
メタアラミド繊維の製造方法
【課題】塩を副生しない合成法により得られたメタアラミドオリゴマーを溶融紡糸してメタアラミド繊維を製造する方法を提供する。
【解決手段】ジアミン成分として3,4’−ジアミノジフェニルエーテルとメタフェニレンジアミンを全ジアミン成分に対し30mol%/70mol%〜70mol%/30mol%の比率で用い、酸成分としてイソフタル酸を用い、これらを加熱することでN−メチル−2−ピロリドン中で測定した溶液対数粘度0.1〜0.4dL/gのメタアラミドオリゴマーを得、これを溶融繊維化するメタアラミド繊維の製造方法。
【解決手段】ジアミン成分として3,4’−ジアミノジフェニルエーテルとメタフェニレンジアミンを全ジアミン成分に対し30mol%/70mol%〜70mol%/30mol%の比率で用い、酸成分としてイソフタル酸を用い、これらを加熱することでN−メチル−2−ピロリドン中で測定した溶液対数粘度0.1〜0.4dL/gのメタアラミドオリゴマーを得、これを溶融繊維化するメタアラミド繊維の製造方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、メタアラミド繊維の製造方法に関するものである。さらに詳しくは、従来の溶液紡糸とは異なり溶融紡糸によるメタアラミド繊維の製造方法に関するものである。
【背景技術】
【0002】
メタアラミド繊維は耐熱繊維として、モーターや変圧器等の電気絶縁紙、焼却炉のバグフィルター、ハニカム構造体、耐熱作業服などに広く利用されている。従来、ジアミンと芳香族ジカルボン酸の酸クロリドを溶液中で反応させる酸クロ法でポリマーを得、これを有機溶媒を用いて溶液紡糸することでメタアラミド繊維を得ていた。しかしながら、酸クロ法ではアルカリの中和のために必ず塩が発生し、これの除去が必要であり製造プロセスが煩雑であった。さらに有機溶媒を用いた溶液紡糸を行うため溶液回収等のコストがかさむばかりか環境負荷も大きいという問題があった(特許文献1)。通常、耐熱繊維として用いられるメタアラミドは熱可塑性が無いため不融という耐熱作業服には適した特性を有しているが、逆に溶融紡糸が不可能であり溶液紡糸を行わざるを得ないものであった。
【0003】
このため、ラクタムをメタアラミドに共存させることで熱可塑性を発現させ、メタアラミドの溶融紡糸を行う努力も過去にされた(特許文献2)が、メタアラミドの合成はジカルボン酸の酸クロリドとラクタムを反応させる酸クロ法の延長であり、ポリマー合成上の問題が解決したわけではなかった。さらに、溶融紡糸時に有害なラクタムがガスとしてポリマー中から発生し、安全・環境上の問題が発生する可能性も懸念された。
【0004】
一方、メタアラミドの合成に酸クロリドを用いない直接重合法の開示がある(特許文献3)。しかしながら、溶融紡糸性に関する知見は開示されていない。
【0005】
もし、塩を副生しない合成法により得られたメタアラミドを溶融紡糸できれば、安全・環境上だけでなく製造設備の簡素化が可能であり、産業的には非常に有用であると考えられる。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特公昭48−17551号公報
【特許文献2】特開平2−145620号公報
【特許文献3】特開2009−256610号公報
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明の課題は、塩を副生しない合成法により得られたメタアラミドを溶融紡糸するメタアラミド繊維の製造方法を提供することにある。
【課題を解決するための手段】
【0008】
上記課題を解決するための本発明に係る製造方法は、以下のとおりである。
(1)ジアミン成分として3,4’−ジアミノジフェニルエーテルとメタフェニレンジアミンを全ジアミン成分に対し30mol%/70mol%〜70mol%/30mol%の比率で用い、酸成分としてイソフタル酸を用い、これらを加熱することでN−メチル−2−ピロリドン中で測定した溶液対数粘度0.1〜0.4dL/gのメタアラミドオリゴマーを得、これを溶融繊維化するメタアラミド繊維の製造方法。
(2)メタアラミドオリゴマーの溶融粘度を100〜2000Pa・sとして溶融繊維化する(1)記載のメタアラミド繊維の製造方法。
(3)メタアラミドオリゴマーの末端の40%以上をアミン末端とする(1)または(2)記載のメタアラミド繊維の製造方法。
(4)(1)〜(3)のいずれかの方法で得られたメタアラミド繊維を固相重合するメタアラミド繊維の製造方法。
【発明の効果】
【0009】
本発明に係るメタアラミド繊維の製造方法によれば、安全・環境上だけでなく製造設備の簡素化が可能となり、メタアラミド繊維のトータルコストダウンが図れる。
【図面の簡単な説明】
【0010】
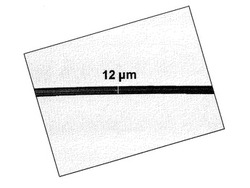
【図1】本発明により得られるメタアラミド繊維の側面写真である。
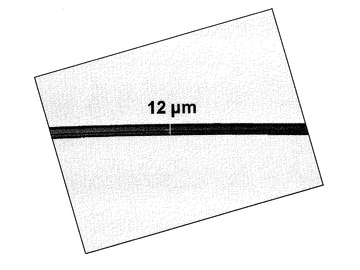
