
ミモシン又はその酸付加塩とミモシン誘導体の製造方法並びにミモシンの品質管理法
【課題】工業的な大量生産に応用し得る、ミモシンをマメ科の植物で亜熱帯地方に広く分布するギンネムから高純度且つ低コストで分離精製できる方法、精製ミモシンの品質管理に有用な光学純度の検定方法、及びミモシン導入ペプチドの製造に有用なミモシン誘導体の製造方法を提供すること。
【解決手段】本発明の精製ミモシン又はその酸付加塩の製造方法は、
(1) ギンネムを水に浸漬してミモシンを抽出する工程、
(2) 抽出液中に陽イオン交換樹脂を添加してミモシンを陽イオン交換樹脂に吸着させる工程、
(3) 前記陽イオン交換樹脂からミモシンを溶出させる工程、
(4) 工程(3)で得られた溶出液からミモシンの粗結晶を得る工程、
(5) 再結晶法により前記粗結晶からミモシンの結晶を得る工程
を含む。
【解決手段】本発明の精製ミモシン又はその酸付加塩の製造方法は、
(1) ギンネムを水に浸漬してミモシンを抽出する工程、
(2) 抽出液中に陽イオン交換樹脂を添加してミモシンを陽イオン交換樹脂に吸着させる工程、
(3) 前記陽イオン交換樹脂からミモシンを溶出させる工程、
(4) 工程(3)で得られた溶出液からミモシンの粗結晶を得る工程、
(5) 再結晶法により前記粗結晶からミモシンの結晶を得る工程
を含む。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、精製ミモシン又はその酸付加塩の製造方法、ミモシンの光学純度の検定方法、保護基が導入されたミモシン誘導体、及びFmoc基等の保護基による保護ミモシンの製造方法に関する。
【背景技術】
【0002】
近年、生物学と化学を融合させ、生体の機能と化合物との関係から有用物質や生体反応に関わる化合物を探索する手法としてケミカルバイオロジーが脚光を浴びている。この流れに先行してコンビナトリアルケミストリーが創薬を中心に有用物質の創出手法として重視されてきた。当該分野では、天然のタンパク質中には存在しない非天然アミノ酸/異常アミノ酸の利用が盛んであるが、化学合成による高純度の立体不斉アミノ酸の製造は容易ではなく、また極めて高価である。特徴のある非天然アミノ酸/異常アミノ酸を効率よく天然物から取得することは、光学純度の点でも有利である。しかしながら精製の過程で起きうるラセミ化によって精製物の光学純度に問題が生じうるため、光学純度を正確に検定することは重要である。
【0003】
数多くの種類の異常アミノ酸が知られているが、その中でもミモシンはチロシナーゼ阻害活性、アレロパシー活性、細胞毒性、あるいは抗腫瘍活性などを有するため、創薬、農薬創製、機能性化粧品開発等の分野での応用が期待される(特許文献1、非特許文献1)。
【0004】
熱帯植物ギンネムは、家畜飼料等では有害とされるミモシンを含むことが知られており、水に浸漬することによりギンネムからミモシンを除去できる方法が知られている(特許文献2)。また、ギンネムからのミモシンの抽出方法としては、非特許文献2に記載されるように、水中にギンネムを浸漬してミモシンを抽出し、該溶液から限外濾過を用いてミモシンを分離する方法が知られている。
【0005】
ミモシンはキラル炭素を有する分子であるため光学異性体が存在するが、通常、生物活性は光学異性体によって異なるため、ミモシンを各種分野で応用するためには光学純度の検定が必要不可欠である。また、ギンネムから抽出や精製作業中にミモシンの一部がラセミ化する可能性があるため、高純度サンプルを保障するための品質管理法は、とりわけ創薬原料などには重要である。
【0006】
上記の通り、ミモシンそのものは化合物として既知であり、また化学合成による製法もその全合成は報告されているが、合成はD,L体混合物であり、その筆者は天然物をラセミ化させて合成の標品としている(非特許文献3)。また、天然物由来として、マメなどの植物由来で抽出され試薬として市販されている。しかしながら現在市販されているミモシンは高価であり、研究目的はともかくとして、大量に使用する工業的なミモシンの利用は行われていない。その理由の一つはミモシンそのものが安定性に乏しいことである。大量に抽出する安価な方法は知られていない。また、大量に誘導体化して例えばペプチド合成に用いるという例もほとんど知られていない。キラリティ(立体不斉)に関しての報告は全くない。実際に、キラルカラムを利用してエナンショマーラベリングという手法でアミノ酸のキラリティを解明する手法が知られているが(非特許文献4〜8)、ミモシンではこの手法、エナンショマーラベリング法を用いることができなかった。この理由はミモシンが誘導体化の過程で分解するからである。また、アミノ酸のキラル分離用のカラムが市販されている(Crownpak(+)、ダイセル化学工業株式会社)。このカラムで分離した場合、天然アミノ酸であるアラニンラセミ体の一方とミモシンの溶出位置とが重なり厳密な判定は不可能であった。ミモシンは分解するとアラニンを生成することが知られている。したがって、これまで抽出されたミモシンのキラリティを正確に報告した例は無い。
【0007】
ミモシンを創薬に用いる場合、ミモシン単体で用いるよりも、ペプチドに導入することで細胞透過効率や分子/細胞認識能などの機能を付与することができればより付加価値が高まる。公知のペプチド合成法では、官能基を保護基で保護することによってペプチド鎖伸張中の副反応を回避できるため、たとえばアミノ酸のアミノ基をFmoc等で保護する必要がある。
【0008】
しかしながら、ギンネムから水で抽出した粗ミモシン溶液から、高純度のミモシンを分離精製するための工業的に有利な手法は知られていないため、ミモシンを創薬等の種々の分野で応用することもできないのが現状である。非特許文献2の方法は、水に抽出した粗ミモシン溶液からミモシンを抽出する方法であるが、限外濾過を用いるものであり、費用もかかることから、工業的なスケールで大量に精製する方法としては不向きである。また、ギンネムから分離精製したミモシンの光学純度を正確に測定できる方法は知られていない。さらに、ミモシンをペプチドへ導入するためには保護基を導入しなければならないが、ミモシンの効率的な誘導体化法とその検定法も知られていない。
【0009】
【特許文献1】特開昭63-295502
【特許文献2】特開昭63-294746
【非特許文献1】W.D. DeWys and T.C. Hall, Europ. J. Cancer Vol. 9, pp. 281-283. 1973.
【非特許文献2】農薬 1994, Vol 41, No 2, page 18-22 日本農薬株式会社
【非特許文献3】R. Adams and J.L. Johnson, Leucenol. VI. A Total Synthesis. J. Am. Chem Soc., 1949, 71, 705-708,
【非特許文献4】Gerhardt, J, Nokihara, K and Yamamoto, R. Design and applications of a novel amino acid analyzer for D/L and quantitative analysis with the use of gas chromatography. In: Smith, JA and Rivier, JE, editors. Peptides: Chemistry, Structure and Biology. Leiden: Escom Science Publishers BV; 1992. p 531-532.
【非特許文献5】Nokihara, K, Yamamoto, R, Nishine, T and Gerhardt, J. Instrumentation and applications of a novel amino acid analyzer, Shimadzu-CAT DLAA-1. In: Takai, K, editor. Frontiers and Horizons in amino acid research. Amsterdam: Elsevier Science Publishers BV; 1992. p 391-395.
【非特許文献6】Frank, H, Nicholson, GJ and Bayer, E. Enantiomer labeling, a method for the quantitative analysis of amino acids. J Chromatogr 1978; 167,187-196.
【非特許文献7】Frank, H, Nicholson, GJ and Bayer, E. Rapid gas chromatographic separation of amino acid enantiomers with a novel chiral stationary phase. J Chromatogr Sci 1977; 15,174-176.
【非特許文献8】Nokihara, K., and Gerhardt, J. Development of an Improved Automated Gas-chromatographic Chiral Analysis System: Application to Non-natural Amino Acids and Natural Protein Hydrolysates, Chirality 2001; 13, 431-434.
