
ムコン酸およびムコン酸塩の異性体を生成するための方法
生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネートを提供すること;cis,cis−ムコネートの実質的にすべてが、cis,trans−ムコネートへと異性化される反応条件下で、上記cis,cis−ムコネートをcis,trans−ムコネートへと異性化すること;上記cis,trans−ムコネートを分離すること;および上記cis,trans−ムコネートを結晶化することによる、ムコネートのcis,trans−異性体およびtrans,trans−異性体を生成するための方法。上記cis,trans−異性体は、上記trans,trans−異性体へとさらに異性化され得る。
【発明の詳細な説明】
【技術分野】
【0001】
(関連出願)
本願は、2010年1月8日に出願された米国仮特許出願第61/335,638号の利益および優先権を主張し、この米国仮特許出願の開示は、その全体が本明細書中に参考として援用される。
【0002】
(発明の分野)
本発明は、一般に、再生可能な供給材料からムコネートを生物学的に生成することに関する。本発明は、より詳細には、再生可能なバイオマス由来炭素源から、ムコネート異性体、ならびにその前駆物質および誘導体を生成することに関する。
【背景技術】
【0003】
(発明の背景)
ジメチルテレフタレート(DMT)の世界的消費は、2012年で平均して397万トン(メートル法の トン)まで突出している。DMTは、テレフタル酸およびメタノールのエステルであり、ポリエステル(ポリエチレンテレフタレートおよびポリトリメチレンテレフタレートが挙げられる)の生成において使用される。DMTはまた、産業用プラスチック、自動車部品、フィルム、釣り糸、および食品パッケージ材料の製造において使用される主要な成分である。
【0004】
伝統的には、DMT生成は、テレフタル酸を、パラキシレンの触媒による均一酸化(homogeneous oxidation)によって生成されるメタノールでエステル化することを利用する。例えば、液体パラキシレンは、コバルト塩触媒の存在下で空気によって酸化されて、オキシデート(oxidate)含有p−トルイル酸およびモノメチルテレフタレートを形成し得、エステル化は、メタノールの存在下で行われて、DMTを形成し得る。
【0005】
トリメリト酸(TMA)は、粉体塗料、インク、エナメル線(wire enamel)、低揮発性の高性能可塑剤、および高温適用のための産業用ポリマーのための樹脂を含む、化学産業における中間体としての適用を有する別の商業的に重要な生成物である。TMAはまた、脱水されて、トリメリト酸無水物を生成し得、これは、ポリマーおよび化学的中間体の生成のための、別の商業的に重要な出発材料である。
【0006】
伝統的には、TMAは、プソイドクメン(1,2,4−トリメチルベンゼン)の酸化によって生成される。テレフタル酸およびイソフタル酸は、溶媒としての酢酸および触媒系(コバルト、マンガンおよび臭素が挙げられる)の存在下で、p−キシレンもしくはm−キシレンの液相酸化によって商業的に生成され得る。
【0007】
これらプロセスは、多くの他の商業的に重要な化学前駆物質、中間体、および生成物を生成するためのプロセスのように、環境的に鋭敏な(environmentally sensitive)かつ非再生可能な供給材料(例えば、石油供給材料)に大きく依存すること、およびそれらの望ましくない副生成物(例えば、温室効果ガス、重金属、ハロゲン、発癌性炭化水素)を生成する傾向に起因して、望ましくない可能性がある。よって、再生可能な供給材料を利用して、DMT、TMA、および他の化学生成物を生成する改善された方法およびシステムが必要である。
【0008】
特許文献1(Frostらによる)および特許文献2(Frostらによる)に記載されるように(これら出願の開示はともに、それらの全体が本明細書に参考として援用される)、DMTおよびTMAは、ムコン酸から生成され得る。さらに、ムコン酸(その二重結合および二酸の官能性に起因して、2,4−ヘキサジエン二酸としても公知)は、広く種々の反応を受け得る。多くのムコン酸誘導体は公知であり、これらとしては、ラクトン、スルホン、ポリアミド、ポリエステル、チオエステル、付加ポリマー、および他の化合物が挙げられる。このような化合物は、広く種々の用途を有し、上記用途としては、界面活性剤、難燃剤、UV光安定化剤、熱硬化性プラスチック、熱プラスチック(thermoplastic)およびコーティングとしての用途が挙げられる。従って、再生可能な供給材料からムコン酸もしくはムコネートを生物学的に生成するための改善された方法は、DMT、TMAおよび他の化学物質を生成するために非常に望ましい。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】米国公開出願公開第2010/0314243号明細書
【特許文献2】国際公開第2010/148049号
【発明の概要】
【課題を解決するための手段】
【0010】
本発明の記載では、用語「ムコネート」および「ムコン酸」を使用する。用語「ムコン酸」とは、両方のカルボン酸官能基がプロトン化され、その分子が形式上中性種である化学種に言及する。ムコン酸は、化学式HOOC−CH=CH−CH=CH−−COOHを有する。用語「ムコネート」とは、カルボン酸官能基のうちの一方もしくは両方が脱プロトン化されて、生理学的pH値において優勢な化学種であるアニオン性もしくは二重アニオン性の形態を与える、対応する脱プロトン化化学種に言及する。しかし、用語「ムコン酸」および「ムコネート」とは、同じ分子のプロトン化形態もしくは脱プロトン化形態に言及するので、上記分子のプロトン化と脱プロトン化(例えば、非イオン化とイオン化)形態との間の差異が有用に区別されない場合に、上記用語は同義語として使用される。
【0011】
本発明は、バイオマス由来炭素源から、ムコネートの3種の異性体(すなわち、cis,cis異性体;cis,trans異性体;およびtrans,trans異性体)、ならびにその前駆物質および誘導体を生成するための方法を提供する。上記異性体は、その2個の二重結合の周りの幾何的配置により、構造的に異なる。さらに、上記異性体は、異なる物理的特性(例えば、融点)および化学反応性を有し得る。上記方法は、芳香族アミノ酸生合成の共通経路を有する微生物においてエリスロース 4−ホスフェート(E4P)およびホスホエノールピルビン酸(PEP)へと生体触媒による変換を行う能力のある、容易に入手可能な炭素源からの生成物の微生物生合成を含み得る。
【0012】
1つの好ましい炭素源は、D−グルコースである。有利なことには、本発明と関連して使用可能なD−グルコースおよび他の炭素源は、非毒性である。さらに、このような炭素源は、再生可能であり、デンプン、セルロース、ならびにトウモロコシ、サトウキビ、砂糖大根、木材パルプ、および他のバイオマス資源において見いだされる糖に由来する。
【0013】
本発明における種々の工程を促進するために適した宿主微生物は、芳香族アミノ酸生合成の内因性共通経路を有する属から選択され得る。好ましい宿主生物としては、Klebsiella pneumoniaeおよびAcinetobacter calcoaceticusに内因性の選択された遺伝子を発現するように遺伝子操作されたEscherichia coliの変異株が挙げられる。本発明における使用のための1つの好ましいE.coli変異体は、E.coli AB2834であり、これは、酵素シキミ酸デヒドロゲナーゼをコードするaroE遺伝子座における変異に起因して、3−デヒドロシキメート(DHS)(芳香族アミノ酸生合成の共通経路に沿った中間体)をシキミ酸へと変換するのを触媒できない栄養要求性変異体である。
【0014】
芳香族アミノ酸生合成の共通経路は、細菌および植物において、芳香族アミノ酸であるフェニルアラニン、チロシン、およびトリプトファンを生成する。上記共通経路は、分子コリスメートで終了し、これは、その後、3つの別個の最終経路によってフェニルアラニン、チロシン、およびトリプトファンへと変換される。
【0015】
上記共通する芳香族アミノ酸生合成経路の生成効率を増大させるためのアプローチとしては、米国特許第5,168,056号(1992年12月1日発行)、米国特許第5,616,496号(1997年4月1日発行)、および米国特許出願第07/994194号(1992年12月21日出願、現在、放棄されている)(これらすべての開示は、それら全体が本明細書において参考として援用される)に記載されるものが挙げられる。
【0016】
上記遺伝子操作された宿主生物を使用するにあたって、芳香族アミノ酸生合成へと指向される炭素の流れは、上記共通経路に沿って進行して、DHSの上昇した細胞内レベルを生じ得る。DHSは、上記芳香族アミノ酸生合成の共通経路に沿った変異(これは、DHSからコリスメートへの変換を妨げる)に起因して蓄積する。上記DHSは、酵素3−デヒドロシキミ酸脱水酵素(aroZ)のための基質として働き、DHSに対するこの酵素の作用により、プロトカテクエートを生じる。プロトカテクエートは、その後、プロトカテク酸デカルボキシラーゼ(aroY)として公知の別の酵素を介して、カテコールへと変換される。このように形成された上記カテコールは、続いて、酵素カテコール 1,2−ジオキシゲナーゼ(catA)の作用によって、cis,cis−ムコン酸へと変換される。
【0017】
DHSからcis,cis−ムコネートの生合成を触媒する3つの酵素(すなわち、aroZ、aroY、およびcatA)は、適切なプロモーターの制御下でこれら3つの酵素をコードする遺伝子を含む組換えDNAを使用して、宿主細胞において発現され得る。炭素の流れは、それによって、芳香族アミノ酸生合成の経路から離れて、cis,cis−ムコネートを生成する分岐した経路へと押しやられ得る。このように形成されたcis,cis−ムコン酸は、細胞外培地(medium)へと蓄積され得、これは、遠心分離、濾過、もしくは当該分野で公知の他の方法によって上記細胞から分離され得る。上記単離されたcis,cis−ムコン酸は、その後、化学的に水素化されて、アジピン酸を生じ得る。
【0018】
本発明の種々の実施形態において、上記cis,cis−ムコネートが生成された後、これは、引き続いて、cis,trans−ムコネートもしくはtrans,trans−ムコネートに異性化され得る。上記cis,trans−ムコネートもしくはtrans,trans−ムコネートはともに、異なる物理的特性および化学反応性を有し、これらは、cis,cis−ムコネートのものとは異なるかもしくはこれを超える有用性を与え得る。例えば、cis,trans−異性体は、水性培地および/もしくは有機培地においてcis,cis−ムコネートより高い溶解性を有し得、このことは、有利な回収および加工処理を可能にする。さらなる例として、上記trans,trans−異性体は、Diels−Alder反応における反応物として上記cis,cis−異性体を超える特有の有用性を有し得る。
【0019】
一局面において、本発明は、cis,trans−ムコネートを生成するための方法を特徴とする。上記方法は、生体触媒による変換(例えば、上記aroZ、aroY、およびcatA酵素を利用する)を介して再生可能炭素源から生成されたcis,cis−ムコネートを提供する工程、上記cis,cis−ムコネートの実質的にすべてがcis,trans−ムコネートへと異性化される反応条件下で、cis,cis−ムコネートをcis,trans−ムコネートへと異性化する工程、上記cis,trans−ムコネートを分離する工程、および上記分離されたcis,trans−ムコネートを結晶化する工程(例えば、上記プロトン化cis,trans−ムコン酸として)を包含する。
【0020】
別の局面において、本発明は、cis,trans−ムコネートを生成するための方法を特徴とする。上記方法は、生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネートを含む発酵ブロスを提供する工程;上記cis,cis−ムコネートの実質的にすべてがcis,trans−ムコネートへと異性化される反応条件下で、cis,cis−ムコネートをcis,trans−ムコネートへと異性化する工程;上記ブロスから上記cis,trans−ムコネートを分離する工程;および上記cis,trans−ムコネートを結晶化する工程を包含する。
【0021】
なお別の局面において、本発明は、本発明の特徴である方法によって生成されるcis,trans−ムコネートを特徴とする。上記cis,trans−ムコネートは、塩(例えば、ムコン酸ナトリウム、ムコン酸カルシウム、もしくはムコン酸アンモニウムなどの無機塩)として回収され得る。
【0022】
なお別の局面において、本発明は、trans,trans−ムコネートを生成するための方法を特徴とし、上記方法は、上記cis,cis−ムコネートの実質的にすべてがtrans,trans−ムコネートへと異性化される反応条件下で、生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネートをtrans,trans−ムコネートへと異性化する工程を包含する。例えば、上記異性化反応は、貴金属水素化触媒によって、スポンジメタル水素化触媒によって、もしくは骨格水素化触媒(skeletal hydrogenation catalyst)によって触媒され得る。
【0023】
なお別の局面において、本発明は、trans,trans−ムコネートを生成するための方法を特徴とし、上記方法は、上記cis,trans−ムコネートのうちの実質的にすべてがtrans,trans−ムコネートへと異性化される反応条件下で、生体触媒による変換を介して再生可能炭素源から生成されたcis,trans−ムコネートをtrans,trans−ムコネートへと異性化する工程を包含する。例えば、上記異性化反応は、貴金属水素化触媒によって、スポンジメタル水素化触媒によって、もしくは骨格水素化触媒によって触媒され得る。
【0024】
さらに別の局面において、本発明は、本発明が特徴とする方法によって生成されたtrans,trans−ムコネート(例えば、再生可能なtrans,trans−ムコネート)を特徴とする。
【0025】
他の例において、上記局面のうちのいずれか、または本明細書に記載される任意の方法、器具、もしくは組成物は、以下の特徴のうちの1つ以上を含み得る。
【0026】
種々の実施形態において、上記方法は、3−デヒドロシキミ酸脱水酵素(例えば、aroZ)、プロトカテク酸デカルボキシラーゼ(例えば、aroY)およびカテコール 1,2−ジオキシゲナーゼ(例えば、catA)を発現する組換え細胞を、再生可能炭素源を含む培地中で、かつこのような再生可能炭素源が上記細胞の芳香族アミノ酸生合成の共通経路において見いだされた酵素類によってDHSに変換される条件下で、培養する工程を包含し、得られたDHSは、cis,cis−ムコネートへと生体触媒により変換される。
【0027】
上記再生可能炭素源の発酵によるcis,cis−ムコネートの生成は、組換え細胞および細胞外cis,cis−ムコネートを含むブロスを生成し得る。上記生成はまた、上記組換え細胞、細胞砕片、不溶性タンパク質および他の望ましくない固体を、上記ブロスから分離して、上記発酵によって形成されたcis,cis−ムコネートの実質的にすべてもしくはその大部分を含む清澄にした発酵ブロスを与える工程を包含し得る。次いで、上記cis,cis−ムコネートは、上記清澄にした発酵ブロス中においてcis,trans−ムコネートへと異性化され得る。
【0028】
特定の実施形態において、生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネートを含む発酵ブロスは、cis,trans−ムコネートもしくはtrans,trans−ムコネートを生成するために提供され得る。上記発酵ブロスは、3−デヒドロシキミ酸脱水酵素、プロトカテク酸デカルボキシラーゼおよびカテコール 1,2−ジオキシゲナーゼを発現する組換え細胞を含み得る。いくつかの実施形態において、上記発酵ブロスは、容器中で提供され、上記異性化反応は、上記容器中で行われる。上記容器は、発酵容器であり得る。いくつかの例において、3−デヒドロシキミ酸脱水酵素、プロトカテク酸デカルボキシラーゼおよびカテコール1,2−ジオキシゲナーゼを発現する組換え細胞は、上記再生可能炭素源を含む培地中で、かつ上記再生可能炭素源が、上記細胞の芳香族アミノ酸生合成の共通経路における酵素類によって3−デヒドロシキメートへと変換され、かつ、上記3−デヒドロシキメートは、生体触媒によりcis,cis−ムコネートに変換される条件下で、培養され得る。例えば、上記組換え細胞は、上記発酵容器中で培養され得、それによって、上記発酵ブロスを生成することができる。さらに、上記組換え細胞は、所望の通り、上記発酵ブロスから除去され得る。
【0029】
いくつかの実施形態において、上記異性化反応は、酸によって触媒される。上記酸は、無機酸(例えば、鉱酸)もしくは有機酸であり得る。酸は、水和形態もしくは無水形態のいずれかで上記プロセスに適用され得る。一例において、塩の副生成物は、硫酸アンモニウムであり得る。これは、その後、例えば、肥料として使用され得る。上記異性化反応は、約1.5〜約6.5の間(例えば、1.5、1.75、2、2.25、2.5、2.75、3、3.25、3.5、3.75、4、4.25、4.5、4.75、5、5.25、5.5、5.75、6、6.25、6.5)のpHにおいて行われ得る。好ましくは、上記異性化反応は、約3.5〜約4.5の間のpHにおいて行われ得る。
【0030】
特定の実施形態において、上記異性化反応は、約47℃以上の(例えば、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、もしくはより高い)温度で行われる。好ましくは、上記異性化反応は、約60℃以上の温度で行われ得る。上記異性化反応は、8時間以内、7.75時間以内、7.5時間以内、7.25時間以内、7時間以内、6.75時間以内、6.5時間以内、6.25時間以内、6時間以内、5.75時間以内、5.5時間以内、5.25時間以内、5時間以内、4.75時間以内、4.5時間以内、4.25時間以内、4時間以内、3.75時間以内、3.5時間以内、3.25時間以内、3時間以内、2.75時間以内、2.5時間以内、2.25時間以内、2時間以内、1.75時間以内、1.5時間以内、1.25時間以内、1時間以内、0.75時間以内、0.5時間以内、もしくは0.25時間以内で実質的に完了し得る。
【0031】
種々の実施形態において、上記異性化反応は、その反応混合物からcis,trans−ムコネートを実質的に沈殿させることなく進められる。特定の実施形態において、上記異性化反応は、cis,cis−ムコネートをcis,trans−ムコネートへと異性化することをモニターする工程を包含する。いくつかの実施形態において、上記異性化反応は、ほぼ大気圧より高い圧力で行われる。
【0032】
種々の実施形態において、異性化後、上記cis,trans−ムコネートは、上記cis,trans−ムコン酸を沈殿させるのに十分な、さらなる酸性化によって、溶液、培地、ブロス、もしくは発酵ブロスから分離され得る。上記ブロスは、約3.0未満の(例えば、2.9、2.8、2.7、2.6、2.5、2.4、2.3、2.2、2.1、2、もしくはこれより低い)pHへと酸性化され得る。上記ブロスは、約2未満のpHへとさらに酸性化され得る。
【0033】
特定の実施形態において、上記分離する工程は、約37℃未満、約25℃未満、約−4℃未満、もしくは約−20℃未満の温度へと溶液を冷却する工程を包含する。
【0034】
特定の実施形態において、上記分離する工程は、沈殿させられたcis,trans−ムコン酸を分離するために、遠心分離、濾過、もしくは他の物理的プロセスを含む。種々の実施形態において、上記分離する工程は、有機溶媒を使用して、上記cis,trans−ムコネートを上記発酵ブロスから抽出する工程を包含する。上記有機溶媒は、メタノール、エタノール、プロパノール、イソプロパノール、ブタノール、酢酸、アセトニトリル、アセトン、およびテトラヒドロフラン、tert−ブチルメチルエーテル、メチルテトラヒドロフラン、シクロヘキサノンもしくはシクロヘキサノール、またはこれらの混合物のうちの1種以上を含み得る。一実施形態において、上記抽出は、上記cis,trans−ムコン酸の顕著な沈殿なく、約7〜4の間の(例えば、約7、6.75、6.5、6.25、6、5.75、5.5、5.25、5、5.75、5.5、5.25、4)pHにおいて行われ得、酸の自動化された添加を使用して、上記抽出が進むにつれて、この領域に上記pHを維持することを包含し得る。別の実施形態において、上記抽出する工程は、上記有機溶媒によって溶解される、沈殿したcis,trans−ムコン酸の存在下で、約4未満の(例えば、約4、3.75、3.5、3.25、3、2.75、2.5、2.25、2)pHにおいて行われ得る。さらに別の実施形態において、上記抽出する工程は、細胞、細胞砕片、タンパク質、もしくは他の望ましくない物質を、上記発酵ブロスから最初に除去せずに、行われ得る。なお別の実施形態において、上記抽出する工程は、膜によって媒介され得る。
【0035】
特定の実施形態において、上記cis,cis−ムコネートは、上記発酵ブロスから最初に除去され得、次いで、上記異性化、分離、および精製工程に供され得る。このような除去は、抽出、沈殿、イオン交換クロマトグラフィー、選択的膜分離、電気透析、もしくは当該分野で公知の他の方法によって達成され得る。
【0036】
いくつかの実施形態において、上記cis,trans−ムコン酸は、有機溶媒を使用する結晶化によって精製される。上記有機溶媒は、メタノール、エタノール、プロパノール、イソプロパノール、ブタノール、酢酸、アセトニトリル、アセトン、およびテトラヒドロフランのうちの1種以上を含み得る。
【0037】
いくつかの実施形態において、上記結晶化は、上記発酵ブロスからの回収後に、沈殿したcis,trans−ムコン酸を乾燥させることなく行われ得る。特定の実施形態において、上記結晶化は、分離されたcis,trans−ムコン酸から望ましくない塩を除去する工程を包含する。種々の実施形態において、上記結晶化は、cis,trans−ムコン酸の第1の収穫物を集めた後に、上記結晶化培池を濃縮する工程、およびcis,trans−ムコン酸の第2の収穫物を上記濃縮した培池から集める工程を包含する。
【0038】
特定の実施形態において、trans,trans−ムコネートを生成するための方法は、cis,trans−ムコネートの生成、上記cis,trans−ムコネートのうちの少なくとも約65%をtrans,trans−ムコネートへと異性化する工程、および上記trans,trans−ムコネートを単離する工程を包含する。上記方法は、上記cis,trans−ムコネートのうちの少なくとも約65%、約66%、約67%、約68%、約69%、約70%、約71%、約72%、約73%、約74%、約75%、約76%、約77%、約78%、約79%、約80%、約81%、約82%、約83%、約84%、約85%、約86%、約87%、約88%、約89%、約90%、約91%、約92%、約93%、約94%、約95%、約96%、約97%、約98%、約99%、もしくは約100%を、trans,trans−ムコネートへと異性化する工程を包含し得る。あるいは、trans,trans−ムコネートは、適切な条件(例えば、pH、温度、触媒など)下でcis,cis−ムコネートから生成もしくは異性化され得る。
【0039】
種々の実施形態において、上記異性化反応は、I2によって、貴金属水素化触媒によって、スポンジメタル水素化触媒によって、もしくは骨格水素化触媒によって、触媒される。上記貴金属は、水素化触媒として機能する任意の貴金属(例えば、白金、パラジウムなど)であり得る。上記スポンジメタルもしくは骨格触媒は、ニッケル−アルミニウム合金(例えば、W.R.Grace and Companyから入手可能なRANEY(登録商標)ニッケル触媒)であり得る。上記金属触媒は、不均一触媒(例えば、粒子)もしくは担持触媒(supported catalyst)(例えば、シリカ、アルミナ、炭素などのような支持体上)の形態で存在し得る。
【0040】
いくつかの実施形態において、生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネートを提供する工程は、酵素3−デヒドロシキミ酸脱水酵素およびプロトカテク酸デカルボキシラーゼを発現するKlebsiella pneumoniae、ならびに酵素カテコール 1,2−ジオキシゲナーゼを発現するAcinetobacter calcoaceticusに由来する異種構造遺伝子で形質転換された細菌細胞を使用する。ここで上記細菌細胞の培養物は、約88時間以内に、1.38M グルコースを少なくとも約0.42M cis,cis−ムコン酸へと変換するに少なくとも十分な速度で、グルコースをcis,cis−ムコン酸へと生体触媒により変換する。上記細菌細胞形質転換体は、酵素3−デオキシ−D−アラビノ−ヘプツロソン酸 7−リン酸シンターゼおよび3−デヒドロキナ酸シンターゼを発現する異種DNA配列を含み得る。上記細菌細胞形質転換体は、酵素トランスケトラーゼ、3−デオキシ−D−アラビノ−ヘプツロソン酸 7−リン酸シンターゼおよび3−デヒドロキナ酸シンターゼを発現する異種DNA配列を含み得る。上記細菌細胞は、3−デヒドロシキメートをコリスメートに変換することをブロックする、芳香族アミノ酸生合成の共通経路において変異を有する変異細胞系から選択され得る。上記細菌細胞は、3−デヒドロシキメートをコリスメートに変換することをブロックする、上記芳香族アミノ酸生合成の共通経路において変異を有する変異細胞系から選択される。
【0041】
特定の実施形態において、生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネートを提供する工程は、酵素種3−デヒドロシキミ酸脱水酵素およびプロトカテク酸デカルボキシラーゼを発現するKlebsiella pneumoniaeに由来する構造遺伝子、ならびに酵素種カテコール 1,2−ジオキシゲナーゼを発現するAcinetobacter calcoaceticusに由来する構造遺伝子で形質転換された細菌細胞を、上記細胞の芳香族アミノ酸生合成の共通経路における上記酵素類によって3−デヒドロシキメートへと変換される炭素源を含む培地中で培養して、少なくとも約0.95ミリモル/リットル/時間の速度で、3−デヒドロシキメートの生体触媒による変換によって、cis,cis−ムコン酸を生成する工程を包含する。他の実施形態において、cis,cis−ムコン酸は、少なくとも約0.97ミリモル/リットル/時間以上、1.0ミリモル/リットル/時間以上、1.2ミリモル/リットル/時間以上、1.4ミリモル/リットル/時間以上、1.6ミリモル/リットル/時間以上、1.8ミリモル/リットル/時間以上、2.0ミリモル/リットル/時間以上の速度で生成される。
【0042】
種々の実施形態において、生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネートを提供する工程は、3−デヒドロシキミ酸脱水酵素、プロトカテク酸デカルボキシラーゼ、カテコール 1,2−ジオキシゲナーゼ、トランスケトラーゼ、3−デオキシ−D−アラビノ−ヘプツロソン酸 7−リン酸シンターゼ、および3−デヒドロキナ酸シンターゼをコードする異種構造遺伝子を発現する形質転換された細菌細胞を、上記細胞の芳香族アミノ酸生合成の共通経路における上記酵素類によって3−デヒドロシキメートに変換される炭素源を含む培地中で培養して、3−デヒドロシキメートの生体触媒による変換によって、少なくとも約0.95ミリモル/リットル/時間の速度で、cis,cis−ムコン酸を生成する工程を包含する。他の実施形態において、cis,cis−ムコン酸は、少なくとも約0.97ミリモル/リットル/時間以上、1.0ミリモル/リットル/時間以上、1.2ミリモル/リットル/時間以上、1.4ミリモル/リットル/時間以上、1.6ミリモル/リットル/時間以上、1.8ミリモル/リットル/時間以上、2.0ミリモル/リットル/時間以上の速度で生成される。
