
メラノコルチン−4受容体モジュレーターとしての置換ヘテロアリールピペリジン誘導体
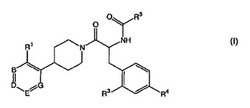
本発明は、メラノコルチン−4受容体モジュレーターとしての置換ヘテロアリールピペリジン誘導体(I)に関する。その構造及び立体化学構造によって、本発明の化合物は、ヒトメラノコルチン−4受容体(MC−4R)の選択的アゴニスト又は選択的アンタゴニストである。アゴニストは、肥満症、糖尿病及び性的機能不全などの障害及び疾患の治療のために使用することができ、一方アンタゴニストは、癌悪液質、筋肉疲労、食欲不振症、筋萎縮性側索硬化症(ALS)、不安神経症及びうつなどの障害及び疾患の治療のために有用である。一般に、MC−4Rの調節が関与する全ての疾患及び障害は、本発明の化合物で治療することができる。
【発明の詳細な説明】
【発明の詳細な説明】
【0001】
[発明の分野]
本発明は、メラノコルチン−4受容体モジュレーターとしての置換ヘテロアリールピペリジン誘導体に関する。構造及び立体化学構造によって、本発明の化合物は、ヒトメラノコルチン−4受容体(MC−4R)の選択的アゴニスト又は選択的アンタゴニストである。アゴニストは、肥満症、糖尿病及び性的機能不全などの障害及び疾患の治療のために使用することができ、一方アンタゴニストは、癌悪液質、筋肉疲労、食欲不振症、筋萎縮性側索硬化症、不安神経症及びうつなどの障害及び疾患の治療のために有用である。一般に、MC−4Rの調節が関与している全ての疾患及び障害は、本発明の化合物で治療することができる。
【0002】
[発明の背景]
メラノコルチン(MC)は、タンパク質分解的切断を介してプロオピオメラノコルチン(POMC)から生じる。これらのペプチド、副腎皮質刺激ホルモン(ACTH)、α−メラニン細胞刺激ホルモン(α−MSH)、β−MSH及びγ−MSHは、12〜39個のアミノ酸でありサイズに幅がある。中枢MC−4R活性化についての最も重要な内因性アゴニストは、トリデカペプチドα−MSHであるようである。MCの中で、α−MSHは脳において神経伝達物質又は神経調節物質として作用することが報告された。MCペプチド、特にα−MSHは、摂食行動、色素沈着及び外分泌機能を含めた生物学的機能について広範囲の作用を有する。α−MSHの生物学的作用は、メラノコルチン受容体(MC−R)と称される7回膜貫通Gタンパク質共役受容体のサブファミリーを介してもたらされる。これらのMC−Rのいずれかの活性化は、cAMP形成の刺激をもたらす。
【0003】
現在まで、MCについての5種の別個のタイプの受容体サブタイプ(MC−1RからMC−5R)が同定されており、これらは異なる組織で発現している。
【0004】
MC−1Rは、メラニン形成細胞において最初に見出された。動物におけるMC−1Rの天然不活性変異体は、チロシナーゼの制御を介してフェオメラニンのユーメラニンへの変換を制御することによって色素沈着の変化及びそれに続くより薄い毛色を生じさせることが示された。これら及び他の研究から、MC−1Rは、動物におけるメラニン生成及び毛色並びにヒトにおける皮膚色の重要な制御因子であること明らかである。
【0005】
MC−2Rは、ACTH受容体を代表する副腎において発現する。MC−2Rは、α−MSHの受容体ではないが、副腎皮質刺激ホルモンI(ACTH I)の受容体である。
【0006】
MC−3Rは、脳(主に、視床下部にある)並びに腸及び胎盤などの末梢組織において発現し、ノックアウト研究は、MC−3Rが、摂食行動、体重及び熱産生の変化に関与することがあることを明らかにした。
【0007】
MC−4Rは、脳内で主として発現する。圧倒的なデータは、エネルギー恒常性におけるMC−4Rの役割を指示する。動物におけるMC−4Rの遺伝子ノックアウト及び薬理的操作は、MC−4Rをアゴナイズすることは体重減少をもたらし、MC−4Rをアンタゴナイズすることは体重増加をもたらすことを示した(A.Kaskら、「Selective antagonist for the melanocortin−4 receptor(HS014)increases food intake in free−feeding rats」、Biochem.Biophys.Res.Commun.、245:90〜93(1998))。
【0008】
MC−5Rは、白色脂肪、胎盤を含めた多くの末梢組織において遍在性に発現し、低レベルの発現もまた脳において観察される。しかし、その発現は、外分泌腺において最も多い。マウスにおけるこの受容体の遺伝子ノックアウトは、外分泌腺機能の調節の変化をもたらし、水の反発及び体温調節の変化をもたらす。MC−5Rノックアウトマウスはまた、皮脂腺脂質産生が減少することが明らかとなった(Chenら、Cell、91:789〜798(1997))。
【0009】
MC−3R及びMC−4Rモジュレーター並びに肥満症及び食欲不振症などの体重障害の治療におけるそれらの使用の研究についての注目が集まってきた。しかし、MCペプチドは、色素沈着、摂食行動及び外分泌機能の調節におけるそれらの役割に加えて、強力な生理学的効果を有するという証拠が示されてきた。特に、α−MSHは、炎症性腸疾患、腎虚血/再潅流傷害及び内毒素誘発肝炎を含めた炎症の急性及び慢性モデルの両方において強力な抗炎症効果を誘発することが最近示された。これらのモデルにおけるα−MSHの投与は、炎症が媒介する組織の損傷の実質的な減少、白血球浸潤の有意な減少、並びにサイトカイン及び他の伝達物質の上昇したレベルのベースライン値近くへの劇的な減少をもたらす。最近の研究は、α−MSHの消炎作用はMC−1Rによって媒介されることを示した。MC−1Rのアゴニズムが抗炎症反応をもたらす機構は、炎症誘発性転写活性化因子であるNF−κBの阻害を介していると思われる。NF−κBは、炎症誘発性カスケードの中心的な構成要素であり、その活性化は、多くの炎症性疾患の開始における中心事象である。さらに、α−MSHの消炎作用は、部分的に、MC−3R及び/又はMC−5Rのアゴニズムによって媒介されることがある。
【0010】
MC−4Rシグナル伝達が摂食行動の媒介において重要であるという証拠が示されてきたが、肥満症の管理のために標的となることができる特定の単一のMC−Rはまだ同定されていない(S.Q.Giraudoら、「Feeding effects of hypothalamic injection of melanocortin−4 receptor ligands」、Brain Research、80:302〜306(1998))。肥満症におけるMC−Rの関与についてのさらなる証拠には、下記が含まれる。1)MC−1R、MC−3R及びMC−4Rのアンタゴニストを異所的に発現しているアグーチ(Avy)マウスは肥満であり、これらの3種のMC−Rの作用を遮断することは過食症及び代謝障害をもたらす場合があることをこれは示す。2)MC−4Rノックアウトマウス(D.Huszarら、Cell、88:131〜141(1997))は、アグーチマウスの表現型を再現し、これらのマウスは肥満である。3)げっ歯類において側脳室内(ICV)に注射した環状ヘプタペプチドメラノタニンII(MT−II)(非選択的MC−1R、−3R、−4R、及び−5Rアゴニスト)は、いくつかの動物飼育モデル(NPY、ob/ob、アグーチ、絶食)において食物摂取量を減少させ、一方ICV注射したSHU−9119(MC−3R及び4Rアンタゴニスト;MC−1R及び−5Rアゴニスト)は、この作用を逆転させ、過食症を誘発することができる。4)ズッカー糖尿病肥満ラットのα−NDP−MSH誘導体(HP−228)による慢性腹腔内処理は、MC−1R、−3R、−4R、及び−5Rを活性化し、12週間の期間に亘り食物摂取量及び体重増加を減少させることが報告されてきた(I.Corcosら、「HP−228 is a potent agonist of melanocortin receptor−4 and significantly attenuates obesity and diabetes in Zucker fatty rats」、Society for Neuroscience Abstracts、23:673(1997))。
【0011】
MC−4Rは、他の生理機能、すなわちグルーミング行動の制御、勃起及び血圧の制御においてもまた役割を果たしているように思われる。勃起不全は、性交を成功させるのに十分な陰茎勃起を達成することができない病状を意味する。「インポテンス」という用語は、この蔓延している状態を説明するために用いられることが多い。合成メラノコルチン受容体アゴニストは、心因性の勃起不全を有する男性において勃起を起こすことが見出された(H.Wessellsら、「Synthetic Melanotropic Peptide Initiates Erections in Men With Psychogenic Erectile Dysfunction:Double−Blind,Placebo Controlled Crossover Study」、J.Urol.、160:389〜393、(1998))。脳のメラノコルチン受容体の活性化は、性的刺激の通常の刺激をもたらすように思われる。男性及び/又は女性の性的機能不全におけるMC−Rの関与についての証拠は、国際公開第00/74679号パンフレットにおいて詳述されている。
【0012】
糖尿病は、哺乳動物においてグルコースを筋肉及び肝細胞に貯蔵するためグリコーゲンに変換する能力が衰えるため、血液におけるグルコースレベルを制御する哺乳動物の能力が損なわれる疾患である。I型糖尿病において、グルコースを貯蔵するこの能力の減少は、インスリン産生の減少に起因する。「II型糖尿病」又は「インスリン非依存性真性糖尿病」(NIDDM)は、主要なインスリン感受性組織である筋肉、肝臓及び脂肪組織においてグルコース及び脂質代謝についてのインスリン刺激又は調節作用に対する深刻な耐性に起因する糖尿病の形態である。このインスリン応答性に対する耐性は、グルコース取込み、酸化及び筋肉における貯蔵の不十分なインスリン活性化、並びに脂肪組織における脂肪分解並びに肝臓におけるグルコース産生及び分泌の不適切なインスリン抑制をもたらす。これらの細胞がインスリンに対して脱感作される場合、体は異常に高レベルのインスリンを産生することによって代償しようとし、高インスリン血症が起こる。高インスリン血症は、高血圧症及び体重の増加と関連する。インスリンは、インスリン感受性細胞による血液からのグルコース、アミノ酸及びトリグリセリドの細胞取込みの促進に関与するため、インスリン非感受性は、心血管疾患における危険因子であるトリグリセリド及びLDLのレベルの上昇をもたらす場合がある。高血圧症、体重増加、トリグリセリド増加及びLDL増加と併せた高インスリン血症を含む症状の一群は、症候群Xとして知られている。MC−4Rアゴニストは、NIDDM及び症候群Xの治療において有用であり得る。
【0013】
下記の知見に基づくように、MC受容体サブタイプの中で、ストレスとの関係及び情動行動の調節に関してMC4受容体はまた興味深い。ストレスは、内分泌、生化学及び行動事象を含めた複雑な反応のカスケードを開始させる。これらの反応の多くは、コルチコトロピン放出因子(CRF)の放出によって開始される(M.J.Owen及びC.B.Nemeroff、「Physiology and pharmacology of corticotrophin releasing factor」、Pharmacol.Rev.43:425〜473(1991))。脳CRF系の活性化に加えて、酵素処理によってプロオピオメラノコルチンから生じるメラノコルチン(MC)は、ストレスへの重要な行動反応及び生化学的反応、結果的に、不安神経症及びうつなどのストレス誘発性の障害を媒介するといういくつかの証拠がある(Shigeyuki Chakiら、「Anxiolytic−Like and Antidepressant−Like Activities of MCL0129(1−[(S)−2−(4−Fluorophenyl)−2−(4−isopropylpiperadin−1−yl)ethyl]−4−[4−(2−methoxynaphthalen−1−yl)butyl]piperazine), a Novel and Potent Nonpeptide Antagonist of the Melanocortin−4 Receptor」、J.Pharm.Exp.Ther.304(2)、818〜826(2003))。
【0014】
悪性腫癌又は感染などの慢性疾患は、食欲の減少及び除脂肪体重の減少の併発からもたらされる悪液質と関連する場合が多い。除脂肪体重の大幅な減少は、炎症過程によって引き起こされる場合が多く、脳におけるα−MSHの産生を増加させるサイトカイン(例えば、TNF−α)の血漿レベルの増加と通常関連する。α−MSHによる視床下部におけるMC4受容体の活性化は、食欲を減少させ、エネルギーの消費を増加させる。腫瘍マウスにおける実験上の証拠は、遺伝子MC4受容体ノックアウト又はMC4受容体遮断によって、悪液質は予防又は回復に向かうことができることを示す。処理したマウスにおける体重の増加は、より多量の除脂肪体重(主として骨格筋からなる)に起因する(D.L.Marksら、「Role of the central melanocortin system in cachexia」、Cancer Res.61:1432〜1438(2001))。
【0015】
臨床所見は、筋萎縮性側索硬化症(ALS)の進行が体重と逆相関することがあることを示している(例えば、Ludolph AC、Neuromuscul Disord.(2006)16(8):530〜8)。したがって、MC−4R阻害剤は、ALS患者の治療に使用することができる。
【0016】
メラノコルチン受容体のモジュレーターは、文献からすでに公知である。国際公開第2004/024720A1号パンフレットは、ヒトメラノコルチン−4受容体の選択的アゴニストであるピペラジン尿素誘導体について記載しており、したがってそれらは、肥満症に関連する障害の予防の治療において有用であると主張されている。
【0017】
国際公開第2005/047253A1号パンフレットは、メラノコルチン受容体アゴニストとして機能すると主張されている4,4−二置換ピペリジン誘導体について記載している。
【0018】
置換ピペリジン誘導体はまた、カルボン酸及びエステル(ピペリジン環又はピペラジン環を分子の中核として有し、その核がパラ位において5〜7員の複素環、フェニル環、ピリジン環又はチアゾール環でさらに置換されている)に関するDE 103 00973において記載されている。前記環は、エステル基で任意選択で置換されている。この化合物は、頭痛、インスリン非依存性真性糖尿病(NIDDM)、心血管疾患、モルヒネ耐性、皮膚疾患、炎症、アレルギー性鼻炎、喘息;血管拡張を伴い、その結果として組織内の血液循環の減少を伴う疾患の治療;エストロゲン欠乏の女性の閉経期ホットフラッシュの急性期治療若しくは予防的治療、又は疼痛の治療のための医薬の調製において使用される。
【0019】
上記のような様々な疾患及び障害の治療における未解決の欠陥を考慮して、本発明の目的は、癌悪液質、筋肉疲労、食欲不振症、不安神経症、うつ、肥満症、糖尿病、性的機能不全、筋萎縮性側索硬化症、及びMC−4Rの関与を伴う他の疾患を治療するためのメラノコルチン−4受容体モジュレーターとして有用な、血液脳関門を通過する改善された能力を有する新規な置換ヘテロアリールピペリジン誘導体を提供することにある。
【0020】
[発明の概要]
本発明は、構造式(I)の置換ヘテロアリールピペリジン誘導体に関し、
【化1】

式中、R1、R3、R4、R5、B、D、E及びGは、下記で説明するように定義される。
【0021】
構造式(I)のヘテロアリールピペリジン誘導体は、メラノコルチン受容体モジュレーターとして有効であり、選択的メラノコルチン−4受容体(MC−4R)モジュレーターとして特に有効である。したがって、それらは、MC−4Rの活性化又は不活性化が関与している障害の治療に有用である。アゴニストは、肥満症、糖尿病及び性的機能不全などの障害及び疾患の治療のために使用することができ、一方、アンタゴニストは、癌悪液質、筋肉疲労、食欲不振症、筋萎縮性側索硬化症(ALS)、不安神経症及びうつなどの障害及び疾患の治療のために有用である。
【0022】
したがって、本発明は、癌悪液質、筋肉疲労、食欲不振症、筋萎縮性側索硬化症(ALS)、不安神経症、うつ、肥満症、真性糖尿病、男性若しくは女性の性的機能不全及び勃起不全の治療及び/又は予防のための式(I)の化合物に関する。
【0023】
さらなる態様において、本発明は、癌悪液質、筋肉疲労、食欲不振症、筋萎縮性側索硬化症(ALS)、不安神経症、うつ、肥満症、真性糖尿病、男性若しくは女性の性的機能不全及び勃起不全の治療及び/又は予防のための医薬品の調製のための式(I)の化合物の使用に関する。
【0024】
本発明はまた、本発明の化合物と薬学的に許容される担体とを含む医薬組成物に関する。
【0025】
[発明の詳細な説明]
本発明は、メラノコルチン受容体モジュレーター、特に、選択的MC−4Rアゴニスト及びMC−4Rアンタゴニストとして有用な置換ヘテロアリールピペリジン誘導体に関する。
【0026】
本発明の化合物は、構造式(I)
【化2】
並びにそのエナンチオマー、ジアステレオマー、互変異性体、溶媒和物及び薬学的に許容される塩によって表され、
式中、
R1は、−N(R10)−(C(R6)2)m−T、
−(C(R6)2)l−T、又は
−O−(C(R6)2)m−Tであり、
R6は、
H、
F、
OH、
OCH3、
C1〜6−アルキル(ハロゲン、CN、OH及びOCH3から選択される1〜3個の置換基で任意選択で置換されている)、並びに
C3〜6−シクロアルキル(ハロゲン、CN、OH及びOCH3から選択される1〜3個の置換基で任意選択で置換されている)から独立に選択され、
Tは、NR7R8、
【化3】
であり、
R7及びR8は、互いに独立に、
H、
C1〜6−アルキル、
C2〜6−アルケニル、
C2〜6−アルキニル、及び
C2〜6−アルキレン−O−C1〜6−アルキルから選択され、
各アルキル、アルケニル及びアルキニルは、1個若しくは複数のハロゲン原子、CN又はOHで任意選択で置換されており、
R9は、
ハロゲン、
CN、
OH、
C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、並びに
O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
C1〜6−アルキレン−O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、又は
NR12R13から独立に選択され、
R10は、H又は
C1〜6−アルキルであり、
R11は、
ハロゲン、
CN、
OH、
C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
C2〜6−アルケニル、
C2〜6−アルキニル、
O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
C1〜6−アルキレン−O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
C0〜6−アルキル−C3〜6−シクロアルキル、
−OC(O)C1〜6−アルキル、
−NH2、
−NH(C1〜6−アルキル)、及び
−N(C1〜6−アルキル)2から独立に選択され、
R12及びR13は、互いに独立に、
C1〜6−アルキル(OHで任意選択で置換されている)、
C2〜6−アルケニル、
C2〜6−アルキニル、
C2〜6−アルキレン−O−C1〜6−アルキル、及び
C2〜6−アルキレン−N−(C1〜6−アルキル)2から選択され、
Wは、CH、O又はNR10であり、
Xは、CH又はNであり、
Yは、CH又はNであり、
Zは、CH又はNであり、
Aは、0〜2個の窒素原子を含有する3〜7員の飽和、不飽和又は芳香族環であり、
Bは、CR2又はNであり、
Gは、CR2又はNであり、
Dは、CR2又はNであり、
Eは、CR2又はNであり、
ただし、可変部分B、G、D及びEの1つ又は2つは、Nでなくてはならず、
R2は、
H、
F、
Cl、
CH3、
OCH3、及び
CF3から独立に選択され、
R3は、H、
Cl、
F、又は
CH3であり、
R4は、Cl、
F、又は
CH3であり、
R5は、
【化4】
モルホリン(1〜3個の同じ若しくは異なる置換基R14で任意選択で置換されている)、
環中に1個の窒素原子と任意選択でO、N及びSから選択されるさらなるヘテロ原子とを含有する4〜7員の飽和又は部分不飽和複素環(複素環は、1〜4個の同じ若しくは異なる置換基R11で任意選択で置換されている)、又は
NR12R13であり、
R14は、C1〜6−アルキル、
C1〜6−アルキレン−O−C1〜6−アルキル、
C1〜6−アルキレン−OH、
C1〜6−アルキレン−NH2、
C1〜6−アルキレン−NH−C1〜6−アルキル、又は
C1〜6−アルキレン−N(C1〜6−アルキル)2であり、
R15は、H又は
C1〜6−アルキルであり、
lは、0、1、2、3又は4であり、
mは、0、1、2、3又は4であり、
oは、0、1又は2であり、
pは、0、1、2、3又は4であり、
rは、0、1、2、3又は4であり、
sは、1又は2であり、
tは、0又は1である。
【0027】
好ましい実施形態では、式(I)の化合物は、下記のように定義される。
R1は、−N(R10)−(C(R6)2)m−T、
−(C(R6)2)l−T、又は
−O−(C(R6)2)m−Tであり、
R6は、
H、
F、
OH、
OCH3、
C1〜6−アルキル(ハロゲン、CN、OH及びOCH3から選択される1〜3個の置換基で任意選択で置換されている)、並びに
C3〜6−シクロアルキル(ハロゲン、CN、OH及びOCH3から選択される1〜3個の置換基で任意選択で置換されている)から独立に選択され、
Tは、NR7R8、
モルホリン、
【化5】
であり、
R7及びR8は、互いに独立に、
H、
C1〜6−アルキル、
C2〜6−アルケニル、
C2〜6−アルキニル、及び
C2〜6−アルキレン−O−C1〜6−アルキルから選択され、
各アルキル、アルケニル及びアルキニルは、1個若しくは複数のハロゲン原子、CN又はOHで任意選択で置換されており、
R9は、
ハロゲン、
CN、
OH、
C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、並びに
O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
C1〜6−アルキレン−O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)から独立に選択され、
R10は、H又は
C1〜C6−アルキルであり、
R11は、
ハロゲン、
CN、
OH、
C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
C1〜6−アルキレン−O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
−NH2、
−NH(C1〜6−アルキル)、及び
−N(C1〜6−アルキル)2から独立に選択され、
Xは、CH又はNであり、
Yは、CH又はNであり、
Zは、CH又はNであり、
Aは、0〜2個の窒素原子を含有する3〜7員の飽和、不飽和又は芳香族環であり、
Bは、CR2又はNであり、
Gは、CR2又はNであり、
Dは、CR2又はNであり、
Eは、CR2又はNであり、
ただし、可変部分B、G、D及びEの1つ又は2つは、Nでなくてはならず、
R2は、
H、
F、
Cl、
CH3、
OCH3、及び
CF3から独立に選択され、
R3は、H、
Cl、
F、又は
CH3であり、
R4は、Cl又はFであり、
R5は、
【化6】
モルホリン(1〜3個の同じ若しくは異なる置換基R14で任意選択で置換されている)、又は
NR12R13であり、
R12及びR13は、互いに独立に、
C1〜6−アルキル、
C2〜6−アルケニル、
C2〜6−アルキニル、
C2〜6−アルキレン−O−C1〜6−アルキル、及び
C2〜6−アルキレン−N−(C1〜6−アルキル)2から選択され、
R14は、C1〜6−アルキル、
C1〜6−アルキレン−O−C1〜6−アルキル、
C1〜6−アルキレン−OH、
C1〜6−アルキレン−NH2、
C1〜6−アルキレン−NH−C1〜6−アルキル、又は
C1〜6−アルキレン−N(C1〜6−アルキル)2であり、
lは、0、1、2、3又は4であり、
mは、0、1、2、3又は4であり、
oは、0、1又は2であり、
pは、0、1、2、3又は4であり、
qは、0、1、2、又は3であり、
rは、0、1、2、3又は4であり、
sは、1又は2である。
【0028】
好ましくは、式(I)による化合物は、下記の立体異性体式(I’)の構造的立体配座をとり、
【化7】
式中、B、G、D、E、R1、R3、R4及びR5は、上記定義の通りである。
【0029】
一般式(I)中の部分
【化8】
は、下記の構造から選択される。
【化9】
【0030】
その中で、可変部分R2は、上記のように定義される。本発明の好ましい実施形態では、R2は、H、Cl、F又はCH3を表す。さらに好ましくは、R2は、H又はCH3を表す。
【0031】
この部分
【化10】
の好ましい実施形態は、下記の構造である。
【化11】
【0032】
本発明の好ましい実施形態では、可変部分R1は、−N(R10)−(C(R6)2)m−T、−(C(R6)2)l−T、又は−O−(C(R6)2)m−T(式中、R6及びR10は、好ましくはH及びC1〜6−アルキルからなる群から独立に選択される)を表す。
【0033】
R3は、H、Cl、又はCH3、さらに好ましくはClを表すことがさらに好ましい。他の実施形態において、R3は、好ましくはFを表す。
【0034】
好ましくは、R4は、Clを表す。
【0035】
本発明の好ましい実施形態では、可変部分R5は、
【化12】
を表し、
式中、rは、好ましくは0又は1であり、qは、好ましくは数字1又は2をとる。
【0036】
さらなる好ましい実施形態では、R7及びR8の少なくとも1つは、C1〜6−アルキル、C2〜6−アルケニル、C2〜6−アルキニル及びC2〜6−アルキレン−O−C1〜6−アルキルから、さらに好ましくはC2〜6−アルケニル、C2〜6−アルキニル及びC2〜6−アルキレン−O−C1〜6−アルキルからなる群から選択される。
【0037】
R9は、ハロゲン、CN、OH、C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、並びにO−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)からなる群から独立に選択されることが好ましい。
【0038】
可変部分lは、好ましくは2又は3から選択される。
【0039】
可変部分mは、好ましくは2、3又は4から、さらに好ましくは2又は3から選択される。
【0040】
式(I)の化合物について、Tは、好ましくは下記の基
【化13】
【化14】
【化15】
【化16】
【化17】
【化18】
からなる群から選択される。
【0041】
好ましい実施形態では、R5は、好ましくは
【化19】
【化20】
【化21】
からなる群から選択される。
【0042】
上記の基のいくつか若しくは全てが好ましい又はより好ましい意味を有する式(I)の化合物もまた、本発明の1つの目的である。
【0043】
上記及び下記において用いられる用語は、下記で説明するような意味を有する。
【0044】
アルキルは、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、tert−ブチル、n−ペンチル、イソペンチル、ネオペンチル、又はヘキシルなどの、1〜6個の炭素原子を有する直鎖又は分岐状のアルキルである。
【0045】
アルケニルは、ビニル、アリル、1−プロペニル、2−ブテニル、2−メチル−2−ブテニル、イソプロペニル、ペンテニル、又はヘキセニルなどの、2〜6個の炭素原子を有し、少なくとも1つの炭素−炭素二重結合を含有する直鎖又は分岐状のアルキルである。
【0046】
アルキニルは、エチニル、1−プロピニル、1−ブチニル、2−ブチニル、ペンチニル又はヘキシニルなどの、2〜6個の炭素原子を有し、少なくとも1つの炭素−炭素三重結合を含有する直鎖又は分岐状のアルキルである。
【0047】
0〜2個の窒素原子を含有する3〜7員の飽和、不飽和又は芳香族環は、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル又はシクロヘプチルなどの、3〜7員の飽和炭素環を包含する。前記用語は、シクロブテニル、シクロペンテニル、シクロヘキセニル、シクロヘプテニル、シクロヘキサ−1,4−ジエン若しくはシクロヘプタジエン、又はベンゼンなどの芳香環などの、3〜7員の不飽和炭素環をさらに包含する。窒素含有3〜7員の飽和、不飽和又は芳香族の複素環が、上記の用語にさらに包含される。この例には、アゼチジン、ピロリジン、ピペリジン、アゼパン、ピペラジン、ピリジン、ピリミジン、ピラジン、ピロール、イミダゾール、及びピラゾールが挙げられる。
【0048】
構造式(I)の化合物は、メラノコルチン受容体モジュレーターとして有効であり、MC−4Rの選択的モジュレーターとして特に有効である。それらはしたがって、癌悪液質、筋肉疲労、食欲不振症、筋萎縮性側索硬化症、不安神経症、うつ、肥満症、糖尿病、性的機能不全、及びMC−4Rの関与を伴う他の疾患などのMC−4Rの活性化及び不活性化に対して応答性の障害の治療及び/又は予防のために有用である。
【0049】
構造式(I)の化合物は、MC−4Rのアンタゴニストとして特に有用である。したがって、それらは、好ましくは癌悪液質、筋肉疲労、食欲不振症、筋萎縮性側索硬化症、不安神経症及びうつの治療及び/又は予防のための医薬品の調製のために使用される。
【0050】
光学異性体−ジアステレオマー−幾何異性体−互変異性体
構造式(I)の化合物は、1つ又は複数の不斉中心を含有し、ラセミ化合物及びラセミ混合物、単一のエナンチオマー、ジアステレオマー混合物及び個々のジアステレオマーとして生じる場合がある。本発明は、構造式(I)の化合物の全てのこのような異性体形態を包含することを意味する。
【0051】
構造式(I)の化合物は、例えば、適切な溶媒、例えば、メタノール又は酢酸エチル又はこれらの混合物からの分別結晶によって、或いは光学活性な固定相を使用したキラルクロマトグラフィーを介して、これらの個々のジアステレオマーに分離することができる。絶対立体配置は、公知の絶対配置の不斉中心を含有する試薬で必要に応じて誘導体化した結晶性生成物又は結晶性中間体のX線結晶構造解析によって決定することができる。
【0052】
代わりに、一般式(I)の化合物の任意の立体異性体は、光学的に純粋な出発物質又は公知の絶対配置の試薬を使用して立体特異的合成によって得ることができる。
【0053】
塩
「薬学的に許容される塩」という用語は、無機塩基又は有機塩基及び無機酸又は有機酸を含めた、薬学的に許容される無毒性塩基又は酸から調製される塩を意味する。無機塩基に由来する塩には、アルミニウム、アンモニウム、カルシウム、銅、第二鉄、第一鉄、リチウム、マグネシウム、マンガン塩、マンガン(II)、カリウム、ナトリウム、亜鉛などが挙げられる。特に好ましい塩は、アンモニウム、カルシウム、リチウム、マグネシウム、カリウム及びナトリウム塩である。薬学的に許容される無毒有機の塩基に由来する塩には、第一級、第二級及び第三級アミン、天然の置換アミンを含めた置換アミン、環状アミン及び塩基性イオン交換樹脂の塩(アルギニン、ベタイン、カフェイン、コリン、N,N’−ジベンジルエチレンジアミン、ジエチルアミン、2−ジエチルアミノエタノール、2−ジメチルアミノ−エタノール、エタノールアミン、エチレンジアミン、N−エチルモルホリン、N−エチルピペリジン、グルカミン、グルコサミン、ヒスチジン、ヒドラバミン、イソプロピルアミン、リシン、メチルグルカミン、モルホリン、ピペラジン、ピペリジン、ポリアミン樹脂、プロカイン、プリン、テオブロミン、トリエチルアミン、トリメチルアミン、トリプロピルアミン、トロメタミンなど)が挙げられる。
【0054】
本発明の化合物が塩基性である場合、塩は、無機酸及び有機酸を含めた薬学的に許容される無毒性酸から調製することができる。このような酸には、酢酸、ベンゼンスルホン酸、安息香酸、カンファースルホン酸、クエン酸、エタンスルホン酸、ギ酸、フマル酸、グルコン酸、グルタミン酸、臭化水素酸、塩酸、イセチオン酸、乳酸、マレイン酸、リンゴ酸、マンデル酸、メタンスルホン酸、マロン酸、粘液酸、硝酸、パモン酸、パントテン酸、リン酸、プロピオン酸、コハク酸、硫酸、酒石酸、p−トルエンスルホン酸、トリフルオロ酢酸などが挙げられる。特に好ましい酸は、クエン酸、フマル酸、臭化水素酸、塩酸、マレイン酸、リン酸、硫酸及び酒石酸である。
【0055】
本明細書で使用する場合、式(I)の化合物への適用は、薬学的に許容される塩もまた含むことを意味することが理解されるであろう。
【0056】
有用性
式(I)の化合物は、メラノコルチン受容体モジュレーターであり、したがって、それだけに限らないが、MC−1R、MC−2R、MC−3R、MC−4R又はMC−5Rが挙げられるメラノコルチン受容体の1つ又は複数の不活性化に対して応答性の疾患、障害又は状態の治療、制御又は予防において有用である。このような疾患、障害又は状態には、それだけに限らないが、癌悪液質、筋肉疲労、食欲不振症、不安神経症、うつ、肥満症(食欲を減少させ、代謝率を増加させ、脂肪摂取を減少させ、又は炭水化物への欲求を減少させることによって)、真性糖尿病(耐糖能を増強させ、インスリン抵抗性を減少させることによって)、並びに男性及び女性の性的機能不全(インポテンス、性的衝動の喪失及び勃起不全を含めた)が挙げられる。
【0057】
式(I)の化合物は、高血圧症、高脂血症、骨関節炎、癌、胆嚢疾患、睡眠時無呼吸、強迫行為、ノイローゼ、不眠/睡眠障害、薬物乱用、疼痛、発熱、炎症、免疫修飾、関節リウマチ、皮膚日焼け、ざ瘡及び他の皮膚障害;アルツハイマー病の治療を含めた神経保護及び認識及び記憶増強の治療、制御又は予防においてさらに使用することができる。
【0058】
投与及び用量範囲
任意の適切な投与経路が、哺乳動物、特にヒトに、有効量の本発明の化合物を提供するために用いることができる。例えば、経口、直腸、局所、非経口、眼球、肺、経鼻などを用いることができる。剤形には、錠剤、トローチ剤、分散剤、懸濁剤、溶液剤、カプセル剤、クリーム剤、軟膏、エアゾール剤などが挙げられる。好ましくは、式(I)の化合物は、経口又は局所で投与される。
【0059】
用いられる有効量の活性成分は、用いられる特定化合物、投与方法、治療される状態及び治療される状態の重篤度によって変化することがある。このような投与量は、当業者によって容易に確認することができる。
【0060】
癌悪液質、筋肉疲労、筋萎縮性側索硬化症又は食欲不振症を治療する場合、本発明の化合物が、約0.001ミリグラム〜約100ミリグラム/体重キログラムの1日投与量で投与されると(好ましくは単回用量、若しくは1日2〜6回の分割用量で、又は持続放出形態で与えられる)、一般に満足のいく結果が得られる。70kgの成人したヒトの場合、総1日用量は一般に、約0.07ミリグラム〜約3500ミリグラムであろう。この投与計画は、最適な治療反応を得るために調節してもよい。
【0061】
糖尿病及び/若しくは高血糖症と合わせて、又は単独で肥満症を治療する場合、本発明の化合物が、約0.001ミリグラム〜約100ミリグラム/体重キログラムの1日投与量で投与されると(好ましくは単回用量、若しくは1日2〜6回の分割用量で、又は持続放出形態で与えられる)、一般に満足のいく結果が得られる。70kgの成人したヒトの場合、総1日用量は一般に、約0.07ミリグラム〜約3500ミリグラムであろう。この投与計画は、最適な治療反応を得るために調節してもよい。
【0062】
真性糖尿病及び/又は高血糖症、並びに式(I)の化合物が有用な他の疾患又は障害を治療する場合、本発明の化合物が、約0.001ミリグラム〜約100ミリグラム/動物体重キログラムの1日投与量で投与される場合(好ましくは単回用量、若しくは1日2〜6回の分割用量で、又は持続放出形態で与えられる)、一般に満足のいく結果が得られる。70kgの成人したヒトの場合、総1日用量は一般に、約0.07ミリグラム〜約3500ミリグラムであろう。この投与計画、最適な治療反応を得るために調節してもよい。
【0063】
性的機能不全の治療のために、本発明の化合物は、0.001ミリグラム〜約100ミリグラム/体重キログラムの範囲の1用量単位で、好ましくは経口の単回用量又は鼻用スプレーとして与えられる。
【0064】
製剤
式(I)の化合物は、好ましくは投与の前に剤形に製剤される。したがって、本発明はまた、式(I)の化合物と適切な医薬担体とを含む医薬組成物を含む。
【0065】
本発明の医薬組成物は、周知の容易に利用できる成分を使用して、公知の手順によって調製される。本発明の製剤の作製において、活性成分(式(I)の化合物)は通常、担体と混合され、又は担体によって希釈され、又は担体中に封入されるが、これは、カプセル剤、サシェ剤、紙又は他の容器の形態中のことがある。担体が希釈剤としての機能を果たす場合、それは、活性成分のためのビヒクル、賦形剤又は媒質として作用する固体、半固体又は液体物質であることがある。したがって、組成物は、錠剤、丸剤、散剤、ロゼンジ、サシェ剤、カシェ剤、エリキシル剤、懸濁剤、乳剤、溶液剤、シロップ剤、エアロゾル剤(固体として又は液体媒体中)、軟質及び硬質ゼラチンカプセル剤、坐薬、無菌注射剤、並びに包装された無菌散剤の形態の場合がある。
【0066】
適切な担体、賦形剤及び希釈剤のいくつかの例には、ラクトース、デキストロース、スクロース、ソルビトール、マンニトール、デンプン、アラビアゴム、リン酸カルシウム、アルギネート、トラガカント、ゼラチン、ケイ酸カルシウム、微結晶性セルロース、ポリビニルピロリドン、セルロース、水、シロップ、メチルセルロース、メチル及びプロピルヒドロキシベンゾエート、タルク、ステアリン酸マグネシウム及び鉱油が挙げられる。製剤は、滑沢剤、湿潤剤、乳化剤及び懸濁化剤、保存料、甘味剤又は香味剤をさらに含むことができる。本発明の組成物は、患者に投与した後、活性成分の即時放出、持続放出又は遅延放出を実現するように製剤されることがある。
【0067】
本発明の化合物の調製
本発明の式(I)の化合物の調製を説明するとき、「A部分」、「B部分」及び「C部分」という用語が下記で使用される。この部分という概念を下記で例示する。
【化22】
【0068】
本発明の化合物の調製は、順次又は収束的合成経路によって行ってもよい。当業者であれば、一般に、式(I)の化合物のA及びB部分は、アミド結合を介して結合していることを理解するであろう。当業者はしたがって、標準的なペプチドカップリング反応条件を介して2つの部分を結合する多数の経路及び方法を容易に予見することができる。
【0069】
「標準的なペプチドカップリング反応条件」という表現は、HOBtなどの触媒の存在下で、DCMなどの不活性溶媒中、EDCI、ジシクロヘキシルカルボジイミド及びベンゾトリアゾール−1−イルオキシトリス(ジメチルアミノ)−ホスホニウムヘキサフルオロホスフェートなどの酸活性化剤を使用した、カルボン酸とアミンとのカップリングを意味する。所望の反応を促進し、望ましくない反応を最小化するための、アミン及びカルボン酸についての保護基の使用については、これまでにもたくさん記述されている。存在し得る保護基の除去に必要な条件は、Greeneら、Protective Groups in Organic Synthesis、John Wiley & Sons、Inc.、New York、NY 1991において見出すことができる。
【0070】
Z、Boc及びFmocなどの保護基は、合成において広範に使用されており、それらの除去条件は当業者には周知である。例えば、Z基の除去は、エタノールなどのプロトン性溶媒中、活性炭担持パラジウムなどの貴金属又はその酸化物の存在下で、水素による接触水素化によって行うことができる。他の潜在的に反応性の官能基の存在によって接触水素化が禁忌である場合、Zの除去はまた、臭化水素の酢酸溶液による処理によって、又はTFAと硫化ジメチルとの混合物による処理によって行うことができる。Boc保護基の除去は、塩化メチレン、メタノール又は酢酸エチルなどの溶媒中、TFA又はHCl又は塩化水素ガスなどの強酸によって行われる。
【0071】
式(I)の化合物のB及びC部分は、尿素官能基を介して共に結合している。したがって、当業者であれば、異なる周知の方法を使用した2つの部分を結合する多数の経路及び方法を容易に想定することができる。
【0072】
式(I)の化合物は、ジアステレオマー混合物として存在する場合、メタノール、酢酸エチル又はこれらの混合物などの適切な溶媒からの分別結晶によって、エナンチオマーのジアステレオマー対に分離することができる。このようにして得られたエナンチオマー対は、光学活性な酸を分割剤として使用することにより従来の手段によって個々の立体異性体に分離することができる。代わりに、式(I)の化合物の任意のエナンチオマーは、光学的に純粋な出発物質又は公知の立体配置の試薬を使用して立体特異的合成によって得ることができる。
【0073】
本発明の式(I)の化合物は、適切な材料を使用して下記のスキーム及び実施例の手順によって調製することができ、下記の具体例によってさらに例示される。さらに、当技術分野で通常の技術と併せて本明細書に記載する手順を用いることによって、ここで特許請求されている本発明のさらなる化合物を、容易に調製することができる。しかし、実施例において例示した化合物は、本発明として考えられる唯一の種類を形成するものとして解釈されない。実施例は、本発明の化合物の調製についての詳細をさらに例示する。下記の調製手順の条件及び方法の公知の変形形態を、これらの化合物を調製するために使用することができることは当業者であれば容易に理解するであろう。本化合物は一般に、従前に記載したものなどの薬学的に許容されるそれらの塩の形態で単離される。単離した塩に相当する遊離アミン塩基は、水性炭酸水素ナトリウム、炭酸ナトリウム、水酸化ナトリウム及び水酸化カリウムなどの適切な塩基による中和、及び遊離したアミン遊離塩基の有機溶媒への抽出、それに続くエバポレーション(evaporation)によって生成することができる。この方法で単離されたアミン遊離塩基は、有機溶媒への溶解、続いて適切な酸の添加、それに続くエバポレーション、沈殿又は結晶化によって他の薬学的に許容される塩にさらに変換することができる。全ての温度は、摂氏温度である。
【0074】
下記のスキーム、調製及び実施例において、様々な試薬の記号及び略語は、下記の意味を有する。
Ac2O 無水酢酸
AcOH 酢酸
Boc tert−ブトキシカルボニル
Bu ブチル
BuLi ブチルリチウム
DBU 1,8−ジアザビシクロ[5.4.0]ウンデス−7−エン
DCC N,N’−ジシクロヘキシルカルボジイミド
DCE 1,2−ジクロロエタン
DCM ジクロロメタン
DEAD アゾジカルボン酸ジエチル
DIAD アゾジカルボン酸ジイソプロピル
DIC N,N’−ジイソプロピルカルボジイミド
DIEA エチル−ジイソプロピルアミン
DMA N,N−ジメチルアセトアミド
DMAP 4−ジメチルアミノピリジン
DMF N,N−ジメチルホルムアミド
DMS 硫化ジメチル
DMSO ジメチルスルホキシド
dppf 1,1’−ビス(ジフェニルホスフィノ)−フェロセン
EDCI 1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド塩酸塩
Et2O ジエチルエーテル
EtOAc 酢酸エチル
EtOH エタノール
Fmoc 9−フルオレニルメチルオキシカルボニル
h 時間(複数可)
HOBt 1−ヒドロキシベンゾトリアゾール水和物
HTMP 2,2,6,6−テトラメチルピペリジン
MeCN アセトニトリル
MeOH メタノール
mp. 融点
NMM N−メチルモルホリン
PG 保護基
PPh3 トリフェニルホスフィン
RT 室温
TEA トリエチルアミン
TFA トリフルオロ酢酸
THF テトラヒドロフラン
Z ベンジルオキシカルボニル
Z−OSu N−(ベンジルオキシカルボニルオキシ)スクシンイミド
【0075】
下記のアミノ酸誘導体は、PepTech Corporation(20 Mall Road、Suite 460、Burlington、MA 01803、USA)によってカスタム合成された(D−2−クロロ−4−フルオロフェニルアラニンメチルエステル塩酸塩、D−4−クロロ−2−フルオロフェニルアラニンメチルエステル塩酸塩、及びD−2,4−ジフルオロ−フェニルアラニンメチルエステル塩酸塩)。D−2−クロロ−4−メチルフェニルアラニンメチルエステル塩酸塩及びD−4−クロロ−2−メチルフェニルアラニンメチルエステル塩酸塩は、NetChem、Inc.(100 Jersey Ave、Suite A211、New Brunswick、NJ 08901、USA)から得た。
【0076】
cis−3−アザ−ビシクロ[3.1.0]ヘキサン塩酸塩は、米国特許第4,183,857号明細書において記載されているように調製した。2−フルオロ−3−ヨード−ピラジンは、Tetrahedron1998、54、4899〜4912において記載されているように調製した。4−フルオロ−3−ヨード−ピリジンは、Tetrahedron1993、49、49〜64において記載されているように調製した。
【0077】
反応スキーム1:
配向性オルト−メタル化
【化23】
ヘテロアリールピペリジン、オルト−フルオロ−ヨードピリジン、−ピリダジン及び−ピラジンの合成のための出発物質は、反応スキーム1において示されているように調製することができる。フルオロ置換ヘテロアリールは、適切な温度にてTHFなどの適切な溶媒中、n−ブチルリチウム及びアミン(ジイソプロピルアミンなど)、又は2,2,6,6−テトラメチルピペリジンなどの試薬から調製されるアミドでメタル化することができる。このように得られたリチオ誘導体は、引き続いてヨウ素と反応し、所望の化合物を得ることができる。
【0078】
反応スキーム2:
アセチル化
【化24】
ヘテロアリールピペリジン、オルト−アセトキシ−ブロモピリジン、−ピリダジン及び−ピラジンの合成のための他の出発物質は、反応スキーム2において示されているように調製することができる。ブロモ原子に対してオルト位においてヒドロキシ基を含有するブロモ又はクロロヘテロアリールは、ピリジンなどの適切な塩基の存在下で、DCMなどの適切な溶媒中で適切な温度にて、無水酢酸などのアセチル化試薬と反応することができる。
【0079】
反応スキーム3:
ブロモ又はヨード置換ヘテロアリールの根岸カップリング反応
【化25】
【化26】
