
ムスカリン性受容体M1拮抗薬による肥満の処置
【課題】肥満症の治療および体重減少の助長のための有効な新規薬物および方法の提供。
【解決手段】優先的なムスカリン性アセチルコリン受容体M1拮抗薬を、任意選択で選択的ムスカリン性アセチルコリン受容体M1拮抗薬以外の少なくとも1つの抗うつ薬と共に投与することによって、肥満症を治療し、所望の減量をもたらし、または望ましくない体重増加を予防する方法。優先的なムスカリン性アセチルコリン受容体M1拮抗薬は、任意選択で抗肥満剤、例えば食欲減退薬と共に投与することができる。少なくとも1つの選択的ムスカリン性アセチルコリン受容体M1拮抗薬を選択的ムスカリン性アセチルコリン受容体M1拮抗薬以外の少なくとも1つの抗うつ薬と組み合わせて投与するための医薬組成物およびキット。
【解決手段】優先的なムスカリン性アセチルコリン受容体M1拮抗薬を、任意選択で選択的ムスカリン性アセチルコリン受容体M1拮抗薬以外の少なくとも1つの抗うつ薬と共に投与することによって、肥満症を治療し、所望の減量をもたらし、または望ましくない体重増加を予防する方法。優先的なムスカリン性アセチルコリン受容体M1拮抗薬は、任意選択で抗肥満剤、例えば食欲減退薬と共に投与することができる。少なくとも1つの選択的ムスカリン性アセチルコリン受容体M1拮抗薬を選択的ムスカリン性アセチルコリン受容体M1拮抗薬以外の少なくとも1つの抗うつ薬と組み合わせて投与するための医薬組成物およびキット。
【発明の詳細な説明】
【技術分野】
【0001】
関連出願への相互参照
この出願は、2006年6月16日に出願された米国仮出願第60/805,066号および2006年10月12日に出願された米国仮出願第60/829,225号(これらの両方の全体の開示は、参考として本明細書に援用される)の利益を主張する。
【0002】
連邦政府による資金提供を受けた研究開発の下になされた発明の権利に関する記載
該当なし
発明の分野
本発明は、選択的M1ムスカリン性受容体(M1R)拮抗薬の単独投与または抗うつ薬と組み合わせた投与による肥満症の治療および体重減少の助長に関する。
【背景技術】
【0003】
発明の背景
神経伝達物質アセチルコリン(ACh)は、エフェクター細胞膜において2種類の受容体:リガンド開口型イオンチャンネルであるニコチン性受容体(nAChR)、およびGタンパク質共役受容体であるムスカリン性受容体(mAChR)と相互作用する。哺乳類では、M1〜M5と呼ばれるmAChRの5つのサブタイプが同定されている。M1ムスカリン性受容体(M1R)は、中枢神経系と末梢神経系の両方、具体的には大脳皮質および交感神経節で見られる。M1Rによって媒介されるムスカリン様効果は、M1R−選択的拮抗薬の使用、およびさらに最近ではM1R−ヌルマウスの開発により大いに研究されている。
【0004】
現在既知のmAChR拮抗薬は、単一のムスカリン性受容体サブタイプに対して絶対的な選択性を示さないが、薬物のピレンゼピンおよびテレンゼピンは、M1Rに対して高い相対的親和性を示し、したがってM1R−選択的であるとみなされることが多い。ピレンゼピンは、欧州、日本、およびカナダで消化性潰瘍疾患を治療するために使用される。テレンゼピンは、同じ適応症の臨床試験で試験されている。治療用量で、これらは、非選択的mAChR拮抗薬がするように平滑筋の活動を抑制することなく、胃酸およびペプシンの分泌を適度に低減する。
【0005】
M1Rサブタイプが、うつ病性障害および不安のある種の様相に関与し得ることを示唆する証拠がいくつかある。ピレンゼピンをラット前脳の側坐核に直接注射すると、抗うつ活性の一般的尺度であるPorsolt水泳試験における水泳時間の増大が起こった(非特許文献1を参照のこと)。M1R−ヌルマウスも、Porsolt水泳試験での水泳時間の増大、および社会的相互作用試験における社会的接触の増大を示した(非特許文献2を参照のこと)。
【0006】
ピレンゼピンおよびテレンゼピンは、イミプラミンなどの三環系抗うつ薬に構造的に類似しているが、消化性潰瘍疾患の治療で経口投与した場合に向精神効果を有することは知られていない。さらに、マウスおよびラットに関してより早い段階の研究において、ピレンゼピンの全身投与では、いずれの行動効果も導き出すことができなかった(非特許文献3を参照のこと)。このような効果の欠如は、ピレンゼピンが、げっ歯類およびヒトを含めて、様々な種において顕著な血液脳関門透過性を示さないという観察により説明することができる(非特許文献4;非特許文献5を参照のこと)。そういう理由で、Porsolt水泳試験でのピレンゼピンの効果に関する上記の研究では、薬物を試験動物の脳に直接注射することを採用した。
【0007】
選択的M1R拮抗薬を使用して、脂質代謝を変更し、体脂肪貯蔵量を低減することも開示されている。例えば、特許文献1を参照のこと。しかし、所望の結果を実現するためには、24時間の期間において所定の時刻にM1R拮抗薬を投与することが必要であった。さらに、非特許文献6は、インスリン非依存性糖尿病(NIDDM)と診断された非肥満および肥満ヒト患者にピレンゼピンを投与することを開示する。Bevanは、ピレンゼピンの投与の時間を食事の時間と関連付けたが、ピレンゼピンの脂質生成に対する感受性に干渉する能力、または体重減少を助長する際のその使用を開示も示唆もしていない。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】米国特許第5,668,155号明細書
【非特許文献】
【0009】
【非特許文献1】Chau,D.T.ら,Neuroscience,2001年,104巻,3号,p.791−8
【非特許文献2】Miyakawa,T.ら,J Neurosci.,2001年,21巻,14号,p.5239−50
【非特許文献3】Rogoz,Z.,Skuza,G.,Sowinska,H.,Pol.J.Pharmacol.Pharm.,1981年,31巻,p.615−26
【非特許文献4】Hammer,R.,Koss,F.W.,Scand.J.Gastroenterol,Suppl,1979年,14巻,57号,p.1−6
【非特許文献5】Bymaster,F.P.ら,J.Pharmacol.Exp.Ther.,1993年,267巻,1号,p.16−24
【非特許文献6】Bevanら,Clinical Endocrinology,(1991年),36巻,p.85−91
【発明の概要】
【発明が解決しようとする課題】
【0010】
肥満症の治療および体重減少の助長のための有効な新規薬物が求められている。本発明は、これとその他の必要性に対処する。
【課題を解決するための手段】
【0011】
要旨
本発明は、治療上有効量の1つまたは複数のM1R−選択的拮抗薬を投与することによって、肥満症を治療し、食欲を抑制し、体重減少を助長または促進し、所望の体重の維持を助長または促進し、望ましくない体重増加を予防または低減するための方法を提供する。本方法を実施する際に、1つもしくは複数のM1R−選択的拮抗薬を、他の薬理作用剤なしに、または他の薬理作用剤、例えばM1R−選択的拮抗薬以外の1つもしくは複数の抗うつ薬と組み合わせて投与することができる。いくつかの実施形態では、1つまたは複数のM1R−選択的拮抗薬を1つまたは複数の抗肥満剤と共投与することができる。いくつかの実施形態では、M1R−選択的拮抗薬以外の1つまたは複数の抗うつ薬と組み合わせた1つまたは複数のM1R−選択的拮抗薬を、1つまたは複数の抗肥満剤と共投与することができる。
【0012】
1つまたは複数のM1R−選択的拮抗薬は、24時間の期間において所定の時刻に投与する必要はない。1つまたは複数のM1R−選択的拮抗薬の投与をホルモンの日周性振動の最上点または最下点と相関させることなく、有効な結果を実現することができる。1つまたは複数のM1R−選択的拮抗薬の投与は、一日のうちの特定の時間または食事と相関
させる必要がない。いくつかの実施形態では、1つまたは複数のM1R−選択的拮抗薬の投与の時間は、食事、例えば食事前、中、後と相関するように定められる。
したがって、第1の態様では、本発明は、体重減少を促進するか、または安定な体重の維持を助長するための方法を提供し、この方法は、それを必要とする肥満または過体重の個体に、治療上有効量の1つまたは複数の体重減少を達成するためのM1R−選択的拮抗薬を投与することを含み、それによって体重減少が促進され、または安定な体重の維持が助長される。
【0013】
別の態様では、本発明は、治療上有効量の1つまたは複数のM1R−選択的拮抗薬および1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬の混合物を含む医薬組成物を提供する。いくつかの実施形態では、医薬組成物は、治療上有効量の1つまたは複数のM1R−選択的拮抗薬および1つまたは複数の抗肥満剤の混合物を含む。いくつかの実施形態では、医薬組成物は、治療上有効量の1つまたは複数のM1R−選択的拮抗薬、M1R−選択的拮抗薬以外の1つまたは複数の抗うつ薬、および1つまたは複数の抗肥満剤の混合物を含む。
【0014】
別の態様では、本発明は、治療上有効量の1つまたは複数のM1R−選択的拮抗薬および1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬の混合物を含むキットを提供する。いくつかの実施形態では、キットは、治療上有効量の1つまたは複数のM1R−選択的拮抗薬および1つまたは複数の抗肥満剤の混合物を含む。いくつかの実施形態では、キットは、治療上有効量の1つまたは複数のM1R−選択的拮抗薬、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬、および1つまたは複数の抗肥満剤の混合物を含む。
【0015】
本方法を実施するための実施形態、ならびに医薬組成物およびキットのための実施形態に関して、一実施形態では、1つまたは複数のM1R−選択的拮抗薬は、ピレンゼピン、テレンゼピン、およびそれらの組合せからなる群より選択される。一実施形態では、M1R−選択的拮抗薬はテレンゼピン(ラセミ体または光学異性体)である。一実施形態では、M1R−選択的拮抗薬はピレンゼピンである。
【0016】
一実施形態では、1つまたは複数のM1R−選択的拮抗薬は、第2の薬理作用剤なしに投与される。
【0017】
一実施形態では、1つもしくは複数のM1R−選択的拮抗薬は、組合せで、または1つもしくは複数のM1R−選択的拮抗薬以外の抗うつ薬と組み合わせて投与される。一実施形態では、抗うつ薬は、選択的セロトニン再取り込み阻害薬(SSRI)および選択的セロトニン−ノルエピネフリン再取り込み阻害薬(SNRI)からなる群より選択される。
【0018】
一実施形態では、抗うつ薬はSSRIである。一実施形態では、SSRIは、シタロプラム、エシタロプラム、フルオキセチン、フルボキサミン、パロキセチン、およびセルトラリンからなる群より選択される。一実施形態では、SSRIは、シタロプラム、セルトラリン、パロキセチン、およびフルオキセチンからなる群より選択される。
【0019】
一実施形態では、抗うつ薬はSNRIである。一実施形態では、SNRIは、ミルナシプラン、ミルタザピン、ベンラファキシン、デュロキセチン、デスベンラファキシン、およびシブトラミンからなる群より選択される。一実施形態では、SNRIはベンラファキシンである。
【0020】
これらの方法は、抗肥満剤の投与を必要とすることなく、例えば食欲減退薬の投与を必要とすることなく、有効な結果を実現する。しかし、いくつかの実施形態では、1つまたは複数のM1R−選択的拮抗薬は、組合せで、または1つもしくは複数の抗肥満剤、例え
ば1つもしくは複数の食欲減退薬と組み合わせて投与される。
【0021】
さらに、1つまたは複数のM1R−選択的拮抗薬の投与の時間を定めることなく、有効な結果を実現することができる。抗うつ薬、抗肥満剤、および食欲減退薬を含めて、共投与された活性剤も、投与の時間を定めることなく、有効な結果をもたらす。
【0022】
一実施形態では、1つまたは複数のM1R−選択的拮抗薬は、テレンゼピン(ラセミ体または光学異性体)であり、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬はフルオキセチン(ラセミ体または光学異性体)である。
【0023】
一実施形態では、1つまたは複数のM1R−選択的拮抗薬はテレンゼピン(ラセミ体または光学異性体)であり、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬はフルボキサミンである。
【0024】
一実施形態では、1つまたは複数のM1R−選択的拮抗薬はテレンゼピン(ラセミ体または光学異性体)であり、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬はセルトラリンまたはそのS−エナンチオマー、Zoloft(登録商標)である。
【0025】
一実施形態では、1つまたは複数のM1R−選択的拮抗薬はテレンゼピン(ラセミ体または光学異性体)であり、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬はシタロプラム(またはエシタロプラム)である。
【0026】
一実施形態では、1つまたは複数のM1R−選択的拮抗薬はテレンゼピン(ラセミ体または光学異性体)であり、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬はパロキセチンである。
【0027】
一実施形態では、1つまたは複数のM1R−選択的拮抗薬はテレンゼピン(ラセミ体または光学異性体)であり、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬はベンラファキシン(ラセミ体または光学異性体)である。
【0028】
一実施形態では、1つまたは複数のM1R−選択的拮抗薬はテレンゼピン(ラセミ体または光学異性体)であり、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬はデスベンラファキシンである。
【0029】
一実施形態では、1つまたは複数のM1R−選択的拮抗薬はテレンゼピン(ラセミ体または光学異性体)であり、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬はデュロキセチンである。
【0030】
一実施形態では、1つまたは複数のM1R−選択的拮抗薬はテレンゼピン(ラセミ体または光学異性体)であり、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬はシブトラミンである。
【0031】
一実施形態では、1つまたは複数のM1R−選択的拮抗薬はテレンゼピン(ラセミ体または光学異性体)であり、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬はミルナシプランである。
【0032】
一実施形態では、1つまたは複数のM1R−選択的拮抗薬はテレンゼピン(ラセミ体または光学異性体)であり、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬はミルタザピンである。
【0033】
一実施形態では、1つまたは複数のM1R−選択的拮抗薬はテレンゼピン(ラセミ体または光学異性体)であり、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬はブプロピオンである。
【0034】
一実施形態では、1つまたは複数のM1R−選択的拮抗薬はテレンゼピン(ラセミ体または光学異性体)であり、1つまたは複数の抗肥満剤はフェンテルミンである。
【0035】
関連する態様では、本発明は、肥満症を治療し、食欲を抑制し、体重減少を助長または促進し、所望の体重の維持を助長または促進し、望ましくない体重増加を予防または低減するための医薬品を調製するための方法、またはその使用を提供し、この医薬品は、治療上有効量の1つまたは複数のM1R−選択的拮抗薬を含有する。医薬品は、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬も任意選択で含有することができる。医薬品は、1つまたは複数の抗肥満剤も任意選択で含有することができる。医薬品の実施形態は、本明細書に記載する通りである。
【0036】
いくつかの実施形態では、本発明の方法および組成物は、本明細書に記載する薬理作用剤の組合せを含む。いくつかの実施形態では、本発明の方法および組成物は、本質的に本明細書に記載する薬理作用剤の組合せからなる。
定義
用語「肥満の」または「肥満症」は、過剰の脂肪組織のためボディマス指数(BMI)が30kg/m2以上である個体を意味する。肥満症は、体脂肪含量を基準として定義することもできる:男性の場合、25%を超える体脂肪含量、または女性の場合、30%を超える体脂肪含量。「病的に肥満の」個体のボディマス指数は、35kg/m2を超える。
【0037】
用語「過体重」は、ボディマス指数が25kg/m2以上であるが、30kg/m2未満である個体を意味する。
【0038】
用語「ボディマス指数」または「BMI」は、個体の体重が身長に対して適切であるかどうかを推定する体重対身長比の測定を意味する。本明細書では、個体のボディマス指数は、以下の通り算出される。
BMI=(ポンド×700)/(身長、単位インチ)2
または
BMI=(キログラム)/(身長、単位メートル)2
用語「ベースライン体重」は、治療の開始時に個体によって提示された体重を意味する。
【0039】
本明細書では、「投与すること」は、経口(「po」)投与、坐剤としての投与、局所的接触、静脈内(「iv」)、腹腔内(「ip」)、筋肉内(「im」)、病巣内、鼻腔内、もしくは皮下(「sc」)投与、またはスローリリースの装置、例えばミニ浸透圧ポンプの対象者への移植を意味する。投与は、非経口および経粘膜{例えば、経口、経鼻、腟内、直腸、または経皮)を含めて、任意の経路による。非経口投与としては、例えば静脈内、筋肉内、細動脈内、皮内、皮下、腹腔内、脳室内、および頭蓋内投与が挙げられる。送達の他のモードとしては、リポソーム製剤、静脈内注入、経皮貼付剤などの使用が挙げられるが、これらに限定されない。
【0040】
用語「全身投与」および「全身投与した」は、化合物または組成物が、薬剤作用の標的部位を含めて、体の部位に循環器系を介して送達されるように化合物または組成物を哺乳類に投与する方法を意味する。全身投与としては、経口、鼻腔内、直腸、および非経口(すなわち、消化管を経由する以外、具体的には筋肉内、静脈内、動脈内、経皮、および皮
下)投与が挙げられるが、これらに限定されない。ただし、本明細書では、全身投与は、循環器系を経由する以外の手段、具体的には髄腔内注射および頭蓋内投与による脳領域への直接投与を含まないことを条件にする。
用語「共投与する」は、個体の血中に2つの活性剤が同時に存在することを意味する。共投与された活性剤は、同時または連続的に送達され得る。
【0041】
本明細書では、用語「治療すること」および「治療」は、用語が適用される疾患もしくは病態、またはこのような疾患もしくは病態の1つもしくは複数の症状の発症を遅延させ、それらの進行を遅延もしくは逆行させ、またはそれらを軽減もしくは予防することを意味する。
【0042】
本明細書では、用語「選択的ムスカリン性受容体M1拮抗薬」および「M1R−選択的拮抗薬」は、ムスカリン性受容体サブタイプM2およびM3に比べて、優先的にムスカリン性受容体M1サブタイプと相互作用を示すムスカリン性アセチルコリン受容体拮抗薬を意味する。M1R−選択的拮抗薬としては、例えばピレンゼピンおよびテレンゼピンが挙げられるが、これらに限定されない。優先的な結合は完全である必要はない。例えば、ピレンゼピンは、M1およびM4受容体サブタイプに対して親和性が類似しているにもかかわらず、M1R−選択的拮抗薬として分類される。
【0043】
M1R−選択的拮抗薬の優先的な結合は、競合的置換アッセイで測定することができる。M1R−選択的拮抗薬は、既知のM2選択的リガンド(例えば、トリピトラミン、ヒンバシン、メトクトラミン)およびM3(例えば、ダリフェナシン、ヘキサヒドロシラジフェニドール)に比べて、優先的に既知のM1R−選択的リガンド(例えば、ピレンゼピンおよび/またはテレンゼピン)に置換する。あるいは、M1R−選択的拮抗薬は、非選択的ムスカリン性リガンドのM2およびM3受容体サブタイプへの結合に置換するのに比べて、優先的に非選択的ムスカリン性リガンド(例えば、キヌクリジニルベンジラート(QNB)、N−メチルスコポラミン(NMS))のM1受容体サブタイプへの結合に置換する。放射性標識競合体の置換に対する相対的効力は、競合体の50%が置換される濃度(IC50)、または平衡解離定数(Kd)によって表すことができる。IC50値および/または平衡解離定数は、利用可能なソフトウェアを使用して、漸増量の非標識試験化合物の存在下での標識リガンドの検出値を入力することによって算出することができる(例えば、LIGAND(Munson,p.J.,およびRodbard,D.,Anal.Biochem.(1980年)107巻,p.220−39またはDATAPLOT,National Technical Information Services)。M1R−選択的拮抗薬は、M1受容体サブタイプへの結合に関するIC50値またはKd値が、M2およびM3受容体サブタイプへの結合に関するそのIC50値またはKd値より少なくとも約3倍低く、好ましくは少なくとも約10倍低く、より好ましくは少なくとも約30倍低い。放射性標識NMSまたはQNBを使用する適用可能な放射性リガンド結合アッセイは、Buckleyら,Molecular Pharmacology(1989年)35巻,p.469−76およびBoldenら,J Pharmacol Exp Ther.(1992年)260巻,p.576−80に開示される。
【0044】
本明細書では、用語「抗肥満剤」は、体重減少をもたらすことを主目的とする薬剤を意味する。抗肥満剤としては、例えば食欲減退薬、ドーパミン作動薬、H3−ヒスタミン拮抗薬、5−HT2c受容体作動薬、β−3アドレナリン受容体作動薬、コレシストキニン作動薬、抗てんかん剤、レプチン、レプチン類似体、およびレプチン受容体作動薬、ニューロペプチドY(NPY)受容体拮抗薬およびモジュレーター、ペプチド−YY(PYY)受容体作動薬、毛様体神経栄養因子、甲状腺ホルモン受容体−β作動薬、カンナビノイドCB1受容体拮抗薬、メラニン凝集ホルモン受容体拮抗薬、膵および胃液リパーゼ阻害薬、メラノコルチン−4受容体作動薬、およびそれらの組合せが挙げられるが、これらに
限定されない。本明細書では、用語「抗肥満剤」は、具体的にM1R選択的ムスカリン拮抗薬および抗うつ薬を除外する。
【0045】
本明細書では、用語「食欲減退薬」または「食欲抑制薬」は同義的に、食欲の抑制を主な所期効果とする薬剤を意味する。食欲減退薬としては、交感神経刺激アミン類が挙げられるが、これらに限定されない。交感神経刺激アミン類は周知であり、例えば、参照により本明細書に組み込まれるGoodman and Gilman’s The Pharmacological Basis of Therapeutics,第11版,Brunton,LazoおよびParker編,McGraw−Hill(2006年),第10章,p.237−263に詳細に述べられている。本明細書では、用語「食欲減退薬」または「食欲抑制薬」は、具体的にM1R選択的ムスカリン拮抗薬および抗うつ薬を除外する。
【0046】
本明細書では、フレーズ「からなる本質的になる」は、方法または組成物に含まれる有効薬剤の属または種、およびこれらの方法または組成物の所期目的に対して不活性である任意の賦形剤を意味する。いくつかの実施形態では、フレーズ「から本質的になる」は、M1R−選択的拮抗薬および抗うつ薬以外の1つまたは複数の追加の活性剤が包含されることを明らかに除外する。いくつかの実施形態では、除外することができる追加の活性剤としては、プロラクチン阻害薬、プロラクチン刺激剤、5−HT受容体拮抗薬、5−HT受容体作動薬、NK−1受容体拮抗薬、および/またはジペプチジルペプチダーゼIV阻害薬の1つまたは複数が挙げられる。
【0047】
用語「放出制御」、「徐放」、「持続放出」、および「時限放出」は同義的に、薬物の放出が即時型でない任意の薬物含有製剤を意味するよう意図されている。すなわち、「放出制御」製剤を使用した場合、経口投与によって、薬物の吸収プールへの即時放出は起こらない。これらの用語は、Remington:The Science and Practice of Pharmacy,第21版,Lippencott Williams & Wilkins(2006年)で定義される「非即時放出」と同義的に使用される。そこで述べられるように、即時および非即時放出の動態は、下記の式を参照することによって定義することができる。
【0048】
【化1】

「吸収プール」は、特定の吸収部位での投与薬物の溶液を表し、kr、ka、およびkeはそれぞれ、(1)製剤からの薬物放出、(2)吸収、および(3)排泄の一次速度定数である。即放性剤形については、薬物放出の速度定数krが吸収速度定数kaよりはるかに大きい。放出制御製剤については、その逆、すなわちkr≪kaが真であり、したがって剤形からの薬物放出速度が、標的領域への薬物送達における律速段階である。
【0049】
用語「徐放」および「持続放出」は、長期間、例えば12時間以上にわたって、薬物の漸進的放出をもたらし、好ましくは、必ずというわけではないが長期にわたって実質的に定常状態の血中薬物レベルをもたらす薬物製剤を意味するように通常の意味で使用される。
【0050】
本明細書では、用語「遅延放出」は、変化しないままで胃を通過し、小腸で溶解する医薬調製物を意味する。
【0051】
本明細書では、「相乗効果」または「相乗効果の」は同義的に、2つの活性剤の相加効果より強いそれらの複合効果を意味する。相乗効果は、無効な用量の2つの活性剤を合わせて有効な効果を生じることによって実現することができる。相乗効果の尺度は、統計的有意性とは無関係である。
本発明はまた、以下の項目を提供する。
(項目1)
体重減少を促進するか、または安定な体重の維持を助長するための方法であって、該方法は、それを必要とする肥満または過体重の個体に、テレンゼピン、ならびに選択的セロトニン再取り込み阻害薬(SSRI)および選択的セロトニン−ノルエピネフリン再取り込み阻害薬(SNRI)からなる群より選択される抗うつ薬を体重減少を達成するための治療上有効量で投与することを含み、それによって体重減少が促進されるか、または安定な体重の維持が助長される、方法。
(項目2)
上記抗うつ薬がSSRIである、項目1に記載の方法。
(項目3)
上記SSRIが、シタロプラム、エシタロプラム、フルオキセチン、フルボキサミン、パロキセチン、およびセルトラリンからなる群より選択される、項目2に記載の方法。
(項目4)
上記SSRIがシタロプラムである、項目3に記載の方法。
(項目5)
上記SSRIがセルトラリンである、項目3に記載の方法。
(項目6)
上記抗うつ薬がSNRIである、項目1に記載の方法。
(項目7)
上記SNRIが、ミルナシプラン、ミルタザピン、ベンラファキシン、デュロキセチン、デスベンラファキシン、およびシブトラミンからなる群より選択される、項目6に記載の方法。
(項目8)
上記SNRIがベンラファキシンである、項目7に記載の方法。
(項目9)
さらに、1つまたは複数の抗肥満剤の投与を含む、項目1に記載の方法。
(項目10)
さらに、1つまたは複数の食欲減退薬の投与を含む、項目1に記載の方法。
(項目11)
体重減少を促進するか、または安定な体重の維持を助長するための方法であって、該方法は、それを必要とする肥満または過体重の個体に、治療上有効量の体重減少を達成するための1つまたは複数のM1R−選択的拮抗薬を投与することを含み、それによって体重減少が促進されるか、または安定な体重の維持が助長される、方法。
(項目12)
上記1つまたは複数のM1R−選択的拮抗薬が、ピレンゼピン、テレンゼピン、およびそれらの組合せからなる群より選択される、項目11に記載の方法。
(項目13)
さらに、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬の投与を含む、項目11に記載の方法。
(項目14)
上記1つまたは複数の抗うつ薬が、三環系抗うつ薬およびその類似体、セロトニン再取り込み阻害薬、セロトニン−ノルエピネフリン再取り込み阻害薬、ノルエピネフリン再取り込み阻害薬、ドーパミン再取り込み阻害薬、ノルエピネフリン−ドーパミン再取り込み阻害薬、セロトニン−ノルエピネフリン−ドーパミン再取り込み阻害薬、セロトニン再取り込み促進薬、セロトニン作動薬およびそのプロドラッグ、モノアミンオキシダーゼ阻害薬、およびそれらの混合物からなる群より選択される、項目13に記載の方法。
(項目15)