【0043】
いくつかの実施形態において、生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネートを提供する工程は、酵素種3−デヒドロシキミ酸脱水酵素およびプロトカテク酸デカルボキシラーゼを発現するKlebsiella pneumoniaeに由来する構造遺伝子、ならびに酵素カテコール 1,2−ジオキシゲナーゼを発現するAcinetobacter calcoaceticusに由来する構造遺伝子で形質転換された細菌細胞を、炭素源を含む培地中で、上記炭素源が、少なくとも約0.95ミリモル/リットル/時間の速度でcis,cis−ムコン酸へと生体触媒により変換される条件下で培養する工程を包含する。他の実施形態において、cis,cis−ムコン酸は、少なくとも約0.97ミリモル/リットル/時間以上、1.0ミリモル/リットル/時間以上、1.2ミリモル/リットル/時間以上、1.4ミリモル/リットル/時間以上、1.6ミリモル/リットル/時間以上、1.8ミリモル/リットル/時間以上、2.0ミリモル/リットル/時間以上の速度で生成される。
【0044】
本発明の他の局面および利点は、以下の図面および説明から明らかになり、これらのうちのすべては、本発明の原理を、例示によってのみ示す。
【図面の簡単な説明】
【0045】
上記に記載される発明の利点は、さらなる利点とともに、添付の図面とともに考慮される以下の説明に言及することによって、よりよく理解され得る。図面は、必ずしも一定の大きさに比例しているわけではなく、代わりに、一般に、本発明の原理を図示する際に強調される。
【図1−1】図1は、芳香族アミノ酸生合成の共通経路およびcis,cis−ムコン酸を3−デヒドロシキメートから合成する分岐経路を示す。
【図1−2】図1は、芳香族アミノ酸生合成の共通経路およびcis,cis−ムコン酸を3−デヒドロシキメートから合成する分岐経路を示す。
【図2】図2は、プラスミドp2−47のプラスミドマップを示し、プラスミドpKD8.243Aが、プラスミドp2−47、pSU1−31、およびpSUaroZY 157−27からどのように生成され得るかを図示する。
【図3】図3は、pKD8.292のプラスミドマップを示し、プラスミドpKD8.292が、プラスミドpIB1345およびpCL1920からどのように生成され得るかを図示する。
【図4】図4Aおよび図4Bは、異なる培地中での、cis,trans−ムコン酸の1H NMRスペクトルの例を示す。
【図5】図5は、pH7におけるムコネート異性化反応についての1H NMR図形の例を示す。
【図6】図6は、pH4におけるムコネート異性化反応についての1H NMR図形の例を示す。
【図7】図7は、pH7におけるムコネート異性化反応についてのHPLC図形の例を示す。
【図8】図8は、pH4におけるムコネート異性化反応についてのHPLC図形の例を示す。
【図9】図9は、pH7におけるムコネート異性化反応についての経時変化の例を示す。
【図10】図10は、pH4におけるムコネート異性化反応についての経時変化の例を示す。
【図11】図11は、ムコン酸のバッチ培養生成を示す。
【発明を実施するための形態】
【0046】
本発明は、芳香族アミノ酸生合成の共通経路(例えば、上記中間体DHSを介して機能するもの)を有し、酵素aroZ、aroY、およびcatAを発現する能力を有する宿主細胞によって使用され得る発酵可能な炭素源から、cis,trans−ムコン酸およびtrans,trans−ムコン酸を生成するための方法を包含する。1つの好ましい実施形態において、上記方法は、cis,cis−ムコン酸を生成するための、発酵可能な炭素源の存在下で、上記宿主細胞を培養する工程、および上記cis,cis−ムコン酸を異性化して、cis,trans−ムコン酸もしくはtrans,trans−ムコン酸を生成する工程を包含する。
【0047】
発酵可能な炭素源は、D−エリスロース 4−ホスフェート(E4P)およびホスホエノールピルベート(PEP)(芳香族アミノ酸生合成の共通経路への2つの前駆化合物)へと生体触媒により変換され得る、本質的に任意の炭素源を含み得る。適切な炭素源としては、バイオマス由来の源、もしくは再生可能な源(例えば、デンプン、セルロース、およびグルコース、ペントース、およびフルクトースのような糖部分、ならびに微生物代謝を支持し得る他の炭素源(例えば、一酸化炭素)が挙げられるが、これらに限定されない。一実施形態において、D−グルコースは、上記バイオマス由来炭素源として使用され得る。
【0048】
本発明において使用するのに適した宿主細胞は、所望の芳香族化合物の生合成による生成のために利用され得る属のメンバーを含む。いくつかの実施形態において、このような宿主細胞は、所望の芳香族化合物の産業スケールの生合成による生成に適している。特に、適切な宿主細胞は、少なくともDHSの生成に対して機能的である芳香族アミノ酸生合成の内因性の共通経路を有し得る。共通する芳香族経路は、広く種々の微生物において内因性であり、種々の芳香族化合物の生成のために使用され得る。米国特許第5,168,056号および同第5,616,496号(その両方の開示は、それら全体が本明細書に参考として援用される)において記載されるように、例えば、微生物の芳香族アミノ酸生合成経路は、本発明において利用され得る。
【0049】
図1は、芳香族アミノ酸生合成の共通経路およびこの経路において多くの中間体とともにE4PおよびPEPからコリスミ酸をもたらす上記共通する芳香族経路を介してcis,cis−ムコン酸を3−デヒドロシキメートから合成する分岐経路を示す。E4Pの利用性は、tkt遺伝子によってコードされるペントースリン酸経路酵素であるトランスケトラーゼによって増大され得る。上記経路における中間体としては、3−デオキシ−D−アラビノ−ヘプツロソン酸 7−ホスフェート(DAHP)、3−デヒドロキネート(DHQ)、3−デヒドロシキメート(DHS)、シキミ酸、シキメート3−ホスフェート(S3P)、および5−エノールピルビルシキメート(enolpyruvoylshikimate)−3−ホスフェート(EPSP)が挙げられる。上記共通経路における酵素としては、DAHPシンターゼ(aroF)、DHQシンターゼ(aroB)、DHQデヒドラターゼ(aroD)、シキミ酸デヒドロゲナーゼ(aroE)、シキミ酸キナーゼ(aroL、aroK)、EPSPシンターゼ(aroA)およびコリスミ酸シンターゼ(aroC)が挙げられる。
【0050】
このタイプの共通経路を含む宿主細胞としては、Escherichia属、Klebsiella属、Corynebacterium属、Brevibacterium属、Arthrobacter属、Bacillus属、Pseudomonas属、Streptomyces属、Staphylococcus属、およびSerratia属に属する原核生物が挙げられる。真核生物宿主細胞もまた、例えば、Saccharomyces属もしくはSchizosaccharomyces属の酵母に関して利用され得る。
【0051】
より具体的には、原核生物宿主細胞は、Escherichia coli、Klebsiella pneumonia、Corynebacterium glutamicum、Corynebacterium herculis、Brevibacterium divaricatum、Brevibacterium lactofermentum、Brevibacterium flavum、Bacillus brevis、Bacillus cereus、Bacillus circulans、Bacillus coagulans、Bacillus lichenformis、Bacillus megaterium、Bacillus mesentericus、Bacillus pumilis、Bacillus subtilis、Pseudomonas aeruginosa、Pseudomonas angulata、Pseudomonas fluorescens、Pseudomonas tabaci、Streptomyces aureofaciens、Streptomyces avermitilis、Streptomyces coelicolor、Streptomyces griseus、Streptomyces kasugensis、Streptomyces lavendulae、Streptomyces lipmanii、Streptomyces lividans、Staphylococcus epidermis、Staphylococcus saprophyticus、およびSerratia marcescensを含む種に由来し得る。真核生物宿主細胞の例としては、Saccharomyces cerevisiaeおよびSaccharomyces carlsbergensisが挙げられる。
【0052】
宿主細胞は、DHSを分岐点分子であるコリスメートへと変換するのをブロックする変異を有する栄養要求性変異細胞系を含み得る。このような変異体は、シキミ酸デヒドロゲナーゼ、シキミ酸キナーゼ、EPSPシンターゼおよびコリスミ酸シンターゼをコードする遺伝子のうちの1種以上における変異に起因して、3−デヒドロシキメート(DHS)をコリスメートへ変換するのを触媒できず、そしてこのようにして、上昇した細胞内レベルのDHSを蓄積する。このような変異細胞系の例としては、Escherichia coli株AB2834、AB2829およびAB2849が挙げられる。
【0053】
E.coli AB2834は、シキミ酸デヒドロゲナーゼをコードするaroE遺伝子座における変異に起因して、3−デヒドロシキメート(DHS)をシキミ酸へ変換することを触媒できない。E.coli AB2834の使用は、芳香族アミノ酸生合成へ指向される炭素の流れが、DHSを超えて進行しないことを確実にし得る。同様に、E.coli AB2829(これは、EPSPシンターゼをコードするaroA遺伝子座における変異に起因して、シキメート 3−ホスフェート(S3P)を5−エノールピルビルシキメート(enoipyruvylshikimate)−3−ホスフェート(EPSP)へ変換することを触媒できない)およびE.coli AB2849(これは、コリスミ酸シンターゼをコードするaroC遺伝子座における変異に起因して、EPSPをコリスミ酸へ変換することを触媒できない)はまた、増大した細胞内レベルのDHSを生じる。
【0054】
宿主細胞は、細胞内DHSが、カテコールへの生体触媒による変換の基質として使用され得るように、形質転換され得、そのカテコールは、その後、ムコン酸へと変換され得る。例えば、宿主細胞は、組換えDNAで形質転換されて、DHSが生成された後に、炭素の流れを芳香族アミノ酸生合成の共通経路から離れて、ムコン酸を生成するための分岐経路へと押しやり得る。
【0055】
図1に示されるように、上記分岐経路における中間体は、プロトカテクエート、カテコール、およびcis,cis−ムコン酸である。DHSをプロトカテクエートに生体触媒により変換することを担う酵素は、図1において「aroZ」と表示される酵素3−デヒドロシキミ酸脱水酵素である。カテコールを形成するためのプロトカテクエートの脱炭酸を担う酵素は、図1において「aroY」と表示されるプロトカテク酸デカルボキシラーゼである。最後に、カテコールの酸化を触媒してcis,cis−ムコン酸を生成する酵素は、図1において「catA」と表示されるカテコール 1,2−ジオキシゲナーゼである。標準的表記法に従って、これら酵素の発現のための遺伝子は、斜体を使用して示され、よって、それぞれ、aroZ、aroY、およびcatAである。上記cis,cis−ムコン酸は、その後、異性化され得る(示されない)。本発明の一実施形態において、宿主細胞は、上記遺伝子aroZ、aroY、およびcatAの構成的発現を示し得る。別の実施形態において、宿主細胞は、上記遺伝子aroZ、aroYおよびcatAのうちのいずれか1種以上;またはこれらのうちの任意の2種の組み合わせの構成的発現を示し得る。さらに別の実施形態において、宿主細胞は、aroZ、aroYおよびcatAのうちのいずれの構成的発現も示さない場合がある。
【0056】
上記酵素3−デヒドロシキミ酸脱水酵素およびプロトカテク酸デカルボキシラーゼは、微生物(例えば、Neurospora、Aspergillus、Acinetobacter、Klebsiella、およびPseudomonas)が芳香族類(aromatics)(ベンゾエートおよびp−ヒドロキシベンゾエート)ならびにヒドロ芳香族類(hydroaromatics)(シキメートおよびキネート)を増殖のための唯一の炭素源として使用することを可能にするオルト切断経路から補充される。DHSデヒドラターゼは、キナ酸およびシキミ酸の微生物異化作用において重要な役割を果たす。Patelは、Klebsiella aerogenesによるp−ヒドロキシベンゾエートの異化作用の間に、プロトカテクエートのカテコールへの変換を触媒するように、プロトカテク酸デカルボキシラーゼを処方した。近年、Ornstonは、Patel株(現在、Enterobacter aerogenesといわれる)の再検査[(a)Grant,D.J.W.;Patel,J.C.Antonie van Leewenhoek 1969,35,325. (b)Grant,D.J.W.Antonie van Leewenhoek 1970,36,161]により、プロトカテク酸デカルボキシラーゼが、p−ヒドロキシベンゾエートの異化作用において代謝的に重要ではなかったと結論づけた[Doten,R.C.;Ornston,N. J.Bacteriol.1987,169,5827]。
【0057】
上記分岐経路へと炭素の流れを指向するように上記宿主細胞を形質転換するための機構は、3−デヒドロシキミ酸脱水酵素、プロトカテク酸デカルボキシラーゼ、およびカテコール 1,2−ジオキシゲナーゼをコードする発現可能な配列を含む遺伝的エレメントの挿入を包含し得る。利用される正確な機構にかかわらず、これら酵素活性の発現は、上記宿主細胞へと組換え遺伝的エレメントの移入によって行われるかもしくは媒介されることが企図される。本明細書で定義される場合、遺伝的エレメントは、生成物(例えば、タンパク質、アポタンパク質、もしくはアンチセンスRNA)についての発現可能なコード配列を有する核酸(一般に、DNAおよびRNA)を含み、これらは、経路酵素機能を発揮し得るかもしくは制御し得る。上記発現される生成物は、酵素として機能し得るか、酵素活性を抑制もしくは抑制解除し得るか、または酵素の発現を制御し得る。これら発現可能な配列をコードする核酸は、染色体性(例えば、宿主細胞染色体へ挿入もしくは組み込まれる)もしくは染色体外性(例えば、プラスミド、コスミドなどによって運ばれる)のいずれかであり得る。
【0058】
本発明の遺伝的エレメントは、プラスミド、コスミド、ファージ、酵母人工染色体もしくは上記遺伝的エレメントの、宿主細胞への移入を媒介する他のベクターによって、宿主細胞に導入され得る。これらベクターは、上記ベクターの複製を制御するcis作用制御エレメントおよび上記ベクターによって運ばれる遺伝的エレメントとともに、複製起点を含み得る。選択マーカーは、上記遺伝的エレメントが導入された宿主細胞の同定を補助するために、上記ベクターに存在し得る。例えば、選択マーカーは、特定の抗生物質(例えば、テトラサイクリン、アンピシリン、クロラムフェニコール、カナマイシン、もしくはネオマイシン)に対する耐性を付与する遺伝子であり得る。
【0059】
遺伝的エレメントを宿主細胞に導入することは、遺伝的エレメントが挿入される染色体外マルチコピープラスミドベクターを利用し得る。プラスミドが媒介する上記遺伝的エレメントの宿主細胞への導入は、制限酵素でのプラスミドの最初の切断、続いて、本発明に従う上記プラスミドおよび遺伝的エレメントの連結を包含する。連結された組換えプラスミドの再環化の際に、プラスミド移入のための形質導入もしくは他の機構(例えば、エレクトロポレーション、マイクロインジェクションなど)は、上記プラスミドを上記宿主細胞に移入するために利用される。遺伝的エレメントを上記宿主細胞に挿入するために適したプラスミドとしては、pBR322およびその誘導体(例えば、pAT153ベクター、pXf3ベクター、pBR325ベクター、pBr327ベクター、pUCベクター)、pACYCおよびその誘導体、pSC101およびその誘導体、ならびにColE1が挙げられるが、これらに限定されない。さらに、コスミドベクター(例えば、pLAFR3)はまた、遺伝的エレメントを宿主細胞に挿入するために適している。プラスミド構築物の例としては、p2−47、pKD8.243A、pKD8.243B、およびpSUaroZY157−27が挙げられるが、これらに限定されない。これらは、3−デヒドロシキミ酸脱水酵素およびプロトカテク酸デカルボキシラーゼをそれぞれコードするKlebsiella pneumoniaeから単離されたaroZおよびaroY遺伝子座を有する。プラスミド構築物のさらなる例としては、カテコール 1,2−ジオキシゲナーゼをコードするAcinetobacter calcoaceticus catAに対して内因性の遺伝的フラグメントを有するpKD8.292が挙げられる。
【0060】
宿主細胞を形質転換するための方法はまた、芳香族アミノ酸生合成の共通経路への炭素の拘束(commitment)を増大させる酵素をコードする遺伝子の挿入を包含し得る。遺伝子の発現は、主に、それ自体のプロモーターによって誘導されるが、任意選択の(optional)発現制御配列(例えば、リプレッサー、およびエンハンサー)を含む他の遺伝的エレメントは、タンパク質、アポタンパク質、もしくはアンチセンスRNAのコード配列の発現もしくは抑制解除を制御するために含められ得る。さらに、組換えDNA構築物が生成され得、それによって、上記遺伝子の天然のプロモーターは、上記遺伝子生成物の発現を増大させるために、代替のプロモーターで置き換えられる。プロモーターは、構成性もしくは誘導性のいずれかであり得る。構成性プロモーターは、細胞の寿命の間に一定の速度で遺伝子の転写を制御するのに対して、誘導性プロモーターの活性は、特定の誘導因子の存在(もしくは非存在)によって決定される場合に変動する。例えば、制御配列は、上記宿主細胞ゲノム中に既にコードされる選択された酵素の過剰発現を促進するために、野生型宿主細胞に挿入され得るか、または代わりに、染色体外でコードされた酵素の合成を制御するために使用され得る。
【0061】
DHSの過剰生成を促進する制御配列が使用され得る。先に示されるように、DHSは、ペントースリン酸経路酵素であるトランスケトラーゼ(tktによってコードされる)とともに、チロシン感受性アイソザイム3−デオキシ−D−アラビノヘプツロソン酸 7−リン酸(DAHP)シンターゼ(aroFによってコードされる)および3−デヒドロキノ酸(DHQ)シンターゼ(aroBによってコードされる)の逐次的触媒活性によって、上記共通経路において合成される。これら生合成酵素の発現は、D−グルコースをDHSへ変換するのを増大させるために増幅され得る。上記共通経路の第1の酵素であるDAHPシンターゼのインビボ触媒活性を増大させると、芳香族生合成へ向かうD−グルコース等価物の流れが増大する。しかし、DAHPシンターゼ触媒活性のレベルは、芳香族生合成に関係するD−グルコースのパーセンテージにおいてさらなる改善は達成されないレベル以上に達する。芳香族アミノ酸生合成のこの制限レベルにおいて、上記ペントースリン酸経路酵素であるトランスケトラーゼの上記触媒レベルの増幅は、上記経路へと吸い上げられるD−グルコースのパーセンテージのかなりの増大を達成する。
【0062】
増幅されたトランスケトラーゼ活性は、D−エリスロース 4−ホスフェート濃度を増大させ得る。DAHPシンターゼの2つの基質のうちの一方として、制限されたD−エリスロース 4−ホスフェートの利用可能性は、DAHPシンターゼ触媒活性を制限し得る。従って、DAHPシンターゼ、DHQシンターゼおよびDHQデヒドラターゼの触媒活性を増幅するための1つの方法は、これらの酵素をコードする組換えDNA配列で微生物触媒を形質転換することによってその酵素種を過剰発現することである。
【0063】
DAHPシンターゼおよびトランスケトラーゼの増幅された発現は、この経路へ向かう通常の炭素の流れが超過した状態にある、芳香族アミノ酸生合成の共通経路へ向かう炭素の流れのサージ(surge)を作り得る。共通の芳香族アミノ酸経路の個々の酵素によって触媒される、基質の生成物への変換の個々の速度が、DAHPシンターゼの速度より低い場合、これら律速酵素の基質は、細胞内に蓄積し得る。
【0064】
微生物(例えば、E.coli)は、頻繁に、外部環境(例えば、バルク発酵培地)へこのような基質を輸送する(export)ことによって、蓄積された基質に対処する。このことは、上記共通経路を介する炭素の流れの喪失を生じる。なぜなら、輸送された基質は、代表的には、微生物の代謝に影響を受けない。DHQシンターゼは、律速共通経路酵素の例である。DHQシンターゼの増幅された発現は、この酵素の律速特徴を除去し、DAHPおよびその非リン酸化アナログであるDAHの蓄積を妨げる。DHQデヒドラターゼは、律速ではない。従って、aroFがコードするDAHPシンターゼ、tktがコードするトランスケトラーゼおよびaroBがコードするDHQシンターゼの増幅された発現は、DHSの生成を増大させ、上記DHSは、DHSデヒドラターゼおよびプロトカテク酸デカルボキシラーゼの存在下で、カテコールに変換され、上記カテコールは、その後、cis,cis−ムコン酸へと生体触媒により変換され、上記cis,cis−ムコン酸は、その後、異性化され得る。
【0065】
上記炭素源とDHSとの間の共通経路に沿った炭素の流れの効率を促進し得る1つのプラスミドは、プラスミドpKD136であり、これは、上記aroF、tktおよびaroB遺伝子をコードする。プラスミドpKD136は、DAHPシンターゼ(aroFによってコードされる)およびトランスケトラーゼ(tktによってコードされる)の増幅された発現に起因して、芳香族生合成への炭素の流れのサージを誘導する。次いで、炭素の流れのこのサージは、DHQシンターゼ(aroBによってコードされる)の増幅された発現に起因して、pKD136によるDHS合成へとそのまま送達される。
【0066】
従って、本発明の好ましい実施形態として、DHSデヒドラターゼ、プロトカテク酸デカルボキシラーゼ、およびカテコール 1,2−ジオキシゲナーゼをコードするEscherichia coli発現遺伝子の異種由来の株を構築し、D−グルコースをcis,cis−ムコン酸へ生体触媒により変換することを可能にした。D−グルコースの、DHSへの効率的変換を、上記宿主細胞をpKD136で形質転換した際に達成した。次いで、上記株E.coli AB2834/pKD136を、プラスミドpKD8.243AおよびpKD8.292で形質転換した。結果は、酵素3−デヒドロシキミ酸脱水酵素(aroZ)、プロトカテク酸デカルボキシラーゼ(aroY)およびカテコール 1,2−ジオキシゲナーゼ(catA)を発現するE.coli AB2834/pKD136/pKD8.243A/pKD8.292であった。この細菌細胞系は、アメリカンタイプカルチャーコレクション(12301 Parklawn Drive,Rockville MD 20852)に、1995年8月1日に寄託し、アクセッション番号69875を割り当てられた。
【0067】
別の実施形態において、E.coli AB2834/pKD136は、E.coli AB2834/pKD136/p2−47/pKD8.292を生成するために、プラスミドp2−47およびpKD8.292で形質転換される。別の実施形態において、E.coli AB2834/pKD136は、E.coli AB2834/pKD136/p2−47/pKD8.292を生成するために、プラスミドpKD8.243BおよびpKD8.292で形質転換される。これら異種宿主細胞系の各々は、D−グルコースをcis,cis−ムコン酸に変換することを触媒する。合成されたcis,cis−ムコン酸は、細胞外に蓄積し、上記細胞から分離され得る。その後、上記cis,cis−ムコン酸は、cis,trans−ムコン酸へと異性化され得、所望される場合、さらにtrans,trans−ムコン酸へと異性化され得る。
【0068】
本発明は、従って、芳香族アミノ酸生合成の内因性共通経路を有する宿主細胞の形質転換体に関する。上記形質転換体は、3−デヒドロシキミ酸脱水酵素、プロトカテク酸デカルボキシラーゼ、およびカテコール 1,2−ジオキシゲナーゼをコードする異種遺伝子の構成的発現によって特徴付けられる。一実施形態において、上記細胞形質転換体は、酵素トランスケトラーゼ、DAHPシンターゼ、およびDHQシンターゼをコードする発現可能な組換えDNA配列でさらに形質転換される。別の実施形態において、上記宿主細胞は、3−デヒドロシキメートをコリスメートに変換するのをブロックするアミノ酸生合成の共通経路における変異を有する、変異を含む変異細胞系の群から選択される。なお別の実施形態において、3−デヒドロシキミ酸脱水酵素およびプロトカテク酸デカルボキシラーゼをコードする遺伝子は、Klebsiella pneumoniaeに対して内因性である。さらなる実施形態において、カテコール 1,2−ジオキシゲナーゼをコードする異種遺伝子は、Acinetobacter calcoaceticusに対して内因性である。
【0069】
(再生可能なムコネート)
再生可能な、生物学的に得られる炭素源から生成されるムコン酸は、植物によって組み込まれた大気の二酸化炭素に由来する炭素から構成される(例えば、グルコース、スクロース、グリセリン、もしくは植物油のような炭素源に由来する)。従って、このようなムコン酸は、それらの分子構造において、化石燃料ベースもしくは石油ベースの炭素ではなく、再生可能な炭素を含む。よって、この特許の主題である上記生合成によるムコネート、および関連する派生生成物は、従来法によって生成されるムコネートより小さな炭素痕跡(footprint)および関連生成物を有する。なぜなら、それらは、化石燃料を枯渇させないか、石油が確保され、それらは、炭素サイクルにおける炭素量を増大させない(例えば、ライフサイクル分析は、世界的な炭素バランスに対して正味の炭素増大を示さない)からである。
【0070】
上記生合成によるムコネートおよび関連生成物は、当該分野で公知の方法(例えば、二重炭素同位体フィンガープリント法(dual carbon−isotopic finger printing))によって、化石燃料もしくは石油化学炭素源から生成されるムコネートおよび関連生成物から区別され得る。この方法は、化学的に同一の材料を別の方法で区別し得、14Cと13Cとの異性体比を使用して、供給源によって(すなわち、非生物学的に対して生物学的)上記材料中の炭素原子を区別する。上記炭素同位体14Cは、不安定であり、5730年の半減期を有する。安定な13C同位体に対する上記不安定な14C同位体の相対量を測定することは、化石(長期間活動をとめた(long dead))供給材料と生物圏の(生きている、よって、再生可能な)供給材料との間の標本炭素を区別することを可能にする(Currie,L.A. 「Source Apportionment of Atmospheric Particles」,Characterization of Environmental Particles, J.Buffle and H.P.van Leeuwen,Eds., 1 of Vol. I of the IUPAC Environmental Analytical Chemistry Series(Lewis Publishers,Inc)(1992)3−74を参照のこと)。放射性炭素の年代測定における基本的仮定は、大気中の14C濃度の定常性が、生きた生物における14Cの定常性をもたらすということである。
【0071】
