
ラクタムタキキニン受容体拮抗薬の製造法
本発明は、ニューロキニン−1(NK−1)受容体拮抗薬として、及びタキキニン、とりわけサブスタンスPのインヒビターとして有用な特定のα,α−二置換γ−ラクタム誘導体の製造法を目的とする。これらの化合物はある種の障害、例えば、嘔吐、尿失禁、うつ状態、及び不安などの治療に有用である。

【発明の詳細な説明】
【背景技術】
【0001】
ヒト・ニューロキニン−1(hNK−1)はG−タンパク質結合受容体であり、中枢神経系及び胃腸管組織に集中している。参照:Nicoll,R.A.;Schenker,C.;Leeman,S.E.Annu.Rev.Neurosci.1980,3,227。神経ペプチド・サブスタンスP(SP)は、hNK−1受容体に対する好適なリガンドであり、多くの生物過程の適正化に関わっている。参照:(a)Guard,S.;Watson,S.P.Neurochem.Int.1991,18,149.(b)Takeuchi,Y.;Shands,E.F.B.;Beusen,D.D.;Marshall,G.R.J.Med.Chem.1998,41,3609及びその引用文献。SP及びhNK−1間の相互作用の制御は、重要な臨床分野、例えば、うつ状態、不安、炎症性腸疾患、及び疼痛などを包含する多様な一連の医学的障害の治療に関与してきた。参照:(a)Quatara,L.;Maggi,C.A.Neuropeptides 1998,32,1.(b)Rupniak,N.M.J.;Kramer,M.S.Trends Pharmacol.Sci.1999,20,485。結果として、有力な治療薬として強力かつ選択的なhNK−1受容体拮抗薬を同定するために、熱心な医薬研究が進められている。参照:(a)Owen,S.N.;Seward,E.M.;Swain,C.J.;Williams,B.J.U.S.Patent6,458,830B1,2001.(b)Finke,P.E.MacCoss,M.;Meurer,L.C.;Mills,S.G.;Caldwell,C.G.;Chen,P.;Durette,P.L.;Hale,J.;Holson,E.;Kopka,I.;Robichaud,A.PCT Int.Appl.WO9714671,1997.(c)Hale,J.J.;Mills,S.G.;MacCoss,M.;Finke,P.E.;Cascieri,M.A.;Sadowski,S.;Ber,E.;Chicchi,G.G.;Kurtz,M.;Metzger,J.;Elermann,G.;Tsou,N.N.;Tattersall D.;Rupniak,N.M.J.;Williams,A.R.;Rycroft,W.;Hargreaves,R.;MacIntyre,D.E.J.Med.Chem.1998,41,4607。
【0002】
本出願は、特定のラクタムhNK−1受容体拮抗薬の製造法を目的とする。この部類の化合物、さらにはこの部類の化合物の代替的な製造法が、WO2006/002117(2006年1月5日公開)及びUS2005−0282886(2005年12月22日公開)に開示されている。本発明は、ラクタムhNK−1受容体拮抗薬の収束的立体制御不斉合成を目的とする。
【発明の概要】
【0003】
発明の要旨
本発明は、ニューロキニン−1(NK−1)受容体拮抗薬として、及びタキキニン、とりわけサブスタンスPのインヒビターとして有用な特定のα,α−二置換γ−ラクタム誘導体の製造法を目的とする。これらの化合物はある種の障害、例えば、嘔吐、尿失禁、うつ状態、及び不安などの治療に有用である。
【0004】
発明の詳細な記載
一つの側面において、本発明は、式(I):
【0005】
【化1】
【0006】
[式中、
R2は以下からなる群より選択され:
(1)水素、及び
(2)C1−6アルキル;
Rは以下からなる群より選択され:
(1)未置換であるか、又はR11、R12及びR13の1個以上により置換されたフェニル;
(2)未置換であるか、又は以下から選択される1個以上の置換基で置換されたC1−8アルキル:
(a)ヒドロキシ、
(b)オキソ、
(c)C1−6アルコキシ、
(d)フェニル−C1−3アルコキシ、
(e)フェニル、
(f)−CN、
(g)ハロ、
(h)−NR9R10(ここで、R9及びR10は、独立して、
(1)水素、
(2)C1−6アルキル、
(3)ヒドロキシ−C1−6アルキル、及び
(4)フェニル、から選択される)、
(i)−NR9COR10(ここで、R9及びR10は上記定義のとおり)、
(j)−NR9CO2R10(ここで、R9及びR10は上記定義のとおり)、
(k)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(l)−COR9(ここで、R9は上記定義のとおり)、及び
(m)−CO2R9(ここで、R9は上記定義のとおり);
【0007】
(3)未置換であるか、又は以下から選択される1個以上の置換基で置換されたC2−6アルケニル:
(a)ヒドロキシ、
(b)オキソ、
(c)C1−6アルコキシ、
(d)フェニル−C1−3アルコキシ、
(e)フェニル、
(f)−CN、
(g)ハロ、
(h)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(i)−COR9(ここで、R9は上記定義のとおり)、及び
(j)−CO2R9(ここで、R9は上記定義のとおり);
【0008】
(4)へテロ環(ここで、該ヘテロ環は、
(A)ベンゾイミダゾリル、
(B)ベンゾフラニル、
(C)ベンゾチオフェニル、
(D)ベンゾオキサゾリル、
(E)フラニル、
(F)イミダゾリル、
(G)インドリル、
(H)イソオキサゾリル、
(I)イソチアゾリル、
(J)オキサジアゾリル、
(K)オキサゾリル、
(L)ピラジニル、
(M)ピラゾリル、
(N)ピリジル、
(O)ピリミジル、
(P)ピロリル、
(Q)キノリル、
(R)テトラゾリル、
(S)チアジアゾリル、
(T)チアゾリル、
(U)チエニル、
(V)トリアゾリル、
(W)アゼチジニル、
(X)1,4−ジオキサニル、
(Y)ヘキサヒドロアゼピニル、
(Z)ピペラジニル、
(AA)ピペリジニル、
(AB)ピロリジニル、
(AC)テトラヒドロフラニル、及び
(AD)テトラヒドロチエニル、
からなる群より選択され、また、該へテロ環は未置換であるか、又は
【0009】
(i)未置換であるか、又はハロ、−CF3、−OCH3、若しくはフェニルで置換されたC1−6アルキル、
(ii)C1−6アルコキシ、
(iii)オキソ、
(iv)ヒドロキシ、
(v)チオキソ、
(vi)−SR9(ここで、R9は上記定義のとおり)、
(vii)ハロ、
(viii)シアノ、
(ix)フェニル、
(x)トリフルオロメチル、
(xi)−(CH2)m−NR9R10(ここで、mは0、1又は2であり;R9及びR10は上記定義のとおり)、
(xii)−NR9COR10(ここで、R9及びR10は上記定義のとおり)、
(xiii)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(xiv)−CO2R9(ここで、R9は上記定義のとおり)、及び
(xv)−(CH2)m−OR9(ここで、m及びR9は上記定義のとおり)
から選択される1個以上の置換基で置換されている);
【0010】
R1は以下からなる群より選択され:
(1)
【0011】
【化2】
【0012】
(2)−C1−8アルキル(ここで、アルキルは未置換であるか、又は以下から選択される1個以上の置換基で置換されている:
(a)ヒドロキシ、
(b)オキソ、
(c)C1−6アルコキシ、
(d)フェニル−C1−3アルコキシ、
(e)フェニル、
(f)−CN、
(g)ハロ、
(h)−NR9R10(ここで、R9及びR10は上記定義のとおり)、
(i)−NR9COR10(ここで、R9及びR10は上記定義のとおり)、
(j)−NR9CO2R10(ここで、R9及びR10は上記定義のとおり)、
(k)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(l)−COR9(ここで、R9は上記定義のとおり)、及び
(m)−CO2R9(ここで、R9は上記定義のとおり));
【0013】
(3)−C2−6アルケニル(ここで、アルケニルは未置換であるか、又は以下から選択される1個以上の置換基で置換されている:
(a)ヒドロキシ、
(b)オキソ、
(c)C1−6アルコキシ、
(d)フェニル−C1−3アルコキシ、
(e)フェニル、
(f)−CN、
(g)ハロ、
(h)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(i)−COR9(ここで、R9は上記定義のとおり)、
(j)−CO2R9(ここで、R9は上記定義のとおり));
(4)−(CO)−フェニル(ここで、該フェニルは未置換であるか、又はR6、R7及びR8の1個以上により置換されている);
【0014】
R6、R7及びR8は、独立して、以下からなる群より選択され:
(1)水素;
(2)未置換であるか、又は以下から選択される1個以上の置換基で置換されたC1−6アルキル:
(a)ヒドロキシ、
(b)オキソ、
(c)C1−6アルコキシ、
(d)フェニル−C1−3アルコキシ、
(e)フェニル、
(f)−CN、
(g)ハロ、
(h)−NR9R10(ここで、R9及びR10は上記定義のとおり)、
(i)−NR9COR10(ここで、R9及びR10は上記定義のとおり)、
(j)−NR9CO2R10(ここで、R9及びR10は上記定義のとおり)、
(k)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(l)−COR9(ここで、R9は上記定義のとおり)、及び
(m)−CO2R9(ここで、R9は上記定義のとおり);
【0015】
(3)未置換であるか、又は以下から選択される1個以上の置換基で置換されたC2−6アルケニル:
(a)ヒドロキシ、
(b)オキソ、
(c)C1−6アルコキシ、
(d)フェニル−C1−3アルコキシ、
(e)フェニル、
(f)−CN、
(g)ハロ、
(h)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(i)−COR9(ここで、R9は上記定義のとおり)、
(j)−CO2R9(ここで、R9は上記定義のとおり);
(4)C2−6アルキニル;
【0016】
(5)未置換であるか、又は以下から選択される1個以上の置換基で置換されたフェニル:
(a)ヒドロキシ、
(b)C1−6アルコキシ、
(c)C1−6アルキル、
(d)C2−5アルケニル、
(e)ハロ、
(f)−CN、
(g)−NO2、
(h)−CF3、
(i)−(CH2)m−NR9R10(ここで、m、R9及びR10は上記定義のとおり)、
(j)−NR9COR10(ここで、R9及びR10は上記定義のとおり)、
(k)−NR9CO2R10(ここで、R9及びR10は上記定義のとおり)、
(l)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(m)−CO2NR9R10(ここで、R9及びR10は上記定義のとおり)、
(n)−COR9(ここで、R9は上記定義のとおり)、
(o)−CO2R9(ここで、R9は上記定義のとおり);
【0017】
(6)ハロ、
(7)−CN、
(8)−CF3、
(9)−NO2、
(10)−SR14(ここで、R14は水素又はC1−5アルキルである)、
(11)−SOR14(ここで、R14は上記定義のとおり)、
(12)−SO2R14(ここで、R14は上記定義のとおり)、
(13)NR9COR10(ここで、R9及びR10は上記定義のとおり)、
(14)CONR9COR10(ここで、R9及びR10は上記定義のとおり)、
(15)NR9R10(ここで、R9及びR10は上記定義のとおり)、
(16)NR9CO2R10(ここで、R9及びR10は上記定義のとおり)、
(17)ヒドロキシ、
(18)C1−6アルコキシ、
(19)COR9(ここで、R9は上記定義のとおり)、
(20)CO2R9(ここで、R9は上記定義のとおり)、
(21)2−ピリジル、
(22)3−ピリジル、
(23)4−ピリジル、
(24)5−テトラゾリル、
(25)2−オキサゾリル、及び
(26)2−チアゾリル;
【0018】
R11、R12及びR13は、独立して、R6、R7及びR8の定義から選択され;そして
Zは、
(1)水素、
(2)C1−6アルキル、及び
(3)ヒドロキシル
から選択される]で示されるラクタムタキキニン受容体拮抗薬又はその薬学的に許容され得る塩の製造法であって、
式(A):
【0019】
【化3】
【0020】
[式中、R14はR6から選択される]
で示される化合物と還元剤とを酸性条件下で反応させて、式(B):
【0021】
【化4】
【0022】
で示される化合物とし、次いで、式(B)の化合物と強酸とを反応させて式(I)で示される化合物とすることを含んでなり、次いで、所望により、式(I)で示される化合物の薬学的に許容され得る塩を形成するために、式(I)で示される化合物と対応する塩の酸とを反応させて、式(I)で示される化合物の薬学的に許容され得る塩を形成することを含んでなる製造法を包含する。
また、本発明は式(A)の化合物を調製するために、式(C):
【0023】
【化5】
【0024】
で示される化合物と酸とを反応させることにより、式(A)で示される化合物を調製することを含んでなる上記の製造法を包含する。
また、本発明は式(C)の化合物を得るために、式(D):
【0025】
【化6】
【0026】
[式中、X1はC1−6アルキル又はフェニルである]
で示される化合物と、アンモニア又はその塩とを反応させることにより、式(C)で示される化合物を調製することを含んでなる上記の製造法を包含する。本発明はまたさらに、式(D)の化合物を得るために、式(E):
【0027】
【化7】
【0028】
[式中、Yはハロゲンである]
で示される化合物と、式(F):
【0029】
【化8】
【0030】
で示される化合物、及び式M1N(R15)2又はM1N(Si(R15)3)2(式中、M1はLi、Na、K又はMgであり、各R15は、独立して、C1−4アルキルから選択される)で示される金属アミドとを、第一の非プロトン性有機溶媒中で反応させることにより、式(D)で示される化合物を調製することを含んでなる上記の製造法を包含する。
また、本発明はさらに、式(E)の化合物を得るために、式(G):
【0031】
【化9】
【0032】
で示される化合物と、ハロゲン化剤とを反応させることにより、式(E)で示される化合物を調製することを含んでなる上記の製造法を包含する。
また、本発明はさらに、式(G)の化合物を得るために、式(H):
【0033】
【化10】
【0034】
で示される化合物と、R−M2(ここで、M2は金属である)とを、第一の遷移金属触媒及びルイス酸の存在下で反応させることにより、式(G)で示される化合物を調製することを含んでなる上記の製造法を包含する。
また、本発明はさらに、式(H)の化合物を得るために、式(J):
【0035】
【化11】
【0036】
で示される化合物と、CH3Liとを、tert−ブチルメチルエーテル(MTBE)中で反応させることにより、式(H)で示される化合物を調製することを含んでなる上記の製造法を包含する。
本発明はまたさらに、式(J)の化合物を得るために、式(K):
【0037】
【化12】
【0038】
[式中、X2はRから選択される]
で示される化合物と、R1−OHとを、第二の遷移金属触媒、リガンド及び亜鉛付加物の存在下で反応させることにより、式(J)で示される化合物を調製することを含んでなる上記の製造法を包含する。
また、本発明はさらに、式(K)の化合物を得るために、式(L):
【0039】
【化13】
【0040】
で示される化合物を酵素的に還元し、次いで、その生成物と、X2−COClとを反応させることにより、式(K)で示される化合物を調製することを含んでなる上記の製造法を包含する。
また、本発明はさらに、式(L)の化合物を得るために、式(M):
【0041】
【化14】
【0042】
で示される化合物とブロム化剤とを反応させ、次いでバッファーの存在下でシアノ化剤を反応させることにより、式(L)で示される化合物を調製することを含んでなる上記の製造法を包含する。
また、本発明は、YがIであり、さらに、式(E)の化合物を得るために、式(N):
【0043】
【化15】
【0044】
で示される化合物と、M3−I(ここで、M3はLi、Na又はKである)とを反応させることにより、式(E)で示される化合物を調製することを含んでなる上記の製造法を包含する。
また、本発明はさらに、式(N)の化合物を得るために、式(O):
【0045】
【化16】
【0046】
で示される化合物と、ClCH2CO2H、ClCH2I、又はClCH2Br及び式M4N(R16)2又はM4N(Si(R16)3)2(ここで、M4はLi、Na、K、又はMgであり、各R16は、独立して、C1−4アルキルから選択される)で示される金属アミドとを、第二の非プロトン性有機溶媒中、約−20℃ないし約40℃の範囲の温度で反応させることにより、式(N)で示される化合物を調製することを含んでなる上記の製造法を包含する。
また、本発明はさらに、式(O)の化合物を得るために、式(P):
【0047】
【化17】
【0048】
で示される化合物又はそのトリエチルアミン塩とメチル化剤とを反応させることにより、式(O)で示される化合物を調製することを含んでなる上記の製造法を包含する。
また、本発明はさらに、式(P)の化合物又はそのトリエチルアミン塩を得るために、式(Q):
【0049】
【化18】
【0050】
で示される化合物と、R−M5(ここで、M5はM2同様の金属である)とを、第一の遷移金属触媒及びルイス酸の存在下で反応させ、次いで、所望により、トリエチルアミドを反応させて塩を形成することにより、式(P)で示される化合物を調製することを含んでなる上記の製造法を包含する。
また、本発明は、式(Q)の化合物を得るために、式(R):
【0051】
【化19】
【0052】
[式中、X3はRから選択される]
で示される化合物とR1−OHとを、第二の遷移金属触媒、リガンド及び亜鉛付加物の存在下で反応させることにより、式(Q)で示される化合物が製造されることを特徴とする上記の製造法を包含する。
また、本発明はさらに、式(R)の化合物を得るために、式(S):
【0053】
【化20】
【0054】
で示される化合物を酵素的に還元し、次いで、その生成物とX3−COClとを反応させることにより、式(R)で示される化合物を調製することを含んでなる上記の製造法を包含する。
また、本発明はさらに、式(S)の化合物を得るために、式(T):
【0055】
【化21】
【0056】
で示される化合物と酸化剤とを反応させることにより、式(S)で示される化合物を調製することを含んでなる上記の製造法を包含する。
また、本発明は、R1が
【0057】
【化22】
【0058】
である上記の製造法を包含する。
また、本発明は、Rが
【0059】
【化23】
【0060】
である上記の製造法を包含する。
また、本発明は、R2がメチルである上記の製造法を包含する。
また、本発明は、式(I)で示される化合物が式(Ia):
【0061】
【化24】
【0062】
によって表されるものである上記の製造法を包含する。
また、本発明は式(Ia)の化合物が薬学的に許容され得る塩である上記の製造法を包含する。
また、本発明は該薬学的に許容され得る塩がベンゼンスルホン酸塩であり、該塩の相当する酸がベンゼンスルホン酸である上記の製造法を包含する。
また、本発明は、式(Ia):
【0063】
【化25】
【0064】
で示される化合物のベンゼンスルホン酸塩を包含する。
また、本発明は、I型で示され、約21.2、9.1、及び8.5オングストロームのd−間隔に相当する特徴的な回折ピークを示す当該ベンゼンスルホン酸塩の無水結晶形を包含する。無水I型はさらに、13.5、10.9及び5.5オングストロームのd−間隔により特徴づけられる。無水I型はなおさらに、4.5、4.3及び4.2オングストロームのd−間隔により特徴づけられる。
【0065】
「第一の非プロトン性有機溶媒」及び「第二の非プロトン性有機溶媒」という用語は、例えば、THF、MTBE、ジメトキシエタン、DMF、DMAc及びジオキサンを意味する。
「ハロゲン化剤」という用語は、例えば、Br2、I2及びIClを意味する。
「第一の遷移金属触媒」及び「第二の遷移金属触媒」という用語は、例えば、[CODRh(OH)]2又はCuX若しくはCuX2(ここで、XはBr、Cl又はI)又はPd(OAc)2などのパラジウム触媒を意味する。
【0066】
「リガンド」という用語は、例えば、1,3−ビス(ジフェニルホスフィノ)プロパンなどのホスフィンリガンドを意味する。
「亜鉛付加物」という用語は、例えば、Et2Znを意味する。
「酵素的に還元する」という用語は、例えば、アルコール・デヒドロゲナーゼ(ADH RE)、NADH、グルコース及びグルコース・デヒドロゲナーゼ(GDH103)により還元することを意味する。
「ブロム化剤」という用語は、例えば、触媒HBr存在下のBr2を意味する。
「シアノ化剤」という用語は、例えば、NaCN及びKCNを意味する。
「バッファー」という用語は、例えば、酢酸及びNH4Clを意味する。
【0067】
「メチル化剤」という用語は、例えば、MeI及びM6CO3を意味する;この場合、M6はDMF、DMAc、DMSO、アセトンなどの極性溶媒中、0〜60℃での、又はH2SO4、TsOH、MsOH、又はPhSO3Hなどの酸触媒存在下、MeOH中、外気温度ないし還流下での、Li、Na、K、Ca、又はCsである。
「還元剤」という用語は、例えば、(C1−4アルキル)3SiHを意味する。
「ルイス酸」という用語は、例えば、TMSClを意味する。
「金属」という用語は、例えば、(OH)2、BF3、MgBr及びLiを意味する。
【0068】
式(I)で示される化合物は不斉中心を有し、本発明は光学異性体のすべてとその混合物を包含する。
さらに、炭素−炭素二重結合をもつ化合物は、Z−及びE−型として存在可能であり、該化合物の異性体型のすべてが本発明に包含される。
いずれかの変数(例えば、アルキル、アリール、R6、R7、R8、R9、R10、R11、R12、R13など)が、いずれかの変数又は式(I)中に一度ならず出現する場合、各出現に際しての定義は、その出現ごとの定義から独立したものである。
【0069】