さらに、1つまたは複数の抗肥満剤の投与を含む、項目11に記載の方法。
(項目16)
さらに、1つまたは複数の食欲減退薬の投与を含む、項目11に記載の方法。
(項目17)
治療上有効量のM1R−選択的拮抗薬と、SSRIおよびSNRIからなる群より選択されるM1R−選択的拮抗薬以外の抗うつ薬との混合物を含む医薬組成物。
(項目18)
上記M1R−選択的拮抗薬がテレンゼピンであり、上記M1R−選択的拮抗薬以外の抗うつ薬がセルトラリンである、項目17に記載の医薬組成物。
(項目19)
上記M1R−選択的拮抗薬がテレンゼピンであり、上記M1R−選択的拮抗薬以外の抗うつ薬がベンラファキシンである、項目17に記載の医薬組成物。
(項目20)
上記M1R−選択的拮抗薬がテレンゼピンであり、上記M1R−選択的拮抗薬以外の抗うつ薬がシタロプラムである、項目17に記載の医薬組成物。
(項目21)
上記M1R−選択的拮抗薬および上記M1R−選択的拮抗薬以外の抗うつ薬の送達が徐放である、項目17に記載の医薬組成物。
(項目22)
治療上有効量のM1R−選択的拮抗薬とM1R−選択的拮抗薬以外の抗うつ薬の組合せを含むキット。
(項目23)
上記M1R−選択的拮抗薬がテレンゼピンであり、上記M1R−選択的拮抗薬以外の抗うつ薬がセルトラリンである、項目22に記載のキット。
(項目24)
上記M1R−選択的拮抗薬がテレンゼピンであり、上記M1R−選択的拮抗薬以外の抗うつ薬がベンラファキシンである、項目22に記載のキット。
(項目25)
上記M1R−選択的拮抗薬がテレンゼピンであり、上記M1R−選択的拮抗薬以外の抗うつ薬がシタロプラムである、項目22に記載のキット。
【図面の簡単な説明】
【0052】
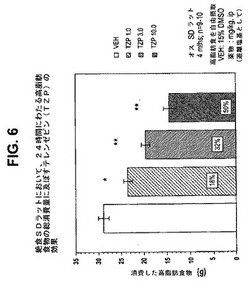
【図1】絶食ラットにおいて、4時間にわたる高脂肪食物の総消費量に及ぼすテレンゼピン(TZP)の効果を示す図である。下記の実施例に記載されるように、4月齢のオスSprague−Dawleyラット(n=9匹〜10匹/群)に、テレンゼピンを1.0mg/kg、3.0mg/kg、または10.0mg/kgの用量で腹腔内投与した。活性剤はすべて、遊離塩基の形で送達した。対照ラット(VEH)には15%のDMSOを投与した。*は、VEHに対してp<0.05を示す。*は、VEHに対してp<0.01を示す。
【図2】絶食ラットにおいて、4時間にわたる高脂肪食物の総消費量に及ぼすテレンゼピン(TZP)単独、シタロプラム(CIT)単独、およびテレンゼピンとシタロプラムの組合せの効果を示す図である。下記の実施例に記載されるように、3月齢のオスSprague−Dawleyラット(n=10匹/群)に、テレンゼピン単独(3mg/kg)、シタロプラム単独(25mg/kg)、または共投与のテレンゼピン(3mg/kg)とシタロプラム(25mg/kg)を腹腔内投与した。活性剤はすべて、遊離塩基の形で送達した。対照ラット(VEH)には37%のDMSOを投与した。*は、VEHに対してp<0.05を示す。*は、VEHに対してp<0.001を示す。「a」は、TZPに対してp<0.05を示す。「b」は、CITに対してp<0.001を示す。
【図3】絶食ラットにおいて、4時間にわたる高脂肪食物の総消費量に及ぼすテレンゼピン(TZP)単独、セルトラリン(SRT)単独、およびテレンゼピンとセルトラリンの組合せの効果を示す図である。下記の実施例に記載されるように、3月齢のオスSprague−Dawleyラット(n=10匹/群)に、テレンゼピン単独(3mg/kg)、セルトラリン単独(10mg/kgまたは30mg/kg)、または共投与のテレンゼピン(3mg/kg)とセルトラリン(10mg/kgまたは30mg/kg)を腹腔内投与した。活性剤はすべて、遊離塩基の形で送達した。対照ラット(VEH)には37%のDMSOを投与した。*は、VEHに対してp<0.05を示す。*は、VEHに対してp<0.001を示す。「a」は、TZPに対してp<0.001を示す。「b」は、TZPに対してp<0.05を示す。
【図4】絶食ラットにおいて、4時間にわたる高脂肪食物の総消費量に及ぼすテレンゼピン(TZP)単独、シブトラミン(SBR)単独、およびテレンゼピンとシブトラミンの組合せの効果を示す図である。下記の実施例に記載されるように、4月齢のオスSprague−Dawleyラット(n=10匹/群)に、テレンゼピン単独(30mg/kg)、シブトラミン単独(1.0mg/kg)、または共投与のテレンゼピン(30mg/kg)とシブトラミン(1.0mg/kg)を経口投与した。活性剤はすべて、遊離塩基の形で送達した。対照ラット(VEH)には10%のDMSOを投与した。**は、VEHに対してp<0.01を示す。「aa」は、SBRに対してp<0.01を示す。
【図5】絶食ラットにおいて、4時間にわたる高脂肪食物の総消費量に及ぼすテレンゼピン(TZP)単独、ベンラファキシン(VEN)単独、およびテレンゼピンとベンラファキシンの組合せの効果を示す図である。下記の実施例に記載されるように、2月/3月齢のオスSprague−Dawleyラット(n=11匹/群)に、テレンゼピン単独(1mg/kgまたは3mg/kg)、ベンラファキシン単独(30mg/kg)、または共投与のテレンゼピン(1mg/kgまたは3mg/kg)とベンラファキシン(30mg/kg)を腹腔内投与した。活性剤はすべて、遊離塩基の形で送達した。対照ラット(VEH)には33%のDMSOを投与した。**は、VEHに対してp<0.001を示す。「a」は、TZPに対してp<0.05を示す。「aa」は、TZPに対してp<0.01を示す。「b」は、VENに対してp<0.05を示す。「bb」は、VENに対してp<0.01を示す。
【図6】絶食ラットにおいて、24時間にわたる高脂肪食物の総消費量に及ぼすテレンゼピン(TZP)の効果を示す図である。下記の実施例に記載されるように、4月齢のオスSprague−Dawleyラット(n=9匹〜10匹/群)に、テレンゼピンを1.0mg/kg、3.0mg/kg、または10.0mg/kgの用量で腹腔内投与した。活性剤はすべて、遊離塩基の形で送達した。対照ラット(VEH)には15%のDMSOを投与した。*は、VEHに対してp<0.05を示す。**は、VEHに対してp<0.01を示す。
【図7】絶食ラットにおいて、24時間にわたる高脂肪食物の総消費量に及ぼすテレンゼピン(TZP)単独、シタロプラム(CIT)単独、およびテレンゼピンとシタロプラムの組合せの効果を示す図である。下記の実施例に記載されるように、3月齢のオスSprague−Dawleyラット(n=10匹/群)に、テレンゼピン単独(3mg/kg)、シタロプラム単独(25mg/kg)、または共投与のテレンゼピン(3mg/kg)とシタロプラム(25mg/kg)を腹腔内投与した。活性剤はすべて、遊離塩基の形で送達した。対照ラット(VEH)には37%のDMSOを投与した。*は、VEHに対してp<0.01を示す。**は、VEHに対してp<0.001を示す。「a」は、TZPに対してp<0.001を示す。「b」は、CITに対してp<0.001を示す。
【図8】絶食ラットにおいて、24時間にわたる高脂肪食物の総消費量に及ぼすテレンゼピン(TZP)単独、セルトラリン(SRT)単独、およびテレンゼピンとセルトラリンの組合せの効果を示す図である。下記の実施例に記載されるように、3月齢のオスSprague−Dawleyラット(n=10匹/群)に、テレンゼピン単独(3mg/kg)、セルトラリン単独(10mg/kgまたは30mg/kg)、または共投与のテレンゼピン(3mg/kg)とセルトラリン(10mg/kgまたは30mg/kg)を腹腔内投与した。活性剤はすべて、遊離塩基の形で送達した。対照ラット(VEH)には37%のDMSOを投与した。*は、VEHに対してp<0.01を示す。**は、VEHに対してp<0.001を示す。「a」は、TZPに対してp<0.05を示す。「aa」は、TZPに対してp<0.001を示す。「b」は、SRTに対してp<0.001を示す。
【図9】絶食ラットにおいて、24時間にわたる高脂肪食物の総消費量に及ぼすテレンゼピン(TZP)単独、シブトラミン(SBR)単独、およびテレンゼピンとシブトラミンの組合せの効果を示す図である。下記の実施例に記載されるように、4月齢のオスSprague−Dawleyラット(n=10匹/群)に、テレンゼピン単独(30mg/kg)、シブトラミン単独(1.0mg/kg)、または共投与のテレンゼピン(30mg/kg)とシブトラミン(1.0mg/kg)を経口投与した。活性剤はすべて、遊離塩基の形で送達した。対照ラット(VEH)には10%のDMSOを投与した。***は、VEHに対してp<0.001を示す。「aaa」は、SBRに対してp<0.001を示す。
【図10】絶食ラットにおいて、24時間にわたる高脂肪食物の総消費量に及ぼすテレンゼピン(TZP)単独、ベンラファキシン(VEN)単独、およびテレンゼピンとベンラファキシンの組合せの効果を示す図である。下記の実施例に記載されるように、2月/3月齢のオスSprague−Dawleyラット(n=11匹/群)に、テレンゼピン単独(1mg/kgまたは3mg/kg)、ベンラファキシン単独(30mg/kg)、または共投与のテレンゼピン(1mg/kgまたは3mg/kg)とベンラファキシン(30mg/kg)を腹腔内投与した。活性剤はすべて、遊離塩基の形で送達した。対照ラット(VEH)には33%のDMSOを投与した。**は、VEHに対してp<0.001を示す。「a」は、TZPに対してp<0.05を示す。「aa」は、TZPに対してp<0.01を示す。「b」は、VENに対してp<0.05を示す。「bb」は、VENに対してp<0.01を示す。
【図11】高脂肪食を摂取しているラットの体重に及ぼすテレンゼピン(TZP)、シタロプラム(CIT)、およびテレンゼピンとシタロプラムの組合せの慢性投与の効果を示す図である。下記の実施例に記載されるように、約15週齢のオスSprague−Dawleyラット(n=10匹/群)に、テレンゼピン単独(3mg/kg)、シタロプラム単独(15mg/kg)、または共投与のテレンゼピン(3mg/kg)とシタロプラム(15mg/kg)を皮下注入した。活性剤はすべて、遊離塩基の形で送達した。対照ラット(VEH)には10%のDMSOを投与した。*は、VEHに対してp<0.05を示す。**は、VEHに対してp<0.01を示す。***は、VEHに対してp<0.001を示す。「aa」は、CITに対してp<0.01を示す。「aaa」は、CITに対してp<0.001を示す。「b」は、TZPに対してp<0.05を示す。斜線部分は治療間隔を示す。
【図12】高脂肪食を摂取しているラットの体重に及ぼすテレンゼピン(TZP)、セルトラリン(SRT)、およびテレンゼピンとセルトラリンの組合せの慢性投与の効果を示す図である。下記の実施例に記載されるように、約15週齢のオスSprague−Dawleyラット(n=10匹/群)に、テレンゼピン単独(3mg/kg)、セルトラリン単独(15mg/kg)、または共投与のテレンゼピン(3mg/kg)とセルトラリン(15mg/kg)を皮下注入した。活性剤はすべて、遊離塩基の形で送達した。対照ラットには、10%のDMSO(VEHl)またはN−メチル−2−ピロリドン+DMSO+水(1:2:2)(「NMP:DMSO:水」またはVEH2)を投与した。*は、VEHに対してp<0.05を示す。**は、VEHに対してp<0.01を示す。***は、VEHに対してp<0.001を示す。「a」は、SRTに対してp<0.05を示す。「aa」は、SRTに対してp<0.01を示す。「aaa」は、SRTに対してp<0.001を示す。「b」は、TZPに対してp<0.05を示す。「bb」は、TZPに対してp<0.01を示す。「bbb」は、TZPに対してp<0.001を示す。斜線部分は治療間隔を示す。
【図13】高脂肪食を摂取しているラットの体重に及ぼすテレンゼピン(TZP)、シブトラミン(SBR)、およびテレンゼピンとシブトラミンの組合せの慢性投与の効果を示す図である。下記の実施例に記載されるように、約15週齢のオスSprague−Dawleyラット(n=10匹/群)に、テレンゼピン単独(1.0mg/kg)、シブトラミン単独(0.3mg/kg)、または共投与のテレンゼピン(1.0mg/kg)とシブトラミン(0.3mg/kg)を経口投与した。活性剤はすべて、遊離塩基の形で送達した。対照ラット(VEH)には水を投与した。*は、VEHに対してp<0.05を示す。**は、VEHに対してp<0.01を示す。「aa」は、SBRに対してp<0.05を示す。「b」は、TZPに対してp<0.05を示す。斜線部分は治療間隔を示す。
【図14】高脂肪食を摂取しているラットの体重に及ぼすテレンゼピン(TZP)、ベンラファキシン(VEN)、およびテレンゼピンとベンラファキシンの組合せの慢性投与の効果を示す図である。下記の実施例に記載されるように、約15週齢のオスSprague−Dawleyラット(n=10匹/群)に、テレンゼピン単独(3mg/kg)、ベンラファキシン単独(30mg/kg)、または共投与のテレンゼピン(3mg/kg)とベンラファキシン(30mg/kg)を皮下注入した。活性剤はすべて、遊離塩基の形で送達した。対照ラット(VEH)には10%のDMSOを投与した。*は、VEHに対してp<0.05を示す。**は、VEHに対してp<0.01を示す。***は、VEHに対してp<0.001を示す。「a」は、VENに対してp<0.05を示す。「aa」は、VENに対してp<0.01を示す。「aaa」は、VENに対してp<0.001を示す。「b」は、TZPに対してp<0.05を示す。「bb」は、TZPに対してp<0.01を示す。「bbb」は、TZPに対してp<0.001を示す。斜線部分は治療間隔を示す。
【図15】高脂肪食を摂取しているラットの体重に及ぼすテレンゼピン(TZP)、フェンテルミン(PHN)、およびテレンゼピンとフェンテルミンの組合せの慢性投与の効果を示す図である。下記の実施例に記載されるように、約15週齢のオスSprague−Dawleyラット(n=10匹/群)に、テレンゼピン単独(0.3mg/kg)、フェンテルミン単独(4.0mg/kg)、または共投与のテレンゼピン(0.3mg/kg)とフェンテルミン(4.0mg/kg)を皮下注入した。活性剤はすべて、遊離塩基の形で送達した。対照ラット(VEH)には生理食塩水を投与した。*は、VEHに対してp<0.05を示す。**は、VEHに対してp<0.01を示す。「a」は、PHNに対してp<0.05を示す。「b」は、TZPに対してp<0.05を示す。斜線部分は治療間隔を示す。
【発明を実施するための形態】
【0053】
1.序論
上述したように、ラットおよびマウスにおけるより早い段階の研究は、ピレンゼピンの全身投与では、いずれの行動効果も導き出すことができなかったこと(Rogoz,Z.,Skuza,G.,Sowinska,H.,Pol.J.Pharmacol.Pharm.,1981年,31巻,p.615−26を参照のこと)、およびピレンゼピンが、げっ歯類およびヒトを含めて、様々な種において顕著な血液脳関門透過性を示さないこと(Hammer,R.,Koss,F.W.,Scand.J.Gastroenterol.,Suppl,1979年,14巻,57号,p.1−6;Bymaster,F.P.ら,J.Pharmacol Exp.Ther.,1993年,267巻,1号,p.16−24を参照のこと)を示した。驚くべきことに、刊行された文献に反して、本発明は、ピレンゼピンおよびテレンゼピンを含めて、M1R−選択的拮抗薬が、治療量で血液脳関門を通過することができ、したがって食欲抑制を含めて、有用な薬理作用を有することを実証する。さらに、以前の知識に反して、M1R−選択的拮抗薬は、24時間の期間において所定の時刻に投与することなく、有効な効果を生じることができる。
【0054】
本発明は、M1R−選択的拮抗薬とある種の他の治療剤を組み合わせ使用によって、肥満症の治療および体重減少の促進を含めて、薬剤用途に有利な予想外の相乗効果が生じることも実証する。
【0055】
本発明は、過体重または肥満の個体において所望の体重減少を実現し、長期間にわたって体重減少の継続および体重維持を達成する有効な薬理学的治療を提供する。M1R−選択的拮抗薬の投与は、予想外に体重減少または体重増加の低減をもたらす。さらに、1つまたは複数のM1R−選択的拮抗薬およびM1R−選択的拮抗薬以外の1つまたは複数の抗うつ剤の共投与は、特に一般的に抗うつ薬の長期投与に伴う体重増加副作用を考えると、予想外にこれらのカテゴリーの薬物のいずれかを単独投与することによって達成されるより多い量の体重減少または体重増加の低減をもたらす(例えば、MasandおよびGupta,Ann.Clin.Psych.14巻,p.175(2002年);ならびにDeshmukhおよびFranco,Cleve.Clin.J.Med.70巻,p.614(2003年))。いくつかの実施形態では、M1R−選択的拮抗薬は、抗肥満剤と共投与することができる。
【0056】
2.肥満症の治療方法および/または体重減少の達成方法
a.治療に供する病態
本方法および組成物は、体重に関連する障害の治療に使用される。本方法および組成物によって治療可能な障害の一般カテゴリーの例としては、とりわけ望ましくないまたは過剰な体重を保持する肥満症、飽満感の欠如が挙げられるが、これらに限定されない。
【0057】
中枢神経系におけるムスカリン性受容体に対するアセチルコリンの作用は、認知、洞察、覚醒、情動、感覚運動ゲーティング、および反射的運動と定方向運動を含めて、多様な行動に影響を及ぼす(Bymasterら,Curr Drug Targets CNS Neurol Disord(2002年)1巻,p.163−181)。ムスカリ
ン性受容体は、コリン作動性ニューロンとの相互作用によるだけでなく、前脳/中脳のドーパミン作動性、GABA作動性およびグルタミン酸作動性ニューロンの活性の調節によっても、これらの機能に影響を及ぼす。神経局在化および微小透析の研究によって、ムスカリン性受容体およびその作動薬または拮抗薬はこれらの系に影響を及ぼし、調節の方向性(興奮/抑制)は特定の受容体サブタイプに依存することが確認された。具体的には、M1/M4を好む拮抗薬であるピレンゼピンの局所微量注射によって、線条体においてドーパミン流出の低減が起こる(Smoldersら,J Neurochem(1997年)68巻,p.1942−1948)。同様に、中脳に直接注入すると、M1/M4受容体を好む拮抗薬であるテレンゼピンは、GABA流出の低減を引き起こす(Smoldersら,1997年、上述)。同様に、スコポラミンなどの非サブタイプ選択的拮抗薬は、前脳においてアセチルコリンレベルの上昇を引き起こす(Izurieta−Sanchezら,Eur J Pharmacol(2000年)399巻,p.151−160)。
【0058】
満足不可能性および強迫的過食を含めて、依存性の障害に関して、中脳辺縁系ドーパミン回路は、嗜癖行動の形成および永続化において重要な役割を果たすと思われる(BerridgeおよびRobinson,Brain Res Brain Res Rev(1998年)28巻,p.309−369;Crespoら,J Neurosci(2006年)26巻,p.6004−6010;Di ChiaraおよびImperato,Proc Natl Acad Sd USA(1988年)85巻,p.5274−5278;HernandezおよびHoebd,Life Sci(1988年)42巻,p.1705−1712).げっ歯類を用いた研究から、線条体における特定の構造である側坐核(NAc)が報酬および嫌悪の制御に関与することが明らかである。NAcは、内側腹側線条体に存在し、さらにシェル、コア、および吻極のサブ領域に分解することができる(ZahmおよびBrog,Neuroscience(1992年)50巻,p.751−767)。
【0059】
ラットは、ドーパミン作動薬をNAcに自己投与し(Hoebelら,Psychopharmacology (Berl)(1983年)81巻,p.158−163)、ヒトにおいて乱用および習慣性を引き起こすことが知られている多数の薬物は、NAcにおいて細胞外ドーパミンレベルを上昇させることがわかっている(Di ChiaraおよびImperato,1988年,上述;HernandezおよびHoebel,1988年,上述;Radaら,Pharmacol Biochem Behav(1996年)53巻,p.809−816)。逆に、側坐核における細胞外ドーパミンの低減は、モルヒネ誘導性およびニコチン誘導性離脱中の嫌悪に随伴することが観察されている(AcquasおよびDi Chiara,(1992年)J Neurochem 58巻,p.1620−1625;Dianaら,J Pharmacol Exp Ther(1995年)272巻,p.781−785;Pothosら,Brain Res(1991年)566巻,p.348−350;Radaら,Psychopharmacology (Berl)(2001年)157巻,p.105−110)。ドーパミンの効果は、受容体サブタイプDlおよびD2によって媒介されるようである。ドーパミンDlまたはD2作動薬をNAcのコアではなくシェルに注射すると、コカインを求めてレバーを押すようオペラント条件付けされたが、次いでコカインを生理食塩水に置換することによってその行動を消去されたラットにおいて、薬物探索行動を復活させることがわかっている(Schmidtら,Eur J Neurosci(2006年)23巻,p.219−228)。
【0060】
NAc内では、コリン作動性とドーパミン作動性の回路は薬理学的に逆のあるようである。アトロピン(非特異的ムスカリン拮抗薬)またはメカミルアミン(非特異的ニコチン拮抗薬)の局所側坐核内投与は、オピオイド強化の獲得を遮断することが報告されている
(Crespoら、2006年,上述)が、モルヒネはNAcにおいてアセチルコリンレベルを低減し(Fiserovaら,Psychopharmacology (Berl)(1999年)142巻,p.85−94;Radaら,Neuropharmacology(1991年)30巻,p.1133−1136)、ナロキソン誘導性オピオイド離脱はアセチルコリンレベルを上昇させる(Fiserovaら,1999年,上述;Radaら,1991年,上述;Radaら,1996年,上述)。同様の現象が、ニコチン依存性ラットにおけるメカミルアミン誘導性離脱に関連して観察されている(Radaら,2001年,上述)。AChの上昇と不快状態の広範な一般的関連性を支持するものとして、AChは、条件付け味覚嫌悪(Markら,Brain Res(1995年)688巻,p.184−188)、脳刺激嫌悪(RadaおよびHoebel,Brain Res(2001年)888巻,p.60−65)、およびジアゼパムからの離脱(RadaおよびHoebel、Eur J Pharmacol(2005年)508巻,p.131−138)、アルコール(Radaら、Pharmacol Biochem Behav(2004年)79巻,p.599−605)または砂糖からの離脱(Colantuoniら,Obes Res(2002年)10巻,p.478−488)によって、NAcにおいて放出される。したがって、コリン作動性伝達の減衰は、嗜癖および習慣性の障害の治療の治療上魅力的な手法である。このような障害は、スクロース離脱に関する知見が例示するように、純粋に薬理学的である必要はない。
【0061】
したがって、優先的にM1ムスカリン性受容体を調節する能力を有する化合物の神経精神医学での用途は広範囲である。したがって、本方法は、障害のあるi)認知処理、ii)情動処理、および/またはiii)欲求動機付けによって生ずる病態を含めて、種々の病態を治療する際に使用(適用)される@@〜に使用される。これらのカテゴリー内の病態としては、肥満症または過剰および/もしくは望ましくない体脂肪の保持を生じる衝動制御障害および食欲障害が挙げられる。
【0062】
したがって、本方法および組成物は、肥満症を治療し、食欲を抑制し、望ましい体重減少を促進し、所望の体重の維持を助長し、望ましくない体重増加を予防または低減する際に使用される。
【0063】
b.薬理作用剤
本方法および組成物で使用する薬理作用剤には、下記に詳細に記載される1つまたは複数の活性剤が、その任意の医薬として許容できる塩、プロドラッグ、ラセミ混合物、配座および/または光学異性体、結晶多形、ならびに同位体を含む任意の医薬として許容できる形で含まれる。
【0064】
i.選択的ムスカリン性受容体M1拮抗薬
本方法は、肥満症を治療し、体重減少および食欲抑制を促進することを必要とする個体に、治療量の1つまたは複数の選択的ムスカリン性受容体M1拮抗薬を投与することによって、肥満症を治療し、体重減少および食欲抑制を促進する。ムスカリン拮抗薬は、参照により本明細書に組み込まれる上述のGoodman and Gilman’s The Pharmacological Basis of Therapeuticsの第7章に概説されている。選択的ムスカリン性受容体M1拮抗薬としては、例えばピレンゼピンおよびテレンゼピンが挙げられる。これらの構造を下記に示す。
【0065】
【化2】
ピレンゼピン(5,11−ジヒドロ−ll−[(4−メチル−l−ピペラジニル)アセチル]−6H−ピリド[2,3−b][l,4]ベンゾジアゼピン−6−オン)は、Azupharma(ドイツ シュツットガルト(Stuttgart,Germany))、Boehringer Ingelheim(ドイツ インゲルハイム(Ingelheim,Germany);Gastrozepin(登録商標)として販売)、Dolorgiet(ドイツ ボン(Bonn,Germany))を含めて、複数の製薬会社によって、ピレンゼピン二塩酸塩として製造および販売されている。ピレンゼピンは、約50mg/日〜約200mg/日、例えば約100〜150mg/日、または50、100、150、もしくは200mg/日の用量で投与することができる。あるいは、ピレンゼピンは、約0.1mg/kg/日〜約10mg/kg/日、通常約0.7mg/kg/日〜約5mg/kg/日の用量で投与することができる。ピレンゼピンの類似体も、本方法を実施する際に使用される。ピレンゼピンの化学的類似体は、例えば米国特許第3,660,380号;第3,743,734号;および第5,324,832号に開示され、それぞれの開示は、全ての目的で参照によりその全体が本明細書に組み込まれる。さらに、ピレンゼピンの用法は、例えば米国特許第5,668,155号に開示される。
【0066】
テレンゼピン(4,9−ジヒドロ−3−メチル−4−[(4−メチル−l−ピペラジニル)アセチル]−10H−チエノ[3,4−b][l,5]ベンゾジアゼピン−10−オン)は、例えばTocris Bioscience(米国ミズーリ州エリスビィル(Ellisville,MO))およびSigma−Aldrich,Inc.(米国ミズーリ州セントルイス(St.Louis,MO))からテレンゼピン二塩酸塩として市販されている。さらに、テレンゼピンの合成は、参照により本明細書に組み込まれる米国特許第4,381,301号に開示される。テレンゼピンは、約0.5mg/日〜約10mg/日、例えば約1〜5mg/日、または0.5、1、2、3、4、5、6、7、8、9、もしくは10mg/日の用量で投与することができる。テレンゼピンの類似体も、本方法を実施する際に使用される。テレンゼピンの化学的類似体およびエナンチオマーは、例えば米国特許第3,953,430号;第4,168,269号;第4,172,831号;第4,381,301号;第5,140,025号、および第5,324,832号に開示され、それぞれの開示は、全ての目的で参照によりその全体が本明細書に組み込まれる。
【0067】
いくつかの実施形態では、テレンゼピンの(+)および(−)エナンチオマーの混合物を含有するラセミ体調製物が投与される。いくつかの実施形態では、テレンゼピンの(+)または(−)エナンチオマーが投与される。テレンゼピンは、35.5kcal/molの活性化障壁によって分離される2つの異なるキラル状態で存在する(Eveleighら,MoI Pharmacol(1989年)35巻,p.477−483;およびSchudtら,Eur J Pharmacol(1989年)165巻,p.87−96)。(+)型のテレンゼピンは、強力な抗ムスカリン様活性を有するが、(−)型はかなり活性が低い。テレンゼピンの選択性は、様々な解剖学的部位で異なるようであり、皮質受容体には、(+)型は(−)異性体に比べて、400倍有効であり;心臓の受容体
には、選択性は低く、(+)型は(−)型より50倍強力である(Eveleighら,上述)。2つの型は、ゆっくり、かつ半減時間約200時間、90度で相互変換する(Eveleighら,上述)。複数の研究では、2つの型が異なる活性を有すると主張されている(Eltze,Eur J Pharmacol(1990年)180巻,p.161−168;Eveleighら,上述;Feifelら,Eur J Pharmacol(1991年)195巻,p.115−123;Kilianら,Agents Actions Suppl 34巻,p.131−147;Schudtら,上述)。
