
モノアミン再取り込み阻害薬としてのフェニル置換シクロアルキルアミン
フェニル置換シクロヘキシルアミン誘導体、ならびにそれらの合成方法および特徴付けを開示する。神経障害を治療/予防するためのこれらの化合物の使用およびそれらの合成方法を本明細書に記載する。本発明の例示的な化合物は、(例えば、シナプス間隙からの)ドーパミン、セロトニンおよびノルエピネフリン等の内因性モノアミンの再取り込みを阻害し、1つまたは複数のモノアミン輸送体を調節する。また、本発明の化合物を組み込む医薬品製剤も提供する。

【発明の詳細な説明】
【技術分野】
【0001】
関連出願の相互参照
[0001] 本出願は、全ての目的のためにその全体が参照により本明細書に組み込まれている2007年5月31日出願の米国仮特許出願第60/941,242号についての優先権を主張する。
【0002】
[0002] 本発明は、神経障害の治療のための化合物および組成物に関する。
【背景技術】
【0003】
[0003] 精神障害は、認知、情動、気分または情緒の異常となる同定可能な症状によって特徴付けられる脳の病理学的な状態である。これらの障害は、症状の重症度、持続時間および機能障害の点で多様であり得る。精神障害は、世界全体で数百万人の人々を苦しめ、その結果、多大な人的苦痛をもたらし、生産性の損失および依存的ケアにより経済的負担を生じる。
【0004】
[0004] 過去数十年にわたり、精神障害を治療するための薬理学作用のある物質の使用が、主として神経科学および分子生物学の両方における研究の進展により大幅に増加している。さらに、より少ない副作用を有するより有効な治療剤であり、精神的な障害を伴う生化学的な変化を矯正することを目指す化合物を創出する化学者の技術レベルも一層高まっている。
【0005】
[0005] しかし、多くの進展がもたらされているにもかかわらず、多くの精神疾患は、治療されず放置されているか、または現在の医薬品を用いて不適切に治療されている状態にある。さらに、現在の薬剤の多くは、精神疾患には関与しない分子標的とも相互作用する。この無差別な結合の結果、療法の総合的な治療成果に大きな影響を及ぼす恐れがある副作用が生じる場合がある。場合によっては、副作用が非常に大きいので、療法を中断する必要がある。
【0006】
[0006] うつ病は、情動障害であり、その病原は、いずれの単一の原因および理論でも説明することができない。うつ病は、持続性の気分の落ち込みまたは周囲に対する関心の低下によって特徴付けられ、少なくとも1つの以下の症状を伴う:エネルギーおよび意欲の低下、集中困難、睡眠および食欲の変化、ならびに時には自殺念慮(American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders, ed. 4. Washington, American Psychiatric Association, 1994)。大うつ病には、高い罹患率および死亡率が伴い、自殺率は、10〜25%である(Kaplan H I, Sadock B J (編): Synopsis of Psychiatry. Baltimore, Williams & Wilkins, 1998, p. 866)。また、二重再取り込み阻害薬(dual reuptake inhibitor)を使用して、うつ病に通常伴う疲労を低下させる場合もある(例えば、"Bupropion augmentation in the treatment of chronic fatigue syndrome with coexistent major depression episode" Schonfeldt-Lecuonaら, Pharmacopsychiatry 39(4):152-4, 2006;"Dysthymia: clinical picture, extent of overlap with chronic fatigue syndrome, neuropharmacological considerations, and new therapeutic vistas" Brunelloら, J. Affect. Disord. 52(1-3):275-90, 1999;"Chronic fatigue syndrome and seasonal affective disorder: comorbidity, diagnostic overlap, and implications for treatment" Termanら, Am. J. Med. 105(3A):115S-124S, 1998を参照されたい)。
【0007】
[0007] うつ病は、ノルアドレナリン作動性またはセロトニン作動性の系における機能不全、より具体的には、機能的に重要なアドレナリン作動性またはセロトニン作動性の受容体における特定の神経伝達物質(NT)の欠乏の結果生じると考えられている。
【0008】
[0008] 神経伝達物質は、特異的な受容体との相互作用の結果として効果を発揮する。ノルエピネフリン(NE)および/またはセロトニン(5−ヒドロキシトリプタミンまたは5-HT)を含めて、神経伝達物質は、脳の神経細胞において合成され、小胞中に保管される。神経インパルス時に、NTは、シナプス間隙中に放出され、そこで、神経伝達物質は、種々のシナプス後受容体と相互作用する。5−HTおよび/またはNEのシナプスのレベルの領域的な欠乏が、うつ病の病因、覚醒および注意力に関与すると考えられている。
【0009】
[0009] ノルエピネフリンは、覚醒状態、夢見および気分の調整に関与する。ノルエピネフリンはまた、血管を収縮させ、心拍数を増加させることによって、血圧の調整に寄与することもできる。
【0010】
[0010] セロトニン(5-HT)は、種々の障害の病因または治療に関係している。5−HTの最も広く研究されている効果は、CNSに対する効果である。5−HTの機能は多数あり、食欲、睡眠、記憶および学習の制御、体温調整、気分、(性行動および幻覚行動を含めた)行動、心血管機能、平滑筋収縮ならびに内分泌調整を含む。末梢では、5−HTは、血小板のホメオスタシスおよびGI管の運動性において主要な役割を担っているようである。5−HTの作用は、3つの主要な機構、すなわち、拡散;代謝;および再取り込みによって止まる。5−HTの作用を止める主要な機構は、シナプス前膜を介しての再取り込みによるものである。5−HTは、種々のシナプス後受容体に対して作用した後、特異的な膜輸送体が関与する取り込み機構によって、その他の生体アミンの取り込み機構に類似する様式で、シナプス間隙から除去され、神経末端に戻る。この取り込みを選択的に阻害する薬剤が、シナプス後受容体における5−HTの濃度を増加させ、種々の精神障害、特にうつ病を治療する場合に有用であることが見い出されている。
【0011】
[0011] 長年にわたって、うつ病の治療へのアプローチには、NEおよび5−HTのレベルを、それらの代謝の阻害(例えば、モノアミンオキシダーゼ阻害薬)または再取り込みの阻害(例えば、三環系抗うつ薬もしくは選択的セロトニン再取り込み阻害薬(SSRI))のいずれかによって増加させる薬剤の使用が関与している。
【0012】
[0012] 米国内で入手可能な、承認されている抗うつ薬が、20種以上ある。現在入手可能な、古典的な三環系抗うつ薬(TCA)は、主としてNEの取り込みを遮断し、かつまた、二級アミンまたは三級アミンのいずれであるかに依存して、様々な程度で5−HTの取り込みも遮断する。イミプラミンおよびアミトリプチリン等の三級アミンは、デシプラミン等の二級アミンと比較すると、カテコールアミンに対してよりも5−HTに対して選択性を示す取り込み阻害薬である。
【0013】
[0013] 選択的セロトニン再取り込み阻害薬は、有望な抗うつ薬として調べられてきた。フルオキセチン(PROZAC(登録商標))、セルトラリン(ZOLOFT(登録商標))およびパロキセチン(PAXIL(登録商標))が、米国で現在市販されているSSRIの3つの例である。これらの薬剤は、TCAを上回る効能を有するようではなく、一般に、作用出現までの時間がより短いわけでもない。しかし、これらは、副作用を生じにくいという利点は有する。これら3つのSSRIのうち、パロキセチンが、5−HTの取り込みの最も強力な阻害薬であり、フルオキセチンが最も弱い。セルトラリンが、NEと比して5−HTを最も選択的に取り込み、フルオキセチンの選択性が最も低い。フルオキセチンおよびセルトラリンは、活性な代謝産物をもたらし、一方、パロキセチンは、不活性な代謝産物に代謝される。SSRIは、一般に、セロトニンの取り込みに限って影響を及ぼし、ムスカリン性受容体、アドレナリン作動性受容体、ドーパミン受容体およびヒスタミン受容体を含む種々の受容体の系に対して親和性を示すことはほとんどまたは全くない。
【0014】
[0014] うつ病の治療に加え、SSRIについて、いくつかのその他の治療上の適用例が調査されている。それらには、アルツハイマー病、攻撃行動、月経前症候群、糖尿病性ニューロパシー、慢性疼痛、線維筋痛症およびアルコール乱用の治療がある。例えば、フルオキセチンは、強迫性障害(OCD)の治療について承認されている。特に意義があるのは、5−HTは、アンフェタミン様の薬物に伴う乱用傾向の行動的影響をもたらすことなく、食事が引き起こす満腹感を高め、空腹感を抑えることによって、食物の消費を減少させることが観察されている点である。したがって、肥満治療におけるSSRIの使用に対して関心が寄せられている。
【0015】
[0015] ベンラファキシン(EFFEXOR(登録商標))は、5−HTおよびNEの両方の取り込みの強力な阻害薬として作用することから、古典的なTCAおよびSSRIとは化学的および薬理学的に異なる二重再取り込み抗うつ薬である。ベンラファキシンもその主要な代謝産物も、アドレナリン作動性アルファ−1受容体に対しては、顕著な親和性を示さない。ベンラファキシンは、TCAの効能と同等な効能、およびSSRIの副作用プロファイルに類似する、穏和な副作用プロファイルを有する。
【0016】
[0016] ドーパミンは、精神病、およびパーキンソン病等の特定の神経変性疾患において主要な役割を担っていると仮定されている。これらにおいては、ドーパミン作動性の神経細胞の欠乏が、根底にある病態であると考えられている。ドーパミンは、動作、情動応答、ならびに喜びおよび疼痛を体験する能力を制御する脳のプロセスに影響を及ぼす。DAの調整は、我々の精神的および肉体的な健康における重大な役割を担っている。特定の薬物が、DAの再取り込みを阻止し、シナプス中により多くのDAを残すことによって、DAの濃度を増加させる。例として、小児の多動、および統合失調症の症状を治療するために治療上使用されるメチルフェニデート(RITALIN(登録商標))がある。ドーパミンの異常は、急性統合失調症患者において主として見られる注意異常のうちの一部の根底にあると考えられている。
【0017】
[0017] 治療の遅れが、これらの薬物の使用には伴う。臨床的に意味のある症状の軽減が実現するまでに、患者は薬物を少なくとも3週間は服用しなければならない。さらに、顕著な数の患者が、現在の療法に全く応答しない。例えば、現在のところ、うつ病と臨床的に診断された症例の30パーセント(30%)までが、現在の薬物療法の全ての形態に対して抵抗性であると推定されている。
【発明の概要】
【課題を解決するための手段】
【0018】
[0018] 本発明は、新規なシクロアルキルアミンおよびそれらの塩に関する。また、本発明は、新規な医薬組成物、ならびに障害および状態の治療におけるそれらの使用にも関する。本発明の化合物の例示的な適応症として、うつ病(例えば、大うつ病性障害、双極性障害)、線維筋痛症、疼痛(例えば、神経因性疼痛)、睡眠時無呼吸、注意欠陥障害(ADD)、注意欠陥多動性障害(ADHD)、下肢静止不能症候群、統合失調症、不安症、強迫性障害、心的外傷後ストレス障害、季節性感情障害(SAD)、月経前不安等の神経学的障害、および神経変性疾患(例えば、パーキンソン病、アルツハイマー病)が挙げられる。また、本発明の化合物は、肥満を治療もしくは予防するためにも、またはこれらに限定されないが、ニコチンおよびコカインの乱用、依存もしくは耽溺を含む物質乱用、依存もしくは耽溺を治療するためにも有用である。
【0019】
[0019] したがって、第1の態様では、本発明は、以下の式を有する化合物を提供する。
【0020】
【化1】
式中、指数nは、0〜2からなる群から選択される整数であり;sは、0〜2からなる群から選択される整数である。Aは、H、置換または非置換アルキル、ハロゲン、および置換または非置換ハロアルキルからなる群から選択されるメンバーである。Xは、H、ハロゲン、置換または非置換アルキル、置換または非置換アリール、置換または非置換ハロアルキル、およびOR5(ただし、R5は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、アシル、およびS(O)2R5a(ただし、R5aは、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルからなる群から選択されるメンバーである)からなる群から選択されるメンバーである)からなる群から選択されるメンバーである。
【0021】
[0020] YおよびZは、ハロゲン、CF3、CN、OR9、SR9、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、置換または非置換ヘテロシクロアルキル、NR10R11、およびNO2からなる群から独立して選択されるメンバーである。R9は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、または置換または非置換ヘテロシクロアルキルを示す。基であるR10およびR11は独立して、H、OR12、アシル、S(O)2R13、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、または置換または非置換ヘテロシクロアルキルを示す。R10とR11とは、それらが結合している窒素と一緒になって任意選択により結合して、R10とR11とが結合する窒素に加えて、1〜3個のヘテロ原子を任意選択により有する3員環〜7員環を形成する。
【0022】
[0021] 記号R12は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、または置換または非置換ヘテロシクロアルキルから選択されるメンバーである。R13は、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルから選択されるメンバーである。
【0023】
[0022] YとZとは、それらが結合している原子と一緒になって任意選択により結合して、5員環〜7員環を形成し、この環はその中に、1〜3個のヘテロ原子を任意選択により有する。当業者には明らかであろうが、YとZとが結合して環をなす場合、環内に組み込まれた原子上の置換基(例えば、R9、R10およびR11)が、これらの置換基が結合している原子の原子価を満たす必要に応じて、存在する(例えば、環の環状構造内に組み込まれる)場合、または存在しない場合があるであろう。
【0024】
[0023] R1およびR2は、H、ハロゲン、CN、CF3、OR6、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、または置換または非置換ヘテロシクロアルキルから独立して選択されるメンバーである。R6は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、または置換または非置換ヘテロシクロアルキルから選択されるメンバーである。
【0025】
[0024] R3およびR4は、H、OR7、アシル、S(O)2R8、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、または置換または非置換ヘテロシクロアルキルから独立して選択されるメンバーである。R7は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、または置換または非置換ヘテロシクロアルキルから選択されるメンバーである。R8は、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルから選択されるメンバーである。
【0026】
[0025] R1、R2、R3およびR4のうちの2つ以上が、それらが結合している原子と一緒になって任意選択により結合して、3員環〜7員環を形成し、この環は、1〜4、好ましくは1〜3個のヘテロ原子を任意選択により含む。
【0027】
[0026] 任意の薬学的に許容できる塩、溶媒和化合物、鏡像異性体、ジアステレオマー、ラセミ混合物、鏡像異性体に富む(enatiomerically enriched)混合物、および上記に記載した化合物の鏡像異性的に純粋な形態が、本発明の範囲内に属する。
【0028】
[0027] 第2の態様では、本発明は、本発明の化合物またはその薬学的に許容できる塩もしくは溶媒和化合物、および薬学的に許容できる担体を含む医薬組成物を提供する。
【0029】
[0028] 第3の態様では、本発明は、モノアミン輸送体リガンドの、セロトニン輸送体、ドーパミン輸送体およびノルエピネフリン輸送体等のモノアミン輸送体に対する結合を阻害する方法を提供する。この方法は、モノアミン輸送体と、本発明の化合物とを接触させることを含む。例示的な実施形態では、モノアミン輸送体リガンドは、セロトニン、ドーパミンおよびノルエピネフリン等のモノアミンである。
【0030】
[0029] 第4の態様では、本発明は、セロトニン輸送体、ドーパミン輸送体およびノルエピネフリン輸送体等、少なくとも1つのモノアミン輸送体の活性を阻害する方法を提供する。この方法は、モノアミン輸送体と本発明の化合物とを接触させることを含む。
【0031】
[0030] 別の態様では、本発明は、セロトニン、ドーパミンおよびノルエピネフリン等、少なくとも1つのモノアミンの細胞による取り込みを阻害する方法を提供する。この方法は、細胞を本発明の化合物と接触させることを含む。例示的な実施形態では、細胞は、神経細胞またはグリア細胞等の脳細胞である。
【0032】
[0031] さらに別の態様では、本発明は、うつ病を、少なくとも1つのモノアミン輸送体の活性を阻害することによって治療する方法を提供する。この方法は、本発明の化合物を哺乳動物の対象に投与することを含む。例示的な実施形態では、本発明の化合物は、少なくとも2つの異なるモノアミン輸送体の活性を阻害する。別の好ましい実施形態では、哺乳動物の対象はヒトである。
【0033】
[0032] さらなる態様では、本発明は、神経障害を治療する方法を提供する。この方法は、治療有効量の本発明の化合物またはその薬学的に許容できる塩もしくは溶媒和化合物を、その治療を必要とする対象に投与することを含む。例示的な実施形態では、対象はヒトである。
【図面の簡単な説明】
【0034】
【図1A】[0033]本発明の例示的な化合物の表である。
【図1B】本発明の例示的な化合物の表である。
【図1C】本発明の例示的な化合物の表である。
【図1D】本発明の例示的な化合物の表である。
【図1E】本発明の例示的な化合物の表である。
【図2】[0034]レセルピン投与ラットにおける、化合物4のベースライン自発運動に対する効果を示すグラフである。
【図3】[0035]レセルピン投与ラットにおける、化合物4のロータロッド(rotarod)における動きに対する効果を示すグラフである。
【図4】[0036]レセルピン投与ラットにおける、化合物4のカタレプシー(catalepsy)に対する効果を示すグラフである。
【図5】[0037]高い用量のL−DOPAと比較した、ロータロッドにおける動きに対する化合物4と低い用量のL−DOPAとの併用の効果を示すグラフである。
【図6】[0038]6−OHDA病変ラットにおける、L−DOPAと化合物4との併用効果を示すグラフである。
【発明を実施するための形態】
【0035】
I.定義
[0039] 「アルキル」という用語は、それ自体でまたは別の置換基の一部として、別段の言及がない限り、直鎖もしくは分枝鎖または環状の炭化水素基、あるいはそれらの組合せを意味し、これは、完全な飽和であっても、または不飽和、例えば、モノ不飽和もしくはポリ不飽和であってもよく、二価および多価の基を含むことができる。アルキル基は、言及される範囲内におけるいくつかの炭素を有するとして任意選択により指定される。すなわち、C1〜C10は、1〜10個の炭素を有する、置換または非置換アルキル部分を意味する。飽和炭化水素基の例として、これらに限定されないが、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、t−ブチル、イソブチル、sec−ブチル、例えば、n−ペンチル、n−ヘキシル、n−ヘプチル、n−オクチルの相同体および異性体;環状アルキル、例えば、シクロヘキシル、(シクロヘキシル)メチル、シクロプロピルメチル、例えば、縮合シクロアルキル(例えば、デカリン)等を含めた縮合環の種等の基が挙げられる。不飽和アルキル基は、1つまたは複数の二重結合または三重結合を有する基、例えば、「アルケニル」および「アルキニル」である。不飽和アルキル基の例として、これらに限定されないが、ビニル、2−プロペニル、クロチル、2−イソペンテニル、2−(ブタジエニル)、2,4−ペンタジエニル、3−(1,4−ペンタジエニル)、エチニル、1−および3−プロピニル、3−ブチニル、ならびに高級な相同体および異性体が挙げられる。「アルキル」という用語はまた、別段に特筆しない限り、以下でより詳細に定義するアルキル誘導体、すなわち、「ヘテロアルキル」等も含むことも意味する。炭化水素基に限定されるアルキル基は、「ホモアルキル」と呼ばれる。「置換アルキル」部分上に見い出される例示的な置換基を、以下に記載する。
【0036】
[0040] 「アルキレン」という用語は、それ自体でまたは別の置換基の一部として、アルカンから誘導された二価の基を意味し、例として、これに限定されないが、−CH2CH2CH2CH2−が挙げられ、「ヘテロアルキレン」として以下に記載する基もさらに含まれる。典型的には、アルキル(またはアルキレン)基は、1〜24個の炭素原子を有し、本発明においては、10個以下の炭素原子を有する基が好ましい。「低級アルキル」または「低級アルキレン」は、より短い鎖のアルキル基またはアルキレン基であり、一般に、8個以下の炭素原子を有する。
【0037】
[0041] 「アルコキシ」、「アルキルアミノ」および「アルキルチオ」(またはチオアルコキシ)という用語を、それらの従来の意味で使用し、分子の残部に、それぞれ酸素原子、アミノ基または硫黄原子を介して結合しているアルキル基を指す。
【0038】
[0042] 「ヘテロアルキル」という用語は、それ自体でまたは別の用語と組み合わせて、別段の言及がない限り、上記に記載した「アルキル」の亜属、すなわち、直鎖もしくは分枝鎖または環状のアルキル基、あるいはそれらの組合せ、飽和または不飽和のアルキル基であって、(任意選択により言及される)いくつかの炭素原子、ならびに好ましくはB、O、N、P、SiおよびSから選択される少なくとも1つのヘテロ原子からなり、窒素原子および硫黄原子は、任意選択により酸化されていてよく、ただし、窒素へテロ原子は、任意選択により四級化されていてもよいアルキル基を意味する。(1つまたは複数の)ヘテロ原子B、O、N、P、SiおよびSは、ヘテロアルキル基の任意の内部の位置にあっても、またはヘテロアルキル基が分子の残部に結合している位置にあっても、もしくはその正反対の末端にあってもよい。例として、これらに限定されないが、−CH2−CH2−O−CH3、−CH2−CH2−NH−CH3、−CH2−CH2−N(CH3)−CH3、−CH2−S−CH2−CH3、−CH2−CH2,−S(O)−CH3、−CH2−CH2−S(O)2−CH3、−CH=CH−O−CH3、−Si(CH3)3、−CH2−CH=N−OCH3および−CH=CH−N(CH3)−CH3が挙げられる。例えば、−CH2−NH−OCH3および−CH2−O−Si(CH3)3等、2個以上のヘテロ原子が連続し得る。同様に、「ヘテロアルキレン」という用語は、それ自体でまたは別の置換基の一部として、ヘテロアルキルから誘導された二価の基を意味し、例として、これらに限定されないが、−CH2−CH2−S−CH2−CH2−および−CH2−S−CH2−CH2−NH−CH2−が挙げられる。ヘテロアルキレン基については、ヘテロ原子はまた、鎖の末端の一方または両方を占めることもできる(例えば、アルキレンオキシ、アルキレンジオキシ、アルキレンアミノ、アルキレンジアミノ等)。その上さらに、アルキレン連結基およびヘテロアルキレン連結基については、連結基の式が記載されている方向によって、連結基の方向性が示されることにはならない。例えば、式−CO2R’−は、−C(O)OR’および−OC(O)R’の両方を任意選択により示す。
【0039】
[0043] 「シクロアルキル」および「ヘテロシクロアルキル」という用語は、それら自体でまたはその他の用語と組み合わせて、別段の言及がない限り、それぞれ「アルキル」および「ヘテロアルキル」の環状型を示す。さらに、ヘテロシクロアルキルについては、ヘテロ原子は、ヘテロ環が分子の残部に結合している位置を占めることができる。シクロアルキルの例として、これらに限定されないが、シクロペンチル、シクロヘキシル、1−シクロヘキセニル、3−シクロヘキセニル、シクロヘプチル等が挙げられる。ヘテロシクロアルキルの例として、これらに限定されないが、1−(1,2,5,6−テトラヒドロピリジル)、1−ピペリジニル、2−ピペリジニル、3−ピペリジニル、4−モルホリニル、3−モルホリニル、テトラヒドロフラン−2−イル、テトラヒドロフラン−3−イル、テトラヒドロチエン−2−イル、テトラヒドロチエン−3−イル、1−ピペラジニル、2−ピペラジニル等が挙げられる。
【0040】
[0044] 「ハロ」または「ハロゲン」という用語は、それら自体でまたは別の置換基の一部として、別段の言及がない限り、フッ素原子、塩素原子、臭素原子またはヨウ素原子を意味する。さらに、「ハロアルキル」等の用語は、モノハロアルキルおよびポリハロアルキルを含むことを意味する。例えば、用語「ハロ(C1〜C4)アルキル」という用語は、これらに限定されないが、トリフルオロメチル、2,2,2−トリフルオロエチル、4−クロロブチル、3−ブロモプロピル等を含むこと意味する。
【0041】
[0045] 「アリール」という用語 は、別段の言及がない限り、単環であっても、または一緒になって縮合しているか、もしくは共有結合によって連結している多環であってもよい(好ましくは、単環〜3員環)、ポリ不飽和、芳香族置換基を意味する。「アリール」の種は、シクロアルキル環、ヘテロシクロアルキル環およびヘテロアリール環と縮合しているアリール環を含む構造を含む。「ヘテロアリール」という用語は、「アリール」の亜属であり、好ましくはB、O、N、P、SiおよびSから選択される1〜4個のヘテロ原子を含有するアリール基を指し、ただし、窒素原子および硫黄原子は、任意選択により酸化されており、(1つまたは複数の)窒素原子は、任意選択により四級化されている。ヘテロアリール基は、ヘテロ原子を介して、分子の残部につながることができる。アリール基およびヘテロアリール基の非限定手的な例として、フェニル、1−ナフチル、2−ナフチル、4−ビフェニル、1−ピロリル、2−ピロリル、3−ピロリル、3−ピラゾリル、2−イミダゾリル、4−イミダゾリル、ピラジニル、2−オキサゾリル、4−オキサゾリル、2−フェニル−4−オキサゾリル、5−オキサゾリル、3−イソオキサゾリル、4−イソオキサゾリル、5−イソオキサゾリル、2−チアゾリル、4−チアゾリル、5−チアゾリル、2−フリル、3−フリル、2−チエニル、3−チエニル、2−ピリジル、3−ピリジル、4−ピリジル、2−ピリミジル、4−ピリミジル、5−ベンゾチアゾリル、プリニル、2−ベンゾイミダゾリル、5−インドリル、1−イソキノリル、5−イソキノリル、2−キノキサリニル、5−キノキサリニル、3−キノリルおよび6−キノリルが挙げられる。上記に特筆したアリール環系およびヘテロアリール環系のそれぞれについての置換基は、以下に記載する許容できる置換基の群から選択される。
【0042】
[0046] 簡潔にするために、「アリール」という用語は、置換基(例えば、アリールオキシ、アリールチオキシ、アリールアルキル)を定義するために使用する場合には、上記で定義したアリール環およびヘテロアリール環の両方を任意選択により含む。したがって、「アリールアルキル」という用語は、アリール基が、アルキル基に結合している基(例えば、ベンジル、フェネチル、ピリジルメチル等)を含むことを意味し、それらには、炭素原子(例えば、メチレン基)が、例えば、酸素原子によって置換アルキル基(例えば、フェノキシメチル、2−ピリジルオキシメチル、3−(1−ナフチルオキシ)プロピル等)が含まれる。
【0043】
[0047] 上記の用語(例えば、「アルキル」、「ヘテロアルキル」、「アリール」および「ヘテロアリール」)のそれぞれは、記述されている基の置換形態および非置換形態の両方を含むことを意味する。各タイプの基の好ましい置換基を以下に示す。
【0044】
[0048] アルキル基およびヘテロアルキル基(アルキレン、アルケニル、ヘテロアルキレン、ヘテロアルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、シクロアルケニルおよびヘテロシクロアルケニルとしばしば呼ばれる基を含む)についての置換基は、「アルキル基置換基」と一般に呼ばれ、それらは、これらに限定されないが、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、置換または非置換ヘテロシクロアルキル、−OR’、=O、=NR’、=N−OR’、−NR’R’’、−SR’、−ハロゲン、−SiR’R’’R’’’、−OC(O)R’、−C(O)R’、−CO2R’、−CONR’R’’、−OC(O)NR’R’’、−NR’’C(O)R’、−NR’−C(O)NR’’R’’’、−NR’’C(O)2R’、−NR−C(NR’R’’R’’’)=NR’’’’、−NR−C(NR’R’’)=NR’’’、−S(O)R’、−S(O)2R’、−S(O)2NR’R’’、−NRSO2R’、−CNおよび−NO2から、好ましくはゼロ〜(2m’+1)(ただし、m'は、そのような基中の炭素原子の総数である)の範囲の数で選択される、多様な基のうちの1種または複数であってよい。R’、R’’、R’’’およびR’’’’はそれぞれ、好ましくは、独立して、水素、置換または非置換ヘテロアルキル、置換または非置換アリール、例えば、1〜3個のハロゲンで置換アリール、置換または非置換アルキル、アルコキシもしくはチオアルコキシの基またはアリールアルキル基を指す。本発明の化合物が2つ以上のR基を含む場合には、例えば、R基のそれぞれが独立して、R’基、R’’基、R’’’基およびR’’’’基のそれぞれであるとして選択され、この場合、これらの基のうちの2つ以上が存在する。R’およびR’’が、同一の窒素原子に結合している場合には、窒素原子と組み合わさって、5員環、6員環または7員環を形成することができる。例えば、−NR’R’’は、これらに限定されないが、1−ピロリジニルおよび4−モルホリニルを含むことを意味する。置換基の上記の論述から、当業者であれば、「アルキル」という用語は、水素基以外の基、すなわち、ハロアルキル(例えば、−CF3および−CH2CF3)ならびにアシル(例えば、−C(O)CH3、−C(O)CF3、−C(O)CH2OCH3等)等に結合している炭素原子を含む基を含むことを意味することを理解するであろう。
【0045】
[0049] アルキル基について記載した置換基と同様に、アリール基およびヘテロアリール基についての置換基は、「アリール基置換基」と一般に呼ばれる。こうした置換基は、例えば、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、置換または非置換ヘテロシクロアルキル、−OR’、=O、=NR’、=N−OR’、−NR’R’’、−SR’、−ハロゲン、−SiR’R’’R’’’、−OC(O)R’、−C(O)R’、−CO2R’、−CONR’R’’、−OC(O)NR’R’’、−NR’’C(O)R’、−NR’−C(O)NR’’R’’’、−NR’’C(O)2R’、−NR−C(NR’R’’R’’’)=NR’’’’、−NR−C(NR’R’’)=NR’’’、−S(O)R’、−S(O)2R’、−S(O)2NR’R’’、−NRSO2R’、−CNおよび−NO2、−R’、−N3、−CH(Ph)2、フルオロ(C1〜C4)アルコキシおよびフルオロ(C1〜C4)アルキルから、ゼロから芳香環系上の空いている原子価の総数までの範囲の数で選択され;R’、R’’、R’’’およびR’’’’は、好ましくは、独立して、水素、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、および置換または非置換ヘテロアリールから選択される。本発明の化合物が2つ以上のR基を含む場合には、例えば、R基のそれぞれが独立して、R’基、R’’基、R’’’基およびR’’’’基のそれぞれであるとして選択され、この場合、これらの基のうちの2つ以上が存在する。
【0046】
[0050] アリール環またはヘテロアリール環の隣接する原子上の置換基のうちの2つを、式−T−C(O)−(CRR’)q−U−(式中、TおよびUは独立して、−NR−、−O−、−CRR’−または一重結合であり、qは、0〜3の整数である)の置換基で任意選択により置換することができる。あるいは、アリール環またはヘテロアリール環の隣接する原子上の置換基のうちの2つを、式−A−(CH2)r−D−(式中、AおよびDは独立して、−CRR’−、−O−、−NR−、−S−、−S(O)−、−S(O)2−、−S(O)2NR’−または一重結合であり、rは、1〜4の整数である)の置換基で任意選択により置換することもできる。このようにして形成された新しい環の一重結合のうちの1つを、二重結合で任意選択により置換することができる。あるいは、アリール環またはヘテロアリール環の隣接する原子上の置換基のうちの2つを、式−(CRR’)s−X’’−(CR’’R’’’)d−(式中、sおよびdは独立して、0〜3の整数であり、X’’は、−O−、−NR’−、−S−、−S(O)−、−S(O)2−または−S(O)2NR’−である)の置換基で任意選択により置換することもできる。置換基R’、R’’、R’’’およびR’’’’は、好ましくは、独立して、水素、または置換または非置換(C1〜C6)アルキルから選択される。
【0047】
[0051] 本明細書で使用する場合、「アシル」という用語は、カルボニル残基C(O)Rを含有する置換基を記載する。Rについての例示的な種として、H、ハロゲン、置換または非置換アルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルが挙げられる。
【0048】
[0052] 本明細書で使用する場合、「縮合環系」という用語は、少なくとも2つの環であって、各環が、別の環と共有する少なくとも2個の原子を有する環を意味する。「縮合環系は、芳香環(すなわち、アリールまたはヘテロアリール)、および飽和または不飽和の非芳香環(すなわち、シクロアルキル、ヘテロシクロアルキル)を含むことができる。「縮合環系」の例には、ナフタレン、インドール、キノリン、クロメン、デカリン等がある。
【0049】
[0053] 本明細書で使用する場合、「ヘテロ原子」という用語は、酸素(O)、窒素(N)、硫黄(S)、ケイ素(Si)、臭素(B)およびリン(P)を含む。
【0050】
[0054] 「R」という記号は、「アルキル基置換基」または「アリール基置換基」(例えば、置換または非置換アルキル基、置換または非置換ヘテロアルキル基、置換または非置換アリール基、置換または非置換ヘテロアリール基、および置換または非置換ヘテロシクロアルキル基から選択される置換基)を示す一般的な略語である。
【0051】
[0055] 「治療有効量」という句は、本明細書で使用する場合、(例えば、哺乳動物のシナプス間隙からのモノアミンの取り込みを阻害し、それによって、治療する生物において、その経路の生物学的帰結を調節することによって)ある所望の治療効果をいずれの医学的治療にも適用できる妥当な利益/リスクの比でもたらすために有効である、本発明の化合物、または本発明の化合物を含む組成物の量を意味する。
【0052】
[0056] 「薬学的に許容できる」という句は、本明細書で利用する場合、理にかなった医学的判断の範囲内において、ヒトおよび動物における使用に適しており、過剰な毒性、刺激、アレルギー応答またはその他の問題もしくは合併症を伴わず、相応の妥当な利益/リスクの比を有する化合物、材料、組成物および/または投与剤型を指す。
【0053】
[0057] 「薬学的に許容できる担体」という句は、本明細書で使用する場合、任意の薬学的に許容できる材料を意味し、これは、液体であっても、または固体であってもよい。例示的な担体として、ビヒクル、希釈剤、添加剤、液体および固体の充填剤、賦形剤、溶媒、溶媒を被包する材料が挙げられる。各担体は、処方のその他の成分に適合しており、患者に対して障害性がないという意味で「許容でき」なければならない。薬学的に許容できる担体として役立ち得る材料のいくつかの例として、(1)糖、すなわち、ラクトース、グルコースおよびスクロース等;(2)デンプン、すなわち、トウモロコシデンプンおよびバレイショデンプン等;(3)セルロースおよびその誘導体、すなわち、カルボキシメチルセルロースナトリウム、エチルセルロースおよび酢酸セルロース;(4)粉末トラガカント;(5)麦芽;(6)ゼラチン;(7)タルク;(8)賦形剤、すなわち、カカオ脂および坐剤用ろう等;(9)油、すなわち、落花生油、綿実油、ベニバナ油、ゴマ油、オリーブ油、トウモロコシ油および大豆油等;(10)グリコール、すなわち、プロピレングリコール等;(11)ポリオール、すなわち、グリセリン、ソルビトール、マンニトールおよびポリエチレングリコール等;(12)エステル、すなわち、オレイン酸エチルおよびラウリン酸エチル等;(13)寒天;(14)緩衝剤、すなわち、水酸化マグネシウムおよび水酸化アルミニウム等;(15)アルギン酸;(16)発熱物質を含まない水;(17)等張食塩水;(18)リンゲル液;(19)エチルアルコール;(20)pH緩衝溶液;(21)ポリエステル、ポリカーボネートおよび/またはポリ酸無水物:ならびに(22)医薬品製剤中で利用するのに適合する、その他の無毒性の物質が挙げられる。
【0054】
[0058] 上記に記載したように、本発明の化合物の特定の実施形態は、アミノまたはアルキルアミノ等の塩基性の官能基を含有することができ、したがって、薬学的に許容できる酸と薬学的に許容できる塩を形成することが可能である。この場合の「薬学的に許容できる塩」という用語は、本発明の化合物の比較的無毒性の無機および有機の酸付加塩を指す。これらの塩は、投与ビヒクル中もしくは投与剤型の製造過程においてin situでまたは別個に、精製された本発明の化合物をその遊離の塩基の形態で、適切な有機もしくは無機の酸と反応させ、このようにして形成した塩をそれに続く精製の間に単離することによって調製することができる。代表的な塩として、臭化水素酸塩、塩酸塩、硫酸塩、スルファミン酸塩、重硫酸塩、リン酸塩、硝酸塩、酢酸塩、吉草酸塩、オレイン酸塩、パルミチン酸塩、ステアリン酸塩、ラウリン酸塩、安息香酸塩、乳酸塩、トシレート、クエン酸塩、マレイン酸塩、アスコルビン酸塩、パルミチン酸塩、フマル酸塩、コハク酸塩、酒石酸塩、ナフチル酸塩、メシル酸塩、ヒドロキシマレイン酸塩(hydroxymaleate)、フェニル酢酸塩、グルタミン酸塩、グルコヘプトン酸塩、サリチル酸塩、スルファニル酸塩、2−アセトキシ安息香酸塩、メタンスルホン酸塩、エタンジスルホン酸塩、シュウ酸塩、イソチオネート、ラクトビオン酸塩およびラウリルスルホン酸塩等が挙げられる。例えば、Bergeら(1977)“Pharmaceutical Salts”,J.Pharm.Sci.66:1−19を参照されたい。
【0055】