単離されたサンプルを扱う場合、サンプルの年代は、関係性t=(−5730/0.693)ln(A/Ao)によってほぼ導き出され得る。ここでt=年代であり、5730年は、上記不安定な14C同位体の半減期であり、AおよびAoは、それぞれ、サンプルおよび現代の標準物質の特定の14C活性である(Hsieh,Y.,Soil ScL Soc.Am J.,56,460,(1992))。しかし、1950年以来の大気圏核実験および1850年以来の化石燃料の燃焼が原因で、14Cは、第2の地球化学的時間の特徴を獲得した。大気中CO2中のその濃度、そして従って、生きている生物圏における濃度は、1960年代半ばには、核実験のピーク時にほぼ2倍になった。上記濃度は、それ以来、概算の緩和半減期(relaxation half−life)約7〜10年で、定常状態宇宙線による(大気中)ベースライン同位体率(14C/12C)約1.2×10−12へと徐々に戻った。(この後者の半減期は、同位体半減期とは区別されなければならない(すなわち、核時代の始まり以来の大気中および生物圏の14Cの変動を追跡するために、詳細な大気核投入/崩壊関数(atmospheric nuclear input/decay function)を使用しなければならない)。それは、近年の生物圏炭素の年ごとの年代測定を約束するこの後者の生物圏14C時間の特徴である。14Cは、加速器質量分析(AMS)によって測定され得、結果は、現代の炭素に対する割合の単位(fM)で与えられる。fMは、米国国立標準技術研究所(National Institute of Standards and Technology)(NIST)の標準参照物質(Standard Reference Materials)(SRMs) 4990Bおよび4990C(それぞれ、シュウ酸標準物質HOxIおよびHOxIIとして公知)によって定義される。上記基本定義は、HOxIの14C/12C同位体比の0.95倍(AD 1950年を基準として)に関連する。現在の生きている生物圏(植物材料)では、fMは約1.1である。
【0072】
安定な炭素同位体である13Cおよび12Cの比は、供給源の識別および配分(apportionment)に対する補完の経路を提供する。所定の生体供給源の材料における13C/12C比は、上記二酸化炭素が固定され、正確な代謝経路をも反映するときには、大気中二酸化炭素における13C/12C比の結果である。局部的なバリエーションもまた存在する。石油、C3植物(広葉樹)、C4植物(禾本(the grasses))、および海洋カーボネート(marine carbonate)はすべて、13C/12Cおよび対応するδ 13C値において顕著な差異を示す。さらに、C3植物およびC4植物の脂質物質は、その代謝経路の結果として、同じ植物の炭水化物成分に由来する物質とは異なって分析される。測定の正確さの範囲内で、13Cは、同位体分別効果に起因して大きなバリエーションを示し、本発明に関してその最も顕著なものは、光合成機構である。植物における炭素同位体比が異なる大きな原因は、上記植物における光合成炭素代謝の経路、特に、第1のカルボキシル化(例えば、大気中CO2の最初の固定)の間に起こる反応の差異と密接に関連する。草木の2つの大きなクラスは、C3(もしくはカルビン−ベンソン)光合成回路を組み込むものと、C4(もしくはハッチ−スラック)光合成回路を組み込むものである。C3植物(例えば、堅木および針葉樹)は、温暖気候帯域において優勢である。C3植物においては、第1のCO2固定もしくはカルボキシル化反応は、酵素リブロース−1,5−二リン酸カルボキシラーゼを必要とし、第1の安定な生成物は、3個の炭素の化合物である。他方で、C4植物は、熱帯地方の禾本、トウモロコシおよびサトウキビのような植物を含む。C4植物において、別の酵素であるホスホエノール−ピルビン酸カルボキシラーゼを必要とするさらなるカルボキシル化反応が、第1のカルボキシル化反応である。その第1の安定な炭素化合物は、4個の炭素の酸であり、これは、その後、脱炭酸される。このようにして放出されたCO2は、上記C3回路によって再固定される。
【0073】
C4植物およびC3植物はともに、ある13C/12C同位体比の範囲を示すが、代表的な値は、約−10〜−14‰(C4)および−21〜−26‰(C3)である(Weberら,J.Agric.Food Chem.,45,2942(1997))。石炭および石油は、一般に、この後者の範囲に入る。上記13C測定スケールは、本来は、pee deeベレムナイト(PDB)石灰岩によって設定されるゼロによって定義され、ここで値は、この材料からのppt偏差(parts per thousand deviations)単位で与えられる。上記δ13C値は、ppt(パーミル)単位にあり、‰と省略され、以下のように計算される:
δ13C≡(13C/12C)サンプル−(13C/12C)標準物質/(13C/12C)標準物質×1000‰
上記PDB標準物質(RM)は枯渇してしまったので、一連の代替のRMが、IAEA、USGS、NIST、および他の選択された国際同位体実験機関と協同して開発された。PDBからの‰偏差の表示法が、δ13Cである。質量44、45および46の分子イオンに関して、高性能安定比質量分析(high precision stable ratio mass spectrometry)(IRMS)によってCO2に対する測定を行う。
【0074】
従って、上記生合成されたムコネートおよび生合成されたムコネートを含む組成物は、14C(fM)および二重炭素同位体フィンガープリント法に基づいて、それらの化石燃料および石油化学由来対応物から区別され得、それにより、新たな組成物が示される(例えば、米国特許第7,169,588号、同第7,531,593号、および同第6,428,767号)。これら生成物を区別する能力は、流通においてこれら物質を追跡するにあたって有益である。例えば、新たなおよび古い炭素同位体プロフィールを両方含む生成物は、古い物質からのみ作製される生成物から区別され得る。従って、生合成によるムコネートおよび誘導体物質は、それらの特有のプロフィールに基づいて、流通において追跡され得る。
【実施例】
【0075】
(実施例1:aroZ遺伝子のクローニング)
DHSデヒドラターゼをコードする遺伝子(aroZと称される)を、Klebsiella pneumoniae DNAのゲノムライブラリーから単離した。ゲノムDNAを、K.pneumoniae株A170−40から精製し、BamH Iで部分的に消化して、15kb〜30kbの範囲のフラグメントを生成した。得られたDNAフラグメントを、BamH Iで予め消化し、その後、ウシ腸アルカリホスファターゼで処理したコスミドpLAFR3に連結した。pLAFR3は、RK2レプリコンを有するテトラサイクリン耐性コスミドである。連結したDNAを、Packagene Packaging System(Promega)を使用してパッケージし、得られたファージ粒子を使用して、E.coli DH5α/pKD136に感染させた。プラスミドpKD136は、トランスケトラーゼ(tkt)、DAHPシンターゼ(aroF)、およびDHQシンターゼ(aroB)をコードする遺伝子、ならびにアンピシリン耐性遺伝子を含むpBR325ベースのベクター(pMB1複製起点)である。テトラサイクリンおよびアンピシリンの両方に耐性であるコロニーを、その後、D−グルコース(4g L)、シキミ酸(0.04g L)、クエン酸第二鉄(0.07g L)、p−トルイジン(1.9g L)、アンピシリン(0.05g L)、およびテトラサイクリン(0.013g L)を含む色素生成性最小培地(M9)プレートにプレートした。37℃において48時間にわたってインキュベートした後、コロニー5−87を取り囲む増殖培地は、プロトカテク酸が上記プレート上にスポットされる場合に起こる、培地の黒ずみ(darkening)に似た、褐色に見えた。DNAは、コロニー5−87の培養物から精製し、pKD136およびテトラサイクリン耐性コスミド(p5−87といわれる)からなった。コスミドp5−87は、BamH Iで完全に消化した場合に、DNAの4個の検出可能なフラグメントを生成する14kb BamH Iフラグメントを含んだ。
【0076】
(実施例2:aroZ遺伝子のクローニングの確認)
コスミドp5−87が上記aroZ遺伝子を含むという確認は、代表的には、D−グルコースをDHSへ変換するE.coli株の形質転換が、DHSをプロトカテク酸へさらに変換し得るという事実に依拠した。E.coli AB2834は、シキミ酸デヒドロゲナーゼをコードする上記aroE遺伝子における変異に起因して、培養上清中にDHSを蓄積する。D−グルコースをDHSに変換することは、AB2834がpKD136で形質転換される場合に最大化される。AB2834を、pKD136およびp5−87で同時形質転換して、アンピシリンおよびテトラサイクリンの両方に耐性であるコロニーを生成した。1LのLB培地(4L 三角フラスコ)に、AB2834/pKD136/p5−87の一晩培養物(5mL)を接種した。上記培養物を、37℃において8時間にわたって撹拌しながら(250rpm)増殖させた。次いで、上記細胞を採取し、グルコース(10g L)、シキミ酸(0.04g L)、アンピシリン(0.05g L)、およびテトラサイクリン(0.013g L)を含む1L(4L 三角フラスコ)の最小M9培地中で再懸濁した。上記培養物を、37℃インキュベーションへと戻した。上記培養物のアリコートを、24時間後および64時間後に取り出し、細胞を除去するために遠心分離にかけた。単離された上清5mLを、各サンプルから集め、水を真空中で除去した。サンプルをD2O中に再溶解し、真空中で濃縮した。この手順の反復は、残りの水の、D2Oでの交換を生じ、サンプルを1H NMRに因る分析に適したものにした。内部標準として3−(トリメチルシリル)プロピオン−2,2,3,3−d4酸のナトリウム塩を使用して、約9mM プロトカテク酸が、培養上清中に蓄積したことが決定された。δ6.94(d, 7 Hz, 1H)およびδ 7.48 (d, 7 Hz, 2H)における診断的共鳴は、プロトカテク酸を示した。DHSは、上記培養上清中に検出されなかった。この実験から、DHSデヒドラターゼ(aroZ)をコードする遺伝子が、プラスミドp5−87上に位置することが結論づけられた。
【0077】
(実施例3:aroZ遺伝子のサブクローニング)
上記aroZコード挿入物のサイズを最小限にしようとする試みにおいて、プラスミドp5−87をBamH Iで消化し、得られたフラグメントを、BamH Iで予め消化し、ホスファターゼで処理したベクターpSU19に連結した。プラスミドpSU19は、上記p15Aレプリコンおよびクロラムフェニコールに対する耐性を付与する遺伝子を含む。E.coli DH5α/pKD136への上記連結生成物の形質転換後、得られたアンピシリンおよびクロラムフェニコール耐性コロニーを、p−トルイジンおよびクエン酸第二鉄を含む色素生成性最小培地アガロースプレートを褐色にする能力について、実施例1に記載されるようにスクリーニングした。この技術を使用して、プラスミドpSU1−31を単離した。これは、pSU19中に含まれる3.5kb BamH I挿入物からなった。AB2834/pKD136/pSU1−31を、実施例1に記載されるものに類似の条件下で、1Lスケールで増殖させた場合、上記培養上清の1H NMR分析は、11mM プロトカテク酸が細胞外に蓄積することを示した。
【0078】
(実施例4:aroY遺伝子のクローニング)
上記aroY遺伝子を含むDNAのフラグメントを、プロトカテクエートを通常合成する株が、代わりに、触媒的に活性なプロトカテク酸デカルボキシラーゼの存在下でカテコールを合成するという事実に基づいて単離した。コスミドp4−20を調製した。これは、pLAFR3に位置した3.5kb BamH I aroZフラグメントを含んだ。EcoR Iで消化したKlebsiella pneumoniae DNAのライブラリーを、pLAFR3中で先に構築したものに類似のコスミドp4−20虫に調製した。λファージ頭部にパッケージしたDNAを使用して、E.coli DH5α/pKD136に感染させ、その結果、アンピシリンおよびテトラサイクリンの両方に対して耐性のコロニーを生じた。コロニーを、p−トルイジンおよびクエン酸第二鉄を含む色素生成性最小培地アガロースプレート上でスクリーニングした。色素生成性最小培地にカテコールを添加すると、等量のプロトカテク酸の添加よりも、周りを囲むアガロースの黒ずみがより濃くなるので、カテコールを合成するそれらコロニーが、プロトカテクエートを合成するコロニーの背景から選択され得ることが予測された。37℃で約24時間にわたってインキュベートした後、コロニー2−47は、すべての他のコロニーが欠いている局所的な褐色の領域を生じていた。
【0079】
コロニー2−47からDNAを単離したところ、プラスミドpKD136およびプラスミドp2−47が得られた。これらプラスミドを、その後、コンピテント細胞に同時形質転換して、E. coli AB2834/pKD136/p2−47を生成した。上記AB2834/pKD136/p2−47の培養上清を、実施例2に記載されるように、1H NMRによって分析した。最小培地中で48時間後に、56mM D−グルコースの溶液は、AB2834/pKD136/p2−47によって20mM カテコールの溶液へと変換されていた。
【0080】
(実施例5:aroY遺伝子のサブクローニング)
プロトカテク酸デカルボキシラーゼをコードするDNAの単離のための、もとのストラテジーと同様に、aroY EcoR Iフラグメントの、その最小サイズへのサブクローニングはまた、DHSデヒドラターゼの存在下でのaroE宿主株によるカテコールの合成に依拠した。EcoR Iで完全になるまでのp2−47の消化は、上記aroY挿入物が、約8kbおよび11.9kbの2個のEcoR Iフラグメントからなることを示した。pSU1−31に上記11.9kb EcoR Iフラグメントが位置することで、プラスミドpSUaroZY157−27を得た。実施例2に記載されるものに類似の条件下で、1L スケールで増殖させた場合、E.coli AB2834/pKD136/pSUaroZY157−27は、56mM D−グルコースを供給した場合に、培養上清中に16mM カテコールを蓄積させた。さらなるサブクローニングに関連した上記11.9kb EcoR Iフラグメントのマッピングから、上記aroY遺伝子が、上記11.9kb フラグメントの中央部近くに位置しているようであることが示された。Hind IIIでのpSUaroZY157−27の消化は、2.3kb Hind IIIフラグメントを生じた。これを、pSU1−31に挿入して、プラスミドpKD8.243Aを得た(図2)。上記2.3kb Hind IIIフラグメントが上記ベクターに対して反対の配向にあるプラスミドpKD8.243Bをまた、単離した。これらプラスミドの各々を、AB2834へと、プラスミドpKD136で同時形質転換した。実施例2に記載されるものに類似の条件下で、1L スケールで増殖させた場合、AB2834/pKD136/pKD8.243Aは、56mM D−グルコースから48時間以内に16mM カテコールを合成したのに対して、AB2834/pKD136/pKD8.243Bは、10mM カテコールを合成した。プロトカテク酸(<4mM)もまた、上記培養上清のうちのいくらかで検出されたが、一貫したベースではなく、微生物合成の最後に常にあるわけではなかった。細菌細胞系AB2834/pKD136/pKD8.243A(これは、酵素種3−デヒドロシキミ酸脱水酵素およびプロトカテク酸デカルボキシラーゼを発現する)を、アメリカンタイプカルチャーコレクション(12301 Parklawn Drive, Rockville MD 20852)に、1996年3月19日に寄託し、アクセッション番号98014が割り当てられた。
【0081】
(実施例6:DHSデヒドラターゼ、プロトカテク酸デカルボキシラーゼ、およびカテコール 1,2−ジオキシゲナーゼの酵素活性)
D−グルコースをカテコールへ変換するのを触媒し得る生物におけるカテコール 1,2−ジオキシゲナーゼの発現は、cis,cis−ムコン酸の微生物合成をもたらすと予測した。プラスミドpIB 1345を得た。これは、上記宿主ベクターpUC19によって供給されたlacプロモーターから発現されるAcinetobacter calcoaceticus catA遺伝子を含む。3つのプラスミドシステムを、D−グルコースからcis,cis−ムコネートを微生物合成するために設計した。プラスミドpKD136(pMB1由来,アンピシリン耐性)およびpKD8.243A(p15A由来,クロラムフェニコール耐性は、使用される増殖条件下で安定に維持されることが見いだされた。第3のプラスミドであるpCL1920を、カテコール 1,2−ジオキシゲナーゼの発現のために選択した。プラスミドpCL1920は、上記pSC101複製起点、およびスペクチノマイシンに対する耐性を付与する遺伝子を含む低コピーベクターである。Sal IおよびKpn IでのpIB1345の消化は、1.5kbフラグメントを生じた。このフラグメントを、その後、pCL1920に配置して、pKD8.292を生成した(図3)。ここでカテコール 1,2−ジオキシゲナーゼは、上記ベクターがコードしたlacプロモーターから発現された。pKD8.243AおよびpKD8.292でのAB2834/pKD136の形質転換は、アンピシリン、クロラムフェニコール、およびスペクチノマイシンに対して耐性であるコロニーを生じた。
【0082】
E.coli AB2834/pKD136/pKD8.243A/pKD8.292が、DHSをcis,cis−ムコネートへ変換するのに必要なオルト切断経路からの遺伝子の各々を発現していることを確認するために、酵素活性を決定した。AB2834/pKD136/pKD8.243A/pKD8.292の培養物を、IPTG(0.2mM)、アンピシリン(0.05g)、クロラムフェニコール(0.02g)およびスペクチノマイシン(0.05g)を含むLB(1L)中、10時間にわたって37℃、250rpmにおいて増殖させた。細胞を採取し、100mM Tris HCl(pH7.5)、2.5mM MgCl2中に再懸濁した。フレンチプレス細胞破砕機(16,000psi)を2回通した後、その溶解物を、遠心分離(40000g,30分,4℃)によって清澄にした。DHSデヒドラターゼ活性を測定するために、各アッセイは、(最終容積1mL) 100mM Tris HCl(pH7.5)、25mM MgCl2、 mM DHS、および細胞溶解物を含んだ。DHSの添加後、プロトカテクエートの形成(ε=3890 M1 cm1)を、290nmで数分間にわたってモニターした。AB2834/pKD136/pKD8.243A/pKD8−292の3個のサンプルについて測定したDHSデヒドラターゼ活性は、0.078ユニットmg±0.009であると決定された。ここで1ユニットは、1μmolのDHSをプロトカテク酸に1分間で変換するのに必要な酵素量である。
【0083】
カテコール 1,2−ジオキシゲナーゼの比活性を、上記で生成した同じ細胞溶解物を使用して決定した。各アッセイは、100mM リン酸カリウム(pH7.5)、0.2mM カテコール、および細胞溶解物を含んだ。cis,cis−ムコネートの形成を、260nmの吸光度での増加を追跡することによってモニターした。上記アッセイの条件下でcis,cis−ムコネートとカテコールとの間のモル吸光係数の差異が、16,000 M1 cm1であると仮定すると、AB2834/pKD136/pKD8.243A/PKD8−292におけるカテコール 1,2−ジオキシゲナーゼ活性は、0.25ユニットmg±0.03であると決定された。ここで1ユニットは、1分あたりの1μmolのcis,cis−ムコネートの形成に対応する。
【0084】
プロトカテク酸デカルボキシラーゼの活性を決定するために、AB2834/pKD136/pKD8.243A/pKD8.292を、実施例6において先に記載されるように増殖させた。細胞を採取し、75mM リン酸緩衝液(pH7.1)中に再懸濁した。フレンチプレス細胞破砕機(16000psi)を通過させて破壊した後、その溶解物を、遠心分離(40000g,30分,4℃)によって清澄にした。プロトカテク酸デカルボキシラーゼ活性を、プロトカテク酸の消費を追跡することによって決定した。各アッセイ(最終容積1mL)は、75mM リン酸ナトリウム(pH6.0)、0.3mM プロトカテク酸、および細胞溶解物を含んだ。290nmでの吸光度の喪失を、経時的にモニターした。AB2834/pKD136/pKD8.243A/pKD8.292におけるプロトカテク酸デカルボキシラーゼ活性は、0.028ユニットmg±0.009であると決定された。ここで1ユニットは、1分あたりの1μmolのプロトカテク酸の酸化に対応する。
【0085】
(実施例7:D−グルコースの、cis,cis−ムコネートへの変換)
E.coli AB2834/pKD136/pKD8.243A/pKD8.292を利用する、D−グルコースからのcis,cis−ムコネートの微生物合成を、以下のように進めた。IPTG(0.2mM)、アンピシリン(0.05g)、クロラムフェニコール(0.02g)およびスペクチノマイシン(0.05g)を含む1LのLB培地(4L 三角震盪フラスコ中)に、AB2834/pKD136/pKD8.243A/pKD8.292の一晩培養物10mLを接種した。細胞を、250rpmにおいて10時間にわたって37℃で増殖させた。上記細胞を採取し、56mM D−グルコース、シキミ酸(0.04g)、IPTG(0.2mM)、アンピシリン(0.05g)、クロラムフェニコール(0.02g)およびスペクチノマイシン(0.05g)を含む1LのM9最小培地中に再懸濁した。上記培養物を、37℃インキュベーションへと戻した。最小培地中での再懸濁後、上記培養物のpHを、特に、最初の12時間にわたって厳密にモニターした。上記培養物がpH6.5に達したとき、5N NaOHを添加して、上記pHが約6.8に戻るように調節した。48時間の蓄積期間にわたって、上記培養物を、pH6.3より下にさせないようにした。最小培地中で24時間後に、実施例2に記載される方法を使用して、23mM D−グルコースとともにその培養上清中において、12mM cis,cis−ムコネートおよび1mM プロトカテクエートを検出した。最小培地中で48時間後に、AB2834/pKD136/pKD8.243A/pKD8.292は、56mM D−グルコースを17mM cis,cis−ムコネートに置き換えた。
【0086】
(実施例7A:20Lスケールでのグルコースの、cis,cis−ムコネートへの変換)
図7Aは、cis,trans−ムコン酸の生成についてWN1/pWN2.248の20Lバッチ培養の結果を示す。その培養物を、6時間ごとに、IPTG(100mM,10mL)を使用してOD600=33において誘導した。約88時間後に、上記ムコン酸タイター(titer)は、59g/L(30%収率)であった。合成されたムコン酸の総量は、1475gであった。これは、約88時間において、約1.38M グルコースを0.42M cis,trans−ムコン酸に変換したことに対応する。表1は、培養を通じての種々の時間での上記cis,trans−ムコン酸生成速度を示す(上記表は、時間の関数としての誘導後生産性を示すことに注意のこと。遠くの(outlying)データ点が排除される(接種後48時間および接種後58時間)場合、平均速度は、1.1g/L/時間である)。IPTG(例えば、酢酸エチル中)の再結晶化が上記ムコン酸タイターを増大させ得ることもまた見いだされた。例えば、いくつかの実験から、20L スケールで約55〜60g/L ムコン酸のタイターが示された。これは、IPTGの再結晶化なしに観察された約50g/L生成に対して約17%増加である(例えば、収率24%に対して収率約30%)。
【0087】
表1.発酵におけるcis,cis−ムコネート生成速度
【0088】
【表1】

グルコースもしくは他の発酵可能な炭素源からのcis,cis−ムコネートの生成に従って、cis,trans−ムコネートを生成するための方法は、(i)生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネートを提供する工程;(ii)cis,cis−ムコネートの実質的にすべてがcis,trans−ムコネートに異性化される反応条件下で、上記cis,cis−ムコネートをcis,trans−ムコネートに異性化する工程;および(iii)上記cis,trans−ムコネートを分離し、上記cis,trans−ムコネートを結晶化する工程を包含する。
【0089】
上記異性化反応は、酸(例えば、無機酸)によって触媒され得る。上記異性化反応は、pH7未満のpHにおいて、好ましくは、約4以下のpHにおいて溶液中で行われ得る。いくつかの例において、上記異性化のpHは、上記値より上であり得、そこでは、cis,cis−ムコネート、cis,trans−ムコネート、およびtrans,trans−ムコネートのうちの1つ以上が溶液から沈殿する。
【0090】
上記異性化反応は、室温より高いかもしくは発酵槽温度より高い温度で行われ得る。例えば、上記異性化反応は、約30℃以上、および好ましくは、60℃以上の温度で行われ得る。
【0091】
上記分離する工程は、溶液を酸性化することによって、上記溶液から上記cis,trans−ムコネートを沈殿させる工程を包含し得る。好ましくは、上記溶液は、約3未満のpHに酸性化され得る。上記分離する工程は、上記溶液を冷却する工程を包含し得る。上記溶液は、約30℃未満、好ましくは0℃未満の温度へと冷却され得る。
【0092】
再結晶化は、有機溶媒を使用し得る。上記有機溶媒は、極性非プロトン性溶媒(例えば、酢酸、ブタノール、イソプロパノール、プロパノール、エタノール、メタノール、ギ酸、水)、極性プロトン性溶媒(例えば、ジオキサン、テトラヒドロフラン、ジクロロメタン、アセトン、アセトニトリル、ジメチルホルムアミド、ジメチルスルホキシド)、および非極性溶媒(例えば、ヘキサン、ベンゼン、トルエン、ジエチルエーテル、クロロホルム、酢酸エチル)のうちの1種以上を含み得る。
【0093】
特定の実施形態において、上記方法は、上記分離されたcis,trans−ムコネートから塩を除去する工程を包含する。上記塩は、無機塩を含み得る。
【0094】
特定の実施形態において、上記方法は、上記cis,trans−ムコネートのうちの少なくとも約50%を、trans,trans−ムコネートに、および好ましくは、95%より多くを異性化する工程を包含する。
【0095】
図11は、6時間ごとに、IPTG(100mM,10mL)を使用してOD600=33において誘導したcis,trans−ムコン酸の生成についての、WN1/pWN2.248の20Lバッチ培養の結果を示す。約88時間後に、上記ムコン酸タイターは、59g/L(30%収率)であった。合成されたムコン酸の総量は、1475gであった。これは、約88時間で約1.38M グルコースを0.42M cis,trans−ムコン酸に変換するのに対応する。
【0096】
(実施例8:ファーメンター中でのcis,cis−ムコネートの、cis,trans−ムコネートへのインサイチュ異性化)
生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネート(例えば、実施例7および実施例7Aの方法に従って)を提供した。上記cis,cis−ムコネートを含むその発酵培養物を、60℃に加温した。上記加温した発酵培養物を、0.5時間にわたって2N 硫酸を添加することによって、pH4に調節した。上記酸性化した培養物を、3.5時間にわたって反応させた。
【0097】
上記反応を、1H NMRおよびPrevail Organic Acid Column(150mm×4.6mm)を備えたHPLCによってモニターして、上記反応のエンドポイントを決定した。これらデータを、図7〜10において、中性pHにおけるコントロール実験とともに示す。一般に、このような異性化反応は、適切な反応パラメーター(例えば、時間、温度、pHなど)を決定するためにモニターし得る。
【0098】
