
メシル酸サキナビル経口投与剤型
【課題】メシル酸サキナビルの均一で迅速な溶解特性を有する固形単位経口投与剤型の提供。
【解決手段】遊離塩基に換算して250〜800mg量の微粉化メシル酸サキナビル、ならびに薬剤的に許容しうる結合剤、崩壊剤および水溶性担体を含んで提供される。メシル酸サキナビルの固形単位剤型が、メシル酸塩を基準にして60〜80%の微粉化メシル酸サキナビル、4〜8%の水溶性結合剤、崩壊剤および担体(それぞれのパーセントは、核剤重量の内訳パーセントである)を含んで提供される。
【解決手段】遊離塩基に換算して250〜800mg量の微粉化メシル酸サキナビル、ならびに薬剤的に許容しうる結合剤、崩壊剤および水溶性担体を含んで提供される。メシル酸サキナビルの固形単位剤型が、メシル酸塩を基準にして60〜80%の微粉化メシル酸サキナビル、4〜8%の水溶性結合剤、崩壊剤および担体(それぞれのパーセントは、核剤重量の内訳パーセントである)を含んで提供される。
【発明の詳細な説明】
【技術分野】
【0001】
メシル酸サキナビルは、ウイルスの複製を制限し、HIV感染個体における免疫機能を改善させるために用いられる数種のプロテアーゼ阻害剤の一つである。メシル酸サキナビルは、200mgカプセル(サキナビル遊離塩基に換算して)として市販されている。これは、INVIRASE(登録商標)の名称でHoffmann-La Roche,Inc.によって販売されており、進行性ヒト免疫不全ウイルス(HIV)感染症の、選ばれた患者群における治療に、抗レトロウイルス性ヌクレオシド同族体と組み合わせて使用するように指示されている。
【0002】
メシル酸サキナビルは、分子量766.96を有する白色〜灰色がかった白色の、非常に微細な結晶性粉末である。遊離塩基の分子量は670.86である。この薬物は高度に疎水性である。INVIRASE(登録商標)(メシル酸サキナビル)の200mgカプセルは、低い経口バイオアベイラビリティーを有するが、これは不完全な吸収および大きな初回通過代謝によるものと考えられている。[Physician's Desk Reference, 57th Ed.(2003)。]メシル酸サキナビルは、非常に低い水溶解性を有する(すなわち、25℃では、水中で2.2mg/mL、模擬胃液中で0.08mg/mL、そして模擬腸液中では事実上不溶性である)。その上、この薬物はpH依存溶解特性を示し、模擬胃液中では限られた溶解性を有する一方、模擬腸液中では事実上不溶である。高脂肪朝食後にサキナビル600mg量(3×200mgカプセル)を1回投与した健康な有志者8人における絶対バイオアベイラビリティーは、平均で4%(CV73%、範囲:1〜9%)であった。より高カロリーの食事を摂った後のサキナビル24時間AUCおよびCmax(n=6)は、より低カロリーの食事を摂った後より、平均で2倍高かった。食事の影響は、2時間後まで持続することが示された。ヒトで観察される、メシル酸サキナビルの対象間変動および食物効果の最小化を企図するには、メシル酸サキナビルの均一で迅速な溶解特性を有する固形経口投与剤型が望まれる。
【0003】
粒子サイズを小さくすることは、対象間変動を最小化し、そしてバイオアベイラビリティーを改善するための一手段である。微粉化は、粒子サイズを減少させるために用いられる一つのアプローチである。しかしながらメシル酸サキナビルは、微粉化に際し、凝塊化して溶解媒体と接触する一次粒子表面積を減少させてしまう傾向がある。微粉化したメシル酸サキナビルは、遅い溶解速度を示す。
【0004】
成人患者用の、INVIRASE(登録商標)の、ヌクレオシド同族体との組み合わせにおける推奨投与量は、3×200mgカプセルを1日3回、食後2〜4時間以内の服用である。この投与則から見て、患者がこれを遵守するかどうかが真の懸念材料である。治療の成功は、たとえば1日に服用すべき単位数を減らすというように、よりよい遵守を促すことによって、よりよく改善できるかもしれない。したがって、サキナビルのより多い量を含有する単位剤型が有用であろうという期待はある。しかしながら、微粉化メシル酸サキナビルの薬充填量を増やすと、薬剤凝塊の問題は悪化する。
【0005】
本発明は、メシル酸塩を基準にして約60〜約80%の微粉化メシル酸サキナビル、約4〜約8%の薬剤的に許容しうる水溶性結合剤、薬剤的に許容しうる崩壊剤、および薬剤的に許容しうる担体(それぞれのパーセントはその剤型の核剤重量の内訳パーセントである)を含む、メシル酸サキナビルの固形単位経口投与剤型を提供するものである。(微粉化メシル酸サキナビル約60〜約80%は、サキナビル遊離塩基に換算して約200〜約800mg量のメシル酸サキナビルに相当する。)
【0006】
好ましい態様において、メシル酸サキナビルは、固形単位経口投与剤型の核剤重量の約70〜75%である。
【0007】
本発明は、遊離塩基に換算して約250〜約800mg量の微粉化メシル酸サキナビル、薬剤的に許容しうる結合剤、薬剤的に許容しうる崩壊剤、および薬剤的に許容しうる水溶性担体を含む、メシル酸サキナビルの固形単位経口投与剤型を提供する。
【図面の簡単な説明】
【0008】
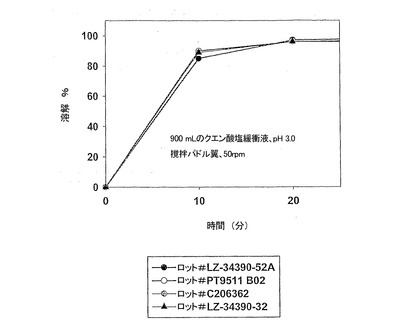
【図1】実施例1の錠剤剤型(遊離塩基換算で500mg)における、本発明製剤のロット間再現性を示す溶解特性を示す。
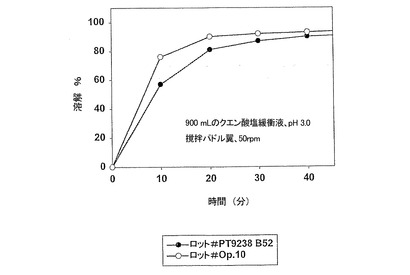
【図2】実施例2のカプセル剤型(遊離塩基換算で200mg)における、現行市販製剤のロット間バラツキを示す溶解特性を示す。
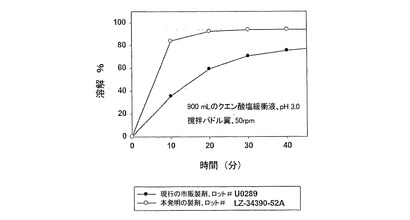
【図3】遊離塩基換算でサキナビル1,000mgの投与量において、錠剤剤型における本発明製剤(実施例1)の溶解特性を、カプセル剤型における現行市販製剤(実施例2)と比較して示す。
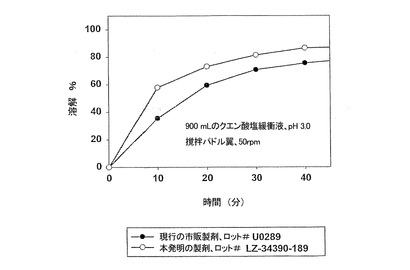
【図4】遊離塩基換算でサキナビル1,000mgの投与量において、カプセル剤型における本発明製剤(実施例3)の溶解特性を、カプセル剤型における現行市販製剤(実施例2)と比較して示す。
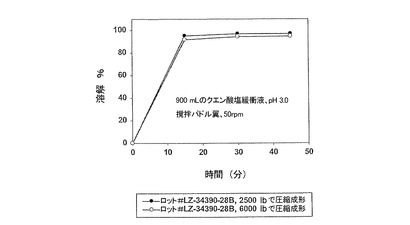
【図5】加えた圧縮力に関係なく、迅速で、高度に再現性のある溶解特性が得られることを表す、実施例1の錠剤剤型(サキナビル遊離塩基換算で500mg)における本発明製剤の溶解特性を示す。
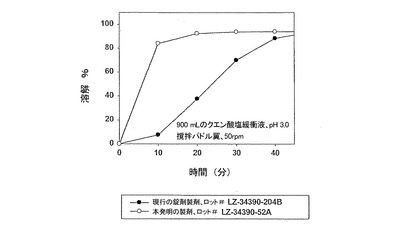
【図6】遊離塩基換算でサキナビル1,000mg投与量において、錠剤剤型における本発明製剤(実施例1)の溶解特性を、従来の錠剤製剤(実施例4)と比較して示す。
