
ムチン−免疫グロブリン融合タンパク質
【課題】超急性を処置または予防するための組成物および方法を提供する。
【解決手段】本発明は、ムチン免疫グロブリン融合ポリペプチド、その生産方法、および超急性拒絶の処置または予防のための方法に関するもので、特に融合ポリペプチドのムチン部分がアルファ1,3−ガラクトシル基転移酵素とベータ1,6N−アセチルグルコサミニル転移酵素とによってグリコシル化されている。本発明は、ひとつには、ムチン型タンパク質主鎖上の異なるコア糖鎖によって炭水化物エピトープGalα1,3Gal(αGal)が特異的に高密度で発現しうるという発見にもとづいている。このような高密度のαGalエピトープによって、遊離糖類と比較して、抗αGalの結合または除去(すなわち、吸着)の増大が生じるか、または固相に結合したαGal決定基をもたらす。本明細書ではポリペプチドをαGal融合ポリペプチドと称する。
【解決手段】本発明は、ムチン免疫グロブリン融合ポリペプチド、その生産方法、および超急性拒絶の処置または予防のための方法に関するもので、特に融合ポリペプチドのムチン部分がアルファ1,3−ガラクトシル基転移酵素とベータ1,6N−アセチルグルコサミニル転移酵素とによってグリコシル化されている。本発明は、ひとつには、ムチン型タンパク質主鎖上の異なるコア糖鎖によって炭水化物エピトープGalα1,3Gal(αGal)が特異的に高密度で発現しうるという発見にもとづいている。このような高密度のαGalエピトープによって、遊離糖類と比較して、抗αGalの結合または除去(すなわち、吸着)の増大が生じるか、または固相に結合したαGal決定基をもたらす。本明細書ではポリペプチドをαGal融合ポリペプチドと称する。
【発明の詳細な説明】
【技術分野】
【0001】
(発明の分野)
本発明は、概して超急性を処置または予防するための組成物および方法に関し、より詳しくは、炭水化物エピトープGalα1,3Galを含む融合ポリペプチドを含有する組成物に関する。
【背景技術】
【0002】
(発明の背景)
同種移植用のドナー臓器が慢性的に欠乏していることは、動物からヒトへの臓器または組織、すなわち異種移植が実施できれば解決することが可能である。ブタは、ヒトの異種移植とって最も適したドナー種であると考えられている。しかし、それを常套手段として用いる前に解決すべきいくつかの問題がある。最初の免疫性障害は、炭水化物エピトープGalα1,3Gal(α−Gal)(ブタ等の他の哺乳類に存在)に特異的なヒト、類人猿、および旧大陸サルの異種反応性(xenoreactive)天然抗体(XNAb)によって、引き起こされる。ブタ血管内皮細胞上のα−Galエピトープに対するXNAbsの結合によって、数分から数時間のうちに移植片機能損失に至る一連のイベントが開始される(非特許文献1)。
【先行技術文献】
【非特許文献】
【0003】
【非特許文献1】Galili, U., 1993; Oriol, R. et al., 1993
【発明の概要】
【課題を解決するための手段】
【0004】
(発明の要旨)
本発明は、ひとつには、ムチン型タンパク質主鎖上の異なるコア糖鎖によって炭水化物エピトープGalα1,3Gal(αGal)が特異的に高密度で発現しうるという発見にもとづいている。このような高密度のαGalエピトープは、遊離糖類または固相に結合したαGal決定基と比較して、抗αGalの結合または除去(すなわち、吸着)の増大をもたらす。本明細書ではポリペプチドをαGal融合ポリペプチドと称する。
【0005】
本発明は、Galα1,3Gal特異的抗体に結合する融合ポリペプチドを特徴とする。融合ポリペプチドとして、ムチン・ポリペプチドと免疫グロブリン・ポリペプチドとが挙げられる。ムチン・ポリペプチドは、以下の配列Hex−HexNol−HexN−Hex−Hex、NeuAc−Hex−HexNol−HexN−Hex−Hex、およびNeuGc−Hex−HexNol−HexN−Hex−Hexを含むグリカン・レパートリーを有する。
【0006】
また、本発明は、第2のポリペプチドに結合したβ1,6,N−アセチルグリコサミン転移酵素とα1,3ガラクトシル基転移酵素とによってグリコシル化されたムチン・ポリペプチドの少なくとも一領域を含む第1のポリペプチドを含む融合ポリペプチドである特徴とする。
【0007】
また、本発明によって提供されるものは、融合ポリペプチドを生産する方法である。融合ポリペプチドの生産を、免疫グロブリン・ポリペプチドの少なくとも一部分をコードしている核酸に対して操作可能に結合したムチン・ポリペプチドをコードする核酸と、α1,3,ガラクトシル基転移酵素ポリペプチドをコードする核酸と、β1,6,−N−アセチルグルコサミン転移酵素ポリペプチドをコードする核酸とを含む細胞を与えることでおこなう。あるいは、融合ポリペプチドの生産を、免疫グロブリン・ポリペプチドの少なくとも一部分をコードしている核酸に対して操作可能に結合したムチン・ポリペプチドをコードする核酸、α1,3,ガラクトシル基転移酵素ポリペプチドをコードする核酸、およびβ1,6,−N−アセチルグルコサミン転移酵素ポリペプチドをコードする核酸を、細胞に対して導入すること(例えば、トランスフェクションまたは形質転換)によって、おこなう。融合ポリペプチドの生産を可能とする条件下で上記細胞を培養し、該培養から融合ポリペプチドを単離する。融合ポリペプチドの単離は、既知の方法を用いておこなわれる。例えば、プロテインAまたはプロテインGクロマトグラフィを用いて、上記融合ポリペプチドを単離する。
【0008】
上記細胞は、真核細胞または原核細胞(例えば、細菌細胞)である。例えば、真核細胞は、ほ乳類動物細胞、昆虫細胞、または酵母細胞である。典型的な真核細胞として、CHO細胞、COS細胞、または293細胞が挙げられる。
【0009】
ムチン・ポリペプチドは、例えばPSGL−1である。好ましくは、ムチン・ポリペプチドは、PSGL−1の細胞外部分である。好ましい実施形態では、第2のポリペプチドは、免疫グロブリン・ポリペプチドの少なくとも一領域を含む。例えば、第2のポリペプチドは、免疫グロブリン重鎖ポリペプチドの少なくとも一領域を含む。あるいは、第2のポリペプチドは、免疫グロブリン重鎖のFC領域を含む。
【0010】
上記融合ポリペプチドは、多量体である。好ましくは、上記融合ポリペプチドは二量体である。
【0011】
また、本発明は、本明細書に記載するαGal融合ポリペプチド・コード化核酸を含むベクターと同様に、αGal融合ポリペプチドをコードする核酸と、本明細書に記載するベクターまたは核酸を含む細胞とを包含する。あるいは、ベクターは、α1,3ガラクトシル基転移酵素および/または2β1,6−N−アセチルグルコサミニル転移酵素をコードする核酸をさらに含む。本発明はまた、αGal融合ポリペプチドを発現させるために遺伝子操作された宿主細胞(例えばCHO細胞)も包含する。
【0012】
別の態様では、本発明は被験体での抗体媒介拒絶(例えば、超急性拒絶)を処置する方法を提供する。この方法は、生体試料(例えば、被験体由来の全血または血漿)を本発明のαGal融合ポリペプチドと接触させて、融合ポリペプチド−抗体複合体を形成することを含む。この複合体を生体試料から除去し、該生体試料を被験体に再び融合させる。
【0013】
また、本発明に包含されるものは、本発明のαGal融合ペプチドに試料を接触させて抗体−融合ペプチド複合体を形成させ、該複合体を生体試料から除去することで、その試料から抗体を除去する方法である。
【0014】
また、本発明に包含されるものは、αGal融合ポリペプチドを含む医薬品組成物である。
【0015】
特に定義しない限り、本明細書中で使用される全ての技術用語および科学用語は、この発明が属する当該技術分野の当業者が一般に理解するものと同じ意味を持つ。本明細書中で記載されるそれらに対する方法と類似の材料または等価物が本発明の実行またはテストで使われることができるにもかかわらず、適した方法と材料は後述する。本明細書中で言及される全ての刊行物、特許出願、特許、および他の参考文献を、全体として本明細書で援用する。矛盾する場合、定義を含む本明細書がコントロールする。また、材料、方法、および実施例は例証のためのものであって、限定することを意図したものではない。
【0016】
本発明はまた、以下の項目を提供する。
(項目1)
融合ポリペプチドであって、
(a) i) Hex−HexNol−HexN−Hex−Hex、
ii) NeuAc−Hex−HexNol−HexN−Hex−Hex、および
iii) NeuGc−Hex−HexNol−HexN−Hex−Hex
の配列を含むグリカン・レパートリーを持つムチン・ポリペプチドと、
(b) 免疫グロブリンポリペプチドの少なくとも一領域とを有し、
該融合ポリペプチドは、Galα1,3Gal特異的抗体に結合することを特徴とする融合ポリペプチド。
(項目2)
上記ムチン・ポリペプチドが、PSGL−1、MUC1、MUC2、MUC3、MUC4、MUC5a、MUC5b、MUC5c、MUC6、MUC11、MUC12、CD34、CD43、CD45、CD96、GlyCAM−1、およびMAdCAM3またはそれらのフラグメントからなる群から選択されることを特徴とする項目1に記載の融合ポリペプチド。
(項目3)
上記ムチン・ポリペプチドが、P−セレクチン糖タンパク質リガンド−1の少なくとも一領域を含むことを特徴とする項目1に記載の融合ポリペプチド。
(項目4)
上記ムチン・ポリペプチドが、P−セレクチン糖タンパク質リガンド−1の細胞外部分を含むことを特徴とする項目1に記載の融合ポリペプチド。
(項目5)
上記第2のポリペプチドが、H鎖免疫グロブリン・ポリペプチドの一領域を含むことを特徴とする項目1に記載の融合ポリペプチド。
(項目6)
上記免疫グロブリンポリペプチドが、免疫グロブリンH鎖のFc領域を含むことを特徴とする項目1に記載の融合ポリペプチド。
(項目7)
上記融合ポリペプチドが二量体であることを特徴とする項目1に記載の融合ポリペプチド。
(項目8)
項目1の融合ポリペプチドを発現するように遺伝子操作されたことを特徴とする宿主細胞。
(項目9)
上記宿主細胞がCHO細胞であることを特徴とする項目8に記載の宿主細胞。
(項目10)
第2のポリペプチドに結合した第1のポリペプチドを含む融合ポリペプチドであって、該第1のポリペプチドは、
(a)ムチン・ポリペプチドであり、そして
(b)α1,3−ガラクトシル基転移酵素およびβ1,6,N−アセチルグリコサミニル転移酵素であり、そして
該第2のポリペプチドは免疫グロブリン・ポリペプチドの少なくとも一領域を含むことを特徴とする融合ポリペプチド。
(項目11)
上記ムチン・ポリペプチドがヒト・ポリペプチドであることを特徴とする項目10に記載の融合ポリペプチド。
(項目12)
上記ムチン・ポリペプチドが、PSGL−1、MUC1、MUC2、MUC3、MUC4、MUC5a、MUC5b、MUC5c、MUC6、MUC11、MUC12、CD34、CD43、CD45、CD96、GlyCAM−1、およびMAdCAM3またはそれらのフラグメントからなる群から選択されることを特徴とする項目10に記載の融合ポリペプチド。
(項目13)
上記ムチン・ポリペプチドが、P−セレクチン糖タンパク質リガンド−1の少なくとも一領域を含むことを特徴とする項目10に記載の融合ポリペプチド。
(項目14)
上記ムチン・ポリペプチドが、P−セレクチン糖タンパク質リガンド−1の細胞外部分を含むことを特徴とする項目10に記載の融合ポリペプチド。
(項目15)
上記第2のポリペプチドが、H鎖免疫グロブリン・ポリペプチドの一領域を含むことを特徴とする項目10に記載の融合ポリペプチド。
(項目16)
上記第2のポリペプチドが、免疫グロブリンH鎖のFc領域を含むことを特徴とする項目10に記載の融合ポリペプチド。
(項目17)
上記融合タンパク質が二量体であることを特徴とする項目10に記載の融合ポリペプチド。
(項目18)
融合ポリペプチドを生産する方法であって、
(a)i)免疫グロブリン・ポリペプチドの少なくとも一部分をコードする核酸に、操作可能に結合したムチン・ポリペプチドをコードする核酸、
ii)α1,3ガラクトシル基転移酵素ポリペプチドをコードする核酸、および
iii)β1,6−N−アセチルグルコサミニル転移酵素ポリペプチドをコードする核酸、
を有する細胞を提供するステップと、
(b)該融合ポリペプチドの生産を可能とする条件下で細胞を培養するステップと、
(c)該融合ポリペプチドを単離するステップと、
を包含することを特徴とする融合ポリペプチドの生産方法。
(項目19)
上記ムチン・ポリペプチドが、PSGL−1、MUC1、MUC2、MUC3、MUC4、MUC5a、MUC5b、MUC5c、MUC6、MUC11、MUC12、CD34、CD43、CD45、CD96、GlyCAM−1、およびMAdCAM3またはそれらのフラグメントからなる群から選択されることを特徴とする項目18に記載の方法。
(項目20)
上記ムチン・ポリペプチドが、P−セレクチン糖タンパク質リガンド−1の少なくとも一領域を含むことを特徴とする項目18に記載の方法。
(項目21)
上記ムチン・ポリペプチドが、P−セレクチン糖タンパク質リガンド−1の細胞外部分を含むことを特徴とする項目18に記載の方法。
(項目22)
上記免疫グロブリン・ポリペプチドが、H鎖免疫グロブリン・ポリペプチドの一領域を含むことを特徴とする項目18に記載の方法。
(項目23)
上記免疫グロブリン・ポリペプチドが、免疫グロブリンH鎖のFc領域を含むことを特徴とする項目18に記載の方法。
(項目24)
上記細胞が真核細胞または原核細胞であること特徴とする項目18に記載の方法。
(項目25)
上記真核細胞がほ乳類細胞、昆虫細胞、または酵母細胞であることを特徴とする項目24に記載の方法。
(項目26)
上記原核細胞が細菌細胞であることを特徴とする項目24に記載の方法。
(項目27)
上記真核細胞がCHO細胞、COS細胞、または293細胞であることを特徴とする項目24に記載の方法。
(項目28)
融合ポリペプチドを生産する方法であって、
(a)細胞に対して、
(i)免疫グロブリン・ポリペプチドの少なくとも一部分をコードする核酸に操作可能に結合したムチン・ポリペプチドをコードする核酸、
(ii)α1,3−ガラクトシル基転移酵素ポリペプチドをコードする核酸、および
(iii)β1,6−N−アセチルグルコサミニル転移酵素ポリペプチドをコードする核酸を導入するステップと、
(b)上記融合ポリペプチドの生産を可能とする条件下で細胞を培養するステップと、
(c)上記融合ポリペプチドを単離するステップと、
を包含することを特徴とする融合ポリペプチドの生産方法。
(項目29)
項目28の方法によって生産されることを特徴とする融合ポリペプチド。
(項目30)
細胞であって、
(a)免疫グロブリン・ポリペプチドの少なくとも一部分をコードする核酸に操作可能に結合したムチン・ポリペプチドをコードする核酸と、
(b)α1,3−ガラクトシル基転移酵素ポリペプチドをコードする核酸と、
(c)β1,6−N−アセチルグルコサミニル転移酵素をコードする核酸と、
を含むことを特徴とする細胞。
(項目31)
上記細胞がCHO細胞であることを特徴とする項目30に記載の細胞。
本発明の他の特徴および効果は、以下の詳細な説明から、さらに特許請求の範囲から、明らかである。
【図面の簡単な説明】
【0017】
【図1】図1は、ベクター単独(CDM8)、PSGL1/mIgG2b、またはPSGL1/mIgG2bおよびブタα1,3GT発現プラスミドによって、トランスフェクションしたCOS細胞の上清から単離されたタンパク質のSDS−PAGEの写真である。これらを、続いてペルオキシダーゼ共役バンデイレイア・シンプリシフォリア(Bandeireia simplicifolia)イソレクチンB4レクチンでプローブ化して、化学発光による視覚化で免疫精製タンパク質上のGalα1,3Galエピトープを検出した。
【図2】図2Aは、50μl抗マウスIgGアガロース・ビーズ上での吸着前後にトランスフェクションしたCOS細胞上清の容量を増加させる際に、PSFGL1/mIgG2bの抗マウスIgGのFcをELISAにより定量化した結果を示す棒グラフである。試料を三重分析した。図2Bは、トランスフェクションしたCOS細胞上清の容量を増加させる際に、PSGL1/mIgG2b融合タンパク質濃度を示すゲルの写真である。
【図3】図3は、51Cr遊離測定で推定される約300ngのGalα1,3Gal置換または非置換PSGL1/mIgG2bを担持する抗マウスIgGアガロース・ビーズ50μl上への吸着後の異なる容量のヒトAB血清による抗体依存型補体媒介PEC−A細胞毒性を示す折れ線グラフである。
【図4】図4は、免疫親和性精製ヒトヒトIgG、IgM、およびIgAのSDS−PAGEゲルの写真である。各試料の4μgを還元および非還元条件下で流し、銀染色でタンパク質を視覚化した。
【図5】図5は、Galα1,3Gal置換PSGL1/mIgG2b上での吸着を51Cr遊離測定で調べた前後における免疫親和性精製ヒトIgG、IgM、およびIgAの抗体依存型補体媒介PEC−A細胞毒性を示す折れ線グラフである(右手側Y軸;%致死)。
【図6】図6は、熱不活性化ヒトAB血清の添加による致死に対する直接的相乗効果を4時間51Cr遊離測定で調べたK562、RajiおよびPEC−A細胞に対するヒトPBMCの直接的細胞毒性効果(血清無し条件)を示す折れ線グラフである(グラフA)。PEC−A細胞の抗体依存型細胞性細胞毒性に対する熱不活化ヒトAB血清の効果を、Galα1,3置換および非置換PSGL1/mIgIgG22b上への吸着前後での4時間51Cr遊離測定で測定した(グラフB)。
【図7】図7は、PSGL−1/mIgG2bcDNAを単独(−)で、またはブタα1,3ガラクトシル基転移酵素cDNA(+)とともに用いて、安定的にトランスフェクションしたCHO−K1、COS、および293T細胞の上清から精製したPSGL−l/mIgG2b融合タンパク免疫親和性を示すウエスタンブロットの写真である。
【図8】図8は、CHO−K1、COS、および293T細胞によって発現されたPSGL−1/mIgG2b上の相対的α−Galエピトープを示す棒グラフである。ブタα1,3ガラクトシル基転移酵素(GalT)の共発現なし(白色棒)または共発現あり(黒色棒)、さらにCHO−K1に関してはコア2β1,6N−アセチルグロコサミニル転移酵素(C2 GnTI)(灰色棒)で、CHO−K1、COS、または293Tで生産されたP−セレクチン糖タンパク質リガンド−1−マウス免疫グロブリンFc融合タンパク質(PSL−1/mIgG2b)上の相対的α−Galエピトープ密度。
【図9】図9は、異なる宿主細胞で生産されたPSGL−1/mIgG2b上に吸着したヒト血液型AB血清のブタ大動脈血管内皮細胞細胞毒性を示す折れ線グラフである。
【図10】図10は、安定的に形質移入されたCHO−K1細胞の上清から精製されたPSGL−1/mIgG2b融合タンパク質のウエスタンブロット分析の写真である。
【図11】図11は、アフィニティー・クロマトグラフィおよびゲル濾過によって精製されたPSGL−1mIgG2bのSDS−PAGEおよびウエスタンブロット分析の写真である。
【図12】図12は、CHOクローン5L4−1に作られたPSGL−1/mIgG2bから遊離したO−グリカンのエレクトロスプレー・イオントラップ・マススペクトル分析を示す。
【図13】図13は、CHOクローンC2−1−9に作られたPSGL−1/mIgG2bから遊離したO−グリカンのエレクトロスプレー・イオントラップ・マススペクトル分析を示す。
【図14】図14は、CHOクローンC2−1−9で作られたPSL−1/mIgG2bから放出されたO−グリカンのマザー・スペクトルに見られる顕著なピークのMS/MS分析を示す一連の説明図である。
【発明を実施するための形態】
【0018】
(発明の詳細な説明)
本発明は、ひとつには、炭水化物エピトープ Galα1,3Gal (αGal)が高密度で、かつムチン型タンパク質主鎖上の異なるコア糖鎖によって、特異的に発現しうるということの発見にもとづいている。より具体的には、本発明は、ムチン型タンパク質主鎖のαGalエピトープの発現が該ポリペプチドを発現する細胞株に依存するという驚くべき発見にもとづいている。さらに、ムチンのグリカン・レパートリーを外来α1,3ガラクトシル基転移酵素およびコア2分岐酵素の共発現によって修飾することができる。この修飾によって、αGalエピトープの密度がよりいっそう高くなり、遊離糖類、固相に結合したαGal決定基、またはα1,3ガラクトシル基転移酵素単独によりトランスフェクションした細胞と比較して、抗αGal抗体の結合または除去(すなわち、吸着)の増加が生ずる。
【0019】
COS細胞でのPSGL−1/mIgG2b融合タンパク質およびブタα1,3ガラクトシル基転移酵素(α1,3GalT)の一時的トランスフェクションは、α−Galエピトープにより大幅に置換された二量体融合タンパク質を生ずる。この融合タンパク質は、大体、1つの二量体あたり複数の末端α−Galエピトープを有するとともに、アガロース・ビーズに固定されたブタ・チログロブリンよりも20倍高く(炭水化物モル基準で)、Galα1,3Gal共役アガロースおよびマクロ孔ガラス・ビーズよりもそれぞれ5,000倍および30,000倍高い異種反応性天然抗体(XNAb)吸着効率を有する。
【0020】
PSGL−1/mlgG2b上のαGalエピトープ密度およびPSGL−1/mlgG2bの抗ブタ抗体吸着効率に対する宿主細胞の重要性を調べるために、そのタンパク質を、ブタα1,3GalTとともに、CHO、COS、および293T細胞で安定的に発現させた。PSGL−1/mIgG2b上でのα−Gal置換のレベルとその抗ブタ抗体吸着能は、宿主細胞に依存した。COS細胞で作られたPSGL−1/mIgG2bでは、α1,3GalTなしでCOS細胞で作られたPSGL−1/mIgG2bと比較して、相対的O.D(GSA−反応性/抗マウスIgG反応性)が5.3倍増加した(図2)。同様に、293T細胞で作られたPSGL−1/mIgG2bでは、相対的O.Dが3.1倍増加した。それとは対照的に、CHO細胞で作られたPSGL−1/mIgG2bでは、1.8倍増加したのみであった(図2)。異なる宿主細胞で作られたPSGL−1/mIgG2bの抗ブタ抗体吸着効率は、そのαGal置換の度合いと相関関係があった。
【0021】
驚くべきことに、CHO細胞でのコア2β1,6GalcNAc転移酵素(C2 GnTI)の共発現は、PSGL−1/mIgG2bαGal エピトープ密度および抗ブタ抗体吸着効率を改善した。さらに、PSGL−1/mIgG2bは、ブタα1,3GalTとともにCHO細胞で発現し、C2 GnTIは、末端Gal−Galと一致する配列を持つ3つの異なるO−グリカンを担持した(図2)。それとは対照的に、末端Gal−Galエピトープは、C2 GnTIなしのCHO細胞で発現されたPSGL−1/mIgG2b上のO−グリカンでは、検出されなかった。図2に示すように、C2 GnTIおよびα1,3GaITの両方を発現しているCHO細胞で生産された融合タンパク質上のα−Galのレベルは、著しく増加し、外来α1,3GalTのみを発現するCOSおよび293T細胞で作られた融合タンパク質のα−Galエピトープ・レベルを超えた。さらに、融合タンパク質の抗ブタ抗体吸着能が、COSおよび293T細胞と比べて、CHO細胞で増加した(図3)。マススペクトル分析によって確認されたことは、α−Galエピトープ密度増加の原因が、C2 FnTIおよびα1,3GalTの両方を発現させるために操作されたCHO細胞で作られたPSGL−1/mIgG2b のO−グリカン上でのコア2分岐およびラクトサミン伸張にあるということであった(図7および図8、ならびに表II)。
【0022】
炭水化物エピトープに対する抗体の結合は、提示された糖類の立体構造(コンホメーション)に依存している。組織血液型抗原が溶液中で異なる立体構造をとることが示されており((Imberty, A. et al., 1995)、また抗血液型抗体が、立体構造に依存する炭水化物決定基(所謂マイクロ・エピトープ)の異なる領域を認識することも示されている(Imberty, A. et al., 1996)。同様に、研究によって、レセプターが、リガンドに結合するときに、よりエネルギー的に好ましくない立体構造を誘導することができるということも示されている(Imberty, A. et al., 1993)。2種類の異種抗原(αGal−LacNAcおよびαGal−Lewis x)に対する研究によって明らかにされたことは、たとえ後者のエピトープのフコース残基がラクトサミン構造に対して立体構造的規制を誘導した場合であっても、末端Galα1,3Gal二糖類がかなりフレキシブルであった(Corzana, F. et al., 2002)。したがって、ともに所謂天然抗体によって認識された組織血液型抗原および異種抗原は、いくつかの異なる立体構造を採用することができ、これらの抗体が認識するエピトープの立体構造を予測することが困難かもしれない。さらに、オリゴ糖の内側コア構造は、タンパク質−炭水化物複合体の結合に直接関係するものではないが、その結合親和性に影響することが示された(Maaheimo, H. et al., 1995)。α1,3GalTおよびC2 GnTIを共発現するCHO細胞上で発現したO−グリカンの構造的分析によって、α−Galエピトープが3つの異なるオリゴ糖で発現されることが明らかになった(図7および表II)。したがって、コア2およびラクトサミン構造の効果とは別に、オリゴ糖のコア2分岐のうちの1つの分岐上に位置したN−アセチルおよびN−グリコリル・ノイラミン酸がそれらの異種抗原によってとられた立体構造に影響を及ぼす可能性があった。また、シアル酸は結合エピトープの一部である可能性がある。したがって、一部の異種反応性抗体レパートリーは、それらの分岐エピトープを、Galili抗原を認識するものとは異なる結合特異性で認識する((Galili, U. et al., 1985)。さらにまた、N−グリコリルノイラミン酸は、ブタで発現される (Bouhours, D. et al., 1996; Malykh, Y. N. et al., 2003; Malykh, Y. N. et al., 2001)。しかし、N−グリコリルノイラミン酸の発現は、ヒトではみとめられず(Varki, A., 2001)、ヒト異種反応性抗体によって認識されることが明らかになっている(Zhu, A. et al., 2002)。したがって、N−グリコリルノイラミン酸含有グリカンは、異種反応性抗体のさらに別のグループと結合する可能性がある。
【0023】
本発明は、抗αGal抗体の吸着剤として有用な多数のαGalエピトープを含むムチン免疫グロブリン融合タンパク質(以下、「αGal融合タンパク質」という)を提供する。例えば、αGal融合タンパク質は、異種移植に先立って血液または血漿からレシピエント抗αGal抗体を取り除くことに有用である。αGal融合タンパク質は、レシピエントの血液または血漿から10%、25%、50%、60%、70%、80%、90%、95%、98%、または100%の抗αGal抗体を吸着する。
【0024】
αGal融合ペプチドは、炭水化物モル基準で、野生型αGal決定基の遊離糖類と比較して、抗血液型抗体の除去または結合に対して、よりいっそう効率的である。αGal融合タンパク質が結合する抗αGal抗体の数は、等量の野生型αGal決定基の遊離糖類と比べると、2倍、4倍、6倍、10倍、20倍、50倍、80倍、100倍、またはそれ以上多い。
【0025】
(融合ポリペプチド)
種々の態様では、本発明は、第2のポリペプチドに結合した糖タンパク質(例えば、ムチン・ポリペプチド)の少なくとも一部分を含む第1のポリペプチドを有する融合タンパク質を提供する。本明細書で用いられるように、「融合タンパク質(fusion protein)」または「キメラ・タンパク質(chimeric protein)」は、非ムチン・ポリペプチドに対して操作可能に結合したムチン・ポリペプチドの少なくとも一部分を含む。「非ムチン・ポリペプチド(non−mucin polypeptide)」は、その質量の少なくとも40%未満がグリカンに起因するポリペプチドをいう。
【0026】
「ムチン・ポリペプチド(mucin polypeptide)」は、ムチン・ドメインを持つポリペプチドをいう。このムチン・ポリペプチドのムチン・ドメイン数は、1、2、3、5、10、20、またはそれ以上である。ムチン・ポリペプチドは、O−グリカンによって置換されたアミノ酸配列によって特徴づけられる任意の糖タンパク質である。例えば、ムチン・ポリペプチドは2つ目または3つ目のアミノ酸ごとにセリンまたはスレオニンを有する。ムチン・ポリペプチドは、分泌されたタンパク質である。あるいは、ムチン・ポリペプチドは細胞表面タンパク質である。
【0027】
ムチン・ドメインは、アミノ酸のスレオニン、セリン、およびプロリンが豊富であり、そこでN−アセチルガラクトサミンを介してオリゴ糖がヒドロキシアミノ酸(O−グリカン)に結合する。ムチン・ドメインは、O−結合グリコシル化部位を含む、あるいは代わりに、該O−結合グリコシル化部位からなる。ムチン・ドメインのO−グリコシル化部位の数は、1個、2個、3個、5個、10個、20個、50個、100個、またはそれ以上である。あるいは、ムチン・ドメインは、N−結合グリコシル化部位を含む、あるいは代わりに、該N−結合グリコシル化部位からなる。ムチン・ポリペプチドは、その質量の50%、60%、80%、90%、95%、または100%がグリカンに起因する。ムチン・ポリペプチドは、MUC遺伝子(すなわち、MUC1、MUC2、MUC3、MUC4、MUC5a、MUC5b、MUC5c、MUC6、MUC11、またはMUC12)によってコードされた任意のポリペプチドである。あるいは、ムチン・ポリペプチドは、Pセレクチン糖タンパク質リガンド1(PSGL−1)、CD34、CD43、CD45、CD96、G1yCAM−1、MAdCAM、赤血球グリコホリン、グリコカリシン、グリコホリン、シアロフォリン、ロイコ・シアリン、低密度リポ蛋白(LDL)−R、ZP3、およびエピグリカニンである。好ましくは、ムチンは、PSGL−1である。PSGL−1は、各々が402個のアミノ酸を含む、1型トランスメンブレン・トポロジーの2つのジスルフィド結合120kDaサブユニットを持ち、該サブユニットの各々が402のアミノ酸を含むホモ二量体糖タンパク質である。細胞外ドメインには、10アミノ酸コンセンサス配列A Q(M) T T P(Q) P(LT) A A(PG) T(M) Eが15回繰り返して存在し、該配列はO−結合オリゴ糖を付加するための潜在的部位を3または4カ所有する。PSGL−1は、各モノマーあたり、O−結合グリコシル化のための部位数が53を上回り、またN−結合グリコシル化のための部位を3つ有すると予測される。
【0028】
ムチン・ポリペプチドは、ムチン・タンパク質の全てまたは一部を含む。あるいは、ムチン・タンパク質は、該ポリペプチドの細胞外部分を含む。例えば、ムチン・ポリペプチドは、PSGL−1の細胞外部分またはその一部を含む(例えば、GenBank寄託番号A57468に開示されるアミノ酸19−319)。ムチン・ポリペプチドは、PSGL−1のシグナル配列部分(例えば、アミノ酸1−18)、膜貫通(トランスメンブラン)ドメイン(例えば、アミノ酸320−343)、および細胞質ドメイン(例えば、アミノ酸344−412)を含む。
【0029】
本発明のαGal融合タンパク質内で、ムチン・ポリペプチドはムチン・タンパク質の全てまたは一部分に一致する。例えば、αGal融合タンパク質は、ムチン・タンパク質の少なくとも一部分を含む。「少なくとも一部分(at least a portion)」は、ムチン・ポリペプチドが少なくとも1つのムチン・ドメイン(例えば、O−結合グリコシル化部位)を含むことを意味している。任意に、ムチン・タンパク質は、ポリペプチドの細胞外部分から構成される。例えば、ムチン・ポリペプチドは、PSGL−1の細胞外部分から構成される。
【0030】
ムチン・ポリペプチドは、表2に示すようにグリカン・レパートリーで装飾される。例えば、ムチン・ポリペプチドは、表2に示す炭水化物配列を1つ、2つ、3つ、4つ、5つ、またはそれ以上有する。例えば、ムチン・ポリペプチドはグリカン・レパートリーを有し、該グリカン・レパートリーとしてHex−HexNol−HexN−Hex−Hex、NeuAc−Hex−HexNol−HexN−Hex−Hex、およびNeuGc−Hex−HexNol−HexN−Hex−Hexが挙げられる。ムチン・ポリペプチドは、末端αGal糖を1個、2個、3個、4個、5個、またはそれ以上有する。好ましくは、その末端糖は、2、3、4、5、またはそれ以上の数の異なるオリゴ糖で発現される。任意に、ムチンはN−アセチル・ノイラミン酸、N−グリコリルノイラミン酸、および/またはシアル酸を含む。さらに、ムチンのオリゴ糖として、コア2分岐、コア1分岐、およびラクトサミン伸張が挙げられる。
【0031】
第1のポリペプチドは、1種類または複数の転移酵素によって、グリコシル化される。転移酵素は外来性である。あるいは、転移酵素は内在性である。第1のポリペプチドは、2つ、3つ、5つ、またはそれ以上の転移酵素によってグリコシル化される。グリコシル化は経時的または連続的である。あるいは、グリコシル化は同時、またはランダム、すなわち特に決まった順序もなくおこなわれる。例えば、第1のポリペプチドは、α1,3ガラクトシル基転移酵素によってグリコシル化される。α1,3ガラクトシル基転移酵素のための適当なソースとして、GenBank寄託番号AAA73558、L36150、BAB30163、AK016248、E46583、またはP50127が挙げられ、本明細書ではそれらをそのまま援用する。あるいは、第1のポリペプチドは、コア2分岐酵素またはN−アセチルゴルコサミン転移酵素(例えば、β1,6N−アセチルグルコサミン転移酵素)によって、グリコシル化される。β1,6N−アセチルグルコサミン転移酵素のための適当なソースとして、GenBank寄託番号CAA79610、Z19550、BA−B66024、またはAP001515が挙げられる。好ましくは、第1のポリペプチドは、1,3ガラクトシル基転移酵素およびβ1,6N−アセチルグルコサミン転移酵素の両方によってグリコシル化される。
【0032】
融合タンパク質内では、用語「操作可能に結合(operatively linked)」は、第1のポリペプチドのO−結合グリコシル化を可能とするようにして、第1および第2のポリペプチドが化学的に結合(最も一般的にはペプチド結合等の共有結合を介して)することを示すように意図されている。融合ポリペプチドをコードする核酸を言及するために用いる場合、「操作可能に結合」という用語は、ムチン・ポリペプチドおよび非ムチン・ポリペプチドをコードする核酸が互いにフレーム単位(in−frame)で融合していることを意味している。非ムチン・ポリペプチドは、ムチン・ポリペプチドのN末端またはC末端に融合しうる。
