
マンノシルトランスフェラーゼ阻害剤を使用した異種蛋白質の効率的生産
O−結合型グリコシル化量の低下した蛋白質組成物を製造するための化合物と方法について記載する。前記方法はPmt介在性O−結合型グリコシル化の所定のベンジリデンチアゾリジンジオン阻害剤の存在下で培養した細胞中で蛋白質を生産する段階を含む。
【発明の詳細な説明】
【背景技術】
【0001】
(発明の背景)
糖蛋白質は触媒作用、シグナル伝達、細胞間コミュニケーション、並びに分子認識及び会合等のヒト及び他の哺乳動物における多くの必須機能に介在する。糖蛋白質は真核生物において非サイトゾル蛋白質の大部分を構成する(Lis and Sharon,1993,Eur.J.Biochem.218:1−27)。多数の糖蛋白質が治療目的に利用されており、この二十年間に天然に存在する糖蛋白質の組換え体はバイオテクノロジー産業の主要な部分になっている。治療薬として使用されている組換えグリコシル化蛋白質の例としては、エリスロポエチン(EPO)、治療用モノクローナル抗体(mAb)、組織プラスミノーゲンアクチベーター(tPA)、インターフェロンβ(IFN−β)、顆粒球−マクロファージコロニー刺激因子(GM−CSF)、及びヒト絨毛性ゴナドトロピン(hCH)が挙げられる(Cumming et al.,1991,Glycobiology 1:115−130)。潜在的予防薬及び治療薬として生産された組換え蛋白質が臨床に近付くにつれ、組換え生産された糖蛋白質のグリコシル化パターンの変動は科学界で最近関心の高い話題となっている。
【0002】
一般に、糖蛋白質オリゴ糖のグリコシル化構造はこれらを生産するために使用される細胞の宿主種により異なる。非ヒト宿主細胞で生産される治療用蛋白質はヒトに免疫原性応答を誘発し得る非ヒトグリコシル化を含む可能性が高く、例えば酵母における高マンノシル化(Ballou,1990,Methods Enzymol.185:440−470);植物におけるα(1,3)−フコース及びβ(1,2)−キシロース(Cabanes−Macheteau et al.,1999.Glycobiology,9:365−372);チャイニーズハムスター卵巣細胞におけるN−グリコリルノイラミン酸(Noguchi et al.,1995.J.Biochem.117:5−62);並びにマウスにおけるGalα−1,3Galグリコシル化(Borrebaeck,et al.,1993,Immun.Today,14:477−479)が報告されている。動物細胞中で蛋白質と結合した糖鎖としては、蛋白質中のアスパラギン(Asn)残基と結合したN−グリコシド結合型糖鎖(別称N−グリカン;又はN−結合型グリコシル化)と、蛋白質中のセリン(Ser)又はスレオニン(Thr)残基と結合したO−グリコシド結合型糖鎖(別称O−グリカン;又はO−結合型グリコシル化)が挙げられる。
【0003】
非ヒト哺乳動物細胞により産生される糖蛋白質のオリゴ糖構造はヒト糖蛋白質のオリゴ糖構造と密接に関連する傾向があるため、大半の商業用糖蛋白質は哺乳動物細胞で生産されている。しかし、哺乳動物細胞には蛋白質産生用宿主細胞としていくつかの重大な欠点がある。コストがかかるという点以外に、哺乳動物細胞における蛋白質の生産方法では不均質なグリコフォーム集団を生じ、体積当たりの力価が低く、安定な細胞株を作製するためには持続的なウイルス閉じ込めと相当の時間が同時に必要となる。
【0004】
蛋白質上の特定グリコフォームがその薬物動態、薬力学、受容体相互作用及び組織特異的標的特性を含む蛋白質の性質に大きな影響を与える可能性があることはよく知られている(Graddis et al.,2002.Curr Pharm Biotechnol.3:285−297)。例えば、Igの各種グリコシル化パターンは各種生物学的性質に相関することが示されている(Jefferis and Lund,1997,Antibody Eng.Chem.Immunol,65:111−128;Wright and Morrison,1997,Trends Biotechnol.,15:26−32)。更に、糖蛋白質のガラクトシル化は細胞培養条件により変動し得るため、糖蛋白質組成によっては糖蛋白質上の特定ガラクトースパターンに応じて免疫原性になる場合があることも示されている(Patel et al.,1992.Biochem J.285:839−845)。しかし、どの特定グリコフォームが望ましい生物学的機能に寄与するのか分からないため、糖蛋白質上の特定グリコフォームを増加できるようにすることが非常に望ましい。グリコフォーム毎に異なる生物学性質に相関しているため、特定グリコフォームをもつ糖蛋白質の増加能を使用すれば、糖蛋白質の特定グリコフォームと特定の生物学的機能の関係を解明することができる。また、特定グリコフォームをもつ糖蛋白質を増加できるならば、特定の特異性をもつ治療用糖蛋白質を製造することも可能になる。従って、特定グリコフォームを増加した糖蛋白質組成物の製造が非常に望ましい。
【0005】
N−結合型グリコシル化の経路は多くの分析の対象となっているが、O−結合型グリコシル化のプロセスと機能はよく分かっていない。しかし、N−結合型グリコシル化とは対照的に、O−グリコシル化はシスゴルジで生じる翻訳後イベントであることが知られている(Varki,1993,Glycobiol.,3:97−130)。N−結合型グリコシル化と同様のO−結合型グリコシル化のコンセンサスアクセプター配列は存在しないと思われるが、数種の糖蛋白質の多数のO−結合型グリコシル化部位の周囲のアミノ酸配列を比較すると、グリコシル化残基に対して−1及び+3位のプロリン残基の頻度が高く、セリン、スレオニン及びアラニン残基が著しく増加していることが分かる(Wilson et al.,1991,Biochem.J.,275:529−534)。糖蛋白質におけるセリン残基とスレオニン残基の部分もO−グリコシル化の潜在部位であると思われる。
【0006】
O−結合型グリコシル化で役割を果たす1つの遺伝子ファミリーはDol−P−Man:蛋白質(Ser/Thr)マンノシルトランスフェラーゼ(Pmt)をコードする遺伝子である。これらの高度に保存された遺伝子はヒト、齧歯類、昆虫等の高等真核生物と、真菌類等の下等真核生物の両者で同定されている。Saccharomyces cerevisiaeやPichia pastoris等の酵母はPmtホモログをコードする7個までのPMT遺伝子をコードする(Wilier et al.Curr.Opin.Struct.Biol.2003 Oct;13(5):621−30に概説)。酵母において、O−結合型グリコシル化は7個のO−マンノシルトランスフェラーゼ遺伝子の1個によりドリコールリン酸マンノースに由来する初期のマンノースが小胞体の新生糖蛋白質のセリン又はスレオニン残基に付加されることにより開始する。酵母にはPmtホモログをコードする7個のPMT遺伝子が存在すると思われるが、酵母における分泌型真菌及び異種蛋白質のO−マンノシル化はヘテロダイマーとして機能すると思われるPmt1とPmt2をコードする遺伝子に主に依存する。PMT1及びPMT2とその蛋白質産物である夫々Pmt1及びPmt2は種間で高度に保存されていると思われる。
【0007】
TannerらはSaccharomyces cerevisiaeのPMT1及びPMT2遺伝子と、O−結合型グリコシル化の低下した組換え蛋白質が産生されるようにPMT遺伝子の1個以上を遺伝子改変した真菌細胞を使用してO−結合型グリコシル化の低下した組換え蛋白質を作製する方法を米国特許第5,714,377号に記載している。
【0008】
Ngらはアンチセンスもしくは同時抑圧の使用、又はO−結合型グリコシル化に関連する遺伝子、特にPMT遺伝子の1個以上を機能低下突然変異させた酵母宿主株の構築によるOグリコシル化の阻害を米国特許出願公開第20020068325号に開示している。
【0009】
UDP−N−アセチル−α−D−ガラクトサミン:ポリペプチドN−アセチルガラクトサミニルトランスフェラーゼ(GalNAcトランスフェラーゼ)は高等真核生物に認められるムチン型O−結合型グリコシル化に関与している。これらの酵素は蛋白質中の特定のセリン及びスレオニンアミノ酸のヒドロキシ基にN−アミノ酸ガラクトサミンを付加することによりこれらのアミノ酸のO−グリコシル化を開始し、その後、マンノース残基を段階的に付加することができる。ClausenらはUDP−N−アセチル−α−D−ガラクトサミン:ポリペプチドN−アセチルガラクトサミニルトランスフェラーゼ(GalNAcトランスフェラーゼ)をコードする核酸ファミリーを米国特許第5,871,990号及び米国特許出願公開第20050026266号に開示している。ClausenはポリペプチドGalNAcトランスフェラーゼのレクチンを選択的に阻害し、O−グリカン生合成に関与する他のグリコシルトランスフェラーゼの基質として機能させず、O−グリコシル化を阻害するために、GalNAc−β−ベンジルの使用を米国特許出願公開第20030186850号に開示している。
【0010】
O−結合型グリコシル化の阻害剤が記載されている。例えば、Orchardらは米国特許第7,105,554号にベンジリデンチアゾリジンジオンと、抗真菌剤(例えば抗カビ剤)としてのその使用について記載している。これらのベンジリデンチアゾリジンジオンはPmt1酵素を阻害し、O−結合型マンノ蛋白質の形成を防止し、真菌細胞壁の完全性を損なうことが報告されている。結末として、細胞膨潤を招き、最終的に破裂により死に至る。
【0011】
Konradらは組織又は細胞におけるO−結合型蛋白質グリコシル化を薬理的に阻害することにより糖尿病を治療又は予防する方法を米国特許出願公開第20020128235号に開示している。この方法はO−結合型N−アセチルグルコサミントランスフェラーゼと結合することによりO−結合型グリコシル化を阻害する(Z)−1−[N−(3−アンモニオプロピル)−N−(n−プロピル)アミノ]ジアゼニウム−1,2−ジオール酸又はその誘導体による糖尿病個体の治療に依存している。
【0012】
Kojimaらは白血球又は腫瘍細胞によるSLex又はSLeaの発現を阻止し、それにより内皮細胞や血小板へのこれらの細胞の接着を阻害するために、O−α−GalNAcの蓄積に繋がるO−グリコシル化の伸長を阻害するベンジル−α−N−アセチルガラクトサミン等の化合物を使用してO−グリコシル化を阻害するための治療用組成物を米国特許第5,268,364号に開示している。
【0013】
Boimeらはグリコシル化部位のアミノ酸配列を改変することによりO−結合型グリコシル化を改変したホルモン変異体を米国特許第6,103,501号に開示している。
【先行技術文献】
【特許文献】
【0014】
【特許文献1】米国特許第5,714,377号明細書
【特許文献2】米国特許出願公開第20020068325号明細書
【特許文献3】米国特許第5,871,990号明細書
【特許文献4】米国特許出願公開第20050026266号明細書
【特許文献5】米国特許出願公開第20030186850号明細書
【特許文献6】米国特許第7,105,554号明細書
【特許文献7】米国特許出願公開第20020128235号明細書
【特許文献8】米国特許第5,268,364号明細書
【特許文献9】米国特許第6,103,501号明細書
【非特許文献】
【0015】
【非特許文献1】Lis and Sharon,1993,Eur.J.Biochem.218:1−27
【非特許文献2】Cumming et al.,1991,Glycobiology 1:115−130
【非特許文献3】Ballou,1990,Methods Enzymol.185:440−470
【非特許文献4】Cabanes−Macheteau et al.,1999.Glycobiology,9:365−372
【非特許文献5】Noguchi et al.,1995.J.Biochem.117:5−62
【非特許文献6】Borrebaeck,et al.,1993,Immun.Today,14:477−479
【非特許文献7】Graddis et al.,2002.Curr Pharm Biotechnol.3:285−297
【非特許文献8】Jefferis and Lund,1997,Antibody Eng.Chem.Immunol,65:111−128
【非特許文献9】Wright and Morrison,1997,Trends Biotechnol.,15:26−32
【非特許文献10】Patel et al.,1992.Biochem J.285:839−845
【非特許文献11】Varki,1993,Glycobiol.,3:97−130
【非特許文献12】Wilson et al.,1991,Biochem.J.,275:529−534
【非特許文献13】Wilier et al.Curr.Opin.Struct.Biol.2003 Oct;13(5):621−30
【発明の概要】
【発明が解決しようとする課題】
【0016】
本発明はO−結合型グリコシル化の低下した組換え蛋白質の生産に有用なPmt蛋白質の新規阻害剤に関する。この結果、真菌及び酵母細胞から産生される蛋白質のO−結合型グリコシル化を制御することが可能になる。
【課題を解決するための手段】
【0017】
O−結合型グリコシル化量の低下した蛋白質組成物を製造するための化合物と方法について記載する。前記方法はPmt介在性O−結合型グリコシル化の所定のベンジリデンチアゾリジンジオン阻害剤の存在下で培養した細胞中で蛋白質を生産する段階を含む。
【図面の簡単な説明】
【0018】
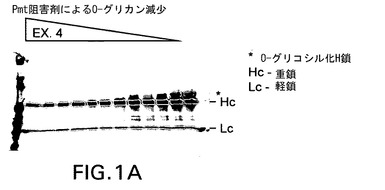
【図1A】実施例4として示す新規阻害剤を含むPmt阻害剤がPichia pastorisにおける分泌型組換えモノクローナル抗体のO−グリコシル化に及ぼす効果を示す。Pmtの化学的阻害剤はO−グリコシル化を用量依存的に低下させた。抗ヒトH+L抗体を使用するウェスタンブロッティングを使用し、増量しながら(左側のレーン方向に高用量)Pmt阻害剤で処理した株の増殖培地中で重鎖(Hc)と軽鎖(Lc)を検出した。移動度の最も遅いバンド(星印で示す)がPmt阻害剤処理により除去されるO−グリコシル化Hcに相当する。
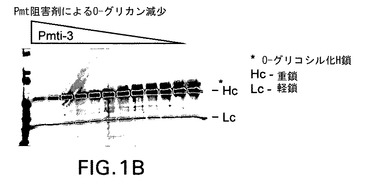
【図1B】実施例4として示す新規阻害剤を含むPmt阻害剤がPichia pastorisにおける分泌型組換えモノクローナル抗体のO−グリコシル化に及ぼす効果を示す。Pmtの化学的阻害剤はO−グリコシル化を用量依存的に低下させた。抗ヒトH+L抗体を使用するウェスタンブロッティングを使用し、増量しながら(左側のレーン方向に高用量)Pmt阻害剤で処理した株の増殖培地中で重鎖(Hc)と軽鎖(Lc)を検出した。移動度の最も遅いバンド(星印で示す)がPmt阻害剤処理により除去されるO−グリコシル化Hcに相当する。
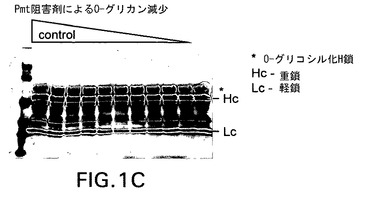
【図1C】実施例4として示す新規阻害剤を含むPmt阻害剤がPichia pastorisにおける分泌型組換えモノクローナル抗体のO−グリコシル化に及ぼす効果を示す。Pmtの化学的阻害剤はO−グリコシル化を用量依存的に低下させた。抗ヒトH+L抗体を使用するウェスタンブロッティングを使用し、増量しながら(左側のレーン方向に高用量)Pmt阻害剤で処理した株の増殖培地中で重鎖(Hc)と軽鎖(Lc)を検出した。移動度の最も遅いバンド(星印で示す)がPmt阻害剤処理により除去されるO−グリコシル化Hcに相当する。
【発明を実施するための形態】
【0019】
本発明は特定宿主細胞中でO−結合型グリコシル化を受け易い蛋白質についてこの細胞型におけるO−結合型グリコシル化量を低下させた(O−結合型グリコシル化ゼロを含む)組換え蛋白質(ポリペプチドと糖蛋白質を含む)を発現させるための新規化合物と方法を提供する。前記方法は該当蛋白質が宿主細胞中でO−結合型グリコシル化を受け易い場合に、前記細胞中における蛋白質のマンノースからセリン又はスレオニン残基への転移に関与するDol−P−Man:蛋白質(Ser/Thr)マンノシルトランスフェラーゼ(Pmt)蛋白質の1種以上の活性の阻害剤である本発明の1種以上の新規化合物の存在下に前記宿主細胞中で前記蛋白質の発現を誘導する段階を含み、場合により前記蛋白質の発現誘導時にBobrowiczらの米国出願公開第2007061631号に記載されているような1種以上のα1,2−マンノシダーゼを併用してもよい。前記阻害剤の存在下で発現される蛋白質は前記阻害剤の不在下で産生させた場合に蛋白質上に存在するようなO−結合型グリコシル化量に比較してO−結合型グリコシル化量が低下している。前記方法は、通常ではO−結合型グリカンをもつ蛋白質を産生する下等真核生物(例えば酵母)や細菌等についてO−結合型グリカン数を低下させた宿主細胞中で蛋白質のO−グリコシル化量を低下させることが望ましい治療関連蛋白質を産生させる手段となるため、特に有用である。なお、本方法は下等真核生物中でO−結合型グリコシル化の低下した蛋白質を発現させるのに特に適しているが、高等真核生物や細菌でも本方法を実施することができる。
【0020】
本発明のPmt阻害剤は以下の群:
【0021】
【化1】

又はその塩から選択される。
【0022】
