
ミオスタチン・アンタゴニストの使用
【課題】性腺機能低下症、リウマチ性悪液質、火傷による悪液質、化学物質の投与による悪液質、糖尿病による悪液質、糖尿病性腎症、プラダー・ウィリー症候群、過剰なTNF-α、ならびに他の筋肉関連、代謝および炎症性傷害から生じる障害を治療するための方法の提供。
【解決手段】フォリスタチン、ミオスタチン・プロドメイン、GDF−11プロドメイン、ミオスタチンに結合するアンタゴニスト性抗体または抗体断片、アクチビンIIB型受容体に結合するアンタゴニスト性抗体または抗体断片、可溶性アクチビンIIB型受容体、可溶性アクチビンIIB型受容体融合タンパク質、可溶性ミオスタチン類似体などのミオスタチン・アンタゴニストの投与。
【解決手段】フォリスタチン、ミオスタチン・プロドメイン、GDF−11プロドメイン、ミオスタチンに結合するアンタゴニスト性抗体または抗体断片、アクチビンIIB型受容体に結合するアンタゴニスト性抗体または抗体断片、可溶性アクチビンIIB型受容体、可溶性アクチビンIIB型受容体融合タンパク質、可溶性ミオスタチン類似体などのミオスタチン・アンタゴニストの投与。
Notice: Undefined index: DEJ in /mnt/www/gzt_disp.php on line 298
【特許請求の範囲】
【請求項1】
性腺機能低下症の影響を治療する必要がある被験体における、こうした影響の治療において使用するための医薬組成物であって、薬学的に許容されうる担体中にミオスタチン・アンタゴニストを含み、ここで該アンタゴニストはフォリスタチン、ミオスタチン・プロドメイン、GDF−11プロドメイン、プロドメイン融合タンパク質、ミオスタチンに結合するアンタゴニスト性抗体または抗体断片、アクチビンIIB型受容体に結合するアンタゴニスト性抗体または抗体断片、可溶性アクチビンIIB型受容体、可溶性アクチビンIIB型受容体融合タンパク質、可溶性ミオスタチン類似体、オリゴヌクレオチド、小分子、ペプチド模倣体、およびミオスタチン結合剤からなる群より選択される、前記医薬組成物。
【請求項2】
性腺機能低下症がアンドロゲン枯渇療法の結果として生じる、請求項1の医薬組成物。
【請求項3】
性腺機能低下症が、性腺機能の加齢に伴う減少から生じる、請求項1の医薬組成物。
【請求項4】
ミオスタチン・アンタゴニストがミオスタチン結合剤であり、そして該結合剤が、配列Cb1b2Wb3WMCPP(配列番号353)、[ここで
b1は、アミノ酸T、I、またはRのいずれか1つから選択され;
b2は、R、S、Qのいずれか1つから選択され;
b3は、P、RおよびQのいずれか1つから選択される]
を含み、そして長さが10〜50アミノ酸の間である、ミオスタチンに結合可能な少なくとも1つのペプチドおよびその生理学的に許容されうる塩を含む、請求項1の医薬組成物。
【請求項5】
ミオスタチン・アンタゴニストがミオスタチン結合剤であり、そして該結合剤が、構造:
(X1)a−F1−(X2)b、またはそのマルチマー;
[ここでF1はビヒクルであり;そしてX1およびX2は、各々独立に
−(L1)c−P1;
−(L1)c−P1−(L2)d−P2;
−(L1)c−P1−(L2)d−P2−(L3)e−P3;
および−(L1)c−P1−(L2)d−P2−(L3)e−P3−(L4)f−P4;
から選択され、
ここでP1、P2、P3、およびP4は、各々ミオスタチンに結合可能なペプチドであり、そして各々は配列Cb1b2Wb3WMCPP(配列番号353)、[ここで
b1は、アミノ酸T、I、またはRのいずれか1つから選択され;
b2は、R、S、Qのいずれか1つから選択され;
