
マキサカルシトール中間体およびその製造方法
【課題】本発明は、マキサカルシトール中間体およびその製造方法に関する。
【解決手段】より詳細には、本発明は、式(I)のキラル化合物、および式(II)のC-20位がR-形またはS-形であるキラル化合物、ならびにこれらのキラル化合物の製造方法を提供する。

【解決手段】より詳細には、本発明は、式(I)のキラル化合物、および式(II)のC-20位がR-形またはS-形であるキラル化合物、ならびにこれらのキラル化合物の製造方法を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ビタミンD類似体、マキサカルシトール、中間体、およびこれらの製造方法に関する。
【背景技術】
【0002】
ビタミンDは、筋肉、免疫系、生殖系、ならびに細胞の増殖および分化に深く影響を及ぼす。実際に、ビタミンD受容体(VDR)を有する細胞は、身体の多くの部分(腸、腎臓、前立腺、骨、骨髄、副甲状腺、皮膚、肝臓、筋肉、およびリンパ系組織などを含む)に見られる。VDRが広範に存在しているため、ビタミンDおよびその類似体は、癌、皮膚、並びに骨の疾患および自己免疫疾患などを含むさまざまな疾患の治療のための化合物として興味が持たれている。
【0003】
何らかの構造的類似性を有するビタミンD類似体類が、これまでに開示されている。例えば、国際出願第87/00834号には、下記式の化合物類:
【化1】
およびその合成方法が記載されている。国際出願第90/09992号には、下記式の化合物類:
【化2】
およびその合成方法が記載されている。これらの化合物類は、異常な細胞増殖および/または細胞分化によって特徴付けられるヒトおよび家畜の疾患の治療に有用である。加えて、Kuboderaらは、1α,-25-ジヒドロキシ-22-オキシビタミンD3類似体の合成およびその分化誘導活性を開示している(Chem. Pharm. Bull. 40 (6) 1494-1499)。
【0004】
ビタミンDおよびその類似体類は、既にSHPT(二次性副甲状腺機能亢進症)の治療に用いられている。パリカルシトール(19-ノル-1,15-ジヒドロキシ-ビタミンD2)およびドキセルカルシフェロール(1α-ヒドロキシ-ビタミンD2)は、米国でs−HPTの治療に推奨されており、22-オキサカルシトール(22-オキサ-1、25(OH)2D3、マキサカルシトール)およびヘキサフルオロ-カルシトリオール(ファレカルシトリオール)は、日本で推奨されている。
【0005】
マキサカルシトールは、いわゆる「非カルセミック」ビタミンD類似体であり、顕著な分化誘導性/抗増殖性を有し、高カルシウム血症を引き起こす能力が低下している。マキサカルシトールは、PTHの強力な抑制剤として開発された。日本では、その使用により慢性透析患者のSHPTの改善が認められた。加えて、尋常性乾癬を含む角化症を有する患者に広く使用され、著しくその症状を改善している。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】国際公開第87/00834号パンフレット
【特許文献2】国際公開第90/09992号パンフレット
【非特許文献】
【0007】
【非特許文献1】Chem. Pharm. Bull. 40 (6) 1494-1499
【発明の概要】
【課題を解決するための手段】
【0008】
本発明は、式(I):
【化3】
のキラル化合物を提供する。
本発明は、さらに、式(II):
【化4】
のC-20位がR-形またはS-形であるキラル化合物を提供する。
【0009】
本発明は、さらに、式(3):
【化5】
を有するマキサカルシトール中間体の製造方法であって、
式(2):
【化6】
の化合物をリチウムアルミニウムハイドライドと反応させる工程を含む方法を提供する。
【図面の簡単な説明】
【0010】
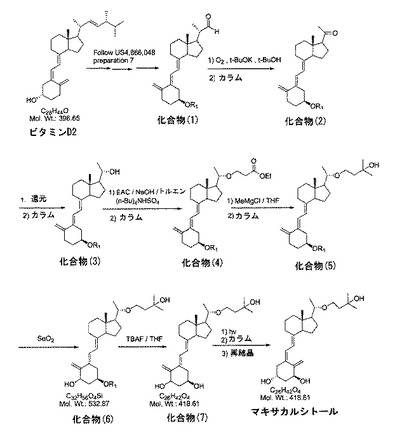
【図1】図1は、マキサカルシトールの合成スキームを説明する。
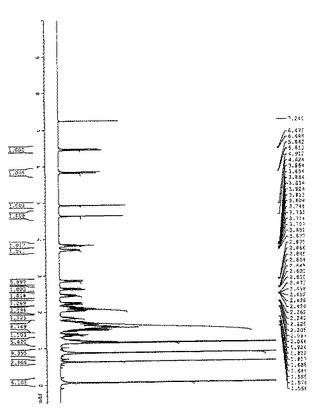
【図2】図2は、化合物(2)の1H NMRの結果を示す: 1H NMR (400 MHz, CDCl3): δ6.38 (1H, d, J=11.6), 5.81 (1H, d, J=11.6), 4.84 (1H, s), 4.57 (1H, s), 3.82 (1H, m), 2.84 (1H, m), 2.64 (1H, m), 2.51 (1H, m), 2.42 (1H, m), 2.25 (1H, dd, J=7.6, 13.6), 2.15 (1H, m), 2.09 (2H, m), 2.06 (3H, s), 1.97 (2H, m), 1.66 (10H, m) 1.18 (1H, t, J=7.2), 0.81 (12H, m), 0.44 (3H, s).
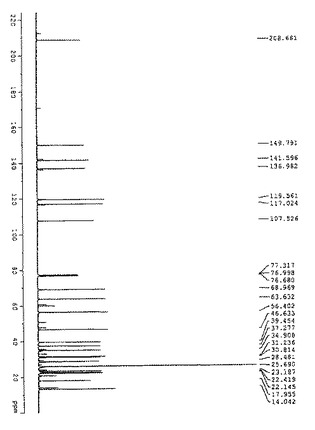
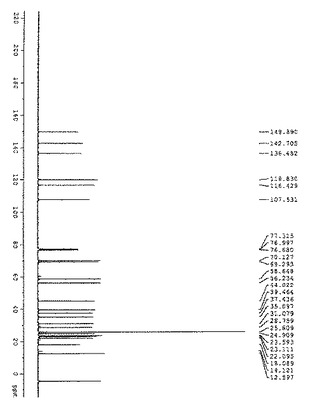
【図3】図3は、化合物(2)の13C NMRの結果を示す: 13C NMR(400MHz, CDCl3): δ208.6, 149.7, 141.5, 136.9, 119.5, 117.0, 107.5, 68.9, 63.6, 56.4, 46.6, 39.4, 37.2, 34.9, 31.2, 30.8, 28.4, 25.6, 23.1, 22.4, 22.1, 17.9, 14.0, 13.2.