【図2】本発明により得られるメタアラミド繊維の一例である。
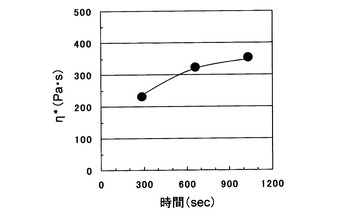
【図3】本発明で用いるメタアラミドオリゴマーの溶融粘度の時間依存性を示す図である。
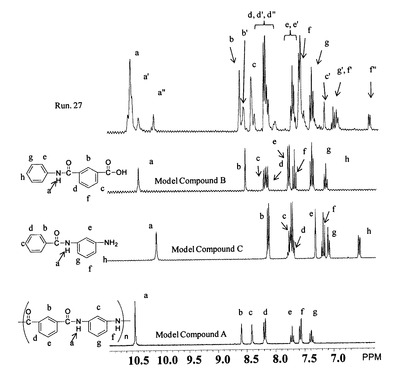
【図4】本発明の実施例および比較例において測定された1H NMRスペクトルの例を示すスペクトル図である。
【発明を実施するための形態】
【0011】
以下に、本発明について、望ましい実施の形態とともに詳細に説明する。
本発明でいうメタアラミドとは、芳香族ジアミンと芳香族ジカルボン酸からなる全芳香族ポリアミドにおいて、ジアミン成分、ジカルボン酸成分ともメタ位にアミン、カルボン酸が結合しているものである。本発明では従来技術とは異なり、酸クロリドを用いずジアミンとジカルボン酸の直接重合によりメタアラミドを得ることが重要であり、これより酸クロ法とは異なり塩の副生が無く、これを除くためのプロセスや設備が不要となるため従来に比べ設備を簡素化しコストダウンに寄与することが可能となる。
【0012】
本発明は3,4’−ジアミノジフェニルエーテル(3,4’−DPE)とメタフェニレンジアミン(mPDA)を30mol%/70mol%〜70mol%/30mol%の比率で用い、ジカルボン酸成分としてイソフタル酸(IPA)としたメタアラミドを溶融繊維化するものであるが、これは従来技術とは異なる3つの重要な発見に基づいている。一つ目は、ジアミン成分としてメタアラミドに3,4’−DPEを共重合することで、メタアラミドに熱可塑性が発現することである。二つ目は、3,4’−DPEが共重合されたメタアラミドでは溶液対数粘度が0.1〜0.4dL/gのオリゴマー(低分子量体)であっても溶融曳糸性を発現することである。3つ目は、3,4’−DPE/mPDA/IPA系メタアラミドオリゴマーでは溶融曳糸性を有するものの、溶液対数粘度が1.0dL/g以上のポリマー(高分子量体)となると熱可塑性を失い不融となることである。
【0013】
これらより、3,4’−DPE/mPDA/IPA系メタアラミドはオリゴマー状態で溶融紡糸を行い、その後高分子量化することで不融繊維とでき、従来のmPDA/IPA系メタアラミドと同様に扱うことができるという大きなメリットがある。
【0014】
このようなメリットを最大限に発揮するためにはメタアラミドを以下のような組成とすることが重要である。すなわち、3,4’−DPEとmPDAを全ジアミン成分に対して30mol%/70mol%〜70mol%/30mol%の比率で用い、ジカルボン酸成分としてIPAを用いることである。ジアミン成分中の3,4’−DPE比率は多い方がメタアラミドの熱可塑性が顕著となるが、30mol%以上とすることでメタアラミドオリゴマーを溶融繊維化するための十分な熱可塑性を付与することができる。好ましくは40mol%以上である。一方、3,4’−DPE比率が高すぎるとメタアラミドを高分子量化しても不融化し難くなるため70mol%以下とすることが重要である。mPDA比率は30mol%以上とすることでメタアラミドを高分子量化した時に十分な不融性を付与することができる。mPDA比率は好ましくは40mol%以上である。また、mPDA比率を多くする方がメタアラミドを高分子量化した時のガラス転移温度を高くできるため好ましい。この観点からもmPDA比率は30mol%以上であることが好ましい。発明者らの検討によると、高分子量化したメタアラミドのガラス転移温度は、mPDA100mol%では276℃、75mol%で264℃、50mol%で253℃、30mol%で244℃、20mol%で240℃であった。なお、ガラス転移温度の測定法は以下のとおりであった。すなわち、セイコーインスツルメント社製ロボットDSC RDC6220を用い、サンプル重量を5.0mg±0.4mgとし、昇温条件を25℃→350℃→25℃→350℃(昇温速度10℃/分)として、2nd runにてガラス転移温度を求めた。
【0015】
また、メタアラミドオリゴマーの分子量は溶液粘度で規定する。本発明ではN−メチル−2−ピロリドンにメタアラミドを濃度0.5g/dLで溶解し、30℃にて測定した溶液対数粘度が0.1〜0.4dL/gのメタアラミドオリゴマーを用いることが重要である。溶液対数粘度をこの範囲とすることで十分な溶融曳糸性が発現させることができる。好ましくは0.10〜0.20dL/gであればメタアラミドオリゴマーの溶融粘度を通常のポリエステル(ポリエチレンテレフタレート)並みとすることができ、既存のポリエステルの溶融紡糸装置を流用できる可能性が有り、産業的意義は大きい。
【0016】