[0059] 「薬学的に許容できる塩」という用語は、本明細書に記載する化合物上に見出される特定の置換基に依存して、比較的無毒性の酸または塩基を用いて調製する、活性な化合物の塩を含む。本発明の化合物が、比較的酸性の官能基を含有する場合には、そのような化合物の中性の形態を、十分な量の所望の塩基と、そのまままたは適切な不活性な溶媒中のいずれかで接触させることによって塩基付加塩を得ることができる。薬学的に許容できる塩基付加塩の例として、ナトリウム塩、カリウム塩、カルシウム塩、アンモニウム塩、有機アミノ塩もしくはマグネシウム塩または類似の塩が挙げられる。本発明の化合物が、比較的塩基性の官能基を含有する場合には、そのような化合物の中性の形態を、十分な量の所望の酸と、そのまままたは適切な不活性な溶媒中のいずれかで接触させることによって酸付加塩を得ることができる。薬学的に許容できる酸付加塩の例として、塩酸、臭化水素酸、硝酸、炭酸、一水素炭酸(monohydrogencarbonic acid)、リン酸、一水素リン酸(monohydrogenphosphoric acid)、二水素リン酸(dihydrogenphosphoric acid)、硫酸、一水素硫酸(monohydrogensulfuric acid)、ヨウ化水素酸または亜リン酸等のような無機酸から誘導した塩、ならびに酢酸、プロピオン酸、イソ酪産、マレイン酸、マロン酸、安息香酸、コハク酸、スベリン酸、フマル酸、乳酸、マンデル酸、フタル酸、ベンゼンスルホン酸、p−トリルスルホン酸、クエン酸、酒石酸、メタンスルホン酸等のような比較的無毒性の有機酸から誘導した塩が挙げられる。また、アルギン酸塩等のアミノ酸の塩、ならびにグルクロン酸またはガラクツロン酸等のような有機酸の塩も挙げられる(例えば、Bergeら, Journal of Pharmaceutical Science, 66: 1-19 (1977)を参照されたい)。特定の特異的な本発明の化合物は、化合物を塩基付加塩または酸付加塩のいずれにも変換することを可能にする、塩基性官能基および酸性官能基の両方を含有する。
【0056】
[0060] 化合物の中性の形態を、好ましくは、塩を塩基または酸と接触させ、従来の様式で親化合物を単離することによって再生する。化合物の親の形態は、極性溶媒中の溶解性等の特定の物性において種々の塩の形態と異なるが、そうでなければ、塩は、本発明の目的に関しては、化合物の親の形態と同等である。
【0057】
[0061] 塩の形態に加えて、本発明は、プロドラッグの形態である化合物も提供する。本明細書に記載する化合物のプロドラッグは、生理的条件下で化学変化を容易に受けて、本発明の化合物をもたらす化合物である。さらに、プロドラッグは、ex vivo環境下で化学的または生化学的な方法によって本発明の化合物に変換することもできる。例えば、プロドラッグを、経皮パッチリザーバ中に適切な酵素または化学試薬と共に置いて、本発明の化合物に緩慢に変換させることができる。
【0058】
[0062] 本発明の特定の化合物は、非溶媒和の形態でも、水和した形態を含む溶媒和の形態でも存在することができる。一般に、溶媒和の形態は、非溶媒和の形態と同等であり、本発明の範囲内に包含される。本発明の特定の化合物は、複数の結晶形態または非結晶の形態で存在することができる。一般に、全ての物理的形態は、本発明が企図する使用のためには同等であり、本発明の範囲内に属するものとする。「化合物または化合物の薬学的に許容できる塩もしくは溶媒和化合物」は、塩および溶媒和化合物の両方である材料を包含するという点で、「または」の包括的な意味を意図する。
【0059】
[0063] 本発明の特定の化合物は、不斉炭素原子(光学中心)または二重結合を有し;ラセミ体、ジアステレオマー、幾何異性体および個々の異性体は、本発明の範囲内に包含される。光学的に活性な(R)−異性体および(S)−異性体は、キラルシントンもしくはキラル試薬を使用して調製しても、または従来の技法を使用して分割してもよい。本明細書に記載する化合物が、オレフィン二重結合またはその他の幾何学的非対称の中心を含有する場合、別段の記載がない限り、化合物は、E幾何異性体およびZ幾何異性体の両方を含むことを意図する。同様にまた、全ての互変異性の形態も含むことを意図する。
【0060】
[0064] 本明細書で使用するラセミ化合物、アンビスケールミック(ambiscalemic)化合物およびスケールミック(scalemic)化合物、または鏡像異性的に純粋な化合物の図的表現は、Maehr,J.Chem.Ed.,62:114−120(1985)から採用しており、波線は、それが示す結合がもたらすことができるであろう任意の立体化学的な関係の否定を示し;実線のくさび形および破線のくさび形は、提示する相対的な立体配置を示す幾何学的な記載であるが、いずれかの絶対的な立体化学を伝えているわけではなく;くさび形の輪郭線および点線または破線は、不定の絶対配置の鏡像異性的に純粋な化合物を表示する。
【0061】
[0065] 「鏡像異性体過剰」および「ジアステレオ異性体過剰」という用語は、本明細書では互換的に使用する。単一の立体中心を有する化合物を、「鏡像異性体過剰」な状態で存在すると呼び、少なくとも2つの立体中心を有する化合物を、「ジアステレオ異性体過剰」な状態で存在すると呼ぶ。
【0062】
[0066] 本発明の化合物はまた、そのような化合物を構成する原子のうちの1個または複数において、不自然な比率の原子の同位体も含有することもできる。例えば、化合物を、例えば、トリチウム(3H)、ヨウ素−125(125I)または炭素−14(14C)等の放射性同位体を用いて放射標識することができる。放射性であっても、またはそうでなくても、本発明の化合物の全ての同位体の変形形態が、本発明の範囲内に包含されるものとする。
【0063】
[0067] 「モノアミン輸送体リガンド」という用語は、モノアミン輸送体に結合する任意の化合物を指す。リガンドは、所与のモノアミン輸送体の天然のリガンドである内因性モノアミン、ならびに特定のモノアミン輸送体に結合することが知られている合成分子等の薬物分子およびその他の化合物を含む。1つの例では、リガンドは、トリチウム等の放射性同位体を含むか、またはそれ以外の様式で(例えば、蛍光により)標識される。所与のモノアミン輸送体の適切なリガンドを選択することは、当業者の能力の範囲内である。例えば、ドーパミン輸送体の既知のリガンドとしては、ドーパミンおよびWIN35428が挙げられ、セロトニン輸送体の既知のリガンドとしては、5−ヒドロキシトリプタミン(セロトニン)およびシタロプラムが挙げられ、ノルエピネフリン輸送体のリガンドとしては、ノルエピネフリンおよびニソキセチンが挙げられる。
【0064】
[0068] 「摂食障害」という用語は、食べることを回避する異常な衝動強迫または異常に多くの量の食物を消費する制御できない衝撃を指す。これらの障害は、罹患者の社会的な生活状態のみならず、また身体的な生活状態にも影響を及ぼす。摂食障害の例として、神経性食欲不振症、過食症および過食が挙げられる。
【0065】
[0069] 「神経障害」という用語は、哺乳動物の中枢神経系または抹消神経系の任意の状態を指す。「神経障害」という用語は、神経変性疾患(例えば、アルツハイマー病、パーキンソン病および筋萎縮性側索硬化症)、神経精神疾患(例えば、統合失調症および全般性不安障害等の不安症)を含む。例示的な神経障害として、MLS(小脳失調症)、ハンチントン病、ダウン症、多発脳梗塞性認知症、てんかん重積症、打撲傷(例えば、脊髄損傷および頭部損傷)、ウイルス感染が誘発する神経変性(例えば、AIDS、脳症)、てんかん、温和なもの忘れ、閉鎖性頭部損傷、睡眠障害、うつ病(例えば、双極性障害)、認知症、運動障害、精神障害、アルコール依存症、心的外傷後ストレス障害等が挙げられる。「神経障害」はまた、こうした障害に伴う任意の状態も含む。例えば、神経変性障害を治療する方法は、神経変性障害に伴う記憶の喪失および/または認知の喪失を治療する方法を含む。例示的な方法としてはまた、神経変性障害に特徴的な神経細胞の機能の喪失を治療または予防することも挙げられるであろう。「神経障害」はまた、モノアミン(例えば、ノルエピネフリン)シグナル伝達経路に少なくとも部分的に関係する任意の疾患または状態(例えば、心血管疾患)も含む。
【0066】
[0070] 「疼痛」は、不快な感覚および情動の経験である。疼痛は、持続期間、病因または病態生理、機構、強度および症状に基づいて分類されている。「疼痛」という用語は、本明細書で使用する場合、全てのカテゴリーの疼痛を指し、刺激または神経の応答の観点から説明される疼痛、例えば、体性痛(侵害性刺激に対する正常な神経の応答)および神経障害性疼痛(損傷または変質した感覚経路の異常な応答であり、明確な侵害性の入力を伴わないことが多い);時間的に類別される疼痛、例えば、慢性疼痛および急性疼痛;重症度、例えば、軽度、中等度または重度の観点から類別される疼痛;ならびに症状であるか、または疾患の状況もしくは症候群の結果である疼痛、例えば、炎症性疼痛、癌性疼痛、AIDS性疼痛、関節症、偏頭痛、三叉神経痛、心虚血および糖尿病性末梢神経因性疼痛を含む(例えば、それぞれの全体が本明細書に参照により組み込まれている、Harrison's Principles of Internal Medicine, pp. 93-98(Wilsonら編, 12th ed. 1991);Williamsら, J. of Med. Chem. 42: 1481-1485(1999)を参照されたい)。「疼痛」はまた、混合性病因の疼痛、二重機構の疼痛、異痛症、灼熱痛、中枢性疼痛、知覚過敏、ヒペルパチー、異常感覚および痛覚過敏を含むことも意味する。
【0067】
[0071] 上記に記載した「体性」痛は、侵害性刺激、すなわち、損傷もしくは疾病等、例えば、外傷、熱傷、感染、炎症に対する、または疾患の過程、すなわち、癌等に対する正常な神経の応答を指し、皮膚性の疼痛(例えば、皮膚、筋肉または関節に起因する)および内臓性の疼痛(例えば、器官に起因する)の両方を含む。
【0068】
[0072] 「神経因性疼痛」は、神経系に対する傷害の結果生じる神経学的な状態の不均一な群である。上記に記載した「神経因性」疼痛は、末梢および/または中枢の感覚経路に対する損傷またはその機能不全、ならびに神経系の機能不全の結果生じる疼痛を指し、こうした疼痛は、明らかな侵害性の入力を伴うことなく発生または持続することが多い。これには、末梢性ニューロパシーおよび中枢性ニューロパシーに関連する疼痛が含まれる。末梢性の神経因性疼痛の一般的な型には、糖尿病性ニューロパシー(また、糖尿病性末梢神経因性疼痛、またはDN、DPNもしくはDPNPとも呼ばれる)、ヘルペス後神経痛(PHN)および三叉神経痛(TGN)がある。脳または脊髄に対する傷害が関与する中枢性の神経因性疼痛は、脳卒中、脊髄損傷に続いて、および多発性硬化症の結果として生じる場合がある。神経因性疼痛の定義に含まれることを意味するその他の型の疼痛には、神経障害性の癌性疼痛、HIV/AIDS に起因する疼痛、幻肢痛および複合性局所疼痛症候群からの疼痛がある。例示的な実施形態では、本発明の化合物は、神経因性疼痛の治療のために有用である。
【0069】
[0073] 神経因性疼痛の一般的な臨床的特徴には、感覚の喪失、異痛症(非侵害性の刺激によって生じる疼痛)、痛覚過敏、およびヒペルパチー(遅延性の感受、加重、および有痛性の残感覚)がある。疼痛は、侵害受容型と神経障害型との、例えば、機械的な脊髄性疼痛と神経根症または脊髄症との組合せであることが多い。
【0070】
[0074] 「急性疼痛」は、典型的には侵襲性の手順、外傷および疾患に伴う侵害性の化学的、熱的または機械的な刺激に対する正常な、予測される生理的な応答である。これは、一般に、時限的であり、組織を脅かし、および/または組織の損傷をもたらす刺激に対する適切な応答として見ることができる。上記に記載した「急性疼痛」は、短い持続時間または突然の開始によって特徴付けられる疼痛を指す。
【0071】
[0075] 「慢性疼痛」は、広い範囲の障害、例えば、外傷、悪性腫瘍、および関節リウマチ等の慢性炎症性疾患において生じる。慢性疼痛は通常、6ヵ月以上持続する。さらに、慢性疼痛の強度は、侵害性刺激または根底にある過程の強度とは不釣合いである場合がある。上記に記載した「慢性疼痛」は、慢性の障害にともなう疼痛、または根底にある障害の回復もしくは損傷の治癒を過ぎても持続し、根底にある過程から予測されるであろうものよりも激烈であることが多い疼痛を指す。慢性疼痛は、頻繁に再発する場合がある。
【0072】
[0076] 「炎症性疼痛」は、組織の損傷およびその結果としての炎症過程に対して応答した疼痛である。炎症性疼痛は、治癒を促す生理的な応答を惹起するという点で適応性である。しかし、炎症はまた、神経細胞の機能にも影響を及ぼす。COX2酵素が誘発するPGE2、ブラジキニンおよびその他の物質を含めた炎症メディエーターは、疼痛伝達神経細胞上の受容体に結合し、それらの機能を変化させ、興奮性を高め、したがって、疼痛感覚が高まる。多くの慢性疼痛は、炎症性の構成要素を有する。上記に記載した「炎症性疼痛」は、症状として、または炎症もしくは免疫系の障害の結果として生じる疼痛を指す。
【0073】
[0077] 上記に記載した「内臓性の疼痛」は、内臓器官内に位置する疼痛を指す。
【0074】
[0078] 上記に記載した「混合性病因」の疼痛は、炎症性の構成要素および神経障害性の構成要素の両方を含有する疼痛を指す。
【0075】
[0079] 上記に記載した「二重機構」の疼痛は、末梢性の感作および中枢性の感作の両方によって増幅および維持される疼痛を指す。
【0076】
[0080] 上記に記載した「灼熱痛」は、外傷性の神経病変後の持続的な灼熱感、異痛症およびヒペルパチーの症候群を指し、血管運動性および発汗促進性の機能不全、ならびに後には栄養性の変化と組み合わさることが多い。
【0077】
[0081] 上記に記載した「中枢性」疼痛は、中枢神経系内の一次病巣または機能不全によって開始する疼痛を指す。
【0078】
[0082] 上記に記載した「知覚過敏」は、特別な感覚を除く、刺激に対して高まった感受性を指す。
【0079】
[0083] 上記に記載した「ヒペルパチー」は、刺激、特に反復性の刺激に対して異常に痛いと感じる反応、および閾値の増加によって特徴付けられる有痛性の症候群を指す。ヒペルパチーは、異痛症、知覚過敏、痛覚過敏または異常感覚を伴って生じる場合がある。
【0080】
[0084] 上記に記載した「異常感覚」は、自発性であっても、または誘起されたものであっても、不快で異常な感覚を指す。異常感覚の特別な場合には、痛覚過敏および異痛症がある。
【0081】
[0085] 上記に記載した「痛覚過敏」は、通常痛いと感じる刺激に対する高まった応答を指す。痛覚過敏は、閾値を上回る刺激に対する高まった疼痛を反映する。
【0082】
[0086] 上記に記載した「灼熱痛」は、 通常では疼痛を招くことがない刺激に起因した疼痛を指す。
【0083】
[0087] 「疼痛」という用語は、神経系の機能不全の結果生じる疼痛、すなわち、神経障害性の疼痛および考えられる一般的な病態生理学的機構の臨床的特徴を共有するが、神経系のいずれかの部分における同定可能な病変によって開始されるわけではない有機的な疼痛の状況を含む。
【0084】
[0088] 「糖尿病性末梢神経因性疼痛」(DPNP、また、糖尿病性ニューロパシー、DNまたは糖尿病性末梢性ニューロパシーとも呼ばれる)という用語は、糖尿病に伴うニューロパシーによって引き起こされる疼痛を指す。DPNPは、「灼熱感」または「痛みが走る感じ」としてのみならず、また激しく痛む疼痛としても説明することができる足における疼痛または刺痛と古典的には表現される。あまり一般的ではないが、患者が、疼痛を、痒み、引き裂かれる感じ、または歯痛様と説明する場合がある。この疼痛には、異痛症および痛覚過敏、ならびにしびれ等の症状の不在が伴う場合がある。
【0085】
[0089] 「ヘルペス後神経痛(Post-Herpetic Neuralgia)」という用語はまた、「ヘルペス後神経痛(Postherpetic Neuralgia)」(PHN)とも呼ばれ、神経線維および皮膚に影響を及ぼす有痛性の状態である。ヘルペス後神経痛は、帯状疱疹の合併症、すなわち、水痘帯状疱疹ウイルス(varicella zoster virus)(VZV)の第2の勃発であり、このウイルスは、最初は、水痘を引き起こす。
【0086】
[0090] 「神経障害性の癌性疼痛」という用語は、癌の結果としての末梢性の神経障害性の疼痛を指し、腫瘍による神経の浸潤もしくは圧縮によって直接的に、または放射線療法および化学療法(化学療法誘発性ニューロパシー)等の癌治療によって間接的に引き起こされる場合がある。
【0087】
[0091] 「HIV/AIDS性の末梢性ニューロパシー」または「HIV/AIDS関連ニューロパシー」という用語は、HIV/AIDSによって引き起こされる末梢性ニューロパシー、すなわち、急性または慢性の炎症性脱髄性ニューロパシー(それぞれ、AIDPおよびCIDP)等、ならびにHIV/AIDSを治療するために使用した薬物の副作用として生じる末梢性ニューロパシーを指す。
【0088】
[0092] 「幻肢痛」という用語は、切断した肢がかつてあった場所から生じるように思える疼痛を指す。幻肢痛はまた、麻痺後(例えば、脊髄損傷後)の肢においても生じる場合がある。「幻肢痛」は通常、その性質が慢性である。
【0089】
[0093] 「三叉神経痛」(TN)という用語は、神経の枝が分布する顔部の領域(唇、目、鼻、頭皮、前頭部、上顎および下顎)において、激烈な、突き刺すような、電気ショック様の疼痛のエピソードを引き起こす第5脳(三叉)神経の障害を指す。三叉神経痛はまた、「自殺性疾患」としても知られている。
【0090】
[0094] 「複合性局所疼痛症候群(CRPS)」という用語は、以前は反射性交換神経性ジストロフィー(RSD)として知られており、慢性疼痛の状態である。CRPSの鍵となる症状は、損傷の重症度とは不釣合いな連続的で激烈な疼痛であり、これは、時間の経過と共に軽減するのではなく悪化する。CRPSは、末梢神経ではなく組織の損傷によって引き起こされる状態を含む1型、および症候群が主要な神経の損傷によって招かれ、時に灼熱痛と呼ばれる2型に分かれる。
【0091】
[0095] 「線維筋痛症」という用語は、疲労および様々なその他の症状が伴う、散在するまたは特異的な筋肉、関節または骨の疼痛によって特徴付けられる慢性の状態を指す。以前は、線維筋痛症は、結合組織炎、慢性筋肉痛症候群、心因性リウマチおよび緊張性筋肉痛等のその他の名前によって知られていた。
【0092】
[0096] 「けいれん」という用語は、ある神経障害を指し、「てんかん」と互換的に使用するが、多くの型のてんかんがあり、それらのいくつかは、けいれんの代わりに、軽微または軽度な症状を示す。全ての型のてんかんが、脳における混乱した突然の電気的活動によって引き起こされ得る。けいれんは、迅速で制御できない揺れである。けいれんの間は、筋肉が、繰り返し収縮および弛緩する。
【0093】
[0097] 「うつ病」という用語は、全ての形態のうつ病を含み、これらには、大うつ病性障害(MDD)、双極性障害、季節性感情障害(SAD)および気分変調症がある。「大うつ病性障害」は、本明細書では、「単極性うつ病」および「大うつ病」と互換的に使用する。「うつ病」はまた、うつ病に通常伴う任意の状態、すなわち、疲労の全ての形態(例えば、慢性疲労症候群)および失認等を含む。
【0094】
II.緒言
[0098] 神経障害の有効な療法を開発するための1つの戦略は、セロトニン (5-HT)、ノルエピネフリン(NE)およびドーパミン(DA)等、2つ以上の生体アミンの再取り込みを同時に阻害する広域スペクトルの抗うつ薬の使用である。このアプローチの理論的根拠は、ドーパミン作動性機能の欠乏が、うつ病の中心となる症状である快感消失と相関し得ることを示す臨床および前臨床の証拠に基づく。Baldessarini,R.J.,“Drugs and the Treatment of Psychiatric Disorders:Depression and Mania,in Goodman and Gilman’s The Pharmacological Basis of Therapeutics 431−459(9th ed 1996)Hardmanら編。
【0095】
[0099] 本発明の例示的な化合物および組成物の利点は、少なくとも2つの神経伝達物質(例えば、NE、5-HTおよびDA)の利用能を、例えば、シナプス間隙からの神経伝達物質の二重(再)取り込みを阻害することによって増加させるそれらの能力である。Skolnickらは、DA、NEおよび5−HTのシナプスにおける利用能を同時に増加させる抗うつ薬の治療プロファイルが、NEおよび/または5−HTのみを阻害する化合物とは異なることを示唆する一連の前臨床の証拠について報告している。Skolnick,P.ら,“Antidepressant−like actions of DOV−21,947:a“triple”reuptake inhibitor,”Eur.J.Pharm.2003,461,103。
【0096】
[0100] 例えば、Skolnickらは、化合物DOV21,947((+)−1−(3,4−ジクロロフェニル)−3−アザビシクロ[3.1.0]へキサン)が、セロトニン、ノルエピネフリンおよびドーパミンの再取り込みを、対応するヒト組換え輸送体を発現するヒト胎児腎臓(HEK293)細胞において阻害する(それぞれ、12、23および96nMのIC50値)ことを報告している。Skolnick,P.ら,“Antidepressant−like actions of DOV−21,947:a“triple”reuptake inhibitor,”Eur.J.Pharm.2003,461,99。さらに、DOV21,947は、(ラットの)強制水泳試験においては無動の持続時間を短縮し、かつまた、尾の懸垂試験においては無動の用量依存性の低下をもたらした。追加の証拠を、例えば、米国特許第6,372,919号中、DOV21,947等の新しい三重再取り込み阻害薬についての前臨床データ中に見い出すことができる。この特許では、DOV21,947は、ノルエピネフリンおよびセロトニンの取り込み部位に対して、ラセミ化合物である(±)−1−(3,4−ジクロロフェニル)−3−アザビシクロ[3.1.0]へキサンよりも、顕著に高い親和性を有するとして開示された。
【0097】
[0101] 総合すると、DOV21,947等の化合物についての前臨床データから、二重または三重再取り込み阻害薬には、臨床におけるうつ病の新規な治療としての可能性があることが示されている。
【0098】
III.組成物
A.シクロアルキルアミン
[0102] 例示的な実施形態では、本発明は、以下の式を有する化合物を提供する。
【0099】
【化2】
式中、指数nは、0、1および2からなる群から選択される整数であり;sは、0、1および2からなる群から選択される整数である。Aは、H、置換または非置換アルキル、ハロゲン、および置換または非置換ハロアルキルから選択されるメンバーである。Xは、H、ハロゲン、置換または非置換アルキル、置換または非置換アリール、置換または非置換ハロアルキル、およびOR5(ただし、R5は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、アシル、およびS(O)2R5a(ただし、R5aは、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルから選択される)から選択される)から選択される。
【0100】
[0103] YおよびZは独立して、H、ハロゲン、CF3、CN、OR9、SR9、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、置換または非置換ヘテロシクロアルキル、NR10R11、またはNO2を示す。YとZとは、それらが結合している原子と一緒になって任意選択により結合して、5員環〜7員環を形成し、この環はその中に、1、2または3個のヘテロ原子を任意選択により有することができる。R9は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、または置換または非置換ヘテロシクロアルキルを示す。基であるR10およびR11は独立して、H、OR12、アシル、S(O)2R13、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、または置換または非置換ヘテロシクロアルキルを示す。R10とR11とは、それらが結合している窒素と一緒になって任意選択により結合して、R10とR11とが結合する窒素に加えて、1、2または3個のヘテロ原子を任意選択により有する3員環、4員環、5員環、6員環または7員環を形成する。記号R12は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、または置換または非置換ヘテロシクロアルキルを示す。R13は、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルから選択される。
【0101】
[0104] R1およびR2は独立して、H、ハロゲン、CN、CF3、OR6、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、または置換または非置換ヘテロシクロアルキルである。R6は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルから選択される。
【0102】
[0105] R3およびR4は独立して、H、OR7、アシル、S(O)2R8、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、または置換または非置換ヘテロシクロアルキルを示す。R7は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、または置換または非置換ヘテロシクロアルキルである。R8は、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルから選択されるメンバーである。
【0103】
[0106] R1、R2、R3およびR4のうちの2つ以上が、それらが結合している原子と一緒になって任意選択により結合して、3員環、4員環、5員環、6員環または7員環を形成し、この環は、1、2、3または4個のヘテロ原子を任意選択により含む。
【0104】
[0107] YとZとは、それらが結合している原子と一緒になって任意選択により結合して、5員環、6員環または7員環を形成し、この環はその中に、1、2または3個のヘテロ原子を任意選択により有する。当業者には明らかであろうが、YとZとが結合して環をなす場合、環内に組み込まれた原子上の置換基(例えば、R9、R10およびR11)が、これらの置換基が結合している原子の原子価を満たす必要に応じて、存在する(例えば、環の環状構造内に組み込まれる)場合、または存在しない場合があるであろう。
【0105】
[0108] 例示的な実施形態では、YおよびZは独立して、ハロゲン、CF3、CN、OR9、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、置換または非置換ヘテロシクロアルキル、NR10R11、およびNO2を示す。YとZとは、それらが結合している原子と一緒になって任意選択により結合して、5員環、6員環または7員環を形成し、この環はその中に、1、2または3個のヘテロ原子を任意選択により有することができる。
【0106】
[0109] 例示的な実施形態では、本発明の化合物は、以下の式による構造を有しない。
【0107】
【化3】
【0108】
[0110] 別の例示的な実施形態では、化合物は、以下の式による構造を有しない。
【0109】
【化4】
【0110】
[0111] 例示的な実施形態では、化合物は、YまたはZのいずれかがHである場合には、R9が、Hおよび置換または非置換アルキルから選択されるメンバー以外であるような構造を有する。例示的な実施形態では、化合物は、YまたはZのいずれかがHである場合には、R9が、Hおよび非置換アルキルから選択されるメンバー以外であるような構造を有する。例示的な実施形態では、化合物は、YまたはZのいずれかがHである場合には、R9が、H、メチルおよびエチルから選択されるメンバー以外であるような構造を有する。
【0111】
[0112] 例示的な実施形態では、化合物は、R5が、Hおよび置換または非置換アルキルから選択されるメンバー以外であるような構造を有する。例示的な実施形態では、化合物は、R5が、Hおよび非置換アルキルから選択されるメンバー以外であるような構造を有する。例示的な実施形態では、化合物は、R5が、H、メチルおよびエチルから選択されるメンバー以外であるような構造を有する。
【0112】
[0113] 種々の例示的な実施形態では、指数sは1である。例示的な実施形態では、指数nは1である。種々の実施形態では、sおよびnの両方が1である。
【0113】
[0114] 例示的な実施形態では、YおよびZは独立して、H、ハロゲン、CNおよびCF3から選択される。種々の実施形態では、YおよびZのうちの少なくとも1つが、H以外である。例示的な実施形態では、YおよびZの両方が、H以外である。
【0114】
[0115] 例示的な実施形態では、R3およびR4は、置換または非置換C1〜C4アルキル、および置換または非置換C1〜C4ヘテロアルキルから独立して選択されるメンバーである。例示的な実施形態では、R3およびR4は、置換または非置換アルケニル、置換または非置換アルキニル、および置換または非置換シクロアルキルからなる群から独立して選択されるメンバーである。
【0115】
[0116] 種々の実施形態では、本発明の化合物は、以下からなる群から選択されるメンバーである構造を有する。
【0116】
【化5】
【0117】
[0117] 選択された実施形態では、本発明の化合物は、式IIおよびIIaから選択される構造を有する。
【0118】
【化6】
【0119】
式IIおよびIIaによる例示的な化合物として、以下が挙げられる。
【0120】
【化7】
【0121】
例示的な実施形態では、YおよびZは、H、ハロゲン、CNおよびCF3からなる群から独立して選択されるメンバーである。例示的な実施形態では、YおよびZは、ハロゲンである。例示的な実施形態では、YおよびZは、クロロである。例示的な実施形態では、sは1である。例示的な実施形態では、nは1である。例示的な実施形態では、R1およびR2は、Hである。例示的な実施形態では、AはHである。別の例示的な実施形態では、R1およびR2は、Hであり、AはHである。
【0122】
[0118] 選択された実施形態では、本発明の化合物は、式IIIおよびIIIaから選択される構造を有する。
【0123】
【化8】
【0124】
[0119] 式IIIおよびIIIaによる例示的な化合物として、以下が挙げられる。
【0125】
【化9】
【0126】
[0120] 例示的な実施形態では、YおよびZは、H、ハロゲン、CNおよびCF3からなる群から独立して選択されるメンバーである。例示的な実施形態では、YおよびZは、ハロゲンである。例示的な実施形態では、YおよびZは、クロロである。例示的な実施形態では、sは1である。例示的な実施形態では、nは1である。例示的な実施形態では、R1およびR2は、Hである。例示的な実施形態では、AはHである。別の例示的な実施形態では、R1およびR2は、Hであり、AはHである。
【0127】
[0121] 例示的な実施形態では、化合物は、式(IV)による構造を有する。
【0128】
【化10】
式中、YおよびZは、独立して選択されるハロゲンである。例示的な実施形態では、化合物は、以下による構造を有する。
【0129】
【化11】
【0130】
[0122] 例示的な実施形態では、化合物は、以下による構造を有する。
【0131】
【化12】
【0132】
[0123] 別の実施形態では、Yは、FおよびClから選択されるメンバーである。別の実施形態では、Zは、FおよびClから選択されるメンバーである。別の実施形態では、YはClであり、ZはClである。別の実施形態では、YはFであり、ZはClである。別の実施形態では、YはClであり、ZはFである。
【0133】
[0124] 例示的な実施形態では、化合物は、式(V)による構造を有する。
【0134】
【化13】
式中、YおよびZは、独立して選択されるハロゲンである。例示的な実施形態では、化合物は、以下による構造を有する。
【0135】
【化14】
【0136】
[0125] 例示的な実施形態では、化合物は、以下による構造を有する。
【0137】
【化15】
【0138】
[0126] 別の実施形態では、Yは、FおよびClから選択されるメンバーである。別の実施形態では、Zは、FおよびClから選択されるメンバーである。別の実施形態では、YはClであり、ZはClである。別の実施形態では、YはFであり、ZはClである。別の実施形態では、YはClであり、ZはFである。
【0139】
[0127] 本発明の例示的な化合物は以下の式を有する。
【0140】
【化16】
式中、R4は、HまたはCH3のいずれかである。
【0141】
[0128] 例示的な実施形態では、化合物は、式(VI)による構造を有する。
【0142】
【化17】
【0143】
[0129] 別の例示的な実施形態では、この構造を有する化合物は、YおよびZから選択される少なくとも1つの、ハロゲンであるメンバーを有する。別の例示的な実施形態では、YおよびZは、ハロゲンである。別の実施形態では、Yは、FおよびClから選択されるメンバーである。別の実施形態では、Zは、FおよびClから選択されるメンバーである。別の実施形態では、YはClであり、ZはClである。別の実施形態では、YはFであり、ZはClである。別の実施形態では、YはClであり、ZはFである。
【0144】
[0130] 例示的な実施形態では、化合物は、式(VII)による構造を有する。
【0145】
【化18】
式中、YおよびZは、Hではなく、Aは、置換または非置換アルキルから選択されるメンバーである。別の例示的な実施形態では、この構造を有する化合物は、YおよびZから選択される少なくとも1つの、ハロゲンであるメンバーを有する。別の例示的な実施形態では、YおよびZは、ハロゲンである。別の実施形態では、Yは、FおよびClから選択されるメンバーである。別の実施形態では、Zは、FおよびClから選択されるメンバーである。別の実施形態では、YはClであり、ZはClである。別の実施形態では、YはFであり、ZはClである。別の実施形態では、YはClであり、ZはFである。例示的な実施形態では、Aは、置換または非置換メチルである。例示的な実施形態では、Aはメチルである。
【0146】
[0131] 例示的な実施形態では、化合物は、式(VIII)による構造を有する。
【0147】
【化19】
式中、YおよびZは、Hではなく、R3およびR4はそれぞれ独立して、Hおよび置換または非置換アルキルから選択される。別の例示的な実施形態では、各R3およびR4はそれぞれ独立して、Hおよび置換または非置換メチルから選択される。別の例示的な実施形態では、R3およびR4はそれぞれ独立して、Hおよびメチルから選択される。別の例示的な実施形態では、この構造を有する化合物は、、YおよびZから選択される少なくとも1つの、ハロゲンであるメンバーを有する。別の例示的な実施形態では、YおよびZは、ハロゲンである。別の実施形態では、Yは、FおよびClから選択されるメンバーである。別の実施形態では、Zは、FおよびClから選択されるメンバーである。別の実施形態では、YはClであり、ZはClである。別の実施形態では、YはFであり、ZはClである。別の実施形態では、YはClであり、ZはFである。
【0148】
[0132] 本発明の例示的な化合物は、以下の式による構造を有する。
【0149】
【化20】
式中、A1、A2、A3およびA4はそれぞれ独立して、O、S、N(Rb)bおよびC(Rb)b(Rc)から選択される。指数aは、0、1および2から選択される整数である。指数bは、それが結合している原子の原子価の要件を満たす必要に応じて、0または1である。RbおよびRcは、H、ハロゲン、CF3、CN、OR14、SR14、NR15R16、NR15S(O)2R14、NR15C(O)R14、S(O)2R14、アシル、C(O)OR14、C(O)NR15R16、S(O)2NR15R16、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルから独立して選択されるメンバーである。各R14、R15およびR16は、H、アシル、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、置換または非置換ヘテロシクロアルキルからなる群から独立して選択されるメンバーであり、ただし、R14、R15およびR16のうちの2つが、それらが結合している原子と一緒になって任意選択により結合して、3員環、4員環、5員環、6員環または7員環を形成し、この環は、1、2または3個のヘテロ原子を任意選択により含む。
【0150】
[0133] 例示的な実施形態では、RbおよびRcは、H、ハロゲン、CN、ハロゲンで置換C1〜C4アルキル(例えば、CF3)、およびC1〜C4アルコキシ(例えば、OMe,OEt、OCF3)からなる群から独立して選択されるメンバーである。
【0151】
[0134] 例示的な実施形態では、YおよびZが結合して、5員環、6員環または7員環を有する縮合環系を形成し、1、2または3個のヘテロ原子を任意選択により含む。したがって、1つの実施形態では、フェニル環の置換基が、以下の構造を有する。
【0152】
【化21】
式中、環Lは、置換または非置換、飽和もしくは不飽和のシクロアルキルもしくはヘテロシクロアルキルであるか、または置換または非置換アリールもしくはヘテロアリールである。
【0153】
[0135] 例示的な縮合環系を以下に示す。
【0154】
【化22】
式中のA1、A2、A3およびA4、ならびにaは、本明細書に記載されている。
【0155】
[0136] 本発明の化合物は、アミンの部分(例えば、一級アミン、二級アミンまたは三級アミンの基)を含み、したがって、化合物(例えば、遊離の塩基)を酸と接触させることによって、化合物を塩の形態に変換することができる。例示的な実施形態では、塩の形態を得て、塩以外の形態では油性または粘性の化合物を固体の物質に変換し、取扱いを容易にする。別の例示的な実施形態では、本発明の化合物の遊離の塩基を、対応する塩に変換することによって、化合物の水性媒体中における溶解性を向上させ、これによって、生物学的利用率、薬物動態および薬力学等の生物学的特徴に影響を及ぼすことができる。したがって、本発明の化合物の任意の塩の形態、すなわち、無機酸の塩(例えば、塩酸塩)または有機酸の塩を含めた薬学的に許容できる塩等が、現在の発明の範囲内に属する。また、本発明の化合物の任意のプロドラッグも、本発明の範囲内に属する。例えば、R3およびR4は、in vivoにおいて切断可能であって、アミン、例えば、一級アミンまたは二級アミンを生じる任意の基であってよい。
【0156】
B.立体異性体を含む組成物
[0137] 本発明の化合物は、1つまたは複数の立体中心を含むことができ、特定の幾何学的なまたは立体異性体の形態で存在してよい。化合物は、キラルであっても、ラセミ体であっても、または1つもしくは複数の立体異性体を含む組成物中に存在してもよい。現在の発明は、鏡像異性体、ジアステレオマー、ラセミ混合物、鏡像異性体に富む混合物、およびジアステレオ異性体に富む混合物を包含する。追加の不斉炭素原子も、アルキル基等の置換基中に存在することができる。全てのそのような異性体、およびそれらの混合物が、本発明中に含まれるものとする。
【0157】
