
メキシレチンのアミノ酸およびペプチドプロドラッグ並びにその使用
本発明は、メキシレチン(およびメキシレチンの活性代謝物)とアミノ酸若しくはペプチドとのプロドラッグ並びにかかるプロドラッグを含む医薬組成物に関する。上記プロドラッグを用いて、痛みの緩和、不整脈の治療、メキシレチンに伴う有害な胃腸副作用の低減、メキシレチンの生物学的利用能の向上および薬物動態の再現性の改善の方法も提供する。グリシン残基により直接若しくは間接的に結合したリシンまたはアルギニンを包含するオリゴペプチドもここに記載する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、メキシレンの様々なアミノ酸およびペプチドプロドラッグ、並びにメキシレチンの薬物動態学的一貫性の向上、神経因性疼痛の治療、そしてメキシレチンに通常伴う有害な胃腸(GI)副作用の回避のためのプロドラッグの使用に関する。また、本発明は不整脈の治療に使用し得る。
【背景技術】
【0002】
神経因性疼痛は、世界人口の2.8%から4.7%に影響を与えていると推定される(非特許文献1)。中枢または末梢として広く分類される神経因性疼痛は、火傷または手足の骨折などの外部刺激を起因とする神経系により検出された痛みよりむしろ、神経系への損傷または神経系の疾患によるか、あるいは神経系それ自体への損傷に由来する痛みによりもたらされる。中枢神経因性疼痛は、中枢神経系(CNS)の損傷の結果として発生し、例えば、多発性硬化症、脊髄損傷、卒中または癌に起因する場合がある。末梢神経因性疼痛は、糖尿病、癌、HIV感染症、手根管症候群および疱疹性神経痛、切断術(幻肢痛)、背部損傷、下腿潰瘍および手術による医原性損傷によってもたらされた末梢神経系の損傷から生じる。医薬品主要七市場にわたる最近の報告では、約3760万人の患者が、中枢性神経因性疼痛に苦しんでおり、さらに17000万人の患者が末梢神経因性疼痛に苦しんでいると推定している(非特許文献1)。
【0003】
神経因性疼痛の症状には、灼熱痛、電撃痛、刺痛、感電型感覚が含まれる。他の一般的な神経因性疼痛の症状は、異痛症(通常は痛みを伴わない刺激に起因する痛み)、知覚過敏症(軽度な感触への誇張された応答)、痛覚過敏(痛みの原因が取り除かれた後であっても、持続する痛み)および気分変調(異常で不快な刺痛またはピンや尖った物で刺す感覚)である。
【0004】
神経因性疼痛は、特定の患者集団においてより一般的である。例えば、糖尿病患者の四分の一および癌患者の三分の一もの患者がかかる痛みを経験する。さらに、帯状疱疹を患っている患者の半数以上が疱疹性神経痛になっており、脊髄損傷を伴う患者の三分の一が神経因性疼痛による影響を受けている(非特許文献2)。
【0005】
現在、神経因性疼痛用のいくつかの効果的な治療法がある。プレガバリン、ガバペンチン、デュロキセチン(セロトニン−ノルエピネフリン再取り込み阻害剤(SNRI)である抗うつ剤)、Δ9テトラヒドロカンナジノールおよびリドカインパッチ(疱疹性神経痛の局所治療用)が、現在利用可能な治療の選択肢となっている。しかしそれぞれが、固有の明確な制限がある。例えば、プレガバリンはCNSへの重大で有害な作用を伴う。最も頻繁なプレガバリン使用の中止につながる副作用は、めまいや眠気である。これら二つの副作用は、より高い投与量のプレガバリン(FDAラベリング)で治療を受けた30%もの患者に発生した。ガバペンチンの場合、その経口生物学的利用能が投与量に比例しない、すなわち投与量が増加すると、生物学的利用能が低下する。約60%、47%、34%、33%、および27%の生物学的利用能がガバペンチンの900、1200、2400、3600、および4800mg/日(FDAラベリング)の投与により観察された。デュロキセチンは、治療を受けた患者の20〜40%に吐き気を、また治療を受けた患者の自殺への関心を伴う(FDAラベリング)。Δ9テトラヒドロカンナジノールは明らかな中毒傾向(DEA分類)がある。
【0006】
メキシレチン、すなわち(rac)−1−(2,6−ジメチルフェノキシ)−2−プロパンアミン塩酸塩(以下に構造を示す))は、ナトリウムチャネルブロック剤であり、結果として局所麻酔薬の特性を有する(非特許文献3)。最初、メキシレチンはクラス1Bの抗不整脈薬としての有用性が見出され、不整脈を治療するために今日でも使用されている。現在、薬は150mg、200mgまたは250mgのカプセルとして入手可能で、心室性不整脈の治療に使用されている。メキシレチン投与に伴う最も頻度の高い有害な反応は、上部胃腸障害である(FDAラベリング)。
【化1】

メキシレチン(1−メチル−2−(2,6−キシリルオキシ)−エチルアミン)塩酸塩
【0007】
さらに近年では、メキシレチンは、様々な原因の神経因性疼痛の治療における有用性の増加が見られる。糖尿病性神経障害、急性および慢性神経痛、アルコール性多発ニューロパチー、放射線治療由来の慢性疼痛、視床痛や糖尿病性の体幹の痛みへの使用が報告されている(非特許文献4)。さらにより最近の報告は、肢端紅痛症(EM)、再発性灼熱痛により特徴付けられる稀な不自由な障害、紅斑および影響を受けた領域(例えば、足、耳)の上昇した温度についての治療におけるメキシレチンの有用性を示唆している(非特許文献5)。また、メキシレチンは、慢性特発感覚性多発ニューロパチー、すなわち患者の抹消下肢におけるしびれまたはうずきで表される疾患に有用であることが見出されている(非特許文献6)。
【0008】
しかしながら、メキシレチンの使用は、高い発症率の吐き気、嘔吐および腹部不快感(治療を受けた患者の38%)を伴う(非特許文献7)。このような有害なGI副作用は、貧弱な患者の服薬遵守に間違いなく寄与する。嘔吐はまた、投与された薬物の部分減量となり、その結果効果が減少し予測不可能になる。嘔吐は、メキシレチンの経口投与量を制限する副作用であり、効果的な血漿中薬物濃度の達成を妨げる場合がある(非特許文献8および非特許文献9)。
【0009】
メキシレチンの嘔吐作用のメカニズムへの理解は限定的である。一つの実験的研究では、メキシレチンが生体内でのラットの胃の中で徐波活動を低下し得るが、空腸筋電性活動に効果がないことを示している(非特許文献10)。胃の徐波活動の抑制は、吐き気および嘔吐を誘導する重要な役割を果たすと考えられている。メキシレチンの場合、胃の徐波活動の抑制が局所鎮静(ナトリウムチャネルをブロック)活動によりもたらされる場合がある。ウサギの食道の括約筋を用いた他の生体外での研究は、静脈内麻酔化合物のケタミンおよびミダゾラムのようにメキシレチンが酸化窒素によってもたらされた非アルドレナリン、非コリン性(NANC)弛緩を抑制し得ることを示唆した(非特許文献11)。この研究では、メキシレチンによる下部食道括約筋の平滑筋における内因性酸化窒素の抑圧がメキシレチンの有害なGI作用に寄与し得ると結論付けている。
【0010】
他の局所麻酔薬リグノカインに対して行われた研究は、消化管への直接作用により誘導される該化合物の経口投与に伴う嘔吐の影響に向けられている。抗不整脈薬のイヌへの同等効果の静脈内および経口投与後、それぞれが同等の全身の血中濃度に到達したにもかかわらず、経口投与した薬剤のみが嘔吐を誘発した(非特許文献12)。
【0011】
メキシレチンが固有の胃刺激特性があることを示唆する文献の報告がある。例えば、メキシレチンの経口摂取後に食道炎が繰り返される事例が報告された(非特許文献13、非特許文献14および非特許文献15)。したがって、メキシレチンの催吐効果がより単純には胃への直接的な刺激作用に起因する可能性がある。
【0012】
これら有害な事象のメカニズムを理解する際のかかる進歩があるにもかかわらず、メキシレチン治療に伴う副作用を減少させる必要があり続ける。したがって、痛みの治療において、薬剤分子の固有の薬理学的利点すべてを保持するが、有害なGI副作用を誘発するという制限を克服するメキシレチン製品への現実的必要性がある。本発明は、この必要性に取り組んでいる。
【0013】
任意の製薬化合物を投与する際に有効性および毒性が重要な考慮事項であるが、メキシレチンの場合、催吐特性が実際患者の服薬遵守および適切かつ治療上有効な投与量レベルに対し大きな障壁である。そのため、メキシレチンで治療する際に嘔吐を減少または排除する本発明の化合物により提供される利点は重要であると期待されている。したがって、本発明は、患者および臨床医に対し従来問題があった治療への容易な利用を提供する。
【先行技術文献】
【非特許文献】
【0014】
【非特許文献1】Neuropathic Pain Network and Pfizer Inc.,2006 survey
【非特許文献2】Neuropathic Pain Network and Pfizer Inc.,2007 survey
【非特許文献3】Scholz A(2002)Brit.J Anaethesia 89,52−61
【非特許文献4】Jarvis and Coukell(1998).Drugs 4,691-707
【非特許文献5】Vivas AC et al(2010)Amer.J.Otolaryngology,May
【非特許文献6】Wolfe GI et al(1999)Arch Neurol 56、540−547)
【非特許文献7】Morganroth(1987).Am.J.Cardiol.60,1276−1281
【非特許文献8】Wright et al.(1997)Ann Pharmacother.31,29-34
【非特許文献9】Galer et al.(1996)J Pain Symptom Manage.12,161-167
【非特許文献10】Bielefeldt and Bass(1991).Digestion 48,43-50
【非特許文献11】Kohjitani et al.(2003).Eur.J.Pharmacol.,465,145-151
【非特許文献12】Smith ER et al 1972)Amer.Heart Journal 83 363−372
【非特許文献2】Penalba C(1986)Ann Gastroenterol Hepatol(Pris)22,267−268
【非特許文献14】Seggewiss RR & Seckfort H(1983)Dtsch Med Wochenshr.108 1018−1020
【非特許文献15】Addler JB(1990)Am J Gastroenterol.85 629−630
【発明の概要】
【0015】
本発明の化合物は、メキシレチンの治療効果を付与するが、嘔吐などのGI副作用を低減または排除するアミドプロドラッグ結合体である。
【0016】
本発明は、式Iのメキシレチンおよびp−OHメキシレチンのプロドラッグ
【化2】
式I
【0017】
またはその薬学的に許容し得る塩に関し、
【0018】
ここで式中のR1は水素および
【化3】
から選択され、
【0019】
R2は、水素、
【化4】
から選択され、
【0020】
O1は未結合p−OHメキシレチン中に存在するフェノール系酸素であり、
【0021】
Xは(−NH−)、(−O−)、または欠落であり、
【0022】
RAAのそれぞれの出現は独立して天然または非天然アミノ酸側鎖であり、
【0023】
n1は0〜16から選択した整数であり、
【0024】
n2のそれぞれの出現は独立して1〜9から選択した整数であり、
【0025】
R3のそれぞれの出現は独立して水素、および任意に置換したアルキル基から選択され、
【0026】
R4およびR5のそれぞれの出現は独立して水素、
【化5】
および任意に置換したアルキル基から選択され、
【0027】
ここで、n1により定義した炭素鎖に生ずる二重結合の場合、二重結合を形成する炭素上にR4は存在し、R5は欠落であり、但し常に、
【0028】
少なくともR1は
【化6】
であるか、あるいは少なくともR2は
【化7】
のいずれか一方であり、また本化合物が次の化合物:HBr.グリシン−(rac)メキシレチン;AcOH.アスパラギン−(rac)メキシレチン;TFA.トリプトファン−(rac)メキシレチン;HBr.アラニン−(rac)メキシレチン;AcOH−フェニルアラニン−(rac)メキシレチンおよびTFA.トリプトファン−(rac)メキシレチン.H2Oの一つでない。
【0029】
一実施形態において、R1は水素であり、R2は
【化8】
である。
【0030】
R2は
【化9】
が好ましい。
【0031】
一実施形態において、R2は水素であり、R1は
【化10】
である。
【0032】
一実施形態において、R2が
【化11】
であり、Xは欠落であり、n1は1、2または3である。R2が水素またはヒドロキシ以外であるとき、R1に関するn2の値は、R2に関する値と独立している。一実施形態において、R1について言えば、n2の値は好ましくは1、2、3または4であり、より好ましくはn2が1または2である。もっとも好ましくはn2が1である。
【0033】
一実施形態において、R2について言えば、n2の値は好ましくは1、2、3または4である。より好ましくはn2が1または2であり、もっとも好ましくはn2が1である。
【0034】
本発明に従って用いるアミノ酸残基には、リジン、グリシン、ホモアルギニン、グルタミン酸、メチルメチオニンおよびグルタミンが含まれる。
【0035】
ジカルボン酸リンカーの一実施形態において、n1は0、1、2、3または4である。
【0036】
一実施形態において、n2の各出現は1、2、3、4および5から選択される。
【0037】
好ましい実施形態において、R3の各出現は水素である。別の実施形態において、R3の各出現は独立して非置換C1−6アルキルから選択される。
【0038】
一実施形態において、R3の各出現は独立して水素または任意に置換したC1−10のアルキル基から選択される。好ましくは、アルキル基が存在するときは、C1−6アルキル基であり、いくつかの実施形態ではtBuを含まない。
【0039】
本発明の化合物は、除外された化合物の一つでないという条件で定義されるような式(I)の化合物である。しかし、除外は常に化合物の使用に適用せず、またこれら化合物の使用の一実施形態において、ただし書きは適用しない。明細書全体で用いる略語(rac)は、ラセミ混合物を指す。例えば、(rac)メキシレチンの場合、これはDLメキシレチンを指す。
【0040】
上述したように、式(I)の化合物はまた以下の化合物:HBr.グリシン−(rac)メキシレチン;AcOH.アスパラギン−(rac)メキシレチン;TFA.トリプトファン−(rac)メキシレチン;HBr.アラニン−(rac)メキシレチン;AcOH.−フェニルアラニン−(rac)メキシレチン;TFA.トリプトファン−(rac)メキシレチン.H2Oの一つでない。この実施形態において、アミノ酸は天然の立体配置である。
【0041】
一実施形態において、n1で定義される鎖中に二重結合がある。この場合、n1は2〜16であり、C=C二重結合はこの部分に存在する。あるいはまた、n1が1の場合、XがNのとき二重結合は存在し得る。いずれにしても、二重結合に関与した炭素原子または複数の原子はR4置換基のみを有し、R5置換基を有しない。一実施形態において、n1の数値に関係なく二重結合が存在しない。
【0042】
他の実施形態において、式(I)の化合物は以下の化合物の一つでない。
アラニン−(rac)メキシレチンアミド;
β−メトキシアスパラギン酸−(rac)メキシレチンアミド;
α−メトキシアスパラギン酸−(rac)メキシレチンアミド;
アスパラギン−(rac)メキシレチンアミド;
グリシン−(rac)メキシレチンアミド;
ロイシン−(rac)メキシレチンアミド;
メチオニン−(rac)メキシレチンアミド;
(rac)メチオニン−(rac)メキシレチンアミド;
フェニルアラニン−(rac)メキシレチンアミド;
アラニングリシングリシン−(rac)メキシレチンアミド、または
トリプトファン−(rac)メキシレチン。
【0043】
好ましくはR4の各出現は水素またはC1−4のアルキルであり、より好ましくは各R4が水素である。
【0044】
好ましくはR5の各出現は水素またはC1−4のアルキルであり、より好ましくは各R5が水素である。
【0045】
好ましい実施形態において、本発明の化合物は一つのプロドラッグ部分を有し、該プロドラッグ部分は1つ、2つまたは3つのアミノ酸(すなわちn2が1、2または3)であり、一方R3はHである。
【0046】
かかる任意置換のいずれかの出現で、1、2、3または4個の独立的に選択された任意の置換基がある。より通常には、存在するとき、かかる置換の各可能性で任意置換の0または1つの出現がある。
【0047】
存在し、分子と化学的に相溶する場合、任意の置換基は独立してヒドロキシ、C1−4アルキル、C1−4アルコキシ、C1−4ハロアルコキシ、フェニル、ベンジル、ハロゲン、シアノ、ニトロ、アミノ、アミドおよびチオからなる群から選択される。
【0048】
他の実施形態において、n2の少なくとも一つの出現は1である。他の実施形態において、n2の各出現は1である。さらに他の実施形態において、n2の少なくとも一つの出現は2である。他の実施形態において、n2の各出現は2である。
【0049】
さらに他の実施形態において、n2の各出現は1または2であり、RAAの各出現は独立して天然アミノ酸側鎖である。
【0050】
一実施形態において、本発明のプロドラッグは一つのプロドラッグ部分を有し、アルギニンもしくはリシンのホモポリマーか、またはアルギニンおよびリシンのヘテロポリマーである。さらなる実施形態において、ホモポリマーまたはヘテロポリマー中に2、3、4、5、6もしくは7個のアミノ酸がある(すなわちn2が2、3、4、5、6または7である)。
【0051】
一実施形態において、RAAの各出現は独立してアミノ酸側鎖、すなわち1〜20炭素原子を有するアミノ酸残基であり、または該残基(グリシンの場合に)が水素である。本発明との関連で、用語“アミノ酸”は天然アミノ酸(天然または非天然の立体化学的配置)および合成アミノ酸の両方を含む。天然アミノ酸は、タンパク質生合成に用いる20個のアミノ酸の一つである。該用語はまた、いくつかの実施形態において、翻訳時にタンパク質に取り込まれ得る他のアミノ酸(ピロリジン、オルニチンおよびセレノシステインを含む)も含むことができる。好ましくは、RAAまたはその各々は天然に存在するアミノ酸である。
【0052】
本発明のメキシレチンプロドラッグの組成物もここに提供する。該組成物は、少なくとも一つの本発明のプロドラッグ(例えば、式Iのプロドラッグ)またはその薬学的に許容し得る塩と、少なくとも一つの薬学的に許容し得る賦形剤とを含む。
【0053】
他の態様において、本発明は、神経因性疼痛のような痛みの治療に用いる式Iのメキシレチン結合体を提供する。
【0054】
本発明の一実施形態は、必要とする被検体の疾患をメキシレチンで治療する方法である。該方法は、本発明のメキシレチンプロドラッグ若しくはその薬学的に許容し得る塩またはその組成物を、必要とする被検体または被検体群に経口投与することを備え、ここでメキシレチンプロドラッグが少なくとも一つのアミノ酸または長さ2〜9個のアミノ酸のペプチドに共有結合したメキシレチンまたはメキシレチン代謝物(例えば、パラ−OH(p−OH)メキシレチン、メタ−OH(m−OH)メキシレチンまたはヒドロキシメチルメキシレチン)からなる。メキシレチンプロドラッグは一つまたは二つのプロドラッグ部分を有する。一つのプロドラッグ部分は、ペプチド結合を介してメキシレチンまたはメキシレチン代謝物のアミノ基に結合することができる。あるいは、またはさらに、メキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメキシレチン)は、フェノール系酸素でカルバメートまたはジカルボン酸リンカーを介して結合したプロドラッグ部分を有することができる。メキシレチンプロドラッグの量は、治療有効量(例えば、鎮痛有効量)であるのが好ましい。疾患は、メキシレチンで治療可能なものとすることができる。例えば、疾患は神経因性疼痛または不整脈である。
【0055】
他の実施形態において、本発明のプロドラッグは、親化合物と比較して、有害な胃腸副作用(吐き気や嘔吐など)を低減すると同時に、治療血漿薬剤濃度の達成の速度と一貫性とについて向上する利益を与える。
【0056】
したがって、一実施形態において、本発明は、メキシレチンの投与に通常伴う胃腸副作用を最小限にするための方法に関する。該方法は、本発明のメキシレチンプロドラッグ若しくはその薬学的に許容し得る塩またはその組成物を必要とする被検体に経口投与することを備え、ここでメキシレチンプロドラッグが少なくとも一つのアミノ酸または長さ2〜9個のアミノ酸のペプチドに対し共有結合したメキシレチンまたはメキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメチルメキシレチン)からなり、また経口投与すると、プロドラッグまたは薬学的に許容し得る塩が、完全に避けることができないとしても、未結合メキシレチンの経口投与後に通常見られる胃腸副作用を最小限にする。メキシレチンプロドラッグは一つまたは二つのプロドラッグ部分を有する。一つのプロドラッグ部分は、ペプチド結合を介してメキシレチンまたはメキシレチン代謝物のアミノ基に結合することができる。あるいは、またはさらに、メキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメキシレチン)は、フェノール系酸素でカルバメートまたはジカルボン酸リンカーを介して結合したプロドラッグ部分を有することができる。メキシレチンの量は治療有効量(例えば、鎮痛有効量)であるのが好ましい。
【0057】
さらなる実施形態において、メキシレチンの投与に伴うGI副作用は、限定されないが、嘔吐、吐き気、下痢および腹部不快感から選択される。
【0058】
本発明の他の実施形態は、メキシレチン血清濃度の被検体間および被検体内における変動の低減に関する。これは通常痛みまたは不整脈の治療中のものである。当該方法は、本発明のメキシレチンプロドラッグ若しくはその薬学的に許容し得る塩またはその組成物を必要とする被検体または被検体群に経口投与することを備え、ここでメキシレチンプロドラッグが少なくとも一つのアミノ酸または長さ2〜9個のアミノ酸のペプチドに対し共有結合したメキシレチンまたはメキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメチルメキシレチン)からなる。メキシレチンプロドラッグは一つまたは二つのプロドラッグ部分を有する。一つのプロドラッグ部分は、ペプチド結合を介してメキシレチンまたはメキシレチン代謝物のアミノ基に結合することができる。あるいは、またはさらに、メキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメキシレチン)は、フェノール系酸素でカルバメートまたはジカルボン酸リンカーを介して結合したプロドラッグ部分を有することができる。メキシレチンプロドラッグの量は治療有効量(例えば、鎮痛有効量)であるのが好ましい。
【0059】
また、本発明の他の実施形態は、必要とする被検体におけるメキシレチンの生物学的利用能の再現性を向上させることに関する。該方法は、本発明のメキシレチンプロドラッグ若しくはその薬学的に許容し得る塩またはその組成物を必要とする被検体または被検体群に経口投与することを備え、ここでメキシレチンプロドラッグが少なくとも一つのアミノ酸または長さ2〜9個のアミノ酸のペプチドに対し共有結合したメキシレチンまたはメキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメチルメキシレチン)からなる。メキシレチンプロドラッグは一つまたは二つのプロドラッグ部分を有する。一つのプロドラッグ部分は、ペプチド結合を介してメキシレチンまたはメキシレチン代謝物のアミノ基に結合することができる。あるいは、またはさらに、メキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメキシレチン)は、フェノール系酸素でカルバメートまたはジカルボン酸リンカーを介して結合したプロドラッグ部分を有することができる。メキシレチンプロドラッグの量は治療有効量(例えば、鎮痛有効量)であるのが好ましい。
【0060】
したがって、本発明はメキシチレンおよびその活性代謝物の天然および/または非天然アミノ酸と短鎖ペプチドのプロドラッグに関する。任意特定の理論に拘束されることを望むことなく、化合物のプロドラッグ部分(すなわちアミノ酸および/またはペプチド部分)は、神経因性疼痛の減少または排除若しくは不整脈の治療のために薬理学的に有効な量の薬剤/代謝物を送達しながら、薬剤またはその活性代謝物の局所作用から腸を一時的に保護する働きをする。このような一時的不活性化は、この薬剤の重大で極めて不所望な嘔吐性副作用を低減する。本発明のプロドラッグはまた、最大血漿中濃度の達成の速度、それゆえ痛み発症の緩和を促進するだけでなく、患者内および患者間の両方でのより一貫性のある患者応答を確実にする薬剤の生物学的利用能の再現性を改善する手段を提供する。これらの与えられた属性は、改善された鎮痛効果と、患者のより良い服薬遵守を確実にするように作用する。
【0061】
本発明はまた、メキシレチン自体と同一またはそれより良好な活性を享受する化合物群に関する。この化合物群において、催吐性効果はやはり発生しうるが、治療活性はメキシレチンと同じかまたは通常より良好である。これら化合物は、痛みや不整脈の治療においてメキシレチン自体と同じか、またはメキシレチンより効果があることが期待される。
【0062】
したがって本発明の他の態様によれば、式(IA)
【化12】
式(IA)
を提供するもので、
【0063】
式中のR1が
【化13】
であり
【0064】
R2がHおよび−OHから選択され、
【0065】
RAAの各出現は独立して1〜20の炭素原子を有する天然または非天然アミノ酸側鎖もしくは水素から選択され、
【0066】
n2は1、2または3から選択した整数であり、
【0067】
R3は水素および任意に置換したC1−4のアルキル基から選択され、ただし
【0068】
化合物が次の化合物:HBr.グリシン−(rac)メキシレチン;AcOH.アスパラギン−(rac)メキシレチン;TFA.トリプトファン−(rac)メキシレチン;HBr.アラニン−(rac)メキシレチン;AcOH−フェニルアラニン−(rac)メキシレチンまたはTFA.トリプトファン−(rac)メキシレチン.H2Oの一つでない。上記の除外において、アミノ酸は天然の立体配置である。
【0069】
他の実施形態において、式(IA)の化合物は以下の化合物の一つでない。
アラニン−(rac)メキシレチンアミド;
β−メトキシアスパラギン酸−(rac)メキシレチンアミド;
α−メトキシアスパラギン酸−(rac)メキシレチンアミド;
アスパラギン−(rac)メキシレチンアミド;
グリシン−(rac)メキシレチンアミド;
ロイシン−(rac)メキシレチンアミド;
メチオニン−(rac)メキシレチンアミド;
メチオニン−(rac)メキシレチンアミド;
フェニルアラニン−(rac)メキシレチンアミド;
アラニングリシングリシン−(rac)メキシレチンアミド;または
トリプトファン−(rac)メキシレチン。
【0070】
他の実施形態において、式(IA)の化合物のみにおいて、RAAは天然アミノ酸側鎖である。
【0071】
一実施形態において、n2の値は1または2が好ましく、もっとも好ましくはn2が1である。
【0072】
好ましい実施形態において、R3は水素である。別の実施形態において、R3は非置換のC1−6アルキルである。
【0073】
本発明の化合物は、除外された化合物の一つでないという条件で定義されるような式(IA)の化合物である。しかし、除外は常に化合物の使用に適用せず、これら化合物の使用の一実施形態において、ただし書きは適用しない。
【0074】
他の実施形態において、式(IA)の化合物は
メキシレチンピペリコン酸アミドおよび
メキシレチンジメチルグリシンアミドとを含む群から選択される。
【0075】
本発明のこれらおよび他の実施形態は、以下の詳細な説明に開示されるか、または明らかであり、かつ包含される。
【図面の簡単な説明】
【0076】
【図1】単離されたウサギの胃輪状平滑筋の調製物の電場刺激による収縮に対する(1)メキシレチン、(2)メキシレチンリシンアミドおよび(3)メキシレチングリシンアミドの効果を示すグラフである。
【発明を実施するための形態】
【0077】
本明細書中で使用されているように、
【0078】
用語“ペプチド”は、特記しない限り、2〜9個のアミノ酸からなるアミノ酸鎖を指す。好ましい実施形態において、本発明で用いるペプチドは、長さが2または3つのアミノ酸である。一実施形態において、ペプチドは分枝状ペプチドとすることができる。この実施形態では、ペプチドの少なくとも1つのアミノ酸側鎖が他のアミノ酸に(末端または側鎖のいずれか一つを介して)結合する。
【0079】
用語“アミノ酸”は、天然に存在するアミノ酸と非天然に発生するアミノ酸との両方を指す。本発明のプロドラッグでの使用を意図するアミノ酸は、天然アミノ酸と非天然アミノ酸との両方を含み、好ましくは天然アミノ酸である。側鎖RAAは(R)または(S)の立体配置のいずれかとすることができる。さらに、DおよびL体のアミノ酸の両方が本発明での使用を意図されている。
【0080】
“天然アミノ酸”は、タンパク質生合成に用いる20個のアミノ酸の一つ、並びに翻訳時にタンパク質に組み込むことができる他のアミノ酸(ピロリジンとセレノシステインとを含む)である。天然アミノ酸は、一般に式
【化14】
を有する。RAAはアミノ酸側鎖といわれるか、または天然アミノ酸の場合には天然アミノ酸側鎖といわれる。天然アミノ酸は、グリシン、アラニン、バリン、ロイシン、イソロイシン、アスパラギン酸、グルタミン酸、セリン、トレオニン、グルタミン、アスパラギン、アルギニン、リジン、プロリン、フェニルアラニン、チロシン、トリプトファン、システイン、メチオニンおよびヒスチジンを含む(表1参照。)。
【0081】
一実施形態において、アミノ酸側鎖を他のアミノ酸に結合する。さらなる実施形態において、側鎖をアミノ酸のN末端、C末端または側鎖を介してアミノ酸に結合する。
【0082】
天然アミノ酸側鎖の例としては、水素(グリシン)、メチル(アラニン)、イソプロピル(バリン)、sec−ブチル(イソロイシン)、−CH2CH(CH3)2(ロイシン)、ベンジル(フェニルアラニン)、p−ヒドロキシベンジル(チロシン)、−CH2OH(セリン)、−CH(OH)CH3(トレオニン)、−CH2−3−インドリル(トリプトファン)、−CH2COOH(アスパラギン酸)、−CH2CH2COOH(グルタミン酸)、−CH2C(O)NH2(アスパラギン)、−CH2CH2C(O)NH2(グルタミン)、−CH2SH(システイン)、−CH2CH2SCH3(メチオニン)、−(CH2)4NH2(リジン)、−(CH2)3NHC(=NH)NH2(アルギニン)およびCH2−3−イミダゾイル(ヒスチジン)が挙げられる。
【0083】
【表1】
【0084】
“非天然アミノ酸”は、標準遺伝コードによりコード化されるか、または翻訳時にタンパク質に組み込まれるものの中にない有機化合物である。従って、非天然アミノ酸はタンパク質生合成に使用した20個の天然に存在するアミノ酸以外のアミノ酸またはアミノ酸類似体を含み、限定しないが、アミノ酸のD−立体異性体が挙げられる。
【0085】
非天然アミノ酸の例としては、限定しないが、シトルリン、ホモシトルリン、ヒドロキシプロリン、ホモアルギニン、ホモセリン、ホモチロシン、ホモプロリン、オルニチン、4−アミノ−フェニルアラニン、サルコシン、ビフェニルアラニン、ホモフェニルアラニン、4−ニトロ−フェニルアラニン、4−フルオロ−フェニルアラニン、2,3,4,5,6−ペンタフルオロ−フェニルアラニン、パラ−アミノ安息香酸、ノルロイシン、シクロヘキシルアラニン、α−アミノイソ酪酸、N−メチル−アラニン、N−メチル−グリシン、N−メチル−グルタミン酸、tert−ブチルグリシン、α−アミノ酪酸、2−アミノイソ酪酸、2−アミノインダン−2−カルボン酸、セレノメチオニン、ランチオニン、デヒドロアラニン、γ−アミノ酪酸、ナフチルアラニン、アミノヘキサン酸、フェニルグリシン、ピペコリン酸、2,3−ジアミノプロピオン酸、テトラヒドロイソキノリン−3−カルボン酸、tert−ロイシン、tert−ブチルアラニン、シクロヘキシルグリシン、ジエチルグリシン、ジプロピルグリシンおよびそれらの誘導体が挙げられ、ここでアミン基の窒素はモノ−またはジ−アルキル化されている。
【0086】
したがって、本発明はまた、当業界で既知(例えば、“Protective Groups in Organic Synthesis”TW GreeneおよびPGM Wuts著、John Wiley & Sons社出版(1999)に記載されたような)の単純な合成変換によって官能化された上述のようなアミノ酸誘導体を想定し、ここに参照する。
【0087】
用語“極性アミノ酸”は、生理的pHで荷電されていない側鎖を有する親水性アミノ酸を指すが、これは二つの原子により通常共有された電子対がかかる原子の一つでより密接に保持される少なくとも一つの結合を有する。遺伝的にコード化された極性アミノ酸は、Asn(N)、Gln(Q)、Ser(S)およびThr(T)を含む。
【0088】
用語“非極性アミノ酸”は、生理的pHで荷電されていない側鎖を有する疎水性アミノ酸を指し、これは二つの原子により通常共有された電子対が一般に該二つ原子の各々で等しく保持される結合を有する(すなわち、側鎖が極性でない)。遺伝的にコード化された非極性アミノ酸は、Leu(L)、Val(V)、Ile(I)、Met(M)、Gly(G)およびAla(A)を含む。
【0089】
用語“脂肪族アミノ酸”は、脂肪族炭化水素側鎖を有する疎水性アミノ酸を指す。遺伝的にコード化された脂肪族アミノ酸は、Ala(A)、Val(V)、Leu(L)およびIle(I)を含む。
【0090】
用語“アミノ”は、−NH2基を指す。
【0091】
基としての用語“アルキル”は、特定した数の炭素原子を有する直鎖または分枝鎖の炭化水素鎖を指す。用語“アルキル”を炭素原子数を参照することなく使用する場合、C1−C10アルキルを指すものと理解すべきである。例えば、C1−10アルキルは、少なくとも1個、そして最大で10個の炭素原子を有する直鎖または分枝鎖アルキルを意味する。好適なアルキル基は、C1−6またはC1−4アルキル基をしばしば含む。本明細書で用いる“アルキル”の例は、限定しないが、メチル、エチル、n−プロピル、n−ブチル、n−ペンチル、イソブチル、イソプロピル、t−ブチル、ヘキシル、ヘプチル、オクチル、ノニルおよびデシルを含む。
【0092】
本明細書で用いる用語“置換アルキル”または“任意に置換したアルキル”は、少なくとも一つの水素が一つ以上の置換基、例えば、限定しないが、ヒドロキシ基、アルコキシ基、アリール基(例えば、フェニル基)、複素環、ハロゲン、トリフルオロメチル基、ペンタフルオロエチル基、シアノ基、シアノメチル基、ニトロ基、アミノ基、アミド基(例えば、−C(O)NH−Rで、Rがメチルのようなアルキル基)、アミヂン、アミド基(例えば、−NHC(O)−Rで、Rがメチルのようなアルキル基)、カルボキシアミド基、カルバメート、カーボネート、エステル、アルコキシエステル(例えば、−C(O)O−Rで、Rがメチルのようなアルキル基)およびアシルオキシエステル(例えば、−OC(O)−Rで、Rがメチルのようなアルキル基)により置換されたアルキル基を指す。用語が置換基自体に、または置換基の置換基に適用されているかどうかについて定義は関係がある。
【0093】
用語“複素環”は、炭素原子と、各々の出現で独立して窒素、リン、酸素および硫黄から選択された1〜5個のヘテロ原子とからなる安定な三員から十五員の環基を指す。
【0094】
本明細書で用いる用語“シクロアルキル”基は、例えば、シクロプロピル、シクロブチル、シクロペンチル、シクロへキシルまたはシクロへプチルのような3〜8個の炭素原子の非芳香族単環の炭化水素環を指す。
【0095】
本明細書で用いる“置換シクロアルキル”は、ここに記載したような一つ以上の置換基、例えば、限定しないが、ヒドロキシ基、アルコキシ基、アリール基(例えば、フェニル基)、複素環、ハロゲン、トリフルオロメチル基、ペンタフルオロエチル基、シアノ基、シアノメチル基、ニトロ基、アミノ基、アミド基(例えば、−C(O)NH−Rで、Rがメチルのようなアルキル基)、アミヂン、アミド基(例えば、−NHC(O)−Rで、Rがメチルのようなアルキル基)、カルボキシアミド基、カルバメート、カーボネート、エステル、アルコキシエステル(例えば、−C(O)O−Rで、Rがメチルのようなアルキル基)およびアシルオキシエステル(例えば、−OC(O)−Rで、Rがメチルのようなアルキル基)をさらに有するシクロアルキル基を指す。用語が置換基自体に、または置換基の置換基に適用されているかどうかについて定義は関係がある。
【0096】
用語“ケト”および“オキソ”は同義で、=O基を指す。
【0097】
用語“カルボニル”は、−C(=O)基を指す。
【0098】
用語“カルボキシル”は、−CO2H基を指し、カルボニルとヒドロキシル基とからなる(より具体的には、C(=O)OH)。
【0099】
本明細書で用いる“アミド”は、
【化15】
