
ムコノラクトン、β−ケトアジピン酸及び/又はレブリン酸の発酵生産
【課題】様々な機能性プラスチックス原料や化学製品の原料と成りうる新規なリグニン分解産物を発酵生産する方法の提供。
【解決手段】本発明は、pcaB遺伝子、pcaC遺伝子と、vanA遺伝子及びvanB遺伝子並びにpcaH遺伝子及びpcaG遺伝子、とから成るDNA分子群、そして任意にpcaD遺伝子を含んで成る組換えベクター、当該組換えベクターを含む形質転換体、当該形質転換体のpcaJ遺伝子が破壊された遺伝子破壊株、並びに当該形質転換体又は当該遺伝子破壊株を用いた、ムコノラクトン、β-ケトアジピン酸の発酵生産方法、レブリン酸の製造方法を提供する。
【解決手段】本発明は、pcaB遺伝子、pcaC遺伝子と、vanA遺伝子及びvanB遺伝子並びにpcaH遺伝子及びpcaG遺伝子、とから成るDNA分子群、そして任意にpcaD遺伝子を含んで成る組換えベクター、当該組換えベクターを含む形質転換体、当該形質転換体のpcaJ遺伝子が破壊された遺伝子破壊株、並びに当該形質転換体又は当該遺伝子破壊株を用いた、ムコノラクトン、β-ケトアジピン酸の発酵生産方法、レブリン酸の製造方法を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、植物成分あるいは石油成分由来、あるいは化学的に合成されたバニリン酸及び/又はプロトカテク酸からムコノラクトン、β-ケトアジピン酸を発酵生産するための多段反応プロセスを構成する酵素をコードした遺伝子を含む組換えベクター、当該組換えベクターを導入した形質転換体、β−ケトアジピン酸又はムコノラクトンの分解能を消失せしめた遺伝子破壊株、並びに当該形質転換体又は当該遺伝子破壊株を用いてムコノラクトン、β-ケトアジピン酸を製造する方法、そしてβ-ケトアジピン酸からレブリン酸を製造する方法に関する。
【背景技術】
【0002】
植物主要成分であるリグニンは、芳香族高分子化合物として植物細胞壁に普遍的に含まれており、樹木では30%、イネやトウモロコシ稈の15%を占めるバイオマス資源である。その他にも植物は多様な芳香族化合物を構成成分として含んでいる。しかし、リグニンを主成分とする植物由来の芳香族成分は化学構造が多様な成分で構成されていることや、複雑な高分子構造を持つため、有効な利用技術が開発されていない。従来の利用技術として挙げられるのは、リグニンを主成分とする植物由来の芳香族成分をアルカリ分解などの化学分解で生成する低分子芳香族分解物から、香料原料であるバニリンを分離製造する実用化技術がある。しかし、化学分解で生成するバニリン以外の多量の低分子芳香族物質の有効な利用方法はないのが現状である。そのため製紙工程で大量に生成するリグニンは有効利用される事無く重油の代替え品として燃焼されている。
【0003】
今日の石油化学工業の発展を支えたのは、多様な化学成分の混合物である原油を触媒分解と分溜によってベンゼンやエチレンなどの単一な中間物質に一旦変換し、それらを原料に多様な機能性プラスチックスを製造するという原理である。この石油化学工業の発展を支えた基本原理は、化学構造の多様さや複雑な高分子構造を持つリグニンなど植物芳香族成分の利用技術においても適用可能な普遍的原理である。リグニンを主成分とする植物由来の芳香族成分利用の技術を開発する上で、石油化学の触媒分解に相当するプロセスとして、リグニンなど植物芳香族成分の加水分解や酸化分解、可溶媒分解などの化学的分解法や、超臨界水や超臨界有機溶媒による物理化学的分解法など多くの低分子化技術が既に数多く研究され開発されている。しかし、もう一つの技術である、各種分解法により生成する植物成分由来低分子混合物から、様々な機能性プラスチックス原料や化学製品の原料と成りうる有用な単一の中間物質(石油化学においてはエチレンやベンゼン)への変換分離技術は開発されていなかった。この技術が開発されれば、リグニンを主成分とする植物由来の芳香族成分利用が、石油化学に匹敵する技術として発展する可能性がある。
【0004】
本発明者らは既に、スフィンゴビウム・パウシモビリス(Sphingobium paucimobilis)SYK-6株等から獲得した情報を用いて、リグニンの加水分解産物であるバニリン、シリンガアルデヒド、バニリン酸、シリンガ酸、プロトカテク酸から、生分解性ポリマーの原料として利用可能な2−ピロン−4,6−ジカルボン酸(PDC)を発酵生産することに成功している(特開2005-278549号公報)。更に、本発明者らは、WO2008/018640号公報において、バニリン、バニリン酸、及び/又はプロトカテク酸から3-カルボキシ-cis, cis-ムコン酸及び/又は3-カルボキシムコノラクトンへの発酵生産プロセスについても報告している。この発酵生産プロセスは、3種の酵素、ベンズアルデヒドデヒドロゲナーゼ(LigV)、バニリン酸ディメチラーゼ(VanAB)、プロトカテク酸3,4−ジオキシゲナーゼ(PcaHG)が同調的に酵素反応を進行することで構成される。
【0005】
石油化学工業の発展により、今日では原油から様々な有機材料が得られている。リグニンを主成分とする植物由来の芳香族成分から付加価値の高い有機材料を生成するには、2−ピロン−4,6−ジカルボン酸や3-カルボキシムコノラクトンに加え、新たなリグニン分解産物の取得が必要であると考えられる。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特開2005-278549号公報
【特許文献2】WO2008/018640号公報
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明は、リグニンの新規分解産物を発酵生産する方法を提供することを課題とする。
【課題を解決するための手段】
【0008】
本発明者は、リグニン等の植物芳香族成分の低分子化処理混合物に含まれるバニリン酸、プロトカテク酸から新規の分解産物を発酵生産する方法を検討した結果、ニューロスポラ・クラッサ(Neurospora crassa)野生型740R23-1A株(FGSC 987)由来の3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素(3-カルボキシ-cis,cis-ムコン酸シクラーゼ遺伝子)が3-カルボキシ-cis,cis-ムコン酸から3-カルボキシムコノラクトン(beta-カルボキシムコノラクトン)を生成するのに対して、シュードモナス・プチダ(Pseudomonas putida)由来の3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素(PcaB)を用いることにより4-カルボキシムコノラクトン(γ-カルボキシムコノラクトン)が生成し、γ-カルボキシムコノラクトンデカルボキシラーゼ(PcaC)及びβ-ケトアジピン酸エノールラクトンヒドロラーゼ(PcaD)を作用させることにより、β-ケトアジピン酸が生成することを見出し、本発明を完成させた。また、ムコノラクトン生産用の株としてPcaD破壊株を作成し、β-ケトアジピン酸生産用の株としてPcaJ破壊株を作成するとともに、当該酸をレブリン酸に変換する方法も確立した。
【0009】
すなわち、(1)本発明は、好ましくはプロモーターとターミネーターとの間に、pcaB遺伝子及びpcaC遺伝子と、vanA遺伝子及びvanB遺伝子並びにpcaH遺伝子及びpcaG遺伝子と、から成るDNA分子群を含んで成る組換えベクター:
ここで、
(a) pcaB遺伝子は下記(a-1)〜(a-4)のいずれかのDNA分子であり、
(a-1) 配列番号1記載のDNA分子、
(a-2) 配列番号2記載のアミノ酸配列をコードするDNA分子、
(a-3) 配列番号1記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つ3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素活性を有するポリペプチドをコードするDNA分子、あるいは
(a-4) 配列番号2記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つ3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素活性を有するポリペプチドをコードするDNA分子、
(b) pcaC遺伝子は下記(b-1)〜(b-4)のいずれかのDNA分子であり、
(b-1) 配列番号3記載のDNA分子、
(b-2) 配列番号4記載のアミノ酸配列をコードするDNA分子、
(b-3) 配列番号3記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つγ-カルボキシムコノラクトンデカルボキシラーゼ活性を有するポリペプチドをコードするDNA分子、あるいは
(b-4) 配列番号4記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つγ-カルボキシムコノラクトンデカルボキシラーゼ活性を有するポリペプチドをコードするDNA分子、
(c) vanA遺伝子は下記(c-1)〜(c-4)のいずれかのDNA分子であり、
(c-1) 配列番号7記載のDNA分子、
(c-2) 配列番号8記載のアミノ酸配列をコードするDNA分子、
(c-3) 配列番号7記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(c-4) 配列番号8記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(d) vanB遺伝子は下記(d-1)〜(d-4)のいずれかのDNA分子であり、
(d-1) 配列番号9記載のDNA分子、
(d-2) 配列番号10記載のアミノ酸配列をコードするDNA分子、
(d-3) 配列番号9記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(d-4) 配列番号10記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(e) pcaH遺伝子は下記(e-1)〜(e-4)のいずれかのDNA分子である。
(e-1) 配列番号11記載のDNA分子、
(e-2) 配列番号12記載のアミノ酸配列をコードするDNA分子、
(e-3) 配列番号11記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(e-4) 配列番号12記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(f) pcaG遺伝子は下記(f-1)〜(f-4)のいずれかのDNA分子である。
(f-1) 配列番号13記載のDNA分子、
(f-2) 配列番号14記載のアミノ酸配列をコードするDNA分子、
(f-3) 配列番号13記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(f-4) 配列番号14記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
を提供する。
(2)本発明は、前記DNA分子群が更に以下のpcaD遺伝子を含んで成る、(1)の組換えベクター:
(g) pcaD遺伝子は下記(g-1)〜(g-4)のいずれかのDNA分子である。
(g-1) 配列番号5記載のDNA分子、
(g-2) 配列番号6記載のアミノ酸配列をコードするDNA分子、
(g-3) 配列番号5記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つβ-ケトアジピン酸エノールラクトンヒドロラーゼ活性を有するポリペプチドをコードするDNA分子、あるいは
(g-4) 配列番号6記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つβ-ケトアジピン酸エノールラクトンヒドロラーゼ活性を有するポリペプチドをコードするDNA分子、
を提供する。
(3)本発明は、(1)又は(2)の組換えベクターを有する形質転換体、を提供する
(4)本発明は、(1)の組換えベクターを有する形質転換体であって、β-ケトアジピン酸エノールラクトン代謝・分解能が欠損している、形質転換体、を提供する。
(5)本発明は、染色体DNA上にpcaB遺伝子及びpcaC遺伝子と、vanA遺伝子及びvanB遺伝子並びにpcaH遺伝子及びpcaG遺伝子と、から成るDNA分子群が存在する微生物細胞又は当該DNA分子群を含んで成る組換えベクターを有する形質転換体の、β-ケトアジピン酸エノールラクトン代謝・分解能が欠損している遺伝子破壊株であって、当該染色体DNAに存在するpcaD遺伝子が破壊されている遺伝子破壊株:
ここで、
(a) pcaB遺伝子は下記(a-1)〜(a-4)のいずれかのDNA分子であり、
(a-1) 配列番号1記載のDNA分子、
(a-2) 配列番号2記載のアミノ酸配列をコードするDNA分子、
(a-3) 配列番号1記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つ3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素活性を有するポリペプチドをコードするDNA分子、あるいは
(a-4) 配列番号2記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つ3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素活性を有するポリペプチドをコードするDNA分子、
(b) pcaC遺伝子は下記(b-1)〜(b-4)のいずれかのDNA分子であり、
(b-1) 配列番号3記載のDNA分子、
(b-2) 配列番号4記載のアミノ酸配列をコードするDNA分子、
(b-3) 配列番号3記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つγ-カルボキシムコノラクトンデカルボキシラーゼ活性を有するポリペプチドをコードするDNA分子、あるいは
(b-4) 配列番号4記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つγ-カルボキシムコノラクトンデカルボキシラーゼ活性を有するポリペプチドをコードするDNA分子、
(c) vanA遺伝子は下記(c-1)〜(c-4)のいずれかのDNA分子であり、
(c-1) 配列番号7記載のDNA分子、
(c-2) 配列番号8記載のアミノ酸配列をコードするDNA分子、
(c-3) 配列番号7記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(c-4) 配列番号8記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(d) vanB遺伝子は下記(d-1)〜(d-4)のいずれかのDNA分子であり、
(d-1) 配列番号9記載のDNA分子、
(d-2) 配列番号10記載のアミノ酸配列をコードするDNA分子、
(d-3) 配列番号9記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(d-4) 配列番号10記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(e) pcaH遺伝子は下記(e-1)〜(e-4)のいずれかのDNA分子であり、
(e-1) 配列番号11記載のDNA分子、
(e-2) 配列番号12記載のアミノ酸配列をコードするDNA分子、
(e-3) 配列番号11記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(e-4) 配列番号12記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(f) pcaG遺伝子は下記(f-1)〜(f-4)のいずれかのDNA分子であり、
(f-1) 配列番号13記載のDNA分子、
(f-2) 配列番号14記載のアミノ酸配列をコードするDNA分子、
(f-3) 配列番号13記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(f-4) 配列番号14記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、そして
(g) pcaD遺伝子は下記(c-1)〜(c-4)のいずれかのDNA分子である。
(g-1) 配列番号5記載のDNA分子、
(g-2) 配列番号6記載のアミノ酸配列をコードするDNA分子、
(g-3) 配列番号5記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つβ-ケトアジピン酸エノールラクトンヒドロラーゼ活性を有するポリペプチドをコードするDNA分子、あるいは
(g-4) 配列番号6記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つβ-ケトアジピン酸エノールラクトンヒドロラーゼ活性を有するポリペプチドをコードするDNA分子、
を提供する。
(6)本発明は、(4)の形質転換体、又は(5)の遺伝子破壊株を、バニリン酸及び/又はプロトカテク酸の存在下で培養し、当該培養物からムコノラクトンを採取することを特徴とする、ムコノラクトンの製造方法、を提供する。
(7)本発明は、(2)の組換えベクターを含む形質転換体であって、β-ケトアジピン酸代謝・分解能が欠損している、形質転換体、を提供する。
(8)本発明は、染色体DNA上にpcaB遺伝子、pcaC遺伝子及びpcaD遺伝子と、vanA遺伝子及びvanB遺伝子並びにpcaH遺伝子及びpcaG遺伝子と、から成るDNA分子群が存在する微生物細胞又は当該DNA分子群を含んで成る組換えベクターを有する形質転換体の、β-ケトアジピン酸代謝・分解能が欠損している遺伝子破壊株であって、当該染色体DNAに存在するpcaJ遺伝子が破壊されている遺伝子破壊株:
ここで、
(a) pcaB遺伝子は下記(a-1)〜(a-4)のいずれかのDNA分子であり、
(a-1) 配列番号1記載のDNA分子、
(a-2) 配列番号2記載のアミノ酸配列をコードするDNA分子、
(a-3) 配列番号1記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つ3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素活性を有するポリペプチドをコードするDNA分子、あるいは
(a-4) 配列番号2記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つ3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素活性を有するポリペプチドをコードするDNA分子、
(b) pcaC遺伝子は下記(b-1)〜(b-4)のいずれかのDNA分子であり、
(b-1) 配列番号3記載のDNA分子、
(b-2) 配列番号4記載のアミノ酸配列をコードするDNA分子、
(b-3) 配列番号3記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つγ-カルボキシムコノラクトンデカルボキシラーゼ活性を有するポリペプチドをコードするDNA分子、あるいは
(b-4) 配列番号4記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つγ-カルボキシムコノラクトンデカルボキシラーゼ活性を有するポリペプチドをコードするDNA分子、
(c) vanA遺伝子は下記(c-1)〜(c-4)のいずれかのDNA分子であり、
(c-1) 配列番号7記載のDNA分子、
(c-2) 配列番号8記載のアミノ酸配列をコードするDNA分子、
(c-3) 配列番号7記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(c-4) 配列番号8記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(d) vanB遺伝子は下記(d-1)〜(d-4)のいずれかのDNA分子であり、
(d-1) 配列番号9記載のDNA分子、
(d-2) 配列番号10記載のアミノ酸配列をコードするDNA分子、
(d-3) 配列番号9記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(d-4) 配列番号10記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(e) pcaH遺伝子は下記(e-1)〜(e-4)のいずれかのDNA分子であり、
(e-1) 配列番号11記載のDNA分子、
(e-2) 配列番号12記載のアミノ酸配列をコードするDNA分子、
(e-3) 配列番号11記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(e-4) 配列番号12記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(f) pcaG遺伝子は下記(f-1)〜(f-4)のいずれかのDNA分子であり、
(f-1) 配列番号13記載のDNA分子、
(f-2) 配列番号14記載のアミノ酸配列をコードするDNA分子、
(f-3) 配列番号13記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(f-4) 配列番号14記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、そして
(g) pcaD遺伝子は下記(c-1)〜(c-4)のいずれかのDNA分子であり、
