2
Elisa力価にて処方物化した。BVDウイルス2型を、約3500相対単位/5ml用量のE2 Elisa力価にて処方物化した。相対単位は、合わせる前の不活化後のバルク液のE2
ELISA力価に基づき算出した。Quil−Aとコレステロールの双方を、0.5mg/用量の濃度にて用いた。Thimerosol及びAMPHIGEN(商標登録)処方物を、それぞれ0.009%及び2.5%の最終濃度にて用いた。水酸化アルミニウム(Rehydragel LV)を、2.0%の最終濃度にて用いた。DDAを免疫調節物質として用いた場合、DDAはAMPHIGEN(商標登録)処方物内に含まれた。AMPHIGEN(商標登録)処方物(すなわち、40%Drakeol−レシチンストック溶液)は1.6mg/mlのDDAを含有し、そして適切に希釈された場合、最終的なワクチン調製物は2.5%AMPHIGEN(商標登録)処方物及び0.1mg/mlのDDAを含有した。
【0112】
異なるワクチン処方物の調製において、BVD画分、レプト、Quil−A、コレステロール、Thimerosol、AMPHIGEN(商標登録)処方物、及び増量剤としての生理食塩液を、Silversonホモジナイザーに添加し、そして10,000±500RPMにて15分間混合した。次いで、成分を、200ミクロンのスクリーンを介して、10,000psiにてマイクロ流動化した。
【0113】
ワクチン処方物が水酸化アルミニウムを含有した場合、マイクロ流動化を水酸化アルミニウムなしで行った。マイクロ流動化が完了した後に、水酸化アルミニウムを添加し、そして撹拌子を用いて4℃にて一晩混合した。
【0114】
(実施例12)
曝露試験用BVDウイルスワクチンの調製
この実験に用いられるワクチン調製物は、BVDウイルス1型及びBVDウイルス2型の双方由来の抗原を含有した。BVD1−5960抗原を、gp53に対する不活化後のELISA力価2535RU/用量にて用いた。BVD2−890抗原を、gp53に対する不活化後のELISA力価3290RU/用量にて用いた。Quil
A及びコレステロールを、0.5mg/mlの濃度にて用いた。Thimersol及びAMPHIGEN(商標登録)処方物を、それぞれ0.009%及び2.5%の最終濃度にて用いた。DDAを免疫調節物質として用いた場合、DDAはAMPHIGEN(商標登録)処方物内に含まれた。AMPHIGEN(商標登録)ストック溶液(40%Drakeol−レシチン溶液)は様々な量のDDAを含有し、そして適切に希釈された場合、最終的なワクチン調製物は2.5%AMPHIGEN(商標登録)処方物及び0.5mg/用量〜2.0mg/用量のDDA濃度を含有した。アルミニウムゲル(Rehydragel-LV)を、2%の最終濃度にて用いた。GPI−0100を、2、3及び5mg/用量の範囲で用いた。
【0115】
すべての成分をSilversonホモジナイザーに添加し、そして10,500rpmにて15分間混和し、次いで10,000psiにて200ミクロンのチャンバーに通すことによりマイクロ流動化した。ワクチン調製物が水酸化アルミニウムを含有した場合、マイクロ流動化を水酸化アルミニウムなしで行った。マイクロ流動化が完了した後に、水酸化アルミニウムを添加し、そして撹拌子を用いて4℃にて一晩混合した。
【0116】
(実施例13)
レプトスピラ抗原を伴うマイクロ流動化されたAmphigenワクチン処方物でワクチン接種後の、レプトスピラ曝露に対する防御
【0117】
【表1】
【0118】
表1は、本試験において試験されたワクチン調製物におけるアジュバント処方物の組成を示す。ワクチン調製物を、実施例11に記載のように調製した。各群には、6例の動物がいた。約7ヶ月齢の交雑種未経産牛を、本試験において用いた。ワクチン接種を、第0日及び第21日に、皮下経路を介して5mlのワクチン容量にて行った。曝露を、国立動物疾病センター(NADC、National agricultural Disease Center)から得たL.