基を指す。本発明において、プロドラッグ部分をアミド結合を介してメキシレチンに結合することができる。この実施形態において、−N−は、非結合メキシレチンまたはメキシレチン代謝物中のアミノ基の窒素である。アミド結合は、アミンをカルボン酸と反応させることによって形成することがてきる。これは、ペプチド結合を形成する反応である。
【0100】
用語“カルバメート基”および“カルバメート”は、
【化16】
基に関連し、式中の−O1−が非結合p−OHメキシレチン分子中のフェノール系酸素である。ここに記載したプロドラッグ部分は、そのアミノ酸またはペプチドおよびカルバメート結合に基づいて参照することができる。そのような参照におけるアミノ酸またはぺプチドは、特記しない限り、アミノ酸またはペプチドのアミノ末端を介してカルボニルリンカーおよびp−OHメキシレチンのフェノール系酸素に結合されていると推測すべきである。
【0101】
例えば、valカルバメート(カルバミン酸バリン)は、式
【化17】
を有する。tyr−valカルバメートのようなペプチド関して、特記しない限り、ペプチド中のもっとも左のアミノ酸がペプチドのアミノ末端であり、カルボニルリンカーを介してp−OHメキシレチンに結合してカルバメートプロドラッグを形成すると推測すべきである。
【0102】
用語“ジカルボン酸リンカー”および“ジカルボキシルリンカー”は、本発明の目的においては同義である。ジカルボン酸リンカーは、メキシレチンとアミノ酸/ペプチド部分との間の基、
【化18】
(−(CO)−(CR4R5)n1−(CO)−)を指す。あるいはまた、“ジカルボン酸リンカー”は、次式
【化19】
(−(CO)−(NH)−(CR4R5)n1−(CO)−)、または次式
【化20】
(−(CO)−(O)−(CR4R5)n1−(CO)−)を有することができる。
【0103】
ジカルボン酸リンカーに関して、1つのカルボニル基をp−OHメキシレチン内の酸素原子に結合する一方、第二のカルボニル基をペプチドまたはアミノ酸のN末端、若しくはアミノ酸側鎖のアミノ基に結合する。
【0104】
本明細書に記載したジカルボン酸プロドラッグの部分は、そのアミノ酸またはペプチドおよびジカルボキシル結合に基づいて参照することができる。かかる参照におけるアミノ酸またはペプチドは、アミノ酸またはペプチドのアミノ末端を介してジカルボキシルリンカーの一つのカルボニル基(元来はカルボキシル基の一部)に結合される一方、特記しない限り、他の基がp−OHメキシレチンに付着すると推測すべきである。ジカルボキシルリンカーは、前述したように様々に置換してもまたは置換しなくてもよい。
【0105】
本発明で使用するジカルボン酸の非限定的なリストを表2に示す。表2に示したジカルボン酸は2〜18個の炭素を含むが、より長鎖のジカルボン酸を本発明でリンカーとして使用することができる。また、ジカルボン酸リンカーを1つ以上の位置で置換することができる。適当に活性化したジカルボン酸を活性化アミノ酸またはペプチドと結合し、次いでp−OHメキシレチンと反応させて本発明のプロドラッグを形成することができる。プロドラッグ合成手順を実施例でより詳しく説明する。
【0106】
【表2】
【0107】
本発明のジカルボン酸リンカーは、リンカー構造
【化21】
および
【化22】
をそれぞれ付与するために最初のカルボニル基に結合した窒素または酸素原子を有する、すなわちXが式1中の(−NH−)または(−O−)とすることができる。かかるジカルボン酸リンカーの例を表3に示し、明細書全体に挙げられている。
【0108】
一実施形態において、ジカルボン酸リンカーを置換する。例えば、一つ以上の
【化23】
置換アルキル基、非置換アルキル基が存在し得る(式1により定義されるようなR3)。これらの実施形態において、X(式1により定義されるような−NH−またはO−)は存在してもまたは欠落してもよい。ジカルボン酸リンカーの例を表3に挙げる。
【0109】
一実施形態において、ジカルボン酸リンカー中の炭素鎖
【化24】
は不飽和であり、一つ以上の二重結合を有することができる。これらの実施形態において、n1≧2であり、R5は二重結合を形成する二つの炭素上に存在しない。そのようなリンカーの一例であるフマル酸を、表3に示す。
【0110】
【表3】
【0111】
本発明のジカルボン酸プロドラッグ部分の例は、次式
【化25】
を有するバリンコハク酸エステルを含む。チロシン−バリンコハク酸エステルのようなジペプチドに関して、別に規定しない限り、薬剤、この場合バリンに隣接するアミノ酸がアミノ末端を経由してジカルボン酸リンカーに結合することを推測すべきである。ジペプチドの末端カルボキシル残基(この場合、チロシン)は、C(カルボキシル)末端を形成する。
【0112】
用語“担体”は、希釈剤、賦形剤および/または活性化合物を投与する媒体を指す。本発明の医薬組成物は、二つ以上の担体の組合せを含むことができる。この種の薬剤担体は水、食塩水溶液、水性デキストロース溶液、水性グリセロール溶液のような無菌の液体や、落花生油、大豆油、鉱油、胡麻油などのような石油、動物、野菜または合成起源のものを含む油とすることができる。水または食塩水溶液と、水性デキストロースと、グリセロール溶液とを、特に注射可能な溶液用の担体として使用するのが好ましい。適当な薬剤担体は、“Remington’s Pharmaceutical Sciences”(E.W.Martin著、第18版)に記載されている。
【0113】
“薬学的に許容し得る”という表現は、一般に安全とみなされている分子的実体および組成物を指す。特に、本発明の実施に用いる薬学的に許容し得る担体は生理的に許容されるもので、患者に投与する際に通常アレルギーまたは類似の有害反応(例えば、胃の不調、めまいなど)を生じない。好ましくは、本明細書で用いるような“薬学的に許容し得る”用語は、適切な政府機関の規制当局によって承認、または米国薬局方若しくは動物での、特にヒトにおける使用のための、他の一般的に認められている薬局方に記載されていることを意味する。
【0114】
“薬学的に許容し得る賦形剤”は、一般に安全で、非毒性で、また生物学的または他の状態で望ましくないものでない医薬組成物を調製するのに有用な賦形剤を意味し、獣医学的使用並びにヒトへの医薬的使用に許容できる賦形剤を含む。本発明に使用するような“薬学的に許容し得る賦形剤”は、かかる賦形剤の一つ以上を含む。
【0115】
用語“治療すること”は、(1)状態、疾患または病気に苦しんでいるかまたはなりやすい状況に置かれているかもしれないが、状態、疾患または病気の臨床的なまたは無症状の症状を経験していないまたは示していない動物に発現する状態、疾患または病気の臨床状態の出現を予防または遅延すること、(2)状態、疾患または病気を抑制すること(例えば、その少なくとも一つの臨床的な若しくは無症状の症状の疾病の発生若しくは維持療法の場合にはその再発を停止させ、減少させまたは遅延させること)、および/または(3)疾患を軽減すること(すなわち、その少なくとも一つの臨床的な若しくは無症状の症状の状態、疾患または病気を退化させること)を含む。治療すべき患者に対する利点は、統計学的に有意であるか、または患者若しくは医師に少なくとも認知可能であるかのどちらかである。
【0116】
用語“被検体”は、ヒトや、家畜(例えば、イヌおよびネコ)のような他の哺乳類を含む。
【0117】
“有効量”は、所望の治療応答をもたらすに十分な本発明のプロドラッグまたは組成物の量を意味する。治療応答は、使用者(例えば、臨床医)が治療に効果的な反応として認識するあらゆる応答とすることができる。治療応答は、一般にプロドラッグ中のメキシレチンまたはp−OHメキシレチンを活性な形状で投与する際に存在する一つ以上の胃腸の副作用の症状の鎮痛および/または改善である。さらに、治療応答の評価に基づいて適切な治療期間、適切な投与量およびあらゆる潜在的な併用療法を決定することは当業者の技能の範囲内である。
【0118】
特に記載がない限り、用語“活性成分”は、ここに記載するような、プロドラッグのメキシレチン部分またはメキシレチン代謝物部分、例えば本発明のプロドラッグのp−OHメキシレチン部分を指すものと理解すべきである。
【0119】
本明細書で用いる“メキシレチン代謝物”とは、p−OHメキシレチン(
【化26】
m−OHメキシレチン(
【化27】
【化28】
【0120】
用語“塩”は、酸付加塩または遊離塩基の付加塩を含むことができる。適当な薬学的に許容し得る塩(例えば、アミノ酸またはペプチドのカルボキシル末端の塩)は、限定しないが、ナトリウム、カリウムおよびセシウム塩のような金属塩;カルシウムおよびマグネシウム塩のようなアルカリ土類金属塩;トリエチルアミン、グアニジンおよびN−置換グアニジン塩、アセトアミジン並びにN−置換アセトアミジン、ピリジン、ピコリン、エタノールアミン、トリエタノールアミン、ジシクロヘキシルアミンおよびN,N’−ジベンジルエチレンジアミン塩のような有機アミン塩を含む。薬学的に許容し得る塩(塩基性窒素中心のもの)は、限定しないが、塩酸塩、臭化水素酸塩、硫酸塩、リン酸塩のような無機酸塩;トリフルオロ酢酸塩およびマレイン酸塩のような有機酸塩;メタンスルホネート、エタンスルホネート、ベンゼンスルホネート、p−トルエンスルホネート、カンファースルホネートおよびナフタレンエルホネートのようなスルホン酸塩;アルギン酸塩、グルコン酸塩、ガラクツロン酸塩、アラニン酸塩、アスパラギン酸塩、グルタミン酸塩のようなアミノ酸塩を含む(例えば、 Bergeおよびその他、“Pharmaceutical Salts,”J.Pharma.Sci.1977年、第66編、第1項を参照)。
【0121】
ここで用いるような用語“生物学的利用能”は、一般にメキシレチンまたはメキシレチン代謝物を製剤から吸収し、全身的に、それゆえ作用部位で入手し得る速度および/または度合いを意味する。連邦規則集第21巻第320部第1節(2003年版)を参照。経口投与形態に関して、生物学的利用能は活性成分を経口投与形態から放出し、作用部位に移動するプロセスに関する。特定処方に対する生物学的利用能のデータは、全身循環へ吸収される投与量の割合の推定値を与える。従って、用語“経口生物学的利用能”は、被検体への単独投与後に全身循環へ吸収される経口投与されたメキシレチン投与量の割合を指す。経口生物学的利用能を決定するための好ましい方法は、経口投与されたメキシレチンのAUCを同じ被検体に静脈内投与された同じメキシレチン投与量のAUCで割り、比率を百分率として表現することである。経口生物学的利用能を算出するための他の方法は、当業者によく理解されており、ShargelおよびYu著,“Applied Biopharmaceutics and Pharmacokinetics”、1999年出版、第4版、Appleton & Lange社(Stamford,Conn)出版にさらに詳細に記載されており、その全体をここに参照して援用する。
【0122】
本発明の化合物
【0123】
一実施形態において、本発明のメキシレチンプロドラッグは、アミド結合を介してメキシレチンのアミノ窒素に結合したプロドラッグ部分を有する。あるいは、またはさらに(すなわち、メキシレチンプロドラッグは二つのプロドラッグ部分を有する)、本発明のプロドラッグは、カルバメートまたはジカルボン酸リンカー基のヒドロキシ基の酸素を介して結合したメキシレチン代謝物(p−OH、m−OHまたはヒドロキシメチルメキシレチン)の新規なアミノ酸およびペプチドプロドラッグである。好ましくは、本発明のプロドラッグは、単一アミノ酸または長さが2〜9のアミノ酸からなる短鎖ペプチドに結合したメキシレチンまたはp−OHメキシレチンを含む。
【0124】
ジカルボン酸プロドラッグの実施形態において、メキシレチン代謝物(例えば、p−OH、m−OHメキシレチンまたはヒドロキシメチルメキシレチン)のヒドロキシ基の酸素は、限定されないが、マロン酸、コハク酸、グルタル酸、アジピン酸もしくは他の長鎖ジカルボン酸またはその置換誘導体(ジカルボン酸リンカーの代表的な例については、表2および表3を参照)のようなジカルボン酸でエステル化することができる。次いで、アミノ酸またはペプチドを、ペプチド/アミノ酸のN末端の窒素またはアミノ酸側鎖に存在する窒素(例えば、リジンの側鎖)を介して残りのカルボキシル基に結合することができる。
【0125】
メキシレチンアミドプロドラッグの実施形態において、プロドラッグ部分(すなわち、アミノ酸またはペプチド)をメキシレチンまたはメキシレチン代謝物のアミノ基に結合することができる。アミド結合は、メキシレチンまたはメキシレチン代謝物のアミノ基をカルボン酸と反応させることによって形成し得る。さらなるメキシレチンアミドプロドラッグの実施形態において、プロドラッグはp−OHメキシレチンプロドラッグであり、二つのプロドラッグ部分を有する。第二のプロドラッグ部分を、ジカルボン酸結合またはカルバメート基を介してp−OH、m−OHメキシレチンまたはヒドロキシメチルメキシレチンのヒドロキシ基の酸素に結合することができる。
【0126】
本発明の化合物の利点
【0127】
特定の理論に拘束されることを望むことなく、ここに記載したメキシレチンまたはメキシレチン代謝物プロドラッグのアミノ酸またはペプチド部分は、消化管内に固有に存在するジペプチドおよびトリペプチドトランスポーターPept1を選択的に利用して吸収を行うと考えられる。一度吸収されると、プロドラッグは加水分解を受けて、活性薬剤を全身循環へ放出する。メキシレチンは、その後部分的に血液中もしくは血漿中に存在する肝臓内および肝臓外の加水分解酵素によってアミノ酸またはペプチドプロドラッグから放出されると考えられる。
【0128】
かかるPept1により助長されたプロドラッグの吸収は、より一貫した経口生物学的利用能の結果として可能となる鎮痛応答においてより高い一貫性を提供することができる。このより再現性の高い経口生物学的利用能の結果として、本発明のプロドラッグは、メキシレチン血漿およびCNS濃度の被験者間および被験者内の変動の相当な低減をもたらし、その結果単一患者または患者集団に対し痛み緩和における変動を著しく少なくする。従って、患者の服薬遵守は、この鎮痛応答の高い予測可能性の結果としてさらに改善されるだろう。
【0129】
メキシレチンが吸収されるまで一時的に不活性化され得る場合、メキシレチンの投与に伴う任意の局所的な媒介嘔吐(すなわち腸管内腔内から)が強力に低減され得る。この不活性化は、薬剤の下部食道括約筋および胃への直接的な暴露を排除することができる。吸収後にのみ活性化するメキシレチンの不活性化プロドラッグは、嘔吐性および他の有害なGI効果を排除する一つの方法である。別のアプローチとして、活性なメキシレチン代謝物(例えば、p−OH、m−OHメキシレチンまたはヒドロキシメチルメキシレチン)のプロドラッグを使用することができる。パラ−ヒドロキシメキシレチンは、親分子のナトリウムチャンネル抑制活性の約25%を保持し、従ってそれ自体有用な薬剤とし得ると報告されている(De Bellis et al.(2006).Brit.J.Pharmacol.149,300-310)。メキシレチンに伴う有害なGI副作用(例えば嘔吐)を一時的な不活性化によって十分に克服することができれば、得られた生成物は神経因性疼痛の治療用薬剤の現在限られた医療設備に価値ある追加を提供することができるだろう。
【0130】
さらに、単一のアミノ酸およびペプチドは毒性のリスクを示すと想定されておらず、アミノ酸またはペプチドはまた、全体の構造および享受された水溶性という重大な変化に起因してメキシレチンまたはその活性なメキシレチン代謝物をおそらく一時的に不活性化するだろう。さらに、慎重に選択されたペプチド結合体は、腸管内腔内でトリプシンのようなペプチダーゼによる部分加水分解により長期または継続的な放出という能力を提供する。例えば、直接または間接的(例えば、グリシンのような他のアミノ酸を介して)なC−末端のポリ−アルギニンまたはポリ−リジンフラグメントの薬剤への導入は、腸管内腔での部分的な加水分解をもたらし、よって生成した吸収能力のあるジ−またはトリ−ペプチド擬態性化合物の吸収用送達速度を制御することができる。次いで、かかる吸収は、ジ−およびトリ−ぺプチドに特異的なPept1のような活性トランスポーターにより行われるだろう。
【0131】
本発明のメキシレチンおよびp−ヒドロキシ代謝物のプロドラッグ
【0132】
一実施形態において、本発明は、式Iのメキシレチンおよびp−OHメキシレチンのプロドラッグ
【化29】
式I
【0133】
またはその薬学的に許容し得る塩に関し、
【0134】
ここで式中のR1は水素および
【化30】
から選択され、
【0135】
R2は、水素、
【化31】
から選択され、
【0136】
O1は未結合p−OHメキシレチン中に存在するフェノール系酸素であり、
【0137】
Xは(−NH−)、(−O−)、または欠落であり、
【0138】
RAAの各出現は独立して天然または非天然アミノ酸側鎖であり、
【0139】
n1は0〜16から選ばれる整数であり、
【0140】
n2の各出現は独立して1〜9から選ばれる整数であり、
【0141】
R3の各出現は独立して水素、置換アルキル基または非置換アルキル基から選択され、
【0142】
R4およびR5の各出現は独立して水素、
【化32】
、置換アルキル基および非置換アルキル基から選択され、
【0143】
n1により定義される炭素鎖における二重結合の場合、二重結合を形成する炭素上にR4は存在し、R5は欠落であり、
【0144】
少なくともR1は
【化33】
であるか、あるいは少なくともR2は
【化34】
のいずれか一方である。
【0145】
ジカルボン酸リンカーの一実施形態において、n1は0、1、2、3または4であり、R1はHである。
【0146】
他のジカルボン酸リンカーの実施形態において、n1は0、1、2、3または4であり、R1は
【化35】
であり、n2の各出現は1、2および3であり、R3はHである。
【0147】
一実施形態において、n2の各出現は1、2、3、4または5である。
【0148】
好ましい一実施形態において、本発明の化合物は一個のプロドラッグ部分を有し、該プロドラッグ部分が1つ、2つまたは3つのアミノ酸(すなわちn2が1、2または3である)を有する一方、R3はHである。
【0149】
他の実施形態において、n2は1である。さらに他の実施形態において、n2は2である。
【0150】
さらに他の実施形態において、n2の各出現は1または2であり、RAAの各出現は独立して天然アミノ酸側鎖である。
【0151】
一実施形態において、プロドラッグは一個のプロドラッグ部分を有し、アルギニンもしくはリシンのホモポリマーか、またはアルギニンおよびリシンのヘテロポリマーである。さらなる実施形態において、ホモポリマーまたはヘテロポリマーに2、3、4、5、6または7個のアミノ酸がある(すなわちn2が2、3、4、5、6または7である)。
【0152】
ジカルボン酸リンカーの一実施形態において、n1は0、1、2または3である。さらなる実施形態において、n1は0、1、2または3である一方、R3、R4およびR5の各出現は水素である。
【0153】
一実施形態において、n2の各出現は1、2、3、4または5である。さらなる実施形態において、n2は1、2、3、4または5である一方、R3の各出現は水素である。
【0154】
好ましい実施形態において、本発明の化合物は一つのプロドラッグ部分を有し、該プロドラッグ部分は1つ、2つまたは3つのアミノ酸(すなわちn2が1、2または3)を有する一方、R3はHである。
【0155】
他の実施形態において、n2は1である。さらに他の実施形態において、n2は2である。
【0156】
さらに他の実施形態において、n2の各出現は1または2であり、RAAの各出現は独立して天然アミノ酸側鎖である。
【0157】
一実施形態において、薬物のアミノ官能基に結合したペプチドプロドラッグ部分は、グリシンまたはリジン残基である。さらなる実施形態において、本発明のペプチドプロドラッグ部分は、グリシンまたはリジンのC末端とアルギニンまたはリジンのアミノ末端(または側鎖の窒素)との間のペプチド結合によりグリシンまたはリジンに直接隣り合うアルギニンまたはリジン残基を取り入れる。従って、以下のジペプチド、すなわち(1)グリシン−アルギニン、(2)グリシン−リジン、(3)リジン−アルギニン、(4)リジン−リジンは、ジペプチドプロドラッグ部分として単独で、あるいはプロドラッグ部分の一部としてのどちらか一方で本発明により意図される。これらの実施形態において、列挙された第一のアミノ酸をメキシレチンに結合する。
【0158】
他の実施形態において、ペプチドプロドラッグ部分を、メキシレチンのヒドロキシ代謝物中のp−ヒドロキシ基に結合する。カルバメートまたはジカルボン酸の架橋を介して結合した好ましいアミノ酸は、限定しないが、バリン、ロイシン、イソロイシンまたはメチオニンを含む。
【0159】
天然に存在するアミノ酸、並びに非天然アミノ酸のいずれかを含むペプチドを本発明に使用し得る。非天然アミノ酸をペプチドプロドラッグ部分またはその一部として使用する場合、ペプチドは非天然アミノ酸を単独で、あるいは天然および非天然アミノ酸の組み合わせを含むことができる。
【0160】
本発明で用いるプロドラッグに使用するアミノ酸は、L−体であるのが好ましい。本発明はまた、D−体のアミノ酸からなるか、またはD−体およびL−体のアミノ酸混合物からなる本発明のプロドラッグを意図している。
【0161】
一実施形態において、式IIのアミド結合メキシレチンプロドラッグを提供する。他の実施形態において、式IIIのアミド結合p−OHメキシレチンプロドラッグを提供する。式II−IIIについて、R3、RAAおよびn2は式Iに与えられるものとして定義される。式II−IIIのプロドラッグの薬学的に許容し得る塩も本発明に包含される。
【0162】
この実施形態において、本発明のプロドラッグは、アミド結合体を介してアミノ酸または短鎖ペプチドに共有結合したメキシレチンまたはp−OHメキシレチンを含み、ここでアミド結合体が薬剤のアミン官能基およびアミノ酸(またはペプチドのC末端)のカルボキシ官能基から形成される。上述したように、本発明で用いるアミノ酸は天然または非天然とすることができる。例えば、グリシン、リジン、アルギニン、シトルリン、オルニチンを、いずれも単一アミノ酸として、またはペプチドの一部分としてメキシレチンまたはp−OHメキシレチンに共有結合することができる。
【化36】
式II
【化37】
式III
【0163】
式IIの一実施形態において、n2は1、2、3または4のいずれかであり、R3はHである。式IIの他の実施形態において、n2は1、2、3または4のいずれかであり、R3はアルキル基である。
【0164】
式IIIの一実施形態において、n2は1、2、3または4のいずれかであり、R3はHである。式IIIの他の実施形態において、n2は1、2、3または4のいずれかであり、R3はアルキル基である。
【0165】
式IIおよび/または式IIIの他の実施形態において、n2は1、2、3、4または5である。さらなる実施形態において、n2は1、2、3、4または5であり、一方R3は水素である。式IIまたは式IIIの好ましい実施形態において、プロドラッグ部分は1つ、2つまたは3つのアミノ酸(すなわちn2は1、2または3)を有する一方、R3はHである。他の実施形態において、n2は1である。また他の実施形態において、n2は2である。また他の実施形態において、n2は1または2であり、RAAの各出現は独立して天然アミノ酸側鎖である。
【0166】
本発明の他の実施形態において、式IVのp−OHメキシレチンカルバメートプロドラッグを提供する。式IVについて、O1、R3、RAAおよびn2は式Iに規定されるように定義される。
【化38】
式IV カルバメート結合p−OHメキシレチンプロドラッグ
【0167】
式IVの一実施形態において、n2は1、2、3または4のいずれかであり、R3はHである。式IVのさらなる実施形態において、n2は1、2、3または4のいずれかであり、R3はHである。またさらなる実施形態において、RAAの各出現は天然アミノ酸側鎖である。
【0168】
式IVの他の実施形態において、n2は1、2、3、4または5である。さらなる実施形態において、n2は1、2、3、4または5であり、一方R3は水素である。式IVの好ましい実施形態において、プロドラッグ部分は1つ、2つまたは3つのアミノ酸(すなわちn2は1、2または3)を有する一方、R3はHである。式IVの別の実施形態において、プロドラッグ部分は1つ、2つまたは3つのアミノ酸(すなわちn2は1、2または3)を有する一方、R3はアルキル基である。
【0169】
さらに別の実施形態において、n2は1である。さらに別の実施形態において、n2は2である。さらに別の実施形態において、n2は1または2であり、RAAの各出現は天然アミノ酸側鎖である。他の実施形態において、RAAの少なくとも1つの出現は非天然アミノ酸側鎖である。
【0170】
本発明の他の実施形態は、式Vのジカルボン酸結合p−OHメキシレチンプロドラッグに関する。式Vについて、O1、R3、R5、RAA、−X−、n1およびn2は式Iに規定されるように定義される。
【化39】
式V ジカルボン酸結合p−OHメキシレチンプロドラッグ
【0171】
式Vの一実施形態において、n1は0〜4から選択される整数である。さらなる実施形態において、R3はHであり、n2は1、2または3である。
【0172】
式Vの一実施形態において、n1は0、1、2または3である。さらなる実施形態において、n1は0、1、2または3であり、一方R3、R4およびR5の各出現は水素である。
【0173】
また、さらなる実施形態において、n2は1、2または3である。式Vの他の実施形態において、Xは欠落であり、n1は1、2または3である。また、さらに式Vの実施形態において、Xは欠落であり、n1は1、2または3であり、n2は1または2であり、R3、R4およびR5はそれぞれ水素である。
【0174】
式Vの一実施形態において、Xは−NH−であり、n1は0、1、2または3であり、n2は1、2または3であり、R3、R4およびR5はそれぞれHである。さらなる実施形態において、n1は2である。
【0175】
式Vの一実施形態において、Xは−O−であり、n1は0、1、2または3であり、n2は1、2または3であり、R3、R4およびR5はそれぞれHである。さらなる実施形態において、n1は2である。
【0176】
式Vの他の実施形態において、Xは欠落であり、n1は1、2または3であり、n2は1、2または3である。また、さらに式Vの実施形態において、Xは欠落であり、n1は1または2であり、n2は1、2、3、4または5である。
【0177】
式Vの好ましい実施形態において、本発明のプロドラッグ部分は1つまたは2つのアミノ酸(すなわちn2は1または2である。)を有する。一実施形態において、n1は1または2であり、一方n2は1、2または3である。
【0178】
式Vの一実施形態において、Xは−O−であり、n1は0、1または2であり、n2は1または2であり、R3はHである。さらなる実施形態において、R4の少なくとも1つの出現は
【化40】
である。
【0179】
一実施形態において、Xは−NH−であり、n1は0、1または2であり、n2は1または2であり、R3はHである。さらなる実施形態において、R4の少なくとも1つの出現は
【化41】
である。
【0180】
式Vの好ましい実施形態において、n2は1、2または3であり、一方R3、R4およびR5はHである。他の実施形態において、n2は1である。また他の実施形態において、n2は2である。さらに式Vの他の実施形態において、n2は1または2であり、RAAの各出現は独立して天然アミノ酸側鎖である。
【0181】
本発明の他の実施形態において、本発明のp−OHメキシレチンプロドラッグは式VIまたはVIIのジカルボン酸結合プロドラッグである。式VIおよび式VIIについて、O1、R3、R4、R5、RAA、n1およびn2は式Iに規定されるように定義される。
【化42】
式VI
【化43】
式VII
【0182】
式VIおよび/または式VIIのジカルボン酸リンカーの一実施形態において、n1は0、1、2または3である。さらなる実施形態において、n1は0、1、2または3であり、一方R3、R4およびR5は水素である。
【0183】
また、式VIおよび/または式VIIのさらなる実施形態において、n2は1、2または3である。式VIおよび/または式VIIのさらなる実施形態において、n1は1、2または3である。またさらなる実施形態において、n1は1、2または3であり、n2は1または2であり、R1、R2およびR3はそれぞれ水素である。
【0184】
式VIおよび/または式VIIの一実施形態において、n1は0、1、2または3であり、n2は1、2または3であり、R3、R4およびR5はそれぞれHである。さらなる実施形態において、n1は2である。
【0185】
式VIおよび/または式VIIの好ましい実施形態において、本発明のプロドラッグ部分は1つ、2つまたは3つのアミノ酸(すなわちn2は1、2または3である。)を有する。一実施形態において、n1は1または2であり、一方n2は1、2または3である。
【0186】
式VIおよび/または式VIIの一実施形態において、n1は0、1または2であり、n2は1または2であり、R3はHである。さらなる実施形態において、R4の少なくとも1つの出現は
【化44】
である。
【0187】
式VIおよび/または式VIIの一実施形態において、n1は0、1または2であり、n2は1または2であり、R3はHである。さらなる実施形態において、R4の少なくとも1つの出現は
【化45】
である。
【0188】
式VIおよび/または式VIIの好ましい実施形態において、n2は1、2または3であり、一方R3、R4およびR5はHである。他の実施形態において、n2は1である。また他の実施形態において、n2は2である。さらに式VIおよび/または式VIIの他の実施形態において、n2は1または2であり、RAAの各出現は独立して天然アミノ酸側鎖である。
【0189】
また、本発明の他の実施形態において、式VIIIのp−OHメキシレチンプロドラッグまたはその薬学的に許容し得る塩を提供する。式VIIIについて、O1、R3、RAA、n1およびn2は式Iに規定されるように定義される。
【化46】
式VIII
【0190】
式VIIIの一実施形態において、n2の少なくとも1つの出現は1、2、3または4であり、R3の少なくとも1つの出現はHである。さらなる実施形態において、n2の各出現は1、2、3または4であり、R3の各出現はHである。
【0191】
式VIIIの他の実施形態において、n2の少なくとも1つの出現は1、2、3、4または5である。さらなる実施形態において、n2の少なくとも1つの出現は1、2、3、4または5であり、一方R3の少なくとも1つの出現は水素である。さらなる実施形態において、n2の各出現は1、2、3、4または5であり、一方R3の各出現は水素である。また、式VIIIのさらなる実施形態において、n2の各出現は1、2、3、4または5であり、一方R3の少なくとも1つの出現はアルキル基である。
【0192】
式VIIIの好ましい実施形態において、少なくとも1つのプロドラッグ部分は1つ、2つまたは3つのアミノ酸(すなわちn2は1、2または3である。)を有し、一方R3の少なくとも1つの出現はHである。式VIIIの他の実施形態において、n2の少なくとも1つの出現は1である。また式VIIIの他の実施形態において、n2の少なくとも1つの出現は1または2であり、RAAの各出現は独立して天然アミノ酸側鎖である。式VIIIの他の実施形態において、n2の両方の出現は1、2および3から選択される。
【0193】
本発明の他の実施形態は、式IXのp−OHメキシレチンプロドラッグまたはその薬学的に許容し得る塩に関する。式IXについて、O1、−X−、R3、R4、R5、n1およびn2は式Iに規定されるように定義される。
【化47】
式IX
【0194】
式IXの一実施形態において、n1は0〜4から選択される整数である。
【0195】
式IXのジカルボン酸リンカーの一実施形態において、n1は0、1、2または3である。さらなる実施形態において、n1は0、1、2または3であり、一方R3、R4およびR5の各出現は水素である。
【0196】
また、さらなる実施形態において、n2は1、2または3である。式IXのさらなる実施形態において、−X−は欠落であり、n1は1、2または3である。また式IXのさらなる実施形態において、n2の各出現は1または2であり、R3、R4およびR5はそれぞれ水素である。
【0197】
式IXの一実施形態において、−X−は−NH−であり、n1は0、1、2または3であり、n2は1、2または3であり、R3、R4およびR5はそれぞれHである。さらなる実施形態において、n1は2である。
【0198】
式IXの一実施形態において、−X−は−O−であり、n1は0、1、2または3であり、n2の各出現は1、2、3、4または5であり、R3、R4およびR5はそれぞれHである。さらなる実施形態において、n1は2である。
【0199】
式IXの他の実施形態において、−X−は欠落であり、n1は1、2または3であり、n2は1、2または3である。また、さらに式IXの実施形態において、−X−は欠落であり、n1は1または2であり、n2の各出現は1、2、3、4または5である。
【0200】
式IXの好ましい実施形態において、本発明のプロドラッグ部分は1つまたは2つのアミノ酸(すなわちn2は1または2である)を有する。一実施形態において、n1は1または2であり、一方n2の各出現は1、2または3である。
【0201】
式IXの一実施形態において、−X−は−O−であり、n1は0、1または2であり、n2は1または2であり、R3はHである。さらなる実施形態において、R4の少なくとも1つの出現は
【化48】
である。
【0202】
式IXの他の実施形態において、−X−は−NH−であり、n1は0、1または2であり、n2の各出現は1または2であり、R3はHである。さらなる実施形態において、R4の少なくとも1つの出現は
【化49】
である。
【0203】
式IXの一実施形態において、−X−は−O−であり、n1は0、1または2であり、n2は1または2であり、R3はHである。さらなる実施形態において、R4の少なくとも1つの出現は
【化50】
である。
【0204】
式IXの他の実施形態において、−X−は−NH−であり、n1は0、1または2であり、n2の各出現は1または2であり、R3はHである。さらなる実施形態において、R4の少なくとも1つの出現は
【化51】
である。
【0205】
式IXの好ましい実施形態において、n2は1、2または3であり、一方R1、R2およびR3はHである。他の実施形態において、n2の少なくとも1つの出現は1である。また他の実施形態において、n2は2である。さらに式IXの他の実施形態において、n2は1または2であり、RAAの各出現は独立して天然アミノ酸側鎖である。
【0206】
本発明の他の実施形態は、式Xおよび式XIのp−OHメキシレチンプロドラッグまたはその薬学的に許容し得る塩に関する。式Xおよび式XIについて、O1、R3、R4、R5、RAA、n1およびn2は式Iに規定されるように定義される。
【化52】
式X
【化53】
式XI
【0207】
式Xおよび/または式XIの一実施形態において、n1は0、1、2または3である。さらなる実施形態において、n1は0、1、2または3であり、一方R3、R4およびR5の各出現は水素である。
【0208】
また、式Xおよび/または式XIのさらなる実施形態において、n2は1、2または3である。式Xおよび/または式XIの他の実施形態において、n1は1、2または3である。また、さらなる実施形態において、n1は1、2または3であり、n2の各出現は1、2、3または4であり、R3、R4およびR5はそれぞれ水素である。
【0209】
式Xおよび/または式XIの一実施形態において、n1は0、1、2または3であり、n2の各出現は1、2または3であり、R3、R4およびR5はそれぞれHである。さらなる実施形態において、n1は2である。
【0210】
式Xおよび/または式XIの好ましい実施形態において、本発明の両方のプロドラッグ部分は1つ、2つ、3つまたは4つのアミノ酸(すなわちn2は1、2、3または4である)を有する。一実施形態において、n1は1または2であり、一方n2の各出現は1、2または3である。
【0211】
式Xおよび/または式XIの一実施形態において、n1は0、1または2であり、n2の少なくとも1つの出現は1または2であり、R3はHである。さらなる実施形態において、R4の少なくとも1つの出現は
【化54】
である。
【0212】
式Xおよび/または式XIの一実施形態において、n1は0、1または2であり、n2の各出現は1、2または3であり、R3はHである。さらなる実施形態において、R4の少なくとも1つの出現は
【化55】
である。
【0213】
式Xおよび/または式XIの好ましい実施形態において、n2は1、2または3であり、一方R1、R2およびR3はHである。他の実施形態において、n2の少なくとも1つの出現は1である。また他の実施形態において、n2の少なくとも1つの出現は2である。さらに式Xおよび/または式XIの他の実施形態において、n2の各出現は1または2であり、RAAの各出現は独立して天然アミノ酸側鎖である。
【0214】
メタ−OHメキシレチンのプロドラッグ
【0215】
上述した式I〜XIの実施形態はメキシレチンおよびp−OHメキシレチンプロドラッグに関するが、本開示はまた他のメキシレチンプロドラッグのプロドラッグを包含する。したがって、m−OHメキシレチンおよびヒドロキシメチルメキシレチンのプロドラッグが本開示の範囲内である。