【0033】
任意に、αGal融合タンパク質は、1つ以上の付加的構成成分に結合する。例えば、αGal融合タンパク質はGST融合タンパク質と付加的に結合し、該αGal融合タンパク質配列がGST(すなわち、グルタチオンS−転移酵素)配列のC末端と融合する。そのような融合タンパク質は、αGal融合タンパク質の精製を促進することができる。あるいは、αGal融合タンパク質は、固相に対して付加的に結合する。種々の固相が当業者に知られている。そのような組成物は、抗αGal抗体の除去を促すことができる。例えば、αGal融合タンパク質は、金属化合物、シリカ、ラテックス、高分子材料等から作られた粒子;マイクロタイター・プレート; ニトロセルロース、もしくはナイロン、またはそれらの組み合わせに結合する。固体支持体に結合したαGal融合タンパク質は、生体試料(例えば、血液または血漿)から抗αGal抗体を除去するための吸着剤として用いられる。
【0034】
融合タンパク質は、そのN末端に異種シグナル配列(すなわち、ムチン核酸によってコードされているポリペプチドに存在しないポリペプチド配列)を含む。例えば、天然ムチン・シグナル配列を取り除いて、他のタンパク質由来のシグナル配列と置き換えることができる。ある種の宿主細胞(例えば、ほ乳類の宿主細胞)では、ポリペプチドの発現および/または分泌を、異種シグナル配列の使用を介して増加させることができる。
【0035】
本発明のキメラ・タンパク質および融合タンパク質は、標準的な組み換えDNA技術によって作ることができる。例えば、異なるポリペプチド配列をコードするDNAフラグメントを、従来の技術にもとづいてフレーム単位で連結する(例えば、連結反応のための平滑末端化(blunt−ended)または突出末端化(stagger−ended)された末端、適当な末端を生ずる制限酵素による消化、必要に応じた付着端の充填、不要な結合を避けるためのアルカリ・ホスファターゼ処理、および酵素による連結反応を用いる)。別の実施形態では、融合遺伝子を、自動DNA合成装置等、従来の技術によって合成することができる。あるいは、2つの連続的な遺伝子フラグメントの間に相補的なオーバーハングを生じるアンカー・プライマーを用いて、遺伝子フラグメントのPCR増幅をおこなうことができる。その後、これらの遺伝子フラグメントをアニーリングおよび再増幅することで、キメラ遺伝子配列が得られる(例えば、Ausubel et al. (eds.) CURRENT PROTOCOLS IN MOLECULAR BIOLOGY, John Wiley & Sons, 1992を参照せよ)。さらに、多くの発現ベクターが商業的に入手可能であり、それらは融合構成成分(例えば、免疫グロブリンH鎖のFc領域)をコードする。融合構成成分がフレーム単位で免疫グロブリン・タンパク質に結合されるように、PSGL−1をコードする核酸をそのようなベクターにクローニングすることができる。典型的なPSGL−1発現ベクターは、配列番号21を含む。
【0036】
αGal融合ポリペプチドは、オリゴマーとして存在する。すなわち、二量体、三量体、または五量体として存在する。好ましくは、αGal融合ポリペプチドは、二量体である。
【0037】
第1のポリペプチド、および/または該第1のポリペプチドをコードする核酸は、当業者公知のムチン・コード配列を用いて構築される。ムチン・ポリペプチドおよび該ムチン・ポリペプチドをコードする核酸の適当なソースとして、GenBank寄託番号NP663625および NM145650、CAD10625および AJ417815、XP140694および XM140694、XP006867および XM006867、ならびに NP00331777および NM009151がそれぞれ挙げられ、本明細書ではそれらをそのまま援用する。
【0038】
あるいは、ムチン・ポリペプチド構成成分は、天然ムチン配列(野生型)に、炭水化物含有量の増加(非突然変異配列と比較して)をもたらす突然変異を持つ変異ムチン・ポリペプチドとして、提供される。例えば、変異ムチン・ポリペプチドは、野生型ムチンと比較して、付加的なO−結合グリコシル化部位を含んでいた。あるいは、変異ムチン・ポリペプチドは、野生型ムチン・ポリペプチドと比較して、セリン、スレオニン、またはプロリンの数が増加するアミノ酸配列突然変異を含む。このような炭水化物含有量の増加は、当業者に公知の方法によって、ムチンのタンパク質と炭水化物との比率を決定することで、評価することができる。
【0039】
いくつかの実施形態では、ムチン・ポリペプチド構成成分は、天然ムチン配列(野生型)に、タンパク質分解に対してより高い耐性のムチン配列(非突然変異配列と比較して)をもたらす突然変異を有する変異ムチン・ポリペプチドとして、提供される。
【0040】
第1のポリペプチドは、完全長PSGL−1を含む。あるいは、第1のポリペプチドは、完全長よりも短いPSGL−1ポリペプチド(例えば、PSGL−1の細胞外部分)を含む。例えば、第1のポリペプチドは、長さが400アミノ酸未満、例えば300、250、150、100、50、または25アミノ酸以下である。典型的なPSGL−1ポリペプチドおよび核酸配列として、GenBank寄託番号XP006867、XM006867、XP140694、および XM140694が挙げられる。
【0041】
第2のポリペプチドは、好ましくは可溶性である。第2のポリペプチドは、αGal融合ポリペプチドと第2のムチン・ポリペプチドとの結合を促進する配列を含む。好ましくは、第2のポリペプチドは、少なくとも免疫グロブリン・ポリペプチドの一領域を含む。「少なくとも一領域(at least a region)」とは、免疫グロブリン分子の任意の部分、例えばL鎖、H鎖、FC領域、Fab領域、Fv領域、またはそれらの任意のフラグメントを含むことを意味する。免疫グロブリン融合ポリペプチドは公知であり、例えば米国特許第5,516,964号、第5,225,538号、第5,428,130号、第5,514,582号、第5,714,147号、および第5,455,165号に記載されている。
【0042】
第2のポリペプチドは、完全長免疫グロブリン・ポリペプチドを含む。あるいは、第2のポリペプチドは、完全長よりも短い免疫グロブリン・ポリペプチド(例えば、H鎖、L鎖、Fab、Fab2、Fv、またはFc)を含む。好ましくは、第2のポリペプチドは、免疫グロブリン・ポリペプチドのH鎖を含む。より好ましくは、第2のポリペプチドは免疫グロブリン・ポリペプチドのFc領域を含む。
【0043】
本発明の別の態様では、第2のポリペプチドは、野生型免疫グロブリンH鎖のFc領域のエフェクター機能よりも劣るエフェクター機能を持つ。Fcエフェクター機能は、例えば、Fcレセプター結合、補体固定、およびT細胞減少活性が含まれる(例えば、米国特許第6,136,310号を参照せよ)。T細胞減少活性、Fcエフェクター機能、および抗体安定性を評価する方法は、公知である。一実施形態では、第2のポリペプチドはFcレセプターに対して低親和性を示すか、あるいは親和性を示さない。別の実施形態では、第2のポリペプチドは補体タンパク質C1qに対して低親和性を示すか、あるいは親和性を示さない。
【0044】
本発明の別の態様は、ムチン・ポリペプチド、またはその誘導体、フラグメント、類似体、もしくは相同体(ホモログ)をコードする核酸を含むベクター、好ましくは発現ベクターに関する。種々の態様では、ベクターは免疫グロブリン・ポリペプチドをコードする核酸に操作可能に結合したムチン・ポリペプチド、またはその誘導体、フラグメント、類似体、もしくは相同体(ホモログ)をコードする核酸を含む。さらに、ベクターは、α1,3ガラクトシル基転移酵素、コア1,6,−N−アセチルグルコサミニル転移酵素またはそれらの任意の組み合わせを含む。転移酵素は、αGal融合タンパク質のムチン部分のペプチド主鎖上に対するαGal決定基の付加を促す。典型的なベクターは、配列番号1、11、または21を含む。本明細書で用いられるように、用語「ベクター(vector)」は核酸分子であり、該核酸分子が結合する別の核酸を搬送することが可能なものをいう。ベクターの1つの種類として、「プラスミド(plasmid)」があり、該プラスミドとは追加のDNAセグメントが連結しうる環状二重鎖DNAループのことである。ベクターの別の種類として、ウイルス・ベクターがあり、追加のDNAセグメントがウイルス・ゲノムに連結することができる。ある種のベクターは、該ベクターが導入される宿主細胞で自律複製ができる(例えば、細菌複製開始点を持つ細菌ベクターおよびエピソームほ乳類ベクター)。他のベクター(例えば、非エピソームほ乳類ベクター)は、宿主細胞に導入されると直ちに該宿主細胞のゲノムに組み込まれ、それによってその宿主ゲノムとともに複製される。さらに、ある種のベクターは、それが操作可能に連結している遺伝子の発現を導くことができる。そのようなベクターを本明細書では「発現ベクター(expression vectors)」と称する。一般に、発現ベクターは、しばしばプラスミドの形で、組換えDNA技術で有用である。本明細書では、ベクターの最も一般的な使用形態がプラスミドであることから、「プラスミド」および「ベクター」を互いに置き換えて使用することができる。しかし、本発明は、等価な機能を呈するそのような発現ベクターの他の形態、例えばウイルス・ベクター(例:複製欠損レトロウイルス、アデノウイルス、およびアデノ関連ウイルス)を含むことを意図している。
【0045】
本発明の組換え発現ベクターは、宿主細胞での核酸発現に適した形状の核酸を含む。このことは、その組換え発現ベクターが1種類以上の調節配列を含むことをを意味し、該配列は発現に使用される宿主細胞にもとづいて選択され、発現される核酸配列に操作可能に結合している。組換え発現ベクターの中で、「操作可能に結合(operably−linked)」とは、ヌクレオチド配列の発現(例えば、生体外(in vitro)転写/翻訳系で、または宿主細胞にベクターが導入された場合は宿主細胞で)を可能とするようにして、目的とするヌクレオチド配列が調節配列に結合していることを意味している。
【0046】
用語「調節配列(regulatory sequence)」が意味することには、プロモーター、エンハンサー、および他の発現調節要素(例えば、ポリアデニル化シグナル)が含まれる。そのような調節配列は、例えば、Goeddel, GENE EXPRESSION TECHNOLOGY: METHODS IN ENZYMOLOGY 185, Academic Press, San Diego, Calif.
(1990)に記載されている。調節配列として、多くの種類の宿主細胞でヌクレオチド配列の構成的発現を導くもの、およびある種の宿主細胞のみにあるヌクレオチド配列の発現を導くもの(例えば、組織特異的調節配列)が挙げられる。当業者が容易に理解することは、発現ベクターの設計が、形質転換すべき宿主細胞の選択、所望のタンパク質の発現レベル等のファクターに依存しうることである。本発明の発現ベクターを宿主細胞に導入することで、本明細書に記載した核酸によってコードされる融合タンパク質または融合ペプチド等(例えば、ABO融合ポリペプチド、ABO融合ポリペプチドの変異体、その他)のタンパク質またはペプチドを生産することができる。
【0047】
本発明の組換え発現ベクターを、原核細胞または真核細胞でのαGal融合ポリペプチドの発現用に設計することができる。例えば、αGal融合ポリペプチドは、大腸菌(Escherichia coli)等の細菌参謀、昆虫細胞(バキュロウイルス発現ベクターを用いる)、酵母細胞、またはほ乳類動物細胞で発現することができる。適当な宿主細胞については、Goeddel, GENE EXPRESSION TECHNOLOGY: METHODS IN ENZYMOLOGY 185, Academic
Press, San Diego, Calif. (1990)でさらに論じられている。あるいは、組換え発現ベクターを、例えばT7プロモーター調節配列およびT7ポリメラーゼを用いて、生体外(in vitro)で転写および翻訳することができる。
【0048】
原核生物でのタンパク質発現は、ほとんどの場合、融合タンパク質または非融合タンパク質の発現を目的とした構成的プロモーターまたは誘導的プロモーターを含むベクターを持つ大腸菌(Escherichia coli)でおこなわれる。融合ベクターは、多数のアミノ酸をそこでコードされるタンパク質、通常は組換えタンパク質のアミノ末端に、加える。そのような融合タンパク質は、概して3つの目的、すなわち(i)組換えタン
パク質の発現を高めること、(ii)組換えタンパク質の溶解性を高めること、および(iii)親和性精製でリガンドとして作用することで組換えタンパク質の精製を助けること、にかなう。しばしば、融合発現ベクターでは、融合タンパク質の精製の後の融合構成成分(fusion moiety)からの組換えタンパク質分離を可能とするために、タンパク質分解切断部位が融合構成成分と組換えタンパク質との接続部分で導入される。そのような酵素(および該酵素の同種認識配列)として、第Xa因子、トロンビン、およびエンテロキナーゼが挙げられる。典型的な融合発現ベクターとして、pGEX(Phanmacia Biotech Inc; Smith and Johnson, 1988. Gene 67: 31−40), pMAL (New England Biolabs, Beverly, Mass.)およびpRIT5 (Pharmacia, Piscataway, N.J.) 挙げられ、これらはグルタチオンS−転移酵素(GST)、マルトースE結合タンパク質、またはプロテインAのそれぞれを標的組換えタンパク質に融合させる。
【0049】
適当な誘導可能な非融合大腸菌(E.coli)発現ベクターの例として、pTrc (Amrann et al., (1988) Gene 69:301−315)および pET 11d (Studier et al., GENE EXPRESSION TECHNOLOGY: METHODS IN ENZYMOLOGY 185, Academic Press, San Diego, Calif. (1990) 60−89)が挙げられる。
【0050】
大腸菌(E.coli)で組換えタンパク質発現を最大にする1つの戦略は、組み換えタンパク質をタンパク質分解的に切断する能力に欠けた宿主細菌でそのタンパク質を発現させることである。例えば、Gottesman, GENE EXPRESSION TECHNOLOGY: METHODS IN ENZYMOLOGY 185, Academic Press, San Diego, Calif. (1990) 119−128を参照せよ。別の戦略は、各アミノ酸に対する個々のコドンが大腸菌(E. coli)で優先的に用いられように、発現ベクターに挿入される核酸の核酸配列を改変することである(例えば、Wada, et al., 1992. Nuci. Acids Res. 20: 2111−2118を参照せよ)。本発明の核酸配列のそのような改変は、標準的DNA合成技術によっておこなうことができる。
【0051】
別の実施形態では、αGal融合ポリペプチド発現ベクターは、酵母発現ベクターである。酵母Saccharonryces cerivisaeでの発現用ベクターの例として、pYepSecl (Baldari, et al., 1987. EMBO J. 6: 229−234)、pMFa (Kurjan and Herskowitz, 1982. Cell 30: 933−943)、pJRY88(Schultz et al., 1987. Gene 54: 113−123)、pYES2(Ivitrogen Corporation, San Diego, Calif.)、およびpicZ (InVitrogen Corp, San Diego, Calif.)が挙げられる。
【0052】
あるいは、αGal融合ポリペプチドをバキュロウイルス発現ベクターを用いて昆虫細胞で発現させることができる。 培養昆虫細胞(例えば、SF9細胞)でのタンパク質発現に利用可能なバキュロウイルス・ベクターとして、pAc系(Smith, et al., 1983. Mol. Cell. Biol. 3: 2156−2165)およびpVL系 (Lucklow and Summers, 1989. Virology 170: 31−39)が挙げられる。
【0053】
さらに別の実施形態では、本発明の核酸はほ乳類発現ベクターを用いてほ乳類細胞で発現させることができる。ほ乳類発現ベクターの例として、pCDM8 (Seed, 1987. Nature 329: 840)およびpMT2PC (Kaufinan, et al., 1987. EMBO J. 6: 187−195)が挙げられる。ほ乳類細胞で用いた場合、発現ベクターの調節機能は、しばしばウイルスの調節要素によって提供される。例えば、一般的に用いられるプロモーターは、ポリオーマ、アデノウイルス2、サイトメガロウイルス、およびシミアンウイルス40に由来する。原核生物および真核細胞のための他の適当な発現系に関しては、例えば、Sambrook, et al., MOLECULAR CLONING: A LABORATORY MANUAL. 2nd ed., Cold Spring Harbor Laboratory, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y., 1989の第16章および第17章を参照せよ。
【0054】
本発明の別の態様は、本発明の組換え発現ベクターが導入されている宿主細胞に関する。用語「宿主細胞(host cell)」および「組換え宿主細胞(recombinant host cell)」は、本明細書では相互に置き換え可能なかたちで用いられる。そのような用語が特定の被験体細胞のみならず、そのような細胞の子孫または潜在的子孫にも言及することが理解される。突然変異または外界いずれかの影響によって後続世代で一定の修飾が生ずることから、実際のところ、そのような子孫は親細胞とは同一ではないと思われるが、それでも本明細書で用いられるようにその用語の範囲内である。
【0055】
宿主細胞は、任意の原核生物または真核細胞である。例えば、αGal融合ポリペプチドは、大腸菌(E. coli)等の細菌細胞、昆虫細胞、酵母、またはほ乳類細胞(例えば、ヒト、チャイニーズ・ハムスター卵巣(CHO)細胞もしくはCOS細胞)で発現される。他の適当な宿主細胞は、当業者に公知である。
【0056】
従来の形質転換または形質移入(トランスフェクション)技術によって、ベクターDNA免疫を原核細胞または真核細胞に導入することができる。本明細書で用いられるように、「形質転換(transformation)」および「トランスフェクション(transfection)」は、外来核酸(例えば、DNA)を宿主細胞に導入するための種々の公知技術に言及するものである。上記技術として、リン酸カルシウムまたは塩化カルシウム共沈、DEAE−デキストラン媒介トランスフェクション、リポフェクション、または電気穿孔法が挙げられる。宿主細胞を形質転換またはトランスフェクションさせるための適当な方法は、Sambrook., et al. (MOLECULAR CLONING: A LABORATORY MANUAL. 2nd ed., Cold Spring Harbor Laboratory, Cold Spring
Harbor Laboratory Press, Cold Spring Harbor, N.Y., 1989)および他の研究室マニュアルに見いだすことができる。
【0057】
ほ乳類細胞の安定的なトランスフェクションのために、用いた発現ベクターおよびトランスフェクション技術に依存して、細胞のわずかなフラクションのみが外来DNAを該細胞のゲノムに組み込まれる可能性がある。これらの組み込まれた要素を同定および選択するために、選択マーカー(例えば、抗生物質耐性)をコードする遺伝子が一般に、目的とする遺伝子とともに宿主細胞に導入される。種々の選択可能マーカーとして、薬物(G418、ハイグロマイシン、およびメトトレキセート)に対する耐性を与えるものが挙げられる。選択マーカーをコードする核酸は、糖タンパク質Ibαをコードするものと同じベクター上で宿主細胞に導入されるか、もしくは別のベクター上で導入される。誘導核酸により安定的にトランスフェクションした細胞は、薬物選択によって同定される(例えば、選択マーカー遺伝子が取り込まれた細胞は生存し、他の細胞は致死する)。
【0058】
本発明の宿主細胞(例えば、培養中の原核または真核宿主細胞)を用いてαGalポリペプチドを生産(すなわち、発現)させることができる。したがって、本発明は、該発明の宿主細胞を用いてαGal融合ポリペプチドを生産するための方法を、さらに提供する。一実施形態では、この方法は、本発明の宿主細胞(αGal融合ポリペプチドをコードする組換え発現ベクターが導入されている)を、αGal融合タンパク質が生産されるように、適当な培地で培養することを含む。別の実施形態では、本発明はさらに培地または宿主細胞からαGalポリペプチドを単離することを、さらに含む。
【0059】
αGal融合ポリペプチドを、従来の条件(例えば、抽出、沈殿、クロマトグラフィー、アフィニティー・クロマトグラフィ、泳動、その他)にもとづいて単離および精製してもよい。例えば、免疫グロブリン融合タンパク質は、該融合タンパク質のFe部分に選択的に結合する固定化プロテインAおよびプロテインGを含むカラムに溶液を通すことによって、精製することが可能である。例えば、Reis, K. J., et al., J. Immunol. 132:3098−3102 (1984)および PCT出願公開番号 W087/00329を参照せよ。融合ポリペプチドの溶離は、カオトロピック塩処理または酢酸(1M)水溶液によっておこなうことが可能である。
【0060】
あるいは、本発明にもとづくαGal融合ポリペプチドを、公知の方法を用いて化学的に合成することができる。ポリペプチドの化学合成が記載されており、例えば、種々のタンパク質合成方法が当該技術分野で普及しており、例えばペプチド合成装置を用いた合成が挙げられる。例えば、Peptide Chemistry, A Practical Textbook, Bodasnsky, Ed. Springer−Verlag, 1988、Merrifield, Science 232: 241−247 (1986)、 Barany, et al, Intl. J. Peptide Protein Res. 30: 705−739 (1987)、 Kent, Ann. Rev. Biochem. 57:957−989 (1988)、および Kaiser, et al, Science 243: 187−198 (1989)を参照せよ。ポリペプチドの精製は、標準的なペプチド精製技術を用いて、ポリペプチドが化学物質前駆体または他の化学物質を実質的に含まないようにして、おこなう。「化学物質前駆体または他の化学物質を実質的に含まない」という言い方は、ペプチドの合成に関与する化学物質前駆体または他の化学物質から該ペプチドが分離されるペプチド調製を包含する。一実施形態では、「化学物質前駆体または他の化学物質を実質的に含まない」という言い方は、化学物質前駆体または非ペプチド化学物質が約30%(乾燥重量で)未満、より好ましくは化学物質前駆体または非ペプチド化学物質が約20%未満、さらに好ましくは化学物質前駆体または非ペプチド化学物質が約10%未満、最も好ましくは化学物質前駆体または非ペプチド化学物質が約5%未満であるペプチドの調製を包含する。
【0061】
ポリペプチドの化学合成は、修飾アミノ酸または非天然アミノ酸(D−アミノ酸および他の小有機分子が挙げられる)の取り込みを促進する。対応のD−アミノ酸アイソフォームによるペプチド内の1つ以上のL−アミノ酸の置換は、酵素的加水分解に対するペプチドの耐性を高めるために、また生物学的に活性なペプチドの1種類以上の特性(すなわち、レセプター結合、機能的潜在能力、または作用の持続時間)を高めるために、用いることができる。例えば、Doherty, et al., 1993. J Med. Chem. 36: 2585−2594、Kirby, et al., 1993. J. Med. Chem. 36:3802−3808、Morita, et al., 1994. FEBS Lett. 353: 84−88、 Wang, et al., 1993. Int. J. Pept. Protein Res. 42: 392−399、Fauchere and Thiunieau, 1992. Adv. Drug Res. 23: 127−159を参照せよ。
【0062】
ペプチド配列への共有結合的架橋の導入は、立体構造的およびトポグラフィックにポリペプチド主鎖を抑制することができる。この戦略は、能力、選択性、および安定性が高まった融合ポリペプチドのペプチド類似体の開発に用いることができる。環状ペプチドの立体構造的エントロピーは、その直鎖状の対応物よりも低いので、特異的立体構造の採用は、非環式の類似体に対するエントロピーよりも環状類似体に対するエントロピーにおいてより少ない減少を伴っておこるので、それによって、結合のための自由エネルギーがより好ましいものとなる。大環化(macrocyclization)は、しばしば、ペプチドN末端とC末端との間、側鎖とN末端またはC末端との間[例えば、K3Fe(CN)6、pH8.5](Samson et al., Endocrinology, 137: 5182−5185 (1996))、または2つのアミノ酸側鎖間での、アミド結合によって達成される。例えば、DeGrado, Adv Protein Chem, 39: 51−124 (1988)を参照せよ。ジスルフィド架橋もまた、線形配列に導入されて、該配列の柔軟性を減少させる。例えば、Rose, et al., Adv Protein Chem, 37: 1−109 (1985)、Mosberg et al., Biochein Biophys Res Commun, 106: 505−512 (1982)を参照せよ。さらに、ペニシラミン(Pen、3−メルカプト−(D)バリン)によるシステイン残基の置換が、いくつかのオピオイド・レセプター相互作用の選択性を高めるのに用いられている。Liplcowski and Carr, Peptides: Synthesis, Structures, and Applications, Gutte, ed., Academic Press pp. 287−320(1995)。
【0063】
(超急性拒絶を処置または予防する方法)
本発明は、超急性拒絶(HAR)、例えば異種移植拒絶を処置または予防する方法も含む。そのような移植組織として、限定されるものではないが、そのような移植組織は含む腎臓、肝臓、皮膚、膵臓、角膜、または心臓が挙げられ、HARの意味にはレシピエントによる任意の抗体媒介移植拒絶が含まれる。移植臓器の超急性拒絶は、臓器がレシピエントの血行にさらされてから数秒または数分のうちに生ずる。臓器は、急速に蒼白になって、レシピエントの体内に残存する場合、壊死が生ずる。この超急性反応は、ドナー臓器に対して抗体が予め形成されたため、起こる。HARは、子宮内で多数の非自己抗原にさらされていることから、経産婦で最も一般的である。輸血回数が多いレシピエントも危険にさらされている。
【0064】
この方法は、被験体由来の生体試料を本発明のαGal融合ペプチドと接触させることを含む。生体試料は、例えば血液、すなわち全血または血漿である。試料は、抗体(例えば、抗血液型抗体)を含むことが知られている、または含むと考えられている。いくつかの態様では、生体試料をαGal融合ペプチドに接触させる前に、該生体試料を被験体から回収する。生体試料を、αGal融合ペプチド− 抗血液型抗体複合体の形成を可能とさせる条件下で、融合ペプチドに接触させる。αGal融合ペプチド−複合体を、もし存在するならば、抗血液型抗体を除去するために生体試料から分離し、さらに該生体試料を被験体に再注入する。HARの処置または予防は、被験体に対して本発明のαGal融合ペプチドを投与することによってもおこなわれる。
【0065】
被験体は、例えばいずれかのほ乳類(例えば、ヒト、霊長類、マウス、ラット、イヌ、ネコ、ウシ、ウマ、ブタ)である。被験体が異種移植を受ける前に、上記処置を施す。あるいは、被験体が異種移植を受けた後に処置を施す。
【0066】
当業者に公知の方法で、生体試料をαGal融合タンパク質に接触させる。例えば、血漿分離交換法または体外免疫吸着である。
【0067】
基本的に、HARに病原学的に関連しているどのような疾患でも、予防または処置が受けいられると考える。臓器移植組織の生存率が、本発明の方法によって処置していない臓器移植組織よりも高い場合、HARが処置または予防されている。移植組織の生存率が意味することは、レシピエントによって移植組織が拒絶される前の時間である。例えば、移植組織が該移植後の少なくとも1、2、4、または8週目に生存している場合に、HARが処置または予防される。好ましくは、移植組織は、3、6、13ヶ月生存する。より好ましくは、移植組織は2、3、5、またはそれ以上の年数生存する。
【0068】
(試料からαGal抗体を除去する方法)
本発明はまた、試料から抗αGal抗体を除去または減少させる方法を含む。試料は、血液または血漿等の体液である。あるいは、試料は生体組織、例えば心臓組織、肝臓組織、皮膚、または腎臓組織である。この方法を試料を本発明のαGal融合ペプチドと接触させることを含む。αGal融合ペプチド− 抗αGal抗体複合体の形成を可能とさせる条件下で、その試料をαGal融合ペプチドと接触させる。αGal融合ペプチド−抗体複合体を、もし存在するならば、抗αGal抗体を除去または減少させるために、生体試料から分離する。
【0069】
(αGal融合ポリペプチドまたはそれをコードする核酸を含む医薬組成物)
ABO融合タンパク質、または該融合タンパク質をコードする核酸分子(本明細書では「治療薬(therapeutics)」または「活性化合物(active compounds)」ともいう)ならびにその誘導体、フラグメント、類似体、およびホモログを、投与のために適当な医薬組成物に取り込むことができる。そのような組成物は、概して核酸分子、タンパク質、または抗体および薬学的に許容される担体を含む。本明細書で用いられるように、「薬学的に許容される担体(pharmaceutically acceptable carrier)」は、薬学的投与に適合性の、任意の、および全ての溶媒、分散媒、コーティング、抗細菌剤、抗真菌剤、等張性および吸着性遅延薬剤、その他を含むことを意図しており、薬学的投与(pharmaceutical addministration)と互換性を持つ。適当な担体は、当該分野での標準的参考書であるRemington’s Pharmaceutical Sciencesの最近の版に記載されており、本明細書ではこの文献を援用する。そのような担体または希釈剤の好ましい例として、限定されるものではないが、水、食塩水、フィンガー溶液(finger’s solutions)、デキストロース溶液、および5%ヒト血清アルブミンが挙げられる。不揮発性油のようなリポソームおよび非水溶ビヒクルも使用可能である。薬学的活性物質に対するそのような媒体および薬剤の使用は、当分野では周知である。任意の従来の媒体または薬剤が活性化合物と適合しない範囲を除いて、組成物でのそれらの使用を意図している。追加の活性成分もまた、組成物に取り込むことができる。
【0070】
本明細書に開示した活性剤もリポソームとして処方することができる。リポソームの調製は、公知の方法、例えばEpstein et al., Proc. Natl. Acad. Sci. USA, 82: 3688 (1985)、Hwang et al., Proc. Natl Acad. Sci. USA, 77: 4030 (1980)、ならびに米国特許第 4,485,045号および第4,544,545号に記載された方法でおこなう。循環時間を高めたリポソームが米国特許第5,013,556号に開示されている。
【0071】
特に有用なリポソームの生成は、ホスファチジルコリン、コレステロール、およびPEG誘導化ホスファチジルエタノールアミン(PEG−PE)を含む脂質組成物による逆相蒸着法によって、おこなうことができる。リポソームを所定のポア・サイズのフィルターから押し出されることで、所望の径を持つリポソームが得られる。
【0072】
本発明の医薬組成物は、目的とする投与経路に適合するように処方される。投与経路の例として、非経口(例えば、静脈内、皮内、および皮下)、経口(例えば、吸入)、経皮(例えば、局所)、経粘膜、および直腸投与が挙げられる。非経口、皮内、または皮下適用に用いる溶液または懸濁液は、以下の成分を含むことができる。すなわち、無菌希釈剤(例えば注射用蒸留水、食塩水、不揮発性油、ポリエチレングリコール、グリセリン、プロピレングリコール、または他の合成溶媒)、抗菌剤(例えば、ベンジルアルコールまたはメチル・パラベン)、抗酸化剤(例えば、アスコルビン酸または重硫酸ナトリウム)、キレート剤(例えば、エチレンジアミン四酢酸(EDTA))、緩衝剤(酢酸塩、クエン酸塩、またはリン酸塩)、および張性調整剤(例えば、塩化ナトリウムおよびデキストロース)である。pHは、酸または塩基(例えば塩化水素または水酸化ナトリウム)で調整することができる。非経口製剤を、ガラスまたはプラスチック製のアンプル、使い捨て注射器、または多人数用バイアルに封入することができる。
【0073】
注射可能な用途に適した医薬組成物として、滅菌水溶液(水溶性)もしくは分散液、または注射可能な滅菌溶液もしくは分散液の即席調製用の滅菌粉末が挙げられる。静脈内投与用に関して、適当な担体として、生理的塩類溶液、静菌水、Cremophor EL(商標)(BASF、Parsippany、N.J.)、またはリン酸緩衝食塩水(PBS)が挙げられる。すべての場合において、上記組成物は滅菌されていなければならず、また注射が容易にとなる範囲で流動的でなければならない。それは、製造および貯蔵の条件下に安定でなければならず、また細菌および菌類等の微生物による汚染作用から保護されなければならない。担体を、例えば、水、エタノール、ポリオール(例として、グリセロール、プロピレングリコール、および液体ポリエチレングリコール)を含む溶媒または分散媒体、ならびにそれらの適当な混合物とすることができる。適当な流動性は、例えば、レシチン等のコーティングを使用することによって、分散液の場合は必要な粒径を維持することによって、および界面活性剤を使用することによって維持することができる。微生物の作用を防止することは、種々の抗菌剤および抗かび剤、例えば、パラベン、クロロプタノール、フェノール、アスコルビン酸、およびチメロサールによって達成可能である。多くの場合、等張剤、例えば糖、ポリアルコール(例として、マンニトール、ソルビトール)、塩化ナトリウム等が組成物中に含まれていことが好ましい。注射可能な組成物の長期にわたる吸着は、吸着を遅延させる薬剤(例えばモノステアリン酸アルミニウムおよびゼラチン)を組成物に含有させることによって達成することができる。