本発明では、メタアラミドオリゴマーの溶融粘度は100〜2000Pa・sとして溶融繊維化することが好ましい。溶融繊維化をスムーズに進め、繊維を細くするには低溶融粘度である方が好ましく、溶融粘度は400Pa・s以下であることが好ましい。一方、溶融粘度は低すぎると逆に曳糸性が低下する場合があるので100Pa・s以上であることが好ましい。本発明で言う溶融粘度とは平行平板式のレオメーターを用い、溶融繊維化温度にて、歪0.5deg.、歪速度62.8rad/秒で測定したη*を溶融粘度とする。例えば末端基がアミンとカルボン酸(1:1)である溶液対数粘度0.17の3,4’−DPE/mPDA/IPA=0.5/0.5/1.0のオリゴマーを窒素雰囲気下300℃で12分間貯留した時のη*は300Pa・sであり、衣料用ポリエステル繊維に用いる極限粘度0.63のホモポリエチレンテレフタレートの300℃でのη*(220Pa・s)に近い値である。なお、本発明で用いるメタアラミドオリゴマーはレオメーターでの溶融粘度測定過程で増粘することが認められており、溶融滞留時間と溶融粘度、溶融曳糸性の関係を調べておくことが好ましい。
【0017】
また、メタアラミドオリゴマーの末端基はカルボン酸末端とアミン末端が有り得るが、アミン末端リッチとした方が溶融粘度が低下し、溶融紡糸には適している。この観点から末端基の40%以上がアミン末端であることが好ましい。末端基量は滴定の他、核磁気共鳴(NMR)などにより定量することができる。
【0018】
本発明で得られるメタアラミド繊維は高重合度化し、ガラス転移温度として240℃以上とすることが、耐熱性の観点から好ましい。
【0019】
以上、本発明で用いるメタアラミドの組成について述べてきたが、以下に、本発明のメタアラミド繊維の製造方法を詳述する。
【0020】
まず、メタアラミドオリゴマーの合成であるが、基本的には特許文献3記載の方法で行うことができる。3,4’−DPE、mPDA、IPAを所望の量だけ秤取り、不活性ガス気流下で加熱する。この時、mPDAは昇華性があるため開放系や真空系で合成を行う際にはmPDAをやや過剰(10mol%程度過剰)とすることが好ましい。mPDAの昇華を抑制するため加圧系で合成を行う時には全て等molとしても良い。また、分子鎖末端はジアミン成分とジカルボン酸成分の仕込み比で制御でき、ジアミン成分とジカルボン酸成分が等molであればアミン末端とカルボン酸末端がほぼ1:1であるが、ジカルボン酸成分が過剰の場合にはカルボン酸末端基が、ジアミン成分が過剰の場合にはアミン末端基が多くなる。本発明ではオリゴマー合成から繊維化までを溶融系で行い、溶液による精製工程を含まない方が設備簡素化の観点から好ましい。このため、ジアミン成分とジカルボン酸成分を全て反応させるようアミン成分とジカルボン酸成分の仕込み量を調整することが好ましい。
【0021】
次に合成温度であるが、3,4’−DPE、mPDA、IPAの融点以上であり、さらに合成されるオリゴマーの融点以上とする観点からは240℃以上であることが好ましい。こうすることで原料であるモノマーやオリゴマーを融解後、撹拌することができ、合成反応を効率よく進めることができる。反応時間は所望の溶液相対粘度となるように適宜決めれば良い。
【0022】
次に、このようにして得られるメタアラミドオリゴマーを溶融繊維化するが、この時、溶融繊維化の方法には特に制限は無く、公知の方法・装置を用いることができる。例えば、定量フィーダーから一軸押出混練機あるいは二軸押出混練機にて溶融し、公知の紡糸口金から吐出することができる。この時、ガット切れ防止のため、混練機にてベントによりガス抜きを行ってもよい。また、口金の形状は、丸孔の他に、Y孔、C孔などの異形口金も使用することができる。ただし、先に述べたように溶融粘度は低い方が曳糸性には有利であるため、メタアラミドオリゴマーの分子量、すなわち溶液対数粘度や分子鎖末端基組成を適切に選んだり、紡糸温度を高温化することが有効である。しかし、紡糸温度を高温化するために高温紡糸装置を新設するとかえってコストアップとなる場合も有るので、溶液対数粘度や分子鎖末端基組成を適切に選ぶことが好ましい。
【0023】
本発明により得られるメタアラミド繊維は、従来公知のメタアラミド繊維と同様に使用できるだけでなく、溶融紡糸を行うため、繊維断面を所望の形状(多角形、扁平、中空など)に設計できるため、繊維断面形状制御による様々な機能向上が期待できる。また、繊維直径制御にも自由度ができ、マイクロファイバーやナノファイバー化することも可能である。さらに、スパンボンドやメルトブロー等を行えば、紡糸一発で不織布化できるため、従来の抄紙法やエレクトロスピニング法に比べ大幅なコストダウンも可能となることが期待される。
【実施例】
【0024】
以下、本発明を実施例に基づいて詳細に説明する。なお、実施例中の測定方法は以下の方法を用いた。
【0025】
A.溶液対数粘度(η)
N−メチル−2−ピロリドンにメタアラミドを濃度0.5g/dLで溶解し、30℃にてオストワルド粘度計にて測定した。
【0026】
B.溶融粘度
UBM社製レオメーターであるRHEOSOL−G3000において平行平板を用い、動的粘弾性を測定し、歪速度62.8rad/秒のη*を溶融粘度とした。測定条件は以下のとおり。
平行円板間距離 0.5mm
歪 0.5deg.