本明細書に記載する方法は蛋白質がO−結合型グリコシル化を受け易い宿主細胞中でO−結合型グリコシル化の低下した蛋白質を産生させるための所定の従来技術の方法の改良である。例えば、TannerらはO−結合型グリコシル化の低下した組換え蛋白質を産生するようにPmt蛋白質をコードするPMT遺伝子の1個以上を遺伝子改変した酵母細胞等の真菌細胞を使用してO−結合型グリコシル化の低下した組換え蛋白質を作製する方法を米国特許第5,714,377号に記載している。真菌宿主細胞でPMT1、PMT2又はPMT4遺伝子を欠失させると、真菌宿主細胞でO−結合型グリコシル化の低下した組換え蛋白質の産生が可能になるが、宿主細胞増殖にはPMT遺伝子の発現が重要であり、単独でどれを欠失させても真菌宿主細胞の増殖能は悪化し、O−結合型グリコシル化量の低下した宿主細胞又は組換え蛋白質を十分な量で生産することが困難になる。PMT2と同時にPMT1又はPMT4を欠失させると、真菌宿主細胞に致命的であると思われる。従って、宿主細胞中のPMT遺伝子の遺伝子除去はO−結合型グリコシル化を低下させた組換え蛋白質を生産するためには望ましくない手段であると思われる。
【0023】
これに対して、本発明の方法で使用される宿主細胞中のPMT遺伝子は改変又は欠失されていないため、宿主細胞は細胞増殖に重要なこれらの蛋白質をO−グリコシル化することができ、この時点までPmt蛋白質の活性は阻害される。一般に、この結果として、PMT遺伝子を欠失させた場合に得られるレベルよりも高レベルまで宿主細胞を増殖させることが可能になる。更に、特定態様では、宿主細胞中の組換え蛋白質の発現は誘導性プロモーターにより制御され、組換え蛋白質の発現が誘導されるまで宿主細胞中のPmt活性は阻害されない。このため、組換え蛋白質の発現誘導とPmt阻害剤の添加前に組換え蛋白質をコードする核酸を含む大量の宿主細胞を培養生産することができる。従って、1個以上のPMT遺伝子を欠失させた培養増殖不良な宿主細胞よりも短期間にO−結合型グリコシル化を低下させた組換え蛋白質をより大量に培養生産することが可能になる。
【0024】
本明細書に記載するPmt阻害剤は実施例5に示すように効力が増加するため、Orchardら(米国特許第7,105,554号)及びBobrowiczら(米国出願公開第2007061631号)に従来記載されている阻害剤よりも改善されている。効力の増加により、真菌O−グリコシル化を許容可能なレベルまで低下させるために使用する阻害剤の量を減らすことができ、及び/又はO−グリカンを完全に除去する可能性が高まる。
【0025】
本明細書に記載する方法は主に特定のN−結合型グリカン構造をもつ糖蛋白質を産生するように遺伝子改変されているだけでなく、糖蛋白質をO−グリコシル化する宿主細胞中におけるO−結合型グリコシル化の低下した糖蛋白質の産生を容易にする。主に特定のN−結合型グリコフォームをもつ多様な糖蛋白質の製造方法が米国特許第7,029,872号と、米国出願公開第20050170452号、20050260729号、20040230042号、20050208617号、20050208617号、20040171826号、20060160179号、20060040353号及び20060211085号に開示されている。本明細書に開示する方法を使用し、主に特定のN−結合型グリカン構造をもち、O−結合型グリコシル化の低下した糖蛋白質を産生させるためには、上記特許及び特許出願に記載されている宿主細胞のいずれか1種を使用することができる。主に特定のN−結合型グリカン構造をもつ糖蛋白質を産生するように遺伝子改変した所定の宿主細胞は改変していない宿主細胞ほど良好に特定条件下で培養増殖できないことが判明した。例えば、高マンノシル化に関与する遺伝子を欠失させ、特定の哺乳動物又はヒト様N−結合型グリカン構造を産生させるために必要な他の遺伝子を付加した特定真菌及び酵母細胞は遺伝子改変していない真菌又は酵母細胞ほど良好に増殖することができない。これらの遺伝子改変真菌又は酵母細胞では、PMT遺伝子の欠失を更に導入すると、細胞致死性になるか、又は十分な量まで培養増殖する能力に悪影響を与える場合がある。本明細書に記載する方法は組換え糖蛋白質の発現を誘導する段階と、場合により1種以上のα1,2−マンノシダーゼと共にPmt蛋白質の活性の阻害剤を添加する段階よりも前に細胞を十分な量まで培養増殖させ、主に特定のN−結合型グリカン構造をもち、O−結合型グリコシル化の低下した組換え糖蛋白質を生産することにより、PMT遺伝子を欠失させる潜在的な悪影響を回避する。
【0026】
本明細書に記載する方法の1側面は、O−結合型グリコシル化が低下し、主に特定のN−結合型グリコフォームを含む糖蛋白質組成物を提供することであり、このような組換え糖蛋白質はCHO細胞等の哺乳動物細胞培養から生産される同一糖蛋白質の組成物に比較して生物活性の増加及び/又は望ましくない免疫原性の低下を示すことができる。O−結合型グリコシル化が低下し、主にN−結合型グリコフォームを含む糖蛋白質組成物を生産すると、望ましくない結果を誘導及び/又はより有効なグリコフォームを減弱する恐れのある有害又は不活性なグリコフォームと不均質混合物の生産が避けられるという利点もある。従って、例えば主にMan5GlcNAc2、Man3GlcNAc2、GlcNAcMan5GlcNAc2、GlcNAcMan3GlcNAc2、GlcNAc2Man3GlcNAc2、GalGlcNAcMan5GlcNAc2、Gal(GlcNAc)2Man5GlcNAc2、(GalGlcNAc)2Man5GlcNAc2、NANAGalGlcNAcMan3GlcNAc2、NANA2Gal2GlcNAcMan3GlcNAc2及びGalGlcNAcMan3GlcNAc2グリコフォームを含み、O−結合型グリコシル化の低下した糖蛋白質分子の治療用医薬組成物はより低用量で十分に有効になり、より高い効果/効力が得られると思われる。
【0027】
一般に、O−結合型グリコシル化の低下した蛋白質の生産方法は、O−結合型グリコシル化の低下した蛋白質を生産することが望ましい組換え又は異種蛋白質をコードする核酸で宿主細胞を形質転換する段階を含む。組換え蛋白質をコードする核酸は組換え蛋白質の発現を可能にする調節配列と機能的に連結される。このような調節配列としては誘導性プロモーターと、場合により融合蛋白質をコードする核酸の上流ないし5’側のエンハンサーと、組換え蛋白質をコードする核酸の3’
側ないし下流の転写終結部位が挙げられる。核酸は更に一般にリボソーム結合部位をもつ5’UTR領域と3’非翻訳領域をコードする。核酸は組換え蛋白質を発現させる細胞で複製可能なベクターの成分であることが多い。ベクターは更に形質転換細胞の認識を可能にするためのマーカーを含むことができる。他方、所定の細胞種、特に酵母は外来ベクター配列をもたない核酸で首尾よく形質転換することができる。
【0028】
目的の組換え蛋白質をコードする核酸は数種類の起源から得ることができる。保存領域のプライマーを使用して蛋白質を発現させることが分かっている細胞株からcDNA配列を増幅することができる(例えばMarks et al.,J.Mol.Biol.581−596(1991)参照)。科学文献中の配列に基づいて核酸を新規に合成することもできる。目的の配列に対応するオーバーラップするオリゴヌクレオチドの伸長により核酸を合成することもできる(例えばCaldas et al.,Protein Engineering,13,353−360(2000)参照)。
【0029】
1側面では、所望時に蛋白質の発現を誘導できるように、蛋白質をコードする核酸を誘導性プロモーターと機能的に連結する。別の側面では、蛋白質をコードする核酸を構成性プロモーターと機能的に連結する。発現された蛋白質を単離し易くするために、細胞培養培地中に蛋白質を分泌させた後に単離できるようにするシグナル配列を蛋白質に付加することが一般に好ましい。第1の側面では、Pmt介在性O−結合型グリコシル化の1種以上の阻害剤を培地に添加する前に所望量の蛋白質を産生させるために十分な所望の多重度の宿主細胞を生産するために十分な時間にわたって形質転換宿主細胞を培養する。インデューサーと阻害剤を同時に培地に添加してもよいし、1種以上のPmt阻害剤を添加する前にインデューサーを培地に添加してもよいし、インデューサーを添加する前に1種以上のPmt阻害剤を培地に添加してもよい。O−結合型グリコシル化の低下した誘導蛋白質が産生され、培地から回収することができ、あるいはシグナル配列をもたない蛋白質の場合には溶解により宿主細胞から回収することができる。蛋白質をコードする核酸を構成性プロモーターと機能的に連結する第2の側面では、1種以上のPmt介在性O−結合型グリコシル化阻害剤を培地に添加すると同時に培養株を樹立し、産生されるO−結合型グリコシル化の低下した蛋白質を培地から回収することができ、あるいはシグナル配列をもたない蛋白質の場合には溶解により宿主細胞から回収することができる。
【0030】
本明細書では、O−結合型グリコシル化の低下した蛋白質を生産するのために有用なPmt蛋白質の1種以上の活性を阻害する化合物又は組成物について記載する。宿主細胞が真菌や酵母等の下等真核生物である場合には、阻害剤は少なくともPmt1もしくはPmt2、又はその両方の活性を阻害することが望ましい。高等真核生物では、阻害剤はPmt1又はPmt2に対応する高等真核生物でのホモログの活性を阻害することが望ましい。
【0031】
本発明の化合物は無傷の機能的PMT遺伝子をもつPichia pastoris株でO−結合型グリコシル化の低下した組換え蛋白質を生産するのに有効であることが判明した。表1と実施例5の図1から明らかなように、無傷の機能的PMT遺伝子をもつ組換えPichia pastorisの培養物に添加し、組換え蛋白質の発現時に誘導性プロモーターと機能的に連結した組換えヒト抗Her2抗体蛋白質をコードする核酸で形質転換した上記4種類のPmt化学阻害剤のいずれか1種は、Orchard et al.(Bioorgan & Med Chem Letters(2004)14:3975−3978)及びEP1313471B1を含む同一著者による特許公開とBobrowiczらの米国出願公開第2007061631号に記載されているPmt阻害剤であるPmti−3で処理したPichia pastoris細胞に認められるO−結合型グリコシル化レベルに対して改善された低いO−結合型グリコシル化レベルをもつ組換え蛋白質を生じた。
【0032】
宿主細胞
本明細書で使用する「細胞」なる用語は以下に記載する宿主細胞を意味する。本明細書に記載する方法の宿主細胞としては高等真核細胞と下等真核細胞の両方が挙げられるが、下等真核細胞(例えば糸状菌や酵母細胞)は経済的に培養でき、蛋白質収率が高く、適宜改変すると、適切なグリコシル化パターンをもつ蛋白質を産生することができるので、一般に蛋白質の発現に好ましい。下等真核生物としては、酵母、真菌類、襟鞭毛虫類、微胞子虫類、アルベオラータ(例えば渦鞭毛虫類)、ストラメノパイル(例えば褐藻類、原生動物)、紅藻植物門(例えば紅藻類)、植物(例えば緑藻類、植物細胞、コケ)及び他の原生生物か挙げられる。酵母及び真菌類としては限定されないが、ピキア(Pichia)種(例えばPichia pastoris、Pichia finlandica、Pichia trehalophila、Pichia koclamae、Pichia membranaefaciens、Pichia minuta(Ogataea minuta、Pichia lindneri)、Pichia opuntiae、Pichia thermotolerans、Pichia salictaria、Pichia guercuum、Pichia pijperi、Pichia stiptis、Pichia methanolica)、サッカロミセス(Saccharomyces)種(例えばSaccharomyces cerevisiea)、Hansenula polymorpha、クルイベロマイセス(Kluyveromyces)種(例えばKluyveromyces lactis)、Candida albicans、アスペルギルス(Aspergillus)種(例えばAspergillus nidulans、Aspergillus niger、Aspergillus oryzae)、Trichoderma reesei、Chrysosporium lucknowense、フサリウム(Fusarium)種(例えばFusarium gramineum、Fusarium venenatum)、Physcomitrella patens及びNeurospora crassaが挙げられる。特に酵母は遺伝学が確立しており、迅速な形質転換、試験蛋白質局在ストラテジー及び簡単な遺伝子ノックアウト技術が可能になるため、一般に好ましい。適切なベクターは3−ホスホグリセリン酸キナーゼ又は他のグリコール酸酵素を含むプロモーター等の発現制御配列と、複製起点、終結配列等を必要に応じてもつ。
【0033】
高い細胞密度まで増殖できると共に大量の組換え蛋白質を分泌できるという理由から、K.lactis、Pichia pastoris、Pichia methanolica及びHansenula polymorpha等の各種酵母が細胞培養に一般に好ましい。同様に、Aspergillus niger、フサリウム種、Neurospora crass等の糸状菌を使用して工業規模で組換え蛋白質を生産することもできる。
【0034】
グリコシル化パターンがヒト様又はヒト化型である蛋白質又は糖蛋白質を発現するように、下等真核生物、特に糸状菌と酵母を遺伝子改変することができる。これはGerngrossらにより米国特許第US7029872号と米国特許出願公開第20040018590号、20050170452号、20050260729号、20040230042号、20050208617号、20040171826号、20050208617号、20060160179号、20060040353号及び20060211085号に記載されているように、選択された内在グリコシル化酵素を除去すること及び/又は外来酵素を供給することにより実現することができる。従って、特定のN−グリカン構造の高収率生産を可能にするように1種以上の酵素又は酵素活性を発現するように宿主細胞を付加的又は代替的に操作することができる。例えば通常では酵素に関連しないシグナルペプチドにより酵素が最適活性をもつような宿主細胞内オルガネラをこのような酵素の標的とすることができる。糖ヌクレオチドトランスポーター及び/又はヌクレオチドジホスファターゼ酵素を発現するように宿主細胞を改変することもできる。トランスポーターとジホスファターゼは適切な区画にグリコシル化酵素の適切な基質を提供し、競合的生成物阻害を低減し、ヌクレオチド二リン酸の除去を促進することにより、人工グリコシル化工程の効率を改善する。例えばGerngrossらの米国特許出願公開第20040018590号及びHamilton,2003,Science 301:1244−46並びに上記米国特許及び特許出願参照。
【0035】
例えば、低下させないと糖蛋白質のN−グリカンにマンノース残基を付加する1,6−マンノシルトランスフェラーゼ活性を低下させると共に、30モル%を上回るMan5GlcNAc2N−グリカンをもつ組換え糖蛋白質の産生を可能にするα−1,2マンノシダーゼ活性の異所性発現用核酸を更に含むように宿主細胞(例えば酵母又は真菌)を選択又は操作することができる。本明細書に記載する方法に従って宿主細胞中で糖蛋白質を産生させると、主にMan5GlcNAc2N−グリカン構造をもち、別の方法により細胞中で産生される糖蛋白質に比較してO−グリコシル化の低下した糖蛋白質が宿主細胞により産生される。別の側面では、主にGlcNAcMan5GlcNAc2N−グリカンをもつ糖蛋白質の産生を可能にするように、GlcNAcトランスフェラーゼI活性の異所性発現用核酸を更に含むように宿主細胞を操作する。本明細書に記載する方法に従って宿主細胞中で糖蛋白質を産生させると、主にGlcNAcMan5GlcNAc2N−グリカン構造をもち、別の方法により細胞中で産生される糖蛋白質に比較してO−グリコシル化の低下した糖蛋白質が宿主細胞により産生される。更に別の側面では、主にGlcNAcMan3GlcNAc2N−グリカンをもつ糖蛋白質の産生を可能にするように、マンノシダーゼII活性の異所性発現用核酸を更に含むように宿主細胞を操作する。本明細書に記載する方法に従って宿主細胞中で糖蛋白質を産生させると、主にGlcNAcMan3GlcNAc2N−グリカン構造をもち、別の方法により細胞中で産生される糖蛋白質に比較してO−グリコシル化の低下した糖蛋白質が宿主細胞により産生される。更に別の側面では、主にGlcNAc2Man3GlcNAc2N−グリカンをもつ糖蛋白質の産生を可能にするように、GlcNAcトランスフェラーゼII活性の異所性発現用核酸を更に含むように宿主細胞を操作する。本明細書に記載する方法に従って宿主細胞中で糖蛋白質を産生させると、主にGlcNAc2Man3GlcNAc2N−グリカン構造をもち、別の方法により細胞中で産生される糖蛋白質に比較してO−グリコシル化の低下した糖蛋白質が宿主細胞により産生される。更に他の側面では、任意組合せでN−結合型グリコシル化に関与し、例えばシアリルトランスフェラーゼ活性、クラスII及びIIIマンノシダーゼ活性、GlcNAcトランスフェラーゼII、III、IV、V、VI、IX活性、並びにガラクトーストランスフェラーゼ活性をコードする1種以上の高等真核生物遺伝子を更に含むことにより特定のハイブリッド又は複合N−グリカン又はヒト様N−グリカン構造を産生するように上記宿主細胞を更に操作することができる。細胞はUDP特異的ジホスファターゼ活性、GDP特異的ジホスファターゼ活性及びUDP−GlcNAcトランスポーター活性をコードする核酸の1種以上を更に含むことが一般に好ましい。
【0036】