【発明の開示】
【発明が解決しようとする課題】
【0010】
従って、本発明の目的は、工業的な大量生産に応用し得る、ミモシンをマメ科の植物で亜熱帯地方に広く分布するギンネムから高純度且つ低コストで分離精製できる方法、精製ミモシンの品質管理に有用な光学純度の検定方法、及びミモシン導入ペプチドの製造に有用なミモシン誘導体の製造方法を提供することである。
【課題を解決するための手段】
【0011】
本願発明者らは、鋭意研究の結果、熱湯による抽出、陽イオン交換樹脂を用いる分離と再結晶により、限外濾過を用いることなく、高純度のミモシンをギンネムから低コストで分離精製することができることを見出した。
【0012】
次いで、精製ミモシンの光学純度の検定方法を確立すべく鋭意研究した。厳密な検定は原薬製造において重要な課題であり、精製ミモシンをペプチドに導入して創薬研究を行なうためには厳密な純度検定手法を確立することが重要である。
【0013】
しかしながら、なかなか目的を達成できなかった。生物化学の分野では、精製物の検定には一般に逆相高速液体クロマトグラフィー(RP−HPLC)が用いられる。その充填剤は主にシリカゲルをベースにODS化(オクタドデシル化)したものであり、市販のHPLCカラムの大半はそのようなODS化シリカゲルを充填したものである。しかしながら、ミモシンはこれら市販のHPLCカラムによる厳密な検定ができない。すなわち、ミモシンはカラムにほとんど保持されることが無く、ボイドの近くに溶出される(ほとんど素通りする)。また、物理化学的にミモシン側鎖の安定性は高くなく、一部分解して生じる副生成物の一部とほとんど同位置に溶出する。順相シリカゲルを用いるとミモシンの吸着が激しく分離精製はできなかった。これらの原因は、ミモシンが残存シラノール基と相互作用をし、溶出パターンはブロードとなり厳密な検定を妨げる。また、キラル分離用のHPLCカラムも例えばダイセル化学工業株式会社から市販されているように公知であるが、このカラムで分離した場合、天然アミノ酸であるL-アラニンとミモシンの溶出位置が極めて近く場合によっては重なるという問題がある。しかもミモシン側鎖の安定性は高くなく、側鎖の官能基が分解するとアラニンが生成することも知られており、ミモシンが光学的に純度が低いのか、分解物が混入しているのか、その量まで検定することは不可能であった。
【0014】
そこで、本願発明者らは、鋭意研究の結果、キラル構造を有する分子をミモシンに導入してミモシンをジアステレオマーに変換することで、ミモシンの光学純度を精度良く検定することができることを見出した。
【0015】
さらに、高純度で分離精製されたミモシンを有効利用すべく、ミモシンをペプチドへ導入するための誘導体化法として、現在一般的なペプチドの合成法であるFmoc法に適応可能な方法を開発した。
【0016】
すなわち、本発明は、
(1) ギンネムを水に浸漬してミモシンを抽出する工程、
(2) 抽出液中に陽イオン交換樹脂を添加してミモシンを陽イオン交換樹脂に吸着させる工程、
(3) 前記陽イオン交換樹脂からミモシンを溶出させる工程、
(4) 工程(3)で得られた溶出液からミモシンの粗結晶を得る工程、
(5) 再結晶法により前記粗結晶からミモシンの結晶を得る工程
を含む、精製ミモシン又はその酸付加塩の製造方法を提供する。また、本発明は、1つ以上のキラル炭素を有する分子を、ミモシンのアミノ基又はカルボキシル基に結合させてミモシンをジアステレオマーに変換し、次いで、該ミモシンを逆相HPLCに付し、アセトニトリル濃度勾配を用いたグラジエント法により該ミモシンを分離溶出させ、溶出液中のミモシンを光学検出器にて検出することを含む、ミモシンの光学純度の検定方法を提供する。さらに、本発明は、ミモシンのアミノ基に保護基が導入されたミモシン誘導体を提供する。さらに、本発明は、塩基の存在下でミモシンと保護化試薬とを反応させ、ミモシンのアミノ基に保護基を導入することを含む、ミモシン誘導体の製造方法を提供する。
【発明の効果】
【0017】
本発明により、自生しているギンネム中に含まれているミモシンを効率良く抽出できる、工業的に有利な手法が初めて提供された。また、ミモシン精製品の品質管理に重要な、ミモシンの光学純度を厳密に検定できる方法が初めて提供された。さらに、ミモシンを有効利用するために必要なミモシン誘導体の製造方法が初めて提供された。本発明の方法によれば、簡便な操作でミモシンを大量に分離精製することができ、純度の検定と誘導体化を確実に行うことができる。分離精製物の確認には純度の検定が必要であり、また、ミモシン誘導体を製造するためには高純度でミモシンを分離精製する必要がある。このように、ミモシンの単離精製法、純度の検定及びミモシンの誘導体化は、相互に関連が深く、本願発明はミモシンを創薬、機能性ペプチドのビルディングブロックとして利用する上で大きな利点をもたらす。
【発明を実施するための最良の形態】
【0018】
本発明の精製ミモシン又はその酸付加塩の製造方法(以下、単に「精製ミモシン製造法」ということがある)に供される、ミモシンを含む天然物はギンネムである。ギンネム(英名:Leucaena、学名:Leucaena leucocephala de Wit)は、世界中の熱帯・亜熱帯地域に繁茂しているマメ科の植物であり、国内では沖縄や小笠原諸島などの亜熱帯地域に畑の緑肥用として導入され、現在では、燃料、土壌浸食防止用に、あるいは砂漠化地域の再緑化植物として栽培され、沖縄県内ではほぼ全域に自生している。利用する部位としては、特に季節を問わずに採取可能な茎葉部であることが好ましい。特に葉は採取除去してもすぐにまた生えてくるため、葉の採取は環境へ与えるダメージも少なく、特に好ましい原料である。豆にもミモシンは含まれるが、豆の場合は挽いてから水に浸けなくてはならず、より手間がかかる。なお、ミモシンの化学式は以下の通りである。
【0019】
【化1】

【0020】
本発明の精製ミモシン製造法の工程(1)では、ギンネムからミモシンを単離する。単離抽出は水で行なうが、好ましくは80℃以上での加熱抽出、より好ましくは沸騰水(100℃)中で抽出を行なうことが望ましい。ギンネム茎葉中にはミモシン分解酵素が存在するとの報告があるが、このような高温条件下で抽出を行うことで、分解酵素によるミモシンの分解を抑制することができ、ミモシンの収量をより高めることができる。ギンネムの茎葉部1kgに対して水は1L〜10L、好ましくは3〜7L用いる。抽出時間は特に限定されないが、通常5分間〜24時間程度であり、高温で抽出する場合には5分間〜20分間程度で好ましく抽出することができる。抽出溶液中の不溶物は、適宜濾過等により除去する。
【0021】
工程(2)では、工程(1)で得られた抽出溶液中に陽イオン交換樹脂を添加して、抽出溶液中のミモシンを陽イオン交換樹脂に吸着させることにより、ミモシンを固相抽出する。陽イオン交換樹脂としては、強酸性陽イオン交換樹脂が好ましい。強酸性陽イオン交換樹脂は特に限定されず、市販品を好ましく用いることができる。抽出溶液10Lに対し、陽イオン交換樹脂は500g〜3000g、好ましくは800g〜1500g用いる。
【0022】
工程(3)では、陽イオン交換樹脂に吸着させたミモシンを溶出させる。ミモシンを吸着した陽イオン交換樹脂は、通常、ミモシンの溶出に先立ち、洗浄処理を行なう。洗浄の条件は特に限定されず、例えば、80%エタノール等のアルコール水溶液中に浸漬し、次いで水中に浸漬して洗浄することができる。例えば、陽イオン交換樹脂1kgに対してアルコール水溶液を1〜10L、好ましくは3〜6L用いて、樹脂を6〜24時間、好ましくは10〜15時間浸漬し、次いで、イオン交換樹脂1kgに対して蒸留水を0.5〜5L、好ましくは1〜3L用いて、樹脂を10〜100分、好ましくは30〜40分浸漬することで、樹脂を洗浄することができる。陽イオン交換樹脂からのミモシンの溶出は、塩基性溶媒を用いて行なうことができる。塩基性溶媒としては、アンモニア水、トリエチルアミン水溶液、水酸化ナトリウム水溶液、水酸化カリウム水溶液等が挙げられるが、これらに限定されない。例えば、イオン交換樹脂1kgに対して1〜4N程度のアンモニア水1L〜10L、好ましくは3〜7Lを用いて、3〜10時間、好ましくは5〜7時間樹脂を浸漬することで、樹脂に吸着したミモシンを溶出させることができる。
【0023】
工程(4)では、工程(3)で得られた溶出液中のミモシンを粗結晶として析出させる。溶出液が濃い褐色の場合は、まず活性炭処理を行う。推定ミモシン存在量100gに対して1から50g 好ましくは5〜10gの活性炭を溶出液に加え室温で5から60分間、好ましくは5〜10分間攪拌した後、活性炭を濾過等で除去する。得られた濾液をエバポレーターで減圧濃縮することが好ましい。濃縮後の溶出液に酸を添加し、pH3〜6、好ましくは4.5〜5.0に調整し、0℃〜10℃の低温下で7〜24時間程度維持することで、溶出液中のミモシンを粗結晶として析出させることができる。溶出液に添加する酸としては、例えば1〜4N程度の塩酸、希硫酸、過塩素酸等を用いることができるが、これらに限定されない。塩酸を用いた場合、該工程(4)ではミモシンを塩酸塩の粗結晶として得ることができる。
【0024】
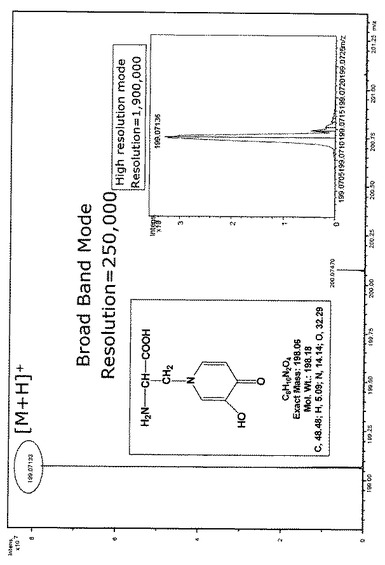
工程(5)では、工程(4)で得られたミモシンの粗結晶から再結晶法によりミモシンの精製を行なう。析出した粗ミモシン結晶を濾過等により溶液から分離した後、強塩基水溶液で溶解し、酸を用いて溶液のpHを3〜6好ましくは4.5〜5.0に調整する。この溶液を0℃〜10℃の低温下で7〜24時間放置することで、純度の高いミモシンを結晶として析出させることができる。粗結晶を溶解する強塩基水溶液としては特に限定されず、水酸化ナトリウム、水酸化カリウム、炭酸カリウム、炭酸ナトリウム等の水溶液を用いることができ、例えば3〜10N程度の水酸化ナトリウム水溶液等を好ましく用いることができる。強塩基水溶液の使用量は、粗ミモシン結晶100gに対し1〜5L、好ましくは2.5〜3.5Lである。pHの調整に用いる酸としては特に限定されず、塩酸、希硫酸、過塩素酸等を用いることができ、例えば3〜10N程度の塩酸等を好ましく用いることができる。塩酸を用いた場合、ミモシンを塩酸塩の結晶として得ることができる。このようにして得られたミモシン又はその塩の純度は、下記実施例に示されるように、HPLC並びにMS分析により高純度であることが確認されている。特に実施例に示すごとく、ここで得られた高純度ミモシンをフーリエ変換イオンサイクロトロン型質量分析計により精密質量分析を行った結果、精密分子量199.07136という値を得た(実施例1、図1)。天然同位体存在比に基づく解析により理論上のミモシンの化学式と一致した。
【0025】
本発明のミモシンの光学純度の検定方法では、まず、ミモシンをジアステレオマーに変換する。ミモシン分子中のアミノ基又はカルボキシル基に、分子中に1つ以上のキラル炭素を有する分子を結合させることで、ミモシンをジアステレオマーに変換することができる。ミモシン分子中に導入する上記キラル炭素を有する分子としては、HPLC用ラベル化剤として市販されている公知の分子を好ましく用いることができる。例えば、イソチオシアン酸2,3,4,6-テトラ-O-アセチル-β-D-グルコピラノシル、イソチオシアン酸2,3,4,6-テトラ-O-ベンゾイル-β-D-グルコピラノシル等を好ましく用いることができるが、これらに限定されない。上記キラル炭素を有する分子のミモシン分子中への導入は、該キラル炭素を有する分子の種類に応じ、この分野で公知の常法により行なうことができる。