【図4】化合物(3)の20R-異性体の1H NMRの結果を示す: 1H NMR (400 MHz, CDCl3): δ6.45 (1H, d, J=11.6), 5.83 (1H, d, J=11.6), 4.91 (1H, s), 4.62 (1H, s), 3.83 (1H, m), 3.71 (1H, m), 2.85 (1H, m), 2.63 (1H, dd, J=4.0, 14.0), 2.45 (1H, m), 2.23 (1H, dd, J=8.0, 13.2), 2.11 (3H, m), 1.82 (1H, m), 1.58 (10H, m), 1.15(4H, m), 0.86 (10H, s), 0.62 (3H, s), 0.05 (6H, s).MS m/z: 431.4 (M+), UV(MeOH) λmax nm : 271.5.
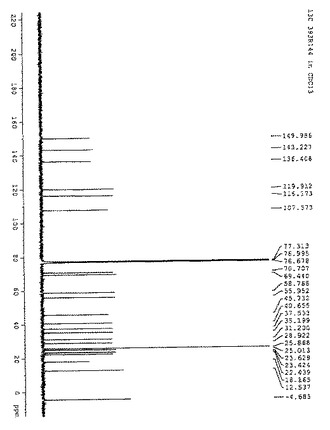
【図5】図5は、化合物(3)の20R-異性体の13C NMRの結果を示す:13C NMR(CDCl3): δ149.9, 143.2, 136.4, 119.9, 116.1, 107.5, 70.7, 69.4, 58.8, 55.9, 45.7, 40.6, 37.5, 35.2, 31.2, 28.9, 25.8, 25.0, 23.6, 23.4, 22.4, 18.1, 12.5.
【図6】図6は、化合物(3)の1H NMRの結果を示す: 1H NMR (400 MHz, CDCl3): δ6.36 (1H, d, J=11.6), 5.84 (1H, d, J=11.6), 4.89 (1H, s), 4.61 (1H, s), 3.83 (1H, m), 3.68 (1H, m), 2.84 (1H, m), 2.60 (1H, dd, J=4.0, 13.6), 2.44 (1H, m), 2.24 (1H, dd, J=8.4, 13.6), 2.12 (1H, m), 1.94 (5H, m), 1.59 (8H, m), 1.20 (5H, m), 0.85 (9H, s), 0.53 (3H, s), 0.03 (6H, s).
【図7】図7は、化合物(3)の13C NMRの結果を示す: 13C NMR(400 MHz, CDCl3): δ149.8, 142.7, 136.4, 119.8, 116.4, 107.5, 70.1, 69.2, 58.6, 56.2, 44.8, 39.4, 37.4, 35.0, 31.0, 28.7, 25.8, 24.9, 23.5, 23.1, 22.0, 18.0, 14.1, 12.5.
【発明を実施するための形態】
【0011】
本発明の方法の記述において、以下の用語は、特に指定しない限り、以下の意味を有する。
【0012】
用語「アルキル」は、直鎖状または分枝状の一価の飽和炭化水素基を意味する。特に指定しない限り、このようなアルキル基は、典型的には1個〜6個の炭素原子を有する。代表的なアルキル基には、例えば、メチル、エチル、n-プロピル、イソプロピル、n-ブチル、sec-ブチル、イソブチル、tert-ブチル、n-ペンチル、n-ヘキシルなどが含まれる。
【0013】
用語「アルコキシ」は、式:(アルキル)-O-(式中、アルキルは、本明細書で定義したとおりである)の一価の基を意味する。代表的なアルキル基には、例えば、メトキシ、エトキシ、n-プロポキシ、イソプロポキシ、n-ブトキシ、sec-ブトキシ、イソブトキシ、tert-ブトキシなどが含まれる。
【0014】
本発明は、式(I):
【化7】
(式中、Rは、OH、O-アシル、O-C1〜8アルキルシリル、またはO-C1〜8アルコキシ-C1〜8アルキルである)
のキラル化合物を提供する。
【0015】
本発明はまた、式(II):
【化8】
(式中、Rは、OH、O-アシル、O-C1〜8アルキルシリル、またはO-C1〜8アルコキシ-C1〜8アルキルである)
のC-20位がR-形またはS-形であるキラル化合物を提供する。
【0016】
用語「キラル」は手を意味するギリシャ語の単語「kheir」に由来し、ここで、手は最もよく知られているキラルな物体であり、左/右対称体が存在することを意味する。例えば、左手と右手は同じではなく、互いに鏡像体であり、したがって「キラル」である。
【0017】
ヒトの手に左と右があるのと同様に、分子にも左と右がある。キラル分子は、その鏡像体と重ね合わせることができない分子である。キラル化合物と、その鏡像体とは、エナンチオマーと呼ばれる。天然のほとんど全てのキラル分子は、単独のエナンチオマーとして存在する。分子を工業的な合成によって製造すると、通常、ラセミ体、すなわち2つのエナンチオマーの50/50組成物の形態で存在する。
【0018】
キラル分子は、光学活性を有し、したがって、エナンチオマーは、時には光学異性体と呼ばれる。各エナンチオマーが偏光面を逆方向に回転させるため、光学活性体と呼ばれる。光を時計方向に回転させるエナンチオマーは、右旋性すなわち(+)であり、逆のエナンチオマーは、左旋性すなわち(−)である。ラセミ混合物は、光学活性を示さない。
【0019】
1948年に、26歳のLouis Pasteurは、ピンセットを用いて、酒石酸塩の右手型および左手型を顕微鏡下で分離した。これらの結晶は、異なる形状を有していた。Pasteurがこれらの結晶を水に溶解すると、一方の結晶は偏光を右に回転させ、他方の結晶は偏光を左に回転させた。したがって、酒石酸塩は、右手型および左手型が分離された最初の分子であり、右手型および左手型は、現在、化学者らによってR(右を意味するラテン語のrectusから)およびS(左を意味するラテン語のsinisterから)と呼ばれている。この実験は、化学者がこの語を使用し始めるおよそ100年前の実験であるが、分子のキラリティーの発見として引き合いに出されることが多い。
【0020】
RおよびSは、分子の絶対配置を表すための記号であり、キラル中心の周りに原子がどのように配置されているかを、優先規則にしたがって示す。
【0021】
本発明の第一の実施態様では、Rは、ヒドロキシル、O-アシル、またはO-tert-ブチルジメチルシリルである。
【0022】
本発明では、これらの化合物を、マキサカルシトールの合成に用いる。
【0023】
本発明は、さらに、化合物(3):
【化9】
の構造を有するマキサカルシトール中間体の製造方法であって、
