
マトリックス支援レーザー脱離イオン化(MALDI)質量分析法(MS)を含む化学分析のための検体の濃縮と分別のために改良された方法と装置
マトリックス支援レーザー脱離イオン化質量分析(MALDIMS)により分析するために血清のような生体サンプルからの検体の事前濃縮と精製のための装置が示されている。

【発明の詳細な説明】
【特許の相互参照】
【0001】
本発明は特許文献1、2、3の特典を主張し、その内容全体を参考用に組込んでいる。
【技術分野】
【0002】
本発明は質量分析法(MS)に、より特定すれば、マトリックス支援レーザー脱離イオン化質量分析法(MALDIMS)により分析するために血清のような生体サンプルからの検体の事前濃縮と精製に関する。
【背景技術】
【0003】
電気泳動濃縮装置のセル(cell)の移動相から固体捕捉相へのタンパク質、ペプチド(peptides)及び他の検体分子の濃縮と捕捉を容易にする装置と方法を開示している。さらに、そのような固体捕捉相は質量分析法での直接分析に適合できる。質量分析法は多数の検体を同時に監視できる。それとは対照的に、他の多くの分析技術は一度に1種のみ、又は、せいぜい1−2種の分子の定量分析を行なえるだけである。低費用計測、使用しやすくすること、高処理量のMALDI法のような質量分析法の最近の進歩が、臨床研究とひいては、全健康産業の変革を約束している。しかしながら、この膨大な可能性を実現するキーは、迅速かつ再現可能に質量分析法のために複雑な生体サンプルを調製できる新しいサンプル調製技術の開発である。そのような技術は、組織のホモジネート、全組織のスライス(slices)、他の固体組織の調製、さらに、全血、血漿、血清、脳脊髄液、唾液、尿等のような液体サンプルを含む多様なサンプルに応じる必要がある。多分、血清が臨床上最も重要な生体液で、数億のサンプルが医療診断のために毎年吸引管により採取されている。血液とリンパ液が病気のバイオマーカー(biomarkers)の豊富な源泉である。なぜなら、血液及びリンパ液の中を循環している天然の血液を発生するタンパク質及びポリペプチド(polypeptides)に加えて、身体組織は血液とリンパ液に追加の細胞要素を放出するからである。それゆえ、これらの循環流体はタンパク質及びポリペプチド(PP)を含む疾患のバイオマーカーを含み、細胞の肥厚化、壊死、アポプトーシス又は新生組織からの抗原の一掃のような病理的状態の指標になる。ここで、PPの用語は、オリゴペプチド(oligopeptides)、又は、2以上のアミノ酸(即ち、約200ダルトン(Dalton))から高分子量のタンパク質(約100万ダルトン以上)までの範囲を含む広い範囲の分子量のタンパク質を示すのに用いられる。
【0004】
血清内の実質的に有望なクラス(class)分野の疾患マーカーは低分子量(LMW)のPP破片で、それが豊富にあり、種々の構造変化をしていることは、大部分ではないが、多くのヒトの疾患を示している(非特許文献1)。LMW血清プロテオーム(proteome)は、サイトカイン(cytokines)、ケモカイン(chemokine)、ペプチド・ホルモン(peptide hormones)、大型タンパク質の分解破片のようないくつかのクラスの生理学的に重要なポリペプチドから作られている。これらのタンパク質分解によるペプチドは、がん、糖尿病、心臓血管系及び感染性の疾患のような病的状態と関連があることが示されている。しかしながら、LMW血清プロテオームの分析は、広範囲のサンプル調製を必要とし、血清中の総タンパク質量を支配する高い比率のアルブミン(albumin)(〜55%)により分析が非常に困難になっている。他の問題としては、他のLMW PP分子の存在度が広く動的な範囲に亘ることや主要な糖タンパク質の膨大な異種性が挙げられる。例えば、ここで臨床的に測定された最も希少なタンパク質は(図1)大きさがアルブミンより小さなものの桁10以上の濃度で存在する(非特許文献2)。しかしながら、これらの希少なタンパク質とペプチドは高い感受性と選択性のある疾患マーカーであり、潜在的な薬物ターゲット(target)になると信じられている。
【0005】
伝統的に、液体クロマトグラフィ(LC)又はアフィニティ(affinity)ベースの方法が適当な分離プロセスを提供するために最大限用いられて来た。LC法による精製にはリンカー(linker)分子をLCカラム(column)内の(機能化固定相を生じる)固定相に化学的に取付けることが含まれる。サンプルがカラムに装填されると、移動相が固定相を通過して流れる。各検体が移動相の代わりに固定相に結合するのに使われ、検体の精製用に提供されているLCカラムを通って(汚染物質及び干渉物質だけでなく)種々の検体の相対的移動速度を決定する。例えば、ペプチド及びタンパク質のような対象検体分子は機能化された固定相に吸収でき、その一方で汚染物質は検体から流出する。次ぎに、移動相を調節して、機能化した固定相から対象となる分子を溶離する。しばしば、アセトニトリル(acetonitrile)と水の混合体のような、MALDI−MSと適合している揮発性緩衝液が、このステップ(step)での移動相として用いられる。このようにして、精製された対象物質がLCカラムから流出してMALDI−MS検体として集められる。ここで、サンプルから塩類及び他の汚染物質を双頭除去しておかないと分析が妨害され、又は、感度が制限される。それゆえ、分析の高い処理方法でのサンプル分析に必要な試薬の分離、濃縮、添加のための装置と手順を改良する必要がある。質量分析法用のサンプル調製技術の最近の見直しでは、これらの方法は時間がかかり、面倒で、高い技術の労働者を必要とし、自動化が困難である(非特許文献3,4)。結果として、1臨床検査の間に分析できるサンプル数は極端に限定され、統計的優位性を実質的に損ない、それゆえ、これら検査の臨床的関連性が損なわれる。結果として、主として、サンプル調製システムが無いことにより、LMW血清プロテオームが、ヒトその他の動物の疾病、疾病治療、遺伝子発現分析のための(質量分析法により検出できる)バイオマーカー(biomaker)が優れているが大きな未開拓の分野になっている。
【0006】
MALDIターゲット・プレート(target plate)上に配置したサンプルのマトリックス支援レーザー脱離イオン化質量分析法((MALDI−MS)による分析が急速に、タンパク質、ペプチドその他の生体分子を分析するための優れた方法になりつつある。MALDI−MS手順は非常に感度の良い分析方法で、多分MS手順が生理的な塩類及びpH緩衝液と特に適合性がよい。さらに、ピコモル以下の量の生物学的巨大分子から高質量イオンを高効率で発生させる能力がこの手法をして巨大分子分析に特に有用なものにしている。しかしながら、血漿又は血清のような未処理生体サンプルのペプチド検体の分析には下記のように質量分析法上特別な問題が生じる。
【0007】
克服すべき第一の問題は高濃度の塩(例えば、ナトリウム、カリウム、塩素、リン酸塩及び炭酸塩)を含むことである。陰イオンは通常のMALDI分析手順を用いることによりペプチド・サンプルのイオン化を抑制するのに特に有効である。陽イオンにもアダクト・スペクトル(adduct spectra)を発生することで問題がある。当該スペクトラは、タンパク質の主要質量ピーク(peaks)を、それぞれが1個の陽イオンの付加的質量を有する多数の付加的質量ピークに分割する。さらに、MALDI−MS分析の成功は、質量分析器に注入する前に、検体と一緒に混合したMALDIマトリックス物質を効果的に結晶化する分析技術者の能力に大幅に依存している。MALDIマトリックス物質は、分析すべきサンプル内の吸着された検体物質と一緒にマトリックスの噴霧化とイオン化に用いられるレーザー光を吸収する必要がある。イオン化された検体分子は、次いで質量分析器内の陽極と陰極の高電圧により、質量分析器イオン検出器内で加速される。(塩又はグリセロール(glycerol)のような)比較的少量の汚染物質が存在するときでさえ、タンパク質とペプチドのような検体の脱離とイオン化を効果的に行うMALDIマトリックスの能力が劇的に低減する。さらに、高い塩濃度がMALDI−MSに必要な限界レーザー光度と(遊離ペプチドのピークを犠牲にして)塩を付加されたペプチドのピーク強度の両方を高める。
【0008】
第二に、ヒトの血清のようなサンプル内で、検体ペプチドは干渉プロテイン(例えば、アルブミン、イムノグロブリン(immunoglobulin)、トランスフェリン(transferin))と比較して、非常に低いコピー(copy)数で頻繁に存在する。対象のペプチドはしばしばリットル当たり丁度1マイクロモルからリットル当たり1ピコモルまで存在する(例えば、ミリリットル当たり1マイクログラムから1ピコグラム)。それと対照的に、総アルブミン及びIgG、IgMのようなガンマ・グロブリン(gamma globulins)が少なくとも、ミリリットル当たり0.01から0.1グラムの範囲の値で存在している。即ち、1×1011倍まで質量で大きい。それゆえ、主要な量が多いタンパク質が混合体のMLADIスペクトルを強く支配する。低強度のピークが主要ピークによりあいまいになるので、少量成分が観察されるのは稀になる。このことはヒトの血清のような生体サンプルでは深刻な問題である。生体サンプル内では、そのように低いコピー数の分子は、干渉タンパク質(例えば、アルブミン、イムノグロブリン及びトランスフェリン)及び塩(例えば、ナトリウム、カリウム、塩素、リン酸塩、炭酸塩)の桁違いに高いモル濃度が存在する中で検出する必要がある。
【0009】
第三に、検体ペプチドの多くは、疎水性であり、血液、血漿、血清に見いだされる主要タンパク質に結合している。特に、アルブミンは疎水性分子に非特異的に不特定に結合する傾向がある。かくして、アルブミンのような望ましくないタンパク質を除去することは、検体ペプチドの喪失にもなる。塩及び合成洗剤のような化学的破壊因子がアルブミンから検体ペプチドの解離を支援することが知られている。しかしながら、これらの因子は積極的にMALDIプロセス(process)を抑制する。例えば、ポリエチレン・グリコール(polyethylene glycol)(PEG)及びトリチオン(Trition)はペプチド及びタンパク質と同じ効率でMALDIによりイオン化と脱離を行う。結果として、これらの種は多くの場合タンパク質とペプチドのイオン化と競合する。それにより、後者からのMALDI−MS信号を抑える。それゆえ、アルブミンからの検体ペプチドを解離するために、化学的破壊因子を加えた後で、分析担当者が検体ペプチドを、その破壊因子のアルブミンと他の汚染タンパク質の両方から分離しなければならない。さらに、マイナー(minor)成分のペプチド検体が分離プロセス中で喪失しないように分離を実行しなければならない。この分離は、検体が疎水性であり、疎水性の面に接合する傾向があるとき、特に困難になる。不幸にして、LC法による生体高分子の精製が、しばしば、30%以上のサンプル減耗を生じ、又、汚染物質(又はサンプル間の信号漏洩)を追加することがある。多くのMALDI−MSユーザーにとって、このサンプルの減耗量は容認できない。第四に、検体のペプチドがそのような低レベルで存在するので、MALDI−MS分析の前に濃縮しなければならない。従来方法によって、先ず、ペプチドの解離、成分の分離である。次いで、濃縮を行うことは退屈で、単調であり、長時間を要し、かつ、労働集約的な多数のステップを必要とする。
【特許文献1】米国特許暫定出願第60/668,337号明細書
【特許文献2】米国特許暫定出願第60/668,794号明細書
【特許文献3】米国特許暫定出願第60/712,255号明細書
【特許文献4】米国特許暫定出願第60/748,771号明細書
【特許文献5】米国特許出願第10/963,336号明細書
【非特許文献1】Tirumalai,R.S.,K.C.Chan,D.A Prieto,H.J.Issaq,T.P.Conrads and T.D.Veenstra,(2003)Characterization of the Low Molecular Weight Human Serum Proteome,Molecular & Cellular Proteomics 2,1096−1103.
【非特許文献2】Anderson N.L.and N.G.Anderson(2002)The Human Plasma Proteome,Molecular and CellIular Proteomics 1,845−867.
【非特許文献3】Westermeier,R.and T.Naven(2002);In:Proteomics in Practice;Wiley−VCH Verlay−GbmH,Weinheim.
【非特許文献4】Hamdan,M.and P.G.Righetti(2005);In:Proteomics Today;John Wiley & Sons,Hoboken,NJ.
【非特許文献5】Schreiner,M,K Strupat,F.Lottspeich and K.Eckerskorn(1996)Ultraviolet Matrix Assisted Laser Desorption ionization−Mass Spectrometry of Electroblotted Proteins,Electrophoresis,17,954961.
【非特許文献6】Bienvenut,W.V.,J.C.Sanchez,A.Karmime,V,Rouge,K.Rose,P.A.Binz and D.F.Hochstrasser(1999)Toward a clinical Molecular Scanner for Proteome Research:Parallel Protein Chemical Processing before and during Western Blot,Alnal Chem..11,4800−4807..
【非特許文献7】Lion,N.,V.Gobry,H.Jensen,J.S.Rossier,H.Girault(2002)Integration of a Membrane−Based Desalting Step in a Microfabricatd Disposable Polymer Injector for Mass Spectrometric Protein Analysis,Electophoresis 23,3483−3588.
【非特許文献8】Muller,M.,F.Gras,P.A.Binz D.F.Hochstrasser and R.D.Appel(2002)Molecular Scanner Experiment with Human plasma:Improving Protein Identification by usmg Intensity Distributions of Matchmg Peptide Masses,Prteomics 2,1413−1425.
【非特許文献9】Scherl,A.,C.G.Zimmermann−Ivol,J.D.Dio,A.R.Vaezzadeh,P.A.Binz,M.Amez−Droz,R.Cochard,J.C.Sanchez,M.Gluckmann and D.F.Hochstrasser(2005)Gold Coating of Non−Conducting Membranes before Matrix−Assisted Laser Desorption/Ionization Tandem Mass Spectrometric Analysis Prevents Charging Effect,Rapid Commun.Mass Spectrom.19,605−610.
【非特許文献10】Moya,W.(2002)Surface Modified Porous Membrane and Process,U.S.Patent Number 6,354,443.
【非特許文献11】Lammerhofer,M.,F.Svec,J.M.J.Frecher and W.Lindner(2000)Monolithic stationary phases for Enantioselective Capillary Electrochromatography,J.Microcolum Separations 12,597−602.
【非特許文献12】Lammerhofer,M.,F.Svec,J.M.J.Frechet and W.Lindner(2001)Capillary Electrochromatorgrapy in Anion−exchange and Normal−Phase Mode using Monolithic Stationary Phases.J.Chromatoglaphy A 925 265−277.
【非特許文献13】Rohr T.,M.,D.F.Ogletree,F.Svec and J.M.J.Frechet(2003)Surface Functionalization of Thermoplastic polymers for the Fabrication of Microfuidic Devices by Photinitiated Grafting,Advanced Functional Materials 13,264−270.
【非特許文献14】Rohr,T.,M.,E.F.Hilder,J.Donovan,F.Svec and J.M.J.Frechet(2003)Photografting and the Control of Surface Chemistry in Three−Dimensional Porous Polymer Monoliths,Macromolecules 36 1677..1684.
【非特許文献15】Stachowiak,T.B.,T.Rohr M.,E.F.Hilder,D.S.Peterson,J.F.Svec,M.Yi and J.M.J Frechet(2003)Fabrication of Porous Polymer Monoliths Covalently Attached to the Walls of Channels in Plastic Microdevices,Electrophorisis 24 3689−3693.
【非特許文献16】16 Peterson,D.S.,T.Rohr,J.F.Svec,M.Yi and J.M.J.Frechet(2003)Dual−Function Microanalytical Device by In Situ Photolighographic Grafting of Porous Polymer Monolith:Integrating Solid−Phase Extraction and Enzymatic Digestion for Peptide Mass Mapping,Analytical Chemistry 75,5328−5335.
【非特許文献17】Peterson,D.S.,Q.Luo,M.,E.F.Hilder,F.Svec,and J.M.J.Frechet(2004)Porous Polymer Monolith for Surface−Enhanced Laser Desorption/Ionization Time−of Flight Mass Spectrometry for small Molecules,Rapid Commun.Mass Spectlom 18,1504−1512.
【非特許文献18】Pucci,V.,M.A.Raggi;F.Svec,and J.M.J.Frechet(2004)Monolithic Columns with a Gradient of Fuctionalities Prepared via Photoinitiated Grafting for Separations using capillary Electrochromatography,J.Sep.279−788.
【非特許文献19】Lee,D.,F.Svec,and J.M.J.Frechet(2004)PhotoPolymerized Monolithic capillary columns for Rapid Micro−High Performance Liquid Chromatographic Separation of Proteins,J.Chromatography A,1051,53−60.
【非特許文献20】Hilder,E.F.,F.Svec,and J.M.J.Frechet(2004)Development and Application of polymeric Monolithic Stationary Phases for Capillary Electrochromatography,J.Chromatography A,1044,3−22
【非特許文献21】Peterson,D.S.,T.Rohr,J.F.Svec and J.M.J.Frechet(2002)Enzymatic Microreactor−on−a−Chip:Protein Mapping Using Trypsin Immobilized on Porous Polymer Monoliths Molded in Channels of Microfluidic Devices,Analytical Chemistry 74,4081−4088.
【非特許文献22】Svec,F.,(2004)Preparation and HPLC Applications of Rigid Macroporous Organic Polymer Monoliths,J.Sep.Sci.,27;747−766.
【非特許文献23】Knochenmuss,R.Anal.Chem 2004;76:3179.
【非特許文献24】Zalluzec,E.J.;et.al.J.Am.Soc.Mass Spectrom 1994;5:230.
【非特許文献25】Andrews,P.C.;et.al.Anal.Chem.1996;68:1910.
【非特許文献26】Andrews,P.C.;et.al.Electrophoresis,1997;18:382.
【非特許文献27】Costello,C.E.;et.al.Rapid Commun.Mass Spectrom.1999;13:1838.
【非特許文献28】Peterson,D.;Rohr,T.;Svec,F.;Frechet,J.M.J.Anal.Chem.2003;75:5328−5335.
【非特許文献29】Frechet,J.M.J.;et.al.Macromolecules 2003;36:1677−1684.
【非特許文献30】Frechet,J.M.J.;et.al.Journal of Chromatography A 2004;1044:3−22.
【非特許文献31】Frechet,J.M.J.;et.al.Electrophoresis 2003;24:3689−3693.
【非特許文献32】Frechet,J.M.J.;et al.Journal of Chromatography A 2004;1051:53−60.
【非特許文献33】Svec,F.J.Sep.Sci.2004;27:747−766.
【非特許文献34】Frechet,J.M.J.;et al.Rapid Commun.Mass Spectrom 2004;18:1504−1512.
【非特許文献35】Ericson,C.;Liao,J.L.;Nakazato,K.;Hjerte'n,S.J.Chromatogr,A 1997;67:33−41.
【非特許文献36】Tempst,P.;et al.Anal.Chem.2004;76:1560−1570.
【発明の開示】
【0010】
それゆえ、本発明の第一の目的は生体サンプルから塩を除去するための方法と装置を提供することである。本発明の第二の目的はタンパク質のような高存在度の分子を生体サンプルから除去し、それにより残留する低存在度の分子の再現性が良く、感受性の高い分析を行なえることである。本発明の第三の目的はアルブミンや他の疎水性タンパク質から検体ペプチドを解離することである。本発明の第四の目的はMALDI質量分析法のための対象となる検体のペプチドとタンパク質を濃縮することである。本発明の第五の目的はサンプルの高処理量を提供するために、前記四目的を簡便で効果的な方法により提供することである。本発明の第六の目的は2以上のサンプルを並行して分析するために多数のサンプルを同時に扱うことである。それにより、本発明の他の目的と組合わせることにより、検査担当者が簡便かつ効果的な方法で生体組織サンプル内のペプチド及びタンパク質の分析を行なうために本発明を使用でき、それにより、検出の感度を高め、サンプルの処理量を高め、分析費用を低減する。最後になるが、分離された検体のペプチド、ポリペプチド、及び、タンパク質の分析を再現性をもって、かつ、定量的に行われることが所望された。それで、本発明の第七の目的は生体サンプル内のペプチド及びタンパク質について再現性があり、定量的なMALDI−MS分析を提供することである。
【0011】
用語PPを用いる場合、小型の2個以上のアミノ酸から100万ダルトン以上の大型タンパク質までの範囲のオリゴペプチドを意味していて、本発明の第八の目的は質量分析法(MS)により、ヒトの血清内のPPのLMW成分検査する分析システムを提供することである。本発明の第九の目的は例えば500ダルトンから500,000ダルトン以上までの広範囲のPPで、かつ、質量分析法(MS)により分析できる十分な融通性のあるPPAS(ペプチド・タンパク質分析システム)を提供することである。本発明の第十の目
的はPPASを改善し、さらに検出感度を高め、1ナノモル(nanomole)から0.1アトモル(attomole)以下までのPPの量を検出でき、MSにより測定された分子量を検出し、定量化できることである。本発明の第十一の目的はMS分析前に低存在度のPPを高存在度のPPから分離して、低存在度PPの検出感度を高めて、ヒトの血清内のPPの分別と分離を改善することである。
【発明を実施するための最良の形態】
【0012】
特許文献5には本発明の分野で用いるための方法と装置を開示していて、本出願書に参考用として、その全体を組込んでいる。さらに、本発明の方法と捕捉スライド(slide)はそこで開示された装置と方法に関連して使用しうる。さらに、本発明の方法と捕捉スライドは特許文献4に開示された装置と関連して使用しうる。特許文献4全体を参考用として本出願書に組込んでいる。
【0013】
本発明の有用な実施例は固相捕捉スライド上への血清のような(又は他の組織からの)生体サンプルに存在する低存在度のタンパク質及びポリペプチドを電気泳動的に分離し、濃縮し、捕捉するペプチド・タンパク質分析システム(PPAS)である。短い洗浄ステップの後で、塩その他の干渉分子が洗い流される。そして、MALDIマトリックス溶液が捕捉スライドに加える。従来技術で良く知られているように、そのようなマトリックス溶液は一般的に有機溶媒を含み、タンパク質を放出してMALDIマトリックス結晶内に組込み、溶媒の乾燥でスライド面上に沈殿する。次ぎに、そのスライドを完全に乾燥して、捕捉したタンパク質の質量と相対的存在度の両方の定量化のために、MALDI−MS測定に直接挿入する。
【0014】
図2及び3に詳細に示すように、PPASは流体サンプルを保持するために、1以上のウエル(well)4を有するカートリッジ(cartridge)2から成っている。カートリッジ2の1実施例には、25個のサンプルを同時に処理するために、25個のサンプル・ウエル(sample well)が含まれる。カートリッジ2の好ましい実施例は96個のサンプルを同時に処理するために8×12の配列で96個のサンプル・ウエルを含む。好ましい実施例では、分離と捕捉を行うのに必要な捕捉スライド42と試薬がカートリッジ2内のサンプル・ウエル4の配列として、事前に配置されている。図3はカートリッジ2を含むサンプル・ウエル4の配列を示している。図2はマルチ・ウエル(multi−well)PPASカートリッジ2の1個のウエルの切断面を示す略図である。
【0015】
各サンプル・ウエル4は上方開口部8、側壁10、下方部分12を有し、ウエル4が広い上方開口部8から狭い下方開口部14に寸法が段々に減少している。サンプル・ウエル4の上方開口部8がサンプル電極20を受入れる。サンプル電極20は図2に示すようにサンプル・ウエル内に配置された電解質サンプルと電気的に接触する。本発明の好ましい実施例で、各サンプル・ウエルが約50μLのサンプル、200μLの電気泳動用緩衝液を保持し、150μLの上部空間を残すように設計されている(全体積400μL)。サンプル電極20は好ましい実施例では、サンプル電極の配列として示されていて、各サンプル・ウエル4の上方開口部8に取外し可能にはめ込まれている。サンプル電極の配列は、それぞれの使用前にDI水又は他の適当な溶媒でアセンブリー(assembly)を洗浄するだけで、再使用可能で清浄にできるよう設計されている。選択肢として、より厳密な浄化が、洗剤、強酸例えばpH2.0以下のもの、又は、強塩基例えばpH12.0以上のもの、又は、有機溶媒例えばメタノール、エタノール、アセトニトリル、アセトン、CS2、ジメチルホルムアミド、ジメチルスルホオキシド、等を用いて行われる。各サンプル・ウエルの下方部分12が、各サンプル・ウエルの下方開口部14の近くの断面積が連続的に低減するように各サンプル・ウエルの下方部分12が成形されている。下方のウエル部分が円錐形で、各サンプル・ウエルの下部で低減した面積の下方開口部14にタンパク質分子が集まるようにしている。下方開口部14の下に分離層30があり、サンプル・ウエル4を捕捉物質40から分離するのに役立つ。分離層30は、サンプル・ウエル4内又は分離層内で選択された第一のサンプル分子を保持し、その一方で、選択された第二のサンプル分子が分離層を通過して、捕捉物質に接触でき、第二のサンプル分子を捕捉し、濃縮できるように機能する。本発明の好ましい実施例では、分離層はポリアクリルアミド・ゲルのようなゲル層から成っている。そのようなゲルは一般に1%から24%のポリアクリルアミドを有する。又、種々の量の架橋剤と重合開始剤も有していて、タンパク質分離の分野の技術者には良く知られている。さらに、好ましい実施例では、サンプル・ウエル4の配列を有し、対応する実質的に同じ分離層30の配列が存在し、好ましくはカートリッジ・ゲル・プレート32内に配置される。そこでは分離層30の配列がカートリッジ・ゲル・プレート(cartridge gel plate)32上に配置された実質的に同じ穴34の配列内に含まれる。一般に、ゲル・プレート32は熱可塑性ポリマー(ポリウレタン、ポリプロピレン等)のような所望の材料から切削、成型又は鋳造により形成される。そのようなゲル・プレートは電気的に絶縁性で、柔軟なポリマー例えば熱硬化性ポリマー、エラストマー(elastomer)、又は、ゴム材料である。一般に、そのような柔軟な材料は良好な液体密封特性を有し、その一方で、サンプル・ウエル4の間で電気的分離も行う。さらに、分離層30もサンプル・ウエル4を1以上の捕捉物質40から分離するのに役立ち、分離層30を通って電気泳動により駆動される検体分子を捕捉し、濃縮するのに役立つ。有利なこととして、分離層30はプレート32に共有結合をしている。そのような共有結合は接合喪失を防ぎ、カートリッジ・アセンブリーの組立を容易にしている。上記のように、液状媒体内でタンパク質の分離に特に有用な分離層はポリアクリルアミドである。それゆえ、ポリアクリルアミドとその支持構造の表面の共有結合が特に有用である。ポリアクリルアミドから固体のポリアクリルアミド支持構造への化学的接合は、物理的に強い複合構造の形成とポリアクリルアミドとその支持構造との間の液体密封部を形成することの両方に役立つ。この場合、特にゲル・プレートの穴34により定義された領域内で、ポリアクリルアミドの分離層30とゲル・プレート32の間で接合が形成される。ポリアクリルアミドによるその支持面へのそのような共有結合を実施する方法で、ポリアクリルアミドの反応混合体がゲル・プレート32内のゲル・プレートの穴34内に堆積する。その後に化学的グラフト・ステップがある。特に頑丈で耐久性のあるポリアクリルアミドの分離層30は基本的に2ステップの反応手順に基づく光グラフト(graft)によりゲル・プレート32に光グラフトされる。両方の反応ステップが支持面と接触したアクリルアミドのモノマー(monomer)とビスアクリルアミド(bis acrylamide)を含む溶液を用いることにより実施できる。(バルク(bulk)反応混合体内での)重合及びポリアクリルアミドの支持構造面例えばゲル・プレート32への接合の両方の開始が紫外線照射又は代わりに化学的開始剤を用いることにより行われる。便宜的には、ほぼ、ゲル・プレートの寸法の物理的保持具を用いて、ゲル・プレート32及びモノマーを含む反応溶液の両方を保持する。さらに、反応混合体は薄いシート(sheet)材料により支持構造に接触して保持される。薄いシート材料は真空クランプ(clamp)のような物理的手段により支持構造で近似的に保持される。
【0016】
好ましい実施例で、ポリアクリルアミドが取付けられる第一の固体面が、光グラフト反応混合体で前処理される。その後、ポリアクリルアミドの支持面への化学的接合及びポリアクリルアミドのバルク混合体の重合反応が同時に行われる。例えば、接合と重合反応の両方が現場での紫外線照射により開始される。好ましい方法では、事前浸漬のステップを用いていて、それは重合前にゲル・プレート材料に光開始剤を吸収させることから成っている。例えば、その事前浸漬ステップは、以下のサブステップ(sub−step)から成る。(a)タイプIIの光開始剤を含む事前浸漬溶液を使用。次いで、(b)例えば、空気のような乾燥気体でゲル・プレートを乾燥。代わりに、その気体を加熱して乾燥に使っても良い。
【0017】
タイプII光開始剤はSigma−Aldrich Companyのような会社から入手できる。一般に、タイプII光開始剤は生体分子と作用し、光開始剤の励起状態が第二の分子(共同開始剤)と相互作用して、遊離基を生じる。例には、ベンゾフェノン/アミン(benzophenones/amines)、チオキサントン/アミン(thioxanthones/amines)が含まれる。
【0018】
タイプII光開始剤の事前浸漬溶液の特定の例は、チオキサンテン−9−オン(thioxanthen−9−one)を0.006%(質量比)のメタノール溶液である。好ましい例で、上記のグラフト及び重合の工程は、表面上に反応混合物を置くことにより実施され、モールド(mold)を真空密封することにより低及び無酸素環境を形成し、ウエルの内面に共有結合している共重合分子を生じるのに十分な時間だけ混合体をUVエネルギーを照射する。一般に、通常のUVエネルギー源(5000−EC ユニット:入手先:Dymax Corporation Torrington,CT,USA:Hランプを装着)を用いて、照射時間を1秒から1時間の間とする。代わりに、非常に強いUV源、又は、瞬間光源を用いて、照射時間を非常に短くする。例えば、1マイクロ秒から1秒以下とする。それでも、他の適当なUV照射源には水銀アーク・ランプが含まれる。
【0019】
他に、ポリアクリルアミド・ゲルが化学的に付加接合できるポリアクリルアミド支持構造として使用するのに適当な別のタイプの材料にはポリウレタン、サントプレン(santoprene)、ポリプロピレン等が含まれる。一般に高分子物質で、その表面、即ち、直鎖又は側鎖部分に抽出可能な水素原子を含む物質なら、いずれも本発明を実施するのに適当なポリマーになる。例えば、抽出可能な水素は、ダブル・アリル(double allylic)水素、アリル水素、第三水素又は第二水素の形になる。特定の例には、以下があるが、それに限定されない。ポリオレフィン、水素化ポリスチレン、環状オレフィン共重合体、ポリ(エチレンテレフタレート)、ナイロン、ポリカーボネート、ポリ塩化ビニル、ポリブチルメタクリレート、ポリスチレン、ポリ(ジメチル・シロキサン)、又は、ポリ(メチルメタクリレート)。接合工程を開始するための追加の光開始剤は一般に当業の技術者に良く知られていて(バルク・ポリアクリルアミド重合反応の混合体の代わりに)グラフトすべき固体ポリマーの表面に対し分配特性有するタイプII光開始剤を含む。
【0020】
好ましい様態で、光グラフトとバルク重合の結合混合体は約68.7%(体積)が水性緩衝液、30%(体積)が19:1比で存在するアクリルアミド(acrylamide)/N,N'−メチレンビスアクリルアミド(N,N'−Methylenebisacrylamide)の40%(W/V)脱イオン水溶液、0.69%(体積)がチオキサンテン−9−オン(Thioxanthen−9−one)の 0.6%(W/V)メタノール溶液、0.41%(体積)が過硫酸アンモニウム(ammonium persulfate)の1%(W/V)脱イオン水溶液、0.20%(体積)が1,2−.ジ(ジメチルアミノ)エタン(TEMED)(又はEDMA)から成っている。反応混合物が混合され、ガラス又はポリマーの下面を有する浅い容器に置かれる。そのような下面は「非付着性」の面例えばTeflon(R)として特に有用である。「非付着性」の面は容器内に含まれる希望の固体支持構造例えばゲル・プレート32にポリアクリルアミドを外しやすくなるように機能する。その手順の中で、ゲル・プレートが光グラフトとバルク重合の結合混合体内に置かれ、UV透過ガラス、又は、薄いポリマープレートのようなUV透過カバーで覆われて、ゲル・プレートの穴34が反応混合物を含むが空気の泡を排除する。UV透過プレート、ポリアクリルアミドの反応混合物を含み、支持構造を機械的に接合している構造が、結合クリップ(clip)、真空クランプ(clamp)又は他のクランプ手段により所定位置に保持される。ゲル・プレートをカバーするUVUV透過プレートの上にフォトマスク(photomask)を使用しうるので、反応混合体と支持構造の希望の部分がUV光源により照射される。それにより、ポリアクリルアミドは事前決定されたパターン(pattern)で支持面に接合される。光重合を開始するために、その構造体をUV照射装置の付近に置き、照射の波長と強度に基づき適当な時間照射する。照射される表面で照射光束が150mW/cm2である場合、照射時間はシステム要因によるが一般的に4分未満である。例えば、Dランプを用いた5000−ECユニット:入手先:Dymax Corporation Torrington,CT,USAをゲル・プレートの表面から約20cmの距離で使用した場合、十分なUV照射が与えられる。UV照射の後で、UV透過カバーを取外し、光グラフトにより固体支持構造(例えば、ゲル・プレート32)に化学的に接合されたポリアクリルアミドを容器から取外し、非重合反応混合体を除去するために、脱イオン水のような適当な洗浄用溶媒で洗浄する。得られたポリアクリルアミド/支持構造の一体部分(例えば、ゲル・プレートに接合したポリアクリルアミド・ゲル)が適当な液体媒体に置かれ、又は、保管のために密封包装を行い、又は、使用のためにカートリッジに直ちに組込む。現場で製作され、支持されたポリアクリルアミドが優れた機械的安定性を示し、その支持材料への良好な接合を示す。上記の同時重合工程は、時間効率が良いように、そのような化学的に接合し、支持されたアクリルアミド構造の製造を行うのに特に便利である。
【0021】
選択肢として、ポリアクリルアミド反応混合体が、追加の有用なリガンド(ligands)例えば、タンパク質、多糖類、DNA、RNA等を含む。そのようなリガンドは便宜的に、重合前にポリアクリルアミド反応混合物に加える。代わりに、隣接する浸漬溶液からリガンドの拡散に十分な時間を与えることにより、又は、浸漬溶液から接着されたポリアクリルアミドへの活発な電気泳動により、重合後にポリアクリルアミドにリガンドを加える。例えば、望ましい場合、ポリアクリルアミドによるアルブミンの保持を高める目的で、改質された炭水化物材料をポリアクリルアミドに加える。そのような材料の例には、ブルー・デキストラン(blue dextran)、タンパク質へのアフィニティを改質したシリカ(silicas)、又はアルブミン結合分野での技術者に知られている他の材料が含まれる。
【0022】
(捕捉スライド)
捕捉物質40が、上面41と下面43を有するカートリッジ捕捉スライド(cartridge capture slides)42に位置しているオリフィス(orifices)50に配置されている。オリフィス50は上面41の上方開口部52、下面43の下方開口部54を有している。さらに、その開口部が捕捉スライド42の内側壁面56から成っている。捕捉材料40は一般にオリフィス50の内側壁面56で捕捉スライド42に取付けられる。その取付は、溶媒、熱、音波、又は、他の溶接手段による溶接を含む接合手段により行われる。代わりに、捕捉材料40は、エポキシ、メタクリレート、シアノアクリレート又は他のタイプの化学的結合材料及び樹脂を用いた化学的共有結合手段によりオリフィス50で捕捉スライド42に取付けられる。
【0023】
捕捉材料40を保持するためにオリフィス50(穴とも言う)を含むカートリッジ捕捉スライド42の好ましい実施例では、カートリッジ捕捉スライド42が長さ約4mmから約6mmの間、幅約3mmから約4mmの間、厚みが約1mmである。より好ましくは、カートリッジ捕捉スライド42が長さ約5.3mm、幅約3.5mm、厚み約1mmである。さらに好ましくはオリフィス50は実質的に円形であり、直径は約0.5mmから約1mmである。同様の寸法が好ましいカートリッジ・ゲル・プレート32にも適用される。
【0024】
図2及び3に示すように、カートリッジ捕捉スライド42の下では、電解質ベースチャンバー60があり、個々のカートリッジ・ウエルをお互いに物理的分離と電気接続をするように機能する。さらに、1以上の共通対極70を1以上の対極チャンバー72に対応している。すぐに使用できるとき、電解質ベースチャンバー60が導電性の電解質ベース媒