本明細書にて使用する場合、「アルキル」という用語は、特定数の炭素原子を有する直鎖、分枝又は環状の配置を有するアルキル基を包含する。「アルキル」の例は、メチル、エチル、プロピル、イソプロピル、ブチル、iso−、sec−及びtert−ブチル、ペンチル、ヘキシル、ヘプチル、3−エチルブチル、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、ノルボルニルなどを包含する。「アルコキシ」は酸素架橋を介して結合する特定数の炭素原子を有するアルキル基を表し、例えば、メトキシ、エトキシ、プロポキシ、ブトキシ及びペントキシである。「アルケニル」は特定数の炭素原子を有する直鎖又は分枝の配置を有し、その鎖のいずれかの位置に少なくとも1つの不飽和が存在し得る炭化水素鎖を包含するものとする;例えば、エテニル、プロペニル、ブテニル、ペンテニル、ジメチルペンチルなどであり、可能な場合、E及びZ型を包含する。「ハロゲン」又は「ハロ」とは、本明細書にて使用する場合、フルオロ、クロロ、ブロモ及びヨードを意味する。
【0070】
「アリール」とはフェニル又はナフチルを意味し、いずれもが未置換であるか、又はハロ、C1−4−アルキル、C1−4−アルコキシ、NO2、CF3、C1−4−アルキルチオ、OH、−N(R6)2、−CO2R6、C1−4−ペルフルオロアルキル、C3−6−ペルフルオロシクロアルキル、及びテトラゾール−5−イルからなる群より選択される1個、2個又は3個の置換基により置換されている。
【0071】
「ヘテロアリール」という用語は、O、N及びSからなる群より選択される1個ないし3個のヘテロ原子を含有してなる未置換、一置換若しくは二置換の5員若しくは6員の芳香族ヘテロ環を意味し、その場合の置換基は、−OH、−SH、−C1−4−アルキル、−C1−4−アルコキシ、−CF3、ハロ、−NO2、−CO2R9、−N(R9R10)及び縮合ベンゾ基からなる群より選択されるメンバーである。
【0072】
当業者が理解するように、薬学的に許容され得る塩は、限定されるものではないが、無機酸との塩、例えば、塩酸塩、硫酸塩、リン酸塩、二リン酸塩、臭化水素酸塩、及び硝酸塩;又は有機酸との塩、例えば、リンゴ酸塩、マレイン酸塩、フマル酸塩、酒石酸塩、コハク酸塩、クエン酸塩、酢酸塩、乳酸塩、メタンスルホン酸塩、p−トルエンスルホン酸塩、2−ヒドロキシエチルスルホン酸塩、パモン酸塩、サリチル酸塩及びステアリン酸塩などである。同様に、薬学的に許容され得るカチオンは、限定されるものではないが、ナトリウム、カリウム、カルシウム、アルミニウム、リチウム及びアンモニウムである。
【0073】
本発明化合物は、過剰なタキキニン、特にサブスタンスPの活性の存在により特徴づけられる多様な臨床症状の予防及び治療に有用である。したがって、例えば、過剰なタキキニン、特にサブスタンスPの活性は、様々な中枢神経系の障害に関わっている。かかる障害は気分障害、例えば、うつ状態、又はより具体的には抑うつ性障害、例えば、単発性エピソード若しくは再発性主抑うつ性障害及び気分変調障害、又は双極性障害、例えば、双極性I障害、双極性II障害及び循環気質障害;不安障害、例えば、広場恐怖症を伴う、若しくは伴わないパニック障害、パニック障害暦を持たない広場恐怖症、特定恐怖症、例えば、特定動物恐怖症、対人恐怖症、強迫反応障害、ストレス障害、例えば、外傷後ストレス障害及び急性ストレス障害、及び全身性不安障害;統合失調症及びその他の精神障害、例えば、統合失調症型障害、分裂感情性障害、妄想性障害、短期精神障害、共有精神障害及び妄想若しくは幻覚を伴う精神障害;せん妄、痴呆、及び健忘症と他の認識若しくは神経変性障害、例えば、アルツハイマー病、老年痴呆、アルツハイマー型痴呆、脈管性痴呆、及び他の痴呆、例えば、HIV疾患、頭部外傷、パーキンソン病、ハンチントン病、ピック病、クロイツフェルト−ヤコブ病による痴呆、若しくは複数の病因による痴呆;パーキンソン病及び他の錐体外路の運動障害、例えば、投薬誘発運動障害、例えば、神経弛緩薬誘発性パーキンソン症、神経弛緩薬誘発性悪性症候群、神経弛緩薬誘発性急性失調症、神経弛緩薬誘発性静座不能、神経弛緩薬誘発性遅発性ジスキネジー、及び神経弛緩薬誘発性姿勢振せん;アルコール、アンフェタミン(又はアンフェタミン様物質)、カフェイン、大麻、コカイン、幻覚剤、吸入とエーロゾル噴射剤、ニコチン、アヘン、フェニルグリシジン誘導体、鎮静剤、催眠薬、及び抗不安薬などの使用から生じる物質関連障害(この物質関連障害は、依存性と乱用、中毒、離脱、中毒性せん妄、離脱性せん妄、持続性痴呆、精神障害、気分障害、不安障害、性機能不全及び睡眠障害である);てんかん;ダウン症候群;MS及びALSなどの髄鞘脱落症及び末梢神経障害などの他の神経病理学的障害、例えば、糖尿病性及び化学療法誘発神経障害、及び帯状疱疹後神経痛、三叉神経痛、分節若しくは肋間神経痛、及びその他の神経痛;及び急性若しくは慢性の脳血管傷害による脳血管障害、例えば、脳梗塞、くも膜下出血若しくは脳浮腫である。
【0074】
タキキニン、とりわけサブスタンスPは、その活性が侵害受容と疼痛にも関与している。本発明化合物は、したがって、以下の柔組織及び末梢の傷害を含む、疼痛を主体とする疾患及び症状の予防又は治療に有用となる:急性外傷、骨関節症、リウマチ様関節炎、筋骨格疼痛、特に外傷後の疼痛、脊髄痛、顔面筋疼痛症候群、頭痛、会陰切開疼痛、及び火傷;深部及び内臓痛、例えば、心臓痛、筋肉痛、眼痛、口顔痛、例えば、歯痛、腹痛、婦人科痛、例えば、月経困難症、及び出産時痛;神経根傷害と関連する疼痛、例えば、末梢神経障害と関連する疼痛、例えば、神経絞扼及び腕神経叢剥離、切断、末梢性神経障害、疼痛性チック、非定型顔面痛、神経根傷害、及びくも膜炎;がん関連疼痛(時に、がん痛と言われる);中枢神経系痛、例えば、脊髄又は脳幹傷害による疼痛;腰痛;坐骨神経痛;強直性脊椎炎、通風;及び瘢痕痛。
【0075】
タキキニン、とりわけサブスタンスPの拮抗薬は、以下の疾患の治療に有用である:呼吸器疾患、特に過剰の粘液分泌と関連する疾患、例えば、慢性閉塞性気道疾患、気管支肺炎、慢性気管支炎、のう胞性線維症と喘息、成人呼吸促迫症候群、及び気管支痙攣;炎症性疾患、例えば、炎症性腸疾患、乾癬、結合組織炎、骨関節症、リウマチ様関節炎、掻痒症及び日焼け;アレルギー、例えば、湿疹と鼻炎;過敏性障害、例えば、ツタウルシ;眼科疾患、例えば、結膜炎、春季結膜炎など;細胞増殖と関連する眼科症状、例えば、増殖性硝子体網膜障害;皮膚疾患、例えば、接触性皮膚炎、アトピー性皮膚炎、蕁麻疹、及びその他の湿疹様皮膚炎。タキキニン、とりわけサブスタンスPのアンタゴニストは、乳房腫瘍、神経節細胞腫、及び小細胞がん、例えば、小細胞肺がんなどの新生物の治療にも有用である。
【0076】
タキキニン、とりわけサブスタンスPの拮抗薬は、以下の胃腸(GI)障害の治療に有用である:GI管の炎症性障害と疾患、例えば、胃炎、胃十二指腸潰瘍、胃がん、胃リンパ腫、内臓の神経性制御と関連する障害、潰瘍性大腸炎、クローン病、過敏性腸症候群及び嘔吐(例えば、化学療法、放射線、毒素、ウイルス若しくは細菌感染、妊娠、前庭障害、例えば、動揺病、真性めまい、めまい及びメニエール病、外科手術、偏頭痛、頭蓋内圧の変化、胃−食道逆流病、胃酸過多、飲食物過食、酸性胃、むしず又は逆流、胸焼け、例えば、エピソード的、夜間又は食事−誘発胸焼け、及び消化不良などにより誘発される嘔吐などの急性、遅延性若しくは予測性嘔吐を含む)。
【0077】
タキキニン、とりわけサブスタンスPの拮抗薬は、以下の多様な他の症状の治療に有用である:ストレス関連体細胞障害;反射性交感神経性ジストロフィ、例えば、肩手症候群;有害免疫反応、例えば、移植組織拒絶反応及び免疫増強若しくは抑制に関係する障害、例えば、全身性紅斑性狼瘡;サイトカイン化学療法から生じる血漿溢血、膀胱機能障害、例えば、膀胱炎、膀胱排尿筋過剰反射、頻尿及び尿失禁(切迫尿失禁、尿意逼迫、及び頻尿の症候をもつ過剰反応膀胱の予防又は治療を含む);線維性及びコラーゲン病、例えば、強皮症及び好酸性肝蛭病;血管拡張及び血管痙攣性疾患を原因とする血流の障害、例えば、アンギナ、血管性頭痛、偏頭痛及びレイノー病;及び前記症状のいずれか、とりわけ偏頭痛の疼痛の伝播に寄与するか、又はそれと関連する疼痛又は侵害受容。本発明化合物は上記症状の合併症の治療、特に、術後疼痛及び術後の悪心と嘔吐の治療にも有益である。
【0078】
本発明の化合物は、特に、化学療法、放射線、毒素、妊娠、前庭障害、動揺病、外科手術、偏頭痛、及び頭蓋内圧の変化などにより誘発される嘔吐などの急性、遅延性若しくは予測性嘔吐を含む嘔吐の予防又は治療に有用である。例えば、本発明化合物は、緩和な、又は高度に嘔吐誘発性のがん化学療法、例えば、高用量投与シスプラチンの開始時及び反復過程と関連する急性及び遅延性の悪心と嘔吐の予防のために、他の嘔吐剤と併用してもよい。とりわけ、本発明化合物は、抗新生物(細胞傷害)剤(例えば、がん化学療法にて常套的に使用される薬剤)を原因とする嘔吐、及び他の薬理学的薬剤、例えば、ロリプラム(rolipram)により誘発される嘔吐の治療に有用である。かかる化学療法剤の例は、アルキル化剤、例えば、エチレンイミン化合物、スルホン酸アルキル及びアルキル化作用をもつ他の化合物、例えば、ニトロソウレア類、シスプラチン及びデカルバジン;代謝拮抗剤、例えば、葉酸、プリン若しくはピリミジンアンタゴニスト;有糸分裂インヒビター、例えば、ヒガンバナアルカロイド及びポドフィロトキシンの誘導体;及び細胞傷害性抗生物質である。化学療法剤の具体例は、例えば、D.J.Stewart in Nausea and Vomiting(悪心及び嘔吐):Recent Research and Clinical Advances(最近の研究及び臨床上の進歩),Eds.J.Kucharczyk et al,CRC Press Inc.,Boca Raton,Florida,USA(1991)pages177−203,特に188ページに記載されている。共通して使用される化学療法剤は、シスプラチン、ダカルバジン(DTIC)、ダクチノマイシン、メクロレタミン、ストレプトゾシン、シクロホスファミド、カルムスチン(BCNU)、ロムスチン(CCNU)、ドキソルビシン(アドリアマイシン)、ダウノルビシン、プロカルバジン、マイトマイシン、シタラビン、エトポシド、メトトレキセート、5−フルオロウラシル、ビンブラスチン、ビンクリスチン、ブレオマイシン及びクロランブシルなどである[R.J.Gralla et al in Cancer Treatment Reports(1984)68(1),163−172]。本発明のさらなる側面は、哺乳動物において、時間生物学(既日リズム位相)的作用を達成し、既日リズム障害を緩和するために、本発明化合物を使用することからなる。
【0079】
【化26】
【0080】
構造的に複雑な(1)は、3種の明確な合成目標に分割できよう:1)二級の立体末端の双方に立体化学を含む立体的に込み合ったエーテル;2)トランス,トランス−1,2,3−三置換シクロペンタン核;及び3)2つの立体中心を含み、その1つが三級アミンであるピロリジノン環(工程図1a)。遠位三級アミンの立体化学に対処するために、我々はケトン(2a)から(1)を製造する戦略を案出した;この戦略はオキサゾリジノン(3a)をヨードケトン(4a)によりジアステレオ選択的にアルキル化することに由来する。(4a)においてシクロペンタン核の相対的立体化学を制御する最も有効な方法は、アリルエーテル(5a)に対するアリール−金属種の基質−制御共役付加を経由して、当該ケトンを熱力学的に好適なジアステレオマーに異性化することである。我々はアリルエーテル(5a)の二級の立体中心双方に対処する最善の方法が、それぞれエナンチオマーとして純粋な形状のアリルアルコール(6a)とアルコール(7a)の収束性の立体特異的カップリング結合を経由することであると想定した。
【0081】
アルコール(7a)は数種のhNK−1拮抗薬に存在する共通の構造的モチーフである:参照:(a)Owen,S.N.;Seward,E.M.;Swain,C.J.;Williams,B.J.U.S.Patent6,458,830B1,2001.(b)Finke,P.E.MacCoss,M.;Meurer,L.C.;Mills,S.G.;Caldwell,C.G.;Chen,P.;Durette,P.L.;Hale,J.;Holson,E.;Kopka,I.;Robichaud,A.PCT Int.Appl.WO9714671,1997.(c)Hale,J.J.;Mills,S.G.;MacCoss,M.;Finke,P.E.;Cascieri,M.A.;Sadowski,S.;Ber,E.;Chicchi,G.G.;Kurtz,M.;Metzger,J.;Elermann,G.;Tsou,N.N.;Tattersall D.;Rupniak,N.M.J.;Williams,A.R.;Rycroft,W.;Hargreaves,R.;MacIntyre,D.E.J.Med.Chem.1998,41,4607;また、このものは対応するアリールメチルケトンの不斉還元を介して容易に入手し得る;参照:Hansen,K.B.;Chilenski,J.R.;Desmond,R.;Devine,P.N.;Grabowski,E.J.J.;Heid,R.;Kubryk,M.;Mathrem D.J.;Varsolona,R.Tetrahedron:Asymmetry 2003,14,3581。
【0082】
対照的に、アリルアルコール(6a)のエナンチオ選択的合成については報告されていない。既存の方法論を3−シアノシクロペンテノン(8a)の不斉還元に適用することは、変わり得る収率で、また穏当なエナンチオ選択性で(6a)を生じた。参照:(a)Ohkuma,T.;Koizumi,M.;Doucet,H.;Pham,T.;Kozawa,M.;Murata,K.;Katayama,E.;Yokozawa,T.;Ikariya,T.;Noyori,R.J.Am.Chem.Soc.1998,120,13529.(b)Corey,E.J.;Guzman−Perez,A.;Lazerwith,S.E.J.Am.Chem.Soc.1997,119,11769.(c)Yun,J.;Buchwald,S.L.;J.Am.Chem.Soc.1999,121,5640.(d)Brown,H.C.;Ramachandran,P.V.Acc.Chem.Res.1992,25,16−24.(e)Midland,M.M.;Tramontano,A.;Kazbubski,A.;Graham,R.S.;Tsai,D.J.S.;Cardin,D.Tetrahedron 1984,40,1371.(f)Noyori,R.;Suzuki,M.Angew.Chem.Int.Ed.Engl.1984,23,847。(8a)に類似のエノンの不斉、生物触媒還元については、先例がなかった[参照:(a)Fonteneau,L.;Rosa,S.;Buisson,D.Tetrahedron:Asymmetry,2002,13,579.(b)Attolini,M.;Bouguir,F.;Iacazio,G.;Peiffer,G.;Maffei,M.Tetrahedron,2001,57,537]が、ケト還元酵素ライブラリーをスクリーニングした。我々は、ロドコッカス・エリスロポリス(Rhodococcus erythropolis)由来のアルコール・デヒドロゲナーゼ(ADH RE)が、高収率(93%)、かつすぐれたエナンチオ選択性(>99%ee)で、エノン(8a)を(S)−アリルアルコール(6a)に効率的に還元することを発見した(工程図2a)。
【0083】
【化27】
a 条件:(a)ADH RE、0.1M K2PO4、グルコース、NAD、pH6.5,93%.(b)2−NapCO2H,2−NapCOCl,DIPEA,DMAP,CH2Cl2,84%,Nap=2−Nap.(c)Et2Zn,6,THF,Pd(OAc)2,dppp,83%
【0084】
高光学純度で調製したアルコール(6a)及び(7a)を用いて、我々は両方の中心でエピマー化を起こさずにこれらのパートナーをカップル結合させる方法を探究した。文献調査した形質転換の立体特異性(η3−アリル金属中間体を経由して進行する)は、Pd−触媒アリルエーテル化を魅力的な選択肢とした;しかし、アルコール(7a)は立体的に過密であり、求核性が乏しいために、このカップリング結合に参画するには不適切な候補であった。参照:Kim,H.;Lee.C.Org.Lett.2002,4369及びその引用文献。我々は、最適とした条件下、化学量論比のナフトエ酸アリルエステル(9a)とアルコール(6a)が、0.5当量のEt2Znの存在下、Pd(OAc)2及びdpppを用いてカップル結合させると、アリルエーテル(10a)が83%アッセイ収率で、かつ両方の立体中心での立体配置が完全に維持されて得られることを見出し、満足した。
【0085】
該ニトリルに対するクープレート共役付加を実施する試行は失敗であったが、我々は5.0当量のアリールホウ酸又は1.5当量のトリフルオロホウ酸アリール(K塩)を用いて、リガンドなしのRh−触媒共役付加(3mol%[CODRh(OH)2]、EtOH、還流)を証明することができた。参照:(a)Batey,R.A.;Thadani,A.N.;Smil,D.V.Org.Letters 1999,1683.(b)Sakai,M.;Hayashi,H.;Miyaura,N.Organometallics 1997,16,4229。ケトンから熱力学的に優位なジアステレオマーへの異性化(NaOMe/MeOH)後に、両方の手法が93%アッセイ収率と高ジアステレオ選択性(>99:1 β−中心、90:10 α−中心)で(11a)を与えた。該ニトリルはMTBE中、MeLiでの処理を経て、容易にメチルケトンに変換され、90%のアッセイ収率で(12a)を供給した(工程図3a)。
【0086】
別法として、メチルケトン(13a)は、ニトリル(10a)から容易に製造され(MeLi、MTBE)、さらに期待どおりに、該ケトンの熱力学的に優位なジアステレオマーへの異性化(NaOMe/MeOH)後に、アリール・グリニヤールのCu−触媒共役付加が、すぐれた収率(95%)、かつ例外的にすぐれたジアステレオ選択性(>99:1 β−中心、98:2 β−中心)で(12a)を提供した。MeOH中、IClによる(13a)の選択的ヨード化は、90%の単離収率でヨードケトン(4a)を生成した。(4a)のX線結晶構造は、この方法により組み立てた4つの立体中心の相対的及び絶対的化学を証明した。
【0087】
【化28】
a条件:(a)4−FPhBF3K、[CODRh(OH)]2、EtOH、90℃、93%.(b)MeLi、MTBE、0℃、90%.(c)NaOMe、MeOH、RT、98%.(d)MeLi、MTBE、0℃、90%.(e)4−FPhMgBr、CuI、TMSCl、THF、−40℃、96%.(f)ICl、MeOH、20℃、90%
【0088】
ヨードケトンを組み立てる収束的高選択的方法(6工程、58%収率)を用いて、我々はピロリジノン環を製造する立体制御方法の開発に注目した。公表されている方法に従い、オキサゾリジノン(3a)をヨードケトン(4a)でアルキル化すると、穏当な収率で(2)を生じた。参照:(a)Karady,S.;Amato,J.;Weinstock,L.Tetrahedron Lett.1984,25,4337.(b)Szumigala,Jr.,R.H.;Onofiok,E.;Karady,S.;Armstrong,III,J.D.;Miller R.A.Tetrahedron Lett.2005,46,4403。90%の収率と>99:1のジアステレオ選択性の最良の結果は、トルエン/DMPU中、低温度で、2.4当量の(3)及び2.5当量のLHMDSにより達成された。水酸化アンモニウムによるオキサゾリジノンの開裂は明らかにアミナール(14a,b)のジアステレオマー混合物を生じた;このものは、(14)から(16)への直接の還元が1当量の水を生成し、それがEt3SiH還元を妨げるので、シラン還元を実施する前に、一旦メタンスルホン酸で脱水して、94%の収率でエナミド(15a,b)のジアステレオ混合物(〜3:1)とした。エナミド(15a,b)は、単離したジアステレオマーとして純粋な異性体(15a)又は(15b)それぞれが、酸性条件下で同じ(15a,b)の3:1混合物に変換されるため、アシルイミニウムカチオン(17)及び(18)の平衡にある。また、アシルイミニウム(18)は熱力学的に(17)よりもより安定である。それ故、Et3SiH/MeSO3Hによる(15a,b)の還元は、優先的に(18)を経由して進行し、(17)を経由するシクロペンタン環上の少量のエピマーとともに、90:10のジアステレオマー混合物として優れた収率で(16)を与えた。(16)の化学選択的脱保護は、HBr/AcOHで遂行した。候補化合物(1)はベンゼンスルホン酸塩として、85%の収率で得た。
【0089】
【化29】
a条件:(a)LiHMDS、トルエン/DMPU、−78℃、90%.(b)NH4OH、THF/H2O、87%.(c)MeSO3H、EtOAc、94%.(d)MeSO3H、Et3SiH、MeCN、95%,(e)HBr/HOAc,(f)PhSO3H、IPAc/IPA/ヘプタン、85%(2工程)
【0090】
結論として、収束的高選択的経路は、強力なhNK−1受容体拮抗薬(1)を合成するために開発されたものである。6ヶ所すべての立体中心は、合計11工程(23%収率)で卓越した選択性をもって巧みに調製された;該方法は7kgの(1)を製造するために使用した。Pd−触媒によるエーテル化と、引き続く(10a)及び(13a)などの環状の基質における基質−制御共役付加の適用方法は、高度に機能化したシクロペンタノイド類の立体制御合成に一般的に適用されている。
上記の方法論は、以下の表に例示するように広く適用されていることが分かっているので、様々なシステムに適用された。
【0091】
【化30】
【0092】
(2b)の実用的な合成を案出するには、2種類の明確な合成目標を扱わねばならない:1)立体的に込み合ったエーテル(二級の立体末端双方に立体化学を含む);及び2)トランス,トランス−1,2,3−三置換シクロペンタン核。我々は、(2b)におけるトランス,トランス−配置が、アリルエーテル(4b)へのアリール−金属種の基質制御共役付加と、それに続くエステルの熱力学的に好適なジアステレオマーへの平衡化を経由して効果的に組み立てられるだろうと想定した(反応工程図2)。(4b)の構築のための最も魅力的な収束的方法は、それぞれエナンチオマーとして純粋な形状のアリルアルコール(5b)とアルコール(6b)との立体特異的カップリングを経ることであろう。上記の逆合成は当該標的構造を3種の類似サイズと複雑さの成分に切断し、これを我々は(2b)のみならず、一連の構造類似体にも適用し得ると想定した。
【0093】
【化31】
【0094】