化合物(2):
【化10】
の構造を金属ハイドライドで還元して化合物(3)を得る工程を含む方法を提供する。
【0024】
本発明の方法では、化合物(2)は、化合物(1):
【化11】
の構造を、金属水酸化物および有機溶媒の存在下、酸素で酸化することによって合成する。
【0025】
本発明の方法では、金属水酸化物は、限定されないが、水酸化カリウムであり、前記有機溶媒は、限定されないが、tert-ブタノールである。
【実施例】
【0026】
以下の実施例は、非限定的なものであり、本発明のさまざまな態様および特徴の単なる代表例である。
【0027】
[合成例1]:化合物(1)の合成
3(R)-(tert-ブチルメチルシリルオキシ)-20(S)-ホルミル-9,20-セコプレグナ- 5(E), 7(E), 10(19)-トリエン
【化12】
化合物 (1)の合成は、米国特許第4,866,048号の製造例1、4、および5〜7に記載された手順に従う。米国特許第4,866,048号に記載された先行技術に従って、1 kgのビタミンD2を用いて、800 gの3(R)-(tert-ブチルメチルシリルオキシ)-20(S)-ホルミル-9,20-セコプレグナ-5(E), 7(E), 10(19)-トリエン(化合物 (1))を、オイル状粘着性生成物として得た。
【0028】
[合成例2]:化合物(2)の合成
(実施例1):
3(R)-(tert-ブチルメチルシリルオキシ)-20(S)-ホルミル-9,20-セコプレグナ-5(E),7(E),10(19)-トリエン(化合物 (1) )(800 g, 1.8 mol)のtert-ブタノール(16L)溶液に、攪拌下、KOH (155 g, 2.76 mol)を添加した。次いで、この溶液に、良好な撹拌下、40℃にて4時間酸素ガスをバブルさせた。
反応が完結した後、tert-ブタノールを蒸発させて除去し、残渣を酢酸エチル(8L)に溶解させ、水で抽出した(8L x 2回)。得られた有機相をMgSO4で無水にした後、濾過した。濾液を、減圧下で濃縮して乾燥させると、オイル状の残渣が得られ、これをカラムカラムクロマトグラフィーで精製(シリカゲル、溶離液はヘキサン中の5%酢酸エチル)して、所望の生成物である化合物(2)を523 g得た(収率67%)。
図2に、化合物(2)の1H NMRの結果を示す。
図3に、化合物(2)の13C NMRの結果を示す。
【0029】
(実施例2):
フラスコに、3(R)-(tert-ブチルメチルシリルオキシ)-20(S)-ホルミル-9,20-セコプレグナ-5(E),7(E),10(19)-トリエン (化合物 (1))(3 g, 6.78 mmol)、N,N-ジメチルホルムアミド(150 ml)、1,4-ジアザビシクロ[2.2.2]オクタン(678 mg, 6 mmol), 酢酸銅一水和物(101 mg, 0.5 mmol)、および2,2’-ビピリジル(82 mg,0.51 mmol)を加えた。この混合物を、40℃にて6日間、良好な撹拌下で空気をバブルさせた。
この反応混合物を酢酸エチル(200 ml)で希釈し、水で抽出し(100 mL x 2)、MgSO4で無水にした。酢酸エチルを蒸発により除去し、オイル状の残渣をカラムクロマトグラフィーで精製(シリカゲル、溶離液はヘキサン中の10%酢酸エチル)して、所望の生成物である化合物(2)を得た。
【0030】
[合成例3]:化合物(3)およびその20R-異性体の合成
(実施例1):
化合物(2)(3 g、7.0 mmol)を、テトラヒドロフラン(140 ml)に溶解し、水素化ホウ素ナトリウム(0.13 g、3.4 mmol)を添加した。次いで、メタノールを、15分かけて滴下により添加した。この反応混合物を、20分間撹拌した後、酢酸エチル(560 ml)で希釈した。この溶液を水(150 mL x 5)および飽和塩化ナトリウム水溶液(150 mL)で抽出し、MgSO4で無水にし、蒸発させて、無色のオイルを得た。このオイル状の残渣をカラムクロマトグラフィーで精製した(シリカゲル、溶離液はヘキサン中の10%酢酸エチル)。最初に留出したものが、化合物(3)の20R-異性体(固体)であった。
図4に、化合物(3)の20R-異性体の1H NMRの結果を示す。
図5に、化合物(3)の20R-異性体の13C NMRの結果を示す。
【0031】
より極性の異性体(化合物(3))を含有するフラクションを蒸発させて、無色のオイル状付加体を得た。
【化13】
図6に、化合物(3)の1H NMRの結果を示す。
図7に、化合物(3)の13C NMRの結果を示す。
【0032】
(実施例2):
化合物(2)(500 g、1.16 mol)をキシレン(10 L)に溶解させ、この反応混合物を100〜130℃に加熱した後、LAH(リチウムアルミニウムハイドライド)(88.5 g、2.33 mol)を添加した。反応を、撹拌下、20分間行い、室温に冷却した。この反応混合物に、飽和硫酸ナトリウム溶液(100 mL)を加えて30分間撹拌した。反応混合物を濾過し、濾液を蒸発させてオイル状の残渣を得た。R/S比は65:35であった。オイル状残渣をカラムクロマトグラフィーで精製(シリカゲル、溶離液はヘキサン中の5%酢酸エチル)して、最初の留出物が化合物(3)の20R-異性体(白色結晶)350 gであり、収率は63.6%であった。
【0033】
より極性の異性体(化合物(3))を含有するフラクションを蒸発させて、無色のオイル状の付加体を得た(123 g、収率24%)。
【0034】
[合成例4]:化合物(4)の合成
【化14】
化合物(3)(123 g、0.28 mol)を、トルエン(6L)および(N-Bu4)NHSO4 (360 mmol)に溶解させ、50% NaOH溶液およびEAC(酢酸エチル、804 mL、7.28 mol)を添加した。反応は、10〜20℃に制御した。反応混合物を5分間撹拌し、次いで、水で希釈した(徐々に添加、4 L)。この溶液を分離し、有機相をMgSO4で無水にし、蒸発させて無色オイルを得た。このオイル状残渣をカラムクロマトグラフィーで精製(シリカゲル、溶離液はヘキサン中の3%酢酸エチル)して、目標化合物(4)をオイル状付加体として得た(112 g、収率73%)。
【0035】
[合成例5]:化合物(5)の合成
【化15】