【0068】
ii.抗うつ薬
本発明で使用するためのM1R−選択的拮抗薬ではない抗うつ剤は、その作用機序によっては限定されず、どのクラスの抗うつ薬でも適用可能である。例えば、三環系抗うつ薬(TCA)およびその類似体、セロトニン再取り込み阻害薬、モノアミンオキシダーゼ阻害薬(MAOI)、セロトニン作動薬およびそのプロドラッグ、ノルエピネフリン再取り込み阻害薬、ドーパミン再取り込み阻害薬、およびセロトニン再取り込み促進薬はすべて、1つまたは複数のM1R−選択的拮抗薬と組み合わせて投与することができる。セロトニン再取り込み阻害薬としては、選択的セロトニン再取り込み阻害薬(SSRI)とセロトニン−ノルエピネフリン再取り込み阻害薬(SNRI)が挙げられる。ノルエピネフリン再取り込み阻害薬としては、特異的ノルエピネフリン再取り込み阻害薬と混合ノルエピネフリン−ドーパミン再取り込み阻害薬(NDRI)が挙げられる。セロトニン−ノルエピネフリン−ドーパミンまたは「三元再取り込み阻害薬」も本発明で使用される。他のカテゴリーの抗うつ薬、例えば四環系抗うつ薬であるマプロチリンもしくはミアンセリン、または作用剤であるトラゾドン、ネファゾドン、もしくははブスピロン;副腎皮質刺激ホルモン放出因子受容体1(CRFl)拮抗薬、ならびにアモキサピン、クロザピン、リスペリドン、オランザピン、クエチアピン、およびアリピプラゾールを含めて、精神病もしくはは双極性障害の状況で活性を有することが発見された化合物も使用することができる。
【0069】
本発明で使用するための三環系抗うつ薬としては、アミネプチン、アミトリプチリン、クロミプラミン、デシプラミン、ドキセピン、ドチエピン、イミプラミン、ノルトリプチリン、プロトリプチリン、トリミプラミン、アモキサピン、および筋弛緩薬であるシクロベンザプリンが挙げられる。他の記載していない三環系抗うつ薬およびその類似体も使用するができる。
【0070】
一実施形態では、有効量の1つまたは複数のM1R−選択的拮抗薬を、有効量の選択的セロトニン再取り込み阻害薬と共投与する。選択的セロトニン再取り込み阻害薬としては、例えばシタロプラム、エシタロプラム、フルオキセチン(ラセミ体または光学異性体)、フルボキサミン、パロキセチン、およびセルトラリン(およびそのS−エナンチオマー、Zoloft(登録商標))が挙げられるが、記載していないSSRIも適用可能である。一実施形態では、シタロプラム(またはエシタロプラム)を、1つまたは複数のM1R−選択的拮抗薬と共投与する。一実施形態では、有効量のフルオキセチン(ラセミ体または光学異性体)を共投与する。一実施形態では、有効量のフルボキサミンを共投与する。一実施形態では、有効量のセルトラリン(またはそのS−エナンチオマー、Zoloft(登録商標))を共投与する。一実施形態では、有効量のパロキセチンを共投与する。一実施形態では、有効量のデュロキセチンを共投与する。
【0071】
一実施形態では、有効量の1つまたは複数のセロトニン−ノルエピネフリン再取り込み阻害薬を、1つまたは複数のM1R−選択的拮抗薬と共投与する。セロトニン−ノルエピネフリン再取り込み阻害薬としては,例えばミルナシプラン、ミルタザピン、ベンラファキシン(ラセミ体または光学異性体)、デュロキセチン、(−)l−(1−ジメチルアミノメチル−5−メトキシベンゾ−シクロブタン−1−イル)シクロヘキサノール(S33
005)、DVS−233(デスベンラファキシン)、DVS−233 SR、およびシブトラミンが挙げられるが、記載していないSNRIも使用される。ミルタザピンの作用機序は、その明らかなセロトニン作動性とノルアドレナリン作動性の二様性作用のため、他のSNRIの作用機序と異なる可能性があるが、本明細書ではSNRIクラスの抗うつ薬の一メンバーと見なされる。一実施形態では、有効量のベンラファキシン(ラセミ体または光学異性体)を共投与する。一実施形態では、有効量のデスベンラファキシンを共投与する。一実施形態では、有効量のシブトラミンを共投与する。一実施形態では、有効量のデュロキセチンを共投与する。一実施形態では、有効量のミルナシプランを共投与する。一実施形態では、有効量のミルタザピンを共投与する。
【0072】
他の実施形態では、有効量の1つまたは複数の選択的ノルエピネフリン再取り込み阻害薬を1つまたは複数のM1R−選択的拮抗薬と共投与する。選択的ノルエピネフリン再取り込み阻害薬としては、例えばレボキセチンおよびアトモキセチンが挙げられる。
【0073】
一実施形態では、有効量の1つまたは複数のノルエピネフリン−ドーパミン再取り込み阻害薬を、1つまたは複数のM1R−選択的拮抗薬と共投与する。ノルエピネフリン−ドーパミン再取り込み阻害薬としては、例えばアミネプチン、モダフィニル、GW353162、およびブプロピオンが挙げられる。ブプロピオンの場合、代謝物はノルアドレナリン作動性再取り込み遮断の原因になると思われる。一実施形態では、有効量のブプロピオンを共投与する。
【0074】
一実施形態では、有効量の1つまたは複数の三元(セロトニン−ノルエピネフリン−ドーパミン)再取り込み阻害薬を、1つまたは複数のM1R−選択的拮抗薬と共投与する。三元再取り込み阻害薬としては、例えばインダトラリン、SEP−225289、DOV
216,303、および(+)−l−(3,4−ジクロロフェニル)−3−アザビシクロ−[3.1.0]ヘキサン塩酸塩(DOV 21,947)が挙げられる。
【0075】
本発明で使用するためのモノアミンオキシダーゼ阻害薬としては、ベフロキサトン、ブロファロミン、デプレニル、イソカルボキサジド、モクロベミド、パルギリン、フェネルジン、セレギリン、およびトラニルシプロミンと、それらの持続性送達および経皮送達の形が挙げられる。
【0076】
M1R−選択的拮抗薬と共投与することができる抗うつ薬としては、マプロチリン、チアネプチン、ネファゾドン、およびトラゾドンが挙げられる。
【0077】
抗うつ薬の適切な投与量は、様々な要因の中でもとりわけ、組成物の選択された投与経路および製剤に依存する。例えば、三環系抗うつ薬は、約25mg/日〜約600mg/日の用量、通常約75mg/日〜約300mg/日の用量で投与される。
【0078】
セロトニン−再取り込み阻害薬は、約5mg/日〜約400mg/日の用量で投与され、通常約20mg/日〜約250mg/日で投与される。特に、本方法を実施する際に、ベンラファキシン(ラセミ体または光学異性体)は、1回の投与当たり約9mg〜約225mgで投与することができ、通常1回の投与当たり約37.5mg、75mg、150mg、または225mgで投与する。ベンラファキシンは、典型的には約25mg/日〜550mg/日,通常約37.5mg/日〜375mg/日、より典型的には約75mg/日−225mg/日、最も典型的には約37.5mg/日、75mg/日、150mg/日、225mg/日、または300mg/日で投与される。個々の患者に応じて、1日のベンラファキシン投与量を分割し、1日1回、2回、3回、4回以上で投与することができる。デスベンラファキシンは、約50mg/日〜600mg/日、例えば約50mg/日、100mg/日、200mg/日、400mg/日、または600mg/日の用量
で投与することができる。セルトラリン(またはそのS−エナンチオマー、Zoloft(登録商標))は、約50mg/日〜200mg/日、通常約100mg/日〜150mg/日の用量で投与することができる。フルオキセチン(ラセミ体または光学異性体)は、約5mg/日〜50mg/日、通常約20mg/日〜40mg/日の用量で投与することができる。フルボキサミンは、約50mg/日〜300mg/日、通常約100mg/日〜200mg/日の用量で投与することができる。パロキセチンは、約10mg/日〜50mg/日、通常約20mg/日〜40mg/日の用量で投与することができる。
【0079】
本方法を実施する際に、シタロプラム(またはエシタロプラム)を、約5mg/日〜60mg/日、好ましくは約10mg/日、20mg/日、または30mg/日で投与することができる。通常、シタロプラムは、1日1回、例えば午前または午後に投与される。しかし、投与量のシタロプラムを1日2回以上で投与される患者もいる。ミルタザピンは、約5mg/日〜100mg/日、例えば約7.5mg/日、15mg/日、30mg/日、45mg/日、または90mg/日の用量で投与することができる。ミルナシプランは、約25mg/日〜200mg/日、例えば約25mg/日、50mg/日、100mg/日、150mg/日、または200mg/日の用量で投与することができる。
【0080】
ブプロピオン、ネファゾドン、およびトラゾドンを含めて、非典型的な抗うつ薬は、約50mg/日〜600mg/日、通常約150mg/日〜400mg/日の用量で投与される。ブプロピオンは、約25mg/日〜300mg/日、例えば約25mg/日、50mg/日、100mg/日、150mg/日、200mg/日、300mg/日の用量で投与することができる。モノアミンオキシダーゼ阻害薬は、典型的には約5mg/日〜90mg/日、通常約10mg/日〜60mg/日の用量で投与される。
【0081】
iii.抗肥満剤
本発明は、有効量の1つまたは複数のM1R−選択的拮抗薬を1つまたは複数の抗肥満剤と組み合わせて投与することも企図する。さらに、本発明は、有効量の1つまたは複数のM1R−選択的拮抗薬と1つまたは複数の抗うつ薬の組合せを、さらに1つまたは複数の抗肥満剤と任意選択で組み合わせて投与することを企図する。(i)1つもしくは複数のM1R−選択的拮抗薬と組み合わせた、または(ii)さらにM1R−選択的拮抗薬と抗うつ薬の組合せと組み合わせた使用に適した抗肥満剤としては、例えば食欲減退薬、ドーパミン作動薬、H3−ヒスタミン拮抗薬、5−HT2c受容体作動薬、β−3アドレナリン受容体作動薬、コレシストキニン作動薬、抗てんかん剤、レプチン、レプチン類似体およびレプチン受容体作動薬、ニューロペプチドY(NPY)受容体拮抗薬およびモジュレーター、ペプチド−YY(PYY)受容体作動薬、毛様体神経栄養因子、甲状腺ホルモン受容体−β作動薬、カンナビノイドCBl受容体拮抗薬、メラニン凝集ホルモン受容体拮抗薬、膵および胃液リパーゼ阻害薬、メラノコルチン−4受容体作動薬、およびそれらの組合せが挙げられる。
【0082】
食欲減退薬としては、例えばアンフェタミン、メタンフェタミン、デキストロアンフェタミン、フェンテルミン、ベンズフェタミン、フェンジメトラジン、フェンメトラジン、ジエチルプロピオン、マジンドール、フェンフルラミン、デキスフェンフルラミン、フェニルプロパノールアミン、エフェドラなどが挙げられる。食欲減退薬は交感神経刺激アミンとすることができる。
【0083】
ドーパミン作動薬としては、例えばER−230、ドプレキシン、メシル酸ブロモクリプチンなどが挙げられる。
【0084】
H3−ヒスタミン拮抗薬としては、例えばイムペンタミン、チオペラミド、シプロキシファン、クロベンプロピット、GT−2331、GT−2394、A−331440など
が挙げられる。
【0085】
5−HT2c受容体作動薬としては、例えば1−(m−クロロフェニル)ピペラジン(m−CPP)、ミルタザピン、APD−356(ロルカセリン)、SCA−136(バビカセリン)、ORG−12962、ORG−37684、ORG−36262、ORG−8484、Ro−60−175、Ro−60−0332、VER−3323、VER−5593、VER−5384、VER−8775、LY−448100、WAY−161503、WAY−470、WAY−163909、MK−212、BVT.933、YM−348、IL−639、IK−264、ATH−88651、ATHX−105などが挙げられる(例えば、Nilsson BM,J.Med.Chem.2006年,49巻,p.4023−4034を参照のこと)。
【0086】
β−3アドレナリン受容体作動薬としては、例えばL−796568、CGP 12177、BRL−28410、SR−58611A、ICI−198157、ZD−2079、BMS−194449、BRL−37344、CP−331679、CP−331648、CP−114271、L−750355、BMS−187413、SR−59062A、BMS−210285、LY−377604、SWR−0342SA、AZ−40140、SB−226552、D−7114、BRL−35135、FR−149175、BRL−26830A、CL−316243、AJ−9677、GW−427353、N−5984、GW−2696などが挙げられる。
【0087】
コレシストキニン作動薬としては、例えばSR−146131、SSR−125180、BP−3.200、A−71623、A−71378、FPL−15849、GI−248573、GW−7178、GI−181771、GW−7854、GW−5823などが挙げられる。
【0088】
膵および胃液リパーゼ阻害薬としては、例えばオルリスタット、セチリスタット(ATL−962)などが挙げられる。
【0089】
抗てんかん剤としては、例えばトピラメート、ゾニサミドなどが挙げられる。
【0090】
有用な他の抗肥満剤としては、レプチン、レプチン類似体、およびレプチン受容体作動薬(LY−355101などを含む)、ニューロペプチドY(NPY)受容体拮抗薬およびモジュレーター(SR−120819−A、PD−160170、NGD−95−1、BIBP−3226、1229−U−91、CGP−71683、BIBO−3304、CP−671906−01、J−115814などを含む)、ペプチド−YY(PYY)受容体作動薬(PYY(3−36)などを含む)、毛様体神経栄養因子(アクソカインなどを含む)、甲状腺ホルモン受容体−β作動薬(KB−141、GC−I、GC−24、GB98/284425などを含む)、カンナビノイドCBl受容体拮抗薬(リモナバン、SR147778、SLV 319などを含む(例えば、Antel Jら,J.Med.Chem.2006年,49巻,p.4008−4016を参照のこと))、メラニン凝集ホルモン受容体拮抗薬 (GlaxoSmithKline 803430X、GlaxoSmithKline 856464、SNAP−7941、T−226296などを含む(例えば、Handlon ALおよびZhou H,J.Med.Chem.2006年,49巻,p.4017−4022を参照のこと))、メラノコルチン−4受容体作動薬(PT−15、Ro27−3225、THIQ、NBI 55886、NBI 56297、NBI 56453、NBI 58702、NBI 58704、MB243などを含む(例えば、Nargund RPら,J.Med.Chem.2006年,49巻,p.4035−4043を参照のこと))、およびそれらの組合せが挙げられる。
【0091】
iv.薬理作用剤の組合せ
いくつかの実施形態では、1つもしくは複数のM1R−選択的拮抗薬を、M1R−選択的拮抗薬ではない1つもしくは複数の抗うつ薬と共投与または同時製剤する。いくつかの実施形態では、1つもしくは複数のM1R−選択的拮抗薬を、1つもしくは複数の抗肥満剤と共投与または共製剤する。いくつかの実施形態では、1つもしくは複数のM1R−選択的拮抗薬を、M1R−選択的拮抗薬ではない1つもしくは複数の抗うつ薬、および1つもしくは複数の抗肥満剤と共投与または共製剤する。M1R−選択的拮抗薬、抗うつ薬、および抗肥満剤は上述する通りである。
【0092】
v.異性体
治療剤のすべての配座異性体(例えば、シスおよびトランス異性体)、ならびにすべての光学異性体(例えば、エナンチオマーおよびジアステレオマー)、このような異性体のラセミ体、ジアステレオマー、および他の混合物、ならびに溶媒和物、水和物、異種同形物、同質多形、および互変異性体は、本発明の範囲内である。
【0093】
vi.同位体
本発明は、1つまたは複数の原子が特定の原子質量または質量数を有する1つまたは複数の原子で置換されている治療剤の同位体標識体も包含する。治療剤およびそのプロドラッグの同位体標識体、ならびに治療剤およびそのプロドラッグの同位体標識した医薬として許容できる塩は、本発明の範囲内である。ある種の状況では、重水素(2H)などのより重い同位体による置換によって、インビボ半減期の延長または必要用量の低減など治療上の利点をもたらす代謝安定性を増大させることができる。本発明の治療剤およびそのプロドラッグの同位体標識体は、一般に当業者に知られている方法に従って、非同位体標識試薬を同位体標識試薬に置換することによって調製することができる。
【0094】
c.投与
i.投与期間
通常、1つまたは複数のM1R−選択的拮抗薬は、長期間にわたって個体に投与される。本方法は、少なくとも20日、いくつかの実施形態では少なくとも40日、60日、80日、または100日、またいくつかの実施形態では少なくとも150日、200日、250日、300日、350日、1年以上実施することができる。本治療方法を、1年より長期間、例えば少なくとも400日、450日、500日、550日、600日、650日、700日、800日、900日、1000日受ける個体もいる。しかし、個体は、本方法での2年、3年、4年以上の治療に成功することができる。
【0095】
通常、本発明に従って治療された対象は、治療の約50日〜70日後に少なくとも約5ポンド〜15ポンド、治療の約90日〜150日後に少なくとも約10ポンド〜25ポンド、また治療の約200日〜400日後に少なくとも約15ポンド〜45ポンド減量することができる。通常は、本発明に従って治療された個体は、治療計画を100日、150日、200日、250日、300日、350日、400日、450日、500日、550日、600日、700日、800日、900日、1000日以上実施することによって、個体のベースライン体重の少なくとも約5%、より一般には少なくとも約10%、15%、または20%を減量することができ、この所望の体重減少を安定に維持することができる。重要なことには、有効量の1つまたは複数の抗うつ薬を長期間にわたって投与することによって、長期間の治療を通して安定な体重状態が助長され、望ましくない体重増加が予防される。本発明の併用治療は、肥満および過体重の個体に特に適切であるが、減量を望み、安定な体重を維持し、または望ましくない体重増加を予防するいずれの個体にも投与することができる。
【0096】
ii.スケジューリング
一般に、本方法を実施する際に、有効量の1つまたは複数のM1R−選択的拮抗薬を単独投与し、あるいは1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬と共投与する。いくつかの実施形態では、有効量の1つまたは複数のM1R−選択的拮抗薬を、1つまたは複数の抗肥満剤と共投与する。いくつかの実施形態では、有効量の1つまたは複数のM1R−選択的拮抗薬を、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬および1つまたは複数の抗肥満剤と共投与する。共投与される薬理作用剤は、一緒にまたは別々に、同時にまたは異なる時間に投与することができる。投与する時、M1R−選択的拮抗薬、抗うつ薬、および抗肥満剤は独立に、必要に応じて1日1回、2回、3回、4回以上、またはそれほど頻繁でなく投与することができる。好ましくは、投与する薬理作用剤を胃日1回投与する。好ましくは、投与する活性剤を、同時1回または複数回、例えば混合物として投与する。薬理作用剤の1つまたは複数を徐放性製剤として投与することができる。
【0097】
ある患者らについては、治療の開始から、1つもしくは複数のM1R−選択的拮抗薬、次いで1つもしくは複数の抗うつ薬、および/または1つもしくは複数の抗肥満剤を同時に投与して、本方法を実施する。ある患者らについては、まず1つもしくは複数のM1R−選択的拮抗薬を投与し、次いで1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤を続いて共投与することによって、本方法を実施する。1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤の投与を開始する前に、3日、5日、7日、10日、14日、20日、または30日の間、最初に患者に1つまたは複数のM1R−選択的拮抗薬を単独投与することができる。
【0098】
体重減少の助長または食欲の抑制の目的で投与する場合、1つまたは複数のM1R−選択的拮抗薬を単独または組み合わせて投与して、予防的には望ましくない体重増加を予防し、または安定な体重を維持し、あるいは治療的には所望の体重減少を実現し、このような体重減少を維持することができる。
【0099】
iii.投与経路
したがって、1つまたは複数のM1R−選択的拮抗薬の単独投与、あるいは1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤と組み合わせた投与は、経口、口腔内;静脈内、皮内、皮下、筋肉内、経皮、経粘膜、鼻腔内を含めて、非経口などの投与を含めて、様々な方法で実現することができる。1つまたは複数のM1R−選択的拮抗薬を1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤と共投与する場合、同じまたは異なる投与経路で投与することができる。
【0100】
いくつかの実施形態では、1つまたは複数のM1R−選択的拮抗薬を単独または組み合わせて、全身投与ではなく、例えばデポーまたは徐放性製剤で局所投与することができる。
【0101】
iv.適切な投与量を決定する方法
M1R−選択的拮抗薬、抗うつ薬、および抗肥満剤のための投与量は、当業者によって実施されている投与量およびスケジューリング計画に従う。本方法で使用するすべての薬理作用剤の適切な投与量の一般指針は、Goodman and Gilman’s The Pharmacological Basis of Therapeutics,第11版,2006年,上述、およびPhysicians’ Desk Reference (PDR)、例えば第59編(2005年)または第60編(2006年)、Thomson PDRに記載され、これらはそれぞれ、参照により本明細書に組み込まれる。発表されたM1R−選択的拮抗薬の投与量は、肥満症を治療し、または体重減少を促進し、または体重増加を抑制するための治療と異なる適応症に関するものである。本発
明の組成物および方法では、本発明を実施するためのM1R−選択的拮抗薬、抗うつ薬、および抗肥満剤の有効な投与量は、他の適応症に関して発表された投与量と等しく、またはそれより低い{例えば、約25%、50%、75%、または100%)ことがあり得る。
【0102】
M1R−選択的拮抗薬、抗うつ薬、および抗肥満剤の適切な投与量は、選択された投与経路、組成物の製剤、患者の応答、病態の重症度、対象の体重、および処方医師の判断を含めて、複数の要因に従って変わる。投与量は、個々の患者によって必要とされるように、経時的に増減することができる。通常、最初に患者に低用量を投与し、次いで患者が忍容できる有効な投与量まで増加させる。
【0103】
有効量の決定は、特に本明細書に記載されている詳細な開示を考慮に入れて、当業者の能力の十分範囲内である。一般に、1つもしくは複数のM1R−選択的拮抗薬および1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤の組合せの効果的または有効な量は、治療された対象において、毒性の副作用が最小限しかまたは全くなく、所望の効果が観察されるまで、まず低用量または少量のM1R−選択的拮抗薬を単独投与し、次いで投与する用量または投与量を徐々に増加し、必要なら第2または第3の医薬品を追加することによって決定される。本発明の組合せの投与のための適切な用量および投与スケジュールを決定するのに適用可能な方法は、例えばGoodman and Gilman’s The Pharmacological Basis of Therapeutics,第1l版,2006,上述;Physicians’ Desk Reference (PDR),上述;Remington: The Science
and Practice of Pharmacy,第21版,2006年,上述;ならびにMartindale:The Complete Drug Reference,Sweetman,2005年,London:Pharmaceutical Press.、およびMartindale,Martindale:The Extra Pharmacopoeia,第31版,1996年,Amer Pharmaceutical Assnに記載され、それぞれ、参照により本明細書に組み込まれる。
【0104】
投与量および間隔は、治療効果を維持するのに十分な活性化合物の血漿中レベルをもたらすように個別に調整することができる。好ましくは、治療上有効な血清レベルは、1日1回の用量を投与することによって実現されるが、有効な1日複数回投与のスケジュールも本発明に包含される。局所投与または選択的取り込みの場合、有効な局所濃度の薬物を血漿中濃度と関係付けることはできない。当業者は、過度の実験なしに治療上有効な局所投与量を最適化することができる。
【0105】
3.医薬組成物
本発明は、さらに治療上有効量の1つもしくは複数のM1R−選択的拮抗薬、および1つもしくは複数の抗うつ薬、および/または1つもしくは複数の抗肥満剤の混合物を含む医薬組成物を提供する。いくつかの実施形態では、M1R−選択的拮抗薬は、テレンゼピン、ピレンゼピン、およびそれらの混合物からなる群より選択される。
【0106】
いくつかの実施形態では、医薬組成物は、選択的セロトニン再取り込み阻害薬(SSRI)、セロトニン−ノルエピネフリン再取り込み阻害薬(SNRI)、ノルエピネフリン再取り込み阻害薬、ドーパミン再取り込み阻害薬、ノルエピネフリン−ドーパミン再取り込み阻害薬(NDRI)、セロトニン−ノルエピネフリン−ドーパミン再取り込み阻害薬、セロトニン再取り込み促進薬、セロトニン作動薬、およびそれらのプロドラッグである1つまたは複数の抗うつ薬を含む。一実施形態では、医薬組成物は、ベンラファキシン(ラセミ体または光学異性体)、デュロキセチン、フルオキセチン(ラセミ体または光学異性体)、シタロプラム、エシタロプラム、フルボキサミン、パロキセチン、S33005
、DVS−233(デスベンラファキシン)、DVS−233 SR、ブプロピオン、GW353162、シブトラミン、アトモキセチン、およびセルトラリン(またはそのS−エナンチオマー、Zoloft(登録商標))からなる群より選択される1つまたは複数の抗うつ薬を含む。
【0107】
一実施形態では、医薬組成物は、治療上有効量のテレンゼピンまたはピレンゼピン、およびSSRIを含む。一実施形態では、医薬組成物は、治療上有効量のテレンゼピンまたはピレンゼピン、およびシタロプラム(またはエシタロプラム)を含む。一実施形態では、医薬組成物は、治療上有効量のテレンゼピンまたはピレンゼピン、およびセルトラリン(またはそのS−エナンチオマー、Zoloft(登録商標))を含む。一実施形態では、医薬組成物は、治療上有効量のテレンゼピンまたはピレンゼピン、およびフルオキセチン(ラセミ体または光学異性体)を含む。一実施形態では、医薬組成物は、治療上有効量のテレンゼピンまたはピレンゼピン、およびフルボキサミンを含む。一実施形態では、医薬組成物は、治療上有効量のテレンゼピンまたはピレンゼピン、およびパロキセチンを含む。
【0108】
一実施形態では、医薬組成物は、治療上有効量のテレンゼピンまたはピレンゼピン、およびSNRIを含む。一実施形態では、医薬組成物は、治療上有効量のテレンゼピンまたはピレンゼピン、およびベンラファキシン(ラセミ体または光学異性体)を含む。一実施形態では、医薬組成物は、治療上有効量のテレンゼピンまたはピレンゼピン、およびデスベンラファキシンを含む。一実施形態では、医薬組成物は、治療上有効量のテレンゼピンまたはピレンゼピン、およびデュロキセチンを含む。一実施形態では、医薬組成物は、治療上有効量のテレンゼピンまたはピレンゼピン、およびミルナシプランを含む。一実施形態では、医薬組成物は、治療上有効量のテレンゼピンまたはピレンゼピン、およびミルタザピンを含む。
【0109】
一実施形態では、医薬組成物は、治療上有効量のテレンゼピンまたはピレンゼピン、およびブプロピオンを含む。
【0110】
1つもしくは複数のM1R−選択的拮抗薬および1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤の組合せは、独立にまたは一緒に、その医薬として許容できる塩の形、またはそれらの化合物がg適切な担体もしくは賦形剤と混合されている医薬組成物の形で、治療上有効量、例えば所望の体重減少もしくは維持をもたらし、または望ましくない体重増加を予防するのに有効な用量で、対象、例えばヒト患者、ネコやイヌなどの家畜に投与することができる。
【0111】
