
マクロライドおよびある特定のシクロデキストリンを含有する、経口、眼部、局所、または非経口使用のための新しい水溶性固体医薬包接複合体およびその水溶液
【課題】新しい水溶性固体医薬包接複合体とその水溶液を提供すること。
【解決手段】マクロライドおよびある特定のシクロデキストリンを含有する、経口、眼科、局所、または非経口使用のための新しい水溶性固体医薬包接複合体とその水溶液。より具体的には、
a)活性成分として、ラパマイシン、ピメクロリムス、テムシロリムス、エベロリムス、またはタクロリムスなどのマクロライドを、非晶質形態で、および任意選択でそれらの多型水和物または溶媒和物の形態で、例えばアセトンまたはエタノールで形成された溶媒和物の形態で、
b)ガンマシクロデキストリンなどの大表面シクロデキストリン
を含有するものであり、前記マクロライドと前記シクロデキストリンとの重量比が1:111から1:333の間の範囲である。
【解決手段】マクロライドおよびある特定のシクロデキストリンを含有する、経口、眼科、局所、または非経口使用のための新しい水溶性固体医薬包接複合体とその水溶液。より具体的には、
a)活性成分として、ラパマイシン、ピメクロリムス、テムシロリムス、エベロリムス、またはタクロリムスなどのマクロライドを、非晶質形態で、および任意選択でそれらの多型水和物または溶媒和物の形態で、例えばアセトンまたはエタノールで形成された溶媒和物の形態で、
b)ガンマシクロデキストリンなどの大表面シクロデキストリン
を含有するものであり、前記マクロライドと前記シクロデキストリンとの重量比が1:111から1:333の間の範囲である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、マクロライドおよびある特定のシクロデキストリンを含有する、経口、眼部(ophthalmic)、局所、または非経口使用のための新しい水溶性固体医薬包接複合体およびその水溶液に関する。
【背景技術】
【0002】
マクロライドは、その活性が、マクロライド環、即ち1種または複数のデオキシ糖が結合されていてもよい大きな大環状ラクトン環、が存在することで生じる薬物(典型的には、抗生物質)の群である。ラクトン環は、通常、14、15、または16員である。マクロライドは、天然物のポリケチドのクラスに属する。このファミリーの中で、登録番号53123−88−9によって特定される、式1のラパマイシンとしても知られるシロリムスと、登録番号104987−11−3によって特定される式2のタクロリムスは、その免疫抑制活性で知られる化合物であり、体内での臓器および骨髄移植の拒絶を予防するのに使用される。
【0003】
【化1】

【0004】
テムシロリムス、エベロリムス、およびピメクロリムス(アスコマイシンの合成誘導体)などのラパマイシン誘導体は、細胞の成長、増殖、および生存の制御に関与する特定のタンパク質(mTOR)に関して、類似した阻害活性を示す。
【0005】
登録番号162635−04−3によって特定される式3のテムシロリムスは、ラパマイシンに構造的に関係しており、進行性腎細胞癌(腎癌の一タイプ)の治療に使用される。このテムシロリムスは、その他のタイプの癌の治療においても研究されている。
【0006】
【化2】
【0007】
登録番号159351−69−6によって特定される式4のエベロリムスは、ある特定のその他の抗癌剤での治療に応答しない進行性腎癌を、治療するのに使用される。このエベロリムスは、ワルデンシュトレームマクログロブリン血症または乳癌のようなその他のタイプの癌の治療においても、研究されている。
【0008】
【化3】
【0009】
登録番号137071−32−0によって特定された式5のピメクロリムス、アスコマイシンの合成誘導体は;アトピー性皮膚炎の治療に使用される。
【0010】
【化4】
【0011】
これら化合物の全ては、0.01から0.000006モル/Lの範囲である、室温での不十分な水溶性を有し、貯蔵後の溶液中では不安定でありかつそのエステル結合の加溶媒分解の結果としてin vitroおよびin vivoの両方において生物活性の損失をもたらすことが報告されている(非特許文献1および2参照)。
【0012】
さらに、固体状態では、これらの化合物は、非晶質または結晶質形態で存在してもよく、この非晶質は非常に不安定で酸化分解する(非特許文献3、4、および5参照)。
【0013】
非晶質形態が、結晶質形態と比較した場合により高い溶解度を有し(結晶質から非晶質材料までの溶解度の上昇は、10から1600倍の間であることが報告されている。)、安定性が低く、分解する傾向があることは、常識である。
【0014】
さらに、低い溶解度は、通常は、体内での不十分な吸収および不十分な利用可能性に関連している。特許文献1は、シクロデキストリンと組み合わせて水にやや溶け難い薬物を溶かした水性非経口溶液が、注射部位または非経口施用後の臓器での薬物沈殿を最小限に抑えることができることを示唆している。
【0015】
特許文献2は、αまたはβシクロデキストリン上のラパマイシンの固体分散体の形をとる、経口投与用の医薬組成物を調製する可能性について記述している。
【0016】
実際に、文献データは、細かく微粉化されたラパマイシンまたは非晶質ラパマイシンを、特許文献3、4、5、および6に記載されているように、ポリブチルメタクリレート(PBMA)、ポリエチレン−co−ビニルアセテート(PEVA)、または硫酸プロタミンおよびセルロースなどの高分子電解質複合体のようないくつかの合成ポリマーの使用を見越した種々のコーティング技法を用いる製剤目的のために安定化させる必要があることを裏付けている。
【先行技術文献】
【特許文献】
【0017】
【特許文献1】米国特許第5024998号明細書
【特許文献2】EP839028
【特許文献3】国際公開第2006026531号パンフレット
【特許文献4】国際公開第2006039237号パンフレット
【特許文献5】国際公開第2007011708号パンフレット
【特許文献6】EP2135601
【非特許文献】
【0018】
【非特許文献1】Yuri V. Il'ichev, Lori Alquier, and Cynthia A. Maryanoff, ARKIVOC, 2007 (XII) 110-131
【非特許文献2】Ping Cai, Rushung Tsao, and Mark E. Ruppen, DMD Fast Forward. May 31, 2007
【非特許文献3】Tetrahedron Letters (1990), 31(34), 4845-8
【非特許文献4】Xenobiotica, 27(9), 869 (1997)
【非特許文献5】J. Org. Chem., 63, 10069, (1998)
【発明の概要】
【発明が解決しようとする課題】
【0019】
しかしこれらのポリマーマトリックスは、難水溶性を示し、または高分子電解質であり、即ち狭いpH範囲でのみ水溶性であってもよいポリマーである。
【0020】
さらに文献データは、これら複合体の安定性に関する定量データを報告していない。
【課題を解決するための手段】
【0021】
上記引用された理由により、安定化非晶質形態をγシクロデキストリンと組み合わせた使用を通してマクロライドの溶解度および安定性を増大させる可能性が、探究された。
【図面の簡単な説明】
【0022】
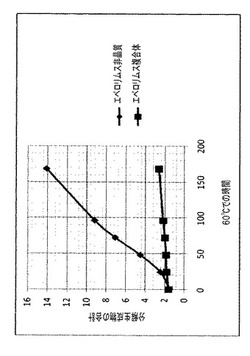
【図1】非晶質ラパマイシンおよび結晶質ラパマイシンのクロマトグラフィ純度(HPLCデータ)の低下を示す図である(60℃で17日間(408時間))。
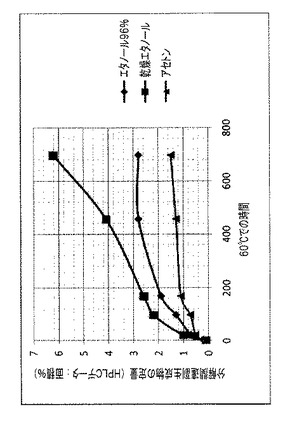
【図2】種々の溶媒:無水エタノール、エタノール96%、およびアセトンを使用して調製されたラパマイシンγCD/複合体の安定性を示す図である。
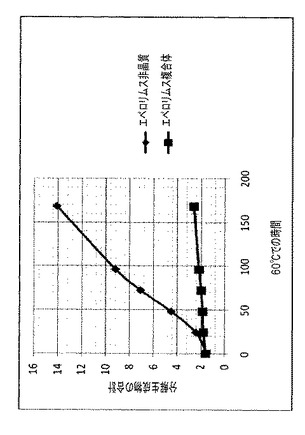
【図3】非晶質エベロリムスに対する、溶媒としてアセトンを使用して調製されたエベロリムスγCD/複合体の安定性を示す図である。
【図4】ラパマイシンγCD複合体のHPLC/MSプロファイルを示す図である。
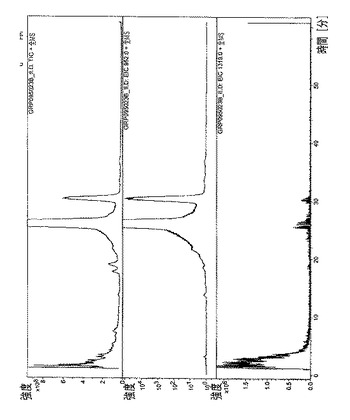
【図5】エベロリムスγCD複合体のHPLC/MSプロファイルを示す図である。
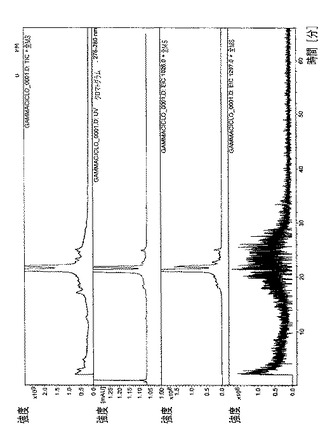
【図6】テムシロリムスγCD複合体のHPLC/MSプロファイルを示す図である。
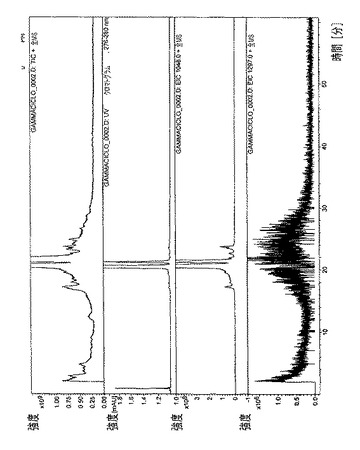