【図7】造粒終点に関係なく、迅速で、高度に再現性のある溶解特性が得られることを表す、実施例1の錠剤剤型(遊離塩基換算で500mg)における本発明製剤の溶解特性を示す。
【0009】
本発明の微粉化メシル酸サキナビル投与剤型は、迅速で高度に再現性のある溶解特性を提供する。本発明のメシル酸サキナビル投与剤型は、HIV感染個体の治療に用いることができる。別の抗レトロウイルス薬、たとえばリトナビルとの共投与が企図される。
【0010】
本発明は、微粉化メシル酸サキナビルを、サキナビル遊離塩基に換算して約200〜約800mg、好ましくは200〜700mg、より好ましくは250〜700mg、さらにより好ましくは500mgの量で含む、メシル酸サキナビルの固形単位経口投与剤型を提供する。
【0011】
メシル酸サキナビルの固形単位経口投与剤型は、さらに、好ましくは水溶性の、そして核剤の約3〜約10重量%、好ましくは約4〜6重量%で存在する薬剤的に許容しうる担体を含む。この水溶性担体は、好都合には、約30〜約200μmの粒子サイズを有する。好ましい水溶性担体は、約100〜150μmの粒子サイズを有するラクトース一水和物である。
【0012】
薬剤的に許容しうる水溶性結合剤は、核剤の約4〜8重量%、好ましくは約4〜6重量%存在する。好ましい水溶性結合剤は、ポリビニルピロリドンである。良好に適合するのは、Povidon、たとえばPovidone K30の商品名で市販されているポリビニルピロリドンである。
【0013】
薬剤的に許容しうる崩壊剤は、核剤の約3〜10重量%、好ましくは約4〜8重量%存在する。好ましい、薬剤的に許容しうる崩壊剤は、クロスカルメロースナトリウムおよびクロスポビドンから選択される。
【0014】
メシル酸サキナビル、担体、結合剤、および崩壊剤の少なくとも一部、からなる粒状物が調製される。この粒状物は、メシル酸サキナビル、担体、結合剤、および崩壊剤の様々な粒子サイズの塊りからなり、そして結果的に生成する粉末は、好都合な圧縮および湿潤特性をもつ自由流動性のものである。単位剤型が錠剤である場合、その錠剤は、部分的にこれらの塊りからつくられる。この錠剤が胃腸液に曝されると崩壊し、微粉化メシル酸サキナビルを放出して迅速溶解に供される。
【0015】
粒状物の調製後、微晶質セルロース(MCC)を、製造する錠剤の機械強度を増すために粒状物外成分として加えてよい。MCCは、好都合には、核剤の約5〜20重量%、好ましくは約5〜15重量%存在する。
【0016】
滑剤、たとえばステアリン酸マグネシウムは、粒状物外成分として、核剤の約0.5〜1.2重量%添加してよい。
【0017】
本発明はまた、メシル酸サキナビルの固形単位経口投与剤型の製造方法をも提供する。この方法は、この薬剤を、崩壊剤および親水性結合剤および担体と一緒に微細造粒する工程を含んでいる。このようにして製造された剤型は、メシル酸サキナビルを結晶形態のまま保持している。
【0018】
本発明の製剤は、錠剤剤型にしても、またはカプセル剤型にしても、現行市販カプセル製剤の溶解特性にくらべ、比較的により速い、そしてより再現性のある溶解特性を示す。さらには、本明細書に開示してある経口投与剤型は、圧縮成形力や造粒終点に関係なく、迅速で高度に再現性のある溶解特性を提供する。本発明の剤型は、好都合には、約400mg〜約1.5gの重さを有する。
【0019】
本発明の固形単位経口投与剤型は、核剤および核剤を内包している部分を有する。核剤は、本発明に従い、メシル酸サキナビル、結合剤、崩壊剤および担体を含む。核剤は、場合により、1種以上の薬剤的に許容しうる賦形剤、たとえばラクトース一水和物を含有する。核剤は、好ましくは、粒状物および粒状物に添加される賦形剤(「粒状物外成分」)の混合物からなる。核剤を内包している部分は、たとえば錠剤のフィルムコーティング、またはカプセルもしくはカプレットのコーティングであってよい。
【0020】
本発明で使用されるメシル酸サキナビルは、小粒子サイズに微粉化されている。微粉化メシル酸サキナビルは、典型的には、約1〜約20μm範囲の粒子を有するメシル酸サキナビルである。固形単位経口投与剤型の調製においては、微粉化メシル酸サキナビルを、メシル酸塩に基づく計算で核剤重量の約60〜約80%量使用する。これは、サキナビル遊離塩基に換算して約200〜約800mg量の微粉化メシル酸サキナビルに相当する。
【0021】
本発明で、場合により使用してよい薬剤的に許容しうる賦形剤には、言及したもの以外のタイプの賦形剤が包含される。したがって、たとえば核剤中の結合剤および崩壊剤のパーセントは、前指定のまま維持される。薬剤的に許容しうる場合により使用してよい賦形剤は、微晶質セルロースおよび滑剤を包含する。
【0022】
滑剤は、たとえば、ステアリン酸マグネシウムおよびタルクを包含する。ステアリン酸マグネシウムがより好まれる。滑剤は、核剤の粒状物外成分として用いることができる。滑剤は、好ましくは、核剤の0.5〜1.2重量%で存在する。
【0023】
摩砕して乾燥した粒状物に加えられる賦形剤は、滑剤、崩壊剤および希釈剤の群から選択できる。製薬上の賦形剤は、たとえば、微晶質セルロース、コーンスターチ、ステアリン酸マグネシウムなどであってよい。錠剤の調製には、造粒、摩砕、混合、潤滑化、圧縮(錠剤成形)、および、典型的には水性フィルムコーティングによって、固形剤型を固形単位経口投与剤型として加工することができる。
【0024】
本発明の剤型は、微粉化メシル酸サキナビルを、崩壊剤および親水性結合剤および担体と一緒に微細造粒し、摩砕することによって調製される。この粒状物は、好ましくは、滑剤と混合されて錠剤化され、そして水性ベースのフィルムでコーティングされる。微細造粒工程の間に微粉化メシル酸サキナビルは解凝塊され、そして親水性担体および結合剤によって湿潤し、それによって溶解媒体との接触の際に一次粒子の表面積が最大化される。
【0025】
微粉化メシル酸サキナビルの固形単位経口投与剤型の製造方法は、遊離塩基に換算して約200〜800mgの微粉化メシル酸サキナビル、薬剤的に許容しうる崩壊剤、および薬剤的に許容しうる水溶性担体の混合物に水溶性結合剤溶液を噴霧し、本発明の均一な粒状物を達成することを含んで提供される。好ましくは、この担体がラクトース一水和物であり、そしてこの崩壊剤がクロスカルメロースナトリウムである。
【0026】
好ましくは、崩壊剤の一部が粒状物に含有され、そして崩壊剤の残部が粒状物外成分として加えられ、そして混合される。粒状物中の崩壊剤と粒状物外成分中の崩壊剤との比は、約3:1〜約1:1である。好ましくは、その比が約2.5:1〜約1.5:1である。より好ましくは、その比が約2:1である。
【0027】
結果として生成する錠剤の機械的強度を増強するため、好ましくは、微晶質セルロースを粒状物外成分として加えて粒状物と混合する。微晶質セルロースは、核剤の約5〜約20重量%、好ましくは約5〜約15重量%存在させる。