hardjo-bovis 203株を用いて行った。曝露は、第57〜59日に、1mlの接種により行った。曝露を、眼及び腟に同時投与した。曝露材料は、5.0X106レプトスピラ/mlを含有した。尿を、レプト培養、FA及びPCRのために一週間毎に採取した。腎臓の採取を、第112日及び第113日の間に行った。
【0119】
【表2】
【0120】
表2は、レプトスピラ曝露試験から得られたデータを示す。曝露された動物におけるレプトスピラ感染率を決定する際に、以下の基準を用いた。腎臓培養が一試料のみで陽性であったならば、動物はレプトスピラに陽性であると考えられる。動物がFAまたはPCRのいずれかに関して一試料のみで陽性であるならば、動物は陰性であると考えられる。試料がFA及びPCRの双方に関して一試料のみで陽性であるならば、レプトスピラに陽性であると考えられた。
【0121】
表2に示す結果は、すべての三つのアッセイに基づくすべてのワクチン群において、尿への排出期間が有意に短かったことを示している。尿及び腎臓のコロニー形成に関する限り、QAC及びDDAを含有するAlOHを含まない処方物の有効性は、同程度であった。AlOHは、本曝露試験においては、QACまたはDDAを含有するワクチンの有効性を改善せず、そしてむしろ低減した。
【0122】
【表3】
【0123】
ワクチン処方物中の双方のレプトスピラ抗原に対する血清学的応答を、ワクチン接種された動物において検出し、そしてピーク応答は第35日にみられた。血清学的応答と曝露に対する防御との間に、相関はなかった。血清学的応答がワクチン中のアルミニウムゲルの存在により増強されたにもかかわらず、ワクチン処方物中のアルミニウムゲルの存在は、防御レベルを低減させた。
【0124】
(実施例14)
AMPHIGEN(商標登録)処方物及びDDAを含有するマイクロ流動化されたワクチン調製物で免疫後の、BVDウイルス抗原に対する免疫応答の誘発、及びBVD
2型ウイルス曝露に対する防御
4〜7ヶ月齢の血清陰性の子ウシを、本実験において用いた。異なる6群があり、そして各群は10例の動物を有した(表4)。第0日及び第21日に、各動物は、2mlのワクチンまたはプラセボの皮下用量を、肩甲骨と頭蓋骨との中ほどにある側頸に1回受けた。
【0125】
【表4】
【0126】
5ml用量の曝露用ウイルス調製物(1鼻孔につき、およそ2.5ml)を、試験第44日に鼻腔内投与した。細胞変性していないBVD
2型ウイルスである単離番号24515(Ellis株)、ロット番号46325−70を、本試験において曝露用株として用いた。保持しておいた曝露用材料の試料を、曝露開始時及びその完了直後に力価測定した(1力価測定につき二つ組で)。5ml用量あたりの平均生存ウイルスは、曝露前は5.3log10FAID50/5ml、そして曝露後は5.4log10FAID50/5mlであった(FAIDは、TCID50に相当する)。
【0127】
動物を、第−3日から第58日まで、毎日モニターした。BVD 2感染に起因する臨床徴候に基づいた0、1、2または3の臨床疾患スコアを、第42日から第58日まで、各動物について行った。第44日におけるスコアは、曝露の前に記録した。BVD
1型及びBVD 2型ウイルス中和抗体の血清力価を測定するために、血液試料(二つの13ml血清分離用チューブ、SST)を、第0、21、35、44及び58日に各動物から採取した。
【0128】
血液試料を、第42日から第58日までの全日、各動物から採取し、そしてバフィーコート細胞におけるBVDウイルスの存在を判定した。第44日に、試料を曝露の前に得た。
【0129】
白血球細胞数を測定するために、血液試料(4ml EDTAチューブ1本)を、第42日から第58日までの全日、各動物から採取した。第44日に、試料を曝露の前に得た。
【0130】
白血球減少症を、40%またはそれを超えるベースライン(曝露2日前及び曝露当日の、曝露前の平均のWBC数)からのWBC数の減少として、定義した。