【0216】
例えば一実施形態において、本発明は式XIIにより包含されるメタ−OHメキシレチンプロドラッグ、
【化56】
式XII
【0217】
またはその薬学的に許容し得る塩に関し、
【0218】
ここで式中のR1は水素および
【化57】
から選択され、
【0219】
R2は、
【化58】
から選択され、
【0220】
O1は未結合メタ−OHメキシレチン中に存在するフェノール系酸素であり、
【0221】
Xは(−NH−)、(−O−)、または欠落であり、
【0222】
RAAの各出現は独立して天然または非天然アミノ酸側鎖であり、
【0223】
n1は0〜16から選ばれる整数であり、
【0224】
n2の各出現は1〜9から選ばれる整数であり、
【0225】
R3の各出現は独立して水素、置換アルキル基または非置換アルキル基から選択され、
【0226】
R4およびR5の各出現は、水素、
【化59】
、置換アルキル基および非置換アルキル基から選択され、
【0227】
n1により定義される炭素鎖における二重結合の場合、二重結合を形成する炭素上にR4は存在し、R5は欠落であり、
【0228】
少なくともR1は
【化60】
であるか、あるいは少なくともR2は
【化61】
である。
【0229】
ジカルボン酸リンカーの一実施形態において、n1は0、1、2、3または4である。
【0230】
一実施形態において、n2の各出現は1、2、3、4および5である。
【0231】
好ましい一実施形態において、本発明の化合物は一つのプロドラッグ部分を有し、該プロドラッグ部分は1つ、2つまたは3つのアミノ酸(すなわちn2が1、2または3である)を有する一方、R3はHである。
【0232】
他の実施形態において、n2は1である。
【0233】
さらに他の実施形態において、n2は2である。
【0234】
さらに他の実施形態において、n2の各出現は1または2であり、RAAの各出現は独立して天然アミノ酸側鎖である。
【0235】
一実施形態において、プロドラッグ部分は、アルギニンもしくはリシンのホモポリマーか、またはアルギニンおよびリシンのヘテロポリマーである。さらなる実施形態において、ホモポリマーまたはヘテロポリマー中に2、3、4、5、6または7のアミノ酸がある(すなわちn2が2、3、4、5、6または7である)。
【0236】
上述した式XIIで提供されるように、本明細書に記載したパラ置換のプロドラッグ部分(例えば、式II〜XIのプロドラッグ)をメタ位にすることもできる。これら実施形態において、パラ位のプロドラッグ部分を水素で置換し、一方プロドラッグ部分をパラ位に隣接する炭素の1つに付加する。カルバメート結合メタ−OHメキシレチンプロドラッグ(式XIII)についての例を以下に示す。
【化62】
【0237】
ヒドロキシメチルメキシレチンのプロドラッグ
【0238】
一実施形態において、本発明は、式XIVにより包含されるヒドロキシメチルメキシレチンプロドラッグ、
【化63】
式XIV
【0239】
またはその薬学的に許容し得る塩に関し、
【0240】
ここで式中のR1は水素および
【化64】
から選択され、
【0241】
R2は、
【化65】
から選択され、
【0242】
O1は未結合ヒドロキシメチルメキシレチン中に存在するフェノール系酸素であり、
【0243】
Xは(−NH−)、(−O−)、または欠落であり、
【0244】
RAAの各出現は独立して天然または非天然アミノ酸側鎖であり、
【0245】
n1は0〜16から選ばれる整数であり、
【0246】
n2の各出現は1〜9から選ばれる整数であり、
【0247】
R3の各出現は独立して水素、置換アルキル基または非置換アルキル基から選択され、
【0248】
R4およびR5の各出現は独立して水素、
【化66】
、置換アルキル基および非置換アルキル基から選択され;二重結合の場合、二重結合を形成する炭素上にR4は存在し、R5は欠落であり、
【0249】
n1により定義される炭素鎖における二重結合の場合、二重結合を形成する炭素上にR4は存在し、R5は欠落であり、
【0250】
少なくともR1は
【化67】
であるか、あるいは少なくともR2は
【化68】
である。
【0251】
ジカルボン酸リンカーの一実施形態において、n1が0、1、2、3または4である。
【0252】
一実施形態において、n2の各出現は1、2、3、4および5である。
【0253】
好ましい一実施形態において、本発明の化合物は一つのプロドラッグ部分を有し、該プロドラッグ部分は1つ、2つまたは3つのアミノ酸(すなわちn2が1、2または3である)を有し、一方R3はHである。
【0254】
他の実施形態において、n2は1である。さらに他の実施形態において、n2は2である。
【0255】
さらに他の実施形態において、n2の各出現は1または2であり、RAAの各出現は独立して天然アミノ酸側鎖である。
【0256】
一実施形態において、プロドラッグ部分は、アルギニンもしくはリシンのホモポリマーか、またはアルギニンおよびリシンのヘテロポリマーである。さらなる実施形態において、ホモポリマーまたはヘテロポリマー中に2、3、4、5、6または7のアミノ酸がある(すなわちn2が2、3、4、5、6または7である)。
【0257】
ここに記載したパラおよびメタ置換のプロドラッグ部分(例えば、式II〜XIIIのプロドラッグ)を、芳香環上のメチル基にあることもできる。これら実施形態において、パラまたはメタ位のプロドラッグ部分を水素で置換し、一方プロドラッグ部分を芳香環上のメチル基に付加する。カルバメート結合ヒドロキシメチルメキシレチンプロドラッグ(式XV)についての例を以下に示す。
【化69】
【0258】
式(II)〜式(XV)のそれぞれについて、式(I)の関連して記載した様々な実施形態もまた適用する(化学的に可能で妥当である場合)が、式(II)〜式(XV)のそれぞれの議論の簡潔さのために単に省略されている。
【0259】
本発明で使用する代表的なアミノ酸およびペプチド
【0260】
以下に記載の代表的なプロドラッグは、p−OHメキシレチンプロドラッグに関する。しかし、同じアミノ酸およびペプチドプロドラッグ部分をm−OHメキシレチンおよびヒドロキシメチルメキシレチンプロドラッグに使用することができる。
【0261】
フェノール系官能基の場合、p−OHメキシレチン代謝物をカルバメートまたはジカルボン酸リンカー(置換または非置換)によりアミノ酸またはペプチドに結合し得る。例えば、マロン酸、コハク酸またはグルタル酸を本発明においてリンカーとして用いることができる。本発明での使用に可能な他のジカルボン酸を表2および表3に示す。p−OHメキシレチン代謝物で用いる好ましいアミノ酸としては、単一のアミノ酸またはジペプチドの一部のいずれかとして結合したバリン、ロイシン若しくはイソロイシンまたは類似物である。例えば、ジペプチドのバリン−バリン、バリン−ロイシン、バリン−イソロイシン、ロイシン−ロイシン、ロイシン−バリン、ロイシン−イソロイシン、イソロイシン−イソロイシン、イソロイシン−バリンおよびイソロイシン−ロイシンを使用することができる。さらにパラ−アミノ安息香酸のような非天然アミノ酸を単独または天然アミノ酸と一緒に使用することができる。
【0262】
特定の理論に拘束されることを望むことなく、メキシレチン若しくはp−OHメキシレチン、m−OHメキシレチンもしくはヒドロキシメチルメキシレチンのプロドラッグのアミノ酸またはペプチド部分は、消化管内で固有のジペプチドおよびトリペプチドトランスポーターPept1を選択的に利用すると考えられる。一度吸収されると、プロドラッグは加水分解を受けて、活性薬剤を全身循環へ放出する。活性薬剤および腸壁間の直接接触の回避は嘔吐のリスクを最小限にする一方、Pept1により助長されたプロドラッグの吸収はより一貫した血漿薬剤濃度を確保する。
【0263】
式Iのメキシレチンプロドラッグの好ましい実施態様は、メキシレチンリジンアミド(慣用名:2,6−ジアミノ−ヘキサン酸[2−(2,6−ジメチルフェノキシ)−1−メチルエチル]−アミド ジトリフルオロ酢酸塩)またはメキシレチンp−OHリジンアミドのトリフルオロ酢酸塩である。
【化70】
メキシレチン−リジンアミドジトリフルオロ酢酸塩
【化71】
p−OHメキシレチン−リジンアミドジトリフルオロ酢酸塩
【0264】
他の実施形態態様において、式Iのメキシレチンプロドラッグはメキシレチングルタミン酸アミドの塩酸塩(慣用名:(S)−4−アミノ−4−[2−(2,6−ジメチル−フェノキシ)−1−メチル−エチルカルバモイル]−酪酸塩酸塩)または対応する活性代謝物p−OHメキシレチンのプロドラッグである。
【化72】
メキシレチングルタミン酸アミド
【0265】
他の実施形態において、式Iのメキシレチンプロドラッグはメキシレチングルタミンアミドの塩酸塩(慣用名:(S)−2−アミノ−ペンタン二酸 5−アミド 1−{[2−(2,6−ジメチル−フェノキシ)−1−メチル−エチル]−アミド}塩酸塩)または対応する活性代謝物p−OHメキシレチンのプロドラッグである。
【化73】
メキシレチングルタミンアミド
【0266】
他の実施形態において、式Iのメキシレチンプロドラッグはメキシレチンホモアルギニンアミドの塩酸塩(慣用名:(S)−2−アミノ−6−グアニジノ−ヘキサン酸[2−(2,6−ジメチル−フェノキシ)−1−メチル−エチル]−アミド二塩酸塩)または対応する活性代謝物p−OHメキシレチンのプロドラッグである。
【化74】
メキシレチンホモアルギニンアミド
【0267】
他の実施形態において、式Iのメキシレチンプロドラッグはメキシレチン塩化メチルメチオニンアミドの塩酸塩(慣用名:((S)−2−アミノ−N−[2−(2,6−ジメチル−フェノキシ)−1−メチル−エチル]−4−(ジメチル−λ4−スルファニルクロライド)−ブチルアミド塩酸塩)または対応する活性代謝物p−OHメキシレチンのプロドラッグである。
【化75】
メキシレチン塩化メチルメチオニンアミド
【0268】
単一のアミノ酸結合体の他の実施形態は、シトルリンまたはオルニチンとのアミド結合体を含む。ジペプチドの好ましい実施形態は、上述したアミノ酸のヘテロまたはホモ二量体を含む結合体である。
【0269】
メキシレチンのオリゴペプチドは、メキシレチンリジンアミド、メキシレチンアルギニンアミド、メキシレチンシトルリンアミドまたはメキシレチンオルニチンアミドへの上述したアミノ酸のいずれかのポリマーの付加により形成することができる。あるいはまた、他の天然または非天然アミノ酸をメキシレチンまたはその活性代謝物に直接結合することができる。メキシレチンプロドラッグのいくつかの例を以下に示す。列挙した第一のアミノ酸は、アミド結合を介してメキシレチンに結合したものである。
【化76】
メキシレチン−Gly−Arg−Arg−Arg
【化77】
メキシレチン−Arg−Arg−Arg−Arg
【化78】
メキシレチン−Lys−Lys−Lys−Lys
【化79】
メキシレチン−Gly−Gly−Arg−Gly
【0270】
p−OH代謝物プロドラッグの好ましい実施形態は、次の2つの単一のアミノ酸(バリンおよびグリシン)の1つまたは両方を有する化合物を含み、以下に示す。
【化80】
4−ヒドロキシメキシレチン−(S)−バリンカルバメートグリシンアミド
【0271】
バリンの代わりの他の単一のアミノ酸は、限定しないが、イソロイシンまたはチロシンを含む一方、グリシンと置き換わるアミノ酸は、限定しないが、オルニチン、シトルリンまたはアルギニンアミドとすることができる。非天然アミノ酸カルバメート結合体は、パラアミノ安息香酸を含む。
【0272】
アミド結合アミノ酸のオリゴペプチドは、上述したように、p−OH代謝物にとってメキシレチン自体に対するものと類似とし得る。
【0273】
好適なアミノ酸は、L−体すべてであるが、本発明はD−体のアミノ酸からなるか、またはD−体およびL−体のアミノ酸混合物からなる式I(または式II〜XV)のプロドラッグも意図している。
【0274】
本発明の化合物の塩、溶媒和物、立体異性体および誘導体
【0275】
以下に記載する代表的な塩は、メキシレチンおよびメキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメチルメキシレチン)のプロドラッグに関する。
【0276】
本発明の方法は、ここに記載したメキシレチン/メキシレチン代謝物のプロドラッグの塩、溶媒和物、立体異性体、例えば、式Iおよび式II〜XVのプロドラッグの塩の使用を包含する。一実施形態において、ここで開示した本発明は、メキシレチンプロドラッグの全ての薬学的に許容し得る塩を包含することを意味する。
【0277】
一般に、本発明の実施に用いるメキシレチンのプロドラッグの薬学的に許容し得る塩は、プロドラッグとそれぞれに見合った所望の酸との反応によって調製される。p−OHメキシレチン代謝物のプロドラッグの場合、これは遊離のカルボキシ官能基の塩を作製することを含むこともできる。かかる塩は、溶液から沈殿させ、濾過により収集するか、または溶媒の蒸発により回収することができる。例えば、塩酸のような酸の水溶液をプロドラッグの水性懸濁液に添加し、生成した混合物を蒸発乾固(凍結乾燥)して酸付加塩を固体として得ることができる。あるいはまた、プロドラッグを適当な溶媒、例えばイソプロパノールのようなアルコールに溶解し、酸を同じ溶媒または他の適当な溶媒に添加することができる。その後、生成した酸付加塩を直接またはジイソプロピルエーテル若しくはヘキサンのようなより低い極性溶媒の添加により沈殿させ、濾過により単離することができる。
【0278】
プロドラッグの酸付加塩は、その遊離塩基形態物を従来方法で塩を生成するに十分な量の所望の酸と接触させることによって調製することができる。遊離塩基形態物は、塩形態物を塩基と接触させ、従来方法で遊離塩基を単離することによって再生することができる。遊離塩基形態物は、それぞれの塩形態物と極性溶媒への溶解性のような特定の物理的性質が若干異なるが、その他の点において塩はそれぞれの遊離塩基と本発明の目的について同等である。
【0279】
p−OHメキシレチン代謝物のプロドラッグの薬学的に許容し得る塩基付加塩は、アルカリ金属およびアルカリ土類金属または有機アミンのような金属またはアミンで形成される。陽イオンとして用いる金属の例としては、ナトリウム、カリウム、マグネシウム、カルシウム等がある。適当なアミンの例としては、N,N'−ジベンジルエチレンジアミン、クロロプロカイン、コリン、ジエタノールアミン、ジシクロヘキシルアミン、エチレンジアミンおよびN−メチルグルカミンがある。
【0280】
酸性化合物の塩基付加塩は、遊離の酸形態物を従来方法で塩を生成するに十分な量の所望の塩基と接触させることによって調製することができる。遊離の酸形態物は、塩形態物を酸と接触させ、遊離酸を単離することによって再生させることができる。
【0281】
本発明の実施に有用なp−OHメキシレチン代謝物の化合物は、塩基性および酸性中心の両方を有し、したがって両性イオンの形態とすることができる。
【0282】
有機化学の当業者は、多くの有機化合物が錯体、すなわち溶媒と反応するかまたは溶媒から沈殿若しくは結晶化した溶媒和物、例えば水との水和物を形成することができることを理解するであろう。本発明に有用な化合物の塩は、有用な水和物などの溶媒和物を形成することができる。溶媒和物の調製技術は当業界で周知である(例えば、Brittain,Polymorphism in Pharmaceutical solids.Marcel Decker,New York(1999)を参照)。本発明の実施に有用な化合物は、一つ以上のキラル中心を持つことができ、また個々の置換基の性質に応じて幾何異性体を有することもできる。
【0283】
ここに記載したメキシレチン(またはメキシレチン代謝物)プロドラッグの個々の異性体は、本発明を実施するために使用することができる。明細書および特許請求の範囲における特定の化合物の記述や命名は、プロドラッグの個々の鏡像異性体並びにプロドラッグの鏡像異性体(ラセミ体またはそれ以外)の混合物の両方を含むことが意図されている。立体化学の決定および立体異性体の溶解のための方法は当業界で周知である。
【0284】
したがって、本発明は式(I)の化合物のいずれの互変異性形態並びに幾何および光学異性体をも包含する。それ故、本発明は式(I)またはその薬学的塩の互変異性体を特に含むことを意図する。
【0285】
本発明の医薬組成物
【0286】
本発明の方法での使用についてプロドラッグをバルク物質として投与し得ることが可能であるが、活性成分を例えば、薬剤が意図された投与の経路および標準的な製薬行為に関して選択された薬学的に許容し得る担体との混合物である医薬製剤で提示するのが好ましい。本発明のいくつかの実施形態において、本発明の組成物は式I〜XVのプロドラッグまたはその薬学的に許容し得る塩から選択したプロドラッグと、薬学的に許容し得る賦形剤とを含む。
【0287】
本発明の製剤は、速放性投薬形態、すなわち、プロドラッグを吸収部位で直ちに放出する投薬形態か、または放出制御投薬形態、すなわち、プロドラッグを所定の期間にわたって放出する投薬形態とすることができる。放出制御投薬形態はあらゆる従来型、例えば、貯蔵若しくはマトリックス型の拡散制御投薬形態;マトリックス、カプセル化若しくは腸溶化の溶解制御投薬形態;または浸透性投形態としてもよい。このような種類の投薬形態は、例えばレミントン「薬学の科学と実務」、第20版、2000年、858−914頁に開示されている。本発明の製剤は、投薬形態および投与量に応じて1日1−6回投与することができる。
【0288】
メキシレチン/p−OHメキシレチン代謝物のアミノ酸およびペプチドプロドラッグの吸収は、Pept1のような活性トランスポーターを経て進展し得る。このトランスポーターは、上部消化管に主に限定されると考えられるので、それ自体消化管の全長に沿って継続的な吸収用の従来の徐放性製剤の有用性を制限する。活性薬剤の継続的な全身発生により血漿中薬剤濃度の持続をもたらさないメキシレチン/p−OHメキシレチン代謝物プロドラッグに対し、プロドラッグの血漿貯蔵、Glumetza(登録商標)またはGluphage XR(登録商標)のようなメトホルミン製品に用いるものと類似の胃保持性または粘膜保持性製剤が有利である。前者は、Gelshield Diffusion Technologyとして既知の薬剤送達システムを活用し、一方後者はいわゆるAcuform送達システムを使用する。いずれにしても、コンセプトは回腸への薬剤伝達を遅らせて、吸収が起こりうる期間を最大化し、血漿中薬剤濃度を効果的に延長することである。消化管に沿った遅延進行を与える他の薬剤送達システムも価値がある。
【0289】
前述の薬剤伝達システムの高度化を必要としないメキシレチンプロドラッグについて、下記のような従来の製剤が適切である。
【0290】
一態様において、本発明は、少なくとも1つの活性な薬学的成分(すなわち、メキシレチンまたはp−OHメキシレチン代謝物のプロドラッグ)またはその薬学的に許容し得る誘導体(例えば、塩または溶媒和物)と、薬学的に許容し得る担体とを含む医薬組成物を提供する。特に、本発明は、治療有効量の本発明の少なくとも一つのプロドラッグまたはその薬学的に許容し得る誘導体と、薬学的に許容し得る担体とを含む医薬組成物を提供する。
【0291】
本発明の方法に対し、本発明で用いるプロドラッグを他の治療法および/または活性剤と組み合わせて使用することができる。したがって、本発明のさらなる態様は、本発明の実施に有用な少なくとも一つの化合物またはその薬学的に許容し得る塩もしくは溶媒和物と、第二の活性剤と、随意的な薬学的に許容し得る担体とを含む医薬組成物を提供する。
【0292】
同じ製剤に組み合わせるとき、2つの化合物が互いにかつ製剤の他の成分の存在下で安定で、また相溶性であると理解されるであろう。別々に剤形化するとき、これらはあらゆる好都合な製剤で、当業界でこの種化合物に既知の方法で都合よく提供することができる。いくつかの実施形態において、2つの化合物は、(1)2つの別のメキシレチンのプロドラッグ、(2)メキシレチンのプロドラッグおよびp−OHメキシレチンのプロドラッグ、(3)2つのp−OHメキシレチンのプロドラッグ、(4)メキシレチンのプロドラッグおよびm−OHメキシレチンのプロドラッグ、(5)メキシレチンのプロドラッグおよびヒドロキシメチルメキシレチンのプロドラッグ、(6)2つのm−OHメキシレチンのプロドラッグ、(7)2つのヒドロキシメチルメキシレチンのプロドラッグ、(8)メタ−OHメキシレチンのプロドラッグおよびp−OHメキシレチンのプロドラッグ、(9)ヒドロキシメチルメキシレチンのプロドラッグおよびp−OHメキシレチンのプロドラッグのいずれかである。他の実施形態において、2つの化合物は式Iのプロドラッグと、別の効能のための他の化合物とを含む。
【0293】
ここで用いるプロドラッグは、ヒト用または獣医用薬での使用にあらゆる簡便な方法で投与するために剤形化することができるので、本発明はヒト用または獣医用薬での使用に適した本発明の化合物を含む医薬組成物を本発明の範囲内で含む。かかる組成物は、一つ以上の適当な担体の援助で従来の方法での使用に提示することができる。治療目的の使用に許容し得る担体は製薬技術分野で周知であり、例えばレミントンの製薬科学(マック出版社、A.R.ジェンナーロ編集、1985年)に記載されている。医薬用担体の選択は、意図する投与経路および標準的な製薬実務に対して行うことができる。加えて、医薬組成物は担体としてあらゆる適当な結合剤、潤滑剤、懸濁剤、コーティング剤および/または可溶化剤を含むことができる。
【0294】
防腐剤、安定剤、染料そしてさらに香料を医薬組成物に付与することができる。防腐剤の例としては、安息香酸ナトリウム、アスコルビン酸およびp−ヒドロキシ安息香酸エステルが挙げられる。抗酸化剤および懸濁化剤を用いることもできる。
【0295】
本発明に用いる化合物は、湿式粉砕のような既知の粉砕手順を用いて粉砕して錠剤形成用およびその他の製剤タイプに適した粒径を得ることができる。化合物の微粉細調製物(ナノ粒子)は、当業界で既知の方法により調製することができ、例えば国際特許出願WO02/00196(SmithklineBeecham)を参照。
【0296】
本発明の化合物および医薬組成物は、経口投与(例えば錠剤、小袋、カプセル、トローチ、丸剤、巨丸剤、散剤、ペースト、顆粒、薬包または予備混合調製、座薬、エリキシル剤、溶液、懸濁液、分散液、ゲル、シロップとしてまたは摂取溶液として)することを意図する。さらに、該化合物は使用前に水または他の適当な媒介物、随意に香料と着色料との構成のために乾燥粉末として存在してもよい。固体および液体組成物は、当業界で周知の方法に従って調製することができる。このような組成物はまた、固体または液体形態が可能な一つ以上の薬学的に許容し得る担体および賦形剤を含むことができる。
【0297】
分散液は、グリセリン、液体ポリエチレングリコール、トリアセチン油およびその混合物のような液体担体または中間体中で調製することができる。液体担体または中間体は、例えば水、エタノール、ポリオール(例えば、グリセロール、プロピレングリコール等)、植物油、非毒性のグリセリンエステルおよびその適当な混合物を含む溶剤または液体分散媒体とすることができる。適当な流動性は、リポソームの発生、分散液の場合適当な粒径の管理によるか、または界面活性剤の添加により維持し得る。
【0298】
錠剤は、微結晶性セルロース、乳糖、クエン酸ナトリウム、炭酸カルシウム、二塩基性リン酸カルシウムおよびグリシンのような賦形剤、デンプン(好ましくはトウモロコシ、ジャガイモまたはタピオカのデンプン)、デンプングリコール酸ナトリウム、クロスカルメロースナトリウムおよび特定の錯体ケイ酸塩のような崩壊剤、並びにポリビニルピロリドン、ヒドロキシプロピルメチルセルロース(HPMC)、ヒドロキシプロピルセルロース(HPC)、ショ糖、ゼラチンおよびアカシアのような顆粒化結合剤を含むことができる。
【0299】
さらに、ステアリン酸マグネシウム、ステアリン酸、ベヘン酸グリセリルおよびタルクのような滑沢剤を含むことができる。
【0300】
本発明に有用な経口組成物用の薬学的に許容し得る崩壊剤の例としては、限定しないが、デンプン、アルファ化デンプン、デンプングリコール酸ナトリウム、カルボキシメチルセルロースナトリウム、クロスカルメロースナトリウム、微結晶性セルロース、アルギン酸塩、樹脂、界面活性剤、発泡性組成物、水溶性ケイ酸アルミニウムおよび架橋ポリビニルピロリドンが挙げられる。
【0301】
本発明に有用な経口組成物用の薬学的に許容し得る結合剤の例としては、限定しないが、アカシア;メチルセルロース、カルボキシメチルセルロース、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロースまたはヒドロキシエチルセルロースのようなセルロース誘導体;ゼラチン、グルコース、デキストロース、キシリトール、ポリメタクリレート、ポリビニルピロリドン、ソルビトール、デンプン、アルファ化デンプン、トラガカント、キサンタン樹脂、アルギン酸塩、ケイ酸マグネシウムアルミニウム、ポリエチレングリコールまたはベントナイトが挙げられる。
【0302】
本発明に有用な経口組成物用の薬学的に許容し得る充填剤の例としては、限定しないが、乳糖、アンヒドロラクトース、ラクトース一水和物、ショ糖、デキストロース、マンニトール、ソルビトール、デンプン、セルロース(特に微結晶性セルロース)、ジヒドロまたは無水リン酸カルシウム、炭酸カルシウムおよび硫酸カルシウムが挙げられる。
【0303】
本発明の組成物に有用な薬学的に許容し得る潤滑剤の例としては、限定しないが、ステアリン酸マグネシウム、タルク、ポリエチレングリコール、エチレンオキサイドのポリマー、ラウリル硫酸ナトリウム、ラウリル硫酸マグネシウム、オレイン酸ナトリウム、フマル酸ステアリルナトリウムおよびコロイド状二酸化ケイ素が挙げられる。
【0304】
経口組成物に適した薬学的に許容し得る匂い物質の例としては、限定しないが、油、花、果物(例えば、バナナ、リンゴ、サワーチェリー、桃)の抽出物およびその組み合わせのような合成芳香油や天然芳香油、ならびに類似の芳香抽出物が挙げられる。その使用は多くの因子に依存するが、医薬組成物を服用する人々のための感覚受容性が最も重要である。
【0305】
経口組成物に適した薬学的に許容し得る染料の例としては、限定しないが、二酸化チタン、β−カロチンおよびグレープフルーツの皮の抽出物のような合成および天然物染料が挙げられる。
【0306】
経口組成物に有用で、通常嚥下を容易にし、放出特性を変更し、外観を向上させ、および/または組成物の味を覆うために用いる薬学的に許容し得るコーティング剤の例としては、限定しないが、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロースおよびアクリル酸−メタクリル酸エステル共重合体が挙げられる。
【0307】
経口組成物に適した薬学的に許容し得る甘味料の例としては、限定しないが、アスパルテーム、サッカリン、サッカリンナトリウム、シクラミン酸ナトリウム、キシリトール、マンニトール、ソルビトール、ラクトースおよびスクロースが挙げられる。
【0308】
本発明に有用な薬学的に許容し得る緩衝液の適当な例としては、限定しないが、クエン酸、クエン酸ナトリウム、重曹、二塩基性リン酸ナトリウム、酸化マグネシウム、炭酸カルシウムおよび水酸化マグネシウムが挙げられる。
【0309】
本発明に有用な薬学的に許容し得る界面活性剤の適当な例としては、限定しないが、ラウリル硫酸ナトリウムおよびポリソルベート類が挙げられる。
【0310】
同様な種類の固体組成物を、ゼラチンカプセル中の充填剤として用いることもできる。これに関して好ましい賦形剤は、ラクトース、デンプン、セルロース、乳糖または高分子量ポリエチレングリコールを含む。水性懸濁液および/またはエリキシル剤の場合、薬剤を種々の甘味剤または芳香剤、着色剤または染料と、乳化剤および/または懸濁化剤と、水、エタノール、プロピレングリコールおよびグリセリンのような希釈剤と、並びにその混合物と組み合わせることができる。
【0311】
薬学的に許容し得る防腐剤の適当な例としては、限定しないが、様々な抗菌性および抗真菌性の薬剤、例えば、エタノール、プロピレングリコール、ベンジルアルコール、クロロブタノール、第四級アンモニウム塩およびパラベン類(例えば、メチルパラベン、エチルパラベン、プロピルパラベン等)のような溶剤が挙げられる。
【0312】
薬学的に許容し得る安定剤や抗酸化剤の適当な例としては、限定しないが、エチレンジアミン四酢酸(EDTA)、チオ尿素、トコフェロールおよびブチルヒドロキシアニソールが挙げられる。
【0313】
本発明の医薬組成物は、体積あたり0.01−99%重量の本発明で包含されるプロドラッグを含有することができる。
【0314】
投薬量
【0315】
特に示されない限り、ここで規定したプロドラッグ投薬量は、メキシレチン遊離塩基の当量を指す。
【0316】
本発明の方法に従って治療すべき適切な被検体は、かかる治療を必要とするあらゆるヒトや動物などである。動物またはヒトが経験する痛みの重篤度を含む痛みの診断と臨床評価の方法は、当業界で周知である。従って、患者が痛みのための治療を必要とするかどうかを判断することは、当業界における通常の施術者(例えば、医師または獣医師)の技量の範囲内である。患者は、好ましくは哺乳動物、より好ましくはヒトであるが、臨床試験またはスクリーニング若しくは動物モデルを用いる活性実験の観点から実験動物を含むあらゆる動物とすることができる。したがって、当業者によって容易に理解されるように、本発明の方法および組成物はあらゆる動物、特に限定しないが、猫科またはイヌ科被検体のような家畜、ウシ、ウマ、ヤギ、ヒツジおよびブタ被検体のような家畜、マウス、ラット、ウサギ、ヤギ、ヒツジ、ブタ、イヌ、ネコのような研究用動物、ニワトリ、七面鳥、小鳥等のような鳥類を含む哺乳動物に投与するのに特に適している。
【0317】
通常、医師は個々の被検体に最も適した実際の投薬量を決定する。任意特定の個人用の具体的な投与度合いと投与頻度は変化し得るもので、使用する特定化合物の活性、該化合物の代謝安定性および作用長さ、年齢、体重、一般的健康、性別、食事、投与の方式および時間、排出速度、薬物の組み合わせ、特定疾患の重篤度並びに個々が受けている治療を含む様々な要因に依存する。
【0318】
FDAが承認したメキシレチン塩酸塩製剤であるMexitil(登録商標)は、150mg、200mgおよび250mgのカプセルで利用可能である。100mgのメキシレチン塩酸塩は83.31mgのメキシレチン塩基に相当する。通常、Mexitil(登録商標)を8時間ごとに投与する。本発明の一実施形態において、プロドラッグ投薬量は、Mexitil(登録商標)の投薬量の1つから選択され、8時間ごとに一回投与することができる。別の実施形態では、プロドラッグ投薬量は、Mexitil(登録商標)の投薬量の1つから選択され、12時間ごとまたは24時間ごとに1回投与することができる。
【0319】
一実施形態において、メキシレチンプロドラッグの有効な毎日の投薬量は、プロドラッグ1mg〜2000mg、好ましくは100mgから2000mgである。例えば、本発明で包含されるプロドラッグは、一日あたり約200mg〜約2000mgのプロドラッグ、好ましくは一日あたり約200mg〜1000mgのプロドラッグを与える投薬形態で製剤化し得る。好ましい実施形態において、本発明のプロドラッグの有効量は、250mg、500mg、750mg/日のいずれかである。
【0320】
別の実施形態において、p−OHメキシレチンプロドラッグの有効な毎日の投薬量は、プロドラッグ4mg〜8000mg、好ましくは400mgから8000mgである。別の実施形態において、p−OHメキシレチンプロドラッグの有効な毎日の投薬量は、1000mgまたは3000mgのいずれかである。
【0321】
治療すべき痛みの重篤度に応じて、当該技術範囲内で容易に決定し得るようで、過度な実験なしに適当な治療に有効かつ安全な投薬量を被検体に投与することができる。ヒトへの経口投与の場合、プロドラッグの毎日の投薬度合いは、単一または分割投与量とすることができる。治療の期間は、当業者によって決定することができ、痛みの性質(例えば、慢性対急性の状態)および/または治療に対する治療応答の速さと程度を反映すべきである。
【0322】
痛みの治療法において、本発明で包含されるプロドラッグを他の治療法と組み合わせておよび/または他の活性薬剤と組み合せて投与することができる。例えば、本発明で包含されるプロドラッグを痛みの管理に用いる他の活性剤と組み合わせて患者に投与することができる。本発明で包含されるプロドラッグと組み合わせて投与すべき活性剤は、例えば、アセトアミノフェンおよびイブプロフェンを含む非ステロイド性抗炎症薬、もしくはオキシコドン、オキシモルフォン、レボルファノールを含むオピオイド、またはオンダンセトロン、ドンペリドン、ヒヨスチンまたはメトクロプラミドのような抗嘔吐剤からなる群から選択した薬剤を含むことかできる。このような併用療法において、本発明で包含されるプロドラッグは、他の治療法および/または活性剤に優先して、同時に、または引き続いて投与しても良い。
【0323】
本発明で包含されるプロドラッグを他の活性剤と組合わせて投与する場合、かかる組み合わせの個々の成分をあらゆる簡便な経路によって個別または一緒にした製剤処方で順次または同時に投与することができる。順次に投与する場合、本発明で包含されるプロドラッグまたは第二の活性剤のいずれかを先に投与することができる。例えば、他の活性剤との併用療法の場合、本発明で包含されるプロドラッグを薬物組み合わせの有益な効果を提供する投薬計画で順次に投与することができる。同時に投与する場合、組み合わせ物を同一または異なる医薬組成物のいずれかで投与することができる。例えば、本発明で包含されるプロドラッグおよび他の活性剤は、これら薬剤の一定割合を有する単一のカプセルもしくは錠剤、または各薬剤に対して複数の別々のカプセルもしくは錠剤のようにして実質的に同時に投与することができる。
【0324】
本発明で包含されるプロドラッグを、痛みの治療方法で活性な他の薬剤と組み合わせて使用する場合、各化合物の投与量は、化合物を単独で使用する場合のものと異なる場合がある。適切な投与量は、当業者により容易に理解されるであろう。
【0325】
本発明の方法
【0326】
本発明の一実施形態は、必要とする被検体の疾患をメキシレチンで治療する方法である。該方法は、本発明のメキシレチンプロドラッグ、その薬学的に許容し得る塩または組成物を必要とする被検体に経口投与することを備え、ここでメキシレチンプロドラッグが少なくとも一つのアミノ酸または長さ2〜9個のアミノ酸のペプチドに共有結合したメキシレチンまたはメキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメチルメキシレチン)からなる。メキシレチンプロドラッグは一または二つのプロドラッグ部分を有する。一つのプロドラッグ部分を、ペプチド結合を介してメキシレチンまたはメキシレチン代謝物のアミノ基に結合することができる。あるいは、またはさらに、メキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメキシレチン)は、フェノール系酸素でカルバメートまたはジカルボン酸リンカーを介して結合したプロドラッグ部分を有することができる。メキシレチンの量は、治療有効量(例えば、鎮痛有効量)であるのが好ましい。疾患は、メキシレチンで治療可能なものとすることができる。例えば、疾患は神経因性疼痛または不整脈であってもよい。
【0327】
本発明のさらなる実施形態において、メキシレチンに伴うGI副作用を誘発することなしに、必要とする被検体の疾患をメキシレチンで治療する方法を提供する。かかる方法は、本発明のメキシレチンプロドラッグ、その薬学的に許容し得る塩または組成物を必要とする被検体に経口投与することを備え、ここでメキシレチンプロドラッグが少なくとも一つのアミノ酸または長さ2〜9個のアミノ酸のペプチドに対し共有結合したメキシレチンまたはメキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメチルメキシレチン)からなり、経口投与するとプロドラッグまたは薬学的に許容し得る塩が、完全に避けることができないとしても、未結合メキシレチンの経口投与後に通常見られる胃腸副作用を最小限にする。メキシレチンプロドラッグは一または二つのプロドラッグ部分を有する。一つのプロドラッグ部分を、ペプチド結合を介してメキシレチンまたはメキシレチン代謝物のアミノ基に結合することができる。あるいは、またはさらに、メキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメキシレチン)は、フェノール系酸素でカルバメートまたはジカルボン酸リンカーを介して結合したプロドラッグ部分を有することができる。メキシレチンの量は、治療有効量(例えば、鎮痛有効量)であるのが好ましい。疾患は、メキシレチンで治療可能なものとすることができる。例えば、疾患は神経因性疼痛または不整脈であってもよい。さらなる実施形態において、メキシレチンの投与に伴うGI副作用は、限定しないが、嘔吐、吐き気、下痢および腹部不快感から選択される。
【0328】
ここに記載したメキシレチンプロドラッグは、メキシレチン塩酸塩のような非プロドラッグのメキシレチン塩形態と比較して、胃腸環境における腸運動に対する有害作用の誘発が統計的に相当に低い(例えば平均値)。