【0026】
次いで、ジアステレオマーに変換したミモシンを逆相HPLCに付して分画する。上記したように、ミモシン分子は、一般にHPLCに用いられているODS化シリカゲル(C18シリカゲル)カラムにはほとんど保持されることが無く、厳密な検定が非常に困難であるが、ミモシンをジアステレオマー化することでカラムに好ましく保持されるようになり、他の成分と分離して溶出させることが可能になる。従って、逆相HPLCの固定相としては、アルキル基修飾シリカゲル、パーフィユージョンクロマトグラフィー充填剤等を用いることができ、例えばHiPep-Intrada(株式会社ハイペップ研究所)や逆相のPorosカラム(アプライドバオシステムズ・ジャパン)等のような市販の逆相HPLC分析用カラムを好ましく用いることができる。特に、粒子径3μm,細孔径30nmでアルキルリガンドを導入し適度に表面極性を持たせた高純度シリカゲル充填剤を用いた市販のカラムHiPep-Intrada(株式会社ハイペップ研究所)によってとりわけ明確なD体、L体の分離が可能である。
【0027】
逆相HPLCの移動相(溶出液)としては、アセトニトリル濃度勾配が用いられ、グラジエント法により分離溶出が行なわれる。グラジエント法は、段階的勾配(ステップバイズグラジエント)でも直線的勾配(リニアグラジエント)でもよい。例えば、A=0.1%トリフルオロ酢酸(TFA)水溶液と、B=0.1%TFA−90%アセトニトリル水溶液を用いて、15分〜1時間程度かけてA:B=99:1→69:31とするリニアグラジエントが好ましい。
【0028】
逆相HPLCにおいて、カラムの温度は常温でよい。送液速度等のパラメーターも常法に準じて設定することができる。溶出液中のミモシンは、UV検出器等の公知の光学検出器を用いて検出することができる。
【0029】
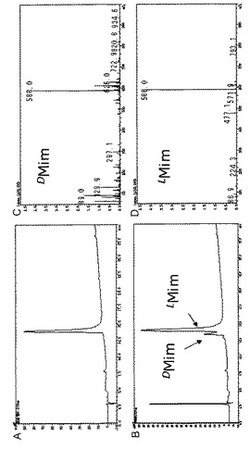
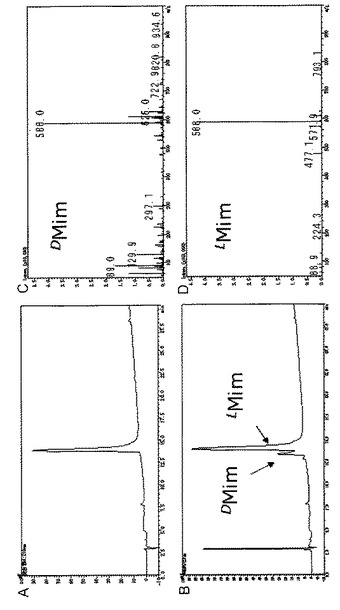
本発明の光学純度の検定方法によれば、ミモシンのピークがインジェクションノイズと重複することなく、ミモシンのピークを明瞭に検出することができる。下記実施例に具体的に示されるように、明らかに一部ラセミ化したミモシンを本発明の検定方法でオンラインによる質量分析計を用いて純度検定すると、D体とL体のミモシンのピークがいずれも明瞭に確認できる(実施例2、図2B)。一方、本発明の精製ミモシン製造法でギンネムから抽出精製されたミモシン精製品を同様に純度検定すると、L体のピークのみが明瞭に確認される(実施例2、図2A)。これらのデータは、本発明の精製ミモシン製造法及び光学純度の検定方法が非常に優れていることを示すものである。
【0030】
ミモシンのジアステレオマー化にイソチオシアン酸2,3,4,6-テトラ-O-アセチル-β-D-グルコピラノシルを用いる場合を例として、具体的に光学純度の検定方法を説明すると、以下の通りである。ただし、この説明は本発明の範囲をイソチオシアン酸2,3,4,6-テトラ-O-アセチル-β-D-グルコピラノシルを用いる方法に限定することを意図するものではない。
【0031】
分離精製ミモシンの飽和水溶液、イソチオシアン酸2,3,4,6-テトラ-O-アセチル-β-D-グルコピラノシル1mM〜100mM、好ましくは5mM〜15mMのアセトニトリル溶液、炭酸水素ナトリウム1mM〜100mM、好ましくは5mM〜15mMの水溶液を等量ずつ混合し、室温にて0.5〜3時間、好ましくは1〜2時間おいて、ミモシンのアミノ基にイソチオシアン酸2,3,4,6-テトラ-O-アセチル-β-D-グルコピラノシルを導入してジアステレオマー化する。この溶液を逆相HPLCで分析する。逆相HPLCの溶出液には0.1%TFA水溶液と0.1%TFA−90%アセトニトリル水溶液を用いて、アセトニトリル濃度勾配によりグラジエント分析する(0.1%TFA水溶液:0.1%TFA−90%アセトニトリル水溶液=99:1 → 69:31、30分間)。検出は200〜280nmの紫外線により行なう。
【0032】
本発明のミモシン誘導体は、ミモシンのアミノ基に保護基を導入したものである。ミモシンをペプチド鎖や他の分子に導入するためには通常、保護ミモシンを用いる。そのようなミモシン誘導体を製造するためには、純度の高いミモシンを得ることが重要であるが、従来はミモシンを高純度で精製する方法や、ミモシンの純度を厳密に検定する方法は知られていなかった。上記ミモシン誘導体は、本願発明者らが開発した精製ミモシン製造法及び純度検定方法により、初めて製造可能になったものである。
【0033】
保護基としては特に限定されず、アミノ酸分子中のアミノ基を保護するために用いられるいかなる保護基であってもよい。例えば、ピペリジン等で切断が可能なFmoc基(9-フルオレニルメトキシ基)、酸により切断が可能なBoc基(tert-ブトキシカルボニル基)、パラジウム触媒により切断が可能なZ基(ベンジルオキシカルボニル基)、薄い酸で切断可能なTrt基(トリチル基)などが挙げられる。これらのうち、切断の簡便性、合成上の観点(特に副生成物の観点)から種々に検討した結果、ピペリジン等で除去可能なFmoc基が好ましい。現在最も一般的なペプチド合成法はFmoc法であり、Fmoc保護ミモシンであればFmoc法に適用可能である。アミノ基にFmoc基が導入されたFmoc保護ミモシンの化学式を以下に示す。
【0034】
【化2】
【0035】
上記したミモシン誘導体は、塩基の存在下でミモシンと保護化試薬とを反応させ、ミモシンのアミノ基に保護基を導入することにより製造することができる。「保護化試薬」とは、アミノ酸分子中のアミノ基に保護基を結合させることができる試薬であり、そのような試薬は種々のものが公知で市販品も多く存在する。本発明においては、公知のいずれの保護化試薬をも用いることができる。公知の保護化試薬の具体例として、Fmoc化試薬としてはFmoc−Cl(クロロギ酸フルオレニルメチル)、Fmoc−OSu(9−フルオレニルメチルスクシンイミジルカルボネート)等が挙げられ、Boc化試薬としてはBoc2O(ジ-tert-ブチルジカルボネート)等が挙げられ、Z化試薬としてはZ-Cl(クロロギ酸ベンジル)等が挙げられ、Trt化試薬としてはTrt-Cl(トリチルクロライド)等が挙げられるが、これらに限定されない。
【0036】
反応系内に共存させる上記塩基としては、炭酸ナトリウム、炭酸カリウム、炭酸水素ナトリウム、炭酸水素カリウム等を用いることができる。塩基及び保護化試薬の使用量は、特に限定されないが、ミモシンに対し2〜5当量の塩基と1〜3当量の保護化試薬とを用いることで満足できる結果が得られる。反応溶媒は水系溶媒であり、用いる保護化試薬の種類に応じて水に適宜極性溶媒を混合して用いることができる。例えば、保護化試薬としてFmoc-OSuを用いる場合は、水:ジオキサン=1:1の混合溶媒中で好ましくFmoc化反応を行なうことができる。反応系内に添加するミモシンの量は、通常、15〜60g/L程度である。下記実施例では、600mLの保護化反応系に対し20gの精製ミモシン塩酸塩が用いられている。反応温度は室温でよい。反応時間は通常6〜24時間、好ましくは10〜15時間である。
【0037】
ミモシンと保護化試薬とを反応させた場合、アミノ基のみならずヒドロキシ基にも一部保護基が導入され得る。保護化反応の後、反応溶液に塩基を添加することで、ヒドロキシ基に導入された保護基を外し、アミノ基のみが保護されたミモシン誘導体の収量を向上させることができる。従って、保護化試薬との反応後、反応溶液に塩基を添加する工程を加えることが好ましい。ここで用いる塩基も、上記と同様に炭酸ナトリウム、炭酸カリウム、炭酸水素ナトリウム、炭酸水素カリウム等を用いることができる。塩基の使用量は、反応開始時のミモシンに対し通常5〜50当量である。例えば、0.1Mの炭酸ナトリウム水溶液を用いる場合、保護化反応後の反応溶液を0.1M炭酸ナトリウム水溶液で2〜5倍希釈すればよい。塩基を添加して得られた溶液は、通常、反応溶液量の1/5〜1/2の酢酸エチルで2〜5回抽出する。水層を1N程度の塩酸でpH3〜6、好ましくはpH4〜5に調整すると、保護基が導入されたミモシン誘導体が析出するので、濾取等により溶液から回収することでミモシン誘導体を得ることができる。
【0038】
こうして得られたミモシン誘導体の純度は、逆相HPLCにより測定することができる。この逆相HPLCの条件は、上記した本発明のミモシンの光学純度の検定方法における逆相HPLCと同様に行なうことができる。
【実施例】
【0039】
以下、本発明を実施例に基づきより具体的に説明する。もっとも、本発明は下記実施例に限定されるものではない。
【0040】
実施例1
ミモシンの大量分離精製
新鮮なギンネムの葉25 Kgを沸騰水100 Lで10分間煮沸抽出した。抽出液約100 Lを室温まで冷却してろ過により不溶物を除去した後、強陽イオン交換樹脂(AMBERLITE(商標) IR120 H(Plus), GFS Chemicals Inc., オハイオ州米国)を10 Kg加えて一晩吸着させた。イオン交換樹脂を80%エタノール25L中に12時間浸漬して洗浄を行い、続けて25 Lの蒸留水中に2時間浸漬して洗浄した。イオン交換樹脂を2Nアンモニア水 40 L中に6時間浸漬してミモシンをイオン交換樹脂から溶出させた。溶出液はまず活性炭処理をした。活性炭の量は当該スケールでは5gを用い室温で10分間攪拌した後活性炭を濾過によって除去した。得られた濾液を減圧濃縮し、濃縮液を6N HClでpH4.5〜5.0に調整し、4℃で一晩置くことで粗ミモシン塩酸塩を析出させ、ミモシン粗結晶を得た(融点175℃分解)。濾取により得られた粗結晶を6N NaOH水溶液1.8 Lに溶解させ、4℃で一晩置くことで再結晶させ、ミモシン塩酸塩の結晶を得た。25 Kgのギンネムから100 gの高純度ミモシンが得られた。
融点225℃、旋光度[α]D 22.2 = -23.2 (C=2.11 mg/1.5 mL 溶媒:水)
【0041】
フーリエ変換イオンサイクロトロン型質量分析計は高分解能な精密質量解析能力によって確実な化学組成の決定が行えることが公知である。ブルカーダルトニクス社の当該質量分析計モデルApex-Q94eを用いて、ここで得られた高純度ミモシンを分析した結果、精密分子量199.07136という値を得た(図1)。天然同位体存在比に基づく解析により元素組成 C8H11N2O4 [M+H]となり、理論上の化学式と一致した。これによっても精製品が高純度であることが示された。
【0042】
実施例2
分離精製したミモシンの光学純度分析
(1) ジアステレオマー化による光学純度分析
ミモシンをジアステレオマー化する化合物として、光学活性HPLC用ラベル化剤A5514(イソチオシアン酸2,3,4,6-テトラ-O-アセチル-β-D-グルコピラノシル、東京化成)を用いた(下記式中の2)。
【0043】
【化3】
【0044】