(g-1) 配列番号5記載のDNA分子、
(g-2) 配列番号6記載のアミノ酸配列をコードするDNA分子、
(g-3) 配列番号5記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つβ-ケトアジピン酸エノールラクトンヒドロラーゼ活性を有するポリペプチドをコードするDNA分子、あるいは
(g-4) 配列番号6記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つβ-ケトアジピン酸エノールラクトンヒドロラーゼ活性を有するポリペプチドをコードするDNA分子、
(h) pcaJ遺伝子は下記(h-1)〜(h-4)のいずれかのDNA分子である。
(h-1) 配列番号21記載のDNA分子、
(h-2) 配列番号22記載のアミノ酸配列をコードするDNA分子、
(h-3) 配列番号21記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つβ-ケトアジピン酸スクシニルCoAトランスフェラーゼ活性を有するポリペプチドをコードするDNA分子、あるいは
(h-4) 配列番号22記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つβ-ケトアジピン酸スクシニルCoAトランスフェラーゼ活性を有するポリペプチドをコードするDNA分子、
を提供する。
(9)本発明は、(7)の形質転換体、又は(8)の遺伝子破壊株を、バニリン酸及び/又はプロトカテク酸の存在下で培養し、当該培養物からβ-ケトアジピン酸を採取することを特徴とする、β-ケトアジピン酸の製造方法、を提供する。
(10)本発明は、(9)の方法に従い製造されたβ-ケトアジピン酸を脱炭酸反応にかけてレブリン酸を得ることを特徴とする、レブリン酸の製造方法、を提供する。
【発明の効果】
【0010】
本発明によれば、バニリン酸及び/又はプロトカテク酸から、ムコノラクトン、β-ケトアジピン酸までの変換に必要な酵素(VanAB, PcaHG, PcaB, PcaC, PcaD)による以下の多段階代謝プロセス:
【化1】

が同調的に進行することで、ムコノラクトン、β-ケトアジピン酸の量産が可能となる。β−ケトアジピン酸は、反応性が高いケトンをβ位に、そしてカルボン酸を両末端に有している。β−ケトアジピン酸と同様に、末端にカルボン酸を2つ有するアジピン酸が66−ナイロンなどの高分子製造原料として利用されていることを考慮すると、β−ケトアジピン酸も高分子材料や化学製品への応用が期待される。また、γ-クロトノラクトン骨格を持つムコノラクトンは、香料、樹脂用溶剤、染み抜き剤、接着剤、塗料剥離剤等への応用が期待される。
【0011】
更に、得られたβ-ケトアジピン酸は、酸処理することでレブリン酸に変換することもできる。レブリン酸は、種々の有用な化成品の原料となる基礎的な化学物質であり、β-アセチルプロピオン酸とも称されている。レブリン酸自体も抗菌剤、燃料組成物等、様々な分野での用途が知られている(特開2003−226641号公報、特表2007−510787号公報等)。
【図面の簡単な説明】
【0012】
【図1A】図1Aは、組換えベクターpVAadi1F6の作製方法を示す図である。
【図1B】図1Bは、組換えベクターpVAaKL1Fの作製方法を示す図である。
【図2】図2は、実施例3で調製した培養液中のOD、基質、変換物の変化を示す図である。
【図3】図3は、実施例3で得られたβ−ケトアジピン酸の結晶の光学顕微鏡写真である。
【図4】図4は、実施例4で調製した培養液中のOD、基質、変換物の変化を示す図である
【図5】図5は、実施例5で調製した培養液中のOD、基質、変換物の変化を示す図である。
【図6】図6は、実施例5で得られたムコノラクトンの結晶の光学顕微鏡写真である。
【図7】図7は、実施例6で調製した培養液中のOD、基質、変換物の変化を示す図である。
【発明を実施するための形態】
【0013】
本発明の組換えベクターは、バニリン酸、プロトカテク酸、これらの混合物等のリグニン分解物からムコノラクトンへの製造プロセス、そしてムコノラクトンからβ−ケトアジピン酸への製造プロセスを触媒するための酵素遺伝子を含む組換えベクターである。本発明の組換えベクターは、具体的な態様において、バニリン酸、プロトカテク酸、これらの混合物等のリグニン分解物からムコノラクトンを製造するプロセスを触媒する、配列番号1記載のDNA分子又は配列番号2記載のアミノ酸配列をコードするDNA分子から成る3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素遺伝子(pcaB)及び配列番号3記載のDNA分子又は配列番号4記載のアミノ酸配列をコードするDNA分子から成るγ-カルボキシムコノラクトンデカルボキシラーゼ遺伝子(pcaC)と、配列番号7記載のDNA分子又は配列番号8記載のアミノ酸配列をコードするDNA分子から成るバニリン酸ディメチラーゼサブユニット遺伝子(vanA)、配列番号9記載のDNA分子又は配列番号10記載のアミノ酸配列をコードするDNA分子から成るバニリン酸ディメチラーゼサブユニット遺伝子(vanB)、配列番号11記載のDNA分子又は配列番号12記載のアミノ酸配列をコードするDNA分子から成るプロトカテク酸3,4−ジオキシゲナーゼサブユニット遺伝子(pcaH)、配列番号13記載のDNA分子又は配列番号14記載のアミノ酸配列をコードするDNA分子から成るプロトカテク酸3,4−ジオキシゲナーゼサブユニット遺伝子(pcaH)と、から成る遺伝子群を含む組換えベクターである。
【0014】
本発明の組換えベクターは、別の具体的な態様において、バニリン酸、プロトカテク酸、これらの混合物等のリグニン分解物からβ−ケトアジピン酸を製造するプロセスを触媒する、配列番号1記載のDNA分子又は配列番号2記載のアミノ酸配列をコードするDNA分子から成る3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素遺伝子(pcaB)、配列番号3記載のDNA分子又は配列番号4記載のアミノ酸配列をコードするDNA分子から成るγ-カルボキシムコノラクトンデカルボキシラーゼ遺伝子(pcaC)、及び配列番号5記載のDNA分子又は配列番号6記載のアミノ酸配列をコードするDNA分子から成るβ-ケトアジピン酸エノールラクトンヒドロラーゼ遺伝子(pcaD)と、配列番号7記載のDNA分子又は配列番号8記載のアミノ酸配列をコードするDNA分子から成るバニリン酸ディメチラーゼサブユニット遺伝子(vanA)、配列番号9記載のDNA分子又は配列番号10記載のアミノ酸配列をコードするDNA分子から成るバニリン酸ディメチラーゼサブユニット遺伝子(vanB)、配列番号11記載のDNA分子又は配列番号12記載のアミノ酸配列をコードするDNA分子から成るプロトカテク酸3,4−ジオキシゲナーゼサブユニット遺伝子(pcaH)、配列番号13記載のDNA分子又は配列番号14記載のアミノ酸配列をコードするDNA分子から成るプロトカテク酸3,4−ジオキシゲナーゼサブユニット遺伝子(pcaH)と、から成る遺伝子群を含む組換えベクターである。
【0015】
本発明の組換えベクターにおいては、上記の特定の遺伝子群がすべて一つの発現ベクター内に含まれるように構成されていてもよく、あるいは当該遺伝子群が2グループ以上に分かれて、それぞれ別個の発現ベクターに含まれるように構成されていてもよい。発現ベクターにおいて、かかる遺伝子群は、プロモーターとターミネーターとの間に挿入することができる。しかしながら、本発明で使用する遺伝子群の配置はこの位置に限定されない。
【0016】
本発明の組換えベクターに含まれる3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素遺伝子(pcaB)、γ-カルボキシムコノラクトンデカルボキシラーゼ遺伝子(pcaC)及びβ-ケトアジピン酸エノールラクトンヒドロラーゼ遺伝子(pcaD)は、シュードモナス・プチダ(Pseudomonas putida)KT2440株から、以下のプライマー及びアニーリング条件を用い、PCR法により獲得した:
pcaB:フォワードプライマー (GATGATGGCGATTGCCTTCC)(配列番号15)、リバースプライマー (GTCTCCTTCAGGCAGTGAAACG)(配列番号16)、アニーリング温度50℃;
pcaC:フォワードプライマー (CTCCTTCTAGACATGGACGAGAAAGAACGTTACGACGC)(配列番号17)、リバースプライマー (AGTGGATCCTTACTGGCGAGAACTCTACGCCAAG)(配列番号18)、アニーリング温度55℃;
pcaD:フォワードプライマー (ATAAGCTTCAACGAGACCGCTGTGGC)(配列番号19)、リバースプライマー (AATCTAGAGCGTTGTCCTCAGTGAG)(配列番号20)、アニーリング温度55℃。ここで、pcaB、pcaC、pcaDのオープンリーディングフレームの塩基配列をそれぞれ配列番号1、3、5に、そして当該配列によってコードされるタンパク質のアミノ酸配列をそれぞれ配列番号2,4、6に記載する。
【0017】
本発明で使用するバニリン酸ディメチラーゼサブユニット遺伝子(vanA及びvanB)は、特開2005-278549号公報(上掲)に記載の、シュードモナス・プチダ(Pseudomonas putida)PpY101株由来のバニリン酸ディメチラーゼ遺伝子(同公報の配列番号1)に相当するが、当該配列は本発明のものと一部異なる(配列番号7及び8)。ここで、バニリン酸ディメチラーゼサブユニット(VanA及びVanB)は、会合してバニリン酸ディメチラーゼ(VanAB)を構成する。
【0018】
バニリン酸ディメチラーゼ(VanAB)は、微生物細胞内で本来半減期の短い不安定ポリペプチドとして生成されている。バニリン酸ディメチラーゼは、特開2005-278549に記載されているように、LacZのαフラグメントに含まれる16アミノ酸からなるオリゴペプチド、或いはヒスチジン6個を含む30アミノ酸からなるオリゴペプチドを一例とする、VanAの安定化に寄与し、かつVanAの酵素機能を妨害しない様々なポリペプチドをVanAのN 末端に付加することにより安定化される。
【0019】
プロトカテク酸3,4−ジオキシゲナーゼサブユニット遺伝子(pcaH及びpcaG)は、WO2008/018640号公報(上掲)に記載のとおり、シュードモナス・プチダKT2440株由来であり、J Bacteriol. 1989 Nov; 171(11): 5915-21にその遺伝子情報が開示されている。プロトカテク酸3,4−ジオキシゲナーゼサブユニット(PcaH及びPcaG)は、会合してプロトカテク酸3,4−ジオキシゲナーゼ(PcaHG)を構成する。
【0020】
本発明のベクターに含まれる遺伝子の取得方法は特に限定されず、例えば、開示されている当該遺伝子の塩基配列の方法に基づいて適当なプローブやライブラリーを調製し、それらを用いて、菌株のcDNAライブラリーやゲノムDNAライブラリーをスクリーニングすることで得ることができる。具体的には、本発明の遺伝子を発現する微生物、例えばシュードモナス・プチダ(Pseudomonas putida)KT2440株等より、常法に従ってゲノムDNAライブラリーを調製し、当該ライブラリーから、本発明遺伝子に特有の適当なプローブ等を用いて所望クローンを選択することにより製造することができる。微生物株からの全RNAの分離、mRNAの分離及び精製、ゲノムDNAの取得及びそのクローニングなどは、いずれも常法に従って行うことができる。
【0021】
上記方法において用いられるプローブとしては、本発明の遺伝子の塩基配列に関する情報をもとにして化学合成されたDNAなどが一般的に使用できる。また、本発明の遺伝子の塩基配列情報に基づき設定したセンス・プライマー及びアンチセンス・プライマーを、スクリーニング用プローブとして用いることができる。
【0022】
本発明の遺伝子の取得に際しては、PCR(Science,230,1350(1985))によるDNA増幅法が好適に利用できる。増幅させたDNA断片の単離精製は、常法に従って行うことができる。例えばゲル電気泳動法などが挙げられる。上記方法に従って得られる本発明の遺伝子は、常法、例えばジデオキシ法(Proc.Natl.Acad.Sci.,USA.,74,5463(1977))、マキサム−ギルバート法(Methods in Enzymology,65,499(1980))などに従って、その塩基配列を決定することができる。また、簡便には、市販のシークエンスキットなどを用いて、その塩基配列を決定することができる。
【0023】
pcaB遺伝子(配列番号1)、pcaC遺伝子(配列番号3)pcaD遺伝子(配列番号5)、vanA遺伝子(配列番号7)及びvanB遺伝子(配列番号9)並びにpcaH遺伝子(配列番号11)及びpcaG遺伝子(配列番号13)は、対応する配列番号によって特定されるDNA分子や、そのアミノ酸配列をコードするDNA分子の他に、そのDNA分子と相補的な塩基配列からなるDNAと高ストリンジェントな条件でハイブリダイズし、かつそのDNA分子と同一の活性を有するポリペプチドをコードするDNA分子も含まれる。
【0024】
ここで、本明細書で使用する場合、「高ストリンジェントな条件」とは、いわゆる特異的なハイブリッドが形成され、非特異的なハイブリッドが形成されない条件をいう。高ストリンジェントな条件としては、同一性が高いDNA同士がハイブリダイズし、それより同一性が低いDNA同士がハイブリダイズしない条件、例えば、Molecular cloning a Laboratory manual 2nd edition(Sambrookら、1989)に記載の条件が挙げられる。具体的には、通常のサザンハイブリダイゼーションにおける洗浄の条件である60℃、1×SSC、0.1%SDS、好ましくは、0.1×SSC、0.1%SDSで相当する塩濃度でハイブリダイズする条件が挙げられる。
【0025】
本発明のベクターに含まれる遺伝子の取得に際しては、PCR法(Science,230,1350(1985))によるDNA増幅法が好適に利用できる。増幅させたDNA断片の単離精製は、常法に従って行うことができる。例えばゲル電気泳動法などが挙げられる。上記方法に従って得られる本発明の遺伝子は、常法、例えばジデオキシ法(Proc.Natl.Acad.Sci.,USA.,74,5463(1977))、マキサム−ギルバート法(Methods in Enzymology,65,499(1980))などに従って、その塩基配列を決定することができる。また、簡便には、市販のシークエンスキットなどを用いて、その塩基配列を決定することができる。
【0026】
高ストリンジェントでハイブリダイズするDNA分子に加え、本発明のベクターに含まれる各酵素のコード遺伝子は、(a) 配列番号2に記載されている3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つ3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素活性を有するポリペプチドをコードするDNA分子、(b) 配列番号4に記載されているγ-カルボキシムコノラクトンデカルボキシラーゼのアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つγ-カルボキシムコノラクトンデカルボキシラーゼ活性を有するポリペプチドをコードするDNA分子、 (c) 配列番号8に記載されているバニリン酸ディメチラーゼサブユニットのアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、(d) 配列番号10に記載されているバニリン酸ディメチラーゼサブユニットのアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、(e) 配列番号12に記載されているプロトカテク酸3,4−ジオキシゲナーゼサブユニットのアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、 (f) 配列番号14に記載されているプロトカテク酸3,4−ジオキシゲナーゼサブユニットのアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、そして(g) 配列番号6に記載されているβ-ケトアジピン酸エノールラクトンヒドロラーゼのアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つβ-ケトアジピン酸エノールラクトンヒドロラーゼ活性を有するポリペプチドをコードするDNA分子、であってもよい。
【0027】
ここで、「1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列」とは、各配列番号によって特定されるアミノ酸配列と比較して、所望の酵素活性を失わない範囲の数のアミノ酸が欠失、置換もしくは付加された変異アミノ酸配列を意味する。
【0028】
PcaB, PcaC, PcaD, VanA若しくはVanB又はPcaH若しくはPcaGをコードする遺伝子と高ストリンジェントな条件でハイブリダイズするDNA分子、あるいはPcaB, PcaC, PcaD, VanA若しくはVanB又はPcaH若しくはPcaGの上記変異アミノ酸配列に対応するタンパク質は、元の遺伝子配列によってコードされる酵素と同じ酵素反応を触媒するものでなくてはならない。例えば、「3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素活性を有するタンパク質」とは、3-カルボキシ-Cis, Cis-ムコン酸を閉環して4-カルボキシムコノラクトン(γ-カルボキシムコノラクトン)を形成する活性を保持しているタンパク質を意味する。また、「γ-カルボキシムコノラクトンデカルボキシラーゼ活性を有するタンパク質」とは、γ-カルボキシムコノラクトンのγ位のカルボキシル基を脱離(脱炭酸)してβ-ケトアジピン酸エノールラクトンを形成する活性を保持しているタンパク質を意味し、そして、「β-ケトアジピン酸エノールラクトンヒドロラーゼ活性を有するタンパク質」とは、β-ケトアジピン酸エノールラクトンのラクトン環を加水分解反応により開環してβ-ケトアジピン酸を生成する活性を保持しているタンパク質を意味する。
【0029】
PcaB, PcaC, PcaD, VanA若しくはVanB又はPcaH若しくはPcaGをコードする遺伝子群を挿入するためのベクターは、宿主中で複製可能なものであれば特に制限されず、例えば、プラスミドDNA、ファージDNAなどが挙げられる。
【0030】
プラスミドDNAとしては、pBR322、pBR325、pUC118、pUC119、pET21、pET28、pGEX−4T、pQE−30、pQE−60などの大腸菌宿主用プラスミド、pUB110、pTP5などの枯草菌用プラスミド、YEp13、YEp24、YCp50などの酵母宿主用プラスミド、pBI221、pBI121などの植物細胞宿主用プラスミドなどが挙げられる。ファージDNAとしてはλファージなどが挙げられる。更に、レトロウイルス又はワクシニアウイルスなどの動物ウイルス、バキュロウイルスなどの昆虫ウイルスベクターを用いることもできる。
【0031】
本発明に係る遺伝子群をベクターに挿入するには、まず、本発明に係る各遺伝子を有する精製されたDNAを適当な制限酵素で切断し、適宜末端を平滑化、脱リン酸化等した後、適当なベクターDNAの制限酵素部位又はマルチクローニングサイトに挿入してベクターに連結する方法などが採用される。本発明のベクター内でのpcaB、pcaC、pcaD、VanA、VanB、PcaH、PcaGの配置は、特に限定されないが、プロモーターとターミネーターの間であることが好ましい。後述するとおり、実施例においては、各遺伝子はLacZのαフラグメントとインフレームで接続され、Lacプロモーターごと切り出された後、連結されている(図1A)。
【0032】
本発明に係る遺伝子群は、その遺伝子群の機能が発揮されるようにベクターに組み込むことができる。すなわち、ベクターは、本発明に係る各遺伝子、プロモーター、所望によりエンハンサーなどのシスエレメント、スプライシングシグナル、ポリA付加シグナル、選択マーカー、リボソーム結合配列(シャイン・ダルガノ配列)などを含むように調製することができる。選択マーカーとしては、例えば、アンピシリン耐性遺伝子、ネオマイシン耐性遺伝子、ジヒドロ葉酸還元酵素遺伝子などを使用することができる。
【0033】
プロモーターとしては、大腸菌などの宿主中で発現できるものであればいずれを用いてもよい。例えばlacプロモーター、trpプロモーター、PLプロモーター、PRプロモーターなどの大腸菌由来のものやT7プロモーターなどのファージ由来のものが用いられる。更に、tacプロモーターなどのように人為的に設計改変されたプロモーターを用いてもよい。
【0034】
pcaB, pcaC, pcaD, VanA, VanB, PcaH, PcaGを含む組換えベクターを、これらの遺伝子群が発現し得るように宿主中に導入することにより、形質転換することができる。形質転換の方法としては、プロトプラスト法、コンピテントセル法、エレクトロポレーション法等が挙げられる。
【0035】