反応スキーム3において示されているように、オルト−フルオロ−ブロモピリジン、ピリダジン及びピラジンは、ヨウ化銅(I)及びジクロロ(1,1’−ビス(ジフェニル−ホスフィノ)−フェロセン)パラジウム(II)DCM付加体の存在下で、DMAなどの不活性溶媒中、(1−tert−ブトキシカルボニルピペリジン−4−イル)(ヨード)亜鉛との根岸カップリングに供され(J.Org.Chem.2004、69、5120〜5123)、その結果として生じるアリールピペリジンを得ることができる。オルト−フルオロ−ヨードピリジン、−ピリダジン及び−ピラジンが、出発物質として使用される場合、同じ生成物が得られる。
【0080】
代わりに、根岸カップリングは、反応スキーム2からのオルト−アセトキシ置換ブロモピリジン、ピリダジン及びピラジンを出発物質として使用して行うことができる。遊離アルコールは、水とメタノールとの混合物などの適切な溶媒中、水酸化リチウムなどの塩基による酢酸エステルのけん化によって得られる。オルト−アセトキシ−クロロピリジン、−ピリダジン及び−ピラジンの出発物質としての使用は、同じ生成物の形成をもたらす。
【0081】
反応スキーム4:
ヘテロアリールとカルボキシアルデヒド又はベンジルアミン官能基との根岸カップリング反応
【化27】
任意選択で置換されているオルト−カルボキシ−ブロモピリジン、ピリダジン及び−ピラジンは、反応スキーム4において示されているように根岸カップリングに供することができる。ヨウ化銅(I)及びジクロロ(1,1’−ビス(ジフェニル−ホスフィノ)−フェロセン)パラジウム(II)DCM付加体の存在下で、DMAなどの不活性溶媒中、(1−tert−ブトキシカルボニルピペリジン−4−イル)(ヨード)亜鉛との反応によって、このように得られたアリールピペリジンがもたらされる。トリアセトキシ水素化ホウ素ナトリウムなどの還元剤の存在下で、1,2−ジクロロエタンなどの適切な溶媒中、キャッピング基T−Hによる還元的アミノ化によって、Boc保護されたA部分がもたらされる。
【0082】
代わりに、任意選択で置換されているオルト−カルボキシ−ブロモピリジン、ピリダジン及びピラジンは、根岸カップリングが行われる前に、還元的アミノ化ステップに最初に供することができる。
【0083】
上記の反応順序はまた、任意選択で置換されているオルト−カルボキシ−クロロピリジン、ピリダジン及び−ピラジンを使用して行うことができる。
【0084】
反応スキーム5:
アミノアルコールの調製
【化28】
N−置換アミノアルコールHO(C(R6)2)m−Tは、反応スキーム5において記載されているように得ることができる。所定の温度にて水などの適切な溶媒中、任意選択で置換されているアミノアルコールHO(C(R6)2)mNH2と、ギ酸及びホルムアルデヒドの混合物との反応によって、相当するN,N−ジメチル化アミノアルコールの形成がもたらされる。このようなアミノアルコールの環状類似体は、炭酸カリウムなどの塩基の存在下で、アセトニトリルなどの適切な溶媒中、任意選択で置換されているアミノアルコールHO(C(R6)2)mNH2と、α,ω−ジブロモアルカンとの反応によって得ることができる。
【0085】
反応スキーム6:
アミノアルコール前駆体としてのエポキシド
【化29】
反応スキーム6において示されているように、任意選択で置換されているエポキシドは、位置選択的方法で、水などの適切な溶媒中で適切なアミンT−Hと反応し、α−置換β−アミノアルコールを形成することができる。
【0086】
反応スキーム7:
求核芳香族置換反応
【化30】
反応スキーム7において示されているように、フルオロ置換ピリジル、ピリダジル及びピラジニルピペリジンは、水素化ナトリウムなどの塩基の存在下で、DMFなどの溶媒中で適切な温度にて、ω−T−キャップされたアルキルアルコールとの求核芳香族置換反応に供され、Boc保護されたA部分を得ることができる。
【0087】
代わりに、フルオロ置換ヘテロアリールピペリジンはまた、BuLi又はDIEAなどの塩基の存在下で、THFなどの適切な溶媒中又は溶媒を伴わず適切な温度にて、ω−T−キャップされた第一級又は第二級アルキルアミンと反応して、Boc保護されたA部分を得ることができる。
【0088】
反応スキーム8
環状アミンを含有するA部分の合成
【化31】
反応スキーム8において示されているように、反応スキーム3からの中間生成物である任意選択で置換されているフルオロピリジン、ピリダジン及びピラジンはまた、水素化ナトリウムなどの塩基の存在下で、DMFなどの適切な溶媒中、環状第三級アミン部分を含有するアルコールとの求核芳香族置換反応に供され、Boc保護されたA部分を得ることができる。
【0089】
同様に、保護された環状第二級アミン部分を含有するアルコールを、上記の条件を使用して構造単位として導入することができる。保護基は、ピペリジンの保護のために使用されるBoc−保護基に対して直交しなくてはならない。A部分とB−C部分とのカップリングの後に、この保護基は、標準的な方法を用いて除去することができる。
【0090】
反応スキーム9:
アルキルエーテルスペーサー(R1=−O(C(R6)2)m−T)によるA部分の代替合成
【化32】
アルキルエーテルスペーサー(R1=−O(C(R6)2)m−T)を担持するA部分の合成は、代わりに反応スキーム9において示されているように行うことができる。Boc保護されたピペリジンは、Cs2CO3又はNaHなどの塩基の存在下で、DMFなどの適切な溶媒中、キャッピング基Tを担持する塩化アルキル又は臭化アルキルと反応し、Boc保護されたA部分が生じる。
【0091】
反応スキーム10:
光延条件を使用したアルキルエーテルスペーサー(R1=−O(C(R6)2)m−T)によるA部分の合成
【化33】
反応スキーム10において示されているように、反応スキーム3からの中間生成物である任意選択で置換されている(ヒドロキシヘテロアリール)ピペリジンはまた、DEAD又はDIADなどの試薬、及びPPh3などのホスフィンの存在下で、THFなどの適切な溶媒中、ω−T−キャップされたアルキルアルコールでアルキル化し、Boc保護されたA部分を得ることができる。
【0092】
同様に、同じ中間体は、上記の反応条件を使用してω−ブロモアルキルアルコールと反応させて、相当するエーテルに接近することができ、それを引き続いてK2CO3又はNaHなどの適切な塩基の存在下で、MeCN、THF、又はDMFなどの適切な溶媒中、適切な温度にて、キャッピング基Tをアルキル化するために使用し、Boc保護されたA部分をもたらすことができる。
【0093】
反応スキーム11:
5−ピペリジン−4−イル−ピリミジン由来A部分の合成
【化34】
5−ピペリジン−4−イル−ピリミジン由来A部分は、反応スキーム11において示されているように合成することができる。Boc−4−ピペリドンは、四塩化チタンなどの試薬及びピリジンなどの塩基の存在下、適切な温度にて、マロン酸ジエステルと反応することができる。この反応の生成物は、水及びメタノールの混合物などの適切な溶媒中、10%炭担持パラジウムなどの触媒を使用して水素化し、相当するBoc−2−ピペリジン−4−イル−マロン酸ジエステルを得ることができる。ナトリウムメトキシドなどの塩基の存在下で、メタノールなどの溶媒中でのアミジン塩酸塩との反応によって、任意選択で置換されている1H−ピリミジン−4,6−ジオンがもたらされ、それはBoc保護基をZ保護基と交換した後、POCl3などの試薬を使用して、sym−コリジンなどの適切な塩基の存在下で、アセトニトリルなどの適切な溶媒中、4,6−ジクロロ−ピリミジンに変換することができる。任意選択で置換されている4,6−ジクロロ−ピリミジンは、水素化ナトリウムなどの塩基の存在下で、THFなどの適切な溶媒中、ω−T−キャップされたアルキルアルコールと反応することができる。それに続く水素化は、酸化カルシウムの存在下で、ジエチルエーテルなどの適切な溶媒中、10%炭担持パラジウムなどの触媒を使用して行われ、5−ピペリジン−4−イル−ピリミジン由来A部分がもたらされる。
【0094】
反応スキーム12
A部分の脱保護
【化35】
一般に、Boc保護されたヘテロアリールピペリジン(A部分)の出発物質は、カップリング手順に供する前に、TFA/CH2Cl2、HCl/EtOAc、HCl/ジオキサン又はMeOH/ジオキサン中のHClの存在下で、硫化ジメチル(DMS)などのカチオンスカベンジャーと共に又は伴わず、脱保護することができる。それはカップリング手順に供する前に遊離塩基に変換することができ、又はある場合には塩として使用することができる。
【0095】
反応スキーム13
グリニャール反応又はルパート試薬を使用したアミンH−R5の合成
【化36】
反応スキーム13は、同じ炭素原子においてヒドロキシ置換基と1つのさらなる置換基とを担持するアミンH−R5の合成を示す。このようなアミンは、適切な保護基で保護されているオキソ置換アミンを使用して合成することができる。適切な保護基には、これらだけに限らないが、Boc、Z、ベンジル、及びベンズヒドリルが挙げられる。保護されたオキソ置換アミンは、THF又はジエチルエーテルなどの不活性溶媒中で適切な温度にてグリニャール試薬と反応して、相当する置換されているアルコールを得ることができる。
【0096】
代わりに、フッ化セシウムなどの試薬の存在下で、THFなどの適切な溶媒中で所定の温度にて、保護されたオキソ置換アミンとルパート試薬(トリフルオロメチルトリメチルシラン)との反応によって、トリフルオロメチル化したアルコールがもたらされる。
【0097】
上記の合成法によって得た保護されたアミンH−R5の保護基は、標準条件を使用して除去することができる。
【0098】
反応スキーム14:
B−C部分の形成
【化37】
B−C部分は、反応スキーム14において示されているように合成することができる。任意選択で置換されているフェニルアラニンは、メタノール中で塩化チオニル又は塩化オキサリルなどの活性化剤を使用して、相当するメチルエステル塩酸塩に変換することができる。アミノ酸メチルエステル塩酸塩は、NaHCO3(水溶液)などの塩基の存在下で、DCMなどの適切な溶媒中、トリホスゲンなどの試薬と反応して、イソシアネートを得ることができ、これは引き続いてDCM又はDMFなどの適切な溶媒中でアミンR5−Hと反応することができる。R5−Hが塩酸塩の形態で使用される場合、DIEAなどの適切な塩基をさらに使用し、遊離アミンR5−Hを遊離する。エステル官能基は、適切な溶媒又は溶媒混合物(水/THF/メタノールなど)中でLiOHなどの塩基で加水分解し、B−C−部分に接近することができる。
【0099】
反応スキーム15:
B−C部分の固相合成
【化38】
代わりに、B−C−部分はまた、反応スキーム15において示されているように固相上で合成することができる。ワング樹脂は、DMAPなどの塩基の存在下で、DMF又はDCMなどの適切な溶媒中、DIC又はDCCなどの試薬を使用して、任意選択で置換されているFmoc−保護されたフェニルアラニンと共に充填することができる。固体支持体上の未反応のOH−基のキャッピングは、DMF又はDCMなどの適切な溶媒中、無水酢酸とのそれに続く反応によって行うことができる。DCMなどの溶媒中でピペリジン又はジエチルアミンなどの塩基によるFmoc−保護基の除去後、遊離アミンは、TEAなどの塩基の存在下で、DCMなどの適切な溶媒中、p−ニトロフェニルクロロホルメートによって活性化したカルバメートに変換することができる。DCMなどの適切な溶媒中での前記p−ニトロフェニルカルバメートのアミンR5−Hとの反応によって、所望のB−C部分が生じる。R5−Hが塩酸塩の形態で使用される場合、DIEAなどの適切な塩基がさらに使用され、遊離アミンR5−Hが遊離される。固体支持体からの切断は、DCM中のTFAによる処理によって行うことができる。この経路によって得られるB−C−部分は、精製又は適切なA部分と直接カップリングすることができる。
【0100】
反応スキーム16:
A部分のB−C部分とのカップリング及び塩の形成
【化39】
反応スキーム16において示されているように、A部分は、EDCI/HOBt、塩基(N−メチルモルホリン(NMM)など)及び溶媒(ジクロロメタン(DCM)など)の存在下で、B−C部分とカップリングすることができる。DCM、DMF、THFなどの適切な溶媒、又は上記の溶媒の混合物を、カップリング手順のために使用することができる。適切な塩基には、トリエチルアミン(TEA)、ジイソプロピルエチルアミン(DIEA)、N−メチルモルホリン(NMM)、コリジン又は2,6−ルチジンが挙げられる。EDCI/HOBtが使用される場合、塩基は必要ないことがある。
【0101】
一般に、反応が完了した後、反応混合物はEtOAc、DCM又はEt2Oなどの適切な有機溶媒で希釈することができ、次いで水、HCl、NaHSO4、炭酸水素塩、NaH2PO4、リン酸緩衝液(pH7)、ブライン又は任意のこれらの組合せなどの水溶液で洗浄する。反応混合物を濃縮し、次いで適切な有機溶媒及び水溶液に分配することができる。反応混合物を濃縮し、水性後処理なしでクロマトグラフィーにかけることができる。
【0102】
溶媒又は溶媒混合物(ジエチルエーテル/アセトンなど)中のHClを使用して、生成物を塩酸塩などの薬学的に許容される塩に転移させることができる。
【0103】
反応スキーム17:
ニトロフェニルホルメート中間体を介した尿素形成
【化40】
3つの部分はまた、反応スキーム17において示されているように段階的に結合させることができる。適切なA部分は、EDCI/HOBt、塩基(N−メチルモルホリン(NMM)など)及び溶媒(ジクロロメタン(DCM)など)の存在下でBoc保護されたB部分とカップリングし、続いてジオキサンとメタノールとの混合物中で、塩化水素の補助によるBoc脱保護が起こる。生成物は、NMMなどの塩基の存在下でDCMなどの適切な溶媒中、4−ニトロフェニルクロロホルメートと反応し、4−ニトロフェニルカルバメートを生じさせることができ、それは引き続いてDIEAなどの塩基の存在下でTHFなどの適切な溶媒中、アミンH−R5で処理し、目的化合物に接近することができる。最終生成物は、上記のような薬学的に許容される塩に変換することができる。
【0104】
反応スキーム18:
試薬として1−メチル−3−(アミノ−1−カルボニル)−3H−イミダゾール−1−イウムヨージドを使用した尿素形成
【化41】
反応スキーム18において示されているように、THFなどの適切な溶媒中で適切な温度にて、1,1’−カルボニルジイミダゾールは、アミンと反応することができる。この反応の生成物は、アセトニトリルなどの適切な溶媒中、ヨウ化メチルとさらに反応し、1−メチル−3−(アミノ−1−カルボニル)−3H−イミダゾール−1−イウムヨージドがもたらされる。この活性化種は、トリエチルアミンなどの塩基の存在下で、THFなどの適切な溶媒中、脱保護されたA−B部分と反応し、最終生成物がもたらされる。最終生成物は、上記のような薬学的に許容される塩に変換することができる。
【0105】
分析的LC−MS
式(I)による本発明の化合物を、分析的LC−MSによって分析した。分析において使用した条件を、下記に要約する。
【0106】
分析の条件の要約:
SPD−M10Avp UV/Visダイオードアレイ検出器及びQP2010 MS−検出器(ESI+モード、214及び254nmでのUV−検出)を備えたLC10Advp−Pump(島津製作所)、
カラム:Waters XTerra MS C18、3.5μm、2.1*100mm、
線形グラジェント、水中のアセトニトリル(0.15%HCOOH)
0.4ml/分の流速;
移動相A:水(0.15%HCOOH+5%アセトニトリル)
移動相B:アセトニトリル(0.15%HCOOH+5%水)
グラジェントA:
開始濃度、1%アセトニトリル(0.15%HCOOH)
9.00分、B.濃度 30
10.00分、B.曲線 3
12.00分、B.濃度 99
15.00分、B.濃度 99
15.20分、B.濃度 1
18.00分、ポンプ 停止
グラジェントB:
開始濃度、10%アセトニトリル(0.15%HCOOH)
10.00分、B.濃度 60
11.00分、B.曲線 2
12.00分、B.濃度 99
15.00分、B.濃度 99
15.20分、B.濃度 10
18.00分、ポンプ 停止
グラジェントC:
線形グラジェント、水中の5%〜95%アセトニトリル(0.15%HCOOH)
0.00分、5%B
5.00分、95%B
5.10分、99%B
6.40分、99%B
6.50分、5%B
8.00分、ポンプ停止
グラジェントD:
線形グラジェント、水中の5%〜80%アセトニトリル(0.15%HCOOH)
0.00分、5%B
5.00分、80%B
5.10分、99%B
6.40分、99%B
6.50分、5%B
8.00分、ポンプ停止
グラジェントE:
線形グラジェント、水中の1%〜70%アセトニトリル(0.15%HCOOH)
0.00分、1%B
5.00分、70%B
5.10分、99%B
6.40分、99%B
6.50分、5%B
8.00分、ポンプ停止
【0107】
下記の表は、反応スキーム1〜18によって調製することができる本発明の詳細な実施例を記載する。しかし、これらの実施例は、いかなる形でも本発明の範囲を限定すると解釈されない。
【0108】
【化42】
【表1】
【表2】
【0109】
【化43】
【表3】
【0110】
【化44】
【表4】
【0111】
【化45】
【表5】
【表6】
【表7】
【表8】
【表9】
【表10】
【0112】
【化46】
【表11】
【0113】
【化47】
【表12】
【0114】
【化48】
【表13】
【0115】
【化49】
【表14】
【0116】
【化50】
【表15】
【0117】
【化51】
【表16】
【0118】
【化52】
【表17】
【表18】
【0119】
【化53】
【表19】
【表20】
【表21】
【0120】
【化54】
【表22】
【0121】
【化55】
【表23】
【0122】
【化56】
【表24】
【0123】
下記の実施例は、本発明を例示するために提示し、本発明の範囲をいかなる形でも限定しない。
【0124】
一般の中間体:
1−tert−ブトキシカルボニルピペリジン−4−イル)(ヨード)亜鉛:
【化57】
亜鉛の活性化
シュレンクフラスコにセライト(1.28g)を入れ、真空中で加熱することによって乾燥させた。次いで、亜鉛粉末(6.51g)及び乾燥N,N−ジメチルアセトアミド(15ml)を加えた。クロロトリメチルシラン(1.14ml)と1,2−ジブロモエタン(0.80ml)との7:5v/v混合物をN,N−ジメチルアセトアミド(1ml)中の溶液として、温度を65℃未満に維持する速度で(約15分)加えている間に、混合物を室温で撹拌した。このように得られたスラリーを15分間熟成させた。
【0125】
亜鉛の挿入
Boc−4−ヨードピペリジン(24.8g)のN,N−ジメチルアセトアミド(39ml)溶液を、温度を65℃未満に維持する速度で(約20分)、上記の混合物にゆっくり加えた。次いで、このように得られた反応混合物を室温で30分間熟成させた。懸濁液をアルゴン下フリットで濾過し、全ての固体を除去した。このように得られた淡黄色の溶液を、アルゴン下室温で保存し、カップリング反応のために直接使用した。
【0126】
3−フルオロ−3’,4’,5’,6’−テトラヒドロ−2’H−[2,4’]ビピリジニル−1’−カルボン酸tert−ブチルエステル:
【化58】
500mlのフラスコに、2−ブロモ−3−フルオロピリジン(10.0g)、[1,1’−ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)のジクロロメタンとの錯体(1.39g)、ヨウ化銅(I)(0.65g)、及び乾燥N,N−ジメチルアセトアミド(80ml)を入れた。このように得られた混合物を交互の真空/アルゴンパージで脱気した。次いで、1−tert−ブトキシカルボニルピペリジン−4−イル)(ヨード)亜鉛(79.7mmol)を加えた。混合物をもう一度脱気し、次いで80℃に一晩加熱した。次いで、N,N−ジメチルアセトアミドの主要部分を蒸発させ、残りをEtOAc及び水の混合物(各々500ml)に溶解した。次いで、これをセライトで濾過し、分液漏斗に移した。相を分離し、水層をEtOAc(3×250ml)で抽出した。合わせた有機層を水及びブライン(各々500ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をクロマトグラフィーによって精製し、茶色の固体の形態で所望の化合物を得た。
【0127】
(S)−1−ピロリジン−1−イル−ブタン−2−オール:
【化59】
ピロリジン(5.76ml)を、(S)−(−)−1,2−エポキシブタン(5.00ml)と水(25ml)との混合物に加えた。反応混合物を室温で一晩激しく撹拌した。さらなる水(25ml)を加え、有機物質をジエチルエーテル(2×100ml)で抽出した。合わせた有機層を乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。揮発性の不純物を除去するために、エーテル及びトルエン(各々2×10ml)との何回かの同時蒸発を行い、無色の液体の形態の所望の化合物を得た。
【0128】
(R)−2−ピロリジン−1−イル−ブタン−1−オール:
【化60】
(R)−(−)−2−アミノ−1−ブタノール(5.00g)及び1,4−ジブロモブタン(6.61ml)のCH3CN(60ml)溶液に、K2CO3(15.48g)を加え、このように得られた懸濁液を還流温度で19時間撹拌した。反応混合物を真空中で蒸発させ、残渣をEtOAc及び水に分割した。有機層を水及びブラインで洗浄した。水層をEtOAcで抽出した。合わせた有機層をNa2SO4上で乾燥させ、真空中で乾燥するまで蒸発させた。粗生成物をクーゲルロール蒸留(12ミリバール、106〜150℃)によって精製し、透明で無色の油を得た。
【0129】
(S)−1−ジメチルアミノ−ブタン−2−オール:
【化61】
(S)−(−)−1,2−エポキシブタン(9.66)と水(40ml)との混合物を、100mlのフラスコ中に入れた。ジメチルアミン(水中の40%溶液、20.3ml)を一度に加え、反応混合物を氷浴で数分間冷却し、一晩室温で撹拌した。固体NaClを飽和するまで加え、混合物を、DCM(3×50ml)で抽出した。合わせた有機層を水及びブライン(各々30ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、注意深く蒸発させ(20ミリバール/25℃)、無色の液体を得た。
【0130】
(R)−2−ジメチルアミノ−ブタン−1−オール:
【化62】
(R)−(−)−2−アミノ−1−ブタノール(5.00g)を、冷却したギ酸(11.0ml)に一滴ずつ加えた。ホルムアルデヒド(36.5%水溶液、10.73ml)を加え、混合物を0℃で1h撹拌した.次いで、それを90℃に4h加熱した。反応物を真空中で蒸発させ、油性残渣をDCM(150ml)及び1NのNaOHに分配した。有機相を1NのNa2CO3及びブラインで洗浄した。DCM相をMgSO4上で乾燥させ、濾過し、蒸発させ、黄色の油を得た。それをクーゲルロール蒸留(60ミリバール、85〜130℃)によって精製した。生成物を、無色の油の形態で得た。
【0131】
2−ジメチルアミノ−2−メチル−プロパン−1−オール:
【化63】
2−アミノ−2−メチル−1−プロパノール(5.00g)、ホルムアルデヒド水溶液(水中36.5%、10.73ml)及び水(20ml)中のギ酸(11.00ml)の混合物を、0℃にて1h撹拌し、次いで90℃に4h加熱した。反応混合物を真空中で濃縮し、残渣を水(20ml)で希釈し、1NのNaOHを加えることによってアルカリ性(pH14)にした。さらに、固体NaClを飽和するまで加えた。水溶液を、ジクロロメタン(3×50ml)で抽出し、合わせた有機層を乾燥させ(Na2SO4)、真空中で濃縮した。減圧(40ミリバール)下での蒸留によって、所望の化合物を無色の油として得た。
【0132】
(S)−1−メチル−ピペリジン−3−オール:
【化64】
(S)−3−ヒドロキシピペリジン塩酸塩(4.00g)、ホルムアルデヒド水溶液(水中36.5%、3.73ml)及び水(20ml)中のギ酸(2.19ml)の混合物の還流を一晩続けた。反応混合物を真空中で濃縮し、残渣を水(20ml)で希釈し、1NのNaOHを加えることによってアルカリ性(pH14)にした。さらに、固体NaClを飽和するまで加えた。水溶液を、ジクロロメタン(3×50ml)で抽出し、合わせた有機層を乾燥させ(Na2SO4)、真空中で濃縮した。減圧(10ミリバール)下での蒸留によって、所望の化合物を無色の液体として得た。
【0133】
3−((R)−2−ジメチルアミノ−ブトキシ)−3’,4’,5’,6’−テトラヒドロ−2’H−[2,4’]ビピリジニル−1’−カルボン酸tert−ブチルエステル:
【化65】
(R)−2−ジメチルアミノ−ブタン−1−オール(1.004g)を、アルゴン下で0℃にて水素化ナトリウム(鉱油中60%、240mg)のDMF(30ml)懸濁液に加えた。冷却槽を取り外し、混合物を室温で2h撹拌した。次いで、3−フルオロ−3’,4’,5’,6’−テトラヒドロ−2’H−[2,4’]ビピリジニル−1’−カルボン酸tert−ブチルエステル(1.200g)を加え、反応混合物を120℃に加熱した(油浴温)。120℃で一晩撹拌した後、反応混合物を真空中で濃縮した。残りの濃褐色の油を酢酸エチル(150ml)に溶解し、混合物を1NのNa2CO3(2×60ml)で洗浄した。合わせた水層をEtOAc(30ml)で再抽出し、合わせた有機層をブライン(50ml)で洗浄し、乾燥させ(MgSO4)、濾過し、蒸発させ、濃褐色の油を得た。それをカラムクロマトグラフィーによって精製した。
【0134】
[(R)−1−(1’,2’、3’,4’,5’,6’−ヘキサヒドロ−[2,4’]ビピリジニル−3−イルオキシメチル)−プロピル]−ジメチルアミン:
【化66】
3−((R)−2−ジメチルアミノ−ブトキシ)−3’,4’,5’,6’−テトラヒドロ−2’H−[2,4’]ビピリジニル−1’−カルボン酸tert−ブチルエステル(788mg)を、ジオキサン(15ml)とメタノール(4ml)との混合物に溶解した。ジオキサン(15ml)中の4NのHClを加え、反応物を室温で1h撹拌した。トルエン(4×30ml)との同時蒸発を含めた全ての揮発性物質の蒸発によって、ベージュ色の半固体を得て、それを高真空中で一晩さらに乾燥させた。残りの固体を10mlの乾燥ジエチルエーテルで粉砕した。溶媒をデカントし、生成物を真空中で乾燥させた。
【0135】
B−C部分の合成:
B−C部分1:
中間体A1):
【化67】
D−2,4−ジクロロフェニルアラニン(10.00g)のメタノール(100ml)懸濁液に、塩化チオニル(9.39ml)を一滴ずつ加えた。添加の間に、透明な溶液が形成され、反応物の還流を始めた。反応混合物の還流を2h続けた。室温に冷却した後、混合物を40℃で乾燥するまで蒸発させた。粗生成物をジエチルエーテル中で粉砕し、不溶性化合物を濾過し、ジエチルエーテルで洗浄し、最終的に真空中室温にてP2O5上で一晩乾燥させた。生成物を、無色の針状晶の形態で得た。
【0136】
中間体B1):
【化68】
350mlの三つ口平底フラスコに機械式撹拌機を搭載し、DCM(80ml)、飽和炭酸水素ナトリウム水溶液(80ml)、及び中間体A1)(5.69g)を入れた。二相性混合物を氷浴中冷却し、トリホスゲン(1.96g)を1回で加える間に機械的に撹拌した。反応混合物を氷浴中45分間撹拌し、次いで250mlの分液漏斗に注いだ。有機層を集め、水層を20mlのDCMで3回抽出した。合わせた有機層を水で洗浄し、Na2SO4上で乾燥させ、濾過し、真空中で乾燥するまで蒸発させ、粗生成物を半固体として得た。残渣をクーゲルロール蒸留によって精製した(200〜240℃、0.04〜0.08ミリバール)。生成物を透明で無色の油として得た。
【0137】
中間体C1):
【化69】
氷冷の中間体B1)(4.99g)のDCM(50ml)溶液に、ピロリジン(4.56ml)を加えた。10分後氷浴を取り外し、撹拌を4h続けた。反応混合物を真空中で蒸発させた。残渣をEtOAcに再溶解し、有機層を1NのHCl、水、飽和Na2CO3、水及びブラインで洗浄した。全ての水層をEtOAcで抽出した。合わせた有機層をNa2SO4上で乾燥させ、真空中で乾燥するまで蒸発させた。
【0138】
B−C部分1:
【化70】
中間体C1)(6.28g)を、MeOH(100ml)及びTHF(30ml)に0℃で溶解した。水酸化リチウム一水和物(1.53g)の水(30ml)溶液を5分に亘って一滴ずつ加えた。混合物を0℃で60分間撹拌し、次いで0.5MのHClを加えることによって酸性化した。反応混合物をEtOAcで2度抽出した。合わせた有機層を水で2度、及びブラインで洗浄し、Na2SO4上で乾燥させ、真空中で蒸発させた。固体残留物をEt2O中で粉砕し、次いで濾過し、Et2Oで洗浄した。生成物を白色の固体として得た。
【0139】
B−C部分2:
中間体A2):
【化71】
氷冷の中間体B1)(200mg)の無水DMF(2.5ml)溶液に、3−ヒドロキシアゼチジン塩酸塩(160mg)及びトリエチルアミン(205μl)の無水DMF(2.5ml)溶液をアルゴン下で加えた。混合物を0℃で4h30分間撹拌させた。反応混合物を真空中で蒸発させた。残渣を冷たいEtOAc(75ml)で希釈し、有機相を0.1MのHCl(2×25ml)及びブラインで洗浄した。水相をEtOAcで再抽出し、有機相をNa2SO4上で乾燥させ、濾過し、蒸発させた。無色の油を得て、それを高真空下にて乾燥させた。
【0140】
B−C部分2:
【化72】
中間体A2)(255mg)を、MeOH(1.5ml)及びTHF(4.0ml)に0℃で溶解した。水酸化リチウム一水和物(62mg)のH2O(1.5ml)溶液を加えた。混合物を0℃で4h30分撹拌した。1NのHClを加えることによって反応混合物を中和し、溶媒を真空中で蒸発させた。次いで、水相を1NのHCl(pH約1〜2)で酸性化した。水相をEtOAc(約1×20ml)で抽出した。有機層を水及びブラインで洗浄した。水相をEtOAcで再抽出し、合わせた有機層をNa2SO4上で乾燥させ、真空中で蒸発させ、無色の固体を得た。
【0141】
B−C部分3:
中間体A3):
【化73】
(R)−3−ピロリジノール(1.43g)を、氷冷の中間体B1)(1.50g)のCH2Cl2(25ml)溶液に加えた。2時間後、氷浴を取り外し、撹拌を2時間続けた。反応混合物を真空中で蒸発させた。残渣をEtOAcに再溶解し、有機層を1NのHCl、水、飽和NaHCO3、水及びブラインで洗浄した。全ての水層をEtOAcで抽出した。合わせた有機層をMgSO4上で乾燥させ、濾過し、真空中で乾燥するまで蒸発させた。
【0142】
B−C部分3:
【化74】
中間体A3)(1.78g)を、MeOH(35ml)及びTHF(9.5ml)に0℃で溶解した。水酸化リチウム一水和物(0.41g)のH2O(9.5ml)溶液を5分に亘って一滴ずつ加えた。混合物を0℃で2時間撹拌した。0.5MのHClを加えることによって反応混合物を酸性化し、次いで、EtOAcで2度抽出した。有機層を水及びブラインで2度洗浄した。合わせた有機層をNa2SO4上で乾燥させ、真空中で蒸発させた。固体残留物をEt2O中で粉砕し、次いで濾過し、Et2Oで洗浄し、無色の結晶性固体を得た。
【0143】
B−C部分4:
中間体A4):
【化75】
(S)−3−ピロリジノール(1.43g)を、氷冷のイソシアネート(1.50g)のCH2Cl2(25ml)溶液に加えた。20分後、氷浴を取り外し、4時間撹拌を続けた。反応混合物を真空中で蒸発させた。残渣をEtOAcに再溶解し、有機層を1NのHCl、水、飽和NaHCO3及びブラインで洗浄した。各水層をEtOAcで再抽出した。合わせた有機層をMgSO4上で乾燥させ、濾過し、真空中で乾燥するまで蒸発させた。生成物を、無色の安定した泡の形態で得た。
【0144】
B−C部分4:
【化76】
中間体A4)(1.97g)を、MeOH(35ml)及びTHF(9.5ml)に0℃で溶解した。水酸化リチウム一水和物(0.45g)のH2O(9.5ml)溶液を5分に亘って一滴ずつ加えた。混合物を0℃で2時間撹拌した。0.5MのHClを加えることによって反応混合物を酸性化し、次いでEtOAcで2度抽出した。有機層を水及びブラインで洗浄した。合わせた有機層をNa2SO4上で乾燥させ、真空中で蒸発させた。残渣をジエチルエーテルに溶解し、再び蒸発させた。残りの無色の泡を真空中で乾燥させた。
【0145】
実施例2の合成:
中間体2a):
【化77】
火炎乾燥シュレンクフラスコに、3−ブロモ−2−フルオロ−6−ピコリン(434mg)、[1,1’−ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)のジクロロメタンとの錯体(56mg)、ヨウ化銅(I)(26mg)、及び乾燥N,N−ジメチルアセトアミド(3ml)を入れた。このように得られた混合物を交互の真空/アルゴンパージで脱気した。次いで、1−tert−ブトキシカルボニルピペリジン−4−イル)(ヨード)亜鉛(3.19mmol、上記のように調製)を加えた。混合物をもう一度脱気し、次いで80℃に一晩加熱した。次いで、N,N−ジメチルアセトアミドの主要部分を蒸発させ、残りをEtOAc及び水の混合物(各々50ml)に溶解した。次いで、混合物をセライトで濾過し、分液漏斗に移した。相を分離し、水層を、EtOAc(2×50ml)で抽出した。合わせた有機層を水及びブライン(各々100ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をカラムクロマトグラフィーによって精製し、無色の油の形態で所望の化合物を得た。
【0146】
中間体2b):
【化78】
密封した管中に、中間体2a)(0.55g)、メチル−(2−ピロリジン−1−イル−エチル)−アミン(2.10g)、及びN,N−ジイソプロピルエチルアミン(286μl)を入れた。反応混合物を150℃に4日間加熱した。揮発性物質の主要部分を蒸発させ、残りをEtOAc(100ml)に溶解した。溶液をNaHCO3(2×25ml)で洗浄し、合わせた水層をEtOAc(25ml)で再抽出した。有機層を混合し、ブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、蒸発させた。粗生成物をカラムクロマトグラフィーによって精製し、未反応の出発物質及び所望の生成物を含有する混合画分を単離した。所望の化合物を、酸性条件下にて分取HPLCによって最終的に茶色がかった樹脂の形態で単離した。
【0147】
中間体2c):
【化79】
DCM(1ml)とMeOH(1ml)との混合物中のBoc保護された中間体2b)(69mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、2ml)を加え、溶液を室温で1h撹拌した。全ての揮発性物質の蒸発、並びにトルエン(2×5ml)、及びアセトン(5ml)との同時蒸発によってベージュ色の固体がもたらされ、それをデシケーター中シカペント(Sicapent)上で一晩さらに乾燥させた。
【0148】
実施例2:
【化80】
中間体2c)(68mg)及びB−C部分1(60mg)、1−ヒドロキシベンゾトリアゾール水和物(32mg)及びN−メチルモルホリン(58μl)を、DMF(3ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(48mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(12μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(70ml)で希釈し、飽和Na2CO3(3×25ml)、H2O及びブライン(各々25ml)で洗浄した。有機層を乾燥させ(Na2SO4)、溶媒を真空中で除去した。カラムクロマトグラフィーによる粗生成物の精製によって黄色がかった樹脂の形態の相当するアミンを得た。これをEtOAc(1ml)に溶解し、塩化水素のジエチルエーテル溶液(1.0M、114μl)で処理した。相当する二塩酸塩の完全な沈殿を得るために、このように得られた懸濁液をヘキサン(5ml)で希釈した。固体を濾過し、ヘキサンで洗浄し、デシケーター中シカペント上で一晩乾燥させ、オフホワイトの固体の形態の所望の化合物を得た。
【0149】
実施例4の合成:
中間体4a):
【化81】
冷却した(0℃)1−メチルピペラジン(257μl)の乾燥THF(2ml)溶液に、ヘキサン(0.50ml)中のn−ブチルリチウム(2.5M)をシリンジによって一滴ずつ加えた。室温で15分間撹拌した後、次いで、このように得られたリチウムアミドを、2a)(387mg)の乾燥THF(2ml)溶液に0℃で一滴ずつ加えた。2.5h後、反応混合物を飽和NH4Cl(2ml)で加水分解し、EtOAc(70ml)で希釈した。有機層のNaHCO3(2×25ml)及びブライン(25ml)による抽出、Na2SO4上での乾燥、濾過及び真空濃縮によって、黄色の油がもたらされた。これをカラムクロマトグラフィーによって精製し、黄色がかった樹脂の形態の所望の化合物を得た。
【0150】
中間体4b):
【化82】
DCM(1ml)とMeOH(1ml)との混合物中のBoc保護された中間体4a)(117mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、2ml)を加え、溶液を室温で1h撹拌した。全ての揮発性物質の蒸発、並びにトルエン(2×5ml)、及びアセトン(5ml)との同時蒸発によってベージュ色の固体がもたらされ、それをデシケーター中シカペント上で一晩さらに乾燥させた。
【0151】
実施例4:
【化83】
中間体4b)(66mg)及びB−C部分1(70mg)、1−ヒドロキシベンゾトリアゾール水和物(37mg)及びN−メチルモルホリン(68μl)を、DMF(3ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(56mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(14μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(70ml)で希釈し、飽和Na2CO3(3×25ml)、H2O及びブライン(各々25ml)で洗浄した。有機層を乾燥させ(Na2SO4)、溶媒を真空中で除去した。カラムクロマトグラフィーによる粗生成物の精製によって黄色がかった固体の形態の相当するアミンを得た。これをEtOAc(1ml)に溶解し、塩化水素のジエチルエーテル溶液(1.0M、214μl)で処理した。相当する二塩酸塩の完全な沈殿を得るために、このように得られた懸濁液をヘキサン(5ml)で希釈した。固体を濾過し、ヘキサンで洗浄し、デシケーター中シカペント上で一晩乾燥させ、白色の固体の形態の所望の化合物を得た。
【0152】
実施例9の合成:
中間体9a):
【化84】
火炎乾燥シュレンクフラスコに、3−ブロモ−2−フルオロ−5−ピコリン(1.00g)、[1,1’−ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)のジクロロメタンとの錯体(129mg)、ヨウ化銅(I)(60mg)、及び乾燥N,N−ジメチルアセトアミド(7ml)を入れた。このように得られた混合物を交互の真空/アルゴンパージで脱気した。次いで、1−tert−ブトキシカルボニルピペリジン−4−イル)(ヨード)亜鉛(7.37mmol、上記のように調製)を加えた。混合物をもう一度脱気し、次いで80℃にて一晩加熱した。次いで、N,N−ジメチルアセトアミドの主要部分を蒸発させ、残りをEtOAc及び水の混合物(各々100ml)に溶解した。次いで、混合物をセライトで濾過し、分液漏斗に移した。相を分離し、水層をEtOAc(3×50ml)で抽出した。合わせた有機層を水及びブライン(各々100ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をカラムクロマトグラフィーによって精製し、白色の固体の形態で所望の化合物を得た。
【0153】
中間体9b):
【化85】
(S)−1−ピロリジン−1−イル−ブタン−2−オール(191mg)のDMF(1ml)溶液を、アルゴン下0℃にて水素化ナトリウム(鉱油(36mg)中の60%分散物)のDMF(1ml)懸濁液に加えた。冷却槽を取り外し、混合物を室温で1h撹拌した。次いで、中間体9a)(158mg)のDMF(1ml)溶液を加え、反応混合物を60℃に1h加熱した。反応混合物を0℃に冷却し、次いで飽和NH4Cl(1ml)で加水分解した。EtOAc(70ml)を加え、混合物をNaHCO3(2×25ml)で洗浄した。合わせた水層をEtOAc(25ml)で再抽出し、次いで合わせた有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物を分取HPLCによって精製し、黄色がかった油の形態の相当するジホルメートを得た。
【0154】
中間体9c):
【化86】
DCM(2ml)とMeOH(0.5ml)との混合物中のBoc保護された中間体9b)(125mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、2ml)を加え、溶液を室温で30分間撹拌した。全ての揮発性物質の蒸発、並びにトルエン、アセトン、及びジエチルエーテル(各々5ml)との同時蒸発によって白色の固体がもたらされ、それをデシケーター中シカペント上で一晩さらに乾燥させた。
【0155】
実施例9:
【化87】
中間体9c)(55mg)及びB−C部分1(55mg)、1−ヒドロキシベンゾトリアゾール水和物(29mg)及びN−メチルモルホリン(54μl)を、DMF(3ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(44mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(11μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(70ml)で希釈し、飽和Na2CO3(3×25ml)、水及びブライン(各々25ml)で洗浄した。有機層を乾燥させ(Na2SO4)、溶媒を真空中で除去した。粗生成物を分取HPLCによって精製し、茶色がかった樹脂の形態の相当するジホルメートを得た。
【0156】
実施例11の合成:
中間体11a):
【化88】
火炎乾燥シュレンクフラスコに、3−フルオロ−4−ヨードピリジン(1.00g)、[1,1’−ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)のジクロロメタンとの錯体(110mg)、ヨウ化銅(I)(51mg)、及び乾燥N,N−ジメチルアセトアミド(6ml)を入れた。このように得られた混合物を交互の真空/アルゴンパージで脱気した。次いで、1−tert−ブトキシカルボニルピペリジン−4−イル)(ヨード)亜鉛(6.28mmol、上記のように調製)を加えた。混合物をもう一度脱気し、次いで80℃に一晩加熱した。次いで、N,N−ジメチルアセトアミドの主要部分を蒸発させ、残りをEtOAc及び水の混合物(各々100ml)に溶解した。次いで、混合物をセライトで濾過し、分液漏斗に移した。相を分離し、水層をEtOAc(3×30ml)で抽出した。合わせた有機層を水及びブライン(各々75ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をカラムクロマトグラフィーによって精製し、茶色がかった固体の形態で所望の化合物を得た。
【0157】
中間体11b):
【化89】
(S)−1−ピロリジン−1−イル−ブタン−2−オール(156mg)のDMF(2ml)溶液を、アルゴン下0℃にて水素化ナトリウム(鉱油(29mg)中の60%分散物)のDMF(1ml)懸濁液に加えた。冷却槽を取り外し、混合物を室温で1h撹拌した。次いで、中間体11a)(102mg)のDMF(2ml)溶液を加え、反応混合物を120℃に一晩加熱した。反応混合物を0℃に冷却し、次いで飽和NH4Cl(1ml)で加水分解した。EtOAc(50ml)を加え、混合物をNaHCO3(2×25ml)で洗浄した。合わせた水層をEtOAc(25ml)で再抽出し、次いで合わせた有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物を分取HPLCによって精製し、黄色がかった樹脂の形態の相当するジホルメートを得た。
【0158】
中間体11c):
【化90】
DCM(1ml)とMeOH(1ml)との混合物中のBoc保護された中間体11b)(111mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、2ml)を加え、溶液を室温で15分撹拌した。全ての揮発性物質の蒸発、並びにトルエン(2×5ml)、及びアセトン(5ml)との同時蒸発によってベージュ色の固体がもたらされ、それをデシケーター中シカペント上で一晩さらに乾燥させた。
【0159】
実施例11:
【化91】
中間体11c)(69mg)及びB−C部分1(58mg)、1−ヒドロキシベンゾトリアゾール水和物(31mg)及びN−メチルモルホリン(57μl)を、DMF(5ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(47mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(12μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(70ml)で希釈し、飽和Na2CO3(3×25ml)、H2O及びブライン(各々25ml)で洗浄した。有機層を乾燥させ(Na2SO4)、溶媒を真空中で除去した。カラムクロマトグラフィーによる粗生成物の精製によって黄色がかった樹脂の形態の相当するアミンを得た。これをEtOAc(1ml)に溶解し、塩化水素のジエチルエーテル溶液(1.0M、143μl)で処理した。相当する二塩酸塩の完全な沈殿を得るために、このように得られた懸濁液をヘキサン(5ml)で希釈した。固体を濾過し、ヘキサンで洗浄し、デシケーター中シカペント上で一晩乾燥させ、ベージュ色の固体の形態の所望の化合物を得た。
【0160】
実施例12の合成:
中間体12a):
【化92】
火炎乾燥シュレンクフラスコに、4−フルオロ−3−ヨード−ピリジン(1.00g)、[1,1’−ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)のジクロロメタンとの錯体(110mg)、ヨウ化銅(I)(51mg)、及び乾燥N,N−ジメチルアセトアミド(6ml)を入れた。このように得られた混合物を交互の真空/アルゴンパージで脱気した。次いで、1−tert−ブトキシカルボニルピペリジン−4−イル)(ヨード)亜鉛(6.28mmol、上記のように調製)を加えた。混合物をもう一度脱気し、次いで80℃に一晩加熱した。次いで、N,N−ジメチルアセトアミドの主要部分を蒸発させ、残りをEtOAc及び水の混合物(各々100ml)に溶解した。次いで、混合物をセライトで濾過し、分液漏斗に移した。相を分離し、水層をEtOAc(3×30ml)で抽出した。合わせた有機層を水及びブライン(各々75ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をカラムクロマトグラフィーによって精製し、茶色がかった固体の形態で所望の化合物を得た。