【0329】
本発明の別の態様において、薬物動態を改善し、必要とする被検体におけるメキシレチンの作用持続時間を延ばす方法を提供する。かかる方法は、本発明のプロドラッグまたはその組成物の有効量を必要とする被検体に投与することを備え、ここで血漿中濃度の時間プロファイルを調整して、メキシレチン濃度の初期の急激な上昇を最小限にし、あらゆる不所望な作用を最小限化する一方、薬剤が血漿中に存続する時間(プロドラッグからの継続的発生に起因する)、それ故作用持続時間を著しく延長する。
【0330】
さらなる態様において、メキシレチン血漿濃度の被検体間または被検体内の変動を低減する方法を提供する。かかる方法は、必要とする被検体または被検体群に本発明のプロドラッグまたはその組成物の有効量を投与することを備える。
【0331】
さらなる実施形態において、p−OHメキシレチンまたはm−OHメキシレチンのプロドラッグは前記方法に用いる。
【0332】
他の実施形態において、神経因性疼痛を除去、低減または治療する方法を提供する。該方法は、本発明のメキシレチンプロドラッグ、その薬学的に許容し得る塩または組成物を必要とする被検体に経口投与することを備え、ここでメキシレチンプロドラッグが少なくとも一つのアミノ酸または長さ2〜9個のアミノ酸のペプチドに対し共有結合したメキシレチンまたはメキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメチルメキシレチン)からなる。メキシレチンプロドラッグは一または二つのプロドラッグ部分を有する。一つのプロドラッグ部分を、ペプチド結合を介してメキシレチンまたはメキシレチン代謝物のアミノ基に結合することができる。あるいは、またはさらに、メキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメキシレチン)は、フェノール系酸素でカルバメートまたはジカルボン酸リンカーを介して結合したプロドラッグ部分を有することができる。メキシレチンの量は、治療有効量(例えば、鎮痛有効量)であるのが好ましい。
【0333】
さらなる実施形態において、p−OHメキシレチンまたはm−OHメキシレチンのプロドラッグを前記方法に用いる。
【0334】
本発明の他の実施形態は、被検体間および被検体内におけるメキシレチン血清濃度の変動を低減することに関する。該方法は、本発明のメキシレチンプロドラッグ、その薬学的に許容し得る塩または組成物を必要とする被検体に経口投与することを備え、ここでメキシレチンプロドラッグが少なくとも一つのアミノ酸または長さ2〜9個のアミノ酸のペプチドに対し共有結合したメキシレチンまたはメキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメチルメキシレチン)からなる。メキシレチンプロドラッグは一または二つのプロドラッグ部分を有する。一つのプロドラッグ部分を、ペプチド結合を介してメキシレチンまたはメキシレチン代謝物のアミノ基に結合することができる。あるいは、またはさらに、メキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメキシレチン)は、フェノール系酸素でカルバメートまたはジカルボン酸リンカーを介して結合したプロドラッグ部分を有することができる。メキシレチンの量は、治療有効量(例えば、鎮痛有効量)であるのが好ましい。
【0335】
また、本発明の他の実施形態は、必要とする被検体におけるメキシレチンの生物学的利用能の再現性を向上させることに関する。該方法は、本発明のメキシレチンプロドラッグ、その薬学的に許容し得る塩または組成物を必要とする被検体に経口投与することを備え、ここでメキシレチンプロドラッグが少なくとも一つのアミノ酸または長さ2〜9個のアミノ酸のペプチドに対し共有結合したメキシレチンまたはメキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメチルメキシレチン)からなる。メキシレチンプロドラッグは一または二つのプロドラッグ部分を有する。一つのプロドラッグ部分を、ペプチド結合を介してメキシレチンまたはメキシレチン代謝物のアミノ基に結合することができる。あるいは、またはさらに、メキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメキシレチン)は、フェノール系酸素でカルバメートまたはジカルボン酸リンカーを介して結合したプロドラッグ部分を有することができる。メキシレチンの量は、治療有効量(例えば、鎮痛有効量)であるのが好ましい。
【0336】
さらなる実施形態において、p−OHメキシレチンまたはm−OHメキシレチンのプロドラッグを前記方法に用いる。
【0337】
本発明はまた、一つ以上の原子を同じ原子番号を有するが、自然界で通常見られる原子質量または質量数と異なる原子質量または質量数を有する原子によって置換する式(1)の全ての薬学的に許容し得る同位体標識化合物の合成を含む。
【0338】
本発明の化合物に包含するのに適した同位体の例としては、2Hおよび3Hのような水素、11C、13Cおよび14Cのような炭素、36Clのような塩素、18Fのようなフッ素、123Iおよび125Iのようなヨウ素、13Nおよび15Nのような窒素、15O、17Oおよび18Oのような酸素、32Pのようなリン、そして35Sのような硫黄の同位体が挙げられる。
【0339】
特定の同位体標識化合物、例えば、放射性同位体を組み込んだものは、薬物および/または基質の組織分布研究に有用である。トリチウム、すなわち3Hおよび炭素−14、すなわち14Cの放射性同位体は、取り込みの容易さおよび検出の素早い手段の点で本目的に特に有用である。
【0340】
重水素、すなわち2Hのようなより重い同位体での置換は、より大きな代謝安定性に由来する特定の治療上の利点、例えば、生体内での半減期の増加または必要投与量の減少を付与することができ、従っていくつかの状況では好まれる。
【0341】
11C、18F、15Oおよび13Nのような陽電子放出同位体での置換は、基質の受容体占有率を検討するための陽電子放射トポグラフィー(PET)の研究に役立つ。
【0342】
同位体標識化合物は、一般に当業者に既知の従来技術、または上記で採用した非標識試薬の代わりに適当な同位体標識試薬を用いて説明されるものと類似の方法によって製造することができる。
【0343】
本明細書の説明および特許請求の範囲を通して、単語“備える”および“含む”ならびにその単語の変形、例えば“備えている”および“備える”は、“限定しないが含む”ことを意味し、および他の部分、添加剤、成分、整数または工程を排除することを意図したものではない(および排除しない)。
【0344】
本明細書の説明および特許請求の範囲を通して、文脈上別の解釈をする必要がある場合を除き、単数形は複数形を包含する。特に、文脈上別の解釈をする必要がある場合を除き、不定冠詞が使用されている場合、本明細書が複数形および単数形を意図していると理解されるべきである。
【0345】
本発明の特定の態様、実施形態または実施例と一緒になって記載される特徴、整数、特性、化合物、化学的部分または基は、それに矛盾がない限り、ここに記載される任意の他の態様、実施形態または実施例も適用可能であると理解されるべきである。
【実施例】
【0346】
本発明を、さらに以下の実施例を参照して説明する。しかしながら、これら実施例は上述した実施形態と同様に例示であり、いかなる意味においても本発明の可能とされる範囲を制限するものとして解釈されるべきではないことに留意すべきである。
【0347】
一般的合成方法
【0348】
本発明のメキシレチンプロドラッグの合成は2つの異なる工程で達成することができる。アミノ酸またはペプチドの活性化エステル、例えば、(S)−リジンの活性化エステルのN,N’−ジ−t−ブチルオキシカルボニル−(S)−リジンスクシンイミドを(rac)−メキシレチン塩酸塩にカップリングして、N−保護化プロドラッグの(rac)−メキシレチン−N,N’−ジ−t−ブチルオキシカルボニル−(S)−リジンを得ることができる。その後、かかる化合物をトリフルオロ酢酸で脱保護してプロドラッグを得ることができる。
【0349】
上述のように、活性化リジンを他の活性化アミノ酸またはペプチドに速やかに置換することができる。
【0350】
実施例1:(rac)−メキシレチン−(S)−リジンジトリフルオロ酢酸塩の合成
【0351】
メキシレチン−(S)−リジンジトリフルオロ酢酸塩の合成を2つの異なる工程で達成する。最初に、(S)−リジンの活性化エステルのN,N’−ジ−t−ブチルオキシカルボニル−(S)−リジンスクシンイミドをN−メチルモルホリン(NMM)の存在下で(rac)−メキシレチン塩酸塩にカップリングし、クロマトグラフィーによる精製後、N−保護化プロドラッグの(rac)−メキシレチン−N,N’−ジ−t−ブチルオキシカルボニル−(S)−リジンを得た(スキーム1)。
【0352】
その後、トリフルオロ酢酸を用いてBOC基の脱保護を達成して、高粘性ガラス状油として所望の(rac)−メキシレチン−(S)−リジンジトリフルオロ酢酸塩を得た。かかる油は、高真空下での乾燥により発泡するが、空気中に放置すると崩壊することを確かめた。明確にするために、メキシレチンの一方のエナンチオマーのみを示す。
【化81】
(rac)−メキシレチン(S)−リジンジトリフルオロ酢酸塩の合成経路
【0353】
1H NMR(DMSO−d6)スペクトル
8.60(m、1H、NH)、8.16(br、3H、NH3+)、7.76(br、3H、NH3+)、7.02(m、2H、ArH)、6.92(m、1H、ArH)、4.22(m、1H、□−CH)、3.67(d、J=4.5Hz、CH2)、2.73(m、2H、NCH2)、2.21(s、6H、2×CH3)、1.73(m、2H、CH2)、1.52(m、2H、CH2)、1.30(m、5H、CH3+CH2)。
【0354】
実施例2:(rac)−メキシレチン−グリシントリフルオロ酢酸塩の合成
【0355】
N−t−ブチルオキシカルボニル−グリシンスクシンイミドをNMMの存在下で(rac)−メキシレチン塩酸塩にカップリングし、クロマトグラフィーによる精製後、良好な収率でN−保護化プロドラッグの(rac)−メキシレチン−N−t−ブチルオキシカルボニル−グリシンを得た(スキーム2)。
【0356】
その後、トリフルオロ酢酸を用いてBOC基の脱保護を達成した。ジエチルエーテルでの粉砕およびろ過により、優れた収率で白色固体として所要の(rac)−グリシンメキシレチントリフルオロ酢酸塩を得た(スキーム2)。明確にするために、メキシレチンの一方のエナンチオマーのみをスキーム2に示すことに留意すべきである。
【化82】
スキーム2 グリシン−(rac)−メキシレチントリフルオロ酢酸塩の合成経路
【0357】
トリフルオロ酢酸を用いてBOC基の脱保護をその後達成し、ジエチルエーテルによるろ過により優れた収率で白色固体としてグリシン−(rac)−メキシレチントリフルオロ酢酸塩を得た。
【0358】
1H NMR(DMSO−d6)スペクトル
8.52(d、J=7.8Hz、1H、NH)、8.03(br、3H、NH3+)、7.01(m、2H、ArH)、6.93(m、1H、ArH)、3.64(m、4H、2×CH2)、2.22(s、6H、2×CH3)、1.28(d、J=6.6Hz、3H、CH3)。
【0359】
実施例3:メキシレチン−(S)−ホモアルギニンアミド二塩酸塩の合成
【0360】
メキシレチン−(S)−ホモアルギニンアミド二塩酸塩の合成は4つの異なる工程で達成した。“活性エステル”のN−BOC−(S)−ホモアルギニン−(NO2)N−ヒドロキシスクシンイミドエステルを、N−ヒドロキシスクシンイミドとN−BOC−(S)−ホモアルギニン−(NO2)との間のDCCカップリングを経由して作成した。メキシレチン塩酸塩との反応に続いて、Biotage製Isolera自動クロマトグラフィーシステムを用いる逆相条件下での精製後、N−保護化プロドラッグのN−BOC−(S)−ホモアルギニン−(NO2)−メキシレチンを良好な収率で得た。
【化83】
メキシレチン−(S)−ホモアルギニンアミド二塩酸塩の合成経路
【0361】
パラジウムを炭素に対して用いる接触水素化によりニトロ基を還元して、N−BOC−(S)−ホモアルギニン−メキシレチンを得た。Boc基の除去をトリフルオロ酢酸で達成した。粗生成物をジエチルエーテル中2M塩化水素で塩交換し、Biotage製Isolera自動クロマトグラフィーシステムを用いる逆相条件下で精製して、メキシレチン−(S)−ホモアルギニンアミド二塩酸塩を白色のガラス状固体として得た。
【0362】
1H NMR(DMSO−d6)スペクトル
8.79(d、J=8.1Hz、1H、NH)、8.35(br、3H、NH3+)、7.92(m、1H、NH)、7.02(d、J=7.5Hz、2H、2×ArH)、6.91(m、1H、ArH)、4.20(m、1H、□−CH)、3.82〜3.65(m、3H、CH+OCH2)、3.09(m、2H、□−CH2)、2.22(s、6H、2×CH3)、1.75(m、2H、CH2)、1.49〜1.37(m、4H、2×CH2)、1.28(m、3H、CH3)。
【0363】
実施例4:メキシレチン−(S)−グルタミン酸アミド塩酸塩の合成
【0364】
メキシレチン−(S)−グルタミン酸アミド塩酸塩の合成は、2つの異なる工程で達成した。最初に、(S)−グルタミン酸の“活性化エステル”であるN−BOC−(S)−グルタミン酸(tert−ブチルエステル)N−ヒドロキシスクシンイミドエステルをメキシレチン塩酸塩にカップリングした。この後クロマトグラフィーにより精製して、良好な収率で保護プロドラッグのN−BOC−(S)−グルタミン酸(tert−ブチルエステル)−メキシレチンを得た。
【化84】
メキシレチン−(S)−グルタミン酸アミド塩酸塩の合成経路
【0365】
4M塩化水素のジオキサン溶液を用いてBocおよびtert−ブチル基の脱保護を達成した。粗生成物を逆相条件下でBiotage製Isolera自動クロマトグラフィーシステムを用いて精製して、所望のメキシレチン−(S)−グルタミン酸アミド塩酸塩をガラス質白色固体として得た。
【0366】
1H NMR(DMSO−d6)スペクトル
8.71(m、1H、NH)、8.31(s、3H、NH3+)、7.01(d、J=7.5Hz、2H、2×ArH)、6.91(m、1H,ArH)、4.21(m、1H、グルタミン酸α−CH)、3.67(m、3H、不明瞭、メキシレチンCH+OCH2)、2.36(m、2H、□−CH2)、2.23(s、3H、CH3)、2.21(s、3H、CH3)、1.99(m、2H、CH2)、1.27(d、J=6.6Hz、3H、CH3)。
【0367】
実施例5:メキシレチン−[(S)−S−メチル−メチオニン塩化]アミド塩酸塩
【0368】
メキシレチン−[(S)−S−メチル−メチオニン塩化]アミド塩酸塩の合成を3つの異なる工程で達成した。第一に、(S)−メチオニンの“活性化エステル”であるN−BOC−(S)−メチオニンN−ヒドロキシスクシンイミドエステルをメキシレチン塩酸塩にカップリングして、良好な収率で保護プロドラッグのN−BOC−(S)−メチオニン−メキシレチンを得た。続いて、S−メチル化をメタノール中でヨウ化メチルを用いて達成し、化合物を逆相クロマトグラフィーにより精製して、[N−BOC−(S)−メチオニン−S−メチル−ヨウ化]−メキシレチンを与えた。
【化85】
メキシレチン−[(S)−S−メチル−メチオニン塩化]アミド塩酸塩の合成経路
【0369】
ジオキサン中4M塩化水素を用いてBoc基の脱保護を行い、続いて逆相クロマトグラフィーにより精製して、所望のメキシレチン−[(S)−S−メチル−メチオニン]アミド塩酸塩を得た。
【0370】
1H NMR(DMSO−d6)スペクトル
9.32(bd、1H、NH)、8.73(br、3H、NH3+)、7.02(d、J=7.2Hz、2H、2×ArH)、6.92(m、1H、ArH)、4.20(m、1H、□−CH)、4.06(m、1H、CH)、3.72(m、4H、CH2+OCH2)、3.01(s、3H、S−CH3)、2.98(s、3H、S−CH3)、2.32(不明瞭、2H、CH2)、2.22(s、6H、2×CH3)、1.29(m、3H、CH3)。
【0371】
実施例6:メキシレチン−(S)−グルタミンアミド塩酸塩
【0372】
メキシレチン−(S)−グルタミンアミド塩酸塩の合成を、メキシレチン−(S)−グルタミン酸アミド塩酸塩のものと同様な方法で達成した。この場合、N−Boc−(S)−グルタミンN−ヒドロキシスクシンイミドエステルをメキシレチン塩酸塩にカップリングして、良好な収率でN−保護プロドラッグのN−Boc−(S)−グルタミン−メキシレチンを得た。
【化86】
メキシレチン−(S)−グルタミンアミド塩酸塩の合成経路
【0373】
その後、ジオキサン中で4M塩化水素を用いてBoc基の脱保護を達成して、メキシレチン−(S)−グルタミンアミド塩酸塩を灰色がかった白色固体として得た。
【0374】
1H NMR(DMSO−d6)スペクトル
8.73(d、J=8.1Hz、1H、NH)、8.35(br、3H、NH3+)、7.50(br、1H、0.5NH2)、7.01(d、J=7.5Hz、2H、2×ArH)、6.92(m、2H、ArH+0.5NH2)、4.20(m、1H、α−CH)、3.66(m、3H、不明瞭、CH2+CH)、2.21(m、6H、2×CH3)、1.97(m、2H、□−CH2)、1.28(d、J=6.6Hz、3H、CH3)。
【0375】
化合物の評価
催吐活性または吐き気は、胃内の局所麻酔効果の直接の結果として生じると考えられる。これは、胃内容排出を促進する胃内の低波動運動(“ハウスキーパー”波)の抑制に起因する。局所麻酔効果は、ナトリウムチャンネルの遮断により媒介される。本発明の化合物は、ナトリウムチャンネルに対し高いIC50値を示すかなり乏しい活性を有することにより嘔吐を低減または排除すると考えられている。したがって、メキシレチンのナトリウムチャンネルのブロック効果は、メキシレチン自体の代わりに本発明の化合物を投与することにより一時的に不活性化される。一度かかる化合物を吸収すると、メキシレチンへ定量的に変換され、それにより低減または排除された嘔吐および/または吐き気のメキシレチンで認識されたすべての治療的効果が得られる。以下の実施例に示したIC50値は、化合物の低減された催吐の可能性を実証する(例えば表3の高いIC50値で示される)。
【0376】
実施例7:哺乳類細胞で発現したクローン化Nav1.1チャンネルに対するメキシレチンおよび様々なメキシレチンのアミノ酸プロドラッグの効果
不活性化(一時的に)され、それゆえ腸内で直接の催吐性効果をもたらすのが少ないメキシレチンのアミノ酸プロドラッグを同定する試みにおいて、一連の結合体をその潜在的な局所麻酔活性について生体外で哺乳類細胞に発現したナトリウム1.1チャンネルへの効果を評価することにより検査した。
【0377】
方法
【0378】
hNav1.1試験方法
【0379】
hNav1.1チャンネルcDNA(SCN1A遺伝子)で安定に形質転換したCHO細胞を使用して、hNav1.1チャンネルの潜在的ブロックをグラフ1に示した刺激電圧パターンにより測定し、電圧電位を表4に示す。パルスパターンを二回繰り返し、TA添加の前および添加5分後に3つの試験パルスでのピーク電流振幅を測定した(ITP1、TP11およびITP12)。
【0380】
【化87】
グラフ1 hNav1.x試験方法用の電圧プロトコル
【0381】
【表4】
【0382】
データ解析
【0383】
データ収集および分析は、IonWorks Quattroシステムオペレーションソフトウェア(バージョン2.0.2、Molecular Devices Corporation、ユニオンシティ、カリフォルニア州)を使用して行った。データは漏洩電流に対し補正した。
【0384】
トニックブロックを次のように算出した。
%ブロック(トニック) = (1 - ITP1,TA / ITP1,Control) × 100%
式中のITP1,ControlおよびITP1,TAは、それぞれコントロールおよび試験物の存在下においてTP1により誘導された内部ピークNa+電流である。
10Hzブロック、すなわち刺激周波数10Hzでの周波数依存ブロックを次のように算出した。
%ブロック(10Hz) = (1 - ITP11,TA / ITP11,Control) × 100%
式中のITP11,ControlおよびITP11,TAは、それぞれコントロールおよび試験物の存在下においてTP11により誘導された内部ピークNa+電流である。
不活性化状態ブロックは、調整脱分極パルス(TP11)による試験パルス(TP12)の電流振幅における減少として定義される。不活性化状態ブロックを次のように算出した。
%ブロック(不活性化状態) = (1-ITP12,TA / ITP12,Control) × 100%
式中のITP12,ControlおよびITP12,TAは、それぞれコントロールおよび試験物の存在下においてTP12により誘導された内部ピークNa+電流である。
ブロックについての濃度応答データを次の方程式に合わせた。
%ブロック = { 1 - 1 / [ 1 + ( [試験] / IC50 ) N ] } × 100%
ここで[試験]は試験物の濃度、IC50は50%阻害を産生する試験物の濃度、Nはヒル係数、%ブロックは試験物の各濃度で阻害されたイオンチャンネル電流の百分率である。非線形最小二乗法を、Excel2000(マイクロソフト、レッドモンド、ワシントン州)用のソルバーアドインを用いて解いた。
【0385】
結果
【0386】
表5に見られるように、試験した22化合物のうち7化合物が親化合物の10倍超のIC50値を有し、4化合物が20倍より高い効能を有した。これらは、ホモアルギニン、アルギニン、グルタミン酸および塩化S−メチルメチオニン複合体であり、メキシレチン自体の38μMと比較すると、それぞれ624、811、>1000μMおよび>1000μM(10Hzブロック)の値であった。効能におけるこのような減少は、胃/腸の上皮表面への直接作用および生成する催吐の可能性を減少すると期待される。
【0387】
【表5−1】
【表5−2】
【0388】
実施例8:イヌにおける様々なメキシレチンプロドラッグ由来のメキシレチンの全身アベイラビリティの評価
【0389】
方法
【0390】
試験物質(すなわち、メキシレチンおよび様々なメキシレチンのアミノ酸プロドラッグ)をイヌ五頭のグループに強制経口投与した。試験動物の特徴を下記の表6に示す。
【0391】
【表6】
【0392】
血液サンプルを投与後様々な時間で採取し、確証されたLC−MS−MSアッセイを用いる親薬物用の分析に提出した。血漿分析データに由来した薬物動態パラメータを、Win Nonlinを用いて求めた。結果を表7に与える。
【0393】
結果
【0394】
データは、試験した様々なアミノ酸プロドラッグ由来のメキシレチンの全身アベイラビリティについて大幅に変動があることを示す。例えば、ニコチン酸アミドおよびイソニコチン酸アミド由来のものについて、メキシレチンに関して無視できるような生物学的利用能であった。一方、グルタミンアミドプロドラッグの経口投与はほぼ完璧な生物学的利用能となった。
【0395】
【表7】
【0396】
実施例9:カニクイザルにおける様々なメキシレチンプロドラッグ由来のメキシレチンの全身アベイラビリティの評価
【0397】
方法
【0398】
試験物質(すなわち、メキシレチンおよび様々なメキシレチンのアミノ酸プロドラッグ)を、5頭のオスのカニクイザルのグループに強制経口投与した。血液サンプルを投与後様々な時間で採取し、確証されたLC−MS−MSアッセイを使用して親薬物用の分析に提出した。血漿分析データから導かれた薬物動態パラメータを、Win Nonlinを用いて求めた。結果を表8に示す。
【0399】
結果
【0400】
イヌにおけるデータのように、試験された様々なアミノ酸プロドラッグ由来のメキシレチンの全身アベイラビリティにおける著しい変動を示す。再びニコチン酸アミドおよびイソニコチン酸アミド由来のものについて、メキシレチンに関し生物学的利用能は無視できるほど小さかったが、グルタミンアミドプロドラッグはほぼ完全な生物学的利用能となった。
【0401】
【表8】
【0402】
実施例10:ウサギの胃平滑筋の収縮に対するメキシレチンおよびメキシレチングリシンおよびリジンアミドの影響
【0403】
低減したナトリウムチャンネルブロック特性を有するメキシレチンの二つの原型アミノ酸結合体(メキシレチングリシンおよびリジンアミド)を用いて、ウサギの胃平滑筋に対するこれら結合体とメキシレチンとの直接的な影響の比較を検討した。かかる直接的な影響の大きさは、メキシレチンに伴う催吐性の決定要因となると期待される。従って、EFSで刺激した胃平滑筋に対する直接的な影響の減少は、より低い催吐応答になると期待される。
【0404】
方法
【0405】
胃の幽門洞領域から切断したウサギの胃平滑筋全層(粘膜も無処理)の細長い一片を白金環電極の間に装着した。組織を約1gの一定張力まで伸ばし、力発生の変化を高感度変換機を用いて記録した。
【0406】
組織を14Hzで0.5ミリ秒のパルス幅の電場刺激(EFS)に置きながら、刺激に対する最適電圧を求めた。その後、パルスの伝達を50秒ごとに20秒間続けた。
【0407】
最適電圧でのEFSをプロトコル(安定した応答=“EFSのベースラインの測定”)の間中に継続した。
【0408】
採用した試験条件は以下のとおりである。
(1)媒体(脱イオン水、試験物と等容量で添加した)
(2)7つの濃度のメキシレチン(10nM、100nM、1mM、3mM、10mM、30mM、100mM)
(3)7つの濃度のメキシレチン−リジン−アミド(10nM、100nM、1mM、3mM、10mM、30mM、100mM)
(4)7つの濃度のメキシレチン−グリシン−アミド(10nM、100nM、1mM、3mM、10mM、30mM、100mM)
【0409】
ベースラインEFSの10分後、試験物または媒体(脱イオン水)の最初の添加を行った。
【0410】
試験濃度は、各添加の間のPBS洗浄で累積的に加えた。
【0411】
試験濃度は、各添加の間のPSS洗浄で非累積的に加えた。次に、TTX(Na+チャネル遮断薬)を試料に添加して、EFS反応が神経刺激により誘発されたことを確認し、並びにナトリウムチャネル遮断薬の活性(メキシレチンと同様な機構)を確認した。その後、EFSを停止した。
【0412】
結果
【0413】
この調査の結果は、ウサギの胃平滑筋に対するメキシレチン自体並びにメキシレチンリジンアミドおよびメキシレチングリシンアミドの影響の明らかな違いを示す図1に要約される。3つの化合物すべてが、EFSで誘発したウサギの胃の収縮を徐々に減少させる一方、プロドラッグ複合体は著しく低い効能であった。計算したED50値は、メキシレチン、メキシレチングリシンアミドおよびメキシレチンリジンアミドについてそれぞれ2.17、9.16および21.83μMであった。この機能解析における効能の減少の大きさは、Nav1.1チャネルの遮蔽の生体外での評価中に観察されたものと一致し、後者が胃上皮への影響の良い指標であることを示唆する。胃の筋肉に対する直接作用の能力の減少は、プロドラッグに対する直接的に媒介される催吐性応答の可能性を最小限に抑えることができる。
【0414】
実施例11:メキシレチンおよびメキシレチン−グリシン−アミドをフェレットへの経口投与した後の催吐性に対する影響の評価
【0415】
ナトリウムチャネルのブロック特性を低減したメキシレチンの原型アミノ酸結合体(メキシレチン−グリシン−アミド)を使用して、フェレットにおけるそれらとメキシレチンとの催吐性に対する影響の比較を検討した。
【0416】
方法
【0417】
オスのフェレット(n=7)をそれぞれの試験日の前日の午後遅くまでペレット飼料に自由に採食できる状態にした。その後、飼料を取り除き、フェレットを一晩飢餓状態にした。飼料を嘔吐観察の終了後まで戻さなかった。試験の朝に、20mg/kgのメキシレチン塩酸塩溶液または等モル量のメキシレチングリシンアミドのいずれかを、5mL/kgの一定投与容積を用いて動物に経口投与した。経口処理後2時間継続的に動物を観察し、吐き気および嘔吐の全ての発生を記録した。
【0418】
結果
【0419】
表9および10に表される結果は、親化合物を投与した後に見られるものと比較して、プロドラッグを与えた後では、嘔吐の頻度および持続時間が著しく減少したことを示す。プロドラッグの投与後の嘔吐の平均値は、親薬物を投与後に観察されるものの30%未満に低下した。同様に、嘔吐の持続時間は、プロドラッグの投与後では大きく低減し、メキシレチン自体の投与後に見られるものの30%未満になった。可能性として、これらのデータは、この原型メキシレチンアミノ酸プロドラッグについて、吐き気および嘔吐を生じさせるような効能が低減することを示し、ヒトにおいて改善された有効性および患者の服薬遵守につながると期待される。
【0420】
【表9】
【0421】
【表10】
【0422】
実施例12:フェレットにおける親薬物由来とメキシレチングリシンアミド由来とのメキシレチンの全身アベイラビリティの評価
【0423】
原型プロドラッグのメキシレチングリシンアミドに伴う催吐性低減への影響が、単純に、薬剤の低全身アベイラビリティの結果でないことを確かめるために、薬物動態の比較を行った。
【0424】
方法
【0425】
試験物質(すなわち、メキシレチンおよび様々なメキシレチンのアミノ酸プロドラッグ)を、6頭のフェレットのグループに強制経口投与した。
【0426】
血液サンプルを投与後様々な時間で採取し、確証されたLC−MS−MSアッセイを使用してプロドラッグおよび親薬物用の分析に提出した。血漿分析データから導かれた薬物動態パラメータを、Win Nonlinを用いて求めた。
【0427】
結果
【0428】
結果を表11に示す。薬剤自体またはグリシンアミドプロドラッグのどちらかを与えた後の薬剤への全身曝露の比較では、メキシレチンへの全暴露が比較可能であることを示した。表11に示すように、グリシンプロドラッグ由来のメキシレチンの生物学的利用能の平均相対値が、親分子を与えた後の場合の94%であり、プロドラッグに伴う嘔吐の減少は薬剤への乏しい全身暴露に起因するものではないことの確信が得られた。
【0429】
【表11】
【0430】
本発明は、上述したように式I
【化88】
式1
のメキシレチンおよびp−OHメキシレチンのプロドラッグに関する。
本発明はまた、式(I)の化合物および薬学的に許容し得る賦形剤を含む組成物に関する。
本発明はまた、メキシレチンに伴うGI副作用を誘発することなく痛みの治療に使用するための式(I)の化合物に関する。
メキシレチンに伴う胃腸副作用は、吐き気、消化不良、嘔吐、下痢、便秘またはこれらの副作用の組み合わせである。
治療する痛みは、糖尿病性神経障害、急性および慢性神経痛、アルコール性多発ニューロパチー、放射線治療由来の慢性疼痛、視床痛や糖尿病性の体幹の痛み、神経痛、肢端紅痛症、慢性特発感覚性多発ニューロパチー若しくはがんおよびその治療に伴う痛みに起因する痛みのような神経因性疼痛である。
プロドラッグは、メキシレチンリジンアミド、メキシレチンホモアルギニンアミド、メキシレチングルタミン酸アミド、メキシレチングルタミンアミドまたはメキシレチンメチルメチオニンアミドとすることができる。
【技術分野】
【0001】
本発明は、メキシレンの様々なアミノ酸およびペプチドプロドラッグ、並びにメキシレチンの薬物動態学的一貫性の向上、神経因性疼痛の治療、そしてメキシレチンに通常伴う有害な胃腸(GI)副作用の回避のためのプロドラッグの使用に関する。また、本発明は不整脈の治療に使用し得る。
【背景技術】
【0002】
神経因性疼痛は、世界人口の2.8%から4.7%に影響を与えていると推定される(非特許文献1)。中枢または末梢として広く分類される神経因性疼痛は、火傷または手足の骨折などの外部刺激を起因とする神経系により検出された痛みよりむしろ、神経系への損傷または神経系の疾患によるか、あるいは神経系それ自体への損傷に由来する痛みによりもたらされる。中枢神経因性疼痛は、中枢神経系(CNS)の損傷の結果として発生し、例えば、多発性硬化症、脊髄損傷、卒中または癌に起因する場合がある。末梢神経因性疼痛は、糖尿病、癌、HIV感染症、手根管症候群および疱疹性神経痛、切断術(幻肢痛)、背部損傷、下腿潰瘍および手術による医原性損傷によってもたらされた末梢神経系の損傷から生じる。医薬品主要七市場にわたる最近の報告では、約3760万人の患者が、中枢性神経因性疼痛に苦しんでおり、さらに17000万人の患者が末梢神経因性疼痛に苦しんでいると推定している(非特許文献1)。
【0003】
神経因性疼痛の症状には、灼熱痛、電撃痛、刺痛、感電型感覚が含まれる。他の一般的な神経因性疼痛の症状は、異痛症(通常は痛みを伴わない刺激に起因する痛み)、知覚過敏症(軽度な感触への誇張された応答)、痛覚過敏(痛みの原因が取り除かれた後であっても、持続する痛み)および気分変調(異常で不快な刺痛またはピンや尖った物で刺す感覚)である。
【0004】
神経因性疼痛は、特定の患者集団においてより一般的である。例えば、糖尿病患者の四分の一および癌患者の三分の一もの患者がかかる痛みを経験する。さらに、帯状疱疹を患っている患者の半数以上が疱疹性神経痛になっており、脊髄損傷を伴う患者の三分の一が神経因性疼痛による影響を受けている(非特許文献2)。
【0005】
現在、神経因性疼痛用のいくつかの効果的な治療法がある。プレガバリン、ガバペンチン、デュロキセチン(セロトニン−ノルエピネフリン再取り込み阻害剤(SNRI)である抗うつ剤)、Δ9テトラヒドロカンナジノールおよびリドカインパッチ(疱疹性神経痛の局所治療用)が、現在利用可能な治療の選択肢となっている。しかしそれぞれが、固有の明確な制限がある。例えば、プレガバリンはCNSへの重大で有害な作用を伴う。最も頻繁なプレガバリン使用の中止につながる副作用は、めまいや眠気である。これら二つの副作用は、より高い投与量のプレガバリン(FDAラベリング)で治療を受けた30%もの患者に発生した。ガバペンチンの場合、その経口生物学的利用能が投与量に比例しない、すなわち投与量が増加すると、生物学的利用能が低下する。約60%、47%、34%、33%、および27%の生物学的利用能がガバペンチンの900、1200、2400、3600、および4800mg/日(FDAラベリング)の投与により観察された。デュロキセチンは、治療を受けた患者の20〜40%に吐き気を、また治療を受けた患者の自殺への関心を伴う(FDAラベリング)。Δ9テトラヒドロカンナジノールは明らかな中毒傾向(DEA分類)がある。
【0006】
メキシレチン、すなわち(rac)−1−(2,6−ジメチルフェノキシ)−2−プロパンアミン塩酸塩(以下に構造を示す))は、ナトリウムチャネルブロック剤であり、結果として局所麻酔薬の特性を有する(非特許文献3)。最初、メキシレチンはクラス1Bの抗不整脈薬としての有用性が見出され、不整脈を治療するために今日でも使用されている。現在、薬は150mg、200mgまたは250mgのカプセルとして入手可能で、心室性不整脈の治療に使用されている。メキシレチン投与に伴う最も頻度の高い有害な反応は、上部胃腸障害である(FDAラベリング)。
【化1】
メキシレチン(1−メチル−2−(2,6−キシリルオキシ)−エチルアミン)塩酸塩
【0007】
さらに近年では、メキシレチンは、様々な原因の神経因性疼痛の治療における有用性の増加が見られる。糖尿病性神経障害、急性および慢性神経痛、アルコール性多発ニューロパチー、放射線治療由来の慢性疼痛、視床痛や糖尿病性の体幹の痛みへの使用が報告されている(非特許文献4)。さらにより最近の報告は、肢端紅痛症(EM)、再発性灼熱痛により特徴付けられる稀な不自由な障害、紅斑および影響を受けた領域(例えば、足、耳)の上昇した温度についての治療におけるメキシレチンの有用性を示唆している(非特許文献5)。また、メキシレチンは、慢性特発感覚性多発ニューロパチー、すなわち患者の抹消下肢におけるしびれまたはうずきで表される疾患に有用であることが見出されている(非特許文献6)。
【0008】
しかしながら、メキシレチンの使用は、高い発症率の吐き気、嘔吐および腹部不快感(治療を受けた患者の38%)を伴う(非特許文献7)。このような有害なGI副作用は、貧弱な患者の服薬遵守に間違いなく寄与する。嘔吐はまた、投与された薬物の部分減量となり、その結果効果が減少し予測不可能になる。嘔吐は、メキシレチンの経口投与量を制限する副作用であり、効果的な血漿中薬物濃度の達成を妨げる場合がある(非特許文献8および非特許文献9)。
【0009】
メキシレチンの嘔吐作用のメカニズムへの理解は限定的である。一つの実験的研究では、メキシレチンが生体内でのラットの胃の中で徐波活動を低下し得るが、空腸筋電性活動に効果がないことを示している(非特許文献10)。胃の徐波活動の抑制は、吐き気および嘔吐を誘導する重要な役割を果たすと考えられている。メキシレチンの場合、胃の徐波活動の抑制が局所鎮静(ナトリウムチャネルをブロック)活動によりもたらされる場合がある。ウサギの食道の括約筋を用いた他の生体外での研究は、静脈内麻酔化合物のケタミンおよびミダゾラムのようにメキシレチンが酸化窒素によってもたらされた非アルドレナリン、非コリン性(NANC)弛緩を抑制し得ることを示唆した(非特許文献11)。この研究では、メキシレチンによる下部食道括約筋の平滑筋における内因性酸化窒素の抑圧がメキシレチンの有害なGI作用に寄与し得ると結論付けている。