本発明の1つもしくは複数のM1R−選択的拮抗薬および1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤の組合せは、治療的投与のための種々の製剤に組み込むことができる。さらに詳細には、本発明の組合せは、一緒にまたは別々に、適切な医薬として許容できる担体または希釈液との製剤によって医薬組成物に製剤することができ、固体、半固体、液体、または気体の形の調製物、具体的には錠剤、カプセル剤、丸剤、散剤、顆粒剤、糖衣錠、ゲル剤、スラリー、軟膏剤、液剤、坐剤、注射剤、吸入剤、およびエアゾール剤に製剤することができる。
【0112】
本発明で使用するための適切な製剤は、例えばRemington:The Science and Practice of Pharmacy,第21版,2006年,上述;Martindale: The Complete Drug Reference,Sweetman,2005年,London:Pharmaceutical Press.;Niazi,Handbook of Pharmaceutical Manufacturing Formulations,2004年,CRC Pre
ss;およびGibson,Pharmaceutical Preformulation and Formulation:A Practical Guide from Candidate Drug Selection to Commercial
Dosage Form,2001年,Interpharm Pressに出ており、これらは参照により本明細書に組み込まれる。本明細書に記載する医薬組成物は、当業者に周知の様式、すなわち通常の混合、溶解、顆粒化、糖衣錠作製、分級、乳化、カプセル化、封入、または凍結乾燥プロセスで製造することができる。下記の方法および賦形剤は、例示するものにすぎず、決して限定するものではない。
【0113】
一実施形態では、1つもしくは複数のM1R−選択的拮抗薬および1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤の組合せは、徐放、放出制御、持続放出、時限放出、または遅延放出製剤、例えば治療剤を含有する固体疎水性ポリマーの半透過性マトリックスでの送達用に調製される。様々なタイプの徐放材料が確立しており、当業者に周知せある。現在の持続放出製剤としては、フィルムコーティング錠剤、マルチパーティキュレートまたはペレット系、親水性または親油性材料を使用したマトリックス技術、および造孔賦形剤を含むワックス系錠剤が挙げられる(例えば、Huangら,Drug Dev.Ind.Pharm.29巻,p.79(2003年);Pearnchobら,Drug Dev.Ind.Pharm.29巻,p.925(2003年);Maggiら,Eur.J.Pharm.Biopharm.55巻,p.99(2003年);Khanvilkarら,Drug Dev.Ind.Pharm.228巻,p.601(2002年);およびSchmidtら,Int.J.Pharm.216巻,p.9(2001年)を参照のこと)。徐放送達システムは、そのデザインに応じて、何時間かまたは何日間かにわたって、例えば4時間、6時間、8時間、10時間、12時間、16時間、20時間、24時間以上にわたって、化合物を放出することができる。通常、徐放性製剤は、天然または合成ポリマー、例えばポリビニルピロリドン(PVP)など重合体のビニルピロリドン類;親水性カルボキシビニルポリマー類;メチルセルロース、エチルセルロース、ヒドロキシプロピルセルロース、およびヒドロキシプロピルメチルセルロースなど疎水性および/または親水性のハイドロコロイド類;ならびにカルボキシポリメチレンを使用して調製することができる。
【0114】
徐放性または持続放出製剤は、二酸化チタン、二酸化ケイ素、酸化亜鉛、および粘土を含めて、鉱物などの天然材料を使用して調製することもできる(参照により本明細書に組み込まれる米国特許第6,638,521号を参照のこと)。本発明の1つもしくは複数のM1R−選択的拮抗薬および1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤の組合せを送達する際に使用することができる持続放出製剤としては、例えば米国特許第6,635,680号;第6,624,200号;第6,613,361号;第6,613,358号、第6,596,308号;第6,589,563号;第6,562,375号;第6,548,084号;第6,541,020号;第6,537,579号;第6,528,080号、および第6,524,621号2機債のものが挙げられ、これらはそれぞれ、参照により本明細書に組み込まれる。特に重要な放出制御製剤としては、米国特許第6,607,751号;第6,599,529号;第6,569,463号;第6,565,883号;第6,482,440号;第6,403,597号;第6,319,919号;第6,150,354号;第6,080,736号;第5,672,356号;第5,472,704号;第5,445,829号;第5,312,817号、および第5,296,483号に記載のものが挙げられ、これらはそれぞれ、参照により本明細書に組み込まれる。当業者は、容易に他の適用可能な徐放性製剤を理解するであろう。
【0115】
経口投与のために、1つもしくは複数のM1R−選択的拮抗薬および1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤の組合せを、当技術分野でよく知
られている医薬として許容できる担体と組み合わせることによって容易に製剤することができる。このような担体によって、化合物を、治療対象の患者が経口摂取するための錠剤、丸剤、糖衣錠、カプセル剤、乳剤、親油性および親水性の懸濁剤、液剤、ゲル剤、シロップ剤、スラリー、懸濁剤などとして製剤することができる。経口使用の医薬調製物は、化合物を固体賦形剤と混合し、得られた混合物を任意選択で中砕し、望むなら適切な佐剤を添加した後、顆粒の混合物を処理して、錠剤または糖衣錠コアを得ることによって得ることができる。適切な賦形剤は、特に充填剤、具体的にはラクトース、スクロース、マンニトール、もしくはソルビトールを含めて、砂糖類;セルロース調製物類、例えばトウモロコシデンプン、小麦デンプン、米デンプン、馬鈴薯デンプン、ゼラチン、トラガカントゴム、メチルセルロース、ヒドロキシプロピルメチルセルロース、カルボキシメチルセルロースナトリウムなど、および/またはポリビニルピロリドン(PVP)である。望むなら、架橋ポリビニルピロリドン、寒天、またはアルギン酸またはその塩、具体的にはアルギン酸ナトリウムなどの崩壊剤を添加することができる。
【0116】
経口使用することができる医薬調製物としては、ゼラチンで作製されたプッシュフィットカプセル剤、ならびにゼラチンおよびグリセロールやソルビトールなどの可塑剤で作製された密封軟カプセル剤が挙げられる。プッシュフィットカプセル剤は、活性成分を、ラクトースなどの充填剤、デンプン類などの結合剤、および/またはタルクもしくはステアリン酸マグネシウムなどの滑沢剤、および任意選択で安定剤と混合して含有することができる。軟カプセル剤では、活性化合物を、脂肪油類、流動パラフィン、または液状ポリエチレングリコール類など適切な液体に溶解または懸濁することができる。さらに、安定剤を添加することができる。経口投与用の製剤はすべて、このような投与に適した投与量であるべきである。
【0117】
糖衣錠コアには適切なコーティングが設けられている。このために、アラビアゴム、タルク、ポリビニルピロリドン、カルボポールゲル、ポリエチレングリコール、および/または二酸化チタンを任意選択で含有することができる濃縮された糖液、ラッカー溶液、ならびに適切な有機溶媒または溶媒混合物を使用することができる。錠剤または糖衣錠コーティングには、識別のためまたは活性化合物の用量の異なる組合せを特徴付けるために、染料または顔料を添加することができる。
【0118】
化合物を、注射、例えばボーラス注射または連続注入による非経口投与用に製剤することができる。注射のために、1つもしくは複数のM1R−選択的拮抗薬および1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤の組合せを、植物油もしくは他の類似の油類、合成脂肪族酸グリセリド類、高級脂肪族酸類のエステル、またはプロピレングリコールなどの水性または非水溶媒中、望むなら、通常の添加剤、具体的には可溶化剤、等張剤、懸濁化剤、乳化剤、安定剤、および防腐剤を含めて、溶解、懸濁、または乳化することによって製剤にすることができる。好ましくは、本発明の組合せを、水溶液、好ましくはハンクス液、リンゲル液、または生理適合性緩衝液などの生理食塩緩衝液中で製剤することができる。注射用製剤は、防腐剤を添加した単位剤形、例えばアンプル、または複数回投与容器で提供することができる。組成物は、油性または水性ビヒクル中の懸濁剤、液剤、または乳剤のような形を取ることができ、懸濁化、安定化、および/または分散化剤などの製剤化剤を含有することができる。
【0119】
非経口投与用医薬製剤としては、水溶性の形の活性化合物水溶液が挙げられる。さらに、活性化合物の懸濁剤は、適切な油性注射懸濁剤として調製することができる。適切な親油性溶媒またはビヒクルとしては、ゴマ油などの脂肪油、またはオレイン酸エチルまたはトリグリセリド類、またはリポソーム類などの合成脂肪酸エステルが挙げられる。水性注射懸濁剤は、その懸濁剤の粘度を増大させる物質、具体的にはカルボキシメチルセルロースナトリウム、ソルビトール、またはデキストランを含有することができる。場合によっ
ては、懸濁剤は、適当な安定剤、または高濃度液剤の調製が可能になるように化合物の溶解性を増大させる作用剤も含有することもできる。あるいは、活性成分は、使用する前に適当なビヒクル、例えば発熱物質を含まない無菌水で構成する散剤とすることができる。
【0120】
全身投与は、経粘膜または経皮手段によるものとすることもできる。経粘膜または経皮投与のために、透過するべき関門に適した浸透剤を製剤で使用する。局所投与のために、作用剤を軟膏剤、クリーム、膏薬、散剤、およびゲル剤に製剤する。一実施形態では、経皮送達剤はDMSOとすることができる。経皮送達システムとしては、例えば貼付剤を挙げることができる。経粘膜投与のために、透過するべき関門に適した浸透剤を製剤で使用する。このような浸透剤は、一般に当技術分野で知られている。本発明で使用することができる経皮送達製剤としては、例えば米国特許第6,589,549号;第6,544,548号;第6,517,864号;第6,512,010号;第6,465,006号;第6,379,696号;第6,312,717号、および第6,310,177号に記載されているものが挙げられ、これらはそれぞれ、参照により本明細書に組み込まれる。
【0121】
口腔投与のために、組成物は、通常の方式で製剤される錠剤またはロゼンジの形とすることができる。
【0122】
上述の製剤に加えて、本発明の1つもしくは複数のM1R−選択的拮抗薬および1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤の組合せは、埋め込み調製物として製剤することもできる。このような長時間作用性製剤は、移植(例えば、皮下または筋肉内)または筋肉内注射によって投与することができる。したがって、例えば化合物は、適当なポリマーまたは疎水性材料(例えば、許容できる油中の乳濁液)またはイオン交換樹脂、またはやや溶けにくい誘導体、例えばやや溶けにくい塩と共に製剤することができる。
【0123】
医薬組成物は、適当な固体相もしくはゲル相担体または賦形剤も含むことができる。このような担体または賦形剤としては、例えば炭酸カルシウム、リン酸カルシウム、様々な砂糖類、デンプン類、セルロース誘導体類、ゼラチン、およびポリエチレングリコール類などのポリマー類が挙げられるが、これらに限定されない。
【0124】
4.キット
本発明の医薬組成物は、キットで提供することができる。いくつかの実施形態では、本発明のキットは、1つもしくは複数のM1R−選択的拮抗薬および1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤を別々の製剤として含む。いくつかの実施形態では、キットは、1つもしくは複数のM1R−選択的拮抗薬および1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤を同じ製剤中に含む。いくつかの実施形態では、キットは、治療の過程を通して、1つもしくは複数のM1R−選択的拮抗薬および1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤を独立に、均一の剤形で提供する。いくつかの実施形態では、キットは、治療の過程にわたって、1つもしくは複数のM1R−選択的拮抗薬および1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤を独立に、個体の要件に従って増加または低減させるが、通常有効な用量レベルまで増加させる漸増減投与量で提供する。
【0125】
一実施形態では、キットは、テレンゼピンおよびピレンゼピンからなる群より選択される1つまたは複数のM1R−選択的拮抗薬を含む1つまたは複数の医薬組成物を含む。
【0126】
いくつかの実施形態では、キットは、選択的セロトニン再取り込み阻害薬(SSRI)、セロトニン−ノルエピネフリン再取り込み阻害薬(SNRI)、ノルエピネフリン再取
り込み阻害薬、ドーパミン再取り込み阻害薬、ノルエピネフリン−ドーパミン再取り込み阻害薬(NDRI)、セロトニン−ノルエピネフリン−ドーパミン再取り込み阻害薬、およびそれらの混合物からなる群より選択される1つまたは複数の抗うつ薬を含む。一実施形態では、キットは、ベンラファキシン(ラセミ体または光学異性体)、フルオキセチン(ラセミ体または光学異性体)、デュロキセチン、パロキセチン、シタロプラム、エシタロプラム、フルボキサミン、S33005、DVS−233(デスベンラファキシン)、DVS−233 SR、ブプロピオン、GW353162、シブトラミン、アトモキセチン、およびセルトラリン(またはそのS−エナンチオマー、Zoloft(登録商標))からなる群より選択される1つまたは複数の抗うつ薬を含む1つまたは複数の医薬組成物を含む。
【0127】
一実施形態では、キットは、治療上有効量のテレンゼピンまたはピレンゼピン、およびSSRIを含む。一実施形態では、キットは、治療上有効量のテレンゼピンまたはピレンゼピン、およびシタロプラム(またはエシタロプラム)を含む。一実施形態では、キットは、治療上有効量のテレンゼピンまたはピレンゼピン、およびセルトラリン(またはそのS−エナンチオマー、Zoloft(登録商標))を含む。一実施形態では、キットは、治療上有効量のテレンゼピンまたはピレンゼピン、およびフルオキセチン(ラセミ体または光学異性体)を含む。一実施形態では、キットは、治療上有効量のテレンゼピンまたはピレンゼピン、およびフルボキサミンを含む。一実施形態では、キットは、治療上有効量のテレンゼピンまたはピレンゼピン、およびパロキセチンを含む。
【0128】
一実施形態では、キットは、治療上有効量のテレンゼピンまたはピレンゼピン、およびSNRIを含む。一実施形態では、キットは、治療上有効量のテレンゼピンまたはピレンゼピン、およびベンラファキシン(ラセミ体または光学異性体)を含む。一実施形態では、キットは、治療上有効量のテレンゼピンまたはピレンゼピン、およびデスベンラファキシンを含む。一実施形態では、キットは、治療上有効量のテレンゼピンまたはピレンゼピン、およびデュロキセチンを含む。一実施形態では、キットは、治療上有効量のテレンゼピンまたはピレンゼピン、およびミルナシプランを含む。一実施形態では、キットは、治療上有効量のテレンゼピンまたはピレンゼピン、およびミルタザピンを含む。
【0129】
一実施形態では、キットは、治療上有効量のテレンゼピンまたはピレンゼピン、およびブプロピオンを含む。
【0130】
いくつかの実施形態では、キットは、食欲減退薬、ドーパミン作動薬、H3−ヒスタミン拮抗薬、5−HT2c受容体作動薬、β−3アドレナリン受容体作動薬、コレシストキニン作動薬、抗てんかん剤、レプチン、レプチン類似体およびレプチン受容体作動薬、ニューロペプチドY(NPY)受容体拮抗薬およびモジュレーター、ペプチド−YY(PYY)受容体作動薬、毛様体神経栄養因子、甲状腺ホルモン受容体−β作動薬、カンナビノイドCB1受容体拮抗薬、メラニン凝集ホルモン受容体拮抗薬、膵および胃液リパーゼ阻害薬、メラノコルチン−4受容体作動薬からなる群より選択される1つまたは複数の抗肥満剤を含む。
【0131】
一実施形態では、キットは、治療上有効量のテレンゼピンまたはピレンゼピン、およびフェンテルミンを含む。
【実施例】
【0132】
下記の実施例は、特許請求した発明を例示するために記載するものであって、限定するために記載するものではない。
【0133】
(実施例1)
食欲抑制:3月齢の(300〜350グラム)オスSprague−Dawleyラット(個別に収容)を用いて、化合物の食欲抑制剤効果を評価した。試験前の2週間、ラットを「高脂肪」食(BioServ Diet #F3282またはResearch Diets #12451)に順化させた(食餌および水の自由摂取)。食餌を翌朝戻したとき摂餌を刺激するために、実験の1日前(5:00PM)に食餌をケージから取り出した(水は、実験全体を通して摂取可能のままであった)。食餌を提供する前に、ラット(n=8〜10匹/投与群)に調査中の化合物を腹腔内(ip)または経口(po)投与し、ホームケージに戻し、直ちに予め計量した食餌を与えた。投与後4時間に、食餌をケージから取り出し、計量し、記録し(4時間消費量)、翌朝までラットに戻した。投与後24時間、残存する食餌を再び計量し、記録した(24時間消費量)。4時間と24時間間隔の累積消費量(グラムで表す)を算出した。このアッセイでは、ビヒクルで処置したラットは、典型的に4時間間隔で約8グラム、および24時間間隔で約25グラムを消費する。
【0134】
処置効果を表1および図1〜5(4時間消費量)、ならびに表2および図6〜10(24時間消費量)に示す。これらの効果を、生の消費量(単位:グラム、±SEM[1 平均値の標準誤差])と4時間または24時間の消費量低減(%)=[1−(薬物処置後の消費量/ビヒクル処置後の消費量)]×100%として示す。表1および2の用量の欄の同様の上付き文字は、(個々の処置と共投与の比較を容易にするため)同じ実験に由来する値を示す。統計分析は、一元ANOVA(分散分析)、続いて全体のαを0.05としたBonferroni多重比較試験で行った。表1および2では、星印(*)は、ビヒクル処置ラットに比べて有意な効果を示し、文字(aまたはb)は、単一化合物で処置したラットに比べて有意な効果(抗うつ薬による有意については「a」、およびテレンゼピンによる有意については「b」)を示す。表1および2では、符号1個はp<0.05を示し、符号2個はp<0.01を示し、符号3個はp<0.001を示す)。統計的有意性を示すために使用される符号は、対応する図で異なることがある。
【0135】
【表1】
【0136】
【表2】
効果の特徴
24時間消費量効果は、作用の持続時間をよりよく反映し、4時間消費量効果よりよい食欲抑制薬効果の例である。試験されたテレンゼピンの用量(ip)はすべて、単独投与したとき消費量を有意に低減させた(図1および6)。また、24時間後、ほぼすべての組合せは、単なる相加から予想される消費量低減より大きな消費量低減をもたらした。
【0137】
例えば、25mg/kgのシタロプラムは、4時間にわたって中程度の消費量低減をもたらした(31%、p<0.05)が、24時間にわたっては本質的に効果がなかった(4%、n.s.)。3mg/kgのテレンゼピンは、4時間にわたってより大きい消費量低減をもたらした(45%、p<0.00l)が、この低減は24時間にわたっても依然として有意であった(27%、p<0.0l)。これらの化合物の共投与は、有意な消費量低減を4時間(73%、p<0.001)および24時間(60%、p<0.001)にわたってもたらした(4時間および24時間の効果について、それぞれ図2および7を参照のこと)。24時間の効果の大きさは、相加効果から予想されるはずの大きさより大きいので相乗効果であることを示している。
【0138】
別の例では、30mg/kgのベンラファキシンは、4時間にわたって有意ではない消費量低減をもたらし(13%)、24時間にわたって本質的に効果がなかった(0%、n.s.)。3mg/kgのテレンゼピンは、4時間にわたってより大きい消費量低減をもたらし(35%、p<0.001)、これは24時間にわたっても依然として有意であった(22%、p<0.001)。これらの化合物の共投与は、有意な消費量低減を4時間(56%、p<0.001)および24時間(38%、p<0.001)にわたってもたらした(4時間および24時間の効果について、それぞれ図5および10を参照のこと)。4時間と24時間の効果の大きさは、相加効果から予想されるはずの大きさより大きいので相乗効果であることを示している。同様に、より低用量のテレンゼピン(1mg/kg)と30mg/kgのベンラファキシンの共投与も、両方の時間間隔にわたって消費量に相乗効果をもたらした。
【0139】
別の例では、30mg/kgのセルトラリンは、4時間にわたって中程度の消費量低減をもたらし(27%、p<0.05)、これは24時間にわたって維持された(28%、p<0.001)が、より低用量(10mg/kg)では、有効性および持続期間が低減した(有意ではない低減は、4時間および24時間にわたってそれぞれ21%および2%であった)。3mg/kgのテレンゼピンは、4時間にわたってより大きい消費量低減をもたらし(45%、p<0.001)、これは24時間にわたっても依然として有意であった(27%、p<0.01)。テレンゼピンおよび「高用量」のセルタリン(sertaline)の共投与は、有意な消費量低減を4時間(73%、p<0.001)および24時間(63%、p<0.001)にわたってもたらした。同様に、テレンゼピンと「低用量」のセルタリン(sertaline)の共投与は、有意な消費量低減を4時間(69%、p<0.001)および24時間(47%、p<0.001)にわたってもたらした(4時間および24時間の効果についてはそれぞれ、図3および8を参照のこと)。4時間と24時間の効果の大きさは、相加効果から予想されるはずの大きさより大きいので相乗効果であることを示している。
【0140】
この実施例のほぼすべての組合せでは、これらの組合せによってもたらされた低減は、ビヒクル処置ラットだけでなく、どちらかの化合物単独の投与の効果とも有意に異なる。ほぼすべての組合せの実験で、消費量低減は、両方の時間間隔にわたって50%を超えた。臨床参考物であるシブトラミンも、単独投与されたとき有効であった。しかし、有意な低減をもたらすためには、経口投与が必要であった(腹腔内投与データは記載せず)。さらに、30mg/kgのテレンゼピンを共投与すると、シブトラミンの有効用量を1mg/kgまで低減することができる。これは、用量の3倍(4時間消費量)〜10倍(24時間消費量)の低減を意味する。
【0141】
(実施例2)
体重増加の低減:
3月齢〜4月齢(475−550グラム)のSprague−Dawleyオスラット(個別に収容)を用いて、化合物の体重増加を予防する能力を評価した。慢性実験の開始時に、ラットに「高脂肪」食(BioServ Diet #F3282またはResearch Diets #12451){自由摂餌)を約1か月続けていた。実験期間を通して、個々の体重および水の消費量を3回/週記録した。データ収集を約2週間した後、ラットを等しい平均体重の処置群となるように均衡した。イソフルオランにより誘導された麻酔下で、ラットに、適切な薬物濃度(平均体重および算出した送達時間に基づく)を含む浸透圧ミニポンプ(Alzet 2ML2)を外科的に移植した(肩甲下の皮下[sc]に留置)。あるいは、経口投与経路を使用した研究のために、ラットに、3mL/kgの体積で14日間にわたって毎日強制経口投与した。データ収集を薬物処置下でさらに約2週間継続した。処置間隔について体重の変化(グラムで表す)を算出した。このア
ッセイでは、ビヒクル処置ラットは、典型的には2週間間隔にわたって約35〜40グラム(+9%)増加する。表3に、処置効果を、ビヒクル群と処置群の体重増加(%)=[(薬物注入の終わりの体重−ベースライン体重)/ベースライン体重]×100%;および低減(%)=(処置群の体重増加(%)−ビヒクル群の体重増加(%))で示す。処置期間全体を通して、体重をベースライン体重の百分率として図11〜15に示す。表3の用量の欄の同様な上付き文字は、(個々の処置と共投与の比較を容易にするため)同じ実験に由来する値を示す。統計分析は、一元ANOVA(分散分析)、続いて全体のαを0.05としたBonferroni多重比較試験で行った。表3では、星印(*)は、ビヒクル処置ラットに比べて有意な効果を示し、文字(aまたはb)は、単一化合物で処置したラットに比べて有意な効果(抗うつ薬による有意については「a」、およびテレンゼピンによる有意については「b」)を示す。表3では、符号1個はp<0.05を示し、符号2個はp<0.01を示し、符号3個はp<0.001を示す)。統計的有意性を示すために使用される符号は、対応する図で異なることがある。
【0142】
【表3】
効果の特徴
テレンゼピン単独の(経口または皮下)投与は、ビヒクル処置対照と突き合わせて比べたとき、高脂肪食を摂取しているラットの体重増加を有意に低減させた(ベースラインから3%〜7%低減)(図11〜15を参照のこと)。臨床参考化合物であるシブトラミンも、ビヒクル処置対照と突き合わせて比べたとき、体重増加を有意に低減させた(ベースラインから6%低減)(図13を参照のこと)。さらに、より高い用量(10mg/kgおよびl5mg/kgであるが、5mg/kgでない)のセルトラリンも、ビヒクル処置対照と突き合わせて比べたとき、体重増加を有意に低減させた(ベースラインから3%〜7%低減)(図12を参照のこと)。組合せの研究で使用された他の化合物/用量はすべ
て、単独投与したとき体重に効果をもたらさなかった。
【0143】
セルトラリン+テレンゼピンおよびベンラファキシン+テレンゼピンの組合せは、相乗的であり、ビヒクル処置で観察された体重の減少だけでなく、どちらかの化合物単独で観察された体重の減少に比べても有意に大きい体重の減少をもたらした。
【0144】
セルトラリン+テレンゼピン(図12)の場合は、15mg/kgのセルトラリン単独は、ビヒクル処置ラットに比べて有意な体重の減少(6.8%、p<0.001)をもたらし、3mg/kgのテレンゼピン単独も、ビヒクル処置ラットに比べて有意な体重の減少(3.8%、p<0.05)をもたらした。しかし、これらと同じ用量のセルトラリンとテレンゼピンの共投与は、ベースラインから11.3%の低減をもたらした。これは、ビヒクル処置(p<0.001)だけでなく、セルトラリン単独(p<0.001)またはテレンゼピン単独(p<0.05)での処置と比べても有意であった。
【0145】
ベンラファキシン+テレンゼピン(図14)の場合は、30mg/kgのベンラファキシン単独では、ビヒクル処置ラットに比べて有意な体重の減少をもたらすことができなかった(0.7%)が、3mg/kgのテレンゼピン単独は、ビヒクル処置ラットに比べて有意な体重の減少(3.8%、p<0.05)をもたらした。しかし、これらと同じ用量のベンラファキシンとテレンゼピンの共投与は、ベースラインから9.1%の低減をもたらした。これは、ビヒクル処置(p<0.001)だけでなく、ベンラファキシン単独(p<0.001)またはテレンゼピン単独(p<0.001)での処置と比べても有意であった。上記の2つの例では、共投与によって全体重の減少の大きさが増大することによって、相乗効果が明らかである。
【0146】
本明細書に記載する実施例および実施態様は例示の目的にしかすぎず、それを踏まえた様々な修正または変更は、当業者に示唆されるものであり、本願の趣旨および範囲、ならびに添付の特許請求の範囲内に包含されるべきであることを理解されたい。本明細書に引用されるすべての刊行物、特許、および特許出願は、全ての目的で参照によりそれらの全体が本明細書に組み込まれる。
【技術分野】
【0001】
関連出願への相互参照
この出願は、2006年6月16日に出願された米国仮出願第60/805,066号および2006年10月12日に出願された米国仮出願第60/829,225号(これらの両方の全体の開示は、参考として本明細書に援用される)の利益を主張する。
【0002】
連邦政府による資金提供を受けた研究開発の下になされた発明の権利に関する記載
該当なし
発明の分野
本発明は、選択的M1ムスカリン性受容体(M1R)拮抗薬の単独投与または抗うつ薬と組み合わせた投与による肥満症の治療および体重減少の助長に関する。