【0161】
中間体12b):
【化93】
(S)−1−ピロリジン−1−イル−ブタン−2−オール(187mg)のDMF(2ml)溶液を、アルゴン下0℃で水素化ナトリウム(鉱油(35mg)中60%分散物)のDMF(1ml)懸濁液に加えた。冷却槽を取り外し、混合物を室温で1h撹拌した。次いで、中間体12a)(122mg)のDMF(2ml)溶液を加え、反応混合物を60℃に1h加熱した。反応混合物を0℃に冷却し、次いで飽和NH4Cl(1ml)で加水分解した。EtOAc(70ml)を加え、混合物をNaHCO3(2×25ml)で洗浄した。合わせた水層をEtOAc(25ml)で再抽出し、次いで合わせた有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物を分取HPLCによって精製し、黄色がかった樹脂の形態の相当するジホルメートを得た。
【0162】
中間体12c):
【化94】
DCM(1ml)とMeOH(1ml)との混合物中のBoc保護された中間体12b)(151mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、2ml)を加え、溶液を室温で2h撹拌した。全ての揮発性物質の蒸発、並びにトルエン(2×5ml)、アセトン、及びエーテル(各々5ml)との同時蒸発によってベージュ色の固体がもたらされ、それをデシケーター中シカペント上で一晩さらに乾燥させた。
【0163】
実施例12:
【化95】
中間体12c)(68mg)及びB−C部分1(61mg)、1−ヒドロキシベンゾトリアゾール水和物(33mg)及びN−メチルモルホリン(59μl)を、DMF(3ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(49mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(12μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(70ml)で希釈し、飽和Na2CO3(3×25ml)、水及びブライン(各々25ml)で洗浄した。有機層を乾燥させ(Na2SO4)、溶媒を真空中で除去した。カラムクロマトグラフィーによる粗生成物の精製によって茶色がかった樹脂の形態の相当するアミンを得た。これをEtOAc(1ml)に溶解し、塩化水素のジエチルエーテル溶液(1.0M、178μl)で処理した。相当する二塩酸塩の完全な沈殿を得るために、このように得られた懸濁液をヘキサン(5ml)で希釈した。固体を濾過し、ヘキサンで洗浄し、デシケーター中シカペント上で一晩乾燥させ、オフホワイトの固体の形態の所望の化合物を得た。
【0164】
実施例14の合成:
中間体14a):
【化96】
2−ブロモ−3−ピリジノール(1.04g)のピリジン(2.00ml)及び乾燥DCM(20ml)溶液を、0℃に冷却した。無水酢酸(1.76ml)を加え、反応混合物を室温で一晩撹拌した。全ての揮発性物質を蒸発させ、残りを水(30ml)に懸濁させた。室温で1h撹拌した後、懸濁液を1NのNaHCO3(50ml)で希釈し、EtOAc(3×50ml)で抽出した。合わせた有機層を1NのNaHCO3、及びブライン(各々50ml)で洗浄し、乾燥させ(MgSO4)、濾過し、蒸発させ、琥珀色の油の形態の所望の化合物を得た。
【0165】
中間体14b):
【化97】
火炎乾燥シュレンクフラスコに、中間体14a)(2.00g)、[1,1’−ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)のジクロロメタンとの錯体(227mg)、ヨウ化銅(I)(106mg)、及び乾燥N,N−ジメチルアセトアミド(10ml)を入れた。このように得られた混合物を交互の真空/アルゴンパージで脱気した。次いで、1−tert−ブトキシカルボニルピペリジン−4−イル)(ヨード)亜鉛(13.0mmol、上記のように調製)を加えた。混合物をもう一度脱気し、次いで80℃に一晩加熱した。次いで、N,N−ジメチルアセトアミドの主要部分を蒸発させ、残りをEtOAc及び水の混合物(各々200ml)に溶解した。次いで、混合物をセライトで濾過し、分液漏斗に移した。相を分離し、水層をEtOAc(3×75ml)で抽出した。合わせた有機層を水及びブライン(各々150ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をカラムクロマトグラフィーによって精製し、茶色がかった固体の形態で所望の化合物を得た。
【0166】
中間体14c):
【化98】
中間体14b)(1.97g)のMeOH(60ml)溶液を、0℃に冷却した。水酸化リチウム(368mg)の水(30ml)溶液を加え、反応混合物を0℃で10分間撹拌した。0.5MのHClを0℃にて一滴ずつ添加することによってpHを約7に調節した。水(100ml)を加え、EtOAc(3×100ml)による抽出を行った。ブライン(150ml)による洗浄、乾燥(Na2SO4)、濾過及び蒸発の後、所望の化合物を茶色がかった発泡体として得た。
【0167】
中間体14d):
【化99】
中間体14c)(207mg)、1−(2−クロロエチル)ピロリジン塩酸塩(190mg)、及び炭酸セシウム(969mg)のDMF(10ml)溶液を、室温で一晩撹拌した。全ての揮発性物質を蒸発させ、残渣をEtOAc及び飽和NaHCO3(各々50ml)に分配した。水層を、EtOAc(2×25ml)で抽出した。合わせた有機層を水及びブライン(各々25ml)で洗浄し、乾燥させ(Na2SO4)、蒸発させ、所望の化合物を茶色がかった樹脂として得た。
【0168】
中間体14e):
【化100】
DCM(2ml)とMeOH(0.5ml)との混合物中のBoc保護された中間体14d)(253mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、2ml)を加え、溶液を室温で15分撹拌した。全ての揮発性物質の蒸発、並びにトルエン、アセトン、及びエーテル(各々5ml)との同時蒸発によってベージュ色の固体がもたらされ、それをデシケーター中シカペント上で一晩さらに乾燥させた。
【0169】
実施例14:
【化101】
中間体14e)(180mg)及びB−C部分1(201mg)、1−ヒドロキシベンゾトリアゾール水和物(107mg)及びN−メチルモルホリン(195μl)を、DMF(5ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(161mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(41μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(100ml)で希釈し、飽和Na2CO3(3×50ml)、水及びブライン(各々50ml)で洗浄した。有機層を乾燥させ(Na2SO4)、溶媒を真空中で除去した。カラムクロマトグラフィーによる粗生成物の精製によって茶色がかった樹脂の形態の相当するアミンを得た。これをEtOAc(1ml)に溶解し、塩化水素のジエチルエーテル溶液(1.0M、564μl)で処理した。相当する二塩酸塩の完全な沈殿を得るために、このように得られた懸濁液をヘキサン(5ml)で希釈した。固体を濾過し、ヘキサンで洗浄し、デシケーター中シカペント上で一晩乾燥させ、白色の固体の形態の所望の化合物を得た。
【0170】
実施例21の合成:
中間体21a):
【化102】
(S)−1−メチル−ピペリジン−3−オール(228mg)のDMF(2ml)溶液を、アルゴン下で0℃にて水素化ナトリウム(鉱油(53mg)中60%分散物)のDMF(1ml)懸濁液に加えた。冷却槽を取り外し、混合物を室温で1h撹拌した。次いで、3−フルオロ−3’,4’,5’,6’−テトラヒドロ−2’H−[2,4’]ビピリジニル−1’−カルボン酸tert−ブチルエステル(185mg)のDMF(2ml)溶液を加え、反応混合物を120℃に一晩加熱した。反応混合物を0℃に冷却し、次いで飽和NH4Cl(1ml)で加水分解した。EtOAc(50ml)を加え、混合物をNaHCO3(2×25ml)で洗浄した。合わせた水層をEtOAc(25ml)で再抽出し、次いで合わせた有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物を分取HPLCによって精製し、黄色がかった樹脂の形態の相当するジホルメートを得た。
【0171】
中間体21b):
【化103】
DCM(1ml)とMeOH(1ml)との混合物中のBoc保護された中間体21a)(148mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、2ml)を加え、溶液を室温で15分撹拌した。全ての揮発性物質の蒸発、並びにトルエン(2×5ml)、及びアセトン(5ml)との同時蒸発によってベージュ色の固体がもたらされ、それをデシケーター中シカペント上で一晩さらに乾燥させた。
【0172】
実施例21:
【化104】
中間体21b)(93mg)及びB−C部分1(83mg)、1−ヒドロキシベンゾトリアゾール水和物(44mg)及びN−メチルモルホリン(81μl)を、DMF(5ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(67mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(17μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(70ml)で希釈し、飽和Na2CO3(3×25ml)、水及びブライン(各々25ml)で洗浄した。有機層を乾燥させ(Na2SO4)、溶媒を真空中で除去した。カラムクロマトグラフィーによる粗生成物の精製によって黄色がかった樹脂の形態の相当するアミンを得た。これをEtOAc(1ml)に溶解し、塩化水素のジエチルエーテル溶液(1.0M、238μl)で処理した。相当する二塩酸塩の完全な沈殿を得るために、このように得られた懸濁液をヘキサン(5ml)で希釈した。固体を濾過し、ヘキサンで洗浄し、デシケーター中シカペント上で一晩乾燥させ、白色の固体の形態の所望の化合物を得た。
【0173】
実施例28の合成:
中間体28a):
【化105】
トリアセトキシ水素化ホウ素ナトリウム(1512mg)を、2−ブロモ−3−ピリジンカルボキシアルデヒド(948mg)及び1−メチルピペラジン(566μl)の1,2−ジクロロエタン(20ml)溶液に加えた。室温で1h撹拌した後、混合物をEtOAc(100ml)で希釈し、飽和NaHCO3(2×50ml)、H2O及びブライン(各々50ml)で洗浄した。Na2SO4上での乾燥、濾過及び溶媒の蒸発の後、茶色がかった油を得た。クロマトグラフィーによる精製によって、黄色がかった油の形態の所望の化合物を得た。
【0174】
中間体28b):
【化106】
火炎乾燥シュレンクフラスコに、中間体28a)(729mg)、[1,1’−ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)のジクロロメタンとの錯体(66mg)、ヨウ化銅(I)(31mg)、及び乾燥N,N−ジメチルアセトアミド(4ml)を入れた。このように得られた混合物を交互の真空/アルゴンパージで脱気した。次いで、1−tert−ブトキシカルボニルピペリジン−4−イル)(ヨード)亜鉛(3.78mmol、上記のように調製)を加えた。混合物をもう一度脱気し、次いで80℃に一晩加熱した。次いで、N,N−ジメチルアセトアミドの主要部分を蒸発させ、残りをEtOAc及び水の混合物(各々50ml)に溶解した。次いで、これをセライトで濾過し、分液漏斗に移した。相を分離し、水層をEtOAc(2×50ml)で抽出した。合わせた有機層をブライン(75ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をクロマトグラフィーによって精製し、茶色の油の形態で所望の化合物を得た。
【0175】
中間体28c):
【化107】
DCM(3ml)とMeOH(3ml)との混合物中のBoc保護された中間体28b)(383mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、6ml)を加え、溶液を室温で1h撹拌した。全ての揮発性物質の蒸発、並びにトルエン及びアセトン(各々20ml)との同時蒸発によって淡褐色の固体がもたらされ、それをデシケーター(シカペント)中で一晩さらに乾燥させた。
【0176】
実施例28:
【化108】
中間体28c)(165mg)及びB−C部分1(152mg)、1−ヒドロキシ−ベンゾトリアゾール水和物(80mg)及びN−メチルモルホリン(147μl)を、DMF(2ml)に溶解した。室温で30分撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(121mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(31μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(70ml)で希釈し、飽和Na2CO3(3×25ml)、水及びブライン(各々25ml)で洗浄した。有機層を乾燥させ(Na2SO4)、溶媒を真空中で除去した。分取HPLCによる粗生成物の精製によって相当するジホルメートを僅かに茶色がかった樹脂として得た。
【0177】
実施例41の合成:
中間体41a):
【化109】
1−ベンズヒドリルアゼタン−3−オール(674mg)のDMF(2ml)溶液を、アルゴン下0℃にて水素化ナトリウム(鉱油(75mg)中60%分散物)のDMF(2ml)懸濁液に加えた。冷却槽を取り外し、混合物を室温で1h撹拌した。次いで、3−フルオロ−3’,4’,5’,6’−テトラヒドロ−2’H−[2,4’]ビピリジニル−1’−カルボン酸tert−ブチルエステル(263mg)のDMF(2ml)溶液を加え、反応混合物を120℃に一晩加熱した。反応混合物を0℃に冷却し、次いで飽和NH4Cl(1ml)で加水分解した。EtOAc(50ml)を加え、混合物をNaHCO3(2×25ml)で洗浄した。合わせた水層をEtOAc(25ml)で再抽出し、次いで合わせた有機層を水及びブライン(各々25ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をクロマトグラフィーによって精製し、所望の生成物を黄色がかった発泡体として得た。
【0178】
中間体41b):
【化110】
MeOH(2ml)とAcOH(0.2ml)との混合物中の炭素担持20重量%水酸化パラジウム(10mg)の存在下で、1バールの水素下で室温にて、一晩中間体41a)(122mg)を水素化した。シリンジフィルターによる濾過及び溶媒の完全な蒸発によって、所望の化合物が無色の油としてもたらされた。
【0179】
中間体41c):
【化111】
トリアセトキシ水素化ホウ素ナトリウム(72mg)を、中間体41b)(137mg)及びホルムアルデヒド(水中36.5%、18.5μl)の1,2−ジクロロエタン(2ml)溶液に加えた。反応混合物を2h室温で撹拌した。次いで、混合物をEtOAc(50ml)で希釈し、飽和NaHCO3(2×25ml)及びH2O(25ml)で洗浄した。水層を混合し、EtOAc(25ml)で再抽出した。全ての有機層を合わせ、ブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、蒸発させた。粗生成物をクロマトグラフィーによって精製し、所望の生成物を無色の油として得た。
【0180】
中間体41d):
【化112】
DCM(1ml)とMeOH(1ml)との混合物中のBoc保護された中間体41c)(50mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、2ml)を加え、溶液を室温で1h撹拌した。全ての揮発性物質の蒸発、並びにトルエン及びアセトン(各々5ml)との同時蒸発によって白色の固体がもたらされ、それをデシケーター(シカペント)中で一晩さらに乾燥させた。
【0181】
実施例41:
【化113】
中間体41d)(51mg)及びB−C部分1(62mg)、1−ヒドロキシ−ベンゾトリアゾール水和物(33mg)及びN−メチルモルホリン(60μl)を、DMF(2ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(50mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(13μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(70ml)で希釈し、飽和Na2CO3(3×25ml)、水及びブライン(各々25ml)で洗浄した。有機層を乾燥させ(Na2SO4)、溶媒を真空中で除去した。分取HPLCによる粗生成物の精製によって相当するジホルメートを無色のガラス状固体として得た。
【0182】
実施例57の合成:
中間体57a):
【化114】
cis−(1S,2R)−2−アミノシクロペンタノール塩酸塩(6.55g)、ホルムアルデヒド水溶液(36.5%、12.2ml)及び水(30ml)中のギ酸(7.18ml)の混合物を、一晩加熱還流した。反応混合物を真空中で濃縮し、1NのNaOHを加えることによって残りをアルカリ性(pH14)にした。さらに、固体NaClを飽和するまで加えた。水相をDCM(3×50ml)で抽出し、合わせた有機層を水及びブライン(各々30ml)で洗浄し、次いで乾燥させ(Na2SO4)、真空中で濃縮し、黄色がかった液体を得た。
【0183】
中間体57b):
【化115】
中間体57a)(1.27g)を無水EtOH(50ml)に溶解し、水素化ホウ素ナトリウム(1.13g)を室温で少量ずつ加えた。一晩撹拌した後、飽和NH4Cl(10ml)を加え、EtOHの主要部分を真空中で除去した。残渣を0.25NのNaOH(50ml)で希釈し、固体NaClを飽和するまで加えた。水相をDCM(3×25ml)で抽出し、合わせた有機層を水及びブライン(各々25ml)で洗浄し、次いで乾燥させ(Na2SO4)、真空中で濃縮した。粗生成物をクロマトグラフィーによって精製し、無色の結晶の形態の所望の生成物を得た。
【0184】
中間体57c):
【化116】
中間体57b)(333mg)のDMF(1ml)溶液を、アルゴン下で0℃にて水素化ナトリウム(鉱油(69mg)中60%分散物)のDMF(3ml)懸濁液に加えた。冷却槽を取り外し、混合物を室温で1h撹拌した。次いで、3−フルオロ−3’,4’,5’,6’−テトラヒドロ−2’H−[2,4’]ビピリジニル−1’−カルボン酸tert−ブチルエステル(241mg)のDMF(1ml)溶液を加え、反応混合物を120℃に一晩加熱した。反応混合物を0℃に冷却し、次いで飽和NH4Cl(1ml)でクエンチした。EtOAc(70ml)を加え、混合物をNaHCO3(2×30ml)で洗浄した。合わせた水層をEtOAc(30ml)で再抽出し、次いで合わせた有機層をブライン(50ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をクロマトグラフィーによって精製し、所望の生成物を茶色がかった油として得た。
【0185】
中間体57d):
【化117】
DCM(1ml)とMeOH(1ml)との混合物中のBoc保護された中間体57c)(270mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、2ml)を加え、溶液を室温で10分間撹拌した。全ての揮発性物質の蒸発、並びにトルエン及びアセトン(各々5ml)との同時蒸発によってベージュ色の固体がもたらされ、それをデシケーター(シカペント)中で一晩さらに乾燥させた。
【0186】
実施例57:
【化118】
中間体57d)(110mg)及びB−C部分1(100mg)、1−ヒドロキシ−ベンゾトリアゾール水和物(53mg)及びN−メチルモルホリン(97μl)を、DMF(2ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(80mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(20μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(70ml)で希釈し、飽和Na2CO3(3×25ml)、水及びブライン(各々25ml)で洗浄した。有機層を乾燥させ(Na2SO4)、溶媒を真空中で除去した。分取HPLCによる粗生成物の精製によって相当するジホルメートを無色のガラス状固体として得た。
【0187】
実施例59の合成:
中間体59a):
【化119】
火炎乾燥シュレンクフラスコに、2−クロロ−5−フルオロ−3−ホルミルピリジン(1596mg)、[1,1’−ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)のジクロロメタンとの錯体(245mg)、ヨウ化銅(I)(114mg)、及び乾燥N,N−ジメチルアセトアミド(14ml)を入れた。このように得られた混合物を交互の真空/アルゴンパージで脱気した。次いで、1−tert−ブトキシカルボニルピペリジン−4−イル)(ヨード)亜鉛(14.0mmol、上記のように調製)を加えた。混合物をもう一度脱気し、次いで80℃に一晩加熱した。次いで、N,N−ジメチルアセトアミドの主要部分を蒸発させ、残りをEtOAc及び水の混合物(各々75ml)に溶解した。次いで、これをセライトで濾過し、分液漏斗に移した。相を分離し、水層をEtOAc(2×100ml)で抽出した。合わせた有機層をブライン(100ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をカラムクロマトグラフィーによって精製し、黄色い樹脂の形態で所望の化合物を得た。
【0188】
中間体59b):
【化120】
トリアセトキシ水素化ホウ素ナトリウム(585mg)を、中間体59a)(607mg)及び1−メチルピペラジン(219μl)の1,2−ジクロロエタン(10ml)溶液に加えた。室温で3h撹拌した後、さらなるトリアセトキシ水素化ホウ素ナトリウム(209mg)を加え、撹拌を一晩続けた。混合物をEtOAc(100ml)で希釈し、飽和NaHCO3(2×50ml)、H2O(50ml)及びブライン(75ml)で洗浄した。Na2SO4上での乾燥、濾過及び溶媒の蒸発の後、茶色がかった樹脂を得た。クロマトグラフィーによる精製によって、茶色がかった油の形態の所望の化合物を得た。
【0189】
中間体59c):
【化121】
DCM(1.5ml)とMeOH(3.5ml)との混合物中のBoc保護された中間体59b)(504mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、3ml)を加え、溶液を室温で1.5h撹拌した。全ての揮発性物質の蒸発、並びにトルエン(2×10ml)及びアセトン(15ml)との同時蒸発によって茶色の固体がもたらされ、それをデシケーター(シカペント)中で一晩さらに乾燥させた。
【0190】
実施例59:
【化122】
中間体59c)(150mg)及びB−C部分1(129mg)、1−ヒドロキシ−ベンゾトリアゾール水和物(69mg)及びN−メチルモルホリン(125μl)を、DMF(5ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(103mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(26μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(50ml)で希釈し、飽和Na2CO3(3×25ml)で洗浄した。合わせた水層をEtOAc(25ml)で再抽出した。次いで、全ての有機層を混合し、ブライン(25ml)で洗浄した。有機層を乾燥させ(Na2SO4)、溶媒を真空中で除去した。分取HPLCによる粗生成物の精製によって相当するホルメートを黄色がかった樹脂として得た。
【0191】
実施例60及び61の合成:
中間体60及び61a):
【化123】
2−クロロ−5−フルオロピリジン−3−オール(2.50g)及びピリジン(5.61ml)のDCM(60ml)溶液を、0℃に冷却した。これに、無水酢酸(4.96ml)を一滴ずつ加え、反応混合物を室温で一晩撹拌した。全ての揮発性物質を蒸発させ、残渣を水(30ml)で希釈し、1h撹拌し、未反応の無水酢酸を加水分解した。この混合物をEtOAc(100ml)及び0.5MのNaHCO3(150ml)で希釈した。相分離後、水層を、EtOAc(2×100ml)で抽出した。合わせた有機層を0.5MのNaHCO3及びブライン(各々100ml)で再抽出し、Na2SO4上で乾燥させ、蒸発させた。クロマトグラフィーによる精製によって、無色の液体の形態の所望の化合物を得た。
【0192】
中間体60及び61b):
【化124】
火炎乾燥シュレンクフラスコに、中間体60及び61a)(2.38g)、[1,1’−ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)のジクロロメタンとの錯体(307mg)、ヨウ化銅(I)(143mg)、及び乾燥N,N−ジメチルアセトアミド(15ml)を入れた。このように得られた混合物を交互の真空/アルゴンパージで脱気した。次いで、1−tert−ブトキシカルボニルピペリジン−4−イル)(ヨード)亜鉛(17.6mmol、上記のように調製)を加えた。混合物をもう一度脱気し、次いで80℃に一晩加熱した。次いで、N,N−ジメチルアセトアミドの主要部分を蒸発させ、残りをEtOAc及び水の混合物(各々75ml)に溶解した。次いで、これをセライトで濾過し、分液漏斗に移した。相を分離し、水層をEtOAc(2×50ml)で抽出した。合わせた有機層を水及びブライン(各々75ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をカラムクロマトグラフィーによって精製し、茶色がかった油の形態で所望の化合物を得た。
【0193】
中間体60及び61c):
【化125】
中間体60及び61b)(1.82g)のMeOH(50ml)溶液を、0℃に冷却した。水酸化リチウム(0.32g)の水(25ml)溶液を一度に加え、0℃で撹拌を続けた。15分後、0.5MのHClを一定ずつ加えることによってpHを約7に調節した。次いで、混合物を水(100ml)で希釈し、EtOAc(3×100ml)で抽出した。合わせた有機層をブライン(100ml)で洗浄し、乾燥させ(Na2SO4)、蒸発させ、茶色がかった発泡体を得た。
【0194】
中間体60及び61d):
【化126】
アルゴン下で、中間体60及び61c)(375mg)、(R)−2−ジメチルアミノ−ブタン−1−オール(297mg)、及びトリフェニルホスフィン(664mg)のTHF(15ml)溶液を、0℃に冷却した。このアゾジカルボン酸ジエチル、40%トルエン溶液(1.16ml)を一滴ずつ加えた。冷却槽を取り外し、撹拌を室温で3h続けた。全ての揮発性物質の蒸発によって茶色の油を得て、これをクロマトグラフィーによって最初に精製し(Ph3POの除去)、次いで分取HPLCによって、異性体を分離した。両方の異性体を茶色がかった樹脂として得た。
【0195】
中間体60e):
【化127】
DCM(1ml)とMeOH(1ml)との混合物中のBoc保護された中間体60及び61d)P1(82mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、2ml)を加え、溶液を室温で10分間撹拌した。全ての揮発性物質の蒸発、並びにトルエン及びアセトン(各々5ml)との同時蒸発によって所望の生成物が白色固体としてもたらされた。
【0196】
実施例60:
【化128】
中間体60e)(95mg)及びB−C部分1(89mg)、1−ヒドロキシ−ベンゾトリアゾール水和物(47mg)及びN−メチルモルホリン(86μl)を、DMF(3ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(71mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(18μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(50ml)で希釈し、飽和Na2CO3(3×25ml)及び水(25ml)で洗浄した。全ての水層を混合し、EtOAc(25ml)で再抽出した。次いで、合わせた有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、真空中で蒸発させた。残渣を分取HPLCによって精製し、相当するアミンを黄色がかった樹脂として得た。これをEtOAc(1ml)に溶解し、塩化水素のジエチルエーテル溶液(1.0M、207μl)で処理した。相当する二塩酸塩の完全な沈殿を得るために、このように得られた懸濁液をヘキサン(5ml)で希釈した。固体を濾過し、ヘキサンで洗浄し、デシケーター(シカペント)中で一晩乾燥させ、白色の固体の形態の所望の化合物を得た。
【0197】
中間体61e):
【化129】
DCM(1ml)とMeOH(1ml)との混合物中のBoc保護された中間体60及び61d)P2(40mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、2ml)を加え、溶液を室温で10分間撹拌した。全ての揮発性物質の蒸発、並びにトルエン及びアセトン(各々5ml)との同時蒸発によって所望の生成物が白色固体としてもたらされた。
【0198】
実施例61:
【化130】
中間体61e)(48mg)及びB−C部分1(43mg)、1−ヒドロキシ−ベンゾトリアゾール水和物(23mg)及びN−メチルモルホリン(42μl)を、DMF(3ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(35mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(9μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(50ml)で希釈し、飽和Na2CO3(3×25ml)及び水(25ml)で洗浄した。全ての水層を混合し、EtOAc(25ml)で再抽出した。次いで、合わせた有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、真空中で蒸発させた。粗生成物を分取HPLCによって精製し、相当するホルメートを無色の樹脂として得た。
【0199】
実施例63の合成:
中間体63a):
【化131】
n−ブチルリチウム(2.5M)のヘキサン(8.97ml)溶液を、アルゴン雰囲気下で冷却し(−50℃)撹拌した乾燥THF(200ml)に加えた。次いで、2,2,6,6−テトラメチルピペリジン(4.13ml)を加えた。混合物を0℃に20分間温めた。次いで、混合物を−78℃とした。この混合物に、フルオロピラジン(2.00g)のTHF(50ml)溶液を加えた。撹拌をこの温度で5分間続けた。次いで、ヨウ素(10.4g)のTHF(50ml)溶液を導入し、撹拌を−78℃で1h続けた。次いで、35%塩酸水溶液(20ml)、EtOH(20ml)及びTHF(50ml)の溶液を使用して加水分解を−78℃で行った。溶液を室温に温め、飽和NaHCO3で僅かに塩基性(pH10)とした。溶液をチオ硫酸ナトリウムで脱色し、蒸発させ、有機溶媒を除去した。残渣を水(150ml)で希釈し、DCM(3×200ml)で抽出した。有機抽出物を乾燥させ(Na2SO4)、蒸発させた。粗生成物をカラムクロマトグラフィーによって精製し、黄色がかった結晶として所望の化合物を得た(mp.41〜44℃)。
【0200】
中間体63b):
【化132】
火炎乾燥シュレンクフラスコに、中間体63a)(1.00g)、[1,1’−ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)のジクロロメタンとの錯体(109mg)、ヨウ化銅(I)(51mg)、及び乾燥N,N−ジメチルアセトアミド(6ml)を入れた。このように得られた混合物を、交互の真空/アルゴンパージで脱気した。次いで、1−tert−ブトキシカルボニルピペリジン−4−イル)(ヨード)亜鉛(6.25mmol、上記のように調製)を加えた。混合物をもう一度脱気し、次いで80℃に一晩加熱した。次いで、N,N−ジメチルアセトアミドの主要部分を蒸発させ、残りをEtOAc及び水の混合物(各々100ml)に溶解した。次いで、混合物をセライトで濾過し、分液漏斗に移した。相を分離し、水層をEtOAc(3×30ml)で抽出した。合わせた有機層を水及びブライン(各々75ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をカラムクロマトグラフィーによって精製し、茶色がかった油の形態で所望の化合物を得た。
【0201】
中間体63c):
【化133】
(S)−1−ピロリジン−1−イル−ブタン−2−オール(211mg)のDMF(2ml)溶液を、アルゴン下で0℃にて水素化ナトリウム(鉱油(39mg)中60%分散物)のDMF(1ml)懸濁液に加えた。冷却槽を取り外し、混合物を室温で1h撹拌した。次いで、中間体63b)(138mg)のDMF(2ml)溶液を加え、反応混合物を60℃に1h加熱した。反応混合物を0℃に冷却し、次いで飽和NH4Cl(1ml)で加水分解した。EtOAc(70ml)を加え、混合物をNaHCO3(2×25ml)で洗浄した。合わせた水層をEtOAc(25ml)で再抽出し、次いで合わせた有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物を分取HPLCによって精製し、黄色がかった樹脂の形態の相当するホルメートを得た。
【0202】
中間体63d):
【化134】
DCM(1ml)とMeOH(1ml)との混合物中のBoc保護された中間体63c)(181mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、2ml)を加え、溶液を室温で2h撹拌した。全ての揮発性物質の蒸発、並びにトルエン(2×5ml)、アセトン(5ml)、及びエーテル(5ml)との同時蒸発によってベージュ色の固体がもたらされ、それをデシケーター中シカペント上で一晩さらに乾燥させた。
【0203】
実施例63:
【化135】
中間体63d)(67mg)及びB−C部分1)(68mg)、1−ヒドロキシベンゾトリアゾール水和物(36mg)及びN−メチルモルホリン(66μl)を、DMF(3ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(54mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(14μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(70ml)で希釈し、飽和Na2CO3(3×25ml)、水及びブライン(各々25ml)で洗浄した。有機層を乾燥させ(Na2SO4)、溶媒を真空中で除去した。カラムクロマトグラフィーによる粗生成物の精製によって茶色がかった樹脂の形態の相当するアミンを得た。これをEtOAc(1ml)に溶解し、塩化水素のジエチルエーテル溶液(1.0M、178μl)で処理した。相当する塩酸塩の完全な沈殿を得るために、このように得られた懸濁液をヘキサン(5ml)で希釈した。固体を濾過し、ヘキサンで洗浄し、デシケーター中シカペント上で一晩乾燥させ、オフホワイトの固体の形態の所望の化合物を得た。
【0204】
実施例64の合成:
中間体64a):
【化136】
内部温度計を備えた三つ口フラスコに乾燥THF(80ml)を入れ、アルゴン雰囲気下で−10℃に冷却した。磁気撹拌子で撹拌する間、四塩化チタンの1M溶液(40ml)を、温度を−10℃〜−5℃に維持するために30分以内で一滴ずつ加えた。さらに30分間−5℃にて撹拌後、マロン酸ジメチル(2.286ml)及びN−Boc−ピペリジン−4−オン(4.384g)を−15℃〜−10℃で加えた。反応混合物は茶色の懸濁液となった。温度を−10℃未満に保つ一方、無水ピリジン(6.632ml)のTHF(14ml)溶液を30分以内で加えた。反応物は透明な茶色の溶液となった。冷却槽を取り外し、撹拌を室温で46h続けた。THFを蒸発させ、残りの油を1NのNaHCO3中で懸濁させた。沈殿しているチタン塩を濾過し、酢酸エチルで洗浄した。濾液を、第2の部分の1NのNaHCO3及びブラインで洗浄した。有機相をMgSO4上で乾燥させ、濾過し、蒸発させ、オレンジ色の油を得た。それをシリカゲルカラムクロマトグラフィーによって精製した。
【0205】
中間体64b):
【化137】
250mlの二つ口フラスコ中で、中間体64a)(3.16g)をメタノール(70ml)と水(0.5ml)との混合物に溶解した。フラスコを空にし、アルゴンでフラッシュした。活性炭担持5%パラジウム(400mg)を加え、フラスコを空にし、水素でフラッシュした。反応混合物を、水素雰囲気下にて(1atm)室温で一晩激しく撹拌した。触媒をセライトで濾過し、フィルターをメタノール(150ml)で洗浄した。濾液を真空中で蒸発させた。粗生成物をDCM(15ml)に溶解し、0.45μmのシリンジフィルターで濾過し、真空中で蒸発させた。透明な無色の油を得て、それを高真空下にて一晩乾燥させた。
【0206】
中間体64c):
【化138】
ナトリウムメトキシドは、ナトリウム(306mg)を冷却したメタノール(6.0ml)に溶解することによって調製した。塩酸アセトアミジン(415mg)をこの混合物に加え、沈殿している塩化ナトリウムを25mmのシリンジフィルターによる濾過によって除去した。中間体64b)(1.40g)をすばやく撹拌されている溶液に加えた。形成された僅かに濁った溶液を2日間室温に保った。溶媒を真空中で蒸発させた。残渣を水(10ml)に溶解し、水相をトルエン(6ml)で抽出した。有機相を廃棄した。水層を0℃に冷却し、pH4に達するまで6NのHClを一滴ずつ加えた。沈殿した無色の固体を吸引濾過によって集め、冷水(4ml)で洗浄した。生成物を真空中で乾燥させた。
【0207】
中間体64d):
【化139】
Boc保護中間体64c)を、ジオキサン(6ml)とメタノール(1ml)との混合物に懸濁させた。ジオキサン(6ml)中の4NのHClを加えた。反応物を室温で1h撹拌した。トルエン(2×20ml)との同時蒸発を含めた全ての揮発性物質の蒸発によって白色の固体を得て、それを高真空中で一晩乾燥させた。粗生成物を乾燥ジエチルエーテル(6ml)で粉砕した。溶媒をデカントし、生成物を真空中で乾燥させた。
【0208】
中間体64e):
【化140】
Z−OSu(546mg)のDMF(10ml)溶液を、トリエチルアミン(0.55ml)で処理した。中間体64d)のDMF(3ml)懸濁液を加え、反応物を室温で15時間撹拌した。溶媒を真空中で蒸発させ、残りのオフホワイトの固体をジエチルエーテル(2×)で粉砕した。生成物を濾過によって単離し、真空中で乾燥させた。
【0209】
中間体64f):
【化141】
中間体64e)をアセトニトリル(4ml)に懸濁させ、sym−コリジン(0.231ml)で処理した。POCl3(0.54ml)のアセトニトリル(2ml)溶液を0℃で一滴ずつ加え、反応物を還流温度で8h撹拌した。溶媒及び過剰な試薬を蒸留によって真空中で除去した。残りの茶色の固体を氷冷の酢酸エチルに溶解し、水、1NのNaHCO3及びブラインで抽出した。有機相をMgSO4上で乾燥させ、濾過し、真空中で蒸発させた。
【0210】
中間体64g):
【化142】
火炎乾燥フラスコ中で、水素化ナトリウム(鉱油中60%、18.9mg)を、アルゴン雰囲気下でTHF(5ml)に懸濁させた。(R)−2−ジメチルアミノ−ブタン−1−オール(110mg)を一滴ずつ加え、混合物を室温で1h撹拌し、相当するナトリウムアルコキシドが形成された。ジクロロピリミジン中間体64f)を、アルゴン雰囲気下にてTHF(3ml)に溶解した。溶液を0℃に冷却し、上記の調製したナトリウムアルコラートを30分以内で一滴ずつ加えた。混合物を室温に温め、室温で5h撹拌した。さらなる0.15当量の上記のアルコキシドを0℃で加え、室温で3h撹拌を続けた。反応混合物を真空中で蒸発させ、琥珀色の粘性油を得て、それを酢酸エチルに溶解した。溶液を1NのNaHCO3及びブラインで洗浄し、MgSO4上で乾燥させ、濾過し、蒸発させた。粗生成物をクロマトグラフィーによって精製した。
【0211】
中間体64h):
【化143】
中間体64g)(115mg)をメタノール(5ml)に溶解した。酸化カルシウム(84mg)、続いて10%パラジウム炭素(100mg)を加えた。混合物を真空中で繰り返し脱気し、アルゴン、最後に水素でフラッシュした。反応物を水素バルーン下で一晩撹拌した。反応物をセライトで濾過し、フィルターを80mlのメタノールで洗浄した。溶媒を蒸発させ、白色混濁状の残渣を酢酸エチルで希釈し、1NのNa2CO3及びブラインで抽出した。有機相をMgSO4上で乾燥させ、濾過し、蒸発させ、所望の生成物を無色の油として得た。
【0212】
実施例64:
【化144】
DMF(2.6ml)中のB−C部分1(84.5mg)、HOBt(42.8mg)、EDCI(76mg)及びN−メチルモルホリン(56.4μl)を、室温で1時間撹拌した。中間体64h)(68.0mg)を鮮黄色の溶液に加え、撹拌を一晩続けた。DMFを真空中で蒸発させ、残りの黄色の油を酢酸エチル(100ml)に溶解した。有機溶液を、1NのNa2CO3(2×30ml)及びブラインで抽出した。各水相を酢酸エチルで一度再抽出した。合わせた有機層を乾燥させ(MgSO4)、濾過し、真空中で蒸発させた。生成物を分取HPLCによって精製した。
【0213】
実施例73の合成:
中間体73a):
【化145】
5−クロロ−3−フルオロ−ピリジン−2−オール(1.04g)をピリジン(13ml)に溶解し、アルゴン下0℃に冷却した。次いで、トリフルオロメタンスルホン酸無水物(1.36ml)を、シリンジを介して一滴ずつ加えた。室温で一晩撹拌した後、反応混合物を真空中で濃縮した。残りを水及びジエチルエーテル(各々100ml)に分配した。水層を、ジエチルエーテル(2×50ml)で抽出し、合わせたエーテル抽出物を水及びブライン(各々50ml)で洗浄した。Na2SO4上での乾燥及び溶媒の蒸発によって、茶色の油を得た。カラムクロマトグラフィーによる精製によって、黄色がかった液体の形態の所望の化合物がもたらされた。
【0214】
中間体73b):
【化146】
火炎乾燥シュレンクフラスコに、中間体73a)(956mg)、[1,1’−ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)のジクロロメタンとの錯体(84mg)、ヨウ化銅(I)(39mg)、及び乾燥N,N−ジメチルアセトアミド(5ml)を入れた。このように得られた混合物を交互の真空/アルゴンパージで脱気した。次いで、1−tert−ブトキシカルボニルピペリジン−4−イル)(ヨード)亜鉛(4.89mmol、上記のように調製)を加えた。混合物をもう一度脱気し、次いで80℃に一晩加熱した。次いで、N,N−ジメチルアセトアミドの主要部分を蒸発させ、残りをEtOAc及び水の混合物(各々75ml)に溶解した。次いで、これをセライトで濾過し、分液漏斗に移した。相を分離し、水層をEtOAc(2×50ml)で抽出した。合わせた有機層を水及びブライン(各々50ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をカラムクロマトグラフィーによって精製し、茶色がかった固体の形態で所望の化合物を得た。
【0215】
中間体73c):
【化147】
(R)−2−ジメチルアミノ−ブタン−1−オール(252mg)のDMF(1ml)溶液を、アルゴン下で0℃にて水素化ナトリウム(鉱油(64mg)中60%分散物)のDMF(2ml)懸濁液に加えた。冷却槽を取り外し、混合物を室温で1h撹拌した。次いで、中間体73b)(338mg)のDMF(2ml)溶液を加え、反応混合物を120℃に一晩加熱した。反応混合物を0℃に冷却し、次いで飽和NH4Cl(1ml)で加水分解した。EtOAc(50ml)を加え、混合物をNaHCO3(2×25ml)で洗浄した。合わせた水層をEtOAc(25ml)で再抽出し、次いで合わせた有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をクロマトグラフィーによって精製し、所望の生成物を黄色がかった油として得た。
【0216】
中間体73d):
【化148】
DCM(1ml)とMeOH(1ml)との混合物中のBoc保護された中間体73c)(380mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、2ml)を加え、溶液を室温で1h撹拌した。全ての揮発性物質の蒸発、並びにトルエン及びアセトン(各々5ml)との同時蒸発によって白色の固体がもたらされ、それをデシケーター(シカペント)中で一晩さらに乾燥させた。
【0217】
実施例73:
【化149】
中間体73d)(166mg)及び(R)−3−(2,4−ジメチル−フェニル)−2−[(ピロリジン−1−カルボニル)−アミノ]−プロピオン酸(130mg)、1−ヒドロキシ−ベンゾトリアゾール水和物(79mg)及びN−メチルモルホリン(143μl)を、DMF(5ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(118mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(30μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(50ml)で希釈し、飽和Na2CO3(3×25ml)及び水(25ml)で洗浄した。全ての水層を混合し、EtOAc(2×25ml)で再抽出した。次いで、合わせた有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、真空中で蒸発させた。残渣を分取HPLCによって精製し、相当するホルメートを黄色がかった樹脂として得た。
【0218】