【0074】
注射可能な滅菌溶液の調製は、必要に応じて上記に列挙される成分の1つまたは組み合わせとともに、適当な溶媒中に活性化合物(例えばABO融合タンパク質)を所望量取り込み、続いて濾過滅菌することによっておこなうことができる。通常、分散液は、塩基性分散媒と上記に列挙したうちの所望の他成分とを含む滅菌ビヒクルに活性化合物を取り入れることによって、調製される。注射可能な滅菌溶液を調製するための滅菌粉末の場合、調製法は、事前に濾過滅菌した溶液から活性成分プラス任意の追加の所望成分の粉末を生ずる真空乾燥および凍結乾燥である。
【0075】
経口組成物は、一般に不活性希釈剤または食用の担体を含む。それをゼラチンカプセルに封入または錠剤に圧縮することができる。経口治療薬投与の目的で、活性化合物を賦形剤とともに取り入れることができ、錠剤、トローチ、またはカプセルの形で使用することができる。経口組成物の調製は、流動性の担体を使用しておこなうこともでき、うがい薬として試用することができる。この場合、流動担体の化合物を経口投与し、口の中を洗い、吐き出すか、または飲み込む。薬学的に互換の結合剤および/または補助材料を組成物の一部として含むことができる。錠剤、ピル、カプセル、トローチ等は、以下の成分のいずれかまたは類似の性質の化合物を含むことができる。すなわち、バインダー(例えば、微結晶性セルロース、トラガカントゴムまたはゼラチン)、賦形剤(例えば、澱粉またはラクトース、崩壊剤(例えばアルギン酸)、プリモゲル(Primogel)、またはトウモロコシ澱粉)、滑沢剤(例えば、ステアリン酸マグネシウムまたはステローツ(Sterotes))、流動促進剤(例えば、コロイド状二酸化ケイ素)、甘味料(例えば、スクロースまたはサッカリン)、あるいは香料(例えば、ペパーミント、メチルサリチレート、またはオレンジ香料)である。
【0076】
吸入投与では、適当な推進体(例えばガス(例えば二酸化炭素)を含む加圧容器もしくはディスペンサ、または噴霧器からのエアゾール・スプレーのかたちで化合物を送達する。
【0077】
全身投与は、経粘膜または経皮手段によってもおこなうことができる。経粘膜もしくは経皮投与のために、透過すべきバリヤーに対する適当な浸透剤が処方に用いられる。そのような浸透剤は周知であって、例えば、経粘膜投与用に、該浸透剤として、界面活性剤、胆汁酸塩、およびフシジン酸誘導体が挙げられる。経粘膜投与は、鼻噴霧または坐薬を用いることにより達成することができる。経皮投与のために、活性化合物は周知の軟膏、塗剤、ゲル類、またはクリームとして処方される。
【0078】
上記化合物を、直腸送達のための坐薬(例えば、カカオ脂および他のグリセリド等、従来の坐剤基剤による)または保持浣腸の形で調製することもできる。
【0079】
一実施形態において、活性化合物は担体とともに調製され、該担体は、身体から化合物が迅速に除去されないように保護するもので、例えばインプラントおよびマイクロカプセル化した送達系等の徐放製剤である。エチレンビニルアセテート、ポリ無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル、およびポリ乳酸等の生分解性の生体適合性ポリマーを使用することができる。そのような製剤の調製のための方法は、当業者にとって明らかである。上記材料は、Alza CorporationおよびNova Pharmaceuticals, Inc.から商業的に入手することができる。リポソーム懸濁液(ウイルス抗原に対する単クローン抗体により感染細胞を標的化したリポソームを含む)を、薬学的に許容される担体として使うこともできる。これらは、米国特許第号4,522,811号に記載されているように、当業者に知られている方法によって調整することができる。
【0080】
いくつかの実施形態では、経口のまたは非経口組成物は、投与の容易さと投薬量の均一性とのために投薬量単位形状で処方される。本明細書で用いられるように投薬量単位の形態は、処置される被験体のための単位投薬量として適している物理的に分離した単位をいう。すなわち、各々の単位は、必須の薬学的担体に関連して所望の治療効果を得るために計算された所定量の活性化合物を含む。本発明の投薬量単位形態の仕様は、活性化合物のユニークな特徴、達成すべき特定の治療効果、および個人の治療のためのそのような活性化合物を合成する当該技術分野固有の限界に指図され、かつ直接的に依存している。
【0081】
本発明の核酸分子をベクターに挿入して遺伝子治療ベクターとして用いることができる。遺伝子治療ベクターは、例えば静脈注射、局所投与(例えば、米国特許第5,328,470号を参照せよ)、または定位的注射(例えば、Chen, et al., 1994. Proc. Natl. Acad. Sci. USA 91: 3054−3057)によって達成することができる。 遺伝子治療ベクターの製剤は、許容可能な希釈剤に遺伝子治療ベクターを含むことができ、あるいは遺伝子送達ビヒクルが包埋される徐放マトリックスを含むことができる。あるいは、完全な遺伝子送達ベクターを、組換え型の細胞(例えばレトロウイルス・ベクター)からインタクトなかたちで生ずることができる場合は、製剤は遺伝子送達系を生ずる1種類以上の細胞を含むことができる。
【0082】
必要に応じて、徐放製剤を調製することができる。徐放製剤の適当な例として、抗体含有固体疎水性ポリマーの半透性マトリックスが挙げられ、該マトリックスは、膜等の造形品の形状、またはマイクロカプセルの形状である。徐放性マトリックスの例として、ポリエステル類、ヒドロゲル(例えば、ポリ(2−ヒドロキシエチルーメタクリレート)、またはポリ(ビニルアルコール))、ポリラクチド(米国特許第3,773,919号)、L−グルタミン酸とγエチル−L−グルタミン酸塩との共重合体、非分解性エチレンビニルアセテート、分解性乳酸−グリコール酸共重合体(例えば、LUPRON DEPOT(商標)(乳酸−グリコール酸共重合体およびロイプロイドアセテートから構成される注射可能なミクロスフェア )、およびポリ−D−(−)−3−ヒドロキシ酪酸が挙げられる。エチレン−ビニルアセテートおよび乳酸グリコール酸のようなポリマーが100日以上にわたって分子の放出を可能にする一方で、特定のヒドロゲルはより短時間のあいだにタンパク質を放出する。
【0083】
医薬組成物を、投与のためのインストラクションと共に、容器、パック、またはディスペンサに入れることができる。
【0084】
(略語)
以下の略語が本明細書で用いられる。すなわち、ADCC、抗体依存性細胞障害; BSA、ウシ血清アルブミン; DXR、遅発性異物性拒否;ELISA、酵素結合免疫吸着検定法; FT、フコシル転移酵素;Gal、D−ガラクトース; GT、ガラクトシル基転移酵素;Glc、D‐グルコース; G1cNAc、D−Nアセチルグルコサミン; G1yCAM−1、グリコシル化依存性細胞接着分子−1;HAR, 超急性拒絶;Ig、免疫グロブリン; MAdCAM、粘膜アドレシン細胞接着分子; PAEC、ブタの大動脈血管内皮細胞; PBMC、末梢血単核細胞; PSGL−1、Pセレクチン糖タンパク質リガンド−1;RBC、赤血球; SDS−PAGE、ドデシル硫酸ナトリウム−ポリアクリルアミドゲル電気泳動法; Hex、ヘキソース; HexNAc、N−アセチル・ヘキソサミン; NeuAc、N−アセチル・ノイラミン酸; NeuGc、N−グリコリルノイラミン酸;およびHexNolはN−アセチル・ヘキソサミンの開いた(環でない)形状である。
【0085】
本発明を、以下の非限定的例で更に説明する。
【実施例】
【0086】
(実施例1:置換組換えPセレクチン糖タンパク質リガンド/免疫グロブリン融合タンパク質の一時的発現)
(一般的方法)
(細胞培養)
COS−7 m6細胞(35)およびSV40ラージT抗原不死化ブタ大動脈内皮細胞株PEC−A(36)を10%ウシ胎児血清(FBS)および25μg/ml硫酸ゲンタマイシンを含むダルベッコの改質イーグル培地(DMEM)で継代した。ヒトの赤血白血病細胞株K562とバーキットリンパ腫細胞株RajiをATCCから入手し、10%FBS、100IU/mlペニシリン、および100μg/mlストレプトマイシンを含むHEPES緩衝RPMI 1640で培養した。
【0087】
(発現ベクターの構築)
ブタα1,3GT(37−39)を、コーディング配列の5′末端、コザック(Kozak)翻訳開始コンセンサス配列、およびHind3制限部位に相補的な6つのコドンを持つフォワード・プライマーと、コーディング配列の3′末端、翻訳停止およびNot1制限部位に相補的な6つのコドンを持つリバース・プライマーとを用いて、ブタ脾臓cDNAからPCR増幅した。増幅1,3GTcDNAを、Hind3およびNot1(35)を用いて、CDM8のポリリンカーにクローニングした。P−セクレチンへの結合を媒介する高度にグリコシル化したムチン型のタンパク質であるPセレクチン糖タンパク質リガンド−1 (PSGL−1)(40)をコードする配列を、HL−60cDNAライブラリーからPCRによって得て、Hind3およびNot1を用いてCDM8にクローニングし、さらにDNAの塩基配列決定(シーケンシング)をおこなって確認した。ムチン免疫グロブリン発現プラスミドは、フレーム単位のPSGL−1の細胞外部分のPCR増幅cDNAをBamH1部位を介して、CDM7(Seed, B. et al)の発現カセットとして担持されたマウスIgG2のFe部分(ヒンジ、CH2およびCH3)に融合させることによって、構築した。
【0088】
(分泌ムチン/免疫グロブリン・キメラの生産および精製)
COSm6細胞を、DEAE−デキストラン・プロトコールおよび1μgのCsSI勾配生成プラスミドDNA/mlトランスフェクション・カクテルを用いてトランスフェクトさせた。COS細胞を、空ベクター(CDM8)、PSGL1/mIgG2bプラスミド単独またはα1,3GTコード化プラスミドとの組み合わせにより、約70%密集度でトランスフェクトさせた。トランスフェクション細胞を、トランスフェクションの翌日にトリプシン処理して新しいフラスコへ移した。約12時間にわたる付着の後で、培地を捨て、細胞をリン酸緩衝食塩水(PBS)で洗い、その後、新たに7日間にわたって無血清AIM−V培地(cat.nr. 12030, Life technologies Inc.)で培養した。培養後、上清を回収し、デブリスを遠心(1,400xg、20分)により沈殿させ、NaN3を0.02%まで添加した。PSGLI/mIgG2b融合タンパク質を、4℃で一晩、ヤギ抗マウスIgGアガロース・ビーズ(A−6531、Sigma)上で転倒回転させて精製した。ビーズをPBSで洗い、続いてSDS−PAGEおよびウエスタンブロット分析、またはヒトAB血清および精製ヒト免疫グロブリンの吸着に用いた。
【0089】
(ヒトIgG、IgM、およびIgAの精製)
ヒトIgG、IgM、およびIgAを、ヤギ抗ヒトIgG(Fe特異的、A−3316,Sigma)、IgM(μ鎖特異的、A−9935、Sigma)、およびIgA(α鎖特異的、A−2692、Sigma)アガロース・ビーズを用いて、ヒトAB血清(20人を上回る人数の健康血液ドナーからプールした)から精製した。手短に言うと、5mlのスラリー(2.5ml充填ビーズ)を、10mm直径のカラムに注入し、PBSで洗浄した。プールしてあるヒトAB血清10mlを、ペリスタル型ポンプを使用して1ml/分で適用し、カラム容量のPBSのPBSで数回洗浄し、1ml/分の流速で0.1Mグリシン、0.15M NaCl、pH2.4により溶離させた。1mlのフラクションを、0.7mlの中和緩衝液(0.2Mトリス/塩酸、pH 9)を含んでいる試験管に回収した。280nmの吸着を分光光度的に読み取り、タンパク質を含んでいる試験管をプールし、1%PBSに対して透析し、さらに凍結乾燥した。凍結乾燥免疫グロブリンを蒸留水に再懸濁し、その濃度を1gGについては16mg/ml、IgAについては4mg/ml、さらにIgMについては2mg/mlに合わせてた。
【0090】
(SDS−PAGEおよびウエスタン・ブロット分析)
SDS−PAGEは、垂直型Mini−Protean II電気泳動システム(Bio−Rad, Herculus, Calif.)(41)を用いて5%の濃縮用ゲルと6または10%の分離ゲルとによるLeammliの方法で、おこなった。Mini Trans−Blot電気泳動トランスファー・セル(Bio−Rad, Herculus, Calif.) (42)を用いて、分離タンパク質をHybond(商標)−Cエクストラ・メンブレン(Amersham)上にブロッティングした。タンパク質ゲルの染色を、製造元の指示(Bio−Rad, Herculus, Calif.)に従って、銀染色キットを使用しておこなった。3%BSA含有PBSで少なくとも2時間にわたってブロッキングした後、上記メンブレンを、室温で、0.2mMCaCl2含有PBS(pH6.8)に1μg/mlの濃度で希釈されたペルオキシダーゼ共役バンデイレイア・シンプリシフォリア(Bandereia simplicifolia)イソレクチンB4(L−5391、Sigma)により、2時間プロービングした。メンブレンをPBS(pH6.8)で5回洗浄し、製造元(Amersham)の指示に従ってECL(商標)キットを用いたケミルミネセンスで結合レクチンを視覚化した。
(抗マウスIgFcELISAによるPSGLb1/mgG2bの定量化)
吸着前後の細胞培養上清の融合タンパク質濃度の測定は、融合タンパク質を親和性精製多クローン性ヤギ抗マウスIgGFc抗体(cat.nr. 55482, Cappel/Organon Teknika, Durham, N.C.)で捕獲する96穴ELISAアッセイによっておこなった。3%BSA含有PBSでブロッキングした後、融合タンパク質を捕獲し、O−フェニレンジアミン二塩化水素化物(Sigma)を基質として用いてペルオキシダーゼ共役親和性精製多クローン性抗マウスIgGFc抗体(cat.nr. 55566, Organon Teknika, Durham, N.C.) で検出した。プレートを492nmで読み取り、AIMV無血清培地に再懸濁した精製マウスIgGFcフラグメント((cat.nr. 015−000−008, Jackson lmmunoResearch Labs., Inc., West Grove, Pa.)の希釈シリーズを用いてELSAのキャリブレーションをおこなった。
【0091】
(ブタ大動脈血管内皮細胞ELISA)
PEC−A細胞を、ゼラチン・コーティング96穴プレート(Nunclon, Denmark)に15,000細胞/ウエルの密度で播種し、AIMV無血清培地で48時間インキュベートした。プレートを、0.15MNaCl2含有0.02%Tween20で5回洗浄し、50μl/ウエルの精製ヒトIgG、IgM、およびIgAを含むPBSで、各々の開始濃度を8、1、および2mg/mlとして、室温で1時間培養した。上記のようにプレートを再び洗浄し、50μlアルカリ・ホスファターゼ共役ヤギ抗ヒトIgG(γ鎖特異的;A3312、Sigma)、IgM(μ鎖特異的;A1067、Sigma)、およびIgA(α鎖特異的;A3062、Sigma)F(ab)’2フラグメント含有PBS(1:200希釈)を添加し、室温で1時間にわたりインキュベートした。プレートを上記のように洗浄し、基質p−ニトロフェニル・リン酸(Sigma104−105)でインキュベートし、405nmで読み取った。
【0092】
(ブタ大動脈血管内皮細胞細胞毒性アッセイ)
PEC−A ELISAについて記載したように、PEC−A細胞を96穴プレートに播種して培養した。48時間培養した後、細胞をNa251CrO4(cat.nr. CJS4、Amersham)1μCi/ウエルで37℃、1時間にわたりロードし、AIMV培地で3回洗浄した。15μlの連続希釈した吸着または非吸着ヒトAB血清または精製ヒトIgG、IgA、もしくはIgM抗体を、補体源としての50μlのウサギ血清(Cat. no. 439665、Biotest AG, Dreieich, Germany)とともに添加した。5%CO2雰囲気下、37℃で4時間インキュベートした後、上清をSkatron上清回収システム(Skatro Instruments, Norway)を用いて収集し、ガンマ・カウンター(1282 Compugamma, LKB Wallac)で分析した。各血清およびIg試料を三重反復分析した。パーセント致死を、最大放出と自然発生的放出との差で割った自然発生的放出で差し引いた測定放出として、計算した。
【0093】
(抗体依存性細胞毒性(ADCC))
ヒトPBMCを、南(South)病院(ストックホルム)の血液バンクで、健康なドナーから調製した新鮮な軟膜 (バフィー・コート)を単離した。6mlのバフィー・コートと、1mg/mlBSAおよび3.35mg/mlEDTAを含む15mlのPBSとを、50mlポリプロピレン管内で混合した。10分間にわたり500gで遠心した後、血小板リッチな上清を捨てて、6mlの下部相を6mlのハンクス液(HBSS)と混合し、さらに6mlLympoprep(商標)(Nycomed Pharma AS)で下層を形成した。遠心後(800xg、20分)、界面を新しい管に移し、HBSSで3回洗浄し、さらに無血清AIMV培地に再懸濁した。エフェクター細胞調製の最終ステップは、37℃および5%CO2で1時間インキュベートしてプラスチック粘着細胞を取り除いた組織培養フラスコに、PBMCを移すことであった。標的細胞は、上記のように保ったK562およびRaji細胞、またはPEC−A細胞であり、これらの細胞をアッセイの前日にトリプシン処理し、その後AIMV無血清培地で培養することでプラスチック表面への再粘着を防いだ。アッセイの時点で、PEC−A細胞をフラスコの底から洗い落とした。標的細胞をNa251CrO4、100μCi/1x106細胞で、37℃で1時間にわたりロードし、HBSSで3回洗浄し、さらにAIMVに再懸濁することで、最終濃度を5x104/mlとした。5、000個の標的細胞を、2倍希釈で50:1から6.25:1の範囲にあるエフェクター細胞(E):標的細胞(T)比で、10%加熱不活性化ヒトAB血清有りまたは無しの200μlAIMV培地に含まれたエフェクター細胞がある各ウエルに加えた。
【0094】
自然発生的放出を、エフェクター細胞無しのAIMV培地200μlでインキュベートした5,000個の標的細胞を有するウエルで読み取り、最大放出を、AIMV培地100μlで5,000個の標的細胞を100μlの5%TritonX−100とともにインキュベートしたウェルで読み取った。各E:T比を三重反復分析した。37℃で4時間インキュベートした後、上清をSkatron上清回収システム(Skatro Instruments, Norway)を用いて収集し、ガンマ・カウンター(LKB Wallac)で分析した。パーセント致死を、最大放出と自然発生的放出との差で割った自然発生的放出で差し引いた測定放出として、計算した。
【0095】
(結果)
(PSGL1/mIgG2b融合タンパク質の発現および特徴付け)
ベクター・プラスミドCDM8、PSGL1/mIgG2bプラスミド、またはブタα1,3GTプラスミドとともにPSGL1/mIgG2bプラスミドによりトランスフェクションしたCOS−7m6細胞由来の上清を、トランスフェクション後、約7日目に回収した。分泌ムチン/Ig融合タンパク質を、抗マウスIgGアガロース・ビーズ上での吸着によって精製し、検出のためにバンデイレイア・シンプリシフォリア(Bandereia simplicifolia)イソレクチンB4 (BSA IB4)を用いたSDS−PAGEおよびウエスタンブロット分析にかけた。図1に示すように、融合タンパク質は、銀による染色が相対的に弱い145kDaの見かけ分子量を持つ広帯域として、還元条件下で移動した。大きさの不均一性(約125ないし165kDa)および低染色性は、高グリコシル化ムチン型タンパク質(43,44)の挙動に関して既におこなわれた観察と一致する。融合タンパク質は、おそらくホモ二量体として生産される。なぜなら、非還元条件下でのSDS−PAGEによって、見かけ分子量が250kDaを上回る二重のバンドが見られたためである。PSGL1/mIgG2bプラスミド単独またはα1,3GTプラスミドとともにトランスフェクションした同一数のCOS−7m6細胞に由来する2種類の上清から親和性精製された融合タンパク質の量は、同じであった。BSA IB4によってエレクトロブロッティング・メンブレンをプロービングすることで、ブタα1,3GTによるトランスフェクション後に得られた融合タンパク質の強い染色が明らかになった(図1)。しかし、COS細胞がシミアン(Simian)サル(α1,3活性が欠けた旧大陸サル)由来であるという事実にもかかわらず、α1,3GT cDNAの同時トランスフェクションなしでCOS−7m6細胞で生産されたPSGL1/mIgG2bもまたBSA IB4による染色が弱かった。このことは、BSA IB4レクチンがまさしくGa1α1,3Galエピトープ(45)よりもわずかに広い特異性を持つことを示している。それにもかかわらず、ブタα1,3GTcDNAの同時トランスフェクションは、高度にGalα1,3Gal置換PSGL1/mIgG2b融合タンパク質の発現を支持した。
【0096】
トランスフェクションしたCOS細胞の上清中、および吸着後のヤギ抗マウスIgGアガロース・ビーズ上でのPSGL1/mIgG2bの定量化。トランスフェクションしたCOS細胞の上清に含まれるPSGL1/mIgG2bの量を定量するために、サンドイッチELISAを用いた。一般に、材料および方法で説明したように、70%コンフルエンスでCOS細胞を含む5本の培養フラスコ(260mlフラスコ、Nunclon(商標))をトランスフェクトさせ、かつインキュベートした。フラスコ1本あたり10mlのAIMV培地中で7日間にわたるインキュベーション期間の後、培地を回収した。そのようなトランスフェクションからの上清に含まれる融合タンパク質の濃度を、抗マウスIgGアガロース・ビーズ(50μl充填ビーズに対応)の100μlゲル・スラリー上での吸着後の異なる容量の上清と同様に、精製マウスIgGFcフラグメントで較正したELISAによって測定した(図2)。上清に含まれるPSGL1/mIgG2bの濃度は、150ないし200ng/μlの範囲内であり、この特定の実験では、その濃度は約160ng/μlであった(図2A、非吸着カラム)。50pl充填抗マウスIgGアガロース・ビーズ上に吸着した後の2、4、および8mlの上清に残るPSGL1/mIgG2bの濃度は、それぞれ32、89、および117ng/μlであった。このことは、2、4、および8mlの上清由来の50μlの充填抗マウスIgGアガロース・ビーズ上に吸着されている260、290、および360ngのPSGL1/mIgG2bに一致する。B.シンプリシフォリア(B.simplicifolia)1134レクチンによるウエスタン・ブロット分析によって、50μlの充填ビーズが1mlの上清からPSGL1/mIgG2b融合タンパク質を吸着しうること、また2mlではやっと気づくほどのレベル(不図示)であることが明らかになった。
【0097】
固定化Galα1,3Gal−置換PSGL1/mIgG2bの吸着能力。PSGL1/mIgG2bプラスミド単独で、またはブタα1,3GTcDNAとともにトランスフェクションしたCOS細胞由来の上清20mlを抗マウスIgGアガロース・ビーズのゲル・スラリー500μlと混合した。しっかりと洗浄した後、ビーズを等量ずつ分け、100μlのゲル・スラリー(50μl充填ビーズ)を0.25、0.5、1.0、2.0、および4.0mlのプールされた補体減少ヒトAB血清と混合し、4℃で4時間にわたり転倒回転させた。PSGL1/mIgG2bおよびGalα1,3Gal置換PSGL1/mIgG2b上での吸着の後、4時間51Cr放出アッセイを用いてウサギ補体の存在下、血清をブタ内皮細胞細胞毒性についてアッセイした(図3)。図3に示すように、各希釈ステップで約300ngのPSGL1/mIgG2b(上記参照)を担持するビーズ100μlが4および2mlのAB血清の細胞毒性を減ずることができ、また1ml以下のヒトAB血清に存在する細胞毒性を完全に吸着することができる。注意すべきことは、非Galα1,3Gal置換PSGL1/mIgG2bの同一量が0.25ml吸着ヒトAB血清の細胞毒性をわずかだけ減少させることである(図3)。
【0098】
(補体依存ブタ内皮細胞細胞毒性および結合に対するGalα1,3Gal置換PSGL1/mIgG2bの効果)
PSGLI/mlgG2bが個々のヒト免疫グロブリンクラスを吸収することができた効率を調べるために、抗ヒトIgG、IgM、およびIgAアガロース・ビード上で、免疫アフィニティークロマトグラフィーによってヒトAB血清から、ヒトIgG、IgM、7およびIgAを精製した。単離後、IgAを抗IgGおよびIgMカラムに通過させ、IgGおよびIgMの痕跡を取り除く。この手順を、同様に他のIgクラスについても実行した。Ig分画純度は、SDS−PAGEでチェックした(図4)。正常血清で見いだされる濃度で、ヒトIgGおよびIgM(IgAではない)をウサギ補体の存在下、PEC−Aに対して細胞毒性を示した(図5)。IgGおよびIgM分画に存在する細胞毒性は、Galα1,3Gal−置換PSGL1/mIgG2b上での吸着によって、完全に除去された。IgA分画によって示される細胞毒性の欠如がPEC−Aに対するヒトIgA抗体の結合の欠如によるものかどうかを調べるために、細胞ELISAを、結合IgG、IgM、およびIgA抗体を検出するために細胞毒性アッセイで使用された同一Ig分画で実施した。アルカリホスファターゼ共役クラス特異的F(ab)′2フラグメントを2次抗体として用いた。IgGおよびIgMの細胞毒性がGalα1,3Gal−置換PSGL1/mIgG2bの吸着によって完全に取り除かれても、IgGに関しては70%(30から70%の範囲)を上回って、IgMに関しては55%(10から55%の範囲)を上回っては、結合は決して減少しなかった(図5)。ヒト免疫グロブリンAは明らかにPEC−Aと結合し、結合はGalα1,3Gal−置換PSGLI/mIgG2b上での吸着後に、わずかに減少しただけだった(29%以下)。したがって、IgA分画の細胞毒性の欠如は、IgAフラクションがPEC−Aに結合できないことによって説明することはできず、補体を活性化させることによると思われる。
【0099】
(ブタ内皮細胞のADCCに対するGalα1,3Gal−置換PSGL1/mIgG2bの効果)
いくつかのアッセイが無血清条件下でおこなわれ、K562およびRaji細胞と比較した場合、PEC−Aは新たに単離したPBMCによって致死を指示する中間の感度を有し、K562はヒトNK細胞による致死に対して感度が高く、Rajiは鈍感であった(図6A)。10%ヒト補体不活性化AB血清の存在下、致死率がほぼ倍となり、以前の公表データ(30)と一致して生体外(in vitro)でのADCC効果が支持される(図6B)。しかし、全てのPEC−A細胞毒性抗体(上記参照)を取り除くことが知られている条件下で、もし血清がGalα1,3Gal−置換PSGL1/mgIgG2bによって吸着されると、致死率は無血清条件下で見られる致死率よりもわずかに低いレベルまで減少する。一方、Galα1,3Galエピトープを持たないPSGL1/mIgG2b融合タンパク質それ自体は、ヒトAB血清存在下で致死率の増加を引き起こすことはできない(図6B)。これらのデータは、Galα1,3Gal特異性による抗ブタ抗体が生体外(in vitro)で抗体依存性細胞性細胞毒性を支持することができるということ、またそれが効果的に補体結合の細胞毒性の抗ブタ抗体を除去するちょうどその時、Galα1,3Gal−置換PSGL1/mIgG2b融合タンパク質はこれらの抗体を効果的に除去すること、という考えを支持する。
【0100】
(参考文献)
【0101】
【数1】

【0102】
【数2】
【0103】
【数3】
【0104】
【数4】
【0105】
【数5】
【0106】
【数6】
(実施例2: 置換された組換えPセレクチン糖タンパク質リガンド/免疫グロブリン融合タンパク質の安定な発現)
(一般法)
(細胞培養)
CHO−K1、COS7m6、293T、およびブタ大動脈内皮細胞株PEC−A(Khodadoust, M. M. et al., 1995)を10%胎児ウシ血清(FBS)および25μg/ml硫酸ゲンタマイシンを含むダルベッコ改変イーグル培地(DMEM)中で培養した。選択培地は、下記に示すように、ピューロマイシン(cat. no. P7255; Sigma, St. Louis, MO. 63178)、ハイグロマイシン(cat. no. 400051; Calbiochem, La Jolla, CA 92039)、およびG418(cat. no. G7034; Sigma, St. Louis, MO 63178)を含有していた。
(発現プラスミドの構築)
記載(Liu, J. et al., 1997)されるように、ブタα1,3Ga1T(Gustafsson, K. et al., 1994)およびPSGL−1/mIgG2b発現プラスミドを構築した。フォワードおよびリバース・プライマーとして、それぞれcgcgggctcgagatgaagatattcaaatgtおよびcgcggggcggccgctcatgatgtggtagtgagatを用いて、HL60 cDNAライブラリからC2 GnTI cDNAをPCRによって増幅した。安定なトランスフェクタントを生成するために用いられるベクターは、両方向性であり、ポリリンカーの上流のEF1αプロモーター、スプライス・ドナーおよびアクセプター部位、ならびにSV40の両方向性ポリ(A)付加シグナルを有していた。この転写ユニットとは反対の配向で、かつ逆方向からのポリ(A)シグナルを用いているのが、HSV TKプロモーターに続いてピューロマイシン・アセチル転移酵素(EF1α/PAC)、ハイグロマイシンb(EF1α/Hyg)、およびネオマイシン(EF1α/Neo)耐性遺伝子(N. Chiu, J. Holgersson and B. Seed)のコーディング配列からなる第2の転写ユニットであった。HindIIIおよびNotIを用いて、ブタα1,3GalTおよびPSGL−l/mIgG2bのcDNAをEF1α/HygおよびEF1α/PACベクターにそれぞれ交換した。XhoIおよびNotIを用いて、C2GnTI遺伝子をEF1α/Neoに交換した。
【0107】
(DNAトランスフェクションおよびクローン選択)
付着CHO−K1、COS7m6、および293T細胞を75cm2T型フラスコに播種し、約24時間後に、70ないし80%の細胞集密(cell confluency)でトランスフェクションした。改変ポリエチレンイミン(PEI)トランスフェクション法をトランスフェクションに対して用いた(Boussif, O. et al., 1995; He, Z. et al., 2001)。トランスフェクション後24時間に、各T型フラスコ中の細胞を5つの100mmペトリ皿に分け、選択培地中でインキュベートした。選択培地中のピューロマイシンの濃度は、CHO−K1、COS7m6、および293T細胞に対して、それぞれ6.0、1.5、および1.0μg/mlであった。CHO−K1、COS7m6、および293T細胞に対して、それぞれ550、50、および100μg/mlのハイグロマイシンb濃度を用い、CHO−K1細胞に対して、900μg/mlのG418濃度を用いた。選択培地を3日毎に変えた。約2週間後に、薬剤耐性クローンが形成した。クローンを顕微鏡下で同定し、ピペットマンを用いて手で採取した。選択したコロニーを、選択薬剤の存在下96穴プレート中でさらに2週間培養した。細胞が80ないし90%集密に達したとき細胞培養上清を回収し、ヤギ抗マウスIgG Fc抗体を用いて、ELISA、SDS−PAGE、およびウエスタン・ブロット法によって、PSGL−1/mIg2bを評価した。最も高いPSGL−1/mIgG2b発現を有するCHO−K1、COS7m6、および293Tクローンを、プラスミドをコードするブタα1,3Ga1Tでトランスフェクトし、ハイグロマイシン含有培地中で選択した。ヤギ抗マウスIgG Fc抗体と、末端αGalを認識するGSA I
1B4−レクチンとの両方を用いて、ELISA、SDS−PAGE、およびウエスタン・ブロットによって、耐性クローンを単離および特徴付けした。PSGL−1/mIgG2b上で相対的に高いαGal発現を有する2つのCHOクローンをC2 GnTIでさらにトランスフェクトして、G418含有培地中で選択した。より多くの複合体O−グリカンを示すPSGL−1/mIgG2bの大きさの増加によって、C2 GnTIの発現が確認された。
【0108】
(SDS−PAGEおよびウエスタン・ブロット法)
垂直型Mini−Protean II電気泳動システム(Bio−Rad, Hercules, CA, USA)を使用して、5%スタッキング・ゲルおよび8%分離ゲルを用いて、Laemmliの方法(Laemmli, U. K., 1970)によってSDS−PAGEをおこなった。還元および非還元条件下で、試料を電気泳動にかけた。分離度を増加するために、4ないし15%勾配ゲル(cat.no. 161−1104; Bio−Rad, Hercules, CA, USA)または4ないし12%勾配gels(cat.no NP0322; Invitrogen, Lidingo, Sweden)を複数の実験で用いた。後者のゲルは、MES緩衝液(cat.noNP0002; Invitrogen)と組み合わせて用いた。高精度タンパク質標準(cat.no RPN756; Amersham Biosciences, Uppsala, Sweden)をタンパク質分子量測定のための基準として適用した。Rubyと組み合わせてPro Q Emerald 300糖タンパク質検出キット(cat.no P21855; Molecular Probes, Leiden, The Netherlands)を用いて、タンパク質ゲルを染色した。CCDカメラを搭載するFlour−S Max MultiImagerで、これらのゲルを可視化した。Mini TransBlot(Bio−Rad)電気泳動トランスファー・セルを用いて、Hybond C外膜(cat.no. RPN203E; Amershain Biosciences)またはニトロセルロース膜(cat.no LC2001; Invitrogen)上に分離されたタンパク質も電気泳動的にブロットした(Towbin, H. et al., 1979)。0.2Tween20を含むPBS中3%BSAで1時間ブロッキングした後、1μg/ml濃度に希釈したペルオキシダーゼ結合GSA I IB4−レクチン(cat.no. L−5391; Sigma)、1:1,000で希釈したペルオキシダーゼ結合ヤギ抗マウスIgG Fc抗体(cat.no. A−9917; Sigma)、および1:1,000で希釈したマウス抗PSGL−1抗体(クローンKPL−1、cat.no 557502; BD PharMingen, San Diego, CA)を用いて、膜を室温で1時間プロービングした。2次抗体は、1:50,000で希釈したペルオキシダーゼ結合ヤギ抗マウスIgG F(ab)′2(cat.no A 2304; Sigma)であった。ブロッキング緩衝液で、全ての希釈をおこなった。インキュベーション間および後で、0.2%Tween20を含有するPBSで膜を3回洗浄した。ECLキット(cat.no. RPN 2106; Amersham Biosciences)を用いて、製造元の指示にしたがい、化学発光によって結合レクチンおよび抗体を可視化した。