歪速度 0.628〜62.8rad/秒までを繰り返し測定した。
なお、所望の温度に到達後3分間放置した後、データ取得を開始し、この時刻を0分とした。
【0027】
C.溶融曳糸性
溶融曳糸性は上記溶融粘度測定終了直後にヒーターブロックを開け、平行円板を引き離し、糸を曳くかどうかで判断した。より具体的には、平行円板全面にわたって4cm以上の曳糸性を示したものを曳糸性良好(◎)、平行円板の縁部のみが曳糸され、その長さが2cm以上のものを曳糸性有り(○)、平行円板の縁部のみが曳糸され、その長さが2cm未満のものを曳糸性無し(△)、全く曳糸せずしかも溶融粘度測定不能のものを熱可塑性無し(×)とした。すなわち、◎および○評価のものを曳糸性有りとした。
【0028】
D.ポリマ末端基量
図4に示すモデル物質A,B,Cを作製し、1H NMRスペクトルを測定した。ここにおいて、aのシグナルはオリゴマー内部のアミド水素、a’ のシグナルはカルボン酸末端のアミド水素、a”のシグナルはアミン末端のアミド水素にそれぞれ対応する。そこで、各サンプルの1H NMRスペクトルを測定し、シグナルの積分比(a’の積分値)/(a’の積分値+a”の積分値)x100(%)を、アミン末端基の量とした。
【0029】
[実施例1]
3,4’−DPEを2.5mmol、mPDAを2.75mmol、IPAを5mmol秤量し、アルゴン置換された試験管に投入した。これをマグネティックスターラーで撹拌しながらマントルヒーターにて室温から260℃まで昇温し、さらに260℃で1時間保持し、3,4’−DPE/mPDA/IPA=0.5/0.5/1.0のメタアラミドオリゴマーを得た。これを冷却後、N−メチル−2−ピロリドンに溶解しメタノールを加えて沈殿させ、これを濾別後乾燥し精製した。このオリゴマーのηを測定したところ0.17dL/gであった。また、アミン末端基数とカルボン酸末端基数の比は1:1(アミン末端基量=50%)であった。
【0030】
そしてこれを0.2g程度秤取り、レオメーターで300℃にて溶融粘度を測定し、測定終了後すぐに測定用の円板を上昇させ溶融曳糸性を調べたところ、平板間で糸を曳き溶融メタアラミド繊維を得ることができた(図2)。この時のデータ取得開始から最後の歪速度62.8rad/秒での測定までの時間は1035秒(17分15秒)で、η*は353Pa・s(表1)であった。また、これを冷却した後、溶液相対粘度を測定したところ0.23dL/gであった(表1)。なお、表1には溶融曳糸試験を行った状態でのη*およびηを記載している。また、溶融粘度の経時変化を調べたところ図3のように、加熱時間とともに増粘傾向を示し、ηの上昇と呼応していた。
【0031】
[実施例2、3]
3,4’−DPE、mPDAの仕込み比を変更し、実施例1と同様の操作で、3,4’−DPE/mPDAの組成比を0.35/0.65(実施例2)および0.65/0.35(実施例3)の精製されたメタアラミドオリゴマーを合成した。これらオリゴマーのηはどちらも0.13dL/gであった。いずれも優れた溶融曳糸性を示し、メタアラミドオリゴマー繊維を得ることができた(表1)。
【0032】
[比較例1〜3]
3,4’−DPE、mPDAの仕込み比を変更し、実施例1と同様の操作で、表1に示した組成のメタアラミドオリゴマーを得た。しかし、mPDAが70mol%を超える水準では溶融曳糸性が不良であった(比較例1、2)。比較例3では溶融曳糸性は認められたものの、mPDA含有量が少ないため、個分子量化したとしても耐熱性が不足すると考えられた。
【0033】
[比較例4]
実施例2で得たメタアラミドオリゴマーをさらに固相重合し、ηが0.41dL/gのメタアラミドオリゴマーを得た。これの溶融曳糸性を320℃で調べたが、溶融曳糸性評価は不良(評価:△)であった。溶融曳糸性評価時のηは、0.44dL/gであった。
【0034】
[実施例4、5]
溶融曳糸性評価を行う温度を280℃および320℃として、実施例1と同様に溶融曳糸性評価を行った。結果を表1に示すが、いずれも溶融曳糸性良好であった。
【0035】
[実施例6]
実施例1で作製したメタアラミドオリゴマーをアルゴン置換した試験管に投入し、マントルヒーターを用い300℃まで昇温後17分間保持した。その後、ステンレス製のニードルを試験管に差込み、これに溶融したメタアラミドオリゴマーを付着させた後、ニードルを一旦試験の口に付着させた後勢いよくニードルを引き、メタアラミドオリゴマーの繊維を数メートル得た。この繊維の側面を光学顕微鏡で観察したところ、繊維の幅が最も狭いところで12μmであった(図1)。図3に示した溶融粘度の時間依存性から溶融曳糸時の溶融粘度は340Pa・s、ηは0.22dL/gと見積もられた。
【0036】
[実施例7]
実施例6で得た細いメタアラミドオリゴマー繊維を金属製のリール状物に巻きつけ、減圧下200℃に1週間保持し固相重合を行った。得られた繊維は不融性を有しており、十分高分子量化できていることが確認できた。
【0037】
[実施例8]
実施例6で得たメタアラミドオリゴマー繊維を空気中(常圧)200℃で無荷重下1000時間処理を行ったが、強度低下は認められず優れた長期耐熱性を示した。
【0038】
【表1】

【産業上の利用可能性】
【0039】
本発明に係るメタアラミド繊維の製造方法はあらゆる用途に適用可能であり、とくに、安全及び環境上の配慮が要求される用途に好適である。
【技術分野】
【0001】
本発明は、メタアラミド繊維の製造方法に関するものである。さらに詳しくは、従来の溶液紡糸とは異なり溶融紡糸によるメタアラミド繊維の製造方法に関するものである。