宿主としては、本発明の遺伝子群を発現できるものであれば特に限定されるものではない。例えば、エッシェリヒア・コリ(Escherichia coli)などのエッシェリヒア属、バチルス・ズブチリス(Bacillus subtilis)などのバチルス属、シュードモナス・プチダ(Pseudomonas putida)などのシュードモナス属、リゾビウム・メリロティ(Rhizobium meliloti)などのリゾビウム属に属する細菌類の他に、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)、シゾサッカロマイセス・ポンベ(Schizosaccharomyces pombe)、ピヒア・パストリス(Pichia pastoris)などの酵母;シロイヌナズナ、タバコ、トウモロコシ、イネ、ニンジンなどから株化した植物細胞や該植物から調製したプロトプラスト;COS細胞、CHO細胞などの動物細胞;及び、Sf9、Sf21などの昆虫細胞が挙げられる。生育速度およびタンパク質の安定性の観点から、シュードモナス属の細菌が好ましい。
【0036】
また、使用する宿主は、意図する最終産物がムコノラクトンである場合、ムコノラクトンをβ-ケトアジピン酸に分解・代謝しない、もしくは当該分解・代謝能を消失せしめた遺伝子破壊株であることが好ましい。当該遺伝子破壊株は、宿主のβ-ケトアジピン酸エノールラクトンヒドロラーゼ活性を欠損させること、例えば染色体上のpcaD遺伝子を、当業者に知られている方法により破壊することで得られる。
【0037】
更に、意図する最終産物がβ-ケトアジピン酸である場合、使用する宿主は、生産物であるβ-ケトアジピン酸を分解・代謝しない、もしくは当該分解・代謝能を消失せしめた遺伝子破壊株が好ましい。当該遺伝子破壊株は、宿主のβ-ケトアジピン酸スクシニルCoAトランスフェラーゼ活性を欠損させること、例えば染色体上のpcaJ遺伝子(配列番号21)を破壊することで得られる。破壊されるpcaJ遺伝子は、配列番号22のアミノ酸配列をコードするDNA分子、そのDNA分子と相補的な塩基配列からなるDNAと高ストリンジェントな条件でハイブリダイズし、かつそのDNA分子と同一の活性を有するポリペプチドをコードするDNA分子、あるいはβ-ケトアジピン酸スクシニルCoAトランスフェラーゼのアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つβ-ケトアジピン酸スクシニルCoAトランスフェラーゼ活性を有するポリペプチドをコードするDNA分子、であってもよい。
【0038】
形質転換体の選択は、形質転換に使用するベクターの選択マーカー、例えば形質転換体のDNA組換えにより獲得する薬剤耐性を指標にすることができる。薬剤耐性マーカーとしては、例えば、カナマイシン耐性遺伝子、アンピシリン耐性遺伝子、テトラサイクリン耐性遺伝子等が挙げられる。これらの形質転換体の中から目的の組換えベクターを含有する形質転換体の選択は、例えば遺伝子の部分的なDNA断片をプローブとして用いたコロニーハイブリダイゼーション法により行うのが好ましい。プローブの標識としては、例えば放射性同位元素、ジゴキシゲニン、酵素等を用いることができる。
【0039】
本発明の製造方法において、上述のように得られた組換えベクターをトランスフェクションした所望の細胞、すなわち形質転換体、あるいは遺伝子破壊株は、発現する酵素の基質であるバニリン酸及び/又はプロトカテク酸や、糖類、窒素源、金属塩、ミネラル、ビタミン等を含む培地を用いて、適当な条件下で培養すればよい。上記基質は、バニリン酸、プロトカテク酸のいずれか1つ以上添加すればよく、それらの混合物でもよい。
【0040】
培地のpHは、形質転換体又は遺伝子破壊株が生育し得る範囲のpHであればよく、pH 6〜8程度に調整するのが好適である。培養は、好気的条件下で、15〜40℃、好ましくは28〜37℃で基質を投入してから少なくとも6時間、典型的には2〜7日間振盪又は通気攪拌培養すればよい。培養後に得られたムコノラクトンやβ-ケトアジピン酸は、適当な溶媒で抽出した後、任意に精製し、そして/あるいは再結晶化してもよい。
【0041】
最終産物の1つであるβ-ケトアジピン酸は、水溶液中や各種溶媒中で脱炭酸されることでレブリン酸に変換される。当該脱炭酸反応は、β-ケトアジピン酸水溶液に適当な酸を加えることにより促進される。生成したレブリン酸は溶媒抽出等により回収することができる。
【0042】
本発明の製造方法によって得られるムコノラクトン、β-ケトアジピン酸及び/又はレブリン酸は、付加価値の高い有機材料を生成する原料としての役割が期待される。
【0043】
以下、具体例を挙げて、本発明を更に具体的に説明する。なお、本発明はこれにより限定されるものではない。
【実施例】
【0044】
実施例1A β-ケトアジピン酸生産のための組換えプラスミドpVAadi1F6の作製
プラスミドppcaHGXは、pcaH遺伝子(配列番号11)及びpcaG遺伝子(配列番号13)をPCRにより増幅し、pT7Blue T-vector (Novagen)にTAクローニングした後、制限酵素XbaIでセルフライゲーションして作製した。プラスミドppcaBは、pcaB遺伝子(配列番号1)をPCRにより増幅し、pBluescriptII SK(+)ベクターに結合することにより作製した。プラスミドppcaCは、pcaC遺伝子(配列番号3)をPCRにより増幅し、pT7Blue T-vector (Novagen)にTAクローニングして作製した。プラスミドppcaDは、pcaD遺伝子(配列番号5)をPCRにより増幅し、pT7Blue T-vector (Novagen)にTAクローニングした後、制限酵素HindIIIでセルフライゲーションして作製した。その後、ppcaDを制限酵素VspIで切断した後、末端平滑化によって得られるDNA断片と、pBluescriptII SK(+)を制限酵素EcoRIとSalIで切断した後、末端平滑化によって得られるDNA断片とを結合することにより、プラスミドppcaD-ESを作製した。
【0045】
ppcaHGXを制限酵素BamHIとEcoRIで切断した後、末端平滑化によって得られるDNA断片と、ppcaBを制限酵素VspIとKpnIで切断した後、末端平滑化によって得られるDNA断片とを結合することにより、プラスミドppcaHGBを作製した。ppcaHGBを制限酵素XhoIで切断した後、末端平滑化によって得られるDNA断片と、ppcaCを制限酵素VspIとEcoRIで切断した後、末端平滑化によって得られるDNA断片とを結合することにより、プラスミドppcaHGBCを作製した。ppcaHGBCを制限酵素KpnIで切断した後、末端平滑化によって得られるDNA断片と、ppcaD-ESを制限酵素PstIとApaIで切断した後、末端平滑化によって得られるDNA断片とを結合することにより、プラスミドppcaHGBCDを作製した。次いで、ppcaHGBCDを制限酵素VspIとXhoIで切断した後、末端平滑化によって得られるDNA断片と、WO2008/018640号公報記載のpDVZ21を制限酵素XbaIで切断した後、末端平滑化によって得られるDNA断片とを結合することにより、プラスミドpVAadi1F6を作製した(図1A)。
【0046】
実施例1B ムコノラクトン生産のための組換えプラスミドpVAaKL1Fの作製
実施例1AのプラスミドpVAadi1F6を制限酵素KpnIで切断して得られるDNA断片を再結合することにより、約500 bpのDNA断片が欠損したプラスミドpVAaKL1Fを作製した(図1B)。
【0047】
実施例2A プラスミドpVAadi1F6を導入するホスト細胞の作製
(1)ホスト細胞の選択
シュードモナス・プチダ(Pseudomonas putida)KT2440株(ATCC Number:47054)のβ-ケトアジピン酸代謝能を欠損させたKT2440dJ株をホストとして使用する。
【0048】
(2)シュードモナス・プチダKT2440dJ株の作製方法
シュードモナス・プチダ KT2440株はβ-ケトアジピン酸代謝能を保持している。そのため、β-ケトアジピン酸スクシニルCoAトランスフェラーゼ (PcaJ)遺伝子を下記の方法で破壊した。
【0049】
プラスミドppcaIJは、pcaIJ遺伝子をPCRにより増幅し、pT7Blue T-vector (Novagen)にTAクローニングして作製した。ppcaIJをpcaJ内部の制限酵素サイトであるApaIで切断した後、末端平滑化によって得られるDNA断片と、pUC6Cがもつクロラムフェニコール耐性遺伝子を制限酵素PstIで切り出した後、末端平滑化によって得られるDNA断片とを結合することにより、pcaJ遺伝子をクロラムフェニコール耐性遺伝子で破壊した。さらに、このプラスミドを制限酵素PstIで切り出した後、pK19mobsacBのPstIサイトに導入することで、pcaJ遺伝子を破壊するためのプラスミドpIJcmを作製した。プラスミドpIJcmをシュードモナス・プチダ KT2440株に導入し、相同組み換えが起こった株を選抜することにより、KT2440株のpcaJ遺伝子が破壊された株であるKT2440dJ株を得た。
【0050】
実施例2B プラスミドpVAaKL1Fを導入するホスト細胞の作製
(1)ホスト細胞の選択
シュードモナス・プチダKT2440株(ATCC Number:47054)のβ-ケトアジピン酸エノールラクトン代謝能を欠損させたKT2440dD株をホストとして使用する。
【0051】
(2)シュードモナス・プチダKT2440dD株の作製方法
シュードモナス・プチダKT2440株はbeta-ケトアジピン酸エノールラクトン代謝能を保持している。そのため、β-ケトアジピン酸エノールラクトンヒドロラーゼ遺伝子(pcaD)を下記の方法で破壊した。プラスミドppcaDは、pcaD遺伝子をPCRにより増幅し、pT7Blue T-vector (Novagen)にTAクローニングして作製した。ppcaDをpcaD内部の制限酵素サイトであるXmaIIIで切断した後、末端平滑化によって得られるDNA断片と、pUC6Cがもつクロラムフェニコール耐性遺伝子を制限酵素PstIで切り出した後、末端平滑化によって得られるDNA断片とを結合することにより、pcaD遺伝子をクロラムフェニコール耐性遺伝子で破壊した。さらに、このプラスミドを制限酵素PstI-EcoRIで切り出した後、pK19mobsacBのPstI-EcoRIサイトに導入することで、pcaD遺伝子を破壊するためのプラスミドpDcmを作製した。プラスミドpDcmをシュードモナス・プチダKT2440株に導入し、相同組み換えが起こった株を選抜することにより、KT2440株のpcaD遺伝子が破壊された株であるKT2440dD株を得た。
【0052】
実施例3A プラスミドpVAadi1F6を導入した形質転換細胞によるプロトカテク酸からβ-ケトアジピン酸への製造
(1)β-ケトアジピン酸を発酵生産するための多段反応プロセスの酵素遺伝子を含む組み換えプラスミドpVAadi1F6(実施例1A)を大腸菌JM109株に形質転換し、25 mg/Lのカナマイシンを含むLB培地(10ml)で、37℃で18時間振とう培養し、増殖した培養細胞から組み換えプラスミドpVAadi1F6を抽出した。
【0053】
(2)植物成分由来、化学合成もしくは石油由来のバニリン、バニリン酸、プロトカテク酸、p-ヒドロキシベンズアルデヒド、p-ヒドロキシ安息香酸の分解代謝酵素機能を消失せしめたシュードモナス・プチダKT2440dJ株(実施例2A)を、LB液体培地200mlで、28℃ 24時間培養し氷中で30分冷却した。4℃ 10分10000rpmで遠心集菌し、200mlの0℃蒸留水で温和に洗浄後再び遠心集菌した。続いて150mlの0℃蒸留水で温和に洗浄後、遠心集菌した。さらに100mlの0℃蒸留水で温和に洗浄後、遠心集菌した。集菌した微生物細胞を、10%グリセロールを含む蒸留水に懸濁し0℃にて保持した。
【0054】
(3)(1)のプラスミドpVAadi1F6を約0.05μgを含む蒸留水4μlを0.2cmのキュベットに入れ、(2)の10%グリセロールを含む蒸留水に懸濁した細胞液40μlを加え、(25μF、2500V、12msec)の条件でエレクトロポレーションにかけた。
【0055】
(4)上記処理した細胞全量を10mlのLB液体培地に接種し、28℃で6時間培養した。培養後遠心によって菌体を集め50 mg/Lのカナマイシンを含むLB平板に塗布し28℃で48時間培養し、プラスミドpVAadi1F6を保持するカナマイシン耐性を示す形質転換株を得た。本菌をシュードモナス・プチダKT2440dJ-VAadi1株と名づけた。
【0056】
(5)KT2440dJ-VAadi1株を、200mlのLB液体培地(50 mg/Lのカナマイシンを含む)に接種し28℃で16時間培養し前培養菌体懸濁液とした。5 Lの無機塩-yeast extract-グルコース液体培地及び消泡剤(Antiform A) 3 mlを10 L容量のジャーファーメンター(発酵槽)を用いて調製し、そこに培養したKT2440dJ-VAadi1株の前培養菌体懸濁液50mlを混合し、28℃ 700 rpm/minの通気攪拌下、OD660=7〜9まで培養した(14時間〜18時間)。
【0057】
(6)OD660=7〜9に達した発酵槽の培養液に、基質であるプロトカテク酸90 gを含む0.1NのNaOH水溶液(pH8.0に調整)500mlを、ペリスタポンプを用いて10〜14時間かけて添加した。反応の進行に伴うβ-ケトアジピン酸の生成により、培養液のpHが低下する。それを防ぐためpHセンサーに連結したペリスタポンプで5NのNaOH溶液を添加して培養液のpHを維持した。基質添加前後のODの変化を図2に示す。
【0058】
(7)反応終了後、発酵槽の培地をプラスチック容器(バケツ)に移した。培養液から遠心分離(6000rpm、20℃)により菌体成分を沈殿除去し、得られた上清を塩化ナトリウムで飽和させた後に塩酸を加えpH 1.5にした。
【0059】
(8)ジャーファーメンターより定期的にサンプリングした培養液(約15 ml)を適当に希釈して、含まれている基質濃度を1〜5 mMに調整した。その際、内標準物質として安息香酸を最終濃度1 mMとなるように添加した。このサンプル3 mlを分取し、12N 塩酸を数滴加えてpHを1に調整した後、3 mlの酢酸エチルを加えて激しく混和した。4,000 rpmにて遠心し、上清の酢酸エチル層を100 μl分取した。そして、酢酸エチルを乾固した後、50 μlのピリジンと50 μlのTMS化剤 (BSTFA: GL-サイエンス)を加えて混合し、室温にて30分反応させた。このサンプル1 μlをガスクロマトグラフィーにて分析し、予め作成済みの検量線データを用いて、ジャーファーメンターサンプル中の各基質濃度を計算した。その結果、図2に示すように反応液中のβ-ケトアジピン酸は、約99mM(収率100%)検出された。
【0060】
(9)β-ケトアジピン酸は、酢酸エチルで抽出後、エバポレーターを用いて乾固した。その後、アセトンに再溶解して、石油エーテルなどの貧溶媒を添加した条件で結晶化させ(図3)、このサンプルをガスクロマトグラフィー・マススペクトロメトリーで分析した。その後、重DMSO中にてNMR測定を行った。
δH (DMSO-d6), 2.4 (t, 2H), 2.7(t, 2H), 3.5(s, 2H)
δC (DMSO-d6), 28.1, 38.0, 49.5, 169.2, 174.0, 203.2
これらの結果から、得られたサンプルは夾雑物がなく、純度が高いβ−ケトアジピン酸であることが分かった。
【0061】
実施例4 プラスミドpVAadi1F6を導入した形質転換細胞によるバニリン酸からβ-ケトアジピン酸への製造
(1)β-ケトアジピン酸を発酵生産するための多段反応プロセスの酵素遺伝子を含む組み換えプラスミドpVAadi1F6(実施例1)を大腸菌JM109株に形質転換し、25 mg/Lのカナマイシンを含むLB培地(10ml)で、37℃で18時間振とう培養し、増殖した培養細胞から組み換えプラスミドpVAadi1F6を抽出した。
【0062】
(2)植物成分由来、化学合成もしくは石油由来のバニリン、バニリン酸、プロトカテク酸、p-ヒドロキシベンズアルデヒド、p-ヒドロキシ安息香酸の分解代謝酵素機能を消失せしめた微生物であるシュードモナス・プチダKT2440dJ株(実施例2)を、LB液体培地200mlで、28℃ 24時間培養し氷中で30分冷却した。4℃ 10分10000rpmで遠心集菌し、200mlの0℃蒸留水で温和に洗浄後再び遠心集菌した。続いて150mlの0℃蒸留水で温和に洗浄後、遠心集菌した。さらに100mlの0℃蒸留水で温和に洗浄後、遠心集菌した。集菌した微生物細胞を、10%グリセロールを含む蒸留水に懸濁し0℃にて保持した。
【0063】
(3)(1)のプラスミドpVAadi1F6を約0.05μgを含む蒸留水4μlを0.2cmのキュベットに入れ、(2)の10%グリセロールを含む蒸留水に懸濁した細胞液40μlを加え、(25μF、2500V、12msec)の条件でエレクトロポレーションにかけた。
【0064】
(4)上記処理した細胞全量を10mlのLB液体培地に接種し、28℃で6時間培養した。培養後遠心によって菌体を集め50 mg/Lのカナマイシンを含むLB平板に塗布し28℃で48時間培養し、プラスミドpVAadi1F6を保持するカナマイシン耐性を示す形質転換株を得た。本菌をシュードモナス・プチダKT2440dJ-VAadi1株と名づけた。
【0065】
(5)KT2440dJ-VAadi1株を、200mlのLB液体培地(50 mg/Lのカナマイシンを含む)に接種し28℃で16時間培養し前培養菌体懸濁液とした。5 Lの無機塩-yeast extract-グルコース液体培地及び消泡剤(Antiform A) 3 mlを10 L容量のジャーファーメンター(発酵槽)を用いて調製し、そこに培養したKT2440dJ-VAadi1株の前培養菌体懸濁液50mlを混合し、28℃ 700 rpm/minの通気攪拌下、OD660=7〜9まで培養した(14時間〜18時間)。
(6)OD660=7〜9に達した発酵槽の培養液に、基質であるバニリン酸28 gを含む0.1NのNaOH水溶液(pH8.0に調整)500mlを、ペリスタポンプを用いて10時間かけて添加した。反応の進行に伴うβ-ケトアジピン酸の生成により、培養液のpHが低下する。それを防ぐためpHセンサーに連結したペリスタポンプで5NのNaOH溶液を添加して培養液のpHを維持した。基質添加前後のODの変化を図4に示す。
【0066】
(7)反応終了後、発酵槽の培地をプラスチック容器(バケツ)に移した。培養液から遠心分離(6000rpm、20℃)により菌体成分を沈殿除去し、得られた上清を塩化ナトリウムで飽和させた後に塩酸を加えpH 1.5にした。反応液中のβ-ケトアジピン酸を実施例3と同様の方法により定量した結果、図4に示すように約33mM(収率100%)検出された。
【0067】
実施例5 プラスミドpVAaKL1Fを導入した形質転換細胞によるプロトカテク酸からムコノラクトンへの製造
(1)ムコノラクトンを発酵生産するための多段反応プロセスの酵素遺伝子を含む組み換えプラスミドpVAaKL1Fを大腸菌JM109株に形質転換し、25 mg/Lのカナマイシンを含むLB培地(10ml)で、37℃で18時間振とう培養し、増殖した培養細胞から組み換えプラスミドpVAaKL1Fを抽出した。
【0068】
(2)植物成分由来、化学合成もしくは石油由来のバニリン、バニリン酸、プロトカテク酸、p-ヒドロキシベンズアルデヒド、p-ヒドロキシ安息香酸の分解代謝酵素機能を消失せしめた微生物であるシュードモナス属細菌(Pseudomonas putida KT2440dD)を、LB液体培地200mlで、28℃ 24時間培養し氷中で30分冷却した。4℃ 10分10000rpmで遠心集菌し、200mlの0℃蒸留水で温和に洗浄後再び遠心集菌した。続いて150mlの0℃蒸留水で温和に洗浄後、遠心集菌した。さらに100mlの0℃蒸留水で温和に洗浄後、遠心集菌した。集菌した微生物細胞を、10%グリセロールを含む蒸留水に懸濁し0℃にて保持した。
【0069】
(3)(1)のプラスミドpVAaKL1Fを約0.05μgを含む蒸留水4μlを0.2cmのキュベットに入れ、(2)の10%グリセロールを含む蒸留水に懸濁した細胞液40μlを加え、(25μF、2500V、12msec)の条件でエレクトロポレーションにかけた。
【0070】
(4)上記処理した細胞全量を10mlのLB液体培地に接種し、28℃で6時間培養した。培養後遠心によって菌体を集め50 mg/Lのカナマイシンを含むLB平板に塗布し28℃で48時間培養し、プラスミドpVAaKL1Fを保持するカナマイシン耐性を示す形質転換株を得た。本菌をシュードモナス・プチダKT2440dD-VAaKL1株と名づけた。
【0071】
(5)KT2440dD-VAaKL1株を、200mlのLB液体培地(50 mg/Lのカナマイシンを含む)に接種し28℃で16時間培養し前培養菌体懸濁液とした。4.3 Lの無機塩-yeast extract-グルコース液体培地及び消泡剤(Antiform A) 3 mlを10 L容量のジャーファーメンター(発酵槽)を用いて調製し、そこに培養したKT2440dD-VAaKL1株の前培養菌体懸濁液200mlを混合し、28℃ 700 rpm/minの通気攪拌下、OD660=7〜9まで培養した(10時間〜16時間)(図5)。
【0072】
(6)OD660=7〜9に達した発酵槽の培養液に、基質であるプロトカテク酸57 gを含む0.1NのNaOH水溶液(pH8.0に調整)500mlを、ペリスタポンプを用いて10時間かけて添加した。反応の進行に伴うムコノラクトンの生成により、培養液のpHが低下する。それを防ぐためpHセンサーに連結したペリスタポンプで5NのNaOH溶液を添加して培養液のpHを維持した。
【0073】
反応の進行は薄層クロマトグラフィー(TLC)によって確認した。添加したプロトカテク酸は基質添加終了後、約17時間で消失することが確認された。
【0074】
(7)反応終了後、発酵槽の培地をプラスチック容器(バケツ)に移した。培養液から遠心分離(6000rpm、20℃)により菌体成分を沈殿除去し、得られた上清を塩化ナトリウムで飽和させた後に塩酸を加えpH 1.5にした。反応液中のムコノラクトン定量の結果は図5に示すように26時間の時点で約61.7 mM(収率95%)検出された。