【0109】
(PSGL−1/mIgG2b上のαGalエピトープ密度および酵素免疫検定法を用いたPSGL−1/mIgG2bの定量)
二抗体サンドイッチELISAによって、細胞培養上清中の組み換えPSGL−1/mIgG2bの濃度と、その相対的α−Galエピトープ密度とを測定した。20μg/mlの濃度で、アフィニティー精製ポリクローナル・ヤギ抗マウスIgG Fc抗体(cat. nr. 55482; Cappel/Organon Teknika, Durham, NC)で96ウェルELISAプレートを一晩4℃で被覆した。PBS中の1%BSAを用いて、該プレートを1時間ブロッキングした。PSGL−1/mIgG2bを含有する上清を4時間インキュベートし、その後、0.5%(v/v)Tween20を含有するPBSで3回洗浄した。洗浄後、1:3,000希釈のペルオキシダーゼ結合抗マウスIgG Fc抗体(cat.no. A−9917; Sigma)または1:2,000で希釈したペルオキシダーゼ結合GSA I IB4−レクチン(cat.no. L−5391;Sigma)でプレートを2時間インキュベートした。3,3′,5,5′−テトラメチルベンジジンジハイドロクロライド(cat. nr. T−3405; Sigma, Sweden)を用いて、結合ペルオキシダーゼ結合抗体またはペルオキシダーゼ結合GSA−レクチンを可視化した。2M H2SO4によって、反応を停止し、450nmでプレートを読み取った。較正に対して、融合タンパク質生成のために用いられる培地中または1%BSAを含有するPBS中で再懸濁した精製マウスIgG Fcフラグメント(cat. Nr. 015−000−008; Jackson ImmunoResearch Labs., Inc., West Grove, PA)の希釈系列を用いて、PSGL−1/mIgG2b濃度を評価した。2つのELISAから得た相対的O.D.(GSA反応性/抗マウスIgG反応性)を比較することによって、α−Galエピトープ密度を測定した。
【0110】
(ブタ大動脈内皮細胞細胞毒性アッセイ)
記載(Liu, J. et al., 2003)されるように、ブタ大動脈内皮細胞(PAEC)細胞毒性アッセイを行った。細胞毒性を最大の40%(y=0.4)に減少させるために各細胞クローンから必要とされるPSGL−1/mIgG2bの量を、各融合タンパク質に対する測定値の直線回帰後に得た曲線を記述する式から推論し、その後対応するx値(マイクログラム吸着体)を算出した。
【0111】
(CHOクローンの撹拌フラスコ・バッチ培養)
6.0x107個の細胞(90ないし100%集密を持つ細胞を含む10個の175cm2T型フラスコを表す)を用いて、各バッチ培養を開始した。トリプシン(0.5mg/ml)・EDTA(0.2mg/ml)での消化後に、細胞を少量の培地中で再懸濁し、200xgで5分間遠心して、過剰なトリプシンを除去した。Burkerチャンバー
中の細胞を計数することによって細胞密度を測定し、培地を添加して、最終濃度3.0x105個細胞/mlにした。細胞懸濁物を1L撹拌フラスコに移し、速度60rpmで培養物を撹拌するために細胞回転装置(Integra Biosciences, Wallisellen, Switzerland)を用いた。単独またはC2 GnTIとの組合せでα1,3GalTを発現するCHO−K1細胞を分泌するPSGL−1/mIgG2bを、それぞれピューロマイシン(200μg/ml)またはピューロマイシン(200μg/ml)およびG418(500μg/ml)の存在下で培養した。細胞を2日毎に計数した。細胞密度が5.0x105個細胞/mlに達したとき、細胞密度がもう1度3.0x105個細胞/mlに等しくなるように新たな培地を添加した。細胞懸濁物量が1,000mlになるまでこれを繰り返した。その後、細胞生存率が50%に減少するまで、細胞を持続的に培養した。
【0112】
(組み換えPSGL−1/mIgG2bの精製)
20分間1,420xgで遠心することによって、堆積物から上清を取り除いた。取り除いた上清を、ヤギ抗マウスIgG(分子全体)−アガロース(cat.no. A 6531; Sigma)10mlを含有するカラムに流速0.5ml/分で通した。PBS120mlでの洗浄後に、3M NaSCN120mlで、結合融合タンパク質を溶出した。検出用の抗マウスIgGを用いたSDS−PAGEおよびウエスタン・ブロット法による分析に続いて、融合タンパク質を含有する管の内容物をプールした。PSGL−1/mIgG2bを含む画分を蒸留水に対して透析し、凍結乾燥し、蒸留H2Oの1ないし2ml中で再懸濁した。融合タンパク質の濃度をELISAによって測定した。より低い分子量の夾雑物を除去するために、FPLC(Pharmacia Biotech, Sweden)を用いて流速0.5ml/分でPBSで溶出させるHiPrep 16/60 Sephacryl S−200 HRカラム(cat.no. 17−1166−01; Amersham Biosciences, Uppsala, Sweden)上でのゲル濾過によって、融合タンパク質をさらに精製した。5ml画分を回収し、280nmでのUV分光測光によって、タンパク質を含有する管を同定した。SDS−PAGEおよびウエスタン・ブロット法によって、プールした画分を再び分析し、蒸留水中でプール、透析、および再懸濁した。
【0113】
(精製PSGL−1/mIgG2bからのO結合グリカンの化学的放出および過メチル化)
記載(Carlstedt, I. et al., 1993)されるように、β脱離によってオリゴ糖を放出させた。放出されたオリゴ糖を45℃窒素流動下で気化し、記載(Hansson, G. C. et al., 1993)されるように、わずかな修飾で、CiucanuおよびKerek(Ciucanu, I. et al., 1984)にしたがって、過メチル化した。
【0114】
(質量分析法)
LCQイオン・トラップ質量分析計(ThermoFinnigan, San Jose, CA)を用いて、陽イオンモードでのエレクトロスプレー・イオン化質量分析法(ESI−MS)を実行した。メタノール:水(1:1)中で試料を溶解し、質量分析計に流速5ないし10μl/分で導入した。窒素を空間電荷層ガスとして用いて、ニードル電圧を4.0kVに設定した。加熱キャピラリーの温度を200℃に設定した。合計10ないし20個のスペクトルを総計し、ESI−MSおよびESI−MS/MSスペクトルを産生した。
【0115】
(結果)
(異なる宿主細胞中でのα−Gal置換P−セレクチン糖タンパク質リガンド−1/マウスIgG2bの安定的な発現)
ピューロマイシンを添加した選択培地中での培養の15ないし20日後に、CHO−K1、COS7m6、および293T細胞の異なる大きさのコロニーを位相差顕微鏡によって同定した。該顕微鏡下で、各細胞型の192個のコロニーをピペットによって採取し、選択下での更なる増殖のために2つの96穴プレートに移した。IgサンドイッチELISAを用いて、個々のクローンの上清中の融合タンパク質濃度を評価し、31個のCHO−K1コロニー、8個のCOS7m6コロニー、および36個の293Tコロニーが抗マウスIgG Fc陽性であった。各細胞株由来のコロニーを分泌する上位5つを24ウェル・プレートに移動し、さらに増殖させた。最もよく発現するCHO−K1、COS7m6、および293Tクローンを、ハイグロマイシンB耐性遺伝子を保持するα1,3GaIT・コード・プラスミドでトランスフェクションした。ピューロマイシンおよびハイグロマイシンを用いて、α1,3GalT遺伝子を安定的に組み込んだPSGL−1/mIgG2b発現細胞を選択した。27個のCHO−K1コロニー、3個のCOS7m6コロニー、および31個の293Tコロニーが選択された。抗マウスIgGおよびグリフォニア・シンプリシフォリア(Griffonia simplicifolia)I IB4レクチンELISAで測定されるような融合タンパク質の濃度と、そのα−Galエピトープ置換の相対的レベルとにもとづいて、増殖すべきコロニーを選択した。ブタα1,3GalT有りまたは無しでCHO−K1(それぞれ、クローン5L4−1および10)、COS7m6(それぞれ、クローン51および2H5)、および293T(それぞれ、クローン14およびC)細胞中で発現するイムノアフィニティー単離PSGL−1/mIgG2bをSDS−PAGEおよびウエスタン・ブロット法によって特徴づけた(表1、図7)。全ての細胞株は、非還元条件下で、約300kDaの抗マウスIgG Fc反応性タンパク質を産生した(図7A)。先行の観察(Liu, J. et al., 1997; Liu, J. et al., 2003)と一致して、還元での半分の大きさへの減少によって示される(図7Aおよび図7Bを比較せよ)ように、PSGL−1/mIgG2bはニ量体として産生された。GSA I IB4レクチンを用いて、異なる細胞型で生成された融合タンパク質上のα−Galエピトープの存在を検出した(図7B)。CHO−K1(クローン5L4−1)、COS7m6(クローン51)、および293T(クローン14)中のα1,3Ga1Tの共発現は、レクチンによって検出されたように、融合タンパク質上のα−Galエピトープの発現に至った。α1,3GalT無しで293T細胞中で生成されたPSGL−1/mIgG2bのレクチン反応性は予想外であり、融合タンパク質上のGalili抗原以外のα−Gal残基の存在を示す(図7B)。α1,3GalTの存在下でCOSおよび293T細胞中で産生された融合タンパク質は、CHO−K1細胞中で産生された融合タンパク質より大きい大きさの糖形態を含有していた(図7B)。
【0116】
(表1 CHO、COS、および293T由来の細胞クローン)
【0117】
【表1】
(PSGL−1/mIgG2b上のα−Galエピトープ密度は、その生成のために用いられる宿主細胞に依存する)
CHO、COS、および293T細胞中で生成されたPSGL−1/mIgG2b上の相対的なα−Galエピトープ密度をELISAによって測定した(図8)。α1,3GalTの存在下でCOS細胞中で生成されたPSGL−1/mIgG2bは、α1,3GalT無しでCOS中で生成されたPSGL−1/mIgG2bと比べて、相対的O.D.(GSA反応性/抗マウスIgG反応性)で5.3倍の増加を示した(図8)。293T細胞に関しては、相対的O.D.で3.1倍の増加があり、CHO細胞に関しては、1.8倍の増加だけであった(図8)。ELISA結果は、イムノアフィニティ精製PSGL−1/mIgG2bのウエスタン・ブロット実験で見られる相対的GSAレクチン染色と一致していた(図7B)。
【0118】
(異なる宿主細胞中で生成されるPSGL−1/mIgG2bの抗ブタ抗体吸着効果は、そのα−Gal置換の程度と相関する)
ブタ大動脈内皮細胞細胞毒性を用いて、ブタα1,3GalTを共発現するCHO−K1(クローン5L4−1)、COS7m6(クローン5I)、および293T(クローン14)細胞中で産生されたPSGL−1/mIgG2bの、ヒト血液型AB血清の抗ブタ反応性抗体を吸着する能力を評価した(図9)。ヒトAB血清PAEC細胞毒性を最大40%に減少させるために、CHO細胞生成PSGL−1/mIgG2b9.1μgが必要であった(図9)。COSおよび293T細胞生成PSGL−1/mIgG2b関しては、PAEC細胞毒性を同レベルまで減少するために、それぞれ16倍および4倍少なく必要であった。さらに、α1,3GalT遺伝子の存在下でCHO−K1細胞中で生成されたPSGL−1/mIgG2bは、血液型AB血清のPAEC細胞毒性を36%をより下に減少することができなかった一方で、12および25%への減少がそれぞれ、非飽和条件下であっても、α1,3GalT発現COSおよび293T細胞中で生成されたPSGL−1/mIgG2bで見られた。
【0119】
(CHO細胞中のコア2β1,6GlcNAc転移酵素の共発現は、PSGL−1/mIgG2bαGalエピトープ密度および抗ブタ抗体吸着効果を向上する)
CHO−K1細胞分泌ムチン/Ig上のα−Galエピトープの数を増加させる試みでは、PSGL−1/mIgG2b、α1,3GalT、およびC2 GnTIを安定的に発現するCHO−K1細胞を樹立し、抗マウスIgG抗体およびGSA I IB4を用いて、ELISA、SDS−PAGE、およびウエスタン・ブロットによって、それらの細胞によって分泌されるPSGL−1/mIgG2bを分析した。PSGL−1/mIgG2bの明らかなMWは、コア2酵素の安定的な発現に続いて増加し、融合タンパク質上のより多くの複合体グリカンを示した(図10)。PSGL−1/mIgG2b上のα−Galエピトープ密度は、α1,3GalT無しでCHO−K1細胞中で生成されたPSGL−1/mIgG2bと比べて13.0倍の増加を示し、α1,3GalT単独を用いて生成されたPSGL−1/mIgG2bと比べて7.4倍の増加を示した(図8)。さらに、α1,3GalTおよびC2 GnTIを安定的に発現するaCHO−K1(CHO−C2−1−9)中で産生されたPSGL−1/mIgG2bの抗ブタ抗体吸着効果は、α1,3GalTを共発現する293TおよびCOS細胞中で産生されたPSGL−1/mIgG2bの吸着効果と同様であり(図9)、CHOクローン5L4−1中で生成されたPSGL−1/mIgG2bと比べて、ヒトAB血清のPAEC細胞毒性を最大40%に減少させるために10倍少ない融合タンパク質が必要であった。
【0120】
(O結合グリカン構造的特徴付けのための組み換えPSGL−1/mIgG2bの精製)
単独、ブタα1,3GalTとの組み合わせ、またはα1,3GalTおよびC2 GnTIとの組み合わせでPSGL−1/mIgG2bを発現する安定的にトランスフェクションしたCHO−K1細胞(それぞれ、クローン10、クローン5L4−1、またはクローンC2−1−9)の1L撹拌フラスコ培養物から、組み換えPSGL−1/mIgG2bを精製した。Oグリカン構造分析を干渉する可能性のある混入するグリコシル化タンパク質を完全に除去するために、抗マウスIgGアフィニティー・クロマトグラフィーおよびゲル濾過を含むツー・ステップ精製法を設定した。各細胞クローンから得た細胞上清2リットルのアフィニティー精製は、ELISAによって評価されたように、CHO−10、5L4−1、およびC2−1−9から、それぞれPSGL−1/mIgG2b2.2mg、1.2mg、および0.95mgを生じた。ゲル濾過カラム上での更なる精製は、それぞれ、0.22mg、0.19mg、および0.29mgの最終PSGL−1/mIgG2b収率を生じた。アフィニティーおよびゲル濾過カラムから溶出した画分をSDS−PAGEおよびウエスタン・ブロット法によって分析した(ここではクローン10に対して示す)。糖タンパク質染色キットをRubyと組み合わせて用いて、グリコシル化タンパク質と、非グリコシル化タンパク質とを検出し(図11Aおよび11B)、抗PSGL−1抗体は、PSGL−1/mIgG2bの存在を確認した(図11C)。この抗体は、PSGL−1/mIgG2b二量体を表す300kDa付近のバンド(図11Cレーン2および4ないし9)と強く結合した。還元形態の融合タンパク質由来の150kDa付近のバンドも見られ(レーン4ないし6)、融合タンパク質分解産物を表す可能性が最も高い60ないし70kDaの弱いバンドも見られる(レーン7ないし9)。図11Aおよび11Bでは、抗PSGL−1抗体で染色されていない300kDaバンドもレーン1および3で見られ、細胞培養培地由来のタンパク質を表す可能性が最も高い。このことは、アフィニティー精製上清中のその存在(レーン3)によっても裏づけされ、該タンパク質が抗Igアフィニティー・カラムに吸着されていないことを示す。しかし、抗PSGL−1抗体に染色されていない50ないし60kDaのMWを持つグリコシル化バンドをアフィニティー精製画分で見ることができる(図11Aレーン4)。このタンパク質も、恐らく細胞培養培地に由来するものであり、融合タンパク質とともにアフィニティー・カラムに吸着される。このタンパク質をゲル濾過によって除去し、該ゲル濾過の間、該タンパク質(図11Aおよび11B,レーン7ないし9)は、融合タンパク質(図11A,11B,および11C、レーン5ないし6)より遅れて溶出した。50ないし70kDa付近の付加的な非グリコシル化タンパク質をゲル濾過によって除去した(図11B、レーン4とレーン7ないし9とを比較せよ)。各クローンに対して、最も多い量の融合タンパク質を含有するゲル濾過画分をオリゴ糖放出のために選択した。クローン10に対して示すように(図11Aおよび11B、レーン5)、この画分は、いずれの有意な量の混入するタンパク質(グリコシル化または非グリコシル化)も含有していなかった。
【0121】
(精製組み換えPSGL−1/mIgG2bから放出される過メチル化オリゴ糖の質量分析法)
クローンCHO−10および5L4−1から放出された過メチル化オリゴ糖は、m/z895.4および1256.5付近のピークの2つの最も多数を占める群を持つ類似のMSスペクトルを与え(図12)、一方で、クローンC2−1−9によって産生されたPSGL−1/mIgG2bから放出されたO−グリカンの質量スペクトルは、より複雑なパターンを示した(図13)。タンデム質量分析法(MS/MS)によって、ESI−MSスペクトル中のイオンのオリゴ糖配列を導き出した。このように得られた配列および仮構造を表2に示す。下記に、我々は、α1,3GalTおよびC2 GnTIも発現するCHO細胞(C2−1−9)中で産生されたPSGL−1/mIgG2b上のO−グリカンのMS/MS分析の結果について記載する。MSおよびMS/MSスペクトル中のイオン全てをナトリウム化イオンとして検出した。
【0122】
(C2−1−9のMS/MS分析)
親スペクトル中の最も強度のピークは、一連のステップのMS/MSによって評価されるように、NeuAc−Hex−HexNol−HexN−Hex−Hex構造を表すm/z1548.7の偽分子イオン([M+Na]+)である(図14)。このイオンのMS2は、m/z951.3([M−NeuAc−Hex−O+NA]+)および1173.5([M−NeuAc+Na]+)の2つのより大きなフラグメント・イオンと、m/z506.1([M−Hex−Hex−HexN−NeuAc+Na]+)、620.2([NeuAc−Hex−O+Na]+)、690.2([Hex−Hex−HexN+Na]+)、751.3([M−Hex−Hex−NeuAc+Na]+または[M−Hex−NeuAc−Hex+Na]+)、881.3([M−Hex−Hex−HexN+Na]+)、969.5([M−NeuAc−Hex+Na]+)、および1330.7([M−Hex+Na]+)の複数のより小さなフラグメント・イオンとを与えた。m/z1173.5のフラグメント・イオンを単離して、MS3によって分析すると、m/z951.4、506.2、690.3、および751.5のフラグメント・イオンを生じた。より大きなピーク、すなわち951.4をMS4によってさらに分析し、m/z445.3([Hex−Hex+Na]+)、463.0([Hex−Hex−O+Na]+)、690.3 、および733.6([M−Hex−NeuAc−Hex−O+Na]+)のフラグメント・イオンを生じた。最終的に、MS4分析の優勢なフラグメント・イオン(690.3)をMS5によって分析した。これによって、末端Hex−Hexを表すm/z415.1および445.3の配列イオンを生じ、前者のイオンは、1つの酸素およびそのメチル基を消失していた。Hex−Hex−O構造も見いだされた(463.0)。さらに、内部Hex−HexN構造が、ヘキソースに結合する1つの酸素を持つもの(472.2)および持たないもの(454.0)で見られた。Hex−Hex−HexN構造からのO−Meの消失(660.1)、C−O−Meの消失(648.2)、およびN−C−O−Meの消失(619.4)も見られ、ここで、最後の1つは、恐らく内部HexNからのN−アセチル基の消失を表す。m/z533.2のより大きなフラグメント・イオンもMS5スペクトル中で見られた。このイオンは、最内側のHexNのクロス・リング・フラグメントに対応し(図14)、ヘキソースが1−4結合中のHexNに結合することを示す。この配列は、2型構造および末端Galで伸張されたシアル酸付加コア2と整合する可能性が最も高い。
【0123】
m/z1548.7のイオンを以外に、Galα1,3Galで終了すると考えられる2つの他の偽分子イオンが、m/z1578.7および1187.6で、クローンC2−1−9のESI−MSスペクトル中で見出された。m/z1578.7のイオンをMS2分析のために単離した。その結果は、NeuGc−Hex−HexNOl−HexN−Hex−Hex構造を示し、m/z1173.5([M−NeuGc+Na]+)、951.5([M−NeuGc−Hex−O+Na]+)、911.3([M−Hex−Hex−HexN+Na]+)、690.3([Hex−Hex−HexN+Na]+)、および676.3([Hex−Hex−HexN−Me+Na]+)のフラグメント・イオンを持つ。1173.5イオンのMS3分析は、m/z951.5のより大きなフラグメント・イオンと、m/z506.1([M−Hex−Hex−HexN−NeuAc+Na]+)、690.2、および969.3([M−NeuAc−Hex+Na]+)の複数のより小さなフラグメント・イオンとを生じた。m/z951.5のフラグメント・イオンを第4のステップ中のMS/MSによって分析し、m/z690.4の1つのより大きなフラグメント・イオンおよびm/z658.2(690.4−O−Me)のより小さなフラグメント・イオンを与えた。しかし、m/z1578.7のイオンのMS2スペクトルでは、m/z981.5および1203.4の未同定のフラグメント・イオンが観察された。m/z1203.4のイオンのMS3およびMS4分析は、m/z981.4、720.4、690.1、506.1、および688.3(720.1−O−Me)のフラグメント・イオンを生じた。MS3およびMS4スペクトル両方で見られるm/z720.4のイオンは、m/z690.1の特徴的なフラグメント・イオンより多い30質量単位であり、Hex−Hex−HexN配列を表す。残念なことに、m/z720.4のイオンの更なるMS/MS分析は可能でなかった。考えられる末端Galα1−3Galを持つESI−MSスペクトル中の他の偽分子イオン(図13)がm/z1187.6で観察された。このイオンのMS2実験は、Hex−HexNol−HexN−Hex−Hexまたは2型伸張および末端Galを持つコア2と整合するm/z969.5([M−Hex+Na]+)、951.4([M−Hex−O+Na]+)、756.2([M−Hex−Hex+Na]+)、690.3([Hex−Hex−HexN+Na]+)、520.3([M−Hex−Hex−HexN+Na]+)、および445.1([Hex−Hex+Na]+)のフラグメント・イオンを生じた(表2)。したがって、末端Galα1,3Galを潜在的に発現する中性およびシアリル酸付加オリゴ糖は、C2−1−9クローンによって産生されるが、シアル酸付加(NeuAc)構造は、最も多量なものであると考えられる。これに加えて、末端Hex−Hex(Galα1−3Gal)を持たない複数のシアル酸付加オリゴ糖を見ることができるが、相対的存在量はより少ない(図13および表2)。
【0124】
(表2 PSGL−1/mIgG2b由来O−グリカンの配列および仮構造)
【0125】
【表2−1】
【0126】
【表2−2】
(実施例3 発現ベクター)
融合ポリペプチドの生成に有用な好例の発現ベクターは次のとおりである。
【0127】
【表3−1】
【0128】
【表3−2】
【0129】
【表3−3】
【0130】
【表3−4】
【0131】
【表4】
【0132】
【表5−1】
【0133】
【表5−2】
【0134】
【表5−3】
【0135】
【表5−4】
【0136】
【表6−1】
【0137】
【表6−2】
(参考文献)
【0138】
【表7−1】
【0139】
【表7−2】
【0140】
【表7−3】
【0141】
【表7−4】
【0142】
【表8−1】
【0143】
【表8−2】
【0144】
【数7】
【0145】
【数8】
【0146】
【数9】
【0147】
【数10】
【0148】
【数11】
【0149】
【数12】
【0150】
【数13】
(他の実施形態)
本発明は、その詳細な説明とともに記載されたが、上記の記載は、添付の請求項の範囲によって定義される本発明の範囲を説明するものであって、限定を意図するものではない。他の態様、利点、および変更は以下の請求項の範囲内にある。
【技術分野】
【0001】
(発明の分野)
本発明は、概して超急性を処置または予防するための組成物および方法に関し、より詳しくは、炭水化物エピトープGalα1,3Galを含む融合ポリペプチドを含有する組成物に関する。
【背景技術】
【0002】
(発明の背景)
同種移植用のドナー臓器が慢性的に欠乏していることは、動物からヒトへの臓器または組織、すなわち異種移植が実施できれば解決することが可能である。ブタは、ヒトの異種移植とって最も適したドナー種であると考えられている。しかし、それを常套手段として用いる前に解決すべきいくつかの問題がある。最初の免疫性障害は、炭水化物エピトープGalα1,3Gal(α−Gal)(ブタ等の他の哺乳類に存在)に特異的なヒト、類人猿、および旧大陸サルの異種反応性(xenoreactive)天然抗体(XNAb)によって、引き起こされる。ブタ血管内皮細胞上のα−Galエピトープに対するXNAbsの結合によって、数分から数時間のうちに移植片機能損失に至る一連のイベントが開始される(非特許文献1)。
【先行技術文献】
【非特許文献】
【0003】
【非特許文献1】Galili, U., 1993; Oriol, R. et al., 1993
【発明の概要】
【課題を解決するための手段】
【0004】
(発明の要旨)
本発明は、ひとつには、ムチン型タンパク質主鎖上の異なるコア糖鎖によって炭水化物エピトープGalα1,3Gal(αGal)が特異的に高密度で発現しうるという発見にもとづいている。このような高密度のαGalエピトープは、遊離糖類または固相に結合したαGal決定基と比較して、抗αGalの結合または除去(すなわち、吸着)の増大をもたらす。本明細書ではポリペプチドをαGal融合ポリペプチドと称する。
【0005】
本発明は、Galα1,3Gal特異的抗体に結合する融合ポリペプチドを特徴とする。融合ポリペプチドとして、ムチン・ポリペプチドと免疫グロブリン・ポリペプチドとが挙げられる。ムチン・ポリペプチドは、以下の配列Hex−HexNol−HexN−Hex−Hex、NeuAc−Hex−HexNol−HexN−Hex−Hex、およびNeuGc−Hex−HexNol−HexN−Hex−Hexを含むグリカン・レパートリーを有する。
【0006】
また、本発明は、第2のポリペプチドに結合したβ1,6,N−アセチルグリコサミン転移酵素とα1,3ガラクトシル基転移酵素とによってグリコシル化されたムチン・ポリペプチドの少なくとも一領域を含む第1のポリペプチドを含む融合ポリペプチドである特徴とする。
【0007】
また、本発明によって提供されるものは、融合ポリペプチドを生産する方法である。融合ポリペプチドの生産を、免疫グロブリン・ポリペプチドの少なくとも一部分をコードしている核酸に対して操作可能に結合したムチン・ポリペプチドをコードする核酸と、α1,3,ガラクトシル基転移酵素ポリペプチドをコードする核酸と、β1,6,−N−アセチルグルコサミン転移酵素ポリペプチドをコードする核酸とを含む細胞を与えることでおこなう。あるいは、融合ポリペプチドの生産を、免疫グロブリン・ポリペプチドの少なくとも一部分をコードしている核酸に対して操作可能に結合したムチン・ポリペプチドをコードする核酸、α1,3,ガラクトシル基転移酵素ポリペプチドをコードする核酸、およびβ1,6,−N−アセチルグルコサミン転移酵素ポリペプチドをコードする核酸を、細胞に対して導入すること(例えば、トランスフェクションまたは形質転換)によって、おこなう。融合ポリペプチドの生産を可能とする条件下で上記細胞を培養し、該培養から融合ポリペプチドを単離する。融合ポリペプチドの単離は、既知の方法を用いておこなわれる。例えば、プロテインAまたはプロテインGクロマトグラフィを用いて、上記融合ポリペプチドを単離する。
【0008】
上記細胞は、真核細胞または原核細胞(例えば、細菌細胞)である。例えば、真核細胞は、ほ乳類動物細胞、昆虫細胞、または酵母細胞である。典型的な真核細胞として、CHO細胞、COS細胞、または293細胞が挙げられる。
【0009】
ムチン・ポリペプチドは、例えばPSGL−1である。好ましくは、ムチン・ポリペプチドは、PSGL−1の細胞外部分である。好ましい実施形態では、第2のポリペプチドは、免疫グロブリン・ポリペプチドの少なくとも一領域を含む。例えば、第2のポリペプチドは、免疫グロブリン重鎖ポリペプチドの少なくとも一領域を含む。あるいは、第2のポリペプチドは、免疫グロブリン重鎖のFC領域を含む。
【0010】
上記融合ポリペプチドは、多量体である。好ましくは、上記融合ポリペプチドは二量体である。
【0011】
また、本発明は、本明細書に記載するαGal融合ポリペプチド・コード化核酸を含むベクターと同様に、αGal融合ポリペプチドをコードする核酸と、本明細書に記載するベクターまたは核酸を含む細胞とを包含する。あるいは、ベクターは、α1,3ガラクトシル基転移酵素および/または2β1,6−N−アセチルグルコサミニル転移酵素をコードする核酸をさらに含む。本発明はまた、αGal融合ポリペプチドを発現させるために遺伝子操作された宿主細胞(例えばCHO細胞)も包含する。
【0012】
別の態様では、本発明は被験体での抗体媒介拒絶(例えば、超急性拒絶)を処置する方法を提供する。この方法は、生体試料(例えば、被験体由来の全血または血漿)を本発明のαGal融合ポリペプチドと接触させて、融合ポリペプチド−抗体複合体を形成することを含む。この複合体を生体試料から除去し、該生体試料を被験体に再び融合させる。
【0013】
また、本発明に包含されるものは、本発明のαGal融合ペプチドに試料を接触させて抗体−融合ペプチド複合体を形成させ、該複合体を生体試料から除去することで、その試料から抗体を除去する方法である。
【0014】
また、本発明に包含されるものは、αGal融合ポリペプチドを含む医薬品組成物である。
【0015】
特に定義しない限り、本明細書中で使用される全ての技術用語および科学用語は、この発明が属する当該技術分野の当業者が一般に理解するものと同じ意味を持つ。本明細書中で記載されるそれらに対する方法と類似の材料または等価物が本発明の実行またはテストで使われることができるにもかかわらず、適した方法と材料は後述する。本明細書中で言及される全ての刊行物、特許出願、特許、および他の参考文献を、全体として本明細書で援用する。矛盾する場合、定義を含む本明細書がコントロールする。また、材料、方法、および実施例は例証のためのものであって、限定することを意図したものではない。
【0016】
本発明はまた、以下の項目を提供する。
(項目1)
融合ポリペプチドであって、
(a) i) Hex−HexNol−HexN−Hex−Hex、
ii) NeuAc−Hex−HexNol−HexN−Hex−Hex、および
iii) NeuGc−Hex−HexNol−HexN−Hex−Hex
の配列を含むグリカン・レパートリーを持つムチン・ポリペプチドと、
(b) 免疫グロブリンポリペプチドの少なくとも一領域とを有し、
該融合ポリペプチドは、Galα1,3Gal特異的抗体に結合することを特徴とする融合ポリペプチド。
(項目2)
上記ムチン・ポリペプチドが、PSGL−1、MUC1、MUC2、MUC3、MUC4、MUC5a、MUC5b、MUC5c、MUC6、MUC11、MUC12、CD34、CD43、CD45、CD96、GlyCAM−1、およびMAdCAM3またはそれらのフラグメントからなる群から選択されることを特徴とする項目1に記載の融合ポリペプチド。
(項目3)
上記ムチン・ポリペプチドが、P−セレクチン糖タンパク質リガンド−1の少なくとも一領域を含むことを特徴とする項目1に記載の融合ポリペプチド。
(項目4)
上記ムチン・ポリペプチドが、P−セレクチン糖タンパク質リガンド−1の細胞外部分を含むことを特徴とする項目1に記載の融合ポリペプチド。
(項目5)
上記第2のポリペプチドが、H鎖免疫グロブリン・ポリペプチドの一領域を含むことを特徴とする項目1に記載の融合ポリペプチド。
(項目6)
上記免疫グロブリンポリペプチドが、免疫グロブリンH鎖のFc領域を含むことを特徴とする項目1に記載の融合ポリペプチド。
(項目7)
上記融合ポリペプチドが二量体であることを特徴とする項目1に記載の融合ポリペプチド。
(項目8)
項目1の融合ポリペプチドを発現するように遺伝子操作されたことを特徴とする宿主細胞。
(項目9)
上記宿主細胞がCHO細胞であることを特徴とする項目8に記載の宿主細胞。
(項目10)
第2のポリペプチドに結合した第1のポリペプチドを含む融合ポリペプチドであって、該第1のポリペプチドは、
(a)ムチン・ポリペプチドであり、そして
(b)α1,3−ガラクトシル基転移酵素およびβ1,6,N−アセチルグリコサミニル転移酵素であり、そして
該第2のポリペプチドは免疫グロブリン・ポリペプチドの少なくとも一領域を含むことを特徴とする融合ポリペプチド。
(項目11)
上記ムチン・ポリペプチドがヒト・ポリペプチドであることを特徴とする項目10に記載の融合ポリペプチド。