【背景技術】
【0002】
メタアラミド繊維は耐熱繊維として、モーターや変圧器等の電気絶縁紙、焼却炉のバグフィルター、ハニカム構造体、耐熱作業服などに広く利用されている。従来、ジアミンと芳香族ジカルボン酸の酸クロリドを溶液中で反応させる酸クロ法でポリマーを得、これを有機溶媒を用いて溶液紡糸することでメタアラミド繊維を得ていた。しかしながら、酸クロ法ではアルカリの中和のために必ず塩が発生し、これの除去が必要であり製造プロセスが煩雑であった。さらに有機溶媒を用いた溶液紡糸を行うため溶液回収等のコストがかさむばかりか環境負荷も大きいという問題があった(特許文献1)。通常、耐熱繊維として用いられるメタアラミドは熱可塑性が無いため不融という耐熱作業服には適した特性を有しているが、逆に溶融紡糸が不可能であり溶液紡糸を行わざるを得ないものであった。
【0003】
このため、ラクタムをメタアラミドに共存させることで熱可塑性を発現させ、メタアラミドの溶融紡糸を行う努力も過去にされた(特許文献2)が、メタアラミドの合成はジカルボン酸の酸クロリドとラクタムを反応させる酸クロ法の延長であり、ポリマー合成上の問題が解決したわけではなかった。さらに、溶融紡糸時に有害なラクタムがガスとしてポリマー中から発生し、安全・環境上の問題が発生する可能性も懸念された。
【0004】
一方、メタアラミドの合成に酸クロリドを用いない直接重合法の開示がある(特許文献3)。しかしながら、溶融紡糸性に関する知見は開示されていない。
【0005】
もし、塩を副生しない合成法により得られたメタアラミドを溶融紡糸できれば、安全・環境上だけでなく製造設備の簡素化が可能であり、産業的には非常に有用であると考えられる。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特公昭48−17551号公報
【特許文献2】特開平2−145620号公報
【特許文献3】特開2009−256610号公報
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明の課題は、塩を副生しない合成法により得られたメタアラミドを溶融紡糸するメタアラミド繊維の製造方法を提供することにある。
【課題を解決するための手段】
【0008】
上記課題を解決するための本発明に係る製造方法は、以下のとおりである。
(1)ジアミン成分として3,4’−ジアミノジフェニルエーテルとメタフェニレンジアミンを全ジアミン成分に対し30mol%/70mol%〜70mol%/30mol%の比率で用い、酸成分としてイソフタル酸を用い、これらを加熱することでN−メチル−2−ピロリドン中で測定した溶液対数粘度0.1〜0.4dL/gのメタアラミドオリゴマーを得、これを溶融繊維化するメタアラミド繊維の製造方法。
(2)メタアラミドオリゴマーの溶融粘度を100〜2000Pa・sとして溶融繊維化する(1)記載のメタアラミド繊維の製造方法。
(3)メタアラミドオリゴマーの末端の40%以上をアミン末端とする(1)または(2)記載のメタアラミド繊維の製造方法。
(4)(1)〜(3)のいずれかの方法で得られたメタアラミド繊維を固相重合するメタアラミド繊維の製造方法。
【発明の効果】
【0009】
本発明に係るメタアラミド繊維の製造方法によれば、安全・環境上だけでなく製造設備の簡素化が可能となり、メタアラミド繊維のトータルコストダウンが図れる。
【図面の簡単な説明】
【0010】
【図1】本発明により得られるメタアラミド繊維の側面写真である。
【図2】本発明により得られるメタアラミド繊維の一例である。
【図3】本発明で用いるメタアラミドオリゴマーの溶融粘度の時間依存性を示す図である。
【図4】本発明の実施例および比較例において測定された1H NMRスペクトルの例を示すスペクトル図である。
【発明を実施するための形態】
【0011】
以下に、本発明について、望ましい実施の形態とともに詳細に説明する。
本発明でいうメタアラミドとは、芳香族ジアミンと芳香族ジカルボン酸からなる全芳香族ポリアミドにおいて、ジアミン成分、ジカルボン酸成分ともメタ位にアミン、カルボン酸が結合しているものである。本発明では従来技術とは異なり、酸クロリドを用いずジアミンとジカルボン酸の直接重合によりメタアラミドを得ることが重要であり、これより酸クロ法とは異なり塩の副生が無く、これを除くためのプロセスや設備が不要となるため従来に比べ設備を簡素化しコストダウンに寄与することが可能となる。
【0012】
本発明は3,4’−ジアミノジフェニルエーテル(3,4’−DPE)とメタフェニレンジアミン(mPDA)を30mol%/70mol%〜70mol%/30mol%の比率で用い、ジカルボン酸成分としてイソフタル酸(IPA)としたメタアラミドを溶融繊維化するものであるが、これは従来技術とは異なる3つの重要な発見に基づいている。一つ目は、ジアミン成分としてメタアラミドに3,4’−DPEを共重合することで、メタアラミドに熱可塑性が発現することである。二つ目は、3,4’−DPEが共重合されたメタアラミドでは溶液対数粘度が0.1〜0.4dL/gのオリゴマー(低分子量体)であっても溶融曳糸性を発現することである。3つ目は、3,4’−DPE/mPDA/IPA系メタアラミドオリゴマーでは溶融曳糸性を有するものの、溶液対数粘度が1.0dL/g以上のポリマー(高分子量体)となると熱可塑性を失い不融となることである。
【0013】
これらより、3,4’−DPE/mPDA/IPA系メタアラミドはオリゴマー状態で溶融紡糸を行い、その後高分子量化することで不融繊維とでき、従来のmPDA/IPA系メタアラミドと同様に扱うことができるという大きなメリットがある。