臨床疾患スコアを、以下のように疾患状態を定義するために用いた:スコアが≦1ならば、疾患=なし;スコアが>2ならば、疾患=あり。
【0131】
表5及び表6に示すように、AMPHIGEN(商標登録)処方物、Quil AまたはDDAとともにBVDウイルス抗原を含有し、かつマイクロ流動化されたワクチンでワクチン接種された群は、BVD
1型とBVD 2型ウイルスの双方に対する有意な血清ウイルス中和力価によって血清転換した。これらの群では、曝露後にウイルス血症を示す動物の率の有意な低減もあったが、対照群では、100%の動物がウイルス血症であった(表7)。加えて、これらのワクチン接種された群において、疾患の頻度もまた有意に低減した(表8)。同様に、白血球減少症を示す動物の率もワクチン群において低減し、そして白血球減少症の低減は、Quil
Aを含有する群よりもDDAを含有する群においてより有意であった(表9)。対照群では、ワクチン接種群と比較した場合に、体重増加の有意な降下があった(表10)。
【0132】
血清学
第0日におけるワクチン接種の前に、試験におけるすべての動物は、BVDウイルス 1型及び2型に対する抗体に血清陰性(SVN
<1:2)であった(データは示さず)。二回目のワクチン接種(第35日)後14日に、プラセボを投与されたすべての動物(T01)は、BVDウイルス1型及び2型に対する抗体に血清陰性のままであり;そして、ITA(研究用被験抗原)でワクチン接種されたすべての動物(T02、T03、T04、T05及びT06)は、BVDウイルス1型及び2型に対する抗体に血清陽性(SVN
≧1:8)であった。AMPHIGEN(商標登録)処方物をアジュバントとして添加したDDA 2mg/用量のワクチンを投与された1例の動物は、第35日に、BVDウイルス2型に対する抗体に対してSVN力価3であった(表11及び表12)。
【0133】
第44日における曝露の前に、1例を除くすべての対照(T01)は、BVDウイルス1型及び2型に対する抗体に血清陰性(SVN
<1:2)であった(データは示さず)。1例の対照(#2497)は、BVDウイルス1型に対する抗体に血清陽性(SVN=10)、そしてBVDウイルス2型に対する抗体に血清陰性であった。曝露後14日に、試験におけるすべての動物は、BVDウイルス1型及び2型に対する抗体に血清陽性であった。
【0134】
【表5】
【0135】
【表6】
【0136】
【表7】
【0137】
【表8】
【0138】
【表9】
【0139】
【表10】
【0140】
ウイルスの単離
表13に示すデータのように、曝露期間(第44日〜第58日)中に、対照(T01)におけるすべての10例の動物は、ウイルス血症であった(BVDウイルスが、1日以上で単離された)。ITA投与群では、ウイルス血症の動物の頻度は、10例の各群(それぞれ、T02、T03、T04、T05及びT06)において1、0、3、2及び2例であった。対照とITA投与群との間の差は、統計学的に有意であった(P≦0.05)。ウイルス血症の最小二乗平均日数もまた、ITA投与群(0.0〜0.5日)と比較して、対照で有意に大きかった(10.4日)。
【0141】
臨床疾患
臨床徴候スコア2または3の動物は、BVD疾患の徴候を示していると考えられた。表14に示すように、BVDウイルス疾患の臨床徴候を伴う動物の頻度は、対照(T01)において10例中9例、そしてITA投与各群(それぞれ、T02、T03、T04、T05及びT06)において、10例中1、2、0、0及び0例であった。対照とITA投与群との間の差は、統計学的に有意であった(P≦0.05)。
【0142】
白血球減少症