【0010】
他の局所麻酔薬リグノカインに対して行われた研究は、消化管への直接作用により誘導される該化合物の経口投与に伴う嘔吐の影響に向けられている。抗不整脈薬のイヌへの同等効果の静脈内および経口投与後、それぞれが同等の全身の血中濃度に到達したにもかかわらず、経口投与した薬剤のみが嘔吐を誘発した(非特許文献12)。
【0011】
メキシレチンが固有の胃刺激特性があることを示唆する文献の報告がある。例えば、メキシレチンの経口摂取後に食道炎が繰り返される事例が報告された(非特許文献13、非特許文献14および非特許文献15)。したがって、メキシレチンの催吐効果がより単純には胃への直接的な刺激作用に起因する可能性がある。
【0012】
これら有害な事象のメカニズムを理解する際のかかる進歩があるにもかかわらず、メキシレチン治療に伴う副作用を減少させる必要があり続ける。したがって、痛みの治療において、薬剤分子の固有の薬理学的利点すべてを保持するが、有害なGI副作用を誘発するという制限を克服するメキシレチン製品への現実的必要性がある。本発明は、この必要性に取り組んでいる。
【0013】
任意の製薬化合物を投与する際に有効性および毒性が重要な考慮事項であるが、メキシレチンの場合、催吐特性が実際患者の服薬遵守および適切かつ治療上有効な投与量レベルに対し大きな障壁である。そのため、メキシレチンで治療する際に嘔吐を減少または排除する本発明の化合物により提供される利点は重要であると期待されている。したがって、本発明は、患者および臨床医に対し従来問題があった治療への容易な利用を提供する。
【先行技術文献】
【非特許文献】
【0014】
【非特許文献1】Neuropathic Pain Network and Pfizer Inc.,2006 survey
【非特許文献2】Neuropathic Pain Network and Pfizer Inc.,2007 survey
【非特許文献3】Scholz A(2002)Brit.J Anaethesia 89,52−61
【非特許文献4】Jarvis and Coukell(1998).Drugs 4,691-707
【非特許文献5】Vivas AC et al(2010)Amer.J.Otolaryngology,May
【非特許文献6】Wolfe GI et al(1999)Arch Neurol 56、540−547)
【非特許文献7】Morganroth(1987).Am.J.Cardiol.60,1276−1281
【非特許文献8】Wright et al.(1997)Ann Pharmacother.31,29-34
【非特許文献9】Galer et al.(1996)J Pain Symptom Manage.12,161-167
【非特許文献10】Bielefeldt and Bass(1991).Digestion 48,43-50
【非特許文献11】Kohjitani et al.(2003).Eur.J.Pharmacol.,465,145-151
【非特許文献12】Smith ER et al 1972)Amer.Heart Journal 83 363−372
【非特許文献2】Penalba C(1986)Ann Gastroenterol Hepatol(Pris)22,267−268
【非特許文献14】Seggewiss RR & Seckfort H(1983)Dtsch Med Wochenshr.108 1018−1020
【非特許文献15】Addler JB(1990)Am J Gastroenterol.85 629−630
【発明の概要】
【0015】
本発明の化合物は、メキシレチンの治療効果を付与するが、嘔吐などのGI副作用を低減または排除するアミドプロドラッグ結合体である。
【0016】
本発明は、式Iのメキシレチンおよびp−OHメキシレチンのプロドラッグ
【化2】
式I
【0017】
またはその薬学的に許容し得る塩に関し、
【0018】
ここで式中のR1は水素および
【化3】
から選択され、
【0019】
R2は、水素、
【化4】
から選択され、
【0020】
O1は未結合p−OHメキシレチン中に存在するフェノール系酸素であり、
【0021】
Xは(−NH−)、(−O−)、または欠落であり、
【0022】
RAAのそれぞれの出現は独立して天然または非天然アミノ酸側鎖であり、
【0023】
n1は0〜16から選択した整数であり、
【0024】
n2のそれぞれの出現は独立して1〜9から選択した整数であり、
【0025】
R3のそれぞれの出現は独立して水素、および任意に置換したアルキル基から選択され、
【0026】
R4およびR5のそれぞれの出現は独立して水素、
【化5】
および任意に置換したアルキル基から選択され、
【0027】
ここで、n1により定義した炭素鎖に生ずる二重結合の場合、二重結合を形成する炭素上にR4は存在し、R5は欠落であり、但し常に、
【0028】
少なくともR1は
【化6】
であるか、あるいは少なくともR2は
【化7】
のいずれか一方であり、また本化合物が次の化合物:HBr.グリシン−(rac)メキシレチン;AcOH.アスパラギン−(rac)メキシレチン;TFA.トリプトファン−(rac)メキシレチン;HBr.アラニン−(rac)メキシレチン;AcOH−フェニルアラニン−(rac)メキシレチンおよびTFA.トリプトファン−(rac)メキシレチン.H2Oの一つでない。
【0029】
一実施形態において、R1は水素であり、R2は
【化8】
である。
【0030】
R2は
【化9】
が好ましい。
【0031】
一実施形態において、R2は水素であり、R1は
【化10】
である。
【0032】
一実施形態において、R2が
【化11】
であり、Xは欠落であり、n1は1、2または3である。R2が水素またはヒドロキシ以外であるとき、R1に関するn2の値は、R2に関する値と独立している。一実施形態において、R1について言えば、n2の値は好ましくは1、2、3または4であり、より好ましくはn2が1または2である。もっとも好ましくはn2が1である。
【0033】
一実施形態において、R2について言えば、n2の値は好ましくは1、2、3または4である。より好ましくはn2が1または2であり、もっとも好ましくはn2が1である。
【0034】
本発明に従って用いるアミノ酸残基には、リジン、グリシン、ホモアルギニン、グルタミン酸、メチルメチオニンおよびグルタミンが含まれる。
【0035】
ジカルボン酸リンカーの一実施形態において、n1は0、1、2、3または4である。
【0036】
一実施形態において、n2の各出現は1、2、3、4および5から選択される。
【0037】
好ましい実施形態において、R3の各出現は水素である。別の実施形態において、R3の各出現は独立して非置換C1−6アルキルから選択される。
【0038】
一実施形態において、R3の各出現は独立して水素または任意に置換したC1−10のアルキル基から選択される。好ましくは、アルキル基が存在するときは、C1−6アルキル基であり、いくつかの実施形態ではtBuを含まない。
【0039】
本発明の化合物は、除外された化合物の一つでないという条件で定義されるような式(I)の化合物である。しかし、除外は常に化合物の使用に適用せず、またこれら化合物の使用の一実施形態において、ただし書きは適用しない。明細書全体で用いる略語(rac)は、ラセミ混合物を指す。例えば、(rac)メキシレチンの場合、これはDLメキシレチンを指す。
【0040】
上述したように、式(I)の化合物はまた以下の化合物:HBr.グリシン−(rac)メキシレチン;AcOH.アスパラギン−(rac)メキシレチン;TFA.トリプトファン−(rac)メキシレチン;HBr.アラニン−(rac)メキシレチン;AcOH.−フェニルアラニン−(rac)メキシレチン;TFA.トリプトファン−(rac)メキシレチン.H2Oの一つでない。この実施形態において、アミノ酸は天然の立体配置である。
【0041】
一実施形態において、n1で定義される鎖中に二重結合がある。この場合、n1は2〜16であり、C=C二重結合はこの部分に存在する。あるいはまた、n1が1の場合、XがNのとき二重結合は存在し得る。いずれにしても、二重結合に関与した炭素原子または複数の原子はR4置換基のみを有し、R5置換基を有しない。一実施形態において、n1の数値に関係なく二重結合が存在しない。
【0042】
他の実施形態において、式(I)の化合物は以下の化合物の一つでない。
アラニン−(rac)メキシレチンアミド;
β−メトキシアスパラギン酸−(rac)メキシレチンアミド;
α−メトキシアスパラギン酸−(rac)メキシレチンアミド;
アスパラギン−(rac)メキシレチンアミド;
グリシン−(rac)メキシレチンアミド;
ロイシン−(rac)メキシレチンアミド;
メチオニン−(rac)メキシレチンアミド;
(rac)メチオニン−(rac)メキシレチンアミド;
フェニルアラニン−(rac)メキシレチンアミド;
アラニングリシングリシン−(rac)メキシレチンアミド、または
トリプトファン−(rac)メキシレチン。
【0043】
好ましくはR4の各出現は水素またはC1−4のアルキルであり、より好ましくは各R4が水素である。
【0044】
好ましくはR5の各出現は水素またはC1−4のアルキルであり、より好ましくは各R5が水素である。
【0045】
好ましい実施形態において、本発明の化合物は一つのプロドラッグ部分を有し、該プロドラッグ部分は1つ、2つまたは3つのアミノ酸(すなわちn2が1、2または3)であり、一方R3はHである。
【0046】
かかる任意置換のいずれかの出現で、1、2、3または4個の独立的に選択された任意の置換基がある。より通常には、存在するとき、かかる置換の各可能性で任意置換の0または1つの出現がある。
【0047】
存在し、分子と化学的に相溶する場合、任意の置換基は独立してヒドロキシ、C1−4アルキル、C1−4アルコキシ、C1−4ハロアルコキシ、フェニル、ベンジル、ハロゲン、シアノ、ニトロ、アミノ、アミドおよびチオからなる群から選択される。
【0048】
他の実施形態において、n2の少なくとも一つの出現は1である。他の実施形態において、n2の各出現は1である。さらに他の実施形態において、n2の少なくとも一つの出現は2である。他の実施形態において、n2の各出現は2である。
【0049】
さらに他の実施形態において、n2の各出現は1または2であり、RAAの各出現は独立して天然アミノ酸側鎖である。
【0050】
一実施形態において、本発明のプロドラッグは一つのプロドラッグ部分を有し、アルギニンもしくはリシンのホモポリマーか、またはアルギニンおよびリシンのヘテロポリマーである。さらなる実施形態において、ホモポリマーまたはヘテロポリマー中に2、3、4、5、6もしくは7個のアミノ酸がある(すなわちn2が2、3、4、5、6または7である)。
【0051】
一実施形態において、RAAの各出現は独立してアミノ酸側鎖、すなわち1〜20炭素原子を有するアミノ酸残基であり、または該残基(グリシンの場合に)が水素である。本発明との関連で、用語“アミノ酸”は天然アミノ酸(天然または非天然の立体化学的配置)および合成アミノ酸の両方を含む。天然アミノ酸は、タンパク質生合成に用いる20個のアミノ酸の一つである。該用語はまた、いくつかの実施形態において、翻訳時にタンパク質に取り込まれ得る他のアミノ酸(ピロリジン、オルニチンおよびセレノシステインを含む)も含むことができる。好ましくは、RAAまたはその各々は天然に存在するアミノ酸である。
【0052】
本発明のメキシレチンプロドラッグの組成物もここに提供する。該組成物は、少なくとも一つの本発明のプロドラッグ(例えば、式Iのプロドラッグ)またはその薬学的に許容し得る塩と、少なくとも一つの薬学的に許容し得る賦形剤とを含む。
【0053】
他の態様において、本発明は、神経因性疼痛のような痛みの治療に用いる式Iのメキシレチン結合体を提供する。
【0054】
本発明の一実施形態は、必要とする被検体の疾患をメキシレチンで治療する方法である。該方法は、本発明のメキシレチンプロドラッグ若しくはその薬学的に許容し得る塩またはその組成物を、必要とする被検体または被検体群に経口投与することを備え、ここでメキシレチンプロドラッグが少なくとも一つのアミノ酸または長さ2〜9個のアミノ酸のペプチドに共有結合したメキシレチンまたはメキシレチン代謝物(例えば、パラ−OH(p−OH)メキシレチン、メタ−OH(m−OH)メキシレチンまたはヒドロキシメチルメキシレチン)からなる。メキシレチンプロドラッグは一つまたは二つのプロドラッグ部分を有する。一つのプロドラッグ部分は、ペプチド結合を介してメキシレチンまたはメキシレチン代謝物のアミノ基に結合することができる。あるいは、またはさらに、メキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメキシレチン)は、フェノール系酸素でカルバメートまたはジカルボン酸リンカーを介して結合したプロドラッグ部分を有することができる。メキシレチンプロドラッグの量は、治療有効量(例えば、鎮痛有効量)であるのが好ましい。疾患は、メキシレチンで治療可能なものとすることができる。例えば、疾患は神経因性疼痛または不整脈である。
【0055】
他の実施形態において、本発明のプロドラッグは、親化合物と比較して、有害な胃腸副作用(吐き気や嘔吐など)を低減すると同時に、治療血漿薬剤濃度の達成の速度と一貫性とについて向上する利益を与える。
【0056】
したがって、一実施形態において、本発明は、メキシレチンの投与に通常伴う胃腸副作用を最小限にするための方法に関する。該方法は、本発明のメキシレチンプロドラッグ若しくはその薬学的に許容し得る塩またはその組成物を必要とする被検体に経口投与することを備え、ここでメキシレチンプロドラッグが少なくとも一つのアミノ酸または長さ2〜9個のアミノ酸のペプチドに対し共有結合したメキシレチンまたはメキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメチルメキシレチン)からなり、また経口投与すると、プロドラッグまたは薬学的に許容し得る塩が、完全に避けることができないとしても、未結合メキシレチンの経口投与後に通常見られる胃腸副作用を最小限にする。メキシレチンプロドラッグは一つまたは二つのプロドラッグ部分を有する。一つのプロドラッグ部分は、ペプチド結合を介してメキシレチンまたはメキシレチン代謝物のアミノ基に結合することができる。あるいは、またはさらに、メキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメキシレチン)は、フェノール系酸素でカルバメートまたはジカルボン酸リンカーを介して結合したプロドラッグ部分を有することができる。メキシレチンの量は治療有効量(例えば、鎮痛有効量)であるのが好ましい。
【0057】
さらなる実施形態において、メキシレチンの投与に伴うGI副作用は、限定されないが、嘔吐、吐き気、下痢および腹部不快感から選択される。
【0058】
本発明の他の実施形態は、メキシレチン血清濃度の被検体間および被検体内における変動の低減に関する。これは通常痛みまたは不整脈の治療中のものである。当該方法は、本発明のメキシレチンプロドラッグ若しくはその薬学的に許容し得る塩またはその組成物を必要とする被検体または被検体群に経口投与することを備え、ここでメキシレチンプロドラッグが少なくとも一つのアミノ酸または長さ2〜9個のアミノ酸のペプチドに対し共有結合したメキシレチンまたはメキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメチルメキシレチン)からなる。メキシレチンプロドラッグは一つまたは二つのプロドラッグ部分を有する。一つのプロドラッグ部分は、ペプチド結合を介してメキシレチンまたはメキシレチン代謝物のアミノ基に結合することができる。あるいは、またはさらに、メキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメキシレチン)は、フェノール系酸素でカルバメートまたはジカルボン酸リンカーを介して結合したプロドラッグ部分を有することができる。メキシレチンプロドラッグの量は治療有効量(例えば、鎮痛有効量)であるのが好ましい。
【0059】
また、本発明の他の実施形態は、必要とする被検体におけるメキシレチンの生物学的利用能の再現性を向上させることに関する。該方法は、本発明のメキシレチンプロドラッグ若しくはその薬学的に許容し得る塩またはその組成物を必要とする被検体または被検体群に経口投与することを備え、ここでメキシレチンプロドラッグが少なくとも一つのアミノ酸または長さ2〜9個のアミノ酸のペプチドに対し共有結合したメキシレチンまたはメキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメチルメキシレチン)からなる。メキシレチンプロドラッグは一つまたは二つのプロドラッグ部分を有する。一つのプロドラッグ部分は、ペプチド結合を介してメキシレチンまたはメキシレチン代謝物のアミノ基に結合することができる。あるいは、またはさらに、メキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメキシレチン)は、フェノール系酸素でカルバメートまたはジカルボン酸リンカーを介して結合したプロドラッグ部分を有することができる。メキシレチンプロドラッグの量は治療有効量(例えば、鎮痛有効量)であるのが好ましい。
【0060】
したがって、本発明はメキシチレンおよびその活性代謝物の天然および/または非天然アミノ酸と短鎖ペプチドのプロドラッグに関する。任意特定の理論に拘束されることを望むことなく、化合物のプロドラッグ部分(すなわちアミノ酸および/またはペプチド部分)は、神経因性疼痛の減少または排除若しくは不整脈の治療のために薬理学的に有効な量の薬剤/代謝物を送達しながら、薬剤またはその活性代謝物の局所作用から腸を一時的に保護する働きをする。このような一時的不活性化は、この薬剤の重大で極めて不所望な嘔吐性副作用を低減する。本発明のプロドラッグはまた、最大血漿中濃度の達成の速度、それゆえ痛み発症の緩和を促進するだけでなく、患者内および患者間の両方でのより一貫性のある患者応答を確実にする薬剤の生物学的利用能の再現性を改善する手段を提供する。これらの与えられた属性は、改善された鎮痛効果と、患者のより良い服薬遵守を確実にするように作用する。
【0061】
本発明はまた、メキシレチン自体と同一またはそれより良好な活性を享受する化合物群に関する。この化合物群において、催吐性効果はやはり発生しうるが、治療活性はメキシレチンと同じかまたは通常より良好である。これら化合物は、痛みや不整脈の治療においてメキシレチン自体と同じか、またはメキシレチンより効果があることが期待される。
【0062】
したがって本発明の他の態様によれば、式(IA)
【化12】
式(IA)
を提供するもので、
【0063】
式中のR1が
【化13】
であり
【0064】
R2がHおよび−OHから選択され、
【0065】
RAAの各出現は独立して1〜20の炭素原子を有する天然または非天然アミノ酸側鎖もしくは水素から選択され、
【0066】
n2は1、2または3から選択した整数であり、
【0067】
R3は水素および任意に置換したC1−4のアルキル基から選択され、ただし
【0068】
化合物が次の化合物:HBr.グリシン−(rac)メキシレチン;AcOH.アスパラギン−(rac)メキシレチン;TFA.トリプトファン−(rac)メキシレチン;HBr.アラニン−(rac)メキシレチン;AcOH−フェニルアラニン−(rac)メキシレチンまたはTFA.トリプトファン−(rac)メキシレチン.H2Oの一つでない。上記の除外において、アミノ酸は天然の立体配置である。
【0069】
他の実施形態において、式(IA)の化合物は以下の化合物の一つでない。
アラニン−(rac)メキシレチンアミド;
β−メトキシアスパラギン酸−(rac)メキシレチンアミド;
α−メトキシアスパラギン酸−(rac)メキシレチンアミド;
アスパラギン−(rac)メキシレチンアミド;
グリシン−(rac)メキシレチンアミド;
ロイシン−(rac)メキシレチンアミド;
メチオニン−(rac)メキシレチンアミド;
メチオニン−(rac)メキシレチンアミド;
フェニルアラニン−(rac)メキシレチンアミド;
アラニングリシングリシン−(rac)メキシレチンアミド;または
トリプトファン−(rac)メキシレチン。
【0070】
他の実施形態において、式(IA)の化合物のみにおいて、RAAは天然アミノ酸側鎖である。
【0071】
一実施形態において、n2の値は1または2が好ましく、もっとも好ましくはn2が1である。
【0072】
好ましい実施形態において、R3は水素である。別の実施形態において、R3は非置換のC1−6アルキルである。
【0073】
本発明の化合物は、除外された化合物の一つでないという条件で定義されるような式(IA)の化合物である。しかし、除外は常に化合物の使用に適用せず、これら化合物の使用の一実施形態において、ただし書きは適用しない。
【0074】
他の実施形態において、式(IA)の化合物は
メキシレチンピペリコン酸アミドおよび
メキシレチンジメチルグリシンアミドとを含む群から選択される。
【0075】
本発明のこれらおよび他の実施形態は、以下の詳細な説明に開示されるか、または明らかであり、かつ包含される。
【図面の簡単な説明】
【0076】
【図1】単離されたウサギの胃輪状平滑筋の調製物の電場刺激による収縮に対する(1)メキシレチン、(2)メキシレチンリシンアミドおよび(3)メキシレチングリシンアミドの効果を示すグラフである。
【発明を実施するための形態】
【0077】
本明細書中で使用されているように、
【0078】
用語“ペプチド”は、特記しない限り、2〜9個のアミノ酸からなるアミノ酸鎖を指す。好ましい実施形態において、本発明で用いるペプチドは、長さが2または3つのアミノ酸である。一実施形態において、ペプチドは分枝状ペプチドとすることができる。この実施形態では、ペプチドの少なくとも1つのアミノ酸側鎖が他のアミノ酸に(末端または側鎖のいずれか一つを介して)結合する。
【0079】
用語“アミノ酸”は、天然に存在するアミノ酸と非天然に発生するアミノ酸との両方を指す。本発明のプロドラッグでの使用を意図するアミノ酸は、天然アミノ酸と非天然アミノ酸との両方を含み、好ましくは天然アミノ酸である。側鎖RAAは(R)または(S)の立体配置のいずれかとすることができる。さらに、DおよびL体のアミノ酸の両方が本発明での使用を意図されている。
【0080】
“天然アミノ酸”は、タンパク質生合成に用いる20個のアミノ酸の一つ、並びに翻訳時にタンパク質に組み込むことができる他のアミノ酸(ピロリジンとセレノシステインとを含む)である。天然アミノ酸は、一般に式
【化14】
を有する。RAAはアミノ酸側鎖といわれるか、または天然アミノ酸の場合には天然アミノ酸側鎖といわれる。天然アミノ酸は、グリシン、アラニン、バリン、ロイシン、イソロイシン、アスパラギン酸、グルタミン酸、セリン、トレオニン、グルタミン、アスパラギン、アルギニン、リジン、プロリン、フェニルアラニン、チロシン、トリプトファン、システイン、メチオニンおよびヒスチジンを含む(表1参照。)。
【0081】
一実施形態において、アミノ酸側鎖を他のアミノ酸に結合する。さらなる実施形態において、側鎖をアミノ酸のN末端、C末端または側鎖を介してアミノ酸に結合する。
【0082】
天然アミノ酸側鎖の例としては、水素(グリシン)、メチル(アラニン)、イソプロピル(バリン)、sec−ブチル(イソロイシン)、−CH2CH(CH3)2(ロイシン)、ベンジル(フェニルアラニン)、p−ヒドロキシベンジル(チロシン)、−CH2OH(セリン)、−CH(OH)CH3(トレオニン)、−CH2−3−インドリル(トリプトファン)、−CH2COOH(アスパラギン酸)、−CH2CH2COOH(グルタミン酸)、−CH2C(O)NH2(アスパラギン)、−CH2CH2C(O)NH2(グルタミン)、−CH2SH(システイン)、−CH2CH2SCH3(メチオニン)、−(CH2)4NH2(リジン)、−(CH2)3NHC(=NH)NH2(アルギニン)およびCH2−3−イミダゾイル(ヒスチジン)が挙げられる。
【0083】
【表1】
【0084】
“非天然アミノ酸”は、標準遺伝コードによりコード化されるか、または翻訳時にタンパク質に組み込まれるものの中にない有機化合物である。従って、非天然アミノ酸はタンパク質生合成に使用した20個の天然に存在するアミノ酸以外のアミノ酸またはアミノ酸類似体を含み、限定しないが、アミノ酸のD−立体異性体が挙げられる。
【0085】
非天然アミノ酸の例としては、限定しないが、シトルリン、ホモシトルリン、ヒドロキシプロリン、ホモアルギニン、ホモセリン、ホモチロシン、ホモプロリン、オルニチン、4−アミノ−フェニルアラニン、サルコシン、ビフェニルアラニン、ホモフェニルアラニン、4−ニトロ−フェニルアラニン、4−フルオロ−フェニルアラニン、2,3,4,5,6−ペンタフルオロ−フェニルアラニン、パラ−アミノ安息香酸、ノルロイシン、シクロヘキシルアラニン、α−アミノイソ酪酸、N−メチル−アラニン、N−メチル−グリシン、N−メチル−グルタミン酸、tert−ブチルグリシン、α−アミノ酪酸、2−アミノイソ酪酸、2−アミノインダン−2−カルボン酸、セレノメチオニン、ランチオニン、デヒドロアラニン、γ−アミノ酪酸、ナフチルアラニン、アミノヘキサン酸、フェニルグリシン、ピペコリン酸、2,3−ジアミノプロピオン酸、テトラヒドロイソキノリン−3−カルボン酸、tert−ロイシン、tert−ブチルアラニン、シクロヘキシルグリシン、ジエチルグリシン、ジプロピルグリシンおよびそれらの誘導体が挙げられ、ここでアミン基の窒素はモノ−またはジ−アルキル化されている。
【0086】
したがって、本発明はまた、当業界で既知(例えば、“Protective Groups in Organic Synthesis”TW GreeneおよびPGM Wuts著、John Wiley & Sons社出版(1999)に記載されたような)の単純な合成変換によって官能化された上述のようなアミノ酸誘導体を想定し、ここに参照する。
【0087】
用語“極性アミノ酸”は、生理的pHで荷電されていない側鎖を有する親水性アミノ酸を指すが、これは二つの原子により通常共有された電子対がかかる原子の一つでより密接に保持される少なくとも一つの結合を有する。遺伝的にコード化された極性アミノ酸は、Asn(N)、Gln(Q)、Ser(S)およびThr(T)を含む。
【0088】
用語“非極性アミノ酸”は、生理的pHで荷電されていない側鎖を有する疎水性アミノ酸を指し、これは二つの原子により通常共有された電子対が一般に該二つ原子の各々で等しく保持される結合を有する(すなわち、側鎖が極性でない)。遺伝的にコード化された非極性アミノ酸は、Leu(L)、Val(V)、Ile(I)、Met(M)、Gly(G)およびAla(A)を含む。
【0089】
用語“脂肪族アミノ酸”は、脂肪族炭化水素側鎖を有する疎水性アミノ酸を指す。遺伝的にコード化された脂肪族アミノ酸は、Ala(A)、Val(V)、Leu(L)およびIle(I)を含む。
【0090】
用語“アミノ”は、−NH2基を指す。
【0091】
基としての用語“アルキル”は、特定した数の炭素原子を有する直鎖または分枝鎖の炭化水素鎖を指す。用語“アルキル”を炭素原子数を参照することなく使用する場合、C1−C10アルキルを指すものと理解すべきである。例えば、C1−10アルキルは、少なくとも1個、そして最大で10個の炭素原子を有する直鎖または分枝鎖アルキルを意味する。好適なアルキル基は、C1−6またはC1−4アルキル基をしばしば含む。本明細書で用いる“アルキル”の例は、限定しないが、メチル、エチル、n−プロピル、n−ブチル、n−ペンチル、イソブチル、イソプロピル、t−ブチル、ヘキシル、ヘプチル、オクチル、ノニルおよびデシルを含む。
【0092】
本明細書で用いる用語“置換アルキル”または“任意に置換したアルキル”は、少なくとも一つの水素が一つ以上の置換基、例えば、限定しないが、ヒドロキシ基、アルコキシ基、アリール基(例えば、フェニル基)、複素環、ハロゲン、トリフルオロメチル基、ペンタフルオロエチル基、シアノ基、シアノメチル基、ニトロ基、アミノ基、アミド基(例えば、−C(O)NH−Rで、Rがメチルのようなアルキル基)、アミヂン、アミド基(例えば、−NHC(O)−Rで、Rがメチルのようなアルキル基)、カルボキシアミド基、カルバメート、カーボネート、エステル、アルコキシエステル(例えば、−C(O)O−Rで、Rがメチルのようなアルキル基)およびアシルオキシエステル(例えば、−OC(O)−Rで、Rがメチルのようなアルキル基)により置換されたアルキル基を指す。用語が置換基自体に、または置換基の置換基に適用されているかどうかについて定義は関係がある。
【0093】
用語“複素環”は、炭素原子と、各々の出現で独立して窒素、リン、酸素および硫黄から選択された1〜5個のヘテロ原子とからなる安定な三員から十五員の環基を指す。
【0094】
本明細書で用いる用語“シクロアルキル”基は、例えば、シクロプロピル、シクロブチル、シクロペンチル、シクロへキシルまたはシクロへプチルのような3〜8個の炭素原子の非芳香族単環の炭化水素環を指す。
【0095】
本明細書で用いる“置換シクロアルキル”は、ここに記載したような一つ以上の置換基、例えば、限定しないが、ヒドロキシ基、アルコキシ基、アリール基(例えば、フェニル基)、複素環、ハロゲン、トリフルオロメチル基、ペンタフルオロエチル基、シアノ基、シアノメチル基、ニトロ基、アミノ基、アミド基(例えば、−C(O)NH−Rで、Rがメチルのようなアルキル基)、アミヂン、アミド基(例えば、−NHC(O)−Rで、Rがメチルのようなアルキル基)、カルボキシアミド基、カルバメート、カーボネート、エステル、アルコキシエステル(例えば、−C(O)O−Rで、Rがメチルのようなアルキル基)およびアシルオキシエステル(例えば、−OC(O)−Rで、Rがメチルのようなアルキル基)をさらに有するシクロアルキル基を指す。用語が置換基自体に、または置換基の置換基に適用されているかどうかについて定義は関係がある。
【0096】
用語“ケト”および“オキソ”は同義で、=O基を指す。
【0097】
用語“カルボニル”は、−C(=O)基を指す。
【0098】
用語“カルボキシル”は、−CO2H基を指し、カルボニルとヒドロキシル基とからなる(より具体的には、C(=O)OH)。
【0099】
本明細書で用いる“アミド”は、
【化15】
基を指す。本発明において、プロドラッグ部分をアミド結合を介してメキシレチンに結合することができる。この実施形態において、−N−は、非結合メキシレチンまたはメキシレチン代謝物中のアミノ基の窒素である。アミド結合は、アミンをカルボン酸と反応させることによって形成することがてきる。これは、ペプチド結合を形成する反応である。
【0100】
用語“カルバメート基”および“カルバメート”は、
【化16】
基に関連し、式中の−O1−が非結合p−OHメキシレチン分子中のフェノール系酸素である。ここに記載したプロドラッグ部分は、そのアミノ酸またはペプチドおよびカルバメート結合に基づいて参照することができる。そのような参照におけるアミノ酸またはぺプチドは、特記しない限り、アミノ酸またはペプチドのアミノ末端を介してカルボニルリンカーおよびp−OHメキシレチンのフェノール系酸素に結合されていると推測すべきである。
【0101】
例えば、valカルバメート(カルバミン酸バリン)は、式
【化17】
を有する。tyr−valカルバメートのようなペプチド関して、特記しない限り、ペプチド中のもっとも左のアミノ酸がペプチドのアミノ末端であり、カルボニルリンカーを介してp−OHメキシレチンに結合してカルバメートプロドラッグを形成すると推測すべきである。
【0102】
用語“ジカルボン酸リンカー”および“ジカルボキシルリンカー”は、本発明の目的においては同義である。ジカルボン酸リンカーは、メキシレチンとアミノ酸/ペプチド部分との間の基、
【化18】
(−(CO)−(CR4R5)n1−(CO)−)を指す。あるいはまた、“ジカルボン酸リンカー”は、次式
【化19】
(−(CO)−(NH)−(CR4R5)n1−(CO)−)、または次式
【化20】
(−(CO)−(O)−(CR4R5)n1−(CO)−)を有することができる。
【0103】
ジカルボン酸リンカーに関して、1つのカルボニル基をp−OHメキシレチン内の酸素原子に結合する一方、第二のカルボニル基をペプチドまたはアミノ酸のN末端、若しくはアミノ酸側鎖のアミノ基に結合する。
【0104】
本明細書に記載したジカルボン酸プロドラッグの部分は、そのアミノ酸またはペプチドおよびジカルボキシル結合に基づいて参照することができる。かかる参照におけるアミノ酸またはペプチドは、アミノ酸またはペプチドのアミノ末端を介してジカルボキシルリンカーの一つのカルボニル基(元来はカルボキシル基の一部)に結合される一方、特記しない限り、他の基がp−OHメキシレチンに付着すると推測すべきである。ジカルボキシルリンカーは、前述したように様々に置換してもまたは置換しなくてもよい。
【0105】
本発明で使用するジカルボン酸の非限定的なリストを表2に示す。表2に示したジカルボン酸は2〜18個の炭素を含むが、より長鎖のジカルボン酸を本発明でリンカーとして使用することができる。また、ジカルボン酸リンカーを1つ以上の位置で置換することができる。適当に活性化したジカルボン酸を活性化アミノ酸またはペプチドと結合し、次いでp−OHメキシレチンと反応させて本発明のプロドラッグを形成することができる。プロドラッグ合成手順を実施例でより詳しく説明する。
【0106】
【表2】
【0107】
本発明のジカルボン酸リンカーは、リンカー構造
【化21】
および
【化22】
をそれぞれ付与するために最初のカルボニル基に結合した窒素または酸素原子を有する、すなわちXが式1中の(−NH−)または(−O−)とすることができる。かかるジカルボン酸リンカーの例を表3に示し、明細書全体に挙げられている。
【0108】
一実施形態において、ジカルボン酸リンカーを置換する。例えば、一つ以上の
【化23】
置換アルキル基、非置換アルキル基が存在し得る(式1により定義されるようなR3)。これらの実施形態において、X(式1により定義されるような−NH−またはO−)は存在してもまたは欠落してもよい。ジカルボン酸リンカーの例を表3に挙げる。
【0109】
一実施形態において、ジカルボン酸リンカー中の炭素鎖
【化24】
は不飽和であり、一つ以上の二重結合を有することができる。これらの実施形態において、n1≧2であり、R5は二重結合を形成する二つの炭素上に存在しない。そのようなリンカーの一例であるフマル酸を、表3に示す。
【0110】
【表3】
【0111】
本発明のジカルボン酸プロドラッグ部分の例は、次式
【化25】
を有するバリンコハク酸エステルを含む。チロシン−バリンコハク酸エステルのようなジペプチドに関して、別に規定しない限り、薬剤、この場合バリンに隣接するアミノ酸がアミノ末端を経由してジカルボン酸リンカーに結合することを推測すべきである。ジペプチドの末端カルボキシル残基(この場合、チロシン)は、C(カルボキシル)末端を形成する。
【0112】
用語“担体”は、希釈剤、賦形剤および/または活性化合物を投与する媒体を指す。本発明の医薬組成物は、二つ以上の担体の組合せを含むことができる。この種の薬剤担体は水、食塩水溶液、水性デキストロース溶液、水性グリセロール溶液のような無菌の液体や、落花生油、大豆油、鉱油、胡麻油などのような石油、動物、野菜または合成起源のものを含む油とすることができる。水または食塩水溶液と、水性デキストロースと、グリセロール溶液とを、特に注射可能な溶液用の担体として使用するのが好ましい。適当な薬剤担体は、“Remington’s Pharmaceutical Sciences”(E.W.Martin著、第18版)に記載されている。
【0113】
“薬学的に許容し得る”という表現は、一般に安全とみなされている分子的実体および組成物を指す。特に、本発明の実施に用いる薬学的に許容し得る担体は生理的に許容されるもので、患者に投与する際に通常アレルギーまたは類似の有害反応(例えば、胃の不調、めまいなど)を生じない。好ましくは、本明細書で用いるような“薬学的に許容し得る”用語は、適切な政府機関の規制当局によって承認、または米国薬局方若しくは動物での、特にヒトにおける使用のための、他の一般的に認められている薬局方に記載されていることを意味する。
【0114】
“薬学的に許容し得る賦形剤”は、一般に安全で、非毒性で、また生物学的または他の状態で望ましくないものでない医薬組成物を調製するのに有用な賦形剤を意味し、獣医学的使用並びにヒトへの医薬的使用に許容できる賦形剤を含む。本発明に使用するような“薬学的に許容し得る賦形剤”は、かかる賦形剤の一つ以上を含む。
【0115】
用語“治療すること”は、(1)状態、疾患または病気に苦しんでいるかまたはなりやすい状況に置かれているかもしれないが、状態、疾患または病気の臨床的なまたは無症状の症状を経験していないまたは示していない動物に発現する状態、疾患または病気の臨床状態の出現を予防または遅延すること、(2)状態、疾患または病気を抑制すること(例えば、その少なくとも一つの臨床的な若しくは無症状の症状の疾病の発生若しくは維持療法の場合にはその再発を停止させ、減少させまたは遅延させること)、および/または(3)疾患を軽減すること(すなわち、その少なくとも一つの臨床的な若しくは無症状の症状の状態、疾患または病気を退化させること)を含む。治療すべき患者に対する利点は、統計学的に有意であるか、または患者若しくは医師に少なくとも認知可能であるかのどちらかである。