実施例1で分離精製したミモシンの飽和水溶液を調整し、10mM光学活性HPLC用ラベル化剤(東京化成A5514)と10mM炭酸水素ナトリウムそれぞれ5μLずつとり混合し、室温で30分おいて反応させた。
【0045】
反応後の混合溶液を10倍に希釈して逆相HPLCにより分析した。分析条件を以下に示す。
カラム:HiPep-Intrada、サイズ3.0X150 mm(株式会社ハイペップ研究所)
カラム温度:25℃
溶出液:A=0.1%TFA水溶液、B=0.1%TFA−90%アセトニトリル水溶液
A:B=99:1→69:31(30分)のリニアグラジエント
流速:0.3 mL/min
検出:210及び254nm UV
【0046】
HPLCによる分析結果(検出:210nm UV)を図2に示す。実施例1の方法でギンネムから抽出、精製されたミモシン精製品(図2A)では、L体ミモシンのピークが明瞭に確認され、純度が非常に高いことが確認できた。比較のために、実施例1のミモシン精製品を文献[R. Adams and J.L. Johnson, Leucenol. VI. A Total Synthesis. J. Am. Chem Soc., 1949, 71, 705-708]に記載の方法で部分的にラセミ化させたミモシンを同様にジアステレオマー化してHPLC分析した結果を図2Bに示す。この場合には、D体とL体のピークが明瞭に確認された(図2B)。これにより、実施例2の検定方法の精度が非常に優れていることも確認できた。カラムの溶出液はオンラインでイオントラップ型質量分析を行ない、目的の化合物を同定した。質量分析データを図2C(D−ミモシン)及び2D(L−ミモシン)に示す。
Calcd.: 587.55, Found: 588.0 (M+H)
【0047】
(2) キラルカラムを用いた光学純度分析
公知のキラル分離用カラムCrownpak(+)(ダイセル化学工業株式会社)を用いて、実施例1で分離精製したミモシンのHPLC分析を行なった。
HPLC分析条件
カラム:ダイセル Crownpak(+) (4.0 id. x 150 mm)
カラム温度:25℃
溶出液:0.1% HClO4定組成溶離in 10 min
流速:0.4 mL/min
検出:210 nm
【0048】
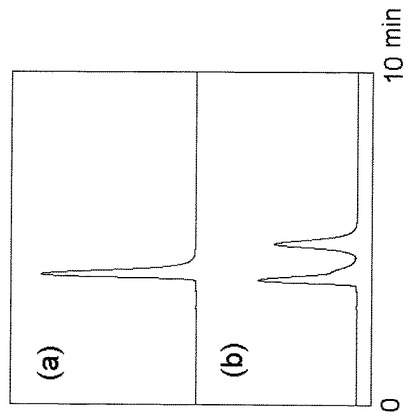
ミモシンの分析結果を図3上段(a)に示す。図3下段(b)には、同一の条件でDL-アラニンを分析した結果を示す。図3に示される通り、キラル分離カラムを用いた場合、ミモシンの溶出位置がL-アラニンの溶出位置と極めて近くなった。ミモシンは分解するとアラニンを生成するため、キラル分離カラムによっては、ミモシンが分解してアラニンを生じているか否かを判別することは困難であり、厳密な光学検定はできなかった。
【0049】
実施例3
ミモシン誘導体の合成
【0050】
【化4】
【0051】
実施例1で得られたミモシン40 gと炭酸ナトリウム42 g(ミモシンに対し2当量)の水溶液600 mLを調製した。そこにFmoc−OSu 100 g(1.5当量)のジオキサン溶液600 mLを滴下した(ジオキサン:H2O=1:1)。6時間撹拌したのち、反応溶液を0.1 M炭酸ナトリウムで3600 mLまで希釈してさらに6時間撹拌した。反応は逆相HPLCにより分析して追跡した。反応溶液を酢酸エチル2000 mLで2回抽出し、水層を1N HClで中和すると固体が析出した。この固体を濾取することでFmoc保護ミモシン(Fmoc-Mim-OH)を50.8 g、収率60%で得た。
質量分析
Calcd.: 420.4, Found; 421.1 (M+H)
核磁気共鳴スペクトル解析
1H NMR (300 MHz, DMSO) δ 8.13-7.85 (m, 4H), 7.60-7.59 (br m, 2H), 7.44-7.29 (m, 4H), 7.03-7.00 (br m, 1H), 4.69 (br s, 1H), 4.55 (br s, 1H), 4.37-4.18 (m, 4H).
旋光度 [α]D 23.4 = -3.09 (C=2.34 mg/1.5 mL 溶媒 メタノール)
【0052】
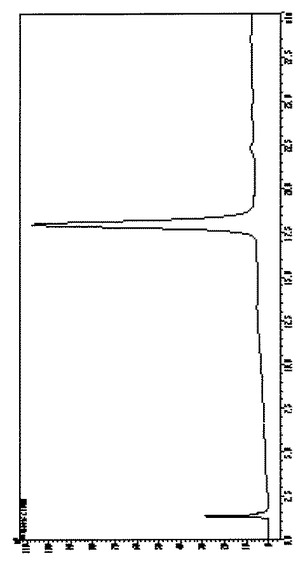
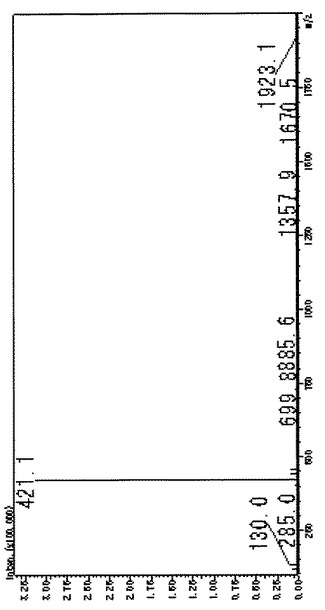
得られたFmoc-Mim-OHを逆相HPLCにより純度検定した。カラムの溶出液はオンラインでイオントラップ質量分析を行なった。HPLCの結果を図4に、質量分析データを図5に示す。図4に示される通り、Fmoc-Mim-OHの明瞭なピークが観察され、純度が非常に高いことが確認できた。なお、上記反応の追跡及び純度検定に用いた逆相HPLCの条件は以下の通りである。
カラム: POROS-R2, 4.6x100 mm (アプライドバオシステムズ・ジャパン)
カラム温度:25℃
移動相:A=0.1% TFA水溶液、B=0.1% TFA-90%アセトニトリル水溶液
B=1%→31% in 30 min(リニアグラジエント)
流速:0.3 mL/min
検出:210nm UV
【図面の簡単な説明】
【0053】
【図1】フーリエ変換イオンサイクロトロン型質量分析計による、実施例1で製造したミモシン精製品の精密質量分析とその解析結果を示す。
【図2】実施例2で行なった、ジアステレオマー化したミモシンの逆相HPLCによる分析データ(検出:210nm UV)(A、B)及び質量分析データ(C、D)である。Aは実施例1で製造したミモシン精製品の分析データ、Bはミモシン精製品を公知の方法で一部ラセミ化したミモシンの分析データを示す。CはB中の左側のピークの化合物(D−ミモシン)の質量分析データ、Dは右側のピークの化合物(L−ミモシン)の質量分析データである。
【図3】公知のキラル分離用カラムを用いてミモシン及びアラニンをHPLC分析した結果を示す。上段(a)は実施例1で製造したミモシン精製品のデータ、下段(b)はDL-アラニンを分析したデータである。
【図4】Fmoc保護ミモシン(Fmoc-Mim-OH)の逆相HPLCによる純度検定結果である。
【図5】Fmoc保護ミモシン(Fmoc-Mim-OH)の質量分析データである。
【技術分野】
【0001】
本発明は、精製ミモシン又はその酸付加塩の製造方法、ミモシンの光学純度の検定方法、保護基が導入されたミモシン誘導体、及びFmoc基等の保護基による保護ミモシンの製造方法に関する。
【背景技術】
【0002】
近年、生物学と化学を融合させ、生体の機能と化合物との関係から有用物質や生体反応に関わる化合物を探索する手法としてケミカルバイオロジーが脚光を浴びている。この流れに先行してコンビナトリアルケミストリーが創薬を中心に有用物質の創出手法として重視されてきた。当該分野では、天然のタンパク質中には存在しない非天然アミノ酸/異常アミノ酸の利用が盛んであるが、化学合成による高純度の立体不斉アミノ酸の製造は容易ではなく、また極めて高価である。特徴のある非天然アミノ酸/異常アミノ酸を効率よく天然物から取得することは、光学純度の点でも有利である。しかしながら精製の過程で起きうるラセミ化によって精製物の光学純度に問題が生じうるため、光学純度を正確に検定することは重要である。
【0003】
数多くの種類の異常アミノ酸が知られているが、その中でもミモシンはチロシナーゼ阻害活性、アレロパシー活性、細胞毒性、あるいは抗腫瘍活性などを有するため、創薬、農薬創製、機能性化粧品開発等の分野での応用が期待される(特許文献1、非特許文献1)。
【0004】
熱帯植物ギンネムは、家畜飼料等では有害とされるミモシンを含むことが知られており、水に浸漬することによりギンネムからミモシンを除去できる方法が知られている(特許文献2)。また、ギンネムからのミモシンの抽出方法としては、非特許文献2に記載されるように、水中にギンネムを浸漬してミモシンを抽出し、該溶液から限外濾過を用いてミモシンを分離する方法が知られている。
【0005】
ミモシンはキラル炭素を有する分子であるため光学異性体が存在するが、通常、生物活性は光学異性体によって異なるため、ミモシンを各種分野で応用するためには光学純度の検定が必要不可欠である。また、ギンネムから抽出や精製作業中にミモシンの一部がラセミ化する可能性があるため、高純度サンプルを保障するための品質管理法は、とりわけ創薬原料などには重要である。
【0006】
上記の通り、ミモシンそのものは化合物として既知であり、また化学合成による製法もその全合成は報告されているが、合成はD,L体混合物であり、その筆者は天然物をラセミ化させて合成の標品としている(非特許文献3)。また、天然物由来として、マメなどの植物由来で抽出され試薬として市販されている。しかしながら現在市販されているミモシンは高価であり、研究目的はともかくとして、大量に使用する工業的なミモシンの利用は行われていない。その理由の一つはミモシンそのものが安定性に乏しいことである。大量に抽出する安価な方法は知られていない。また、大量に誘導体化して例えばペプチド合成に用いるという例もほとんど知られていない。キラリティ(立体不斉)に関しての報告は全くない。実際に、キラルカラムを利用してエナンショマーラベリングという手法でアミノ酸のキラリティを解明する手法が知られているが(非特許文献4〜8)、ミモシンではこの手法、エナンショマーラベリング法を用いることができなかった。この理由はミモシンが誘導体化の過程で分解するからである。また、アミノ酸のキラル分離用のカラムが市販されている(Crownpak(+)、ダイセル化学工業株式会社)。このカラムで分離した場合、天然アミノ酸であるアラニンラセミ体の一方とミモシンの溶出位置とが重なり厳密な判定は不可能であった。ミモシンは分解するとアラニンを生成することが知られている。したがって、これまで抽出されたミモシンのキラリティを正確に報告した例は無い。
【0007】
ミモシンを創薬に用いる場合、ミモシン単体で用いるよりも、ペプチドに導入することで細胞透過効率や分子/細胞認識能などの機能を付与することができればより付加価値が高まる。公知のペプチド合成法では、官能基を保護基で保護することによってペプチド鎖伸張中の副反応を回避できるため、たとえばアミノ酸のアミノ基をFmoc等で保護する必要がある。
【0008】
しかしながら、ギンネムから水で抽出した粗ミモシン溶液から、高純度のミモシンを分離精製するための工業的に有利な手法は知られていないため、ミモシンを創薬等の種々の分野で応用することもできないのが現状である。非特許文献2の方法は、水に抽出した粗ミモシン溶液からミモシンを抽出する方法であるが、限外濾過を用いるものであり、費用もかかることから、工業的なスケールで大量に精製する方法としては不向きである。また、ギンネムから分離精製したミモシンの光学純度を正確に測定できる方法は知られていない。さらに、ミモシンをペプチドへ導入するためには保護基を導入しなければならないが、ミモシンの効率的な誘導体化法とその検定法も知られていない。
【0009】
【特許文献1】特開昭63-295502
【特許文献2】特開昭63-294746
【非特許文献1】W.D. DeWys and T.C. Hall, Europ. J. Cancer Vol. 9, pp. 281-283. 1973.