【背景技術】
【0003】
発明の背景
神経伝達物質アセチルコリン(ACh)は、エフェクター細胞膜において2種類の受容体:リガンド開口型イオンチャンネルであるニコチン性受容体(nAChR)、およびGタンパク質共役受容体であるムスカリン性受容体(mAChR)と相互作用する。哺乳類では、M1〜M5と呼ばれるmAChRの5つのサブタイプが同定されている。M1ムスカリン性受容体(M1R)は、中枢神経系と末梢神経系の両方、具体的には大脳皮質および交感神経節で見られる。M1Rによって媒介されるムスカリン様効果は、M1R−選択的拮抗薬の使用、およびさらに最近ではM1R−ヌルマウスの開発により大いに研究されている。
【0004】
現在既知のmAChR拮抗薬は、単一のムスカリン性受容体サブタイプに対して絶対的な選択性を示さないが、薬物のピレンゼピンおよびテレンゼピンは、M1Rに対して高い相対的親和性を示し、したがってM1R−選択的であるとみなされることが多い。ピレンゼピンは、欧州、日本、およびカナダで消化性潰瘍疾患を治療するために使用される。テレンゼピンは、同じ適応症の臨床試験で試験されている。治療用量で、これらは、非選択的mAChR拮抗薬がするように平滑筋の活動を抑制することなく、胃酸およびペプシンの分泌を適度に低減する。
【0005】
M1Rサブタイプが、うつ病性障害および不安のある種の様相に関与し得ることを示唆する証拠がいくつかある。ピレンゼピンをラット前脳の側坐核に直接注射すると、抗うつ活性の一般的尺度であるPorsolt水泳試験における水泳時間の増大が起こった(非特許文献1を参照のこと)。M1R−ヌルマウスも、Porsolt水泳試験での水泳時間の増大、および社会的相互作用試験における社会的接触の増大を示した(非特許文献2を参照のこと)。
【0006】
ピレンゼピンおよびテレンゼピンは、イミプラミンなどの三環系抗うつ薬に構造的に類似しているが、消化性潰瘍疾患の治療で経口投与した場合に向精神効果を有することは知られていない。さらに、マウスおよびラットに関してより早い段階の研究において、ピレンゼピンの全身投与では、いずれの行動効果も導き出すことができなかった(非特許文献3を参照のこと)。このような効果の欠如は、ピレンゼピンが、げっ歯類およびヒトを含めて、様々な種において顕著な血液脳関門透過性を示さないという観察により説明することができる(非特許文献4;非特許文献5を参照のこと)。そういう理由で、Porsolt水泳試験でのピレンゼピンの効果に関する上記の研究では、薬物を試験動物の脳に直接注射することを採用した。
【0007】
選択的M1R拮抗薬を使用して、脂質代謝を変更し、体脂肪貯蔵量を低減することも開示されている。例えば、特許文献1を参照のこと。しかし、所望の結果を実現するためには、24時間の期間において所定の時刻にM1R拮抗薬を投与することが必要であった。さらに、非特許文献6は、インスリン非依存性糖尿病(NIDDM)と診断された非肥満および肥満ヒト患者にピレンゼピンを投与することを開示する。Bevanは、ピレンゼピンの投与の時間を食事の時間と関連付けたが、ピレンゼピンの脂質生成に対する感受性に干渉する能力、または体重減少を助長する際のその使用を開示も示唆もしていない。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】米国特許第5,668,155号明細書
【非特許文献】
【0009】
【非特許文献1】Chau,D.T.ら,Neuroscience,2001年,104巻,3号,p.791−8
【非特許文献2】Miyakawa,T.ら,J Neurosci.,2001年,21巻,14号,p.5239−50
【非特許文献3】Rogoz,Z.,Skuza,G.,Sowinska,H.,Pol.J.Pharmacol.Pharm.,1981年,31巻,p.615−26
【非特許文献4】Hammer,R.,Koss,F.W.,Scand.J.Gastroenterol,Suppl,1979年,14巻,57号,p.1−6
【非特許文献5】Bymaster,F.P.ら,J.Pharmacol.Exp.Ther.,1993年,267巻,1号,p.16−24
【非特許文献6】Bevanら,Clinical Endocrinology,(1991年),36巻,p.85−91
【発明の概要】
【発明が解決しようとする課題】
【0010】
肥満症の治療および体重減少の助長のための有効な新規薬物が求められている。本発明は、これとその他の必要性に対処する。
【課題を解決するための手段】
【0011】
要旨
本発明は、治療上有効量の1つまたは複数のM1R−選択的拮抗薬を投与することによって、肥満症を治療し、食欲を抑制し、体重減少を助長または促進し、所望の体重の維持を助長または促進し、望ましくない体重増加を予防または低減するための方法を提供する。本方法を実施する際に、1つもしくは複数のM1R−選択的拮抗薬を、他の薬理作用剤なしに、または他の薬理作用剤、例えばM1R−選択的拮抗薬以外の1つもしくは複数の抗うつ薬と組み合わせて投与することができる。いくつかの実施形態では、1つまたは複数のM1R−選択的拮抗薬を1つまたは複数の抗肥満剤と共投与することができる。いくつかの実施形態では、M1R−選択的拮抗薬以外の1つまたは複数の抗うつ薬と組み合わせた1つまたは複数のM1R−選択的拮抗薬を、1つまたは複数の抗肥満剤と共投与することができる。
【0012】
1つまたは複数のM1R−選択的拮抗薬は、24時間の期間において所定の時刻に投与する必要はない。1つまたは複数のM1R−選択的拮抗薬の投与をホルモンの日周性振動の最上点または最下点と相関させることなく、有効な結果を実現することができる。1つまたは複数のM1R−選択的拮抗薬の投与は、一日のうちの特定の時間または食事と相関
させる必要がない。いくつかの実施形態では、1つまたは複数のM1R−選択的拮抗薬の投与の時間は、食事、例えば食事前、中、後と相関するように定められる。
したがって、第1の態様では、本発明は、体重減少を促進するか、または安定な体重の維持を助長するための方法を提供し、この方法は、それを必要とする肥満または過体重の個体に、治療上有効量の1つまたは複数の体重減少を達成するためのM1R−選択的拮抗薬を投与することを含み、それによって体重減少が促進され、または安定な体重の維持が助長される。
【0013】
別の態様では、本発明は、治療上有効量の1つまたは複数のM1R−選択的拮抗薬および1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬の混合物を含む医薬組成物を提供する。いくつかの実施形態では、医薬組成物は、治療上有効量の1つまたは複数のM1R−選択的拮抗薬および1つまたは複数の抗肥満剤の混合物を含む。いくつかの実施形態では、医薬組成物は、治療上有効量の1つまたは複数のM1R−選択的拮抗薬、M1R−選択的拮抗薬以外の1つまたは複数の抗うつ薬、および1つまたは複数の抗肥満剤の混合物を含む。
【0014】
別の態様では、本発明は、治療上有効量の1つまたは複数のM1R−選択的拮抗薬および1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬の混合物を含むキットを提供する。いくつかの実施形態では、キットは、治療上有効量の1つまたは複数のM1R−選択的拮抗薬および1つまたは複数の抗肥満剤の混合物を含む。いくつかの実施形態では、キットは、治療上有効量の1つまたは複数のM1R−選択的拮抗薬、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬、および1つまたは複数の抗肥満剤の混合物を含む。
【0015】
本方法を実施するための実施形態、ならびに医薬組成物およびキットのための実施形態に関して、一実施形態では、1つまたは複数のM1R−選択的拮抗薬は、ピレンゼピン、テレンゼピン、およびそれらの組合せからなる群より選択される。一実施形態では、M1R−選択的拮抗薬はテレンゼピン(ラセミ体または光学異性体)である。一実施形態では、M1R−選択的拮抗薬はピレンゼピンである。
【0016】
一実施形態では、1つまたは複数のM1R−選択的拮抗薬は、第2の薬理作用剤なしに投与される。
【0017】
一実施形態では、1つもしくは複数のM1R−選択的拮抗薬は、組合せで、または1つもしくは複数のM1R−選択的拮抗薬以外の抗うつ薬と組み合わせて投与される。一実施形態では、抗うつ薬は、選択的セロトニン再取り込み阻害薬(SSRI)および選択的セロトニン−ノルエピネフリン再取り込み阻害薬(SNRI)からなる群より選択される。
【0018】
一実施形態では、抗うつ薬はSSRIである。一実施形態では、SSRIは、シタロプラム、エシタロプラム、フルオキセチン、フルボキサミン、パロキセチン、およびセルトラリンからなる群より選択される。一実施形態では、SSRIは、シタロプラム、セルトラリン、パロキセチン、およびフルオキセチンからなる群より選択される。
【0019】
一実施形態では、抗うつ薬はSNRIである。一実施形態では、SNRIは、ミルナシプラン、ミルタザピン、ベンラファキシン、デュロキセチン、デスベンラファキシン、およびシブトラミンからなる群より選択される。一実施形態では、SNRIはベンラファキシンである。
【0020】
これらの方法は、抗肥満剤の投与を必要とすることなく、例えば食欲減退薬の投与を必要とすることなく、有効な結果を実現する。しかし、いくつかの実施形態では、1つまたは複数のM1R−選択的拮抗薬は、組合せで、または1つもしくは複数の抗肥満剤、例え
ば1つもしくは複数の食欲減退薬と組み合わせて投与される。
【0021】
さらに、1つまたは複数のM1R−選択的拮抗薬の投与の時間を定めることなく、有効な結果を実現することができる。抗うつ薬、抗肥満剤、および食欲減退薬を含めて、共投与された活性剤も、投与の時間を定めることなく、有効な結果をもたらす。
【0022】
一実施形態では、1つまたは複数のM1R−選択的拮抗薬は、テレンゼピン(ラセミ体または光学異性体)であり、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬はフルオキセチン(ラセミ体または光学異性体)である。
【0023】
一実施形態では、1つまたは複数のM1R−選択的拮抗薬はテレンゼピン(ラセミ体または光学異性体)であり、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬はフルボキサミンである。
【0024】
一実施形態では、1つまたは複数のM1R−選択的拮抗薬はテレンゼピン(ラセミ体または光学異性体)であり、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬はセルトラリンまたはそのS−エナンチオマー、Zoloft(登録商標)である。
【0025】
一実施形態では、1つまたは複数のM1R−選択的拮抗薬はテレンゼピン(ラセミ体または光学異性体)であり、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬はシタロプラム(またはエシタロプラム)である。
【0026】
一実施形態では、1つまたは複数のM1R−選択的拮抗薬はテレンゼピン(ラセミ体または光学異性体)であり、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬はパロキセチンである。
【0027】
一実施形態では、1つまたは複数のM1R−選択的拮抗薬はテレンゼピン(ラセミ体または光学異性体)であり、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬はベンラファキシン(ラセミ体または光学異性体)である。
【0028】
一実施形態では、1つまたは複数のM1R−選択的拮抗薬はテレンゼピン(ラセミ体または光学異性体)であり、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬はデスベンラファキシンである。
【0029】
一実施形態では、1つまたは複数のM1R−選択的拮抗薬はテレンゼピン(ラセミ体または光学異性体)であり、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬はデュロキセチンである。
【0030】
一実施形態では、1つまたは複数のM1R−選択的拮抗薬はテレンゼピン(ラセミ体または光学異性体)であり、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬はシブトラミンである。
【0031】
一実施形態では、1つまたは複数のM1R−選択的拮抗薬はテレンゼピン(ラセミ体または光学異性体)であり、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬はミルナシプランである。
【0032】
一実施形態では、1つまたは複数のM1R−選択的拮抗薬はテレンゼピン(ラセミ体または光学異性体)であり、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬はミルタザピンである。
【0033】
一実施形態では、1つまたは複数のM1R−選択的拮抗薬はテレンゼピン(ラセミ体または光学異性体)であり、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬はブプロピオンである。
【0034】
一実施形態では、1つまたは複数のM1R−選択的拮抗薬はテレンゼピン(ラセミ体または光学異性体)であり、1つまたは複数の抗肥満剤はフェンテルミンである。
【0035】
関連する態様では、本発明は、肥満症を治療し、食欲を抑制し、体重減少を助長または促進し、所望の体重の維持を助長または促進し、望ましくない体重増加を予防または低減するための医薬品を調製するための方法、またはその使用を提供し、この医薬品は、治療上有効量の1つまたは複数のM1R−選択的拮抗薬を含有する。医薬品は、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬も任意選択で含有することができる。医薬品は、1つまたは複数の抗肥満剤も任意選択で含有することができる。医薬品の実施形態は、本明細書に記載する通りである。
【0036】
いくつかの実施形態では、本発明の方法および組成物は、本明細書に記載する薬理作用剤の組合せを含む。いくつかの実施形態では、本発明の方法および組成物は、本質的に本明細書に記載する薬理作用剤の組合せからなる。
定義
用語「肥満の」または「肥満症」は、過剰の脂肪組織のためボディマス指数(BMI)が30kg/m2以上である個体を意味する。肥満症は、体脂肪含量を基準として定義することもできる:男性の場合、25%を超える体脂肪含量、または女性の場合、30%を超える体脂肪含量。「病的に肥満の」個体のボディマス指数は、35kg/m2を超える。
【0037】
用語「過体重」は、ボディマス指数が25kg/m2以上であるが、30kg/m2未満である個体を意味する。
【0038】
用語「ボディマス指数」または「BMI」は、個体の体重が身長に対して適切であるかどうかを推定する体重対身長比の測定を意味する。本明細書では、個体のボディマス指数は、以下の通り算出される。
BMI=(ポンド×700)/(身長、単位インチ)2
または
BMI=(キログラム)/(身長、単位メートル)2
用語「ベースライン体重」は、治療の開始時に個体によって提示された体重を意味する。
【0039】
本明細書では、「投与すること」は、経口(「po」)投与、坐剤としての投与、局所的接触、静脈内(「iv」)、腹腔内(「ip」)、筋肉内(「im」)、病巣内、鼻腔内、もしくは皮下(「sc」)投与、またはスローリリースの装置、例えばミニ浸透圧ポンプの対象者への移植を意味する。投与は、非経口および経粘膜{例えば、経口、経鼻、腟内、直腸、または経皮)を含めて、任意の経路による。非経口投与としては、例えば静脈内、筋肉内、細動脈内、皮内、皮下、腹腔内、脳室内、および頭蓋内投与が挙げられる。送達の他のモードとしては、リポソーム製剤、静脈内注入、経皮貼付剤などの使用が挙げられるが、これらに限定されない。
【0040】
用語「全身投与」および「全身投与した」は、化合物または組成物が、薬剤作用の標的部位を含めて、体の部位に循環器系を介して送達されるように化合物または組成物を哺乳類に投与する方法を意味する。全身投与としては、経口、鼻腔内、直腸、および非経口(すなわち、消化管を経由する以外、具体的には筋肉内、静脈内、動脈内、経皮、および皮
下)投与が挙げられるが、これらに限定されない。ただし、本明細書では、全身投与は、循環器系を経由する以外の手段、具体的には髄腔内注射および頭蓋内投与による脳領域への直接投与を含まないことを条件にする。
用語「共投与する」は、個体の血中に2つの活性剤が同時に存在することを意味する。共投与された活性剤は、同時または連続的に送達され得る。
【0041】
本明細書では、用語「治療すること」および「治療」は、用語が適用される疾患もしくは病態、またはこのような疾患もしくは病態の1つもしくは複数の症状の発症を遅延させ、それらの進行を遅延もしくは逆行させ、またはそれらを軽減もしくは予防することを意味する。
【0042】
本明細書では、用語「選択的ムスカリン性受容体M1拮抗薬」および「M1R−選択的拮抗薬」は、ムスカリン性受容体サブタイプM2およびM3に比べて、優先的にムスカリン性受容体M1サブタイプと相互作用を示すムスカリン性アセチルコリン受容体拮抗薬を意味する。M1R−選択的拮抗薬としては、例えばピレンゼピンおよびテレンゼピンが挙げられるが、これらに限定されない。優先的な結合は完全である必要はない。例えば、ピレンゼピンは、M1およびM4受容体サブタイプに対して親和性が類似しているにもかかわらず、M1R−選択的拮抗薬として分類される。
【0043】
M1R−選択的拮抗薬の優先的な結合は、競合的置換アッセイで測定することができる。M1R−選択的拮抗薬は、既知のM2選択的リガンド(例えば、トリピトラミン、ヒンバシン、メトクトラミン)およびM3(例えば、ダリフェナシン、ヘキサヒドロシラジフェニドール)に比べて、優先的に既知のM1R−選択的リガンド(例えば、ピレンゼピンおよび/またはテレンゼピン)に置換する。あるいは、M1R−選択的拮抗薬は、非選択的ムスカリン性リガンドのM2およびM3受容体サブタイプへの結合に置換するのに比べて、優先的に非選択的ムスカリン性リガンド(例えば、キヌクリジニルベンジラート(QNB)、N−メチルスコポラミン(NMS))のM1受容体サブタイプへの結合に置換する。放射性標識競合体の置換に対する相対的効力は、競合体の50%が置換される濃度(IC50)、または平衡解離定数(Kd)によって表すことができる。IC50値および/または平衡解離定数は、利用可能なソフトウェアを使用して、漸増量の非標識試験化合物の存在下での標識リガンドの検出値を入力することによって算出することができる(例えば、LIGAND(Munson,p.J.,およびRodbard,D.,Anal.Biochem.(1980年)107巻,p.220−39またはDATAPLOT,National Technical Information Services)。M1R−選択的拮抗薬は、M1受容体サブタイプへの結合に関するIC50値またはKd値が、M2およびM3受容体サブタイプへの結合に関するそのIC50値またはKd値より少なくとも約3倍低く、好ましくは少なくとも約10倍低く、より好ましくは少なくとも約30倍低い。放射性標識NMSまたはQNBを使用する適用可能な放射性リガンド結合アッセイは、Buckleyら,Molecular Pharmacology(1989年)35巻,p.469−76およびBoldenら,J Pharmacol Exp Ther.(1992年)260巻,p.576−80に開示される。
【0044】
本明細書では、用語「抗肥満剤」は、体重減少をもたらすことを主目的とする薬剤を意味する。抗肥満剤としては、例えば食欲減退薬、ドーパミン作動薬、H3−ヒスタミン拮抗薬、5−HT2c受容体作動薬、β−3アドレナリン受容体作動薬、コレシストキニン作動薬、抗てんかん剤、レプチン、レプチン類似体、およびレプチン受容体作動薬、ニューロペプチドY(NPY)受容体拮抗薬およびモジュレーター、ペプチド−YY(PYY)受容体作動薬、毛様体神経栄養因子、甲状腺ホルモン受容体−β作動薬、カンナビノイドCB1受容体拮抗薬、メラニン凝集ホルモン受容体拮抗薬、膵および胃液リパーゼ阻害薬、メラノコルチン−4受容体作動薬、およびそれらの組合せが挙げられるが、これらに
限定されない。本明細書では、用語「抗肥満剤」は、具体的にM1R選択的ムスカリン拮抗薬および抗うつ薬を除外する。
【0045】
本明細書では、用語「食欲減退薬」または「食欲抑制薬」は同義的に、食欲の抑制を主な所期効果とする薬剤を意味する。食欲減退薬としては、交感神経刺激アミン類が挙げられるが、これらに限定されない。交感神経刺激アミン類は周知であり、例えば、参照により本明細書に組み込まれるGoodman and Gilman’s The Pharmacological Basis of Therapeutics,第11版,Brunton,LazoおよびParker編,McGraw−Hill(2006年),第10章,p.237−263に詳細に述べられている。本明細書では、用語「食欲減退薬」または「食欲抑制薬」は、具体的にM1R選択的ムスカリン拮抗薬および抗うつ薬を除外する。
【0046】
本明細書では、フレーズ「からなる本質的になる」は、方法または組成物に含まれる有効薬剤の属または種、およびこれらの方法または組成物の所期目的に対して不活性である任意の賦形剤を意味する。いくつかの実施形態では、フレーズ「から本質的になる」は、M1R−選択的拮抗薬および抗うつ薬以外の1つまたは複数の追加の活性剤が包含されることを明らかに除外する。いくつかの実施形態では、除外することができる追加の活性剤としては、プロラクチン阻害薬、プロラクチン刺激剤、5−HT受容体拮抗薬、5−HT受容体作動薬、NK−1受容体拮抗薬、および/またはジペプチジルペプチダーゼIV阻害薬の1つまたは複数が挙げられる。
【0047】
用語「放出制御」、「徐放」、「持続放出」、および「時限放出」は同義的に、薬物の放出が即時型でない任意の薬物含有製剤を意味するよう意図されている。すなわち、「放出制御」製剤を使用した場合、経口投与によって、薬物の吸収プールへの即時放出は起こらない。これらの用語は、Remington:The Science and Practice of Pharmacy,第21版,Lippencott Williams & Wilkins(2006年)で定義される「非即時放出」と同義的に使用される。そこで述べられるように、即時および非即時放出の動態は、下記の式を参照することによって定義することができる。
【0048】
【化1】
「吸収プール」は、特定の吸収部位での投与薬物の溶液を表し、kr、ka、およびkeはそれぞれ、(1)製剤からの薬物放出、(2)吸収、および(3)排泄の一次速度定数である。即放性剤形については、薬物放出の速度定数krが吸収速度定数kaよりはるかに大きい。放出制御製剤については、その逆、すなわちkr≪kaが真であり、したがって剤形からの薬物放出速度が、標的領域への薬物送達における律速段階である。
【0049】
用語「徐放」および「持続放出」は、長期間、例えば12時間以上にわたって、薬物の漸進的放出をもたらし、好ましくは、必ずというわけではないが長期にわたって実質的に定常状態の血中薬物レベルをもたらす薬物製剤を意味するように通常の意味で使用される。
【0050】
本明細書では、用語「遅延放出」は、変化しないままで胃を通過し、小腸で溶解する医薬調製物を意味する。
【0051】
本明細書では、「相乗効果」または「相乗効果の」は同義的に、2つの活性剤の相加効果より強いそれらの複合効果を意味する。相乗効果は、無効な用量の2つの活性剤を合わせて有効な効果を生じることによって実現することができる。相乗効果の尺度は、統計的有意性とは無関係である。
本発明はまた、以下の項目を提供する。
(項目1)
体重減少を促進するか、または安定な体重の維持を助長するための方法であって、該方法は、それを必要とする肥満または過体重の個体に、テレンゼピン、ならびに選択的セロトニン再取り込み阻害薬(SSRI)および選択的セロトニン−ノルエピネフリン再取り込み阻害薬(SNRI)からなる群より選択される抗うつ薬を体重減少を達成するための治療上有効量で投与することを含み、それによって体重減少が促進されるか、または安定な体重の維持が助長される、方法。
(項目2)
上記抗うつ薬がSSRIである、項目1に記載の方法。
(項目3)
上記SSRIが、シタロプラム、エシタロプラム、フルオキセチン、フルボキサミン、パロキセチン、およびセルトラリンからなる群より選択される、項目2に記載の方法。
(項目4)
上記SSRIがシタロプラムである、項目3に記載の方法。
(項目5)
上記SSRIがセルトラリンである、項目3に記載の方法。
(項目6)
上記抗うつ薬がSNRIである、項目1に記載の方法。
(項目7)
上記SNRIが、ミルナシプラン、ミルタザピン、ベンラファキシン、デュロキセチン、デスベンラファキシン、およびシブトラミンからなる群より選択される、項目6に記載の方法。
(項目8)
上記SNRIがベンラファキシンである、項目7に記載の方法。
(項目9)
さらに、1つまたは複数の抗肥満剤の投与を含む、項目1に記載の方法。
(項目10)
さらに、1つまたは複数の食欲減退薬の投与を含む、項目1に記載の方法。
(項目11)
体重減少を促進するか、または安定な体重の維持を助長するための方法であって、該方法は、それを必要とする肥満または過体重の個体に、治療上有効量の体重減少を達成するための1つまたは複数のM1R−選択的拮抗薬を投与することを含み、それによって体重減少が促進されるか、または安定な体重の維持が助長される、方法。
(項目12)
上記1つまたは複数のM1R−選択的拮抗薬が、ピレンゼピン、テレンゼピン、およびそれらの組合せからなる群より選択される、項目11に記載の方法。
(項目13)
さらに、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬の投与を含む、項目11に記載の方法。
(項目14)
上記1つまたは複数の抗うつ薬が、三環系抗うつ薬およびその類似体、セロトニン再取り込み阻害薬、セロトニン−ノルエピネフリン再取り込み阻害薬、ノルエピネフリン再取り込み阻害薬、ドーパミン再取り込み阻害薬、ノルエピネフリン−ドーパミン再取り込み阻害薬、セロトニン−ノルエピネフリン−ドーパミン再取り込み阻害薬、セロトニン再取り込み促進薬、セロトニン作動薬およびそのプロドラッグ、モノアミンオキシダーゼ阻害薬、およびそれらの混合物からなる群より選択される、項目13に記載の方法。
(項目15)
さらに、1つまたは複数の抗肥満剤の投与を含む、項目11に記載の方法。
(項目16)
さらに、1つまたは複数の食欲減退薬の投与を含む、項目11に記載の方法。
(項目17)
治療上有効量のM1R−選択的拮抗薬と、SSRIおよびSNRIからなる群より選択されるM1R−選択的拮抗薬以外の抗うつ薬との混合物を含む医薬組成物。
(項目18)
上記M1R−選択的拮抗薬がテレンゼピンであり、上記M1R−選択的拮抗薬以外の抗うつ薬がセルトラリンである、項目17に記載の医薬組成物。
(項目19)
上記M1R−選択的拮抗薬がテレンゼピンであり、上記M1R−選択的拮抗薬以外の抗うつ薬がベンラファキシンである、項目17に記載の医薬組成物。
(項目20)
上記M1R−選択的拮抗薬がテレンゼピンであり、上記M1R−選択的拮抗薬以外の抗うつ薬がシタロプラムである、項目17に記載の医薬組成物。
(項目21)
上記M1R−選択的拮抗薬および上記M1R−選択的拮抗薬以外の抗うつ薬の送達が徐放である、項目17に記載の医薬組成物。
(項目22)
治療上有効量のM1R−選択的拮抗薬とM1R−選択的拮抗薬以外の抗うつ薬の組合せを含むキット。
(項目23)
上記M1R−選択的拮抗薬がテレンゼピンであり、上記M1R−選択的拮抗薬以外の抗うつ薬がセルトラリンである、項目22に記載のキット。
(項目24)
上記M1R−選択的拮抗薬がテレンゼピンであり、上記M1R−選択的拮抗薬以外の抗うつ薬がベンラファキシンである、項目22に記載のキット。
(項目25)
上記M1R−選択的拮抗薬がテレンゼピンであり、上記M1R−選択的拮抗薬以外の抗うつ薬がシタロプラムである、項目22に記載のキット。
【図面の簡単な説明】
【0052】
【図1】絶食ラットにおいて、4時間にわたる高脂肪食物の総消費量に及ぼすテレンゼピン(TZP)の効果を示す図である。下記の実施例に記載されるように、4月齢のオスSprague−Dawleyラット(n=9匹〜10匹/群)に、テレンゼピンを1.0mg/kg、3.0mg/kg、または10.0mg/kgの用量で腹腔内投与した。活性剤はすべて、遊離塩基の形で送達した。対照ラット(VEH)には15%のDMSOを投与した。*は、VEHに対してp<0.05を示す。*は、VEHに対してp<0.01を示す。
