
モノアシルグリセロールリパーゼの薬物標的化と関連する方法及び組成物
本発明は、モノアシルグリセロールリパーゼ(MAGL)を選択的に阻害する化合物を提供する。また本発明は、生体内での2−アラキドノイルグリセロール(2−AG)媒介内在性カンナビノイドシグナル伝達を刺激するために、並びに内在性カンナビノイドシグナル伝達する症状を治療するために、MAGLの選択的阻害剤を使用する方法をも提供する。加えて本発明は、MAGLの選択的阻害剤でMAGLを薬物標的にすることよって癌を治療する又は腫瘍の増殖を阻害する方法を提供する。さらに本発明は、改良された生化学的及び薬学的特性を有するMAGL阻害剤のスクリーニング方法をも提供する。
【発明の詳細な説明】
【技術分野】
【0001】
著作権告知
37C.F.R.§1.71(e)に基づき、出願人は、本情報開示の一部が著作権保護を受ける資料を含むことに特に言及しておく。本著作権者は、何人による本特許書類又は本特許の情報開示の複写複製に対しても、それが特許商標局の特許ファイル又は記録中に出現する場合には異議を唱えないが、その他の場合には全ての著作権を留保する。
【0002】
関連出願の相互参照
本特許出願は、米国仮出願第61/199,286号(出願日2008年11月14日)による優先権の利益を主張する。優先権基礎出願の全ての情報開示は、参照することにより、そのまま、すべての目的のために本明細書で援用されている。
【0003】
政府支援に関する陳述
本発明は、一部、国立衛生研究所の助成金第R01−DA017259号及びR01−DA025285号による政府支援により成された。それ故に、米国政府は、本発明に一定の権利を有することができる。
【0004】
発明の分野
本発明は、全体として他の脳セリンヒドロラーゼにモノアシルグリセロールリパーゼ(MAGL)を選択的に阻害するための、並びに被験者における2−アラキドノイルグリセロール(2−AG)媒介内在性カンナビノイドシグナル伝達を刺激するための方法及び組成物に関する。また本発明は、腫瘍細胞の増殖を阻害するための及び癌を治療するための方法にも関連する。
【0005】
発明の背景
カンナビノイド受容体CB1及びCB2は、マリファナの向精神成分、Δ9−テトラヒドロカンナビノールの分子標的である。CB1は主に脳で発現するが、また肺、肝臓及び腎臓でも発現する。CB2は、主に免疫系及び造血細胞で発現する。また、二つの内在性リガンド、言い換えれば「内在性カンナビノイド」、即ちアラキドン酸含有脂質アナンダミド(N−アラキドノイルエタノールアミン、AEA)及び2−アラキドノイルグリセロール(2−AG)も同定されている。内在性カンナビノイド系は、食欲、痛覚、炎症、及び記憶を含むさまざまな生理的プロセスを制御しており、並びにそれは、肥満、慢性疼痛、、及び抑鬱等の障害を治療するために、重要な最新の薬学的興味の中心となっている。
【0006】
内在性カンナビノイドシグナル伝達は、酵素的加水分解によって堅固に制御されている。2−AG又はAEAの加水分解により、アラキドン酸(AA)が産生される。主要なAEA−加水分解酵素は、脂肪酸アミドヒドロラーゼ(FAAH)である。遺伝子的な又は薬理学的なFAAHの破壊により、神経系及び抹消の至る所でAEAレベルが著しく上昇し、結果的に、痛覚、炎症、、及び抑鬱の減少を含む、多発性のCB1−及び/又はCB−2依存性の行動的影響に帰着する。興味深いことに、低体温症及び運動障害等の、直接作用性CB1アゴニストの周知の他の行動的影響の幾つかは、FAAH破壊動物では観察されない(ゴパーラジュら(Goparaju et al.)、「フェブス・レターズ(FEBS Lett.)」、1998年、第422巻:p.69−73;及びカスリアら(Kathuria et. al.)、「ネイチャー・メディスン(Nat Med)」、2003年、第9巻:p.76−81)。これらの動物は、また野生型の2−AG17レベルを有しており、このことは生体内でさらなるCB1制御の挙動プロセスが2−AGによって媒介されているかもしれないことを示唆している。幾つかのエビデンスは、モノアシルグリセロールリパーゼ(MAGL)が神経系における2−AGの加水分解のための主要な酵素であることを示唆している。例えば、ディンら(Dinh et al.)、「プロシーディングス・オブ・ザ・ナショナル・アカデミー・オブ・サイエンシズ・オブ・ザ・ユナイテッド・ステイツ・オブ・アメリカ(Proc.Natl.Acad.Sci.USA)」、2002年、第99巻:p.10819−24;ディンら(Dinh et al.)、「モレキュラー・ファルマコロジー(Mol.Pharmacol.)」、2004年、第66巻:p.1260−4;ブランクマンら(Blankman et al.)、「ケミストリー&バイオロジー(Chem.Biol.)」、2007年、第14巻:p.1347−56;及び野村ら(Nomura et al.)、「ネイチャー・ケミカル・バイオロジー(Nat.Chem.Biol.)」、2008年、第4巻:p373−8、を参照。しかしながら、これら以前の研究のどれも、MAGLが生体内における2−AGの加水分解で果たす役割を明確には検討していない。一方、幾つかのMAGL阻害剤が当該技術分野において開示されているが(ホーマンら(Hohmann et al.)、「ネイチャー(Nature)」、2005年、第435巻:p.1108−12;ヴァーヴェルら(Varvel et al.)、「ジャーナル・オブ・ファルマコロジー・アンド・エクスペリメンタル・セラピューティックス(J.Pharmacol.Esp.Ther.)」、2002年、第301巻:p.915−24;及びサーリオら(Saario et al.)、「ケミストリー&バイオロジー(Chem.Biol.)」、2005年、第12巻:p.649−56)、そのどれも生体内で薬理学的なツールとして一般的に使用するために要求されるレベルの有効性も明確性も示していない。
【0007】
腫瘍及び癌は、多くの人々の生命と健康を脅かす疾患又は症状である。侵襲性の腫瘍細胞の無抑制の増殖が、しばしば結果として悪性腫瘍(癌)を形成する。癌を根治的に治療することのできる有効な医薬又は治療法は未だ存在しない。現時点で腫瘍の治療に使用される主要な方法は、手術、放射線療法、化学療法、生物学的療法、及びホルモン療法、中国の伝統療法、温熱療法、ラジオ波焼灼療法等の他の療法等々である。しかし、利用可能なほとんどの治療法は、被験者の症候及び身体的症候を軽減することができるだけであり、疾患を治療することはできず、しかも通常、各治療法は副作用を含む不利益を有している。
【0008】
当該技術分野において、生体内で強力かつ選択的にMAGLを阻害する化合物を求める、並びに内在性カンナビノイドシグナル伝達活性する症状又は障害を治療するための新規な方法を求めるが存在する。また発明者らには、腫瘍増殖の阻害及び癌治療のための代替可能で効果的な方法を求めるが存在する。本発明は、これら及び当該技術分野において実現されていない他の。
【発明の概要】
【0009】
一態様において、本発明は、被験者における2−アラキドノイルグリセロール媒介内在性カンナビノイドシグナル伝達を刺激するための方法を提供する。当該方法は、モノアシルグリセロールリパーゼ(MAGL)を選択的に阻害する化合物の治療的に有効な量を、被験者に対し投与すること(例えば、腹腔内注射又は経口投与)を。これらの方法の幾つかにおいて、化合物は、脂肪酸アミドヒドロラーゼ(FAAH)にMAGL選択的。例えば、化合物は、FAAHMAGLの阻害に少なくとも100倍の選択性を有することができる。これらの方法の幾つかで用いるMAGL選択的阻害剤は、本明細書に開示する式Iに示す構造を有する。幾つかの他の方法において、MAGL特異的阻害剤は、FAAH及びα/βヒドロラーゼ6(ABHD6)の両方にMAGL選択的である。幾つかの好ましい実施態様においては、MAGL選択的阻害剤は化合物JZL184である。好ましくは、本発明の方法で治療されるべき被験者はである。これらの方法の幾つかにおいて治療されるべき被験者は、例えば疼痛、炎症、抑鬱又は等の、内在性カンナビノイドシグナル伝達によって媒介される又は関連する、症状又は障害を患っている。
【0010】
関連する態様において、本発明は、被験者における、内在性カンナビノイドシグナル伝達する症状、又はその治療効果が増加したカンナビノイド(例えば、2−AG)レベルに由来することができる疾患、を治療又は回復するための方法を提供する。これらの方法は、モノアシルグリセロールリパーゼ(MAGL)を選択的に阻害する化合物の治療的に有効な量を含む医薬組成物を被験者に投与することを含んでいる。これらの方法の幾つかは、2−AGシグナル伝達が役割を果たす、例えば疼痛等の症状又は障害を治療することに向けられる。これらの方法の幾つかは、式Iに示す構造を有するMAGL選択的阻害剤を用いる。当該方法の幾つかにおいて、MAGL選択的阻害剤は、FAAHにモノアシルグリセロールリパーゼ(MAGL)を選択的に阻害する。幾つかの他の方法において、用いた化合物はFAAH及びABHD6の両方にMAGLの阻害に選択的である。幾つかの好ましい実施態様において、用いるMAGL選択的阻害剤は化合物JZL184である。
【0011】
他の態様において、本発明はMAGL選択的阻害剤の化合物を提供する。これらの化合物は、下記の式Iに示す構造を有する:
【化1】

【0012】
式中、XはN又はCH、R1はH又はOHであり、そしてR2及びR3の各々は、下記の構造の有する。
【化2】
【0013】
本発明の好ましいMAGL選択的阻害剤の化合物は化合物JZL184である。その構造は、式Iに示すが、式中XはCH、R1はOHであり、そしてR2及びR3の各々は、下記に示す構造を有する。
【化3】
【0014】
さらに他の態様において、本発明は、改良された性質を有するMAGL阻害剤の化合物を同定するための方法を提供する。当該方法は、(a)本明細書で開示されるMAGL選択的阻害剤(例えば、JZL184)の一以上の構造アナログを合成すること、(b)MAGL選択的阻害剤の比較して改良された生物学的又は薬学的性質を有するアナログを同定すること、を。式Iに包含される任意の化合物も、構造アナログの合成ためのリード化合物として役立つことができる。幾つかの好ましい実施態様において、化合物JZL184が使用される。これらのスクリーニング法において、アナログにおいて同定されるべき、改良された生物学的又は薬学的性質は、例えば、MAGLに対する増強された阻害活性又は脳セリンヒドロラーゼMAGLに対する増加された選択性等であることができる。
【0015】
他の態様において、本発明は、腫瘍細胞、特に侵襲性の腫瘍細胞の増殖を阻害するための方法を提供する。当該方法は、モノアシルグリセロールリパーゼ(MAGL)を特異的に阻害する化合物と接触させることを。幾つかの実施態様において、当該方法は被験者に存在する腫瘍の増殖を阻害することに向けられる。被験者は、癌を有すると診断された者であることができる。また被験者は、癌にかかりやすい又は癌になる危険のある者であことができる。当該方法は、充実性腫瘍及び白血病の細胞を含む、任意の腫瘍細胞の増殖を阻害するために用いることができる。幾つかの実施態様において、当該方法は、乳房腫瘍細胞、卵巣腫瘍細胞、黒色腫細胞、肺腫瘍細胞又は脳腫瘍細胞の増殖を阻害するために使用される。
【0016】
本発明の幾つかの方法において、用いられるMAGL阻害化合物(又は「MAGL拮抗化合物」又は「MAGLアンタゴニスト化合物」)は、脂肪酸アミドヒドロラーゼ(FAAH)にMAGLの阻害に選択的である。例えば、化合物は、本明細書で開示される任意のMAGL選択的阻害剤であることができる。幾つかの他の実施態様において、用いられるMAGL阻害化合物は、MAGLに特異的な阻害ポリヌクレオチドである。このような阻害性ポリヌクレオチドの例には、低分子緩衝RNA(siRNA)、マイクロRNA(miRNA)、低分子ヘアピン型RNA(shRNA)、アンチセンス核酸及び相補DNA(cDNA)が含まれる。
【0017】
関連する態様において、本発明は、被験者の腫瘍の症候を治療又は回復する方法を提供する。これらの方法は、モノアシルグリセロールリパーゼ(MAGL)を特異的に阻害する化合物の治療的に有効な量を含む医薬組成物を被験者に投与することを含んでいる。任意の腫瘍又は癌により苦しめられる被験者も、これらの方法による治療のために回復可能である。例えば、当該方法は、乳腺腫瘍、卵巣腫瘍、黒色腫、肺腫瘍又は脳腫瘍を患う被験者を治療するために使用することができる。幾つかの方法において、用いられるMAGL阻害剤は、脂肪酸アミドヒドロラーゼ(FAAH)にMAGLの阻害に選択的である化合物である。幾つかの他の方法において、MAGLの発現又は細胞内レベルを特異的に減少させる阻害ポリヌクレオチドが使用される。
【0018】
本発明の特徴及び長所に関するより深い理解は、明細書及び特許請求の範囲の残りの部分を参照することによって得ることができるであろう。
【図面の簡単な説明】
【0019】
【図1】図1A−1Bは、MAGL阻害剤の構造及び拮抗ABPPプロファイルを示している。A:MAGL阻害剤の構造;B:マウス脳膜プロテオーム中のセリンヒドロラーゼ活性へのMAGL阻害剤の影響を示す拮抗ABPP。比較のために示したものは、それぞれ選択的FAAH及びABHD6阻害剤、URB597(カスリアら(Kathuria et al.)、「ネイチャー・メディスン(Nat Med)」、2003年、第9巻:p.76−81)及びWWL70(リーら(Li et al.)、「ジャーナル・オブ・ザ・アメリカン・ケミカル・ソサイアティ(J.Amer.Chem.Soc.)」、2007年、第129巻:p.9594−5)である。阻害剤を脳膜と共に30分間インキュベートし、次にセリンヒドロラーゼ指向ABPPプローブFPローダミン(2マイクロM、30分間)で処理し、それからプロテオームをSDS−PAGE及びインゲル蛍光スキャニングによって分析し、阻害された酵素を検出した。対照プロテオームは、DMSOのみで処理した。蛍光ゲルをグレイスケールで示す。文献(例えば、ブランクマンら(Blankman et al.)、「ケミストリー&バイオロジー(Chem.Biol.)」、2007年、第14巻:p.1347−56)に報告された通り、脳MAGLがSDS−PAGEによって35kDaの二重線として移動することに特に言及しておく。
【図2】図2A−2Cは、JZL184の二の生体外キャラクタリゼーションを示している。A:拮抗的ABPPによって定量した、マウス脳膜セリンヒドロラーゼへのJZL184の濃度依存的影響;B:基質アッセイ法(それぞれ2−AG及びオレイン酸アミド)によって定量した、JZL184による脳膜MAGL及びFAAH活性の遮断;C:基質アッセイ法(それぞれ2−AG及びアナンダミド)によって定量した、JZL184による組換え型MAGL及びFAAH活性の遮断。酵素はCOS7細胞中で組換えにより発現させた。JZL184は、組換え型MAGL活性のほぼ完全な遮断を引き起こしたが(>95%)、おそらく他の酵素の活性を反映して、脳膜においては2−AG加水分解活性の〜15%の残存が観察されたことに特に言及しておく。A−Cについては、分析に先立って試料をJZL184で30分間処理した。B及びCについては、データは、三つの独立した実験に対する平均±標準誤差として表した。
【図3】図3A−3Fは、JZL184の生体内でのキャラクタリゼーションを示している。A及びB:明示の投与量(4−40mg/kg、腹腔内投与)でJZL184により4時間処理したマウスから調製した脳膜の、セリンヒドロラーゼ活性プロファイル(A)並びにMAGL及びFAAH活性(B);C:JZL184により処理(16mg/kg、腹腔内投与、4時間)したマウスから調製された脳膜中のセリンヒドロラーゼ活性のABPP−MudPIT分析。MAGL及びFAAHの対照シグナルは、それぞれ赤色及び青色のバーで示す;D−F:明示の投与量(4−40mg/kg、腹腔内投与)でJZL184により4時間処理したマウス由来の、脳の2−AG(D)、アラキドン酸(E)、及びNAE(F)レベル(F)。Fについては、このFAAH阻害剤によって誘導されるNAEの増加を確認するために、URB597で処理(10mg/kg、腹腔内投与)したマウスからのデータも示す。B−Fについては、溶媒処理対照動物と対比した阻害剤処理動物について、*はp<0.05、**はp<0.01である。データは、平均±標準誤差として表した。群当たりn=3−5である。
【図4】図4A−4Eは、生体内でのJZL184の阻害活性の時間経過解析を示している。A及びB:明示の時間、JZL184(16mg/kg、腹腔内投与)により処理したマウスから調製した脳膜のセリンヒドロラーゼ活性プロファイル(A)並びにMAGL及びFAAH活性(B);C−E:明示の時間JZL184により処理(16mg/kg、腹腔内投与)したマウス由来の、脳の2−AG(D)、アラキドン酸(E)、及びNAEレベル。B−Eについては、溶媒処理対照動物と対比した阻害剤処理動物について、*はp<0.05、**はp<0.01である。データは、平均±標準誤差として表した。群当たりn=3−5である。
【図5】図5A−5Eは、JZL184の行動的影響を示している。A及びB:JZL184(16mg/kg)は、熱痛覚の尾浸漬アッセイ法において(A)並びに有害な化学痛覚の酢酸腹腔緊張アッセイ法において(B)鎮痛作用を引き起こした。これらの効果は、CB1アンタゴニスト、リモナバン(3mg/kg)による前処理によって遮断された;C−E:JZL184は、著しい低体温症(C)、運動性低下症(D)、及び反射亢進(E)を引き起こしたが、それらはまたリモナバンによって著しく減弱された。ベースラインの尾浸漬潜伏時間及び直腸温度は、それぞれ0.82±0.02秒及び37.4±0.1℃であった。溶媒−JZL184処理マウスと対比した溶媒−溶媒処理マウスについて、*はp<0.05、**はp<0.01である。溶媒処理マウス、リモナバン-JZL184処理マウス対比した溶媒−JZL184処理マウスについて、#はp<0.05、##はp<0.01である。データは、平均±標準誤差として表した。群当たりn=10−14である。
【図6】図6A−6Bは、化合物WWL98の構造(A)及び化合物JZL184の合成経路(B)を示している。他のカルバミン酸系阻害剤も同様の経路で合成した。
【図7】図7A−7Cは、MAGL及びFAAHのIZL184による経時的な阻害を示している。A:マウス脳膜プロテオーム中のMAGL活性のFPローダミン標識化のJZL184による阻害;B:マウス脳膜プロテオーム中のFAAH活性のFPローダミン標識化のJZL184による阻害;C:JZL184によるMAGL及びFAAH阻害についての計算値kobs/[I]。
【図8】図8A−8Bは、化合物JZL184は、CB1受容体に結合しないこと又は2−AG生合成酵素DAGLβを阻害しないことを示している。A:JZL184は、CB1受容体から3H−リモナバンの結合を置換しなかった;B:JZL184(25μM)は、組換えでCOS−7細胞中に発現されたDAGLβを阻害しなかった。既知のDAGLβ阻害剤テトラヒドロリプスタチン(THL、25μM)を正の対照として使用した。
【図9】図9A−9BはJZL184の用量反応のための脳脂質測定値を示している。JZL184(4−40mg/kg、腹腔内投与)の投与4時間後に脳のC18:1又はC16:0MAG(A)又は遊離脂肪酸(B)レベルにおける著しい変化は測定されないことが認められた。
【図10】図10A−10Eは、JZL184の経口投与の効果を示している。A及びB:明示の投与量(4−40mg/kg、経口投与)でJZL184により4時間処理したマウスから調製した脳膜の、セリンヒドロラーゼ活性プロファイル(A)並びにMAGL及びFAAH活性(B);C及びD:JZL184(1−40mg/kg、経口投与)により4時間処理したマウス由来の、脳の2−AG(C)及びAEA(D)レベル;E:腹腔内又は経口投与によって処理したマウス由来の脳のJZL184の相対レベル。B−Dについては、溶媒処理動物と対比した阻害剤処理動物について、*はp<0.05、**はp<0.01である。データは、平均±標準誤差として表した。群当たりn=3−5である。
【図11】図11A−11Cは、JZL184処理に続くマウスにおける脳脂質測定値の時間経過を示している。JZL184投与(16mg/kg、腹腔内投与)に続く24時間を通して、脳のC18:1又はC16:0のNAE(A)、MAG(B)、又は遊離脂肪酸(C)レベルにおいて著しい変化は認められなかった。
【図12】図12は、マウス脳中のJZL184レベルの時間経過解析を示している。JZL184を16mg/kgで経口投与した。
【図13】図13A−13Dは、侵襲性の癌細胞内でMAGLが増加することを示しているが、ここで当該酵素はモノアシルグリセロール(MAG)及び遊離脂肪酸(FFA)レベルを制御する。(A)非侵襲性(青色)及び侵襲性(赤色)のヒト癌細胞株内のセリンヒドロラーゼ活性のABPP。セリンヒドロラーゼ活性は、細胞全体のプロテオームにおいて活性に基づくプローブ、FPローダミンで標識化し、SDS−PAGE及びインゲル蛍光スキャニング(蛍光ゲルをグレイスケールで示す)によって検出した。赤色枠で強調したのは二の酵素、MAGL及びKIAA1363は、非侵襲性癌細胞と対比して侵襲性癌細胞において一貫して増加している。33及び35kDaのFPローダミン標識バンドがMAGLを表すことを確認するために、プロテオームを、DMSO又は選択的MAGL阻害剤JZL184(1μM、4時間)により前処理した癌細胞からも調製した。(B)JZL184(1μM、4時間)の存在下(赤色バー)又は非存在下(黒色バー)における癌細胞のC20:4MAG加水分解活性。(C、D)MAGLの阻害(JZL184 1μM、4時間)は、侵襲性細胞のMAGレベルを増加し(C)FFAレベルを減少するが(D)、しかし非侵襲性細胞ではそうではない。侵襲性の癌細胞は、それぞれのMAGL活性を反映して、非侵襲性の癌細胞と比較して基本的により高レベルのFFA(及びより低レベルのMAG)を有することに特に言及しておく。DMSO処理対照群と対比したJZL184処理群について、*はp<0.05、**はp<0.01である。非侵襲性癌細胞と対比した侵襲性癌細胞について、#はp<0.05、##はp<0.01である。データは、平均±標準誤差として表した。群当たりn=4−5である。
【図14】図14A−14Fは、MAGLの安定なshRNA媒介ノックダウンが侵襲性癌細胞においてFFAレベルを低下することを示している。(A、D)MAGLを、二の独立した短ヘアピンRNA(shRNA)オリゴヌクレオチド(shMAGL1、shMAGL2)を使用して安定にノックダウンした結果、異なるセリンヒドロラーゼ(DPPIV)を標的とするshRNAを発現するsh対照細胞と比較してC8161及びSKOV3細胞のMAGL活性において>70%が減少した。(B、C、E、F)shMAGL細胞は、MAGレベルの増加(B、E)及びFFAレベルの減少(C、F)を示している。sh対照群と対比したshMAGL群について、*はp<0.05、**はp<0.01である。非侵襲性癌細胞と対比した侵襲性癌細胞について、#はp<0.05、##はp<0.01である。sh対照細胞のMAGL活性並びにMAG及びFFAレベルは、親癌細胞株のそれらからそれほど異なっていなかった。データは、平均±標準誤差として表した。群当たりn=4−5である。
【図15】図15A−15Cは、高悪性度の原発性ヒト卵巣腫瘍が良性腫瘍と比較して増加したMAGL活性及びFFAを有することを示している。(A)個体の腫瘍検体についてのC20:4MAG加水分解活性測定値。JZL184(1μM、30分間)による前処理が、測定された加水分解活性の大部分はMAGLによることを確認した。(B)高悪性度腫瘍と対比した良性腫瘍におけるMAGL活性の概要図であり、ここで各値は、(A)に示した総C20:4MAG加水分解活性のJZL184感受性の部分として表した。(C)高悪性度腫瘍と対比した良性腫瘍におけるFFAレベル。良性腫瘍群と対比した高悪性度群について、**はp<0.01である。データは、平均±標準誤差として表した。群当たりn=10−13である。
【図16】図16A−16Jは、shRNA媒介ノックダウン及びMAGLの薬理学的阻害が癌の侵襲性をことを示している。(A−C、F−G)shMAGL細胞は、sh対照及び非感染の親細胞と比較して、減少した移動(A、F)、浸潤(B、G)、及び細胞生存(C、H)を示す。移動及び浸潤アッセイは、細胞50,000個をコラーゲン(10μg/ml)で被覆した膜又はバイオコートTMマトリゲル(BioCoatTM Matrigel)(登録商標)を含有する8μm孔径を備えたインサートに播種する前に、癌細胞を4時間無血清培地に移植することによってそれぞれ実施した。C8161及びSKOV3の移動時間は、それぞれ5時間及び20時間だった。移動又は浸潤した細胞は、倍率400Xで4視野当たり計数した平均数±標準誤差で言及する。細胞生存アッセイは、細胞20、000個を無血清培地中で96ウェルプレートに播種することによって実施した。生存は、WST−1増殖アッセイを使用して評価した。代表的な移動プレート(倍率400X)をshMAGL細胞と対比したsh対照細胞について示す(A、F)。(D、I)shMAGL細胞は、sh対照細胞及び非感染の親細胞と比較して、SCIDマウスにおいて損傷した腫瘍増殖を示している。C8161又はSKOV3細胞2x106個/100μlを側腹部の皮下に注入し、ノギスを使用して腫瘍増殖を測定した。(E、J)JZL184処理(4μl/gポリエチレングリセロール300溶媒として毎日40mg/kgを経口投与)は、溶媒処理と比較してSCIDマウスにおける異種移植した腫瘍増殖速度を著しく減少する。sh対照群と対比したshMAGL群又は溶媒処理群と対比したJZL184群について、*はp<0.05、**はp<0.01である。sh対照細胞及び親癌細胞は、その移動、細胞生存、浸潤、又は生体内での腫瘍増殖においてそれほど異なっていなかった。データは、平均±標準誤差として表した。群当たりn=5−8である。
【図17】図17A−17Fは、MAGLの異所性発現がFFAレベルを増加し、そしてMUM2C黒色腫細胞の生体外及び生体内の病原性を亢進することを示している。(A)ABPP(上段パネル)、ウェスタンブロット(中断パネル)及びC20:4MAG加水分解活性(下段パネル)によって確認したMUM2C細胞中のMAGLの過剰発現(MAGL−OE、赤色バー)。対照細胞及びS122A細胞(黒色バー)は、それぞれ空のベクター(EV)に感染した癌細胞又は触媒的に不活性なMAGL変異株(S122A)に相当する。ウェスタン分析によりS122A−MAGL変異株の過剰発現を確認したが、当該変異株は、ABPP及びC20:4MAG加水分解アッセイによって評価されたように、いかなる活性も示さなかった。(B、C)MAGL−OE細胞は、EV対照細胞及びS122A細胞と比較してより低いMAGレベル(B)及びより高いFFAレベル(C)を含んでいる。これらの代謝的効果は、生体内原位置でのJZL184による処理(1μM、4時間、栗色のバー)によって逆転された。(D、E)MAGL−OE MUM2C細胞は、EV及びS122A対照細胞と比較して、増加した移動(D)及び浸潤(E)を示している。この亢進した移動及び浸潤は、JZL184(1μM、4時間)によって逆転された。代表的な移動パネルを示す(D)。(F)MAGL−OE MUM2C細胞は、EV又はS122A対照細胞と比較してSCIDマウスにおいて著しく亢進した腫瘍増殖速度を示す(細胞2x106個を正所性に移植)。対照群と対比したMAGL−OE群について、*はp<0.05、**はp<0.01である。データは平均±標準誤差で表す。群当たりn=4−6である。
【図18】図18A−18Dは、外来性脂肪酸での処理によるshMAGL癌細胞の病原性の特性の回復をいる。(A)減少したshMAGL細胞の移動は、パルミチン酸又はステアリン酸による処理(20μM、4時間、斜線赤色バー)によって逆転される。(B)パルミチン酸又はステアリン酸の添加(20μM、4時間)は、MUM2C及びOVCAR3細胞の移動を増加し、そしてJZL184処理MAGL−OE細胞における減少した移動を救出する。(C)shMAGL−C8161細胞の減少した腫瘍増殖は、高脂肪食(HFD)(60kcal%脂肪)による処理によって逆転される。癌細胞を側腹部へ注入する前の2週間、マウスを通常固形飼料(NC)又はHFDで飼い、腫瘍増殖の時間経過を通じてこれらの飼料で養った。挿入図は、時間経過を通じての動物の体重である。HFD群由来の外植されたshMAGL腫瘍(斜線赤色バー)は、NC群由来のshMAGL腫瘍(赤色バー)と比較して増加したFFAレベルを含んでいる。sh対照群と対比したshMAGL群について、*はp<0.05、**はp<0.01である。shMAGL対照群(それそれDMSO処理群及びNC処理群)と対比したパルミチン酸又はステアリン酸又はHFD処理したshMAGL群について、##はp<0.01である。データは、平均±標準誤差として表した。(A)については、群当たりn=4−5;(B、C)については、群当たりn=7−8である。
【図19】図19A−19Iは、MAGLがプロ腫瘍原性シグナル伝達分子に富む脂質代謝ネットワークを制御することを示している。(A、B)EV対照細胞と対比したMAGL−OE(MUM2C及びOVCAR3の両方)(A)及びsh対照細胞と対比したshMAGL(B)の異常MAGL活性に関する癌細胞モデルのリピドーム解析。示した代謝産物は、EV対照細胞と対比したMAGL−OE細胞(MUM2C及びOVCAR3の両方)において増加した(赤色)又は減少した(青色)代謝産物であり(A)、そしてsh対照細胞と対比したshMAGL細胞(C8161及びSKOV3の両方)においては逆の特性を示した(B)。代謝産物の親質量を提供しており、円の大きさは相対的な変化の大きさを示している。(C、D)癌細胞モデルにおけるC16:0LPA及びPGE2レベルの定量。(E)C16:0LPA(100nM、4時間)又はPGE2(100nM、4時間)によるshMAGL癌細胞の処理は、sh対照細胞と比較してそれらの移動障害を救出する。百日咳毒素(PTX)(100ng/ml、4時間)は、MAGL−OE癌細胞の増加した移動を逆転する。(G)MAGL−OE細胞と対比したEV対照細胞におけるLPAによる移動の濃度依存的刺激。LPAによって誘導されるEV対照細胞における移動の最大刺激は、MAGL−OE細胞において見出される亢進した移動と基本的に一致し、そしてLPAはMAGL−OE細胞の移動をそれ以上増加しないことに特に言及しておく。(H)移動損傷パーセンテージで表した、shMAGL癌細胞と対比したsh対照癌細胞のEGFR(上皮成長因子受容体)阻害剤チロホスチンAG−1478に対する感受性。チロホスチンの抗移動効果に関するIC50値をパネル中に提供する。(I)MAGL−FFA経路を他のプロ腫瘍原性脂質へと結びつけることが可能な代謝ネットワークを示す図。(A)及び(B)について、データは比較群間の平均相対変化として表した;群当たりn=4−5。(C−G)について、それぞれの対照細胞群と比較したMAGL−OE細胞群又はshMAGL細胞群(C−G)、DMSO処理対照細胞と対比したJZL184処理細胞(C、D)については、*はP<0.05、**はP<0.01である。侵襲性細胞(C8161、SKOV3)と対比した非侵襲性親細胞(MUM2C、OVCAR3)(C、D)、又はsh対照細胞と対比したLPA/PGE2処理shMAGL細胞(E)については、##はP<0.01である。データは、平均±標準誤差で表した;群当たりn=3−5。
【図20】図20A−20Gは、パネルを通して、非侵襲性と対比した侵襲性の黒色腫細胞株及び卵巣癌細胞株のMAGL、KIAA1363及び脂肪分解プロファイルを示している。MAGL阻害剤JZL184による癌細胞処理によって引き起こされる増加したMAGと減少したFFAとの間に質量バランスの矛盾が存在する。(A−C)侵襲性黒色腫細胞(C8161)及び卵巣細胞(SKOV3)は、それらの非侵襲性カウンターパート(それぞれMUM2C及びOVCAR3細胞)と比較してより大きな移動(A)及び浸潤(B)及びより速い腫瘍増殖速度(C)を示す。(D)癌細胞株の脂肪分解特性は、他の脂肪分解活性と比較して、侵襲性癌細胞において強いMAG加水分解活性を示す。生体外での、種々の脂質基質からの遊離脂肪酸(FFA)の遊離を測定した。細胞を無血清培地中、生体内原位置でDMSO又はJZL184により4時間処理した。細胞溶解物(20μg)を100μM脂質基質と共に室温で1時間インキュベートした。上段及び下段のパネルは、比較及び可視化を容易にするために同じデータを異なったy軸の尺度(MAG加水分解活性の有無)で示している。脂質基質についての略称は下記のとおりである。TAG、トリアシルグリセロール;DAG、ジアシルグリセロール;MAG、モノアシルグリセロール;PC、ホスファチジルコリン;LPC、リゾホスファチジルコリン;PE、ホスファチジルエタノールアミン;LPE、リゾホスファチジルエタノールアミン。非侵襲性癌株群と対比した侵襲性癌株群については、*はp<0.05、**はp<0.01であり、JZL184と対比したDMSOについては、##はp<0.01である。データは、平均±標準誤差として表した;群当たり、n=3−15(G)リゾリン脂質の増加は、MAGL阻害剤JZL184による癌細胞処理によって引き起こされる増加したMAGと減少したFFAの間の質量バランスの矛盾を説明する。(E)JZL184(1μM、4時間)は、C8161及びSKOV3細胞において5500−6100pmol/106のFFAレベルの減少を引き起こす。このFFAレベルの減少は、JZL184処理癌細胞において見出されるリゾホスファチジルコリン(LPC)及びリゾホスファチジルエタノールアミン(LPE)レベルの正味の増加(4100−6500pmol/106細胞)と同様の大きさである。(F)C17:0MAG(20μM、1時間)は、癌細胞によってC17:0LPC及びC17:0LPEに変換されるが、しかもこの変換は癌細胞をJZL184(1μM、4時間)と共にプレインキュベーションすることによってより亢進される。C17:0MAGは、またC17:0LPAにも変換されるが、しかしこの場合には、当該変換はJZL184によって遮断される。これらのデータは、C17:0MAGは、癌細胞中でC17:0LPC及びC17:0LPEに直接転換されるが、一方、C17:0MAGのC17:0LPAへの変換には、C17:0MAGのC17:0FFAへのMAGL依存加水分解が要求されることを示している。C20:4MAG(20μM、1時間)は、プロスタグランジンE2(PGE2)に変換されるが、この変換はJZL184によって遮断される。(G)C16:0及びC20:4FFAは、侵襲性癌細胞によってそれぞれLPA及びPGE2に変換される。C8161及びSKOV3細胞を、無血清培地中、d2−C16:0FFA又はd8−C20:4FFA(10μM、4時間)で処理した。d2−C16:0FFAは、d2−C16:0 LPAに変換され、d8−C20:4FFAは、d8−C20:4PGE2に変換された。処理細胞と対比した対照細胞の比較については、*はp<0.05、**はp<0.01であり、MAG単独処理細胞と対比したJZL184/MAG処理細胞の比較については、#はp<0.05、##はp<0.01である。データは、平均±標準誤差で表した;群当たり、n=3−4。
【図21】図21A−21Iは、MAGLが侵襲性癌細胞株においてMAG、FFA及び癌細胞の病原性を制御することを示している。(A、B)JZL184処理(1μM、4時間)は、231MFP細胞においてMAGレベルを増加し(A)及びFFAレベルを減少する。(C)shMAGLプローブは、活性に基づいたタンパク質プロファイリング(ABPP)によって判断すると、侵襲性癌細胞C8161、SKOV3、及び231MFPにおいて他のセリンヒドロラーゼと比較してMAGLの活性を選択的に減少する。活性に基づいたプローブFPローダミンにより細胞全体のプロテオームにおいてセリンヒドロラーゼ活性を標識化し、SDS−PAGE及びインゲル蛍光スキャニング(蛍光ゲルをグレイスケールで示す)によって検出した。、ABPP(対C8161、SKOV3、及び231MFP)及びC20:4MAG基質アッセイ(対231MFP)によって評価しところ、MAGL活性は両方のshMAGLプローブ(shMAGL1、shMAGL2)により>75%減少した。(D、E)shMAGL細胞は、sh対照又は231MFP親細胞と比較して増加したMAGレベル(D)及び減少したFFAレベル(E)を示す。(F−H)shMAGL細胞は、sh対照又は231MFP親細胞と比較して減少した移動(F)、浸潤(G)、及び細胞生存(H)を示す。移動障害は、C16:0FFA又はC18:0FFAによる処理(20μM、4時間)によって救出された。(I)JZL184によるMAGLの急性遮断により、癌細胞の移動が損傷される。それぞれの対照群と対比したJZL184処理細胞又はshMAGL細胞については、**はp<0.01である。DMSO処理shMAGL群と対比したC16:0又はC18:0FFA処理群については、##はp<0.01である。データは、平均±標準誤差として表した;群当たり、n=4−6。
【図22】図22A−22Bは、生体内でのJZL184処理(経口強制飼養40mg/kg、30日間、4μL/gポリエチレングリコールを毎日1回投与)が腫瘍異種移植片MAGL活性を阻害すること(A)及びshMAGL腫瘍がsh対照細胞と比較してより低いFFAレベルを含んでいたこと(B)を示している。溶媒又はJZL184処理したマウス腫瘍ホモジネートを、100μMC20:4MAGの添加前に、DMSO又はJZL184(1μM、30分間)と共に生体外で30分間インキュベートした。それぞれ溶媒腫瘍又はsh対照腫瘍と対比したJZL184処理腫瘍又はshMAGL腫瘍については、*はp<0.05、**はp<0.01である。データは、平均±標準誤差として表した;グループ当たり、n=4。
【図23】図23A−23Gは、MAGLの異所性発現がFFAレベルを増加し、卵巣癌細胞OVCAR3及びMUM2C並びに黒色腫細胞の生体外及び生体内での病原性を亢進することを示している。(A)ABPP(上段パネル)、ウェスタンブロット(中段パネル)及びC20:4MAG加水分解活性(下段パネル)によって確認した、OVCAR3細胞におけるMAGLの過剰発現(MAGL−OE、赤色バー)。対照及びS122A細胞(黒色バー)は、空ベクターにより感染した癌細胞又は触媒的に不活性なMAGL変異株(S122A)に相当する。ウェスタン分析は、S122A−MAGL変異株の過剰発現を確認したが、当該変異株はABPP及びC20:4MAG加水分解アッセイによる評価においてはいかなる活性をも示さなかった。(B、C)MAGL−OE細胞は、EV対照細胞及びS122A細胞と比較してより低いMAGレベル(B)及びより高いFFAレベル(C)を含んでいる。これらの代謝的効果は、生体内原位置でのJZL184による処理(1μM、4時間、栗色のバー)によって逆転された。(D、E)MAGL−OE OVCAR3細胞は、EV及びS122A対照細胞と比較して増加した移動(D)及び浸潤(E)を示す。この亢進した移動及び浸潤は、JZL184(1μM、4時間)によって逆転された。代表的な移動のパネルを示す(D)。(F)MAGL−OE MUM2C及びOVCAR3細胞は、EV対照細胞と比較して著しく亢進した細胞生存(血清飢餓培養後24時間)を示す。(G)MAGL−OE MUM2C細胞の亢進した腫瘍増殖速度は、侵襲性のC8161黒色腫細胞の腫瘍増殖速度と同様である。C8161及びMUM2Cのデータは、それぞれ図16E及び図17Fに由来する(これらの実験は、腫瘍増殖速度の直接比較を可能にするために同時に実施したことを特に言及しておく)。対照群と対比したMAGL−OE群については、*はp<0.05、**はp<0.01である。データは、平均±標準誤差として表した;群当たり、n=4−6。
【図24】図24A−24Jは、癌の攻撃性における、カンナビノイドのシグナル伝達、エネルギー力学、脂肪酸ベータ酸化又は脂肪酸合成の評価を示している。sh対照及びshMAGL癌細胞におけるFFAレベルは、CB1アンタゴニスト(リモナバン、1μM、4時間)又はCB2アンタゴニスト処理(AM630、1μM、4時間)によって変化しなかった。C16:0及びC20:4FFAについてのデータを示す。他のFFA(C18:0、C18:1)についても同様の結果が見出された。(B)shMAGL癌細胞における移動障害は、CB1アンタゴニストであるリモナバン又はCB2アンタゴニストであるAM630のどちらの処理(1μM、4時間)によっても、又は両方のアンタゴニストによる共同処理によっても救出されなかった。sh対照細胞と対比した各shMAGL群について、**はp<0.01である。データは、平均±標準誤差として表した;群当たり、n=6。(C、D)DNAマイクロアレイ分析によって測定したCB1受容体(C)及びCB2受容体(D)mRNAレベル。左の棒グラフは、HU133APlus2.0アフィメトリクスアレイにより実施されたヒト癌細胞株のNCI60パネルの分析から抽出したデータに相当する(ロスら(Ross et al.)、2000年)。右の棒グラフは、HU133APlus2.0アフィメトリクスアレイにより実施された本研究に使用した侵襲性及び非侵襲性癌細胞株の分析から抽出したデータに相当する。比較のために、高いCB1受容体レベルを含むSNB−19癌細胞株を示す。(E−H)MAGLの増加した発現(MAGL−OE)又は減少した発現(shMAGL)は、癌細胞におけるNAD+(E)、NADH(F)、ピルビン酸(G)、又は乳酸(H)レベルを変化させない。データは、平均±標準誤差として表した;群当たり、n=6。(I)脂肪酸β酸化阻害剤トリメタジジン(3−ケトアシルコエンザイムAチオラーゼ阻害剤、100μM、4時間のプレインキュベーション及び移動に関してはコインキュベーション)又はエトモキシル(CPTI阻害剤、100μM、4時間のプレインキュベーション及び移動に関してはコインキュベーション)は、癌細胞の移動に影響を与えなかった。対照群と対比したMAGL−OE群について、**はp<0.01である。データは、平均±標準誤差として表した、群当たり、n=3。(J)エトモキシル処理(100μM、4時間)は、C20:4FFAを除いては癌細胞中のFFAレベルを変化させなかったが、当該FFAはC8161及びSKOV3細胞において増加した。それぞれの対照群と対比したshMAGL又はMAGL−OE細胞について、*はp<0.05、**はp<0.01である。DMSO処理対照群と対比したエトモキシル処理群については、#はp<0.05である。データは、平均±標準誤差で表した;群当たり、n=3−4。
【図25】図25A−25Dは、EGFRキナーゼ阻害剤チロホスチンの癌細胞への影響を示している。チロホスチンは、shMAGL癌細胞において見出される移動及び細胞生存の障害と同様のレベルで、C8161(A、B)及びSKOV3(C、D)細胞において、移動及び細胞生存(48時間、無血清)を減少した。C8161及びSKOV3shMAGL細胞は、細胞移動及び細胞生存(A、B)においてチロホスチン誘発障害に対する亢進した感受性を示す。溶媒処理sh対照細胞又はshMAGL癌細胞と対比したチロホスチン処理細胞の間では、*はp<0.05、**はP<0.01である;同処理群のsh対照とshMAGL癌細胞との間では、##はp<0.01である。データは、平均±標準誤差として表した、群当たり、n=4。
【0020】
発明の詳細な説明
I.概要
2−アラキドノイルグリセロール(2−AG)及びアナンダミドは、カンナビノイド受容体CB1及びCB2を活性化する脂質伝達物質である。内在性カンナビノイドシグナル伝達は、アナンダミドについては、脂肪酸アミドヒドロラーゼ(FAAH)によって媒介される過程、2−AGについては、モノアシルグリセロールリパーゼ(MAGL)によって基本的に制御されていると考えられている過程である、酵素的加水分解によって終結する。本発明は、MAGLの選択的阻害剤の本発明者らによる合成及びキャラクタリゼーションに一部基づいている。MAGL選択的阻害剤はMAGLのヒドロラーゼ活性を強力に阻害する一方で、FAAH等の他の脳セリンヒドロラーゼにほとんど又は全く阻害活性を有しないことが見出された。加えて、マウスへの腹腔内注射又は経口投与において、選択的MAGL阻害剤は、脳の2−AGレベルを著しく増加させることができたが、アナンダミド又は他のN−アシルエタノールアミン等の他の既知の脂質シグナル伝達分子のレベルを変化。また、阻害剤により処理されたマウスも、痛覚消失症を含む一連のCB1依存的行動的影響を示。
【0021】
発明者らは、MAGL及びその遊離脂肪酸(FFA)産物が、侵襲性のヒト癌細胞及び原発性の腫瘍において上方制御されており、それによって、移動、浸潤、生存、及び生体内での腫瘍増殖を促進する、発癌性のシグナル伝達脂質に富む脂肪酸代謝ネットワークが制御されることをさらに発見した。非侵襲性癌細胞におけるMAGLの過剰発現がこの脂肪酸代謝ネットワークを繰り返し、MAGL阻害剤によって逆転される、病原性−表現型を増加することが見出された。発明者らは、MAGLの遮断が生体外での移動だけでなく生体内での腫瘍増殖をも障害することを見出した。加えて、MAGL遮断に由来する表現型は、外来源のFFAによって救出された。これらのデータは、癌細胞においてFFAを増加するために、並びに移動活性及び腫瘍活性を増加するために、MAGLが必要かつ十分であることを示している。
【0022】
これらの発見に従って、本発明は、MAGL選択的アンタゴニスト化合物又はその薬学的に許容可能な塩を含む治療的な組成物を提供する。これらの組成物は、異所性の又は不十分なカンナビノイドシグナル伝達、特に2−AG媒介内在性カンナビノイドシグナル伝達に連結又は媒介される、被験者の障害又は症状の症候を治療又は回復するために、又は増加したカンナビノイドレベルから有利な効果を引き出すことのできる疾患を治療するために使用することができる。このような障害又は症状には、例えば、疼痛、炎症、抑鬱、、多発性硬化症、緑内障、及びアルツハイマー病が含まれる。これらの特殊な症状又は障害のいずれかを患う被験者における治療応用に加えて、本発明の組成物は、一般的にカンナビノイド依存性行動的影響(例えば、痛覚消失症)を誘導するために、又は2−AG媒介内在性カンナビノイドシグナル伝達活性を刺激又は増大するために、他の被験者においても用いることができる。本明細書で例示するMAGL選択的阻害化合物をリード化合物として使用することにより、本発明は、改良された生物学的活性及び薬学的特性を備えた新規なMAGL阻害剤のためのスクリーニング方法をさらに提供する。当該新規MAGL阻害剤は、異常な又は減少したカンナビノイドシグナル伝達活性によって引き起こされる又は関連する、障害又は症状の治療における増強された特性を備えた治療薬を提供することができる。次節に、本発明の組成物の生成及び使用、並びに本発明の実施方法についてのガイダンスを提供する。
【0023】
II.定義
別途定義が無い限り、本明細書で使用する技術的及び科学的用語は、本発明の属する分野の当業者によって通常に理解されるのと同様の意味を有する。下記の参考文献は、本発明において使用する多くの用語の一般的な定義を当業者に提供する:シングルトンら(Singleton et al.)著、「微生物学及び分子生物学辞典(DICTIONARY OF MICROBIOLOGY AND MOLECULAR BIOLOGY)」(第2版、1994年);「ケンブリッジ科学・技術辞典(THE CAMBRIDGE DICTIONARY OF SCIENCE AND TECHNOLOGY)」、(ウォーカー(Walker)編、1988年);及びヘイル&マーハム(Hale & Marham)著、「ハーパー・コリンズ生物学辞典(THE HARPER COLLINS DICTIONARY OF BIOLOGY)」、(1991年)。加えて、本発明の実施において読者を援助するために下記の定義を提供する。
【0024】
用語「薬剤(agnt)」又は「試験薬(test agent)」には、任意の物質、分子、元素、化合物、実体、又はそれらの組合せが含まれる。限定されるものではないが、例えば、タンパク質、ポリペプチド、有機小分子、多糖類、ポリヌクレオチド、その他等が含まれる。天然物、合成化合物、又は化学物質、又は二以上の物質の組合せであることができる。特に指定のない限り、用語「薬剤(agent)」、「物質(substance)」、及び「化合物(compound)」は、本明細書で互換的に使用する。
【0025】
用語「アナログ(analog)」は、基準分子(例えば、本明細書で開示するMAGL選択的阻害剤)に構造的に類似するが、しかし、代替置換基で基準分子の特異的な置換基を置換することによって、標的化され制御された方法で修飾された分子に言及するために本明細書で使用する。基準分子と比較してアナログは、当業者にとり、同一、類似の、又は改良された有用性が期待されるであろう。改良した特性(標的分子に対する高結合親和性等)を有する、既知化合物の変異型を同定するためのアナログの合成及びスクリーニングは、製薬化学において周知の研究法である。
【0026】
「抗悪性腫瘍剤(antineoplastic agent)」は、の新生物、特に細胞腫、肉腫、リンパ腫、又は白血病等の悪性(癌性)の病変の発達又は進行を阻害する機能特性を有する薬剤に言及するために本明細書で使用する。転移阻害はしばしば抗悪性腫瘍剤の一特性である。
【0027】
化学療法剤は、腫瘍性疾患の治療方法においてポリクローナル抗グリカン抗体と併用して使用することができる。抗グリカン抗体を含む抗体−細胞毒抱合体もまた、標準的な癌治療によって誘導された免疫を追加免疫するために使用することができる。これらの場合には、投与される化学療法剤の服用量を減少することが可能である(モキールら(Mokyr et al.)、「カンサー・リサーチ(Cancer Research)」、1998年、第58巻:5301−5304)。抗グリカン抗体併用の科学理論的な解釈は、ほとんどの化学療法化合物の細胞毒性作用の結果である細胞死が、結果として抗原提示経路において増加した腫瘍抗原レベルに帰着するというものである。このように、抗グリカン抗体は、腫瘍細胞の化学療法放出を刺激して免疫応答を追加免疫することができる。