(項目12)
上記ムチン・ポリペプチドが、PSGL−1、MUC1、MUC2、MUC3、MUC4、MUC5a、MUC5b、MUC5c、MUC6、MUC11、MUC12、CD34、CD43、CD45、CD96、GlyCAM−1、およびMAdCAM3またはそれらのフラグメントからなる群から選択されることを特徴とする項目10に記載の融合ポリペプチド。
(項目13)
上記ムチン・ポリペプチドが、P−セレクチン糖タンパク質リガンド−1の少なくとも一領域を含むことを特徴とする項目10に記載の融合ポリペプチド。
(項目14)
上記ムチン・ポリペプチドが、P−セレクチン糖タンパク質リガンド−1の細胞外部分を含むことを特徴とする項目10に記載の融合ポリペプチド。
(項目15)
上記第2のポリペプチドが、H鎖免疫グロブリン・ポリペプチドの一領域を含むことを特徴とする項目10に記載の融合ポリペプチド。
(項目16)
上記第2のポリペプチドが、免疫グロブリンH鎖のFc領域を含むことを特徴とする項目10に記載の融合ポリペプチド。
(項目17)
上記融合タンパク質が二量体であることを特徴とする項目10に記載の融合ポリペプチド。
(項目18)
融合ポリペプチドを生産する方法であって、
(a)i)免疫グロブリン・ポリペプチドの少なくとも一部分をコードする核酸に、操作可能に結合したムチン・ポリペプチドをコードする核酸、
ii)α1,3ガラクトシル基転移酵素ポリペプチドをコードする核酸、および
iii)β1,6−N−アセチルグルコサミニル転移酵素ポリペプチドをコードする核酸、
を有する細胞を提供するステップと、
(b)該融合ポリペプチドの生産を可能とする条件下で細胞を培養するステップと、
(c)該融合ポリペプチドを単離するステップと、
を包含することを特徴とする融合ポリペプチドの生産方法。
(項目19)
上記ムチン・ポリペプチドが、PSGL−1、MUC1、MUC2、MUC3、MUC4、MUC5a、MUC5b、MUC5c、MUC6、MUC11、MUC12、CD34、CD43、CD45、CD96、GlyCAM−1、およびMAdCAM3またはそれらのフラグメントからなる群から選択されることを特徴とする項目18に記載の方法。
(項目20)
上記ムチン・ポリペプチドが、P−セレクチン糖タンパク質リガンド−1の少なくとも一領域を含むことを特徴とする項目18に記載の方法。
(項目21)
上記ムチン・ポリペプチドが、P−セレクチン糖タンパク質リガンド−1の細胞外部分を含むことを特徴とする項目18に記載の方法。
(項目22)
上記免疫グロブリン・ポリペプチドが、H鎖免疫グロブリン・ポリペプチドの一領域を含むことを特徴とする項目18に記載の方法。
(項目23)
上記免疫グロブリン・ポリペプチドが、免疫グロブリンH鎖のFc領域を含むことを特徴とする項目18に記載の方法。
(項目24)
上記細胞が真核細胞または原核細胞であること特徴とする項目18に記載の方法。
(項目25)
上記真核細胞がほ乳類細胞、昆虫細胞、または酵母細胞であることを特徴とする項目24に記載の方法。
(項目26)
上記原核細胞が細菌細胞であることを特徴とする項目24に記載の方法。
(項目27)
上記真核細胞がCHO細胞、COS細胞、または293細胞であることを特徴とする項目24に記載の方法。
(項目28)
融合ポリペプチドを生産する方法であって、
(a)細胞に対して、
(i)免疫グロブリン・ポリペプチドの少なくとも一部分をコードする核酸に操作可能に結合したムチン・ポリペプチドをコードする核酸、
(ii)α1,3−ガラクトシル基転移酵素ポリペプチドをコードする核酸、および
(iii)β1,6−N−アセチルグルコサミニル転移酵素ポリペプチドをコードする核酸を導入するステップと、
(b)上記融合ポリペプチドの生産を可能とする条件下で細胞を培養するステップと、
(c)上記融合ポリペプチドを単離するステップと、
を包含することを特徴とする融合ポリペプチドの生産方法。
(項目29)
項目28の方法によって生産されることを特徴とする融合ポリペプチド。
(項目30)
細胞であって、
(a)免疫グロブリン・ポリペプチドの少なくとも一部分をコードする核酸に操作可能に結合したムチン・ポリペプチドをコードする核酸と、
(b)α1,3−ガラクトシル基転移酵素ポリペプチドをコードする核酸と、
(c)β1,6−N−アセチルグルコサミニル転移酵素をコードする核酸と、
を含むことを特徴とする細胞。
(項目31)
上記細胞がCHO細胞であることを特徴とする項目30に記載の細胞。
本発明の他の特徴および効果は、以下の詳細な説明から、さらに特許請求の範囲から、明らかである。
【図面の簡単な説明】
【0017】
【図1】図1は、ベクター単独(CDM8)、PSGL1/mIgG2b、またはPSGL1/mIgG2bおよびブタα1,3GT発現プラスミドによって、トランスフェクションしたCOS細胞の上清から単離されたタンパク質のSDS−PAGEの写真である。これらを、続いてペルオキシダーゼ共役バンデイレイア・シンプリシフォリア(Bandeireia simplicifolia)イソレクチンB4レクチンでプローブ化して、化学発光による視覚化で免疫精製タンパク質上のGalα1,3Galエピトープを検出した。
【図2】図2Aは、50μl抗マウスIgGアガロース・ビーズ上での吸着前後にトランスフェクションしたCOS細胞上清の容量を増加させる際に、PSFGL1/mIgG2bの抗マウスIgGのFcをELISAにより定量化した結果を示す棒グラフである。試料を三重分析した。図2Bは、トランスフェクションしたCOS細胞上清の容量を増加させる際に、PSGL1/mIgG2b融合タンパク質濃度を示すゲルの写真である。
【図3】図3は、51Cr遊離測定で推定される約300ngのGalα1,3Gal置換または非置換PSGL1/mIgG2bを担持する抗マウスIgGアガロース・ビーズ50μl上への吸着後の異なる容量のヒトAB血清による抗体依存型補体媒介PEC−A細胞毒性を示す折れ線グラフである。
【図4】図4は、免疫親和性精製ヒトヒトIgG、IgM、およびIgAのSDS−PAGEゲルの写真である。各試料の4μgを還元および非還元条件下で流し、銀染色でタンパク質を視覚化した。
【図5】図5は、Galα1,3Gal置換PSGL1/mIgG2b上での吸着を51Cr遊離測定で調べた前後における免疫親和性精製ヒトIgG、IgM、およびIgAの抗体依存型補体媒介PEC−A細胞毒性を示す折れ線グラフである(右手側Y軸;%致死)。
【図6】図6は、熱不活性化ヒトAB血清の添加による致死に対する直接的相乗効果を4時間51Cr遊離測定で調べたK562、RajiおよびPEC−A細胞に対するヒトPBMCの直接的細胞毒性効果(血清無し条件)を示す折れ線グラフである(グラフA)。PEC−A細胞の抗体依存型細胞性細胞毒性に対する熱不活化ヒトAB血清の効果を、Galα1,3置換および非置換PSGL1/mIgIgG22b上への吸着前後での4時間51Cr遊離測定で測定した(グラフB)。
【図7】図7は、PSGL−1/mIgG2bcDNAを単独(−)で、またはブタα1,3ガラクトシル基転移酵素cDNA(+)とともに用いて、安定的にトランスフェクションしたCHO−K1、COS、および293T細胞の上清から精製したPSGL−l/mIgG2b融合タンパク免疫親和性を示すウエスタンブロットの写真である。
【図8】図8は、CHO−K1、COS、および293T細胞によって発現されたPSGL−1/mIgG2b上の相対的α−Galエピトープを示す棒グラフである。ブタα1,3ガラクトシル基転移酵素(GalT)の共発現なし(白色棒)または共発現あり(黒色棒)、さらにCHO−K1に関してはコア2β1,6N−アセチルグロコサミニル転移酵素(C2 GnTI)(灰色棒)で、CHO−K1、COS、または293Tで生産されたP−セレクチン糖タンパク質リガンド−1−マウス免疫グロブリンFc融合タンパク質(PSL−1/mIgG2b)上の相対的α−Galエピトープ密度。
【図9】図9は、異なる宿主細胞で生産されたPSGL−1/mIgG2b上に吸着したヒト血液型AB血清のブタ大動脈血管内皮細胞細胞毒性を示す折れ線グラフである。
【図10】図10は、安定的に形質移入されたCHO−K1細胞の上清から精製されたPSGL−1/mIgG2b融合タンパク質のウエスタンブロット分析の写真である。
【図11】図11は、アフィニティー・クロマトグラフィおよびゲル濾過によって精製されたPSGL−1mIgG2bのSDS−PAGEおよびウエスタンブロット分析の写真である。
【図12】図12は、CHOクローン5L4−1に作られたPSGL−1/mIgG2bから遊離したO−グリカンのエレクトロスプレー・イオントラップ・マススペクトル分析を示す。
【図13】図13は、CHOクローンC2−1−9に作られたPSGL−1/mIgG2bから遊離したO−グリカンのエレクトロスプレー・イオントラップ・マススペクトル分析を示す。
【図14】図14は、CHOクローンC2−1−9で作られたPSL−1/mIgG2bから放出されたO−グリカンのマザー・スペクトルに見られる顕著なピークのMS/MS分析を示す一連の説明図である。
【発明を実施するための形態】
【0018】
(発明の詳細な説明)
本発明は、ひとつには、炭水化物エピトープ Galα1,3Gal (αGal)が高密度で、かつムチン型タンパク質主鎖上の異なるコア糖鎖によって、特異的に発現しうるということの発見にもとづいている。より具体的には、本発明は、ムチン型タンパク質主鎖のαGalエピトープの発現が該ポリペプチドを発現する細胞株に依存するという驚くべき発見にもとづいている。さらに、ムチンのグリカン・レパートリーを外来α1,3ガラクトシル基転移酵素およびコア2分岐酵素の共発現によって修飾することができる。この修飾によって、αGalエピトープの密度がよりいっそう高くなり、遊離糖類、固相に結合したαGal決定基、またはα1,3ガラクトシル基転移酵素単独によりトランスフェクションした細胞と比較して、抗αGal抗体の結合または除去(すなわち、吸着)の増加が生ずる。
【0019】
COS細胞でのPSGL−1/mIgG2b融合タンパク質およびブタα1,3ガラクトシル基転移酵素(α1,3GalT)の一時的トランスフェクションは、α−Galエピトープにより大幅に置換された二量体融合タンパク質を生ずる。この融合タンパク質は、大体、1つの二量体あたり複数の末端α−Galエピトープを有するとともに、アガロース・ビーズに固定されたブタ・チログロブリンよりも20倍高く(炭水化物モル基準で)、Galα1,3Gal共役アガロースおよびマクロ孔ガラス・ビーズよりもそれぞれ5,000倍および30,000倍高い異種反応性天然抗体(XNAb)吸着効率を有する。
【0020】
PSGL−1/mlgG2b上のαGalエピトープ密度およびPSGL−1/mlgG2bの抗ブタ抗体吸着効率に対する宿主細胞の重要性を調べるために、そのタンパク質を、ブタα1,3GalTとともに、CHO、COS、および293T細胞で安定的に発現させた。PSGL−1/mIgG2b上でのα−Gal置換のレベルとその抗ブタ抗体吸着能は、宿主細胞に依存した。COS細胞で作られたPSGL−1/mIgG2bでは、α1,3GalTなしでCOS細胞で作られたPSGL−1/mIgG2bと比較して、相対的O.D(GSA−反応性/抗マウスIgG反応性)が5.3倍増加した(図2)。同様に、293T細胞で作られたPSGL−1/mIgG2bでは、相対的O.Dが3.1倍増加した。それとは対照的に、CHO細胞で作られたPSGL−1/mIgG2bでは、1.8倍増加したのみであった(図2)。異なる宿主細胞で作られたPSGL−1/mIgG2bの抗ブタ抗体吸着効率は、そのαGal置換の度合いと相関関係があった。
【0021】
驚くべきことに、CHO細胞でのコア2β1,6GalcNAc転移酵素(C2 GnTI)の共発現は、PSGL−1/mIgG2bαGal エピトープ密度および抗ブタ抗体吸着効率を改善した。さらに、PSGL−1/mIgG2bは、ブタα1,3GalTとともにCHO細胞で発現し、C2 GnTIは、末端Gal−Galと一致する配列を持つ3つの異なるO−グリカンを担持した(図2)。それとは対照的に、末端Gal−Galエピトープは、C2 GnTIなしのCHO細胞で発現されたPSGL−1/mIgG2b上のO−グリカンでは、検出されなかった。図2に示すように、C2 GnTIおよびα1,3GaITの両方を発現しているCHO細胞で生産された融合タンパク質上のα−Galのレベルは、著しく増加し、外来α1,3GalTのみを発現するCOSおよび293T細胞で作られた融合タンパク質のα−Galエピトープ・レベルを超えた。さらに、融合タンパク質の抗ブタ抗体吸着能が、COSおよび293T細胞と比べて、CHO細胞で増加した(図3)。マススペクトル分析によって確認されたことは、α−Galエピトープ密度増加の原因が、C2 FnTIおよびα1,3GalTの両方を発現させるために操作されたCHO細胞で作られたPSGL−1/mIgG2b のO−グリカン上でのコア2分岐およびラクトサミン伸張にあるということであった(図7および図8、ならびに表II)。
【0022】
炭水化物エピトープに対する抗体の結合は、提示された糖類の立体構造(コンホメーション)に依存している。組織血液型抗原が溶液中で異なる立体構造をとることが示されており((Imberty, A. et al., 1995)、また抗血液型抗体が、立体構造に依存する炭水化物決定基(所謂マイクロ・エピトープ)の異なる領域を認識することも示されている(Imberty, A. et al., 1996)。同様に、研究によって、レセプターが、リガンドに結合するときに、よりエネルギー的に好ましくない立体構造を誘導することができるということも示されている(Imberty, A. et al., 1993)。2種類の異種抗原(αGal−LacNAcおよびαGal−Lewis x)に対する研究によって明らかにされたことは、たとえ後者のエピトープのフコース残基がラクトサミン構造に対して立体構造的規制を誘導した場合であっても、末端Galα1,3Gal二糖類がかなりフレキシブルであった(Corzana, F. et al., 2002)。したがって、ともに所謂天然抗体によって認識された組織血液型抗原および異種抗原は、いくつかの異なる立体構造を採用することができ、これらの抗体が認識するエピトープの立体構造を予測することが困難かもしれない。さらに、オリゴ糖の内側コア構造は、タンパク質−炭水化物複合体の結合に直接関係するものではないが、その結合親和性に影響することが示された(Maaheimo, H. et al., 1995)。α1,3GalTおよびC2 GnTIを共発現するCHO細胞上で発現したO−グリカンの構造的分析によって、α−Galエピトープが3つの異なるオリゴ糖で発現されることが明らかになった(図7および表II)。したがって、コア2およびラクトサミン構造の効果とは別に、オリゴ糖のコア2分岐のうちの1つの分岐上に位置したN−アセチルおよびN−グリコリル・ノイラミン酸がそれらの異種抗原によってとられた立体構造に影響を及ぼす可能性があった。また、シアル酸は結合エピトープの一部である可能性がある。したがって、一部の異種反応性抗体レパートリーは、それらの分岐エピトープを、Galili抗原を認識するものとは異なる結合特異性で認識する((Galili, U. et al., 1985)。さらにまた、N−グリコリルノイラミン酸は、ブタで発現される (Bouhours, D. et al., 1996; Malykh, Y. N. et al., 2003; Malykh, Y. N. et al., 2001)。しかし、N−グリコリルノイラミン酸の発現は、ヒトではみとめられず(Varki, A., 2001)、ヒト異種反応性抗体によって認識されることが明らかになっている(Zhu, A. et al., 2002)。したがって、N−グリコリルノイラミン酸含有グリカンは、異種反応性抗体のさらに別のグループと結合する可能性がある。
【0023】
本発明は、抗αGal抗体の吸着剤として有用な多数のαGalエピトープを含むムチン免疫グロブリン融合タンパク質(以下、「αGal融合タンパク質」という)を提供する。例えば、αGal融合タンパク質は、異種移植に先立って血液または血漿からレシピエント抗αGal抗体を取り除くことに有用である。αGal融合タンパク質は、レシピエントの血液または血漿から10%、25%、50%、60%、70%、80%、90%、95%、98%、または100%の抗αGal抗体を吸着する。
【0024】
αGal融合ペプチドは、炭水化物モル基準で、野生型αGal決定基の遊離糖類と比較して、抗血液型抗体の除去または結合に対して、よりいっそう効率的である。αGal融合タンパク質が結合する抗αGal抗体の数は、等量の野生型αGal決定基の遊離糖類と比べると、2倍、4倍、6倍、10倍、20倍、50倍、80倍、100倍、またはそれ以上多い。
【0025】
(融合ポリペプチド)
種々の態様では、本発明は、第2のポリペプチドに結合した糖タンパク質(例えば、ムチン・ポリペプチド)の少なくとも一部分を含む第1のポリペプチドを有する融合タンパク質を提供する。本明細書で用いられるように、「融合タンパク質(fusion protein)」または「キメラ・タンパク質(chimeric protein)」は、非ムチン・ポリペプチドに対して操作可能に結合したムチン・ポリペプチドの少なくとも一部分を含む。「非ムチン・ポリペプチド(non−mucin polypeptide)」は、その質量の少なくとも40%未満がグリカンに起因するポリペプチドをいう。
【0026】
「ムチン・ポリペプチド(mucin polypeptide)」は、ムチン・ドメインを持つポリペプチドをいう。このムチン・ポリペプチドのムチン・ドメイン数は、1、2、3、5、10、20、またはそれ以上である。ムチン・ポリペプチドは、O−グリカンによって置換されたアミノ酸配列によって特徴づけられる任意の糖タンパク質である。例えば、ムチン・ポリペプチドは2つ目または3つ目のアミノ酸ごとにセリンまたはスレオニンを有する。ムチン・ポリペプチドは、分泌されたタンパク質である。あるいは、ムチン・ポリペプチドは細胞表面タンパク質である。
【0027】
ムチン・ドメインは、アミノ酸のスレオニン、セリン、およびプロリンが豊富であり、そこでN−アセチルガラクトサミンを介してオリゴ糖がヒドロキシアミノ酸(O−グリカン)に結合する。ムチン・ドメインは、O−結合グリコシル化部位を含む、あるいは代わりに、該O−結合グリコシル化部位からなる。ムチン・ドメインのO−グリコシル化部位の数は、1個、2個、3個、5個、10個、20個、50個、100個、またはそれ以上である。あるいは、ムチン・ドメインは、N−結合グリコシル化部位を含む、あるいは代わりに、該N−結合グリコシル化部位からなる。ムチン・ポリペプチドは、その質量の50%、60%、80%、90%、95%、または100%がグリカンに起因する。ムチン・ポリペプチドは、MUC遺伝子(すなわち、MUC1、MUC2、MUC3、MUC4、MUC5a、MUC5b、MUC5c、MUC6、MUC11、またはMUC12)によってコードされた任意のポリペプチドである。あるいは、ムチン・ポリペプチドは、Pセレクチン糖タンパク質リガンド1(PSGL−1)、CD34、CD43、CD45、CD96、G1yCAM−1、MAdCAM、赤血球グリコホリン、グリコカリシン、グリコホリン、シアロフォリン、ロイコ・シアリン、低密度リポ蛋白(LDL)−R、ZP3、およびエピグリカニンである。好ましくは、ムチンは、PSGL−1である。PSGL−1は、各々が402個のアミノ酸を含む、1型トランスメンブレン・トポロジーの2つのジスルフィド結合120kDaサブユニットを持ち、該サブユニットの各々が402のアミノ酸を含むホモ二量体糖タンパク質である。細胞外ドメインには、10アミノ酸コンセンサス配列A Q(M) T T P(Q) P(LT) A A(PG) T(M) Eが15回繰り返して存在し、該配列はO−結合オリゴ糖を付加するための潜在的部位を3または4カ所有する。PSGL−1は、各モノマーあたり、O−結合グリコシル化のための部位数が53を上回り、またN−結合グリコシル化のための部位を3つ有すると予測される。
【0028】
ムチン・ポリペプチドは、ムチン・タンパク質の全てまたは一部を含む。あるいは、ムチン・タンパク質は、該ポリペプチドの細胞外部分を含む。例えば、ムチン・ポリペプチドは、PSGL−1の細胞外部分またはその一部を含む(例えば、GenBank寄託番号A57468に開示されるアミノ酸19−319)。ムチン・ポリペプチドは、PSGL−1のシグナル配列部分(例えば、アミノ酸1−18)、膜貫通(トランスメンブラン)ドメイン(例えば、アミノ酸320−343)、および細胞質ドメイン(例えば、アミノ酸344−412)を含む。
【0029】
本発明のαGal融合タンパク質内で、ムチン・ポリペプチドはムチン・タンパク質の全てまたは一部分に一致する。例えば、αGal融合タンパク質は、ムチン・タンパク質の少なくとも一部分を含む。「少なくとも一部分(at least a portion)」は、ムチン・ポリペプチドが少なくとも1つのムチン・ドメイン(例えば、O−結合グリコシル化部位)を含むことを意味している。任意に、ムチン・タンパク質は、ポリペプチドの細胞外部分から構成される。例えば、ムチン・ポリペプチドは、PSGL−1の細胞外部分から構成される。
【0030】
ムチン・ポリペプチドは、表2に示すようにグリカン・レパートリーで装飾される。例えば、ムチン・ポリペプチドは、表2に示す炭水化物配列を1つ、2つ、3つ、4つ、5つ、またはそれ以上有する。例えば、ムチン・ポリペプチドはグリカン・レパートリーを有し、該グリカン・レパートリーとしてHex−HexNol−HexN−Hex−Hex、NeuAc−Hex−HexNol−HexN−Hex−Hex、およびNeuGc−Hex−HexNol−HexN−Hex−Hexが挙げられる。ムチン・ポリペプチドは、末端αGal糖を1個、2個、3個、4個、5個、またはそれ以上有する。好ましくは、その末端糖は、2、3、4、5、またはそれ以上の数の異なるオリゴ糖で発現される。任意に、ムチンはN−アセチル・ノイラミン酸、N−グリコリルノイラミン酸、および/またはシアル酸を含む。さらに、ムチンのオリゴ糖として、コア2分岐、コア1分岐、およびラクトサミン伸張が挙げられる。
【0031】
第1のポリペプチドは、1種類または複数の転移酵素によって、グリコシル化される。転移酵素は外来性である。あるいは、転移酵素は内在性である。第1のポリペプチドは、2つ、3つ、5つ、またはそれ以上の転移酵素によってグリコシル化される。グリコシル化は経時的または連続的である。あるいは、グリコシル化は同時、またはランダム、すなわち特に決まった順序もなくおこなわれる。例えば、第1のポリペプチドは、α1,3ガラクトシル基転移酵素によってグリコシル化される。α1,3ガラクトシル基転移酵素のための適当なソースとして、GenBank寄託番号AAA73558、L36150、BAB30163、AK016248、E46583、またはP50127が挙げられ、本明細書ではそれらをそのまま援用する。あるいは、第1のポリペプチドは、コア2分岐酵素またはN−アセチルゴルコサミン転移酵素(例えば、β1,6N−アセチルグルコサミン転移酵素)によって、グリコシル化される。β1,6N−アセチルグルコサミン転移酵素のための適当なソースとして、GenBank寄託番号CAA79610、Z19550、BA−B66024、またはAP001515が挙げられる。好ましくは、第1のポリペプチドは、1,3ガラクトシル基転移酵素およびβ1,6N−アセチルグルコサミン転移酵素の両方によってグリコシル化される。
【0032】
融合タンパク質内では、用語「操作可能に結合(operatively linked)」は、第1のポリペプチドのO−結合グリコシル化を可能とするようにして、第1および第2のポリペプチドが化学的に結合(最も一般的にはペプチド結合等の共有結合を介して)することを示すように意図されている。融合ポリペプチドをコードする核酸を言及するために用いる場合、「操作可能に結合」という用語は、ムチン・ポリペプチドおよび非ムチン・ポリペプチドをコードする核酸が互いにフレーム単位(in−frame)で融合していることを意味している。非ムチン・ポリペプチドは、ムチン・ポリペプチドのN末端またはC末端に融合しうる。
【0033】
任意に、αGal融合タンパク質は、1つ以上の付加的構成成分に結合する。例えば、αGal融合タンパク質はGST融合タンパク質と付加的に結合し、該αGal融合タンパク質配列がGST(すなわち、グルタチオンS−転移酵素)配列のC末端と融合する。そのような融合タンパク質は、αGal融合タンパク質の精製を促進することができる。あるいは、αGal融合タンパク質は、固相に対して付加的に結合する。種々の固相が当業者に知られている。そのような組成物は、抗αGal抗体の除去を促すことができる。例えば、αGal融合タンパク質は、金属化合物、シリカ、ラテックス、高分子材料等から作られた粒子;マイクロタイター・プレート; ニトロセルロース、もしくはナイロン、またはそれらの組み合わせに結合する。固体支持体に結合したαGal融合タンパク質は、生体試料(例えば、血液または血漿)から抗αGal抗体を除去するための吸着剤として用いられる。
【0034】
融合タンパク質は、そのN末端に異種シグナル配列(すなわち、ムチン核酸によってコードされているポリペプチドに存在しないポリペプチド配列)を含む。例えば、天然ムチン・シグナル配列を取り除いて、他のタンパク質由来のシグナル配列と置き換えることができる。ある種の宿主細胞(例えば、ほ乳類の宿主細胞)では、ポリペプチドの発現および/または分泌を、異種シグナル配列の使用を介して増加させることができる。
【0035】
本発明のキメラ・タンパク質および融合タンパク質は、標準的な組み換えDNA技術によって作ることができる。例えば、異なるポリペプチド配列をコードするDNAフラグメントを、従来の技術にもとづいてフレーム単位で連結する(例えば、連結反応のための平滑末端化(blunt−ended)または突出末端化(stagger−ended)された末端、適当な末端を生ずる制限酵素による消化、必要に応じた付着端の充填、不要な結合を避けるためのアルカリ・ホスファターゼ処理、および酵素による連結反応を用いる)。別の実施形態では、融合遺伝子を、自動DNA合成装置等、従来の技術によって合成することができる。あるいは、2つの連続的な遺伝子フラグメントの間に相補的なオーバーハングを生じるアンカー・プライマーを用いて、遺伝子フラグメントのPCR増幅をおこなうことができる。その後、これらの遺伝子フラグメントをアニーリングおよび再増幅することで、キメラ遺伝子配列が得られる(例えば、Ausubel et al. (eds.) CURRENT PROTOCOLS IN MOLECULAR BIOLOGY, John Wiley & Sons, 1992を参照せよ)。さらに、多くの発現ベクターが商業的に入手可能であり、それらは融合構成成分(例えば、免疫グロブリンH鎖のFc領域)をコードする。融合構成成分がフレーム単位で免疫グロブリン・タンパク質に結合されるように、PSGL−1をコードする核酸をそのようなベクターにクローニングすることができる。典型的なPSGL−1発現ベクターは、配列番号21を含む。
【0036】
αGal融合ポリペプチドは、オリゴマーとして存在する。すなわち、二量体、三量体、または五量体として存在する。好ましくは、αGal融合ポリペプチドは、二量体である。
【0037】
第1のポリペプチド、および/または該第1のポリペプチドをコードする核酸は、当業者公知のムチン・コード配列を用いて構築される。ムチン・ポリペプチドおよび該ムチン・ポリペプチドをコードする核酸の適当なソースとして、GenBank寄託番号NP663625および NM145650、CAD10625および AJ417815、XP140694および XM140694、XP006867および XM006867、ならびに NP00331777および NM009151がそれぞれ挙げられ、本明細書ではそれらをそのまま援用する。
【0038】
あるいは、ムチン・ポリペプチド構成成分は、天然ムチン配列(野生型)に、炭水化物含有量の増加(非突然変異配列と比較して)をもたらす突然変異を持つ変異ムチン・ポリペプチドとして、提供される。例えば、変異ムチン・ポリペプチドは、野生型ムチンと比較して、付加的なO−結合グリコシル化部位を含んでいた。あるいは、変異ムチン・ポリペプチドは、野生型ムチン・ポリペプチドと比較して、セリン、スレオニン、またはプロリンの数が増加するアミノ酸配列突然変異を含む。このような炭水化物含有量の増加は、当業者に公知の方法によって、ムチンのタンパク質と炭水化物との比率を決定することで、評価することができる。
【0039】
いくつかの実施形態では、ムチン・ポリペプチド構成成分は、天然ムチン配列(野生型)に、タンパク質分解に対してより高い耐性のムチン配列(非突然変異配列と比較して)をもたらす突然変異を有する変異ムチン・ポリペプチドとして、提供される。
【0040】
第1のポリペプチドは、完全長PSGL−1を含む。あるいは、第1のポリペプチドは、完全長よりも短いPSGL−1ポリペプチド(例えば、PSGL−1の細胞外部分)を含む。例えば、第1のポリペプチドは、長さが400アミノ酸未満、例えば300、250、150、100、50、または25アミノ酸以下である。典型的なPSGL−1ポリペプチドおよび核酸配列として、GenBank寄託番号XP006867、XM006867、XP140694、および XM140694が挙げられる。
【0041】
第2のポリペプチドは、好ましくは可溶性である。第2のポリペプチドは、αGal融合ポリペプチドと第2のムチン・ポリペプチドとの結合を促進する配列を含む。好ましくは、第2のポリペプチドは、少なくとも免疫グロブリン・ポリペプチドの一領域を含む。「少なくとも一領域(at least a region)」とは、免疫グロブリン分子の任意の部分、例えばL鎖、H鎖、FC領域、Fab領域、Fv領域、またはそれらの任意のフラグメントを含むことを意味する。免疫グロブリン融合ポリペプチドは公知であり、例えば米国特許第5,516,964号、第5,225,538号、第5,428,130号、第5,514,582号、第5,714,147号、および第5,455,165号に記載されている。
【0042】
第2のポリペプチドは、完全長免疫グロブリン・ポリペプチドを含む。あるいは、第2のポリペプチドは、完全長よりも短い免疫グロブリン・ポリペプチド(例えば、H鎖、L鎖、Fab、Fab2、Fv、またはFc)を含む。好ましくは、第2のポリペプチドは、免疫グロブリン・ポリペプチドのH鎖を含む。より好ましくは、第2のポリペプチドは免疫グロブリン・ポリペプチドのFc領域を含む。
【0043】
本発明の別の態様では、第2のポリペプチドは、野生型免疫グロブリンH鎖のFc領域のエフェクター機能よりも劣るエフェクター機能を持つ。Fcエフェクター機能は、例えば、Fcレセプター結合、補体固定、およびT細胞減少活性が含まれる(例えば、米国特許第6,136,310号を参照せよ)。T細胞減少活性、Fcエフェクター機能、および抗体安定性を評価する方法は、公知である。一実施形態では、第2のポリペプチドはFcレセプターに対して低親和性を示すか、あるいは親和性を示さない。別の実施形態では、第2のポリペプチドは補体タンパク質C1qに対して低親和性を示すか、あるいは親和性を示さない。
【0044】
本発明の別の態様は、ムチン・ポリペプチド、またはその誘導体、フラグメント、類似体、もしくは相同体(ホモログ)をコードする核酸を含むベクター、好ましくは発現ベクターに関する。種々の態様では、ベクターは免疫グロブリン・ポリペプチドをコードする核酸に操作可能に結合したムチン・ポリペプチド、またはその誘導体、フラグメント、類似体、もしくは相同体(ホモログ)をコードする核酸を含む。さらに、ベクターは、α1,3ガラクトシル基転移酵素、コア1,6,−N−アセチルグルコサミニル転移酵素またはそれらの任意の組み合わせを含む。転移酵素は、αGal融合タンパク質のムチン部分のペプチド主鎖上に対するαGal決定基の付加を促す。典型的なベクターは、配列番号1、11、または21を含む。本明細書で用いられるように、用語「ベクター(vector)」は核酸分子であり、該核酸分子が結合する別の核酸を搬送することが可能なものをいう。ベクターの1つの種類として、「プラスミド(plasmid)」があり、該プラスミドとは追加のDNAセグメントが連結しうる環状二重鎖DNAループのことである。ベクターの別の種類として、ウイルス・ベクターがあり、追加のDNAセグメントがウイルス・ゲノムに連結することができる。ある種のベクターは、該ベクターが導入される宿主細胞で自律複製ができる(例えば、細菌複製開始点を持つ細菌ベクターおよびエピソームほ乳類ベクター)。他のベクター(例えば、非エピソームほ乳類ベクター)は、宿主細胞に導入されると直ちに該宿主細胞のゲノムに組み込まれ、それによってその宿主ゲノムとともに複製される。さらに、ある種のベクターは、それが操作可能に連結している遺伝子の発現を導くことができる。そのようなベクターを本明細書では「発現ベクター(expression vectors)」と称する。一般に、発現ベクターは、しばしばプラスミドの形で、組換えDNA技術で有用である。本明細書では、ベクターの最も一般的な使用形態がプラスミドであることから、「プラスミド」および「ベクター」を互いに置き換えて使用することができる。しかし、本発明は、等価な機能を呈するそのような発現ベクターの他の形態、例えばウイルス・ベクター(例:複製欠損レトロウイルス、アデノウイルス、およびアデノ関連ウイルス)を含むことを意図している。
【0045】
本発明の組換え発現ベクターは、宿主細胞での核酸発現に適した形状の核酸を含む。このことは、その組換え発現ベクターが1種類以上の調節配列を含むことをを意味し、該配列は発現に使用される宿主細胞にもとづいて選択され、発現される核酸配列に操作可能に結合している。組換え発現ベクターの中で、「操作可能に結合(operably−linked)」とは、ヌクレオチド配列の発現(例えば、生体外(in vitro)転写/翻訳系で、または宿主細胞にベクターが導入された場合は宿主細胞で)を可能とするようにして、目的とするヌクレオチド配列が調節配列に結合していることを意味している。
【0046】
用語「調節配列(regulatory sequence)」が意味することには、プロモーター、エンハンサー、および他の発現調節要素(例えば、ポリアデニル化シグナル)が含まれる。そのような調節配列は、例えば、Goeddel, GENE EXPRESSION TECHNOLOGY: METHODS IN ENZYMOLOGY 185, Academic Press, San Diego, Calif.