表15に示すように、曝露期間(第44日〜第58日)中に、対照(T01)におけるすべての10例の動物は、白血球減少症であった(第42〜44日の曝露前ベースラインから、白血球数が40%低減)。白血球減少症を伴う動物の頻度は、ITA投与各群(それぞれ、T02、T03、T04、T05及びT06)において、10例中6、2、4、3及び2例であった。対照と、0.5mg/用量のAMPHIGEN(商標登録)処方物及び水酸化アルミニウムをアジュバントとして添加したワクチン投与群(T03)との間の差は、統計学的に有意であった(P≦0.05)。白血球減少症の最小二乗平均日数は、ITA投与群(0.2〜1.2日)と比較して、対照で有意に大きかった(7.8日)。
【0143】
(実施例15)
GPI−0100を含有するマイクロ流動化されたワクチン調製物で免疫後の、BVDウイルス抗原に対する免疫応答の誘発、及びBVD2型ウイルス曝露に対する防御
実施例14に記載の一連の実験条件にしたがい、そしてQuil AとGPI−0100との直接比較を行った。表11及び表12に示すように、マイクロ流動化されたAMPHIGEN(商標登録)処方物に基づいた、Quil
AまたはGPI−0100のいずれかを含有する調製物に含まれるBVD抗原でワクチン接種された動物は、BVD1型とBVD2型ウイルスの双方に対する有意な抗体価を有した。BVD1型ウイルスに対する抗体価は、BVD2型ウイルスに対するものよりもかなり高かった。しかしながら、それに続くBVD2型ウイルスによる曝露は、強力な防御を示し、そして疾患発生率は、マイクロ流動化されたAMPHIGEN(商標登録)処方物に基づくGPI−0100含有ワクチン調製物でワクチン接種された子ウシにおいて、低減した。
【0144】
【表11】
【0145】
【表12】
【0146】
【表13】
【0147】
【表14】
【0148】
【表15】
【0149】
結論として、各ワクチンの安全性は、ワクチン接種された動物において副作用または死亡がないことにより示された。各ワクチンの効力は、100%のワクチン接種された動物における血清転換(BVD−1及びBVD−2に対するSVN抗体価
>1:8)によって、示された。曝露に対する十分な耐性は、GPI−0100 2mgのみをアジュバントとして添加したワクチンにより示された。
【0150】
(実施例16)
マイクロ流動化された水中油型乳剤中にマイクロカプセル化された抗原を含有するワクチン調製物
3gのトレハロース(Fluka社)を水に加えて、333mg/mlのトレハロース溶液を得た。0.8%SDS溶液中に可溶化した組換えPauA抗原(SDS/rPauA)をトレハロース溶液に加えて、494μg rPauA/mlの最終濃度を得た。次の工程で、10gのポリ乳酸グリコール酸(PLG-Resomer
RE 503H、ベーリンガー・インゲルハイム社)を、200mlの塩化メチレン(MeCl2)に溶解した。得られたPLG/MeCl2溶液を、最初の工程で調製されたSDS−rPauA/トレハロース溶液と組み合わせた。組み合わせた溶液を、(MicrofluidicsモデルM110EHのマイクロ流動化装置)を用いてマイクロ流動化にかけ、そしてマイクロ流動化された調製物を、(テプコ噴霧乾燥機モデルSD−05)を用いて噴霧乾燥した。噴霧乾燥した材料を、500ミクロンのスクリーンを用いて収集した。
【0151】
この噴霧乾燥した材料中のrPauA濃度を、ウエスタンブロット解析を用いて定量した。1.04mgの噴霧乾燥した材料を、50μlのアセトン中に溶解し、そして室温にて10分間、13,200rpmで遠心分離した。上清を、除去した。上清画分及びペレット画分を、生物学的安全フード内で2.5時間乾燥させた。ペレットを、47.43μLの試料溶液(25μlの試料緩衝液+10μlの還元剤+65μlの水)中に再懸濁した。乾燥させた上清画分を、20μlの試料溶液で再懸濁した。ウエスタン解析において、精製PauAを、噴霧乾燥した材料のrPauA含量を定量するための標準品として用いた。
【0152】