【0014】
このようなメリットを最大限に発揮するためにはメタアラミドを以下のような組成とすることが重要である。すなわち、3,4’−DPEとmPDAを全ジアミン成分に対して30mol%/70mol%〜70mol%/30mol%の比率で用い、ジカルボン酸成分としてIPAを用いることである。ジアミン成分中の3,4’−DPE比率は多い方がメタアラミドの熱可塑性が顕著となるが、30mol%以上とすることでメタアラミドオリゴマーを溶融繊維化するための十分な熱可塑性を付与することができる。好ましくは40mol%以上である。一方、3,4’−DPE比率が高すぎるとメタアラミドを高分子量化しても不融化し難くなるため70mol%以下とすることが重要である。mPDA比率は30mol%以上とすることでメタアラミドを高分子量化した時に十分な不融性を付与することができる。mPDA比率は好ましくは40mol%以上である。また、mPDA比率を多くする方がメタアラミドを高分子量化した時のガラス転移温度を高くできるため好ましい。この観点からもmPDA比率は30mol%以上であることが好ましい。発明者らの検討によると、高分子量化したメタアラミドのガラス転移温度は、mPDA100mol%では276℃、75mol%で264℃、50mol%で253℃、30mol%で244℃、20mol%で240℃であった。なお、ガラス転移温度の測定法は以下のとおりであった。すなわち、セイコーインスツルメント社製ロボットDSC RDC6220を用い、サンプル重量を5.0mg±0.4mgとし、昇温条件を25℃→350℃→25℃→350℃(昇温速度10℃/分)として、2nd runにてガラス転移温度を求めた。
【0015】
また、メタアラミドオリゴマーの分子量は溶液粘度で規定する。本発明ではN−メチル−2−ピロリドンにメタアラミドを濃度0.5g/dLで溶解し、30℃にて測定した溶液対数粘度が0.1〜0.4dL/gのメタアラミドオリゴマーを用いることが重要である。溶液対数粘度をこの範囲とすることで十分な溶融曳糸性が発現させることができる。好ましくは0.10〜0.20dL/gであればメタアラミドオリゴマーの溶融粘度を通常のポリエステル(ポリエチレンテレフタレート)並みとすることができ、既存のポリエステルの溶融紡糸装置を流用できる可能性が有り、産業的意義は大きい。
【0016】
本発明では、メタアラミドオリゴマーの溶融粘度は100〜2000Pa・sとして溶融繊維化することが好ましい。溶融繊維化をスムーズに進め、繊維を細くするには低溶融粘度である方が好ましく、溶融粘度は400Pa・s以下であることが好ましい。一方、溶融粘度は低すぎると逆に曳糸性が低下する場合があるので100Pa・s以上であることが好ましい。本発明で言う溶融粘度とは平行平板式のレオメーターを用い、溶融繊維化温度にて、歪0.5deg.、歪速度62.8rad/秒で測定したη*を溶融粘度とする。例えば末端基がアミンとカルボン酸(1:1)である溶液対数粘度0.17の3,4’−DPE/mPDA/IPA=0.5/0.5/1.0のオリゴマーを窒素雰囲気下300℃で12分間貯留した時のη*は300Pa・sであり、衣料用ポリエステル繊維に用いる極限粘度0.63のホモポリエチレンテレフタレートの300℃でのη*(220Pa・s)に近い値である。なお、本発明で用いるメタアラミドオリゴマーはレオメーターでの溶融粘度測定過程で増粘することが認められており、溶融滞留時間と溶融粘度、溶融曳糸性の関係を調べておくことが好ましい。
【0017】
また、メタアラミドオリゴマーの末端基はカルボン酸末端とアミン末端が有り得るが、アミン末端リッチとした方が溶融粘度が低下し、溶融紡糸には適している。この観点から末端基の40%以上がアミン末端であることが好ましい。末端基量は滴定の他、核磁気共鳴(NMR)などにより定量することができる。
【0018】
本発明で得られるメタアラミド繊維は高重合度化し、ガラス転移温度として240℃以上とすることが、耐熱性の観点から好ましい。
【0019】
以上、本発明で用いるメタアラミドの組成について述べてきたが、以下に、本発明のメタアラミド繊維の製造方法を詳述する。
【0020】
まず、メタアラミドオリゴマーの合成であるが、基本的には特許文献3記載の方法で行うことができる。3,4’−DPE、mPDA、IPAを所望の量だけ秤取り、不活性ガス気流下で加熱する。この時、mPDAは昇華性があるため開放系や真空系で合成を行う際にはmPDAをやや過剰(10mol%程度過剰)とすることが好ましい。mPDAの昇華を抑制するため加圧系で合成を行う時には全て等molとしても良い。また、分子鎖末端はジアミン成分とジカルボン酸成分の仕込み比で制御でき、ジアミン成分とジカルボン酸成分が等molであればアミン末端とカルボン酸末端がほぼ1:1であるが、ジカルボン酸成分が過剰の場合にはカルボン酸末端基が、ジアミン成分が過剰の場合にはアミン末端基が多くなる。本発明ではオリゴマー合成から繊維化までを溶融系で行い、溶液による精製工程を含まない方が設備簡素化の観点から好ましい。このため、ジアミン成分とジカルボン酸成分を全て反応させるようアミン成分とジカルボン酸成分の仕込み量を調整することが好ましい。
【0021】
次に合成温度であるが、3,4’−DPE、mPDA、IPAの融点以上であり、さらに合成されるオリゴマーの融点以上とする観点からは240℃以上であることが好ましい。こうすることで原料であるモノマーやオリゴマーを融解後、撹拌することができ、合成反応を効率よく進めることができる。反応時間は所望の溶液相対粘度となるように適宜決めれば良い。