(1990)に記載されている。調節配列として、多くの種類の宿主細胞でヌクレオチド配列の構成的発現を導くもの、およびある種の宿主細胞のみにあるヌクレオチド配列の発現を導くもの(例えば、組織特異的調節配列)が挙げられる。当業者が容易に理解することは、発現ベクターの設計が、形質転換すべき宿主細胞の選択、所望のタンパク質の発現レベル等のファクターに依存しうることである。本発明の発現ベクターを宿主細胞に導入することで、本明細書に記載した核酸によってコードされる融合タンパク質または融合ペプチド等(例えば、ABO融合ポリペプチド、ABO融合ポリペプチドの変異体、その他)のタンパク質またはペプチドを生産することができる。
【0047】
本発明の組換え発現ベクターを、原核細胞または真核細胞でのαGal融合ポリペプチドの発現用に設計することができる。例えば、αGal融合ポリペプチドは、大腸菌(Escherichia coli)等の細菌参謀、昆虫細胞(バキュロウイルス発現ベクターを用いる)、酵母細胞、またはほ乳類動物細胞で発現することができる。適当な宿主細胞については、Goeddel, GENE EXPRESSION TECHNOLOGY: METHODS IN ENZYMOLOGY 185, Academic
Press, San Diego, Calif. (1990)でさらに論じられている。あるいは、組換え発現ベクターを、例えばT7プロモーター調節配列およびT7ポリメラーゼを用いて、生体外(in vitro)で転写および翻訳することができる。
【0048】
原核生物でのタンパク質発現は、ほとんどの場合、融合タンパク質または非融合タンパク質の発現を目的とした構成的プロモーターまたは誘導的プロモーターを含むベクターを持つ大腸菌(Escherichia coli)でおこなわれる。融合ベクターは、多数のアミノ酸をそこでコードされるタンパク質、通常は組換えタンパク質のアミノ末端に、加える。そのような融合タンパク質は、概して3つの目的、すなわち(i)組換えタン
パク質の発現を高めること、(ii)組換えタンパク質の溶解性を高めること、および(iii)親和性精製でリガンドとして作用することで組換えタンパク質の精製を助けること、にかなう。しばしば、融合発現ベクターでは、融合タンパク質の精製の後の融合構成成分(fusion moiety)からの組換えタンパク質分離を可能とするために、タンパク質分解切断部位が融合構成成分と組換えタンパク質との接続部分で導入される。そのような酵素(および該酵素の同種認識配列)として、第Xa因子、トロンビン、およびエンテロキナーゼが挙げられる。典型的な融合発現ベクターとして、pGEX(Phanmacia Biotech Inc; Smith and Johnson, 1988. Gene 67: 31−40), pMAL (New England Biolabs, Beverly, Mass.)およびpRIT5 (Pharmacia, Piscataway, N.J.) 挙げられ、これらはグルタチオンS−転移酵素(GST)、マルトースE結合タンパク質、またはプロテインAのそれぞれを標的組換えタンパク質に融合させる。
【0049】
適当な誘導可能な非融合大腸菌(E.coli)発現ベクターの例として、pTrc (Amrann et al., (1988) Gene 69:301−315)および pET 11d (Studier et al., GENE EXPRESSION TECHNOLOGY: METHODS IN ENZYMOLOGY 185, Academic Press, San Diego, Calif. (1990) 60−89)が挙げられる。
【0050】
大腸菌(E.coli)で組換えタンパク質発現を最大にする1つの戦略は、組み換えタンパク質をタンパク質分解的に切断する能力に欠けた宿主細菌でそのタンパク質を発現させることである。例えば、Gottesman, GENE EXPRESSION TECHNOLOGY: METHODS IN ENZYMOLOGY 185, Academic Press, San Diego, Calif. (1990) 119−128を参照せよ。別の戦略は、各アミノ酸に対する個々のコドンが大腸菌(E. coli)で優先的に用いられように、発現ベクターに挿入される核酸の核酸配列を改変することである(例えば、Wada, et al., 1992. Nuci. Acids Res. 20: 2111−2118を参照せよ)。本発明の核酸配列のそのような改変は、標準的DNA合成技術によっておこなうことができる。
【0051】
別の実施形態では、αGal融合ポリペプチド発現ベクターは、酵母発現ベクターである。酵母Saccharonryces cerivisaeでの発現用ベクターの例として、pYepSecl (Baldari, et al., 1987. EMBO J. 6: 229−234)、pMFa (Kurjan and Herskowitz, 1982. Cell 30: 933−943)、pJRY88(Schultz et al., 1987. Gene 54: 113−123)、pYES2(Ivitrogen Corporation, San Diego, Calif.)、およびpicZ (InVitrogen Corp, San Diego, Calif.)が挙げられる。
【0052】
あるいは、αGal融合ポリペプチドをバキュロウイルス発現ベクターを用いて昆虫細胞で発現させることができる。 培養昆虫細胞(例えば、SF9細胞)でのタンパク質発現に利用可能なバキュロウイルス・ベクターとして、pAc系(Smith, et al., 1983. Mol. Cell. Biol. 3: 2156−2165)およびpVL系 (Lucklow and Summers, 1989. Virology 170: 31−39)が挙げられる。
【0053】
さらに別の実施形態では、本発明の核酸はほ乳類発現ベクターを用いてほ乳類細胞で発現させることができる。ほ乳類発現ベクターの例として、pCDM8 (Seed, 1987. Nature 329: 840)およびpMT2PC (Kaufinan, et al., 1987. EMBO J. 6: 187−195)が挙げられる。ほ乳類細胞で用いた場合、発現ベクターの調節機能は、しばしばウイルスの調節要素によって提供される。例えば、一般的に用いられるプロモーターは、ポリオーマ、アデノウイルス2、サイトメガロウイルス、およびシミアンウイルス40に由来する。原核生物および真核細胞のための他の適当な発現系に関しては、例えば、Sambrook, et al., MOLECULAR CLONING: A LABORATORY MANUAL. 2nd ed., Cold Spring Harbor Laboratory, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y., 1989の第16章および第17章を参照せよ。
【0054】
本発明の別の態様は、本発明の組換え発現ベクターが導入されている宿主細胞に関する。用語「宿主細胞(host cell)」および「組換え宿主細胞(recombinant host cell)」は、本明細書では相互に置き換え可能なかたちで用いられる。そのような用語が特定の被験体細胞のみならず、そのような細胞の子孫または潜在的子孫にも言及することが理解される。突然変異または外界いずれかの影響によって後続世代で一定の修飾が生ずることから、実際のところ、そのような子孫は親細胞とは同一ではないと思われるが、それでも本明細書で用いられるようにその用語の範囲内である。
【0055】
宿主細胞は、任意の原核生物または真核細胞である。例えば、αGal融合ポリペプチドは、大腸菌(E. coli)等の細菌細胞、昆虫細胞、酵母、またはほ乳類細胞(例えば、ヒト、チャイニーズ・ハムスター卵巣(CHO)細胞もしくはCOS細胞)で発現される。他の適当な宿主細胞は、当業者に公知である。
【0056】
従来の形質転換または形質移入(トランスフェクション)技術によって、ベクターDNA免疫を原核細胞または真核細胞に導入することができる。本明細書で用いられるように、「形質転換(transformation)」および「トランスフェクション(transfection)」は、外来核酸(例えば、DNA)を宿主細胞に導入するための種々の公知技術に言及するものである。上記技術として、リン酸カルシウムまたは塩化カルシウム共沈、DEAE−デキストラン媒介トランスフェクション、リポフェクション、または電気穿孔法が挙げられる。宿主細胞を形質転換またはトランスフェクションさせるための適当な方法は、Sambrook., et al. (MOLECULAR CLONING: A LABORATORY MANUAL. 2nd ed., Cold Spring Harbor Laboratory, Cold Spring
Harbor Laboratory Press, Cold Spring Harbor, N.Y., 1989)および他の研究室マニュアルに見いだすことができる。
【0057】
ほ乳類細胞の安定的なトランスフェクションのために、用いた発現ベクターおよびトランスフェクション技術に依存して、細胞のわずかなフラクションのみが外来DNAを該細胞のゲノムに組み込まれる可能性がある。これらの組み込まれた要素を同定および選択するために、選択マーカー(例えば、抗生物質耐性)をコードする遺伝子が一般に、目的とする遺伝子とともに宿主細胞に導入される。種々の選択可能マーカーとして、薬物(G418、ハイグロマイシン、およびメトトレキセート)に対する耐性を与えるものが挙げられる。選択マーカーをコードする核酸は、糖タンパク質Ibαをコードするものと同じベクター上で宿主細胞に導入されるか、もしくは別のベクター上で導入される。誘導核酸により安定的にトランスフェクションした細胞は、薬物選択によって同定される(例えば、選択マーカー遺伝子が取り込まれた細胞は生存し、他の細胞は致死する)。
【0058】
本発明の宿主細胞(例えば、培養中の原核または真核宿主細胞)を用いてαGalポリペプチドを生産(すなわち、発現)させることができる。したがって、本発明は、該発明の宿主細胞を用いてαGal融合ポリペプチドを生産するための方法を、さらに提供する。一実施形態では、この方法は、本発明の宿主細胞(αGal融合ポリペプチドをコードする組換え発現ベクターが導入されている)を、αGal融合タンパク質が生産されるように、適当な培地で培養することを含む。別の実施形態では、本発明はさらに培地または宿主細胞からαGalポリペプチドを単離することを、さらに含む。
【0059】
αGal融合ポリペプチドを、従来の条件(例えば、抽出、沈殿、クロマトグラフィー、アフィニティー・クロマトグラフィ、泳動、その他)にもとづいて単離および精製してもよい。例えば、免疫グロブリン融合タンパク質は、該融合タンパク質のFe部分に選択的に結合する固定化プロテインAおよびプロテインGを含むカラムに溶液を通すことによって、精製することが可能である。例えば、Reis, K. J., et al., J. Immunol. 132:3098−3102 (1984)および PCT出願公開番号 W087/00329を参照せよ。融合ポリペプチドの溶離は、カオトロピック塩処理または酢酸(1M)水溶液によっておこなうことが可能である。
【0060】
あるいは、本発明にもとづくαGal融合ポリペプチドを、公知の方法を用いて化学的に合成することができる。ポリペプチドの化学合成が記載されており、例えば、種々のタンパク質合成方法が当該技術分野で普及しており、例えばペプチド合成装置を用いた合成が挙げられる。例えば、Peptide Chemistry, A Practical Textbook, Bodasnsky, Ed. Springer−Verlag, 1988、Merrifield, Science 232: 241−247 (1986)、 Barany, et al, Intl. J. Peptide Protein Res. 30: 705−739 (1987)、 Kent, Ann. Rev. Biochem. 57:957−989 (1988)、および Kaiser, et al, Science 243: 187−198 (1989)を参照せよ。ポリペプチドの精製は、標準的なペプチド精製技術を用いて、ポリペプチドが化学物質前駆体または他の化学物質を実質的に含まないようにして、おこなう。「化学物質前駆体または他の化学物質を実質的に含まない」という言い方は、ペプチドの合成に関与する化学物質前駆体または他の化学物質から該ペプチドが分離されるペプチド調製を包含する。一実施形態では、「化学物質前駆体または他の化学物質を実質的に含まない」という言い方は、化学物質前駆体または非ペプチド化学物質が約30%(乾燥重量で)未満、より好ましくは化学物質前駆体または非ペプチド化学物質が約20%未満、さらに好ましくは化学物質前駆体または非ペプチド化学物質が約10%未満、最も好ましくは化学物質前駆体または非ペプチド化学物質が約5%未満であるペプチドの調製を包含する。
【0061】
ポリペプチドの化学合成は、修飾アミノ酸または非天然アミノ酸(D−アミノ酸および他の小有機分子が挙げられる)の取り込みを促進する。対応のD−アミノ酸アイソフォームによるペプチド内の1つ以上のL−アミノ酸の置換は、酵素的加水分解に対するペプチドの耐性を高めるために、また生物学的に活性なペプチドの1種類以上の特性(すなわち、レセプター結合、機能的潜在能力、または作用の持続時間)を高めるために、用いることができる。例えば、Doherty, et al., 1993. J Med. Chem. 36: 2585−2594、Kirby, et al., 1993. J. Med. Chem. 36:3802−3808、Morita, et al., 1994. FEBS Lett. 353: 84−88、 Wang, et al., 1993. Int. J. Pept. Protein Res. 42: 392−399、Fauchere and Thiunieau, 1992. Adv. Drug Res. 23: 127−159を参照せよ。
【0062】
ペプチド配列への共有結合的架橋の導入は、立体構造的およびトポグラフィックにポリペプチド主鎖を抑制することができる。この戦略は、能力、選択性、および安定性が高まった融合ポリペプチドのペプチド類似体の開発に用いることができる。環状ペプチドの立体構造的エントロピーは、その直鎖状の対応物よりも低いので、特異的立体構造の採用は、非環式の類似体に対するエントロピーよりも環状類似体に対するエントロピーにおいてより少ない減少を伴っておこるので、それによって、結合のための自由エネルギーがより好ましいものとなる。大環化(macrocyclization)は、しばしば、ペプチドN末端とC末端との間、側鎖とN末端またはC末端との間[例えば、K3Fe(CN)6、pH8.5](Samson et al., Endocrinology, 137: 5182−5185 (1996))、または2つのアミノ酸側鎖間での、アミド結合によって達成される。例えば、DeGrado, Adv Protein Chem, 39: 51−124 (1988)を参照せよ。ジスルフィド架橋もまた、線形配列に導入されて、該配列の柔軟性を減少させる。例えば、Rose, et al., Adv Protein Chem, 37: 1−109 (1985)、Mosberg et al., Biochein Biophys Res Commun, 106: 505−512 (1982)を参照せよ。さらに、ペニシラミン(Pen、3−メルカプト−(D)バリン)によるシステイン残基の置換が、いくつかのオピオイド・レセプター相互作用の選択性を高めるのに用いられている。Liplcowski and Carr, Peptides: Synthesis, Structures, and Applications, Gutte, ed., Academic Press pp. 287−320(1995)。
【0063】
(超急性拒絶を処置または予防する方法)
本発明は、超急性拒絶(HAR)、例えば異種移植拒絶を処置または予防する方法も含む。そのような移植組織として、限定されるものではないが、そのような移植組織は含む腎臓、肝臓、皮膚、膵臓、角膜、または心臓が挙げられ、HARの意味にはレシピエントによる任意の抗体媒介移植拒絶が含まれる。移植臓器の超急性拒絶は、臓器がレシピエントの血行にさらされてから数秒または数分のうちに生ずる。臓器は、急速に蒼白になって、レシピエントの体内に残存する場合、壊死が生ずる。この超急性反応は、ドナー臓器に対して抗体が予め形成されたため、起こる。HARは、子宮内で多数の非自己抗原にさらされていることから、経産婦で最も一般的である。輸血回数が多いレシピエントも危険にさらされている。
【0064】
この方法は、被験体由来の生体試料を本発明のαGal融合ペプチドと接触させることを含む。生体試料は、例えば血液、すなわち全血または血漿である。試料は、抗体(例えば、抗血液型抗体)を含むことが知られている、または含むと考えられている。いくつかの態様では、生体試料をαGal融合ペプチドに接触させる前に、該生体試料を被験体から回収する。生体試料を、αGal融合ペプチド− 抗血液型抗体複合体の形成を可能とさせる条件下で、融合ペプチドに接触させる。αGal融合ペプチド−複合体を、もし存在するならば、抗血液型抗体を除去するために生体試料から分離し、さらに該生体試料を被験体に再注入する。HARの処置または予防は、被験体に対して本発明のαGal融合ペプチドを投与することによってもおこなわれる。
【0065】
被験体は、例えばいずれかのほ乳類(例えば、ヒト、霊長類、マウス、ラット、イヌ、ネコ、ウシ、ウマ、ブタ)である。被験体が異種移植を受ける前に、上記処置を施す。あるいは、被験体が異種移植を受けた後に処置を施す。
【0066】
当業者に公知の方法で、生体試料をαGal融合タンパク質に接触させる。例えば、血漿分離交換法または体外免疫吸着である。
【0067】
基本的に、HARに病原学的に関連しているどのような疾患でも、予防または処置が受けいられると考える。臓器移植組織の生存率が、本発明の方法によって処置していない臓器移植組織よりも高い場合、HARが処置または予防されている。移植組織の生存率が意味することは、レシピエントによって移植組織が拒絶される前の時間である。例えば、移植組織が該移植後の少なくとも1、2、4、または8週目に生存している場合に、HARが処置または予防される。好ましくは、移植組織は、3、6、13ヶ月生存する。より好ましくは、移植組織は2、3、5、またはそれ以上の年数生存する。
【0068】
(試料からαGal抗体を除去する方法)
本発明はまた、試料から抗αGal抗体を除去または減少させる方法を含む。試料は、血液または血漿等の体液である。あるいは、試料は生体組織、例えば心臓組織、肝臓組織、皮膚、または腎臓組織である。この方法を試料を本発明のαGal融合ペプチドと接触させることを含む。αGal融合ペプチド− 抗αGal抗体複合体の形成を可能とさせる条件下で、その試料をαGal融合ペプチドと接触させる。αGal融合ペプチド−抗体複合体を、もし存在するならば、抗αGal抗体を除去または減少させるために、生体試料から分離する。
【0069】
(αGal融合ポリペプチドまたはそれをコードする核酸を含む医薬組成物)
ABO融合タンパク質、または該融合タンパク質をコードする核酸分子(本明細書では「治療薬(therapeutics)」または「活性化合物(active compounds)」ともいう)ならびにその誘導体、フラグメント、類似体、およびホモログを、投与のために適当な医薬組成物に取り込むことができる。そのような組成物は、概して核酸分子、タンパク質、または抗体および薬学的に許容される担体を含む。本明細書で用いられるように、「薬学的に許容される担体(pharmaceutically acceptable carrier)」は、薬学的投与に適合性の、任意の、および全ての溶媒、分散媒、コーティング、抗細菌剤、抗真菌剤、等張性および吸着性遅延薬剤、その他を含むことを意図しており、薬学的投与(pharmaceutical addministration)と互換性を持つ。適当な担体は、当該分野での標準的参考書であるRemington’s Pharmaceutical Sciencesの最近の版に記載されており、本明細書ではこの文献を援用する。そのような担体または希釈剤の好ましい例として、限定されるものではないが、水、食塩水、フィンガー溶液(finger’s solutions)、デキストロース溶液、および5%ヒト血清アルブミンが挙げられる。不揮発性油のようなリポソームおよび非水溶ビヒクルも使用可能である。薬学的活性物質に対するそのような媒体および薬剤の使用は、当分野では周知である。任意の従来の媒体または薬剤が活性化合物と適合しない範囲を除いて、組成物でのそれらの使用を意図している。追加の活性成分もまた、組成物に取り込むことができる。
【0070】
本明細書に開示した活性剤もリポソームとして処方することができる。リポソームの調製は、公知の方法、例えばEpstein et al., Proc. Natl. Acad. Sci. USA, 82: 3688 (1985)、Hwang et al., Proc. Natl Acad. Sci. USA, 77: 4030 (1980)、ならびに米国特許第 4,485,045号および第4,544,545号に記載された方法でおこなう。循環時間を高めたリポソームが米国特許第5,013,556号に開示されている。
【0071】
特に有用なリポソームの生成は、ホスファチジルコリン、コレステロール、およびPEG誘導化ホスファチジルエタノールアミン(PEG−PE)を含む脂質組成物による逆相蒸着法によって、おこなうことができる。リポソームを所定のポア・サイズのフィルターから押し出されることで、所望の径を持つリポソームが得られる。
【0072】
本発明の医薬組成物は、目的とする投与経路に適合するように処方される。投与経路の例として、非経口(例えば、静脈内、皮内、および皮下)、経口(例えば、吸入)、経皮(例えば、局所)、経粘膜、および直腸投与が挙げられる。非経口、皮内、または皮下適用に用いる溶液または懸濁液は、以下の成分を含むことができる。すなわち、無菌希釈剤(例えば注射用蒸留水、食塩水、不揮発性油、ポリエチレングリコール、グリセリン、プロピレングリコール、または他の合成溶媒)、抗菌剤(例えば、ベンジルアルコールまたはメチル・パラベン)、抗酸化剤(例えば、アスコルビン酸または重硫酸ナトリウム)、キレート剤(例えば、エチレンジアミン四酢酸(EDTA))、緩衝剤(酢酸塩、クエン酸塩、またはリン酸塩)、および張性調整剤(例えば、塩化ナトリウムおよびデキストロース)である。pHは、酸または塩基(例えば塩化水素または水酸化ナトリウム)で調整することができる。非経口製剤を、ガラスまたはプラスチック製のアンプル、使い捨て注射器、または多人数用バイアルに封入することができる。
【0073】
注射可能な用途に適した医薬組成物として、滅菌水溶液(水溶性)もしくは分散液、または注射可能な滅菌溶液もしくは分散液の即席調製用の滅菌粉末が挙げられる。静脈内投与用に関して、適当な担体として、生理的塩類溶液、静菌水、Cremophor EL(商標)(BASF、Parsippany、N.J.)、またはリン酸緩衝食塩水(PBS)が挙げられる。すべての場合において、上記組成物は滅菌されていなければならず、また注射が容易にとなる範囲で流動的でなければならない。それは、製造および貯蔵の条件下に安定でなければならず、また細菌および菌類等の微生物による汚染作用から保護されなければならない。担体を、例えば、水、エタノール、ポリオール(例として、グリセロール、プロピレングリコール、および液体ポリエチレングリコール)を含む溶媒または分散媒体、ならびにそれらの適当な混合物とすることができる。適当な流動性は、例えば、レシチン等のコーティングを使用することによって、分散液の場合は必要な粒径を維持することによって、および界面活性剤を使用することによって維持することができる。微生物の作用を防止することは、種々の抗菌剤および抗かび剤、例えば、パラベン、クロロプタノール、フェノール、アスコルビン酸、およびチメロサールによって達成可能である。多くの場合、等張剤、例えば糖、ポリアルコール(例として、マンニトール、ソルビトール)、塩化ナトリウム等が組成物中に含まれていことが好ましい。注射可能な組成物の長期にわたる吸着は、吸着を遅延させる薬剤(例えばモノステアリン酸アルミニウムおよびゼラチン)を組成物に含有させることによって達成することができる。
【0074】
注射可能な滅菌溶液の調製は、必要に応じて上記に列挙される成分の1つまたは組み合わせとともに、適当な溶媒中に活性化合物(例えばABO融合タンパク質)を所望量取り込み、続いて濾過滅菌することによっておこなうことができる。通常、分散液は、塩基性分散媒と上記に列挙したうちの所望の他成分とを含む滅菌ビヒクルに活性化合物を取り入れることによって、調製される。注射可能な滅菌溶液を調製するための滅菌粉末の場合、調製法は、事前に濾過滅菌した溶液から活性成分プラス任意の追加の所望成分の粉末を生ずる真空乾燥および凍結乾燥である。
【0075】
経口組成物は、一般に不活性希釈剤または食用の担体を含む。それをゼラチンカプセルに封入または錠剤に圧縮することができる。経口治療薬投与の目的で、活性化合物を賦形剤とともに取り入れることができ、錠剤、トローチ、またはカプセルの形で使用することができる。経口組成物の調製は、流動性の担体を使用しておこなうこともでき、うがい薬として試用することができる。この場合、流動担体の化合物を経口投与し、口の中を洗い、吐き出すか、または飲み込む。薬学的に互換の結合剤および/または補助材料を組成物の一部として含むことができる。錠剤、ピル、カプセル、トローチ等は、以下の成分のいずれかまたは類似の性質の化合物を含むことができる。すなわち、バインダー(例えば、微結晶性セルロース、トラガカントゴムまたはゼラチン)、賦形剤(例えば、澱粉またはラクトース、崩壊剤(例えばアルギン酸)、プリモゲル(Primogel)、またはトウモロコシ澱粉)、滑沢剤(例えば、ステアリン酸マグネシウムまたはステローツ(Sterotes))、流動促進剤(例えば、コロイド状二酸化ケイ素)、甘味料(例えば、スクロースまたはサッカリン)、あるいは香料(例えば、ペパーミント、メチルサリチレート、またはオレンジ香料)である。
【0076】
吸入投与では、適当な推進体(例えばガス(例えば二酸化炭素)を含む加圧容器もしくはディスペンサ、または噴霧器からのエアゾール・スプレーのかたちで化合物を送達する。
【0077】
全身投与は、経粘膜または経皮手段によってもおこなうことができる。経粘膜もしくは経皮投与のために、透過すべきバリヤーに対する適当な浸透剤が処方に用いられる。そのような浸透剤は周知であって、例えば、経粘膜投与用に、該浸透剤として、界面活性剤、胆汁酸塩、およびフシジン酸誘導体が挙げられる。経粘膜投与は、鼻噴霧または坐薬を用いることにより達成することができる。経皮投与のために、活性化合物は周知の軟膏、塗剤、ゲル類、またはクリームとして処方される。
【0078】
上記化合物を、直腸送達のための坐薬(例えば、カカオ脂および他のグリセリド等、従来の坐剤基剤による)または保持浣腸の形で調製することもできる。
【0079】
一実施形態において、活性化合物は担体とともに調製され、該担体は、身体から化合物が迅速に除去されないように保護するもので、例えばインプラントおよびマイクロカプセル化した送達系等の徐放製剤である。エチレンビニルアセテート、ポリ無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル、およびポリ乳酸等の生分解性の生体適合性ポリマーを使用することができる。そのような製剤の調製のための方法は、当業者にとって明らかである。上記材料は、Alza CorporationおよびNova Pharmaceuticals, Inc.から商業的に入手することができる。リポソーム懸濁液(ウイルス抗原に対する単クローン抗体により感染細胞を標的化したリポソームを含む)を、薬学的に許容される担体として使うこともできる。これらは、米国特許第号4,522,811号に記載されているように、当業者に知られている方法によって調整することができる。
【0080】
いくつかの実施形態では、経口のまたは非経口組成物は、投与の容易さと投薬量の均一性とのために投薬量単位形状で処方される。本明細書で用いられるように投薬量単位の形態は、処置される被験体のための単位投薬量として適している物理的に分離した単位をいう。すなわち、各々の単位は、必須の薬学的担体に関連して所望の治療効果を得るために計算された所定量の活性化合物を含む。本発明の投薬量単位形態の仕様は、活性化合物のユニークな特徴、達成すべき特定の治療効果、および個人の治療のためのそのような活性化合物を合成する当該技術分野固有の限界に指図され、かつ直接的に依存している。
【0081】