【図7】ピメクロリムスγCD複合体のHPLC/MSプロファイルを示す図である。
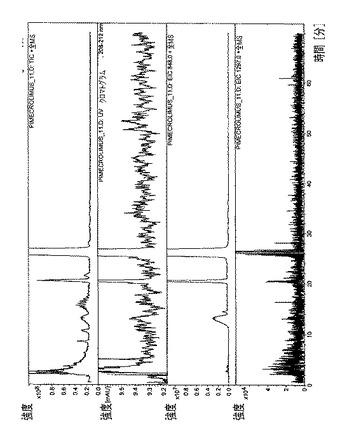
【図8】タクロリムスγCD複合体のHPLC/MSプロファイルを示す図である。
【図9】ラパマイシンのHPLC/UVおよびHPLC/MSプロファイルを示す図である。
【図10】エベロリムスのHPLC/UVおよびHPLC/MSプロファイルを示す図である。
【図11】テムシロリムスのHPLC/UVおよびHPLC/MSプロファイルを示す図である。
【図12】ピメクロリムスのHPLC/UVおよびHPLC/MSプロファイルを示す図である。
【図13】タクロリムスのHPLC/UVおよびHPLC/MSプロファイルを示す図である。
【図14】噴霧乾燥器で処理したラパマイシン(出発材料)のDRXを示す図である。
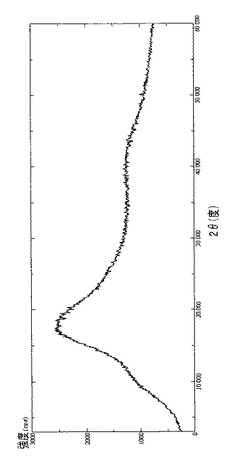
【図15】噴霧乾燥器で処理したピメクロリムス(出発材料)のDRXを示す図である。
【図16】噴霧乾燥器で処理したテムシロリムス(出発材料)のDRXを示す図である。
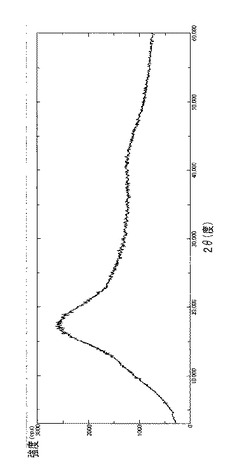
【図17】噴霧乾燥器で処理したタクロリムス(出発材料)のDRXを示す図である。
【図18】噴霧乾燥器で処理したエベロリムス(出発材料)のDRXを示す図である。
【図19】γCD(出発材料)のDRXを示す図である。
【図20】結晶質ラパマイシン(出発材料)のDRXを示す図である。
【図21】ラパマイシンγCDシクロデキストリン複合体のDRXを示す図である。
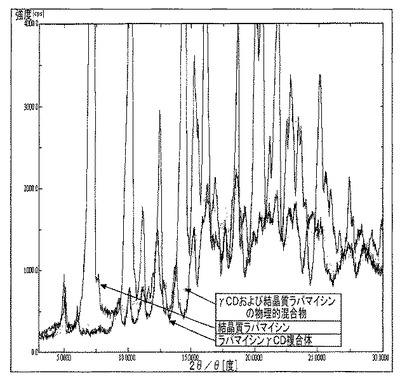
【図22】γCDおよびラパマイシン(0.6%w/w)の物理的混合物のDRXを示す図である。
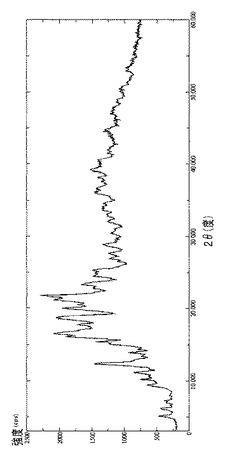
【図23】γCDおよび結晶質ラパマイシンの物理的混合物(0.6%w/w;緑色)と、結晶質ラパマイシンのDRX(赤色)と、ラパマイシンγCD複合体のDRX(青色)との、3から30 2θの範囲における、重ね合わせた拡大DRCスペクトルを示す図である。
【発明を実施するための形態】
【0023】
下記の定義は、本明細書および特許請求の範囲全体を通して使用される。
【0024】
「αシクロデキストリン」または「αCD」という用語は、シクロマルトヘキサオースとも呼ばれる、登録番号10016−20−3で特定された化合物を指す。
【0025】
「βシクロデキストリン」または「βCD」という用語は、シクロマルトヘプタオースとも呼ばれる、登録番号7585−39−9で特定された化合物を指す。「γシクロデキストリン」または「γCD」という用語は、シクロマルトオクタオースとも呼ばれる、登録番号17465−86−0で特定された化合物を指す。
【0026】
「非晶質」という用語は、非結晶質である化合物の固体状態を指す。
【0027】
本明細書で使用される「マクロライド」という用語は、ラパマイシン、テムシロリムス、エベロリムス、ピメクロリムス、およびタクロリムスを指す。
【0028】
本発明の目的は、包接複合体の形成によって、シクロデキストリンに吸着された場合のマクロライドの安定性および溶解度の両方を改善することであった。
【0029】
当業者に周知のように、これらの複合体は、水溶性、安定性、またはバイオアベイラビリティの増大など、ゲスト分子に比べて変化した物理化学的性質をしばしば示す。
【0030】
したがって本発明の目的は、製剤の目的で通常用いられる付随材料(accompanying material)の毒性をおそらくは低下させながら、製剤の目的で使用してもよい、γシクロデキストリンとのマクロライドの包接複合体を提供することである。
【0031】
事実、驚くべきことに、γシクロデキストリンは、マクロライドの安定性に良好な作用を示し、かつ共溶媒として有機溶媒を使用することなく過飽和水溶液からの沈殿を防止することがわかった。
【0032】
したがって、本発明の別の目的は、いかなる有機溶媒も含有しないこれら複合体の水溶液である。
【0033】
本発明の別の目的は、経口、眼部、局所、および注射可能な製剤を調製するためのこれら複合体の使用、ならびに免疫抑制剤としてのそのような複合体および/または製剤の使用である。
【0034】
製剤の研究を進めるために、本発明者らは、微粉化したラパマイシン(10-5ミクロン)および溶液(エタノール)にしたものに対して予備安定性試験を行った。
【0035】
本発明者らにより得られた結果は、細かく微粉化されまた溶液状態になったこの生成物が、不安定であることを裏付け:数時間以内で、本発明者らは感知可能なアッセイ値の低下(−10%)を観察した。
【0036】
分解から、非晶質ラパマイシンまたは細かく微粉化されたラパマイシンを安定化させるため、本発明者らは、いくつかの試みを行ったが成功しなかった:αトコフェロールおよびアスコルビン酸などの抗酸化剤の添加は、不活性雰囲気(窒素)下および冷蔵条件(0〜4℃)での作業であっても、この分解を制御することができなかった。
【0037】
この挙動は、20ミクロンよりも小さい微粉化ラパマイシン、非晶質ラパマイシン、および溶液状態でのみ観察され、より大きな粒度(即ち、>100ミクロン)の結晶質ラパマイシンでは観察されなかった。
【0038】
表1および対応する図1では、非晶質および結晶質(粒度>100ミクロンの生成物)形態のラパマイシンに関して60℃で得られた安定性データを報告し:結晶質ラパマイシンは408時間(17日)後に同じクロマトグラフィ純度を示すが、非晶質ラパマイシンは、クロマトグラフィ純度に−14%の低下を示す。
【0039】
【表1】
【0040】
本発明者らは、ラパマイシンのいくつかの合成誘導体(即ち、テムシロリムスおよびエベロリムス)で、およびピメクロリムスおよびタクロリムスでも同じ分解が生じることを確認した。
【0041】
これら安定性の問題を克服するために、本発明者らは、市販のシクロデキストリンと、このファミリーのいくつかの化合物の中でαシクロデキストリン(αCD)、βシクロデキストリン(βCD)、およびγシクロデキストリン(γCD)との複合体の形成を通して、分解からこれらマクロライドを安定化させる可能性について研究することを決定した。これら環状オリゴ糖は、1→4結合された6、7、および8個のα−D−グルコピラノシド単位からなるという共通の特徴を有する。
【0042】
シクロデキストリンの存在下での、これらマクロライドのHPLCクロマトグラフィ移動度に関する予備研究は、これらマクロライドとシクロデキストリンとの相互作用が非常に弱いことを確認する。
【0043】
本発明者により評価された第1のマクロライドは、αシクロデキストリン(αCD)、βシクロデキストリン(βCD)、およびγシクロデキストリン(γCD)に吸着されたラパマイシンであった。
【0044】
これらの複合体を調製するために、ラパマイシンを、有機溶媒に、好ましくは、アセトン、メタノール、およびエタノールの間から選択された極性有機溶媒に溶解し、次いでこれら溶液をシクロデキストリンと混合した。水溶性複合体を得るために、ラパマイシン、またはその誘導体の1種と、シクロデキストリンとの重量比は、有利には、1:100から1:400の間、より好ましくは1:111から1:333の間に含まれている。次いで得られた不均一混合物を、真空中で慎重に蒸発させて、複合体を固体粉末として得、次いで湿潤固体粉末を真空乾燥した。
【0045】
ラパマイシンおよびシクロデキストリンの不均一混合物の真空蒸発の適切な代替例は、噴霧乾燥器技法で、懸濁液の直接濾過によって、または凍結乾燥によって実現させた。前記濾過は、前記不均一混合物を有機溶媒で、好ましくは非極性有機溶媒で、より好ましくはC5〜C8直鎖状または分岐状炭化水素で希釈することによって、任意選択で行うことができる。
【0046】
得られた複合体を、まず、20〜25℃の溶液中および−20℃で貯蔵された固体粉末として、安定性に関して評価した。
【0047】
溶液(水/アセトニトリル 1/1 v/v混合物)中での安定性データは、評価されたシクロデキストリン複合体の中で、γCD複合体は、時間ゼロでラパマイシンのより高い含量を示し;さらにγCDおよびβCD複合体は、20〜25℃の溶液中で19日後に、初期アッセイ値からの低下を示さないが、同じ貯蔵条件において、αCD複合体は、アッセイ値に−24%の低下を示す(実験セクションの表2)ことを示している。
【0048】