【0022】
次に、このようにして得られるメタアラミドオリゴマーを溶融繊維化するが、この時、溶融繊維化の方法には特に制限は無く、公知の方法・装置を用いることができる。例えば、定量フィーダーから一軸押出混練機あるいは二軸押出混練機にて溶融し、公知の紡糸口金から吐出することができる。この時、ガット切れ防止のため、混練機にてベントによりガス抜きを行ってもよい。また、口金の形状は、丸孔の他に、Y孔、C孔などの異形口金も使用することができる。ただし、先に述べたように溶融粘度は低い方が曳糸性には有利であるため、メタアラミドオリゴマーの分子量、すなわち溶液対数粘度や分子鎖末端基組成を適切に選んだり、紡糸温度を高温化することが有効である。しかし、紡糸温度を高温化するために高温紡糸装置を新設するとかえってコストアップとなる場合も有るので、溶液対数粘度や分子鎖末端基組成を適切に選ぶことが好ましい。
【0023】
本発明により得られるメタアラミド繊維は、従来公知のメタアラミド繊維と同様に使用できるだけでなく、溶融紡糸を行うため、繊維断面を所望の形状(多角形、扁平、中空など)に設計できるため、繊維断面形状制御による様々な機能向上が期待できる。また、繊維直径制御にも自由度ができ、マイクロファイバーやナノファイバー化することも可能である。さらに、スパンボンドやメルトブロー等を行えば、紡糸一発で不織布化できるため、従来の抄紙法やエレクトロスピニング法に比べ大幅なコストダウンも可能となることが期待される。
【実施例】
【0024】
以下、本発明を実施例に基づいて詳細に説明する。なお、実施例中の測定方法は以下の方法を用いた。
【0025】
A.溶液対数粘度(η)
N−メチル−2−ピロリドンにメタアラミドを濃度0.5g/dLで溶解し、30℃にてオストワルド粘度計にて測定した。
【0026】
B.溶融粘度
UBM社製レオメーターであるRHEOSOL−G3000において平行平板を用い、動的粘弾性を測定し、歪速度62.8rad/秒のη*を溶融粘度とした。測定条件は以下のとおり。
平行円板間距離 0.5mm
歪 0.5deg.
歪速度 0.628〜62.8rad/秒までを繰り返し測定した。
なお、所望の温度に到達後3分間放置した後、データ取得を開始し、この時刻を0分とした。
【0027】
C.溶融曳糸性
溶融曳糸性は上記溶融粘度測定終了直後にヒーターブロックを開け、平行円板を引き離し、糸を曳くかどうかで判断した。より具体的には、平行円板全面にわたって4cm以上の曳糸性を示したものを曳糸性良好(◎)、平行円板の縁部のみが曳糸され、その長さが2cm以上のものを曳糸性有り(○)、平行円板の縁部のみが曳糸され、その長さが2cm未満のものを曳糸性無し(△)、全く曳糸せずしかも溶融粘度測定不能のものを熱可塑性無し(×)とした。すなわち、◎および○評価のものを曳糸性有りとした。
【0028】
D.ポリマ末端基量
図4に示すモデル物質A,B,Cを作製し、1H NMRスペクトルを測定した。ここにおいて、aのシグナルはオリゴマー内部のアミド水素、a’ のシグナルはカルボン酸末端のアミド水素、a”のシグナルはアミン末端のアミド水素にそれぞれ対応する。そこで、各サンプルの1H NMRスペクトルを測定し、シグナルの積分比(a’の積分値)/(a’の積分値+a”の積分値)x100(%)を、アミン末端基の量とした。
【0029】
[実施例1]
3,4’−DPEを2.5mmol、mPDAを2.75mmol、IPAを5mmol秤量し、アルゴン置換された試験管に投入した。これをマグネティックスターラーで撹拌しながらマントルヒーターにて室温から260℃まで昇温し、さらに260℃で1時間保持し、3,4’−DPE/mPDA/IPA=0.5/0.5/1.0のメタアラミドオリゴマーを得た。これを冷却後、N−メチル−2−ピロリドンに溶解しメタノールを加えて沈殿させ、これを濾別後乾燥し精製した。このオリゴマーのηを測定したところ0.17dL/gであった。また、アミン末端基数とカルボン酸末端基数の比は1:1(アミン末端基量=50%)であった。
【0030】
そしてこれを0.2g程度秤取り、レオメーターで300℃にて溶融粘度を測定し、測定終了後すぐに測定用の円板を上昇させ溶融曳糸性を調べたところ、平板間で糸を曳き溶融メタアラミド繊維を得ることができた(図2)。この時のデータ取得開始から最後の歪速度62.8rad/秒での測定までの時間は1035秒(17分15秒)で、η*は353Pa・s(表1)であった。また、これを冷却した後、溶液相対粘度を測定したところ0.23dL/gであった(表1)。なお、表1には溶融曳糸試験を行った状態でのη*およびηを記載している。また、溶融粘度の経時変化を調べたところ図3のように、加熱時間とともに増粘傾向を示し、ηの上昇と呼応していた。
【0031】
[実施例2、3]
3,4’−DPE、mPDAの仕込み比を変更し、実施例1と同様の操作で、3,4’−DPE/mPDAの組成比を0.35/0.65(実施例2)および0.65/0.35(実施例3)の精製されたメタアラミドオリゴマーを合成した。これらオリゴマーのηはどちらも0.13dL/gであった。いずれも優れた溶融曳糸性を示し、メタアラミドオリゴマー繊維を得ることができた(表1)。
【0032】
[比較例1〜3]
3,4’−DPE、mPDAの仕込み比を変更し、実施例1と同様の操作で、表1に示した組成のメタアラミドオリゴマーを得た。しかし、mPDAが70mol%を超える水準では溶融曳糸性が不良であった(比較例1、2)。比較例3では溶融曳糸性は認められたものの、mPDA含有量が少ないため、個分子量化したとしても耐熱性が不足すると考えられた。