b3は、P、RおよびQのいずれか1つから選択される]を含み、
そしてここで、L1、L2、L3、およびL4は、各々リンカーであり;
そしてa、b、c、d、eおよびfは、各々独立に0または1である、但しaおよびbの少なくとも1つは1である;]
およびその生理学的に許容されうる塩を有する、請求項1の医薬組成物。
【請求項6】
P1、P2、P3、およびP4が、ミオスタチンに結合可能なペプチドであり、そして独立に、配列番号305〜308、310〜313、315〜331、333および335〜346からなる群より選択される、請求項4または5に記載の医薬組成物。
【請求項7】
aが0およびbが1、またはaが1およびbが0である、請求項5または6に記載の医薬組成物。
【請求項8】
ビヒクルがFcドメインである、請求項5ないし7のいずれか1項に記載の医薬組成物。
【請求項9】
結合剤が、構造:F1−L1−P1;F1−L1−P1−L2−P2;P1−L1−F1;P2−L2−P1−L1−F1;を有し、そしてここで、F1はヒトIgG Fcドメインである、請求項7に記載の医薬組成物。
【請求項10】
結合剤が、構造F1−(L1)−P1またはF1−L1−P1−L2−P2を有し、ここでP1およびP2は、配列番号311、325、326、336または337のいずれか1つから選択され、そしてここでL1は(Gly)5またはAQが続く(Gly)5であり、そしてここで、F1はヒトIgG Fcドメインである、請求項9に記載の医薬組成物。
【請求項11】
糖尿病性腎症を治療する必要がある被験体における、糖尿病性腎症の治療において使用するための医薬組成物であって、薬学的に許容されうる担体中にミオスタチン・アンタゴニストを含み、ここで該アンタゴニストはフォリスタチン、ミオスタチン・プロドメイン、GDF−11プロドメイン、プロドメイン融合タンパク質、ミオスタチンに結合するアンタゴニスト性抗体または抗体断片、アクチビンIIB型受容体に結合するアンタゴニスト性抗体または抗体断片、可溶性アクチビンIIB型受容体、可溶性アクチビンIIB型受容体融合タンパク質、可溶性ミオスタチン類似体、オリゴヌクレオチド、小分子、ペプチド模倣体、およびミオスタチン結合剤からなる群より選択される、前記医薬組成物。
【請求項12】
ミオスタチン・アンタゴニストがミオスタチン結合剤であり、そして該結合剤が、配列Cb1b2Wb3WMCPP(配列番号353)、[ここで
b1は、アミノ酸T、I、またはRのいずれか1つから選択され;
b2は、R、S、Qのいずれか1つから選択され;
b3は、P、RおよびQのいずれか1つから選択される]
を含み、そして長さが10〜50アミノ酸の間である、ミオスタチンに結合可能な少なくとも1つのペプチドおよびその生理学的に許容されうる塩を含む、請求項11の医薬組成物。
【請求項13】
ミオスタチン・アンタゴニストがミオスタチン結合剤であり、そして該結合剤が、構造:
(X1)a−F1−(X2)b、またはそのマルチマー;
[ここでF1はビヒクルであり;そしてX1およびX2は、各々独立に
−(L1)c−P1;
−(L1)c−P1−(L2)d−P2;
−(L1)c−P1−(L2)d−P2−(L3)e−P3;
および−(L1)c−P1−(L2)d−P2−(L3)e−P3−(L4)f−P4;
から選択され、
ここでP1、P2、P3、およびP4は、各々ミオスタチンに結合可能なペプチドであり;そして各々は配列Cb1b2Wb3WMCPP(配列番号353)[ここで
b1は、アミノ酸T、I、またはRのいずれか1つから選択され;
b2は、R、S、Qのいずれか1つから選択され;
b3は、P、RおよびQのいずれか1つから選択される]を含み、