化合物(4) (112 g、0.21 mol)を、窒素下でテトラヒドロフラン(224 mL)に溶解させた後、10℃未満に冷却した。この攪拌した溶液に、メチルマグネシウムクロライド(210 mL, MeMgCl、テトラヒドロフラン中22%、0.63 mol)を滴下により添加した。この反応混合物を、30分攪拌し、水を添加することによってクエンチし(徐々に添加、38 mL)、次いで濾過した。得られた濾液をMgSO4で無水にし、蒸発させて無色オイルを得た。このオイルをカラムクロマトグラフィーで精製(シリカゲル、溶離液はヘキサン中の7%酢酸エチル)して、目標化合物(5)をオイル状付加体として得た(77.9 g、収率71%)。
1H NMR (400 MHz, CDCl3): δ6.43 (1H, d, J=11.6), 5.83 (1H, d, J=11.6), 4.89 (1H, s), 4.61 (1H, s), 3.81 (1H, m), 3.62 (1H, m), 3.46 (1H, m), 3.23 (1H, m), 2.82 (1H, dd, J=3.6, 13.6), 2.62 (1H, dd, J=3.6, 13.6), 2.44 (1H, m), 2.23 (1H, m), 2.13 (1H, m), 1.96 (2H, m), 1.83 (4H, m), 1.71 (6H, m), 1.55 (6H, m), 1.21 (6H, m), 1.16 (5H, m), 0.85 (9H, s), 0.51 (3H, s), 0.04 (6H, s).
13C NMR(CDCl3): δ149.9, 142.7, 136.5, 119.8, 116.4, 107.5, 78.9, 70.4, 69.3, 65.5, 57.1, 56.2, 44.7, 41.5, 39.6, 37.5, 35.1, 31.1, 29.3, 29.0, 25.8, 23.1, 22.1, 18.8, 18.1, 12.6.
【0036】
[合成例6]:化合物(6)の合成
【化16】
化合物(5)(77.9 g、0.15 mol)を、N-メチルモルホリンN-オキシド(30 g、0.25 mol)を含有するジクロロメタン(467 mL)に溶解させた。撹拌したこの溶液を、窒素下で加熱還流させ、二酸化セレン(6.7 g、0.06 mol)のアセトニトリル(233 mL)溶液を速やかに添加した。添加した後、この混合物を約2時間加熱還流させ、次いで冷却し、さらなるジクロロメタンで希釈し、水で洗浄し、MgSO4で無水にし、濃縮して、粗生成物である化合物(6)を得た。次いで、この粗生成物をカラムクロマトグラフィーで精製(シリカゲル、溶離液はヘキサン中の10%酢酸エチル)して、目標化合物(6)をオイル状付加体として得た(43.6 g、収率54%)。
1H NMR (400 MHz, CDCl3): δ6.46 (1H, d, J=11.6), 5.83 (1H, d, J=11.6), 5.03 (1H, s), 4.91 (1H, s), 4.45 (1H, m), 4.16 (1H, m), 3.80 (1H, m), 3.45 (1H, m), 3.22 (1H, m), 2.82 (1H, dd, J=3.6, 13.6), 2.49 (1H, dd, J=3.6, 13.6), 2.37 (1H, m), 1.83 (5H, m), 1.70 (5H, m), 1.53 (3H, m), 1.29 (2H, m), 1.20 (10H, m), 1.16 (4H, m), 0.84 (9H, s), 0.50 (3H, s), 0.04 (6H, s).
13C NMR(CDCl3): δ153.0, 143.3, 134.5, 122.2, 116.5, 107.6, 78.9, 70.4, 66.7, 65.5, 57.0, 56.1, 44.7, 42.8, 41.4, 39.5, 36.9, 29.3, 29.0, 28.8, 25.7, 23.1, 22.1, 18.8, 18.0, 12.5.
【0037】
[合成例7]:化合物(7)の合成
【化17】
化合物(6) (43.6 g、0.08 mol)を、テトラ-n-ブチルアンモニウムフルオライド(40 g、0.13 mol)を含有するテトラヒドロフラン(261 mL)に溶解させた。撹拌したこの溶液を、窒素下で2.5時間加熱還流させた。冷却した後、この反応溶液を、酢酸エチルと2%炭酸水素ナトリウム溶液との間で分配させ、有機相を水で洗浄し、無水にし、さらに濃縮した。残渣をカラムクロマトグラフィーで精製(シリカゲル、溶離液はヘキサン中の50%酢酸エチル)して、化合物(7)を得た(16.2 g、収率47%)。
【0038】
[合成例8]:マキサカルシトールの合成
【化18】
化合物(7)(13.6 g、30 mmol)および9-セチルアントラセン(1.36 g、6.17 mmol)をアセトン(2250 mL)に溶解させた。このアセトン溶液を、アルゴン雰囲気下、約5℃の温度で約4時間、350 nmのUV光によって光照射した。光照射した後、フェニルボロン酸(1.6 g、1.31 mmol)をこの反応混合物に添加し、反応物を3.5時間撹拌した。次いで、この溶液を、濃縮し、カラムクロマトグラフィーに通して精製して、粗マキサカルシトール(9.7 g、収率74.6%)を得た。
【0039】
[合成例9]:マキサカルシトールの結晶化
粗マキサカルシトール(9.7g, 23.2mmol)を、ジエチルエーテル(200mL)に溶解させた。この溶液を冷却し、5〜10℃にて24時間保った。形成された結晶を濾過し、減圧下、室温で乾燥させて、最終生成物であるマキサカルシトールを得た(1.5 g、純度99.8%、収率15.4%、[α]D20D=+44°)。
【技術分野】
【0001】
本発明は、ビタミンD類似体、マキサカルシトール、中間体、およびこれらの製造方法に関する。
【背景技術】
【0002】
ビタミンDは、筋肉、免疫系、生殖系、ならびに細胞の増殖および分化に深く影響を及ぼす。実際に、ビタミンD受容体(VDR)を有する細胞は、身体の多くの部分(腸、腎臓、前立腺、骨、骨髄、副甲状腺、皮膚、肝臓、筋肉、およびリンパ系組織などを含む)に見られる。VDRが広範に存在しているため、ビタミンDおよびその類似体は、癌、皮膚、並びに骨の疾患および自己免疫疾患などを含むさまざまな疾患の治療のための化合物として興味が持たれている。
【0003】
何らかの構造的類似性を有するビタミンD類似体類が、これまでに開示されている。例えば、国際出願第87/00834号には、下記式の化合物類:
【化1】
およびその合成方法が記載されている。国際出願第90/09992号には、下記式の化合物類:
【化2】
およびその合成方法が記載されている。これらの化合物類は、異常な細胞増殖および/または細胞分化によって特徴付けられるヒトおよび家畜の疾患の治療に有用である。加えて、Kuboderaらは、1α,-25-ジヒドロキシ-22-オキシビタミンD3類似体の合成およびその分化誘導活性を開示している(Chem. Pharm. Bull. 40 (6) 1494-1499)。
【0004】
ビタミンDおよびその類似体類は、既にSHPT(二次性副甲状腺機能亢進症)の治療に用いられている。パリカルシトール(19-ノル-1,15-ジヒドロキシ-ビタミンD2)およびドキセルカルシフェロール(1α-ヒドロキシ-ビタミンD2)は、米国でs−HPTの治療に推奨されており、22-オキサカルシトール(22-オキサ-1、25(OH)2D3、マキサカルシトール)およびヘキサフルオロ-カルシトリオール(ファレカルシトリオール)は、日本で推奨されている。
【0005】
マキサカルシトールは、いわゆる「非カルセミック」ビタミンD類似体であり、顕著な分化誘導性/抗増殖性を有し、高カルシウム血症を引き起こす能力が低下している。マキサカルシトールは、PTHの強力な抑制剤として開発された。日本では、その使用により慢性透析患者のSHPTの改善が認められた。加えて、尋常性乾癬を含む角化症を有する患者に広く使用され、著しくその症状を改善している。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】国際公開第87/00834号パンフレット
【特許文献2】国際公開第90/09992号パンフレット
【非特許文献】
【0007】
【非特許文献1】Chem. Pharm. Bull. 40 (6) 1494-1499
【発明の概要】
【課題を解決するための手段】
【0008】
本発明は、式(I):
【化3】
のキラル化合物を提供する。
本発明は、さらに、式(II):
【化4】
のC-20位がR-形またはS-形であるキラル化合物を提供する。
【0009】
本発明は、さらに、式(3):
【化5】
を有するマキサカルシトール中間体の製造方法であって、
式(2):
【化6】
の化合物をリチウムアルミニウムハイドライドと反応させる工程を含む方法を提供する。
【図面の簡単な説明】
【0010】
【図1】図1は、マキサカルシトールの合成スキームを説明する。
【図2】図2は、化合物(2)の1H NMRの結果を示す: 1H NMR (400 MHz, CDCl3): δ6.38 (1H, d, J=11.6), 5.81 (1H, d, J=11.6), 4.84 (1H, s), 4.57 (1H, s), 3.82 (1H, m), 2.84 (1H, m), 2.64 (1H, m), 2.51 (1H, m), 2.42 (1H, m), 2.25 (1H, dd, J=7.6, 13.6), 2.15 (1H, m), 2.09 (2H, m), 2.06 (3H, s), 1.97 (2H, m), 1.66 (10H, m) 1.18 (1H, t, J=7.2), 0.81 (12H, m), 0.44 (3H, s).
【図3】図3は、化合物(2)の13C NMRの結果を示す: 13C NMR(400MHz, CDCl3): δ208.6, 149.7, 141.5, 136.9, 119.5, 117.0, 107.5, 68.9, 63.6, 56.4, 46.6, 39.4, 37.2, 34.9, 31.2, 30.8, 28.4, 25.6, 23.1, 22.4, 22.1, 17.9, 14.0, 13.2.
【図4】化合物(3)の20R-異性体の1H NMRの結果を示す: 1H NMR (400 MHz, CDCl3): δ6.45 (1H, d, J=11.6), 5.83 (1H, d, J=11.6), 4.91 (1H, s), 4.62 (1H, s), 3.83 (1H, m), 3.71 (1H, m), 2.85 (1H, m), 2.63 (1H, dd, J=4.0, 14.0), 2.45 (1H, m), 2.23 (1H, dd, J=8.0, 13.2), 2.11 (3H, m), 1.82 (1H, m), 1.58 (10H, m), 1.15(4H, m), 0.86 (10H, s), 0.62 (3H, s), 0.05 (6H, s).MS m/z: 431.4 (M+), UV(MeOH) λmax nm : 271.5.
【図5】図5は、化合物(3)の20R-異性体の13C NMRの結果を示す:13C NMR(CDCl3): δ149.9, 143.2, 136.4, 119.9, 116.1, 107.5, 70.7, 69.4, 58.8, 55.9, 45.7, 40.6, 37.5, 35.2, 31.2, 28.9, 25.8, 25.0, 23.6, 23.4, 22.4, 18.1, 12.5.
【図6】図6は、化合物(3)の1H NMRの結果を示す: 1H NMR (400 MHz, CDCl3): δ6.36 (1H, d, J=11.6), 5.84 (1H, d, J=11.6), 4.89 (1H, s), 4.61 (1H, s), 3.83 (1H, m), 3.68 (1H, m), 2.84 (1H, m), 2.60 (1H, dd, J=4.0, 13.6), 2.44 (1H, m), 2.24 (1H, dd, J=8.4, 13.6), 2.12 (1H, m), 1.94 (5H, m), 1.59 (8H, m), 1.20 (5H, m), 0.85 (9H, s), 0.53 (3H, s), 0.03 (6H, s).
【図7】図7は、化合物(3)の13C NMRの結果を示す: 13C NMR(400 MHz, CDCl3): δ149.8, 142.7, 136.4, 119.8, 116.4, 107.5, 70.1, 69.2, 58.6, 56.2, 44.8, 39.4, 37.4, 35.0, 31.0, 28.7, 25.8, 24.9, 23.5, 23.1, 22.0, 18.0, 14.1, 12.5.