[0138] 例えば、本発明の化合物の特定の鏡像異性体が所望される場合には、これを、不斉合成によって、またはキラルの補助物質を用いて誘導体化することによって調製することができ、後者の場合、得られたジアステレオマーの混合物を分離し、補助基を切断して、純粋な所望の鏡像異性体を得る。あるいは、分子が、アミノ基等の塩基性の官能基またはカルボキシル基等の酸性の官能基を含有する場合には、ジアステレオマーの塩を、適切な光学的に活性な酸または塩基を用いて形成し、それに続いて、このようにして形成したジアステレオマーを分別結晶または当技術分野で既知のクロマトグラフ的な手段によって分割し、それに続いて、純粋な鏡像異性体を回収することができる。さらに、鏡像異性体とジアステレオマーとの分離はしばしば、キラルの安定している相を利用するクロマトグラフィーを使用して、任意選択により化学的な誘導体化(例えば、アミンからのカルバメートの形成)と組み合わせて達成される。
【0158】
[0139] 本明細書で使用する場合、「鏡像異性体に富む」または「ジアステレオ異性体に富む」という用語は、約50%超、好ましくは約70%超、より好ましくは約90%超で、鏡像異性体過剰(ee)またはジアステレオ異性体過剰(de)を有する化合物を指す。一般に、約90%超の鏡像異性的またはジアステレオ異性的な純度、例えば、約95%超、約97%超および約99%超のeeまたはdeを有する組成物が特に好ましい。
【0159】
[0140] 「鏡像異性体過剰」および「ジアステレオ異性体過剰」という用語は、本明細書では互換的に使用する。単一の立体中心を有する化合物を、「鏡像異性体過剰」の状態で存在すると呼び、少なくとも2つの立体中心を有する化合物を、「ジアステレオ異性体過剰」の状態で存在すると呼ぶ。
【0160】
[0141] 例えば、「鏡像異性体過剰」という用語は、当技術分野では周知であり、以下のように定義される。
【0161】
【数1】
【0162】
[0142] 例えば、「鏡像異性体過剰」という用語は、「光学純度」というより古い用語に関連し、両方が、同一の現象の測定値である。eeの値は、0から100までの数であり、ゼロは、ラセミ体であり、100は、鏡像異性的に純粋である。過去に98%光学的に純粋であるといわれていた化合物は、今では、96%eeによってより正確に特徴づけられる。90%eeは、問題になっている材料中、95%が、1つの鏡像異性体として存在し、かつ5%が、(1つまたは複数の)その他の鏡像異性体として存在することを反映する。
【0163】
[0143] したがって、1つの実施形態では、本発明は、本発明の化合物の第1の立体異性体および少なくとも1つの追加の立体異性体を含む組成物を提供する。第1の立体異性体は、少なくとも約80%、好ましくは少なくとも約90%、より好ましくは少なくとも約95%のジアステレオ異性体過剰または鏡像異性体過剰の状態で存在することができる。特に好ましい実施形態では、第1の立体異性体は、少なくとも約96%、少なくとも約97%、少なくとも約98%、少なくとも約99%、または少なくとも約99.5%のジアステレオ異性体過剰または鏡像異性体過剰の状態で存在する。鏡像異性体過剰またはジアステレオ異性体過剰は、相互の立体異性体を正確に比較して決定してもよく、または少なくとも2つのその他の立体異性体の合計と比較して決定してもよい。例示的な実施形態では、鏡像異性体過剰またはジアステレオ異性体過剰を、混合物中に存在する全てのその他の検出可能な立体異性体と比較して決定する。立体異性体は、解析する混合物中のそのような立体異性体の濃度をキラルHPLC等の一般的な解析方法を使用して決定することができる場合には、検出可能である。
【0164】
C.化合物の合成
1.一般論
[0144] 本発明の化合物は、純粋なシス異性体もしくはラセミ混合物、または2つ以上のジアステレオマーの混合物として合成することができる。立体異性体は、適切な合成段階において、例えば、HPLC等のキラルカラムクロマトグラフィーによって分離して、それぞれの立体異性体の鏡像異性的/ジアステレオ異性体に富む形態または鏡像異性的もしくはジアステレオ異性的に純粋な形態を得ることができる。立体化学を、NMRカップリングのパターンに基づいて、任意選択により文献値と併せて割り当てることができる。絶対配置を、既知の立体配置のキラル前駆体からの合成によって、または結晶化した材料を使用するX線結晶解析による決定によって決定することができる。
【0165】
[0145] 立体化学的配置は、アミンを有する側鎖およびシクロアルキル環上の置換基の相対的な立体配置に従って定義される。2つ以上の置換基が存在する場合には、より高い順位(IUPAC)の置換基を使用して、立体化学的配置を決定する。
【0166】
[0146] 本発明の化合物は、以下に記載するスキームに従って合成することができる。所望の本発明の化合物を合成するために、スキーム中に示す例示的な試薬に代わる、適切な代替の試薬を選択することは、当業者の能力の範囲内である。また、必要に応じて合成の工程を除外または追加することも当業者の能力の範囲内である。
【0167】
2.シクロアルキルアミンの一般的な合成
[0147] 1つの実施形態では、本発明の化合物を、以下のスキーム1に示すように、対応するアミノケトンaから合成した。
【0168】
【化23】
【0169】
[0148] ジメチルアミノメチルシクロヘキサノンaを、アリールグリニャール試薬b〜dと縮合させて、ラセミ体のアミノアルコールを得た。ラセミ体の生成物を、半調製用のchiralpak ADカラム上でキラルクロマトグラフィーによって精製して、鏡像異性体1、24および28、ならびに2、26および27を得た。スキーム1中に開示したRyおよびRzの値の他に、RyおよびRzは、置換または非置換アルキル、Cl、Br、F、NR10R11、OR9、SR9、および置換または非置換アリールから独立して選択されるメンバーである。
【0170】
【化24】
【0171】
[0149] スキーム2を参照して、ラセミ体のシス−2−(アミノメチル)−1−(3,4−ジクロロフェニル)シクロヘキサノールを、キラルHPLCにより構成要素の異性体である8(動きがより速い鏡像異性体)と9(動きがより遅い鏡像異性体)とに分離することによって、2の親である一級アミン、すなわち、化合物9を得た。ギ酸、ホルムアルデヒドおよびシアン水素化ホウ素ナトリウムを用いる還元的アミノ化によって、9を2に変換した。また、ラセミ体のシス−1−(3,4−ジクロロフェニル)−2−((ジメチルアミノ)メチル)シクロヘキサノールのキラルHPLCよる分離後に、2を、動きがより遅い鏡像異性体としても得た;DEADおよび酸性EtOHを用いる2段階工程の手順において2を還元することによって、モノ−メチル誘導体4を得た。
【0172】
D.医薬組成物
[0150] 例示的な態様では、本発明は、本明細書に記載する化合物またはその薬学的に許容できる塩もしくは溶媒和化合物、および少なくとも1つの薬学的に許容できる担体を含む医薬組成物を提供する。種々の実施形態では、化合物は、シス異性体である。例示的な実施形態では、化合物は、式(I)〜(IX)から選択されるメンバーである構造を有する。
【0173】
[0151] 以下に詳細に記載するように、本発明の医薬組成物は、固体または液体の形態で投与するために特別に製剤化することができ、これには、経口投与に適合した形態、例えば、錠剤、水薬(水性もしくは非水性の液剤もしくは懸濁剤)、(静脈内および筋肉内を含めた)非経口投与または硬膜外注射に適合した形態、例えば、無菌の液剤または懸濁剤、あるいは持続放出性の製剤が含まれる。本発明の医薬組成物はまた、経皮投与のために特異的に製剤化することもできる。
【0174】
[0152] 本発明の医薬組成物は、経口的、非経口的、皮下的、経皮的、経鼻的に投与しても、または肛門から坐剤によって投与してもよい。本発明の医薬組成物はまた、制御送達デバイスを使用して投与してもよい。
【0175】
[0153] 本発明の製剤は、経口投与および非経口投与、特に、筋肉内投与、静脈内投与および皮下投与に適した製剤を含む。製剤は、単位投与剤型として好都合に提供することができ、薬学の技術分野で周知の任意の方法によって調製することができる。担体材料と組み合わせて、単一の投与剤型をもたらすことができる活性成分の量は、治療する宿主および特定の投与形態に依存して変化する。担体材料と組み合わせて、単一の投与剤型をもたらすことができる活性成分の量は、一般に、患者に対して毒性を示すことなく、治療効果をもたらす化合物の量である。一般に、この量は、百パーセント中、活性成分が、約1パーセントから約99パーセントまでに及ぶ。
【0176】
[0154] 例示的な単位投与製剤は、活性成分もしくはその薬学的に許容できる塩の有効用量、またはその適切な一部分を含有する製剤である。予防または治療に用いる用量の程度は、典型的には、治療しようとする状態の性質および重症度、ならびに投与経路によって異なる。また、用量、およびおそらく投与頻度も、個々の患者の年齢、体重および応答によって異なる。一般に、1日当たりの総投与量は(単回投与また分割投与として)、1日当たり約1mgから1日当たり約7000mgまで、好ましくは、1日当たり約1mgから1日当たり約100mgまで、より好ましくは、1日当たり約10mgから1日当たり約100mgまで、さらにより好ましくは、約20mgから、約100mgまで、約80mgまで、または約60mgまでの範囲である。いくつかの実施形態では、1日当たりの総投与量は、1日当たり約50mgから約500mgまで、好ましくは、1日当たり約100mgから約500mgまでの範囲であってよい。小児、65歳超の患者、および腎臓または肝臓の機能が損なわれている患者には、最初は、低い用量を投与し、投与量は、個人の応答および/または血中レベルに基づいて設定することがさらに推奨される。当業者には明らかになるであろうが、場合によっては、これらの範囲以外の投与量を使用する必要がある場合がある。さらに、臨床医または担当医であれば、個々の患者の応答に併せて、療法を中断、加減または終了する方法および時期が分かることにも留意されたい。
【0177】
[0155] 特定の実施形態では、本発明の製剤は、シクロデキストリン、リポソーム、ミセル形成物質、例えば、胆汁酸、ならびにポリマー性の担体、例えば、ポリエステルおよびポリ酸無水物からなる群から選択される賦形剤と;本発明の化合物とを含む。特定の実施形態では、上記の製剤によって、本発明の化合物は、経口的に体内に吸収され利用され得るようになる。
【0178】
[0156] これらの製剤または組成物を調製する方法は、本発明の化合物を、担体および任意選択により1つまたは複数の付属の成分と混ぜ合わせる工程を含む。一般に、本発明の化合物を、液体の担体もしくは微粉化した固体の担体または両方と均一かつ密接に混ぜ合わせ、次いで、必要に応じて、製品の形状となすことによって製剤を調製する。
【0179】
[0157] 経口投与に適した本発明の製剤は、カプセル剤、カシェ剤、丸剤、錠剤、カプレット剤、(芳香剤を添加した基剤、通常スクロースおよびアラビアゴムもしくはトラガカントを使用する)ドロップ剤、散剤、顆粒剤の形態であってもよく、あるいは水性もしくは非水性の液体中の液剤もしくは懸濁剤としても、または水中油型もしくは油中水型の乳濁剤としても、またはエリキシル剤もしくはシロップ剤としても、あるいは(ゼラチンおよびグリセリン等の不活性な基剤またはスクロースおよびアラビアゴムを使用する)芳香錠剤(pastille)としてもよく、それぞれが、あらかじめ決定した量の本発明の化合物を、活性成分として含有する。本発明の化合物はまた、ボーラス、舐剤またはペースト剤としても投与することができる。
【0180】
[0158] 経口投与のための本発明の固体の投与剤型(カプセル剤、錠剤、カプレット剤、丸剤、糖衣錠剤、散剤、顆粒剤等)においては、活性成分を、1つもしくは複数の薬学的に許容できる担体、すなわち、クエン酸ナトリウムもしくはリン酸二カルシウム等、ならびに/または以下のいずれかと混合する:(1)充填剤もしくは増量剤、すなわち、デンプン、ラクトース、スクロース、グルコース、マンニトール、シアル酸および/もしくはケイ酸等;(2)結合剤、すなわち、例えば、カルボキシメチルセルロース、アルギン酸、ゼラチン、ポリビニルピロリドン、スクロースおよび/もしくはアラビアゴム等:(3)湿潤剤、すなわち、グリセロール等;(4)崩壊剤、すなわち、寒天、炭酸カルシウム、バレイショデンプンもしくはタピオカデンプン、アルギン酸、特定のケイ酸塩および炭酸ナトリウム等;(5)溶解遅延剤、すなわち、パラフィン等;(b)吸収促進剤、すなわち、四級アンモニウム化合物等;(7)湿潤剤、すなわち、例えば、セチルアルコール、モノステアリン酸グリセロールおよび非イオン性界面活性剤等;(8)吸収剤、すなわち、カオリンおよびベントナイトクレイ等;(9)滑沢剤、すなわち、タルク、ステアリン酸カルシウム、ステアリン酸マグネシウム、固体のポリエチレングリコール、ラウリル硫酸ナトリウムおよびそれらの混合物等;ならびに(10)着色剤。カプセル剤、錠剤および丸剤の場合には、医薬組成物はまた、緩衝剤も含むことができる。また、類似の型の固体の組成および高分子量のポリエチレングリコール等も、ラクトースまたは乳糖等の賦形剤を使用する軟質および硬質の殻のゼラチンカプセル剤中で、充填剤として利用することができる。
【0181】
[0159] 錠剤は、任意選択により1つまたは複数の付属の成分と共に、圧縮または成型によって作製することができる。圧縮錠剤は、結合剤(例えば、ゼラチンまたはヒドロキシプロピルメチルセルロース)、滑沢剤、不活性な希釈剤、保存剤、崩壊剤(例えば、デンプングリコール酸ナトリウムもしくは架橋カルボキシメチルセルロースナトリウム)、表面活性剤または分散剤を使用して調製することができる。成型錠剤は、不活性な液体の希釈剤を用いて湿潤させた粉末化合物の混合物を、適切な機械中で成型することによって作製することができる。
【0182】
[0160] 本発明の医薬組成物の錠剤およびその他の固体の投与剤型、すなわち、糖衣錠剤、カプセル剤、丸剤および顆粒剤等は、任意選択により、腸溶コーティングおよび医薬品製剤技術分野で周知のその他の被覆等の被覆および殻を用いて収容するかまたはそうした被覆および殻を有するように調製することができる。これらの固体の投与剤型はまた、その中の活性成分を緩慢にまたは制御して放出するように、例えば、所望の放出プロファイルをもたらすために様々な比率のヒドロキシプロピルメチルセルロースを使用して、その他のポリマー性マトリックス、リポソームおよび/またはマイクロスフェアを使用して製剤化することもできる。これらの固体の投与剤型は、急速に放出するように、例えば、凍結乾燥して製剤化することができる。これらの固体の投与剤型は、例えば、細菌を保持するフィルターを通すろ過によって、または使用直前に無菌水もしくは何らかのその他の無菌の注射用媒体中に溶解させることができる無菌の固体組成物の形態中に滅菌剤を組み込むことによって滅菌することができる。これらの組成物はまた、任意選択により隠蔽剤を含有することができ、消化管の特定の部分に限ってまたはそこで優先的に、(1つまたは複数の)活性成分を、任意選択により遅延させて放出することもできる。使用することができる組成物を包埋する例として、ポリマー性物質およびろうが挙げられる。活性成分はまた、適切な場合には、1つまたは複数の上記に記載した賦形剤と共に、マイクロカプセルの形態となすこともできる。
【0183】
[0161] 本発明の化合物の経口投与のための液体の投与剤型には、薬学的に許容できる乳剤、マイクロエマルション、液剤、懸濁剤、シロップ剤およびエリキシル剤がある。活性成分に加えて、液体の投与剤型は、当技術分野で一般に使用される不活性な希釈剤、すなわち、例えば、水またはその他の溶媒等、可溶化剤ならびに乳化剤、すなわち、エチルアルコール、イソプロピルアルコール、炭酸エチル、酢酸エチル、ベンジルアルコール、安息香酸ベンジル、プロピレングリコール、1,3−ブチレングリコール、油(特に、綿実油、落花生油、トウモロコシ油、胚芽油、オリーブ油、ヒマシ油およびゴマ油)、グリセロール、テトラヒドロフリルアルコール、ポリエチレングリコールならびにソルビタンの脂肪酸エステル、さらにそれらの混合物等も含有することができる。
【0184】
[0162] 不活性な希釈剤の他に、経口用組成物はまた、アジュバント、すなわち、湿潤剤、乳化剤および懸濁化剤、甘味剤、芳香剤、着色剤、矯臭剤ならびに保存剤等も含むことができる。
【0185】
[0163] 懸濁剤は、活性化合物に加えて、懸濁化剤、例えば、エトキシ化イソステアリルアルコール、ポリオキシエチレンソルビトールおよびソルビタンエステル、微結晶セルロース、メタ水酸化アルミニウム、ベントナイト、寒天ならびにトラガカント、さらにそれらの混合物も含有することができる。
【0186】
[0164] 非経口投与に適した本発明の医薬組成物は、1つまたは複数の本発明の化合物を、1つまたは複数の薬学的に許容できる無菌の等張な水性または非水性の液剤、分散剤、懸濁剤もしくは乳剤、または使用直前に無菌の注射用の液剤もしくは分散剤の中に再構成することができる無菌の散剤と組み合わせて含むことができる。こうした注射用の液剤または分散剤は、糖、アルコール、抗酸化剤、緩衝剤、静菌剤、意図するレシピエントの血液と処方を等張にする溶質、または懸濁化剤もしくは増粘剤を含有する場合がある。
【0187】
[0165] 本発明の医薬組成物において利用することができる、適切な水性および非水性の担体の例として、水、エタノール、ポリオール(グリセロール、プロピレングリコール、ポリエチレングリコール等)およびそれらの適切な混合物、オリーブ油等の植物油、ならびにオレイン酸エチル等の注射用の有機エステルが挙げられる。適切な流動性を、例えば、レシチン等の被覆材料を使用することによって、分散剤の場合には必要な粒子サイズを維持することによって、および界面活性剤を使用することによって維持することができる。
【0188】
[0166] これらの組成物はまた、アジュバント、すなわち、保存剤、湿潤剤、乳化剤および分散剤等も含有することができる。微生物の対象化合物に対する作用を、種々の抗細菌剤および抗真菌剤、例えば、パラベン、クロロブタノール、フェノールソルビン酸等を含めることによって確実に予防することができる。また、糖、塩化ナトリウム等の等張化剤を、組成物中に含めることが望ましい場合もある。さらに、注射用の医薬品の形態の吸収の延長を、モノステアリン酸アルミニウムおよびゼラチン等の吸収を遅延させる薬剤を含めることによってもたらすこともできる。
【0189】
[0167] 場合によっては、薬物の効果を延長させるために、皮下または筋肉内の注射からの薬物の吸収を遅くすることが望ましい。これは、水溶性の低い結晶性または非結晶性の材料を液体中に懸濁させることによって達成することができる。薬物の吸収速度は、この場合、その溶解速度に依存し、当該溶解速度は、今度は、結晶サイズおよび結晶形態に依存し得る。あるいは、非経口投与用の薬物の形態の吸収遅延は、薬物を油性のビヒクル中に溶解または懸濁させることによっても達成される。
【0190】
[0168] 注射用デポーの形態を、対象化合物のマイクロカプセルのマトリックスを、ポリ乳酸−ポリグリコリド等の生分解性ポリマー中に形成することによって作製することができる。薬物のポリマーに対する比および利用する特定のポリマーの性質に依存して、薬物放出の速度を制御することができる。その他の生分解性ポリマーの例として、ポリ(オルトエステル)およびポリ(酸無水物)が挙げられる。また、デポー注射用製剤は、身体組織に適合するリポソームまたはマイクロエマルションの中に薬物を捕捉することによっても調製される。作用延長型の錠剤の形態をとる本発明の医薬組成物または単位投与剤型は、薬物治療を一定期間にわたってもたらすように、薬物物質を放出するように製剤化された圧縮錠剤を含むことができる。薬物物質の放出が、投与後の一定期間にわたりまたは特定の生理的状態が存在するようになるまで阻止される作用遅延型の錠剤を含む、いくつかの錠剤の型がある。薬物物質の完全な用量を消化液に定期的に放出する作用反復型の錠剤を形成することができる。また、含有する薬物物質の一定量を消化液に連続的に放出する徐放性の錠剤も形成することができる。
【0191】
[0169] 本発明の化合物はまた、制御放出手段によっても、または当業者によく知られている送達デバイスによっても投与することができる。例として、これらに限定されないが、米国特許第3,845,770号;第3,916,899号;第3,536、809号;第3,598,123号;および第4,008,719号、第5,674,533号、第5,059,595号、第5,591,767号、第5,120,548号、第5,073,543号、第5,639,476号、第5,354,556号、および第5,733,566号に記載されているものが挙げられる。これらはそれぞれ、参照により本明細書に組み込まれている。そのような投与剤型を使用して、例えば、ヒドロキシプロピルメチルセルロース、その他のポリマーのマトリックス、ゲル、透過性の膜、浸透圧によるシステム、多層被覆、微小粒子、リポソーム、マイクロスフェア、またはそれらの組合せを使用して、様々な比率で所望の放出プロファイルをもたらして、1つまたは複数の活性成分の緩除放出または制御放出を得ることができる。本明細書に記載するものを含めて、当業者に既知の適切な制御放出性の処方を容易に選択して、発明の化合物に関して使用することができる。したがって、本発明は、これらに限定されないが、制御放出するようになされた錠剤、カプセル剤、ジェルキャップ剤およびカプレット剤等の経口投与に適した単一の単位投与剤型を包含する。
【0192】
[0170] 全ての制御放出性の医薬製品は、それらの非制御性の対応物によって達成されたものを上回るように薬物療法を改善するという共通の目標を有する。理想的には、最適に設計された制御放出性の調製物の医学的治療における使用は、最小の薬物物質を利用して、最短時間で状態を治癒または制御することによって特徴付けられる。制御放出性の製剤の利点には、薬物活性の長期化、投与頻度の低下、および患者のコンプライアンスの向上がある。さらに、制御放出性の製剤を使用して、作用の開始時期、または薬物の血中レベル等のその他の特徴に影響を及ぼすことができ、したがって、副作用(例えば、有害作用)の発生率に影響を及ぼすことができる。
【0193】
[0171] 大部分の制御放出性の製剤は、所望の治療効果を即座にもたらす薬物(活性成分)の量を最初に放出し、このレベルの治療または予防の効果を長期の期間にわたって維持するための薬物の別の量を次第にかつ連続的に放出するように設計される。体内でこの一定のレベルの薬物を維持するためには、投与剤型から薬物が、代謝され身体から排泄される薬物の量を置換する速度で放出されなければならない。活性成分の制御放出を、これらに限定されないが、pH、温度、酵素、水もしくはその他の生理的条件を含めた種々の条件、または化合物によって刺激することができる。
【0194】
[0172] 本発明の化合物はまた、経皮から、外用および粘膜からの投与剤型として製剤化することもでき、こうした剤型には、これらに限定されないが、眼科用液剤、スプレー剤、エアロゾル剤、クリーム剤、ローション剤、軟膏剤、ジェル剤、液剤、乳剤、懸濁剤、または当業者に既知のその他の剤型がある。例えば、Remington’s Pharmaceutical Sciences,16thおよび18th eds.,Mack Publishing,Easton PA(1980および1990);ならびにIntroduction to Pharmaceutical Dosage Forms,4th ed.,Lea & Febiger,Philadelphia(1985)を参照されたい。経皮投与剤型には、「リザーバ型」または「マトリックス型」のパッチがあり、これを皮膚に適用し、特定の期間にわたり着用して、所望の量の活性成分を浸透させる。
【0195】
[0173] 本発明が包含する経皮から、外用および粘膜からの投与剤型を得るために使用することができる適切な賦形剤(例えば、担体および希釈剤)ならびにその他の材料は、薬学の技術分野の業者にはよく知られており、所与の医薬組成物または投与剤型が適用される特定の組織によって異なる。
【0196】
[0174] 治療しようとする特異的な組織に依存して、追加の構成成分を、本発明の活性成分を用いる治療の前に、治療に併せて、または治療の後に使用することができる。例えば、浸透増強剤を使用して、活性成分の組織への送達を支援することができる。
【0197】
[0175] また、医薬組成物もしくは投与剤型のpH、または医薬組成物もしくは投与剤型を適用する組織のpHを加減して、1つまたは複数の活性成分の送達を改善することもできる。同様に、溶媒担体の極性、そのイオン強度またはその浸透圧を加減して、送達を改善することもできる。また、ステアリン酸等の化合物を、医薬組成物または投与剤型に添加して、送達を改善するように、1つまたは複数の活性成分の親水性または親油性を好都合に変化させることもできる。この場合、ステアリン酸が、製剤のための脂質ビヒクルとして、乳化剤または界面活性剤として、および送達増強剤または浸透増強剤として役立つ場合がある。活性成分の異なる塩、水和物または溶媒和化合物を使用して、得られた組成物の特性をさらに加減することができる。
【0198】
[0176] 本発明の化合物を医薬品として、ヒトおよび動物に投与する場合、それ自体で、または薬学的に許容できる担体と組み合わせて、例えば、活性成分を0.1〜99.5%を含有する医薬組成物として与えることができる。
【0199】
[0177] 本発明の調製物は、経口的および非経口的に与えることができる。もちろん、これらは、各投与経路に適した剤型で与えられる。それらは、例えば、錠剤またはカプセル剤の形態で、注射により、および静脈内投与により投与される。1つの実施形態では、経口投与が好ましい。
【0200】
[0178] 「非経口投与」および「非経口的に投与した」という句は、本明細書で使用する場合、腸内投与および外用投与以外の通常注射による投与形態を意味し、静脈内、筋肉内、動脈内、くも膜下腔内、関節内、眼窩内、心臓内、皮内、腹腔内、経気管、皮下、角質下、関節腔内、嚢下、くも膜下、脊髄内および胸骨内への注射および注入を非限定的に含む。
【0201】
[0179] 選択する投与量のレベルは、利用される特定の本発明の化合物またはそのエステル、塩もしくはアミドの活性、投与経路、投与の時期、利用される特定の化合物の排泄および代謝の速度、治療期間、利用される特定の化合物と併用するその他の薬物、化合物および/または材料、治療する患者の年齢、性別、体重、状態、全般的な健康状態および過去の病歴、ならびに医学の技術分野で周知の類似の要因を含む多様な要因に依存する。
【0202】
[0180] 当技術分野の通常の技術を有する医師または獣医師であれば、必要とされる医薬組成物の有効量を容易に決定し、処方することができる。例えば、医師または獣医師は、所望の治療効果を達成するために必要な用量よりも低いレベルの、医薬組成物中で利用される本発明の化合物の用量から開始し、投与量を所望の効果を達成するまで次第に増加させることができるであろう。
【0203】
[0181] 一般に、本発明の化合物の適切な1日当たりの用量は、治療効果をもたらすために有効な最も低い用量である化合物の量である。そのような有効用量は一般に、上記に記載した要因に依存する。一般に、本発明の化合物の患者に対する経口、静脈内、側脳室内および皮下への投与量は、1日当たり、体重1キログラム当たり約0.005mgから体重1キログラム当たり約5mgまでの範囲である。例示的な実施形態では、本発明の化合物の経口投与量は、1日当たり約10mgから約300mgまでの範囲である。例示的な実施形態では、本発明の化合物の経口投与量は、1日当たり約20mgから約250mgまでの範囲である。例示的な実施形態では、本発明の化合物の経口投与量は、1日当たり約100mgから約300mgまでの範囲である。例示的な実施形態では、本発明の化合物の経口投与量は、1日当たり約10mgから約100mgまでの範囲である。例示的な実施形態では、本発明の化合物の経口投与量は、1日当たり約25mgから約50mgまでの範囲である。例示的な実施形態では、本発明の化合物の経口投与量は、1日当たり約50mgから約200mgまでの範囲である。上記に列挙した投与量の範囲のそれぞれを、単位投与製剤として製剤化することができる。
【0204】
[0182] 「治療」または「治療する」という用語は、療法、再発の予防および急性症状の緩和を包含するものとする。「治療する」は、症状の緩和および基底状態の消散のいずれかまたは両方を指すことに留意されたい。本発明の状態の多くにおいては、本発明の化合物または組成物の投与が、疾患状況に対して直接的ではなく、むしろ何らかの悪質な症状に対して作用する場合があり、そうした症状の改善が、疾患状況の全般的なかつ望ましい緩和をもたらす。また、本発明の化合物を使用して、疾患を阻止することもできる(予防)。
【0205】
[0183] この治療を受ける患者は、霊長類、特にヒト、ならびにウマ、ウシ、ブタおよびヒツジ等のその他の哺乳動物を、かつ家禽および一般的なペットを含むそれを必要とする任意の動物である。
【0206】
[0184] 本発明の化合物および医薬組成物は、その他の医薬品、例えば、抗菌剤、すなわち、ペニシリン、セファロスポリン、アミノグリコシドおよびグリコペプチド等と併用して投与することができる。したがって、併用療法は、活性化合物を、最初に投与した薬剤の治療効果が、それに続く薬剤を投与するときに、完全には消失していないように、逐次、同時および個別に投与することを含む。
【0207】
[0185] 例示的な実施形態では、本発明の化合物が治療効果を示す適応症を呈する対象は、それ以外のためには、本発明の化合物または本発明の化合物を包含する構造の属に属する化合物を用いた治療を必要としない。
【0208】
IV.方法
A.モノアミン輸送体に対する結合
[0186] 種々の態様では、本発明は、本発明の化合物をモノアミン輸送体に対して結合させる方法を提供する。この方法は、モノアミン輸送体と、本発明の化合物とを接触させることを含む。
【0209】
[0187] 本発明は、モノアミン輸送体リガンドの、モノアミン輸送体(セロトニン輸送体、ドーパミン輸送体およびノルエピネフリン輸送体等)に対する結合を阻害する方法をさらに提供する。この方法は、モノアミン輸送体と、本発明の化合物とを接触させることを含む。例示的な実施形態では、モノアミン輸送体リガンドは、セロトニン、ドーパミンまたはノルエピネフリン等の内因性モノアミンである。別の例示的な実施形態では、リガンドは、モノアミン輸送体に対する結合親和性を有することが知られている薬物分子または別の小型分子である。別の例示的な実施形態では、モノアミン輸送体リガンドは、モノアミン輸送体に対して結合することが知られている、放射性活性物質で標識した化合物である。
【0210】
[0188] 例示的な実施形態では、本明細書に記載するもの等のex vivoにおける結合アッセイを使用して、リガンド結合の阻害を示す。例示的な実施形態では、本発明の化合物は、ビヒクルと比較して、平均結合を、約1%〜約100%、好ましくは約10%〜約100%、より好ましくは約20%〜約90%阻害する。平均結合の阻害は、好ましくは、用量依存性である。
【0211】
B.モノアミン輸送体活性の阻害
[0189] 種々の実施形態では、本発明は、セロトニン輸送体、ドーパミン輸送体およびノルエピネフリン輸送体等、少なくとも1つのモノアミン輸送体の活性を調節する(例えば、阻害する、増大させる)方法を提供する。この方法は、モノアミン輸送体と、本発明の化合物とを接触させることを含む。例示的な実施形態では、治療有効量の本発明の化合物またはその薬学的に許容できる塩もしくは溶媒和化合物を対象に投与することによって、モノアミン輸送体を本発明の化合物と接触させる。対象はヒトであってよい。例示的な実施形態では、モノアミン輸送体は、ドーパミン輸送体(DAT)、セロトニン輸送体(SERT)またはノルエピネフリン輸送体(NET)である。種々の例示的な実施形態では、本発明の化合物は、少なくとも2つの異なるモノアミン輸送体の活性を阻害する。モノアミン輸送体活性の阻害は、当技術分野で既知のアッセイを使用して測定することができる。例示的なアッセイ形式として、in vitroにおける機能的取り込みアッセイが挙げられる。例示的な実施形態では、機能的取り込みアッセイは、所望のモノアミン輸送体を発現する適切な細胞系を活用する。種々の例示的な実施形態では、機能的取り込みアッセイは、適切な生物の脳組織から単離したシナプトソームを活用する。あるいは、モノアミン輸送体活性の阻害は、当技術分野で既知の、例えば、適切な膜の調製物を活用する受容体結合実験を使用して評価することができる。例示的なアッセイでは、本明細書に記載するように、試験対象(例えば、ラット)を、本発明の化合物、および基準化合物を用いて処理し、それに続いて、脳組織を単離し、受容体の占有率をex vivoにおいて解析する。
【0212】
C.モノアミン取り込みの阻害
[0190] 種々の態様では、本発明は、少なくとも1つのモノアミン(例えば、ドーパミン、セロトニン、ノルエピネフリン)の細胞による取り込みを阻害する方法を提供する。この方法は、細胞を本発明の化合物と接触させることを含む。例示的な実施形態では、細胞は、神経細胞またはグリア細胞等の脳細胞である。1つの例では、モノアミン取り込みの阻害をin vivoにおいて起こす。生物中では、ドーパミンまたはセロトニン等のモノアミンの、例えば、シナプス間隙からの神経細胞による取り込み(また、再取り込みとも呼ばれる)が起きる。したがって、1つの実施形態では、神経細胞を、哺乳動物のシナプス間隙と接触させる。別の例示的な実施形態では、モノアミン取り込みの阻害を、in vitroにおいて起こす。これらの方法では、細胞は、脳細胞、すなわち、組換えのモノアミン輸送体を発現する神経細胞または細胞型等であってよい。
【0213】
[0191] 1つの実施形態では、化合物は、少なくとも2つの異なるモノアミンの取り込みを阻害する。これは、例えば、複数の異なるモノアミン輸送体を同時に発現する細胞型(単離したシナプトソーム等)を活用する種々のin vitroにおける機能的取り込みアッセイを実施することによって示すこともでき、またはそれぞれが異なるモノアミン輸送体、すなわち、組換えのドーパミン輸送体等を発現する、2つの異なる細胞型を、適切な、標識したモノアミンと一緒に使用することによって示すこともできる。以下の本明細書に記載するもの等の機能的モノアミン取り込みアッセイにおいて、阻害薬(例えば、本発明の化合物)が、約0.1nM〜約10μM、好ましくは約1nM〜約1μM、より好ましくは約1nM〜約500nM、さらにより好ましくは約1nM〜約100nMのIC50を示す場合に、モノアミン取り込みの阻害が実証される。
【0214】
D.神経障害の治療
[0192] 別の態様では、本発明は、神経障害を、少なくとも1つのモノアミン輸送体の活性を阻害することによって治療する方法を提供する。この方法は、治療有効量の本発明の組成物、あるいは化合物またはその薬学的に許容できる塩もしくは溶媒和化合物を、その治療を必要とする対象に投与することを含む。例示的な実施形態では、哺乳動物の対象はヒトである。別の例示的な実施形態では、本発明の化合物は、少なくとも2つの異なるモノアミン輸送体の活性を阻害する。例えば、本発明の化合物は、セロトニン輸送体、ドーパミン輸送体およびノルエピネフリン輸送体のうちの少なくとも2つの活性を阻害する。モノアミン輸送体の活性の阻害は、以下の本明細書に記載するように、機能的モノアミン取り込みアッセイによって示すことができる。
【0215】
[0193] 化合物の活性を、種々の当技術分野で認識されている動物モデルにおいて実証することができる。例えば、本発明の化合物の抗うつ薬活性は、うつ病の適切な動物モデル、すなわち、ラットの強制水泳試験、マウスの尾の懸垂試験、およびラットの自発運動解析等を活用することによって示すことができる。ラットの強制水泳試験はまた、2つ以上のモノアミン輸送体に対して活性(混合性モノアミン輸送体活性)を有する化合物の解析にも適している。例えば、泳ぐ活動の増加は、セロトニン再取り込みの阻害を示し、一方、登る活動の増加は、ノルエピネフリン再取り込みの阻害を示す。
【0216】
[0194] 種々の実施形態では、本発明の化合物は、少なくとも1つの動物モデルにおいて活性を示し、これを使用して、化合物の活性を測定し、神経障害を治療する場合におけるそれらの効能を推定することができる。例えば、動物モデルが、うつ病(例えば、無動)についてである場合、本発明の化合物は、少なくとも1つの動物モデルにおいて、平均的な無動(immobility)を阻害する場合には、ビヒクルと比較して、約5%〜約90%、好ましくは約10%〜約70%、より好ましくは約10%〜約50%、より好ましくは約15%〜約50%活性である。種々の実施形態では、本発明の化合物は、処理動物とビヒクル投与動物との間において、測定したエンドポイントの類似した差異をもたらす。
【0217】
[0195] 種々の実施形態では、本発明は、抗うつ薬様の効果をもたらす方法を提供する。この方法は、治療有効量の本発明の化合物または組成物またはその薬学的に許容できる塩もしくは溶媒和化合物を、それを必要とする哺乳動物の対象に投与することを含む。抗うつ薬様の効果は、本明細書に記載するもの等の疾患の動物モデルを使用して測定することができる。
【0218】
[0196] 種々の例示的な実施形態では、神経障害は、うつ病(例えば、大うつ病性障害、双極性障害、単極性障害、気分変調症および季節性感情障害)、失認、線維筋痛症、疼痛(例えば、神経因性疼痛)、精神医学的状態によって生じる睡眠障害を含む睡眠関連障害(例えば、睡眠時無呼吸、不眠症、ナルコレプシー、カタプレキシー)、慢性疲労症候群、注意欠陥障害(ADD)、注意欠陥多動性障害(ADHD)、下肢静止不能症候群、統合失調症、不安症(例えば、全般性不安障害、社会不安障害、パニック障害)、強迫性障害、心的外傷後ストレス障害、季節性感情障害(SAD)、月経前不安、閉経後の血管運動性症状(例えば、一過性熱感、寝汗)、ならびに神経変性疾患(例えば、パーキンソン病、アルツハイマー病および筋萎縮性側索硬化症)、躁状態、気分変調性障害、気分循環性障害、肥満、ならびに物質乱用または依存(例えば、コカイン耽溺、ニコチン耽溺)からなる群から選択されるメンバーである。例示的な実施形態では、神経障害は、大うつ病性障害等のうつ病である。例示的な実施形態では、本発明の化合物は、失認とうつ病等、併存症である2つの状態/障害を治療するために有用である。
【0219】
[0197] 神経障害は、老人性認知症、アルツハイマー型認知症、認知、記憶の喪失、健忘症/健忘症候群、てんかん、意識の撹乱、昏睡、注意力の低下、会話障害、レノックス症候群、自閉症および多動症候群を含む脳機能の障害を非限定的に含む。
【0220】
[0198] 神経因性疼痛は、ヘルペス後(帯状疱疹後)神経痛、反射性交換神経性ジストロフィー/灼熱痛または神経の外傷、幻肢痛、手根管症候群および末梢性ニューロパシー(糖尿病性ニューロパシーまたは慢性のアルコールの使用によって生じるニューロパシー等)を非限定的に含む。
【0221】
[0199] 本発明の方法を使用して治療することができるその他の例示的な疾患および状態として、肥満;偏頭痛または偏頭痛性の頭痛;不随意性の排尿、尿の滴下または漏れ、腹圧性尿失禁(SUI)、切迫性尿失禁、労作性尿失禁、反射性失禁、受動性失禁および溢流性失禁を非限定的に含めた、尿失禁;さらに、心理的および/または生理的な要因が引き起こす性機能障害、勃起不全、早漏、膣の乾燥、性的興奮の欠如、オルガスム達成喪失を非限定的に含めた、男性または女性における性機能障害、ならびに抑制された性欲、抑制された性的興奮、抑制された女性のオルガスム、抑制された男性のオルガスム、機能性性交疼痛症、機能性膣痙および非定型性の精神性的機能障害を非限定的に含めた、精神性的機能障害が挙げられる。
【0222】
[0200] 例示的な実施形態では、神経障害は肥満であり、患者に供給する化合物の治療有効量は、前記患者が満腹感を抱くのに十分な量である。
【0223】
[0201] 例示的な実施形態では、本明細書に記載する化合物は、前記化合物に対する耽溺を引き起こすことなく、中枢神経の障害を治療/予防する。
【0224】
[0202] 以下の実施例を、本発明の例示的な特徴を例証するために提供する。
【実施例】
【0225】
[0203] 以下の実施例は、選択した本発明の実施形態を例証するために提供されており、これらを、本発明の範囲を限定するものであると解釈してはならない。