体62で充填され、対極チャンバー72が対極電解質74で充填されている。ベース媒体と対極電解質がイオンを導通させ、対極チャンバー72内の対極70と捕捉スライド42内の捕捉材料40を電気的に接続する。対極チャンバーは側壁76を有し、すぐに使用できるとき、実質的に全表面に対し、少なくとも部分的に垂直になっているので、電解質74上の電極70の作用によって発生する気体の泡(例えば、水素又は酸素)の脱出のために、連続的上昇路を提供する。有利なことに、電解質ベース媒体62が、例えば、一般用の可溶性の陰イオンと陽イオンのペア(pair)を水溶液中0.001から1モルの濃度で含んでいるので、高い導電性である。この汎用の陰イオンと陽イオンのペアは、チャンバー60及び72の材料に適合していれば、実質上、どの可溶性の陰イオンと陽イオンのペアでも良く、例えば、ナトリウム、リチウム、カルシウム、マグネシウム等及び塩化物、フッ化物、硫酸塩、チオシアナイド等の塩である。当該ペアから成る好ましい塩のひとつがKClであるのは、陰イオンと陽イオンが実質的に同じ拡散係数を有し、それにより電解液の濃度が異なる2種類の電解液間の境界での拡散の可能性を最小限にするからである。一般的に、汎用の可溶性の陰イオン及び陽イオンのペアは弱酸でも弱塩基でもない。なぜなら、弱い酸又は塩基の濃度の違い又は導電性の違いを有する2種の電解液間の境界での酸又は塩基の帯電形の移動が、境界での、又は、境界を横切ってpHの変化を生じるからである。しかしながら、これらの電解液はそのような弱い酸又は塩基を含みうる。ただし、それは本出願書の他の部分に示すように、電解質のpHの制御又は修正を生じる手順を用いて入念に選択され、使用される。
【0025】
電解質ベース媒体62を、粘性を高め、漏洩を防止し、又は、気泡を捕捉するために、又は、ゲル化物質を溶解することにより、又は、当業の技術者に良く知られているように、ゲルとして加える。ゲル化物質はデンプン又はアガロース(agarose)又は親水性ポリマーの共重合例えばアクリルアミド又はヒドロキシメチルメタクリレート(hydoroxymethlmethacrylate)のようなものである。さらに、1以上の対極チャンバー72も電解質ベースチャンバー60で用いられるのと同じ組成を有する電解質74を充填している。しかしながら、チャンバー72が捕捉材料40から物理的に分離されているので、電解質74を構成する導電性塩の選択では広い寛容度が可能である。例えば、高濃度の無機塩(例えば、0.1から10モル)、及び、普通の対極70により生じる水素又は水酸化物のイオンのpH緩衝液を提供するために、通常用いられる弱酸又は弱塩基が、随意、対極電解質74を構成しうる。例として、1.0M,pH8.0のトリスヒドロキシメチルアミノメタンクロリド(tris(hydroxymethyl)aminomethane−chloride)、三塩化物((tris)chloride)、又は、1.0M,pH9.2の硼酸カリウム(potassium borate)、又は、1.0M,pH7.0のイミダゾリウムクロライド(imidazolium chloride)等である。しかし、適当な高緩衝溶液であれば事実上どれも十分であり、当業の技術者には良く知られている。
【0026】
好ましい実施例では、カートリッジ2の電解質ベースチャンバー60はゲル化された対極緩衝液74を事前充填される。例えば、ゲル化溶液は1%アガロース・ゲルで良く、又、1.0M KCl,1mM ヒスチジン(histidine),pH7.8から成っている。さらに、好ましい実施例で、カートリッジ・ゲル・プレート32内の分離層30及びカートリッジ2のカートリッジ捕捉スライド42内の多孔性捕捉材料40はイオンによる導電性液体媒体を事前に充電される。例えば、分離層30はポリアクリルアミド・ゲルであり、1 mMから500 mMの無機塩を含む電解液内で重合したポリアクリルアミドを2%から12%含み、又は、15%の高さもある。1実施例では、分離層に事前に充填された電解質は50mMのKCl、100mMのヒスチジン(histidine),pH 7.8である。カートリッジ2のカートリッジ捕捉スライド42内の多孔性捕捉材料40の中に事前充填された電解質の組成は、溶媒に溶解された多様な導電性の塩で良い。その溶媒は水溶液、又は、他の適当な有機溶媒で良く、例えば、メタノール、エタノール、プロパノール等、又は、代わりにアセトニトリル(acetonitrile)又は他の水溶性有機溶媒である。最適には、使用する溶媒は、また、多孔性の捕捉材料の検体を通る適当な導電性を与える1から1Mの有機又は無機の塩を含む。便宜的に、分離層を形成するのに用いられたのと同じ溶液、例えば、10mM KCl,100mM ヒスチジン(histidine),pH8.0を使用しうる。
【0027】
分析システムとして提供するために、電子計測制御要素が使い捨てカートリッジ2と共に用いられる。調節可能な+/−300V電源(即ち、調節範囲は600V)100を使用して、電気泳動法に必要な電場を供給できる。そのような比較的低い電圧の電源で十分なのは、分離距離を1cm未満、一般的に、約0.1から0.5cmにしうるからである。さらに、分離と捕捉のステップ工程を監視するために、各サンプル・ウエルを通過して、サンプル電極20から対極74への電流を別個に監視する。例えば、96個の個別電流計を使用しうる。多数の電流計が電流測定のために単一回路から成っているが、電流値を報告するためのサンプル及び保持の回路を有している(例えば、1Hzの報告周波数)。好ましい方式では、結果はコンピューターのモニターにグラフとして表示される。代わりに、調節可能な一定電流源を電圧源の代わりに用いる。通常、電流源はサンプル・ウエルごとに0から100ミリアンペアを供給する。より一般的には、電流源から0−10ミリアンペアを供給する。有利なことに、コンピューターで制御され、選択可能な電流源/電圧源を使用しうる。好ましい選択可能な供給源とその使用方法は特許文献4に開示されていて、その明細書全体が参考用として本出願書に組込まれている。
【0028】
代わりに、本発明を実施するのに必要な電子要素はより単純なもので良く、例えば、直流電圧源及びサンプル電極の配列を含むだけでよい。この代替的実施例では調節可能な+/−100ボルトの電圧源(通常<25ボルト)を用いて、電気泳動に必要な電場を供給している。例えば、25個のサンプル分析システムでは、25個の電流計を使用し、それぞれが(1Hzの報告周波数での)電流値を報告するためのサンプル及び保持の回路を有している。希望する場合、結果はコンピューターのモニターにグラフとして表示される。代わりに、又、さらに単純にする場合には、電気泳動法を電圧源のみで実行しうる、即ち、電流を監視しないが、事前決定された時間だけ電気泳動法を実施するか、又は、代わりに検出可能な(視覚的、化学的又は電気的)終点に達するまで実施する。
【0029】
以下に示す方法を実施するのに適当な装置には、+/−100V電源、25チャンネルの個別に調節できる電位差計、Agilentモデル34970Aデータ取得/スイッチ、25個のウエルのLexanカートリッジ、ラップトップ(laptop)・コンピューターが含まれる。Agilentデータ取得システムのためのソフトウエアで、25個のサンプル・ウエルのそれぞれについて時間の関数として電圧と電流を記録するように構成しうる。Applied Biosystems Voyager DF and 4700 model MALDI質量分析器を用いて、タンパク質とペプチドを含む検体の質量分析法と定量化を行なえる。
【0030】
システムの操作説明:
1.サンプル分子の第一群と第二群の混合体をサンプル・ウエル4に置く。
2.サンプル電極20をサンプル・ウエル4内のサンプルと電気的に導通させる。
3.サンプル電極20に電圧源100から電気を通じて、サンプル・ウエル4内で誘導電流の反応(即ち、酸化又は還元の反応)を生じさせ、それにより、イオン電流200を電極20からサンプル・ウエル4を通過し、分離層30を通過し、捕捉材料40を通過し、電解質ベース媒体62を通過し、対極チャンバー72内の対極電解質を通過し、最後に、誘導電流による酸化又は還元反応(サンプル・ウエル4内のサンプル電極20で生じるものとは反対)を対極70で生じさせる。
4.イオン電流200が電場を生じ、それが最初に帯電したサンプル分子を電気誘導的に
駆動して分離層30を通過して、カートリッジ捕捉スライド42上のオリフィス50に位置した捕捉材料40上に集まる。
5.同時に第二のサンプル分子は分離層30を通過しない。これは、無帯電か又は第一サンプル分子の帯電とは反対になる結果によるか、又は、第二の分子が分離層30内に留まるか、遅らされる結果による。
6.カートリッジ捕捉スライド42上に第一サンプル分子の捕捉後に、スライドをカートリッジ・ウエル・フレーム6から取外す。
7.カートリッジ捕捉スライド42を脱イオン水で、又は、適当な溶媒で洗浄して、質量分析法のような分析と干渉する塩及び他の物質を除去する。
8.MALDIマトリックス溶液を捕捉スライド42上の捕捉材料40に加える。そして乾燥させる。
9.捕捉材料40に添加された乾燥MALDIマトリックスを有する捕捉スライドをMALDI質量分析器に挿入される。そして、第一検体の質量をMALDI−MSにより分析される。例えば、各(m/z)ピーク高さ又はピーク面積の平均及び標準偏差はサンプル・ウエル4に加えられるサンプル材料の量又は加えられたサンプル材料の発生源の関数として決定される。(例えば、共通の特徴、医学的徴候又は診断を共有するヒトのグループから採取したサンプル)(ここでmは質量、zは単位電荷を意味する)。
【0031】
例えば、ステップ8及び9で、そのように調製されたMALDI捕捉スライドの分析の間に、適当な溶媒に溶解されたMALDIマトリックスの小さな液滴が検体捕捉領域に加えられる。溶媒は検体を溶解でき、溶媒が蒸発すると共に検体が捕捉膜の上面に形成されるMALDIマトリックス結晶内に組込まれる。溶液の蒸発とMALDIマトリックス結晶の形成のために、通常、1分から60分の時間の後で、サンプル・プレートはMALDI質量分析器に導入できる。MALDIサンプル・プレートを質量分析器に挿入する時にMALDIマトリックス結晶が強いUVレーザー光のパルスで照射され、検体分子の一部のイオン化を生じる。これはMALDI−MS分野の技術者に良く知られている。
【0032】
別の例によると、ステップ5で、ポリアクリルアミドが分離層30として使用される。そのようにポリアクリルアミドが用いられるとき、ポリアクリルアミドに含まれるアクリルアミド又はビス・アクリルアミド(bis acrylamide)が十分に高い濃度、架橋結合、厚みになり一定分子量(又はm/z)より小さい分子のみが分離層を通過できる。選択された分子量がタンパク質の場合約30,000ダルトン、即ち、タンパク質のLMW成分である特別の場合、分離層は、生体組織からの30,00ダルトンより大きな高存在度タンパク質を除去するのに使用される。生体組織には、脳、筋肉、肝臓、肺、膵臓、卵巣、睾丸及び特に血漿と血清が含まれる。例えば、血清の場合、分離層がアルブミン、IgG、IgA、ヘモグロビン、ハプトグロビン(haptoglobin)、トランスフェリン(transferin)を除去する。これらは通常この変更組織内のプロテイン総質量の約95%になる。代わりに、1%アガロースのような非ふるい分け(non−sieving)ゲルをカートリッジ・ゲル・プレート32に組込んで、高分子量のタンパク質の除去無しで、分離を実行しうる。カートリッジ捕捉スライド42の1以上の捕捉材料40上に1以上の検体の捕捉後MALDIマトリックスが捕捉材料40及び前記のようにMALDI質量分析器により、第一及び第二の結合された分子について分析された材料に加えられる。
【0033】
本発明の好ましい実施例はサンプル・ウエル4の配列を有し、各ウエルが、上方開口部8、側壁10、下方開口部14を有し、又、カートリッジ・ウエル・フレーム6内に収納されている。この好ましい実施例は、又、各サンプルウエルごとにひとつの対応するサンプル電極20の配列と分離層30の配列を有している。好ましくは、分離層30の配列はカートリッジ・ゲル・プレート32内の配列として含まれ、そこではサンプル・ウエル4の下方開口部14と位置合わせするように、ゲル・プレートが適当なピッチ(pitch
)で間隔を置いている。分析の後で、1以上の捕捉材料40の配列を含むスライド42は、後日、再検査又は検証を実現できる。
【0034】
サンプル・ウエル4の配列を有するカートリッジ捕捉スライド42は、単一の捕捉スライドとして、又は、2以上の直列に積み重ねられたカートリッジ捕捉スライドの積み重ねとして存在し、検体は2以上の捕捉スライド内に存在する各捕捉材料40を直列に通過する。そのような2以上の捕捉スライドの積み重ねが存在するとき、各スライド内の捕捉材料は実質的に同一であり、又は、代わりに、実質的に異なる。有利なこととして、継続的に直列の捕捉スライドの中で、以下に詳細に示すように、実質的に異なる捕捉材料が種々の検体を選択した捕捉スライドに分けるために使用しうる。
【0035】
例として、捕捉スライド42内の捕捉材料40は単一材料、例えばMillipore
Corp.,Billericia,MA(USA)から入手できるImmobilon−P又はImmbilon−PSQとして得られる多孔性ポリビニリデンジフルオルド(polyvinylidene difluorde)(PVDF)から作られる。特許文献5に詳細に示されているように、熱、超音波又はレーザーによる溶接により、多孔性PVDA捕捉材料を捕捉スライド42に取付けて、捕捉層40を形成する。MALDI−MSによる分析のためにポリアクリルアミド・ゲルから電子ブロッティング(blotting)によりタンパク質を捕捉するためにPVDFを用いることについて詳細な文献が存在する(非特許文献5,6,7,8)。さらに、有利なこととして、MALDI−MSによる分析中に、導電材料の薄い層でそのような膜を被覆することはそのようなPVDFの耐電を防止する(非特許文献9)。
【0036】
図2に示すように捕捉スライド42の連続的な層の数を2以上に増加することにより、サンプル検体の分別を高められる。この実施例では、カートリッジ捕捉スライド42を積み重ねて、検体の分子が各カートリッジ捕捉スライドの捕捉材料40を通過する。捕捉スライド42の連続的捕捉材料内のPP分子の分別が、捕捉スライド42の2以上の連続的層のそれぞれに実質的に異なる化学的又は物理的表面特性の捕捉材料40を用いることによりかなり改善されるので、それぞれがサンプル内のPP(タンパク質とポリペプチド)の構造的に異なる分子に対して実質的に異なるアフィニティを有する。
【0037】
捕捉スライド42上の多数の連続的層でサンプル・タンパク質の分別を実施するため、各捕捉スライドは膜から成る捕捉材料40を有する。それで、装置の運転中に、サンプル検体、例えば、タンパク質又はポリペプチドが、連続的に電気泳動的に駆動されて、連続的に2以上の捕捉膜を通過する。有利なこととして、連続的に用いられる各捕捉膜が異なるクラスの検体に対して実質的に異なるアフィニティを有している。異なるアフィニティを持つそのような膜の例はPVDF又は他の多孔性ポリマー、検体に対する膜のアフィニティを変える低分子量の修正材料で被覆された膜が含まれる。例えば、疎水性の膜に段階別濃度の親水性ポリマーで被覆し、次ぎに、高分子量の膜材料に親水性ポリマーを不可逆的に結合する反応ステップを実行する。例えば、多孔性PVDF膜(例えば、Millipore Corp.,Billericia,MA(USA)から入手できるImmobilon−P又はImmbilon−PSQ)は種々の溶液で被覆しうる。この場合、種々の溶液のそれぞれが中立の親水性ポリマーの種々の濃度を含む。そのような低分子量のポリマーの例には以下が含まれる。
1.ポリエチレングリコール(PEG),例:Fluka Cat.No.94646,分子量35,000
2.オリビニールピロリドン(PVP),例:Sigma Cat.No.PVP40T,分子量40,000
3.ポリビニルアルコール(PVA),例:Sigma Cat.No.P8136,分子量 30,000
【0038】
そのような低分子量ポリマーを高分子量ポリマーの膜に被覆し、不可逆結合をするための手順は従来技術で良く知られている。Nafion.RTMのような高帯電ポリマーをPVDF膜に被覆し、不可逆結合をする例示的方法は米国特許に示されている(非特許文献10)。低分子量ポリマーを高分子量PVDFに不可逆結合するために、この方法は、PVDFの溶融温度より低い温度での焼き付けを用いる。直接的であるが、この方法は、膜に被覆用ポリマーのかなりの部分を非共有結合的に結合させている。このゆるく結合された被覆用材料がその後MALDI−MS分析中の検体のイオン化を抑制する。有利なこととして、グルタルアルデヒド(glutaraldehyde)のような種々の化学的架橋結合用試薬を用いて膜にポリマーを非可逆的に共有結合を行う。例えば、その架橋結合用試薬は、従来技術で知られているように、ヘテロ又はホモの二官能価架橋結合試薬である。
【0039】
被覆及び不可逆結合の実施後、電気泳動により駆動される分別の手順の最適化が行われる。行われた実験では小さいが強く帯電したペプチド及びタンパク質が、PVDFベースの捕捉膜上に最初に捕捉されることを示している。分離時間を段々に延ばす(又は、代わりに、印加電圧を高くする)ことにより、段々に大きなタンパク質がPVDFベースの捕捉膜上に捕捉される。さらに、これらの実験は捕捉されたペプチドの一部が捕捉膜から有機溶媒(又は有機溶媒を含むMALDIマトリックス溶液)により溶出でき、MALDI質量分析法により定量的に検出されることを示した。さらに、血清サンプル内に見いだされるタンパク質の連続的分別部分を膜の目標上で捕捉できる。分別手順は以下の方法で最適化できる。
【0040】
(最適化の方法)
1.標準のタンパク質・サンプルの測定された標準体積を、カートリッジ・ウエルのそれぞれに同じ測定された体積をピペット(pipet)で移すことにより(例えば、2μLのヒトの血清・サンプル又は1以上のタンパク質又はポリペプチドの他の適当な標準混合物として)加える。
2.事前決定された実行時間の間、膜を横切るように電流を通過させることにより、膜の面に垂直な電場を加える。例えば、十分な電圧を加え、5から120分の範囲の実行時間の間、膜面積の平方mm当たり0.1から10mAの電流密度が加えられる。電場が加えられる時間中に、捕捉材料40のそれぞれを通過する電流が監視され、又、プロット(plot)され、マルチ・ウエル(multi−well)捕捉スライド42に配置された捕捉材料の配列内の種々の場所に存在する捕捉材料40内の電場で均一性と再現性を確保する。膜を通過する電流が、捕捉材料内の帯電したサンプル検体の電気的濃度を生じる。3.電気濃縮手順が完了した後で、PPASカートリッジから捕捉スライドを外す。
4.捕捉スライドを洗浄し、塩その他の干渉物質を除去する。
5.MALDIマトリックス溶液を捕捉スライド42上の捕捉物質に添加し、乾燥させる。
6.捕捉スライドをMALDI質量分析器に挿入し、MALDIにより分析する。例えば、各ピーク高さの平均と標準偏差を用いられた血清サンプルの量の関数として決定される。
7.ステップ1からステップ6までを少なくとも2回以上繰返すことにより最適化を行う。各回ごとに、電流密度、実行時間、又は、電流密度と実行時間の両方が変る。一般に電子密度は、膜の平方メートル当たり0.1から10ミリアンペアの電流密度(又は、寄り一般的に、捕捉スライド42の穴50の平方ミリメートル当たり0.1から100ミリアンペア)であり、実行時間は5分から120分の間である。標準サンプルから質量分析器により検出された最大数のタンパク質又はポリペプチドのピーク、又は、どれか1以上のピークで最大強度を与える状態(電流密度と実行時間)が「標準最適化状態(standared optimized condition)」として採用される。
【0041】
最適な方法は生体サンプル例えばSigma Chemical Companyから購入した通常のヒト血清(100mL)又は市販の同等品を用いて実行される。代わりに、そのような生体サンプルは、血漿、尿、脳脊髄液、腹水、唾液等のような他の生体液である。他の適当な生体サンプルには、生体組織からの又は細胞培地から得た溶解した細胞を含む。最適化方法は、手順内の1以上の追加パラメーターを変えながら、連続的に1回以上繰返して良い。追加パラメーターは、サンプル要素(例えば、pHと電導度)と体積、電解質緩衝液の要素、時間、電流密度、捕捉材料、MALDIマトリックス溶液の要素又は緩衝液又はサンプルの体積のようなものである。最適化方法でMALDI−MSにより得られたデータは分析され、そして、質量分析法内で認識されたPP検体のピークの数と高さを最適化するように相互に関連付けられる実験パラメーターと分析結果が比較される。
【0042】
上記の一実施例で、ステップ#2で電場を加えている間の電流−電圧の関係を時間の関数として測定される。ステップ#2を終了する時点を決定するために、電流−電圧の関係から捕捉材料40を通過する抵抗での電荷を一定時間に亘って計算する。5分から45分の5分間隔を含む個別期間に、捕捉膜の配列上に種々の電気泳動による動きの部分を捕捉するために、タイム・コース(time−course)を実施する。得られたデータは時間をベースとしたLMWヒト血清の分別に対する有効性を決定するために分析される。この時間ベースの分析は、選ばれた分子量の範囲の血清ペプチド及びタンパク質の分析手順として使用しうる。最初の部分の目標となるペプチドは約1−2,000ダルトン、続く部分は、2−5,5−10,10−15,15−50、50,100,100−200,及び>200の×1000ダルトンである。各分子量範囲の分析に対して、各分子量範囲で単一サンプルを分析し、スペクトル(複数)を結合して、完全なプロテオーム・プロフィル(proteome profile)を提供しうるように、関連する標準運転手順書(SOP)が選択される。
【0043】
分析と最適化を実施する前に、血清を10マイクロリットルから10mlのアリコート(aliquots)、例えば、450μLのアリコートに分割され、そして、−80℃で保存される。例えば、以下の実験を実施して、PPASを用いたpHベースのLMW血清サンプルの分別を実証しうる。サンプル緩衝液内のペプチド/タンパク質の標準を(及び、通常のヒトの血清にスパイク(spike)された標準を用いて)検出するための装置の感度と再現性を、検体が安定するように選択されたpH値で検査しうる。例えば、便宜的にpH7.0のタンパク質及びペプチドが用いられるが、3から11のpH値も用いられる。(捕捉膜が評価しうるほどのイオン交換特性を有さないので、又、未結合の緩衝用の種がMALDI質量分析器により検出される前に捕捉膜から洗い流されるので、緩衝用の種は臨界的でない。)この目的に用いられる最初のペプチド・タンパク質の標準は、ユビキチン(ubiquitin),グラミシジン(gramicidin),シトクローム(cytochrome)C,インスリン酸化B鎖(insulin oxidized B Chain)及びACTH破片(18−39)である。別の適当な標準タンパク質を、PPASによりカバーすべきタンパク質の各分子量の適用範囲に追加しうる。(ノイズより高い標準偏差の3倍として定義された)ヒトの血清内の各標準を検出する感度を決定する。約20のPPASカートリッジを分析して、システムの再現性を決定する。カートリッジの半分は負の電気濃縮モードで処理する(即ち、負に帯電した検体をサンプル・ウエル4から電気泳動的に駆動して捕捉材料40上に濃縮する)。他の半分は正モードで処理する(即ち、正に帯電した検体をサンプル・ウエル4から電気泳動的に駆動して捕捉材料40に濃縮する)。得られた方法は各サンプルを5以上の成分に分別するのに使用しうる。
【0044】
PVDFのような事前成形された膜を用いる代わりに、捕捉材料40に対して実質的に
類似の機能をする捕捉材料を捕捉スライド42内のオリフィス50に投入する。例えば、捕捉材料は疎水性で多孔性のポリメタクリレートとしうる。このポリメタクリレートとしては、ポリ(ブチルメタクリレート)、ポリ(メチルメタクリレート)、ポリ(エチレンジメタクリレート)、ポリ(ベンジルメタクリレート)、又は、これらのポリマーの混合体、例えば、ポリ(ブチルメタクリレート−コ−エチレン−ジメタクリレート)が挙げられる。代わりに、捕捉材料は親水性多孔質のポリメタクリレートであっても良く、そのようなものとしては、ポリ(2−ヒドロキシエチルメタクリレート)、ポリ(グリシジルメタクリレート、ポリ(ジエチレングリコールジイメタクリレート)、又は、これらの混合体である。
さらに、より有利なこととして、捕捉材料が疎水性が正確にある範囲の疎水性から選択されるように親水性ポリマー及び疎水性ポリマーの混合物から形成されうる。当業の技術者に良く知られている多数の手順に基づいて、キャストポリマーを捕捉スライド42のオリフィス50の側壁56に堆積させ、又、付着させ、取付けても良い。(非特許文献ll,12,13,14,15,16,17,18,19,20,2l,22)
【0045】
捕捉スライド42製造の好ましい実施例で、捕捉スライド内のオリフィス50の側壁56が最初にビニル化され(vinylized)、壁56に多孔性のモノリス(monolith)ポリマーの共有結合が可能になる。ビニル化手順では、オリフィス50を最初にアセトンで洗浄し、次ぎに脱イオン水で洗浄し、0.2モル/リットルの水酸化ナトリウムで30分間活性化し、水洗した後、0.2モル/リットルのHCIで30分間処理し、最後にエタノールで洗浄する。次ぎに、酢酸を用いてpHを5に調節し、95%エタノール中20%が3−(トリメトキシシリル)プロピルメタクリレート((3−trimethoxysilyl)propyl methacrylate)の溶液から成っているメタクリレート(methacrylate)の重合混合体は、1mmの深さのモノリスを通って30分間洗い流される。エタノールで洗浄し、窒素流で乾燥した後で、機能化したスライドが24時間室温で放置される。次ぎに、オリフィスがメタクリレートの重合混合体を用いて注意深く一杯になるように充填される。開示されたように、その混合体はメタクリレート・モノマー及びポロゲン(porogen)溶媒を含む(非特許文献ll,12,13,14,15,16,17,18,19,20,2l,22)。
【0046】
疎水性モノマーを重合混合体に組込むことで、疎水性モノリスを製造できる。同様に、親水性モノマーを選択することで、親水性モノリスを製造できる。さらに、任意の事前決定された比での親水性と疎水性のモノマー混合体を用いて、希望の親水性又は疎水性のモノリスを製造できる。これらいずれの場合でも、(赤外線照射を除去するために)水フィルターに組込まれたキセノン・ランプを使用して重合を開始できる。150ワット以上のキセノン・ランプを用いることで、約10cmの距離で約10分間照射後に重合は完了する。重合の後で、重合混合体内のポロゲンとして機能する溶媒を、例えば、シリンジ・ポンプ(syringe pump)により送られるメタノールの加圧流を用いて洗い流す。代わりに、12時間以上ポロゲンを洗浄液に単純に拡散することにより除去しうる。多孔性のモノリシック(monolithic)ポリマーは小ビーズ(beads)から成るポリマーと比較していくつかの利点がある。例えば、モノリシック・ポリマーは活性表面積を有意に増加しうる。さらに、モノリシック・ポリマーはオリフィス50の壁に直接付着できる。
【0047】
分別と捕捉のステップの後で、カートリッジ捕捉スライド42の各層を分離し、ここで示すように質量分析器で個別に解析しうる。2以上の捕捉層にする追加分別により(各捕捉膜に組込まれたアフィニティにより示される)タンパク質について多くの情報を提供し、又、MSによる検出感度も高めている。(なぜなら、各捕捉材料がPPの全分子を比例的に低減していて、実質的に同じPP分子の大きな部分を各捕捉材料40に組込まれるからである。)
【0048】
図4は、Applied Biosystems,Inc./Sciex Voyager DE MALDI TOF質量分析器のための標準スライド・ホルダー90に直接挿入されるカートリッジ捕捉スライドの好ましい実施例を示す。カートリッジ捕捉スライド42は低導電性材料から作られているので、電気泳動の電流の10%以上が、カートリッジ捕捉スライド42内の捕捉材料40も含む穴50内の電解液を通過する。より一般的には、カートリッジ捕捉スライドの導電性は、電流の75%から99.999%がカートリッジ捕捉スライド42内の捕捉材料40も含む穴50内の電解液を通過する程のものである。装置の運転中にこれを達成するために、通常、カートリッジ捕捉スライド42を作るのに用いる材料の体積抵抗は102から1010Ω−cmの間になる。より一般的には、カートリッジ捕捉スライド42の体積抵抗は104から106Ω−cmになる。しかしながら、この材料の僅かな導電性がMALDI−MS分析によるその後の分析中に捕捉された電解液のイオン化の間、捕捉スライド42の帯電を防止する。
【0049】
有利なこととして、カートリッジ捕捉スライド42も非常に平坦で、又は、代替的に、図4に示されるように、MALDIスライド・ホルダー90に挿入することにより+/−50ミクロンの平坦度になるように設計されている。カートリッジ捕捉スライド42がサンプル・ホルダー90に、機械的ガイド92により、又は、代わりに、磁石のような強磁性材料により、取付けられる。例えば、磁石は小さな希土類磁石例えばネオジウム・鉄・ホウ素(NdFeB)の厚み約1mm、直径約2mmの磁石である。強磁性材料が、捕捉膜上のサンプル検体のMS分析中に、下方要素のフレーム部材(及び取付けられた捕捉部材)をMALDIサンプル・プレートに保持するように機能する。この目的のためにこれらの磁石は(#318ステンレス鋼)に十分な力で保持する。
【0050】
運転中に、帯電した可動検体は加えられた電場内を、分析システムのカートリッジ・ウエル・フレーム6内に配置された多数のサンプル・ウエルの配列内にあるサンプル・ウエル4から移動する。各サンプル・ウエル4は1以上のサンプル検体(例えば、PP's)を有するサンプルから成る電解液を保持するのに役立つ。さらに、各サンプル・ウエルは、流体との電気接触を確立するために電解液に挿入するサンプル電極20を受入れるのに役立つ。電圧が共通対極70に対して電極20に加えられるとき、イオン電流がウエル4内の流体を通過し、ウエル4はサンプルとpH緩衝用電解希釈液からなり、それにより、サンプル・ウエル4内の電場を生じる。その電場により、電気泳動的動きを生じて、サンプル・ウエル4内の1以上の電解液を分離する。有利なことに、捕捉材料を含む穴の断面積がサンプル・ウエル2の断面積より実質的に小さく、穴50の中にある捕捉材料40内の検体を電気泳動的に濃縮する。分析システムの好ましい実施例には、1から400μLのサンプル体積を収容するサンプル・ウエル4が含まれる。各ウエルの内径は上方開口部が約6.7mmで、下方開口部が約1.0mmに狭くなり、捕捉スライド42内に捕捉材料40を含む小直径の穴50に、電気泳動によって検体分子を濃縮できる。ウエルはそのウエルの内側側壁10の下方部分12内に狭くなっている。一般に、そのような下方部分12内の側壁はウエルの全体として垂直な中心軸から20度から30度の傾斜になっている。好ましい実施例では、その傾斜がウエル2の中心軸から24度から26度の間である。一般に、サンプル・ウエル2の直径は5−20mmの間にあり、捕捉領域の直径は10ミクロンから1.5mmの間である。
【0051】
サンプル・ウエル2の下方開口部14を分離しているのは、薄い分離層30である。分離層はふるい分け材料(例えばアクリルアミド)から成っていて、イオンによる導電性と流体同士の接触を維持している。ふるい分け材料は事前成形され、組立てられている。又は、現場成形されうる。現場成形の場合、ポリアクリルアミドの層は、液状アクリルアミド・モノマーと架橋剤をウエルの中に希望の厚みになるように注入することにより行われる。次ぎに、液体を、例えば、過硫酸アンモニウムのような遊離基連鎖開始剤を組込むこ
とにより、又は、リボフラビン(riboflavin)のような光線感作物質を添加すること、又、例えば、UV光又はリボフラビンに対しては400−450nmの光のような光線感作物質が吸収する波長の光で照射することにより、組立前に重合させる。さらに、分離層30が1以上の直列配置で積上げ可能なふるい分け又は分離の層として提供できる。例えば、アガロース・ゲル(agarose gel)を、多孔性のポリアクリル・アミド層、多孔性の透析膜又はその両方を直列配置の組合わせで使用しうる。そのような直列配置の組合わせで多孔性のアガロースは最初の前フィルター(filter)として機能し、サンプル検体、又は、高い存在度のタンパク質のような緩衝物質による過剰負荷から多孔性のポリアクリル・アミド層を保持する。その一方で、ポリアクリル・アミドは第二の前フィルターとして、電気泳動による濃縮の際に、透析膜がタンパク質で詰まるのを防止する。
【0052】
一般的に、捕捉材料40が穴50に含まれ、カートリッジ捕捉スライド42を横切るように通過する。帯電した検体が電気泳動により駆動されて、分離層を通過する。次ぎに、1以上のカートリッジ捕捉スライド(CCS)上のアセンブリーの捕捉材料40に捕捉され、連続的なカートリッジ捕捉スライドのオリフィスが同軸的に配置され、検体は各穴内の多孔性捕捉材料40を連続的に通過する。それにより捕捉された検体が、捕捉スライド42内に保持された捕捉材料40の中で、大きな体積のサンプル・ウエル4から小さな体積に濃縮される。さらに、多数の検体が、連続的カートリッジ捕捉スライド内の捕捉材料により分離され、捕捉される。それにより、実質的に検体が別のスライド部分に分別される。カートリッジ捕捉スライド42のアセンブリーが一連の積み重ねられた層として1以上の連続的捕捉スライド、通常、1−10個、より一般的には1−5個の連続的スライドから成り、可能性としては、1−100個以上の連続的スライドから成っている。捕捉スライド42の連続的な層の積み重ねは、運転中にイオン電流がスライド42の各層を、第一の連続的捕捉スライドから、次ぎに、第二の連続的捕捉スライド等と、最後の連続的捕捉スライドまで連続的に通過されるようにして組立てられる。
【0053】
有利なことに、捕捉スライドの穴50内の捕捉材料40は改質された捕捉材料を含み、その改質は選択した検体に対する捕捉材料40のアフィニティを高める。さらに、そのように改質された捕捉スライドを種々の検体に対して差別的に高いアフィニティを持つように改質しうる。さらに、種々の検体に対して差別的に高いアフィニティを持つそのように改質された捕捉スライドは、検体が第一の捕捉スライドに、次ぎに第二、さらに第三の捕捉スライド等に遭遇するように逐次積み重ねられる。各捕捉スライド42は種々の検体に対して、差別的に高いアフィニティを持つ捕捉材料40を有する。第一番の連続的捕捉スライド42は最初に選択した検体に対して高いアフィニティを有している。二番目の捕捉スライド42は二番目に選択した検体に対して高いアフィニティを有している。さらに、三番目に選択された捕捉スライド42の捕捉材料40は三番目に選択した検体に対して高いアフィニティを有している等、順番に行われる。それにより、第一、第二、第三の検体の連続的捕捉スライドへの分別が便利にかつ迅速的に行われる。
【0054】
選んだ捕捉スライドに対して高いアフィニティを有する検体を事前決定する。例えば、ある検体が、検体−反検体の結合ペア(pair)のメンバー(member)であり、捕捉材料40が反検体の接合により修正された場合、事前決定された捕捉スライド42の捕捉材料40が、事前決定されたスライド内の事前決定された検体の特定捕捉になる。例えば、ある検体が抗体により認識できる抗原性エピトープを有しているので、事前決定された連続的捕捉スライド42内の捕捉材料40へその特定抗体を固定することにより、事前決定されたスライド42内の事前決定された検体の特定捕捉になる。抗体の代わりに、結合ペアの相補性メンバーを用いて、相補性検体を結合するのに用いられる。この場合の相補性メンバーはリガンド・レセプター(ligand−receptor)のペアと呼ばれる。リガンド・レセプターのアフィニティは高アフィニティ又は低アフィニティを有
するように選択できる。種々の検体が、異なる検体に対する異なるアフィニティを有する連続的捕捉スライドにより選択的に捕捉される。それにより、検体の分離層への分別が実現される。
【0055】
各捕捉材料40が、剛体の固体支持から成るカートリッジ捕捉スライド42に取付けられ、それにより、捕捉材料のその後の処理を容易にしている。処理には、洗浄、乾燥、MALDIマトリックスの使用、第二の乾燥ステップ、MALDI質量分析器での質量分析法が含まれる。種々の検体に対する同じ又は異なるアフィニティを持つ多数の捕捉材料が、それゆえ、多数の捕捉スライドの穴50内に挿入でき、直列に積上げられ、お互いの穴の位置合わせするように、検体が種々の捕捉材料を連続的に通過するようにしている。
【0056】
例えば、各スライドは、多孔性のポリマー膜を持つ1以上の小さなオリフィスを有するポリプロピレン・フレーム、捕捉領域を含む1以上のオリフィスのそれぞれに対する、モノリス成形、溶接付け、糊付け、その他の取付け部から成っている。
【0057】
捕捉スライド42の穴50内の捕捉材料40がカートリッジ捕捉スライド42の上面44と下面46と導通していて、それに電流が流れている。捕捉スライド42の下面への電気接触が電解質ベースチャンバー60内に含まれる(アガロース・ゲルのような)イオン導通(電解質)媒体から成る電解質ベース媒体62を通過する。電解質ベース媒体は共通対極を収容する対極チャンバー72内に含まれている対極電解質を通じて共通電極70と電気的接触をする。電圧がサンプル電極20と共通電極70の間に印加されたとき、電流が2電極の間に流れるようにシステムが作られる。電極間に配置された電解質内でイオン物質により電流が流れる。それ故に、サンプル・ウエル内に存在する帯電した電解質が電極20又は対極70に向って電気泳動的に駆動される。サンプル電極20と対極70の間に事前決定された極性の電圧が印加されたとき、電極70に向って駆動された電解質が電路内に存在する捕捉材料40内に濃縮される。対極に対して2以上のサンプル電極20に選択した電圧極性を加えることは、捕捉スライド42内の2以上の対応する捕捉材料に別個に、又、同時に濃縮させる。サンプル・ウエルに加えられた電圧は両方が正に、又は、両方が負の極性になるように選択しうる。それゆえ、正に帯電した又は負に帯電した電解質は単独に又は同時に別々に捕捉材料に濃縮される。代わりに、サンプル電極の極性があるウエル内を正に、他のウエルを負に事前決定される。それゆえ、負に帯電した、又、正に帯電した検体を、単一の捕捉スライド42内の2以上の異なる捕捉材料に同時に捕捉する。分析システム300内で、個別電気回路はサンプル電極からサンプル・ウエルを通じて、分離層を通じて、捕捉スライド42内の穴を通じて、電解質ベースチャンバーを通じて、対極チャンバーに含まれ、共通対極70と接触している対極電解質72を通じて接続される。有利なことに、分析ステップには、解離と分離のステップが含まれ、低存在度の検体分子から高存在度の検体分子の枯渇を生じる。そのようなステップは、質量分析法によりペプチド及びタンパク質の検体の高い感度と再現性に有用である。
【0058】
そのような解離及び分離のステップは、非イオン性洗剤の添加又はサンプル・ウエル4内に存在する他の適当な解離要素を用いることにより行われる。例えば、電圧の印加ステップ前に、洗剤を適当なpH緩衝の電解液に加えられる。代わりに、洗剤をサンプルに、又は、サンプル・ウエル4内に存在する他の試薬を加える。非イオン性洗剤は疎水性ペプチドをアルブミン及びIgGのような大きな分子量で高存在度の分子から効果的に解離する。次ぎに、電圧(及びそれによる電流)が、サンプル及び共通対極の間に加えられたとき、(加えられた電圧の符号及び電解液上の電荷の符号により)正極又は負極の方に、サンプル内の選択された電解液の帯電が駆動される。
【0059】
サンプルの選択されたpH値で、正帯電と負帯電の検体の二成分分離が行われる。例えば、サンプルのpH7.8で処理し、共通対極に対して、正電圧のサンプル電極20に正
電圧を加えることで、正の電流がサンプル電極20から共通対極70へ流れる。正の電流は対応するサンプル・ウエル4内の検体を正に帯電させ、ウエル4から移動させ、対応するウエル4の直下の穴50に存在する捕捉材料40内に(捕捉スライド42上に)捕捉させる。(即ち、装置は正モード(mode)で運転と言われる)。逆に、負の電圧をサンプル電極に加えたとき、負の電流が、サンプル電極20から共通対極70に流れる。負の電流はサンプル内の検体を負帯電させ、サンプルから移動し、対応するウエル4直下の穴50に存在する捕捉材料40内に(捕捉スライド42上に)捕捉される。(即ち、装置は正モード(mode)で運転と言われる)。負又は正のモードで、各サンプル電極から流れる電流は通常10マイクロアンペアから10ミリアンペアである。より一般的には、電流は0.2から2.0ミリアンペアである。
【0060】
通常、少なくとも2個のサンプル・ウエルが1サンプルの分別に用いられる。サンプル・ウエルのひとつで、共通対極に対して、サンプル電極が正極性で、他は負極性である。即ち、正モードと負モードの分離が同時に行われている。正帯電と負帯電の検体の分離は、システム・オペレーターにより事前決定されたサンプルpHで同時に生じる。それゆえ、事前決定されたpHで正帯電と負帯電のものへのサンプル検体の分別を同時に行なえる。さらに、単一サンプルを種々の等電点の2以上の部分に分割することは、2種類のpH値を有する2以上のサンプル・ウエル内のサンプル緩衝液を用いることにより可能になる。それにより、本発明の別の実施例で、等電点に基づく検体の分別、濃縮、捕捉が以下に開示しているように、実現しうる。