20%マンニトールストック溶液を、100mgのマンニトール(シグマ社)を500mlの注射用水(WFI)に溶解することにより、調製した。溶液をホットプレート/撹拌機で40℃に加熱し、そして30℃に冷ました。溶液を、0.22ミクロンの無菌フィルター(ミリポア社)を介して無菌ろ過した。2.5%カルボキシメチルセルロース溶液を、12.5gのカルボキシメチルセルロース(シグマ社)を500mlのWFIに溶解することによって調製し、そして4℃にて一晩混合した。溶液を、121℃にて高圧滅菌した。
【0153】
噴霧乾燥により得られた粉末を、5%マンニトール、0.3%カルボキシメチルセルロース及び1:5000のThimerosolを含有する溶液中で再構成した。最終溶液を3mlバイアルに分割し、そして凍結乾燥機(USIFROID)を用いて凍結乾燥した。凍結乾燥粉末は、マイクロカプセル化されたrPauAを表した。マイクロカプセル化されたサブユニットタンパク質抗原を、2mlのAMPHIGEN(商標登録)処方物を含有するマイクロ流動化された水中油型乳剤(実施例20に記載されるマイクロ流動化された乳剤など)中に再懸濁する。
【0154】
(実施例17)
水中油型乳剤中に細菌全細胞抗原と組換えタンパク質抗原の双方を含有するマイクロ流動化されたワクチン処方物の調製
実施例2及び3に記載のように水中油型乳剤に内在的に添加された、組換えStreptococcus
uberis PauAタンパク質と大腸菌細菌細胞の双方を含有する二つのワクチン調製物を、作った。組換えPauA抗原は100μg/用量の濃度であり、そして大腸菌細胞は4X109/用量の最終数であった。二つのワクチン処方物の乳剤アジュバント組成物を、表16に示す。
【0155】
【表16】
【0156】
(実施例18)
水中油型乳剤中にrPauA及び全細胞細菌因子を含有するマイクロ流動化されたワクチンに対する免疫応答
成熟乳牛を、本実験において用いた。動物は、登録時に初回または二回目の授乳が終了したところであった。2mlの各ワクチン処方物を、乾乳時(D−0)に一度、28日後(D=28)、及び分娩後4〜10日(C+4〜C+10)に再び、3回皮下投与した。最初及び三回目の用量を首の左側に投与し、そして二回目の用量を首の右側に投与した。血液を、各ワクチン接種の前、ならびに三回目のワクチン接種後およそ14日及び32日に採取した。大腸菌及びrPauAに対する抗体価を、ELISAにより測定した。図8に示すように、結果は、rPauAに対する抗体価が、GPI−0100を免疫刺激剤として含有するワクチン処方物でワクチン接種された群においてより高く、そして最初のワクチン接種後第70日にピークとなったことを示している。大腸菌抗原に対する抗体価を、図9に示す。免疫刺激剤としてのGPI−0100の存在が、免疫刺激剤としてDDAを伴う処方物と比較した場合に相対的により高い抗体価を誘導したにもかかわらず、大腸菌抗原に対する抗体価は、双方のワクチン処方物で同程度であった。
【0157】
(実施例19)
マイクロ流動化されたAMPHIGEN(商標登録)処方物に基づくワクチン調製物の殺ウイルス活性の分析
マイクロ流動化がウイルスを不活化するかどうかを判定するために、三つのマイクロ流動化されたAMPHIGEN(商標登録)処方物に基づくウイルス調製物の殺ウイルス活性を測定した。この三つの調製物は、三つの異なるウシ感染性ウイルス、すなわちウシヘルペスウイルス(BHV)、パラインフルエンザウイルス3(PI3)及びウシ呼吸器合胞体ウイルス(BRSV)を含有した。
【0158】
三つのワクチン調製物における殺ウイルス活性の検出を、USDA 9CFR.113.35要件にしたがって行った。
表16に示した結果は、AMPHIGEN(商標登録)処方物に基づくワクチン調製物のマイクロ流動化がワクチン調製物のいかなる有意な不活化も引き起こさないことを、示している。
【0159】
【表17】
【0160】
(実施例20)
マイクロ流動化されたAMPHIGEN(商標登録)処方物の調製
AMPHIGEN(商標登録)処方物を、DRAKEOLレシチン油溶液(25%レシチンを伴う軽質鉱油)とTWEEN 80(0.18%の最終濃度で)とSpan