26日後に、−20℃で貯蔵された固体複合体(粉末)で得られた安定性データは、αCDに関してアッセイ値に−12%の低下を示し、βCDに関して−5.4%の低下を示すが、γCD複合体のラパマイシン含量は、同じ実験条件で変化しない(実験セクションの表3)。
【0049】
驚くべきことに、1および2個のα−D−グルコピラノシド単位のみ異なる用いられたシクロデキストリンの化学的類似性にも関わらず、ラパマイシンとのγCD複合体のみが、溶液中および固体形態の両方で良好な安定性データを示した。
【0050】
これら予備データはさらに、固体状態(粉末)で、異なる貯蔵条件:25℃(相対湿度60%)および40℃(相対湿度75%)で5および15日後(実験セクションの表4および5)で確認した。
【0051】
したがって、ラパマイシンγCD複合体が、25℃および40℃の両方でαおよびβCD複合体よりも安定であっただけではなく、40℃でのγCD複合体からのラパマイシンの放出は、ほぼ定量的であることを確認した(CDに投入されたラパマイシンの回収の95%)。
【0052】
これらのデータに基づいて、本発明者らは驚くべきことに、本発明者らの実験条件で、γCDのみが溶液状態および固体状態の両方で分解からラパマイシンを安定化させることができ、一方αおよびβCD複合体とラパマイシンとの複合体は、不安定であることを見出した。
【0053】
文献データは、γCDとの不安定な複合体の形成を通した分解からラパマイシンを安定化させる可能性について、記述していない。さらに、γシクロデキストリンのこの独特の挙動は、構造的観点から非常に類似しておりかつしばしば製剤目的で利用するのに等価であると見なされる親化合物αおよびβCDとは、全く異なっている。
【0054】
γCDとの複合体を通した、分解に対してラパマイシンを安定化させるこの可能性について、本発明者らはエベロリムス、テムシロリムス、タクロリムス、およびピメクロリムスに関してさらに探究し、これら構造的に関連ある化合物は共通する挙動を有することを示しており;一方、非晶質形態または細かく微粉化された(即ち、粒度分布が10-5ミクロンである、)純粋な化合物は不安定であり、対応するγCD複合体は溶液状態および固体状態の両方で安定であることを示している。
【0055】
図3および表7において、本発明者らは、エベロリムス複合体対非晶質エベロリムスの安定性データを報告し:60℃で96時間後の、エベロリムス複合体に関する分解関連副生成物の増加は+36%であり、一方、非晶質エベロリムスに関しては+513%である。類似データが、γCDテムシロリムス複合体およびγCDピメクロリムス複合体に関して得られた。
【0056】
これら複合体に関して行われた分析データは、HPLC/MS、HPLC/UVによる特徴付け(図4〜13参照)およびX線回折データ(DRX;図14〜23参照)を含み:これら分析データは、用いられた吸収技法がマクロライドおよび固体状態のγCDの純度プロファイルを変更せず、一方、溶媒処理後に評価したマクロライドの全ては、大部分が非晶質形態であることを裏付けている。得られた複合体から生成されたDRXデータは、マクロライドが、その大部分を非晶質形態として存在していることを裏付けている。
【0057】
最後に、試験がなされたγCDとのマクロライド複合体の全てに関し、本発明者らは、下記の表8に示されるように、当初のマクロライドからの、水溶性の感知可能な増加を確認した。
【0058】
【表2】
【0059】
実験セクション
材料および方法
ラパマイシン、テムシロリムス、エベロリムス、タクロリムス、およびピメクロリムスを、POLI INDUSTRIA CHIMICA SpAにより調製した。αシクロデキストリン(αCD)、βシクロデキストリン(βCD)、およびγシクロデキストリン(γCD)は、Flukaから購入した。
【0060】
ラパマイシン、テムシロリムス、エベロリムス、タクロリムス、およびピメクロリムスに関するアッセイおよび純度を測定するのに使用されるHPLC法を、本明細書で報告する。
【0061】
ラパマイシンのクロマトグラフィ純度のHPLC測定:カラム:Thermo BDS Hypersil C18;3μm(100×4.6mm)。移動相組成物:定組成溶液
・成分A:アセトニトリル50%
・成分B:酢酸アンモニウム緩衝液pH5.8(酢酸アンモニウムの濃度0.8g/l;最終pH値は、氷酢酸で5.8の最終値にした。)50%
流量:1.0ml/分。カラム温度:55℃。注入体積:100μl。サンプル溶液:ラパマイシン25mgを計量し、アセトニトリル/水 1/1 v/v 100ml(最終濃度0.25mg/ml)に溶解した。UV検出器:278nm
【0062】
ラパマイシンのHPLCアッセイ測定:カラム:Hypersil BDS−C18;3μm(100×4.6mm)。移動相組成物:定組成溶液
・成分A:アセトニトリル58%
・成分B:酢酸アンモニウム緩衝液pH5.8(酢酸アンモニウムの濃度0.8g/l;最終pH値は、氷酢酸で5.8の最終値にした。)42%
流量:1.5ml/分。カラム温度:55℃。注入体積:30μl
サンプルおよび標準溶液:アセトニトリル/水 1/1中、0.25mg/mlの濃度で母液を調製し、次いで移動相で希釈して、20マイクログラム/mlの最終濃度を得た。UV検出器:278nm。
【0063】
テムシロリムスおよびエベロリムスのクロマトグラフィ純度のHPLC測定:カラム:Zorbax SB−C18;3.5μm(75×4.6mm)、プレカラム:Symmetry Shield RP18;5μm(20×3.9mm)。
移動相組成物:
・成分A:水900ml、アセトニトリル100ml、および50%酢酸水溶液50μl・成分B:アセトニトリル1000mlおよび50%酢酸水溶液50μl
勾配溶出:
【0064】
【表3】
【0065】
ピメクロリムスのクロマトグラフィ純度のHPLC測定:カラム:YMC ODS AQ、5μm(250×4.6mm)。移動相組成物:70/30アセトニトリル/0.01Mリン酸緩衝液(pH2.5)で定組成溶出。流量:1.2ml/分。カラム温度:60℃。サンプル温度10℃。注入体積:10μl。サンプル溶液:アセトニトリルに溶かしたピメクロリムスの0.5mg/ml溶液を調製。検出器UV:210nm
【0066】
タクロリムスのクロマトグラフィ純度のHPLC測定:カラム:Symmetry C18;3.5μm(150×2.1mm)。移動相組成物:定組成溶出
・成分A:0.1%酢酸水溶液58%
・成分B:アセトニトリル15%
・成分C:テトラヒドロフラン27%
・流量:0.3ml/分。カラム温度:50℃。注入体積:10μl。
サンプル溶液:タクロリムス25mgを計量し、アセトニトリル/水 1/1 v/v 25ml(最終濃度1mg/ml)に溶解した。検出器UV:278nm
利用した質量分光計は、陽イオン化におけるイオントラップAgilent Mod.6300である。
【0067】
利用した噴霧乾燥装置は、Advance不活性ループB−295機器を備えたBuchiモデルB290である。
【0068】
DRXスペクトル(粉末)を、5000から60000の開始角[1/2 2θ]から、回折計(PW1710 Philips)を使用して記録した。回折ダイアグラムを、サンプルのいかなる処理も行わずにCuアノードを用いて得た(Kα=1.54060ÅおよびKα=1.54439Å)。
【実施例1】
【0069】
乾燥による再構成可能な固体ラパマイシン/シクロデキストリン複合体の調製(溶媒としてエタノール95%)
エタノール(96%)に溶かしたラパマイシンの0.4%w/v溶液を、シクロデキストリン粉末に添加した。ラパマイシンとシクロデキストリンとの最終的な相対比を、表2および3に報告する。
【0070】
この懸濁液を、30分間撹拌しながら20〜25℃に維持し、次いで得られた混合物を、エタノールを除去するために18時間真空乾燥した。乾燥固体粉末を、窒素雰囲気下で−20℃で貯蔵した。
【0071】
表2において、溶液状態(1/1 水/アセトニトリル混合物)にあるラパマイシン/CD複合体のアッセイデータ(HPLCデータ)を、20〜25℃で、t=0および19日後に報告する。
【0072】
【表4】
【0073】
【表5】
【0074】
【表6】
【0075】
【表7】
【実施例2】
【0076】
乾燥による再構成可能な固体ラパマイシン/シクロデキストリン複合体の調製(溶媒としてアセトン)
アセトンにラパマイシンを溶かした0.4%w/v溶液を、γシクロデキストリンに添加した。ラパマイシンとシクロデキストリンとの最終的な相対比は、0.6%w/wであった。得られた懸濁液を、撹拌しながら30分間20〜25℃に維持した。得られた混合物を、アセトンを除去するために18時間真空乾燥した。任意選択で、湿潤固体を、n−ヘプタンを用いた懸濁液の希釈および濾過によって回収してもよく;次いで乾燥器内に移してもよい。