【0116】
用語“被検体”は、ヒトや、家畜(例えば、イヌおよびネコ)のような他の哺乳類を含む。
【0117】
“有効量”は、所望の治療応答をもたらすに十分な本発明のプロドラッグまたは組成物の量を意味する。治療応答は、使用者(例えば、臨床医)が治療に効果的な反応として認識するあらゆる応答とすることができる。治療応答は、一般にプロドラッグ中のメキシレチンまたはp−OHメキシレチンを活性な形状で投与する際に存在する一つ以上の胃腸の副作用の症状の鎮痛および/または改善である。さらに、治療応答の評価に基づいて適切な治療期間、適切な投与量およびあらゆる潜在的な併用療法を決定することは当業者の技能の範囲内である。
【0118】
特に記載がない限り、用語“活性成分”は、ここに記載するような、プロドラッグのメキシレチン部分またはメキシレチン代謝物部分、例えば本発明のプロドラッグのp−OHメキシレチン部分を指すものと理解すべきである。
【0119】
本明細書で用いる“メキシレチン代謝物”とは、p−OHメキシレチン(
【化26】
m−OHメキシレチン(
【化27】
【化28】
【0120】
用語“塩”は、酸付加塩または遊離塩基の付加塩を含むことができる。適当な薬学的に許容し得る塩(例えば、アミノ酸またはペプチドのカルボキシル末端の塩)は、限定しないが、ナトリウム、カリウムおよびセシウム塩のような金属塩;カルシウムおよびマグネシウム塩のようなアルカリ土類金属塩;トリエチルアミン、グアニジンおよびN−置換グアニジン塩、アセトアミジン並びにN−置換アセトアミジン、ピリジン、ピコリン、エタノールアミン、トリエタノールアミン、ジシクロヘキシルアミンおよびN,N’−ジベンジルエチレンジアミン塩のような有機アミン塩を含む。薬学的に許容し得る塩(塩基性窒素中心のもの)は、限定しないが、塩酸塩、臭化水素酸塩、硫酸塩、リン酸塩のような無機酸塩;トリフルオロ酢酸塩およびマレイン酸塩のような有機酸塩;メタンスルホネート、エタンスルホネート、ベンゼンスルホネート、p−トルエンスルホネート、カンファースルホネートおよびナフタレンエルホネートのようなスルホン酸塩;アルギン酸塩、グルコン酸塩、ガラクツロン酸塩、アラニン酸塩、アスパラギン酸塩、グルタミン酸塩のようなアミノ酸塩を含む(例えば、 Bergeおよびその他、“Pharmaceutical Salts,”J.Pharma.Sci.1977年、第66編、第1項を参照)。
【0121】
ここで用いるような用語“生物学的利用能”は、一般にメキシレチンまたはメキシレチン代謝物を製剤から吸収し、全身的に、それゆえ作用部位で入手し得る速度および/または度合いを意味する。連邦規則集第21巻第320部第1節(2003年版)を参照。経口投与形態に関して、生物学的利用能は活性成分を経口投与形態から放出し、作用部位に移動するプロセスに関する。特定処方に対する生物学的利用能のデータは、全身循環へ吸収される投与量の割合の推定値を与える。従って、用語“経口生物学的利用能”は、被検体への単独投与後に全身循環へ吸収される経口投与されたメキシレチン投与量の割合を指す。経口生物学的利用能を決定するための好ましい方法は、経口投与されたメキシレチンのAUCを同じ被検体に静脈内投与された同じメキシレチン投与量のAUCで割り、比率を百分率として表現することである。経口生物学的利用能を算出するための他の方法は、当業者によく理解されており、ShargelおよびYu著,“Applied Biopharmaceutics and Pharmacokinetics”、1999年出版、第4版、Appleton & Lange社(Stamford,Conn)出版にさらに詳細に記載されており、その全体をここに参照して援用する。
【0122】
本発明の化合物
【0123】
一実施形態において、本発明のメキシレチンプロドラッグは、アミド結合を介してメキシレチンのアミノ窒素に結合したプロドラッグ部分を有する。あるいは、またはさらに(すなわち、メキシレチンプロドラッグは二つのプロドラッグ部分を有する)、本発明のプロドラッグは、カルバメートまたはジカルボン酸リンカー基のヒドロキシ基の酸素を介して結合したメキシレチン代謝物(p−OH、m−OHまたはヒドロキシメチルメキシレチン)の新規なアミノ酸およびペプチドプロドラッグである。好ましくは、本発明のプロドラッグは、単一アミノ酸または長さが2〜9のアミノ酸からなる短鎖ペプチドに結合したメキシレチンまたはp−OHメキシレチンを含む。
【0124】
ジカルボン酸プロドラッグの実施形態において、メキシレチン代謝物(例えば、p−OH、m−OHメキシレチンまたはヒドロキシメチルメキシレチン)のヒドロキシ基の酸素は、限定されないが、マロン酸、コハク酸、グルタル酸、アジピン酸もしくは他の長鎖ジカルボン酸またはその置換誘導体(ジカルボン酸リンカーの代表的な例については、表2および表3を参照)のようなジカルボン酸でエステル化することができる。次いで、アミノ酸またはペプチドを、ペプチド/アミノ酸のN末端の窒素またはアミノ酸側鎖に存在する窒素(例えば、リジンの側鎖)を介して残りのカルボキシル基に結合することができる。
【0125】
メキシレチンアミドプロドラッグの実施形態において、プロドラッグ部分(すなわち、アミノ酸またはペプチド)をメキシレチンまたはメキシレチン代謝物のアミノ基に結合することができる。アミド結合は、メキシレチンまたはメキシレチン代謝物のアミノ基をカルボン酸と反応させることによって形成し得る。さらなるメキシレチンアミドプロドラッグの実施形態において、プロドラッグはp−OHメキシレチンプロドラッグであり、二つのプロドラッグ部分を有する。第二のプロドラッグ部分を、ジカルボン酸結合またはカルバメート基を介してp−OH、m−OHメキシレチンまたはヒドロキシメチルメキシレチンのヒドロキシ基の酸素に結合することができる。
【0126】
本発明の化合物の利点
【0127】
特定の理論に拘束されることを望むことなく、ここに記載したメキシレチンまたはメキシレチン代謝物プロドラッグのアミノ酸またはペプチド部分は、消化管内に固有に存在するジペプチドおよびトリペプチドトランスポーターPept1を選択的に利用して吸収を行うと考えられる。一度吸収されると、プロドラッグは加水分解を受けて、活性薬剤を全身循環へ放出する。メキシレチンは、その後部分的に血液中もしくは血漿中に存在する肝臓内および肝臓外の加水分解酵素によってアミノ酸またはペプチドプロドラッグから放出されると考えられる。
【0128】
かかるPept1により助長されたプロドラッグの吸収は、より一貫した経口生物学的利用能の結果として可能となる鎮痛応答においてより高い一貫性を提供することができる。このより再現性の高い経口生物学的利用能の結果として、本発明のプロドラッグは、メキシレチン血漿およびCNS濃度の被験者間および被験者内の変動の相当な低減をもたらし、その結果単一患者または患者集団に対し痛み緩和における変動を著しく少なくする。従って、患者の服薬遵守は、この鎮痛応答の高い予測可能性の結果としてさらに改善されるだろう。
【0129】
メキシレチンが吸収されるまで一時的に不活性化され得る場合、メキシレチンの投与に伴う任意の局所的な媒介嘔吐(すなわち腸管内腔内から)が強力に低減され得る。この不活性化は、薬剤の下部食道括約筋および胃への直接的な暴露を排除することができる。吸収後にのみ活性化するメキシレチンの不活性化プロドラッグは、嘔吐性および他の有害なGI効果を排除する一つの方法である。別のアプローチとして、活性なメキシレチン代謝物(例えば、p−OH、m−OHメキシレチンまたはヒドロキシメチルメキシレチン)のプロドラッグを使用することができる。パラ−ヒドロキシメキシレチンは、親分子のナトリウムチャンネル抑制活性の約25%を保持し、従ってそれ自体有用な薬剤とし得ると報告されている(De Bellis et al.(2006).Brit.J.Pharmacol.149,300-310)。メキシレチンに伴う有害なGI副作用(例えば嘔吐)を一時的な不活性化によって十分に克服することができれば、得られた生成物は神経因性疼痛の治療用薬剤の現在限られた医療設備に価値ある追加を提供することができるだろう。
【0130】
さらに、単一のアミノ酸およびペプチドは毒性のリスクを示すと想定されておらず、アミノ酸またはペプチドはまた、全体の構造および享受された水溶性という重大な変化に起因してメキシレチンまたはその活性なメキシレチン代謝物をおそらく一時的に不活性化するだろう。さらに、慎重に選択されたペプチド結合体は、腸管内腔内でトリプシンのようなペプチダーゼによる部分加水分解により長期または継続的な放出という能力を提供する。例えば、直接または間接的(例えば、グリシンのような他のアミノ酸を介して)なC−末端のポリ−アルギニンまたはポリ−リジンフラグメントの薬剤への導入は、腸管内腔での部分的な加水分解をもたらし、よって生成した吸収能力のあるジ−またはトリ−ペプチド擬態性化合物の吸収用送達速度を制御することができる。次いで、かかる吸収は、ジ−およびトリ−ぺプチドに特異的なPept1のような活性トランスポーターにより行われるだろう。
【0131】
本発明のメキシレチンおよびp−ヒドロキシ代謝物のプロドラッグ
【0132】
一実施形態において、本発明は、式Iのメキシレチンおよびp−OHメキシレチンのプロドラッグ
【化29】
式I
【0133】
またはその薬学的に許容し得る塩に関し、
【0134】
ここで式中のR1は水素および
【化30】
から選択され、
【0135】
R2は、水素、
【化31】
から選択され、
【0136】
O1は未結合p−OHメキシレチン中に存在するフェノール系酸素であり、
【0137】
Xは(−NH−)、(−O−)、または欠落であり、
【0138】
RAAの各出現は独立して天然または非天然アミノ酸側鎖であり、
【0139】
n1は0〜16から選ばれる整数であり、
【0140】
n2の各出現は独立して1〜9から選ばれる整数であり、
【0141】
R3の各出現は独立して水素、置換アルキル基または非置換アルキル基から選択され、
【0142】
R4およびR5の各出現は独立して水素、
【化32】
、置換アルキル基および非置換アルキル基から選択され、
【0143】
n1により定義される炭素鎖における二重結合の場合、二重結合を形成する炭素上にR4は存在し、R5は欠落であり、
【0144】
少なくともR1は
【化33】
であるか、あるいは少なくともR2は
【化34】
のいずれか一方である。
【0145】
ジカルボン酸リンカーの一実施形態において、n1は0、1、2、3または4であり、R1はHである。
【0146】
他のジカルボン酸リンカーの実施形態において、n1は0、1、2、3または4であり、R1は
【化35】
であり、n2の各出現は1、2および3であり、R3はHである。
【0147】
一実施形態において、n2の各出現は1、2、3、4または5である。
【0148】
好ましい一実施形態において、本発明の化合物は一個のプロドラッグ部分を有し、該プロドラッグ部分が1つ、2つまたは3つのアミノ酸(すなわちn2が1、2または3である)を有する一方、R3はHである。
【0149】
他の実施形態において、n2は1である。さらに他の実施形態において、n2は2である。
【0150】
さらに他の実施形態において、n2の各出現は1または2であり、RAAの各出現は独立して天然アミノ酸側鎖である。
【0151】
一実施形態において、プロドラッグは一個のプロドラッグ部分を有し、アルギニンもしくはリシンのホモポリマーか、またはアルギニンおよびリシンのヘテロポリマーである。さらなる実施形態において、ホモポリマーまたはヘテロポリマーに2、3、4、5、6または7個のアミノ酸がある(すなわちn2が2、3、4、5、6または7である)。
【0152】
ジカルボン酸リンカーの一実施形態において、n1は0、1、2または3である。さらなる実施形態において、n1は0、1、2または3である一方、R3、R4およびR5の各出現は水素である。
【0153】
一実施形態において、n2の各出現は1、2、3、4または5である。さらなる実施形態において、n2は1、2、3、4または5である一方、R3の各出現は水素である。
【0154】
好ましい実施形態において、本発明の化合物は一つのプロドラッグ部分を有し、該プロドラッグ部分は1つ、2つまたは3つのアミノ酸(すなわちn2が1、2または3)を有する一方、R3はHである。
【0155】
他の実施形態において、n2は1である。さらに他の実施形態において、n2は2である。
【0156】
さらに他の実施形態において、n2の各出現は1または2であり、RAAの各出現は独立して天然アミノ酸側鎖である。
【0157】
一実施形態において、薬物のアミノ官能基に結合したペプチドプロドラッグ部分は、グリシンまたはリジン残基である。さらなる実施形態において、本発明のペプチドプロドラッグ部分は、グリシンまたはリジンのC末端とアルギニンまたはリジンのアミノ末端(または側鎖の窒素)との間のペプチド結合によりグリシンまたはリジンに直接隣り合うアルギニンまたはリジン残基を取り入れる。従って、以下のジペプチド、すなわち(1)グリシン−アルギニン、(2)グリシン−リジン、(3)リジン−アルギニン、(4)リジン−リジンは、ジペプチドプロドラッグ部分として単独で、あるいはプロドラッグ部分の一部としてのどちらか一方で本発明により意図される。これらの実施形態において、列挙された第一のアミノ酸をメキシレチンに結合する。
【0158】
他の実施形態において、ペプチドプロドラッグ部分を、メキシレチンのヒドロキシ代謝物中のp−ヒドロキシ基に結合する。カルバメートまたはジカルボン酸の架橋を介して結合した好ましいアミノ酸は、限定しないが、バリン、ロイシン、イソロイシンまたはメチオニンを含む。
【0159】
天然に存在するアミノ酸、並びに非天然アミノ酸のいずれかを含むペプチドを本発明に使用し得る。非天然アミノ酸をペプチドプロドラッグ部分またはその一部として使用する場合、ペプチドは非天然アミノ酸を単独で、あるいは天然および非天然アミノ酸の組み合わせを含むことができる。
【0160】
本発明で用いるプロドラッグに使用するアミノ酸は、L−体であるのが好ましい。本発明はまた、D−体のアミノ酸からなるか、またはD−体およびL−体のアミノ酸混合物からなる本発明のプロドラッグを意図している。
【0161】
一実施形態において、式IIのアミド結合メキシレチンプロドラッグを提供する。他の実施形態において、式IIIのアミド結合p−OHメキシレチンプロドラッグを提供する。式II−IIIについて、R3、RAAおよびn2は式Iに与えられるものとして定義される。式II−IIIのプロドラッグの薬学的に許容し得る塩も本発明に包含される。
【0162】
この実施形態において、本発明のプロドラッグは、アミド結合体を介してアミノ酸または短鎖ペプチドに共有結合したメキシレチンまたはp−OHメキシレチンを含み、ここでアミド結合体が薬剤のアミン官能基およびアミノ酸(またはペプチドのC末端)のカルボキシ官能基から形成される。上述したように、本発明で用いるアミノ酸は天然または非天然とすることができる。例えば、グリシン、リジン、アルギニン、シトルリン、オルニチンを、いずれも単一アミノ酸として、またはペプチドの一部分としてメキシレチンまたはp−OHメキシレチンに共有結合することができる。
【化36】
式II
【化37】
式III
【0163】
式IIの一実施形態において、n2は1、2、3または4のいずれかであり、R3はHである。式IIの他の実施形態において、n2は1、2、3または4のいずれかであり、R3はアルキル基である。
【0164】
式IIIの一実施形態において、n2は1、2、3または4のいずれかであり、R3はHである。式IIIの他の実施形態において、n2は1、2、3または4のいずれかであり、R3はアルキル基である。
【0165】
式IIおよび/または式IIIの他の実施形態において、n2は1、2、3、4または5である。さらなる実施形態において、n2は1、2、3、4または5であり、一方R3は水素である。式IIまたは式IIIの好ましい実施形態において、プロドラッグ部分は1つ、2つまたは3つのアミノ酸(すなわちn2は1、2または3)を有する一方、R3はHである。他の実施形態において、n2は1である。また他の実施形態において、n2は2である。また他の実施形態において、n2は1または2であり、RAAの各出現は独立して天然アミノ酸側鎖である。
【0166】
本発明の他の実施形態において、式IVのp−OHメキシレチンカルバメートプロドラッグを提供する。式IVについて、O1、R3、RAAおよびn2は式Iに規定されるように定義される。
【化38】
式IV カルバメート結合p−OHメキシレチンプロドラッグ
【0167】
式IVの一実施形態において、n2は1、2、3または4のいずれかであり、R3はHである。式IVのさらなる実施形態において、n2は1、2、3または4のいずれかであり、R3はHである。またさらなる実施形態において、RAAの各出現は天然アミノ酸側鎖である。
【0168】
式IVの他の実施形態において、n2は1、2、3、4または5である。さらなる実施形態において、n2は1、2、3、4または5であり、一方R3は水素である。式IVの好ましい実施形態において、プロドラッグ部分は1つ、2つまたは3つのアミノ酸(すなわちn2は1、2または3)を有する一方、R3はHである。式IVの別の実施形態において、プロドラッグ部分は1つ、2つまたは3つのアミノ酸(すなわちn2は1、2または3)を有する一方、R3はアルキル基である。
【0169】
さらに別の実施形態において、n2は1である。さらに別の実施形態において、n2は2である。さらに別の実施形態において、n2は1または2であり、RAAの各出現は天然アミノ酸側鎖である。他の実施形態において、RAAの少なくとも1つの出現は非天然アミノ酸側鎖である。
【0170】
本発明の他の実施形態は、式Vのジカルボン酸結合p−OHメキシレチンプロドラッグに関する。式Vについて、O1、R3、R5、RAA、−X−、n1およびn2は式Iに規定されるように定義される。
【化39】
式V ジカルボン酸結合p−OHメキシレチンプロドラッグ
【0171】
式Vの一実施形態において、n1は0〜4から選択される整数である。さらなる実施形態において、R3はHであり、n2は1、2または3である。
【0172】
式Vの一実施形態において、n1は0、1、2または3である。さらなる実施形態において、n1は0、1、2または3であり、一方R3、R4およびR5の各出現は水素である。
【0173】
また、さらなる実施形態において、n2は1、2または3である。式Vの他の実施形態において、Xは欠落であり、n1は1、2または3である。また、さらに式Vの実施形態において、Xは欠落であり、n1は1、2または3であり、n2は1または2であり、R3、R4およびR5はそれぞれ水素である。
【0174】
式Vの一実施形態において、Xは−NH−であり、n1は0、1、2または3であり、n2は1、2または3であり、R3、R4およびR5はそれぞれHである。さらなる実施形態において、n1は2である。
【0175】
式Vの一実施形態において、Xは−O−であり、n1は0、1、2または3であり、n2は1、2または3であり、R3、R4およびR5はそれぞれHである。さらなる実施形態において、n1は2である。
【0176】
式Vの他の実施形態において、Xは欠落であり、n1は1、2または3であり、n2は1、2または3である。また、さらに式Vの実施形態において、Xは欠落であり、n1は1または2であり、n2は1、2、3、4または5である。
【0177】
式Vの好ましい実施形態において、本発明のプロドラッグ部分は1つまたは2つのアミノ酸(すなわちn2は1または2である。)を有する。一実施形態において、n1は1または2であり、一方n2は1、2または3である。
【0178】
式Vの一実施形態において、Xは−O−であり、n1は0、1または2であり、n2は1または2であり、R3はHである。さらなる実施形態において、R4の少なくとも1つの出現は
【化40】
である。
【0179】
一実施形態において、Xは−NH−であり、n1は0、1または2であり、n2は1または2であり、R3はHである。さらなる実施形態において、R4の少なくとも1つの出現は
【化41】
である。
【0180】
式Vの好ましい実施形態において、n2は1、2または3であり、一方R3、R4およびR5はHである。他の実施形態において、n2は1である。また他の実施形態において、n2は2である。さらに式Vの他の実施形態において、n2は1または2であり、RAAの各出現は独立して天然アミノ酸側鎖である。
【0181】
本発明の他の実施形態において、本発明のp−OHメキシレチンプロドラッグは式VIまたはVIIのジカルボン酸結合プロドラッグである。式VIおよび式VIIについて、O1、R3、R4、R5、RAA、n1およびn2は式Iに規定されるように定義される。
【化42】
式VI
【化43】
式VII
【0182】
式VIおよび/または式VIIのジカルボン酸リンカーの一実施形態において、n1は0、1、2または3である。さらなる実施形態において、n1は0、1、2または3であり、一方R3、R4およびR5は水素である。
【0183】
また、式VIおよび/または式VIIのさらなる実施形態において、n2は1、2または3である。式VIおよび/または式VIIのさらなる実施形態において、n1は1、2または3である。またさらなる実施形態において、n1は1、2または3であり、n2は1または2であり、R1、R2およびR3はそれぞれ水素である。
【0184】
式VIおよび/または式VIIの一実施形態において、n1は0、1、2または3であり、n2は1、2または3であり、R3、R4およびR5はそれぞれHである。さらなる実施形態において、n1は2である。
【0185】
式VIおよび/または式VIIの好ましい実施形態において、本発明のプロドラッグ部分は1つ、2つまたは3つのアミノ酸(すなわちn2は1、2または3である。)を有する。一実施形態において、n1は1または2であり、一方n2は1、2または3である。
【0186】
式VIおよび/または式VIIの一実施形態において、n1は0、1または2であり、n2は1または2であり、R3はHである。さらなる実施形態において、R4の少なくとも1つの出現は
【化44】
である。
【0187】
式VIおよび/または式VIIの一実施形態において、n1は0、1または2であり、n2は1または2であり、R3はHである。さらなる実施形態において、R4の少なくとも1つの出現は
【化45】
である。
【0188】
式VIおよび/または式VIIの好ましい実施形態において、n2は1、2または3であり、一方R3、R4およびR5はHである。他の実施形態において、n2は1である。また他の実施形態において、n2は2である。さらに式VIおよび/または式VIIの他の実施形態において、n2は1または2であり、RAAの各出現は独立して天然アミノ酸側鎖である。
【0189】
また、本発明の他の実施形態において、式VIIIのp−OHメキシレチンプロドラッグまたはその薬学的に許容し得る塩を提供する。式VIIIについて、O1、R3、RAA、n1およびn2は式Iに規定されるように定義される。
【化46】
式VIII
【0190】
式VIIIの一実施形態において、n2の少なくとも1つの出現は1、2、3または4であり、R3の少なくとも1つの出現はHである。さらなる実施形態において、n2の各出現は1、2、3または4であり、R3の各出現はHである。
【0191】
式VIIIの他の実施形態において、n2の少なくとも1つの出現は1、2、3、4または5である。さらなる実施形態において、n2の少なくとも1つの出現は1、2、3、4または5であり、一方R3の少なくとも1つの出現は水素である。さらなる実施形態において、n2の各出現は1、2、3、4または5であり、一方R3の各出現は水素である。また、式VIIIのさらなる実施形態において、n2の各出現は1、2、3、4または5であり、一方R3の少なくとも1つの出現はアルキル基である。
【0192】
式VIIIの好ましい実施形態において、少なくとも1つのプロドラッグ部分は1つ、2つまたは3つのアミノ酸(すなわちn2は1、2または3である。)を有し、一方R3の少なくとも1つの出現はHである。式VIIIの他の実施形態において、n2の少なくとも1つの出現は1である。また式VIIIの他の実施形態において、n2の少なくとも1つの出現は1または2であり、RAAの各出現は独立して天然アミノ酸側鎖である。式VIIIの他の実施形態において、n2の両方の出現は1、2および3から選択される。
【0193】
本発明の他の実施形態は、式IXのp−OHメキシレチンプロドラッグまたはその薬学的に許容し得る塩に関する。式IXについて、O1、−X−、R3、R4、R5、n1およびn2は式Iに規定されるように定義される。
【化47】
式IX
【0194】
式IXの一実施形態において、n1は0〜4から選択される整数である。
【0195】
式IXのジカルボン酸リンカーの一実施形態において、n1は0、1、2または3である。さらなる実施形態において、n1は0、1、2または3であり、一方R3、R4およびR5の各出現は水素である。
【0196】
また、さらなる実施形態において、n2は1、2または3である。式IXのさらなる実施形態において、−X−は欠落であり、n1は1、2または3である。また式IXのさらなる実施形態において、n2の各出現は1または2であり、R3、R4およびR5はそれぞれ水素である。
【0197】
式IXの一実施形態において、−X−は−NH−であり、n1は0、1、2または3であり、n2は1、2または3であり、R3、R4およびR5はそれぞれHである。さらなる実施形態において、n1は2である。
【0198】
式IXの一実施形態において、−X−は−O−であり、n1は0、1、2または3であり、n2の各出現は1、2、3、4または5であり、R3、R4およびR5はそれぞれHである。さらなる実施形態において、n1は2である。
【0199】
式IXの他の実施形態において、−X−は欠落であり、n1は1、2または3であり、n2は1、2または3である。また、さらに式IXの実施形態において、−X−は欠落であり、n1は1または2であり、n2の各出現は1、2、3、4または5である。
【0200】
式IXの好ましい実施形態において、本発明のプロドラッグ部分は1つまたは2つのアミノ酸(すなわちn2は1または2である)を有する。一実施形態において、n1は1または2であり、一方n2の各出現は1、2または3である。
【0201】
式IXの一実施形態において、−X−は−O−であり、n1は0、1または2であり、n2は1または2であり、R3はHである。さらなる実施形態において、R4の少なくとも1つの出現は
【化48】
である。
【0202】
式IXの他の実施形態において、−X−は−NH−であり、n1は0、1または2であり、n2の各出現は1または2であり、R3はHである。さらなる実施形態において、R4の少なくとも1つの出現は
【化49】
である。
【0203】
式IXの一実施形態において、−X−は−O−であり、n1は0、1または2であり、n2は1または2であり、R3はHである。さらなる実施形態において、R4の少なくとも1つの出現は
【化50】
である。
【0204】
式IXの他の実施形態において、−X−は−NH−であり、n1は0、1または2であり、n2の各出現は1または2であり、R3はHである。さらなる実施形態において、R4の少なくとも1つの出現は
【化51】
である。
【0205】
式IXの好ましい実施形態において、n2は1、2または3であり、一方R1、R2およびR3はHである。他の実施形態において、n2の少なくとも1つの出現は1である。また他の実施形態において、n2は2である。さらに式IXの他の実施形態において、n2は1または2であり、RAAの各出現は独立して天然アミノ酸側鎖である。
【0206】
本発明の他の実施形態は、式Xおよび式XIのp−OHメキシレチンプロドラッグまたはその薬学的に許容し得る塩に関する。式Xおよび式XIについて、O1、R3、R4、R5、RAA、n1およびn2は式Iに規定されるように定義される。
【化52】
式X
【化53】
式XI
【0207】
式Xおよび/または式XIの一実施形態において、n1は0、1、2または3である。さらなる実施形態において、n1は0、1、2または3であり、一方R3、R4およびR5の各出現は水素である。
【0208】
また、式Xおよび/または式XIのさらなる実施形態において、n2は1、2または3である。式Xおよび/または式XIの他の実施形態において、n1は1、2または3である。また、さらなる実施形態において、n1は1、2または3であり、n2の各出現は1、2、3または4であり、R3、R4およびR5はそれぞれ水素である。
【0209】
式Xおよび/または式XIの一実施形態において、n1は0、1、2または3であり、n2の各出現は1、2または3であり、R3、R4およびR5はそれぞれHである。さらなる実施形態において、n1は2である。
【0210】
式Xおよび/または式XIの好ましい実施形態において、本発明の両方のプロドラッグ部分は1つ、2つ、3つまたは4つのアミノ酸(すなわちn2は1、2、3または4である)を有する。一実施形態において、n1は1または2であり、一方n2の各出現は1、2または3である。
【0211】
式Xおよび/または式XIの一実施形態において、n1は0、1または2であり、n2の少なくとも1つの出現は1または2であり、R3はHである。さらなる実施形態において、R4の少なくとも1つの出現は
【化54】
である。
【0212】
式Xおよび/または式XIの一実施形態において、n1は0、1または2であり、n2の各出現は1、2または3であり、R3はHである。さらなる実施形態において、R4の少なくとも1つの出現は
【化55】
である。
【0213】
式Xおよび/または式XIの好ましい実施形態において、n2は1、2または3であり、一方R1、R2およびR3はHである。他の実施形態において、n2の少なくとも1つの出現は1である。また他の実施形態において、n2の少なくとも1つの出現は2である。さらに式Xおよび/または式XIの他の実施形態において、n2の各出現は1または2であり、RAAの各出現は独立して天然アミノ酸側鎖である。
【0214】
メタ−OHメキシレチンのプロドラッグ
【0215】
上述した式I〜XIの実施形態はメキシレチンおよびp−OHメキシレチンプロドラッグに関するが、本開示はまた他のメキシレチンプロドラッグのプロドラッグを包含する。したがって、m−OHメキシレチンおよびヒドロキシメチルメキシレチンのプロドラッグが本開示の範囲内である。
【0216】
例えば一実施形態において、本発明は式XIIにより包含されるメタ−OHメキシレチンプロドラッグ、
【化56】
式XII
【0217】
またはその薬学的に許容し得る塩に関し、
【0218】
ここで式中のR1は水素および
【化57】
から選択され、
【0219】
R2は、
【化58】
から選択され、
【0220】
O1は未結合メタ−OHメキシレチン中に存在するフェノール系酸素であり、
【0221】
Xは(−NH−)、(−O−)、または欠落であり、
【0222】
RAAの各出現は独立して天然または非天然アミノ酸側鎖であり、
【0223】
n1は0〜16から選ばれる整数であり、
【0224】
n2の各出現は1〜9から選ばれる整数であり、
【0225】
R3の各出現は独立して水素、置換アルキル基または非置換アルキル基から選択され、
【0226】
R4およびR5の各出現は、水素、
【化59】
、置換アルキル基および非置換アルキル基から選択され、
【0227】
n1により定義される炭素鎖における二重結合の場合、二重結合を形成する炭素上にR4は存在し、R5は欠落であり、
【0228】
少なくともR1は
【化60】
であるか、あるいは少なくともR2は
【化61】
である。
【0229】
ジカルボン酸リンカーの一実施形態において、n1は0、1、2、3または4である。
【0230】
一実施形態において、n2の各出現は1、2、3、4および5である。
【0231】
好ましい一実施形態において、本発明の化合物は一つのプロドラッグ部分を有し、該プロドラッグ部分は1つ、2つまたは3つのアミノ酸(すなわちn2が1、2または3である)を有する一方、R3はHである。
【0232】
他の実施形態において、n2は1である。
【0233】
さらに他の実施形態において、n2は2である。
【0234】
さらに他の実施形態において、n2の各出現は1または2であり、RAAの各出現は独立して天然アミノ酸側鎖である。
【0235】
一実施形態において、プロドラッグ部分は、アルギニンもしくはリシンのホモポリマーか、またはアルギニンおよびリシンのヘテロポリマーである。さらなる実施形態において、ホモポリマーまたはヘテロポリマー中に2、3、4、5、6または7のアミノ酸がある(すなわちn2が2、3、4、5、6または7である)。
【0236】
上述した式XIIで提供されるように、本明細書に記載したパラ置換のプロドラッグ部分(例えば、式II〜XIのプロドラッグ)をメタ位にすることもできる。これら実施形態において、パラ位のプロドラッグ部分を水素で置換し、一方プロドラッグ部分をパラ位に隣接する炭素の1つに付加する。カルバメート結合メタ−OHメキシレチンプロドラッグ(式XIII)についての例を以下に示す。
【化62】
【0237】
ヒドロキシメチルメキシレチンのプロドラッグ
【0238】
一実施形態において、本発明は、式XIVにより包含されるヒドロキシメチルメキシレチンプロドラッグ、
【化63】
式XIV
【0239】
またはその薬学的に許容し得る塩に関し、
【0240】
ここで式中のR1は水素および
【化64】
から選択され、
【0241】
R2は、
【化65】
から選択され、
【0242】
O1は未結合ヒドロキシメチルメキシレチン中に存在するフェノール系酸素であり、
【0243】
Xは(−NH−)、(−O−)、または欠落であり、
【0244】
RAAの各出現は独立して天然または非天然アミノ酸側鎖であり、
【0245】
n1は0〜16から選ばれる整数であり、
【0246】
n2の各出現は1〜9から選ばれる整数であり、
【0247】
R3の各出現は独立して水素、置換アルキル基または非置換アルキル基から選択され、
【0248】
R4およびR5の各出現は独立して水素、
【化66】
、置換アルキル基および非置換アルキル基から選択され;二重結合の場合、二重結合を形成する炭素上にR4は存在し、R5は欠落であり、
【0249】
n1により定義される炭素鎖における二重結合の場合、二重結合を形成する炭素上にR4は存在し、R5は欠落であり、
【0250】
少なくともR1は
【化67】
であるか、あるいは少なくともR2は
【化68】
である。
【0251】
ジカルボン酸リンカーの一実施形態において、n1が0、1、2、3または4である。
【0252】
一実施形態において、n2の各出現は1、2、3、4および5である。
【0253】
好ましい一実施形態において、本発明の化合物は一つのプロドラッグ部分を有し、該プロドラッグ部分は1つ、2つまたは3つのアミノ酸(すなわちn2が1、2または3である)を有し、一方R3はHである。
【0254】
他の実施形態において、n2は1である。さらに他の実施形態において、n2は2である。
【0255】
さらに他の実施形態において、n2の各出現は1または2であり、RAAの各出現は独立して天然アミノ酸側鎖である。
【0256】
一実施形態において、プロドラッグ部分は、アルギニンもしくはリシンのホモポリマーか、またはアルギニンおよびリシンのヘテロポリマーである。さらなる実施形態において、ホモポリマーまたはヘテロポリマー中に2、3、4、5、6または7のアミノ酸がある(すなわちn2が2、3、4、5、6または7である)。
【0257】
ここに記載したパラおよびメタ置換のプロドラッグ部分(例えば、式II〜XIIIのプロドラッグ)を、芳香環上のメチル基にあることもできる。これら実施形態において、パラまたはメタ位のプロドラッグ部分を水素で置換し、一方プロドラッグ部分を芳香環上のメチル基に付加する。カルバメート結合ヒドロキシメチルメキシレチンプロドラッグ(式XV)についての例を以下に示す。
【化69】
【0258】
式(II)〜式(XV)のそれぞれについて、式(I)の関連して記載した様々な実施形態もまた適用する(化学的に可能で妥当である場合)が、式(II)〜式(XV)のそれぞれの議論の簡潔さのために単に省略されている。
【0259】
本発明で使用する代表的なアミノ酸およびペプチド
【0260】
以下に記載の代表的なプロドラッグは、p−OHメキシレチンプロドラッグに関する。しかし、同じアミノ酸およびペプチドプロドラッグ部分をm−OHメキシレチンおよびヒドロキシメチルメキシレチンプロドラッグに使用することができる。
【0261】
フェノール系官能基の場合、p−OHメキシレチン代謝物をカルバメートまたはジカルボン酸リンカー(置換または非置換)によりアミノ酸またはペプチドに結合し得る。例えば、マロン酸、コハク酸またはグルタル酸を本発明においてリンカーとして用いることができる。本発明での使用に可能な他のジカルボン酸を表2および表3に示す。p−OHメキシレチン代謝物で用いる好ましいアミノ酸としては、単一のアミノ酸またはジペプチドの一部のいずれかとして結合したバリン、ロイシン若しくはイソロイシンまたは類似物である。例えば、ジペプチドのバリン−バリン、バリン−ロイシン、バリン−イソロイシン、ロイシン−ロイシン、ロイシン−バリン、ロイシン−イソロイシン、イソロイシン−イソロイシン、イソロイシン−バリンおよびイソロイシン−ロイシンを使用することができる。さらにパラ−アミノ安息香酸のような非天然アミノ酸を単独または天然アミノ酸と一緒に使用することができる。