【0028】
好ましくは、滑剤、たとえばステアリン酸マグネシウムを、圧縮成形の際、打錠パンチに適切な潤滑性を提供するため、粒状物の外側に加える。この滑剤は、核剤の約0.5〜約1.2重量%存在させる。
【0029】
本発明の一態様において、錠剤は以下のようにして製造される:
a)単位投与量あたり、(遊離塩基に換算して)約200〜約800mg量の微粉化メシル 酸サキナビルと、親水性担体および崩壊剤とを、高せん断造粒機中で混合する。
b)撹拌しながら、結合剤水溶液を、工程(a)からの粉末混合物に噴霧するか、または ゆっくり添加して最適造粒終点を達成させる。
c)工程(b)からの湿潤粒状物を、解砕して均一な粒状物にする。
d)工程(c)からの湿潤粒状物を、40〜50℃に設定した強制空気循環炉中で、または入口空気温度50〜60℃の流動床乾燥機中で、粒状物の湿分が1.5〜2%になるまで乾燥する。
e)工程(d)からの乾燥粒状物を摩砕する。
f)工程(e)からの摩砕粒状物を、適切な錠剤希釈剤、たとえば微晶質セルロース、お よび崩壊剤と混合する。
g)工程(f)からの混合物を、適切な滑剤、たとえばステアリン酸マグネシウムで潤滑 する。
h)工程(g)からの最終混合物を、錠剤プレスで圧縮成形する。
i)工程(h)からの錠剤を、水性フィルムでコーティングする。
【実施例】
【0030】
以下の実施例において、最適造粒終点は、造粒工程が、もはやさらなる検知可能な粒子サイズ変化をもたらさない時点として目視検査により決定した。
【0031】
実施例1
【0032】
【表1】

【0033】
I)核剤の製造
A)微粉化メシル酸サキナビル、ラクトース一水和物、およびクロスカルメロースナトリトリウムの一部(クロスカルメロースナトリウム全量の約66.7%)を、高せん断造粒機中において、低速のインペラーおよび低速のアジテーターを用い、5分間混合した。
B)20重量%Povidone K30溶液の調製:ステンレス鋼容器の中で、Povidone K30を純 水(160mg/錠剤)にゆっくり加え、そしてプロペラミキサーを用いて混合した。
Povidone K30が完全に溶解するまで混合撹拌を続けた。
C)(1)高せん断造粒機中で、工程Aからの粉末混合物に、工程Bからの20重量%Povidone K30溶液を噴霧し、そして低速のインペラーおよび低速のアジテーターを用い、8〜10分間、継続的に混合することによって粉末混合物を造粒した。
C)(2)追加的な純水(約180mg/錠剤)を、工程C(1)からの粉末混合物に噴霧し、低速のインペラーおよび低速のアジテーターを用いて8〜10分間、継続的に混合した。粒状物の追加的な混練を実施して、最適造粒終点に到達させた。湿潤粒状物を、低速のインペラーおよび低速のアジテーターを用い、ポリエチレン内張り容器中に取り出した。
D)この湿潤粒状物を、19.05mm円形開口スクリーン(#750Q)を備えたCo-milに1,350rpmで通すか、または10mm円形開口スクリーンを備えたFrewitt回転ふるいに1,000〜2,000rpmで通して解砕した。
E)工程Dからの解砕された湿潤粒状物を、入口空気温度が65±10℃に設定されている流動床乾燥機中で、粒状物の湿分が1.8%未満(90℃に設定したOmnimark湿分分析計を用いた乾燥減量により測定)になるまで乾燥した。
F)工程Eからの乾燥粒状物を、1.27mm円形格子開口スクリーン(#050G)を備えたCo-milに4,500rpmで通すか、または2.0mm円形スクリーンを備えたFrewittハンマーミルに、粉砕刃を3,170rpm前進で用いて通し、粉砕した。
G)工程Fからの摩砕された粒状物、Avicel PH101、およびクロスカルメロースナトリウムの残部(クロスカルメロースナトリウム全体の約33.3%)を、PKブレンダーまたは等価の装置の中で10分間混合した。
H)工程GのPKブレンダーからの粒状物の約50%を取り除いた。
I)ステアリン酸マグネシウム(#30メッシュステンレス鋼スクリーンを通したもの)を、工程HのPKブレンダーに加えた。工程Hからの取り除いておいた粒状物をPKブレンダーに戻し、そして5分間混合した。
J)工程Iからの粒状物を、以下の仕様を用いて圧縮成形した:
パンチサイズ:長円形、8.74mm×18.75mm、標準凹面
錠剤重量: 800mg(760〜840mg)
錠剤硬さ: 30SCU(25〜35SCU)または210N(175〜245N)
【0034】
II)フィルムコートの塗布
A)フィルムコーティング用懸濁液の調製
ステンレス鋼容器の中で、トリアセチンおよびAquacoat ECD-30を、プロペラミキサーを用いて純水中に分散させ、45分間混合撹拌した。
ヒドロキシプロピルメチルセルロース2910(6mPa・s{6cP})、タルク、二酸化チタン、酸化鉄イエロー、および酸化鉄レッドの粉末混合物を上記の分散液に加え、空気を巻き込まないように穏やかに混合した。混合撹拌をさらに60分間、または均一な懸濁液が得られるまで継続した。
【0035】
B)フィルムコートの塗布
I)(核剤の製造)の工程Jからの核剤を、穴のあいたコーティングパンの中に仕込んだ。入口空気温度を60±10℃まで徐々に上げて核剤を温め、断続的に転動させながら、出口温度を40±5℃に到達させた。
入口空気温度を60±10℃に保ったまま、核剤がパン内部で十分転動するようにパン回転速度を上げた。核剤に、上記II)Aからのフィルムコーティング用懸濁液を噴霧し、そしてエアスプレー系を継続的に使用しながら核剤を転動させた。製造物温度は45±5℃に維持した。錠剤1個あたり、乾燥基準でフィルムコート20mg(17〜23mgの範囲)を塗布した。
入口空気温度を50±5℃に、そしてパン回転速度を4±2rpmに下げた。コーティングされた錠剤の乾燥を、2〜4分間継続した。
入口空気温度を40±5℃に下げ、そしてコーティングされた錠剤を、錠剤湿分が2.0%未満(90℃に設定したOmnimark湿分分析計を用いた乾燥減量により測定)になるまで乾燥した。加熱を止め、そして錠剤を時々転動させることによって室温に冷却した。
【0036】
実施例2
【0037】
【表2】
【0038】
A)微粉化メシル酸サキナビル、無水ラクトース、微晶質セルロース、およびグリコール酸デンプンナトリウムの一部(グリコール酸デンプンナトリウム全量の56.25%を、高せん断造粒機中で混合した。
B)Povidone K30溶液を、高せん断造粒機中で粉末混合物(工程A)に加え、そして継続的に混合してその粉末混合物を造粒した。
C)追加的な純水を、工程Bからの粉末混合物に加え、最適造粒終点が得られるまで継続的に混合した。湿潤粒状物をポリエチレン内張り容器中へ取り出した。
D)工程Cからの湿潤粒状物を、摩砕機を通して解砕した。