【0061】
等電点に基づく検体の電荷ベースの分別に加えて、選択肢として、ふるい分け材料がサンプルと捕捉材料の間の分離層で使用される。サンプル・ウエル4から対応する捕捉材料40に向って電気泳動的に駆動された検体がふるい分けによる分離層30を最初に通過しなければならない。それゆえ、ゲル電気泳動法の分野の技術者に良く知られているように、一定のm/z値を持つ低分子量の検体に対して、同じm/z値を持つ高分子量の検体の移動を遅らせることにより、追加分別を行うのに役立つ。(洗剤(例えば、ドデシル硫酸ナトリウム)が分子量にほぼ比例してタンパク質に結合するように存在して、それゆえ、全タンパク質に同様のm/z値を与えるとき、ポリアクリル・アミド・ゲルを通過するタンパク質の移動時間はタンパク質の分子量の対数にほぼ比例していることが良く知られている。)それにより、種々の分子サイズのタンパク質に対してm/z値が類似しているとき、ふるい分け材料がLMWプロテオームを分離するのに使用しうる。ふるい分けによるそのような分離で、事前決定された範囲の分子量でタンパク質の最適分離を行うため、事前決定された電圧又は電流の使用時間が選ばれる。低分子量(即ち高い移動性)の検体がふるい分け層を迅速に通過する。それゆえ、低い移動性の検体より前に、捕捉材料40上に捕捉される。それゆえ、本発明により機械的分離が行われる。そのような高い移動性の検体は単一の捕捉材料に濃縮され、又は、一連の2以上の積み重ね可能な捕捉スライドを通過することにより、その後の分離は組合わせで行われ、2以上のスライドのそれぞれが少なくとも1個の穴が隣接する捕捉スライドの他の穴と同軸に配置されている。さらに、順番に、各穴が種々の事前決定された捕捉材料を有している。それにより、アフィニティに基づいて検体の分離を行い、又、それにより、最少数の捕捉スライドを用いて、最大の分別を行う。例えば、種々の捕捉材料はその捕捉材料の疎水性の違いから成っている。別の例によると、サンプル・ウエルに最も近い位置にある上方の捕捉材料は、最小の疎水性であり、サンプル・ウエルから直列配置で最も遠い捕捉材料が最も疎水性である。それにより、疎水性の勾配が、その疎水性に基づき検体の分離と分析を行なえるように、作られる。アフィニティにより行われた連続的スライドによるそのような分別を用いて、次ぎに、分子量とアフィニティの両方による分離を組合わせて、又、組合わせて同時に行われる。多様な2以上のサンプルが個別に、及び、カートリッジ2内で同時に分離される。複数のサンプルがそのような多様なやり方により同時に分離される。
【0062】
有利なこととして、分離と捕捉の層が比較的薄いことにより、分別と捕捉のステップは、分離と捕捉のステップを比較的迅速に実施できる。この目的のために、通常、分離と捕捉の層は厚みが通常20ミクロンから20mmの間になる。より一般的には、分離と捕捉の層の厚みは200ミクロンから5mmの間である。薄い分離と捕捉の層に対して、例えば、500ミクロンから2.0mmの厚みに対して、分別ステップは10秒から100分かかる。通例、分離と捕捉は1時間未満に生じる。より一般的には、分離と捕捉は1分から約10分の間で行われる。捕捉ステップの後で、PPAS装置が分解され(図3に示すように)、カートリッジ捕捉スライドのそれぞれが短時間洗浄され、MALDI又はエレクトロスプレー(electrospray)質量分析法による検出に干渉する塩又は他の化学種を除去する。例えば、捕捉スライドは脱イオン水で単純に洗浄される。洗浄ステップの後で、MALDIマトリックス溶液が各捕捉スライドの捕捉領域のそれぞれに添加される。そして、マトリックスを乾燥できる。乾燥ステップの後で、捕捉された検体の質量分析法のために、質量分析器(例えばMALDI−TOF MS)に直接スライドを挿入する。代わりに、質量分析器で捕捉材料40から直接捕捉された検体を検出するために、検体を最初捕捉材料から溶出させ、MULDI−MS、エレクトロスプレー、酵素例えばトリプシン(trypsin)によるタンパク質分解消化を含む種々の手段により検出される。そして、得られたペプチド破片の分析、即ち、タンパク質の識別と分析の分野の技術者に良く知られている「ペプチド・マップ(peptide map)又は他の分析手段を構成することによる。
【0063】
カートリッジ捕捉スライドは、カーボン・ドープ(carbon dope)されたポリプロピレンから射出成形され、電荷の拡散無しに直接MALDI解析を可能にしている(非特許文献23、24、25、26、27)。捕捉材料40は、適当な手段により、例えば、接着剤を用いることにより、又は、捕捉材料、スライド又はその両方に溶剤添加又は加熱をする溶接により捕捉スライド材料に取付けたポリビニリジンジフルオリド(PVDF)のような疎水性膜から形成しうる。代わりに、捕捉スライド内のオリフィスに捕捉材料を注入しうる。例えば、捕捉材料は多孔性のポリ(ブチルメタクリレート−コ−エチレンヂメタクリレート)のポリマー・モノリスとしうる。そのようなモノリスはSvecらにより開示された手順(非特許文献28−34)に基づく重合により成形しうる。密に結合した捕捉材料を有する頑丈な捕捉スライドのために、スライド・オリフィスの内壁面は最初にビニール化(vinylized)して、そのモノリスを壁に共有結合できる(非特許文献35)。オリフィスをアセトンと水で洗浄して、0.2mol/Lの水酸化ナトリウムで30分間活性化し、脱イオン水で短時間洗浄し、その後、0.2mol/LのHClにより30分間、最後にエタノールで短時間洗浄する。95%エタノール中の20%が3−(トリメトキシシリル)プロピルメタクリレート溶液、pH5(例えば、0.1から1.0%の酢酸を含むエタノール)で、厚さ約1mmのモノリスを30分間洗い流す。エタノールによる洗浄と窒素流内での乾燥の後で、機能化されたスライドを室温で約24時間放置する。モノマーの適切な選択によりモノリスの親水性を選択できる。次ぎに、オリフィスに注意深く過充填になるようにメタクリレート重合混合体で充填し、蒸発防止のためのカバーをし、重合せしめた。標準としては、キセノン・ランプに水フィルターを取付けて、光重合を開始するのに用いている。重合は約10cmの距離から10分間の照射の後で完了する。そして、そのモノリスをシリンジ・ポンプ(syringe pump)又は比較的遅い連続した流れを供給する他の適当な手段が送るメタノールを用いて、そのモノリスを約12時間洗浄する。多孔性のモノリシック(monolithic)ポリマーは、ビーズ(beads)から成るポリマーと比較して、有効表面積を有意に高められる。代わりに、以下に好ましい実施例として開示したように、そのような多孔性のモノリシック重合体とクロマトグラフィ(chromatography)粒子の混合体が捕捉材料40として用いられる。
【0064】
PPAS装置内で調製されたカートリッジ2内の捕捉スライド42上の捕捉材料40上
に捕捉されたサンプル検体のMALDI−MS分析のために、適当な溶媒に溶解したMALDIマトリックスの小さな液滴をその検体捕捉領域に添加する。その溶媒は検体を溶解でき、その溶媒の蒸発と共に、その検体は、捕捉膜の上面上に形成するMALDIマトリックスの結晶内に組込まれる。溶媒の液体を蒸発させ、MALDIマトリックスの結晶を形成するための時間を与えた後、そのサンプル・プレートはMALDI質量分析器に導入できるようになる。例として、図4はカートリッジ捕捉スライドと標準のApplied Biosystems,Inc/Sciex Voyager DE MALDI TOF 質量分析器用スライド・ホルダーへの直接挿入を示している。MALDIサンプル・プレートの質量分析器への挿入時にMALDIマトリックスの結晶を強いUVレーザー光のパルスで照射し、その結果、検体分子の破片のイオン化を生じる。これはMALDI質量分析法分野の技術者には良く知られている。
【0065】
(選択的洗浄の構成と方法による干渉性化学種の除去)
捕捉スライド42の穴50内に保持された捕捉材料40上への検体捕捉後に、捕捉スライドはカートリッジ2の分解により取外される。次ぎに、塩、及び、無機及び有機のpH干渉性の種を捕捉スライド及びそのスライドの穴内に保持される捕捉材料から洗い出される。しかしながら、洗浄処理中に捕捉材料上に対象検体を保持するために、洗浄剤の組成を注意深く選択する。タンパク質及びペプチドのようなPP検体を保持するために、疎水性捕捉材料のそのような選択的洗浄には、通常、実質的な水性溶媒を用いている。洗浄は、拡散、圧力に駆動された流れ、電気泳動(即ち帯電した干渉物質)によるか、又は、代わりに、電気浸透により、又は、そのような2以上の方法の組合わせにより行われる。
【0066】
例えば、洗浄溶液の圧力に駆動された流れを、捕捉スライド42を横断して差圧が加わるように設計された図4aに示すような装置により行われる。そのような差圧は、例えば、スライドの片側にある真空マニフォールド200により真空を生じて、反対側の流体槽から真空マニフォールド200に向ってスライド内の捕捉材料40を通過する流れを生じさせることによる。代わりに、正圧は正圧マニフォールド202により、スライドの流体槽を有する側に加えられ、それにより、実質的に同じ圧力で駆動された洗浄液を捕捉材料を横断して流す。どちらの場合も、捕捉スライド42が、密封用のゴム又は軟質ポリマー・ガスケット(gasket)、又は、Oリングのようなスライド密封手段206と関連して機能する圧力保持用サポート204により支持されている。有利なこととして、負又は正の圧力を用いて、捕捉スライド42内の2以上の穴50内に多様な2以上の捕捉材料40を横断して実質的に同時に流体を流れさせる。洗浄手順に用いられる流体は、例えば、脱イオン水(DI)、又は、代わりに、DI中の0.1%のトリフルオル酢酸(TFA)のような「MALDIフレンドリー(friendly)」イオン含有溶液にできて、捕捉材料40から干渉塩を排除する。一方で、捕捉材料に結合した希望のPP検体を保持することができる。そのような「MALDIフレンドリー」イオンは特徴として、陽子の減少又は増加により中性化したとき、かなりの蒸気圧を有し、質量分析法の真空チャンバー(chamber)内を迅速に「排出」できる。「MALDIフレンドリー」の材料は特定pH値でイオン化するもので、酢酸、アンモニア、蟻酸、プロピオン(propionic)酸、ピペリジン(piperizine)、ピリジン(pyridine)等であり、MALDI−質量分析法用のサンプル作成分野の技術者に良く知られている。
【0067】
代わりに、図4bに示すように、電気泳動装置300はスライド42内の捕捉材料(40)を横断した電場を加えるために用いられる。電気泳動装置は、電源302、陽極と陰極として使うための電極ペア306を有している流体槽とスライド・ホルダー304、及び、陽極を陰極から分離するように機能する隔壁308から成っているので捕捉スライド42内の穴50を電流が通過しなければならない。
【0068】
無機塩及び無機と有機のpH緩衝物質が無い捕捉スライド上の捕捉材料を最も有効な電
気泳動により洗浄する場合、以下の原則が単独で又は組合わせで用いられる。
【0069】
A.疎水性イオン交換
第一のイオン交換ステップが、MALDIフレンドリーのイオンに対するMALDI非フレンドリーの干渉物質を交換するのに使用されるが、pHが調節されるとき、MALDIフレンドリーのイオンが上記のようにかなりの蒸気圧を有する。捕捉スライド上の捕捉材料が疎水性イオンに対するアフィニティを有するとき、例えば、「逆相」クロマトグラフィの原則が捕捉検体に用いられるとき、疎水性干渉物質が捕捉材料にも結合する。従って、第一ステップで、そのような疎水性干渉物質を同様の電荷の疎水性イオンと交換する(即ち、正電荷又は負電荷の物質)。例えば、ヒスチジン(histidine)緩衝液(等電点7.8)が用いる場合、(ツビッターイオンヒスチジン(zwitter ionic histidine)は極端に“MALDI−非フレンドリー”である)、ヒスチジンを、ヒスチジンが負帯電になるpHで負帯電のトリフルオロアセテート(trifluoracetate)イオンと、ヒスチジンが負帯電するpHで、即ち、7.8を超えるpHで、交換しうる。通例として、pH9.0で0.1Mのトリフルオロ酢酸がこの目的のために用いられる。1ミリアンペアの電流が捕捉スライド42の1mmの穴を5分間通過することがこの目的に特に有効であることが見いだされている。負帯電又は正帯電の他の干渉物質は、緩衝物質HEPES,TES,HEPPS,CAPS,CHES,ACES,ADA,BES,MES,MOPS,PIPESのように、これらの負帯電のイオン(陰イオン)をトリフルオロ酢酸の陰イオンと同様に交換することにより、個別の等電点より高くに選択されたpHで個別に除去できる。代わりに、例として、ヒスチジンを正帯電ピリジン・イオン(陽イオン)と、電気泳動法による洗浄を行うことにより交換できる。この洗浄は比較的低いpHで、即ち、pH<7.8で、ヒスチジンとピリジンの両方が例えば、pH4.0で正帯電している。
【0070】
B.高電場電気泳動
この方法により、電解液の導電性が、例えば、蒸留水内での実質的希釈により低減する。そして、大きな電場が捕捉スライド42内の捕捉材料40を横断して置かれる。この方法で、ゆるく結合した疎水性緩衝イオンがモノリスから解離する。それらが再結合する前に、高い電場により掃き出される。この方法で、pHは3−11の範囲になり、より一般的には、最適性能のために、又、導電性を比較的低く保つために、4−10の範囲にする。高電場を加えて、過大な電流を生じないために低い導電度を必要とする。上記に開示された装置を用いて、ウエル当たり1ミリアンペアを超す電流は捕捉スライド42の穴50内で過大なジュール(Joule)加熱を生じる。そのようなジュール加熱は従来技術で良く知られていて、電流の平方即ちI2Rに比例する。Iは電流を、Rは抵抗を示す。
【0071】
C.電気浸透(EEO)の流れ
EEOにより発生した流れはモノリスを横断した電場に比例していて、ゼータ(zeta)電位(即ち、モノリスの固体相からモノリス内の液体への剪断面を横断する電位の低下)の関数でもある。帯電した疎水性物質は捕捉材料40から洗い流されるので、ゼータはゼロになる。それゆえ、洗浄ステップが完了すると共に流れも減少する。高い電場が高いEEOのために最適である。それゆえ、上記の高い電場の電気泳動に最適なのと実質的に同じ条件がEEOの流れにとっても最適である。
【0072】
D.クーロン反発力
これは最も単純な機構である。捕捉スライド42が、結合された緩衝イオンが帯電されるpHでの蒸留水又は脱イオン水のような薄い電解質内に置かれる。イオンのクーロン反発力がそれらをミクロリス(microlith)から押出す。好ましくは、洗浄溶液のイオン力が低く、溶解イオンが1ミリモル未満である。さらに、捕捉材料を洗い流すべき干渉性のイオンを含む種とペアを組む疎水性の種を避けるべきである。例えば、正帯電し
たヒスチジンが干渉物質(即ち、ヒスチジンが7.8より低いpHで結合している)であれば、酢酸塩、蟻酸塩、TFA、HEPES、PiPES、又は、他の疎水性陰イオンを避けるべきである。
【0073】
(例:電気泳動による洗浄手順)
捕捉材料40から、(4−10の範囲の中性のpHの中で)、干渉する疎水性陰イオン例えばTFA,HEPES,PiPESを除去するために、以下のステップを実行する。1.pH9(+/−0.5pH単位)で0.1%のトリフルオロ酢酸の洗浄溶液を用いて、5分間、捕捉スライド内の穴を通して開口面積平方ミリメートル当たり1ミリアンペアを供給する。これでイオン交換を実現する。
2.ステップ#1を実施した後で、洗浄溶液を蒸留水又は脱イオン水で約1/100に希釈する。電圧は、開口面積平方mm当たり約0.125ミリアンペアになるように高める。(又は、電源で利用できる最大値のどちらか低い方)。この洗浄ステップの実行で、さらに5分間を使い、又、原則BからDに基づいた洗浄を行う。
【0074】
干渉する疎水性陰イオンが捕捉材料40に結合している干渉物質として存在するとき、同上のステップを実施するが、MALDIフレンドリーの陽イオン物質を用いる代わりに、例えば、トリフルオロアセテートの代わりに、ピリジニウム(pyridinium)を用いる。ピリジニウム・トリフルオロアセテートは4.0から5.0のpHで特に有効である。
【0075】
どちらの場合でも、上記2種類の電気式洗浄ステップの組合わせが、どちらの電気式洗浄ステップ単独よりも優れている。
【0076】
(カートリッジ捕捉スライドからの捕捉された検体の溶離及びMALDI質量分析法による分析のためのMALDIマトリックスの添加)
検体が濃縮され、カートリッジ捕捉スライド42の穴50により保持された捕捉材料40上に捕捉されると、潜在的MALDI干渉物質が除去されて、捕捉された検体を分析する。例えば、MALDI−TOF質量分析法による分析が行われる。標準のMALDI−MSマトリックスの組成と方法を用いて捕捉されたタンパク質を溶解し、MALDI質量分析器での分析のためにMALDIマトリックス結晶内にそれらを堆積する。そのような標準の手順は質量分析法の分野で良く知られていて、文献に良く記録されている(非特許文献36)。
【0077】
例示的標準のMALDIマトリックス及び手順は、アセトニトリル内の20mg/mlシナピン酸(cynapinic acid)の1部と水中の0.1%(v/v)トリフルオロ酢酸1部の混合体(即ち、シナピン酸の最終濃度は10mg/ml)から成るマトリックス溶液を用いるべきである。カートリッジ捕捉スライド内に保持されて、マトリックス溶液の混合体0.25マイクロリットルをサンプル・サイド(sample side)の各捕捉材料40に注意深く添加する。それで、(周囲のスライド材料に広がるよりはむしろ)大部分の溶液が材料上に留まる。空気中での乾燥後、マトリックス溶液の第二の等体積添加が同じように行われる。そして、カートリッジ捕捉スライド42の乾燥が、空気乾燥により、又は、乾燥器内に用いた真空により行われる。アセトニトリルと水の溶媒を除去した後で、MALDI−MSによる測定と分析がMALDI質量分析法で行われる。便宜的に、捕捉スライド42がそのスライドを保持するために機械的ガイド92を有するスライド・ホルダー90に挿入される。Applied Biosystems Voyater MALDI質量分析器で使用するのに適したスライド・ホルダーの例が図4に示されている。そのような方法による分析で、捕捉スライド42の上面41に露出した捕捉材料にMALDIマトリックス溶液を用いることにより最適の結果が得られる。その後、スライド42の上面41も質量分析器のサンプル・ホルダーに位置してMALDI
質量分析法のために、捕捉スライドと同じ面を、MALDI質量分析器のレーザー・ビーム(laser beam)で調べる。それにより、検体のイオンを放出し、質量分析器と共にあるイオン電流検知器により検出される。
【0078】
好ましい代替分析手順で、検体溶出用溶媒がサンプル・スライド42の下面43に(即ち、サンプル・ウエル4の反対側の面に)最初に加えられる。この手順により、検体分子が捕捉材料から溶出し、MALDIマトリックスの結晶を形成する前に、(及び、内部に検体分子が組込まれる前に、)捕捉スライドの上面41に濃縮される。この手順により、溶離処理の感度が高くなり、検体の変形が少なくなり、検体を捕捉している捕捉材料内の深さへの分析の依存性が低減する。MALDIマトリックスは、溶出用溶媒と共にスライド42の下面43に、代わりに、検体の溶出処理が完了した後で、上面41に加えられる。
【0079】
例示的方法は以下の通りである。
1.1サンプル又は複数のサンプルをカートリッジ2のサンプル・ウエルに置く。
2.サンプル電極20により、事前決定された電圧又は事前決定された電流を各サンプル・ウエルに加える。
3.事前決定された電荷の検体分子が分離層30を通じて他の検体から電気泳動的に分離され、多孔性の捕捉材料40を有する部位で捕捉スライド42の上面を通じて濃縮され、捕捉される。
4.捕捉スライド42にアクセス(access)できるように、カートリッジを分解する。
5.干渉物質を除去する洗浄手順の後で、検体は多孔性捕捉材料から捕捉スライドの分析側に、好ましい態様では、上面41に溶出される。
6.MALDIマトリックスがスライドの同じ分析側に、好ましい態様では、上面41に加えられる。
7.MALDIマトリックスを空気中で、他の乾燥気体で、又は真空で乾燥し、その捕捉スライドをMALDI質量検出器に分析のために挿入する。それで、分析面をレーザー・ビーム・プローブ及びMALDI質量分析器のイオン検出器に露出する。本発明の好ましい実施例では、捕捉スライドの上面41をそのように曝露する。
8.捕捉スライド上に捕捉された検体を質量(より正確には、そのm/z値)及びその相対的存在度を求めて分析する。
【0080】
好ましい態様では、ステップ#5及び#6を以下のように結合する。
【0081】
カートリッジ捕捉スライドが(1−10cm/secの空気速度のファン(fan)のような)乾燥装置上で反転する。アセトニトリルと脱イオン水の溶液(典型的に9:1 v/v)を各捕捉スライド42の下面43で曝露された捕捉材料40に加える。数分間、多孔性捕捉材料を通じて溶出用溶媒の吸引後、このステップの後で、第二の溶出ステップではMALDIマトリックス例えばメタノール内の濃縮したシナピン酸(例えば、9.0−90mM、pH7.0−8.0の脱イオン水)が含まれる。このステップの後で第三ステップがあり、その中で、pHを酸性になるように調節する。典型的に、アセトニトリル9部と脱イオン水内の0.1%トリフルオロ酢酸1部に調節される。MALDIマトリックス(例えば、シナピン酸)を乾燥器内で真空乾燥を行なえるようにする。十分な乾燥後、MALDI−MSの測定と分析を、例えば、Bruker Autoflex model又はApplied Biosystems Voyager modelのようなMALI質量分析器内で行う。このMALDIマトリックス添加の方法はスライドの上側への生体分子の溶出、さらに、そのようなMALDI−MS測定内の検体分子の検出限界を低減することに対応している。
【0082】
(好ましいカートリッジ捕捉スライドの構成、捕捉材料及びその製造方法)
カートリッジ捕捉スライド42は穴50、オリフィス内に保持される捕捉材料40を有している。好ましい実施例では捕捉スライドが96個の穴を有し、8×12の長方形配列に配置されている(即ち、12列8行)。その中で、各穴の中心は最も近い4個の隣接穴のそれぞれから9.00mm離れて配置される(即ち、9.00ミリのピッチを有する)。好ましい実施例では、穴は直径約1mm、深さ約1mmである。製造では、全体として平坦な捕捉スライドで、厚みの変動は非常に小さく(典型的には、約+/−50ミクロン未満)、オリフィスを有するが、そのオリフィスは、ポリマー装置製造分野の技術者には好く知られている機械加工(例えば、レーザー又は機械的穴開けによる)成形又は鋳造により形成される。好ましい実施例では、捕捉スライドの材料はバルク(bulk)及び表面の導電性を最適化するように選択される。前述のように、カートリッジ捕捉スライド42の導電性は、サンプル電極20により捕捉スライドに加えられた電流の75%から99.999%が、(バルク・スライド材料を通過する代わりに)穴50内の電解質を通過する。装置の運転中にこの状態を実現するためには、通常、カートリッジ捕捉スライド42を作るのに用いられる材料の体積抵抗は、102から1010オーム・センチメートルの間である。より一般的にはカートリッジ捕捉スライド42を作るのに用いられる材料の体積抵抗は、104から108オーム・センチメートルの間である。この捕捉スライド材料の導電度が、MALDI−MS分析によるその後の分析で捕捉された検体のイオン化の間に捕捉スライド42の帯電を防止する。代わりに、カートリッジ捕捉スライドのバルク導電度が多少導電性になり、相当量の導電度を与えるために、表面の抵抗コーティング(coating)を行うことにより、希望の状態を実現するように表面導電度を調節する。
【0083】
捕捉スライド42が穴50と共に形成されると、溶剤又は加熱による溶接、穴への材料の鋳造又は他の取付手段による膜の取付けのようないくつかの手段によって穴内に捕捉材料40を堆積し、取付ける。好ましい態様では、鋳造は現場での2種類の光重合反応による接合を行うことにより与えられる。両方の反応は光重合を開始するために紫外線照射を用いることにより真空テーブル(table)上の一体成形で行われる。その方法で、適当な鋳造型が機械加工による熱可塑性、熱硬化性、又は、金属、又は、他の方法による成形型により形成される。成形は捕捉スライド42の穴50内に捕捉材料40を保持しなければならない。そして、有利なこととして、当業界の技術者に好く知られているように、ラジカル重合反応を終わらせるように機能する酸素を排除する。例えば、成形型は薄いシート(sheet)材料例えばポリエチレン又は“Saran Wrap”から成り、そのような成形手順の分野の熟練者に好く知られているようにスライドを真空テーブル上に保持しながら、そのシートを真空によりスライドの穴に対して所定の場所に保持する。
【0084】
第一の光重合ステップが二重結合又はビニール(vinyl)基を穴50の壁に光グラフトする。光グラフト処理では、光グラフト用溶液を穴内に置き、穴50を囲む捕捉スライド材料に共有結合する共重合分子を生じるのに必要な時間UV光を照射する。UV照射をDymax Corporation,Torrington,CT,USAからの5000−ECユニットにHランプを用いて行うとき、必要な照射時間は一般的に1−5分の長さになる。
【0085】
適当な光グラフト反応の混合体は、メチルメタクリレート(MMA)48.5質量%、エチレングリコール・ジメタクリレート(EDMA)48.5質量%、ベンゾフェノン3質量%から成っている。その反応混合物を秤量し、酸素を追い出すためにアルゴン、ヘリウム又は窒素のような気体を混合し、散布する。捕捉スライドを混合体内に浸漬し、次ぎに、過剰物を除去することにより、各ウエルにピペッティング(pipetting)することにより、又は、他の方法で反応溶液をウエルの内部に供給することにより、その散布された反応混合物をウエル内に置く。次ぎに、捕捉スライドをモールド内に置き、そのモールドを真空テーブル(Pharmacia Fine Chemicals,Mod
el GSD−4)の上に置き、その真空を動作させる。次ぎに、UV透過性プラスチック・シートをモールドの充填された穴上に置かれる。そのようなプラスチック・シートはSaran Wrapのような市販のプラスチック・ラップ(plastic wrap)から、ポリジメチルシロキサンのシート、又は、適当なUV透過性気体遮断材料のようなシート・ゴムから供給できる。真空テーブル内に保持された捕捉スライドに対して所定の位置に、プラスチック・シートを、手操作で保持する。その後、光グラフト反応を水銀アーク・ランプ付のUV照射装置に配置して、一定時間照射する。照射時間はシステム因子によるが、一般的に1分未満で、照射された表面積の平方センチメートル当たり100 mWの照射フラックス(flux)である。例えば、十分なUV照射は捕捉スライド面から約20cmの距離で動作する400W水銀ランプにより与えられる。UV照射のあとで、プラスチック・カバーが取外され、光グラフトが行われた捕捉スライドがモールドから除去され、アセトン(acetone)で洗浄される。光グラフトが行われたスライドがアセトン内に置かれ、一定時間そこに保持され、痕跡量のモノマーと、捕捉スライド42の面に未接合のままである共重合体の破片を除去する。
【0086】
第二の光グラフトステップは、固体の現場形成から成るが、多孔性のモノリス材料が、以前のステップで取付けられた光グラフトされた共重合体により捕捉スライド42に部分的に取付けられている。今回の方法で、モノリス形成がウエル内に反応混合体を置くことにより行われ、モールドを真空密封して、モノマーとポロゲン(porogens)の混合体からモノリスを発生するため、一定時間UV照射することにより低酸素及び無酸素の環境を形成する。そのようなポロゲンは当業界で知られていて、反応剤と混合して、その後に、固体相を形成するとき、多孔性の固体形成を促進する。代わりに、UV照射は、Dランプ付で、Dymax Corporation Torrington CT,USAからの5000−ECユニットにより行われる。
【0087】
好ましい方法で、モノリス「反応混合体A(“reaction mixture A”)」が用いられる。「反応混合体A」には、1−デカノール(decanol)5グラム、n−ブチル・メタクリレート(butyl methacrylate)2.4グラム、EDMA 1.6グラム、シクロヘキサノール(cyclohexanol)1グラムが開始剤と共に含まれる。ジメチル・アセトフェノン(DMAP)は開始剤としてモノマーの全質量を1%として比例値が用いられる。それゆえ、この場合、0.4グラムのDMAPが用いられる。反応混合体を秤量し、DMAPが完全に溶解するまで混合し、酸素を追い出すために、アルゴン、ヘリウム又は窒素のような気体を散布する。約10mLの反応混合体をモールドに最初に充填し、次ぎに、そのスライドをモールド内に置くことにより、散布された反応混合体がウエル内に置かれる。代わりに、反応混合体を各ウエル内にピペットで注入する。又は、他の方法で、ウエルの内部に送られる。次ぎに、捕捉スライドをモールド内に置く。そのモールドを真空テーブル(入手先:Pharmacia Fine Chemicals,Model GSD−4)上に置き、真空を機能させる。そして、捕捉スライドの上に十分なプラスチック・シートを供給してモールドをカバーする。そのようなプラスチック・シートはSaran wrapのような市販のプラスチック・ラップから、ポリジメチルシロキサン・シートのようなシート・ゴムから、又は、適当なカバリング(covering)材料から得られる。そして、モールドと含まれる捕捉スライドを真空テーブルに固定するために、十分な真空を生じるまで、真空テーブルに保持することにより、プラスチック・シートを所定位置に保持する。次ぎに、捕捉スライドの一部をキセノン又はメタルハライド・アーク・ランプ付のUV照射装置内に置き、ある時間照射する。照射時間はシステム因子によるが一般的に照射表面積が平方センチメートル当たり150mVの 照射光束で、4分未満である。十分なUV照射が例えば捕捉スライド面(入手先:SLM Instruments,Champain,IL,USA)から約20cmの距離で動作する400W キセノン・ランプにより与えられる。UV照射の後で、プラスチック・カバーが外される。モノリスで充填された捕捉スライドがモールドから注意深く外されて、メタノールで洗浄される。モノリスで充填されたスライドを約10ボリューム(volumes)のメタノール内に1−24時間置かれて、メタノールが高級アルコールを置換し、かつ、残留未反応のモノマーを除去する。新規のメタノールを用いて各連続バッチ(batch)を洗浄し、適切な清浄度にする。
【0088】
当業界の技術者に一般的に知られているように、(その方法と反応混合体Aの両方の)多くの適切な変更が存在する。非特許文献11−22が例を示している。実験を通じて、2以上の異なる捕捉材料の異種結合により形成された捕捉材料42は、単独使用時の純粋なモノリシック(monolithic)捕捉材料より優れていることを我々は見いだしている。一般に、異種の組合わせは、固体、事前形成された、粒状のクロマトグラフィ・メディア(chromatography media)から成り、固体又は多孔質のコア(core)粒子から成っている。粒子はいわゆる「逆相」粒状のクロマトグラフィ・メディア(即ち、疎水性粒子、又は、代わりに、陽イオン又は陰イオンの「イオン交換メディア」)である。そのような材料の例には、高純度シリカ、タンパク質アフィニティを修正したシリカ、重合クロマトグラフィー用の多孔質又は固体のビーズ(beads)又は他の固体粒状の材料が含まれ、クロマトグラフィーの分野の技術者又はそのような材料の技術者によく知られている。代わりに、混合体又は2以上のそのようなメディアの混合体又は交互層を用いる。各場合に、粒状のクロマトグラフィー・メディアは適当なマトリックスにより所定場所に保持される。その適当なマトリックスは、捕捉スライド42の表面に十分接着するのに、又、クロマトグラフィ・メディアを所定場所に確実に捕捉するのに適当な材料である。例えば、便宜上、上記の、又、非特許文献11−22に一般的に示されている同じ組成がそのようなマトリックスに用いられる。一般的に、好まれた「逆相」の異種の組合わせが、生体サンプルからのタンパク質とペプチドの捕捉のために、いわゆる「逆相」の粒状クロマトグラフィ・メディアとモノリシック固体相の間で作られている。モノリシック材料は上記に引用したもの(及び非特許文献11−22に一般的に)示されている。
【0089】
特に、生体のペプチド及びタンパク質の捕捉のために好ましい実施例は、「反応混合体A」の33%をAlltech SPEバルク溶剤C8(入手先:Alltech Associates,Deerfield IL,Cat No 211504)で置換される。純粋な反応混合体Aを用いるために上記の一般手順が混合体と共に用いられる。さらに、好ましくは、粒状材料が、モノマー、架橋剤及び開始剤を含む反応混合体の粘度を高めるのに役立つ。粘度の増加は鋳造の際に穴から重合反応混合体の漏洩を防止するのに役立つ。それにより、事前決定された粒状物の組成を、一般に1%の粒子から99%の粒状材料の範囲で、選択することにより希望の値に調節しうる。より一般的には、粒子材料は10%から90%の範囲とする。さらに、より一般的には、粒子材料は25%から50%の範囲とする。タンパク質とポリペプチドを捕捉するための好ましい捕捉材料を製造するのに用いられる粒状のクロマトグラフィ粒子の別の例を表1に示す。
【0090】
表1 「ミクロリス」捕捉用化学物質
捕捉機構 例
順相 シリカ、アルミナ
逆相 C2−C18、重合体樹脂、モノリス
イオン交換 SCX,SAX,WAX,WCX
固定化された金属のアフィニティ Ni,Fe,Ga
抗体の捕捉 Protain G,Protein A,Step
tavidin,カスタム抗体
小さな分子のアフィニティ Blue Sepharose/Dextran,C
ustom ligand libraries
【0091】
これらのクロマトグラフィ材料はバルク量でのいくつかの製造により供給され、0.2−500ミクロンの範囲の粒子寸法を有している。単位体積当たり結合能力が大きいことにより多孔質粒子が好まれるけれども、多孔質又は非多孔質の粒子が用いられる。別の例によると、製造企業には、Agilent,Alltech,Applied Byosystems,Phenomonex,Supelco,Watersが含まれる。本発明の好ましい実施例に含まれるのはC8逆相樹脂、特に、Alltec(部品番号#206250)で、既に記したように捕捉材料としてメタクリレート樹脂と共に結合されている。これらの粒子樹脂の組合わせが、生体高分子の捕捉で高い効用を示している。その生体高分子には、特にタンパク質とペプチド、及び、より一般的には、炭水化物、多糖類、オリゴヌクレオチド(oligonucleotides)が含まれ、質量分析法を用いることにより、その後の脱離/イオン化に対応する。
【0092】
非重合の樹脂と事前重合を行った粒子をそのように結合することは、その後に、ここでは1単位として重合したとき、「ミクロリス」結合と呼ぶ。そのようなミクロリスは、事前成形され、通例、MALDI適合樹脂により薄い捕捉スライドの構成に保持された市販のクロマトグラフィ・メディアから成っている。そのようなミクロリスの個別構成を埋込みのために選ばれたクロマトグラフィ・メディアの構成に基づいて事前決定される。そのような構成には若干の例の名前を出すと、順相、逆相、イオン交換、固定化された金属のアフィニティ、小さな分子のリガンド(ligand)のアフィニティが含まれ、抗体捕捉のアフィニティとレクチン(lectin)捕捉のアフィニティを含めたクロマトグラフィ・メディアが含まれるが、それに限定されない。他の例は生体分子のクロマトグラフィック分離とそのような目的のための市販メディア選択分野の技術者によく知られている。
【0093】
多孔のモノリシック材料と粒状媒体の混合体のひとつの予想外の特性は、その組合わせが固体相の捕捉材料の多孔性を高めることである。それゆえ、ここに記された方法に基づいて作られたモノリスを通過する圧力で駆動された流れが、入手できる文献及びここで述べた文の両方に示されている「モノリシック捕捉材料」を通過するより大きくなる。多孔性モノリシック樹脂と事前重合した粒子状クロマトグラフィ・メディアの組合わせ(即ち、混合体)は、その組合わせは得られた捕捉材料の引張り力を高める。それゆえ、(100%)純粋な多孔性モノリシック捕捉材料(例えば、100%反応混合体Aから成形)が、真空乾燥時に、直径1mmの穴への鋳造に割れを生じて、捕捉スライド面に接着しなくなる。それと対照的に、Alltech SPE Bulk Sorbent C8の33%の組込みで、そのような割れと接着不能を防止する。そのような異種組成を一般に「ミクロリス(microliths)」と呼ばれ、上記の方法により作られ、優れた機械的強度、安定性を有することが認められ、かつ、捕捉スライド面で優れた接着性を有する。さらに、我々は、そのような「ミクロリス」が電気泳動装置内でタンパク質、ペプチド及び他の検体分子を捕捉する能力を有することを見いだしている。さらに、そのようなミクロリスが十分に平坦に(例えば、+/−50ミクロン)鋳造して、マトリックス支援レーザー脱離/イオン化質量分析法(MALDI−MS)を用いたその後の分析のために、優れた面を提供する。
【0094】
(血清から高存在度のタンパク質の解離と除去)
臨床上重要な血液、血漿又は血清の低存在度ペプチド分析に伴う主要な問題は高存在度のタンパク質が低存在度のタンパク質及びペプチドの外観を隠すことである。しかしながら、血液、血漿又は血清から高存在度のタンパク質のアフィニティ除去はかなりの数の低存在度・疎水性ペプチドも除去されると想定されている。分離と分析の方法例の好ましい実施例では、血清サンプルは最初MALDIと適合性のある洗剤で処理して解離を促進し、その後に分子量による分別で、高存在度で高分子量のタンパク質を除去する。
【0095】
全てのサンプルをステンレス鋼のサンプル・プレート又は導電面を有する平坦な重合材料から作られた使い捨て捕捉材料に加えた。例えば、2マイクロリットル(μl)のサンプル体積を溶液の液滴として、直接添加するか、又は、モノリシック捕捉材料上に電気泳動で捕捉する。乾燥後、MALDI質量分析法に直接置かれる。別の例では、0.5マイクロリットルのMALDIマトリックス溶液をピペットでサンプル・スポット(sample spot)上に添加し、乾燥させる。タンパク質とペプチドは一般的にMALDIマトリックスとして用いられたアルファシアノ−4−ヒドロキシナミック・アシッド(CHCA)(alphacyano−4−hydroxycinamic acid)と共に分析される。それは一般に1,000から15,000ダルトン(Da)の低分子量のペプチド及びポリペプチドに対して、MALDI質量分析法の結果で、最良の信号対ノイズ(noise)の比を与えるからである。CHCAマトリックス溶液の組成は以下の通りである:CHCAは、50%アセトニトリルと水性0.1%のトリフロロ酢酸の混合体の中に飽和している。MALDIマトリックス溶液の全ての材料はSigma Chemical Co.(St.Louis,MO,USA)から入手しうる。全てのMALDI−MS分析はABI Voyager DE MALDI−TOF及び社内設計のQGEN_PR2法により行われる。典型的な分光計の設定は:加速電圧20kV、グリッド電圧(grid voltage)94.1%、ガイド・ワイヤ(guide wire)電圧0.050%、遅延時間110ns、レーザー設定3000、平均走査64、1.1E−6torr、低質量ゲート511、陰イオンをoff。
【0096】
デモンストレーション(demonstration)のために、2種のポリペプチド標準、例えば、ACTHの破片(18−39)及びインスリン酸化B鎖をウシ血清アルブミン(BSA)と共に混合し、ステンレス鋼のMALDIターゲット・プレート上に直接ピペット添加する。図5は、〜127pmolのBSAが存在する中であるけれども、1ピコモル(pmol)に希釈され、MALDI質量分析法用プレート上にスポットを再現するために加えられ、乾燥されて、又、分子の質量と強度を別個に分析された2種のそのようなポリペプチド標準を示している。図5は1pmolのACTHの破片と1pmolのインスリンを明確に識別しうることを明確に示している。同様に、図6に示すように、ペプチドの破片の量が10倍未満即ち0.1pmolの2標準であり、同じBSAの濃度と量であるとき、図5−7に示すようにMALDI質量分析法による分析中にイオン化を実質的に抑制する。
【0097】
図7に示すのは、図6に示された結果に対して用いられた同じサンプルのMALDI−TOF帯域である。しかしながら、図6に示した結果について、サンプルを最初に捕捉スライド上の電気泳動で濃縮と捕捉を行う。その手順では、単一層のカートリッジ捕捉スライドを用いる。その手順で、2μLのサンプルを、250mMの水性L−ヒスチジン・バッファ(L−histidine buffer)と組合わせて、単一モノリス捕捉スライドを用いることにより処理する。捕捉は約1ミリアンペアの電流を十分な時間流して、伝達された全電荷が約1クーロン(coulomb)になるようにすることにより行われる。その結果は、サンプル混合体からの干渉としてシステムが効果的にBSAを除去することを示している。これらの結果は、装置と手順を組合わせて用いたとき、アルブミンのような大型タンパク質(即ち、30,000ダルトン超)により生じた低分子量(即ち、30,000ダルトン未満)のタンパク質とポリペプチドの検出から実質的信号干渉を効果的に除去し、それにより、低分子量の分子から得た質量分析法の信号を劇的に改善する。