【0262】
特定の理論に拘束されることを望むことなく、メキシレチン若しくはp−OHメキシレチン、m−OHメキシレチンもしくはヒドロキシメチルメキシレチンのプロドラッグのアミノ酸またはペプチド部分は、消化管内で固有のジペプチドおよびトリペプチドトランスポーターPept1を選択的に利用すると考えられる。一度吸収されると、プロドラッグは加水分解を受けて、活性薬剤を全身循環へ放出する。活性薬剤および腸壁間の直接接触の回避は嘔吐のリスクを最小限にする一方、Pept1により助長されたプロドラッグの吸収はより一貫した血漿薬剤濃度を確保する。
【0263】
式Iのメキシレチンプロドラッグの好ましい実施態様は、メキシレチンリジンアミド(慣用名:2,6−ジアミノ−ヘキサン酸[2−(2,6−ジメチルフェノキシ)−1−メチルエチル]−アミド ジトリフルオロ酢酸塩)またはメキシレチンp−OHリジンアミドのトリフルオロ酢酸塩である。
【化70】
メキシレチン−リジンアミドジトリフルオロ酢酸塩
【化71】
p−OHメキシレチン−リジンアミドジトリフルオロ酢酸塩
【0264】
他の実施形態態様において、式Iのメキシレチンプロドラッグはメキシレチングルタミン酸アミドの塩酸塩(慣用名:(S)−4−アミノ−4−[2−(2,6−ジメチル−フェノキシ)−1−メチル−エチルカルバモイル]−酪酸塩酸塩)または対応する活性代謝物p−OHメキシレチンのプロドラッグである。
【化72】
メキシレチングルタミン酸アミド
【0265】
他の実施形態において、式Iのメキシレチンプロドラッグはメキシレチングルタミンアミドの塩酸塩(慣用名:(S)−2−アミノ−ペンタン二酸 5−アミド 1−{[2−(2,6−ジメチル−フェノキシ)−1−メチル−エチル]−アミド}塩酸塩)または対応する活性代謝物p−OHメキシレチンのプロドラッグである。
【化73】
メキシレチングルタミンアミド
【0266】
他の実施形態において、式Iのメキシレチンプロドラッグはメキシレチンホモアルギニンアミドの塩酸塩(慣用名:(S)−2−アミノ−6−グアニジノ−ヘキサン酸[2−(2,6−ジメチル−フェノキシ)−1−メチル−エチル]−アミド二塩酸塩)または対応する活性代謝物p−OHメキシレチンのプロドラッグである。
【化74】
メキシレチンホモアルギニンアミド
【0267】
他の実施形態において、式Iのメキシレチンプロドラッグはメキシレチン塩化メチルメチオニンアミドの塩酸塩(慣用名:((S)−2−アミノ−N−[2−(2,6−ジメチル−フェノキシ)−1−メチル−エチル]−4−(ジメチル−λ4−スルファニルクロライド)−ブチルアミド塩酸塩)または対応する活性代謝物p−OHメキシレチンのプロドラッグである。
【化75】
メキシレチン塩化メチルメチオニンアミド
【0268】
単一のアミノ酸結合体の他の実施形態は、シトルリンまたはオルニチンとのアミド結合体を含む。ジペプチドの好ましい実施形態は、上述したアミノ酸のヘテロまたはホモ二量体を含む結合体である。
【0269】
メキシレチンのオリゴペプチドは、メキシレチンリジンアミド、メキシレチンアルギニンアミド、メキシレチンシトルリンアミドまたはメキシレチンオルニチンアミドへの上述したアミノ酸のいずれかのポリマーの付加により形成することができる。あるいはまた、他の天然または非天然アミノ酸をメキシレチンまたはその活性代謝物に直接結合することができる。メキシレチンプロドラッグのいくつかの例を以下に示す。列挙した第一のアミノ酸は、アミド結合を介してメキシレチンに結合したものである。
【化76】
メキシレチン−Gly−Arg−Arg−Arg
【化77】
メキシレチン−Arg−Arg−Arg−Arg
【化78】
メキシレチン−Lys−Lys−Lys−Lys
【化79】
メキシレチン−Gly−Gly−Arg−Gly
【0270】
p−OH代謝物プロドラッグの好ましい実施形態は、次の2つの単一のアミノ酸(バリンおよびグリシン)の1つまたは両方を有する化合物を含み、以下に示す。
【化80】
4−ヒドロキシメキシレチン−(S)−バリンカルバメートグリシンアミド
【0271】
バリンの代わりの他の単一のアミノ酸は、限定しないが、イソロイシンまたはチロシンを含む一方、グリシンと置き換わるアミノ酸は、限定しないが、オルニチン、シトルリンまたはアルギニンアミドとすることができる。非天然アミノ酸カルバメート結合体は、パラアミノ安息香酸を含む。
【0272】
アミド結合アミノ酸のオリゴペプチドは、上述したように、p−OH代謝物にとってメキシレチン自体に対するものと類似とし得る。
【0273】
好適なアミノ酸は、L−体すべてであるが、本発明はD−体のアミノ酸からなるか、またはD−体およびL−体のアミノ酸混合物からなる式I(または式II〜XV)のプロドラッグも意図している。
【0274】
本発明の化合物の塩、溶媒和物、立体異性体および誘導体
【0275】
以下に記載する代表的な塩は、メキシレチンおよびメキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメチルメキシレチン)のプロドラッグに関する。
【0276】
本発明の方法は、ここに記載したメキシレチン/メキシレチン代謝物のプロドラッグの塩、溶媒和物、立体異性体、例えば、式Iおよび式II〜XVのプロドラッグの塩の使用を包含する。一実施形態において、ここで開示した本発明は、メキシレチンプロドラッグの全ての薬学的に許容し得る塩を包含することを意味する。
【0277】
一般に、本発明の実施に用いるメキシレチンのプロドラッグの薬学的に許容し得る塩は、プロドラッグとそれぞれに見合った所望の酸との反応によって調製される。p−OHメキシレチン代謝物のプロドラッグの場合、これは遊離のカルボキシ官能基の塩を作製することを含むこともできる。かかる塩は、溶液から沈殿させ、濾過により収集するか、または溶媒の蒸発により回収することができる。例えば、塩酸のような酸の水溶液をプロドラッグの水性懸濁液に添加し、生成した混合物を蒸発乾固(凍結乾燥)して酸付加塩を固体として得ることができる。あるいはまた、プロドラッグを適当な溶媒、例えばイソプロパノールのようなアルコールに溶解し、酸を同じ溶媒または他の適当な溶媒に添加することができる。その後、生成した酸付加塩を直接またはジイソプロピルエーテル若しくはヘキサンのようなより低い極性溶媒の添加により沈殿させ、濾過により単離することができる。
【0278】
プロドラッグの酸付加塩は、その遊離塩基形態物を従来方法で塩を生成するに十分な量の所望の酸と接触させることによって調製することができる。遊離塩基形態物は、塩形態物を塩基と接触させ、従来方法で遊離塩基を単離することによって再生することができる。遊離塩基形態物は、それぞれの塩形態物と極性溶媒への溶解性のような特定の物理的性質が若干異なるが、その他の点において塩はそれぞれの遊離塩基と本発明の目的について同等である。
【0279】
p−OHメキシレチン代謝物のプロドラッグの薬学的に許容し得る塩基付加塩は、アルカリ金属およびアルカリ土類金属または有機アミンのような金属またはアミンで形成される。陽イオンとして用いる金属の例としては、ナトリウム、カリウム、マグネシウム、カルシウム等がある。適当なアミンの例としては、N,N'−ジベンジルエチレンジアミン、クロロプロカイン、コリン、ジエタノールアミン、ジシクロヘキシルアミン、エチレンジアミンおよびN−メチルグルカミンがある。
【0280】
酸性化合物の塩基付加塩は、遊離の酸形態物を従来方法で塩を生成するに十分な量の所望の塩基と接触させることによって調製することができる。遊離の酸形態物は、塩形態物を酸と接触させ、遊離酸を単離することによって再生させることができる。
【0281】
本発明の実施に有用なp−OHメキシレチン代謝物の化合物は、塩基性および酸性中心の両方を有し、したがって両性イオンの形態とすることができる。
【0282】
有機化学の当業者は、多くの有機化合物が錯体、すなわち溶媒と反応するかまたは溶媒から沈殿若しくは結晶化した溶媒和物、例えば水との水和物を形成することができることを理解するであろう。本発明に有用な化合物の塩は、有用な水和物などの溶媒和物を形成することができる。溶媒和物の調製技術は当業界で周知である(例えば、Brittain,Polymorphism in Pharmaceutical solids.Marcel Decker,New York(1999)を参照)。本発明の実施に有用な化合物は、一つ以上のキラル中心を持つことができ、また個々の置換基の性質に応じて幾何異性体を有することもできる。
【0283】
ここに記載したメキシレチン(またはメキシレチン代謝物)プロドラッグの個々の異性体は、本発明を実施するために使用することができる。明細書および特許請求の範囲における特定の化合物の記述や命名は、プロドラッグの個々の鏡像異性体並びにプロドラッグの鏡像異性体(ラセミ体またはそれ以外)の混合物の両方を含むことが意図されている。立体化学の決定および立体異性体の溶解のための方法は当業界で周知である。
【0284】
したがって、本発明は式(I)の化合物のいずれの互変異性形態並びに幾何および光学異性体をも包含する。それ故、本発明は式(I)またはその薬学的塩の互変異性体を特に含むことを意図する。
【0285】
本発明の医薬組成物
【0286】
本発明の方法での使用についてプロドラッグをバルク物質として投与し得ることが可能であるが、活性成分を例えば、薬剤が意図された投与の経路および標準的な製薬行為に関して選択された薬学的に許容し得る担体との混合物である医薬製剤で提示するのが好ましい。本発明のいくつかの実施形態において、本発明の組成物は式I〜XVのプロドラッグまたはその薬学的に許容し得る塩から選択したプロドラッグと、薬学的に許容し得る賦形剤とを含む。
【0287】
本発明の製剤は、速放性投薬形態、すなわち、プロドラッグを吸収部位で直ちに放出する投薬形態か、または放出制御投薬形態、すなわち、プロドラッグを所定の期間にわたって放出する投薬形態とすることができる。放出制御投薬形態はあらゆる従来型、例えば、貯蔵若しくはマトリックス型の拡散制御投薬形態;マトリックス、カプセル化若しくは腸溶化の溶解制御投薬形態;または浸透性投形態としてもよい。このような種類の投薬形態は、例えばレミントン「薬学の科学と実務」、第20版、2000年、858−914頁に開示されている。本発明の製剤は、投薬形態および投与量に応じて1日1−6回投与することができる。
【0288】
メキシレチン/p−OHメキシレチン代謝物のアミノ酸およびペプチドプロドラッグの吸収は、Pept1のような活性トランスポーターを経て進展し得る。このトランスポーターは、上部消化管に主に限定されると考えられるので、それ自体消化管の全長に沿って継続的な吸収用の従来の徐放性製剤の有用性を制限する。活性薬剤の継続的な全身発生により血漿中薬剤濃度の持続をもたらさないメキシレチン/p−OHメキシレチン代謝物プロドラッグに対し、プロドラッグの血漿貯蔵、Glumetza(登録商標)またはGluphage XR(登録商標)のようなメトホルミン製品に用いるものと類似の胃保持性または粘膜保持性製剤が有利である。前者は、Gelshield Diffusion Technologyとして既知の薬剤送達システムを活用し、一方後者はいわゆるAcuform送達システムを使用する。いずれにしても、コンセプトは回腸への薬剤伝達を遅らせて、吸収が起こりうる期間を最大化し、血漿中薬剤濃度を効果的に延長することである。消化管に沿った遅延進行を与える他の薬剤送達システムも価値がある。
【0289】
前述の薬剤伝達システムの高度化を必要としないメキシレチンプロドラッグについて、下記のような従来の製剤が適切である。
【0290】
一態様において、本発明は、少なくとも1つの活性な薬学的成分(すなわち、メキシレチンまたはp−OHメキシレチン代謝物のプロドラッグ)またはその薬学的に許容し得る誘導体(例えば、塩または溶媒和物)と、薬学的に許容し得る担体とを含む医薬組成物を提供する。特に、本発明は、治療有効量の本発明の少なくとも一つのプロドラッグまたはその薬学的に許容し得る誘導体と、薬学的に許容し得る担体とを含む医薬組成物を提供する。
【0291】
本発明の方法に対し、本発明で用いるプロドラッグを他の治療法および/または活性剤と組み合わせて使用することができる。したがって、本発明のさらなる態様は、本発明の実施に有用な少なくとも一つの化合物またはその薬学的に許容し得る塩もしくは溶媒和物と、第二の活性剤と、随意的な薬学的に許容し得る担体とを含む医薬組成物を提供する。
【0292】
同じ製剤に組み合わせるとき、2つの化合物が互いにかつ製剤の他の成分の存在下で安定で、また相溶性であると理解されるであろう。別々に剤形化するとき、これらはあらゆる好都合な製剤で、当業界でこの種化合物に既知の方法で都合よく提供することができる。いくつかの実施形態において、2つの化合物は、(1)2つの別のメキシレチンのプロドラッグ、(2)メキシレチンのプロドラッグおよびp−OHメキシレチンのプロドラッグ、(3)2つのp−OHメキシレチンのプロドラッグ、(4)メキシレチンのプロドラッグおよびm−OHメキシレチンのプロドラッグ、(5)メキシレチンのプロドラッグおよびヒドロキシメチルメキシレチンのプロドラッグ、(6)2つのm−OHメキシレチンのプロドラッグ、(7)2つのヒドロキシメチルメキシレチンのプロドラッグ、(8)メタ−OHメキシレチンのプロドラッグおよびp−OHメキシレチンのプロドラッグ、(9)ヒドロキシメチルメキシレチンのプロドラッグおよびp−OHメキシレチンのプロドラッグのいずれかである。他の実施形態において、2つの化合物は式Iのプロドラッグと、別の効能のための他の化合物とを含む。
【0293】
ここで用いるプロドラッグは、ヒト用または獣医用薬での使用にあらゆる簡便な方法で投与するために剤形化することができるので、本発明はヒト用または獣医用薬での使用に適した本発明の化合物を含む医薬組成物を本発明の範囲内で含む。かかる組成物は、一つ以上の適当な担体の援助で従来の方法での使用に提示することができる。治療目的の使用に許容し得る担体は製薬技術分野で周知であり、例えばレミントンの製薬科学(マック出版社、A.R.ジェンナーロ編集、1985年)に記載されている。医薬用担体の選択は、意図する投与経路および標準的な製薬実務に対して行うことができる。加えて、医薬組成物は担体としてあらゆる適当な結合剤、潤滑剤、懸濁剤、コーティング剤および/または可溶化剤を含むことができる。
【0294】
防腐剤、安定剤、染料そしてさらに香料を医薬組成物に付与することができる。防腐剤の例としては、安息香酸ナトリウム、アスコルビン酸およびp−ヒドロキシ安息香酸エステルが挙げられる。抗酸化剤および懸濁化剤を用いることもできる。
【0295】
本発明に用いる化合物は、湿式粉砕のような既知の粉砕手順を用いて粉砕して錠剤形成用およびその他の製剤タイプに適した粒径を得ることができる。化合物の微粉細調製物(ナノ粒子)は、当業界で既知の方法により調製することができ、例えば国際特許出願WO02/00196(SmithklineBeecham)を参照。
【0296】
本発明の化合物および医薬組成物は、経口投与(例えば錠剤、小袋、カプセル、トローチ、丸剤、巨丸剤、散剤、ペースト、顆粒、薬包または予備混合調製、座薬、エリキシル剤、溶液、懸濁液、分散液、ゲル、シロップとしてまたは摂取溶液として)することを意図する。さらに、該化合物は使用前に水または他の適当な媒介物、随意に香料と着色料との構成のために乾燥粉末として存在してもよい。固体および液体組成物は、当業界で周知の方法に従って調製することができる。このような組成物はまた、固体または液体形態が可能な一つ以上の薬学的に許容し得る担体および賦形剤を含むことができる。
【0297】
分散液は、グリセリン、液体ポリエチレングリコール、トリアセチン油およびその混合物のような液体担体または中間体中で調製することができる。液体担体または中間体は、例えば水、エタノール、ポリオール(例えば、グリセロール、プロピレングリコール等)、植物油、非毒性のグリセリンエステルおよびその適当な混合物を含む溶剤または液体分散媒体とすることができる。適当な流動性は、リポソームの発生、分散液の場合適当な粒径の管理によるか、または界面活性剤の添加により維持し得る。
【0298】
錠剤は、微結晶性セルロース、乳糖、クエン酸ナトリウム、炭酸カルシウム、二塩基性リン酸カルシウムおよびグリシンのような賦形剤、デンプン(好ましくはトウモロコシ、ジャガイモまたはタピオカのデンプン)、デンプングリコール酸ナトリウム、クロスカルメロースナトリウムおよび特定の錯体ケイ酸塩のような崩壊剤、並びにポリビニルピロリドン、ヒドロキシプロピルメチルセルロース(HPMC)、ヒドロキシプロピルセルロース(HPC)、ショ糖、ゼラチンおよびアカシアのような顆粒化結合剤を含むことができる。
【0299】
さらに、ステアリン酸マグネシウム、ステアリン酸、ベヘン酸グリセリルおよびタルクのような滑沢剤を含むことができる。
【0300】
本発明に有用な経口組成物用の薬学的に許容し得る崩壊剤の例としては、限定しないが、デンプン、アルファ化デンプン、デンプングリコール酸ナトリウム、カルボキシメチルセルロースナトリウム、クロスカルメロースナトリウム、微結晶性セルロース、アルギン酸塩、樹脂、界面活性剤、発泡性組成物、水溶性ケイ酸アルミニウムおよび架橋ポリビニルピロリドンが挙げられる。
【0301】
本発明に有用な経口組成物用の薬学的に許容し得る結合剤の例としては、限定しないが、アカシア;メチルセルロース、カルボキシメチルセルロース、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロースまたはヒドロキシエチルセルロースのようなセルロース誘導体;ゼラチン、グルコース、デキストロース、キシリトール、ポリメタクリレート、ポリビニルピロリドン、ソルビトール、デンプン、アルファ化デンプン、トラガカント、キサンタン樹脂、アルギン酸塩、ケイ酸マグネシウムアルミニウム、ポリエチレングリコールまたはベントナイトが挙げられる。
【0302】
本発明に有用な経口組成物用の薬学的に許容し得る充填剤の例としては、限定しないが、乳糖、アンヒドロラクトース、ラクトース一水和物、ショ糖、デキストロース、マンニトール、ソルビトール、デンプン、セルロース(特に微結晶性セルロース)、ジヒドロまたは無水リン酸カルシウム、炭酸カルシウムおよび硫酸カルシウムが挙げられる。
【0303】
本発明の組成物に有用な薬学的に許容し得る潤滑剤の例としては、限定しないが、ステアリン酸マグネシウム、タルク、ポリエチレングリコール、エチレンオキサイドのポリマー、ラウリル硫酸ナトリウム、ラウリル硫酸マグネシウム、オレイン酸ナトリウム、フマル酸ステアリルナトリウムおよびコロイド状二酸化ケイ素が挙げられる。
【0304】
経口組成物に適した薬学的に許容し得る匂い物質の例としては、限定しないが、油、花、果物(例えば、バナナ、リンゴ、サワーチェリー、桃)の抽出物およびその組み合わせのような合成芳香油や天然芳香油、ならびに類似の芳香抽出物が挙げられる。その使用は多くの因子に依存するが、医薬組成物を服用する人々のための感覚受容性が最も重要である。
【0305】
経口組成物に適した薬学的に許容し得る染料の例としては、限定しないが、二酸化チタン、β−カロチンおよびグレープフルーツの皮の抽出物のような合成および天然物染料が挙げられる。
【0306】
経口組成物に有用で、通常嚥下を容易にし、放出特性を変更し、外観を向上させ、および/または組成物の味を覆うために用いる薬学的に許容し得るコーティング剤の例としては、限定しないが、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロースおよびアクリル酸−メタクリル酸エステル共重合体が挙げられる。
【0307】
経口組成物に適した薬学的に許容し得る甘味料の例としては、限定しないが、アスパルテーム、サッカリン、サッカリンナトリウム、シクラミン酸ナトリウム、キシリトール、マンニトール、ソルビトール、ラクトースおよびスクロースが挙げられる。
【0308】
本発明に有用な薬学的に許容し得る緩衝液の適当な例としては、限定しないが、クエン酸、クエン酸ナトリウム、重曹、二塩基性リン酸ナトリウム、酸化マグネシウム、炭酸カルシウムおよび水酸化マグネシウムが挙げられる。
【0309】
本発明に有用な薬学的に許容し得る界面活性剤の適当な例としては、限定しないが、ラウリル硫酸ナトリウムおよびポリソルベート類が挙げられる。
【0310】
同様な種類の固体組成物を、ゼラチンカプセル中の充填剤として用いることもできる。これに関して好ましい賦形剤は、ラクトース、デンプン、セルロース、乳糖または高分子量ポリエチレングリコールを含む。水性懸濁液および/またはエリキシル剤の場合、薬剤を種々の甘味剤または芳香剤、着色剤または染料と、乳化剤および/または懸濁化剤と、水、エタノール、プロピレングリコールおよびグリセリンのような希釈剤と、並びにその混合物と組み合わせることができる。
【0311】
薬学的に許容し得る防腐剤の適当な例としては、限定しないが、様々な抗菌性および抗真菌性の薬剤、例えば、エタノール、プロピレングリコール、ベンジルアルコール、クロロブタノール、第四級アンモニウム塩およびパラベン類(例えば、メチルパラベン、エチルパラベン、プロピルパラベン等)のような溶剤が挙げられる。
【0312】
薬学的に許容し得る安定剤や抗酸化剤の適当な例としては、限定しないが、エチレンジアミン四酢酸(EDTA)、チオ尿素、トコフェロールおよびブチルヒドロキシアニソールが挙げられる。
【0313】
本発明の医薬組成物は、体積あたり0.01−99%重量の本発明で包含されるプロドラッグを含有することができる。
【0314】
投薬量
【0315】
特に示されない限り、ここで規定したプロドラッグ投薬量は、メキシレチン遊離塩基の当量を指す。
【0316】
本発明の方法に従って治療すべき適切な被検体は、かかる治療を必要とするあらゆるヒトや動物などである。動物またはヒトが経験する痛みの重篤度を含む痛みの診断と臨床評価の方法は、当業界で周知である。従って、患者が痛みのための治療を必要とするかどうかを判断することは、当業界における通常の施術者(例えば、医師または獣医師)の技量の範囲内である。患者は、好ましくは哺乳動物、より好ましくはヒトであるが、臨床試験またはスクリーニング若しくは動物モデルを用いる活性実験の観点から実験動物を含むあらゆる動物とすることができる。したがって、当業者によって容易に理解されるように、本発明の方法および組成物はあらゆる動物、特に限定しないが、猫科またはイヌ科被検体のような家畜、ウシ、ウマ、ヤギ、ヒツジおよびブタ被検体のような家畜、マウス、ラット、ウサギ、ヤギ、ヒツジ、ブタ、イヌ、ネコのような研究用動物、ニワトリ、七面鳥、小鳥等のような鳥類を含む哺乳動物に投与するのに特に適している。
【0317】
通常、医師は個々の被検体に最も適した実際の投薬量を決定する。任意特定の個人用の具体的な投与度合いと投与頻度は変化し得るもので、使用する特定化合物の活性、該化合物の代謝安定性および作用長さ、年齢、体重、一般的健康、性別、食事、投与の方式および時間、排出速度、薬物の組み合わせ、特定疾患の重篤度並びに個々が受けている治療を含む様々な要因に依存する。
【0318】
FDAが承認したメキシレチン塩酸塩製剤であるMexitil(登録商標)は、150mg、200mgおよび250mgのカプセルで利用可能である。100mgのメキシレチン塩酸塩は83.31mgのメキシレチン塩基に相当する。通常、Mexitil(登録商標)を8時間ごとに投与する。本発明の一実施形態において、プロドラッグ投薬量は、Mexitil(登録商標)の投薬量の1つから選択され、8時間ごとに一回投与することができる。別の実施形態では、プロドラッグ投薬量は、Mexitil(登録商標)の投薬量の1つから選択され、12時間ごとまたは24時間ごとに1回投与することができる。
【0319】
一実施形態において、メキシレチンプロドラッグの有効な毎日の投薬量は、プロドラッグ1mg〜2000mg、好ましくは100mgから2000mgである。例えば、本発明で包含されるプロドラッグは、一日あたり約200mg〜約2000mgのプロドラッグ、好ましくは一日あたり約200mg〜1000mgのプロドラッグを与える投薬形態で製剤化し得る。好ましい実施形態において、本発明のプロドラッグの有効量は、250mg、500mg、750mg/日のいずれかである。
【0320】
別の実施形態において、p−OHメキシレチンプロドラッグの有効な毎日の投薬量は、プロドラッグ4mg〜8000mg、好ましくは400mgから8000mgである。別の実施形態において、p−OHメキシレチンプロドラッグの有効な毎日の投薬量は、1000mgまたは3000mgのいずれかである。
【0321】
治療すべき痛みの重篤度に応じて、当該技術範囲内で容易に決定し得るようで、過度な実験なしに適当な治療に有効かつ安全な投薬量を被検体に投与することができる。ヒトへの経口投与の場合、プロドラッグの毎日の投薬度合いは、単一または分割投与量とすることができる。治療の期間は、当業者によって決定することができ、痛みの性質(例えば、慢性対急性の状態)および/または治療に対する治療応答の速さと程度を反映すべきである。
【0322】
痛みの治療法において、本発明で包含されるプロドラッグを他の治療法と組み合わせておよび/または他の活性薬剤と組み合せて投与することができる。例えば、本発明で包含されるプロドラッグを痛みの管理に用いる他の活性剤と組み合わせて患者に投与することができる。本発明で包含されるプロドラッグと組み合わせて投与すべき活性剤は、例えば、アセトアミノフェンおよびイブプロフェンを含む非ステロイド性抗炎症薬、もしくはオキシコドン、オキシモルフォン、レボルファノールを含むオピオイド、またはオンダンセトロン、ドンペリドン、ヒヨスチンまたはメトクロプラミドのような抗嘔吐剤からなる群から選択した薬剤を含むことかできる。このような併用療法において、本発明で包含されるプロドラッグは、他の治療法および/または活性剤に優先して、同時に、または引き続いて投与しても良い。
【0323】
本発明で包含されるプロドラッグを他の活性剤と組合わせて投与する場合、かかる組み合わせの個々の成分をあらゆる簡便な経路によって個別または一緒にした製剤処方で順次または同時に投与することができる。順次に投与する場合、本発明で包含されるプロドラッグまたは第二の活性剤のいずれかを先に投与することができる。例えば、他の活性剤との併用療法の場合、本発明で包含されるプロドラッグを薬物組み合わせの有益な効果を提供する投薬計画で順次に投与することができる。同時に投与する場合、組み合わせ物を同一または異なる医薬組成物のいずれかで投与することができる。例えば、本発明で包含されるプロドラッグおよび他の活性剤は、これら薬剤の一定割合を有する単一のカプセルもしくは錠剤、または各薬剤に対して複数の別々のカプセルもしくは錠剤のようにして実質的に同時に投与することができる。
【0324】
本発明で包含されるプロドラッグを、痛みの治療方法で活性な他の薬剤と組み合わせて使用する場合、各化合物の投与量は、化合物を単独で使用する場合のものと異なる場合がある。適切な投与量は、当業者により容易に理解されるであろう。
【0325】
本発明の方法
【0326】
本発明の一実施形態は、必要とする被検体の疾患をメキシレチンで治療する方法である。該方法は、本発明のメキシレチンプロドラッグ、その薬学的に許容し得る塩または組成物を必要とする被検体に経口投与することを備え、ここでメキシレチンプロドラッグが少なくとも一つのアミノ酸または長さ2〜9個のアミノ酸のペプチドに共有結合したメキシレチンまたはメキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメチルメキシレチン)からなる。メキシレチンプロドラッグは一または二つのプロドラッグ部分を有する。一つのプロドラッグ部分を、ペプチド結合を介してメキシレチンまたはメキシレチン代謝物のアミノ基に結合することができる。あるいは、またはさらに、メキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメキシレチン)は、フェノール系酸素でカルバメートまたはジカルボン酸リンカーを介して結合したプロドラッグ部分を有することができる。メキシレチンの量は、治療有効量(例えば、鎮痛有効量)であるのが好ましい。疾患は、メキシレチンで治療可能なものとすることができる。例えば、疾患は神経因性疼痛または不整脈であってもよい。
【0327】
本発明のさらなる実施形態において、メキシレチンに伴うGI副作用を誘発することなしに、必要とする被検体の疾患をメキシレチンで治療する方法を提供する。かかる方法は、本発明のメキシレチンプロドラッグ、その薬学的に許容し得る塩または組成物を必要とする被検体に経口投与することを備え、ここでメキシレチンプロドラッグが少なくとも一つのアミノ酸または長さ2〜9個のアミノ酸のペプチドに対し共有結合したメキシレチンまたはメキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメチルメキシレチン)からなり、経口投与するとプロドラッグまたは薬学的に許容し得る塩が、完全に避けることができないとしても、未結合メキシレチンの経口投与後に通常見られる胃腸副作用を最小限にする。メキシレチンプロドラッグは一または二つのプロドラッグ部分を有する。一つのプロドラッグ部分を、ペプチド結合を介してメキシレチンまたはメキシレチン代謝物のアミノ基に結合することができる。あるいは、またはさらに、メキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメキシレチン)は、フェノール系酸素でカルバメートまたはジカルボン酸リンカーを介して結合したプロドラッグ部分を有することができる。メキシレチンの量は、治療有効量(例えば、鎮痛有効量)であるのが好ましい。疾患は、メキシレチンで治療可能なものとすることができる。例えば、疾患は神経因性疼痛または不整脈であってもよい。さらなる実施形態において、メキシレチンの投与に伴うGI副作用は、限定しないが、嘔吐、吐き気、下痢および腹部不快感から選択される。
【0328】
ここに記載したメキシレチンプロドラッグは、メキシレチン塩酸塩のような非プロドラッグのメキシレチン塩形態と比較して、胃腸環境における腸運動に対する有害作用の誘発が統計的に相当に低い(例えば平均値)。
【0329】
本発明の別の態様において、薬物動態を改善し、必要とする被検体におけるメキシレチンの作用持続時間を延ばす方法を提供する。かかる方法は、本発明のプロドラッグまたはその組成物の有効量を必要とする被検体に投与することを備え、ここで血漿中濃度の時間プロファイルを調整して、メキシレチン濃度の初期の急激な上昇を最小限にし、あらゆる不所望な作用を最小限化する一方、薬剤が血漿中に存続する時間(プロドラッグからの継続的発生に起因する)、それ故作用持続時間を著しく延長する。
【0330】
さらなる態様において、メキシレチン血漿濃度の被検体間または被検体内の変動を低減する方法を提供する。かかる方法は、必要とする被検体または被検体群に本発明のプロドラッグまたはその組成物の有効量を投与することを備える。
【0331】
さらなる実施形態において、p−OHメキシレチンまたはm−OHメキシレチンのプロドラッグは前記方法に用いる。
【0332】
他の実施形態において、神経因性疼痛を除去、低減または治療する方法を提供する。該方法は、本発明のメキシレチンプロドラッグ、その薬学的に許容し得る塩または組成物を必要とする被検体に経口投与することを備え、ここでメキシレチンプロドラッグが少なくとも一つのアミノ酸または長さ2〜9個のアミノ酸のペプチドに対し共有結合したメキシレチンまたはメキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメチルメキシレチン)からなる。メキシレチンプロドラッグは一または二つのプロドラッグ部分を有する。一つのプロドラッグ部分を、ペプチド結合を介してメキシレチンまたはメキシレチン代謝物のアミノ基に結合することができる。あるいは、またはさらに、メキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメキシレチン)は、フェノール系酸素でカルバメートまたはジカルボン酸リンカーを介して結合したプロドラッグ部分を有することができる。メキシレチンの量は、治療有効量(例えば、鎮痛有効量)であるのが好ましい。
【0333】
さらなる実施形態において、p−OHメキシレチンまたはm−OHメキシレチンのプロドラッグを前記方法に用いる。
【0334】
本発明の他の実施形態は、被検体間および被検体内におけるメキシレチン血清濃度の変動を低減することに関する。該方法は、本発明のメキシレチンプロドラッグ、その薬学的に許容し得る塩または組成物を必要とする被検体に経口投与することを備え、ここでメキシレチンプロドラッグが少なくとも一つのアミノ酸または長さ2〜9個のアミノ酸のペプチドに対し共有結合したメキシレチンまたはメキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメチルメキシレチン)からなる。メキシレチンプロドラッグは一または二つのプロドラッグ部分を有する。一つのプロドラッグ部分を、ペプチド結合を介してメキシレチンまたはメキシレチン代謝物のアミノ基に結合することができる。あるいは、またはさらに、メキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメキシレチン)は、フェノール系酸素でカルバメートまたはジカルボン酸リンカーを介して結合したプロドラッグ部分を有することができる。メキシレチンの量は、治療有効量(例えば、鎮痛有効量)であるのが好ましい。
【0335】
また、本発明の他の実施形態は、必要とする被検体におけるメキシレチンの生物学的利用能の再現性を向上させることに関する。該方法は、本発明のメキシレチンプロドラッグ、その薬学的に許容し得る塩または組成物を必要とする被検体に経口投与することを備え、ここでメキシレチンプロドラッグが少なくとも一つのアミノ酸または長さ2〜9個のアミノ酸のペプチドに対し共有結合したメキシレチンまたはメキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメチルメキシレチン)からなる。メキシレチンプロドラッグは一または二つのプロドラッグ部分を有する。一つのプロドラッグ部分を、ペプチド結合を介してメキシレチンまたはメキシレチン代謝物のアミノ基に結合することができる。あるいは、またはさらに、メキシレチン代謝物(例えば、p−OHメキシレチン、m−OHメキシレチンまたはヒドロキシメキシレチン)は、フェノール系酸素でカルバメートまたはジカルボン酸リンカーを介して結合したプロドラッグ部分を有することができる。メキシレチンの量は、治療有効量(例えば、鎮痛有効量)であるのが好ましい。
【0336】
さらなる実施形態において、p−OHメキシレチンまたはm−OHメキシレチンのプロドラッグを前記方法に用いる。
【0337】
本発明はまた、一つ以上の原子を同じ原子番号を有するが、自然界で通常見られる原子質量または質量数と異なる原子質量または質量数を有する原子によって置換する式(1)の全ての薬学的に許容し得る同位体標識化合物の合成を含む。
【0338】
本発明の化合物に包含するのに適した同位体の例としては、2Hおよび3Hのような水素、11C、13Cおよび14Cのような炭素、36Clのような塩素、18Fのようなフッ素、123Iおよび125Iのようなヨウ素、13Nおよび15Nのような窒素、15O、17Oおよび18Oのような酸素、32Pのようなリン、そして35Sのような硫黄の同位体が挙げられる。
【0339】
特定の同位体標識化合物、例えば、放射性同位体を組み込んだものは、薬物および/または基質の組織分布研究に有用である。トリチウム、すなわち3Hおよび炭素−14、すなわち14Cの放射性同位体は、取り込みの容易さおよび検出の素早い手段の点で本目的に特に有用である。
【0340】
重水素、すなわち2Hのようなより重い同位体での置換は、より大きな代謝安定性に由来する特定の治療上の利点、例えば、生体内での半減期の増加または必要投与量の減少を付与することができ、従っていくつかの状況では好まれる。
【0341】
11C、18F、15Oおよび13Nのような陽電子放出同位体での置換は、基質の受容体占有率を検討するための陽電子放射トポグラフィー(PET)の研究に役立つ。
【0342】
同位体標識化合物は、一般に当業者に既知の従来技術、または上記で採用した非標識試薬の代わりに適当な同位体標識試薬を用いて説明されるものと類似の方法によって製造することができる。
【0343】
本明細書の説明および特許請求の範囲を通して、単語“備える”および“含む”ならびにその単語の変形、例えば“備えている”および“備える”は、“限定しないが含む”ことを意味し、および他の部分、添加剤、成分、整数または工程を排除することを意図したものではない(および排除しない)。
【0344】