E)工程Dからの解砕された湿潤粒状物を、入口空気温度が65±10℃に設定された流動床乾燥機中で、粒状物の湿分が1.8%未満(90℃に設定したOmnimark湿分分析計を用いた乾燥減量により測定)になるまで乾燥した。
F)工程Eからの乾燥粒状物を、摩砕機に通した。
G)工程Fからの摩砕された粒状物を、ブレンダー中でグリコール酸デンプンナトリウムの一部(グリコール酸デンプンナトリウム全量の43.75%)、タルクおよびステアリン酸マグネシウムと混合した。
H)工程Gからの最終混合物を、カプセル充填機を用い、カプセル(#0)の中に、目標充填重量408mgで封入した。
【0039】
実施例3
【0040】
【表3】
【0041】
工程A〜Iについては、実施例1に詳述したものと同じ手順に従った。
工程J)工程Iからの最終混合物を、カプセル充填機を用い、カプセル(#0)の中に、目標充填重量320mgで封入した。
【0042】
実施例4
【0043】
【表4】
【0044】
工程A〜Gについては、実施例2に詳述したものと同じ手順に従った。
工程H)工程Gからの粒状物を、以下の仕様を用いて圧縮成形した:
パンチサイズ:長円形、9.28mm×20.02mm、標準凹面
錠剤重量: 1,200mg(1,140〜1,260mg)
錠剤硬さ: 25〜35SCUまたは175〜245N
【0045】
溶解試験:
メシル酸サキナビルを含有する経口投与剤型(実施例1〜4)につき、37±0.5℃で平衡化された、クエン酸緩衝液(pH3)の900mL中で、パドル法(USP Apparatus 2)(50rpm)を用いて溶解性を評価した。異なる時間間隔で試料を分取し、紫外分光測光法により分析した。
【技術分野】
【0001】
メシル酸サキナビルは、ウイルスの複製を制限し、HIV感染個体における免疫機能を改善させるために用いられる数種のプロテアーゼ阻害剤の一つである。メシル酸サキナビルは、200mgカプセル(サキナビル遊離塩基に換算して)として市販されている。これは、INVIRASE(登録商標)の名称でHoffmann-La Roche,Inc.によって販売されており、進行性ヒト免疫不全ウイルス(HIV)感染症の、選ばれた患者群における治療に、抗レトロウイルス性ヌクレオシド同族体と組み合わせて使用するように指示されている。
【0002】
メシル酸サキナビルは、分子量766.96を有する白色〜灰色がかった白色の、非常に微細な結晶性粉末である。遊離塩基の分子量は670.86である。この薬物は高度に疎水性である。INVIRASE(登録商標)(メシル酸サキナビル)の200mgカプセルは、低い経口バイオアベイラビリティーを有するが、これは不完全な吸収および大きな初回通過代謝によるものと考えられている。[Physician's Desk Reference, 57th Ed.(2003)。]メシル酸サキナビルは、非常に低い水溶解性を有する(すなわち、25℃では、水中で2.2mg/mL、模擬胃液中で0.08mg/mL、そして模擬腸液中では事実上不溶性である)。その上、この薬物はpH依存溶解特性を示し、模擬胃液中では限られた溶解性を有する一方、模擬腸液中では事実上不溶である。高脂肪朝食後にサキナビル600mg量(3×200mgカプセル)を1回投与した健康な有志者8人における絶対バイオアベイラビリティーは、平均で4%(CV73%、範囲:1〜9%)であった。より高カロリーの食事を摂った後のサキナビル24時間AUCおよびCmax(n=6)は、より低カロリーの食事を摂った後より、平均で2倍高かった。食事の影響は、2時間後まで持続することが示された。ヒトで観察される、メシル酸サキナビルの対象間変動および食物効果の最小化を企図するには、メシル酸サキナビルの均一で迅速な溶解特性を有する固形経口投与剤型が望まれる。
【0003】
粒子サイズを小さくすることは、対象間変動を最小化し、そしてバイオアベイラビリティーを改善するための一手段である。微粉化は、粒子サイズを減少させるために用いられる一つのアプローチである。しかしながらメシル酸サキナビルは、微粉化に際し、凝塊化して溶解媒体と接触する一次粒子表面積を減少させてしまう傾向がある。微粉化したメシル酸サキナビルは、遅い溶解速度を示す。
【0004】
成人患者用の、INVIRASE(登録商標)の、ヌクレオシド同族体との組み合わせにおける推奨投与量は、3×200mgカプセルを1日3回、食後2〜4時間以内の服用である。この投与則から見て、患者がこれを遵守するかどうかが真の懸念材料である。治療の成功は、たとえば1日に服用すべき単位数を減らすというように、よりよい遵守を促すことによって、よりよく改善できるかもしれない。したがって、サキナビルのより多い量を含有する単位剤型が有用であろうという期待はある。しかしながら、微粉化メシル酸サキナビルの薬充填量を増やすと、薬剤凝塊の問題は悪化する。
【0005】
本発明は、メシル酸塩を基準にして約60〜約80%の微粉化メシル酸サキナビル、約4〜約8%の薬剤的に許容しうる水溶性結合剤、薬剤的に許容しうる崩壊剤、および薬剤的に許容しうる担体(それぞれのパーセントはその剤型の核剤重量の内訳パーセントである)を含む、メシル酸サキナビルの固形単位経口投与剤型を提供するものである。(微粉化メシル酸サキナビル約60〜約80%は、サキナビル遊離塩基に換算して約200〜約800mg量のメシル酸サキナビルに相当する。)
【0006】
好ましい態様において、メシル酸サキナビルは、固形単位経口投与剤型の核剤重量の約70〜75%である。
【0007】
本発明は、遊離塩基に換算して約250〜約800mg量の微粉化メシル酸サキナビル、薬剤的に許容しうる結合剤、薬剤的に許容しうる崩壊剤、および薬剤的に許容しうる水溶性担体を含む、メシル酸サキナビルの固形単位経口投与剤型を提供する。
【図面の簡単な説明】
【0008】
【図1】実施例1の錠剤剤型(遊離塩基換算で500mg)における、本発明製剤のロット間再現性を示す溶解特性を示す。
【図2】実施例2のカプセル剤型(遊離塩基換算で200mg)における、現行市販製剤のロット間バラツキを示す溶解特性を示す。
【図3】遊離塩基換算でサキナビル1,000mgの投与量において、錠剤剤型における本発明製剤(実施例1)の溶解特性を、カプセル剤型における現行市販製剤(実施例2)と比較して示す。
【図4】遊離塩基換算でサキナビル1,000mgの投与量において、カプセル剤型における本発明製剤(実施例3)の溶解特性を、カプセル剤型における現行市販製剤(実施例2)と比較して示す。
【図5】加えた圧縮力に関係なく、迅速で、高度に再現性のある溶解特性が得られることを表す、実施例1の錠剤剤型(サキナビル遊離塩基換算で500mg)における本発明製剤の溶解特性を示す。