【0098】
(LMWヒトの血清分析)
単一のカートリッジ捕捉スライドを用いることにより本発明の実施例を用いて実験が行われた。本発明の手順に基づいてMALDI MSにより低分子量のタンパク質/ペプチドのプロファイリング(profiling)のためにヒトの血清の調製の実行可能性を
決定するためにそのような研究が行われた。例えば洗剤で処理した血清サンプルはEppendorfのミクロチューブ(microtube)(500μLの体積)内の100μLのヒト血清(Sigma Chemical Co.から入手)に10μg/μLのオクチル−b−D−グリコピラノシド(octyl−b−D−glucopyranoside)(OG)を添加することにより作られる。サンプルは、洗剤で処理した血清の10μLのアリコート(aliquot)、250mMのヒスチジン・バッファ(histidine buffer)の内の100μL、1μLのTexas Red labeled−Leu Enkephalen(250mMのヒスチジン・バッファの内のトレーサー(tracer)として)、0.5μLのグリセロール(glycerol)から作られる。得られたサンプル混合体を約1000gで1分間遠心力を加え、混合液滴を得る。
【0099】
サンプル成分の分離と結合を実行するために、調製されたサンプルの10μLのアリコートをサンプル・ウエルに加えられる。例えば、プラチナから作られた陰極がサンプル・ウエル内に直接配置される。反対の電極、プラチナの陽極が対極チャンバーに置かれた対極電解液に接触するように配置される。
【0100】
プラチナの陽極と陰極の電極は、電位計(Princeton Applied Research ,model 273)に接続され、電極間に約1mAの電流が加えられる。電圧をゼロに設定し、電極への電線を切離す前に約20分間、分離を進めた。
【0101】
次ぎに、試作カートリッジを分解し、ゲルと捕捉層を蛍光でチェックする。分析システムは十分に機能し、そのときに、捕捉材料40に結合するために選択されたタンパク質とペプチドから実質的に全ての蛍光発光が観察され、捕捉部位に結合していることが観察される。捕捉スライドを約5分間脱イオン水に浸漬する。蛍光による捕捉部位の目視検査の後で、スライドを完全に空気で乾燥させる。次ぎに、MALDIマトリックスの0.5μLのアリコートを捕捉スポットの上側に添加する。その捕捉スポットをVoyager DE MALDI MSで直接分析を行う。図8はMALDIマトリックスとしてCHCAを用いることによりサンプルから得られた質量分析法を示している。図はヒトの血清からの低分子量ポリペプチドの検出で、良好な信号対ノイズ比を示している。
【0102】
上記の例で示したのと同様のパラメーターを用いるとき、血清をPPASカートリッジ内の2個所のウエルに添加する。タンパク質、又は、他からの解離を促進するために洗剤を用いて、1以上のサンプルを処理する。そうすることで、洗剤で処理したサンプルを250μLのL−ヒスチジンと結合させ、pH6.8に調節し、サンプルと接触したサンプル電極の極性により1.0mAの電流を加えた。他の洗剤で処理したサンプルを250μLのL−ヒスチジンと結合し、pH7.0に調節し、同様に、サンプル電極によるバイアス(biased)で−1.0mAの電流を供給する。図に示すように、2個の反対極性のサンプル・ウエルから観察されたスペクトルが完全に異なる相補性のタンパク質とペプチドのピークを示している。これらのデータは同じサンプルの2成分pH分別を明確に実証している。
【0103】
(別の例と方法)
(材料)
全ての材料を市販業者から入手可能で、含まれるのは:アセトニトリル、トリフルオロ酢酸(TFA)、n−オクトグルコシド、CHCA、L−ヒスチジン、ポリアクリルイミドである。血清の調製はSigma Co.からの0.5mLポリプロピレン管内で行われる。C−18コーテッド超常磁性ビーズはBruker Daltonicsから購入しうる。
【0104】
(血清サンプル)
悪性疾患が知られていない志願被験者から、又、前立腺がん(グリーソン得点6−7)が確認された同意患者からの血液サンプルを8.5mLのガラス・バキュテナー(Vacutainer)管で供給される。1時間までの室温で凝固させ、4℃、5分間、1000rpmの遠心分離にかけた。血清は等分され、−80℃で冷凍保存された。患者及び対照の血清がVanderbilt University Medical Centerが承認した臨床手順に従って集められる。
【0105】
(クロマトグラフィによる分離)
選択された場合で、血清を逆相磁気ビーズを用いることにより、又は、ここで述べたPPAS装置により分別する。磁気ビーズ・クロマトグラフィでは、血清を、超常磁性で、多孔質シリカ・ベースの粒子(<1μmの直径、80%酸化鉄)、C18による表面誘導体化により培養する。C18/K磁気粒子の懸濁液(500,000粒子/μg;50μg/μLのDD水)を均一分散を得るための渦巻き運動により2分間完全に混合する。次ぎに、50μLビーズ溶液を50μLの血清に加え、5回前後のピペット操作によりゆっくりと混合する。そして、磁石を用いて、ビーズを管の側面に引き寄せ、上澄みをピペットにより除去して、廃棄する。次ぎにビーズを水中0.1%TFAの200μLで完全に洗浄する。最後に、10回前後のピペット操作により、20%次ぎに70%のアセトニトリルの体積5μLを用いて、粒子からペプチドを段階的に溶出する。そして、3μLの溶出液を他の管に移し、6μLのMALDIマトリックス溶液と混合する。そして、1μLをMS分析のために置く。
【0106】
PPAS Device(TM)を用いることによるサンプル検体のモノリシック分別/濃縮:
タンパク質とペプチドの検体を上記の一般手順を用いることにより分析する。到着時点で、血清アリコートを直ちに−80℃で保管する。例えば、血清サンプルを、16mMの重炭酸アンモニウム250μLと250mMのL−ヒスチジンを、Eppendorf microtube(500μLの体積)内の1μg/μLのオクチル−b−D−グルコピラノシド(OG)、0.5μLグリセロール、10μLのヒト血清に加えることにより調製しうる。得られたサンプル混合体を約1000gで1分間遠心力を加え、混合液滴を得る。サンプルの半分をpH7.0に他の半分をpH6.8に調節する。160mMの重炭酸アンモニウム及び250mMのL−ヒスチジン緩衝液がカートリッジ・リザーバー・バッファ(cartridge reservoir buffer)(モノリスの下)に用いられる。全サンプルを5回の反復処理で分析しうる。
【0107】
捕捉スライド42内に捕捉材料40の鋳造のために、カーボン・ドープド・ポリプロピレン(carbon doped polypropylen)(〜50,000オーム/cm)の複数の貫通穴を含むスライドが射出成形される。そして、そのスライドは2枚の軟質シリコン・ゴムのガスケット(gasket)と2枚の石英プレートの間にサンドイッチ(sandwich)になっている。前記の機能化溶液が各貫通穴にピペットにより置かれる。そして、水フィルターを組込んだキセノン・アーク・ランプを用いて照射される。次ぎに、そのサンドイッチから基盤を除去し、ブチル・メタクリレート及び2−ヒドロキシエチル・メタクリレートを含むモノリス溶液を各貫通穴に加える。前記のように、サンドイッチを再構成し、15分間照射する。鋳造手順の後で、スライドを106mMの重炭酸アンモニウム及び250mMのL−ヒスチジンの中に30分間浸漬する。最後に、そのモノリス・スライドを脱イオン水により完全に洗浄する。プラチナで作られた陰極をサンプル・ウエルに直接置く。プラチナの陽極を緩衝液槽に接触するように置く。プラチナの電極を特別設計の多重化電位計に接続し、約1mAの電流をウエルに加える。電圧をゼロに設定し、電極への電線を切断する前に、処理を20分間続行した。サンプルの半分は+1mAで、残りの半分は−1mAで、約20分間処理する。各分析の過程中に各ウエルの電流を測定し、プロットする。電気濃縮手順が完了した後で、PPASカートリッジ2を分解し、カートリッジ捕捉スライド42を洗浄してpH緩衝液及び塩のような干渉物質を除去する。最後に、CHCA MALDIマトリックスを用いて、そのスライドをMALDI−MSにより直接分析する。
【0108】
PPAS装置手順及び磁気ビーズの両方について、30 fmol(ペプチド当たり)と500 fmol(タンパク質当たり)の市販の校正標準(Bruker Daltonics)もCHCAマトリックスと混合し、ターゲット・プレート上に別個に加える。ターゲット・プレートは3×2のパターンに配置されている6個の隣接血清サンプルの中央に位置している。a)単一装置の単一ウエル、b)同じ装置の異なるウエル、c)異なる装置の同じウエル、d)異なる装置の異なるウエルの中の変動性を評価するために再現性を決定する。要因分析を用いて、ウエルの位置、又は、ウエル間の相互作用(又は他の変数)の影響を決定する。
【0109】
(質量分析法)
ペプチドのプロフィール(profiles)を、Applied Biosystems Voyager DE及び4700 model MALDI質量分析器により、典型的手順を用いて分析する:加速電圧20kV、グリッド電圧94.1%、ガイド・ワイア電圧0.050%、遅延時間110ns、レーザー設定3000、平均走査64、1.1E−6torr、低質量ゲート511、陰イオン・オフ。周波数帯は直線モードで取得する。
【0110】
MS測定のソフトウエアにより取得したスペクトルは、市販のEfectaソフトウエア(Efecta Technologies,Corp.,Steamboad Springs,CO.)このソフトウエアは取得中の自動化、平滑化、ベースライン修正、ピーク指定を行う。全てのデータ操作はTempstら(非特許文献36)が発表した技術に基づいて行われる。手動操作の外部校正の後で、ピーク(即ち、m/z)のリスト(lists)を、その後の統計分析に必要なテキスト・ファイル・フォーマット(text file format)のファイルに保存される(以下参照)。
【0111】
ピークのリストは一連のデータ変換のためのデータベースに導入される。最初のパターン(pattern)分析のため単純二進法を最初に作るために、ピーク強度を低減して、特定サンプル内で観察されたペプチドの得られたビン(bins)のいずれかの中での存在又は不存在を示す。
【0112】
次ぎに、ペプチドの質量(例えば、1500ppm)と比例して拡大するウインドウ(window)内のビンニング(binning)により特定セット(set)内の全てのサンプルに亘ってピークの位置合わせをする。ビンニングは、全てのサンプルからの全てのm/zの値をひとつの長いリストに合わせることにより行われ、値を増加することにより分類される。最初の質量を“real(実)”と表示し、隣接する質量分類と比較する。ユーザーが定義したウインドウ内の隣接質量が“duplicate(複製)”と呼ばれる。プロセス(process)は、まだ表示されていない次ぎに大きなm/z値について、分類リスト内の全質量に“real(実)”又は“duplicate(複製)”と標識を付けるまで繰返される。次ぎに、“duplicate(複製)”の質量を廃棄する。現在の適用で、実験により、許容範囲は2Da又は1500ppm(0.15%)になる。“real(実)”質量が恣意的で、ビンの指名にのみ用いられるので、ここで留意すべきは各ビンの質量内の最初のm/z値の割付けである。m/z値をビンに配置されると、結果と共に、表計算ソフトが自動的に出力される。最初の列はビンニング・プロセス(binning process)を生き延びた“real(実)”質量全部のリストを示す。残りの列はサンプルを、又、対応する“real(実)”の質量と共に、ビンに配置されたピークを有するかどうかを示している。
【0113】
(統計的データ分析)
研究セットの全サンプルに亘って、m/zピークのビンニングの後で、Efecktaソフトウエアを使用してプロテオミック(proteomic)データを評価する。集団を代表するために、ソフトウエア内で仮想「実験」が作られる。全てのサンプル内に見いだされたユビキチン(ubiquitin)と少なくとも他のひとつのペプチド・ピークによりデータが正規化される。実験のパラメータ・セクションで、サンプルに癌性か正常かの標識を付ける。翻訳セクションで、分析モードを“比率の対数”と用いられた全測定値に設定する。サンプル名は不連続パラメーターとして表示される。実験が作られると、一方向ANOVAノンパラメトリック(nonparametric)検定(Mann−Whitney U test)とp<0.05での無多重検定補正をフィルター(filter)として用いる。この検定は多数のサンプルを有する2種類のグループに亘って有意に変化しない集団はフィルターを通さないことを意味する。そのフィルターは前立腺がんと対照のグループ間で重要な変化を示した集団を後方に残す。その変化を2種の技術を用いることにより確認する:クラスタリング(clustering)とクラス予測、
第一の技術については、Effectaのクラスタリング・ツール(clustering tool)を用いて、結果を決定樹として表示する。x軸について、類似したサンプルを決定樹上のお互いの近くに置く。サンプルの類似性について、Pearsonの相関性により評価する。類似しないサンプルはお互いから離して置かれる。y軸についても、類似性の検定にPearsonの相関性を用いて、同じように集団のグループ化を行う。クラスタリングの方法では半分のサンプルについてデータがない集団を廃棄した。
【0114】
第二の確認のため、ノンパラメトリック検定からフィルターを通過したペプチド集団を、k−ニアレスト・ネイバー(k−nearest neighbor)と呼ばれるクラス予測アルゴリズム(algorithm)によっても分析する。クラス予測の正確性を学ぶために、“leave−one−out”(非特許文献6、19)として知られる相互検証法を実行する。それはクラス予測アルゴリズムの訓練セットとしてN−1のサンプルを取る。そして、N番目のサンプルをテスト・セットとして使用し、処理をN回繰返して、全サンプルを一度はテスト・セットとして用いるようにする。
【0115】
MALDI MS分析の場合、中央のカートリッジ・アセンブリーを適当なMALDI質量分析法用サンプル・プレートに取付けて、質量分析器に導入する。そして、適当な溶媒に溶解したMALDIマトリックスの小液滴を捕捉膜の検体捕捉領域に加える。溶媒は捕捉膜上の捕捉部位に存在する検体を溶解させる。溶解が蒸発すると共に、検体はMALDIマトリックスの結晶に組込まれて、捕捉膜の上面上に形成される。溶媒液の蒸発と、MALDIマトリックスの結晶形成のための時間を与えた後で、サンプル・プレートをMALDI質量分析器への導入準備ができる。MALDIサンプル・プレートの質量分析器への挿入時に、MALDIマトリックスの結晶を強力なUVレーザー光のパルスで照射し、検体分子の破片的イオン化を生じる。この一部分からのイオンを検出器への飛行時間に基づいて測定し、質量対電荷の比及び強度に基づいてプロットする。
【実施例1】
【0116】
血清に存在するタンパク質の分析
図3及び4はPPASの25ウエル型を用いた結果を示していて、捕捉膜が1枚で、その後の分析はMALDI質量分析法による。試作版のPPASを用いて、血清サンプルの配列からの分離を比較的高速(60分以内)で同時に行う。その後の分離層の厚みの低減は約5mmから約1.0mm以下になり、分離層の両側に加えられる電圧を約1.0から10ボルトが、約10から100ボルトに上昇して、10分以内での分離、濃縮、捕捉を可能にしている。使い捨てMALDIプレート上に直接選択した部分の電気泳動による濃縮が、MALDI−MSの感度を高め、プロファイリングの差の表現を早めるという追加
の利点を与えている。臨床的に重要で、血液、血漿又は血清の中の低存在度のペプチドを分析することに伴う主要な問題は、高存在度のタンパク質が低存在度のペプチドの外観を隠すことである。しかしながら、血液、血漿又は血清のサンプルからの高存在度タンパク質のアフィニティ除去はかなりの数の低存在度で疎水性のペプチドも除去すると想定されていた。これらの研究で血清サンプルは解離とその後のPPASを用いた分離と濃縮を促進し、MALDI−MSによる検出のために、MALDI適合洗剤で処理された。そのようなMALDI適合洗剤の例は、Trinton X−100、octyl glucoside、NP−40等のような中性の物質である。そのような中性洗剤はPP捕捉層内の電気泳動的濃縮をしない。
【0117】
PP混合体のMS分析からの結果を精製したPP標準(例えば、ユビキチン、シトクロームC、インスリン及び1%TFAのみを含むサンプル)と比較する。その標準サンプルは0.1%TFAに直接希釈しうる(800フェムトモル/μL又は10フェムトモル/μLに)。MALDI−MSによる分析の前に、溶媒を蒸発した後で存在する干渉物質が無いかほとんど無いようにする。代わりに、発色性又は蛍光性の標識を付けたPPが標準として組み込める。例えば、蛍光性の分子の場合、タンパク質変更の分野での技術者に良く知られている試薬と方法を用いて、Fluorescein(F)、Texas Red(TR)、Rhodamine(Rh)、Marina Blue(MB)と標識を付けられる。それゆえ、0.2μLのTR−ユビキチン、MBウシ血清アルブミン(MB−BSA)、それぞれが1−2μg/μLで、25%(w/v)グリセロールと共に250mM水性L−ヒスチジン緩衝液を含む2μLのサンプルに組込まれる。
【0118】
図3及び4に示された結果は、ヒトの血清から高分子量・高存在度のタンパク質を除去するのに用いられたポリアクリル・アミド層により得られた。図3及び4に示されたスペクトルにはアルブミンが観察されなかった(68,000のm/zで)。これらの結果は、MALDIによる干渉抑圧により混合体から血清のアルブミンを効果的に除去していることを示している。しかしながら、電気泳動の実施時間が1時間以上に延長されたときに、アルブミン信号の始まりが観察された。付随的に、他の捕捉されたタンパク質の強度低下が観察された。多分、良く知られているように、高存在度アルブミンの存在の中で低存在度タンパク質のイオン化が抑制されたことによる。PPAS内の高分子量タンパク質を分析するために、ポリアクリル・アミド層を非ふるい分け(non−sieving)アガロース層により(及び、例えば、アフィニティ・クロマトグラフィによる交互処理により除去された高存在度タンパク質により)置換しうる。
【0119】
本発明のPPASが、固体相捕捉膜上にタンパク質とポリペプチドを捕捉し、塩及び他の干渉分子を洗い流すことができる。そして、MALDIマトリックス溶液の膜への添加時に、タンパク質が放出され、膜面に沈殿するMALDIマトリックスの結晶に組込まれる。MALDIマトリックス添加後に、膜を乾燥し、付着したタンパク質の質量と相対的存在度の定量化のために、MALDI−MS計測に直接挿入する。
【0120】
PPASは、選択された分離pHで正電荷又は負電荷の分子の(限定的にのみの)分別に1枚の捕捉膜を用いるだけでよい。1枚の捕捉膜によるPPASを高存在度タンパク質の除去に提供する(組込まれたふるい分け層によるか、又は、予備ステップで実行)。他の分別を実行する必要がない。選択肢として、さらに分別を強化するために、2以上の捕捉膜を直列に使用しうる。MALDI−MSは高存在度分子によりサンプルのイオン化抑制を受けるので、そのような分別の増加は実行した分別にほぼ比例して感度を高める。
【0121】
本発明の基本例はアルファ・プロトタイプ・システム(alpha prototype system)と共に示されている。プロトタイプ・システムは25個の捕捉ウエルの5×5配列を有し、1個のカートリッジ内で電気泳動による分離と捕捉を同時に行う。
図10に示す質量分析法の結果の場合、サンプルのpHは7.8であり、各ウエル内の電流は示された時間に対して、1mAに設定されていた。タンパク質の捕捉後、膜は脱イオン水により洗浄され、次ぎに、MALDIマトリックス溶液の添加により放出された。そのマトリックス溶液は、アルファシアノ−4−ヒドロキシナミック・アシッド(CHCA)で飽和された0.1%トリフルオロ酢酸溶液の1体積とアセトニトリルの1体積から成っている。次ぎに、そのマトリックスは乾燥され、分析のためにMALDI質量分析器内に置かれた。タイム・コースは、20分と40分の間に最初の正帯電タンパク質が到着する必要があること及び追加のタンパク質が40分と60分の間に到着する必要があることを示している。示されていないのは、60分以降に捕捉されたタンパク質に実質的変化が観察されないことを示すデータである。全てのMALDI−MS分析が、ABI/Perceptive Biosystems Voyager DE(MALDI−TOF)を用いた社内設計のQGEN_PR2法を用いることにより行われた。CHCAマトリックス溶液と共に用いるために、典型的な分析器の設定は:加速電圧20kV、グリッド電圧94.1%、ガイド・ワイア電圧0.050%、遅延時間110ns、レーザー設定3000、平均走査64、1.1E−6torr、低質量ゲート511、陰イオン・オフ。
【0122】
図11に示したMALDI質量分析法の結果の場合、電極の極性を逆にすることを除いて、手順と分析は図10に示したものと同様である。それゆえ、(極性反転の)2条件に基づいて観察されるタンパク質が、サンプルの事前決定されたpH(即ち、この場合7.8)で両スペクトルで観察されたタンパク質の負帯電が逆になるという事実に基づいて明らかに異なる。正帯電したタンパク質による結果と同様に(図10)、負帯電のタンパク質を捕捉するためのタイム・コースは、最初の負帯電のタンパク質が到着するのに必要なのは40分から80分の間であること、及び、追加のタンパク質の到着は80分から120分の間であることを示している。さらに、120分以降に観察された捕捉されているタンパク質に実質的変化が無いことを示すデータは示されていない。(図10に示された実験で用いられる電流は図11に示す実験で用いられた電流の2倍であることに留意されたい。逆に、図10に示された電気泳動の時間は図11で示した値の半分である。即ち、(期間の2倍に対する)電気泳動中に用いられる電荷移動のクーロン数は、図10に示す時間の半分の値と同じである。(それゆえ、示された2実験に移動した電荷)。
【0123】
(MALDI質量分析法のための対象発明の別の利用方法)
MALDIマトリックスは、以前に発表した方法を用いて調製し、その後に以下の一般的のひとつを用いることによりカートリッジ捕捉スライド42に加えられる:1)手操作のピペットによる添加、2)市販の液体取扱い用ワークステーション(workstation)を用いた添加、3)スプレー・コーティング、4)マトリックス溶液内へのカートリッジ捕捉スライドの浸漬、濃縮されたマトリックス溶液はMALDI分析に容認できるマトリックス対検体の比を実現するように添加される。
【0124】
特に有用なひとつの適用は、モノリス表面上にシナピン酸溶液(50:50のアセトニトリル/0.1%トリフルオロ酢酸で20mg/mL)を堆積させることから成っている。この溶液の体積0.25μLをカートリッジ捕捉スライドの上面にマイクロピペットを用いて添加する。この溶液を約5分の経過に亘って室内状態で乾燥する。その時点で、追加のマトリックス0.25μLを添加する。そのスライドを室内状態で又は真空乾燥器の中で乾燥させる。
【0125】
通例、MALDIマトリックスの堆積と乾燥の後で、捕捉スライドを、計器メーカーの仕様書に基づいて、質量分析器に導入する。スライドは、カートリッジ捕捉スライドをMALDI質量分析器のx−yサンプル・ステージに適合させる特別設計のスレッド(sled)にはめ込むように設計されている。そのスレッドは以下の条件に適合するように設
計されている:A)カートリッジ捕捉スライドを質量分析器内のイオン抽出軸に垂直に保持しなければならない。B)スレッドとカートリッジ捕捉スライドの境界は、カートリッジ捕捉スライドの表面電荷を消失させる通路を与えるようにしなければならない。C)カートリッジ捕捉スライドの表面の高さは各計器標準のサンプル・キャリア(sample carrier)のそれと一致しなければならない。D)そのスレッドに対する各モノリスの位置は常に同じでなければならない。これらの各要件は質量分析法の分野の技術者に知られている。
【0126】
捕捉スライド42上に捕捉された検体の質量分析は、市販されていて、そのような分析分野の技術者にはよく知られているツール(tool)のセットを用いることにより標準方式で処理される。例えば、ベースライン(baseline)減算、正規化、ピーク検出、スペクトルの位置合わせは、ProTS−Data(Efecta Technologies,Inc.;Steamboat Spring,CO;Version 1.1.1.0)として市販されているソフトウエアを用いて行われる。そのデータ解析を要約すると以下の通りである:
1.バックグラウンドの推定と減算:バックグラウンド信号は頑丈で、ローカル(local)で、統計的な推定機能により算定される。バックグラウンドは実質的に“ノイズ”であり、生物学的関連情報を含まず、スペクトルごとに異なっているので、各スペクトルからバックグラウンド値を減算することにより比較可能性を高めるのに、振幅情報が必要である。
2.正規化:イオン化されたサンプルの量はスペクトルごとに、レーザー出力の変化、イオン化可能なサンプルの量の変化、MALDIプレート上のレーザー位置の変化により変動しうる。ピーク振幅に関する定量的情報の信頼度を高めるために、スペクトルを全イオン電流に正規化する。
3.ピーク・ピッキング(peak picking):スペクトル内のピークを特定し、かつ、その信号/ノイズの比の信頼できる推定値を割付けるために、ノイズ算定機能が計算され、使用される。組織サンプルからの典型的なMALDIスペクトルに対して、我々は典型的に信号/ノイズ比のカットオフ(cutoff)3を用いて100から200の間のピークを検出する。
4.スペクトルの位置合わせ:ひとつのスペクトルの絶対質量スケール(scale)がかなり変動しうる。そして、共通ピークの選択は、共通のm/zスケールにスペクトルを登録するのに使用できる。
【0127】
(クラシファイア(classifier)の発生と検証)
標準の質量分析法の分析内で、質量ピークのリスト(重心の値及び正規化した強度を含む)が作成され、個別データ・ファイルに送られる。種々のソフトウエア・ツール/セットが、これらのデータにより質量分析からのバイオマーカー検出を容易にしている。同時にそのソフトウエアは、通常の変動を有する種々の集団について統計的有意性を評価するための有用なツールを提供している。機能のランキングは、グループを差別するための機能の重要性について、ある程度のアイデア(idea)を与えるけれども、より完全な分析には指導付き学習手順内で機能を使用することが必要である。指導付き学習では、訓練セット内の各インスタンス(instance)(即ち、各スペクトル)に対して、カテゴリー・ラベル(category label)を与える。そして、誤分類の数の低減を求める。指導付き学習問題を扱うために、多様な手順が開発されている。指導付き分類のアルゴリズムの出力が一般的に、新インスタンス又はスペクトルのためのクラス・ラベルを発生する(訓練セットにより)クラスファイアとして使用しうる。事例Aとして,Webb,A.John Wiley & Sons Ltd.,2002,Statistical pattern recognitionを、又、事例Bとして、Duda,O.R.,Hart,P.E.,Stork,D.G.,Wiley & Sons Ltd.,2001 ,Pattern Classification.を参照された
い。
【0128】
本発明は種々の具体的で好ましい実施例と技術を参照して記されている。しかしながら本発明の精神と範囲の中にあっても多くの変形と修正を行なえることを理解すべきである。
【図面の簡単な説明】
【0129】
【図1】ヒト血漿のプロテオームが血清内に存在するタンパク質とポリペプチドの分析例を示す。濃度範囲を10以上の桁数で示す。(図面は参考文献2に適合させている)
【図2】分析システムの1個のウエルの切断略図である。分析システムの好ましい実施例では分析システムはカートリッジ内に含まれる96個のサンプル・ウエルの8×12の配列を有している。
【図3】分析システムの好ましい実施例内のカートリッジから成るサンプル・ウエルの配列の略図である。
【図4】機械的案内92を有するMALDIスライド・ホルダー90に挿入された穴50を示す捕捉スライド42の好ましい実施例である。
【図4a】圧力で駆動され、捕捉スライドを横断する流体の流れを加えるためのスライド洗浄マニフォールドである。
【図4b】捕捉スライド上の捕捉材料と電解液の接触を維持するための、又、捕捉物質を横切る電解液に電場を与えるための電気泳動スライド洗浄装置である。
【図5】鋼製MALDIターゲット・プレート上の1pmolのポリペプチド標準及び〜127pmolのBSAのプロットである。
【図6】鋼製MALDIターゲット・プレート上の0.1pmolのポリペプチド標準及び〜127pmolのBSAのプロットである。
【図7】PPAS装置内のアルブミン枯渇液で濃縮された0.1pmolのポリペプチド標準及び〜127pmolのBSAのプロットである。
【図8】血清タンパク質のMALDI質量分析の例である。
【図9】PPAS装置を用いた二成分pH分別である。
【図10】捕捉材料として単一捕捉膜を有するタンパク質/ポリペプチド分析システム(PPAS)のアルファ・プロトタイプを用いて得られたヒト血清内の正帯電LMWタンパク質分析からの質量分析結果である。
【図11】捕捉材料として単一捕捉膜を有するタンパク質/ポリペプチド分析システム(PPAS)のアルファ・プロトタイプを用いて得られたヒト血清内の負帯電LMWタンパク質分析からの質量分析結果である。
【特許の相互参照】
【0001】
本発明は特許文献1、2、3の特典を主張し、その内容全体を参考用に組込んでいる。
【技術分野】
【0002】
本発明は質量分析法(MS)に、より特定すれば、マトリックス支援レーザー脱離イオン化質量分析法(MALDIMS)により分析するために血清のような生体サンプルからの検体の事前濃縮と精製に関する。
【背景技術】
【0003】
電気泳動濃縮装置のセル(cell)の移動相から固体捕捉相へのタンパク質、ペプチド(peptides)及び他の検体分子の濃縮と捕捉を容易にする装置と方法を開示している。さらに、そのような固体捕捉相は質量分析法での直接分析に適合できる。質量分析法は多数の検体を同時に監視できる。それとは対照的に、他の多くの分析技術は一度に1種のみ、又は、せいぜい1−2種の分子の定量分析を行なえるだけである。低費用計測、使用しやすくすること、高処理量のMALDI法のような質量分析法の最近の進歩が、臨床研究とひいては、全健康産業の変革を約束している。しかしながら、この膨大な可能性を実現するキーは、迅速かつ再現可能に質量分析法のために複雑な生体サンプルを調製できる新しいサンプル調製技術の開発である。そのような技術は、組織のホモジネート、全組織のスライス(slices)、他の固体組織の調製、さらに、全血、血漿、血清、脳脊髄液、唾液、尿等のような液体サンプルを含む多様なサンプルに応じる必要がある。多分、血清が臨床上最も重要な生体液で、数億のサンプルが医療診断のために毎年吸引管により採取されている。血液とリンパ液が病気のバイオマーカー(biomarkers)の豊富な源泉である。なぜなら、血液及びリンパ液の中を循環している天然の血液を発生するタンパク質及びポリペプチド(polypeptides)に加えて、身体組織は血液とリンパ液に追加の細胞要素を放出するからである。それゆえ、これらの循環流体はタンパク質及びポリペプチド(PP)を含む疾患のバイオマーカーを含み、細胞の肥厚化、壊死、アポプトーシス又は新生組織からの抗原の一掃のような病理的状態の指標になる。ここで、PPの用語は、オリゴペプチド(oligopeptides)、又は、2以上のアミノ酸(即ち、約200ダルトン(Dalton))から高分子量のタンパク質(約100万ダルトン以上)までの範囲を含む広い範囲の分子量のタンパク質を示すのに用いられる。
【0004】
血清内の実質的に有望なクラス(class)分野の疾患マーカーは低分子量(LMW)のPP破片で、それが豊富にあり、種々の構造変化をしていることは、大部分ではないが、多くのヒトの疾患を示している(非特許文献1)。LMW血清プロテオーム(proteome)は、サイトカイン(cytokines)、ケモカイン(chemokine)、ペプチド・ホルモン(peptide hormones)、大型タンパク質の分解破片のようないくつかのクラスの生理学的に重要なポリペプチドから作られている。これらのタンパク質分解によるペプチドは、がん、糖尿病、心臓血管系及び感染性の疾患のような病的状態と関連があることが示されている。しかしながら、LMW血清プロテオームの分析は、広範囲のサンプル調製を必要とし、血清中の総タンパク質量を支配する高い比率のアルブミン(albumin)(〜55%)により分析が非常に困難になっている。他の問題としては、他のLMW PP分子の存在度が広く動的な範囲に亘ることや主要な糖タンパク質の膨大な異種性が挙げられる。例えば、ここで臨床的に測定された最も希少なタンパク質は(図1)大きさがアルブミンより小さなものの桁10以上の濃度で存在する(非特許文献2)。しかしながら、これらの希少なタンパク質とペプチドは高い感受性と選択性のある疾患マーカーであり、潜在的な薬物ターゲット(target)になると信じられている。
【0005】
伝統的に、液体クロマトグラフィ(LC)又はアフィニティ(affinity)ベースの方法が適当な分離プロセスを提供するために最大限用いられて来た。LC法による精製にはリンカー(linker)分子をLCカラム(column)内の(機能化固定相を生じる)固定相に化学的に取付けることが含まれる。サンプルがカラムに装填されると、移動相が固定相を通過して流れる。各検体が移動相の代わりに固定相に結合するのに使われ、検体の精製用に提供されているLCカラムを通って(汚染物質及び干渉物質だけでなく)種々の検体の相対的移動速度を決定する。例えば、ペプチド及びタンパク質のような対象検体分子は機能化された固定相に吸収でき、その一方で汚染物質は検体から流出する。次ぎに、移動相を調節して、機能化した固定相から対象となる分子を溶離する。しばしば、アセトニトリル(acetonitrile)と水の混合体のような、MALDI−MSと適合している揮発性緩衝液が、このステップ(step)での移動相として用いられる。このようにして、精製された対象物質がLCカラムから流出してMALDI−MS検体として集められる。ここで、サンプルから塩類及び他の汚染物質を双頭除去しておかないと分析が妨害され、又は、感度が制限される。それゆえ、分析の高い処理方法でのサンプル分析に必要な試薬の分離、濃縮、添加のための装置と手順を改良する必要がある。質量分析法用のサンプル調製技術の最近の見直しでは、これらの方法は時間がかかり、面倒で、高い技術の労働者を必要とし、自動化が困難である(非特許文献3,4)。結果として、1臨床検査の間に分析できるサンプル数は極端に限定され、統計的優位性を実質的に損ない、それゆえ、これら検査の臨床的関連性が損なわれる。結果として、主として、サンプル調製システムが無いことにより、LMW血清プロテオームが、ヒトその他の動物の疾病、疾病治療、遺伝子発現分析のための(質量分析法により検出できる)バイオマーカー(biomaker)が優れているが大きな未開拓の分野になっている。
【0006】
MALDIターゲット・プレート(target plate)上に配置したサンプルのマトリックス支援レーザー脱離イオン化質量分析法((MALDI−MS)による分析が急速に、タンパク質、ペプチドその他の生体分子を分析するための優れた方法になりつつある。MALDI−MS手順は非常に感度の良い分析方法で、多分MS手順が生理的な塩類及びpH緩衝液と特に適合性がよい。さらに、ピコモル以下の量の生物学的巨大分子から高質量イオンを高効率で発生させる能力がこの手法をして巨大分子分析に特に有用なものにしている。しかしながら、血漿又は血清のような未処理生体サンプルのペプチド検体の分析には下記のように質量分析法上特別な問題が生じる。
【0007】
克服すべき第一の問題は高濃度の塩(例えば、ナトリウム、カリウム、塩素、リン酸塩及び炭酸塩)を含むことである。陰イオンは通常のMALDI分析手順を用いることによりペプチド・サンプルのイオン化を抑制するのに特に有効である。陽イオンにもアダクト・スペクトル(adduct spectra)を発生することで問題がある。当該スペクトラは、タンパク質の主要質量ピーク(peaks)を、それぞれが1個の陽イオンの付加的質量を有する多数の付加的質量ピークに分割する。さらに、MALDI−MS分析の成功は、質量分析器に注入する前に、検体と一緒に混合したMALDIマトリックス物質を効果的に結晶化する分析技術者の能力に大幅に依存している。MALDIマトリックス物質は、分析すべきサンプル内の吸着された検体物質と一緒にマトリックスの噴霧化とイオン化に用いられるレーザー光を吸収する必要がある。イオン化された検体分子は、次いで質量分析器内の陽極と陰極の高電圧により、質量分析器イオン検出器内で加速される。(塩又はグリセロール(glycerol)のような)比較的少量の汚染物質が存在するときでさえ、タンパク質とペプチドのような検体の脱離とイオン化を効果的に行うMALDIマトリックスの能力が劇的に低減する。さらに、高い塩濃度がMALDI−MSに必要な限界レーザー光度と(遊離ペプチドのピークを犠牲にして)塩を付加されたペプチドのピーク強度の両方を高める。
【0008】
第二に、ヒトの血清のようなサンプル内で、検体ペプチドは干渉プロテイン(例えば、アルブミン、イムノグロブリン(immunoglobulin)、トランスフェリン(transferin))と比較して、非常に低いコピー(copy)数で頻繁に存在する。対象のペプチドはしばしばリットル当たり丁度1マイクロモルからリットル当たり1ピコモルまで存在する(例えば、ミリリットル当たり1マイクログラムから1ピコグラム)。それと対照的に、総アルブミン及びIgG、IgMのようなガンマ・グロブリン(gamma globulins)が少なくとも、ミリリットル当たり0.01から0.1グラムの範囲の値で存在している。即ち、1×1011倍まで質量で大きい。それゆえ、主要な量が多いタンパク質が混合体のMLADIスペクトルを強く支配する。低強度のピークが主要ピークによりあいまいになるので、少量成分が観察されるのは稀になる。このことはヒトの血清のような生体サンプルでは深刻な問題である。生体サンプル内では、そのように低いコピー数の分子は、干渉タンパク質(例えば、アルブミン、イムノグロブリン及びトランスフェリン)及び塩(例えば、ナトリウム、カリウム、塩素、リン酸塩、炭酸塩)の桁違いに高いモル濃度が存在する中で検出する必要がある。
【0009】
第三に、検体ペプチドの多くは、疎水性であり、血液、血漿、血清に見いだされる主要タンパク質に結合している。特に、アルブミンは疎水性分子に非特異的に不特定に結合する傾向がある。かくして、アルブミンのような望ましくないタンパク質を除去することは、検体ペプチドの喪失にもなる。塩及び合成洗剤のような化学的破壊因子がアルブミンから検体ペプチドの解離を支援することが知られている。しかしながら、これらの因子は積極的にMALDIプロセス(process)を抑制する。例えば、ポリエチレン・グリコール(polyethylene glycol)(PEG)及びトリチオン(Trition)はペプチド及びタンパク質と同じ効率でMALDIによりイオン化と脱離を行う。結果として、これらの種は多くの場合タンパク質とペプチドのイオン化と競合する。それにより、後者からのMALDI−MS信号を抑える。それゆえ、アルブミンからの検体ペプチドを解離するために、化学的破壊因子を加えた後で、分析担当者が検体ペプチドを、その破壊因子のアルブミンと他の汚染タンパク質の両方から分離しなければならない。さらに、マイナー(minor)成分のペプチド検体が分離プロセス中で喪失しないように分離を実行しなければならない。この分離は、検体が疎水性であり、疎水性の面に接合する傾向があるとき、特に困難になる。不幸にして、LC法による生体高分子の精製が、しばしば、30%以上のサンプル減耗を生じ、又、汚染物質(又はサンプル間の信号漏洩)を追加することがある。多くのMALDI−MSユーザーにとって、このサンプルの減耗量は容認できない。第四に、検体のペプチドがそのような低レベルで存在するので、MALDI−MS分析の前に濃縮しなければならない。従来方法によって、先ず、ペプチドの解離、成分の分離である。次いで、濃縮を行うことは退屈で、単調であり、長時間を要し、かつ、労働集約的な多数のステップを必要とする。
【特許文献1】米国特許暫定出願第60/668,337号明細書
【特許文献2】米国特許暫定出願第60/668,794号明細書
【特許文献3】米国特許暫定出願第60/712,255号明細書
【特許文献4】米国特許暫定出願第60/748,771号明細書
【特許文献5】米国特許出願第10/963,336号明細書
【非特許文献1】Tirumalai,R.S.,K.C.Chan,D.A Prieto,H.J.Issaq,T.P.Conrads and T.D.Veenstra,(2003)Characterization of the Low Molecular Weight Human Serum Proteome,Molecular & Cellular Proteomics 2,1096−1103.