そしてここで、L1、L2、L3、およびL4は、各々リンカーであり;
そしてa、b、c、d、eおよびfは、各々独立に0または1である、但しaおよびbの少なくとも1つは1である]、
およびその生理学的に許容されうる塩を有する、請求項11の医薬組成物。
【請求項14】
P1、P2、P3、およびP4は、ミオスタチンに結合可能なペプチドであり、そして独立に、配列番号305〜308、310〜313、315〜331、333および335〜346からなる群より選択される、請求項12または13に記載の医薬組成物。
【請求項15】
aが0およびbが1、またはaが1およびbが0である、請求項13または14に記載の医薬組成物。
【請求項16】
ビヒクルがFcドメインである、請求項13ないし15のいずれか1項に記載の医薬組成物。
【請求項17】
結合剤が、構造:F1−L1−P1;F1−L1−P1−L2−P2;P1−L1−F1;P2−L2−P1−L1−F1;を有し、そしてここで、F1はヒトIgG Fcドメインである、請求項15に記載の医薬組成物。
【請求項18】
結合剤が、構造:F1−(L1)−P1またはF1−L1−P1−L2−P2を有し、ここでP1およびP2は、配列番号311、325、326、336または337のいずれか1つから選択され、そしてここでL1は(Gly)5またはAQが続く(Gly)5であり、そしてここで、F1はヒトIgG Fcドメインである、請求項17に記載の医薬組成物。
【請求項19】
過剰なTNF−αを治療する必要がある被験体における、過剰なTNF−αの治療において使用するための医薬組成物であって、薬学的に許容されうる担体中にミオスタチン・アンタゴニストを含み、ここで該アンタゴニストはフォリスタチン、ミオスタチン・プロドメイン、GDF−11プロドメイン、プロドメイン融合タンパク質、ミオスタチンに結合するアンタゴニスト性抗体または抗体断片、アクチビンIIB型受容体に結合するアンタゴニスト性抗体または抗体断片、可溶性アクチビンIIB型受容体、可溶性アクチビン
IIB型受容体融合タンパク質、可溶性ミオスタチン類似体、オリゴヌクレオチド、小分子、ペプチド模倣体、およびミオスタチン結合剤からなる群より選択される、前記医薬組成物。
【請求項20】
ミオスタチン・アンタゴニストがミオスタチン結合剤であり、そして該結合剤が、配列Cb1b2Wb3WMCPP(配列番号353)、[ここで
b1は、アミノ酸T、I、またはRのいずれか1つから選択され;
b2は、R、S、Qのいずれか1つから選択され;
b3は、P、RおよびQのいずれか1つから選択される]
を含み、そして長さが10〜50アミノ酸の間である、ミオスタチンに結合可能な少なくとも1つのペプチドおよびその生理学的に許容されうる塩を含む、請求項19の医薬組成物。
【請求項21】
ミオスタチン・アンタゴニストがミオスタチン結合剤であり、そして該結合剤が、構造:
(X1)a−F1−(X2)b、またはそのマルチマー;
[ここでF1はビヒクルであり;そしてX1およびX2は、各々独立に
−(L1)c−P1;
−(L1)c−P1−(L2)d−P2;
−(L1)c−P1−(L2)d−P2−(L3)e−P3;
および−(L1)c−P1−(L2)d−P2−(L3)e−P3−(L4)f−P4;
から選択され、
ここでP1、P2、P3、およびP4は、各々ミオスタチンに結合可能なペプチドであり、そして各々は配列Cb1b2Wb3WMCPP(配列番号353)、[ここで
b1は、アミノ酸T、I、またはRのいずれか1つから選択され;
b2は、R、S、Qのいずれか1つから選択され;
b3は、P、RおよびQのいずれか1つから選択される]を含み、