【0028】
本明細書で使用する場合、「接触(contacting)」は、その通常の意味を有するが、二以上の分子の組み合わせ又は分子と細胞の組み合わせ(例えば、一化合物と一細胞)をも意味する。接触は、生体外で、例えば、二以上の薬剤を組み合わせて又は一薬剤と一細胞若しくは一細胞溶解物を組み合わせて、試験管又は他の容器中で引き起こすことができる。接触は、生体内でも、例えば、被験者の体内のタンパク質又はポリペプチドを特異的に標的とする薬剤を被験者に投与することによって引き起こすことができる。
【0029】
カンナビノイドシグナル伝達と関連する症状又は障害とは、異所性の又は抑制された内在性カンナビノイドシグナル伝達に連結、媒介又は関連する障害をいう。このように、当該障害には、減少した内在性カンナビノイドレベル(例えば、2−AG)又は内在性カンナビノイドを分解する過剰な酵素活性によって引き起こされる症状が包含される。カンナビノイドシグナル伝達と関連する症状又は障害には、内在性カンナビノイドシグナル伝達の増大によって有益な効果(例えば、疼痛)を誘導することのできる症状が含まれる。当該用語には広く、治療効果が増加したカンナビノイドレベル又は減少したアラキドン酸レベルから獲得することのできる疾病も含まれる(例えば、アルツハイマー病又は多発性硬化症)。本発明の幾つかの好ましい実施態様において、当該症状又は障害は、2−AGシグナル伝達と関連又は付随する(例えば、異所性の又は抑制された2−AGシグナル伝達)症状・障害であり、又は2−AGシグナル伝達活性を刺激することによって有益な効果を誘導することのできる症状・障害である。このような症状又は障害の例には、疼痛、抑鬱、、心血管障害、代謝障害、炎症性症候群及び卒中が含まれる。
【0030】
本発明において使用する場合、心血管障害は、心臓又は血管(動脈及び静脈)に関わる疾患のクラスをいう。それは、心臓血管系に悪影響を及ぼす任意の疾患を含むが、しかし特にアテローム性動脈硬化症(動脈疾患)と関連するこれらの症状をいう。これらの症状は同様の原因、機序、及び治療法を有する。通常の心血管障害の例には、動脈硬化症(動脈の硬化)、アテローム性動脈硬化症(血管壁上のコレステロール蓄積等の粥腫)、リウマチ性心疾患、全身性高血圧症及び卒中が含まれる。本明細書で使用する場合、炎症性疾患とは、異常な、過剰な又は調節解除された炎症に付随する疾患又は症状である。それらには、の種々の疾患の根底にある、、無な疾患群含まれる。代謝障害とは、正常な成長発育に必須のある代謝反応が起きないときに発生する健康状態をいう。これらには、アミノ酸代謝障害(例えば、メープルシロップ尿症及びホモシスチン尿症)、有機酸代謝障害(例えば、メチルマロン酸尿症)、脂肪酸ベータ酸化障害(例えば、MCAD欠損症)、テイ・サックス病及びゴーシェ病等の脂質代謝障害(脂質蓄積症)、ミトコンドリア障害(例えば、ミトコンドリア心筋症)、及びペルオキソーム病(例えば、ツェルベルガー症候群)含まれる。
【0031】
カンナビノイドの行動的影響(又はCB1依存性及び/又はCB2依存性行動的影響)とは、カンナビノイドシグナル伝達活性の結果として被験者によって示される生理学的及び行動上の応答をいう。カンナビノイドの行動的影響の幾つかは、2−AGシグナル伝達に関与するが、本明細書では2−AG媒介行動的影響(又は2−AG依存行動的影響)と。下記の実施例において詳述する通り、2−AG媒介行動的影響には無痛覚症(減少した痛覚)が含まれる。特に指定のない限り、カンナビノイドの行動影響には、また被験者に存する又は被験者によって経験される、炎症、又は抑鬱に付随する症候の軽減又は回復も包含される。
【0032】
内在性カンナビノイド系とは、食欲、痛覚、気分、及び記憶を含む、種々の生理学的過程に関与する、神経調節物質及びその受容体の一群をいう。カンナビノイド受容体(カンナビスの向精神効果を媒介する受容体と同一)に結合する内在性の脂質がカンナビノイド命名される。、内在性カンナビノイド系には(1)それぞれ主に中枢神経系及び抹消神経に位置する2のGタンパク質共役型受容体である;(2)内在性アラキドン酸に基づく脂質である、アナンダミド(N-アラキドニルエタノールアミン又はAEA)及び2−アラキドノイルグリセロール(2−AG)集合的に「内在性カンナビノイド」と;及び(3)内在性カンナビノイドアナンダミド及び2−AGを合成及び分解する酵素。内在性カンナビノイド系は、遺伝的及び薬理学的方法を使用して研究されてきた。これらの研究は、神経調節物質放出、運動学習、シナプス可塑性、食欲、及び痛覚を含む、様々な生理学的過程における内在性カンナビノイドシグナル伝達についての広範な役割を明らかにした。
【0033】
痛覚過敏は、被験者(例えば、温血動物)が疼痛に対し増加した感受性を示す症状である。それは、身体への物理的損傷、例えば、手術によって不可避的に生じた受傷に付随することが知られている。疼痛は、関節炎及びリウマチ性疾患等のの炎症症状に付随することも知られている。痛覚過敏には、限定されるものではないが、炎症(例えば、リウマチ性関節炎及び変形性関節症)と関連する疼痛、術後痛、後分娩痛、歯科症状(齲蝕及び歯肉炎)と関連する疼痛、限定されるものではないが、日焼け、擦過傷、挫傷その他を含む熱症と関連する疼痛、スポーツ損傷及び捻挫、限定されるものではないが、ツタウルシ、アレルギー性皮疹及び皮膚炎を含む皮膚症状と関連する疼痛、及び軽い刺激に対する感受性を増加する有害冷感等の他の疼痛等の軽中度の疼痛から激痛までが含まれる。
【0034】
基準分子(例えば、MAGLポリペプチド)又は細胞(例えば、腫瘍細胞)の活性に関する用語「調節(modulate)」とは、活性の上方制御(、活性化又は刺激)又は下方制御(、阻害又は抑制)をいう。ポリペプチド等の基準分子の調節には、分子の発現又は細胞内レベルの変化も含まれる。生物学的活性(例えば、MAGL発現又はリパーゼ活性、又は腫瘍細胞増殖)に関する用語「阻害する(inhibit)」又は「阻害(inhibition)」とは、活性の抑制又は下方制御をいう。阻害の機序は、例えば基準分子又は細胞への結合によって直接的であことができる。阻害は、例えば、別の方法で基準分子又は細胞に結合し調節する他の分子への結合及び/又は修飾によって間接的であことができる。
【0035】
「ポリヌクレオチド」又は「核酸配列」とは、ヌクレオチドのをいう。幾つかの例において、ポリヌクレオチドとは、それが由来するところの生物体のゲノム中において、それがすぐ近くに隣接しているどちらのコード配列ともすぐ近くに隣接していない配列をいう。それ故に、当該用語には、例えば、ベクター:自己複製プラスミド若しくはウイルス;又は原核生物若しくは真核生物のゲノムDNAに取り込まれた、又は他の配列とは別個の独立した分子(例えば、cDNA)として存在する組換えDNAが含まれる。ポリヌクレオチドは、リボヌクレオチド、デオキシリボヌクレオチド、又は各々のヌクレオチドの修飾型であることができる。
【0036】
ポリペプチド又はタンパク質とは、単量体がアミド結合によって結合したアミノ酸残基である重合体をいう。アミノ酸がアルファ-アミノ酸である場合、L−光学異性体又はD−光学異性体のどちらでも使用することができるが、L−異性体が典型的である。ポリペプチド又はタンパク質フラグメントは、自然界に見出されるタンパク質のものと同じ又は実質的に同一のアミノ酸配列を有することができる。実質的に同一の配列を有するポリペプチド又はタンパク質とは、アミノ酸配列が完全ではないが大部分同じであり、それが関連する配列の機能活性が維持されていることを意味する。
【0037】
用語「被験者」には、他の非動物、例えば、ウマ、イヌ、ネコ、マウス及びラットが含まれる。
【0038】
基準分子(例えば、MAGL選択的阻害剤)の変異型とは、基準分子全体又はそのフラグメントのどちらかと構造及び生物学的活性が実質的に類似する分子をいうことを示している。このように、二つの分子が類似の活性を有するならば、一方の分子の成分又は構造が他の分子に見出されるものと同じでなくても、又はアミノ酸残基の配列が同じでなくても、それらは当該用語が本明細書で使用するところの変異型とみなされる。
【0039】
侵襲性癌又は侵襲性腫瘍とは、迅速に増殖及び拡散する癌又は腫瘍をいう。いかなる起源の癌又は腫瘍も適した条件下では侵襲性となることができる。従って、侵襲性腫瘍には、例えば、侵襲性乳癌、侵襲性前立腺癌、侵襲性脳腫瘍、侵襲性子宮頚部癌、侵襲性結腸癌、侵襲性肺癌、侵襲性胃癌、侵襲性肝癌、侵襲性腎臓癌、侵襲性黒色腫及び侵襲性卵巣癌が含まれる。腫瘍が侵襲性となるためには、血管からはるか遠くにあるその腫瘤の中央の細胞に栄養を与えることができなければならない。これは血管新生によって達成される。突然変異によって、わずかな癌細胞は、血管新生成長因子を産生する能力を獲得できるかもしれない。これらの成長因子は、腫瘍によって近くの組織に放出されるタンパク質であり、そこでは、それらが新生血管を刺激して腫瘍内に成長させる。このことが、腫瘍が腫瘤中で急速に拡大し組織を囲んで浸潤することを可能にしている。それは、癌細胞のために新生血管中へ漏出し及び身体中を循環する経路をも提供するが、そこでそれらは転移を形成しながら他の器官に停止する。
【0040】
III.MAGL選択的阻害剤
本発明は、新規なMAGL阻害剤を提供する。MAGL阻害剤には、MAGLの発現、修飾、制御若しくは活性を妨害する化合物、又はMAGLの正常な生物学的活性(例えば、そのセリンヒドロラーゼ活性)の一以上を下方制御する化合物が含まれる。特に本発明のMAGL阻害剤は、MAGL酵素活性を遮断又は阻害する。上記で言及したように、MAGLは、神経系における、カンナビノイド受容体リガンド、2−AGの加水分解の原因となる主要な酵素である。MAGL阻害剤又はアンタゴニスト化合物は、痛覚消失症等のカンナビノイド依存行動的影響を誘発するのに有用である。MAGL酵素活性を特異的に阻害又は抑制する化合物は、異常な(例えば、抑制又は減少した)カンナビノイドシグナル伝達活性によって引き起こされる又はする障害又は症状を治療するうえで種々の治療的又は予防的応用を有することができる。MAGL酵素活性を阻害するいかなる分子も、カンナビノイド依存行動的影響を誘発する、又は抑制されたカンナビノイドシグナル伝達活性に連結した症状を治療することができるかもしれない。しかしならが、他の脳セリンヒドロラーゼ(例えば、FAAH)をも阻害するMAGLアンタゴニスト化合物は、これらの他の酵素によって媒介される様々な生物学的機能により妨害されるかもしれない。このような非選択的MAGL阻害剤は、抑制されたカンナビノイドシグナル伝達活性に連結した障害(例えば、疼痛消失)を治療することができるが、多くの望まれない副作用を有しがちである。従って、このような治療的な応用においてはMAGL酵素活性を選択的に阻害する分子が好ましい。MAGL酵素活性を特異的に阻害することによって、一方では他の脳セリンヒドロラーゼ(FAAH)に著しい影響を生じずに、不十分な又は抑制された2−AGシグナル伝達活性と関連した又は連結した症候を治療又は回復することができる。
【0041】
従って本発明は、他の脳セリンヒドロラーゼ(例えば、FAAH又はABHD6)の一以上の酵素活性が著しくは悪影響を受けない濃度で、MAGLの酵素活性を実質的に遮断又は阻害する、新規なMAGL−選択的阻害剤(又は「MAGL−特異的阻害剤」)を提供する。幾つかの好ましい実施態様において、MAGL阻害剤は、MAGLによる2−AGの加水分解を遮断する。特に、本発明のMAGL選択的阻害剤又はMAGL選択的アンタゴニストは、FAAH阻害に比較してMAGLの阻害に対し選択性を示す化合物である。他の脳セリンヒドロラーゼ(例えば、FAAH)にまさったMAGLに対する化合物の選択性は、当該化合物の存在下にに関する各酵素の酵素活性を分析することによって測定することができる。明細書の実施例で詳述する通り、選択性は、脳膜プロテオームにおける化合物による基質(例えば、MAGLに対しては2−AG及びFAAHには対してはオレイン酸アミド)の加水分解阻害のIC50値を測定することによって決定することができる。IC50値を測定するのに加えて、FAAHにMAGLに対する阻害剤の選択性は、化合物存在下での酵素的触媒加水分解反応の擬一次速度定数(kobs/[I])を測定することによっても決定することができる。これは、下記の実施例に記載した通り、脳膜プロテオーム中に存在する酵素により生体外で実施することもできる。脳膜プロテオームに存在する加水分解酵素を使用する以外に、酵素活性アッセイは、組換え酵素によっても実施することができる。例えば、他の脳セリンヒドロラーゼと同様にMAGL、FAAHは、本明細書に記載した方法又は当該技術分野で報告された方法に従って組換えで容易に得ることができる(例えば、ブランクら(Blank et al.)、「ケミストリー&バイオロジー(Chem.Biol.)」、2007年、第14巻:p.1347−56)。典型的には、MAGL選択的阻害剤は、FAAHに対するそのIC50値よりも少なくとも20倍、好ましくは少なくとも50、100、250又は500倍低いMAGLに対するIC50値を有すべきである。kobs/[I]により測定するならば、MAGL選択的阻害剤は、通常、FAAHに対するそれよりも少なくとも10倍、好ましくは少なくとも25、50、75、100又は300倍高いMAGLに対する速度定数を有するだろう。
【0042】
本発明のMAGL選択的阻害剤の幾つかは、FAAHに加えて、また一以上の他の脳セリンヒドロラーゼMAGLに対して選択的でもある。例えば、化合物は、ABHD6及び/又はABHD12MAGLに対して選択的であることができる。ABHD6及びABHD12は、マウス脳において2−AGヒドロラーゼ活性を示すことが明らかにされた他の二つの脳酵素であり、明確な細胞内分布を示している(例えば、ブランクら(Blank et al.)、「ケミストリー&バイオロジー(Chem.Biol.)」、2007年、第14巻:p.1347−56を参照)。これらの酵素を阻害しないMAGL選択的アンタゴニストは、内在性カンナビノイドシグナル伝達と関連した現在は未知の機能を研究及び証明するために有用である。
【0043】
本発明においては、種々のMAGL選択的アンタゴニストを使用することができる。本発明によって合成されたこれらMAGL選択的阻害剤の幾つか、下記の実施例に記載する。これらの化合物は、下記に示す式Iの構造を有する。これらの化合物は、本明細書の実施例において開示した合成スキームに従って容易に調製することができる。
【化4】
【0044】
式中、XはN又はCH、R1はH又はOHであり、そしてR2及びR3の各々は、下記の構造の一つを有する。
【化5】
【0045】
例示したMAGL−特異的阻害剤の幾つかは、FAAHにMAGL選択的。実施例において詳述する通り、図1Aに示す化合物WWL152、WWL162、JZL175及びJZL184の全ては、FAAHMAGLに対して選択性を示す。化合物の幾つかは、FAAHに加えて他の脳セリンヒドロラーゼにもまさってMAGLに対して選択的である。これらには、式Iによって包含される化合物JZL184及びJZL175が含まれる。これらの二つの化合物は、特異的にMAGLを阻害し、ABHD6と同様にFAAHに関して全く又は非常に減少した阻害活性しか有しない。
【0046】
これらのMAGL選択的阻害剤のいずれも、下記の実施例に詳述する合成スキームに従って生産することができる。例示したMAGL選択的アンタゴニスト以外に、追加のMAGL選択的阻害剤(例えば、JZL184又はJZL175の機能的な誘導体又はアナログ)は、本明細書に記載した方法又は当該技術分野において記載された方法を使用して容易に同定することができる。本明細書に記載する通り、例示するMAGL選択的阻害剤のアナログ化合物のライブラリーを作成することができる。アナログ化合物は、それからMAGL阻害の特異的活性及び他の脳セリンヒドロラーゼにまさった選択性に関してスクリーニングすることができる。
【0047】
IV.内在性カンナビノイドシグナル伝達の亢進及びそれに付随する症状の治療
本発明は、このような治療を必要とする被験者において、内在性カンナビノイドシグナル伝達(特に2−AGシグナル伝達活性)を刺激若しくは増大する、又はカンナビノイドの加水分解(例えば、2−AGの加水分解)を阻害する方法を提供する。下記の実施例に明らかにする通り、本発明のMAGL特異的阻害剤によるMAGLの阻害は、生体内での2−AG媒介内在性カンナビノイドシグナル伝達を増大するために十分であるが、一方、脳セリンヒドロラーゼの非選択的阻害により別に生じるかもしれない、いかなる意図しない結果も制限している。本発明者らは、幾つかのカンナビノイド依存性の行動的影響がMAGLの選択的阻害に由来こともした。これらの行動的影響(2−AG媒介行動的影響)には、例えば、痛覚消失症、運動性低下症、低温低下、及び反射亢進が含まれる。種々の状況下で、ある被験者においては一定の2−AG媒介行動的影響(例えば、痛覚消失症)を誘発することが望ましいかもしれない。例えば、疼痛を患う被験者は、MAGL選択的阻害によって誘発される痛覚消失から治療的利益を得ることができる。被験者における2−AGシグナル伝達活性を刺激又は増大するために、当該方法は、本明細書で開示するMAGL選択的阻害剤の治療的に有効な量を被験者に投与することを含んでいる。いくつかの関連する方法は、本発明のMAGL選択的阻害剤を被験者に投与することによって、CB1及び/又はCB2依存性の行動的影響を被験者に誘発することに向けられている。いくつかの方法において、被験者は、例えば、2−AGによって媒介される行動的影響、例えば痛覚消失を誘発することができるように、化合物を投与される。
【0048】
本明細書で開示するMAGL選択的阻害剤は、また一般的に異常なカンナビノイドシグナル伝達(特に2−AGシグナル伝達活性)により引き起こされる又は関連する障害、又は2−AGシグナル伝達活性を増大又は刺激することによって有益な効果を得ることができる症状を治療するためにも適している。従って、MAGL選択的阻害剤を用いる場合、本発明は、カンナビノイドシグナル伝達に連結する又は関連する障害又は症状を患う被験者を治療するための方法をさらに提供する。当該方法は、本明細書で開示するMAGL選択的阻害剤の治療的に有効な量を、治療を必要とする被験者に投与することを必要とする。治療されるべき被験者には、異常な(不十分又は抑制された)カンナビノイドシグナル伝達により媒介される又は関連する障害又は症状を患う被験者が含まれる。治療されるべき被験者には、また増大したカンナビノイドシグナル伝達活性から有益な効果を得ることができる症状を有する被験者でもあることができる。いくつかの好ましい実施態様において、当該方法は、2−AGシグナル伝達と関連する症状を患う被験者において、2−AG媒介シグナル伝達活性を刺激することに向けられる。これらの障害及び症状の例には、疼痛、炎症、、抑鬱、心血管障害、代謝障害、及び卒中が含まれる。本発明の方法に適した多くの炎症性障害が存在する。これらの障害は、例えば、喘息、自己免疫疾患、慢性炎症、慢性前立腺炎、糸球体腎炎、過敏症、炎症性腸疾患、骨盤内炎症性疾患、再灌流障害、リウマチ性関節炎、移植拒絶及び脈管炎等、当該技術分野において周知である。卒中に加えて、本発明の治療方法に適した心血管障害には、また動脈瘤、狭心症、アテローム性動脈硬化症、脳血管疾患、うっ血性心不全、冠動脈疾患、及び心筋梗塞(心臓発作)も含まれる。同様に、増大する内在性カンナビノイドシグナル伝達(例えば、2−AG媒介シグナル伝達活性)から利益を売ることのできる代謝障害は当該技術分野で周知である。このような障害の例には、フェニルケトン尿症、アルカプトン尿症、地中海貧血症、ポルフィリン症、テイ・サックス病、ハーラー症候群、ゴーシェ病、ガラクトース血症、クッシング症候群、真性糖尿病、甲状腺機能亢進症、及び甲状腺機能低下症が含まれる。
【0049】
本発明のMAGL選択的阻害剤は、又そのカンナビノイドの治療効果が報告されている他の症状を治療するためにも利用することができる。このような症状又は障害の例には、緑内障、多発性硬化症及び抑制された食欲によって引き起こされる消耗疾患が含まれる。消耗(wasting)とは、筋肉及び脂肪組織を衰弱させる衰弱性の疾患(例えば、細菌又はウイルス感染)又は症状の経過をいう。カンナビノイドがこれらの疾患又は障害に対する一定の治療的効果を提供することができると報告されている。例えば、ニューウェルら(Newell et al.)、「トランスアクションズ・オブ・ザ・オフタルモロジカル・ソサイアティ・オブ・ザ・ユナイテッド・キングダム(Trans.Ophthalmol.Soc.UK)」、1979年、第99巻:p.269−71;ブッシュヴァルトら(Wushwald et al.)、「ディー・ファルマジー(Pharmazie.)」、2002年、第57巻:p.108−14;クロックスフォードら(Croxford et al.)、「ドラッグス・オブ・トゥデイ(Drugs Today)」、2004年、第40巻:p.663−76;ウィラン(Whelan)、「ドラッグ・ディスカバリー・トゥデイ(Drug Discov.Today)」、2002年、第7巻:p.745−6;及びコスチニユックら(Costiniuk et al.)、「ザ・カナディアン・ジャーナル・オブ・ガストロエンテロロジー(Can.J.Gastroenterol.)」、2008年、第22巻:p.376−80を参照。本発明のMAGL選択的阻害剤は、2−AGの加水分解を阻害することによって治療効果を促進することができる。本発明のMAGL選択的阻害剤は、アルツハイマー病を患う被験者に治療的利益を提供するためにさらに用いることができる。アルツハイマー被験者の脳に増加したアラキドン酸が存在することが報告されており、それはアルツハイマー病上の発達と進行におけるアラキドン酸の役割を示唆している。例えば、ロスら(Ross et al.)、「ジャーナル・オブ・ニューロケミストリー(J.Neurochem.)」、1998年、第70巻、p.786−93;チャールイモイックら(Charlimoniuk et al.)、「ニューロケミストリー・インターナショナル(Neruochem.Int.)」、2006年、第48巻:p.1−8;及びラポポートプロスタグランジン・ロイコット・エッセント・(Prostaglandins Leukot Essent Fatty Acids)」2008年10月28日、参照。本発明のMAGL阻害剤によりMAGL酵素活性を阻害することによって、2−AGの加水分解及び結果として生じるアラキドン酸産物を抑制又は減少することができる。
【0050】
典型的には、本発明の治療方法には、本発明のMAGL特異的阻害剤を含有する医薬組成物を治療の必要な被験者に投与することが含まれる。MAGL特異的阻害剤は、単独で又は障害に適した他の既知の治療薬と併用して使用することができる。例えば、疼痛を有する被験者には、疼痛を緩和する他の鎮痛剤を追加的に投与することができる。このような既知の鎮痛剤には、モルヒネ及びモクソニジンが含まれる(米国特許第6,117,879号を参照)。幾つかの他の方法において、本発明のMAGL特異的阻害剤は、被験者の内在性カンナビノイドシグナル伝達活性を亢進するためにFAAH選択的阻害剤と併用して使用することもできる。MAGL阻害剤単独又はFAAH阻害単独と比較して、MAGL選択的阻害剤及びFAAH選択阻害剤両方の投与は、望むカンナビノイド依存性の行動的影響、例えば疼痛の減少又は炎症、、及び抑鬱に付随する症候の回復を被験者に強力により良く誘発することができた。FAAH選択的阻害剤は当該技術分野で既知である(例えば、フェグリーら(Fegley et al.))、「ジャーナル・オブ・ファルマコロジー・アンド・エクスペリメンタル・セラピューティクス(J.Pharmacol.Exp.Ther.)」、2005年、第313巻:p.352−8)。例えば、FAAH選択的阻害剤URB597は、ケイマンケミカル社(Cayman Chemicals)(アナーバー、ミシガン州)等から容易に市販品を入手することができる。
【0051】
例として、本発明のMAGL選択的阻害剤は、疼痛を患う被験者の症候を治療又は緩和するために使用することができる。疼痛は、多くの医的障害に存する。例えば、炎症は疼痛を誘発することができる。炎症性疾患の例には、変形性関節症、大腸炎、心臓炎、皮膚炎、筋炎、神経炎、リウマチ性関節炎及び狼瘡等の膠原病性脈管疾患が含まれる。これらの症状のどれかを有する被験者は、しばしば亢進した痛覚を体験する。過度の疼痛を引き起こすかもしれない他の病状又は医療行為には、外傷、手術、切断、膿瘍、灼熱痛、脱髄疾患、三叉神経痛、慢性アルコール中毒、卒中、視床痛症候群、糖尿病、癌ウイルス感染、及び化学療法が含まれる。
【0052】
一般的に、治療は、被験者が薬理学的及び/又は生理学的な効果を獲得することを可能にする。当該効果は、カンナビノイドシグナル伝達と関連する症状若しくは障害又はその前兆又はその症候から被験者を完全に又は部分的に防止するかとの点からは、予防的であってもよい。それはまた、カンナビノイドシグナル伝達及び/又は障害に有害作用(例えば、疼痛)と関連する症状又は疾患に対する部分的か又は完全な治癒の点からは治療的であることができる。被験者がである場合、人が知覚する疼痛のレベルは、被験者当人に疼痛を述べること又は疼痛を他の痛みの経験と比較することを頼むことによって評価することができる。あるいは、疼痛のレベルは、ストレス関連因子の放出又は抹消神経系若しくはCNS(中枢神経系)中の疼痛伝達神経の活動等の、被験者の疼痛に対する身体反応を測定することによって較正することができる。又、疼痛レベルは、人が疼痛がないと報告するのに要求される、又は被験者が疼痛の症候を示すことを停止するのに要求される、十分に特性化された鎮痛剤の量を測定することによっても較正することができる。
【0053】
本発明の治療法に適した被験者は、の被験者、以外の哺乳動物及びMAGLを発現する他の動物であることができる。被験者は、現在疼痛を引き起こしておりそして疼痛を引き起こし続けそうな進行中の症状を有していてもよい。また被験者は、通常痛みの結果を伴う術式又は事象を耐えてきてもよいし又は耐えることになってもよい。例えば、被験者は、糖尿病性神経因性疼痛又は膠原病性脈管疾患等の慢性の疼痛症状を有していてもよい。被験者は、また炎症、神経損傷、又は毒暴露(化学療法薬への暴露を含む)を有していてもよい。治療又は治療介入は、被験者の疼痛を減少又は和らげ、その結果、被験者が知覚する疼痛レベルが、治療しなかったとしたならば被験者が知覚したであろう疼痛レベルと比較して減少することを意図している。
【0054】
幾つかの実施態様において、神経性疼痛を有する被験者の治療を意図している。これらの被験者は、神経根障害、単神経障害、多発性単神経障害、多発性神経障害又は神経叢障害に分類される神経障害を有していてもよい。これらの種類の疾患は、限定されないが、外傷、卒中、脱髄疾患、膿瘍、手術、切断、神経の炎症性疾患、灼熱痛、糖尿病、膠原病性脈管疾患、三叉神経痛、リウマチ性関節炎、毒、癌(直接的又は間接的に(例えば、腫瘍随伴性)神経損傷を引き起こすことのできるもの)、慢性アルコール中毒、ヘルペス感染症、エイズ、及び化学療法を含む、さまざまな神経損傷症状又は術式によって引き起こされていてもよい。疼痛を引き起こす神経損傷は、末梢又はCNS神経の中にあってもよい。
【0055】
本発明の幾つかの実施態様において、痛覚過敏の治療又は緩和を必要とする被験者には、MAGLの阻害剤を一以上の追加の疼痛緩和剤と併用した組成物が投与される。これは、個々の鎮痛剤はしばしば疼痛緩和効果を部分的にのみ提供するからであるが、というのもそれは多くのなかのまさに一つの疼痛伝達経路を妨害するからである。しかしながら、疾患又は病状と関連する疼痛には、しばしば多様な受容器及び異なったシグナル伝達経路が関与している。それ故に、これらの状況下で侵害受容器を緩和するために一以上の疼痛軽減剤が必要であるかもしれない。幾つかの他の応用において、MAGL阻害剤は疼痛知覚経路中の異なる箇所に作用する鎮痛剤と併用して投与することができる。例えば、非ステロイド性抗炎症薬(NSAID)(例えば、アセトアミノフェン、イブブロフェン、及びインドメタシン)等の一クラスの鎮痛剤は、侵害受容器によって検出される刺激の化学伝達物質を下方制御する。オピオイド等の他のクラスの薬剤は、CNSにおける侵害受容情報の処理を変化させる。また、抗痙攣薬及び抗鬱薬を含む局所麻酔薬等の他の鎮痛剤が含まれていてもよい。MAGL阻害剤に加え、一以上のクラスの薬剤を投与することにより、より効果的な疼痛緩和を提供することができる。
【0056】
V.新規MAGL阻害剤のスクリーニング
本明細書に記載する特異的MAGL阻害剤に加えて、本発明は、また改良された特性を有する新規MAGL阻害剤のスクリーニング方法をも提供する。薬剤発見過程における重要な段階は、化学アナログリード化学鋳型の選択である。一定の分子標的のためのリード化学鋳型を同定する過程には、典型的には、機能アッセイにおける多数の化合物(しばしば100、000以上)のスクリーニング、活性を確認するための二次アッセイの試験用のある任意の活性閾値に基づく亜集合の選抜、並びに次に残った活性化合物の化学的合成の適合性の評価、が含まれる。本明細書に記載する新規MAGL阻害剤、例えば、式Iの化合物は、改良された生物学的又は薬学的特性を有する関連化合物を探索するためのリード化合物を提供する。例えば、これらMAGL阻害剤のアナログ又は誘導体は、MAGLに対するより高い親和性を有する又は皮膚により浸透性である化合物を同定することによって選別することができる。このような改良された特性を有する化合物は、種々の薬学的応用のために適している。
【0057】
本発明のスクリーニング方法には、典型的には最初に本明細書で例示するMAGL選択的阻害剤(例えば、JZL184又はJZL175)のアナログ、誘導体又は変形型の合成が含まれる。スクリーニングのために一定のMAGL阻害剤の構造的アナログのライブラリーを作成すると、次に、当該アナログ又は変異型が由来するMAGL阻害剤の当該特性と比較して改良された生物学的特性を有するアナログ又は誘導体を同定するために機能アッセイを実施する。例えば、現有するMAGL選択的阻害剤のアナログ化合物は、その選択性を維持しつつ、MAGLに対する高められた阻害活性(例えば、減少したIC50)を求めて選別することができる。あるいは、アナログは、FAAH及び/又はABHD6にMAGLに対する亢進した選択性を求めて選別することができる。それらは、また追加の脳セリンヒドロラーゼ(例えば、ABHD12)にMAGLに対する選択性を求めて選別することもできる。幾つかの他の実施態様において、アナログは、MAGLポリペプチドに対する亢進した結合親和性を求めて選別することができる。さらに、アナログ化合物のライブラリーは、例えば、より良い皮膚浸透性等の薬学的な性質又は薬物動態学的な性質を有する化合物を同定することにより評価することができる。
【0058】
さらに幾つかの実施態様において、本発明のスクリーニング法は、FAAH及びMAGLの両方を選択的に阻害することのできる治療薬を同定することにも向けられる。AEA及び2−AGの両方によって制御される行動的過程(例えば、痛覚)は、MAGL−FAAH二重阻害剤によってより強力に影響を受けることができるであろう。実施例で証明する通り、MAGL選択的阻害剤(例えば、JZL184)は、高濃度において他の脳セリンヒドロラーゼに著しい影響を与えずにFAAHを阻害することもできる。それ故に、これらの化合物は、MAGL及びFAAHの二重阻害剤の開発のリード化合物の骨格構造として役立つことができる。
【0059】
本明細書で例示するMAGL選択的阻害剤の化学的骨格に基づくアナログ又は誘導体を合成するためには、当該技術分野で周知の有機化学方法がだけである。例えば、既知化合物の化学アナログのコンビナトリアルライブラリーは、段階的な方法で合成することができる。化合物の大きなコンビナトリアルライブラリーは、WO第95/12608号、WO第93/06121号、WO第94/08051号、WO第95/35503号及びWO第95/30642号に記載された、エンコードされた合成ライブラリー(ESL)法によって構築することができる。種々のアナログの他の合成方法は、例えば、オーバーマン(Overman)著、「有機反応(Organic Reactions)」、第1−62巻、ウィリー・インターサイエンス(Wiley−Interscience)、2003年;ブルームら(Broom et al.)、「フェデラル・プロキュアメント(FedProc.)」、1986年、第45巻:p.2779−83;ベン−メナームら(Ben−Menahem et al.)、「リーセント・プログレス・イン・ホルモン・リサーチ(RecentProg.Horm.Res.)」、1999年、第54巻:p.271−88;シュラムら(Schramm et al.)、「アニュアル・レビュー・オブ・バイオケミストリー(Annu.Rev.Biochem)」、1998年、第67巻:p.693−720;ボーリンら(Bolin et al.)、「バイオポリマーズ(Biopolymers)」、1995年、第37巻:p.57−66;カールテンら(Karten et al.)、「エンドクライン・レビューズ(EndocrRev.)」、1986年、第7巻:p.44−66;ホら(Ho et al.)著、「有機合成戦略(Tactics of Organic Synthsis)」、ウィリー・インターサイエンス(Wiley−Interscience)、1994年;及びシャイトら(Scheit et al)著、「ヌクレオチド・アナログ:合成及び生物学的機能(Nucleotide Analogs:Synthesis and Biological Function)」、ジョンウィリー&サンズ(John Wiley&Sons)、1980年、に記載されている。
【0060】
のMAGL選択的阻害剤のアナログ化合物のライブラリー亢進した生物学的活性又は薬学的特性は、当該技術分野で周知の機能アッセイによっても容易に選別することができる。例えば、MAGL及び他の脳セリンヒドロラーゼの化合物による阻害は、例えば、脳膜プロテオーム中に存在する酵素によって同族基質の加水分解を定量すること等、本明細書で開示する又は当該技術分野で周知の酵素活性アッセイにより試験することができる。又は他の種由来の他の脳セリンヒドロラーゼと同様にMAGL、FAAHも当該技術分野で周知である。その塩基配列のクローニング並びにその酵素的及び他の生物学的活性のキャラクタリゼーションは、当該技術分野で全て報告されている。例えば、ブランクマンら(Blankman et al.)、「ケミストリー&バイオロジー(Chem.Biol)」、2007年、第14巻:p.1347−56;ソンマ・デルペロら(Somma−Delpero et al.)、「ザ・バイオケミカル・ジャーナル(Biochem J.)」、1995年、第312巻:p.519−25;ディンら(Dinh et al.)、「プロシーディングス・オブ・ザ・ナショナル・アカデミー・オブ・サイエンシズ・オブ・ザ・ユナイテッド・ステイツ・オブ・アメリカ(Proc.Natl.Acad.Sci.USA)、2002年、第99巻、p.10819−9;ディンら(Dinh et al.)、「モレキュラー・ファルマコロジー(Mol.Pharmacol.)」、2004年、第66巻:p.1260−4;ムッソリーら(Muccioli et al.)、「ザ・ジャーナル・オブ・ニューロサイエンス(J.Neurosci.)」、2007年、第27巻:p.2883−9;ザンら(Giang et al.)、「プロシーディングス・オブ・ザ・ナショナル・アカデミー・オブ・サイエンシズ・オブ・ザ・ユナイテッド・ステイツ・オブ・アメリカ(Proc.Natl.Acad.Sci.USA)、1997年、第94巻、p.2238−2242;ワンら(Wan et al.)、「ゲノミクス(Genomics)」、1998年、第54巻:p.408−414;パトリセリら(Patricelli et al.)、「バイオオーガニック&メディシナル・ケミストリー・レターズ(Bioorg.Med.Chem.Lett.)」、1998年、第8巻:p.613−618;及びゴーパルディ(Goparaju et al.)、「バイオヒミカ・エト・バイオフィジカ・アクタ(Biochim.Biophys.Acta)」、1999年、第1441巻:p.77−84、を参照。
【0061】
化合物の他の生物学的又は生化学的活性の改良(例えば、MAGLに対する亢進した結合親和性)は、当該技術分野で方法により試験することができる。一般的な概要については、例えば、サンブルックら(Sambrook et al.)著、「モレキュラー・クローニング:実験マニュアル(Molecular Cloning:A Laboratory Manual)」、コールドスプリング・ハーバー・プレス(Cold Spring Harbor Press)、ニューヨーク(N.Y.)、第3版(2000年);アウスベルら(Ausubel et al.)、「分子生物学カレントプロトコル(Current Protocols in Molecular Biology)」、ジョンウィリー&サンズ、インク(John Wiley&Sons、Inc.)、ニューヨーク(New York)(1999年);及びバーガー及びキンメル(Bergr and Kimmel)著、「酵素学における方法(Methods In Enzymology)」、サンディエゴ(San Dego)、アカデミックプレス、インク(Academic Prss,Inc)を参照。また本発明のスクリーニング法において用いることのできる追加の生化学的又は薬学的アッセイ法も当該技術分野で周知であり日常的に実施されている。例えば、MAGL選択的阻害剤のアナログ化合物の皮膚浸透特性は、例えば、レミントン著「薬学の科学と実践(The Science and Practice of Pharmacy)」、ジェンナロ(Gennaro)編、リッピンコット・ウィリアムズ&ウィルキンス(Lippincott Williamas&Wilkins)、(第20版、2003年)、等に記載された方法を使用して分析することができる。
【0062】
VI.MAGL標的化による癌治療及び腫瘍増殖阻害
本発明は、腫瘍細胞増殖阻害ための方法及び組成物を提供する。典型的には、これら本発明の治療応用には、モノアシルグリセロールリパーゼ(MAGL)と特異的に拮抗又は阻害する化合物が用いられる。腫瘍細胞の増殖は、当該腫瘍細胞をMAGL拮抗化合物の治療的に有効な量と接触させることによって阻害、抑制又は遅延される。幾つかの実施態様において、腫瘍細胞は、例えば癌発症の危険を有する又は危険にある被験者に存在する。これらの治療応用の幾つかの好ましい実施態様は、侵襲性の腫瘍又は腫瘍細胞の増殖を阻害することに向けられる。
【0063】
幾つかの関連する実施態様において、本発明は、腫瘍細胞の増殖を阻害することによって被験者体内の癌を治療するための方法を提供する。当該方法は、また被験者体内の腫瘍形成を防止するためにも有用である。典型的には、当該方法には、治療を必要とする被験者に本明細書で開示するMAGL拮抗化合物を含有する医薬組成物を投与することが含まれる。MAGL拮抗化合物は、単独で、又は被験者体内で相乗効果を提供するために、他の既知の抗癌剤と併用して使用することができる。
【0064】
本発明で用いるMAGL拮抗化合物には、MAGLの細胞レベルを下方制御する又は生物学的活性(例えば、リパーゼ活性)を阻害するいかなる薬剤も含まれる。適したMAGL拮抗化合物には、以下に記載するスクリーニングに従って同定することができる新規薬剤、例えば追加の小分子化合物又は抗体(例えば、アンタゴニスト抗体)等も含まれる。幾つかの実施態様において、腫瘍増殖を阻害する又は癌を治療するために用いられるMAGL拮抗化合物は、特異的にMAGL発現を阻害する又はその細胞レベルを下方制御する薬剤(例えば、阻害ポリヌクレオチド)である。例えば、MAGLを標的にする阻害ポリヌクレオチドには、低分子干渉RNA(siRNA)、マイクロRNA(miRNA)、低分子ヘアピン型RNA(shRNA)、アンチセンス核酸、又は相補DNAが含まれる。これらの核酸薬剤を標的遺伝子の発現を特異的に停止させるために使用することは、周知であり当該技術分野において。MAGLを特異的に標的とするこのような核酸薬剤は、当該技術分野で周知の方法を使用して調製することができる。例として、MAGL遺伝子を標的とするshRNA及びsiRNAは、下記の実施例で明らかなとおり、MAGL発現レベルを下方制御するために使用することができる。
【0065】
二本鎖RNAによる内在性遺伝子の機能及び発現との干渉は、例えば、ファイアーら(Fire et al.)、「ネイチャー(Nature)」、1998年、第391巻:p.806−811、に記載された線虫;例えば、ケンネーデルら(Kennerdell et al.)、「セル(Cell)」、1998年、第95巻:p.1017−1026、に記載されたショウジョウバエ;及び例えば、ウィアニら(Wianni et al.)、「ネイチャー・セル・バイオロジー(Nat.CellBiol.)」、2000年、第2巻:p.70−75、に記載されたマウス胎仔等の種々の生物で示されている。このような二本鎖RNAは、鋳型の両方向から読み取る一本鎖RNAの生体外転写並びにセンス及びアンチセンスRNA鎖の生体外アニーリングによって合成することができる。二本鎖RNAは、またMAGL遺伝子が逆向き反復配列によって引き離された反対方向にクローンされたcDNAベクター構築物から合成することもできる。細胞へ遺伝子導入された後、当該RNAは転写され相補鎖と再アニールされる。MAGL遺伝子を標的とする二本鎖RNAは、適当な構築物の形質移入によって細胞(例えば、腫瘍細胞)に導入することができる。例として、MAGL発現を下方制御する特異的なsiRNA及びshRNAのアンチセンス及びセンス鎖の配列を本明細書で開示する。MAGLを標的とする他の型の阻害ポリヌクレオチドと同様に追加のsiRNA又はshRNA分子は、当該技術分野で周知に標準的技術に従って容易に作成することができる。
【0066】
幾つかの他の実施態様において、本発明の治療応用は、MAGLの生物学的活性を阻害するMAGL拮抗化合物を用いる。これらには、MAGLの酵素活性と拮抗する本明細書に開示するMAGL選択的阻害剤が含まれる。適したMAGL拮抗化合物には、またMAGLポリペプチドに特異的に結合しそのリパーゼ活性と拮抗するアンタゴニスト抗体も含まれる。この点については、これらの中でモノクローナル抗体薬剤が最も好ましい。抗MAGLアンタゴニスト抗体は、例えば、「モノクローナル抗体−生産、工学及び臨床応用(Monoclonal Antibodies‐‐Production Engineerign And Clinical Applications)」、リッターら(Ritter et al.)編、ケンブリッジ大学プレス(Cambridge University Press)、ケンブリッジ(Cambridge)、英国(UK)、1995年;及びハーロー及びレイン(Harlow and Lane)著、「抗体、実験マニュアル(Antibodies、A Laboratory Mannual)」、コールドスプリング・ハーバー・プレス(Cold Spring Harbor Press)、第3版、2000年、等の当該技術分野で周知であり日常的に実施されている方法を使用して作成することができる。癌治療用の放射標識モノクローナル抗体は特に周知であり例えば、「放射標識抗体による癌治療(Cancer Therapy With Radiolabelled Antibodies)」、DMゴールデンベルク(D.M.Goldenberg)編、CRCプレス(CRC Press)、ボカラトン(Boca Raton)、フロリダ(Fla)、1995年、に記載されている。
【0067】
MAGL発現又はその酵素活性を阻害又は下方制御する化合物は、他の治療法と併用して使用することができる。例えば、手術及び放射線療法受けている被験者には、本発明の医薬組成物を投与することもできる。加えて、また化学療法、ホルモン療養及び凍結療法は、癌を患う被験者を治療するために、本発明の治療応用と併用することもできる。MAGL拮抗化合物は、これらの疾患の治療又は防止するための他の治療化合物の投与と共に、腫瘍増殖を防止するため又は癌を治療するために被験者に使用することもできる。MAGL拮抗化合物を他の抗癌剤と共に投与する場合、両者はどちらの順番でも又は同時に投与することができる。これらの治療化合物は、化学療法剤、焼灼術又は他の治療用ホルモン、抗腫瘍薬、癌に有用なモノクローナル抗体及び血管新生阻害剤であっても良い。
【0068】
当該技術分野で知られた多数の抗癌剤があり、例えば、「癌治療学:試験薬及び臨床薬(Cancer Therapeutics:Experimental and Clinical Agents)」、タイヒャー(Teicher)(編)、ヒューマナ・プレス(Humana Press)(第1版、1997年);及び「グッドマン及びギルマンズの薬物療法学の薬理学的基礎(Goodman and Gilman‘s The Pharmacological Basis of Therapeutics)」、ハルドマンら(Hardman et al.)(編)、マッグローヒル・プロフェッショナル(McGraw−Hill Professional)(第10版、2001年)、に記載されている通りである。適した抗癌剤の例には、5−フルオロウラシル、硫酸ビンブラスチン、リン酸エストラムスチン、スラミン及びストロンチウム−89が含まれる。適した化学療法薬の例には、アスパラギナーゼ、硫酸ブレオマイシン、シスプラチン、シタラビン、リン酸フルダラビン、マイトマイシン、及びストレプトゾシンが含まれる。本発明と併用して使用されるかもしれないホルモンは、ジエチルスチルベストロール(DES)、ロイプロリド、フルタミド、酢酸シプロテロン、ケトコナゾール及びアミノグルテチミドである。
【0069】
本発明の方法による治療に適した被験者は、種々の型の癌を患っている被験者、又は癌発症の危険にある又はを有する被験者である。本発明の方法は、多数の腫瘍細胞の増殖を阻害するために用いることができる。治療を施すことのできる腫瘍又は癌の例には、肺、皮膚、乳房、脳、胃腸、尿生殖路(例えば、腎臓、膀胱及び尿道、前立腺、睾丸)、血管、神経系、骨及び肝臓由来の腫瘍が含まれる。それらは、充実性腫瘍、白血病及び転移性腫瘍を包含する。本発明に適した充実性腫瘍には、例えば、肉腫、黒色腫、癌腫、又は他の充実性腫瘍癌が含まれる。
【0070】
肉腫は、胚性結合組織のような物質から構成される腫瘍を包含し、一般的に原線維又は均質な物質中に包埋したぎっしりと詰まった細胞から構成される。肉腫には、限定されるものではないが、軟骨肉腫、線維肉腫、リンパ肉腫、黒色肉腫、粘液肉腫、骨肉腫、アベメシー肉腫、脂肪性肉腫、脂肪肉腫、胞巣状軟部肉腫、エナメル上皮肉腫、ブドウ状肉腫、緑色腫、絨毛癌、胎児性肉腫、ウィルム肉腫、子宮内膜肉腫、間質性肉腫、ユーイング肉腫、筋膜肉腫、線維芽肉腫、巨細胞肉腫、顆粒球性肉腫、ホジキン肉腫、特発性多発性色素性出血性肉腫、B細胞免疫芽球肉腫、リンパ腫、T細胞免疫芽球肉腫、イエンセン肉腫、カポジ肉腫、クッパー星細胞肉腫、血管肉腫、白血肉腫、悪性間葉肉腫、傍骨性骨肉腫、細網肉腫、ラウス肉腫、漿液嚢胞性肉腫、滑膜肉腫、及び血管拡張型肉腫が含まれる。
【0071】
黒色腫とは、皮膚及び他の器官のメラニン細胞系から生じる腫瘍をいう。黒色腫には、例えば、末端黒子型黒色腫、メラニン欠乏性黒色腫、良性若年性黒色腫、クラウドマン黒色腫、S91黒色腫、ハーディング・パッセー黒色腫、若年性黒色腫、悪性黒子型黒色腫、悪性黒色腫、小結節性黒色腫、爪下黒子性黒色腫、及び表在拡大型黒色腫が含まれる。
【0072】
癌腫とは、周囲の組織に浸潤し転移を起こす、上皮細胞から構成される悪性の新生物をいう。代表的な癌腫には、例えば、上皮癌、結腸直腸癌、胃癌、口腔癌、膵癌、卵巣癌、又は腎細胞癌がさらに含まれる。代表的な癌腫には、例えば、腺房癌、小葉癌、腺嚢癌腫、腺様嚢胞癌、腺癌、副腎皮質癌、肺胞癌、肺胞上皮癌、基底細胞癌(carcinoma basocellulare)、基底細胞癌(basaloid carcinoma)、基底有棘細胞癌、気管支肺胞上皮癌、細気管支癌、気管支原性肺癌、セレブリフォルム癌、胆管細胞癌、絨毛膜癌腫、粘液性癌腫、面疱癌、子宮体癌、篩状癌、鎧状癌、皮膚癌、円柱癌、円柱細胞癌、腺管癌、デュラム癌腫、胎児性癌、髄癌、類表皮癌腫、腺様上皮癌、外向方発育癌、潰瘍癌、癌腫血管腫、膠様癌、粘液性癌腫、巨細胞癌腫、巨細胞癌、腺癌、顆粒膜細胞腫、毛母癌、血性癌腫、肝細胞癌、ハースル細胞癌、卵黄嚢腫瘍、副腎癌、小児胎児性癌、上皮内癌(carcinoma in situ)、表皮内癌、上皮内癌(intraepithelial carcinoma)、
Krompecher‘s癌、クルチツキー細胞癌、大細胞癌、レンズ状癌(lenticular carcinoma)、レンズ状癌(carcinoma lenticulare)、脂肪腫、リンパ上皮性癌、髄様癌(carcinoma medullare)、髄様癌(medllary carinoma)、黒色癌、軟部癌腫、粘液性癌(mucinous carcinoma)、粘液性癌(carcinoma muciparum)、粘液細胞性癌、粘表皮癌、粘液癌(carinoba mucosum)、粘液性癌(mucous carcinoma)、粘液腫状癌、鼻咽頭癌腫、燕麦細胞癌、類骨癌(carcinoma ossificans)、類骨癌腫(osteoid carcinoma)、乳頭癌、門脈周囲癌、前浸潤癌、有棘細胞癌、のリ状癌、腎細胞癌、予備細胞癌、肉腫状癌、シュナイダー癌腫、硬性癌、陰嚢癌、印環細胞癌、単純癌、小細胞癌、ソラノイド癌腫、回転楕円面細胞癌腫、紡錘細胞癌、海綿様癌、扁平上皮癌(squamous carcinoma)、扁平上皮細胞癌(squamous cell carcinoma)、ストリング癌、毛血管拡張性癌(cacinoma telangiectaticum)、毛細血管拡張症様癌(carcinoma telangiectodes)、移行上皮癌、結節癌(arcinoma tuberosum)、結節性癌(tuberous carcinoma)、いぼ状癌、及び絨毛癌が含まれる。