【非特許文献2】Anderson N.L.and N.G.Anderson(2002)The Human Plasma Proteome,Molecular and CellIular Proteomics 1,845−867.
【非特許文献3】Westermeier,R.and T.Naven(2002);In:Proteomics in Practice;Wiley−VCH Verlay−GbmH,Weinheim.
【非特許文献4】Hamdan,M.and P.G.Righetti(2005);In:Proteomics Today;John Wiley & Sons,Hoboken,NJ.
【非特許文献5】Schreiner,M,K Strupat,F.Lottspeich and K.Eckerskorn(1996)Ultraviolet Matrix Assisted Laser Desorption ionization−Mass Spectrometry of Electroblotted Proteins,Electrophoresis,17,954961.
【非特許文献6】Bienvenut,W.V.,J.C.Sanchez,A.Karmime,V,Rouge,K.Rose,P.A.Binz and D.F.Hochstrasser(1999)Toward a clinical Molecular Scanner for Proteome Research:Parallel Protein Chemical Processing before and during Western Blot,Alnal Chem..11,4800−4807..
【非特許文献7】Lion,N.,V.Gobry,H.Jensen,J.S.Rossier,H.Girault(2002)Integration of a Membrane−Based Desalting Step in a Microfabricatd Disposable Polymer Injector for Mass Spectrometric Protein Analysis,Electophoresis 23,3483−3588.
【非特許文献8】Muller,M.,F.Gras,P.A.Binz D.F.Hochstrasser and R.D.Appel(2002)Molecular Scanner Experiment with Human plasma:Improving Protein Identification by usmg Intensity Distributions of Matchmg Peptide Masses,Prteomics 2,1413−1425.
【非特許文献9】Scherl,A.,C.G.Zimmermann−Ivol,J.D.Dio,A.R.Vaezzadeh,P.A.Binz,M.Amez−Droz,R.Cochard,J.C.Sanchez,M.Gluckmann and D.F.Hochstrasser(2005)Gold Coating of Non−Conducting Membranes before Matrix−Assisted Laser Desorption/Ionization Tandem Mass Spectrometric Analysis Prevents Charging Effect,Rapid Commun.Mass Spectrom.19,605−610.
【非特許文献10】Moya,W.(2002)Surface Modified Porous Membrane and Process,U.S.Patent Number 6,354,443.
【非特許文献11】Lammerhofer,M.,F.Svec,J.M.J.Frecher and W.Lindner(2000)Monolithic stationary phases for Enantioselective Capillary Electrochromatography,J.Microcolum Separations 12,597−602.
【非特許文献12】Lammerhofer,M.,F.Svec,J.M.J.Frechet and W.Lindner(2001)Capillary Electrochromatorgrapy in Anion−exchange and Normal−Phase Mode using Monolithic Stationary Phases.J.Chromatoglaphy A 925 265−277.
【非特許文献13】Rohr T.,M.,D.F.Ogletree,F.Svec and J.M.J.Frechet(2003)Surface Functionalization of Thermoplastic polymers for the Fabrication of Microfuidic Devices by Photinitiated Grafting,Advanced Functional Materials 13,264−270.
【非特許文献14】Rohr,T.,M.,E.F.Hilder,J.Donovan,F.Svec and J.M.J.Frechet(2003)Photografting and the Control of Surface Chemistry in Three−Dimensional Porous Polymer Monoliths,Macromolecules 36 1677..1684.
【非特許文献15】Stachowiak,T.B.,T.Rohr M.,E.F.Hilder,D.S.Peterson,J.F.Svec,M.Yi and J.M.J Frechet(2003)Fabrication of Porous Polymer Monoliths Covalently Attached to the Walls of Channels in Plastic Microdevices,Electrophorisis 24 3689−3693.
【非特許文献16】16 Peterson,D.S.,T.Rohr,J.F.Svec,M.Yi and J.M.J.Frechet(2003)Dual−Function Microanalytical Device by In Situ Photolighographic Grafting of Porous Polymer Monolith:Integrating Solid−Phase Extraction and Enzymatic Digestion for Peptide Mass Mapping,Analytical Chemistry 75,5328−5335.
【非特許文献17】Peterson,D.S.,Q.Luo,M.,E.F.Hilder,F.Svec,and J.M.J.Frechet(2004)Porous Polymer Monolith for Surface−Enhanced Laser Desorption/Ionization Time−of Flight Mass Spectrometry for small Molecules,Rapid Commun.Mass Spectlom 18,1504−1512.
【非特許文献18】Pucci,V.,M.A.Raggi;F.Svec,and J.M.J.Frechet(2004)Monolithic Columns with a Gradient of Fuctionalities Prepared via Photoinitiated Grafting for Separations using capillary Electrochromatography,J.Sep.279−788.
【非特許文献19】Lee,D.,F.Svec,and J.M.J.Frechet(2004)PhotoPolymerized Monolithic capillary columns for Rapid Micro−High Performance Liquid Chromatographic Separation of Proteins,J.Chromatography A,1051,53−60.
【非特許文献20】Hilder,E.F.,F.Svec,and J.M.J.Frechet(2004)Development and Application of polymeric Monolithic Stationary Phases for Capillary Electrochromatography,J.Chromatography A,1044,3−22
【非特許文献21】Peterson,D.S.,T.Rohr,J.F.Svec and J.M.J.Frechet(2002)Enzymatic Microreactor−on−a−Chip:Protein Mapping Using Trypsin Immobilized on Porous Polymer Monoliths Molded in Channels of Microfluidic Devices,Analytical Chemistry 74,4081−4088.
【非特許文献22】Svec,F.,(2004)Preparation and HPLC Applications of Rigid Macroporous Organic Polymer Monoliths,J.Sep.Sci.,27;747−766.
【非特許文献23】Knochenmuss,R.Anal.Chem 2004;76:3179.
【非特許文献24】Zalluzec,E.J.;et.al.J.Am.Soc.Mass Spectrom 1994;5:230.
【非特許文献25】Andrews,P.C.;et.al.Anal.Chem.1996;68:1910.
【非特許文献26】Andrews,P.C.;et.al.Electrophoresis,1997;18:382.
【非特許文献27】Costello,C.E.;et.al.Rapid Commun.Mass Spectrom.1999;13:1838.
【非特許文献28】Peterson,D.;Rohr,T.;Svec,F.;Frechet,J.M.J.Anal.Chem.2003;75:5328−5335.
【非特許文献29】Frechet,J.M.J.;et.al.Macromolecules 2003;36:1677−1684.
【非特許文献30】Frechet,J.M.J.;et.al.Journal of Chromatography A 2004;1044:3−22.
【非特許文献31】Frechet,J.M.J.;et.al.Electrophoresis 2003;24:3689−3693.
【非特許文献32】Frechet,J.M.J.;et al.Journal of Chromatography A 2004;1051:53−60.
【非特許文献33】Svec,F.J.Sep.Sci.2004;27:747−766.
【非特許文献34】Frechet,J.M.J.;et al.Rapid Commun.Mass Spectrom 2004;18:1504−1512.
【非特許文献35】Ericson,C.;Liao,J.L.;Nakazato,K.;Hjerte'n,S.J.Chromatogr,A 1997;67:33−41.
【非特許文献36】Tempst,P.;et al.Anal.Chem.2004;76:1560−1570.
【発明の開示】
【0010】
それゆえ、本発明の第一の目的は生体サンプルから塩を除去するための方法と装置を提供することである。本発明の第二の目的はタンパク質のような高存在度の分子を生体サンプルから除去し、それにより残留する低存在度の分子の再現性が良く、感受性の高い分析を行なえることである。本発明の第三の目的はアルブミンや他の疎水性タンパク質から検体ペプチドを解離することである。本発明の第四の目的はMALDI質量分析法のための対象となる検体のペプチドとタンパク質を濃縮することである。本発明の第五の目的はサンプルの高処理量を提供するために、前記四目的を簡便で効果的な方法により提供することである。本発明の第六の目的は2以上のサンプルを並行して分析するために多数のサンプルを同時に扱うことである。それにより、本発明の他の目的と組合わせることにより、検査担当者が簡便かつ効果的な方法で生体組織サンプル内のペプチド及びタンパク質の分析を行なうために本発明を使用でき、それにより、検出の感度を高め、サンプルの処理量を高め、分析費用を低減する。最後になるが、分離された検体のペプチド、ポリペプチド、及び、タンパク質の分析を再現性をもって、かつ、定量的に行われることが所望された。それで、本発明の第七の目的は生体サンプル内のペプチド及びタンパク質について再現性があり、定量的なMALDI−MS分析を提供することである。
【0011】
用語PPを用いる場合、小型の2個以上のアミノ酸から100万ダルトン以上の大型タンパク質までの範囲のオリゴペプチドを意味していて、本発明の第八の目的は質量分析法(MS)により、ヒトの血清内のPPのLMW成分検査する分析システムを提供することである。本発明の第九の目的は例えば500ダルトンから500,000ダルトン以上までの広範囲のPPで、かつ、質量分析法(MS)により分析できる十分な融通性のあるPPAS(ペプチド・タンパク質分析システム)を提供することである。本発明の第十の目
的はPPASを改善し、さらに検出感度を高め、1ナノモル(nanomole)から0.1アトモル(attomole)以下までのPPの量を検出でき、MSにより測定された分子量を検出し、定量化できることである。本発明の第十一の目的はMS分析前に低存在度のPPを高存在度のPPから分離して、低存在度PPの検出感度を高めて、ヒトの血清内のPPの分別と分離を改善することである。
【発明を実施するための最良の形態】
【0012】
特許文献5には本発明の分野で用いるための方法と装置を開示していて、本出願書に参考用として、その全体を組込んでいる。さらに、本発明の方法と捕捉スライド(slide)はそこで開示された装置と方法に関連して使用しうる。さらに、本発明の方法と捕捉スライドは特許文献4に開示された装置と関連して使用しうる。特許文献4全体を参考用として本出願書に組込んでいる。
【0013】
本発明の有用な実施例は固相捕捉スライド上への血清のような(又は他の組織からの)生体サンプルに存在する低存在度のタンパク質及びポリペプチドを電気泳動的に分離し、濃縮し、捕捉するペプチド・タンパク質分析システム(PPAS)である。短い洗浄ステップの後で、塩その他の干渉分子が洗い流される。そして、MALDIマトリックス溶液が捕捉スライドに加える。従来技術で良く知られているように、そのようなマトリックス溶液は一般的に有機溶媒を含み、タンパク質を放出してMALDIマトリックス結晶内に組込み、溶媒の乾燥でスライド面上に沈殿する。次ぎに、そのスライドを完全に乾燥して、捕捉したタンパク質の質量と相対的存在度の両方の定量化のために、MALDI−MS測定に直接挿入する。
【0014】
図2及び3に詳細に示すように、PPASは流体サンプルを保持するために、1以上のウエル(well)4を有するカートリッジ(cartridge)2から成っている。カートリッジ2の1実施例には、25個のサンプルを同時に処理するために、25個のサンプル・ウエル(sample well)が含まれる。カートリッジ2の好ましい実施例は96個のサンプルを同時に処理するために8×12の配列で96個のサンプル・ウエルを含む。好ましい実施例では、分離と捕捉を行うのに必要な捕捉スライド42と試薬がカートリッジ2内のサンプル・ウエル4の配列として、事前に配置されている。図3はカートリッジ2を含むサンプル・ウエル4の配列を示している。図2はマルチ・ウエル(multi−well)PPASカートリッジ2の1個のウエルの切断面を示す略図である。
【0015】
各サンプル・ウエル4は上方開口部8、側壁10、下方部分12を有し、ウエル4が広い上方開口部8から狭い下方開口部14に寸法が段々に減少している。サンプル・ウエル4の上方開口部8がサンプル電極20を受入れる。サンプル電極20は図2に示すようにサンプル・ウエル内に配置された電解質サンプルと電気的に接触する。本発明の好ましい実施例で、各サンプル・ウエルが約50μLのサンプル、200μLの電気泳動用緩衝液を保持し、150μLの上部空間を残すように設計されている(全体積400μL)。サンプル電極20は好ましい実施例では、サンプル電極の配列として示されていて、各サンプル・ウエル4の上方開口部8に取外し可能にはめ込まれている。サンプル電極の配列は、それぞれの使用前にDI水又は他の適当な溶媒でアセンブリー(assembly)を洗浄するだけで、再使用可能で清浄にできるよう設計されている。選択肢として、より厳密な浄化が、洗剤、強酸例えばpH2.0以下のもの、又は、強塩基例えばpH12.0以上のもの、又は、有機溶媒例えばメタノール、エタノール、アセトニトリル、アセトン、CS2、ジメチルホルムアミド、ジメチルスルホオキシド、等を用いて行われる。各サンプル・ウエルの下方部分12が、各サンプル・ウエルの下方開口部14の近くの断面積が連続的に低減するように各サンプル・ウエルの下方部分12が成形されている。下方のウエル部分が円錐形で、各サンプル・ウエルの下部で低減した面積の下方開口部14にタンパク質分子が集まるようにしている。下方開口部14の下に分離層30があり、サンプル・ウエル4を捕捉物質40から分離するのに役立つ。分離層30は、サンプル・ウエル4内又は分離層内で選択された第一のサンプル分子を保持し、その一方で、選択された第二のサンプル分子が分離層を通過して、捕捉物質に接触でき、第二のサンプル分子を捕捉し、濃縮できるように機能する。本発明の好ましい実施例では、分離層はポリアクリルアミド・ゲルのようなゲル層から成っている。そのようなゲルは一般に1%から24%のポリアクリルアミドを有する。又、種々の量の架橋剤と重合開始剤も有していて、タンパク質分離の分野の技術者には良く知られている。さらに、好ましい実施例では、サンプル・ウエル4の配列を有し、対応する実質的に同じ分離層30の配列が存在し、好ましくはカートリッジ・ゲル・プレート32内に配置される。そこでは分離層30の配列がカートリッジ・ゲル・プレート(cartridge gel plate)32上に配置された実質的に同じ穴34の配列内に含まれる。一般に、ゲル・プレート32は熱可塑性ポリマー(ポリウレタン、ポリプロピレン等)のような所望の材料から切削、成型又は鋳造により形成される。そのようなゲル・プレートは電気的に絶縁性で、柔軟なポリマー例えば熱硬化性ポリマー、エラストマー(elastomer)、又は、ゴム材料である。一般に、そのような柔軟な材料は良好な液体密封特性を有し、その一方で、サンプル・ウエル4の間で電気的分離も行う。さらに、分離層30もサンプル・ウエル4を1以上の捕捉物質40から分離するのに役立ち、分離層30を通って電気泳動により駆動される検体分子を捕捉し、濃縮するのに役立つ。有利なこととして、分離層30はプレート32に共有結合をしている。そのような共有結合は接合喪失を防ぎ、カートリッジ・アセンブリーの組立を容易にしている。上記のように、液状媒体内でタンパク質の分離に特に有用な分離層はポリアクリルアミドである。それゆえ、ポリアクリルアミドとその支持構造の表面の共有結合が特に有用である。ポリアクリルアミドから固体のポリアクリルアミド支持構造への化学的接合は、物理的に強い複合構造の形成とポリアクリルアミドとその支持構造との間の液体密封部を形成することの両方に役立つ。この場合、特にゲル・プレートの穴34により定義された領域内で、ポリアクリルアミドの分離層30とゲル・プレート32の間で接合が形成される。ポリアクリルアミドによるその支持面へのそのような共有結合を実施する方法で、ポリアクリルアミドの反応混合体がゲル・プレート32内のゲル・プレートの穴34内に堆積する。その後に化学的グラフト・ステップがある。特に頑丈で耐久性のあるポリアクリルアミドの分離層30は基本的に2ステップの反応手順に基づく光グラフト(graft)によりゲル・プレート32に光グラフトされる。両方の反応ステップが支持面と接触したアクリルアミドのモノマー(monomer)とビスアクリルアミド(bis acrylamide)を含む溶液を用いることにより実施できる。(バルク(bulk)反応混合体内での)重合及びポリアクリルアミドの支持構造面例えばゲル・プレート32への接合の両方の開始が紫外線照射又は代わりに化学的開始剤を用いることにより行われる。便宜的には、ほぼ、ゲル・プレートの寸法の物理的保持具を用いて、ゲル・プレート32及びモノマーを含む反応溶液の両方を保持する。さらに、反応混合体は薄いシート(sheet)材料により支持構造に接触して保持される。薄いシート材料は真空クランプ(clamp)のような物理的手段により支持構造で近似的に保持される。
【0016】
好ましい実施例で、ポリアクリルアミドが取付けられる第一の固体面が、光グラフト反応混合体で前処理される。その後、ポリアクリルアミドの支持面への化学的接合及びポリアクリルアミドのバルク混合体の重合反応が同時に行われる。例えば、接合と重合反応の両方が現場での紫外線照射により開始される。好ましい方法では、事前浸漬のステップを用いていて、それは重合前にゲル・プレート材料に光開始剤を吸収させることから成っている。例えば、その事前浸漬ステップは、以下のサブステップ(sub−step)から成る。(a)タイプIIの光開始剤を含む事前浸漬溶液を使用。次いで、(b)例えば、空気のような乾燥気体でゲル・プレートを乾燥。代わりに、その気体を加熱して乾燥に使っても良い。
【0017】
タイプII光開始剤はSigma−Aldrich Companyのような会社から入手できる。一般に、タイプII光開始剤は生体分子と作用し、光開始剤の励起状態が第二の分子(共同開始剤)と相互作用して、遊離基を生じる。例には、ベンゾフェノン/アミン(benzophenones/amines)、チオキサントン/アミン(thioxanthones/amines)が含まれる。
【0018】
タイプII光開始剤の事前浸漬溶液の特定の例は、チオキサンテン−9−オン(thioxanthen−9−one)を0.006%(質量比)のメタノール溶液である。好ましい例で、上記のグラフト及び重合の工程は、表面上に反応混合物を置くことにより実施され、モールド(mold)を真空密封することにより低及び無酸素環境を形成し、ウエルの内面に共有結合している共重合分子を生じるのに十分な時間だけ混合体をUVエネルギーを照射する。一般に、通常のUVエネルギー源(5000−EC ユニット:入手先:Dymax Corporation Torrington,CT,USA:Hランプを装着)を用いて、照射時間を1秒から1時間の間とする。代わりに、非常に強いUV源、又は、瞬間光源を用いて、照射時間を非常に短くする。例えば、1マイクロ秒から1秒以下とする。それでも、他の適当なUV照射源には水銀アーク・ランプが含まれる。
【0019】
他に、ポリアクリルアミド・ゲルが化学的に付加接合できるポリアクリルアミド支持構造として使用するのに適当な別のタイプの材料にはポリウレタン、サントプレン(santoprene)、ポリプロピレン等が含まれる。一般に高分子物質で、その表面、即ち、直鎖又は側鎖部分に抽出可能な水素原子を含む物質なら、いずれも本発明を実施するのに適当なポリマーになる。例えば、抽出可能な水素は、ダブル・アリル(double allylic)水素、アリル水素、第三水素又は第二水素の形になる。特定の例には、以下があるが、それに限定されない。ポリオレフィン、水素化ポリスチレン、環状オレフィン共重合体、ポリ(エチレンテレフタレート)、ナイロン、ポリカーボネート、ポリ塩化ビニル、ポリブチルメタクリレート、ポリスチレン、ポリ(ジメチル・シロキサン)、又は、ポリ(メチルメタクリレート)。接合工程を開始するための追加の光開始剤は一般に当業の技術者に良く知られていて(バルク・ポリアクリルアミド重合反応の混合体の代わりに)グラフトすべき固体ポリマーの表面に対し分配特性有するタイプII光開始剤を含む。
【0020】
好ましい様態で、光グラフトとバルク重合の結合混合体は約68.7%(体積)が水性緩衝液、30%(体積)が19:1比で存在するアクリルアミド(acrylamide)/N,N'−メチレンビスアクリルアミド(N,N'−Methylenebisacrylamide)の40%(W/V)脱イオン水溶液、0.69%(体積)がチオキサンテン−9−オン(Thioxanthen−9−one)の 0.6%(W/V)メタノール溶液、0.41%(体積)が過硫酸アンモニウム(ammonium persulfate)の1%(W/V)脱イオン水溶液、0.20%(体積)が1,2−.ジ(ジメチルアミノ)エタン(TEMED)(又はEDMA)から成っている。反応混合物が混合され、ガラス又はポリマーの下面を有する浅い容器に置かれる。そのような下面は「非付着性」の面例えばTeflon(R)として特に有用である。「非付着性」の面は容器内に含まれる希望の固体支持構造例えばゲル・プレート32にポリアクリルアミドを外しやすくなるように機能する。その手順の中で、ゲル・プレートが光グラフトとバルク重合の結合混合体内に置かれ、UV透過ガラス、又は、薄いポリマープレートのようなUV透過カバーで覆われて、ゲル・プレートの穴34が反応混合物を含むが空気の泡を排除する。UV透過プレート、ポリアクリルアミドの反応混合物を含み、支持構造を機械的に接合している構造が、結合クリップ(clip)、真空クランプ(clamp)又は他のクランプ手段により所定位置に保持される。ゲル・プレートをカバーするUVUV透過プレートの上にフォトマスク(photomask)を使用しうるので、反応混合体と支持構造の希望の部分がUV光源により照射される。それにより、ポリアクリルアミドは事前決定されたパターン(pattern)で支持面に接合される。光重合を開始するために、その構造体をUV照射装置の付近に置き、照射の波長と強度に基づき適当な時間照射する。照射される表面で照射光束が150mW/cm2である場合、照射時間はシステム要因によるが一般的に4分未満である。例えば、Dランプを用いた5000−ECユニット:入手先:Dymax Corporation Torrington,CT,USAをゲル・プレートの表面から約20cmの距離で使用した場合、十分なUV照射が与えられる。UV照射の後で、UV透過カバーを取外し、光グラフトにより固体支持構造(例えば、ゲル・プレート32)に化学的に接合されたポリアクリルアミドを容器から取外し、非重合反応混合体を除去するために、脱イオン水のような適当な洗浄用溶媒で洗浄する。得られたポリアクリルアミド/支持構造の一体部分(例えば、ゲル・プレートに接合したポリアクリルアミド・ゲル)が適当な液体媒体に置かれ、又は、保管のために密封包装を行い、又は、使用のためにカートリッジに直ちに組込む。現場で製作され、支持されたポリアクリルアミドが優れた機械的安定性を示し、その支持材料への良好な接合を示す。上記の同時重合工程は、時間効率が良いように、そのような化学的に接合し、支持されたアクリルアミド構造の製造を行うのに特に便利である。
【0021】
選択肢として、ポリアクリルアミド反応混合体が、追加の有用なリガンド(ligands)例えば、タンパク質、多糖類、DNA、RNA等を含む。そのようなリガンドは便宜的に、重合前にポリアクリルアミド反応混合物に加える。代わりに、隣接する浸漬溶液からリガンドの拡散に十分な時間を与えることにより、又は、浸漬溶液から接着されたポリアクリルアミドへの活発な電気泳動により、重合後にポリアクリルアミドにリガンドを加える。例えば、望ましい場合、ポリアクリルアミドによるアルブミンの保持を高める目的で、改質された炭水化物材料をポリアクリルアミドに加える。そのような材料の例には、ブルー・デキストラン(blue dextran)、タンパク質へのアフィニティを改質したシリカ(silicas)、又はアルブミン結合分野での技術者に知られている他の材料が含まれる。
【0022】
(捕捉スライド)
捕捉物質40が、上面41と下面43を有するカートリッジ捕捉スライド(cartridge capture slides)42に位置しているオリフィス(orifices)50に配置されている。オリフィス50は上面41の上方開口部52、下面43の下方開口部54を有している。さらに、その開口部が捕捉スライド42の内側壁面56から成っている。捕捉材料40は一般にオリフィス50の内側壁面56で捕捉スライド42に取付けられる。その取付は、溶媒、熱、音波、又は、他の溶接手段による溶接を含む接合手段により行われる。代わりに、捕捉材料40は、エポキシ、メタクリレート、シアノアクリレート又は他のタイプの化学的結合材料及び樹脂を用いた化学的共有結合手段によりオリフィス50で捕捉スライド42に取付けられる。
【0023】
捕捉材料40を保持するためにオリフィス50(穴とも言う)を含むカートリッジ捕捉スライド42の好ましい実施例では、カートリッジ捕捉スライド42が長さ約4mmから約6mmの間、幅約3mmから約4mmの間、厚みが約1mmである。より好ましくは、カートリッジ捕捉スライド42が長さ約5.3mm、幅約3.5mm、厚み約1mmである。さらに好ましくはオリフィス50は実質的に円形であり、直径は約0.5mmから約1mmである。同様の寸法が好ましいカートリッジ・ゲル・プレート32にも適用される。
【0024】
図2及び3に示すように、カートリッジ捕捉スライド42の下では、電解質ベースチャンバー60があり、個々のカートリッジ・ウエルをお互いに物理的分離と電気接続をするように機能する。さらに、1以上の共通対極70を1以上の対極チャンバー72に対応している。すぐに使用できるとき、電解質ベースチャンバー60が導電性の電解質ベース媒
体62で充填され、対極チャンバー72が対極電解質74で充填されている。ベース媒体と対極電解質がイオンを導通させ、対極チャンバー72内の対極70と捕捉スライド42内の捕捉材料40を電気的に接続する。対極チャンバーは側壁76を有し、すぐに使用できるとき、実質的に全表面に対し、少なくとも部分的に垂直になっているので、電解質74上の電極70の作用によって発生する気体の泡(例えば、水素又は酸素)の脱出のために、連続的上昇路を提供する。有利なことに、電解質ベース媒体62が、例えば、一般用の可溶性の陰イオンと陽イオンのペア(pair)を水溶液中0.001から1モルの濃度で含んでいるので、高い導電性である。この汎用の陰イオンと陽イオンのペアは、チャンバー60及び72の材料に適合していれば、実質上、どの可溶性の陰イオンと陽イオンのペアでも良く、例えば、ナトリウム、リチウム、カルシウム、マグネシウム等及び塩化物、フッ化物、硫酸塩、チオシアナイド等の塩である。当該ペアから成る好ましい塩のひとつがKClであるのは、陰イオンと陽イオンが実質的に同じ拡散係数を有し、それにより電解液の濃度が異なる2種類の電解液間の境界での拡散の可能性を最小限にするからである。一般的に、汎用の可溶性の陰イオン及び陽イオンのペアは弱酸でも弱塩基でもない。なぜなら、弱い酸又は塩基の濃度の違い又は導電性の違いを有する2種の電解液間の境界での酸又は塩基の帯電形の移動が、境界での、又は、境界を横切ってpHの変化を生じるからである。しかしながら、これらの電解液はそのような弱い酸又は塩基を含みうる。ただし、それは本出願書の他の部分に示すように、電解質のpHの制御又は修正を生じる手順を用いて入念に選択され、使用される。
【0025】
電解質ベース媒体62を、粘性を高め、漏洩を防止し、又は、気泡を捕捉するために、又は、ゲル化物質を溶解することにより、又は、当業の技術者に良く知られているように、ゲルとして加える。ゲル化物質はデンプン又はアガロース(agarose)又は親水性ポリマーの共重合例えばアクリルアミド又はヒドロキシメチルメタクリレート(hydoroxymethlmethacrylate)のようなものである。さらに、1以上の対極チャンバー72も電解質ベースチャンバー60で用いられるのと同じ組成を有する電解質74を充填している。しかしながら、チャンバー72が捕捉材料40から物理的に分離されているので、電解質74を構成する導電性塩の選択では広い寛容度が可能である。例えば、高濃度の無機塩(例えば、0.1から10モル)、及び、普通の対極70により生じる水素又は水酸化物のイオンのpH緩衝液を提供するために、通常用いられる弱酸又は弱塩基が、随意、対極電解質74を構成しうる。例として、1.0M,pH8.0のトリスヒドロキシメチルアミノメタンクロリド(tris(hydroxymethyl)aminomethane−chloride)、三塩化物((tris)chloride)、又は、1.0M,pH9.2の硼酸カリウム(potassium borate)、又は、1.0M,pH7.0のイミダゾリウムクロライド(imidazolium chloride)等である。しかし、適当な高緩衝溶液であれば事実上どれも十分であり、当業の技術者には良く知られている。
【0026】
好ましい実施例では、カートリッジ2の電解質ベースチャンバー60はゲル化された対極緩衝液74を事前充填される。例えば、ゲル化溶液は1%アガロース・ゲルで良く、又、1.0M KCl,1mM ヒスチジン(histidine),pH7.8から成っている。さらに、好ましい実施例で、カートリッジ・ゲル・プレート32内の分離層30及びカートリッジ2のカートリッジ捕捉スライド42内の多孔性捕捉材料40はイオンによる導電性液体媒体を事前に充電される。例えば、分離層30はポリアクリルアミド・ゲルであり、1 mMから500 mMの無機塩を含む電解液内で重合したポリアクリルアミドを2%から12%含み、又は、15%の高さもある。1実施例では、分離層に事前に充填された電解質は50mMのKCl、100mMのヒスチジン(histidine),pH 7.8である。カートリッジ2のカートリッジ捕捉スライド42内の多孔性捕捉材料40の中に事前充填された電解質の組成は、溶媒に溶解された多様な導電性の塩で良い。その溶媒は水溶液、又は、他の適当な有機溶媒で良く、例えば、メタノール、エタノール、プロパノール等、又は、代わりにアセトニトリル(acetonitrile)又は他の水溶性有機溶媒である。最適には、使用する溶媒は、また、多孔性の捕捉材料の検体を通る適当な導電性を与える1から1Mの有機又は無機の塩を含む。便宜的に、分離層を形成するのに用いられたのと同じ溶液、例えば、10mM KCl,100mM ヒスチジン(histidine),pH8.0を使用しうる。
【0027】
分析システムとして提供するために、電子計測制御要素が使い捨てカートリッジ2と共に用いられる。調節可能な+/−300V電源(即ち、調節範囲は600V)100を使用して、電気泳動法に必要な電場を供給できる。そのような比較的低い電圧の電源で十分なのは、分離距離を1cm未満、一般的に、約0.1から0.5cmにしうるからである。さらに、分離と捕捉のステップ工程を監視するために、各サンプル・ウエルを通過して、サンプル電極20から対極74への電流を別個に監視する。例えば、96個の個別電流計を使用しうる。多数の電流計が電流測定のために単一回路から成っているが、電流値を報告するためのサンプル及び保持の回路を有している(例えば、1Hzの報告周波数)。好ましい方式では、結果はコンピューターのモニターにグラフとして表示される。代わりに、調節可能な一定電流源を電圧源の代わりに用いる。通常、電流源はサンプル・ウエルごとに0から100ミリアンペアを供給する。より一般的には、電流源から0−10ミリアンペアを供給する。有利なことに、コンピューターで制御され、選択可能な電流源/電圧源を使用しうる。好ましい選択可能な供給源とその使用方法は特許文献4に開示されていて、その明細書全体が参考用として本出願書に組込まれている。
【0028】
代わりに、本発明を実施するのに必要な電子要素はより単純なもので良く、例えば、直流電圧源及びサンプル電極の配列を含むだけでよい。この代替的実施例では調節可能な+/−100ボルトの電圧源(通常<25ボルト)を用いて、電気泳動に必要な電場を供給している。例えば、25個のサンプル分析システムでは、25個の電流計を使用し、それぞれが(1Hzの報告周波数での)電流値を報告するためのサンプル及び保持の回路を有している。希望する場合、結果はコンピューターのモニターにグラフとして表示される。代わりに、又、さらに単純にする場合には、電気泳動法を電圧源のみで実行しうる、即ち、電流を監視しないが、事前決定された時間だけ電気泳動法を実施するか、又は、代わりに検出可能な(視覚的、化学的又は電気的)終点に達するまで実施する。
【0029】
以下に示す方法を実施するのに適当な装置には、+/−100V電源、25チャンネルの個別に調節できる電位差計、Agilentモデル34970Aデータ取得/スイッチ、25個のウエルのLexanカートリッジ、ラップトップ(laptop)・コンピューターが含まれる。Agilentデータ取得システムのためのソフトウエアで、25個のサンプル・ウエルのそれぞれについて時間の関数として電圧と電流を記録するように構成しうる。Applied Biosystems Voyager DF and 4700 model MALDI質量分析器を用いて、タンパク質とペプチドを含む検体の質量分析法と定量化を行なえる。
【0030】
システムの操作説明:
1.サンプル分子の第一群と第二群の混合体をサンプル・ウエル4に置く。
2.サンプル電極20をサンプル・ウエル4内のサンプルと電気的に導通させる。
3.サンプル電極20に電圧源100から電気を通じて、サンプル・ウエル4内で誘導電流の反応(即ち、酸化又は還元の反応)を生じさせ、それにより、イオン電流200を電極20からサンプル・ウエル4を通過し、分離層30を通過し、捕捉材料40を通過し、電解質ベース媒体62を通過し、対極チャンバー72内の対極電解質を通過し、最後に、誘導電流による酸化又は還元反応(サンプル・ウエル4内のサンプル電極20で生じるものとは反対)を対極70で生じさせる。
4.イオン電流200が電場を生じ、それが最初に帯電したサンプル分子を電気誘導的に
駆動して分離層30を通過して、カートリッジ捕捉スライド42上のオリフィス50に位置した捕捉材料40上に集まる。
5.同時に第二のサンプル分子は分離層30を通過しない。これは、無帯電か又は第一サンプル分子の帯電とは反対になる結果によるか、又は、第二の分子が分離層30内に留まるか、遅らされる結果による。
6.カートリッジ捕捉スライド42上に第一サンプル分子の捕捉後に、スライドをカートリッジ・ウエル・フレーム6から取外す。
7.カートリッジ捕捉スライド42を脱イオン水で、又は、適当な溶媒で洗浄して、質量分析法のような分析と干渉する塩及び他の物質を除去する。
8.MALDIマトリックス溶液を捕捉スライド42上の捕捉材料40に加える。そして乾燥させる。
9.捕捉材料40に添加された乾燥MALDIマトリックスを有する捕捉スライドをMALDI質量分析器に挿入される。そして、第一検体の質量をMALDI−MSにより分析される。例えば、各(m/z)ピーク高さ又はピーク面積の平均及び標準偏差はサンプル・ウエル4に加えられるサンプル材料の量又は加えられたサンプル材料の発生源の関数として決定される。(例えば、共通の特徴、医学的徴候又は診断を共有するヒトのグループから採取したサンプル)(ここでmは質量、zは単位電荷を意味する)。
【0031】