【図2】絶食ラットにおいて、4時間にわたる高脂肪食物の総消費量に及ぼすテレンゼピン(TZP)単独、シタロプラム(CIT)単独、およびテレンゼピンとシタロプラムの組合せの効果を示す図である。下記の実施例に記載されるように、3月齢のオスSprague−Dawleyラット(n=10匹/群)に、テレンゼピン単独(3mg/kg)、シタロプラム単独(25mg/kg)、または共投与のテレンゼピン(3mg/kg)とシタロプラム(25mg/kg)を腹腔内投与した。活性剤はすべて、遊離塩基の形で送達した。対照ラット(VEH)には37%のDMSOを投与した。*は、VEHに対してp<0.05を示す。*は、VEHに対してp<0.001を示す。「a」は、TZPに対してp<0.05を示す。「b」は、CITに対してp<0.001を示す。
【図3】絶食ラットにおいて、4時間にわたる高脂肪食物の総消費量に及ぼすテレンゼピン(TZP)単独、セルトラリン(SRT)単独、およびテレンゼピンとセルトラリンの組合せの効果を示す図である。下記の実施例に記載されるように、3月齢のオスSprague−Dawleyラット(n=10匹/群)に、テレンゼピン単独(3mg/kg)、セルトラリン単独(10mg/kgまたは30mg/kg)、または共投与のテレンゼピン(3mg/kg)とセルトラリン(10mg/kgまたは30mg/kg)を腹腔内投与した。活性剤はすべて、遊離塩基の形で送達した。対照ラット(VEH)には37%のDMSOを投与した。*は、VEHに対してp<0.05を示す。*は、VEHに対してp<0.001を示す。「a」は、TZPに対してp<0.001を示す。「b」は、TZPに対してp<0.05を示す。
【図4】絶食ラットにおいて、4時間にわたる高脂肪食物の総消費量に及ぼすテレンゼピン(TZP)単独、シブトラミン(SBR)単独、およびテレンゼピンとシブトラミンの組合せの効果を示す図である。下記の実施例に記載されるように、4月齢のオスSprague−Dawleyラット(n=10匹/群)に、テレンゼピン単独(30mg/kg)、シブトラミン単独(1.0mg/kg)、または共投与のテレンゼピン(30mg/kg)とシブトラミン(1.0mg/kg)を経口投与した。活性剤はすべて、遊離塩基の形で送達した。対照ラット(VEH)には10%のDMSOを投与した。**は、VEHに対してp<0.01を示す。「aa」は、SBRに対してp<0.01を示す。
【図5】絶食ラットにおいて、4時間にわたる高脂肪食物の総消費量に及ぼすテレンゼピン(TZP)単独、ベンラファキシン(VEN)単独、およびテレンゼピンとベンラファキシンの組合せの効果を示す図である。下記の実施例に記載されるように、2月/3月齢のオスSprague−Dawleyラット(n=11匹/群)に、テレンゼピン単独(1mg/kgまたは3mg/kg)、ベンラファキシン単独(30mg/kg)、または共投与のテレンゼピン(1mg/kgまたは3mg/kg)とベンラファキシン(30mg/kg)を腹腔内投与した。活性剤はすべて、遊離塩基の形で送達した。対照ラット(VEH)には33%のDMSOを投与した。**は、VEHに対してp<0.001を示す。「a」は、TZPに対してp<0.05を示す。「aa」は、TZPに対してp<0.01を示す。「b」は、VENに対してp<0.05を示す。「bb」は、VENに対してp<0.01を示す。
【図6】絶食ラットにおいて、24時間にわたる高脂肪食物の総消費量に及ぼすテレンゼピン(TZP)の効果を示す図である。下記の実施例に記載されるように、4月齢のオスSprague−Dawleyラット(n=9匹〜10匹/群)に、テレンゼピンを1.0mg/kg、3.0mg/kg、または10.0mg/kgの用量で腹腔内投与した。活性剤はすべて、遊離塩基の形で送達した。対照ラット(VEH)には15%のDMSOを投与した。*は、VEHに対してp<0.05を示す。**は、VEHに対してp<0.01を示す。
【図7】絶食ラットにおいて、24時間にわたる高脂肪食物の総消費量に及ぼすテレンゼピン(TZP)単独、シタロプラム(CIT)単独、およびテレンゼピンとシタロプラムの組合せの効果を示す図である。下記の実施例に記載されるように、3月齢のオスSprague−Dawleyラット(n=10匹/群)に、テレンゼピン単独(3mg/kg)、シタロプラム単独(25mg/kg)、または共投与のテレンゼピン(3mg/kg)とシタロプラム(25mg/kg)を腹腔内投与した。活性剤はすべて、遊離塩基の形で送達した。対照ラット(VEH)には37%のDMSOを投与した。*は、VEHに対してp<0.01を示す。**は、VEHに対してp<0.001を示す。「a」は、TZPに対してp<0.001を示す。「b」は、CITに対してp<0.001を示す。
【図8】絶食ラットにおいて、24時間にわたる高脂肪食物の総消費量に及ぼすテレンゼピン(TZP)単独、セルトラリン(SRT)単独、およびテレンゼピンとセルトラリンの組合せの効果を示す図である。下記の実施例に記載されるように、3月齢のオスSprague−Dawleyラット(n=10匹/群)に、テレンゼピン単独(3mg/kg)、セルトラリン単独(10mg/kgまたは30mg/kg)、または共投与のテレンゼピン(3mg/kg)とセルトラリン(10mg/kgまたは30mg/kg)を腹腔内投与した。活性剤はすべて、遊離塩基の形で送達した。対照ラット(VEH)には37%のDMSOを投与した。*は、VEHに対してp<0.01を示す。**は、VEHに対してp<0.001を示す。「a」は、TZPに対してp<0.05を示す。「aa」は、TZPに対してp<0.001を示す。「b」は、SRTに対してp<0.001を示す。
【図9】絶食ラットにおいて、24時間にわたる高脂肪食物の総消費量に及ぼすテレンゼピン(TZP)単独、シブトラミン(SBR)単独、およびテレンゼピンとシブトラミンの組合せの効果を示す図である。下記の実施例に記載されるように、4月齢のオスSprague−Dawleyラット(n=10匹/群)に、テレンゼピン単独(30mg/kg)、シブトラミン単独(1.0mg/kg)、または共投与のテレンゼピン(30mg/kg)とシブトラミン(1.0mg/kg)を経口投与した。活性剤はすべて、遊離塩基の形で送達した。対照ラット(VEH)には10%のDMSOを投与した。***は、VEHに対してp<0.001を示す。「aaa」は、SBRに対してp<0.001を示す。
【図10】絶食ラットにおいて、24時間にわたる高脂肪食物の総消費量に及ぼすテレンゼピン(TZP)単独、ベンラファキシン(VEN)単独、およびテレンゼピンとベンラファキシンの組合せの効果を示す図である。下記の実施例に記載されるように、2月/3月齢のオスSprague−Dawleyラット(n=11匹/群)に、テレンゼピン単独(1mg/kgまたは3mg/kg)、ベンラファキシン単独(30mg/kg)、または共投与のテレンゼピン(1mg/kgまたは3mg/kg)とベンラファキシン(30mg/kg)を腹腔内投与した。活性剤はすべて、遊離塩基の形で送達した。対照ラット(VEH)には33%のDMSOを投与した。**は、VEHに対してp<0.001を示す。「a」は、TZPに対してp<0.05を示す。「aa」は、TZPに対してp<0.01を示す。「b」は、VENに対してp<0.05を示す。「bb」は、VENに対してp<0.01を示す。
【図11】高脂肪食を摂取しているラットの体重に及ぼすテレンゼピン(TZP)、シタロプラム(CIT)、およびテレンゼピンとシタロプラムの組合せの慢性投与の効果を示す図である。下記の実施例に記載されるように、約15週齢のオスSprague−Dawleyラット(n=10匹/群)に、テレンゼピン単独(3mg/kg)、シタロプラム単独(15mg/kg)、または共投与のテレンゼピン(3mg/kg)とシタロプラム(15mg/kg)を皮下注入した。活性剤はすべて、遊離塩基の形で送達した。対照ラット(VEH)には10%のDMSOを投与した。*は、VEHに対してp<0.05を示す。**は、VEHに対してp<0.01を示す。***は、VEHに対してp<0.001を示す。「aa」は、CITに対してp<0.01を示す。「aaa」は、CITに対してp<0.001を示す。「b」は、TZPに対してp<0.05を示す。斜線部分は治療間隔を示す。
【図12】高脂肪食を摂取しているラットの体重に及ぼすテレンゼピン(TZP)、セルトラリン(SRT)、およびテレンゼピンとセルトラリンの組合せの慢性投与の効果を示す図である。下記の実施例に記載されるように、約15週齢のオスSprague−Dawleyラット(n=10匹/群)に、テレンゼピン単独(3mg/kg)、セルトラリン単独(15mg/kg)、または共投与のテレンゼピン(3mg/kg)とセルトラリン(15mg/kg)を皮下注入した。活性剤はすべて、遊離塩基の形で送達した。対照ラットには、10%のDMSO(VEHl)またはN−メチル−2−ピロリドン+DMSO+水(1:2:2)(「NMP:DMSO:水」またはVEH2)を投与した。*は、VEHに対してp<0.05を示す。**は、VEHに対してp<0.01を示す。***は、VEHに対してp<0.001を示す。「a」は、SRTに対してp<0.05を示す。「aa」は、SRTに対してp<0.01を示す。「aaa」は、SRTに対してp<0.001を示す。「b」は、TZPに対してp<0.05を示す。「bb」は、TZPに対してp<0.01を示す。「bbb」は、TZPに対してp<0.001を示す。斜線部分は治療間隔を示す。
【図13】高脂肪食を摂取しているラットの体重に及ぼすテレンゼピン(TZP)、シブトラミン(SBR)、およびテレンゼピンとシブトラミンの組合せの慢性投与の効果を示す図である。下記の実施例に記載されるように、約15週齢のオスSprague−Dawleyラット(n=10匹/群)に、テレンゼピン単独(1.0mg/kg)、シブトラミン単独(0.3mg/kg)、または共投与のテレンゼピン(1.0mg/kg)とシブトラミン(0.3mg/kg)を経口投与した。活性剤はすべて、遊離塩基の形で送達した。対照ラット(VEH)には水を投与した。*は、VEHに対してp<0.05を示す。**は、VEHに対してp<0.01を示す。「aa」は、SBRに対してp<0.05を示す。「b」は、TZPに対してp<0.05を示す。斜線部分は治療間隔を示す。
【図14】高脂肪食を摂取しているラットの体重に及ぼすテレンゼピン(TZP)、ベンラファキシン(VEN)、およびテレンゼピンとベンラファキシンの組合せの慢性投与の効果を示す図である。下記の実施例に記載されるように、約15週齢のオスSprague−Dawleyラット(n=10匹/群)に、テレンゼピン単独(3mg/kg)、ベンラファキシン単独(30mg/kg)、または共投与のテレンゼピン(3mg/kg)とベンラファキシン(30mg/kg)を皮下注入した。活性剤はすべて、遊離塩基の形で送達した。対照ラット(VEH)には10%のDMSOを投与した。*は、VEHに対してp<0.05を示す。**は、VEHに対してp<0.01を示す。***は、VEHに対してp<0.001を示す。「a」は、VENに対してp<0.05を示す。「aa」は、VENに対してp<0.01を示す。「aaa」は、VENに対してp<0.001を示す。「b」は、TZPに対してp<0.05を示す。「bb」は、TZPに対してp<0.01を示す。「bbb」は、TZPに対してp<0.001を示す。斜線部分は治療間隔を示す。
【図15】高脂肪食を摂取しているラットの体重に及ぼすテレンゼピン(TZP)、フェンテルミン(PHN)、およびテレンゼピンとフェンテルミンの組合せの慢性投与の効果を示す図である。下記の実施例に記載されるように、約15週齢のオスSprague−Dawleyラット(n=10匹/群)に、テレンゼピン単独(0.3mg/kg)、フェンテルミン単独(4.0mg/kg)、または共投与のテレンゼピン(0.3mg/kg)とフェンテルミン(4.0mg/kg)を皮下注入した。活性剤はすべて、遊離塩基の形で送達した。対照ラット(VEH)には生理食塩水を投与した。*は、VEHに対してp<0.05を示す。**は、VEHに対してp<0.01を示す。「a」は、PHNに対してp<0.05を示す。「b」は、TZPに対してp<0.05を示す。斜線部分は治療間隔を示す。
【発明を実施するための形態】
【0053】
1.序論
上述したように、ラットおよびマウスにおけるより早い段階の研究は、ピレンゼピンの全身投与では、いずれの行動効果も導き出すことができなかったこと(Rogoz,Z.,Skuza,G.,Sowinska,H.,Pol.J.Pharmacol.Pharm.,1981年,31巻,p.615−26を参照のこと)、およびピレンゼピンが、げっ歯類およびヒトを含めて、様々な種において顕著な血液脳関門透過性を示さないこと(Hammer,R.,Koss,F.W.,Scand.J.Gastroenterol.,Suppl,1979年,14巻,57号,p.1−6;Bymaster,F.P.ら,J.Pharmacol Exp.Ther.,1993年,267巻,1号,p.16−24を参照のこと)を示した。驚くべきことに、刊行された文献に反して、本発明は、ピレンゼピンおよびテレンゼピンを含めて、M1R−選択的拮抗薬が、治療量で血液脳関門を通過することができ、したがって食欲抑制を含めて、有用な薬理作用を有することを実証する。さらに、以前の知識に反して、M1R−選択的拮抗薬は、24時間の期間において所定の時刻に投与することなく、有効な効果を生じることができる。
【0054】
本発明は、M1R−選択的拮抗薬とある種の他の治療剤を組み合わせ使用によって、肥満症の治療および体重減少の促進を含めて、薬剤用途に有利な予想外の相乗効果が生じることも実証する。
【0055】
本発明は、過体重または肥満の個体において所望の体重減少を実現し、長期間にわたって体重減少の継続および体重維持を達成する有効な薬理学的治療を提供する。M1R−選択的拮抗薬の投与は、予想外に体重減少または体重増加の低減をもたらす。さらに、1つまたは複数のM1R−選択的拮抗薬およびM1R−選択的拮抗薬以外の1つまたは複数の抗うつ剤の共投与は、特に一般的に抗うつ薬の長期投与に伴う体重増加副作用を考えると、予想外にこれらのカテゴリーの薬物のいずれかを単独投与することによって達成されるより多い量の体重減少または体重増加の低減をもたらす(例えば、MasandおよびGupta,Ann.Clin.Psych.14巻,p.175(2002年);ならびにDeshmukhおよびFranco,Cleve.Clin.J.Med.70巻,p.614(2003年))。いくつかの実施形態では、M1R−選択的拮抗薬は、抗肥満剤と共投与することができる。
【0056】
2.肥満症の治療方法および/または体重減少の達成方法
a.治療に供する病態
本方法および組成物は、体重に関連する障害の治療に使用される。本方法および組成物によって治療可能な障害の一般カテゴリーの例としては、とりわけ望ましくないまたは過剰な体重を保持する肥満症、飽満感の欠如が挙げられるが、これらに限定されない。
【0057】
中枢神経系におけるムスカリン性受容体に対するアセチルコリンの作用は、認知、洞察、覚醒、情動、感覚運動ゲーティング、および反射的運動と定方向運動を含めて、多様な行動に影響を及ぼす(Bymasterら,Curr Drug Targets CNS Neurol Disord(2002年)1巻,p.163−181)。ムスカリ
ン性受容体は、コリン作動性ニューロンとの相互作用によるだけでなく、前脳/中脳のドーパミン作動性、GABA作動性およびグルタミン酸作動性ニューロンの活性の調節によっても、これらの機能に影響を及ぼす。神経局在化および微小透析の研究によって、ムスカリン性受容体およびその作動薬または拮抗薬はこれらの系に影響を及ぼし、調節の方向性(興奮/抑制)は特定の受容体サブタイプに依存することが確認された。具体的には、M1/M4を好む拮抗薬であるピレンゼピンの局所微量注射によって、線条体においてドーパミン流出の低減が起こる(Smoldersら,J Neurochem(1997年)68巻,p.1942−1948)。同様に、中脳に直接注入すると、M1/M4受容体を好む拮抗薬であるテレンゼピンは、GABA流出の低減を引き起こす(Smoldersら,1997年、上述)。同様に、スコポラミンなどの非サブタイプ選択的拮抗薬は、前脳においてアセチルコリンレベルの上昇を引き起こす(Izurieta−Sanchezら,Eur J Pharmacol(2000年)399巻,p.151−160)。
【0058】
満足不可能性および強迫的過食を含めて、依存性の障害に関して、中脳辺縁系ドーパミン回路は、嗜癖行動の形成および永続化において重要な役割を果たすと思われる(BerridgeおよびRobinson,Brain Res Brain Res Rev(1998年)28巻,p.309−369;Crespoら,J Neurosci(2006年)26巻,p.6004−6010;Di ChiaraおよびImperato,Proc Natl Acad Sd USA(1988年)85巻,p.5274−5278;HernandezおよびHoebd,Life Sci(1988年)42巻,p.1705−1712).げっ歯類を用いた研究から、線条体における特定の構造である側坐核(NAc)が報酬および嫌悪の制御に関与することが明らかである。NAcは、内側腹側線条体に存在し、さらにシェル、コア、および吻極のサブ領域に分解することができる(ZahmおよびBrog,Neuroscience(1992年)50巻,p.751−767)。
【0059】
ラットは、ドーパミン作動薬をNAcに自己投与し(Hoebelら,Psychopharmacology (Berl)(1983年)81巻,p.158−163)、ヒトにおいて乱用および習慣性を引き起こすことが知られている多数の薬物は、NAcにおいて細胞外ドーパミンレベルを上昇させることがわかっている(Di ChiaraおよびImperato,1988年,上述;HernandezおよびHoebel,1988年,上述;Radaら,Pharmacol Biochem Behav(1996年)53巻,p.809−816)。逆に、側坐核における細胞外ドーパミンの低減は、モルヒネ誘導性およびニコチン誘導性離脱中の嫌悪に随伴することが観察されている(AcquasおよびDi Chiara,(1992年)J Neurochem 58巻,p.1620−1625;Dianaら,J Pharmacol Exp Ther(1995年)272巻,p.781−785;Pothosら,Brain Res(1991年)566巻,p.348−350;Radaら,Psychopharmacology (Berl)(2001年)157巻,p.105−110)。ドーパミンの効果は、受容体サブタイプDlおよびD2によって媒介されるようである。ドーパミンDlまたはD2作動薬をNAcのコアではなくシェルに注射すると、コカインを求めてレバーを押すようオペラント条件付けされたが、次いでコカインを生理食塩水に置換することによってその行動を消去されたラットにおいて、薬物探索行動を復活させることがわかっている(Schmidtら,Eur J Neurosci(2006年)23巻,p.219−228)。
【0060】
NAc内では、コリン作動性とドーパミン作動性の回路は薬理学的に逆のあるようである。アトロピン(非特異的ムスカリン拮抗薬)またはメカミルアミン(非特異的ニコチン拮抗薬)の局所側坐核内投与は、オピオイド強化の獲得を遮断することが報告されている
(Crespoら、2006年,上述)が、モルヒネはNAcにおいてアセチルコリンレベルを低減し(Fiserovaら,Psychopharmacology (Berl)(1999年)142巻,p.85−94;Radaら,Neuropharmacology(1991年)30巻,p.1133−1136)、ナロキソン誘導性オピオイド離脱はアセチルコリンレベルを上昇させる(Fiserovaら,1999年,上述;Radaら,1991年,上述;Radaら,1996年,上述)。同様の現象が、ニコチン依存性ラットにおけるメカミルアミン誘導性離脱に関連して観察されている(Radaら,2001年,上述)。AChの上昇と不快状態の広範な一般的関連性を支持するものとして、AChは、条件付け味覚嫌悪(Markら,Brain Res(1995年)688巻,p.184−188)、脳刺激嫌悪(RadaおよびHoebel,Brain Res(2001年)888巻,p.60−65)、およびジアゼパムからの離脱(RadaおよびHoebel、Eur J Pharmacol(2005年)508巻,p.131−138)、アルコール(Radaら、Pharmacol Biochem Behav(2004年)79巻,p.599−605)または砂糖からの離脱(Colantuoniら,Obes Res(2002年)10巻,p.478−488)によって、NAcにおいて放出される。したがって、コリン作動性伝達の減衰は、嗜癖および習慣性の障害の治療の治療上魅力的な手法である。このような障害は、スクロース離脱に関する知見が例示するように、純粋に薬理学的である必要はない。
【0061】
したがって、優先的にM1ムスカリン性受容体を調節する能力を有する化合物の神経精神医学での用途は広範囲である。したがって、本方法は、障害のあるi)認知処理、ii)情動処理、および/またはiii)欲求動機付けによって生ずる病態を含めて、種々の病態を治療する際に使用(適用)される@@〜に使用される。これらのカテゴリー内の病態としては、肥満症または過剰および/もしくは望ましくない体脂肪の保持を生じる衝動制御障害および食欲障害が挙げられる。
【0062】
したがって、本方法および組成物は、肥満症を治療し、食欲を抑制し、望ましい体重減少を促進し、所望の体重の維持を助長し、望ましくない体重増加を予防または低減する際に使用される。
【0063】
b.薬理作用剤
本方法および組成物で使用する薬理作用剤には、下記に詳細に記載される1つまたは複数の活性剤が、その任意の医薬として許容できる塩、プロドラッグ、ラセミ混合物、配座および/または光学異性体、結晶多形、ならびに同位体を含む任意の医薬として許容できる形で含まれる。
【0064】
i.選択的ムスカリン性受容体M1拮抗薬
本方法は、肥満症を治療し、体重減少および食欲抑制を促進することを必要とする個体に、治療量の1つまたは複数の選択的ムスカリン性受容体M1拮抗薬を投与することによって、肥満症を治療し、体重減少および食欲抑制を促進する。ムスカリン拮抗薬は、参照により本明細書に組み込まれる上述のGoodman and Gilman’s The Pharmacological Basis of Therapeuticsの第7章に概説されている。選択的ムスカリン性受容体M1拮抗薬としては、例えばピレンゼピンおよびテレンゼピンが挙げられる。これらの構造を下記に示す。
【0065】
【化2】
ピレンゼピン(5,11−ジヒドロ−ll−[(4−メチル−l−ピペラジニル)アセチル]−6H−ピリド[2,3−b][l,4]ベンゾジアゼピン−6−オン)は、Azupharma(ドイツ シュツットガルト(Stuttgart,Germany))、Boehringer Ingelheim(ドイツ インゲルハイム(Ingelheim,Germany);Gastrozepin(登録商標)として販売)、Dolorgiet(ドイツ ボン(Bonn,Germany))を含めて、複数の製薬会社によって、ピレンゼピン二塩酸塩として製造および販売されている。ピレンゼピンは、約50mg/日〜約200mg/日、例えば約100〜150mg/日、または50、100、150、もしくは200mg/日の用量で投与することができる。あるいは、ピレンゼピンは、約0.1mg/kg/日〜約10mg/kg/日、通常約0.7mg/kg/日〜約5mg/kg/日の用量で投与することができる。ピレンゼピンの類似体も、本方法を実施する際に使用される。ピレンゼピンの化学的類似体は、例えば米国特許第3,660,380号;第3,743,734号;および第5,324,832号に開示され、それぞれの開示は、全ての目的で参照によりその全体が本明細書に組み込まれる。さらに、ピレンゼピンの用法は、例えば米国特許第5,668,155号に開示される。
【0066】
テレンゼピン(4,9−ジヒドロ−3−メチル−4−[(4−メチル−l−ピペラジニル)アセチル]−10H−チエノ[3,4−b][l,5]ベンゾジアゼピン−10−オン)は、例えばTocris Bioscience(米国ミズーリ州エリスビィル(Ellisville,MO))およびSigma−Aldrich,Inc.(米国ミズーリ州セントルイス(St.Louis,MO))からテレンゼピン二塩酸塩として市販されている。さらに、テレンゼピンの合成は、参照により本明細書に組み込まれる米国特許第4,381,301号に開示される。テレンゼピンは、約0.5mg/日〜約10mg/日、例えば約1〜5mg/日、または0.5、1、2、3、4、5、6、7、8、9、もしくは10mg/日の用量で投与することができる。テレンゼピンの類似体も、本方法を実施する際に使用される。テレンゼピンの化学的類似体およびエナンチオマーは、例えば米国特許第3,953,430号;第4,168,269号;第4,172,831号;第4,381,301号;第5,140,025号、および第5,324,832号に開示され、それぞれの開示は、全ての目的で参照によりその全体が本明細書に組み込まれる。
【0067】
いくつかの実施形態では、テレンゼピンの(+)および(−)エナンチオマーの混合物を含有するラセミ体調製物が投与される。いくつかの実施形態では、テレンゼピンの(+)または(−)エナンチオマーが投与される。テレンゼピンは、35.5kcal/molの活性化障壁によって分離される2つの異なるキラル状態で存在する(Eveleighら,MoI Pharmacol(1989年)35巻,p.477−483;およびSchudtら,Eur J Pharmacol(1989年)165巻,p.87−96)。(+)型のテレンゼピンは、強力な抗ムスカリン様活性を有するが、(−)型はかなり活性が低い。テレンゼピンの選択性は、様々な解剖学的部位で異なるようであり、皮質受容体には、(+)型は(−)異性体に比べて、400倍有効であり;心臓の受容体
には、選択性は低く、(+)型は(−)型より50倍強力である(Eveleighら,上述)。2つの型は、ゆっくり、かつ半減時間約200時間、90度で相互変換する(Eveleighら,上述)。複数の研究では、2つの型が異なる活性を有すると主張されている(Eltze,Eur J Pharmacol(1990年)180巻,p.161−168;Eveleighら,上述;Feifelら,Eur J Pharmacol(1991年)195巻,p.115−123;Kilianら,Agents Actions Suppl 34巻,p.131−147;Schudtら,上述)。
【0068】
ii.抗うつ薬
本発明で使用するためのM1R−選択的拮抗薬ではない抗うつ剤は、その作用機序によっては限定されず、どのクラスの抗うつ薬でも適用可能である。例えば、三環系抗うつ薬(TCA)およびその類似体、セロトニン再取り込み阻害薬、モノアミンオキシダーゼ阻害薬(MAOI)、セロトニン作動薬およびそのプロドラッグ、ノルエピネフリン再取り込み阻害薬、ドーパミン再取り込み阻害薬、およびセロトニン再取り込み促進薬はすべて、1つまたは複数のM1R−選択的拮抗薬と組み合わせて投与することができる。セロトニン再取り込み阻害薬としては、選択的セロトニン再取り込み阻害薬(SSRI)とセロトニン−ノルエピネフリン再取り込み阻害薬(SNRI)が挙げられる。ノルエピネフリン再取り込み阻害薬としては、特異的ノルエピネフリン再取り込み阻害薬と混合ノルエピネフリン−ドーパミン再取り込み阻害薬(NDRI)が挙げられる。セロトニン−ノルエピネフリン−ドーパミンまたは「三元再取り込み阻害薬」も本発明で使用される。他のカテゴリーの抗うつ薬、例えば四環系抗うつ薬であるマプロチリンもしくはミアンセリン、または作用剤であるトラゾドン、ネファゾドン、もしくははブスピロン;副腎皮質刺激ホルモン放出因子受容体1(CRFl)拮抗薬、ならびにアモキサピン、クロザピン、リスペリドン、オランザピン、クエチアピン、およびアリピプラゾールを含めて、精神病もしくはは双極性障害の状況で活性を有することが発見された化合物も使用することができる。
【0069】
本発明で使用するための三環系抗うつ薬としては、アミネプチン、アミトリプチリン、クロミプラミン、デシプラミン、ドキセピン、ドチエピン、イミプラミン、ノルトリプチリン、プロトリプチリン、トリミプラミン、アモキサピン、および筋弛緩薬であるシクロベンザプリンが挙げられる。他の記載していない三環系抗うつ薬およびその類似体も使用するができる。
【0070】