【図6】遊離塩基換算でサキナビル1,000mg投与量において、錠剤剤型における本発明製剤(実施例1)の溶解特性を、従来の錠剤製剤(実施例4)と比較して示す。
【図7】造粒終点に関係なく、迅速で、高度に再現性のある溶解特性が得られることを表す、実施例1の錠剤剤型(遊離塩基換算で500mg)における本発明製剤の溶解特性を示す。
【0009】
本発明の微粉化メシル酸サキナビル投与剤型は、迅速で高度に再現性のある溶解特性を提供する。本発明のメシル酸サキナビル投与剤型は、HIV感染個体の治療に用いることができる。別の抗レトロウイルス薬、たとえばリトナビルとの共投与が企図される。
【0010】
本発明は、微粉化メシル酸サキナビルを、サキナビル遊離塩基に換算して約200〜約800mg、好ましくは200〜700mg、より好ましくは250〜700mg、さらにより好ましくは500mgの量で含む、メシル酸サキナビルの固形単位経口投与剤型を提供する。
【0011】
メシル酸サキナビルの固形単位経口投与剤型は、さらに、好ましくは水溶性の、そして核剤の約3〜約10重量%、好ましくは約4〜6重量%で存在する薬剤的に許容しうる担体を含む。この水溶性担体は、好都合には、約30〜約200μmの粒子サイズを有する。好ましい水溶性担体は、約100〜150μmの粒子サイズを有するラクトース一水和物である。
【0012】
薬剤的に許容しうる水溶性結合剤は、核剤の約4〜8重量%、好ましくは約4〜6重量%存在する。好ましい水溶性結合剤は、ポリビニルピロリドンである。良好に適合するのは、Povidon、たとえばPovidone K30の商品名で市販されているポリビニルピロリドンである。
【0013】
薬剤的に許容しうる崩壊剤は、核剤の約3〜10重量%、好ましくは約4〜8重量%存在する。好ましい、薬剤的に許容しうる崩壊剤は、クロスカルメロースナトリウムおよびクロスポビドンから選択される。
【0014】
メシル酸サキナビル、担体、結合剤、および崩壊剤の少なくとも一部、からなる粒状物が調製される。この粒状物は、メシル酸サキナビル、担体、結合剤、および崩壊剤の様々な粒子サイズの塊りからなり、そして結果的に生成する粉末は、好都合な圧縮および湿潤特性をもつ自由流動性のものである。単位剤型が錠剤である場合、その錠剤は、部分的にこれらの塊りからつくられる。この錠剤が胃腸液に曝されると崩壊し、微粉化メシル酸サキナビルを放出して迅速溶解に供される。
【0015】
粒状物の調製後、微晶質セルロース(MCC)を、製造する錠剤の機械強度を増すために粒状物外成分として加えてよい。MCCは、好都合には、核剤の約5〜20重量%、好ましくは約5〜15重量%存在する。
【0016】
滑剤、たとえばステアリン酸マグネシウムは、粒状物外成分として、核剤の約0.5〜1.2重量%添加してよい。
【0017】
本発明はまた、メシル酸サキナビルの固形単位経口投与剤型の製造方法をも提供する。この方法は、この薬剤を、崩壊剤および親水性結合剤および担体と一緒に微細造粒する工程を含んでいる。このようにして製造された剤型は、メシル酸サキナビルを結晶形態のまま保持している。
【0018】
本発明の製剤は、錠剤剤型にしても、またはカプセル剤型にしても、現行市販カプセル製剤の溶解特性にくらべ、比較的により速い、そしてより再現性のある溶解特性を示す。さらには、本明細書に開示してある経口投与剤型は、圧縮成形力や造粒終点に関係なく、迅速で高度に再現性のある溶解特性を提供する。本発明の剤型は、好都合には、約400mg〜約1.5gの重さを有する。
【0019】
本発明の固形単位経口投与剤型は、核剤および核剤を内包している部分を有する。核剤は、本発明に従い、メシル酸サキナビル、結合剤、崩壊剤および担体を含む。核剤は、場合により、1種以上の薬剤的に許容しうる賦形剤、たとえばラクトース一水和物を含有する。核剤は、好ましくは、粒状物および粒状物に添加される賦形剤(「粒状物外成分」)の混合物からなる。核剤を内包している部分は、たとえば錠剤のフィルムコーティング、またはカプセルもしくはカプレットのコーティングであってよい。
【0020】
本発明で使用されるメシル酸サキナビルは、小粒子サイズに微粉化されている。微粉化メシル酸サキナビルは、典型的には、約1〜約20μm範囲の粒子を有するメシル酸サキナビルである。固形単位経口投与剤型の調製においては、微粉化メシル酸サキナビルを、メシル酸塩に基づく計算で核剤重量の約60〜約80%量使用する。これは、サキナビル遊離塩基に換算して約200〜約800mg量の微粉化メシル酸サキナビルに相当する。
【0021】
本発明で、場合により使用してよい薬剤的に許容しうる賦形剤には、言及したもの以外のタイプの賦形剤が包含される。したがって、たとえば核剤中の結合剤および崩壊剤のパーセントは、前指定のまま維持される。薬剤的に許容しうる場合により使用してよい賦形剤は、微晶質セルロースおよび滑剤を包含する。
【0022】
滑剤は、たとえば、ステアリン酸マグネシウムおよびタルクを包含する。ステアリン酸マグネシウムがより好まれる。滑剤は、核剤の粒状物外成分として用いることができる。滑剤は、好ましくは、核剤の0.5〜1.2重量%で存在する。
【0023】
摩砕して乾燥した粒状物に加えられる賦形剤は、滑剤、崩壊剤および希釈剤の群から選択できる。製薬上の賦形剤は、たとえば、微晶質セルロース、コーンスターチ、ステアリン酸マグネシウムなどであってよい。錠剤の調製には、造粒、摩砕、混合、潤滑化、圧縮(錠剤成形)、および、典型的には水性フィルムコーティングによって、固形剤型を固形単位経口投与剤型として加工することができる。
【0024】
本発明の剤型は、微粉化メシル酸サキナビルを、崩壊剤および親水性結合剤および担体と一緒に微細造粒し、摩砕することによって調製される。この粒状物は、好ましくは、滑剤と混合されて錠剤化され、そして水性ベースのフィルムでコーティングされる。微細造粒工程の間に微粉化メシル酸サキナビルは解凝塊され、そして親水性担体および結合剤によって湿潤し、それによって溶解媒体との接触の際に一次粒子の表面積が最大化される。
【0025】
微粉化メシル酸サキナビルの固形単位経口投与剤型の製造方法は、遊離塩基に換算して約200〜800mgの微粉化メシル酸サキナビル、薬剤的に許容しうる崩壊剤、および薬剤的に許容しうる水溶性担体の混合物に水溶性結合剤溶液を噴霧し、本発明の均一な粒状物を達成することを含んで提供される。好ましくは、この担体がラクトース一水和物であり、そしてこの崩壊剤がクロスカルメロースナトリウムである。
【0026】
好ましくは、崩壊剤の一部が粒状物に含有され、そして崩壊剤の残部が粒状物外成分として加えられ、そして混合される。粒状物中の崩壊剤と粒状物外成分中の崩壊剤との比は、約3:1〜約1:1である。好ましくは、その比が約2.5:1〜約1.5:1である。より好ましくは、その比が約2:1である。
【0027】