本明細書に教示するようなO−結合型グリコシル化の低下した蛋白質及び糖蛋白質を発現させるためには植物及び植物細胞培養物を使用することができる(例えばLarrick & Fry,1991,Hum.Antibodies Hybridomas 2:172−89;Benvenuto et al.,1991,Plant Mol.Biol.17:865−74;Durin et al.,1990,Plant Mol.Biol.15:281−93;Hiatt et al.,1989,Nature 342:76−8参照)。好ましい植物宿主としては、例えばアブラナ(Arabidopsis)、タバコ(Nicotiana tabacum)、マルバタバコ(Nicotiana rustica)及びジャガイモ(Solanum tuberosum)が挙げられる。
【0037】
本明細書に教示するようなO−結合型グリコシル化の低下した蛋白質及び糖蛋白質を産生させるためには、例えばバキュロウイルス系発現システム等の昆虫細胞培養物を使用することもできる(例えばPutlitz et al.,1990,Bio/Technology 8:651−654参照)。
【0038】
本明細書に教示するようなO−結合型グリコシル化の低下した蛋白質及び糖蛋白質を発現及び産生させるためには、一般に下等真核生物や原核生物ほど経済的に培養できないが、哺乳動物組織細胞培養物を使用することもできる(Winnacker,From Genes to Clones(VCH Publishers,NY,1987)参照)。適切な宿主としては、CHO細胞株、各種COS細胞株、HeLa細胞、好ましくはミエローマ細胞株等、又は形質転換B細胞もしくはハイブリドーマが挙げられる。これらの細胞の発現ベクターは複製起点、プロモーター、エンハンサー等の発現制御配列(Queen et al.,1986,Immunol.Rev.89:49−68)と、リボソーム結合部位、RNAスプライス部位、ポリアデニル化部位及び転写ターミネーター配列等の必要なプロセシング情報部位を含むことができる。発現制御配列は免疫グロブリン遺伝子、SV40、アデノウイルス、ウシパピローマウイルス、サイトメガロウイルス等に由来するプロモーターである。一般に、発現ベクターにはneoR発現カセット等の選択マーカーが含まれる。
【0039】
発現させようとする蛋白質をコードする核酸は細胞宿主の種類により異なる従来方法により宿主細胞に導入することができる。例えば、リン酸カルシウム処理、プロトプラスト融合、自然交配、リポフェクション、バイオリスティクス法、ウイルス形質導入又はエレクトロポレーションを細胞宿主に使用することができる。植物細胞及び組織にはタングステン粒子バリスイック遺伝子導入が好ましい(一般に、Maniatis et al.,Molecular Cloning:A Laboratory Manual(Cold Spring Harbor Press,1982)参照)。
【0040】
発現後、硫安沈殿、アフィニティーカラム、カラムクロマトグラフィー、ゲル電気泳動等を含む当分野の標準手順によりO−結合型グリコシル化の低下した蛋白質又は糖蛋白質を精製することができる(一般にScopes,R.,Protein Purification(Springer−Verlag,N.Y.,1982)参照)。医薬用には、均質度が少なくとも約90〜95%の実質的に純粋な糖蛋白質が好ましく、98〜99%又はそれ以上の均質度が最も好ましい。必要に応じて部分的又は均質まで精製後、(体外を含む)治療又はアッセイ法、免疫蛍光染色等の開発及び実施に蛋白質を使用することができる。(一般に、Immunological Methods,Vols.I and II(Lefkovits and Pernis,eds.,Academic Press,NY,1979 and 1981参照))。
【0041】
従って、主要種としてN−グリカン構造を含み、Pmt介在性O−結合型グリコシル化阻害剤又は2個以上のマンノース残基をグリカン構造から除去することが可能なα−1,2−マンノシダーゼ又はその両者の存在下でインキュベートしてしない宿主細胞で産生された糖蛋白質の組成物に比較してO−結合型グリコシル化の低下した糖蛋白質組成物も提供する。特定側面において、糖蛋白質組成物はMan5GlcNAc2、Man3GlcNAc2、GlcNAcMan5GlcNAc2、GlcNAcMan3GlcNAc2、GlcNAc2Man3GlcNAc2、GalGlcNAcMan5GlcNAc2、Gal(GlcNAc)2Man5GlcNAc2、(GalGlcNAc)2Man5GlcNAc2、NANAGalGlcNAcMan3GlcNAc2、NANA2Gal2GlcNAcMan3GlcNAc2、及びGalGlcNAcMan3GlcNAc2グリコフォームから構成される群から選択される主要なN−グリカン構造をもつ糖蛋白質を含む。
【0042】
医薬組成物
活性治療剤としての糖蛋白質と医薬的に許容可能な他の各種成分を含有する医薬組成物にO−結合型グリコシル化の低下した蛋白質及び糖蛋白質を配合することができる(Remington’s Pharmaceutical Science(15th ed.,Mack Publishing Company,Easton,Pennsylvania,1980)参照)。好ましい剤形は所期投与方法と治療用途により異なる。組成物には目的の製剤に応じて動物又はヒト投与用医薬組成物を製剤化するために一般に使用される賦形剤として定義される医薬的に許容可能な非毒性キャリヤー又は希釈剤を更に添加することができる。希釈剤は組成物の生物活性に影響を与えないように選択される。このような希釈剤の例は蒸留水、リン酸緩衝生理食塩水、リンゲル液、ブドウ糖液及びハンクス液である。更に、医薬組成物又は製剤には他のキャリヤー、アジュバント、又は非毒性非治療用非免疫原性安定剤等も加えることができる。
【0043】
非経口投与用医薬組成物は無菌で実質的に等張且つパイロジェンフリーであり、FDA又は同様の機関のGMPに従って製造される。水、油類、食塩水、グリセロール又はエタノール等の滅菌液とすることができる医薬キャリヤーと共に生理的に許容可能な希釈剤に薬剤を溶解又は懸濁した注射剤として糖蛋白質を投与することができる。更に、湿潤剤又は乳化剤、界面活性剤、pH緩衝物質等の助剤を組成物に加えることができる。医薬組成物の他の成分は石油、動物、植物又は合成由来成分であり、例えば落花生油、大豆油及び鉱物油が挙げられる。一般に、プロピレングリコールやポリエチレングリコール等のグリコール類が特に注射溶液用の好ましい液体キャリヤーである。活性成分の持続放出を可能にするように製剤化することができるデポー注射剤やインプラント製剤として糖蛋白質を投与することもできる。一般に、組成物は液体溶液又は懸濁液としての注射剤として製造され、注射前に液体賦形剤に溶解又は懸濁するのに適した固体製剤を製造することもできる。上記のようなアジュバント効果を高めるために、製剤をポリラクチド、ポリグリコリド又はコポリマー等のリポソーム又は微粒子で乳化又はカプセル化することもできる(Langer,Science 249,1527(1990)及びHanes,Advanced Drug Delivery Reviews 28,97−119(1997)参照)。
【0044】
「又はその塩」なる用語は無機又は有機塩基を含む許容可能な塩基から製造される塩を意味する。無機塩基から誘導される塩としてはアルミニウム、アンモニウム、カルシウム、銅、三価鉄、二価鉄、リチウム、マグネシウム、三価マンガン、二価マンガン、カリウム、ナトリウム、亜鉛等の塩が挙げられる。固体形態の塩は2種類以上の結晶構造で存在してもよいし、水和物形態でもよい。塩基から誘導される塩としては第一級、第二級及び第三級アミン、置換アミン(天然置換アミンを含む)、環状アミン、並びに塩基性イオン交換樹脂(例えばアルギニン、ベタイン、カフェイン、コリン、N,N’−ジベンジルエチレンジアミン、ジエチルアミン、2−ジエチルアミノエタノール、2−ジメチルアミノエタノール、エタノールアミン、エチレンジアミン、N−エチルモルホリン、N−エチルピペリジン、グルカミン、グルコサミン、ヒスチジン、ヒドラバミン、イソプロピルアミン、リジン、メチルグルカミン、モルホリン、ピペラジン、ピペリジン、ポリアミン樹脂、プロカイン、プリン、テオブロミン、トリエチルアミン、トリメチルアミン、トリプロピルアミン、トロメタミン等)の塩が挙げられる。
【0045】
本発明の組成物は1個以上の不斉中心を含む場合があり、従って、ラセミ体及びラセミ混合物、単一エナンチオマー、ジアステレオマー混合物及び個々のジアステレオマーとして存在することができる。本発明はこれらの化合物のこのような全異性体を含むものとする。
【0046】
本明細書に特に定義しない限り、本発明に関して使用する全科学技術用語及び術語は当業者に通常理解されている通りの意味である。更に、内容からそうでないことが必要とされる場合を除き、単数形の用語は複数形を含み、複数形の用語は単数形を含む。一般に、本明細書に記載する生化学、酵素学、分子及び細胞生物学、微生物学、遺伝学並びに蛋白質及び核酸化学とハイブリダイゼーションに関して使用する術語とその技術は当分野で周知であり、広く使用されているものである。特に指定しない限り、本発明の方法及び技術は本明細書の随所に引用及び記載する各種一般文献及び特殊文献に記載されているような当分野で周知の慣用方法に従って一般に実施される。例えばSambrook et al.Molecular Cloning:A Laboratory Manual,2d ed.,Cold Spring Harbor Laboratory Press,Cold Spring Harbor,N.Y.(1989);Ausubel et al.,Current Protocols in Molecular Biology,Greene Publishing Associates(1992及び2002補遺);Harlow and Lane,Antibodies:A Laboratory Manual,Cold Spring Harbor Laboratory Press,Cold Spring Harbor,N.Y.(1990);Taylor and Drickamer,Introduction to Glycobiology,Oxford Univ.Press(2003);Worthington Enzyme Manual,Worthington Biochemical Corp.,Freehold,NJ;Handbook of Biochemistry:Section A Proteins,Vol I,CRC Press(1976);Handbook of Biochemistry:Section A Proteins,Vol II,CRC Press(1976);Essentials of Glycobiology,Cold Spring Harbor Laboratory Press(1999)参照。
【0047】
以下、実施例により本発明の各種Pmt阻害剤の製造方法について記載する。
【実施例1】
【0048】
【化2】
【0049】
ステップA
【0050】
【化3】
【0051】
1−1(5.90g,25mmol)のTHF(200mL)溶液にNaH(2.0g,50mmol)を少量ずつ加えた。得られた混合物を0℃で30分間撹拌した。次にNBS(4.86g,27.5mmol)を加え、15分間撹拌した。白色固体を濾過し、濾液を濃縮して残渣を得、CHCl3に溶解し、Na2SO4で乾燥した。溶媒を蒸発させて1−2(5g,収率63%)を得、それ以上精製せずに次段階で使用した。
【0052】
ステップB
【0053】
【化4】
【0054】
1−2(1.8g,6mmol)と1−3(1.3g,5mmol)の無水DMF(5mL)溶液にNaH(2.4gg,10mmol)を加えた後、室温で一晩撹拌した。得られた混合物を水とEtOAに分配した。有機層を合わせてブラインで洗浄し、硫酸ナトリウムで乾燥し、濃縮した。残渣を分取TLCにより精製し、1−4(800mg,収率40%)を得た。1H−NMR(400MHz,CDCl3)δ9.66(s,1H),7.74〜7.71(m,2H),7.49〜7.46(m,1H),7.37〜7.33(m,5H),7.20(d,J=2.02Hz,1H),6.97〜6.92(m,3H),4.28〜4.19(m,6H),3.16(t,J=6.6Hz,2H),1.11(m,6H)。
【0055】
ステップC
【0056】
【化5】
【0057】
LAH(380mg,10mmol)のTHF(50mL)溶液に0℃で1−4(1.3g,2.6mmol)を加え、得られた混合物を室温で30分間撹拌した。混合物を希HCl水溶液で注意深く処理した後、水とEtOAに分配した。有機層をブラインで洗浄し、硫酸ナトリウムで乾燥し、濃縮した。残渣を分取TLCにより精製し、1−5(260mg,収率20%)を得た。
【0058】
ステップD
【0059】
【化6】
【0060】
1−5(300mg,0.54mmol)の2,2−ジメトキシプロパン(1mL)溶液に触媒量のTsOH・H2O(〜10mg)を加えた後、得られた混合物を室温で1時間撹拌した。混合物を分取TLCにより直接精製し、1−6(100mg,収率28%)を得た。1H−NMR(400MHz,MeOD)δppm 7.52(d,J=8.05Hz,2H),7.40〜7.15(m,5H),6.90〜6.85(m,2H),6.80〜6.75(m,2H),4.25(s,2H),4.23(m,6H),3.0(m,2H),1.48(t,6H)。
【0061】
ステップE
【0062】
【化7】
【0063】
1−6(100mg,0.22mmol)の無水DCM(25mL)溶液にMnO2(174mg,2mmol)を一度に加え、得られた混合物を約2時間加熱還流した。室温まで冷却後、混合物を濾過し、濾液を濃縮し、残渣を分取TLCにより精製し、1−7(60mg,収率60%)を得た。1H−NMR(400MHz,MeOD)δppm 9.5(s,1H),7.52(d,J=7.2Hz,1H),7.48(d,J=2.8Hz,2H),7.30(m,3H),7.25(m,6H),7.0(t,J=8.6Hz,2H),6.89(m,J=8.6Hz,2H),6.76(d,J=8Hz,1H),4.23(m,6H),3.0(m,2H),1.48(t,6H)。
【0064】
ステップF
【0065】
【化8】
【0066】
1−7(80mg,0.17mmol)のTHF溶液に6N HClを加え、得られた混合物を室温で2時間撹拌した。溶媒を減圧除去し、粗生成物1−8(60mg,収率82%)を得、それ以上精製せずに次段階に供した。
【0067】
ステップG
【0068】
【化9】
【0069】
1−8(60mg,0.146mmol)のDMF(6mL)溶液に℃でNaH(24mg,1mmol)を加え、得られた混合物を同一温度で約30分間撹拌後、CH3I(0.2mL)を加えた。混合物を同一温度で更に1時間撹拌した。次に混合物をH2OとEtOAcに分配した。有機層を合わせてブラインで洗浄し、硫酸ナトリウムで乾燥し、濃縮した。残渣を分取TLCにより精製し、1−9(40mg,収率63.0%)を得た。1H−NMR(400MHz,MeOD)δppm 9.6(s,1H),7.52(d,J=8.08Hz,2H),7.43(m,1H),7.36〜7.30(m,5H),7.16(d,J=2.02Hz,1H),7.0(t,J=8.6Hz,2H),6.89(m,J=8.6Hz,2H),4.23(t,J=6.56Hz,2H),3.95〜3.85(m,4H),3.30(s,6H),3.13(dd,J1=6.26Hz,J2=6.26Hz,2H)。
【0070】
ステップH
【0071】
【化10】
【0072】
1−9(40mg,0.09mmol)、1−10(19mg,0.09mmol)及びNH4OAc(70mg,0.9mmol)をトルエン(10mL)に加えた混合物を窒素下で約3時間加熱還流した。室温まで冷却後、得られた混合物をpH=4〜5まで希HCl水溶液により処理した後、H2OとEtOAcに分配した。有機層を合わせてブラインで洗浄し、Na2SO4で乾燥し、濃縮した。残渣を分取HPLCにより精製し、実施例1(30mg,収率50%)を黄色い固体として得た。1H−NMR(400MHz,MeOD)δppm 7.46〜7.53(m,3H),7.31〜7.40(m,5H),7.15〜7.18(m,1H),6.92〜7.06(m,4H),4.71(s,2H),4.25〜4.30(m,2H),3.85〜3.94(m,4H),3.26〜3.29(m,6H),3.10〜3.13(dd,J=6.26Hz,J=6.26Hz,2H)。MS m/z 612(M+1)+。
【実施例2】
【0073】
【化11】
【0074】
ステップA
【0075】
【化12】
【0076】
化合物2−1(150mg,0.58mmol,1.0当量)、化合物2−2(214mg,0.87mmol,1.5当量)のDMF(10mL)溶液にCs2CO3(161mg,0.49mmol,0.85当量)を加えた。次に混合物を室温で一晩撹拌後、80℃まで1.5時間加熱した。得られた混合物を次にH2OとEtOAcに分配した。有機層を合わせてブラインで洗浄し、Na2SO4で乾燥し、濃縮し、分取TLCにより精製し、2−3(122mg,収率49.4%)を得た。
【0077】
ステップB
【0078】
【化13】
【0079】
化合物2−3(122mg,0.29mmol,1.0当量)、2−4(57mg,0.30mmol,1.05当量)及びNH4OAc(223mg.2.90mmol,10.0当量)をトルエン(15mL)に加えた混合物を窒素下に約3時間加熱還流した。得られた混合物を10%塩酸で処理してpH=4〜5とした後、H2OとEtOAcに分配した。有機層を合わせてブラインで洗浄し、Na2SO4で乾燥し、濃縮し、分取TLCにより精製し、実施例2(52mg,収率29.9%)を得た。1H−NMR(400MHz,MeOD)δ7.41−7.47(m,3H),7.19−7.35(m,10H),6.99−7.07(m,3H),6.87−6.92(m,2H),6.22(s,1H),4.75(s,2H),4.24−4.29(m,2H),3.72−3.13(m,2H)。MS m/z 600(M+l)+。