【0073】
白血病は、造血器官の進行性の悪性疾患を包含し、血管及び骨髄中の白血球及びその前駆体の歪んだ増殖及び生育によって一般的に特徴づけられる。白血病は、臨床的には一般的に(1)疾患の継続期間及び特性‐‐急性又は慢性;(2)関与する細胞の型;骨髄系(骨髄性)、リンパ系(リンパ向性の)、又は単球系;並びに(3)血液中の異常細胞数の増加又は非増加‐白血病性又は無白血病性(亜白血病性)、に基づいて分類される。白血病には、例えば、急性非リンパ球性白血病、慢性リンパ球性白血病、急性顆粒球白血病、慢性顆粒球白血病、急性前骨髄球白血病、急性T細胞白血病、非白血病性白血病、白血球血症白血病、好塩基球性白血病、芽球細胞性白血病、ウシ白血病、慢性骨髄性白血病、皮膚白血病、胎児性白血病、好酸球性白血病、グロス白血病、有毛状細胞性白血病、肝細胞性白血病(hemoblastic leukemia)、血球芽細胞性白血病、組織球白血病、肝細胞白血病(stem cell leukemia)、急性単球性白血病、白血球減少性白血病、リンパ性白血病(lymphatic leukemia)、リンパ芽球性白血病、リンパ球性白血病、リンパ性白血病(lymphogenous leukemia)、リンパ性白血病(lymphoid leuemia)、リンパ肉腫細胞性白血病、肥満細胞白血病、巨核球性白血病、小骨髄芽球性白血病、単球性白血病、骨髄芽球性白血病、骨髄性白血病、骨髄顆粒球性白血病、骨髄単球性白血病、ネーゲリ白血病、形質細胞白血病(plasma cell leukemia)、形質細胞性白血病(plasmacytic leukemia)、前骨髄球性白血病、リーダー細胞白血病、シリング白血病、肝細胞白血病(stem cell leukemia)、亜白血性白血病、及び未分化細胞白血病が含まれる。
【0074】
本発明の方法により治療することのできる他の癌又は腫瘍には、例えば、ホジキン病、非ホジキンリンパ腫、多発性骨髄腫、神経芽腫、乳癌、卵巣癌、肺癌、横紋筋肉腫、原発性血小板症、原発性マクログロブリン血症、小細胞肺腫瘍、原発性脳腫瘍、胃癌、結腸癌、悪性膵島細胞腺腫、悪性類癌腫、膀胱癌、前癌皮膚障害、精巣癌、リンパ腫、甲状腺癌、神経芽腫、食道癌、泌尿生殖器癌、悪性高カルシウム血症、子宮頚部癌、子宮内膜癌、副腎皮質癌、及び前立腺癌が含まれる。
【0075】
VII.医薬組成物及び投与
本発明は、内在性カンナビノイドシグナル伝達によって媒介される又は関連する症状又は障害を治療するための薬剤の製造においてMAGLアンタゴニスト化合物(例えば、MAGL選択的阻害剤)の使用を提供する。幾つかの関連する応用において、MAGL拮抗化合物は、腫瘍細胞増殖阻害及び癌治療のために使用される。これらの治療応用において、治療を必要とする被験者には、MAGLアンタゴニスト化合物を単独で投与することができる。しかしながら、MAGLアンタゴニスト化合物又はその薬学的に許容可能な塩を含む医薬組成物の投与の方がより好ましい。医薬組成物に用いることのできるMAGLアンタゴニスト化合物の例には、小分子有機化合物であるJZL184、JZL175、WWL152及びWWL162と同様に下記の実施例に記載する阻害ポリヌクレオチドが含まれる。本発明のスクリーニング方法に従って同定するこのできる新規のMAGL阻害剤もまた使用することができる。本発明は、例えば、キット等の医薬品の組み合せも提供する。このような医薬品の組み合せは、本明細書で開示するMAGLアンタゴニスト化合物である活性薬剤と同様に、どのような形態又は組成物であっても、少なくとも一つを併用組成物と、薬剤の投与説明書を含むことができる。
【0076】
MAGL選択的阻害剤を含む医薬組成物は種々の形態で調製することができる。適した固形又は液体製剤形態は、例えば、顆粒、粉末、錠剤、被覆錠剤、(マイクロ)カプセル、座薬、シロップ剤、乳剤、懸濁剤、クリーム剤、エアロゾル剤、滴剤又はアンプル形態の注射液であり、並びに活性化合物の遅延性放出に関する製剤である。それらは、当該技術分野で周知の標準的なプロトコル、例えば、レミントン(Remington)著「薬学の科学と実践(The Science and Practice of Pharmacy)」、ジェンナロ(Gennaro)編、リッピンコット・ウィリアムズ&ウィルキンス(Lippincott Williams & Wilkins)(第20版、2003年)、に従って調製することができる。医薬組成物は、典型的には腫瘍増殖を阻害するために、又は2−AGシグナル伝達に関連する又は媒介される障害又は症状の症候を減じる又は回復するために十分なMAGLアンタゴニスト化合物の有効な量を含んでいる。MAGLアンタゴニスト化合物に加えて、医薬組成物は、組成物を高める若しくは安定させる、又は組成物の調製を促進する一定の薬学的に許容可能な担体を含むこともできる。例えば、MAGLアンタゴニスト化合物は、安定性又は薬理学的特性を高めるために、その投与前に卵白アルブミン又は血清アルブミン等の担体タンパク質と複合体を形成することができる。医薬組成物の種々の形態は、また崩壊剤、結合剤、被覆剤、膨潤剤、滑沢剤、着香料、甘味剤及び精製水等の通常当該技術分野で日常的に使用される不活性な希釈剤を含むエリキシル剤等の賦形剤及び添加物及び/又は補助剤を含むこともできる。
【0077】
薬学的に許容可能な担体は、部分的には投与される特定の成分と同様に組成物の特定の投与方法によって決定される。それらは、他の成分と適合しかつ被験者に有害でないという意味において、薬学的及び生理学的の両方においても許容可能でなければならない。担体は、例えば経口、舌下、直腸、経鼻、静脈内、非経口等の投与に望ましい製剤形態に依存して様々な形態をとってもよい。例えば、非水系の溶媒の例は、プロピレングリコール、ポリエチレングリコール、オリーブオイル等の植物油、オレイン酸エチル等の注射可能な有機エステルである。密封包帯用の担体は、皮膚透過性を増すため及び吸収を高めるために使用することができる。経口投与用の液体剤形は、一般的に液体剤形を含有するリポソーム溶液を含んでいてもよい。
【0078】
MAGL阻害化合物を含有する医薬組成物は、治療的に有効な量又は服用量で局所的に又は全身的に投与することができる。それらは、非経口的に、腸管的に、注射、迅速点滴、鼻咽頭吸収、経皮吸収によって、直腸的に及び経口的に投与することができる。治療的に有効な量とは、被験者の治療すべき障害又は症状の症候(例えば、侵害受容性疼痛)を減少又は阻害するために十分な量を意味する。このような有効な量は、被験者の疼痛に対する常態の感受性、その身長、体重、年齢、及び健康状態、疼痛の起源、MAGLアンタゴニスト化合物の投与方法、投与された特定の阻害剤、及び他の要因に依存して被験者ごとに異なるであろう。結果として、特定の状況下において特定の被験者に対する有効な量を経験的に決めるのが賢明である。
【0079】
のMAGLアンタゴニスト化合物について、当業者であれば薬学的な方法を使用して、容易に腫瘍増殖を阻害し及び癌を治療する当該化合物の有効量、又はカンナビノイドシグナル伝達と関連する症状を治療する化合物の有効量を同定することができる。典型的には、生体外で使用される投与量は、医薬組成物の生体内原位置での投与のための有用な量に関する役立つガイダンスを提供することができ、並びに動物モデルはの被験者における特定の障害の治療のための有効な投与量を決定するために使用することができる。よりしばしば、適した薬用量は、通常のに関する、最大許容用量及び安全投与量を決めるために、哺乳動物種に関する臨床研究によって決定することができる。より高い投与量が要求される特定の状況を除いて、MAGLアンタゴニスト化合物の好ましい投与量は普通には、一日当たり約0.001から約1000mg、より普通には約0.01から約500mgの範囲にある。一般に、投与されるMAGLアンタゴニスト化合物の分量は、被験者の症状を有効に及び確実に防止又は最小化する最少の投与量である。それ故に、上記の投与量の範囲は、本明細書での教示のための一般的なガイダンス及び補助を提供することを意図するものであり、本発明の範囲を限定することを意図するものではない。本発明の医薬組成物の調製及び投与に関する追加のガイダンスもまた当該技術分野で記載されている。例えば、「グッドマン及びギルマンズの薬物療法学の薬理学的基礎(Goodman & Gilman‘s The Pharmacological Bases of Therapeutics)」、ハルドマン(Hardman)ら編、マッグローヒル・プロフェッショナル(McGraw−Hill Professional(第10版、2001年);レミントン(Rmington)著「薬学の科学と実践(The Science and Practice of Pharmacy)」、ジェンナロ(Gennaro)編、リッピンコット・ウィリアムズ&ウィルキンス(Lippincott Williams&Wilkins)(第20版、2003年);及び「製剤投与形態及び薬剤デリバリーシステム(Pharmaceutical Dosage Forms and Drug Delivery Systems)」、アンセルら(Ansel et al.)編、リッピンコット・ウィリアムズ&ウィルキンス(Lippincott Williams&Wilkins)(第7版、1999年)を参照。
【0080】
実施例
下記の実施例は、例示するために提供するものであり、本発明を限定するものではない。
【0081】
実施例1.MAGL選択的阻害剤の開発
セリンヒドロラーゼに対する選択的阻害剤の探究には、この酵素クラスに特別な多様な特徴から利益を得ることができるという可能性がある。第一に、セリンヒドロラーゼは、他の酵素クラスとは交差反応性をほとんど又は全く示さない特異的な化学基による共有結合性の不活性化に感受性である。これらの反応性のケモタイプの間で主要なのはカルバメートであるが、それは、これらの酵素の保存セリン求核剤との共有結合性反応(カルバミル化)の後の、その穏やかな求電子性及び加水分解安定性のために、選択的で不可逆的なセリンヒドロラーゼ阻害剤の設計のために特権的な骨格構造として同定された。第二に、セリンヒドロラーゼの機能状態は、活性に基づいたタンパク質プロファイリング(ABPP)法を使用して天然の生物システムにおいて集合的にプロファイルすることができる。セリンヒドロラーゼのABPPには、この大きく多様な種類の酵素のための一般的な活性に基づくプローブとして役立つ、レポータータグ付フルオロホスホネート(FP)を使用する(リォウら(Liu et al.)、「プロシーディングス・オブ・ザ・ナショナル・アカデミー・オブ・サイエンシズ・オブ・ザ・ユナイテッド・ステイツ・オブ・アメリカ(Proc.Natl.Acad.Sci.U.S.A.)」、1999年、第96巻:p.14694−9)。拮抗的な方法で実施する場合、ABPPは、小分子の酵素阻害剤の潜在能力と選択性を複合プロテオーム中で直接評価するための強力な選別法として役立つことができる。選別法は、当該技術分野で、例えば、リォウら(Liu et al.)、「プロシーディングス・オブ・ザ・ナショナル・アカデミー・オブ・サイエンシズ・オブ・ザ・ユナイテッド・ステイツ・オブ・アメリカ(Proc.Natl.Acad.Sci.U.S.A.)」、1999年、第96巻:p.1494−9;リーら(Li et al.)、「ジャーナル・オブ・ジ・アメリカン・ケミカル・ソサイアティ(J.Am.Chem.Soc.)」、2007年、第129巻:p.9594−5;ロイングら(Leung et al.)、「ネイチャー・バイオテクノロジー(Nat.Biotech.)」、2003年、第21巻:p.687−91;及びグリーンバウムら(Greenbaum et al.)、「モレキュラー&セルラー・プロテオミクス(Mol.Cell.Proteomics)」、2002年、第1巻:p.60−68、等に既述の通り実施することができる。
【0082】
構造的に多様なカルバメートのライブラリーについて拮抗的ABPP選別を実施している間に、発明者らは、FAAH、MAGL及び、ABHD6(図1B)を含む脳セリンヒドロラーゼの特定の亜集合を阻害する化合物WWL98(図6)を同定した。発明者らは、次にMAGLの潜在能力及び選択性を改良するためにWWL98を修飾することに注力した。WWL152及びWWL162(図1A)で代表される通り、ピペラジンのスペーサ及び末端ビスアリールモチーフの取り込は、MAGL及びABHD6に関する潜在能力を維持しつつ、一方FAAHに関する活性を顕著に減少すること(図1B)が発見された。ピペラジンをピペリジニルメタノール骨格構造で置換すると、FAAH及びABHD6の両方にMAGLに対する選択性を示す化合物である、JZL175(図1B)を産出した。最後に、追加の選択性の増強は、ピペリジニルメタノール骨格構造上へのビス(メチレン−3,4−ジオキシフェニル)の包接によって行いJZL184(図1A及びB)を与えた。
【0083】
実施例2.脳プロテオーム中のMAGL選択的阻害剤活性
マウス脳膜プロテオームの50nMの低濃度のJZL184による30分間のプレインキュベーションの後に、拮抗的ABPPによってMAGL活性のほぼ完全な遮断が認められた(図2A)。対照的に、他の酵素(FAAH、ABHD6)の阻害は、より高濃度のJZL184(10μM)まで認められなかった。基質アッセイにより、JZL184が脳膜中において2−AG及びオレイン酸アミド(FAAH基質)の加水分解の遮断に対しそれぞれ8nM及び4μMのIC50値を示した(図2B)通りの選択性度を確認した。COS7細胞で発現した場合に(図2C)、組換えMAGL及びFAAHに関して同等の阻害効果が認められた。興味深いことに、脳膜は、試験したJZL184の最高濃度においてさえも2−AG加水分解活性の〜15%を維持したが、おそらくJZL184に非感受性の他の2−AG加水分解酵素の寄与を反映している。JZL184は、MAGLの時間依存的な阻害(図7)を示したが、それは共有結合型の不活性化機序を暗示している。そこで発明者らは、またMAGL及びFAAHに対するJZL184の相対活性をkobs/[I]値を測定することによって評価したが、当該値よりMAGLの阻害にたいする選択性が>300倍であることが(それぞれ4400及び13M−1s−1)確認された。最後に発明者らは、内在性カンナビノイド系の他の成分に関するJZL184の活性を試験した。当該化合物は、CB1受容体と相互作用せず、又は2−AG生合成酵素であるジアシルグリセロールリパーゼ−βを阻害しなかった(図8)。まとめると、これらのデータは、JZL184がマウス脳プロテオーム中でMAGLを強力かつ選択的に阻害することを示している。
【0084】
実施例3.選択的阻害活性剤の生体内活性
生体内でのJZL184のMAGL遮断活性を評価するために、C57B1/6雄マウスにJZL184を投与し(4−40mg/kg、腹腔内)、分析のために4時間後に屠殺した。脳膜プロテオームの拮抗的ABPPは、試験したJZL184の最少投与量(4mg/kg)における、FAAHを含む他の脳セリンヒドロラーゼに最少の影響をともなった、MAGLの75%阻害を明らかにした(図3A)。また、これらのデータは、MAGL及びFAAHに対しても基質アッセイによって確認した(図3B)。脳2−AG加水分解の残存活性は、JZL184の4から16mg/kgへの投与量の増加によって対照値の25%から15%にさらに減少することができたが(図3B)、そのことは拮抗的ABPPによって定量された通りMAGL活性のほぼ完全な遮断と関連していた(図3A)。FAAHは、また用量依存的様式でも阻害されたが、しかし16mg/kgのJZL184においても、拮抗的ABPP(図3A)又は基質アッセイ(図3B)によって定量した通り、FAAH活性の実質的な部分(〜35%)は無傷で残存していた。
【0085】
発明者らのゲルに基づくABPP分析は既に生体内でのMAGLに対する高選択性を示唆したが、本法の分解能の制限により、JZL184処理動物中の脳セリンヒドロラーゼの機能状態の完全な評価が妨げられていた。そこで発明者らは、ゲルに基づくABPPと比較して高い分解能及び感受性を示す、ABPP−MudPITと命名した、高性能液体クロマトグラフィー−質量分析(LC−MS)プラットフォーム(ジェサニーら(Jessani et al.)、「ネイチャー・メソッズ(Nat.Methods)」、2005年、第2巻:p.691−7、を参照)を使用して脳プロテオームを試験した。ABPP−MudPITは、トランスポーター及び生合成酵素を含む、内在性カンナビノイド系の他の成分の阻害剤に対する予期しない標的を同定するために以前使用した。手短に言えば、JZL184又は溶媒で処理したマウス由来の脳膜プロテオームを、ビオチン標識したFPプローブであるFP−ビオチンと共に拮抗的ABPPにかけた。次にFPビオチン標識タンパク質をアビジンで増強し、ビーズ上でトリプシンにより消化し、多次元LC−MSで分析し、探索アルゴリズムSEQUESTを使用して同定した。ABPP−MudPITは、JZL184(16mg/kg、4時間)がほぼ完全なMAGL活性の遮断及びFAAHの部分的な阻害を引き起こしたことを確認した(図3C)。印象的なことに、ABPP−MudPITによってプロファイルされた他の〜40の脳セリンヒドロラーゼのいずれもJZL184によって著しい影響を受けなかった(図3C及び表1)。これらのデータは、高投与量で(16mg/kg)長時間(4時間)投与された場合でさえ、JZL184が脳内でMAGLに対する優れた選択性を維持することを証明している。
【表1】
生体内でのJZL184の選択性に関するプロファイルを確立した後、発明者らは、次にMAGL及びFAAHに対する内在性の基質及び産物の候補の脳レベルを測定した。試験したJZL184の最低投与量(4mg/kg)においてさえ、2−AGレベルは処理4時間後に5倍に増加し、阻害剤のより高投与量においてはベースラインより8−10倍に増加することができた(図3d)。予想通り、脳2−AGの増加には、アラキドン酸レベルの著しい減少が伴った(図3E)。モノ−パルミトイルグリセロール(PG)及びモノ−オレオイルグリセロール等の他のMAG、及びその相応する遊離脂肪酸(それぞれパルミチン酸及びオレイン酸)は、JZL184処理動物由来の脳で著しくは変化しなかった(図9)。重要なことには、また脳アナンダミドレベルも試験された全ての投与量のJZL184によって影響を受けなかった(図3F)。N−パルミトイルエタノールアミン(PEA)、及びN−オレオイルエタノールアミン(OEA)等の他のN−アシルエタノールアミン(NAE)は、試験された最高投与量のJZL184(40mg/kg)を除き変化しなかったが、そこでは、これらの脂質のわずかな増加(〜2倍)が認められた。NAEレベルへの影響の欠如は、生体内のNAEレベルを高めるためにはFAAHの部分的な破壊では不十分であることを明らかにした公表された研究と一致している。JZL184処理マウスの脂質プロファイルは、脳FAAH活性を完全に阻害し、この組織中の2−AG又はアラキドン酸レベルを変えずにアナンダミド及び他のNAEレベルの著しい増加を引き起こす、選択的FAAH阻害剤URB597によって誘発されたプロファイルとは著しく違っていた。同様なデータがまたJZL184の経口投与の後でも得られたが(図10)、このことは生体内のMAGLの選択的遮断を実施するために本化合物は、多様な経路によって投与することができることを示している。
【0086】
実施例4.JZL184処理マウスにおける迅速持続的MAGL阻害
生体内でのJZL184阻害の経時変化を測定するために、マウスにJZL184を投与し(16mg/kg、腹腔内)、分析のために0.5、1、2、4、8、及び24時間に屠殺した。脳膜プロテオームのABPP(図4A)と同様に2−AG及びオレイン酸アミド加水分解アッセイ(図4B)は、MAGL阻害が、処理後30分以内に最大速度を達成するほど迅速であり、2−AG加水分解活性の>80%阻害を少なくとも24時間持続するほど長期持続性であることを示した(図4A)。対照的に、FAAH遮断は、よりゆっくりと起こり、試験のどの時点においても決して70%を超えなかった(図4B)。MAGLの迅速阻害に付随してJZL184処理は、脳で2−AG蓄積を即座に引き起こした。30分間のうちに脳2−AGレベルは既に7倍に増加し、少なくとも8時間は対照レベルの7−9倍が残存した(図4C)。興味深いことに、MAGLの活性の>80%がこの時点で阻害されたままであるのに、2−AGレベルは24時間のうちにほぼ対照レベルに回復した。2−AGの増加は、アラキドン酸の急速かつ持続性の減少と一致した(図4D)。アナンアミドを含むNAEと同様に他のMAG及び遊離脂肪酸も、JZL184処理マウスの分析時間経過を通して変化しなかった(図4E及び図11)。
【0087】
実施例5.MAGL選択的阻害剤によって誘発されるマウスの行動的影響
JZL184によって引き起こされる劇的かつ持続性の脳2−AGレベルの増加は、本阻害剤が内在性カンナビノイド媒介行動的影響を誘発するかもしれないことを示唆した。直接的なCB1アゴニストは、げっ歯類において、痛覚消失症、運動性低下症、低体温症、及び強硬症(あわせてカンナビノイド活性の四つ組み試験と呼ばれる)を含む多様な行動的影響を促進することが知られている。興味深いことに、FAAH阻害剤は、四つ組み試験において、痛覚消失症を引き起こすが他のカンナビノイド行動的表現型を引きおこさず、大部分は不活性である(リヒトマンら(Lichtman et al.)、「ザ・ジャーナル・オブ・ファルマコロジー・アンド・エクスペリメンタル・セラピューティクス(J.Pharmacol.Exp.Ther.)」、2004年、第311巻:p.441−8)。JZL184(16mg/kg、2時間)は、また急性熱痛覚の尾浸漬試験(図5A)及び内臓痛の酢酸苦悶試験(図5B)を含む、多数の疼痛アッセイにおいて著しい鎮痛作用を示すことも見出された。両方の場合において、JZL184の効果は、CB1アンタゴニストであるリモナバンでの前処理(3mg/kg)によって遮断された(図5A及びB)。FAAH阻害剤とは著しく対照的にJZL184は、また低体温症(図5C)及び運動性低下症(図5D)をも顕著に促進した。JZL184処理は強硬症を誘発しなかった一方で、バー試験で評価した場合に、処理マウス14匹中7匹が反射亢進又は「ポップコーン」行動(ペイテルら(Patelet al.)、「ジャーナル・オブ・ファルマコロジー・アンド・エクスペリメンタル・セラピューティックス(J.Pharmacol.Exp.Ther.)」、2001年、第297巻:p.629−37)を示した(図5E)。低体温及び反射亢進の効果は、リモナバンによって完全に遮断されたが、そのことは、それらがCB1受容体によって媒介されることを示している。発明者らは、JZL184の運動性低下効果に対するCB1受容体の寄与を明確に決定することができなかったが、というのはリモナバンが独自に運動性の顕著な減少を誘発することを見出したからである(図5D)。しかしながら、JZL184誘発性不動症の部分的遮断がリモナバンにより認められ(図5D)、そのことは本表現型に対するCB1受容体の寄与を示唆している。
【0088】
まとめると、これらのデータは、JZL184が直接的CB1アゴニストの薬理学的なプロファイルに定性的に似た広い一連のカンナビノイドの行動的影響をげっ歯類において誘発することを示している。加えて、これらの表現型の幾つか(例えば、低体温症及び反射亢進)は、生体内でのMAGL阻害の大きさを用量設定することによって、より有益な効果(例えば、痛覚消失)から薬理学的に脱共役することができた。実際に、より低い投与量はMAGL活性のより低い遮断に帰着したけれども、発明者らは、本研究で試験したJZL184の全投与量範囲(4−40mg/kg)にわたって顕著な2−AGの増加を認めた。これらのデータは、このようにMAGLの部分的な阻害であっても生体内の2−AG媒介内在性カンナビノイドシグナル伝達の増大にとって十分であるかもしれないことを示している。
【0089】
実施例6.MAGL阻害剤の合成
合成法一般特に断りがない限り、全ての試薬は、より市販品を購入しそれ以上精製せずに使用した。脱水溶媒は、市販の予備脱水した酸素不含の製剤を活性化アルミナカラムに通すことによって得た。他に断らない限り、全ての反応は、乾燥器で乾燥したガラス器具を使用して窒素雰囲気下で実施した。フラッシュクロマトグラフィーは230−400メッシュのシリカゲルを使用して実施した。NMRスペクトルは、CDCl3中バリアン・イノバ-400(Varian Inova−400)又はブルカーDMX−600(Bruker DMX−600)スペクトルメータで記録し、トリメチルシラン(TMS)又は残存溶媒のピークに対しリファレンス設定した。化学シフトはTMSに対し相対的にppmで報告し、J値はHzで報告する。高分解能質量分析実験は、スクリップス研究所質量分析コア部(The Scrips Research Institute Mass Spectrometry Core)において、エレクトロンスプレーイオン化飛行時間(ESI−TOF)を使用してアジレント(Agilent)質量分析計で実施した。
【0090】
JZL184の合成撹拌する4−ブロモ−1,2−メチレンジオキシベンゼン(2.01g、10mmol)の無水THF溶液(30ml)にn−BuLi(n−ブチルリチウム)(3.8ml、トルエン中2.6M、9.9mmol)を−78℃で滴下し添加した。同温度で1.5時間撹拌した後、エチルN−Cbz−イソニペコチン酸(1.02g、3.5mmol)の無水THF溶液(10ml)を滴下して添加し、移行を定量化するために追加分のTHF(3ml)を使用した。試薬の添加が終了した後、冷却浴槽を取り除いたところ、懸濁液は徐々に透明溶液に変わった。3時間後、反応を水で停止しEtOAc(酢酸エチル)で希釈した。層は分離し、有機層を水で2回、食塩水で1回洗浄し、Na2SO4で脱水し、減圧下で濃縮した。粗製油をフラッシュクロマトグラフィー(ヘキサン:酢酸エチル=6:1その後4:1)で精製して白色固形物として中間体アルコール(0.81g、収率47%)を得た:1H NMR(CDCl3、400 MHz)δ 1.29(qd、J=4、12 Hz、2H)、1.51(bs、2H)、2.38(tt、J=2.8、12 Hz、1H)、2.58(s、1H)、2.75(t、J=12 Hz、2H)、4.19(bs、2H)、5.03(d、J=5 Hz、2H)、5.85(s、4H)、6.70(d、J=8 Hz、2H)、6.88−6.92(m、4H)、7.24−7.33(m、5H);13C NMR(CDCl3、100 MHz)δ 26.6、44.4、44.6、67.1、79.4、101.1、106.9、107.9、118.9、127.9、128.0、128.5、136.9、140.1、146.2、147.7、155.2;C28H27NNaO7[M+Na]+に対する計算上のHRMS512.1680は512.1670に見出された。本中間体の一部分(0.72g、1.5mmol)をCH2Cl2/EtOH(1:1 v/v、40ml)に溶解し、Pd/C(パラジウム炭素)(10%、0.20g)を一度に添加した。反応液はH2下(1気圧)で一晩撹拌した。出発原料が完全に消費された後(12−24時間)、反応液をろ過し減圧下で濃縮した。生じた粗製油をCH2Cl2(30mL)に溶解し、それにEt3N(トリエチルアミン)(2ml、15mmol)及びクロロギ酸4−ニトロフェニル(460mg、2.3mmol)を順次添加した。反応液を一晩撹拌した。翌朝、反応を水で停止しEtOAcで希釈した。層は分離し、有機層を2N NaOHで2回、食塩水で1回洗浄し、Na2SO4で脱水し、減圧下で濃縮した。粗製油をフラッシュクロマトグラフィー(ヘキサン:酢酸エチル=6:1その後4:1)で精製して、減圧下で放置すると黄色固形物に変わる、淡黄色の油としてJZL184(550mg、2段階収率71%)を得た。
【0091】
JZL175の合成撹拌するエチルN−Cbz−イソニペコチン酸(0.84g、2.9mmol)の無水THF溶液(10ml)にブロモ(4−メトキシフェニル)マグネシウム(15ml、THF中0.5M)を室温で滴下し添加した。試薬の添加終了後に、反応液を加熱して一晩還流した。翌朝、反応を水で停止しEtOAcで希釈した。層は分離し、有機層を水で2回、食塩水で1回洗浄し、Na2SO4で脱水し、減圧下で濃縮した。粗製油をフラッシュクロマトグラフィー(ヘキサン:酢酸エチル=6:1その後2:1)で精製して淡黄色固形物として中間体アルコール(0.90g、収率69%)を得た:1H NMR(CDCl3、400 MHz)δ 1.24−1.33(m、2H)、1.54(bs、2H)、2.14−2.18(m、1H)、2.46(t、J=12 Hz、1H)、2.77(t、J=11 Hz、2H)、3.75(s、6H)、4.21(bs、2H)、5.06(s、2H)、6.81(d、J=9 Hz、4H)、7.27−7.34(m、9H)。この物質は、これ以上のキャラクタリゼーションをせずに使用した。本中間体の一部(55mg、0.18mmol)を、JZL184の合成と同様の方法で、Pd/C及びクロロギ酸4−ニトロフェニルによる2段階手順によって処理した。粗製油をフラッシュクロマトグラフィー(ヘキサン:酢酸エチル=6:1その後2:1)で精製し、減圧下に放置すると黄色固形物に変化する、淡黄色油としてJZL175(55mg、2段階収率62%)を得た:1H NMR(CDCl3、400 MHz)δ 1.40(m、2H)、1.65(t、J=11 Hz、2H)、2.26(bs、1H)、2.55(t、J=12 Hz、1H)、2.87(t、J=12 Hz、1H)、3.00(t、J=13 Hz、1H)、3.76(s、6H)、4.27(t、J=13 Hz、2H)、6.84(d、J=9 Hz、4H)、7.24(d、J=9 Hz、2H)、7.34(d、J=9 Hz、4H)、8.19(d、J=9 Hz、2H);13C NMR(CDCl3、100 MHz)δ 26.6、26.9、44.4、45.2、55.4、79.2、113.6、113.7、122.4、125.1、127.3、137.8、137.9、144.8、152.2、156.5、158.4;C27H27N2O6[M−H2O+H]+に対する計算上のHRMS475.1864は475.1864に見出された。
【0092】
WWL152の合成撹拌するN−Bocピペラジン(100mg、0.54mmol)のCH2Cl2溶液(5ml)にクロロギ酸4−ニトロフェニル(109mg、0.54mmol)及びEt3N(75μl、0.54mmol)を室温で順次添加した。3時間後、反応液を減圧下で濃縮した。粗製混合物をフラッシュクロマトグラフィー(ヘキサン:酢酸エチル=3:1)で精製して白色固形物としてBoc保護中間体(100mg、収率52%)を得た:1H NMR(CDCl3、400 MHz)δ 8.24(d、J=9 Hz、2H)、7.31(d、J=9 Hz、2H)、3.67(bs、2H)、3.54(bs、6H),1.50(s、9H)。当該中間体(100mg、0.28mmol)に1:1 v/v TFA:CH2Cl2(2ml)を室温で添加した。2時間後、反応液を減圧下で濃縮した。粗製物はそれ以上精製せずに使用した。撹拌する4−メトキシベンゾフェノン(100mg、0.5mmol)EtOH溶液(5ml)にNaBH4(38mg、1mmol)を室温で添加した。翌朝、反応液を水(10ml)上に注ぎ込み、1時間撹拌し、次に生成物をフィルターで除き減圧下で濃縮した。CH2Cl2(5ml)中の粗製アルコールに塩化オキサリル(40μL、0.46mmol)を室温で滴下して添加した。粗製物をCH3CN(2ml)に再溶解し、それに4−ニトロフェニルピペラジン-1−カルボキシレート(58mg、0.23mmol)及びEt3N(32μl、0.23mmol)を順次添加した。試薬の添加終了後に、反応液を加熱し一晩還流した。翌朝、反応液を減圧下で濃縮した。粗製混合物をフラッシュクロマトグラフィー(ヘキサン:酢酸エチル=1:1)で精製して白色固形物としてWWL152(201mg、3段階収率84%)を得た:1H NMR(CDCl3、600 MHz)δ 8.24(d、J=9 Hz、2H)、7.32(d、J=8 Hz、4H)、7.27(d、J=9 Hz、2H)、6.84(d、J=8 Hz、4H)、4.22(s、1H)、3.76(s、6H)、3.66(s、2H)、3.57(s、2H)、2.45(s、4H);。C26H27N3O6[M+H]+に対する計算上のHRMS478.1973は、478.1977に見出された。
【0093】
WWL162の合成CH2Cl2(2mL)中の1−(ビス(4−フルオロフェニル)メチル)ピペラジン(29mg、0.1mmol)にクロロギ酸4−ニトロフェニル(20mg、0.1mmol)及びEt3N(14μl、0.1mmol)を室温で順次添加した。3時間後、反応液を減圧下で濃縮した。粗製混合物をフラッシュクロマトグラフィー(ヘキサン:酢酸エチル=3:1)で精製して白色固形物としてWWL162(42mg、収率92%)を得た:1H NMR(CDCl3、600 MHz)δ 8.24(d、J=9 Hz、2H)、7.36(m、4H)、7.28(d、J=9 Hz、2H)、7.00(m、4H)、4.29(s、1H)、3.68(s、2H)、3.59(s、2H)、2.44(s、4H);C24H21F2N3O4[M+H]+に対する計算上のHRMS454.1573は、454.1578に見出された。
【0094】
実施例7.他の方法及び実験材料
実験材料2−AG、ペンタデカン酸(PDA)、AEA、及びURB597は、ケイマンケミカル社(アナーバー、ミシガン州)(Cayman Chemicals(Ann Arbor、MI))から購入した。テトラヒドロリプスタチン(THL)は、シグマ社(セントルイス、ミズーリ州)(Sigma(St.Louis、MO))から購入した。FPローダミン及びFPビオチンは、既述の通り合成した(リォウら(Liu et al.)、「プロシーディングス・オブ・ザ・ナショナル・アカデミー・オブ・サイエンシズ・オブ・ザ・ユナイテッド・ステイツ・オブ・アメリカ(Proc.Natl.Acad.Sci.U.S.A.)」、1999年、第96巻:p.14694−9);及びパトリセリら(Patricelli et al.)、「プロテオミクス(Proteomics)」、2001年、第1巻:p.1067−71)。リモナバンはNIDA(ロックヴィル、メリーランド州(Rockville、MD))から購入し、容量比18:1:1の食塩水、エタノール、及びalkamuls-620(ローヌプーラン社製、プリンストン、ニュージャージー州(Rhone−Poulenic、Princeton、NJ))からなる溶媒に溶解した。13Cオレイン酸アミド(13C18H35NO)は、クラヴァットら(Cravatt et al.)、「サイエンス(Science)」、1995年、第268巻、p.1506−9、に記載された方法に従って13Cオレイン酸(スペクトラ・ステイブル・アイソトープ社製、コロンビア、メリーランド州(Spectra Stable Isotopes、Collumbia、MD))から合成した。
【0095】
マウス組織プロテオームの調製マウス脳をpH7.5のPBS中、Dounceホモジナイザーで均質化し、続いて壊死組織片を取り除くために低速回転(1,400g、5分間)した。それから、細胞質画分を上清に、膜画分を沈渣として得るために、上清を遠心(64,000g、45分間)にかけた。沈渣を洗浄し超音波処理によってPBS緩衝液に再懸濁した。各画分の総タンパク質濃度は、タンパク質アッセイキット(バイオラド社製(Bio−Rad))を使用して定量した。試料は、使用するまで−80℃で貯蔵した。
【0096】
COS7又はHEK293細胞における組換え体の発現手短にいえば、マウスのセリンヒドロラーゼをコードする完全長cDNAは、オープン・バイオ・システム社(ハンツビル、アラバマ州)(OpenBioSystems(Huntsville、AL))から購入した。cDNAは、直接形質移入するか(利用可能ならば真核細胞発現ベクター中に)又はpcDNA3(インビトロジェン社製(Invitrogen))にサブクローニングした。COS7細胞は、完全培地(L−グルタミン、非必須アミノ酸、ピルビン酸ナトリウム、及びFCS添加DMEM)、37℃及び5%CO2で10cmディッシュ中、〜70%集密まで培養した。当該細胞は、適切なcDNA又は空ベクター対照(「ニセの」)及び製造者のプロトコルに従って形質移入試薬FUGENE6(ロシュ・アプライド・サイエンス社製(Roche Applied Science))を使用することによって一過性に形質移入を行った。48時間後、細胞はリン酸緩衝食塩水(PBS)で2回洗浄し、擦過により採取し、PBS0.25mlに再懸濁し、超音波処理によって溶解した。細胞溶解物は、アッセイにおいて細胞全体のホモジネートとして使用した。
【0097】
酵素活性アッセイ手短に言うと、2−AG(100μM)を、マウス脳膜(20μg)又はCOS7細胞中の組換えMAGL(0.1μg)と、PBS(200μl)中、室温で10分間インキュベートした。容量比2:1のCHCl3:MeOH500μLの添加によって反応を停止し、PDA0.5nmolを投与し、ボルテックスし、次に相を分離するために遠心分離(1,400g、3分間)した。結果として生じた有機相の30μlをアジレント1100シリーズLC−MSD SL装置(Agilent 1100 series LC−MSD SL instrument)に注入した。LC分離は、プレカラム(C18、3.5μm、2mmx20mm)と共にジェミニ(Gemini)逆相C18カラム(5μm、4.6mmx50mm、フェノモネクス社製(Phenomonex))によって行った。移動相Aは、容量比95:5のH2O:MeOHから構成し、移動相Bは、容量比60:35:5のi−PrOH:MeOH:H2Oから構成した。陰イオン化法におけるイオン形成を補助するために0.1%の水酸化アンモニウムを含有させた。流束は0.5ml/分であり、濃度勾配は、1.5分間は0%B、5分間かけて直線的に100%Bに増加し、続いて3.5分間は100%Bの均一濃度勾配とし、2分間かけて0%Bに平衡化した。MS分析は、エレクトロスプレーイオン源により実行した。キャピラリ電圧は3.0kVに設定し、フラグメンター電圧は100Vに設定した。乾燥ガス温度は350℃、乾燥ガス流束は10 l/分、及びネブライザ圧力は35psiであった。加水分解産物は、PDA標準と比較したピーク下の面積を測定して定量した。13Cオレイン酸アミド又はAEA加水分解活性は、PBS(100μl)中、それぞれマウス脳膜(50μg)又はCOS7細胞(5μg)中の組換えFAAHを使用して測定した。反応は、インキュベーション時間を37℃で20分間とし、反応を停止するために容量比2:1のCHCl3:MeOH300μlを使用した以外、2−AG加水分解のために記載したのと同様の方法で実施した。DAGLβ加水分解活性は、PBS(100μl)中でHEK細胞(50μg)中の組換えDAGLβを使用して測定した。反応は、インキュベーション時間を37℃で30分間とした以外、オレイン酸アミド加水分解のために記載したのと同様の方法で実施した。
【0098】
[3H]リモナバン結合によるJZL184置換上記の通り膜を調製した。膜タンパク質(8μg)を、非標識リモナバン5μMの存在下又は非存在下に(非特異的結合を定量するため)、0.2-3nM[3H]リモナバン含有50mMトリス塩酸、3mM MgCl2、0.2mM EGTA、100mM NaCl、 1.25g/l 、と30℃で90分間インキュベートした。2番目のセットの試料も、JZL184濃度を変化させる(0.001−10μM)以外は、同様のプロトコルを使用して調製した。反応は、リットル当たりトリス緩衝液5gを含有するBSA(トリスBSA)に予浸したワットマン(Whatman)GF/Bガラス繊維フィルタ減圧ろ過によって停止し、続いて4℃のトリスBSAで3回洗浄した。結合した放射活性は、シンチレーション液であるサイニット・セイフ、エコノ1(ScinitSafe Econo 1)で抽出後に45%の効率で液体シンチレーション分光光度法によって測定した。
【0099】
脳脂質測定脳半分の重量を量り、続いて、N−アシルエタノールアミン(NAE)、モノアシルグリセロール(MAG)又は遊離脂肪酸(FFA)測定用の標準品(200pmolのd4−OEA、20pmolのd4−AEA、400pmolのC15:0−MAG、及び4nmolのPDA)を含む、容量比2:1:1のCHCl3:MeOH:トリスpH8.0(8ml)、Dounceホモジナイザーで均質化した。混合物をボルテックスし、次に遠心分離した(1,400g、10分間)。有機層を取り除き、終容量が再び8mlになるまでCHCl3を添加し、抽出を繰り返した。有機抽出液を併せN2気流下で乾燥し、容量比2:1のCHCl3:MeOH(120μl)に再可溶化した。再可溶化脂質の30μL及び15μLを、それぞれポジティブモード測定(MAG及びNAE)及びネガティブモード測定(遊離脂肪酸)のために注入した。脂質測定は、上記と同様の装置及び溶媒を使用してLC−MSによって実施した。0.1%ギ酸又は0.1%水酸化アンモニウム等の溶媒改良剤を、それぞれポジティブ及びネガティブイオン化モードにおいてイオン形成を補助するために含有させた。ポジティブモード測定のためには、各操作とも流速0.1ml/分、5分間で開始した。濃度勾配は、0%Bで開始し、流速0.4ml/分で40分間かけて直線的に100%Bに増加し、続いて7分間は100%Bの均一濃度勾配とし、次に流速0.5ml/分で8分間かけて0%Bに平衡化した。ネガティブモード測定のためには、各操作とも流速0.1ml/分、3分間で開始した。濃度勾配は、0%Bで開始し、流速0.4ml/分で17分間かけて直線的に100%Bに増加し、続いて7分間は100%Bの均一濃度勾配とし、次に流速0.5ml/分で3分間かけて0%Bに平衡化した。MAG、NAE、及びFFAは、C15:0又は重水素化標準品と比較したピーク下面積を測定することによって定量した。標的LC−MS測定は、選択イオン検出(SIM)を使用して実施した。
【0100】
生体内でJZL184の標的となるSHのABPP−MudPIT解析上記の通り、JZL184又は溶媒で処理したマウス由来の脳膜プロテオームの一部(1ml、mlPBS当たり1mg)を、5μMのFPビオチンにより室温で1時間標識し、Lys−C消化段階を省略した以外は、当該技術分野で既述の通りABPP−MudTIT分析のために調製した(グリーンバウムら、「モレキュラー&セルラー・プロテオミクス(Mol.Cell.Proteomics)」、2002年、第1巻:p.60−68;及びアレキサンダーら(Alexander et al.)、「ジャーナル・オブ・ジ・アメリカン・ケミカル・ソサイアティ(J.Am.Chem.Soc.)」、2006年、第128巻:p.9699−704)。溶出したペプチドのMudPIT解析は、直結型アジレント100LC−テルモフィニガンLTQ−MS装置(a coupled Agilent 1100 LC−ThermoFinnigan LTQ−MS instrument)に関し既述した通り実施した。全データセットは、SEQUEST検索アルゴリズムを使用してマウスのIPIデータベースで検索し、結果はDTASELECTによりフィルター処理しグループ分けした。1.8(+1)、2.5(+2)、3.5(+3)以上大きな相互相関スコア及び0.08以上大きなデルタCNスコアを有するペプチドを、スペクトラル・カウンティング分析に含めた。対照試料の中で平均して5以上のスペクトル・カウントが同定されたタンパク質のみを、比較分析の考慮に入れた。特に、プローブ標識タンパク質は、「無プローブ」対照の操作(ビオチン標識FP不含の上述の通り実施した実験)において認められるよりも少なくとも5倍大きなスペクトル数を有するFP処理試料の中に存在することによって更に同定される。スペクトル・カウントは、平均値の標準誤差(SEM)を付随する三試料の平均として報告する。
【0101】
行動的研究リモナバンと対比したJZL184の薬理学的効果を下記の指標を使用して評価した:自発運動活性、ホットプレート試験及び尾浸漬試験による侵害受容、バー試験による強硬症、及び低体温症。群当たり14匹のマウスのサンプルサイズで、全4グループのマウスを使用した。注射前に、尾浸漬試験におけるベースライン応答、並びに直腸温度に関する評価を行った。被験者にリモナバン又は溶媒を腹腔内注射し、続いて10分後にJZL184又はそれぞれの溶媒を腹腔内注射した。自発運動活性はJZL184又は溶媒投与の120分後に評価し、退避反応潜時は、2回目の注射135分後に評価し、並びに強硬症及び直腸温度は注射145分後に評価した。運動性低下を測定するために、各マウスを透明なプレキシガラス製箱(42.7x21.0x20.4cm)に評価時間の10分間放置し、エニメイズ(Anymaze)ソフトウェアを使用して(ステールティング社製、ウッドデール、イリノイ州(Stoelting、Wood Dale、Illinois))静止時間のパーセントを測定した。被験者の侵害受容は、尾浸漬アッセイにより評価した。頭を先にし尾をバッグから外に状態で、各マウスを吸収パッドで製作された小さなバッグ(VWRサイエンティフィック・プロダクツ社製(VWR Scientific Products);直径4cm、長さ11cm)に配置した。実験者はマウスを優しく抱き、尾の先約1cmを56.0℃に維持した温浴に浸漬した。10秒以内の遮断時間にマウスがその尾を温浴から退かせるための潜時を評価した。強硬症は、各被験者の前足を、床面から4.5cm高い棒(直径0.75cm)の上に載せるバー試験を使用して評価した。その足を棒の上に10秒間置き静止したままのマウス(呼吸運動を除外)を強硬症と評価した。本研究においていずれのマウスも強硬症でなかったにもかかわらず、JZL184で単独処理したマウスの半数が、前足を棒の上に載せられた時に、跳躍又は「ポップコーン」行動と例示される、反射亢進を示した。直腸温度は、熱電対探針を直腸に2.0cm挿入して測定し、温度は遠隔時期温度計によって得た。
【0102】
未処理の被験者の第二群(群当たりn=10−11匹マウス)を酢酸ストレッチングアッセイで評価した。被験者にリモナバン又はその溶媒を皮下注射し、続いて10分後にJZL184又は溶媒を皮下注射した。二番目の注射の120分後に、酢酸(0.6%)を10μl/g体重の量で腹腔内に注入した。酢酸投与後の20分間のマウス当たりのストレッチング(腹部の収斂、体幹の回転(ねじれ)及び全身及び後肢の伸展)回数を計数した。
【0103】
拮抗的活性に基づいたタンパク質プロファイリング(ABPP)実験生体外での阻害活性の選択性を当該技術分野で記載された拮抗的ABPP法を使用して試験した(例えば、リーら、「ジャーナル・オブ・ジ・アメリカン・ケミカル・ソサイアティ(J.Am.Chem.Soc.)」、2007年、第129巻:p.9594−5;及びロイングら(Leung et al.)、「ネイチャー・バイオテクノロジー(Nat.Biotech.)」、2003年:第21巻、p.687−91)。手短に言えば、本明細書に記載の通りに調製したマウスの脳膜プロテオームを、PBSで1mg/mlに希釈し、全反応液量50μl中、終濃度2μMでのFPローダミンの添加前に、種々の濃度の阻害剤(1nMから10μM)と37℃で30分間プレインキュベートした。室温で30分後、反応液は、4xSDS−PAGE用ローディング緩衝液により反応停止し、90℃で5分間煮沸し、SDS−PAGEに負荷し、フラットベッド蛍光スキャナを使用してゲル中で可視化した。
【0104】
JZL184による生体外研究標準アッセイは、基質又はABPPプローブの添加前に、タンパク質試料をJZL184と37℃で30分間プレインキュベートすることによって実施した。基質アッセイから濃度依存阻害曲線が得られ、当該曲線は、プリズム(Prism)ソフトウェア(グラフパッド(GraphPad))を使用して95%の信頼区間を有するEC50値を得るために適していた。kobs/[I]値の測定のために、脳膜プロテオーム(1mg/ml、全量300μl)をJZL184とインキュベートした(0.01−15μM、10−40分間、37℃)。10分ごとに反応液の50μlを取り出し、FPローダミン(2μM)で2分間処理し、4xSDS−PAGEローディング緩衝液で反応停止し、90℃で5分間煮沸した。混合性の反応液をSDS−PAGEに負荷し、フラットベット蛍光スキャナを使用してゲル中で可視化した。残存活性のパーセントは、MAGL又はFAAHのバンドに相当する統合した光学密度を測定することによって決定したが、その結果は擬一次速度定数を決定する指数曲線と適合した。
【0105】
JZL184による生体内研究JZL184(純品)をボルテックスし、超音波処理し、及び穏やかに加熱することによって容量比4:1のPEG300:ツイーン80に直接溶解した(10、4、2、又は1mg/g)。C57B1/6J雄マウス(6−8週令、20−26g)にJZL184又はJZL184無添加の容量比4:1のPEG300:ツイーン80溶媒を4μ/g体重の量で腹腔内投与した(上記希釈によって40、16、8、又は4mg/kg)。明示した時間後に、マウスをイソフルランで麻酔し、断頭によって屠殺した。脳を取り除き、正中矢状面に沿って半側切断し、次に各半分を液体N2で瞬間凍結した。脳の半分は上記の通りタンパク質分析用に調製し、他の半分は代謝産物分析用に使用した。URB597を超音波によって容量比18:1:1の食塩水:エマルファオー(emulphor):エタノールに溶解したのと10μl/g体重(最終投与量10mg/kg)の量で腹腔内投与したのを除き、URB597によるFAAHの選択的阻害も上記と同様の方法で行った。