アルコール(6b)は数種の薬物候補に存在する共通の構造要素であり[参照:a)Nelson,T.D.;Rosen,J.D.;Smitrovich,J.H.;Payack,J.;Craig,B.;Matty,L.;Huffman,M.A.;McNamara,J.Org.Lett.2005,55.b)Zhao,M.M.;McNamara,J.M.;Ho,G.−J.;Emerson,K.M.;Song,Z.J.;Tschaen,D.M.;Brands,K.M.J.;Dolling,U.−H.;Grabowski,E.J.J.;Reider,P.J.;Cottrell,I.F.;Ashwood,M.S.;Bishop,B.C.J.Org.Chem.2002,6743]、アリールメチルケトンの不斉還元を経て容易に調製した。参照:Hansen,K.B.;Chilenski,J.R.;Desmond,R.;Devine,P.N.;Grabowski,E.J.J.;Heid,R.;Kubryk,M.;Mathre,D.;Varsolona,R.Tetrahedron:Asymmetry 2003,3581。対照的に、アリルアルコール(5b)とその構造類似体のエナンチオ選択的合成については報告がなかった。(5b)への最も直接的な経路は、3−カルボキシメチルシクロペンテノン(7b)の不斉合成を経由することであろう。参照:a)Catino,A.J.;Forslund,R.E.;Doyle,M.P.J.Am.Chem.Soc.2004,13622−13623.b)Yu,J−Q.;Corey,E.J.J.Am.Chem.Soc.2003,3232−3233。(7b)の不斉還元に既存の方法論を適用すると、相応の収率、かつ並みのエナンチオ選択性で、アリルアルコール(6b)が得られた(表1)。参照:a)Ohkuma,T.;Koizumi,M.;Doucet、H.;Pham,T.;Kozawa,M.;Murata,K.;Katayama,E.;Yokozawa,T.;Ikariya,T.;Noyori,R.J.Am.Chem.Soc.1998,120,13529.b)Corey,E.J.;Guzman−Perez,A.;Lazerwith,S.E.J.Am.Chem.Soc.1997,119,11769.c)Yun,J.;Buchwald,S.L.;J.Am.Chem.Soc.1999,121,5640.d)Brown,H.C.;Ramachandran,P.V.Acc.Chem.Res.1992,25,16−24.e)Midland,M.M.;Tramontano,A.;Kazbubski,A.;Graham,R.S.;Tsai,D.J.S.;Cardin,D.Tetrahedron 1984,40,1371.f)Noyori,R.;Suzuki,M.Angew.Chem.Int.Ed.Engl.1984,23,847。
【0095】
(7b)の生物触媒還元の実行可能性を決定するために(参照;a)Fonteneau,L.;Rosa,S.;Buisson,D.Tetrahedron:Asymmetry,2002,13,579.b)Attolini,M.;Bouguir,F.;Iacazio,G.;Peiffer,G.;Maffei,M.Tetrahedron,2001,57,537)、ケトリダクターゼ・ライブラリーのスクリーニングを実施した。我々は、ロドコッカス・エリスロポリス(Rhodococcus erythropolis)からのアルコール・デヒドロゲナーゼ(ADH RE)が、好収率(83%)、かつ所望の(S)−エナンチオマーに対してすぐれたエナンチオ選択性(>99%ee)で、(7b)を(5b)に効率的に還元することを発見し、満足した(表1;第4項目)。この方法の信頼性の証拠として、3−シアノシクロペンテノン(8b)(このものはRu−触媒転移水素化における遂行能力が極端に乏しい)が、高収率、かつ優れたエナンチオ選択性でアリルアルコール(9b)に還元された。
【0096】
【表1】
【0097】
手元にある光学的に純粋なアリルアルコール(5b)により、我々はいずれの立体中心をも混交させることなく、(5b)をアルコール(6b)にカップル結合させる方法を探索した。η3−アリル金属中間体を経て進行する反応の文献に記載された立体特異性は、Pd−触媒によるアリルエーテル化を魅力的な選択とした。参照:a)Shu,C.;Hartwig,J.F.Angew.Chem.Int.Ed.2004,4794.b)Kim,H.;Lee.C.Org.Lett.2002,4369.b)Evans,P.A.;Leahy,D.K.J.Am.Chem.Soc.2002,7882。しかし、アルコール(6b)は立体的に密集し、求核性に乏しいために、このカップル結合に関与させるのに適切な候補ではなかった。驚くべきことに、(6b)の亜鉛アルコキシドは、Pd触媒によるエーテル化について報告されている標準的な条件下で、酢酸アリル(10b)に容易にカップル結合して、50%の収率でアリルエーテル(4)を単一のジアステレオマーとして生成した。エナンチオマー過剰率レベルの異なる(10b)を光学的に純粋な(6b)とのエーテル化に付したとき、それぞれの量のジアステレオマー(4ab)が観察され、高度の立体特異性を示した(表2)。
【0098】
【表2】
【0099】
エーテル化における穏当な収率は、この反応条件下で分解を受けてしまうアリルエステルの非常に反応性の高いことの結果であった。この反応性の減弱は、安息香酸エステル又はナフトエ酸エステルなどの弱い脱離基の選択により達成され、(4b)の収率を73%まで改善した。ナフトエ酸アリルエステル(11b)は結晶性固体であり、さらなる開発のために選択した。広範なリガンドのスクリーニングは、ブッフワルド(Buchwald)ビアリールジホスフィンがPd−触媒によるエーテル化において有効であったが、あまり高価ではなく、より簡単に入手し得る1,3−ジフェニルホスフィノプロパン(dppp)によっても同等の結果が得られることを明らかにした。さらなる改善として、アルコール(6b)の量は生成物のアッセイ収率に影響を与えることなく、1.0当量まで減少させることができた。最適な条件下、アリルエーテル(4b)は、収率80%、ジアステレオマー比>99:1で調製した(反応工程図3)。
【0100】
【化32】
【0101】
Pd−触媒によるアリルエーテル化のために開発された条件は、様々なアルコールに寛容であり、反応性中心での立体化学を完全に保持したまま、多様な一連のアリルエーテルを提供した(表3)。アリルエステル(11b)とアルコール(6b)の両方のエナンチオマーとのカップル結合においては、僅かな反応性の差異が観察されただけであった;しかし、立体的により妨害されたアルコールの場合には、明らかな「マッチ」及び「ミスマッチ」の対合が観察された。さらに、アリルエステルとアキラルアルコールとのカップル結合は、高エナンチオマー比で、期待どおりのアリルエーテルを提供した。この反応はエステル、ニトリル、さらにはケトンをも含む多様な電子求引性基に寛容であった;しかし、3−位に電子求引性基を欠く環状アリルエステルは、アリルエーテル化反応に不活性であった。
【0102】
【表3】
【0103】
アリルエーテル(4b)の高度に収束した効率的な調製は、アリール−金属種のジアステレオ選択的共役付加を経てシクロペンタン(2b)へ接近する手段を提供した。グリニヤール又はクプラート法により共役付加を達成する試行は、少なからぬ量の1,2−付加生成物と共に、様々な量の所望の共役付加生成物に導いた。アリールホウ酸誘導体のエナンチオ選択的Rh−触媒共役付加における最近の進歩が、我々に、我々の基質に対してこの方法を試みることを促した。参照:Hayashi,T.;Yamasaki,K.Chem Rev.2003,2829及びその引用文献。典型的に、エナンチオ選択的変異体と関連する問題は、「リガンドのない」背景反応の高い反応速度である。結果として、我々はリガンドの不在下に、基質(4b)を標準的な条件(5当量のホウ酸、3mol%[CODRh(OH)]2、ジオキサン/水)に付した。我々は、完全な立体制御とともに所望の共役付加産物を、89%のアッセイ収率で観察したことに満足した。ホウ酸量の有意な削減は、アリールトリフルオロホウ酸カリウム塩(参照:a)Batey,R.;Thanadi,A.N.;Smil,D.V.Org.Lett.1999,1683.b)Darses,S.;Genet,J.P.;Brayer,J.L.;Demoute,J.P.Tetrahedron Lett.1997,4393;このものは完全な変換を達成するために、1.5当量を必要とする)を用いることにより達成することができた。また、我々は、エタノールが溶媒系としてジオキサン/水の優れた代替物であり、より急速な変換、良好な収率、及びより汚染のない反応を提供することを発見した。最適化した条件下、熱力学的に好適な異性体へ平衡化した後、95:5のエピマー混合物として93%の収率で(15b)を得た。この混合物は容易に酸(2b)に加水分解され、また優れた収率でトリエチルアミン塩として単離され、不所望のエピマーは好都合に回避された(反応工程図4)。
【0104】
【化33】
【0105】
(2b)の効率的な製造法を証明するために、我々はこの方法の範囲と領域を決定しようとした。様々な電子豊富及び電子欠乏アリールホウ酸が、ジアステレオ選択Rh−触媒による共役付加において適切な求核試薬であり、それぞれの場合に、高収率と完全なジアステレオ選択性を提供した(表4)。この方法論は適度に障害のあるアリールホウ酸にも寛容であった;しかし、2,6−二置換アリールホウ酸での共役付加生成物は低収率であった。ヘテロ環状ホウ酸及びビニルホウ酸は、Rh−触媒によるプロセスでは全く無効であり、すべての事例において所望の共役付加物の収率は<5%であった。この方法論はまたα,β−不飽和ケトンとニトリルには有効であり、すぐれた収率とジアステレオ選択性でそれぞれの生成物を提供した。すべての基質(エステル、ニトリル、及びケトン)は、熱力学的平衡後に、高エピマー比を与えた(それぞれ、95:5、92:8、及び98:2)。
【0106】
【表4】
【0107】
Rh−触媒による共役付加方法はα,β−不飽和ケトンxxには有効であるが、エノレート・トラップとして塩化トリメチルシリルを用いる臭化4−フルオロフェニルマグネシウムのCu−触媒共役付加(参照:Varchi,G.;Ricci,A.;Cahiez,G.;Knochel,P.Tetrahedron,2000,2727)は、ケトンが熱力学的に好ましい異性体に平衡化した後、98:2のエピマー混合物として実際上94%の収率でxxを提供した。Cu−触媒による共役付加は、Rh−触媒による共役付加方法では達成し得なかったアリール、ビニル、ヘテロ芳香族、及びアルキル金属種の付加についても実証された。
【0108】
結論として、我々は非常にすくない工程により傑出した選択性で、高度に機能化されたシクロペンタノイド構造を途切れることなく構築する方法を報告してきた。この方法の収束性は、それを、構造的な複雑さを迅速に組み上げるための価値ある手段とし、そのルートの組み立てユニットの性質は、構造類似体の効率的生産を可能とする。本明細書に記載した例は、多様な一連の複雑な中間体が、この化学によって接近可能となることを示している。
次に、我々はキラル・オキサゾリジノン(3a)を、シクロペンタン酸(2b)から誘導されるハロメチルケトンでジアステレオ選択的にアルキル化することを試みた。
【0109】
【化34】
【0110】
クロロメチルケトン(4c)は、2工程経由で酸中間体(2b)から調製した:i)(2b)のメチルエステル化であって、(2b)を、極性溶媒、例えば、DMF、DMAc、DMSO若しくはアセトンなど中、約0℃ないし約60℃の範囲の温度で、ヨウ化メチル及びLi、Na、K、Ca若しくはCsなどの炭酸アルカリ金属により処理することからなる。また(2b)を、酸触媒、例えば、硫酸、TsOH、MsOH、又はベンゼンスルホン酸などの存在下、約50℃ないし還流の温度範囲で、MeOHにより処理することによりメチル化し得る;ii)メチルエステル(2c)のクロロメチル化は(2c)を、ClCH2CO2H及び式M4N(R16)2又はM4N(Si(R16)3)2(ここで、M4はLi、Na、K、又はMg(Mgは二価であり、変化する要あり)であり、各R16は、独立して、C1−4アルキルから選択された)の金属アミドにより、非プロトン性有機溶媒、例えば、THF中、約−20℃ないし約40℃の温度範囲で処理することから構成された。ClCH2I又はClCH2Brはまたこの反応で使用され得る。
【0111】
化合物(4a)は、極性有機溶媒、例えば、DMF、DMAc、DMSO、又はアセトンなどの中、約0℃ないし約60℃の温度範囲で、ヨウ化アルカリ金属若しくはアンモニア、例えば、LiI、NaI、KI、R4NI(ここで、RはH又はC1−4アルキルから選択される)による処理によって調製された。無水の条件がより良好な収率を与えた。
【0112】
【化35】
【0113】
キーとなる中間体(2a)の合成法は、本発明の実施態様を形成した。本方法は、式MN(R)2又はMN(SiR3)2(ここで、MはLi、Na、又はKであり、各Rは、独立して、C1−4アルキルから選択された)のアルカリ金属アミドの条件下、トルエン若しくはTHFなどの有機溶媒中、非プロトン性極性溶媒又はアミン付加体(例えば、DMPU、DMI、又はTMEDA)の存在又は非存在下で、約−78℃ないし約−40℃の温度範囲で、(3a)のエノレートをα−ハロケトン(4)に付加させることにより、四級キラル炭素中心を含有して構成された。極性の低い溶媒と非プロトン性極性溶媒との低い反応温度での組み合わせは、より良好な収率を与えた。
【0114】
トルエンなどの有機溶媒中、(2a)の溶液中の残余過剰量の(3a)は、水性LiOHでの選択的加水分解と、引き続く、約0℃ないし約40℃の温度範囲での水性NaHSO3処理により除去することができた。
【0115】
【化36】
【0116】
アルキル化されたオキサゾリジノン(2a)の、有機溶媒(例えば、THF、DME、メタノール、エタノール又はイソプロパノール)中、水性若しくは有機性NH3による約0℃ないし約60℃の温度範囲での加アンモニア分解は、直接に、アミナル(14a,b)のジアステレオマー混合物を与えた。
【0117】
【化37】
【0118】
(14a,b)のヒドロキシ基を還元して(16)とするために、酸、例えば、トリフルオロ酢酸、メタンスルホン酸、トリフルオロボラン・エーテレートなどの存在下、アセトニトリル、トルエン、酢酸、ニトロメタン、若しくはニトロエタンなどの有機溶媒中、又はそのままの状態で、約−40℃ないし約60℃の温度で、式R3SiH(ここで、RはH又はC1−4アルキルから選択される)で示されるシランにより、反応が完結するまで(通常、約2〜24時間)処理した。この還元は、(14a,b)からエナミン中間体(15a,b)を経由して、有機溶媒、例えば、アセトニトリル、トルエン、酢酸エチル、酢酸、ニトロメタン、若しくはニトロエタンなど中、又はそのままの状態で、約−40℃ないし約60℃の温度で、酸、例えば、トリフルオロ酢酸、メタンスルホン酸、トリフルオロボラン・エーテレートなどで処理することにより進行することが判明した。化合物(16)は同様の条件下で、(14a,b)及び(15a,b)の両方から製造し、そのジアステレオマー(16a)との混合物として得た。より良好な収率とジアステレオ選択性は、アセトニトリル中、エナミン(15a,b)から低温で還元により得られた。
【0119】
【化38】
【0120】
(16)のベンジルオキシカルボニル(Cbz)基を遷移貴金属、例えば、Pd/C、Ph(OH)2/C、Pt/Cなどによる接触水添により、又は酸性溶媒和により切断して(1)とした;接触水添はメタノール、エタノールなどのアルコール系溶媒中で、また溶媒和はHCl、HBr若しくはHIなどのハロゲン化水素、又はハロゲン化金属若しくはアンモニウムと酸(例えば、トリフルオロ酢酸、メタンスルホン酸など)との組み合わせなどの等価物により、有機溶媒(例えば、アセトニトリル、トルエン、又は酢酸)中、約0℃ないし約60℃の温度で実施した。このCbz基の脱保護を酸性条件下で実施した場合、(1)は(14a,b)又は(15a,b)からワンポット方式で得られた。Cbz基の脱保護の副産物であるハロゲン化ベンジルは、ヘプタンによる逆抽出により、反応混合物より容易に除去された。(1)の遊離塩基はその両親媒性物性のために、水層から有機層に抽出することが困難であった。t−BuOHとMTBEの混合溶媒は、水層から遊離塩基を抽出するために有効であることが判明した。粗製の遊離塩基は、IPA−IPAc−ヘプタン溶媒系からベンゼンスルホン酸塩として結晶化することにより精製した。ベンゼンスルホン酸塩は2つの結晶型、I型又はII型を有する。II型はI型よりも熱力学的により安定である。ベンゼンスルホン酸塩は、要すれば、アセトニトリル及びアルコール(例えば、メタノール、エタノール、又はイソプロパノール)から再結晶し得る。
【0121】
【化39】
【0122】
ラクタムhNK−1受容体拮抗薬(1)の、シクロペンタン核の高度に収束的、効率的、さらに完全にジアステレオ選択的、エナンチオ選択的合成が確認されている。本明細書に記載されている一般的方法は、酸中間体(2)及びヨードケトン中間体(3)両方の合成に適用された;これら中間体は共に好収率及び受容可能な純度で(1)を生成することが実証されている。
【0123】
【化40】
【0124】
(1)を作製するための初期の開発研究は、主として(2)が成功裏に(1)に変換されたことのある唯一の中間体であったために、(2)の効率的な合成に向けられていた。我々は高度に収束的なジアステレオ選択的、エナンチオ選択的様式で、迅速に(2)を製造する新しい方法を成功裏に開発した(反応工程図1)。この合成のハイライトは、3−カルボキシメチルシクロペンテノン(5)の高度にエナンチオ選択的酵素還元、立体特異的Pd−触媒によるエーテル化、及びジアステレオ選択的Rh−触媒による共役付加である。
【0125】
【化41】
【0126】
この合成法は既存の(2)の製造法よりも改善されていたが、このルートは(5)の調製において、規模的に、またコスト的に、Rh−触媒による共役付加方法を困難なものとした。最終的にこの化学における改良法は、(2)が7工程で(1)に変換し得ることを証明した(反応工程図2)。この改良されたルートを試験すると、(2)を(3)に変換するためには数工程が必要であることから、(3)が(1)を収束的に合成するためのより好適な合成標的であることが判明した。
【0127】
【化42】
【0128】
(2)を合成するために開発された一般的方法は、(3)の製造に必要な基質の変化に適応させるために修飾した(反応工程図3)。(3)を直接的に標的とすることにより、全工程は、大量の出発原料を製造する使用可能な方法を提供し、Rh−触媒による共役付加をCu−媒介共役付加に転換し、該方法の数工程を除くことにより、改善された。最適化された(1)の合成はシアノケトン出発原料から10工程(ヨードケトンから4工程)に縮減された。30gスケールでの(2)と(3)両方に対するこの方法の発見と立証について以下に記載する。
【0129】
【化43】
【0130】
【表5】
【0131】
基本的な溶媒の選抜では、当初のエーテル化条件にて使用したTHF(表3)よりも良好な溶媒は見つからなかった。他の配位エーテル型溶媒(又は多分三級アミン付加体)が改善に役立ち得るとの見込みが未だ存在する。このことは未だ検討されていない。
【0132】
【表6】
【0133】
リガンドを徹底的にスクリーニングして(表4)、ブッフワルド・ビアリールホスフィン・リガンドのジシクロヘキシルホスフィノ変異体が、対応するジ−tert−ブチルホスフィノ体よりもはるかに良好に機能することを明らかにした。このことは、比較的に障害の少ないホスフィン類がPd−触媒によるエーテル化に好適であることを物語っていた。一般に、ブッフワルド・ビアリールホスフィン・リガンドは、dpppなどのジホスフィンをキレート化する(典型的には、室温で2〜24時間)よりも僅かに高い収率とより速い反応(通常、0℃で2時間)を与えた。それにもかかわらず、dpppリガンドはコストが低いという有利性と、良好な利用可能性を有していた。キレート化ジホスフィン・リガンドに関しては、dpppがdppe或いはdppbよりもより効果的であった。触媒負荷をPd2%及びリガンド3%(ブッフワルド・ビアリールホスフィン・リガンドを使用)に低下させると、p−ニトロ安息香酸エステルのエーテル化が僅かに遅くなったが、アッセイ収率は実質的に変らなかった(0℃で7時間後に96%の変換率と70%の収率)。
【0134】
【表7】
【0135】
さらなる改良として、ナフトエ酸基質(R=2−Nap)の場合には、(9)のアッセイ収率に有意な影響を与えることなく、ベンジルアルコール(8)の量を1.2当量に減量できることが判明した。ナフトエ酸エステルとdpppリガンドの組み合わせは、通算収率とコスト効率の意味で最善の反応を提供し、さらなる開発のために選択された。
【0136】
反応濃度の影響についても簡単に検討した。THF中、0.8Mと0.3Mの反応の間に小さな差(78%対75%のアッセイ収率)が観察された(本明細書で、Mは、mLの溶媒に対するmmol(ミリモル)のエステル基質と定義した;Et2Zn溶液が供給するヘプタン溶媒を含む)。しかし、幾分製造率の高い0.8Mの反応は、攪拌の困難な濃厚油を形成することが問題となった。妥協点として、最終決定した手法では0.5Mの濃度を選択した。
【0137】
ナフトエ酸エステル(7)をエーテル化のための理想的な基質として同定し、製法をその形成のために開発した。アリルアルコール(6)のアシル化が反応性に富む反応であり、塩化ナフトイルでの処理が所望のアリルエステルのアッセイ収率を低下させることは銘記すべきである。対照的に、無水ナフトエ酸のその場での形成(酸塩化物と酸から)と、引き続くアルコールでの処理が、好収率(91%)でアリルエステル(7)を生成した。結晶化による単離は、77%の収率と>99%のLCAPで(7)を提供した。
【0138】
エーテル化反応のための3gでの先行試行は、1.2当量のアルコール(8)、3%Pd及び4%dpppリガンドを用いて実施した。78%のアッセイ収率と75%の単離収率(95%LCAP、98%アッセイ)が得られたが、反応は比較的停滞ぎみであり、>99%の完結までに2日を要した。それ故、4%Pdと6%のリガンドを20gでの実証のために使用した。
【0139】
20gスケールでの反応は、1時間の導入期間を示したが、反応が加速し始めたときにも有意な発熱は観察されなかった。アリルエーテル(9)は、ダルコ(Darco)KB−Bでの処理(Pdを除去するため)、溶媒のMeOHへの交換、及びMeOH−水からの結晶化によって単離された。20gスケールでは母液の損失(6%)の増加のために、幾分低い単離収率を観察したが、3gでの先行試行に比較して、有意により純度の高い生成物を得た。最終的に、20gの(9)は、71%の収率と>98%の純度で結晶化により単離した(反応工程図4)。