【実施例3】
【0080】
【化14】
【0081】
ステップA
【0082】
【化15】
【0083】
3−1(1g,6.37mmol)の無水THF(20mL)溶液に−70℃でBuLi(3mL,7.68mmol)を滴下した後、混合物を室温で30分間撹拌した。再び−70℃まで冷却後、シクロヘキサンカルボアルデヒド(0.72g,6.43mmol)の無水THF(5mL)溶液を滴下した。30分間撹拌後、飽和NH4Cl水溶液を混合物に加え、得られた混合物をEtOAcで抽出した。有機層を合わせてNa2SO4で乾燥し、濃縮し、3−2(1.52g,14.3%)を粗製油状物として得た。
【0084】
ステップB
【0085】
【化16】
【0086】
3−2(600mg,3.1mmol)を無水DCM(15mL)に加えた混合物に0℃でSOCl2(2mL)を滴下した。次に得られた混合物を2時間室温で撹拌した。混合物を減圧濃縮後、EtOAcで希釈した。有機層を飽和NaHCO3水溶液、ブラインで洗浄し、Na2SO4で乾燥後、濃縮し、3−3(500mg,粗生成物)を得た。
【0087】
ステップC
【0088】
【化17】
【0089】
3−3(450mg,粗生成物)と3−4(375mg,1.44mmol)のDMF(10mL)溶液にCs2CO3(399mg,1.22mmol)を一度に加えた後、混合物を110℃まで18時間加熱した。混合物をH2Oで希釈し、EtOAcで抽出した。有機層を合わせてNa2SO4で乾燥し、濃縮した。残渣をTLCにより精製し、3−5(130mg)を油状物として得、次段階でそのまま使用した。
【0090】
ステップD
【0091】
【化18】
【0092】
3−5(130mg,0.3mmol)と3−6(60mg,0.31mmol)をトルエン(15mL)に加えた混合物にNH4OAc(180mg,2.34mmol)を一度に加えた後、混合物を3.5時間加熱還流した。次に混合物を室温まで冷却し、10% HCl水溶液を加えてpH=5とした。混合物をEtOAcで抽出した。有機層を合わせてNa2SO4で乾燥し、濃縮し、黄色い油状物を得た。残渣を分取TLCにより精製し、実施例3(150mg,収率82.4%)を得た。1H−NMR(300MHz,MeOD)δ7.60(s,1H),7.19〜7.49(m,11H),6.88(s,1H),6.81(s,1H),5.05(s,1H),4.65(s,2H),4.25〜4.39(m,2H),3.15(m,2H),1.60〜1.98(m,5H),1.00〜1.46(m,6H)。MS m/z:607(M+1)+。
【実施例4】
【0093】
【化19】
【0094】
ステップA
【0095】
【化20】
【0096】
ナトリウム(8.5mg,0.37mmol)を40℃で2−メチルプロパン−1−オール(1.3g,17.5mmol)に溶解した後、化合物4−1(2g,16.7mmol)を50℃で30分間かけて滴下した。次に反応混合物を70℃まで加熱し、一晩撹拌した。得られた混合物をH2OとEtOAcに分配した。有機層をブラインで洗浄し、硫酸ナトリウムで乾燥し、濃縮し、4−2(1.0g,粗生成物)を得、次段階でそのまま使用した。
【0097】
ステップB
【0098】
【化21】
【0099】
4−2(1.0g,粗生成物)と4−3(400mg,1.64mmol,1.0当量)とPPh3(516mg,1.96mmol,1.2当量)のTHF(20mL)溶液に0℃でDIAD(406mg,1.96mmol,1.2当量)を滴下した。得られた混合物を室温で一晩撹拌後、H2OとEtOAcに分配した。有機層をブラインで洗浄し、Na2SO4で乾燥し、濃縮し、分取TLCにより精製し、4(90mg,収率12.0%)を得た。1H−NMR(400MHz,MeOD)δ9.72(s,1H),7.28−7.40(m,9H),6.98−7.05(t,J=8.78Hz,2H),6.89−6.92(d,J=8.28Hz,1H),5.42−5.46(m,1H),4.22−4.32(m,2H),3.86−3.92(m,1H),3.68−3.74(m,1H),3.24−3.34(m,2H),3.12−3.18(m,2H),1.82−1.90(m,1H),0.84−0.88(m,6H)。
【0100】
ステップC
【0101】
【化22】
【0102】
実施例4の製造は実施例3の製造と同様である。1H−NMR(400MHz,MeOD)δ7.49(s,1H),7.24−7.40(m,7H),6.98−7.05(m,4H),6.90−6.91(m,1H),5.37−5.40(m,1H),4.75(s,2H),4.21−4.32(m,2H),3.83−3.88(m,1H),3.69−3.72(m,1H),3.33−3.35(m,2H),3.09−3.13(t,J=6.57Hz,2H),1.79−1.87(m,1H),0.86−0.88(m,6H)。
【実施例5】
【0103】
本実施例はヒト抗Her2抗体の重鎖(Hc)及び軽鎖(Lc)をコードする発現ベクターで形質転換し、本明細書に記載する新規Pmt阻害剤で処理したPichia pastorisがO−グリコシル化の低下した糖蛋白質を産生したことを示す。
【0104】
発現/組込みプラスミドベクターpGLY2988は抗Her2の重鎖(Hc)及び軽鎖(Lc)をコードするメタノール誘導型Pichia pastoris AOX1プロモーターの制御下におかれた発現カセットを含む。α−MATプレシグナルペプチド(配列番号1及び2)のN末端に融合した抗Her2 Hc及びLcをGene Art AGにより合成した。ユニークな5’EcoR1及び3’Fse1部位をもつように各々を合成した。抗Her2 Hcのヌクレオチド配列とアミノ酸配列を夫々配列番号3及び4に示す。抗Her2 Lcのヌクレオチド配列とアミノ酸配列を夫々配列番号5及び6に示す。Pichia pastoris TRP2標的核酸とゼオシン耐性マーカーを含み、AOX1プロモーターとSaccharomyces cerevisiae CYCターミネーターの制御下におかれた発現カセットを作製できる発現プラスミドベクターpGLY2198に、ユニークな5’EcoR1及び3’Fse1部位を使用して、α−MATプレシグナルペプチドに融合したHc及びLc蛋白質をコードする両方の核酸フラグメントを夫々別々にサブクローニングし、夫々プラスミドベクターpGLY2987及びpGLY2338を形成した。次にBamHIとNotIで消化することによりLc発現カセットをプラスミドベクターpGLY2338から切出し、BamH1とNot1で消化しておいたプラスミドベクターpGLY2987にサブクローニングし、最終発現プラスミドベクターpGLY2988を作製した。
【0105】
抗Her2発現株yGLY4280を以下のように構築した。TRP2標的領域を切断する制限酵素Spe1で消化した5μgのpGLY2988を使用して株yGLY22−1を形質転換した。従来記載されている方法(Nett and Gerngross,Yeast 20:1279(2003);Choi et al.,PNAS USA 100:5022(2003);Hamilton et al.,Science 301:1244(2003))を使用して株yGLY22−1(och1Δ::lacZbmt2Δ::lacZ/KlMNN2−2/mnn4L1Δ::lacZ/MmSLC35A3pno1Δmnn4Δ::lacZmet16Δ::lacZ)を構築した。
【0106】
yGLY22−1の形質転換は原則的に以下のように実施した。ODが約0.2〜6になるまでYGLY22−1をYPD培地(酵母エキス(1%)、ペプトン(2%)、デキストロース(2%))50mL中で一晩培養した。氷上で30分間インキュベーション後、細胞を2500〜3000rpmで5分間遠心することによりペレット化した。培地を除去し、細胞を氷冷滅菌1Mソルビトールで3回洗浄後、氷冷滅菌1Mソルビトール0.5mlに再懸濁した。直鎖化DNA10μL(10ug)と細胞懸濁液100μLをエレクトロポレーションキュベットに加え、氷上で5分間インキュベートした。プリセットされたPichia pastorisプロトコル(2kV,25μF,200Ω)に従ってBio−Rad GenePulser Xcellでエレクトロポレーションを行った直後にYPDS回復培地(YPD培地+1Mソルビトール)1mLを加えた。形質転換細胞を室温(26℃)で4時間〜一晩回復させた後、細胞を選択培地に撒いた。ゼオシンを加えた培地で選択後、形質転換細胞を小規模発現分析によりスクリーニングし、抗Her2発現を検出した。高レベル抗Her2発現に基づいて株yGLY4280を選択した。
【0107】
1%酵母エキス、2%ペプトン、100mMリン酸カリウム緩衝液(pH6.0),1.34%酵母窒素塩基、4×10−5%ビオチン、及び1%グリセロールから構成される緩衝グリセロール複合培地(BMGY)を使用して振盪フラスコで24℃にて株yGLY4280の抗Her2蛋白質発現を実施した。蛋白質発現誘導培地はBMGYのグリセロールを1%メタノールに代えた緩衝メタノール複合培地(BMMY)とした。誘導培地添加時に100%メタノール中のPmt阻害剤を増殖培地に終濃度0.15ug/mLまで加えた。これはPmt阻害剤で処理しない場合に観測される値の約50%までO−グリコシル化を低下させるために十分な中間用量であり、従って各種阻害剤の効力の比較を可能にする。誘導培地で24時間更に増殖後、培養液を回収し、2,000rpmで5分間遠心し、上清から細胞を除去した。
【0108】
Dionex−HPLC(HPAEC−PAD)を使用してO−グリカン測定を以下のように実施した。O−グリコシル化低下を測定するために、プロテインAクロマトグラフィー(Li et al.Nat.Biotechnol.24(2):210−5(2006))を使用して蛋白質を増殖培地から精製し、アルカリ除去(β除去)によりO−グリカンを蛋白質から遊離及び分離させた(Harvey,Mass Spectrometry Reviews 18:349−451(1999))。この方法は更に、遊離したO−グリカン(オリゴマンノース又はマンノース)の新たに形成された還元末端をマンニトールに還元する。従って、各O−グリカンのユニークな指標としてマンニトール基を利用することができる。β除去にはPBS緩衝液100μL容量に含まれる0.5nmol以上の蛋白質が必要であった。サンプルを水素化ホウ素アルカリ試薬25μLで処理し、50℃で16時間インキュベートした。アラビトール内部標準約20uLを加えた後、氷酢酸10μLを加えた。次にSEPABEADSとAG 50W−X8樹脂の両方を充填したMilliporeフィルターでサンプルを遠心し、水洗した。洗浄液を含むサンプルをプラスチックオートサンプラーに移し、遠心蒸発器で蒸発乾涸した。1% AcOH/MeOH 150μLをサンプルに加え、サンプルを遠心蒸発器で蒸発乾涸した。この最後の段階を更に5回繰返した。水200μLを加え、高pH陰イオン交換クロマトグラフィーとパルス式電気化学検出法を組合せたDionex HPLC(HPAEC−PAD)によりサンプル100μLを分析した。マンニトール回収量に基づいて平均O−グリカン率を求めた。
【0109】
結果を表1に要約する通り、4種類の新規Pmt阻害剤である実施例1〜4はOrchard et al.(Bioorgan & Med Chem Letters(2004)14:3975−3978)及びEP1313471B1を含む同一著者による特許公開とBobrowiczらの米国出願公開第2007061631号に記載されているようなPmt阻害剤であるPmti−3で観測されるレベルよりも低レベルまでO−グリコシル化を低下させることが明らかである。Pmti−3は以下の化学構造をもつ。
【0110】
【化23】
【0111】
【表1】
【0112】
図1は新規Pmt阻害剤がPichiaにより産生される組換え蛋白質のO−グリコシル化を有効に低下させることを実証する画像であり、抗Her2重鎖(Hc)のO−グリコシル化に及ぼすPmt阻害剤の増量の効果を示す。ウェル当たりBMGY培地0.5mlを加えた96穴ディープウェルプレート(Qiagen,Valencia,CA)に株yGLY4280を接種した。激しく振盪しながら24時間増殖後に96ウェルプレートを2,000rpmで5分間遠心し、細胞をペレット化した。培地を除去し、BMMY培地0.5mLによる洗浄段階後、Pmt阻害剤を第1行から最終行まで(11穴)2倍ずつ希釈したBMMY培地0.2mLに細胞を再懸濁した。ウェル#1には阻害剤5ug/mLを加え、ウェル#2には2.5ug/mLを加え、以下同様にしてウェル#10に0.009ng/mLを加え、#11には阻害剤を加えなかった。激しく振盪しながら更に24時間増殖後にプレートを2,000rpmで5分間遠心し、細胞をペレット化し、清澄化した上清をウェスタンブロット分析し、抗Her2発現を検出した。ウェスタンブロットは以下のように実施した。Laemmli,U.K.(1970)Nature 227,680−685に従って還元ポリアクリルアミドゲル電気泳動(SDS−PAGE)により上清7μLを分離後、ニトロセルロースメンブレン(Schleicher & Schuell,現Whatman,Inc.,Florham Park,NJ)にエレクトロブロットした。ペルオキシダーゼ標識抗ヒトHc及びLc抗体(Calbiochem/EMD Biosciences,La Jolla,CA)を使用してウェスタンブロットで抗Her2抗体鎖を検出し、ImmunoPure金属増感型DAB基質キット(Pierce Biotechnology,Rockford,IL)を使用して展開した。図1は実施例4からの新規Pmt阻害剤(EX.4,図1A)をOrchard et al.からのPmti−3(図1B)と対照としての不活性化合物(図1C)に対比試験したこのような分析の1例の結果を示す。図1A及びBに示すように、実施例4とPmti−3は0.018ug/mLまでの低濃度で抗Her2 HcのO−グリコシル化を有効に低下させた。これに対して不活性対照化合物(図1C)はHc O−グリコシル化の低下を示さなかった。実施例1、2及び3に示す阻害剤でも同様の結果が得られた。これらの結果をまとめると、実施例1〜4に示す新規Pmt阻害剤は真菌O−グリコシル化の有効な阻害剤であることが分かる。
【0113】
本明細書では例示態様について本発明を説明するが、当然のことながら本発明はこれらの態様に限定されない。本明細書の教示を受けた当業者は本発明の範囲に含まれる他の変形及び態様も認識しよう。従って、本発明は以下の特許請求の範囲のみにより限定される。
【背景技術】
【0001】
(発明の背景)
糖蛋白質は触媒作用、シグナル伝達、細胞間コミュニケーション、並びに分子認識及び会合等のヒト及び他の哺乳動物における多くの必須機能に介在する。糖蛋白質は真核生物において非サイトゾル蛋白質の大部分を構成する(Lis and Sharon,1993,Eur.J.Biochem.218:1−27)。多数の糖蛋白質が治療目的に利用されており、この二十年間に天然に存在する糖蛋白質の組換え体はバイオテクノロジー産業の主要な部分になっている。治療薬として使用されている組換えグリコシル化蛋白質の例としては、エリスロポエチン(EPO)、治療用モノクローナル抗体(mAb)、組織プラスミノーゲンアクチベーター(tPA)、インターフェロンβ(IFN−β)、顆粒球−マクロファージコロニー刺激因子(GM−CSF)、及びヒト絨毛性ゴナドトロピン(hCH)が挙げられる(Cumming et al.,1991,Glycobiology 1:115−130)。潜在的予防薬及び治療薬として生産された組換え蛋白質が臨床に近付くにつれ、組換え生産された糖蛋白質のグリコシル化パターンの変動は科学界で最近関心の高い話題となっている。
【0002】
一般に、糖蛋白質オリゴ糖のグリコシル化構造はこれらを生産するために使用される細胞の宿主種により異なる。非ヒト宿主細胞で生産される治療用蛋白質はヒトに免疫原性応答を誘発し得る非ヒトグリコシル化を含む可能性が高く、例えば酵母における高マンノシル化(Ballou,1990,Methods Enzymol.185:440−470);植物におけるα(1,3)−フコース及びβ(1,2)−キシロース(Cabanes−Macheteau et al.,1999.Glycobiology,9:365−372);チャイニーズハムスター卵巣細胞におけるN−グリコリルノイラミン酸(Noguchi et al.,1995.J.Biochem.117:5−62);並びにマウスにおけるGalα−1,3Galグリコシル化(Borrebaeck,et al.,1993,Immun.Today,14:477−479)が報告されている。動物細胞中で蛋白質と結合した糖鎖としては、蛋白質中のアスパラギン(Asn)残基と結合したN−グリコシド結合型糖鎖(別称N−グリカン;又はN−結合型グリコシル化)と、蛋白質中のセリン(Ser)又はスレオニン(Thr)残基と結合したO−グリコシド結合型糖鎖(別称O−グリカン;又はO−結合型グリコシル化)が挙げられる。
【0003】
非ヒト哺乳動物細胞により産生される糖蛋白質のオリゴ糖構造はヒト糖蛋白質のオリゴ糖構造と密接に関連する傾向があるため、大半の商業用糖蛋白質は哺乳動物細胞で生産されている。しかし、哺乳動物細胞には蛋白質産生用宿主細胞としていくつかの重大な欠点がある。コストがかかるという点以外に、哺乳動物細胞における蛋白質の生産方法では不均質なグリコフォーム集団を生じ、体積当たりの力価が低く、安定な細胞株を作製するためには持続的なウイルス閉じ込めと相当の時間が同時に必要となる。
【0004】