【0106】
実施例8.癌発症機序における増加MAG活性の同定
癌発発症機序に寄与する酵素活性を同定するために、発明者らは、黒色腫[侵襲性(C8161、MUM2B)、非侵襲性(MUM2C)]、卵巣癌[侵襲性(SKOV3)、非侵襲性(OVCAR3)]、及び乳癌[侵襲性(231MFP)、非侵襲性(MCF7)]を含む、多様な起源の腫瘍由来の、侵襲性及び非侵襲性のヒト癌細胞株のパネル(調査対照群)の機能的なプロテオーム解析を行った。侵襲性癌細胞株は、その非侵襲性のカウンターパートと比較して、より大きな生体外での移動及び生体内での腫瘍増殖速度を示すことが確認された。これらの癌細胞株由来のプロテオームを、セリンヒドロラーゼ指向フルオロホスホネート(FP)活性に基づいたプローブを使用して、活性に基づいたタンパク質プロファイリング(ABPP)によって選別した。本研究の目的は、これらの保存酵素の変化が癌細胞の病態に対し寄与する高い可能性を有するであろうとの仮説の下に研究しつつ、非侵襲性と比較して侵襲性癌細胞株において一貫して変化する加水分解酵素活性を同定することにあった。
【0107】
スペクトラル・カウンティングによって判断したところ、この分析で検出された50以上のセリンヒドロラーゼの中で、二の酵素、KIAA1363及びMAGLが非侵襲性のカウンターパートと比較して侵襲性癌細胞において一貫して増加することが見出された。発明者らは、侵襲性癌細胞におけるKIAA1363及びMAGLの増加をゲルに基づくABPPによって確認したが、そこではプロテオームをローダミンタグFPで処理し、1D−SDS−PAGE及びインゲル蛍光スキャニングによって分析する(図13A)。どちらの場合も、KIAA1363についての差別的グリコシル化のため、及びおそらくMAGLについての選択的スプライシングのために(カールソンら(Karlsson et al.)、「ジーン(Gene)」、2001年、第272巻:p.11−18)、各酵素の二つの型が検出された(図13A)。襲性癌細胞におけるMAGLの高められた活性は、基質C20:4MAGを使用して侵確認した(図13B)。数種類の酵素がMAG加水分解活性を示すことが明らかにされてきたので(ブランクマンら(Blankman et al.)、「ケミストリー&バイオロジー(ChemBiol)」、2007年、第14巻:p1347−1356)、発明者らは、強力で選択的なMAGL阻害剤JZL184を使用して、MAGLが癌細胞中のこの過程に成す寄与を確認した。JZL184(1μM、4時間)は癌細胞のMAG加水分解活性を劇的に減少し(図13B)、MAGLの33及び35kDA型の両方のFPローダミンシグナルを選択的に遮断した(図13A)。対照的に、発明者らは、JZL184処理が癌細胞によって示されるジアシルグリセロール、トリアシルグリセロール、リゾリン脂質、及びリン脂質を含む、数種類の追加のクラスの脂質に対する加水分解活性を変化させないことを見出した。これらのデータは、侵襲性癌細胞が極めて増加したMAG活性を示し、例え全てではないにしても、この活性の殆どがMAGL酵素に基づくことを明らかにしている。
【0108】
研究は、MAGLが侵襲性癌細胞の遊離脂肪酸(FFA)レベルを制御することを明らかにしている。MAGLの既知の機能と調和して、発明者らは、JZL184によるMAGLの遮断が(1μM、4時間)、各々の侵襲性癌細胞株において、2−AGを含む数種類のモノアシルグリセロール(MAG)レベルの著しい増加を引き起こすことを見出した(図13C)。興味深いことに、しかしながら、MAGL阻害はまた侵襲性癌細胞におけるFFAレベルの著しい減少も引き起こした(図13D)。この驚くべき発見は正常細胞におけるMAGLの機能とは対照をなしている。この酵素が一般的にはFFAレベルを制御しないことは知られていた(野村ら(Nomura et al.)、「ネイチャー・ケミカル・バイオロジー(NatChemBio)」、2008年、第4巻、p.373−378)。発明者らは、またC20:4FFA及びMAGを除いては、FFAの減少の大きさが相応するMAGの増加を大いに上回る(C16:0、C18:0、及びC18:1脂質に対し、それぞれ〜5000−6100pmol及び41−75pmol)ことにも注目した。質量収支におけるこの明らかな矛盾が、JZL184処理癌細胞における増加したMAGの他の代謝産物への変換によって説明することは可能かもしれない。この前提と一致して、発明者らは、リピドーム解析がJZL184処理癌細胞においてリゾリン脂質の二の主要なクラス、リゾホスファチジルコリン(LPC)及びリゾホスファチジルエタノールアミン(LPE)の著しい増加を明らかにすることを見出した。これらのリゾリン脂質の増加の累積的大きさは(4100−6500pmol)、JZL184処理細胞において認められるFFAの減少とぴったりと一致した。とりわけ、発明者らは、癌細胞においてC20:4リゾリン脂質を検出しなかったが、これは、なぜJZL184がそれぞれC20:4MAG及びFFAの同様の大きさの増加及び減少を引き起こしたかの、ありそうな説明を提供している。非天然C17:0−MAGを使用した代謝標識研究は、MAGが侵襲誠意癌細胞によってLPC及びLPEに変換されること、並びにこの代謝変換がJZL184処理によって著しく亢進されること確認した。最後に、発明者らは、JZL184処理が非侵襲性癌細胞株におけるMAG及びFFAレベルに影響を与えなかったことを見出したが(図13C、13D)、これはこれらの細胞における無視できるMAG発現と一致する(図13A、13B)。
【0109】
発明者らは、次に、二の独立したshRNAプローブを使用するRNA干渉技術によってMAGLの発現を安定してノックダウンしたが、その両方とも侵襲性癌細胞株においてMAGL活性の70−80%を減少した、(図14A、図14及び図21)。他のセリンヒドロラーゼ活性は、shMAGLプローブによっては影響されなかったが(図14A、図14及び図21)、このことはこれらの試薬の特異性を確認している。両方のshMAGLプローブとも、侵襲性黒色腫(図14B、14C)、卵巣癌(図14E、14F)、及び乳癌細胞(図21)において、著しいMAGの増加及び相応するFFAの減少を引き起こした。あわせて、これらのデータは、急性の(薬理学的)及び安定した(shRNA)MAGLの両方の遮断が侵襲性癌細胞においてMAGの増加及びFFAの減少を引き起こすことを明らかにしている。さらに、侵襲性癌細胞が非侵襲性癌細胞よりも高い基礎レベルのFFAを発現すること、並びにこの変化した代謝プロファイルがMAGL阻害によってほとんど根絶されることに注目すべきである。これらの興味ある発見は、MAGLが侵襲性癌細胞におけるFFAレベルの主要な制御因子であることを示している。最後に、発明者らは、MAGL活性(図15A、15B)及びFFAレベル(図15C)が、良性又は低悪性度腫瘍と比較して高悪性度の原発性ヒト卵巣腫瘍においても増加することを確認した。このように、MAGL−FFA経路の高められた発現は、侵襲性ヒト癌細胞株及び原発性腫瘍両方の顕著な特徴である。
【0110】
実施例9.MAGL調節による癌の病原性制御
発明者らは、最初にMAGL発現の破壊の影響及び癌の病原性に関する活性を特定した。病原性の変化について、一組の生体外及び生体内アッセイを使用して、shMAGL癌細胞株を試験した。shMAGL黒色腫細胞(C8161)、卵巣癌細胞(SKOV3)、及び乳癌細胞(231MFP)は、著しく減少した生体外の移動(図16A、図16F及び図21)、浸潤(図16B、図16G及び図21)、及び血清飢餓条件下の細胞生存(図16C、図16H及び図21)を示した。JZL184によるMAGLの急性の薬理学的遮断もまた癌細胞の移動を減少し(図21)、生存を減少しなかったが、これは、おそらく癌の病原力を最大に障害するには、持続性のMAGL阻害が要求されることを示している。
【0111】
shMAGL−C8161及びSKOV3癌細胞は、免疫不全マウスで実施した皮下異種移植片移植の研究において(図16D、16I)、著しく減少した腫瘍増殖速度を示した。同様の腫瘍増殖速度の障害が、腫瘍中のMAGL活性を遮断することが確認された治療レジームに従って(データ不掲載)、JZL184を一日一回投与した(40mg/kg、経口投与)C8161及びSKOV3異種移植片移植マウスにおいても認められた(図16E、16J)。とりわけ、MAGL破壊腫瘍は、かなり低いFFAレベルを有したが、このことは、MAGLが生体内で増殖する癌細胞内の脂肪酸代謝に対する制御を維持することを示している。まとめると、これらの生体外及び生体内での研究は、MAGL活性が悪性の癌細胞によって示される侵襲性の特性の幾つかを支持することを証明している。
【0112】
発明者らは、次に非侵襲性癌細胞におけるMAGLの発現がその脂質代謝プロファイル及び病原性を変化させることができるか否かを。MUM2C及びOVCAR3細胞の安定なMAGL過発現変異体(MAGL−OE)並びに対照変異体[空ベクター発現体又はセリン求核試薬がアラニンに変異された(S122A)触媒的に不活性な変異体]をレトロウイルス感染によって作成し、それらのそれぞれのMAGL活性を、ABPP及びC20:4MAG基質アッセイによって評価した。両アッセイにより、MAGL−OE細胞が対照細胞と比較して10倍以上大きなMAGL活性の増加を有することを確認した(図17A)。MAGL−OE細胞は、また著しいMAGの減少(図17B)とFFAの増加(図17C)も示した。この変化した代謝プロファイルは、MAGL−OE細胞において増加した移動(図17D)、浸潤(図17E)、及び生存(データ不掲載)を付随した。これらの効果のいずれもS122A MAGL変異体を発現する癌細胞において認められなかったが、そのことは、それらがMAGL活性を要求することを示している。また、この前提と一致して、MAGL−OE細胞において認められた代謝及び病原性効果の両方ともJZL184の単回処理によって逆転された。最後に、MAGL−OE MUM2C細胞は、また対照細胞と比較して生体内での増加した腫瘍増殖も示した(図17F)。とりわけ、MAGL−OE MUM2C細胞の増加した腫瘍増殖速度は侵襲性C8161細胞のそれにほぼ匹敵したが(データ不掲載)、このことは、MAGLが黒色腫細胞において高度に腫瘍原性の表現型を誘発するのに充分であることを示している。まとめるとこれらのデータは、非侵襲性癌細胞における異所的なMAGLの発現が、生体外及び生体内の両方でそのFFAレベルを増加し及び病原性を促進するのに充分であることを示している。
【0113】
実施例10.MAGL−癌細胞の障害病原性の代謝レスキュー
MAGLは、そのMAG基質レベルを減少することによって、又はそのFFA産物レベルを増加することによって、又はその両方によって、癌細胞の侵襲性を支持することができた。MAGの中で、主要なシグナル伝達分子は内在性カンナビノイドである2−AGであり、それはCB1及びCB2受容体を活性化する。そこで発明者らは、亢進した内在性カンナビノイドシグナル伝達(増加した2−AGレベルから生じる)が、MAGL破壊癌細胞において認められる抗移動効果を媒介することができるか否かを試験した。しかしながら、発明者らは、CB1アンタゴンストもCB2アンタゴニストもshMAGL癌細胞の移動障害を救出しないことを見出した。CB1及びCB2アンタゴニストは、また癌細胞中のMAG又はFFAレベルにも影響をあたえなかった(データ不掲載)。これらの発見は、侵襲性癌細胞における低いCB1及びCB2受容体の発現レベルと結びついて(データ不掲載)、MAGLの癌の侵襲性に関する効果は内在性カンナビノイドシグナル伝達によって媒介されなかったことを主張している。
【0114】
MGAL触媒反応の産物に注目して、発明者らは、仮にMAGLの前腫瘍形成性効果がFFA(又はその二次代謝産物)によって媒介されるとするならば、shMAGL癌細胞の病原性の障害は、外来起源の脂肪酸による処理によって救出されるかもしれないと推論した。この仮説を支持するが、侵襲性癌細胞におけるMAGLによって制御される二の主要なFFA、パルミチン酸又はステアリン酸のshMAGL又はJZL184処理MAGL−OE癌細胞への添加は(それぞれC16:0及びC18:0FFA;20μM、4時間)、当該細胞の移動活性を完全に回復させた(図18A、18B)。C16:0及びC18:0FFAは、また非侵襲性癌細胞MUM2C及びOVCAR3の移動活性を刺激することも見出された(図18B)。発明者らは、次に増加したFFA送達量が生体内でshMAGLについて見出される腫瘍増殖障害を修正することができるか否かを特定した。免疫不全マウスを異種移植片腫瘍増殖実験の期間を通して通常の固形飼料か又は高脂肪食で飼育した。とりわけ、shMAGL−C8161細胞の障害した腫瘍増殖速度は、高脂肪食を与えられたマウスにおいて完全に救出された。対照的に、sh対照C8161細胞は、通常飼料と高脂肪食とにおいて同等の腫瘍増殖速度を示した。高脂肪食群のshMAGL−C8161細胞に対する腫瘍増殖の回復は、切除した腫瘍中の著しく増加したFFAレベルと相互に関連があった(図18D)。
【0115】
まとめると、これらの結果は、MAGLが増加したFFAレベルを持続的に維持することによって癌細胞の病原性の特性を支持することを示している。発明者らは、次にこの代謝プロファイルが侵襲性癌細胞の大きな脂質代謝ネットワークに影響することができるか否かを尋ねた。
【0116】
実施例11.MAGLは前腫瘍形成性シグナルに富んだ脂肪酸代謝ネットワークを制御する
発明者らは、最初にMAGL−FFA経路が、解糖系に燃料を供給するためにNAD+を再生する(連続的な脂肪アシルグリセリド/FFA再循環による)ための方法として貢献することができるか否かを検討したが、それは、癌における増加した中性脂質ヒドララーゼ活性に対する必要性を説明するために仮定されたものである(プルジビッツコウスキーら(Przybytkowski et al.)、「バイオケミストリー&セル・バイオロジー(Biochem.CellBiol.)」、2007年、第85巻:p.301−310)。このモデルの議論をするうちに、発明者らは、対照細胞と比較してshMAGL及びMAGL−OE細胞中においてNAD+/NADHの比率並びにピルビン酸及び乳酸レベルが不変であることを見出した。増加した脂肪分解に対する他の可能な理由は、癌細胞の重要なエネルギー源として役立つことができる、β酸化のためのFFA基質を産生することであるかもしれなかった。しかしながら、発明者らは、β酸化の律速段階を触媒する、カルニチンパルミトイルトランスフェラーゼ1(CPT1)の阻害剤が、癌細胞(sh対照、shMAGL、又はMAGL−OE)の移動活性に影響を与えないことを見出した。CPT1遮断は、sh対照細胞においてのみ増加したC20:4FFAを除き、癌細胞中のFFAレベルを変化することもできなかった。さらなる研究により侵襲性癌細胞における減少したCPT1発現が明らかとなったが、そのことは、MAGL脂肪酸代謝経路の病原性効果の下流の伝達物質としてのβ酸化に関する役割に対するエビデンスを提供している。
【0117】
FFAが膜構造物及びシグナル伝達分子の産生及び再構築のための基礎的な構成要素であることを考慮すると、MAGLにおける攪乱が悪性腫瘍にとって重要な幾つかの脂肪依存性生化学的ネットワークに影響を与えることを期待できるかもしれない。この仮説を検証するために、発明者らは、1)対照癌細胞と比較したMAGL−OE(OVCAR3、MUM2C)、及び2)sh対照癌細胞と比較したshMAGL(SKOV3、C8161)の比較を含む、変更したMAGL活性を有する癌細胞モデルのリピドーム解析を行った。癌細胞の有機抽出物を、数種の主要な脂質ファミリーをプロファイルする、非標的液体クロマトグラフィー−質量分析(LC−MS)プラットホームを使用して分析し、比較群の間で顕著に変化したレベルを有する代謝産物を、XCMSソフトウェアを使用して同定した。全体的なプロファイルを補完するために、発明者らは、標準的なリピドミック方法による検出にとっては低すぎる存在量である、特別な生理活性脂質(例えば、プロスタグランジン)の標的測定も行った。結果として得られたデータセットは、次にMAGLによって制御される脂質代謝産物の共通のサインを同定するために採掘されたが、発明者らは、当該産物を、MAGL−OE細胞において著しく増加し又は減少し、並びにそのそれぞれの対照群と比較してshMAGL細胞において逆の変化を示した代謝産物と定義した(図19A、19B)。
【0118】
幾つかのリゾリン脂質(LPC、LPA、LPE)、エーテル脂質(MAGE、アルキルLPE)、ホスファチジン酸(PA)、及びプロスタグランジンE2(PGE2)を含む、MAGL脂肪酸代謝ネットワーク中の脂質の殆どは、MAGL−OE及びshMAGL細胞において、それぞれ一貫して増加及び減少するというFFAにたいする同様のプロファイルを示した。MAGのみが、逆のプロファイルを示すことが見出された(shMAGL及びMAGL−OE細胞において、それぞれ増加及び減少する)。興味深いことに、この全体のリピドミックなサインは、その非侵襲性のカウンターパートと比較したときに侵襲性癌細胞においても実質的に認められた(例えば、それぞれMUM2Cと比較したC8161及びOVCAR3と比較したSKOV3)。これらの発見は、MAGLが侵襲性癌細胞において、FFA及びMAGだけでなく多数の二次脂質代謝産物からも成る脂質代謝ネットワークを制御することを証明している。発明者らは、JZL184によるMAGLの急性阻害が、特徴的な二次リピドミックなサインを引き起こすことを見出したことにさらに興味をそそられた。shMAGL細胞において認められた通りJZL184処理は、高MAGLレベルを発現する各癌細胞株においてLPA及びPGE2の減少を引き起こした(図19C、19D)。対照的に、以前にも説明した通り、LPC及びLPEの(減少というより)増加がJZL184処理細胞において認められた(図20)。これらのデータは、急性及び慢性のMAGL遮断が癌細胞において明確なメタボロミック効果を引き起こすことを示しているが、おそらくFFAの長期枯渇と対比した短期枯渇の特異的な成果を反映している。最後に、ホスファチジルコリン、ホスファチジルエタノールアミン、セラミド、スフィンゴシン−1−リン酸、コレステロール、ジアシルグリセロール、及びトリアシルグリセロールを含む幾つかのクラスの脂質は、MAGL活性の減少又は増加によって影響を受けなかったが、このことは、それらが、この酵素により制御される脂肪酸代謝ネットワークの制限成分であることを強調している。
【0119】
MAGL脂肪酸代謝ネットワーク内にLPA及びPGE2を含む数種類の前腫瘍形成性脂質メッセンジャーが存在するが、それらは癌細胞の侵襲性を促進することが報告されている。代謝標識研究により、侵襲性癌細胞がMAG及びFFAの両方をLPA及びPGE2に変換することができ、MAGに関しては、この変換がJZL184によって遮断されることが確認された(データ不掲載)。興味深いことに、LPA又はPGE2どちらかの処理は、sh対照細胞の移動に影響を与えない濃度において、shMAGL癌細胞の障害した移動を救出した(図19E)。逆に、MAGL−OE癌細胞の増加した移動活性は、Gi/Go阻害剤である百日咳毒素によって完全に遮断された(図19F)。最後に、MAGL−OE細胞において認められた刺激された移動の程度は、外からのLPA添加によって引き起こされる最大効果に匹敵した(図19G)。まとめると、これらのデータは、Gタンパク質共役型受容体に作用して高い移動活性を促進する生理活性脂質の産生を増加させることによって少なくとも部分的に、MAGLが癌の病原性に寄与することを示唆している。MAGL脂肪酸代謝ネットワークが他の前腫瘍形成性シグナル伝達系と相互作用するか否かを評価するために、発明者らは、shMAGL及びsh対照細胞を上皮増殖因子受容体(EGFR)の阻害剤で処理した。shMAGL細胞は、EGFR遮断の抗移動効果(図19H)及び抗生存効果(データ不掲載)に対する著しく高められた感受性を示す。これらのデータは、以前の発見と一致しており、このことは、癌細胞におけるLPAとEGFRシグナル伝達経路との間の実質的なクロストーク(相互干渉)を示している。
【0120】
実施例12.腫瘍発症機序におけるMAGL研究のための実験材料及び方法
実験材料C8161、MUM−2B、MUM−2C、及び231MFPを除き、全ての細胞株は、NCI60癌細胞株パネルの部分であり、米国国立癌研究所開発治療プログラムから入手した。C8161、MUM−2B、及びMUM−2C細胞株は、マリー・ヘンドリックス(Mary Hendrix)から提供を受けた。231MFP細胞は、既述の通り(ジェサニーら、2004年;前掲書)、MDA−MB−231細胞の外植した異種移植片腫瘍から作成した。C15:0、C16:0、C18:0及びC18:1 FFA、C18:1 FFA−OH及びC16:0、C18:0、C18:1、及びC20:4MAGは、シグマ社から購入した。C12:0MAGE及びC15:0MAGは、それぞれアレッキス・バイオケミカルズ社(Alexis Biochemicals)及びヌ・チェック・プリプ社(Nu−check Prep)から購入した。C20:4FFA及びPGE2は、ケイマン・ケミカルズ社から購入した。他の代表的な種々のクラスの脂質の脂質標準品は、シグマ社、ケイマン・ケミカルズ社、ヌ・チェック・プリプ社、又はアヴァンチー・リピッズ社(Avanti Lipids)から購入した。FPローダミン及びFPビオチンは、既述の方法に従って合成した。JZL184は、以前詳述した通り合成した。ヒトMAGL抗体(抗N末端アミノ酸1−121)は、ノヴァス・バイオロジカルズ社(Novus Biologicals)から購入した。
【0121】
癌細胞におけるMAGLの薬理学的阻害薬理学的阻害研究は、既述の通り行った(チャングら(Chiang et al.)、「ケミストリー&バイオロジー(Chem.Biol.)」、2006年、第13巻:p.1041−1050)。癌細胞は、ウシ胎児血清10%(v/v)添加RPMI培地1640中、5%CO2/95%空気の加湿した雰囲気で維持した。集密度80%で細胞をトリプシン処理し、血球計数板を使用して計数し、そして1x106個の細胞を6cmのディッシュに播いた(亜集密的)。播種の総20時間後に、細胞をPBSで2回洗浄し、阻害剤を無血清RPMI培地中、0.1%でJZL184(1μM)又は溶媒(DMSO)とインキュベートした。4時間のインキュベーション後、細胞を採取しABPP又はLC−MSによって分析した。
【0122】
ABPP−MudPITによる癌細胞プロテオーム由来のセリンヒドロラーゼ活性の同定及び比較定量各々のヒト癌細胞株由来の可溶性及び不溶性のプロテオーム画分を既述の通り作成し(ジェサニーら、2002年;前掲書)、ABPP−MudPITによって分析した(ジェサニーら、2005年;前掲書)。FPビオチンンによるプロテオームの標識化反応の標準的な条件は下記の通りであった:プロテオームを最終タンパク質濃度1.0μg/μlに調節し、プロテオーム1000μgを5μMのFPビオチンにより室温(RT)で2時間標識した。インキュベーション後、不溶性プロテオームを4℃で1時間回転して1%トリトンXにより可溶化した。可溶性及び不溶性プロテオーム画分由来のFP標識タンパク質の増強は、既述の通り行った(キッドら(Kidd et al.)、「バイオケミストリー(Biochemistry)」、2001年、第40巻:p.4005−15)。アビジン強化プロテオームを、1)1%SDS、2)6M尿素、3)50mMトリスpH8.0で8分間2回洗浄し、最終的に200μlの8M尿素50mMトリスpH8.0に再懸濁した。次に、ビーズ上の消化をするための試料を10mMのTCEPによる室温、30分間での還元によって調製し、当該試料を12mMのヨードアセトアミドにより暗所下、室温で30分間アルキル化した。消化は、試料を50mMトリスpH8.0で2M尿素に希釈後に、トリプシン(2mMのCaCl2存在下5.5μg/μlの3μL)により37℃で12時間行った。最後に、ペプチド試料を5%ギ酸の最終濃度に酸性化した。
【0123】
消化したペプチド混合物を二相性(強陽イオン交換/逆相)のキャピラリ―カラムにし、タンデム型質量分析法を組み合わせた二次元液体クロマトグラフィー(2D−LC)分離によって分析した。ペプチドは、5段階のMudPIT実験で溶出し(0、10、25、80及び100%の塩濃度段階(salt bumps)を使用)、データは、ダイナミック排出点灯(60秒)でデータ依存性獲得モードに設定した、イオントラップ型質量分析計LTQ(サーモ・サイエンティフィック社製(Thermo Scientific))で収集した。特に、1のMSサーベイ(ms1)フルスキャンには、7のms2のスキャンが続いた。ms2のスペクトルデータは、ロー・エックストラクター(1.9.1版)(RAW Xtractor(version1.9.1))を使用して未加工のファイルから抽出した。ms2スペクトルデータは、結果として総数22935のユニークな収録項目に帰着した、各集合遺伝子の識別子に関連する3.26版のヒトIPIデータベース由来の最も長期にわたる収録項目を含む、注文製のデータベースを対照してセクウェスト・アルゴリズム(3版)(SEQUEST(Version 3.0))を使用して検索した。加えて、これらの収録項目を置き換え、フォースル・ディスカバリー・レート(false−discovery rate)の評価のためにデータベースに付加した。SEQUEST検索は、メチオニン残基(16Da)の酸化、システイン残基(アルキル化のため57Da)のstatic修飾、非酵素的特異性及び前駆質量に対する±1.5Da及び生成物イオン質量に対する±0.5Daの質量許容誤差を考慮した。一致するms2スペクトルの結果を集合し、DTAセレクト(2.0.27版)(DTASelect(version2.0.27))を使用してフィルター処理した。ペプチドの最大偽陽性率0.1%を達成するために、二次判別分析を使用した。
【0124】
得られた全プロテオミックのデータは、最初にセリンヒドロラーゼに関して、既知のこのファミリーのメンバーに相応する、インタープロ(InterPro)ドメイン識別子(IPR000120、IPR000379、IPR001031、IPR001087、IPR001466、IPR002641、IPR002642、IPR002921、IPR004177、IPR005181、IPR007751、IPR008262、IPR010662)の手作業で集められたリストを使用してフィルター処理した。セリンヒドロラーゼの結果のセットを、次に、試験した少なくとも一の癌細胞株について、「無プローブ」対照プロテオームと比較してFPローダミン処理プロテオームにおいて、>10倍の高いスペクトルカウントを示す酵素に対してフィルター処理した。これらの計算のために、膜及び可溶性プロテオーム由来のスペクトルカウント値を、各癌細胞株由来の各セリンヒドロラーゼについて合併した。この分析は、〜50のセリンヒドロラーゼ活性のリストに帰着した。以前の研究は、比較定量が、比較中の二のグループの少なくとも一グループにおいて、平均≧10のスペクトルカウントを示すタンパク質にかなり限定されることを明らかにしている(ジェサニーら(Jessani et al.)、2005年;前掲書)。このフィルター処理を使用して(ここで、比較グループは、その非侵襲性カウンターパートと比較した侵襲性癌細胞株と定義した)、発明者らは、比較定量のための〜35のセリンヒドロラーゼを同定した。これらのセリンヒドロラーゼの中で、KIAA1363及びMAGLが侵襲性関連ヒドロラーゼ用に定義された下記の判定基準に適った:1)その非侵襲性カウンターパートと比較して三の全ての侵襲性癌細胞株における平均スペクトルカウントが>2倍である、並びに2)侵襲性及び非侵襲性癌細胞株対の間の較差に対するp値が<0.01である。
【0125】
癌細胞プロテオームのABPP及び加水分解活性アッセイABPP−MudPIT又はゲルに基づくABPPによる癌細胞プロテオーム由来のセリンヒドロラーゼ活性の同定及び比較定量は、既述の通りそれぞれFPビオチン(5μM)及びFTローダミン(2μM)を使用して行った(ジェサニーら(Jessani et al.)、「プロシーディングス・オブ・ザ・ナショナル・アカデミー・オブ・サイエンシズ・オブ・ザ・ユナイテッド・ステイツ・オブ・アメリカ(Proc.Natl.Acad.Sci.U.S.A.)」、2002年、第99巻:p.10335−10240;及びジェサニーら(Jessani et al.)、「ネイチャー・メソッズ(Nat.Methods)」、2005年、第2巻:p.691−697)。C20:4MAG加水分解活性アッセイは、既述の通り行った(ブランクマンら(Blankman et a.)、「ケミストリー&バイオロジー(Chem.Biol.)」、2007年、第14巻:p.1347−1356)。ABPP実験に関しては、細胞可溶化プロテオームを2μM FPローダミンにより室温で30分間処理した(全反応液量50μl)。反応を1容量の標準4xSDS/PAGEローディング緩衝液で停止し(還元)、SDS/PAGE(アクリルアミド10%)によって分離し、日立FMBio IIeフラットベッド蛍光スキャナ(Hitachi FMBイオIIe flatbed fluororescence scanner)(ミライバイオ社製(MiraiBio))によりインゲルで可視化した。統合したバンドの強度を標識タンパク質に対し計算し、各酵素活性のレベルを測定するために独立した細胞三試料から平均をとった。IC50値は、各阻害剤濃度での3回の試行からの得た用量反応曲線から、シグマプロット10.0(SigmaPlot 10.0)を使用して決定した。MAGL発現もまた標準法を使用してウェスタンブロッティングによって評価した。
【0126】
加水分解活性アッセイのために、擦過により細胞を採取する前に、細胞を生体内原位置で無血清RPMI培地中JZL184(1μM)により4時間処理した。次に、トリス緩衝液中の細胞溶解物(20μg)を脂質(100μM、例えばMAGL活性に対してはC20:4MAG)と室温で30分間、200μl容量でインキュベートした。反応を600μlの2:1クロロホルム:メタノールにより停止し、内部標準のC15:0FFA又はC12:0MAGE10nmolを添加した。産物は有機層に抽出され、当該有機層を抽出してLC−MSに直接注入した。LC−MSの設定は、既述の通りとした(ブランクマンら(Blankman et al.)、2007年;前掲書)。産物レベルは(例えば、MAGL活性に対するC20:4FFA)、内部標準レベルと比較して定量し、一定の内部標準レベルと変化する脂質濃度の間の標準曲線を作成した。酵素反応の直線位相の間で比活性を決定した(例えば、基質20%未満利用)。
【0127】
ヒト原発性卵巣腫瘍被験者は、診断され、ブリガム・アンド・ウィメンズ病院(Brigham and Women‘s Hospital)及びダナファーバー癌センター(Dana−Farber Cancer Center)(ボストン、マサチューセッツ州(Boston、MA))で卵巣腫瘍の治療を受けていた。すべて被験者由来の生物学的な検体を収集し、ブリガム・アンド・ウィメンズ病院の被験者委員会によって承認されたプロトコルに従って保存記録した。病理組織学的診断は、ブリガム・アンド・ウィメンズ病院の婦人科の病理医によって判定された。腫瘍を分類し、産婦人科の国際連盟システム(FIGO)に従って悪性度の類別をした。本研究に関し、10の良性及び13の高悪性度の悪性卵巣腫瘍試料をMAGL活性及び代謝産物測定のために使用した。良性の症例には、良性嚢腫、卵巣線維腫及び良性漿液性嚢胞腺腫が含まれ、一方悪性の症例は、全て高悪性度の漿液性乳頭状癌であった。新鮮な腫瘍組織をメスで2−5mm片に切り離し、個々にアルミ箔に包み、液体窒素中で瞬間凍結(snap frozen)し、−80℃の冷凍庫に保存した。MAGL活性及びFFAレベルは上述の通り測定した。
【0128】
ヒト癌細胞株におけるRNA干渉研究RNA干渉の研究は、既述の通り行った(チャングら(Chiang et al.)、2006年)。手短に言えば、低分子ヘアピン型RNA構築物をpLP−レトロQ(pLP−RetroQ)アクセプターシステムにサブクローンし、アンフォパック(AmphoPack)−293細胞株(クロンテック社製(Clontech))を使用してレトロウイルスを作成した。RNA干渉の研究に利用したヘアピン型オリゴヌクレオチドは;MAGL用には(shMAGL1)、5‘−CAACTTTCAAGGTCCTTGC−3’(配列番号3)及び(shMAGL2)、5‘−AGACTACCCTGGGCTTCC−3’(配列番号4);sh対照用には(shDPPIV)5‘−GATTCTTCTGGGACTGCTG−3’(配列番号5)であった。sh対照構築物は、侵襲性と非侵襲性癌細胞(DPPIV)の間で一貫して変化しない他と別なセリンヒドロラーゼを標的とするよう設計した。内在性タンパク質を標的とするsh対照構築物(非機能的なスクランブルド構築物よりは)を使用することが、RNA干渉機構の一般的な活性による非特異的な効果を制御する。培養1−6日の上清を含むウイルスを収集し、超遠心分離で濃縮し、10μg/mlのポリブレンの存在下に48時間安定して細胞を感染するために使用した。感染に続いて、ピューロマイシン(C8161、MUM2C、SKOV3、及び231MFPに対しては1μg/ml、OVCAR3に対しては0.3μg/ml)を含む培地で3日間選抜を行ったが、それは、レトロウイルスベクターがこの選択マーカーを含んでいたからである。感染した細胞を増殖し、ABPPによって酵素活性の損失及びC20:4MAG加水分解活性に関し試験を行った。
【0129】
ヒト癌細胞株における過剰発現の研究安定したMAGL過剰発現は、RNA干渉研究に関し上述した通りAmphoPack−293細胞株を使用してレトロウイルスを作成し、MAGL遺伝子をpMSCVpuroベクター(pMSCVpuro vector)(クロンテック社製)にサブクローニングすることによって達成した。ヒトMAGL構築物は、プライマー、5’−GCTCTCGAGGCCGCCATGCCAGAGGAAAGTTCC−3’(配列番号6)及び5’−AGCTGAATTCTCAGGGTGGGGACGCAGTTCCTG−3’(配列番号7)によるPCRによって作成した。PCR産物は、XhoI及びEcoRIの制限サイトを使用してpMSCVCpuro(クロンテック社製)にサブクローニングした。
【0130】
細胞移動、細胞生存、及び浸潤の研究移動アッセイは、コラーゲン10μg/mlで被覆した孔径8μmの膜を有するトランスウェル・チャンバー(コーニング社製)(Transwell chamber(Corning))中で37℃で4時間行った。移動アッセイ開始前、全24時間に、癌細胞株を6cmのディッシュ当たり1x106細胞の濃度で播いた。移動アッセイ開始時に、細胞をPBSで2回洗浄して採取し、次に無血清培地で4時間、血清飢餓とした。この4時間の間に、阻害剤、脂質又は溶媒(0.1%DMSO)を細胞とプレインキュベートした。血清飢餓細胞をトリプシン処理し、1400gで3分間回転し、再懸濁して計数した。(阻害剤、脂質又は溶媒を含有する)無血清培地200μl中の50,000個の細胞をトランスウェルの上部室(chamber)に播種した。阻害剤、脂質又は溶媒を下部室にも添加し、細胞を5時間(C8161及び231MFP細胞)又は20時間(SKOV3、MUM2C及びOVCAR3細胞)移動させた。次にフィルターを固定し、ディフ・クイック(デイド・ベーリング社製)(Diff−Quik(Dade Behring))で染色した。室を通って移動しなかった細胞を綿球で取り除いた。移動した細胞を倍率400xで計数し、各移動室から4視野を独立して計数した。1移動室についての4視野における細胞の平均をn=1と表す。
【0131】
細胞生存アッセイは、細胞増殖試薬WST−1(ロッシュ社製)を使用して既述の通り行った(ロカら(Roca et al.)、「ザ・ジャーナル・オブ・バイオロジカル・ケミストリー(J.Biol.Chem.)」、2008年、第283巻、p.25057−73;及びシッディキュイら(Siddiqui et al.)、「ブレスト・カンサー・リサーチ(Breast Cancer Res)」、2005年、第7巻、p.R645−654)。細胞をPBSで2回洗浄し、トリプシン処理によって採取し、PBSで2回洗浄し、1400gで遠心分離し、無血清培地に再懸濁した。WST−1(20μl)の添加前、0、24又は48時間に、20,000細胞を96ウェルプレートの無血清培地200μl中で37℃/5%CO2で4時間放置した。分光光度計を使用して450nmで吸光度を測定した。浸潤アッセイは、BDマトリゲルインベージョンチャンバー(BD Mtrigel Invasion Chambers)を使用して製造者のプロトコルに従って行った。
【0132】
腫瘍異種移植研究ヒト癌異種移植片は、癌細胞株をC.B17SCIDマウス(タコニック・ファームス社製(Taconic Farms))の側腹部に異所的に移植することによって確立した。手短に言えば、細胞をPBSで2回洗浄し、トリプシン処理し、そして血清を含有する培地で採取した。次に、採取した細胞を無血清培地で2回洗浄し、濃度2.0x104細胞/μlで再懸濁し、100μlを注入した。腫瘍の増殖は、3日ごとにカリパスで測定した。HFD研究のために、マウスを癌細胞注入の2週間前に60kcal%脂肪食(調査食)の状況に置いた。3−6日ごとにマウスの体重を量った。慢性JZL184処理研究のために、ポリエチレングリコール300(4μL/g)の経口胃内投与によって、マウスをJZL184又は溶媒で日に一回(毎日ほぼ同時間)処理した。処理は、癌細胞の異所的注入後直ちに開始した。
【0133】
癌細胞中の脂質測定脂質測定は、既述のプロトコルと同様に行った(チャングら(Chiang et al.)、2006年;前掲書)。癌細胞を、細胞プロファイルへの血清由来脂質の寄与を最小にするために、無血清培地で4時間生育した。細胞を(1x106細胞/6cmディッシュ、集密度80%)、リン酸緩衝生理食塩水(PBS)で2回洗浄し、1、400gで遠心分離によって分離し、クロロホルム:メタノール:トリス緩衝液の2:1:1混合液4ml中Dounceホモジナイザーで均質化した。試料は、次の合成標準品の存在下に均質化した:C12:0MAGE(10nmol)、C15:0FFA(10nmol)。有機層及び水層は、2000g、5分間の遠心分離によって分別し、有機層を収集した。水層を5%ギ酸の添加によって酸性化し(LPA等の代謝産物に対し)、続いてクロロホルム2mlを添加した。混合液をボルテックスし、有機層を併せてN2下で完全に乾燥し、クロロホルム100μlに溶解し、その30μlをLC−MSによって分析した。
【0134】
LC−MS分析は、アジレント1100LC−MSD SL装置(Agilent 1100 LC−MSD SL instrument)を使用して行った。LC分離は、プレカラム(C18、3.5μm、2mmx20mm)と共にフェノモネックス社製(Phenomonex)のジェミニ(Gemini)逆相C18カラム(直径5μmの粒子、50mmx4.6mm)によって行った。移動相Aは、95:5の比の水:メタノールから構成し、移動相Bは、60:35:5の比の2−プロパノール、メタノール、及び水から構成した。0.1%ギ酸及び0.1%水酸化アンモニウム等の溶媒改良剤を、それぞれポジティブ及びネガティブイオン化モードにおいて、LC分離能を改善するためと同様にイオン形成を補助するために、使用した。各操作の流速は、クロロホルム注入に付随する背圧を軽減するために、5分間、0.1ml/分で開始した。濃度勾配は、0%Bで開始し、流速0.4ml/分で45分間かけて直線的に100%Bに増加し、続いて17分間は流速0.5ml/分の100%Bの均一濃度勾配とし、次に流速0.5ml/分で8分間かけて0%Bに平衡化した。MS分析は、エレクトロンスプレーイオン化(ESI)源によって行った。キャピラリ電圧は3.0kVに設定し、フラグメンター電圧は100Vに設定した。乾燥ガス温度は350℃、乾燥ガス流束は10L/分、及びネブライザ圧力は35psiであった。
【0135】
リピドーム解析は、標的及び非標的モードの両方でC−MS分析によって行った。標準的な脂質クラスは、選択イオン検出(SIM)を使用して標的化によって定量し(非標的化分析によって定量した高存在量の代謝産物を除く)、代謝産物の同一性は、代謝産物のそれぞれの標準品との共溶出、及びアジレント6520精密質量QTOF−MS−MS(Agilent 6520 Accurate Mass QTOF−MAS−MS)による精密質量分析により測定される10ppm以内の質量精度によって確認した。これらの脂質は、ピーク下の面積を測定することによって定量し、内部標準に対し規準化した(ポジティブモードにおけるC12:0MAGE又はネガティブモードにおけるC15:0FFA)。各々の脂質種のpmoles/1x106細胞における絶対定量は、10nmolのC12:0MAGE及び/又はC15:0FFAと対照して分析した各クラスのそれぞれの脂質の標準曲線の作成に基づいたが、そこでは脂質及び標準品の相対抽出効率も考慮に入れた。MAGは、内部標準C15:0MAG(C12:0MAGEの代わりに)に基づいても定量したが、同一の結果が得られた。MSインターフェースにおいてMAGの脂肪酸アシルアニオン種への5%未満の分解が存在した。発明者らは、非天然のC15:0MAG種(これもまた、内在性MAG種と同様の分解パターンを有する)と対照してMAGレベルを定量したので、インターフェースにおける極小のMAG分解レベルは、MAGの絶対定量を妨げなかった。MAG及びFFA定量のための動的線形範囲は、それぞれ0.6−10,000pmoles及び1−10,000pmolesであった。非標的化データを質量範囲200−1200Daを使用してアジレント1100LC−MSD SL装置に収集し、計算的な分析のためにコモン・データ・フォーマット(CDF)として転送した。試料対の間の違った発現代謝産物は、多数のLC−MSトレースからの質量ピークの相対シグナル強度を一列に並べ定量する、XCMS分析物プロファイリングソフトウェアを使用することによって同定した。重要な阻害剤、shMAGL又はMAGL−OE感受性のピーク変化は、内部標準に対し規準化したピーク下面積を使用することにより手作業の定量によって確認した。
【0136】
本明細書で記載された実施例及び実施態様は、例示的な目的のためだけにあること、並びに当該実施例等に照らしての種々の修正又は変更は、当業者に提案されており、そして本出願及び別記の特許請求の範囲の精神及び範囲内に含まれることを理解されたい。本明細書に記載された方法及び材料と類似の又は同等の如何なる方法及び材料も本発明の実施又は試験に使用することができるのであるが、好ましい方法及び材料が記載されている。
【0137】
本明細書で引用した、全ての刊行物、ジェンバンク配列、ATCC寄託、特許及び特許出願は、これによってそっくりそのまま及び全ての目的のために、各々が個々にそのように表示されているかのような言及によって明確に援用される。
【技術分野】
【0001】
著作権告知
37C.F.R.§1.71(e)に基づき、出願人は、本情報開示の一部が著作権保護を受ける資料を含むことに特に言及しておく。本著作権者は、何人による本特許書類又は本特許の情報開示の複写複製に対しても、それが特許商標局の特許ファイル又は記録中に出現する場合には異議を唱えないが、その他の場合には全ての著作権を留保する。
【0002】
関連出願の相互参照
本特許出願は、米国仮出願第61/199,286号(出願日2008年11月14日)による優先権の利益を主張する。優先権基礎出願の全ての情報開示は、参照することにより、そのまま、すべての目的のために本明細書で援用されている。
【0003】
政府支援に関する陳述
本発明は、一部、国立衛生研究所の助成金第R01−DA017259号及びR01−DA025285号による政府支援により成された。それ故に、米国政府は、本発明に一定の権利を有することができる。
【0004】
発明の分野
本発明は、全体として他の脳セリンヒドロラーゼにモノアシルグリセロールリパーゼ(MAGL)を選択的に阻害するための、並びに被験者における2−アラキドノイルグリセロール(2−AG)媒介内在性カンナビノイドシグナル伝達を刺激するための方法及び組成物に関する。また本発明は、腫瘍細胞の増殖を阻害するための及び癌を治療するための方法にも関連する。
【0005】
発明の背景
カンナビノイド受容体CB1及びCB2は、マリファナの向精神成分、Δ9−テトラヒドロカンナビノールの分子標的である。CB1は主に脳で発現するが、また肺、肝臓及び腎臓でも発現する。CB2は、主に免疫系及び造血細胞で発現する。また、二つの内在性リガンド、言い換えれば「内在性カンナビノイド」、即ちアラキドン酸含有脂質アナンダミド(N−アラキドノイルエタノールアミン、AEA)及び2−アラキドノイルグリセロール(2−AG)も同定されている。内在性カンナビノイド系は、食欲、痛覚、炎症、及び記憶を含むさまざまな生理的プロセスを制御しており、並びにそれは、肥満、慢性疼痛、、及び抑鬱等の障害を治療するために、重要な最新の薬学的興味の中心となっている。
【0006】
内在性カンナビノイドシグナル伝達は、酵素的加水分解によって堅固に制御されている。2−AG又はAEAの加水分解により、アラキドン酸(AA)が産生される。主要なAEA−加水分解酵素は、脂肪酸アミドヒドロラーゼ(FAAH)である。遺伝子的な又は薬理学的なFAAHの破壊により、神経系及び抹消の至る所でAEAレベルが著しく上昇し、結果的に、痛覚、炎症、、及び抑鬱の減少を含む、多発性のCB1−及び/又はCB−2依存性の行動的影響に帰着する。興味深いことに、低体温症及び運動障害等の、直接作用性CB1アゴニストの周知の他の行動的影響の幾つかは、FAAH破壊動物では観察されない(ゴパーラジュら(Goparaju et al.)、「フェブス・レターズ(FEBS Lett.)」、1998年、第422巻:p.69−73;及びカスリアら(Kathuria et. al.)、「ネイチャー・メディスン(Nat Med)」、2003年、第9巻:p.76−81)。これらの動物は、また野生型の2−AG17レベルを有しており、このことは生体内でさらなるCB1制御の挙動プロセスが2−AGによって媒介されているかもしれないことを示唆している。幾つかのエビデンスは、モノアシルグリセロールリパーゼ(MAGL)が神経系における2−AGの加水分解のための主要な酵素であることを示唆している。例えば、ディンら(Dinh et al.)、「プロシーディングス・オブ・ザ・ナショナル・アカデミー・オブ・サイエンシズ・オブ・ザ・ユナイテッド・ステイツ・オブ・アメリカ(Proc.Natl.Acad.Sci.USA)」、2002年、第99巻:p.10819−24;ディンら(Dinh et al.)、「モレキュラー・ファルマコロジー(Mol.Pharmacol.)」、2004年、第66巻:p.1260−4;ブランクマンら(Blankman et al.)、「ケミストリー&バイオロジー(Chem.Biol.)」、2007年、第14巻:p.1347−56;及び野村ら(Nomura et al.)、「ネイチャー・ケミカル・バイオロジー(Nat.Chem.Biol.)」、2008年、第4巻:p373−8、を参照。しかしながら、これら以前の研究のどれも、MAGLが生体内における2−AGの加水分解で果たす役割を明確には検討していない。一方、幾つかのMAGL阻害剤が当該技術分野において開示されているが(ホーマンら(Hohmann et al.)、「ネイチャー(Nature)」、2005年、第435巻:p.1108−12;ヴァーヴェルら(Varvel et al.)、「ジャーナル・オブ・ファルマコロジー・アンド・エクスペリメンタル・セラピューティックス(J.Pharmacol.Esp.Ther.)」、2002年、第301巻:p.915−24;及びサーリオら(Saario et al.)、「ケミストリー&バイオロジー(Chem.Biol.)」、2005年、第12巻:p.649−56)、そのどれも生体内で薬理学的なツールとして一般的に使用するために要求されるレベルの有効性も明確性も示していない。
【0007】
腫瘍及び癌は、多くの人々の生命と健康を脅かす疾患又は症状である。侵襲性の腫瘍細胞の無抑制の増殖が、しばしば結果として悪性腫瘍(癌)を形成する。癌を根治的に治療することのできる有効な医薬又は治療法は未だ存在しない。現時点で腫瘍の治療に使用される主要な方法は、手術、放射線療法、化学療法、生物学的療法、及びホルモン療法、中国の伝統療法、温熱療法、ラジオ波焼灼療法等の他の療法等々である。しかし、利用可能なほとんどの治療法は、被験者の症候及び身体的症候を軽減することができるだけであり、疾患を治療することはできず、しかも通常、各治療法は副作用を含む不利益を有している。
【0008】
当該技術分野において、生体内で強力かつ選択的にMAGLを阻害する化合物を求める、並びに内在性カンナビノイドシグナル伝達活性する症状又は障害を治療するための新規な方法を求めるが存在する。また発明者らには、腫瘍増殖の阻害及び癌治療のための代替可能で効果的な方法を求めるが存在する。本発明は、これら及び当該技術分野において実現されていない他の。
【発明の概要】
【0009】
一態様において、本発明は、被験者における2−アラキドノイルグリセロール媒介内在性カンナビノイドシグナル伝達を刺激するための方法を提供する。当該方法は、モノアシルグリセロールリパーゼ(MAGL)を選択的に阻害する化合物の治療的に有効な量を、被験者に対し投与すること(例えば、腹腔内注射又は経口投与)を。これらの方法の幾つかにおいて、化合物は、脂肪酸アミドヒドロラーゼ(FAAH)にMAGL選択的。例えば、化合物は、FAAHMAGLの阻害に少なくとも100倍の選択性を有することができる。これらの方法の幾つかで用いるMAGL選択的阻害剤は、本明細書に開示する式Iに示す構造を有する。幾つかの他の方法において、MAGL特異的阻害剤は、FAAH及びα/βヒドロラーゼ6(ABHD6)の両方にMAGL選択的である。幾つかの好ましい実施態様においては、MAGL選択的阻害剤は化合物JZL184である。好ましくは、本発明の方法で治療されるべき被験者はである。これらの方法の幾つかにおいて治療されるべき被験者は、例えば疼痛、炎症、抑鬱又は等の、内在性カンナビノイドシグナル伝達によって媒介される又は関連する、症状又は障害を患っている。