そしてここで、L1、L2、L3、およびL4は、各々リンカーであり;
そしてa、b、c、d、eおよびfは、各々独立に0または1である、但しaおよびbの少なくとも1つは1である;]
およびその生理学的に許容されうる塩を有する、請求項19の医薬組成物。
【請求項22】
P1、P2、P3、およびP4が、ミオスタチンに結合可能なペプチドであり、そして独立に、配列番号305〜308、310〜313、315〜331、333および335〜346からなる群より選択される、
請求項20または21に記載の医薬組成物。
【請求項23】
aが0およびbが1、またはaが1およびbが0である、請求項21または22に記載の医薬組成物。
【請求項24】
ビヒクルがFcドメインである、請求項21ないし23のいずれか1項に記載の医薬組成物。
【請求項25】
結合剤が、構造:F1−L1−P1;F1−L1−P1−L2−P2;P1−L1−F1;P2−L2−P1−L1−F1;を有し、そしてここで、F1はヒトIgG Fcドメインである、請求項23に記載の医薬組成物。
【請求項26】
結合剤が、構造F1−(L1)−P1またはF1−L1−P1−L2−P2を有し、ここでP1およびP2は、配列番号311、325、326、336または337のいずれ
か1つから選択され、そしてここでL1は(Gly)5またはAQが続く(Gly)5であり、そしてここで、F1はヒトIgG Fcドメインである、請求項25に記載の医薬組成物。
【請求項1】
性腺機能低下症の影響を治療する必要がある被験体における、こうした影響の治療において使用するための医薬組成物であって、薬学的に許容されうる担体中にミオスタチン・アンタゴニストを含み、ここで該アンタゴニストはフォリスタチン、ミオスタチン・プロドメイン、GDF−11プロドメイン、プロドメイン融合タンパク質、ミオスタチンに結合するアンタゴニスト性抗体または抗体断片、アクチビンIIB型受容体に結合するアンタゴニスト性抗体または抗体断片、可溶性アクチビンIIB型受容体、可溶性アクチビンIIB型受容体融合タンパク質、可溶性ミオスタチン類似体、オリゴヌクレオチド、小分子、ペプチド模倣体、およびミオスタチン結合剤からなる群より選択される、前記医薬組成物。
【請求項2】
性腺機能低下症がアンドロゲン枯渇療法の結果として生じる、請求項1の医薬組成物。
【請求項3】
性腺機能低下症が、性腺機能の加齢に伴う減少から生じる、請求項1の医薬組成物。
【請求項4】
ミオスタチン・アンタゴニストがミオスタチン結合剤であり、そして該結合剤が、配列Cb1b2Wb3WMCPP(配列番号353)、[ここで
b1は、アミノ酸T、I、またはRのいずれか1つから選択され;
b2は、R、S、Qのいずれか1つから選択され;
b3は、P、RおよびQのいずれか1つから選択される]
を含み、そして長さが10〜50アミノ酸の間である、ミオスタチンに結合可能な少なくとも1つのペプチドおよびその生理学的に許容されうる塩を含む、請求項1の医薬組成物。
【請求項5】
ミオスタチン・アンタゴニストがミオスタチン結合剤であり、そして該結合剤が、構造:
(X1)a−F1−(X2)b、またはそのマルチマー;
[ここでF1はビヒクルであり;そしてX1およびX2は、各々独立に
−(L1)c−P1;
−(L1)c−P1−(L2)d−P2;
−(L1)c−P1−(L2)d−P2−(L3)e−P3;
および−(L1)c−P1−(L2)d−P2−(L3)e−P3−(L4)f−P4;
から選択され、
ここでP1、P2、P3、およびP4は、各々ミオスタチンに結合可能なペプチドであり、そして各々は配列Cb1b2Wb3WMCPP(配列番号353)、[ここで