【0226】
(実施例1)
1a.手順
[0204] 以下の実施例中、別段に特筆しない限り、以下の一般的な実験手順を使用した。全ての市販の試薬は、さらなる精製なしで使用した。無水反応を、火炎乾燥したガラス器具中、N2下で実施した。NMRスペクトルを、Varian製400MHz分光計上、トリメチルシラン(TMS)を内部標準として有する重クロロホルムまたはメタノール−d4中で記録した。ISCO Combiflashシステムを使用し、254nmにおいて検出するか、またはISCOの順相シリカゲルカートリッジを使用して、シリカゲルカラムクロマトグラフィーを実施した。
【0227】
1b.解析HPLC
[0205] 解析HPLCを、Agilent製Zorbax RX−C18 5μm、4.6×250mmカラムに接続したHewlett Packard Series 1100ポンプ上で実施し、Hewlett Packard Series 1100 UV/Vis検出器上で、214nmおよび254nmでモニターして検出した。典型的な流速=1ml/分。3本の異なるHPLCカラム、および種々の溶出プロトコールを使用した。例えば、(1)直線的勾配で流す、Agilent製Zorbax RX−C18 5μm、4.6×250mmカラム。溶媒A=0.05%TFAを有するH2O、溶媒B=0.05%TFAを有するMeCN。時間0分=5%溶媒B、時間4分=40%溶媒B、時間8分=100%溶媒B、12分=5%溶媒B、20分=5%溶媒B;(2)3分で5→100%でB(アセトニトリル/0.1%ギ酸)および溶媒A(水/0.1%ギ酸)の勾配を流す、Phenomenex 3μ C18カラム;(3)5分で5→100%でBの勾配を流す(溶媒B(アセトニトリル/0.1%ギ酸)および溶媒A(水/0.1%ギ酸))、Phenomenex 5μ C18カラム。
【0228】
1c.逆相HPLCによる精製
[0206] 逆相HPLCによる精製を、Gilson製のシステム上で、Phenomenex 5μ C18(50×21.2mm)カラムを使用して実施した。標準的な分離方法は、溶媒A(水/0.1%ギ酸)中、10→100%B(アセトニトリル/0.1%ギ酸)の10分の勾配であった。典型的には、粗試料を、MeOH中に溶解させた。画分を、Genovac(低圧における遠心分離)によって濃縮した。
【0229】
1d.GC−MS
[0207] ガスクロマトグラフィーを、Hewlett Packard 6890 Series GC System上で、Hewlett Packard 5973 Series Mass Selective Detectorに連結したHP1カラム(30メートル、0.15μの膜の厚さ)を用いて実施した。以下の直線的温度勾配を使用した:100℃5分間、次いで、20℃/分で320℃まで。320℃で10分間保持。
【0230】
1e.LCMS
[0208] LCMSを、Micromass Platform LCに接続したAgilent 1100 Seriesシステム上で実施した。以下のカラムおよび勾配を使用した:カラム:Luna C18(2)、3μm粒子サイズ、30×2.0mmのカラム寸法。流速=0.5mL/分、溶媒A=95%H2O、5%MeOH中の0.1M NH4Ac、pH6.0、溶媒B=MeOH中の溶媒B:0.1M NH4Ac。6回注入する直線的勾配:時間0分=100%溶媒A、時間10分=100%溶媒B、時間12分=100%溶媒B、時間12分10秒=100%溶媒A、時間14分=100%溶媒A、時間14分20秒=100%溶媒A。
【0231】
1f.マイクロ波(μW)による再結晶
[0209] 粗塩(例えば、HCl塩)を、撹拌棒を有するマイクロ波用の槽に入れた。再結晶用溶媒を添加し、槽を、標的温度で所与の時間にわたり加熱した。反応器中で、槽を50℃まで冷却し、次いで、取り出し、RTまでゆっくり冷却した。典型的には、N,N−ジメチルアミンを、EtOAcまたはEtOAc:CH3CN(2:1)中で再結晶させた。典型的には、N−Meまたは一級アミンは、CH3CN中で再結晶した。
【0232】
(実施例2)
2a.実験手順および特徴付けのデータ
【0233】
【化25】
[0210] THF(20mL)中のケトンa(2.0g、13mmol)の溶液に、−78℃で、3,4−ジクロロフェニルマグネシウムブロミド(THF中、0.5M、38mL、19mmol)を添加した。反応混合液を−78℃で30分間撹拌してから、30分かけて0℃まで加温した。NH4Clの飽和溶液(30mL)を、反応混合液に添加して、反応をクエンチした。得られた生成物を、ジエチルエーテルを用いて抽出した(2×100mL)。組み合わせた抽出物を乾燥および濃縮した。残渣に対して、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン/Et3N=1:10:0.1)を行って、1と2とのラセミ混合物を得た(3.5g、90%)。このラセミ混合物を、キラルADカラム(溶離液として、ヘキサン/iPrOH/DEA=95/5/0.1)によって分離して、純粋な1(動きがより速い鏡像異性体)と2(動きがより遅い鏡像異性体)とを得た。
【0234】
2a1.1/2についてのデータ
[0211] 1H NMR(400MHz,CD3OD)δ7.73(d,J=1.6Hz,1H)、7.52(d,J=8.4Hz,1H)、7.47(dd,J=1.6,8.4Hz,1H)、3.01(dd,J=13.2,10.4Hz,IH)、2.76(s,3H)、2.65(s,3H)、2.57(dd,J=2.0,13.2Hz,1H)、2.28(m,2H)、1.9(m,2H)、1.70(m,2H)、1.6(m,2H);13C NMR(100MHz,CD3OD)δ148.86、132.32、130.32、127.49、125.08、74.11、60.22、45.03、41.22、40.29、25.71、24.64、21.16;ESI MS m/z 302.1、304.0。
【0235】
2b.シクロアルキルアミンの脱アルキル反応
【0236】
【化26】
[0212] トルエン中の2(0.8g、2.65mmol)の溶液に、DEAD(0.69g、0.63mL、3.96mmol)を添加した。反応混合液を100℃で4時間加熱してから、濃縮した。残渣をEtOH30mL中に溶解させ、NH4Clの飽和溶液(30mL)を添加した。反応混合液を50℃で6時間撹拌してから、濃縮した。NaOH溶液(2M、10mL)を、得られた混合液に添加し、生成物を、ジエチルエーテルを用いて抽出した(2×80mL)。組み合わせた抽出物を乾燥および濃縮した。残渣を、逆相カラムクロマトグラフィー(CH3CN/H2O=5/95〜95/5)により精製して、4を得た(0.32g、42%)。
【0237】
2b1.4についてのデータ
[0213] 1H NMR(400MHz,CD3OD)δ7.60(s,1H)、7.37(m,2H)、2.57(dd,J=2.0,12.4Hz,1H)、2.28(dd,J=2.8,12.4Hz,1H)、2.23(s,3H)、1.88(m,2H)、1.78(m,2H)、1.62(m,1H)、1.56(m,2H)、1.40(m,1H);13C NMR(100MHz,CD3OD)δ150.86、132.40、130.21、130.09、127.56、124.77、77.03、53.54、43.95、40.82、36.81、26.45、26.05、22.05;ESI MS m/z 288.1。
【0238】
2c.シクロヘキサノンからのシクロヘキシルアミンの合成
【0239】
【化27】
[0214] H2O(50mL)中のシクロヘキサノン(23.7g、25.0mL、0.242モル)の溶液に、HCHO(37%、37.5mL、0.46モル)およびK2CO3(0.52g、3.76mmol)を添加した。反応混合液を60℃で3時間撹拌した。次いで、生成物を、ジエチルエーテルを用いて抽出した(2×300mL)。組み合わせた抽出物を乾燥および濃縮した。残渣を、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン=l:7〜1:2)により精製して、kを得た(10.8g、35%)。
【0240】
[0215] THF(60mL)中のk(3.2g、25mmol)の溶液に、−20℃で、3,4−ジクロロフェニルマグネシウムブロミド溶液(0.5M、100mL、50mmol)を添加した。反応混合液を30分間撹拌してから、NH4Clの溶液(20mL)によりクエンチした。次いで、生成物を、ジエチルエーテルを用いて抽出した(2×100mL)。組み合わせた抽出物を乾燥および濃縮した。残渣を、シリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=l:7〜1:2)により精製して、mを得た(2.1g、31%)。
【0241】
[0216] THF(40mL)中のm(1.6g、5.8mmol)の溶液に、r.t.で、PPh3(1.8g、7.0mmol)、DEAD(1.2g、7.0mmol)およびジフェニルホスホラジデート(DPPA)(1.9g、7.0mmol)を添加した。得られた黄色の溶液を、一晩撹拌してから、濃縮した。残渣に対して、シリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)=1:10〜1:1を行って、所望の生成物nを得た(1.32g、74%)。
【0242】
[0217] THF(30mL)中のn(1.00g、3.34mmol)の溶液に、PPh3(1.75g、6.68mmol)を添加した。反応混合液を、24時間撹拌してから、H2O(10mLを添加した。得られた混合液を、さらに2日間撹拌してから、濃縮した。残渣に対して、逆相カラムクロマトグラフィー(CH3CN/H2O=5/95〜95/5)を行って、所望の生成物oを得た(0.75g、82%)。このラセミ混合物を、キラルADカラムにより、(エタノール/メタノール/へキサン/DEA=3/2/95/0.1)を用いて分離して、純粋な鏡像異性体8(動きがより速い鏡像異性体)と9(動きがより遅い鏡像異性体)とを得た。
【0243】
【化28】
【0244】
2c1. 8/9についてのデータ
[0218] 1H NMR(400MHz,CD3OD)δ7.73(広幅,1H)、7.40(d,J=8.4Hz,1H)、7.30(m,2H)、2.69(dd,J=2.0,13.2Hz,1H)、2.56(dd,J=2.8,13.2Hz,1H)、2.20(m,2H)、1.80(m,2H)、2.28(m,2H)、1.60(m,2H)、1.50(m,3H);13C NMR(100MHz,CD3OD)δ150.75、132.38、130.19、130.06、127.64、124.81、77.28、43.63、43.45、41.16、26.38、25.34、22.06;ESI MS m/z 274.1、276.0。
【0245】
[0219] 化合物9を、ホルムアルデヒドを用いる還元的アミノ化によって2に変換した。
【0246】
【化29】
【0247】
(実施例3)
3a.実験手順
【0248】
【化30】
[0220] これらの合成について活用した実験条件は、実施例1および2において利用したものと同様の実験条件であった。
【0249】
【化31】
【0250】
[0221] これらの合成について活用した実験条件は、実施例1および2において利用したものと同様の実験条件であった。
【0251】
(実施例4)
4a.in vitroにおけるヒト5−HT/NE/DAの再取り込み阻害のデータ
化合物を、セロトニン(5-HT)、ノルエピネフリン(NE)およびドーパミン(DA)の機能的取り込みの阻害について、ラットの全脳、視床下部もしくは線条体のそれぞれから調製したシナプトソームにおいて、および/または組換えヒト輸送体を使用して試験した。このアッセイの詳細は、参照により組み込まれているUS2007/0203111Alに記載されている。ヒト再取り込み輸送体についての機能的取り込みアッセイに関する結果を以下に示す。
【0252】
【表1】
【0253】
4b.in vitroにおけるPKデータ(ヒトにおける代謝安定性、CYP450酵素の阻害、HERG電流の阻害)
【0254】
【表2】
【0255】
4c.尾の懸垂試験、自発運動(locomotor activity)試験および強制水泳試験
4c1.マウスの尾の懸垂試験
[0222] 抗うつ薬活性を検出する、この方法は、Steruら(Psychopharmacology, 85: 367-370(1985))により記載された方法に従う。げっ歯類は、尾を懸垂することにより、素早く無動になる。抗うつ薬は、無動の持続時間を短縮する。
【0256】
[0223] 動物の行動を、Steruら(Prog. Neuropsychopharmacol. Exp. Psychiatry 11: 659-671 (1987))によって開発された装置に類似する、コンピュータ化した装置(Med-Associates Inc.製)を使用して、5分間自動的に記録した。10〜12匹のマウスを、各1群として試験した。典型的には、化合物を3種の用量(1〜30mg/kg)において、1回、すなわち、試験30〜60分前に経口投与して評価し、ビヒクルの対照群と比較した。デシプラミン(100mg/kg)を、同一の実験条件下で投与し、正の基準物質として使用した。
【0257】
[0224] データを、一元配置分散分析(ANOVA)、それに続いて、適切な場合には、事後比較によって解析した。p<0.05の場合、効果を有意であるとみなした。データを、平均および平均に対する標準誤差(s.e.m)として示す。
【0258】
4c2.自発運動
[0225] 化合物の無動時間に対する効果が、ベースラインの運動活性に対する一般的な刺激効果とは関連がないことを確認するために、自発運動を、光電セルによりモニターするケージ(Med-Associates Inc.製)を使用して評価した。各試験チャンバーは、動物の動きを測定するための赤外光電セルビームを装備していた。水平および垂直な活動を測定した。
【0259】
[0226] ラットまたはマウスを、ビヒクルまたは試験化合物を用いて前処理し、元々のケージ中に戻し、それに続いて、自発運動用ケージ中に個別に入れ、活動を1〜5分の間、最長60分の期間にわたりモニターした。
【0260】
[0227] データを、一元配置分散分析(ANOVA)、それに続いて、適切な場合には、事後比較によって解析した。p<0.05の場合、効果を有意であるとみなした。データを、平均および平均に対する標準誤差(s.e.m)として示す。
【0261】
4c3.結果の概要
[0228] 本発明の化合物の効果を、マウスの尾の懸垂試験および自発運動試験において評価した。結果から、試験した全ての化合物が、抗うつ薬様のプロファイル(すなわち、無動時間の有意な短縮)を呈し、3〜30mg/kg、POの範囲のMEDを示すことが示された。尾の懸垂試験において活性を示す用量においては、ベースラインの運動活性は変化または低下しないことが観察され、このことから、抗うつ薬様活性は一般的な刺激効果には起因しないことが示された。
【0262】
[0229] また、本発明の化合物の効果を、ラットの強制水泳試験および自発運動試験においても評価した。全ての化合物が、抗うつ薬様の効果を呈し、10〜30mg/kg、POの範囲のMEDを示した。これらの化合物によって生じた無動の低下は、泳ぐ行動および登る行動の増加に起因するように思われ、このことから、混合性の輸送体活性(すなわち、SNRIプロファイル)が示された。また、マウスの尾の懸垂の結果と同様に、ラットの強制水泳試験も、この化合物について抗うつ薬様の活性を示した。
【0263】
【表3】
【0264】
[0230] TST後、各処理群からの4匹の代表的なマウスから、脳および血漿の試料を採取し、これらの組織における2および4対する暴露レベルを解析した。この試験において、2は、無動の用量依存性の低下を呈した(上記を参照されたい)。2の経口投与に続いて、顕著なレベルの4つの代謝産物が血漿および脳のレベルにおいて見出された。
【0265】
4c4.ラットの強制水泳試験
[0231] 抗うつ薬活性を検出する、この方法は、Porsoltら(Eur. J. Pharmacol., 47: 379-391(1978))によって記載され、Luckiら(Psychopharm, 121: 66-72(1995))によって改変された方法に従った。素早く脱出することができない状況において泳ぐことを強制されたラットは、無動になる。抗うつ薬は、無動の持続時間を短縮する。さらに、この試験においては、活性を示す行動の明確に異なるパターンが、ノルエピネフリン(NE)およびセロトニン(5-HT)の取り込みを選択的に阻害する抗うつ薬によって生じる。選択的NE再取り込み阻害薬は、登る行動を増加させることによって、無動を低下させ、一方、選択的5−HT再取り込み阻害薬は、泳ぐ行動を増加させることによって、無動を低下させる。
【0266】
[0232] 実験の第1日に(セッション1)に、ラットを個別に、22cmの水(25℃)を含有する円筒(高さ=40cm;直径=20cm)の中に、15分間入れ、次いで、24時間後に、水の中に、5分の試験の間(セッション2)戻した。これらのセッションを、テープに録画し、5分の試験の間、無動の持続時間、ならびに泳ぐ行動および登る行動を測定した。12匹のラットを、各1群として試験した。試験は、盲検で実施した。典型的には、化合物を、3種の用量(1〜30mg/kg)において、2回、すなわち、試験(セッション2)の24時間前および30〜60分前に経口投与して評価し、ビヒクルの対照群と比較した。デシプラミン(20mg/kg i.p.)を、同一の実験条件下で投与し、正の基準物質として使用した。
【0267】
[0233] データを、一元配置分散分析(ANOVA)、それに続いて、適切な場合には、事後比較によって解析した。p<0.05の場合、効果を有意であるとみなす。データを、平均および平均に対する標準誤差(s.e.m)として示す。
【0268】
【表4】
【0269】
(実施例5)
5a.ex vivoにおける結合アッセイ
[0234] 末梢からの化合物の投与後の、中枢における、ノルアドレナリン(NA)輸送体部位、5−HT輸送体部位およびドーパミン(DA)輸送体部位の受容体占有率を、それぞれ、[3H]ニソキセチン、[3H]シタロプラムおよび[3H]WIN35428の結合を使用して決定した。液体シンチレーション測定を使用して、放射活性を定量化した。
【0270】
[0235] C57BL/6マウス(25〜30g)に、ビヒクルまたは化合物のいずれかを、4種の用量レベルで経口投与した。処理60分後に、マウスを堵殺した。全脳を取り出し、皮質および線状体を解体して取り出してから、ドライアイス上で凍結した。脳組織は、アッセイの当日まで−20℃で保管した。各半球からの皮質を個別に凍結した。一方を、NA輸送体部位の占有率を決定するために使用し、他方を、5−HT輸送体部位の占有率を決定するために使用した。線状体は、DA輸送体部位の占有率を決定するために使用した。
【0271】
[0236] 各半球からの前頭皮質、または線状体を個別に、密閉可能なガラス製/テフロン(登録商標)製のホモジナイザーを使用して、氷冷したアッセイ用緩衝液中でホモジナイズし、直ちに、結合アッセイにおいて使用した。
【0272】
5b.マウス脳における、[3H]シタロプラムの、5−HT輸送体(SERT)部位に対する結合
[0237] 皮質膜(400μL;組織の1.25mg湿潤重量/チューブに相当する)を、1.3nMの単一の濃度の[3H]シタロプラム50μl、および緩衝液50μl(全結合)またはパロキセチン50μl(0.5μM;非特異的結合)のいずれかと共に、27℃で1時間インキュベートした。各動物について、3本のチューブを使用して全結合を決定し、3本のチューブを使用して非特異的結合を決定した。
【0273】
5c.マウス脳における、[3H]ニソキセチンの、ノルエピネフリン輸送体(NET)部位に対する結合
[0238] 皮質膜(400μL;組織の6.0mg湿潤重量/チューブに相当する)を、0.6nMの単一の濃度の[3H]ニソキセチン50μL、および緩衝液50μL(全結合)またはマジンドール50μL(1μM;非特異的結合)のいずれかと共に、4℃で4時間インキュベートした。各動物について、3本のチューブを使用して全結合を決定し、3本のチューブを使用して非特異的結合を決定した。
【0274】
5d.マウス脳における、[3H]WIN35428の、DA輸送体(DAT)部位に対する結合
[0239] 線状体膜(200μL;組織の2mg湿潤重量/チューブに相当する)を、24nMの単一の濃度の[3H]WIN35428 25μL、および緩衝液25μL(全結合)またはGBR12935 25μL(1μM;非特異的結合)のいずれかと共に、4℃で2時間インキュベートした。各動物について、2本のチューブを使用して全結合を決定し、2本のチューブを使用して非特異的結合を決定した。
【0275】
[0240] 膜に結合している放射活性を、Skatron製細胞ハーベスターを使用して、真空下で、0.5%PEI中にあらかじめ浸漬したSkatron11731フィルターを通してろ過することによって回収した。フィルターを、氷冷したリン酸緩衝液を用いて素早く洗浄し、放射活性(dpm)を、液体シンチレーション測定(1mL Packard MV Goldシンチレーター)によって決定した。
【0276】
5e.データ解析
[0241] 特異的結合の値(dpm)を、各動物について、平均全結合(dpm)から、平均非特異的結合(dpm)を引くことによって得た。データは、平均特異的結合(dpm)、およびビヒクル処理対照を100%とみなし、そのパーセントとして示す。
【0277】
5f.結果の概要
[0242] 2についてのex vivoにおけるSERT、NETおよびDATの結合/受容体占有率のデータを得た。
【0278】
【表5】
【0279】
(実施例6)
【0280】
【表6】
【0281】
(実施例7)
7a.レセルピンのラットモデル
[0243] レセルピン処理ラットパーキンソン病モデルにおいて、化合物4の単独でのまたはL−DOPAと組み合わせた効果を評価した。抗パーキンソ病活性(運動の欠損およびアキネジアの逆転)を検出する、この方法は、Johnstonら(Exp Neurol, 191, 243-250, 2005)によって記載されている方法に従う。行動試験の18時間前に、ラットにイソフルランを用いて軽く麻酔を施し、レセルピン(3mg/kg、sc)を、脱水を予防するための食塩水(50ml/kg)と併せて注射した。
【0282】
7b.行動の評価
[0244] 加速ロータロッド:加速ロータロッド上における動きを、4段階のラット用ロータロッド(MedAssociates製、米国)を使用して評価した。ロータロッドの回転スピードを、3.5〜35rpmに5分かけて増加させ、動物がロッド上に留まる時間を、3回のトライアルの平均として決定した。
【0283】
[0245] カタレプシー試験:カタレプシーを、ラットの前肢をベンチ表面から6cm上に懸垂させた水平な木製のロッドの上部の上に置くことによって評価した。ロッドから両方の肢を離すまでに要する時間を、最長120秒まで記録した。各動物につき3回のトライアルを実施した。
【0284】
[0246] オープンフィールド:オープンフィールド領域における活動を、自動化活動モニター(Linton Instrumentation製、英国)を使用して評価した。ラットを、活動用の箱の中に入れ、自発運動を、240分の期間にわたり記録した。
【0285】
[0247] 薬物投与:単独療法実験の場合、5つの異なる治療の効果を評価した:1)ビヒクル(無菌水、PO)、2)化合物4、3mg/kg(PO)、3)化合物4、10mg/kg(PO)、4)化合物4、30mg/kg、および5)正の基準物質であるL−DOPA(80mg/kg)、80mg/kg(IP)。併用実験の場合、5つの異なる治療を評価した:1)化合物4ビヒクル(PO)+L−DOPAビヒクル(IP)、2)化合物4(10mg/kg、PO)+L−DOPAビヒクル(IP)、3)化合物4ビヒクル(PO)+LDOPA(30mg/kg、IP)、4)化合物4(10mg/kg、PO)+L−DOPA(30mg/kg、IP)、および5)化合物4ビヒクル(PO)+L−DOPA(80mg/kg、IP)。治療を無作為化して施し、各動物に、全ての治療条件を与えた。化合物4は、行動の評価60分前に投与し、L−DOPAは、行動試験の直前に投与した。
【0286】
[0248] 結果から、多様な行動試験において、化合物4(3〜30mg/kg、PO)単独によって、動きが用量依存性に改善されることが示された(ベースライン自発運動、図2、ロータロッド、図3、およびカタレプシー、図4)。行動試験に依存して、化合物4の効果は、L−DOPA(80mg/kg)を用いた場合に観察された効果と類似する場合またはそれより弱い場合を示した。併用実験では、化合物4(10mg/kg、PO)と低い用量のL−DOPA(30mg/kg)との組合せが、程度の点では等しく、かつより高い用量のL−DOPAの投与(80mg/kg)によりもたらされた効果より持続時間の長い効果を示した、図5。
【0287】
[0249] データから、化合物4は単独療法としては、何らかの抗パーキンソン病作用を有し得るが、効果はL−DOPAほど強力ではないことが示唆されている。化合物4と低い用量のL−DOPAとの併用実験からは、程度の点では等しく、かつより高い用量のL−DOPAによりもたらされた作用より長い持続時間の抗パーキンソン病作用が示された。したがって、化合物4は、「L−DOPA節約性」と記載することができるであろう。
【0288】
(実施例8)
8a.ラット片側性6−ヒドロキシドーパミン(6-OHDA)病変モデル
[0250] げっ歯類6−OHDA病変ラットパーキンソン病モデルにおいて、化合物4の単独でのまたはL−DOPAと組み合わせた効果を評価した。抗パーキンソ病活性(運動の欠損およびアキネジアの逆転)を検出する、この方法は、Henryら(Exp Neurol, 151(2): 334-42, 1998)によって記載されている方法に従う。
【0289】
[0251] 動物の準備:手術前に、ラットにパルギリン(5mg/kg、ip)およびデシプラミン(25mg/kg ip)を投与して、ドーパミン作動性神経細胞に対して、それに続く6−OHDAの利用能を最適化し、毒性についての特異性を増加させた。次いで、ラットにイソフルランを用いて麻酔を施し、ラットを定位固定枠中に入れた。十字縫合を暴露させた後、(PaxinosおよびWatson, 1986の図譜に従って)頭蓋骨中、右内側前脳束上、十字縫合に対して2.8mm後側および2mm外側の座標において、頭蓋穿孔をドリルで開けた。次いで、28GのHamilton製の針を、頭蓋骨の9mm下まで下ろした。次いで、6−OHDA(2.5μl中の12.5μg)を注射した(1μl/分)。次いで、針を現位置に4分間維持して、確実に、溶液を完全に吸収させた。注射針をゆっくり取り出した後、創傷を閉鎖し、動物に、食塩水(50ml/kg、sc)、鎮痛薬(ケトプロフェン、0.5mg/kg)および広域スペクトルの抗生物質(エンロフロキサシン、75mg/kg)を投与した。手術後、動物を未処置の状態で3週間維持し、病変を発展および安定化させてから、行動の評価を開始した。
【0290】
8b.行動の評価
[0252] 肢の設置試験:肢の設置試験によって、感覚刺激に応答したそれぞれの前肢の正確な設置を評価する。ラットの胴体を軽く保持し、一方の前肢は、親指と人差し指との間で拘束し、他方の肢は、保持せず自由にさせた。次いで、ラットを、テーブルの縁部に対して平行に保持し、自由な前肢は、当該縁部に隣接させた状態にした。次いで、動物をテーブルに向かって移動させ、感覚毛をテーブルの縁部に対して軽く接触させて、自由な肢からの前肢設置応答を惹起した。全部で10回のトライアルを、素早く連続して実施してから、他方の肢について、この手順を繰り返した。試験を、6−OHDA病変の側とは反対側の肢の良好な設置応答のパーセントとして定量化した。病変の側と同側の肢の設置は、全ての動物において100%の事例で良好であった。
【0291】
[0253] 薬物投与:単独療法実験の場合、5つの異なる治療の効果を評価した:1)ビヒクル(無菌水、PO)、2)化合物4、3mg/kg(PO)、3)化合物4、10mg/kg(PO)、4)化合物4、30mg/kg、および5)正の基準物質であるL−DOPA、6.5mg/kg(IP)。併用実験の場合、5つの異なる治療を評価した:1)化合物4ビヒクル(PO)+L−DOPAビヒクル(IP)、2)化合物4(10mg/kg、PO)+L−DOPAビヒクル(IP)、3)化合物4ビヒクル(PO)+L−DOPA(2mg/kg、IP)、4)化合物4(10mg/kg、PO)+L−DOPA(2mg/kg、IP)、および5)化合物4ビヒクル(PO)+L−DOPA(6.5mg/kg、IP)。治療を無作為化して施し、各動物に、それぞれの治療条件を与えた。化合物4は、行動の評価60分前に投与し、L−DOPAは、行動試験の直前に投与した。
【0292】
[0254] 結果から、化合物4(3〜30mg/kg、PO)は単独では、肢設置タスクにおける動きの改善をほとんどまたは全くもたらさないことが示された。化合物4(10mg/kg、PO)と低い用量のL−DOPAとの併用により、肢設置の動きが有意に向上し、このことから、化合物4とL−DOPAとの作用の間には、相乗効果があるという何らかの証拠を得た(図6)。
【0293】
[0255] 本発明は、実施例において開示した特定の実施形態によって、その範囲が限定されるわけではない。これらの実施例は、本発明のいくつかの態様を例証するためのものであり、機能的に同等である実施形態はいずれも、本発明の範囲に属する。まさに、本明細書において示し、記載したものに加えて、本発明の種々の改変形態が当業者には明らかになるであるろうし、それらは、添付の特許請求の範囲に属するものとする。
【0294】
[0256] 全ての特許、特許出願および本出願に引用したその他の刊行物は、全ての目的のために、それらの全体が参照により本明細書に組み込まれている。
【技術分野】
【0001】
関連出願の相互参照
[0001] 本出願は、全ての目的のためにその全体が参照により本明細書に組み込まれている2007年5月31日出願の米国仮特許出願第60/941,242号についての優先権を主張する。
【0002】
[0002] 本発明は、神経障害の治療のための化合物および組成物に関する。
【背景技術】
【0003】
[0003] 精神障害は、認知、情動、気分または情緒の異常となる同定可能な症状によって特徴付けられる脳の病理学的な状態である。これらの障害は、症状の重症度、持続時間および機能障害の点で多様であり得る。精神障害は、世界全体で数百万人の人々を苦しめ、その結果、多大な人的苦痛をもたらし、生産性の損失および依存的ケアにより経済的負担を生じる。
【0004】
[0004] 過去数十年にわたり、精神障害を治療するための薬理学作用のある物質の使用が、主として神経科学および分子生物学の両方における研究の進展により大幅に増加している。さらに、より少ない副作用を有するより有効な治療剤であり、精神的な障害を伴う生化学的な変化を矯正することを目指す化合物を創出する化学者の技術レベルも一層高まっている。
【0005】
[0005] しかし、多くの進展がもたらされているにもかかわらず、多くの精神疾患は、治療されず放置されているか、または現在の医薬品を用いて不適切に治療されている状態にある。さらに、現在の薬剤の多くは、精神疾患には関与しない分子標的とも相互作用する。この無差別な結合の結果、療法の総合的な治療成果に大きな影響を及ぼす恐れがある副作用が生じる場合がある。場合によっては、副作用が非常に大きいので、療法を中断する必要がある。
【0006】
[0006] うつ病は、情動障害であり、その病原は、いずれの単一の原因および理論でも説明することができない。うつ病は、持続性の気分の落ち込みまたは周囲に対する関心の低下によって特徴付けられ、少なくとも1つの以下の症状を伴う:エネルギーおよび意欲の低下、集中困難、睡眠および食欲の変化、ならびに時には自殺念慮(American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders, ed. 4. Washington, American Psychiatric Association, 1994)。大うつ病には、高い罹患率および死亡率が伴い、自殺率は、10〜25%である(Kaplan H I, Sadock B J (編): Synopsis of Psychiatry. Baltimore, Williams & Wilkins, 1998, p. 866)。また、二重再取り込み阻害薬(dual reuptake inhibitor)を使用して、うつ病に通常伴う疲労を低下させる場合もある(例えば、"Bupropion augmentation in the treatment of chronic fatigue syndrome with coexistent major depression episode" Schonfeldt-Lecuonaら, Pharmacopsychiatry 39(4):152-4, 2006;"Dysthymia: clinical picture, extent of overlap with chronic fatigue syndrome, neuropharmacological considerations, and new therapeutic vistas" Brunelloら, J. Affect. Disord. 52(1-3):275-90, 1999;"Chronic fatigue syndrome and seasonal affective disorder: comorbidity, diagnostic overlap, and implications for treatment" Termanら, Am. J. Med. 105(3A):115S-124S, 1998を参照されたい)。
【0007】
[0007] うつ病は、ノルアドレナリン作動性またはセロトニン作動性の系における機能不全、より具体的には、機能的に重要なアドレナリン作動性またはセロトニン作動性の受容体における特定の神経伝達物質(NT)の欠乏の結果生じると考えられている。
【0008】
[0008] 神経伝達物質は、特異的な受容体との相互作用の結果として効果を発揮する。ノルエピネフリン(NE)および/またはセロトニン(5−ヒドロキシトリプタミンまたは5-HT)を含めて、神経伝達物質は、脳の神経細胞において合成され、小胞中に保管される。神経インパルス時に、NTは、シナプス間隙中に放出され、そこで、神経伝達物質は、種々のシナプス後受容体と相互作用する。5−HTおよび/またはNEのシナプスのレベルの領域的な欠乏が、うつ病の病因、覚醒および注意力に関与すると考えられている。
【0009】
[0009] ノルエピネフリンは、覚醒状態、夢見および気分の調整に関与する。ノルエピネフリンはまた、血管を収縮させ、心拍数を増加させることによって、血圧の調整に寄与することもできる。
【0010】
[0010] セロトニン(5-HT)は、種々の障害の病因または治療に関係している。5−HTの最も広く研究されている効果は、CNSに対する効果である。5−HTの機能は多数あり、食欲、睡眠、記憶および学習の制御、体温調整、気分、(性行動および幻覚行動を含めた)行動、心血管機能、平滑筋収縮ならびに内分泌調整を含む。末梢では、5−HTは、血小板のホメオスタシスおよびGI管の運動性において主要な役割を担っているようである。5−HTの作用は、3つの主要な機構、すなわち、拡散;代謝;および再取り込みによって止まる。5−HTの作用を止める主要な機構は、シナプス前膜を介しての再取り込みによるものである。5−HTは、種々のシナプス後受容体に対して作用した後、特異的な膜輸送体が関与する取り込み機構によって、その他の生体アミンの取り込み機構に類似する様式で、シナプス間隙から除去され、神経末端に戻る。この取り込みを選択的に阻害する薬剤が、シナプス後受容体における5−HTの濃度を増加させ、種々の精神障害、特にうつ病を治療する場合に有用であることが見い出されている。
【0011】
[0011] 長年にわたって、うつ病の治療へのアプローチには、NEおよび5−HTのレベルを、それらの代謝の阻害(例えば、モノアミンオキシダーゼ阻害薬)または再取り込みの阻害(例えば、三環系抗うつ薬もしくは選択的セロトニン再取り込み阻害薬(SSRI))のいずれかによって増加させる薬剤の使用が関与している。
【0012】
[0012] 米国内で入手可能な、承認されている抗うつ薬が、20種以上ある。現在入手可能な、古典的な三環系抗うつ薬(TCA)は、主としてNEの取り込みを遮断し、かつまた、二級アミンまたは三級アミンのいずれであるかに依存して、様々な程度で5−HTの取り込みも遮断する。イミプラミンおよびアミトリプチリン等の三級アミンは、デシプラミン等の二級アミンと比較すると、カテコールアミンに対してよりも5−HTに対して選択性を示す取り込み阻害薬である。
【0013】
[0013] 選択的セロトニン再取り込み阻害薬は、有望な抗うつ薬として調べられてきた。フルオキセチン(PROZAC(登録商標))、セルトラリン(ZOLOFT(登録商標))およびパロキセチン(PAXIL(登録商標))が、米国で現在市販されているSSRIの3つの例である。これらの薬剤は、TCAを上回る効能を有するようではなく、一般に、作用出現までの時間がより短いわけでもない。しかし、これらは、副作用を生じにくいという利点は有する。これら3つのSSRIのうち、パロキセチンが、5−HTの取り込みの最も強力な阻害薬であり、フルオキセチンが最も弱い。セルトラリンが、NEと比して5−HTを最も選択的に取り込み、フルオキセチンの選択性が最も低い。フルオキセチンおよびセルトラリンは、活性な代謝産物をもたらし、一方、パロキセチンは、不活性な代謝産物に代謝される。SSRIは、一般に、セロトニンの取り込みに限って影響を及ぼし、ムスカリン性受容体、アドレナリン作動性受容体、ドーパミン受容体およびヒスタミン受容体を含む種々の受容体の系に対して親和性を示すことはほとんどまたは全くない。
【0014】
[0014] うつ病の治療に加え、SSRIについて、いくつかのその他の治療上の適用例が調査されている。それらには、アルツハイマー病、攻撃行動、月経前症候群、糖尿病性ニューロパシー、慢性疼痛、線維筋痛症およびアルコール乱用の治療がある。例えば、フルオキセチンは、強迫性障害(OCD)の治療について承認されている。特に意義があるのは、5−HTは、アンフェタミン様の薬物に伴う乱用傾向の行動的影響をもたらすことなく、食事が引き起こす満腹感を高め、空腹感を抑えることによって、食物の消費を減少させることが観察されている点である。したがって、肥満治療におけるSSRIの使用に対して関心が寄せられている。
【0015】
[0015] ベンラファキシン(EFFEXOR(登録商標))は、5−HTおよびNEの両方の取り込みの強力な阻害薬として作用することから、古典的なTCAおよびSSRIとは化学的および薬理学的に異なる二重再取り込み抗うつ薬である。ベンラファキシンもその主要な代謝産物も、アドレナリン作動性アルファ−1受容体に対しては、顕著な親和性を示さない。ベンラファキシンは、TCAの効能と同等な効能、およびSSRIの副作用プロファイルに類似する、穏和な副作用プロファイルを有する。
【0016】
[0016] ドーパミンは、精神病、およびパーキンソン病等の特定の神経変性疾患において主要な役割を担っていると仮定されている。これらにおいては、ドーパミン作動性の神経細胞の欠乏が、根底にある病態であると考えられている。ドーパミンは、動作、情動応答、ならびに喜びおよび疼痛を体験する能力を制御する脳のプロセスに影響を及ぼす。DAの調整は、我々の精神的および肉体的な健康における重大な役割を担っている。特定の薬物が、DAの再取り込みを阻止し、シナプス中により多くのDAを残すことによって、DAの濃度を増加させる。例として、小児の多動、および統合失調症の症状を治療するために治療上使用されるメチルフェニデート(RITALIN(登録商標))がある。ドーパミンの異常は、急性統合失調症患者において主として見られる注意異常のうちの一部の根底にあると考えられている。