【0077】
乾燥固体粉末を、窒素雰囲気下で−20℃で貯蔵した。
【0078】
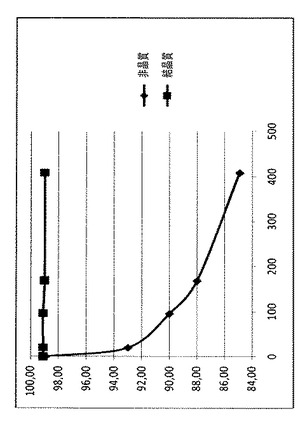
60℃での、得られたラパマイシン/γCD複合体(粉末)の安定性データを、乾燥エタノールおよびエタノール96%を溶媒として使用して得られたものと比較し;得られたデータを、図2および表6にまとめた。特に、図2では、60℃で696時間、粉末に関して行われた安定性試験中、分解によって生じた関連ある副生成物の合計(HPLCデータ)を報告する。
【0079】
【表8】
【実施例3】
【0080】
噴霧乾燥器による再構成可能な固体ラパマイシン/シクロデキストリン複合体の調製(溶媒としてアセトン)
アセトンに溶かしたラパマイシンの0.1%w/v溶液を、γシクロデキストリンに添加した。
【0081】
ラパマイシンとシクロデキストリンとの最終的な相対比は、0.6%w/wであった。得られた懸濁液を、撹拌しながら30分間20〜25℃に維持した。
【0082】
得られた懸濁液を、下記の実験条件で噴霧乾燥器で処理した:
・不活性窒素温度=70℃
・供給速度=17ml/分
・出口窒素温度=54℃
・アスピレーター速度=80%
・ノズルクリーニングの頻度=2ショット/秒
【0083】
得られた固体粉末を30℃で8時間乾燥し、次いで窒素雰囲気下で−20℃で貯蔵した。この複合体の特徴は、実施例2で得られた生成物に関して記述したものと同じである。
【0084】
γCDエベロリムス複合体、γCDテムシロリムス複合体、γCDタクロリムス複合体、およびγCDピメクロリムス複合体の調製
これらの複合体を、実施例2および3のそれぞれに従い調製した。
【0085】
γCDラパマイシン複合体、γCDエベロリムス複合体、γCDテムシロリムス複合体、γCDピメクロリムス複合体、γCDタクロリムス複合体、および出発材料として利用される、対応するマクロライドのHPLC UVおよびMSデータ
全ての複合体のHPLCプロファイル(HPLCおよびUV検出器)を、図4〜8で収集した。各複合体ごとに、本発明者らは、実験セクションで記述された特定のHPLCクロマトグラフィ条件を利用した。
・図4.ラパマイシンγCD複合体=全イオン電流(TIC)、m/z=952での単イオン(カリウムとのラパマイシンの分子イオンの付加体;m/z=952)、m/z=1319での単イオン(ナトリウムとのγシクロデキストリンの付加体;m/z=1319)
・図5.エベロリムスγCD複合体=全イオン電流(TIC)、スペクトルUV(278nm)、m/z=1026での単イオン(カリウムとのエベロリムスの分子イオンの付加体)、m/z=1297での単イオン(γシクロデキストリンの分子イオン)
・図6.テムシロリムスγCD複合体=全イオン電流(TIC)、スペクトルUV(278nm)、m/z=1048での単イオン(アンモニアとのテムシロリムスの分子イオンの付加体)、m/z=1297での単イオン(γシクロデキストリンの分子イオン)
・図7.ピメクロリムスγCD複合体=全イオン電流(TIC)、スペクトルUV(210nm)、m/z=848での単イオン(カリウムとのピメクロリムスの分子イオンの付加体)、m/z=1297での単イオン(γシクロデキストリンの分子イオン)
・図8.タクロリムスγCD複合体=全イオン電流(TIC)、スペクトルUV(220nm)、m/z=1297での単イオン(γシクロデキストリンの分子イオン)、m/z=822での単イオン(水とのタクロリムスの分子イオンの付加体)
【0086】
t=0での、これらマクロライド複合体のHPLC UVおよびMSクロマトグラフィプロファイルは、出発材料として利用された対応するマクロライドとは変わらないことがわかった。
【0087】
CD複合体の調製で出発材料として利用された各マクロライドのHPLC/MSおよびHPLC/UV分析を、本明細書に報告する。
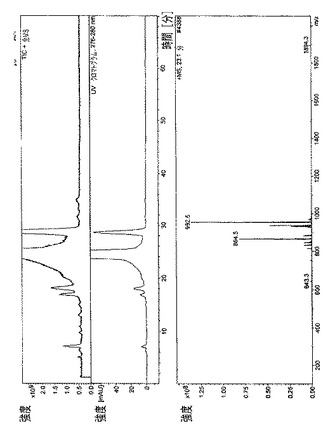
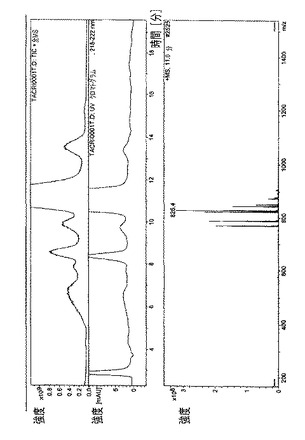
・図9.ラパマイシン=23分で溶出された主なラパマイシン異性体の、全イオン電流(TIC)、HPLC/UVプロファイル、および質量スペクトル。
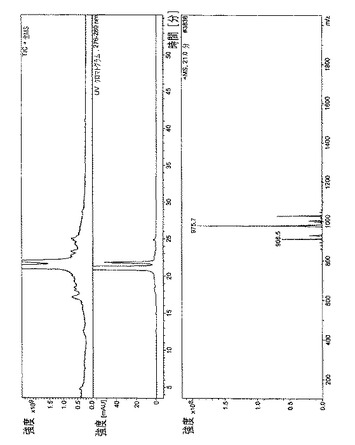
・図10.エベロリムス=21分で溶出された主なエベロリムス異性体の、全イオン電流(TIC)、HPLC/UVプロファイル、および質量スペクトル。
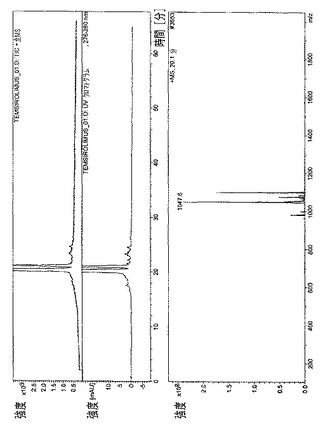
・図11.テムシロリムス=20分で溶出された主なテムシロリムス異性体の、全イオン電流(TIC)、HPLC/UVプロファイル、および質量スペクトル。
・図12.ピメクロリムス=27分で溶出された主なピメクロリムス異性体の、全イオン電流(TIC)、HPLC/UVプロファイル、および質量スペクトル。
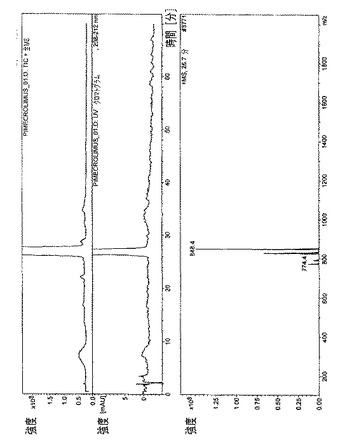
・図13.タクロリムス=11分0秒で溶出された主なタクロリムス異性体の、全イオン電流(TIC)、HPLC/UVプロファイル、および質量スペクトル(同じく掲載のスペクトルにおいてm/z=822で水付加体およびm/z=826でナトリウムとの付加体が確認できる)。
【0088】
γCDとのエベロリムス複合体の安定性
実施例2および3のそれぞれにより調製されたCDとのエベロリムス複合体に関する安定性データを、図3および表7に報告する。
【0089】
特に、図3には、60℃で96時間、粉末で行われた安定性試験中の、分解によって生じた関連副生成物の合計(面積%を単位とするHPLCデータ)が報告されている。
【0090】
【表9】
【0091】
γラパマイシン複合体、γCDエベロリムス複合体、γCDテムシロリムス複合体、γCDタクロリムス複合体、およびγCDピメクロリムス複合体の固体状態
ラパマイシン結晶質を噴霧乾燥器で処理した後(またはエタノールもしくはアセトン溶液から蒸発させた後)の、またγCD(出発材料)、ラパマイシンγCD複合体、結晶質ラパマイシン(出発材料)と、ピメクロリムス、テムシロリムス、タクロリムス、およびエベロリムスを噴霧乾燥器で処理した後(またはエタノールもしくはアセトン溶液から蒸発させた後)の固体状態を、図14〜23に報告する。これらの分析は、エタノールもしくはアセトン溶液からの蒸発後、これらマクロライドが非晶質形態にあることを裏付けている。γCDとの複合体のDRXスペクトルは、図22および下記の表9の両方で明示されるように、γCDの結晶質相が変化していないことを裏付けており、用いられた技法の分解能によれば、結晶質形態には微量のマクロライドも存在しない。
【0092】
【表10】
【技術分野】
【0001】
本発明は、マクロライドおよびある特定のシクロデキストリンを含有する、経口、眼部(ophthalmic)、局所、または非経口使用のための新しい水溶性固体医薬包接複合体およびその水溶液に関する。
【背景技術】
【0002】
マクロライドは、その活性が、マクロライド環、即ち1種または複数のデオキシ糖が結合されていてもよい大きな大環状ラクトン環、が存在することで生じる薬物(典型的には、抗生物質)の群である。ラクトン環は、通常、14、15、または16員である。マクロライドは、天然物のポリケチドのクラスに属する。このファミリーの中で、登録番号53123−88−9によって特定される、式1のラパマイシンとしても知られるシロリムスと、登録番号104987−11−3によって特定される式2のタクロリムスは、その免疫抑制活性で知られる化合物であり、体内での臓器および骨髄移植の拒絶を予防するのに使用される。
【0003】
【化1】
【0004】
テムシロリムス、エベロリムス、およびピメクロリムス(アスコマイシンの合成誘導体)などのラパマイシン誘導体は、細胞の成長、増殖、および生存の制御に関与する特定のタンパク質(mTOR)に関して、類似した阻害活性を示す。
【0005】
登録番号162635−04−3によって特定される式3のテムシロリムスは、ラパマイシンに構造的に関係しており、進行性腎細胞癌(腎癌の一タイプ)の治療に使用される。このテムシロリムスは、その他のタイプの癌の治療においても研究されている。
【0006】
【化2】
【0007】
登録番号159351−69−6によって特定される式4のエベロリムスは、ある特定のその他の抗癌剤での治療に応答しない進行性腎癌を、治療するのに使用される。このエベロリムスは、ワルデンシュトレームマクログロブリン血症または乳癌のようなその他のタイプの癌の治療においても、研究されている。