80(0.08%の最終濃度で)とを組み合わせて、36±1℃にて8〜22時間混合することにより、調製した。次いで、Ross(Hauppauge、ニューヨーク州、11788)乳化装置をおよそ3400rpmで用いて、油混合物を生理食塩液に加えた。続いて、該混合物を、200μmの相互作用チャンバーを伴うマイクロ流動化装置に、4500±500psiにて一度通した。図10A及び図10Bは、マイクロ流動化されたAMPHIGEN(商標登録)処方物の安定性を示す。レーザー回折により測定された、最初の開始時点における粒子サイズ分布(図10A)は、22ヶ月間の4℃保管後の粒子サイズ分布(図10B)とほとんど同一であった。
【0161】
(実施例21)
QuilA−コレステロール免疫原性複合体の電子顕微鏡による分析
Quil A及びコレステロールにより形成された免疫原性複合体の性質を判定するため、これらの二成分の混合物を、抗原の存在下及び非存在下のいずれかで作った。
【0162】
50mlのKR−Hals緩衝液を含有するビーカーに、溶液を磁気撹拌子で攪拌しながら、BVD I型抗原を加えた。その後、Quil
Aの濃縮ストック溶液(50mg/ml)を、溶液を攪拌しながら1滴ずつ加えて、50μg/mlの最終濃度に到達させた。Quil
Aの添加後に、コレステロール・エタノール溶液(18mg/ml)を50μg/mlの最終濃度となるように加えた。
【0163】
二番目のビーカーには、Quil A及びコレステロールを、BVD I型抗原を全く含まない50mlの緩衝液に同じ方法で加えた。
透過型電子顕微鏡用に、10μlの各試料を、formvar/炭素担体プラットホームを伴う400メッシュ銅グリッド(Electron
Microscopy Sciences, Inc.、フォートワシントン、ペンシルバニア州)上に吸着させた。該試料を、ろ過したpH5.2の2%リンタングステン酸10μlを造影剤として用いて、ネガティブ染色した。該試料を、80kVの加速電圧にてJEOL
1230透過型電子顕微鏡(JEOL Inc.、東京、日本)を用いて、検討した。デジタルイメージングを、Gatan BioScan 792カメラで行った。顕微鏡撮影を、4489 EMフィルムで行い、そしてコダブロムII RC F3紙(Eastman Kodak Company、ロチェスター、ニューヨーク州)に印刷した。
【0164】
BVD I型抗原を全く含まない、コレステロール及びQuil Aのみを含有する溶液では、らせん状ミセルが、Quil Aミセル及びコレステロール結晶とともに検出された(図11)。らせん状ミセルは、絡み合い、そしてメッシュのようにみえた。BVD
I型抗原を含有する試料では、らせん状ミセルは、ランダムに特定の高密度領域を取り囲むことがみられた(図12)。該高密度領域はBVD1型抗原を表し、そしてQuil
Aとコレステロールの会合により生じるらせん状免疫原性複合体は、BVD I型抗原の表面に吸着することが見いだされている。
【図面の簡単な説明】
【0165】
【図1】図1は、マイクロ流動化されていないワクチン組成物のバッチ調製のための過程を示す。この過程では、種々のワクチン成分を、左の添加容器に添加し、そして最終的には、成分を単純な機械的手段により混ぜ合わせる混合容器へと投入する。
【図2】図2は、内在的に取り込まれた抗原を含有するマイクロ流動化されたワクチン組成物を調製するための過程を示す。種々のワクチン成分を、添加容器に添加し、そして単純な機械的手段により混合するためのプレ乳剤混和ユニットに移す。
【図3】図3は、マイクロ流動化されていないAMPHIGEN(商標登録)処方物に基づくワクチン、マイクロ流動化されたAMPHIGEN(商標登録)処方物に基づくワクチン、及びベンチ混和ワクチン調製物の液滴サイズの分布を、示す。
【図4】図4は、マイクロ流動化されたワクチン調製物では相分離しないことを示す。