例えば、ステップ8及び9で、そのように調製されたMALDI捕捉スライドの分析の間に、適当な溶媒に溶解されたMALDIマトリックスの小さな液滴が検体捕捉領域に加えられる。溶媒は検体を溶解でき、溶媒が蒸発すると共に検体が捕捉膜の上面に形成されるMALDIマトリックス結晶内に組込まれる。溶液の蒸発とMALDIマトリックス結晶の形成のために、通常、1分から60分の時間の後で、サンプル・プレートはMALDI質量分析器に導入できる。MALDIサンプル・プレートを質量分析器に挿入する時にMALDIマトリックス結晶が強いUVレーザー光のパルスで照射され、検体分子の一部のイオン化を生じる。これはMALDI−MS分野の技術者に良く知られている。
【0032】
別の例によると、ステップ5で、ポリアクリルアミドが分離層30として使用される。そのようにポリアクリルアミドが用いられるとき、ポリアクリルアミドに含まれるアクリルアミド又はビス・アクリルアミド(bis acrylamide)が十分に高い濃度、架橋結合、厚みになり一定分子量(又はm/z)より小さい分子のみが分離層を通過できる。選択された分子量がタンパク質の場合約30,000ダルトン、即ち、タンパク質のLMW成分である特別の場合、分離層は、生体組織からの30,00ダルトンより大きな高存在度タンパク質を除去するのに使用される。生体組織には、脳、筋肉、肝臓、肺、膵臓、卵巣、睾丸及び特に血漿と血清が含まれる。例えば、血清の場合、分離層がアルブミン、IgG、IgA、ヘモグロビン、ハプトグロビン(haptoglobin)、トランスフェリン(transferin)を除去する。これらは通常この変更組織内のプロテイン総質量の約95%になる。代わりに、1%アガロースのような非ふるい分け(non−sieving)ゲルをカートリッジ・ゲル・プレート32に組込んで、高分子量のタンパク質の除去無しで、分離を実行しうる。カートリッジ捕捉スライド42の1以上の捕捉材料40上に1以上の検体の捕捉後MALDIマトリックスが捕捉材料40及び前記のようにMALDI質量分析器により、第一及び第二の結合された分子について分析された材料に加えられる。
【0033】
本発明の好ましい実施例はサンプル・ウエル4の配列を有し、各ウエルが、上方開口部8、側壁10、下方開口部14を有し、又、カートリッジ・ウエル・フレーム6内に収納されている。この好ましい実施例は、又、各サンプルウエルごとにひとつの対応するサンプル電極20の配列と分離層30の配列を有している。好ましくは、分離層30の配列はカートリッジ・ゲル・プレート32内の配列として含まれ、そこではサンプル・ウエル4の下方開口部14と位置合わせするように、ゲル・プレートが適当なピッチ(pitch
)で間隔を置いている。分析の後で、1以上の捕捉材料40の配列を含むスライド42は、後日、再検査又は検証を実現できる。
【0034】
サンプル・ウエル4の配列を有するカートリッジ捕捉スライド42は、単一の捕捉スライドとして、又は、2以上の直列に積み重ねられたカートリッジ捕捉スライドの積み重ねとして存在し、検体は2以上の捕捉スライド内に存在する各捕捉材料40を直列に通過する。そのような2以上の捕捉スライドの積み重ねが存在するとき、各スライド内の捕捉材料は実質的に同一であり、又は、代わりに、実質的に異なる。有利なこととして、継続的に直列の捕捉スライドの中で、以下に詳細に示すように、実質的に異なる捕捉材料が種々の検体を選択した捕捉スライドに分けるために使用しうる。
【0035】
例として、捕捉スライド42内の捕捉材料40は単一材料、例えばMillipore
Corp.,Billericia,MA(USA)から入手できるImmobilon−P又はImmbilon−PSQとして得られる多孔性ポリビニリデンジフルオルド(polyvinylidene difluorde)(PVDF)から作られる。特許文献5に詳細に示されているように、熱、超音波又はレーザーによる溶接により、多孔性PVDA捕捉材料を捕捉スライド42に取付けて、捕捉層40を形成する。MALDI−MSによる分析のためにポリアクリルアミド・ゲルから電子ブロッティング(blotting)によりタンパク質を捕捉するためにPVDFを用いることについて詳細な文献が存在する(非特許文献5,6,7,8)。さらに、有利なこととして、MALDI−MSによる分析中に、導電材料の薄い層でそのような膜を被覆することはそのようなPVDFの耐電を防止する(非特許文献9)。
【0036】
図2に示すように捕捉スライド42の連続的な層の数を2以上に増加することにより、サンプル検体の分別を高められる。この実施例では、カートリッジ捕捉スライド42を積み重ねて、検体の分子が各カートリッジ捕捉スライドの捕捉材料40を通過する。捕捉スライド42の連続的捕捉材料内のPP分子の分別が、捕捉スライド42の2以上の連続的層のそれぞれに実質的に異なる化学的又は物理的表面特性の捕捉材料40を用いることによりかなり改善されるので、それぞれがサンプル内のPP(タンパク質とポリペプチド)の構造的に異なる分子に対して実質的に異なるアフィニティを有する。
【0037】
捕捉スライド42上の多数の連続的層でサンプル・タンパク質の分別を実施するため、各捕捉スライドは膜から成る捕捉材料40を有する。それで、装置の運転中に、サンプル検体、例えば、タンパク質又はポリペプチドが、連続的に電気泳動的に駆動されて、連続的に2以上の捕捉膜を通過する。有利なこととして、連続的に用いられる各捕捉膜が異なるクラスの検体に対して実質的に異なるアフィニティを有している。異なるアフィニティを持つそのような膜の例はPVDF又は他の多孔性ポリマー、検体に対する膜のアフィニティを変える低分子量の修正材料で被覆された膜が含まれる。例えば、疎水性の膜に段階別濃度の親水性ポリマーで被覆し、次ぎに、高分子量の膜材料に親水性ポリマーを不可逆的に結合する反応ステップを実行する。例えば、多孔性PVDF膜(例えば、Millipore Corp.,Billericia,MA(USA)から入手できるImmobilon−P又はImmbilon−PSQ)は種々の溶液で被覆しうる。この場合、種々の溶液のそれぞれが中立の親水性ポリマーの種々の濃度を含む。そのような低分子量のポリマーの例には以下が含まれる。
1.ポリエチレングリコール(PEG),例:Fluka Cat.No.94646,分子量35,000
2.オリビニールピロリドン(PVP),例:Sigma Cat.No.PVP40T,分子量40,000
3.ポリビニルアルコール(PVA),例:Sigma Cat.No.P8136,分子量 30,000
【0038】
そのような低分子量ポリマーを高分子量ポリマーの膜に被覆し、不可逆結合をするための手順は従来技術で良く知られている。Nafion.RTMのような高帯電ポリマーをPVDF膜に被覆し、不可逆結合をする例示的方法は米国特許に示されている(非特許文献10)。低分子量ポリマーを高分子量PVDFに不可逆結合するために、この方法は、PVDFの溶融温度より低い温度での焼き付けを用いる。直接的であるが、この方法は、膜に被覆用ポリマーのかなりの部分を非共有結合的に結合させている。このゆるく結合された被覆用材料がその後MALDI−MS分析中の検体のイオン化を抑制する。有利なこととして、グルタルアルデヒド(glutaraldehyde)のような種々の化学的架橋結合用試薬を用いて膜にポリマーを非可逆的に共有結合を行う。例えば、その架橋結合用試薬は、従来技術で知られているように、ヘテロ又はホモの二官能価架橋結合試薬である。
【0039】
被覆及び不可逆結合の実施後、電気泳動により駆動される分別の手順の最適化が行われる。行われた実験では小さいが強く帯電したペプチド及びタンパク質が、PVDFベースの捕捉膜上に最初に捕捉されることを示している。分離時間を段々に延ばす(又は、代わりに、印加電圧を高くする)ことにより、段々に大きなタンパク質がPVDFベースの捕捉膜上に捕捉される。さらに、これらの実験は捕捉されたペプチドの一部が捕捉膜から有機溶媒(又は有機溶媒を含むMALDIマトリックス溶液)により溶出でき、MALDI質量分析法により定量的に検出されることを示した。さらに、血清サンプル内に見いだされるタンパク質の連続的分別部分を膜の目標上で捕捉できる。分別手順は以下の方法で最適化できる。
【0040】
(最適化の方法)
1.標準のタンパク質・サンプルの測定された標準体積を、カートリッジ・ウエルのそれぞれに同じ測定された体積をピペット(pipet)で移すことにより(例えば、2μLのヒトの血清・サンプル又は1以上のタンパク質又はポリペプチドの他の適当な標準混合物として)加える。
2.事前決定された実行時間の間、膜を横切るように電流を通過させることにより、膜の面に垂直な電場を加える。例えば、十分な電圧を加え、5から120分の範囲の実行時間の間、膜面積の平方mm当たり0.1から10mAの電流密度が加えられる。電場が加えられる時間中に、捕捉材料40のそれぞれを通過する電流が監視され、又、プロット(plot)され、マルチ・ウエル(multi−well)捕捉スライド42に配置された捕捉材料の配列内の種々の場所に存在する捕捉材料40内の電場で均一性と再現性を確保する。膜を通過する電流が、捕捉材料内の帯電したサンプル検体の電気的濃度を生じる。3.電気濃縮手順が完了した後で、PPASカートリッジから捕捉スライドを外す。
4.捕捉スライドを洗浄し、塩その他の干渉物質を除去する。
5.MALDIマトリックス溶液を捕捉スライド42上の捕捉物質に添加し、乾燥させる。
6.捕捉スライドをMALDI質量分析器に挿入し、MALDIにより分析する。例えば、各ピーク高さの平均と標準偏差を用いられた血清サンプルの量の関数として決定される。
7.ステップ1からステップ6までを少なくとも2回以上繰返すことにより最適化を行う。各回ごとに、電流密度、実行時間、又は、電流密度と実行時間の両方が変る。一般に電子密度は、膜の平方メートル当たり0.1から10ミリアンペアの電流密度(又は、寄り一般的に、捕捉スライド42の穴50の平方ミリメートル当たり0.1から100ミリアンペア)であり、実行時間は5分から120分の間である。標準サンプルから質量分析器により検出された最大数のタンパク質又はポリペプチドのピーク、又は、どれか1以上のピークで最大強度を与える状態(電流密度と実行時間)が「標準最適化状態(standared optimized condition)」として採用される。
【0041】
最適な方法は生体サンプル例えばSigma Chemical Companyから購入した通常のヒト血清(100mL)又は市販の同等品を用いて実行される。代わりに、そのような生体サンプルは、血漿、尿、脳脊髄液、腹水、唾液等のような他の生体液である。他の適当な生体サンプルには、生体組織からの又は細胞培地から得た溶解した細胞を含む。最適化方法は、手順内の1以上の追加パラメーターを変えながら、連続的に1回以上繰返して良い。追加パラメーターは、サンプル要素(例えば、pHと電導度)と体積、電解質緩衝液の要素、時間、電流密度、捕捉材料、MALDIマトリックス溶液の要素又は緩衝液又はサンプルの体積のようなものである。最適化方法でMALDI−MSにより得られたデータは分析され、そして、質量分析法内で認識されたPP検体のピークの数と高さを最適化するように相互に関連付けられる実験パラメーターと分析結果が比較される。
【0042】
上記の一実施例で、ステップ#2で電場を加えている間の電流−電圧の関係を時間の関数として測定される。ステップ#2を終了する時点を決定するために、電流−電圧の関係から捕捉材料40を通過する抵抗での電荷を一定時間に亘って計算する。5分から45分の5分間隔を含む個別期間に、捕捉膜の配列上に種々の電気泳動による動きの部分を捕捉するために、タイム・コース(time−course)を実施する。得られたデータは時間をベースとしたLMWヒト血清の分別に対する有効性を決定するために分析される。この時間ベースの分析は、選ばれた分子量の範囲の血清ペプチド及びタンパク質の分析手順として使用しうる。最初の部分の目標となるペプチドは約1−2,000ダルトン、続く部分は、2−5,5−10,10−15,15−50、50,100,100−200,及び>200の×1000ダルトンである。各分子量範囲の分析に対して、各分子量範囲で単一サンプルを分析し、スペクトル(複数)を結合して、完全なプロテオーム・プロフィル(proteome profile)を提供しうるように、関連する標準運転手順書(SOP)が選択される。
【0043】
分析と最適化を実施する前に、血清を10マイクロリットルから10mlのアリコート(aliquots)、例えば、450μLのアリコートに分割され、そして、−80℃で保存される。例えば、以下の実験を実施して、PPASを用いたpHベースのLMW血清サンプルの分別を実証しうる。サンプル緩衝液内のペプチド/タンパク質の標準を(及び、通常のヒトの血清にスパイク(spike)された標準を用いて)検出するための装置の感度と再現性を、検体が安定するように選択されたpH値で検査しうる。例えば、便宜的にpH7.0のタンパク質及びペプチドが用いられるが、3から11のpH値も用いられる。(捕捉膜が評価しうるほどのイオン交換特性を有さないので、又、未結合の緩衝用の種がMALDI質量分析器により検出される前に捕捉膜から洗い流されるので、緩衝用の種は臨界的でない。)この目的に用いられる最初のペプチド・タンパク質の標準は、ユビキチン(ubiquitin),グラミシジン(gramicidin),シトクローム(cytochrome)C,インスリン酸化B鎖(insulin oxidized B Chain)及びACTH破片(18−39)である。別の適当な標準タンパク質を、PPASによりカバーすべきタンパク質の各分子量の適用範囲に追加しうる。(ノイズより高い標準偏差の3倍として定義された)ヒトの血清内の各標準を検出する感度を決定する。約20のPPASカートリッジを分析して、システムの再現性を決定する。カートリッジの半分は負の電気濃縮モードで処理する(即ち、負に帯電した検体をサンプル・ウエル4から電気泳動的に駆動して捕捉材料40上に濃縮する)。他の半分は正モードで処理する(即ち、正に帯電した検体をサンプル・ウエル4から電気泳動的に駆動して捕捉材料40に濃縮する)。得られた方法は各サンプルを5以上の成分に分別するのに使用しうる。
【0044】
PVDFのような事前成形された膜を用いる代わりに、捕捉材料40に対して実質的に
類似の機能をする捕捉材料を捕捉スライド42内のオリフィス50に投入する。例えば、捕捉材料は疎水性で多孔性のポリメタクリレートとしうる。このポリメタクリレートとしては、ポリ(ブチルメタクリレート)、ポリ(メチルメタクリレート)、ポリ(エチレンジメタクリレート)、ポリ(ベンジルメタクリレート)、又は、これらのポリマーの混合体、例えば、ポリ(ブチルメタクリレート−コ−エチレン−ジメタクリレート)が挙げられる。代わりに、捕捉材料は親水性多孔質のポリメタクリレートであっても良く、そのようなものとしては、ポリ(2−ヒドロキシエチルメタクリレート)、ポリ(グリシジルメタクリレート、ポリ(ジエチレングリコールジイメタクリレート)、又は、これらの混合体である。
さらに、より有利なこととして、捕捉材料が疎水性が正確にある範囲の疎水性から選択されるように親水性ポリマー及び疎水性ポリマーの混合物から形成されうる。当業の技術者に良く知られている多数の手順に基づいて、キャストポリマーを捕捉スライド42のオリフィス50の側壁56に堆積させ、又、付着させ、取付けても良い。(非特許文献ll,12,13,14,15,16,17,18,19,20,2l,22)
【0045】
捕捉スライド42製造の好ましい実施例で、捕捉スライド内のオリフィス50の側壁56が最初にビニル化され(vinylized)、壁56に多孔性のモノリス(monolith)ポリマーの共有結合が可能になる。ビニル化手順では、オリフィス50を最初にアセトンで洗浄し、次ぎに脱イオン水で洗浄し、0.2モル/リットルの水酸化ナトリウムで30分間活性化し、水洗した後、0.2モル/リットルのHCIで30分間処理し、最後にエタノールで洗浄する。次ぎに、酢酸を用いてpHを5に調節し、95%エタノール中20%が3−(トリメトキシシリル)プロピルメタクリレート((3−trimethoxysilyl)propyl methacrylate)の溶液から成っているメタクリレート(methacrylate)の重合混合体は、1mmの深さのモノリスを通って30分間洗い流される。エタノールで洗浄し、窒素流で乾燥した後で、機能化したスライドが24時間室温で放置される。次ぎに、オリフィスがメタクリレートの重合混合体を用いて注意深く一杯になるように充填される。開示されたように、その混合体はメタクリレート・モノマー及びポロゲン(porogen)溶媒を含む(非特許文献ll,12,13,14,15,16,17,18,19,20,2l,22)。
【0046】
疎水性モノマーを重合混合体に組込むことで、疎水性モノリスを製造できる。同様に、親水性モノマーを選択することで、親水性モノリスを製造できる。さらに、任意の事前決定された比での親水性と疎水性のモノマー混合体を用いて、希望の親水性又は疎水性のモノリスを製造できる。これらいずれの場合でも、(赤外線照射を除去するために)水フィルターに組込まれたキセノン・ランプを使用して重合を開始できる。150ワット以上のキセノン・ランプを用いることで、約10cmの距離で約10分間照射後に重合は完了する。重合の後で、重合混合体内のポロゲンとして機能する溶媒を、例えば、シリンジ・ポンプ(syringe pump)により送られるメタノールの加圧流を用いて洗い流す。代わりに、12時間以上ポロゲンを洗浄液に単純に拡散することにより除去しうる。多孔性のモノリシック(monolithic)ポリマーは小ビーズ(beads)から成るポリマーと比較していくつかの利点がある。例えば、モノリシック・ポリマーは活性表面積を有意に増加しうる。さらに、モノリシック・ポリマーはオリフィス50の壁に直接付着できる。
【0047】
分別と捕捉のステップの後で、カートリッジ捕捉スライド42の各層を分離し、ここで示すように質量分析器で個別に解析しうる。2以上の捕捉層にする追加分別により(各捕捉膜に組込まれたアフィニティにより示される)タンパク質について多くの情報を提供し、又、MSによる検出感度も高めている。(なぜなら、各捕捉材料がPPの全分子を比例的に低減していて、実質的に同じPP分子の大きな部分を各捕捉材料40に組込まれるからである。)
【0048】
図4は、Applied Biosystems,Inc./Sciex Voyager DE MALDI TOF質量分析器のための標準スライド・ホルダー90に直接挿入されるカートリッジ捕捉スライドの好ましい実施例を示す。カートリッジ捕捉スライド42は低導電性材料から作られているので、電気泳動の電流の10%以上が、カートリッジ捕捉スライド42内の捕捉材料40も含む穴50内の電解液を通過する。より一般的には、カートリッジ捕捉スライドの導電性は、電流の75%から99.999%がカートリッジ捕捉スライド42内の捕捉材料40も含む穴50内の電解液を通過する程のものである。装置の運転中にこれを達成するために、通常、カートリッジ捕捉スライド42を作るのに用いる材料の体積抵抗は102から1010Ω−cmの間になる。より一般的には、カートリッジ捕捉スライド42の体積抵抗は104から106Ω−cmになる。しかしながら、この材料の僅かな導電性がMALDI−MS分析によるその後の分析中に捕捉された電解液のイオン化の間、捕捉スライド42の帯電を防止する。
【0049】
有利なこととして、カートリッジ捕捉スライド42も非常に平坦で、又は、代替的に、図4に示されるように、MALDIスライド・ホルダー90に挿入することにより+/−50ミクロンの平坦度になるように設計されている。カートリッジ捕捉スライド42がサンプル・ホルダー90に、機械的ガイド92により、又は、代わりに、磁石のような強磁性材料により、取付けられる。例えば、磁石は小さな希土類磁石例えばネオジウム・鉄・ホウ素(NdFeB)の厚み約1mm、直径約2mmの磁石である。強磁性材料が、捕捉膜上のサンプル検体のMS分析中に、下方要素のフレーム部材(及び取付けられた捕捉部材)をMALDIサンプル・プレートに保持するように機能する。この目的のためにこれらの磁石は(#318ステンレス鋼)に十分な力で保持する。
【0050】
運転中に、帯電した可動検体は加えられた電場内を、分析システムのカートリッジ・ウエル・フレーム6内に配置された多数のサンプル・ウエルの配列内にあるサンプル・ウエル4から移動する。各サンプル・ウエル4は1以上のサンプル検体(例えば、PP's)を有するサンプルから成る電解液を保持するのに役立つ。さらに、各サンプル・ウエルは、流体との電気接触を確立するために電解液に挿入するサンプル電極20を受入れるのに役立つ。電圧が共通対極70に対して電極20に加えられるとき、イオン電流がウエル4内の流体を通過し、ウエル4はサンプルとpH緩衝用電解希釈液からなり、それにより、サンプル・ウエル4内の電場を生じる。その電場により、電気泳動的動きを生じて、サンプル・ウエル4内の1以上の電解液を分離する。有利なことに、捕捉材料を含む穴の断面積がサンプル・ウエル2の断面積より実質的に小さく、穴50の中にある捕捉材料40内の検体を電気泳動的に濃縮する。分析システムの好ましい実施例には、1から400μLのサンプル体積を収容するサンプル・ウエル4が含まれる。各ウエルの内径は上方開口部が約6.7mmで、下方開口部が約1.0mmに狭くなり、捕捉スライド42内に捕捉材料40を含む小直径の穴50に、電気泳動によって検体分子を濃縮できる。ウエルはそのウエルの内側側壁10の下方部分12内に狭くなっている。一般に、そのような下方部分12内の側壁はウエルの全体として垂直な中心軸から20度から30度の傾斜になっている。好ましい実施例では、その傾斜がウエル2の中心軸から24度から26度の間である。一般に、サンプル・ウエル2の直径は5−20mmの間にあり、捕捉領域の直径は10ミクロンから1.5mmの間である。
【0051】
サンプル・ウエル2の下方開口部14を分離しているのは、薄い分離層30である。分離層はふるい分け材料(例えばアクリルアミド)から成っていて、イオンによる導電性と流体同士の接触を維持している。ふるい分け材料は事前成形され、組立てられている。又は、現場成形されうる。現場成形の場合、ポリアクリルアミドの層は、液状アクリルアミド・モノマーと架橋剤をウエルの中に希望の厚みになるように注入することにより行われる。次ぎに、液体を、例えば、過硫酸アンモニウムのような遊離基連鎖開始剤を組込むこ
とにより、又は、リボフラビン(riboflavin)のような光線感作物質を添加すること、又、例えば、UV光又はリボフラビンに対しては400−450nmの光のような光線感作物質が吸収する波長の光で照射することにより、組立前に重合させる。さらに、分離層30が1以上の直列配置で積上げ可能なふるい分け又は分離の層として提供できる。例えば、アガロース・ゲル(agarose gel)を、多孔性のポリアクリル・アミド層、多孔性の透析膜又はその両方を直列配置の組合わせで使用しうる。そのような直列配置の組合わせで多孔性のアガロースは最初の前フィルター(filter)として機能し、サンプル検体、又は、高い存在度のタンパク質のような緩衝物質による過剰負荷から多孔性のポリアクリル・アミド層を保持する。その一方で、ポリアクリル・アミドは第二の前フィルターとして、電気泳動による濃縮の際に、透析膜がタンパク質で詰まるのを防止する。
【0052】
一般的に、捕捉材料40が穴50に含まれ、カートリッジ捕捉スライド42を横切るように通過する。帯電した検体が電気泳動により駆動されて、分離層を通過する。次ぎに、1以上のカートリッジ捕捉スライド(CCS)上のアセンブリーの捕捉材料40に捕捉され、連続的なカートリッジ捕捉スライドのオリフィスが同軸的に配置され、検体は各穴内の多孔性捕捉材料40を連続的に通過する。それにより捕捉された検体が、捕捉スライド42内に保持された捕捉材料40の中で、大きな体積のサンプル・ウエル4から小さな体積に濃縮される。さらに、多数の検体が、連続的カートリッジ捕捉スライド内の捕捉材料により分離され、捕捉される。それにより、実質的に検体が別のスライド部分に分別される。カートリッジ捕捉スライド42のアセンブリーが一連の積み重ねられた層として1以上の連続的捕捉スライド、通常、1−10個、より一般的には1−5個の連続的スライドから成り、可能性としては、1−100個以上の連続的スライドから成っている。捕捉スライド42の連続的な層の積み重ねは、運転中にイオン電流がスライド42の各層を、第一の連続的捕捉スライドから、次ぎに、第二の連続的捕捉スライド等と、最後の連続的捕捉スライドまで連続的に通過されるようにして組立てられる。
【0053】
有利なことに、捕捉スライドの穴50内の捕捉材料40は改質された捕捉材料を含み、その改質は選択した検体に対する捕捉材料40のアフィニティを高める。さらに、そのように改質された捕捉スライドを種々の検体に対して差別的に高いアフィニティを持つように改質しうる。さらに、種々の検体に対して差別的に高いアフィニティを持つそのように改質された捕捉スライドは、検体が第一の捕捉スライドに、次ぎに第二、さらに第三の捕捉スライド等に遭遇するように逐次積み重ねられる。各捕捉スライド42は種々の検体に対して、差別的に高いアフィニティを持つ捕捉材料40を有する。第一番の連続的捕捉スライド42は最初に選択した検体に対して高いアフィニティを有している。二番目の捕捉スライド42は二番目に選択した検体に対して高いアフィニティを有している。さらに、三番目に選択された捕捉スライド42の捕捉材料40は三番目に選択した検体に対して高いアフィニティを有している等、順番に行われる。それにより、第一、第二、第三の検体の連続的捕捉スライドへの分別が便利にかつ迅速的に行われる。
【0054】
選んだ捕捉スライドに対して高いアフィニティを有する検体を事前決定する。例えば、ある検体が、検体−反検体の結合ペア(pair)のメンバー(member)であり、捕捉材料40が反検体の接合により修正された場合、事前決定された捕捉スライド42の捕捉材料40が、事前決定されたスライド内の事前決定された検体の特定捕捉になる。例えば、ある検体が抗体により認識できる抗原性エピトープを有しているので、事前決定された連続的捕捉スライド42内の捕捉材料40へその特定抗体を固定することにより、事前決定されたスライド42内の事前決定された検体の特定捕捉になる。抗体の代わりに、結合ペアの相補性メンバーを用いて、相補性検体を結合するのに用いられる。この場合の相補性メンバーはリガンド・レセプター(ligand−receptor)のペアと呼ばれる。リガンド・レセプターのアフィニティは高アフィニティ又は低アフィニティを有
するように選択できる。種々の検体が、異なる検体に対する異なるアフィニティを有する連続的捕捉スライドにより選択的に捕捉される。それにより、検体の分離層への分別が実現される。
【0055】
各捕捉材料40が、剛体の固体支持から成るカートリッジ捕捉スライド42に取付けられ、それにより、捕捉材料のその後の処理を容易にしている。処理には、洗浄、乾燥、MALDIマトリックスの使用、第二の乾燥ステップ、MALDI質量分析器での質量分析法が含まれる。種々の検体に対する同じ又は異なるアフィニティを持つ多数の捕捉材料が、それゆえ、多数の捕捉スライドの穴50内に挿入でき、直列に積上げられ、お互いの穴の位置合わせするように、検体が種々の捕捉材料を連続的に通過するようにしている。
【0056】
例えば、各スライドは、多孔性のポリマー膜を持つ1以上の小さなオリフィスを有するポリプロピレン・フレーム、捕捉領域を含む1以上のオリフィスのそれぞれに対する、モノリス成形、溶接付け、糊付け、その他の取付け部から成っている。
【0057】
捕捉スライド42の穴50内の捕捉材料40がカートリッジ捕捉スライド42の上面44と下面46と導通していて、それに電流が流れている。捕捉スライド42の下面への電気接触が電解質ベースチャンバー60内に含まれる(アガロース・ゲルのような)イオン導通(電解質)媒体から成る電解質ベース媒体62を通過する。電解質ベース媒体は共通対極を収容する対極チャンバー72内に含まれている対極電解質を通じて共通電極70と電気的接触をする。電圧がサンプル電極20と共通電極70の間に印加されたとき、電流が2電極の間に流れるようにシステムが作られる。電極間に配置された電解質内でイオン物質により電流が流れる。それ故に、サンプル・ウエル内に存在する帯電した電解質が電極20又は対極70に向って電気泳動的に駆動される。サンプル電極20と対極70の間に事前決定された極性の電圧が印加されたとき、電極70に向って駆動された電解質が電路内に存在する捕捉材料40内に濃縮される。対極に対して2以上のサンプル電極20に選択した電圧極性を加えることは、捕捉スライド42内の2以上の対応する捕捉材料に別個に、又、同時に濃縮させる。サンプル・ウエルに加えられた電圧は両方が正に、又は、両方が負の極性になるように選択しうる。それゆえ、正に帯電した又は負に帯電した電解質は単独に又は同時に別々に捕捉材料に濃縮される。代わりに、サンプル電極の極性があるウエル内を正に、他のウエルを負に事前決定される。それゆえ、負に帯電した、又、正に帯電した検体を、単一の捕捉スライド42内の2以上の異なる捕捉材料に同時に捕捉する。分析システム300内で、個別電気回路はサンプル電極からサンプル・ウエルを通じて、分離層を通じて、捕捉スライド42内の穴を通じて、電解質ベースチャンバーを通じて、対極チャンバーに含まれ、共通対極70と接触している対極電解質72を通じて接続される。有利なことに、分析ステップには、解離と分離のステップが含まれ、低存在度の検体分子から高存在度の検体分子の枯渇を生じる。そのようなステップは、質量分析法によりペプチド及びタンパク質の検体の高い感度と再現性に有用である。
【0058】
そのような解離及び分離のステップは、非イオン性洗剤の添加又はサンプル・ウエル4内に存在する他の適当な解離要素を用いることにより行われる。例えば、電圧の印加ステップ前に、洗剤を適当なpH緩衝の電解液に加えられる。代わりに、洗剤をサンプルに、又は、サンプル・ウエル4内に存在する他の試薬を加える。非イオン性洗剤は疎水性ペプチドをアルブミン及びIgGのような大きな分子量で高存在度の分子から効果的に解離する。次ぎに、電圧(及びそれによる電流)が、サンプル及び共通対極の間に加えられたとき、(加えられた電圧の符号及び電解液上の電荷の符号により)正極又は負極の方に、サンプル内の選択された電解液の帯電が駆動される。
【0059】
サンプルの選択されたpH値で、正帯電と負帯電の検体の二成分分離が行われる。例えば、サンプルのpH7.8で処理し、共通対極に対して、正電圧のサンプル電極20に正
電圧を加えることで、正の電流がサンプル電極20から共通対極70へ流れる。正の電流は対応するサンプル・ウエル4内の検体を正に帯電させ、ウエル4から移動させ、対応するウエル4の直下の穴50に存在する捕捉材料40内に(捕捉スライド42上に)捕捉させる。(即ち、装置は正モード(mode)で運転と言われる)。逆に、負の電圧をサンプル電極に加えたとき、負の電流が、サンプル電極20から共通対極70に流れる。負の電流はサンプル内の検体を負帯電させ、サンプルから移動し、対応するウエル4直下の穴50に存在する捕捉材料40内に(捕捉スライド42上に)捕捉される。(即ち、装置は正モード(mode)で運転と言われる)。負又は正のモードで、各サンプル電極から流れる電流は通常10マイクロアンペアから10ミリアンペアである。より一般的には、電流は0.2から2.0ミリアンペアである。
【0060】
通常、少なくとも2個のサンプル・ウエルが1サンプルの分別に用いられる。サンプル・ウエルのひとつで、共通対極に対して、サンプル電極が正極性で、他は負極性である。即ち、正モードと負モードの分離が同時に行われている。正帯電と負帯電の検体の分離は、システム・オペレーターにより事前決定されたサンプルpHで同時に生じる。それゆえ、事前決定されたpHで正帯電と負帯電のものへのサンプル検体の分別を同時に行なえる。さらに、単一サンプルを種々の等電点の2以上の部分に分割することは、2種類のpH値を有する2以上のサンプル・ウエル内のサンプル緩衝液を用いることにより可能になる。それにより、本発明の別の実施例で、等電点に基づく検体の分別、濃縮、捕捉が以下に開示しているように、実現しうる。
【0061】
等電点に基づく検体の電荷ベースの分別に加えて、選択肢として、ふるい分け材料がサンプルと捕捉材料の間の分離層で使用される。サンプル・ウエル4から対応する捕捉材料40に向って電気泳動的に駆動された検体がふるい分けによる分離層30を最初に通過しなければならない。それゆえ、ゲル電気泳動法の分野の技術者に良く知られているように、一定のm/z値を持つ低分子量の検体に対して、同じm/z値を持つ高分子量の検体の移動を遅らせることにより、追加分別を行うのに役立つ。(洗剤(例えば、ドデシル硫酸ナトリウム)が分子量にほぼ比例してタンパク質に結合するように存在して、それゆえ、全タンパク質に同様のm/z値を与えるとき、ポリアクリル・アミド・ゲルを通過するタンパク質の移動時間はタンパク質の分子量の対数にほぼ比例していることが良く知られている。)それにより、種々の分子サイズのタンパク質に対してm/z値が類似しているとき、ふるい分け材料がLMWプロテオームを分離するのに使用しうる。ふるい分けによるそのような分離で、事前決定された範囲の分子量でタンパク質の最適分離を行うため、事前決定された電圧又は電流の使用時間が選ばれる。低分子量(即ち高い移動性)の検体がふるい分け層を迅速に通過する。それゆえ、低い移動性の検体より前に、捕捉材料40上に捕捉される。それゆえ、本発明により機械的分離が行われる。そのような高い移動性の検体は単一の捕捉材料に濃縮され、又は、一連の2以上の積み重ね可能な捕捉スライドを通過することにより、その後の分離は組合わせで行われ、2以上のスライドのそれぞれが少なくとも1個の穴が隣接する捕捉スライドの他の穴と同軸に配置されている。さらに、順番に、各穴が種々の事前決定された捕捉材料を有している。それにより、アフィニティに基づいて検体の分離を行い、又、それにより、最少数の捕捉スライドを用いて、最大の分別を行う。例えば、種々の捕捉材料はその捕捉材料の疎水性の違いから成っている。別の例によると、サンプル・ウエルに最も近い位置にある上方の捕捉材料は、最小の疎水性であり、サンプル・ウエルから直列配置で最も遠い捕捉材料が最も疎水性である。それにより、疎水性の勾配が、その疎水性に基づき検体の分離と分析を行なえるように、作られる。アフィニティにより行われた連続的スライドによるそのような分別を用いて、次ぎに、分子量とアフィニティの両方による分離を組合わせて、又、組合わせて同時に行われる。多様な2以上のサンプルが個別に、及び、カートリッジ2内で同時に分離される。複数のサンプルがそのような多様なやり方により同時に分離される。
【0062】
有利なこととして、分離と捕捉の層が比較的薄いことにより、分別と捕捉のステップは、分離と捕捉のステップを比較的迅速に実施できる。この目的のために、通常、分離と捕捉の層は厚みが通常20ミクロンから20mmの間になる。より一般的には、分離と捕捉の層の厚みは200ミクロンから5mmの間である。薄い分離と捕捉の層に対して、例えば、500ミクロンから2.0mmの厚みに対して、分別ステップは10秒から100分かかる。通例、分離と捕捉は1時間未満に生じる。より一般的には、分離と捕捉は1分から約10分の間で行われる。捕捉ステップの後で、PPAS装置が分解され(図3に示すように)、カートリッジ捕捉スライドのそれぞれが短時間洗浄され、MALDI又はエレクトロスプレー(electrospray)質量分析法による検出に干渉する塩又は他の化学種を除去する。例えば、捕捉スライドは脱イオン水で単純に洗浄される。洗浄ステップの後で、MALDIマトリックス溶液が各捕捉スライドの捕捉領域のそれぞれに添加される。そして、マトリックスを乾燥できる。乾燥ステップの後で、捕捉された検体の質量分析法のために、質量分析器(例えばMALDI−TOF MS)に直接スライドを挿入する。代わりに、質量分析器で捕捉材料40から直接捕捉された検体を検出するために、検体を最初捕捉材料から溶出させ、MULDI−MS、エレクトロスプレー、酵素例えばトリプシン(trypsin)によるタンパク質分解消化を含む種々の手段により検出される。そして、得られたペプチド破片の分析、即ち、タンパク質の識別と分析の分野の技術者に良く知られている「ペプチド・マップ(peptide map)又は他の分析手段を構成することによる。
【0063】
カートリッジ捕捉スライドは、カーボン・ドープ(carbon dope)されたポリプロピレンから射出成形され、電荷の拡散無しに直接MALDI解析を可能にしている(非特許文献23、24、25、26、27)。捕捉材料40は、適当な手段により、例えば、接着剤を用いることにより、又は、捕捉材料、スライド又はその両方に溶剤添加又は加熱をする溶接により捕捉スライド材料に取付けたポリビニリジンジフルオリド(PVDF)のような疎水性膜から形成しうる。代わりに、捕捉スライド内のオリフィスに捕捉材料を注入しうる。例えば、捕捉材料は多孔性のポリ(ブチルメタクリレート−コ−エチレンヂメタクリレート)のポリマー・モノリスとしうる。そのようなモノリスはSvecらにより開示された手順(非特許文献28−34)に基づく重合により成形しうる。密に結合した捕捉材料を有する頑丈な捕捉スライドのために、スライド・オリフィスの内壁面は最初にビニール化(vinylized)して、そのモノリスを壁に共有結合できる(非特許文献35)。オリフィスをアセトンと水で洗浄して、0.2mol/Lの水酸化ナトリウムで30分間活性化し、脱イオン水で短時間洗浄し、その後、0.2mol/LのHClにより30分間、最後にエタノールで短時間洗浄する。95%エタノール中の20%が3−(トリメトキシシリル)プロピルメタクリレート溶液、pH5(例えば、0.1から1.0%の酢酸を含むエタノール)で、厚さ約1mmのモノリスを30分間洗い流す。エタノールによる洗浄と窒素流内での乾燥の後で、機能化されたスライドを室温で約24時間放置する。モノマーの適切な選択によりモノリスの親水性を選択できる。次ぎに、オリフィスに注意深く過充填になるようにメタクリレート重合混合体で充填し、蒸発防止のためのカバーをし、重合せしめた。標準としては、キセノン・ランプに水フィルターを取付けて、光重合を開始するのに用いている。重合は約10cmの距離から10分間の照射の後で完了する。そして、そのモノリスをシリンジ・ポンプ(syringe pump)又は比較的遅い連続した流れを供給する他の適当な手段が送るメタノールを用いて、そのモノリスを約12時間洗浄する。多孔性のモノリシック(monolithic)ポリマーは、ビーズ(beads)から成るポリマーと比較して、有効表面積を有意に高められる。代わりに、以下に好ましい実施例として開示したように、そのような多孔性のモノリシック重合体とクロマトグラフィ(chromatography)粒子の混合体が捕捉材料40として用いられる。
【0064】
PPAS装置内で調製されたカートリッジ2内の捕捉スライド42上の捕捉材料40上
に捕捉されたサンプル検体のMALDI−MS分析のために、適当な溶媒に溶解したMALDIマトリックスの小さな液滴をその検体捕捉領域に添加する。その溶媒は検体を溶解でき、その溶媒の蒸発と共に、その検体は、捕捉膜の上面上に形成するMALDIマトリックスの結晶内に組込まれる。溶媒の液体を蒸発させ、MALDIマトリックスの結晶を形成するための時間を与えた後、そのサンプル・プレートはMALDI質量分析器に導入できるようになる。例として、図4はカートリッジ捕捉スライドと標準のApplied Biosystems,Inc/Sciex Voyager DE MALDI TOF 質量分析器用スライド・ホルダーへの直接挿入を示している。MALDIサンプル・プレートの質量分析器への挿入時にMALDIマトリックスの結晶を強いUVレーザー光のパルスで照射し、その結果、検体分子の破片のイオン化を生じる。これはMALDI質量分析法分野の技術者には良く知られている。