【0008】
【化3】
【0009】
登録番号137071−32−0によって特定された式5のピメクロリムス、アスコマイシンの合成誘導体は;アトピー性皮膚炎の治療に使用される。
【0010】
【化4】
【0011】
これら化合物の全ては、0.01から0.000006モル/Lの範囲である、室温での不十分な水溶性を有し、貯蔵後の溶液中では不安定でありかつそのエステル結合の加溶媒分解の結果としてin vitroおよびin vivoの両方において生物活性の損失をもたらすことが報告されている(非特許文献1および2参照)。
【0012】
さらに、固体状態では、これらの化合物は、非晶質または結晶質形態で存在してもよく、この非晶質は非常に不安定で酸化分解する(非特許文献3、4、および5参照)。
【0013】
非晶質形態が、結晶質形態と比較した場合により高い溶解度を有し(結晶質から非晶質材料までの溶解度の上昇は、10から1600倍の間であることが報告されている。)、安定性が低く、分解する傾向があることは、常識である。
【0014】
さらに、低い溶解度は、通常は、体内での不十分な吸収および不十分な利用可能性に関連している。特許文献1は、シクロデキストリンと組み合わせて水にやや溶け難い薬物を溶かした水性非経口溶液が、注射部位または非経口施用後の臓器での薬物沈殿を最小限に抑えることができることを示唆している。
【0015】
特許文献2は、αまたはβシクロデキストリン上のラパマイシンの固体分散体の形をとる、経口投与用の医薬組成物を調製する可能性について記述している。
【0016】
実際に、文献データは、細かく微粉化されたラパマイシンまたは非晶質ラパマイシンを、特許文献3、4、5、および6に記載されているように、ポリブチルメタクリレート(PBMA)、ポリエチレン−co−ビニルアセテート(PEVA)、または硫酸プロタミンおよびセルロースなどの高分子電解質複合体のようないくつかの合成ポリマーの使用を見越した種々のコーティング技法を用いる製剤目的のために安定化させる必要があることを裏付けている。
【先行技術文献】
【特許文献】
【0017】
【特許文献1】米国特許第5024998号明細書
【特許文献2】EP839028
【特許文献3】国際公開第2006026531号パンフレット
【特許文献4】国際公開第2006039237号パンフレット
【特許文献5】国際公開第2007011708号パンフレット
【特許文献6】EP2135601
【非特許文献】
【0018】
【非特許文献1】Yuri V. Il'ichev, Lori Alquier, and Cynthia A. Maryanoff, ARKIVOC, 2007 (XII) 110-131
【非特許文献2】Ping Cai, Rushung Tsao, and Mark E. Ruppen, DMD Fast Forward. May 31, 2007
【非特許文献3】Tetrahedron Letters (1990), 31(34), 4845-8
【非特許文献4】Xenobiotica, 27(9), 869 (1997)
【非特許文献5】J. Org. Chem., 63, 10069, (1998)
【発明の概要】
【発明が解決しようとする課題】
【0019】
しかしこれらのポリマーマトリックスは、難水溶性を示し、または高分子電解質であり、即ち狭いpH範囲でのみ水溶性であってもよいポリマーである。
【0020】
さらに文献データは、これら複合体の安定性に関する定量データを報告していない。
【課題を解決するための手段】
【0021】
上記引用された理由により、安定化非晶質形態をγシクロデキストリンと組み合わせた使用を通してマクロライドの溶解度および安定性を増大させる可能性が、探究された。
【図面の簡単な説明】
【0022】
【図1】非晶質ラパマイシンおよび結晶質ラパマイシンのクロマトグラフィ純度(HPLCデータ)の低下を示す図である(60℃で17日間(408時間))。
【図2】種々の溶媒:無水エタノール、エタノール96%、およびアセトンを使用して調製されたラパマイシンγCD/複合体の安定性を示す図である。
【図3】非晶質エベロリムスに対する、溶媒としてアセトンを使用して調製されたエベロリムスγCD/複合体の安定性を示す図である。
【図4】ラパマイシンγCD複合体のHPLC/MSプロファイルを示す図である。
【図5】エベロリムスγCD複合体のHPLC/MSプロファイルを示す図である。
【図6】テムシロリムスγCD複合体のHPLC/MSプロファイルを示す図である。
【図7】ピメクロリムスγCD複合体のHPLC/MSプロファイルを示す図である。
【図8】タクロリムスγCD複合体のHPLC/MSプロファイルを示す図である。
【図9】ラパマイシンのHPLC/UVおよびHPLC/MSプロファイルを示す図である。
【図10】エベロリムスのHPLC/UVおよびHPLC/MSプロファイルを示す図である。
【図11】テムシロリムスのHPLC/UVおよびHPLC/MSプロファイルを示す図である。
【図12】ピメクロリムスのHPLC/UVおよびHPLC/MSプロファイルを示す図である。
【図13】タクロリムスのHPLC/UVおよびHPLC/MSプロファイルを示す図である。
【図14】噴霧乾燥器で処理したラパマイシン(出発材料)のDRXを示す図である。
【図15】噴霧乾燥器で処理したピメクロリムス(出発材料)のDRXを示す図である。
【図16】噴霧乾燥器で処理したテムシロリムス(出発材料)のDRXを示す図である。
【図17】噴霧乾燥器で処理したタクロリムス(出発材料)のDRXを示す図である。
【図18】噴霧乾燥器で処理したエベロリムス(出発材料)のDRXを示す図である。
【図19】γCD(出発材料)のDRXを示す図である。
【図20】結晶質ラパマイシン(出発材料)のDRXを示す図である。
【図21】ラパマイシンγCDシクロデキストリン複合体のDRXを示す図である。
【図22】γCDおよびラパマイシン(0.6%w/w)の物理的混合物のDRXを示す図である。
【図23】γCDおよび結晶質ラパマイシンの物理的混合物(0.6%w/w;緑色)と、結晶質ラパマイシンのDRX(赤色)と、ラパマイシンγCD複合体のDRX(青色)との、3から30 2θの範囲における、重ね合わせた拡大DRCスペクトルを示す図である。
【発明を実施するための形態】
【0023】
下記の定義は、本明細書および特許請求の範囲全体を通して使用される。
【0024】
「αシクロデキストリン」または「αCD」という用語は、シクロマルトヘキサオースとも呼ばれる、登録番号10016−20−3で特定された化合物を指す。
【0025】
「βシクロデキストリン」または「βCD」という用語は、シクロマルトヘプタオースとも呼ばれる、登録番号7585−39−9で特定された化合物を指す。「γシクロデキストリン」または「γCD」という用語は、シクロマルトオクタオースとも呼ばれる、登録番号17465−86−0で特定された化合物を指す。
【0026】
「非晶質」という用語は、非結晶質である化合物の固体状態を指す。
【0027】
本明細書で使用される「マクロライド」という用語は、ラパマイシン、テムシロリムス、エベロリムス、ピメクロリムス、およびタクロリムスを指す。
【0028】
本発明の目的は、包接複合体の形成によって、シクロデキストリンに吸着された場合のマクロライドの安定性および溶解度の両方を改善することであった。
【0029】
当業者に周知のように、これらの複合体は、水溶性、安定性、またはバイオアベイラビリティの増大など、ゲスト分子に比べて変化した物理化学的性質をしばしば示す。
【0030】
したがって本発明の目的は、製剤の目的で通常用いられる付随材料(accompanying material)の毒性をおそらくは低下させながら、製剤の目的で使用してもよい、γシクロデキストリンとのマクロライドの包接複合体を提供することである。
【0031】
事実、驚くべきことに、γシクロデキストリンは、マクロライドの安定性に良好な作用を示し、かつ共溶媒として有機溶媒を使用することなく過飽和水溶液からの沈殿を防止することがわかった。
【0032】
したがって、本発明の別の目的は、いかなる有機溶媒も含有しないこれら複合体の水溶液である。
【0033】
本発明の別の目的は、経口、眼部、局所、および注射可能な製剤を調製するためのこれら複合体の使用、ならびに免疫抑制剤としてのそのような複合体および/または製剤の使用である。
【0034】