図5は、粗製の発酵ブロス中で、pH7におけるcis,cis−ムコネートからcis,trans−ムコネートへの異性化反応の1H NMRによる追跡を示す。0〜1.25時間および3.25時間までの時間追跡を、中性pH(例えば、これは、実際の発酵の間における近似のpHレベルである)においてcis,cis ムコン酸からcis,trans ムコン酸への異性化が本質的にないことを実証する。従って、cis,cis−ムコネートの異性化は、実際の発酵の間には全くもしくは無視できる程度にしか起こっていない。
【0099】
図6は、粗製発酵ブロスにおけるpH4でのムコネート異性化反応についての1H NMRによる追跡を示す。0〜1.25時間および3.25時間までの時間追跡は、cis,cis ムコン酸からcis,trans−ムコン酸への異性化が、酸性pHにおいて急速に進み、上記異性化は、約1.25時間後に本質的に完了することを実証する。
【0100】
図7は、pH7におけるムコネート異性化反応についてのHPLCによる追跡を示す。図9は、pH7におけるムコネート異性化反応の時間経過を示す。1H NMRによる追跡と同様に、これらHPLCによる追跡および時間経過は、中性pHにおけるcis,cis ムコン酸からcis,trans−ムコン酸への異性化が本質的にないことを実証する。
【0101】
図8は、pH4におけるムコネート異性化反応についてのHPLCによる追跡を示す。図10は、pH4におけるムコネート異性化反応の時間経過を示す。1H NMRによる追跡と同様に、これらHPLCによる追跡および時間経過は、cis,cis−ムコン酸からcis,trans−ムコン酸への異性化は、酸性pHにおいて急速に進み、上記異性化は、約1.25時間後に本質的に完了することを実証する。
【0102】
(実施例9:酸性化、沈殿、および濾過による発酵ブロスからのcis,trans−ムコン酸の分離)
異性化後に(例えば、実施例8におけるように)、cis,trans−ムコン酸を含むブロスをほぼ周囲温度へと冷却し、上記細胞、細胞砕片および沈殿した固体を、遠心分離によって培養ブロスから除去した。あるいは、このような固体を、濾過によって除去し得る(例えば、100kD SARTOCON(登録商標) Sliceカセットを介して)。次いで、上記無細胞ブロスを濾過によって清澄にして、タンパク質を除去した(例えば、10kD SARTOCON(登録商標) Sliceカセットを介して)。
【0103】
濾過後に、上記清澄にしたブロスのpHを、濃硫酸を添加することによってpH1.5に調節した。種々のpH値において沈殿するcis,trans−ムコネートの量を、表2に示す。
【0104】
表2.異なるpH値におけるcis,trans−ムコネートの沈殿
【0105】
【表2】
上記酸性化したブロスを、撹拌せずに1.5時間にわたって4℃へと冷却したところ、その時間の間に、粗製cis,trans−ムコン酸は、わずかに黄色の固体として沈殿した。この物質を濾過によって回収したところ、上記清澄化したブロスに存在する上記cis,trans−ムコン酸のうちの約60%を構成した。上記沈殿は、より長い期間(例えば、一晩)にわたって、そして/またはより低温(例えば、−20℃)において継続し、塩の夾雑を減少させながら生成物の回収を増大させられ得る。
【0106】
上記濾液は、さらなるcis,trans−ムコン酸を含んだ。上記さらなるcis,trans−ムコン酸を回収するために、上記濾液を減圧下でエバポレートして、その容積を約50%減らした。上記濃縮した濾液を、−20℃へと一晩冷却したところ、その時間の間に、粗製cis,trans−ムコン酸の第2の収穫物が沈殿した。上記沈殿物を、濾過により再び回収した。
【0107】
上記粗製cis,trans−ムコン酸固体を合わせ、アセトニトリルを使用して結晶化して、精製されたcis,trans−ムコネートを白色固体として生成させた。メタノールを使用する結晶化は、同様の収率を提供した。メタノールはまた、上記精製された生成物における塩の夾雑を減らした。
【0108】
図4Aは、結晶化したcis,trans−ムコン酸の1H NMRスペクトルを示す。図4Bは、グルコースを欠く最小塩類培地中に再懸濁した結晶化したcis,trans−ムコン酸の1H NMRスペクトルを示す。上記図4Bのスペクトルは、上記発酵ブロス中の他の成分によって引き起こされるcis,trans−ムコン酸のNMRスペクトルシフトに近似し、従って、上記発酵ブロス中での上記cis,cisからcis,transへの異性化反応を比較するためモニターすることを可能にする。
【0109】
(実施例10:有機溶媒を使用する発酵ブロスからのcis,trans−ムコン酸の抽出)
有利なことには、cis,trans−ムコン酸は、驚くべきことにかつ予測外に、上記cis,cis異性体もしくはtrans,trans異性体のいずれよりも、有機溶媒中で可溶性である。従って、分離する工程(例えば、実施例8の分離する工程)は、上記cis,trans−ムコネートを溶液(例えば、発酵ブロス)から、有機溶媒を使用して抽出する工程を包含し得る。
【0110】
上記cis,trans−ムコネートが抽出される上記溶液は、培養発酵ブロス全体もしくは、無細胞でタンパク質を含まない発酵ブロスであり得る。例えば、濾過(例えば、上記ブロスを、0.1μM 中空ファイバー濾過ユニットに通過させる)によって、細胞をブロスから除去し得る。例えば、濾過によって(例えば、例えば、SARTOCON(登録商標)から入手可能な10kD 接線流濾過システムを介して)、タンパク質は、ブロスから除去され得る。
【0111】
抽出用の上記有機溶媒(例えば、水相と非混和性の溶媒)としては、例えば、メチルイソブチルケトン(MIBK)、酢酸エチル、酢酸イソプロピル(酢酸プロピル)、ヘプタン(混合物)、メチル tert−ブチルエーテル、キシレン、塩化メチレン、シクロヘキサノール、デカリン、テトラリン、テトラロン、シクロヘキサン、酢酸ブチル、メチルテトラヒドロフラン(THF)、シクロヘキサノン/シクロヘキサノール(市販の混合物)、1−オクタノール、イソアミルアルコール、および2−エチルヘキサノールのうちの1種以上が挙げられ得る。
【0112】
水相に添加され得、上記cis,trans−ムコネートの抽出および同時もしくはその後のエステル化をともに促進する他の有機溶媒としては、例えば、メタノール、エタノール、プロパノール、イソプロパノール、酢酸、アセトニトリル、およびアセトン、ならびにブタノール(例えば、1−ブタノールおよびイソブタノール)および水と完全には混和性でない他のアルコールのうちの1種以上が挙げられる。
【0113】
上記溶媒抽出は、約4未満のpHにおいて(例えば、上記cis,trans−ムコン酸が十分にプロトン化され、上記抽出に使用される上記有機溶媒へと分配されるpHにおいて)行われ得る。沈殿を誘導するに十分低いpHレベルですら、上記プロトン化したcis,trans−ムコン酸の画分は、溶液中に残り得る。例えば、および上記実施例9の表2に示されるように、pH3において、本来は上記溶液中にある上記cis,trans−ムコン酸のうちの約60%は、沈殿し、濾過によって分離され得る。しかし、溶液に残っている上記cis,trans−ムコン酸のうちの約40%は、濾過によって分離できないが、抽出によって回収できる。よって、溶媒抽出は、有機溶媒を使用して溶液から上記cis,trans−ムコン酸を抽出する工程を包含しない方法と比較して、cis,trans−ムコネートの単離収率を増大させ得る。cis,trans−ムコネートのいくらかの部分が沈殿し、上記水溶液中にないとしても、上記cis,trans−ムコネートの抽出を進め得ることはまた、実施例9の表2中のデータから明らかである。
【0114】
上記溶媒抽出はまた、上記cis,trans−ムコネートを無機塩(例えば、硫酸アンモニウム、硫酸カルシウム)から分離する工程を包含し得る。さらに、溶媒抽出は、やはり無機塩の沈殿を引き起こし、従って無機塩との夾雑を引き起こし得る沈殿より純粋なcis,trans−ムコネートを生成し得る。
【0115】
溶媒および/もしくはpHパラメーターの選択は、一連の単純な測定によって促進され得る。各可能性のある溶媒について、3種すべてのムコン酸異性体(cis,cis−異性体、cis,trans−異性体、およびtrans,trans−異性体)に対して抽出が行われ得る(例えば、分配係数を測定するために)。各異性体はまた、約pH1〜pH7未満の間の増分(例えば、0.5)を使用したpH値の範囲に対して試験され得る。
【0116】
(実施例11:溶媒抽出による発酵ブロスからのcis,trans−ムコン酸の分離)
cis,trans−ムコン酸に異性化した発酵ブロスを得、これを、pH約3へと酸性化した。上記固体cis,trans−ムコン酸を濾過によって取り出して、約5〜10g/Lのcis,trans−ムコン酸を含む酸性化した発酵ブロスにした。
【0117】
15mLの上記濾過したブロスを各々含む個々の50mLコニカル遠心チューブに、以下の表に列挙される各溶媒15mLを添加した。各チューブを、2分間にわたって撹拌し、その有機相および水相を、分離させた。上記水相をピペットによって上記溶媒層から分離し、新しい50mLコニカルチューブに入れた。各抽出の上記水相および溶媒相の両方を、HPLCを使用してムコン酸について分析した。第2の抽出を、新鮮な溶媒を使用して、上記分離した水層の各々に対して行った。再び、上記サンプルを撹拌し、沈殿させ、上記水相および有機相を分離し、分析した。
【0118】
結果を、以下の表3、表4および表5に示す。cis,trans−ムコン酸の異なる量を有する異なるブロスサンプルを使用して、各表における結果を得た。
【0119】
表3. 9.82g/L cis,trans−ムコン酸を含むブロス
【0120】
【表3】
表4.10.69g/L cis,trans−ムコン酸を含むブロス
【0121】
【表4】
表5.4.86g/L cis,trans−ムコン酸を含むブロス
【0122】
【表5】
上記cis,trans−ムコン酸の十分量が沈殿した十分に低いレベルへと酸性化した後に、溶媒抽出がcis,trans−ムコン酸を回収する能力を試験するために、M9塩の溶液中のcis,trans−ムコン酸の溶液を使用して、発酵ブロスを模倣した。15gのcis,trans−ムコン酸を、約60g/Lのタイターを与える250mLのM9塩に添加した。これは、水酸化ナトリウムを使用して、上記pHを7.0へと上昇させることによって達成した。次いで、上記ブロスを、硫酸でpH3へと酸性化したところ、上記cis,trans−ムコン酸の沈殿が生じた。上記固体沈殿物を上記酸性混合物中に残し、スラリー全体を、上記に記載されるように、溶媒で2回抽出した。結果を、表6に示す。
【0123】
表6.沈殿したムコン酸を含め、63.34g/L cis,trans−ムコン酸を含むブロス
【0124】
【表6】
(実施例12:ヨウ素によって触媒される、cis,trans−ムコン酸の、trans,trans−ムコン酸への異性化)
cis,trans−ムコン酸(1.00g)、触媒量のI2(53mg)、およびMeCN(35ml)を含む混合物を加熱して、11時間にわたって還流した。室温へと冷却した後、その沈殿した固体を濾過し、冷MeCNで洗浄した。真空下で乾燥させた後、0.80g(80%収率)の精製trans,trans−ムコン酸は、黄褐色粉末として存在した。この手順によって得られた物質は、1Hおよび13C NMR分光法によって、trans,trans−ムコン酸であると確認された。上記異性化反応は、多くの他の試験した溶媒中よりも、非極性溶媒(例えば、THF)中でより進行した。
【0125】
(実施例13:水素化触媒によって触媒される、cis,trans−ムコン酸の、trans,trans−ムコン酸への異性化)
cis,trans−ムコン酸(1.00g)および触媒量のパラジウム担持炭素(Pd/C,5%)を含む混合物を、50mLのメタノール中に調製する。上記メタノール反応混合物を、1時間にわたって還流させ、室温へと冷却し、次いで、上記担持されたパラジウム触媒を、濾過によって除去する。残った反応溶液を、元の容積の約1/2へとエバポレートし、次いで、1容積のMeCNで希釈する。減圧下でのエバポレーションを、上記メタノールが除去されかつ上記trans,trans−ムコン酸が溶液から落ち始める(begins to fall out)まで継続する。得られた固体を濾過し、冷MeCNで洗浄する。真空下で乾燥させた後、約0.80g(80%収率)の精製trans,trans−ムコン酸が、黄褐色粉末として存在し得る。この手順によって得られる物質は、1Hおよび13C NMR分光法によって、trans,trans−ムコン酸であると確認され得る。
【0126】
(実施例14:水素化触媒によって触媒される、cis,cis−ムコン酸の、trans,trans−ムコン酸への異性化)
cis,cis−ムコン酸(1.00g)および触媒量のパラジウム担持炭素(Pd/C,5%)を含む混合物を、50mLのメタノール中に調製する。上記メタノール反応混合物を、1時間にわたって還流させ、室温へと冷却し、次いで、上記担持されたパラジウム触媒を、濾過によって除去する。残った反応溶液を、元の容積の約1/2になるまでエバポレートし、次いで、1容積のMeCNで希釈する。減圧下でのエバポレーションを、上記メタノールが除去されかつ上記trans,trans−ムコン酸が溶液から落ち始めるまで継続する。得られた固体を濾過し、冷MeCNで洗浄する。真空下で乾燥させた後、約0.80g(80%収率)の精製trans,trans−ムコン酸が、黄褐色粉末として存在し得る。この手順によって得られる物質は、1Hおよび13C NMR分光法によって、trans,trans−ムコン酸であると確認され得る。
【0127】
本発明は、具体的に示され、そして特定の実施形態を参照して記載されてきたが、形態および詳細における種々の変更が、添付の特許請求の範囲によって定義される発明の趣旨および範囲から逸脱することなくなされ得ることは、当業者によって理解されるべきである。
【図4A】
【図4B】
【技術分野】
【0001】
(関連出願)
本願は、2010年1月8日に出願された米国仮特許出願第61/335,638号の利益および優先権を主張し、この米国仮特許出願の開示は、その全体が本明細書中に参考として援用される。
【0002】
(発明の分野)
本発明は、一般に、再生可能な供給材料からムコネートを生物学的に生成することに関する。本発明は、より詳細には、再生可能なバイオマス由来炭素源から、ムコネート異性体、ならびにその前駆物質および誘導体を生成することに関する。
【背景技術】
【0003】
(発明の背景)
ジメチルテレフタレート(DMT)の世界的消費は、2012年で平均して397万トン(メートル法の トン)まで突出している。DMTは、テレフタル酸およびメタノールのエステルであり、ポリエステル(ポリエチレンテレフタレートおよびポリトリメチレンテレフタレートが挙げられる)の生成において使用される。DMTはまた、産業用プラスチック、自動車部品、フィルム、釣り糸、および食品パッケージ材料の製造において使用される主要な成分である。
【0004】
伝統的には、DMT生成は、テレフタル酸を、パラキシレンの触媒による均一酸化(homogeneous oxidation)によって生成されるメタノールでエステル化することを利用する。例えば、液体パラキシレンは、コバルト塩触媒の存在下で空気によって酸化されて、オキシデート(oxidate)含有p−トルイル酸およびモノメチルテレフタレートを形成し得、エステル化は、メタノールの存在下で行われて、DMTを形成し得る。
【0005】
トリメリト酸(TMA)は、粉体塗料、インク、エナメル線(wire enamel)、低揮発性の高性能可塑剤、および高温適用のための産業用ポリマーのための樹脂を含む、化学産業における中間体としての適用を有する別の商業的に重要な生成物である。TMAはまた、脱水されて、トリメリト酸無水物を生成し得、これは、ポリマーおよび化学的中間体の生成のための、別の商業的に重要な出発材料である。
【0006】
伝統的には、TMAは、プソイドクメン(1,2,4−トリメチルベンゼン)の酸化によって生成される。テレフタル酸およびイソフタル酸は、溶媒としての酢酸および触媒系(コバルト、マンガンおよび臭素が挙げられる)の存在下で、p−キシレンもしくはm−キシレンの液相酸化によって商業的に生成され得る。
【0007】
これらプロセスは、多くの他の商業的に重要な化学前駆物質、中間体、および生成物を生成するためのプロセスのように、環境的に鋭敏な(environmentally sensitive)かつ非再生可能な供給材料(例えば、石油供給材料)に大きく依存すること、およびそれらの望ましくない副生成物(例えば、温室効果ガス、重金属、ハロゲン、発癌性炭化水素)を生成する傾向に起因して、望ましくない可能性がある。よって、再生可能な供給材料を利用して、DMT、TMA、および他の化学生成物を生成する改善された方法およびシステムが必要である。
【0008】
特許文献1(Frostらによる)および特許文献2(Frostらによる)に記載されるように(これら出願の開示はともに、それらの全体が本明細書に参考として援用される)、DMTおよびTMAは、ムコン酸から生成され得る。さらに、ムコン酸(その二重結合および二酸の官能性に起因して、2,4−ヘキサジエン二酸としても公知)は、広く種々の反応を受け得る。多くのムコン酸誘導体は公知であり、これらとしては、ラクトン、スルホン、ポリアミド、ポリエステル、チオエステル、付加ポリマー、および他の化合物が挙げられる。このような化合物は、広く種々の用途を有し、上記用途としては、界面活性剤、難燃剤、UV光安定化剤、熱硬化性プラスチック、熱プラスチック(thermoplastic)およびコーティングとしての用途が挙げられる。従って、再生可能な供給材料からムコン酸もしくはムコネートを生物学的に生成するための改善された方法は、DMT、TMAおよび他の化学物質を生成するために非常に望ましい。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】米国公開出願公開第2010/0314243号明細書
【特許文献2】国際公開第2010/148049号
【発明の概要】
【課題を解決するための手段】
【0010】
本発明の記載では、用語「ムコネート」および「ムコン酸」を使用する。用語「ムコン酸」とは、両方のカルボン酸官能基がプロトン化され、その分子が形式上中性種である化学種に言及する。ムコン酸は、化学式HOOC−CH=CH−CH=CH−−COOHを有する。用語「ムコネート」とは、カルボン酸官能基のうちの一方もしくは両方が脱プロトン化されて、生理学的pH値において優勢な化学種であるアニオン性もしくは二重アニオン性の形態を与える、対応する脱プロトン化化学種に言及する。しかし、用語「ムコン酸」および「ムコネート」とは、同じ分子のプロトン化形態もしくは脱プロトン化形態に言及するので、上記分子のプロトン化と脱プロトン化(例えば、非イオン化とイオン化)形態との間の差異が有用に区別されない場合に、上記用語は同義語として使用される。
【0011】
本発明は、バイオマス由来炭素源から、ムコネートの3種の異性体(すなわち、cis,cis異性体;cis,trans異性体;およびtrans,trans異性体)、ならびにその前駆物質および誘導体を生成するための方法を提供する。上記異性体は、その2個の二重結合の周りの幾何的配置により、構造的に異なる。さらに、上記異性体は、異なる物理的特性(例えば、融点)および化学反応性を有し得る。上記方法は、芳香族アミノ酸生合成の共通経路を有する微生物においてエリスロース 4−ホスフェート(E4P)およびホスホエノールピルビン酸(PEP)へと生体触媒による変換を行う能力のある、容易に入手可能な炭素源からの生成物の微生物生合成を含み得る。
【0012】
1つの好ましい炭素源は、D−グルコースである。有利なことには、本発明と関連して使用可能なD−グルコースおよび他の炭素源は、非毒性である。さらに、このような炭素源は、再生可能であり、デンプン、セルロース、ならびにトウモロコシ、サトウキビ、砂糖大根、木材パルプ、および他のバイオマス資源において見いだされる糖に由来する。
【0013】
本発明における種々の工程を促進するために適した宿主微生物は、芳香族アミノ酸生合成の内因性共通経路を有する属から選択され得る。好ましい宿主生物としては、Klebsiella pneumoniaeおよびAcinetobacter calcoaceticusに内因性の選択された遺伝子を発現するように遺伝子操作されたEscherichia coliの変異株が挙げられる。本発明における使用のための1つの好ましいE.coli変異体は、E.coli AB2834であり、これは、酵素シキミ酸デヒドロゲナーゼをコードするaroE遺伝子座における変異に起因して、3−デヒドロシキメート(DHS)(芳香族アミノ酸生合成の共通経路に沿った中間体)をシキミ酸へと変換するのを触媒できない栄養要求性変異体である。
【0014】
芳香族アミノ酸生合成の共通経路は、細菌および植物において、芳香族アミノ酸であるフェニルアラニン、チロシン、およびトリプトファンを生成する。上記共通経路は、分子コリスメートで終了し、これは、その後、3つの別個の最終経路によってフェニルアラニン、チロシン、およびトリプトファンへと変換される。
【0015】
上記共通する芳香族アミノ酸生合成経路の生成効率を増大させるためのアプローチとしては、米国特許第5,168,056号(1992年12月1日発行)、米国特許第5,616,496号(1997年4月1日発行)、および米国特許出願第07/994194号(1992年12月21日出願、現在、放棄されている)(これらすべての開示は、それら全体が本明細書において参考として援用される)に記載されるものが挙げられる。
【0016】
上記遺伝子操作された宿主生物を使用するにあたって、芳香族アミノ酸生合成へと指向される炭素の流れは、上記共通経路に沿って進行して、DHSの上昇した細胞内レベルを生じ得る。DHSは、上記芳香族アミノ酸生合成の共通経路に沿った変異(これは、DHSからコリスメートへの変換を妨げる)に起因して蓄積する。上記DHSは、酵素3−デヒドロシキミ酸脱水酵素(aroZ)のための基質として働き、DHSに対するこの酵素の作用により、プロトカテクエートを生じる。プロトカテクエートは、その後、プロトカテク酸デカルボキシラーゼ(aroY)として公知の別の酵素を介して、カテコールへと変換される。このように形成された上記カテコールは、続いて、酵素カテコール 1,2−ジオキシゲナーゼ(catA)の作用によって、cis,cis−ムコン酸へと変換される。
【0017】
DHSからcis,cis−ムコネートの生合成を触媒する3つの酵素(すなわち、aroZ、aroY、およびcatA)は、適切なプロモーターの制御下でこれら3つの酵素をコードする遺伝子を含む組換えDNAを使用して、宿主細胞において発現され得る。炭素の流れは、それによって、芳香族アミノ酸生合成の経路から離れて、cis,cis−ムコネートを生成する分岐した経路へと押しやられ得る。このように形成されたcis,cis−ムコン酸は、細胞外培地(medium)へと蓄積され得、これは、遠心分離、濾過、もしくは当該分野で公知の他の方法によって上記細胞から分離され得る。上記単離されたcis,cis−ムコン酸は、その後、化学的に水素化されて、アジピン酸を生じ得る。
【0018】
本発明の種々の実施形態において、上記cis,cis−ムコネートが生成された後、これは、引き続いて、cis,trans−ムコネートもしくはtrans,trans−ムコネートに異性化され得る。上記cis,trans−ムコネートもしくはtrans,trans−ムコネートはともに、異なる物理的特性および化学反応性を有し、これらは、cis,cis−ムコネートのものとは異なるかもしくはこれを超える有用性を与え得る。例えば、cis,trans−異性体は、水性培地および/もしくは有機培地においてcis,cis−ムコネートより高い溶解性を有し得、このことは、有利な回収および加工処理を可能にする。さらなる例として、上記trans,trans−異性体は、Diels−Alder反応における反応物として上記cis,cis−異性体を超える特有の有用性を有し得る。
【0019】
一局面において、本発明は、cis,trans−ムコネートを生成するための方法を特徴とする。上記方法は、生体触媒による変換(例えば、上記aroZ、aroY、およびcatA酵素を利用する)を介して再生可能炭素源から生成されたcis,cis−ムコネートを提供する工程、上記cis,cis−ムコネートの実質的にすべてがcis,trans−ムコネートへと異性化される反応条件下で、cis,cis−ムコネートをcis,trans−ムコネートへと異性化する工程、上記cis,trans−ムコネートを分離する工程、および上記分離されたcis,trans−ムコネートを結晶化する工程(例えば、上記プロトン化cis,trans−ムコン酸として)を包含する。
【0020】
別の局面において、本発明は、cis,trans−ムコネートを生成するための方法を特徴とする。上記方法は、生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネートを含む発酵ブロスを提供する工程;上記cis,cis−ムコネートの実質的にすべてがcis,trans−ムコネートへと異性化される反応条件下で、cis,cis−ムコネートをcis,trans−ムコネートへと異性化する工程;上記ブロスから上記cis,trans−ムコネートを分離する工程;および上記cis,trans−ムコネートを結晶化する工程を包含する。
【0021】
なお別の局面において、本発明は、本発明の特徴である方法によって生成されるcis,trans−ムコネートを特徴とする。上記cis,trans−ムコネートは、塩(例えば、ムコン酸ナトリウム、ムコン酸カルシウム、もしくはムコン酸アンモニウムなどの無機塩)として回収され得る。
【0022】
なお別の局面において、本発明は、trans,trans−ムコネートを生成するための方法を特徴とし、上記方法は、上記cis,cis−ムコネートの実質的にすべてがtrans,trans−ムコネートへと異性化される反応条件下で、生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネートをtrans,trans−ムコネートへと異性化する工程を包含する。例えば、上記異性化反応は、貴金属水素化触媒によって、スポンジメタル水素化触媒によって、もしくは骨格水素化触媒(skeletal hydrogenation catalyst)によって触媒され得る。
【0023】
なお別の局面において、本発明は、trans,trans−ムコネートを生成するための方法を特徴とし、上記方法は、上記cis,trans−ムコネートのうちの実質的にすべてがtrans,trans−ムコネートへと異性化される反応条件下で、生体触媒による変換を介して再生可能炭素源から生成されたcis,trans−ムコネートをtrans,trans−ムコネートへと異性化する工程を包含する。例えば、上記異性化反応は、貴金属水素化触媒によって、スポンジメタル水素化触媒によって、もしくは骨格水素化触媒によって触媒され得る。
【0024】
さらに別の局面において、本発明は、本発明が特徴とする方法によって生成されたtrans,trans−ムコネート(例えば、再生可能なtrans,trans−ムコネート)を特徴とする。
【0025】
他の例において、上記局面のうちのいずれか、または本明細書に記載される任意の方法、器具、もしくは組成物は、以下の特徴のうちの1つ以上を含み得る。
【0026】
種々の実施形態において、上記方法は、3−デヒドロシキミ酸脱水酵素(例えば、aroZ)、プロトカテク酸デカルボキシラーゼ(例えば、aroY)およびカテコール 1,2−ジオキシゲナーゼ(例えば、catA)を発現する組換え細胞を、再生可能炭素源を含む培地中で、かつこのような再生可能炭素源が上記細胞の芳香族アミノ酸生合成の共通経路において見いだされた酵素類によってDHSに変換される条件下で、培養する工程を包含し、得られたDHSは、cis,cis−ムコネートへと生体触媒により変換される。