【非特許文献2】農薬 1994, Vol 41, No 2, page 18-22 日本農薬株式会社
【非特許文献3】R. Adams and J.L. Johnson, Leucenol. VI. A Total Synthesis. J. Am. Chem Soc., 1949, 71, 705-708,
【非特許文献4】Gerhardt, J, Nokihara, K and Yamamoto, R. Design and applications of a novel amino acid analyzer for D/L and quantitative analysis with the use of gas chromatography. In: Smith, JA and Rivier, JE, editors. Peptides: Chemistry, Structure and Biology. Leiden: Escom Science Publishers BV; 1992. p 531-532.
【非特許文献5】Nokihara, K, Yamamoto, R, Nishine, T and Gerhardt, J. Instrumentation and applications of a novel amino acid analyzer, Shimadzu-CAT DLAA-1. In: Takai, K, editor. Frontiers and Horizons in amino acid research. Amsterdam: Elsevier Science Publishers BV; 1992. p 391-395.
【非特許文献6】Frank, H, Nicholson, GJ and Bayer, E. Enantiomer labeling, a method for the quantitative analysis of amino acids. J Chromatogr 1978; 167,187-196.
【非特許文献7】Frank, H, Nicholson, GJ and Bayer, E. Rapid gas chromatographic separation of amino acid enantiomers with a novel chiral stationary phase. J Chromatogr Sci 1977; 15,174-176.
【非特許文献8】Nokihara, K., and Gerhardt, J. Development of an Improved Automated Gas-chromatographic Chiral Analysis System: Application to Non-natural Amino Acids and Natural Protein Hydrolysates, Chirality 2001; 13, 431-434.
【発明の開示】
【発明が解決しようとする課題】
【0010】
従って、本発明の目的は、工業的な大量生産に応用し得る、ミモシンをマメ科の植物で亜熱帯地方に広く分布するギンネムから高純度且つ低コストで分離精製できる方法、精製ミモシンの品質管理に有用な光学純度の検定方法、及びミモシン導入ペプチドの製造に有用なミモシン誘導体の製造方法を提供することである。
【課題を解決するための手段】
【0011】
本願発明者らは、鋭意研究の結果、熱湯による抽出、陽イオン交換樹脂を用いる分離と再結晶により、限外濾過を用いることなく、高純度のミモシンをギンネムから低コストで分離精製することができることを見出した。
【0012】
次いで、精製ミモシンの光学純度の検定方法を確立すべく鋭意研究した。厳密な検定は原薬製造において重要な課題であり、精製ミモシンをペプチドに導入して創薬研究を行なうためには厳密な純度検定手法を確立することが重要である。
【0013】
しかしながら、なかなか目的を達成できなかった。生物化学の分野では、精製物の検定には一般に逆相高速液体クロマトグラフィー(RP−HPLC)が用いられる。その充填剤は主にシリカゲルをベースにODS化(オクタドデシル化)したものであり、市販のHPLCカラムの大半はそのようなODS化シリカゲルを充填したものである。しかしながら、ミモシンはこれら市販のHPLCカラムによる厳密な検定ができない。すなわち、ミモシンはカラムにほとんど保持されることが無く、ボイドの近くに溶出される(ほとんど素通りする)。また、物理化学的にミモシン側鎖の安定性は高くなく、一部分解して生じる副生成物の一部とほとんど同位置に溶出する。順相シリカゲルを用いるとミモシンの吸着が激しく分離精製はできなかった。これらの原因は、ミモシンが残存シラノール基と相互作用をし、溶出パターンはブロードとなり厳密な検定を妨げる。また、キラル分離用のHPLCカラムも例えばダイセル化学工業株式会社から市販されているように公知であるが、このカラムで分離した場合、天然アミノ酸であるL-アラニンとミモシンの溶出位置が極めて近く場合によっては重なるという問題がある。しかもミモシン側鎖の安定性は高くなく、側鎖の官能基が分解するとアラニンが生成することも知られており、ミモシンが光学的に純度が低いのか、分解物が混入しているのか、その量まで検定することは不可能であった。
【0014】
そこで、本願発明者らは、鋭意研究の結果、キラル構造を有する分子をミモシンに導入してミモシンをジアステレオマーに変換することで、ミモシンの光学純度を精度良く検定することができることを見出した。
【0015】
さらに、高純度で分離精製されたミモシンを有効利用すべく、ミモシンをペプチドへ導入するための誘導体化法として、現在一般的なペプチドの合成法であるFmoc法に適応可能な方法を開発した。
【0016】
すなわち、本発明は、
(1) ギンネムを水に浸漬してミモシンを抽出する工程、
(2) 抽出液中に陽イオン交換樹脂を添加してミモシンを陽イオン交換樹脂に吸着させる工程、
(3) 前記陽イオン交換樹脂からミモシンを溶出させる工程、
(4) 工程(3)で得られた溶出液からミモシンの粗結晶を得る工程、
(5) 再結晶法により前記粗結晶からミモシンの結晶を得る工程
を含む、精製ミモシン又はその酸付加塩の製造方法を提供する。また、本発明は、1つ以上のキラル炭素を有する分子を、ミモシンのアミノ基又はカルボキシル基に結合させてミモシンをジアステレオマーに変換し、次いで、該ミモシンを逆相HPLCに付し、アセトニトリル濃度勾配を用いたグラジエント法により該ミモシンを分離溶出させ、溶出液中のミモシンを光学検出器にて検出することを含む、ミモシンの光学純度の検定方法を提供する。さらに、本発明は、ミモシンのアミノ基に保護基が導入されたミモシン誘導体を提供する。さらに、本発明は、塩基の存在下でミモシンと保護化試薬とを反応させ、ミモシンのアミノ基に保護基を導入することを含む、ミモシン誘導体の製造方法を提供する。
【発明の効果】
【0017】
本発明により、自生しているギンネム中に含まれているミモシンを効率良く抽出できる、工業的に有利な手法が初めて提供された。また、ミモシン精製品の品質管理に重要な、ミモシンの光学純度を厳密に検定できる方法が初めて提供された。さらに、ミモシンを有効利用するために必要なミモシン誘導体の製造方法が初めて提供された。本発明の方法によれば、簡便な操作でミモシンを大量に分離精製することができ、純度の検定と誘導体化を確実に行うことができる。分離精製物の確認には純度の検定が必要であり、また、ミモシン誘導体を製造するためには高純度でミモシンを分離精製する必要がある。このように、ミモシンの単離精製法、純度の検定及びミモシンの誘導体化は、相互に関連が深く、本願発明はミモシンを創薬、機能性ペプチドのビルディングブロックとして利用する上で大きな利点をもたらす。
【発明を実施するための最良の形態】
【0018】
本発明の精製ミモシン又はその酸付加塩の製造方法(以下、単に「精製ミモシン製造法」ということがある)に供される、ミモシンを含む天然物はギンネムである。ギンネム(英名:Leucaena、学名:Leucaena leucocephala de Wit)は、世界中の熱帯・亜熱帯地域に繁茂しているマメ科の植物であり、国内では沖縄や小笠原諸島などの亜熱帯地域に畑の緑肥用として導入され、現在では、燃料、土壌浸食防止用に、あるいは砂漠化地域の再緑化植物として栽培され、沖縄県内ではほぼ全域に自生している。利用する部位としては、特に季節を問わずに採取可能な茎葉部であることが好ましい。特に葉は採取除去してもすぐにまた生えてくるため、葉の採取は環境へ与えるダメージも少なく、特に好ましい原料である。豆にもミモシンは含まれるが、豆の場合は挽いてから水に浸けなくてはならず、より手間がかかる。なお、ミモシンの化学式は以下の通りである。
【0019】
【化1】
【0020】
本発明の精製ミモシン製造法の工程(1)では、ギンネムからミモシンを単離する。単離抽出は水で行なうが、好ましくは80℃以上での加熱抽出、より好ましくは沸騰水(100℃)中で抽出を行なうことが望ましい。ギンネム茎葉中にはミモシン分解酵素が存在するとの報告があるが、このような高温条件下で抽出を行うことで、分解酵素によるミモシンの分解を抑制することができ、ミモシンの収量をより高めることができる。ギンネムの茎葉部1kgに対して水は1L〜10L、好ましくは3〜7L用いる。抽出時間は特に限定されないが、通常5分間〜24時間程度であり、高温で抽出する場合には5分間〜20分間程度で好ましく抽出することができる。抽出溶液中の不溶物は、適宜濾過等により除去する。
【0021】