製剤の研究を進めるために、本発明者らは、微粉化したラパマイシン(10-5ミクロン)および溶液(エタノール)にしたものに対して予備安定性試験を行った。
【0035】
本発明者らにより得られた結果は、細かく微粉化されまた溶液状態になったこの生成物が、不安定であることを裏付け:数時間以内で、本発明者らは感知可能なアッセイ値の低下(−10%)を観察した。
【0036】
分解から、非晶質ラパマイシンまたは細かく微粉化されたラパマイシンを安定化させるため、本発明者らは、いくつかの試みを行ったが成功しなかった:αトコフェロールおよびアスコルビン酸などの抗酸化剤の添加は、不活性雰囲気(窒素)下および冷蔵条件(0〜4℃)での作業であっても、この分解を制御することができなかった。
【0037】
この挙動は、20ミクロンよりも小さい微粉化ラパマイシン、非晶質ラパマイシン、および溶液状態でのみ観察され、より大きな粒度(即ち、>100ミクロン)の結晶質ラパマイシンでは観察されなかった。
【0038】
表1および対応する図1では、非晶質および結晶質(粒度>100ミクロンの生成物)形態のラパマイシンに関して60℃で得られた安定性データを報告し:結晶質ラパマイシンは408時間(17日)後に同じクロマトグラフィ純度を示すが、非晶質ラパマイシンは、クロマトグラフィ純度に−14%の低下を示す。
【0039】
【表1】
【0040】
本発明者らは、ラパマイシンのいくつかの合成誘導体(即ち、テムシロリムスおよびエベロリムス)で、およびピメクロリムスおよびタクロリムスでも同じ分解が生じることを確認した。
【0041】
これら安定性の問題を克服するために、本発明者らは、市販のシクロデキストリンと、このファミリーのいくつかの化合物の中でαシクロデキストリン(αCD)、βシクロデキストリン(βCD)、およびγシクロデキストリン(γCD)との複合体の形成を通して、分解からこれらマクロライドを安定化させる可能性について研究することを決定した。これら環状オリゴ糖は、1→4結合された6、7、および8個のα−D−グルコピラノシド単位からなるという共通の特徴を有する。
【0042】
シクロデキストリンの存在下での、これらマクロライドのHPLCクロマトグラフィ移動度に関する予備研究は、これらマクロライドとシクロデキストリンとの相互作用が非常に弱いことを確認する。
【0043】
本発明者により評価された第1のマクロライドは、αシクロデキストリン(αCD)、βシクロデキストリン(βCD)、およびγシクロデキストリン(γCD)に吸着されたラパマイシンであった。
【0044】
これらの複合体を調製するために、ラパマイシンを、有機溶媒に、好ましくは、アセトン、メタノール、およびエタノールの間から選択された極性有機溶媒に溶解し、次いでこれら溶液をシクロデキストリンと混合した。水溶性複合体を得るために、ラパマイシン、またはその誘導体の1種と、シクロデキストリンとの重量比は、有利には、1:100から1:400の間、より好ましくは1:111から1:333の間に含まれている。次いで得られた不均一混合物を、真空中で慎重に蒸発させて、複合体を固体粉末として得、次いで湿潤固体粉末を真空乾燥した。
【0045】
ラパマイシンおよびシクロデキストリンの不均一混合物の真空蒸発の適切な代替例は、噴霧乾燥器技法で、懸濁液の直接濾過によって、または凍結乾燥によって実現させた。前記濾過は、前記不均一混合物を有機溶媒で、好ましくは非極性有機溶媒で、より好ましくはC5〜C8直鎖状または分岐状炭化水素で希釈することによって、任意選択で行うことができる。
【0046】
得られた複合体を、まず、20〜25℃の溶液中および−20℃で貯蔵された固体粉末として、安定性に関して評価した。
【0047】
溶液(水/アセトニトリル 1/1 v/v混合物)中での安定性データは、評価されたシクロデキストリン複合体の中で、γCD複合体は、時間ゼロでラパマイシンのより高い含量を示し;さらにγCDおよびβCD複合体は、20〜25℃の溶液中で19日後に、初期アッセイ値からの低下を示さないが、同じ貯蔵条件において、αCD複合体は、アッセイ値に−24%の低下を示す(実験セクションの表2)ことを示している。
【0048】
26日後に、−20℃で貯蔵された固体複合体(粉末)で得られた安定性データは、αCDに関してアッセイ値に−12%の低下を示し、βCDに関して−5.4%の低下を示すが、γCD複合体のラパマイシン含量は、同じ実験条件で変化しない(実験セクションの表3)。
【0049】
驚くべきことに、1および2個のα−D−グルコピラノシド単位のみ異なる用いられたシクロデキストリンの化学的類似性にも関わらず、ラパマイシンとのγCD複合体のみが、溶液中および固体形態の両方で良好な安定性データを示した。
【0050】
これら予備データはさらに、固体状態(粉末)で、異なる貯蔵条件:25℃(相対湿度60%)および40℃(相対湿度75%)で5および15日後(実験セクションの表4および5)で確認した。
【0051】
したがって、ラパマイシンγCD複合体が、25℃および40℃の両方でαおよびβCD複合体よりも安定であっただけではなく、40℃でのγCD複合体からのラパマイシンの放出は、ほぼ定量的であることを確認した(CDに投入されたラパマイシンの回収の95%)。
【0052】
これらのデータに基づいて、本発明者らは驚くべきことに、本発明者らの実験条件で、γCDのみが溶液状態および固体状態の両方で分解からラパマイシンを安定化させることができ、一方αおよびβCD複合体とラパマイシンとの複合体は、不安定であることを見出した。
【0053】
文献データは、γCDとの不安定な複合体の形成を通した分解からラパマイシンを安定化させる可能性について、記述していない。さらに、γシクロデキストリンのこの独特の挙動は、構造的観点から非常に類似しておりかつしばしば製剤目的で利用するのに等価であると見なされる親化合物αおよびβCDとは、全く異なっている。
【0054】
γCDとの複合体を通した、分解に対してラパマイシンを安定化させるこの可能性について、本発明者らはエベロリムス、テムシロリムス、タクロリムス、およびピメクロリムスに関してさらに探究し、これら構造的に関連ある化合物は共通する挙動を有することを示しており;一方、非晶質形態または細かく微粉化された(即ち、粒度分布が10-5ミクロンである、)純粋な化合物は不安定であり、対応するγCD複合体は溶液状態および固体状態の両方で安定であることを示している。
【0055】
図3および表7において、本発明者らは、エベロリムス複合体対非晶質エベロリムスの安定性データを報告し:60℃で96時間後の、エベロリムス複合体に関する分解関連副生成物の増加は+36%であり、一方、非晶質エベロリムスに関しては+513%である。類似データが、γCDテムシロリムス複合体およびγCDピメクロリムス複合体に関して得られた。
【0056】
これら複合体に関して行われた分析データは、HPLC/MS、HPLC/UVによる特徴付け(図4〜13参照)およびX線回折データ(DRX;図14〜23参照)を含み:これら分析データは、用いられた吸収技法がマクロライドおよび固体状態のγCDの純度プロファイルを変更せず、一方、溶媒処理後に評価したマクロライドの全ては、大部分が非晶質形態であることを裏付けている。得られた複合体から生成されたDRXデータは、マクロライドが、その大部分を非晶質形態として存在していることを裏付けている。
【0057】
最後に、試験がなされたγCDとのマクロライド複合体の全てに関し、本発明者らは、下記の表8に示されるように、当初のマクロライドからの、水溶性の感知可能な増加を確認した。
【0058】
【表2】
【0059】
実験セクション
材料および方法
ラパマイシン、テムシロリムス、エベロリムス、タクロリムス、およびピメクロリムスを、POLI INDUSTRIA CHIMICA SpAにより調製した。αシクロデキストリン(αCD)、βシクロデキストリン(βCD)、およびγシクロデキストリン(γCD)は、Flukaから購入した。
【0060】
ラパマイシン、テムシロリムス、エベロリムス、タクロリムス、およびピメクロリムスに関するアッセイおよび純度を測定するのに使用されるHPLC法を、本明細書で報告する。
【0061】
ラパマイシンのクロマトグラフィ純度のHPLC測定:カラム:Thermo BDS Hypersil C18;3μm(100×4.6mm)。移動相組成物:定組成溶液
・成分A:アセトニトリル50%
・成分B:酢酸アンモニウム緩衝液pH5.8(酢酸アンモニウムの濃度0.8g/l;最終pH値は、氷酢酸で5.8の最終値にした。)50%
流量:1.0ml/分。カラム温度:55℃。注入体積:100μl。サンプル溶液:ラパマイシン25mgを計量し、アセトニトリル/水 1/1 v/v 100ml(最終濃度0.25mg/ml)に溶解した。UV検出器:278nm