【0027】
上記再生可能炭素源の発酵によるcis,cis−ムコネートの生成は、組換え細胞および細胞外cis,cis−ムコネートを含むブロスを生成し得る。上記生成はまた、上記組換え細胞、細胞砕片、不溶性タンパク質および他の望ましくない固体を、上記ブロスから分離して、上記発酵によって形成されたcis,cis−ムコネートの実質的にすべてもしくはその大部分を含む清澄にした発酵ブロスを与える工程を包含し得る。次いで、上記cis,cis−ムコネートは、上記清澄にした発酵ブロス中においてcis,trans−ムコネートへと異性化され得る。
【0028】
特定の実施形態において、生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネートを含む発酵ブロスは、cis,trans−ムコネートもしくはtrans,trans−ムコネートを生成するために提供され得る。上記発酵ブロスは、3−デヒドロシキミ酸脱水酵素、プロトカテク酸デカルボキシラーゼおよびカテコール 1,2−ジオキシゲナーゼを発現する組換え細胞を含み得る。いくつかの実施形態において、上記発酵ブロスは、容器中で提供され、上記異性化反応は、上記容器中で行われる。上記容器は、発酵容器であり得る。いくつかの例において、3−デヒドロシキミ酸脱水酵素、プロトカテク酸デカルボキシラーゼおよびカテコール1,2−ジオキシゲナーゼを発現する組換え細胞は、上記再生可能炭素源を含む培地中で、かつ上記再生可能炭素源が、上記細胞の芳香族アミノ酸生合成の共通経路における酵素類によって3−デヒドロシキメートへと変換され、かつ、上記3−デヒドロシキメートは、生体触媒によりcis,cis−ムコネートに変換される条件下で、培養され得る。例えば、上記組換え細胞は、上記発酵容器中で培養され得、それによって、上記発酵ブロスを生成することができる。さらに、上記組換え細胞は、所望の通り、上記発酵ブロスから除去され得る。
【0029】
いくつかの実施形態において、上記異性化反応は、酸によって触媒される。上記酸は、無機酸(例えば、鉱酸)もしくは有機酸であり得る。酸は、水和形態もしくは無水形態のいずれかで上記プロセスに適用され得る。一例において、塩の副生成物は、硫酸アンモニウムであり得る。これは、その後、例えば、肥料として使用され得る。上記異性化反応は、約1.5〜約6.5の間(例えば、1.5、1.75、2、2.25、2.5、2.75、3、3.25、3.5、3.75、4、4.25、4.5、4.75、5、5.25、5.5、5.75、6、6.25、6.5)のpHにおいて行われ得る。好ましくは、上記異性化反応は、約3.5〜約4.5の間のpHにおいて行われ得る。
【0030】
特定の実施形態において、上記異性化反応は、約47℃以上の(例えば、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、もしくはより高い)温度で行われる。好ましくは、上記異性化反応は、約60℃以上の温度で行われ得る。上記異性化反応は、8時間以内、7.75時間以内、7.5時間以内、7.25時間以内、7時間以内、6.75時間以内、6.5時間以内、6.25時間以内、6時間以内、5.75時間以内、5.5時間以内、5.25時間以内、5時間以内、4.75時間以内、4.5時間以内、4.25時間以内、4時間以内、3.75時間以内、3.5時間以内、3.25時間以内、3時間以内、2.75時間以内、2.5時間以内、2.25時間以内、2時間以内、1.75時間以内、1.5時間以内、1.25時間以内、1時間以内、0.75時間以内、0.5時間以内、もしくは0.25時間以内で実質的に完了し得る。
【0031】
種々の実施形態において、上記異性化反応は、その反応混合物からcis,trans−ムコネートを実質的に沈殿させることなく進められる。特定の実施形態において、上記異性化反応は、cis,cis−ムコネートをcis,trans−ムコネートへと異性化することをモニターする工程を包含する。いくつかの実施形態において、上記異性化反応は、ほぼ大気圧より高い圧力で行われる。
【0032】
種々の実施形態において、異性化後、上記cis,trans−ムコネートは、上記cis,trans−ムコン酸を沈殿させるのに十分な、さらなる酸性化によって、溶液、培地、ブロス、もしくは発酵ブロスから分離され得る。上記ブロスは、約3.0未満の(例えば、2.9、2.8、2.7、2.6、2.5、2.4、2.3、2.2、2.1、2、もしくはこれより低い)pHへと酸性化され得る。上記ブロスは、約2未満のpHへとさらに酸性化され得る。
【0033】
特定の実施形態において、上記分離する工程は、約37℃未満、約25℃未満、約−4℃未満、もしくは約−20℃未満の温度へと溶液を冷却する工程を包含する。
【0034】
特定の実施形態において、上記分離する工程は、沈殿させられたcis,trans−ムコン酸を分離するために、遠心分離、濾過、もしくは他の物理的プロセスを含む。種々の実施形態において、上記分離する工程は、有機溶媒を使用して、上記cis,trans−ムコネートを上記発酵ブロスから抽出する工程を包含する。上記有機溶媒は、メタノール、エタノール、プロパノール、イソプロパノール、ブタノール、酢酸、アセトニトリル、アセトン、およびテトラヒドロフラン、tert−ブチルメチルエーテル、メチルテトラヒドロフラン、シクロヘキサノンもしくはシクロヘキサノール、またはこれらの混合物のうちの1種以上を含み得る。一実施形態において、上記抽出は、上記cis,trans−ムコン酸の顕著な沈殿なく、約7〜4の間の(例えば、約7、6.75、6.5、6.25、6、5.75、5.5、5.25、5、5.75、5.5、5.25、4)pHにおいて行われ得、酸の自動化された添加を使用して、上記抽出が進むにつれて、この領域に上記pHを維持することを包含し得る。別の実施形態において、上記抽出する工程は、上記有機溶媒によって溶解される、沈殿したcis,trans−ムコン酸の存在下で、約4未満の(例えば、約4、3.75、3.5、3.25、3、2.75、2.5、2.25、2)pHにおいて行われ得る。さらに別の実施形態において、上記抽出する工程は、細胞、細胞砕片、タンパク質、もしくは他の望ましくない物質を、上記発酵ブロスから最初に除去せずに、行われ得る。なお別の実施形態において、上記抽出する工程は、膜によって媒介され得る。
【0035】
特定の実施形態において、上記cis,cis−ムコネートは、上記発酵ブロスから最初に除去され得、次いで、上記異性化、分離、および精製工程に供され得る。このような除去は、抽出、沈殿、イオン交換クロマトグラフィー、選択的膜分離、電気透析、もしくは当該分野で公知の他の方法によって達成され得る。
【0036】
いくつかの実施形態において、上記cis,trans−ムコン酸は、有機溶媒を使用する結晶化によって精製される。上記有機溶媒は、メタノール、エタノール、プロパノール、イソプロパノール、ブタノール、酢酸、アセトニトリル、アセトン、およびテトラヒドロフランのうちの1種以上を含み得る。
【0037】
いくつかの実施形態において、上記結晶化は、上記発酵ブロスからの回収後に、沈殿したcis,trans−ムコン酸を乾燥させることなく行われ得る。特定の実施形態において、上記結晶化は、分離されたcis,trans−ムコン酸から望ましくない塩を除去する工程を包含する。種々の実施形態において、上記結晶化は、cis,trans−ムコン酸の第1の収穫物を集めた後に、上記結晶化培池を濃縮する工程、およびcis,trans−ムコン酸の第2の収穫物を上記濃縮した培池から集める工程を包含する。
【0038】
特定の実施形態において、trans,trans−ムコネートを生成するための方法は、cis,trans−ムコネートの生成、上記cis,trans−ムコネートのうちの少なくとも約65%をtrans,trans−ムコネートへと異性化する工程、および上記trans,trans−ムコネートを単離する工程を包含する。上記方法は、上記cis,trans−ムコネートのうちの少なくとも約65%、約66%、約67%、約68%、約69%、約70%、約71%、約72%、約73%、約74%、約75%、約76%、約77%、約78%、約79%、約80%、約81%、約82%、約83%、約84%、約85%、約86%、約87%、約88%、約89%、約90%、約91%、約92%、約93%、約94%、約95%、約96%、約97%、約98%、約99%、もしくは約100%を、trans,trans−ムコネートへと異性化する工程を包含し得る。あるいは、trans,trans−ムコネートは、適切な条件(例えば、pH、温度、触媒など)下でcis,cis−ムコネートから生成もしくは異性化され得る。
【0039】
種々の実施形態において、上記異性化反応は、I2によって、貴金属水素化触媒によって、スポンジメタル水素化触媒によって、もしくは骨格水素化触媒によって、触媒される。上記貴金属は、水素化触媒として機能する任意の貴金属(例えば、白金、パラジウムなど)であり得る。上記スポンジメタルもしくは骨格触媒は、ニッケル−アルミニウム合金(例えば、W.R.Grace and Companyから入手可能なRANEY(登録商標)ニッケル触媒)であり得る。上記金属触媒は、不均一触媒(例えば、粒子)もしくは担持触媒(supported catalyst)(例えば、シリカ、アルミナ、炭素などのような支持体上)の形態で存在し得る。
【0040】
いくつかの実施形態において、生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネートを提供する工程は、酵素3−デヒドロシキミ酸脱水酵素およびプロトカテク酸デカルボキシラーゼを発現するKlebsiella pneumoniae、ならびに酵素カテコール 1,2−ジオキシゲナーゼを発現するAcinetobacter calcoaceticusに由来する異種構造遺伝子で形質転換された細菌細胞を使用する。ここで上記細菌細胞の培養物は、約88時間以内に、1.38M グルコースを少なくとも約0.42M cis,cis−ムコン酸へと変換するに少なくとも十分な速度で、グルコースをcis,cis−ムコン酸へと生体触媒により変換する。上記細菌細胞形質転換体は、酵素3−デオキシ−D−アラビノ−ヘプツロソン酸 7−リン酸シンターゼおよび3−デヒドロキナ酸シンターゼを発現する異種DNA配列を含み得る。上記細菌細胞形質転換体は、酵素トランスケトラーゼ、3−デオキシ−D−アラビノ−ヘプツロソン酸 7−リン酸シンターゼおよび3−デヒドロキナ酸シンターゼを発現する異種DNA配列を含み得る。上記細菌細胞は、3−デヒドロシキメートをコリスメートに変換することをブロックする、芳香族アミノ酸生合成の共通経路において変異を有する変異細胞系から選択され得る。上記細菌細胞は、3−デヒドロシキメートをコリスメートに変換することをブロックする、上記芳香族アミノ酸生合成の共通経路において変異を有する変異細胞系から選択される。
【0041】
特定の実施形態において、生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネートを提供する工程は、酵素種3−デヒドロシキミ酸脱水酵素およびプロトカテク酸デカルボキシラーゼを発現するKlebsiella pneumoniaeに由来する構造遺伝子、ならびに酵素種カテコール 1,2−ジオキシゲナーゼを発現するAcinetobacter calcoaceticusに由来する構造遺伝子で形質転換された細菌細胞を、上記細胞の芳香族アミノ酸生合成の共通経路における上記酵素類によって3−デヒドロシキメートへと変換される炭素源を含む培地中で培養して、少なくとも約0.95ミリモル/リットル/時間の速度で、3−デヒドロシキメートの生体触媒による変換によって、cis,cis−ムコン酸を生成する工程を包含する。他の実施形態において、cis,cis−ムコン酸は、少なくとも約0.97ミリモル/リットル/時間以上、1.0ミリモル/リットル/時間以上、1.2ミリモル/リットル/時間以上、1.4ミリモル/リットル/時間以上、1.6ミリモル/リットル/時間以上、1.8ミリモル/リットル/時間以上、2.0ミリモル/リットル/時間以上の速度で生成される。
【0042】
種々の実施形態において、生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネートを提供する工程は、3−デヒドロシキミ酸脱水酵素、プロトカテク酸デカルボキシラーゼ、カテコール 1,2−ジオキシゲナーゼ、トランスケトラーゼ、3−デオキシ−D−アラビノ−ヘプツロソン酸 7−リン酸シンターゼ、および3−デヒドロキナ酸シンターゼをコードする異種構造遺伝子を発現する形質転換された細菌細胞を、上記細胞の芳香族アミノ酸生合成の共通経路における上記酵素類によって3−デヒドロシキメートに変換される炭素源を含む培地中で培養して、3−デヒドロシキメートの生体触媒による変換によって、少なくとも約0.95ミリモル/リットル/時間の速度で、cis,cis−ムコン酸を生成する工程を包含する。他の実施形態において、cis,cis−ムコン酸は、少なくとも約0.97ミリモル/リットル/時間以上、1.0ミリモル/リットル/時間以上、1.2ミリモル/リットル/時間以上、1.4ミリモル/リットル/時間以上、1.6ミリモル/リットル/時間以上、1.8ミリモル/リットル/時間以上、2.0ミリモル/リットル/時間以上の速度で生成される。
【0043】
いくつかの実施形態において、生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネートを提供する工程は、酵素種3−デヒドロシキミ酸脱水酵素およびプロトカテク酸デカルボキシラーゼを発現するKlebsiella pneumoniaeに由来する構造遺伝子、ならびに酵素カテコール 1,2−ジオキシゲナーゼを発現するAcinetobacter calcoaceticusに由来する構造遺伝子で形質転換された細菌細胞を、炭素源を含む培地中で、上記炭素源が、少なくとも約0.95ミリモル/リットル/時間の速度でcis,cis−ムコン酸へと生体触媒により変換される条件下で培養する工程を包含する。他の実施形態において、cis,cis−ムコン酸は、少なくとも約0.97ミリモル/リットル/時間以上、1.0ミリモル/リットル/時間以上、1.2ミリモル/リットル/時間以上、1.4ミリモル/リットル/時間以上、1.6ミリモル/リットル/時間以上、1.8ミリモル/リットル/時間以上、2.0ミリモル/リットル/時間以上の速度で生成される。
【0044】
本発明の他の局面および利点は、以下の図面および説明から明らかになり、これらのうちのすべては、本発明の原理を、例示によってのみ示す。
【図面の簡単な説明】
【0045】
上記に記載される発明の利点は、さらなる利点とともに、添付の図面とともに考慮される以下の説明に言及することによって、よりよく理解され得る。図面は、必ずしも一定の大きさに比例しているわけではなく、代わりに、一般に、本発明の原理を図示する際に強調される。
【図1−1】図1は、芳香族アミノ酸生合成の共通経路およびcis,cis−ムコン酸を3−デヒドロシキメートから合成する分岐経路を示す。
【図1−2】図1は、芳香族アミノ酸生合成の共通経路およびcis,cis−ムコン酸を3−デヒドロシキメートから合成する分岐経路を示す。
【図2】図2は、プラスミドp2−47のプラスミドマップを示し、プラスミドpKD8.243Aが、プラスミドp2−47、pSU1−31、およびpSUaroZY 157−27からどのように生成され得るかを図示する。
【図3】図3は、pKD8.292のプラスミドマップを示し、プラスミドpKD8.292が、プラスミドpIB1345およびpCL1920からどのように生成され得るかを図示する。
【図4】図4Aおよび図4Bは、異なる培地中での、cis,trans−ムコン酸の1H NMRスペクトルの例を示す。
【図5】図5は、pH7におけるムコネート異性化反応についての1H NMR図形の例を示す。
【図6】図6は、pH4におけるムコネート異性化反応についての1H NMR図形の例を示す。
【図7】図7は、pH7におけるムコネート異性化反応についてのHPLC図形の例を示す。
【図8】図8は、pH4におけるムコネート異性化反応についてのHPLC図形の例を示す。
【図9】図9は、pH7におけるムコネート異性化反応についての経時変化の例を示す。
【図10】図10は、pH4におけるムコネート異性化反応についての経時変化の例を示す。
【図11】図11は、ムコン酸のバッチ培養生成を示す。
【発明を実施するための形態】
【0046】
本発明は、芳香族アミノ酸生合成の共通経路(例えば、上記中間体DHSを介して機能するもの)を有し、酵素aroZ、aroY、およびcatAを発現する能力を有する宿主細胞によって使用され得る発酵可能な炭素源から、cis,trans−ムコン酸およびtrans,trans−ムコン酸を生成するための方法を包含する。1つの好ましい実施形態において、上記方法は、cis,cis−ムコン酸を生成するための、発酵可能な炭素源の存在下で、上記宿主細胞を培養する工程、および上記cis,cis−ムコン酸を異性化して、cis,trans−ムコン酸もしくはtrans,trans−ムコン酸を生成する工程を包含する。
【0047】
発酵可能な炭素源は、D−エリスロース 4−ホスフェート(E4P)およびホスホエノールピルベート(PEP)(芳香族アミノ酸生合成の共通経路への2つの前駆化合物)へと生体触媒により変換され得る、本質的に任意の炭素源を含み得る。適切な炭素源としては、バイオマス由来の源、もしくは再生可能な源(例えば、デンプン、セルロース、およびグルコース、ペントース、およびフルクトースのような糖部分、ならびに微生物代謝を支持し得る他の炭素源(例えば、一酸化炭素)が挙げられるが、これらに限定されない。一実施形態において、D−グルコースは、上記バイオマス由来炭素源として使用され得る。
【0048】
本発明において使用するのに適した宿主細胞は、所望の芳香族化合物の生合成による生成のために利用され得る属のメンバーを含む。いくつかの実施形態において、このような宿主細胞は、所望の芳香族化合物の産業スケールの生合成による生成に適している。特に、適切な宿主細胞は、少なくともDHSの生成に対して機能的である芳香族アミノ酸生合成の内因性の共通経路を有し得る。共通する芳香族経路は、広く種々の微生物において内因性であり、種々の芳香族化合物の生成のために使用され得る。米国特許第5,168,056号および同第5,616,496号(その両方の開示は、それら全体が本明細書に参考として援用される)において記載されるように、例えば、微生物の芳香族アミノ酸生合成経路は、本発明において利用され得る。
【0049】
図1は、芳香族アミノ酸生合成の共通経路およびこの経路において多くの中間体とともにE4PおよびPEPからコリスミ酸をもたらす上記共通する芳香族経路を介してcis,cis−ムコン酸を3−デヒドロシキメートから合成する分岐経路を示す。E4Pの利用性は、tkt遺伝子によってコードされるペントースリン酸経路酵素であるトランスケトラーゼによって増大され得る。上記経路における中間体としては、3−デオキシ−D−アラビノ−ヘプツロソン酸 7−ホスフェート(DAHP)、3−デヒドロキネート(DHQ)、3−デヒドロシキメート(DHS)、シキミ酸、シキメート3−ホスフェート(S3P)、および5−エノールピルビルシキメート(enolpyruvoylshikimate)−3−ホスフェート(EPSP)が挙げられる。上記共通経路における酵素としては、DAHPシンターゼ(aroF)、DHQシンターゼ(aroB)、DHQデヒドラターゼ(aroD)、シキミ酸デヒドロゲナーゼ(aroE)、シキミ酸キナーゼ(aroL、aroK)、EPSPシンターゼ(aroA)およびコリスミ酸シンターゼ(aroC)が挙げられる。
【0050】
このタイプの共通経路を含む宿主細胞としては、Escherichia属、Klebsiella属、Corynebacterium属、Brevibacterium属、Arthrobacter属、Bacillus属、Pseudomonas属、Streptomyces属、Staphylococcus属、およびSerratia属に属する原核生物が挙げられる。真核生物宿主細胞もまた、例えば、Saccharomyces属もしくはSchizosaccharomyces属の酵母に関して利用され得る。
【0051】
より具体的には、原核生物宿主細胞は、Escherichia coli、Klebsiella pneumonia、Corynebacterium glutamicum、Corynebacterium herculis、Brevibacterium divaricatum、Brevibacterium lactofermentum、Brevibacterium flavum、Bacillus brevis、Bacillus cereus、Bacillus circulans、Bacillus coagulans、Bacillus lichenformis、Bacillus megaterium、Bacillus mesentericus、Bacillus pumilis、Bacillus subtilis、Pseudomonas aeruginosa、Pseudomonas angulata、Pseudomonas fluorescens、Pseudomonas tabaci、Streptomyces aureofaciens、Streptomyces avermitilis、Streptomyces coelicolor、Streptomyces griseus、Streptomyces kasugensis、Streptomyces lavendulae、Streptomyces lipmanii、Streptomyces lividans、Staphylococcus epidermis、Staphylococcus saprophyticus、およびSerratia marcescensを含む種に由来し得る。真核生物宿主細胞の例としては、Saccharomyces cerevisiaeおよびSaccharomyces carlsbergensisが挙げられる。
【0052】
宿主細胞は、DHSを分岐点分子であるコリスメートへと変換するのをブロックする変異を有する栄養要求性変異細胞系を含み得る。このような変異体は、シキミ酸デヒドロゲナーゼ、シキミ酸キナーゼ、EPSPシンターゼおよびコリスミ酸シンターゼをコードする遺伝子のうちの1種以上における変異に起因して、3−デヒドロシキメート(DHS)をコリスメートへ変換するのを触媒できず、そしてこのようにして、上昇した細胞内レベルのDHSを蓄積する。このような変異細胞系の例としては、Escherichia coli株AB2834、AB2829およびAB2849が挙げられる。
【0053】
E.coli AB2834は、シキミ酸デヒドロゲナーゼをコードするaroE遺伝子座における変異に起因して、3−デヒドロシキメート(DHS)をシキミ酸へ変換することを触媒できない。E.coli AB2834の使用は、芳香族アミノ酸生合成へ指向される炭素の流れが、DHSを超えて進行しないことを確実にし得る。同様に、E.coli AB2829(これは、EPSPシンターゼをコードするaroA遺伝子座における変異に起因して、シキメート 3−ホスフェート(S3P)を5−エノールピルビルシキメート(enoipyruvylshikimate)−3−ホスフェート(EPSP)へ変換することを触媒できない)およびE.coli AB2849(これは、コリスミ酸シンターゼをコードするaroC遺伝子座における変異に起因して、EPSPをコリスミ酸へ変換することを触媒できない)はまた、増大した細胞内レベルのDHSを生じる。
【0054】
宿主細胞は、細胞内DHSが、カテコールへの生体触媒による変換の基質として使用され得るように、形質転換され得、そのカテコールは、その後、ムコン酸へと変換され得る。例えば、宿主細胞は、組換えDNAで形質転換されて、DHSが生成された後に、炭素の流れを芳香族アミノ酸生合成の共通経路から離れて、ムコン酸を生成するための分岐経路へと押しやり得る。
【0055】
図1に示されるように、上記分岐経路における中間体は、プロトカテクエート、カテコール、およびcis,cis−ムコン酸である。DHSをプロトカテクエートに生体触媒により変換することを担う酵素は、図1において「aroZ」と表示される酵素3−デヒドロシキミ酸脱水酵素である。カテコールを形成するためのプロトカテクエートの脱炭酸を担う酵素は、図1において「aroY」と表示されるプロトカテク酸デカルボキシラーゼである。最後に、カテコールの酸化を触媒してcis,cis−ムコン酸を生成する酵素は、図1において「catA」と表示されるカテコール 1,2−ジオキシゲナーゼである。標準的表記法に従って、これら酵素の発現のための遺伝子は、斜体を使用して示され、よって、それぞれ、aroZ、aroY、およびcatAである。上記cis,cis−ムコン酸は、その後、異性化され得る(示されない)。本発明の一実施形態において、宿主細胞は、上記遺伝子aroZ、aroY、およびcatAの構成的発現を示し得る。別の実施形態において、宿主細胞は、上記遺伝子aroZ、aroYおよびcatAのうちのいずれか1種以上;またはこれらのうちの任意の2種の組み合わせの構成的発現を示し得る。さらに別の実施形態において、宿主細胞は、aroZ、aroYおよびcatAのうちのいずれの構成的発現も示さない場合がある。
【0056】
上記酵素3−デヒドロシキミ酸脱水酵素およびプロトカテク酸デカルボキシラーゼは、微生物(例えば、Neurospora、Aspergillus、Acinetobacter、Klebsiella、およびPseudomonas)が芳香族類(aromatics)(ベンゾエートおよびp−ヒドロキシベンゾエート)ならびにヒドロ芳香族類(hydroaromatics)(シキメートおよびキネート)を増殖のための唯一の炭素源として使用することを可能にするオルト切断経路から補充される。DHSデヒドラターゼは、キナ酸およびシキミ酸の微生物異化作用において重要な役割を果たす。Patelは、Klebsiella aerogenesによるp−ヒドロキシベンゾエートの異化作用の間に、プロトカテクエートのカテコールへの変換を触媒するように、プロトカテク酸デカルボキシラーゼを処方した。近年、Ornstonは、Patel株(現在、Enterobacter aerogenesといわれる)の再検査[(a)Grant,D.J.W.;Patel,J.C.Antonie van Leewenhoek 1969,35,325. (b)Grant,D.J.W.Antonie van Leewenhoek 1970,36,161]により、プロトカテク酸デカルボキシラーゼが、p−ヒドロキシベンゾエートの異化作用において代謝的に重要ではなかったと結論づけた[Doten,R.C.;Ornston,N. J.Bacteriol.1987,169,5827]。
【0057】
上記分岐経路へと炭素の流れを指向するように上記宿主細胞を形質転換するための機構は、3−デヒドロシキミ酸脱水酵素、プロトカテク酸デカルボキシラーゼ、およびカテコール 1,2−ジオキシゲナーゼをコードする発現可能な配列を含む遺伝的エレメントの挿入を包含し得る。利用される正確な機構にかかわらず、これら酵素活性の発現は、上記宿主細胞へと組換え遺伝的エレメントの移入によって行われるかもしくは媒介されることが企図される。本明細書で定義される場合、遺伝的エレメントは、生成物(例えば、タンパク質、アポタンパク質、もしくはアンチセンスRNA)についての発現可能なコード配列を有する核酸(一般に、DNAおよびRNA)を含み、これらは、経路酵素機能を発揮し得るかもしくは制御し得る。上記発現される生成物は、酵素として機能し得るか、酵素活性を抑制もしくは抑制解除し得るか、または酵素の発現を制御し得る。これら発現可能な配列をコードする核酸は、染色体性(例えば、宿主細胞染色体へ挿入もしくは組み込まれる)もしくは染色体外性(例えば、プラスミド、コスミドなどによって運ばれる)のいずれかであり得る。
【0058】