工程(2)では、工程(1)で得られた抽出溶液中に陽イオン交換樹脂を添加して、抽出溶液中のミモシンを陽イオン交換樹脂に吸着させることにより、ミモシンを固相抽出する。陽イオン交換樹脂としては、強酸性陽イオン交換樹脂が好ましい。強酸性陽イオン交換樹脂は特に限定されず、市販品を好ましく用いることができる。抽出溶液10Lに対し、陽イオン交換樹脂は500g〜3000g、好ましくは800g〜1500g用いる。
【0022】
工程(3)では、陽イオン交換樹脂に吸着させたミモシンを溶出させる。ミモシンを吸着した陽イオン交換樹脂は、通常、ミモシンの溶出に先立ち、洗浄処理を行なう。洗浄の条件は特に限定されず、例えば、80%エタノール等のアルコール水溶液中に浸漬し、次いで水中に浸漬して洗浄することができる。例えば、陽イオン交換樹脂1kgに対してアルコール水溶液を1〜10L、好ましくは3〜6L用いて、樹脂を6〜24時間、好ましくは10〜15時間浸漬し、次いで、イオン交換樹脂1kgに対して蒸留水を0.5〜5L、好ましくは1〜3L用いて、樹脂を10〜100分、好ましくは30〜40分浸漬することで、樹脂を洗浄することができる。陽イオン交換樹脂からのミモシンの溶出は、塩基性溶媒を用いて行なうことができる。塩基性溶媒としては、アンモニア水、トリエチルアミン水溶液、水酸化ナトリウム水溶液、水酸化カリウム水溶液等が挙げられるが、これらに限定されない。例えば、イオン交換樹脂1kgに対して1〜4N程度のアンモニア水1L〜10L、好ましくは3〜7Lを用いて、3〜10時間、好ましくは5〜7時間樹脂を浸漬することで、樹脂に吸着したミモシンを溶出させることができる。
【0023】
工程(4)では、工程(3)で得られた溶出液中のミモシンを粗結晶として析出させる。溶出液が濃い褐色の場合は、まず活性炭処理を行う。推定ミモシン存在量100gに対して1から50g 好ましくは5〜10gの活性炭を溶出液に加え室温で5から60分間、好ましくは5〜10分間攪拌した後、活性炭を濾過等で除去する。得られた濾液をエバポレーターで減圧濃縮することが好ましい。濃縮後の溶出液に酸を添加し、pH3〜6、好ましくは4.5〜5.0に調整し、0℃〜10℃の低温下で7〜24時間程度維持することで、溶出液中のミモシンを粗結晶として析出させることができる。溶出液に添加する酸としては、例えば1〜4N程度の塩酸、希硫酸、過塩素酸等を用いることができるが、これらに限定されない。塩酸を用いた場合、該工程(4)ではミモシンを塩酸塩の粗結晶として得ることができる。
【0024】
工程(5)では、工程(4)で得られたミモシンの粗結晶から再結晶法によりミモシンの精製を行なう。析出した粗ミモシン結晶を濾過等により溶液から分離した後、強塩基水溶液で溶解し、酸を用いて溶液のpHを3〜6好ましくは4.5〜5.0に調整する。この溶液を0℃〜10℃の低温下で7〜24時間放置することで、純度の高いミモシンを結晶として析出させることができる。粗結晶を溶解する強塩基水溶液としては特に限定されず、水酸化ナトリウム、水酸化カリウム、炭酸カリウム、炭酸ナトリウム等の水溶液を用いることができ、例えば3〜10N程度の水酸化ナトリウム水溶液等を好ましく用いることができる。強塩基水溶液の使用量は、粗ミモシン結晶100gに対し1〜5L、好ましくは2.5〜3.5Lである。pHの調整に用いる酸としては特に限定されず、塩酸、希硫酸、過塩素酸等を用いることができ、例えば3〜10N程度の塩酸等を好ましく用いることができる。塩酸を用いた場合、ミモシンを塩酸塩の結晶として得ることができる。このようにして得られたミモシン又はその塩の純度は、下記実施例に示されるように、HPLC並びにMS分析により高純度であることが確認されている。特に実施例に示すごとく、ここで得られた高純度ミモシンをフーリエ変換イオンサイクロトロン型質量分析計により精密質量分析を行った結果、精密分子量199.07136という値を得た(実施例1、図1)。天然同位体存在比に基づく解析により理論上のミモシンの化学式と一致した。
【0025】
本発明のミモシンの光学純度の検定方法では、まず、ミモシンをジアステレオマーに変換する。ミモシン分子中のアミノ基又はカルボキシル基に、分子中に1つ以上のキラル炭素を有する分子を結合させることで、ミモシンをジアステレオマーに変換することができる。ミモシン分子中に導入する上記キラル炭素を有する分子としては、HPLC用ラベル化剤として市販されている公知の分子を好ましく用いることができる。例えば、イソチオシアン酸2,3,4,6-テトラ-O-アセチル-β-D-グルコピラノシル、イソチオシアン酸2,3,4,6-テトラ-O-ベンゾイル-β-D-グルコピラノシル等を好ましく用いることができるが、これらに限定されない。上記キラル炭素を有する分子のミモシン分子中への導入は、該キラル炭素を有する分子の種類に応じ、この分野で公知の常法により行なうことができる。
【0026】
次いで、ジアステレオマーに変換したミモシンを逆相HPLCに付して分画する。上記したように、ミモシン分子は、一般にHPLCに用いられているODS化シリカゲル(C18シリカゲル)カラムにはほとんど保持されることが無く、厳密な検定が非常に困難であるが、ミモシンをジアステレオマー化することでカラムに好ましく保持されるようになり、他の成分と分離して溶出させることが可能になる。従って、逆相HPLCの固定相としては、アルキル基修飾シリカゲル、パーフィユージョンクロマトグラフィー充填剤等を用いることができ、例えばHiPep-Intrada(株式会社ハイペップ研究所)や逆相のPorosカラム(アプライドバオシステムズ・ジャパン)等のような市販の逆相HPLC分析用カラムを好ましく用いることができる。特に、粒子径3μm,細孔径30nmでアルキルリガンドを導入し適度に表面極性を持たせた高純度シリカゲル充填剤を用いた市販のカラムHiPep-Intrada(株式会社ハイペップ研究所)によってとりわけ明確なD体、L体の分離が可能である。
【0027】
逆相HPLCの移動相(溶出液)としては、アセトニトリル濃度勾配が用いられ、グラジエント法により分離溶出が行なわれる。グラジエント法は、段階的勾配(ステップバイズグラジエント)でも直線的勾配(リニアグラジエント)でもよい。例えば、A=0.1%トリフルオロ酢酸(TFA)水溶液と、B=0.1%TFA−90%アセトニトリル水溶液を用いて、15分〜1時間程度かけてA:B=99:1→69:31とするリニアグラジエントが好ましい。
【0028】
逆相HPLCにおいて、カラムの温度は常温でよい。送液速度等のパラメーターも常法に準じて設定することができる。溶出液中のミモシンは、UV検出器等の公知の光学検出器を用いて検出することができる。
【0029】
本発明の光学純度の検定方法によれば、ミモシンのピークがインジェクションノイズと重複することなく、ミモシンのピークを明瞭に検出することができる。下記実施例に具体的に示されるように、明らかに一部ラセミ化したミモシンを本発明の検定方法でオンラインによる質量分析計を用いて純度検定すると、D体とL体のミモシンのピークがいずれも明瞭に確認できる(実施例2、図2B)。一方、本発明の精製ミモシン製造法でギンネムから抽出精製されたミモシン精製品を同様に純度検定すると、L体のピークのみが明瞭に確認される(実施例2、図2A)。これらのデータは、本発明の精製ミモシン製造法及び光学純度の検定方法が非常に優れていることを示すものである。
【0030】
ミモシンのジアステレオマー化にイソチオシアン酸2,3,4,6-テトラ-O-アセチル-β-D-グルコピラノシルを用いる場合を例として、具体的に光学純度の検定方法を説明すると、以下の通りである。ただし、この説明は本発明の範囲をイソチオシアン酸2,3,4,6-テトラ-O-アセチル-β-D-グルコピラノシルを用いる方法に限定することを意図するものではない。
【0031】
分離精製ミモシンの飽和水溶液、イソチオシアン酸2,3,4,6-テトラ-O-アセチル-β-D-グルコピラノシル1mM〜100mM、好ましくは5mM〜15mMのアセトニトリル溶液、炭酸水素ナトリウム1mM〜100mM、好ましくは5mM〜15mMの水溶液を等量ずつ混合し、室温にて0.5〜3時間、好ましくは1〜2時間おいて、ミモシンのアミノ基にイソチオシアン酸2,3,4,6-テトラ-O-アセチル-β-D-グルコピラノシルを導入してジアステレオマー化する。この溶液を逆相HPLCで分析する。逆相HPLCの溶出液には0.1%TFA水溶液と0.1%TFA−90%アセトニトリル水溶液を用いて、アセトニトリル濃度勾配によりグラジエント分析する(0.1%TFA水溶液:0.1%TFA−90%アセトニトリル水溶液=99:1 → 69:31、30分間)。検出は200〜280nmの紫外線により行なう。
【0032】
本発明のミモシン誘導体は、ミモシンのアミノ基に保護基を導入したものである。ミモシンをペプチド鎖や他の分子に導入するためには通常、保護ミモシンを用いる。そのようなミモシン誘導体を製造するためには、純度の高いミモシンを得ることが重要であるが、従来はミモシンを高純度で精製する方法や、ミモシンの純度を厳密に検定する方法は知られていなかった。上記ミモシン誘導体は、本願発明者らが開発した精製ミモシン製造法及び純度検定方法により、初めて製造可能になったものである。
【0033】
保護基としては特に限定されず、アミノ酸分子中のアミノ基を保護するために用いられるいかなる保護基であってもよい。例えば、ピペリジン等で切断が可能なFmoc基(9-フルオレニルメトキシ基)、酸により切断が可能なBoc基(tert-ブトキシカルボニル基)、パラジウム触媒により切断が可能なZ基(ベンジルオキシカルボニル基)、薄い酸で切断可能なTrt基(トリチル基)などが挙げられる。これらのうち、切断の簡便性、合成上の観点(特に副生成物の観点)から種々に検討した結果、ピペリジン等で除去可能なFmoc基が好ましい。現在最も一般的なペプチド合成法はFmoc法であり、Fmoc保護ミモシンであればFmoc法に適用可能である。アミノ基にFmoc基が導入されたFmoc保護ミモシンの化学式を以下に示す。