【0075】
(8)ムコノラクトンは、酢酸エチルで抽出後、エバポレーターを用いて乾固した。その後、酢酸エチルに再度溶解して、結晶化した(図6)。
その後、重アセトン中にてNMR測定を行った。
δH (acetone-d6), 2.7(m, 1H), 2.8(m, 1H), 5.4(m, 1H), 6.1(m, 1H), 7.8(m, 1H)
δC (acetone-d6)、 37.5, 79.4, 121.2, 165.5, 169.5, 170.1
これらの結果から、得られたサンプルは夾雑物がなく、純度が高いムコノラクトンであることが分かった。
【0076】
実施例6 プラスミドpVAaKL1Fを導入した形質転換細胞によるバニリン酸からムコノラクトンへの製造
(1)ムコノラクトンを発酵生産するための多段反応プロセスの酵素遺伝子を含む組み換えプラスミドpVAaKL1Fを大腸菌JM109株に形質転換し、25 mg/Lのカナマイシンを含むLB培地(10ml)で、37℃で18時間振とう培養し、増殖した培養細胞から組み換えプラスミドpVAaKL1Fを抽出した。
【0077】
(2)植物成分由来、化学合成もしくは石油由来のバニリン、バニリン酸、プロトカテク酸、p-ヒドロキシベンズアルデヒド、p-ヒドロキシ安息香酸の分解代謝酵素機能を消失せしめた微生物であるシュードモナス属細菌(Pseudomonas putida KT2440dD)を、LB液体培地200mlで、28℃ 24時間培養し氷中で30分冷却した。4℃ 10分10000rpmで遠心集菌し、200mlの0℃蒸留水で温和に洗浄後再び遠心集菌した。続いて150mlの0℃蒸留水で温和に洗浄後、遠心集菌した。さらに100mlの0℃蒸留水で温和に洗浄後、遠心集菌した。集菌した微生物細胞を、10%グリセロールを含む蒸留水に懸濁し0℃にて保持した。
【0078】
(3)(1)のプラスミドpVAaKL1Fを約0.05μgを含む蒸留水4μlを0.2cmのキュベットに入れ、(2)の10%グリセロールを含む蒸留水に懸濁した細胞液40μlを加え、(25μF、2500V、12msec)の条件でエレクトロポレーションにかけた。
【0079】
(4)上記処理した細胞全量を10mlのLB液体培地に接種し、28℃で6時間培養した。培養後遠心によって菌体を集め50 mg/Lのカナマイシンを含むLB平板に塗布し28℃で48時間培養し、プラスミドpVAaKL1Fを保持するカナマイシン耐性を示す形質転換株を得た。本菌をシュードモナス・プチダ KT2440dD-VAaKL1株と名づけた。
【0080】
(5)KT2440dD-VAaKL1株を、200mlのLB液体培地(50 mg/Lのカナマイシンを含む)に接種し28℃で16時間培養し前培養菌体懸濁液とした。4.3 Lの無機塩-yeast extract-グルコース液体培地及び消泡剤(Antiform A) 3 mlを10 L容量のジャーファーメンター(発酵槽)を用いて調製し、そこに培養したKT2440dD-VAaKL1株の前培養菌体懸濁液200mlを混合し、28℃ 700 rpm/minの通気攪拌下、OD660=7〜9まで培養した(10時間〜16時間)(図7)。
【0081】
(6)OD660=7〜9に達した発酵槽の培養液に、基質であるバニリン酸28 gを含む0.1NのNaOH水溶液(pH8.0に調整)500mlを、ペリスタポンプを用いて10時間かけて添加した。反応の進行に伴うムコノラクトンの生成により、培養液のpHが低下する。それを防ぐためpHセンサーに連結したペリスタポンプで5NのNaOH溶液を添加して培養液のpHを維持した。
【0082】
反応の進行は薄層クロマトグラフィー(TLC)によって確認した。添加したバニリン酸は基質添加終了後、約26時間で消失することが確認された。
【0083】
(7)反応終了後、発酵槽の培地をプラスチック容器(バケツ)に移した。培養液から遠心分離(6000rpm、20℃)により菌体成分を沈殿除去し、得られた上清を塩化ナトリウムで飽和させた後に塩酸を加えpH 1.5にした。反応液中のムコノラクトン定量の結果は図7に示すように約28.5 mM(収率95%)検出された。
【0084】
(8)ムコノラクトンは、酢酸エチルで抽出後、エバポレーターを用いて乾固した。その後、酢酸エチルに再度溶解して、結晶化した。
【0085】
実施例7 β-ケトアジピン酸の酸処理によるレブリン酸の製造
実施例3、4により得られたβ-ケトアジピン酸粉末を水に溶解し塩酸を少量添加し、酢酸エチルで抽出後、重DMSOに溶解しNMR測定を行った。
δH (DMSO-d6), 2.0(s, 3H), 2.3(t, 2H), 2.6(t, 2H)
δC (DMSO-d6), 28.2, 30.5, 38.0, 174.2, 207.4
NMR測定の結果から上記抽出物はレブリン酸であることがわかった。
【技術分野】
【0001】
本発明は、植物成分あるいは石油成分由来、あるいは化学的に合成されたバニリン酸及び/又はプロトカテク酸からムコノラクトン、β-ケトアジピン酸を発酵生産するための多段反応プロセスを構成する酵素をコードした遺伝子を含む組換えベクター、当該組換えベクターを導入した形質転換体、β−ケトアジピン酸又はムコノラクトンの分解能を消失せしめた遺伝子破壊株、並びに当該形質転換体又は当該遺伝子破壊株を用いてムコノラクトン、β-ケトアジピン酸を製造する方法、そしてβ-ケトアジピン酸からレブリン酸を製造する方法に関する。
【背景技術】
【0002】
植物主要成分であるリグニンは、芳香族高分子化合物として植物細胞壁に普遍的に含まれており、樹木では30%、イネやトウモロコシ稈の15%を占めるバイオマス資源である。その他にも植物は多様な芳香族化合物を構成成分として含んでいる。しかし、リグニンを主成分とする植物由来の芳香族成分は化学構造が多様な成分で構成されていることや、複雑な高分子構造を持つため、有効な利用技術が開発されていない。従来の利用技術として挙げられるのは、リグニンを主成分とする植物由来の芳香族成分をアルカリ分解などの化学分解で生成する低分子芳香族分解物から、香料原料であるバニリンを分離製造する実用化技術がある。しかし、化学分解で生成するバニリン以外の多量の低分子芳香族物質の有効な利用方法はないのが現状である。そのため製紙工程で大量に生成するリグニンは有効利用される事無く重油の代替え品として燃焼されている。
【0003】
今日の石油化学工業の発展を支えたのは、多様な化学成分の混合物である原油を触媒分解と分溜によってベンゼンやエチレンなどの単一な中間物質に一旦変換し、それらを原料に多様な機能性プラスチックスを製造するという原理である。この石油化学工業の発展を支えた基本原理は、化学構造の多様さや複雑な高分子構造を持つリグニンなど植物芳香族成分の利用技術においても適用可能な普遍的原理である。リグニンを主成分とする植物由来の芳香族成分利用の技術を開発する上で、石油化学の触媒分解に相当するプロセスとして、リグニンなど植物芳香族成分の加水分解や酸化分解、可溶媒分解などの化学的分解法や、超臨界水や超臨界有機溶媒による物理化学的分解法など多くの低分子化技術が既に数多く研究され開発されている。しかし、もう一つの技術である、各種分解法により生成する植物成分由来低分子混合物から、様々な機能性プラスチックス原料や化学製品の原料と成りうる有用な単一の中間物質(石油化学においてはエチレンやベンゼン)への変換分離技術は開発されていなかった。この技術が開発されれば、リグニンを主成分とする植物由来の芳香族成分利用が、石油化学に匹敵する技術として発展する可能性がある。
【0004】
本発明者らは既に、スフィンゴビウム・パウシモビリス(Sphingobium paucimobilis)SYK-6株等から獲得した情報を用いて、リグニンの加水分解産物であるバニリン、シリンガアルデヒド、バニリン酸、シリンガ酸、プロトカテク酸から、生分解性ポリマーの原料として利用可能な2−ピロン−4,6−ジカルボン酸(PDC)を発酵生産することに成功している(特開2005-278549号公報)。更に、本発明者らは、WO2008/018640号公報において、バニリン、バニリン酸、及び/又はプロトカテク酸から3-カルボキシ-cis, cis-ムコン酸及び/又は3-カルボキシムコノラクトンへの発酵生産プロセスについても報告している。この発酵生産プロセスは、3種の酵素、ベンズアルデヒドデヒドロゲナーゼ(LigV)、バニリン酸ディメチラーゼ(VanAB)、プロトカテク酸3,4−ジオキシゲナーゼ(PcaHG)が同調的に酵素反応を進行することで構成される。
【0005】
石油化学工業の発展により、今日では原油から様々な有機材料が得られている。リグニンを主成分とする植物由来の芳香族成分から付加価値の高い有機材料を生成するには、2−ピロン−4,6−ジカルボン酸や3-カルボキシムコノラクトンに加え、新たなリグニン分解産物の取得が必要であると考えられる。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特開2005-278549号公報
【特許文献2】WO2008/018640号公報
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明は、リグニンの新規分解産物を発酵生産する方法を提供することを課題とする。
【課題を解決するための手段】
【0008】
本発明者は、リグニン等の植物芳香族成分の低分子化処理混合物に含まれるバニリン酸、プロトカテク酸から新規の分解産物を発酵生産する方法を検討した結果、ニューロスポラ・クラッサ(Neurospora crassa)野生型740R23-1A株(FGSC 987)由来の3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素(3-カルボキシ-cis,cis-ムコン酸シクラーゼ遺伝子)が3-カルボキシ-cis,cis-ムコン酸から3-カルボキシムコノラクトン(beta-カルボキシムコノラクトン)を生成するのに対して、シュードモナス・プチダ(Pseudomonas putida)由来の3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素(PcaB)を用いることにより4-カルボキシムコノラクトン(γ-カルボキシムコノラクトン)が生成し、γ-カルボキシムコノラクトンデカルボキシラーゼ(PcaC)及びβ-ケトアジピン酸エノールラクトンヒドロラーゼ(PcaD)を作用させることにより、β-ケトアジピン酸が生成することを見出し、本発明を完成させた。また、ムコノラクトン生産用の株としてPcaD破壊株を作成し、β-ケトアジピン酸生産用の株としてPcaJ破壊株を作成するとともに、当該酸をレブリン酸に変換する方法も確立した。
【0009】
すなわち、(1)本発明は、好ましくはプロモーターとターミネーターとの間に、pcaB遺伝子及びpcaC遺伝子と、vanA遺伝子及びvanB遺伝子並びにpcaH遺伝子及びpcaG遺伝子と、から成るDNA分子群を含んで成る組換えベクター:
ここで、
(a) pcaB遺伝子は下記(a-1)〜(a-4)のいずれかのDNA分子であり、
(a-1) 配列番号1記載のDNA分子、
(a-2) 配列番号2記載のアミノ酸配列をコードするDNA分子、
(a-3) 配列番号1記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つ3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素活性を有するポリペプチドをコードするDNA分子、あるいは
(a-4) 配列番号2記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つ3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素活性を有するポリペプチドをコードするDNA分子、
(b) pcaC遺伝子は下記(b-1)〜(b-4)のいずれかのDNA分子であり、
(b-1) 配列番号3記載のDNA分子、
(b-2) 配列番号4記載のアミノ酸配列をコードするDNA分子、
(b-3) 配列番号3記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つγ-カルボキシムコノラクトンデカルボキシラーゼ活性を有するポリペプチドをコードするDNA分子、あるいは
(b-4) 配列番号4記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つγ-カルボキシムコノラクトンデカルボキシラーゼ活性を有するポリペプチドをコードするDNA分子、
(c) vanA遺伝子は下記(c-1)〜(c-4)のいずれかのDNA分子であり、
(c-1) 配列番号7記載のDNA分子、
(c-2) 配列番号8記載のアミノ酸配列をコードするDNA分子、
(c-3) 配列番号7記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(c-4) 配列番号8記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(d) vanB遺伝子は下記(d-1)〜(d-4)のいずれかのDNA分子であり、
(d-1) 配列番号9記載のDNA分子、
(d-2) 配列番号10記載のアミノ酸配列をコードするDNA分子、
(d-3) 配列番号9記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(d-4) 配列番号10記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(e) pcaH遺伝子は下記(e-1)〜(e-4)のいずれかのDNA分子である。
(e-1) 配列番号11記載のDNA分子、
(e-2) 配列番号12記載のアミノ酸配列をコードするDNA分子、
(e-3) 配列番号11記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(e-4) 配列番号12記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(f) pcaG遺伝子は下記(f-1)〜(f-4)のいずれかのDNA分子である。
(f-1) 配列番号13記載のDNA分子、
(f-2) 配列番号14記載のアミノ酸配列をコードするDNA分子、
(f-3) 配列番号13記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(f-4) 配列番号14記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
を提供する。
(2)本発明は、前記DNA分子群が更に以下のpcaD遺伝子を含んで成る、(1)の組換えベクター:
(g) pcaD遺伝子は下記(g-1)〜(g-4)のいずれかのDNA分子である。
(g-1) 配列番号5記載のDNA分子、
(g-2) 配列番号6記載のアミノ酸配列をコードするDNA分子、
(g-3) 配列番号5記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つβ-ケトアジピン酸エノールラクトンヒドロラーゼ活性を有するポリペプチドをコードするDNA分子、あるいは
(g-4) 配列番号6記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つβ-ケトアジピン酸エノールラクトンヒドロラーゼ活性を有するポリペプチドをコードするDNA分子、
を提供する。
(3)本発明は、(1)又は(2)の組換えベクターを有する形質転換体、を提供する
(4)本発明は、(1)の組換えベクターを有する形質転換体であって、β-ケトアジピン酸エノールラクトン代謝・分解能が欠損している、形質転換体、を提供する。
(5)本発明は、染色体DNA上にpcaB遺伝子及びpcaC遺伝子と、vanA遺伝子及びvanB遺伝子並びにpcaH遺伝子及びpcaG遺伝子と、から成るDNA分子群が存在する微生物細胞又は当該DNA分子群を含んで成る組換えベクターを有する形質転換体の、β-ケトアジピン酸エノールラクトン代謝・分解能が欠損している遺伝子破壊株であって、当該染色体DNAに存在するpcaD遺伝子が破壊されている遺伝子破壊株:
ここで、
(a) pcaB遺伝子は下記(a-1)〜(a-4)のいずれかのDNA分子であり、
(a-1) 配列番号1記載のDNA分子、
(a-2) 配列番号2記載のアミノ酸配列をコードするDNA分子、
(a-3) 配列番号1記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つ3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素活性を有するポリペプチドをコードするDNA分子、あるいは
(a-4) 配列番号2記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つ3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素活性を有するポリペプチドをコードするDNA分子、
(b) pcaC遺伝子は下記(b-1)〜(b-4)のいずれかのDNA分子であり、
(b-1) 配列番号3記載のDNA分子、
(b-2) 配列番号4記載のアミノ酸配列をコードするDNA分子、
(b-3) 配列番号3記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つγ-カルボキシムコノラクトンデカルボキシラーゼ活性を有するポリペプチドをコードするDNA分子、あるいは
(b-4) 配列番号4記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つγ-カルボキシムコノラクトンデカルボキシラーゼ活性を有するポリペプチドをコードするDNA分子、
(c) vanA遺伝子は下記(c-1)〜(c-4)のいずれかのDNA分子であり、
(c-1) 配列番号7記載のDNA分子、
(c-2) 配列番号8記載のアミノ酸配列をコードするDNA分子、
(c-3) 配列番号7記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(c-4) 配列番号8記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(d) vanB遺伝子は下記(d-1)〜(d-4)のいずれかのDNA分子であり、
(d-1) 配列番号9記載のDNA分子、
(d-2) 配列番号10記載のアミノ酸配列をコードするDNA分子、
(d-3) 配列番号9記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(d-4) 配列番号10記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(e) pcaH遺伝子は下記(e-1)〜(e-4)のいずれかのDNA分子であり、
(e-1) 配列番号11記載のDNA分子、
(e-2) 配列番号12記載のアミノ酸配列をコードするDNA分子、
(e-3) 配列番号11記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(e-4) 配列番号12記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(f) pcaG遺伝子は下記(f-1)〜(f-4)のいずれかのDNA分子であり、
(f-1) 配列番号13記載のDNA分子、
(f-2) 配列番号14記載のアミノ酸配列をコードするDNA分子、
(f-3) 配列番号13記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(f-4) 配列番号14記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、そして
(g) pcaD遺伝子は下記(c-1)〜(c-4)のいずれかのDNA分子である。
(g-1) 配列番号5記載のDNA分子、
(g-2) 配列番号6記載のアミノ酸配列をコードするDNA分子、
(g-3) 配列番号5記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つβ-ケトアジピン酸エノールラクトンヒドロラーゼ活性を有するポリペプチドをコードするDNA分子、あるいは
(g-4) 配列番号6記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つβ-ケトアジピン酸エノールラクトンヒドロラーゼ活性を有するポリペプチドをコードするDNA分子、
を提供する。
(6)本発明は、(4)の形質転換体、又は(5)の遺伝子破壊株を、バニリン酸及び/又はプロトカテク酸の存在下で培養し、当該培養物からムコノラクトンを採取することを特徴とする、ムコノラクトンの製造方法、を提供する。
(7)本発明は、(2)の組換えベクターを含む形質転換体であって、β-ケトアジピン酸代謝・分解能が欠損している、形質転換体、を提供する。
(8)本発明は、染色体DNA上にpcaB遺伝子、pcaC遺伝子及びpcaD遺伝子と、vanA遺伝子及びvanB遺伝子並びにpcaH遺伝子及びpcaG遺伝子と、から成るDNA分子群が存在する微生物細胞又は当該DNA分子群を含んで成る組換えベクターを有する形質転換体の、β-ケトアジピン酸代謝・分解能が欠損している遺伝子破壊株であって、当該染色体DNAに存在するpcaJ遺伝子が破壊されている遺伝子破壊株:
ここで、
(a) pcaB遺伝子は下記(a-1)〜(a-4)のいずれかのDNA分子であり、