本発明の遺伝的エレメントは、プラスミド、コスミド、ファージ、酵母人工染色体もしくは上記遺伝的エレメントの、宿主細胞への移入を媒介する他のベクターによって、宿主細胞に導入され得る。これらベクターは、上記ベクターの複製を制御するcis作用制御エレメントおよび上記ベクターによって運ばれる遺伝的エレメントとともに、複製起点を含み得る。選択マーカーは、上記遺伝的エレメントが導入された宿主細胞の同定を補助するために、上記ベクターに存在し得る。例えば、選択マーカーは、特定の抗生物質(例えば、テトラサイクリン、アンピシリン、クロラムフェニコール、カナマイシン、もしくはネオマイシン)に対する耐性を付与する遺伝子であり得る。
【0059】
遺伝的エレメントを宿主細胞に導入することは、遺伝的エレメントが挿入される染色体外マルチコピープラスミドベクターを利用し得る。プラスミドが媒介する上記遺伝的エレメントの宿主細胞への導入は、制限酵素でのプラスミドの最初の切断、続いて、本発明に従う上記プラスミドおよび遺伝的エレメントの連結を包含する。連結された組換えプラスミドの再環化の際に、プラスミド移入のための形質導入もしくは他の機構(例えば、エレクトロポレーション、マイクロインジェクションなど)は、上記プラスミドを上記宿主細胞に移入するために利用される。遺伝的エレメントを上記宿主細胞に挿入するために適したプラスミドとしては、pBR322およびその誘導体(例えば、pAT153ベクター、pXf3ベクター、pBR325ベクター、pBr327ベクター、pUCベクター)、pACYCおよびその誘導体、pSC101およびその誘導体、ならびにColE1が挙げられるが、これらに限定されない。さらに、コスミドベクター(例えば、pLAFR3)はまた、遺伝的エレメントを宿主細胞に挿入するために適している。プラスミド構築物の例としては、p2−47、pKD8.243A、pKD8.243B、およびpSUaroZY157−27が挙げられるが、これらに限定されない。これらは、3−デヒドロシキミ酸脱水酵素およびプロトカテク酸デカルボキシラーゼをそれぞれコードするKlebsiella pneumoniaeから単離されたaroZおよびaroY遺伝子座を有する。プラスミド構築物のさらなる例としては、カテコール 1,2−ジオキシゲナーゼをコードするAcinetobacter calcoaceticus catAに対して内因性の遺伝的フラグメントを有するpKD8.292が挙げられる。
【0060】
宿主細胞を形質転換するための方法はまた、芳香族アミノ酸生合成の共通経路への炭素の拘束(commitment)を増大させる酵素をコードする遺伝子の挿入を包含し得る。遺伝子の発現は、主に、それ自体のプロモーターによって誘導されるが、任意選択の(optional)発現制御配列(例えば、リプレッサー、およびエンハンサー)を含む他の遺伝的エレメントは、タンパク質、アポタンパク質、もしくはアンチセンスRNAのコード配列の発現もしくは抑制解除を制御するために含められ得る。さらに、組換えDNA構築物が生成され得、それによって、上記遺伝子の天然のプロモーターは、上記遺伝子生成物の発現を増大させるために、代替のプロモーターで置き換えられる。プロモーターは、構成性もしくは誘導性のいずれかであり得る。構成性プロモーターは、細胞の寿命の間に一定の速度で遺伝子の転写を制御するのに対して、誘導性プロモーターの活性は、特定の誘導因子の存在(もしくは非存在)によって決定される場合に変動する。例えば、制御配列は、上記宿主細胞ゲノム中に既にコードされる選択された酵素の過剰発現を促進するために、野生型宿主細胞に挿入され得るか、または代わりに、染色体外でコードされた酵素の合成を制御するために使用され得る。
【0061】
DHSの過剰生成を促進する制御配列が使用され得る。先に示されるように、DHSは、ペントースリン酸経路酵素であるトランスケトラーゼ(tktによってコードされる)とともに、チロシン感受性アイソザイム3−デオキシ−D−アラビノヘプツロソン酸 7−リン酸(DAHP)シンターゼ(aroFによってコードされる)および3−デヒドロキノ酸(DHQ)シンターゼ(aroBによってコードされる)の逐次的触媒活性によって、上記共通経路において合成される。これら生合成酵素の発現は、D−グルコースをDHSへ変換するのを増大させるために増幅され得る。上記共通経路の第1の酵素であるDAHPシンターゼのインビボ触媒活性を増大させると、芳香族生合成へ向かうD−グルコース等価物の流れが増大する。しかし、DAHPシンターゼ触媒活性のレベルは、芳香族生合成に関係するD−グルコースのパーセンテージにおいてさらなる改善は達成されないレベル以上に達する。芳香族アミノ酸生合成のこの制限レベルにおいて、上記ペントースリン酸経路酵素であるトランスケトラーゼの上記触媒レベルの増幅は、上記経路へと吸い上げられるD−グルコースのパーセンテージのかなりの増大を達成する。
【0062】
増幅されたトランスケトラーゼ活性は、D−エリスロース 4−ホスフェート濃度を増大させ得る。DAHPシンターゼの2つの基質のうちの一方として、制限されたD−エリスロース 4−ホスフェートの利用可能性は、DAHPシンターゼ触媒活性を制限し得る。従って、DAHPシンターゼ、DHQシンターゼおよびDHQデヒドラターゼの触媒活性を増幅するための1つの方法は、これらの酵素をコードする組換えDNA配列で微生物触媒を形質転換することによってその酵素種を過剰発現することである。
【0063】
DAHPシンターゼおよびトランスケトラーゼの増幅された発現は、この経路へ向かう通常の炭素の流れが超過した状態にある、芳香族アミノ酸生合成の共通経路へ向かう炭素の流れのサージ(surge)を作り得る。共通の芳香族アミノ酸経路の個々の酵素によって触媒される、基質の生成物への変換の個々の速度が、DAHPシンターゼの速度より低い場合、これら律速酵素の基質は、細胞内に蓄積し得る。
【0064】
微生物(例えば、E.coli)は、頻繁に、外部環境(例えば、バルク発酵培地)へこのような基質を輸送する(export)ことによって、蓄積された基質に対処する。このことは、上記共通経路を介する炭素の流れの喪失を生じる。なぜなら、輸送された基質は、代表的には、微生物の代謝に影響を受けない。DHQシンターゼは、律速共通経路酵素の例である。DHQシンターゼの増幅された発現は、この酵素の律速特徴を除去し、DAHPおよびその非リン酸化アナログであるDAHの蓄積を妨げる。DHQデヒドラターゼは、律速ではない。従って、aroFがコードするDAHPシンターゼ、tktがコードするトランスケトラーゼおよびaroBがコードするDHQシンターゼの増幅された発現は、DHSの生成を増大させ、上記DHSは、DHSデヒドラターゼおよびプロトカテク酸デカルボキシラーゼの存在下で、カテコールに変換され、上記カテコールは、その後、cis,cis−ムコン酸へと生体触媒により変換され、上記cis,cis−ムコン酸は、その後、異性化され得る。
【0065】
上記炭素源とDHSとの間の共通経路に沿った炭素の流れの効率を促進し得る1つのプラスミドは、プラスミドpKD136であり、これは、上記aroF、tktおよびaroB遺伝子をコードする。プラスミドpKD136は、DAHPシンターゼ(aroFによってコードされる)およびトランスケトラーゼ(tktによってコードされる)の増幅された発現に起因して、芳香族生合成への炭素の流れのサージを誘導する。次いで、炭素の流れのこのサージは、DHQシンターゼ(aroBによってコードされる)の増幅された発現に起因して、pKD136によるDHS合成へとそのまま送達される。
【0066】
従って、本発明の好ましい実施形態として、DHSデヒドラターゼ、プロトカテク酸デカルボキシラーゼ、およびカテコール 1,2−ジオキシゲナーゼをコードするEscherichia coli発現遺伝子の異種由来の株を構築し、D−グルコースをcis,cis−ムコン酸へ生体触媒により変換することを可能にした。D−グルコースの、DHSへの効率的変換を、上記宿主細胞をpKD136で形質転換した際に達成した。次いで、上記株E.coli AB2834/pKD136を、プラスミドpKD8.243AおよびpKD8.292で形質転換した。結果は、酵素3−デヒドロシキミ酸脱水酵素(aroZ)、プロトカテク酸デカルボキシラーゼ(aroY)およびカテコール 1,2−ジオキシゲナーゼ(catA)を発現するE.coli AB2834/pKD136/pKD8.243A/pKD8.292であった。この細菌細胞系は、アメリカンタイプカルチャーコレクション(12301 Parklawn Drive,Rockville MD 20852)に、1995年8月1日に寄託し、アクセッション番号69875を割り当てられた。
【0067】
別の実施形態において、E.coli AB2834/pKD136は、E.coli AB2834/pKD136/p2−47/pKD8.292を生成するために、プラスミドp2−47およびpKD8.292で形質転換される。別の実施形態において、E.coli AB2834/pKD136は、E.coli AB2834/pKD136/p2−47/pKD8.292を生成するために、プラスミドpKD8.243BおよびpKD8.292で形質転換される。これら異種宿主細胞系の各々は、D−グルコースをcis,cis−ムコン酸に変換することを触媒する。合成されたcis,cis−ムコン酸は、細胞外に蓄積し、上記細胞から分離され得る。その後、上記cis,cis−ムコン酸は、cis,trans−ムコン酸へと異性化され得、所望される場合、さらにtrans,trans−ムコン酸へと異性化され得る。
【0068】
本発明は、従って、芳香族アミノ酸生合成の内因性共通経路を有する宿主細胞の形質転換体に関する。上記形質転換体は、3−デヒドロシキミ酸脱水酵素、プロトカテク酸デカルボキシラーゼ、およびカテコール 1,2−ジオキシゲナーゼをコードする異種遺伝子の構成的発現によって特徴付けられる。一実施形態において、上記細胞形質転換体は、酵素トランスケトラーゼ、DAHPシンターゼ、およびDHQシンターゼをコードする発現可能な組換えDNA配列でさらに形質転換される。別の実施形態において、上記宿主細胞は、3−デヒドロシキメートをコリスメートに変換するのをブロックするアミノ酸生合成の共通経路における変異を有する、変異を含む変異細胞系の群から選択される。なお別の実施形態において、3−デヒドロシキミ酸脱水酵素およびプロトカテク酸デカルボキシラーゼをコードする遺伝子は、Klebsiella pneumoniaeに対して内因性である。さらなる実施形態において、カテコール 1,2−ジオキシゲナーゼをコードする異種遺伝子は、Acinetobacter calcoaceticusに対して内因性である。
【0069】
(再生可能なムコネート)
再生可能な、生物学的に得られる炭素源から生成されるムコン酸は、植物によって組み込まれた大気の二酸化炭素に由来する炭素から構成される(例えば、グルコース、スクロース、グリセリン、もしくは植物油のような炭素源に由来する)。従って、このようなムコン酸は、それらの分子構造において、化石燃料ベースもしくは石油ベースの炭素ではなく、再生可能な炭素を含む。よって、この特許の主題である上記生合成によるムコネート、および関連する派生生成物は、従来法によって生成されるムコネートより小さな炭素痕跡(footprint)および関連生成物を有する。なぜなら、それらは、化石燃料を枯渇させないか、石油が確保され、それらは、炭素サイクルにおける炭素量を増大させない(例えば、ライフサイクル分析は、世界的な炭素バランスに対して正味の炭素増大を示さない)からである。
【0070】
上記生合成によるムコネートおよび関連生成物は、当該分野で公知の方法(例えば、二重炭素同位体フィンガープリント法(dual carbon−isotopic finger printing))によって、化石燃料もしくは石油化学炭素源から生成されるムコネートおよび関連生成物から区別され得る。この方法は、化学的に同一の材料を別の方法で区別し得、14Cと13Cとの異性体比を使用して、供給源によって(すなわち、非生物学的に対して生物学的)上記材料中の炭素原子を区別する。上記炭素同位体14Cは、不安定であり、5730年の半減期を有する。安定な13C同位体に対する上記不安定な14C同位体の相対量を測定することは、化石(長期間活動をとめた(long dead))供給材料と生物圏の(生きている、よって、再生可能な)供給材料との間の標本炭素を区別することを可能にする(Currie,L.A. 「Source Apportionment of Atmospheric Particles」,Characterization of Environmental Particles, J.Buffle and H.P.van Leeuwen,Eds., 1 of Vol. I of the IUPAC Environmental Analytical Chemistry Series(Lewis Publishers,Inc)(1992)3−74を参照のこと)。放射性炭素の年代測定における基本的仮定は、大気中の14C濃度の定常性が、生きた生物における14Cの定常性をもたらすということである。
【0071】
単離されたサンプルを扱う場合、サンプルの年代は、関係性t=(−5730/0.693)ln(A/Ao)によってほぼ導き出され得る。ここでt=年代であり、5730年は、上記不安定な14C同位体の半減期であり、AおよびAoは、それぞれ、サンプルおよび現代の標準物質の特定の14C活性である(Hsieh,Y.,Soil ScL Soc.Am J.,56,460,(1992))。しかし、1950年以来の大気圏核実験および1850年以来の化石燃料の燃焼が原因で、14Cは、第2の地球化学的時間の特徴を獲得した。大気中CO2中のその濃度、そして従って、生きている生物圏における濃度は、1960年代半ばには、核実験のピーク時にほぼ2倍になった。上記濃度は、それ以来、概算の緩和半減期(relaxation half−life)約7〜10年で、定常状態宇宙線による(大気中)ベースライン同位体率(14C/12C)約1.2×10−12へと徐々に戻った。(この後者の半減期は、同位体半減期とは区別されなければならない(すなわち、核時代の始まり以来の大気中および生物圏の14Cの変動を追跡するために、詳細な大気核投入/崩壊関数(atmospheric nuclear input/decay function)を使用しなければならない)。それは、近年の生物圏炭素の年ごとの年代測定を約束するこの後者の生物圏14C時間の特徴である。14Cは、加速器質量分析(AMS)によって測定され得、結果は、現代の炭素に対する割合の単位(fM)で与えられる。fMは、米国国立標準技術研究所(National Institute of Standards and Technology)(NIST)の標準参照物質(Standard Reference Materials)(SRMs) 4990Bおよび4990C(それぞれ、シュウ酸標準物質HOxIおよびHOxIIとして公知)によって定義される。上記基本定義は、HOxIの14C/12C同位体比の0.95倍(AD 1950年を基準として)に関連する。現在の生きている生物圏(植物材料)では、fMは約1.1である。
【0072】
安定な炭素同位体である13Cおよび12Cの比は、供給源の識別および配分(apportionment)に対する補完の経路を提供する。所定の生体供給源の材料における13C/12C比は、上記二酸化炭素が固定され、正確な代謝経路をも反映するときには、大気中二酸化炭素における13C/12C比の結果である。局部的なバリエーションもまた存在する。石油、C3植物(広葉樹)、C4植物(禾本(the grasses))、および海洋カーボネート(marine carbonate)はすべて、13C/12Cおよび対応するδ 13C値において顕著な差異を示す。さらに、C3植物およびC4植物の脂質物質は、その代謝経路の結果として、同じ植物の炭水化物成分に由来する物質とは異なって分析される。測定の正確さの範囲内で、13Cは、同位体分別効果に起因して大きなバリエーションを示し、本発明に関してその最も顕著なものは、光合成機構である。植物における炭素同位体比が異なる大きな原因は、上記植物における光合成炭素代謝の経路、特に、第1のカルボキシル化(例えば、大気中CO2の最初の固定)の間に起こる反応の差異と密接に関連する。草木の2つの大きなクラスは、C3(もしくはカルビン−ベンソン)光合成回路を組み込むものと、C4(もしくはハッチ−スラック)光合成回路を組み込むものである。C3植物(例えば、堅木および針葉樹)は、温暖気候帯域において優勢である。C3植物においては、第1のCO2固定もしくはカルボキシル化反応は、酵素リブロース−1,5−二リン酸カルボキシラーゼを必要とし、第1の安定な生成物は、3個の炭素の化合物である。他方で、C4植物は、熱帯地方の禾本、トウモロコシおよびサトウキビのような植物を含む。C4植物において、別の酵素であるホスホエノール−ピルビン酸カルボキシラーゼを必要とするさらなるカルボキシル化反応が、第1のカルボキシル化反応である。その第1の安定な炭素化合物は、4個の炭素の酸であり、これは、その後、脱炭酸される。このようにして放出されたCO2は、上記C3回路によって再固定される。
【0073】
C4植物およびC3植物はともに、ある13C/12C同位体比の範囲を示すが、代表的な値は、約−10〜−14‰(C4)および−21〜−26‰(C3)である(Weberら,J.Agric.Food Chem.,45,2942(1997))。石炭および石油は、一般に、この後者の範囲に入る。上記13C測定スケールは、本来は、pee deeベレムナイト(PDB)石灰岩によって設定されるゼロによって定義され、ここで値は、この材料からのppt偏差(parts per thousand deviations)単位で与えられる。上記δ13C値は、ppt(パーミル)単位にあり、‰と省略され、以下のように計算される:
δ13C≡(13C/12C)サンプル−(13C/12C)標準物質/(13C/12C)標準物質×1000‰
上記PDB標準物質(RM)は枯渇してしまったので、一連の代替のRMが、IAEA、USGS、NIST、および他の選択された国際同位体実験機関と協同して開発された。PDBからの‰偏差の表示法が、δ13Cである。質量44、45および46の分子イオンに関して、高性能安定比質量分析(high precision stable ratio mass spectrometry)(IRMS)によってCO2に対する測定を行う。
【0074】
従って、上記生合成されたムコネートおよび生合成されたムコネートを含む組成物は、14C(fM)および二重炭素同位体フィンガープリント法に基づいて、それらの化石燃料および石油化学由来対応物から区別され得、それにより、新たな組成物が示される(例えば、米国特許第7,169,588号、同第7,531,593号、および同第6,428,767号)。これら生成物を区別する能力は、流通においてこれら物質を追跡するにあたって有益である。例えば、新たなおよび古い炭素同位体プロフィールを両方含む生成物は、古い物質からのみ作製される生成物から区別され得る。従って、生合成によるムコネートおよび誘導体物質は、それらの特有のプロフィールに基づいて、流通において追跡され得る。
【実施例】
【0075】
(実施例1:aroZ遺伝子のクローニング)
DHSデヒドラターゼをコードする遺伝子(aroZと称される)を、Klebsiella pneumoniae DNAのゲノムライブラリーから単離した。ゲノムDNAを、K.pneumoniae株A170−40から精製し、BamH Iで部分的に消化して、15kb〜30kbの範囲のフラグメントを生成した。得られたDNAフラグメントを、BamH Iで予め消化し、その後、ウシ腸アルカリホスファターゼで処理したコスミドpLAFR3に連結した。pLAFR3は、RK2レプリコンを有するテトラサイクリン耐性コスミドである。連結したDNAを、Packagene Packaging System(Promega)を使用してパッケージし、得られたファージ粒子を使用して、E.coli DH5α/pKD136に感染させた。プラスミドpKD136は、トランスケトラーゼ(tkt)、DAHPシンターゼ(aroF)、およびDHQシンターゼ(aroB)をコードする遺伝子、ならびにアンピシリン耐性遺伝子を含むpBR325ベースのベクター(pMB1複製起点)である。テトラサイクリンおよびアンピシリンの両方に耐性であるコロニーを、その後、D−グルコース(4g L)、シキミ酸(0.04g L)、クエン酸第二鉄(0.07g L)、p−トルイジン(1.9g L)、アンピシリン(0.05g L)、およびテトラサイクリン(0.013g L)を含む色素生成性最小培地(M9)プレートにプレートした。37℃において48時間にわたってインキュベートした後、コロニー5−87を取り囲む増殖培地は、プロトカテク酸が上記プレート上にスポットされる場合に起こる、培地の黒ずみ(darkening)に似た、褐色に見えた。DNAは、コロニー5−87の培養物から精製し、pKD136およびテトラサイクリン耐性コスミド(p5−87といわれる)からなった。コスミドp5−87は、BamH Iで完全に消化した場合に、DNAの4個の検出可能なフラグメントを生成する14kb BamH Iフラグメントを含んだ。
【0076】
(実施例2:aroZ遺伝子のクローニングの確認)
コスミドp5−87が上記aroZ遺伝子を含むという確認は、代表的には、D−グルコースをDHSへ変換するE.coli株の形質転換が、DHSをプロトカテク酸へさらに変換し得るという事実に依拠した。E.coli AB2834は、シキミ酸デヒドロゲナーゼをコードする上記aroE遺伝子における変異に起因して、培養上清中にDHSを蓄積する。D−グルコースをDHSに変換することは、AB2834がpKD136で形質転換される場合に最大化される。AB2834を、pKD136およびp5−87で同時形質転換して、アンピシリンおよびテトラサイクリンの両方に耐性であるコロニーを生成した。1LのLB培地(4L 三角フラスコ)に、AB2834/pKD136/p5−87の一晩培養物(5mL)を接種した。上記培養物を、37℃において8時間にわたって撹拌しながら(250rpm)増殖させた。次いで、上記細胞を採取し、グルコース(10g L)、シキミ酸(0.04g L)、アンピシリン(0.05g L)、およびテトラサイクリン(0.013g L)を含む1L(4L 三角フラスコ)の最小M9培地中で再懸濁した。上記培養物を、37℃インキュベーションへと戻した。上記培養物のアリコートを、24時間後および64時間後に取り出し、細胞を除去するために遠心分離にかけた。単離された上清5mLを、各サンプルから集め、水を真空中で除去した。サンプルをD2O中に再溶解し、真空中で濃縮した。この手順の反復は、残りの水の、D2Oでの交換を生じ、サンプルを1H NMRに因る分析に適したものにした。内部標準として3−(トリメチルシリル)プロピオン−2,2,3,3−d4酸のナトリウム塩を使用して、約9mM プロトカテク酸が、培養上清中に蓄積したことが決定された。δ6.94(d, 7 Hz, 1H)およびδ 7.48 (d, 7 Hz, 2H)における診断的共鳴は、プロトカテク酸を示した。DHSは、上記培養上清中に検出されなかった。この実験から、DHSデヒドラターゼ(aroZ)をコードする遺伝子が、プラスミドp5−87上に位置することが結論づけられた。
【0077】
(実施例3:aroZ遺伝子のサブクローニング)
上記aroZコード挿入物のサイズを最小限にしようとする試みにおいて、プラスミドp5−87をBamH Iで消化し、得られたフラグメントを、BamH Iで予め消化し、ホスファターゼで処理したベクターpSU19に連結した。プラスミドpSU19は、上記p15Aレプリコンおよびクロラムフェニコールに対する耐性を付与する遺伝子を含む。E.coli DH5α/pKD136への上記連結生成物の形質転換後、得られたアンピシリンおよびクロラムフェニコール耐性コロニーを、p−トルイジンおよびクエン酸第二鉄を含む色素生成性最小培地アガロースプレートを褐色にする能力について、実施例1に記載されるようにスクリーニングした。この技術を使用して、プラスミドpSU1−31を単離した。これは、pSU19中に含まれる3.5kb BamH I挿入物からなった。AB2834/pKD136/pSU1−31を、実施例1に記載されるものに類似の条件下で、1Lスケールで増殖させた場合、上記培養上清の1H NMR分析は、11mM プロトカテク酸が細胞外に蓄積することを示した。
【0078】
(実施例4:aroY遺伝子のクローニング)
上記aroY遺伝子を含むDNAのフラグメントを、プロトカテクエートを通常合成する株が、代わりに、触媒的に活性なプロトカテク酸デカルボキシラーゼの存在下でカテコールを合成するという事実に基づいて単離した。コスミドp4−20を調製した。これは、pLAFR3に位置した3.5kb BamH I aroZフラグメントを含んだ。EcoR Iで消化したKlebsiella pneumoniae DNAのライブラリーを、pLAFR3中で先に構築したものに類似のコスミドp4−20虫に調製した。λファージ頭部にパッケージしたDNAを使用して、E.coli DH5α/pKD136に感染させ、その結果、アンピシリンおよびテトラサイクリンの両方に対して耐性のコロニーを生じた。コロニーを、p−トルイジンおよびクエン酸第二鉄を含む色素生成性最小培地アガロースプレート上でスクリーニングした。色素生成性最小培地にカテコールを添加すると、等量のプロトカテク酸の添加よりも、周りを囲むアガロースの黒ずみがより濃くなるので、カテコールを合成するそれらコロニーが、プロトカテクエートを合成するコロニーの背景から選択され得ることが予測された。37℃で約24時間にわたってインキュベートした後、コロニー2−47は、すべての他のコロニーが欠いている局所的な褐色の領域を生じていた。
【0079】
コロニー2−47からDNAを単離したところ、プラスミドpKD136およびプラスミドp2−47が得られた。これらプラスミドを、その後、コンピテント細胞に同時形質転換して、E. coli AB2834/pKD136/p2−47を生成した。上記AB2834/pKD136/p2−47の培養上清を、実施例2に記載されるように、1H NMRによって分析した。最小培地中で48時間後に、56mM D−グルコースの溶液は、AB2834/pKD136/p2−47によって20mM カテコールの溶液へと変換されていた。
【0080】
(実施例5:aroY遺伝子のサブクローニング)
プロトカテク酸デカルボキシラーゼをコードするDNAの単離のための、もとのストラテジーと同様に、aroY EcoR Iフラグメントの、その最小サイズへのサブクローニングはまた、DHSデヒドラターゼの存在下でのaroE宿主株によるカテコールの合成に依拠した。EcoR Iで完全になるまでのp2−47の消化は、上記aroY挿入物が、約8kbおよび11.9kbの2個のEcoR Iフラグメントからなることを示した。pSU1−31に上記11.9kb EcoR Iフラグメントが位置することで、プラスミドpSUaroZY157−27を得た。実施例2に記載されるものに類似の条件下で、1L スケールで増殖させた場合、E.coli AB2834/pKD136/pSUaroZY157−27は、56mM D−グルコースを供給した場合に、培養上清中に16mM カテコールを蓄積させた。さらなるサブクローニングに関連した上記11.9kb EcoR Iフラグメントのマッピングから、上記aroY遺伝子が、上記11.9kb フラグメントの中央部近くに位置しているようであることが示された。Hind IIIでのpSUaroZY157−27の消化は、2.3kb Hind IIIフラグメントを生じた。これを、pSU1−31に挿入して、プラスミドpKD8.243Aを得た(図2)。上記2.3kb Hind IIIフラグメントが上記ベクターに対して反対の配向にあるプラスミドpKD8.243Bをまた、単離した。これらプラスミドの各々を、AB2834へと、プラスミドpKD136で同時形質転換した。実施例2に記載されるものに類似の条件下で、1L スケールで増殖させた場合、AB2834/pKD136/pKD8.243Aは、56mM D−グルコースから48時間以内に16mM カテコールを合成したのに対して、AB2834/pKD136/pKD8.243Bは、10mM カテコールを合成した。プロトカテク酸(<4mM)もまた、上記培養上清のうちのいくらかで検出されたが、一貫したベースではなく、微生物合成の最後に常にあるわけではなかった。細菌細胞系AB2834/pKD136/pKD8.243A(これは、酵素種3−デヒドロシキミ酸脱水酵素およびプロトカテク酸デカルボキシラーゼを発現する)を、アメリカンタイプカルチャーコレクション(12301 Parklawn Drive, Rockville MD 20852)に、1996年3月19日に寄託し、アクセッション番号98014が割り当てられた。