実施例85の合成:
中間体85a):
【化150】
ワング樹脂(500mg、Wang resin co.、100〜200メッシュ(Nova、01−64−5027)、1.1mmol/g)を、DMF(5ml)中のDMAP(5mg)の存在下で、DIC(255μl)で活性化したFmoc−D−2,4−ジクロロフェニルアラニン(753mg)の溶液で処理した。混合物を一晩反応させた。過剰の試薬を濾過によって除去した。樹脂をDMF(3×4ml)で洗浄した。DMF(3ml)、続いて無水酢酸(260μl)を加えた。混合物を1h反応させた。過剰の試薬を濾過によって除去した。樹脂結合中間体を、DMF(3×4ml)、MeOH(3×4ml)、THF(3×4ml)、DCM(3×4ml)及びジエチルエーテル(3×4ml)で連続的に洗浄した。樹脂を減圧下で乾燥させた。
【0219】
中間体85b):
【化151】
中間体85a)(200mg)を、20%ピペリジンのDCM(4ml)溶液で処理した。混合物を30分間反応させた。過剰の試薬を濾過によって除去した。樹脂結合中間体を、DMF(3×2ml)、MeOH(3×2ml)、THF(3×2ml)、DCM(3×2ml)及びジエチルエーテル(3×2ml)で連続的に洗浄した。樹脂を減圧下で乾燥させた。
【0220】
中間体85c):
【化152】
中間体85b)(200mg)を、冷たい4−ニトロフェニルクロロホルメート(222mg)及びトリエチルアミン(216μl)のDCM(2.5ml)溶液で処理した。混合物を室温で2h反応させた。過剰の試薬を濾過によって除去した。樹脂結合中間体を、DCM(3×2ml)、THF(3×2ml)、DCM(3×2ml)及びジエチルエーテル(3×2ml)で連続的に洗浄した。樹脂を減圧下で乾燥させた。
【0221】
中間体85d):
【化153】
中間体85c)(200mg)を、ビス(2−メトキシエチル)−アミン(322μl)のDCM(2ml)溶液で処理した。混合物を室温で2h反応させた。過剰の試薬を濾過によって除去した。樹脂結合中間体を、DMF(3×3ml)、MeOH(3×3ml)、THF(3×3ml)、DCM(3×3ml)及びジエチルエーテル(3×3ml)で連続的に洗浄した。樹脂を減圧下で乾燥させた。DCM中の20%TFA(3ml)を樹脂に加え、混合物を30分間反応させた。生成物を濾過し、溶媒を除去した。
【0222】
実施例85:
【化154】
DMF(2ml)中の中間体85d)(95mg)に、中間体3−((R)−2−ピロリジン−1−イル−ブトキシ)−1’,2’,3’,4’,5’,6’−ヘキサヒドロ−[2,4’]ビピリジニル三塩酸塩(91mg)、N−メチルモルホリン(109μl)、及びHOBt(37mg)を加え、混合物を20分間撹拌した。EDCI(46mg)を加え、撹拌を一晩続けた。反応混合物をブライン(20ml)に注ぎ、酢酸エチルで希釈し、有機相を分離した。水相を酢酸エチルで2度抽出した。合わせた有機相を飽和炭酸水素ナトリウム溶液で2度、水及びブラインで2度洗浄し、Na2SO4上で乾燥させ、濃縮した。粗生成物を分取LC−MSで精製し、引き続いて80%tBuOH水溶液から凍結乾燥し、淡黄色の固体を得た。
【0223】
実施例88の合成:
中間体88a):
【化155】
中間体85c)(200mg)を、チアゾリジン(172μl)のDCM(2ml)溶液で処理した。混合物を室温で3h反応させた。DBU(164μl)を加え、混合物を室温で1h反応させた。過剰の試薬を濾過によって除去した。樹脂結合中間体を、DMF(3×3ml)、MeOH(3×3ml)、THF(3×3ml)、DCM(3×3ml)及びジエチルエーテル(3×3ml)で連続的に洗浄した。樹脂を減圧下で乾燥させた。DCM中の20%TFA(3ml)を樹脂に加え、混合物を30分間反応させた。生成物を濾過し、溶媒を除去した。
【0224】
実施例88:
【化156】
DMF(2ml)中の中間体88a)(60mg)に、3−((R)−2−ピロリジン−1−イル−ブトキシ)−1’,2’,3’,4’,5’,6’−ヘキサヒドロ−[2,4’]ビピリジニル三塩酸塩(66mg)、N−メチルモルホリン(79μl)、及びHOBt(28mg)を加え、混合物を20分間撹拌した。EDCI(35mg)を加え、撹拌を一晩続けた。反応混合物をブライン(20ml)に注ぎ、酢酸エチルで希釈し、有機相を分離した。水相を酢酸エチルで2度抽出した。合わせた有機相を飽和炭酸水素ナトリウム溶液で2度、水及びブラインで2度洗浄し、Na2SO4上で乾燥させ、濃縮した。粗生成物を分取LC−MSで精製し、引き続いて80%tBuOH水溶液から凍結乾燥し、白色の固体を得た。
【0225】
実施例99の合成:
中間体99a):
【化157】
火炎乾燥フラスコ中で、3−オキソ−アゼチジン−1−カルボン酸tert−ブチルエステル(500mg)をアルゴン下でTHF(6ml)に溶解し、溶液を0℃に冷却した。臭化メチルマグネシウム(ジエチルエーテル中3M溶液、1.95ml)を一滴ずつ加え、ミルク状懸濁液を3h撹拌した。反応物を飽和水性NH4Clで注意深く加水分解し、EtOAc(80ml)で抽出した。有機相をブラインで洗浄し、Na2SO4上で乾燥させ、濾過し、蒸発させ、生成物を無色の固体として得た。
【0226】
中間体99b):
【化158】
トリフルオロ酢酸(4ml)を、中間体99a)のDCM(40ml)溶液に室温で一滴ずつ加えた。混合物を90分間撹拌した。溶媒の半分を蒸発させ、次いでトルエンを加え、蒸発を続けた。全ての溶媒を蒸発させる前に、この同時蒸発手順を2度繰り返した。残りの生成物を高真空下にて一晩乾燥させた。
【0227】
中間体99c):
【化159】
氷冷の中間体99b)(4.451g)及びトリエチルアミン(2.34ml)のDMF(30ml)溶液に、(R)−3−(2,4−ジクロロ−フェニル)−2−イソシアナト−プロピオン酸メチルエステル(3.048g)の無水DMF(50ml)溶液をアルゴン下で加えた。混合物を0℃で4h撹拌させた。反応混合物を真空中で蒸発させた。残渣をEtOAc(300ml)中で希釈し、有機相を0.1NのHCl(2×90ml)、0.1NのNaHCO3及びブラインで洗浄した。水相をEtOAcで再抽出し、合わせた有機相をNa2SO4下で乾燥させ、濾過し、蒸発させた。粗化合物をカラムクロマトグラフィーで精製し、無色の安定した発泡体を得た。
【0228】
中間体99d):
【化160】
中間体99c)をMeOH(2ml)とTHF(6ml)との混合物に0℃で溶解した。水酸化リチウム一水和物(73mg)のH2O(2ml)溶液を加えた。混合物を0℃で3h撹拌した。1NのHClを加えることによって反応混合物を中和させ、MeOH及びTHFを真空中で蒸発させた。1NのHCl(pH約1〜2)で水相を酸性化した。水相をEtOAc(30ml)で抽出した。有機層を水及びブラインで洗浄した。水相をEtOAcで再抽出し、合わせた有機層をNa2SO4上で乾燥させ、濾過し、蒸発させた。粗生成物を高真空下にて乾燥させ白色の発泡体を得た。
【0229】
実施例99:
【化161】
DMF(50ml)中の中間体99d)(2.523g)、HOBt(1.113g)、EDCI(1.974g)及びNMM(1.47ml)を、1時間室温で撹拌した。3−((R)−2−ピロリジン−1−イル−ブトキシ)−1’,2’,3’,4’,5’,6’−ヘキサヒドロ−[2,4’]ビピリジニル三塩酸塩(2.050g)及び第2の部分のNMM(1.33ml)を加え、撹拌を一晩続けた。DMFを真空中で蒸発させ、残りのオレンジ色の油を酢酸エチル(300ml)に溶解した。有機溶液を1NのNa2CO3(2×150ml)及びブラインで洗浄した。各水相を酢酸エチルで一度再抽出した。有機層を乾燥させ(Na2SO4)、濾過し、真空中で蒸発させた。粗生成物を分取HPLCによって精製した。
【0230】
実施例125の合成:
中間体125a):
【化162】
酢酸水銀(1.625g)を水(6ml)に溶解した。3−メチレン−N−Boc−ピペリジン(0.986g)のTHF(6ml)溶液を室温にて一滴ずつ加えた。20分後、酢酸水銀を溶解し、当初黄色の溶液は透明及び無色になった。混合物をさらに40分室温で撹拌した。反応物を氷水中で冷却し、3Nの水酸化ナトリウム溶液(5ml)を加えた。茶色の色が現れた。次いで、水素化ホウ素ナトリウム(0.19g)のNaOH(3N)(5ml)溶液を加え、混合物を15分間撹拌した。pH6となるまで酸性酸を加え、水素発生が止まった。混合物を沈殿した水銀からデカントし、蒸発させた。残渣を水及び酢酸エチルに分配した。有機相を水及びブラインで洗浄し、MgSO4上で乾燥させ、濾過し、蒸発させた。淡黄色の油が残り、それをカラムクロマトグラフィーによって精製した。生成物を、透明で無色の粘性油の形態で得て、それを高真空中で乾燥させた。
【0231】
中間体125b):
【化163】
中間体125a)(846mg)をジオキサン(30ml)に溶解した。ジオキサン(30ml)中の4NのHClを加え、反応物を室温で3.5時間撹拌した。溶媒を蒸発させ、残渣をトルエンと数回同時蒸発させた。残りの淡黄色の固体を洗浄し、高真空下にて一晩乾燥させた。
【0232】
中間体125c):
【化164】
氷冷の(R)−3−(2,4−ジクロロ−フェニル)−2−イソシアナト−プロピオン酸メチルエステル(271mg)のDMF(2.5ml)溶液に、中間体125b)(300mg)及びEt3N(279μl)のDMF(3ml)溶液を加えた。混合物を0℃で4h撹拌させた。反応混合物を真空中で蒸発させた。残渣をEtOAc(50ml)で希釈し、有機相を0.1NのHCl(2×20ml)及びブラインで洗浄した。水相をEtOAcで再抽出し、合わせた有機相をNa2SO4上で乾燥させ、濾過し、蒸発させた。
【0233】
中間体125d):
【化165】
中間体125c)(404mg)を、MeOH(1.5ml)及びTHF(5.0ml)に0℃で溶解した。水酸化リチウム一水和物(83mg)のH2O(1.5ml)溶液を加えた。混合物を0℃で1.5h撹拌した。1NのHClを加えることによって反応混合物を中和させ、MeOH及びTHFを真空中で蒸発させた。1NのHCl(pH約1〜2)で水相を酸性化した。水相を酢酸エチル(50ml)で抽出した。有機層を水及びブラインで洗浄した。水相をEtOAcで再抽出し、合わせた有機層をNa2SO4上で乾燥させ、真空中で蒸発させ、無色の発泡体を得た。
【0234】
実施例125:
【化166】
DMF(3ml)中の中間体125d)(190mg)、HOBt(64.7mg)、EDCI(138mg)及びNMM(102μl)を、室温で1時間撹拌した。[(R)−1−(1’,2’,3’,4’,5’,6’−ヘキサヒドロ−[2,4’]ビピリジニル−3−イルオキシメチル)−プロピル]−ジメチル−アミン三塩酸塩(163mg)及び第2の部分のNMM(93μl)を加え、撹拌を一晩続けた。DMFを真空中で蒸発させ、残りの黄色の油を酢酸エチル(100ml)に溶解した。有機溶液を1NのNa2CO3(2×30ml)及びブラインで洗浄した。各水相を酢酸エチルで一度再抽出した。有機層を乾燥させ(Na2SO4)、濾過し、真空中で蒸発させた。残渣を分取HPLCによって精製した。
【0235】
実施例127の合成:
中間体127a):
【化167】
DCM(20ml)中のBoc−(S)−1−アゼチジン−2−イル−メタノール(J.Med.Chem.2005、48、7637〜7647)(1.47g)に、TFA(10ml)を0℃で加え、溶液を前記温度で1.5h撹拌した。全ての揮発性物質の蒸発、並びにトルエン(2×20ml)との同時蒸発によって、所望の化合物が濁った黄色の油としてもたらされた。
【0236】
中間体127b):
【化168】
氷冷の(R)−3−(2,4−ジクロロ−フェニル)−2−イソシアナト−プロピオン酸メチルエステル(上記の合成)(587mg)の無水DCM(6ml)溶液に、中間体127a)(1.51g)及びトリエチルアミン(0.90ml)の無水DMF(4ml)溶液をアルゴン下で加えた。全ての揮発性物質を蒸発させる前に、混合物を0℃で5h撹拌した。残りをEtOAc(100ml)で希釈し、有機相を0.1MのHCl(2×50ml)及び水(50ml)で洗浄した。全ての水相を混合し、EtOAc(2×50ml)で再抽出した。合わせた有機層をブライン(50ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をクロマトグラフィーによって精製し、所望のエステルを白色固体として得た。
【0237】
中間体127c):
【化169】
水酸化リチウム(36mg)の水(0.9ml)溶液を、アルゴン下で0℃にて中間体127b)(274mg)のMeOH(2.0ml)及びTHF(4.3ml)溶液に加えた。反応混合物を0℃で2h撹拌した。1MのHClを加えることによって混合物を中和させ、MeOH及びTHFの主要部分を真空中で除去した。次いで、残りを1MのHClによってpH3〜4に酸性化し、混合物をEtOAc(2×25ml)で抽出した。有機層を水及びブライン(各々25ml)で洗浄した。水相をEtOAc(25ml)で再抽出し、合わせた有機層を乾燥させ(Na2SO4)、蒸発させ、相当する酸を無色の発泡体として得た。
【0238】
実施例127:
【化170】
[(R)−1−(1’,2’,3’,4’,5’,6’−ヘキサヒドロ−[2,4’]ビピリジニル−3−イルオキシメチル)−プロピル]−ジメチル−アミン三塩酸塩(125mg)及び中間体127c)(123mg)、1−ヒドロキシ−ベンゾトリアゾール水和物(62mg)及びN−メチルモルホリン(114μl)を、DMF(5ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(94mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(24μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(50ml)で希釈し、飽和NaHCO3(3×25ml)で洗浄した。全ての水層を混合し、EtOAc(25ml)で再抽出した。次いで、合わせた有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、真空中で蒸発させた。残渣を分取HPLCによって精製し、所望のアミンを黄色がかった樹脂として得た。
【0239】
実施例129の合成:
中間体129a):
【化171】
トリフルオロメチルトリメチルシラン(0.93ml)及びフッ化セシウム(0.96g)を、ベンズヒドリル−アゼチジン−3−オン(1.00g)のTHF(12.5ml)溶液に加えた。室温で1h撹拌した後、飽和NH4Cl(12.5ml)及びフッ化テトラブチルアンモニウム(498mg)を加え、撹拌をさらに6h続けた。混合物を、ジエチルエーテル(3×50ml)で抽出し、有機層をMgSO4上で乾燥させた。真空濃縮によってオレンジ色の油を得て、それをクロマトグラフィーによって精製し、黄色の油の形態の所望のアルコールを得た。
【0240】
中間体129b):
【化172】
炭素担持20重量%水酸化パラジウム(202mg)及び1MのHCl(1.52ml)を、中間体129a)(466mg)のMeOH溶液に加えた。反応混合物を、1バールの水素下で室温にて3h水素化した。触媒をセライトで濾過し、メタノールで洗浄した。濾液を濃縮し、真空下で乾燥させた。次いで、残渣をヘキサンで洗浄した。有機層をデカントし、ベージュ色の固体を得た。
【0241】
中間体129c):
【化173】
氷冷の(R)−3−(2,4−ジクロロ−フェニル)−2−イソシアナト−プロピオン酸メチルエステル(上記の合成)(178mg)の無水DCM(3ml)溶液に、中間体129b)(230mg)及びトリエチルアミン(0.27ml)の無水DMF(2ml)溶液をアルゴン下で加えた。混合物を0℃で3h撹拌させた。次いで、混合物をEtOAc(50ml)で希釈し、有機相を0.1MのHCl(2×25ml)及び水(25ml)で洗浄した。全ての水相を混合し、EtOAc(2×50ml)で再抽出した。次いで、合わせた有機層をブライン(50ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をクロマトグラフィーによって精製し、所望のエステルを無色の発泡体として得た。
【0242】
中間体129d):
【化174】
水酸化リチウム(17mg)の水(0.5ml)溶液を、アルゴン下で0℃にて中間体129c)(151mg)のMeOH(0.9ml)及びTHF(2.0ml)溶液に加えた。反応混合物を0℃で3h撹拌した。1MのHClを加えることによって混合物を中和させ、MeOH及びTHFの主要部分を真空中で除去した。次いで、残りを1MのHClによってpH3〜4に酸性化し、混合物をEtOAc(2×25ml)で抽出した。有機層を水及びブライン(各々25ml)で洗浄し、乾燥させ(Na2SO4)、蒸発させ、相当する酸を無色の樹脂として得た。
【0243】
実施例129:
【化175】
[(R)−1−(1’,2’,3’,4’,5’,6’−ヘキサヒドロ−[2,4’]ビピリジニル−3−イルオキシメチル)−プロピル]−ジメチル−アミン三塩酸塩(111mg)及び中間体129d)(125mg)、1−ヒドロキシ−ベンゾトリアゾール水和物(55mg)及びN−メチルモルホリン(100μl)を、DMF(5ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(82mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(100μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(50ml)で希釈し、飽和NaHCO3(3×25ml)で洗浄した。全ての水層を混合し、EtOAc(25ml)で再抽出した。次いで、合わせた有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、真空中で蒸発させた。残渣を分取HPLCによって精製し、所望のアミンを黄色がかった樹脂として得た。
【0244】
実施例130の合成:
中間体130a):
【化176】
火炎乾燥フラスコ中で、トリメチルスルホキソニウムヨージド(2.57g)及び水素化ナトリウム(鉱油中60%、476mg)を、DMSO(8.8ml)に10℃で懸濁させた。混合物を室温で1.5時間撹拌した。tert−ブチル−3−オキソピロリジン−1−カルボキシレート(2.00g)のDMSO(2.2ml)溶液を15分以内で一滴ずつ加えた。撹拌を1時間続けた。氷水(50ml)及びブライン(50ml)を加えることによって反応物を加水分解した。水性懸濁液を、ジエチルエーテル(2×50ml)で抽出した。合わせたエーテル溶液をブラインで洗浄し、MgSO4上で乾燥させ、濾過した。濾液を真空中で蒸発させ、粘性の茶色の油を得て、それをクーゲルロール蒸留によって精製した。生成物を、無色の油として得た。
【0245】
中間体130b):
【化177】
火炎乾燥フラスコ中、シアン化銅(I)(105mg)を、無水THF(2ml)に懸濁させた。懸濁液を−76℃に冷却し、シクロプロピルマグネシウムブロミド(THF中0.5M、9.34ml)を加えた。混合物を1時間撹拌し、続いて中間体130a)(155mg)の乾燥THF(1ml)溶液をゆっくりと加えた。反応物を室温にゆっくりと温め、一晩撹拌した。混合物を飽和水性NH4Cl及びジエチルエーテルに分配した。エーテル相を水及びブラインで洗浄した。各水相をジエチルエーテルで一度再抽出した。合わせた有機相をMgSO4上で乾燥させ、濾過し、蒸発させた。粗生成物をカラムクロマトグラフィーによって精製した。
【0246】
中間体130c):
【化178】
中間体130b)(116mg)をジオキサン(6ml)に溶解した。ジオキサン(6ml)中の4NのHClを加え、反応物を室温で1.5時間撹拌した。溶媒を蒸発させ、残渣をトルエンで数回同時蒸発した。残りの淡赤色の固体を高真空下にて乾燥させた。
【0247】
中間体130d):
【化179】
氷冷の(R)−3−(2,4−ジクロロ−フェニル)−2−イソシアナト−プロピオン酸メチルエステル(125mg)のDMF(4ml)溶液に、中間体130c)(89mg)及びトリエチルアミン(134μl)の無水DMF(2ml)溶液をアルゴン下で加えた。混合物を0℃で2時間、室温でさらに2時間撹拌させた。反応混合物を真空中で蒸発させた。残渣をDCM(80ml)で希釈し、有機相を0.1NのHCl(20ml)、1NのNaHCO3(20ml)及びブラインで洗浄した。各水相をDCMで再抽出し、次いで合わせた有機相をNa2SO4上で乾燥させ、濾過し、蒸発させた。薄茶色の油を得て、高真空下にて乾燥させた。粗生成物をカラムクロマトグラフィーによって精製した。
【0248】
中間体130e):
【化180】
中間体130d)(171mg)を、MeOH(1ml)及びTHF(5ml)に0℃で溶解した。水酸化リチウム一水和物(34.6mg)のH2O(1ml)溶液を加えた。混合物を0℃で4h撹拌した。1NのHClを加えることによって反応混合物を中和させ、MeOH及びTHFを真空中で蒸発させた。1NのHCl(pH約1〜2)で水相を酸性化し、EtOAc(1×20ml)で抽出した。有機層を水及びブラインで洗浄した。水相をEtOAcで再抽出し、合わせた有機層をNa2SO4上で乾燥させ、真空中で蒸発させ、白色の発泡体を得た。粗生成物をカラムクロマトグラフィーによって精製した。
【0249】
実施例130:
【化181】
[(R)−1−(1’,2’,3’,4’,5’,6’−ヘキサヒドロ−[2,4’]ビピリジニル−3−イルオキシメチル)−プロピル]−ジメチル−アミン三塩酸塩(123mg)、中間体130e)(165mg)及びHOBt(73mg)を、DCM(6ml)中で混合した。EDCI(73mg)及びNMM(132μl)を加えた。反応物を室温で1時間撹拌した。第2の部分のNMM(28μl)を加え、撹拌を一晩続けた。混合物をEtOAc(50ml)で希釈し、1NのNa2CO3(3×25ml)及びブラインで洗浄した。全ての水相をEtOAcで一度再抽出した。合わせた有機層を乾燥させ(Na2SO4)、濾過し、真空中で蒸発させた。粗生成物を分取HPLCによって精製した。
【0250】
実施例133の合成:
中間体133a):
【化182】
トリアセトキシ水素化ホウ素ナトリウム(5.88g)を、3−オキセタノン(1.00g)及びN−ベンジル−メチルアミン(2.52g、2.7ml)の1,2−ジクロロエタン(300ml)溶液に加え、直ちに続いて酢酸(1.6ml)を加えた。室温で18h激しく撹拌した後、このように得られた微細懸濁液を0℃に冷却し、200mlの水を注意深く加えることによって反応物をクエンチした。系を1h撹拌させ、残った試薬は完全な加水分解に達した。次いで、有機相を集め、水相をDCM(2×50ml)でさらに抽出した。次いで、合わせた有機相を飽和NaHCO3(2×50ml)、H2O(50ml)及びブライン(75ml)で洗浄した。Na2SO4上での乾燥、濾過及び溶媒の蒸発の後、化合物をオレンジ色の粗シロップとして得た。クロマトグラフィーによる精製によって、黄色がかった油の形態の所望の化合物を得た。
【0251】
中間体133b):
【化183】
炭素担持20重量%水酸化パラジウム(195mg)の存在下で1バールの水素下で室温にて、EtOAc(25ml)中の溶液中の中間体133a)(1.94g)を、一晩水素化した。上清のシリンジフィルターによる濾過及びEtOAc溶液(5ml)による触媒のさらなる洗浄によって、30mlのEtOAc中の所望の化合物のニート溶液を得た。
【0252】
中間体133c):
【化184】
氷冷の(R)−3−(2,4−ジクロロ−フェニル)−2−イソシアナト−プロピオン酸メチルエステル(上記のような合成)(450mg)の無水DCM(10ml)溶液に、中間体133b)のEtOAc(6ml;2.62mmol)溶液を加えた。混合物を0℃で1h撹拌させ、次いで室温に17hで戻らせた。この時点で全ての揮発性物質を蒸発させ、このように得られた無色のワックスを真空中で十分に乾燥させ、過剰の中間体133b)を除去し、所望のエステルを純粋な形態で得た。
【0253】
中間体133d):
【化185】
水酸化リチウム(80mg)の水(1.5ml)溶液を、アルゴン下で0℃にて中間体133c)(698mg)のMeOH(9.0ml)及びTHF(1.5ml)溶液に加えた。反応混合物を0℃で2h撹拌した。トリフルオロ酢酸(380mg;260μl)を加えることによって混合物を中和させ、MeOH及びTHFの主要部分を真空中で除去した。次いで、残りを水(10ml)で希釈し、EtOAc(3×25ml)で抽出した。有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、蒸発させ、高真空下での十分な乾燥の後、相当する酸を無色の発泡体として得た。
【0254】
実施例133:
【化186】
[(R)−1−(1’,2’,3’,4’,5’,6’−ヘキサヒドロ−[2,4’]ビピリジニル−3−イルオキシメチル)−プロピル]−ジメチル−アミン三塩酸塩(120mg)及び中間体133d)(153mg)、1−ヒドロキシ−ベンゾトリアゾール水和物(61mg)及びN−メチルモルホリン(119μl)を、DCM(15ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(88mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(26μl)を加え、撹拌を一晩続けた。揮発性物質を蒸発させ、残渣をEtOAc(50ml)及び飽和NaHCO3(25ml)に分配した。有機相を集め、飽和NaHCO3(2×25ml)でさらに洗浄した。水層を混合し、DCM(10ml)で抽出した。次いで、合わせた有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、真空中で蒸発させた。残渣を分取HPLCによって精製し、所望の誘導体の二ギ酸塩を黄色がかった樹脂として得た。
【0255】
実施例134の合成:
中間体134a):
【化187】
N−ベンジル−メチルアミン(1.05g、1.12ml)を、固体無水Na2CO3(2.00g)の存在下で3−ヒドロキシメチル−3−メチルオキセタンp−トシレート(2.23g)のアセトニトリル(35ml)溶液に加えた。次いで、懸濁液を室温で6日間激しく撹拌し、その時点で変換のさらなる進展は顕著ではなかった。全ての揮発性物質を蒸発させ、残渣をEtOAc(50ml)及び水(50ml)に分配した。有機相を集め、水及びブライン(各々25ml)でさらに洗浄した。合わせた水相をDCM(25ml)で抽出した。次いで、合わせた有機相をNa2SO4上で乾燥させ、濾過し、蒸発させ、部分的に結晶化した粗シロップを得た。クロマトグラフィーによる精製によって、黄色がかった油の形態の所望の化合物を得た。
【0256】
中間体134b):
【化188】
炭素担持20重量%水酸化パラジウム(60mg)の存在下、1バールの水素下で室温にて一晩、EtOAc(10ml)中の溶液中の中間体134a)(360mg)を水素化した。シリンジフィルターによる濾過及びEtOAc(5ml)による触媒のさらなる洗浄によって、15mlのEtOAc中の所望の化合物のニート溶液を得た。
【0257】
中間体134c):
【化189】
氷冷の(R)−3−(2,4−ジクロロ−フェニル)−2−イソシアナト−プロピオン酸メチルエステル(上記のような合成)(300mg)の無水DCM(9ml)溶液に、中間体134b)のEtOAc(15ml;1.75mmol)溶液を加えた。混合物を0℃で1h撹拌させ、次いで17hで室温に戻らせた。この時点で、全ての揮発性物質を蒸発させ、淡黄色のシロップ/ワックスを得た。粗生成物をクロマトグラフィーによって精製し、所望のエステルを、室温で保存すると結晶化する透明なシロップとして得た。
【0258】
中間体134d):
【化190】
水酸化リチウム(42mg)の水(0.8ml)溶液を、アルゴン下で0℃にて中間体134c)(305mg)のMeOH(5.0ml)及びTHF(0.8ml)溶液に加えた。反応混合物を0℃で2h撹拌した。トリフルオロ酢酸(200mg;135μl)を加えることによって混合物を中和させ、MeOH及びTHFの主要部分を真空中で除去した。次いで、残りを水(10ml)で希釈し、EtOAc(3×25ml)で抽出した。有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、蒸発させ、相当する酸を、高真空中で不安定な泡を形成する無色の粘性シロップとして得た。
【0259】
実施例134:
【化191】
[(R)−1−(1’,2’,3’,4’,5’,6’−ヘキサヒドロ−[2,4’]ビピリジニル−3−イルオキシメチル)−プロピル]−ジメチル−アミン三塩酸塩(100mg)及び中間体134d)(115mg)、1−ヒドロキシ−ベンゾトリアゾール水和物(52mg)及びN−メチルモルホリン(100μl)を、DCM(15ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(75mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(22μl)を加え、撹拌を一晩続けた。揮発性物質を蒸発させ、残渣をEtOAc(50ml)及び飽和NaHCO3(25ml)に分配した。有機相を集め、飽和NaHCO3(2×25ml)でさらに洗浄した。水層を混合し、DCM(10ml)で抽出した。次いで、合わせた有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、真空中で蒸発させた。残渣を分取HPLCによって精製し、所望の誘導体の二ギ酸塩を黄色がかった樹脂として得た。
【0260】
さらなる実施例を下記に例示する。
【表25】
【0261】
生物学的アッセイ
A 結合アッセイ
膜結合アッセイを使用して、ヒトメラノコルチン受容体を発現するHEK293細胞膜調製物に結合している蛍光標識NDP−α−MSHの競争的阻害剤を同定する。
【0262】
試験化合物又は非標識NDP−α−MSHを、様々な濃度で384ウェルマイクロタイタープレートに分注する。蛍光標識NDP−α−MSHを、単一の濃度で分注し、続いて膜調製物を加える。プレートを室温で5hインキュベートする。
【0263】
蛍光偏光の程度を蛍光偏光マイクロプレートリーダーで決定する。
【0264】
B 機能アッセイ
ヒトメラノコルチン受容体のアゴニスト活性を、均質膜をベースとするアッセイにおいて決定する。cAMP特異抗体上の結合部位の限られた数についての、非標識cAMPと一定量の蛍光標識cAMPとの間の競合は、蛍光偏光によって明らかになる。
【0265】
試験化合物又は非標識NDP−α−MSHを、様々な濃度で384ウェルマイクロタイタープレートに分注する。ヒトメラノコルチン受容体を発現するHEK293細胞からの膜調製物を加える。短いプレインキュベーション期間後、適切な量のATP、GTP及びcAMP抗体を加え、蛍光標識cAMPコンジュゲートを分注する前に、プレートをさらにインキュベートする。プレートを、蛍光偏光マイクロプレートリーダーで読み取る前に、4℃で2hインキュベートする。試験化合物への反応として生成されたcAMPの量を、NDP−α−MSHによる刺激からもたらされるcAMPの生成と比較する。
【0266】
本発明の代表的な化合物を試験し、メラノコルチン−4受容体に結合することを見出した。これらの化合物は一般に、2μM未満のIC50値を有することが見出された。
【表26】
【表27】
【表28】
【表29】
【表30】
【表31】
【0267】
C In vivo食物摂取モデル
1 自発的摂食パラダイム
試験化合物のi.p.、s.c.又はp.o.投与後に、ラットにおける食物摂取量を測定する(例えば、A.S.Chenら、Transgenic Res 2000 Apr;9(2):145〜154を参照されたい)。
【0268】
2 LPS誘発食欲不振症及び腫瘍誘発悪液質のモデル
試験化合物のラットへのi.p.又はp.o.投与によって、リポ多糖(LPS)投与により誘発される食欲不振症又は腫瘍増殖によって誘発される悪液質の予防又は寛解を決定する(例えば、D.L.Marks、N.Ling、及びR.D.Cone、Cancer Res 2001 Feb 15;61(4):1432〜1438を参照されたい)。
【0269】
D ラット交尾外アッセイ
性的に成熟している雄性帝王切開由来Sprague−Dawley(CD)ラット(60日齢超)(交尾外評価の間に陰茎が陰茎鞘への収縮するのを防止するために提靭帯を外科的に切除する)を使用する。動物に食物及び水を自由に与え、通常の明/暗サイクルで維持する。試験は明サイクルの間に行う。
【0270】
1 交尾外反射試験のための仰臥位拘束への条件付け
この条件付けは約4日かかる。1日目、動物を暗い拘束器内に入れ、15〜30分間放置する。2日目、動物を拘束器中にて仰臥位で15〜30分間拘束する。3日目、動物の陰茎鞘を収縮させて仰臥位で15〜30分間拘束する。4日目、動物の陰茎反応が観察されるまで陰茎鞘を収縮させて仰臥位で拘束する。何匹かの動物は、手順に完全に順応する前に、さらに何日かの条件付けが必要となる。非応答動物はさらなる評価から除外する。いかなる取り扱い又は評価の後も、正の強化を確実にするため、動物に褒美を与える。
【0271】
2 交尾外反射試験
ラットを、通常の頭及び足のグルーミングができる適切なサイズの円筒内に胴体腹側を入れて仰臥位で軽く拘束する。400〜500グラムのラットでは、円筒の直径は約8cmである。胴体下部及び後肢を、非粘着性材料(ベトラップ)で拘束する。陰茎亀頭を通す孔を設けたさらなる断片のベトラップで動物を固定し、包皮鞘を収縮した位置に保つ。典型的には交尾外性器反射試験と称される陰茎反応を観察する。典型的には、鞘収縮後数分内に一連の陰茎勃起が自発的に起こるであろう。通常の反射発生性勃起反応のタイプには、伸長、怒張、カップ及びフリップが挙げられる。伸長は、陰茎本体の延長として分類される。怒張は陰茎亀頭の拡張である。カップとは、陰茎亀頭の遠心縁が瞬間的に張ってカップを形成する強度の勃起として定義される。フリップは陰茎本体の背屈である。
【0272】
ベースライン及び又はビヒクル評価を行って、動物がどのように反応するか、及び反応するかどうかを判定する。動物の中には最初の反応までに長時間かかるものもあれば、一方他に全くの非応答動物がある。このベースライン評価の間に、最初の反応までの潜時、反応の回数及びタイプを記録する。試験時間枠は、最初の反応後15分である。
【0273】
評価と評価の間に最低1日おいた後、これらの同じ動物に、試験化合物を20mg/kgで投与し、陰茎反射を評価する。全ての評価を録画し、後で点数をつける。データを集め、対応のある両側t検定を使用して分析し、個々の動物についてベースライン及び/又はビヒクル評価を薬物処理評価と比較する。最小4匹の動物群を用いて、ばらつきを抑える。
【0274】
正の参照対照を各試験に含め、試験の有効性を確保する。動物には、実施する試験の特性によっていくつかの投与経路で投与することができる。投与経路には、静脈内(IV)、腹腔内(IP)、皮下(SC)及び脳内室(ICV)が挙げられる。
【0275】
E 雌性性的機能不全のモデル
雌の性受容性に関連するげっ歯類アッセイには、ロードシスの行動モデル及び交尾活動の直接観察が挙げられる。雄性及び雌性ラットの両方におけるオルガスムを測定するための、麻酔下の脊髄離断ラットにおける尿道性器反射モデルもまたある。これら及び雌性性的機能不全の他の確立した動物モデルは、K.E.McKennaら、A Model For The Study of Sexual Function In Anesthetized Male And Female Rats、Am.J.Physiol.(Regulatory Integrative Comp.Physiol 30):R1276〜R1285、1991;K.E.McKennaら、Modulation By Peripheral Serotonin of The Threshold For Sexual Reflexes In Female Rats、Pharm.Bioch.Behav.、40:151〜156、1991;及びL.K.Takahashiら、Dual Estradiol Action In The Diencephalon And The Regulation of Sociosexual Behavior In Female Golden Hamsters、Brain Res.、359:194〜207、1985において記載されている。
【0276】
医薬組成物の例
本発明の化合物の経口組成物の特定の実施形態として、十分に微粉化したラクトースと共に23mgの実施例6を製剤し、0号硬質ゼラチンカプセルを満たす580〜590mgの総量を提供する。
【0277】
本発明の化合物の経口組成物の他の特定の実施形態として、十分に微粉化したラクトースと共に28mgの実施例14を製剤し、0号硬質ゼラチンカプセルを満たす580〜590mgの総量を提供する。
【0278】
その特定の好ましい実施形態に関して本発明を説明し例示してきたが、本発明の趣旨及び範囲から逸脱することなく様々な変更、修正及び置換をその中で行うことができることを当業者なら理解するであろう。例えば、有効量は、以上の説明から明らかなような好ましい用量以外に、観察される特定の薬理学的反応の結果として適用可能であることがあり、それは選択した特定の活性化合物、並びに製剤及び用いる投与方法のタイプによって変化することがあり、結果におけるこのような予測される変動又は差異は、本発明の目的及び実施に従って意図されている。したがって、本発明は下記の特許請求の範囲によってのみ限定され、このような特許請求の範囲は妥当であればできるだけ広く解釈されることを意図する。
【発明の詳細な説明】
【0001】
[発明の分野]
本発明は、メラノコルチン−4受容体モジュレーターとしての置換ヘテロアリールピペリジン誘導体に関する。構造及び立体化学構造によって、本発明の化合物は、ヒトメラノコルチン−4受容体(MC−4R)の選択的アゴニスト又は選択的アンタゴニストである。アゴニストは、肥満症、糖尿病及び性的機能不全などの障害及び疾患の治療のために使用することができ、一方アンタゴニストは、癌悪液質、筋肉疲労、食欲不振症、筋萎縮性側索硬化症、不安神経症及びうつなどの障害及び疾患の治療のために有用である。一般に、MC−4Rの調節が関与している全ての疾患及び障害は、本発明の化合物で治療することができる。
【0002】
[発明の背景]
メラノコルチン(MC)は、タンパク質分解的切断を介してプロオピオメラノコルチン(POMC)から生じる。これらのペプチド、副腎皮質刺激ホルモン(ACTH)、α−メラニン細胞刺激ホルモン(α−MSH)、β−MSH及びγ−MSHは、12〜39個のアミノ酸でありサイズに幅がある。中枢MC−4R活性化についての最も重要な内因性アゴニストは、トリデカペプチドα−MSHであるようである。MCの中で、α−MSHは脳において神経伝達物質又は神経調節物質として作用することが報告された。MCペプチド、特にα−MSHは、摂食行動、色素沈着及び外分泌機能を含めた生物学的機能について広範囲の作用を有する。α−MSHの生物学的作用は、メラノコルチン受容体(MC−R)と称される7回膜貫通Gタンパク質共役受容体のサブファミリーを介してもたらされる。これらのMC−Rのいずれかの活性化は、cAMP形成の刺激をもたらす。
【0003】
現在まで、MCについての5種の別個のタイプの受容体サブタイプ(MC−1RからMC−5R)が同定されており、これらは異なる組織で発現している。
【0004】
MC−1Rは、メラニン形成細胞において最初に見出された。動物におけるMC−1Rの天然不活性変異体は、チロシナーゼの制御を介してフェオメラニンのユーメラニンへの変換を制御することによって色素沈着の変化及びそれに続くより薄い毛色を生じさせることが示された。これら及び他の研究から、MC−1Rは、動物におけるメラニン生成及び毛色並びにヒトにおける皮膚色の重要な制御因子であること明らかである。
【0005】
MC−2Rは、ACTH受容体を代表する副腎において発現する。MC−2Rは、α−MSHの受容体ではないが、副腎皮質刺激ホルモンI(ACTH I)の受容体である。
【0006】
MC−3Rは、脳(主に、視床下部にある)並びに腸及び胎盤などの末梢組織において発現し、ノックアウト研究は、MC−3Rが、摂食行動、体重及び熱産生の変化に関与することがあることを明らかにした。
【0007】
MC−4Rは、脳内で主として発現する。圧倒的なデータは、エネルギー恒常性におけるMC−4Rの役割を指示する。動物におけるMC−4Rの遺伝子ノックアウト及び薬理的操作は、MC−4Rをアゴナイズすることは体重減少をもたらし、MC−4Rをアンタゴナイズすることは体重増加をもたらすことを示した(A.Kaskら、「Selective antagonist for the melanocortin−4 receptor(HS014)increases food intake in free−feeding rats」、Biochem.Biophys.Res.Commun.、245:90〜93(1998))。
【0008】
MC−5Rは、白色脂肪、胎盤を含めた多くの末梢組織において遍在性に発現し、低レベルの発現もまた脳において観察される。しかし、その発現は、外分泌腺において最も多い。マウスにおけるこの受容体の遺伝子ノックアウトは、外分泌腺機能の調節の変化をもたらし、水の反発及び体温調節の変化をもたらす。MC−5Rノックアウトマウスはまた、皮脂腺脂質産生が減少することが明らかとなった(Chenら、Cell、91:789〜798(1997))。
【0009】
MC−3R及びMC−4Rモジュレーター並びに肥満症及び食欲不振症などの体重障害の治療におけるそれらの使用の研究についての注目が集まってきた。しかし、MCペプチドは、色素沈着、摂食行動及び外分泌機能の調節におけるそれらの役割に加えて、強力な生理学的効果を有するという証拠が示されてきた。特に、α−MSHは、炎症性腸疾患、腎虚血/再潅流傷害及び内毒素誘発肝炎を含めた炎症の急性及び慢性モデルの両方において強力な抗炎症効果を誘発することが最近示された。これらのモデルにおけるα−MSHの投与は、炎症が媒介する組織の損傷の実質的な減少、白血球浸潤の有意な減少、並びにサイトカイン及び他の伝達物質の上昇したレベルのベースライン値近くへの劇的な減少をもたらす。最近の研究は、α−MSHの消炎作用はMC−1Rによって媒介されることを示した。MC−1Rのアゴニズムが抗炎症反応をもたらす機構は、炎症誘発性転写活性化因子であるNF−κBの阻害を介していると思われる。NF−κBは、炎症誘発性カスケードの中心的な構成要素であり、その活性化は、多くの炎症性疾患の開始における中心事象である。さらに、α−MSHの消炎作用は、部分的に、MC−3R及び/又はMC−5Rのアゴニズムによって媒介されることがある。
【0010】
MC−4Rシグナル伝達が摂食行動の媒介において重要であるという証拠が示されてきたが、肥満症の管理のために標的となることができる特定の単一のMC−Rはまだ同定されていない(S.Q.Giraudoら、「Feeding effects of hypothalamic injection of melanocortin−4 receptor ligands」、Brain Research、80:302〜306(1998))。肥満症におけるMC−Rの関与についてのさらなる証拠には、下記が含まれる。1)MC−1R、MC−3R及びMC−4Rのアンタゴニストを異所的に発現しているアグーチ(Avy)マウスは肥満であり、これらの3種のMC−Rの作用を遮断することは過食症及び代謝障害をもたらす場合があることをこれは示す。2)MC−4Rノックアウトマウス(D.Huszarら、Cell、88:131〜141(1997))は、アグーチマウスの表現型を再現し、これらのマウスは肥満である。3)げっ歯類において側脳室内(ICV)に注射した環状ヘプタペプチドメラノタニンII(MT−II)(非選択的MC−1R、−3R、−4R、及び−5Rアゴニスト)は、いくつかの動物飼育モデル(NPY、ob/ob、アグーチ、絶食)において食物摂取量を減少させ、一方ICV注射したSHU−9119(MC−3R及び4Rアンタゴニスト;MC−1R及び−5Rアゴニスト)は、この作用を逆転させ、過食症を誘発することができる。4)ズッカー糖尿病肥満ラットのα−NDP−MSH誘導体(HP−228)による慢性腹腔内処理は、MC−1R、−3R、−4R、及び−5Rを活性化し、12週間の期間に亘り食物摂取量及び体重増加を減少させることが報告されてきた(I.Corcosら、「HP−228 is a potent agonist of melanocortin receptor−4 and significantly attenuates obesity and diabetes in Zucker fatty rats」、Society for Neuroscience Abstracts、23:673(1997))。
【0011】
MC−4Rは、他の生理機能、すなわちグルーミング行動の制御、勃起及び血圧の制御においてもまた役割を果たしているように思われる。勃起不全は、性交を成功させるのに十分な陰茎勃起を達成することができない病状を意味する。「インポテンス」という用語は、この蔓延している状態を説明するために用いられることが多い。合成メラノコルチン受容体アゴニストは、心因性の勃起不全を有する男性において勃起を起こすことが見出された(H.Wessellsら、「Synthetic Melanotropic Peptide Initiates Erections in Men With Psychogenic Erectile Dysfunction:Double−Blind,Placebo Controlled Crossover Study」、J.Urol.、160:389〜393、(1998))。脳のメラノコルチン受容体の活性化は、性的刺激の通常の刺激をもたらすように思われる。男性及び/又は女性の性的機能不全におけるMC−Rの関与についての証拠は、国際公開第00/74679号パンフレットにおいて詳述されている。
【0012】