【0010】
関連する態様において、本発明は、被験者における、内在性カンナビノイドシグナル伝達する症状、又はその治療効果が増加したカンナビノイド(例えば、2−AG)レベルに由来することができる疾患、を治療又は回復するための方法を提供する。これらの方法は、モノアシルグリセロールリパーゼ(MAGL)を選択的に阻害する化合物の治療的に有効な量を含む医薬組成物を被験者に投与することを含んでいる。これらの方法の幾つかは、2−AGシグナル伝達が役割を果たす、例えば疼痛等の症状又は障害を治療することに向けられる。これらの方法の幾つかは、式Iに示す構造を有するMAGL選択的阻害剤を用いる。当該方法の幾つかにおいて、MAGL選択的阻害剤は、FAAHにモノアシルグリセロールリパーゼ(MAGL)を選択的に阻害する。幾つかの他の方法において、用いた化合物はFAAH及びABHD6の両方にMAGLの阻害に選択的である。幾つかの好ましい実施態様において、用いるMAGL選択的阻害剤は化合物JZL184である。
【0011】
他の態様において、本発明はMAGL選択的阻害剤の化合物を提供する。これらの化合物は、下記の式Iに示す構造を有する:
【化1】
【0012】
式中、XはN又はCH、R1はH又はOHであり、そしてR2及びR3の各々は、下記の構造の有する。
【化2】
【0013】
本発明の好ましいMAGL選択的阻害剤の化合物は化合物JZL184である。その構造は、式Iに示すが、式中XはCH、R1はOHであり、そしてR2及びR3の各々は、下記に示す構造を有する。
【化3】
【0014】
さらに他の態様において、本発明は、改良された性質を有するMAGL阻害剤の化合物を同定するための方法を提供する。当該方法は、(a)本明細書で開示されるMAGL選択的阻害剤(例えば、JZL184)の一以上の構造アナログを合成すること、(b)MAGL選択的阻害剤の比較して改良された生物学的又は薬学的性質を有するアナログを同定すること、を。式Iに包含される任意の化合物も、構造アナログの合成ためのリード化合物として役立つことができる。幾つかの好ましい実施態様において、化合物JZL184が使用される。これらのスクリーニング法において、アナログにおいて同定されるべき、改良された生物学的又は薬学的性質は、例えば、MAGLに対する増強された阻害活性又は脳セリンヒドロラーゼMAGLに対する増加された選択性等であることができる。
【0015】
他の態様において、本発明は、腫瘍細胞、特に侵襲性の腫瘍細胞の増殖を阻害するための方法を提供する。当該方法は、モノアシルグリセロールリパーゼ(MAGL)を特異的に阻害する化合物と接触させることを。幾つかの実施態様において、当該方法は被験者に存在する腫瘍の増殖を阻害することに向けられる。被験者は、癌を有すると診断された者であることができる。また被験者は、癌にかかりやすい又は癌になる危険のある者であことができる。当該方法は、充実性腫瘍及び白血病の細胞を含む、任意の腫瘍細胞の増殖を阻害するために用いることができる。幾つかの実施態様において、当該方法は、乳房腫瘍細胞、卵巣腫瘍細胞、黒色腫細胞、肺腫瘍細胞又は脳腫瘍細胞の増殖を阻害するために使用される。
【0016】
本発明の幾つかの方法において、用いられるMAGL阻害化合物(又は「MAGL拮抗化合物」又は「MAGLアンタゴニスト化合物」)は、脂肪酸アミドヒドロラーゼ(FAAH)にMAGLの阻害に選択的である。例えば、化合物は、本明細書で開示される任意のMAGL選択的阻害剤であることができる。幾つかの他の実施態様において、用いられるMAGL阻害化合物は、MAGLに特異的な阻害ポリヌクレオチドである。このような阻害性ポリヌクレオチドの例には、低分子緩衝RNA(siRNA)、マイクロRNA(miRNA)、低分子ヘアピン型RNA(shRNA)、アンチセンス核酸及び相補DNA(cDNA)が含まれる。
【0017】
関連する態様において、本発明は、被験者の腫瘍の症候を治療又は回復する方法を提供する。これらの方法は、モノアシルグリセロールリパーゼ(MAGL)を特異的に阻害する化合物の治療的に有効な量を含む医薬組成物を被験者に投与することを含んでいる。任意の腫瘍又は癌により苦しめられる被験者も、これらの方法による治療のために回復可能である。例えば、当該方法は、乳腺腫瘍、卵巣腫瘍、黒色腫、肺腫瘍又は脳腫瘍を患う被験者を治療するために使用することができる。幾つかの方法において、用いられるMAGL阻害剤は、脂肪酸アミドヒドロラーゼ(FAAH)にMAGLの阻害に選択的である化合物である。幾つかの他の方法において、MAGLの発現又は細胞内レベルを特異的に減少させる阻害ポリヌクレオチドが使用される。
【0018】
本発明の特徴及び長所に関するより深い理解は、明細書及び特許請求の範囲の残りの部分を参照することによって得ることができるであろう。
【図面の簡単な説明】
【0019】
【図1】図1A−1Bは、MAGL阻害剤の構造及び拮抗ABPPプロファイルを示している。A:MAGL阻害剤の構造;B:マウス脳膜プロテオーム中のセリンヒドロラーゼ活性へのMAGL阻害剤の影響を示す拮抗ABPP。比較のために示したものは、それぞれ選択的FAAH及びABHD6阻害剤、URB597(カスリアら(Kathuria et al.)、「ネイチャー・メディスン(Nat Med)」、2003年、第9巻:p.76−81)及びWWL70(リーら(Li et al.)、「ジャーナル・オブ・ザ・アメリカン・ケミカル・ソサイアティ(J.Amer.Chem.Soc.)」、2007年、第129巻:p.9594−5)である。阻害剤を脳膜と共に30分間インキュベートし、次にセリンヒドロラーゼ指向ABPPプローブFPローダミン(2マイクロM、30分間)で処理し、それからプロテオームをSDS−PAGE及びインゲル蛍光スキャニングによって分析し、阻害された酵素を検出した。対照プロテオームは、DMSOのみで処理した。蛍光ゲルをグレイスケールで示す。文献(例えば、ブランクマンら(Blankman et al.)、「ケミストリー&バイオロジー(Chem.Biol.)」、2007年、第14巻:p.1347−56)に報告された通り、脳MAGLがSDS−PAGEによって35kDaの二重線として移動することに特に言及しておく。
【図2】図2A−2Cは、JZL184の二の生体外キャラクタリゼーションを示している。A:拮抗的ABPPによって定量した、マウス脳膜セリンヒドロラーゼへのJZL184の濃度依存的影響;B:基質アッセイ法(それぞれ2−AG及びオレイン酸アミド)によって定量した、JZL184による脳膜MAGL及びFAAH活性の遮断;C:基質アッセイ法(それぞれ2−AG及びアナンダミド)によって定量した、JZL184による組換え型MAGL及びFAAH活性の遮断。酵素はCOS7細胞中で組換えにより発現させた。JZL184は、組換え型MAGL活性のほぼ完全な遮断を引き起こしたが(>95%)、おそらく他の酵素の活性を反映して、脳膜においては2−AG加水分解活性の〜15%の残存が観察されたことに特に言及しておく。A−Cについては、分析に先立って試料をJZL184で30分間処理した。B及びCについては、データは、三つの独立した実験に対する平均±標準誤差として表した。
【図3】図3A−3Fは、JZL184の生体内でのキャラクタリゼーションを示している。A及びB:明示の投与量(4−40mg/kg、腹腔内投与)でJZL184により4時間処理したマウスから調製した脳膜の、セリンヒドロラーゼ活性プロファイル(A)並びにMAGL及びFAAH活性(B);C:JZL184により処理(16mg/kg、腹腔内投与、4時間)したマウスから調製された脳膜中のセリンヒドロラーゼ活性のABPP−MudPIT分析。MAGL及びFAAHの対照シグナルは、それぞれ赤色及び青色のバーで示す;D−F:明示の投与量(4−40mg/kg、腹腔内投与)でJZL184により4時間処理したマウス由来の、脳の2−AG(D)、アラキドン酸(E)、及びNAE(F)レベル(F)。Fについては、このFAAH阻害剤によって誘導されるNAEの増加を確認するために、URB597で処理(10mg/kg、腹腔内投与)したマウスからのデータも示す。B−Fについては、溶媒処理対照動物と対比した阻害剤処理動物について、*はp<0.05、**はp<0.01である。データは、平均±標準誤差として表した。群当たりn=3−5である。
【図4】図4A−4Eは、生体内でのJZL184の阻害活性の時間経過解析を示している。A及びB:明示の時間、JZL184(16mg/kg、腹腔内投与)により処理したマウスから調製した脳膜のセリンヒドロラーゼ活性プロファイル(A)並びにMAGL及びFAAH活性(B);C−E:明示の時間JZL184により処理(16mg/kg、腹腔内投与)したマウス由来の、脳の2−AG(D)、アラキドン酸(E)、及びNAEレベル。B−Eについては、溶媒処理対照動物と対比した阻害剤処理動物について、*はp<0.05、**はp<0.01である。データは、平均±標準誤差として表した。群当たりn=3−5である。
【図5】図5A−5Eは、JZL184の行動的影響を示している。A及びB:JZL184(16mg/kg)は、熱痛覚の尾浸漬アッセイ法において(A)並びに有害な化学痛覚の酢酸腹腔緊張アッセイ法において(B)鎮痛作用を引き起こした。これらの効果は、CB1アンタゴニスト、リモナバン(3mg/kg)による前処理によって遮断された;C−E:JZL184は、著しい低体温症(C)、運動性低下症(D)、及び反射亢進(E)を引き起こしたが、それらはまたリモナバンによって著しく減弱された。ベースラインの尾浸漬潜伏時間及び直腸温度は、それぞれ0.82±0.02秒及び37.4±0.1℃であった。溶媒−JZL184処理マウスと対比した溶媒−溶媒処理マウスについて、*はp<0.05、**はp<0.01である。溶媒処理マウス、リモナバン-JZL184処理マウス対比した溶媒−JZL184処理マウスについて、#はp<0.05、##はp<0.01である。データは、平均±標準誤差として表した。群当たりn=10−14である。
【図6】図6A−6Bは、化合物WWL98の構造(A)及び化合物JZL184の合成経路(B)を示している。他のカルバミン酸系阻害剤も同様の経路で合成した。
【図7】図7A−7Cは、MAGL及びFAAHのIZL184による経時的な阻害を示している。A:マウス脳膜プロテオーム中のMAGL活性のFPローダミン標識化のJZL184による阻害;B:マウス脳膜プロテオーム中のFAAH活性のFPローダミン標識化のJZL184による阻害;C:JZL184によるMAGL及びFAAH阻害についての計算値kobs/[I]。
【図8】図8A−8Bは、化合物JZL184は、CB1受容体に結合しないこと又は2−AG生合成酵素DAGLβを阻害しないことを示している。A:JZL184は、CB1受容体から3H−リモナバンの結合を置換しなかった;B:JZL184(25μM)は、組換えでCOS−7細胞中に発現されたDAGLβを阻害しなかった。既知のDAGLβ阻害剤テトラヒドロリプスタチン(THL、25μM)を正の対照として使用した。
【図9】図9A−9BはJZL184の用量反応のための脳脂質測定値を示している。JZL184(4−40mg/kg、腹腔内投与)の投与4時間後に脳のC18:1又はC16:0MAG(A)又は遊離脂肪酸(B)レベルにおける著しい変化は測定されないことが認められた。
【図10】図10A−10Eは、JZL184の経口投与の効果を示している。A及びB:明示の投与量(4−40mg/kg、経口投与)でJZL184により4時間処理したマウスから調製した脳膜の、セリンヒドロラーゼ活性プロファイル(A)並びにMAGL及びFAAH活性(B);C及びD:JZL184(1−40mg/kg、経口投与)により4時間処理したマウス由来の、脳の2−AG(C)及びAEA(D)レベル;E:腹腔内又は経口投与によって処理したマウス由来の脳のJZL184の相対レベル。B−Dについては、溶媒処理動物と対比した阻害剤処理動物について、*はp<0.05、**はp<0.01である。データは、平均±標準誤差として表した。群当たりn=3−5である。
【図11】図11A−11Cは、JZL184処理に続くマウスにおける脳脂質測定値の時間経過を示している。JZL184投与(16mg/kg、腹腔内投与)に続く24時間を通して、脳のC18:1又はC16:0のNAE(A)、MAG(B)、又は遊離脂肪酸(C)レベルにおいて著しい変化は認められなかった。
【図12】図12は、マウス脳中のJZL184レベルの時間経過解析を示している。JZL184を16mg/kgで経口投与した。
【図13】図13A−13Dは、侵襲性の癌細胞内でMAGLが増加することを示しているが、ここで当該酵素はモノアシルグリセロール(MAG)及び遊離脂肪酸(FFA)レベルを制御する。(A)非侵襲性(青色)及び侵襲性(赤色)のヒト癌細胞株内のセリンヒドロラーゼ活性のABPP。セリンヒドロラーゼ活性は、細胞全体のプロテオームにおいて活性に基づくプローブ、FPローダミンで標識化し、SDS−PAGE及びインゲル蛍光スキャニング(蛍光ゲルをグレイスケールで示す)によって検出した。赤色枠で強調したのは二の酵素、MAGL及びKIAA1363は、非侵襲性癌細胞と対比して侵襲性癌細胞において一貫して増加している。33及び35kDaのFPローダミン標識バンドがMAGLを表すことを確認するために、プロテオームを、DMSO又は選択的MAGL阻害剤JZL184(1μM、4時間)により前処理した癌細胞からも調製した。(B)JZL184(1μM、4時間)の存在下(赤色バー)又は非存在下(黒色バー)における癌細胞のC20:4MAG加水分解活性。(C、D)MAGLの阻害(JZL184 1μM、4時間)は、侵襲性細胞のMAGレベルを増加し(C)FFAレベルを減少するが(D)、しかし非侵襲性細胞ではそうではない。侵襲性の癌細胞は、それぞれのMAGL活性を反映して、非侵襲性の癌細胞と比較して基本的により高レベルのFFA(及びより低レベルのMAG)を有することに特に言及しておく。DMSO処理対照群と対比したJZL184処理群について、*はp<0.05、**はp<0.01である。非侵襲性癌細胞と対比した侵襲性癌細胞について、#はp<0.05、##はp<0.01である。データは、平均±標準誤差として表した。群当たりn=4−5である。
【図14】図14A−14Fは、MAGLの安定なshRNA媒介ノックダウンが侵襲性癌細胞においてFFAレベルを低下することを示している。(A、D)MAGLを、二の独立した短ヘアピンRNA(shRNA)オリゴヌクレオチド(shMAGL1、shMAGL2)を使用して安定にノックダウンした結果、異なるセリンヒドロラーゼ(DPPIV)を標的とするshRNAを発現するsh対照細胞と比較してC8161及びSKOV3細胞のMAGL活性において>70%が減少した。(B、C、E、F)shMAGL細胞は、MAGレベルの増加(B、E)及びFFAレベルの減少(C、F)を示している。sh対照群と対比したshMAGL群について、*はp<0.05、**はp<0.01である。非侵襲性癌細胞と対比した侵襲性癌細胞について、#はp<0.05、##はp<0.01である。sh対照細胞のMAGL活性並びにMAG及びFFAレベルは、親癌細胞株のそれらからそれほど異なっていなかった。データは、平均±標準誤差として表した。群当たりn=4−5である。
【図15】図15A−15Cは、高悪性度の原発性ヒト卵巣腫瘍が良性腫瘍と比較して増加したMAGL活性及びFFAを有することを示している。(A)個体の腫瘍検体についてのC20:4MAG加水分解活性測定値。JZL184(1μM、30分間)による前処理が、測定された加水分解活性の大部分はMAGLによることを確認した。(B)高悪性度腫瘍と対比した良性腫瘍におけるMAGL活性の概要図であり、ここで各値は、(A)に示した総C20:4MAG加水分解活性のJZL184感受性の部分として表した。(C)高悪性度腫瘍と対比した良性腫瘍におけるFFAレベル。良性腫瘍群と対比した高悪性度群について、**はp<0.01である。データは、平均±標準誤差として表した。群当たりn=10−13である。
【図16】図16A−16Jは、shRNA媒介ノックダウン及びMAGLの薬理学的阻害が癌の侵襲性をことを示している。(A−C、F−G)shMAGL細胞は、sh対照及び非感染の親細胞と比較して、減少した移動(A、F)、浸潤(B、G)、及び細胞生存(C、H)を示す。移動及び浸潤アッセイは、細胞50,000個をコラーゲン(10μg/ml)で被覆した膜又はバイオコートTMマトリゲル(BioCoatTM Matrigel)(登録商標)を含有する8μm孔径を備えたインサートに播種する前に、癌細胞を4時間無血清培地に移植することによってそれぞれ実施した。C8161及びSKOV3の移動時間は、それぞれ5時間及び20時間だった。移動又は浸潤した細胞は、倍率400Xで4視野当たり計数した平均数±標準誤差で言及する。細胞生存アッセイは、細胞20、000個を無血清培地中で96ウェルプレートに播種することによって実施した。生存は、WST−1増殖アッセイを使用して評価した。代表的な移動プレート(倍率400X)をshMAGL細胞と対比したsh対照細胞について示す(A、F)。(D、I)shMAGL細胞は、sh対照細胞及び非感染の親細胞と比較して、SCIDマウスにおいて損傷した腫瘍増殖を示している。C8161又はSKOV3細胞2x106個/100μlを側腹部の皮下に注入し、ノギスを使用して腫瘍増殖を測定した。(E、J)JZL184処理(4μl/gポリエチレングリセロール300溶媒として毎日40mg/kgを経口投与)は、溶媒処理と比較してSCIDマウスにおける異種移植した腫瘍増殖速度を著しく減少する。sh対照群と対比したshMAGL群又は溶媒処理群と対比したJZL184群について、*はp<0.05、**はp<0.01である。sh対照細胞及び親癌細胞は、その移動、細胞生存、浸潤、又は生体内での腫瘍増殖においてそれほど異なっていなかった。データは、平均±標準誤差として表した。群当たりn=5−8である。
【図17】図17A−17Fは、MAGLの異所性発現がFFAレベルを増加し、そしてMUM2C黒色腫細胞の生体外及び生体内の病原性を亢進することを示している。(A)ABPP(上段パネル)、ウェスタンブロット(中断パネル)及びC20:4MAG加水分解活性(下段パネル)によって確認したMUM2C細胞中のMAGLの過剰発現(MAGL−OE、赤色バー)。対照細胞及びS122A細胞(黒色バー)は、それぞれ空のベクター(EV)に感染した癌細胞又は触媒的に不活性なMAGL変異株(S122A)に相当する。ウェスタン分析によりS122A−MAGL変異株の過剰発現を確認したが、当該変異株は、ABPP及びC20:4MAG加水分解アッセイによって評価されたように、いかなる活性も示さなかった。(B、C)MAGL−OE細胞は、EV対照細胞及びS122A細胞と比較してより低いMAGレベル(B)及びより高いFFAレベル(C)を含んでいる。これらの代謝的効果は、生体内原位置でのJZL184による処理(1μM、4時間、栗色のバー)によって逆転された。(D、E)MAGL−OE MUM2C細胞は、EV及びS122A対照細胞と比較して、増加した移動(D)及び浸潤(E)を示している。この亢進した移動及び浸潤は、JZL184(1μM、4時間)によって逆転された。代表的な移動パネルを示す(D)。(F)MAGL−OE MUM2C細胞は、EV又はS122A対照細胞と比較してSCIDマウスにおいて著しく亢進した腫瘍増殖速度を示す(細胞2x106個を正所性に移植)。対照群と対比したMAGL−OE群について、*はp<0.05、**はp<0.01である。データは平均±標準誤差で表す。群当たりn=4−6である。
【図18】図18A−18Dは、外来性脂肪酸での処理によるshMAGL癌細胞の病原性の特性の回復をいる。(A)減少したshMAGL細胞の移動は、パルミチン酸又はステアリン酸による処理(20μM、4時間、斜線赤色バー)によって逆転される。(B)パルミチン酸又はステアリン酸の添加(20μM、4時間)は、MUM2C及びOVCAR3細胞の移動を増加し、そしてJZL184処理MAGL−OE細胞における減少した移動を救出する。(C)shMAGL−C8161細胞の減少した腫瘍増殖は、高脂肪食(HFD)(60kcal%脂肪)による処理によって逆転される。癌細胞を側腹部へ注入する前の2週間、マウスを通常固形飼料(NC)又はHFDで飼い、腫瘍増殖の時間経過を通じてこれらの飼料で養った。挿入図は、時間経過を通じての動物の体重である。HFD群由来の外植されたshMAGL腫瘍(斜線赤色バー)は、NC群由来のshMAGL腫瘍(赤色バー)と比較して増加したFFAレベルを含んでいる。sh対照群と対比したshMAGL群について、*はp<0.05、**はp<0.01である。shMAGL対照群(それそれDMSO処理群及びNC処理群)と対比したパルミチン酸又はステアリン酸又はHFD処理したshMAGL群について、##はp<0.01である。データは、平均±標準誤差として表した。(A)については、群当たりn=4−5;(B、C)については、群当たりn=7−8である。
【図19】図19A−19Iは、MAGLがプロ腫瘍原性シグナル伝達分子に富む脂質代謝ネットワークを制御することを示している。(A、B)EV対照細胞と対比したMAGL−OE(MUM2C及びOVCAR3の両方)(A)及びsh対照細胞と対比したshMAGL(B)の異常MAGL活性に関する癌細胞モデルのリピドーム解析。示した代謝産物は、EV対照細胞と対比したMAGL−OE細胞(MUM2C及びOVCAR3の両方)において増加した(赤色)又は減少した(青色)代謝産物であり(A)、そしてsh対照細胞と対比したshMAGL細胞(C8161及びSKOV3の両方)においては逆の特性を示した(B)。代謝産物の親質量を提供しており、円の大きさは相対的な変化の大きさを示している。(C、D)癌細胞モデルにおけるC16:0LPA及びPGE2レベルの定量。(E)C16:0LPA(100nM、4時間)又はPGE2(100nM、4時間)によるshMAGL癌細胞の処理は、sh対照細胞と比較してそれらの移動障害を救出する。百日咳毒素(PTX)(100ng/ml、4時間)は、MAGL−OE癌細胞の増加した移動を逆転する。(G)MAGL−OE細胞と対比したEV対照細胞におけるLPAによる移動の濃度依存的刺激。LPAによって誘導されるEV対照細胞における移動の最大刺激は、MAGL−OE細胞において見出される亢進した移動と基本的に一致し、そしてLPAはMAGL−OE細胞の移動をそれ以上増加しないことに特に言及しておく。(H)移動損傷パーセンテージで表した、shMAGL癌細胞と対比したsh対照癌細胞のEGFR(上皮成長因子受容体)阻害剤チロホスチンAG−1478に対する感受性。チロホスチンの抗移動効果に関するIC50値をパネル中に提供する。(I)MAGL−FFA経路を他のプロ腫瘍原性脂質へと結びつけることが可能な代謝ネットワークを示す図。(A)及び(B)について、データは比較群間の平均相対変化として表した;群当たりn=4−5。(C−G)について、それぞれの対照細胞群と比較したMAGL−OE細胞群又はshMAGL細胞群(C−G)、DMSO処理対照細胞と対比したJZL184処理細胞(C、D)については、*はP<0.05、**はP<0.01である。侵襲性細胞(C8161、SKOV3)と対比した非侵襲性親細胞(MUM2C、OVCAR3)(C、D)、又はsh対照細胞と対比したLPA/PGE2処理shMAGL細胞(E)については、##はP<0.01である。データは、平均±標準誤差で表した;群当たりn=3−5。
【図20】図20A−20Gは、パネルを通して、非侵襲性と対比した侵襲性の黒色腫細胞株及び卵巣癌細胞株のMAGL、KIAA1363及び脂肪分解プロファイルを示している。MAGL阻害剤JZL184による癌細胞処理によって引き起こされる増加したMAGと減少したFFAとの間に質量バランスの矛盾が存在する。(A−C)侵襲性黒色腫細胞(C8161)及び卵巣細胞(SKOV3)は、それらの非侵襲性カウンターパート(それぞれMUM2C及びOVCAR3細胞)と比較してより大きな移動(A)及び浸潤(B)及びより速い腫瘍増殖速度(C)を示す。(D)癌細胞株の脂肪分解特性は、他の脂肪分解活性と比較して、侵襲性癌細胞において強いMAG加水分解活性を示す。生体外での、種々の脂質基質からの遊離脂肪酸(FFA)の遊離を測定した。細胞を無血清培地中、生体内原位置でDMSO又はJZL184により4時間処理した。細胞溶解物(20μg)を100μM脂質基質と共に室温で1時間インキュベートした。上段及び下段のパネルは、比較及び可視化を容易にするために同じデータを異なったy軸の尺度(MAG加水分解活性の有無)で示している。脂質基質についての略称は下記のとおりである。TAG、トリアシルグリセロール;DAG、ジアシルグリセロール;MAG、モノアシルグリセロール;PC、ホスファチジルコリン;LPC、リゾホスファチジルコリン;PE、ホスファチジルエタノールアミン;LPE、リゾホスファチジルエタノールアミン。非侵襲性癌株群と対比した侵襲性癌株群については、*はp<0.05、**はp<0.01であり、JZL184と対比したDMSOについては、##はp<0.01である。データは、平均±標準誤差として表した;群当たり、n=3−15(G)リゾリン脂質の増加は、MAGL阻害剤JZL184による癌細胞処理によって引き起こされる増加したMAGと減少したFFAの間の質量バランスの矛盾を説明する。(E)JZL184(1μM、4時間)は、C8161及びSKOV3細胞において5500−6100pmol/106のFFAレベルの減少を引き起こす。このFFAレベルの減少は、JZL184処理癌細胞において見出されるリゾホスファチジルコリン(LPC)及びリゾホスファチジルエタノールアミン(LPE)レベルの正味の増加(4100−6500pmol/106細胞)と同様の大きさである。(F)C17:0MAG(20μM、1時間)は、癌細胞によってC17:0LPC及びC17:0LPEに変換されるが、しかもこの変換は癌細胞をJZL184(1μM、4時間)と共にプレインキュベーションすることによってより亢進される。C17:0MAGは、またC17:0LPAにも変換されるが、しかしこの場合には、当該変換はJZL184によって遮断される。これらのデータは、C17:0MAGは、癌細胞中でC17:0LPC及びC17:0LPEに直接転換されるが、一方、C17:0MAGのC17:0LPAへの変換には、C17:0MAGのC17:0FFAへのMAGL依存加水分解が要求されることを示している。C20:4MAG(20μM、1時間)は、プロスタグランジンE2(PGE2)に変換されるが、この変換はJZL184によって遮断される。(G)C16:0及びC20:4FFAは、侵襲性癌細胞によってそれぞれLPA及びPGE2に変換される。C8161及びSKOV3細胞を、無血清培地中、d2−C16:0FFA又はd8−C20:4FFA(10μM、4時間)で処理した。d2−C16:0FFAは、d2−C16:0 LPAに変換され、d8−C20:4FFAは、d8−C20:4PGE2に変換された。処理細胞と対比した対照細胞の比較については、*はp<0.05、**はp<0.01であり、MAG単独処理細胞と対比したJZL184/MAG処理細胞の比較については、#はp<0.05、##はp<0.01である。データは、平均±標準誤差で表した;群当たり、n=3−4。
【図21】図21A−21Iは、MAGLが侵襲性癌細胞株においてMAG、FFA及び癌細胞の病原性を制御することを示している。(A、B)JZL184処理(1μM、4時間)は、231MFP細胞においてMAGレベルを増加し(A)及びFFAレベルを減少する。(C)shMAGLプローブは、活性に基づいたタンパク質プロファイリング(ABPP)によって判断すると、侵襲性癌細胞C8161、SKOV3、及び231MFPにおいて他のセリンヒドロラーゼと比較してMAGLの活性を選択的に減少する。活性に基づいたプローブFPローダミンにより細胞全体のプロテオームにおいてセリンヒドロラーゼ活性を標識化し、SDS−PAGE及びインゲル蛍光スキャニング(蛍光ゲルをグレイスケールで示す)によって検出した。、ABPP(対C8161、SKOV3、及び231MFP)及びC20:4MAG基質アッセイ(対231MFP)によって評価しところ、MAGL活性は両方のshMAGLプローブ(shMAGL1、shMAGL2)により>75%減少した。(D、E)shMAGL細胞は、sh対照又は231MFP親細胞と比較して増加したMAGレベル(D)及び減少したFFAレベル(E)を示す。(F−H)shMAGL細胞は、sh対照又は231MFP親細胞と比較して減少した移動(F)、浸潤(G)、及び細胞生存(H)を示す。移動障害は、C16:0FFA又はC18:0FFAによる処理(20μM、4時間)によって救出された。(I)JZL184によるMAGLの急性遮断により、癌細胞の移動が損傷される。それぞれの対照群と対比したJZL184処理細胞又はshMAGL細胞については、**はp<0.01である。DMSO処理shMAGL群と対比したC16:0又はC18:0FFA処理群については、##はp<0.01である。データは、平均±標準誤差として表した;群当たり、n=4−6。
【図22】図22A−22Bは、生体内でのJZL184処理(経口強制飼養40mg/kg、30日間、4μL/gポリエチレングリコールを毎日1回投与)が腫瘍異種移植片MAGL活性を阻害すること(A)及びshMAGL腫瘍がsh対照細胞と比較してより低いFFAレベルを含んでいたこと(B)を示している。溶媒又はJZL184処理したマウス腫瘍ホモジネートを、100μMC20:4MAGの添加前に、DMSO又はJZL184(1μM、30分間)と共に生体外で30分間インキュベートした。それぞれ溶媒腫瘍又はsh対照腫瘍と対比したJZL184処理腫瘍又はshMAGL腫瘍については、*はp<0.05、**はp<0.01である。データは、平均±標準誤差として表した;グループ当たり、n=4。
【図23】図23A−23Gは、MAGLの異所性発現がFFAレベルを増加し、卵巣癌細胞OVCAR3及びMUM2C並びに黒色腫細胞の生体外及び生体内での病原性を亢進することを示している。(A)ABPP(上段パネル)、ウェスタンブロット(中段パネル)及びC20:4MAG加水分解活性(下段パネル)によって確認した、OVCAR3細胞におけるMAGLの過剰発現(MAGL−OE、赤色バー)。対照及びS122A細胞(黒色バー)は、空ベクターにより感染した癌細胞又は触媒的に不活性なMAGL変異株(S122A)に相当する。ウェスタン分析は、S122A−MAGL変異株の過剰発現を確認したが、当該変異株はABPP及びC20:4MAG加水分解アッセイによる評価においてはいかなる活性をも示さなかった。(B、C)MAGL−OE細胞は、EV対照細胞及びS122A細胞と比較してより低いMAGレベル(B)及びより高いFFAレベル(C)を含んでいる。これらの代謝的効果は、生体内原位置でのJZL184による処理(1μM、4時間、栗色のバー)によって逆転された。(D、E)MAGL−OE OVCAR3細胞は、EV及びS122A対照細胞と比較して増加した移動(D)及び浸潤(E)を示す。この亢進した移動及び浸潤は、JZL184(1μM、4時間)によって逆転された。代表的な移動のパネルを示す(D)。(F)MAGL−OE MUM2C及びOVCAR3細胞は、EV対照細胞と比較して著しく亢進した細胞生存(血清飢餓培養後24時間)を示す。(G)MAGL−OE MUM2C細胞の亢進した腫瘍増殖速度は、侵襲性のC8161黒色腫細胞の腫瘍増殖速度と同様である。C8161及びMUM2Cのデータは、それぞれ図16E及び図17Fに由来する(これらの実験は、腫瘍増殖速度の直接比較を可能にするために同時に実施したことを特に言及しておく)。対照群と対比したMAGL−OE群については、*はp<0.05、**はp<0.01である。データは、平均±標準誤差として表した;群当たり、n=4−6。
【図24】図24A−24Jは、癌の攻撃性における、カンナビノイドのシグナル伝達、エネルギー力学、脂肪酸ベータ酸化又は脂肪酸合成の評価を示している。sh対照及びshMAGL癌細胞におけるFFAレベルは、CB1アンタゴニスト(リモナバン、1μM、4時間)又はCB2アンタゴニスト処理(AM630、1μM、4時間)によって変化しなかった。C16:0及びC20:4FFAについてのデータを示す。他のFFA(C18:0、C18:1)についても同様の結果が見出された。(B)shMAGL癌細胞における移動障害は、CB1アンタゴニストであるリモナバン又はCB2アンタゴニストであるAM630のどちらの処理(1μM、4時間)によっても、又は両方のアンタゴニストによる共同処理によっても救出されなかった。sh対照細胞と対比した各shMAGL群について、**はp<0.01である。データは、平均±標準誤差として表した;群当たり、n=6。(C、D)DNAマイクロアレイ分析によって測定したCB1受容体(C)及びCB2受容体(D)mRNAレベル。左の棒グラフは、HU133APlus2.0アフィメトリクスアレイにより実施されたヒト癌細胞株のNCI60パネルの分析から抽出したデータに相当する(ロスら(Ross et al.)、2000年)。右の棒グラフは、HU133APlus2.0アフィメトリクスアレイにより実施された本研究に使用した侵襲性及び非侵襲性癌細胞株の分析から抽出したデータに相当する。比較のために、高いCB1受容体レベルを含むSNB−19癌細胞株を示す。(E−H)MAGLの増加した発現(MAGL−OE)又は減少した発現(shMAGL)は、癌細胞におけるNAD+(E)、NADH(F)、ピルビン酸(G)、又は乳酸(H)レベルを変化させない。データは、平均±標準誤差として表した;群当たり、n=6。(I)脂肪酸β酸化阻害剤トリメタジジン(3−ケトアシルコエンザイムAチオラーゼ阻害剤、100μM、4時間のプレインキュベーション及び移動に関してはコインキュベーション)又はエトモキシル(CPTI阻害剤、100μM、4時間のプレインキュベーション及び移動に関してはコインキュベーション)は、癌細胞の移動に影響を与えなかった。対照群と対比したMAGL−OE群について、**はp<0.01である。データは、平均±標準誤差として表した、群当たり、n=3。(J)エトモキシル処理(100μM、4時間)は、C20:4FFAを除いては癌細胞中のFFAレベルを変化させなかったが、当該FFAはC8161及びSKOV3細胞において増加した。それぞれの対照群と対比したshMAGL又はMAGL−OE細胞について、*はp<0.05、**はp<0.01である。DMSO処理対照群と対比したエトモキシル処理群については、#はp<0.05である。データは、平均±標準誤差で表した;群当たり、n=3−4。
【図25】図25A−25Dは、EGFRキナーゼ阻害剤チロホスチンの癌細胞への影響を示している。チロホスチンは、shMAGL癌細胞において見出される移動及び細胞生存の障害と同様のレベルで、C8161(A、B)及びSKOV3(C、D)細胞において、移動及び細胞生存(48時間、無血清)を減少した。C8161及びSKOV3shMAGL細胞は、細胞移動及び細胞生存(A、B)においてチロホスチン誘発障害に対する亢進した感受性を示す。溶媒処理sh対照細胞又はshMAGL癌細胞と対比したチロホスチン処理細胞の間では、*はp<0.05、**はP<0.01である;同処理群のsh対照とshMAGL癌細胞との間では、##はp<0.01である。データは、平均±標準誤差として表した、群当たり、n=4。
【0020】
発明の詳細な説明
I.概要
2−アラキドノイルグリセロール(2−AG)及びアナンダミドは、カンナビノイド受容体CB1及びCB2を活性化する脂質伝達物質である。内在性カンナビノイドシグナル伝達は、アナンダミドについては、脂肪酸アミドヒドロラーゼ(FAAH)によって媒介される過程、2−AGについては、モノアシルグリセロールリパーゼ(MAGL)によって基本的に制御されていると考えられている過程である、酵素的加水分解によって終結する。本発明は、MAGLの選択的阻害剤の本発明者らによる合成及びキャラクタリゼーションに一部基づいている。MAGL選択的阻害剤はMAGLのヒドロラーゼ活性を強力に阻害する一方で、FAAH等の他の脳セリンヒドロラーゼにほとんど又は全く阻害活性を有しないことが見出された。加えて、マウスへの腹腔内注射又は経口投与において、選択的MAGL阻害剤は、脳の2−AGレベルを著しく増加させることができたが、アナンダミド又は他のN−アシルエタノールアミン等の他の既知の脂質シグナル伝達分子のレベルを変化。また、阻害剤により処理されたマウスも、痛覚消失症を含む一連のCB1依存的行動的影響を示。
【0021】
発明者らは、MAGL及びその遊離脂肪酸(FFA)産物が、侵襲性のヒト癌細胞及び原発性の腫瘍において上方制御されており、それによって、移動、浸潤、生存、及び生体内での腫瘍増殖を促進する、発癌性のシグナル伝達脂質に富む脂肪酸代謝ネットワークが制御されることをさらに発見した。非侵襲性癌細胞におけるMAGLの過剰発現がこの脂肪酸代謝ネットワークを繰り返し、MAGL阻害剤によって逆転される、病原性−表現型を増加することが見出された。発明者らは、MAGLの遮断が生体外での移動だけでなく生体内での腫瘍増殖をも障害することを見出した。加えて、MAGL遮断に由来する表現型は、外来源のFFAによって救出された。これらのデータは、癌細胞においてFFAを増加するために、並びに移動活性及び腫瘍活性を増加するために、MAGLが必要かつ十分であることを示している。
【0022】
これらの発見に従って、本発明は、MAGL選択的アンタゴニスト化合物又はその薬学的に許容可能な塩を含む治療的な組成物を提供する。これらの組成物は、異所性の又は不十分なカンナビノイドシグナル伝達、特に2−AG媒介内在性カンナビノイドシグナル伝達に連結又は媒介される、被験者の障害又は症状の症候を治療又は回復するために、又は増加したカンナビノイドレベルから有利な効果を引き出すことのできる疾患を治療するために使用することができる。このような障害又は症状には、例えば、疼痛、炎症、抑鬱、、多発性硬化症、緑内障、及びアルツハイマー病が含まれる。これらの特殊な症状又は障害のいずれかを患う被験者における治療応用に加えて、本発明の組成物は、一般的にカンナビノイド依存性行動的影響(例えば、痛覚消失症)を誘導するために、又は2−AG媒介内在性カンナビノイドシグナル伝達活性を刺激又は増大するために、他の被験者においても用いることができる。本明細書で例示するMAGL選択的阻害化合物をリード化合物として使用することにより、本発明は、改良された生物学的活性及び薬学的特性を備えた新規なMAGL阻害剤のためのスクリーニング方法をさらに提供する。当該新規MAGL阻害剤は、異常な又は減少したカンナビノイドシグナル伝達活性によって引き起こされる又は関連する、障害又は症状の治療における増強された特性を備えた治療薬を提供することができる。次節に、本発明の組成物の生成及び使用、並びに本発明の実施方法についてのガイダンスを提供する。
【0023】
II.定義
別途定義が無い限り、本明細書で使用する技術的及び科学的用語は、本発明の属する分野の当業者によって通常に理解されるのと同様の意味を有する。下記の参考文献は、本発明において使用する多くの用語の一般的な定義を当業者に提供する:シングルトンら(Singleton et al.)著、「微生物学及び分子生物学辞典(DICTIONARY OF MICROBIOLOGY AND MOLECULAR BIOLOGY)」(第2版、1994年);「ケンブリッジ科学・技術辞典(THE CAMBRIDGE DICTIONARY OF SCIENCE AND TECHNOLOGY)」、(ウォーカー(Walker)編、1988年);及びヘイル&マーハム(Hale & Marham)著、「ハーパー・コリンズ生物学辞典(THE HARPER COLLINS DICTIONARY OF BIOLOGY)」、(1991年)。加えて、本発明の実施において読者を援助するために下記の定義を提供する。
【0024】
用語「薬剤(agnt)」又は「試験薬(test agent)」には、任意の物質、分子、元素、化合物、実体、又はそれらの組合せが含まれる。限定されるものではないが、例えば、タンパク質、ポリペプチド、有機小分子、多糖類、ポリヌクレオチド、その他等が含まれる。天然物、合成化合物、又は化学物質、又は二以上の物質の組合せであることができる。特に指定のない限り、用語「薬剤(agent)」、「物質(substance)」、及び「化合物(compound)」は、本明細書で互換的に使用する。
【0025】
用語「アナログ(analog)」は、基準分子(例えば、本明細書で開示するMAGL選択的阻害剤)に構造的に類似するが、しかし、代替置換基で基準分子の特異的な置換基を置換することによって、標的化され制御された方法で修飾された分子に言及するために本明細書で使用する。基準分子と比較してアナログは、当業者にとり、同一、類似の、又は改良された有用性が期待されるであろう。改良した特性(標的分子に対する高結合親和性等)を有する、既知化合物の変異型を同定するためのアナログの合成及びスクリーニングは、製薬化学において周知の研究法である。
【0026】
「抗悪性腫瘍剤(antineoplastic agent)」は、の新生物、特に細胞腫、肉腫、リンパ腫、又は白血病等の悪性(癌性)の病変の発達又は進行を阻害する機能特性を有する薬剤に言及するために本明細書で使用する。転移阻害はしばしば抗悪性腫瘍剤の一特性である。
【0027】
化学療法剤は、腫瘍性疾患の治療方法においてポリクローナル抗グリカン抗体と併用して使用することができる。抗グリカン抗体を含む抗体−細胞毒抱合体もまた、標準的な癌治療によって誘導された免疫を追加免疫するために使用することができる。これらの場合には、投与される化学療法剤の服用量を減少することが可能である(モキールら(Mokyr et al.)、「カンサー・リサーチ(Cancer Research)」、1998年、第58巻:5301−5304)。抗グリカン抗体併用の科学理論的な解釈は、ほとんどの化学療法化合物の細胞毒性作用の結果である細胞死が、結果として抗原提示経路において増加した腫瘍抗原レベルに帰着するというものである。このように、抗グリカン抗体は、腫瘍細胞の化学療法放出を刺激して免疫応答を追加免疫することができる。
【0028】
本明細書で使用する場合、「接触(contacting)」は、その通常の意味を有するが、二以上の分子の組み合わせ又は分子と細胞の組み合わせ(例えば、一化合物と一細胞)をも意味する。接触は、生体外で、例えば、二以上の薬剤を組み合わせて又は一薬剤と一細胞若しくは一細胞溶解物を組み合わせて、試験管又は他の容器中で引き起こすことができる。接触は、生体内でも、例えば、被験者の体内のタンパク質又はポリペプチドを特異的に標的とする薬剤を被験者に投与することによって引き起こすことができる。
【0029】
カンナビノイドシグナル伝達と関連する症状又は障害とは、異所性の又は抑制された内在性カンナビノイドシグナル伝達に連結、媒介又は関連する障害をいう。このように、当該障害には、減少した内在性カンナビノイドレベル(例えば、2−AG)又は内在性カンナビノイドを分解する過剰な酵素活性によって引き起こされる症状が包含される。カンナビノイドシグナル伝達と関連する症状又は障害には、内在性カンナビノイドシグナル伝達の増大によって有益な効果(例えば、疼痛)を誘導することのできる症状が含まれる。当該用語には広く、治療効果が増加したカンナビノイドレベル又は減少したアラキドン酸レベルから獲得することのできる疾病も含まれる(例えば、アルツハイマー病又は多発性硬化症)。本発明の幾つかの好ましい実施態様において、当該症状又は障害は、2−AGシグナル伝達と関連又は付随する(例えば、異所性の又は抑制された2−AGシグナル伝達)症状・障害であり、又は2−AGシグナル伝達活性を刺激することによって有益な効果を誘導することのできる症状・障害である。このような症状又は障害の例には、疼痛、抑鬱、、心血管障害、代謝障害、炎症性症候群及び卒中が含まれる。
【0030】
本発明において使用する場合、心血管障害は、心臓又は血管(動脈及び静脈)に関わる疾患のクラスをいう。それは、心臓血管系に悪影響を及ぼす任意の疾患を含むが、しかし特にアテローム性動脈硬化症(動脈疾患)と関連するこれらの症状をいう。これらの症状は同様の原因、機序、及び治療法を有する。通常の心血管障害の例には、動脈硬化症(動脈の硬化)、アテローム性動脈硬化症(血管壁上のコレステロール蓄積等の粥腫)、リウマチ性心疾患、全身性高血圧症及び卒中が含まれる。本明細書で使用する場合、炎症性疾患とは、異常な、過剰な又は調節解除された炎症に付随する疾患又は症状である。それらには、の種々の疾患の根底にある、、無な疾患群含まれる。代謝障害とは、正常な成長発育に必須のある代謝反応が起きないときに発生する健康状態をいう。これらには、アミノ酸代謝障害(例えば、メープルシロップ尿症及びホモシスチン尿症)、有機酸代謝障害(例えば、メチルマロン酸尿症)、脂肪酸ベータ酸化障害(例えば、MCAD欠損症)、テイ・サックス病及びゴーシェ病等の脂質代謝障害(脂質蓄積症)、ミトコンドリア障害(例えば、ミトコンドリア心筋症)、及びペルオキソーム病(例えば、ツェルベルガー症候群)含まれる。
【0031】
カンナビノイドの行動的影響(又はCB1依存性及び/又はCB2依存性行動的影響)とは、カンナビノイドシグナル伝達活性の結果として被験者によって示される生理学的及び行動上の応答をいう。カンナビノイドの行動的影響の幾つかは、2−AGシグナル伝達に関与するが、本明細書では2−AG媒介行動的影響(又は2−AG依存行動的影響)と。下記の実施例において詳述する通り、2−AG媒介行動的影響には無痛覚症(減少した痛覚)が含まれる。特に指定のない限り、カンナビノイドの行動影響には、また被験者に存する又は被験者によって経験される、炎症、又は抑鬱に付随する症候の軽減又は回復も包含される。
【0032】
内在性カンナビノイド系とは、食欲、痛覚、気分、及び記憶を含む、種々の生理学的過程に関与する、神経調節物質及びその受容体の一群をいう。カンナビノイド受容体(カンナビスの向精神効果を媒介する受容体と同一)に結合する内在性の脂質がカンナビノイド命名される。、内在性カンナビノイド系には(1)それぞれ主に中枢神経系及び抹消神経に位置する2のGタンパク質共役型受容体である;(2)内在性アラキドン酸に基づく脂質である、アナンダミド(N-アラキドニルエタノールアミン又はAEA)及び2−アラキドノイルグリセロール(2−AG)集合的に「内在性カンナビノイド」と;及び(3)内在性カンナビノイドアナンダミド及び2−AGを合成及び分解する酵素。内在性カンナビノイド系は、遺伝的及び薬理学的方法を使用して研究されてきた。これらの研究は、神経調節物質放出、運動学習、シナプス可塑性、食欲、及び痛覚を含む、様々な生理学的過程における内在性カンナビノイドシグナル伝達についての広範な役割を明らかにした。
【0033】
痛覚過敏は、被験者(例えば、温血動物)が疼痛に対し増加した感受性を示す症状である。それは、身体への物理的損傷、例えば、手術によって不可避的に生じた受傷に付随することが知られている。疼痛は、関節炎及びリウマチ性疾患等のの炎症症状に付随することも知られている。痛覚過敏には、限定されるものではないが、炎症(例えば、リウマチ性関節炎及び変形性関節症)と関連する疼痛、術後痛、後分娩痛、歯科症状(齲蝕及び歯肉炎)と関連する疼痛、限定されるものではないが、日焼け、擦過傷、挫傷その他を含む熱症と関連する疼痛、スポーツ損傷及び捻挫、限定されるものではないが、ツタウルシ、アレルギー性皮疹及び皮膚炎を含む皮膚症状と関連する疼痛、及び軽い刺激に対する感受性を増加する有害冷感等の他の疼痛等の軽中度の疼痛から激痛までが含まれる。
【0034】
基準分子(例えば、MAGLポリペプチド)又は細胞(例えば、腫瘍細胞)の活性に関する用語「調節(modulate)」とは、活性の上方制御(、活性化又は刺激)又は下方制御(、阻害又は抑制)をいう。ポリペプチド等の基準分子の調節には、分子の発現又は細胞内レベルの変化も含まれる。生物学的活性(例えば、MAGL発現又はリパーゼ活性、又は腫瘍細胞増殖)に関する用語「阻害する(inhibit)」又は「阻害(inhibition)」とは、活性の抑制又は下方制御をいう。阻害の機序は、例えば基準分子又は細胞への結合によって直接的であことができる。阻害は、例えば、別の方法で基準分子又は細胞に結合し調節する他の分子への結合及び/又は修飾によって間接的であことができる。
【0035】
「ポリヌクレオチド」又は「核酸配列」とは、ヌクレオチドのをいう。幾つかの例において、ポリヌクレオチドとは、それが由来するところの生物体のゲノム中において、それがすぐ近くに隣接しているどちらのコード配列ともすぐ近くに隣接していない配列をいう。それ故に、当該用語には、例えば、ベクター:自己複製プラスミド若しくはウイルス;又は原核生物若しくは真核生物のゲノムDNAに取り込まれた、又は他の配列とは別個の独立した分子(例えば、cDNA)として存在する組換えDNAが含まれる。ポリヌクレオチドは、リボヌクレオチド、デオキシリボヌクレオチド、又は各々のヌクレオチドの修飾型であることができる。
【0036】
ポリペプチド又はタンパク質とは、単量体がアミド結合によって結合したアミノ酸残基である重合体をいう。アミノ酸がアルファ-アミノ酸である場合、L−光学異性体又はD−光学異性体のどちらでも使用することができるが、L−異性体が典型的である。ポリペプチド又はタンパク質フラグメントは、自然界に見出されるタンパク質のものと同じ又は実質的に同一のアミノ酸配列を有することができる。実質的に同一の配列を有するポリペプチド又はタンパク質とは、アミノ酸配列が完全ではないが大部分同じであり、それが関連する配列の機能活性が維持されていることを意味する。