結果として生成する錠剤の機械的強度を増強するため、好ましくは、微晶質セルロースを粒状物外成分として加えて粒状物と混合する。微晶質セルロースは、核剤の約5〜約20重量%、好ましくは約5〜約15重量%存在させる。
【0028】
好ましくは、滑剤、たとえばステアリン酸マグネシウムを、圧縮成形の際、打錠パンチに適切な潤滑性を提供するため、粒状物の外側に加える。この滑剤は、核剤の約0.5〜約1.2重量%存在させる。
【0029】
本発明の一態様において、錠剤は以下のようにして製造される:
a)単位投与量あたり、(遊離塩基に換算して)約200〜約800mg量の微粉化メシル 酸サキナビルと、親水性担体および崩壊剤とを、高せん断造粒機中で混合する。
b)撹拌しながら、結合剤水溶液を、工程(a)からの粉末混合物に噴霧するか、または ゆっくり添加して最適造粒終点を達成させる。
c)工程(b)からの湿潤粒状物を、解砕して均一な粒状物にする。
d)工程(c)からの湿潤粒状物を、40〜50℃に設定した強制空気循環炉中で、または入口空気温度50〜60℃の流動床乾燥機中で、粒状物の湿分が1.5〜2%になるまで乾燥する。
e)工程(d)からの乾燥粒状物を摩砕する。
f)工程(e)からの摩砕粒状物を、適切な錠剤希釈剤、たとえば微晶質セルロース、お よび崩壊剤と混合する。
g)工程(f)からの混合物を、適切な滑剤、たとえばステアリン酸マグネシウムで潤滑 する。
h)工程(g)からの最終混合物を、錠剤プレスで圧縮成形する。
i)工程(h)からの錠剤を、水性フィルムでコーティングする。
【実施例】
【0030】
以下の実施例において、最適造粒終点は、造粒工程が、もはやさらなる検知可能な粒子サイズ変化をもたらさない時点として目視検査により決定した。
【0031】
実施例1
【0032】
【表1】
【0033】
I)核剤の製造
A)微粉化メシル酸サキナビル、ラクトース一水和物、およびクロスカルメロースナトリトリウムの一部(クロスカルメロースナトリウム全量の約66.7%)を、高せん断造粒機中において、低速のインペラーおよび低速のアジテーターを用い、5分間混合した。
B)20重量%Povidone K30溶液の調製:ステンレス鋼容器の中で、Povidone K30を純 水(160mg/錠剤)にゆっくり加え、そしてプロペラミキサーを用いて混合した。
Povidone K30が完全に溶解するまで混合撹拌を続けた。
C)(1)高せん断造粒機中で、工程Aからの粉末混合物に、工程Bからの20重量%Povidone K30溶液を噴霧し、そして低速のインペラーおよび低速のアジテーターを用い、8〜10分間、継続的に混合することによって粉末混合物を造粒した。
C)(2)追加的な純水(約180mg/錠剤)を、工程C(1)からの粉末混合物に噴霧し、低速のインペラーおよび低速のアジテーターを用いて8〜10分間、継続的に混合した。粒状物の追加的な混練を実施して、最適造粒終点に到達させた。湿潤粒状物を、低速のインペラーおよび低速のアジテーターを用い、ポリエチレン内張り容器中に取り出した。
D)この湿潤粒状物を、19.05mm円形開口スクリーン(#750Q)を備えたCo-milに1,350rpmで通すか、または10mm円形開口スクリーンを備えたFrewitt回転ふるいに1,000〜2,000rpmで通して解砕した。
E)工程Dからの解砕された湿潤粒状物を、入口空気温度が65±10℃に設定されている流動床乾燥機中で、粒状物の湿分が1.8%未満(90℃に設定したOmnimark湿分分析計を用いた乾燥減量により測定)になるまで乾燥した。
F)工程Eからの乾燥粒状物を、1.27mm円形格子開口スクリーン(#050G)を備えたCo-milに4,500rpmで通すか、または2.0mm円形スクリーンを備えたFrewittハンマーミルに、粉砕刃を3,170rpm前進で用いて通し、粉砕した。
G)工程Fからの摩砕された粒状物、Avicel PH101、およびクロスカルメロースナトリウムの残部(クロスカルメロースナトリウム全体の約33.3%)を、PKブレンダーまたは等価の装置の中で10分間混合した。
H)工程GのPKブレンダーからの粒状物の約50%を取り除いた。
I)ステアリン酸マグネシウム(#30メッシュステンレス鋼スクリーンを通したもの)を、工程HのPKブレンダーに加えた。工程Hからの取り除いておいた粒状物をPKブレンダーに戻し、そして5分間混合した。
J)工程Iからの粒状物を、以下の仕様を用いて圧縮成形した:
パンチサイズ:長円形、8.74mm×18.75mm、標準凹面
錠剤重量: 800mg(760〜840mg)
錠剤硬さ: 30SCU(25〜35SCU)または210N(175〜245N)
【0034】
II)フィルムコートの塗布
A)フィルムコーティング用懸濁液の調製
ステンレス鋼容器の中で、トリアセチンおよびAquacoat ECD-30を、プロペラミキサーを用いて純水中に分散させ、45分間混合撹拌した。
ヒドロキシプロピルメチルセルロース2910(6mPa・s{6cP})、タルク、二酸化チタン、酸化鉄イエロー、および酸化鉄レッドの粉末混合物を上記の分散液に加え、空気を巻き込まないように穏やかに混合した。混合撹拌をさらに60分間、または均一な懸濁液が得られるまで継続した。
【0035】
B)フィルムコートの塗布
I)(核剤の製造)の工程Jからの核剤を、穴のあいたコーティングパンの中に仕込んだ。入口空気温度を60±10℃まで徐々に上げて核剤を温め、断続的に転動させながら、出口温度を40±5℃に到達させた。
入口空気温度を60±10℃に保ったまま、核剤がパン内部で十分転動するようにパン回転速度を上げた。核剤に、上記II)Aからのフィルムコーティング用懸濁液を噴霧し、そしてエアスプレー系を継続的に使用しながら核剤を転動させた。製造物温度は45±5℃に維持した。錠剤1個あたり、乾燥基準でフィルムコート20mg(17〜23mgの範囲)を塗布した。
入口空気温度を50±5℃に、そしてパン回転速度を4±2rpmに下げた。コーティングされた錠剤の乾燥を、2〜4分間継続した。
入口空気温度を40±5℃に下げ、そしてコーティングされた錠剤を、錠剤湿分が2.0%未満(90℃に設定したOmnimark湿分分析計を用いた乾燥減量により測定)になるまで乾燥した。加熱を止め、そして錠剤を時々転動させることによって室温に冷却した。
【0036】
実施例2
【0037】
【表2】
【0038】
A)微粉化メシル酸サキナビル、無水ラクトース、微晶質セルロース、およびグリコール酸デンプンナトリウムの一部(グリコール酸デンプンナトリウム全量の56.25%を、高せん断造粒機中で混合した。
B)Povidone K30溶液を、高せん断造粒機中で粉末混合物(工程A)に加え、そして継続的に混合してその粉末混合物を造粒した。