【0065】
(選択的洗浄の構成と方法による干渉性化学種の除去)
捕捉スライド42の穴50内に保持された捕捉材料40上への検体捕捉後に、捕捉スライドはカートリッジ2の分解により取外される。次ぎに、塩、及び、無機及び有機のpH干渉性の種を捕捉スライド及びそのスライドの穴内に保持される捕捉材料から洗い出される。しかしながら、洗浄処理中に捕捉材料上に対象検体を保持するために、洗浄剤の組成を注意深く選択する。タンパク質及びペプチドのようなPP検体を保持するために、疎水性捕捉材料のそのような選択的洗浄には、通常、実質的な水性溶媒を用いている。洗浄は、拡散、圧力に駆動された流れ、電気泳動(即ち帯電した干渉物質)によるか、又は、代わりに、電気浸透により、又は、そのような2以上の方法の組合わせにより行われる。
【0066】
例えば、洗浄溶液の圧力に駆動された流れを、捕捉スライド42を横断して差圧が加わるように設計された図4aに示すような装置により行われる。そのような差圧は、例えば、スライドの片側にある真空マニフォールド200により真空を生じて、反対側の流体槽から真空マニフォールド200に向ってスライド内の捕捉材料40を通過する流れを生じさせることによる。代わりに、正圧は正圧マニフォールド202により、スライドの流体槽を有する側に加えられ、それにより、実質的に同じ圧力で駆動された洗浄液を捕捉材料を横断して流す。どちらの場合も、捕捉スライド42が、密封用のゴム又は軟質ポリマー・ガスケット(gasket)、又は、Oリングのようなスライド密封手段206と関連して機能する圧力保持用サポート204により支持されている。有利なこととして、負又は正の圧力を用いて、捕捉スライド42内の2以上の穴50内に多様な2以上の捕捉材料40を横断して実質的に同時に流体を流れさせる。洗浄手順に用いられる流体は、例えば、脱イオン水(DI)、又は、代わりに、DI中の0.1%のトリフルオル酢酸(TFA)のような「MALDIフレンドリー(friendly)」イオン含有溶液にできて、捕捉材料40から干渉塩を排除する。一方で、捕捉材料に結合した希望のPP検体を保持することができる。そのような「MALDIフレンドリー」イオンは特徴として、陽子の減少又は増加により中性化したとき、かなりの蒸気圧を有し、質量分析法の真空チャンバー(chamber)内を迅速に「排出」できる。「MALDIフレンドリー」の材料は特定pH値でイオン化するもので、酢酸、アンモニア、蟻酸、プロピオン(propionic)酸、ピペリジン(piperizine)、ピリジン(pyridine)等であり、MALDI−質量分析法用のサンプル作成分野の技術者に良く知られている。
【0067】
代わりに、図4bに示すように、電気泳動装置300はスライド42内の捕捉材料(40)を横断した電場を加えるために用いられる。電気泳動装置は、電源302、陽極と陰極として使うための電極ペア306を有している流体槽とスライド・ホルダー304、及び、陽極を陰極から分離するように機能する隔壁308から成っているので捕捉スライド42内の穴50を電流が通過しなければならない。
【0068】
無機塩及び無機と有機のpH緩衝物質が無い捕捉スライド上の捕捉材料を最も有効な電
気泳動により洗浄する場合、以下の原則が単独で又は組合わせで用いられる。
【0069】
A.疎水性イオン交換
第一のイオン交換ステップが、MALDIフレンドリーのイオンに対するMALDI非フレンドリーの干渉物質を交換するのに使用されるが、pHが調節されるとき、MALDIフレンドリーのイオンが上記のようにかなりの蒸気圧を有する。捕捉スライド上の捕捉材料が疎水性イオンに対するアフィニティを有するとき、例えば、「逆相」クロマトグラフィの原則が捕捉検体に用いられるとき、疎水性干渉物質が捕捉材料にも結合する。従って、第一ステップで、そのような疎水性干渉物質を同様の電荷の疎水性イオンと交換する(即ち、正電荷又は負電荷の物質)。例えば、ヒスチジン(histidine)緩衝液(等電点7.8)が用いる場合、(ツビッターイオンヒスチジン(zwitter ionic histidine)は極端に“MALDI−非フレンドリー”である)、ヒスチジンを、ヒスチジンが負帯電になるpHで負帯電のトリフルオロアセテート(trifluoracetate)イオンと、ヒスチジンが負帯電するpHで、即ち、7.8を超えるpHで、交換しうる。通例として、pH9.0で0.1Mのトリフルオロ酢酸がこの目的のために用いられる。1ミリアンペアの電流が捕捉スライド42の1mmの穴を5分間通過することがこの目的に特に有効であることが見いだされている。負帯電又は正帯電の他の干渉物質は、緩衝物質HEPES,TES,HEPPS,CAPS,CHES,ACES,ADA,BES,MES,MOPS,PIPESのように、これらの負帯電のイオン(陰イオン)をトリフルオロ酢酸の陰イオンと同様に交換することにより、個別の等電点より高くに選択されたpHで個別に除去できる。代わりに、例として、ヒスチジンを正帯電ピリジン・イオン(陽イオン)と、電気泳動法による洗浄を行うことにより交換できる。この洗浄は比較的低いpHで、即ち、pH<7.8で、ヒスチジンとピリジンの両方が例えば、pH4.0で正帯電している。
【0070】
B.高電場電気泳動
この方法により、電解液の導電性が、例えば、蒸留水内での実質的希釈により低減する。そして、大きな電場が捕捉スライド42内の捕捉材料40を横断して置かれる。この方法で、ゆるく結合した疎水性緩衝イオンがモノリスから解離する。それらが再結合する前に、高い電場により掃き出される。この方法で、pHは3−11の範囲になり、より一般的には、最適性能のために、又、導電性を比較的低く保つために、4−10の範囲にする。高電場を加えて、過大な電流を生じないために低い導電度を必要とする。上記に開示された装置を用いて、ウエル当たり1ミリアンペアを超す電流は捕捉スライド42の穴50内で過大なジュール(Joule)加熱を生じる。そのようなジュール加熱は従来技術で良く知られていて、電流の平方即ちI2Rに比例する。Iは電流を、Rは抵抗を示す。
【0071】
C.電気浸透(EEO)の流れ
EEOにより発生した流れはモノリスを横断した電場に比例していて、ゼータ(zeta)電位(即ち、モノリスの固体相からモノリス内の液体への剪断面を横断する電位の低下)の関数でもある。帯電した疎水性物質は捕捉材料40から洗い流されるので、ゼータはゼロになる。それゆえ、洗浄ステップが完了すると共に流れも減少する。高い電場が高いEEOのために最適である。それゆえ、上記の高い電場の電気泳動に最適なのと実質的に同じ条件がEEOの流れにとっても最適である。
【0072】
D.クーロン反発力
これは最も単純な機構である。捕捉スライド42が、結合された緩衝イオンが帯電されるpHでの蒸留水又は脱イオン水のような薄い電解質内に置かれる。イオンのクーロン反発力がそれらをミクロリス(microlith)から押出す。好ましくは、洗浄溶液のイオン力が低く、溶解イオンが1ミリモル未満である。さらに、捕捉材料を洗い流すべき干渉性のイオンを含む種とペアを組む疎水性の種を避けるべきである。例えば、正帯電し
たヒスチジンが干渉物質(即ち、ヒスチジンが7.8より低いpHで結合している)であれば、酢酸塩、蟻酸塩、TFA、HEPES、PiPES、又は、他の疎水性陰イオンを避けるべきである。
【0073】
(例:電気泳動による洗浄手順)
捕捉材料40から、(4−10の範囲の中性のpHの中で)、干渉する疎水性陰イオン例えばTFA,HEPES,PiPESを除去するために、以下のステップを実行する。1.pH9(+/−0.5pH単位)で0.1%のトリフルオロ酢酸の洗浄溶液を用いて、5分間、捕捉スライド内の穴を通して開口面積平方ミリメートル当たり1ミリアンペアを供給する。これでイオン交換を実現する。
2.ステップ#1を実施した後で、洗浄溶液を蒸留水又は脱イオン水で約1/100に希釈する。電圧は、開口面積平方mm当たり約0.125ミリアンペアになるように高める。(又は、電源で利用できる最大値のどちらか低い方)。この洗浄ステップの実行で、さらに5分間を使い、又、原則BからDに基づいた洗浄を行う。
【0074】
干渉する疎水性陰イオンが捕捉材料40に結合している干渉物質として存在するとき、同上のステップを実施するが、MALDIフレンドリーの陽イオン物質を用いる代わりに、例えば、トリフルオロアセテートの代わりに、ピリジニウム(pyridinium)を用いる。ピリジニウム・トリフルオロアセテートは4.0から5.0のpHで特に有効である。
【0075】
どちらの場合でも、上記2種類の電気式洗浄ステップの組合わせが、どちらの電気式洗浄ステップ単独よりも優れている。
【0076】
(カートリッジ捕捉スライドからの捕捉された検体の溶離及びMALDI質量分析法による分析のためのMALDIマトリックスの添加)
検体が濃縮され、カートリッジ捕捉スライド42の穴50により保持された捕捉材料40上に捕捉されると、潜在的MALDI干渉物質が除去されて、捕捉された検体を分析する。例えば、MALDI−TOF質量分析法による分析が行われる。標準のMALDI−MSマトリックスの組成と方法を用いて捕捉されたタンパク質を溶解し、MALDI質量分析器での分析のためにMALDIマトリックス結晶内にそれらを堆積する。そのような標準の手順は質量分析法の分野で良く知られていて、文献に良く記録されている(非特許文献36)。
【0077】
例示的標準のMALDIマトリックス及び手順は、アセトニトリル内の20mg/mlシナピン酸(cynapinic acid)の1部と水中の0.1%(v/v)トリフルオロ酢酸1部の混合体(即ち、シナピン酸の最終濃度は10mg/ml)から成るマトリックス溶液を用いるべきである。カートリッジ捕捉スライド内に保持されて、マトリックス溶液の混合体0.25マイクロリットルをサンプル・サイド(sample side)の各捕捉材料40に注意深く添加する。それで、(周囲のスライド材料に広がるよりはむしろ)大部分の溶液が材料上に留まる。空気中での乾燥後、マトリックス溶液の第二の等体積添加が同じように行われる。そして、カートリッジ捕捉スライド42の乾燥が、空気乾燥により、又は、乾燥器内に用いた真空により行われる。アセトニトリルと水の溶媒を除去した後で、MALDI−MSによる測定と分析がMALDI質量分析法で行われる。便宜的に、捕捉スライド42がそのスライドを保持するために機械的ガイド92を有するスライド・ホルダー90に挿入される。Applied Biosystems Voyater MALDI質量分析器で使用するのに適したスライド・ホルダーの例が図4に示されている。そのような方法による分析で、捕捉スライド42の上面41に露出した捕捉材料にMALDIマトリックス溶液を用いることにより最適の結果が得られる。その後、スライド42の上面41も質量分析器のサンプル・ホルダーに位置してMALDI
質量分析法のために、捕捉スライドと同じ面を、MALDI質量分析器のレーザー・ビーム(laser beam)で調べる。それにより、検体のイオンを放出し、質量分析器と共にあるイオン電流検知器により検出される。
【0078】
好ましい代替分析手順で、検体溶出用溶媒がサンプル・スライド42の下面43に(即ち、サンプル・ウエル4の反対側の面に)最初に加えられる。この手順により、検体分子が捕捉材料から溶出し、MALDIマトリックスの結晶を形成する前に、(及び、内部に検体分子が組込まれる前に、)捕捉スライドの上面41に濃縮される。この手順により、溶離処理の感度が高くなり、検体の変形が少なくなり、検体を捕捉している捕捉材料内の深さへの分析の依存性が低減する。MALDIマトリックスは、溶出用溶媒と共にスライド42の下面43に、代わりに、検体の溶出処理が完了した後で、上面41に加えられる。
【0079】
例示的方法は以下の通りである。
1.1サンプル又は複数のサンプルをカートリッジ2のサンプル・ウエルに置く。
2.サンプル電極20により、事前決定された電圧又は事前決定された電流を各サンプル・ウエルに加える。
3.事前決定された電荷の検体分子が分離層30を通じて他の検体から電気泳動的に分離され、多孔性の捕捉材料40を有する部位で捕捉スライド42の上面を通じて濃縮され、捕捉される。
4.捕捉スライド42にアクセス(access)できるように、カートリッジを分解する。
5.干渉物質を除去する洗浄手順の後で、検体は多孔性捕捉材料から捕捉スライドの分析側に、好ましい態様では、上面41に溶出される。
6.MALDIマトリックスがスライドの同じ分析側に、好ましい態様では、上面41に加えられる。
7.MALDIマトリックスを空気中で、他の乾燥気体で、又は真空で乾燥し、その捕捉スライドをMALDI質量検出器に分析のために挿入する。それで、分析面をレーザー・ビーム・プローブ及びMALDI質量分析器のイオン検出器に露出する。本発明の好ましい実施例では、捕捉スライドの上面41をそのように曝露する。
8.捕捉スライド上に捕捉された検体を質量(より正確には、そのm/z値)及びその相対的存在度を求めて分析する。
【0080】
好ましい態様では、ステップ#5及び#6を以下のように結合する。
【0081】
カートリッジ捕捉スライドが(1−10cm/secの空気速度のファン(fan)のような)乾燥装置上で反転する。アセトニトリルと脱イオン水の溶液(典型的に9:1 v/v)を各捕捉スライド42の下面43で曝露された捕捉材料40に加える。数分間、多孔性捕捉材料を通じて溶出用溶媒の吸引後、このステップの後で、第二の溶出ステップではMALDIマトリックス例えばメタノール内の濃縮したシナピン酸(例えば、9.0−90mM、pH7.0−8.0の脱イオン水)が含まれる。このステップの後で第三ステップがあり、その中で、pHを酸性になるように調節する。典型的に、アセトニトリル9部と脱イオン水内の0.1%トリフルオロ酢酸1部に調節される。MALDIマトリックス(例えば、シナピン酸)を乾燥器内で真空乾燥を行なえるようにする。十分な乾燥後、MALDI−MSの測定と分析を、例えば、Bruker Autoflex model又はApplied Biosystems Voyager modelのようなMALI質量分析器内で行う。このMALDIマトリックス添加の方法はスライドの上側への生体分子の溶出、さらに、そのようなMALDI−MS測定内の検体分子の検出限界を低減することに対応している。
【0082】
(好ましいカートリッジ捕捉スライドの構成、捕捉材料及びその製造方法)
カートリッジ捕捉スライド42は穴50、オリフィス内に保持される捕捉材料40を有している。好ましい実施例では捕捉スライドが96個の穴を有し、8×12の長方形配列に配置されている(即ち、12列8行)。その中で、各穴の中心は最も近い4個の隣接穴のそれぞれから9.00mm離れて配置される(即ち、9.00ミリのピッチを有する)。好ましい実施例では、穴は直径約1mm、深さ約1mmである。製造では、全体として平坦な捕捉スライドで、厚みの変動は非常に小さく(典型的には、約+/−50ミクロン未満)、オリフィスを有するが、そのオリフィスは、ポリマー装置製造分野の技術者には好く知られている機械加工(例えば、レーザー又は機械的穴開けによる)成形又は鋳造により形成される。好ましい実施例では、捕捉スライドの材料はバルク(bulk)及び表面の導電性を最適化するように選択される。前述のように、カートリッジ捕捉スライド42の導電性は、サンプル電極20により捕捉スライドに加えられた電流の75%から99.999%が、(バルク・スライド材料を通過する代わりに)穴50内の電解質を通過する。装置の運転中にこの状態を実現するためには、通常、カートリッジ捕捉スライド42を作るのに用いられる材料の体積抵抗は、102から1010オーム・センチメートルの間である。より一般的にはカートリッジ捕捉スライド42を作るのに用いられる材料の体積抵抗は、104から108オーム・センチメートルの間である。この捕捉スライド材料の導電度が、MALDI−MS分析によるその後の分析で捕捉された検体のイオン化の間に捕捉スライド42の帯電を防止する。代わりに、カートリッジ捕捉スライドのバルク導電度が多少導電性になり、相当量の導電度を与えるために、表面の抵抗コーティング(coating)を行うことにより、希望の状態を実現するように表面導電度を調節する。
【0083】
捕捉スライド42が穴50と共に形成されると、溶剤又は加熱による溶接、穴への材料の鋳造又は他の取付手段による膜の取付けのようないくつかの手段によって穴内に捕捉材料40を堆積し、取付ける。好ましい態様では、鋳造は現場での2種類の光重合反応による接合を行うことにより与えられる。両方の反応は光重合を開始するために紫外線照射を用いることにより真空テーブル(table)上の一体成形で行われる。その方法で、適当な鋳造型が機械加工による熱可塑性、熱硬化性、又は、金属、又は、他の方法による成形型により形成される。成形は捕捉スライド42の穴50内に捕捉材料40を保持しなければならない。そして、有利なこととして、当業界の技術者に好く知られているように、ラジカル重合反応を終わらせるように機能する酸素を排除する。例えば、成形型は薄いシート(sheet)材料例えばポリエチレン又は“Saran Wrap”から成り、そのような成形手順の分野の熟練者に好く知られているようにスライドを真空テーブル上に保持しながら、そのシートを真空によりスライドの穴に対して所定の場所に保持する。
【0084】
第一の光重合ステップが二重結合又はビニール(vinyl)基を穴50の壁に光グラフトする。光グラフト処理では、光グラフト用溶液を穴内に置き、穴50を囲む捕捉スライド材料に共有結合する共重合分子を生じるのに必要な時間UV光を照射する。UV照射をDymax Corporation,Torrington,CT,USAからの5000−ECユニットにHランプを用いて行うとき、必要な照射時間は一般的に1−5分の長さになる。
【0085】
適当な光グラフト反応の混合体は、メチルメタクリレート(MMA)48.5質量%、エチレングリコール・ジメタクリレート(EDMA)48.5質量%、ベンゾフェノン3質量%から成っている。その反応混合物を秤量し、酸素を追い出すためにアルゴン、ヘリウム又は窒素のような気体を混合し、散布する。捕捉スライドを混合体内に浸漬し、次ぎに、過剰物を除去することにより、各ウエルにピペッティング(pipetting)することにより、又は、他の方法で反応溶液をウエルの内部に供給することにより、その散布された反応混合物をウエル内に置く。次ぎに、捕捉スライドをモールド内に置き、そのモールドを真空テーブル(Pharmacia Fine Chemicals,Mod
el GSD−4)の上に置き、その真空を動作させる。次ぎに、UV透過性プラスチック・シートをモールドの充填された穴上に置かれる。そのようなプラスチック・シートはSaran Wrapのような市販のプラスチック・ラップ(plastic wrap)から、ポリジメチルシロキサンのシート、又は、適当なUV透過性気体遮断材料のようなシート・ゴムから供給できる。真空テーブル内に保持された捕捉スライドに対して所定の位置に、プラスチック・シートを、手操作で保持する。その後、光グラフト反応を水銀アーク・ランプ付のUV照射装置に配置して、一定時間照射する。照射時間はシステム因子によるが、一般的に1分未満で、照射された表面積の平方センチメートル当たり100 mWの照射フラックス(flux)である。例えば、十分なUV照射は捕捉スライド面から約20cmの距離で動作する400W水銀ランプにより与えられる。UV照射のあとで、プラスチック・カバーが取外され、光グラフトが行われた捕捉スライドがモールドから除去され、アセトン(acetone)で洗浄される。光グラフトが行われたスライドがアセトン内に置かれ、一定時間そこに保持され、痕跡量のモノマーと、捕捉スライド42の面に未接合のままである共重合体の破片を除去する。
【0086】
第二の光グラフトステップは、固体の現場形成から成るが、多孔性のモノリス材料が、以前のステップで取付けられた光グラフトされた共重合体により捕捉スライド42に部分的に取付けられている。今回の方法で、モノリス形成がウエル内に反応混合体を置くことにより行われ、モールドを真空密封して、モノマーとポロゲン(porogens)の混合体からモノリスを発生するため、一定時間UV照射することにより低酸素及び無酸素の環境を形成する。そのようなポロゲンは当業界で知られていて、反応剤と混合して、その後に、固体相を形成するとき、多孔性の固体形成を促進する。代わりに、UV照射は、Dランプ付で、Dymax Corporation Torrington CT,USAからの5000−ECユニットにより行われる。
【0087】
好ましい方法で、モノリス「反応混合体A(“reaction mixture A”)」が用いられる。「反応混合体A」には、1−デカノール(decanol)5グラム、n−ブチル・メタクリレート(butyl methacrylate)2.4グラム、EDMA 1.6グラム、シクロヘキサノール(cyclohexanol)1グラムが開始剤と共に含まれる。ジメチル・アセトフェノン(DMAP)は開始剤としてモノマーの全質量を1%として比例値が用いられる。それゆえ、この場合、0.4グラムのDMAPが用いられる。反応混合体を秤量し、DMAPが完全に溶解するまで混合し、酸素を追い出すために、アルゴン、ヘリウム又は窒素のような気体を散布する。約10mLの反応混合体をモールドに最初に充填し、次ぎに、そのスライドをモールド内に置くことにより、散布された反応混合体がウエル内に置かれる。代わりに、反応混合体を各ウエル内にピペットで注入する。又は、他の方法で、ウエルの内部に送られる。次ぎに、捕捉スライドをモールド内に置く。そのモールドを真空テーブル(入手先:Pharmacia Fine Chemicals,Model GSD−4)上に置き、真空を機能させる。そして、捕捉スライドの上に十分なプラスチック・シートを供給してモールドをカバーする。そのようなプラスチック・シートはSaran wrapのような市販のプラスチック・ラップから、ポリジメチルシロキサン・シートのようなシート・ゴムから、又は、適当なカバリング(covering)材料から得られる。そして、モールドと含まれる捕捉スライドを真空テーブルに固定するために、十分な真空を生じるまで、真空テーブルに保持することにより、プラスチック・シートを所定位置に保持する。次ぎに、捕捉スライドの一部をキセノン又はメタルハライド・アーク・ランプ付のUV照射装置内に置き、ある時間照射する。照射時間はシステム因子によるが一般的に照射表面積が平方センチメートル当たり150mVの 照射光束で、4分未満である。十分なUV照射が例えば捕捉スライド面(入手先:SLM Instruments,Champain,IL,USA)から約20cmの距離で動作する400W キセノン・ランプにより与えられる。UV照射の後で、プラスチック・カバーが外される。モノリスで充填された捕捉スライドがモールドから注意深く外されて、メタノールで洗浄される。モノリスで充填されたスライドを約10ボリューム(volumes)のメタノール内に1−24時間置かれて、メタノールが高級アルコールを置換し、かつ、残留未反応のモノマーを除去する。新規のメタノールを用いて各連続バッチ(batch)を洗浄し、適切な清浄度にする。
【0088】
当業界の技術者に一般的に知られているように、(その方法と反応混合体Aの両方の)多くの適切な変更が存在する。非特許文献11−22が例を示している。実験を通じて、2以上の異なる捕捉材料の異種結合により形成された捕捉材料42は、単独使用時の純粋なモノリシック(monolithic)捕捉材料より優れていることを我々は見いだしている。一般に、異種の組合わせは、固体、事前形成された、粒状のクロマトグラフィ・メディア(chromatography media)から成り、固体又は多孔質のコア(core)粒子から成っている。粒子はいわゆる「逆相」粒状のクロマトグラフィ・メディア(即ち、疎水性粒子、又は、代わりに、陽イオン又は陰イオンの「イオン交換メディア」)である。そのような材料の例には、高純度シリカ、タンパク質アフィニティを修正したシリカ、重合クロマトグラフィー用の多孔質又は固体のビーズ(beads)又は他の固体粒状の材料が含まれ、クロマトグラフィーの分野の技術者又はそのような材料の技術者によく知られている。代わりに、混合体又は2以上のそのようなメディアの混合体又は交互層を用いる。各場合に、粒状のクロマトグラフィー・メディアは適当なマトリックスにより所定場所に保持される。その適当なマトリックスは、捕捉スライド42の表面に十分接着するのに、又、クロマトグラフィ・メディアを所定場所に確実に捕捉するのに適当な材料である。例えば、便宜上、上記の、又、非特許文献11−22に一般的に示されている同じ組成がそのようなマトリックスに用いられる。一般的に、好まれた「逆相」の異種の組合わせが、生体サンプルからのタンパク質とペプチドの捕捉のために、いわゆる「逆相」の粒状クロマトグラフィ・メディアとモノリシック固体相の間で作られている。モノリシック材料は上記に引用したもの(及び非特許文献11−22に一般的に)示されている。
【0089】
特に、生体のペプチド及びタンパク質の捕捉のために好ましい実施例は、「反応混合体A」の33%をAlltech SPEバルク溶剤C8(入手先:Alltech Associates,Deerfield IL,Cat No 211504)で置換される。純粋な反応混合体Aを用いるために上記の一般手順が混合体と共に用いられる。さらに、好ましくは、粒状材料が、モノマー、架橋剤及び開始剤を含む反応混合体の粘度を高めるのに役立つ。粘度の増加は鋳造の際に穴から重合反応混合体の漏洩を防止するのに役立つ。それにより、事前決定された粒状物の組成を、一般に1%の粒子から99%の粒状材料の範囲で、選択することにより希望の値に調節しうる。より一般的には、粒子材料は10%から90%の範囲とする。さらに、より一般的には、粒子材料は25%から50%の範囲とする。タンパク質とポリペプチドを捕捉するための好ましい捕捉材料を製造するのに用いられる粒状のクロマトグラフィ粒子の別の例を表1に示す。
【0090】
表1 「ミクロリス」捕捉用化学物質
捕捉機構 例
順相 シリカ、アルミナ
逆相 C2−C18、重合体樹脂、モノリス
イオン交換 SCX,SAX,WAX,WCX
固定化された金属のアフィニティ Ni,Fe,Ga
抗体の捕捉 Protain G,Protein A,Step
tavidin,カスタム抗体
小さな分子のアフィニティ Blue Sepharose/Dextran,C
ustom ligand libraries
【0091】
これらのクロマトグラフィ材料はバルク量でのいくつかの製造により供給され、0.2−500ミクロンの範囲の粒子寸法を有している。単位体積当たり結合能力が大きいことにより多孔質粒子が好まれるけれども、多孔質又は非多孔質の粒子が用いられる。別の例によると、製造企業には、Agilent,Alltech,Applied Byosystems,Phenomonex,Supelco,Watersが含まれる。本発明の好ましい実施例に含まれるのはC8逆相樹脂、特に、Alltec(部品番号#206250)で、既に記したように捕捉材料としてメタクリレート樹脂と共に結合されている。これらの粒子樹脂の組合わせが、生体高分子の捕捉で高い効用を示している。その生体高分子には、特にタンパク質とペプチド、及び、より一般的には、炭水化物、多糖類、オリゴヌクレオチド(oligonucleotides)が含まれ、質量分析法を用いることにより、その後の脱離/イオン化に対応する。
【0092】
非重合の樹脂と事前重合を行った粒子をそのように結合することは、その後に、ここでは1単位として重合したとき、「ミクロリス」結合と呼ぶ。そのようなミクロリスは、事前成形され、通例、MALDI適合樹脂により薄い捕捉スライドの構成に保持された市販のクロマトグラフィ・メディアから成っている。そのようなミクロリスの個別構成を埋込みのために選ばれたクロマトグラフィ・メディアの構成に基づいて事前決定される。そのような構成には若干の例の名前を出すと、順相、逆相、イオン交換、固定化された金属のアフィニティ、小さな分子のリガンド(ligand)のアフィニティが含まれ、抗体捕捉のアフィニティとレクチン(lectin)捕捉のアフィニティを含めたクロマトグラフィ・メディアが含まれるが、それに限定されない。他の例は生体分子のクロマトグラフィック分離とそのような目的のための市販メディア選択分野の技術者によく知られている。
【0093】
多孔のモノリシック材料と粒状媒体の混合体のひとつの予想外の特性は、その組合わせが固体相の捕捉材料の多孔性を高めることである。それゆえ、ここに記された方法に基づいて作られたモノリスを通過する圧力で駆動された流れが、入手できる文献及びここで述べた文の両方に示されている「モノリシック捕捉材料」を通過するより大きくなる。多孔性モノリシック樹脂と事前重合した粒子状クロマトグラフィ・メディアの組合わせ(即ち、混合体)は、その組合わせは得られた捕捉材料の引張り力を高める。それゆえ、(100%)純粋な多孔性モノリシック捕捉材料(例えば、100%反応混合体Aから成形)が、真空乾燥時に、直径1mmの穴への鋳造に割れを生じて、捕捉スライド面に接着しなくなる。それと対照的に、Alltech SPE Bulk Sorbent C8の33%の組込みで、そのような割れと接着不能を防止する。そのような異種組成を一般に「ミクロリス(microliths)」と呼ばれ、上記の方法により作られ、優れた機械的強度、安定性を有することが認められ、かつ、捕捉スライド面で優れた接着性を有する。さらに、我々は、そのような「ミクロリス」が電気泳動装置内でタンパク質、ペプチド及び他の検体分子を捕捉する能力を有することを見いだしている。さらに、そのようなミクロリスが十分に平坦に(例えば、+/−50ミクロン)鋳造して、マトリックス支援レーザー脱離/イオン化質量分析法(MALDI−MS)を用いたその後の分析のために、優れた面を提供する。
【0094】
(血清から高存在度のタンパク質の解離と除去)
臨床上重要な血液、血漿又は血清の低存在度ペプチド分析に伴う主要な問題は高存在度のタンパク質が低存在度のタンパク質及びペプチドの外観を隠すことである。しかしながら、血液、血漿又は血清から高存在度のタンパク質のアフィニティ除去はかなりの数の低存在度・疎水性ペプチドも除去されると想定されている。分離と分析の方法例の好ましい実施例では、血清サンプルは最初MALDIと適合性のある洗剤で処理して解離を促進し、その後に分子量による分別で、高存在度で高分子量のタンパク質を除去する。
【0095】
全てのサンプルをステンレス鋼のサンプル・プレート又は導電面を有する平坦な重合材料から作られた使い捨て捕捉材料に加えた。例えば、2マイクロリットル(μl)のサンプル体積を溶液の液滴として、直接添加するか、又は、モノリシック捕捉材料上に電気泳動で捕捉する。乾燥後、MALDI質量分析法に直接置かれる。別の例では、0.5マイクロリットルのMALDIマトリックス溶液をピペットでサンプル・スポット(sample spot)上に添加し、乾燥させる。タンパク質とペプチドは一般的にMALDIマトリックスとして用いられたアルファシアノ−4−ヒドロキシナミック・アシッド(CHCA)(alphacyano−4−hydroxycinamic acid)と共に分析される。それは一般に1,000から15,000ダルトン(Da)の低分子量のペプチド及びポリペプチドに対して、MALDI質量分析法の結果で、最良の信号対ノイズ(noise)の比を与えるからである。CHCAマトリックス溶液の組成は以下の通りである:CHCAは、50%アセトニトリルと水性0.1%のトリフロロ酢酸の混合体の中に飽和している。MALDIマトリックス溶液の全ての材料はSigma Chemical Co.(St.Louis,MO,USA)から入手しうる。全てのMALDI−MS分析はABI Voyager DE MALDI−TOF及び社内設計のQGEN_PR2法により行われる。典型的な分光計の設定は:加速電圧20kV、グリッド電圧(grid voltage)94.1%、ガイド・ワイヤ(guide wire)電圧0.050%、遅延時間110ns、レーザー設定3000、平均走査64、1.1E−6torr、低質量ゲート511、陰イオンをoff。
【0096】
デモンストレーション(demonstration)のために、2種のポリペプチド標準、例えば、ACTHの破片(18−39)及びインスリン酸化B鎖をウシ血清アルブミン(BSA)と共に混合し、ステンレス鋼のMALDIターゲット・プレート上に直接ピペット添加する。図5は、〜127pmolのBSAが存在する中であるけれども、1ピコモル(pmol)に希釈され、MALDI質量分析法用プレート上にスポットを再現するために加えられ、乾燥されて、又、分子の質量と強度を別個に分析された2種のそのようなポリペプチド標準を示している。図5は1pmolのACTHの破片と1pmolのインスリンを明確に識別しうることを明確に示している。同様に、図6に示すように、ペプチドの破片の量が10倍未満即ち0.1pmolの2標準であり、同じBSAの濃度と量であるとき、図5−7に示すようにMALDI質量分析法による分析中にイオン化を実質的に抑制する。
【0097】
図7に示すのは、図6に示された結果に対して用いられた同じサンプルのMALDI−TOF帯域である。しかしながら、図6に示した結果について、サンプルを最初に捕捉スライド上の電気泳動で濃縮と捕捉を行う。その手順では、単一層のカートリッジ捕捉スライドを用いる。その手順で、2μLのサンプルを、250mMの水性L−ヒスチジン・バッファ(L−histidine buffer)と組合わせて、単一モノリス捕捉スライドを用いることにより処理する。捕捉は約1ミリアンペアの電流を十分な時間流して、伝達された全電荷が約1クーロン(coulomb)になるようにすることにより行われる。その結果は、サンプル混合体からの干渉としてシステムが効果的にBSAを除去することを示している。これらの結果は、装置と手順を組合わせて用いたとき、アルブミンのような大型タンパク質(即ち、30,000ダルトン超)により生じた低分子量(即ち、30,000ダルトン未満)のタンパク質とポリペプチドの検出から実質的信号干渉を効果的に除去し、それにより、低分子量の分子から得た質量分析法の信号を劇的に改善する。
【0098】
(LMWヒトの血清分析)
単一のカートリッジ捕捉スライドを用いることにより本発明の実施例を用いて実験が行われた。本発明の手順に基づいてMALDI MSにより低分子量のタンパク質/ペプチドのプロファイリング(profiling)のためにヒトの血清の調製の実行可能性を
決定するためにそのような研究が行われた。例えば洗剤で処理した血清サンプルはEppendorfのミクロチューブ(microtube)(500μLの体積)内の100μLのヒト血清(Sigma Chemical Co.から入手)に10μg/μLのオクチル−b−D−グリコピラノシド(octyl−b−D−glucopyranoside)(OG)を添加することにより作られる。サンプルは、洗剤で処理した血清の10μLのアリコート(aliquot)、250mMのヒスチジン・バッファ(histidine buffer)の内の100μL、1μLのTexas Red labeled−Leu Enkephalen(250mMのヒスチジン・バッファの内のトレーサー(tracer)として)、0.5μLのグリセロール(glycerol)から作られる。得られたサンプル混合体を約1000gで1分間遠心力を加え、混合液滴を得る。
【0099】
サンプル成分の分離と結合を実行するために、調製されたサンプルの10μLのアリコートをサンプル・ウエルに加えられる。例えば、プラチナから作られた陰極がサンプル・ウエル内に直接配置される。反対の電極、プラチナの陽極が対極チャンバーに置かれた対極電解液に接触するように配置される。
【0100】
プラチナの陽極と陰極の電極は、電位計(Princeton Applied Research ,model 273)に接続され、電極間に約1mAの電流が加えられる。電圧をゼロに設定し、電極への電線を切離す前に約20分間、分離を進めた。
【0101】
次ぎに、試作カートリッジを分解し、ゲルと捕捉層を蛍光でチェックする。分析システムは十分に機能し、そのときに、捕捉材料40に結合するために選択されたタンパク質とペプチドから実質的に全ての蛍光発光が観察され、捕捉部位に結合していることが観察される。捕捉スライドを約5分間脱イオン水に浸漬する。蛍光による捕捉部位の目視検査の後で、スライドを完全に空気で乾燥させる。次ぎに、MALDIマトリックスの0.5μLのアリコートを捕捉スポットの上側に添加する。その捕捉スポットをVoyager DE MALDI MSで直接分析を行う。図8はMALDIマトリックスとしてCHCAを用いることによりサンプルから得られた質量分析法を示している。図はヒトの血清からの低分子量ポリペプチドの検出で、良好な信号対ノイズ比を示している。
【0102】
上記の例で示したのと同様のパラメーターを用いるとき、血清をPPASカートリッジ内の2個所のウエルに添加する。タンパク質、又は、他からの解離を促進するために洗剤を用いて、1以上のサンプルを処理する。そうすることで、洗剤で処理したサンプルを250μLのL−ヒスチジンと結合させ、pH6.8に調節し、サンプルと接触したサンプル電極の極性により1.0mAの電流を加えた。他の洗剤で処理したサンプルを250μLのL−ヒスチジンと結合し、pH7.0に調節し、同様に、サンプル電極によるバイアス(biased)で−1.0mAの電流を供給する。図に示すように、2個の反対極性のサンプル・ウエルから観察されたスペクトルが完全に異なる相補性のタンパク質とペプチドのピークを示している。これらのデータは同じサンプルの2成分pH分別を明確に実証している。
【0103】
(別の例と方法)
(材料)
全ての材料を市販業者から入手可能で、含まれるのは:アセトニトリル、トリフルオロ酢酸(TFA)、n−オクトグルコシド、CHCA、L−ヒスチジン、ポリアクリルイミドである。血清の調製はSigma Co.からの0.5mLポリプロピレン管内で行われる。C−18コーテッド超常磁性ビーズはBruker Daltonicsから購入しうる。
【0104】
(血清サンプル)
悪性疾患が知られていない志願被験者から、又、前立腺がん(グリーソン得点6−7)が確認された同意患者からの血液サンプルを8.5mLのガラス・バキュテナー(Vacutainer)管で供給される。1時間までの室温で凝固させ、4℃、5分間、1000rpmの遠心分離にかけた。血清は等分され、−80℃で冷凍保存された。患者及び対照の血清がVanderbilt University Medical Centerが承認した臨床手順に従って集められる。
【0105】
(クロマトグラフィによる分離)