【0034】
【化2】
【0035】
上記したミモシン誘導体は、塩基の存在下でミモシンと保護化試薬とを反応させ、ミモシンのアミノ基に保護基を導入することにより製造することができる。「保護化試薬」とは、アミノ酸分子中のアミノ基に保護基を結合させることができる試薬であり、そのような試薬は種々のものが公知で市販品も多く存在する。本発明においては、公知のいずれの保護化試薬をも用いることができる。公知の保護化試薬の具体例として、Fmoc化試薬としてはFmoc−Cl(クロロギ酸フルオレニルメチル)、Fmoc−OSu(9−フルオレニルメチルスクシンイミジルカルボネート)等が挙げられ、Boc化試薬としてはBoc2O(ジ-tert-ブチルジカルボネート)等が挙げられ、Z化試薬としてはZ-Cl(クロロギ酸ベンジル)等が挙げられ、Trt化試薬としてはTrt-Cl(トリチルクロライド)等が挙げられるが、これらに限定されない。
【0036】
反応系内に共存させる上記塩基としては、炭酸ナトリウム、炭酸カリウム、炭酸水素ナトリウム、炭酸水素カリウム等を用いることができる。塩基及び保護化試薬の使用量は、特に限定されないが、ミモシンに対し2〜5当量の塩基と1〜3当量の保護化試薬とを用いることで満足できる結果が得られる。反応溶媒は水系溶媒であり、用いる保護化試薬の種類に応じて水に適宜極性溶媒を混合して用いることができる。例えば、保護化試薬としてFmoc-OSuを用いる場合は、水:ジオキサン=1:1の混合溶媒中で好ましくFmoc化反応を行なうことができる。反応系内に添加するミモシンの量は、通常、15〜60g/L程度である。下記実施例では、600mLの保護化反応系に対し20gの精製ミモシン塩酸塩が用いられている。反応温度は室温でよい。反応時間は通常6〜24時間、好ましくは10〜15時間である。
【0037】
ミモシンと保護化試薬とを反応させた場合、アミノ基のみならずヒドロキシ基にも一部保護基が導入され得る。保護化反応の後、反応溶液に塩基を添加することで、ヒドロキシ基に導入された保護基を外し、アミノ基のみが保護されたミモシン誘導体の収量を向上させることができる。従って、保護化試薬との反応後、反応溶液に塩基を添加する工程を加えることが好ましい。ここで用いる塩基も、上記と同様に炭酸ナトリウム、炭酸カリウム、炭酸水素ナトリウム、炭酸水素カリウム等を用いることができる。塩基の使用量は、反応開始時のミモシンに対し通常5〜50当量である。例えば、0.1Mの炭酸ナトリウム水溶液を用いる場合、保護化反応後の反応溶液を0.1M炭酸ナトリウム水溶液で2〜5倍希釈すればよい。塩基を添加して得られた溶液は、通常、反応溶液量の1/5〜1/2の酢酸エチルで2〜5回抽出する。水層を1N程度の塩酸でpH3〜6、好ましくはpH4〜5に調整すると、保護基が導入されたミモシン誘導体が析出するので、濾取等により溶液から回収することでミモシン誘導体を得ることができる。
【0038】
こうして得られたミモシン誘導体の純度は、逆相HPLCにより測定することができる。この逆相HPLCの条件は、上記した本発明のミモシンの光学純度の検定方法における逆相HPLCと同様に行なうことができる。
【実施例】
【0039】
以下、本発明を実施例に基づきより具体的に説明する。もっとも、本発明は下記実施例に限定されるものではない。
【0040】
実施例1
ミモシンの大量分離精製
新鮮なギンネムの葉25 Kgを沸騰水100 Lで10分間煮沸抽出した。抽出液約100 Lを室温まで冷却してろ過により不溶物を除去した後、強陽イオン交換樹脂(AMBERLITE(商標) IR120 H(Plus), GFS Chemicals Inc., オハイオ州米国)を10 Kg加えて一晩吸着させた。イオン交換樹脂を80%エタノール25L中に12時間浸漬して洗浄を行い、続けて25 Lの蒸留水中に2時間浸漬して洗浄した。イオン交換樹脂を2Nアンモニア水 40 L中に6時間浸漬してミモシンをイオン交換樹脂から溶出させた。溶出液はまず活性炭処理をした。活性炭の量は当該スケールでは5gを用い室温で10分間攪拌した後活性炭を濾過によって除去した。得られた濾液を減圧濃縮し、濃縮液を6N HClでpH4.5〜5.0に調整し、4℃で一晩置くことで粗ミモシン塩酸塩を析出させ、ミモシン粗結晶を得た(融点175℃分解)。濾取により得られた粗結晶を6N NaOH水溶液1.8 Lに溶解させ、4℃で一晩置くことで再結晶させ、ミモシン塩酸塩の結晶を得た。25 Kgのギンネムから100 gの高純度ミモシンが得られた。
融点225℃、旋光度[α]D 22.2 = -23.2 (C=2.11 mg/1.5 mL 溶媒:水)
【0041】
フーリエ変換イオンサイクロトロン型質量分析計は高分解能な精密質量解析能力によって確実な化学組成の決定が行えることが公知である。ブルカーダルトニクス社の当該質量分析計モデルApex-Q94eを用いて、ここで得られた高純度ミモシンを分析した結果、精密分子量199.07136という値を得た(図1)。天然同位体存在比に基づく解析により元素組成 C8H11N2O4 [M+H]となり、理論上の化学式と一致した。これによっても精製品が高純度であることが示された。
【0042】
実施例2
分離精製したミモシンの光学純度分析
(1) ジアステレオマー化による光学純度分析
ミモシンをジアステレオマー化する化合物として、光学活性HPLC用ラベル化剤A5514(イソチオシアン酸2,3,4,6-テトラ-O-アセチル-β-D-グルコピラノシル、東京化成)を用いた(下記式中の2)。
【0043】
【化3】
【0044】
実施例1で分離精製したミモシンの飽和水溶液を調整し、10mM光学活性HPLC用ラベル化剤(東京化成A5514)と10mM炭酸水素ナトリウムそれぞれ5μLずつとり混合し、室温で30分おいて反応させた。
【0045】
反応後の混合溶液を10倍に希釈して逆相HPLCにより分析した。分析条件を以下に示す。
カラム:HiPep-Intrada、サイズ3.0X150 mm(株式会社ハイペップ研究所)
カラム温度:25℃
溶出液:A=0.1%TFA水溶液、B=0.1%TFA−90%アセトニトリル水溶液
A:B=99:1→69:31(30分)のリニアグラジエント
流速:0.3 mL/min
検出:210及び254nm UV
【0046】
HPLCによる分析結果(検出:210nm UV)を図2に示す。実施例1の方法でギンネムから抽出、精製されたミモシン精製品(図2A)では、L体ミモシンのピークが明瞭に確認され、純度が非常に高いことが確認できた。比較のために、実施例1のミモシン精製品を文献[R. Adams and J.L. Johnson, Leucenol. VI. A Total Synthesis. J. Am. Chem Soc., 1949, 71, 705-708]に記載の方法で部分的にラセミ化させたミモシンを同様にジアステレオマー化してHPLC分析した結果を図2Bに示す。この場合には、D体とL体のピークが明瞭に確認された(図2B)。これにより、実施例2の検定方法の精度が非常に優れていることも確認できた。カラムの溶出液はオンラインでイオントラップ型質量分析を行ない、目的の化合物を同定した。質量分析データを図2C(D−ミモシン)及び2D(L−ミモシン)に示す。
Calcd.: 587.55, Found: 588.0 (M+H)
【0047】
(2) キラルカラムを用いた光学純度分析
公知のキラル分離用カラムCrownpak(+)(ダイセル化学工業株式会社)を用いて、実施例1で分離精製したミモシンのHPLC分析を行なった。
HPLC分析条件
カラム:ダイセル Crownpak(+) (4.0 id. x 150 mm)
カラム温度:25℃
溶出液:0.1% HClO4定組成溶離in 10 min
流速:0.4 mL/min
検出:210 nm
【0048】
ミモシンの分析結果を図3上段(a)に示す。図3下段(b)には、同一の条件でDL-アラニンを分析した結果を示す。図3に示される通り、キラル分離カラムを用いた場合、ミモシンの溶出位置がL-アラニンの溶出位置と極めて近くなった。ミモシンは分解するとアラニンを生成するため、キラル分離カラムによっては、ミモシンが分解してアラニンを生じているか否かを判別することは困難であり、厳密な光学検定はできなかった。
【0049】
実施例3
ミモシン誘導体の合成
【0050】
【化4】
【0051】
実施例1で得られたミモシン40 gと炭酸ナトリウム42 g(ミモシンに対し2当量)の水溶液600 mLを調製した。そこにFmoc−OSu 100 g(1.5当量)のジオキサン溶液600 mLを滴下した(ジオキサン:H2O=1:1)。6時間撹拌したのち、反応溶液を0.1 M炭酸ナトリウムで3600 mLまで希釈してさらに6時間撹拌した。反応は逆相HPLCにより分析して追跡した。反応溶液を酢酸エチル2000 mLで2回抽出し、水層を1N HClで中和すると固体が析出した。この固体を濾取することでFmoc保護ミモシン(Fmoc-Mim-OH)を50.8 g、収率60%で得た。
質量分析
Calcd.: 420.4, Found; 421.1 (M+H)
核磁気共鳴スペクトル解析
1H NMR (300 MHz, DMSO) δ 8.13-7.85 (m, 4H), 7.60-7.59 (br m, 2H), 7.44-7.29 (m, 4H), 7.03-7.00 (br m, 1H), 4.69 (br s, 1H), 4.55 (br s, 1H), 4.37-4.18 (m, 4H).