【0033】
[比較例4]
実施例2で得たメタアラミドオリゴマーをさらに固相重合し、ηが0.41dL/gのメタアラミドオリゴマーを得た。これの溶融曳糸性を320℃で調べたが、溶融曳糸性評価は不良(評価:△)であった。溶融曳糸性評価時のηは、0.44dL/gであった。
【0034】
[実施例4、5]
溶融曳糸性評価を行う温度を280℃および320℃として、実施例1と同様に溶融曳糸性評価を行った。結果を表1に示すが、いずれも溶融曳糸性良好であった。
【0035】
[実施例6]
実施例1で作製したメタアラミドオリゴマーをアルゴン置換した試験管に投入し、マントルヒーターを用い300℃まで昇温後17分間保持した。その後、ステンレス製のニードルを試験管に差込み、これに溶融したメタアラミドオリゴマーを付着させた後、ニードルを一旦試験の口に付着させた後勢いよくニードルを引き、メタアラミドオリゴマーの繊維を数メートル得た。この繊維の側面を光学顕微鏡で観察したところ、繊維の幅が最も狭いところで12μmであった(図1)。図3に示した溶融粘度の時間依存性から溶融曳糸時の溶融粘度は340Pa・s、ηは0.22dL/gと見積もられた。
【0036】
[実施例7]
実施例6で得た細いメタアラミドオリゴマー繊維を金属製のリール状物に巻きつけ、減圧下200℃に1週間保持し固相重合を行った。得られた繊維は不融性を有しており、十分高分子量化できていることが確認できた。
【0037】
[実施例8]
実施例6で得たメタアラミドオリゴマー繊維を空気中(常圧)200℃で無荷重下1000時間処理を行ったが、強度低下は認められず優れた長期耐熱性を示した。
【0038】
【表1】
【産業上の利用可能性】
【0039】
本発明に係るメタアラミド繊維の製造方法はあらゆる用途に適用可能であり、とくに、安全及び環境上の配慮が要求される用途に好適である。
【特許請求の範囲】
【請求項1】
ジアミン成分として3,4’−ジアミノジフェニルエーテルとメタフェニレンジアミンを全ジアミン成分に対し30mol%/70mol%〜70mol%/30mol%の比率で用い、酸成分としてイソフタル酸を用い、これらを加熱することでN−メチル−2−ピロリドン中で測定した溶液対数粘度0.1〜0.4dL/gのメタアラミドオリゴマーを得、これを溶融繊維化することを特徴とするメタアラミド繊維の製造方法。
【請求項2】
メタアラミドオリゴマーの溶融粘度を100〜2000Pa・sとして溶融繊維化する、請求項1に記載のメタアラミド繊維の製造方法。
【請求項3】
メタアラミドオリゴマーの末端の40%以上をアミン末端とする、請求項1または2に記載のメタアラミド繊維の製造方法。
【請求項4】
請求項1〜3のいずれかの方法で得られたメタアラミド繊維を固相重合するメタアラミド繊維の製造方法。
【請求項1】
ジアミン成分として3,4’−ジアミノジフェニルエーテルとメタフェニレンジアミンを全ジアミン成分に対し30mol%/70mol%〜70mol%/30mol%の比率で用い、酸成分としてイソフタル酸を用い、これらを加熱することでN−メチル−2−ピロリドン中で測定した溶液対数粘度0.1〜0.4dL/gのメタアラミドオリゴマーを得、これを溶融繊維化することを特徴とするメタアラミド繊維の製造方法。
【請求項2】
メタアラミドオリゴマーの溶融粘度を100〜2000Pa・sとして溶融繊維化する、請求項1に記載のメタアラミド繊維の製造方法。
【請求項3】
メタアラミドオリゴマーの末端の40%以上をアミン末端とする、請求項1または2に記載のメタアラミド繊維の製造方法。
【請求項4】
請求項1〜3のいずれかの方法で得られたメタアラミド繊維を固相重合するメタアラミド繊維の製造方法。
【図1】

【図2】

【図3】
【図4】
【図2】
【図3】
【図4】
【公開番号】特開2012−167387(P2012−167387A)
【公開日】平成24年9月6日(2012.9.6)
【国際特許分類】
【出願番号】特願2011−26916(P2011−26916)
【出願日】平成23年2月10日(2011.2.10)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成21年度、独立行政法人科学技術振興機構、研究成果最適展開支援事業、フィージビリティスタディ 可能性発掘タイプ(シーズ顕在化)「アラミドの固相重合および溶融成形可能性の研究」、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(304021417)国立大学法人東京工業大学 (1,821)
【出願人】(000003159)東レ株式会社 (7,677)
【Fターム(参考)】
【公開日】平成24年9月6日(2012.9.6)
【国際特許分類】
【出願日】平成23年2月10日(2011.2.10)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成21年度、独立行政法人科学技術振興機構、研究成果最適展開支援事業、フィージビリティスタディ 可能性発掘タイプ(シーズ顕在化)「アラミドの固相重合および溶融成形可能性の研究」、産業技術力強化法第19条の適用を受ける特許出願
【出願人】(304021417)国立大学法人東京工業大学 (1,821)
【出願人】(000003159)東レ株式会社 (7,677)
【Fターム(参考)】
[ Back to top ]