選択された場合で、血清を逆相磁気ビーズを用いることにより、又は、ここで述べたPPAS装置により分別する。磁気ビーズ・クロマトグラフィでは、血清を、超常磁性で、多孔質シリカ・ベースの粒子(<1μmの直径、80%酸化鉄)、C18による表面誘導体化により培養する。C18/K磁気粒子の懸濁液(500,000粒子/μg;50μg/μLのDD水)を均一分散を得るための渦巻き運動により2分間完全に混合する。次ぎに、50μLビーズ溶液を50μLの血清に加え、5回前後のピペット操作によりゆっくりと混合する。そして、磁石を用いて、ビーズを管の側面に引き寄せ、上澄みをピペットにより除去して、廃棄する。次ぎにビーズを水中0.1%TFAの200μLで完全に洗浄する。最後に、10回前後のピペット操作により、20%次ぎに70%のアセトニトリルの体積5μLを用いて、粒子からペプチドを段階的に溶出する。そして、3μLの溶出液を他の管に移し、6μLのMALDIマトリックス溶液と混合する。そして、1μLをMS分析のために置く。
【0106】
PPAS Device(TM)を用いることによるサンプル検体のモノリシック分別/濃縮:
タンパク質とペプチドの検体を上記の一般手順を用いることにより分析する。到着時点で、血清アリコートを直ちに−80℃で保管する。例えば、血清サンプルを、16mMの重炭酸アンモニウム250μLと250mMのL−ヒスチジンを、Eppendorf microtube(500μLの体積)内の1μg/μLのオクチル−b−D−グルコピラノシド(OG)、0.5μLグリセロール、10μLのヒト血清に加えることにより調製しうる。得られたサンプル混合体を約1000gで1分間遠心力を加え、混合液滴を得る。サンプルの半分をpH7.0に他の半分をpH6.8に調節する。160mMの重炭酸アンモニウム及び250mMのL−ヒスチジン緩衝液がカートリッジ・リザーバー・バッファ(cartridge reservoir buffer)(モノリスの下)に用いられる。全サンプルを5回の反復処理で分析しうる。
【0107】
捕捉スライド42内に捕捉材料40の鋳造のために、カーボン・ドープド・ポリプロピレン(carbon doped polypropylen)(〜50,000オーム/cm)の複数の貫通穴を含むスライドが射出成形される。そして、そのスライドは2枚の軟質シリコン・ゴムのガスケット(gasket)と2枚の石英プレートの間にサンドイッチ(sandwich)になっている。前記の機能化溶液が各貫通穴にピペットにより置かれる。そして、水フィルターを組込んだキセノン・アーク・ランプを用いて照射される。次ぎに、そのサンドイッチから基盤を除去し、ブチル・メタクリレート及び2−ヒドロキシエチル・メタクリレートを含むモノリス溶液を各貫通穴に加える。前記のように、サンドイッチを再構成し、15分間照射する。鋳造手順の後で、スライドを106mMの重炭酸アンモニウム及び250mMのL−ヒスチジンの中に30分間浸漬する。最後に、そのモノリス・スライドを脱イオン水により完全に洗浄する。プラチナで作られた陰極をサンプル・ウエルに直接置く。プラチナの陽極を緩衝液槽に接触するように置く。プラチナの電極を特別設計の多重化電位計に接続し、約1mAの電流をウエルに加える。電圧をゼロに設定し、電極への電線を切断する前に、処理を20分間続行した。サンプルの半分は+1mAで、残りの半分は−1mAで、約20分間処理する。各分析の過程中に各ウエルの電流を測定し、プロットする。電気濃縮手順が完了した後で、PPASカートリッジ2を分解し、カートリッジ捕捉スライド42を洗浄してpH緩衝液及び塩のような干渉物質を除去する。最後に、CHCA MALDIマトリックスを用いて、そのスライドをMALDI−MSにより直接分析する。
【0108】
PPAS装置手順及び磁気ビーズの両方について、30 fmol(ペプチド当たり)と500 fmol(タンパク質当たり)の市販の校正標準(Bruker Daltonics)もCHCAマトリックスと混合し、ターゲット・プレート上に別個に加える。ターゲット・プレートは3×2のパターンに配置されている6個の隣接血清サンプルの中央に位置している。a)単一装置の単一ウエル、b)同じ装置の異なるウエル、c)異なる装置の同じウエル、d)異なる装置の異なるウエルの中の変動性を評価するために再現性を決定する。要因分析を用いて、ウエルの位置、又は、ウエル間の相互作用(又は他の変数)の影響を決定する。
【0109】
(質量分析法)
ペプチドのプロフィール(profiles)を、Applied Biosystems Voyager DE及び4700 model MALDI質量分析器により、典型的手順を用いて分析する:加速電圧20kV、グリッド電圧94.1%、ガイド・ワイア電圧0.050%、遅延時間110ns、レーザー設定3000、平均走査64、1.1E−6torr、低質量ゲート511、陰イオン・オフ。周波数帯は直線モードで取得する。
【0110】
MS測定のソフトウエアにより取得したスペクトルは、市販のEfectaソフトウエア(Efecta Technologies,Corp.,Steamboad Springs,CO.)このソフトウエアは取得中の自動化、平滑化、ベースライン修正、ピーク指定を行う。全てのデータ操作はTempstら(非特許文献36)が発表した技術に基づいて行われる。手動操作の外部校正の後で、ピーク(即ち、m/z)のリスト(lists)を、その後の統計分析に必要なテキスト・ファイル・フォーマット(text file format)のファイルに保存される(以下参照)。
【0111】
ピークのリストは一連のデータ変換のためのデータベースに導入される。最初のパターン(pattern)分析のため単純二進法を最初に作るために、ピーク強度を低減して、特定サンプル内で観察されたペプチドの得られたビン(bins)のいずれかの中での存在又は不存在を示す。
【0112】
次ぎに、ペプチドの質量(例えば、1500ppm)と比例して拡大するウインドウ(window)内のビンニング(binning)により特定セット(set)内の全てのサンプルに亘ってピークの位置合わせをする。ビンニングは、全てのサンプルからの全てのm/zの値をひとつの長いリストに合わせることにより行われ、値を増加することにより分類される。最初の質量を“real(実)”と表示し、隣接する質量分類と比較する。ユーザーが定義したウインドウ内の隣接質量が“duplicate(複製)”と呼ばれる。プロセス(process)は、まだ表示されていない次ぎに大きなm/z値について、分類リスト内の全質量に“real(実)”又は“duplicate(複製)”と標識を付けるまで繰返される。次ぎに、“duplicate(複製)”の質量を廃棄する。現在の適用で、実験により、許容範囲は2Da又は1500ppm(0.15%)になる。“real(実)”質量が恣意的で、ビンの指名にのみ用いられるので、ここで留意すべきは各ビンの質量内の最初のm/z値の割付けである。m/z値をビンに配置されると、結果と共に、表計算ソフトが自動的に出力される。最初の列はビンニング・プロセス(binning process)を生き延びた“real(実)”質量全部のリストを示す。残りの列はサンプルを、又、対応する“real(実)”の質量と共に、ビンに配置されたピークを有するかどうかを示している。
【0113】
(統計的データ分析)
研究セットの全サンプルに亘って、m/zピークのビンニングの後で、Efecktaソフトウエアを使用してプロテオミック(proteomic)データを評価する。集団を代表するために、ソフトウエア内で仮想「実験」が作られる。全てのサンプル内に見いだされたユビキチン(ubiquitin)と少なくとも他のひとつのペプチド・ピークによりデータが正規化される。実験のパラメータ・セクションで、サンプルに癌性か正常かの標識を付ける。翻訳セクションで、分析モードを“比率の対数”と用いられた全測定値に設定する。サンプル名は不連続パラメーターとして表示される。実験が作られると、一方向ANOVAノンパラメトリック(nonparametric)検定(Mann−Whitney U test)とp<0.05での無多重検定補正をフィルター(filter)として用いる。この検定は多数のサンプルを有する2種類のグループに亘って有意に変化しない集団はフィルターを通さないことを意味する。そのフィルターは前立腺がんと対照のグループ間で重要な変化を示した集団を後方に残す。その変化を2種の技術を用いることにより確認する:クラスタリング(clustering)とクラス予測、
第一の技術については、Effectaのクラスタリング・ツール(clustering tool)を用いて、結果を決定樹として表示する。x軸について、類似したサンプルを決定樹上のお互いの近くに置く。サンプルの類似性について、Pearsonの相関性により評価する。類似しないサンプルはお互いから離して置かれる。y軸についても、類似性の検定にPearsonの相関性を用いて、同じように集団のグループ化を行う。クラスタリングの方法では半分のサンプルについてデータがない集団を廃棄した。
【0114】
第二の確認のため、ノンパラメトリック検定からフィルターを通過したペプチド集団を、k−ニアレスト・ネイバー(k−nearest neighbor)と呼ばれるクラス予測アルゴリズム(algorithm)によっても分析する。クラス予測の正確性を学ぶために、“leave−one−out”(非特許文献6、19)として知られる相互検証法を実行する。それはクラス予測アルゴリズムの訓練セットとしてN−1のサンプルを取る。そして、N番目のサンプルをテスト・セットとして使用し、処理をN回繰返して、全サンプルを一度はテスト・セットとして用いるようにする。
【0115】
MALDI MS分析の場合、中央のカートリッジ・アセンブリーを適当なMALDI質量分析法用サンプル・プレートに取付けて、質量分析器に導入する。そして、適当な溶媒に溶解したMALDIマトリックスの小液滴を捕捉膜の検体捕捉領域に加える。溶媒は捕捉膜上の捕捉部位に存在する検体を溶解させる。溶解が蒸発すると共に、検体はMALDIマトリックスの結晶に組込まれて、捕捉膜の上面上に形成される。溶媒液の蒸発と、MALDIマトリックスの結晶形成のための時間を与えた後で、サンプル・プレートをMALDI質量分析器への導入準備ができる。MALDIサンプル・プレートの質量分析器への挿入時に、MALDIマトリックスの結晶を強力なUVレーザー光のパルスで照射し、検体分子の破片的イオン化を生じる。この一部分からのイオンを検出器への飛行時間に基づいて測定し、質量対電荷の比及び強度に基づいてプロットする。
【実施例1】
【0116】
血清に存在するタンパク質の分析
図3及び4はPPASの25ウエル型を用いた結果を示していて、捕捉膜が1枚で、その後の分析はMALDI質量分析法による。試作版のPPASを用いて、血清サンプルの配列からの分離を比較的高速(60分以内)で同時に行う。その後の分離層の厚みの低減は約5mmから約1.0mm以下になり、分離層の両側に加えられる電圧を約1.0から10ボルトが、約10から100ボルトに上昇して、10分以内での分離、濃縮、捕捉を可能にしている。使い捨てMALDIプレート上に直接選択した部分の電気泳動による濃縮が、MALDI−MSの感度を高め、プロファイリングの差の表現を早めるという追加
の利点を与えている。臨床的に重要で、血液、血漿又は血清の中の低存在度のペプチドを分析することに伴う主要な問題は、高存在度のタンパク質が低存在度のペプチドの外観を隠すことである。しかしながら、血液、血漿又は血清のサンプルからの高存在度タンパク質のアフィニティ除去はかなりの数の低存在度で疎水性のペプチドも除去すると想定されていた。これらの研究で血清サンプルは解離とその後のPPASを用いた分離と濃縮を促進し、MALDI−MSによる検出のために、MALDI適合洗剤で処理された。そのようなMALDI適合洗剤の例は、Trinton X−100、octyl glucoside、NP−40等のような中性の物質である。そのような中性洗剤はPP捕捉層内の電気泳動的濃縮をしない。
【0117】
PP混合体のMS分析からの結果を精製したPP標準(例えば、ユビキチン、シトクロームC、インスリン及び1%TFAのみを含むサンプル)と比較する。その標準サンプルは0.1%TFAに直接希釈しうる(800フェムトモル/μL又は10フェムトモル/μLに)。MALDI−MSによる分析の前に、溶媒を蒸発した後で存在する干渉物質が無いかほとんど無いようにする。代わりに、発色性又は蛍光性の標識を付けたPPが標準として組み込める。例えば、蛍光性の分子の場合、タンパク質変更の分野での技術者に良く知られている試薬と方法を用いて、Fluorescein(F)、Texas Red(TR)、Rhodamine(Rh)、Marina Blue(MB)と標識を付けられる。それゆえ、0.2μLのTR−ユビキチン、MBウシ血清アルブミン(MB−BSA)、それぞれが1−2μg/μLで、25%(w/v)グリセロールと共に250mM水性L−ヒスチジン緩衝液を含む2μLのサンプルに組込まれる。
【0118】
図3及び4に示された結果は、ヒトの血清から高分子量・高存在度のタンパク質を除去するのに用いられたポリアクリル・アミド層により得られた。図3及び4に示されたスペクトルにはアルブミンが観察されなかった(68,000のm/zで)。これらの結果は、MALDIによる干渉抑圧により混合体から血清のアルブミンを効果的に除去していることを示している。しかしながら、電気泳動の実施時間が1時間以上に延長されたときに、アルブミン信号の始まりが観察された。付随的に、他の捕捉されたタンパク質の強度低下が観察された。多分、良く知られているように、高存在度アルブミンの存在の中で低存在度タンパク質のイオン化が抑制されたことによる。PPAS内の高分子量タンパク質を分析するために、ポリアクリル・アミド層を非ふるい分け(non−sieving)アガロース層により(及び、例えば、アフィニティ・クロマトグラフィによる交互処理により除去された高存在度タンパク質により)置換しうる。
【0119】
本発明のPPASが、固体相捕捉膜上にタンパク質とポリペプチドを捕捉し、塩及び他の干渉分子を洗い流すことができる。そして、MALDIマトリックス溶液の膜への添加時に、タンパク質が放出され、膜面に沈殿するMALDIマトリックスの結晶に組込まれる。MALDIマトリックス添加後に、膜を乾燥し、付着したタンパク質の質量と相対的存在度の定量化のために、MALDI−MS計測に直接挿入する。
【0120】
PPASは、選択された分離pHで正電荷又は負電荷の分子の(限定的にのみの)分別に1枚の捕捉膜を用いるだけでよい。1枚の捕捉膜によるPPASを高存在度タンパク質の除去に提供する(組込まれたふるい分け層によるか、又は、予備ステップで実行)。他の分別を実行する必要がない。選択肢として、さらに分別を強化するために、2以上の捕捉膜を直列に使用しうる。MALDI−MSは高存在度分子によりサンプルのイオン化抑制を受けるので、そのような分別の増加は実行した分別にほぼ比例して感度を高める。
【0121】
本発明の基本例はアルファ・プロトタイプ・システム(alpha prototype system)と共に示されている。プロトタイプ・システムは25個の捕捉ウエルの5×5配列を有し、1個のカートリッジ内で電気泳動による分離と捕捉を同時に行う。
図10に示す質量分析法の結果の場合、サンプルのpHは7.8であり、各ウエル内の電流は示された時間に対して、1mAに設定されていた。タンパク質の捕捉後、膜は脱イオン水により洗浄され、次ぎに、MALDIマトリックス溶液の添加により放出された。そのマトリックス溶液は、アルファシアノ−4−ヒドロキシナミック・アシッド(CHCA)で飽和された0.1%トリフルオロ酢酸溶液の1体積とアセトニトリルの1体積から成っている。次ぎに、そのマトリックスは乾燥され、分析のためにMALDI質量分析器内に置かれた。タイム・コースは、20分と40分の間に最初の正帯電タンパク質が到着する必要があること及び追加のタンパク質が40分と60分の間に到着する必要があることを示している。示されていないのは、60分以降に捕捉されたタンパク質に実質的変化が観察されないことを示すデータである。全てのMALDI−MS分析が、ABI/Perceptive Biosystems Voyager DE(MALDI−TOF)を用いた社内設計のQGEN_PR2法を用いることにより行われた。CHCAマトリックス溶液と共に用いるために、典型的な分析器の設定は:加速電圧20kV、グリッド電圧94.1%、ガイド・ワイア電圧0.050%、遅延時間110ns、レーザー設定3000、平均走査64、1.1E−6torr、低質量ゲート511、陰イオン・オフ。
【0122】
図11に示したMALDI質量分析法の結果の場合、電極の極性を逆にすることを除いて、手順と分析は図10に示したものと同様である。それゆえ、(極性反転の)2条件に基づいて観察されるタンパク質が、サンプルの事前決定されたpH(即ち、この場合7.8)で両スペクトルで観察されたタンパク質の負帯電が逆になるという事実に基づいて明らかに異なる。正帯電したタンパク質による結果と同様に(図10)、負帯電のタンパク質を捕捉するためのタイム・コースは、最初の負帯電のタンパク質が到着するのに必要なのは40分から80分の間であること、及び、追加のタンパク質の到着は80分から120分の間であることを示している。さらに、120分以降に観察された捕捉されているタンパク質に実質的変化が無いことを示すデータは示されていない。(図10に示された実験で用いられる電流は図11に示す実験で用いられた電流の2倍であることに留意されたい。逆に、図10に示された電気泳動の時間は図11で示した値の半分である。即ち、(期間の2倍に対する)電気泳動中に用いられる電荷移動のクーロン数は、図10に示す時間の半分の値と同じである。(それゆえ、示された2実験に移動した電荷)。
【0123】
(MALDI質量分析法のための対象発明の別の利用方法)
MALDIマトリックスは、以前に発表した方法を用いて調製し、その後に以下の一般的のひとつを用いることによりカートリッジ捕捉スライド42に加えられる:1)手操作のピペットによる添加、2)市販の液体取扱い用ワークステーション(workstation)を用いた添加、3)スプレー・コーティング、4)マトリックス溶液内へのカートリッジ捕捉スライドの浸漬、濃縮されたマトリックス溶液はMALDI分析に容認できるマトリックス対検体の比を実現するように添加される。
【0124】
特に有用なひとつの適用は、モノリス表面上にシナピン酸溶液(50:50のアセトニトリル/0.1%トリフルオロ酢酸で20mg/mL)を堆積させることから成っている。この溶液の体積0.25μLをカートリッジ捕捉スライドの上面にマイクロピペットを用いて添加する。この溶液を約5分の経過に亘って室内状態で乾燥する。その時点で、追加のマトリックス0.25μLを添加する。そのスライドを室内状態で又は真空乾燥器の中で乾燥させる。
【0125】
通例、MALDIマトリックスの堆積と乾燥の後で、捕捉スライドを、計器メーカーの仕様書に基づいて、質量分析器に導入する。スライドは、カートリッジ捕捉スライドをMALDI質量分析器のx−yサンプル・ステージに適合させる特別設計のスレッド(sled)にはめ込むように設計されている。そのスレッドは以下の条件に適合するように設
計されている:A)カートリッジ捕捉スライドを質量分析器内のイオン抽出軸に垂直に保持しなければならない。B)スレッドとカートリッジ捕捉スライドの境界は、カートリッジ捕捉スライドの表面電荷を消失させる通路を与えるようにしなければならない。C)カートリッジ捕捉スライドの表面の高さは各計器標準のサンプル・キャリア(sample carrier)のそれと一致しなければならない。D)そのスレッドに対する各モノリスの位置は常に同じでなければならない。これらの各要件は質量分析法の分野の技術者に知られている。
【0126】
捕捉スライド42上に捕捉された検体の質量分析は、市販されていて、そのような分析分野の技術者にはよく知られているツール(tool)のセットを用いることにより標準方式で処理される。例えば、ベースライン(baseline)減算、正規化、ピーク検出、スペクトルの位置合わせは、ProTS−Data(Efecta Technologies,Inc.;Steamboat Spring,CO;Version 1.1.1.0)として市販されているソフトウエアを用いて行われる。そのデータ解析を要約すると以下の通りである:
1.バックグラウンドの推定と減算:バックグラウンド信号は頑丈で、ローカル(local)で、統計的な推定機能により算定される。バックグラウンドは実質的に“ノイズ”であり、生物学的関連情報を含まず、スペクトルごとに異なっているので、各スペクトルからバックグラウンド値を減算することにより比較可能性を高めるのに、振幅情報が必要である。
2.正規化:イオン化されたサンプルの量はスペクトルごとに、レーザー出力の変化、イオン化可能なサンプルの量の変化、MALDIプレート上のレーザー位置の変化により変動しうる。ピーク振幅に関する定量的情報の信頼度を高めるために、スペクトルを全イオン電流に正規化する。
3.ピーク・ピッキング(peak picking):スペクトル内のピークを特定し、かつ、その信号/ノイズの比の信頼できる推定値を割付けるために、ノイズ算定機能が計算され、使用される。組織サンプルからの典型的なMALDIスペクトルに対して、我々は典型的に信号/ノイズ比のカットオフ(cutoff)3を用いて100から200の間のピークを検出する。
4.スペクトルの位置合わせ:ひとつのスペクトルの絶対質量スケール(scale)がかなり変動しうる。そして、共通ピークの選択は、共通のm/zスケールにスペクトルを登録するのに使用できる。
【0127】
(クラシファイア(classifier)の発生と検証)
標準の質量分析法の分析内で、質量ピークのリスト(重心の値及び正規化した強度を含む)が作成され、個別データ・ファイルに送られる。種々のソフトウエア・ツール/セットが、これらのデータにより質量分析からのバイオマーカー検出を容易にしている。同時にそのソフトウエアは、通常の変動を有する種々の集団について統計的有意性を評価するための有用なツールを提供している。機能のランキングは、グループを差別するための機能の重要性について、ある程度のアイデア(idea)を与えるけれども、より完全な分析には指導付き学習手順内で機能を使用することが必要である。指導付き学習では、訓練セット内の各インスタンス(instance)(即ち、各スペクトル)に対して、カテゴリー・ラベル(category label)を与える。そして、誤分類の数の低減を求める。指導付き学習問題を扱うために、多様な手順が開発されている。指導付き分類のアルゴリズムの出力が一般的に、新インスタンス又はスペクトルのためのクラス・ラベルを発生する(訓練セットにより)クラスファイアとして使用しうる。事例Aとして,Webb,A.John Wiley & Sons Ltd.,2002,Statistical pattern recognitionを、又、事例Bとして、Duda,O.R.,Hart,P.E.,Stork,D.G.,Wiley & Sons Ltd.,2001 ,Pattern Classification.を参照された
い。
【0128】
本発明は種々の具体的で好ましい実施例と技術を参照して記されている。しかしながら本発明の精神と範囲の中にあっても多くの変形と修正を行なえることを理解すべきである。
【図面の簡単な説明】
【0129】
【図1】ヒト血漿のプロテオームが血清内に存在するタンパク質とポリペプチドの分析例を示す。濃度範囲を10以上の桁数で示す。(図面は参考文献2に適合させている)
【図2】分析システムの1個のウエルの切断略図である。分析システムの好ましい実施例では分析システムはカートリッジ内に含まれる96個のサンプル・ウエルの8×12の配列を有している。
【図3】分析システムの好ましい実施例内のカートリッジから成るサンプル・ウエルの配列の略図である。
【図4】機械的案内92を有するMALDIスライド・ホルダー90に挿入された穴50を示す捕捉スライド42の好ましい実施例である。
【図4a】圧力で駆動され、捕捉スライドを横断する流体の流れを加えるためのスライド洗浄マニフォールドである。
【図4b】捕捉スライド上の捕捉材料と電解液の接触を維持するための、又、捕捉物質を横切る電解液に電場を与えるための電気泳動スライド洗浄装置である。
【図5】鋼製MALDIターゲット・プレート上の1pmolのポリペプチド標準及び〜127pmolのBSAのプロットである。
【図6】鋼製MALDIターゲット・プレート上の0.1pmolのポリペプチド標準及び〜127pmolのBSAのプロットである。
【図7】PPAS装置内のアルブミン枯渇液で濃縮された0.1pmolのポリペプチド標準及び〜127pmolのBSAのプロットである。
【図8】血清タンパク質のMALDI質量分析の例である。
【図9】PPAS装置を用いた二成分pH分別である。
【図10】捕捉材料として単一捕捉膜を有するタンパク質/ポリペプチド分析システム(PPAS)のアルファ・プロトタイプを用いて得られたヒト血清内の正帯電LMWタンパク質分析からの質量分析結果である。
【図11】捕捉材料として単一捕捉膜を有するタンパク質/ポリペプチド分析システム(PPAS)のアルファ・プロトタイプを用いて得られたヒト血清内の負帯電LMWタンパク質分析からの質量分析結果である。
【特許請求の範囲】
【請求項1】
電解液内に流体サンプルを保持するためのサンプル・ウエル、
拡散性イオンのための通路及びウエルとの流体の導通を与える分離層、
拡散性イオンのための通路及び分離層との流体の導通を与える捕捉層、
から成るサンプル内の検体を電気泳動的に分離し、濃縮し、捕捉するための装置。
【請求項2】
サンプル・ウエルが上方開口部、側壁、下方開口部から成り、サンプル・ウエルの上方開口部から下方開口部まで、寸法が徐々に低減している請求項1の装置。
【請求項3】
サンプル・ウエルが約400μLまでの内部の全体積を有する請求項1の装置。
【請求項4】
サンプル・ウエルの配列として配置された複数のサンプル・ウエルから成る請求項1の装置。
【請求項5】
分離層がゲルから成る請求項1の装置。
【請求項6】
ゲルがポリアクリル・アミドのゲルである請求項5の装置。
【請求項7】
分離層がカートリッジ・ゲル・プレート内に配置された穴の中に配置されている請求項1の装置。
【請求項8】
実質的に同一の分離層の配列がカートリッジ・ゲル・プレート内に配置された実質的に同じ穴の配列内に配置されている請求項1の装置。
【請求項9】
カートリッジ・ゲル・プレートが電気的に絶縁性の熱可塑性ポリマーから作られている請求項8の装置。
【請求項10】
分離層がカートリッジ・ゲル・プレートの穴でカートリッジ・ゲル・プレートに共有結合されている請求項8の装置。
【請求項11】
捕捉層が多孔質の材料である請求項1の装置。
【請求項12】
捕捉層が、疎水性で多孔質のポリマー、親水性で多孔質のポリマー、又は、親水性と疎水性のポリマーの混合体である請求項1の装置。
【請求項13】
捕捉層が多孔質のポリビニリデン・ジフルオリド(poly(vinylidene difuoride))の材料である請求項1の装置。
【請求項14】
捕捉層がカートリッジ捕捉スライド内に配置された穴内に配置されている請求項1の装置。
【請求項15】
実質的に同じ捕捉層が、カートリッジ捕捉スライド内に配置された実質的に同じ穴の配列内に配置されている請求項1の装置。
【請求項16】
静電荷消失を行うために、カートリッジ捕捉スライドが低い電導度を有している請求項15の装置。
【請求項17】
カートリッジ捕捉スライドが、約104から約108Ω−cmの間の体積抵抗率を有している請求項15の装置。
【請求項18】
カートリッジ捕捉スライドは、MALDI質量分析器内に挿入されたとき、+/−50ミクロンの平坦度である又は平坦度にできる請求項15の装置。
【請求項19】
カートリッジ捕捉スライドが長さ約5.3mm、幅約3.5mm、厚み約1mmである請求項15の装置。
【請求項20】
捕捉スライド内の実質的に同じ穴が実質的に円形であり、直径が約0.5−1mmである請求項15の装置。
【請求項21】
1枚のカートリッジ捕捉スライド内の捕捉層が、隣接するカートリッジ捕捉スライド内の対応する捕捉層と位置合わせするように、2枚以上のカートリッジ捕捉スライドが直列に積み重なった請求項15の装置。
【請求項22】
1枚のカートリッジ捕捉スライド内の捕捉層が隣接する捕捉スライド内の対応する捕捉層と流体及び電気の導通が行われる請求項21の装置。
【請求項23】
その中に配置された複数の穴を含むカートリッジ捕捉スライド、
その複数の穴の中に配置された複数の捕捉層、
から成る質量分析器での分析のためのサンプル検体を捕捉するための装置。
【請求項24】
捕捉層が多孔質の材料から作られている請求項23の装置。
【請求項25】
その多孔質の材料が疎水性多孔質ポリマー、親水性多孔質ポリマー、又は、親水性と疎水性の材料の混合体である請求項24の装置。
【請求項26】
捕捉層が多孔質のポリビニリデン・ジフルオリドである請求項23の装置。
【請求項27】
複数の捕捉層を含む複数の穴が配列として配置されている請求項23の装置。
【請求項28】
その配列が96個所の穴から成っている請求項27の装置。
【請求項29】
静電荷消失を行うために、カートリッジ捕捉スライドが低い電導度を有している請求項23の装置
【請求項30】
カートリッジ捕捉スライドが、約104から約108Ω−cmの間の体積抵抗率を有している請求項23の装置。
【請求項31】
カートリッジ捕捉スライドが、MALDI質量分析器内に挿入されたとき、+/−50ミクロンの平坦度である又は平坦度にできる請求項23の装置。
【請求項32】
カートリッジ捕捉スライドが長さ約5.3mm、幅約3.5mm、厚み約1mmである請求項23の装置。
【請求項33】
カートリッジ捕捉スライドがその捕捉スライド内の複数の穴が実質的に円形であり、直径が約0.5−1mmである請求項23の装置。
【請求項34】
請求項1の装置を用意すること、
サンプル・ウエル内に検体を含むサンプル流体を置くこと、
分離層を通って、捕捉層上への検体の電気的輸送を生じるようにサンプル流体に電流を
加えること、
質量分析器内の捕捉層上で検体の質量を特定すること、
から成る質量分析器での分析により検体を特定する方法。
【請求項35】
カートリッジ捕捉スライドに配置された穴の配列、
穴の配列内に配置された捕捉層の配列、
この場合、カートリッジ捕捉スライドが、サンプル・ウエル及びカートリッジ・ゲル・プレートの配列を含む装置内に組込まれていること、及び、その場合、カートリッジ・ゲル・プレートが、分離層の配列に配置されている穴の配列を含んでいること、
から成る質量分析器で用いるように適合できるカートリッジ捕捉スライド。
【請求項36】
約104から約108Ω−cmの間の体積抵抗率を有している請求項35に基づくカートリッジ捕捉スライド。
【請求項37】
MALDI質量分析器内に挿入したとき、+/−50ミクロンの平坦度である又は平坦度にできる請求項35に基づくカートリッジ捕捉スライド。
【請求項38】
長さ約5.3mm、幅約3.5mm、厚み約1mmである請求項35に基づくカートリッジ捕捉スライド。
【請求項39】
複数の穴が実質的に円形であり、直径が約0.5−1mmである請求項35に基づくカートリッジ捕捉スライド。
【請求項1】
電解液内に流体サンプルを保持するためのサンプル・ウエル、
拡散性イオンのための通路及びウエルとの流体の導通を与える分離層、
拡散性イオンのための通路及び分離層との流体の導通を与える捕捉層、
から成るサンプル内の検体を電気泳動的に分離し、濃縮し、捕捉するための装置。
【請求項2】
サンプル・ウエルが上方開口部、側壁、下方開口部から成り、サンプル・ウエルの上方開口部から下方開口部まで、寸法が徐々に低減している請求項1の装置。
【請求項3】
サンプル・ウエルが約400μLまでの内部の全体積を有する請求項1の装置。
【請求項4】
サンプル・ウエルの配列として配置された複数のサンプル・ウエルから成る請求項1の装置。
【請求項5】
分離層がゲルから成る請求項1の装置。
【請求項6】
ゲルがポリアクリル・アミドのゲルである請求項5の装置。
【請求項7】
分離層がカートリッジ・ゲル・プレート内に配置された穴の中に配置されている請求項1の装置。
【請求項8】
実質的に同一の分離層の配列がカートリッジ・ゲル・プレート内に配置された実質的に同じ穴の配列内に配置されている請求項1の装置。
【請求項9】
カートリッジ・ゲル・プレートが電気的に絶縁性の熱可塑性ポリマーから作られている請求項8の装置。
【請求項10】
分離層がカートリッジ・ゲル・プレートの穴でカートリッジ・ゲル・プレートに共有結合されている請求項8の装置。
【請求項11】
捕捉層が多孔質の材料である請求項1の装置。
【請求項12】
捕捉層が、疎水性で多孔質のポリマー、親水性で多孔質のポリマー、又は、親水性と疎水性のポリマーの混合体である請求項1の装置。
【請求項13】
捕捉層が多孔質のポリビニリデン・ジフルオリド(poly(vinylidene difuoride))の材料である請求項1の装置。
【請求項14】
捕捉層がカートリッジ捕捉スライド内に配置された穴内に配置されている請求項1の装置。
【請求項15】
実質的に同じ捕捉層が、カートリッジ捕捉スライド内に配置された実質的に同じ穴の配列内に配置されている請求項1の装置。
【請求項16】
静電荷消失を行うために、カートリッジ捕捉スライドが低い電導度を有している請求項15の装置。
【請求項17】
カートリッジ捕捉スライドが、約104から約108Ω−cmの間の体積抵抗率を有している請求項15の装置。
【請求項18】
カートリッジ捕捉スライドは、MALDI質量分析器内に挿入されたとき、+/−50ミクロンの平坦度である又は平坦度にできる請求項15の装置。
【請求項19】
カートリッジ捕捉スライドが長さ約5.3mm、幅約3.5mm、厚み約1mmである請求項15の装置。
【請求項20】
捕捉スライド内の実質的に同じ穴が実質的に円形であり、直径が約0.5−1mmである請求項15の装置。
【請求項21】
1枚のカートリッジ捕捉スライド内の捕捉層が、隣接するカートリッジ捕捉スライド内の対応する捕捉層と位置合わせするように、2枚以上のカートリッジ捕捉スライドが直列に積み重なった請求項15の装置。
【請求項22】
1枚のカートリッジ捕捉スライド内の捕捉層が隣接する捕捉スライド内の対応する捕捉層と流体及び電気の導通が行われる請求項21の装置。
【請求項23】
その中に配置された複数の穴を含むカートリッジ捕捉スライド、
その複数の穴の中に配置された複数の捕捉層、
から成る質量分析器での分析のためのサンプル検体を捕捉するための装置。
【請求項24】
捕捉層が多孔質の材料から作られている請求項23の装置。
【請求項25】
その多孔質の材料が疎水性多孔質ポリマー、親水性多孔質ポリマー、又は、親水性と疎水性の材料の混合体である請求項24の装置。
【請求項26】
捕捉層が多孔質のポリビニリデン・ジフルオリドである請求項23の装置。
【請求項27】
複数の捕捉層を含む複数の穴が配列として配置されている請求項23の装置。
【請求項28】
その配列が96個所の穴から成っている請求項27の装置。
【請求項29】
静電荷消失を行うために、カートリッジ捕捉スライドが低い電導度を有している請求項23の装置
【請求項30】
カートリッジ捕捉スライドが、約104から約108Ω−cmの間の体積抵抗率を有している請求項23の装置。
【請求項31】
カートリッジ捕捉スライドが、MALDI質量分析器内に挿入されたとき、+/−50ミクロンの平坦度である又は平坦度にできる請求項23の装置。
【請求項32】
カートリッジ捕捉スライドが長さ約5.3mm、幅約3.5mm、厚み約1mmである請求項23の装置。
【請求項33】
カートリッジ捕捉スライドがその捕捉スライド内の複数の穴が実質的に円形であり、直径が約0.5−1mmである請求項23の装置。
【請求項34】
請求項1の装置を用意すること、
サンプル・ウエル内に検体を含むサンプル流体を置くこと、
分離層を通って、捕捉層上への検体の電気的輸送を生じるようにサンプル流体に電流を
加えること、
質量分析器内の捕捉層上で検体の質量を特定すること、
から成る質量分析器での分析により検体を特定する方法。
【請求項35】
カートリッジ捕捉スライドに配置された穴の配列、
穴の配列内に配置された捕捉層の配列、
この場合、カートリッジ捕捉スライドが、サンプル・ウエル及びカートリッジ・ゲル・プレートの配列を含む装置内に組込まれていること、及び、その場合、カートリッジ・ゲル・プレートが、分離層の配列に配置されている穴の配列を含んでいること、
から成る質量分析器で用いるように適合できるカートリッジ捕捉スライド。
【請求項36】
約104から約108Ω−cmの間の体積抵抗率を有している請求項35に基づくカートリッジ捕捉スライド。
【請求項37】
MALDI質量分析器内に挿入したとき、+/−50ミクロンの平坦度である又は平坦度にできる請求項35に基づくカートリッジ捕捉スライド。
【請求項38】
長さ約5.3mm、幅約3.5mm、厚み約1mmである請求項35に基づくカートリッジ捕捉スライド。
【請求項39】
複数の穴が実質的に円形であり、直径が約0.5−1mmである請求項35に基づくカートリッジ捕捉スライド。
【図1】

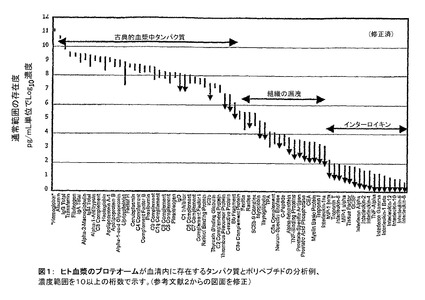
【図2】
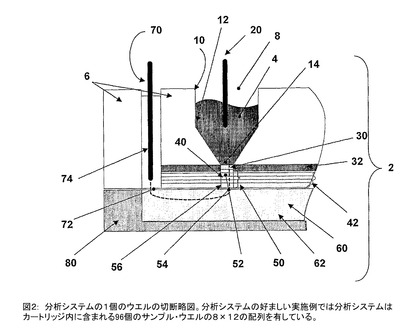
【図3】
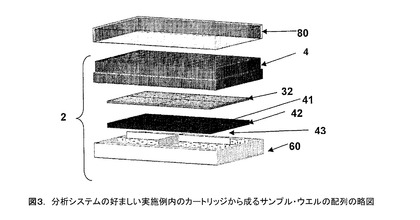
【図4】
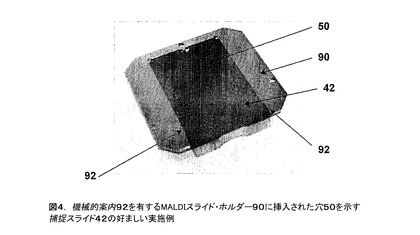
【図4a】
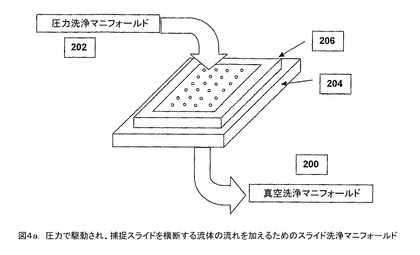
【図4b】
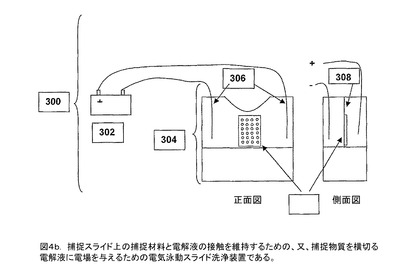
【図5】
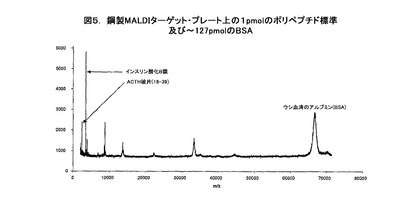
【図6】
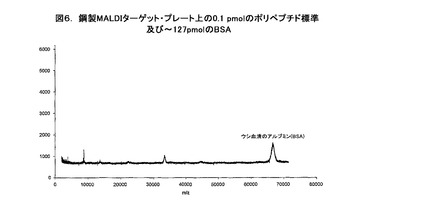
【図7】
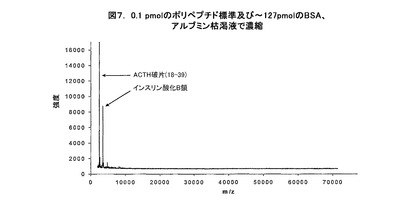
【図8】
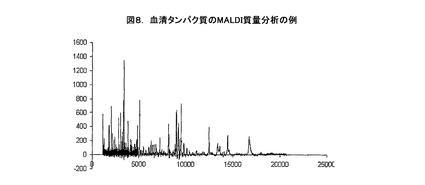
【図9】
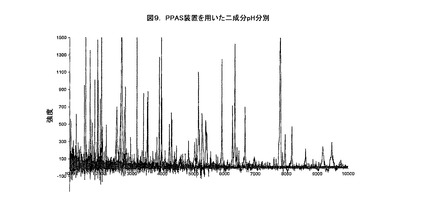
【図10】
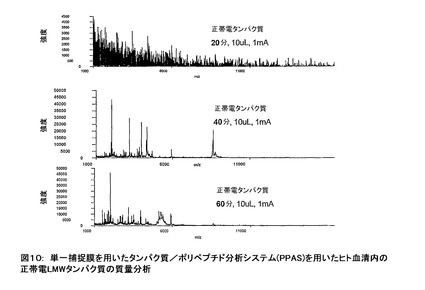
【図11】
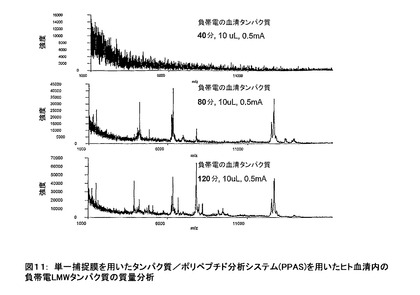
【図2】
【図3】
【図4】
【図4a】
【図4b】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【公表番号】特表2008−534987(P2008−534987A)
【公表日】平成20年8月28日(2008.8.28)
【国際特許分類】
【出願番号】特願2008−505627(P2008−505627)
【出願日】平成18年4月5日(2006.4.5)
【国際出願番号】PCT/US2006/013300
【国際公開番号】WO2006/110644
【国際公開日】平成18年10月19日(2006.10.19)
【出願人】(507330899)プロテイン・デイスカバリー・インコーポレーテツド (3)
【Fターム(参考)】
【公表日】平成20年8月28日(2008.8.28)
【国際特許分類】
【出願日】平成18年4月5日(2006.4.5)
【国際出願番号】PCT/US2006/013300
【国際公開番号】WO2006/110644
【国際公開日】平成18年10月19日(2006.10.19)
【出願人】(507330899)プロテイン・デイスカバリー・インコーポレーテツド (3)
【Fターム(参考)】
[ Back to top ]