旋光度 [α]D 23.4 = -3.09 (C=2.34 mg/1.5 mL 溶媒 メタノール)
【0052】
得られたFmoc-Mim-OHを逆相HPLCにより純度検定した。カラムの溶出液はオンラインでイオントラップ質量分析を行なった。HPLCの結果を図4に、質量分析データを図5に示す。図4に示される通り、Fmoc-Mim-OHの明瞭なピークが観察され、純度が非常に高いことが確認できた。なお、上記反応の追跡及び純度検定に用いた逆相HPLCの条件は以下の通りである。
カラム: POROS-R2, 4.6x100 mm (アプライドバオシステムズ・ジャパン)
カラム温度:25℃
移動相:A=0.1% TFA水溶液、B=0.1% TFA-90%アセトニトリル水溶液
B=1%→31% in 30 min(リニアグラジエント)
流速:0.3 mL/min
検出:210nm UV
【図面の簡単な説明】
【0053】
【図1】フーリエ変換イオンサイクロトロン型質量分析計による、実施例1で製造したミモシン精製品の精密質量分析とその解析結果を示す。
【図2】実施例2で行なった、ジアステレオマー化したミモシンの逆相HPLCによる分析データ(検出:210nm UV)(A、B)及び質量分析データ(C、D)である。Aは実施例1で製造したミモシン精製品の分析データ、Bはミモシン精製品を公知の方法で一部ラセミ化したミモシンの分析データを示す。CはB中の左側のピークの化合物(D−ミモシン)の質量分析データ、Dは右側のピークの化合物(L−ミモシン)の質量分析データである。
【図3】公知のキラル分離用カラムを用いてミモシン及びアラニンをHPLC分析した結果を示す。上段(a)は実施例1で製造したミモシン精製品のデータ、下段(b)はDL-アラニンを分析したデータである。
【図4】Fmoc保護ミモシン(Fmoc-Mim-OH)の逆相HPLCによる純度検定結果である。
【図5】Fmoc保護ミモシン(Fmoc-Mim-OH)の質量分析データである。
【特許請求の範囲】
【請求項1】
(1) ギンネムを水に浸漬してミモシンを抽出する工程、
(2) 抽出液中に陽イオン交換樹脂を添加してミモシンを陽イオン交換樹脂に吸着させる工程、
(3) 前記陽イオン交換樹脂からミモシンを溶出させる工程、
(4) 工程(3)で得られた溶出液からミモシンの粗結晶を得る工程、
(5) 再結晶法により前記粗結晶からミモシンの結晶を得る工程
を含む、精製ミモシン又はその酸付加塩の製造方法。
【請求項2】
前記ギンネムはギンネムの茎葉部である請求項1記載の方法。
【請求項3】
前記工程(1)は80℃以上の加熱抽出により行われる請求項1又は2記載の方法。
【請求項4】
前記工程(2)で添加する陽イオン交換樹脂は強酸性陽イオン交換樹脂である請求項1ないし3のいずれか1項に記載の方法。
【請求項5】
前記工程(3)におけるミモシンの溶出は塩基性溶媒を用いて行なわれる請求項1ないし4のいずれか1項に記載の方法。
【請求項6】
前記塩基性溶媒は、アンモニア水、トリエチルアミン水溶液、水酸化ナトリウム水溶液又は水酸化カリウム水溶液である請求項5記載の方法。
【請求項7】
前記工程(4)は、前記溶出液を濃縮後、酸を添加して溶出液をpH3〜6に調整し、次いで該溶出液を0℃〜10℃で7〜24時間維持して粗結晶を析出させることを含む、請求項1ないし6のいずれか1項に記載の方法。
【請求項8】
前記酸は塩酸、希硫酸又は過塩素酸である請求項7記載の方法。
【請求項9】
前記工程(5)は、前記粗結晶100gに対し1〜5Lの強塩基水溶液を用いて前記粗結晶を強塩基水溶液中に溶解させ、得られた水溶液を酸でpH3〜6に調整し、次いで該水溶液を0℃〜10℃で7〜24時間維持して結晶を析出させることを含む、請求項1ないし8のいずれか1項に記載の方法。
【請求項10】
前記強塩基は水酸化ナトリウム、水酸化カリウム、炭酸カリウム又は炭酸ナトリウムである請求項9記載の方法。
【請求項11】
1つ以上のキラル炭素を有する分子を、ミモシンのアミノ基又はカルボキシル基に結合させてミモシンをジアステレオマーに変換し、次いで、該ミモシンを逆相HPLCに付し、アセトニトリル濃度勾配を用いたグラジエント法により該ミモシンを分離溶出させ、溶出液中のミモシンを光学検出器にて検出することを含む、ミモシンの光学純度の検定方法。
【請求項12】
前記1つ以上のキラル炭素を有する分子は、イソチオシアン酸2,3,4,6-テトラ-O-アセチル-β-D-グルコピラノシル又はイソチオシアン酸2,3,4,6-テトラ-O-ベンゾイル-β-D-グルコピラノシルである請求項11記載の方法。
【請求項13】
前記グラジエント法は、0.1%トリフルオロ酢酸水溶液:0.1%トリフルオロ酢酸−90%アセトニトリル水溶液=99:1から69:31への30分間直線濃度勾配を用いる請求項11又は12記載の方法。
【請求項14】
ミモシンのアミノ基に保護基が導入されたミモシン誘導体。
【請求項15】
前記保護基がFmoc基、Boc基、Z基又はトリチル基である請求項14記載のミモシン誘導体。
【請求項16】
前記保護基がFmoc基である請求項15記載のミモシン誘導体。
【請求項17】
下記式で示されるFmoc保護ミモシンである請求項16記載のミモシン誘導体。
【化1】
【請求項18】
塩基の存在下でミモシンと保護化試薬とを反応させ、ミモシンのアミノ基に保護基を導入することを含む、ミモシン誘導体の製造方法。
【請求項19】
ミモシンに対し2〜5当量の塩基と1〜3当量の保護化試薬とを用いる請求項18記載の方法。
【請求項20】
前記保護化試薬がFmoc化試薬である請求項18又は19記載の方法。
【請求項21】
前記Fmoc化試薬がFmoc−OSu(9−フルオレニルメチルスクシンイミジルカルボネート)である請求項20記載の方法。
【請求項22】
ジオキサンと水の混合溶媒中でミモシンとFmoc−OSuとを反応させる請求項21記載の方法。
【請求項23】
前記塩基が炭酸ナトリウム、炭酸カリウム、炭酸水素ナトリウム又は炭酸水素カリウムである請求項18ないし22のいずれか1項に記載の方法。
【請求項24】
ミモシンと保護化試薬とを反応させた後、得られた反応溶液に、前記ミモシンに対し1〜5当量の塩基を添加し、次いで、得られた溶液に酸を添加して中和し、アミノ基に保護基が導入されたミモシンの固体を析出させ、該固体を回収することをさらに含む、請求項18ないし23のいずれか1項に記載の方法。
【請求項25】
前記反応溶液に添加する前記塩基が、炭酸ナトリウム、炭酸カリウム、炭酸水素ナトリウム又は炭酸水素カリウムである請求項24記載の方法。
【請求項1】
(1) ギンネムを水に浸漬してミモシンを抽出する工程、
(2) 抽出液中に陽イオン交換樹脂を添加してミモシンを陽イオン交換樹脂に吸着させる工程、
(3) 前記陽イオン交換樹脂からミモシンを溶出させる工程、
(4) 工程(3)で得られた溶出液からミモシンの粗結晶を得る工程、
(5) 再結晶法により前記粗結晶からミモシンの結晶を得る工程
を含む、精製ミモシン又はその酸付加塩の製造方法。
【請求項2】
前記ギンネムはギンネムの茎葉部である請求項1記載の方法。
【請求項3】
前記工程(1)は80℃以上の加熱抽出により行われる請求項1又は2記載の方法。
【請求項4】
前記工程(2)で添加する陽イオン交換樹脂は強酸性陽イオン交換樹脂である請求項1ないし3のいずれか1項に記載の方法。
【請求項5】
前記工程(3)におけるミモシンの溶出は塩基性溶媒を用いて行なわれる請求項1ないし4のいずれか1項に記載の方法。
【請求項6】
前記塩基性溶媒は、アンモニア水、トリエチルアミン水溶液、水酸化ナトリウム水溶液又は水酸化カリウム水溶液である請求項5記載の方法。
【請求項7】
前記工程(4)は、前記溶出液を濃縮後、酸を添加して溶出液をpH3〜6に調整し、次いで該溶出液を0℃〜10℃で7〜24時間維持して粗結晶を析出させることを含む、請求項1ないし6のいずれか1項に記載の方法。
【請求項8】
前記酸は塩酸、希硫酸又は過塩素酸である請求項7記載の方法。
【請求項9】
前記工程(5)は、前記粗結晶100gに対し1〜5Lの強塩基水溶液を用いて前記粗結晶を強塩基水溶液中に溶解させ、得られた水溶液を酸でpH3〜6に調整し、次いで該水溶液を0℃〜10℃で7〜24時間維持して結晶を析出させることを含む、請求項1ないし8のいずれか1項に記載の方法。
【請求項10】
前記強塩基は水酸化ナトリウム、水酸化カリウム、炭酸カリウム又は炭酸ナトリウムである請求項9記載の方法。
【請求項11】
1つ以上のキラル炭素を有する分子を、ミモシンのアミノ基又はカルボキシル基に結合させてミモシンをジアステレオマーに変換し、次いで、該ミモシンを逆相HPLCに付し、アセトニトリル濃度勾配を用いたグラジエント法により該ミモシンを分離溶出させ、溶出液中のミモシンを光学検出器にて検出することを含む、ミモシンの光学純度の検定方法。
【請求項12】
前記1つ以上のキラル炭素を有する分子は、イソチオシアン酸2,3,4,6-テトラ-O-アセチル-β-D-グルコピラノシル又はイソチオシアン酸2,3,4,6-テトラ-O-ベンゾイル-β-D-グルコピラノシルである請求項11記載の方法。
【請求項13】
前記グラジエント法は、0.1%トリフルオロ酢酸水溶液:0.1%トリフルオロ酢酸−90%アセトニトリル水溶液=99:1から69:31への30分間直線濃度勾配を用いる請求項11又は12記載の方法。
【請求項14】
ミモシンのアミノ基に保護基が導入されたミモシン誘導体。
【請求項15】
前記保護基がFmoc基、Boc基、Z基又はトリチル基である請求項14記載のミモシン誘導体。
【請求項16】
前記保護基がFmoc基である請求項15記載のミモシン誘導体。
【請求項17】
下記式で示されるFmoc保護ミモシンである請求項16記載のミモシン誘導体。
【化1】
【請求項18】
塩基の存在下でミモシンと保護化試薬とを反応させ、ミモシンのアミノ基に保護基を導入することを含む、ミモシン誘導体の製造方法。
【請求項19】
ミモシンに対し2〜5当量の塩基と1〜3当量の保護化試薬とを用いる請求項18記載の方法。
【請求項20】
前記保護化試薬がFmoc化試薬である請求項18又は19記載の方法。
【請求項21】
前記Fmoc化試薬がFmoc−OSu(9−フルオレニルメチルスクシンイミジルカルボネート)である請求項20記載の方法。
【請求項22】
ジオキサンと水の混合溶媒中でミモシンとFmoc−OSuとを反応させる請求項21記載の方法。
【請求項23】
前記塩基が炭酸ナトリウム、炭酸カリウム、炭酸水素ナトリウム又は炭酸水素カリウムである請求項18ないし22のいずれか1項に記載の方法。
【請求項24】
ミモシンと保護化試薬とを反応させた後、得られた反応溶液に、前記ミモシンに対し1〜5当量の塩基を添加し、次いで、得られた溶液に酸を添加して中和し、アミノ基に保護基が導入されたミモシンの固体を析出させ、該固体を回収することをさらに含む、請求項18ないし23のいずれか1項に記載の方法。
【請求項25】
前記反応溶液に添加する前記塩基が、炭酸ナトリウム、炭酸カリウム、炭酸水素ナトリウム又は炭酸水素カリウムである請求項24記載の方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公開番号】特開2010−47487(P2010−47487A)
【公開日】平成22年3月4日(2010.3.4)
【国際特許分類】
【出願番号】特願2008−210592(P2008−210592)
【出願日】平成20年8月19日(2008.8.19)
【出願人】(502249851)株式会社ハイペップ研究所 (11)
【Fターム(参考)】
【公開日】平成22年3月4日(2010.3.4)
【国際特許分類】
【出願日】平成20年8月19日(2008.8.19)
【出願人】(502249851)株式会社ハイペップ研究所 (11)
【Fターム(参考)】
[ Back to top ]