本明細書の説明および特許請求の範囲を通して、文脈上別の解釈をする必要がある場合を除き、単数形は複数形を包含する。特に、文脈上別の解釈をする必要がある場合を除き、不定冠詞が使用されている場合、本明細書が複数形および単数形を意図していると理解されるべきである。
【0345】
本発明の特定の態様、実施形態または実施例と一緒になって記載される特徴、整数、特性、化合物、化学的部分または基は、それに矛盾がない限り、ここに記載される任意の他の態様、実施形態または実施例も適用可能であると理解されるべきである。
【実施例】
【0346】
本発明を、さらに以下の実施例を参照して説明する。しかしながら、これら実施例は上述した実施形態と同様に例示であり、いかなる意味においても本発明の可能とされる範囲を制限するものとして解釈されるべきではないことに留意すべきである。
【0347】
一般的合成方法
【0348】
本発明のメキシレチンプロドラッグの合成は2つの異なる工程で達成することができる。アミノ酸またはペプチドの活性化エステル、例えば、(S)−リジンの活性化エステルのN,N’−ジ−t−ブチルオキシカルボニル−(S)−リジンスクシンイミドを(rac)−メキシレチン塩酸塩にカップリングして、N−保護化プロドラッグの(rac)−メキシレチン−N,N’−ジ−t−ブチルオキシカルボニル−(S)−リジンを得ることができる。その後、かかる化合物をトリフルオロ酢酸で脱保護してプロドラッグを得ることができる。
【0349】
上述のように、活性化リジンを他の活性化アミノ酸またはペプチドに速やかに置換することができる。
【0350】
実施例1:(rac)−メキシレチン−(S)−リジンジトリフルオロ酢酸塩の合成
【0351】
メキシレチン−(S)−リジンジトリフルオロ酢酸塩の合成を2つの異なる工程で達成する。最初に、(S)−リジンの活性化エステルのN,N’−ジ−t−ブチルオキシカルボニル−(S)−リジンスクシンイミドをN−メチルモルホリン(NMM)の存在下で(rac)−メキシレチン塩酸塩にカップリングし、クロマトグラフィーによる精製後、N−保護化プロドラッグの(rac)−メキシレチン−N,N’−ジ−t−ブチルオキシカルボニル−(S)−リジンを得た(スキーム1)。
【0352】
その後、トリフルオロ酢酸を用いてBOC基の脱保護を達成して、高粘性ガラス状油として所望の(rac)−メキシレチン−(S)−リジンジトリフルオロ酢酸塩を得た。かかる油は、高真空下での乾燥により発泡するが、空気中に放置すると崩壊することを確かめた。明確にするために、メキシレチンの一方のエナンチオマーのみを示す。
【化81】
(rac)−メキシレチン(S)−リジンジトリフルオロ酢酸塩の合成経路
【0353】
1H NMR(DMSO−d6)スペクトル
8.60(m、1H、NH)、8.16(br、3H、NH3+)、7.76(br、3H、NH3+)、7.02(m、2H、ArH)、6.92(m、1H、ArH)、4.22(m、1H、□−CH)、3.67(d、J=4.5Hz、CH2)、2.73(m、2H、NCH2)、2.21(s、6H、2×CH3)、1.73(m、2H、CH2)、1.52(m、2H、CH2)、1.30(m、5H、CH3+CH2)。
【0354】
実施例2:(rac)−メキシレチン−グリシントリフルオロ酢酸塩の合成
【0355】
N−t−ブチルオキシカルボニル−グリシンスクシンイミドをNMMの存在下で(rac)−メキシレチン塩酸塩にカップリングし、クロマトグラフィーによる精製後、良好な収率でN−保護化プロドラッグの(rac)−メキシレチン−N−t−ブチルオキシカルボニル−グリシンを得た(スキーム2)。
【0356】
その後、トリフルオロ酢酸を用いてBOC基の脱保護を達成した。ジエチルエーテルでの粉砕およびろ過により、優れた収率で白色固体として所要の(rac)−グリシンメキシレチントリフルオロ酢酸塩を得た(スキーム2)。明確にするために、メキシレチンの一方のエナンチオマーのみをスキーム2に示すことに留意すべきである。
【化82】
スキーム2 グリシン−(rac)−メキシレチントリフルオロ酢酸塩の合成経路
【0357】
トリフルオロ酢酸を用いてBOC基の脱保護をその後達成し、ジエチルエーテルによるろ過により優れた収率で白色固体としてグリシン−(rac)−メキシレチントリフルオロ酢酸塩を得た。
【0358】
1H NMR(DMSO−d6)スペクトル
8.52(d、J=7.8Hz、1H、NH)、8.03(br、3H、NH3+)、7.01(m、2H、ArH)、6.93(m、1H、ArH)、3.64(m、4H、2×CH2)、2.22(s、6H、2×CH3)、1.28(d、J=6.6Hz、3H、CH3)。
【0359】
実施例3:メキシレチン−(S)−ホモアルギニンアミド二塩酸塩の合成
【0360】
メキシレチン−(S)−ホモアルギニンアミド二塩酸塩の合成は4つの異なる工程で達成した。“活性エステル”のN−BOC−(S)−ホモアルギニン−(NO2)N−ヒドロキシスクシンイミドエステルを、N−ヒドロキシスクシンイミドとN−BOC−(S)−ホモアルギニン−(NO2)との間のDCCカップリングを経由して作成した。メキシレチン塩酸塩との反応に続いて、Biotage製Isolera自動クロマトグラフィーシステムを用いる逆相条件下での精製後、N−保護化プロドラッグのN−BOC−(S)−ホモアルギニン−(NO2)−メキシレチンを良好な収率で得た。
【化83】
メキシレチン−(S)−ホモアルギニンアミド二塩酸塩の合成経路
【0361】
パラジウムを炭素に対して用いる接触水素化によりニトロ基を還元して、N−BOC−(S)−ホモアルギニン−メキシレチンを得た。Boc基の除去をトリフルオロ酢酸で達成した。粗生成物をジエチルエーテル中2M塩化水素で塩交換し、Biotage製Isolera自動クロマトグラフィーシステムを用いる逆相条件下で精製して、メキシレチン−(S)−ホモアルギニンアミド二塩酸塩を白色のガラス状固体として得た。
【0362】
1H NMR(DMSO−d6)スペクトル
8.79(d、J=8.1Hz、1H、NH)、8.35(br、3H、NH3+)、7.92(m、1H、NH)、7.02(d、J=7.5Hz、2H、2×ArH)、6.91(m、1H、ArH)、4.20(m、1H、□−CH)、3.82〜3.65(m、3H、CH+OCH2)、3.09(m、2H、□−CH2)、2.22(s、6H、2×CH3)、1.75(m、2H、CH2)、1.49〜1.37(m、4H、2×CH2)、1.28(m、3H、CH3)。
【0363】
実施例4:メキシレチン−(S)−グルタミン酸アミド塩酸塩の合成
【0364】
メキシレチン−(S)−グルタミン酸アミド塩酸塩の合成は、2つの異なる工程で達成した。最初に、(S)−グルタミン酸の“活性化エステル”であるN−BOC−(S)−グルタミン酸(tert−ブチルエステル)N−ヒドロキシスクシンイミドエステルをメキシレチン塩酸塩にカップリングした。この後クロマトグラフィーにより精製して、良好な収率で保護プロドラッグのN−BOC−(S)−グルタミン酸(tert−ブチルエステル)−メキシレチンを得た。
【化84】
メキシレチン−(S)−グルタミン酸アミド塩酸塩の合成経路
【0365】
4M塩化水素のジオキサン溶液を用いてBocおよびtert−ブチル基の脱保護を達成した。粗生成物を逆相条件下でBiotage製Isolera自動クロマトグラフィーシステムを用いて精製して、所望のメキシレチン−(S)−グルタミン酸アミド塩酸塩をガラス質白色固体として得た。
【0366】
1H NMR(DMSO−d6)スペクトル
8.71(m、1H、NH)、8.31(s、3H、NH3+)、7.01(d、J=7.5Hz、2H、2×ArH)、6.91(m、1H,ArH)、4.21(m、1H、グルタミン酸α−CH)、3.67(m、3H、不明瞭、メキシレチンCH+OCH2)、2.36(m、2H、□−CH2)、2.23(s、3H、CH3)、2.21(s、3H、CH3)、1.99(m、2H、CH2)、1.27(d、J=6.6Hz、3H、CH3)。
【0367】
実施例5:メキシレチン−[(S)−S−メチル−メチオニン塩化]アミド塩酸塩
【0368】
メキシレチン−[(S)−S−メチル−メチオニン塩化]アミド塩酸塩の合成を3つの異なる工程で達成した。第一に、(S)−メチオニンの“活性化エステル”であるN−BOC−(S)−メチオニンN−ヒドロキシスクシンイミドエステルをメキシレチン塩酸塩にカップリングして、良好な収率で保護プロドラッグのN−BOC−(S)−メチオニン−メキシレチンを得た。続いて、S−メチル化をメタノール中でヨウ化メチルを用いて達成し、化合物を逆相クロマトグラフィーにより精製して、[N−BOC−(S)−メチオニン−S−メチル−ヨウ化]−メキシレチンを与えた。
【化85】
メキシレチン−[(S)−S−メチル−メチオニン塩化]アミド塩酸塩の合成経路
【0369】
ジオキサン中4M塩化水素を用いてBoc基の脱保護を行い、続いて逆相クロマトグラフィーにより精製して、所望のメキシレチン−[(S)−S−メチル−メチオニン]アミド塩酸塩を得た。
【0370】
1H NMR(DMSO−d6)スペクトル
9.32(bd、1H、NH)、8.73(br、3H、NH3+)、7.02(d、J=7.2Hz、2H、2×ArH)、6.92(m、1H、ArH)、4.20(m、1H、□−CH)、4.06(m、1H、CH)、3.72(m、4H、CH2+OCH2)、3.01(s、3H、S−CH3)、2.98(s、3H、S−CH3)、2.32(不明瞭、2H、CH2)、2.22(s、6H、2×CH3)、1.29(m、3H、CH3)。
【0371】
実施例6:メキシレチン−(S)−グルタミンアミド塩酸塩
【0372】
メキシレチン−(S)−グルタミンアミド塩酸塩の合成を、メキシレチン−(S)−グルタミン酸アミド塩酸塩のものと同様な方法で達成した。この場合、N−Boc−(S)−グルタミンN−ヒドロキシスクシンイミドエステルをメキシレチン塩酸塩にカップリングして、良好な収率でN−保護プロドラッグのN−Boc−(S)−グルタミン−メキシレチンを得た。
【化86】
メキシレチン−(S)−グルタミンアミド塩酸塩の合成経路
【0373】
その後、ジオキサン中で4M塩化水素を用いてBoc基の脱保護を達成して、メキシレチン−(S)−グルタミンアミド塩酸塩を灰色がかった白色固体として得た。
【0374】
1H NMR(DMSO−d6)スペクトル
8.73(d、J=8.1Hz、1H、NH)、8.35(br、3H、NH3+)、7.50(br、1H、0.5NH2)、7.01(d、J=7.5Hz、2H、2×ArH)、6.92(m、2H、ArH+0.5NH2)、4.20(m、1H、α−CH)、3.66(m、3H、不明瞭、CH2+CH)、2.21(m、6H、2×CH3)、1.97(m、2H、□−CH2)、1.28(d、J=6.6Hz、3H、CH3)。
【0375】
化合物の評価
催吐活性または吐き気は、胃内の局所麻酔効果の直接の結果として生じると考えられる。これは、胃内容排出を促進する胃内の低波動運動(“ハウスキーパー”波)の抑制に起因する。局所麻酔効果は、ナトリウムチャンネルの遮断により媒介される。本発明の化合物は、ナトリウムチャンネルに対し高いIC50値を示すかなり乏しい活性を有することにより嘔吐を低減または排除すると考えられている。したがって、メキシレチンのナトリウムチャンネルのブロック効果は、メキシレチン自体の代わりに本発明の化合物を投与することにより一時的に不活性化される。一度かかる化合物を吸収すると、メキシレチンへ定量的に変換され、それにより低減または排除された嘔吐および/または吐き気のメキシレチンで認識されたすべての治療的効果が得られる。以下の実施例に示したIC50値は、化合物の低減された催吐の可能性を実証する(例えば表3の高いIC50値で示される)。
【0376】
実施例7:哺乳類細胞で発現したクローン化Nav1.1チャンネルに対するメキシレチンおよび様々なメキシレチンのアミノ酸プロドラッグの効果
不活性化(一時的に)され、それゆえ腸内で直接の催吐性効果をもたらすのが少ないメキシレチンのアミノ酸プロドラッグを同定する試みにおいて、一連の結合体をその潜在的な局所麻酔活性について生体外で哺乳類細胞に発現したナトリウム1.1チャンネルへの効果を評価することにより検査した。
【0377】
方法
【0378】
hNav1.1試験方法
【0379】
hNav1.1チャンネルcDNA(SCN1A遺伝子)で安定に形質転換したCHO細胞を使用して、hNav1.1チャンネルの潜在的ブロックをグラフ1に示した刺激電圧パターンにより測定し、電圧電位を表4に示す。パルスパターンを二回繰り返し、TA添加の前および添加5分後に3つの試験パルスでのピーク電流振幅を測定した(ITP1、TP11およびITP12)。
【0380】
【化87】
グラフ1 hNav1.x試験方法用の電圧プロトコル
【0381】
【表4】
【0382】
データ解析
【0383】
データ収集および分析は、IonWorks Quattroシステムオペレーションソフトウェア(バージョン2.0.2、Molecular Devices Corporation、ユニオンシティ、カリフォルニア州)を使用して行った。データは漏洩電流に対し補正した。
【0384】
トニックブロックを次のように算出した。
%ブロック(トニック) = (1 - ITP1,TA / ITP1,Control) × 100%
式中のITP1,ControlおよびITP1,TAは、それぞれコントロールおよび試験物の存在下においてTP1により誘導された内部ピークNa+電流である。
10Hzブロック、すなわち刺激周波数10Hzでの周波数依存ブロックを次のように算出した。
%ブロック(10Hz) = (1 - ITP11,TA / ITP11,Control) × 100%
式中のITP11,ControlおよびITP11,TAは、それぞれコントロールおよび試験物の存在下においてTP11により誘導された内部ピークNa+電流である。
不活性化状態ブロックは、調整脱分極パルス(TP11)による試験パルス(TP12)の電流振幅における減少として定義される。不活性化状態ブロックを次のように算出した。
%ブロック(不活性化状態) = (1-ITP12,TA / ITP12,Control) × 100%
式中のITP12,ControlおよびITP12,TAは、それぞれコントロールおよび試験物の存在下においてTP12により誘導された内部ピークNa+電流である。
ブロックについての濃度応答データを次の方程式に合わせた。
%ブロック = { 1 - 1 / [ 1 + ( [試験] / IC50 ) N ] } × 100%
ここで[試験]は試験物の濃度、IC50は50%阻害を産生する試験物の濃度、Nはヒル係数、%ブロックは試験物の各濃度で阻害されたイオンチャンネル電流の百分率である。非線形最小二乗法を、Excel2000(マイクロソフト、レッドモンド、ワシントン州)用のソルバーアドインを用いて解いた。
【0385】
結果
【0386】
表5に見られるように、試験した22化合物のうち7化合物が親化合物の10倍超のIC50値を有し、4化合物が20倍より高い効能を有した。これらは、ホモアルギニン、アルギニン、グルタミン酸および塩化S−メチルメチオニン複合体であり、メキシレチン自体の38μMと比較すると、それぞれ624、811、>1000μMおよび>1000μM(10Hzブロック)の値であった。効能におけるこのような減少は、胃/腸の上皮表面への直接作用および生成する催吐の可能性を減少すると期待される。
【0387】
【表5−1】
【表5−2】
【0388】
実施例8:イヌにおける様々なメキシレチンプロドラッグ由来のメキシレチンの全身アベイラビリティの評価
【0389】
方法
【0390】
試験物質(すなわち、メキシレチンおよび様々なメキシレチンのアミノ酸プロドラッグ)をイヌ五頭のグループに強制経口投与した。試験動物の特徴を下記の表6に示す。
【0391】
【表6】
【0392】
血液サンプルを投与後様々な時間で採取し、確証されたLC−MS−MSアッセイを用いる親薬物用の分析に提出した。血漿分析データに由来した薬物動態パラメータを、Win Nonlinを用いて求めた。結果を表7に与える。
【0393】
結果
【0394】
データは、試験した様々なアミノ酸プロドラッグ由来のメキシレチンの全身アベイラビリティについて大幅に変動があることを示す。例えば、ニコチン酸アミドおよびイソニコチン酸アミド由来のものについて、メキシレチンに関して無視できるような生物学的利用能であった。一方、グルタミンアミドプロドラッグの経口投与はほぼ完璧な生物学的利用能となった。
【0395】
【表7】
【0396】
実施例9:カニクイザルにおける様々なメキシレチンプロドラッグ由来のメキシレチンの全身アベイラビリティの評価
【0397】
方法
【0398】
試験物質(すなわち、メキシレチンおよび様々なメキシレチンのアミノ酸プロドラッグ)を、5頭のオスのカニクイザルのグループに強制経口投与した。血液サンプルを投与後様々な時間で採取し、確証されたLC−MS−MSアッセイを使用して親薬物用の分析に提出した。血漿分析データから導かれた薬物動態パラメータを、Win Nonlinを用いて求めた。結果を表8に示す。
【0399】
結果
【0400】
イヌにおけるデータのように、試験された様々なアミノ酸プロドラッグ由来のメキシレチンの全身アベイラビリティにおける著しい変動を示す。再びニコチン酸アミドおよびイソニコチン酸アミド由来のものについて、メキシレチンに関し生物学的利用能は無視できるほど小さかったが、グルタミンアミドプロドラッグはほぼ完全な生物学的利用能となった。
【0401】
【表8】
【0402】
実施例10:ウサギの胃平滑筋の収縮に対するメキシレチンおよびメキシレチングリシンおよびリジンアミドの影響
【0403】
低減したナトリウムチャンネルブロック特性を有するメキシレチンの二つの原型アミノ酸結合体(メキシレチングリシンおよびリジンアミド)を用いて、ウサギの胃平滑筋に対するこれら結合体とメキシレチンとの直接的な影響の比較を検討した。かかる直接的な影響の大きさは、メキシレチンに伴う催吐性の決定要因となると期待される。従って、EFSで刺激した胃平滑筋に対する直接的な影響の減少は、より低い催吐応答になると期待される。
【0404】
方法
【0405】
胃の幽門洞領域から切断したウサギの胃平滑筋全層(粘膜も無処理)の細長い一片を白金環電極の間に装着した。組織を約1gの一定張力まで伸ばし、力発生の変化を高感度変換機を用いて記録した。
【0406】
組織を14Hzで0.5ミリ秒のパルス幅の電場刺激(EFS)に置きながら、刺激に対する最適電圧を求めた。その後、パルスの伝達を50秒ごとに20秒間続けた。
【0407】
最適電圧でのEFSをプロトコル(安定した応答=“EFSのベースラインの測定”)の間中に継続した。
【0408】
採用した試験条件は以下のとおりである。
(1)媒体(脱イオン水、試験物と等容量で添加した)
(2)7つの濃度のメキシレチン(10nM、100nM、1mM、3mM、10mM、30mM、100mM)
(3)7つの濃度のメキシレチン−リジン−アミド(10nM、100nM、1mM、3mM、10mM、30mM、100mM)
(4)7つの濃度のメキシレチン−グリシン−アミド(10nM、100nM、1mM、3mM、10mM、30mM、100mM)
【0409】
ベースラインEFSの10分後、試験物または媒体(脱イオン水)の最初の添加を行った。
【0410】
試験濃度は、各添加の間のPBS洗浄で累積的に加えた。
【0411】
試験濃度は、各添加の間のPSS洗浄で非累積的に加えた。次に、TTX(Na+チャネル遮断薬)を試料に添加して、EFS反応が神経刺激により誘発されたことを確認し、並びにナトリウムチャネル遮断薬の活性(メキシレチンと同様な機構)を確認した。その後、EFSを停止した。
【0412】
結果
【0413】
この調査の結果は、ウサギの胃平滑筋に対するメキシレチン自体並びにメキシレチンリジンアミドおよびメキシレチングリシンアミドの影響の明らかな違いを示す図1に要約される。3つの化合物すべてが、EFSで誘発したウサギの胃の収縮を徐々に減少させる一方、プロドラッグ複合体は著しく低い効能であった。計算したED50値は、メキシレチン、メキシレチングリシンアミドおよびメキシレチンリジンアミドについてそれぞれ2.17、9.16および21.83μMであった。この機能解析における効能の減少の大きさは、Nav1.1チャネルの遮蔽の生体外での評価中に観察されたものと一致し、後者が胃上皮への影響の良い指標であることを示唆する。胃の筋肉に対する直接作用の能力の減少は、プロドラッグに対する直接的に媒介される催吐性応答の可能性を最小限に抑えることができる。
【0414】
実施例11:メキシレチンおよびメキシレチン−グリシン−アミドをフェレットへの経口投与した後の催吐性に対する影響の評価
【0415】
ナトリウムチャネルのブロック特性を低減したメキシレチンの原型アミノ酸結合体(メキシレチン−グリシン−アミド)を使用して、フェレットにおけるそれらとメキシレチンとの催吐性に対する影響の比較を検討した。
【0416】
方法
【0417】
オスのフェレット(n=7)をそれぞれの試験日の前日の午後遅くまでペレット飼料に自由に採食できる状態にした。その後、飼料を取り除き、フェレットを一晩飢餓状態にした。飼料を嘔吐観察の終了後まで戻さなかった。試験の朝に、20mg/kgのメキシレチン塩酸塩溶液または等モル量のメキシレチングリシンアミドのいずれかを、5mL/kgの一定投与容積を用いて動物に経口投与した。経口処理後2時間継続的に動物を観察し、吐き気および嘔吐の全ての発生を記録した。
【0418】
結果
【0419】
表9および10に表される結果は、親化合物を投与した後に見られるものと比較して、プロドラッグを与えた後では、嘔吐の頻度および持続時間が著しく減少したことを示す。プロドラッグの投与後の嘔吐の平均値は、親薬物を投与後に観察されるものの30%未満に低下した。同様に、嘔吐の持続時間は、プロドラッグの投与後では大きく低減し、メキシレチン自体の投与後に見られるものの30%未満になった。可能性として、これらのデータは、この原型メキシレチンアミノ酸プロドラッグについて、吐き気および嘔吐を生じさせるような効能が低減することを示し、ヒトにおいて改善された有効性および患者の服薬遵守につながると期待される。
【0420】
【表9】
【0421】
【表10】
【0422】
実施例12:フェレットにおける親薬物由来とメキシレチングリシンアミド由来とのメキシレチンの全身アベイラビリティの評価
【0423】
原型プロドラッグのメキシレチングリシンアミドに伴う催吐性低減への影響が、単純に、薬剤の低全身アベイラビリティの結果でないことを確かめるために、薬物動態の比較を行った。
【0424】
方法
【0425】
試験物質(すなわち、メキシレチンおよび様々なメキシレチンのアミノ酸プロドラッグ)を、6頭のフェレットのグループに強制経口投与した。
【0426】
血液サンプルを投与後様々な時間で採取し、確証されたLC−MS−MSアッセイを使用してプロドラッグおよび親薬物用の分析に提出した。血漿分析データから導かれた薬物動態パラメータを、Win Nonlinを用いて求めた。
【0427】
結果
【0428】
結果を表11に示す。薬剤自体またはグリシンアミドプロドラッグのどちらかを与えた後の薬剤への全身曝露の比較では、メキシレチンへの全暴露が比較可能であることを示した。表11に示すように、グリシンプロドラッグ由来のメキシレチンの生物学的利用能の平均相対値が、親分子を与えた後の場合の94%であり、プロドラッグに伴う嘔吐の減少は薬剤への乏しい全身暴露に起因するものではないことの確信が得られた。
【0429】
【表11】
【0430】
本発明は、上述したように式I
【化88】
式1
のメキシレチンおよびp−OHメキシレチンのプロドラッグに関する。
本発明はまた、式(I)の化合物および薬学的に許容し得る賦形剤を含む組成物に関する。
本発明はまた、メキシレチンに伴うGI副作用を誘発することなく痛みの治療に使用するための式(I)の化合物に関する。
メキシレチンに伴う胃腸副作用は、吐き気、消化不良、嘔吐、下痢、便秘またはこれらの副作用の組み合わせである。
治療する痛みは、糖尿病性神経障害、急性および慢性神経痛、アルコール性多発ニューロパチー、放射線治療由来の慢性疼痛、視床痛や糖尿病性の体幹の痛み、神経痛、肢端紅痛症、慢性特発感覚性多発ニューロパチー若しくはがんおよびその治療に伴う痛みに起因する痛みのような神経因性疼痛である。
プロドラッグは、メキシレチンリジンアミド、メキシレチンホモアルギニンアミド、メキシレチングルタミン酸アミド、メキシレチングルタミンアミドまたはメキシレチンメチルメチオニンアミドとすることができる。
【特許請求の範囲】
【請求項1】
次式(I)
【化1】
式I
(式中のR1は水素および
【化2】
から選択され、
R2は水素、
【化3】
から選択され、
O1は未結合p−OHメキシレチン中に存在するフェノール系酸素であり、
Xは(−NH−)および(−O−)から選択されるか、またはXは欠落であり、
RAAの各出現は独立して天然または非天然アミノ酸側鎖であり、
n1は0〜16から選択される整数であり、
n2の各出現は独立して1〜9から選択される整数であり、
R3の各出現は独立して水素および任意に置換したアルキル基から選択され、
R4およびR5の各出現は独立して水素、
【化4】
および任意に置換したアルキル基から選択され、
ここでn1により定義された炭素鎖に生ずる二重結合の場合、二重結合を形成する炭素上にR4は存在し、R5は欠落であり、
但し少なくともR1は
【化5】
であるか、あるいは少なくともR2は
【化6】
のいずれか一方であり、次の化合物:HBr.グリシン−(rac)メキシレチン;AcOH.アスパラギン−(rac)メキシレチン;TFA.トリプトファン−(rac)メキシレチン;HBr.アラニン−(rac)メキシレチン;AcOH−フェニルアラニン−(rac)メキシレチンまたはTFA.トリプトファン−(rac)メキシレチン.H2Oの一つでない)の化合物またはその薬学的に許容し得る塩。
【請求項2】
前記R1が水素であり、R2が
【化7】
からなる群より選択される請求項1に記載の化合物。
【請求項3】
前記R2が
【化8】
である請求項2に記載の化合物。
【請求項4】
前記R2が水素であり、R1が
【化9】
である請求項1に記載の化合物。
【請求項5】
前記R3の各出現が水素である請求項1〜4のいずれかに記載の化合物。
【請求項6】
前記R3の各出現が独立して非置換C1−6アルキルから選択される請求項1〜4のいずれかに記載の化合物。
【請求項7】
前記R4が、各出現で水素またはC1−4アルキルである請求項4〜6のいずれかに記載の化合物。
【請求項8】
前記R5が、各出現で水素またはC1−4アルキルである請求項4〜6のいずれかに記載の化合物。
【請求項9】
前記各R5が水素である請求項8に記載の化合物。
【請求項10】
前記式(I)の化合物が、次の化合物:
アラニン−(rac)メキシレチンアミド;
β−メトキシアスパラギン酸−(rac)メキシレチンアミド;
α−メトキシアスパラギン酸−(rac)メキシレチンアミド;
アスパラギン−(rac)メキシレチンアミド;
グリシン−(rac)メキシレチンアミド;
ロイシン−(rac)メキシレチンアミド;
メチオニン−(rac)メキシレチンアミド;
(rac)メチオニン−(rac)メキシレチンアミド;
フェニルアラニン−(rac)メキシレチンアミド;
アラニングリシングリシン−(rac)メキシレチンアミドまたは
トリプトファン−(rac)メキシレチンの一つでない前記請求項のいずれかに記載の化合物。
【請求項11】
前記式(I)の化合物が一つのプロドラッグ部分を有し、該プロドラッグ部分が1つ、2つまたは3つのアミノ酸を有し、式中のn2が1、2または3である前記請求項のいずれかに記載の化合物。
【請求項12】
前記RAAの各出現が独立してアミノ酸側鎖であり、アミノ酸残基が1〜20個の炭素原子有するか、または該残基が水素である前記請求項のいずれかに記載の化合物。
【請求項13】
前記RAAが天然に存在するアミノ酸である請求項12に記載の化合物。
【請求項14】
メキシレチンサルコシンアミド;
メキシレチンスレオニンアミド;
メキシレチンヒスチジンアミド;
メキシレチンセリンアミド;
メキシレチン2−メチルβアラニンアミド;
メキシレチングリシンアミド;
メキシレチングルタミンアミド;
メキシレチンシクロプロピルグリシンアミド;
メキシレチンβアミノアラニンアミド;
メキシレチンニコチン酸アミド;
メキシレチンシトルリンアミド;
メキシレチンイソニコチン酸アミド;
メキシレチンホモアルギニンアミド;
メキシレチンアルギニンアミド;
メキシレチングルタミン酸アミドおよび
メキシレチンS−メチル−塩化メチオニンアミドからなる群より選択される式(I)の化合物。
【請求項15】
医薬に用いる式(I)の化合物。
【請求項16】
メキシレチンと比較して同時に低減した胃腸副作用を有する痛みおよび/または不整脈の治療用の式(I)の化合物。
【請求項17】
前記メキシレチンの投与に伴う胃腸副作用が、嘔吐、吐き気、下痢および腹部不快感からなる群より選択される請求項16に記載の化合物。
【請求項18】
式(I)の化合物と、一つ以上の医薬賦形剤とを含む組成物。
【請求項19】
次式(IA)
【化10】
式IA
(式中のR1が
【化11】
であり、
R2が水素および
【化12】
からなる群より選択され、
RAAの各出現が独立して1〜20個の炭素原子を有する天然または非天然アミノ酸側鎖若しくは水素であり、
n2が1、2または3から選択される整数であり、
R3が水素および任意に置換したC1〜4アルキル基からなる群より選択され、
但し次の化合物:HBr.グリシン−(rac)メキシレチン、AcOH;アスパラギン−(rac)メキシレチン;TFA.トリプトファン−(rac)メキシレチン;HBr.アラニン−(rac)メキシレチン;AcOH.−フェニルアラニン−(rac)メキシレチンおよびTFA.トリプトファン−(rac)メキシレチン.H2Oの一つでない)の化合物またはその薬学的に許容し得る塩。
【請求項20】
前記n2が1である請求項19に記載の式(IA)の化合物。
【請求項21】
前記RAAが天然アミノ酸側鎖である請求項19または20に記載の式(IA)の化合物。
【請求項22】
メキシレチンピペコリン酸アミドおよびメキシレチンジメチルグリシンアミドからなる群より選択される式(IA)の化合物。
【請求項23】
痛みおよび/または不整脈の治療用の請求項19〜22のいずれかに記載の式(IA)の化合物。
【請求項1】
次式(I)
【化1】
式I
(式中のR1は水素および
【化2】
から選択され、
R2は水素、
【化3】
から選択され、
O1は未結合p−OHメキシレチン中に存在するフェノール系酸素であり、
Xは(−NH−)および(−O−)から選択されるか、またはXは欠落であり、
RAAの各出現は独立して天然または非天然アミノ酸側鎖であり、
n1は0〜16から選択される整数であり、
n2の各出現は独立して1〜9から選択される整数であり、
R3の各出現は独立して水素および任意に置換したアルキル基から選択され、
R4およびR5の各出現は独立して水素、
【化4】
および任意に置換したアルキル基から選択され、
ここでn1により定義された炭素鎖に生ずる二重結合の場合、二重結合を形成する炭素上にR4は存在し、R5は欠落であり、
但し少なくともR1は
【化5】
であるか、あるいは少なくともR2は
【化6】
のいずれか一方であり、次の化合物:HBr.グリシン−(rac)メキシレチン;AcOH.アスパラギン−(rac)メキシレチン;TFA.トリプトファン−(rac)メキシレチン;HBr.アラニン−(rac)メキシレチン;AcOH−フェニルアラニン−(rac)メキシレチンまたはTFA.トリプトファン−(rac)メキシレチン.H2Oの一つでない)の化合物またはその薬学的に許容し得る塩。
【請求項2】
前記R1が水素であり、R2が
【化7】
からなる群より選択される請求項1に記載の化合物。
【請求項3】
前記R2が
【化8】
である請求項2に記載の化合物。
【請求項4】
前記R2が水素であり、R1が
【化9】
である請求項1に記載の化合物。
【請求項5】
前記R3の各出現が水素である請求項1〜4のいずれかに記載の化合物。
【請求項6】
前記R3の各出現が独立して非置換C1−6アルキルから選択される請求項1〜4のいずれかに記載の化合物。
【請求項7】
前記R4が、各出現で水素またはC1−4アルキルである請求項4〜6のいずれかに記載の化合物。
【請求項8】
前記R5が、各出現で水素またはC1−4アルキルである請求項4〜6のいずれかに記載の化合物。
【請求項9】
前記各R5が水素である請求項8に記載の化合物。
【請求項10】
前記式(I)の化合物が、次の化合物:
アラニン−(rac)メキシレチンアミド;
β−メトキシアスパラギン酸−(rac)メキシレチンアミド;
α−メトキシアスパラギン酸−(rac)メキシレチンアミド;
アスパラギン−(rac)メキシレチンアミド;
グリシン−(rac)メキシレチンアミド;
ロイシン−(rac)メキシレチンアミド;
メチオニン−(rac)メキシレチンアミド;
(rac)メチオニン−(rac)メキシレチンアミド;
フェニルアラニン−(rac)メキシレチンアミド;
アラニングリシングリシン−(rac)メキシレチンアミドまたは
トリプトファン−(rac)メキシレチンの一つでない前記請求項のいずれかに記載の化合物。
【請求項11】
前記式(I)の化合物が一つのプロドラッグ部分を有し、該プロドラッグ部分が1つ、2つまたは3つのアミノ酸を有し、式中のn2が1、2または3である前記請求項のいずれかに記載の化合物。
【請求項12】
前記RAAの各出現が独立してアミノ酸側鎖であり、アミノ酸残基が1〜20個の炭素原子有するか、または該残基が水素である前記請求項のいずれかに記載の化合物。
【請求項13】
前記RAAが天然に存在するアミノ酸である請求項12に記載の化合物。
【請求項14】
メキシレチンサルコシンアミド;
メキシレチンスレオニンアミド;
メキシレチンヒスチジンアミド;
メキシレチンセリンアミド;
メキシレチン2−メチルβアラニンアミド;
メキシレチングリシンアミド;
メキシレチングルタミンアミド;
メキシレチンシクロプロピルグリシンアミド;
メキシレチンβアミノアラニンアミド;
メキシレチンニコチン酸アミド;
メキシレチンシトルリンアミド;
メキシレチンイソニコチン酸アミド;
メキシレチンホモアルギニンアミド;
メキシレチンアルギニンアミド;
メキシレチングルタミン酸アミドおよび
メキシレチンS−メチル−塩化メチオニンアミドからなる群より選択される式(I)の化合物。
【請求項15】
医薬に用いる式(I)の化合物。
【請求項16】
メキシレチンと比較して同時に低減した胃腸副作用を有する痛みおよび/または不整脈の治療用の式(I)の化合物。
【請求項17】
前記メキシレチンの投与に伴う胃腸副作用が、嘔吐、吐き気、下痢および腹部不快感からなる群より選択される請求項16に記載の化合物。
【請求項18】
式(I)の化合物と、一つ以上の医薬賦形剤とを含む組成物。
【請求項19】
次式(IA)
【化10】
式IA
(式中のR1が
【化11】
であり、
R2が水素および
【化12】
からなる群より選択され、
RAAの各出現が独立して1〜20個の炭素原子を有する天然または非天然アミノ酸側鎖若しくは水素であり、
n2が1、2または3から選択される整数であり、
R3が水素および任意に置換したC1〜4アルキル基からなる群より選択され、
但し次の化合物:HBr.グリシン−(rac)メキシレチン、AcOH;アスパラギン−(rac)メキシレチン;TFA.トリプトファン−(rac)メキシレチン;HBr.アラニン−(rac)メキシレチン;AcOH.−フェニルアラニン−(rac)メキシレチンおよびTFA.トリプトファン−(rac)メキシレチン.H2Oの一つでない)の化合物またはその薬学的に許容し得る塩。
【請求項20】
前記n2が1である請求項19に記載の式(IA)の化合物。
【請求項21】
前記RAAが天然アミノ酸側鎖である請求項19または20に記載の式(IA)の化合物。
【請求項22】
メキシレチンピペコリン酸アミドおよびメキシレチンジメチルグリシンアミドからなる群より選択される式(IA)の化合物。
【請求項23】
痛みおよび/または不整脈の治療用の請求項19〜22のいずれかに記載の式(IA)の化合物。
【図1】

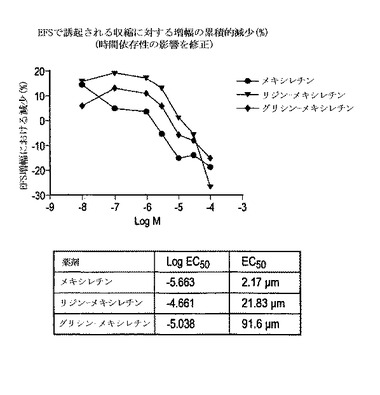
【公表番号】特表2012−530764(P2012−530764A)
【公表日】平成24年12月6日(2012.12.6)
【国際特許分類】
【出願番号】特願2012−516754(P2012−516754)
【出願日】平成22年6月24日(2010.6.24)
【国際出願番号】PCT/EP2010/059039
【国際公開番号】WO2010/149760
【国際公開日】平成22年12月29日(2010.12.29)
【出願人】(507055017)シャイア エルエルシー (29)
【Fターム(参考)】
【公表日】平成24年12月6日(2012.12.6)
【国際特許分類】
【出願日】平成22年6月24日(2010.6.24)
【国際出願番号】PCT/EP2010/059039
【国際公開番号】WO2010/149760
【国際公開日】平成22年12月29日(2010.12.29)
【出願人】(507055017)シャイア エルエルシー (29)
【Fターム(参考)】
[ Back to top ]