【0017】
[0017] 治療の遅れが、これらの薬物の使用には伴う。臨床的に意味のある症状の軽減が実現するまでに、患者は薬物を少なくとも3週間は服用しなければならない。さらに、顕著な数の患者が、現在の療法に全く応答しない。例えば、現在のところ、うつ病と臨床的に診断された症例の30パーセント(30%)までが、現在の薬物療法の全ての形態に対して抵抗性であると推定されている。
【発明の概要】
【課題を解決するための手段】
【0018】
[0018] 本発明は、新規なシクロアルキルアミンおよびそれらの塩に関する。また、本発明は、新規な医薬組成物、ならびに障害および状態の治療におけるそれらの使用にも関する。本発明の化合物の例示的な適応症として、うつ病(例えば、大うつ病性障害、双極性障害)、線維筋痛症、疼痛(例えば、神経因性疼痛)、睡眠時無呼吸、注意欠陥障害(ADD)、注意欠陥多動性障害(ADHD)、下肢静止不能症候群、統合失調症、不安症、強迫性障害、心的外傷後ストレス障害、季節性感情障害(SAD)、月経前不安等の神経学的障害、および神経変性疾患(例えば、パーキンソン病、アルツハイマー病)が挙げられる。また、本発明の化合物は、肥満を治療もしくは予防するためにも、またはこれらに限定されないが、ニコチンおよびコカインの乱用、依存もしくは耽溺を含む物質乱用、依存もしくは耽溺を治療するためにも有用である。
【0019】
[0019] したがって、第1の態様では、本発明は、以下の式を有する化合物を提供する。
【0020】
【化1】
式中、指数nは、0〜2からなる群から選択される整数であり;sは、0〜2からなる群から選択される整数である。Aは、H、置換または非置換アルキル、ハロゲン、および置換または非置換ハロアルキルからなる群から選択されるメンバーである。Xは、H、ハロゲン、置換または非置換アルキル、置換または非置換アリール、置換または非置換ハロアルキル、およびOR5(ただし、R5は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、アシル、およびS(O)2R5a(ただし、R5aは、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルからなる群から選択されるメンバーである)からなる群から選択されるメンバーである)からなる群から選択されるメンバーである。
【0021】
[0020] YおよびZは、ハロゲン、CF3、CN、OR9、SR9、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、置換または非置換ヘテロシクロアルキル、NR10R11、およびNO2からなる群から独立して選択されるメンバーである。R9は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、または置換または非置換ヘテロシクロアルキルを示す。基であるR10およびR11は独立して、H、OR12、アシル、S(O)2R13、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、または置換または非置換ヘテロシクロアルキルを示す。R10とR11とは、それらが結合している窒素と一緒になって任意選択により結合して、R10とR11とが結合する窒素に加えて、1〜3個のヘテロ原子を任意選択により有する3員環〜7員環を形成する。
【0022】
[0021] 記号R12は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、または置換または非置換ヘテロシクロアルキルから選択されるメンバーである。R13は、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルから選択されるメンバーである。
【0023】
[0022] YとZとは、それらが結合している原子と一緒になって任意選択により結合して、5員環〜7員環を形成し、この環はその中に、1〜3個のヘテロ原子を任意選択により有する。当業者には明らかであろうが、YとZとが結合して環をなす場合、環内に組み込まれた原子上の置換基(例えば、R9、R10およびR11)が、これらの置換基が結合している原子の原子価を満たす必要に応じて、存在する(例えば、環の環状構造内に組み込まれる)場合、または存在しない場合があるであろう。
【0024】
[0023] R1およびR2は、H、ハロゲン、CN、CF3、OR6、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、または置換または非置換ヘテロシクロアルキルから独立して選択されるメンバーである。R6は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、または置換または非置換ヘテロシクロアルキルから選択されるメンバーである。
【0025】
[0024] R3およびR4は、H、OR7、アシル、S(O)2R8、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、または置換または非置換ヘテロシクロアルキルから独立して選択されるメンバーである。R7は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、または置換または非置換ヘテロシクロアルキルから選択されるメンバーである。R8は、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルから選択されるメンバーである。
【0026】
[0025] R1、R2、R3およびR4のうちの2つ以上が、それらが結合している原子と一緒になって任意選択により結合して、3員環〜7員環を形成し、この環は、1〜4、好ましくは1〜3個のヘテロ原子を任意選択により含む。
【0027】
[0026] 任意の薬学的に許容できる塩、溶媒和化合物、鏡像異性体、ジアステレオマー、ラセミ混合物、鏡像異性体に富む(enatiomerically enriched)混合物、および上記に記載した化合物の鏡像異性的に純粋な形態が、本発明の範囲内に属する。
【0028】
[0027] 第2の態様では、本発明は、本発明の化合物またはその薬学的に許容できる塩もしくは溶媒和化合物、および薬学的に許容できる担体を含む医薬組成物を提供する。
【0029】
[0028] 第3の態様では、本発明は、モノアミン輸送体リガンドの、セロトニン輸送体、ドーパミン輸送体およびノルエピネフリン輸送体等のモノアミン輸送体に対する結合を阻害する方法を提供する。この方法は、モノアミン輸送体と、本発明の化合物とを接触させることを含む。例示的な実施形態では、モノアミン輸送体リガンドは、セロトニン、ドーパミンおよびノルエピネフリン等のモノアミンである。
【0030】
[0029] 第4の態様では、本発明は、セロトニン輸送体、ドーパミン輸送体およびノルエピネフリン輸送体等、少なくとも1つのモノアミン輸送体の活性を阻害する方法を提供する。この方法は、モノアミン輸送体と本発明の化合物とを接触させることを含む。
【0031】
[0030] 別の態様では、本発明は、セロトニン、ドーパミンおよびノルエピネフリン等、少なくとも1つのモノアミンの細胞による取り込みを阻害する方法を提供する。この方法は、細胞を本発明の化合物と接触させることを含む。例示的な実施形態では、細胞は、神経細胞またはグリア細胞等の脳細胞である。
【0032】
[0031] さらに別の態様では、本発明は、うつ病を、少なくとも1つのモノアミン輸送体の活性を阻害することによって治療する方法を提供する。この方法は、本発明の化合物を哺乳動物の対象に投与することを含む。例示的な実施形態では、本発明の化合物は、少なくとも2つの異なるモノアミン輸送体の活性を阻害する。別の好ましい実施形態では、哺乳動物の対象はヒトである。
【0033】
[0032] さらなる態様では、本発明は、神経障害を治療する方法を提供する。この方法は、治療有効量の本発明の化合物またはその薬学的に許容できる塩もしくは溶媒和化合物を、その治療を必要とする対象に投与することを含む。例示的な実施形態では、対象はヒトである。
【図面の簡単な説明】
【0034】
【図1A】[0033]本発明の例示的な化合物の表である。
【図1B】本発明の例示的な化合物の表である。
【図1C】本発明の例示的な化合物の表である。
【図1D】本発明の例示的な化合物の表である。
【図1E】本発明の例示的な化合物の表である。
【図2】[0034]レセルピン投与ラットにおける、化合物4のベースライン自発運動に対する効果を示すグラフである。
【図3】[0035]レセルピン投与ラットにおける、化合物4のロータロッド(rotarod)における動きに対する効果を示すグラフである。
【図4】[0036]レセルピン投与ラットにおける、化合物4のカタレプシー(catalepsy)に対する効果を示すグラフである。
【図5】[0037]高い用量のL−DOPAと比較した、ロータロッドにおける動きに対する化合物4と低い用量のL−DOPAとの併用の効果を示すグラフである。
【図6】[0038]6−OHDA病変ラットにおける、L−DOPAと化合物4との併用効果を示すグラフである。
【発明を実施するための形態】
【0035】
I.定義
[0039] 「アルキル」という用語は、それ自体でまたは別の置換基の一部として、別段の言及がない限り、直鎖もしくは分枝鎖または環状の炭化水素基、あるいはそれらの組合せを意味し、これは、完全な飽和であっても、または不飽和、例えば、モノ不飽和もしくはポリ不飽和であってもよく、二価および多価の基を含むことができる。アルキル基は、言及される範囲内におけるいくつかの炭素を有するとして任意選択により指定される。すなわち、C1〜C10は、1〜10個の炭素を有する、置換または非置換アルキル部分を意味する。飽和炭化水素基の例として、これらに限定されないが、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、t−ブチル、イソブチル、sec−ブチル、例えば、n−ペンチル、n−ヘキシル、n−ヘプチル、n−オクチルの相同体および異性体;環状アルキル、例えば、シクロヘキシル、(シクロヘキシル)メチル、シクロプロピルメチル、例えば、縮合シクロアルキル(例えば、デカリン)等を含めた縮合環の種等の基が挙げられる。不飽和アルキル基は、1つまたは複数の二重結合または三重結合を有する基、例えば、「アルケニル」および「アルキニル」である。不飽和アルキル基の例として、これらに限定されないが、ビニル、2−プロペニル、クロチル、2−イソペンテニル、2−(ブタジエニル)、2,4−ペンタジエニル、3−(1,4−ペンタジエニル)、エチニル、1−および3−プロピニル、3−ブチニル、ならびに高級な相同体および異性体が挙げられる。「アルキル」という用語はまた、別段に特筆しない限り、以下でより詳細に定義するアルキル誘導体、すなわち、「ヘテロアルキル」等も含むことも意味する。炭化水素基に限定されるアルキル基は、「ホモアルキル」と呼ばれる。「置換アルキル」部分上に見い出される例示的な置換基を、以下に記載する。
【0036】
[0040] 「アルキレン」という用語は、それ自体でまたは別の置換基の一部として、アルカンから誘導された二価の基を意味し、例として、これに限定されないが、−CH2CH2CH2CH2−が挙げられ、「ヘテロアルキレン」として以下に記載する基もさらに含まれる。典型的には、アルキル(またはアルキレン)基は、1〜24個の炭素原子を有し、本発明においては、10個以下の炭素原子を有する基が好ましい。「低級アルキル」または「低級アルキレン」は、より短い鎖のアルキル基またはアルキレン基であり、一般に、8個以下の炭素原子を有する。
【0037】
[0041] 「アルコキシ」、「アルキルアミノ」および「アルキルチオ」(またはチオアルコキシ)という用語を、それらの従来の意味で使用し、分子の残部に、それぞれ酸素原子、アミノ基または硫黄原子を介して結合しているアルキル基を指す。
【0038】
[0042] 「ヘテロアルキル」という用語は、それ自体でまたは別の用語と組み合わせて、別段の言及がない限り、上記に記載した「アルキル」の亜属、すなわち、直鎖もしくは分枝鎖または環状のアルキル基、あるいはそれらの組合せ、飽和または不飽和のアルキル基であって、(任意選択により言及される)いくつかの炭素原子、ならびに好ましくはB、O、N、P、SiおよびSから選択される少なくとも1つのヘテロ原子からなり、窒素原子および硫黄原子は、任意選択により酸化されていてよく、ただし、窒素へテロ原子は、任意選択により四級化されていてもよいアルキル基を意味する。(1つまたは複数の)ヘテロ原子B、O、N、P、SiおよびSは、ヘテロアルキル基の任意の内部の位置にあっても、またはヘテロアルキル基が分子の残部に結合している位置にあっても、もしくはその正反対の末端にあってもよい。例として、これらに限定されないが、−CH2−CH2−O−CH3、−CH2−CH2−NH−CH3、−CH2−CH2−N(CH3)−CH3、−CH2−S−CH2−CH3、−CH2−CH2,−S(O)−CH3、−CH2−CH2−S(O)2−CH3、−CH=CH−O−CH3、−Si(CH3)3、−CH2−CH=N−OCH3および−CH=CH−N(CH3)−CH3が挙げられる。例えば、−CH2−NH−OCH3および−CH2−O−Si(CH3)3等、2個以上のヘテロ原子が連続し得る。同様に、「ヘテロアルキレン」という用語は、それ自体でまたは別の置換基の一部として、ヘテロアルキルから誘導された二価の基を意味し、例として、これらに限定されないが、−CH2−CH2−S−CH2−CH2−および−CH2−S−CH2−CH2−NH−CH2−が挙げられる。ヘテロアルキレン基については、ヘテロ原子はまた、鎖の末端の一方または両方を占めることもできる(例えば、アルキレンオキシ、アルキレンジオキシ、アルキレンアミノ、アルキレンジアミノ等)。その上さらに、アルキレン連結基およびヘテロアルキレン連結基については、連結基の式が記載されている方向によって、連結基の方向性が示されることにはならない。例えば、式−CO2R’−は、−C(O)OR’および−OC(O)R’の両方を任意選択により示す。
【0039】
[0043] 「シクロアルキル」および「ヘテロシクロアルキル」という用語は、それら自体でまたはその他の用語と組み合わせて、別段の言及がない限り、それぞれ「アルキル」および「ヘテロアルキル」の環状型を示す。さらに、ヘテロシクロアルキルについては、ヘテロ原子は、ヘテロ環が分子の残部に結合している位置を占めることができる。シクロアルキルの例として、これらに限定されないが、シクロペンチル、シクロヘキシル、1−シクロヘキセニル、3−シクロヘキセニル、シクロヘプチル等が挙げられる。ヘテロシクロアルキルの例として、これらに限定されないが、1−(1,2,5,6−テトラヒドロピリジル)、1−ピペリジニル、2−ピペリジニル、3−ピペリジニル、4−モルホリニル、3−モルホリニル、テトラヒドロフラン−2−イル、テトラヒドロフラン−3−イル、テトラヒドロチエン−2−イル、テトラヒドロチエン−3−イル、1−ピペラジニル、2−ピペラジニル等が挙げられる。
【0040】
[0044] 「ハロ」または「ハロゲン」という用語は、それら自体でまたは別の置換基の一部として、別段の言及がない限り、フッ素原子、塩素原子、臭素原子またはヨウ素原子を意味する。さらに、「ハロアルキル」等の用語は、モノハロアルキルおよびポリハロアルキルを含むことを意味する。例えば、用語「ハロ(C1〜C4)アルキル」という用語は、これらに限定されないが、トリフルオロメチル、2,2,2−トリフルオロエチル、4−クロロブチル、3−ブロモプロピル等を含むこと意味する。
【0041】
[0045] 「アリール」という用語 は、別段の言及がない限り、単環であっても、または一緒になって縮合しているか、もしくは共有結合によって連結している多環であってもよい(好ましくは、単環〜3員環)、ポリ不飽和、芳香族置換基を意味する。「アリール」の種は、シクロアルキル環、ヘテロシクロアルキル環およびヘテロアリール環と縮合しているアリール環を含む構造を含む。「ヘテロアリール」という用語は、「アリール」の亜属であり、好ましくはB、O、N、P、SiおよびSから選択される1〜4個のヘテロ原子を含有するアリール基を指し、ただし、窒素原子および硫黄原子は、任意選択により酸化されており、(1つまたは複数の)窒素原子は、任意選択により四級化されている。ヘテロアリール基は、ヘテロ原子を介して、分子の残部につながることができる。アリール基およびヘテロアリール基の非限定手的な例として、フェニル、1−ナフチル、2−ナフチル、4−ビフェニル、1−ピロリル、2−ピロリル、3−ピロリル、3−ピラゾリル、2−イミダゾリル、4−イミダゾリル、ピラジニル、2−オキサゾリル、4−オキサゾリル、2−フェニル−4−オキサゾリル、5−オキサゾリル、3−イソオキサゾリル、4−イソオキサゾリル、5−イソオキサゾリル、2−チアゾリル、4−チアゾリル、5−チアゾリル、2−フリル、3−フリル、2−チエニル、3−チエニル、2−ピリジル、3−ピリジル、4−ピリジル、2−ピリミジル、4−ピリミジル、5−ベンゾチアゾリル、プリニル、2−ベンゾイミダゾリル、5−インドリル、1−イソキノリル、5−イソキノリル、2−キノキサリニル、5−キノキサリニル、3−キノリルおよび6−キノリルが挙げられる。上記に特筆したアリール環系およびヘテロアリール環系のそれぞれについての置換基は、以下に記載する許容できる置換基の群から選択される。
【0042】
[0046] 簡潔にするために、「アリール」という用語は、置換基(例えば、アリールオキシ、アリールチオキシ、アリールアルキル)を定義するために使用する場合には、上記で定義したアリール環およびヘテロアリール環の両方を任意選択により含む。したがって、「アリールアルキル」という用語は、アリール基が、アルキル基に結合している基(例えば、ベンジル、フェネチル、ピリジルメチル等)を含むことを意味し、それらには、炭素原子(例えば、メチレン基)が、例えば、酸素原子によって置換アルキル基(例えば、フェノキシメチル、2−ピリジルオキシメチル、3−(1−ナフチルオキシ)プロピル等)が含まれる。
【0043】
[0047] 上記の用語(例えば、「アルキル」、「ヘテロアルキル」、「アリール」および「ヘテロアリール」)のそれぞれは、記述されている基の置換形態および非置換形態の両方を含むことを意味する。各タイプの基の好ましい置換基を以下に示す。
【0044】
[0048] アルキル基およびヘテロアルキル基(アルキレン、アルケニル、ヘテロアルキレン、ヘテロアルケニル、アルキニル、シクロアルキル、ヘテロシクロアルキル、シクロアルケニルおよびヘテロシクロアルケニルとしばしば呼ばれる基を含む)についての置換基は、「アルキル基置換基」と一般に呼ばれ、それらは、これらに限定されないが、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、置換または非置換ヘテロシクロアルキル、−OR’、=O、=NR’、=N−OR’、−NR’R’’、−SR’、−ハロゲン、−SiR’R’’R’’’、−OC(O)R’、−C(O)R’、−CO2R’、−CONR’R’’、−OC(O)NR’R’’、−NR’’C(O)R’、−NR’−C(O)NR’’R’’’、−NR’’C(O)2R’、−NR−C(NR’R’’R’’’)=NR’’’’、−NR−C(NR’R’’)=NR’’’、−S(O)R’、−S(O)2R’、−S(O)2NR’R’’、−NRSO2R’、−CNおよび−NO2から、好ましくはゼロ〜(2m’+1)(ただし、m'は、そのような基中の炭素原子の総数である)の範囲の数で選択される、多様な基のうちの1種または複数であってよい。R’、R’’、R’’’およびR’’’’はそれぞれ、好ましくは、独立して、水素、置換または非置換ヘテロアルキル、置換または非置換アリール、例えば、1〜3個のハロゲンで置換アリール、置換または非置換アルキル、アルコキシもしくはチオアルコキシの基またはアリールアルキル基を指す。本発明の化合物が2つ以上のR基を含む場合には、例えば、R基のそれぞれが独立して、R’基、R’’基、R’’’基およびR’’’’基のそれぞれであるとして選択され、この場合、これらの基のうちの2つ以上が存在する。R’およびR’’が、同一の窒素原子に結合している場合には、窒素原子と組み合わさって、5員環、6員環または7員環を形成することができる。例えば、−NR’R’’は、これらに限定されないが、1−ピロリジニルおよび4−モルホリニルを含むことを意味する。置換基の上記の論述から、当業者であれば、「アルキル」という用語は、水素基以外の基、すなわち、ハロアルキル(例えば、−CF3および−CH2CF3)ならびにアシル(例えば、−C(O)CH3、−C(O)CF3、−C(O)CH2OCH3等)等に結合している炭素原子を含む基を含むことを意味することを理解するであろう。
【0045】
[0049] アルキル基について記載した置換基と同様に、アリール基およびヘテロアリール基についての置換基は、「アリール基置換基」と一般に呼ばれる。こうした置換基は、例えば、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、置換または非置換ヘテロシクロアルキル、−OR’、=O、=NR’、=N−OR’、−NR’R’’、−SR’、−ハロゲン、−SiR’R’’R’’’、−OC(O)R’、−C(O)R’、−CO2R’、−CONR’R’’、−OC(O)NR’R’’、−NR’’C(O)R’、−NR’−C(O)NR’’R’’’、−NR’’C(O)2R’、−NR−C(NR’R’’R’’’)=NR’’’’、−NR−C(NR’R’’)=NR’’’、−S(O)R’、−S(O)2R’、−S(O)2NR’R’’、−NRSO2R’、−CNおよび−NO2、−R’、−N3、−CH(Ph)2、フルオロ(C1〜C4)アルコキシおよびフルオロ(C1〜C4)アルキルから、ゼロから芳香環系上の空いている原子価の総数までの範囲の数で選択され;R’、R’’、R’’’およびR’’’’は、好ましくは、独立して、水素、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、および置換または非置換ヘテロアリールから選択される。本発明の化合物が2つ以上のR基を含む場合には、例えば、R基のそれぞれが独立して、R’基、R’’基、R’’’基およびR’’’’基のそれぞれであるとして選択され、この場合、これらの基のうちの2つ以上が存在する。
【0046】
[0050] アリール環またはヘテロアリール環の隣接する原子上の置換基のうちの2つを、式−T−C(O)−(CRR’)q−U−(式中、TおよびUは独立して、−NR−、−O−、−CRR’−または一重結合であり、qは、0〜3の整数である)の置換基で任意選択により置換することができる。あるいは、アリール環またはヘテロアリール環の隣接する原子上の置換基のうちの2つを、式−A−(CH2)r−D−(式中、AおよびDは独立して、−CRR’−、−O−、−NR−、−S−、−S(O)−、−S(O)2−、−S(O)2NR’−または一重結合であり、rは、1〜4の整数である)の置換基で任意選択により置換することもできる。このようにして形成された新しい環の一重結合のうちの1つを、二重結合で任意選択により置換することができる。あるいは、アリール環またはヘテロアリール環の隣接する原子上の置換基のうちの2つを、式−(CRR’)s−X’’−(CR’’R’’’)d−(式中、sおよびdは独立して、0〜3の整数であり、X’’は、−O−、−NR’−、−S−、−S(O)−、−S(O)2−または−S(O)2NR’−である)の置換基で任意選択により置換することもできる。置換基R’、R’’、R’’’およびR’’’’は、好ましくは、独立して、水素、または置換または非置換(C1〜C6)アルキルから選択される。
【0047】
[0051] 本明細書で使用する場合、「アシル」という用語は、カルボニル残基C(O)Rを含有する置換基を記載する。Rについての例示的な種として、H、ハロゲン、置換または非置換アルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルが挙げられる。
【0048】
[0052] 本明細書で使用する場合、「縮合環系」という用語は、少なくとも2つの環であって、各環が、別の環と共有する少なくとも2個の原子を有する環を意味する。「縮合環系は、芳香環(すなわち、アリールまたはヘテロアリール)、および飽和または不飽和の非芳香環(すなわち、シクロアルキル、ヘテロシクロアルキル)を含むことができる。「縮合環系」の例には、ナフタレン、インドール、キノリン、クロメン、デカリン等がある。
【0049】
[0053] 本明細書で使用する場合、「ヘテロ原子」という用語は、酸素(O)、窒素(N)、硫黄(S)、ケイ素(Si)、臭素(B)およびリン(P)を含む。
【0050】
[0054] 「R」という記号は、「アルキル基置換基」または「アリール基置換基」(例えば、置換または非置換アルキル基、置換または非置換ヘテロアルキル基、置換または非置換アリール基、置換または非置換ヘテロアリール基、および置換または非置換ヘテロシクロアルキル基から選択される置換基)を示す一般的な略語である。
【0051】
[0055] 「治療有効量」という句は、本明細書で使用する場合、(例えば、哺乳動物のシナプス間隙からのモノアミンの取り込みを阻害し、それによって、治療する生物において、その経路の生物学的帰結を調節することによって)ある所望の治療効果をいずれの医学的治療にも適用できる妥当な利益/リスクの比でもたらすために有効である、本発明の化合物、または本発明の化合物を含む組成物の量を意味する。
【0052】
[0056] 「薬学的に許容できる」という句は、本明細書で利用する場合、理にかなった医学的判断の範囲内において、ヒトおよび動物における使用に適しており、過剰な毒性、刺激、アレルギー応答またはその他の問題もしくは合併症を伴わず、相応の妥当な利益/リスクの比を有する化合物、材料、組成物および/または投与剤型を指す。
【0053】
[0057] 「薬学的に許容できる担体」という句は、本明細書で使用する場合、任意の薬学的に許容できる材料を意味し、これは、液体であっても、または固体であってもよい。例示的な担体として、ビヒクル、希釈剤、添加剤、液体および固体の充填剤、賦形剤、溶媒、溶媒を被包する材料が挙げられる。各担体は、処方のその他の成分に適合しており、患者に対して障害性がないという意味で「許容でき」なければならない。薬学的に許容できる担体として役立ち得る材料のいくつかの例として、(1)糖、すなわち、ラクトース、グルコースおよびスクロース等;(2)デンプン、すなわち、トウモロコシデンプンおよびバレイショデンプン等;(3)セルロースおよびその誘導体、すなわち、カルボキシメチルセルロースナトリウム、エチルセルロースおよび酢酸セルロース;(4)粉末トラガカント;(5)麦芽;(6)ゼラチン;(7)タルク;(8)賦形剤、すなわち、カカオ脂および坐剤用ろう等;(9)油、すなわち、落花生油、綿実油、ベニバナ油、ゴマ油、オリーブ油、トウモロコシ油および大豆油等;(10)グリコール、すなわち、プロピレングリコール等;(11)ポリオール、すなわち、グリセリン、ソルビトール、マンニトールおよびポリエチレングリコール等;(12)エステル、すなわち、オレイン酸エチルおよびラウリン酸エチル等;(13)寒天;(14)緩衝剤、すなわち、水酸化マグネシウムおよび水酸化アルミニウム等;(15)アルギン酸;(16)発熱物質を含まない水;(17)等張食塩水;(18)リンゲル液;(19)エチルアルコール;(20)pH緩衝溶液;(21)ポリエステル、ポリカーボネートおよび/またはポリ酸無水物:ならびに(22)医薬品製剤中で利用するのに適合する、その他の無毒性の物質が挙げられる。
【0054】
[0058] 上記に記載したように、本発明の化合物の特定の実施形態は、アミノまたはアルキルアミノ等の塩基性の官能基を含有することができ、したがって、薬学的に許容できる酸と薬学的に許容できる塩を形成することが可能である。この場合の「薬学的に許容できる塩」という用語は、本発明の化合物の比較的無毒性の無機および有機の酸付加塩を指す。これらの塩は、投与ビヒクル中もしくは投与剤型の製造過程においてin situでまたは別個に、精製された本発明の化合物をその遊離の塩基の形態で、適切な有機もしくは無機の酸と反応させ、このようにして形成した塩をそれに続く精製の間に単離することによって調製することができる。代表的な塩として、臭化水素酸塩、塩酸塩、硫酸塩、スルファミン酸塩、重硫酸塩、リン酸塩、硝酸塩、酢酸塩、吉草酸塩、オレイン酸塩、パルミチン酸塩、ステアリン酸塩、ラウリン酸塩、安息香酸塩、乳酸塩、トシレート、クエン酸塩、マレイン酸塩、アスコルビン酸塩、パルミチン酸塩、フマル酸塩、コハク酸塩、酒石酸塩、ナフチル酸塩、メシル酸塩、ヒドロキシマレイン酸塩(hydroxymaleate)、フェニル酢酸塩、グルタミン酸塩、グルコヘプトン酸塩、サリチル酸塩、スルファニル酸塩、2−アセトキシ安息香酸塩、メタンスルホン酸塩、エタンジスルホン酸塩、シュウ酸塩、イソチオネート、ラクトビオン酸塩およびラウリルスルホン酸塩等が挙げられる。例えば、Bergeら(1977)“Pharmaceutical Salts”,J.Pharm.Sci.66:1−19を参照されたい。
【0055】
[0059] 「薬学的に許容できる塩」という用語は、本明細書に記載する化合物上に見出される特定の置換基に依存して、比較的無毒性の酸または塩基を用いて調製する、活性な化合物の塩を含む。本発明の化合物が、比較的酸性の官能基を含有する場合には、そのような化合物の中性の形態を、十分な量の所望の塩基と、そのまままたは適切な不活性な溶媒中のいずれかで接触させることによって塩基付加塩を得ることができる。薬学的に許容できる塩基付加塩の例として、ナトリウム塩、カリウム塩、カルシウム塩、アンモニウム塩、有機アミノ塩もしくはマグネシウム塩または類似の塩が挙げられる。本発明の化合物が、比較的塩基性の官能基を含有する場合には、そのような化合物の中性の形態を、十分な量の所望の酸と、そのまままたは適切な不活性な溶媒中のいずれかで接触させることによって酸付加塩を得ることができる。薬学的に許容できる酸付加塩の例として、塩酸、臭化水素酸、硝酸、炭酸、一水素炭酸(monohydrogencarbonic acid)、リン酸、一水素リン酸(monohydrogenphosphoric acid)、二水素リン酸(dihydrogenphosphoric acid)、硫酸、一水素硫酸(monohydrogensulfuric acid)、ヨウ化水素酸または亜リン酸等のような無機酸から誘導した塩、ならびに酢酸、プロピオン酸、イソ酪産、マレイン酸、マロン酸、安息香酸、コハク酸、スベリン酸、フマル酸、乳酸、マンデル酸、フタル酸、ベンゼンスルホン酸、p−トリルスルホン酸、クエン酸、酒石酸、メタンスルホン酸等のような比較的無毒性の有機酸から誘導した塩が挙げられる。また、アルギン酸塩等のアミノ酸の塩、ならびにグルクロン酸またはガラクツロン酸等のような有機酸の塩も挙げられる(例えば、Bergeら, Journal of Pharmaceutical Science, 66: 1-19 (1977)を参照されたい)。特定の特異的な本発明の化合物は、化合物を塩基付加塩または酸付加塩のいずれにも変換することを可能にする、塩基性官能基および酸性官能基の両方を含有する。
【0056】
[0060] 化合物の中性の形態を、好ましくは、塩を塩基または酸と接触させ、従来の様式で親化合物を単離することによって再生する。化合物の親の形態は、極性溶媒中の溶解性等の特定の物性において種々の塩の形態と異なるが、そうでなければ、塩は、本発明の目的に関しては、化合物の親の形態と同等である。
【0057】
[0061] 塩の形態に加えて、本発明は、プロドラッグの形態である化合物も提供する。本明細書に記載する化合物のプロドラッグは、生理的条件下で化学変化を容易に受けて、本発明の化合物をもたらす化合物である。さらに、プロドラッグは、ex vivo環境下で化学的または生化学的な方法によって本発明の化合物に変換することもできる。例えば、プロドラッグを、経皮パッチリザーバ中に適切な酵素または化学試薬と共に置いて、本発明の化合物に緩慢に変換させることができる。
【0058】
[0062] 本発明の特定の化合物は、非溶媒和の形態でも、水和した形態を含む溶媒和の形態でも存在することができる。一般に、溶媒和の形態は、非溶媒和の形態と同等であり、本発明の範囲内に包含される。本発明の特定の化合物は、複数の結晶形態または非結晶の形態で存在することができる。一般に、全ての物理的形態は、本発明が企図する使用のためには同等であり、本発明の範囲内に属するものとする。「化合物または化合物の薬学的に許容できる塩もしくは溶媒和化合物」は、塩および溶媒和化合物の両方である材料を包含するという点で、「または」の包括的な意味を意図する。
【0059】
[0063] 本発明の特定の化合物は、不斉炭素原子(光学中心)または二重結合を有し;ラセミ体、ジアステレオマー、幾何異性体および個々の異性体は、本発明の範囲内に包含される。光学的に活性な(R)−異性体および(S)−異性体は、キラルシントンもしくはキラル試薬を使用して調製しても、または従来の技法を使用して分割してもよい。本明細書に記載する化合物が、オレフィン二重結合またはその他の幾何学的非対称の中心を含有する場合、別段の記載がない限り、化合物は、E幾何異性体およびZ幾何異性体の両方を含むことを意図する。同様にまた、全ての互変異性の形態も含むことを意図する。
【0060】
[0064] 本明細書で使用するラセミ化合物、アンビスケールミック(ambiscalemic)化合物およびスケールミック(scalemic)化合物、または鏡像異性的に純粋な化合物の図的表現は、Maehr,J.Chem.Ed.,62:114−120(1985)から採用しており、波線は、それが示す結合がもたらすことができるであろう任意の立体化学的な関係の否定を示し;実線のくさび形および破線のくさび形は、提示する相対的な立体配置を示す幾何学的な記載であるが、いずれかの絶対的な立体化学を伝えているわけではなく;くさび形の輪郭線および点線または破線は、不定の絶対配置の鏡像異性的に純粋な化合物を表示する。
【0061】
[0065] 「鏡像異性体過剰」および「ジアステレオ異性体過剰」という用語は、本明細書では互換的に使用する。単一の立体中心を有する化合物を、「鏡像異性体過剰」な状態で存在すると呼び、少なくとも2つの立体中心を有する化合物を、「ジアステレオ異性体過剰」な状態で存在すると呼ぶ。
【0062】
[0066] 本発明の化合物はまた、そのような化合物を構成する原子のうちの1個または複数において、不自然な比率の原子の同位体も含有することもできる。例えば、化合物を、例えば、トリチウム(3H)、ヨウ素−125(125I)または炭素−14(14C)等の放射性同位体を用いて放射標識することができる。放射性であっても、またはそうでなくても、本発明の化合物の全ての同位体の変形形態が、本発明の範囲内に包含されるものとする。
【0063】
[0067] 「モノアミン輸送体リガンド」という用語は、モノアミン輸送体に結合する任意の化合物を指す。リガンドは、所与のモノアミン輸送体の天然のリガンドである内因性モノアミン、ならびに特定のモノアミン輸送体に結合することが知られている合成分子等の薬物分子およびその他の化合物を含む。1つの例では、リガンドは、トリチウム等の放射性同位体を含むか、またはそれ以外の様式で(例えば、蛍光により)標識される。所与のモノアミン輸送体の適切なリガンドを選択することは、当業者の能力の範囲内である。例えば、ドーパミン輸送体の既知のリガンドとしては、ドーパミンおよびWIN35428が挙げられ、セロトニン輸送体の既知のリガンドとしては、5−ヒドロキシトリプタミン(セロトニン)およびシタロプラムが挙げられ、ノルエピネフリン輸送体のリガンドとしては、ノルエピネフリンおよびニソキセチンが挙げられる。
【0064】
[0068] 「摂食障害」という用語は、食べることを回避する異常な衝動強迫または異常に多くの量の食物を消費する制御できない衝撃を指す。これらの障害は、罹患者の社会的な生活状態のみならず、また身体的な生活状態にも影響を及ぼす。摂食障害の例として、神経性食欲不振症、過食症および過食が挙げられる。
【0065】
[0069] 「神経障害」という用語は、哺乳動物の中枢神経系または抹消神経系の任意の状態を指す。「神経障害」という用語は、神経変性疾患(例えば、アルツハイマー病、パーキンソン病および筋萎縮性側索硬化症)、神経精神疾患(例えば、統合失調症および全般性不安障害等の不安症)を含む。例示的な神経障害として、MLS(小脳失調症)、ハンチントン病、ダウン症、多発脳梗塞性認知症、てんかん重積症、打撲傷(例えば、脊髄損傷および頭部損傷)、ウイルス感染が誘発する神経変性(例えば、AIDS、脳症)、てんかん、温和なもの忘れ、閉鎖性頭部損傷、睡眠障害、うつ病(例えば、双極性障害)、認知症、運動障害、精神障害、アルコール依存症、心的外傷後ストレス障害等が挙げられる。「神経障害」はまた、こうした障害に伴う任意の状態も含む。例えば、神経変性障害を治療する方法は、神経変性障害に伴う記憶の喪失および/または認知の喪失を治療する方法を含む。例示的な方法としてはまた、神経変性障害に特徴的な神経細胞の機能の喪失を治療または予防することも挙げられるであろう。「神経障害」はまた、モノアミン(例えば、ノルエピネフリン)シグナル伝達経路に少なくとも部分的に関係する任意の疾患または状態(例えば、心血管疾患)も含む。
【0066】
[0070] 「疼痛」は、不快な感覚および情動の経験である。疼痛は、持続期間、病因または病態生理、機構、強度および症状に基づいて分類されている。「疼痛」という用語は、本明細書で使用する場合、全てのカテゴリーの疼痛を指し、刺激または神経の応答の観点から説明される疼痛、例えば、体性痛(侵害性刺激に対する正常な神経の応答)および神経障害性疼痛(損傷または変質した感覚経路の異常な応答であり、明確な侵害性の入力を伴わないことが多い);時間的に類別される疼痛、例えば、慢性疼痛および急性疼痛;重症度、例えば、軽度、中等度または重度の観点から類別される疼痛;ならびに症状であるか、または疾患の状況もしくは症候群の結果である疼痛、例えば、炎症性疼痛、癌性疼痛、AIDS性疼痛、関節症、偏頭痛、三叉神経痛、心虚血および糖尿病性末梢神経因性疼痛を含む(例えば、それぞれの全体が本明細書に参照により組み込まれている、Harrison's Principles of Internal Medicine, pp. 93-98(Wilsonら編, 12th ed. 1991);Williamsら, J. of Med. Chem. 42: 1481-1485(1999)を参照されたい)。「疼痛」はまた、混合性病因の疼痛、二重機構の疼痛、異痛症、灼熱痛、中枢性疼痛、知覚過敏、ヒペルパチー、異常感覚および痛覚過敏を含むことも意味する。
【0067】
[0071] 上記に記載した「体性」痛は、侵害性刺激、すなわち、損傷もしくは疾病等、例えば、外傷、熱傷、感染、炎症に対する、または疾患の過程、すなわち、癌等に対する正常な神経の応答を指し、皮膚性の疼痛(例えば、皮膚、筋肉または関節に起因する)および内臓性の疼痛(例えば、器官に起因する)の両方を含む。
【0068】
[0072] 「神経因性疼痛」は、神経系に対する傷害の結果生じる神経学的な状態の不均一な群である。上記に記載した「神経因性」疼痛は、末梢および/または中枢の感覚経路に対する損傷またはその機能不全、ならびに神経系の機能不全の結果生じる疼痛を指し、こうした疼痛は、明らかな侵害性の入力を伴うことなく発生または持続することが多い。これには、末梢性ニューロパシーおよび中枢性ニューロパシーに関連する疼痛が含まれる。末梢性の神経因性疼痛の一般的な型には、糖尿病性ニューロパシー(また、糖尿病性末梢神経因性疼痛、またはDN、DPNもしくはDPNPとも呼ばれる)、ヘルペス後神経痛(PHN)および三叉神経痛(TGN)がある。脳または脊髄に対する傷害が関与する中枢性の神経因性疼痛は、脳卒中、脊髄損傷に続いて、および多発性硬化症の結果として生じる場合がある。神経因性疼痛の定義に含まれることを意味するその他の型の疼痛には、神経障害性の癌性疼痛、HIV/AIDS に起因する疼痛、幻肢痛および複合性局所疼痛症候群からの疼痛がある。例示的な実施形態では、本発明の化合物は、神経因性疼痛の治療のために有用である。