(a-1) 配列番号1記載のDNA分子、
(a-2) 配列番号2記載のアミノ酸配列をコードするDNA分子、
(a-3) 配列番号1記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つ3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素活性を有するポリペプチドをコードするDNA分子、あるいは
(a-4) 配列番号2記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つ3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素活性を有するポリペプチドをコードするDNA分子、
(b) pcaC遺伝子は下記(b-1)〜(b-4)のいずれかのDNA分子であり、
(b-1) 配列番号3記載のDNA分子、
(b-2) 配列番号4記載のアミノ酸配列をコードするDNA分子、
(b-3) 配列番号3記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つγ-カルボキシムコノラクトンデカルボキシラーゼ活性を有するポリペプチドをコードするDNA分子、あるいは
(b-4) 配列番号4記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つγ-カルボキシムコノラクトンデカルボキシラーゼ活性を有するポリペプチドをコードするDNA分子、
(c) vanA遺伝子は下記(c-1)〜(c-4)のいずれかのDNA分子であり、
(c-1) 配列番号7記載のDNA分子、
(c-2) 配列番号8記載のアミノ酸配列をコードするDNA分子、
(c-3) 配列番号7記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(c-4) 配列番号8記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(d) vanB遺伝子は下記(d-1)〜(d-4)のいずれかのDNA分子であり、
(d-1) 配列番号9記載のDNA分子、
(d-2) 配列番号10記載のアミノ酸配列をコードするDNA分子、
(d-3) 配列番号9記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(d-4) 配列番号10記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(e) pcaH遺伝子は下記(e-1)〜(e-4)のいずれかのDNA分子であり、
(e-1) 配列番号11記載のDNA分子、
(e-2) 配列番号12記載のアミノ酸配列をコードするDNA分子、
(e-3) 配列番号11記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(e-4) 配列番号12記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(f) pcaG遺伝子は下記(f-1)〜(f-4)のいずれかのDNA分子であり、
(f-1) 配列番号13記載のDNA分子、
(f-2) 配列番号14記載のアミノ酸配列をコードするDNA分子、
(f-3) 配列番号13記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(f-4) 配列番号14記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、そして
(g) pcaD遺伝子は下記(c-1)〜(c-4)のいずれかのDNA分子であり、
(g-1) 配列番号5記載のDNA分子、
(g-2) 配列番号6記載のアミノ酸配列をコードするDNA分子、
(g-3) 配列番号5記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つβ-ケトアジピン酸エノールラクトンヒドロラーゼ活性を有するポリペプチドをコードするDNA分子、あるいは
(g-4) 配列番号6記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つβ-ケトアジピン酸エノールラクトンヒドロラーゼ活性を有するポリペプチドをコードするDNA分子、
(h) pcaJ遺伝子は下記(h-1)〜(h-4)のいずれかのDNA分子である。
(h-1) 配列番号21記載のDNA分子、
(h-2) 配列番号22記載のアミノ酸配列をコードするDNA分子、
(h-3) 配列番号21記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つβ-ケトアジピン酸スクシニルCoAトランスフェラーゼ活性を有するポリペプチドをコードするDNA分子、あるいは
(h-4) 配列番号22記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つβ-ケトアジピン酸スクシニルCoAトランスフェラーゼ活性を有するポリペプチドをコードするDNA分子、
を提供する。
(9)本発明は、(7)の形質転換体、又は(8)の遺伝子破壊株を、バニリン酸及び/又はプロトカテク酸の存在下で培養し、当該培養物からβ-ケトアジピン酸を採取することを特徴とする、β-ケトアジピン酸の製造方法、を提供する。
(10)本発明は、(9)の方法に従い製造されたβ-ケトアジピン酸を脱炭酸反応にかけてレブリン酸を得ることを特徴とする、レブリン酸の製造方法、を提供する。
【発明の効果】
【0010】
本発明によれば、バニリン酸及び/又はプロトカテク酸から、ムコノラクトン、β-ケトアジピン酸までの変換に必要な酵素(VanAB, PcaHG, PcaB, PcaC, PcaD)による以下の多段階代謝プロセス:
【化1】
が同調的に進行することで、ムコノラクトン、β-ケトアジピン酸の量産が可能となる。β−ケトアジピン酸は、反応性が高いケトンをβ位に、そしてカルボン酸を両末端に有している。β−ケトアジピン酸と同様に、末端にカルボン酸を2つ有するアジピン酸が66−ナイロンなどの高分子製造原料として利用されていることを考慮すると、β−ケトアジピン酸も高分子材料や化学製品への応用が期待される。また、γ-クロトノラクトン骨格を持つムコノラクトンは、香料、樹脂用溶剤、染み抜き剤、接着剤、塗料剥離剤等への応用が期待される。
【0011】
更に、得られたβ-ケトアジピン酸は、酸処理することでレブリン酸に変換することもできる。レブリン酸は、種々の有用な化成品の原料となる基礎的な化学物質であり、β-アセチルプロピオン酸とも称されている。レブリン酸自体も抗菌剤、燃料組成物等、様々な分野での用途が知られている(特開2003−226641号公報、特表2007−510787号公報等)。
【図面の簡単な説明】
【0012】
【図1A】図1Aは、組換えベクターpVAadi1F6の作製方法を示す図である。
【図1B】図1Bは、組換えベクターpVAaKL1Fの作製方法を示す図である。
【図2】図2は、実施例3で調製した培養液中のOD、基質、変換物の変化を示す図である。
【図3】図3は、実施例3で得られたβ−ケトアジピン酸の結晶の光学顕微鏡写真である。
【図4】図4は、実施例4で調製した培養液中のOD、基質、変換物の変化を示す図である
【図5】図5は、実施例5で調製した培養液中のOD、基質、変換物の変化を示す図である。
【図6】図6は、実施例5で得られたムコノラクトンの結晶の光学顕微鏡写真である。
【図7】図7は、実施例6で調製した培養液中のOD、基質、変換物の変化を示す図である。
【発明を実施するための形態】
【0013】
本発明の組換えベクターは、バニリン酸、プロトカテク酸、これらの混合物等のリグニン分解物からムコノラクトンへの製造プロセス、そしてムコノラクトンからβ−ケトアジピン酸への製造プロセスを触媒するための酵素遺伝子を含む組換えベクターである。本発明の組換えベクターは、具体的な態様において、バニリン酸、プロトカテク酸、これらの混合物等のリグニン分解物からムコノラクトンを製造するプロセスを触媒する、配列番号1記載のDNA分子又は配列番号2記載のアミノ酸配列をコードするDNA分子から成る3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素遺伝子(pcaB)及び配列番号3記載のDNA分子又は配列番号4記載のアミノ酸配列をコードするDNA分子から成るγ-カルボキシムコノラクトンデカルボキシラーゼ遺伝子(pcaC)と、配列番号7記載のDNA分子又は配列番号8記載のアミノ酸配列をコードするDNA分子から成るバニリン酸ディメチラーゼサブユニット遺伝子(vanA)、配列番号9記載のDNA分子又は配列番号10記載のアミノ酸配列をコードするDNA分子から成るバニリン酸ディメチラーゼサブユニット遺伝子(vanB)、配列番号11記載のDNA分子又は配列番号12記載のアミノ酸配列をコードするDNA分子から成るプロトカテク酸3,4−ジオキシゲナーゼサブユニット遺伝子(pcaH)、配列番号13記載のDNA分子又は配列番号14記載のアミノ酸配列をコードするDNA分子から成るプロトカテク酸3,4−ジオキシゲナーゼサブユニット遺伝子(pcaH)と、から成る遺伝子群を含む組換えベクターである。
【0014】
本発明の組換えベクターは、別の具体的な態様において、バニリン酸、プロトカテク酸、これらの混合物等のリグニン分解物からβ−ケトアジピン酸を製造するプロセスを触媒する、配列番号1記載のDNA分子又は配列番号2記載のアミノ酸配列をコードするDNA分子から成る3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素遺伝子(pcaB)、配列番号3記載のDNA分子又は配列番号4記載のアミノ酸配列をコードするDNA分子から成るγ-カルボキシムコノラクトンデカルボキシラーゼ遺伝子(pcaC)、及び配列番号5記載のDNA分子又は配列番号6記載のアミノ酸配列をコードするDNA分子から成るβ-ケトアジピン酸エノールラクトンヒドロラーゼ遺伝子(pcaD)と、配列番号7記載のDNA分子又は配列番号8記載のアミノ酸配列をコードするDNA分子から成るバニリン酸ディメチラーゼサブユニット遺伝子(vanA)、配列番号9記載のDNA分子又は配列番号10記載のアミノ酸配列をコードするDNA分子から成るバニリン酸ディメチラーゼサブユニット遺伝子(vanB)、配列番号11記載のDNA分子又は配列番号12記載のアミノ酸配列をコードするDNA分子から成るプロトカテク酸3,4−ジオキシゲナーゼサブユニット遺伝子(pcaH)、配列番号13記載のDNA分子又は配列番号14記載のアミノ酸配列をコードするDNA分子から成るプロトカテク酸3,4−ジオキシゲナーゼサブユニット遺伝子(pcaH)と、から成る遺伝子群を含む組換えベクターである。
【0015】
本発明の組換えベクターにおいては、上記の特定の遺伝子群がすべて一つの発現ベクター内に含まれるように構成されていてもよく、あるいは当該遺伝子群が2グループ以上に分かれて、それぞれ別個の発現ベクターに含まれるように構成されていてもよい。発現ベクターにおいて、かかる遺伝子群は、プロモーターとターミネーターとの間に挿入することができる。しかしながら、本発明で使用する遺伝子群の配置はこの位置に限定されない。
【0016】
本発明の組換えベクターに含まれる3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素遺伝子(pcaB)、γ-カルボキシムコノラクトンデカルボキシラーゼ遺伝子(pcaC)及びβ-ケトアジピン酸エノールラクトンヒドロラーゼ遺伝子(pcaD)は、シュードモナス・プチダ(Pseudomonas putida)KT2440株から、以下のプライマー及びアニーリング条件を用い、PCR法により獲得した:
pcaB:フォワードプライマー (GATGATGGCGATTGCCTTCC)(配列番号15)、リバースプライマー (GTCTCCTTCAGGCAGTGAAACG)(配列番号16)、アニーリング温度50℃;
pcaC:フォワードプライマー (CTCCTTCTAGACATGGACGAGAAAGAACGTTACGACGC)(配列番号17)、リバースプライマー (AGTGGATCCTTACTGGCGAGAACTCTACGCCAAG)(配列番号18)、アニーリング温度55℃;
pcaD:フォワードプライマー (ATAAGCTTCAACGAGACCGCTGTGGC)(配列番号19)、リバースプライマー (AATCTAGAGCGTTGTCCTCAGTGAG)(配列番号20)、アニーリング温度55℃。ここで、pcaB、pcaC、pcaDのオープンリーディングフレームの塩基配列をそれぞれ配列番号1、3、5に、そして当該配列によってコードされるタンパク質のアミノ酸配列をそれぞれ配列番号2,4、6に記載する。
【0017】
本発明で使用するバニリン酸ディメチラーゼサブユニット遺伝子(vanA及びvanB)は、特開2005-278549号公報(上掲)に記載の、シュードモナス・プチダ(Pseudomonas putida)PpY101株由来のバニリン酸ディメチラーゼ遺伝子(同公報の配列番号1)に相当するが、当該配列は本発明のものと一部異なる(配列番号7及び8)。ここで、バニリン酸ディメチラーゼサブユニット(VanA及びVanB)は、会合してバニリン酸ディメチラーゼ(VanAB)を構成する。
【0018】
バニリン酸ディメチラーゼ(VanAB)は、微生物細胞内で本来半減期の短い不安定ポリペプチドとして生成されている。バニリン酸ディメチラーゼは、特開2005-278549に記載されているように、LacZのαフラグメントに含まれる16アミノ酸からなるオリゴペプチド、或いはヒスチジン6個を含む30アミノ酸からなるオリゴペプチドを一例とする、VanAの安定化に寄与し、かつVanAの酵素機能を妨害しない様々なポリペプチドをVanAのN 末端に付加することにより安定化される。
【0019】
プロトカテク酸3,4−ジオキシゲナーゼサブユニット遺伝子(pcaH及びpcaG)は、WO2008/018640号公報(上掲)に記載のとおり、シュードモナス・プチダKT2440株由来であり、J Bacteriol. 1989 Nov; 171(11): 5915-21にその遺伝子情報が開示されている。プロトカテク酸3,4−ジオキシゲナーゼサブユニット(PcaH及びPcaG)は、会合してプロトカテク酸3,4−ジオキシゲナーゼ(PcaHG)を構成する。
【0020】
本発明のベクターに含まれる遺伝子の取得方法は特に限定されず、例えば、開示されている当該遺伝子の塩基配列の方法に基づいて適当なプローブやライブラリーを調製し、それらを用いて、菌株のcDNAライブラリーやゲノムDNAライブラリーをスクリーニングすることで得ることができる。具体的には、本発明の遺伝子を発現する微生物、例えばシュードモナス・プチダ(Pseudomonas putida)KT2440株等より、常法に従ってゲノムDNAライブラリーを調製し、当該ライブラリーから、本発明遺伝子に特有の適当なプローブ等を用いて所望クローンを選択することにより製造することができる。微生物株からの全RNAの分離、mRNAの分離及び精製、ゲノムDNAの取得及びそのクローニングなどは、いずれも常法に従って行うことができる。
【0021】
上記方法において用いられるプローブとしては、本発明の遺伝子の塩基配列に関する情報をもとにして化学合成されたDNAなどが一般的に使用できる。また、本発明の遺伝子の塩基配列情報に基づき設定したセンス・プライマー及びアンチセンス・プライマーを、スクリーニング用プローブとして用いることができる。
【0022】
本発明の遺伝子の取得に際しては、PCR(Science,230,1350(1985))によるDNA増幅法が好適に利用できる。増幅させたDNA断片の単離精製は、常法に従って行うことができる。例えばゲル電気泳動法などが挙げられる。上記方法に従って得られる本発明の遺伝子は、常法、例えばジデオキシ法(Proc.Natl.Acad.Sci.,USA.,74,5463(1977))、マキサム−ギルバート法(Methods in Enzymology,65,499(1980))などに従って、その塩基配列を決定することができる。また、簡便には、市販のシークエンスキットなどを用いて、その塩基配列を決定することができる。
【0023】
pcaB遺伝子(配列番号1)、pcaC遺伝子(配列番号3)pcaD遺伝子(配列番号5)、vanA遺伝子(配列番号7)及びvanB遺伝子(配列番号9)並びにpcaH遺伝子(配列番号11)及びpcaG遺伝子(配列番号13)は、対応する配列番号によって特定されるDNA分子や、そのアミノ酸配列をコードするDNA分子の他に、そのDNA分子と相補的な塩基配列からなるDNAと高ストリンジェントな条件でハイブリダイズし、かつそのDNA分子と同一の活性を有するポリペプチドをコードするDNA分子も含まれる。
【0024】
ここで、本明細書で使用する場合、「高ストリンジェントな条件」とは、いわゆる特異的なハイブリッドが形成され、非特異的なハイブリッドが形成されない条件をいう。高ストリンジェントな条件としては、同一性が高いDNA同士がハイブリダイズし、それより同一性が低いDNA同士がハイブリダイズしない条件、例えば、Molecular cloning a Laboratory manual 2nd edition(Sambrookら、1989)に記載の条件が挙げられる。具体的には、通常のサザンハイブリダイゼーションにおける洗浄の条件である60℃、1×SSC、0.1%SDS、好ましくは、0.1×SSC、0.1%SDSで相当する塩濃度でハイブリダイズする条件が挙げられる。
【0025】
本発明のベクターに含まれる遺伝子の取得に際しては、PCR法(Science,230,1350(1985))によるDNA増幅法が好適に利用できる。増幅させたDNA断片の単離精製は、常法に従って行うことができる。例えばゲル電気泳動法などが挙げられる。上記方法に従って得られる本発明の遺伝子は、常法、例えばジデオキシ法(Proc.Natl.Acad.Sci.,USA.,74,5463(1977))、マキサム−ギルバート法(Methods in Enzymology,65,499(1980))などに従って、その塩基配列を決定することができる。また、簡便には、市販のシークエンスキットなどを用いて、その塩基配列を決定することができる。
【0026】
高ストリンジェントでハイブリダイズするDNA分子に加え、本発明のベクターに含まれる各酵素のコード遺伝子は、(a) 配列番号2に記載されている3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つ3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素活性を有するポリペプチドをコードするDNA分子、(b) 配列番号4に記載されているγ-カルボキシムコノラクトンデカルボキシラーゼのアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つγ-カルボキシムコノラクトンデカルボキシラーゼ活性を有するポリペプチドをコードするDNA分子、 (c) 配列番号8に記載されているバニリン酸ディメチラーゼサブユニットのアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、(d) 配列番号10に記載されているバニリン酸ディメチラーゼサブユニットのアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、(e) 配列番号12に記載されているプロトカテク酸3,4−ジオキシゲナーゼサブユニットのアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、 (f) 配列番号14に記載されているプロトカテク酸3,4−ジオキシゲナーゼサブユニットのアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、そして(g) 配列番号6に記載されているβ-ケトアジピン酸エノールラクトンヒドロラーゼのアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つβ-ケトアジピン酸エノールラクトンヒドロラーゼ活性を有するポリペプチドをコードするDNA分子、であってもよい。
【0027】
ここで、「1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列」とは、各配列番号によって特定されるアミノ酸配列と比較して、所望の酵素活性を失わない範囲の数のアミノ酸が欠失、置換もしくは付加された変異アミノ酸配列を意味する。
【0028】
PcaB, PcaC, PcaD, VanA若しくはVanB又はPcaH若しくはPcaGをコードする遺伝子と高ストリンジェントな条件でハイブリダイズするDNA分子、あるいはPcaB, PcaC, PcaD, VanA若しくはVanB又はPcaH若しくはPcaGの上記変異アミノ酸配列に対応するタンパク質は、元の遺伝子配列によってコードされる酵素と同じ酵素反応を触媒するものでなくてはならない。例えば、「3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素活性を有するタンパク質」とは、3-カルボキシ-Cis, Cis-ムコン酸を閉環して4-カルボキシムコノラクトン(γ-カルボキシムコノラクトン)を形成する活性を保持しているタンパク質を意味する。また、「γ-カルボキシムコノラクトンデカルボキシラーゼ活性を有するタンパク質」とは、γ-カルボキシムコノラクトンのγ位のカルボキシル基を脱離(脱炭酸)してβ-ケトアジピン酸エノールラクトンを形成する活性を保持しているタンパク質を意味し、そして、「β-ケトアジピン酸エノールラクトンヒドロラーゼ活性を有するタンパク質」とは、β-ケトアジピン酸エノールラクトンのラクトン環を加水分解反応により開環してβ-ケトアジピン酸を生成する活性を保持しているタンパク質を意味する。