蛋白質上の特定グリコフォームがその薬物動態、薬力学、受容体相互作用及び組織特異的標的特性を含む蛋白質の性質に大きな影響を与える可能性があることはよく知られている(Graddis et al.,2002.Curr Pharm Biotechnol.3:285−297)。例えば、Igの各種グリコシル化パターンは各種生物学的性質に相関することが示されている(Jefferis and Lund,1997,Antibody Eng.Chem.Immunol,65:111−128;Wright and Morrison,1997,Trends Biotechnol.,15:26−32)。更に、糖蛋白質のガラクトシル化は細胞培養条件により変動し得るため、糖蛋白質組成によっては糖蛋白質上の特定ガラクトースパターンに応じて免疫原性になる場合があることも示されている(Patel et al.,1992.Biochem J.285:839−845)。しかし、どの特定グリコフォームが望ましい生物学的機能に寄与するのか分からないため、糖蛋白質上の特定グリコフォームを増加できるようにすることが非常に望ましい。グリコフォーム毎に異なる生物学性質に相関しているため、特定グリコフォームをもつ糖蛋白質の増加能を使用すれば、糖蛋白質の特定グリコフォームと特定の生物学的機能の関係を解明することができる。また、特定グリコフォームをもつ糖蛋白質を増加できるならば、特定の特異性をもつ治療用糖蛋白質を製造することも可能になる。従って、特定グリコフォームを増加した糖蛋白質組成物の製造が非常に望ましい。
【0005】
N−結合型グリコシル化の経路は多くの分析の対象となっているが、O−結合型グリコシル化のプロセスと機能はよく分かっていない。しかし、N−結合型グリコシル化とは対照的に、O−グリコシル化はシスゴルジで生じる翻訳後イベントであることが知られている(Varki,1993,Glycobiol.,3:97−130)。N−結合型グリコシル化と同様のO−結合型グリコシル化のコンセンサスアクセプター配列は存在しないと思われるが、数種の糖蛋白質の多数のO−結合型グリコシル化部位の周囲のアミノ酸配列を比較すると、グリコシル化残基に対して−1及び+3位のプロリン残基の頻度が高く、セリン、スレオニン及びアラニン残基が著しく増加していることが分かる(Wilson et al.,1991,Biochem.J.,275:529−534)。糖蛋白質におけるセリン残基とスレオニン残基の部分もO−グリコシル化の潜在部位であると思われる。
【0006】
O−結合型グリコシル化で役割を果たす1つの遺伝子ファミリーはDol−P−Man:蛋白質(Ser/Thr)マンノシルトランスフェラーゼ(Pmt)をコードする遺伝子である。これらの高度に保存された遺伝子はヒト、齧歯類、昆虫等の高等真核生物と、真菌類等の下等真核生物の両者で同定されている。Saccharomyces cerevisiaeやPichia pastoris等の酵母はPmtホモログをコードする7個までのPMT遺伝子をコードする(Wilier et al.Curr.Opin.Struct.Biol.2003 Oct;13(5):621−30に概説)。酵母において、O−結合型グリコシル化は7個のO−マンノシルトランスフェラーゼ遺伝子の1個によりドリコールリン酸マンノースに由来する初期のマンノースが小胞体の新生糖蛋白質のセリン又はスレオニン残基に付加されることにより開始する。酵母にはPmtホモログをコードする7個のPMT遺伝子が存在すると思われるが、酵母における分泌型真菌及び異種蛋白質のO−マンノシル化はヘテロダイマーとして機能すると思われるPmt1とPmt2をコードする遺伝子に主に依存する。PMT1及びPMT2とその蛋白質産物である夫々Pmt1及びPmt2は種間で高度に保存されていると思われる。
【0007】
TannerらはSaccharomyces cerevisiaeのPMT1及びPMT2遺伝子と、O−結合型グリコシル化の低下した組換え蛋白質が産生されるようにPMT遺伝子の1個以上を遺伝子改変した真菌細胞を使用してO−結合型グリコシル化の低下した組換え蛋白質を作製する方法を米国特許第5,714,377号に記載している。
【0008】
Ngらはアンチセンスもしくは同時抑圧の使用、又はO−結合型グリコシル化に関連する遺伝子、特にPMT遺伝子の1個以上を機能低下突然変異させた酵母宿主株の構築によるOグリコシル化の阻害を米国特許出願公開第20020068325号に開示している。
【0009】
UDP−N−アセチル−α−D−ガラクトサミン:ポリペプチドN−アセチルガラクトサミニルトランスフェラーゼ(GalNAcトランスフェラーゼ)は高等真核生物に認められるムチン型O−結合型グリコシル化に関与している。これらの酵素は蛋白質中の特定のセリン及びスレオニンアミノ酸のヒドロキシ基にN−アミノ酸ガラクトサミンを付加することによりこれらのアミノ酸のO−グリコシル化を開始し、その後、マンノース残基を段階的に付加することができる。ClausenらはUDP−N−アセチル−α−D−ガラクトサミン:ポリペプチドN−アセチルガラクトサミニルトランスフェラーゼ(GalNAcトランスフェラーゼ)をコードする核酸ファミリーを米国特許第5,871,990号及び米国特許出願公開第20050026266号に開示している。ClausenはポリペプチドGalNAcトランスフェラーゼのレクチンを選択的に阻害し、O−グリカン生合成に関与する他のグリコシルトランスフェラーゼの基質として機能させず、O−グリコシル化を阻害するために、GalNAc−β−ベンジルの使用を米国特許出願公開第20030186850号に開示している。
【0010】
O−結合型グリコシル化の阻害剤が記載されている。例えば、Orchardらは米国特許第7,105,554号にベンジリデンチアゾリジンジオンと、抗真菌剤(例えば抗カビ剤)としてのその使用について記載している。これらのベンジリデンチアゾリジンジオンはPmt1酵素を阻害し、O−結合型マンノ蛋白質の形成を防止し、真菌細胞壁の完全性を損なうことが報告されている。結末として、細胞膨潤を招き、最終的に破裂により死に至る。
【0011】
Konradらは組織又は細胞におけるO−結合型蛋白質グリコシル化を薬理的に阻害することにより糖尿病を治療又は予防する方法を米国特許出願公開第20020128235号に開示している。この方法はO−結合型N−アセチルグルコサミントランスフェラーゼと結合することによりO−結合型グリコシル化を阻害する(Z)−1−[N−(3−アンモニオプロピル)−N−(n−プロピル)アミノ]ジアゼニウム−1,2−ジオール酸又はその誘導体による糖尿病個体の治療に依存している。
【0012】
Kojimaらは白血球又は腫瘍細胞によるSLex又はSLeaの発現を阻止し、それにより内皮細胞や血小板へのこれらの細胞の接着を阻害するために、O−α−GalNAcの蓄積に繋がるO−グリコシル化の伸長を阻害するベンジル−α−N−アセチルガラクトサミン等の化合物を使用してO−グリコシル化を阻害するための治療用組成物を米国特許第5,268,364号に開示している。
【0013】
Boimeらはグリコシル化部位のアミノ酸配列を改変することによりO−結合型グリコシル化を改変したホルモン変異体を米国特許第6,103,501号に開示している。
【先行技術文献】
【特許文献】
【0014】
【特許文献1】米国特許第5,714,377号明細書
【特許文献2】米国特許出願公開第20020068325号明細書
【特許文献3】米国特許第5,871,990号明細書
【特許文献4】米国特許出願公開第20050026266号明細書
【特許文献5】米国特許出願公開第20030186850号明細書
【特許文献6】米国特許第7,105,554号明細書
【特許文献7】米国特許出願公開第20020128235号明細書
【特許文献8】米国特許第5,268,364号明細書
【特許文献9】米国特許第6,103,501号明細書
【非特許文献】
【0015】
【非特許文献1】Lis and Sharon,1993,Eur.J.Biochem.218:1−27
【非特許文献2】Cumming et al.,1991,Glycobiology 1:115−130
【非特許文献3】Ballou,1990,Methods Enzymol.185:440−470
【非特許文献4】Cabanes−Macheteau et al.,1999.Glycobiology,9:365−372
【非特許文献5】Noguchi et al.,1995.J.Biochem.117:5−62
【非特許文献6】Borrebaeck,et al.,1993,Immun.Today,14:477−479
【非特許文献7】Graddis et al.,2002.Curr Pharm Biotechnol.3:285−297
【非特許文献8】Jefferis and Lund,1997,Antibody Eng.Chem.Immunol,65:111−128
【非特許文献9】Wright and Morrison,1997,Trends Biotechnol.,15:26−32
【非特許文献10】Patel et al.,1992.Biochem J.285:839−845
【非特許文献11】Varki,1993,Glycobiol.,3:97−130
【非特許文献12】Wilson et al.,1991,Biochem.J.,275:529−534
【非特許文献13】Wilier et al.Curr.Opin.Struct.Biol.2003 Oct;13(5):621−30
【発明の概要】
【発明が解決しようとする課題】
【0016】
本発明はO−結合型グリコシル化の低下した組換え蛋白質の生産に有用なPmt蛋白質の新規阻害剤に関する。この結果、真菌及び酵母細胞から産生される蛋白質のO−結合型グリコシル化を制御することが可能になる。
【課題を解決するための手段】
【0017】
O−結合型グリコシル化量の低下した蛋白質組成物を製造するための化合物と方法について記載する。前記方法はPmt介在性O−結合型グリコシル化の所定のベンジリデンチアゾリジンジオン阻害剤の存在下で培養した細胞中で蛋白質を生産する段階を含む。
【図面の簡単な説明】
【0018】
【図1A】実施例4として示す新規阻害剤を含むPmt阻害剤がPichia pastorisにおける分泌型組換えモノクローナル抗体のO−グリコシル化に及ぼす効果を示す。Pmtの化学的阻害剤はO−グリコシル化を用量依存的に低下させた。抗ヒトH+L抗体を使用するウェスタンブロッティングを使用し、増量しながら(左側のレーン方向に高用量)Pmt阻害剤で処理した株の増殖培地中で重鎖(Hc)と軽鎖(Lc)を検出した。移動度の最も遅いバンド(星印で示す)がPmt阻害剤処理により除去されるO−グリコシル化Hcに相当する。
【図1B】実施例4として示す新規阻害剤を含むPmt阻害剤がPichia pastorisにおける分泌型組換えモノクローナル抗体のO−グリコシル化に及ぼす効果を示す。Pmtの化学的阻害剤はO−グリコシル化を用量依存的に低下させた。抗ヒトH+L抗体を使用するウェスタンブロッティングを使用し、増量しながら(左側のレーン方向に高用量)Pmt阻害剤で処理した株の増殖培地中で重鎖(Hc)と軽鎖(Lc)を検出した。移動度の最も遅いバンド(星印で示す)がPmt阻害剤処理により除去されるO−グリコシル化Hcに相当する。
【図1C】実施例4として示す新規阻害剤を含むPmt阻害剤がPichia pastorisにおける分泌型組換えモノクローナル抗体のO−グリコシル化に及ぼす効果を示す。Pmtの化学的阻害剤はO−グリコシル化を用量依存的に低下させた。抗ヒトH+L抗体を使用するウェスタンブロッティングを使用し、増量しながら(左側のレーン方向に高用量)Pmt阻害剤で処理した株の増殖培地中で重鎖(Hc)と軽鎖(Lc)を検出した。移動度の最も遅いバンド(星印で示す)がPmt阻害剤処理により除去されるO−グリコシル化Hcに相当する。
【発明を実施するための形態】
【0019】
本発明は特定宿主細胞中でO−結合型グリコシル化を受け易い蛋白質についてこの細胞型におけるO−結合型グリコシル化量を低下させた(O−結合型グリコシル化ゼロを含む)組換え蛋白質(ポリペプチドと糖蛋白質を含む)を発現させるための新規化合物と方法を提供する。前記方法は該当蛋白質が宿主細胞中でO−結合型グリコシル化を受け易い場合に、前記細胞中における蛋白質のマンノースからセリン又はスレオニン残基への転移に関与するDol−P−Man:蛋白質(Ser/Thr)マンノシルトランスフェラーゼ(Pmt)蛋白質の1種以上の活性の阻害剤である本発明の1種以上の新規化合物の存在下に前記宿主細胞中で前記蛋白質の発現を誘導する段階を含み、場合により前記蛋白質の発現誘導時にBobrowiczらの米国出願公開第2007061631号に記載されているような1種以上のα1,2−マンノシダーゼを併用してもよい。前記阻害剤の存在下で発現される蛋白質は前記阻害剤の不在下で産生させた場合に蛋白質上に存在するようなO−結合型グリコシル化量に比較してO−結合型グリコシル化量が低下している。前記方法は、通常ではO−結合型グリカンをもつ蛋白質を産生する下等真核生物(例えば酵母)や細菌等についてO−結合型グリカン数を低下させた宿主細胞中で蛋白質のO−グリコシル化量を低下させることが望ましい治療関連蛋白質を産生させる手段となるため、特に有用である。なお、本方法は下等真核生物中でO−結合型グリコシル化の低下した蛋白質を発現させるのに特に適しているが、高等真核生物や細菌でも本方法を実施することができる。
【0020】
本発明のPmt阻害剤は以下の群:
【0021】
【化1】
又はその塩から選択される。
【0022】
本明細書に記載する方法は蛋白質がO−結合型グリコシル化を受け易い宿主細胞中でO−結合型グリコシル化の低下した蛋白質を産生させるための所定の従来技術の方法の改良である。例えば、TannerらはO−結合型グリコシル化の低下した組換え蛋白質を産生するようにPmt蛋白質をコードするPMT遺伝子の1個以上を遺伝子改変した酵母細胞等の真菌細胞を使用してO−結合型グリコシル化の低下した組換え蛋白質を作製する方法を米国特許第5,714,377号に記載している。真菌宿主細胞でPMT1、PMT2又はPMT4遺伝子を欠失させると、真菌宿主細胞でO−結合型グリコシル化の低下した組換え蛋白質の産生が可能になるが、宿主細胞増殖にはPMT遺伝子の発現が重要であり、単独でどれを欠失させても真菌宿主細胞の増殖能は悪化し、O−結合型グリコシル化量の低下した宿主細胞又は組換え蛋白質を十分な量で生産することが困難になる。PMT2と同時にPMT1又はPMT4を欠失させると、真菌宿主細胞に致命的であると思われる。従って、宿主細胞中のPMT遺伝子の遺伝子除去はO−結合型グリコシル化を低下させた組換え蛋白質を生産するためには望ましくない手段であると思われる。
【0023】
これに対して、本発明の方法で使用される宿主細胞中のPMT遺伝子は改変又は欠失されていないため、宿主細胞は細胞増殖に重要なこれらの蛋白質をO−グリコシル化することができ、この時点までPmt蛋白質の活性は阻害される。一般に、この結果として、PMT遺伝子を欠失させた場合に得られるレベルよりも高レベルまで宿主細胞を増殖させることが可能になる。更に、特定態様では、宿主細胞中の組換え蛋白質の発現は誘導性プロモーターにより制御され、組換え蛋白質の発現が誘導されるまで宿主細胞中のPmt活性は阻害されない。このため、組換え蛋白質の発現誘導とPmt阻害剤の添加前に組換え蛋白質をコードする核酸を含む大量の宿主細胞を培養生産することができる。従って、1個以上のPMT遺伝子を欠失させた培養増殖不良な宿主細胞よりも短期間にO−結合型グリコシル化を低下させた組換え蛋白質をより大量に培養生産することが可能になる。
【0024】
本明細書に記載するPmt阻害剤は実施例5に示すように効力が増加するため、Orchardら(米国特許第7,105,554号)及びBobrowiczら(米国出願公開第2007061631号)に従来記載されている阻害剤よりも改善されている。効力の増加により、真菌O−グリコシル化を許容可能なレベルまで低下させるために使用する阻害剤の量を減らすことができ、及び/又はO−グリカンを完全に除去する可能性が高まる。
【0025】
本明細書に記載する方法は主に特定のN−結合型グリカン構造をもつ糖蛋白質を産生するように遺伝子改変されているだけでなく、糖蛋白質をO−グリコシル化する宿主細胞中におけるO−結合型グリコシル化の低下した糖蛋白質の産生を容易にする。主に特定のN−結合型グリコフォームをもつ多様な糖蛋白質の製造方法が米国特許第7,029,872号と、米国出願公開第20050170452号、20050260729号、20040230042号、20050208617号、20050208617号、20040171826号、20060160179号、20060040353号及び20060211085号に開示されている。本明細書に開示する方法を使用し、主に特定のN−結合型グリカン構造をもち、O−結合型グリコシル化の低下した糖蛋白質を産生させるためには、上記特許及び特許出願に記載されている宿主細胞のいずれか1種を使用することができる。主に特定のN−結合型グリカン構造をもつ糖蛋白質を産生するように遺伝子改変した所定の宿主細胞は改変していない宿主細胞ほど良好に特定条件下で培養増殖できないことが判明した。例えば、高マンノシル化に関与する遺伝子を欠失させ、特定の哺乳動物又はヒト様N−結合型グリカン構造を産生させるために必要な他の遺伝子を付加した特定真菌及び酵母細胞は遺伝子改変していない真菌又は酵母細胞ほど良好に増殖することができない。これらの遺伝子改変真菌又は酵母細胞では、PMT遺伝子の欠失を更に導入すると、細胞致死性になるか、又は十分な量まで培養増殖する能力に悪影響を与える場合がある。本明細書に記載する方法は組換え糖蛋白質の発現を誘導する段階と、場合により1種以上のα1,2−マンノシダーゼと共にPmt蛋白質の活性の阻害剤を添加する段階よりも前に細胞を十分な量まで培養増殖させ、主に特定のN−結合型グリカン構造をもち、O−結合型グリコシル化の低下した組換え糖蛋白質を生産することにより、PMT遺伝子を欠失させる潜在的な悪影響を回避する。