【0081】
(実施例6:DHSデヒドラターゼ、プロトカテク酸デカルボキシラーゼ、およびカテコール 1,2−ジオキシゲナーゼの酵素活性)
D−グルコースをカテコールへ変換するのを触媒し得る生物におけるカテコール 1,2−ジオキシゲナーゼの発現は、cis,cis−ムコン酸の微生物合成をもたらすと予測した。プラスミドpIB 1345を得た。これは、上記宿主ベクターpUC19によって供給されたlacプロモーターから発現されるAcinetobacter calcoaceticus catA遺伝子を含む。3つのプラスミドシステムを、D−グルコースからcis,cis−ムコネートを微生物合成するために設計した。プラスミドpKD136(pMB1由来,アンピシリン耐性)およびpKD8.243A(p15A由来,クロラムフェニコール耐性は、使用される増殖条件下で安定に維持されることが見いだされた。第3のプラスミドであるpCL1920を、カテコール 1,2−ジオキシゲナーゼの発現のために選択した。プラスミドpCL1920は、上記pSC101複製起点、およびスペクチノマイシンに対する耐性を付与する遺伝子を含む低コピーベクターである。Sal IおよびKpn IでのpIB1345の消化は、1.5kbフラグメントを生じた。このフラグメントを、その後、pCL1920に配置して、pKD8.292を生成した(図3)。ここでカテコール 1,2−ジオキシゲナーゼは、上記ベクターがコードしたlacプロモーターから発現された。pKD8.243AおよびpKD8.292でのAB2834/pKD136の形質転換は、アンピシリン、クロラムフェニコール、およびスペクチノマイシンに対して耐性であるコロニーを生じた。
【0082】
E.coli AB2834/pKD136/pKD8.243A/pKD8.292が、DHSをcis,cis−ムコネートへ変換するのに必要なオルト切断経路からの遺伝子の各々を発現していることを確認するために、酵素活性を決定した。AB2834/pKD136/pKD8.243A/pKD8.292の培養物を、IPTG(0.2mM)、アンピシリン(0.05g)、クロラムフェニコール(0.02g)およびスペクチノマイシン(0.05g)を含むLB(1L)中、10時間にわたって37℃、250rpmにおいて増殖させた。細胞を採取し、100mM Tris HCl(pH7.5)、2.5mM MgCl2中に再懸濁した。フレンチプレス細胞破砕機(16,000psi)を2回通した後、その溶解物を、遠心分離(40000g,30分,4℃)によって清澄にした。DHSデヒドラターゼ活性を測定するために、各アッセイは、(最終容積1mL) 100mM Tris HCl(pH7.5)、25mM MgCl2、 mM DHS、および細胞溶解物を含んだ。DHSの添加後、プロトカテクエートの形成(ε=3890 M1 cm1)を、290nmで数分間にわたってモニターした。AB2834/pKD136/pKD8.243A/pKD8−292の3個のサンプルについて測定したDHSデヒドラターゼ活性は、0.078ユニットmg±0.009であると決定された。ここで1ユニットは、1μmolのDHSをプロトカテク酸に1分間で変換するのに必要な酵素量である。
【0083】
カテコール 1,2−ジオキシゲナーゼの比活性を、上記で生成した同じ細胞溶解物を使用して決定した。各アッセイは、100mM リン酸カリウム(pH7.5)、0.2mM カテコール、および細胞溶解物を含んだ。cis,cis−ムコネートの形成を、260nmの吸光度での増加を追跡することによってモニターした。上記アッセイの条件下でcis,cis−ムコネートとカテコールとの間のモル吸光係数の差異が、16,000 M1 cm1であると仮定すると、AB2834/pKD136/pKD8.243A/PKD8−292におけるカテコール 1,2−ジオキシゲナーゼ活性は、0.25ユニットmg±0.03であると決定された。ここで1ユニットは、1分あたりの1μmolのcis,cis−ムコネートの形成に対応する。
【0084】
プロトカテク酸デカルボキシラーゼの活性を決定するために、AB2834/pKD136/pKD8.243A/pKD8.292を、実施例6において先に記載されるように増殖させた。細胞を採取し、75mM リン酸緩衝液(pH7.1)中に再懸濁した。フレンチプレス細胞破砕機(16000psi)を通過させて破壊した後、その溶解物を、遠心分離(40000g,30分,4℃)によって清澄にした。プロトカテク酸デカルボキシラーゼ活性を、プロトカテク酸の消費を追跡することによって決定した。各アッセイ(最終容積1mL)は、75mM リン酸ナトリウム(pH6.0)、0.3mM プロトカテク酸、および細胞溶解物を含んだ。290nmでの吸光度の喪失を、経時的にモニターした。AB2834/pKD136/pKD8.243A/pKD8.292におけるプロトカテク酸デカルボキシラーゼ活性は、0.028ユニットmg±0.009であると決定された。ここで1ユニットは、1分あたりの1μmolのプロトカテク酸の酸化に対応する。
【0085】
(実施例7:D−グルコースの、cis,cis−ムコネートへの変換)
E.coli AB2834/pKD136/pKD8.243A/pKD8.292を利用する、D−グルコースからのcis,cis−ムコネートの微生物合成を、以下のように進めた。IPTG(0.2mM)、アンピシリン(0.05g)、クロラムフェニコール(0.02g)およびスペクチノマイシン(0.05g)を含む1LのLB培地(4L 三角震盪フラスコ中)に、AB2834/pKD136/pKD8.243A/pKD8.292の一晩培養物10mLを接種した。細胞を、250rpmにおいて10時間にわたって37℃で増殖させた。上記細胞を採取し、56mM D−グルコース、シキミ酸(0.04g)、IPTG(0.2mM)、アンピシリン(0.05g)、クロラムフェニコール(0.02g)およびスペクチノマイシン(0.05g)を含む1LのM9最小培地中に再懸濁した。上記培養物を、37℃インキュベーションへと戻した。最小培地中での再懸濁後、上記培養物のpHを、特に、最初の12時間にわたって厳密にモニターした。上記培養物がpH6.5に達したとき、5N NaOHを添加して、上記pHが約6.8に戻るように調節した。48時間の蓄積期間にわたって、上記培養物を、pH6.3より下にさせないようにした。最小培地中で24時間後に、実施例2に記載される方法を使用して、23mM D−グルコースとともにその培養上清中において、12mM cis,cis−ムコネートおよび1mM プロトカテクエートを検出した。最小培地中で48時間後に、AB2834/pKD136/pKD8.243A/pKD8.292は、56mM D−グルコースを17mM cis,cis−ムコネートに置き換えた。
【0086】
(実施例7A:20Lスケールでのグルコースの、cis,cis−ムコネートへの変換)
図7Aは、cis,trans−ムコン酸の生成についてWN1/pWN2.248の20Lバッチ培養の結果を示す。その培養物を、6時間ごとに、IPTG(100mM,10mL)を使用してOD600=33において誘導した。約88時間後に、上記ムコン酸タイター(titer)は、59g/L(30%収率)であった。合成されたムコン酸の総量は、1475gであった。これは、約88時間において、約1.38M グルコースを0.42M cis,trans−ムコン酸に変換したことに対応する。表1は、培養を通じての種々の時間での上記cis,trans−ムコン酸生成速度を示す(上記表は、時間の関数としての誘導後生産性を示すことに注意のこと。遠くの(outlying)データ点が排除される(接種後48時間および接種後58時間)場合、平均速度は、1.1g/L/時間である)。IPTG(例えば、酢酸エチル中)の再結晶化が上記ムコン酸タイターを増大させ得ることもまた見いだされた。例えば、いくつかの実験から、20L スケールで約55〜60g/L ムコン酸のタイターが示された。これは、IPTGの再結晶化なしに観察された約50g/L生成に対して約17%増加である(例えば、収率24%に対して収率約30%)。
【0087】
表1.発酵におけるcis,cis−ムコネート生成速度
【0088】
【表1】
グルコースもしくは他の発酵可能な炭素源からのcis,cis−ムコネートの生成に従って、cis,trans−ムコネートを生成するための方法は、(i)生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネートを提供する工程;(ii)cis,cis−ムコネートの実質的にすべてがcis,trans−ムコネートに異性化される反応条件下で、上記cis,cis−ムコネートをcis,trans−ムコネートに異性化する工程;および(iii)上記cis,trans−ムコネートを分離し、上記cis,trans−ムコネートを結晶化する工程を包含する。
【0089】
上記異性化反応は、酸(例えば、無機酸)によって触媒され得る。上記異性化反応は、pH7未満のpHにおいて、好ましくは、約4以下のpHにおいて溶液中で行われ得る。いくつかの例において、上記異性化のpHは、上記値より上であり得、そこでは、cis,cis−ムコネート、cis,trans−ムコネート、およびtrans,trans−ムコネートのうちの1つ以上が溶液から沈殿する。
【0090】
上記異性化反応は、室温より高いかもしくは発酵槽温度より高い温度で行われ得る。例えば、上記異性化反応は、約30℃以上、および好ましくは、60℃以上の温度で行われ得る。
【0091】
上記分離する工程は、溶液を酸性化することによって、上記溶液から上記cis,trans−ムコネートを沈殿させる工程を包含し得る。好ましくは、上記溶液は、約3未満のpHに酸性化され得る。上記分離する工程は、上記溶液を冷却する工程を包含し得る。上記溶液は、約30℃未満、好ましくは0℃未満の温度へと冷却され得る。
【0092】
再結晶化は、有機溶媒を使用し得る。上記有機溶媒は、極性非プロトン性溶媒(例えば、酢酸、ブタノール、イソプロパノール、プロパノール、エタノール、メタノール、ギ酸、水)、極性プロトン性溶媒(例えば、ジオキサン、テトラヒドロフラン、ジクロロメタン、アセトン、アセトニトリル、ジメチルホルムアミド、ジメチルスルホキシド)、および非極性溶媒(例えば、ヘキサン、ベンゼン、トルエン、ジエチルエーテル、クロロホルム、酢酸エチル)のうちの1種以上を含み得る。
【0093】
特定の実施形態において、上記方法は、上記分離されたcis,trans−ムコネートから塩を除去する工程を包含する。上記塩は、無機塩を含み得る。
【0094】
特定の実施形態において、上記方法は、上記cis,trans−ムコネートのうちの少なくとも約50%を、trans,trans−ムコネートに、および好ましくは、95%より多くを異性化する工程を包含する。
【0095】
図11は、6時間ごとに、IPTG(100mM,10mL)を使用してOD600=33において誘導したcis,trans−ムコン酸の生成についての、WN1/pWN2.248の20Lバッチ培養の結果を示す。約88時間後に、上記ムコン酸タイターは、59g/L(30%収率)であった。合成されたムコン酸の総量は、1475gであった。これは、約88時間で約1.38M グルコースを0.42M cis,trans−ムコン酸に変換するのに対応する。
【0096】
(実施例8:ファーメンター中でのcis,cis−ムコネートの、cis,trans−ムコネートへのインサイチュ異性化)
生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネート(例えば、実施例7および実施例7Aの方法に従って)を提供した。上記cis,cis−ムコネートを含むその発酵培養物を、60℃に加温した。上記加温した発酵培養物を、0.5時間にわたって2N 硫酸を添加することによって、pH4に調節した。上記酸性化した培養物を、3.5時間にわたって反応させた。
【0097】
上記反応を、1H NMRおよびPrevail Organic Acid Column(150mm×4.6mm)を備えたHPLCによってモニターして、上記反応のエンドポイントを決定した。これらデータを、図7〜10において、中性pHにおけるコントロール実験とともに示す。一般に、このような異性化反応は、適切な反応パラメーター(例えば、時間、温度、pHなど)を決定するためにモニターし得る。
【0098】
図5は、粗製の発酵ブロス中で、pH7におけるcis,cis−ムコネートからcis,trans−ムコネートへの異性化反応の1H NMRによる追跡を示す。0〜1.25時間および3.25時間までの時間追跡を、中性pH(例えば、これは、実際の発酵の間における近似のpHレベルである)においてcis,cis ムコン酸からcis,trans ムコン酸への異性化が本質的にないことを実証する。従って、cis,cis−ムコネートの異性化は、実際の発酵の間には全くもしくは無視できる程度にしか起こっていない。
【0099】
図6は、粗製発酵ブロスにおけるpH4でのムコネート異性化反応についての1H NMRによる追跡を示す。0〜1.25時間および3.25時間までの時間追跡は、cis,cis ムコン酸からcis,trans−ムコン酸への異性化が、酸性pHにおいて急速に進み、上記異性化は、約1.25時間後に本質的に完了することを実証する。
【0100】
図7は、pH7におけるムコネート異性化反応についてのHPLCによる追跡を示す。図9は、pH7におけるムコネート異性化反応の時間経過を示す。1H NMRによる追跡と同様に、これらHPLCによる追跡および時間経過は、中性pHにおけるcis,cis ムコン酸からcis,trans−ムコン酸への異性化が本質的にないことを実証する。
【0101】
図8は、pH4におけるムコネート異性化反応についてのHPLCによる追跡を示す。図10は、pH4におけるムコネート異性化反応の時間経過を示す。1H NMRによる追跡と同様に、これらHPLCによる追跡および時間経過は、cis,cis−ムコン酸からcis,trans−ムコン酸への異性化は、酸性pHにおいて急速に進み、上記異性化は、約1.25時間後に本質的に完了することを実証する。
【0102】
(実施例9:酸性化、沈殿、および濾過による発酵ブロスからのcis,trans−ムコン酸の分離)
異性化後に(例えば、実施例8におけるように)、cis,trans−ムコン酸を含むブロスをほぼ周囲温度へと冷却し、上記細胞、細胞砕片および沈殿した固体を、遠心分離によって培養ブロスから除去した。あるいは、このような固体を、濾過によって除去し得る(例えば、100kD SARTOCON(登録商標) Sliceカセットを介して)。次いで、上記無細胞ブロスを濾過によって清澄にして、タンパク質を除去した(例えば、10kD SARTOCON(登録商標) Sliceカセットを介して)。
【0103】
濾過後に、上記清澄にしたブロスのpHを、濃硫酸を添加することによってpH1.5に調節した。種々のpH値において沈殿するcis,trans−ムコネートの量を、表2に示す。
【0104】
表2.異なるpH値におけるcis,trans−ムコネートの沈殿
【0105】
【表2】
上記酸性化したブロスを、撹拌せずに1.5時間にわたって4℃へと冷却したところ、その時間の間に、粗製cis,trans−ムコン酸は、わずかに黄色の固体として沈殿した。この物質を濾過によって回収したところ、上記清澄化したブロスに存在する上記cis,trans−ムコン酸のうちの約60%を構成した。上記沈殿は、より長い期間(例えば、一晩)にわたって、そして/またはより低温(例えば、−20℃)において継続し、塩の夾雑を減少させながら生成物の回収を増大させられ得る。
【0106】
上記濾液は、さらなるcis,trans−ムコン酸を含んだ。上記さらなるcis,trans−ムコン酸を回収するために、上記濾液を減圧下でエバポレートして、その容積を約50%減らした。上記濃縮した濾液を、−20℃へと一晩冷却したところ、その時間の間に、粗製cis,trans−ムコン酸の第2の収穫物が沈殿した。上記沈殿物を、濾過により再び回収した。
【0107】
上記粗製cis,trans−ムコン酸固体を合わせ、アセトニトリルを使用して結晶化して、精製されたcis,trans−ムコネートを白色固体として生成させた。メタノールを使用する結晶化は、同様の収率を提供した。メタノールはまた、上記精製された生成物における塩の夾雑を減らした。
【0108】
図4Aは、結晶化したcis,trans−ムコン酸の1H NMRスペクトルを示す。図4Bは、グルコースを欠く最小塩類培地中に再懸濁した結晶化したcis,trans−ムコン酸の1H NMRスペクトルを示す。上記図4Bのスペクトルは、上記発酵ブロス中の他の成分によって引き起こされるcis,trans−ムコン酸のNMRスペクトルシフトに近似し、従って、上記発酵ブロス中での上記cis,cisからcis,transへの異性化反応を比較するためモニターすることを可能にする。
【0109】
(実施例10:有機溶媒を使用する発酵ブロスからのcis,trans−ムコン酸の抽出)
有利なことには、cis,trans−ムコン酸は、驚くべきことにかつ予測外に、上記cis,cis異性体もしくはtrans,trans異性体のいずれよりも、有機溶媒中で可溶性である。従って、分離する工程(例えば、実施例8の分離する工程)は、上記cis,trans−ムコネートを溶液(例えば、発酵ブロス)から、有機溶媒を使用して抽出する工程を包含し得る。
【0110】
上記cis,trans−ムコネートが抽出される上記溶液は、培養発酵ブロス全体もしくは、無細胞でタンパク質を含まない発酵ブロスであり得る。例えば、濾過(例えば、上記ブロスを、0.1μM 中空ファイバー濾過ユニットに通過させる)によって、細胞をブロスから除去し得る。例えば、濾過によって(例えば、例えば、SARTOCON(登録商標)から入手可能な10kD 接線流濾過システムを介して)、タンパク質は、ブロスから除去され得る。
【0111】
抽出用の上記有機溶媒(例えば、水相と非混和性の溶媒)としては、例えば、メチルイソブチルケトン(MIBK)、酢酸エチル、酢酸イソプロピル(酢酸プロピル)、ヘプタン(混合物)、メチル tert−ブチルエーテル、キシレン、塩化メチレン、シクロヘキサノール、デカリン、テトラリン、テトラロン、シクロヘキサン、酢酸ブチル、メチルテトラヒドロフラン(THF)、シクロヘキサノン/シクロヘキサノール(市販の混合物)、1−オクタノール、イソアミルアルコール、および2−エチルヘキサノールのうちの1種以上が挙げられ得る。
【0112】
水相に添加され得、上記cis,trans−ムコネートの抽出および同時もしくはその後のエステル化をともに促進する他の有機溶媒としては、例えば、メタノール、エタノール、プロパノール、イソプロパノール、酢酸、アセトニトリル、およびアセトン、ならびにブタノール(例えば、1−ブタノールおよびイソブタノール)および水と完全には混和性でない他のアルコールのうちの1種以上が挙げられる。
【0113】
上記溶媒抽出は、約4未満のpHにおいて(例えば、上記cis,trans−ムコン酸が十分にプロトン化され、上記抽出に使用される上記有機溶媒へと分配されるpHにおいて)行われ得る。沈殿を誘導するに十分低いpHレベルですら、上記プロトン化したcis,trans−ムコン酸の画分は、溶液中に残り得る。例えば、および上記実施例9の表2に示されるように、pH3において、本来は上記溶液中にある上記cis,trans−ムコン酸のうちの約60%は、沈殿し、濾過によって分離され得る。しかし、溶液に残っている上記cis,trans−ムコン酸のうちの約40%は、濾過によって分離できないが、抽出によって回収できる。よって、溶媒抽出は、有機溶媒を使用して溶液から上記cis,trans−ムコン酸を抽出する工程を包含しない方法と比較して、cis,trans−ムコネートの単離収率を増大させ得る。cis,trans−ムコネートのいくらかの部分が沈殿し、上記水溶液中にないとしても、上記cis,trans−ムコネートの抽出を進め得ることはまた、実施例9の表2中のデータから明らかである。
【0114】
上記溶媒抽出はまた、上記cis,trans−ムコネートを無機塩(例えば、硫酸アンモニウム、硫酸カルシウム)から分離する工程を包含し得る。さらに、溶媒抽出は、やはり無機塩の沈殿を引き起こし、従って無機塩との夾雑を引き起こし得る沈殿より純粋なcis,trans−ムコネートを生成し得る。
【0115】
溶媒および/もしくはpHパラメーターの選択は、一連の単純な測定によって促進され得る。各可能性のある溶媒について、3種すべてのムコン酸異性体(cis,cis−異性体、cis,trans−異性体、およびtrans,trans−異性体)に対して抽出が行われ得る(例えば、分配係数を測定するために)。各異性体はまた、約pH1〜pH7未満の間の増分(例えば、0.5)を使用したpH値の範囲に対して試験され得る。
【0116】
(実施例11:溶媒抽出による発酵ブロスからのcis,trans−ムコン酸の分離)
cis,trans−ムコン酸に異性化した発酵ブロスを得、これを、pH約3へと酸性化した。上記固体cis,trans−ムコン酸を濾過によって取り出して、約5〜10g/Lのcis,trans−ムコン酸を含む酸性化した発酵ブロスにした。
【0117】
15mLの上記濾過したブロスを各々含む個々の50mLコニカル遠心チューブに、以下の表に列挙される各溶媒15mLを添加した。各チューブを、2分間にわたって撹拌し、その有機相および水相を、分離させた。上記水相をピペットによって上記溶媒層から分離し、新しい50mLコニカルチューブに入れた。各抽出の上記水相および溶媒相の両方を、HPLCを使用してムコン酸について分析した。第2の抽出を、新鮮な溶媒を使用して、上記分離した水層の各々に対して行った。再び、上記サンプルを撹拌し、沈殿させ、上記水相および有機相を分離し、分析した。
【0118】
結果を、以下の表3、表4および表5に示す。cis,trans−ムコン酸の異なる量を有する異なるブロスサンプルを使用して、各表における結果を得た。
【0119】
表3. 9.82g/L cis,trans−ムコン酸を含むブロス
【0120】
【表3】
表4.10.69g/L cis,trans−ムコン酸を含むブロス
【0121】
【表4】
表5.4.86g/L cis,trans−ムコン酸を含むブロス
【0122】
【表5】
上記cis,trans−ムコン酸の十分量が沈殿した十分に低いレベルへと酸性化した後に、溶媒抽出がcis,trans−ムコン酸を回収する能力を試験するために、M9塩の溶液中のcis,trans−ムコン酸の溶液を使用して、発酵ブロスを模倣した。15gのcis,trans−ムコン酸を、約60g/Lのタイターを与える250mLのM9塩に添加した。これは、水酸化ナトリウムを使用して、上記pHを7.0へと上昇させることによって達成した。次いで、上記ブロスを、硫酸でpH3へと酸性化したところ、上記cis,trans−ムコン酸の沈殿が生じた。上記固体沈殿物を上記酸性混合物中に残し、スラリー全体を、上記に記載されるように、溶媒で2回抽出した。結果を、表6に示す。
【0123】
表6.沈殿したムコン酸を含め、63.34g/L cis,trans−ムコン酸を含むブロス
【0124】
【表6】
(実施例12:ヨウ素によって触媒される、cis,trans−ムコン酸の、trans,trans−ムコン酸への異性化)
cis,trans−ムコン酸(1.00g)、触媒量のI2(53mg)、およびMeCN(35ml)を含む混合物を加熱して、11時間にわたって還流した。室温へと冷却した後、その沈殿した固体を濾過し、冷MeCNで洗浄した。真空下で乾燥させた後、0.80g(80%収率)の精製trans,trans−ムコン酸は、黄褐色粉末として存在した。この手順によって得られた物質は、1Hおよび13C NMR分光法によって、trans,trans−ムコン酸であると確認された。上記異性化反応は、多くの他の試験した溶媒中よりも、非極性溶媒(例えば、THF)中でより進行した。
【0125】
(実施例13:水素化触媒によって触媒される、cis,trans−ムコン酸の、trans,trans−ムコン酸への異性化)
cis,trans−ムコン酸(1.00g)および触媒量のパラジウム担持炭素(Pd/C,5%)を含む混合物を、50mLのメタノール中に調製する。上記メタノール反応混合物を、1時間にわたって還流させ、室温へと冷却し、次いで、上記担持されたパラジウム触媒を、濾過によって除去する。残った反応溶液を、元の容積の約1/2へとエバポレートし、次いで、1容積のMeCNで希釈する。減圧下でのエバポレーションを、上記メタノールが除去されかつ上記trans,trans−ムコン酸が溶液から落ち始める(begins to fall out)まで継続する。得られた固体を濾過し、冷MeCNで洗浄する。真空下で乾燥させた後、約0.80g(80%収率)の精製trans,trans−ムコン酸が、黄褐色粉末として存在し得る。この手順によって得られる物質は、1Hおよび13C NMR分光法によって、trans,trans−ムコン酸であると確認され得る。
【0126】
(実施例14:水素化触媒によって触媒される、cis,cis−ムコン酸の、trans,trans−ムコン酸への異性化)
cis,cis−ムコン酸(1.00g)および触媒量のパラジウム担持炭素(Pd/C,5%)を含む混合物を、50mLのメタノール中に調製する。上記メタノール反応混合物を、1時間にわたって還流させ、室温へと冷却し、次いで、上記担持されたパラジウム触媒を、濾過によって除去する。残った反応溶液を、元の容積の約1/2になるまでエバポレートし、次いで、1容積のMeCNで希釈する。減圧下でのエバポレーションを、上記メタノールが除去されかつ上記trans,trans−ムコン酸が溶液から落ち始めるまで継続する。得られた固体を濾過し、冷MeCNで洗浄する。真空下で乾燥させた後、約0.80g(80%収率)の精製trans,trans−ムコン酸が、黄褐色粉末として存在し得る。この手順によって得られる物質は、1Hおよび13C NMR分光法によって、trans,trans−ムコン酸であると確認され得る。
【0127】
本発明は、具体的に示され、そして特定の実施形態を参照して記載されてきたが、形態および詳細における種々の変更が、添付の特許請求の範囲によって定義される発明の趣旨および範囲から逸脱することなくなされ得ることは、当業者によって理解されるべきである。
【図4A】
【図4B】
【特許請求の範囲】
【請求項1】
cis,trans−ムコネートを生成するための方法であって、該方法は、
生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネートを提供する工程;
該cis,cis−ムコネートの実質的にすべてがcis,trans−ムコネートへと異性化される反応条件下で、cis,cis−ムコネートをcis,trans−ムコネートへと異性化する工程;
該cis,trans−ムコネートを分離する工程;および
該cis,trans−ムコネートを結晶化する工程
を包含する、方法。
【請求項2】
3−デヒドロシキミ酸脱水酵素、プロトカテク酸デカルボキシラーゼおよびカテコール 1,2−ジオキシゲナーゼを発現する組換え細胞を、前記再生可能炭素源を含む培地中で、かつ該再生可能炭素源が、該細胞の芳香族アミノ酸生合成の共通経路における酵素類によって3−デヒドロシキメートに変換され、かつ、該3−デヒドロシキメートがcis,cis−ムコネートへと生体触媒により変換される条件下で培養する工程、