【0069】
[0073] 神経因性疼痛の一般的な臨床的特徴には、感覚の喪失、異痛症(非侵害性の刺激によって生じる疼痛)、痛覚過敏、およびヒペルパチー(遅延性の感受、加重、および有痛性の残感覚)がある。疼痛は、侵害受容型と神経障害型との、例えば、機械的な脊髄性疼痛と神経根症または脊髄症との組合せであることが多い。
【0070】
[0074] 「急性疼痛」は、典型的には侵襲性の手順、外傷および疾患に伴う侵害性の化学的、熱的または機械的な刺激に対する正常な、予測される生理的な応答である。これは、一般に、時限的であり、組織を脅かし、および/または組織の損傷をもたらす刺激に対する適切な応答として見ることができる。上記に記載した「急性疼痛」は、短い持続時間または突然の開始によって特徴付けられる疼痛を指す。
【0071】
[0075] 「慢性疼痛」は、広い範囲の障害、例えば、外傷、悪性腫瘍、および関節リウマチ等の慢性炎症性疾患において生じる。慢性疼痛は通常、6ヵ月以上持続する。さらに、慢性疼痛の強度は、侵害性刺激または根底にある過程の強度とは不釣合いである場合がある。上記に記載した「慢性疼痛」は、慢性の障害にともなう疼痛、または根底にある障害の回復もしくは損傷の治癒を過ぎても持続し、根底にある過程から予測されるであろうものよりも激烈であることが多い疼痛を指す。慢性疼痛は、頻繁に再発する場合がある。
【0072】
[0076] 「炎症性疼痛」は、組織の損傷およびその結果としての炎症過程に対して応答した疼痛である。炎症性疼痛は、治癒を促す生理的な応答を惹起するという点で適応性である。しかし、炎症はまた、神経細胞の機能にも影響を及ぼす。COX2酵素が誘発するPGE2、ブラジキニンおよびその他の物質を含めた炎症メディエーターは、疼痛伝達神経細胞上の受容体に結合し、それらの機能を変化させ、興奮性を高め、したがって、疼痛感覚が高まる。多くの慢性疼痛は、炎症性の構成要素を有する。上記に記載した「炎症性疼痛」は、症状として、または炎症もしくは免疫系の障害の結果として生じる疼痛を指す。
【0073】
[0077] 上記に記載した「内臓性の疼痛」は、内臓器官内に位置する疼痛を指す。
【0074】
[0078] 上記に記載した「混合性病因」の疼痛は、炎症性の構成要素および神経障害性の構成要素の両方を含有する疼痛を指す。
【0075】
[0079] 上記に記載した「二重機構」の疼痛は、末梢性の感作および中枢性の感作の両方によって増幅および維持される疼痛を指す。
【0076】
[0080] 上記に記載した「灼熱痛」は、外傷性の神経病変後の持続的な灼熱感、異痛症およびヒペルパチーの症候群を指し、血管運動性および発汗促進性の機能不全、ならびに後には栄養性の変化と組み合わさることが多い。
【0077】
[0081] 上記に記載した「中枢性」疼痛は、中枢神経系内の一次病巣または機能不全によって開始する疼痛を指す。
【0078】
[0082] 上記に記載した「知覚過敏」は、特別な感覚を除く、刺激に対して高まった感受性を指す。
【0079】
[0083] 上記に記載した「ヒペルパチー」は、刺激、特に反復性の刺激に対して異常に痛いと感じる反応、および閾値の増加によって特徴付けられる有痛性の症候群を指す。ヒペルパチーは、異痛症、知覚過敏、痛覚過敏または異常感覚を伴って生じる場合がある。
【0080】
[0084] 上記に記載した「異常感覚」は、自発性であっても、または誘起されたものであっても、不快で異常な感覚を指す。異常感覚の特別な場合には、痛覚過敏および異痛症がある。
【0081】
[0085] 上記に記載した「痛覚過敏」は、通常痛いと感じる刺激に対する高まった応答を指す。痛覚過敏は、閾値を上回る刺激に対する高まった疼痛を反映する。
【0082】
[0086] 上記に記載した「灼熱痛」は、 通常では疼痛を招くことがない刺激に起因した疼痛を指す。
【0083】
[0087] 「疼痛」という用語は、神経系の機能不全の結果生じる疼痛、すなわち、神経障害性の疼痛および考えられる一般的な病態生理学的機構の臨床的特徴を共有するが、神経系のいずれかの部分における同定可能な病変によって開始されるわけではない有機的な疼痛の状況を含む。
【0084】
[0088] 「糖尿病性末梢神経因性疼痛」(DPNP、また、糖尿病性ニューロパシー、DNまたは糖尿病性末梢性ニューロパシーとも呼ばれる)という用語は、糖尿病に伴うニューロパシーによって引き起こされる疼痛を指す。DPNPは、「灼熱感」または「痛みが走る感じ」としてのみならず、また激しく痛む疼痛としても説明することができる足における疼痛または刺痛と古典的には表現される。あまり一般的ではないが、患者が、疼痛を、痒み、引き裂かれる感じ、または歯痛様と説明する場合がある。この疼痛には、異痛症および痛覚過敏、ならびにしびれ等の症状の不在が伴う場合がある。
【0085】
[0089] 「ヘルペス後神経痛(Post-Herpetic Neuralgia)」という用語はまた、「ヘルペス後神経痛(Postherpetic Neuralgia)」(PHN)とも呼ばれ、神経線維および皮膚に影響を及ぼす有痛性の状態である。ヘルペス後神経痛は、帯状疱疹の合併症、すなわち、水痘帯状疱疹ウイルス(varicella zoster virus)(VZV)の第2の勃発であり、このウイルスは、最初は、水痘を引き起こす。
【0086】
[0090] 「神経障害性の癌性疼痛」という用語は、癌の結果としての末梢性の神経障害性の疼痛を指し、腫瘍による神経の浸潤もしくは圧縮によって直接的に、または放射線療法および化学療法(化学療法誘発性ニューロパシー)等の癌治療によって間接的に引き起こされる場合がある。
【0087】
[0091] 「HIV/AIDS性の末梢性ニューロパシー」または「HIV/AIDS関連ニューロパシー」という用語は、HIV/AIDSによって引き起こされる末梢性ニューロパシー、すなわち、急性または慢性の炎症性脱髄性ニューロパシー(それぞれ、AIDPおよびCIDP)等、ならびにHIV/AIDSを治療するために使用した薬物の副作用として生じる末梢性ニューロパシーを指す。
【0088】
[0092] 「幻肢痛」という用語は、切断した肢がかつてあった場所から生じるように思える疼痛を指す。幻肢痛はまた、麻痺後(例えば、脊髄損傷後)の肢においても生じる場合がある。「幻肢痛」は通常、その性質が慢性である。
【0089】
[0093] 「三叉神経痛」(TN)という用語は、神経の枝が分布する顔部の領域(唇、目、鼻、頭皮、前頭部、上顎および下顎)において、激烈な、突き刺すような、電気ショック様の疼痛のエピソードを引き起こす第5脳(三叉)神経の障害を指す。三叉神経痛はまた、「自殺性疾患」としても知られている。
【0090】
[0094] 「複合性局所疼痛症候群(CRPS)」という用語は、以前は反射性交換神経性ジストロフィー(RSD)として知られており、慢性疼痛の状態である。CRPSの鍵となる症状は、損傷の重症度とは不釣合いな連続的で激烈な疼痛であり、これは、時間の経過と共に軽減するのではなく悪化する。CRPSは、末梢神経ではなく組織の損傷によって引き起こされる状態を含む1型、および症候群が主要な神経の損傷によって招かれ、時に灼熱痛と呼ばれる2型に分かれる。
【0091】
[0095] 「線維筋痛症」という用語は、疲労および様々なその他の症状が伴う、散在するまたは特異的な筋肉、関節または骨の疼痛によって特徴付けられる慢性の状態を指す。以前は、線維筋痛症は、結合組織炎、慢性筋肉痛症候群、心因性リウマチおよび緊張性筋肉痛等のその他の名前によって知られていた。
【0092】
[0096] 「けいれん」という用語は、ある神経障害を指し、「てんかん」と互換的に使用するが、多くの型のてんかんがあり、それらのいくつかは、けいれんの代わりに、軽微または軽度な症状を示す。全ての型のてんかんが、脳における混乱した突然の電気的活動によって引き起こされ得る。けいれんは、迅速で制御できない揺れである。けいれんの間は、筋肉が、繰り返し収縮および弛緩する。
【0093】
[0097] 「うつ病」という用語は、全ての形態のうつ病を含み、これらには、大うつ病性障害(MDD)、双極性障害、季節性感情障害(SAD)および気分変調症がある。「大うつ病性障害」は、本明細書では、「単極性うつ病」および「大うつ病」と互換的に使用する。「うつ病」はまた、うつ病に通常伴う任意の状態、すなわち、疲労の全ての形態(例えば、慢性疲労症候群)および失認等を含む。
【0094】
II.緒言
[0098] 神経障害の有効な療法を開発するための1つの戦略は、セロトニン (5-HT)、ノルエピネフリン(NE)およびドーパミン(DA)等、2つ以上の生体アミンの再取り込みを同時に阻害する広域スペクトルの抗うつ薬の使用である。このアプローチの理論的根拠は、ドーパミン作動性機能の欠乏が、うつ病の中心となる症状である快感消失と相関し得ることを示す臨床および前臨床の証拠に基づく。Baldessarini,R.J.,“Drugs and the Treatment of Psychiatric Disorders:Depression and Mania,in Goodman and Gilman’s The Pharmacological Basis of Therapeutics 431−459(9th ed 1996)Hardmanら編。
【0095】
[0099] 本発明の例示的な化合物および組成物の利点は、少なくとも2つの神経伝達物質(例えば、NE、5-HTおよびDA)の利用能を、例えば、シナプス間隙からの神経伝達物質の二重(再)取り込みを阻害することによって増加させるそれらの能力である。Skolnickらは、DA、NEおよび5−HTのシナプスにおける利用能を同時に増加させる抗うつ薬の治療プロファイルが、NEおよび/または5−HTのみを阻害する化合物とは異なることを示唆する一連の前臨床の証拠について報告している。Skolnick,P.ら,“Antidepressant−like actions of DOV−21,947:a“triple”reuptake inhibitor,”Eur.J.Pharm.2003,461,103。
【0096】
[0100] 例えば、Skolnickらは、化合物DOV21,947((+)−1−(3,4−ジクロロフェニル)−3−アザビシクロ[3.1.0]へキサン)が、セロトニン、ノルエピネフリンおよびドーパミンの再取り込みを、対応するヒト組換え輸送体を発現するヒト胎児腎臓(HEK293)細胞において阻害する(それぞれ、12、23および96nMのIC50値)ことを報告している。Skolnick,P.ら,“Antidepressant−like actions of DOV−21,947:a“triple”reuptake inhibitor,”Eur.J.Pharm.2003,461,99。さらに、DOV21,947は、(ラットの)強制水泳試験においては無動の持続時間を短縮し、かつまた、尾の懸垂試験においては無動の用量依存性の低下をもたらした。追加の証拠を、例えば、米国特許第6,372,919号中、DOV21,947等の新しい三重再取り込み阻害薬についての前臨床データ中に見い出すことができる。この特許では、DOV21,947は、ノルエピネフリンおよびセロトニンの取り込み部位に対して、ラセミ化合物である(±)−1−(3,4−ジクロロフェニル)−3−アザビシクロ[3.1.0]へキサンよりも、顕著に高い親和性を有するとして開示された。
【0097】
[0101] 総合すると、DOV21,947等の化合物についての前臨床データから、二重または三重再取り込み阻害薬には、臨床におけるうつ病の新規な治療としての可能性があることが示されている。
【0098】
III.組成物
A.シクロアルキルアミン
[0102] 例示的な実施形態では、本発明は、以下の式を有する化合物を提供する。
【0099】
【化2】
式中、指数nは、0、1および2からなる群から選択される整数であり;sは、0、1および2からなる群から選択される整数である。Aは、H、置換または非置換アルキル、ハロゲン、および置換または非置換ハロアルキルから選択されるメンバーである。Xは、H、ハロゲン、置換または非置換アルキル、置換または非置換アリール、置換または非置換ハロアルキル、およびOR5(ただし、R5は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、アシル、およびS(O)2R5a(ただし、R5aは、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルから選択される)から選択される)から選択される。
【0100】
[0103] YおよびZは独立して、H、ハロゲン、CF3、CN、OR9、SR9、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、置換または非置換ヘテロシクロアルキル、NR10R11、またはNO2を示す。YとZとは、それらが結合している原子と一緒になって任意選択により結合して、5員環〜7員環を形成し、この環はその中に、1、2または3個のヘテロ原子を任意選択により有することができる。R9は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、または置換または非置換ヘテロシクロアルキルを示す。基であるR10およびR11は独立して、H、OR12、アシル、S(O)2R13、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、または置換または非置換ヘテロシクロアルキルを示す。R10とR11とは、それらが結合している窒素と一緒になって任意選択により結合して、R10とR11とが結合する窒素に加えて、1、2または3個のヘテロ原子を任意選択により有する3員環、4員環、5員環、6員環または7員環を形成する。記号R12は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、または置換または非置換ヘテロシクロアルキルを示す。R13は、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルから選択される。
【0101】
[0104] R1およびR2は独立して、H、ハロゲン、CN、CF3、OR6、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、または置換または非置換ヘテロシクロアルキルである。R6は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルから選択される。
【0102】
[0105] R3およびR4は独立して、H、OR7、アシル、S(O)2R8、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、または置換または非置換ヘテロシクロアルキルを示す。R7は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、または置換または非置換ヘテロシクロアルキルである。R8は、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルから選択されるメンバーである。
【0103】
[0106] R1、R2、R3およびR4のうちの2つ以上が、それらが結合している原子と一緒になって任意選択により結合して、3員環、4員環、5員環、6員環または7員環を形成し、この環は、1、2、3または4個のヘテロ原子を任意選択により含む。
【0104】
[0107] YとZとは、それらが結合している原子と一緒になって任意選択により結合して、5員環、6員環または7員環を形成し、この環はその中に、1、2または3個のヘテロ原子を任意選択により有する。当業者には明らかであろうが、YとZとが結合して環をなす場合、環内に組み込まれた原子上の置換基(例えば、R9、R10およびR11)が、これらの置換基が結合している原子の原子価を満たす必要に応じて、存在する(例えば、環の環状構造内に組み込まれる)場合、または存在しない場合があるであろう。
【0105】
[0108] 例示的な実施形態では、YおよびZは独立して、ハロゲン、CF3、CN、OR9、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、置換または非置換ヘテロシクロアルキル、NR10R11、およびNO2を示す。YとZとは、それらが結合している原子と一緒になって任意選択により結合して、5員環、6員環または7員環を形成し、この環はその中に、1、2または3個のヘテロ原子を任意選択により有することができる。
【0106】
[0109] 例示的な実施形態では、本発明の化合物は、以下の式による構造を有しない。
【0107】
【化3】
【0108】
[0110] 別の例示的な実施形態では、化合物は、以下の式による構造を有しない。
【0109】
【化4】
【0110】
[0111] 例示的な実施形態では、化合物は、YまたはZのいずれかがHである場合には、R9が、Hおよび置換または非置換アルキルから選択されるメンバー以外であるような構造を有する。例示的な実施形態では、化合物は、YまたはZのいずれかがHである場合には、R9が、Hおよび非置換アルキルから選択されるメンバー以外であるような構造を有する。例示的な実施形態では、化合物は、YまたはZのいずれかがHである場合には、R9が、H、メチルおよびエチルから選択されるメンバー以外であるような構造を有する。
【0111】
[0112] 例示的な実施形態では、化合物は、R5が、Hおよび置換または非置換アルキルから選択されるメンバー以外であるような構造を有する。例示的な実施形態では、化合物は、R5が、Hおよび非置換アルキルから選択されるメンバー以外であるような構造を有する。例示的な実施形態では、化合物は、R5が、H、メチルおよびエチルから選択されるメンバー以外であるような構造を有する。
【0112】
[0113] 種々の例示的な実施形態では、指数sは1である。例示的な実施形態では、指数nは1である。種々の実施形態では、sおよびnの両方が1である。
【0113】
[0114] 例示的な実施形態では、YおよびZは独立して、H、ハロゲン、CNおよびCF3から選択される。種々の実施形態では、YおよびZのうちの少なくとも1つが、H以外である。例示的な実施形態では、YおよびZの両方が、H以外である。
【0114】
[0115] 例示的な実施形態では、R3およびR4は、置換または非置換C1〜C4アルキル、および置換または非置換C1〜C4ヘテロアルキルから独立して選択されるメンバーである。例示的な実施形態では、R3およびR4は、置換または非置換アルケニル、置換または非置換アルキニル、および置換または非置換シクロアルキルからなる群から独立して選択されるメンバーである。
【0115】
[0116] 種々の実施形態では、本発明の化合物は、以下からなる群から選択されるメンバーである構造を有する。
【0116】
【化5】
【0117】
[0117] 選択された実施形態では、本発明の化合物は、式IIおよびIIaから選択される構造を有する。
【0118】
【化6】
【0119】
式IIおよびIIaによる例示的な化合物として、以下が挙げられる。
【0120】
【化7】
【0121】
例示的な実施形態では、YおよびZは、H、ハロゲン、CNおよびCF3からなる群から独立して選択されるメンバーである。例示的な実施形態では、YおよびZは、ハロゲンである。例示的な実施形態では、YおよびZは、クロロである。例示的な実施形態では、sは1である。例示的な実施形態では、nは1である。例示的な実施形態では、R1およびR2は、Hである。例示的な実施形態では、AはHである。別の例示的な実施形態では、R1およびR2は、Hであり、AはHである。
【0122】
[0118] 選択された実施形態では、本発明の化合物は、式IIIおよびIIIaから選択される構造を有する。
【0123】
【化8】
【0124】
[0119] 式IIIおよびIIIaによる例示的な化合物として、以下が挙げられる。
【0125】
【化9】
【0126】
[0120] 例示的な実施形態では、YおよびZは、H、ハロゲン、CNおよびCF3からなる群から独立して選択されるメンバーである。例示的な実施形態では、YおよびZは、ハロゲンである。例示的な実施形態では、YおよびZは、クロロである。例示的な実施形態では、sは1である。例示的な実施形態では、nは1である。例示的な実施形態では、R1およびR2は、Hである。例示的な実施形態では、AはHである。別の例示的な実施形態では、R1およびR2は、Hであり、AはHである。
【0127】
[0121] 例示的な実施形態では、化合物は、式(IV)による構造を有する。
【0128】
【化10】
式中、YおよびZは、独立して選択されるハロゲンである。例示的な実施形態では、化合物は、以下による構造を有する。
【0129】
【化11】
【0130】
[0122] 例示的な実施形態では、化合物は、以下による構造を有する。
【0131】
【化12】
【0132】
[0123] 別の実施形態では、Yは、FおよびClから選択されるメンバーである。別の実施形態では、Zは、FおよびClから選択されるメンバーである。別の実施形態では、YはClであり、ZはClである。別の実施形態では、YはFであり、ZはClである。別の実施形態では、YはClであり、ZはFである。
【0133】
[0124] 例示的な実施形態では、化合物は、式(V)による構造を有する。
【0134】
【化13】
式中、YおよびZは、独立して選択されるハロゲンである。例示的な実施形態では、化合物は、以下による構造を有する。
【0135】
【化14】
【0136】
[0125] 例示的な実施形態では、化合物は、以下による構造を有する。
【0137】
【化15】
【0138】
[0126] 別の実施形態では、Yは、FおよびClから選択されるメンバーである。別の実施形態では、Zは、FおよびClから選択されるメンバーである。別の実施形態では、YはClであり、ZはClである。別の実施形態では、YはFであり、ZはClである。別の実施形態では、YはClであり、ZはFである。
【0139】
[0127] 本発明の例示的な化合物は以下の式を有する。
【0140】
【化16】
式中、R4は、HまたはCH3のいずれかである。
【0141】
[0128] 例示的な実施形態では、化合物は、式(VI)による構造を有する。
【0142】
【化17】
【0143】
[0129] 別の例示的な実施形態では、この構造を有する化合物は、YおよびZから選択される少なくとも1つの、ハロゲンであるメンバーを有する。別の例示的な実施形態では、YおよびZは、ハロゲンである。別の実施形態では、Yは、FおよびClから選択されるメンバーである。別の実施形態では、Zは、FおよびClから選択されるメンバーである。別の実施形態では、YはClであり、ZはClである。別の実施形態では、YはFであり、ZはClである。別の実施形態では、YはClであり、ZはFである。
【0144】
[0130] 例示的な実施形態では、化合物は、式(VII)による構造を有する。
【0145】
【化18】
式中、YおよびZは、Hではなく、Aは、置換または非置換アルキルから選択されるメンバーである。別の例示的な実施形態では、この構造を有する化合物は、YおよびZから選択される少なくとも1つの、ハロゲンであるメンバーを有する。別の例示的な実施形態では、YおよびZは、ハロゲンである。別の実施形態では、Yは、FおよびClから選択されるメンバーである。別の実施形態では、Zは、FおよびClから選択されるメンバーである。別の実施形態では、YはClであり、ZはClである。別の実施形態では、YはFであり、ZはClである。別の実施形態では、YはClであり、ZはFである。例示的な実施形態では、Aは、置換または非置換メチルである。例示的な実施形態では、Aはメチルである。
【0146】
[0131] 例示的な実施形態では、化合物は、式(VIII)による構造を有する。
【0147】
【化19】
式中、YおよびZは、Hではなく、R3およびR4はそれぞれ独立して、Hおよび置換または非置換アルキルから選択される。別の例示的な実施形態では、各R3およびR4はそれぞれ独立して、Hおよび置換または非置換メチルから選択される。別の例示的な実施形態では、R3およびR4はそれぞれ独立して、Hおよびメチルから選択される。別の例示的な実施形態では、この構造を有する化合物は、、YおよびZから選択される少なくとも1つの、ハロゲンであるメンバーを有する。別の例示的な実施形態では、YおよびZは、ハロゲンである。別の実施形態では、Yは、FおよびClから選択されるメンバーである。別の実施形態では、Zは、FおよびClから選択されるメンバーである。別の実施形態では、YはClであり、ZはClである。別の実施形態では、YはFであり、ZはClである。別の実施形態では、YはClであり、ZはFである。
【0148】
[0132] 本発明の例示的な化合物は、以下の式による構造を有する。
【0149】
【化20】
式中、A1、A2、A3およびA4はそれぞれ独立して、O、S、N(Rb)bおよびC(Rb)b(Rc)から選択される。指数aは、0、1および2から選択される整数である。指数bは、それが結合している原子の原子価の要件を満たす必要に応じて、0または1である。RbおよびRcは、H、ハロゲン、CF3、CN、OR14、SR14、NR15R16、NR15S(O)2R14、NR15C(O)R14、S(O)2R14、アシル、C(O)OR14、C(O)NR15R16、S(O)2NR15R16、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルから独立して選択されるメンバーである。各R14、R15およびR16は、H、アシル、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、置換または非置換ヘテロシクロアルキルからなる群から独立して選択されるメンバーであり、ただし、R14、R15およびR16のうちの2つが、それらが結合している原子と一緒になって任意選択により結合して、3員環、4員環、5員環、6員環または7員環を形成し、この環は、1、2または3個のヘテロ原子を任意選択により含む。
【0150】
[0133] 例示的な実施形態では、RbおよびRcは、H、ハロゲン、CN、ハロゲンで置換C1〜C4アルキル(例えば、CF3)、およびC1〜C4アルコキシ(例えば、OMe,OEt、OCF3)からなる群から独立して選択されるメンバーである。
【0151】
[0134] 例示的な実施形態では、YおよびZが結合して、5員環、6員環または7員環を有する縮合環系を形成し、1、2または3個のヘテロ原子を任意選択により含む。したがって、1つの実施形態では、フェニル環の置換基が、以下の構造を有する。
【0152】
【化21】
式中、環Lは、置換または非置換、飽和もしくは不飽和のシクロアルキルもしくはヘテロシクロアルキルであるか、または置換または非置換アリールもしくはヘテロアリールである。
【0153】
[0135] 例示的な縮合環系を以下に示す。
【0154】
【化22】
式中のA1、A2、A3およびA4、ならびにaは、本明細書に記載されている。
【0155】
[0136] 本発明の化合物は、アミンの部分(例えば、一級アミン、二級アミンまたは三級アミンの基)を含み、したがって、化合物(例えば、遊離の塩基)を酸と接触させることによって、化合物を塩の形態に変換することができる。例示的な実施形態では、塩の形態を得て、塩以外の形態では油性または粘性の化合物を固体の物質に変換し、取扱いを容易にする。別の例示的な実施形態では、本発明の化合物の遊離の塩基を、対応する塩に変換することによって、化合物の水性媒体中における溶解性を向上させ、これによって、生物学的利用率、薬物動態および薬力学等の生物学的特徴に影響を及ぼすことができる。したがって、本発明の化合物の任意の塩の形態、すなわち、無機酸の塩(例えば、塩酸塩)または有機酸の塩を含めた薬学的に許容できる塩等が、現在の発明の範囲内に属する。また、本発明の化合物の任意のプロドラッグも、本発明の範囲内に属する。例えば、R3およびR4は、in vivoにおいて切断可能であって、アミン、例えば、一級アミンまたは二級アミンを生じる任意の基であってよい。
【0156】
B.立体異性体を含む組成物
[0137] 本発明の化合物は、1つまたは複数の立体中心を含むことができ、特定の幾何学的なまたは立体異性体の形態で存在してよい。化合物は、キラルであっても、ラセミ体であっても、または1つもしくは複数の立体異性体を含む組成物中に存在してもよい。現在の発明は、鏡像異性体、ジアステレオマー、ラセミ混合物、鏡像異性体に富む混合物、およびジアステレオ異性体に富む混合物を包含する。追加の不斉炭素原子も、アルキル基等の置換基中に存在することができる。全てのそのような異性体、およびそれらの混合物が、本発明中に含まれるものとする。
【0157】
[0138] 例えば、本発明の化合物の特定の鏡像異性体が所望される場合には、これを、不斉合成によって、またはキラルの補助物質を用いて誘導体化することによって調製することができ、後者の場合、得られたジアステレオマーの混合物を分離し、補助基を切断して、純粋な所望の鏡像異性体を得る。あるいは、分子が、アミノ基等の塩基性の官能基またはカルボキシル基等の酸性の官能基を含有する場合には、ジアステレオマーの塩を、適切な光学的に活性な酸または塩基を用いて形成し、それに続いて、このようにして形成したジアステレオマーを分別結晶または当技術分野で既知のクロマトグラフ的な手段によって分割し、それに続いて、純粋な鏡像異性体を回収することができる。さらに、鏡像異性体とジアステレオマーとの分離はしばしば、キラルの安定している相を利用するクロマトグラフィーを使用して、任意選択により化学的な誘導体化(例えば、アミンからのカルバメートの形成)と組み合わせて達成される。
【0158】
[0139] 本明細書で使用する場合、「鏡像異性体に富む」または「ジアステレオ異性体に富む」という用語は、約50%超、好ましくは約70%超、より好ましくは約90%超で、鏡像異性体過剰(ee)またはジアステレオ異性体過剰(de)を有する化合物を指す。一般に、約90%超の鏡像異性的またはジアステレオ異性的な純度、例えば、約95%超、約97%超および約99%超のeeまたはdeを有する組成物が特に好ましい。
【0159】
[0140] 「鏡像異性体過剰」および「ジアステレオ異性体過剰」という用語は、本明細書では互換的に使用する。単一の立体中心を有する化合物を、「鏡像異性体過剰」の状態で存在すると呼び、少なくとも2つの立体中心を有する化合物を、「ジアステレオ異性体過剰」の状態で存在すると呼ぶ。
【0160】
[0141] 例えば、「鏡像異性体過剰」という用語は、当技術分野では周知であり、以下のように定義される。
【0161】
【数1】
【0162】
[0142] 例えば、「鏡像異性体過剰」という用語は、「光学純度」というより古い用語に関連し、両方が、同一の現象の測定値である。eeの値は、0から100までの数であり、ゼロは、ラセミ体であり、100は、鏡像異性的に純粋である。過去に98%光学的に純粋であるといわれていた化合物は、今では、96%eeによってより正確に特徴づけられる。90%eeは、問題になっている材料中、95%が、1つの鏡像異性体として存在し、かつ5%が、(1つまたは複数の)その他の鏡像異性体として存在することを反映する。
【0163】
[0143] したがって、1つの実施形態では、本発明は、本発明の化合物の第1の立体異性体および少なくとも1つの追加の立体異性体を含む組成物を提供する。第1の立体異性体は、少なくとも約80%、好ましくは少なくとも約90%、より好ましくは少なくとも約95%のジアステレオ異性体過剰または鏡像異性体過剰の状態で存在することができる。特に好ましい実施形態では、第1の立体異性体は、少なくとも約96%、少なくとも約97%、少なくとも約98%、少なくとも約99%、または少なくとも約99.5%のジアステレオ異性体過剰または鏡像異性体過剰の状態で存在する。鏡像異性体過剰またはジアステレオ異性体過剰は、相互の立体異性体を正確に比較して決定してもよく、または少なくとも2つのその他の立体異性体の合計と比較して決定してもよい。例示的な実施形態では、鏡像異性体過剰またはジアステレオ異性体過剰を、混合物中に存在する全てのその他の検出可能な立体異性体と比較して決定する。立体異性体は、解析する混合物中のそのような立体異性体の濃度をキラルHPLC等の一般的な解析方法を使用して決定することができる場合には、検出可能である。
【0164】
C.化合物の合成
1.一般論
[0144] 本発明の化合物は、純粋なシス異性体もしくはラセミ混合物、または2つ以上のジアステレオマーの混合物として合成することができる。立体異性体は、適切な合成段階において、例えば、HPLC等のキラルカラムクロマトグラフィーによって分離して、それぞれの立体異性体の鏡像異性的/ジアステレオ異性体に富む形態または鏡像異性的もしくはジアステレオ異性的に純粋な形態を得ることができる。立体化学を、NMRカップリングのパターンに基づいて、任意選択により文献値と併せて割り当てることができる。絶対配置を、既知の立体配置のキラル前駆体からの合成によって、または結晶化した材料を使用するX線結晶解析による決定によって決定することができる。
【0165】
[0145] 立体化学的配置は、アミンを有する側鎖およびシクロアルキル環上の置換基の相対的な立体配置に従って定義される。2つ以上の置換基が存在する場合には、より高い順位(IUPAC)の置換基を使用して、立体化学的配置を決定する。
【0166】
[0146] 本発明の化合物は、以下に記載するスキームに従って合成することができる。所望の本発明の化合物を合成するために、スキーム中に示す例示的な試薬に代わる、適切な代替の試薬を選択することは、当業者の能力の範囲内である。また、必要に応じて合成の工程を除外または追加することも当業者の能力の範囲内である。
【0167】
2.シクロアルキルアミンの一般的な合成
[0147] 1つの実施形態では、本発明の化合物を、以下のスキーム1に示すように、対応するアミノケトンaから合成した。
【0168】
【化23】
【0169】
[0148] ジメチルアミノメチルシクロヘキサノンaを、アリールグリニャール試薬b〜dと縮合させて、ラセミ体のアミノアルコールを得た。ラセミ体の生成物を、半調製用のchiralpak ADカラム上でキラルクロマトグラフィーによって精製して、鏡像異性体1、24および28、ならびに2、26および27を得た。スキーム1中に開示したRyおよびRzの値の他に、RyおよびRzは、置換または非置換アルキル、Cl、Br、F、NR10R11、OR9、SR9、および置換または非置換アリールから独立して選択されるメンバーである。
【0170】
【化24】
【0171】
[0149] スキーム2を参照して、ラセミ体のシス−2−(アミノメチル)−1−(3,4−ジクロロフェニル)シクロヘキサノールを、キラルHPLCにより構成要素の異性体である8(動きがより速い鏡像異性体)と9(動きがより遅い鏡像異性体)とに分離することによって、2の親である一級アミン、すなわち、化合物9を得た。ギ酸、ホルムアルデヒドおよびシアン水素化ホウ素ナトリウムを用いる還元的アミノ化によって、9を2に変換した。また、ラセミ体のシス−1−(3,4−ジクロロフェニル)−2−((ジメチルアミノ)メチル)シクロヘキサノールのキラルHPLCよる分離後に、2を、動きがより遅い鏡像異性体としても得た;DEADおよび酸性EtOHを用いる2段階工程の手順において2を還元することによって、モノ−メチル誘導体4を得た。
【0172】
D.医薬組成物
[0150] 例示的な態様では、本発明は、本明細書に記載する化合物またはその薬学的に許容できる塩もしくは溶媒和化合物、および少なくとも1つの薬学的に許容できる担体を含む医薬組成物を提供する。種々の実施形態では、化合物は、シス異性体である。例示的な実施形態では、化合物は、式(I)〜(IX)から選択されるメンバーである構造を有する。
【0173】
[0151] 以下に詳細に記載するように、本発明の医薬組成物は、固体または液体の形態で投与するために特別に製剤化することができ、これには、経口投与に適合した形態、例えば、錠剤、水薬(水性もしくは非水性の液剤もしくは懸濁剤)、(静脈内および筋肉内を含めた)非経口投与または硬膜外注射に適合した形態、例えば、無菌の液剤または懸濁剤、あるいは持続放出性の製剤が含まれる。本発明の医薬組成物はまた、経皮投与のために特異的に製剤化することもできる。
【0174】
[0152] 本発明の医薬組成物は、経口的、非経口的、皮下的、経皮的、経鼻的に投与しても、または肛門から坐剤によって投与してもよい。本発明の医薬組成物はまた、制御送達デバイスを使用して投与してもよい。
【0175】
[0153] 本発明の製剤は、経口投与および非経口投与、特に、筋肉内投与、静脈内投与および皮下投与に適した製剤を含む。製剤は、単位投与剤型として好都合に提供することができ、薬学の技術分野で周知の任意の方法によって調製することができる。担体材料と組み合わせて、単一の投与剤型をもたらすことができる活性成分の量は、治療する宿主および特定の投与形態に依存して変化する。担体材料と組み合わせて、単一の投与剤型をもたらすことができる活性成分の量は、一般に、患者に対して毒性を示すことなく、治療効果をもたらす化合物の量である。一般に、この量は、百パーセント中、活性成分が、約1パーセントから約99パーセントまでに及ぶ。
【0176】
[0154] 例示的な単位投与製剤は、活性成分もしくはその薬学的に許容できる塩の有効用量、またはその適切な一部分を含有する製剤である。予防または治療に用いる用量の程度は、典型的には、治療しようとする状態の性質および重症度、ならびに投与経路によって異なる。また、用量、およびおそらく投与頻度も、個々の患者の年齢、体重および応答によって異なる。一般に、1日当たりの総投与量は(単回投与また分割投与として)、1日当たり約1mgから1日当たり約7000mgまで、好ましくは、1日当たり約1mgから1日当たり約100mgまで、より好ましくは、1日当たり約10mgから1日当たり約100mgまで、さらにより好ましくは、約20mgから、約100mgまで、約80mgまで、または約60mgまでの範囲である。いくつかの実施形態では、1日当たりの総投与量は、1日当たり約50mgから約500mgまで、好ましくは、1日当たり約100mgから約500mgまでの範囲であってよい。小児、65歳超の患者、および腎臓または肝臓の機能が損なわれている患者には、最初は、低い用量を投与し、投与量は、個人の応答および/または血中レベルに基づいて設定することがさらに推奨される。当業者には明らかになるであろうが、場合によっては、これらの範囲以外の投与量を使用する必要がある場合がある。さらに、臨床医または担当医であれば、個々の患者の応答に併せて、療法を中断、加減または終了する方法および時期が分かることにも留意されたい。
【0177】
[0155] 特定の実施形態では、本発明の製剤は、シクロデキストリン、リポソーム、ミセル形成物質、例えば、胆汁酸、ならびにポリマー性の担体、例えば、ポリエステルおよびポリ酸無水物からなる群から選択される賦形剤と;本発明の化合物とを含む。特定の実施形態では、上記の製剤によって、本発明の化合物は、経口的に体内に吸収され利用され得るようになる。
【0178】
[0156] これらの製剤または組成物を調製する方法は、本発明の化合物を、担体および任意選択により1つまたは複数の付属の成分と混ぜ合わせる工程を含む。一般に、本発明の化合物を、液体の担体もしくは微粉化した固体の担体または両方と均一かつ密接に混ぜ合わせ、次いで、必要に応じて、製品の形状となすことによって製剤を調製する。
【0179】