【0029】
PcaB, PcaC, PcaD, VanA若しくはVanB又はPcaH若しくはPcaGをコードする遺伝子群を挿入するためのベクターは、宿主中で複製可能なものであれば特に制限されず、例えば、プラスミドDNA、ファージDNAなどが挙げられる。
【0030】
プラスミドDNAとしては、pBR322、pBR325、pUC118、pUC119、pET21、pET28、pGEX−4T、pQE−30、pQE−60などの大腸菌宿主用プラスミド、pUB110、pTP5などの枯草菌用プラスミド、YEp13、YEp24、YCp50などの酵母宿主用プラスミド、pBI221、pBI121などの植物細胞宿主用プラスミドなどが挙げられる。ファージDNAとしてはλファージなどが挙げられる。更に、レトロウイルス又はワクシニアウイルスなどの動物ウイルス、バキュロウイルスなどの昆虫ウイルスベクターを用いることもできる。
【0031】
本発明に係る遺伝子群をベクターに挿入するには、まず、本発明に係る各遺伝子を有する精製されたDNAを適当な制限酵素で切断し、適宜末端を平滑化、脱リン酸化等した後、適当なベクターDNAの制限酵素部位又はマルチクローニングサイトに挿入してベクターに連結する方法などが採用される。本発明のベクター内でのpcaB、pcaC、pcaD、VanA、VanB、PcaH、PcaGの配置は、特に限定されないが、プロモーターとターミネーターの間であることが好ましい。後述するとおり、実施例においては、各遺伝子はLacZのαフラグメントとインフレームで接続され、Lacプロモーターごと切り出された後、連結されている(図1A)。
【0032】
本発明に係る遺伝子群は、その遺伝子群の機能が発揮されるようにベクターに組み込むことができる。すなわち、ベクターは、本発明に係る各遺伝子、プロモーター、所望によりエンハンサーなどのシスエレメント、スプライシングシグナル、ポリA付加シグナル、選択マーカー、リボソーム結合配列(シャイン・ダルガノ配列)などを含むように調製することができる。選択マーカーとしては、例えば、アンピシリン耐性遺伝子、ネオマイシン耐性遺伝子、ジヒドロ葉酸還元酵素遺伝子などを使用することができる。
【0033】
プロモーターとしては、大腸菌などの宿主中で発現できるものであればいずれを用いてもよい。例えばlacプロモーター、trpプロモーター、PLプロモーター、PRプロモーターなどの大腸菌由来のものやT7プロモーターなどのファージ由来のものが用いられる。更に、tacプロモーターなどのように人為的に設計改変されたプロモーターを用いてもよい。
【0034】
pcaB, pcaC, pcaD, VanA, VanB, PcaH, PcaGを含む組換えベクターを、これらの遺伝子群が発現し得るように宿主中に導入することにより、形質転換することができる。形質転換の方法としては、プロトプラスト法、コンピテントセル法、エレクトロポレーション法等が挙げられる。
【0035】
宿主としては、本発明の遺伝子群を発現できるものであれば特に限定されるものではない。例えば、エッシェリヒア・コリ(Escherichia coli)などのエッシェリヒア属、バチルス・ズブチリス(Bacillus subtilis)などのバチルス属、シュードモナス・プチダ(Pseudomonas putida)などのシュードモナス属、リゾビウム・メリロティ(Rhizobium meliloti)などのリゾビウム属に属する細菌類の他に、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)、シゾサッカロマイセス・ポンベ(Schizosaccharomyces pombe)、ピヒア・パストリス(Pichia pastoris)などの酵母;シロイヌナズナ、タバコ、トウモロコシ、イネ、ニンジンなどから株化した植物細胞や該植物から調製したプロトプラスト;COS細胞、CHO細胞などの動物細胞;及び、Sf9、Sf21などの昆虫細胞が挙げられる。生育速度およびタンパク質の安定性の観点から、シュードモナス属の細菌が好ましい。
【0036】
また、使用する宿主は、意図する最終産物がムコノラクトンである場合、ムコノラクトンをβ-ケトアジピン酸に分解・代謝しない、もしくは当該分解・代謝能を消失せしめた遺伝子破壊株であることが好ましい。当該遺伝子破壊株は、宿主のβ-ケトアジピン酸エノールラクトンヒドロラーゼ活性を欠損させること、例えば染色体上のpcaD遺伝子を、当業者に知られている方法により破壊することで得られる。
【0037】
更に、意図する最終産物がβ-ケトアジピン酸である場合、使用する宿主は、生産物であるβ-ケトアジピン酸を分解・代謝しない、もしくは当該分解・代謝能を消失せしめた遺伝子破壊株が好ましい。当該遺伝子破壊株は、宿主のβ-ケトアジピン酸スクシニルCoAトランスフェラーゼ活性を欠損させること、例えば染色体上のpcaJ遺伝子(配列番号21)を破壊することで得られる。破壊されるpcaJ遺伝子は、配列番号22のアミノ酸配列をコードするDNA分子、そのDNA分子と相補的な塩基配列からなるDNAと高ストリンジェントな条件でハイブリダイズし、かつそのDNA分子と同一の活性を有するポリペプチドをコードするDNA分子、あるいはβ-ケトアジピン酸スクシニルCoAトランスフェラーゼのアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つβ-ケトアジピン酸スクシニルCoAトランスフェラーゼ活性を有するポリペプチドをコードするDNA分子、であってもよい。
【0038】
形質転換体の選択は、形質転換に使用するベクターの選択マーカー、例えば形質転換体のDNA組換えにより獲得する薬剤耐性を指標にすることができる。薬剤耐性マーカーとしては、例えば、カナマイシン耐性遺伝子、アンピシリン耐性遺伝子、テトラサイクリン耐性遺伝子等が挙げられる。これらの形質転換体の中から目的の組換えベクターを含有する形質転換体の選択は、例えば遺伝子の部分的なDNA断片をプローブとして用いたコロニーハイブリダイゼーション法により行うのが好ましい。プローブの標識としては、例えば放射性同位元素、ジゴキシゲニン、酵素等を用いることができる。
【0039】
本発明の製造方法において、上述のように得られた組換えベクターをトランスフェクションした所望の細胞、すなわち形質転換体、あるいは遺伝子破壊株は、発現する酵素の基質であるバニリン酸及び/又はプロトカテク酸や、糖類、窒素源、金属塩、ミネラル、ビタミン等を含む培地を用いて、適当な条件下で培養すればよい。上記基質は、バニリン酸、プロトカテク酸のいずれか1つ以上添加すればよく、それらの混合物でもよい。
【0040】
培地のpHは、形質転換体又は遺伝子破壊株が生育し得る範囲のpHであればよく、pH 6〜8程度に調整するのが好適である。培養は、好気的条件下で、15〜40℃、好ましくは28〜37℃で基質を投入してから少なくとも6時間、典型的には2〜7日間振盪又は通気攪拌培養すればよい。培養後に得られたムコノラクトンやβ-ケトアジピン酸は、適当な溶媒で抽出した後、任意に精製し、そして/あるいは再結晶化してもよい。
【0041】
最終産物の1つであるβ-ケトアジピン酸は、水溶液中や各種溶媒中で脱炭酸されることでレブリン酸に変換される。当該脱炭酸反応は、β-ケトアジピン酸水溶液に適当な酸を加えることにより促進される。生成したレブリン酸は溶媒抽出等により回収することができる。
【0042】
本発明の製造方法によって得られるムコノラクトン、β-ケトアジピン酸及び/又はレブリン酸は、付加価値の高い有機材料を生成する原料としての役割が期待される。
【0043】
以下、具体例を挙げて、本発明を更に具体的に説明する。なお、本発明はこれにより限定されるものではない。
【実施例】
【0044】
実施例1A β-ケトアジピン酸生産のための組換えプラスミドpVAadi1F6の作製
プラスミドppcaHGXは、pcaH遺伝子(配列番号11)及びpcaG遺伝子(配列番号13)をPCRにより増幅し、pT7Blue T-vector (Novagen)にTAクローニングした後、制限酵素XbaIでセルフライゲーションして作製した。プラスミドppcaBは、pcaB遺伝子(配列番号1)をPCRにより増幅し、pBluescriptII SK(+)ベクターに結合することにより作製した。プラスミドppcaCは、pcaC遺伝子(配列番号3)をPCRにより増幅し、pT7Blue T-vector (Novagen)にTAクローニングして作製した。プラスミドppcaDは、pcaD遺伝子(配列番号5)をPCRにより増幅し、pT7Blue T-vector (Novagen)にTAクローニングした後、制限酵素HindIIIでセルフライゲーションして作製した。その後、ppcaDを制限酵素VspIで切断した後、末端平滑化によって得られるDNA断片と、pBluescriptII SK(+)を制限酵素EcoRIとSalIで切断した後、末端平滑化によって得られるDNA断片とを結合することにより、プラスミドppcaD-ESを作製した。
【0045】
ppcaHGXを制限酵素BamHIとEcoRIで切断した後、末端平滑化によって得られるDNA断片と、ppcaBを制限酵素VspIとKpnIで切断した後、末端平滑化によって得られるDNA断片とを結合することにより、プラスミドppcaHGBを作製した。ppcaHGBを制限酵素XhoIで切断した後、末端平滑化によって得られるDNA断片と、ppcaCを制限酵素VspIとEcoRIで切断した後、末端平滑化によって得られるDNA断片とを結合することにより、プラスミドppcaHGBCを作製した。ppcaHGBCを制限酵素KpnIで切断した後、末端平滑化によって得られるDNA断片と、ppcaD-ESを制限酵素PstIとApaIで切断した後、末端平滑化によって得られるDNA断片とを結合することにより、プラスミドppcaHGBCDを作製した。次いで、ppcaHGBCDを制限酵素VspIとXhoIで切断した後、末端平滑化によって得られるDNA断片と、WO2008/018640号公報記載のpDVZ21を制限酵素XbaIで切断した後、末端平滑化によって得られるDNA断片とを結合することにより、プラスミドpVAadi1F6を作製した(図1A)。
【0046】
実施例1B ムコノラクトン生産のための組換えプラスミドpVAaKL1Fの作製
実施例1AのプラスミドpVAadi1F6を制限酵素KpnIで切断して得られるDNA断片を再結合することにより、約500 bpのDNA断片が欠損したプラスミドpVAaKL1Fを作製した(図1B)。
【0047】
実施例2A プラスミドpVAadi1F6を導入するホスト細胞の作製
(1)ホスト細胞の選択
シュードモナス・プチダ(Pseudomonas putida)KT2440株(ATCC Number:47054)のβ-ケトアジピン酸代謝能を欠損させたKT2440dJ株をホストとして使用する。
【0048】
(2)シュードモナス・プチダKT2440dJ株の作製方法
シュードモナス・プチダ KT2440株はβ-ケトアジピン酸代謝能を保持している。そのため、β-ケトアジピン酸スクシニルCoAトランスフェラーゼ (PcaJ)遺伝子を下記の方法で破壊した。
【0049】
プラスミドppcaIJは、pcaIJ遺伝子をPCRにより増幅し、pT7Blue T-vector (Novagen)にTAクローニングして作製した。ppcaIJをpcaJ内部の制限酵素サイトであるApaIで切断した後、末端平滑化によって得られるDNA断片と、pUC6Cがもつクロラムフェニコール耐性遺伝子を制限酵素PstIで切り出した後、末端平滑化によって得られるDNA断片とを結合することにより、pcaJ遺伝子をクロラムフェニコール耐性遺伝子で破壊した。さらに、このプラスミドを制限酵素PstIで切り出した後、pK19mobsacBのPstIサイトに導入することで、pcaJ遺伝子を破壊するためのプラスミドpIJcmを作製した。プラスミドpIJcmをシュードモナス・プチダ KT2440株に導入し、相同組み換えが起こった株を選抜することにより、KT2440株のpcaJ遺伝子が破壊された株であるKT2440dJ株を得た。
【0050】
実施例2B プラスミドpVAaKL1Fを導入するホスト細胞の作製
(1)ホスト細胞の選択
シュードモナス・プチダKT2440株(ATCC Number:47054)のβ-ケトアジピン酸エノールラクトン代謝能を欠損させたKT2440dD株をホストとして使用する。
【0051】
(2)シュードモナス・プチダKT2440dD株の作製方法
シュードモナス・プチダKT2440株はbeta-ケトアジピン酸エノールラクトン代謝能を保持している。そのため、β-ケトアジピン酸エノールラクトンヒドロラーゼ遺伝子(pcaD)を下記の方法で破壊した。プラスミドppcaDは、pcaD遺伝子をPCRにより増幅し、pT7Blue T-vector (Novagen)にTAクローニングして作製した。ppcaDをpcaD内部の制限酵素サイトであるXmaIIIで切断した後、末端平滑化によって得られるDNA断片と、pUC6Cがもつクロラムフェニコール耐性遺伝子を制限酵素PstIで切り出した後、末端平滑化によって得られるDNA断片とを結合することにより、pcaD遺伝子をクロラムフェニコール耐性遺伝子で破壊した。さらに、このプラスミドを制限酵素PstI-EcoRIで切り出した後、pK19mobsacBのPstI-EcoRIサイトに導入することで、pcaD遺伝子を破壊するためのプラスミドpDcmを作製した。プラスミドpDcmをシュードモナス・プチダKT2440株に導入し、相同組み換えが起こった株を選抜することにより、KT2440株のpcaD遺伝子が破壊された株であるKT2440dD株を得た。
【0052】
実施例3A プラスミドpVAadi1F6を導入した形質転換細胞によるプロトカテク酸からβ-ケトアジピン酸への製造
(1)β-ケトアジピン酸を発酵生産するための多段反応プロセスの酵素遺伝子を含む組み換えプラスミドpVAadi1F6(実施例1A)を大腸菌JM109株に形質転換し、25 mg/Lのカナマイシンを含むLB培地(10ml)で、37℃で18時間振とう培養し、増殖した培養細胞から組み換えプラスミドpVAadi1F6を抽出した。
【0053】
(2)植物成分由来、化学合成もしくは石油由来のバニリン、バニリン酸、プロトカテク酸、p-ヒドロキシベンズアルデヒド、p-ヒドロキシ安息香酸の分解代謝酵素機能を消失せしめたシュードモナス・プチダKT2440dJ株(実施例2A)を、LB液体培地200mlで、28℃ 24時間培養し氷中で30分冷却した。4℃ 10分10000rpmで遠心集菌し、200mlの0℃蒸留水で温和に洗浄後再び遠心集菌した。続いて150mlの0℃蒸留水で温和に洗浄後、遠心集菌した。さらに100mlの0℃蒸留水で温和に洗浄後、遠心集菌した。集菌した微生物細胞を、10%グリセロールを含む蒸留水に懸濁し0℃にて保持した。
【0054】
(3)(1)のプラスミドpVAadi1F6を約0.05μgを含む蒸留水4μlを0.2cmのキュベットに入れ、(2)の10%グリセロールを含む蒸留水に懸濁した細胞液40μlを加え、(25μF、2500V、12msec)の条件でエレクトロポレーションにかけた。
【0055】
(4)上記処理した細胞全量を10mlのLB液体培地に接種し、28℃で6時間培養した。培養後遠心によって菌体を集め50 mg/Lのカナマイシンを含むLB平板に塗布し28℃で48時間培養し、プラスミドpVAadi1F6を保持するカナマイシン耐性を示す形質転換株を得た。本菌をシュードモナス・プチダKT2440dJ-VAadi1株と名づけた。
【0056】
(5)KT2440dJ-VAadi1株を、200mlのLB液体培地(50 mg/Lのカナマイシンを含む)に接種し28℃で16時間培養し前培養菌体懸濁液とした。5 Lの無機塩-yeast extract-グルコース液体培地及び消泡剤(Antiform A) 3 mlを10 L容量のジャーファーメンター(発酵槽)を用いて調製し、そこに培養したKT2440dJ-VAadi1株の前培養菌体懸濁液50mlを混合し、28℃ 700 rpm/minの通気攪拌下、OD660=7〜9まで培養した(14時間〜18時間)。
【0057】
(6)OD660=7〜9に達した発酵槽の培養液に、基質であるプロトカテク酸90 gを含む0.1NのNaOH水溶液(pH8.0に調整)500mlを、ペリスタポンプを用いて10〜14時間かけて添加した。反応の進行に伴うβ-ケトアジピン酸の生成により、培養液のpHが低下する。それを防ぐためpHセンサーに連結したペリスタポンプで5NのNaOH溶液を添加して培養液のpHを維持した。基質添加前後のODの変化を図2に示す。
【0058】
(7)反応終了後、発酵槽の培地をプラスチック容器(バケツ)に移した。培養液から遠心分離(6000rpm、20℃)により菌体成分を沈殿除去し、得られた上清を塩化ナトリウムで飽和させた後に塩酸を加えpH 1.5にした。
【0059】
(8)ジャーファーメンターより定期的にサンプリングした培養液(約15 ml)を適当に希釈して、含まれている基質濃度を1〜5 mMに調整した。その際、内標準物質として安息香酸を最終濃度1 mMとなるように添加した。このサンプル3 mlを分取し、12N 塩酸を数滴加えてpHを1に調整した後、3 mlの酢酸エチルを加えて激しく混和した。4,000 rpmにて遠心し、上清の酢酸エチル層を100 μl分取した。そして、酢酸エチルを乾固した後、50 μlのピリジンと50 μlのTMS化剤 (BSTFA: GL-サイエンス)を加えて混合し、室温にて30分反応させた。このサンプル1 μlをガスクロマトグラフィーにて分析し、予め作成済みの検量線データを用いて、ジャーファーメンターサンプル中の各基質濃度を計算した。その結果、図2に示すように反応液中のβ-ケトアジピン酸は、約99mM(収率100%)検出された。
【0060】
(9)β-ケトアジピン酸は、酢酸エチルで抽出後、エバポレーターを用いて乾固した。その後、アセトンに再溶解して、石油エーテルなどの貧溶媒を添加した条件で結晶化させ(図3)、このサンプルをガスクロマトグラフィー・マススペクトロメトリーで分析した。その後、重DMSO中にてNMR測定を行った。
δH (DMSO-d6), 2.4 (t, 2H), 2.7(t, 2H), 3.5(s, 2H)
δC (DMSO-d6), 28.1, 38.0, 49.5, 169.2, 174.0, 203.2
これらの結果から、得られたサンプルは夾雑物がなく、純度が高いβ−ケトアジピン酸であることが分かった。
【0061】
実施例4 プラスミドpVAadi1F6を導入した形質転換細胞によるバニリン酸からβ-ケトアジピン酸への製造
(1)β-ケトアジピン酸を発酵生産するための多段反応プロセスの酵素遺伝子を含む組み換えプラスミドpVAadi1F6(実施例1)を大腸菌JM109株に形質転換し、25 mg/Lのカナマイシンを含むLB培地(10ml)で、37℃で18時間振とう培養し、増殖した培養細胞から組み換えプラスミドpVAadi1F6を抽出した。
【0062】
(2)植物成分由来、化学合成もしくは石油由来のバニリン、バニリン酸、プロトカテク酸、p-ヒドロキシベンズアルデヒド、p-ヒドロキシ安息香酸の分解代謝酵素機能を消失せしめた微生物であるシュードモナス・プチダKT2440dJ株(実施例2)を、LB液体培地200mlで、28℃ 24時間培養し氷中で30分冷却した。4℃ 10分10000rpmで遠心集菌し、200mlの0℃蒸留水で温和に洗浄後再び遠心集菌した。続いて150mlの0℃蒸留水で温和に洗浄後、遠心集菌した。さらに100mlの0℃蒸留水で温和に洗浄後、遠心集菌した。集菌した微生物細胞を、10%グリセロールを含む蒸留水に懸濁し0℃にて保持した。
【0063】
(3)(1)のプラスミドpVAadi1F6を約0.05μgを含む蒸留水4μlを0.2cmのキュベットに入れ、(2)の10%グリセロールを含む蒸留水に懸濁した細胞液40μlを加え、(25μF、2500V、12msec)の条件でエレクトロポレーションにかけた。
【0064】
(4)上記処理した細胞全量を10mlのLB液体培地に接種し、28℃で6時間培養した。培養後遠心によって菌体を集め50 mg/Lのカナマイシンを含むLB平板に塗布し28℃で48時間培養し、プラスミドpVAadi1F6を保持するカナマイシン耐性を示す形質転換株を得た。本菌をシュードモナス・プチダKT2440dJ-VAadi1株と名づけた。
【0065】
(5)KT2440dJ-VAadi1株を、200mlのLB液体培地(50 mg/Lのカナマイシンを含む)に接種し28℃で16時間培養し前培養菌体懸濁液とした。5 Lの無機塩-yeast extract-グルコース液体培地及び消泡剤(Antiform A) 3 mlを10 L容量のジャーファーメンター(発酵槽)を用いて調製し、そこに培養したKT2440dJ-VAadi1株の前培養菌体懸濁液50mlを混合し、28℃ 700 rpm/minの通気攪拌下、OD660=7〜9まで培養した(14時間〜18時間)。
(6)OD660=7〜9に達した発酵槽の培養液に、基質であるバニリン酸28 gを含む0.1NのNaOH水溶液(pH8.0に調整)500mlを、ペリスタポンプを用いて10時間かけて添加した。反応の進行に伴うβ-ケトアジピン酸の生成により、培養液のpHが低下する。それを防ぐためpHセンサーに連結したペリスタポンプで5NのNaOH溶液を添加して培養液のpHを維持した。基質添加前後のODの変化を図4に示す。
【0066】