【0026】
本明細書に記載する方法の1側面は、O−結合型グリコシル化が低下し、主に特定のN−結合型グリコフォームを含む糖蛋白質組成物を提供することであり、このような組換え糖蛋白質はCHO細胞等の哺乳動物細胞培養から生産される同一糖蛋白質の組成物に比較して生物活性の増加及び/又は望ましくない免疫原性の低下を示すことができる。O−結合型グリコシル化が低下し、主にN−結合型グリコフォームを含む糖蛋白質組成物を生産すると、望ましくない結果を誘導及び/又はより有効なグリコフォームを減弱する恐れのある有害又は不活性なグリコフォームと不均質混合物の生産が避けられるという利点もある。従って、例えば主にMan5GlcNAc2、Man3GlcNAc2、GlcNAcMan5GlcNAc2、GlcNAcMan3GlcNAc2、GlcNAc2Man3GlcNAc2、GalGlcNAcMan5GlcNAc2、Gal(GlcNAc)2Man5GlcNAc2、(GalGlcNAc)2Man5GlcNAc2、NANAGalGlcNAcMan3GlcNAc2、NANA2Gal2GlcNAcMan3GlcNAc2及びGalGlcNAcMan3GlcNAc2グリコフォームを含み、O−結合型グリコシル化の低下した糖蛋白質分子の治療用医薬組成物はより低用量で十分に有効になり、より高い効果/効力が得られると思われる。
【0027】
一般に、O−結合型グリコシル化の低下した蛋白質の生産方法は、O−結合型グリコシル化の低下した蛋白質を生産することが望ましい組換え又は異種蛋白質をコードする核酸で宿主細胞を形質転換する段階を含む。組換え蛋白質をコードする核酸は組換え蛋白質の発現を可能にする調節配列と機能的に連結される。このような調節配列としては誘導性プロモーターと、場合により融合蛋白質をコードする核酸の上流ないし5’側のエンハンサーと、組換え蛋白質をコードする核酸の3’
側ないし下流の転写終結部位が挙げられる。核酸は更に一般にリボソーム結合部位をもつ5’UTR領域と3’非翻訳領域をコードする。核酸は組換え蛋白質を発現させる細胞で複製可能なベクターの成分であることが多い。ベクターは更に形質転換細胞の認識を可能にするためのマーカーを含むことができる。他方、所定の細胞種、特に酵母は外来ベクター配列をもたない核酸で首尾よく形質転換することができる。
【0028】
目的の組換え蛋白質をコードする核酸は数種類の起源から得ることができる。保存領域のプライマーを使用して蛋白質を発現させることが分かっている細胞株からcDNA配列を増幅することができる(例えばMarks et al.,J.Mol.Biol.581−596(1991)参照)。科学文献中の配列に基づいて核酸を新規に合成することもできる。目的の配列に対応するオーバーラップするオリゴヌクレオチドの伸長により核酸を合成することもできる(例えばCaldas et al.,Protein Engineering,13,353−360(2000)参照)。
【0029】
1側面では、所望時に蛋白質の発現を誘導できるように、蛋白質をコードする核酸を誘導性プロモーターと機能的に連結する。別の側面では、蛋白質をコードする核酸を構成性プロモーターと機能的に連結する。発現された蛋白質を単離し易くするために、細胞培養培地中に蛋白質を分泌させた後に単離できるようにするシグナル配列を蛋白質に付加することが一般に好ましい。第1の側面では、Pmt介在性O−結合型グリコシル化の1種以上の阻害剤を培地に添加する前に所望量の蛋白質を産生させるために十分な所望の多重度の宿主細胞を生産するために十分な時間にわたって形質転換宿主細胞を培養する。インデューサーと阻害剤を同時に培地に添加してもよいし、1種以上のPmt阻害剤を添加する前にインデューサーを培地に添加してもよいし、インデューサーを添加する前に1種以上のPmt阻害剤を培地に添加してもよい。O−結合型グリコシル化の低下した誘導蛋白質が産生され、培地から回収することができ、あるいはシグナル配列をもたない蛋白質の場合には溶解により宿主細胞から回収することができる。蛋白質をコードする核酸を構成性プロモーターと機能的に連結する第2の側面では、1種以上のPmt介在性O−結合型グリコシル化阻害剤を培地に添加すると同時に培養株を樹立し、産生されるO−結合型グリコシル化の低下した蛋白質を培地から回収することができ、あるいはシグナル配列をもたない蛋白質の場合には溶解により宿主細胞から回収することができる。
【0030】
本明細書では、O−結合型グリコシル化の低下した蛋白質を生産するのために有用なPmt蛋白質の1種以上の活性を阻害する化合物又は組成物について記載する。宿主細胞が真菌や酵母等の下等真核生物である場合には、阻害剤は少なくともPmt1もしくはPmt2、又はその両方の活性を阻害することが望ましい。高等真核生物では、阻害剤はPmt1又はPmt2に対応する高等真核生物でのホモログの活性を阻害することが望ましい。
【0031】
本発明の化合物は無傷の機能的PMT遺伝子をもつPichia pastoris株でO−結合型グリコシル化の低下した組換え蛋白質を生産するのに有効であることが判明した。表1と実施例5の図1から明らかなように、無傷の機能的PMT遺伝子をもつ組換えPichia pastorisの培養物に添加し、組換え蛋白質の発現時に誘導性プロモーターと機能的に連結した組換えヒト抗Her2抗体蛋白質をコードする核酸で形質転換した上記4種類のPmt化学阻害剤のいずれか1種は、Orchard et al.(Bioorgan & Med Chem Letters(2004)14:3975−3978)及びEP1313471B1を含む同一著者による特許公開とBobrowiczらの米国出願公開第2007061631号に記載されているPmt阻害剤であるPmti−3で処理したPichia pastoris細胞に認められるO−結合型グリコシル化レベルに対して改善された低いO−結合型グリコシル化レベルをもつ組換え蛋白質を生じた。
【0032】
宿主細胞
本明細書で使用する「細胞」なる用語は以下に記載する宿主細胞を意味する。本明細書に記載する方法の宿主細胞としては高等真核細胞と下等真核細胞の両方が挙げられるが、下等真核細胞(例えば糸状菌や酵母細胞)は経済的に培養でき、蛋白質収率が高く、適宜改変すると、適切なグリコシル化パターンをもつ蛋白質を産生することができるので、一般に蛋白質の発現に好ましい。下等真核生物としては、酵母、真菌類、襟鞭毛虫類、微胞子虫類、アルベオラータ(例えば渦鞭毛虫類)、ストラメノパイル(例えば褐藻類、原生動物)、紅藻植物門(例えば紅藻類)、植物(例えば緑藻類、植物細胞、コケ)及び他の原生生物か挙げられる。酵母及び真菌類としては限定されないが、ピキア(Pichia)種(例えばPichia pastoris、Pichia finlandica、Pichia trehalophila、Pichia koclamae、Pichia membranaefaciens、Pichia minuta(Ogataea minuta、Pichia lindneri)、Pichia opuntiae、Pichia thermotolerans、Pichia salictaria、Pichia guercuum、Pichia pijperi、Pichia stiptis、Pichia methanolica)、サッカロミセス(Saccharomyces)種(例えばSaccharomyces cerevisiea)、Hansenula polymorpha、クルイベロマイセス(Kluyveromyces)種(例えばKluyveromyces lactis)、Candida albicans、アスペルギルス(Aspergillus)種(例えばAspergillus nidulans、Aspergillus niger、Aspergillus oryzae)、Trichoderma reesei、Chrysosporium lucknowense、フサリウム(Fusarium)種(例えばFusarium gramineum、Fusarium venenatum)、Physcomitrella patens及びNeurospora crassaが挙げられる。特に酵母は遺伝学が確立しており、迅速な形質転換、試験蛋白質局在ストラテジー及び簡単な遺伝子ノックアウト技術が可能になるため、一般に好ましい。適切なベクターは3−ホスホグリセリン酸キナーゼ又は他のグリコール酸酵素を含むプロモーター等の発現制御配列と、複製起点、終結配列等を必要に応じてもつ。
【0033】
高い細胞密度まで増殖できると共に大量の組換え蛋白質を分泌できるという理由から、K.lactis、Pichia pastoris、Pichia methanolica及びHansenula polymorpha等の各種酵母が細胞培養に一般に好ましい。同様に、Aspergillus niger、フサリウム種、Neurospora crass等の糸状菌を使用して工業規模で組換え蛋白質を生産することもできる。
【0034】
グリコシル化パターンがヒト様又はヒト化型である蛋白質又は糖蛋白質を発現するように、下等真核生物、特に糸状菌と酵母を遺伝子改変することができる。これはGerngrossらにより米国特許第US7029872号と米国特許出願公開第20040018590号、20050170452号、20050260729号、20040230042号、20050208617号、20040171826号、20050208617号、20060160179号、20060040353号及び20060211085号に記載されているように、選択された内在グリコシル化酵素を除去すること及び/又は外来酵素を供給することにより実現することができる。従って、特定のN−グリカン構造の高収率生産を可能にするように1種以上の酵素又は酵素活性を発現するように宿主細胞を付加的又は代替的に操作することができる。例えば通常では酵素に関連しないシグナルペプチドにより酵素が最適活性をもつような宿主細胞内オルガネラをこのような酵素の標的とすることができる。糖ヌクレオチドトランスポーター及び/又はヌクレオチドジホスファターゼ酵素を発現するように宿主細胞を改変することもできる。トランスポーターとジホスファターゼは適切な区画にグリコシル化酵素の適切な基質を提供し、競合的生成物阻害を低減し、ヌクレオチド二リン酸の除去を促進することにより、人工グリコシル化工程の効率を改善する。例えばGerngrossらの米国特許出願公開第20040018590号及びHamilton,2003,Science 301:1244−46並びに上記米国特許及び特許出願参照。
【0035】
例えば、低下させないと糖蛋白質のN−グリカンにマンノース残基を付加する1,6−マンノシルトランスフェラーゼ活性を低下させると共に、30モル%を上回るMan5GlcNAc2N−グリカンをもつ組換え糖蛋白質の産生を可能にするα−1,2マンノシダーゼ活性の異所性発現用核酸を更に含むように宿主細胞(例えば酵母又は真菌)を選択又は操作することができる。本明細書に記載する方法に従って宿主細胞中で糖蛋白質を産生させると、主にMan5GlcNAc2N−グリカン構造をもち、別の方法により細胞中で産生される糖蛋白質に比較してO−グリコシル化の低下した糖蛋白質が宿主細胞により産生される。別の側面では、主にGlcNAcMan5GlcNAc2N−グリカンをもつ糖蛋白質の産生を可能にするように、GlcNAcトランスフェラーゼI活性の異所性発現用核酸を更に含むように宿主細胞を操作する。本明細書に記載する方法に従って宿主細胞中で糖蛋白質を産生させると、主にGlcNAcMan5GlcNAc2N−グリカン構造をもち、別の方法により細胞中で産生される糖蛋白質に比較してO−グリコシル化の低下した糖蛋白質が宿主細胞により産生される。更に別の側面では、主にGlcNAcMan3GlcNAc2N−グリカンをもつ糖蛋白質の産生を可能にするように、マンノシダーゼII活性の異所性発現用核酸を更に含むように宿主細胞を操作する。本明細書に記載する方法に従って宿主細胞中で糖蛋白質を産生させると、主にGlcNAcMan3GlcNAc2N−グリカン構造をもち、別の方法により細胞中で産生される糖蛋白質に比較してO−グリコシル化の低下した糖蛋白質が宿主細胞により産生される。更に別の側面では、主にGlcNAc2Man3GlcNAc2N−グリカンをもつ糖蛋白質の産生を可能にするように、GlcNAcトランスフェラーゼII活性の異所性発現用核酸を更に含むように宿主細胞を操作する。本明細書に記載する方法に従って宿主細胞中で糖蛋白質を産生させると、主にGlcNAc2Man3GlcNAc2N−グリカン構造をもち、別の方法により細胞中で産生される糖蛋白質に比較してO−グリコシル化の低下した糖蛋白質が宿主細胞により産生される。更に他の側面では、任意組合せでN−結合型グリコシル化に関与し、例えばシアリルトランスフェラーゼ活性、クラスII及びIIIマンノシダーゼ活性、GlcNAcトランスフェラーゼII、III、IV、V、VI、IX活性、並びにガラクトーストランスフェラーゼ活性をコードする1種以上の高等真核生物遺伝子を更に含むことにより特定のハイブリッド又は複合N−グリカン又はヒト様N−グリカン構造を産生するように上記宿主細胞を更に操作することができる。細胞はUDP特異的ジホスファターゼ活性、GDP特異的ジホスファターゼ活性及びUDP−GlcNAcトランスポーター活性をコードする核酸の1種以上を更に含むことが一般に好ましい。
【0036】
本明細書に教示するようなO−結合型グリコシル化の低下した蛋白質及び糖蛋白質を発現させるためには植物及び植物細胞培養物を使用することができる(例えばLarrick & Fry,1991,Hum.Antibodies Hybridomas 2:172−89;Benvenuto et al.,1991,Plant Mol.Biol.17:865−74;Durin et al.,1990,Plant Mol.Biol.15:281−93;Hiatt et al.,1989,Nature 342:76−8参照)。好ましい植物宿主としては、例えばアブラナ(Arabidopsis)、タバコ(Nicotiana tabacum)、マルバタバコ(Nicotiana rustica)及びジャガイモ(Solanum tuberosum)が挙げられる。
【0037】
本明細書に教示するようなO−結合型グリコシル化の低下した蛋白質及び糖蛋白質を産生させるためには、例えばバキュロウイルス系発現システム等の昆虫細胞培養物を使用することもできる(例えばPutlitz et al.,1990,Bio/Technology 8:651−654参照)。
【0038】
本明細書に教示するようなO−結合型グリコシル化の低下した蛋白質及び糖蛋白質を発現及び産生させるためには、一般に下等真核生物や原核生物ほど経済的に培養できないが、哺乳動物組織細胞培養物を使用することもできる(Winnacker,From Genes to Clones(VCH Publishers,NY,1987)参照)。適切な宿主としては、CHO細胞株、各種COS細胞株、HeLa細胞、好ましくはミエローマ細胞株等、又は形質転換B細胞もしくはハイブリドーマが挙げられる。これらの細胞の発現ベクターは複製起点、プロモーター、エンハンサー等の発現制御配列(Queen et al.,1986,Immunol.Rev.89:49−68)と、リボソーム結合部位、RNAスプライス部位、ポリアデニル化部位及び転写ターミネーター配列等の必要なプロセシング情報部位を含むことができる。発現制御配列は免疫グロブリン遺伝子、SV40、アデノウイルス、ウシパピローマウイルス、サイトメガロウイルス等に由来するプロモーターである。一般に、発現ベクターにはneoR発現カセット等の選択マーカーが含まれる。
【0039】
発現させようとする蛋白質をコードする核酸は細胞宿主の種類により異なる従来方法により宿主細胞に導入することができる。例えば、リン酸カルシウム処理、プロトプラスト融合、自然交配、リポフェクション、バイオリスティクス法、ウイルス形質導入又はエレクトロポレーションを細胞宿主に使用することができる。植物細胞及び組織にはタングステン粒子バリスイック遺伝子導入が好ましい(一般に、Maniatis et al.,Molecular Cloning:A Laboratory Manual(Cold Spring Harbor Press,1982)参照)。
【0040】
発現後、硫安沈殿、アフィニティーカラム、カラムクロマトグラフィー、ゲル電気泳動等を含む当分野の標準手順によりO−結合型グリコシル化の低下した蛋白質又は糖蛋白質を精製することができる(一般にScopes,R.,Protein Purification(Springer−Verlag,N.Y.,1982)参照)。医薬用には、均質度が少なくとも約90〜95%の実質的に純粋な糖蛋白質が好ましく、98〜99%又はそれ以上の均質度が最も好ましい。必要に応じて部分的又は均質まで精製後、(体外を含む)治療又はアッセイ法、免疫蛍光染色等の開発及び実施に蛋白質を使用することができる。(一般に、Immunological Methods,Vols.I and II(Lefkovits and Pernis,eds.,Academic Press,NY,1979 and 1981参照))。
【0041】