一実施形態では、有効量の1つまたは複数のM1R−選択的拮抗薬を、有効量の選択的セロトニン再取り込み阻害薬と共投与する。選択的セロトニン再取り込み阻害薬としては、例えばシタロプラム、エシタロプラム、フルオキセチン(ラセミ体または光学異性体)、フルボキサミン、パロキセチン、およびセルトラリン(およびそのS−エナンチオマー、Zoloft(登録商標))が挙げられるが、記載していないSSRIも適用可能である。一実施形態では、シタロプラム(またはエシタロプラム)を、1つまたは複数のM1R−選択的拮抗薬と共投与する。一実施形態では、有効量のフルオキセチン(ラセミ体または光学異性体)を共投与する。一実施形態では、有効量のフルボキサミンを共投与する。一実施形態では、有効量のセルトラリン(またはそのS−エナンチオマー、Zoloft(登録商標))を共投与する。一実施形態では、有効量のパロキセチンを共投与する。一実施形態では、有効量のデュロキセチンを共投与する。
【0071】
一実施形態では、有効量の1つまたは複数のセロトニン−ノルエピネフリン再取り込み阻害薬を、1つまたは複数のM1R−選択的拮抗薬と共投与する。セロトニン−ノルエピネフリン再取り込み阻害薬としては,例えばミルナシプラン、ミルタザピン、ベンラファキシン(ラセミ体または光学異性体)、デュロキセチン、(−)l−(1−ジメチルアミノメチル−5−メトキシベンゾ−シクロブタン−1−イル)シクロヘキサノール(S33
005)、DVS−233(デスベンラファキシン)、DVS−233 SR、およびシブトラミンが挙げられるが、記載していないSNRIも使用される。ミルタザピンの作用機序は、その明らかなセロトニン作動性とノルアドレナリン作動性の二様性作用のため、他のSNRIの作用機序と異なる可能性があるが、本明細書ではSNRIクラスの抗うつ薬の一メンバーと見なされる。一実施形態では、有効量のベンラファキシン(ラセミ体または光学異性体)を共投与する。一実施形態では、有効量のデスベンラファキシンを共投与する。一実施形態では、有効量のシブトラミンを共投与する。一実施形態では、有効量のデュロキセチンを共投与する。一実施形態では、有効量のミルナシプランを共投与する。一実施形態では、有効量のミルタザピンを共投与する。
【0072】
他の実施形態では、有効量の1つまたは複数の選択的ノルエピネフリン再取り込み阻害薬を1つまたは複数のM1R−選択的拮抗薬と共投与する。選択的ノルエピネフリン再取り込み阻害薬としては、例えばレボキセチンおよびアトモキセチンが挙げられる。
【0073】
一実施形態では、有効量の1つまたは複数のノルエピネフリン−ドーパミン再取り込み阻害薬を、1つまたは複数のM1R−選択的拮抗薬と共投与する。ノルエピネフリン−ドーパミン再取り込み阻害薬としては、例えばアミネプチン、モダフィニル、GW353162、およびブプロピオンが挙げられる。ブプロピオンの場合、代謝物はノルアドレナリン作動性再取り込み遮断の原因になると思われる。一実施形態では、有効量のブプロピオンを共投与する。
【0074】
一実施形態では、有効量の1つまたは複数の三元(セロトニン−ノルエピネフリン−ドーパミン)再取り込み阻害薬を、1つまたは複数のM1R−選択的拮抗薬と共投与する。三元再取り込み阻害薬としては、例えばインダトラリン、SEP−225289、DOV
216,303、および(+)−l−(3,4−ジクロロフェニル)−3−アザビシクロ−[3.1.0]ヘキサン塩酸塩(DOV 21,947)が挙げられる。
【0075】
本発明で使用するためのモノアミンオキシダーゼ阻害薬としては、ベフロキサトン、ブロファロミン、デプレニル、イソカルボキサジド、モクロベミド、パルギリン、フェネルジン、セレギリン、およびトラニルシプロミンと、それらの持続性送達および経皮送達の形が挙げられる。
【0076】
M1R−選択的拮抗薬と共投与することができる抗うつ薬としては、マプロチリン、チアネプチン、ネファゾドン、およびトラゾドンが挙げられる。
【0077】
抗うつ薬の適切な投与量は、様々な要因の中でもとりわけ、組成物の選択された投与経路および製剤に依存する。例えば、三環系抗うつ薬は、約25mg/日〜約600mg/日の用量、通常約75mg/日〜約300mg/日の用量で投与される。
【0078】
セロトニン−再取り込み阻害薬は、約5mg/日〜約400mg/日の用量で投与され、通常約20mg/日〜約250mg/日で投与される。特に、本方法を実施する際に、ベンラファキシン(ラセミ体または光学異性体)は、1回の投与当たり約9mg〜約225mgで投与することができ、通常1回の投与当たり約37.5mg、75mg、150mg、または225mgで投与する。ベンラファキシンは、典型的には約25mg/日〜550mg/日,通常約37.5mg/日〜375mg/日、より典型的には約75mg/日−225mg/日、最も典型的には約37.5mg/日、75mg/日、150mg/日、225mg/日、または300mg/日で投与される。個々の患者に応じて、1日のベンラファキシン投与量を分割し、1日1回、2回、3回、4回以上で投与することができる。デスベンラファキシンは、約50mg/日〜600mg/日、例えば約50mg/日、100mg/日、200mg/日、400mg/日、または600mg/日の用量
で投与することができる。セルトラリン(またはそのS−エナンチオマー、Zoloft(登録商標))は、約50mg/日〜200mg/日、通常約100mg/日〜150mg/日の用量で投与することができる。フルオキセチン(ラセミ体または光学異性体)は、約5mg/日〜50mg/日、通常約20mg/日〜40mg/日の用量で投与することができる。フルボキサミンは、約50mg/日〜300mg/日、通常約100mg/日〜200mg/日の用量で投与することができる。パロキセチンは、約10mg/日〜50mg/日、通常約20mg/日〜40mg/日の用量で投与することができる。
【0079】
本方法を実施する際に、シタロプラム(またはエシタロプラム)を、約5mg/日〜60mg/日、好ましくは約10mg/日、20mg/日、または30mg/日で投与することができる。通常、シタロプラムは、1日1回、例えば午前または午後に投与される。しかし、投与量のシタロプラムを1日2回以上で投与される患者もいる。ミルタザピンは、約5mg/日〜100mg/日、例えば約7.5mg/日、15mg/日、30mg/日、45mg/日、または90mg/日の用量で投与することができる。ミルナシプランは、約25mg/日〜200mg/日、例えば約25mg/日、50mg/日、100mg/日、150mg/日、または200mg/日の用量で投与することができる。
【0080】
ブプロピオン、ネファゾドン、およびトラゾドンを含めて、非典型的な抗うつ薬は、約50mg/日〜600mg/日、通常約150mg/日〜400mg/日の用量で投与される。ブプロピオンは、約25mg/日〜300mg/日、例えば約25mg/日、50mg/日、100mg/日、150mg/日、200mg/日、300mg/日の用量で投与することができる。モノアミンオキシダーゼ阻害薬は、典型的には約5mg/日〜90mg/日、通常約10mg/日〜60mg/日の用量で投与される。
【0081】
iii.抗肥満剤
本発明は、有効量の1つまたは複数のM1R−選択的拮抗薬を1つまたは複数の抗肥満剤と組み合わせて投与することも企図する。さらに、本発明は、有効量の1つまたは複数のM1R−選択的拮抗薬と1つまたは複数の抗うつ薬の組合せを、さらに1つまたは複数の抗肥満剤と任意選択で組み合わせて投与することを企図する。(i)1つもしくは複数のM1R−選択的拮抗薬と組み合わせた、または(ii)さらにM1R−選択的拮抗薬と抗うつ薬の組合せと組み合わせた使用に適した抗肥満剤としては、例えば食欲減退薬、ドーパミン作動薬、H3−ヒスタミン拮抗薬、5−HT2c受容体作動薬、β−3アドレナリン受容体作動薬、コレシストキニン作動薬、抗てんかん剤、レプチン、レプチン類似体およびレプチン受容体作動薬、ニューロペプチドY(NPY)受容体拮抗薬およびモジュレーター、ペプチド−YY(PYY)受容体作動薬、毛様体神経栄養因子、甲状腺ホルモン受容体−β作動薬、カンナビノイドCBl受容体拮抗薬、メラニン凝集ホルモン受容体拮抗薬、膵および胃液リパーゼ阻害薬、メラノコルチン−4受容体作動薬、およびそれらの組合せが挙げられる。
【0082】
食欲減退薬としては、例えばアンフェタミン、メタンフェタミン、デキストロアンフェタミン、フェンテルミン、ベンズフェタミン、フェンジメトラジン、フェンメトラジン、ジエチルプロピオン、マジンドール、フェンフルラミン、デキスフェンフルラミン、フェニルプロパノールアミン、エフェドラなどが挙げられる。食欲減退薬は交感神経刺激アミンとすることができる。
【0083】
ドーパミン作動薬としては、例えばER−230、ドプレキシン、メシル酸ブロモクリプチンなどが挙げられる。
【0084】
H3−ヒスタミン拮抗薬としては、例えばイムペンタミン、チオペラミド、シプロキシファン、クロベンプロピット、GT−2331、GT−2394、A−331440など
が挙げられる。
【0085】
5−HT2c受容体作動薬としては、例えば1−(m−クロロフェニル)ピペラジン(m−CPP)、ミルタザピン、APD−356(ロルカセリン)、SCA−136(バビカセリン)、ORG−12962、ORG−37684、ORG−36262、ORG−8484、Ro−60−175、Ro−60−0332、VER−3323、VER−5593、VER−5384、VER−8775、LY−448100、WAY−161503、WAY−470、WAY−163909、MK−212、BVT.933、YM−348、IL−639、IK−264、ATH−88651、ATHX−105などが挙げられる(例えば、Nilsson BM,J.Med.Chem.2006年,49巻,p.4023−4034を参照のこと)。
【0086】
β−3アドレナリン受容体作動薬としては、例えばL−796568、CGP 12177、BRL−28410、SR−58611A、ICI−198157、ZD−2079、BMS−194449、BRL−37344、CP−331679、CP−331648、CP−114271、L−750355、BMS−187413、SR−59062A、BMS−210285、LY−377604、SWR−0342SA、AZ−40140、SB−226552、D−7114、BRL−35135、FR−149175、BRL−26830A、CL−316243、AJ−9677、GW−427353、N−5984、GW−2696などが挙げられる。
【0087】
コレシストキニン作動薬としては、例えばSR−146131、SSR−125180、BP−3.200、A−71623、A−71378、FPL−15849、GI−248573、GW−7178、GI−181771、GW−7854、GW−5823などが挙げられる。
【0088】
膵および胃液リパーゼ阻害薬としては、例えばオルリスタット、セチリスタット(ATL−962)などが挙げられる。
【0089】
抗てんかん剤としては、例えばトピラメート、ゾニサミドなどが挙げられる。
【0090】
有用な他の抗肥満剤としては、レプチン、レプチン類似体、およびレプチン受容体作動薬(LY−355101などを含む)、ニューロペプチドY(NPY)受容体拮抗薬およびモジュレーター(SR−120819−A、PD−160170、NGD−95−1、BIBP−3226、1229−U−91、CGP−71683、BIBO−3304、CP−671906−01、J−115814などを含む)、ペプチド−YY(PYY)受容体作動薬(PYY(3−36)などを含む)、毛様体神経栄養因子(アクソカインなどを含む)、甲状腺ホルモン受容体−β作動薬(KB−141、GC−I、GC−24、GB98/284425などを含む)、カンナビノイドCBl受容体拮抗薬(リモナバン、SR147778、SLV 319などを含む(例えば、Antel Jら,J.Med.Chem.2006年,49巻,p.4008−4016を参照のこと))、メラニン凝集ホルモン受容体拮抗薬 (GlaxoSmithKline 803430X、GlaxoSmithKline 856464、SNAP−7941、T−226296などを含む(例えば、Handlon ALおよびZhou H,J.Med.Chem.2006年,49巻,p.4017−4022を参照のこと))、メラノコルチン−4受容体作動薬(PT−15、Ro27−3225、THIQ、NBI 55886、NBI 56297、NBI 56453、NBI 58702、NBI 58704、MB243などを含む(例えば、Nargund RPら,J.Med.Chem.2006年,49巻,p.4035−4043を参照のこと))、およびそれらの組合せが挙げられる。
【0091】
iv.薬理作用剤の組合せ
いくつかの実施形態では、1つもしくは複数のM1R−選択的拮抗薬を、M1R−選択的拮抗薬ではない1つもしくは複数の抗うつ薬と共投与または同時製剤する。いくつかの実施形態では、1つもしくは複数のM1R−選択的拮抗薬を、1つもしくは複数の抗肥満剤と共投与または共製剤する。いくつかの実施形態では、1つもしくは複数のM1R−選択的拮抗薬を、M1R−選択的拮抗薬ではない1つもしくは複数の抗うつ薬、および1つもしくは複数の抗肥満剤と共投与または共製剤する。M1R−選択的拮抗薬、抗うつ薬、および抗肥満剤は上述する通りである。
【0092】
v.異性体
治療剤のすべての配座異性体(例えば、シスおよびトランス異性体)、ならびにすべての光学異性体(例えば、エナンチオマーおよびジアステレオマー)、このような異性体のラセミ体、ジアステレオマー、および他の混合物、ならびに溶媒和物、水和物、異種同形物、同質多形、および互変異性体は、本発明の範囲内である。
【0093】
vi.同位体
本発明は、1つまたは複数の原子が特定の原子質量または質量数を有する1つまたは複数の原子で置換されている治療剤の同位体標識体も包含する。治療剤およびそのプロドラッグの同位体標識体、ならびに治療剤およびそのプロドラッグの同位体標識した医薬として許容できる塩は、本発明の範囲内である。ある種の状況では、重水素(2H)などのより重い同位体による置換によって、インビボ半減期の延長または必要用量の低減など治療上の利点をもたらす代謝安定性を増大させることができる。本発明の治療剤およびそのプロドラッグの同位体標識体は、一般に当業者に知られている方法に従って、非同位体標識試薬を同位体標識試薬に置換することによって調製することができる。
【0094】
c.投与
i.投与期間
通常、1つまたは複数のM1R−選択的拮抗薬は、長期間にわたって個体に投与される。本方法は、少なくとも20日、いくつかの実施形態では少なくとも40日、60日、80日、または100日、またいくつかの実施形態では少なくとも150日、200日、250日、300日、350日、1年以上実施することができる。本治療方法を、1年より長期間、例えば少なくとも400日、450日、500日、550日、600日、650日、700日、800日、900日、1000日受ける個体もいる。しかし、個体は、本方法での2年、3年、4年以上の治療に成功することができる。
【0095】
通常、本発明に従って治療された対象は、治療の約50日〜70日後に少なくとも約5ポンド〜15ポンド、治療の約90日〜150日後に少なくとも約10ポンド〜25ポンド、また治療の約200日〜400日後に少なくとも約15ポンド〜45ポンド減量することができる。通常は、本発明に従って治療された個体は、治療計画を100日、150日、200日、250日、300日、350日、400日、450日、500日、550日、600日、700日、800日、900日、1000日以上実施することによって、個体のベースライン体重の少なくとも約5%、より一般には少なくとも約10%、15%、または20%を減量することができ、この所望の体重減少を安定に維持することができる。重要なことには、有効量の1つまたは複数の抗うつ薬を長期間にわたって投与することによって、長期間の治療を通して安定な体重状態が助長され、望ましくない体重増加が予防される。本発明の併用治療は、肥満および過体重の個体に特に適切であるが、減量を望み、安定な体重を維持し、または望ましくない体重増加を予防するいずれの個体にも投与することができる。
【0096】
ii.スケジューリング
一般に、本方法を実施する際に、有効量の1つまたは複数のM1R−選択的拮抗薬を単独投与し、あるいは1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬と共投与する。いくつかの実施形態では、有効量の1つまたは複数のM1R−選択的拮抗薬を、1つまたは複数の抗肥満剤と共投与する。いくつかの実施形態では、有効量の1つまたは複数のM1R−選択的拮抗薬を、1つまたは複数のM1R−選択的拮抗薬以外の抗うつ薬および1つまたは複数の抗肥満剤と共投与する。共投与される薬理作用剤は、一緒にまたは別々に、同時にまたは異なる時間に投与することができる。投与する時、M1R−選択的拮抗薬、抗うつ薬、および抗肥満剤は独立に、必要に応じて1日1回、2回、3回、4回以上、またはそれほど頻繁でなく投与することができる。好ましくは、投与する薬理作用剤を胃日1回投与する。好ましくは、投与する活性剤を、同時1回または複数回、例えば混合物として投与する。薬理作用剤の1つまたは複数を徐放性製剤として投与することができる。
【0097】
ある患者らについては、治療の開始から、1つもしくは複数のM1R−選択的拮抗薬、次いで1つもしくは複数の抗うつ薬、および/または1つもしくは複数の抗肥満剤を同時に投与して、本方法を実施する。ある患者らについては、まず1つもしくは複数のM1R−選択的拮抗薬を投与し、次いで1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤を続いて共投与することによって、本方法を実施する。1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤の投与を開始する前に、3日、5日、7日、10日、14日、20日、または30日の間、最初に患者に1つまたは複数のM1R−選択的拮抗薬を単独投与することができる。
【0098】
体重減少の助長または食欲の抑制の目的で投与する場合、1つまたは複数のM1R−選択的拮抗薬を単独または組み合わせて投与して、予防的には望ましくない体重増加を予防し、または安定な体重を維持し、あるいは治療的には所望の体重減少を実現し、このような体重減少を維持することができる。
【0099】
iii.投与経路
したがって、1つまたは複数のM1R−選択的拮抗薬の単独投与、あるいは1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤と組み合わせた投与は、経口、口腔内;静脈内、皮内、皮下、筋肉内、経皮、経粘膜、鼻腔内を含めて、非経口などの投与を含めて、様々な方法で実現することができる。1つまたは複数のM1R−選択的拮抗薬を1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤と共投与する場合、同じまたは異なる投与経路で投与することができる。
【0100】
いくつかの実施形態では、1つまたは複数のM1R−選択的拮抗薬を単独または組み合わせて、全身投与ではなく、例えばデポーまたは徐放性製剤で局所投与することができる。
【0101】
iv.適切な投与量を決定する方法
M1R−選択的拮抗薬、抗うつ薬、および抗肥満剤のための投与量は、当業者によって実施されている投与量およびスケジューリング計画に従う。本方法で使用するすべての薬理作用剤の適切な投与量の一般指針は、Goodman and Gilman’s The Pharmacological Basis of Therapeutics,第11版,2006年,上述、およびPhysicians’ Desk Reference (PDR)、例えば第59編(2005年)または第60編(2006年)、Thomson PDRに記載され、これらはそれぞれ、参照により本明細書に組み込まれる。発表されたM1R−選択的拮抗薬の投与量は、肥満症を治療し、または体重減少を促進し、または体重増加を抑制するための治療と異なる適応症に関するものである。本発
明の組成物および方法では、本発明を実施するためのM1R−選択的拮抗薬、抗うつ薬、および抗肥満剤の有効な投与量は、他の適応症に関して発表された投与量と等しく、またはそれより低い{例えば、約25%、50%、75%、または100%)ことがあり得る。
【0102】
M1R−選択的拮抗薬、抗うつ薬、および抗肥満剤の適切な投与量は、選択された投与経路、組成物の製剤、患者の応答、病態の重症度、対象の体重、および処方医師の判断を含めて、複数の要因に従って変わる。投与量は、個々の患者によって必要とされるように、経時的に増減することができる。通常、最初に患者に低用量を投与し、次いで患者が忍容できる有効な投与量まで増加させる。
【0103】
有効量の決定は、特に本明細書に記載されている詳細な開示を考慮に入れて、当業者の能力の十分範囲内である。一般に、1つもしくは複数のM1R−選択的拮抗薬および1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤の組合せの効果的または有効な量は、治療された対象において、毒性の副作用が最小限しかまたは全くなく、所望の効果が観察されるまで、まず低用量または少量のM1R−選択的拮抗薬を単独投与し、次いで投与する用量または投与量を徐々に増加し、必要なら第2または第3の医薬品を追加することによって決定される。本発明の組合せの投与のための適切な用量および投与スケジュールを決定するのに適用可能な方法は、例えばGoodman and Gilman’s The Pharmacological Basis of Therapeutics,第1l版,2006,上述;Physicians’ Desk Reference (PDR),上述;Remington: The Science
and Practice of Pharmacy,第21版,2006年,上述;ならびにMartindale:The Complete Drug Reference,Sweetman,2005年,London:Pharmaceutical Press.、およびMartindale,Martindale:The Extra Pharmacopoeia,第31版,1996年,Amer Pharmaceutical Assnに記載され、それぞれ、参照により本明細書に組み込まれる。
【0104】
投与量および間隔は、治療効果を維持するのに十分な活性化合物の血漿中レベルをもたらすように個別に調整することができる。好ましくは、治療上有効な血清レベルは、1日1回の用量を投与することによって実現されるが、有効な1日複数回投与のスケジュールも本発明に包含される。局所投与または選択的取り込みの場合、有効な局所濃度の薬物を血漿中濃度と関係付けることはできない。当業者は、過度の実験なしに治療上有効な局所投与量を最適化することができる。
【0105】
3.医薬組成物
本発明は、さらに治療上有効量の1つもしくは複数のM1R−選択的拮抗薬、および1つもしくは複数の抗うつ薬、および/または1つもしくは複数の抗肥満剤の混合物を含む医薬組成物を提供する。いくつかの実施形態では、M1R−選択的拮抗薬は、テレンゼピン、ピレンゼピン、およびそれらの混合物からなる群より選択される。
【0106】
いくつかの実施形態では、医薬組成物は、選択的セロトニン再取り込み阻害薬(SSRI)、セロトニン−ノルエピネフリン再取り込み阻害薬(SNRI)、ノルエピネフリン再取り込み阻害薬、ドーパミン再取り込み阻害薬、ノルエピネフリン−ドーパミン再取り込み阻害薬(NDRI)、セロトニン−ノルエピネフリン−ドーパミン再取り込み阻害薬、セロトニン再取り込み促進薬、セロトニン作動薬、およびそれらのプロドラッグである1つまたは複数の抗うつ薬を含む。一実施形態では、医薬組成物は、ベンラファキシン(ラセミ体または光学異性体)、デュロキセチン、フルオキセチン(ラセミ体または光学異性体)、シタロプラム、エシタロプラム、フルボキサミン、パロキセチン、S33005
、DVS−233(デスベンラファキシン)、DVS−233 SR、ブプロピオン、GW353162、シブトラミン、アトモキセチン、およびセルトラリン(またはそのS−エナンチオマー、Zoloft(登録商標))からなる群より選択される1つまたは複数の抗うつ薬を含む。
【0107】
一実施形態では、医薬組成物は、治療上有効量のテレンゼピンまたはピレンゼピン、およびSSRIを含む。一実施形態では、医薬組成物は、治療上有効量のテレンゼピンまたはピレンゼピン、およびシタロプラム(またはエシタロプラム)を含む。一実施形態では、医薬組成物は、治療上有効量のテレンゼピンまたはピレンゼピン、およびセルトラリン(またはそのS−エナンチオマー、Zoloft(登録商標))を含む。一実施形態では、医薬組成物は、治療上有効量のテレンゼピンまたはピレンゼピン、およびフルオキセチン(ラセミ体または光学異性体)を含む。一実施形態では、医薬組成物は、治療上有効量のテレンゼピンまたはピレンゼピン、およびフルボキサミンを含む。一実施形態では、医薬組成物は、治療上有効量のテレンゼピンまたはピレンゼピン、およびパロキセチンを含む。
【0108】
一実施形態では、医薬組成物は、治療上有効量のテレンゼピンまたはピレンゼピン、およびSNRIを含む。一実施形態では、医薬組成物は、治療上有効量のテレンゼピンまたはピレンゼピン、およびベンラファキシン(ラセミ体または光学異性体)を含む。一実施形態では、医薬組成物は、治療上有効量のテレンゼピンまたはピレンゼピン、およびデスベンラファキシンを含む。一実施形態では、医薬組成物は、治療上有効量のテレンゼピンまたはピレンゼピン、およびデュロキセチンを含む。一実施形態では、医薬組成物は、治療上有効量のテレンゼピンまたはピレンゼピン、およびミルナシプランを含む。一実施形態では、医薬組成物は、治療上有効量のテレンゼピンまたはピレンゼピン、およびミルタザピンを含む。
【0109】
一実施形態では、医薬組成物は、治療上有効量のテレンゼピンまたはピレンゼピン、およびブプロピオンを含む。
【0110】
1つもしくは複数のM1R−選択的拮抗薬および1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤の組合せは、独立にまたは一緒に、その医薬として許容できる塩の形、またはそれらの化合物がg適切な担体もしくは賦形剤と混合されている医薬組成物の形で、治療上有効量、例えば所望の体重減少もしくは維持をもたらし、または望ましくない体重増加を予防するのに有効な用量で、対象、例えばヒト患者、ネコやイヌなどの家畜に投与することができる。
【0111】
本発明の1つもしくは複数のM1R−選択的拮抗薬および1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤の組合せは、治療的投与のための種々の製剤に組み込むことができる。さらに詳細には、本発明の組合せは、一緒にまたは別々に、適切な医薬として許容できる担体または希釈液との製剤によって医薬組成物に製剤することができ、固体、半固体、液体、または気体の形の調製物、具体的には錠剤、カプセル剤、丸剤、散剤、顆粒剤、糖衣錠、ゲル剤、スラリー、軟膏剤、液剤、坐剤、注射剤、吸入剤、およびエアゾール剤に製剤することができる。
【0112】
本発明で使用するための適切な製剤は、例えばRemington:The Science and Practice of Pharmacy,第21版,2006年,上述;Martindale: The Complete Drug Reference,Sweetman,2005年,London:Pharmaceutical Press.;Niazi,Handbook of Pharmaceutical Manufacturing Formulations,2004年,CRC Pre
ss;およびGibson,Pharmaceutical Preformulation and Formulation:A Practical Guide from Candidate Drug Selection to Commercial
Dosage Form,2001年,Interpharm Pressに出ており、これらは参照により本明細書に組み込まれる。本明細書に記載する医薬組成物は、当業者に周知の様式、すなわち通常の混合、溶解、顆粒化、糖衣錠作製、分級、乳化、カプセル化、封入、または凍結乾燥プロセスで製造することができる。下記の方法および賦形剤は、例示するものにすぎず、決して限定するものではない。
【0113】