【発明を実施するための形態】
【0011】
本発明の方法の記述において、以下の用語は、特に指定しない限り、以下の意味を有する。
【0012】
用語「アルキル」は、直鎖状または分枝状の一価の飽和炭化水素基を意味する。特に指定しない限り、このようなアルキル基は、典型的には1個〜6個の炭素原子を有する。代表的なアルキル基には、例えば、メチル、エチル、n-プロピル、イソプロピル、n-ブチル、sec-ブチル、イソブチル、tert-ブチル、n-ペンチル、n-ヘキシルなどが含まれる。
【0013】
用語「アルコキシ」は、式:(アルキル)-O-(式中、アルキルは、本明細書で定義したとおりである)の一価の基を意味する。代表的なアルキル基には、例えば、メトキシ、エトキシ、n-プロポキシ、イソプロポキシ、n-ブトキシ、sec-ブトキシ、イソブトキシ、tert-ブトキシなどが含まれる。
【0014】
本発明は、式(I):
【化7】
(式中、Rは、OH、O-アシル、O-C1〜8アルキルシリル、またはO-C1〜8アルコキシ-C1〜8アルキルである)
のキラル化合物を提供する。
【0015】
本発明はまた、式(II):
【化8】
(式中、Rは、OH、O-アシル、O-C1〜8アルキルシリル、またはO-C1〜8アルコキシ-C1〜8アルキルである)
のC-20位がR-形またはS-形であるキラル化合物を提供する。
【0016】
用語「キラル」は手を意味するギリシャ語の単語「kheir」に由来し、ここで、手は最もよく知られているキラルな物体であり、左/右対称体が存在することを意味する。例えば、左手と右手は同じではなく、互いに鏡像体であり、したがって「キラル」である。
【0017】
ヒトの手に左と右があるのと同様に、分子にも左と右がある。キラル分子は、その鏡像体と重ね合わせることができない分子である。キラル化合物と、その鏡像体とは、エナンチオマーと呼ばれる。天然のほとんど全てのキラル分子は、単独のエナンチオマーとして存在する。分子を工業的な合成によって製造すると、通常、ラセミ体、すなわち2つのエナンチオマーの50/50組成物の形態で存在する。
【0018】
キラル分子は、光学活性を有し、したがって、エナンチオマーは、時には光学異性体と呼ばれる。各エナンチオマーが偏光面を逆方向に回転させるため、光学活性体と呼ばれる。光を時計方向に回転させるエナンチオマーは、右旋性すなわち(+)であり、逆のエナンチオマーは、左旋性すなわち(−)である。ラセミ混合物は、光学活性を示さない。
【0019】
1948年に、26歳のLouis Pasteurは、ピンセットを用いて、酒石酸塩の右手型および左手型を顕微鏡下で分離した。これらの結晶は、異なる形状を有していた。Pasteurがこれらの結晶を水に溶解すると、一方の結晶は偏光を右に回転させ、他方の結晶は偏光を左に回転させた。したがって、酒石酸塩は、右手型および左手型が分離された最初の分子であり、右手型および左手型は、現在、化学者らによってR(右を意味するラテン語のrectusから)およびS(左を意味するラテン語のsinisterから)と呼ばれている。この実験は、化学者がこの語を使用し始めるおよそ100年前の実験であるが、分子のキラリティーの発見として引き合いに出されることが多い。
【0020】
RおよびSは、分子の絶対配置を表すための記号であり、キラル中心の周りに原子がどのように配置されているかを、優先規則にしたがって示す。
【0021】
本発明の第一の実施態様では、Rは、ヒドロキシル、O-アシル、またはO-tert-ブチルジメチルシリルである。
【0022】
本発明では、これらの化合物を、マキサカルシトールの合成に用いる。
【0023】
本発明は、さらに、化合物(3):
【化9】
の構造を有するマキサカルシトール中間体の製造方法であって、
化合物(2):
【化10】
の構造を金属ハイドライドで還元して化合物(3)を得る工程を含む方法を提供する。
【0024】
本発明の方法では、化合物(2)は、化合物(1):
【化11】
の構造を、金属水酸化物および有機溶媒の存在下、酸素で酸化することによって合成する。
【0025】
本発明の方法では、金属水酸化物は、限定されないが、水酸化カリウムであり、前記有機溶媒は、限定されないが、tert-ブタノールである。
【実施例】
【0026】
以下の実施例は、非限定的なものであり、本発明のさまざまな態様および特徴の単なる代表例である。
【0027】
[合成例1]:化合物(1)の合成
3(R)-(tert-ブチルメチルシリルオキシ)-20(S)-ホルミル-9,20-セコプレグナ- 5(E), 7(E), 10(19)-トリエン
【化12】
化合物 (1)の合成は、米国特許第4,866,048号の製造例1、4、および5〜7に記載された手順に従う。米国特許第4,866,048号に記載された先行技術に従って、1 kgのビタミンD2を用いて、800 gの3(R)-(tert-ブチルメチルシリルオキシ)-20(S)-ホルミル-9,20-セコプレグナ-5(E), 7(E), 10(19)-トリエン(化合物 (1))を、オイル状粘着性生成物として得た。
【0028】
[合成例2]:化合物(2)の合成
(実施例1):
3(R)-(tert-ブチルメチルシリルオキシ)-20(S)-ホルミル-9,20-セコプレグナ-5(E),7(E),10(19)-トリエン(化合物 (1) )(800 g, 1.8 mol)のtert-ブタノール(16L)溶液に、攪拌下、KOH (155 g, 2.76 mol)を添加した。次いで、この溶液に、良好な撹拌下、40℃にて4時間酸素ガスをバブルさせた。
反応が完結した後、tert-ブタノールを蒸発させて除去し、残渣を酢酸エチル(8L)に溶解させ、水で抽出した(8L x 2回)。得られた有機相をMgSO4で無水にした後、濾過した。濾液を、減圧下で濃縮して乾燥させると、オイル状の残渣が得られ、これをカラムカラムクロマトグラフィーで精製(シリカゲル、溶離液はヘキサン中の5%酢酸エチル)して、所望の生成物である化合物(2)を523 g得た(収率67%)。
図2に、化合物(2)の1H NMRの結果を示す。
図3に、化合物(2)の13C NMRの結果を示す。
【0029】
(実施例2):
フラスコに、3(R)-(tert-ブチルメチルシリルオキシ)-20(S)-ホルミル-9,20-セコプレグナ-5(E),7(E),10(19)-トリエン (化合物 (1))(3 g, 6.78 mmol)、N,N-ジメチルホルムアミド(150 ml)、1,4-ジアザビシクロ[2.2.2]オクタン(678 mg, 6 mmol), 酢酸銅一水和物(101 mg, 0.5 mmol)、および2,2’-ビピリジル(82 mg,0.51 mmol)を加えた。この混合物を、40℃にて6日間、良好な撹拌下で空気をバブルさせた。