糖尿病は、哺乳動物においてグルコースを筋肉及び肝細胞に貯蔵するためグリコーゲンに変換する能力が衰えるため、血液におけるグルコースレベルを制御する哺乳動物の能力が損なわれる疾患である。I型糖尿病において、グルコースを貯蔵するこの能力の減少は、インスリン産生の減少に起因する。「II型糖尿病」又は「インスリン非依存性真性糖尿病」(NIDDM)は、主要なインスリン感受性組織である筋肉、肝臓及び脂肪組織においてグルコース及び脂質代謝についてのインスリン刺激又は調節作用に対する深刻な耐性に起因する糖尿病の形態である。このインスリン応答性に対する耐性は、グルコース取込み、酸化及び筋肉における貯蔵の不十分なインスリン活性化、並びに脂肪組織における脂肪分解並びに肝臓におけるグルコース産生及び分泌の不適切なインスリン抑制をもたらす。これらの細胞がインスリンに対して脱感作される場合、体は異常に高レベルのインスリンを産生することによって代償しようとし、高インスリン血症が起こる。高インスリン血症は、高血圧症及び体重の増加と関連する。インスリンは、インスリン感受性細胞による血液からのグルコース、アミノ酸及びトリグリセリドの細胞取込みの促進に関与するため、インスリン非感受性は、心血管疾患における危険因子であるトリグリセリド及びLDLのレベルの上昇をもたらす場合がある。高血圧症、体重増加、トリグリセリド増加及びLDL増加と併せた高インスリン血症を含む症状の一群は、症候群Xとして知られている。MC−4Rアゴニストは、NIDDM及び症候群Xの治療において有用であり得る。
【0013】
下記の知見に基づくように、MC受容体サブタイプの中で、ストレスとの関係及び情動行動の調節に関してMC4受容体はまた興味深い。ストレスは、内分泌、生化学及び行動事象を含めた複雑な反応のカスケードを開始させる。これらの反応の多くは、コルチコトロピン放出因子(CRF)の放出によって開始される(M.J.Owen及びC.B.Nemeroff、「Physiology and pharmacology of corticotrophin releasing factor」、Pharmacol.Rev.43:425〜473(1991))。脳CRF系の活性化に加えて、酵素処理によってプロオピオメラノコルチンから生じるメラノコルチン(MC)は、ストレスへの重要な行動反応及び生化学的反応、結果的に、不安神経症及びうつなどのストレス誘発性の障害を媒介するといういくつかの証拠がある(Shigeyuki Chakiら、「Anxiolytic−Like and Antidepressant−Like Activities of MCL0129(1−[(S)−2−(4−Fluorophenyl)−2−(4−isopropylpiperadin−1−yl)ethyl]−4−[4−(2−methoxynaphthalen−1−yl)butyl]piperazine), a Novel and Potent Nonpeptide Antagonist of the Melanocortin−4 Receptor」、J.Pharm.Exp.Ther.304(2)、818〜826(2003))。
【0014】
悪性腫癌又は感染などの慢性疾患は、食欲の減少及び除脂肪体重の減少の併発からもたらされる悪液質と関連する場合が多い。除脂肪体重の大幅な減少は、炎症過程によって引き起こされる場合が多く、脳におけるα−MSHの産生を増加させるサイトカイン(例えば、TNF−α)の血漿レベルの増加と通常関連する。α−MSHによる視床下部におけるMC4受容体の活性化は、食欲を減少させ、エネルギーの消費を増加させる。腫瘍マウスにおける実験上の証拠は、遺伝子MC4受容体ノックアウト又はMC4受容体遮断によって、悪液質は予防又は回復に向かうことができることを示す。処理したマウスにおける体重の増加は、より多量の除脂肪体重(主として骨格筋からなる)に起因する(D.L.Marksら、「Role of the central melanocortin system in cachexia」、Cancer Res.61:1432〜1438(2001))。
【0015】
臨床所見は、筋萎縮性側索硬化症(ALS)の進行が体重と逆相関することがあることを示している(例えば、Ludolph AC、Neuromuscul Disord.(2006)16(8):530〜8)。したがって、MC−4R阻害剤は、ALS患者の治療に使用することができる。
【0016】
メラノコルチン受容体のモジュレーターは、文献からすでに公知である。国際公開第2004/024720A1号パンフレットは、ヒトメラノコルチン−4受容体の選択的アゴニストであるピペラジン尿素誘導体について記載しており、したがってそれらは、肥満症に関連する障害の予防の治療において有用であると主張されている。
【0017】
国際公開第2005/047253A1号パンフレットは、メラノコルチン受容体アゴニストとして機能すると主張されている4,4−二置換ピペリジン誘導体について記載している。
【0018】
置換ピペリジン誘導体はまた、カルボン酸及びエステル(ピペリジン環又はピペラジン環を分子の中核として有し、その核がパラ位において5〜7員の複素環、フェニル環、ピリジン環又はチアゾール環でさらに置換されている)に関するDE 103 00973において記載されている。前記環は、エステル基で任意選択で置換されている。この化合物は、頭痛、インスリン非依存性真性糖尿病(NIDDM)、心血管疾患、モルヒネ耐性、皮膚疾患、炎症、アレルギー性鼻炎、喘息;血管拡張を伴い、その結果として組織内の血液循環の減少を伴う疾患の治療;エストロゲン欠乏の女性の閉経期ホットフラッシュの急性期治療若しくは予防的治療、又は疼痛の治療のための医薬の調製において使用される。
【0019】
上記のような様々な疾患及び障害の治療における未解決の欠陥を考慮して、本発明の目的は、癌悪液質、筋肉疲労、食欲不振症、不安神経症、うつ、肥満症、糖尿病、性的機能不全、筋萎縮性側索硬化症、及びMC−4Rの関与を伴う他の疾患を治療するためのメラノコルチン−4受容体モジュレーターとして有用な、血液脳関門を通過する改善された能力を有する新規な置換ヘテロアリールピペリジン誘導体を提供することにある。
【0020】
[発明の概要]
本発明は、構造式(I)の置換ヘテロアリールピペリジン誘導体に関し、
【化1】
式中、R1、R3、R4、R5、B、D、E及びGは、下記で説明するように定義される。
【0021】
構造式(I)のヘテロアリールピペリジン誘導体は、メラノコルチン受容体モジュレーターとして有効であり、選択的メラノコルチン−4受容体(MC−4R)モジュレーターとして特に有効である。したがって、それらは、MC−4Rの活性化又は不活性化が関与している障害の治療に有用である。アゴニストは、肥満症、糖尿病及び性的機能不全などの障害及び疾患の治療のために使用することができ、一方、アンタゴニストは、癌悪液質、筋肉疲労、食欲不振症、筋萎縮性側索硬化症(ALS)、不安神経症及びうつなどの障害及び疾患の治療のために有用である。
【0022】
したがって、本発明は、癌悪液質、筋肉疲労、食欲不振症、筋萎縮性側索硬化症(ALS)、不安神経症、うつ、肥満症、真性糖尿病、男性若しくは女性の性的機能不全及び勃起不全の治療及び/又は予防のための式(I)の化合物に関する。
【0023】
さらなる態様において、本発明は、癌悪液質、筋肉疲労、食欲不振症、筋萎縮性側索硬化症(ALS)、不安神経症、うつ、肥満症、真性糖尿病、男性若しくは女性の性的機能不全及び勃起不全の治療及び/又は予防のための医薬品の調製のための式(I)の化合物の使用に関する。
【0024】
本発明はまた、本発明の化合物と薬学的に許容される担体とを含む医薬組成物に関する。
【0025】
[発明の詳細な説明]
本発明は、メラノコルチン受容体モジュレーター、特に、選択的MC−4Rアゴニスト及びMC−4Rアンタゴニストとして有用な置換ヘテロアリールピペリジン誘導体に関する。
【0026】
本発明の化合物は、構造式(I)
【化2】
並びにそのエナンチオマー、ジアステレオマー、互変異性体、溶媒和物及び薬学的に許容される塩によって表され、
式中、
R1は、−N(R10)−(C(R6)2)m−T、
−(C(R6)2)l−T、又は
−O−(C(R6)2)m−Tであり、
R6は、
H、
F、
OH、
OCH3、
C1〜6−アルキル(ハロゲン、CN、OH及びOCH3から選択される1〜3個の置換基で任意選択で置換されている)、並びに
C3〜6−シクロアルキル(ハロゲン、CN、OH及びOCH3から選択される1〜3個の置換基で任意選択で置換されている)から独立に選択され、
Tは、NR7R8、
【化3】
であり、
R7及びR8は、互いに独立に、
H、
C1〜6−アルキル、
C2〜6−アルケニル、
C2〜6−アルキニル、及び
C2〜6−アルキレン−O−C1〜6−アルキルから選択され、
各アルキル、アルケニル及びアルキニルは、1個若しくは複数のハロゲン原子、CN又はOHで任意選択で置換されており、
R9は、
ハロゲン、
CN、
OH、
C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、並びに
O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
C1〜6−アルキレン−O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、又は
NR12R13から独立に選択され、
R10は、H又は
C1〜6−アルキルであり、
R11は、
ハロゲン、
CN、
OH、
C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
C2〜6−アルケニル、
C2〜6−アルキニル、
O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
C1〜6−アルキレン−O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
C0〜6−アルキル−C3〜6−シクロアルキル、
−OC(O)C1〜6−アルキル、
−NH2、
−NH(C1〜6−アルキル)、及び
−N(C1〜6−アルキル)2から独立に選択され、
R12及びR13は、互いに独立に、
C1〜6−アルキル(OHで任意選択で置換されている)、
C2〜6−アルケニル、
C2〜6−アルキニル、
C2〜6−アルキレン−O−C1〜6−アルキル、及び
C2〜6−アルキレン−N−(C1〜6−アルキル)2から選択され、
Wは、CH、O又はNR10であり、
Xは、CH又はNであり、
Yは、CH又はNであり、
Zは、CH又はNであり、
Aは、0〜2個の窒素原子を含有する3〜7員の飽和、不飽和又は芳香族環であり、
Bは、CR2又はNであり、
Gは、CR2又はNであり、
Dは、CR2又はNであり、
Eは、CR2又はNであり、
ただし、可変部分B、G、D及びEの1つ又は2つは、Nでなくてはならず、
R2は、
H、
F、
Cl、
CH3、
OCH3、及び
CF3から独立に選択され、
R3は、H、
Cl、
F、又は
CH3であり、
R4は、Cl、
F、又は
CH3であり、
R5は、
【化4】
モルホリン(1〜3個の同じ若しくは異なる置換基R14で任意選択で置換されている)、
環中に1個の窒素原子と任意選択でO、N及びSから選択されるさらなるヘテロ原子とを含有する4〜7員の飽和又は部分不飽和複素環(複素環は、1〜4個の同じ若しくは異なる置換基R11で任意選択で置換されている)、又は
NR12R13であり、
R14は、C1〜6−アルキル、
C1〜6−アルキレン−O−C1〜6−アルキル、
C1〜6−アルキレン−OH、
C1〜6−アルキレン−NH2、
C1〜6−アルキレン−NH−C1〜6−アルキル、又は
C1〜6−アルキレン−N(C1〜6−アルキル)2であり、
R15は、H又は
C1〜6−アルキルであり、
lは、0、1、2、3又は4であり、
mは、0、1、2、3又は4であり、
oは、0、1又は2であり、
pは、0、1、2、3又は4であり、
rは、0、1、2、3又は4であり、
sは、1又は2であり、
tは、0又は1である。
【0027】
好ましい実施形態では、式(I)の化合物は、下記のように定義される。
R1は、−N(R10)−(C(R6)2)m−T、
−(C(R6)2)l−T、又は
−O−(C(R6)2)m−Tであり、
R6は、
H、
F、
OH、
OCH3、
C1〜6−アルキル(ハロゲン、CN、OH及びOCH3から選択される1〜3個の置換基で任意選択で置換されている)、並びに
C3〜6−シクロアルキル(ハロゲン、CN、OH及びOCH3から選択される1〜3個の置換基で任意選択で置換されている)から独立に選択され、
Tは、NR7R8、
モルホリン、
【化5】
であり、
R7及びR8は、互いに独立に、
H、
C1〜6−アルキル、
C2〜6−アルケニル、
C2〜6−アルキニル、及び
C2〜6−アルキレン−O−C1〜6−アルキルから選択され、
各アルキル、アルケニル及びアルキニルは、1個若しくは複数のハロゲン原子、CN又はOHで任意選択で置換されており、
R9は、
ハロゲン、
CN、
OH、
C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、並びに
O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
C1〜6−アルキレン−O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)から独立に選択され、
R10は、H又は
C1〜C6−アルキルであり、
R11は、
ハロゲン、
CN、
OH、
C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
C1〜6−アルキレン−O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
−NH2、
−NH(C1〜6−アルキル)、及び
−N(C1〜6−アルキル)2から独立に選択され、
Xは、CH又はNであり、
Yは、CH又はNであり、
Zは、CH又はNであり、
Aは、0〜2個の窒素原子を含有する3〜7員の飽和、不飽和又は芳香族環であり、
Bは、CR2又はNであり、
Gは、CR2又はNであり、
Dは、CR2又はNであり、
Eは、CR2又はNであり、
ただし、可変部分B、G、D及びEの1つ又は2つは、Nでなくてはならず、
R2は、
H、
F、
Cl、
CH3、
OCH3、及び
CF3から独立に選択され、
R3は、H、
Cl、
F、又は
CH3であり、
R4は、Cl又はFであり、
R5は、
【化6】
モルホリン(1〜3個の同じ若しくは異なる置換基R14で任意選択で置換されている)、又は
NR12R13であり、
R12及びR13は、互いに独立に、
C1〜6−アルキル、
C2〜6−アルケニル、
C2〜6−アルキニル、
C2〜6−アルキレン−O−C1〜6−アルキル、及び
C2〜6−アルキレン−N−(C1〜6−アルキル)2から選択され、
R14は、C1〜6−アルキル、
C1〜6−アルキレン−O−C1〜6−アルキル、
C1〜6−アルキレン−OH、
C1〜6−アルキレン−NH2、
C1〜6−アルキレン−NH−C1〜6−アルキル、又は
C1〜6−アルキレン−N(C1〜6−アルキル)2であり、
lは、0、1、2、3又は4であり、
mは、0、1、2、3又は4であり、
oは、0、1又は2であり、
pは、0、1、2、3又は4であり、
qは、0、1、2、又は3であり、
rは、0、1、2、3又は4であり、
sは、1又は2である。
【0028】
好ましくは、式(I)による化合物は、下記の立体異性体式(I’)の構造的立体配座をとり、
【化7】
式中、B、G、D、E、R1、R3、R4及びR5は、上記定義の通りである。
【0029】
一般式(I)中の部分
【化8】
は、下記の構造から選択される。
【化9】
【0030】
その中で、可変部分R2は、上記のように定義される。本発明の好ましい実施形態では、R2は、H、Cl、F又はCH3を表す。さらに好ましくは、R2は、H又はCH3を表す。
【0031】
この部分
【化10】
の好ましい実施形態は、下記の構造である。
【化11】
【0032】
本発明の好ましい実施形態では、可変部分R1は、−N(R10)−(C(R6)2)m−T、−(C(R6)2)l−T、又は−O−(C(R6)2)m−T(式中、R6及びR10は、好ましくはH及びC1〜6−アルキルからなる群から独立に選択される)を表す。
【0033】
R3は、H、Cl、又はCH3、さらに好ましくはClを表すことがさらに好ましい。他の実施形態において、R3は、好ましくはFを表す。
【0034】
好ましくは、R4は、Clを表す。
【0035】
本発明の好ましい実施形態では、可変部分R5は、
【化12】
を表し、
式中、rは、好ましくは0又は1であり、qは、好ましくは数字1又は2をとる。
【0036】
さらなる好ましい実施形態では、R7及びR8の少なくとも1つは、C1〜6−アルキル、C2〜6−アルケニル、C2〜6−アルキニル及びC2〜6−アルキレン−O−C1〜6−アルキルから、さらに好ましくはC2〜6−アルケニル、C2〜6−アルキニル及びC2〜6−アルキレン−O−C1〜6−アルキルからなる群から選択される。
【0037】
R9は、ハロゲン、CN、OH、C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、並びにO−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)からなる群から独立に選択されることが好ましい。
【0038】
可変部分lは、好ましくは2又は3から選択される。
【0039】
可変部分mは、好ましくは2、3又は4から、さらに好ましくは2又は3から選択される。
【0040】
式(I)の化合物について、Tは、好ましくは下記の基
【化13】
【化14】
【化15】
【化16】
【化17】
【化18】
からなる群から選択される。
【0041】
好ましい実施形態では、R5は、好ましくは
【化19】
【化20】
【化21】
からなる群から選択される。
【0042】
上記の基のいくつか若しくは全てが好ましい又はより好ましい意味を有する式(I)の化合物もまた、本発明の1つの目的である。
【0043】
上記及び下記において用いられる用語は、下記で説明するような意味を有する。
【0044】
アルキルは、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、tert−ブチル、n−ペンチル、イソペンチル、ネオペンチル、又はヘキシルなどの、1〜6個の炭素原子を有する直鎖又は分岐状のアルキルである。
【0045】
アルケニルは、ビニル、アリル、1−プロペニル、2−ブテニル、2−メチル−2−ブテニル、イソプロペニル、ペンテニル、又はヘキセニルなどの、2〜6個の炭素原子を有し、少なくとも1つの炭素−炭素二重結合を含有する直鎖又は分岐状のアルキルである。
【0046】
アルキニルは、エチニル、1−プロピニル、1−ブチニル、2−ブチニル、ペンチニル又はヘキシニルなどの、2〜6個の炭素原子を有し、少なくとも1つの炭素−炭素三重結合を含有する直鎖又は分岐状のアルキルである。
【0047】
0〜2個の窒素原子を含有する3〜7員の飽和、不飽和又は芳香族環は、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル又はシクロヘプチルなどの、3〜7員の飽和炭素環を包含する。前記用語は、シクロブテニル、シクロペンテニル、シクロヘキセニル、シクロヘプテニル、シクロヘキサ−1,4−ジエン若しくはシクロヘプタジエン、又はベンゼンなどの芳香環などの、3〜7員の不飽和炭素環をさらに包含する。窒素含有3〜7員の飽和、不飽和又は芳香族の複素環が、上記の用語にさらに包含される。この例には、アゼチジン、ピロリジン、ピペリジン、アゼパン、ピペラジン、ピリジン、ピリミジン、ピラジン、ピロール、イミダゾール、及びピラゾールが挙げられる。
【0048】
構造式(I)の化合物は、メラノコルチン受容体モジュレーターとして有効であり、MC−4Rの選択的モジュレーターとして特に有効である。それらはしたがって、癌悪液質、筋肉疲労、食欲不振症、筋萎縮性側索硬化症、不安神経症、うつ、肥満症、糖尿病、性的機能不全、及びMC−4Rの関与を伴う他の疾患などのMC−4Rの活性化及び不活性化に対して応答性の障害の治療及び/又は予防のために有用である。
【0049】
構造式(I)の化合物は、MC−4Rのアンタゴニストとして特に有用である。したがって、それらは、好ましくは癌悪液質、筋肉疲労、食欲不振症、筋萎縮性側索硬化症、不安神経症及びうつの治療及び/又は予防のための医薬品の調製のために使用される。
【0050】
光学異性体−ジアステレオマー−幾何異性体−互変異性体
構造式(I)の化合物は、1つ又は複数の不斉中心を含有し、ラセミ化合物及びラセミ混合物、単一のエナンチオマー、ジアステレオマー混合物及び個々のジアステレオマーとして生じる場合がある。本発明は、構造式(I)の化合物の全てのこのような異性体形態を包含することを意味する。
【0051】
構造式(I)の化合物は、例えば、適切な溶媒、例えば、メタノール又は酢酸エチル又はこれらの混合物からの分別結晶によって、或いは光学活性な固定相を使用したキラルクロマトグラフィーを介して、これらの個々のジアステレオマーに分離することができる。絶対立体配置は、公知の絶対配置の不斉中心を含有する試薬で必要に応じて誘導体化した結晶性生成物又は結晶性中間体のX線結晶構造解析によって決定することができる。
【0052】
代わりに、一般式(I)の化合物の任意の立体異性体は、光学的に純粋な出発物質又は公知の絶対配置の試薬を使用して立体特異的合成によって得ることができる。
【0053】
塩
「薬学的に許容される塩」という用語は、無機塩基又は有機塩基及び無機酸又は有機酸を含めた、薬学的に許容される無毒性塩基又は酸から調製される塩を意味する。無機塩基に由来する塩には、アルミニウム、アンモニウム、カルシウム、銅、第二鉄、第一鉄、リチウム、マグネシウム、マンガン塩、マンガン(II)、カリウム、ナトリウム、亜鉛などが挙げられる。特に好ましい塩は、アンモニウム、カルシウム、リチウム、マグネシウム、カリウム及びナトリウム塩である。薬学的に許容される無毒有機の塩基に由来する塩には、第一級、第二級及び第三級アミン、天然の置換アミンを含めた置換アミン、環状アミン及び塩基性イオン交換樹脂の塩(アルギニン、ベタイン、カフェイン、コリン、N,N’−ジベンジルエチレンジアミン、ジエチルアミン、2−ジエチルアミノエタノール、2−ジメチルアミノ−エタノール、エタノールアミン、エチレンジアミン、N−エチルモルホリン、N−エチルピペリジン、グルカミン、グルコサミン、ヒスチジン、ヒドラバミン、イソプロピルアミン、リシン、メチルグルカミン、モルホリン、ピペラジン、ピペリジン、ポリアミン樹脂、プロカイン、プリン、テオブロミン、トリエチルアミン、トリメチルアミン、トリプロピルアミン、トロメタミンなど)が挙げられる。
【0054】
本発明の化合物が塩基性である場合、塩は、無機酸及び有機酸を含めた薬学的に許容される無毒性酸から調製することができる。このような酸には、酢酸、ベンゼンスルホン酸、安息香酸、カンファースルホン酸、クエン酸、エタンスルホン酸、ギ酸、フマル酸、グルコン酸、グルタミン酸、臭化水素酸、塩酸、イセチオン酸、乳酸、マレイン酸、リンゴ酸、マンデル酸、メタンスルホン酸、マロン酸、粘液酸、硝酸、パモン酸、パントテン酸、リン酸、プロピオン酸、コハク酸、硫酸、酒石酸、p−トルエンスルホン酸、トリフルオロ酢酸などが挙げられる。特に好ましい酸は、クエン酸、フマル酸、臭化水素酸、塩酸、マレイン酸、リン酸、硫酸及び酒石酸である。
【0055】
本明細書で使用する場合、式(I)の化合物への適用は、薬学的に許容される塩もまた含むことを意味することが理解されるであろう。
【0056】
有用性
式(I)の化合物は、メラノコルチン受容体モジュレーターであり、したがって、それだけに限らないが、MC−1R、MC−2R、MC−3R、MC−4R又はMC−5Rが挙げられるメラノコルチン受容体の1つ又は複数の不活性化に対して応答性の疾患、障害又は状態の治療、制御又は予防において有用である。このような疾患、障害又は状態には、それだけに限らないが、癌悪液質、筋肉疲労、食欲不振症、不安神経症、うつ、肥満症(食欲を減少させ、代謝率を増加させ、脂肪摂取を減少させ、又は炭水化物への欲求を減少させることによって)、真性糖尿病(耐糖能を増強させ、インスリン抵抗性を減少させることによって)、並びに男性及び女性の性的機能不全(インポテンス、性的衝動の喪失及び勃起不全を含めた)が挙げられる。
【0057】
式(I)の化合物は、高血圧症、高脂血症、骨関節炎、癌、胆嚢疾患、睡眠時無呼吸、強迫行為、ノイローゼ、不眠/睡眠障害、薬物乱用、疼痛、発熱、炎症、免疫修飾、関節リウマチ、皮膚日焼け、ざ瘡及び他の皮膚障害;アルツハイマー病の治療を含めた神経保護及び認識及び記憶増強の治療、制御又は予防においてさらに使用することができる。
【0058】
投与及び用量範囲
任意の適切な投与経路が、哺乳動物、特にヒトに、有効量の本発明の化合物を提供するために用いることができる。例えば、経口、直腸、局所、非経口、眼球、肺、経鼻などを用いることができる。剤形には、錠剤、トローチ剤、分散剤、懸濁剤、溶液剤、カプセル剤、クリーム剤、軟膏、エアゾール剤などが挙げられる。好ましくは、式(I)の化合物は、経口又は局所で投与される。
【0059】
用いられる有効量の活性成分は、用いられる特定化合物、投与方法、治療される状態及び治療される状態の重篤度によって変化することがある。このような投与量は、当業者によって容易に確認することができる。
【0060】
癌悪液質、筋肉疲労、筋萎縮性側索硬化症又は食欲不振症を治療する場合、本発明の化合物が、約0.001ミリグラム〜約100ミリグラム/体重キログラムの1日投与量で投与されると(好ましくは単回用量、若しくは1日2〜6回の分割用量で、又は持続放出形態で与えられる)、一般に満足のいく結果が得られる。70kgの成人したヒトの場合、総1日用量は一般に、約0.07ミリグラム〜約3500ミリグラムであろう。この投与計画は、最適な治療反応を得るために調節してもよい。
【0061】
糖尿病及び/若しくは高血糖症と合わせて、又は単独で肥満症を治療する場合、本発明の化合物が、約0.001ミリグラム〜約100ミリグラム/体重キログラムの1日投与量で投与されると(好ましくは単回用量、若しくは1日2〜6回の分割用量で、又は持続放出形態で与えられる)、一般に満足のいく結果が得られる。70kgの成人したヒトの場合、総1日用量は一般に、約0.07ミリグラム〜約3500ミリグラムであろう。この投与計画は、最適な治療反応を得るために調節してもよい。
【0062】
真性糖尿病及び/又は高血糖症、並びに式(I)の化合物が有用な他の疾患又は障害を治療する場合、本発明の化合物が、約0.001ミリグラム〜約100ミリグラム/動物体重キログラムの1日投与量で投与される場合(好ましくは単回用量、若しくは1日2〜6回の分割用量で、又は持続放出形態で与えられる)、一般に満足のいく結果が得られる。70kgの成人したヒトの場合、総1日用量は一般に、約0.07ミリグラム〜約3500ミリグラムであろう。この投与計画、最適な治療反応を得るために調節してもよい。
【0063】
性的機能不全の治療のために、本発明の化合物は、0.001ミリグラム〜約100ミリグラム/体重キログラムの範囲の1用量単位で、好ましくは経口の単回用量又は鼻用スプレーとして与えられる。
【0064】
製剤
式(I)の化合物は、好ましくは投与の前に剤形に製剤される。したがって、本発明はまた、式(I)の化合物と適切な医薬担体とを含む医薬組成物を含む。
【0065】
本発明の医薬組成物は、周知の容易に利用できる成分を使用して、公知の手順によって調製される。本発明の製剤の作製において、活性成分(式(I)の化合物)は通常、担体と混合され、又は担体によって希釈され、又は担体中に封入されるが、これは、カプセル剤、サシェ剤、紙又は他の容器の形態中のことがある。担体が希釈剤としての機能を果たす場合、それは、活性成分のためのビヒクル、賦形剤又は媒質として作用する固体、半固体又は液体物質であることがある。したがって、組成物は、錠剤、丸剤、散剤、ロゼンジ、サシェ剤、カシェ剤、エリキシル剤、懸濁剤、乳剤、溶液剤、シロップ剤、エアロゾル剤(固体として又は液体媒体中)、軟質及び硬質ゼラチンカプセル剤、坐薬、無菌注射剤、並びに包装された無菌散剤の形態の場合がある。
【0066】
適切な担体、賦形剤及び希釈剤のいくつかの例には、ラクトース、デキストロース、スクロース、ソルビトール、マンニトール、デンプン、アラビアゴム、リン酸カルシウム、アルギネート、トラガカント、ゼラチン、ケイ酸カルシウム、微結晶性セルロース、ポリビニルピロリドン、セルロース、水、シロップ、メチルセルロース、メチル及びプロピルヒドロキシベンゾエート、タルク、ステアリン酸マグネシウム及び鉱油が挙げられる。製剤は、滑沢剤、湿潤剤、乳化剤及び懸濁化剤、保存料、甘味剤又は香味剤をさらに含むことができる。本発明の組成物は、患者に投与した後、活性成分の即時放出、持続放出又は遅延放出を実現するように製剤されることがある。
【0067】
本発明の化合物の調製
本発明の式(I)の化合物の調製を説明するとき、「A部分」、「B部分」及び「C部分」という用語が下記で使用される。この部分という概念を下記で例示する。
【化22】
【0068】
本発明の化合物の調製は、順次又は収束的合成経路によって行ってもよい。当業者であれば、一般に、式(I)の化合物のA及びB部分は、アミド結合を介して結合していることを理解するであろう。当業者はしたがって、標準的なペプチドカップリング反応条件を介して2つの部分を結合する多数の経路及び方法を容易に予見することができる。
【0069】
「標準的なペプチドカップリング反応条件」という表現は、HOBtなどの触媒の存在下で、DCMなどの不活性溶媒中、EDCI、ジシクロヘキシルカルボジイミド及びベンゾトリアゾール−1−イルオキシトリス(ジメチルアミノ)−ホスホニウムヘキサフルオロホスフェートなどの酸活性化剤を使用した、カルボン酸とアミンとのカップリングを意味する。所望の反応を促進し、望ましくない反応を最小化するための、アミン及びカルボン酸についての保護基の使用については、これまでにもたくさん記述されている。存在し得る保護基の除去に必要な条件は、Greeneら、Protective Groups in Organic Synthesis、John Wiley & Sons、Inc.、New York、NY 1991において見出すことができる。
【0070】
Z、Boc及びFmocなどの保護基は、合成において広範に使用されており、それらの除去条件は当業者には周知である。例えば、Z基の除去は、エタノールなどのプロトン性溶媒中、活性炭担持パラジウムなどの貴金属又はその酸化物の存在下で、水素による接触水素化によって行うことができる。他の潜在的に反応性の官能基の存在によって接触水素化が禁忌である場合、Zの除去はまた、臭化水素の酢酸溶液による処理によって、又はTFAと硫化ジメチルとの混合物による処理によって行うことができる。Boc保護基の除去は、塩化メチレン、メタノール又は酢酸エチルなどの溶媒中、TFA又はHCl又は塩化水素ガスなどの強酸によって行われる。
【0071】
式(I)の化合物のB及びC部分は、尿素官能基を介して共に結合している。したがって、当業者であれば、異なる周知の方法を使用した2つの部分を結合する多数の経路及び方法を容易に想定することができる。
【0072】
式(I)の化合物は、ジアステレオマー混合物として存在する場合、メタノール、酢酸エチル又はこれらの混合物などの適切な溶媒からの分別結晶によって、エナンチオマーのジアステレオマー対に分離することができる。このようにして得られたエナンチオマー対は、光学活性な酸を分割剤として使用することにより従来の手段によって個々の立体異性体に分離することができる。代わりに、式(I)の化合物の任意のエナンチオマーは、光学的に純粋な出発物質又は公知の立体配置の試薬を使用して立体特異的合成によって得ることができる。
【0073】
本発明の式(I)の化合物は、適切な材料を使用して下記のスキーム及び実施例の手順によって調製することができ、下記の具体例によってさらに例示される。さらに、当技術分野で通常の技術と併せて本明細書に記載する手順を用いることによって、ここで特許請求されている本発明のさらなる化合物を、容易に調製することができる。しかし、実施例において例示した化合物は、本発明として考えられる唯一の種類を形成するものとして解釈されない。実施例は、本発明の化合物の調製についての詳細をさらに例示する。下記の調製手順の条件及び方法の公知の変形形態を、これらの化合物を調製するために使用することができることは当業者であれば容易に理解するであろう。本化合物は一般に、従前に記載したものなどの薬学的に許容されるそれらの塩の形態で単離される。単離した塩に相当する遊離アミン塩基は、水性炭酸水素ナトリウム、炭酸ナトリウム、水酸化ナトリウム及び水酸化カリウムなどの適切な塩基による中和、及び遊離したアミン遊離塩基の有機溶媒への抽出、それに続くエバポレーション(evaporation)によって生成することができる。この方法で単離されたアミン遊離塩基は、有機溶媒への溶解、続いて適切な酸の添加、それに続くエバポレーション、沈殿又は結晶化によって他の薬学的に許容される塩にさらに変換することができる。全ての温度は、摂氏温度である。
【0074】
下記のスキーム、調製及び実施例において、様々な試薬の記号及び略語は、下記の意味を有する。
Ac2O 無水酢酸
AcOH 酢酸
Boc tert−ブトキシカルボニル
Bu ブチル
BuLi ブチルリチウム
DBU 1,8−ジアザビシクロ[5.4.0]ウンデス−7−エン
DCC N,N’−ジシクロヘキシルカルボジイミド
DCE 1,2−ジクロロエタン
DCM ジクロロメタン
DEAD アゾジカルボン酸ジエチル
DIAD アゾジカルボン酸ジイソプロピル
DIC N,N’−ジイソプロピルカルボジイミド
DIEA エチル−ジイソプロピルアミン
DMA N,N−ジメチルアセトアミド
DMAP 4−ジメチルアミノピリジン
DMF N,N−ジメチルホルムアミド
DMS 硫化ジメチル
DMSO ジメチルスルホキシド
dppf 1,1’−ビス(ジフェニルホスフィノ)−フェロセン
EDCI 1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド塩酸塩
Et2O ジエチルエーテル
EtOAc 酢酸エチル
EtOH エタノール
Fmoc 9−フルオレニルメチルオキシカルボニル
h 時間(複数可)
HOBt 1−ヒドロキシベンゾトリアゾール水和物
HTMP 2,2,6,6−テトラメチルピペリジン
MeCN アセトニトリル
MeOH メタノール
mp. 融点
NMM N−メチルモルホリン
PG 保護基
PPh3 トリフェニルホスフィン
RT 室温
TEA トリエチルアミン
TFA トリフルオロ酢酸
THF テトラヒドロフラン
Z ベンジルオキシカルボニル
Z−OSu N−(ベンジルオキシカルボニルオキシ)スクシンイミド
【0075】
下記のアミノ酸誘導体は、PepTech Corporation(20 Mall Road、Suite 460、Burlington、MA 01803、USA)によってカスタム合成された(D−2−クロロ−4−フルオロフェニルアラニンメチルエステル塩酸塩、D−4−クロロ−2−フルオロフェニルアラニンメチルエステル塩酸塩、及びD−2,4−ジフルオロ−フェニルアラニンメチルエステル塩酸塩)。D−2−クロロ−4−メチルフェニルアラニンメチルエステル塩酸塩及びD−4−クロロ−2−メチルフェニルアラニンメチルエステル塩酸塩は、NetChem、Inc.(100 Jersey Ave、Suite A211、New Brunswick、NJ 08901、USA)から得た。
【0076】
cis−3−アザ−ビシクロ[3.1.0]ヘキサン塩酸塩は、米国特許第4,183,857号明細書において記載されているように調製した。2−フルオロ−3−ヨード−ピラジンは、Tetrahedron1998、54、4899〜4912において記載されているように調製した。4−フルオロ−3−ヨード−ピリジンは、Tetrahedron1993、49、49〜64において記載されているように調製した。
【0077】
反応スキーム1:
配向性オルト−メタル化
【化23】
ヘテロアリールピペリジン、オルト−フルオロ−ヨードピリジン、−ピリダジン及び−ピラジンの合成のための出発物質は、反応スキーム1において示されているように調製することができる。フルオロ置換ヘテロアリールは、適切な温度にてTHFなどの適切な溶媒中、n−ブチルリチウム及びアミン(ジイソプロピルアミンなど)、又は2,2,6,6−テトラメチルピペリジンなどの試薬から調製されるアミドでメタル化することができる。このように得られたリチオ誘導体は、引き続いてヨウ素と反応し、所望の化合物を得ることができる。
【0078】
反応スキーム2:
アセチル化
【化24】
ヘテロアリールピペリジン、オルト−アセトキシ−ブロモピリジン、−ピリダジン及び−ピラジンの合成のための他の出発物質は、反応スキーム2において示されているように調製することができる。ブロモ原子に対してオルト位においてヒドロキシ基を含有するブロモ又はクロロヘテロアリールは、ピリジンなどの適切な塩基の存在下で、DCMなどの適切な溶媒中で適切な温度にて、無水酢酸などのアセチル化試薬と反応することができる。
【0079】
反応スキーム3:
ブロモ又はヨード置換ヘテロアリールの根岸カップリング反応
【化25】
【化26】
反応スキーム3において示されているように、オルト−フルオロ−ブロモピリジン、ピリダジン及びピラジンは、ヨウ化銅(I)及びジクロロ(1,1’−ビス(ジフェニル−ホスフィノ)−フェロセン)パラジウム(II)DCM付加体の存在下で、DMAなどの不活性溶媒中、(1−tert−ブトキシカルボニルピペリジン−4−イル)(ヨード)亜鉛との根岸カップリングに供され(J.Org.Chem.2004、69、5120〜5123)、その結果として生じるアリールピペリジンを得ることができる。オルト−フルオロ−ヨードピリジン、−ピリダジン及び−ピラジンが、出発物質として使用される場合、同じ生成物が得られる。
【0080】
代わりに、根岸カップリングは、反応スキーム2からのオルト−アセトキシ置換ブロモピリジン、ピリダジン及びピラジンを出発物質として使用して行うことができる。遊離アルコールは、水とメタノールとの混合物などの適切な溶媒中、水酸化リチウムなどの塩基による酢酸エステルのけん化によって得られる。オルト−アセトキシ−クロロピリジン、−ピリダジン及び−ピラジンの出発物質としての使用は、同じ生成物の形成をもたらす。
【0081】
反応スキーム4:
ヘテロアリールとカルボキシアルデヒド又はベンジルアミン官能基との根岸カップリング反応
【化27】
任意選択で置換されているオルト−カルボキシ−ブロモピリジン、ピリダジン及び−ピラジンは、反応スキーム4において示されているように根岸カップリングに供することができる。ヨウ化銅(I)及びジクロロ(1,1’−ビス(ジフェニル−ホスフィノ)−フェロセン)パラジウム(II)DCM付加体の存在下で、DMAなどの不活性溶媒中、(1−tert−ブトキシカルボニルピペリジン−4−イル)(ヨード)亜鉛との反応によって、このように得られたアリールピペリジンがもたらされる。トリアセトキシ水素化ホウ素ナトリウムなどの還元剤の存在下で、1,2−ジクロロエタンなどの適切な溶媒中、キャッピング基T−Hによる還元的アミノ化によって、Boc保護されたA部分がもたらされる。
【0082】
代わりに、任意選択で置換されているオルト−カルボキシ−ブロモピリジン、ピリダジン及びピラジンは、根岸カップリングが行われる前に、還元的アミノ化ステップに最初に供することができる。
【0083】
上記の反応順序はまた、任意選択で置換されているオルト−カルボキシ−クロロピリジン、ピリダジン及び−ピラジンを使用して行うことができる。
【0084】
反応スキーム5:
アミノアルコールの調製
【化28】
N−置換アミノアルコールHO(C(R6)2)m−Tは、反応スキーム5において記載されているように得ることができる。所定の温度にて水などの適切な溶媒中、任意選択で置換されているアミノアルコールHO(C(R6)2)mNH2と、ギ酸及びホルムアルデヒドの混合物との反応によって、相当するN,N−ジメチル化アミノアルコールの形成がもたらされる。このようなアミノアルコールの環状類似体は、炭酸カリウムなどの塩基の存在下で、アセトニトリルなどの適切な溶媒中、任意選択で置換されているアミノアルコールHO(C(R6)2)mNH2と、α,ω−ジブロモアルカンとの反応によって得ることができる。
【0085】
反応スキーム6:
アミノアルコール前駆体としてのエポキシド
【化29】
反応スキーム6において示されているように、任意選択で置換されているエポキシドは、位置選択的方法で、水などの適切な溶媒中で適切なアミンT−Hと反応し、α−置換β−アミノアルコールを形成することができる。
【0086】
反応スキーム7:
求核芳香族置換反応
【化30】
反応スキーム7において示されているように、フルオロ置換ピリジル、ピリダジル及びピラジニルピペリジンは、水素化ナトリウムなどの塩基の存在下で、DMFなどの溶媒中で適切な温度にて、ω−T−キャップされたアルキルアルコールとの求核芳香族置換反応に供され、Boc保護されたA部分を得ることができる。
【0087】
代わりに、フルオロ置換ヘテロアリールピペリジンはまた、BuLi又はDIEAなどの塩基の存在下で、THFなどの適切な溶媒中又は溶媒を伴わず適切な温度にて、ω−T−キャップされた第一級又は第二級アルキルアミンと反応して、Boc保護されたA部分を得ることができる。
【0088】
反応スキーム8
環状アミンを含有するA部分の合成
【化31】
反応スキーム8において示されているように、反応スキーム3からの中間生成物である任意選択で置換されているフルオロピリジン、ピリダジン及びピラジンはまた、水素化ナトリウムなどの塩基の存在下で、DMFなどの適切な溶媒中、環状第三級アミン部分を含有するアルコールとの求核芳香族置換反応に供され、Boc保護されたA部分を得ることができる。
【0089】
同様に、保護された環状第二級アミン部分を含有するアルコールを、上記の条件を使用して構造単位として導入することができる。保護基は、ピペリジンの保護のために使用されるBoc−保護基に対して直交しなくてはならない。A部分とB−C部分とのカップリングの後に、この保護基は、標準的な方法を用いて除去することができる。
【0090】
反応スキーム9:
アルキルエーテルスペーサー(R1=−O(C(R6)2)m−T)によるA部分の代替合成
【化32】
アルキルエーテルスペーサー(R1=−O(C(R6)2)m−T)を担持するA部分の合成は、代わりに反応スキーム9において示されているように行うことができる。Boc保護されたピペリジンは、Cs2CO3又はNaHなどの塩基の存在下で、DMFなどの適切な溶媒中、キャッピング基Tを担持する塩化アルキル又は臭化アルキルと反応し、Boc保護されたA部分が生じる。
【0091】
反応スキーム10:
光延条件を使用したアルキルエーテルスペーサー(R1=−O(C(R6)2)m−T)によるA部分の合成
【化33】
反応スキーム10において示されているように、反応スキーム3からの中間生成物である任意選択で置換されている(ヒドロキシヘテロアリール)ピペリジンはまた、DEAD又はDIADなどの試薬、及びPPh3などのホスフィンの存在下で、THFなどの適切な溶媒中、ω−T−キャップされたアルキルアルコールでアルキル化し、Boc保護されたA部分を得ることができる。
【0092】
同様に、同じ中間体は、上記の反応条件を使用してω−ブロモアルキルアルコールと反応させて、相当するエーテルに接近することができ、それを引き続いてK2CO3又はNaHなどの適切な塩基の存在下で、MeCN、THF、又はDMFなどの適切な溶媒中、適切な温度にて、キャッピング基Tをアルキル化するために使用し、Boc保護されたA部分をもたらすことができる。
【0093】
反応スキーム11:
5−ピペリジン−4−イル−ピリミジン由来A部分の合成
【化34】
5−ピペリジン−4−イル−ピリミジン由来A部分は、反応スキーム11において示されているように合成することができる。Boc−4−ピペリドンは、四塩化チタンなどの試薬及びピリジンなどの塩基の存在下、適切な温度にて、マロン酸ジエステルと反応することができる。この反応の生成物は、水及びメタノールの混合物などの適切な溶媒中、10%炭担持パラジウムなどの触媒を使用して水素化し、相当するBoc−2−ピペリジン−4−イル−マロン酸ジエステルを得ることができる。ナトリウムメトキシドなどの塩基の存在下で、メタノールなどの溶媒中でのアミジン塩酸塩との反応によって、任意選択で置換されている1H−ピリミジン−4,6−ジオンがもたらされ、それはBoc保護基をZ保護基と交換した後、POCl3などの試薬を使用して、sym−コリジンなどの適切な塩基の存在下で、アセトニトリルなどの適切な溶媒中、4,6−ジクロロ−ピリミジンに変換することができる。任意選択で置換されている4,6−ジクロロ−ピリミジンは、水素化ナトリウムなどの塩基の存在下で、THFなどの適切な溶媒中、ω−T−キャップされたアルキルアルコールと反応することができる。それに続く水素化は、酸化カルシウムの存在下で、ジエチルエーテルなどの適切な溶媒中、10%炭担持パラジウムなどの触媒を使用して行われ、5−ピペリジン−4−イル−ピリミジン由来A部分がもたらされる。
【0094】
反応スキーム12
A部分の脱保護
【化35】
一般に、Boc保護されたヘテロアリールピペリジン(A部分)の出発物質は、カップリング手順に供する前に、TFA/CH2Cl2、HCl/EtOAc、HCl/ジオキサン又はMeOH/ジオキサン中のHClの存在下で、硫化ジメチル(DMS)などのカチオンスカベンジャーと共に又は伴わず、脱保護することができる。それはカップリング手順に供する前に遊離塩基に変換することができ、又はある場合には塩として使用することができる。
【0095】
反応スキーム13
グリニャール反応又はルパート試薬を使用したアミンH−R5の合成
【化36】
反応スキーム13は、同じ炭素原子においてヒドロキシ置換基と1つのさらなる置換基とを担持するアミンH−R5の合成を示す。このようなアミンは、適切な保護基で保護されているオキソ置換アミンを使用して合成することができる。適切な保護基には、これらだけに限らないが、Boc、Z、ベンジル、及びベンズヒドリルが挙げられる。保護されたオキソ置換アミンは、THF又はジエチルエーテルなどの不活性溶媒中で適切な温度にてグリニャール試薬と反応して、相当する置換されているアルコールを得ることができる。
【0096】
代わりに、フッ化セシウムなどの試薬の存在下で、THFなどの適切な溶媒中で所定の温度にて、保護されたオキソ置換アミンとルパート試薬(トリフルオロメチルトリメチルシラン)との反応によって、トリフルオロメチル化したアルコールがもたらされる。
【0097】
上記の合成法によって得た保護されたアミンH−R5の保護基は、標準条件を使用して除去することができる。
【0098】
反応スキーム14:
B−C部分の形成
【化37】
B−C部分は、反応スキーム14において示されているように合成することができる。任意選択で置換されているフェニルアラニンは、メタノール中で塩化チオニル又は塩化オキサリルなどの活性化剤を使用して、相当するメチルエステル塩酸塩に変換することができる。アミノ酸メチルエステル塩酸塩は、NaHCO3(水溶液)などの塩基の存在下で、DCMなどの適切な溶媒中、トリホスゲンなどの試薬と反応して、イソシアネートを得ることができ、これは引き続いてDCM又はDMFなどの適切な溶媒中でアミンR5−Hと反応することができる。R5−Hが塩酸塩の形態で使用される場合、DIEAなどの適切な塩基をさらに使用し、遊離アミンR5−Hを遊離する。エステル官能基は、適切な溶媒又は溶媒混合物(水/THF/メタノールなど)中でLiOHなどの塩基で加水分解し、B−C−部分に接近することができる。