をさらに包含する、請求項1に記載の方法。
【請求項3】
前記異性化反応は、酸により触媒され、ここで該酸は、無機酸もしくは有機酸であり得る、請求項1に記載の方法。
【請求項4】
前記異性化反応は、約3.0〜約6.5の間、もしくは約3.5〜約4.5の間のpHにおいて溶液中で行われる、請求項1に記載の方法。
【請求項5】
前記異性化反応は、47℃以上の温度、もしくは約60℃以上の温度において行われる、請求項1に記載の方法。
【請求項6】
前記異性化反応は、8時間内に、7.75時間内に、7.5時間内に、7.25時間内に、7時間内に、6.75時間内に、6.5時間内に、6.25時間内に、6時間内に、5.75時間内に、5.5時間内に、5.25時間内に、5時間内に、4.75時間内に、4.5時間内に、4.25時間内に、4時間内に、3.75時間内に、3.5時間内に、3.25時間内に、3時間内に、2.75時間内に、2.5時間内に、2.25時間内に、2時間内に、1.75時間内に、1.5時間内に、1.25時間内に、1時間内に、0.75時間内に、0.5時間内に、もしくは0.25時間内に実質的に完了する、請求項1に記載の方法。
【請求項7】
前記異性化反応は、実質的にcis,trans−ムコネートの沈殿なしで進行する、請求項1に記載の方法。
【請求項8】
請求項2に記載の方法であって、ここで、前記培養する工程は、前記組換え細胞および細胞外cis,trans−ムコネートを含むブロスを生成し、かつ、該組換え細胞を該ブロスから除去する工程をさらに包含する、方法。
【請求項9】
前記分離する工程は、前記cis,trans−ムコネートを酸性化によって溶液から沈殿させる工程を包含し、ここで好ましくは、該溶液は、約3.0未満、もしくは約2.0未満のpHへと酸性化される、請求項1に記載の方法。
【請求項10】
前記分離する工程は、約37℃未満、約25℃未満、約−4℃未満、もしくは約−20℃未満の温度へと前記溶液を冷却する工程をさらに包含する、請求項9に記載の方法。
【請求項11】
cis,cis−ムコネートのcis,trans−ムコネートへの前記異性化をモニターする工程をさらに包含する、請求項1に記載の方法。
【請求項12】
前記cis,trans−ムコネートは、有機溶媒で結晶化され、ここで好ましくは、該有機溶媒は、メタノール、エタノール、プロパノール、イソプロパノール、ブタノール、酢酸、アセトニトリル、アセトン、およびテトラヒドロフランのうちの1種以上を含む、請求項1に記載の方法。
【請求項13】
前記分離されたcis,trans−ムコネートから塩を除去する工程をさらに包含し、ここで好ましくは、該塩は、無機塩を含む、請求項1に記載の方法。
【請求項14】
cis,trans−ムコネートの第1の収穫物を沈殿させた後に、前記溶液を濃縮する工程;および
cis,trans−ムコネートの第2の収穫物を、濃縮した溶液から沈殿させる工程、
をさらに包含する、請求項9に記載の方法。
【請求項15】
前記異性化反応は、ほぼ大気圧より高い圧力で行われる、請求項1に記載の方法。
【請求項16】
前記cis,trans−ムコネートのうちの少なくとも約65%をtrans,trans−ムコネートへと、該cis,trans−ムコネートのうちの少なくとも約75%をtrans,trans−ムコネートへと、該cis,trans−ムコネートのうちの少なくとも約85%をtrans,trans−ムコネートへと、もしくは該cis,trans−ムコネートのうちの少なくとも約95%をtrans,trans−ムコネートへと異性化する工程をさらに包含する、請求項1に記載の方法。
【請求項17】
前記cis,trans−ムコネートは、I2によって、貴金属水素化触媒によって、スポンジメタル水素化触媒によって、もしくは骨格水素化触媒によって触媒される反応において、trans,trans−ムコネートに異性化される、請求項16に記載の方法。
【請求項18】
cis,trans−ムコネートを生成するための方法であって、該方法は、
生体触媒による変換を介して再生可能炭素源から生成されるcis,cis−ムコネートを含む発酵ブロスを提供する工程;
cis,cis−ムコネートのうちの実質的にすべてが、cis,trans−ムコネートへと異性化される反応条件下で、該cis,cis−ムコネートをcis,trans−ムコネートへと異性化する工程;
該cis,trans−ムコネートを該ブロスから分離する工程;および
該cis,trans−ムコネートを結晶化する工程、
を包含する、方法。
【請求項19】
前記発酵ブロスは、3−デヒドロシキミ酸脱水酵素、プロトカテク酸デカルボキシラーゼおよびカテコール 1,2−ジオキシゲナーゼを発現する組換え細胞を含む、請求項18に記載の方法。
【請求項20】
前記組換え細胞を前記発酵ブロスから除去する工程をさらに包含する、請求項19に記載の方法。
【請求項21】
前記発酵ブロスは容器中に提供され、ここで、前記異性化反応は、該容器中で行われ、ここで好ましくは、該容器は、発酵槽容器である、請求項18に記載の方法。
【請求項22】
3−デヒドロシキミ酸脱水酵素、プロトカテク酸デカルボキシラーゼおよびカテコール 1,2−ジオキシゲナーゼを発現する組換え細胞を、前記再生可能炭素源を含む培地中で、かつ該再生可能炭素源が、該細胞の芳香族アミノ酸生合成の共通経路における酵素類によって3−デヒドロシキメートへと変換され、かつ、該3−デヒドロシキメートは、cis,cis−ムコネートへと生体触媒により変換される条件下で、培養する工程、
をさらに包含し、
ここで該組換え細胞は、前記発酵槽容器中で培養され、それによって、前記発酵ブロスを生成する、
請求項21に記載の方法。
【請求項23】
前記分離する工程は、前記cis,trans−ムコネートを、有機溶媒を使用して溶液から抽出する工程を包含する、請求項1または18に記載の方法。
【請求項24】
前記有機溶媒は、メタノール、エタノール、プロパノール、イソプロパノール、ブタノール、酢酸、アセトニトリル、アセトン、およびテトラヒドロフランのうちの1種以上を含む、請求項23に記載の方法。
【請求項25】
前記分離する工程は、約7未満、約6.75未満、約6.5未満、約6.25未満、約6未満、約5.75未満、約5.5未満、約5.25未満、約5未満、約4.75未満、約4.5未満、約4.25未満、約4未満、約3.75未満、約3.5未満、約3.25未満、もしくは約3未満のpHにおいて行われる、請求項23に記載の方法。
【請求項26】
前記分離する工程は、前記cis,trans−ムコネートを無機塩から分離する工程をさらに包含する、請求項23に記載の方法。
【請求項27】
前記分離する工程は、有機溶媒を使用して溶液から前記cis,trans−ムコネートを抽出する工程を含まない方法と比較して、cis,trans−ムコネートの単離収率を増大させる、請求項23に記載の方法。
【請求項28】
生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネートを提供する工程は、酵素3−デヒドロシキミ酸脱水酵素およびプロトカテク酸デカルボキシラーゼを発現するKlebsiella pneumoniaeに由来する異種構造遺伝子、ならびに酵素カテコール 1,2−ジオキシゲナーゼを発現するAcinetobacter calcoaceticusに由来する異種構造遺伝子で形質転換された細菌細胞を使用し、ここで該細菌細胞の培養は、約88時間以内に、少なくとも約1.38M グルコースを少なくとも約0.42M cis,cis−ムコン酸へと生体触媒により変換する、請求項1に記載の方法。
【請求項29】
酵素3−デオキシ−D−アラビノ−ヘプツロソン酸 7−リン酸シンターゼおよび3−デヒドロキナ酸シンターゼを発現する異種DNA配列をさらに含む、請求項28に記載の細菌細胞形質転換体。
【請求項30】
酵素トランスケトラーゼを発現する異種DNA配列をさらに含む、請求項29に記載の細菌細胞形質転換体。
【請求項31】
前記細菌細胞は、芳香族アミノ酸生合成の共通経路における変異を有し、3−デヒドロシキメートのコリスメートへの変換をブロックする変異細胞系から選択される、請求項30に記載の細菌細胞形質転換体。
【請求項32】
前記細菌細胞は、芳香族アミノ酸生合成の共通経路における変異を有し、3−デヒドロシキメートのコリスメートへの変換をブロックする変異細胞系から選択される、請求項28に記載の細菌細胞形質転換体。
【請求項33】
生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネートを提供する工程は、酵素種3−デヒドロシキミ酸脱水酵素およびプロトカテク酸デカルボキシラーゼを発現するKlebsiella pneumoniaeに由来する構造遺伝子、ならびに酵素種カテコール 1,2−ジオキシゲナーゼを発現するAcinetobacter calcoaceticusに由来する構造遺伝子で形質転換された細菌細胞を、該細胞の芳香族アミノ酸生合成の共通経路における前記酵素類によって3−デヒドロシキメートへと変換される炭素源を含む培地中で培養して、3−デヒドロシキメートの生体触媒による変換によって少なくとも約0.95ミリモル/リットル/時間の速度でcis,cis−ムコン酸を生成する工程を包含する、請求項1に記載の方法。
【請求項34】
生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネートを提供する工程は、3−デヒドロシキミ酸脱水酵素、プロトカテク酸デカルボキシラーゼ、カテコール 1,2−ジオキシゲナーゼ、トランスケトラーゼ、3−デオキシ−D−アラビノ−ヘプツロソン酸 7−リン酸シンターゼ、および3−デヒドロキナ酸シンターゼをコードする異種構造遺伝子を発現する形質転換された細菌細胞を、該細胞の芳香族アミノ酸生合成の共通経路における前記酵素類によって3−デヒドロシキメートに変換される炭素源を含む培地中で培養して、3−デヒドロシキメートの生体触媒による変換によって少なくとも約0.95ミリモル/リットル/時間の速度でcis,cis−ムコン酸を生成する工程を包含する、請求項1に記載の方法。
【請求項35】
生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネートを提供する工程は、酵素種3−デヒドロシキミ酸脱水酵素およびプロトカテク酸デカルボキシラーゼを発現するKlebsiella pneumoniaeに由来する構造遺伝子、ならびに酵素カテコール 1,2−ジオキシゲナーゼを発現するAcinetobacter calcoaceticusに由来する構造遺伝子で形質転換された細菌細胞を、炭素源を含む培地中で、該炭素源が、少なくとも約0.95ミリモル/リットル/時間の速度でcis,cis−ムコン酸へと生体触媒により変換される条件下で、培養する工程を包含する、請求項1に記載の方法。
【請求項36】
請求項1〜15および18〜35のいずれか1項に記載の方法によって生成される、再生可能なcis,trans−ムコネート。
【請求項37】
trans,trans−ムコネートを生成するための方法であって、該方法は、
生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネートを、該cis,cis−ムコネートのうちの実質的にすべてがtrans,trans−ムコネートへと異性化される反応条件下で、trans,trans−ムコネートへと異性化する工程
を包含する、方法。
【請求項38】
前記異性化反応は、貴金属水素化触媒によって、スポンジメタル水素化触媒によって、もしくは骨格水素化触媒によって触媒される、請求項37に記載の方法。
【請求項39】
trans,trans−ムコネートを生成するための方法であって、該方法は、
生体触媒による変換を介して再生可能炭素源から生成されたcis,trans−ムコネートを、該cis,trans−ムコネートのうちの実質的にすべてがtrans,trans−ムコネートへと異性化される反応条件下で、trans,trans−ムコネートへと異性化する工程、
を包含する、方法。
【請求項40】
前記異性化反応は、貴金属水素化触媒によって、スポンジメタル水素化触媒によって、もしくは骨格水素化触媒によって触媒される、請求項39に記載の方法。
【請求項41】
請求項16〜17および37〜40のいずれか1項に記載の方法によって生成された、再生可能なtrans,trans−ムコネート。
【請求項1】
cis,trans−ムコネートを生成するための方法であって、該方法は、
生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネートを提供する工程;
該cis,cis−ムコネートの実質的にすべてがcis,trans−ムコネートへと異性化される反応条件下で、cis,cis−ムコネートをcis,trans−ムコネートへと異性化する工程;
該cis,trans−ムコネートを分離する工程;および
該cis,trans−ムコネートを結晶化する工程
を包含する、方法。
【請求項2】
3−デヒドロシキミ酸脱水酵素、プロトカテク酸デカルボキシラーゼおよびカテコール 1,2−ジオキシゲナーゼを発現する組換え細胞を、前記再生可能炭素源を含む培地中で、かつ該再生可能炭素源が、該細胞の芳香族アミノ酸生合成の共通経路における酵素類によって3−デヒドロシキメートに変換され、かつ、該3−デヒドロシキメートがcis,cis−ムコネートへと生体触媒により変換される条件下で培養する工程、
をさらに包含する、請求項1に記載の方法。
【請求項3】
前記異性化反応は、酸により触媒され、ここで該酸は、無機酸もしくは有機酸であり得る、請求項1に記載の方法。
【請求項4】
前記異性化反応は、約3.0〜約6.5の間、もしくは約3.5〜約4.5の間のpHにおいて溶液中で行われる、請求項1に記載の方法。
【請求項5】
前記異性化反応は、47℃以上の温度、もしくは約60℃以上の温度において行われる、請求項1に記載の方法。
【請求項6】
前記異性化反応は、8時間内に、7.75時間内に、7.5時間内に、7.25時間内に、7時間内に、6.75時間内に、6.5時間内に、6.25時間内に、6時間内に、5.75時間内に、5.5時間内に、5.25時間内に、5時間内に、4.75時間内に、4.5時間内に、4.25時間内に、4時間内に、3.75時間内に、3.5時間内に、3.25時間内に、3時間内に、2.75時間内に、2.5時間内に、2.25時間内に、2時間内に、1.75時間内に、1.5時間内に、1.25時間内に、1時間内に、0.75時間内に、0.5時間内に、もしくは0.25時間内に実質的に完了する、請求項1に記載の方法。
【請求項7】
前記異性化反応は、実質的にcis,trans−ムコネートの沈殿なしで進行する、請求項1に記載の方法。
【請求項8】
請求項2に記載の方法であって、ここで、前記培養する工程は、前記組換え細胞および細胞外cis,trans−ムコネートを含むブロスを生成し、かつ、該組換え細胞を該ブロスから除去する工程をさらに包含する、方法。
【請求項9】
前記分離する工程は、前記cis,trans−ムコネートを酸性化によって溶液から沈殿させる工程を包含し、ここで好ましくは、該溶液は、約3.0未満、もしくは約2.0未満のpHへと酸性化される、請求項1に記載の方法。
【請求項10】
前記分離する工程は、約37℃未満、約25℃未満、約−4℃未満、もしくは約−20℃未満の温度へと前記溶液を冷却する工程をさらに包含する、請求項9に記載の方法。
【請求項11】
cis,cis−ムコネートのcis,trans−ムコネートへの前記異性化をモニターする工程をさらに包含する、請求項1に記載の方法。
【請求項12】
前記cis,trans−ムコネートは、有機溶媒で結晶化され、ここで好ましくは、該有機溶媒は、メタノール、エタノール、プロパノール、イソプロパノール、ブタノール、酢酸、アセトニトリル、アセトン、およびテトラヒドロフランのうちの1種以上を含む、請求項1に記載の方法。
【請求項13】
前記分離されたcis,trans−ムコネートから塩を除去する工程をさらに包含し、ここで好ましくは、該塩は、無機塩を含む、請求項1に記載の方法。
【請求項14】
cis,trans−ムコネートの第1の収穫物を沈殿させた後に、前記溶液を濃縮する工程;および
cis,trans−ムコネートの第2の収穫物を、濃縮した溶液から沈殿させる工程、
をさらに包含する、請求項9に記載の方法。
【請求項15】
前記異性化反応は、ほぼ大気圧より高い圧力で行われる、請求項1に記載の方法。
【請求項16】
前記cis,trans−ムコネートのうちの少なくとも約65%をtrans,trans−ムコネートへと、該cis,trans−ムコネートのうちの少なくとも約75%をtrans,trans−ムコネートへと、該cis,trans−ムコネートのうちの少なくとも約85%をtrans,trans−ムコネートへと、もしくは該cis,trans−ムコネートのうちの少なくとも約95%をtrans,trans−ムコネートへと異性化する工程をさらに包含する、請求項1に記載の方法。
【請求項17】
前記cis,trans−ムコネートは、I2によって、貴金属水素化触媒によって、スポンジメタル水素化触媒によって、もしくは骨格水素化触媒によって触媒される反応において、trans,trans−ムコネートに異性化される、請求項16に記載の方法。
【請求項18】
cis,trans−ムコネートを生成するための方法であって、該方法は、
生体触媒による変換を介して再生可能炭素源から生成されるcis,cis−ムコネートを含む発酵ブロスを提供する工程;
cis,cis−ムコネートのうちの実質的にすべてが、cis,trans−ムコネートへと異性化される反応条件下で、該cis,cis−ムコネートをcis,trans−ムコネートへと異性化する工程;
該cis,trans−ムコネートを該ブロスから分離する工程;および
該cis,trans−ムコネートを結晶化する工程、
を包含する、方法。
【請求項19】
前記発酵ブロスは、3−デヒドロシキミ酸脱水酵素、プロトカテク酸デカルボキシラーゼおよびカテコール 1,2−ジオキシゲナーゼを発現する組換え細胞を含む、請求項18に記載の方法。
【請求項20】
前記組換え細胞を前記発酵ブロスから除去する工程をさらに包含する、請求項19に記載の方法。
【請求項21】
前記発酵ブロスは容器中に提供され、ここで、前記異性化反応は、該容器中で行われ、ここで好ましくは、該容器は、発酵槽容器である、請求項18に記載の方法。
【請求項22】
3−デヒドロシキミ酸脱水酵素、プロトカテク酸デカルボキシラーゼおよびカテコール 1,2−ジオキシゲナーゼを発現する組換え細胞を、前記再生可能炭素源を含む培地中で、かつ該再生可能炭素源が、該細胞の芳香族アミノ酸生合成の共通経路における酵素類によって3−デヒドロシキメートへと変換され、かつ、該3−デヒドロシキメートは、cis,cis−ムコネートへと生体触媒により変換される条件下で、培養する工程、
をさらに包含し、
ここで該組換え細胞は、前記発酵槽容器中で培養され、それによって、前記発酵ブロスを生成する、
請求項21に記載の方法。
【請求項23】
前記分離する工程は、前記cis,trans−ムコネートを、有機溶媒を使用して溶液から抽出する工程を包含する、請求項1または18に記載の方法。
【請求項24】
前記有機溶媒は、メタノール、エタノール、プロパノール、イソプロパノール、ブタノール、酢酸、アセトニトリル、アセトン、およびテトラヒドロフランのうちの1種以上を含む、請求項23に記載の方法。
【請求項25】
前記分離する工程は、約7未満、約6.75未満、約6.5未満、約6.25未満、約6未満、約5.75未満、約5.5未満、約5.25未満、約5未満、約4.75未満、約4.5未満、約4.25未満、約4未満、約3.75未満、約3.5未満、約3.25未満、もしくは約3未満のpHにおいて行われる、請求項23に記載の方法。
【請求項26】
前記分離する工程は、前記cis,trans−ムコネートを無機塩から分離する工程をさらに包含する、請求項23に記載の方法。
【請求項27】
前記分離する工程は、有機溶媒を使用して溶液から前記cis,trans−ムコネートを抽出する工程を含まない方法と比較して、cis,trans−ムコネートの単離収率を増大させる、請求項23に記載の方法。
【請求項28】
生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネートを提供する工程は、酵素3−デヒドロシキミ酸脱水酵素およびプロトカテク酸デカルボキシラーゼを発現するKlebsiella pneumoniaeに由来する異種構造遺伝子、ならびに酵素カテコール 1,2−ジオキシゲナーゼを発現するAcinetobacter calcoaceticusに由来する異種構造遺伝子で形質転換された細菌細胞を使用し、ここで該細菌細胞の培養は、約88時間以内に、少なくとも約1.38M グルコースを少なくとも約0.42M cis,cis−ムコン酸へと生体触媒により変換する、請求項1に記載の方法。
【請求項29】
酵素3−デオキシ−D−アラビノ−ヘプツロソン酸 7−リン酸シンターゼおよび3−デヒドロキナ酸シンターゼを発現する異種DNA配列をさらに含む、請求項28に記載の細菌細胞形質転換体。
【請求項30】
酵素トランスケトラーゼを発現する異種DNA配列をさらに含む、請求項29に記載の細菌細胞形質転換体。
【請求項31】
前記細菌細胞は、芳香族アミノ酸生合成の共通経路における変異を有し、3−デヒドロシキメートのコリスメートへの変換をブロックする変異細胞系から選択される、請求項30に記載の細菌細胞形質転換体。
【請求項32】
前記細菌細胞は、芳香族アミノ酸生合成の共通経路における変異を有し、3−デヒドロシキメートのコリスメートへの変換をブロックする変異細胞系から選択される、請求項28に記載の細菌細胞形質転換体。
【請求項33】
生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネートを提供する工程は、酵素種3−デヒドロシキミ酸脱水酵素およびプロトカテク酸デカルボキシラーゼを発現するKlebsiella pneumoniaeに由来する構造遺伝子、ならびに酵素種カテコール 1,2−ジオキシゲナーゼを発現するAcinetobacter calcoaceticusに由来する構造遺伝子で形質転換された細菌細胞を、該細胞の芳香族アミノ酸生合成の共通経路における前記酵素類によって3−デヒドロシキメートへと変換される炭素源を含む培地中で培養して、3−デヒドロシキメートの生体触媒による変換によって少なくとも約0.95ミリモル/リットル/時間の速度でcis,cis−ムコン酸を生成する工程を包含する、請求項1に記載の方法。
【請求項34】
生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネートを提供する工程は、3−デヒドロシキミ酸脱水酵素、プロトカテク酸デカルボキシラーゼ、カテコール 1,2−ジオキシゲナーゼ、トランスケトラーゼ、3−デオキシ−D−アラビノ−ヘプツロソン酸 7−リン酸シンターゼ、および3−デヒドロキナ酸シンターゼをコードする異種構造遺伝子を発現する形質転換された細菌細胞を、該細胞の芳香族アミノ酸生合成の共通経路における前記酵素類によって3−デヒドロシキメートに変換される炭素源を含む培地中で培養して、3−デヒドロシキメートの生体触媒による変換によって少なくとも約0.95ミリモル/リットル/時間の速度でcis,cis−ムコン酸を生成する工程を包含する、請求項1に記載の方法。
【請求項35】
生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネートを提供する工程は、酵素種3−デヒドロシキミ酸脱水酵素およびプロトカテク酸デカルボキシラーゼを発現するKlebsiella pneumoniaeに由来する構造遺伝子、ならびに酵素カテコール 1,2−ジオキシゲナーゼを発現するAcinetobacter calcoaceticusに由来する構造遺伝子で形質転換された細菌細胞を、炭素源を含む培地中で、該炭素源が、少なくとも約0.95ミリモル/リットル/時間の速度でcis,cis−ムコン酸へと生体触媒により変換される条件下で、培養する工程を包含する、請求項1に記載の方法。
【請求項36】
請求項1〜15および18〜35のいずれか1項に記載の方法によって生成される、再生可能なcis,trans−ムコネート。
【請求項37】
trans,trans−ムコネートを生成するための方法であって、該方法は、
生体触媒による変換を介して再生可能炭素源から生成されたcis,cis−ムコネートを、該cis,cis−ムコネートのうちの実質的にすべてがtrans,trans−ムコネートへと異性化される反応条件下で、trans,trans−ムコネートへと異性化する工程
を包含する、方法。
【請求項38】
前記異性化反応は、貴金属水素化触媒によって、スポンジメタル水素化触媒によって、もしくは骨格水素化触媒によって触媒される、請求項37に記載の方法。
【請求項39】
trans,trans−ムコネートを生成するための方法であって、該方法は、
生体触媒による変換を介して再生可能炭素源から生成されたcis,trans−ムコネートを、該cis,trans−ムコネートのうちの実質的にすべてがtrans,trans−ムコネートへと異性化される反応条件下で、trans,trans−ムコネートへと異性化する工程、
を包含する、方法。
【請求項40】
前記異性化反応は、貴金属水素化触媒によって、スポンジメタル水素化触媒によって、もしくは骨格水素化触媒によって触媒される、請求項39に記載の方法。
【請求項41】
請求項16〜17および37〜40のいずれか1項に記載の方法によって生成された、再生可能なtrans,trans−ムコネート。
【図1−1】

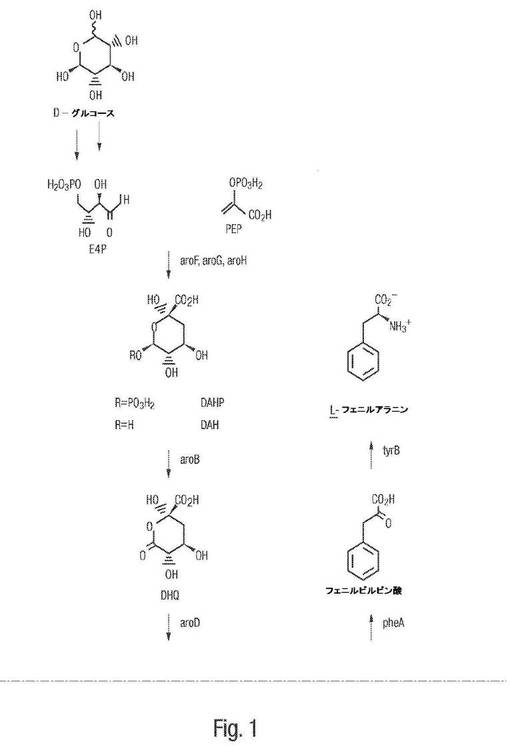
【図1−2】
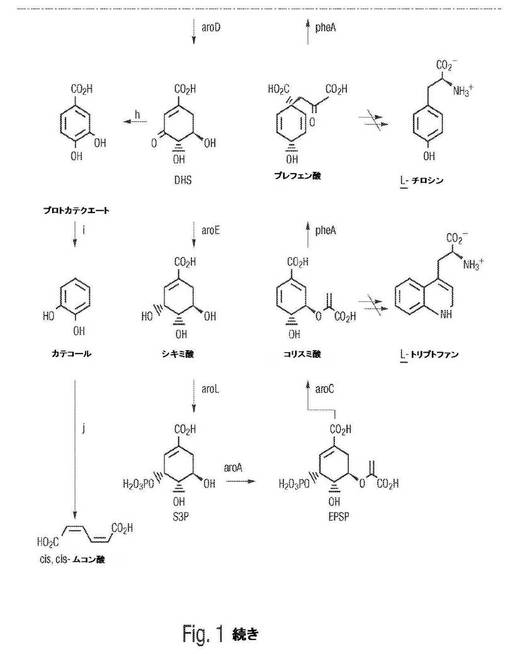
【図2】
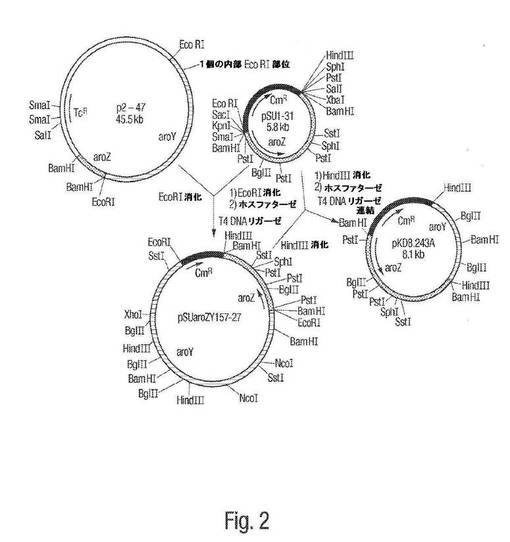
【図3】
【図5】
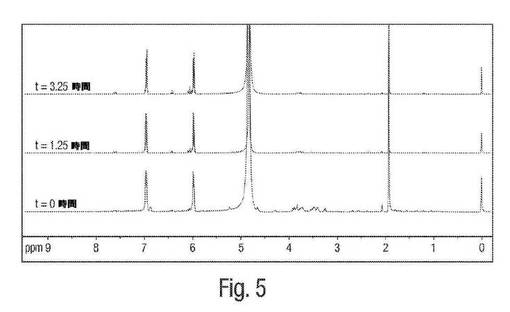
【図6】
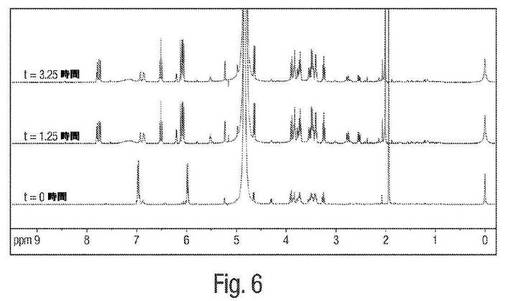
【図7】
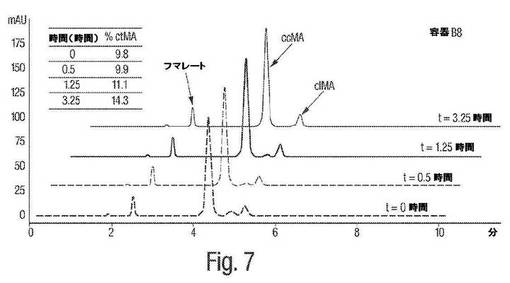
【図8】
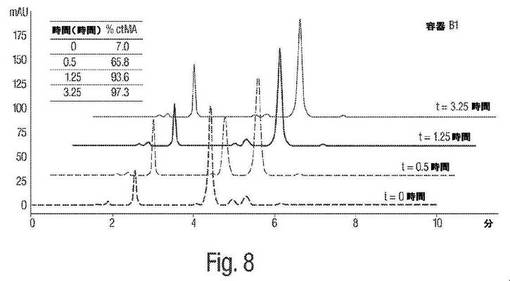
【図9】
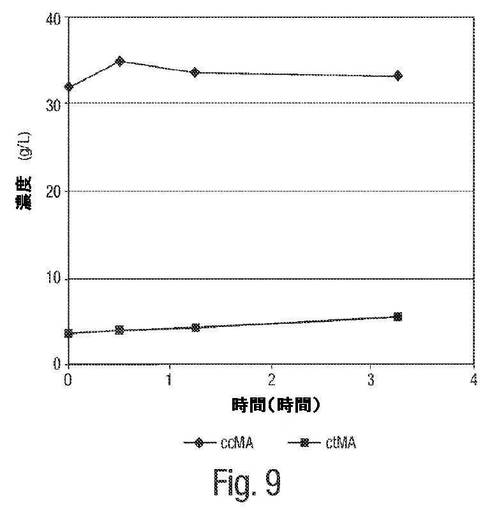
【図10】
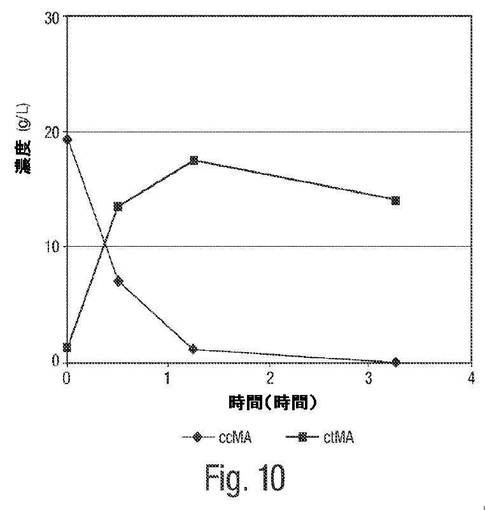
【図11】
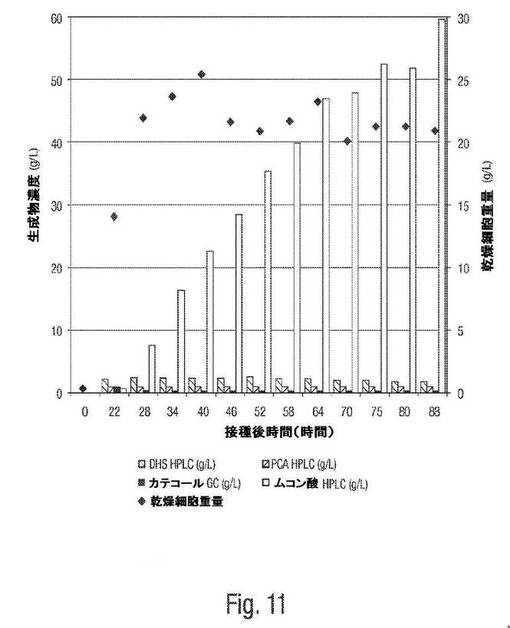
【図1−2】
【図2】
【図3】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【公表番号】特表2013−516196(P2013−516196A)
【公表日】平成25年5月13日(2013.5.13)
【国際特許分類】
【出願番号】特願2012−548206(P2012−548206)
【出願日】平成23年1月10日(2011.1.10)
【国際出願番号】PCT/US2011/020681
【国際公開番号】WO2011/085311
【国際公開日】平成23年7月14日(2011.7.14)
【出願人】(511289736)アミリス, インコーポレイテッド (4)
【Fターム(参考)】
【公表日】平成25年5月13日(2013.5.13)
【国際特許分類】
【出願日】平成23年1月10日(2011.1.10)
【国際出願番号】PCT/US2011/020681
【国際公開番号】WO2011/085311
【国際公開日】平成23年7月14日(2011.7.14)
【出願人】(511289736)アミリス, インコーポレイテッド (4)
【Fターム(参考)】
[ Back to top ]