(7)反応終了後、発酵槽の培地をプラスチック容器(バケツ)に移した。培養液から遠心分離(6000rpm、20℃)により菌体成分を沈殿除去し、得られた上清を塩化ナトリウムで飽和させた後に塩酸を加えpH 1.5にした。反応液中のβ-ケトアジピン酸を実施例3と同様の方法により定量した結果、図4に示すように約33mM(収率100%)検出された。
【0067】
実施例5 プラスミドpVAaKL1Fを導入した形質転換細胞によるプロトカテク酸からムコノラクトンへの製造
(1)ムコノラクトンを発酵生産するための多段反応プロセスの酵素遺伝子を含む組み換えプラスミドpVAaKL1Fを大腸菌JM109株に形質転換し、25 mg/Lのカナマイシンを含むLB培地(10ml)で、37℃で18時間振とう培養し、増殖した培養細胞から組み換えプラスミドpVAaKL1Fを抽出した。
【0068】
(2)植物成分由来、化学合成もしくは石油由来のバニリン、バニリン酸、プロトカテク酸、p-ヒドロキシベンズアルデヒド、p-ヒドロキシ安息香酸の分解代謝酵素機能を消失せしめた微生物であるシュードモナス属細菌(Pseudomonas putida KT2440dD)を、LB液体培地200mlで、28℃ 24時間培養し氷中で30分冷却した。4℃ 10分10000rpmで遠心集菌し、200mlの0℃蒸留水で温和に洗浄後再び遠心集菌した。続いて150mlの0℃蒸留水で温和に洗浄後、遠心集菌した。さらに100mlの0℃蒸留水で温和に洗浄後、遠心集菌した。集菌した微生物細胞を、10%グリセロールを含む蒸留水に懸濁し0℃にて保持した。
【0069】
(3)(1)のプラスミドpVAaKL1Fを約0.05μgを含む蒸留水4μlを0.2cmのキュベットに入れ、(2)の10%グリセロールを含む蒸留水に懸濁した細胞液40μlを加え、(25μF、2500V、12msec)の条件でエレクトロポレーションにかけた。
【0070】
(4)上記処理した細胞全量を10mlのLB液体培地に接種し、28℃で6時間培養した。培養後遠心によって菌体を集め50 mg/Lのカナマイシンを含むLB平板に塗布し28℃で48時間培養し、プラスミドpVAaKL1Fを保持するカナマイシン耐性を示す形質転換株を得た。本菌をシュードモナス・プチダKT2440dD-VAaKL1株と名づけた。
【0071】
(5)KT2440dD-VAaKL1株を、200mlのLB液体培地(50 mg/Lのカナマイシンを含む)に接種し28℃で16時間培養し前培養菌体懸濁液とした。4.3 Lの無機塩-yeast extract-グルコース液体培地及び消泡剤(Antiform A) 3 mlを10 L容量のジャーファーメンター(発酵槽)を用いて調製し、そこに培養したKT2440dD-VAaKL1株の前培養菌体懸濁液200mlを混合し、28℃ 700 rpm/minの通気攪拌下、OD660=7〜9まで培養した(10時間〜16時間)(図5)。
【0072】
(6)OD660=7〜9に達した発酵槽の培養液に、基質であるプロトカテク酸57 gを含む0.1NのNaOH水溶液(pH8.0に調整)500mlを、ペリスタポンプを用いて10時間かけて添加した。反応の進行に伴うムコノラクトンの生成により、培養液のpHが低下する。それを防ぐためpHセンサーに連結したペリスタポンプで5NのNaOH溶液を添加して培養液のpHを維持した。
【0073】
反応の進行は薄層クロマトグラフィー(TLC)によって確認した。添加したプロトカテク酸は基質添加終了後、約17時間で消失することが確認された。
【0074】
(7)反応終了後、発酵槽の培地をプラスチック容器(バケツ)に移した。培養液から遠心分離(6000rpm、20℃)により菌体成分を沈殿除去し、得られた上清を塩化ナトリウムで飽和させた後に塩酸を加えpH 1.5にした。反応液中のムコノラクトン定量の結果は図5に示すように26時間の時点で約61.7 mM(収率95%)検出された。
【0075】
(8)ムコノラクトンは、酢酸エチルで抽出後、エバポレーターを用いて乾固した。その後、酢酸エチルに再度溶解して、結晶化した(図6)。
その後、重アセトン中にてNMR測定を行った。
δH (acetone-d6), 2.7(m, 1H), 2.8(m, 1H), 5.4(m, 1H), 6.1(m, 1H), 7.8(m, 1H)
δC (acetone-d6)、 37.5, 79.4, 121.2, 165.5, 169.5, 170.1
これらの結果から、得られたサンプルは夾雑物がなく、純度が高いムコノラクトンであることが分かった。
【0076】
実施例6 プラスミドpVAaKL1Fを導入した形質転換細胞によるバニリン酸からムコノラクトンへの製造
(1)ムコノラクトンを発酵生産するための多段反応プロセスの酵素遺伝子を含む組み換えプラスミドpVAaKL1Fを大腸菌JM109株に形質転換し、25 mg/Lのカナマイシンを含むLB培地(10ml)で、37℃で18時間振とう培養し、増殖した培養細胞から組み換えプラスミドpVAaKL1Fを抽出した。
【0077】
(2)植物成分由来、化学合成もしくは石油由来のバニリン、バニリン酸、プロトカテク酸、p-ヒドロキシベンズアルデヒド、p-ヒドロキシ安息香酸の分解代謝酵素機能を消失せしめた微生物であるシュードモナス属細菌(Pseudomonas putida KT2440dD)を、LB液体培地200mlで、28℃ 24時間培養し氷中で30分冷却した。4℃ 10分10000rpmで遠心集菌し、200mlの0℃蒸留水で温和に洗浄後再び遠心集菌した。続いて150mlの0℃蒸留水で温和に洗浄後、遠心集菌した。さらに100mlの0℃蒸留水で温和に洗浄後、遠心集菌した。集菌した微生物細胞を、10%グリセロールを含む蒸留水に懸濁し0℃にて保持した。
【0078】
(3)(1)のプラスミドpVAaKL1Fを約0.05μgを含む蒸留水4μlを0.2cmのキュベットに入れ、(2)の10%グリセロールを含む蒸留水に懸濁した細胞液40μlを加え、(25μF、2500V、12msec)の条件でエレクトロポレーションにかけた。
【0079】
(4)上記処理した細胞全量を10mlのLB液体培地に接種し、28℃で6時間培養した。培養後遠心によって菌体を集め50 mg/Lのカナマイシンを含むLB平板に塗布し28℃で48時間培養し、プラスミドpVAaKL1Fを保持するカナマイシン耐性を示す形質転換株を得た。本菌をシュードモナス・プチダ KT2440dD-VAaKL1株と名づけた。
【0080】
(5)KT2440dD-VAaKL1株を、200mlのLB液体培地(50 mg/Lのカナマイシンを含む)に接種し28℃で16時間培養し前培養菌体懸濁液とした。4.3 Lの無機塩-yeast extract-グルコース液体培地及び消泡剤(Antiform A) 3 mlを10 L容量のジャーファーメンター(発酵槽)を用いて調製し、そこに培養したKT2440dD-VAaKL1株の前培養菌体懸濁液200mlを混合し、28℃ 700 rpm/minの通気攪拌下、OD660=7〜9まで培養した(10時間〜16時間)(図7)。
【0081】
(6)OD660=7〜9に達した発酵槽の培養液に、基質であるバニリン酸28 gを含む0.1NのNaOH水溶液(pH8.0に調整)500mlを、ペリスタポンプを用いて10時間かけて添加した。反応の進行に伴うムコノラクトンの生成により、培養液のpHが低下する。それを防ぐためpHセンサーに連結したペリスタポンプで5NのNaOH溶液を添加して培養液のpHを維持した。
【0082】
反応の進行は薄層クロマトグラフィー(TLC)によって確認した。添加したバニリン酸は基質添加終了後、約26時間で消失することが確認された。
【0083】
(7)反応終了後、発酵槽の培地をプラスチック容器(バケツ)に移した。培養液から遠心分離(6000rpm、20℃)により菌体成分を沈殿除去し、得られた上清を塩化ナトリウムで飽和させた後に塩酸を加えpH 1.5にした。反応液中のムコノラクトン定量の結果は図7に示すように約28.5 mM(収率95%)検出された。
【0084】
(8)ムコノラクトンは、酢酸エチルで抽出後、エバポレーターを用いて乾固した。その後、酢酸エチルに再度溶解して、結晶化した。
【0085】
実施例7 β-ケトアジピン酸の酸処理によるレブリン酸の製造
実施例3、4により得られたβ-ケトアジピン酸粉末を水に溶解し塩酸を少量添加し、酢酸エチルで抽出後、重DMSOに溶解しNMR測定を行った。
δH (DMSO-d6), 2.0(s, 3H), 2.3(t, 2H), 2.6(t, 2H)
δC (DMSO-d6), 28.2, 30.5, 38.0, 174.2, 207.4
NMR測定の結果から上記抽出物はレブリン酸であることがわかった。
【特許請求の範囲】
【請求項1】
pcaB遺伝子及びpcaC遺伝子と、vanA遺伝子及びvanB遺伝子並びにpcaH遺伝子及びpcaG遺伝子と、から成るDNA分子群を含んで成る組換えベクター:
ここで、
(a) pcaB遺伝子は下記(a-1)〜(a-4)のいずれかのDNA分子であり、
(a-1) 配列番号1記載のDNA分子、
(a-2) 配列番号2記載のアミノ酸配列をコードするDNA分子、
(a-3) 配列番号1記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つ3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素活性を有するポリペプチドをコードするDNA分子、あるいは
(a-4) 配列番号2記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つ3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素活性を有するポリペプチドをコードするDNA分子、
(b) pcaC遺伝子は下記(b-1)〜(b-4)のいずれかのDNA分子であり、
(b-1) 配列番号3記載のDNA分子、
(b-2) 配列番号4記載のアミノ酸配列をコードするDNA分子、
(b-3) 配列番号3記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つγ-カルボキシムコノラクトンデカルボキシラーゼ活性を有するポリペプチドをコードするDNA分子、あるいは
(b-4) 配列番号4記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つγ-カルボキシムコノラクトンデカルボキシラーゼ活性を有するポリペプチドをコードするDNA分子、
(c) vanA遺伝子は下記(c-1)〜(c-4)のいずれかのDNA分子であり、
(c-1) 配列番号7記載のDNA分子、
(c-2) 配列番号8記載のアミノ酸配列をコードするDNA分子、
(c-3) 配列番号7記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(c-4) 配列番号8記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(d) vanB遺伝子は下記(d-1)〜(d-4)のいずれかのDNA分子であり、
(d-1) 配列番号9記載のDNA分子、
(d-2) 配列番号10記載のアミノ酸配列をコードするDNA分子、
(d-3) 配列番号9記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(d-4) 配列番号10記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(e) pcaH遺伝子は下記(e-1)〜(e-4)のいずれかのDNA分子である。
(e-1) 配列番号11記載のDNA分子、
(e-2) 配列番号12記載のアミノ酸配列をコードするDNA分子、
(e-3) 配列番号11記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(e-4) 配列番号12記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(f) pcaG遺伝子は下記(f-1)〜(f-4)のいずれかのDNA分子である。
(f-1) 配列番号13記載のDNA分子、
(f-2) 配列番号14記載のアミノ酸配列をコードするDNA分子、
(f-3) 配列番号13記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(f-4) 配列番号14記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子。
【請求項2】
前記DNA分子群が更に以下のpcaD遺伝子を含んで成る、請求項1に記載の組換えベクター:
(g) pcaD遺伝子は下記(g-1)〜(g-4)のいずれかのDNA分子である。
(g-1) 配列番号5記載のDNA分子、
(g-2) 配列番号6記載のアミノ酸配列をコードするDNA分子、
(g-3) 配列番号5記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つβ-ケトアジピン酸エノールラクトンヒドロラーゼ活性を有するポリペプチドをコードするDNA分子、あるいは
(g-4) 配列番号6記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つβ-ケトアジピン酸エノールラクトンヒドロラーゼ活性を有するポリペプチドをコードするDNA分子。
【請求項3】
請求項1又は2に記載の組換えベクターを有する形質転換体。
【請求項4】
請求項1に記載の組換えベクターを有する形質転換体であって、β-ケトアジピン酸エノールラクトン代謝・分解能が欠損している、形質転換体。
【請求項5】
染色体DNA上にpcaB遺伝子及びpcaC遺伝子と、vanA遺伝子及びvanB遺伝子並びにpcaH遺伝子及びpcaG遺伝子と、から成るDNA分子群が存在する微生物細胞又は当該DNA分子群を含んで成る組換えベクターを有する形質転換体の、β-ケトアジピン酸エノールラクトン代謝・分解能が欠損している遺伝子破壊株であって、当該染色体DNAに存在するpcaD遺伝子が破壊されている遺伝子破壊株:
ここで、
(a) pcaB遺伝子は下記(a-1)〜(a-4)のいずれかのDNA分子であり、
(a-1) 配列番号1記載のDNA分子、
(a-2) 配列番号2記載のアミノ酸配列をコードするDNA分子、
(a-3) 配列番号1記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つ3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素活性を有するポリペプチドをコードするDNA分子、あるいは
(a-4) 配列番号2記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つ3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素活性を有するポリペプチドをコードするDNA分子、
(b) pcaC遺伝子は下記(b-1)〜(b-4)のいずれかのDNA分子であり、
(b-1) 配列番号3記載のDNA分子、
(b-2) 配列番号4記載のアミノ酸配列をコードするDNA分子、
(b-3) 配列番号3記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つγ-カルボキシムコノラクトンデカルボキシラーゼ活性を有するポリペプチドをコードするDNA分子、あるいは
(b-4) 配列番号4記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つγ-カルボキシムコノラクトンデカルボキシラーゼ活性を有するポリペプチドをコードするDNA分子、
(c) vanA遺伝子は下記(c-1)〜(c-4)のいずれかのDNA分子であり、
(c-1) 配列番号7記載のDNA分子、
(c-2) 配列番号8記載のアミノ酸配列をコードするDNA分子、
(c-3) 配列番号7記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(c-4) 配列番号8記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(d) vanB遺伝子は下記(d-1)〜(d-4)のいずれかのDNA分子であり、
(d-1) 配列番号9記載のDNA分子、
(d-2) 配列番号10記載のアミノ酸配列をコードするDNA分子、
(d-3) 配列番号9記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(d-4) 配列番号10記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(e) pcaH遺伝子は下記(e-1)〜(e-4)のいずれかのDNA分子であり、
(e-1) 配列番号11記載のDNA分子、
(e-2) 配列番号12記載のアミノ酸配列をコードするDNA分子、
(e-3) 配列番号11記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(e-4) 配列番号12記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(f) pcaG遺伝子は下記(f-1)〜(f-4)のいずれかのDNA分子であり、
(f-1) 配列番号13記載のDNA分子、
(f-2) 配列番号14記載のアミノ酸配列をコードするDNA分子、
(f-3) 配列番号13記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(f-4) 配列番号14記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、そして
(g) pcaD遺伝子は下記(c-1)〜(c-4)のいずれかのDNA分子である。
(g-1) 配列番号5記載のDNA分子、
(g-2) 配列番号6記載のアミノ酸配列をコードするDNA分子、
(g-3) 配列番号5記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つβ-ケトアジピン酸エノールラクトンヒドロラーゼ活性を有するポリペプチドをコードするDNA分子、あるいは
(g-4) 配列番号6記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つβ-ケトアジピン酸エノールラクトンヒドロラーゼ活性を有するポリペプチドをコードするDNA分子。
【請求項6】
請求項4に記載の形質転換体、又は請求項5に記載の遺伝子破壊株を、バニリン酸及び/又はプロトカテク酸の存在下で培養し、当該培養物からムコノラクトンを採取することを特徴とする、ムコノラクトンの製造方法。
【請求項7】
請求項2に記載の組換えベクターを含む形質転換体であって、β−ケトアジピン酸代謝・分解能が欠損している、形質転換体。
【請求項8】
染色体DNA上にpcaB遺伝子、pcaC遺伝子及びpcaD遺伝子と、vanA遺伝子及びvanB遺伝子並びにpcaH遺伝子及びpcaG遺伝子と、から成るDNA分子群が存在する微生物細胞又は当該DNA分子群を含んで成る組換えベクターを有する形質転換体の、β-ケトアジピン酸代謝・分解能が欠損している遺伝子破壊株であって、当該染色体DNAに存在するpcaJ遺伝子が破壊されている遺伝子破壊株:
ここで、
(a) pcaB遺伝子は下記(a-1)〜(a-4)のいずれかのDNA分子であり、
(a-1) 配列番号1記載のDNA分子、
(a-2) 配列番号2記載のアミノ酸配列をコードするDNA分子、
(a-3) 配列番号1記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つ3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素活性を有するポリペプチドをコードするDNA分子、あるいは
(a-4) 配列番号2記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つ3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素活性を有するポリペプチドをコードするDNA分子、
(b) pcaC遺伝子は下記(b-1)〜(b-4)のいずれかのDNA分子であり、
(b-1) 配列番号3記載のDNA分子、
(b-2) 配列番号4記載のアミノ酸配列をコードするDNA分子、
(b-3) 配列番号3記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つγ-カルボキシムコノラクトンデカルボキシラーゼ活性を有するポリペプチドをコードするDNA分子、あるいは
(b-4) 配列番号4記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つγ-カルボキシムコノラクトンデカルボキシラーゼ活性を有するポリペプチドをコードするDNA分子、
(c) vanA遺伝子は下記(c-1)〜(c-4)のいずれかのDNA分子であり、
(c-1) 配列番号7記載のDNA分子、
(c-2) 配列番号8記載のアミノ酸配列をコードするDNA分子、
(c-3) 配列番号7記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(c-4) 配列番号8記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(d) vanB遺伝子は下記(d-1)〜(d-4)のいずれかのDNA分子であり、
(d-1) 配列番号9記載のDNA分子、
(d-2) 配列番号10記載のアミノ酸配列をコードするDNA分子、