C)追加的な純水を、工程Bからの粉末混合物に加え、最適造粒終点が得られるまで継続的に混合した。湿潤粒状物をポリエチレン内張り容器中へ取り出した。
D)工程Cからの湿潤粒状物を、摩砕機を通して解砕した。
E)工程Dからの解砕された湿潤粒状物を、入口空気温度が65±10℃に設定された流動床乾燥機中で、粒状物の湿分が1.8%未満(90℃に設定したOmnimark湿分分析計を用いた乾燥減量により測定)になるまで乾燥した。
F)工程Eからの乾燥粒状物を、摩砕機に通した。
G)工程Fからの摩砕された粒状物を、ブレンダー中でグリコール酸デンプンナトリウムの一部(グリコール酸デンプンナトリウム全量の43.75%)、タルクおよびステアリン酸マグネシウムと混合した。
H)工程Gからの最終混合物を、カプセル充填機を用い、カプセル(#0)の中に、目標充填重量408mgで封入した。
【0039】
実施例3
【0040】
【表3】
【0041】
工程A〜Iについては、実施例1に詳述したものと同じ手順に従った。
工程J)工程Iからの最終混合物を、カプセル充填機を用い、カプセル(#0)の中に、目標充填重量320mgで封入した。
【0042】
実施例4
【0043】
【表4】
【0044】
工程A〜Gについては、実施例2に詳述したものと同じ手順に従った。
工程H)工程Gからの粒状物を、以下の仕様を用いて圧縮成形した:
パンチサイズ:長円形、9.28mm×20.02mm、標準凹面
錠剤重量: 1,200mg(1,140〜1,260mg)
錠剤硬さ: 25〜35SCUまたは175〜245N
【0045】
溶解試験:
メシル酸サキナビルを含有する経口投与剤型(実施例1〜4)につき、37±0.5℃で平衡化された、クエン酸緩衝液(pH3)の900mL中で、パドル法(USP Apparatus 2)(50rpm)を用いて溶解性を評価した。異なる時間間隔で試料を分取し、紫外分光測光法により分析した。
【特許請求の範囲】
【請求項1】
メシル酸塩を基準にして約60〜約80%の微粉化メシル酸サキナビル、約4〜約8%の薬剤的に許容しうる水溶性結合剤、薬剤的に許容しうる崩壊剤、および薬剤的に許容しうる担体を含み、ここでそれぞれのパーセントは、その剤型の核剤重量の内訳パーセントである、メシル酸サキナビルの固形単位経口投与剤型。
【請求項2】
メシル酸サキナビルが、剤型の核剤重量の約70〜約75%である、請求項1記載の剤型。
【請求項3】
剤型中のメシル酸サキナビルが、サキナビル遊離塩基に換算して約200〜約700mgの量で存在する、請求項1または2記載の剤型。
【請求項4】
剤型中のメシル酸サキナビルが、サキナビル遊離塩基に換算して約250〜約700mgの量で存在する、請求項1〜3のいずれか1項記載の剤型。
【請求項5】
剤型中のメシル酸サキナビルが、サキナビル遊離塩基に換算して約500mgの量で存在する、請求項1〜4のいずれか1項記載の剤型。
【請求項6】
薬剤的に許容しうる担体が水溶性である、請求項1記載の剤型。
【請求項7】
薬剤的に許容しうる担体が、核剤重量の約3〜約10%存在する、請求項6記載の剤型。
【請求項8】
薬剤的に許容しうる担体が、約30〜約200μmの粒子サイズを有している、請求項6または7記載の剤型。
【請求項9】
薬剤的に許容しうる担体が、ラクトース一水和物である、請求項6〜8のいずれか1項記載の剤型。
【請求項10】
ラクトース一水和物が、約100〜約150μmの粒子サイズを有する、請求項9記載の剤型。
【請求項11】
薬剤的に許容しうる水溶性結合剤が、核剤重量の約4〜約8%存在する、請求項1記載の剤型。
【請求項12】
薬剤的に許容しうる水溶性結合剤が、ポリビニルピロリドンである、請求項11記載の剤型。
【請求項13】
薬剤的に許容しうる崩壊剤が、核剤重量の約3〜約10%存在する、請求項1記載の剤型。
【請求項14】
崩壊剤が、クロスカルメロースナトリウムおよびクロスポビドンから選択される、請求項13記載の剤型。
【請求項15】
さらに微晶質セルロースを含む、請求項1記載の剤型。
【請求項16】
微晶質セルロースが、核剤重量の約5〜約20%存在する、請求項15記載の剤型。
【請求項17】
剤型が、さらに滑剤を含む、請求項1記載の剤型。
【請求項18】
滑剤がステアリン酸マグネシウムである、請求項17記載の剤型。
【請求項19】
剤型の核剤が、粒状物および粒状物外成分の混合物からなり、ここで、該粒状物は微粉化メシル酸サキナビル、結合剤、担体、および崩壊剤を含み、そして該粒状物外成分は崩壊剤を含み、粒状物中の崩壊剤と粒状物外成分中の崩壊剤との比が約3:1〜約1:1である、請求項1記載の剤型。
【請求項20】
さらに、核剤の粒状物外成分中に含まれる滑剤を含む、請求項19記載の剤型。
【請求項21】
錠剤、カプセルおよびカプレットからなる群より選択される、請求項1〜20のいずれか1項記載の剤型。
【請求項22】
約400mg〜約1.5gの重さを有する、請求項1〜21のいずれか1項記載の剤型。
【請求項23】
遊離塩基に換算して約200〜約800mg量の微粉化メシル酸サキナビル、薬剤的に許容しうる水溶性結合剤、薬剤的に許容しうる崩壊剤、および薬剤的に許容しうる担体を含む、メシル酸サキナビルの固形単位経口投与剤型。
【請求項24】
微粉化メシル酸サキナビルが250〜約700mgの量で存在する、請求項23記載の固形単位経口投与剤型。
【請求項25】
メシル酸サキナビルが結晶形態で存在する、請求項1〜24のいずれか1項記載の固形単位経口投与剤型。
【請求項26】
微粉化メシル酸サキナビル、薬剤的に許容しうる水溶性結合剤、薬剤的に許容しうる担体および医薬活性崩壊剤の混合物を微細造粒することを含み、そしてそれに続く摩砕工程を含む、請求項1〜25のいずれか1項記載の固形単位経口投与剤型の製造方法。
【請求項27】
工程:
a)単位投与量あたり、(遊離塩基に換算して)約200〜約800mg量の微粉化メシル酸サキナビルと、親水性担体および崩壊剤とを、高せん断造粒機中で混合し、
b)撹拌しながら、工程(a)からの粉末混合物に結合剤水溶液を噴霧するか、またはゆっくり添加して最適な造粒終点を達成させ、
c)工程(b)からの湿潤粒状物を解砕して均一な粒状物にし、
d)工程(c)からの湿潤粒状物を、40〜50℃に設定した強制空気循環炉の中で、または入口空気温度50〜60℃の流動床乾燥機の中で、粒状物の湿分が1.5〜2%になるまで乾燥し、
e)工程(d)からの乾燥粒状物を摩砕し、
f)工程(e)からの摩砕粒状物を、適切な錠剤希釈剤と混合し、
g)工程(f)からの混合物を、適切な滑剤で潤滑し、
h)工程(g)からの最終混合物を、錠剤プレス上で圧縮成形し、
i)工程(h)からの錠剤を、水性フィルムでコーティングする;
を含む、請求項26記載の方法。
【請求項28】
治療に用いるための、請求項1〜25のいずれか1項記載の固形単位経口投与剤型。