本発明の核酸分子をベクターに挿入して遺伝子治療ベクターとして用いることができる。遺伝子治療ベクターは、例えば静脈注射、局所投与(例えば、米国特許第5,328,470号を参照せよ)、または定位的注射(例えば、Chen, et al., 1994. Proc. Natl. Acad. Sci. USA 91: 3054−3057)によって達成することができる。 遺伝子治療ベクターの製剤は、許容可能な希釈剤に遺伝子治療ベクターを含むことができ、あるいは遺伝子送達ビヒクルが包埋される徐放マトリックスを含むことができる。あるいは、完全な遺伝子送達ベクターを、組換え型の細胞(例えばレトロウイルス・ベクター)からインタクトなかたちで生ずることができる場合は、製剤は遺伝子送達系を生ずる1種類以上の細胞を含むことができる。
【0082】
必要に応じて、徐放製剤を調製することができる。徐放製剤の適当な例として、抗体含有固体疎水性ポリマーの半透性マトリックスが挙げられ、該マトリックスは、膜等の造形品の形状、またはマイクロカプセルの形状である。徐放性マトリックスの例として、ポリエステル類、ヒドロゲル(例えば、ポリ(2−ヒドロキシエチルーメタクリレート)、またはポリ(ビニルアルコール))、ポリラクチド(米国特許第3,773,919号)、L−グルタミン酸とγエチル−L−グルタミン酸塩との共重合体、非分解性エチレンビニルアセテート、分解性乳酸−グリコール酸共重合体(例えば、LUPRON DEPOT(商標)(乳酸−グリコール酸共重合体およびロイプロイドアセテートから構成される注射可能なミクロスフェア )、およびポリ−D−(−)−3−ヒドロキシ酪酸が挙げられる。エチレン−ビニルアセテートおよび乳酸グリコール酸のようなポリマーが100日以上にわたって分子の放出を可能にする一方で、特定のヒドロゲルはより短時間のあいだにタンパク質を放出する。
【0083】
医薬組成物を、投与のためのインストラクションと共に、容器、パック、またはディスペンサに入れることができる。
【0084】
(略語)
以下の略語が本明細書で用いられる。すなわち、ADCC、抗体依存性細胞障害; BSA、ウシ血清アルブミン; DXR、遅発性異物性拒否;ELISA、酵素結合免疫吸着検定法; FT、フコシル転移酵素;Gal、D−ガラクトース; GT、ガラクトシル基転移酵素;Glc、D‐グルコース; G1cNAc、D−Nアセチルグルコサミン; G1yCAM−1、グリコシル化依存性細胞接着分子−1;HAR, 超急性拒絶;Ig、免疫グロブリン; MAdCAM、粘膜アドレシン細胞接着分子; PAEC、ブタの大動脈血管内皮細胞; PBMC、末梢血単核細胞; PSGL−1、Pセレクチン糖タンパク質リガンド−1;RBC、赤血球; SDS−PAGE、ドデシル硫酸ナトリウム−ポリアクリルアミドゲル電気泳動法; Hex、ヘキソース; HexNAc、N−アセチル・ヘキソサミン; NeuAc、N−アセチル・ノイラミン酸; NeuGc、N−グリコリルノイラミン酸;およびHexNolはN−アセチル・ヘキソサミンの開いた(環でない)形状である。
【0085】
本発明を、以下の非限定的例で更に説明する。
【実施例】
【0086】
(実施例1:置換組換えPセレクチン糖タンパク質リガンド/免疫グロブリン融合タンパク質の一時的発現)
(一般的方法)
(細胞培養)
COS−7 m6細胞(35)およびSV40ラージT抗原不死化ブタ大動脈内皮細胞株PEC−A(36)を10%ウシ胎児血清(FBS)および25μg/ml硫酸ゲンタマイシンを含むダルベッコの改質イーグル培地(DMEM)で継代した。ヒトの赤血白血病細胞株K562とバーキットリンパ腫細胞株RajiをATCCから入手し、10%FBS、100IU/mlペニシリン、および100μg/mlストレプトマイシンを含むHEPES緩衝RPMI 1640で培養した。
【0087】
(発現ベクターの構築)
ブタα1,3GT(37−39)を、コーディング配列の5′末端、コザック(Kozak)翻訳開始コンセンサス配列、およびHind3制限部位に相補的な6つのコドンを持つフォワード・プライマーと、コーディング配列の3′末端、翻訳停止およびNot1制限部位に相補的な6つのコドンを持つリバース・プライマーとを用いて、ブタ脾臓cDNAからPCR増幅した。増幅1,3GTcDNAを、Hind3およびNot1(35)を用いて、CDM8のポリリンカーにクローニングした。P−セクレチンへの結合を媒介する高度にグリコシル化したムチン型のタンパク質であるPセレクチン糖タンパク質リガンド−1 (PSGL−1)(40)をコードする配列を、HL−60cDNAライブラリーからPCRによって得て、Hind3およびNot1を用いてCDM8にクローニングし、さらにDNAの塩基配列決定(シーケンシング)をおこなって確認した。ムチン免疫グロブリン発現プラスミドは、フレーム単位のPSGL−1の細胞外部分のPCR増幅cDNAをBamH1部位を介して、CDM7(Seed, B. et al)の発現カセットとして担持されたマウスIgG2のFe部分(ヒンジ、CH2およびCH3)に融合させることによって、構築した。
【0088】
(分泌ムチン/免疫グロブリン・キメラの生産および精製)
COSm6細胞を、DEAE−デキストラン・プロトコールおよび1μgのCsSI勾配生成プラスミドDNA/mlトランスフェクション・カクテルを用いてトランスフェクトさせた。COS細胞を、空ベクター(CDM8)、PSGL1/mIgG2bプラスミド単独またはα1,3GTコード化プラスミドとの組み合わせにより、約70%密集度でトランスフェクトさせた。トランスフェクション細胞を、トランスフェクションの翌日にトリプシン処理して新しいフラスコへ移した。約12時間にわたる付着の後で、培地を捨て、細胞をリン酸緩衝食塩水(PBS)で洗い、その後、新たに7日間にわたって無血清AIM−V培地(cat.nr. 12030, Life technologies Inc.)で培養した。培養後、上清を回収し、デブリスを遠心(1,400xg、20分)により沈殿させ、NaN3を0.02%まで添加した。PSGLI/mIgG2b融合タンパク質を、4℃で一晩、ヤギ抗マウスIgGアガロース・ビーズ(A−6531、Sigma)上で転倒回転させて精製した。ビーズをPBSで洗い、続いてSDS−PAGEおよびウエスタンブロット分析、またはヒトAB血清および精製ヒト免疫グロブリンの吸着に用いた。
【0089】
(ヒトIgG、IgM、およびIgAの精製)
ヒトIgG、IgM、およびIgAを、ヤギ抗ヒトIgG(Fe特異的、A−3316,Sigma)、IgM(μ鎖特異的、A−9935、Sigma)、およびIgA(α鎖特異的、A−2692、Sigma)アガロース・ビーズを用いて、ヒトAB血清(20人を上回る人数の健康血液ドナーからプールした)から精製した。手短に言うと、5mlのスラリー(2.5ml充填ビーズ)を、10mm直径のカラムに注入し、PBSで洗浄した。プールしてあるヒトAB血清10mlを、ペリスタル型ポンプを使用して1ml/分で適用し、カラム容量のPBSのPBSで数回洗浄し、1ml/分の流速で0.1Mグリシン、0.15M NaCl、pH2.4により溶離させた。1mlのフラクションを、0.7mlの中和緩衝液(0.2Mトリス/塩酸、pH 9)を含んでいる試験管に回収した。280nmの吸着を分光光度的に読み取り、タンパク質を含んでいる試験管をプールし、1%PBSに対して透析し、さらに凍結乾燥した。凍結乾燥免疫グロブリンを蒸留水に再懸濁し、その濃度を1gGについては16mg/ml、IgAについては4mg/ml、さらにIgMについては2mg/mlに合わせてた。
【0090】
(SDS−PAGEおよびウエスタン・ブロット分析)
SDS−PAGEは、垂直型Mini−Protean II電気泳動システム(Bio−Rad, Herculus, Calif.)(41)を用いて5%の濃縮用ゲルと6または10%の分離ゲルとによるLeammliの方法で、おこなった。Mini Trans−Blot電気泳動トランスファー・セル(Bio−Rad, Herculus, Calif.) (42)を用いて、分離タンパク質をHybond(商標)−Cエクストラ・メンブレン(Amersham)上にブロッティングした。タンパク質ゲルの染色を、製造元の指示(Bio−Rad, Herculus, Calif.)に従って、銀染色キットを使用しておこなった。3%BSA含有PBSで少なくとも2時間にわたってブロッキングした後、上記メンブレンを、室温で、0.2mMCaCl2含有PBS(pH6.8)に1μg/mlの濃度で希釈されたペルオキシダーゼ共役バンデイレイア・シンプリシフォリア(Bandereia simplicifolia)イソレクチンB4(L−5391、Sigma)により、2時間プロービングした。メンブレンをPBS(pH6.8)で5回洗浄し、製造元(Amersham)の指示に従ってECL(商標)キットを用いたケミルミネセンスで結合レクチンを視覚化した。
(抗マウスIgFcELISAによるPSGLb1/mgG2bの定量化)
吸着前後の細胞培養上清の融合タンパク質濃度の測定は、融合タンパク質を親和性精製多クローン性ヤギ抗マウスIgGFc抗体(cat.nr. 55482, Cappel/Organon Teknika, Durham, N.C.)で捕獲する96穴ELISAアッセイによっておこなった。3%BSA含有PBSでブロッキングした後、融合タンパク質を捕獲し、O−フェニレンジアミン二塩化水素化物(Sigma)を基質として用いてペルオキシダーゼ共役親和性精製多クローン性抗マウスIgGFc抗体(cat.nr. 55566, Organon Teknika, Durham, N.C.) で検出した。プレートを492nmで読み取り、AIMV無血清培地に再懸濁した精製マウスIgGFcフラグメント((cat.nr. 015−000−008, Jackson lmmunoResearch Labs., Inc., West Grove, Pa.)の希釈シリーズを用いてELSAのキャリブレーションをおこなった。
【0091】
(ブタ大動脈血管内皮細胞ELISA)
PEC−A細胞を、ゼラチン・コーティング96穴プレート(Nunclon, Denmark)に15,000細胞/ウエルの密度で播種し、AIMV無血清培地で48時間インキュベートした。プレートを、0.15MNaCl2含有0.02%Tween20で5回洗浄し、50μl/ウエルの精製ヒトIgG、IgM、およびIgAを含むPBSで、各々の開始濃度を8、1、および2mg/mlとして、室温で1時間培養した。上記のようにプレートを再び洗浄し、50μlアルカリ・ホスファターゼ共役ヤギ抗ヒトIgG(γ鎖特異的;A3312、Sigma)、IgM(μ鎖特異的;A1067、Sigma)、およびIgA(α鎖特異的;A3062、Sigma)F(ab)’2フラグメント含有PBS(1:200希釈)を添加し、室温で1時間にわたりインキュベートした。プレートを上記のように洗浄し、基質p−ニトロフェニル・リン酸(Sigma104−105)でインキュベートし、405nmで読み取った。
【0092】
(ブタ大動脈血管内皮細胞細胞毒性アッセイ)
PEC−A ELISAについて記載したように、PEC−A細胞を96穴プレートに播種して培養した。48時間培養した後、細胞をNa251CrO4(cat.nr. CJS4、Amersham)1μCi/ウエルで37℃、1時間にわたりロードし、AIMV培地で3回洗浄した。15μlの連続希釈した吸着または非吸着ヒトAB血清または精製ヒトIgG、IgA、もしくはIgM抗体を、補体源としての50μlのウサギ血清(Cat. no. 439665、Biotest AG, Dreieich, Germany)とともに添加した。5%CO2雰囲気下、37℃で4時間インキュベートした後、上清をSkatron上清回収システム(Skatro Instruments, Norway)を用いて収集し、ガンマ・カウンター(1282 Compugamma, LKB Wallac)で分析した。各血清およびIg試料を三重反復分析した。パーセント致死を、最大放出と自然発生的放出との差で割った自然発生的放出で差し引いた測定放出として、計算した。
【0093】
(抗体依存性細胞毒性(ADCC))
ヒトPBMCを、南(South)病院(ストックホルム)の血液バンクで、健康なドナーから調製した新鮮な軟膜 (バフィー・コート)を単離した。6mlのバフィー・コートと、1mg/mlBSAおよび3.35mg/mlEDTAを含む15mlのPBSとを、50mlポリプロピレン管内で混合した。10分間にわたり500gで遠心した後、血小板リッチな上清を捨てて、6mlの下部相を6mlのハンクス液(HBSS)と混合し、さらに6mlLympoprep(商標)(Nycomed Pharma AS)で下層を形成した。遠心後(800xg、20分)、界面を新しい管に移し、HBSSで3回洗浄し、さらに無血清AIMV培地に再懸濁した。エフェクター細胞調製の最終ステップは、37℃および5%CO2で1時間インキュベートしてプラスチック粘着細胞を取り除いた組織培養フラスコに、PBMCを移すことであった。標的細胞は、上記のように保ったK562およびRaji細胞、またはPEC−A細胞であり、これらの細胞をアッセイの前日にトリプシン処理し、その後AIMV無血清培地で培養することでプラスチック表面への再粘着を防いだ。アッセイの時点で、PEC−A細胞をフラスコの底から洗い落とした。標的細胞をNa251CrO4、100μCi/1x106細胞で、37℃で1時間にわたりロードし、HBSSで3回洗浄し、さらにAIMVに再懸濁することで、最終濃度を5x104/mlとした。5、000個の標的細胞を、2倍希釈で50:1から6.25:1の範囲にあるエフェクター細胞(E):標的細胞(T)比で、10%加熱不活性化ヒトAB血清有りまたは無しの200μlAIMV培地に含まれたエフェクター細胞がある各ウエルに加えた。
【0094】
自然発生的放出を、エフェクター細胞無しのAIMV培地200μlでインキュベートした5,000個の標的細胞を有するウエルで読み取り、最大放出を、AIMV培地100μlで5,000個の標的細胞を100μlの5%TritonX−100とともにインキュベートしたウェルで読み取った。各E:T比を三重反復分析した。37℃で4時間インキュベートした後、上清をSkatron上清回収システム(Skatro Instruments, Norway)を用いて収集し、ガンマ・カウンター(LKB Wallac)で分析した。パーセント致死を、最大放出と自然発生的放出との差で割った自然発生的放出で差し引いた測定放出として、計算した。
【0095】
(結果)
(PSGL1/mIgG2b融合タンパク質の発現および特徴付け)
ベクター・プラスミドCDM8、PSGL1/mIgG2bプラスミド、またはブタα1,3GTプラスミドとともにPSGL1/mIgG2bプラスミドによりトランスフェクションしたCOS−7m6細胞由来の上清を、トランスフェクション後、約7日目に回収した。分泌ムチン/Ig融合タンパク質を、抗マウスIgGアガロース・ビーズ上での吸着によって精製し、検出のためにバンデイレイア・シンプリシフォリア(Bandereia simplicifolia)イソレクチンB4 (BSA IB4)を用いたSDS−PAGEおよびウエスタンブロット分析にかけた。図1に示すように、融合タンパク質は、銀による染色が相対的に弱い145kDaの見かけ分子量を持つ広帯域として、還元条件下で移動した。大きさの不均一性(約125ないし165kDa)および低染色性は、高グリコシル化ムチン型タンパク質(43,44)の挙動に関して既におこなわれた観察と一致する。融合タンパク質は、おそらくホモ二量体として生産される。なぜなら、非還元条件下でのSDS−PAGEによって、見かけ分子量が250kDaを上回る二重のバンドが見られたためである。PSGL1/mIgG2bプラスミド単独またはα1,3GTプラスミドとともにトランスフェクションした同一数のCOS−7m6細胞に由来する2種類の上清から親和性精製された融合タンパク質の量は、同じであった。BSA IB4によってエレクトロブロッティング・メンブレンをプロービングすることで、ブタα1,3GTによるトランスフェクション後に得られた融合タンパク質の強い染色が明らかになった(図1)。しかし、COS細胞がシミアン(Simian)サル(α1,3活性が欠けた旧大陸サル)由来であるという事実にもかかわらず、α1,3GT cDNAの同時トランスフェクションなしでCOS−7m6細胞で生産されたPSGL1/mIgG2bもまたBSA IB4による染色が弱かった。このことは、BSA IB4レクチンがまさしくGa1α1,3Galエピトープ(45)よりもわずかに広い特異性を持つことを示している。それにもかかわらず、ブタα1,3GTcDNAの同時トランスフェクションは、高度にGalα1,3Gal置換PSGL1/mIgG2b融合タンパク質の発現を支持した。
【0096】
トランスフェクションしたCOS細胞の上清中、および吸着後のヤギ抗マウスIgGアガロース・ビーズ上でのPSGL1/mIgG2bの定量化。トランスフェクションしたCOS細胞の上清に含まれるPSGL1/mIgG2bの量を定量するために、サンドイッチELISAを用いた。一般に、材料および方法で説明したように、70%コンフルエンスでCOS細胞を含む5本の培養フラスコ(260mlフラスコ、Nunclon(商標))をトランスフェクトさせ、かつインキュベートした。フラスコ1本あたり10mlのAIMV培地中で7日間にわたるインキュベーション期間の後、培地を回収した。そのようなトランスフェクションからの上清に含まれる融合タンパク質の濃度を、抗マウスIgGアガロース・ビーズ(50μl充填ビーズに対応)の100μlゲル・スラリー上での吸着後の異なる容量の上清と同様に、精製マウスIgGFcフラグメントで較正したELISAによって測定した(図2)。上清に含まれるPSGL1/mIgG2bの濃度は、150ないし200ng/μlの範囲内であり、この特定の実験では、その濃度は約160ng/μlであった(図2A、非吸着カラム)。50pl充填抗マウスIgGアガロース・ビーズ上に吸着した後の2、4、および8mlの上清に残るPSGL1/mIgG2bの濃度は、それぞれ32、89、および117ng/μlであった。このことは、2、4、および8mlの上清由来の50μlの充填抗マウスIgGアガロース・ビーズ上に吸着されている260、290、および360ngのPSGL1/mIgG2bに一致する。B.シンプリシフォリア(B.simplicifolia)1134レクチンによるウエスタン・ブロット分析によって、50μlの充填ビーズが1mlの上清からPSGL1/mIgG2b融合タンパク質を吸着しうること、また2mlではやっと気づくほどのレベル(不図示)であることが明らかになった。
【0097】
固定化Galα1,3Gal−置換PSGL1/mIgG2bの吸着能力。PSGL1/mIgG2bプラスミド単独で、またはブタα1,3GTcDNAとともにトランスフェクションしたCOS細胞由来の上清20mlを抗マウスIgGアガロース・ビーズのゲル・スラリー500μlと混合した。しっかりと洗浄した後、ビーズを等量ずつ分け、100μlのゲル・スラリー(50μl充填ビーズ)を0.25、0.5、1.0、2.0、および4.0mlのプールされた補体減少ヒトAB血清と混合し、4℃で4時間にわたり転倒回転させた。PSGL1/mIgG2bおよびGalα1,3Gal置換PSGL1/mIgG2b上での吸着の後、4時間51Cr放出アッセイを用いてウサギ補体の存在下、血清をブタ内皮細胞細胞毒性についてアッセイした(図3)。図3に示すように、各希釈ステップで約300ngのPSGL1/mIgG2b(上記参照)を担持するビーズ100μlが4および2mlのAB血清の細胞毒性を減ずることができ、また1ml以下のヒトAB血清に存在する細胞毒性を完全に吸着することができる。注意すべきことは、非Galα1,3Gal置換PSGL1/mIgG2bの同一量が0.25ml吸着ヒトAB血清の細胞毒性をわずかだけ減少させることである(図3)。
【0098】
(補体依存ブタ内皮細胞細胞毒性および結合に対するGalα1,3Gal置換PSGL1/mIgG2bの効果)
PSGLI/mlgG2bが個々のヒト免疫グロブリンクラスを吸収することができた効率を調べるために、抗ヒトIgG、IgM、およびIgAアガロース・ビード上で、免疫アフィニティークロマトグラフィーによってヒトAB血清から、ヒトIgG、IgM、7およびIgAを精製した。単離後、IgAを抗IgGおよびIgMカラムに通過させ、IgGおよびIgMの痕跡を取り除く。この手順を、同様に他のIgクラスについても実行した。Ig分画純度は、SDS−PAGEでチェックした(図4)。正常血清で見いだされる濃度で、ヒトIgGおよびIgM(IgAではない)をウサギ補体の存在下、PEC−Aに対して細胞毒性を示した(図5)。IgGおよびIgM分画に存在する細胞毒性は、Galα1,3Gal−置換PSGL1/mIgG2b上での吸着によって、完全に除去された。IgA分画によって示される細胞毒性の欠如がPEC−Aに対するヒトIgA抗体の結合の欠如によるものかどうかを調べるために、細胞ELISAを、結合IgG、IgM、およびIgA抗体を検出するために細胞毒性アッセイで使用された同一Ig分画で実施した。アルカリホスファターゼ共役クラス特異的F(ab)′2フラグメントを2次抗体として用いた。IgGおよびIgMの細胞毒性がGalα1,3Gal−置換PSGL1/mIgG2bの吸着によって完全に取り除かれても、IgGに関しては70%(30から70%の範囲)を上回って、IgMに関しては55%(10から55%の範囲)を上回っては、結合は決して減少しなかった(図5)。ヒト免疫グロブリンAは明らかにPEC−Aと結合し、結合はGalα1,3Gal−置換PSGLI/mIgG2b上での吸着後に、わずかに減少しただけだった(29%以下)。したがって、IgA分画の細胞毒性の欠如は、IgAフラクションがPEC−Aに結合できないことによって説明することはできず、補体を活性化させることによると思われる。
【0099】
(ブタ内皮細胞のADCCに対するGalα1,3Gal−置換PSGL1/mIgG2bの効果)
いくつかのアッセイが無血清条件下でおこなわれ、K562およびRaji細胞と比較した場合、PEC−Aは新たに単離したPBMCによって致死を指示する中間の感度を有し、K562はヒトNK細胞による致死に対して感度が高く、Rajiは鈍感であった(図6A)。10%ヒト補体不活性化AB血清の存在下、致死率がほぼ倍となり、以前の公表データ(30)と一致して生体外(in vitro)でのADCC効果が支持される(図6B)。しかし、全てのPEC−A細胞毒性抗体(上記参照)を取り除くことが知られている条件下で、もし血清がGalα1,3Gal−置換PSGL1/mgIgG2bによって吸着されると、致死率は無血清条件下で見られる致死率よりもわずかに低いレベルまで減少する。一方、Galα1,3Galエピトープを持たないPSGL1/mIgG2b融合タンパク質それ自体は、ヒトAB血清存在下で致死率の増加を引き起こすことはできない(図6B)。これらのデータは、Galα1,3Gal特異性による抗ブタ抗体が生体外(in vitro)で抗体依存性細胞性細胞毒性を支持することができるということ、またそれが効果的に補体結合の細胞毒性の抗ブタ抗体を除去するちょうどその時、Galα1,3Gal−置換PSGL1/mIgG2b融合タンパク質はこれらの抗体を効果的に除去すること、という考えを支持する。
【0100】
(参考文献)
【0101】
【数1】
【0102】
【数2】
【0103】
【数3】
【0104】
【数4】
【0105】
【数5】
【0106】
【数6】
(実施例2: 置換された組換えPセレクチン糖タンパク質リガンド/免疫グロブリン融合タンパク質の安定な発現)
(一般法)
(細胞培養)
CHO−K1、COS7m6、293T、およびブタ大動脈内皮細胞株PEC−A(Khodadoust, M. M. et al., 1995)を10%胎児ウシ血清(FBS)および25μg/ml硫酸ゲンタマイシンを含むダルベッコ改変イーグル培地(DMEM)中で培養した。選択培地は、下記に示すように、ピューロマイシン(cat. no. P7255; Sigma, St. Louis, MO. 63178)、ハイグロマイシン(cat. no. 400051; Calbiochem, La Jolla, CA 92039)、およびG418(cat. no. G7034; Sigma, St. Louis, MO 63178)を含有していた。
(発現プラスミドの構築)
記載(Liu, J. et al., 1997)されるように、ブタα1,3Ga1T(Gustafsson, K. et al., 1994)およびPSGL−1/mIgG2b発現プラスミドを構築した。フォワードおよびリバース・プライマーとして、それぞれcgcgggctcgagatgaagatattcaaatgtおよびcgcggggcggccgctcatgatgtggtagtgagatを用いて、HL60 cDNAライブラリからC2 GnTI cDNAをPCRによって増幅した。安定なトランスフェクタントを生成するために用いられるベクターは、両方向性であり、ポリリンカーの上流のEF1αプロモーター、スプライス・ドナーおよびアクセプター部位、ならびにSV40の両方向性ポリ(A)付加シグナルを有していた。この転写ユニットとは反対の配向で、かつ逆方向からのポリ(A)シグナルを用いているのが、HSV TKプロモーターに続いてピューロマイシン・アセチル転移酵素(EF1α/PAC)、ハイグロマイシンb(EF1α/Hyg)、およびネオマイシン(EF1α/Neo)耐性遺伝子(N. Chiu, J. Holgersson and B. Seed)のコーディング配列からなる第2の転写ユニットであった。HindIIIおよびNotIを用いて、ブタα1,3GalTおよびPSGL−l/mIgG2bのcDNAをEF1α/HygおよびEF1α/PACベクターにそれぞれ交換した。XhoIおよびNotIを用いて、C2GnTI遺伝子をEF1α/Neoに交換した。
【0107】
(DNAトランスフェクションおよびクローン選択)
付着CHO−K1、COS7m6、および293T細胞を75cm2T型フラスコに播種し、約24時間後に、70ないし80%の細胞集密(cell confluency)でトランスフェクションした。改変ポリエチレンイミン(PEI)トランスフェクション法をトランスフェクションに対して用いた(Boussif, O. et al., 1995; He, Z. et al., 2001)。トランスフェクション後24時間に、各T型フラスコ中の細胞を5つの100mmペトリ皿に分け、選択培地中でインキュベートした。選択培地中のピューロマイシンの濃度は、CHO−K1、COS7m6、および293T細胞に対して、それぞれ6.0、1.5、および1.0μg/mlであった。CHO−K1、COS7m6、および293T細胞に対して、それぞれ550、50、および100μg/mlのハイグロマイシンb濃度を用い、CHO−K1細胞に対して、900μg/mlのG418濃度を用いた。選択培地を3日毎に変えた。約2週間後に、薬剤耐性クローンが形成した。クローンを顕微鏡下で同定し、ピペットマンを用いて手で採取した。選択したコロニーを、選択薬剤の存在下96穴プレート中でさらに2週間培養した。細胞が80ないし90%集密に達したとき細胞培養上清を回収し、ヤギ抗マウスIgG Fc抗体を用いて、ELISA、SDS−PAGE、およびウエスタン・ブロット法によって、PSGL−1/mIg2bを評価した。最も高いPSGL−1/mIgG2b発現を有するCHO−K1、COS7m6、および293Tクローンを、プラスミドをコードするブタα1,3Ga1Tでトランスフェクトし、ハイグロマイシン含有培地中で選択した。ヤギ抗マウスIgG Fc抗体と、末端αGalを認識するGSA I
1B4−レクチンとの両方を用いて、ELISA、SDS−PAGE、およびウエスタン・ブロットによって、耐性クローンを単離および特徴付けした。PSGL−1/mIgG2b上で相対的に高いαGal発現を有する2つのCHOクローンをC2 GnTIでさらにトランスフェクトして、G418含有培地中で選択した。より多くの複合体O−グリカンを示すPSGL−1/mIgG2bの大きさの増加によって、C2 GnTIの発現が確認された。
【0108】
(SDS−PAGEおよびウエスタン・ブロット法)