【0140】
【化44】
【0141】
【化45】
【0142】
シクロペンタン核の合成を完成するためには、ジアステレオ選択的共役付加が必要であった。我々は、嵩だかいエーテル置換基が侵入してくる求核試薬の面選択性を有効に制御して、共役付加生成物に所望のトランス立体配置を与え得るであろうと想定した。熱力学的条件下でのエステルの異性化がトランス配置を強力に支持し、完全にジアステレオ選択的様式でトランス,トランス−1,2,3−三置換シクロペンタン(10)を与えた。
【0143】
基質(9)への最適な共役付加には、基質への4−フルオロフェニル・グリニヤール試薬の付加が関与しよう。同様の形質変換についての文献調査では、所望の共役付加生成物の中庸ないし低収率(20〜60%)を与える入り混じった結果を明らかにした。実際、(9)に関して、CuI及びTMSClの存在下で、臭化4−フルオロフェニルマグネシウムとの共役付加を実施した場合、約45%の(10)を観察した(eq.7)。さらに分析すると、共役付加生成物はさらにグリニヤールとの1,2−付加を受けて、ジアリールカルビノール(27)を生じた;このものは40%の収率で得られた。付加物、溶媒、又は温度によるこの過剰反応を回避する試行は失敗に終わった。事実、必要量に満たないグリニヤール試薬で反応を実施したとしても、等モル比の(10)と(27)が観察された。
【0144】
【化46】
【0145】
グリニヤール共役付加に対する別法は、Rh−触媒によるアリールホウ酸との共役付加であった。この分野の文献報告の殆どが、共役付加生成物において不斉誘導を生じるキラルリガンドの使用が関わっている。この反応の不斉変異体を開発することと関連する問題の一つは、「リガンドなし」のRh−触媒による共役付加を遂行することの容易さである。我々は、ジアステレオ選択的なRh−を触媒とする共役付加をリガンドの不存在下で容易に実施し得るとすることに疑いを持っていた。実際には、標準的な条件下で、[CODRh(OH)]2の存在下、(9)を4−フルオロフェニルホウ酸と反応させると、良好な選択性(>99:1)で所望の共役付加生成物(10)が得られた。銘記すべきは、共役付加ステップのアッセイ収率が、反応に加えたホウ酸の量に強く影響されるということである(表5)。1.5当量限りのホウ酸を負荷した場合、32%のアッセイ収率が得られただけであった。対照的に、5当量のホウ酸を負荷した場合には、89%アッセイ収率の所望の生成物が観察された。このことは、ホウ酸のヒドロ脱ボリル化が競合的副反応であるとする文献の報告と矛盾がない。
【0146】
【表8】
【0147】
Rh−触媒による共役付加にアリール亜鉛試薬を使用する試みは不成功であり、多くの不純物と最小量の所望生成物(<10%)に導いた。対照的に、アリールトリフルオロホウ酸塩に置き換えると、非常に上手くいき、アッセイ収率の損失なしに、フルオロホウ酸塩の添加を1.5当量に減量し得る。この反応はジオキサン/H2Oで観察された性能に比べてEtOH中での性能が改善されることが判明し、1.5当量のアリールフルオロホウ酸塩を使用するだけで、93%のアッセイ収率で(10)が得られた(eq.8)。
【0148】
【化47】
【0149】
興味深いことに、(10)と(10a)のジアステレオマー比は、グリニヤール共役付加で観察される比よりもかなり高かった(85:15に対し55:45)が、これは恐らく反応条件下での部分的異性化によるものである。(10)と(10a)の混合物を異性化条件(NaOMe、MeOH、50℃)に付すと、その比が95:5の平衡に達した(反応工程図5)。さらに加熱しても、あるいは塩基性としても、その比は変らなかった。以前に開発した方法では、(1)の合成を完結させるために、(10)を必要とした;しかし、固形物の単離の容易さを示すために、また標準品と比較してその構造と不純物プロフィールを検証するために、(10)を(2)に変換した。
【0150】
【化48】
【0151】
異性化の後、エステル(10)を同じ反応容器内で加水分解し、酸(2)とすることができた。水性HClで中和した後、対応する酸を抽出して、NEt3半溶媒和物として単離した。共役付加/異性化方法は20gで実証し、同じ容器中で加水分解して、(2)を得た。19.7gの(2)をNEt3半溶媒和物として単離したが、その物質の純度は以前の方法で得られたものと同様であった;98.2LCAP(1.3面積%;10a由来のエピマー)。
【0152】
要約すると、酵素還元/Pd−エーテル化/Rh−共役付加ルートが、酸中間体(2)の迅速な接近のための効率的な方法として立証された。この合成はこのフラグメントの当初の合成よりも有意に短く(5工程対>12工程)、従来の合成法で要求された変わりやすいエーテル化方法を回避し、同様の純度と改善された通算収率(35%対<25%)で(2)を生じる。
【図面の簡単な説明】
【0153】
【図1】図1は式(Ia)で示される化合物のベンゼンスルホン酸塩の無水結晶形I型の特徴的なX線回折パターンである。
【図2】図2は式(Ia)で示される化合物のベンゼンスルホン酸塩の無水結晶形I型の典型的なDSC曲線である。
【発明を実施するための形態】
【実施例】
【0154】
実験手法:第1部
工程1−カルボキシメチルシクロペンテンの酸化:
【0155】
【化49】
【0156】
手法:
磁気攪拌バーと内部温度測定用電極を備え、冷却(0℃)した2L容量の丸底フラスコに、無水酢酸(615g、570mL、6.02mol)を加えた。三酸化クロム(214g、2.14mol)を分割して加え、その間、攪拌を一定に保ち、発熱を制御した。得られる血液様赤色溶液を、温度が20℃に冷えるまで攪拌して三酸化クロムを溶解した。5L容量の三頚フラスコに滴下漏斗、上部からの攪拌装置、窒素送入口及び内部温度測定用電極を取り付け、CH2Cl2(1.4L)中の(4)(100g、101mL、0.793mol)を加えた。三酸化クロムと無水酢酸からなる酸化溶液を滴下漏斗に入れ、10ないし14℃の内部温度を維持しながら、反応混合物に滴下した。当初黄色であった溶液が酸化剤を最初の2〜3滴加えた後、暗色となった。
【0157】
反応は、実験室での容器サイズに制限があるため、同じサイズのバッチで2回実施した。各バッチは以下のように正確に同じ方法で処理した。暗色の均一溶液は、上部からの攪拌装置を備えた4Lビーカーに注意深く注入した。反応フラスコを250mLのCH2Cl2ですすいだ。500mLのH2Oを加え、次いで、10gのNaHCO3を加えると、ガスが発生した。追加のNaHCO3(830g、10mol)を分割して加え、その間、粘稠混合物の攪拌速度を500rpmに維持した。得られる暗緑色の懸濁液を1LのH2Oで希釈し、ソルカフロックの1cmパッドを容れた3Lの硝子フィルター付き漏斗で濾過した。二相溶液をCH2Cl2(3×1L)で抽出し、併合した有機相をMgSO4で乾燥し、次いで濾過し、得られる溶液を減圧濃縮して淡緑色油を得た。30cmビグローカラムで蒸留し、次いでMTBE:ヘキサン(1:10、55mL全量)から再結晶し、38.4gの(5)を白色結晶性固体として得た(35%)。
【0158】
工程2−酵素還元:
【0159】
【化50】
【0160】
手法:
カリウム二塩基性バッファー(100mM、pH7.0、2L)に、ギ酸ナトリウム(120g)及びニコチンアミド・アデニン・ジヌクレオチド(NAD、8グラム)を加えると、バッファーpHが6.7に下がった。このバッファーに、酵素、アルコールデヒドロゲナーゼRE(5g、185KU)、ギ酸デヒドロゲナーゼ(20g、94KU)を加えた。基質(5)(10g、0.071mol)を粉末としてそのまま加え、温度を25℃に調整し、2N硫酸でpHを6.5に調節した。反応を24時間熟成し、次いで、酢酸エチル(2倍容量抽出)で抽出し、さらに減圧濃縮した。(6)の通算収率は83%、抽出による損失は2%、残余エノンは<3%であった。残りの13%の質量バランスは、水溶液中での不安定性によるエノンの損失によると判断した。
【0161】
工程3−アリルアルコールのアシル化
【0162】
【化51】
【0163】
手法:
ジクロロメタン(200mL)中、2−ナフトエ酸(24.6g、143mmol)及び塩化2−ナフトイル(27.2g、143mmol)の懸濁液を氷浴にて+5℃の内部温度に冷却した。ジイソプロピルエチルアミン(89mL、511mmol)を加え、その間、内部温度を24℃以下に維持した。発熱が収まったところで、氷浴を室温水浴に置き換えた。最初に褐色の澄明な溶液が形成され、それが次第に微細なスラリーに変るので、それを室温で30分間攪拌した。ジクロロメタン(50mL)中の(6)(14.4gアッセイ、102mmol、>99%ee)及びDMAP(1.25g、10.2mmol、0.10当量)からなる溶液を一度で加えた。非常に穏やかな発熱が観察され、温度が23℃で最大に達した。反応混合物を室温で2.5時間攪拌した。水(10mL)を加え、反応混合物を室温で2.5時間攪拌した。この時点でのHPLC分析は、過剰の無水ナフトエ酸が完全に加水分解されていることを示した。反応混合物をMTBE(500mL)と併合し、飽和のNaHCO3水(2×500mL)、水(500mL)、1M−HCl水(500mL)及び最後に水(4×500mL)で洗浄した。暗褐色の濁りのある有機相をソルカフロックの短いパッドで濾過し、濾液中に91%のアッセイ収率を得た。この溶液を200mL容量まで濃縮し、ヘプタン(200mL)を加え、この混合物を〜10gのシリカゲルで濾過し、ヘプタン−MTBE(2:1)100mLで溶出した。濾液を200mL容量まで濃縮し、種結晶を加えた。さらに50mLの溶媒を40℃で除去し、残りの懸濁液は室温にもどしながら2時間攪拌した。室温で15時間後、懸濁液を濾過し、濾過ケーキを50mLのヘプタンで洗い(母液損失4%)、23.2gの(7)を微細白色粉末として、77%収率及び99%LCAPで得た。
【0164】
工程4−Pd−触媒によるエーテル化反応:
【0165】
【化52】
【0166】
手法:
250mL容量の丸底フラスコに(8)(23.3g、90.3mmol、1.2当量)を容れ、排気してから窒素を充填した。THF(75mL)を加え、得られる溶液を氷浴で+5℃に冷やした。1.0M−Et2Zn/ヘプタン(45mL、45mmol)を加えると、緩やかに発熱し、15℃となった。冷却浴を除き、得られる澄明な溶液を室温で1時間攪拌した。
【0167】
別個の500mL丸底フラスコに、(7)(22.3g、75.3mmol)、Pd(OAc)2(676mg、3.01mmol、4mol%)、1,3−ビス(ジフェニルホスフィノ)プロパン(1.86g、4.51mmol、6mol%)及びL−トリプトファン(1.54g、7.54mmol、10mol%)を装入した。次いで、フラスコを排気してから窒素で充満させ、氷浴にて冷却した。第一のフラスコからのアルコキシド溶液はカニューレを介して第二のフラスコに移した。冷浴を除去し、得られる緑褐色懸濁液を室温で16時間攪拌した。この時点でのHPLC分析は、ナフトエ酸エステルの完全な変換を示していた。淡褐色懸濁液を氷浴で冷却し、1M−HCl水(100mL)及びMTBE(100mL)と併合すると発熱して15℃となった。この懸濁液を5℃で15分間攪拌し、次いでソルカフロックで濾過してMTBE(200mL)で溶出した。淡黄色濾液を水(3×200mL)、5%NaHCO3水(2×300mL)、及び水(2×200mL)で洗浄した。有機相をHPLCで分析すると、生成物を78%のアッセイ収率で得た。ダルコ(Darco)KB−B(15g)を有機相に加え、懸濁液を室温で3時間攪拌し、次いで、ソルカフロックで濾過した。ほぼ無色の濾液をMeOHと溶媒交換し、最終容量120mLとした。水(5mL)を加え、この溶液に種結晶を加え、さらに水(70mL)を15分間で滴下した。スラリーを室温で1時間攪拌し、次いで濾過した。得られる白色結晶を真空乾燥し、20.4gの(9)を71%の収率及び98%のLCAPで得た。
【0168】
工程5−Rh−触媒による共役付加/加水分解/酸単離:
【0169】
【化53】
【0170】
手法:
磁気攪拌機、熱電対、及び窒素送入口を備えた250mL容量三頚丸底フラスコに、(9)(19.0g、0.0497mol)、NaHCO3(1.94g、0.023mol)、フルオロホウ酸塩(16.1g、0.080mol)、及び[CODRh(OH)]2(646mg、0.0015mol)を加えた。フラスコを密封し、1時間窒素を送入した。1時間窒素で脱ガスしてあったエタノール(150mL)を、固形物を容れた反応容器にカニューレを介して供給し、その反応混合物を90℃に加熱した。HPLC分析によると、反応は3時間以内に完結した。混合物を50℃に冷却し、11mLの25wt%NaOMe/MeOHを供給した。この混合物を50℃で4時間熟成した;このものは95:5のジアステレオマー比を示し、完全な平衡にあった。混合物を40℃に冷やし、3N−KOH(40mL)を供給し、40℃で1時間熟成すると、HPLC分析で完全な加水分解を示した。混合物を室温に冷やし、水270mLとヘプタン270mLで希釈した。ヘプタン層を除去し、水層をヘプタン300mLと混合し、50mLの濃HClで中和した。層分離し、ヘプタン層を水(200mL)で2回洗浄した。ヘプタン溶液をアッセイすると、(10)が18.4g(83%)であることを示した。ヘプタン溶液を共沸蒸留させてKf<500とし、次いで、150mLの容量に濃縮した。この溶液をMTBE(15mL)で希釈し、45℃に加温し、NEt3(3.0mL、0.0215mol)を加えた。このバッチを15分間熟成した後、この混合物に種結晶(10mg)を加え、バッチを2時間かけて室温までゆっくりと冷やした。バッチを1時間熟成し、上清をアッセイし(10.8mg/mL)、溶融硝子漏斗にて濾過した。得られる結晶性固体をヘプタン:MTBE(10:1)50mLで洗い、19.4gの(2)(76%)を得た。母液への損失=1.95g(7.6%)。単離した固体を分析すると、98.0wt%の純度、98.2LCAPを示し、最大の不純物は(10a)由来のジアステレオマーであった(1.3面積%)。ベンジル性ジアステレオマーも0.3面積%で存在した。
【0171】
工程6−メチルエステル化/クロロメチル化/ヨード化:
【0172】
【化54】
【0173】
手法:
DMF(480mL)中、K2CO3(63.5g、460mmol)及び(2b)(160g、98.6w%、306mmol)の懸濁液に、ヨウ化メチル(38.2mL、613mmol)を23℃で30分かけて加えた。混合物を室温で20時間攪拌した。この反応混合物に、MTBE(1280mL)、10%食塩水(800ml)、及びH2O(800ml)を加えた。有機層を分離し、0.5Nリン酸バッファー(800ml、pH6.8)及び10%食塩水(800ml)で洗った。有機層(1240ml)を40℃でのMTBEとの共沸処理により乾燥し、次いで、溶媒を40℃でTHFと置き換えた。THF溶液中のこの粗製の(2c)(480ml、136gアッセイ、283mmol、92.5%、KF<400ppm)は、さらに生成することなしに次工程で使用した。
【0174】
1.9M−n−BuMgCl/THF(6当量、1700mmol、895mL)の溶液に、ジイソプロピルアミン(6.6当量、1870mmol、262mL)を25〜30℃で加え、得られるスラリーを2時間20〜25℃で攪拌した。THF(678mL)中、(2c)(136g、238mmol)及びクロロ酢酸(3当量、850mmol、80.3g)の混合物をこのスラリーに1時間かけて、15℃以下で滴下した。反応混合物を20〜25℃で16時間攪拌し、次いで、6N−HCl水(15当量、4.25mol、709mL)及びMTBE(1360mL)からなる混合物に15℃以下で、30分かけて移行した。有機層を分離し、0.5Nリン酸バッファー(800ml、pH6.8)により、水層のpHが>6.0となるまで洗い、次いで、23%食塩水で洗った。有機層を溶液のKFが<500ppmとなるまで、MTBEとの共沸処理により乾燥し、次いで、全容量を1440mLに調整した。(4c)のHPLCによるアッセイ収率は125g(251mmol、88.7%)であった。
【0175】
MTBE(1440mL)中の(4c)(125g、251mmol)の溶液をアセトンと溶媒置換し、全容量を1120mLに調整した。この溶液にNaI(56.5g、377mmol、1.5当量)を加え、さらにアセトン(125ml)を室温で加えた。混合物を外気温で16時間攪拌した。反応混合物を(4a)の種結晶(125mg)と水(2120mL)の懸濁液に1時間かけて滴下し、反応容器をアセトン(125ml)ですすいだ。2時間の熟成の後、(4a)の結晶を濾取し、濾液がpH>4.5となるまで水洗した。乾燥後、(4a)を黄色結晶(153g、93.8wt%、99.2%収率)として得た。
【0176】
工程7−オキサゾリジノンのカップリング結合:
【0177】
【化55】
【0178】
手法:
トルエン(100mL)中、DMPU(15.0mL)及びDMI(15.0mL)の混合物に、1.0M−LHMDS/ヘキサン(40.8mL、40.8mmol、2.4当量)を20〜25℃で加えた。得られる混合物を5分間以上攪拌し、次いで、−75℃以下に冷却した。この溶液に、トルエン(100mL)中のオキサゾリジノン(3a)(13.2g、42.5mmol、2.5当量)を−70℃以下で1時間かけてゆっくりと加え、得られる溶液を−70℃以下で30分間攪拌した。次いで、上記溶液に、トルエン(50.0mL)中の(4a)(10.0g、17.0mmol)の溶液を−70℃以下で1時間かけてゆっくりと加えた。−70℃以下で30分間攪拌した後、反応混合物に10%クエン酸水(100mL)を加えて反応停止し、外気温まで加温した。有機層を分離し、10%NaHCO3水(50mL)で洗い、次いで、外気温で18時間、1N−LiOH水(100mL、100mmol、5.9当量)で処理した。水層を分離した後、有機層を1N−NaHSO3水(100mL、100mmol、5.9当量)及び10%食塩水(50.0mL)で洗浄した。生成物(2a)をトルエン溶液(9.44g、72%、254mL)として得、さらに精製することなく次工程で使用した。
【0179】
工程8−エナミン形成:
【0180】
【化56】
【0181】
手法:
(2a)(2.02gA、2.62mmol)のトルエン溶液を40℃でTHF(約23.0mL)と溶媒交換し、次いで、外気温で24時間、25%NH3水(20.6当量、4.04mL、54.0mmol)で処理した。反応混合物に5%食塩水(10.1mL)及びAcOEt(10.1mL)を加えた。有機層を分離し、10%NaHSO3水(10.1mL×2)、10%K2HPO4水(10.1mL)、及び20%食塩水(10.1mL)で洗った。アミナール(14a,b)をAcOEt溶液(1.23g、1.80mmol、70.8%、31.8mL)として得た。
【0182】
(14a,b)(4.04g、KF4.6%)のAcOEt溶液をAcOEtにより共沸乾燥し、40℃で20.2mL(KF<0.3%)まで濃縮した。次いで、この混合物に0〜5℃でMsOH(0.31mL、1当量)を加え、反応混合物を0〜5℃で1時間攪拌した。反応混合物に水(20.2mL)を加え、有機層を分離し、0〜10℃で10%K2HPO4水(20.2mL)により、15〜25℃で20%NaCl水(20.2mL)により順次洗い、次いで、24〜27℃で活性炭(シラサギP:312mg及びダルコKB−B:312mg)により30分間処理した。混合物を濾過し、AcOEt(12.1mL)ですすいだ。濾液と洗液を併合し、CH3CNと溶媒交換し、15.9mLに濃縮した。CH3CN中、エナミン(15a,b)を位置異性体の混合物(3.19g、100%、異性体比76:24)として得た。
【0183】
工程9−最終段階(シラン還元/脱保護/結晶化):
【0184】
【化57】
【0185】
手法:
MsOH(5.85mL、80.3mmol、8.7当量)及びEt3SiH(1.93mL、12.1mmol、1.3当量)を、0℃以下で連続して、CH3CN中のエナミン(15a,b)の溶液(30mL、6.15g、9.25mmol、1当量)に加え、次いで、得られる混合物を−5〜0℃で4時間攪拌した。(15a,b)が完全に消費された後、30%HBr/AcOH(3.20mL、16.1mmol、3.5当量)を5℃以下で滴下した。この混合物を38〜42℃に加温し、同温度で一夜攪拌し、次いで、0℃に冷却した。この反応混合物に、水(30.8mL)及び活性炭(シラサギP、1.23g)を連続して加えた。得られた混合物を1時間攪拌した後、濾過し、CH3CN/H2O=1/1(18.5mL)ですすいだ。濾液と洗液を併合し、ヘプタン(61.5mL×3)で洗浄した。水溶液を20℃以下で5N−NaOH水(17mL)によりpH3〜4に調整した。この反応混合物にMTBE/t−BuOH=2/1(30.8mL)を加え、得られた混合物を20℃以下で5N−NaOH水(19.5mL)によりpH=9〜10の塩基性とした。有機層を分離した後、水層をMTBE/t−BuOH=2/1(30.8mL)で抽出した。容器をMTBE/t−BuOH=2/1(12.3mL)ですすいだ。有機層を併合し、12%リン酸バッファー(pH=6.5;30.8mL)及び23%食塩水(30.8mL)により順次洗浄した。有機溶液はIPAcと溶媒交換(KF=444ppm)すると、不均一系となった。IPAc懸濁液を濾過し、濾過固体をIPAc(12.3mL)で洗った。併合した濾液を35mLに濃縮した。IPAc中、有機塩基としての化合物(1)を褐色溶液として得た(4.93g、100%)。
【0186】
IPAc中、(1)遊離塩基の溶液(221mg/mL、5.0mL、1.11g、2.08mmol、1当量)をIPAc(9.43mL)で希釈した。この溶液に、PhSO3H・H2O/IPA(1.5M、1.38mL、2.08mmol、1.0当量)を2時間かけて40℃で滴下した。得られたスラリーを40℃で一夜攪拌し、このスラリーにヘプタン(16.7mL)を1時間で添加した。40℃で21時間攪拌した後、このスラリーを外気温まで冷やした。生成物を濾集し、IPAc/ヘプタン(1/1:8.3mL×2)で洗い、減圧下、40℃で一夜乾燥した。化合物(1)はベンゼンスルホン酸塩の無色固体(1.21g、99wt%、84.6%、I型)として得た。
【0187】
実験手法−第2部:
工程1−シクロペンテノンの臭素化:
【0188】
【化58】
【0189】
手法:
磁気攪拌バー、窒素送入口、滴下漏斗及び内部温度測定用電極を備えた3L容量の丸底フラスコに、1.25LのCH2Cl2中の2−シクロペンテノン(100g、1225mmol)を装入し、−20℃に冷却した。HBr(27.3mL、245mmol)を加え、その淡黄色溶液を5分間攪拌した。滴下漏斗に臭素(61.9mL、1225mmol)を容れ、内部温度を−24ないし−20℃に維持しながら、1時間かけてそれを滴下した。添加の間に、臭素は急速に脱色した。この黄色溶液を、TLCが出発エノンの消失を示すまで、30分間、−20℃で攪拌した。内部温度を−20℃に維持しながら、ピリジン(149mL、1837mmol)を滴下した。滴下終了後、この溶液を0℃で1時間攪拌した。1M−Na2S2O3(1L)を加えて反応を停止し、MTBE(2L)で希釈した。有機相を1M−HCl(2×1L)で洗い、次いで、H2O(1L)で洗った。暗色の有機相をNa2SO4で乾燥し、濾過した。CH2Cl2の残部をMTBEと溶媒交換し、MTBE:CH2Cl2の最終の比が8:1になるようにした。この暗色溶液を30%(60g)ダルコKB−Bと一夜攪拌した。ダルコはソルカフロックのショートパッドで濾去し、無色の溶液を得た。減圧下で溶媒を除去し、170gの(15)を白色結晶性固体(95.83wt%;87%単離収率)として得た。
【0190】
工程2−シアノ化/脱離:
【0191】
【化59】
【0192】
手法:
MeOH(400mL)中、(15)(62.5gアッセイ;0.388mol)とAcOH(22mL、0.384mol)の溶液に、15℃(吸熱溶解による)で、固形のNaCN(取り扱い注意;猛毒性;28.5g、0.582mol、1.5当量)を加えた。温度は5分間で15℃から30℃に上昇し、その時点でフラスコを氷水浴で冷却した。内部温度が18℃まで下がったところで冷却浴を除いた。1.5時間室温で攪拌した(TLCによると変換は不完全)。次いで、追加の固形NaCN(9.5g、0.194mol)を加え、室温で2時間攪拌した(TLCによると完全に変換)。
【0193】
フラスコの底に溶けずに残っていた約5gの固形NaCNから傾斜して、褐色の反応混合物を3Lの分液漏斗に移した。この溶液を水(1L)と併合し、CH2Cl2(1L+400mL+400mL)で抽出した。3つの有機相中の生成物をアッセイ(HPLC)すると、それぞれ、76%、4%、及び0.6%であった。3度目の抽出後の水相の生成物の損失は0.3%であった。
【0194】
第一及び第二の有機相(合計アッセイ収率80%)を組み合わせ、シリカ(約100gのシリカ)の短いプラグで濾過し、濾液を47.1gの重量(HPLCにより66wt%純度)に濃縮した。暗褐色油の20.6gアリコートを1mmHg、60〜70℃で蒸留し、12.7gの(16)を淡黄色液体として得た(94wt%純度、このアリコートにもとづく収率66%)。
【0195】
工程3−酵素還元:
【0196】
【化60】
【0197】
カリウム二塩基性バッファー(0.1M、pH7.0、1L)に、グルコース(100g)及びニコチンアミド・アデニン・ジヌクレオチド(NAD、4g)を加え、バッファーのpHを6.7に低下させた。このバッファーに酵素、アルコール・デヒドロゲナーゼRE(1g、37KU)、グルコース・デヒドロゲナーゼ103(1g、67KU)を加えた。35℃の温度及び400rpmの攪拌による1Lの反応のために、2つの500mL反応機を使用した。基質(16)(20g、0.19mol)を各反応機に10gずつ直接加えた。2.5M炭酸カリウムによりpHを6.5に調整した。基質(16)はpH>8.0で不安定であることが知られており、そのため、上記の表面添加により、また通常使用される2N−NaOHの替わりに弱い塩基(2.5M炭酸カリウム)を用いて、塩基との接触を最小とした。反応を20時間熟成させ、その時点での変換率は95%より高かった。この変換はグルコン酸の形成から生じるpH変化を調節する塩基の消費量により容易にモニターすることができる。補助因子リサイクルからのグルコン酸の形成は、形成されるアリルアルコール生成物の量に正比例した。反応液を酢酸エチル又はイソプロピルアルコールで抽出(2倍容量の抽出)し、次いで、減圧濃縮した。(17)の通算収率は92%であり、抽出による損失は1%、残余エノンは<2%であった。5%の質量バランスの損失は、反応条件下での(16)の分解によるものであった。
【0198】
工程4−アシル化:
【0199】
【化61】
【0200】
手法:
磁気攪拌バー及び窒素送入口を備えた1L容量の一頚丸底フラスコに、2−ナフトエ酸(20.98g、122mmol)及び塩化2−ナフトイル(23.49g、122mmol)及びジクロロメタン(135mL)を装入した。フラスコを内部温度0℃に冷却した。ジイソプロピルエチルアミン(76mL、436mmol)を加え、その間、内部温度を<5℃に維持した。得られた濁りのある褐色溶液を室温に加温し、30分間攪拌した。(S)−1−シアノ−1−シクロペンテン−3−オール(17、9.50g、87.1mmol)及びDMAP(1.07g、8.71mmol)をジクロロメタン(50mL)に溶かした。この溶液を反応混合物に一度に加え、室温で3時間攪拌した。水(8.50mL、472mmol)を加え、反応液を室温で90分間攪拌した。反応液をMTBE(400mL)で希釈し、飽和NaHCO3(2×400mL)で洗浄した。次いで、有機層をH2O(400mL)、1M−HCl(400mL)及びH2O(4×400mL)で洗った。合計水相損失は1.5%であった。暗色有機層をダルコKB−B(5.7g)とともに3時間攪拌した。この溶液をソルカフロックのパッドで濾過し、減圧下、40℃で濃縮し、淡黄色固体を得た。この固体をMTBE(300mL)に溶かし、40℃で減圧下で一定容量のヘプタンと溶媒交換した。この固体を濾過し、ヘプタンで洗い、19.24gの(18)を淡黄色固体として得た(100.0wt%、84%単離収率)。3.4%の損失が母液で起こった。
【0201】
工程5−Pd−触媒によるエーテル化:
【0202】
【化62】
【0203】
手法:
熱電対及び窒素送入口を備えた500mL三頚丸底フラスコを排気してから、窒素を充満させた。次いで、フラスコに、空気との接触を最小としながら、注意深く、アルコール(8)(31.0g、120mmol、1.0当量)を装入し、隔壁で密封し、THF(150mL)をシリンジで装入した。この溶液を氷浴にて+5℃に冷却した。1.0M−Et2Zn/ヘキサン(63mL、63mmol)を加えると、おだやかに発熱し、13℃となった。この溶液を氷浴中で30分間攪拌し、この反応混合物に、ニトリル(18)(31.6g、120mmol)、Pd(OAc)2(1.35g、6.01mmol、5mol%)、1,3−ビス(ジフェニルホスフィノ)プロパン(3.71g、9.00mmol、7.5mol%)、及びL−トリプトファン(2.45g、12.0mmol、10mol%)を、空気との接触を最小としながら、注意深く、固形物として加えた。15分後、氷浴を除去し、反応混合物を放置して室温とした。1時間後、緩和な発熱反応が始まり、内部温度が27℃に達した(外部冷却は適用されず)。さらに2時間後、HPLCにて分析すると、(18)の完全な変換を示した。薄い懸濁液を1Lのフラスコに移し、15gのソルカフロックを加え、次いで、激しく攪拌しながらMTBE(300mL)を加えた。懸濁液を30分間攪拌し、ソルカフロックで濾過し、濾液をジクロロメタン(200mL)と併合し、1M−HCl水(2×500mL)、水(500mL)、5%Na2CO3水(3×500mL)、及び水(2×500mL)で洗った。有機相をHPLC分析すると、(19)のアッセイ収率は84%であることを示した。有機相にダルコKB−B(15g)を加え、その懸濁液を室温で18時間攪拌し、ソルカフロックで濾過した。ほぼ無色の濾液を油状まで濃縮し、81wt%の(19)(77%単離収率)、14wt%のアルコール(8)、及び0.7%のエチルエーテル(30)を含む淡黄褐色の油39.78gを得た。
【0204】
工程6−メチルケトン形成:
【0205】
【化63】
【0206】
手法:
窒素送入口、熱電対及び磁気攪拌機を備えた1Lの三頚丸底フラスコに、MTBE(530mL)を加えた。この溶液を0℃に冷却し、MeLi溶液(1.6M/Et2O、123mL、0.196mol)を加えた。MTBE(160mL)中、(19)(41.33g、83wt%、34.3gアッセイ量、0.098mol)の溶液を、バッチ温度を5℃以下(tmax=4℃)に維持しながら、滴下漏斗経由で添加した。添加終了後、バッチを0℃で1時間熟成し、−70℃に冷却し、次いで、トリフルオロ酢酸(24mL、0.32mol)を一度で装入すると、温度が−40℃に上昇した。10%H3PO4溶液(250mL)を加え、得られる混合物を室温まで上昇させ、30分間熟成した。2相混合物を200mLのMTBEと200mLの10%H3PO4に加えた。有機相を水(500mL)で、次いで、1M−Na2CO3(500mL)、次いで、水(500mL)で、次いで、水(250mL)で洗った。有機相を硫酸ナトリウムで乾燥し、アッセイすると、34.5gの(20)を示した(96%アッセイ収率、87.9LCAP、Pd−エーテル化からのベンジルアルコール(8)10.6%)。
【0207】
炭酸ナトリウムでの洗浄は次工程の共役付加の成功にとって最重要である。この洗浄がない場合、〜20%の変換が観察されたのみであった。水洗が何回必要であるかは未知である。
【0208】
工程7−共役付加/異性化:
【0209】
【化64】
【0210】
手法:
窒素送入口、熱電対及び磁気攪拌機を備えた1Lの三頚丸底フラスコに、CuI(8.8g、0.046mol)を加えた。フラスコに窒素を1時間送入した。フラスコにTHF(255ml)を装入し、スラリーを0℃に冷却した。グリニヤール溶液(2.0M/Et2O、68.2mL、0.136mol)を0℃で加え、その温度を10℃以下に保持した。30分間熟成した後、その混合物を−70℃に冷却し、TMSCl(32.0mL、0.252mol)を加え、次いで、THF(180mL+70mL洗浄)中(20)(41.4g、79wt%、32.7gアッセイ量、0.089mol)の溶液を0℃で加えた。この添加の間、反応温度は−40℃を超えないようにした。1時間の間に、反応液は−40℃から−20℃に上昇させ、HPLC分析すると、変換率98.4%を示した。次いで、反応液を0℃に加温し、1時間熟成させると、完全な変換を示した。この混合物に0℃で1M−HCl(255mL)を加えて反応停止させると、27℃まで発熱した。この混合物を室温で1時間熟成させ、MTBE(500mL)に移行させ、層分離した。有機層を1M−HCl(500mL)と、次いで水(500mL)で洗った。この時点で、溶液から固形物が沈殿した。混合物をソルカフロック床(MTBEで湿潤)で濾過し、床を250mLのMTBEで洗った。層分離し、有機層を250mLの水で洗った。有機層を硫酸ナトリウムで乾燥し、〜600mLに濃縮し、アッセイすると(21)のアッセイ収量は39.52gであった(ジアステレオマーの15:85混合物)。
【0211】
MTBE中、生成物の粗製溶液を濃縮して油とし、窒素送入口、熱電対及び磁気攪拌機を備えた1Lの三頚丸底フラスコ中、MeOH400mLで希釈した。フラスコを20℃の水浴に浸漬し、NaOMe(25wt%/MeOH、10mL、0.044mol)を反応液にゆっくり加え、温度を25℃未満に維持した。1時間だけ熟成した後、ジアステレオマーの比は15:85から98:2に上昇した。反応液を0℃に冷やし、600mLのヘプタンで希釈し、1M−HCl(500mL)で反応停止させた。二相性混合物を静置し、有機層を分離した(水層への損失2%)。有機層を2回水洗(各250mL)し、硫酸ナトリウムで乾燥し、濃縮して、(21)を油として得た(47.9g、80.8wt%の(21)、98%アッセイ収率、79.8LCAP、10.5面積%ベンジルアルコール(8))。
【0212】
工程8−ヨードケトン形成:
【0213】
【化65】
【0214】
手法:
窒素送入口、熱電対及び磁気攪拌機を備えた500mLの三頚丸底フラスコに、メタノール(230mL)中の(21)(24.6g、80.8wt%、19.8gアッセイ量、0.0429mol)を加えた。この溶液を20℃の水浴に浸漬し、ICl(1.0M/CH2Cl2、77.5mL、0.0775mol)を20分間で滴下し、その温度を25℃以下に保持した。反応液を室温で2時間熟成させ、その時点で完全な変換がHPLC分析により観察された。この混合物をMTBE(250mL)と10%Na2S2O3/5%NaHCO3(250mL)で−10℃にて反応停止させた。この混合物をさらにMTBE(200mL)と10%Na2S2O3/5%NaHCO3(200mL)で希釈し、層分離した。有機層を2回水洗し(各300mL)、硫酸ナトリウムで乾燥し、アッセイすると、22.0gのヨードケトン(3)(87%収率)を示した。有機溶液を濃縮して固体とし、メタノール(170mL)で希釈した。容器に50mgのヨードケトンを播種し、この反応混合物に2時間かけて水(37.5mL)を滴下した。この混合物を一夜熟成させ、その上清(5.9mg/mL)についてアッセイし、濾過し、70mLのMeOH:H2O(70:30)で洗浄し、22.4gの(3)を白色固体として得た(93wt%ヨードケトン、20.8アッセイ収量、83%;97.3LCAP、0.9%Cl−ケトン、1.4%ケトン異性体)。
【0215】
塩:
式(Ia)で示される化合物の種々結晶性塩を調製し、評価した。
【0216】
【化66】
【0217】
これらの塩についての物理化学的データを以下の表に示す。
【0218】
【表9】
【0219】
ベシル酸塩の無水結晶形I型は物理的及び化学的共に安定であり、熱力学的にII型よりも安定であり、また通常非吸湿性であった。
【0220】
X線粉末回折での検討は、分子構造、結晶化度、及び多形を特性化するために広く使用される。ベシル酸塩の無水結晶形I型のX線粉末回折パターンは、PW3040/60コンソールを有するフィリップス分析用X′パートPRO−X線回折システムにて生成させた。PW3373/00セラミックCu LEF X線チューブK−アルファ放射を線源として用いた。
【0221】
図1はベシル酸塩の無水結晶形I型のX線回折パターンを示す。当該無水I型は、21.2、9.1、及び8.5オングストロームのd−間隔に相当する特徴的な回折ピークを示した。当該無水I型はさらに、13,5、10.9及び5.5オングストロームのd−間隔より特徴づけられた。当該無水I型はなおさらに、4.5、4.3、及び4.2オングストロームのd−間隔により特徴づけられた。
【0222】
DSCデータはTAインストルメンツDSC2910又は同等の装置を使用して、閉鎖したパン中、窒素気流下に、10℃/分の加熱速度で取得した。
【0223】
図2はベシル酸塩の無水結晶形I型の示差熱量走査を示す。当該無水結晶形I型は、273.2℃の開始温度で融解するために吸熱性を示し、ピーク温度は274.4℃であり、エンタルピー変化は61.3J/gである。
【背景技術】
【0001】
ヒト・ニューロキニン−1(hNK−1)はG−タンパク質結合受容体であり、中枢神経系及び胃腸管組織に集中している。参照:Nicoll,R.A.;Schenker,C.;Leeman,S.E.Annu.Rev.Neurosci.1980,3,227。神経ペプチド・サブスタンスP(SP)は、hNK−1受容体に対する好適なリガンドであり、多くの生物過程の適正化に関わっている。参照:(a)Guard,S.;Watson,S.P.Neurochem.Int.1991,18,149.(b)Takeuchi,Y.;Shands,E.F.B.;Beusen,D.D.;Marshall,G.R.J.Med.Chem.1998,41,3609及びその引用文献。SP及びhNK−1間の相互作用の制御は、重要な臨床分野、例えば、うつ状態、不安、炎症性腸疾患、及び疼痛などを包含する多様な一連の医学的障害の治療に関与してきた。参照:(a)Quatara,L.;Maggi,C.A.Neuropeptides 1998,32,1.(b)Rupniak,N.M.J.;Kramer,M.S.Trends Pharmacol.Sci.1999,20,485。結果として、有力な治療薬として強力かつ選択的なhNK−1受容体拮抗薬を同定するために、熱心な医薬研究が進められている。参照:(a)Owen,S.N.;Seward,E.M.;Swain,C.J.;Williams,B.J.U.S.Patent6,458,830B1,2001.(b)Finke,P.E.MacCoss,M.;Meurer,L.C.;Mills,S.G.;Caldwell,C.G.;Chen,P.;Durette,P.L.;Hale,J.;Holson,E.;Kopka,I.;Robichaud,A.PCT Int.Appl.WO9714671,1997.(c)Hale,J.J.;Mills,S.G.;MacCoss,M.;Finke,P.E.;Cascieri,M.A.;Sadowski,S.;Ber,E.;Chicchi,G.G.;Kurtz,M.;Metzger,J.;Elermann,G.;Tsou,N.N.;Tattersall D.;Rupniak,N.M.J.;Williams,A.R.;Rycroft,W.;Hargreaves,R.;MacIntyre,D.E.J.Med.Chem.1998,41,4607。
【0002】
本出願は、特定のラクタムhNK−1受容体拮抗薬の製造法を目的とする。この部類の化合物、さらにはこの部類の化合物の代替的な製造法が、WO2006/002117(2006年1月5日公開)及びUS2005−0282886(2005年12月22日公開)に開示されている。本発明は、ラクタムhNK−1受容体拮抗薬の収束的立体制御不斉合成を目的とする。
【発明の概要】
【0003】
発明の要旨
本発明は、ニューロキニン−1(NK−1)受容体拮抗薬として、及びタキキニン、とりわけサブスタンスPのインヒビターとして有用な特定のα,α−二置換γ−ラクタム誘導体の製造法を目的とする。これらの化合物はある種の障害、例えば、嘔吐、尿失禁、うつ状態、及び不安などの治療に有用である。
【0004】
発明の詳細な記載
一つの側面において、本発明は、式(I):
【0005】
【化1】
【0006】
[式中、
R2は以下からなる群より選択され:
(1)水素、及び
(2)C1−6アルキル;
Rは以下からなる群より選択され:
(1)未置換であるか、又はR11、R12及びR13の1個以上により置換されたフェニル;
(2)未置換であるか、又は以下から選択される1個以上の置換基で置換されたC1−8アルキル:
(a)ヒドロキシ、
(b)オキソ、
(c)C1−6アルコキシ、
(d)フェニル−C1−3アルコキシ、
(e)フェニル、
(f)−CN、
(g)ハロ、
(h)−NR9R10(ここで、R9及びR10は、独立して、
(1)水素、
(2)C1−6アルキル、
(3)ヒドロキシ−C1−6アルキル、及び
(4)フェニル、から選択される)、
(i)−NR9COR10(ここで、R9及びR10は上記定義のとおり)、
(j)−NR9CO2R10(ここで、R9及びR10は上記定義のとおり)、
(k)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(l)−COR9(ここで、R9は上記定義のとおり)、及び
(m)−CO2R9(ここで、R9は上記定義のとおり);
【0007】
(3)未置換であるか、又は以下から選択される1個以上の置換基で置換されたC2−6アルケニル:
(a)ヒドロキシ、
(b)オキソ、
(c)C1−6アルコキシ、
(d)フェニル−C1−3アルコキシ、
(e)フェニル、
(f)−CN、
(g)ハロ、
(h)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(i)−COR9(ここで、R9は上記定義のとおり)、及び
(j)−CO2R9(ここで、R9は上記定義のとおり);
【0008】
(4)へテロ環(ここで、該ヘテロ環は、
(A)ベンゾイミダゾリル、
(B)ベンゾフラニル、
(C)ベンゾチオフェニル、
(D)ベンゾオキサゾリル、
(E)フラニル、
(F)イミダゾリル、
(G)インドリル、
(H)イソオキサゾリル、
(I)イソチアゾリル、
(J)オキサジアゾリル、
(K)オキサゾリル、
(L)ピラジニル、
(M)ピラゾリル、
(N)ピリジル、
(O)ピリミジル、
(P)ピロリル、
(Q)キノリル、
(R)テトラゾリル、
(S)チアジアゾリル、
(T)チアゾリル、
(U)チエニル、
(V)トリアゾリル、
(W)アゼチジニル、
(X)1,4−ジオキサニル、
(Y)ヘキサヒドロアゼピニル、
(Z)ピペラジニル、
(AA)ピペリジニル、
(AB)ピロリジニル、
(AC)テトラヒドロフラニル、及び
(AD)テトラヒドロチエニル、
からなる群より選択され、また、該へテロ環は未置換であるか、又は
【0009】
(i)未置換であるか、又はハロ、−CF3、−OCH3、若しくはフェニルで置換されたC1−6アルキル、
(ii)C1−6アルコキシ、
(iii)オキソ、
(iv)ヒドロキシ、
(v)チオキソ、
(vi)−SR9(ここで、R9は上記定義のとおり)、
(vii)ハロ、
(viii)シアノ、
(ix)フェニル、
(x)トリフルオロメチル、
(xi)−(CH2)m−NR9R10(ここで、mは0、1又は2であり;R9及びR10は上記定義のとおり)、
(xii)−NR9COR10(ここで、R9及びR10は上記定義のとおり)、
(xiii)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(xiv)−CO2R9(ここで、R9は上記定義のとおり)、及び
(xv)−(CH2)m−OR9(ここで、m及びR9は上記定義のとおり)
から選択される1個以上の置換基で置換されている);
【0010】
R1は以下からなる群より選択され:
(1)
【0011】
【化2】
【0012】
(2)−C1−8アルキル(ここで、アルキルは未置換であるか、又は以下から選択される1個以上の置換基で置換されている:
(a)ヒドロキシ、
(b)オキソ、
(c)C1−6アルコキシ、
(d)フェニル−C1−3アルコキシ、
(e)フェニル、
(f)−CN、
(g)ハロ、
(h)−NR9R10(ここで、R9及びR10は上記定義のとおり)、
(i)−NR9COR10(ここで、R9及びR10は上記定義のとおり)、
(j)−NR9CO2R10(ここで、R9及びR10は上記定義のとおり)、
(k)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(l)−COR9(ここで、R9は上記定義のとおり)、及び
(m)−CO2R9(ここで、R9は上記定義のとおり));
【0013】
(3)−C2−6アルケニル(ここで、アルケニルは未置換であるか、又は以下から選択される1個以上の置換基で置換されている:
(a)ヒドロキシ、
(b)オキソ、
(c)C1−6アルコキシ、
(d)フェニル−C1−3アルコキシ、