(d-3) 配列番号9記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(d-4) 配列番号10記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(e) pcaH遺伝子は下記(e-1)〜(e-4)のいずれかのDNA分子であり、
(e-1) 配列番号11記載のDNA分子、
(e-2) 配列番号12記載のアミノ酸配列をコードするDNA分子、
(e-3) 配列番号11記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(e-4) 配列番号12記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(f) pcaG遺伝子は下記(f-1)〜(f-4)のいずれかのDNA分子であり、
(f-1) 配列番号13記載のDNA分子、
(f-2) 配列番号14記載のアミノ酸配列をコードするDNA分子、
(f-3) 配列番号13記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(f-4) 配列番号14記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、そして
(g) pcaD遺伝子は下記(c-1)〜(c-4)のいずれかのDNA分子であり、
(g-1) 配列番号5記載のDNA分子、
(g-2) 配列番号6記載のアミノ酸配列をコードするDNA分子、
(g-3) 配列番号5記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つβ-ケトアジピン酸エノールラクトンヒドロラーゼ活性を有するポリペプチドをコードするDNA分子、あるいは
(g-4) 配列番号6記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つβ-ケトアジピン酸エノールラクトンヒドロラーゼ活性を有するポリペプチドをコードするDNA分子、
(h) pcaJ遺伝子は下記(h-1)〜(h-4)のいずれかのDNA分子である。
(h-1) 配列番号21記載のDNA分子、
(h-2) 配列番号22記載のアミノ酸配列をコードするDNA分子、
(h-3) 配列番号21記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つβ-ケトアジピン酸スクシニルCoAトランスフェラーゼ活性を有するポリペプチドをコードするDNA分子、あるいは
(h-4) 配列番号22記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つβ-ケトアジピン酸スクシニルCoAトランスフェラーゼ活性を有するポリペプチドをコードするDNA分子。
【請求項9】
請求項7に記載の形質転換体、又は請求項8に記載の遺伝子破壊株を、バニリン酸及び/又はプロトカテク酸の存在下で培養し、当該培養物からβ-ケトアジピン酸を採取することを特徴とする、β-ケトアジピン酸の製造方法。
【請求項10】
請求項9に記載の方法に従い製造されたβ-ケトアジピン酸を脱炭酸反応にかけてレブリン酸を得ることを特徴とする、レブリン酸の製造方法。
【請求項1】
pcaB遺伝子及びpcaC遺伝子と、vanA遺伝子及びvanB遺伝子並びにpcaH遺伝子及びpcaG遺伝子と、から成るDNA分子群を含んで成る組換えベクター:
ここで、
(a) pcaB遺伝子は下記(a-1)〜(a-4)のいずれかのDNA分子であり、
(a-1) 配列番号1記載のDNA分子、
(a-2) 配列番号2記載のアミノ酸配列をコードするDNA分子、
(a-3) 配列番号1記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つ3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素活性を有するポリペプチドをコードするDNA分子、あるいは
(a-4) 配列番号2記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つ3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素活性を有するポリペプチドをコードするDNA分子、
(b) pcaC遺伝子は下記(b-1)〜(b-4)のいずれかのDNA分子であり、
(b-1) 配列番号3記載のDNA分子、
(b-2) 配列番号4記載のアミノ酸配列をコードするDNA分子、
(b-3) 配列番号3記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つγ-カルボキシムコノラクトンデカルボキシラーゼ活性を有するポリペプチドをコードするDNA分子、あるいは
(b-4) 配列番号4記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つγ-カルボキシムコノラクトンデカルボキシラーゼ活性を有するポリペプチドをコードするDNA分子、
(c) vanA遺伝子は下記(c-1)〜(c-4)のいずれかのDNA分子であり、
(c-1) 配列番号7記載のDNA分子、
(c-2) 配列番号8記載のアミノ酸配列をコードするDNA分子、
(c-3) 配列番号7記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(c-4) 配列番号8記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(d) vanB遺伝子は下記(d-1)〜(d-4)のいずれかのDNA分子であり、
(d-1) 配列番号9記載のDNA分子、
(d-2) 配列番号10記載のアミノ酸配列をコードするDNA分子、
(d-3) 配列番号9記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(d-4) 配列番号10記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(e) pcaH遺伝子は下記(e-1)〜(e-4)のいずれかのDNA分子である。
(e-1) 配列番号11記載のDNA分子、
(e-2) 配列番号12記載のアミノ酸配列をコードするDNA分子、
(e-3) 配列番号11記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(e-4) 配列番号12記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(f) pcaG遺伝子は下記(f-1)〜(f-4)のいずれかのDNA分子である。
(f-1) 配列番号13記載のDNA分子、
(f-2) 配列番号14記載のアミノ酸配列をコードするDNA分子、
(f-3) 配列番号13記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(f-4) 配列番号14記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子。
【請求項2】
前記DNA分子群が更に以下のpcaD遺伝子を含んで成る、請求項1に記載の組換えベクター:
(g) pcaD遺伝子は下記(g-1)〜(g-4)のいずれかのDNA分子である。
(g-1) 配列番号5記載のDNA分子、
(g-2) 配列番号6記載のアミノ酸配列をコードするDNA分子、
(g-3) 配列番号5記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つβ-ケトアジピン酸エノールラクトンヒドロラーゼ活性を有するポリペプチドをコードするDNA分子、あるいは
(g-4) 配列番号6記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つβ-ケトアジピン酸エノールラクトンヒドロラーゼ活性を有するポリペプチドをコードするDNA分子。
【請求項3】
請求項1又は2に記載の組換えベクターを有する形質転換体。
【請求項4】
請求項1に記載の組換えベクターを有する形質転換体であって、β-ケトアジピン酸エノールラクトン代謝・分解能が欠損している、形質転換体。
【請求項5】
染色体DNA上にpcaB遺伝子及びpcaC遺伝子と、vanA遺伝子及びvanB遺伝子並びにpcaH遺伝子及びpcaG遺伝子と、から成るDNA分子群が存在する微生物細胞又は当該DNA分子群を含んで成る組換えベクターを有する形質転換体の、β-ケトアジピン酸エノールラクトン代謝・分解能が欠損している遺伝子破壊株であって、当該染色体DNAに存在するpcaD遺伝子が破壊されている遺伝子破壊株:
ここで、
(a) pcaB遺伝子は下記(a-1)〜(a-4)のいずれかのDNA分子であり、
(a-1) 配列番号1記載のDNA分子、
(a-2) 配列番号2記載のアミノ酸配列をコードするDNA分子、
(a-3) 配列番号1記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つ3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素活性を有するポリペプチドをコードするDNA分子、あるいは
(a-4) 配列番号2記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つ3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素活性を有するポリペプチドをコードするDNA分子、
(b) pcaC遺伝子は下記(b-1)〜(b-4)のいずれかのDNA分子であり、
(b-1) 配列番号3記載のDNA分子、
(b-2) 配列番号4記載のアミノ酸配列をコードするDNA分子、
(b-3) 配列番号3記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つγ-カルボキシムコノラクトンデカルボキシラーゼ活性を有するポリペプチドをコードするDNA分子、あるいは
(b-4) 配列番号4記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つγ-カルボキシムコノラクトンデカルボキシラーゼ活性を有するポリペプチドをコードするDNA分子、
(c) vanA遺伝子は下記(c-1)〜(c-4)のいずれかのDNA分子であり、
(c-1) 配列番号7記載のDNA分子、
(c-2) 配列番号8記載のアミノ酸配列をコードするDNA分子、
(c-3) 配列番号7記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(c-4) 配列番号8記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(d) vanB遺伝子は下記(d-1)〜(d-4)のいずれかのDNA分子であり、
(d-1) 配列番号9記載のDNA分子、
(d-2) 配列番号10記載のアミノ酸配列をコードするDNA分子、
(d-3) 配列番号9記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(d-4) 配列番号10記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(e) pcaH遺伝子は下記(e-1)〜(e-4)のいずれかのDNA分子であり、
(e-1) 配列番号11記載のDNA分子、
(e-2) 配列番号12記載のアミノ酸配列をコードするDNA分子、
(e-3) 配列番号11記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(e-4) 配列番号12記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(f) pcaG遺伝子は下記(f-1)〜(f-4)のいずれかのDNA分子であり、
(f-1) 配列番号13記載のDNA分子、
(f-2) 配列番号14記載のアミノ酸配列をコードするDNA分子、
(f-3) 配列番号13記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(f-4) 配列番号14記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、そして
(g) pcaD遺伝子は下記(c-1)〜(c-4)のいずれかのDNA分子である。
(g-1) 配列番号5記載のDNA分子、
(g-2) 配列番号6記載のアミノ酸配列をコードするDNA分子、
(g-3) 配列番号5記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つβ-ケトアジピン酸エノールラクトンヒドロラーゼ活性を有するポリペプチドをコードするDNA分子、あるいは
(g-4) 配列番号6記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つβ-ケトアジピン酸エノールラクトンヒドロラーゼ活性を有するポリペプチドをコードするDNA分子。
【請求項6】
請求項4に記載の形質転換体、又は請求項5に記載の遺伝子破壊株を、バニリン酸及び/又はプロトカテク酸の存在下で培養し、当該培養物からムコノラクトンを採取することを特徴とする、ムコノラクトンの製造方法。
【請求項7】
請求項2に記載の組換えベクターを含む形質転換体であって、β−ケトアジピン酸代謝・分解能が欠損している、形質転換体。
【請求項8】
染色体DNA上にpcaB遺伝子、pcaC遺伝子及びpcaD遺伝子と、vanA遺伝子及びvanB遺伝子並びにpcaH遺伝子及びpcaG遺伝子と、から成るDNA分子群が存在する微生物細胞又は当該DNA分子群を含んで成る組換えベクターを有する形質転換体の、β-ケトアジピン酸代謝・分解能が欠損している遺伝子破壊株であって、当該染色体DNAに存在するpcaJ遺伝子が破壊されている遺伝子破壊株:
ここで、
(a) pcaB遺伝子は下記(a-1)〜(a-4)のいずれかのDNA分子であり、
(a-1) 配列番号1記載のDNA分子、
(a-2) 配列番号2記載のアミノ酸配列をコードするDNA分子、
(a-3) 配列番号1記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つ3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素活性を有するポリペプチドをコードするDNA分子、あるいは
(a-4) 配列番号2記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つ3-カルボキシ-cis,cis-ムコン酸ラクトン化酵素活性を有するポリペプチドをコードするDNA分子、
(b) pcaC遺伝子は下記(b-1)〜(b-4)のいずれかのDNA分子であり、
(b-1) 配列番号3記載のDNA分子、
(b-2) 配列番号4記載のアミノ酸配列をコードするDNA分子、
(b-3) 配列番号3記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つγ-カルボキシムコノラクトンデカルボキシラーゼ活性を有するポリペプチドをコードするDNA分子、あるいは
(b-4) 配列番号4記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つγ-カルボキシムコノラクトンデカルボキシラーゼ活性を有するポリペプチドをコードするDNA分子、
(c) vanA遺伝子は下記(c-1)〜(c-4)のいずれかのDNA分子であり、
(c-1) 配列番号7記載のDNA分子、
(c-2) 配列番号8記載のアミノ酸配列をコードするDNA分子、
(c-3) 配列番号7記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(c-4) 配列番号8記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(d) vanB遺伝子は下記(d-1)〜(d-4)のいずれかのDNA分子であり、
(d-1) 配列番号9記載のDNA分子、
(d-2) 配列番号10記載のアミノ酸配列をコードするDNA分子、
(d-3) 配列番号9記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(d-4) 配列番号10記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つバニリン酸ディメチラーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(e) pcaH遺伝子は下記(e-1)〜(e-4)のいずれかのDNA分子であり、
(e-1) 配列番号11記載のDNA分子、
(e-2) 配列番号12記載のアミノ酸配列をコードするDNA分子、
(e-3) 配列番号11記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(e-4) 配列番号12記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、
(f) pcaG遺伝子は下記(f-1)〜(f-4)のいずれかのDNA分子であり、
(f-1) 配列番号13記載のDNA分子、
(f-2) 配列番号14記載のアミノ酸配列をコードするDNA分子、
(f-3) 配列番号13記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、あるいは
(f-4) 配列番号14記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つプロトカテク酸3,4−ジオキシゲナーゼ活性を有するポリペプチドのサブユニットをコードするDNA分子、そして
(g) pcaD遺伝子は下記(c-1)〜(c-4)のいずれかのDNA分子であり、
(g-1) 配列番号5記載のDNA分子、
(g-2) 配列番号6記載のアミノ酸配列をコードするDNA分子、
(g-3) 配列番号5記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つβ-ケトアジピン酸エノールラクトンヒドロラーゼ活性を有するポリペプチドをコードするDNA分子、あるいは
(g-4) 配列番号6記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つβ-ケトアジピン酸エノールラクトンヒドロラーゼ活性を有するポリペプチドをコードするDNA分子、
(h) pcaJ遺伝子は下記(h-1)〜(h-4)のいずれかのDNA分子である。
(h-1) 配列番号21記載のDNA分子、
(h-2) 配列番号22記載のアミノ酸配列をコードするDNA分子、
(h-3) 配列番号21記載のDNA分子又はその相補配列から成るDNA分子と高ストリンジェントな条件下でハイブリダイズし、且つβ-ケトアジピン酸スクシニルCoAトランスフェラーゼ活性を有するポリペプチドをコードするDNA分子、あるいは
(h-4) 配列番号22記載のアミノ酸配列の1若しくは数個のアミノ酸が欠失、置換及び/又は付加されたアミノ酸配列から成り、且つβ-ケトアジピン酸スクシニルCoAトランスフェラーゼ活性を有するポリペプチドをコードするDNA分子。
【請求項9】
請求項7に記載の形質転換体、又は請求項8に記載の遺伝子破壊株を、バニリン酸及び/又はプロトカテク酸の存在下で培養し、当該培養物からβ-ケトアジピン酸を採取することを特徴とする、β-ケトアジピン酸の製造方法。
【請求項10】
請求項9に記載の方法に従い製造されたβ-ケトアジピン酸を脱炭酸反応にかけてレブリン酸を得ることを特徴とする、レブリン酸の製造方法。
【図1A】

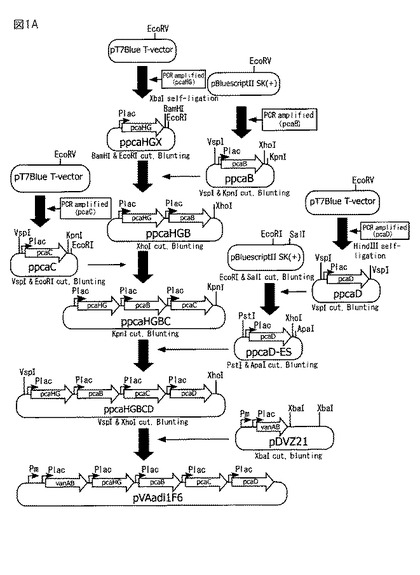
【図1B】
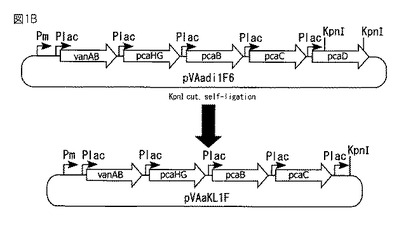
【図2】
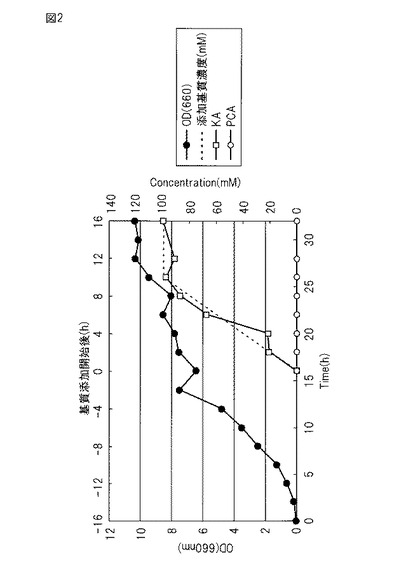
【図4】
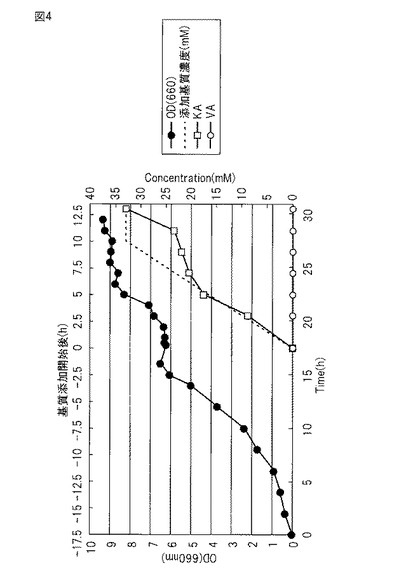
【図5】
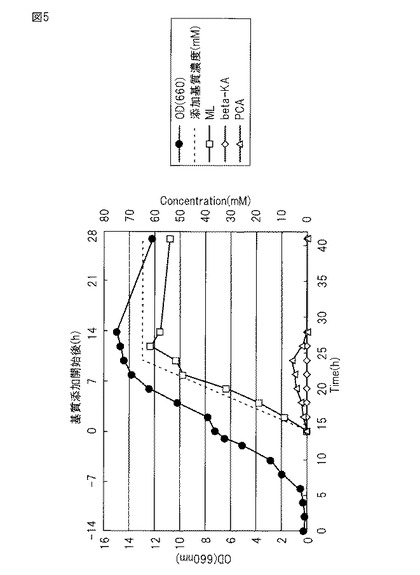
【図7】
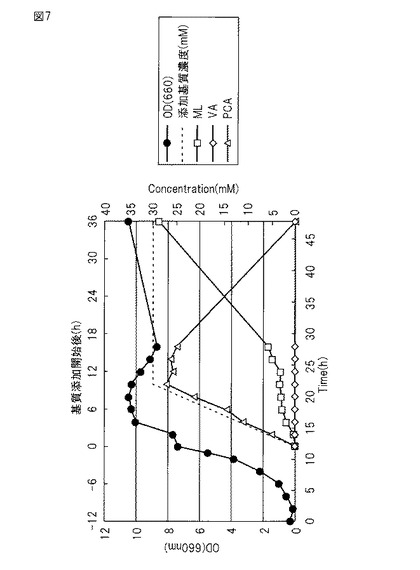
【図3】
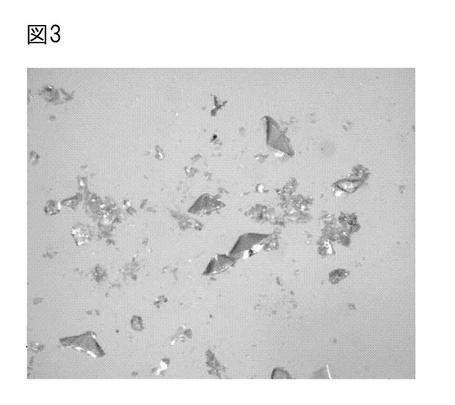
【図6】

【図1B】
【図2】
【図4】
【図5】
【図7】
【図3】
【図6】
【公開番号】特開2012−59(P2012−59A)
【公開日】平成24年1月5日(2012.1.5)
【国際特許分類】
【出願番号】特願2010−138548(P2010−138548)
【出願日】平成22年6月17日(2010.6.17)
【出願人】(000003218)株式会社豊田自動織機 (4,162)
【出願人】(504132881)国立大学法人東京農工大学 (595)
【出願人】(304021288)国立大学法人長岡技術科学大学 (458)
【出願人】(501186173)独立行政法人森林総合研究所 (91)
【Fターム(参考)】
【公開日】平成24年1月5日(2012.1.5)
【国際特許分類】
【出願日】平成22年6月17日(2010.6.17)
【出願人】(000003218)株式会社豊田自動織機 (4,162)
【出願人】(504132881)国立大学法人東京農工大学 (595)
【出願人】(304021288)国立大学法人長岡技術科学大学 (458)
【出願人】(501186173)独立行政法人森林総合研究所 (91)
【Fターム(参考)】
[ Back to top ]