【0099】
反応スキーム15:
B−C部分の固相合成
【化38】
代わりに、B−C−部分はまた、反応スキーム15において示されているように固相上で合成することができる。ワング樹脂は、DMAPなどの塩基の存在下で、DMF又はDCMなどの適切な溶媒中、DIC又はDCCなどの試薬を使用して、任意選択で置換されているFmoc−保護されたフェニルアラニンと共に充填することができる。固体支持体上の未反応のOH−基のキャッピングは、DMF又はDCMなどの適切な溶媒中、無水酢酸とのそれに続く反応によって行うことができる。DCMなどの溶媒中でピペリジン又はジエチルアミンなどの塩基によるFmoc−保護基の除去後、遊離アミンは、TEAなどの塩基の存在下で、DCMなどの適切な溶媒中、p−ニトロフェニルクロロホルメートによって活性化したカルバメートに変換することができる。DCMなどの適切な溶媒中での前記p−ニトロフェニルカルバメートのアミンR5−Hとの反応によって、所望のB−C部分が生じる。R5−Hが塩酸塩の形態で使用される場合、DIEAなどの適切な塩基がさらに使用され、遊離アミンR5−Hが遊離される。固体支持体からの切断は、DCM中のTFAによる処理によって行うことができる。この経路によって得られるB−C−部分は、精製又は適切なA部分と直接カップリングすることができる。
【0100】
反応スキーム16:
A部分のB−C部分とのカップリング及び塩の形成
【化39】
反応スキーム16において示されているように、A部分は、EDCI/HOBt、塩基(N−メチルモルホリン(NMM)など)及び溶媒(ジクロロメタン(DCM)など)の存在下で、B−C部分とカップリングすることができる。DCM、DMF、THFなどの適切な溶媒、又は上記の溶媒の混合物を、カップリング手順のために使用することができる。適切な塩基には、トリエチルアミン(TEA)、ジイソプロピルエチルアミン(DIEA)、N−メチルモルホリン(NMM)、コリジン又は2,6−ルチジンが挙げられる。EDCI/HOBtが使用される場合、塩基は必要ないことがある。
【0101】
一般に、反応が完了した後、反応混合物はEtOAc、DCM又はEt2Oなどの適切な有機溶媒で希釈することができ、次いで水、HCl、NaHSO4、炭酸水素塩、NaH2PO4、リン酸緩衝液(pH7)、ブライン又は任意のこれらの組合せなどの水溶液で洗浄する。反応混合物を濃縮し、次いで適切な有機溶媒及び水溶液に分配することができる。反応混合物を濃縮し、水性後処理なしでクロマトグラフィーにかけることができる。
【0102】
溶媒又は溶媒混合物(ジエチルエーテル/アセトンなど)中のHClを使用して、生成物を塩酸塩などの薬学的に許容される塩に転移させることができる。
【0103】
反応スキーム17:
ニトロフェニルホルメート中間体を介した尿素形成
【化40】
3つの部分はまた、反応スキーム17において示されているように段階的に結合させることができる。適切なA部分は、EDCI/HOBt、塩基(N−メチルモルホリン(NMM)など)及び溶媒(ジクロロメタン(DCM)など)の存在下でBoc保護されたB部分とカップリングし、続いてジオキサンとメタノールとの混合物中で、塩化水素の補助によるBoc脱保護が起こる。生成物は、NMMなどの塩基の存在下でDCMなどの適切な溶媒中、4−ニトロフェニルクロロホルメートと反応し、4−ニトロフェニルカルバメートを生じさせることができ、それは引き続いてDIEAなどの塩基の存在下でTHFなどの適切な溶媒中、アミンH−R5で処理し、目的化合物に接近することができる。最終生成物は、上記のような薬学的に許容される塩に変換することができる。
【0104】
反応スキーム18:
試薬として1−メチル−3−(アミノ−1−カルボニル)−3H−イミダゾール−1−イウムヨージドを使用した尿素形成
【化41】
反応スキーム18において示されているように、THFなどの適切な溶媒中で適切な温度にて、1,1’−カルボニルジイミダゾールは、アミンと反応することができる。この反応の生成物は、アセトニトリルなどの適切な溶媒中、ヨウ化メチルとさらに反応し、1−メチル−3−(アミノ−1−カルボニル)−3H−イミダゾール−1−イウムヨージドがもたらされる。この活性化種は、トリエチルアミンなどの塩基の存在下で、THFなどの適切な溶媒中、脱保護されたA−B部分と反応し、最終生成物がもたらされる。最終生成物は、上記のような薬学的に許容される塩に変換することができる。
【0105】
分析的LC−MS
式(I)による本発明の化合物を、分析的LC−MSによって分析した。分析において使用した条件を、下記に要約する。
【0106】
分析の条件の要約:
SPD−M10Avp UV/Visダイオードアレイ検出器及びQP2010 MS−検出器(ESI+モード、214及び254nmでのUV−検出)を備えたLC10Advp−Pump(島津製作所)、
カラム:Waters XTerra MS C18、3.5μm、2.1*100mm、
線形グラジェント、水中のアセトニトリル(0.15%HCOOH)
0.4ml/分の流速;
移動相A:水(0.15%HCOOH+5%アセトニトリル)
移動相B:アセトニトリル(0.15%HCOOH+5%水)
グラジェントA:
開始濃度、1%アセトニトリル(0.15%HCOOH)
9.00分、B.濃度 30
10.00分、B.曲線 3
12.00分、B.濃度 99
15.00分、B.濃度 99
15.20分、B.濃度 1
18.00分、ポンプ 停止
グラジェントB:
開始濃度、10%アセトニトリル(0.15%HCOOH)
10.00分、B.濃度 60
11.00分、B.曲線 2
12.00分、B.濃度 99
15.00分、B.濃度 99
15.20分、B.濃度 10
18.00分、ポンプ 停止
グラジェントC:
線形グラジェント、水中の5%〜95%アセトニトリル(0.15%HCOOH)
0.00分、5%B
5.00分、95%B
5.10分、99%B
6.40分、99%B
6.50分、5%B
8.00分、ポンプ停止
グラジェントD:
線形グラジェント、水中の5%〜80%アセトニトリル(0.15%HCOOH)
0.00分、5%B
5.00分、80%B
5.10分、99%B
6.40分、99%B
6.50分、5%B
8.00分、ポンプ停止
グラジェントE:
線形グラジェント、水中の1%〜70%アセトニトリル(0.15%HCOOH)
0.00分、1%B
5.00分、70%B
5.10分、99%B
6.40分、99%B
6.50分、5%B
8.00分、ポンプ停止
【0107】
下記の表は、反応スキーム1〜18によって調製することができる本発明の詳細な実施例を記載する。しかし、これらの実施例は、いかなる形でも本発明の範囲を限定すると解釈されない。
【0108】
【化42】
【表1】
【表2】
【0109】
【化43】
【表3】
【0110】
【化44】
【表4】
【0111】
【化45】
【表5】
【表6】
【表7】
【表8】
【表9】
【表10】
【0112】
【化46】
【表11】
【0113】
【化47】
【表12】
【0114】
【化48】
【表13】
【0115】
【化49】
【表14】
【0116】
【化50】
【表15】
【0117】
【化51】
【表16】
【0118】
【化52】
【表17】
【表18】
【0119】
【化53】
【表19】
【表20】
【表21】
【0120】
【化54】
【表22】
【0121】
【化55】
【表23】
【0122】
【化56】
【表24】
【0123】
下記の実施例は、本発明を例示するために提示し、本発明の範囲をいかなる形でも限定しない。
【0124】
一般の中間体:
1−tert−ブトキシカルボニルピペリジン−4−イル)(ヨード)亜鉛:
【化57】
亜鉛の活性化
シュレンクフラスコにセライト(1.28g)を入れ、真空中で加熱することによって乾燥させた。次いで、亜鉛粉末(6.51g)及び乾燥N,N−ジメチルアセトアミド(15ml)を加えた。クロロトリメチルシラン(1.14ml)と1,2−ジブロモエタン(0.80ml)との7:5v/v混合物をN,N−ジメチルアセトアミド(1ml)中の溶液として、温度を65℃未満に維持する速度で(約15分)加えている間に、混合物を室温で撹拌した。このように得られたスラリーを15分間熟成させた。
【0125】
亜鉛の挿入
Boc−4−ヨードピペリジン(24.8g)のN,N−ジメチルアセトアミド(39ml)溶液を、温度を65℃未満に維持する速度で(約20分)、上記の混合物にゆっくり加えた。次いで、このように得られた反応混合物を室温で30分間熟成させた。懸濁液をアルゴン下フリットで濾過し、全ての固体を除去した。このように得られた淡黄色の溶液を、アルゴン下室温で保存し、カップリング反応のために直接使用した。
【0126】
3−フルオロ−3’,4’,5’,6’−テトラヒドロ−2’H−[2,4’]ビピリジニル−1’−カルボン酸tert−ブチルエステル:
【化58】
500mlのフラスコに、2−ブロモ−3−フルオロピリジン(10.0g)、[1,1’−ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)のジクロロメタンとの錯体(1.39g)、ヨウ化銅(I)(0.65g)、及び乾燥N,N−ジメチルアセトアミド(80ml)を入れた。このように得られた混合物を交互の真空/アルゴンパージで脱気した。次いで、1−tert−ブトキシカルボニルピペリジン−4−イル)(ヨード)亜鉛(79.7mmol)を加えた。混合物をもう一度脱気し、次いで80℃に一晩加熱した。次いで、N,N−ジメチルアセトアミドの主要部分を蒸発させ、残りをEtOAc及び水の混合物(各々500ml)に溶解した。次いで、これをセライトで濾過し、分液漏斗に移した。相を分離し、水層をEtOAc(3×250ml)で抽出した。合わせた有機層を水及びブライン(各々500ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をクロマトグラフィーによって精製し、茶色の固体の形態で所望の化合物を得た。
【0127】
(S)−1−ピロリジン−1−イル−ブタン−2−オール:
【化59】
ピロリジン(5.76ml)を、(S)−(−)−1,2−エポキシブタン(5.00ml)と水(25ml)との混合物に加えた。反応混合物を室温で一晩激しく撹拌した。さらなる水(25ml)を加え、有機物質をジエチルエーテル(2×100ml)で抽出した。合わせた有機層を乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。揮発性の不純物を除去するために、エーテル及びトルエン(各々2×10ml)との何回かの同時蒸発を行い、無色の液体の形態の所望の化合物を得た。
【0128】
(R)−2−ピロリジン−1−イル−ブタン−1−オール:
【化60】
(R)−(−)−2−アミノ−1−ブタノール(5.00g)及び1,4−ジブロモブタン(6.61ml)のCH3CN(60ml)溶液に、K2CO3(15.48g)を加え、このように得られた懸濁液を還流温度で19時間撹拌した。反応混合物を真空中で蒸発させ、残渣をEtOAc及び水に分割した。有機層を水及びブラインで洗浄した。水層をEtOAcで抽出した。合わせた有機層をNa2SO4上で乾燥させ、真空中で乾燥するまで蒸発させた。粗生成物をクーゲルロール蒸留(12ミリバール、106〜150℃)によって精製し、透明で無色の油を得た。
【0129】
(S)−1−ジメチルアミノ−ブタン−2−オール:
【化61】
(S)−(−)−1,2−エポキシブタン(9.66)と水(40ml)との混合物を、100mlのフラスコ中に入れた。ジメチルアミン(水中の40%溶液、20.3ml)を一度に加え、反応混合物を氷浴で数分間冷却し、一晩室温で撹拌した。固体NaClを飽和するまで加え、混合物を、DCM(3×50ml)で抽出した。合わせた有機層を水及びブライン(各々30ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、注意深く蒸発させ(20ミリバール/25℃)、無色の液体を得た。
【0130】
(R)−2−ジメチルアミノ−ブタン−1−オール:
【化62】
(R)−(−)−2−アミノ−1−ブタノール(5.00g)を、冷却したギ酸(11.0ml)に一滴ずつ加えた。ホルムアルデヒド(36.5%水溶液、10.73ml)を加え、混合物を0℃で1h撹拌した.次いで、それを90℃に4h加熱した。反応物を真空中で蒸発させ、油性残渣をDCM(150ml)及び1NのNaOHに分配した。有機相を1NのNa2CO3及びブラインで洗浄した。DCM相をMgSO4上で乾燥させ、濾過し、蒸発させ、黄色の油を得た。それをクーゲルロール蒸留(60ミリバール、85〜130℃)によって精製した。生成物を、無色の油の形態で得た。
【0131】
2−ジメチルアミノ−2−メチル−プロパン−1−オール:
【化63】
2−アミノ−2−メチル−1−プロパノール(5.00g)、ホルムアルデヒド水溶液(水中36.5%、10.73ml)及び水(20ml)中のギ酸(11.00ml)の混合物を、0℃にて1h撹拌し、次いで90℃に4h加熱した。反応混合物を真空中で濃縮し、残渣を水(20ml)で希釈し、1NのNaOHを加えることによってアルカリ性(pH14)にした。さらに、固体NaClを飽和するまで加えた。水溶液を、ジクロロメタン(3×50ml)で抽出し、合わせた有機層を乾燥させ(Na2SO4)、真空中で濃縮した。減圧(40ミリバール)下での蒸留によって、所望の化合物を無色の油として得た。
【0132】
(S)−1−メチル−ピペリジン−3−オール:
【化64】
(S)−3−ヒドロキシピペリジン塩酸塩(4.00g)、ホルムアルデヒド水溶液(水中36.5%、3.73ml)及び水(20ml)中のギ酸(2.19ml)の混合物の還流を一晩続けた。反応混合物を真空中で濃縮し、残渣を水(20ml)で希釈し、1NのNaOHを加えることによってアルカリ性(pH14)にした。さらに、固体NaClを飽和するまで加えた。水溶液を、ジクロロメタン(3×50ml)で抽出し、合わせた有機層を乾燥させ(Na2SO4)、真空中で濃縮した。減圧(10ミリバール)下での蒸留によって、所望の化合物を無色の液体として得た。
【0133】
3−((R)−2−ジメチルアミノ−ブトキシ)−3’,4’,5’,6’−テトラヒドロ−2’H−[2,4’]ビピリジニル−1’−カルボン酸tert−ブチルエステル:
【化65】
(R)−2−ジメチルアミノ−ブタン−1−オール(1.004g)を、アルゴン下で0℃にて水素化ナトリウム(鉱油中60%、240mg)のDMF(30ml)懸濁液に加えた。冷却槽を取り外し、混合物を室温で2h撹拌した。次いで、3−フルオロ−3’,4’,5’,6’−テトラヒドロ−2’H−[2,4’]ビピリジニル−1’−カルボン酸tert−ブチルエステル(1.200g)を加え、反応混合物を120℃に加熱した(油浴温)。120℃で一晩撹拌した後、反応混合物を真空中で濃縮した。残りの濃褐色の油を酢酸エチル(150ml)に溶解し、混合物を1NのNa2CO3(2×60ml)で洗浄した。合わせた水層をEtOAc(30ml)で再抽出し、合わせた有機層をブライン(50ml)で洗浄し、乾燥させ(MgSO4)、濾過し、蒸発させ、濃褐色の油を得た。それをカラムクロマトグラフィーによって精製した。
【0134】
[(R)−1−(1’,2’、3’,4’,5’,6’−ヘキサヒドロ−[2,4’]ビピリジニル−3−イルオキシメチル)−プロピル]−ジメチルアミン:
【化66】
3−((R)−2−ジメチルアミノ−ブトキシ)−3’,4’,5’,6’−テトラヒドロ−2’H−[2,4’]ビピリジニル−1’−カルボン酸tert−ブチルエステル(788mg)を、ジオキサン(15ml)とメタノール(4ml)との混合物に溶解した。ジオキサン(15ml)中の4NのHClを加え、反応物を室温で1h撹拌した。トルエン(4×30ml)との同時蒸発を含めた全ての揮発性物質の蒸発によって、ベージュ色の半固体を得て、それを高真空中で一晩さらに乾燥させた。残りの固体を10mlの乾燥ジエチルエーテルで粉砕した。溶媒をデカントし、生成物を真空中で乾燥させた。
【0135】
B−C部分の合成:
B−C部分1:
中間体A1):
【化67】
D−2,4−ジクロロフェニルアラニン(10.00g)のメタノール(100ml)懸濁液に、塩化チオニル(9.39ml)を一滴ずつ加えた。添加の間に、透明な溶液が形成され、反応物の還流を始めた。反応混合物の還流を2h続けた。室温に冷却した後、混合物を40℃で乾燥するまで蒸発させた。粗生成物をジエチルエーテル中で粉砕し、不溶性化合物を濾過し、ジエチルエーテルで洗浄し、最終的に真空中室温にてP2O5上で一晩乾燥させた。生成物を、無色の針状晶の形態で得た。
【0136】
中間体B1):
【化68】
350mlの三つ口平底フラスコに機械式撹拌機を搭載し、DCM(80ml)、飽和炭酸水素ナトリウム水溶液(80ml)、及び中間体A1)(5.69g)を入れた。二相性混合物を氷浴中冷却し、トリホスゲン(1.96g)を1回で加える間に機械的に撹拌した。反応混合物を氷浴中45分間撹拌し、次いで250mlの分液漏斗に注いだ。有機層を集め、水層を20mlのDCMで3回抽出した。合わせた有機層を水で洗浄し、Na2SO4上で乾燥させ、濾過し、真空中で乾燥するまで蒸発させ、粗生成物を半固体として得た。残渣をクーゲルロール蒸留によって精製した(200〜240℃、0.04〜0.08ミリバール)。生成物を透明で無色の油として得た。
【0137】
中間体C1):
【化69】
氷冷の中間体B1)(4.99g)のDCM(50ml)溶液に、ピロリジン(4.56ml)を加えた。10分後氷浴を取り外し、撹拌を4h続けた。反応混合物を真空中で蒸発させた。残渣をEtOAcに再溶解し、有機層を1NのHCl、水、飽和Na2CO3、水及びブラインで洗浄した。全ての水層をEtOAcで抽出した。合わせた有機層をNa2SO4上で乾燥させ、真空中で乾燥するまで蒸発させた。
【0138】
B−C部分1:
【化70】
中間体C1)(6.28g)を、MeOH(100ml)及びTHF(30ml)に0℃で溶解した。水酸化リチウム一水和物(1.53g)の水(30ml)溶液を5分に亘って一滴ずつ加えた。混合物を0℃で60分間撹拌し、次いで0.5MのHClを加えることによって酸性化した。反応混合物をEtOAcで2度抽出した。合わせた有機層を水で2度、及びブラインで洗浄し、Na2SO4上で乾燥させ、真空中で蒸発させた。固体残留物をEt2O中で粉砕し、次いで濾過し、Et2Oで洗浄した。生成物を白色の固体として得た。
【0139】
B−C部分2:
中間体A2):
【化71】
氷冷の中間体B1)(200mg)の無水DMF(2.5ml)溶液に、3−ヒドロキシアゼチジン塩酸塩(160mg)及びトリエチルアミン(205μl)の無水DMF(2.5ml)溶液をアルゴン下で加えた。混合物を0℃で4h30分間撹拌させた。反応混合物を真空中で蒸発させた。残渣を冷たいEtOAc(75ml)で希釈し、有機相を0.1MのHCl(2×25ml)及びブラインで洗浄した。水相をEtOAcで再抽出し、有機相をNa2SO4上で乾燥させ、濾過し、蒸発させた。無色の油を得て、それを高真空下にて乾燥させた。
【0140】
B−C部分2:
【化72】
中間体A2)(255mg)を、MeOH(1.5ml)及びTHF(4.0ml)に0℃で溶解した。水酸化リチウム一水和物(62mg)のH2O(1.5ml)溶液を加えた。混合物を0℃で4h30分撹拌した。1NのHClを加えることによって反応混合物を中和し、溶媒を真空中で蒸発させた。次いで、水相を1NのHCl(pH約1〜2)で酸性化した。水相をEtOAc(約1×20ml)で抽出した。有機層を水及びブラインで洗浄した。水相をEtOAcで再抽出し、合わせた有機層をNa2SO4上で乾燥させ、真空中で蒸発させ、無色の固体を得た。
【0141】
B−C部分3:
中間体A3):
【化73】
(R)−3−ピロリジノール(1.43g)を、氷冷の中間体B1)(1.50g)のCH2Cl2(25ml)溶液に加えた。2時間後、氷浴を取り外し、撹拌を2時間続けた。反応混合物を真空中で蒸発させた。残渣をEtOAcに再溶解し、有機層を1NのHCl、水、飽和NaHCO3、水及びブラインで洗浄した。全ての水層をEtOAcで抽出した。合わせた有機層をMgSO4上で乾燥させ、濾過し、真空中で乾燥するまで蒸発させた。
【0142】
B−C部分3:
【化74】
中間体A3)(1.78g)を、MeOH(35ml)及びTHF(9.5ml)に0℃で溶解した。水酸化リチウム一水和物(0.41g)のH2O(9.5ml)溶液を5分に亘って一滴ずつ加えた。混合物を0℃で2時間撹拌した。0.5MのHClを加えることによって反応混合物を酸性化し、次いで、EtOAcで2度抽出した。有機層を水及びブラインで2度洗浄した。合わせた有機層をNa2SO4上で乾燥させ、真空中で蒸発させた。固体残留物をEt2O中で粉砕し、次いで濾過し、Et2Oで洗浄し、無色の結晶性固体を得た。
【0143】
B−C部分4:
中間体A4):
【化75】
(S)−3−ピロリジノール(1.43g)を、氷冷のイソシアネート(1.50g)のCH2Cl2(25ml)溶液に加えた。20分後、氷浴を取り外し、4時間撹拌を続けた。反応混合物を真空中で蒸発させた。残渣をEtOAcに再溶解し、有機層を1NのHCl、水、飽和NaHCO3及びブラインで洗浄した。各水層をEtOAcで再抽出した。合わせた有機層をMgSO4上で乾燥させ、濾過し、真空中で乾燥するまで蒸発させた。生成物を、無色の安定した泡の形態で得た。
【0144】
B−C部分4:
【化76】
中間体A4)(1.97g)を、MeOH(35ml)及びTHF(9.5ml)に0℃で溶解した。水酸化リチウム一水和物(0.45g)のH2O(9.5ml)溶液を5分に亘って一滴ずつ加えた。混合物を0℃で2時間撹拌した。0.5MのHClを加えることによって反応混合物を酸性化し、次いでEtOAcで2度抽出した。有機層を水及びブラインで洗浄した。合わせた有機層をNa2SO4上で乾燥させ、真空中で蒸発させた。残渣をジエチルエーテルに溶解し、再び蒸発させた。残りの無色の泡を真空中で乾燥させた。
【0145】
実施例2の合成:
中間体2a):
【化77】
火炎乾燥シュレンクフラスコに、3−ブロモ−2−フルオロ−6−ピコリン(434mg)、[1,1’−ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)のジクロロメタンとの錯体(56mg)、ヨウ化銅(I)(26mg)、及び乾燥N,N−ジメチルアセトアミド(3ml)を入れた。このように得られた混合物を交互の真空/アルゴンパージで脱気した。次いで、1−tert−ブトキシカルボニルピペリジン−4−イル)(ヨード)亜鉛(3.19mmol、上記のように調製)を加えた。混合物をもう一度脱気し、次いで80℃に一晩加熱した。次いで、N,N−ジメチルアセトアミドの主要部分を蒸発させ、残りをEtOAc及び水の混合物(各々50ml)に溶解した。次いで、混合物をセライトで濾過し、分液漏斗に移した。相を分離し、水層を、EtOAc(2×50ml)で抽出した。合わせた有機層を水及びブライン(各々100ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をカラムクロマトグラフィーによって精製し、無色の油の形態で所望の化合物を得た。
【0146】
中間体2b):
【化78】
密封した管中に、中間体2a)(0.55g)、メチル−(2−ピロリジン−1−イル−エチル)−アミン(2.10g)、及びN,N−ジイソプロピルエチルアミン(286μl)を入れた。反応混合物を150℃に4日間加熱した。揮発性物質の主要部分を蒸発させ、残りをEtOAc(100ml)に溶解した。溶液をNaHCO3(2×25ml)で洗浄し、合わせた水層をEtOAc(25ml)で再抽出した。有機層を混合し、ブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、蒸発させた。粗生成物をカラムクロマトグラフィーによって精製し、未反応の出発物質及び所望の生成物を含有する混合画分を単離した。所望の化合物を、酸性条件下にて分取HPLCによって最終的に茶色がかった樹脂の形態で単離した。
【0147】
中間体2c):
【化79】
DCM(1ml)とMeOH(1ml)との混合物中のBoc保護された中間体2b)(69mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、2ml)を加え、溶液を室温で1h撹拌した。全ての揮発性物質の蒸発、並びにトルエン(2×5ml)、及びアセトン(5ml)との同時蒸発によってベージュ色の固体がもたらされ、それをデシケーター中シカペント(Sicapent)上で一晩さらに乾燥させた。
【0148】
実施例2:
【化80】
中間体2c)(68mg)及びB−C部分1(60mg)、1−ヒドロキシベンゾトリアゾール水和物(32mg)及びN−メチルモルホリン(58μl)を、DMF(3ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(48mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(12μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(70ml)で希釈し、飽和Na2CO3(3×25ml)、H2O及びブライン(各々25ml)で洗浄した。有機層を乾燥させ(Na2SO4)、溶媒を真空中で除去した。カラムクロマトグラフィーによる粗生成物の精製によって黄色がかった樹脂の形態の相当するアミンを得た。これをEtOAc(1ml)に溶解し、塩化水素のジエチルエーテル溶液(1.0M、114μl)で処理した。相当する二塩酸塩の完全な沈殿を得るために、このように得られた懸濁液をヘキサン(5ml)で希釈した。固体を濾過し、ヘキサンで洗浄し、デシケーター中シカペント上で一晩乾燥させ、オフホワイトの固体の形態の所望の化合物を得た。
【0149】
実施例4の合成:
中間体4a):
【化81】
冷却した(0℃)1−メチルピペラジン(257μl)の乾燥THF(2ml)溶液に、ヘキサン(0.50ml)中のn−ブチルリチウム(2.5M)をシリンジによって一滴ずつ加えた。室温で15分間撹拌した後、次いで、このように得られたリチウムアミドを、2a)(387mg)の乾燥THF(2ml)溶液に0℃で一滴ずつ加えた。2.5h後、反応混合物を飽和NH4Cl(2ml)で加水分解し、EtOAc(70ml)で希釈した。有機層のNaHCO3(2×25ml)及びブライン(25ml)による抽出、Na2SO4上での乾燥、濾過及び真空濃縮によって、黄色の油がもたらされた。これをカラムクロマトグラフィーによって精製し、黄色がかった樹脂の形態の所望の化合物を得た。
【0150】
中間体4b):
【化82】
DCM(1ml)とMeOH(1ml)との混合物中のBoc保護された中間体4a)(117mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、2ml)を加え、溶液を室温で1h撹拌した。全ての揮発性物質の蒸発、並びにトルエン(2×5ml)、及びアセトン(5ml)との同時蒸発によってベージュ色の固体がもたらされ、それをデシケーター中シカペント上で一晩さらに乾燥させた。
【0151】
実施例4:
【化83】
中間体4b)(66mg)及びB−C部分1(70mg)、1−ヒドロキシベンゾトリアゾール水和物(37mg)及びN−メチルモルホリン(68μl)を、DMF(3ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(56mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(14μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(70ml)で希釈し、飽和Na2CO3(3×25ml)、H2O及びブライン(各々25ml)で洗浄した。有機層を乾燥させ(Na2SO4)、溶媒を真空中で除去した。カラムクロマトグラフィーによる粗生成物の精製によって黄色がかった固体の形態の相当するアミンを得た。これをEtOAc(1ml)に溶解し、塩化水素のジエチルエーテル溶液(1.0M、214μl)で処理した。相当する二塩酸塩の完全な沈殿を得るために、このように得られた懸濁液をヘキサン(5ml)で希釈した。固体を濾過し、ヘキサンで洗浄し、デシケーター中シカペント上で一晩乾燥させ、白色の固体の形態の所望の化合物を得た。
【0152】
実施例9の合成:
中間体9a):
【化84】
火炎乾燥シュレンクフラスコに、3−ブロモ−2−フルオロ−5−ピコリン(1.00g)、[1,1’−ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)のジクロロメタンとの錯体(129mg)、ヨウ化銅(I)(60mg)、及び乾燥N,N−ジメチルアセトアミド(7ml)を入れた。このように得られた混合物を交互の真空/アルゴンパージで脱気した。次いで、1−tert−ブトキシカルボニルピペリジン−4−イル)(ヨード)亜鉛(7.37mmol、上記のように調製)を加えた。混合物をもう一度脱気し、次いで80℃にて一晩加熱した。次いで、N,N−ジメチルアセトアミドの主要部分を蒸発させ、残りをEtOAc及び水の混合物(各々100ml)に溶解した。次いで、混合物をセライトで濾過し、分液漏斗に移した。相を分離し、水層をEtOAc(3×50ml)で抽出した。合わせた有機層を水及びブライン(各々100ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をカラムクロマトグラフィーによって精製し、白色の固体の形態で所望の化合物を得た。
【0153】
中間体9b):
【化85】
(S)−1−ピロリジン−1−イル−ブタン−2−オール(191mg)のDMF(1ml)溶液を、アルゴン下0℃にて水素化ナトリウム(鉱油(36mg)中の60%分散物)のDMF(1ml)懸濁液に加えた。冷却槽を取り外し、混合物を室温で1h撹拌した。次いで、中間体9a)(158mg)のDMF(1ml)溶液を加え、反応混合物を60℃に1h加熱した。反応混合物を0℃に冷却し、次いで飽和NH4Cl(1ml)で加水分解した。EtOAc(70ml)を加え、混合物をNaHCO3(2×25ml)で洗浄した。合わせた水層をEtOAc(25ml)で再抽出し、次いで合わせた有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物を分取HPLCによって精製し、黄色がかった油の形態の相当するジホルメートを得た。
【0154】
中間体9c):
【化86】
DCM(2ml)とMeOH(0.5ml)との混合物中のBoc保護された中間体9b)(125mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、2ml)を加え、溶液を室温で30分間撹拌した。全ての揮発性物質の蒸発、並びにトルエン、アセトン、及びジエチルエーテル(各々5ml)との同時蒸発によって白色の固体がもたらされ、それをデシケーター中シカペント上で一晩さらに乾燥させた。
【0155】
実施例9:
【化87】
中間体9c)(55mg)及びB−C部分1(55mg)、1−ヒドロキシベンゾトリアゾール水和物(29mg)及びN−メチルモルホリン(54μl)を、DMF(3ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(44mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(11μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(70ml)で希釈し、飽和Na2CO3(3×25ml)、水及びブライン(各々25ml)で洗浄した。有機層を乾燥させ(Na2SO4)、溶媒を真空中で除去した。粗生成物を分取HPLCによって精製し、茶色がかった樹脂の形態の相当するジホルメートを得た。
【0156】
実施例11の合成:
中間体11a):
【化88】
火炎乾燥シュレンクフラスコに、3−フルオロ−4−ヨードピリジン(1.00g)、[1,1’−ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)のジクロロメタンとの錯体(110mg)、ヨウ化銅(I)(51mg)、及び乾燥N,N−ジメチルアセトアミド(6ml)を入れた。このように得られた混合物を交互の真空/アルゴンパージで脱気した。次いで、1−tert−ブトキシカルボニルピペリジン−4−イル)(ヨード)亜鉛(6.28mmol、上記のように調製)を加えた。混合物をもう一度脱気し、次いで80℃に一晩加熱した。次いで、N,N−ジメチルアセトアミドの主要部分を蒸発させ、残りをEtOAc及び水の混合物(各々100ml)に溶解した。次いで、混合物をセライトで濾過し、分液漏斗に移した。相を分離し、水層をEtOAc(3×30ml)で抽出した。合わせた有機層を水及びブライン(各々75ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をカラムクロマトグラフィーによって精製し、茶色がかった固体の形態で所望の化合物を得た。
【0157】
中間体11b):
【化89】
(S)−1−ピロリジン−1−イル−ブタン−2−オール(156mg)のDMF(2ml)溶液を、アルゴン下0℃にて水素化ナトリウム(鉱油(29mg)中の60%分散物)のDMF(1ml)懸濁液に加えた。冷却槽を取り外し、混合物を室温で1h撹拌した。次いで、中間体11a)(102mg)のDMF(2ml)溶液を加え、反応混合物を120℃に一晩加熱した。反応混合物を0℃に冷却し、次いで飽和NH4Cl(1ml)で加水分解した。EtOAc(50ml)を加え、混合物をNaHCO3(2×25ml)で洗浄した。合わせた水層をEtOAc(25ml)で再抽出し、次いで合わせた有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物を分取HPLCによって精製し、黄色がかった樹脂の形態の相当するジホルメートを得た。
【0158】
中間体11c):
【化90】
DCM(1ml)とMeOH(1ml)との混合物中のBoc保護された中間体11b)(111mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、2ml)を加え、溶液を室温で15分撹拌した。全ての揮発性物質の蒸発、並びにトルエン(2×5ml)、及びアセトン(5ml)との同時蒸発によってベージュ色の固体がもたらされ、それをデシケーター中シカペント上で一晩さらに乾燥させた。
【0159】
実施例11:
【化91】
中間体11c)(69mg)及びB−C部分1(58mg)、1−ヒドロキシベンゾトリアゾール水和物(31mg)及びN−メチルモルホリン(57μl)を、DMF(5ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(47mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(12μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(70ml)で希釈し、飽和Na2CO3(3×25ml)、H2O及びブライン(各々25ml)で洗浄した。有機層を乾燥させ(Na2SO4)、溶媒を真空中で除去した。カラムクロマトグラフィーによる粗生成物の精製によって黄色がかった樹脂の形態の相当するアミンを得た。これをEtOAc(1ml)に溶解し、塩化水素のジエチルエーテル溶液(1.0M、143μl)で処理した。相当する二塩酸塩の完全な沈殿を得るために、このように得られた懸濁液をヘキサン(5ml)で希釈した。固体を濾過し、ヘキサンで洗浄し、デシケーター中シカペント上で一晩乾燥させ、ベージュ色の固体の形態の所望の化合物を得た。
【0160】
実施例12の合成:
中間体12a):
【化92】
火炎乾燥シュレンクフラスコに、4−フルオロ−3−ヨード−ピリジン(1.00g)、[1,1’−ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)のジクロロメタンとの錯体(110mg)、ヨウ化銅(I)(51mg)、及び乾燥N,N−ジメチルアセトアミド(6ml)を入れた。このように得られた混合物を交互の真空/アルゴンパージで脱気した。次いで、1−tert−ブトキシカルボニルピペリジン−4−イル)(ヨード)亜鉛(6.28mmol、上記のように調製)を加えた。混合物をもう一度脱気し、次いで80℃に一晩加熱した。次いで、N,N−ジメチルアセトアミドの主要部分を蒸発させ、残りをEtOAc及び水の混合物(各々100ml)に溶解した。次いで、混合物をセライトで濾過し、分液漏斗に移した。相を分離し、水層をEtOAc(3×30ml)で抽出した。合わせた有機層を水及びブライン(各々75ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をカラムクロマトグラフィーによって精製し、茶色がかった固体の形態で所望の化合物を得た。
【0161】
中間体12b):
【化93】
(S)−1−ピロリジン−1−イル−ブタン−2−オール(187mg)のDMF(2ml)溶液を、アルゴン下0℃で水素化ナトリウム(鉱油(35mg)中60%分散物)のDMF(1ml)懸濁液に加えた。冷却槽を取り外し、混合物を室温で1h撹拌した。次いで、中間体12a)(122mg)のDMF(2ml)溶液を加え、反応混合物を60℃に1h加熱した。反応混合物を0℃に冷却し、次いで飽和NH4Cl(1ml)で加水分解した。EtOAc(70ml)を加え、混合物をNaHCO3(2×25ml)で洗浄した。合わせた水層をEtOAc(25ml)で再抽出し、次いで合わせた有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物を分取HPLCによって精製し、黄色がかった樹脂の形態の相当するジホルメートを得た。
【0162】
中間体12c):
【化94】
DCM(1ml)とMeOH(1ml)との混合物中のBoc保護された中間体12b)(151mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、2ml)を加え、溶液を室温で2h撹拌した。全ての揮発性物質の蒸発、並びにトルエン(2×5ml)、アセトン、及びエーテル(各々5ml)との同時蒸発によってベージュ色の固体がもたらされ、それをデシケーター中シカペント上で一晩さらに乾燥させた。
【0163】
実施例12:
【化95】
中間体12c)(68mg)及びB−C部分1(61mg)、1−ヒドロキシベンゾトリアゾール水和物(33mg)及びN−メチルモルホリン(59μl)を、DMF(3ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(49mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(12μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(70ml)で希釈し、飽和Na2CO3(3×25ml)、水及びブライン(各々25ml)で洗浄した。有機層を乾燥させ(Na2SO4)、溶媒を真空中で除去した。カラムクロマトグラフィーによる粗生成物の精製によって茶色がかった樹脂の形態の相当するアミンを得た。これをEtOAc(1ml)に溶解し、塩化水素のジエチルエーテル溶液(1.0M、178μl)で処理した。相当する二塩酸塩の完全な沈殿を得るために、このように得られた懸濁液をヘキサン(5ml)で希釈した。固体を濾過し、ヘキサンで洗浄し、デシケーター中シカペント上で一晩乾燥させ、オフホワイトの固体の形態の所望の化合物を得た。
【0164】
実施例14の合成:
中間体14a):
【化96】
2−ブロモ−3−ピリジノール(1.04g)のピリジン(2.00ml)及び乾燥DCM(20ml)溶液を、0℃に冷却した。無水酢酸(1.76ml)を加え、反応混合物を室温で一晩撹拌した。全ての揮発性物質を蒸発させ、残りを水(30ml)に懸濁させた。室温で1h撹拌した後、懸濁液を1NのNaHCO3(50ml)で希釈し、EtOAc(3×50ml)で抽出した。合わせた有機層を1NのNaHCO3、及びブライン(各々50ml)で洗浄し、乾燥させ(MgSO4)、濾過し、蒸発させ、琥珀色の油の形態の所望の化合物を得た。
【0165】
中間体14b):
【化97】
火炎乾燥シュレンクフラスコに、中間体14a)(2.00g)、[1,1’−ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)のジクロロメタンとの錯体(227mg)、ヨウ化銅(I)(106mg)、及び乾燥N,N−ジメチルアセトアミド(10ml)を入れた。このように得られた混合物を交互の真空/アルゴンパージで脱気した。次いで、1−tert−ブトキシカルボニルピペリジン−4−イル)(ヨード)亜鉛(13.0mmol、上記のように調製)を加えた。混合物をもう一度脱気し、次いで80℃に一晩加熱した。次いで、N,N−ジメチルアセトアミドの主要部分を蒸発させ、残りをEtOAc及び水の混合物(各々200ml)に溶解した。次いで、混合物をセライトで濾過し、分液漏斗に移した。相を分離し、水層をEtOAc(3×75ml)で抽出した。合わせた有機層を水及びブライン(各々150ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をカラムクロマトグラフィーによって精製し、茶色がかった固体の形態で所望の化合物を得た。
【0166】
中間体14c):
【化98】
中間体14b)(1.97g)のMeOH(60ml)溶液を、0℃に冷却した。水酸化リチウム(368mg)の水(30ml)溶液を加え、反応混合物を0℃で10分間撹拌した。0.5MのHClを0℃にて一滴ずつ添加することによってpHを約7に調節した。水(100ml)を加え、EtOAc(3×100ml)による抽出を行った。ブライン(150ml)による洗浄、乾燥(Na2SO4)、濾過及び蒸発の後、所望の化合物を茶色がかった発泡体として得た。
【0167】
中間体14d):
【化99】
中間体14c)(207mg)、1−(2−クロロエチル)ピロリジン塩酸塩(190mg)、及び炭酸セシウム(969mg)のDMF(10ml)溶液を、室温で一晩撹拌した。全ての揮発性物質を蒸発させ、残渣をEtOAc及び飽和NaHCO3(各々50ml)に分配した。水層を、EtOAc(2×25ml)で抽出した。合わせた有機層を水及びブライン(各々25ml)で洗浄し、乾燥させ(Na2SO4)、蒸発させ、所望の化合物を茶色がかった樹脂として得た。
【0168】
中間体14e):
【化100】
DCM(2ml)とMeOH(0.5ml)との混合物中のBoc保護された中間体14d)(253mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、2ml)を加え、溶液を室温で15分撹拌した。全ての揮発性物質の蒸発、並びにトルエン、アセトン、及びエーテル(各々5ml)との同時蒸発によってベージュ色の固体がもたらされ、それをデシケーター中シカペント上で一晩さらに乾燥させた。
【0169】
実施例14:
【化101】
中間体14e)(180mg)及びB−C部分1(201mg)、1−ヒドロキシベンゾトリアゾール水和物(107mg)及びN−メチルモルホリン(195μl)を、DMF(5ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(161mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(41μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(100ml)で希釈し、飽和Na2CO3(3×50ml)、水及びブライン(各々50ml)で洗浄した。有機層を乾燥させ(Na2SO4)、溶媒を真空中で除去した。カラムクロマトグラフィーによる粗生成物の精製によって茶色がかった樹脂の形態の相当するアミンを得た。これをEtOAc(1ml)に溶解し、塩化水素のジエチルエーテル溶液(1.0M、564μl)で処理した。相当する二塩酸塩の完全な沈殿を得るために、このように得られた懸濁液をヘキサン(5ml)で希釈した。固体を濾過し、ヘキサンで洗浄し、デシケーター中シカペント上で一晩乾燥させ、白色の固体の形態の所望の化合物を得た。
【0170】
実施例21の合成:
中間体21a):
【化102】
(S)−1−メチル−ピペリジン−3−オール(228mg)のDMF(2ml)溶液を、アルゴン下で0℃にて水素化ナトリウム(鉱油(53mg)中60%分散物)のDMF(1ml)懸濁液に加えた。冷却槽を取り外し、混合物を室温で1h撹拌した。次いで、3−フルオロ−3’,4’,5’,6’−テトラヒドロ−2’H−[2,4’]ビピリジニル−1’−カルボン酸tert−ブチルエステル(185mg)のDMF(2ml)溶液を加え、反応混合物を120℃に一晩加熱した。反応混合物を0℃に冷却し、次いで飽和NH4Cl(1ml)で加水分解した。EtOAc(50ml)を加え、混合物をNaHCO3(2×25ml)で洗浄した。合わせた水層をEtOAc(25ml)で再抽出し、次いで合わせた有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物を分取HPLCによって精製し、黄色がかった樹脂の形態の相当するジホルメートを得た。
【0171】
中間体21b):
【化103】
DCM(1ml)とMeOH(1ml)との混合物中のBoc保護された中間体21a)(148mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、2ml)を加え、溶液を室温で15分撹拌した。全ての揮発性物質の蒸発、並びにトルエン(2×5ml)、及びアセトン(5ml)との同時蒸発によってベージュ色の固体がもたらされ、それをデシケーター中シカペント上で一晩さらに乾燥させた。
【0172】
実施例21:
【化104】