【図5】図5は、マイクロ流動化されたAMPHIGEN(商標登録)処方物に基づくワクチン調製物(A907505)、及び対照である三つのマイクロ流動化されていないAMPHIGEN(商標登録)処方物に基づくワクチン調製物(A904369、A904370及びA904371)中に内在的に取り込まれた抗原の安定性の比較を、示す。すべての四つのワクチン調製物を、4℃にて2年間保管した。保管中の異なる時点(0、6、12または24ヶ月)で、すべての四つの処方物を用いて、3ヶ月齢のウシをワクチン接種した。ワクチン接種を、2mlのワクチン用量で第0日及び第21日に行い、そして二回目のワクチン接種後2週間に、血清を採取した。BVDII型ウイルスに対する中和抗体価を、血清試料のそれぞれにおいて測定した。データを、5例の動物の幾何平均として示した。
【図6】図6は、マイクロ流動化されたワクチン及びマイクロ流動化されていないワクチンの投与前及び投与後のウシの直腸温の最小二乗平均を示す。T01:プラセボ群−1倍用量;T02:プラセボ群−2倍用量;T03:マイクロ流動化されていない処方物−1倍用量;T04:マイクロ流動化されていない処方物−2倍用量;T05:マイクロ流動化された処方物−1倍用量;T06:マイクロ流動化された処方物−2倍用量。
【図7】図7は、マイクロ流動化されていないワクチン処方物及びマイクロ流動化されたワクチン処方物の投与後にウシにおいて観察される注射部位反応体積の最小二乗平均を示す。T03:マイクロ流動化されていない処方物−1倍用量;T04:マイクロ流動化されていない処方物−2倍用量;T05:マイクロ流動化された処方物−1倍用量;T06:マイクロ流動化された処方物−2倍用量。
【図8】図8は、組換えPauA抗原と大腸菌全細胞抗原の双方を含有する種々のワクチン処方物でワクチン接種後の、Streptococcus uberis由来組換えPauA抗原のIgG力価の幾何平均を示す。
【図9】図9は、組換えPauA抗原と大腸菌全細胞抗原の双方を含有する種々のワクチン処方物でワクチン接種後の、Streptococcus uberis由来大腸菌全細胞抗原のIgG力価の幾何平均を示す。
【図10A】図10A及び図10Bは、最初の生成時(図10A)及び生成後22ヶ月(図10B)におけるマイクロ流動化されたAmphigen処方物の粒子サイズ分布を示す。
【図10B】図10A及び図10Bは、最初の生成時(図10A)及び生成後22ヶ月(図10B)におけるマイクロ流動化されたAmphigen処方物の粒子サイズ分布を示す。
【図11】図11は、Quil Aミセル及びコレステロール結晶とともに形成されたらせん状ミセルを示す電子顕微鏡写真である。
【図12】図12は、BVD I型抗原の表面にQuil A及びコレステロールにより形成されたらせん状免疫原性複合体を示す電子顕微鏡写真である。
【特許請求の範囲】
【請求項1】
サポニン配糖体、ステロール及び抗原を含んでなるワクチンであって、前記サポニン配糖体及び前記ステロールが互いに会合してらせん状ミセルの形状の複合体を形成し、かつ前記抗原が前記らせん状ミセルと混合されているが、しかしその内部には組み込まれていない、上記ワクチン。
【請求項2】
獣医学的に許容可能なキャリアーをさらに含んでなる、請求項1に記載のワクチン組成物。
【請求項3】
前記の獣医学的に許容可能なキャリアーが水中油型乳剤である、請求項2に記載のワクチン組成物。
【請求項4】
前記サポニン配糖体がトリテルペノイドである、請求項1に記載のワクチン組成物。
【請求項5】
前記トリテルペノイドがQuil Aである、請求項4に記載のワクチン組成物。
【請求項6】
前記ステロールがコレステロールである、請求項1に記載のワクチン組成物。
【請求項7】
前記抗原がウイルス抗原、細菌抗原またはその組み合わせを含んでなる、請求項1に記載のワクチン組成物。
【請求項8】
前記抗原がウシウイルス性下痢ウイルス(BVDV)1型またはBVDV2型抗原を含んでなる、請求項7に記載のワクチン組成物。
【請求項9】
請求項1に記載のワクチンの製造方法であって、