【0062】
ラパマイシンのHPLCアッセイ測定:カラム:Hypersil BDS−C18;3μm(100×4.6mm)。移動相組成物:定組成溶液
・成分A:アセトニトリル58%
・成分B:酢酸アンモニウム緩衝液pH5.8(酢酸アンモニウムの濃度0.8g/l;最終pH値は、氷酢酸で5.8の最終値にした。)42%
流量:1.5ml/分。カラム温度:55℃。注入体積:30μl
サンプルおよび標準溶液:アセトニトリル/水 1/1中、0.25mg/mlの濃度で母液を調製し、次いで移動相で希釈して、20マイクログラム/mlの最終濃度を得た。UV検出器:278nm。
【0063】
テムシロリムスおよびエベロリムスのクロマトグラフィ純度のHPLC測定:カラム:Zorbax SB−C18;3.5μm(75×4.6mm)、プレカラム:Symmetry Shield RP18;5μm(20×3.9mm)。
移動相組成物:
・成分A:水900ml、アセトニトリル100ml、および50%酢酸水溶液50μl・成分B:アセトニトリル1000mlおよび50%酢酸水溶液50μl
勾配溶出:
【0064】
【表3】
【0065】
ピメクロリムスのクロマトグラフィ純度のHPLC測定:カラム:YMC ODS AQ、5μm(250×4.6mm)。移動相組成物:70/30アセトニトリル/0.01Mリン酸緩衝液(pH2.5)で定組成溶出。流量:1.2ml/分。カラム温度:60℃。サンプル温度10℃。注入体積:10μl。サンプル溶液:アセトニトリルに溶かしたピメクロリムスの0.5mg/ml溶液を調製。検出器UV:210nm
【0066】
タクロリムスのクロマトグラフィ純度のHPLC測定:カラム:Symmetry C18;3.5μm(150×2.1mm)。移動相組成物:定組成溶出
・成分A:0.1%酢酸水溶液58%
・成分B:アセトニトリル15%
・成分C:テトラヒドロフラン27%
・流量:0.3ml/分。カラム温度:50℃。注入体積:10μl。
サンプル溶液:タクロリムス25mgを計量し、アセトニトリル/水 1/1 v/v 25ml(最終濃度1mg/ml)に溶解した。検出器UV:278nm
利用した質量分光計は、陽イオン化におけるイオントラップAgilent Mod.6300である。
【0067】
利用した噴霧乾燥装置は、Advance不活性ループB−295機器を備えたBuchiモデルB290である。
【0068】
DRXスペクトル(粉末)を、5000から60000の開始角[1/2 2θ]から、回折計(PW1710 Philips)を使用して記録した。回折ダイアグラムを、サンプルのいかなる処理も行わずにCuアノードを用いて得た(Kα=1.54060ÅおよびKα=1.54439Å)。
【実施例1】
【0069】
乾燥による再構成可能な固体ラパマイシン/シクロデキストリン複合体の調製(溶媒としてエタノール95%)
エタノール(96%)に溶かしたラパマイシンの0.4%w/v溶液を、シクロデキストリン粉末に添加した。ラパマイシンとシクロデキストリンとの最終的な相対比を、表2および3に報告する。
【0070】
この懸濁液を、30分間撹拌しながら20〜25℃に維持し、次いで得られた混合物を、エタノールを除去するために18時間真空乾燥した。乾燥固体粉末を、窒素雰囲気下で−20℃で貯蔵した。
【0071】
表2において、溶液状態(1/1 水/アセトニトリル混合物)にあるラパマイシン/CD複合体のアッセイデータ(HPLCデータ)を、20〜25℃で、t=0および19日後に報告する。
【0072】
【表4】
【0073】
【表5】
【0074】
【表6】
【0075】
【表7】
【実施例2】
【0076】
乾燥による再構成可能な固体ラパマイシン/シクロデキストリン複合体の調製(溶媒としてアセトン)
アセトンにラパマイシンを溶かした0.4%w/v溶液を、γシクロデキストリンに添加した。ラパマイシンとシクロデキストリンとの最終的な相対比は、0.6%w/wであった。得られた懸濁液を、撹拌しながら30分間20〜25℃に維持した。得られた混合物を、アセトンを除去するために18時間真空乾燥した。任意選択で、湿潤固体を、n−ヘプタンを用いた懸濁液の希釈および濾過によって回収してもよく;次いで乾燥器内に移してもよい。
【0077】
乾燥固体粉末を、窒素雰囲気下で−20℃で貯蔵した。
【0078】
60℃での、得られたラパマイシン/γCD複合体(粉末)の安定性データを、乾燥エタノールおよびエタノール96%を溶媒として使用して得られたものと比較し;得られたデータを、図2および表6にまとめた。特に、図2では、60℃で696時間、粉末に関して行われた安定性試験中、分解によって生じた関連ある副生成物の合計(HPLCデータ)を報告する。
【0079】
【表8】
【実施例3】
【0080】
噴霧乾燥器による再構成可能な固体ラパマイシン/シクロデキストリン複合体の調製(溶媒としてアセトン)
アセトンに溶かしたラパマイシンの0.1%w/v溶液を、γシクロデキストリンに添加した。
【0081】
ラパマイシンとシクロデキストリンとの最終的な相対比は、0.6%w/wであった。得られた懸濁液を、撹拌しながら30分間20〜25℃に維持した。
【0082】
得られた懸濁液を、下記の実験条件で噴霧乾燥器で処理した:
・不活性窒素温度=70℃
・供給速度=17ml/分
・出口窒素温度=54℃
・アスピレーター速度=80%
・ノズルクリーニングの頻度=2ショット/秒
【0083】
得られた固体粉末を30℃で8時間乾燥し、次いで窒素雰囲気下で−20℃で貯蔵した。この複合体の特徴は、実施例2で得られた生成物に関して記述したものと同じである。
【0084】
γCDエベロリムス複合体、γCDテムシロリムス複合体、γCDタクロリムス複合体、およびγCDピメクロリムス複合体の調製
これらの複合体を、実施例2および3のそれぞれに従い調製した。
【0085】
γCDラパマイシン複合体、γCDエベロリムス複合体、γCDテムシロリムス複合体、γCDピメクロリムス複合体、γCDタクロリムス複合体、および出発材料として利用される、対応するマクロライドのHPLC UVおよびMSデータ
全ての複合体のHPLCプロファイル(HPLCおよびUV検出器)を、図4〜8で収集した。各複合体ごとに、本発明者らは、実験セクションで記述された特定のHPLCクロマトグラフィ条件を利用した。
・図4.ラパマイシンγCD複合体=全イオン電流(TIC)、m/z=952での単イオン(カリウムとのラパマイシンの分子イオンの付加体;m/z=952)、m/z=1319での単イオン(ナトリウムとのγシクロデキストリンの付加体;m/z=1319)
・図5.エベロリムスγCD複合体=全イオン電流(TIC)、スペクトルUV(278nm)、m/z=1026での単イオン(カリウムとのエベロリムスの分子イオンの付加体)、m/z=1297での単イオン(γシクロデキストリンの分子イオン)
・図6.テムシロリムスγCD複合体=全イオン電流(TIC)、スペクトルUV(278nm)、m/z=1048での単イオン(アンモニアとのテムシロリムスの分子イオンの付加体)、m/z=1297での単イオン(γシクロデキストリンの分子イオン)
・図7.ピメクロリムスγCD複合体=全イオン電流(TIC)、スペクトルUV(210nm)、m/z=848での単イオン(カリウムとのピメクロリムスの分子イオンの付加体)、m/z=1297での単イオン(γシクロデキストリンの分子イオン)
・図8.タクロリムスγCD複合体=全イオン電流(TIC)、スペクトルUV(220nm)、m/z=1297での単イオン(γシクロデキストリンの分子イオン)、m/z=822での単イオン(水とのタクロリムスの分子イオンの付加体)
【0086】
t=0での、これらマクロライド複合体のHPLC UVおよびMSクロマトグラフィプロファイルは、出発材料として利用された対応するマクロライドとは変わらないことがわかった。
【0087】
CD複合体の調製で出発材料として利用された各マクロライドのHPLC/MSおよびHPLC/UV分析を、本明細書に報告する。
・図9.ラパマイシン=23分で溶出された主なラパマイシン異性体の、全イオン電流(TIC)、HPLC/UVプロファイル、および質量スペクトル。
・図10.エベロリムス=21分で溶出された主なエベロリムス異性体の、全イオン電流(TIC)、HPLC/UVプロファイル、および質量スペクトル。
・図11.テムシロリムス=20分で溶出された主なテムシロリムス異性体の、全イオン電流(TIC)、HPLC/UVプロファイル、および質量スペクトル。