この反応混合物を酢酸エチル(200 ml)で希釈し、水で抽出し(100 mL x 2)、MgSO4で無水にした。酢酸エチルを蒸発により除去し、オイル状の残渣をカラムクロマトグラフィーで精製(シリカゲル、溶離液はヘキサン中の10%酢酸エチル)して、所望の生成物である化合物(2)を得た。
【0030】
[合成例3]:化合物(3)およびその20R-異性体の合成
(実施例1):
化合物(2)(3 g、7.0 mmol)を、テトラヒドロフラン(140 ml)に溶解し、水素化ホウ素ナトリウム(0.13 g、3.4 mmol)を添加した。次いで、メタノールを、15分かけて滴下により添加した。この反応混合物を、20分間撹拌した後、酢酸エチル(560 ml)で希釈した。この溶液を水(150 mL x 5)および飽和塩化ナトリウム水溶液(150 mL)で抽出し、MgSO4で無水にし、蒸発させて、無色のオイルを得た。このオイル状の残渣をカラムクロマトグラフィーで精製した(シリカゲル、溶離液はヘキサン中の10%酢酸エチル)。最初に留出したものが、化合物(3)の20R-異性体(固体)であった。
図4に、化合物(3)の20R-異性体の1H NMRの結果を示す。
図5に、化合物(3)の20R-異性体の13C NMRの結果を示す。
【0031】
より極性の異性体(化合物(3))を含有するフラクションを蒸発させて、無色のオイル状付加体を得た。
【化13】
図6に、化合物(3)の1H NMRの結果を示す。
図7に、化合物(3)の13C NMRの結果を示す。
【0032】
(実施例2):
化合物(2)(500 g、1.16 mol)をキシレン(10 L)に溶解させ、この反応混合物を100〜130℃に加熱した後、LAH(リチウムアルミニウムハイドライド)(88.5 g、2.33 mol)を添加した。反応を、撹拌下、20分間行い、室温に冷却した。この反応混合物に、飽和硫酸ナトリウム溶液(100 mL)を加えて30分間撹拌した。反応混合物を濾過し、濾液を蒸発させてオイル状の残渣を得た。R/S比は65:35であった。オイル状残渣をカラムクロマトグラフィーで精製(シリカゲル、溶離液はヘキサン中の5%酢酸エチル)して、最初の留出物が化合物(3)の20R-異性体(白色結晶)350 gであり、収率は63.6%であった。
【0033】
より極性の異性体(化合物(3))を含有するフラクションを蒸発させて、無色のオイル状の付加体を得た(123 g、収率24%)。
【0034】
[合成例4]:化合物(4)の合成
【化14】
化合物(3)(123 g、0.28 mol)を、トルエン(6L)および(N-Bu4)NHSO4 (360 mmol)に溶解させ、50% NaOH溶液およびEAC(酢酸エチル、804 mL、7.28 mol)を添加した。反応は、10〜20℃に制御した。反応混合物を5分間撹拌し、次いで、水で希釈した(徐々に添加、4 L)。この溶液を分離し、有機相をMgSO4で無水にし、蒸発させて無色オイルを得た。このオイル状残渣をカラムクロマトグラフィーで精製(シリカゲル、溶離液はヘキサン中の3%酢酸エチル)して、目標化合物(4)をオイル状付加体として得た(112 g、収率73%)。
【0035】
[合成例5]:化合物(5)の合成
【化15】
化合物(4) (112 g、0.21 mol)を、窒素下でテトラヒドロフラン(224 mL)に溶解させた後、10℃未満に冷却した。この攪拌した溶液に、メチルマグネシウムクロライド(210 mL, MeMgCl、テトラヒドロフラン中22%、0.63 mol)を滴下により添加した。この反応混合物を、30分攪拌し、水を添加することによってクエンチし(徐々に添加、38 mL)、次いで濾過した。得られた濾液をMgSO4で無水にし、蒸発させて無色オイルを得た。このオイルをカラムクロマトグラフィーで精製(シリカゲル、溶離液はヘキサン中の7%酢酸エチル)して、目標化合物(5)をオイル状付加体として得た(77.9 g、収率71%)。
1H NMR (400 MHz, CDCl3): δ6.43 (1H, d, J=11.6), 5.83 (1H, d, J=11.6), 4.89 (1H, s), 4.61 (1H, s), 3.81 (1H, m), 3.62 (1H, m), 3.46 (1H, m), 3.23 (1H, m), 2.82 (1H, dd, J=3.6, 13.6), 2.62 (1H, dd, J=3.6, 13.6), 2.44 (1H, m), 2.23 (1H, m), 2.13 (1H, m), 1.96 (2H, m), 1.83 (4H, m), 1.71 (6H, m), 1.55 (6H, m), 1.21 (6H, m), 1.16 (5H, m), 0.85 (9H, s), 0.51 (3H, s), 0.04 (6H, s).
13C NMR(CDCl3): δ149.9, 142.7, 136.5, 119.8, 116.4, 107.5, 78.9, 70.4, 69.3, 65.5, 57.1, 56.2, 44.7, 41.5, 39.6, 37.5, 35.1, 31.1, 29.3, 29.0, 25.8, 23.1, 22.1, 18.8, 18.1, 12.6.
【0036】
[合成例6]:化合物(6)の合成
【化16】
化合物(5)(77.9 g、0.15 mol)を、N-メチルモルホリンN-オキシド(30 g、0.25 mol)を含有するジクロロメタン(467 mL)に溶解させた。撹拌したこの溶液を、窒素下で加熱還流させ、二酸化セレン(6.7 g、0.06 mol)のアセトニトリル(233 mL)溶液を速やかに添加した。添加した後、この混合物を約2時間加熱還流させ、次いで冷却し、さらなるジクロロメタンで希釈し、水で洗浄し、MgSO4で無水にし、濃縮して、粗生成物である化合物(6)を得た。次いで、この粗生成物をカラムクロマトグラフィーで精製(シリカゲル、溶離液はヘキサン中の10%酢酸エチル)して、目標化合物(6)をオイル状付加体として得た(43.6 g、収率54%)。
1H NMR (400 MHz, CDCl3): δ6.46 (1H, d, J=11.6), 5.83 (1H, d, J=11.6), 5.03 (1H, s), 4.91 (1H, s), 4.45 (1H, m), 4.16 (1H, m), 3.80 (1H, m), 3.45 (1H, m), 3.22 (1H, m), 2.82 (1H, dd, J=3.6, 13.6), 2.49 (1H, dd, J=3.6, 13.6), 2.37 (1H, m), 1.83 (5H, m), 1.70 (5H, m), 1.53 (3H, m), 1.29 (2H, m), 1.20 (10H, m), 1.16 (4H, m), 0.84 (9H, s), 0.50 (3H, s), 0.04 (6H, s).