[0157] 経口投与に適した本発明の製剤は、カプセル剤、カシェ剤、丸剤、錠剤、カプレット剤、(芳香剤を添加した基剤、通常スクロースおよびアラビアゴムもしくはトラガカントを使用する)ドロップ剤、散剤、顆粒剤の形態であってもよく、あるいは水性もしくは非水性の液体中の液剤もしくは懸濁剤としても、または水中油型もしくは油中水型の乳濁剤としても、またはエリキシル剤もしくはシロップ剤としても、あるいは(ゼラチンおよびグリセリン等の不活性な基剤またはスクロースおよびアラビアゴムを使用する)芳香錠剤(pastille)としてもよく、それぞれが、あらかじめ決定した量の本発明の化合物を、活性成分として含有する。本発明の化合物はまた、ボーラス、舐剤またはペースト剤としても投与することができる。
【0180】
[0158] 経口投与のための本発明の固体の投与剤型(カプセル剤、錠剤、カプレット剤、丸剤、糖衣錠剤、散剤、顆粒剤等)においては、活性成分を、1つもしくは複数の薬学的に許容できる担体、すなわち、クエン酸ナトリウムもしくはリン酸二カルシウム等、ならびに/または以下のいずれかと混合する:(1)充填剤もしくは増量剤、すなわち、デンプン、ラクトース、スクロース、グルコース、マンニトール、シアル酸および/もしくはケイ酸等;(2)結合剤、すなわち、例えば、カルボキシメチルセルロース、アルギン酸、ゼラチン、ポリビニルピロリドン、スクロースおよび/もしくはアラビアゴム等:(3)湿潤剤、すなわち、グリセロール等;(4)崩壊剤、すなわち、寒天、炭酸カルシウム、バレイショデンプンもしくはタピオカデンプン、アルギン酸、特定のケイ酸塩および炭酸ナトリウム等;(5)溶解遅延剤、すなわち、パラフィン等;(b)吸収促進剤、すなわち、四級アンモニウム化合物等;(7)湿潤剤、すなわち、例えば、セチルアルコール、モノステアリン酸グリセロールおよび非イオン性界面活性剤等;(8)吸収剤、すなわち、カオリンおよびベントナイトクレイ等;(9)滑沢剤、すなわち、タルク、ステアリン酸カルシウム、ステアリン酸マグネシウム、固体のポリエチレングリコール、ラウリル硫酸ナトリウムおよびそれらの混合物等;ならびに(10)着色剤。カプセル剤、錠剤および丸剤の場合には、医薬組成物はまた、緩衝剤も含むことができる。また、類似の型の固体の組成および高分子量のポリエチレングリコール等も、ラクトースまたは乳糖等の賦形剤を使用する軟質および硬質の殻のゼラチンカプセル剤中で、充填剤として利用することができる。
【0181】
[0159] 錠剤は、任意選択により1つまたは複数の付属の成分と共に、圧縮または成型によって作製することができる。圧縮錠剤は、結合剤(例えば、ゼラチンまたはヒドロキシプロピルメチルセルロース)、滑沢剤、不活性な希釈剤、保存剤、崩壊剤(例えば、デンプングリコール酸ナトリウムもしくは架橋カルボキシメチルセルロースナトリウム)、表面活性剤または分散剤を使用して調製することができる。成型錠剤は、不活性な液体の希釈剤を用いて湿潤させた粉末化合物の混合物を、適切な機械中で成型することによって作製することができる。
【0182】
[0160] 本発明の医薬組成物の錠剤およびその他の固体の投与剤型、すなわち、糖衣錠剤、カプセル剤、丸剤および顆粒剤等は、任意選択により、腸溶コーティングおよび医薬品製剤技術分野で周知のその他の被覆等の被覆および殻を用いて収容するかまたはそうした被覆および殻を有するように調製することができる。これらの固体の投与剤型はまた、その中の活性成分を緩慢にまたは制御して放出するように、例えば、所望の放出プロファイルをもたらすために様々な比率のヒドロキシプロピルメチルセルロースを使用して、その他のポリマー性マトリックス、リポソームおよび/またはマイクロスフェアを使用して製剤化することもできる。これらの固体の投与剤型は、急速に放出するように、例えば、凍結乾燥して製剤化することができる。これらの固体の投与剤型は、例えば、細菌を保持するフィルターを通すろ過によって、または使用直前に無菌水もしくは何らかのその他の無菌の注射用媒体中に溶解させることができる無菌の固体組成物の形態中に滅菌剤を組み込むことによって滅菌することができる。これらの組成物はまた、任意選択により隠蔽剤を含有することができ、消化管の特定の部分に限ってまたはそこで優先的に、(1つまたは複数の)活性成分を、任意選択により遅延させて放出することもできる。使用することができる組成物を包埋する例として、ポリマー性物質およびろうが挙げられる。活性成分はまた、適切な場合には、1つまたは複数の上記に記載した賦形剤と共に、マイクロカプセルの形態となすこともできる。
【0183】
[0161] 本発明の化合物の経口投与のための液体の投与剤型には、薬学的に許容できる乳剤、マイクロエマルション、液剤、懸濁剤、シロップ剤およびエリキシル剤がある。活性成分に加えて、液体の投与剤型は、当技術分野で一般に使用される不活性な希釈剤、すなわち、例えば、水またはその他の溶媒等、可溶化剤ならびに乳化剤、すなわち、エチルアルコール、イソプロピルアルコール、炭酸エチル、酢酸エチル、ベンジルアルコール、安息香酸ベンジル、プロピレングリコール、1,3−ブチレングリコール、油(特に、綿実油、落花生油、トウモロコシ油、胚芽油、オリーブ油、ヒマシ油およびゴマ油)、グリセロール、テトラヒドロフリルアルコール、ポリエチレングリコールならびにソルビタンの脂肪酸エステル、さらにそれらの混合物等も含有することができる。
【0184】
[0162] 不活性な希釈剤の他に、経口用組成物はまた、アジュバント、すなわち、湿潤剤、乳化剤および懸濁化剤、甘味剤、芳香剤、着色剤、矯臭剤ならびに保存剤等も含むことができる。
【0185】
[0163] 懸濁剤は、活性化合物に加えて、懸濁化剤、例えば、エトキシ化イソステアリルアルコール、ポリオキシエチレンソルビトールおよびソルビタンエステル、微結晶セルロース、メタ水酸化アルミニウム、ベントナイト、寒天ならびにトラガカント、さらにそれらの混合物も含有することができる。
【0186】
[0164] 非経口投与に適した本発明の医薬組成物は、1つまたは複数の本発明の化合物を、1つまたは複数の薬学的に許容できる無菌の等張な水性または非水性の液剤、分散剤、懸濁剤もしくは乳剤、または使用直前に無菌の注射用の液剤もしくは分散剤の中に再構成することができる無菌の散剤と組み合わせて含むことができる。こうした注射用の液剤または分散剤は、糖、アルコール、抗酸化剤、緩衝剤、静菌剤、意図するレシピエントの血液と処方を等張にする溶質、または懸濁化剤もしくは増粘剤を含有する場合がある。
【0187】
[0165] 本発明の医薬組成物において利用することができる、適切な水性および非水性の担体の例として、水、エタノール、ポリオール(グリセロール、プロピレングリコール、ポリエチレングリコール等)およびそれらの適切な混合物、オリーブ油等の植物油、ならびにオレイン酸エチル等の注射用の有機エステルが挙げられる。適切な流動性を、例えば、レシチン等の被覆材料を使用することによって、分散剤の場合には必要な粒子サイズを維持することによって、および界面活性剤を使用することによって維持することができる。
【0188】
[0166] これらの組成物はまた、アジュバント、すなわち、保存剤、湿潤剤、乳化剤および分散剤等も含有することができる。微生物の対象化合物に対する作用を、種々の抗細菌剤および抗真菌剤、例えば、パラベン、クロロブタノール、フェノールソルビン酸等を含めることによって確実に予防することができる。また、糖、塩化ナトリウム等の等張化剤を、組成物中に含めることが望ましい場合もある。さらに、注射用の医薬品の形態の吸収の延長を、モノステアリン酸アルミニウムおよびゼラチン等の吸収を遅延させる薬剤を含めることによってもたらすこともできる。
【0189】
[0167] 場合によっては、薬物の効果を延長させるために、皮下または筋肉内の注射からの薬物の吸収を遅くすることが望ましい。これは、水溶性の低い結晶性または非結晶性の材料を液体中に懸濁させることによって達成することができる。薬物の吸収速度は、この場合、その溶解速度に依存し、当該溶解速度は、今度は、結晶サイズおよび結晶形態に依存し得る。あるいは、非経口投与用の薬物の形態の吸収遅延は、薬物を油性のビヒクル中に溶解または懸濁させることによっても達成される。
【0190】
[0168] 注射用デポーの形態を、対象化合物のマイクロカプセルのマトリックスを、ポリ乳酸−ポリグリコリド等の生分解性ポリマー中に形成することによって作製することができる。薬物のポリマーに対する比および利用する特定のポリマーの性質に依存して、薬物放出の速度を制御することができる。その他の生分解性ポリマーの例として、ポリ(オルトエステル)およびポリ(酸無水物)が挙げられる。また、デポー注射用製剤は、身体組織に適合するリポソームまたはマイクロエマルションの中に薬物を捕捉することによっても調製される。作用延長型の錠剤の形態をとる本発明の医薬組成物または単位投与剤型は、薬物治療を一定期間にわたってもたらすように、薬物物質を放出するように製剤化された圧縮錠剤を含むことができる。薬物物質の放出が、投与後の一定期間にわたりまたは特定の生理的状態が存在するようになるまで阻止される作用遅延型の錠剤を含む、いくつかの錠剤の型がある。薬物物質の完全な用量を消化液に定期的に放出する作用反復型の錠剤を形成することができる。また、含有する薬物物質の一定量を消化液に連続的に放出する徐放性の錠剤も形成することができる。
【0191】
[0169] 本発明の化合物はまた、制御放出手段によっても、または当業者によく知られている送達デバイスによっても投与することができる。例として、これらに限定されないが、米国特許第3,845,770号;第3,916,899号;第3,536、809号;第3,598,123号;および第4,008,719号、第5,674,533号、第5,059,595号、第5,591,767号、第5,120,548号、第5,073,543号、第5,639,476号、第5,354,556号、および第5,733,566号に記載されているものが挙げられる。これらはそれぞれ、参照により本明細書に組み込まれている。そのような投与剤型を使用して、例えば、ヒドロキシプロピルメチルセルロース、その他のポリマーのマトリックス、ゲル、透過性の膜、浸透圧によるシステム、多層被覆、微小粒子、リポソーム、マイクロスフェア、またはそれらの組合せを使用して、様々な比率で所望の放出プロファイルをもたらして、1つまたは複数の活性成分の緩除放出または制御放出を得ることができる。本明細書に記載するものを含めて、当業者に既知の適切な制御放出性の処方を容易に選択して、発明の化合物に関して使用することができる。したがって、本発明は、これらに限定されないが、制御放出するようになされた錠剤、カプセル剤、ジェルキャップ剤およびカプレット剤等の経口投与に適した単一の単位投与剤型を包含する。
【0192】
[0170] 全ての制御放出性の医薬製品は、それらの非制御性の対応物によって達成されたものを上回るように薬物療法を改善するという共通の目標を有する。理想的には、最適に設計された制御放出性の調製物の医学的治療における使用は、最小の薬物物質を利用して、最短時間で状態を治癒または制御することによって特徴付けられる。制御放出性の製剤の利点には、薬物活性の長期化、投与頻度の低下、および患者のコンプライアンスの向上がある。さらに、制御放出性の製剤を使用して、作用の開始時期、または薬物の血中レベル等のその他の特徴に影響を及ぼすことができ、したがって、副作用(例えば、有害作用)の発生率に影響を及ぼすことができる。
【0193】
[0171] 大部分の制御放出性の製剤は、所望の治療効果を即座にもたらす薬物(活性成分)の量を最初に放出し、このレベルの治療または予防の効果を長期の期間にわたって維持するための薬物の別の量を次第にかつ連続的に放出するように設計される。体内でこの一定のレベルの薬物を維持するためには、投与剤型から薬物が、代謝され身体から排泄される薬物の量を置換する速度で放出されなければならない。活性成分の制御放出を、これらに限定されないが、pH、温度、酵素、水もしくはその他の生理的条件を含めた種々の条件、または化合物によって刺激することができる。
【0194】
[0172] 本発明の化合物はまた、経皮から、外用および粘膜からの投与剤型として製剤化することもでき、こうした剤型には、これらに限定されないが、眼科用液剤、スプレー剤、エアロゾル剤、クリーム剤、ローション剤、軟膏剤、ジェル剤、液剤、乳剤、懸濁剤、または当業者に既知のその他の剤型がある。例えば、Remington’s Pharmaceutical Sciences,16thおよび18th eds.,Mack Publishing,Easton PA(1980および1990);ならびにIntroduction to Pharmaceutical Dosage Forms,4th ed.,Lea & Febiger,Philadelphia(1985)を参照されたい。経皮投与剤型には、「リザーバ型」または「マトリックス型」のパッチがあり、これを皮膚に適用し、特定の期間にわたり着用して、所望の量の活性成分を浸透させる。
【0195】
[0173] 本発明が包含する経皮から、外用および粘膜からの投与剤型を得るために使用することができる適切な賦形剤(例えば、担体および希釈剤)ならびにその他の材料は、薬学の技術分野の業者にはよく知られており、所与の医薬組成物または投与剤型が適用される特定の組織によって異なる。
【0196】
[0174] 治療しようとする特異的な組織に依存して、追加の構成成分を、本発明の活性成分を用いる治療の前に、治療に併せて、または治療の後に使用することができる。例えば、浸透増強剤を使用して、活性成分の組織への送達を支援することができる。
【0197】
[0175] また、医薬組成物もしくは投与剤型のpH、または医薬組成物もしくは投与剤型を適用する組織のpHを加減して、1つまたは複数の活性成分の送達を改善することもできる。同様に、溶媒担体の極性、そのイオン強度またはその浸透圧を加減して、送達を改善することもできる。また、ステアリン酸等の化合物を、医薬組成物または投与剤型に添加して、送達を改善するように、1つまたは複数の活性成分の親水性または親油性を好都合に変化させることもできる。この場合、ステアリン酸が、製剤のための脂質ビヒクルとして、乳化剤または界面活性剤として、および送達増強剤または浸透増強剤として役立つ場合がある。活性成分の異なる塩、水和物または溶媒和化合物を使用して、得られた組成物の特性をさらに加減することができる。
【0198】
[0176] 本発明の化合物を医薬品として、ヒトおよび動物に投与する場合、それ自体で、または薬学的に許容できる担体と組み合わせて、例えば、活性成分を0.1〜99.5%を含有する医薬組成物として与えることができる。
【0199】
[0177] 本発明の調製物は、経口的および非経口的に与えることができる。もちろん、これらは、各投与経路に適した剤型で与えられる。それらは、例えば、錠剤またはカプセル剤の形態で、注射により、および静脈内投与により投与される。1つの実施形態では、経口投与が好ましい。
【0200】
[0178] 「非経口投与」および「非経口的に投与した」という句は、本明細書で使用する場合、腸内投与および外用投与以外の通常注射による投与形態を意味し、静脈内、筋肉内、動脈内、くも膜下腔内、関節内、眼窩内、心臓内、皮内、腹腔内、経気管、皮下、角質下、関節腔内、嚢下、くも膜下、脊髄内および胸骨内への注射および注入を非限定的に含む。
【0201】
[0179] 選択する投与量のレベルは、利用される特定の本発明の化合物またはそのエステル、塩もしくはアミドの活性、投与経路、投与の時期、利用される特定の化合物の排泄および代謝の速度、治療期間、利用される特定の化合物と併用するその他の薬物、化合物および/または材料、治療する患者の年齢、性別、体重、状態、全般的な健康状態および過去の病歴、ならびに医学の技術分野で周知の類似の要因を含む多様な要因に依存する。
【0202】
[0180] 当技術分野の通常の技術を有する医師または獣医師であれば、必要とされる医薬組成物の有効量を容易に決定し、処方することができる。例えば、医師または獣医師は、所望の治療効果を達成するために必要な用量よりも低いレベルの、医薬組成物中で利用される本発明の化合物の用量から開始し、投与量を所望の効果を達成するまで次第に増加させることができるであろう。
【0203】
[0181] 一般に、本発明の化合物の適切な1日当たりの用量は、治療効果をもたらすために有効な最も低い用量である化合物の量である。そのような有効用量は一般に、上記に記載した要因に依存する。一般に、本発明の化合物の患者に対する経口、静脈内、側脳室内および皮下への投与量は、1日当たり、体重1キログラム当たり約0.005mgから体重1キログラム当たり約5mgまでの範囲である。例示的な実施形態では、本発明の化合物の経口投与量は、1日当たり約10mgから約300mgまでの範囲である。例示的な実施形態では、本発明の化合物の経口投与量は、1日当たり約20mgから約250mgまでの範囲である。例示的な実施形態では、本発明の化合物の経口投与量は、1日当たり約100mgから約300mgまでの範囲である。例示的な実施形態では、本発明の化合物の経口投与量は、1日当たり約10mgから約100mgまでの範囲である。例示的な実施形態では、本発明の化合物の経口投与量は、1日当たり約25mgから約50mgまでの範囲である。例示的な実施形態では、本発明の化合物の経口投与量は、1日当たり約50mgから約200mgまでの範囲である。上記に列挙した投与量の範囲のそれぞれを、単位投与製剤として製剤化することができる。
【0204】
[0182] 「治療」または「治療する」という用語は、療法、再発の予防および急性症状の緩和を包含するものとする。「治療する」は、症状の緩和および基底状態の消散のいずれかまたは両方を指すことに留意されたい。本発明の状態の多くにおいては、本発明の化合物または組成物の投与が、疾患状況に対して直接的ではなく、むしろ何らかの悪質な症状に対して作用する場合があり、そうした症状の改善が、疾患状況の全般的なかつ望ましい緩和をもたらす。また、本発明の化合物を使用して、疾患を阻止することもできる(予防)。
【0205】
[0183] この治療を受ける患者は、霊長類、特にヒト、ならびにウマ、ウシ、ブタおよびヒツジ等のその他の哺乳動物を、かつ家禽および一般的なペットを含むそれを必要とする任意の動物である。
【0206】
[0184] 本発明の化合物および医薬組成物は、その他の医薬品、例えば、抗菌剤、すなわち、ペニシリン、セファロスポリン、アミノグリコシドおよびグリコペプチド等と併用して投与することができる。したがって、併用療法は、活性化合物を、最初に投与した薬剤の治療効果が、それに続く薬剤を投与するときに、完全には消失していないように、逐次、同時および個別に投与することを含む。
【0207】
[0185] 例示的な実施形態では、本発明の化合物が治療効果を示す適応症を呈する対象は、それ以外のためには、本発明の化合物または本発明の化合物を包含する構造の属に属する化合物を用いた治療を必要としない。
【0208】
IV.方法
A.モノアミン輸送体に対する結合
[0186] 種々の態様では、本発明は、本発明の化合物をモノアミン輸送体に対して結合させる方法を提供する。この方法は、モノアミン輸送体と、本発明の化合物とを接触させることを含む。
【0209】
[0187] 本発明は、モノアミン輸送体リガンドの、モノアミン輸送体(セロトニン輸送体、ドーパミン輸送体およびノルエピネフリン輸送体等)に対する結合を阻害する方法をさらに提供する。この方法は、モノアミン輸送体と、本発明の化合物とを接触させることを含む。例示的な実施形態では、モノアミン輸送体リガンドは、セロトニン、ドーパミンまたはノルエピネフリン等の内因性モノアミンである。別の例示的な実施形態では、リガンドは、モノアミン輸送体に対する結合親和性を有することが知られている薬物分子または別の小型分子である。別の例示的な実施形態では、モノアミン輸送体リガンドは、モノアミン輸送体に対して結合することが知られている、放射性活性物質で標識した化合物である。
【0210】
[0188] 例示的な実施形態では、本明細書に記載するもの等のex vivoにおける結合アッセイを使用して、リガンド結合の阻害を示す。例示的な実施形態では、本発明の化合物は、ビヒクルと比較して、平均結合を、約1%〜約100%、好ましくは約10%〜約100%、より好ましくは約20%〜約90%阻害する。平均結合の阻害は、好ましくは、用量依存性である。
【0211】
B.モノアミン輸送体活性の阻害
[0189] 種々の実施形態では、本発明は、セロトニン輸送体、ドーパミン輸送体およびノルエピネフリン輸送体等、少なくとも1つのモノアミン輸送体の活性を調節する(例えば、阻害する、増大させる)方法を提供する。この方法は、モノアミン輸送体と、本発明の化合物とを接触させることを含む。例示的な実施形態では、治療有効量の本発明の化合物またはその薬学的に許容できる塩もしくは溶媒和化合物を対象に投与することによって、モノアミン輸送体を本発明の化合物と接触させる。対象はヒトであってよい。例示的な実施形態では、モノアミン輸送体は、ドーパミン輸送体(DAT)、セロトニン輸送体(SERT)またはノルエピネフリン輸送体(NET)である。種々の例示的な実施形態では、本発明の化合物は、少なくとも2つの異なるモノアミン輸送体の活性を阻害する。モノアミン輸送体活性の阻害は、当技術分野で既知のアッセイを使用して測定することができる。例示的なアッセイ形式として、in vitroにおける機能的取り込みアッセイが挙げられる。例示的な実施形態では、機能的取り込みアッセイは、所望のモノアミン輸送体を発現する適切な細胞系を活用する。種々の例示的な実施形態では、機能的取り込みアッセイは、適切な生物の脳組織から単離したシナプトソームを活用する。あるいは、モノアミン輸送体活性の阻害は、当技術分野で既知の、例えば、適切な膜の調製物を活用する受容体結合実験を使用して評価することができる。例示的なアッセイでは、本明細書に記載するように、試験対象(例えば、ラット)を、本発明の化合物、および基準化合物を用いて処理し、それに続いて、脳組織を単離し、受容体の占有率をex vivoにおいて解析する。
【0212】
C.モノアミン取り込みの阻害
[0190] 種々の態様では、本発明は、少なくとも1つのモノアミン(例えば、ドーパミン、セロトニン、ノルエピネフリン)の細胞による取り込みを阻害する方法を提供する。この方法は、細胞を本発明の化合物と接触させることを含む。例示的な実施形態では、細胞は、神経細胞またはグリア細胞等の脳細胞である。1つの例では、モノアミン取り込みの阻害をin vivoにおいて起こす。生物中では、ドーパミンまたはセロトニン等のモノアミンの、例えば、シナプス間隙からの神経細胞による取り込み(また、再取り込みとも呼ばれる)が起きる。したがって、1つの実施形態では、神経細胞を、哺乳動物のシナプス間隙と接触させる。別の例示的な実施形態では、モノアミン取り込みの阻害を、in vitroにおいて起こす。これらの方法では、細胞は、脳細胞、すなわち、組換えのモノアミン輸送体を発現する神経細胞または細胞型等であってよい。
【0213】
[0191] 1つの実施形態では、化合物は、少なくとも2つの異なるモノアミンの取り込みを阻害する。これは、例えば、複数の異なるモノアミン輸送体を同時に発現する細胞型(単離したシナプトソーム等)を活用する種々のin vitroにおける機能的取り込みアッセイを実施することによって示すこともでき、またはそれぞれが異なるモノアミン輸送体、すなわち、組換えのドーパミン輸送体等を発現する、2つの異なる細胞型を、適切な、標識したモノアミンと一緒に使用することによって示すこともできる。以下の本明細書に記載するもの等の機能的モノアミン取り込みアッセイにおいて、阻害薬(例えば、本発明の化合物)が、約0.1nM〜約10μM、好ましくは約1nM〜約1μM、より好ましくは約1nM〜約500nM、さらにより好ましくは約1nM〜約100nMのIC50を示す場合に、モノアミン取り込みの阻害が実証される。
【0214】
D.神経障害の治療
[0192] 別の態様では、本発明は、神経障害を、少なくとも1つのモノアミン輸送体の活性を阻害することによって治療する方法を提供する。この方法は、治療有効量の本発明の組成物、あるいは化合物またはその薬学的に許容できる塩もしくは溶媒和化合物を、その治療を必要とする対象に投与することを含む。例示的な実施形態では、哺乳動物の対象はヒトである。別の例示的な実施形態では、本発明の化合物は、少なくとも2つの異なるモノアミン輸送体の活性を阻害する。例えば、本発明の化合物は、セロトニン輸送体、ドーパミン輸送体およびノルエピネフリン輸送体のうちの少なくとも2つの活性を阻害する。モノアミン輸送体の活性の阻害は、以下の本明細書に記載するように、機能的モノアミン取り込みアッセイによって示すことができる。
【0215】
[0193] 化合物の活性を、種々の当技術分野で認識されている動物モデルにおいて実証することができる。例えば、本発明の化合物の抗うつ薬活性は、うつ病の適切な動物モデル、すなわち、ラットの強制水泳試験、マウスの尾の懸垂試験、およびラットの自発運動解析等を活用することによって示すことができる。ラットの強制水泳試験はまた、2つ以上のモノアミン輸送体に対して活性(混合性モノアミン輸送体活性)を有する化合物の解析にも適している。例えば、泳ぐ活動の増加は、セロトニン再取り込みの阻害を示し、一方、登る活動の増加は、ノルエピネフリン再取り込みの阻害を示す。
【0216】
[0194] 種々の実施形態では、本発明の化合物は、少なくとも1つの動物モデルにおいて活性を示し、これを使用して、化合物の活性を測定し、神経障害を治療する場合におけるそれらの効能を推定することができる。例えば、動物モデルが、うつ病(例えば、無動)についてである場合、本発明の化合物は、少なくとも1つの動物モデルにおいて、平均的な無動(immobility)を阻害する場合には、ビヒクルと比較して、約5%〜約90%、好ましくは約10%〜約70%、より好ましくは約10%〜約50%、より好ましくは約15%〜約50%活性である。種々の実施形態では、本発明の化合物は、処理動物とビヒクル投与動物との間において、測定したエンドポイントの類似した差異をもたらす。
【0217】
[0195] 種々の実施形態では、本発明は、抗うつ薬様の効果をもたらす方法を提供する。この方法は、治療有効量の本発明の化合物または組成物またはその薬学的に許容できる塩もしくは溶媒和化合物を、それを必要とする哺乳動物の対象に投与することを含む。抗うつ薬様の効果は、本明細書に記載するもの等の疾患の動物モデルを使用して測定することができる。
【0218】
[0196] 種々の例示的な実施形態では、神経障害は、うつ病(例えば、大うつ病性障害、双極性障害、単極性障害、気分変調症および季節性感情障害)、失認、線維筋痛症、疼痛(例えば、神経因性疼痛)、精神医学的状態によって生じる睡眠障害を含む睡眠関連障害(例えば、睡眠時無呼吸、不眠症、ナルコレプシー、カタプレキシー)、慢性疲労症候群、注意欠陥障害(ADD)、注意欠陥多動性障害(ADHD)、下肢静止不能症候群、統合失調症、不安症(例えば、全般性不安障害、社会不安障害、パニック障害)、強迫性障害、心的外傷後ストレス障害、季節性感情障害(SAD)、月経前不安、閉経後の血管運動性症状(例えば、一過性熱感、寝汗)、ならびに神経変性疾患(例えば、パーキンソン病、アルツハイマー病および筋萎縮性側索硬化症)、躁状態、気分変調性障害、気分循環性障害、肥満、ならびに物質乱用または依存(例えば、コカイン耽溺、ニコチン耽溺)からなる群から選択されるメンバーである。例示的な実施形態では、神経障害は、大うつ病性障害等のうつ病である。例示的な実施形態では、本発明の化合物は、失認とうつ病等、併存症である2つの状態/障害を治療するために有用である。
【0219】
[0197] 神経障害は、老人性認知症、アルツハイマー型認知症、認知、記憶の喪失、健忘症/健忘症候群、てんかん、意識の撹乱、昏睡、注意力の低下、会話障害、レノックス症候群、自閉症および多動症候群を含む脳機能の障害を非限定的に含む。
【0220】
[0198] 神経因性疼痛は、ヘルペス後(帯状疱疹後)神経痛、反射性交換神経性ジストロフィー/灼熱痛または神経の外傷、幻肢痛、手根管症候群および末梢性ニューロパシー(糖尿病性ニューロパシーまたは慢性のアルコールの使用によって生じるニューロパシー等)を非限定的に含む。
【0221】
[0199] 本発明の方法を使用して治療することができるその他の例示的な疾患および状態として、肥満;偏頭痛または偏頭痛性の頭痛;不随意性の排尿、尿の滴下または漏れ、腹圧性尿失禁(SUI)、切迫性尿失禁、労作性尿失禁、反射性失禁、受動性失禁および溢流性失禁を非限定的に含めた、尿失禁;さらに、心理的および/または生理的な要因が引き起こす性機能障害、勃起不全、早漏、膣の乾燥、性的興奮の欠如、オルガスム達成喪失を非限定的に含めた、男性または女性における性機能障害、ならびに抑制された性欲、抑制された性的興奮、抑制された女性のオルガスム、抑制された男性のオルガスム、機能性性交疼痛症、機能性膣痙および非定型性の精神性的機能障害を非限定的に含めた、精神性的機能障害が挙げられる。
【0222】
[0200] 例示的な実施形態では、神経障害は肥満であり、患者に供給する化合物の治療有効量は、前記患者が満腹感を抱くのに十分な量である。
【0223】
[0201] 例示的な実施形態では、本明細書に記載する化合物は、前記化合物に対する耽溺を引き起こすことなく、中枢神経の障害を治療/予防する。
【0224】
[0202] 以下の実施例を、本発明の例示的な特徴を例証するために提供する。
【実施例】
【0225】
[0203] 以下の実施例は、選択した本発明の実施形態を例証するために提供されており、これらを、本発明の範囲を限定するものであると解釈してはならない。
【0226】
(実施例1)
1a.手順
[0204] 以下の実施例中、別段に特筆しない限り、以下の一般的な実験手順を使用した。全ての市販の試薬は、さらなる精製なしで使用した。無水反応を、火炎乾燥したガラス器具中、N2下で実施した。NMRスペクトルを、Varian製400MHz分光計上、トリメチルシラン(TMS)を内部標準として有する重クロロホルムまたはメタノール−d4中で記録した。ISCO Combiflashシステムを使用し、254nmにおいて検出するか、またはISCOの順相シリカゲルカートリッジを使用して、シリカゲルカラムクロマトグラフィーを実施した。
【0227】
1b.解析HPLC
[0205] 解析HPLCを、Agilent製Zorbax RX−C18 5μm、4.6×250mmカラムに接続したHewlett Packard Series 1100ポンプ上で実施し、Hewlett Packard Series 1100 UV/Vis検出器上で、214nmおよび254nmでモニターして検出した。典型的な流速=1ml/分。3本の異なるHPLCカラム、および種々の溶出プロトコールを使用した。例えば、(1)直線的勾配で流す、Agilent製Zorbax RX−C18 5μm、4.6×250mmカラム。溶媒A=0.05%TFAを有するH2O、溶媒B=0.05%TFAを有するMeCN。時間0分=5%溶媒B、時間4分=40%溶媒B、時間8分=100%溶媒B、12分=5%溶媒B、20分=5%溶媒B;(2)3分で5→100%でB(アセトニトリル/0.1%ギ酸)および溶媒A(水/0.1%ギ酸)の勾配を流す、Phenomenex 3μ C18カラム;(3)5分で5→100%でBの勾配を流す(溶媒B(アセトニトリル/0.1%ギ酸)および溶媒A(水/0.1%ギ酸))、Phenomenex 5μ C18カラム。
【0228】
1c.逆相HPLCによる精製
[0206] 逆相HPLCによる精製を、Gilson製のシステム上で、Phenomenex 5μ C18(50×21.2mm)カラムを使用して実施した。標準的な分離方法は、溶媒A(水/0.1%ギ酸)中、10→100%B(アセトニトリル/0.1%ギ酸)の10分の勾配であった。典型的には、粗試料を、MeOH中に溶解させた。画分を、Genovac(低圧における遠心分離)によって濃縮した。
【0229】
1d.GC−MS
[0207] ガスクロマトグラフィーを、Hewlett Packard 6890 Series GC System上で、Hewlett Packard 5973 Series Mass Selective Detectorに連結したHP1カラム(30メートル、0.15μの膜の厚さ)を用いて実施した。以下の直線的温度勾配を使用した:100℃5分間、次いで、20℃/分で320℃まで。320℃で10分間保持。
【0230】
1e.LCMS
[0208] LCMSを、Micromass Platform LCに接続したAgilent 1100 Seriesシステム上で実施した。以下のカラムおよび勾配を使用した:カラム:Luna C18(2)、3μm粒子サイズ、30×2.0mmのカラム寸法。流速=0.5mL/分、溶媒A=95%H2O、5%MeOH中の0.1M NH4Ac、pH6.0、溶媒B=MeOH中の溶媒B:0.1M NH4Ac。6回注入する直線的勾配:時間0分=100%溶媒A、時間10分=100%溶媒B、時間12分=100%溶媒B、時間12分10秒=100%溶媒A、時間14分=100%溶媒A、時間14分20秒=100%溶媒A。
【0231】
1f.マイクロ波(μW)による再結晶
[0209] 粗塩(例えば、HCl塩)を、撹拌棒を有するマイクロ波用の槽に入れた。再結晶用溶媒を添加し、槽を、標的温度で所与の時間にわたり加熱した。反応器中で、槽を50℃まで冷却し、次いで、取り出し、RTまでゆっくり冷却した。典型的には、N,N−ジメチルアミンを、EtOAcまたはEtOAc:CH3CN(2:1)中で再結晶させた。典型的には、N−Meまたは一級アミンは、CH3CN中で再結晶した。
【0232】
(実施例2)
2a.実験手順および特徴付けのデータ
【0233】
【化25】
[0210] THF(20mL)中のケトンa(2.0g、13mmol)の溶液に、−78℃で、3,4−ジクロロフェニルマグネシウムブロミド(THF中、0.5M、38mL、19mmol)を添加した。反応混合液を−78℃で30分間撹拌してから、30分かけて0℃まで加温した。NH4Clの飽和溶液(30mL)を、反応混合液に添加して、反応をクエンチした。得られた生成物を、ジエチルエーテルを用いて抽出した(2×100mL)。組み合わせた抽出物を乾燥および濃縮した。残渣に対して、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン/Et3N=1:10:0.1)を行って、1と2とのラセミ混合物を得た(3.5g、90%)。このラセミ混合物を、キラルADカラム(溶離液として、ヘキサン/iPrOH/DEA=95/5/0.1)によって分離して、純粋な1(動きがより速い鏡像異性体)と2(動きがより遅い鏡像異性体)とを得た。
【0234】
2a1.1/2についてのデータ
[0211] 1H NMR(400MHz,CD3OD)δ7.73(d,J=1.6Hz,1H)、7.52(d,J=8.4Hz,1H)、7.47(dd,J=1.6,8.4Hz,1H)、3.01(dd,J=13.2,10.4Hz,IH)、2.76(s,3H)、2.65(s,3H)、2.57(dd,J=2.0,13.2Hz,1H)、2.28(m,2H)、1.9(m,2H)、1.70(m,2H)、1.6(m,2H);13C NMR(100MHz,CD3OD)δ148.86、132.32、130.32、127.49、125.08、74.11、60.22、45.03、41.22、40.29、25.71、24.64、21.16;ESI MS m/z 302.1、304.0。
【0235】
2b.シクロアルキルアミンの脱アルキル反応
【0236】
【化26】
[0212] トルエン中の2(0.8g、2.65mmol)の溶液に、DEAD(0.69g、0.63mL、3.96mmol)を添加した。反応混合液を100℃で4時間加熱してから、濃縮した。残渣をEtOH30mL中に溶解させ、NH4Clの飽和溶液(30mL)を添加した。反応混合液を50℃で6時間撹拌してから、濃縮した。NaOH溶液(2M、10mL)を、得られた混合液に添加し、生成物を、ジエチルエーテルを用いて抽出した(2×80mL)。組み合わせた抽出物を乾燥および濃縮した。残渣を、逆相カラムクロマトグラフィー(CH3CN/H2O=5/95〜95/5)により精製して、4を得た(0.32g、42%)。
【0237】
2b1.4についてのデータ
[0213] 1H NMR(400MHz,CD3OD)δ7.60(s,1H)、7.37(m,2H)、2.57(dd,J=2.0,12.4Hz,1H)、2.28(dd,J=2.8,12.4Hz,1H)、2.23(s,3H)、1.88(m,2H)、1.78(m,2H)、1.62(m,1H)、1.56(m,2H)、1.40(m,1H);13C NMR(100MHz,CD3OD)δ150.86、132.40、130.21、130.09、127.56、124.77、77.03、53.54、43.95、40.82、36.81、26.45、26.05、22.05;ESI MS m/z 288.1。
【0238】
2c.シクロヘキサノンからのシクロヘキシルアミンの合成
【0239】
【化27】
[0214] H2O(50mL)中のシクロヘキサノン(23.7g、25.0mL、0.242モル)の溶液に、HCHO(37%、37.5mL、0.46モル)およびK2CO3(0.52g、3.76mmol)を添加した。反応混合液を60℃で3時間撹拌した。次いで、生成物を、ジエチルエーテルを用いて抽出した(2×300mL)。組み合わせた抽出物を乾燥および濃縮した。残渣を、シリカゲルカラムクロマトグラフィー(酢酸エチル/ヘキサン=l:7〜1:2)により精製して、kを得た(10.8g、35%)。
【0240】
[0215] THF(60mL)中のk(3.2g、25mmol)の溶液に、−20℃で、3,4−ジクロロフェニルマグネシウムブロミド溶液(0.5M、100mL、50mmol)を添加した。反応混合液を30分間撹拌してから、NH4Clの溶液(20mL)によりクエンチした。次いで、生成物を、ジエチルエーテルを用いて抽出した(2×100mL)。組み合わせた抽出物を乾燥および濃縮した。残渣を、シリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル=l:7〜1:2)により精製して、mを得た(2.1g、31%)。
【0241】
[0216] THF(40mL)中のm(1.6g、5.8mmol)の溶液に、r.t.で、PPh3(1.8g、7.0mmol)、DEAD(1.2g、7.0mmol)およびジフェニルホスホラジデート(DPPA)(1.9g、7.0mmol)を添加した。得られた黄色の溶液を、一晩撹拌してから、濃縮した。残渣に対して、シリカゲルカラムクロマトグラフィー(ヘキサン/酢酸エチル)=1:10〜1:1を行って、所望の生成物nを得た(1.32g、74%)。
【0242】
[0217] THF(30mL)中のn(1.00g、3.34mmol)の溶液に、PPh3(1.75g、6.68mmol)を添加した。反応混合液を、24時間撹拌してから、H2O(10mLを添加した。得られた混合液を、さらに2日間撹拌してから、濃縮した。残渣に対して、逆相カラムクロマトグラフィー(CH3CN/H2O=5/95〜95/5)を行って、所望の生成物oを得た(0.75g、82%)。このラセミ混合物を、キラルADカラムにより、(エタノール/メタノール/へキサン/DEA=3/2/95/0.1)を用いて分離して、純粋な鏡像異性体8(動きがより速い鏡像異性体)と9(動きがより遅い鏡像異性体)とを得た。
【0243】
【化28】
【0244】
2c1. 8/9についてのデータ
[0218] 1H NMR(400MHz,CD3OD)δ7.73(広幅,1H)、7.40(d,J=8.4Hz,1H)、7.30(m,2H)、2.69(dd,J=2.0,13.2Hz,1H)、2.56(dd,J=2.8,13.2Hz,1H)、2.20(m,2H)、1.80(m,2H)、2.28(m,2H)、1.60(m,2H)、1.50(m,3H);13C NMR(100MHz,CD3OD)δ150.75、132.38、130.19、130.06、127.64、124.81、77.28、43.63、43.45、41.16、26.38、25.34、22.06;ESI MS m/z 274.1、276.0。