・図12.ピメクロリムス=27分で溶出された主なピメクロリムス異性体の、全イオン電流(TIC)、HPLC/UVプロファイル、および質量スペクトル。
・図13.タクロリムス=11分0秒で溶出された主なタクロリムス異性体の、全イオン電流(TIC)、HPLC/UVプロファイル、および質量スペクトル(同じく掲載のスペクトルにおいてm/z=822で水付加体およびm/z=826でナトリウムとの付加体が確認できる)。
【0088】
γCDとのエベロリムス複合体の安定性
実施例2および3のそれぞれにより調製されたCDとのエベロリムス複合体に関する安定性データを、図3および表7に報告する。
【0089】
特に、図3には、60℃で96時間、粉末で行われた安定性試験中の、分解によって生じた関連副生成物の合計(面積%を単位とするHPLCデータ)が報告されている。
【0090】
【表9】
【0091】
γラパマイシン複合体、γCDエベロリムス複合体、γCDテムシロリムス複合体、γCDタクロリムス複合体、およびγCDピメクロリムス複合体の固体状態
ラパマイシン結晶質を噴霧乾燥器で処理した後(またはエタノールもしくはアセトン溶液から蒸発させた後)の、またγCD(出発材料)、ラパマイシンγCD複合体、結晶質ラパマイシン(出発材料)と、ピメクロリムス、テムシロリムス、タクロリムス、およびエベロリムスを噴霧乾燥器で処理した後(またはエタノールもしくはアセトン溶液から蒸発させた後)の固体状態を、図14〜23に報告する。これらの分析は、エタノールもしくはアセトン溶液からの蒸発後、これらマクロライドが非晶質形態にあることを裏付けている。γCDとの複合体のDRXスペクトルは、図22および下記の表9の両方で明示されるように、γCDの結晶質相が変化していないことを裏付けており、用いられた技法の分解能によれば、結晶質形態には微量のマクロライドも存在しない。
【0092】
【表10】
【特許請求の範囲】
【請求項1】
γシクロデキストリンとのマクロライドの包接複合体であって、マクロライドとγシクロデキストリンとの重量比が、1:100〜1:400に含まれることを特徴とする複合体。
【請求項2】
前記マクロライドとγデキストリンとの重量比は、1:111〜1:333に含まれることを特徴とする請求項1に記載の複合体。
【請求項3】
前記マクロライドは、ラパマイシン、ピメクロリムス、テムシロリムス、エベロリムス、タクロリムスから選択されることを特徴とする請求項1または2に記載の複合体。
【請求項4】
下記のステップ:
a.マクロライドを有機溶媒に溶解して溶液を得るステップ;
b.前記溶液をγシクロデキストリンに添加するステップ;
c.前記混合物を蒸発させるステップ;
を含むことを特徴とする請求項1から3のいずれか一項に記載の複合体を調製するための方法。
【請求項5】
前記マクロライドとγシクロデキストリンとの重量比は、1:100〜1:400に含まれることを特徴とする請求項4に記載の方法。
【請求項6】
前記マクロライドとγシクロデキストリンとの重量比は、1:111〜1:333に含まれることを特徴とする請求項5に記載の方法。
【請求項7】
前記有機溶媒は、極性有機溶媒であることを特徴とする請求項4に記載の方法。
【請求項8】
前記極性有機溶媒は、アセトン、メタノール、および/またはエタノールから選択されることを特徴とする請求項7に記載の方法。
【請求項9】
前記蒸発は、真空、噴霧乾燥、濾過、および/または凍結乾燥の下で行われることを特徴とする請求項4に記載の方法。
【請求項10】
前記濾過は、有機溶媒、好ましくは非極性有機溶媒、より好ましくはC5〜C8直鎖状または分岐状炭化水素を用いた希釈によって行われることを特徴とする請求項9に記載の方法。
【請求項11】
請求項4から10に記載の方法によって得ることが可能であることを特徴とする包接複合体。
【請求項12】
請求項1、2、3、または11のいずれか一項に記載の複合体と、少なくとも1種の薬学的に許容される賦形剤とを含有することを特徴とする医薬組成物。
【請求項13】
前記組成物は、固体剤形であることを特徴とする請求項12に記載の組成物。
【請求項14】
前記組成物は、水溶液であることを特徴とする請求項12に記載の組成物。
【請求項15】
前記水溶液は、いかなる有機溶媒も含有しないことを特徴とする請求項14に記載の組成物。
【請求項16】
経口、眼部、局所、または非経口使用のための請求項12に記載の組成物。
【請求項1】
γシクロデキストリンとのマクロライドの包接複合体であって、マクロライドとγシクロデキストリンとの重量比が、1:100〜1:400に含まれることを特徴とする複合体。
【請求項2】
前記マクロライドとγデキストリンとの重量比は、1:111〜1:333に含まれることを特徴とする請求項1に記載の複合体。
【請求項3】
前記マクロライドは、ラパマイシン、ピメクロリムス、テムシロリムス、エベロリムス、タクロリムスから選択されることを特徴とする請求項1または2に記載の複合体。
【請求項4】
下記のステップ:
a.マクロライドを有機溶媒に溶解して溶液を得るステップ;
b.前記溶液をγシクロデキストリンに添加するステップ;
c.前記混合物を蒸発させるステップ;
を含むことを特徴とする請求項1から3のいずれか一項に記載の複合体を調製するための方法。
【請求項5】
前記マクロライドとγシクロデキストリンとの重量比は、1:100〜1:400に含まれることを特徴とする請求項4に記載の方法。
【請求項6】
前記マクロライドとγシクロデキストリンとの重量比は、1:111〜1:333に含まれることを特徴とする請求項5に記載の方法。
【請求項7】
前記有機溶媒は、極性有機溶媒であることを特徴とする請求項4に記載の方法。
【請求項8】
前記極性有機溶媒は、アセトン、メタノール、および/またはエタノールから選択されることを特徴とする請求項7に記載の方法。
【請求項9】
前記蒸発は、真空、噴霧乾燥、濾過、および/または凍結乾燥の下で行われることを特徴とする請求項4に記載の方法。
【請求項10】
前記濾過は、有機溶媒、好ましくは非極性有機溶媒、より好ましくはC5〜C8直鎖状または分岐状炭化水素を用いた希釈によって行われることを特徴とする請求項9に記載の方法。
【請求項11】
請求項4から10に記載の方法によって得ることが可能であることを特徴とする包接複合体。
【請求項12】
請求項1、2、3、または11のいずれか一項に記載の複合体と、少なくとも1種の薬学的に許容される賦形剤とを含有することを特徴とする医薬組成物。
【請求項13】
前記組成物は、固体剤形であることを特徴とする請求項12に記載の組成物。
【請求項14】
前記組成物は、水溶液であることを特徴とする請求項12に記載の組成物。
【請求項15】
前記水溶液は、いかなる有機溶媒も含有しないことを特徴とする請求項14に記載の組成物。
【請求項16】
経口、眼部、局所、または非経口使用のための請求項12に記載の組成物。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
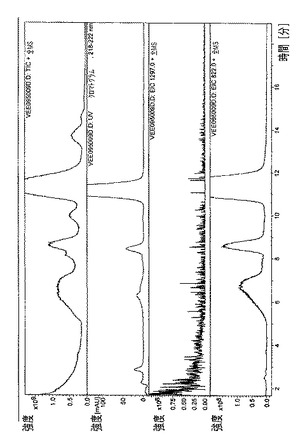
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】

【図16】
【図17】

【図18】
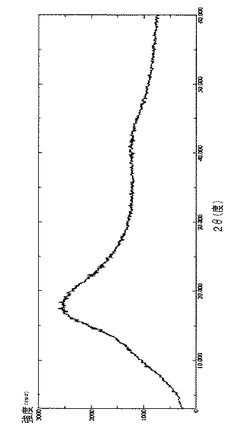
【図19】
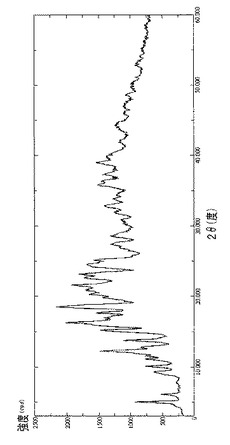
【図20】
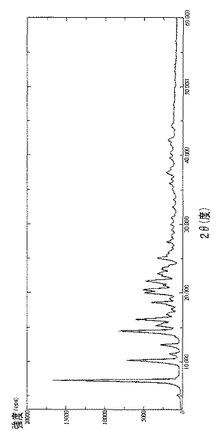
【図21】
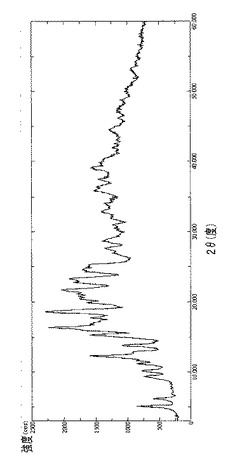
【図22】
【図23】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【公開番号】特開2012−31160(P2012−31160A)
【公開日】平成24年2月16日(2012.2.16)
【国際特許分類】
【外国語出願】
【出願番号】特願2011−145736(P2011−145736)
【出願日】平成23年6月30日(2011.6.30)
【出願人】(511159934)ユーティカルズ ソシエタ ペル アチオニ (4)
【Fターム(参考)】
【公開日】平成24年2月16日(2012.2.16)
【国際特許分類】
【出願番号】特願2011−145736(P2011−145736)
【出願日】平成23年6月30日(2011.6.30)
【出願人】(511159934)ユーティカルズ ソシエタ ペル アチオニ (4)
【Fターム(参考)】
[ Back to top ]