中間体21b)(93mg)及びB−C部分1(83mg)、1−ヒドロキシベンゾトリアゾール水和物(44mg)及びN−メチルモルホリン(81μl)を、DMF(5ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(67mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(17μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(70ml)で希釈し、飽和Na2CO3(3×25ml)、水及びブライン(各々25ml)で洗浄した。有機層を乾燥させ(Na2SO4)、溶媒を真空中で除去した。カラムクロマトグラフィーによる粗生成物の精製によって黄色がかった樹脂の形態の相当するアミンを得た。これをEtOAc(1ml)に溶解し、塩化水素のジエチルエーテル溶液(1.0M、238μl)で処理した。相当する二塩酸塩の完全な沈殿を得るために、このように得られた懸濁液をヘキサン(5ml)で希釈した。固体を濾過し、ヘキサンで洗浄し、デシケーター中シカペント上で一晩乾燥させ、白色の固体の形態の所望の化合物を得た。
【0173】
実施例28の合成:
中間体28a):
【化105】
トリアセトキシ水素化ホウ素ナトリウム(1512mg)を、2−ブロモ−3−ピリジンカルボキシアルデヒド(948mg)及び1−メチルピペラジン(566μl)の1,2−ジクロロエタン(20ml)溶液に加えた。室温で1h撹拌した後、混合物をEtOAc(100ml)で希釈し、飽和NaHCO3(2×50ml)、H2O及びブライン(各々50ml)で洗浄した。Na2SO4上での乾燥、濾過及び溶媒の蒸発の後、茶色がかった油を得た。クロマトグラフィーによる精製によって、黄色がかった油の形態の所望の化合物を得た。
【0174】
中間体28b):
【化106】
火炎乾燥シュレンクフラスコに、中間体28a)(729mg)、[1,1’−ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)のジクロロメタンとの錯体(66mg)、ヨウ化銅(I)(31mg)、及び乾燥N,N−ジメチルアセトアミド(4ml)を入れた。このように得られた混合物を交互の真空/アルゴンパージで脱気した。次いで、1−tert−ブトキシカルボニルピペリジン−4−イル)(ヨード)亜鉛(3.78mmol、上記のように調製)を加えた。混合物をもう一度脱気し、次いで80℃に一晩加熱した。次いで、N,N−ジメチルアセトアミドの主要部分を蒸発させ、残りをEtOAc及び水の混合物(各々50ml)に溶解した。次いで、これをセライトで濾過し、分液漏斗に移した。相を分離し、水層をEtOAc(2×50ml)で抽出した。合わせた有機層をブライン(75ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をクロマトグラフィーによって精製し、茶色の油の形態で所望の化合物を得た。
【0175】
中間体28c):
【化107】
DCM(3ml)とMeOH(3ml)との混合物中のBoc保護された中間体28b)(383mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、6ml)を加え、溶液を室温で1h撹拌した。全ての揮発性物質の蒸発、並びにトルエン及びアセトン(各々20ml)との同時蒸発によって淡褐色の固体がもたらされ、それをデシケーター(シカペント)中で一晩さらに乾燥させた。
【0176】
実施例28:
【化108】
中間体28c)(165mg)及びB−C部分1(152mg)、1−ヒドロキシ−ベンゾトリアゾール水和物(80mg)及びN−メチルモルホリン(147μl)を、DMF(2ml)に溶解した。室温で30分撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(121mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(31μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(70ml)で希釈し、飽和Na2CO3(3×25ml)、水及びブライン(各々25ml)で洗浄した。有機層を乾燥させ(Na2SO4)、溶媒を真空中で除去した。分取HPLCによる粗生成物の精製によって相当するジホルメートを僅かに茶色がかった樹脂として得た。
【0177】
実施例41の合成:
中間体41a):
【化109】
1−ベンズヒドリルアゼタン−3−オール(674mg)のDMF(2ml)溶液を、アルゴン下0℃にて水素化ナトリウム(鉱油(75mg)中60%分散物)のDMF(2ml)懸濁液に加えた。冷却槽を取り外し、混合物を室温で1h撹拌した。次いで、3−フルオロ−3’,4’,5’,6’−テトラヒドロ−2’H−[2,4’]ビピリジニル−1’−カルボン酸tert−ブチルエステル(263mg)のDMF(2ml)溶液を加え、反応混合物を120℃に一晩加熱した。反応混合物を0℃に冷却し、次いで飽和NH4Cl(1ml)で加水分解した。EtOAc(50ml)を加え、混合物をNaHCO3(2×25ml)で洗浄した。合わせた水層をEtOAc(25ml)で再抽出し、次いで合わせた有機層を水及びブライン(各々25ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をクロマトグラフィーによって精製し、所望の生成物を黄色がかった発泡体として得た。
【0178】
中間体41b):
【化110】
MeOH(2ml)とAcOH(0.2ml)との混合物中の炭素担持20重量%水酸化パラジウム(10mg)の存在下で、1バールの水素下で室温にて、一晩中間体41a)(122mg)を水素化した。シリンジフィルターによる濾過及び溶媒の完全な蒸発によって、所望の化合物が無色の油としてもたらされた。
【0179】
中間体41c):
【化111】
トリアセトキシ水素化ホウ素ナトリウム(72mg)を、中間体41b)(137mg)及びホルムアルデヒド(水中36.5%、18.5μl)の1,2−ジクロロエタン(2ml)溶液に加えた。反応混合物を2h室温で撹拌した。次いで、混合物をEtOAc(50ml)で希釈し、飽和NaHCO3(2×25ml)及びH2O(25ml)で洗浄した。水層を混合し、EtOAc(25ml)で再抽出した。全ての有機層を合わせ、ブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、蒸発させた。粗生成物をクロマトグラフィーによって精製し、所望の生成物を無色の油として得た。
【0180】
中間体41d):
【化112】
DCM(1ml)とMeOH(1ml)との混合物中のBoc保護された中間体41c)(50mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、2ml)を加え、溶液を室温で1h撹拌した。全ての揮発性物質の蒸発、並びにトルエン及びアセトン(各々5ml)との同時蒸発によって白色の固体がもたらされ、それをデシケーター(シカペント)中で一晩さらに乾燥させた。
【0181】
実施例41:
【化113】
中間体41d)(51mg)及びB−C部分1(62mg)、1−ヒドロキシ−ベンゾトリアゾール水和物(33mg)及びN−メチルモルホリン(60μl)を、DMF(2ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(50mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(13μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(70ml)で希釈し、飽和Na2CO3(3×25ml)、水及びブライン(各々25ml)で洗浄した。有機層を乾燥させ(Na2SO4)、溶媒を真空中で除去した。分取HPLCによる粗生成物の精製によって相当するジホルメートを無色のガラス状固体として得た。
【0182】
実施例57の合成:
中間体57a):
【化114】
cis−(1S,2R)−2−アミノシクロペンタノール塩酸塩(6.55g)、ホルムアルデヒド水溶液(36.5%、12.2ml)及び水(30ml)中のギ酸(7.18ml)の混合物を、一晩加熱還流した。反応混合物を真空中で濃縮し、1NのNaOHを加えることによって残りをアルカリ性(pH14)にした。さらに、固体NaClを飽和するまで加えた。水相をDCM(3×50ml)で抽出し、合わせた有機層を水及びブライン(各々30ml)で洗浄し、次いで乾燥させ(Na2SO4)、真空中で濃縮し、黄色がかった液体を得た。
【0183】
中間体57b):
【化115】
中間体57a)(1.27g)を無水EtOH(50ml)に溶解し、水素化ホウ素ナトリウム(1.13g)を室温で少量ずつ加えた。一晩撹拌した後、飽和NH4Cl(10ml)を加え、EtOHの主要部分を真空中で除去した。残渣を0.25NのNaOH(50ml)で希釈し、固体NaClを飽和するまで加えた。水相をDCM(3×25ml)で抽出し、合わせた有機層を水及びブライン(各々25ml)で洗浄し、次いで乾燥させ(Na2SO4)、真空中で濃縮した。粗生成物をクロマトグラフィーによって精製し、無色の結晶の形態の所望の生成物を得た。
【0184】
中間体57c):
【化116】
中間体57b)(333mg)のDMF(1ml)溶液を、アルゴン下で0℃にて水素化ナトリウム(鉱油(69mg)中60%分散物)のDMF(3ml)懸濁液に加えた。冷却槽を取り外し、混合物を室温で1h撹拌した。次いで、3−フルオロ−3’,4’,5’,6’−テトラヒドロ−2’H−[2,4’]ビピリジニル−1’−カルボン酸tert−ブチルエステル(241mg)のDMF(1ml)溶液を加え、反応混合物を120℃に一晩加熱した。反応混合物を0℃に冷却し、次いで飽和NH4Cl(1ml)でクエンチした。EtOAc(70ml)を加え、混合物をNaHCO3(2×30ml)で洗浄した。合わせた水層をEtOAc(30ml)で再抽出し、次いで合わせた有機層をブライン(50ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をクロマトグラフィーによって精製し、所望の生成物を茶色がかった油として得た。
【0185】
中間体57d):
【化117】
DCM(1ml)とMeOH(1ml)との混合物中のBoc保護された中間体57c)(270mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、2ml)を加え、溶液を室温で10分間撹拌した。全ての揮発性物質の蒸発、並びにトルエン及びアセトン(各々5ml)との同時蒸発によってベージュ色の固体がもたらされ、それをデシケーター(シカペント)中で一晩さらに乾燥させた。
【0186】
実施例57:
【化118】
中間体57d)(110mg)及びB−C部分1(100mg)、1−ヒドロキシ−ベンゾトリアゾール水和物(53mg)及びN−メチルモルホリン(97μl)を、DMF(2ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(80mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(20μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(70ml)で希釈し、飽和Na2CO3(3×25ml)、水及びブライン(各々25ml)で洗浄した。有機層を乾燥させ(Na2SO4)、溶媒を真空中で除去した。分取HPLCによる粗生成物の精製によって相当するジホルメートを無色のガラス状固体として得た。
【0187】
実施例59の合成:
中間体59a):
【化119】
火炎乾燥シュレンクフラスコに、2−クロロ−5−フルオロ−3−ホルミルピリジン(1596mg)、[1,1’−ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)のジクロロメタンとの錯体(245mg)、ヨウ化銅(I)(114mg)、及び乾燥N,N−ジメチルアセトアミド(14ml)を入れた。このように得られた混合物を交互の真空/アルゴンパージで脱気した。次いで、1−tert−ブトキシカルボニルピペリジン−4−イル)(ヨード)亜鉛(14.0mmol、上記のように調製)を加えた。混合物をもう一度脱気し、次いで80℃に一晩加熱した。次いで、N,N−ジメチルアセトアミドの主要部分を蒸発させ、残りをEtOAc及び水の混合物(各々75ml)に溶解した。次いで、これをセライトで濾過し、分液漏斗に移した。相を分離し、水層をEtOAc(2×100ml)で抽出した。合わせた有機層をブライン(100ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をカラムクロマトグラフィーによって精製し、黄色い樹脂の形態で所望の化合物を得た。
【0188】
中間体59b):
【化120】
トリアセトキシ水素化ホウ素ナトリウム(585mg)を、中間体59a)(607mg)及び1−メチルピペラジン(219μl)の1,2−ジクロロエタン(10ml)溶液に加えた。室温で3h撹拌した後、さらなるトリアセトキシ水素化ホウ素ナトリウム(209mg)を加え、撹拌を一晩続けた。混合物をEtOAc(100ml)で希釈し、飽和NaHCO3(2×50ml)、H2O(50ml)及びブライン(75ml)で洗浄した。Na2SO4上での乾燥、濾過及び溶媒の蒸発の後、茶色がかった樹脂を得た。クロマトグラフィーによる精製によって、茶色がかった油の形態の所望の化合物を得た。
【0189】
中間体59c):
【化121】
DCM(1.5ml)とMeOH(3.5ml)との混合物中のBoc保護された中間体59b)(504mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、3ml)を加え、溶液を室温で1.5h撹拌した。全ての揮発性物質の蒸発、並びにトルエン(2×10ml)及びアセトン(15ml)との同時蒸発によって茶色の固体がもたらされ、それをデシケーター(シカペント)中で一晩さらに乾燥させた。
【0190】
実施例59:
【化122】
中間体59c)(150mg)及びB−C部分1(129mg)、1−ヒドロキシ−ベンゾトリアゾール水和物(69mg)及びN−メチルモルホリン(125μl)を、DMF(5ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(103mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(26μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(50ml)で希釈し、飽和Na2CO3(3×25ml)で洗浄した。合わせた水層をEtOAc(25ml)で再抽出した。次いで、全ての有機層を混合し、ブライン(25ml)で洗浄した。有機層を乾燥させ(Na2SO4)、溶媒を真空中で除去した。分取HPLCによる粗生成物の精製によって相当するホルメートを黄色がかった樹脂として得た。
【0191】
実施例60及び61の合成:
中間体60及び61a):
【化123】
2−クロロ−5−フルオロピリジン−3−オール(2.50g)及びピリジン(5.61ml)のDCM(60ml)溶液を、0℃に冷却した。これに、無水酢酸(4.96ml)を一滴ずつ加え、反応混合物を室温で一晩撹拌した。全ての揮発性物質を蒸発させ、残渣を水(30ml)で希釈し、1h撹拌し、未反応の無水酢酸を加水分解した。この混合物をEtOAc(100ml)及び0.5MのNaHCO3(150ml)で希釈した。相分離後、水層を、EtOAc(2×100ml)で抽出した。合わせた有機層を0.5MのNaHCO3及びブライン(各々100ml)で再抽出し、Na2SO4上で乾燥させ、蒸発させた。クロマトグラフィーによる精製によって、無色の液体の形態の所望の化合物を得た。
【0192】
中間体60及び61b):
【化124】
火炎乾燥シュレンクフラスコに、中間体60及び61a)(2.38g)、[1,1’−ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)のジクロロメタンとの錯体(307mg)、ヨウ化銅(I)(143mg)、及び乾燥N,N−ジメチルアセトアミド(15ml)を入れた。このように得られた混合物を交互の真空/アルゴンパージで脱気した。次いで、1−tert−ブトキシカルボニルピペリジン−4−イル)(ヨード)亜鉛(17.6mmol、上記のように調製)を加えた。混合物をもう一度脱気し、次いで80℃に一晩加熱した。次いで、N,N−ジメチルアセトアミドの主要部分を蒸発させ、残りをEtOAc及び水の混合物(各々75ml)に溶解した。次いで、これをセライトで濾過し、分液漏斗に移した。相を分離し、水層をEtOAc(2×50ml)で抽出した。合わせた有機層を水及びブライン(各々75ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をカラムクロマトグラフィーによって精製し、茶色がかった油の形態で所望の化合物を得た。
【0193】
中間体60及び61c):
【化125】
中間体60及び61b)(1.82g)のMeOH(50ml)溶液を、0℃に冷却した。水酸化リチウム(0.32g)の水(25ml)溶液を一度に加え、0℃で撹拌を続けた。15分後、0.5MのHClを一定ずつ加えることによってpHを約7に調節した。次いで、混合物を水(100ml)で希釈し、EtOAc(3×100ml)で抽出した。合わせた有機層をブライン(100ml)で洗浄し、乾燥させ(Na2SO4)、蒸発させ、茶色がかった発泡体を得た。
【0194】
中間体60及び61d):
【化126】
アルゴン下で、中間体60及び61c)(375mg)、(R)−2−ジメチルアミノ−ブタン−1−オール(297mg)、及びトリフェニルホスフィン(664mg)のTHF(15ml)溶液を、0℃に冷却した。このアゾジカルボン酸ジエチル、40%トルエン溶液(1.16ml)を一滴ずつ加えた。冷却槽を取り外し、撹拌を室温で3h続けた。全ての揮発性物質の蒸発によって茶色の油を得て、これをクロマトグラフィーによって最初に精製し(Ph3POの除去)、次いで分取HPLCによって、異性体を分離した。両方の異性体を茶色がかった樹脂として得た。
【0195】
中間体60e):
【化127】
DCM(1ml)とMeOH(1ml)との混合物中のBoc保護された中間体60及び61d)P1(82mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、2ml)を加え、溶液を室温で10分間撹拌した。全ての揮発性物質の蒸発、並びにトルエン及びアセトン(各々5ml)との同時蒸発によって所望の生成物が白色固体としてもたらされた。
【0196】
実施例60:
【化128】
中間体60e)(95mg)及びB−C部分1(89mg)、1−ヒドロキシ−ベンゾトリアゾール水和物(47mg)及びN−メチルモルホリン(86μl)を、DMF(3ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(71mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(18μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(50ml)で希釈し、飽和Na2CO3(3×25ml)及び水(25ml)で洗浄した。全ての水層を混合し、EtOAc(25ml)で再抽出した。次いで、合わせた有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、真空中で蒸発させた。残渣を分取HPLCによって精製し、相当するアミンを黄色がかった樹脂として得た。これをEtOAc(1ml)に溶解し、塩化水素のジエチルエーテル溶液(1.0M、207μl)で処理した。相当する二塩酸塩の完全な沈殿を得るために、このように得られた懸濁液をヘキサン(5ml)で希釈した。固体を濾過し、ヘキサンで洗浄し、デシケーター(シカペント)中で一晩乾燥させ、白色の固体の形態の所望の化合物を得た。
【0197】
中間体61e):
【化129】
DCM(1ml)とMeOH(1ml)との混合物中のBoc保護された中間体60及び61d)P2(40mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、2ml)を加え、溶液を室温で10分間撹拌した。全ての揮発性物質の蒸発、並びにトルエン及びアセトン(各々5ml)との同時蒸発によって所望の生成物が白色固体としてもたらされた。
【0198】
実施例61:
【化130】
中間体61e)(48mg)及びB−C部分1(43mg)、1−ヒドロキシ−ベンゾトリアゾール水和物(23mg)及びN−メチルモルホリン(42μl)を、DMF(3ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(35mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(9μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(50ml)で希釈し、飽和Na2CO3(3×25ml)及び水(25ml)で洗浄した。全ての水層を混合し、EtOAc(25ml)で再抽出した。次いで、合わせた有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、真空中で蒸発させた。粗生成物を分取HPLCによって精製し、相当するホルメートを無色の樹脂として得た。
【0199】
実施例63の合成:
中間体63a):
【化131】
n−ブチルリチウム(2.5M)のヘキサン(8.97ml)溶液を、アルゴン雰囲気下で冷却し(−50℃)撹拌した乾燥THF(200ml)に加えた。次いで、2,2,6,6−テトラメチルピペリジン(4.13ml)を加えた。混合物を0℃に20分間温めた。次いで、混合物を−78℃とした。この混合物に、フルオロピラジン(2.00g)のTHF(50ml)溶液を加えた。撹拌をこの温度で5分間続けた。次いで、ヨウ素(10.4g)のTHF(50ml)溶液を導入し、撹拌を−78℃で1h続けた。次いで、35%塩酸水溶液(20ml)、EtOH(20ml)及びTHF(50ml)の溶液を使用して加水分解を−78℃で行った。溶液を室温に温め、飽和NaHCO3で僅かに塩基性(pH10)とした。溶液をチオ硫酸ナトリウムで脱色し、蒸発させ、有機溶媒を除去した。残渣を水(150ml)で希釈し、DCM(3×200ml)で抽出した。有機抽出物を乾燥させ(Na2SO4)、蒸発させた。粗生成物をカラムクロマトグラフィーによって精製し、黄色がかった結晶として所望の化合物を得た(mp.41〜44℃)。
【0200】
中間体63b):
【化132】
火炎乾燥シュレンクフラスコに、中間体63a)(1.00g)、[1,1’−ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)のジクロロメタンとの錯体(109mg)、ヨウ化銅(I)(51mg)、及び乾燥N,N−ジメチルアセトアミド(6ml)を入れた。このように得られた混合物を、交互の真空/アルゴンパージで脱気した。次いで、1−tert−ブトキシカルボニルピペリジン−4−イル)(ヨード)亜鉛(6.25mmol、上記のように調製)を加えた。混合物をもう一度脱気し、次いで80℃に一晩加熱した。次いで、N,N−ジメチルアセトアミドの主要部分を蒸発させ、残りをEtOAc及び水の混合物(各々100ml)に溶解した。次いで、混合物をセライトで濾過し、分液漏斗に移した。相を分離し、水層をEtOAc(3×30ml)で抽出した。合わせた有機層を水及びブライン(各々75ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をカラムクロマトグラフィーによって精製し、茶色がかった油の形態で所望の化合物を得た。
【0201】
中間体63c):
【化133】
(S)−1−ピロリジン−1−イル−ブタン−2−オール(211mg)のDMF(2ml)溶液を、アルゴン下で0℃にて水素化ナトリウム(鉱油(39mg)中60%分散物)のDMF(1ml)懸濁液に加えた。冷却槽を取り外し、混合物を室温で1h撹拌した。次いで、中間体63b)(138mg)のDMF(2ml)溶液を加え、反応混合物を60℃に1h加熱した。反応混合物を0℃に冷却し、次いで飽和NH4Cl(1ml)で加水分解した。EtOAc(70ml)を加え、混合物をNaHCO3(2×25ml)で洗浄した。合わせた水層をEtOAc(25ml)で再抽出し、次いで合わせた有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物を分取HPLCによって精製し、黄色がかった樹脂の形態の相当するホルメートを得た。
【0202】
中間体63d):
【化134】
DCM(1ml)とMeOH(1ml)との混合物中のBoc保護された中間体63c)(181mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、2ml)を加え、溶液を室温で2h撹拌した。全ての揮発性物質の蒸発、並びにトルエン(2×5ml)、アセトン(5ml)、及びエーテル(5ml)との同時蒸発によってベージュ色の固体がもたらされ、それをデシケーター中シカペント上で一晩さらに乾燥させた。
【0203】
実施例63:
【化135】
中間体63d)(67mg)及びB−C部分1)(68mg)、1−ヒドロキシベンゾトリアゾール水和物(36mg)及びN−メチルモルホリン(66μl)を、DMF(3ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(54mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(14μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(70ml)で希釈し、飽和Na2CO3(3×25ml)、水及びブライン(各々25ml)で洗浄した。有機層を乾燥させ(Na2SO4)、溶媒を真空中で除去した。カラムクロマトグラフィーによる粗生成物の精製によって茶色がかった樹脂の形態の相当するアミンを得た。これをEtOAc(1ml)に溶解し、塩化水素のジエチルエーテル溶液(1.0M、178μl)で処理した。相当する塩酸塩の完全な沈殿を得るために、このように得られた懸濁液をヘキサン(5ml)で希釈した。固体を濾過し、ヘキサンで洗浄し、デシケーター中シカペント上で一晩乾燥させ、オフホワイトの固体の形態の所望の化合物を得た。
【0204】
実施例64の合成:
中間体64a):
【化136】
内部温度計を備えた三つ口フラスコに乾燥THF(80ml)を入れ、アルゴン雰囲気下で−10℃に冷却した。磁気撹拌子で撹拌する間、四塩化チタンの1M溶液(40ml)を、温度を−10℃〜−5℃に維持するために30分以内で一滴ずつ加えた。さらに30分間−5℃にて撹拌後、マロン酸ジメチル(2.286ml)及びN−Boc−ピペリジン−4−オン(4.384g)を−15℃〜−10℃で加えた。反応混合物は茶色の懸濁液となった。温度を−10℃未満に保つ一方、無水ピリジン(6.632ml)のTHF(14ml)溶液を30分以内で加えた。反応物は透明な茶色の溶液となった。冷却槽を取り外し、撹拌を室温で46h続けた。THFを蒸発させ、残りの油を1NのNaHCO3中で懸濁させた。沈殿しているチタン塩を濾過し、酢酸エチルで洗浄した。濾液を、第2の部分の1NのNaHCO3及びブラインで洗浄した。有機相をMgSO4上で乾燥させ、濾過し、蒸発させ、オレンジ色の油を得た。それをシリカゲルカラムクロマトグラフィーによって精製した。
【0205】
中間体64b):
【化137】
250mlの二つ口フラスコ中で、中間体64a)(3.16g)をメタノール(70ml)と水(0.5ml)との混合物に溶解した。フラスコを空にし、アルゴンでフラッシュした。活性炭担持5%パラジウム(400mg)を加え、フラスコを空にし、水素でフラッシュした。反応混合物を、水素雰囲気下にて(1atm)室温で一晩激しく撹拌した。触媒をセライトで濾過し、フィルターをメタノール(150ml)で洗浄した。濾液を真空中で蒸発させた。粗生成物をDCM(15ml)に溶解し、0.45μmのシリンジフィルターで濾過し、真空中で蒸発させた。透明な無色の油を得て、それを高真空下にて一晩乾燥させた。
【0206】
中間体64c):
【化138】
ナトリウムメトキシドは、ナトリウム(306mg)を冷却したメタノール(6.0ml)に溶解することによって調製した。塩酸アセトアミジン(415mg)をこの混合物に加え、沈殿している塩化ナトリウムを25mmのシリンジフィルターによる濾過によって除去した。中間体64b)(1.40g)をすばやく撹拌されている溶液に加えた。形成された僅かに濁った溶液を2日間室温に保った。溶媒を真空中で蒸発させた。残渣を水(10ml)に溶解し、水相をトルエン(6ml)で抽出した。有機相を廃棄した。水層を0℃に冷却し、pH4に達するまで6NのHClを一滴ずつ加えた。沈殿した無色の固体を吸引濾過によって集め、冷水(4ml)で洗浄した。生成物を真空中で乾燥させた。
【0207】
中間体64d):
【化139】
Boc保護中間体64c)を、ジオキサン(6ml)とメタノール(1ml)との混合物に懸濁させた。ジオキサン(6ml)中の4NのHClを加えた。反応物を室温で1h撹拌した。トルエン(2×20ml)との同時蒸発を含めた全ての揮発性物質の蒸発によって白色の固体を得て、それを高真空中で一晩乾燥させた。粗生成物を乾燥ジエチルエーテル(6ml)で粉砕した。溶媒をデカントし、生成物を真空中で乾燥させた。
【0208】
中間体64e):
【化140】
Z−OSu(546mg)のDMF(10ml)溶液を、トリエチルアミン(0.55ml)で処理した。中間体64d)のDMF(3ml)懸濁液を加え、反応物を室温で15時間撹拌した。溶媒を真空中で蒸発させ、残りのオフホワイトの固体をジエチルエーテル(2×)で粉砕した。生成物を濾過によって単離し、真空中で乾燥させた。
【0209】
中間体64f):
【化141】
中間体64e)をアセトニトリル(4ml)に懸濁させ、sym−コリジン(0.231ml)で処理した。POCl3(0.54ml)のアセトニトリル(2ml)溶液を0℃で一滴ずつ加え、反応物を還流温度で8h撹拌した。溶媒及び過剰な試薬を蒸留によって真空中で除去した。残りの茶色の固体を氷冷の酢酸エチルに溶解し、水、1NのNaHCO3及びブラインで抽出した。有機相をMgSO4上で乾燥させ、濾過し、真空中で蒸発させた。
【0210】
中間体64g):
【化142】
火炎乾燥フラスコ中で、水素化ナトリウム(鉱油中60%、18.9mg)を、アルゴン雰囲気下でTHF(5ml)に懸濁させた。(R)−2−ジメチルアミノ−ブタン−1−オール(110mg)を一滴ずつ加え、混合物を室温で1h撹拌し、相当するナトリウムアルコキシドが形成された。ジクロロピリミジン中間体64f)を、アルゴン雰囲気下にてTHF(3ml)に溶解した。溶液を0℃に冷却し、上記の調製したナトリウムアルコラートを30分以内で一滴ずつ加えた。混合物を室温に温め、室温で5h撹拌した。さらなる0.15当量の上記のアルコキシドを0℃で加え、室温で3h撹拌を続けた。反応混合物を真空中で蒸発させ、琥珀色の粘性油を得て、それを酢酸エチルに溶解した。溶液を1NのNaHCO3及びブラインで洗浄し、MgSO4上で乾燥させ、濾過し、蒸発させた。粗生成物をクロマトグラフィーによって精製した。
【0211】
中間体64h):
【化143】
中間体64g)(115mg)をメタノール(5ml)に溶解した。酸化カルシウム(84mg)、続いて10%パラジウム炭素(100mg)を加えた。混合物を真空中で繰り返し脱気し、アルゴン、最後に水素でフラッシュした。反応物を水素バルーン下で一晩撹拌した。反応物をセライトで濾過し、フィルターを80mlのメタノールで洗浄した。溶媒を蒸発させ、白色混濁状の残渣を酢酸エチルで希釈し、1NのNa2CO3及びブラインで抽出した。有機相をMgSO4上で乾燥させ、濾過し、蒸発させ、所望の生成物を無色の油として得た。
【0212】
実施例64:
【化144】
DMF(2.6ml)中のB−C部分1(84.5mg)、HOBt(42.8mg)、EDCI(76mg)及びN−メチルモルホリン(56.4μl)を、室温で1時間撹拌した。中間体64h)(68.0mg)を鮮黄色の溶液に加え、撹拌を一晩続けた。DMFを真空中で蒸発させ、残りの黄色の油を酢酸エチル(100ml)に溶解した。有機溶液を、1NのNa2CO3(2×30ml)及びブラインで抽出した。各水相を酢酸エチルで一度再抽出した。合わせた有機層を乾燥させ(MgSO4)、濾過し、真空中で蒸発させた。生成物を分取HPLCによって精製した。
【0213】
実施例73の合成:
中間体73a):
【化145】
5−クロロ−3−フルオロ−ピリジン−2−オール(1.04g)をピリジン(13ml)に溶解し、アルゴン下0℃に冷却した。次いで、トリフルオロメタンスルホン酸無水物(1.36ml)を、シリンジを介して一滴ずつ加えた。室温で一晩撹拌した後、反応混合物を真空中で濃縮した。残りを水及びジエチルエーテル(各々100ml)に分配した。水層を、ジエチルエーテル(2×50ml)で抽出し、合わせたエーテル抽出物を水及びブライン(各々50ml)で洗浄した。Na2SO4上での乾燥及び溶媒の蒸発によって、茶色の油を得た。カラムクロマトグラフィーによる精製によって、黄色がかった液体の形態の所望の化合物がもたらされた。
【0214】
中間体73b):
【化146】
火炎乾燥シュレンクフラスコに、中間体73a)(956mg)、[1,1’−ビス(ジフェニルホスフィノ)フェロセン]ジクロロパラジウム(II)のジクロロメタンとの錯体(84mg)、ヨウ化銅(I)(39mg)、及び乾燥N,N−ジメチルアセトアミド(5ml)を入れた。このように得られた混合物を交互の真空/アルゴンパージで脱気した。次いで、1−tert−ブトキシカルボニルピペリジン−4−イル)(ヨード)亜鉛(4.89mmol、上記のように調製)を加えた。混合物をもう一度脱気し、次いで80℃に一晩加熱した。次いで、N,N−ジメチルアセトアミドの主要部分を蒸発させ、残りをEtOAc及び水の混合物(各々75ml)に溶解した。次いで、これをセライトで濾過し、分液漏斗に移した。相を分離し、水層をEtOAc(2×50ml)で抽出した。合わせた有機層を水及びブライン(各々50ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をカラムクロマトグラフィーによって精製し、茶色がかった固体の形態で所望の化合物を得た。
【0215】
中間体73c):
【化147】
(R)−2−ジメチルアミノ−ブタン−1−オール(252mg)のDMF(1ml)溶液を、アルゴン下で0℃にて水素化ナトリウム(鉱油(64mg)中60%分散物)のDMF(2ml)懸濁液に加えた。冷却槽を取り外し、混合物を室温で1h撹拌した。次いで、中間体73b)(338mg)のDMF(2ml)溶液を加え、反応混合物を120℃に一晩加熱した。反応混合物を0℃に冷却し、次いで飽和NH4Cl(1ml)で加水分解した。EtOAc(50ml)を加え、混合物をNaHCO3(2×25ml)で洗浄した。合わせた水層をEtOAc(25ml)で再抽出し、次いで合わせた有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をクロマトグラフィーによって精製し、所望の生成物を黄色がかった油として得た。
【0216】
中間体73d):
【化148】
DCM(1ml)とMeOH(1ml)との混合物中のBoc保護された中間体73c)(380mg)に、塩化水素の1,4−ジオキサン溶液(4.0M、2ml)を加え、溶液を室温で1h撹拌した。全ての揮発性物質の蒸発、並びにトルエン及びアセトン(各々5ml)との同時蒸発によって白色の固体がもたらされ、それをデシケーター(シカペント)中で一晩さらに乾燥させた。
【0217】
実施例73:
【化149】
中間体73d)(166mg)及び(R)−3−(2,4−ジメチル−フェニル)−2−[(ピロリジン−1−カルボニル)−アミノ]−プロピオン酸(130mg)、1−ヒドロキシ−ベンゾトリアゾール水和物(79mg)及びN−メチルモルホリン(143μl)を、DMF(5ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(118mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(30μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(50ml)で希釈し、飽和Na2CO3(3×25ml)及び水(25ml)で洗浄した。全ての水層を混合し、EtOAc(2×25ml)で再抽出した。次いで、合わせた有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、真空中で蒸発させた。残渣を分取HPLCによって精製し、相当するホルメートを黄色がかった樹脂として得た。
【0218】
実施例85の合成:
中間体85a):
【化150】
ワング樹脂(500mg、Wang resin co.、100〜200メッシュ(Nova、01−64−5027)、1.1mmol/g)を、DMF(5ml)中のDMAP(5mg)の存在下で、DIC(255μl)で活性化したFmoc−D−2,4−ジクロロフェニルアラニン(753mg)の溶液で処理した。混合物を一晩反応させた。過剰の試薬を濾過によって除去した。樹脂をDMF(3×4ml)で洗浄した。DMF(3ml)、続いて無水酢酸(260μl)を加えた。混合物を1h反応させた。過剰の試薬を濾過によって除去した。樹脂結合中間体を、DMF(3×4ml)、MeOH(3×4ml)、THF(3×4ml)、DCM(3×4ml)及びジエチルエーテル(3×4ml)で連続的に洗浄した。樹脂を減圧下で乾燥させた。
【0219】
中間体85b):
【化151】
中間体85a)(200mg)を、20%ピペリジンのDCM(4ml)溶液で処理した。混合物を30分間反応させた。過剰の試薬を濾過によって除去した。樹脂結合中間体を、DMF(3×2ml)、MeOH(3×2ml)、THF(3×2ml)、DCM(3×2ml)及びジエチルエーテル(3×2ml)で連続的に洗浄した。樹脂を減圧下で乾燥させた。
【0220】
中間体85c):
【化152】
中間体85b)(200mg)を、冷たい4−ニトロフェニルクロロホルメート(222mg)及びトリエチルアミン(216μl)のDCM(2.5ml)溶液で処理した。混合物を室温で2h反応させた。過剰の試薬を濾過によって除去した。樹脂結合中間体を、DCM(3×2ml)、THF(3×2ml)、DCM(3×2ml)及びジエチルエーテル(3×2ml)で連続的に洗浄した。樹脂を減圧下で乾燥させた。
【0221】
中間体85d):
【化153】
中間体85c)(200mg)を、ビス(2−メトキシエチル)−アミン(322μl)のDCM(2ml)溶液で処理した。混合物を室温で2h反応させた。過剰の試薬を濾過によって除去した。樹脂結合中間体を、DMF(3×3ml)、MeOH(3×3ml)、THF(3×3ml)、DCM(3×3ml)及びジエチルエーテル(3×3ml)で連続的に洗浄した。樹脂を減圧下で乾燥させた。DCM中の20%TFA(3ml)を樹脂に加え、混合物を30分間反応させた。生成物を濾過し、溶媒を除去した。
【0222】
実施例85:
【化154】
DMF(2ml)中の中間体85d)(95mg)に、中間体3−((R)−2−ピロリジン−1−イル−ブトキシ)−1’,2’,3’,4’,5’,6’−ヘキサヒドロ−[2,4’]ビピリジニル三塩酸塩(91mg)、N−メチルモルホリン(109μl)、及びHOBt(37mg)を加え、混合物を20分間撹拌した。EDCI(46mg)を加え、撹拌を一晩続けた。反応混合物をブライン(20ml)に注ぎ、酢酸エチルで希釈し、有機相を分離した。水相を酢酸エチルで2度抽出した。合わせた有機相を飽和炭酸水素ナトリウム溶液で2度、水及びブラインで2度洗浄し、Na2SO4上で乾燥させ、濃縮した。粗生成物を分取LC−MSで精製し、引き続いて80%tBuOH水溶液から凍結乾燥し、淡黄色の固体を得た。
【0223】
実施例88の合成:
中間体88a):
【化155】
中間体85c)(200mg)を、チアゾリジン(172μl)のDCM(2ml)溶液で処理した。混合物を室温で3h反応させた。DBU(164μl)を加え、混合物を室温で1h反応させた。過剰の試薬を濾過によって除去した。樹脂結合中間体を、DMF(3×3ml)、MeOH(3×3ml)、THF(3×3ml)、DCM(3×3ml)及びジエチルエーテル(3×3ml)で連続的に洗浄した。樹脂を減圧下で乾燥させた。DCM中の20%TFA(3ml)を樹脂に加え、混合物を30分間反応させた。生成物を濾過し、溶媒を除去した。
【0224】
実施例88:
【化156】
DMF(2ml)中の中間体88a)(60mg)に、3−((R)−2−ピロリジン−1−イル−ブトキシ)−1’,2’,3’,4’,5’,6’−ヘキサヒドロ−[2,4’]ビピリジニル三塩酸塩(66mg)、N−メチルモルホリン(79μl)、及びHOBt(28mg)を加え、混合物を20分間撹拌した。EDCI(35mg)を加え、撹拌を一晩続けた。反応混合物をブライン(20ml)に注ぎ、酢酸エチルで希釈し、有機相を分離した。水相を酢酸エチルで2度抽出した。合わせた有機相を飽和炭酸水素ナトリウム溶液で2度、水及びブラインで2度洗浄し、Na2SO4上で乾燥させ、濃縮した。粗生成物を分取LC−MSで精製し、引き続いて80%tBuOH水溶液から凍結乾燥し、白色の固体を得た。
【0225】
実施例99の合成:
中間体99a):
【化157】
火炎乾燥フラスコ中で、3−オキソ−アゼチジン−1−カルボン酸tert−ブチルエステル(500mg)をアルゴン下でTHF(6ml)に溶解し、溶液を0℃に冷却した。臭化メチルマグネシウム(ジエチルエーテル中3M溶液、1.95ml)を一滴ずつ加え、ミルク状懸濁液を3h撹拌した。反応物を飽和水性NH4Clで注意深く加水分解し、EtOAc(80ml)で抽出した。有機相をブラインで洗浄し、Na2SO4上で乾燥させ、濾過し、蒸発させ、生成物を無色の固体として得た。
【0226】
中間体99b):
【化158】
トリフルオロ酢酸(4ml)を、中間体99a)のDCM(40ml)溶液に室温で一滴ずつ加えた。混合物を90分間撹拌した。溶媒の半分を蒸発させ、次いでトルエンを加え、蒸発を続けた。全ての溶媒を蒸発させる前に、この同時蒸発手順を2度繰り返した。残りの生成物を高真空下にて一晩乾燥させた。
【0227】
中間体99c):
【化159】
氷冷の中間体99b)(4.451g)及びトリエチルアミン(2.34ml)のDMF(30ml)溶液に、(R)−3−(2,4−ジクロロ−フェニル)−2−イソシアナト−プロピオン酸メチルエステル(3.048g)の無水DMF(50ml)溶液をアルゴン下で加えた。混合物を0℃で4h撹拌させた。反応混合物を真空中で蒸発させた。残渣をEtOAc(300ml)中で希釈し、有機相を0.1NのHCl(2×90ml)、0.1NのNaHCO3及びブラインで洗浄した。水相をEtOAcで再抽出し、合わせた有機相をNa2SO4下で乾燥させ、濾過し、蒸発させた。粗化合物をカラムクロマトグラフィーで精製し、無色の安定した発泡体を得た。
【0228】
中間体99d):
【化160】
中間体99c)をMeOH(2ml)とTHF(6ml)との混合物に0℃で溶解した。水酸化リチウム一水和物(73mg)のH2O(2ml)溶液を加えた。混合物を0℃で3h撹拌した。1NのHClを加えることによって反応混合物を中和させ、MeOH及びTHFを真空中で蒸発させた。1NのHCl(pH約1〜2)で水相を酸性化した。水相をEtOAc(30ml)で抽出した。有機層を水及びブラインで洗浄した。水相をEtOAcで再抽出し、合わせた有機層をNa2SO4上で乾燥させ、濾過し、蒸発させた。粗生成物を高真空下にて乾燥させ白色の発泡体を得た。
【0229】
実施例99:
【化161】
DMF(50ml)中の中間体99d)(2.523g)、HOBt(1.113g)、EDCI(1.974g)及びNMM(1.47ml)を、1時間室温で撹拌した。3−((R)−2−ピロリジン−1−イル−ブトキシ)−1’,2’,3’,4’,5’,6’−ヘキサヒドロ−[2,4’]ビピリジニル三塩酸塩(2.050g)及び第2の部分のNMM(1.33ml)を加え、撹拌を一晩続けた。DMFを真空中で蒸発させ、残りのオレンジ色の油を酢酸エチル(300ml)に溶解した。有機溶液を1NのNa2CO3(2×150ml)及びブラインで洗浄した。各水相を酢酸エチルで一度再抽出した。有機層を乾燥させ(Na2SO4)、濾過し、真空中で蒸発させた。粗生成物を分取HPLCによって精製した。
【0230】
実施例125の合成:
中間体125a):
【化162】
酢酸水銀(1.625g)を水(6ml)に溶解した。3−メチレン−N−Boc−ピペリジン(0.986g)のTHF(6ml)溶液を室温にて一滴ずつ加えた。20分後、酢酸水銀を溶解し、当初黄色の溶液は透明及び無色になった。混合物をさらに40分室温で撹拌した。反応物を氷水中で冷却し、3Nの水酸化ナトリウム溶液(5ml)を加えた。茶色の色が現れた。次いで、水素化ホウ素ナトリウム(0.19g)のNaOH(3N)(5ml)溶液を加え、混合物を15分間撹拌した。pH6となるまで酸性酸を加え、水素発生が止まった。混合物を沈殿した水銀からデカントし、蒸発させた。残渣を水及び酢酸エチルに分配した。有機相を水及びブラインで洗浄し、MgSO4上で乾燥させ、濾過し、蒸発させた。淡黄色の油が残り、それをカラムクロマトグラフィーによって精製した。生成物を、透明で無色の粘性油の形態で得て、それを高真空中で乾燥させた。
【0231】
中間体125b):
【化163】
中間体125a)(846mg)をジオキサン(30ml)に溶解した。ジオキサン(30ml)中の4NのHClを加え、反応物を室温で3.5時間撹拌した。溶媒を蒸発させ、残渣をトルエンと数回同時蒸発させた。残りの淡黄色の固体を洗浄し、高真空下にて一晩乾燥させた。
【0232】
中間体125c):
【化164】
氷冷の(R)−3−(2,4−ジクロロ−フェニル)−2−イソシアナト−プロピオン酸メチルエステル(271mg)のDMF(2.5ml)溶液に、中間体125b)(300mg)及びEt3N(279μl)のDMF(3ml)溶液を加えた。混合物を0℃で4h撹拌させた。反応混合物を真空中で蒸発させた。残渣をEtOAc(50ml)で希釈し、有機相を0.1NのHCl(2×20ml)及びブラインで洗浄した。水相をEtOAcで再抽出し、合わせた有機相をNa2SO4上で乾燥させ、濾過し、蒸発させた。
【0233】
中間体125d):
【化165】
中間体125c)(404mg)を、MeOH(1.5ml)及びTHF(5.0ml)に0℃で溶解した。水酸化リチウム一水和物(83mg)のH2O(1.5ml)溶液を加えた。混合物を0℃で1.5h撹拌した。1NのHClを加えることによって反応混合物を中和させ、MeOH及びTHFを真空中で蒸発させた。1NのHCl(pH約1〜2)で水相を酸性化した。水相を酢酸エチル(50ml)で抽出した。有機層を水及びブラインで洗浄した。水相をEtOAcで再抽出し、合わせた有機層をNa2SO4上で乾燥させ、真空中で蒸発させ、無色の発泡体を得た。