【0037】
用語「被験者」には、他の非動物、例えば、ウマ、イヌ、ネコ、マウス及びラットが含まれる。
【0038】
基準分子(例えば、MAGL選択的阻害剤)の変異型とは、基準分子全体又はそのフラグメントのどちらかと構造及び生物学的活性が実質的に類似する分子をいうことを示している。このように、二つの分子が類似の活性を有するならば、一方の分子の成分又は構造が他の分子に見出されるものと同じでなくても、又はアミノ酸残基の配列が同じでなくても、それらは当該用語が本明細書で使用するところの変異型とみなされる。
【0039】
侵襲性癌又は侵襲性腫瘍とは、迅速に増殖及び拡散する癌又は腫瘍をいう。いかなる起源の癌又は腫瘍も適した条件下では侵襲性となることができる。従って、侵襲性腫瘍には、例えば、侵襲性乳癌、侵襲性前立腺癌、侵襲性脳腫瘍、侵襲性子宮頚部癌、侵襲性結腸癌、侵襲性肺癌、侵襲性胃癌、侵襲性肝癌、侵襲性腎臓癌、侵襲性黒色腫及び侵襲性卵巣癌が含まれる。腫瘍が侵襲性となるためには、血管からはるか遠くにあるその腫瘤の中央の細胞に栄養を与えることができなければならない。これは血管新生によって達成される。突然変異によって、わずかな癌細胞は、血管新生成長因子を産生する能力を獲得できるかもしれない。これらの成長因子は、腫瘍によって近くの組織に放出されるタンパク質であり、そこでは、それらが新生血管を刺激して腫瘍内に成長させる。このことが、腫瘍が腫瘤中で急速に拡大し組織を囲んで浸潤することを可能にしている。それは、癌細胞のために新生血管中へ漏出し及び身体中を循環する経路をも提供するが、そこでそれらは転移を形成しながら他の器官に停止する。
【0040】
III.MAGL選択的阻害剤
本発明は、新規なMAGL阻害剤を提供する。MAGL阻害剤には、MAGLの発現、修飾、制御若しくは活性を妨害する化合物、又はMAGLの正常な生物学的活性(例えば、そのセリンヒドロラーゼ活性)の一以上を下方制御する化合物が含まれる。特に本発明のMAGL阻害剤は、MAGL酵素活性を遮断又は阻害する。上記で言及したように、MAGLは、神経系における、カンナビノイド受容体リガンド、2−AGの加水分解の原因となる主要な酵素である。MAGL阻害剤又はアンタゴニスト化合物は、痛覚消失症等のカンナビノイド依存行動的影響を誘発するのに有用である。MAGL酵素活性を特異的に阻害又は抑制する化合物は、異常な(例えば、抑制又は減少した)カンナビノイドシグナル伝達活性によって引き起こされる又はする障害又は症状を治療するうえで種々の治療的又は予防的応用を有することができる。MAGL酵素活性を阻害するいかなる分子も、カンナビノイド依存行動的影響を誘発する、又は抑制されたカンナビノイドシグナル伝達活性に連結した症状を治療することができるかもしれない。しかしならが、他の脳セリンヒドロラーゼ(例えば、FAAH)をも阻害するMAGLアンタゴニスト化合物は、これらの他の酵素によって媒介される様々な生物学的機能により妨害されるかもしれない。このような非選択的MAGL阻害剤は、抑制されたカンナビノイドシグナル伝達活性に連結した障害(例えば、疼痛消失)を治療することができるが、多くの望まれない副作用を有しがちである。従って、このような治療的な応用においてはMAGL酵素活性を選択的に阻害する分子が好ましい。MAGL酵素活性を特異的に阻害することによって、一方では他の脳セリンヒドロラーゼ(FAAH)に著しい影響を生じずに、不十分な又は抑制された2−AGシグナル伝達活性と関連した又は連結した症候を治療又は回復することができる。
【0041】
従って本発明は、他の脳セリンヒドロラーゼ(例えば、FAAH又はABHD6)の一以上の酵素活性が著しくは悪影響を受けない濃度で、MAGLの酵素活性を実質的に遮断又は阻害する、新規なMAGL−選択的阻害剤(又は「MAGL−特異的阻害剤」)を提供する。幾つかの好ましい実施態様において、MAGL阻害剤は、MAGLによる2−AGの加水分解を遮断する。特に、本発明のMAGL選択的阻害剤又はMAGL選択的アンタゴニストは、FAAH阻害に比較してMAGLの阻害に対し選択性を示す化合物である。他の脳セリンヒドロラーゼ(例えば、FAAH)にまさったMAGLに対する化合物の選択性は、当該化合物の存在下にに関する各酵素の酵素活性を分析することによって測定することができる。明細書の実施例で詳述する通り、選択性は、脳膜プロテオームにおける化合物による基質(例えば、MAGLに対しては2−AG及びFAAHには対してはオレイン酸アミド)の加水分解阻害のIC50値を測定することによって決定することができる。IC50値を測定するのに加えて、FAAHにMAGLに対する阻害剤の選択性は、化合物存在下での酵素的触媒加水分解反応の擬一次速度定数(kobs/[I])を測定することによっても決定することができる。これは、下記の実施例に記載した通り、脳膜プロテオーム中に存在する酵素により生体外で実施することもできる。脳膜プロテオームに存在する加水分解酵素を使用する以外に、酵素活性アッセイは、組換え酵素によっても実施することができる。例えば、他の脳セリンヒドロラーゼと同様にMAGL、FAAHは、本明細書に記載した方法又は当該技術分野で報告された方法に従って組換えで容易に得ることができる(例えば、ブランクら(Blank et al.)、「ケミストリー&バイオロジー(Chem.Biol.)」、2007年、第14巻:p.1347−56)。典型的には、MAGL選択的阻害剤は、FAAHに対するそのIC50値よりも少なくとも20倍、好ましくは少なくとも50、100、250又は500倍低いMAGLに対するIC50値を有すべきである。kobs/[I]により測定するならば、MAGL選択的阻害剤は、通常、FAAHに対するそれよりも少なくとも10倍、好ましくは少なくとも25、50、75、100又は300倍高いMAGLに対する速度定数を有するだろう。
【0042】
本発明のMAGL選択的阻害剤の幾つかは、FAAHに加えて、また一以上の他の脳セリンヒドロラーゼMAGLに対して選択的でもある。例えば、化合物は、ABHD6及び/又はABHD12MAGLに対して選択的であることができる。ABHD6及びABHD12は、マウス脳において2−AGヒドロラーゼ活性を示すことが明らかにされた他の二つの脳酵素であり、明確な細胞内分布を示している(例えば、ブランクら(Blank et al.)、「ケミストリー&バイオロジー(Chem.Biol.)」、2007年、第14巻:p.1347−56を参照)。これらの酵素を阻害しないMAGL選択的アンタゴニストは、内在性カンナビノイドシグナル伝達と関連した現在は未知の機能を研究及び証明するために有用である。
【0043】
本発明においては、種々のMAGL選択的アンタゴニストを使用することができる。本発明によって合成されたこれらMAGL選択的阻害剤の幾つか、下記の実施例に記載する。これらの化合物は、下記に示す式Iの構造を有する。これらの化合物は、本明細書の実施例において開示した合成スキームに従って容易に調製することができる。
【化4】
【0044】
式中、XはN又はCH、R1はH又はOHであり、そしてR2及びR3の各々は、下記の構造の一つを有する。
【化5】
【0045】
例示したMAGL−特異的阻害剤の幾つかは、FAAHにMAGL選択的。実施例において詳述する通り、図1Aに示す化合物WWL152、WWL162、JZL175及びJZL184の全ては、FAAHMAGLに対して選択性を示す。化合物の幾つかは、FAAHに加えて他の脳セリンヒドロラーゼにもまさってMAGLに対して選択的である。これらには、式Iによって包含される化合物JZL184及びJZL175が含まれる。これらの二つの化合物は、特異的にMAGLを阻害し、ABHD6と同様にFAAHに関して全く又は非常に減少した阻害活性しか有しない。
【0046】
これらのMAGL選択的阻害剤のいずれも、下記の実施例に詳述する合成スキームに従って生産することができる。例示したMAGL選択的アンタゴニスト以外に、追加のMAGL選択的阻害剤(例えば、JZL184又はJZL175の機能的な誘導体又はアナログ)は、本明細書に記載した方法又は当該技術分野において記載された方法を使用して容易に同定することができる。本明細書に記載する通り、例示するMAGL選択的阻害剤のアナログ化合物のライブラリーを作成することができる。アナログ化合物は、それからMAGL阻害の特異的活性及び他の脳セリンヒドロラーゼにまさった選択性に関してスクリーニングすることができる。
【0047】
IV.内在性カンナビノイドシグナル伝達の亢進及びそれに付随する症状の治療
本発明は、このような治療を必要とする被験者において、内在性カンナビノイドシグナル伝達(特に2−AGシグナル伝達活性)を刺激若しくは増大する、又はカンナビノイドの加水分解(例えば、2−AGの加水分解)を阻害する方法を提供する。下記の実施例に明らかにする通り、本発明のMAGL特異的阻害剤によるMAGLの阻害は、生体内での2−AG媒介内在性カンナビノイドシグナル伝達を増大するために十分であるが、一方、脳セリンヒドロラーゼの非選択的阻害により別に生じるかもしれない、いかなる意図しない結果も制限している。本発明者らは、幾つかのカンナビノイド依存性の行動的影響がMAGLの選択的阻害に由来こともした。これらの行動的影響(2−AG媒介行動的影響)には、例えば、痛覚消失症、運動性低下症、低温低下、及び反射亢進が含まれる。種々の状況下で、ある被験者においては一定の2−AG媒介行動的影響(例えば、痛覚消失症)を誘発することが望ましいかもしれない。例えば、疼痛を患う被験者は、MAGL選択的阻害によって誘発される痛覚消失から治療的利益を得ることができる。被験者における2−AGシグナル伝達活性を刺激又は増大するために、当該方法は、本明細書で開示するMAGL選択的阻害剤の治療的に有効な量を被験者に投与することを含んでいる。いくつかの関連する方法は、本発明のMAGL選択的阻害剤を被験者に投与することによって、CB1及び/又はCB2依存性の行動的影響を被験者に誘発することに向けられている。いくつかの方法において、被験者は、例えば、2−AGによって媒介される行動的影響、例えば痛覚消失を誘発することができるように、化合物を投与される。
【0048】
本明細書で開示するMAGL選択的阻害剤は、また一般的に異常なカンナビノイドシグナル伝達(特に2−AGシグナル伝達活性)により引き起こされる又は関連する障害、又は2−AGシグナル伝達活性を増大又は刺激することによって有益な効果を得ることができる症状を治療するためにも適している。従って、MAGL選択的阻害剤を用いる場合、本発明は、カンナビノイドシグナル伝達に連結する又は関連する障害又は症状を患う被験者を治療するための方法をさらに提供する。当該方法は、本明細書で開示するMAGL選択的阻害剤の治療的に有効な量を、治療を必要とする被験者に投与することを必要とする。治療されるべき被験者には、異常な(不十分又は抑制された)カンナビノイドシグナル伝達により媒介される又は関連する障害又は症状を患う被験者が含まれる。治療されるべき被験者には、また増大したカンナビノイドシグナル伝達活性から有益な効果を得ることができる症状を有する被験者でもあることができる。いくつかの好ましい実施態様において、当該方法は、2−AGシグナル伝達と関連する症状を患う被験者において、2−AG媒介シグナル伝達活性を刺激することに向けられる。これらの障害及び症状の例には、疼痛、炎症、、抑鬱、心血管障害、代謝障害、及び卒中が含まれる。本発明の方法に適した多くの炎症性障害が存在する。これらの障害は、例えば、喘息、自己免疫疾患、慢性炎症、慢性前立腺炎、糸球体腎炎、過敏症、炎症性腸疾患、骨盤内炎症性疾患、再灌流障害、リウマチ性関節炎、移植拒絶及び脈管炎等、当該技術分野において周知である。卒中に加えて、本発明の治療方法に適した心血管障害には、また動脈瘤、狭心症、アテローム性動脈硬化症、脳血管疾患、うっ血性心不全、冠動脈疾患、及び心筋梗塞(心臓発作)も含まれる。同様に、増大する内在性カンナビノイドシグナル伝達(例えば、2−AG媒介シグナル伝達活性)から利益を売ることのできる代謝障害は当該技術分野で周知である。このような障害の例には、フェニルケトン尿症、アルカプトン尿症、地中海貧血症、ポルフィリン症、テイ・サックス病、ハーラー症候群、ゴーシェ病、ガラクトース血症、クッシング症候群、真性糖尿病、甲状腺機能亢進症、及び甲状腺機能低下症が含まれる。
【0049】
本発明のMAGL選択的阻害剤は、又そのカンナビノイドの治療効果が報告されている他の症状を治療するためにも利用することができる。このような症状又は障害の例には、緑内障、多発性硬化症及び抑制された食欲によって引き起こされる消耗疾患が含まれる。消耗(wasting)とは、筋肉及び脂肪組織を衰弱させる衰弱性の疾患(例えば、細菌又はウイルス感染)又は症状の経過をいう。カンナビノイドがこれらの疾患又は障害に対する一定の治療的効果を提供することができると報告されている。例えば、ニューウェルら(Newell et al.)、「トランスアクションズ・オブ・ザ・オフタルモロジカル・ソサイアティ・オブ・ザ・ユナイテッド・キングダム(Trans.Ophthalmol.Soc.UK)」、1979年、第99巻:p.269−71;ブッシュヴァルトら(Wushwald et al.)、「ディー・ファルマジー(Pharmazie.)」、2002年、第57巻:p.108−14;クロックスフォードら(Croxford et al.)、「ドラッグス・オブ・トゥデイ(Drugs Today)」、2004年、第40巻:p.663−76;ウィラン(Whelan)、「ドラッグ・ディスカバリー・トゥデイ(Drug Discov.Today)」、2002年、第7巻:p.745−6;及びコスチニユックら(Costiniuk et al.)、「ザ・カナディアン・ジャーナル・オブ・ガストロエンテロロジー(Can.J.Gastroenterol.)」、2008年、第22巻:p.376−80を参照。本発明のMAGL選択的阻害剤は、2−AGの加水分解を阻害することによって治療効果を促進することができる。本発明のMAGL選択的阻害剤は、アルツハイマー病を患う被験者に治療的利益を提供するためにさらに用いることができる。アルツハイマー被験者の脳に増加したアラキドン酸が存在することが報告されており、それはアルツハイマー病上の発達と進行におけるアラキドン酸の役割を示唆している。例えば、ロスら(Ross et al.)、「ジャーナル・オブ・ニューロケミストリー(J.Neurochem.)」、1998年、第70巻、p.786−93;チャールイモイックら(Charlimoniuk et al.)、「ニューロケミストリー・インターナショナル(Neruochem.Int.)」、2006年、第48巻:p.1−8;及びラポポートプロスタグランジン・ロイコット・エッセント・(Prostaglandins Leukot Essent Fatty Acids)」2008年10月28日、参照。本発明のMAGL阻害剤によりMAGL酵素活性を阻害することによって、2−AGの加水分解及び結果として生じるアラキドン酸産物を抑制又は減少することができる。
【0050】
典型的には、本発明の治療方法には、本発明のMAGL特異的阻害剤を含有する医薬組成物を治療の必要な被験者に投与することが含まれる。MAGL特異的阻害剤は、単独で又は障害に適した他の既知の治療薬と併用して使用することができる。例えば、疼痛を有する被験者には、疼痛を緩和する他の鎮痛剤を追加的に投与することができる。このような既知の鎮痛剤には、モルヒネ及びモクソニジンが含まれる(米国特許第6,117,879号を参照)。幾つかの他の方法において、本発明のMAGL特異的阻害剤は、被験者の内在性カンナビノイドシグナル伝達活性を亢進するためにFAAH選択的阻害剤と併用して使用することもできる。MAGL阻害剤単独又はFAAH阻害単独と比較して、MAGL選択的阻害剤及びFAAH選択阻害剤両方の投与は、望むカンナビノイド依存性の行動的影響、例えば疼痛の減少又は炎症、、及び抑鬱に付随する症候の回復を被験者に強力により良く誘発することができた。FAAH選択的阻害剤は当該技術分野で既知である(例えば、フェグリーら(Fegley et al.))、「ジャーナル・オブ・ファルマコロジー・アンド・エクスペリメンタル・セラピューティクス(J.Pharmacol.Exp.Ther.)」、2005年、第313巻:p.352−8)。例えば、FAAH選択的阻害剤URB597は、ケイマンケミカル社(Cayman Chemicals)(アナーバー、ミシガン州)等から容易に市販品を入手することができる。
【0051】
例として、本発明のMAGL選択的阻害剤は、疼痛を患う被験者の症候を治療又は緩和するために使用することができる。疼痛は、多くの医的障害に存する。例えば、炎症は疼痛を誘発することができる。炎症性疾患の例には、変形性関節症、大腸炎、心臓炎、皮膚炎、筋炎、神経炎、リウマチ性関節炎及び狼瘡等の膠原病性脈管疾患が含まれる。これらの症状のどれかを有する被験者は、しばしば亢進した痛覚を体験する。過度の疼痛を引き起こすかもしれない他の病状又は医療行為には、外傷、手術、切断、膿瘍、灼熱痛、脱髄疾患、三叉神経痛、慢性アルコール中毒、卒中、視床痛症候群、糖尿病、癌ウイルス感染、及び化学療法が含まれる。
【0052】
一般的に、治療は、被験者が薬理学的及び/又は生理学的な効果を獲得することを可能にする。当該効果は、カンナビノイドシグナル伝達と関連する症状若しくは障害又はその前兆又はその症候から被験者を完全に又は部分的に防止するかとの点からは、予防的であってもよい。それはまた、カンナビノイドシグナル伝達及び/又は障害に有害作用(例えば、疼痛)と関連する症状又は疾患に対する部分的か又は完全な治癒の点からは治療的であることができる。被験者がである場合、人が知覚する疼痛のレベルは、被験者当人に疼痛を述べること又は疼痛を他の痛みの経験と比較することを頼むことによって評価することができる。あるいは、疼痛のレベルは、ストレス関連因子の放出又は抹消神経系若しくはCNS(中枢神経系)中の疼痛伝達神経の活動等の、被験者の疼痛に対する身体反応を測定することによって較正することができる。又、疼痛レベルは、人が疼痛がないと報告するのに要求される、又は被験者が疼痛の症候を示すことを停止するのに要求される、十分に特性化された鎮痛剤の量を測定することによっても較正することができる。
【0053】
本発明の治療法に適した被験者は、の被験者、以外の哺乳動物及びMAGLを発現する他の動物であることができる。被験者は、現在疼痛を引き起こしておりそして疼痛を引き起こし続けそうな進行中の症状を有していてもよい。また被験者は、通常痛みの結果を伴う術式又は事象を耐えてきてもよいし又は耐えることになってもよい。例えば、被験者は、糖尿病性神経因性疼痛又は膠原病性脈管疾患等の慢性の疼痛症状を有していてもよい。被験者は、また炎症、神経損傷、又は毒暴露(化学療法薬への暴露を含む)を有していてもよい。治療又は治療介入は、被験者の疼痛を減少又は和らげ、その結果、被験者が知覚する疼痛レベルが、治療しなかったとしたならば被験者が知覚したであろう疼痛レベルと比較して減少することを意図している。
【0054】
幾つかの実施態様において、神経性疼痛を有する被験者の治療を意図している。これらの被験者は、神経根障害、単神経障害、多発性単神経障害、多発性神経障害又は神経叢障害に分類される神経障害を有していてもよい。これらの種類の疾患は、限定されないが、外傷、卒中、脱髄疾患、膿瘍、手術、切断、神経の炎症性疾患、灼熱痛、糖尿病、膠原病性脈管疾患、三叉神経痛、リウマチ性関節炎、毒、癌(直接的又は間接的に(例えば、腫瘍随伴性)神経損傷を引き起こすことのできるもの)、慢性アルコール中毒、ヘルペス感染症、エイズ、及び化学療法を含む、さまざまな神経損傷症状又は術式によって引き起こされていてもよい。疼痛を引き起こす神経損傷は、末梢又はCNS神経の中にあってもよい。
【0055】
本発明の幾つかの実施態様において、痛覚過敏の治療又は緩和を必要とする被験者には、MAGLの阻害剤を一以上の追加の疼痛緩和剤と併用した組成物が投与される。これは、個々の鎮痛剤はしばしば疼痛緩和効果を部分的にのみ提供するからであるが、というのもそれは多くのなかのまさに一つの疼痛伝達経路を妨害するからである。しかしながら、疾患又は病状と関連する疼痛には、しばしば多様な受容器及び異なったシグナル伝達経路が関与している。それ故に、これらの状況下で侵害受容器を緩和するために一以上の疼痛軽減剤が必要であるかもしれない。幾つかの他の応用において、MAGL阻害剤は疼痛知覚経路中の異なる箇所に作用する鎮痛剤と併用して投与することができる。例えば、非ステロイド性抗炎症薬(NSAID)(例えば、アセトアミノフェン、イブブロフェン、及びインドメタシン)等の一クラスの鎮痛剤は、侵害受容器によって検出される刺激の化学伝達物質を下方制御する。オピオイド等の他のクラスの薬剤は、CNSにおける侵害受容情報の処理を変化させる。また、抗痙攣薬及び抗鬱薬を含む局所麻酔薬等の他の鎮痛剤が含まれていてもよい。MAGL阻害剤に加え、一以上のクラスの薬剤を投与することにより、より効果的な疼痛緩和を提供することができる。
【0056】
V.新規MAGL阻害剤のスクリーニング
本明細書に記載する特異的MAGL阻害剤に加えて、本発明は、また改良された特性を有する新規MAGL阻害剤のスクリーニング方法をも提供する。薬剤発見過程における重要な段階は、化学アナログリード化学鋳型の選択である。一定の分子標的のためのリード化学鋳型を同定する過程には、典型的には、機能アッセイにおける多数の化合物(しばしば100、000以上)のスクリーニング、活性を確認するための二次アッセイの試験用のある任意の活性閾値に基づく亜集合の選抜、並びに次に残った活性化合物の化学的合成の適合性の評価、が含まれる。本明細書に記載する新規MAGL阻害剤、例えば、式Iの化合物は、改良された生物学的又は薬学的特性を有する関連化合物を探索するためのリード化合物を提供する。例えば、これらMAGL阻害剤のアナログ又は誘導体は、MAGLに対するより高い親和性を有する又は皮膚により浸透性である化合物を同定することによって選別することができる。このような改良された特性を有する化合物は、種々の薬学的応用のために適している。
【0057】
本発明のスクリーニング方法には、典型的には最初に本明細書で例示するMAGL選択的阻害剤(例えば、JZL184又はJZL175)のアナログ、誘導体又は変形型の合成が含まれる。スクリーニングのために一定のMAGL阻害剤の構造的アナログのライブラリーを作成すると、次に、当該アナログ又は変異型が由来するMAGL阻害剤の当該特性と比較して改良された生物学的特性を有するアナログ又は誘導体を同定するために機能アッセイを実施する。例えば、現有するMAGL選択的阻害剤のアナログ化合物は、その選択性を維持しつつ、MAGLに対する高められた阻害活性(例えば、減少したIC50)を求めて選別することができる。あるいは、アナログは、FAAH及び/又はABHD6にMAGLに対する亢進した選択性を求めて選別することができる。それらは、また追加の脳セリンヒドロラーゼ(例えば、ABHD12)にMAGLに対する選択性を求めて選別することもできる。幾つかの他の実施態様において、アナログは、MAGLポリペプチドに対する亢進した結合親和性を求めて選別することができる。さらに、アナログ化合物のライブラリーは、例えば、より良い皮膚浸透性等の薬学的な性質又は薬物動態学的な性質を有する化合物を同定することにより評価することができる。
【0058】
さらに幾つかの実施態様において、本発明のスクリーニング法は、FAAH及びMAGLの両方を選択的に阻害することのできる治療薬を同定することにも向けられる。AEA及び2−AGの両方によって制御される行動的過程(例えば、痛覚)は、MAGL−FAAH二重阻害剤によってより強力に影響を受けることができるであろう。実施例で証明する通り、MAGL選択的阻害剤(例えば、JZL184)は、高濃度において他の脳セリンヒドロラーゼに著しい影響を与えずにFAAHを阻害することもできる。それ故に、これらの化合物は、MAGL及びFAAHの二重阻害剤の開発のリード化合物の骨格構造として役立つことができる。
【0059】
本明細書で例示するMAGL選択的阻害剤の化学的骨格に基づくアナログ又は誘導体を合成するためには、当該技術分野で周知の有機化学方法がだけである。例えば、既知化合物の化学アナログのコンビナトリアルライブラリーは、段階的な方法で合成することができる。化合物の大きなコンビナトリアルライブラリーは、WO第95/12608号、WO第93/06121号、WO第94/08051号、WO第95/35503号及びWO第95/30642号に記載された、エンコードされた合成ライブラリー(ESL)法によって構築することができる。種々のアナログの他の合成方法は、例えば、オーバーマン(Overman)著、「有機反応(Organic Reactions)」、第1−62巻、ウィリー・インターサイエンス(Wiley−Interscience)、2003年;ブルームら(Broom et al.)、「フェデラル・プロキュアメント(FedProc.)」、1986年、第45巻:p.2779−83;ベン−メナームら(Ben−Menahem et al.)、「リーセント・プログレス・イン・ホルモン・リサーチ(RecentProg.Horm.Res.)」、1999年、第54巻:p.271−88;シュラムら(Schramm et al.)、「アニュアル・レビュー・オブ・バイオケミストリー(Annu.Rev.Biochem)」、1998年、第67巻:p.693−720;ボーリンら(Bolin et al.)、「バイオポリマーズ(Biopolymers)」、1995年、第37巻:p.57−66;カールテンら(Karten et al.)、「エンドクライン・レビューズ(EndocrRev.)」、1986年、第7巻:p.44−66;ホら(Ho et al.)著、「有機合成戦略(Tactics of Organic Synthsis)」、ウィリー・インターサイエンス(Wiley−Interscience)、1994年;及びシャイトら(Scheit et al)著、「ヌクレオチド・アナログ:合成及び生物学的機能(Nucleotide Analogs:Synthesis and Biological Function)」、ジョンウィリー&サンズ(John Wiley&Sons)、1980年、に記載されている。
【0060】
のMAGL選択的阻害剤のアナログ化合物のライブラリー亢進した生物学的活性又は薬学的特性は、当該技術分野で周知の機能アッセイによっても容易に選別することができる。例えば、MAGL及び他の脳セリンヒドロラーゼの化合物による阻害は、例えば、脳膜プロテオーム中に存在する酵素によって同族基質の加水分解を定量すること等、本明細書で開示する又は当該技術分野で周知の酵素活性アッセイにより試験することができる。又は他の種由来の他の脳セリンヒドロラーゼと同様にMAGL、FAAHも当該技術分野で周知である。その塩基配列のクローニング並びにその酵素的及び他の生物学的活性のキャラクタリゼーションは、当該技術分野で全て報告されている。例えば、ブランクマンら(Blankman et al.)、「ケミストリー&バイオロジー(Chem.Biol)」、2007年、第14巻:p.1347−56;ソンマ・デルペロら(Somma−Delpero et al.)、「ザ・バイオケミカル・ジャーナル(Biochem J.)」、1995年、第312巻:p.519−25;ディンら(Dinh et al.)、「プロシーディングス・オブ・ザ・ナショナル・アカデミー・オブ・サイエンシズ・オブ・ザ・ユナイテッド・ステイツ・オブ・アメリカ(Proc.Natl.Acad.Sci.USA)、2002年、第99巻、p.10819−9;ディンら(Dinh et al.)、「モレキュラー・ファルマコロジー(Mol.Pharmacol.)」、2004年、第66巻:p.1260−4;ムッソリーら(Muccioli et al.)、「ザ・ジャーナル・オブ・ニューロサイエンス(J.Neurosci.)」、2007年、第27巻:p.2883−9;ザンら(Giang et al.)、「プロシーディングス・オブ・ザ・ナショナル・アカデミー・オブ・サイエンシズ・オブ・ザ・ユナイテッド・ステイツ・オブ・アメリカ(Proc.Natl.Acad.Sci.USA)、1997年、第94巻、p.2238−2242;ワンら(Wan et al.)、「ゲノミクス(Genomics)」、1998年、第54巻:p.408−414;パトリセリら(Patricelli et al.)、「バイオオーガニック&メディシナル・ケミストリー・レターズ(Bioorg.Med.Chem.Lett.)」、1998年、第8巻:p.613−618;及びゴーパルディ(Goparaju et al.)、「バイオヒミカ・エト・バイオフィジカ・アクタ(Biochim.Biophys.Acta)」、1999年、第1441巻:p.77−84、を参照。
【0061】
化合物の他の生物学的又は生化学的活性の改良(例えば、MAGLに対する亢進した結合親和性)は、当該技術分野で方法により試験することができる。一般的な概要については、例えば、サンブルックら(Sambrook et al.)著、「モレキュラー・クローニング:実験マニュアル(Molecular Cloning:A Laboratory Manual)」、コールドスプリング・ハーバー・プレス(Cold Spring Harbor Press)、ニューヨーク(N.Y.)、第3版(2000年);アウスベルら(Ausubel et al.)、「分子生物学カレントプロトコル(Current Protocols in Molecular Biology)」、ジョンウィリー&サンズ、インク(John Wiley&Sons、Inc.)、ニューヨーク(New York)(1999年);及びバーガー及びキンメル(Bergr and Kimmel)著、「酵素学における方法(Methods In Enzymology)」、サンディエゴ(San Dego)、アカデミックプレス、インク(Academic Prss,Inc)を参照。また本発明のスクリーニング法において用いることのできる追加の生化学的又は薬学的アッセイ法も当該技術分野で周知であり日常的に実施されている。例えば、MAGL選択的阻害剤のアナログ化合物の皮膚浸透特性は、例えば、レミントン著「薬学の科学と実践(The Science and Practice of Pharmacy)」、ジェンナロ(Gennaro)編、リッピンコット・ウィリアムズ&ウィルキンス(Lippincott Williamas&Wilkins)、(第20版、2003年)、等に記載された方法を使用して分析することができる。
【0062】
VI.MAGL標的化による癌治療及び腫瘍増殖阻害
本発明は、腫瘍細胞増殖阻害ための方法及び組成物を提供する。典型的には、これら本発明の治療応用には、モノアシルグリセロールリパーゼ(MAGL)と特異的に拮抗又は阻害する化合物が用いられる。腫瘍細胞の増殖は、当該腫瘍細胞をMAGL拮抗化合物の治療的に有効な量と接触させることによって阻害、抑制又は遅延される。幾つかの実施態様において、腫瘍細胞は、例えば癌発症の危険を有する又は危険にある被験者に存在する。これらの治療応用の幾つかの好ましい実施態様は、侵襲性の腫瘍又は腫瘍細胞の増殖を阻害することに向けられる。
【0063】
幾つかの関連する実施態様において、本発明は、腫瘍細胞の増殖を阻害することによって被験者体内の癌を治療するための方法を提供する。当該方法は、また被験者体内の腫瘍形成を防止するためにも有用である。典型的には、当該方法には、治療を必要とする被験者に本明細書で開示するMAGL拮抗化合物を含有する医薬組成物を投与することが含まれる。MAGL拮抗化合物は、単独で、又は被験者体内で相乗効果を提供するために、他の既知の抗癌剤と併用して使用することができる。
【0064】
本発明で用いるMAGL拮抗化合物には、MAGLの細胞レベルを下方制御する又は生物学的活性(例えば、リパーゼ活性)を阻害するいかなる薬剤も含まれる。適したMAGL拮抗化合物には、以下に記載するスクリーニングに従って同定することができる新規薬剤、例えば追加の小分子化合物又は抗体(例えば、アンタゴニスト抗体)等も含まれる。幾つかの実施態様において、腫瘍増殖を阻害する又は癌を治療するために用いられるMAGL拮抗化合物は、特異的にMAGL発現を阻害する又はその細胞レベルを下方制御する薬剤(例えば、阻害ポリヌクレオチド)である。例えば、MAGLを標的にする阻害ポリヌクレオチドには、低分子干渉RNA(siRNA)、マイクロRNA(miRNA)、低分子ヘアピン型RNA(shRNA)、アンチセンス核酸、又は相補DNAが含まれる。これらの核酸薬剤を標的遺伝子の発現を特異的に停止させるために使用することは、周知であり当該技術分野において。MAGLを特異的に標的とするこのような核酸薬剤は、当該技術分野で周知の方法を使用して調製することができる。例として、MAGL遺伝子を標的とするshRNA及びsiRNAは、下記の実施例で明らかなとおり、MAGL発現レベルを下方制御するために使用することができる。
【0065】
二本鎖RNAによる内在性遺伝子の機能及び発現との干渉は、例えば、ファイアーら(Fire et al.)、「ネイチャー(Nature)」、1998年、第391巻:p.806−811、に記載された線虫;例えば、ケンネーデルら(Kennerdell et al.)、「セル(Cell)」、1998年、第95巻:p.1017−1026、に記載されたショウジョウバエ;及び例えば、ウィアニら(Wianni et al.)、「ネイチャー・セル・バイオロジー(Nat.CellBiol.)」、2000年、第2巻:p.70−75、に記載されたマウス胎仔等の種々の生物で示されている。このような二本鎖RNAは、鋳型の両方向から読み取る一本鎖RNAの生体外転写並びにセンス及びアンチセンスRNA鎖の生体外アニーリングによって合成することができる。二本鎖RNAは、またMAGL遺伝子が逆向き反復配列によって引き離された反対方向にクローンされたcDNAベクター構築物から合成することもできる。細胞へ遺伝子導入された後、当該RNAは転写され相補鎖と再アニールされる。MAGL遺伝子を標的とする二本鎖RNAは、適当な構築物の形質移入によって細胞(例えば、腫瘍細胞)に導入することができる。例として、MAGL発現を下方制御する特異的なsiRNA及びshRNAのアンチセンス及びセンス鎖の配列を本明細書で開示する。MAGLを標的とする他の型の阻害ポリヌクレオチドと同様に追加のsiRNA又はshRNA分子は、当該技術分野で周知に標準的技術に従って容易に作成することができる。
【0066】
幾つかの他の実施態様において、本発明の治療応用は、MAGLの生物学的活性を阻害するMAGL拮抗化合物を用いる。これらには、MAGLの酵素活性と拮抗する本明細書に開示するMAGL選択的阻害剤が含まれる。適したMAGL拮抗化合物には、またMAGLポリペプチドに特異的に結合しそのリパーゼ活性と拮抗するアンタゴニスト抗体も含まれる。この点については、これらの中でモノクローナル抗体薬剤が最も好ましい。抗MAGLアンタゴニスト抗体は、例えば、「モノクローナル抗体−生産、工学及び臨床応用(Monoclonal Antibodies‐‐Production Engineerign And Clinical Applications)」、リッターら(Ritter et al.)編、ケンブリッジ大学プレス(Cambridge University Press)、ケンブリッジ(Cambridge)、英国(UK)、1995年;及びハーロー及びレイン(Harlow and Lane)著、「抗体、実験マニュアル(Antibodies、A Laboratory Mannual)」、コールドスプリング・ハーバー・プレス(Cold Spring Harbor Press)、第3版、2000年、等の当該技術分野で周知であり日常的に実施されている方法を使用して作成することができる。癌治療用の放射標識モノクローナル抗体は特に周知であり例えば、「放射標識抗体による癌治療(Cancer Therapy With Radiolabelled Antibodies)」、DMゴールデンベルク(D.M.Goldenberg)編、CRCプレス(CRC Press)、ボカラトン(Boca Raton)、フロリダ(Fla)、1995年、に記載されている。
【0067】
MAGL発現又はその酵素活性を阻害又は下方制御する化合物は、他の治療法と併用して使用することができる。例えば、手術及び放射線療法受けている被験者には、本発明の医薬組成物を投与することもできる。加えて、また化学療法、ホルモン療養及び凍結療法は、癌を患う被験者を治療するために、本発明の治療応用と併用することもできる。MAGL拮抗化合物は、これらの疾患の治療又は防止するための他の治療化合物の投与と共に、腫瘍増殖を防止するため又は癌を治療するために被験者に使用することもできる。MAGL拮抗化合物を他の抗癌剤と共に投与する場合、両者はどちらの順番でも又は同時に投与することができる。これらの治療化合物は、化学療法剤、焼灼術又は他の治療用ホルモン、抗腫瘍薬、癌に有用なモノクローナル抗体及び血管新生阻害剤であっても良い。
【0068】
当該技術分野で知られた多数の抗癌剤があり、例えば、「癌治療学:試験薬及び臨床薬(Cancer Therapeutics:Experimental and Clinical Agents)」、タイヒャー(Teicher)(編)、ヒューマナ・プレス(Humana Press)(第1版、1997年);及び「グッドマン及びギルマンズの薬物療法学の薬理学的基礎(Goodman and Gilman‘s The Pharmacological Basis of Therapeutics)」、ハルドマンら(Hardman et al.)(編)、マッグローヒル・プロフェッショナル(McGraw−Hill Professional)(第10版、2001年)、に記載されている通りである。適した抗癌剤の例には、5−フルオロウラシル、硫酸ビンブラスチン、リン酸エストラムスチン、スラミン及びストロンチウム−89が含まれる。適した化学療法薬の例には、アスパラギナーゼ、硫酸ブレオマイシン、シスプラチン、シタラビン、リン酸フルダラビン、マイトマイシン、及びストレプトゾシンが含まれる。本発明と併用して使用されるかもしれないホルモンは、ジエチルスチルベストロール(DES)、ロイプロリド、フルタミド、酢酸シプロテロン、ケトコナゾール及びアミノグルテチミドである。
【0069】
本発明の方法による治療に適した被験者は、種々の型の癌を患っている被験者、又は癌発症の危険にある又はを有する被験者である。本発明の方法は、多数の腫瘍細胞の増殖を阻害するために用いることができる。治療を施すことのできる腫瘍又は癌の例には、肺、皮膚、乳房、脳、胃腸、尿生殖路(例えば、腎臓、膀胱及び尿道、前立腺、睾丸)、血管、神経系、骨及び肝臓由来の腫瘍が含まれる。それらは、充実性腫瘍、白血病及び転移性腫瘍を包含する。本発明に適した充実性腫瘍には、例えば、肉腫、黒色腫、癌腫、又は他の充実性腫瘍癌が含まれる。
【0070】
肉腫は、胚性結合組織のような物質から構成される腫瘍を包含し、一般的に原線維又は均質な物質中に包埋したぎっしりと詰まった細胞から構成される。肉腫には、限定されるものではないが、軟骨肉腫、線維肉腫、リンパ肉腫、黒色肉腫、粘液肉腫、骨肉腫、アベメシー肉腫、脂肪性肉腫、脂肪肉腫、胞巣状軟部肉腫、エナメル上皮肉腫、ブドウ状肉腫、緑色腫、絨毛癌、胎児性肉腫、ウィルム肉腫、子宮内膜肉腫、間質性肉腫、ユーイング肉腫、筋膜肉腫、線維芽肉腫、巨細胞肉腫、顆粒球性肉腫、ホジキン肉腫、特発性多発性色素性出血性肉腫、B細胞免疫芽球肉腫、リンパ腫、T細胞免疫芽球肉腫、イエンセン肉腫、カポジ肉腫、クッパー星細胞肉腫、血管肉腫、白血肉腫、悪性間葉肉腫、傍骨性骨肉腫、細網肉腫、ラウス肉腫、漿液嚢胞性肉腫、滑膜肉腫、及び血管拡張型肉腫が含まれる。
【0071】
黒色腫とは、皮膚及び他の器官のメラニン細胞系から生じる腫瘍をいう。黒色腫には、例えば、末端黒子型黒色腫、メラニン欠乏性黒色腫、良性若年性黒色腫、クラウドマン黒色腫、S91黒色腫、ハーディング・パッセー黒色腫、若年性黒色腫、悪性黒子型黒色腫、悪性黒色腫、小結節性黒色腫、爪下黒子性黒色腫、及び表在拡大型黒色腫が含まれる。
【0072】
癌腫とは、周囲の組織に浸潤し転移を起こす、上皮細胞から構成される悪性の新生物をいう。代表的な癌腫には、例えば、上皮癌、結腸直腸癌、胃癌、口腔癌、膵癌、卵巣癌、又は腎細胞癌がさらに含まれる。代表的な癌腫には、例えば、腺房癌、小葉癌、腺嚢癌腫、腺様嚢胞癌、腺癌、副腎皮質癌、肺胞癌、肺胞上皮癌、基底細胞癌(carcinoma basocellulare)、基底細胞癌(basaloid carcinoma)、基底有棘細胞癌、気管支肺胞上皮癌、細気管支癌、気管支原性肺癌、セレブリフォルム癌、胆管細胞癌、絨毛膜癌腫、粘液性癌腫、面疱癌、子宮体癌、篩状癌、鎧状癌、皮膚癌、円柱癌、円柱細胞癌、腺管癌、デュラム癌腫、胎児性癌、髄癌、類表皮癌腫、腺様上皮癌、外向方発育癌、潰瘍癌、癌腫血管腫、膠様癌、粘液性癌腫、巨細胞癌腫、巨細胞癌、腺癌、顆粒膜細胞腫、毛母癌、血性癌腫、肝細胞癌、ハースル細胞癌、卵黄嚢腫瘍、副腎癌、小児胎児性癌、上皮内癌(carcinoma in situ)、表皮内癌、上皮内癌(intraepithelial carcinoma)、
Krompecher‘s癌、クルチツキー細胞癌、大細胞癌、レンズ状癌(lenticular carcinoma)、レンズ状癌(carcinoma lenticulare)、脂肪腫、リンパ上皮性癌、髄様癌(carcinoma medullare)、髄様癌(medllary carinoma)、黒色癌、軟部癌腫、粘液性癌(mucinous carcinoma)、粘液性癌(carcinoma muciparum)、粘液細胞性癌、粘表皮癌、粘液癌(carinoba mucosum)、粘液性癌(mucous carcinoma)、粘液腫状癌、鼻咽頭癌腫、燕麦細胞癌、類骨癌(carcinoma ossificans)、類骨癌腫(osteoid carcinoma)、乳頭癌、門脈周囲癌、前浸潤癌、有棘細胞癌、のリ状癌、腎細胞癌、予備細胞癌、肉腫状癌、シュナイダー癌腫、硬性癌、陰嚢癌、印環細胞癌、単純癌、小細胞癌、ソラノイド癌腫、回転楕円面細胞癌腫、紡錘細胞癌、海綿様癌、扁平上皮癌(squamous carcinoma)、扁平上皮細胞癌(squamous cell carcinoma)、ストリング癌、毛血管拡張性癌(cacinoma telangiectaticum)、毛細血管拡張症様癌(carcinoma telangiectodes)、移行上皮癌、結節癌(arcinoma tuberosum)、結節性癌(tuberous carcinoma)、いぼ状癌、及び絨毛癌が含まれる。
【0073】
白血病は、造血器官の進行性の悪性疾患を包含し、血管及び骨髄中の白血球及びその前駆体の歪んだ増殖及び生育によって一般的に特徴づけられる。白血病は、臨床的には一般的に(1)疾患の継続期間及び特性‐‐急性又は慢性;(2)関与する細胞の型;骨髄系(骨髄性)、リンパ系(リンパ向性の)、又は単球系;並びに(3)血液中の異常細胞数の増加又は非増加‐白血病性又は無白血病性(亜白血病性)、に基づいて分類される。白血病には、例えば、急性非リンパ球性白血病、慢性リンパ球性白血病、急性顆粒球白血病、慢性顆粒球白血病、急性前骨髄球白血病、急性T細胞白血病、非白血病性白血病、白血球血症白血病、好塩基球性白血病、芽球細胞性白血病、ウシ白血病、慢性骨髄性白血病、皮膚白血病、胎児性白血病、好酸球性白血病、グロス白血病、有毛状細胞性白血病、肝細胞性白血病(hemoblastic leukemia)、血球芽細胞性白血病、組織球白血病、肝細胞白血病(stem cell leukemia)、急性単球性白血病、白血球減少性白血病、リンパ性白血病(lymphatic leukemia)、リンパ芽球性白血病、リンパ球性白血病、リンパ性白血病(lymphogenous leukemia)、リンパ性白血病(lymphoid leuemia)、リンパ肉腫細胞性白血病、肥満細胞白血病、巨核球性白血病、小骨髄芽球性白血病、単球性白血病、骨髄芽球性白血病、骨髄性白血病、骨髄顆粒球性白血病、骨髄単球性白血病、ネーゲリ白血病、形質細胞白血病(plasma cell leukemia)、形質細胞性白血病(plasmacytic leukemia)、前骨髄球性白血病、リーダー細胞白血病、シリング白血病、肝細胞白血病(stem cell leukemia)、亜白血性白血病、及び未分化細胞白血病が含まれる。
【0074】
本発明の方法により治療することのできる他の癌又は腫瘍には、例えば、ホジキン病、非ホジキンリンパ腫、多発性骨髄腫、神経芽腫、乳癌、卵巣癌、肺癌、横紋筋肉腫、原発性血小板症、原発性マクログロブリン血症、小細胞肺腫瘍、原発性脳腫瘍、胃癌、結腸癌、悪性膵島細胞腺腫、悪性類癌腫、膀胱癌、前癌皮膚障害、精巣癌、リンパ腫、甲状腺癌、神経芽腫、食道癌、泌尿生殖器癌、悪性高カルシウム血症、子宮頚部癌、子宮内膜癌、副腎皮質癌、及び前立腺癌が含まれる。
【0075】
VII.医薬組成物及び投与
本発明は、内在性カンナビノイドシグナル伝達によって媒介される又は関連する症状又は障害を治療するための薬剤の製造においてMAGLアンタゴニスト化合物(例えば、MAGL選択的阻害剤)の使用を提供する。幾つかの関連する応用において、MAGL拮抗化合物は、腫瘍細胞増殖阻害及び癌治療のために使用される。これらの治療応用において、治療を必要とする被験者には、MAGLアンタゴニスト化合物を単独で投与することができる。しかしながら、MAGLアンタゴニスト化合物又はその薬学的に許容可能な塩を含む医薬組成物の投与の方がより好ましい。医薬組成物に用いることのできるMAGLアンタゴニスト化合物の例には、小分子有機化合物であるJZL184、JZL175、WWL152及びWWL162と同様に下記の実施例に記載する阻害ポリヌクレオチドが含まれる。本発明のスクリーニング方法に従って同定するこのできる新規のMAGL阻害剤もまた使用することができる。本発明は、例えば、キット等の医薬品の組み合せも提供する。このような医薬品の組み合せは、本明細書で開示するMAGLアンタゴニスト化合物である活性薬剤と同様に、どのような形態又は組成物であっても、少なくとも一つを併用組成物と、薬剤の投与説明書を含むことができる。
【0076】
MAGL選択的阻害剤を含む医薬組成物は種々の形態で調製することができる。適した固形又は液体製剤形態は、例えば、顆粒、粉末、錠剤、被覆錠剤、(マイクロ)カプセル、座薬、シロップ剤、乳剤、懸濁剤、クリーム剤、エアロゾル剤、滴剤又はアンプル形態の注射液であり、並びに活性化合物の遅延性放出に関する製剤である。それらは、当該技術分野で周知の標準的なプロトコル、例えば、レミントン(Remington)著「薬学の科学と実践(The Science and Practice of Pharmacy)」、ジェンナロ(Gennaro)編、リッピンコット・ウィリアムズ&ウィルキンス(Lippincott Williams & Wilkins)(第20版、2003年)、に従って調製することができる。医薬組成物は、典型的には腫瘍増殖を阻害するために、又は2−AGシグナル伝達に関連する又は媒介される障害又は症状の症候を減じる又は回復するために十分なMAGLアンタゴニスト化合物の有効な量を含んでいる。MAGLアンタゴニスト化合物に加えて、医薬組成物は、組成物を高める若しくは安定させる、又は組成物の調製を促進する一定の薬学的に許容可能な担体を含むこともできる。例えば、MAGLアンタゴニスト化合物は、安定性又は薬理学的特性を高めるために、その投与前に卵白アルブミン又は血清アルブミン等の担体タンパク質と複合体を形成することができる。医薬組成物の種々の形態は、また崩壊剤、結合剤、被覆剤、膨潤剤、滑沢剤、着香料、甘味剤及び精製水等の通常当該技術分野で日常的に使用される不活性な希釈剤を含むエリキシル剤等の賦形剤及び添加物及び/又は補助剤を含むこともできる。
【0077】
薬学的に許容可能な担体は、部分的には投与される特定の成分と同様に組成物の特定の投与方法によって決定される。それらは、他の成分と適合しかつ被験者に有害でないという意味において、薬学的及び生理学的の両方においても許容可能でなければならない。担体は、例えば経口、舌下、直腸、経鼻、静脈内、非経口等の投与に望ましい製剤形態に依存して様々な形態をとってもよい。例えば、非水系の溶媒の例は、プロピレングリコール、ポリエチレングリコール、オリーブオイル等の植物油、オレイン酸エチル等の注射可能な有機エステルである。密封包帯用の担体は、皮膚透過性を増すため及び吸収を高めるために使用することができる。経口投与用の液体剤形は、一般的に液体剤形を含有するリポソーム溶液を含んでいてもよい。
【0078】
MAGL阻害化合物を含有する医薬組成物は、治療的に有効な量又は服用量で局所的に又は全身的に投与することができる。それらは、非経口的に、腸管的に、注射、迅速点滴、鼻咽頭吸収、経皮吸収によって、直腸的に及び経口的に投与することができる。治療的に有効な量とは、被験者の治療すべき障害又は症状の症候(例えば、侵害受容性疼痛)を減少又は阻害するために十分な量を意味する。このような有効な量は、被験者の疼痛に対する常態の感受性、その身長、体重、年齢、及び健康状態、疼痛の起源、MAGLアンタゴニスト化合物の投与方法、投与された特定の阻害剤、及び他の要因に依存して被験者ごとに異なるであろう。結果として、特定の状況下において特定の被験者に対する有効な量を経験的に決めるのが賢明である。
【0079】