垂直型Mini−Protean II電気泳動システム(Bio−Rad, Hercules, CA, USA)を使用して、5%スタッキング・ゲルおよび8%分離ゲルを用いて、Laemmliの方法(Laemmli, U. K., 1970)によってSDS−PAGEをおこなった。還元および非還元条件下で、試料を電気泳動にかけた。分離度を増加するために、4ないし15%勾配ゲル(cat.no. 161−1104; Bio−Rad, Hercules, CA, USA)または4ないし12%勾配gels(cat.no NP0322; Invitrogen, Lidingo, Sweden)を複数の実験で用いた。後者のゲルは、MES緩衝液(cat.noNP0002; Invitrogen)と組み合わせて用いた。高精度タンパク質標準(cat.no RPN756; Amersham Biosciences, Uppsala, Sweden)をタンパク質分子量測定のための基準として適用した。Rubyと組み合わせてPro Q Emerald 300糖タンパク質検出キット(cat.no P21855; Molecular Probes, Leiden, The Netherlands)を用いて、タンパク質ゲルを染色した。CCDカメラを搭載するFlour−S Max MultiImagerで、これらのゲルを可視化した。Mini TransBlot(Bio−Rad)電気泳動トランスファー・セルを用いて、Hybond C外膜(cat.no. RPN203E; Amershain Biosciences)またはニトロセルロース膜(cat.no LC2001; Invitrogen)上に分離されたタンパク質も電気泳動的にブロットした(Towbin, H. et al., 1979)。0.2Tween20を含むPBS中3%BSAで1時間ブロッキングした後、1μg/ml濃度に希釈したペルオキシダーゼ結合GSA I IB4−レクチン(cat.no. L−5391; Sigma)、1:1,000で希釈したペルオキシダーゼ結合ヤギ抗マウスIgG Fc抗体(cat.no. A−9917; Sigma)、および1:1,000で希釈したマウス抗PSGL−1抗体(クローンKPL−1、cat.no 557502; BD PharMingen, San Diego, CA)を用いて、膜を室温で1時間プロービングした。2次抗体は、1:50,000で希釈したペルオキシダーゼ結合ヤギ抗マウスIgG F(ab)′2(cat.no A 2304; Sigma)であった。ブロッキング緩衝液で、全ての希釈をおこなった。インキュベーション間および後で、0.2%Tween20を含有するPBSで膜を3回洗浄した。ECLキット(cat.no. RPN 2106; Amersham Biosciences)を用いて、製造元の指示にしたがい、化学発光によって結合レクチンおよび抗体を可視化した。
【0109】
(PSGL−1/mIgG2b上のαGalエピトープ密度および酵素免疫検定法を用いたPSGL−1/mIgG2bの定量)
二抗体サンドイッチELISAによって、細胞培養上清中の組み換えPSGL−1/mIgG2bの濃度と、その相対的α−Galエピトープ密度とを測定した。20μg/mlの濃度で、アフィニティー精製ポリクローナル・ヤギ抗マウスIgG Fc抗体(cat. nr. 55482; Cappel/Organon Teknika, Durham, NC)で96ウェルELISAプレートを一晩4℃で被覆した。PBS中の1%BSAを用いて、該プレートを1時間ブロッキングした。PSGL−1/mIgG2bを含有する上清を4時間インキュベートし、その後、0.5%(v/v)Tween20を含有するPBSで3回洗浄した。洗浄後、1:3,000希釈のペルオキシダーゼ結合抗マウスIgG Fc抗体(cat.no. A−9917; Sigma)または1:2,000で希釈したペルオキシダーゼ結合GSA I IB4−レクチン(cat.no. L−5391;Sigma)でプレートを2時間インキュベートした。3,3′,5,5′−テトラメチルベンジジンジハイドロクロライド(cat. nr. T−3405; Sigma, Sweden)を用いて、結合ペルオキシダーゼ結合抗体またはペルオキシダーゼ結合GSA−レクチンを可視化した。2M H2SO4によって、反応を停止し、450nmでプレートを読み取った。較正に対して、融合タンパク質生成のために用いられる培地中または1%BSAを含有するPBS中で再懸濁した精製マウスIgG Fcフラグメント(cat. Nr. 015−000−008; Jackson ImmunoResearch Labs., Inc., West Grove, PA)の希釈系列を用いて、PSGL−1/mIgG2b濃度を評価した。2つのELISAから得た相対的O.D.(GSA反応性/抗マウスIgG反応性)を比較することによって、α−Galエピトープ密度を測定した。
【0110】
(ブタ大動脈内皮細胞細胞毒性アッセイ)
記載(Liu, J. et al., 2003)されるように、ブタ大動脈内皮細胞(PAEC)細胞毒性アッセイを行った。細胞毒性を最大の40%(y=0.4)に減少させるために各細胞クローンから必要とされるPSGL−1/mIgG2bの量を、各融合タンパク質に対する測定値の直線回帰後に得た曲線を記述する式から推論し、その後対応するx値(マイクログラム吸着体)を算出した。
【0111】
(CHOクローンの撹拌フラスコ・バッチ培養)
6.0x107個の細胞(90ないし100%集密を持つ細胞を含む10個の175cm2T型フラスコを表す)を用いて、各バッチ培養を開始した。トリプシン(0.5mg/ml)・EDTA(0.2mg/ml)での消化後に、細胞を少量の培地中で再懸濁し、200xgで5分間遠心して、過剰なトリプシンを除去した。Burkerチャンバー
中の細胞を計数することによって細胞密度を測定し、培地を添加して、最終濃度3.0x105個細胞/mlにした。細胞懸濁物を1L撹拌フラスコに移し、速度60rpmで培養物を撹拌するために細胞回転装置(Integra Biosciences, Wallisellen, Switzerland)を用いた。単独またはC2 GnTIとの組合せでα1,3GalTを発現するCHO−K1細胞を分泌するPSGL−1/mIgG2bを、それぞれピューロマイシン(200μg/ml)またはピューロマイシン(200μg/ml)およびG418(500μg/ml)の存在下で培養した。細胞を2日毎に計数した。細胞密度が5.0x105個細胞/mlに達したとき、細胞密度がもう1度3.0x105個細胞/mlに等しくなるように新たな培地を添加した。細胞懸濁物量が1,000mlになるまでこれを繰り返した。その後、細胞生存率が50%に減少するまで、細胞を持続的に培養した。
【0112】
(組み換えPSGL−1/mIgG2bの精製)
20分間1,420xgで遠心することによって、堆積物から上清を取り除いた。取り除いた上清を、ヤギ抗マウスIgG(分子全体)−アガロース(cat.no. A 6531; Sigma)10mlを含有するカラムに流速0.5ml/分で通した。PBS120mlでの洗浄後に、3M NaSCN120mlで、結合融合タンパク質を溶出した。検出用の抗マウスIgGを用いたSDS−PAGEおよびウエスタン・ブロット法による分析に続いて、融合タンパク質を含有する管の内容物をプールした。PSGL−1/mIgG2bを含む画分を蒸留水に対して透析し、凍結乾燥し、蒸留H2Oの1ないし2ml中で再懸濁した。融合タンパク質の濃度をELISAによって測定した。より低い分子量の夾雑物を除去するために、FPLC(Pharmacia Biotech, Sweden)を用いて流速0.5ml/分でPBSで溶出させるHiPrep 16/60 Sephacryl S−200 HRカラム(cat.no. 17−1166−01; Amersham Biosciences, Uppsala, Sweden)上でのゲル濾過によって、融合タンパク質をさらに精製した。5ml画分を回収し、280nmでのUV分光測光によって、タンパク質を含有する管を同定した。SDS−PAGEおよびウエスタン・ブロット法によって、プールした画分を再び分析し、蒸留水中でプール、透析、および再懸濁した。
【0113】
(精製PSGL−1/mIgG2bからのO結合グリカンの化学的放出および過メチル化)
記載(Carlstedt, I. et al., 1993)されるように、β脱離によってオリゴ糖を放出させた。放出されたオリゴ糖を45℃窒素流動下で気化し、記載(Hansson, G. C. et al., 1993)されるように、わずかな修飾で、CiucanuおよびKerek(Ciucanu, I. et al., 1984)にしたがって、過メチル化した。
【0114】
(質量分析法)
LCQイオン・トラップ質量分析計(ThermoFinnigan, San Jose, CA)を用いて、陽イオンモードでのエレクトロスプレー・イオン化質量分析法(ESI−MS)を実行した。メタノール:水(1:1)中で試料を溶解し、質量分析計に流速5ないし10μl/分で導入した。窒素を空間電荷層ガスとして用いて、ニードル電圧を4.0kVに設定した。加熱キャピラリーの温度を200℃に設定した。合計10ないし20個のスペクトルを総計し、ESI−MSおよびESI−MS/MSスペクトルを産生した。
【0115】
(結果)
(異なる宿主細胞中でのα−Gal置換P−セレクチン糖タンパク質リガンド−1/マウスIgG2bの安定的な発現)
ピューロマイシンを添加した選択培地中での培養の15ないし20日後に、CHO−K1、COS7m6、および293T細胞の異なる大きさのコロニーを位相差顕微鏡によって同定した。該顕微鏡下で、各細胞型の192個のコロニーをピペットによって採取し、選択下での更なる増殖のために2つの96穴プレートに移した。IgサンドイッチELISAを用いて、個々のクローンの上清中の融合タンパク質濃度を評価し、31個のCHO−K1コロニー、8個のCOS7m6コロニー、および36個の293Tコロニーが抗マウスIgG Fc陽性であった。各細胞株由来のコロニーを分泌する上位5つを24ウェル・プレートに移動し、さらに増殖させた。最もよく発現するCHO−K1、COS7m6、および293Tクローンを、ハイグロマイシンB耐性遺伝子を保持するα1,3GaIT・コード・プラスミドでトランスフェクションした。ピューロマイシンおよびハイグロマイシンを用いて、α1,3GalT遺伝子を安定的に組み込んだPSGL−1/mIgG2b発現細胞を選択した。27個のCHO−K1コロニー、3個のCOS7m6コロニー、および31個の293Tコロニーが選択された。抗マウスIgGおよびグリフォニア・シンプリシフォリア(Griffonia simplicifolia)I IB4レクチンELISAで測定されるような融合タンパク質の濃度と、そのα−Galエピトープ置換の相対的レベルとにもとづいて、増殖すべきコロニーを選択した。ブタα1,3GalT有りまたは無しでCHO−K1(それぞれ、クローン5L4−1および10)、COS7m6(それぞれ、クローン51および2H5)、および293T(それぞれ、クローン14およびC)細胞中で発現するイムノアフィニティー単離PSGL−1/mIgG2bをSDS−PAGEおよびウエスタン・ブロット法によって特徴づけた(表1、図7)。全ての細胞株は、非還元条件下で、約300kDaの抗マウスIgG Fc反応性タンパク質を産生した(図7A)。先行の観察(Liu, J. et al., 1997; Liu, J. et al., 2003)と一致して、還元での半分の大きさへの減少によって示される(図7Aおよび図7Bを比較せよ)ように、PSGL−1/mIgG2bはニ量体として産生された。GSA I IB4レクチンを用いて、異なる細胞型で生成された融合タンパク質上のα−Galエピトープの存在を検出した(図7B)。CHO−K1(クローン5L4−1)、COS7m6(クローン51)、および293T(クローン14)中のα1,3Ga1Tの共発現は、レクチンによって検出されたように、融合タンパク質上のα−Galエピトープの発現に至った。α1,3GalT無しで293T細胞中で生成されたPSGL−1/mIgG2bのレクチン反応性は予想外であり、融合タンパク質上のGalili抗原以外のα−Gal残基の存在を示す(図7B)。α1,3GalTの存在下でCOSおよび293T細胞中で産生された融合タンパク質は、CHO−K1細胞中で産生された融合タンパク質より大きい大きさの糖形態を含有していた(図7B)。
【0116】
(表1 CHO、COS、および293T由来の細胞クローン)
【0117】
【表1】
(PSGL−1/mIgG2b上のα−Galエピトープ密度は、その生成のために用いられる宿主細胞に依存する)
CHO、COS、および293T細胞中で生成されたPSGL−1/mIgG2b上の相対的なα−Galエピトープ密度をELISAによって測定した(図8)。α1,3GalTの存在下でCOS細胞中で生成されたPSGL−1/mIgG2bは、α1,3GalT無しでCOS中で生成されたPSGL−1/mIgG2bと比べて、相対的O.D.(GSA反応性/抗マウスIgG反応性)で5.3倍の増加を示した(図8)。293T細胞に関しては、相対的O.D.で3.1倍の増加があり、CHO細胞に関しては、1.8倍の増加だけであった(図8)。ELISA結果は、イムノアフィニティ精製PSGL−1/mIgG2bのウエスタン・ブロット実験で見られる相対的GSAレクチン染色と一致していた(図7B)。
【0118】
(異なる宿主細胞中で生成されるPSGL−1/mIgG2bの抗ブタ抗体吸着効果は、そのα−Gal置換の程度と相関する)
ブタ大動脈内皮細胞細胞毒性を用いて、ブタα1,3GalTを共発現するCHO−K1(クローン5L4−1)、COS7m6(クローン5I)、および293T(クローン14)細胞中で産生されたPSGL−1/mIgG2bの、ヒト血液型AB血清の抗ブタ反応性抗体を吸着する能力を評価した(図9)。ヒトAB血清PAEC細胞毒性を最大40%に減少させるために、CHO細胞生成PSGL−1/mIgG2b9.1μgが必要であった(図9)。COSおよび293T細胞生成PSGL−1/mIgG2b関しては、PAEC細胞毒性を同レベルまで減少するために、それぞれ16倍および4倍少なく必要であった。さらに、α1,3GalT遺伝子の存在下でCHO−K1細胞中で生成されたPSGL−1/mIgG2bは、血液型AB血清のPAEC細胞毒性を36%をより下に減少することができなかった一方で、12および25%への減少がそれぞれ、非飽和条件下であっても、α1,3GalT発現COSおよび293T細胞中で生成されたPSGL−1/mIgG2bで見られた。
【0119】
(CHO細胞中のコア2β1,6GlcNAc転移酵素の共発現は、PSGL−1/mIgG2bαGalエピトープ密度および抗ブタ抗体吸着効果を向上する)
CHO−K1細胞分泌ムチン/Ig上のα−Galエピトープの数を増加させる試みでは、PSGL−1/mIgG2b、α1,3GalT、およびC2 GnTIを安定的に発現するCHO−K1細胞を樹立し、抗マウスIgG抗体およびGSA I IB4を用いて、ELISA、SDS−PAGE、およびウエスタン・ブロットによって、それらの細胞によって分泌されるPSGL−1/mIgG2bを分析した。PSGL−1/mIgG2bの明らかなMWは、コア2酵素の安定的な発現に続いて増加し、融合タンパク質上のより多くの複合体グリカンを示した(図10)。PSGL−1/mIgG2b上のα−Galエピトープ密度は、α1,3GalT無しでCHO−K1細胞中で生成されたPSGL−1/mIgG2bと比べて13.0倍の増加を示し、α1,3GalT単独を用いて生成されたPSGL−1/mIgG2bと比べて7.4倍の増加を示した(図8)。さらに、α1,3GalTおよびC2 GnTIを安定的に発現するaCHO−K1(CHO−C2−1−9)中で産生されたPSGL−1/mIgG2bの抗ブタ抗体吸着効果は、α1,3GalTを共発現する293TおよびCOS細胞中で産生されたPSGL−1/mIgG2bの吸着効果と同様であり(図9)、CHOクローン5L4−1中で生成されたPSGL−1/mIgG2bと比べて、ヒトAB血清のPAEC細胞毒性を最大40%に減少させるために10倍少ない融合タンパク質が必要であった。
【0120】
(O結合グリカン構造的特徴付けのための組み換えPSGL−1/mIgG2bの精製)
単独、ブタα1,3GalTとの組み合わせ、またはα1,3GalTおよびC2 GnTIとの組み合わせでPSGL−1/mIgG2bを発現する安定的にトランスフェクションしたCHO−K1細胞(それぞれ、クローン10、クローン5L4−1、またはクローンC2−1−9)の1L撹拌フラスコ培養物から、組み換えPSGL−1/mIgG2bを精製した。Oグリカン構造分析を干渉する可能性のある混入するグリコシル化タンパク質を完全に除去するために、抗マウスIgGアフィニティー・クロマトグラフィーおよびゲル濾過を含むツー・ステップ精製法を設定した。各細胞クローンから得た細胞上清2リットルのアフィニティー精製は、ELISAによって評価されたように、CHO−10、5L4−1、およびC2−1−9から、それぞれPSGL−1/mIgG2b2.2mg、1.2mg、および0.95mgを生じた。ゲル濾過カラム上での更なる精製は、それぞれ、0.22mg、0.19mg、および0.29mgの最終PSGL−1/mIgG2b収率を生じた。アフィニティーおよびゲル濾過カラムから溶出した画分をSDS−PAGEおよびウエスタン・ブロット法によって分析した(ここではクローン10に対して示す)。糖タンパク質染色キットをRubyと組み合わせて用いて、グリコシル化タンパク質と、非グリコシル化タンパク質とを検出し(図11Aおよび11B)、抗PSGL−1抗体は、PSGL−1/mIgG2bの存在を確認した(図11C)。この抗体は、PSGL−1/mIgG2b二量体を表す300kDa付近のバンド(図11Cレーン2および4ないし9)と強く結合した。還元形態の融合タンパク質由来の150kDa付近のバンドも見られ(レーン4ないし6)、融合タンパク質分解産物を表す可能性が最も高い60ないし70kDaの弱いバンドも見られる(レーン7ないし9)。図11Aおよび11Bでは、抗PSGL−1抗体で染色されていない300kDaバンドもレーン1および3で見られ、細胞培養培地由来のタンパク質を表す可能性が最も高い。このことは、アフィニティー精製上清中のその存在(レーン3)によっても裏づけされ、該タンパク質が抗Igアフィニティー・カラムに吸着されていないことを示す。しかし、抗PSGL−1抗体に染色されていない50ないし60kDaのMWを持つグリコシル化バンドをアフィニティー精製画分で見ることができる(図11Aレーン4)。このタンパク質も、恐らく細胞培養培地に由来するものであり、融合タンパク質とともにアフィニティー・カラムに吸着される。このタンパク質をゲル濾過によって除去し、該ゲル濾過の間、該タンパク質(図11Aおよび11B,レーン7ないし9)は、融合タンパク質(図11A,11B,および11C、レーン5ないし6)より遅れて溶出した。50ないし70kDa付近の付加的な非グリコシル化タンパク質をゲル濾過によって除去した(図11B、レーン4とレーン7ないし9とを比較せよ)。各クローンに対して、最も多い量の融合タンパク質を含有するゲル濾過画分をオリゴ糖放出のために選択した。クローン10に対して示すように(図11Aおよび11B、レーン5)、この画分は、いずれの有意な量の混入するタンパク質(グリコシル化または非グリコシル化)も含有していなかった。
【0121】
(精製組み換えPSGL−1/mIgG2bから放出される過メチル化オリゴ糖の質量分析法)
クローンCHO−10および5L4−1から放出された過メチル化オリゴ糖は、m/z895.4および1256.5付近のピークの2つの最も多数を占める群を持つ類似のMSスペクトルを与え(図12)、一方で、クローンC2−1−9によって産生されたPSGL−1/mIgG2bから放出されたO−グリカンの質量スペクトルは、より複雑なパターンを示した(図13)。タンデム質量分析法(MS/MS)によって、ESI−MSスペクトル中のイオンのオリゴ糖配列を導き出した。このように得られた配列および仮構造を表2に示す。下記に、我々は、α1,3GalTおよびC2 GnTIも発現するCHO細胞(C2−1−9)中で産生されたPSGL−1/mIgG2b上のO−グリカンのMS/MS分析の結果について記載する。MSおよびMS/MSスペクトル中のイオン全てをナトリウム化イオンとして検出した。
【0122】
(C2−1−9のMS/MS分析)
親スペクトル中の最も強度のピークは、一連のステップのMS/MSによって評価されるように、NeuAc−Hex−HexNol−HexN−Hex−Hex構造を表すm/z1548.7の偽分子イオン([M+Na]+)である(図14)。このイオンのMS2は、m/z951.3([M−NeuAc−Hex−O+NA]+)および1173.5([M−NeuAc+Na]+)の2つのより大きなフラグメント・イオンと、m/z506.1([M−Hex−Hex−HexN−NeuAc+Na]+)、620.2([NeuAc−Hex−O+Na]+)、690.2([Hex−Hex−HexN+Na]+)、751.3([M−Hex−Hex−NeuAc+Na]+または[M−Hex−NeuAc−Hex+Na]+)、881.3([M−Hex−Hex−HexN+Na]+)、969.5([M−NeuAc−Hex+Na]+)、および1330.7([M−Hex+Na]+)の複数のより小さなフラグメント・イオンとを与えた。m/z1173.5のフラグメント・イオンを単離して、MS3によって分析すると、m/z951.4、506.2、690.3、および751.5のフラグメント・イオンを生じた。より大きなピーク、すなわち951.4をMS4によってさらに分析し、m/z445.3([Hex−Hex+Na]+)、463.0([Hex−Hex−O+Na]+)、690.3 、および733.6([M−Hex−NeuAc−Hex−O+Na]+)のフラグメント・イオンを生じた。最終的に、MS4分析の優勢なフラグメント・イオン(690.3)をMS5によって分析した。これによって、末端Hex−Hexを表すm/z415.1および445.3の配列イオンを生じ、前者のイオンは、1つの酸素およびそのメチル基を消失していた。Hex−Hex−O構造も見いだされた(463.0)。さらに、内部Hex−HexN構造が、ヘキソースに結合する1つの酸素を持つもの(472.2)および持たないもの(454.0)で見られた。Hex−Hex−HexN構造からのO−Meの消失(660.1)、C−O−Meの消失(648.2)、およびN−C−O−Meの消失(619.4)も見られ、ここで、最後の1つは、恐らく内部HexNからのN−アセチル基の消失を表す。m/z533.2のより大きなフラグメント・イオンもMS5スペクトル中で見られた。このイオンは、最内側のHexNのクロス・リング・フラグメントに対応し(図14)、ヘキソースが1−4結合中のHexNに結合することを示す。この配列は、2型構造および末端Galで伸張されたシアル酸付加コア2と整合する可能性が最も高い。
【0123】
m/z1548.7のイオンを以外に、Galα1,3Galで終了すると考えられる2つの他の偽分子イオンが、m/z1578.7および1187.6で、クローンC2−1−9のESI−MSスペクトル中で見出された。m/z1578.7のイオンをMS2分析のために単離した。その結果は、NeuGc−Hex−HexNOl−HexN−Hex−Hex構造を示し、m/z1173.5([M−NeuGc+Na]+)、951.5([M−NeuGc−Hex−O+Na]+)、911.3([M−Hex−Hex−HexN+Na]+)、690.3([Hex−Hex−HexN+Na]+)、および676.3([Hex−Hex−HexN−Me+Na]+)のフラグメント・イオンを持つ。1173.5イオンのMS3分析は、m/z951.5のより大きなフラグメント・イオンと、m/z506.1([M−Hex−Hex−HexN−NeuAc+Na]+)、690.2、および969.3([M−NeuAc−Hex+Na]+)の複数のより小さなフラグメント・イオンとを生じた。m/z951.5のフラグメント・イオンを第4のステップ中のMS/MSによって分析し、m/z690.4の1つのより大きなフラグメント・イオンおよびm/z658.2(690.4−O−Me)のより小さなフラグメント・イオンを与えた。しかし、m/z1578.7のイオンのMS2スペクトルでは、m/z981.5および1203.4の未同定のフラグメント・イオンが観察された。m/z1203.4のイオンのMS3およびMS4分析は、m/z981.4、720.4、690.1、506.1、および688.3(720.1−O−Me)のフラグメント・イオンを生じた。MS3およびMS4スペクトル両方で見られるm/z720.4のイオンは、m/z690.1の特徴的なフラグメント・イオンより多い30質量単位であり、Hex−Hex−HexN配列を表す。残念なことに、m/z720.4のイオンの更なるMS/MS分析は可能でなかった。考えられる末端Galα1−3Galを持つESI−MSスペクトル中の他の偽分子イオン(図13)がm/z1187.6で観察された。このイオンのMS2実験は、Hex−HexNol−HexN−Hex−Hexまたは2型伸張および末端Galを持つコア2と整合するm/z969.5([M−Hex+Na]+)、951.4([M−Hex−O+Na]+)、756.2([M−Hex−Hex+Na]+)、690.3([Hex−Hex−HexN+Na]+)、520.3([M−Hex−Hex−HexN+Na]+)、および445.1([Hex−Hex+Na]+)のフラグメント・イオンを生じた(表2)。したがって、末端Galα1,3Galを潜在的に発現する中性およびシアリル酸付加オリゴ糖は、C2−1−9クローンによって産生されるが、シアル酸付加(NeuAc)構造は、最も多量なものであると考えられる。これに加えて、末端Hex−Hex(Galα1−3Gal)を持たない複数のシアル酸付加オリゴ糖を見ることができるが、相対的存在量はより少ない(図13および表2)。
【0124】
(表2 PSGL−1/mIgG2b由来O−グリカンの配列および仮構造)
【0125】
【表2−1】
【0126】
【表2−2】
(実施例3 発現ベクター)
融合ポリペプチドの生成に有用な好例の発現ベクターは次のとおりである。
【0127】
【表3−1】
【0128】
【表3−2】
【0129】
【表3−3】
【0130】
【表3−4】
【0131】
【表4】
【0132】
【表5−1】
【0133】
【表5−2】
【0134】
【表5−3】
【0135】
【表5−4】
【0136】
【表6−1】
【0137】
【表6−2】
(参考文献)
【0138】
【表7−1】
【0139】
【表7−2】
【0140】
【表7−3】
【0141】
【表7−4】
【0142】
【表8−1】
【0143】
【表8−2】
【0144】
【数7】
【0145】
【数8】
【0146】
【数9】
【0147】
【数10】
【0148】
【数11】
【0149】
【数12】
【0150】
【数13】
(他の実施形態)
本発明は、その詳細な説明とともに記載されたが、上記の記載は、添付の請求項の範囲によって定義される本発明の範囲を説明するものであって、限定を意図するものではない。他の態様、利点、および変更は以下の請求項の範囲内にある。
【特許請求の範囲】
【請求項1】
明細書中に記載の発明。
【請求項1】
明細書中に記載の発明。
【図1】

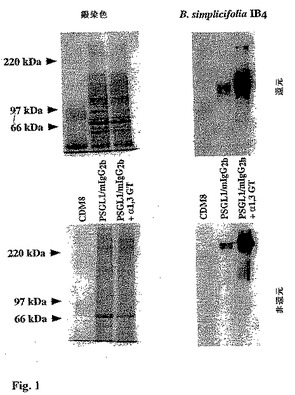
【図2】
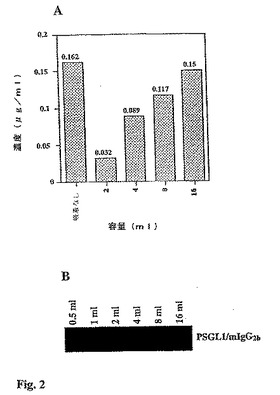
【図3】
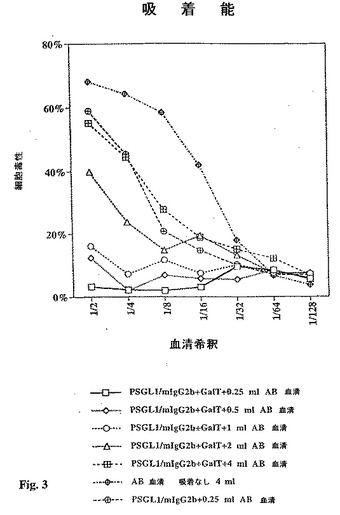
【図4】
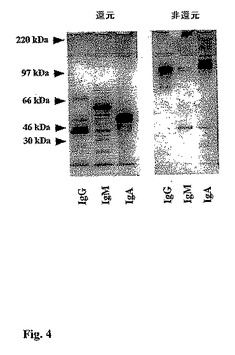
【図5】
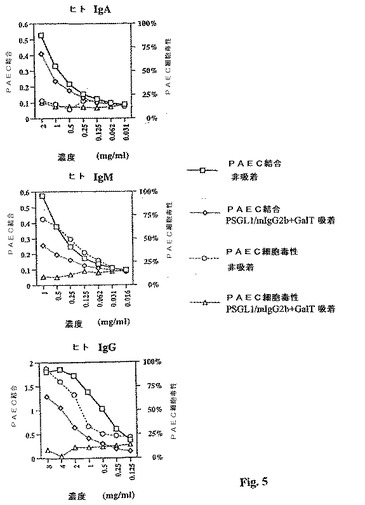
【図6】
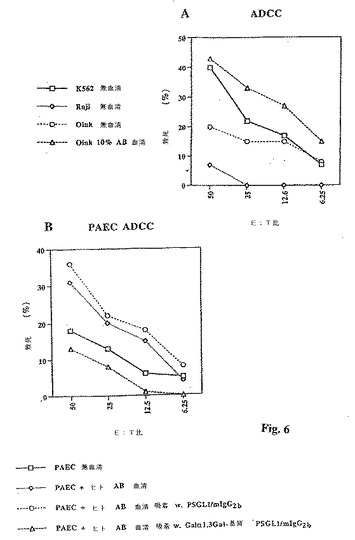
【図7】
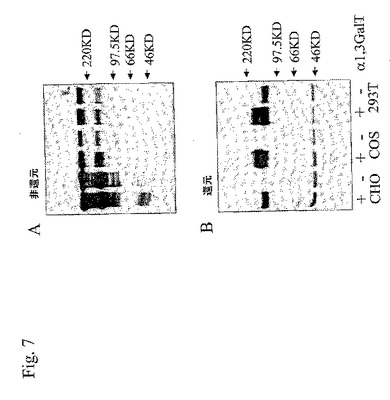
【図8】
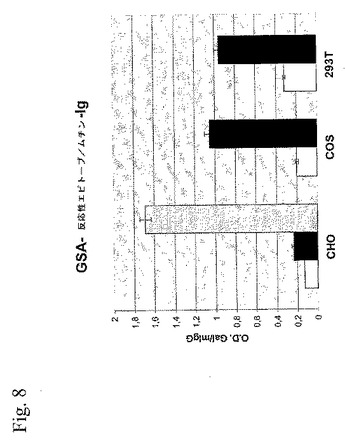
【図9】
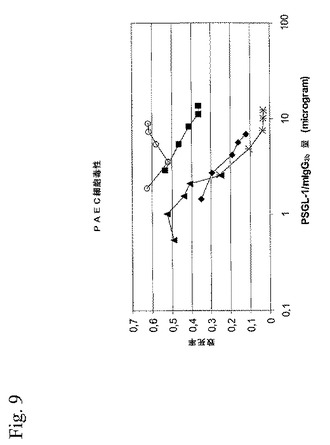
【図10】
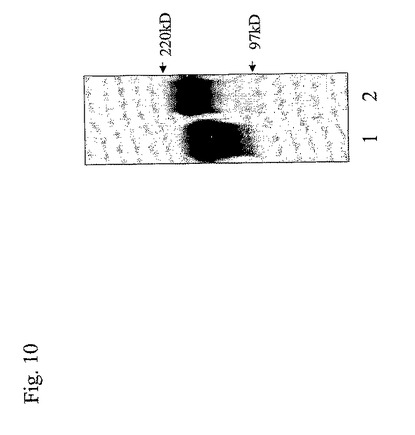
【図11】
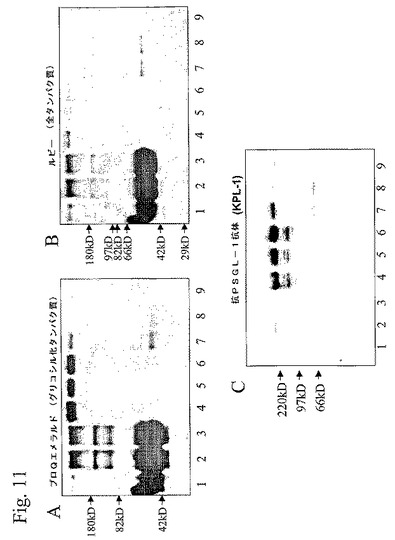
【図12】

【図13】

【図14】
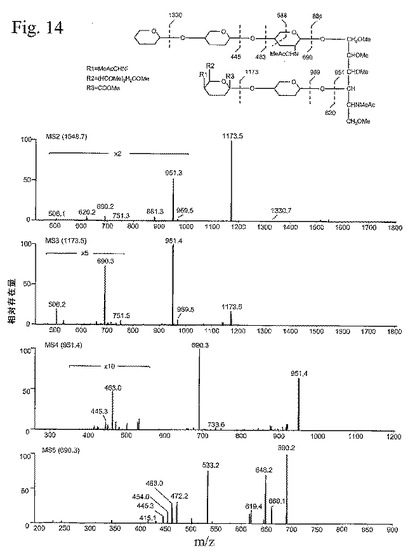
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【公開番号】特開2010−235614(P2010−235614A)
【公開日】平成22年10月21日(2010.10.21)
【国際特許分類】
【出願番号】特願2010−113707(P2010−113707)
【出願日】平成22年5月17日(2010.5.17)
【分割の表示】特願2004−527246(P2004−527246)の分割
【原出願日】平成15年8月11日(2003.8.11)
【出願人】(505049076)レコファーマ アーベー (6)
【Fターム(参考)】
【公開日】平成22年10月21日(2010.10.21)
【国際特許分類】
【出願日】平成22年5月17日(2010.5.17)
【分割の表示】特願2004−527246(P2004−527246)の分割
【原出願日】平成15年8月11日(2003.8.11)
【出願人】(505049076)レコファーマ アーベー (6)
【Fターム(参考)】
[ Back to top ]