(e)フェニル、
(f)−CN、
(g)ハロ、
(h)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(i)−COR9(ここで、R9は上記定義のとおり)、
(j)−CO2R9(ここで、R9は上記定義のとおり));
(4)−(CO)−フェニル(ここで、該フェニルは未置換であるか、又はR6、R7及びR8の1個以上により置換されている);
【0014】
R6、R7及びR8は、独立して、以下からなる群より選択され:
(1)水素;
(2)未置換であるか、又は以下から選択される1個以上の置換基で置換されたC1−6アルキル:
(a)ヒドロキシ、
(b)オキソ、
(c)C1−6アルコキシ、
(d)フェニル−C1−3アルコキシ、
(e)フェニル、
(f)−CN、
(g)ハロ、
(h)−NR9R10(ここで、R9及びR10は上記定義のとおり)、
(i)−NR9COR10(ここで、R9及びR10は上記定義のとおり)、
(j)−NR9CO2R10(ここで、R9及びR10は上記定義のとおり)、
(k)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(l)−COR9(ここで、R9は上記定義のとおり)、及び
(m)−CO2R9(ここで、R9は上記定義のとおり);
【0015】
(3)未置換であるか、又は以下から選択される1個以上の置換基で置換されたC2−6アルケニル:
(a)ヒドロキシ、
(b)オキソ、
(c)C1−6アルコキシ、
(d)フェニル−C1−3アルコキシ、
(e)フェニル、
(f)−CN、
(g)ハロ、
(h)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(i)−COR9(ここで、R9は上記定義のとおり)、
(j)−CO2R9(ここで、R9は上記定義のとおり);
(4)C2−6アルキニル;
【0016】
(5)未置換であるか、又は以下から選択される1個以上の置換基で置換されたフェニル:
(a)ヒドロキシ、
(b)C1−6アルコキシ、
(c)C1−6アルキル、
(d)C2−5アルケニル、
(e)ハロ、
(f)−CN、
(g)−NO2、
(h)−CF3、
(i)−(CH2)m−NR9R10(ここで、m、R9及びR10は上記定義のとおり)、
(j)−NR9COR10(ここで、R9及びR10は上記定義のとおり)、
(k)−NR9CO2R10(ここで、R9及びR10は上記定義のとおり)、
(l)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(m)−CO2NR9R10(ここで、R9及びR10は上記定義のとおり)、
(n)−COR9(ここで、R9は上記定義のとおり)、
(o)−CO2R9(ここで、R9は上記定義のとおり);
【0017】
(6)ハロ、
(7)−CN、
(8)−CF3、
(9)−NO2、
(10)−SR14(ここで、R14は水素又はC1−5アルキルである)、
(11)−SOR14(ここで、R14は上記定義のとおり)、
(12)−SO2R14(ここで、R14は上記定義のとおり)、
(13)NR9COR10(ここで、R9及びR10は上記定義のとおり)、
(14)CONR9COR10(ここで、R9及びR10は上記定義のとおり)、
(15)NR9R10(ここで、R9及びR10は上記定義のとおり)、
(16)NR9CO2R10(ここで、R9及びR10は上記定義のとおり)、
(17)ヒドロキシ、
(18)C1−6アルコキシ、
(19)COR9(ここで、R9は上記定義のとおり)、
(20)CO2R9(ここで、R9は上記定義のとおり)、
(21)2−ピリジル、
(22)3−ピリジル、
(23)4−ピリジル、
(24)5−テトラゾリル、
(25)2−オキサゾリル、及び
(26)2−チアゾリル;
【0018】
R11、R12及びR13は、独立して、R6、R7及びR8の定義から選択され;そして
Zは、
(1)水素、
(2)C1−6アルキル、及び
(3)ヒドロキシル
から選択される]で示されるラクタムタキキニン受容体拮抗薬又はその薬学的に許容され得る塩の製造法であって、
式(A):
【0019】
【化3】
【0020】
[式中、R14はR6から選択される]
で示される化合物と還元剤とを酸性条件下で反応させて、式(B):
【0021】
【化4】
【0022】
で示される化合物とし、次いで、式(B)の化合物と強酸とを反応させて式(I)で示される化合物とすることを含んでなり、次いで、所望により、式(I)で示される化合物の薬学的に許容され得る塩を形成するために、式(I)で示される化合物と対応する塩の酸とを反応させて、式(I)で示される化合物の薬学的に許容され得る塩を形成することを含んでなる製造法を包含する。
また、本発明は式(A)の化合物を調製するために、式(C):
【0023】
【化5】
【0024】
で示される化合物と酸とを反応させることにより、式(A)で示される化合物を調製することを含んでなる上記の製造法を包含する。
また、本発明は式(C)の化合物を得るために、式(D):
【0025】
【化6】
【0026】
[式中、X1はC1−6アルキル又はフェニルである]
で示される化合物と、アンモニア又はその塩とを反応させることにより、式(C)で示される化合物を調製することを含んでなる上記の製造法を包含する。本発明はまたさらに、式(D)の化合物を得るために、式(E):
【0027】
【化7】
【0028】
[式中、Yはハロゲンである]
で示される化合物と、式(F):
【0029】
【化8】
【0030】
で示される化合物、及び式M1N(R15)2又はM1N(Si(R15)3)2(式中、M1はLi、Na、K又はMgであり、各R15は、独立して、C1−4アルキルから選択される)で示される金属アミドとを、第一の非プロトン性有機溶媒中で反応させることにより、式(D)で示される化合物を調製することを含んでなる上記の製造法を包含する。
また、本発明はさらに、式(E)の化合物を得るために、式(G):
【0031】
【化9】
【0032】
で示される化合物と、ハロゲン化剤とを反応させることにより、式(E)で示される化合物を調製することを含んでなる上記の製造法を包含する。
また、本発明はさらに、式(G)の化合物を得るために、式(H):
【0033】
【化10】
【0034】
で示される化合物と、R−M2(ここで、M2は金属である)とを、第一の遷移金属触媒及びルイス酸の存在下で反応させることにより、式(G)で示される化合物を調製することを含んでなる上記の製造法を包含する。
また、本発明はさらに、式(H)の化合物を得るために、式(J):
【0035】
【化11】
【0036】
で示される化合物と、CH3Liとを、tert−ブチルメチルエーテル(MTBE)中で反応させることにより、式(H)で示される化合物を調製することを含んでなる上記の製造法を包含する。
本発明はまたさらに、式(J)の化合物を得るために、式(K):
【0037】
【化12】
【0038】
[式中、X2はRから選択される]
で示される化合物と、R1−OHとを、第二の遷移金属触媒、リガンド及び亜鉛付加物の存在下で反応させることにより、式(J)で示される化合物を調製することを含んでなる上記の製造法を包含する。
また、本発明はさらに、式(K)の化合物を得るために、式(L):
【0039】
【化13】
【0040】
で示される化合物を酵素的に還元し、次いで、その生成物と、X2−COClとを反応させることにより、式(K)で示される化合物を調製することを含んでなる上記の製造法を包含する。
また、本発明はさらに、式(L)の化合物を得るために、式(M):
【0041】
【化14】
【0042】
で示される化合物とブロム化剤とを反応させ、次いでバッファーの存在下でシアノ化剤を反応させることにより、式(L)で示される化合物を調製することを含んでなる上記の製造法を包含する。
また、本発明は、YがIであり、さらに、式(E)の化合物を得るために、式(N):
【0043】
【化15】
【0044】
で示される化合物と、M3−I(ここで、M3はLi、Na又はKである)とを反応させることにより、式(E)で示される化合物を調製することを含んでなる上記の製造法を包含する。
また、本発明はさらに、式(N)の化合物を得るために、式(O):
【0045】
【化16】
【0046】
で示される化合物と、ClCH2CO2H、ClCH2I、又はClCH2Br及び式M4N(R16)2又はM4N(Si(R16)3)2(ここで、M4はLi、Na、K、又はMgであり、各R16は、独立して、C1−4アルキルから選択される)で示される金属アミドとを、第二の非プロトン性有機溶媒中、約−20℃ないし約40℃の範囲の温度で反応させることにより、式(N)で示される化合物を調製することを含んでなる上記の製造法を包含する。
また、本発明はさらに、式(O)の化合物を得るために、式(P):
【0047】
【化17】
【0048】
で示される化合物又はそのトリエチルアミン塩とメチル化剤とを反応させることにより、式(O)で示される化合物を調製することを含んでなる上記の製造法を包含する。
また、本発明はさらに、式(P)の化合物又はそのトリエチルアミン塩を得るために、式(Q):
【0049】
【化18】
【0050】
で示される化合物と、R−M5(ここで、M5はM2同様の金属である)とを、第一の遷移金属触媒及びルイス酸の存在下で反応させ、次いで、所望により、トリエチルアミドを反応させて塩を形成することにより、式(P)で示される化合物を調製することを含んでなる上記の製造法を包含する。
また、本発明は、式(Q)の化合物を得るために、式(R):
【0051】
【化19】
【0052】
[式中、X3はRから選択される]
で示される化合物とR1−OHとを、第二の遷移金属触媒、リガンド及び亜鉛付加物の存在下で反応させることにより、式(Q)で示される化合物が製造されることを特徴とする上記の製造法を包含する。
また、本発明はさらに、式(R)の化合物を得るために、式(S):
【0053】
【化20】
【0054】
で示される化合物を酵素的に還元し、次いで、その生成物とX3−COClとを反応させることにより、式(R)で示される化合物を調製することを含んでなる上記の製造法を包含する。
また、本発明はさらに、式(S)の化合物を得るために、式(T):
【0055】
【化21】
【0056】
で示される化合物と酸化剤とを反応させることにより、式(S)で示される化合物を調製することを含んでなる上記の製造法を包含する。
また、本発明は、R1が
【0057】
【化22】
【0058】
である上記の製造法を包含する。
また、本発明は、Rが
【0059】
【化23】
【0060】
である上記の製造法を包含する。
また、本発明は、R2がメチルである上記の製造法を包含する。
また、本発明は、式(I)で示される化合物が式(Ia):
【0061】
【化24】
【0062】
によって表されるものである上記の製造法を包含する。
また、本発明は式(Ia)の化合物が薬学的に許容され得る塩である上記の製造法を包含する。
また、本発明は該薬学的に許容され得る塩がベンゼンスルホン酸塩であり、該塩の相当する酸がベンゼンスルホン酸である上記の製造法を包含する。
また、本発明は、式(Ia):
【0063】
【化25】
【0064】
で示される化合物のベンゼンスルホン酸塩を包含する。
また、本発明は、I型で示され、約21.2、9.1、及び8.5オングストロームのd−間隔に相当する特徴的な回折ピークを示す当該ベンゼンスルホン酸塩の無水結晶形を包含する。無水I型はさらに、13.5、10.9及び5.5オングストロームのd−間隔により特徴づけられる。無水I型はなおさらに、4.5、4.3及び4.2オングストロームのd−間隔により特徴づけられる。
【0065】
「第一の非プロトン性有機溶媒」及び「第二の非プロトン性有機溶媒」という用語は、例えば、THF、MTBE、ジメトキシエタン、DMF、DMAc及びジオキサンを意味する。
「ハロゲン化剤」という用語は、例えば、Br2、I2及びIClを意味する。
「第一の遷移金属触媒」及び「第二の遷移金属触媒」という用語は、例えば、[CODRh(OH)]2又はCuX若しくはCuX2(ここで、XはBr、Cl又はI)又はPd(OAc)2などのパラジウム触媒を意味する。
【0066】
「リガンド」という用語は、例えば、1,3−ビス(ジフェニルホスフィノ)プロパンなどのホスフィンリガンドを意味する。
「亜鉛付加物」という用語は、例えば、Et2Znを意味する。
「酵素的に還元する」という用語は、例えば、アルコール・デヒドロゲナーゼ(ADH RE)、NADH、グルコース及びグルコース・デヒドロゲナーゼ(GDH103)により還元することを意味する。
「ブロム化剤」という用語は、例えば、触媒HBr存在下のBr2を意味する。
「シアノ化剤」という用語は、例えば、NaCN及びKCNを意味する。
「バッファー」という用語は、例えば、酢酸及びNH4Clを意味する。
【0067】
「メチル化剤」という用語は、例えば、MeI及びM6CO3を意味する;この場合、M6はDMF、DMAc、DMSO、アセトンなどの極性溶媒中、0〜60℃での、又はH2SO4、TsOH、MsOH、又はPhSO3Hなどの酸触媒存在下、MeOH中、外気温度ないし還流下での、Li、Na、K、Ca、又はCsである。
「還元剤」という用語は、例えば、(C1−4アルキル)3SiHを意味する。
「ルイス酸」という用語は、例えば、TMSClを意味する。
「金属」という用語は、例えば、(OH)2、BF3、MgBr及びLiを意味する。
【0068】
式(I)で示される化合物は不斉中心を有し、本発明は光学異性体のすべてとその混合物を包含する。
さらに、炭素−炭素二重結合をもつ化合物は、Z−及びE−型として存在可能であり、該化合物の異性体型のすべてが本発明に包含される。
いずれかの変数(例えば、アルキル、アリール、R6、R7、R8、R9、R10、R11、R12、R13など)が、いずれかの変数又は式(I)中に一度ならず出現する場合、各出現に際しての定義は、その出現ごとの定義から独立したものである。
【0069】
本明細書にて使用する場合、「アルキル」という用語は、特定数の炭素原子を有する直鎖、分枝又は環状の配置を有するアルキル基を包含する。「アルキル」の例は、メチル、エチル、プロピル、イソプロピル、ブチル、iso−、sec−及びtert−ブチル、ペンチル、ヘキシル、ヘプチル、3−エチルブチル、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、ノルボルニルなどを包含する。「アルコキシ」は酸素架橋を介して結合する特定数の炭素原子を有するアルキル基を表し、例えば、メトキシ、エトキシ、プロポキシ、ブトキシ及びペントキシである。「アルケニル」は特定数の炭素原子を有する直鎖又は分枝の配置を有し、その鎖のいずれかの位置に少なくとも1つの不飽和が存在し得る炭化水素鎖を包含するものとする;例えば、エテニル、プロペニル、ブテニル、ペンテニル、ジメチルペンチルなどであり、可能な場合、E及びZ型を包含する。「ハロゲン」又は「ハロ」とは、本明細書にて使用する場合、フルオロ、クロロ、ブロモ及びヨードを意味する。
【0070】
「アリール」とはフェニル又はナフチルを意味し、いずれもが未置換であるか、又はハロ、C1−4−アルキル、C1−4−アルコキシ、NO2、CF3、C1−4−アルキルチオ、OH、−N(R6)2、−CO2R6、C1−4−ペルフルオロアルキル、C3−6−ペルフルオロシクロアルキル、及びテトラゾール−5−イルからなる群より選択される1個、2個又は3個の置換基により置換されている。
【0071】
「ヘテロアリール」という用語は、O、N及びSからなる群より選択される1個ないし3個のヘテロ原子を含有してなる未置換、一置換若しくは二置換の5員若しくは6員の芳香族ヘテロ環を意味し、その場合の置換基は、−OH、−SH、−C1−4−アルキル、−C1−4−アルコキシ、−CF3、ハロ、−NO2、−CO2R9、−N(R9R10)及び縮合ベンゾ基からなる群より選択されるメンバーである。
【0072】
当業者が理解するように、薬学的に許容され得る塩は、限定されるものではないが、無機酸との塩、例えば、塩酸塩、硫酸塩、リン酸塩、二リン酸塩、臭化水素酸塩、及び硝酸塩;又は有機酸との塩、例えば、リンゴ酸塩、マレイン酸塩、フマル酸塩、酒石酸塩、コハク酸塩、クエン酸塩、酢酸塩、乳酸塩、メタンスルホン酸塩、p−トルエンスルホン酸塩、2−ヒドロキシエチルスルホン酸塩、パモン酸塩、サリチル酸塩及びステアリン酸塩などである。同様に、薬学的に許容され得るカチオンは、限定されるものではないが、ナトリウム、カリウム、カルシウム、アルミニウム、リチウム及びアンモニウムである。
【0073】
本発明化合物は、過剰なタキキニン、特にサブスタンスPの活性の存在により特徴づけられる多様な臨床症状の予防及び治療に有用である。したがって、例えば、過剰なタキキニン、特にサブスタンスPの活性は、様々な中枢神経系の障害に関わっている。かかる障害は気分障害、例えば、うつ状態、又はより具体的には抑うつ性障害、例えば、単発性エピソード若しくは再発性主抑うつ性障害及び気分変調障害、又は双極性障害、例えば、双極性I障害、双極性II障害及び循環気質障害;不安障害、例えば、広場恐怖症を伴う、若しくは伴わないパニック障害、パニック障害暦を持たない広場恐怖症、特定恐怖症、例えば、特定動物恐怖症、対人恐怖症、強迫反応障害、ストレス障害、例えば、外傷後ストレス障害及び急性ストレス障害、及び全身性不安障害;統合失調症及びその他の精神障害、例えば、統合失調症型障害、分裂感情性障害、妄想性障害、短期精神障害、共有精神障害及び妄想若しくは幻覚を伴う精神障害;せん妄、痴呆、及び健忘症と他の認識若しくは神経変性障害、例えば、アルツハイマー病、老年痴呆、アルツハイマー型痴呆、脈管性痴呆、及び他の痴呆、例えば、HIV疾患、頭部外傷、パーキンソン病、ハンチントン病、ピック病、クロイツフェルト−ヤコブ病による痴呆、若しくは複数の病因による痴呆;パーキンソン病及び他の錐体外路の運動障害、例えば、投薬誘発運動障害、例えば、神経弛緩薬誘発性パーキンソン症、神経弛緩薬誘発性悪性症候群、神経弛緩薬誘発性急性失調症、神経弛緩薬誘発性静座不能、神経弛緩薬誘発性遅発性ジスキネジー、及び神経弛緩薬誘発性姿勢振せん;アルコール、アンフェタミン(又はアンフェタミン様物質)、カフェイン、大麻、コカイン、幻覚剤、吸入とエーロゾル噴射剤、ニコチン、アヘン、フェニルグリシジン誘導体、鎮静剤、催眠薬、及び抗不安薬などの使用から生じる物質関連障害(この物質関連障害は、依存性と乱用、中毒、離脱、中毒性せん妄、離脱性せん妄、持続性痴呆、精神障害、気分障害、不安障害、性機能不全及び睡眠障害である);てんかん;ダウン症候群;MS及びALSなどの髄鞘脱落症及び末梢神経障害などの他の神経病理学的障害、例えば、糖尿病性及び化学療法誘発神経障害、及び帯状疱疹後神経痛、三叉神経痛、分節若しくは肋間神経痛、及びその他の神経痛;及び急性若しくは慢性の脳血管傷害による脳血管障害、例えば、脳梗塞、くも膜下出血若しくは脳浮腫である。
【0074】
タキキニン、とりわけサブスタンスPは、その活性が侵害受容と疼痛にも関与している。本発明化合物は、したがって、以下の柔組織及び末梢の傷害を含む、疼痛を主体とする疾患及び症状の予防又は治療に有用となる:急性外傷、骨関節症、リウマチ様関節炎、筋骨格疼痛、特に外傷後の疼痛、脊髄痛、顔面筋疼痛症候群、頭痛、会陰切開疼痛、及び火傷;深部及び内臓痛、例えば、心臓痛、筋肉痛、眼痛、口顔痛、例えば、歯痛、腹痛、婦人科痛、例えば、月経困難症、及び出産時痛;神経根傷害と関連する疼痛、例えば、末梢神経障害と関連する疼痛、例えば、神経絞扼及び腕神経叢剥離、切断、末梢性神経障害、疼痛性チック、非定型顔面痛、神経根傷害、及びくも膜炎;がん関連疼痛(時に、がん痛と言われる);中枢神経系痛、例えば、脊髄又は脳幹傷害による疼痛;腰痛;坐骨神経痛;強直性脊椎炎、通風;及び瘢痕痛。
【0075】
タキキニン、とりわけサブスタンスPの拮抗薬は、以下の疾患の治療に有用である:呼吸器疾患、特に過剰の粘液分泌と関連する疾患、例えば、慢性閉塞性気道疾患、気管支肺炎、慢性気管支炎、のう胞性線維症と喘息、成人呼吸促迫症候群、及び気管支痙攣;炎症性疾患、例えば、炎症性腸疾患、乾癬、結合組織炎、骨関節症、リウマチ様関節炎、掻痒症及び日焼け;アレルギー、例えば、湿疹と鼻炎;過敏性障害、例えば、ツタウルシ;眼科疾患、例えば、結膜炎、春季結膜炎など;細胞増殖と関連する眼科症状、例えば、増殖性硝子体網膜障害;皮膚疾患、例えば、接触性皮膚炎、アトピー性皮膚炎、蕁麻疹、及びその他の湿疹様皮膚炎。タキキニン、とりわけサブスタンスPのアンタゴニストは、乳房腫瘍、神経節細胞腫、及び小細胞がん、例えば、小細胞肺がんなどの新生物の治療にも有用である。
【0076】
タキキニン、とりわけサブスタンスPの拮抗薬は、以下の胃腸(GI)障害の治療に有用である:GI管の炎症性障害と疾患、例えば、胃炎、胃十二指腸潰瘍、胃がん、胃リンパ腫、内臓の神経性制御と関連する障害、潰瘍性大腸炎、クローン病、過敏性腸症候群及び嘔吐(例えば、化学療法、放射線、毒素、ウイルス若しくは細菌感染、妊娠、前庭障害、例えば、動揺病、真性めまい、めまい及びメニエール病、外科手術、偏頭痛、頭蓋内圧の変化、胃−食道逆流病、胃酸過多、飲食物過食、酸性胃、むしず又は逆流、胸焼け、例えば、エピソード的、夜間又は食事−誘発胸焼け、及び消化不良などにより誘発される嘔吐などの急性、遅延性若しくは予測性嘔吐を含む)。
【0077】
タキキニン、とりわけサブスタンスPの拮抗薬は、以下の多様な他の症状の治療に有用である:ストレス関連体細胞障害;反射性交感神経性ジストロフィ、例えば、肩手症候群;有害免疫反応、例えば、移植組織拒絶反応及び免疫増強若しくは抑制に関係する障害、例えば、全身性紅斑性狼瘡;サイトカイン化学療法から生じる血漿溢血、膀胱機能障害、例えば、膀胱炎、膀胱排尿筋過剰反射、頻尿及び尿失禁(切迫尿失禁、尿意逼迫、及び頻尿の症候をもつ過剰反応膀胱の予防又は治療を含む);線維性及びコラーゲン病、例えば、強皮症及び好酸性肝蛭病;血管拡張及び血管痙攣性疾患を原因とする血流の障害、例えば、アンギナ、血管性頭痛、偏頭痛及びレイノー病;及び前記症状のいずれか、とりわけ偏頭痛の疼痛の伝播に寄与するか、又はそれと関連する疼痛又は侵害受容。本発明化合物は上記症状の合併症の治療、特に、術後疼痛及び術後の悪心と嘔吐の治療にも有益である。
【0078】