のMAGLアンタゴニスト化合物について、当業者であれば薬学的な方法を使用して、容易に腫瘍増殖を阻害し及び癌を治療する当該化合物の有効量、又はカンナビノイドシグナル伝達と関連する症状を治療する化合物の有効量を同定することができる。典型的には、生体外で使用される投与量は、医薬組成物の生体内原位置での投与のための有用な量に関する役立つガイダンスを提供することができ、並びに動物モデルはの被験者における特定の障害の治療のための有効な投与量を決定するために使用することができる。よりしばしば、適した薬用量は、通常のに関する、最大許容用量及び安全投与量を決めるために、哺乳動物種に関する臨床研究によって決定することができる。より高い投与量が要求される特定の状況を除いて、MAGLアンタゴニスト化合物の好ましい投与量は普通には、一日当たり約0.001から約1000mg、より普通には約0.01から約500mgの範囲にある。一般に、投与されるMAGLアンタゴニスト化合物の分量は、被験者の症状を有効に及び確実に防止又は最小化する最少の投与量である。それ故に、上記の投与量の範囲は、本明細書での教示のための一般的なガイダンス及び補助を提供することを意図するものであり、本発明の範囲を限定することを意図するものではない。本発明の医薬組成物の調製及び投与に関する追加のガイダンスもまた当該技術分野で記載されている。例えば、「グッドマン及びギルマンズの薬物療法学の薬理学的基礎(Goodman & Gilman‘s The Pharmacological Bases of Therapeutics)」、ハルドマン(Hardman)ら編、マッグローヒル・プロフェッショナル(McGraw−Hill Professional(第10版、2001年);レミントン(Rmington)著「薬学の科学と実践(The Science and Practice of Pharmacy)」、ジェンナロ(Gennaro)編、リッピンコット・ウィリアムズ&ウィルキンス(Lippincott Williams&Wilkins)(第20版、2003年);及び「製剤投与形態及び薬剤デリバリーシステム(Pharmaceutical Dosage Forms and Drug Delivery Systems)」、アンセルら(Ansel et al.)編、リッピンコット・ウィリアムズ&ウィルキンス(Lippincott Williams&Wilkins)(第7版、1999年)を参照。
【0080】
実施例
下記の実施例は、例示するために提供するものであり、本発明を限定するものではない。
【0081】
実施例1.MAGL選択的阻害剤の開発
セリンヒドロラーゼに対する選択的阻害剤の探究には、この酵素クラスに特別な多様な特徴から利益を得ることができるという可能性がある。第一に、セリンヒドロラーゼは、他の酵素クラスとは交差反応性をほとんど又は全く示さない特異的な化学基による共有結合性の不活性化に感受性である。これらの反応性のケモタイプの間で主要なのはカルバメートであるが、それは、これらの酵素の保存セリン求核剤との共有結合性反応(カルバミル化)の後の、その穏やかな求電子性及び加水分解安定性のために、選択的で不可逆的なセリンヒドロラーゼ阻害剤の設計のために特権的な骨格構造として同定された。第二に、セリンヒドロラーゼの機能状態は、活性に基づいたタンパク質プロファイリング(ABPP)法を使用して天然の生物システムにおいて集合的にプロファイルすることができる。セリンヒドロラーゼのABPPには、この大きく多様な種類の酵素のための一般的な活性に基づくプローブとして役立つ、レポータータグ付フルオロホスホネート(FP)を使用する(リォウら(Liu et al.)、「プロシーディングス・オブ・ザ・ナショナル・アカデミー・オブ・サイエンシズ・オブ・ザ・ユナイテッド・ステイツ・オブ・アメリカ(Proc.Natl.Acad.Sci.U.S.A.)」、1999年、第96巻:p.14694−9)。拮抗的な方法で実施する場合、ABPPは、小分子の酵素阻害剤の潜在能力と選択性を複合プロテオーム中で直接評価するための強力な選別法として役立つことができる。選別法は、当該技術分野で、例えば、リォウら(Liu et al.)、「プロシーディングス・オブ・ザ・ナショナル・アカデミー・オブ・サイエンシズ・オブ・ザ・ユナイテッド・ステイツ・オブ・アメリカ(Proc.Natl.Acad.Sci.U.S.A.)」、1999年、第96巻:p.1494−9;リーら(Li et al.)、「ジャーナル・オブ・ジ・アメリカン・ケミカル・ソサイアティ(J.Am.Chem.Soc.)」、2007年、第129巻:p.9594−5;ロイングら(Leung et al.)、「ネイチャー・バイオテクノロジー(Nat.Biotech.)」、2003年、第21巻:p.687−91;及びグリーンバウムら(Greenbaum et al.)、「モレキュラー&セルラー・プロテオミクス(Mol.Cell.Proteomics)」、2002年、第1巻:p.60−68、等に既述の通り実施することができる。
【0082】
構造的に多様なカルバメートのライブラリーについて拮抗的ABPP選別を実施している間に、発明者らは、FAAH、MAGL及び、ABHD6(図1B)を含む脳セリンヒドロラーゼの特定の亜集合を阻害する化合物WWL98(図6)を同定した。発明者らは、次にMAGLの潜在能力及び選択性を改良するためにWWL98を修飾することに注力した。WWL152及びWWL162(図1A)で代表される通り、ピペラジンのスペーサ及び末端ビスアリールモチーフの取り込は、MAGL及びABHD6に関する潜在能力を維持しつつ、一方FAAHに関する活性を顕著に減少すること(図1B)が発見された。ピペラジンをピペリジニルメタノール骨格構造で置換すると、FAAH及びABHD6の両方にMAGLに対する選択性を示す化合物である、JZL175(図1B)を産出した。最後に、追加の選択性の増強は、ピペリジニルメタノール骨格構造上へのビス(メチレン−3,4−ジオキシフェニル)の包接によって行いJZL184(図1A及びB)を与えた。
【0083】
実施例2.脳プロテオーム中のMAGL選択的阻害剤活性
マウス脳膜プロテオームの50nMの低濃度のJZL184による30分間のプレインキュベーションの後に、拮抗的ABPPによってMAGL活性のほぼ完全な遮断が認められた(図2A)。対照的に、他の酵素(FAAH、ABHD6)の阻害は、より高濃度のJZL184(10μM)まで認められなかった。基質アッセイにより、JZL184が脳膜中において2−AG及びオレイン酸アミド(FAAH基質)の加水分解の遮断に対しそれぞれ8nM及び4μMのIC50値を示した(図2B)通りの選択性度を確認した。COS7細胞で発現した場合に(図2C)、組換えMAGL及びFAAHに関して同等の阻害効果が認められた。興味深いことに、脳膜は、試験したJZL184の最高濃度においてさえも2−AG加水分解活性の〜15%を維持したが、おそらくJZL184に非感受性の他の2−AG加水分解酵素の寄与を反映している。JZL184は、MAGLの時間依存的な阻害(図7)を示したが、それは共有結合型の不活性化機序を暗示している。そこで発明者らは、またMAGL及びFAAHに対するJZL184の相対活性をkobs/[I]値を測定することによって評価したが、当該値よりMAGLの阻害にたいする選択性が>300倍であることが(それぞれ4400及び13M−1s−1)確認された。最後に発明者らは、内在性カンナビノイド系の他の成分に関するJZL184の活性を試験した。当該化合物は、CB1受容体と相互作用せず、又は2−AG生合成酵素であるジアシルグリセロールリパーゼ−βを阻害しなかった(図8)。まとめると、これらのデータは、JZL184がマウス脳プロテオーム中でMAGLを強力かつ選択的に阻害することを示している。
【0084】
実施例3.選択的阻害活性剤の生体内活性
生体内でのJZL184のMAGL遮断活性を評価するために、C57B1/6雄マウスにJZL184を投与し(4−40mg/kg、腹腔内)、分析のために4時間後に屠殺した。脳膜プロテオームの拮抗的ABPPは、試験したJZL184の最少投与量(4mg/kg)における、FAAHを含む他の脳セリンヒドロラーゼに最少の影響をともなった、MAGLの75%阻害を明らかにした(図3A)。また、これらのデータは、MAGL及びFAAHに対しても基質アッセイによって確認した(図3B)。脳2−AG加水分解の残存活性は、JZL184の4から16mg/kgへの投与量の増加によって対照値の25%から15%にさらに減少することができたが(図3B)、そのことは拮抗的ABPPによって定量された通りMAGL活性のほぼ完全な遮断と関連していた(図3A)。FAAHは、また用量依存的様式でも阻害されたが、しかし16mg/kgのJZL184においても、拮抗的ABPP(図3A)又は基質アッセイ(図3B)によって定量した通り、FAAH活性の実質的な部分(〜35%)は無傷で残存していた。
【0085】
発明者らのゲルに基づくABPP分析は既に生体内でのMAGLに対する高選択性を示唆したが、本法の分解能の制限により、JZL184処理動物中の脳セリンヒドロラーゼの機能状態の完全な評価が妨げられていた。そこで発明者らは、ゲルに基づくABPPと比較して高い分解能及び感受性を示す、ABPP−MudPITと命名した、高性能液体クロマトグラフィー−質量分析(LC−MS)プラットフォーム(ジェサニーら(Jessani et al.)、「ネイチャー・メソッズ(Nat.Methods)」、2005年、第2巻:p.691−7、を参照)を使用して脳プロテオームを試験した。ABPP−MudPITは、トランスポーター及び生合成酵素を含む、内在性カンナビノイド系の他の成分の阻害剤に対する予期しない標的を同定するために以前使用した。手短に言えば、JZL184又は溶媒で処理したマウス由来の脳膜プロテオームを、ビオチン標識したFPプローブであるFP−ビオチンと共に拮抗的ABPPにかけた。次にFPビオチン標識タンパク質をアビジンで増強し、ビーズ上でトリプシンにより消化し、多次元LC−MSで分析し、探索アルゴリズムSEQUESTを使用して同定した。ABPP−MudPITは、JZL184(16mg/kg、4時間)がほぼ完全なMAGL活性の遮断及びFAAHの部分的な阻害を引き起こしたことを確認した(図3C)。印象的なことに、ABPP−MudPITによってプロファイルされた他の〜40の脳セリンヒドロラーゼのいずれもJZL184によって著しい影響を受けなかった(図3C及び表1)。これらのデータは、高投与量で(16mg/kg)長時間(4時間)投与された場合でさえ、JZL184が脳内でMAGLに対する優れた選択性を維持することを証明している。
【表1】
生体内でのJZL184の選択性に関するプロファイルを確立した後、発明者らは、次にMAGL及びFAAHに対する内在性の基質及び産物の候補の脳レベルを測定した。試験したJZL184の最低投与量(4mg/kg)においてさえ、2−AGレベルは処理4時間後に5倍に増加し、阻害剤のより高投与量においてはベースラインより8−10倍に増加することができた(図3d)。予想通り、脳2−AGの増加には、アラキドン酸レベルの著しい減少が伴った(図3E)。モノ−パルミトイルグリセロール(PG)及びモノ−オレオイルグリセロール等の他のMAG、及びその相応する遊離脂肪酸(それぞれパルミチン酸及びオレイン酸)は、JZL184処理動物由来の脳で著しくは変化しなかった(図9)。重要なことには、また脳アナンダミドレベルも試験された全ての投与量のJZL184によって影響を受けなかった(図3F)。N−パルミトイルエタノールアミン(PEA)、及びN−オレオイルエタノールアミン(OEA)等の他のN−アシルエタノールアミン(NAE)は、試験された最高投与量のJZL184(40mg/kg)を除き変化しなかったが、そこでは、これらの脂質のわずかな増加(〜2倍)が認められた。NAEレベルへの影響の欠如は、生体内のNAEレベルを高めるためにはFAAHの部分的な破壊では不十分であることを明らかにした公表された研究と一致している。JZL184処理マウスの脂質プロファイルは、脳FAAH活性を完全に阻害し、この組織中の2−AG又はアラキドン酸レベルを変えずにアナンダミド及び他のNAEレベルの著しい増加を引き起こす、選択的FAAH阻害剤URB597によって誘発されたプロファイルとは著しく違っていた。同様なデータがまたJZL184の経口投与の後でも得られたが(図10)、このことは生体内のMAGLの選択的遮断を実施するために本化合物は、多様な経路によって投与することができることを示している。
【0086】
実施例4.JZL184処理マウスにおける迅速持続的MAGL阻害
生体内でのJZL184阻害の経時変化を測定するために、マウスにJZL184を投与し(16mg/kg、腹腔内)、分析のために0.5、1、2、4、8、及び24時間に屠殺した。脳膜プロテオームのABPP(図4A)と同様に2−AG及びオレイン酸アミド加水分解アッセイ(図4B)は、MAGL阻害が、処理後30分以内に最大速度を達成するほど迅速であり、2−AG加水分解活性の>80%阻害を少なくとも24時間持続するほど長期持続性であることを示した(図4A)。対照的に、FAAH遮断は、よりゆっくりと起こり、試験のどの時点においても決して70%を超えなかった(図4B)。MAGLの迅速阻害に付随してJZL184処理は、脳で2−AG蓄積を即座に引き起こした。30分間のうちに脳2−AGレベルは既に7倍に増加し、少なくとも8時間は対照レベルの7−9倍が残存した(図4C)。興味深いことに、MAGLの活性の>80%がこの時点で阻害されたままであるのに、2−AGレベルは24時間のうちにほぼ対照レベルに回復した。2−AGの増加は、アラキドン酸の急速かつ持続性の減少と一致した(図4D)。アナンアミドを含むNAEと同様に他のMAG及び遊離脂肪酸も、JZL184処理マウスの分析時間経過を通して変化しなかった(図4E及び図11)。
【0087】
実施例5.MAGL選択的阻害剤によって誘発されるマウスの行動的影響
JZL184によって引き起こされる劇的かつ持続性の脳2−AGレベルの増加は、本阻害剤が内在性カンナビノイド媒介行動的影響を誘発するかもしれないことを示唆した。直接的なCB1アゴニストは、げっ歯類において、痛覚消失症、運動性低下症、低体温症、及び強硬症(あわせてカンナビノイド活性の四つ組み試験と呼ばれる)を含む多様な行動的影響を促進することが知られている。興味深いことに、FAAH阻害剤は、四つ組み試験において、痛覚消失症を引き起こすが他のカンナビノイド行動的表現型を引きおこさず、大部分は不活性である(リヒトマンら(Lichtman et al.)、「ザ・ジャーナル・オブ・ファルマコロジー・アンド・エクスペリメンタル・セラピューティクス(J.Pharmacol.Exp.Ther.)」、2004年、第311巻:p.441−8)。JZL184(16mg/kg、2時間)は、また急性熱痛覚の尾浸漬試験(図5A)及び内臓痛の酢酸苦悶試験(図5B)を含む、多数の疼痛アッセイにおいて著しい鎮痛作用を示すことも見出された。両方の場合において、JZL184の効果は、CB1アンタゴニストであるリモナバンでの前処理(3mg/kg)によって遮断された(図5A及びB)。FAAH阻害剤とは著しく対照的にJZL184は、また低体温症(図5C)及び運動性低下症(図5D)をも顕著に促進した。JZL184処理は強硬症を誘発しなかった一方で、バー試験で評価した場合に、処理マウス14匹中7匹が反射亢進又は「ポップコーン」行動(ペイテルら(Patelet al.)、「ジャーナル・オブ・ファルマコロジー・アンド・エクスペリメンタル・セラピューティックス(J.Pharmacol.Exp.Ther.)」、2001年、第297巻:p.629−37)を示した(図5E)。低体温及び反射亢進の効果は、リモナバンによって完全に遮断されたが、そのことは、それらがCB1受容体によって媒介されることを示している。発明者らは、JZL184の運動性低下効果に対するCB1受容体の寄与を明確に決定することができなかったが、というのはリモナバンが独自に運動性の顕著な減少を誘発することを見出したからである(図5D)。しかしながら、JZL184誘発性不動症の部分的遮断がリモナバンにより認められ(図5D)、そのことは本表現型に対するCB1受容体の寄与を示唆している。
【0088】
まとめると、これらのデータは、JZL184が直接的CB1アゴニストの薬理学的なプロファイルに定性的に似た広い一連のカンナビノイドの行動的影響をげっ歯類において誘発することを示している。加えて、これらの表現型の幾つか(例えば、低体温症及び反射亢進)は、生体内でのMAGL阻害の大きさを用量設定することによって、より有益な効果(例えば、痛覚消失)から薬理学的に脱共役することができた。実際に、より低い投与量はMAGL活性のより低い遮断に帰着したけれども、発明者らは、本研究で試験したJZL184の全投与量範囲(4−40mg/kg)にわたって顕著な2−AGの増加を認めた。これらのデータは、このようにMAGLの部分的な阻害であっても生体内の2−AG媒介内在性カンナビノイドシグナル伝達の増大にとって十分であるかもしれないことを示している。
【0089】
実施例6.MAGL阻害剤の合成
合成法一般特に断りがない限り、全ての試薬は、より市販品を購入しそれ以上精製せずに使用した。脱水溶媒は、市販の予備脱水した酸素不含の製剤を活性化アルミナカラムに通すことによって得た。他に断らない限り、全ての反応は、乾燥器で乾燥したガラス器具を使用して窒素雰囲気下で実施した。フラッシュクロマトグラフィーは230−400メッシュのシリカゲルを使用して実施した。NMRスペクトルは、CDCl3中バリアン・イノバ-400(Varian Inova−400)又はブルカーDMX−600(Bruker DMX−600)スペクトルメータで記録し、トリメチルシラン(TMS)又は残存溶媒のピークに対しリファレンス設定した。化学シフトはTMSに対し相対的にppmで報告し、J値はHzで報告する。高分解能質量分析実験は、スクリップス研究所質量分析コア部(The Scrips Research Institute Mass Spectrometry Core)において、エレクトロンスプレーイオン化飛行時間(ESI−TOF)を使用してアジレント(Agilent)質量分析計で実施した。
【0090】
JZL184の合成撹拌する4−ブロモ−1,2−メチレンジオキシベンゼン(2.01g、10mmol)の無水THF溶液(30ml)にn−BuLi(n−ブチルリチウム)(3.8ml、トルエン中2.6M、9.9mmol)を−78℃で滴下し添加した。同温度で1.5時間撹拌した後、エチルN−Cbz−イソニペコチン酸(1.02g、3.5mmol)の無水THF溶液(10ml)を滴下して添加し、移行を定量化するために追加分のTHF(3ml)を使用した。試薬の添加が終了した後、冷却浴槽を取り除いたところ、懸濁液は徐々に透明溶液に変わった。3時間後、反応を水で停止しEtOAc(酢酸エチル)で希釈した。層は分離し、有機層を水で2回、食塩水で1回洗浄し、Na2SO4で脱水し、減圧下で濃縮した。粗製油をフラッシュクロマトグラフィー(ヘキサン:酢酸エチル=6:1その後4:1)で精製して白色固形物として中間体アルコール(0.81g、収率47%)を得た:1H NMR(CDCl3、400 MHz)δ 1.29(qd、J=4、12 Hz、2H)、1.51(bs、2H)、2.38(tt、J=2.8、12 Hz、1H)、2.58(s、1H)、2.75(t、J=12 Hz、2H)、4.19(bs、2H)、5.03(d、J=5 Hz、2H)、5.85(s、4H)、6.70(d、J=8 Hz、2H)、6.88−6.92(m、4H)、7.24−7.33(m、5H);13C NMR(CDCl3、100 MHz)δ 26.6、44.4、44.6、67.1、79.4、101.1、106.9、107.9、118.9、127.9、128.0、128.5、136.9、140.1、146.2、147.7、155.2;C28H27NNaO7[M+Na]+に対する計算上のHRMS512.1680は512.1670に見出された。本中間体の一部分(0.72g、1.5mmol)をCH2Cl2/EtOH(1:1 v/v、40ml)に溶解し、Pd/C(パラジウム炭素)(10%、0.20g)を一度に添加した。反応液はH2下(1気圧)で一晩撹拌した。出発原料が完全に消費された後(12−24時間)、反応液をろ過し減圧下で濃縮した。生じた粗製油をCH2Cl2(30mL)に溶解し、それにEt3N(トリエチルアミン)(2ml、15mmol)及びクロロギ酸4−ニトロフェニル(460mg、2.3mmol)を順次添加した。反応液を一晩撹拌した。翌朝、反応を水で停止しEtOAcで希釈した。層は分離し、有機層を2N NaOHで2回、食塩水で1回洗浄し、Na2SO4で脱水し、減圧下で濃縮した。粗製油をフラッシュクロマトグラフィー(ヘキサン:酢酸エチル=6:1その後4:1)で精製して、減圧下で放置すると黄色固形物に変わる、淡黄色の油としてJZL184(550mg、2段階収率71%)を得た。
【0091】
JZL175の合成撹拌するエチルN−Cbz−イソニペコチン酸(0.84g、2.9mmol)の無水THF溶液(10ml)にブロモ(4−メトキシフェニル)マグネシウム(15ml、THF中0.5M)を室温で滴下し添加した。試薬の添加終了後に、反応液を加熱して一晩還流した。翌朝、反応を水で停止しEtOAcで希釈した。層は分離し、有機層を水で2回、食塩水で1回洗浄し、Na2SO4で脱水し、減圧下で濃縮した。粗製油をフラッシュクロマトグラフィー(ヘキサン:酢酸エチル=6:1その後2:1)で精製して淡黄色固形物として中間体アルコール(0.90g、収率69%)を得た:1H NMR(CDCl3、400 MHz)δ 1.24−1.33(m、2H)、1.54(bs、2H)、2.14−2.18(m、1H)、2.46(t、J=12 Hz、1H)、2.77(t、J=11 Hz、2H)、3.75(s、6H)、4.21(bs、2H)、5.06(s、2H)、6.81(d、J=9 Hz、4H)、7.27−7.34(m、9H)。この物質は、これ以上のキャラクタリゼーションをせずに使用した。本中間体の一部(55mg、0.18mmol)を、JZL184の合成と同様の方法で、Pd/C及びクロロギ酸4−ニトロフェニルによる2段階手順によって処理した。粗製油をフラッシュクロマトグラフィー(ヘキサン:酢酸エチル=6:1その後2:1)で精製し、減圧下に放置すると黄色固形物に変化する、淡黄色油としてJZL175(55mg、2段階収率62%)を得た:1H NMR(CDCl3、400 MHz)δ 1.40(m、2H)、1.65(t、J=11 Hz、2H)、2.26(bs、1H)、2.55(t、J=12 Hz、1H)、2.87(t、J=12 Hz、1H)、3.00(t、J=13 Hz、1H)、3.76(s、6H)、4.27(t、J=13 Hz、2H)、6.84(d、J=9 Hz、4H)、7.24(d、J=9 Hz、2H)、7.34(d、J=9 Hz、4H)、8.19(d、J=9 Hz、2H);13C NMR(CDCl3、100 MHz)δ 26.6、26.9、44.4、45.2、55.4、79.2、113.6、113.7、122.4、125.1、127.3、137.8、137.9、144.8、152.2、156.5、158.4;C27H27N2O6[M−H2O+H]+に対する計算上のHRMS475.1864は475.1864に見出された。
【0092】
WWL152の合成撹拌するN−Bocピペラジン(100mg、0.54mmol)のCH2Cl2溶液(5ml)にクロロギ酸4−ニトロフェニル(109mg、0.54mmol)及びEt3N(75μl、0.54mmol)を室温で順次添加した。3時間後、反応液を減圧下で濃縮した。粗製混合物をフラッシュクロマトグラフィー(ヘキサン:酢酸エチル=3:1)で精製して白色固形物としてBoc保護中間体(100mg、収率52%)を得た:1H NMR(CDCl3、400 MHz)δ 8.24(d、J=9 Hz、2H)、7.31(d、J=9 Hz、2H)、3.67(bs、2H)、3.54(bs、6H),1.50(s、9H)。当該中間体(100mg、0.28mmol)に1:1 v/v TFA:CH2Cl2(2ml)を室温で添加した。2時間後、反応液を減圧下で濃縮した。粗製物はそれ以上精製せずに使用した。撹拌する4−メトキシベンゾフェノン(100mg、0.5mmol)EtOH溶液(5ml)にNaBH4(38mg、1mmol)を室温で添加した。翌朝、反応液を水(10ml)上に注ぎ込み、1時間撹拌し、次に生成物をフィルターで除き減圧下で濃縮した。CH2Cl2(5ml)中の粗製アルコールに塩化オキサリル(40μL、0.46mmol)を室温で滴下して添加した。粗製物をCH3CN(2ml)に再溶解し、それに4−ニトロフェニルピペラジン-1−カルボキシレート(58mg、0.23mmol)及びEt3N(32μl、0.23mmol)を順次添加した。試薬の添加終了後に、反応液を加熱し一晩還流した。翌朝、反応液を減圧下で濃縮した。粗製混合物をフラッシュクロマトグラフィー(ヘキサン:酢酸エチル=1:1)で精製して白色固形物としてWWL152(201mg、3段階収率84%)を得た:1H NMR(CDCl3、600 MHz)δ 8.24(d、J=9 Hz、2H)、7.32(d、J=8 Hz、4H)、7.27(d、J=9 Hz、2H)、6.84(d、J=8 Hz、4H)、4.22(s、1H)、3.76(s、6H)、3.66(s、2H)、3.57(s、2H)、2.45(s、4H);。C26H27N3O6[M+H]+に対する計算上のHRMS478.1973は、478.1977に見出された。
【0093】
WWL162の合成CH2Cl2(2mL)中の1−(ビス(4−フルオロフェニル)メチル)ピペラジン(29mg、0.1mmol)にクロロギ酸4−ニトロフェニル(20mg、0.1mmol)及びEt3N(14μl、0.1mmol)を室温で順次添加した。3時間後、反応液を減圧下で濃縮した。粗製混合物をフラッシュクロマトグラフィー(ヘキサン:酢酸エチル=3:1)で精製して白色固形物としてWWL162(42mg、収率92%)を得た:1H NMR(CDCl3、600 MHz)δ 8.24(d、J=9 Hz、2H)、7.36(m、4H)、7.28(d、J=9 Hz、2H)、7.00(m、4H)、4.29(s、1H)、3.68(s、2H)、3.59(s、2H)、2.44(s、4H);C24H21F2N3O4[M+H]+に対する計算上のHRMS454.1573は、454.1578に見出された。
【0094】
実施例7.他の方法及び実験材料
実験材料2−AG、ペンタデカン酸(PDA)、AEA、及びURB597は、ケイマンケミカル社(アナーバー、ミシガン州)(Cayman Chemicals(Ann Arbor、MI))から購入した。テトラヒドロリプスタチン(THL)は、シグマ社(セントルイス、ミズーリ州)(Sigma(St.Louis、MO))から購入した。FPローダミン及びFPビオチンは、既述の通り合成した(リォウら(Liu et al.)、「プロシーディングス・オブ・ザ・ナショナル・アカデミー・オブ・サイエンシズ・オブ・ザ・ユナイテッド・ステイツ・オブ・アメリカ(Proc.Natl.Acad.Sci.U.S.A.)」、1999年、第96巻:p.14694−9);及びパトリセリら(Patricelli et al.)、「プロテオミクス(Proteomics)」、2001年、第1巻:p.1067−71)。リモナバンはNIDA(ロックヴィル、メリーランド州(Rockville、MD))から購入し、容量比18:1:1の食塩水、エタノール、及びalkamuls-620(ローヌプーラン社製、プリンストン、ニュージャージー州(Rhone−Poulenic、Princeton、NJ))からなる溶媒に溶解した。13Cオレイン酸アミド(13C18H35NO)は、クラヴァットら(Cravatt et al.)、「サイエンス(Science)」、1995年、第268巻、p.1506−9、に記載された方法に従って13Cオレイン酸(スペクトラ・ステイブル・アイソトープ社製、コロンビア、メリーランド州(Spectra Stable Isotopes、Collumbia、MD))から合成した。
【0095】
マウス組織プロテオームの調製マウス脳をpH7.5のPBS中、Dounceホモジナイザーで均質化し、続いて壊死組織片を取り除くために低速回転(1,400g、5分間)した。それから、細胞質画分を上清に、膜画分を沈渣として得るために、上清を遠心(64,000g、45分間)にかけた。沈渣を洗浄し超音波処理によってPBS緩衝液に再懸濁した。各画分の総タンパク質濃度は、タンパク質アッセイキット(バイオラド社製(Bio−Rad))を使用して定量した。試料は、使用するまで−80℃で貯蔵した。
【0096】
COS7又はHEK293細胞における組換え体の発現手短にいえば、マウスのセリンヒドロラーゼをコードする完全長cDNAは、オープン・バイオ・システム社(ハンツビル、アラバマ州)(OpenBioSystems(Huntsville、AL))から購入した。cDNAは、直接形質移入するか(利用可能ならば真核細胞発現ベクター中に)又はpcDNA3(インビトロジェン社製(Invitrogen))にサブクローニングした。COS7細胞は、完全培地(L−グルタミン、非必須アミノ酸、ピルビン酸ナトリウム、及びFCS添加DMEM)、37℃及び5%CO2で10cmディッシュ中、〜70%集密まで培養した。当該細胞は、適切なcDNA又は空ベクター対照(「ニセの」)及び製造者のプロトコルに従って形質移入試薬FUGENE6(ロシュ・アプライド・サイエンス社製(Roche Applied Science))を使用することによって一過性に形質移入を行った。48時間後、細胞はリン酸緩衝食塩水(PBS)で2回洗浄し、擦過により採取し、PBS0.25mlに再懸濁し、超音波処理によって溶解した。細胞溶解物は、アッセイにおいて細胞全体のホモジネートとして使用した。
【0097】
酵素活性アッセイ手短に言うと、2−AG(100μM)を、マウス脳膜(20μg)又はCOS7細胞中の組換えMAGL(0.1μg)と、PBS(200μl)中、室温で10分間インキュベートした。容量比2:1のCHCl3:MeOH500μLの添加によって反応を停止し、PDA0.5nmolを投与し、ボルテックスし、次に相を分離するために遠心分離(1,400g、3分間)した。結果として生じた有機相の30μlをアジレント1100シリーズLC−MSD SL装置(Agilent 1100 series LC−MSD SL instrument)に注入した。LC分離は、プレカラム(C18、3.5μm、2mmx20mm)と共にジェミニ(Gemini)逆相C18カラム(5μm、4.6mmx50mm、フェノモネクス社製(Phenomonex))によって行った。移動相Aは、容量比95:5のH2O:MeOHから構成し、移動相Bは、容量比60:35:5のi−PrOH:MeOH:H2Oから構成した。陰イオン化法におけるイオン形成を補助するために0.1%の水酸化アンモニウムを含有させた。流束は0.5ml/分であり、濃度勾配は、1.5分間は0%B、5分間かけて直線的に100%Bに増加し、続いて3.5分間は100%Bの均一濃度勾配とし、2分間かけて0%Bに平衡化した。MS分析は、エレクトロスプレーイオン源により実行した。キャピラリ電圧は3.0kVに設定し、フラグメンター電圧は100Vに設定した。乾燥ガス温度は350℃、乾燥ガス流束は10 l/分、及びネブライザ圧力は35psiであった。加水分解産物は、PDA標準と比較したピーク下の面積を測定して定量した。13Cオレイン酸アミド又はAEA加水分解活性は、PBS(100μl)中、それぞれマウス脳膜(50μg)又はCOS7細胞(5μg)中の組換えFAAHを使用して測定した。反応は、インキュベーション時間を37℃で20分間とし、反応を停止するために容量比2:1のCHCl3:MeOH300μlを使用した以外、2−AG加水分解のために記載したのと同様の方法で実施した。DAGLβ加水分解活性は、PBS(100μl)中でHEK細胞(50μg)中の組換えDAGLβを使用して測定した。反応は、インキュベーション時間を37℃で30分間とした以外、オレイン酸アミド加水分解のために記載したのと同様の方法で実施した。
【0098】
[3H]リモナバン結合によるJZL184置換上記の通り膜を調製した。膜タンパク質(8μg)を、非標識リモナバン5μMの存在下又は非存在下に(非特異的結合を定量するため)、0.2-3nM[3H]リモナバン含有50mMトリス塩酸、3mM MgCl2、0.2mM EGTA、100mM NaCl、 1.25g/l 、と30℃で90分間インキュベートした。2番目のセットの試料も、JZL184濃度を変化させる(0.001−10μM)以外は、同様のプロトコルを使用して調製した。反応は、リットル当たりトリス緩衝液5gを含有するBSA(トリスBSA)に予浸したワットマン(Whatman)GF/Bガラス繊維フィルタ減圧ろ過によって停止し、続いて4℃のトリスBSAで3回洗浄した。結合した放射活性は、シンチレーション液であるサイニット・セイフ、エコノ1(ScinitSafe Econo 1)で抽出後に45%の効率で液体シンチレーション分光光度法によって測定した。
【0099】
脳脂質測定脳半分の重量を量り、続いて、N−アシルエタノールアミン(NAE)、モノアシルグリセロール(MAG)又は遊離脂肪酸(FFA)測定用の標準品(200pmolのd4−OEA、20pmolのd4−AEA、400pmolのC15:0−MAG、及び4nmolのPDA)を含む、容量比2:1:1のCHCl3:MeOH:トリスpH8.0(8ml)、Dounceホモジナイザーで均質化した。混合物をボルテックスし、次に遠心分離した(1,400g、10分間)。有機層を取り除き、終容量が再び8mlになるまでCHCl3を添加し、抽出を繰り返した。有機抽出液を併せN2気流下で乾燥し、容量比2:1のCHCl3:MeOH(120μl)に再可溶化した。再可溶化脂質の30μL及び15μLを、それぞれポジティブモード測定(MAG及びNAE)及びネガティブモード測定(遊離脂肪酸)のために注入した。脂質測定は、上記と同様の装置及び溶媒を使用してLC−MSによって実施した。0.1%ギ酸又は0.1%水酸化アンモニウム等の溶媒改良剤を、それぞれポジティブ及びネガティブイオン化モードにおいてイオン形成を補助するために含有させた。ポジティブモード測定のためには、各操作とも流速0.1ml/分、5分間で開始した。濃度勾配は、0%Bで開始し、流速0.4ml/分で40分間かけて直線的に100%Bに増加し、続いて7分間は100%Bの均一濃度勾配とし、次に流速0.5ml/分で8分間かけて0%Bに平衡化した。ネガティブモード測定のためには、各操作とも流速0.1ml/分、3分間で開始した。濃度勾配は、0%Bで開始し、流速0.4ml/分で17分間かけて直線的に100%Bに増加し、続いて7分間は100%Bの均一濃度勾配とし、次に流速0.5ml/分で3分間かけて0%Bに平衡化した。MAG、NAE、及びFFAは、C15:0又は重水素化標準品と比較したピーク下面積を測定することによって定量した。標的LC−MS測定は、選択イオン検出(SIM)を使用して実施した。
【0100】
生体内でJZL184の標的となるSHのABPP−MudPIT解析上記の通り、JZL184又は溶媒で処理したマウス由来の脳膜プロテオームの一部(1ml、mlPBS当たり1mg)を、5μMのFPビオチンにより室温で1時間標識し、Lys−C消化段階を省略した以外は、当該技術分野で既述の通りABPP−MudTIT分析のために調製した(グリーンバウムら、「モレキュラー&セルラー・プロテオミクス(Mol.Cell.Proteomics)」、2002年、第1巻:p.60−68;及びアレキサンダーら(Alexander et al.)、「ジャーナル・オブ・ジ・アメリカン・ケミカル・ソサイアティ(J.Am.Chem.Soc.)」、2006年、第128巻:p.9699−704)。溶出したペプチドのMudPIT解析は、直結型アジレント100LC−テルモフィニガンLTQ−MS装置(a coupled Agilent 1100 LC−ThermoFinnigan LTQ−MS instrument)に関し既述した通り実施した。全データセットは、SEQUEST検索アルゴリズムを使用してマウスのIPIデータベースで検索し、結果はDTASELECTによりフィルター処理しグループ分けした。1.8(+1)、2.5(+2)、3.5(+3)以上大きな相互相関スコア及び0.08以上大きなデルタCNスコアを有するペプチドを、スペクトラル・カウンティング分析に含めた。対照試料の中で平均して5以上のスペクトル・カウントが同定されたタンパク質のみを、比較分析の考慮に入れた。特に、プローブ標識タンパク質は、「無プローブ」対照の操作(ビオチン標識FP不含の上述の通り実施した実験)において認められるよりも少なくとも5倍大きなスペクトル数を有するFP処理試料の中に存在することによって更に同定される。スペクトル・カウントは、平均値の標準誤差(SEM)を付随する三試料の平均として報告する。
【0101】
行動的研究リモナバンと対比したJZL184の薬理学的効果を下記の指標を使用して評価した:自発運動活性、ホットプレート試験及び尾浸漬試験による侵害受容、バー試験による強硬症、及び低体温症。群当たり14匹のマウスのサンプルサイズで、全4グループのマウスを使用した。注射前に、尾浸漬試験におけるベースライン応答、並びに直腸温度に関する評価を行った。被験者にリモナバン又は溶媒を腹腔内注射し、続いて10分後にJZL184又はそれぞれの溶媒を腹腔内注射した。自発運動活性はJZL184又は溶媒投与の120分後に評価し、退避反応潜時は、2回目の注射135分後に評価し、並びに強硬症及び直腸温度は注射145分後に評価した。運動性低下を測定するために、各マウスを透明なプレキシガラス製箱(42.7x21.0x20.4cm)に評価時間の10分間放置し、エニメイズ(Anymaze)ソフトウェアを使用して(ステールティング社製、ウッドデール、イリノイ州(Stoelting、Wood Dale、Illinois))静止時間のパーセントを測定した。被験者の侵害受容は、尾浸漬アッセイにより評価した。頭を先にし尾をバッグから外に状態で、各マウスを吸収パッドで製作された小さなバッグ(VWRサイエンティフィック・プロダクツ社製(VWR Scientific Products);直径4cm、長さ11cm)に配置した。実験者はマウスを優しく抱き、尾の先約1cmを56.0℃に維持した温浴に浸漬した。10秒以内の遮断時間にマウスがその尾を温浴から退かせるための潜時を評価した。強硬症は、各被験者の前足を、床面から4.5cm高い棒(直径0.75cm)の上に載せるバー試験を使用して評価した。その足を棒の上に10秒間置き静止したままのマウス(呼吸運動を除外)を強硬症と評価した。本研究においていずれのマウスも強硬症でなかったにもかかわらず、JZL184で単独処理したマウスの半数が、前足を棒の上に載せられた時に、跳躍又は「ポップコーン」行動と例示される、反射亢進を示した。直腸温度は、熱電対探針を直腸に2.0cm挿入して測定し、温度は遠隔時期温度計によって得た。
【0102】
未処理の被験者の第二群(群当たりn=10−11匹マウス)を酢酸ストレッチングアッセイで評価した。被験者にリモナバン又はその溶媒を皮下注射し、続いて10分後にJZL184又は溶媒を皮下注射した。二番目の注射の120分後に、酢酸(0.6%)を10μl/g体重の量で腹腔内に注入した。酢酸投与後の20分間のマウス当たりのストレッチング(腹部の収斂、体幹の回転(ねじれ)及び全身及び後肢の伸展)回数を計数した。
【0103】
拮抗的活性に基づいたタンパク質プロファイリング(ABPP)実験生体外での阻害活性の選択性を当該技術分野で記載された拮抗的ABPP法を使用して試験した(例えば、リーら、「ジャーナル・オブ・ジ・アメリカン・ケミカル・ソサイアティ(J.Am.Chem.Soc.)」、2007年、第129巻:p.9594−5;及びロイングら(Leung et al.)、「ネイチャー・バイオテクノロジー(Nat.Biotech.)」、2003年:第21巻、p.687−91)。手短に言えば、本明細書に記載の通りに調製したマウスの脳膜プロテオームを、PBSで1mg/mlに希釈し、全反応液量50μl中、終濃度2μMでのFPローダミンの添加前に、種々の濃度の阻害剤(1nMから10μM)と37℃で30分間プレインキュベートした。室温で30分後、反応液は、4xSDS−PAGE用ローディング緩衝液により反応停止し、90℃で5分間煮沸し、SDS−PAGEに負荷し、フラットベッド蛍光スキャナを使用してゲル中で可視化した。
【0104】
JZL184による生体外研究標準アッセイは、基質又はABPPプローブの添加前に、タンパク質試料をJZL184と37℃で30分間プレインキュベートすることによって実施した。基質アッセイから濃度依存阻害曲線が得られ、当該曲線は、プリズム(Prism)ソフトウェア(グラフパッド(GraphPad))を使用して95%の信頼区間を有するEC50値を得るために適していた。kobs/[I]値の測定のために、脳膜プロテオーム(1mg/ml、全量300μl)をJZL184とインキュベートした(0.01−15μM、10−40分間、37℃)。10分ごとに反応液の50μlを取り出し、FPローダミン(2μM)で2分間処理し、4xSDS−PAGEローディング緩衝液で反応停止し、90℃で5分間煮沸した。混合性の反応液をSDS−PAGEに負荷し、フラットベット蛍光スキャナを使用してゲル中で可視化した。残存活性のパーセントは、MAGL又はFAAHのバンドに相当する統合した光学密度を測定することによって決定したが、その結果は擬一次速度定数を決定する指数曲線と適合した。
【0105】
JZL184による生体内研究JZL184(純品)をボルテックスし、超音波処理し、及び穏やかに加熱することによって容量比4:1のPEG300:ツイーン80に直接溶解した(10、4、2、又は1mg/g)。C57B1/6J雄マウス(6−8週令、20−26g)にJZL184又はJZL184無添加の容量比4:1のPEG300:ツイーン80溶媒を4μ/g体重の量で腹腔内投与した(上記希釈によって40、16、8、又は4mg/kg)。明示した時間後に、マウスをイソフルランで麻酔し、断頭によって屠殺した。脳を取り除き、正中矢状面に沿って半側切断し、次に各半分を液体N2で瞬間凍結した。脳の半分は上記の通りタンパク質分析用に調製し、他の半分は代謝産物分析用に使用した。URB597を超音波によって容量比18:1:1の食塩水:エマルファオー(emulphor):エタノールに溶解したのと10μl/g体重(最終投与量10mg/kg)の量で腹腔内投与したのを除き、URB597によるFAAHの選択的阻害も上記と同様の方法で行った。
【0106】
実施例8.癌発症機序における増加MAG活性の同定
癌発発症機序に寄与する酵素活性を同定するために、発明者らは、黒色腫[侵襲性(C8161、MUM2B)、非侵襲性(MUM2C)]、卵巣癌[侵襲性(SKOV3)、非侵襲性(OVCAR3)]、及び乳癌[侵襲性(231MFP)、非侵襲性(MCF7)]を含む、多様な起源の腫瘍由来の、侵襲性及び非侵襲性のヒト癌細胞株のパネル(調査対照群)の機能的なプロテオーム解析を行った。侵襲性癌細胞株は、その非侵襲性のカウンターパートと比較して、より大きな生体外での移動及び生体内での腫瘍増殖速度を示すことが確認された。これらの癌細胞株由来のプロテオームを、セリンヒドロラーゼ指向フルオロホスホネート(FP)活性に基づいたプローブを使用して、活性に基づいたタンパク質プロファイリング(ABPP)によって選別した。本研究の目的は、これらの保存酵素の変化が癌細胞の病態に対し寄与する高い可能性を有するであろうとの仮説の下に研究しつつ、非侵襲性と比較して侵襲性癌細胞株において一貫して変化する加水分解酵素活性を同定することにあった。
【0107】
スペクトラル・カウンティングによって判断したところ、この分析で検出された50以上のセリンヒドロラーゼの中で、二の酵素、KIAA1363及びMAGLが非侵襲性のカウンターパートと比較して侵襲性癌細胞において一貫して増加することが見出された。発明者らは、侵襲性癌細胞におけるKIAA1363及びMAGLの増加をゲルに基づくABPPによって確認したが、そこではプロテオームをローダミンタグFPで処理し、1D−SDS−PAGE及びインゲル蛍光スキャニングによって分析する(図13A)。どちらの場合も、KIAA1363についての差別的グリコシル化のため、及びおそらくMAGLについての選択的スプライシングのために(カールソンら(Karlsson et al.)、「ジーン(Gene)」、2001年、第272巻:p.11−18)、各酵素の二つの型が検出された(図13A)。襲性癌細胞におけるMAGLの高められた活性は、基質C20:4MAGを使用して侵確認した(図13B)。数種類の酵素がMAG加水分解活性を示すことが明らかにされてきたので(ブランクマンら(Blankman et al.)、「ケミストリー&バイオロジー(ChemBiol)」、2007年、第14巻:p1347−1356)、発明者らは、強力で選択的なMAGL阻害剤JZL184を使用して、MAGLが癌細胞中のこの過程に成す寄与を確認した。JZL184(1μM、4時間)は癌細胞のMAG加水分解活性を劇的に減少し(図13B)、MAGLの33及び35kDA型の両方のFPローダミンシグナルを選択的に遮断した(図13A)。対照的に、発明者らは、JZL184処理が癌細胞によって示されるジアシルグリセロール、トリアシルグリセロール、リゾリン脂質、及びリン脂質を含む、数種類の追加のクラスの脂質に対する加水分解活性を変化させないことを見出した。これらのデータは、侵襲性癌細胞が極めて増加したMAG活性を示し、例え全てではないにしても、この活性の殆どがMAGL酵素に基づくことを明らかにしている。
【0108】
研究は、MAGLが侵襲性癌細胞の遊離脂肪酸(FFA)レベルを制御することを明らかにしている。MAGLの既知の機能と調和して、発明者らは、JZL184によるMAGLの遮断が(1μM、4時間)、各々の侵襲性癌細胞株において、2−AGを含む数種類のモノアシルグリセロール(MAG)レベルの著しい増加を引き起こすことを見出した(図13C)。興味深いことに、しかしながら、MAGL阻害はまた侵襲性癌細胞におけるFFAレベルの著しい減少も引き起こした(図13D)。この驚くべき発見は正常細胞におけるMAGLの機能とは対照をなしている。この酵素が一般的にはFFAレベルを制御しないことは知られていた(野村ら(Nomura et al.)、「ネイチャー・ケミカル・バイオロジー(NatChemBio)」、2008年、第4巻、p.373−378)。発明者らは、またC20:4FFA及びMAGを除いては、FFAの減少の大きさが相応するMAGの増加を大いに上回る(C16:0、C18:0、及びC18:1脂質に対し、それぞれ〜5000−6100pmol及び41−75pmol)ことにも注目した。質量収支におけるこの明らかな矛盾が、JZL184処理癌細胞における増加したMAGの他の代謝産物への変換によって説明することは可能かもしれない。この前提と一致して、発明者らは、リピドーム解析がJZL184処理癌細胞においてリゾリン脂質の二の主要なクラス、リゾホスファチジルコリン(LPC)及びリゾホスファチジルエタノールアミン(LPE)の著しい増加を明らかにすることを見出した。これらのリゾリン脂質の増加の累積的大きさは(4100−6500pmol)、JZL184処理細胞において認められるFFAの減少とぴったりと一致した。とりわけ、発明者らは、癌細胞においてC20:4リゾリン脂質を検出しなかったが、これは、なぜJZL184がそれぞれC20:4MAG及びFFAの同様の大きさの増加及び減少を引き起こしたかの、ありそうな説明を提供している。非天然C17:0−MAGを使用した代謝標識研究は、MAGが侵襲誠意癌細胞によってLPC及びLPEに変換されること、並びにこの代謝変換がJZL184処理によって著しく亢進されること確認した。最後に、発明者らは、JZL184処理が非侵襲性癌細胞株におけるMAG及びFFAレベルに影響を与えなかったことを見出したが(図13C、13D)、これはこれらの細胞における無視できるMAG発現と一致する(図13A、13B)。
【0109】
発明者らは、次に、二の独立したshRNAプローブを使用するRNA干渉技術によってMAGLの発現を安定してノックダウンしたが、その両方とも侵襲性癌細胞株においてMAGL活性の70−80%を減少した、(図14A、図14及び図21)。他のセリンヒドロラーゼ活性は、shMAGLプローブによっては影響されなかったが(図14A、図14及び図21)、このことはこれらの試薬の特異性を確認している。両方のshMAGLプローブとも、侵襲性黒色腫(図14B、14C)、卵巣癌(図14E、14F)、及び乳癌細胞(図21)において、著しいMAGの増加及び相応するFFAの減少を引き起こした。あわせて、これらのデータは、急性の(薬理学的)及び安定した(shRNA)MAGLの両方の遮断が侵襲性癌細胞においてMAGの増加及びFFAの減少を引き起こすことを明らかにしている。さらに、侵襲性癌細胞が非侵襲性癌細胞よりも高い基礎レベルのFFAを発現すること、並びにこの変化した代謝プロファイルがMAGL阻害によってほとんど根絶されることに注目すべきである。これらの興味ある発見は、MAGLが侵襲性癌細胞におけるFFAレベルの主要な制御因子であることを示している。最後に、発明者らは、MAGL活性(図15A、15B)及びFFAレベル(図15C)が、良性又は低悪性度腫瘍と比較して高悪性度の原発性ヒト卵巣腫瘍においても増加することを確認した。このように、MAGL−FFA経路の高められた発現は、侵襲性ヒト癌細胞株及び原発性腫瘍両方の顕著な特徴である。
【0110】
実施例9.MAGL調節による癌の病原性制御
発明者らは、最初にMAGL発現の破壊の影響及び癌の病原性に関する活性を特定した。病原性の変化について、一組の生体外及び生体内アッセイを使用して、shMAGL癌細胞株を試験した。shMAGL黒色腫細胞(C8161)、卵巣癌細胞(SKOV3)、及び乳癌細胞(231MFP)は、著しく減少した生体外の移動(図16A、図16F及び図21)、浸潤(図16B、図16G及び図21)、及び血清飢餓条件下の細胞生存(図16C、図16H及び図21)を示した。JZL184によるMAGLの急性の薬理学的遮断もまた癌細胞の移動を減少し(図21)、生存を減少しなかったが、これは、おそらく癌の病原力を最大に障害するには、持続性のMAGL阻害が要求されることを示している。