13C NMR(CDCl3): δ153.0, 143.3, 134.5, 122.2, 116.5, 107.6, 78.9, 70.4, 66.7, 65.5, 57.0, 56.1, 44.7, 42.8, 41.4, 39.5, 36.9, 29.3, 29.0, 28.8, 25.7, 23.1, 22.1, 18.8, 18.0, 12.5.
【0037】
[合成例7]:化合物(7)の合成
【化17】
化合物(6) (43.6 g、0.08 mol)を、テトラ-n-ブチルアンモニウムフルオライド(40 g、0.13 mol)を含有するテトラヒドロフラン(261 mL)に溶解させた。撹拌したこの溶液を、窒素下で2.5時間加熱還流させた。冷却した後、この反応溶液を、酢酸エチルと2%炭酸水素ナトリウム溶液との間で分配させ、有機相を水で洗浄し、無水にし、さらに濃縮した。残渣をカラムクロマトグラフィーで精製(シリカゲル、溶離液はヘキサン中の50%酢酸エチル)して、化合物(7)を得た(16.2 g、収率47%)。
【0038】
[合成例8]:マキサカルシトールの合成
【化18】
化合物(7)(13.6 g、30 mmol)および9-セチルアントラセン(1.36 g、6.17 mmol)をアセトン(2250 mL)に溶解させた。このアセトン溶液を、アルゴン雰囲気下、約5℃の温度で約4時間、350 nmのUV光によって光照射した。光照射した後、フェニルボロン酸(1.6 g、1.31 mmol)をこの反応混合物に添加し、反応物を3.5時間撹拌した。次いで、この溶液を、濃縮し、カラムクロマトグラフィーに通して精製して、粗マキサカルシトール(9.7 g、収率74.6%)を得た。
【0039】
[合成例9]:マキサカルシトールの結晶化
粗マキサカルシトール(9.7g, 23.2mmol)を、ジエチルエーテル(200mL)に溶解させた。この溶液を冷却し、5〜10℃にて24時間保った。形成された結晶を濾過し、減圧下、室温で乾燥させて、最終生成物であるマキサカルシトールを得た(1.5 g、純度99.8%、収率15.4%、[α]D20D=+44°)。
【特許請求の範囲】
【請求項1】
式(I):
【化1】
(式中、Rは、OH、O-アシル、O-C1〜8アルキルシリル、またはO-C1〜8アルキルオキシ-C1〜8アルキルである)
を有するキラル化合物。
【請求項2】
Rが、ヒドロキシル、O-アシル、またはO-tert-ブチルジメチルシリルである、請求項1に記載のキラル化合物。
【請求項3】
式(II):
【化2】
(式中、Rは、OH、O-アシル、O-C1〜8アルキルシリル、またはO-C1〜8アルキルオキシ-C1〜8アルキルである)
のC-20位がR-形またはS-形であるキラル化合物。
【請求項4】
Rが、ヒドロキシル、O-アシル、またはO-tert-ブチルジメチルシリルである、請求項3に記載のキラル化合物。
【請求項5】
マキサカルシトールの合成に用いるための、請求項1または3のキラル化合物。
【請求項6】
下記構造を有する化合物(3):
【化3】
であるマキサカルシトールの中間体の製造方法であって、
下記構造の化合物(2):
【化4】
を金属ハイドライドで還元して化合物(3)を得る工程を含む、方法。
【請求項7】
化合物(2)を、下記構造の化合物(1):
【化5】
を、金属水酸化物および有機溶媒の存在下、酸素で酸化することによって合成する、請求項6に記載の方法。
【請求項8】
前記金属水酸化物がKOHであり、前記有機溶媒がtert-ブタノールである、請求項7に記載の方法。
【請求項1】
式(I):
【化1】
(式中、Rは、OH、O-アシル、O-C1〜8アルキルシリル、またはO-C1〜8アルキルオキシ-C1〜8アルキルである)
を有するキラル化合物。
【請求項2】
Rが、ヒドロキシル、O-アシル、またはO-tert-ブチルジメチルシリルである、請求項1に記載のキラル化合物。
【請求項3】
式(II):
【化2】
(式中、Rは、OH、O-アシル、O-C1〜8アルキルシリル、またはO-C1〜8アルキルオキシ-C1〜8アルキルである)
のC-20位がR-形またはS-形であるキラル化合物。
【請求項4】
Rが、ヒドロキシル、O-アシル、またはO-tert-ブチルジメチルシリルである、請求項3に記載のキラル化合物。
【請求項5】
マキサカルシトールの合成に用いるための、請求項1または3のキラル化合物。
【請求項6】
下記構造を有する化合物(3):
【化3】
であるマキサカルシトールの中間体の製造方法であって、
下記構造の化合物(2):
【化4】
を金属ハイドライドで還元して化合物(3)を得る工程を含む、方法。
【請求項7】
化合物(2)を、下記構造の化合物(1):
【化5】
を、金属水酸化物および有機溶媒の存在下、酸素で酸化することによって合成する、請求項6に記載の方法。
【請求項8】
前記金属水酸化物がKOHであり、前記有機溶媒がtert-ブタノールである、請求項7に記載の方法。
【図1】

【図2】

【図3】
【図4】
【図5】
【図6】

【図7】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2011−157326(P2011−157326A)
【公開日】平成23年8月18日(2011.8.18)
【国際特許分類】
【出願番号】特願2010−22200(P2010−22200)
【出願日】平成22年2月3日(2010.2.3)
【出願人】(510031512)フォーモサ・ラボラトリーズ・インコーポレーテッド (2)
【Fターム(参考)】
【公開日】平成23年8月18日(2011.8.18)
【国際特許分類】
【出願日】平成22年2月3日(2010.2.3)
【出願人】(510031512)フォーモサ・ラボラトリーズ・インコーポレーテッド (2)
【Fターム(参考)】
[ Back to top ]