一実施形態では、1つもしくは複数のM1R−選択的拮抗薬および1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤の組合せは、徐放、放出制御、持続放出、時限放出、または遅延放出製剤、例えば治療剤を含有する固体疎水性ポリマーの半透過性マトリックスでの送達用に調製される。様々なタイプの徐放材料が確立しており、当業者に周知せある。現在の持続放出製剤としては、フィルムコーティング錠剤、マルチパーティキュレートまたはペレット系、親水性または親油性材料を使用したマトリックス技術、および造孔賦形剤を含むワックス系錠剤が挙げられる(例えば、Huangら,Drug Dev.Ind.Pharm.29巻,p.79(2003年);Pearnchobら,Drug Dev.Ind.Pharm.29巻,p.925(2003年);Maggiら,Eur.J.Pharm.Biopharm.55巻,p.99(2003年);Khanvilkarら,Drug Dev.Ind.Pharm.228巻,p.601(2002年);およびSchmidtら,Int.J.Pharm.216巻,p.9(2001年)を参照のこと)。徐放送達システムは、そのデザインに応じて、何時間かまたは何日間かにわたって、例えば4時間、6時間、8時間、10時間、12時間、16時間、20時間、24時間以上にわたって、化合物を放出することができる。通常、徐放性製剤は、天然または合成ポリマー、例えばポリビニルピロリドン(PVP)など重合体のビニルピロリドン類;親水性カルボキシビニルポリマー類;メチルセルロース、エチルセルロース、ヒドロキシプロピルセルロース、およびヒドロキシプロピルメチルセルロースなど疎水性および/または親水性のハイドロコロイド類;ならびにカルボキシポリメチレンを使用して調製することができる。
【0114】
徐放性または持続放出製剤は、二酸化チタン、二酸化ケイ素、酸化亜鉛、および粘土を含めて、鉱物などの天然材料を使用して調製することもできる(参照により本明細書に組み込まれる米国特許第6,638,521号を参照のこと)。本発明の1つもしくは複数のM1R−選択的拮抗薬および1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤の組合せを送達する際に使用することができる持続放出製剤としては、例えば米国特許第6,635,680号;第6,624,200号;第6,613,361号;第6,613,358号、第6,596,308号;第6,589,563号;第6,562,375号;第6,548,084号;第6,541,020号;第6,537,579号;第6,528,080号、および第6,524,621号2機債のものが挙げられ、これらはそれぞれ、参照により本明細書に組み込まれる。特に重要な放出制御製剤としては、米国特許第6,607,751号;第6,599,529号;第6,569,463号;第6,565,883号;第6,482,440号;第6,403,597号;第6,319,919号;第6,150,354号;第6,080,736号;第5,672,356号;第5,472,704号;第5,445,829号;第5,312,817号、および第5,296,483号に記載のものが挙げられ、これらはそれぞれ、参照により本明細書に組み込まれる。当業者は、容易に他の適用可能な徐放性製剤を理解するであろう。
【0115】
経口投与のために、1つもしくは複数のM1R−選択的拮抗薬および1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤の組合せを、当技術分野でよく知
られている医薬として許容できる担体と組み合わせることによって容易に製剤することができる。このような担体によって、化合物を、治療対象の患者が経口摂取するための錠剤、丸剤、糖衣錠、カプセル剤、乳剤、親油性および親水性の懸濁剤、液剤、ゲル剤、シロップ剤、スラリー、懸濁剤などとして製剤することができる。経口使用の医薬調製物は、化合物を固体賦形剤と混合し、得られた混合物を任意選択で中砕し、望むなら適切な佐剤を添加した後、顆粒の混合物を処理して、錠剤または糖衣錠コアを得ることによって得ることができる。適切な賦形剤は、特に充填剤、具体的にはラクトース、スクロース、マンニトール、もしくはソルビトールを含めて、砂糖類;セルロース調製物類、例えばトウモロコシデンプン、小麦デンプン、米デンプン、馬鈴薯デンプン、ゼラチン、トラガカントゴム、メチルセルロース、ヒドロキシプロピルメチルセルロース、カルボキシメチルセルロースナトリウムなど、および/またはポリビニルピロリドン(PVP)である。望むなら、架橋ポリビニルピロリドン、寒天、またはアルギン酸またはその塩、具体的にはアルギン酸ナトリウムなどの崩壊剤を添加することができる。
【0116】
経口使用することができる医薬調製物としては、ゼラチンで作製されたプッシュフィットカプセル剤、ならびにゼラチンおよびグリセロールやソルビトールなどの可塑剤で作製された密封軟カプセル剤が挙げられる。プッシュフィットカプセル剤は、活性成分を、ラクトースなどの充填剤、デンプン類などの結合剤、および/またはタルクもしくはステアリン酸マグネシウムなどの滑沢剤、および任意選択で安定剤と混合して含有することができる。軟カプセル剤では、活性化合物を、脂肪油類、流動パラフィン、または液状ポリエチレングリコール類など適切な液体に溶解または懸濁することができる。さらに、安定剤を添加することができる。経口投与用の製剤はすべて、このような投与に適した投与量であるべきである。
【0117】
糖衣錠コアには適切なコーティングが設けられている。このために、アラビアゴム、タルク、ポリビニルピロリドン、カルボポールゲル、ポリエチレングリコール、および/または二酸化チタンを任意選択で含有することができる濃縮された糖液、ラッカー溶液、ならびに適切な有機溶媒または溶媒混合物を使用することができる。錠剤または糖衣錠コーティングには、識別のためまたは活性化合物の用量の異なる組合せを特徴付けるために、染料または顔料を添加することができる。
【0118】
化合物を、注射、例えばボーラス注射または連続注入による非経口投与用に製剤することができる。注射のために、1つもしくは複数のM1R−選択的拮抗薬および1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤の組合せを、植物油もしくは他の類似の油類、合成脂肪族酸グリセリド類、高級脂肪族酸類のエステル、またはプロピレングリコールなどの水性または非水溶媒中、望むなら、通常の添加剤、具体的には可溶化剤、等張剤、懸濁化剤、乳化剤、安定剤、および防腐剤を含めて、溶解、懸濁、または乳化することによって製剤にすることができる。好ましくは、本発明の組合せを、水溶液、好ましくはハンクス液、リンゲル液、または生理適合性緩衝液などの生理食塩緩衝液中で製剤することができる。注射用製剤は、防腐剤を添加した単位剤形、例えばアンプル、または複数回投与容器で提供することができる。組成物は、油性または水性ビヒクル中の懸濁剤、液剤、または乳剤のような形を取ることができ、懸濁化、安定化、および/または分散化剤などの製剤化剤を含有することができる。
【0119】
非経口投与用医薬製剤としては、水溶性の形の活性化合物水溶液が挙げられる。さらに、活性化合物の懸濁剤は、適切な油性注射懸濁剤として調製することができる。適切な親油性溶媒またはビヒクルとしては、ゴマ油などの脂肪油、またはオレイン酸エチルまたはトリグリセリド類、またはリポソーム類などの合成脂肪酸エステルが挙げられる。水性注射懸濁剤は、その懸濁剤の粘度を増大させる物質、具体的にはカルボキシメチルセルロースナトリウム、ソルビトール、またはデキストランを含有することができる。場合によっ
ては、懸濁剤は、適当な安定剤、または高濃度液剤の調製が可能になるように化合物の溶解性を増大させる作用剤も含有することもできる。あるいは、活性成分は、使用する前に適当なビヒクル、例えば発熱物質を含まない無菌水で構成する散剤とすることができる。
【0120】
全身投与は、経粘膜または経皮手段によるものとすることもできる。経粘膜または経皮投与のために、透過するべき関門に適した浸透剤を製剤で使用する。局所投与のために、作用剤を軟膏剤、クリーム、膏薬、散剤、およびゲル剤に製剤する。一実施形態では、経皮送達剤はDMSOとすることができる。経皮送達システムとしては、例えば貼付剤を挙げることができる。経粘膜投与のために、透過するべき関門に適した浸透剤を製剤で使用する。このような浸透剤は、一般に当技術分野で知られている。本発明で使用することができる経皮送達製剤としては、例えば米国特許第6,589,549号;第6,544,548号;第6,517,864号;第6,512,010号;第6,465,006号;第6,379,696号;第6,312,717号、および第6,310,177号に記載されているものが挙げられ、これらはそれぞれ、参照により本明細書に組み込まれる。
【0121】
口腔投与のために、組成物は、通常の方式で製剤される錠剤またはロゼンジの形とすることができる。
【0122】
上述の製剤に加えて、本発明の1つもしくは複数のM1R−選択的拮抗薬および1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤の組合せは、埋め込み調製物として製剤することもできる。このような長時間作用性製剤は、移植(例えば、皮下または筋肉内)または筋肉内注射によって投与することができる。したがって、例えば化合物は、適当なポリマーまたは疎水性材料(例えば、許容できる油中の乳濁液)またはイオン交換樹脂、またはやや溶けにくい誘導体、例えばやや溶けにくい塩と共に製剤することができる。
【0123】
医薬組成物は、適当な固体相もしくはゲル相担体または賦形剤も含むことができる。このような担体または賦形剤としては、例えば炭酸カルシウム、リン酸カルシウム、様々な砂糖類、デンプン類、セルロース誘導体類、ゼラチン、およびポリエチレングリコール類などのポリマー類が挙げられるが、これらに限定されない。
【0124】
4.キット
本発明の医薬組成物は、キットで提供することができる。いくつかの実施形態では、本発明のキットは、1つもしくは複数のM1R−選択的拮抗薬および1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤を別々の製剤として含む。いくつかの実施形態では、キットは、1つもしくは複数のM1R−選択的拮抗薬および1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤を同じ製剤中に含む。いくつかの実施形態では、キットは、治療の過程を通して、1つもしくは複数のM1R−選択的拮抗薬および1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤を独立に、均一の剤形で提供する。いくつかの実施形態では、キットは、治療の過程にわたって、1つもしくは複数のM1R−選択的拮抗薬および1つもしくは複数の抗うつ薬および/または1つもしくは複数の抗肥満剤を独立に、個体の要件に従って増加または低減させるが、通常有効な用量レベルまで増加させる漸増減投与量で提供する。
【0125】
一実施形態では、キットは、テレンゼピンおよびピレンゼピンからなる群より選択される1つまたは複数のM1R−選択的拮抗薬を含む1つまたは複数の医薬組成物を含む。
【0126】
いくつかの実施形態では、キットは、選択的セロトニン再取り込み阻害薬(SSRI)、セロトニン−ノルエピネフリン再取り込み阻害薬(SNRI)、ノルエピネフリン再取
り込み阻害薬、ドーパミン再取り込み阻害薬、ノルエピネフリン−ドーパミン再取り込み阻害薬(NDRI)、セロトニン−ノルエピネフリン−ドーパミン再取り込み阻害薬、およびそれらの混合物からなる群より選択される1つまたは複数の抗うつ薬を含む。一実施形態では、キットは、ベンラファキシン(ラセミ体または光学異性体)、フルオキセチン(ラセミ体または光学異性体)、デュロキセチン、パロキセチン、シタロプラム、エシタロプラム、フルボキサミン、S33005、DVS−233(デスベンラファキシン)、DVS−233 SR、ブプロピオン、GW353162、シブトラミン、アトモキセチン、およびセルトラリン(またはそのS−エナンチオマー、Zoloft(登録商標))からなる群より選択される1つまたは複数の抗うつ薬を含む1つまたは複数の医薬組成物を含む。
【0127】
一実施形態では、キットは、治療上有効量のテレンゼピンまたはピレンゼピン、およびSSRIを含む。一実施形態では、キットは、治療上有効量のテレンゼピンまたはピレンゼピン、およびシタロプラム(またはエシタロプラム)を含む。一実施形態では、キットは、治療上有効量のテレンゼピンまたはピレンゼピン、およびセルトラリン(またはそのS−エナンチオマー、Zoloft(登録商標))を含む。一実施形態では、キットは、治療上有効量のテレンゼピンまたはピレンゼピン、およびフルオキセチン(ラセミ体または光学異性体)を含む。一実施形態では、キットは、治療上有効量のテレンゼピンまたはピレンゼピン、およびフルボキサミンを含む。一実施形態では、キットは、治療上有効量のテレンゼピンまたはピレンゼピン、およびパロキセチンを含む。
【0128】
一実施形態では、キットは、治療上有効量のテレンゼピンまたはピレンゼピン、およびSNRIを含む。一実施形態では、キットは、治療上有効量のテレンゼピンまたはピレンゼピン、およびベンラファキシン(ラセミ体または光学異性体)を含む。一実施形態では、キットは、治療上有効量のテレンゼピンまたはピレンゼピン、およびデスベンラファキシンを含む。一実施形態では、キットは、治療上有効量のテレンゼピンまたはピレンゼピン、およびデュロキセチンを含む。一実施形態では、キットは、治療上有効量のテレンゼピンまたはピレンゼピン、およびミルナシプランを含む。一実施形態では、キットは、治療上有効量のテレンゼピンまたはピレンゼピン、およびミルタザピンを含む。
【0129】
一実施形態では、キットは、治療上有効量のテレンゼピンまたはピレンゼピン、およびブプロピオンを含む。
【0130】
いくつかの実施形態では、キットは、食欲減退薬、ドーパミン作動薬、H3−ヒスタミン拮抗薬、5−HT2c受容体作動薬、β−3アドレナリン受容体作動薬、コレシストキニン作動薬、抗てんかん剤、レプチン、レプチン類似体およびレプチン受容体作動薬、ニューロペプチドY(NPY)受容体拮抗薬およびモジュレーター、ペプチド−YY(PYY)受容体作動薬、毛様体神経栄養因子、甲状腺ホルモン受容体−β作動薬、カンナビノイドCB1受容体拮抗薬、メラニン凝集ホルモン受容体拮抗薬、膵および胃液リパーゼ阻害薬、メラノコルチン−4受容体作動薬からなる群より選択される1つまたは複数の抗肥満剤を含む。
【0131】
一実施形態では、キットは、治療上有効量のテレンゼピンまたはピレンゼピン、およびフェンテルミンを含む。
【実施例】
【0132】
下記の実施例は、特許請求した発明を例示するために記載するものであって、限定するために記載するものではない。
【0133】
(実施例1)
食欲抑制:3月齢の(300〜350グラム)オスSprague−Dawleyラット(個別に収容)を用いて、化合物の食欲抑制剤効果を評価した。試験前の2週間、ラットを「高脂肪」食(BioServ Diet #F3282またはResearch Diets #12451)に順化させた(食餌および水の自由摂取)。食餌を翌朝戻したとき摂餌を刺激するために、実験の1日前(5:00PM)に食餌をケージから取り出した(水は、実験全体を通して摂取可能のままであった)。食餌を提供する前に、ラット(n=8〜10匹/投与群)に調査中の化合物を腹腔内(ip)または経口(po)投与し、ホームケージに戻し、直ちに予め計量した食餌を与えた。投与後4時間に、食餌をケージから取り出し、計量し、記録し(4時間消費量)、翌朝までラットに戻した。投与後24時間、残存する食餌を再び計量し、記録した(24時間消費量)。4時間と24時間間隔の累積消費量(グラムで表す)を算出した。このアッセイでは、ビヒクルで処置したラットは、典型的に4時間間隔で約8グラム、および24時間間隔で約25グラムを消費する。
【0134】
処置効果を表1および図1〜5(4時間消費量)、ならびに表2および図6〜10(24時間消費量)に示す。これらの効果を、生の消費量(単位:グラム、±SEM[1 平均値の標準誤差])と4時間または24時間の消費量低減(%)=[1−(薬物処置後の消費量/ビヒクル処置後の消費量)]×100%として示す。表1および2の用量の欄の同様の上付き文字は、(個々の処置と共投与の比較を容易にするため)同じ実験に由来する値を示す。統計分析は、一元ANOVA(分散分析)、続いて全体のαを0.05としたBonferroni多重比較試験で行った。表1および2では、星印(*)は、ビヒクル処置ラットに比べて有意な効果を示し、文字(aまたはb)は、単一化合物で処置したラットに比べて有意な効果(抗うつ薬による有意については「a」、およびテレンゼピンによる有意については「b」)を示す。表1および2では、符号1個はp<0.05を示し、符号2個はp<0.01を示し、符号3個はp<0.001を示す)。統計的有意性を示すために使用される符号は、対応する図で異なることがある。
【0135】
【表1】
【0136】
【表2】
効果の特徴
24時間消費量効果は、作用の持続時間をよりよく反映し、4時間消費量効果よりよい食欲抑制薬効果の例である。試験されたテレンゼピンの用量(ip)はすべて、単独投与したとき消費量を有意に低減させた(図1および6)。また、24時間後、ほぼすべての組合せは、単なる相加から予想される消費量低減より大きな消費量低減をもたらした。
【0137】
例えば、25mg/kgのシタロプラムは、4時間にわたって中程度の消費量低減をもたらした(31%、p<0.05)が、24時間にわたっては本質的に効果がなかった(4%、n.s.)。3mg/kgのテレンゼピンは、4時間にわたってより大きい消費量低減をもたらした(45%、p<0.00l)が、この低減は24時間にわたっても依然として有意であった(27%、p<0.0l)。これらの化合物の共投与は、有意な消費量低減を4時間(73%、p<0.001)および24時間(60%、p<0.001)にわたってもたらした(4時間および24時間の効果について、それぞれ図2および7を参照のこと)。24時間の効果の大きさは、相加効果から予想されるはずの大きさより大きいので相乗効果であることを示している。
【0138】
別の例では、30mg/kgのベンラファキシンは、4時間にわたって有意ではない消費量低減をもたらし(13%)、24時間にわたって本質的に効果がなかった(0%、n.s.)。3mg/kgのテレンゼピンは、4時間にわたってより大きい消費量低減をもたらし(35%、p<0.001)、これは24時間にわたっても依然として有意であった(22%、p<0.001)。これらの化合物の共投与は、有意な消費量低減を4時間(56%、p<0.001)および24時間(38%、p<0.001)にわたってもたらした(4時間および24時間の効果について、それぞれ図5および10を参照のこと)。4時間と24時間の効果の大きさは、相加効果から予想されるはずの大きさより大きいので相乗効果であることを示している。同様に、より低用量のテレンゼピン(1mg/kg)と30mg/kgのベンラファキシンの共投与も、両方の時間間隔にわたって消費量に相乗効果をもたらした。
【0139】
別の例では、30mg/kgのセルトラリンは、4時間にわたって中程度の消費量低減をもたらし(27%、p<0.05)、これは24時間にわたって維持された(28%、p<0.001)が、より低用量(10mg/kg)では、有効性および持続期間が低減した(有意ではない低減は、4時間および24時間にわたってそれぞれ21%および2%であった)。3mg/kgのテレンゼピンは、4時間にわたってより大きい消費量低減をもたらし(45%、p<0.001)、これは24時間にわたっても依然として有意であった(27%、p<0.01)。テレンゼピンおよび「高用量」のセルタリン(sertaline)の共投与は、有意な消費量低減を4時間(73%、p<0.001)および24時間(63%、p<0.001)にわたってもたらした。同様に、テレンゼピンと「低用量」のセルタリン(sertaline)の共投与は、有意な消費量低減を4時間(69%、p<0.001)および24時間(47%、p<0.001)にわたってもたらした(4時間および24時間の効果についてはそれぞれ、図3および8を参照のこと)。4時間と24時間の効果の大きさは、相加効果から予想されるはずの大きさより大きいので相乗効果であることを示している。
【0140】
この実施例のほぼすべての組合せでは、これらの組合せによってもたらされた低減は、ビヒクル処置ラットだけでなく、どちらかの化合物単独の投与の効果とも有意に異なる。ほぼすべての組合せの実験で、消費量低減は、両方の時間間隔にわたって50%を超えた。臨床参考物であるシブトラミンも、単独投与されたとき有効であった。しかし、有意な低減をもたらすためには、経口投与が必要であった(腹腔内投与データは記載せず)。さらに、30mg/kgのテレンゼピンを共投与すると、シブトラミンの有効用量を1mg/kgまで低減することができる。これは、用量の3倍(4時間消費量)〜10倍(24時間消費量)の低減を意味する。
【0141】
(実施例2)
体重増加の低減:
3月齢〜4月齢(475−550グラム)のSprague−Dawleyオスラット(個別に収容)を用いて、化合物の体重増加を予防する能力を評価した。慢性実験の開始時に、ラットに「高脂肪」食(BioServ Diet #F3282またはResearch Diets #12451){自由摂餌)を約1か月続けていた。実験期間を通して、個々の体重および水の消費量を3回/週記録した。データ収集を約2週間した後、ラットを等しい平均体重の処置群となるように均衡した。イソフルオランにより誘導された麻酔下で、ラットに、適切な薬物濃度(平均体重および算出した送達時間に基づく)を含む浸透圧ミニポンプ(Alzet 2ML2)を外科的に移植した(肩甲下の皮下[sc]に留置)。あるいは、経口投与経路を使用した研究のために、ラットに、3mL/kgの体積で14日間にわたって毎日強制経口投与した。データ収集を薬物処置下でさらに約2週間継続した。処置間隔について体重の変化(グラムで表す)を算出した。このア
ッセイでは、ビヒクル処置ラットは、典型的には2週間間隔にわたって約35〜40グラム(+9%)増加する。表3に、処置効果を、ビヒクル群と処置群の体重増加(%)=[(薬物注入の終わりの体重−ベースライン体重)/ベースライン体重]×100%;および低減(%)=(処置群の体重増加(%)−ビヒクル群の体重増加(%))で示す。処置期間全体を通して、体重をベースライン体重の百分率として図11〜15に示す。表3の用量の欄の同様な上付き文字は、(個々の処置と共投与の比較を容易にするため)同じ実験に由来する値を示す。統計分析は、一元ANOVA(分散分析)、続いて全体のαを0.05としたBonferroni多重比較試験で行った。表3では、星印(*)は、ビヒクル処置ラットに比べて有意な効果を示し、文字(aまたはb)は、単一化合物で処置したラットに比べて有意な効果(抗うつ薬による有意については「a」、およびテレンゼピンによる有意については「b」)を示す。表3では、符号1個はp<0.05を示し、符号2個はp<0.01を示し、符号3個はp<0.001を示す)。統計的有意性を示すために使用される符号は、対応する図で異なることがある。
【0142】
【表3】
効果の特徴
テレンゼピン単独の(経口または皮下)投与は、ビヒクル処置対照と突き合わせて比べたとき、高脂肪食を摂取しているラットの体重増加を有意に低減させた(ベースラインから3%〜7%低減)(図11〜15を参照のこと)。臨床参考化合物であるシブトラミンも、ビヒクル処置対照と突き合わせて比べたとき、体重増加を有意に低減させた(ベースラインから6%低減)(図13を参照のこと)。さらに、より高い用量(10mg/kgおよびl5mg/kgであるが、5mg/kgでない)のセルトラリンも、ビヒクル処置対照と突き合わせて比べたとき、体重増加を有意に低減させた(ベースラインから3%〜7%低減)(図12を参照のこと)。組合せの研究で使用された他の化合物/用量はすべ
て、単独投与したとき体重に効果をもたらさなかった。
【0143】
セルトラリン+テレンゼピンおよびベンラファキシン+テレンゼピンの組合せは、相乗的であり、ビヒクル処置で観察された体重の減少だけでなく、どちらかの化合物単独で観察された体重の減少に比べても有意に大きい体重の減少をもたらした。
【0144】
セルトラリン+テレンゼピン(図12)の場合は、15mg/kgのセルトラリン単独は、ビヒクル処置ラットに比べて有意な体重の減少(6.8%、p<0.001)をもたらし、3mg/kgのテレンゼピン単独も、ビヒクル処置ラットに比べて有意な体重の減少(3.8%、p<0.05)をもたらした。しかし、これらと同じ用量のセルトラリンとテレンゼピンの共投与は、ベースラインから11.3%の低減をもたらした。これは、ビヒクル処置(p<0.001)だけでなく、セルトラリン単独(p<0.001)またはテレンゼピン単独(p<0.05)での処置と比べても有意であった。
【0145】
ベンラファキシン+テレンゼピン(図14)の場合は、30mg/kgのベンラファキシン単独では、ビヒクル処置ラットに比べて有意な体重の減少をもたらすことができなかった(0.7%)が、3mg/kgのテレンゼピン単独は、ビヒクル処置ラットに比べて有意な体重の減少(3.8%、p<0.05)をもたらした。しかし、これらと同じ用量のベンラファキシンとテレンゼピンの共投与は、ベースラインから9.1%の低減をもたらした。これは、ビヒクル処置(p<0.001)だけでなく、ベンラファキシン単独(p<0.001)またはテレンゼピン単独(p<0.001)での処置と比べても有意であった。上記の2つの例では、共投与によって全体重の減少の大きさが増大することによって、相乗効果が明らかである。
【0146】
本明細書に記載する実施例および実施態様は例示の目的にしかすぎず、それを踏まえた様々な修正または変更は、当業者に示唆されるものであり、本願の趣旨および範囲、ならびに添付の特許請求の範囲内に包含されるべきであることを理解されたい。本明細書に引用されるすべての刊行物、特許、および特許出願は、全ての目的で参照によりそれらの全体が本明細書に組み込まれる。
【特許請求の範囲】
【請求項1】
本明細書の一部に記載の発明。
【請求項1】
本明細書の一部に記載の発明。
【図1】

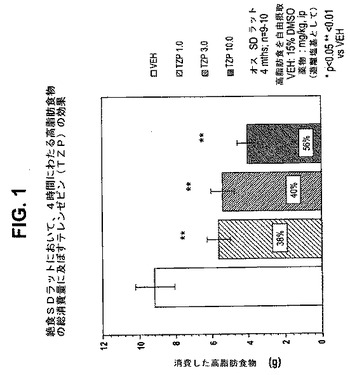
【図2】
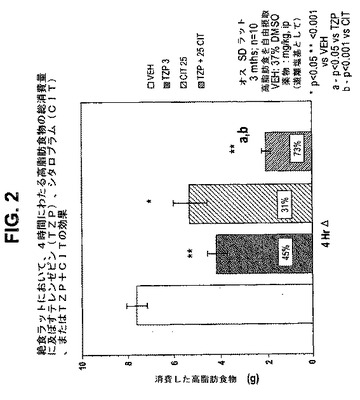
【図3】
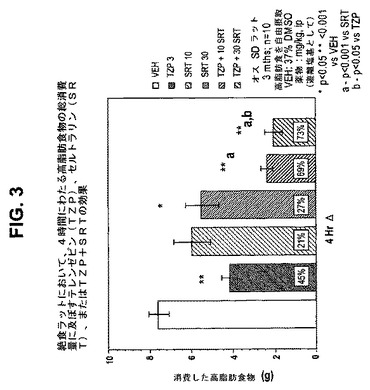
【図4】
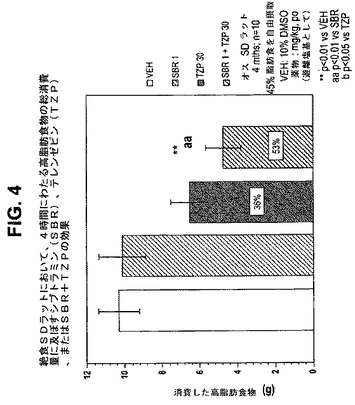
【図5】
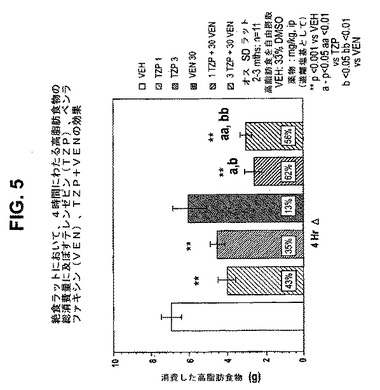
【図6】
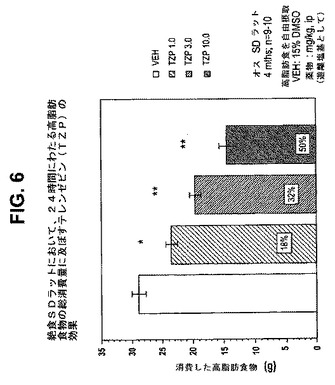
【図7】
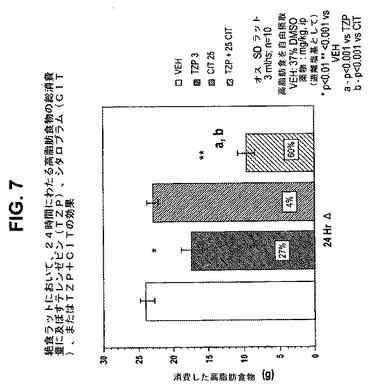
【図8】
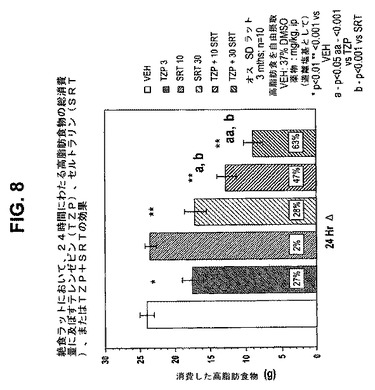
【図9】
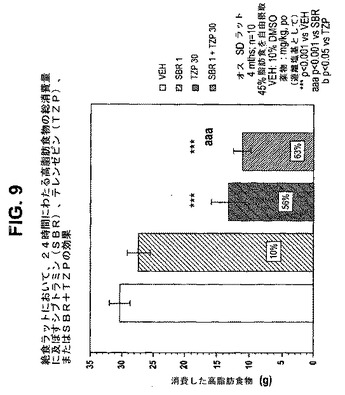
【図10】
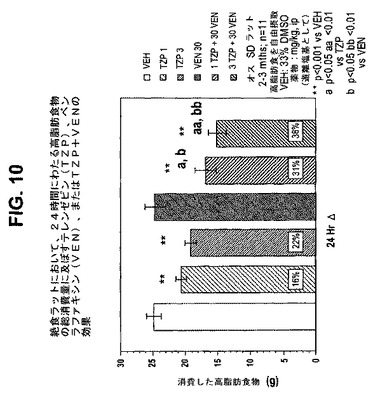
【図11】
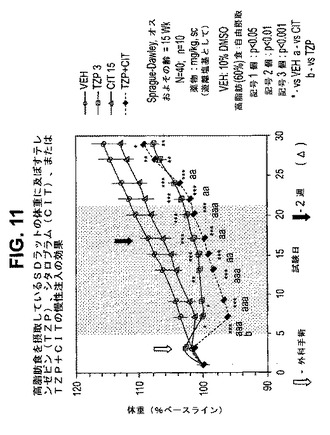
【図12】
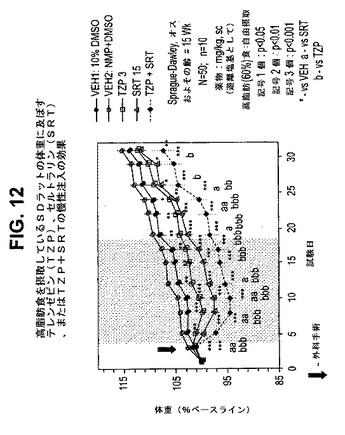
【図13】
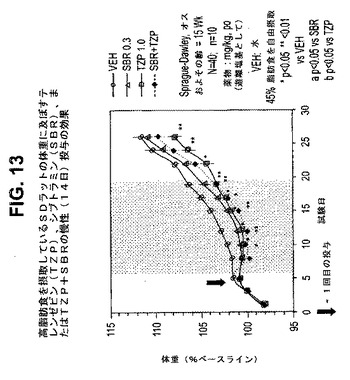
【図14】
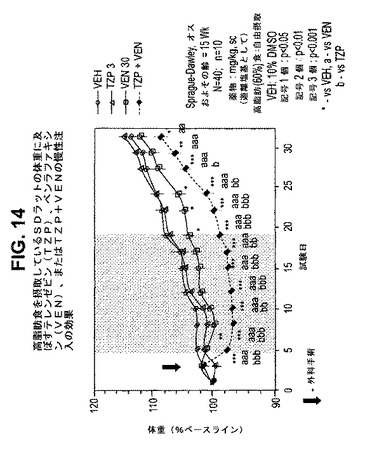
【図15】
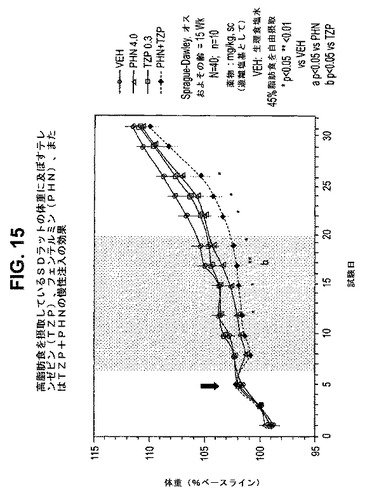
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【公開番号】特開2013−79277(P2013−79277A)
【公開日】平成25年5月2日(2013.5.2)
【国際特許分類】
【外国語出願】
【出願番号】特願2013−1003(P2013−1003)
【出願日】平成25年1月8日(2013.1.8)
【分割の表示】特願2009−530701(P2009−530701)の分割
【原出願日】平成19年6月15日(2007.6.15)
【出願人】(505413679)テラコス・インコーポレイテッド (7)
【Fターム(参考)】
【公開日】平成25年5月2日(2013.5.2)
【国際特許分類】
【出願番号】特願2013−1003(P2013−1003)
【出願日】平成25年1月8日(2013.1.8)
【分割の表示】特願2009−530701(P2009−530701)の分割
【原出願日】平成19年6月15日(2007.6.15)
【出願人】(505413679)テラコス・インコーポレイテッド (7)
【Fターム(参考)】
[ Back to top ]