a)緩衝液中に抗原組成物を調製し;
b)サポニンをステップaの組成物に加え;そして
c)アルコール中のステロールをステップbの組成物に加える、
ステップを含む製造方法。
【請求項1】
サポニン配糖体、ステロール及び抗原を含んでなるワクチンであって、前記サポニン配糖体及び前記ステロールが互いに会合してらせん状ミセルの形状の複合体を形成し、かつ前記抗原が前記らせん状ミセルと混合されているが、しかしその内部には組み込まれていない、上記ワクチン。
【請求項2】
獣医学的に許容可能なキャリアーをさらに含んでなる、請求項1に記載のワクチン組成物。
【請求項3】
前記の獣医学的に許容可能なキャリアーが水中油型乳剤である、請求項2に記載のワクチン組成物。
【請求項4】
前記サポニン配糖体がトリテルペノイドである、請求項1に記載のワクチン組成物。
【請求項5】
前記トリテルペノイドがQuil Aである、請求項4に記載のワクチン組成物。
【請求項6】
前記ステロールがコレステロールである、請求項1に記載のワクチン組成物。
【請求項7】
前記抗原がウイルス抗原、細菌抗原またはその組み合わせを含んでなる、請求項1に記載のワクチン組成物。
【請求項8】
前記抗原がウシウイルス性下痢ウイルス(BVDV)1型またはBVDV2型抗原を含んでなる、請求項7に記載のワクチン組成物。
【請求項9】
請求項1に記載のワクチンの製造方法であって、
a)緩衝液中に抗原組成物を調製し;
b)サポニンをステップaの組成物に加え;そして
c)アルコール中のステロールをステップbの組成物に加える、
ステップを含む製造方法。
【図1】

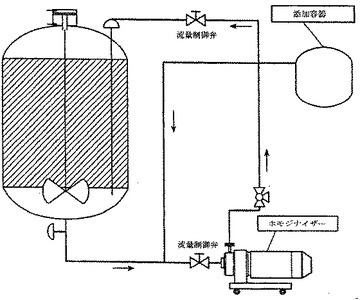
【図2】
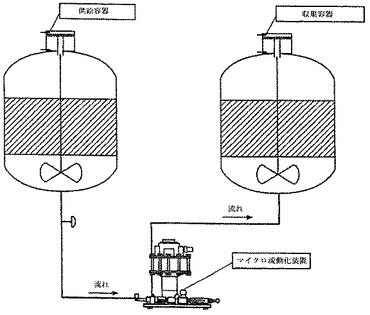
【図3】
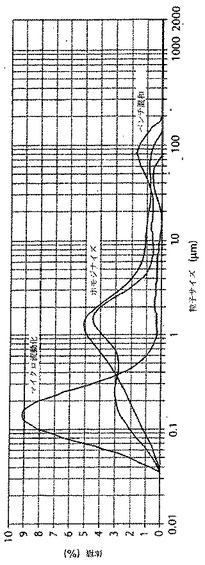
【図4】

【図5】
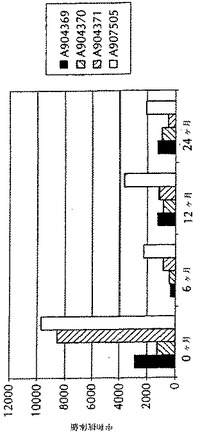
【図6】
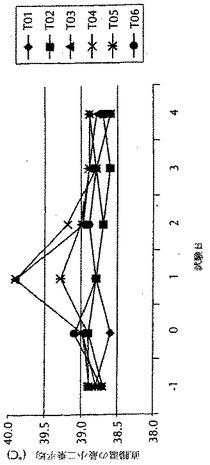
【図7】
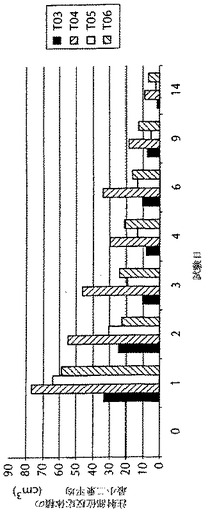
【図8】
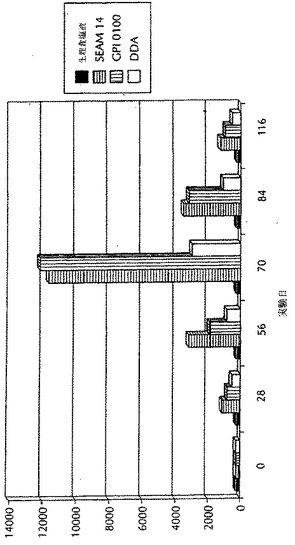
【図9】
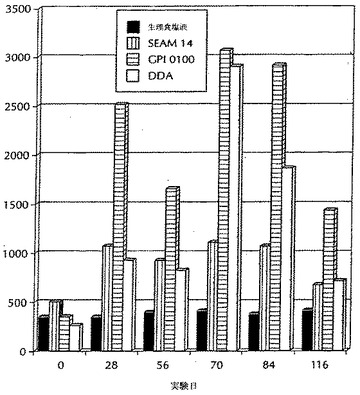
【図10A】
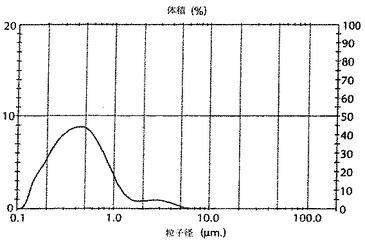
【図10B】
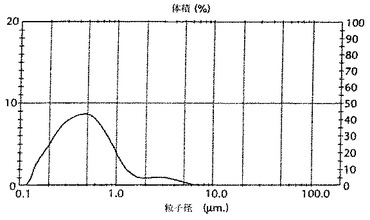
【図11】
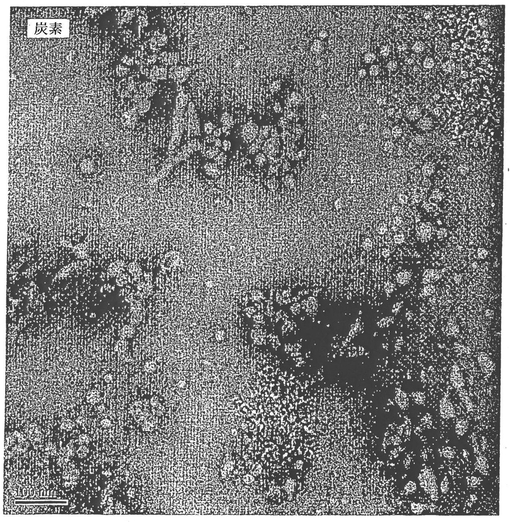
【図12】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10A】
【図10B】
【図11】
【図12】
【公開番号】特開2011−168605(P2011−168605A)
【公開日】平成23年9月1日(2011.9.1)
【国際特許分類】
【出願番号】特願2011−89018(P2011−89018)
【出願日】平成23年4月13日(2011.4.13)
【分割の表示】特願2007−506850(P2007−506850)の分割
【原出願日】平成17年3月24日(2005.3.24)
【出願人】(397067152)ファイザー・プロダクツ・インク (504)
【Fターム(参考)】
【公開日】平成23年9月1日(2011.9.1)
【国際特許分類】
【出願日】平成23年4月13日(2011.4.13)
【分割の表示】特願2007−506850(P2007−506850)の分割
【原出願日】平成17年3月24日(2005.3.24)
【出願人】(397067152)ファイザー・プロダクツ・インク (504)
【Fターム(参考)】
[ Back to top ]