本発明の化合物は、特に、化学療法、放射線、毒素、妊娠、前庭障害、動揺病、外科手術、偏頭痛、及び頭蓋内圧の変化などにより誘発される嘔吐などの急性、遅延性若しくは予測性嘔吐を含む嘔吐の予防又は治療に有用である。例えば、本発明化合物は、緩和な、又は高度に嘔吐誘発性のがん化学療法、例えば、高用量投与シスプラチンの開始時及び反復過程と関連する急性及び遅延性の悪心と嘔吐の予防のために、他の嘔吐剤と併用してもよい。とりわけ、本発明化合物は、抗新生物(細胞傷害)剤(例えば、がん化学療法にて常套的に使用される薬剤)を原因とする嘔吐、及び他の薬理学的薬剤、例えば、ロリプラム(rolipram)により誘発される嘔吐の治療に有用である。かかる化学療法剤の例は、アルキル化剤、例えば、エチレンイミン化合物、スルホン酸アルキル及びアルキル化作用をもつ他の化合物、例えば、ニトロソウレア類、シスプラチン及びデカルバジン;代謝拮抗剤、例えば、葉酸、プリン若しくはピリミジンアンタゴニスト;有糸分裂インヒビター、例えば、ヒガンバナアルカロイド及びポドフィロトキシンの誘導体;及び細胞傷害性抗生物質である。化学療法剤の具体例は、例えば、D.J.Stewart in Nausea and Vomiting(悪心及び嘔吐):Recent Research and Clinical Advances(最近の研究及び臨床上の進歩),Eds.J.Kucharczyk et al,CRC Press Inc.,Boca Raton,Florida,USA(1991)pages177−203,特に188ページに記載されている。共通して使用される化学療法剤は、シスプラチン、ダカルバジン(DTIC)、ダクチノマイシン、メクロレタミン、ストレプトゾシン、シクロホスファミド、カルムスチン(BCNU)、ロムスチン(CCNU)、ドキソルビシン(アドリアマイシン)、ダウノルビシン、プロカルバジン、マイトマイシン、シタラビン、エトポシド、メトトレキセート、5−フルオロウラシル、ビンブラスチン、ビンクリスチン、ブレオマイシン及びクロランブシルなどである[R.J.Gralla et al in Cancer Treatment Reports(1984)68(1),163−172]。本発明のさらなる側面は、哺乳動物において、時間生物学(既日リズム位相)的作用を達成し、既日リズム障害を緩和するために、本発明化合物を使用することからなる。
【0079】
【化26】
【0080】
構造的に複雑な(1)は、3種の明確な合成目標に分割できよう:1)二級の立体末端の双方に立体化学を含む立体的に込み合ったエーテル;2)トランス,トランス−1,2,3−三置換シクロペンタン核;及び3)2つの立体中心を含み、その1つが三級アミンであるピロリジノン環(工程図1a)。遠位三級アミンの立体化学に対処するために、我々はケトン(2a)から(1)を製造する戦略を案出した;この戦略はオキサゾリジノン(3a)をヨードケトン(4a)によりジアステレオ選択的にアルキル化することに由来する。(4a)においてシクロペンタン核の相対的立体化学を制御する最も有効な方法は、アリルエーテル(5a)に対するアリール−金属種の基質−制御共役付加を経由して、当該ケトンを熱力学的に好適なジアステレオマーに異性化することである。我々はアリルエーテル(5a)の二級の立体中心双方に対処する最善の方法が、それぞれエナンチオマーとして純粋な形状のアリルアルコール(6a)とアルコール(7a)の収束性の立体特異的カップリング結合を経由することであると想定した。
【0081】
アルコール(7a)は数種のhNK−1拮抗薬に存在する共通の構造的モチーフである:参照:(a)Owen,S.N.;Seward,E.M.;Swain,C.J.;Williams,B.J.U.S.Patent6,458,830B1,2001.(b)Finke,P.E.MacCoss,M.;Meurer,L.C.;Mills,S.G.;Caldwell,C.G.;Chen,P.;Durette,P.L.;Hale,J.;Holson,E.;Kopka,I.;Robichaud,A.PCT Int.Appl.WO9714671,1997.(c)Hale,J.J.;Mills,S.G.;MacCoss,M.;Finke,P.E.;Cascieri,M.A.;Sadowski,S.;Ber,E.;Chicchi,G.G.;Kurtz,M.;Metzger,J.;Elermann,G.;Tsou,N.N.;Tattersall D.;Rupniak,N.M.J.;Williams,A.R.;Rycroft,W.;Hargreaves,R.;MacIntyre,D.E.J.Med.Chem.1998,41,4607;また、このものは対応するアリールメチルケトンの不斉還元を介して容易に入手し得る;参照:Hansen,K.B.;Chilenski,J.R.;Desmond,R.;Devine,P.N.;Grabowski,E.J.J.;Heid,R.;Kubryk,M.;Mathrem D.J.;Varsolona,R.Tetrahedron:Asymmetry 2003,14,3581。
【0082】
対照的に、アリルアルコール(6a)のエナンチオ選択的合成については報告されていない。既存の方法論を3−シアノシクロペンテノン(8a)の不斉還元に適用することは、変わり得る収率で、また穏当なエナンチオ選択性で(6a)を生じた。参照:(a)Ohkuma,T.;Koizumi,M.;Doucet,H.;Pham,T.;Kozawa,M.;Murata,K.;Katayama,E.;Yokozawa,T.;Ikariya,T.;Noyori,R.J.Am.Chem.Soc.1998,120,13529.(b)Corey,E.J.;Guzman−Perez,A.;Lazerwith,S.E.J.Am.Chem.Soc.1997,119,11769.(c)Yun,J.;Buchwald,S.L.;J.Am.Chem.Soc.1999,121,5640.(d)Brown,H.C.;Ramachandran,P.V.Acc.Chem.Res.1992,25,16−24.(e)Midland,M.M.;Tramontano,A.;Kazbubski,A.;Graham,R.S.;Tsai,D.J.S.;Cardin,D.Tetrahedron 1984,40,1371.(f)Noyori,R.;Suzuki,M.Angew.Chem.Int.Ed.Engl.1984,23,847。(8a)に類似のエノンの不斉、生物触媒還元については、先例がなかった[参照:(a)Fonteneau,L.;Rosa,S.;Buisson,D.Tetrahedron:Asymmetry,2002,13,579.(b)Attolini,M.;Bouguir,F.;Iacazio,G.;Peiffer,G.;Maffei,M.Tetrahedron,2001,57,537]が、ケト還元酵素ライブラリーをスクリーニングした。我々は、ロドコッカス・エリスロポリス(Rhodococcus erythropolis)由来のアルコール・デヒドロゲナーゼ(ADH RE)が、高収率(93%)、かつすぐれたエナンチオ選択性(>99%ee)で、エノン(8a)を(S)−アリルアルコール(6a)に効率的に還元することを発見した(工程図2a)。
【0083】
【化27】
a 条件:(a)ADH RE、0.1M K2PO4、グルコース、NAD、pH6.5,93%.(b)2−NapCO2H,2−NapCOCl,DIPEA,DMAP,CH2Cl2,84%,Nap=2−Nap.(c)Et2Zn,6,THF,Pd(OAc)2,dppp,83%
【0084】
高光学純度で調製したアルコール(6a)及び(7a)を用いて、我々は両方の中心でエピマー化を起こさずにこれらのパートナーをカップル結合させる方法を探究した。文献調査した形質転換の立体特異性(η3−アリル金属中間体を経由して進行する)は、Pd−触媒アリルエーテル化を魅力的な選択肢とした;しかし、アルコール(7a)は立体的に過密であり、求核性が乏しいために、このカップリング結合に参画するには不適切な候補であった。参照:Kim,H.;Lee.C.Org.Lett.2002,4369及びその引用文献。我々は、最適とした条件下、化学量論比のナフトエ酸アリルエステル(9a)とアルコール(6a)が、0.5当量のEt2Znの存在下、Pd(OAc)2及びdpppを用いてカップル結合させると、アリルエーテル(10a)が83%アッセイ収率で、かつ両方の立体中心での立体配置が完全に維持されて得られることを見出し、満足した。
【0085】
該ニトリルに対するクープレート共役付加を実施する試行は失敗であったが、我々は5.0当量のアリールホウ酸又は1.5当量のトリフルオロホウ酸アリール(K塩)を用いて、リガンドなしのRh−触媒共役付加(3mol%[CODRh(OH)2]、EtOH、還流)を証明することができた。参照:(a)Batey,R.A.;Thadani,A.N.;Smil,D.V.Org.Letters 1999,1683.(b)Sakai,M.;Hayashi,H.;Miyaura,N.Organometallics 1997,16,4229。ケトンから熱力学的に優位なジアステレオマーへの異性化(NaOMe/MeOH)後に、両方の手法が93%アッセイ収率と高ジアステレオ選択性(>99:1 β−中心、90:10 α−中心)で(11a)を与えた。該ニトリルはMTBE中、MeLiでの処理を経て、容易にメチルケトンに変換され、90%のアッセイ収率で(12a)を供給した(工程図3a)。
【0086】
別法として、メチルケトン(13a)は、ニトリル(10a)から容易に製造され(MeLi、MTBE)、さらに期待どおりに、該ケトンの熱力学的に優位なジアステレオマーへの異性化(NaOMe/MeOH)後に、アリール・グリニヤールのCu−触媒共役付加が、すぐれた収率(95%)、かつ例外的にすぐれたジアステレオ選択性(>99:1 β−中心、98:2 β−中心)で(12a)を提供した。MeOH中、IClによる(13a)の選択的ヨード化は、90%の単離収率でヨードケトン(4a)を生成した。(4a)のX線結晶構造は、この方法により組み立てた4つの立体中心の相対的及び絶対的化学を証明した。
【0087】
【化28】
a条件:(a)4−FPhBF3K、[CODRh(OH)]2、EtOH、90℃、93%.(b)MeLi、MTBE、0℃、90%.(c)NaOMe、MeOH、RT、98%.(d)MeLi、MTBE、0℃、90%.(e)4−FPhMgBr、CuI、TMSCl、THF、−40℃、96%.(f)ICl、MeOH、20℃、90%
【0088】
ヨードケトンを組み立てる収束的高選択的方法(6工程、58%収率)を用いて、我々はピロリジノン環を製造する立体制御方法の開発に注目した。公表されている方法に従い、オキサゾリジノン(3a)をヨードケトン(4a)でアルキル化すると、穏当な収率で(2)を生じた。参照:(a)Karady,S.;Amato,J.;Weinstock,L.Tetrahedron Lett.1984,25,4337.(b)Szumigala,Jr.,R.H.;Onofiok,E.;Karady,S.;Armstrong,III,J.D.;Miller R.A.Tetrahedron Lett.2005,46,4403。90%の収率と>99:1のジアステレオ選択性の最良の結果は、トルエン/DMPU中、低温度で、2.4当量の(3)及び2.5当量のLHMDSにより達成された。水酸化アンモニウムによるオキサゾリジノンの開裂は明らかにアミナール(14a,b)のジアステレオマー混合物を生じた;このものは、(14)から(16)への直接の還元が1当量の水を生成し、それがEt3SiH還元を妨げるので、シラン還元を実施する前に、一旦メタンスルホン酸で脱水して、94%の収率でエナミド(15a,b)のジアステレオ混合物(〜3:1)とした。エナミド(15a,b)は、単離したジアステレオマーとして純粋な異性体(15a)又は(15b)それぞれが、酸性条件下で同じ(15a,b)の3:1混合物に変換されるため、アシルイミニウムカチオン(17)及び(18)の平衡にある。また、アシルイミニウム(18)は熱力学的に(17)よりもより安定である。それ故、Et3SiH/MeSO3Hによる(15a,b)の還元は、優先的に(18)を経由して進行し、(17)を経由するシクロペンタン環上の少量のエピマーとともに、90:10のジアステレオマー混合物として優れた収率で(16)を与えた。(16)の化学選択的脱保護は、HBr/AcOHで遂行した。候補化合物(1)はベンゼンスルホン酸塩として、85%の収率で得た。
【0089】
【化29】
a条件:(a)LiHMDS、トルエン/DMPU、−78℃、90%.(b)NH4OH、THF/H2O、87%.(c)MeSO3H、EtOAc、94%.(d)MeSO3H、Et3SiH、MeCN、95%,(e)HBr/HOAc,(f)PhSO3H、IPAc/IPA/ヘプタン、85%(2工程)
【0090】
結論として、収束的高選択的経路は、強力なhNK−1受容体拮抗薬(1)を合成するために開発されたものである。6ヶ所すべての立体中心は、合計11工程(23%収率)で卓越した選択性をもって巧みに調製された;該方法は7kgの(1)を製造するために使用した。Pd−触媒によるエーテル化と、引き続く(10a)及び(13a)などの環状の基質における基質−制御共役付加の適用方法は、高度に機能化したシクロペンタノイド類の立体制御合成に一般的に適用されている。
上記の方法論は、以下の表に例示するように広く適用されていることが分かっているので、様々なシステムに適用された。
【0091】
【化30】
【0092】
(2b)の実用的な合成を案出するには、2種類の明確な合成目標を扱わねばならない:1)立体的に込み合ったエーテル(二級の立体末端双方に立体化学を含む);及び2)トランス,トランス−1,2,3−三置換シクロペンタン核。我々は、(2b)におけるトランス,トランス−配置が、アリルエーテル(4b)へのアリール−金属種の基質制御共役付加と、それに続くエステルの熱力学的に好適なジアステレオマーへの平衡化を経由して効果的に組み立てられるだろうと想定した(反応工程図2)。(4b)の構築のための最も魅力的な収束的方法は、それぞれエナンチオマーとして純粋な形状のアリルアルコール(5b)とアルコール(6b)との立体特異的カップリングを経ることであろう。上記の逆合成は当該標的構造を3種の類似サイズと複雑さの成分に切断し、これを我々は(2b)のみならず、一連の構造類似体にも適用し得ると想定した。
【0093】
【化31】
【0094】
アルコール(6b)は数種の薬物候補に存在する共通の構造要素であり[参照:a)Nelson,T.D.;Rosen,J.D.;Smitrovich,J.H.;Payack,J.;Craig,B.;Matty,L.;Huffman,M.A.;McNamara,J.Org.Lett.2005,55.b)Zhao,M.M.;McNamara,J.M.;Ho,G.−J.;Emerson,K.M.;Song,Z.J.;Tschaen,D.M.;Brands,K.M.J.;Dolling,U.−H.;Grabowski,E.J.J.;Reider,P.J.;Cottrell,I.F.;Ashwood,M.S.;Bishop,B.C.J.Org.Chem.2002,6743]、アリールメチルケトンの不斉還元を経て容易に調製した。参照:Hansen,K.B.;Chilenski,J.R.;Desmond,R.;Devine,P.N.;Grabowski,E.J.J.;Heid,R.;Kubryk,M.;Mathre,D.;Varsolona,R.Tetrahedron:Asymmetry 2003,3581。対照的に、アリルアルコール(5b)とその構造類似体のエナンチオ選択的合成については報告がなかった。(5b)への最も直接的な経路は、3−カルボキシメチルシクロペンテノン(7b)の不斉合成を経由することであろう。参照:a)Catino,A.J.;Forslund,R.E.;Doyle,M.P.J.Am.Chem.Soc.2004,13622−13623.b)Yu,J−Q.;Corey,E.J.J.Am.Chem.Soc.2003,3232−3233。(7b)の不斉還元に既存の方法論を適用すると、相応の収率、かつ並みのエナンチオ選択性で、アリルアルコール(6b)が得られた(表1)。参照:a)Ohkuma,T.;Koizumi,M.;Doucet、H.;Pham,T.;Kozawa,M.;Murata,K.;Katayama,E.;Yokozawa,T.;Ikariya,T.;Noyori,R.J.Am.Chem.Soc.1998,120,13529.b)Corey,E.J.;Guzman−Perez,A.;Lazerwith,S.E.J.Am.Chem.Soc.1997,119,11769.c)Yun,J.;Buchwald,S.L.;J.Am.Chem.Soc.1999,121,5640.d)Brown,H.C.;Ramachandran,P.V.Acc.Chem.Res.1992,25,16−24.e)Midland,M.M.;Tramontano,A.;Kazbubski,A.;Graham,R.S.;Tsai,D.J.S.;Cardin,D.Tetrahedron 1984,40,1371.f)Noyori,R.;Suzuki,M.Angew.Chem.Int.Ed.Engl.1984,23,847。
【0095】
(7b)の生物触媒還元の実行可能性を決定するために(参照;a)Fonteneau,L.;Rosa,S.;Buisson,D.Tetrahedron:Asymmetry,2002,13,579.b)Attolini,M.;Bouguir,F.;Iacazio,G.;Peiffer,G.;Maffei,M.Tetrahedron,2001,57,537)、ケトリダクターゼ・ライブラリーのスクリーニングを実施した。我々は、ロドコッカス・エリスロポリス(Rhodococcus erythropolis)からのアルコール・デヒドロゲナーゼ(ADH RE)が、好収率(83%)、かつ所望の(S)−エナンチオマーに対してすぐれたエナンチオ選択性(>99%ee)で、(7b)を(5b)に効率的に還元することを発見し、満足した(表1;第4項目)。この方法の信頼性の証拠として、3−シアノシクロペンテノン(8b)(このものはRu−触媒転移水素化における遂行能力が極端に乏しい)が、高収率、かつ優れたエナンチオ選択性でアリルアルコール(9b)に還元された。
【0096】
【表1】
【0097】
手元にある光学的に純粋なアリルアルコール(5b)により、我々はいずれの立体中心をも混交させることなく、(5b)をアルコール(6b)にカップル結合させる方法を探索した。η3−アリル金属中間体を経て進行する反応の文献に記載された立体特異性は、Pd−触媒によるアリルエーテル化を魅力的な選択とした。参照:a)Shu,C.;Hartwig,J.F.Angew.Chem.Int.Ed.2004,4794.b)Kim,H.;Lee.C.Org.Lett.2002,4369.b)Evans,P.A.;Leahy,D.K.J.Am.Chem.Soc.2002,7882。しかし、アルコール(6b)は立体的に密集し、求核性に乏しいために、このカップル結合に関与させるのに適切な候補ではなかった。驚くべきことに、(6b)の亜鉛アルコキシドは、Pd触媒によるエーテル化について報告されている標準的な条件下で、酢酸アリル(10b)に容易にカップル結合して、50%の収率でアリルエーテル(4)を単一のジアステレオマーとして生成した。エナンチオマー過剰率レベルの異なる(10b)を光学的に純粋な(6b)とのエーテル化に付したとき、それぞれの量のジアステレオマー(4ab)が観察され、高度の立体特異性を示した(表2)。
【0098】
【表2】
【0099】
エーテル化における穏当な収率は、この反応条件下で分解を受けてしまうアリルエステルの非常に反応性の高いことの結果であった。この反応性の減弱は、安息香酸エステル又はナフトエ酸エステルなどの弱い脱離基の選択により達成され、(4b)の収率を73%まで改善した。ナフトエ酸アリルエステル(11b)は結晶性固体であり、さらなる開発のために選択した。広範なリガンドのスクリーニングは、ブッフワルド(Buchwald)ビアリールジホスフィンがPd−触媒によるエーテル化において有効であったが、あまり高価ではなく、より簡単に入手し得る1,3−ジフェニルホスフィノプロパン(dppp)によっても同等の結果が得られることを明らかにした。さらなる改善として、アルコール(6b)の量は生成物のアッセイ収率に影響を与えることなく、1.0当量まで減少させることができた。最適な条件下、アリルエーテル(4b)は、収率80%、ジアステレオマー比>99:1で調製した(反応工程図3)。
【0100】
【化32】
【0101】
Pd−触媒によるアリルエーテル化のために開発された条件は、様々なアルコールに寛容であり、反応性中心での立体化学を完全に保持したまま、多様な一連のアリルエーテルを提供した(表3)。アリルエステル(11b)とアルコール(6b)の両方のエナンチオマーとのカップル結合においては、僅かな反応性の差異が観察されただけであった;しかし、立体的により妨害されたアルコールの場合には、明らかな「マッチ」及び「ミスマッチ」の対合が観察された。さらに、アリルエステルとアキラルアルコールとのカップル結合は、高エナンチオマー比で、期待どおりのアリルエーテルを提供した。この反応はエステル、ニトリル、さらにはケトンをも含む多様な電子求引性基に寛容であった;しかし、3−位に電子求引性基を欠く環状アリルエステルは、アリルエーテル化反応に不活性であった。
【0102】
【表3】
【0103】
アリルエーテル(4b)の高度に収束した効率的な調製は、アリール−金属種のジアステレオ選択的共役付加を経てシクロペンタン(2b)へ接近する手段を提供した。グリニヤール又はクプラート法により共役付加を達成する試行は、少なからぬ量の1,2−付加生成物と共に、様々な量の所望の共役付加生成物に導いた。アリールホウ酸誘導体のエナンチオ選択的Rh−触媒共役付加における最近の進歩が、我々に、我々の基質に対してこの方法を試みることを促した。参照:Hayashi,T.;Yamasaki,K.Chem Rev.2003,2829及びその引用文献。典型的に、エナンチオ選択的変異体と関連する問題は、「リガンドのない」背景反応の高い反応速度である。結果として、我々はリガンドの不在下に、基質(4b)を標準的な条件(5当量のホウ酸、3mol%[CODRh(OH)]2、ジオキサン/水)に付した。我々は、完全な立体制御とともに所望の共役付加産物を、89%のアッセイ収率で観察したことに満足した。ホウ酸量の有意な削減は、アリールトリフルオロホウ酸カリウム塩(参照:a)Batey,R.;Thanadi,A.N.;Smil,D.V.Org.Lett.1999,1683.b)Darses,S.;Genet,J.P.;Brayer,J.L.;Demoute,J.P.Tetrahedron Lett.1997,4393;このものは完全な変換を達成するために、1.5当量を必要とする)を用いることにより達成することができた。また、我々は、エタノールが溶媒系としてジオキサン/水の優れた代替物であり、より急速な変換、良好な収率、及びより汚染のない反応を提供することを発見した。最適化した条件下、熱力学的に好適な異性体へ平衡化した後、95:5のエピマー混合物として93%の収率で(15b)を得た。この混合物は容易に酸(2b)に加水分解され、また優れた収率でトリエチルアミン塩として単離され、不所望のエピマーは好都合に回避された(反応工程図4)。