従って、主要種としてN−グリカン構造を含み、Pmt介在性O−結合型グリコシル化阻害剤又は2個以上のマンノース残基をグリカン構造から除去することが可能なα−1,2−マンノシダーゼ又はその両者の存在下でインキュベートしてしない宿主細胞で産生された糖蛋白質の組成物に比較してO−結合型グリコシル化の低下した糖蛋白質組成物も提供する。特定側面において、糖蛋白質組成物はMan5GlcNAc2、Man3GlcNAc2、GlcNAcMan5GlcNAc2、GlcNAcMan3GlcNAc2、GlcNAc2Man3GlcNAc2、GalGlcNAcMan5GlcNAc2、Gal(GlcNAc)2Man5GlcNAc2、(GalGlcNAc)2Man5GlcNAc2、NANAGalGlcNAcMan3GlcNAc2、NANA2Gal2GlcNAcMan3GlcNAc2、及びGalGlcNAcMan3GlcNAc2グリコフォームから構成される群から選択される主要なN−グリカン構造をもつ糖蛋白質を含む。
【0042】
医薬組成物
活性治療剤としての糖蛋白質と医薬的に許容可能な他の各種成分を含有する医薬組成物にO−結合型グリコシル化の低下した蛋白質及び糖蛋白質を配合することができる(Remington’s Pharmaceutical Science(15th ed.,Mack Publishing Company,Easton,Pennsylvania,1980)参照)。好ましい剤形は所期投与方法と治療用途により異なる。組成物には目的の製剤に応じて動物又はヒト投与用医薬組成物を製剤化するために一般に使用される賦形剤として定義される医薬的に許容可能な非毒性キャリヤー又は希釈剤を更に添加することができる。希釈剤は組成物の生物活性に影響を与えないように選択される。このような希釈剤の例は蒸留水、リン酸緩衝生理食塩水、リンゲル液、ブドウ糖液及びハンクス液である。更に、医薬組成物又は製剤には他のキャリヤー、アジュバント、又は非毒性非治療用非免疫原性安定剤等も加えることができる。
【0043】
非経口投与用医薬組成物は無菌で実質的に等張且つパイロジェンフリーであり、FDA又は同様の機関のGMPに従って製造される。水、油類、食塩水、グリセロール又はエタノール等の滅菌液とすることができる医薬キャリヤーと共に生理的に許容可能な希釈剤に薬剤を溶解又は懸濁した注射剤として糖蛋白質を投与することができる。更に、湿潤剤又は乳化剤、界面活性剤、pH緩衝物質等の助剤を組成物に加えることができる。医薬組成物の他の成分は石油、動物、植物又は合成由来成分であり、例えば落花生油、大豆油及び鉱物油が挙げられる。一般に、プロピレングリコールやポリエチレングリコール等のグリコール類が特に注射溶液用の好ましい液体キャリヤーである。活性成分の持続放出を可能にするように製剤化することができるデポー注射剤やインプラント製剤として糖蛋白質を投与することもできる。一般に、組成物は液体溶液又は懸濁液としての注射剤として製造され、注射前に液体賦形剤に溶解又は懸濁するのに適した固体製剤を製造することもできる。上記のようなアジュバント効果を高めるために、製剤をポリラクチド、ポリグリコリド又はコポリマー等のリポソーム又は微粒子で乳化又はカプセル化することもできる(Langer,Science 249,1527(1990)及びHanes,Advanced Drug Delivery Reviews 28,97−119(1997)参照)。
【0044】
「又はその塩」なる用語は無機又は有機塩基を含む許容可能な塩基から製造される塩を意味する。無機塩基から誘導される塩としてはアルミニウム、アンモニウム、カルシウム、銅、三価鉄、二価鉄、リチウム、マグネシウム、三価マンガン、二価マンガン、カリウム、ナトリウム、亜鉛等の塩が挙げられる。固体形態の塩は2種類以上の結晶構造で存在してもよいし、水和物形態でもよい。塩基から誘導される塩としては第一級、第二級及び第三級アミン、置換アミン(天然置換アミンを含む)、環状アミン、並びに塩基性イオン交換樹脂(例えばアルギニン、ベタイン、カフェイン、コリン、N,N’−ジベンジルエチレンジアミン、ジエチルアミン、2−ジエチルアミノエタノール、2−ジメチルアミノエタノール、エタノールアミン、エチレンジアミン、N−エチルモルホリン、N−エチルピペリジン、グルカミン、グルコサミン、ヒスチジン、ヒドラバミン、イソプロピルアミン、リジン、メチルグルカミン、モルホリン、ピペラジン、ピペリジン、ポリアミン樹脂、プロカイン、プリン、テオブロミン、トリエチルアミン、トリメチルアミン、トリプロピルアミン、トロメタミン等)の塩が挙げられる。
【0045】
本発明の組成物は1個以上の不斉中心を含む場合があり、従って、ラセミ体及びラセミ混合物、単一エナンチオマー、ジアステレオマー混合物及び個々のジアステレオマーとして存在することができる。本発明はこれらの化合物のこのような全異性体を含むものとする。
【0046】
本明細書に特に定義しない限り、本発明に関して使用する全科学技術用語及び術語は当業者に通常理解されている通りの意味である。更に、内容からそうでないことが必要とされる場合を除き、単数形の用語は複数形を含み、複数形の用語は単数形を含む。一般に、本明細書に記載する生化学、酵素学、分子及び細胞生物学、微生物学、遺伝学並びに蛋白質及び核酸化学とハイブリダイゼーションに関して使用する術語とその技術は当分野で周知であり、広く使用されているものである。特に指定しない限り、本発明の方法及び技術は本明細書の随所に引用及び記載する各種一般文献及び特殊文献に記載されているような当分野で周知の慣用方法に従って一般に実施される。例えばSambrook et al.Molecular Cloning:A Laboratory Manual,2d ed.,Cold Spring Harbor Laboratory Press,Cold Spring Harbor,N.Y.(1989);Ausubel et al.,Current Protocols in Molecular Biology,Greene Publishing Associates(1992及び2002補遺);Harlow and Lane,Antibodies:A Laboratory Manual,Cold Spring Harbor Laboratory Press,Cold Spring Harbor,N.Y.(1990);Taylor and Drickamer,Introduction to Glycobiology,Oxford Univ.Press(2003);Worthington Enzyme Manual,Worthington Biochemical Corp.,Freehold,NJ;Handbook of Biochemistry:Section A Proteins,Vol I,CRC Press(1976);Handbook of Biochemistry:Section A Proteins,Vol II,CRC Press(1976);Essentials of Glycobiology,Cold Spring Harbor Laboratory Press(1999)参照。
【0047】
以下、実施例により本発明の各種Pmt阻害剤の製造方法について記載する。
【実施例1】
【0048】
【化2】
【0049】
ステップA
【0050】
【化3】
【0051】
1−1(5.90g,25mmol)のTHF(200mL)溶液にNaH(2.0g,50mmol)を少量ずつ加えた。得られた混合物を0℃で30分間撹拌した。次にNBS(4.86g,27.5mmol)を加え、15分間撹拌した。白色固体を濾過し、濾液を濃縮して残渣を得、CHCl3に溶解し、Na2SO4で乾燥した。溶媒を蒸発させて1−2(5g,収率63%)を得、それ以上精製せずに次段階で使用した。
【0052】
ステップB
【0053】
【化4】
【0054】
1−2(1.8g,6mmol)と1−3(1.3g,5mmol)の無水DMF(5mL)溶液にNaH(2.4gg,10mmol)を加えた後、室温で一晩撹拌した。得られた混合物を水とEtOAに分配した。有機層を合わせてブラインで洗浄し、硫酸ナトリウムで乾燥し、濃縮した。残渣を分取TLCにより精製し、1−4(800mg,収率40%)を得た。1H−NMR(400MHz,CDCl3)δ9.66(s,1H),7.74〜7.71(m,2H),7.49〜7.46(m,1H),7.37〜7.33(m,5H),7.20(d,J=2.02Hz,1H),6.97〜6.92(m,3H),4.28〜4.19(m,6H),3.16(t,J=6.6Hz,2H),1.11(m,6H)。
【0055】
ステップC
【0056】
【化5】
【0057】
LAH(380mg,10mmol)のTHF(50mL)溶液に0℃で1−4(1.3g,2.6mmol)を加え、得られた混合物を室温で30分間撹拌した。混合物を希HCl水溶液で注意深く処理した後、水とEtOAに分配した。有機層をブラインで洗浄し、硫酸ナトリウムで乾燥し、濃縮した。残渣を分取TLCにより精製し、1−5(260mg,収率20%)を得た。
【0058】
ステップD
【0059】
【化6】
【0060】
1−5(300mg,0.54mmol)の2,2−ジメトキシプロパン(1mL)溶液に触媒量のTsOH・H2O(〜10mg)を加えた後、得られた混合物を室温で1時間撹拌した。混合物を分取TLCにより直接精製し、1−6(100mg,収率28%)を得た。1H−NMR(400MHz,MeOD)δppm 7.52(d,J=8.05Hz,2H),7.40〜7.15(m,5H),6.90〜6.85(m,2H),6.80〜6.75(m,2H),4.25(s,2H),4.23(m,6H),3.0(m,2H),1.48(t,6H)。
【0061】
ステップE
【0062】
【化7】
【0063】
1−6(100mg,0.22mmol)の無水DCM(25mL)溶液にMnO2(174mg,2mmol)を一度に加え、得られた混合物を約2時間加熱還流した。室温まで冷却後、混合物を濾過し、濾液を濃縮し、残渣を分取TLCにより精製し、1−7(60mg,収率60%)を得た。1H−NMR(400MHz,MeOD)δppm 9.5(s,1H),7.52(d,J=7.2Hz,1H),7.48(d,J=2.8Hz,2H),7.30(m,3H),7.25(m,6H),7.0(t,J=8.6Hz,2H),6.89(m,J=8.6Hz,2H),6.76(d,J=8Hz,1H),4.23(m,6H),3.0(m,2H),1.48(t,6H)。
【0064】
ステップF
【0065】
【化8】
【0066】
1−7(80mg,0.17mmol)のTHF溶液に6N HClを加え、得られた混合物を室温で2時間撹拌した。溶媒を減圧除去し、粗生成物1−8(60mg,収率82%)を得、それ以上精製せずに次段階に供した。
【0067】
ステップG
【0068】
【化9】
【0069】
1−8(60mg,0.146mmol)のDMF(6mL)溶液に℃でNaH(24mg,1mmol)を加え、得られた混合物を同一温度で約30分間撹拌後、CH3I(0.2mL)を加えた。混合物を同一温度で更に1時間撹拌した。次に混合物をH2OとEtOAcに分配した。有機層を合わせてブラインで洗浄し、硫酸ナトリウムで乾燥し、濃縮した。残渣を分取TLCにより精製し、1−9(40mg,収率63.0%)を得た。1H−NMR(400MHz,MeOD)δppm 9.6(s,1H),7.52(d,J=8.08Hz,2H),7.43(m,1H),7.36〜7.30(m,5H),7.16(d,J=2.02Hz,1H),7.0(t,J=8.6Hz,2H),6.89(m,J=8.6Hz,2H),4.23(t,J=6.56Hz,2H),3.95〜3.85(m,4H),3.30(s,6H),3.13(dd,J1=6.26Hz,J2=6.26Hz,2H)。
【0070】
ステップH
【0071】
【化10】
【0072】
1−9(40mg,0.09mmol)、1−10(19mg,0.09mmol)及びNH4OAc(70mg,0.9mmol)をトルエン(10mL)に加えた混合物を窒素下で約3時間加熱還流した。室温まで冷却後、得られた混合物をpH=4〜5まで希HCl水溶液により処理した後、H2OとEtOAcに分配した。有機層を合わせてブラインで洗浄し、Na2SO4で乾燥し、濃縮した。残渣を分取HPLCにより精製し、実施例1(30mg,収率50%)を黄色い固体として得た。1H−NMR(400MHz,MeOD)δppm 7.46〜7.53(m,3H),7.31〜7.40(m,5H),7.15〜7.18(m,1H),6.92〜7.06(m,4H),4.71(s,2H),4.25〜4.30(m,2H),3.85〜3.94(m,4H),3.26〜3.29(m,6H),3.10〜3.13(dd,J=6.26Hz,J=6.26Hz,2H)。MS m/z 612(M+1)+。
【実施例2】
【0073】
【化11】
【0074】
ステップA
【0075】
【化12】
【0076】
化合物2−1(150mg,0.58mmol,1.0当量)、化合物2−2(214mg,0.87mmol,1.5当量)のDMF(10mL)溶液にCs2CO3(161mg,0.49mmol,0.85当量)を加えた。次に混合物を室温で一晩撹拌後、80℃まで1.5時間加熱した。得られた混合物を次にH2OとEtOAcに分配した。有機層を合わせてブラインで洗浄し、Na2SO4で乾燥し、濃縮し、分取TLCにより精製し、2−3(122mg,収率49.4%)を得た。
【0077】
ステップB
【0078】
【化13】
【0079】
化合物2−3(122mg,0.29mmol,1.0当量)、2−4(57mg,0.30mmol,1.05当量)及びNH4OAc(223mg.2.90mmol,10.0当量)をトルエン(15mL)に加えた混合物を窒素下に約3時間加熱還流した。得られた混合物を10%塩酸で処理してpH=4〜5とした後、H2OとEtOAcに分配した。有機層を合わせてブラインで洗浄し、Na2SO4で乾燥し、濃縮し、分取TLCにより精製し、実施例2(52mg,収率29.9%)を得た。1H−NMR(400MHz,MeOD)δ7.41−7.47(m,3H),7.19−7.35(m,10H),6.99−7.07(m,3H),6.87−6.92(m,2H),6.22(s,1H),4.75(s,2H),4.24−4.29(m,2H),3.72−3.13(m,2H)。MS m/z 600(M+l)+。
【実施例3】
【0080】
【化14】
【0081】
ステップA
【0082】
【化15】
【0083】
3−1(1g,6.37mmol)の無水THF(20mL)溶液に−70℃でBuLi(3mL,7.68mmol)を滴下した後、混合物を室温で30分間撹拌した。再び−70℃まで冷却後、シクロヘキサンカルボアルデヒド(0.72g,6.43mmol)の無水THF(5mL)溶液を滴下した。30分間撹拌後、飽和NH4Cl水溶液を混合物に加え、得られた混合物をEtOAcで抽出した。有機層を合わせてNa2SO4で乾燥し、濃縮し、3−2(1.52g,14.3%)を粗製油状物として得た。
【0084】
ステップB
【0085】
【化16】
【0086】
3−2(600mg,3.1mmol)を無水DCM(15mL)に加えた混合物に0℃でSOCl2(2mL)を滴下した。次に得られた混合物を2時間室温で撹拌した。混合物を減圧濃縮後、EtOAcで希釈した。有機層を飽和NaHCO3水溶液、ブラインで洗浄し、Na2SO4で乾燥後、濃縮し、3−3(500mg,粗生成物)を得た。
【0087】
ステップC
【0088】
【化17】
【0089】
3−3(450mg,粗生成物)と3−4(375mg,1.44mmol)のDMF(10mL)溶液にCs2CO3(399mg,1.22mmol)を一度に加えた後、混合物を110℃まで18時間加熱した。混合物をH2Oで希釈し、EtOAcで抽出した。有機層を合わせてNa2SO4で乾燥し、濃縮した。残渣をTLCにより精製し、3−5(130mg)を油状物として得、次段階でそのまま使用した。
【0090】
ステップD
【0091】
【化18】
【0092】
3−5(130mg,0.3mmol)と3−6(60mg,0.31mmol)をトルエン(15mL)に加えた混合物にNH4OAc(180mg,2.34mmol)を一度に加えた後、混合物を3.5時間加熱還流した。次に混合物を室温まで冷却し、10% HCl水溶液を加えてpH=5とした。混合物をEtOAcで抽出した。有機層を合わせてNa2SO4で乾燥し、濃縮し、黄色い油状物を得た。残渣を分取TLCにより精製し、実施例3(150mg,収率82.4%)を得た。1H−NMR(300MHz,MeOD)δ7.60(s,1H),7.19〜7.49(m,11H),6.88(s,1H),6.81(s,1H),5.05(s,1H),4.65(s,2H),4.25〜4.39(m,2H),3.15(m,2H),1.60〜1.98(m,5H),1.00〜1.46(m,6H)。MS m/z:607(M+1)+。
【実施例4】
【0093】
【化19】
【0094】
ステップA
【0095】
【化20】
【0096】
ナトリウム(8.5mg,0.37mmol)を40℃で2−メチルプロパン−1−オール(1.3g,17.5mmol)に溶解した後、化合物4−1(2g,16.7mmol)を50℃で30分間かけて滴下した。次に反応混合物を70℃まで加熱し、一晩撹拌した。得られた混合物をH2OとEtOAcに分配した。有機層をブラインで洗浄し、硫酸ナトリウムで乾燥し、濃縮し、4−2(1.0g,粗生成物)を得、次段階でそのまま使用した。
【0097】
ステップB
【0098】
【化21】
【0099】