【0234】
実施例125:
【化166】
DMF(3ml)中の中間体125d)(190mg)、HOBt(64.7mg)、EDCI(138mg)及びNMM(102μl)を、室温で1時間撹拌した。[(R)−1−(1’,2’,3’,4’,5’,6’−ヘキサヒドロ−[2,4’]ビピリジニル−3−イルオキシメチル)−プロピル]−ジメチル−アミン三塩酸塩(163mg)及び第2の部分のNMM(93μl)を加え、撹拌を一晩続けた。DMFを真空中で蒸発させ、残りの黄色の油を酢酸エチル(100ml)に溶解した。有機溶液を1NのNa2CO3(2×30ml)及びブラインで洗浄した。各水相を酢酸エチルで一度再抽出した。有機層を乾燥させ(Na2SO4)、濾過し、真空中で蒸発させた。残渣を分取HPLCによって精製した。
【0235】
実施例127の合成:
中間体127a):
【化167】
DCM(20ml)中のBoc−(S)−1−アゼチジン−2−イル−メタノール(J.Med.Chem.2005、48、7637〜7647)(1.47g)に、TFA(10ml)を0℃で加え、溶液を前記温度で1.5h撹拌した。全ての揮発性物質の蒸発、並びにトルエン(2×20ml)との同時蒸発によって、所望の化合物が濁った黄色の油としてもたらされた。
【0236】
中間体127b):
【化168】
氷冷の(R)−3−(2,4−ジクロロ−フェニル)−2−イソシアナト−プロピオン酸メチルエステル(上記の合成)(587mg)の無水DCM(6ml)溶液に、中間体127a)(1.51g)及びトリエチルアミン(0.90ml)の無水DMF(4ml)溶液をアルゴン下で加えた。全ての揮発性物質を蒸発させる前に、混合物を0℃で5h撹拌した。残りをEtOAc(100ml)で希釈し、有機相を0.1MのHCl(2×50ml)及び水(50ml)で洗浄した。全ての水相を混合し、EtOAc(2×50ml)で再抽出した。合わせた有機層をブライン(50ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をクロマトグラフィーによって精製し、所望のエステルを白色固体として得た。
【0237】
中間体127c):
【化169】
水酸化リチウム(36mg)の水(0.9ml)溶液を、アルゴン下で0℃にて中間体127b)(274mg)のMeOH(2.0ml)及びTHF(4.3ml)溶液に加えた。反応混合物を0℃で2h撹拌した。1MのHClを加えることによって混合物を中和させ、MeOH及びTHFの主要部分を真空中で除去した。次いで、残りを1MのHClによってpH3〜4に酸性化し、混合物をEtOAc(2×25ml)で抽出した。有機層を水及びブライン(各々25ml)で洗浄した。水相をEtOAc(25ml)で再抽出し、合わせた有機層を乾燥させ(Na2SO4)、蒸発させ、相当する酸を無色の発泡体として得た。
【0238】
実施例127:
【化170】
[(R)−1−(1’,2’,3’,4’,5’,6’−ヘキサヒドロ−[2,4’]ビピリジニル−3−イルオキシメチル)−プロピル]−ジメチル−アミン三塩酸塩(125mg)及び中間体127c)(123mg)、1−ヒドロキシ−ベンゾトリアゾール水和物(62mg)及びN−メチルモルホリン(114μl)を、DMF(5ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(94mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(24μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(50ml)で希釈し、飽和NaHCO3(3×25ml)で洗浄した。全ての水層を混合し、EtOAc(25ml)で再抽出した。次いで、合わせた有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、真空中で蒸発させた。残渣を分取HPLCによって精製し、所望のアミンを黄色がかった樹脂として得た。
【0239】
実施例129の合成:
中間体129a):
【化171】
トリフルオロメチルトリメチルシラン(0.93ml)及びフッ化セシウム(0.96g)を、ベンズヒドリル−アゼチジン−3−オン(1.00g)のTHF(12.5ml)溶液に加えた。室温で1h撹拌した後、飽和NH4Cl(12.5ml)及びフッ化テトラブチルアンモニウム(498mg)を加え、撹拌をさらに6h続けた。混合物を、ジエチルエーテル(3×50ml)で抽出し、有機層をMgSO4上で乾燥させた。真空濃縮によってオレンジ色の油を得て、それをクロマトグラフィーによって精製し、黄色の油の形態の所望のアルコールを得た。
【0240】
中間体129b):
【化172】
炭素担持20重量%水酸化パラジウム(202mg)及び1MのHCl(1.52ml)を、中間体129a)(466mg)のMeOH溶液に加えた。反応混合物を、1バールの水素下で室温にて3h水素化した。触媒をセライトで濾過し、メタノールで洗浄した。濾液を濃縮し、真空下で乾燥させた。次いで、残渣をヘキサンで洗浄した。有機層をデカントし、ベージュ色の固体を得た。
【0241】
中間体129c):
【化173】
氷冷の(R)−3−(2,4−ジクロロ−フェニル)−2−イソシアナト−プロピオン酸メチルエステル(上記の合成)(178mg)の無水DCM(3ml)溶液に、中間体129b)(230mg)及びトリエチルアミン(0.27ml)の無水DMF(2ml)溶液をアルゴン下で加えた。混合物を0℃で3h撹拌させた。次いで、混合物をEtOAc(50ml)で希釈し、有機相を0.1MのHCl(2×25ml)及び水(25ml)で洗浄した。全ての水相を混合し、EtOAc(2×50ml)で再抽出した。次いで、合わせた有機層をブライン(50ml)で洗浄し、乾燥させ(Na2SO4)、濾過し、真空中で濃縮した。粗生成物をクロマトグラフィーによって精製し、所望のエステルを無色の発泡体として得た。
【0242】
中間体129d):
【化174】
水酸化リチウム(17mg)の水(0.5ml)溶液を、アルゴン下で0℃にて中間体129c)(151mg)のMeOH(0.9ml)及びTHF(2.0ml)溶液に加えた。反応混合物を0℃で3h撹拌した。1MのHClを加えることによって混合物を中和させ、MeOH及びTHFの主要部分を真空中で除去した。次いで、残りを1MのHClによってpH3〜4に酸性化し、混合物をEtOAc(2×25ml)で抽出した。有機層を水及びブライン(各々25ml)で洗浄し、乾燥させ(Na2SO4)、蒸発させ、相当する酸を無色の樹脂として得た。
【0243】
実施例129:
【化175】
[(R)−1−(1’,2’,3’,4’,5’,6’−ヘキサヒドロ−[2,4’]ビピリジニル−3−イルオキシメチル)−プロピル]−ジメチル−アミン三塩酸塩(111mg)及び中間体129d)(125mg)、1−ヒドロキシ−ベンゾトリアゾール水和物(55mg)及びN−メチルモルホリン(100μl)を、DMF(5ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(82mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(100μl)を加え、撹拌を一晩続けた。反応混合物をEtOAc(50ml)で希釈し、飽和NaHCO3(3×25ml)で洗浄した。全ての水層を混合し、EtOAc(25ml)で再抽出した。次いで、合わせた有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、真空中で蒸発させた。残渣を分取HPLCによって精製し、所望のアミンを黄色がかった樹脂として得た。
【0244】
実施例130の合成:
中間体130a):
【化176】
火炎乾燥フラスコ中で、トリメチルスルホキソニウムヨージド(2.57g)及び水素化ナトリウム(鉱油中60%、476mg)を、DMSO(8.8ml)に10℃で懸濁させた。混合物を室温で1.5時間撹拌した。tert−ブチル−3−オキソピロリジン−1−カルボキシレート(2.00g)のDMSO(2.2ml)溶液を15分以内で一滴ずつ加えた。撹拌を1時間続けた。氷水(50ml)及びブライン(50ml)を加えることによって反応物を加水分解した。水性懸濁液を、ジエチルエーテル(2×50ml)で抽出した。合わせたエーテル溶液をブラインで洗浄し、MgSO4上で乾燥させ、濾過した。濾液を真空中で蒸発させ、粘性の茶色の油を得て、それをクーゲルロール蒸留によって精製した。生成物を、無色の油として得た。
【0245】
中間体130b):
【化177】
火炎乾燥フラスコ中、シアン化銅(I)(105mg)を、無水THF(2ml)に懸濁させた。懸濁液を−76℃に冷却し、シクロプロピルマグネシウムブロミド(THF中0.5M、9.34ml)を加えた。混合物を1時間撹拌し、続いて中間体130a)(155mg)の乾燥THF(1ml)溶液をゆっくりと加えた。反応物を室温にゆっくりと温め、一晩撹拌した。混合物を飽和水性NH4Cl及びジエチルエーテルに分配した。エーテル相を水及びブラインで洗浄した。各水相をジエチルエーテルで一度再抽出した。合わせた有機相をMgSO4上で乾燥させ、濾過し、蒸発させた。粗生成物をカラムクロマトグラフィーによって精製した。
【0246】
中間体130c):
【化178】
中間体130b)(116mg)をジオキサン(6ml)に溶解した。ジオキサン(6ml)中の4NのHClを加え、反応物を室温で1.5時間撹拌した。溶媒を蒸発させ、残渣をトルエンで数回同時蒸発した。残りの淡赤色の固体を高真空下にて乾燥させた。
【0247】
中間体130d):
【化179】
氷冷の(R)−3−(2,4−ジクロロ−フェニル)−2−イソシアナト−プロピオン酸メチルエステル(125mg)のDMF(4ml)溶液に、中間体130c)(89mg)及びトリエチルアミン(134μl)の無水DMF(2ml)溶液をアルゴン下で加えた。混合物を0℃で2時間、室温でさらに2時間撹拌させた。反応混合物を真空中で蒸発させた。残渣をDCM(80ml)で希釈し、有機相を0.1NのHCl(20ml)、1NのNaHCO3(20ml)及びブラインで洗浄した。各水相をDCMで再抽出し、次いで合わせた有機相をNa2SO4上で乾燥させ、濾過し、蒸発させた。薄茶色の油を得て、高真空下にて乾燥させた。粗生成物をカラムクロマトグラフィーによって精製した。
【0248】
中間体130e):
【化180】
中間体130d)(171mg)を、MeOH(1ml)及びTHF(5ml)に0℃で溶解した。水酸化リチウム一水和物(34.6mg)のH2O(1ml)溶液を加えた。混合物を0℃で4h撹拌した。1NのHClを加えることによって反応混合物を中和させ、MeOH及びTHFを真空中で蒸発させた。1NのHCl(pH約1〜2)で水相を酸性化し、EtOAc(1×20ml)で抽出した。有機層を水及びブラインで洗浄した。水相をEtOAcで再抽出し、合わせた有機層をNa2SO4上で乾燥させ、真空中で蒸発させ、白色の発泡体を得た。粗生成物をカラムクロマトグラフィーによって精製した。
【0249】
実施例130:
【化181】
[(R)−1−(1’,2’,3’,4’,5’,6’−ヘキサヒドロ−[2,4’]ビピリジニル−3−イルオキシメチル)−プロピル]−ジメチル−アミン三塩酸塩(123mg)、中間体130e)(165mg)及びHOBt(73mg)を、DCM(6ml)中で混合した。EDCI(73mg)及びNMM(132μl)を加えた。反応物を室温で1時間撹拌した。第2の部分のNMM(28μl)を加え、撹拌を一晩続けた。混合物をEtOAc(50ml)で希釈し、1NのNa2CO3(3×25ml)及びブラインで洗浄した。全ての水相をEtOAcで一度再抽出した。合わせた有機層を乾燥させ(Na2SO4)、濾過し、真空中で蒸発させた。粗生成物を分取HPLCによって精製した。
【0250】
実施例133の合成:
中間体133a):
【化182】
トリアセトキシ水素化ホウ素ナトリウム(5.88g)を、3−オキセタノン(1.00g)及びN−ベンジル−メチルアミン(2.52g、2.7ml)の1,2−ジクロロエタン(300ml)溶液に加え、直ちに続いて酢酸(1.6ml)を加えた。室温で18h激しく撹拌した後、このように得られた微細懸濁液を0℃に冷却し、200mlの水を注意深く加えることによって反応物をクエンチした。系を1h撹拌させ、残った試薬は完全な加水分解に達した。次いで、有機相を集め、水相をDCM(2×50ml)でさらに抽出した。次いで、合わせた有機相を飽和NaHCO3(2×50ml)、H2O(50ml)及びブライン(75ml)で洗浄した。Na2SO4上での乾燥、濾過及び溶媒の蒸発の後、化合物をオレンジ色の粗シロップとして得た。クロマトグラフィーによる精製によって、黄色がかった油の形態の所望の化合物を得た。
【0251】
中間体133b):
【化183】
炭素担持20重量%水酸化パラジウム(195mg)の存在下で1バールの水素下で室温にて、EtOAc(25ml)中の溶液中の中間体133a)(1.94g)を、一晩水素化した。上清のシリンジフィルターによる濾過及びEtOAc溶液(5ml)による触媒のさらなる洗浄によって、30mlのEtOAc中の所望の化合物のニート溶液を得た。
【0252】
中間体133c):
【化184】
氷冷の(R)−3−(2,4−ジクロロ−フェニル)−2−イソシアナト−プロピオン酸メチルエステル(上記のような合成)(450mg)の無水DCM(10ml)溶液に、中間体133b)のEtOAc(6ml;2.62mmol)溶液を加えた。混合物を0℃で1h撹拌させ、次いで室温に17hで戻らせた。この時点で全ての揮発性物質を蒸発させ、このように得られた無色のワックスを真空中で十分に乾燥させ、過剰の中間体133b)を除去し、所望のエステルを純粋な形態で得た。
【0253】
中間体133d):
【化185】
水酸化リチウム(80mg)の水(1.5ml)溶液を、アルゴン下で0℃にて中間体133c)(698mg)のMeOH(9.0ml)及びTHF(1.5ml)溶液に加えた。反応混合物を0℃で2h撹拌した。トリフルオロ酢酸(380mg;260μl)を加えることによって混合物を中和させ、MeOH及びTHFの主要部分を真空中で除去した。次いで、残りを水(10ml)で希釈し、EtOAc(3×25ml)で抽出した。有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、蒸発させ、高真空下での十分な乾燥の後、相当する酸を無色の発泡体として得た。
【0254】
実施例133:
【化186】
[(R)−1−(1’,2’,3’,4’,5’,6’−ヘキサヒドロ−[2,4’]ビピリジニル−3−イルオキシメチル)−プロピル]−ジメチル−アミン三塩酸塩(120mg)及び中間体133d)(153mg)、1−ヒドロキシ−ベンゾトリアゾール水和物(61mg)及びN−メチルモルホリン(119μl)を、DCM(15ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(88mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(26μl)を加え、撹拌を一晩続けた。揮発性物質を蒸発させ、残渣をEtOAc(50ml)及び飽和NaHCO3(25ml)に分配した。有機相を集め、飽和NaHCO3(2×25ml)でさらに洗浄した。水層を混合し、DCM(10ml)で抽出した。次いで、合わせた有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、真空中で蒸発させた。残渣を分取HPLCによって精製し、所望の誘導体の二ギ酸塩を黄色がかった樹脂として得た。
【0255】
実施例134の合成:
中間体134a):
【化187】
N−ベンジル−メチルアミン(1.05g、1.12ml)を、固体無水Na2CO3(2.00g)の存在下で3−ヒドロキシメチル−3−メチルオキセタンp−トシレート(2.23g)のアセトニトリル(35ml)溶液に加えた。次いで、懸濁液を室温で6日間激しく撹拌し、その時点で変換のさらなる進展は顕著ではなかった。全ての揮発性物質を蒸発させ、残渣をEtOAc(50ml)及び水(50ml)に分配した。有機相を集め、水及びブライン(各々25ml)でさらに洗浄した。合わせた水相をDCM(25ml)で抽出した。次いで、合わせた有機相をNa2SO4上で乾燥させ、濾過し、蒸発させ、部分的に結晶化した粗シロップを得た。クロマトグラフィーによる精製によって、黄色がかった油の形態の所望の化合物を得た。
【0256】
中間体134b):
【化188】
炭素担持20重量%水酸化パラジウム(60mg)の存在下、1バールの水素下で室温にて一晩、EtOAc(10ml)中の溶液中の中間体134a)(360mg)を水素化した。シリンジフィルターによる濾過及びEtOAc(5ml)による触媒のさらなる洗浄によって、15mlのEtOAc中の所望の化合物のニート溶液を得た。
【0257】
中間体134c):
【化189】
氷冷の(R)−3−(2,4−ジクロロ−フェニル)−2−イソシアナト−プロピオン酸メチルエステル(上記のような合成)(300mg)の無水DCM(9ml)溶液に、中間体134b)のEtOAc(15ml;1.75mmol)溶液を加えた。混合物を0℃で1h撹拌させ、次いで17hで室温に戻らせた。この時点で、全ての揮発性物質を蒸発させ、淡黄色のシロップ/ワックスを得た。粗生成物をクロマトグラフィーによって精製し、所望のエステルを、室温で保存すると結晶化する透明なシロップとして得た。
【0258】
中間体134d):
【化190】
水酸化リチウム(42mg)の水(0.8ml)溶液を、アルゴン下で0℃にて中間体134c)(305mg)のMeOH(5.0ml)及びTHF(0.8ml)溶液に加えた。反応混合物を0℃で2h撹拌した。トリフルオロ酢酸(200mg;135μl)を加えることによって混合物を中和させ、MeOH及びTHFの主要部分を真空中で除去した。次いで、残りを水(10ml)で希釈し、EtOAc(3×25ml)で抽出した。有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、蒸発させ、相当する酸を、高真空中で不安定な泡を形成する無色の粘性シロップとして得た。
【0259】
実施例134:
【化191】
[(R)−1−(1’,2’,3’,4’,5’,6’−ヘキサヒドロ−[2,4’]ビピリジニル−3−イルオキシメチル)−プロピル]−ジメチル−アミン三塩酸塩(100mg)及び中間体134d)(115mg)、1−ヒドロキシ−ベンゾトリアゾール水和物(52mg)及びN−メチルモルホリン(100μl)を、DCM(15ml)に溶解した。室温で30分間撹拌した後、N−(3−ジメチルアミノプロピル)−N−エチルカルボジイミド塩酸塩(75mg)を加え、撹拌をさらに1時間続けた。さらなる量のN−メチルモルホリン(22μl)を加え、撹拌を一晩続けた。揮発性物質を蒸発させ、残渣をEtOAc(50ml)及び飽和NaHCO3(25ml)に分配した。有機相を集め、飽和NaHCO3(2×25ml)でさらに洗浄した。水層を混合し、DCM(10ml)で抽出した。次いで、合わせた有機層をブライン(25ml)で洗浄し、乾燥させ(Na2SO4)、真空中で蒸発させた。残渣を分取HPLCによって精製し、所望の誘導体の二ギ酸塩を黄色がかった樹脂として得た。
【0260】
さらなる実施例を下記に例示する。
【表25】
【0261】
生物学的アッセイ
A 結合アッセイ
膜結合アッセイを使用して、ヒトメラノコルチン受容体を発現するHEK293細胞膜調製物に結合している蛍光標識NDP−α−MSHの競争的阻害剤を同定する。
【0262】
試験化合物又は非標識NDP−α−MSHを、様々な濃度で384ウェルマイクロタイタープレートに分注する。蛍光標識NDP−α−MSHを、単一の濃度で分注し、続いて膜調製物を加える。プレートを室温で5hインキュベートする。
【0263】
蛍光偏光の程度を蛍光偏光マイクロプレートリーダーで決定する。
【0264】
B 機能アッセイ
ヒトメラノコルチン受容体のアゴニスト活性を、均質膜をベースとするアッセイにおいて決定する。cAMP特異抗体上の結合部位の限られた数についての、非標識cAMPと一定量の蛍光標識cAMPとの間の競合は、蛍光偏光によって明らかになる。
【0265】
試験化合物又は非標識NDP−α−MSHを、様々な濃度で384ウェルマイクロタイタープレートに分注する。ヒトメラノコルチン受容体を発現するHEK293細胞からの膜調製物を加える。短いプレインキュベーション期間後、適切な量のATP、GTP及びcAMP抗体を加え、蛍光標識cAMPコンジュゲートを分注する前に、プレートをさらにインキュベートする。プレートを、蛍光偏光マイクロプレートリーダーで読み取る前に、4℃で2hインキュベートする。試験化合物への反応として生成されたcAMPの量を、NDP−α−MSHによる刺激からもたらされるcAMPの生成と比較する。
【0266】
本発明の代表的な化合物を試験し、メラノコルチン−4受容体に結合することを見出した。これらの化合物は一般に、2μM未満のIC50値を有することが見出された。
【表26】
【表27】
【表28】
【表29】
【表30】
【表31】
【0267】
C In vivo食物摂取モデル
1 自発的摂食パラダイム
試験化合物のi.p.、s.c.又はp.o.投与後に、ラットにおける食物摂取量を測定する(例えば、A.S.Chenら、Transgenic Res 2000 Apr;9(2):145〜154を参照されたい)。
【0268】
2 LPS誘発食欲不振症及び腫瘍誘発悪液質のモデル
試験化合物のラットへのi.p.又はp.o.投与によって、リポ多糖(LPS)投与により誘発される食欲不振症又は腫瘍増殖によって誘発される悪液質の予防又は寛解を決定する(例えば、D.L.Marks、N.Ling、及びR.D.Cone、Cancer Res 2001 Feb 15;61(4):1432〜1438を参照されたい)。
【0269】
D ラット交尾外アッセイ
性的に成熟している雄性帝王切開由来Sprague−Dawley(CD)ラット(60日齢超)(交尾外評価の間に陰茎が陰茎鞘への収縮するのを防止するために提靭帯を外科的に切除する)を使用する。動物に食物及び水を自由に与え、通常の明/暗サイクルで維持する。試験は明サイクルの間に行う。
【0270】
1 交尾外反射試験のための仰臥位拘束への条件付け
この条件付けは約4日かかる。1日目、動物を暗い拘束器内に入れ、15〜30分間放置する。2日目、動物を拘束器中にて仰臥位で15〜30分間拘束する。3日目、動物の陰茎鞘を収縮させて仰臥位で15〜30分間拘束する。4日目、動物の陰茎反応が観察されるまで陰茎鞘を収縮させて仰臥位で拘束する。何匹かの動物は、手順に完全に順応する前に、さらに何日かの条件付けが必要となる。非応答動物はさらなる評価から除外する。いかなる取り扱い又は評価の後も、正の強化を確実にするため、動物に褒美を与える。
【0271】
2 交尾外反射試験
ラットを、通常の頭及び足のグルーミングができる適切なサイズの円筒内に胴体腹側を入れて仰臥位で軽く拘束する。400〜500グラムのラットでは、円筒の直径は約8cmである。胴体下部及び後肢を、非粘着性材料(ベトラップ)で拘束する。陰茎亀頭を通す孔を設けたさらなる断片のベトラップで動物を固定し、包皮鞘を収縮した位置に保つ。典型的には交尾外性器反射試験と称される陰茎反応を観察する。典型的には、鞘収縮後数分内に一連の陰茎勃起が自発的に起こるであろう。通常の反射発生性勃起反応のタイプには、伸長、怒張、カップ及びフリップが挙げられる。伸長は、陰茎本体の延長として分類される。怒張は陰茎亀頭の拡張である。カップとは、陰茎亀頭の遠心縁が瞬間的に張ってカップを形成する強度の勃起として定義される。フリップは陰茎本体の背屈である。
【0272】
ベースライン及び又はビヒクル評価を行って、動物がどのように反応するか、及び反応するかどうかを判定する。動物の中には最初の反応までに長時間かかるものもあれば、一方他に全くの非応答動物がある。このベースライン評価の間に、最初の反応までの潜時、反応の回数及びタイプを記録する。試験時間枠は、最初の反応後15分である。
【0273】
評価と評価の間に最低1日おいた後、これらの同じ動物に、試験化合物を20mg/kgで投与し、陰茎反射を評価する。全ての評価を録画し、後で点数をつける。データを集め、対応のある両側t検定を使用して分析し、個々の動物についてベースライン及び/又はビヒクル評価を薬物処理評価と比較する。最小4匹の動物群を用いて、ばらつきを抑える。
【0274】
正の参照対照を各試験に含め、試験の有効性を確保する。動物には、実施する試験の特性によっていくつかの投与経路で投与することができる。投与経路には、静脈内(IV)、腹腔内(IP)、皮下(SC)及び脳内室(ICV)が挙げられる。
【0275】
E 雌性性的機能不全のモデル
雌の性受容性に関連するげっ歯類アッセイには、ロードシスの行動モデル及び交尾活動の直接観察が挙げられる。雄性及び雌性ラットの両方におけるオルガスムを測定するための、麻酔下の脊髄離断ラットにおける尿道性器反射モデルもまたある。これら及び雌性性的機能不全の他の確立した動物モデルは、K.E.McKennaら、A Model For The Study of Sexual Function In Anesthetized Male And Female Rats、Am.J.Physiol.(Regulatory Integrative Comp.Physiol 30):R1276〜R1285、1991;K.E.McKennaら、Modulation By Peripheral Serotonin of The Threshold For Sexual Reflexes In Female Rats、Pharm.Bioch.Behav.、40:151〜156、1991;及びL.K.Takahashiら、Dual Estradiol Action In The Diencephalon And The Regulation of Sociosexual Behavior In Female Golden Hamsters、Brain Res.、359:194〜207、1985において記載されている。
【0276】
医薬組成物の例
本発明の化合物の経口組成物の特定の実施形態として、十分に微粉化したラクトースと共に23mgの実施例6を製剤し、0号硬質ゼラチンカプセルを満たす580〜590mgの総量を提供する。
【0277】
本発明の化合物の経口組成物の他の特定の実施形態として、十分に微粉化したラクトースと共に28mgの実施例14を製剤し、0号硬質ゼラチンカプセルを満たす580〜590mgの総量を提供する。
【0278】
その特定の好ましい実施形態に関して本発明を説明し例示してきたが、本発明の趣旨及び範囲から逸脱することなく様々な変更、修正及び置換をその中で行うことができることを当業者なら理解するであろう。例えば、有効量は、以上の説明から明らかなような好ましい用量以外に、観察される特定の薬理学的反応の結果として適用可能であることがあり、それは選択した特定の活性化合物、並びに製剤及び用いる投与方法のタイプによって変化することがあり、結果におけるこのような予測される変動又は差異は、本発明の目的及び実施に従って意図されている。したがって、本発明は下記の特許請求の範囲によってのみ限定され、このような特許請求の範囲は妥当であればできるだけ広く解釈されることを意図する。
【特許請求の範囲】
【請求項1】
式(I)による化合物、
【化1】
並びにそのエナンチオマー、ジアステレオマー、互変異性体、溶媒和物及び薬学的に許容される塩
[式中、
R1は、−N(R10)−(C(R6)2)m−T、
−(C(R6)2)l−T、又は
−O−(C(R6)2)m−Tであり、
R6は、
H、
F、
OH、
OCH3、
C1〜6−アルキル(ハロゲン、CN、OH及びOCH3から選択される1〜3個の置換基で任意選択で置換されている)、並びに
C3〜6−シクロアルキル(ハロゲン、CN、OH及びOCH3から選択される1〜3個の置換基で任意選択で置換されている)から独立に選択され、
Tは、NR7R8、
【化2】
であり、
R7及びR8は、互いに独立に、
H、
C1〜6−アルキル、
C2〜6−アルケニル、
C2〜6−アルキニル、及び
C2〜6−アルキレン−O−C1〜6−アルキルから選択され、
各アルキル、アルケニル及びアルキニルは、1個若しくは複数のハロゲン原子、CN又はOHで任意選択で置換されており、
R9は、
ハロゲン、
CN、
OH、
C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、並びに
O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
C1〜6−アルキレン−O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、又は
NR12R13から独立に選択され、
R10は、H又は
C1〜6−アルキルであり、
R11は、
ハロゲン、
CN、
OH、
C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
C2〜6−アルケニル、
C2〜6−アルキニル、
O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
C1〜6−アルキレン−O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
C0〜6−アルキル−C3〜6−シクロアルキル、
−OC(O)C1〜6−アルキル、
−NH2、
−NH(C1〜6−アルキル)、及び
−N(C1〜6−アルキル)2から独立に選択され、
R12及びR13は、互いに独立に、
C1〜6−アルキル(OHで任意選択で置換されている)、
C2〜6−アルケニル、
C2〜6−アルキニル、
C2〜6−アルキレン−O−C1〜6−アルキル、及び
C2〜6−アルキレン−N−(C1〜6−アルキル)2から選択され、
Wは、CH、O又はNR10であり、
Xは、CH又はNであり、
Yは、CH又はNであり、
Zは、CH又はNであり、
Aは、0〜2個の窒素原子を含有する3〜7員の飽和、不飽和又は芳香族環であり、
Bは、CR2又はNであり、
Gは、CR2又はNであり、
Dは、CR2又はNであり、
Eは、CR2又はNであり、
ただし、可変部分B、G、D及びEの1つ又は2つは、Nでなければならず、
R2は、
H、
F、
Cl、
CH3、
OCH3、及び
CF3から独立に選択され、
R3は、H、
Cl、
F、又は
CH3であり、
R4は、Cl、
F、又は
CH3であり、
R5は、
【化3】
モルホリン(1〜3個の同じ若しくは異なる置換基R14で任意選択で置換されている)、
環中に1個の窒素原子と任意選択でO、N及びSから選択されるさらなるヘテロ原子とを含有する4〜7員の飽和又は部分不飽和複素環(複素環は、1〜4個の同じ若しくは異なる置換基R11で任意選択で置換されている)、又は
NR12R13であり、
R14は、C1〜6−アルキル、
C1〜6−アルキレン−O−C1〜6−アルキル、
C1〜6−アルキレン−OH、
C1〜6−アルキレン−NH2、
C1〜6−アルキレン−NH−C1〜6−アルキル、又は
C1〜6−アルキレン−N(C1〜6−アルキル)2であり、
R15は、H又は
C1〜6−アルキルであり、
lは、0、1、2、3又は4であり、
mは、0、1、2、3又は4であり、
oは、0、1又は2であり、
pは、0、1、2、3又は4であり、
rは、0、1、2、3又は4であり、
sは、1又は2であり、
tは、0又は1である]。
【請求項2】
R1が、−N(R10)−(C(R6)2)m−T、
−(C(R6)2)l−T、又は
−O−(C(R6)2)m−Tであり、
R6が、
H、
F、
OH、
OCH3、
C1〜6−アルキル(ハロゲン、CN、OH及びOCH3から選択される1〜3個の置換基で任意選択で置換されている)、並びに
C3〜6−シクロアルキル(ハロゲン、CN、OH及びOCH3から選択される1〜3個の置換基で任意選択で置換されている)から独立に選択され、
Tが、NR7R8、
モルホリン、
【化4】
であり、
R7及びR8が、互いに独立に、
H、
C1〜6−アルキル、
C2〜6−アルケニル、
C2〜6−アルキニル、及び
C2〜6−アルキレン−O−C1〜6−アルキルから選択され、
各アルキル、アルケニル及びアルキニルが、1個若しくは複数のハロゲン原子、CN又はOHで任意選択で置換されており、
R9が、
ハロゲン、
CN、
OH、
C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、並びに
O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
C1〜6−アルキレン−O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)から独立に選択され、
R10が、H又は
C1〜C6−アルキルであり、
R11が、
ハロゲン、
CN、
OH、
C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
C1〜6−アルキレン−O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
−NH2、
−NH(C1〜6−アルキル)、及び
−N(C1〜6−アルキル)2から独立に選択され、
Xが、CH又はNであり、
Yが、CH又はNであり、
Zが、CH又はNであり、
Aが、0〜2個の窒素原子を含有する3〜7員の飽和、不飽和又は芳香族環であり、
Bが、CR2又はNであり、
Gが、CR2又はNであり、
Dが、CR2又はNであり、
Eが、CR2又はNであり、
ただし、可変部分B、G、D及びEの1つ又は2つが、Nでなければならず、
R2が、
H、
F、
Cl、
CH3、
OCH3、及び
CF3から独立に選択され、
R3が、H、
Cl、
F、又は
CH3であり、
R4が、Cl又はFであり、
R5が、
【化5】
モルホリン(1〜3個の同じ若しくは異なる置換基R14で任意選択で置換されている)、又は
NR12R13であり、
R12及びR13が、互いに独立に、
C1〜6−アルキル、
C2〜6−アルケニル、
C2〜6−アルキニル、
C2〜6−アルキレン−O−C1〜6−アルキル、及び
C2〜6−アルキレン−N−(C1〜6−アルキル)2から選択され、
R14が、C1〜6−アルキル、
C1〜6−アルキレン−O−C1〜6−アルキル、
C1〜6−アルキレン−OH、
C1〜6−アルキレン−NH2、
C1〜6−アルキレン−NH−C1〜6−アルキル、又は
C1〜6−アルキレン−N(C1〜6−アルキル)2であり、
lが、0、1、2、3又は4であり、
mが、0、1、2、3又は4であり、
oが、0、1又は2であり、
pが、0、1、2、3又は4であり、
qが、0、1、2、又は3であり、
rが、0、1、2、3又は4であり、
sが、1又は2である、請求項1に記載の化合物。
【請求項3】
式(I’)による、請求項1又は2に記載の化合物
【化6】
(式中、B、G、D、E、R1、R3、R4及びR5は、請求項1又は2に記載の通りである)。
【請求項4】
R7及びR8の少なくとも1つが、
C2〜6−アルケニル、
C2〜6−アルキニル、及び
C2〜6−アルキレン−O−C1〜6−アルキルから選択される、請求項1〜3のいずれか一項に記載の化合物。
【請求項5】
R2が、H、F、Cl及びCH3から選択される、請求項1〜4のいずれか一項に記載の化合物。
【請求項6】
lが、2又は3であり、又は
mが、2又は3である、請求項1〜5のいずれか一項に記載の化合物。
【請求項7】
医薬品としての請求項1〜6のいずれか一項に記載の化合物。
【請求項8】
哺乳動物においてメラノコルチン−4受容体の不活性化又は活性化に対して応答性の障害、疾患又は状態の治療又は予防のための、請求項1〜6のいずれか一項に記載の化合物。
【請求項9】
癌悪液質、筋肉疲労、食欲不振症、筋萎縮性側索硬化症(ALS)、不安神経症及び/又はうつの治療又は予防のための、請求項8に記載の化合物。
【請求項10】
肥満症、真性糖尿病、男性若しくは女性の性的機能不全及び/又は勃起不全の治療又は予防のための、請求項8に記載の化合物。
【請求項11】
哺乳動物においてメラノコルチン−4受容体の不活性化又は活性化に対して応答性の障害、疾患又は状態の治療又は予防のための医薬品の調製のための、請求項1〜6のいずれか一項に記載の化合物の使用。
【請求項12】
請求項1〜6のいずれか一項に記載の化合物と、薬学的に許容される担体とを含む医薬組成物。
【請求項1】
式(I)による化合物、
【化1】
並びにそのエナンチオマー、ジアステレオマー、互変異性体、溶媒和物及び薬学的に許容される塩
[式中、
R1は、−N(R10)−(C(R6)2)m−T、
−(C(R6)2)l−T、又は
−O−(C(R6)2)m−Tであり、
R6は、
H、
F、
OH、
OCH3、
C1〜6−アルキル(ハロゲン、CN、OH及びOCH3から選択される1〜3個の置換基で任意選択で置換されている)、並びに
C3〜6−シクロアルキル(ハロゲン、CN、OH及びOCH3から選択される1〜3個の置換基で任意選択で置換されている)から独立に選択され、
Tは、NR7R8、
【化2】
であり、
R7及びR8は、互いに独立に、
H、
C1〜6−アルキル、
C2〜6−アルケニル、
C2〜6−アルキニル、及び
C2〜6−アルキレン−O−C1〜6−アルキルから選択され、
各アルキル、アルケニル及びアルキニルは、1個若しくは複数のハロゲン原子、CN又はOHで任意選択で置換されており、
R9は、
ハロゲン、
CN、
OH、
C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、並びに
O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
C1〜6−アルキレン−O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、又は
NR12R13から独立に選択され、
R10は、H又は
C1〜6−アルキルであり、
R11は、
ハロゲン、
CN、
OH、
C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
C2〜6−アルケニル、
C2〜6−アルキニル、
O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
C1〜6−アルキレン−O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
C0〜6−アルキル−C3〜6−シクロアルキル、
−OC(O)C1〜6−アルキル、
−NH2、
−NH(C1〜6−アルキル)、及び
−N(C1〜6−アルキル)2から独立に選択され、
R12及びR13は、互いに独立に、
C1〜6−アルキル(OHで任意選択で置換されている)、
C2〜6−アルケニル、
C2〜6−アルキニル、
C2〜6−アルキレン−O−C1〜6−アルキル、及び
C2〜6−アルキレン−N−(C1〜6−アルキル)2から選択され、
Wは、CH、O又はNR10であり、
Xは、CH又はNであり、
Yは、CH又はNであり、
Zは、CH又はNであり、
Aは、0〜2個の窒素原子を含有する3〜7員の飽和、不飽和又は芳香族環であり、
Bは、CR2又はNであり、
Gは、CR2又はNであり、
Dは、CR2又はNであり、
Eは、CR2又はNであり、
ただし、可変部分B、G、D及びEの1つ又は2つは、Nでなければならず、
R2は、
H、
F、
Cl、
CH3、
OCH3、及び
CF3から独立に選択され、
R3は、H、
Cl、
F、又は
CH3であり、
R4は、Cl、
F、又は
CH3であり、
R5は、
【化3】
モルホリン(1〜3個の同じ若しくは異なる置換基R14で任意選択で置換されている)、
環中に1個の窒素原子と任意選択でO、N及びSから選択されるさらなるヘテロ原子とを含有する4〜7員の飽和又は部分不飽和複素環(複素環は、1〜4個の同じ若しくは異なる置換基R11で任意選択で置換されている)、又は
NR12R13であり、
R14は、C1〜6−アルキル、
C1〜6−アルキレン−O−C1〜6−アルキル、
C1〜6−アルキレン−OH、
C1〜6−アルキレン−NH2、
C1〜6−アルキレン−NH−C1〜6−アルキル、又は
C1〜6−アルキレン−N(C1〜6−アルキル)2であり、
R15は、H又は
C1〜6−アルキルであり、
lは、0、1、2、3又は4であり、
mは、0、1、2、3又は4であり、
oは、0、1又は2であり、
pは、0、1、2、3又は4であり、
rは、0、1、2、3又は4であり、
sは、1又は2であり、
tは、0又は1である]。
【請求項2】
R1が、−N(R10)−(C(R6)2)m−T、
−(C(R6)2)l−T、又は
−O−(C(R6)2)m−Tであり、
R6が、
H、
F、
OH、
OCH3、
C1〜6−アルキル(ハロゲン、CN、OH及びOCH3から選択される1〜3個の置換基で任意選択で置換されている)、並びに
C3〜6−シクロアルキル(ハロゲン、CN、OH及びOCH3から選択される1〜3個の置換基で任意選択で置換されている)から独立に選択され、
Tが、NR7R8、
モルホリン、
【化4】
であり、
R7及びR8が、互いに独立に、
H、
C1〜6−アルキル、
C2〜6−アルケニル、
C2〜6−アルキニル、及び
C2〜6−アルキレン−O−C1〜6−アルキルから選択され、
各アルキル、アルケニル及びアルキニルが、1個若しくは複数のハロゲン原子、CN又はOHで任意選択で置換されており、
R9が、
ハロゲン、
CN、
OH、
C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、並びに
O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
C1〜6−アルキレン−O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)から独立に選択され、
R10が、H又は
C1〜C6−アルキルであり、
R11が、
ハロゲン、
CN、
OH、
C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
C1〜6−アルキレン−O−C1〜6−アルキル(ハロゲン、CN及びOHから選択される1〜3個の置換基で任意選択で置換されている)、
−NH2、
−NH(C1〜6−アルキル)、及び
−N(C1〜6−アルキル)2から独立に選択され、
Xが、CH又はNであり、
Yが、CH又はNであり、
Zが、CH又はNであり、
Aが、0〜2個の窒素原子を含有する3〜7員の飽和、不飽和又は芳香族環であり、
Bが、CR2又はNであり、
Gが、CR2又はNであり、
Dが、CR2又はNであり、
Eが、CR2又はNであり、
ただし、可変部分B、G、D及びEの1つ又は2つが、Nでなければならず、
R2が、
H、
F、
Cl、
CH3、
OCH3、及び
CF3から独立に選択され、
R3が、H、
Cl、
F、又は
CH3であり、
R4が、Cl又はFであり、
R5が、
【化5】
モルホリン(1〜3個の同じ若しくは異なる置換基R14で任意選択で置換されている)、又は
NR12R13であり、
R12及びR13が、互いに独立に、
C1〜6−アルキル、
C2〜6−アルケニル、
C2〜6−アルキニル、
C2〜6−アルキレン−O−C1〜6−アルキル、及び
C2〜6−アルキレン−N−(C1〜6−アルキル)2から選択され、
R14が、C1〜6−アルキル、
C1〜6−アルキレン−O−C1〜6−アルキル、
C1〜6−アルキレン−OH、
C1〜6−アルキレン−NH2、
C1〜6−アルキレン−NH−C1〜6−アルキル、又は
C1〜6−アルキレン−N(C1〜6−アルキル)2であり、
lが、0、1、2、3又は4であり、
mが、0、1、2、3又は4であり、
oが、0、1又は2であり、
pが、0、1、2、3又は4であり、
qが、0、1、2、又は3であり、
rが、0、1、2、3又は4であり、
sが、1又は2である、請求項1に記載の化合物。
【請求項3】
式(I’)による、請求項1又は2に記載の化合物
【化6】
(式中、B、G、D、E、R1、R3、R4及びR5は、請求項1又は2に記載の通りである)。
【請求項4】
R7及びR8の少なくとも1つが、
C2〜6−アルケニル、
C2〜6−アルキニル、及び
C2〜6−アルキレン−O−C1〜6−アルキルから選択される、請求項1〜3のいずれか一項に記載の化合物。
【請求項5】
R2が、H、F、Cl及びCH3から選択される、請求項1〜4のいずれか一項に記載の化合物。
【請求項6】
lが、2又は3であり、又は
mが、2又は3である、請求項1〜5のいずれか一項に記載の化合物。
【請求項7】
医薬品としての請求項1〜6のいずれか一項に記載の化合物。
【請求項8】
哺乳動物においてメラノコルチン−4受容体の不活性化又は活性化に対して応答性の障害、疾患又は状態の治療又は予防のための、請求項1〜6のいずれか一項に記載の化合物。
【請求項9】
癌悪液質、筋肉疲労、食欲不振症、筋萎縮性側索硬化症(ALS)、不安神経症及び/又はうつの治療又は予防のための、請求項8に記載の化合物。
【請求項10】
肥満症、真性糖尿病、男性若しくは女性の性的機能不全及び/又は勃起不全の治療又は予防のための、請求項8に記載の化合物。
【請求項11】
哺乳動物においてメラノコルチン−4受容体の不活性化又は活性化に対して応答性の障害、疾患又は状態の治療又は予防のための医薬品の調製のための、請求項1〜6のいずれか一項に記載の化合物の使用。
【請求項12】
請求項1〜6のいずれか一項に記載の化合物と、薬学的に許容される担体とを含む医薬組成物。
【公表番号】特表2010−533666(P2010−533666A)
【公表日】平成22年10月28日(2010.10.28)
【国際特許分類】
【出願番号】特願2010−516425(P2010−516425)
【出願日】平成20年7月18日(2008.7.18)
【国際出願番号】PCT/EP2008/005913
【国際公開番号】WO2009/010299
【国際公開日】平成21年1月22日(2009.1.22)
【出願人】(505336024)サンセラ ファーマシューティカルズ (シュバイツ) アーゲー (21)
【Fターム(参考)】
【公表日】平成22年10月28日(2010.10.28)
【国際特許分類】
【出願日】平成20年7月18日(2008.7.18)
【国際出願番号】PCT/EP2008/005913
【国際公開番号】WO2009/010299
【国際公開日】平成21年1月22日(2009.1.22)
【出願人】(505336024)サンセラ ファーマシューティカルズ (シュバイツ) アーゲー (21)
【Fターム(参考)】
[ Back to top ]