b1は、アミノ酸T、I、またはRのいずれか1つから選択され;
b2は、R、S、Qのいずれか1つから選択され;
b3は、P、RおよびQのいずれか1つから選択される]を含み、
そしてここで、L1、L2、L3、およびL4は、各々リンカーであり;
そしてa、b、c、d、eおよびfは、各々独立に0または1である、但しaおよびbの少なくとも1つは1である;]
およびその生理学的に許容されうる塩を有する、請求項1の医薬組成物。
【請求項6】
P1、P2、P3、およびP4が、ミオスタチンに結合可能なペプチドであり、そして独立に、配列番号305〜308、310〜313、315〜331、333および335〜346からなる群より選択される、請求項4または5に記載の医薬組成物。
【請求項7】
aが0およびbが1、またはaが1およびbが0である、請求項5または6に記載の医薬組成物。
【請求項8】
ビヒクルがFcドメインである、請求項5ないし7のいずれか1項に記載の医薬組成物。
【請求項9】
結合剤が、構造:F1−L1−P1;F1−L1−P1−L2−P2;P1−L1−F1;P2−L2−P1−L1−F1;を有し、そしてここで、F1はヒトIgG Fcドメインである、請求項7に記載の医薬組成物。
【請求項10】
結合剤が、構造F1−(L1)−P1またはF1−L1−P1−L2−P2を有し、ここでP1およびP2は、配列番号311、325、326、336または337のいずれか1つから選択され、そしてここでL1は(Gly)5またはAQが続く(Gly)5であり、そしてここで、F1はヒトIgG Fcドメインである、請求項9に記載の医薬組成物。
【請求項11】
糖尿病性腎症を治療する必要がある被験体における、糖尿病性腎症の治療において使用するための医薬組成物であって、薬学的に許容されうる担体中にミオスタチン・アンタゴニストを含み、ここで該アンタゴニストはフォリスタチン、ミオスタチン・プロドメイン、GDF−11プロドメイン、プロドメイン融合タンパク質、ミオスタチンに結合するアンタゴニスト性抗体または抗体断片、アクチビンIIB型受容体に結合するアンタゴニスト性抗体または抗体断片、可溶性アクチビンIIB型受容体、可溶性アクチビンIIB型受容体融合タンパク質、可溶性ミオスタチン類似体、オリゴヌクレオチド、小分子、ペプチド模倣体、およびミオスタチン結合剤からなる群より選択される、前記医薬組成物。
【請求項12】
ミオスタチン・アンタゴニストがミオスタチン結合剤であり、そして該結合剤が、配列Cb1b2Wb3WMCPP(配列番号353)、[ここで
b1は、アミノ酸T、I、またはRのいずれか1つから選択され;
b2は、R、S、Qのいずれか1つから選択され;
b3は、P、RおよびQのいずれか1つから選択される]
を含み、そして長さが10〜50アミノ酸の間である、ミオスタチンに結合可能な少なくとも1つのペプチドおよびその生理学的に許容されうる塩を含む、請求項11の医薬組成物。
【請求項13】
ミオスタチン・アンタゴニストがミオスタチン結合剤であり、そして該結合剤が、構造:
(X1)a−F1−(X2)b、またはそのマルチマー;
[ここでF1はビヒクルであり;そしてX1およびX2は、各々独立に
−(L1)c−P1;
−(L1)c−P1−(L2)d−P2;
−(L1)c−P1−(L2)d−P2−(L3)e−P3;
および−(L1)c−P1−(L2)d−P2−(L3)e−P3−(L4)f−P4;
から選択され、
ここでP1、P2、P3、およびP4は、各々ミオスタチンに結合可能なペプチドであり;そして各々は配列Cb1b2Wb3WMCPP(配列番号353)[ここで