4−2(1.0g,粗生成物)と4−3(400mg,1.64mmol,1.0当量)とPPh3(516mg,1.96mmol,1.2当量)のTHF(20mL)溶液に0℃でDIAD(406mg,1.96mmol,1.2当量)を滴下した。得られた混合物を室温で一晩撹拌後、H2OとEtOAcに分配した。有機層をブラインで洗浄し、Na2SO4で乾燥し、濃縮し、分取TLCにより精製し、4(90mg,収率12.0%)を得た。1H−NMR(400MHz,MeOD)δ9.72(s,1H),7.28−7.40(m,9H),6.98−7.05(t,J=8.78Hz,2H),6.89−6.92(d,J=8.28Hz,1H),5.42−5.46(m,1H),4.22−4.32(m,2H),3.86−3.92(m,1H),3.68−3.74(m,1H),3.24−3.34(m,2H),3.12−3.18(m,2H),1.82−1.90(m,1H),0.84−0.88(m,6H)。
【0100】
ステップC
【0101】
【化22】
【0102】
実施例4の製造は実施例3の製造と同様である。1H−NMR(400MHz,MeOD)δ7.49(s,1H),7.24−7.40(m,7H),6.98−7.05(m,4H),6.90−6.91(m,1H),5.37−5.40(m,1H),4.75(s,2H),4.21−4.32(m,2H),3.83−3.88(m,1H),3.69−3.72(m,1H),3.33−3.35(m,2H),3.09−3.13(t,J=6.57Hz,2H),1.79−1.87(m,1H),0.86−0.88(m,6H)。
【実施例5】
【0103】
本実施例はヒト抗Her2抗体の重鎖(Hc)及び軽鎖(Lc)をコードする発現ベクターで形質転換し、本明細書に記載する新規Pmt阻害剤で処理したPichia pastorisがO−グリコシル化の低下した糖蛋白質を産生したことを示す。
【0104】
発現/組込みプラスミドベクターpGLY2988は抗Her2の重鎖(Hc)及び軽鎖(Lc)をコードするメタノール誘導型Pichia pastoris AOX1プロモーターの制御下におかれた発現カセットを含む。α−MATプレシグナルペプチド(配列番号1及び2)のN末端に融合した抗Her2 Hc及びLcをGene Art AGにより合成した。ユニークな5’EcoR1及び3’Fse1部位をもつように各々を合成した。抗Her2 Hcのヌクレオチド配列とアミノ酸配列を夫々配列番号3及び4に示す。抗Her2 Lcのヌクレオチド配列とアミノ酸配列を夫々配列番号5及び6に示す。Pichia pastoris TRP2標的核酸とゼオシン耐性マーカーを含み、AOX1プロモーターとSaccharomyces cerevisiae CYCターミネーターの制御下におかれた発現カセットを作製できる発現プラスミドベクターpGLY2198に、ユニークな5’EcoR1及び3’Fse1部位を使用して、α−MATプレシグナルペプチドに融合したHc及びLc蛋白質をコードする両方の核酸フラグメントを夫々別々にサブクローニングし、夫々プラスミドベクターpGLY2987及びpGLY2338を形成した。次にBamHIとNotIで消化することによりLc発現カセットをプラスミドベクターpGLY2338から切出し、BamH1とNot1で消化しておいたプラスミドベクターpGLY2987にサブクローニングし、最終発現プラスミドベクターpGLY2988を作製した。
【0105】
抗Her2発現株yGLY4280を以下のように構築した。TRP2標的領域を切断する制限酵素Spe1で消化した5μgのpGLY2988を使用して株yGLY22−1を形質転換した。従来記載されている方法(Nett and Gerngross,Yeast 20:1279(2003);Choi et al.,PNAS USA 100:5022(2003);Hamilton et al.,Science 301:1244(2003))を使用して株yGLY22−1(och1Δ::lacZbmt2Δ::lacZ/KlMNN2−2/mnn4L1Δ::lacZ/MmSLC35A3pno1Δmnn4Δ::lacZmet16Δ::lacZ)を構築した。
【0106】
yGLY22−1の形質転換は原則的に以下のように実施した。ODが約0.2〜6になるまでYGLY22−1をYPD培地(酵母エキス(1%)、ペプトン(2%)、デキストロース(2%))50mL中で一晩培養した。氷上で30分間インキュベーション後、細胞を2500〜3000rpmで5分間遠心することによりペレット化した。培地を除去し、細胞を氷冷滅菌1Mソルビトールで3回洗浄後、氷冷滅菌1Mソルビトール0.5mlに再懸濁した。直鎖化DNA10μL(10ug)と細胞懸濁液100μLをエレクトロポレーションキュベットに加え、氷上で5分間インキュベートした。プリセットされたPichia pastorisプロトコル(2kV,25μF,200Ω)に従ってBio−Rad GenePulser Xcellでエレクトロポレーションを行った直後にYPDS回復培地(YPD培地+1Mソルビトール)1mLを加えた。形質転換細胞を室温(26℃)で4時間〜一晩回復させた後、細胞を選択培地に撒いた。ゼオシンを加えた培地で選択後、形質転換細胞を小規模発現分析によりスクリーニングし、抗Her2発現を検出した。高レベル抗Her2発現に基づいて株yGLY4280を選択した。
【0107】
1%酵母エキス、2%ペプトン、100mMリン酸カリウム緩衝液(pH6.0),1.34%酵母窒素塩基、4×10−5%ビオチン、及び1%グリセロールから構成される緩衝グリセロール複合培地(BMGY)を使用して振盪フラスコで24℃にて株yGLY4280の抗Her2蛋白質発現を実施した。蛋白質発現誘導培地はBMGYのグリセロールを1%メタノールに代えた緩衝メタノール複合培地(BMMY)とした。誘導培地添加時に100%メタノール中のPmt阻害剤を増殖培地に終濃度0.15ug/mLまで加えた。これはPmt阻害剤で処理しない場合に観測される値の約50%までO−グリコシル化を低下させるために十分な中間用量であり、従って各種阻害剤の効力の比較を可能にする。誘導培地で24時間更に増殖後、培養液を回収し、2,000rpmで5分間遠心し、上清から細胞を除去した。
【0108】
Dionex−HPLC(HPAEC−PAD)を使用してO−グリカン測定を以下のように実施した。O−グリコシル化低下を測定するために、プロテインAクロマトグラフィー(Li et al.Nat.Biotechnol.24(2):210−5(2006))を使用して蛋白質を増殖培地から精製し、アルカリ除去(β除去)によりO−グリカンを蛋白質から遊離及び分離させた(Harvey,Mass Spectrometry Reviews 18:349−451(1999))。この方法は更に、遊離したO−グリカン(オリゴマンノース又はマンノース)の新たに形成された還元末端をマンニトールに還元する。従って、各O−グリカンのユニークな指標としてマンニトール基を利用することができる。β除去にはPBS緩衝液100μL容量に含まれる0.5nmol以上の蛋白質が必要であった。サンプルを水素化ホウ素アルカリ試薬25μLで処理し、50℃で16時間インキュベートした。アラビトール内部標準約20uLを加えた後、氷酢酸10μLを加えた。次にSEPABEADSとAG 50W−X8樹脂の両方を充填したMilliporeフィルターでサンプルを遠心し、水洗した。洗浄液を含むサンプルをプラスチックオートサンプラーに移し、遠心蒸発器で蒸発乾涸した。1% AcOH/MeOH 150μLをサンプルに加え、サンプルを遠心蒸発器で蒸発乾涸した。この最後の段階を更に5回繰返した。水200μLを加え、高pH陰イオン交換クロマトグラフィーとパルス式電気化学検出法を組合せたDionex HPLC(HPAEC−PAD)によりサンプル100μLを分析した。マンニトール回収量に基づいて平均O−グリカン率を求めた。
【0109】
結果を表1に要約する通り、4種類の新規Pmt阻害剤である実施例1〜4はOrchard et al.(Bioorgan & Med Chem Letters(2004)14:3975−3978)及びEP1313471B1を含む同一著者による特許公開とBobrowiczらの米国出願公開第2007061631号に記載されているようなPmt阻害剤であるPmti−3で観測されるレベルよりも低レベルまでO−グリコシル化を低下させることが明らかである。Pmti−3は以下の化学構造をもつ。
【0110】
【化23】
【0111】
【表1】
【0112】
図1は新規Pmt阻害剤がPichiaにより産生される組換え蛋白質のO−グリコシル化を有効に低下させることを実証する画像であり、抗Her2重鎖(Hc)のO−グリコシル化に及ぼすPmt阻害剤の増量の効果を示す。ウェル当たりBMGY培地0.5mlを加えた96穴ディープウェルプレート(Qiagen,Valencia,CA)に株yGLY4280を接種した。激しく振盪しながら24時間増殖後に96ウェルプレートを2,000rpmで5分間遠心し、細胞をペレット化した。培地を除去し、BMMY培地0.5mLによる洗浄段階後、Pmt阻害剤を第1行から最終行まで(11穴)2倍ずつ希釈したBMMY培地0.2mLに細胞を再懸濁した。ウェル#1には阻害剤5ug/mLを加え、ウェル#2には2.5ug/mLを加え、以下同様にしてウェル#10に0.009ng/mLを加え、#11には阻害剤を加えなかった。激しく振盪しながら更に24時間増殖後にプレートを2,000rpmで5分間遠心し、細胞をペレット化し、清澄化した上清をウェスタンブロット分析し、抗Her2発現を検出した。ウェスタンブロットは以下のように実施した。Laemmli,U.K.(1970)Nature 227,680−685に従って還元ポリアクリルアミドゲル電気泳動(SDS−PAGE)により上清7μLを分離後、ニトロセルロースメンブレン(Schleicher & Schuell,現Whatman,Inc.,Florham Park,NJ)にエレクトロブロットした。ペルオキシダーゼ標識抗ヒトHc及びLc抗体(Calbiochem/EMD Biosciences,La Jolla,CA)を使用してウェスタンブロットで抗Her2抗体鎖を検出し、ImmunoPure金属増感型DAB基質キット(Pierce Biotechnology,Rockford,IL)を使用して展開した。図1は実施例4からの新規Pmt阻害剤(EX.4,図1A)をOrchard et al.からのPmti−3(図1B)と対照としての不活性化合物(図1C)に対比試験したこのような分析の1例の結果を示す。図1A及びBに示すように、実施例4とPmti−3は0.018ug/mLまでの低濃度で抗Her2 HcのO−グリコシル化を有効に低下させた。これに対して不活性対照化合物(図1C)はHc O−グリコシル化の低下を示さなかった。実施例1、2及び3に示す阻害剤でも同様の結果が得られた。これらの結果をまとめると、実施例1〜4に示す新規Pmt阻害剤は真菌O−グリコシル化の有効な阻害剤であることが分かる。
【0113】
本明細書では例示態様について本発明を説明するが、当然のことながら本発明はこれらの態様に限定されない。本明細書の教示を受けた当業者は本発明の範囲に含まれる他の変形及び態様も認識しよう。従って、本発明は以下の特許請求の範囲のみにより限定される。
【特許請求の範囲】
【請求項1】
以下の群:
【化1】
から選択される化合物又はその塩。
【請求項2】
O−結合型グリコシル化の低下した蛋白質の生産方法であって、
(a)前記蛋白質を産生する培養液中で細胞を増殖させる段階と;
(b)Pmt介在性O−結合型グリコシル化を阻害する請求項1に記載の1種以上の化合物と培養液を接触させる段階と;
(c)宿主細胞により産生された蛋白質を単離する段階を含む前記方法。
【請求項3】
(a)蛋白質をコードする核酸を準備する段階と;
(b)核酸を細胞に導入する段階
により細胞の培養液を準備する請求項2に記載の方法。
【請求項4】
Pmt介在性O−結合型グリコシル化を阻害する1種以上の化合物のいずれかと培養液を接触させる前に、核酸を導入した多数の細胞を得るために十分な時間にわたって培養液を増殖させる請求項3に記載の方法。
【請求項5】
Pmt介在性O−結合型グリコシル化を阻害する1種以上の化合物のいずれかの存在下で培養液を増殖させる請求項3に記載の方法。
【請求項6】
核酸を誘導性プロモーターと機能的に連結する請求項3に記載の方法。
【請求項7】
Pmt介在性O−結合型グリコシル化を阻害する前記1種以上の化合物とプロモーターのインデューサーとに培養液を接触させて蛋白質の発現を誘導し、前記1種以上の阻害剤とインデューサーの存在下で細胞により産生される蛋白質を単離してO−結合型グリコシル化の低下した蛋白質を生産する前に、核酸を導入した多数の細胞を得るために十分な時間にわたって培養液を増殖させる請求項6に記載の方法。
【請求項8】
Pmt介在性O−結合型グリコシル化を阻害する前記1種以上の化合物と培養液を接触させ、Pmt介在性O−結合型グリコシル化を阻害する前記1種以上の化合物とインデューサーの存在下で細胞により産生される蛋白質を単離してO−結合型グリコシル化の低下した蛋白質を生産する前に、培養液をプロモーターのインデューサーと所定時間接触させて蛋白質の発現を誘導する請求項6に記載の方法。
【請求項9】
細胞が真菌細胞である請求項3に記載の方法。
【請求項10】
細胞が酵母細胞である請求項3に記載の方法。
【請求項11】
細胞がK.lactis、Pichia pastoris、Pichia methanolica及びHansenulaから構成される群から選択される請求項3に記載の方法。
【請求項12】
細胞がPichia pastorisである請求項3に記載の方法。
【請求項13】
細胞がN−グリカングリコフォームを主体とする糖蛋白質を産生するように遺伝子改変された酵母又は糸状菌細胞である請求項3に記載の方法。
【請求項14】
N−グリコシル化パターンがヒト様又はヒト化型である糖蛋白質を産生するように細胞が遺伝子改変されている請求項3に記載の方法。
【請求項15】
培地1リットル当たり少なくとも100mgの収率で蛋白質を生産する請求項3に記載の方法。
【請求項1】
以下の群:
【化1】
から選択される化合物又はその塩。
【請求項2】
O−結合型グリコシル化の低下した蛋白質の生産方法であって、
(a)前記蛋白質を産生する培養液中で細胞を増殖させる段階と;
(b)Pmt介在性O−結合型グリコシル化を阻害する請求項1に記載の1種以上の化合物と培養液を接触させる段階と;
(c)宿主細胞により産生された蛋白質を単離する段階を含む前記方法。
【請求項3】
(a)蛋白質をコードする核酸を準備する段階と;
(b)核酸を細胞に導入する段階
により細胞の培養液を準備する請求項2に記載の方法。
【請求項4】
Pmt介在性O−結合型グリコシル化を阻害する1種以上の化合物のいずれかと培養液を接触させる前に、核酸を導入した多数の細胞を得るために十分な時間にわたって培養液を増殖させる請求項3に記載の方法。
【請求項5】
Pmt介在性O−結合型グリコシル化を阻害する1種以上の化合物のいずれかの存在下で培養液を増殖させる請求項3に記載の方法。
【請求項6】
核酸を誘導性プロモーターと機能的に連結する請求項3に記載の方法。
【請求項7】
Pmt介在性O−結合型グリコシル化を阻害する前記1種以上の化合物とプロモーターのインデューサーとに培養液を接触させて蛋白質の発現を誘導し、前記1種以上の阻害剤とインデューサーの存在下で細胞により産生される蛋白質を単離してO−結合型グリコシル化の低下した蛋白質を生産する前に、核酸を導入した多数の細胞を得るために十分な時間にわたって培養液を増殖させる請求項6に記載の方法。
【請求項8】
Pmt介在性O−結合型グリコシル化を阻害する前記1種以上の化合物と培養液を接触させ、Pmt介在性O−結合型グリコシル化を阻害する前記1種以上の化合物とインデューサーの存在下で細胞により産生される蛋白質を単離してO−結合型グリコシル化の低下した蛋白質を生産する前に、培養液をプロモーターのインデューサーと所定時間接触させて蛋白質の発現を誘導する請求項6に記載の方法。
【請求項9】
細胞が真菌細胞である請求項3に記載の方法。
【請求項10】
細胞が酵母細胞である請求項3に記載の方法。
【請求項11】
細胞がK.lactis、Pichia pastoris、Pichia methanolica及びHansenulaから構成される群から選択される請求項3に記載の方法。
【請求項12】
細胞がPichia pastorisである請求項3に記載の方法。
【請求項13】
細胞がN−グリカングリコフォームを主体とする糖蛋白質を産生するように遺伝子改変された酵母又は糸状菌細胞である請求項3に記載の方法。
【請求項14】
N−グリコシル化パターンがヒト様又はヒト化型である糖蛋白質を産生するように細胞が遺伝子改変されている請求項3に記載の方法。
【請求項15】
培地1リットル当たり少なくとも100mgの収率で蛋白質を生産する請求項3に記載の方法。
【図1A】

【図1B】
【図1C】
【図1B】
【図1C】
【公表番号】特表2011−520968(P2011−520968A)
【公表日】平成23年7月21日(2011.7.21)
【国際特許分類】
【出願番号】特願2011−510611(P2011−510611)
【出願日】平成21年5月18日(2009.5.18)
【国際出願番号】PCT/US2009/044297
【国際公開番号】WO2009/143041
【国際公開日】平成21年11月26日(2009.11.26)
【出願人】(390023526)メルク・シャープ・エンド・ドーム・コーポレイション (924)
【Fターム(参考)】
【公表日】平成23年7月21日(2011.7.21)
【国際特許分類】
【出願日】平成21年5月18日(2009.5.18)
【国際出願番号】PCT/US2009/044297
【国際公開番号】WO2009/143041
【国際公開日】平成21年11月26日(2009.11.26)
【出願人】(390023526)メルク・シャープ・エンド・ドーム・コーポレイション (924)
【Fターム(参考)】
[ Back to top ]