【請求項29】
HIV介在疾患の治療に用いるための、請求項1〜25のいずれか1項記載の固形単位経口投与剤型。
【請求項30】
HIV介在疾患の治療用薬剤を製造するための、請求項1〜25のいずれか1項記載の固形単位経口投与剤型の使用。
【請求項1】
メシル酸塩を基準にして約60〜約80%の微粉化メシル酸サキナビル、約4〜約8%の薬剤的に許容しうる水溶性結合剤、薬剤的に許容しうる崩壊剤、および薬剤的に許容しうる担体を含み、ここでそれぞれのパーセントは、その剤型の核剤重量の内訳パーセントである、メシル酸サキナビルの固形単位経口投与剤型。
【請求項2】
メシル酸サキナビルが、剤型の核剤重量の約70〜約75%である、請求項1記載の剤型。
【請求項3】
剤型中のメシル酸サキナビルが、サキナビル遊離塩基に換算して約200〜約700mgの量で存在する、請求項1または2記載の剤型。
【請求項4】
剤型中のメシル酸サキナビルが、サキナビル遊離塩基に換算して約250〜約700mgの量で存在する、請求項1〜3のいずれか1項記載の剤型。
【請求項5】
剤型中のメシル酸サキナビルが、サキナビル遊離塩基に換算して約500mgの量で存在する、請求項1〜4のいずれか1項記載の剤型。
【請求項6】
薬剤的に許容しうる担体が水溶性である、請求項1記載の剤型。
【請求項7】
薬剤的に許容しうる担体が、核剤重量の約3〜約10%存在する、請求項6記載の剤型。
【請求項8】
薬剤的に許容しうる担体が、約30〜約200μmの粒子サイズを有している、請求項6または7記載の剤型。
【請求項9】
薬剤的に許容しうる担体が、ラクトース一水和物である、請求項6〜8のいずれか1項記載の剤型。
【請求項10】
ラクトース一水和物が、約100〜約150μmの粒子サイズを有する、請求項9記載の剤型。
【請求項11】
薬剤的に許容しうる水溶性結合剤が、核剤重量の約4〜約8%存在する、請求項1記載の剤型。
【請求項12】
薬剤的に許容しうる水溶性結合剤が、ポリビニルピロリドンである、請求項11記載の剤型。
【請求項13】
薬剤的に許容しうる崩壊剤が、核剤重量の約3〜約10%存在する、請求項1記載の剤型。
【請求項14】
崩壊剤が、クロスカルメロースナトリウムおよびクロスポビドンから選択される、請求項13記載の剤型。
【請求項15】
さらに微晶質セルロースを含む、請求項1記載の剤型。
【請求項16】
微晶質セルロースが、核剤重量の約5〜約20%存在する、請求項15記載の剤型。
【請求項17】
剤型が、さらに滑剤を含む、請求項1記載の剤型。
【請求項18】
滑剤がステアリン酸マグネシウムである、請求項17記載の剤型。
【請求項19】
剤型の核剤が、粒状物および粒状物外成分の混合物からなり、ここで、該粒状物は微粉化メシル酸サキナビル、結合剤、担体、および崩壊剤を含み、そして該粒状物外成分は崩壊剤を含み、粒状物中の崩壊剤と粒状物外成分中の崩壊剤との比が約3:1〜約1:1である、請求項1記載の剤型。
【請求項20】
さらに、核剤の粒状物外成分中に含まれる滑剤を含む、請求項19記載の剤型。
【請求項21】
錠剤、カプセルおよびカプレットからなる群より選択される、請求項1〜20のいずれか1項記載の剤型。
【請求項22】
約400mg〜約1.5gの重さを有する、請求項1〜21のいずれか1項記載の剤型。
【請求項23】
遊離塩基に換算して約200〜約800mg量の微粉化メシル酸サキナビル、薬剤的に許容しうる水溶性結合剤、薬剤的に許容しうる崩壊剤、および薬剤的に許容しうる担体を含む、メシル酸サキナビルの固形単位経口投与剤型。
【請求項24】
微粉化メシル酸サキナビルが250〜約700mgの量で存在する、請求項23記載の固形単位経口投与剤型。
【請求項25】
メシル酸サキナビルが結晶形態で存在する、請求項1〜24のいずれか1項記載の固形単位経口投与剤型。
【請求項26】
微粉化メシル酸サキナビル、薬剤的に許容しうる水溶性結合剤、薬剤的に許容しうる担体および医薬活性崩壊剤の混合物を微細造粒することを含み、そしてそれに続く摩砕工程を含む、請求項1〜25のいずれか1項記載の固形単位経口投与剤型の製造方法。
【請求項27】
工程:
a)単位投与量あたり、(遊離塩基に換算して)約200〜約800mg量の微粉化メシル酸サキナビルと、親水性担体および崩壊剤とを、高せん断造粒機中で混合し、
b)撹拌しながら、工程(a)からの粉末混合物に結合剤水溶液を噴霧するか、またはゆっくり添加して最適な造粒終点を達成させ、
c)工程(b)からの湿潤粒状物を解砕して均一な粒状物にし、
d)工程(c)からの湿潤粒状物を、40〜50℃に設定した強制空気循環炉の中で、または入口空気温度50〜60℃の流動床乾燥機の中で、粒状物の湿分が1.5〜2%になるまで乾燥し、
e)工程(d)からの乾燥粒状物を摩砕し、
f)工程(e)からの摩砕粒状物を、適切な錠剤希釈剤と混合し、
g)工程(f)からの混合物を、適切な滑剤で潤滑し、
h)工程(g)からの最終混合物を、錠剤プレス上で圧縮成形し、
i)工程(h)からの錠剤を、水性フィルムでコーティングする;
を含む、請求項26記載の方法。
【請求項28】
治療に用いるための、請求項1〜25のいずれか1項記載の固形単位経口投与剤型。
【請求項29】
HIV介在疾患の治療に用いるための、請求項1〜25のいずれか1項記載の固形単位経口投与剤型。
【請求項30】
HIV介在疾患の治療用薬剤を製造するための、請求項1〜25のいずれか1項記載の固形単位経口投与剤型の使用。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2010−280707(P2010−280707A)
【公開日】平成22年12月16日(2010.12.16)
【国際特許分類】
【出願番号】特願2010−181350(P2010−181350)
【出願日】平成22年8月13日(2010.8.13)
【分割の表示】特願2006−519808(P2006−519808)の分割
【原出願日】平成16年7月5日(2004.7.5)
【出願人】(591003013)エフ.ホフマン−ラ ロシュ アーゲー (1,754)
【氏名又は名称原語表記】F. HOFFMANN−LA ROCHE AKTIENGESELLSCHAFT
【Fターム(参考)】
【公開日】平成22年12月16日(2010.12.16)
【国際特許分類】
【出願日】平成22年8月13日(2010.8.13)
【分割の表示】特願2006−519808(P2006−519808)の分割
【原出願日】平成16年7月5日(2004.7.5)
【出願人】(591003013)エフ.ホフマン−ラ ロシュ アーゲー (1,754)
【氏名又は名称原語表記】F. HOFFMANN−LA ROCHE AKTIENGESELLSCHAFT
【Fターム(参考)】
[ Back to top ]