【0245】
[0219] 化合物9を、ホルムアルデヒドを用いる還元的アミノ化によって2に変換した。
【0246】
【化29】
【0247】
(実施例3)
3a.実験手順
【0248】
【化30】
[0220] これらの合成について活用した実験条件は、実施例1および2において利用したものと同様の実験条件であった。
【0249】
【化31】
【0250】
[0221] これらの合成について活用した実験条件は、実施例1および2において利用したものと同様の実験条件であった。
【0251】
(実施例4)
4a.in vitroにおけるヒト5−HT/NE/DAの再取り込み阻害のデータ
化合物を、セロトニン(5-HT)、ノルエピネフリン(NE)およびドーパミン(DA)の機能的取り込みの阻害について、ラットの全脳、視床下部もしくは線条体のそれぞれから調製したシナプトソームにおいて、および/または組換えヒト輸送体を使用して試験した。このアッセイの詳細は、参照により組み込まれているUS2007/0203111Alに記載されている。ヒト再取り込み輸送体についての機能的取り込みアッセイに関する結果を以下に示す。
【0252】
【表1】
【0253】
4b.in vitroにおけるPKデータ(ヒトにおける代謝安定性、CYP450酵素の阻害、HERG電流の阻害)
【0254】
【表2】
【0255】
4c.尾の懸垂試験、自発運動(locomotor activity)試験および強制水泳試験
4c1.マウスの尾の懸垂試験
[0222] 抗うつ薬活性を検出する、この方法は、Steruら(Psychopharmacology, 85: 367-370(1985))により記載された方法に従う。げっ歯類は、尾を懸垂することにより、素早く無動になる。抗うつ薬は、無動の持続時間を短縮する。
【0256】
[0223] 動物の行動を、Steruら(Prog. Neuropsychopharmacol. Exp. Psychiatry 11: 659-671 (1987))によって開発された装置に類似する、コンピュータ化した装置(Med-Associates Inc.製)を使用して、5分間自動的に記録した。10〜12匹のマウスを、各1群として試験した。典型的には、化合物を3種の用量(1〜30mg/kg)において、1回、すなわち、試験30〜60分前に経口投与して評価し、ビヒクルの対照群と比較した。デシプラミン(100mg/kg)を、同一の実験条件下で投与し、正の基準物質として使用した。
【0257】
[0224] データを、一元配置分散分析(ANOVA)、それに続いて、適切な場合には、事後比較によって解析した。p<0.05の場合、効果を有意であるとみなした。データを、平均および平均に対する標準誤差(s.e.m)として示す。
【0258】
4c2.自発運動
[0225] 化合物の無動時間に対する効果が、ベースラインの運動活性に対する一般的な刺激効果とは関連がないことを確認するために、自発運動を、光電セルによりモニターするケージ(Med-Associates Inc.製)を使用して評価した。各試験チャンバーは、動物の動きを測定するための赤外光電セルビームを装備していた。水平および垂直な活動を測定した。
【0259】
[0226] ラットまたはマウスを、ビヒクルまたは試験化合物を用いて前処理し、元々のケージ中に戻し、それに続いて、自発運動用ケージ中に個別に入れ、活動を1〜5分の間、最長60分の期間にわたりモニターした。
【0260】
[0227] データを、一元配置分散分析(ANOVA)、それに続いて、適切な場合には、事後比較によって解析した。p<0.05の場合、効果を有意であるとみなした。データを、平均および平均に対する標準誤差(s.e.m)として示す。
【0261】
4c3.結果の概要
[0228] 本発明の化合物の効果を、マウスの尾の懸垂試験および自発運動試験において評価した。結果から、試験した全ての化合物が、抗うつ薬様のプロファイル(すなわち、無動時間の有意な短縮)を呈し、3〜30mg/kg、POの範囲のMEDを示すことが示された。尾の懸垂試験において活性を示す用量においては、ベースラインの運動活性は変化または低下しないことが観察され、このことから、抗うつ薬様活性は一般的な刺激効果には起因しないことが示された。
【0262】
[0229] また、本発明の化合物の効果を、ラットの強制水泳試験および自発運動試験においても評価した。全ての化合物が、抗うつ薬様の効果を呈し、10〜30mg/kg、POの範囲のMEDを示した。これらの化合物によって生じた無動の低下は、泳ぐ行動および登る行動の増加に起因するように思われ、このことから、混合性の輸送体活性(すなわち、SNRIプロファイル)が示された。また、マウスの尾の懸垂の結果と同様に、ラットの強制水泳試験も、この化合物について抗うつ薬様の活性を示した。
【0263】
【表3】
【0264】
[0230] TST後、各処理群からの4匹の代表的なマウスから、脳および血漿の試料を採取し、これらの組織における2および4対する暴露レベルを解析した。この試験において、2は、無動の用量依存性の低下を呈した(上記を参照されたい)。2の経口投与に続いて、顕著なレベルの4つの代謝産物が血漿および脳のレベルにおいて見出された。
【0265】
4c4.ラットの強制水泳試験
[0231] 抗うつ薬活性を検出する、この方法は、Porsoltら(Eur. J. Pharmacol., 47: 379-391(1978))によって記載され、Luckiら(Psychopharm, 121: 66-72(1995))によって改変された方法に従った。素早く脱出することができない状況において泳ぐことを強制されたラットは、無動になる。抗うつ薬は、無動の持続時間を短縮する。さらに、この試験においては、活性を示す行動の明確に異なるパターンが、ノルエピネフリン(NE)およびセロトニン(5-HT)の取り込みを選択的に阻害する抗うつ薬によって生じる。選択的NE再取り込み阻害薬は、登る行動を増加させることによって、無動を低下させ、一方、選択的5−HT再取り込み阻害薬は、泳ぐ行動を増加させることによって、無動を低下させる。
【0266】
[0232] 実験の第1日に(セッション1)に、ラットを個別に、22cmの水(25℃)を含有する円筒(高さ=40cm;直径=20cm)の中に、15分間入れ、次いで、24時間後に、水の中に、5分の試験の間(セッション2)戻した。これらのセッションを、テープに録画し、5分の試験の間、無動の持続時間、ならびに泳ぐ行動および登る行動を測定した。12匹のラットを、各1群として試験した。試験は、盲検で実施した。典型的には、化合物を、3種の用量(1〜30mg/kg)において、2回、すなわち、試験(セッション2)の24時間前および30〜60分前に経口投与して評価し、ビヒクルの対照群と比較した。デシプラミン(20mg/kg i.p.)を、同一の実験条件下で投与し、正の基準物質として使用した。
【0267】
[0233] データを、一元配置分散分析(ANOVA)、それに続いて、適切な場合には、事後比較によって解析した。p<0.05の場合、効果を有意であるとみなす。データを、平均および平均に対する標準誤差(s.e.m)として示す。
【0268】
【表4】
【0269】
(実施例5)
5a.ex vivoにおける結合アッセイ
[0234] 末梢からの化合物の投与後の、中枢における、ノルアドレナリン(NA)輸送体部位、5−HT輸送体部位およびドーパミン(DA)輸送体部位の受容体占有率を、それぞれ、[3H]ニソキセチン、[3H]シタロプラムおよび[3H]WIN35428の結合を使用して決定した。液体シンチレーション測定を使用して、放射活性を定量化した。
【0270】
[0235] C57BL/6マウス(25〜30g)に、ビヒクルまたは化合物のいずれかを、4種の用量レベルで経口投与した。処理60分後に、マウスを堵殺した。全脳を取り出し、皮質および線状体を解体して取り出してから、ドライアイス上で凍結した。脳組織は、アッセイの当日まで−20℃で保管した。各半球からの皮質を個別に凍結した。一方を、NA輸送体部位の占有率を決定するために使用し、他方を、5−HT輸送体部位の占有率を決定するために使用した。線状体は、DA輸送体部位の占有率を決定するために使用した。
【0271】
[0236] 各半球からの前頭皮質、または線状体を個別に、密閉可能なガラス製/テフロン(登録商標)製のホモジナイザーを使用して、氷冷したアッセイ用緩衝液中でホモジナイズし、直ちに、結合アッセイにおいて使用した。
【0272】
5b.マウス脳における、[3H]シタロプラムの、5−HT輸送体(SERT)部位に対する結合
[0237] 皮質膜(400μL;組織の1.25mg湿潤重量/チューブに相当する)を、1.3nMの単一の濃度の[3H]シタロプラム50μl、および緩衝液50μl(全結合)またはパロキセチン50μl(0.5μM;非特異的結合)のいずれかと共に、27℃で1時間インキュベートした。各動物について、3本のチューブを使用して全結合を決定し、3本のチューブを使用して非特異的結合を決定した。
【0273】
5c.マウス脳における、[3H]ニソキセチンの、ノルエピネフリン輸送体(NET)部位に対する結合
[0238] 皮質膜(400μL;組織の6.0mg湿潤重量/チューブに相当する)を、0.6nMの単一の濃度の[3H]ニソキセチン50μL、および緩衝液50μL(全結合)またはマジンドール50μL(1μM;非特異的結合)のいずれかと共に、4℃で4時間インキュベートした。各動物について、3本のチューブを使用して全結合を決定し、3本のチューブを使用して非特異的結合を決定した。
【0274】
5d.マウス脳における、[3H]WIN35428の、DA輸送体(DAT)部位に対する結合
[0239] 線状体膜(200μL;組織の2mg湿潤重量/チューブに相当する)を、24nMの単一の濃度の[3H]WIN35428 25μL、および緩衝液25μL(全結合)またはGBR12935 25μL(1μM;非特異的結合)のいずれかと共に、4℃で2時間インキュベートした。各動物について、2本のチューブを使用して全結合を決定し、2本のチューブを使用して非特異的結合を決定した。
【0275】
[0240] 膜に結合している放射活性を、Skatron製細胞ハーベスターを使用して、真空下で、0.5%PEI中にあらかじめ浸漬したSkatron11731フィルターを通してろ過することによって回収した。フィルターを、氷冷したリン酸緩衝液を用いて素早く洗浄し、放射活性(dpm)を、液体シンチレーション測定(1mL Packard MV Goldシンチレーター)によって決定した。
【0276】
5e.データ解析
[0241] 特異的結合の値(dpm)を、各動物について、平均全結合(dpm)から、平均非特異的結合(dpm)を引くことによって得た。データは、平均特異的結合(dpm)、およびビヒクル処理対照を100%とみなし、そのパーセントとして示す。
【0277】
5f.結果の概要
[0242] 2についてのex vivoにおけるSERT、NETおよびDATの結合/受容体占有率のデータを得た。
【0278】
【表5】
【0279】
(実施例6)
【0280】
【表6】
【0281】
(実施例7)
7a.レセルピンのラットモデル
[0243] レセルピン処理ラットパーキンソン病モデルにおいて、化合物4の単独でのまたはL−DOPAと組み合わせた効果を評価した。抗パーキンソ病活性(運動の欠損およびアキネジアの逆転)を検出する、この方法は、Johnstonら(Exp Neurol, 191, 243-250, 2005)によって記載されている方法に従う。行動試験の18時間前に、ラットにイソフルランを用いて軽く麻酔を施し、レセルピン(3mg/kg、sc)を、脱水を予防するための食塩水(50ml/kg)と併せて注射した。
【0282】
7b.行動の評価
[0244] 加速ロータロッド:加速ロータロッド上における動きを、4段階のラット用ロータロッド(MedAssociates製、米国)を使用して評価した。ロータロッドの回転スピードを、3.5〜35rpmに5分かけて増加させ、動物がロッド上に留まる時間を、3回のトライアルの平均として決定した。
【0283】
[0245] カタレプシー試験:カタレプシーを、ラットの前肢をベンチ表面から6cm上に懸垂させた水平な木製のロッドの上部の上に置くことによって評価した。ロッドから両方の肢を離すまでに要する時間を、最長120秒まで記録した。各動物につき3回のトライアルを実施した。
【0284】
[0246] オープンフィールド:オープンフィールド領域における活動を、自動化活動モニター(Linton Instrumentation製、英国)を使用して評価した。ラットを、活動用の箱の中に入れ、自発運動を、240分の期間にわたり記録した。
【0285】
[0247] 薬物投与:単独療法実験の場合、5つの異なる治療の効果を評価した:1)ビヒクル(無菌水、PO)、2)化合物4、3mg/kg(PO)、3)化合物4、10mg/kg(PO)、4)化合物4、30mg/kg、および5)正の基準物質であるL−DOPA(80mg/kg)、80mg/kg(IP)。併用実験の場合、5つの異なる治療を評価した:1)化合物4ビヒクル(PO)+L−DOPAビヒクル(IP)、2)化合物4(10mg/kg、PO)+L−DOPAビヒクル(IP)、3)化合物4ビヒクル(PO)+LDOPA(30mg/kg、IP)、4)化合物4(10mg/kg、PO)+L−DOPA(30mg/kg、IP)、および5)化合物4ビヒクル(PO)+L−DOPA(80mg/kg、IP)。治療を無作為化して施し、各動物に、全ての治療条件を与えた。化合物4は、行動の評価60分前に投与し、L−DOPAは、行動試験の直前に投与した。
【0286】
[0248] 結果から、多様な行動試験において、化合物4(3〜30mg/kg、PO)単独によって、動きが用量依存性に改善されることが示された(ベースライン自発運動、図2、ロータロッド、図3、およびカタレプシー、図4)。行動試験に依存して、化合物4の効果は、L−DOPA(80mg/kg)を用いた場合に観察された効果と類似する場合またはそれより弱い場合を示した。併用実験では、化合物4(10mg/kg、PO)と低い用量のL−DOPA(30mg/kg)との組合せが、程度の点では等しく、かつより高い用量のL−DOPAの投与(80mg/kg)によりもたらされた効果より持続時間の長い効果を示した、図5。
【0287】
[0249] データから、化合物4は単独療法としては、何らかの抗パーキンソン病作用を有し得るが、効果はL−DOPAほど強力ではないことが示唆されている。化合物4と低い用量のL−DOPAとの併用実験からは、程度の点では等しく、かつより高い用量のL−DOPAによりもたらされた作用より長い持続時間の抗パーキンソン病作用が示された。したがって、化合物4は、「L−DOPA節約性」と記載することができるであろう。
【0288】
(実施例8)
8a.ラット片側性6−ヒドロキシドーパミン(6-OHDA)病変モデル
[0250] げっ歯類6−OHDA病変ラットパーキンソン病モデルにおいて、化合物4の単独でのまたはL−DOPAと組み合わせた効果を評価した。抗パーキンソ病活性(運動の欠損およびアキネジアの逆転)を検出する、この方法は、Henryら(Exp Neurol, 151(2): 334-42, 1998)によって記載されている方法に従う。
【0289】
[0251] 動物の準備:手術前に、ラットにパルギリン(5mg/kg、ip)およびデシプラミン(25mg/kg ip)を投与して、ドーパミン作動性神経細胞に対して、それに続く6−OHDAの利用能を最適化し、毒性についての特異性を増加させた。次いで、ラットにイソフルランを用いて麻酔を施し、ラットを定位固定枠中に入れた。十字縫合を暴露させた後、(PaxinosおよびWatson, 1986の図譜に従って)頭蓋骨中、右内側前脳束上、十字縫合に対して2.8mm後側および2mm外側の座標において、頭蓋穿孔をドリルで開けた。次いで、28GのHamilton製の針を、頭蓋骨の9mm下まで下ろした。次いで、6−OHDA(2.5μl中の12.5μg)を注射した(1μl/分)。次いで、針を現位置に4分間維持して、確実に、溶液を完全に吸収させた。注射針をゆっくり取り出した後、創傷を閉鎖し、動物に、食塩水(50ml/kg、sc)、鎮痛薬(ケトプロフェン、0.5mg/kg)および広域スペクトルの抗生物質(エンロフロキサシン、75mg/kg)を投与した。手術後、動物を未処置の状態で3週間維持し、病変を発展および安定化させてから、行動の評価を開始した。
【0290】
8b.行動の評価
[0252] 肢の設置試験:肢の設置試験によって、感覚刺激に応答したそれぞれの前肢の正確な設置を評価する。ラットの胴体を軽く保持し、一方の前肢は、親指と人差し指との間で拘束し、他方の肢は、保持せず自由にさせた。次いで、ラットを、テーブルの縁部に対して平行に保持し、自由な前肢は、当該縁部に隣接させた状態にした。次いで、動物をテーブルに向かって移動させ、感覚毛をテーブルの縁部に対して軽く接触させて、自由な肢からの前肢設置応答を惹起した。全部で10回のトライアルを、素早く連続して実施してから、他方の肢について、この手順を繰り返した。試験を、6−OHDA病変の側とは反対側の肢の良好な設置応答のパーセントとして定量化した。病変の側と同側の肢の設置は、全ての動物において100%の事例で良好であった。
【0291】
[0253] 薬物投与:単独療法実験の場合、5つの異なる治療の効果を評価した:1)ビヒクル(無菌水、PO)、2)化合物4、3mg/kg(PO)、3)化合物4、10mg/kg(PO)、4)化合物4、30mg/kg、および5)正の基準物質であるL−DOPA、6.5mg/kg(IP)。併用実験の場合、5つの異なる治療を評価した:1)化合物4ビヒクル(PO)+L−DOPAビヒクル(IP)、2)化合物4(10mg/kg、PO)+L−DOPAビヒクル(IP)、3)化合物4ビヒクル(PO)+L−DOPA(2mg/kg、IP)、4)化合物4(10mg/kg、PO)+L−DOPA(2mg/kg、IP)、および5)化合物4ビヒクル(PO)+L−DOPA(6.5mg/kg、IP)。治療を無作為化して施し、各動物に、それぞれの治療条件を与えた。化合物4は、行動の評価60分前に投与し、L−DOPAは、行動試験の直前に投与した。
【0292】
[0254] 結果から、化合物4(3〜30mg/kg、PO)は単独では、肢設置タスクにおける動きの改善をほとんどまたは全くもたらさないことが示された。化合物4(10mg/kg、PO)と低い用量のL−DOPAとの併用により、肢設置の動きが有意に向上し、このことから、化合物4とL−DOPAとの作用の間には、相乗効果があるという何らかの証拠を得た(図6)。
【0293】
[0255] 本発明は、実施例において開示した特定の実施形態によって、その範囲が限定されるわけではない。これらの実施例は、本発明のいくつかの態様を例証するためのものであり、機能的に同等である実施形態はいずれも、本発明の範囲に属する。まさに、本明細書において示し、記載したものに加えて、本発明の種々の改変形態が当業者には明らかになるであるろうし、それらは、添付の特許請求の範囲に属するものとする。
【0294】
[0256] 全ての特許、特許出願および本出願に引用したその他の刊行物は、全ての目的のために、それらの全体が参照により本明細書に組み込まれている。
【特許請求の範囲】
【請求項1】
【化32】
(式中、
nは、0〜2からなる群から選択される整数であり;
sは、0〜2からなる群から選択される整数であり;
Aは、H、置換または非置換アルキル、ハロゲン、および置換または非置換ハロアルキルからなる群から選択されるメンバーであり、
Xは、H、ハロゲン、置換または非置換アルキル、置換または非置換アリール、置換または非置換ハロアルキル、およびOR5からなる群から選択されるメンバーであり、
ただし、
R5は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、アシル、およびS(O)2R5aからなる群から選択されるメンバーであり、
ただし、
R5aは、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルからなる群から選択されるメンバーであり;
YおよびZは、ハロゲン、CF3、CN、OR9、SR9、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、置換または非置換ヘテロシクロアルキル、NR10R11、およびNO2からなる群から独立して選択されるメンバーであり、
ただし、YとZとは、それらが結合している原子と一緒になって任意選択により結合して、1〜3個のヘテロ原子を任意選択により含む、5員環〜7員環を形成し、
ただし、
R9は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、置換または非置換ヘテロシクロアルキルからなる群から選択されるメンバーであり;かつ
R10およびR11は、H、OR12、アシル、S(O)2R13、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルからなる群から独立して選択されるメンバーであり、
ただし、
R12は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルからなる群から選択されるメンバーであり;かつ
R13は、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルからなる群から選択されるメンバーであり、
ただし、R10とR11とは、それらが結合している原子と一緒になって任意選択により結合して、1〜3個のヘテロ原子を任意選択により含む、3員環〜7員環を形成し;
R1およびR2は、H、ハロゲン、CN、CF3、OR6、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルからなる群から独立して選択されるメンバーであり、
ただし、
R6は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルからなる群から選択されるメンバーであり;かつ
R3およびR4は、H、OR7、アシル、S(O)2R8、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルからなる群から独立して選択されるメンバーであり、
ただし、
R7は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルからなる群から選択されるメンバーであり;
R8は、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルからなる群から選択されるメンバーであり;
ただし、R1、R2、R3およびR4のうちの少なくとも2つが、それらが結合している原子と一緒になって任意選択により結合して、1〜3個のヘテロ原子を任意選択により含む、3員環〜7員環を形成する。)
からなる群から選択されるメンバーである構造を有する化合物。
【請求項2】
R3およびR4が、置換または非置換C1〜C4アルキル、および置換または非置換C1〜C4ヘテロアルキルからなる群から独立して選択されるメンバーである、請求項1に記載の化合物。
【請求項3】
R3およびR4が、置換または非置換アルケニル、置換または非置換アルキニル、および置換または非置換シクロアルキルからなる群から独立して選択されるメンバーである、請求項1に記載の化合物。
【請求項4】
【化33】
(式中、AはHである。)
からなる群から選択されるメンバーである構造を有する、請求項1に記載の化合物。
【請求項5】
【化34】
からなる群から選択されるメンバーである構造を有する、請求項4に記載の化合物。
【請求項6】
YおよびZが、H、ハロゲン、CNおよびCF3からなる群から独立して選択されるメンバーである、請求項4に記載の化合物。
【請求項7】
YおよびZが、クロロである、請求項6に記載の化合物。
【請求項8】
sが1である、請求項7に記載の化合物。
【請求項9】
nが1である、請求項8に記載の化合物。
【請求項10】
【化35】
(式中、R4は、HおよびCH3からなる群から選択されるメンバーである。)
からなる群から選択されるメンバーである構造を有する、請求項9に記載の化合物。
【請求項11】
【化36】
(式中、
A1、A2、A3およびA4はそれぞれ独立して、O、S、N(Rb)bおよびC(Rb)b(Rc)からなる群から選択され;
aは、0〜2からなる群から選択される整数であり;
bは、0〜1からなる群から選択される整数であり;
RbおよびRcは、H、ハロゲン、CF3、CN、OR14、SR14、NR15R16、NR15S(O)2R14、NR15C(O)R14、S(O)2R14、アシル、C(O)OR14、C(O)NR15R16、S(O)2NR15R16、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルからなる群から独立して選択されるメンバーであり;
各R14、R15およびR16は、H、アシル、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、置換または非置換ヘテロシクロアルキルからなる群から独立して選択されるメンバーであり、ただし、R14、R15およびR16のうちの2つが、それらが結合している原子と一緒になって任意選択により結合して、3員環〜7員環を形成し、この環は、1〜3個のヘテロ原子を任意選択により含む。)
からなる群から選択されるメンバーである構造を有する、請求項4に記載の化合物。
【請求項12】
請求項1に記載の化合物またはその薬学的に許容できる塩もしくは溶媒和化合物、および薬学的に許容できる担体、ビヒクルまたは希釈剤を含む医薬組成物。
【請求項13】
神経障害を治療または予防するための方法であって、治療有効量の請求項1に記載の化合物またはその薬学的に許容できる塩もしくは溶媒和化合物を、その治療または予防を必要とする対象に投与することを含む方法。
【請求項14】
前記神経障害が、物質乱用、線維筋痛症、疼痛、睡眠障害、注意欠陥障害(ADD)、注意欠陥多動性障害(ADHD)、下肢静止不能症候群、うつ病、統合失調症、不安症、強迫性障害、パニック障害、心的外傷後ストレス障害、月経前不安および神経変性疾患からなる群から選択されるメンバーである、請求項13に記載の方法。
【請求項15】
前記睡眠障害が、睡眠時無呼吸である、請求項14に記載の方法。
【請求項16】
前記疼痛が、神経因性疼痛である、請求項14に記載の方法。
【請求項17】
摂食障害を治療または予防するための方法であって、治療有効量の請求項1に記載の化合物またはその薬学的に許容できる塩もしくは溶媒和化合物を、その治療または予防を必要とする対象に投与することを含む方法。
【請求項18】
肥満を治療または予防するための方法であって、治療有効量の請求項1に記載の化合物またはその薬学的に許容できる塩もしくは溶媒和化合物を、その治療または予防を必要とする対象に投与することを含む方法。
【請求項19】
細胞からの少なくとも1つのモノアミンの再取り込みを阻害する方法であって、請求項1に記載の化合物またはその薬学的に許容できる塩もしくは溶媒和化合物を、哺乳動物の対象に投与することを含む方法。
【請求項20】
前記モノアミンが、セロトニン、ドーパミンおよびノルエピネフリンからなる群から選択されるメンバーである、請求項19に記載の方法。
【請求項21】
1つまたは複数のモノアミン輸送体を調節する方法であって、請求項1に記載の化合物またはその薬学的に許容できる塩もしくは溶媒和化合物を、哺乳動物の対象に投与することを含む方法。
【請求項22】
前記モノアミン輸送体が、セロトニン輸送体(SERT)、ドーパミン輸送体(DAT)およびノルエピネフリン(NET)輸送体からなる群から選択されるメンバーである、請求項21に記載の方法。
【請求項23】
前記物質乱用が、コカイン、ニコチンまたはそれらの組合せから選択されるメンバーの乱用である、請求項14に記載の方法。
【請求項24】
前記神経変性疾患がパーキンソン病である、請求項14に記載の方法。
【請求項25】
【化37】
の式を有する化合物。
【請求項26】
請求項25に記載の化合物を、薬学的に許容できる賦形剤と組み合わせて含む医薬品製剤。
【請求項27】
【化38】
の式を有する化合物。
【請求項28】
請求項27に記載の化合物を、薬学的に許容できる賦形剤と組み合わせて含む医薬品製剤。
【請求項29】
【化39】
の式を有する化合物。
【請求項30】
請求項29に記載の化合物を、薬学的に許容できる賦形剤と組み合わせて含む医薬品製剤。
【請求項1】
【化32】
(式中、
nは、0〜2からなる群から選択される整数であり;
sは、0〜2からなる群から選択される整数であり;
Aは、H、置換または非置換アルキル、ハロゲン、および置換または非置換ハロアルキルからなる群から選択されるメンバーであり、
Xは、H、ハロゲン、置換または非置換アルキル、置換または非置換アリール、置換または非置換ハロアルキル、およびOR5からなる群から選択されるメンバーであり、
ただし、
R5は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、アシル、およびS(O)2R5aからなる群から選択されるメンバーであり、
ただし、
R5aは、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルからなる群から選択されるメンバーであり;
YおよびZは、ハロゲン、CF3、CN、OR9、SR9、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、置換または非置換ヘテロシクロアルキル、NR10R11、およびNO2からなる群から独立して選択されるメンバーであり、
ただし、YとZとは、それらが結合している原子と一緒になって任意選択により結合して、1〜3個のヘテロ原子を任意選択により含む、5員環〜7員環を形成し、
ただし、
R9は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、置換または非置換ヘテロシクロアルキルからなる群から選択されるメンバーであり;かつ
R10およびR11は、H、OR12、アシル、S(O)2R13、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルからなる群から独立して選択されるメンバーであり、
ただし、
R12は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルからなる群から選択されるメンバーであり;かつ
R13は、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルからなる群から選択されるメンバーであり、
ただし、R10とR11とは、それらが結合している原子と一緒になって任意選択により結合して、1〜3個のヘテロ原子を任意選択により含む、3員環〜7員環を形成し;
R1およびR2は、H、ハロゲン、CN、CF3、OR6、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルからなる群から独立して選択されるメンバーであり、
ただし、
R6は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルからなる群から選択されるメンバーであり;かつ
R3およびR4は、H、OR7、アシル、S(O)2R8、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルからなる群から独立して選択されるメンバーであり、
ただし、
R7は、H、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルからなる群から選択されるメンバーであり;
R8は、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルからなる群から選択されるメンバーであり;
ただし、R1、R2、R3およびR4のうちの少なくとも2つが、それらが結合している原子と一緒になって任意選択により結合して、1〜3個のヘテロ原子を任意選択により含む、3員環〜7員環を形成する。)
からなる群から選択されるメンバーである構造を有する化合物。
【請求項2】
R3およびR4が、置換または非置換C1〜C4アルキル、および置換または非置換C1〜C4ヘテロアルキルからなる群から独立して選択されるメンバーである、請求項1に記載の化合物。
【請求項3】
R3およびR4が、置換または非置換アルケニル、置換または非置換アルキニル、および置換または非置換シクロアルキルからなる群から独立して選択されるメンバーである、請求項1に記載の化合物。
【請求項4】
【化33】
(式中、AはHである。)
からなる群から選択されるメンバーである構造を有する、請求項1に記載の化合物。
【請求項5】
【化34】
からなる群から選択されるメンバーである構造を有する、請求項4に記載の化合物。
【請求項6】
YおよびZが、H、ハロゲン、CNおよびCF3からなる群から独立して選択されるメンバーである、請求項4に記載の化合物。
【請求項7】
YおよびZが、クロロである、請求項6に記載の化合物。
【請求項8】
sが1である、請求項7に記載の化合物。
【請求項9】
nが1である、請求項8に記載の化合物。
【請求項10】
【化35】
(式中、R4は、HおよびCH3からなる群から選択されるメンバーである。)
からなる群から選択されるメンバーである構造を有する、請求項9に記載の化合物。
【請求項11】
【化36】
(式中、
A1、A2、A3およびA4はそれぞれ独立して、O、S、N(Rb)bおよびC(Rb)b(Rc)からなる群から選択され;
aは、0〜2からなる群から選択される整数であり;
bは、0〜1からなる群から選択される整数であり;
RbおよびRcは、H、ハロゲン、CF3、CN、OR14、SR14、NR15R16、NR15S(O)2R14、NR15C(O)R14、S(O)2R14、アシル、C(O)OR14、C(O)NR15R16、S(O)2NR15R16、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、および置換または非置換ヘテロシクロアルキルからなる群から独立して選択されるメンバーであり;
各R14、R15およびR16は、H、アシル、置換または非置換アルキル、置換または非置換ヘテロアルキル、置換または非置換アリール、置換または非置換ヘテロアリール、置換または非置換ヘテロシクロアルキルからなる群から独立して選択されるメンバーであり、ただし、R14、R15およびR16のうちの2つが、それらが結合している原子と一緒になって任意選択により結合して、3員環〜7員環を形成し、この環は、1〜3個のヘテロ原子を任意選択により含む。)
からなる群から選択されるメンバーである構造を有する、請求項4に記載の化合物。
【請求項12】
請求項1に記載の化合物またはその薬学的に許容できる塩もしくは溶媒和化合物、および薬学的に許容できる担体、ビヒクルまたは希釈剤を含む医薬組成物。
【請求項13】
神経障害を治療または予防するための方法であって、治療有効量の請求項1に記載の化合物またはその薬学的に許容できる塩もしくは溶媒和化合物を、その治療または予防を必要とする対象に投与することを含む方法。
【請求項14】
前記神経障害が、物質乱用、線維筋痛症、疼痛、睡眠障害、注意欠陥障害(ADD)、注意欠陥多動性障害(ADHD)、下肢静止不能症候群、うつ病、統合失調症、不安症、強迫性障害、パニック障害、心的外傷後ストレス障害、月経前不安および神経変性疾患からなる群から選択されるメンバーである、請求項13に記載の方法。
【請求項15】
前記睡眠障害が、睡眠時無呼吸である、請求項14に記載の方法。
【請求項16】
前記疼痛が、神経因性疼痛である、請求項14に記載の方法。
【請求項17】
摂食障害を治療または予防するための方法であって、治療有効量の請求項1に記載の化合物またはその薬学的に許容できる塩もしくは溶媒和化合物を、その治療または予防を必要とする対象に投与することを含む方法。
【請求項18】
肥満を治療または予防するための方法であって、治療有効量の請求項1に記載の化合物またはその薬学的に許容できる塩もしくは溶媒和化合物を、その治療または予防を必要とする対象に投与することを含む方法。
【請求項19】
細胞からの少なくとも1つのモノアミンの再取り込みを阻害する方法であって、請求項1に記載の化合物またはその薬学的に許容できる塩もしくは溶媒和化合物を、哺乳動物の対象に投与することを含む方法。
【請求項20】
前記モノアミンが、セロトニン、ドーパミンおよびノルエピネフリンからなる群から選択されるメンバーである、請求項19に記載の方法。
【請求項21】
1つまたは複数のモノアミン輸送体を調節する方法であって、請求項1に記載の化合物またはその薬学的に許容できる塩もしくは溶媒和化合物を、哺乳動物の対象に投与することを含む方法。
【請求項22】
前記モノアミン輸送体が、セロトニン輸送体(SERT)、ドーパミン輸送体(DAT)およびノルエピネフリン(NET)輸送体からなる群から選択されるメンバーである、請求項21に記載の方法。
【請求項23】
前記物質乱用が、コカイン、ニコチンまたはそれらの組合せから選択されるメンバーの乱用である、請求項14に記載の方法。
【請求項24】
前記神経変性疾患がパーキンソン病である、請求項14に記載の方法。
【請求項25】
【化37】
の式を有する化合物。
【請求項26】
請求項25に記載の化合物を、薬学的に許容できる賦形剤と組み合わせて含む医薬品製剤。
【請求項27】
【化38】
の式を有する化合物。
【請求項28】
請求項27に記載の化合物を、薬学的に許容できる賦形剤と組み合わせて含む医薬品製剤。
【請求項29】
【化39】
の式を有する化合物。
【請求項30】
請求項29に記載の化合物を、薬学的に許容できる賦形剤と組み合わせて含む医薬品製剤。
【図1A】

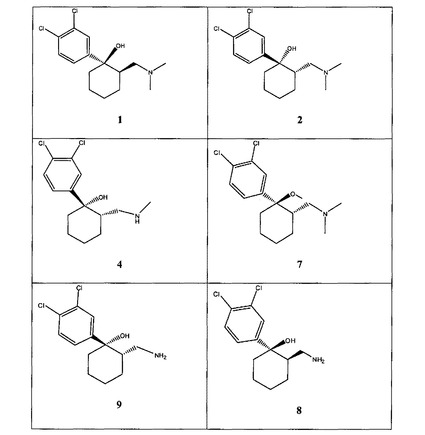
【図1B】
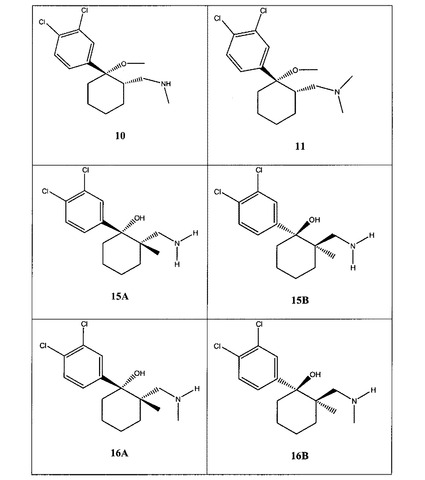
【図1C】
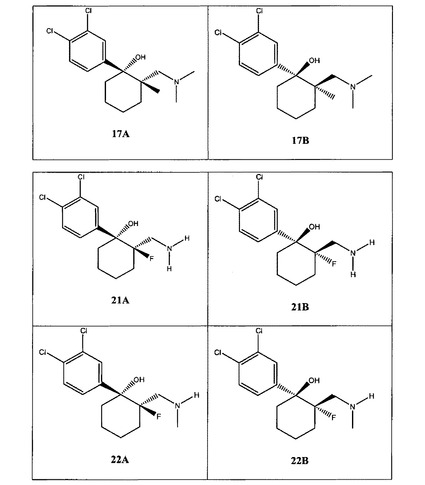
【図1D】
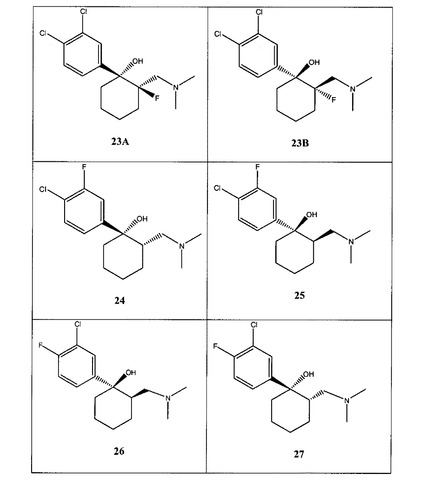
【図1E】
【図2】
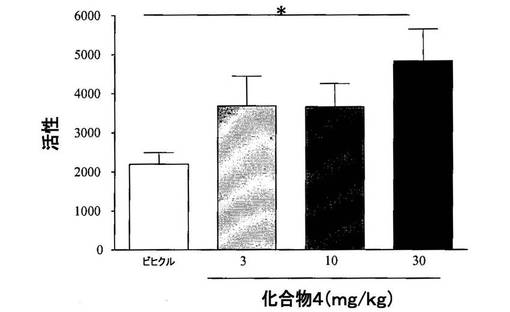
【図3】
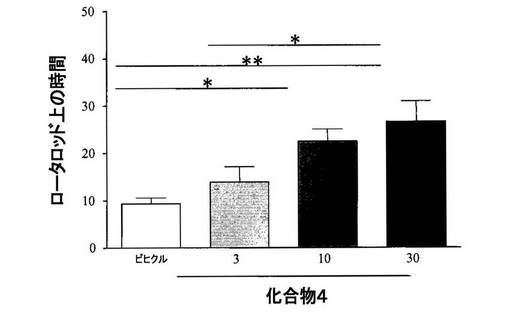
【図4】
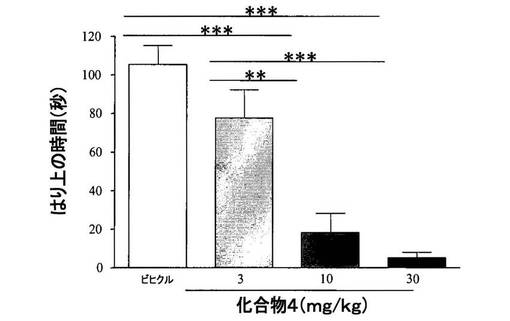
【図5】
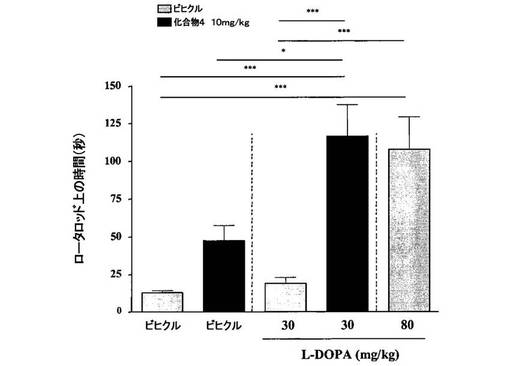
【図6】
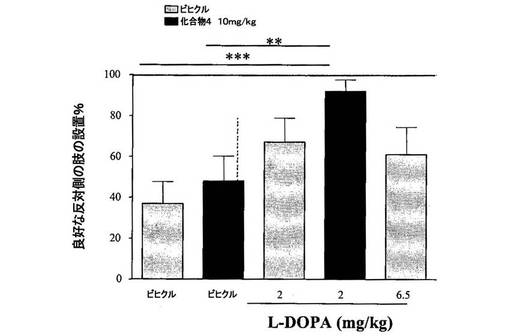
【図1B】
【図1C】
【図1D】
【図1E】
【図2】
【図3】
【図4】
【図5】
【図6】
【公表番号】特表2010−529045(P2010−529045A)
【公表日】平成22年8月26日(2010.8.26)
【国際特許分類】
【出願番号】特願2010−510560(P2010−510560)
【出願日】平成20年6月2日(2008.6.2)
【国際出願番号】PCT/US2008/065571
【国際公開番号】WO2008/151156
【国際公開日】平成20年12月11日(2008.12.11)
【出願人】(500114922)セプラコール インク. (14)
【氏名又は名称原語表記】Sepracor Inc.
【Fターム(参考)】
【公表日】平成22年8月26日(2010.8.26)
【国際特許分類】
【出願日】平成20年6月2日(2008.6.2)
【国際出願番号】PCT/US2008/065571
【国際公開番号】WO2008/151156
【国際公開日】平成20年12月11日(2008.12.11)
【出願人】(500114922)セプラコール インク. (14)
【氏名又は名称原語表記】Sepracor Inc.
【Fターム(参考)】
[ Back to top ]