b1は、アミノ酸T、I、またはRのいずれか1つから選択され;
b2は、R、S、Qのいずれか1つから選択され;
b3は、P、RおよびQのいずれか1つから選択される]を含み、
そしてここで、L1、L2、L3、およびL4は、各々リンカーであり;
そしてa、b、c、d、eおよびfは、各々独立に0または1である、但しaおよびbの少なくとも1つは1である]、
およびその生理学的に許容されうる塩を有する、請求項11の医薬組成物。
【請求項14】
P1、P2、P3、およびP4は、ミオスタチンに結合可能なペプチドであり、そして独立に、配列番号305〜308、310〜313、315〜331、333および335〜346からなる群より選択される、請求項12または13に記載の医薬組成物。
【請求項15】
aが0およびbが1、またはaが1およびbが0である、請求項13または14に記載の医薬組成物。
【請求項16】
ビヒクルがFcドメインである、請求項13ないし15のいずれか1項に記載の医薬組成物。
【請求項17】
結合剤が、構造:F1−L1−P1;F1−L1−P1−L2−P2;P1−L1−F1;P2−L2−P1−L1−F1;を有し、そしてここで、F1はヒトIgG Fcドメインである、請求項15に記載の医薬組成物。
【請求項18】
結合剤が、構造:F1−(L1)−P1またはF1−L1−P1−L2−P2を有し、ここでP1およびP2は、配列番号311、325、326、336または337のいずれか1つから選択され、そしてここでL1は(Gly)5またはAQが続く(Gly)5であり、そしてここで、F1はヒトIgG Fcドメインである、請求項17に記載の医薬組成物。
【請求項19】
過剰なTNF−αを治療する必要がある被験体における、過剰なTNF−αの治療において使用するための医薬組成物であって、薬学的に許容されうる担体中にミオスタチン・アンタゴニストを含み、ここで該アンタゴニストはフォリスタチン、ミオスタチン・プロドメイン、GDF−11プロドメイン、プロドメイン融合タンパク質、ミオスタチンに結合するアンタゴニスト性抗体または抗体断片、アクチビンIIB型受容体に結合するアンタゴニスト性抗体または抗体断片、可溶性アクチビンIIB型受容体、可溶性アクチビン
IIB型受容体融合タンパク質、可溶性ミオスタチン類似体、オリゴヌクレオチド、小分子、ペプチド模倣体、およびミオスタチン結合剤からなる群より選択される、前記医薬組成物。
【請求項20】
ミオスタチン・アンタゴニストがミオスタチン結合剤であり、そして該結合剤が、配列Cb1b2Wb3WMCPP(配列番号353)、[ここで
b1は、アミノ酸T、I、またはRのいずれか1つから選択され;
b2は、R、S、Qのいずれか1つから選択され;
b3は、P、RおよびQのいずれか1つから選択される]
を含み、そして長さが10〜50アミノ酸の間である、ミオスタチンに結合可能な少なくとも1つのペプチドおよびその生理学的に許容されうる塩を含む、請求項19の医薬組成物。
【請求項21】
ミオスタチン・アンタゴニストがミオスタチン結合剤であり、そして該結合剤が、構造:
(X1)a−F1−(X2)b、またはそのマルチマー;
[ここでF1はビヒクルであり;そしてX1およびX2は、各々独立に
−(L1)c−P1;
−(L1)c−P1−(L2)d−P2;
−(L1)c−P1−(L2)d−P2−(L3)e−P3;
および−(L1)c−P1−(L2)d−P2−(L3)e−P3−(L4)f−P4;
から選択され、
ここでP1、P2、P3、およびP4は、各々ミオスタチンに結合可能なペプチドであり、そして各々は配列Cb1b2Wb3WMCPP(配列番号353)、[ここで