【0111】
shMAGL−C8161及びSKOV3癌細胞は、免疫不全マウスで実施した皮下異種移植片移植の研究において(図16D、16I)、著しく減少した腫瘍増殖速度を示した。同様の腫瘍増殖速度の障害が、腫瘍中のMAGL活性を遮断することが確認された治療レジームに従って(データ不掲載)、JZL184を一日一回投与した(40mg/kg、経口投与)C8161及びSKOV3異種移植片移植マウスにおいても認められた(図16E、16J)。とりわけ、MAGL破壊腫瘍は、かなり低いFFAレベルを有したが、このことは、MAGLが生体内で増殖する癌細胞内の脂肪酸代謝に対する制御を維持することを示している。まとめると、これらの生体外及び生体内での研究は、MAGL活性が悪性の癌細胞によって示される侵襲性の特性の幾つかを支持することを証明している。
【0112】
発明者らは、次に非侵襲性癌細胞におけるMAGLの発現がその脂質代謝プロファイル及び病原性を変化させることができるか否かを。MUM2C及びOVCAR3細胞の安定なMAGL過発現変異体(MAGL−OE)並びに対照変異体[空ベクター発現体又はセリン求核試薬がアラニンに変異された(S122A)触媒的に不活性な変異体]をレトロウイルス感染によって作成し、それらのそれぞれのMAGL活性を、ABPP及びC20:4MAG基質アッセイによって評価した。両アッセイにより、MAGL−OE細胞が対照細胞と比較して10倍以上大きなMAGL活性の増加を有することを確認した(図17A)。MAGL−OE細胞は、また著しいMAGの減少(図17B)とFFAの増加(図17C)も示した。この変化した代謝プロファイルは、MAGL−OE細胞において増加した移動(図17D)、浸潤(図17E)、及び生存(データ不掲載)を付随した。これらの効果のいずれもS122A MAGL変異体を発現する癌細胞において認められなかったが、そのことは、それらがMAGL活性を要求することを示している。また、この前提と一致して、MAGL−OE細胞において認められた代謝及び病原性効果の両方ともJZL184の単回処理によって逆転された。最後に、MAGL−OE MUM2C細胞は、また対照細胞と比較して生体内での増加した腫瘍増殖も示した(図17F)。とりわけ、MAGL−OE MUM2C細胞の増加した腫瘍増殖速度は侵襲性C8161細胞のそれにほぼ匹敵したが(データ不掲載)、このことは、MAGLが黒色腫細胞において高度に腫瘍原性の表現型を誘発するのに充分であることを示している。まとめるとこれらのデータは、非侵襲性癌細胞における異所的なMAGLの発現が、生体外及び生体内の両方でそのFFAレベルを増加し及び病原性を促進するのに充分であることを示している。
【0113】
実施例10.MAGL−癌細胞の障害病原性の代謝レスキュー
MAGLは、そのMAG基質レベルを減少することによって、又はそのFFA産物レベルを増加することによって、又はその両方によって、癌細胞の侵襲性を支持することができた。MAGの中で、主要なシグナル伝達分子は内在性カンナビノイドである2−AGであり、それはCB1及びCB2受容体を活性化する。そこで発明者らは、亢進した内在性カンナビノイドシグナル伝達(増加した2−AGレベルから生じる)が、MAGL破壊癌細胞において認められる抗移動効果を媒介することができるか否かを試験した。しかしながら、発明者らは、CB1アンタゴンストもCB2アンタゴニストもshMAGL癌細胞の移動障害を救出しないことを見出した。CB1及びCB2アンタゴニストは、また癌細胞中のMAG又はFFAレベルにも影響をあたえなかった(データ不掲載)。これらの発見は、侵襲性癌細胞における低いCB1及びCB2受容体の発現レベルと結びついて(データ不掲載)、MAGLの癌の侵襲性に関する効果は内在性カンナビノイドシグナル伝達によって媒介されなかったことを主張している。
【0114】
MGAL触媒反応の産物に注目して、発明者らは、仮にMAGLの前腫瘍形成性効果がFFA(又はその二次代謝産物)によって媒介されるとするならば、shMAGL癌細胞の病原性の障害は、外来起源の脂肪酸による処理によって救出されるかもしれないと推論した。この仮説を支持するが、侵襲性癌細胞におけるMAGLによって制御される二の主要なFFA、パルミチン酸又はステアリン酸のshMAGL又はJZL184処理MAGL−OE癌細胞への添加は(それぞれC16:0及びC18:0FFA;20μM、4時間)、当該細胞の移動活性を完全に回復させた(図18A、18B)。C16:0及びC18:0FFAは、また非侵襲性癌細胞MUM2C及びOVCAR3の移動活性を刺激することも見出された(図18B)。発明者らは、次に増加したFFA送達量が生体内でshMAGLについて見出される腫瘍増殖障害を修正することができるか否かを特定した。免疫不全マウスを異種移植片腫瘍増殖実験の期間を通して通常の固形飼料か又は高脂肪食で飼育した。とりわけ、shMAGL−C8161細胞の障害した腫瘍増殖速度は、高脂肪食を与えられたマウスにおいて完全に救出された。対照的に、sh対照C8161細胞は、通常飼料と高脂肪食とにおいて同等の腫瘍増殖速度を示した。高脂肪食群のshMAGL−C8161細胞に対する腫瘍増殖の回復は、切除した腫瘍中の著しく増加したFFAレベルと相互に関連があった(図18D)。
【0115】
まとめると、これらの結果は、MAGLが増加したFFAレベルを持続的に維持することによって癌細胞の病原性の特性を支持することを示している。発明者らは、次にこの代謝プロファイルが侵襲性癌細胞の大きな脂質代謝ネットワークに影響することができるか否かを尋ねた。
【0116】
実施例11.MAGLは前腫瘍形成性シグナルに富んだ脂肪酸代謝ネットワークを制御する
発明者らは、最初にMAGL−FFA経路が、解糖系に燃料を供給するためにNAD+を再生する(連続的な脂肪アシルグリセリド/FFA再循環による)ための方法として貢献することができるか否かを検討したが、それは、癌における増加した中性脂質ヒドララーゼ活性に対する必要性を説明するために仮定されたものである(プルジビッツコウスキーら(Przybytkowski et al.)、「バイオケミストリー&セル・バイオロジー(Biochem.CellBiol.)」、2007年、第85巻:p.301−310)。このモデルの議論をするうちに、発明者らは、対照細胞と比較してshMAGL及びMAGL−OE細胞中においてNAD+/NADHの比率並びにピルビン酸及び乳酸レベルが不変であることを見出した。増加した脂肪分解に対する他の可能な理由は、癌細胞の重要なエネルギー源として役立つことができる、β酸化のためのFFA基質を産生することであるかもしれなかった。しかしながら、発明者らは、β酸化の律速段階を触媒する、カルニチンパルミトイルトランスフェラーゼ1(CPT1)の阻害剤が、癌細胞(sh対照、shMAGL、又はMAGL−OE)の移動活性に影響を与えないことを見出した。CPT1遮断は、sh対照細胞においてのみ増加したC20:4FFAを除き、癌細胞中のFFAレベルを変化することもできなかった。さらなる研究により侵襲性癌細胞における減少したCPT1発現が明らかとなったが、そのことは、MAGL脂肪酸代謝経路の病原性効果の下流の伝達物質としてのβ酸化に関する役割に対するエビデンスを提供している。
【0117】
FFAが膜構造物及びシグナル伝達分子の産生及び再構築のための基礎的な構成要素であることを考慮すると、MAGLにおける攪乱が悪性腫瘍にとって重要な幾つかの脂肪依存性生化学的ネットワークに影響を与えることを期待できるかもしれない。この仮説を検証するために、発明者らは、1)対照癌細胞と比較したMAGL−OE(OVCAR3、MUM2C)、及び2)sh対照癌細胞と比較したshMAGL(SKOV3、C8161)の比較を含む、変更したMAGL活性を有する癌細胞モデルのリピドーム解析を行った。癌細胞の有機抽出物を、数種の主要な脂質ファミリーをプロファイルする、非標的液体クロマトグラフィー−質量分析(LC−MS)プラットホームを使用して分析し、比較群の間で顕著に変化したレベルを有する代謝産物を、XCMSソフトウェアを使用して同定した。全体的なプロファイルを補完するために、発明者らは、標準的なリピドミック方法による検出にとっては低すぎる存在量である、特別な生理活性脂質(例えば、プロスタグランジン)の標的測定も行った。結果として得られたデータセットは、次にMAGLによって制御される脂質代謝産物の共通のサインを同定するために採掘されたが、発明者らは、当該産物を、MAGL−OE細胞において著しく増加し又は減少し、並びにそのそれぞれの対照群と比較してshMAGL細胞において逆の変化を示した代謝産物と定義した(図19A、19B)。
【0118】
幾つかのリゾリン脂質(LPC、LPA、LPE)、エーテル脂質(MAGE、アルキルLPE)、ホスファチジン酸(PA)、及びプロスタグランジンE2(PGE2)を含む、MAGL脂肪酸代謝ネットワーク中の脂質の殆どは、MAGL−OE及びshMAGL細胞において、それぞれ一貫して増加及び減少するというFFAにたいする同様のプロファイルを示した。MAGのみが、逆のプロファイルを示すことが見出された(shMAGL及びMAGL−OE細胞において、それぞれ増加及び減少する)。興味深いことに、この全体のリピドミックなサインは、その非侵襲性のカウンターパートと比較したときに侵襲性癌細胞においても実質的に認められた(例えば、それぞれMUM2Cと比較したC8161及びOVCAR3と比較したSKOV3)。これらの発見は、MAGLが侵襲性癌細胞において、FFA及びMAGだけでなく多数の二次脂質代謝産物からも成る脂質代謝ネットワークを制御することを証明している。発明者らは、JZL184によるMAGLの急性阻害が、特徴的な二次リピドミックなサインを引き起こすことを見出したことにさらに興味をそそられた。shMAGL細胞において認められた通りJZL184処理は、高MAGLレベルを発現する各癌細胞株においてLPA及びPGE2の減少を引き起こした(図19C、19D)。対照的に、以前にも説明した通り、LPC及びLPEの(減少というより)増加がJZL184処理細胞において認められた(図20)。これらのデータは、急性及び慢性のMAGL遮断が癌細胞において明確なメタボロミック効果を引き起こすことを示しているが、おそらくFFAの長期枯渇と対比した短期枯渇の特異的な成果を反映している。最後に、ホスファチジルコリン、ホスファチジルエタノールアミン、セラミド、スフィンゴシン−1−リン酸、コレステロール、ジアシルグリセロール、及びトリアシルグリセロールを含む幾つかのクラスの脂質は、MAGL活性の減少又は増加によって影響を受けなかったが、このことは、それらが、この酵素により制御される脂肪酸代謝ネットワークの制限成分であることを強調している。
【0119】
MAGL脂肪酸代謝ネットワーク内にLPA及びPGE2を含む数種類の前腫瘍形成性脂質メッセンジャーが存在するが、それらは癌細胞の侵襲性を促進することが報告されている。代謝標識研究により、侵襲性癌細胞がMAG及びFFAの両方をLPA及びPGE2に変換することができ、MAGに関しては、この変換がJZL184によって遮断されることが確認された(データ不掲載)。興味深いことに、LPA又はPGE2どちらかの処理は、sh対照細胞の移動に影響を与えない濃度において、shMAGL癌細胞の障害した移動を救出した(図19E)。逆に、MAGL−OE癌細胞の増加した移動活性は、Gi/Go阻害剤である百日咳毒素によって完全に遮断された(図19F)。最後に、MAGL−OE細胞において認められた刺激された移動の程度は、外からのLPA添加によって引き起こされる最大効果に匹敵した(図19G)。まとめると、これらのデータは、Gタンパク質共役型受容体に作用して高い移動活性を促進する生理活性脂質の産生を増加させることによって少なくとも部分的に、MAGLが癌の病原性に寄与することを示唆している。MAGL脂肪酸代謝ネットワークが他の前腫瘍形成性シグナル伝達系と相互作用するか否かを評価するために、発明者らは、shMAGL及びsh対照細胞を上皮増殖因子受容体(EGFR)の阻害剤で処理した。shMAGL細胞は、EGFR遮断の抗移動効果(図19H)及び抗生存効果(データ不掲載)に対する著しく高められた感受性を示す。これらのデータは、以前の発見と一致しており、このことは、癌細胞におけるLPAとEGFRシグナル伝達経路との間の実質的なクロストーク(相互干渉)を示している。
【0120】
実施例12.腫瘍発症機序におけるMAGL研究のための実験材料及び方法
実験材料C8161、MUM−2B、MUM−2C、及び231MFPを除き、全ての細胞株は、NCI60癌細胞株パネルの部分であり、米国国立癌研究所開発治療プログラムから入手した。C8161、MUM−2B、及びMUM−2C細胞株は、マリー・ヘンドリックス(Mary Hendrix)から提供を受けた。231MFP細胞は、既述の通り(ジェサニーら、2004年;前掲書)、MDA−MB−231細胞の外植した異種移植片腫瘍から作成した。C15:0、C16:0、C18:0及びC18:1 FFA、C18:1 FFA−OH及びC16:0、C18:0、C18:1、及びC20:4MAGは、シグマ社から購入した。C12:0MAGE及びC15:0MAGは、それぞれアレッキス・バイオケミカルズ社(Alexis Biochemicals)及びヌ・チェック・プリプ社(Nu−check Prep)から購入した。C20:4FFA及びPGE2は、ケイマン・ケミカルズ社から購入した。他の代表的な種々のクラスの脂質の脂質標準品は、シグマ社、ケイマン・ケミカルズ社、ヌ・チェック・プリプ社、又はアヴァンチー・リピッズ社(Avanti Lipids)から購入した。FPローダミン及びFPビオチンは、既述の方法に従って合成した。JZL184は、以前詳述した通り合成した。ヒトMAGL抗体(抗N末端アミノ酸1−121)は、ノヴァス・バイオロジカルズ社(Novus Biologicals)から購入した。
【0121】
癌細胞におけるMAGLの薬理学的阻害薬理学的阻害研究は、既述の通り行った(チャングら(Chiang et al.)、「ケミストリー&バイオロジー(Chem.Biol.)」、2006年、第13巻:p.1041−1050)。癌細胞は、ウシ胎児血清10%(v/v)添加RPMI培地1640中、5%CO2/95%空気の加湿した雰囲気で維持した。集密度80%で細胞をトリプシン処理し、血球計数板を使用して計数し、そして1x106個の細胞を6cmのディッシュに播いた(亜集密的)。播種の総20時間後に、細胞をPBSで2回洗浄し、阻害剤を無血清RPMI培地中、0.1%でJZL184(1μM)又は溶媒(DMSO)とインキュベートした。4時間のインキュベーション後、細胞を採取しABPP又はLC−MSによって分析した。
【0122】
ABPP−MudPITによる癌細胞プロテオーム由来のセリンヒドロラーゼ活性の同定及び比較定量各々のヒト癌細胞株由来の可溶性及び不溶性のプロテオーム画分を既述の通り作成し(ジェサニーら、2002年;前掲書)、ABPP−MudPITによって分析した(ジェサニーら、2005年;前掲書)。FPビオチンンによるプロテオームの標識化反応の標準的な条件は下記の通りであった:プロテオームを最終タンパク質濃度1.0μg/μlに調節し、プロテオーム1000μgを5μMのFPビオチンにより室温(RT)で2時間標識した。インキュベーション後、不溶性プロテオームを4℃で1時間回転して1%トリトンXにより可溶化した。可溶性及び不溶性プロテオーム画分由来のFP標識タンパク質の増強は、既述の通り行った(キッドら(Kidd et al.)、「バイオケミストリー(Biochemistry)」、2001年、第40巻:p.4005−15)。アビジン強化プロテオームを、1)1%SDS、2)6M尿素、3)50mMトリスpH8.0で8分間2回洗浄し、最終的に200μlの8M尿素50mMトリスpH8.0に再懸濁した。次に、ビーズ上の消化をするための試料を10mMのTCEPによる室温、30分間での還元によって調製し、当該試料を12mMのヨードアセトアミドにより暗所下、室温で30分間アルキル化した。消化は、試料を50mMトリスpH8.0で2M尿素に希釈後に、トリプシン(2mMのCaCl2存在下5.5μg/μlの3μL)により37℃で12時間行った。最後に、ペプチド試料を5%ギ酸の最終濃度に酸性化した。
【0123】
消化したペプチド混合物を二相性(強陽イオン交換/逆相)のキャピラリ―カラムにし、タンデム型質量分析法を組み合わせた二次元液体クロマトグラフィー(2D−LC)分離によって分析した。ペプチドは、5段階のMudPIT実験で溶出し(0、10、25、80及び100%の塩濃度段階(salt bumps)を使用)、データは、ダイナミック排出点灯(60秒)でデータ依存性獲得モードに設定した、イオントラップ型質量分析計LTQ(サーモ・サイエンティフィック社製(Thermo Scientific))で収集した。特に、1のMSサーベイ(ms1)フルスキャンには、7のms2のスキャンが続いた。ms2のスペクトルデータは、ロー・エックストラクター(1.9.1版)(RAW Xtractor(version1.9.1))を使用して未加工のファイルから抽出した。ms2スペクトルデータは、結果として総数22935のユニークな収録項目に帰着した、各集合遺伝子の識別子に関連する3.26版のヒトIPIデータベース由来の最も長期にわたる収録項目を含む、注文製のデータベースを対照してセクウェスト・アルゴリズム(3版)(SEQUEST(Version 3.0))を使用して検索した。加えて、これらの収録項目を置き換え、フォースル・ディスカバリー・レート(false−discovery rate)の評価のためにデータベースに付加した。SEQUEST検索は、メチオニン残基(16Da)の酸化、システイン残基(アルキル化のため57Da)のstatic修飾、非酵素的特異性及び前駆質量に対する±1.5Da及び生成物イオン質量に対する±0.5Daの質量許容誤差を考慮した。一致するms2スペクトルの結果を集合し、DTAセレクト(2.0.27版)(DTASelect(version2.0.27))を使用してフィルター処理した。ペプチドの最大偽陽性率0.1%を達成するために、二次判別分析を使用した。
【0124】
得られた全プロテオミックのデータは、最初にセリンヒドロラーゼに関して、既知のこのファミリーのメンバーに相応する、インタープロ(InterPro)ドメイン識別子(IPR000120、IPR000379、IPR001031、IPR001087、IPR001466、IPR002641、IPR002642、IPR002921、IPR004177、IPR005181、IPR007751、IPR008262、IPR010662)の手作業で集められたリストを使用してフィルター処理した。セリンヒドロラーゼの結果のセットを、次に、試験した少なくとも一の癌細胞株について、「無プローブ」対照プロテオームと比較してFPローダミン処理プロテオームにおいて、>10倍の高いスペクトルカウントを示す酵素に対してフィルター処理した。これらの計算のために、膜及び可溶性プロテオーム由来のスペクトルカウント値を、各癌細胞株由来の各セリンヒドロラーゼについて合併した。この分析は、〜50のセリンヒドロラーゼ活性のリストに帰着した。以前の研究は、比較定量が、比較中の二のグループの少なくとも一グループにおいて、平均≧10のスペクトルカウントを示すタンパク質にかなり限定されることを明らかにしている(ジェサニーら(Jessani et al.)、2005年;前掲書)。このフィルター処理を使用して(ここで、比較グループは、その非侵襲性カウンターパートと比較した侵襲性癌細胞株と定義した)、発明者らは、比較定量のための〜35のセリンヒドロラーゼを同定した。これらのセリンヒドロラーゼの中で、KIAA1363及びMAGLが侵襲性関連ヒドロラーゼ用に定義された下記の判定基準に適った:1)その非侵襲性カウンターパートと比較して三の全ての侵襲性癌細胞株における平均スペクトルカウントが>2倍である、並びに2)侵襲性及び非侵襲性癌細胞株対の間の較差に対するp値が<0.01である。
【0125】
癌細胞プロテオームのABPP及び加水分解活性アッセイABPP−MudPIT又はゲルに基づくABPPによる癌細胞プロテオーム由来のセリンヒドロラーゼ活性の同定及び比較定量は、既述の通りそれぞれFPビオチン(5μM)及びFTローダミン(2μM)を使用して行った(ジェサニーら(Jessani et al.)、「プロシーディングス・オブ・ザ・ナショナル・アカデミー・オブ・サイエンシズ・オブ・ザ・ユナイテッド・ステイツ・オブ・アメリカ(Proc.Natl.Acad.Sci.U.S.A.)」、2002年、第99巻:p.10335−10240;及びジェサニーら(Jessani et al.)、「ネイチャー・メソッズ(Nat.Methods)」、2005年、第2巻:p.691−697)。C20:4MAG加水分解活性アッセイは、既述の通り行った(ブランクマンら(Blankman et a.)、「ケミストリー&バイオロジー(Chem.Biol.)」、2007年、第14巻:p.1347−1356)。ABPP実験に関しては、細胞可溶化プロテオームを2μM FPローダミンにより室温で30分間処理した(全反応液量50μl)。反応を1容量の標準4xSDS/PAGEローディング緩衝液で停止し(還元)、SDS/PAGE(アクリルアミド10%)によって分離し、日立FMBio IIeフラットベッド蛍光スキャナ(Hitachi FMBイオIIe flatbed fluororescence scanner)(ミライバイオ社製(MiraiBio))によりインゲルで可視化した。統合したバンドの強度を標識タンパク質に対し計算し、各酵素活性のレベルを測定するために独立した細胞三試料から平均をとった。IC50値は、各阻害剤濃度での3回の試行からの得た用量反応曲線から、シグマプロット10.0(SigmaPlot 10.0)を使用して決定した。MAGL発現もまた標準法を使用してウェスタンブロッティングによって評価した。
【0126】
加水分解活性アッセイのために、擦過により細胞を採取する前に、細胞を生体内原位置で無血清RPMI培地中JZL184(1μM)により4時間処理した。次に、トリス緩衝液中の細胞溶解物(20μg)を脂質(100μM、例えばMAGL活性に対してはC20:4MAG)と室温で30分間、200μl容量でインキュベートした。反応を600μlの2:1クロロホルム:メタノールにより停止し、内部標準のC15:0FFA又はC12:0MAGE10nmolを添加した。産物は有機層に抽出され、当該有機層を抽出してLC−MSに直接注入した。LC−MSの設定は、既述の通りとした(ブランクマンら(Blankman et al.)、2007年;前掲書)。産物レベルは(例えば、MAGL活性に対するC20:4FFA)、内部標準レベルと比較して定量し、一定の内部標準レベルと変化する脂質濃度の間の標準曲線を作成した。酵素反応の直線位相の間で比活性を決定した(例えば、基質20%未満利用)。
【0127】
ヒト原発性卵巣腫瘍被験者は、診断され、ブリガム・アンド・ウィメンズ病院(Brigham and Women‘s Hospital)及びダナファーバー癌センター(Dana−Farber Cancer Center)(ボストン、マサチューセッツ州(Boston、MA))で卵巣腫瘍の治療を受けていた。すべて被験者由来の生物学的な検体を収集し、ブリガム・アンド・ウィメンズ病院の被験者委員会によって承認されたプロトコルに従って保存記録した。病理組織学的診断は、ブリガム・アンド・ウィメンズ病院の婦人科の病理医によって判定された。腫瘍を分類し、産婦人科の国際連盟システム(FIGO)に従って悪性度の類別をした。本研究に関し、10の良性及び13の高悪性度の悪性卵巣腫瘍試料をMAGL活性及び代謝産物測定のために使用した。良性の症例には、良性嚢腫、卵巣線維腫及び良性漿液性嚢胞腺腫が含まれ、一方悪性の症例は、全て高悪性度の漿液性乳頭状癌であった。新鮮な腫瘍組織をメスで2−5mm片に切り離し、個々にアルミ箔に包み、液体窒素中で瞬間凍結(snap frozen)し、−80℃の冷凍庫に保存した。MAGL活性及びFFAレベルは上述の通り測定した。
【0128】
ヒト癌細胞株におけるRNA干渉研究RNA干渉の研究は、既述の通り行った(チャングら(Chiang et al.)、2006年)。手短に言えば、低分子ヘアピン型RNA構築物をpLP−レトロQ(pLP−RetroQ)アクセプターシステムにサブクローンし、アンフォパック(AmphoPack)−293細胞株(クロンテック社製(Clontech))を使用してレトロウイルスを作成した。RNA干渉の研究に利用したヘアピン型オリゴヌクレオチドは;MAGL用には(shMAGL1)、5‘−CAACTTTCAAGGTCCTTGC−3’(配列番号3)及び(shMAGL2)、5‘−AGACTACCCTGGGCTTCC−3’(配列番号4);sh対照用には(shDPPIV)5‘−GATTCTTCTGGGACTGCTG−3’(配列番号5)であった。sh対照構築物は、侵襲性と非侵襲性癌細胞(DPPIV)の間で一貫して変化しない他と別なセリンヒドロラーゼを標的とするよう設計した。内在性タンパク質を標的とするsh対照構築物(非機能的なスクランブルド構築物よりは)を使用することが、RNA干渉機構の一般的な活性による非特異的な効果を制御する。培養1−6日の上清を含むウイルスを収集し、超遠心分離で濃縮し、10μg/mlのポリブレンの存在下に48時間安定して細胞を感染するために使用した。感染に続いて、ピューロマイシン(C8161、MUM2C、SKOV3、及び231MFPに対しては1μg/ml、OVCAR3に対しては0.3μg/ml)を含む培地で3日間選抜を行ったが、それは、レトロウイルスベクターがこの選択マーカーを含んでいたからである。感染した細胞を増殖し、ABPPによって酵素活性の損失及びC20:4MAG加水分解活性に関し試験を行った。
【0129】
ヒト癌細胞株における過剰発現の研究安定したMAGL過剰発現は、RNA干渉研究に関し上述した通りAmphoPack−293細胞株を使用してレトロウイルスを作成し、MAGL遺伝子をpMSCVpuroベクター(pMSCVpuro vector)(クロンテック社製)にサブクローニングすることによって達成した。ヒトMAGL構築物は、プライマー、5’−GCTCTCGAGGCCGCCATGCCAGAGGAAAGTTCC−3’(配列番号6)及び5’−AGCTGAATTCTCAGGGTGGGGACGCAGTTCCTG−3’(配列番号7)によるPCRによって作成した。PCR産物は、XhoI及びEcoRIの制限サイトを使用してpMSCVCpuro(クロンテック社製)にサブクローニングした。
【0130】
細胞移動、細胞生存、及び浸潤の研究移動アッセイは、コラーゲン10μg/mlで被覆した孔径8μmの膜を有するトランスウェル・チャンバー(コーニング社製)(Transwell chamber(Corning))中で37℃で4時間行った。移動アッセイ開始前、全24時間に、癌細胞株を6cmのディッシュ当たり1x106細胞の濃度で播いた。移動アッセイ開始時に、細胞をPBSで2回洗浄して採取し、次に無血清培地で4時間、血清飢餓とした。この4時間の間に、阻害剤、脂質又は溶媒(0.1%DMSO)を細胞とプレインキュベートした。血清飢餓細胞をトリプシン処理し、1400gで3分間回転し、再懸濁して計数した。(阻害剤、脂質又は溶媒を含有する)無血清培地200μl中の50,000個の細胞をトランスウェルの上部室(chamber)に播種した。阻害剤、脂質又は溶媒を下部室にも添加し、細胞を5時間(C8161及び231MFP細胞)又は20時間(SKOV3、MUM2C及びOVCAR3細胞)移動させた。次にフィルターを固定し、ディフ・クイック(デイド・ベーリング社製)(Diff−Quik(Dade Behring))で染色した。室を通って移動しなかった細胞を綿球で取り除いた。移動した細胞を倍率400xで計数し、各移動室から4視野を独立して計数した。1移動室についての4視野における細胞の平均をn=1と表す。
【0131】
細胞生存アッセイは、細胞増殖試薬WST−1(ロッシュ社製)を使用して既述の通り行った(ロカら(Roca et al.)、「ザ・ジャーナル・オブ・バイオロジカル・ケミストリー(J.Biol.Chem.)」、2008年、第283巻、p.25057−73;及びシッディキュイら(Siddiqui et al.)、「ブレスト・カンサー・リサーチ(Breast Cancer Res)」、2005年、第7巻、p.R645−654)。細胞をPBSで2回洗浄し、トリプシン処理によって採取し、PBSで2回洗浄し、1400gで遠心分離し、無血清培地に再懸濁した。WST−1(20μl)の添加前、0、24又は48時間に、20,000細胞を96ウェルプレートの無血清培地200μl中で37℃/5%CO2で4時間放置した。分光光度計を使用して450nmで吸光度を測定した。浸潤アッセイは、BDマトリゲルインベージョンチャンバー(BD Mtrigel Invasion Chambers)を使用して製造者のプロトコルに従って行った。
【0132】
腫瘍異種移植研究ヒト癌異種移植片は、癌細胞株をC.B17SCIDマウス(タコニック・ファームス社製(Taconic Farms))の側腹部に異所的に移植することによって確立した。手短に言えば、細胞をPBSで2回洗浄し、トリプシン処理し、そして血清を含有する培地で採取した。次に、採取した細胞を無血清培地で2回洗浄し、濃度2.0x104細胞/μlで再懸濁し、100μlを注入した。腫瘍の増殖は、3日ごとにカリパスで測定した。HFD研究のために、マウスを癌細胞注入の2週間前に60kcal%脂肪食(調査食)の状況に置いた。3−6日ごとにマウスの体重を量った。慢性JZL184処理研究のために、ポリエチレングリコール300(4μL/g)の経口胃内投与によって、マウスをJZL184又は溶媒で日に一回(毎日ほぼ同時間)処理した。処理は、癌細胞の異所的注入後直ちに開始した。
【0133】
癌細胞中の脂質測定脂質測定は、既述のプロトコルと同様に行った(チャングら(Chiang et al.)、2006年;前掲書)。癌細胞を、細胞プロファイルへの血清由来脂質の寄与を最小にするために、無血清培地で4時間生育した。細胞を(1x106細胞/6cmディッシュ、集密度80%)、リン酸緩衝生理食塩水(PBS)で2回洗浄し、1、400gで遠心分離によって分離し、クロロホルム:メタノール:トリス緩衝液の2:1:1混合液4ml中Dounceホモジナイザーで均質化した。試料は、次の合成標準品の存在下に均質化した:C12:0MAGE(10nmol)、C15:0FFA(10nmol)。有機層及び水層は、2000g、5分間の遠心分離によって分別し、有機層を収集した。水層を5%ギ酸の添加によって酸性化し(LPA等の代謝産物に対し)、続いてクロロホルム2mlを添加した。混合液をボルテックスし、有機層を併せてN2下で完全に乾燥し、クロロホルム100μlに溶解し、その30μlをLC−MSによって分析した。
【0134】
LC−MS分析は、アジレント1100LC−MSD SL装置(Agilent 1100 LC−MSD SL instrument)を使用して行った。LC分離は、プレカラム(C18、3.5μm、2mmx20mm)と共にフェノモネックス社製(Phenomonex)のジェミニ(Gemini)逆相C18カラム(直径5μmの粒子、50mmx4.6mm)によって行った。移動相Aは、95:5の比の水:メタノールから構成し、移動相Bは、60:35:5の比の2−プロパノール、メタノール、及び水から構成した。0.1%ギ酸及び0.1%水酸化アンモニウム等の溶媒改良剤を、それぞれポジティブ及びネガティブイオン化モードにおいて、LC分離能を改善するためと同様にイオン形成を補助するために、使用した。各操作の流速は、クロロホルム注入に付随する背圧を軽減するために、5分間、0.1ml/分で開始した。濃度勾配は、0%Bで開始し、流速0.4ml/分で45分間かけて直線的に100%Bに増加し、続いて17分間は流速0.5ml/分の100%Bの均一濃度勾配とし、次に流速0.5ml/分で8分間かけて0%Bに平衡化した。MS分析は、エレクトロンスプレーイオン化(ESI)源によって行った。キャピラリ電圧は3.0kVに設定し、フラグメンター電圧は100Vに設定した。乾燥ガス温度は350℃、乾燥ガス流束は10L/分、及びネブライザ圧力は35psiであった。
【0135】
リピドーム解析は、標的及び非標的モードの両方でC−MS分析によって行った。標準的な脂質クラスは、選択イオン検出(SIM)を使用して標的化によって定量し(非標的化分析によって定量した高存在量の代謝産物を除く)、代謝産物の同一性は、代謝産物のそれぞれの標準品との共溶出、及びアジレント6520精密質量QTOF−MS−MS(Agilent 6520 Accurate Mass QTOF−MAS−MS)による精密質量分析により測定される10ppm以内の質量精度によって確認した。これらの脂質は、ピーク下の面積を測定することによって定量し、内部標準に対し規準化した(ポジティブモードにおけるC12:0MAGE又はネガティブモードにおけるC15:0FFA)。各々の脂質種のpmoles/1x106細胞における絶対定量は、10nmolのC12:0MAGE及び/又はC15:0FFAと対照して分析した各クラスのそれぞれの脂質の標準曲線の作成に基づいたが、そこでは脂質及び標準品の相対抽出効率も考慮に入れた。MAGは、内部標準C15:0MAG(C12:0MAGEの代わりに)に基づいても定量したが、同一の結果が得られた。MSインターフェースにおいてMAGの脂肪酸アシルアニオン種への5%未満の分解が存在した。発明者らは、非天然のC15:0MAG種(これもまた、内在性MAG種と同様の分解パターンを有する)と対照してMAGレベルを定量したので、インターフェースにおける極小のMAG分解レベルは、MAGの絶対定量を妨げなかった。MAG及びFFA定量のための動的線形範囲は、それぞれ0.6−10,000pmoles及び1−10,000pmolesであった。非標的化データを質量範囲200−1200Daを使用してアジレント1100LC−MSD SL装置に収集し、計算的な分析のためにコモン・データ・フォーマット(CDF)として転送した。試料対の間の違った発現代謝産物は、多数のLC−MSトレースからの質量ピークの相対シグナル強度を一列に並べ定量する、XCMS分析物プロファイリングソフトウェアを使用することによって同定した。重要な阻害剤、shMAGL又はMAGL−OE感受性のピーク変化は、内部標準に対し規準化したピーク下面積を使用することにより手作業の定量によって確認した。
【0136】
本明細書で記載された実施例及び実施態様は、例示的な目的のためだけにあること、並びに当該実施例等に照らしての種々の修正又は変更は、当業者に提案されており、そして本出願及び別記の特許請求の範囲の精神及び範囲内に含まれることを理解されたい。本明細書に記載された方法及び材料と類似の又は同等の如何なる方法及び材料も本発明の実施又は試験に使用することができるのであるが、好ましい方法及び材料が記載されている。
【0137】
本明細書で引用した、全ての刊行物、ジェンバンク配列、ATCC寄託、特許及び特許出願は、これによってそっくりそのまま及び全ての目的のために、各々が個々にそのように表示されているかのような言及によって明確に援用される。
【特許請求の範囲】
【請求項1】
2−アラキドノイルグリセロール(2−AG)媒介内在性カンナビノイドシグナル伝達を刺激するための方法。
【請求項2】
化合物が脂肪酸アミドヒドロラーゼ(FAAH)にMAGLの阻害に対して選択的である請求項1記載の方法。
【請求項3】
化合物がFAAHにMAGLの阻害に対して少なくとも100倍の選択性を有する請求項2記載の方法。
【請求項4】
がである請求項1記載の方法。
【請求項5】
化合物が下記に示す式Iの構造を有する請求項1記載の方法:
【化1】
式中、XはN又はCH、R1はH又はOHであり、そしてR2及びR3の各々は、下記の構造の一つを有する。
【化2】
【請求項6】
化合物が脂肪酸アミドヒドロラーゼ(FAAH)及びα/βヒドロラーゼ6(ABHD6)の両方にMAGLの阻害に対して選択的である請求項1記載の方法。
【請求項7】
化合物が下記に示す式Iの構造を有する請求項6記載の方法:
【化3】
式中、XはCH、R1はOHであり、そしてR2及びR3の各々は、下記の構造の一つを有する。
【化4】
【請求項8】
がである請求項1記載の方法。
【請求項9】
が疼痛、炎症、抑鬱又は不安を患っている請求項1記載の方法。
【請求項10】
内在性カンナビノイドシグナル伝達するの症状を治療又は回復する方法。
【請求項11】
症状が、疼痛、抑鬱、、炎症性症候群、心血管障害、代謝障害、卒中、アルツハイマー病、多発性硬化症又は緑内障である請求項10記載の方法。
【請求項12】
症状が2−AGシグナル伝達により媒介される又は関連する請求項10記載の方法。
【請求項13】
化合物が下記に示す式Iの構造を有する請求項10記載の方法:
【化5】
式中、XはN又はCH、R1はH又はOHであり、そしてR2及びR3の各々は、下記の構造の一つを有する。
【化6】
【請求項14】
式中、XがCH、R1がOHであり、そしてR2及びR3の各々が、下記に示す構造を有する請求項13記載の方法。
【化7】
【請求項15】
化合物がFAAHにMAGLの阻害に対して選択的である請求項10記載の方法。
【請求項16】
化合物がFAAH及びABHD6の両方にMAGLの阻害に対して選択的である請求項10記載の方法。
【請求項17】
がである請求項10記載の方法。
【請求項18】
下記に示す式Iの化合物:
【化8】
式中、XはN又はCH、R1はH又はOHであり、そしてR2及びR3の各々は、下記の構造の一つを有する。
【化9】
【請求項19】
式中、XがCH、R1がOHであり、そしてR2及びR3の各々が、下記に示す構造を有する請求項18記載の方法。
【化10】
【請求項20】
改良された特性を有するモノアシルグリセロールリパーゼ(MAGL)の阻害剤同定方法:
(a)既知のMAGL阻害剤の一以上の構造アナログ合成;
(b)MAGL阻害剤と比較して改良された生物学的又は薬学的特性を有するアナログを同定するためのアナログに関する機能アッセイ実施;それに改良された特性を有するMAGL阻害剤同定;ここで既知のMAGL阻害剤は、下記に示す式Iの化合物である
【化11】
式中、XはN又はCH、R1はH又はOHであり、そしてR2及びR3の各々は、下記の構造の一つを有する。
【化12】
【請求項21】
式中、XがCH、R1がOHであり、そしてR2及びR3の各々が、下記に示す構造を有する請求項20記載の方法。
【化13】
【請求項22】
改良された生物学的又は薬学的特性がMAGLに対する亢進した阻害活性である請求項20記載の方法。
【請求項23】
改良された生物学的又は薬学的特性が他の脳セリンヒドロラーゼにMAGLに対する増加した選択性である請求項20記載の方法。
【請求項24】
侵襲性の腫瘍細胞の増殖を阻害するための方法。
【請求項25】
腫瘍細胞が癌を有する又は癌になる危険のあるに存在する請求項24記載の方法。
【請求項26】
腫瘍細胞が、乳腺腫瘍細胞、卵巣腫瘍細胞、黒色腫細胞、肺腫瘍細胞又は脳腫瘍細胞である請求項25記載の方法。
【請求項27】
化合物が脂肪酸アミドヒドロラーゼ(FAAH)にMAGLの阻害に対して選択的である請求項24記載の方法。
【請求項28】
化合物が下記に示す式Iの構造を有する請求項27記載の方法:
【化14】
式中、XはN又はCH、R1はH又はOHであり、そしてR2及びR3の各々は、下記の構造の一つを有する。
【化15】
【請求項29】
化合物がMAGLに特異的な阻害ポリヌクレオチドである請求項24記載の方法。
【請求項30】
阻害ヌクレオチドが低分子緩衝RNA(siRNA)、マイクロRNA(miRNA)、低分子ヘアピン型RNA(shRNA)、アンチセンス核酸及び相補DNA(cDNA)である請求項29記載の方法。
【請求項31】
腫瘍の症候を治療又は回復するための方法。
【請求項32】
腫瘍が、乳腺腫瘍、卵巣腫瘍、黒色腫、肺腫瘍又は脳腫瘍である請求項31記載の方法。
【請求項33】
化合物が脂肪酸アミドヒドロラーゼ(FAAH)にMAGLの阻害に対して選択的である請求項31記載の方法。
【請求項34】
化合物が下記に示す式Iの構造を有する請求項33記載の方法:
【化16】
式中、XはN又はCH、R1はH又はOHであり、そしてR2及びR3の各々は、下記の構造の一つを有する。
【化17】
【請求項35】
化合物がMAGLに特異的な阻害ポリヌクレオチドである請求項31記載の方法。
【請求項36】
阻害ヌクレオチドが低分子緩衝RNA(siRNA)、マイクロRNA(miRNA)、低分子ヘアピン型RNA(shRNA)、アンチセンス核酸及び相補DNA(cDNA)である請求項35記載の方法。
【請求項1】
2−アラキドノイルグリセロール(2−AG)媒介内在性カンナビノイドシグナル伝達を刺激するための方法。
【請求項2】
化合物が脂肪酸アミドヒドロラーゼ(FAAH)にMAGLの阻害に対して選択的である請求項1記載の方法。
【請求項3】
化合物がFAAHにMAGLの阻害に対して少なくとも100倍の選択性を有する請求項2記載の方法。
【請求項4】
がである請求項1記載の方法。
【請求項5】
化合物が下記に示す式Iの構造を有する請求項1記載の方法:
【化1】
式中、XはN又はCH、R1はH又はOHであり、そしてR2及びR3の各々は、下記の構造の一つを有する。
【化2】
【請求項6】
化合物が脂肪酸アミドヒドロラーゼ(FAAH)及びα/βヒドロラーゼ6(ABHD6)の両方にMAGLの阻害に対して選択的である請求項1記載の方法。
【請求項7】
化合物が下記に示す式Iの構造を有する請求項6記載の方法:
【化3】
式中、XはCH、R1はOHであり、そしてR2及びR3の各々は、下記の構造の一つを有する。
【化4】
【請求項8】
がである請求項1記載の方法。
【請求項9】
が疼痛、炎症、抑鬱又は不安を患っている請求項1記載の方法。
【請求項10】
内在性カンナビノイドシグナル伝達するの症状を治療又は回復する方法。
【請求項11】
症状が、疼痛、抑鬱、、炎症性症候群、心血管障害、代謝障害、卒中、アルツハイマー病、多発性硬化症又は緑内障である請求項10記載の方法。
【請求項12】
症状が2−AGシグナル伝達により媒介される又は関連する請求項10記載の方法。
【請求項13】
化合物が下記に示す式Iの構造を有する請求項10記載の方法:
【化5】
式中、XはN又はCH、R1はH又はOHであり、そしてR2及びR3の各々は、下記の構造の一つを有する。
【化6】
【請求項14】
式中、XがCH、R1がOHであり、そしてR2及びR3の各々が、下記に示す構造を有する請求項13記載の方法。
【化7】
【請求項15】
化合物がFAAHにMAGLの阻害に対して選択的である請求項10記載の方法。
【請求項16】
化合物がFAAH及びABHD6の両方にMAGLの阻害に対して選択的である請求項10記載の方法。
【請求項17】
がである請求項10記載の方法。
【請求項18】
下記に示す式Iの化合物:
【化8】
式中、XはN又はCH、R1はH又はOHであり、そしてR2及びR3の各々は、下記の構造の一つを有する。
【化9】
【請求項19】
式中、XがCH、R1がOHであり、そしてR2及びR3の各々が、下記に示す構造を有する請求項18記載の方法。
【化10】
【請求項20】
改良された特性を有するモノアシルグリセロールリパーゼ(MAGL)の阻害剤同定方法:
(a)既知のMAGL阻害剤の一以上の構造アナログ合成;
(b)MAGL阻害剤と比較して改良された生物学的又は薬学的特性を有するアナログを同定するためのアナログに関する機能アッセイ実施;それに改良された特性を有するMAGL阻害剤同定;ここで既知のMAGL阻害剤は、下記に示す式Iの化合物である
【化11】
式中、XはN又はCH、R1はH又はOHであり、そしてR2及びR3の各々は、下記の構造の一つを有する。
【化12】
【請求項21】
式中、XがCH、R1がOHであり、そしてR2及びR3の各々が、下記に示す構造を有する請求項20記載の方法。
【化13】
【請求項22】
改良された生物学的又は薬学的特性がMAGLに対する亢進した阻害活性である請求項20記載の方法。
【請求項23】
改良された生物学的又は薬学的特性が他の脳セリンヒドロラーゼにMAGLに対する増加した選択性である請求項20記載の方法。
【請求項24】
侵襲性の腫瘍細胞の増殖を阻害するための方法。
【請求項25】
腫瘍細胞が癌を有する又は癌になる危険のあるに存在する請求項24記載の方法。
【請求項26】
腫瘍細胞が、乳腺腫瘍細胞、卵巣腫瘍細胞、黒色腫細胞、肺腫瘍細胞又は脳腫瘍細胞である請求項25記載の方法。
【請求項27】
化合物が脂肪酸アミドヒドロラーゼ(FAAH)にMAGLの阻害に対して選択的である請求項24記載の方法。
【請求項28】
化合物が下記に示す式Iの構造を有する請求項27記載の方法:
【化14】
式中、XはN又はCH、R1はH又はOHであり、そしてR2及びR3の各々は、下記の構造の一つを有する。
【化15】
【請求項29】
化合物がMAGLに特異的な阻害ポリヌクレオチドである請求項24記載の方法。
【請求項30】
阻害ヌクレオチドが低分子緩衝RNA(siRNA)、マイクロRNA(miRNA)、低分子ヘアピン型RNA(shRNA)、アンチセンス核酸及び相補DNA(cDNA)である請求項29記載の方法。
【請求項31】
腫瘍の症候を治療又は回復するための方法。
【請求項32】
腫瘍が、乳腺腫瘍、卵巣腫瘍、黒色腫、肺腫瘍又は脳腫瘍である請求項31記載の方法。
【請求項33】
化合物が脂肪酸アミドヒドロラーゼ(FAAH)にMAGLの阻害に対して選択的である請求項31記載の方法。
【請求項34】
化合物が下記に示す式Iの構造を有する請求項33記載の方法:
【化16】
式中、XはN又はCH、R1はH又はOHであり、そしてR2及びR3の各々は、下記の構造の一つを有する。
【化17】
【請求項35】
化合物がMAGLに特異的な阻害ポリヌクレオチドである請求項31記載の方法。
【請求項36】
阻害ヌクレオチドが低分子緩衝RNA(siRNA)、マイクロRNA(miRNA)、低分子ヘアピン型RNA(shRNA)、アンチセンス核酸及び相補DNA(cDNA)である請求項35記載の方法。
【図1】

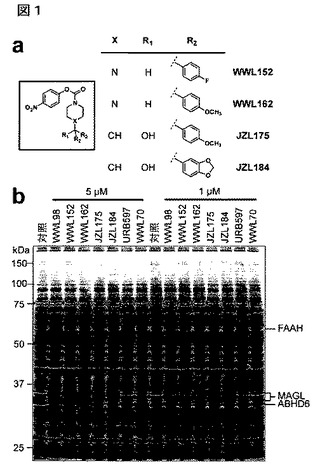
【図2】
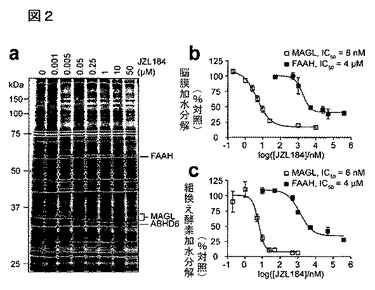
【図3】
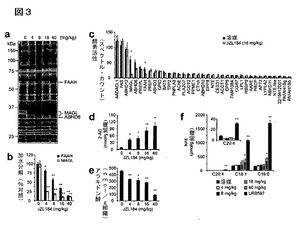
【図4】
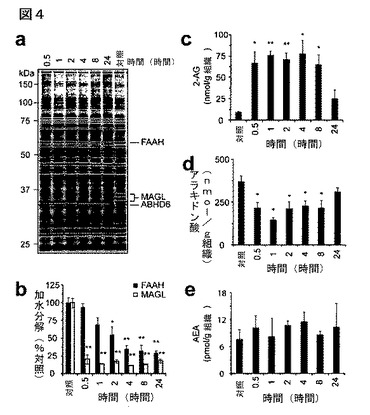
【図5】
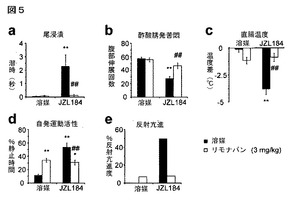
【図6】
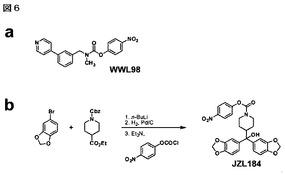
【図7】
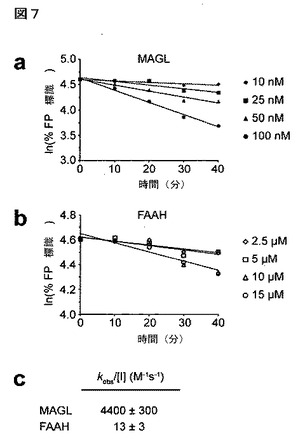
【図8】
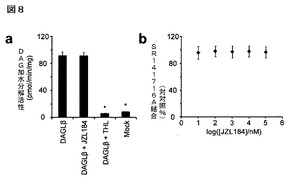
【図9】
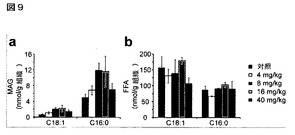
【図10】
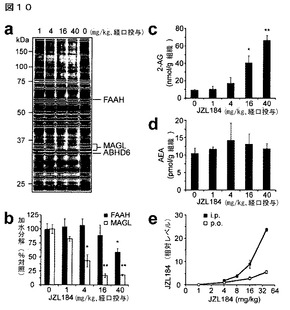
【図11】
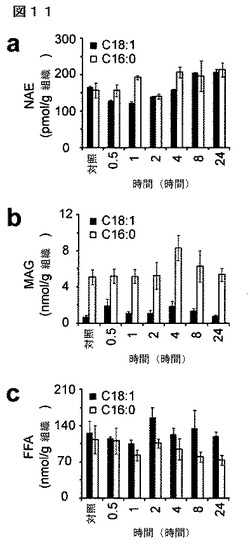
【図12】
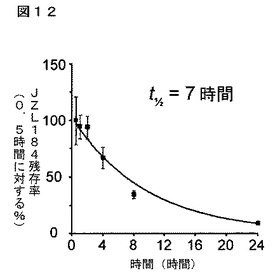
【図13】
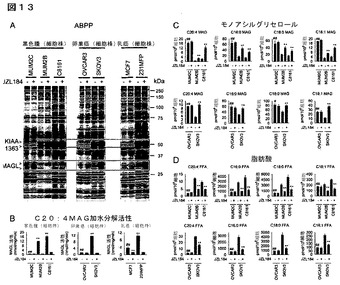
【図14】
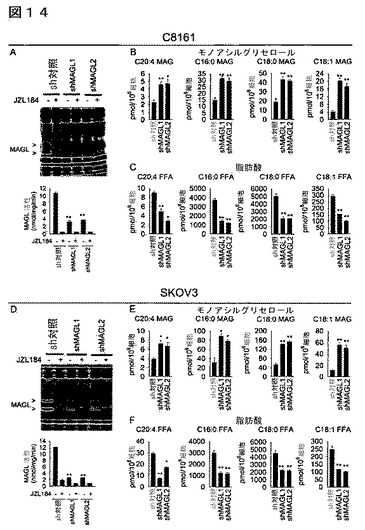
【図15】
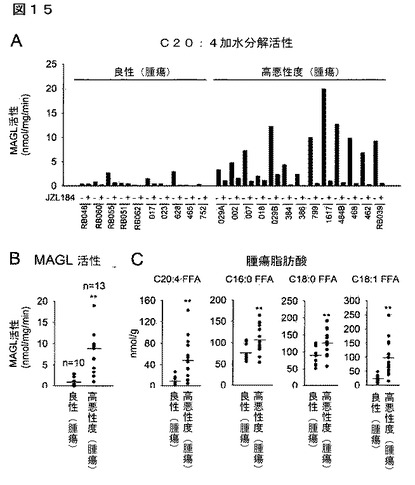
【図16】
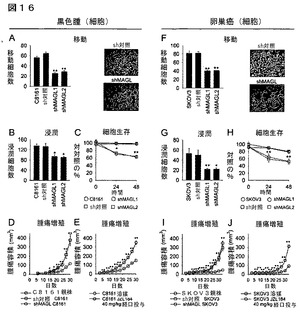
【図17】
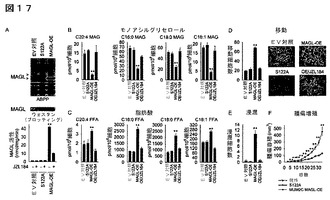
【図18】

【図19】
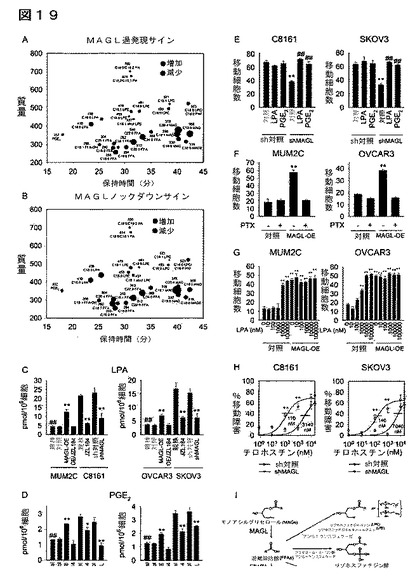
【図20】
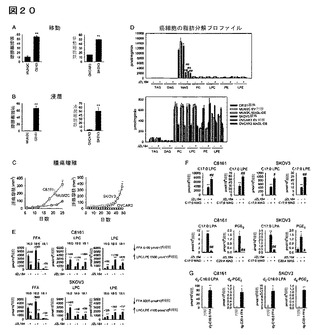
【図21】
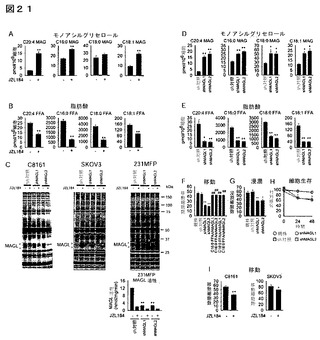
【図22】
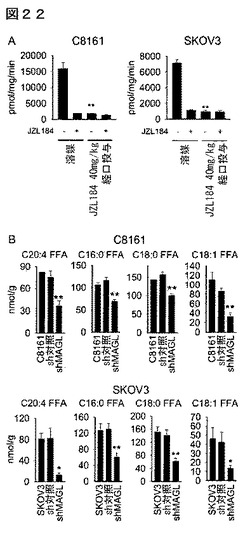
【図23】
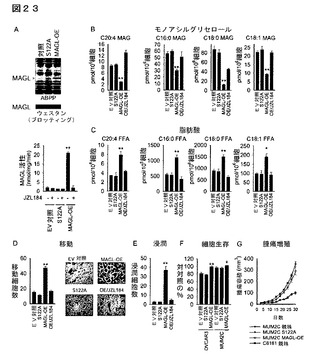
【図24】
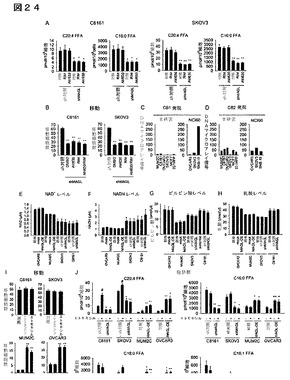
【図25】
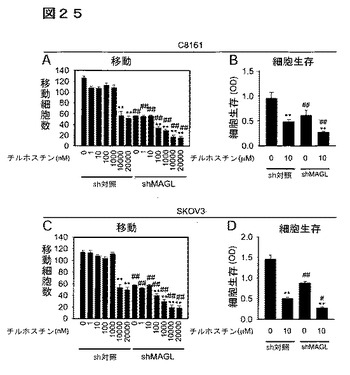
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【公表番号】特表2012−508737(P2012−508737A)
【公表日】平成24年4月12日(2012.4.12)
【国際特許分類】
【出願番号】特願2011−536316(P2011−536316)
【出願日】平成21年11月10日(2009.11.10)
【国際出願番号】PCT/US2009/006045
【国際公開番号】WO2010/056309
【国際公開日】平成22年5月20日(2010.5.20)
【出願人】(399038620)ザ スクリプス リサーチ インスティチュート (51)
【Fターム(参考)】
【公表日】平成24年4月12日(2012.4.12)
【国際特許分類】
【出願日】平成21年11月10日(2009.11.10)
【国際出願番号】PCT/US2009/006045
【国際公開番号】WO2010/056309
【国際公開日】平成22年5月20日(2010.5.20)
【出願人】(399038620)ザ スクリプス リサーチ インスティチュート (51)
【Fターム(参考)】
[ Back to top ]