b1は、アミノ酸T、I、またはRのいずれか1つから選択され;
b2は、R、S、Qのいずれか1つから選択され;
b3は、P、RおよびQのいずれか1つから選択される]を含み、
そしてここで、L1、L2、L3、およびL4は、各々リンカーであり;
そしてa、b、c、d、eおよびfは、各々独立に0または1である、但しaおよびbの少なくとも1つは1である;]
およびその生理学的に許容されうる塩を有する、請求項19の医薬組成物。
【請求項22】
P1、P2、P3、およびP4が、ミオスタチンに結合可能なペプチドであり、そして独立に、配列番号305〜308、310〜313、315〜331、333および335〜346からなる群より選択される、
請求項20または21に記載の医薬組成物。
【請求項23】
aが0およびbが1、またはaが1およびbが0である、請求項21または22に記載の医薬組成物。
【請求項24】
ビヒクルがFcドメインである、請求項21ないし23のいずれか1項に記載の医薬組成物。
【請求項25】
結合剤が、構造:F1−L1−P1;F1−L1−P1−L2−P2;P1−L1−F1;P2−L2−P1−L1−F1;を有し、そしてここで、F1はヒトIgG Fcドメインである、請求項23に記載の医薬組成物。
【請求項26】
結合剤が、構造F1−(L1)−P1またはF1−L1−P1−L2−P2を有し、ここでP1およびP2は、配列番号311、325、326、336または337のいずれ
か1つから選択され、そしてここでL1は(Gly)5またはAQが続く(Gly)5であり、そしてここで、F1はヒトIgG Fcドメインである、請求項25に記載の医薬組成物。
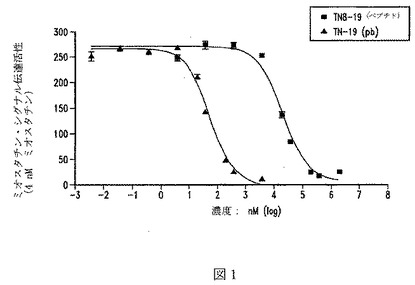
【図1】


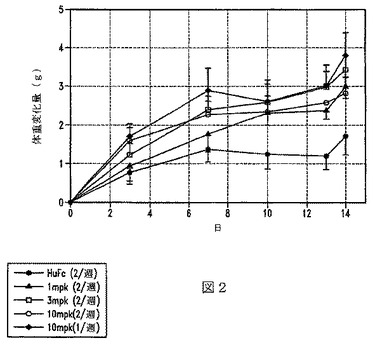
【図2】
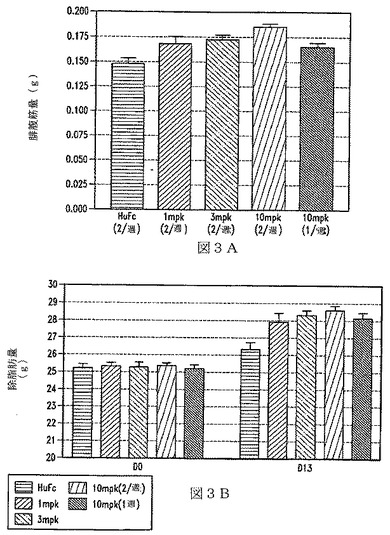
【図3】
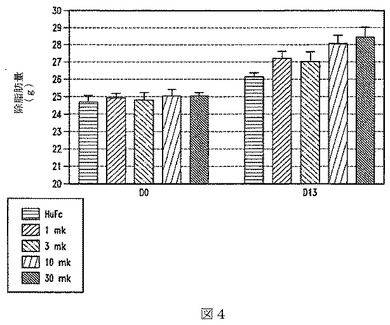
【図4】
【図5】
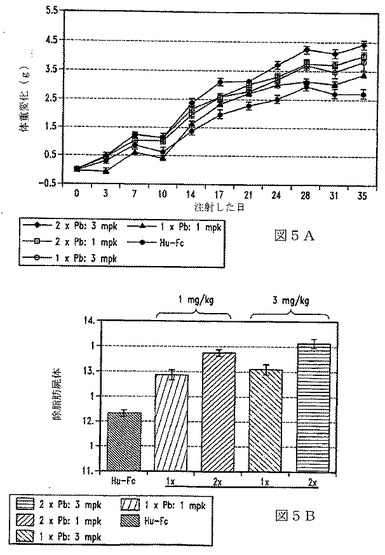
【図6】
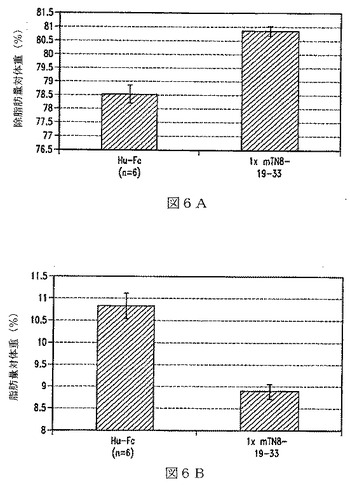
【図7】
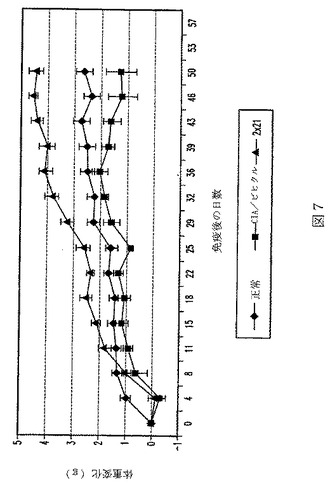
【図8】
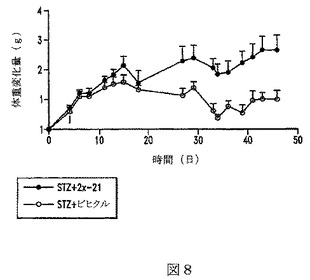
【図9】
【図10】
【図11】
【図12】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【公開番号】特開2013−28620(P2013−28620A)
【公開日】平成25年2月7日(2013.2.7)
【国際特許分類】
【出願番号】特願2012−200485(P2012−200485)
【出願日】平成24年9月12日(2012.9.12)
【分割の表示】特願2008−544468(P2008−544468)の分割
【原出願日】平成18年12月6日(2006.12.6)
【出願人】(500203709)アムジェン インコーポレイテッド (76)
【Fターム(参考)】
【公開日】平成25年2月7日(2013.2.7)
【国際特許分類】
【出願日】平成24年9月12日(2012.9.12)
【分割の表示】特願2008−544468(P2008−544468)の分割
【原出願日】平成18年12月6日(2006.12.6)
【出願人】(500203709)アムジェン インコーポレイテッド (76)
【Fターム(参考)】
[ Back to top ]