【0104】
【化33】
【0105】
(2b)の効率的な製造法を証明するために、我々はこの方法の範囲と領域を決定しようとした。様々な電子豊富及び電子欠乏アリールホウ酸が、ジアステレオ選択Rh−触媒による共役付加において適切な求核試薬であり、それぞれの場合に、高収率と完全なジアステレオ選択性を提供した(表4)。この方法論は適度に障害のあるアリールホウ酸にも寛容であった;しかし、2,6−二置換アリールホウ酸での共役付加生成物は低収率であった。ヘテロ環状ホウ酸及びビニルホウ酸は、Rh−触媒によるプロセスでは全く無効であり、すべての事例において所望の共役付加物の収率は<5%であった。この方法論はまたα,β−不飽和ケトンとニトリルには有効であり、すぐれた収率とジアステレオ選択性でそれぞれの生成物を提供した。すべての基質(エステル、ニトリル、及びケトン)は、熱力学的平衡後に、高エピマー比を与えた(それぞれ、95:5、92:8、及び98:2)。
【0106】
【表4】
【0107】
Rh−触媒による共役付加方法はα,β−不飽和ケトンxxには有効であるが、エノレート・トラップとして塩化トリメチルシリルを用いる臭化4−フルオロフェニルマグネシウムのCu−触媒共役付加(参照:Varchi,G.;Ricci,A.;Cahiez,G.;Knochel,P.Tetrahedron,2000,2727)は、ケトンが熱力学的に好ましい異性体に平衡化した後、98:2のエピマー混合物として実際上94%の収率でxxを提供した。Cu−触媒による共役付加は、Rh−触媒による共役付加方法では達成し得なかったアリール、ビニル、ヘテロ芳香族、及びアルキル金属種の付加についても実証された。
【0108】
結論として、我々は非常にすくない工程により傑出した選択性で、高度に機能化されたシクロペンタノイド構造を途切れることなく構築する方法を報告してきた。この方法の収束性は、それを、構造的な複雑さを迅速に組み上げるための価値ある手段とし、そのルートの組み立てユニットの性質は、構造類似体の効率的生産を可能とする。本明細書に記載した例は、多様な一連の複雑な中間体が、この化学によって接近可能となることを示している。
次に、我々はキラル・オキサゾリジノン(3a)を、シクロペンタン酸(2b)から誘導されるハロメチルケトンでジアステレオ選択的にアルキル化することを試みた。
【0109】
【化34】
【0110】
クロロメチルケトン(4c)は、2工程経由で酸中間体(2b)から調製した:i)(2b)のメチルエステル化であって、(2b)を、極性溶媒、例えば、DMF、DMAc、DMSO若しくはアセトンなど中、約0℃ないし約60℃の範囲の温度で、ヨウ化メチル及びLi、Na、K、Ca若しくはCsなどの炭酸アルカリ金属により処理することからなる。また(2b)を、酸触媒、例えば、硫酸、TsOH、MsOH、又はベンゼンスルホン酸などの存在下、約50℃ないし還流の温度範囲で、MeOHにより処理することによりメチル化し得る;ii)メチルエステル(2c)のクロロメチル化は(2c)を、ClCH2CO2H及び式M4N(R16)2又はM4N(Si(R16)3)2(ここで、M4はLi、Na、K、又はMg(Mgは二価であり、変化する要あり)であり、各R16は、独立して、C1−4アルキルから選択された)の金属アミドにより、非プロトン性有機溶媒、例えば、THF中、約−20℃ないし約40℃の温度範囲で処理することから構成された。ClCH2I又はClCH2Brはまたこの反応で使用され得る。
【0111】
化合物(4a)は、極性有機溶媒、例えば、DMF、DMAc、DMSO、又はアセトンなどの中、約0℃ないし約60℃の温度範囲で、ヨウ化アルカリ金属若しくはアンモニア、例えば、LiI、NaI、KI、R4NI(ここで、RはH又はC1−4アルキルから選択される)による処理によって調製された。無水の条件がより良好な収率を与えた。
【0112】
【化35】
【0113】
キーとなる中間体(2a)の合成法は、本発明の実施態様を形成した。本方法は、式MN(R)2又はMN(SiR3)2(ここで、MはLi、Na、又はKであり、各Rは、独立して、C1−4アルキルから選択された)のアルカリ金属アミドの条件下、トルエン若しくはTHFなどの有機溶媒中、非プロトン性極性溶媒又はアミン付加体(例えば、DMPU、DMI、又はTMEDA)の存在又は非存在下で、約−78℃ないし約−40℃の温度範囲で、(3a)のエノレートをα−ハロケトン(4)に付加させることにより、四級キラル炭素中心を含有して構成された。極性の低い溶媒と非プロトン性極性溶媒との低い反応温度での組み合わせは、より良好な収率を与えた。
【0114】
トルエンなどの有機溶媒中、(2a)の溶液中の残余過剰量の(3a)は、水性LiOHでの選択的加水分解と、引き続く、約0℃ないし約40℃の温度範囲での水性NaHSO3処理により除去することができた。
【0115】
【化36】
【0116】
アルキル化されたオキサゾリジノン(2a)の、有機溶媒(例えば、THF、DME、メタノール、エタノール又はイソプロパノール)中、水性若しくは有機性NH3による約0℃ないし約60℃の温度範囲での加アンモニア分解は、直接に、アミナル(14a,b)のジアステレオマー混合物を与えた。
【0117】
【化37】
【0118】
(14a,b)のヒドロキシ基を還元して(16)とするために、酸、例えば、トリフルオロ酢酸、メタンスルホン酸、トリフルオロボラン・エーテレートなどの存在下、アセトニトリル、トルエン、酢酸、ニトロメタン、若しくはニトロエタンなどの有機溶媒中、又はそのままの状態で、約−40℃ないし約60℃の温度で、式R3SiH(ここで、RはH又はC1−4アルキルから選択される)で示されるシランにより、反応が完結するまで(通常、約2〜24時間)処理した。この還元は、(14a,b)からエナミン中間体(15a,b)を経由して、有機溶媒、例えば、アセトニトリル、トルエン、酢酸エチル、酢酸、ニトロメタン、若しくはニトロエタンなど中、又はそのままの状態で、約−40℃ないし約60℃の温度で、酸、例えば、トリフルオロ酢酸、メタンスルホン酸、トリフルオロボラン・エーテレートなどで処理することにより進行することが判明した。化合物(16)は同様の条件下で、(14a,b)及び(15a,b)の両方から製造し、そのジアステレオマー(16a)との混合物として得た。より良好な収率とジアステレオ選択性は、アセトニトリル中、エナミン(15a,b)から低温で還元により得られた。
【0119】
【化38】
【0120】
(16)のベンジルオキシカルボニル(Cbz)基を遷移貴金属、例えば、Pd/C、Ph(OH)2/C、Pt/Cなどによる接触水添により、又は酸性溶媒和により切断して(1)とした;接触水添はメタノール、エタノールなどのアルコール系溶媒中で、また溶媒和はHCl、HBr若しくはHIなどのハロゲン化水素、又はハロゲン化金属若しくはアンモニウムと酸(例えば、トリフルオロ酢酸、メタンスルホン酸など)との組み合わせなどの等価物により、有機溶媒(例えば、アセトニトリル、トルエン、又は酢酸)中、約0℃ないし約60℃の温度で実施した。このCbz基の脱保護を酸性条件下で実施した場合、(1)は(14a,b)又は(15a,b)からワンポット方式で得られた。Cbz基の脱保護の副産物であるハロゲン化ベンジルは、ヘプタンによる逆抽出により、反応混合物より容易に除去された。(1)の遊離塩基はその両親媒性物性のために、水層から有機層に抽出することが困難であった。t−BuOHとMTBEの混合溶媒は、水層から遊離塩基を抽出するために有効であることが判明した。粗製の遊離塩基は、IPA−IPAc−ヘプタン溶媒系からベンゼンスルホン酸塩として結晶化することにより精製した。ベンゼンスルホン酸塩は2つの結晶型、I型又はII型を有する。II型はI型よりも熱力学的により安定である。ベンゼンスルホン酸塩は、要すれば、アセトニトリル及びアルコール(例えば、メタノール、エタノール、又はイソプロパノール)から再結晶し得る。
【0121】
【化39】
【0122】
ラクタムhNK−1受容体拮抗薬(1)の、シクロペンタン核の高度に収束的、効率的、さらに完全にジアステレオ選択的、エナンチオ選択的合成が確認されている。本明細書に記載されている一般的方法は、酸中間体(2)及びヨードケトン中間体(3)両方の合成に適用された;これら中間体は共に好収率及び受容可能な純度で(1)を生成することが実証されている。
【0123】
【化40】
【0124】
(1)を作製するための初期の開発研究は、主として(2)が成功裏に(1)に変換されたことのある唯一の中間体であったために、(2)の効率的な合成に向けられていた。我々は高度に収束的なジアステレオ選択的、エナンチオ選択的様式で、迅速に(2)を製造する新しい方法を成功裏に開発した(反応工程図1)。この合成のハイライトは、3−カルボキシメチルシクロペンテノン(5)の高度にエナンチオ選択的酵素還元、立体特異的Pd−触媒によるエーテル化、及びジアステレオ選択的Rh−触媒による共役付加である。
【0125】
【化41】
【0126】
この合成法は既存の(2)の製造法よりも改善されていたが、このルートは(5)の調製において、規模的に、またコスト的に、Rh−触媒による共役付加方法を困難なものとした。最終的にこの化学における改良法は、(2)が7工程で(1)に変換し得ることを証明した(反応工程図2)。この改良されたルートを試験すると、(2)を(3)に変換するためには数工程が必要であることから、(3)が(1)を収束的に合成するためのより好適な合成標的であることが判明した。
【0127】
【化42】
【0128】
(2)を合成するために開発された一般的方法は、(3)の製造に必要な基質の変化に適応させるために修飾した(反応工程図3)。(3)を直接的に標的とすることにより、全工程は、大量の出発原料を製造する使用可能な方法を提供し、Rh−触媒による共役付加をCu−媒介共役付加に転換し、該方法の数工程を除くことにより、改善された。最適化された(1)の合成はシアノケトン出発原料から10工程(ヨードケトンから4工程)に縮減された。30gスケールでの(2)と(3)両方に対するこの方法の発見と立証について以下に記載する。
【0129】
【化43】
【0130】
【表5】
【0131】
基本的な溶媒の選抜では、当初のエーテル化条件にて使用したTHF(表3)よりも良好な溶媒は見つからなかった。他の配位エーテル型溶媒(又は多分三級アミン付加体)が改善に役立ち得るとの見込みが未だ存在する。このことは未だ検討されていない。
【0132】
【表6】
【0133】
リガンドを徹底的にスクリーニングして(表4)、ブッフワルド・ビアリールホスフィン・リガンドのジシクロヘキシルホスフィノ変異体が、対応するジ−tert−ブチルホスフィノ体よりもはるかに良好に機能することを明らかにした。このことは、比較的に障害の少ないホスフィン類がPd−触媒によるエーテル化に好適であることを物語っていた。一般に、ブッフワルド・ビアリールホスフィン・リガンドは、dpppなどのジホスフィンをキレート化する(典型的には、室温で2〜24時間)よりも僅かに高い収率とより速い反応(通常、0℃で2時間)を与えた。それにもかかわらず、dpppリガンドはコストが低いという有利性と、良好な利用可能性を有していた。キレート化ジホスフィン・リガンドに関しては、dpppがdppe或いはdppbよりもより効果的であった。触媒負荷をPd2%及びリガンド3%(ブッフワルド・ビアリールホスフィン・リガンドを使用)に低下させると、p−ニトロ安息香酸エステルのエーテル化が僅かに遅くなったが、アッセイ収率は実質的に変らなかった(0℃で7時間後に96%の変換率と70%の収率)。
【0134】
【表7】
【0135】
さらなる改良として、ナフトエ酸基質(R=2−Nap)の場合には、(9)のアッセイ収率に有意な影響を与えることなく、ベンジルアルコール(8)の量を1.2当量に減量できることが判明した。ナフトエ酸エステルとdpppリガンドの組み合わせは、通算収率とコスト効率の意味で最善の反応を提供し、さらなる開発のために選択された。
【0136】
反応濃度の影響についても簡単に検討した。THF中、0.8Mと0.3Mの反応の間に小さな差(78%対75%のアッセイ収率)が観察された(本明細書で、Mは、mLの溶媒に対するmmol(ミリモル)のエステル基質と定義した;Et2Zn溶液が供給するヘプタン溶媒を含む)。しかし、幾分製造率の高い0.8Mの反応は、攪拌の困難な濃厚油を形成することが問題となった。妥協点として、最終決定した手法では0.5Mの濃度を選択した。
【0137】
ナフトエ酸エステル(7)をエーテル化のための理想的な基質として同定し、製法をその形成のために開発した。アリルアルコール(6)のアシル化が反応性に富む反応であり、塩化ナフトイルでの処理が所望のアリルエステルのアッセイ収率を低下させることは銘記すべきである。対照的に、無水ナフトエ酸のその場での形成(酸塩化物と酸から)と、引き続くアルコールでの処理が、好収率(91%)でアリルエステル(7)を生成した。結晶化による単離は、77%の収率と>99%のLCAPで(7)を提供した。
【0138】
エーテル化反応のための3gでの先行試行は、1.2当量のアルコール(8)、3%Pd及び4%dpppリガンドを用いて実施した。78%のアッセイ収率と75%の単離収率(95%LCAP、98%アッセイ)が得られたが、反応は比較的停滞ぎみであり、>99%の完結までに2日を要した。それ故、4%Pdと6%のリガンドを20gでの実証のために使用した。
【0139】
20gスケールでの反応は、1時間の導入期間を示したが、反応が加速し始めたときにも有意な発熱は観察されなかった。アリルエーテル(9)は、ダルコ(Darco)KB−Bでの処理(Pdを除去するため)、溶媒のMeOHへの交換、及びMeOH−水からの結晶化によって単離された。20gスケールでは母液の損失(6%)の増加のために、幾分低い単離収率を観察したが、3gでの先行試行に比較して、有意により純度の高い生成物を得た。最終的に、20gの(9)は、71%の収率と>98%の純度で結晶化により単離した(反応工程図4)。
【0140】
【化44】
【0141】
【化45】
【0142】
シクロペンタン核の合成を完成するためには、ジアステレオ選択的共役付加が必要であった。我々は、嵩だかいエーテル置換基が侵入してくる求核試薬の面選択性を有効に制御して、共役付加生成物に所望のトランス立体配置を与え得るであろうと想定した。熱力学的条件下でのエステルの異性化がトランス配置を強力に支持し、完全にジアステレオ選択的様式でトランス,トランス−1,2,3−三置換シクロペンタン(10)を与えた。
【0143】
基質(9)への最適な共役付加には、基質への4−フルオロフェニル・グリニヤール試薬の付加が関与しよう。同様の形質変換についての文献調査では、所望の共役付加生成物の中庸ないし低収率(20〜60%)を与える入り混じった結果を明らかにした。実際、(9)に関して、CuI及びTMSClの存在下で、臭化4−フルオロフェニルマグネシウムとの共役付加を実施した場合、約45%の(10)を観察した(eq.7)。さらに分析すると、共役付加生成物はさらにグリニヤールとの1,2−付加を受けて、ジアリールカルビノール(27)を生じた;このものは40%の収率で得られた。付加物、溶媒、又は温度によるこの過剰反応を回避する試行は失敗に終わった。事実、必要量に満たないグリニヤール試薬で反応を実施したとしても、等モル比の(10)と(27)が観察された。
【0144】
【化46】
【0145】
グリニヤール共役付加に対する別法は、Rh−触媒によるアリールホウ酸との共役付加であった。この分野の文献報告の殆どが、共役付加生成物において不斉誘導を生じるキラルリガンドの使用が関わっている。この反応の不斉変異体を開発することと関連する問題の一つは、「リガンドなし」のRh−触媒による共役付加を遂行することの容易さである。我々は、ジアステレオ選択的なRh−を触媒とする共役付加をリガンドの不存在下で容易に実施し得るとすることに疑いを持っていた。実際には、標準的な条件下で、[CODRh(OH)]2の存在下、(9)を4−フルオロフェニルホウ酸と反応させると、良好な選択性(>99:1)で所望の共役付加生成物(10)が得られた。銘記すべきは、共役付加ステップのアッセイ収率が、反応に加えたホウ酸の量に強く影響されるということである(表5)。1.5当量限りのホウ酸を負荷した場合、32%のアッセイ収率が得られただけであった。対照的に、5当量のホウ酸を負荷した場合には、89%アッセイ収率の所望の生成物が観察された。このことは、ホウ酸のヒドロ脱ボリル化が競合的副反応であるとする文献の報告と矛盾がない。
【0146】
【表8】
【0147】
Rh−触媒による共役付加にアリール亜鉛試薬を使用する試みは不成功であり、多くの不純物と最小量の所望生成物(<10%)に導いた。対照的に、アリールトリフルオロホウ酸塩に置き換えると、非常に上手くいき、アッセイ収率の損失なしに、フルオロホウ酸塩の添加を1.5当量に減量し得る。この反応はジオキサン/H2Oで観察された性能に比べてEtOH中での性能が改善されることが判明し、1.5当量のアリールフルオロホウ酸塩を使用するだけで、93%のアッセイ収率で(10)が得られた(eq.8)。
【0148】
【化47】
【0149】
興味深いことに、(10)と(10a)のジアステレオマー比は、グリニヤール共役付加で観察される比よりもかなり高かった(85:15に対し55:45)が、これは恐らく反応条件下での部分的異性化によるものである。(10)と(10a)の混合物を異性化条件(NaOMe、MeOH、50℃)に付すと、その比が95:5の平衡に達した(反応工程図5)。さらに加熱しても、あるいは塩基性としても、その比は変らなかった。以前に開発した方法では、(1)の合成を完結させるために、(10)を必要とした;しかし、固形物の単離の容易さを示すために、また標準品と比較してその構造と不純物プロフィールを検証するために、(10)を(2)に変換した。
【0150】
【化48】
【0151】
異性化の後、エステル(10)を同じ反応容器内で加水分解し、酸(2)とすることができた。水性HClで中和した後、対応する酸を抽出して、NEt3半溶媒和物として単離した。共役付加/異性化方法は20gで実証し、同じ容器中で加水分解して、(2)を得た。19.7gの(2)をNEt3半溶媒和物として単離したが、その物質の純度は以前の方法で得られたものと同様であった;98.2LCAP(1.3面積%;10a由来のエピマー)。
【0152】
要約すると、酵素還元/Pd−エーテル化/Rh−共役付加ルートが、酸中間体(2)の迅速な接近のための効率的な方法として立証された。この合成はこのフラグメントの当初の合成よりも有意に短く(5工程対>12工程)、従来の合成法で要求された変わりやすいエーテル化方法を回避し、同様の純度と改善された通算収率(35%対<25%)で(2)を生じる。
【図面の簡単な説明】
【0153】
【図1】図1は式(Ia)で示される化合物のベンゼンスルホン酸塩の無水結晶形I型の特徴的なX線回折パターンである。
【図2】図2は式(Ia)で示される化合物のベンゼンスルホン酸塩の無水結晶形I型の典型的なDSC曲線である。
【発明を実施するための形態】
【実施例】
【0154】
実験手法:第1部
工程1−カルボキシメチルシクロペンテンの酸化:
【0155】
【化49】
【0156】
手法:
磁気攪拌バーと内部温度測定用電極を備え、冷却(0℃)した2L容量の丸底フラスコに、無水酢酸(615g、570mL、6.02mol)を加えた。三酸化クロム(214g、2.14mol)を分割して加え、その間、攪拌を一定に保ち、発熱を制御した。得られる血液様赤色溶液を、温度が20℃に冷えるまで攪拌して三酸化クロムを溶解した。5L容量の三頚フラスコに滴下漏斗、上部からの攪拌装置、窒素送入口及び内部温度測定用電極を取り付け、CH2Cl2(1.4L)中の(4)(100g、101mL、0.793mol)を加えた。三酸化クロムと無水酢酸からなる酸化溶液を滴下漏斗に入れ、10ないし14℃の内部温度を維持しながら、反応混合物に滴下した。当初黄色であった溶液が酸化剤を最初の2〜3滴加えた後、暗色となった。
【0157】
反応は、実験室での容器サイズに制限があるため、同じサイズのバッチで2回実施した。各バッチは以下のように正確に同じ方法で処理した。暗色の均一溶液は、上部からの攪拌装置を備えた4Lビーカーに注意深く注入した。反応フラスコを250mLのCH2Cl2ですすいだ。500mLのH2Oを加え、次いで、10gのNaHCO3を加えると、ガスが発生した。追加のNaHCO3(830g、10mol)を分割して加え、その間、粘稠混合物の攪拌速度を500rpmに維持した。得られる暗緑色の懸濁液を1LのH2Oで希釈し、ソルカフロックの1cmパッドを容れた3Lの硝子フィルター付き漏斗で濾過した。二相溶液をCH2Cl2(3×1L)で抽出し、併合した有機相をMgSO4で乾燥し、次いで濾過し、得られる溶液を減圧濃縮して淡緑色油を得た。30cmビグローカラムで蒸留し、次いでMTBE:ヘキサン(1:10、55mL全量)から再結晶し、38.4gの(5)を白色結晶性固体として得た(35%)。
【0158】
工程2−酵素還元:
【0159】
【化50】
【0160】
手法:
カリウム二塩基性バッファー(100mM、pH7.0、2L)に、ギ酸ナトリウム(120g)及びニコチンアミド・アデニン・ジヌクレオチド(NAD、8グラム)を加えると、バッファーpHが6.7に下がった。このバッファーに、酵素、アルコールデヒドロゲナーゼRE(5g、185KU)、ギ酸デヒドロゲナーゼ(20g、94KU)を加えた。基質(5)(10g、0.071mol)を粉末としてそのまま加え、温度を25℃に調整し、2N硫酸でpHを6.5に調節した。反応を24時間熟成し、次いで、酢酸エチル(2倍容量抽出)で抽出し、さらに減圧濃縮した。(6)の通算収率は83%、抽出による損失は2%、残余エノンは<3%であった。残りの13%の質量バランスは、水溶液中での不安定性によるエノンの損失によると判断した。
【0161】
工程3−アリルアルコールのアシル化
【0162】
【化51】
【0163】
手法:
ジクロロメタン(200mL)中、2−ナフトエ酸(24.6g、143mmol)及び塩化2−ナフトイル(27.2g、143mmol)の懸濁液を氷浴にて+5℃の内部温度に冷却した。ジイソプロピルエチルアミン(89mL、511mmol)を加え、その間、内部温度を24℃以下に維持した。発熱が収まったところで、氷浴を室温水浴に置き換えた。最初に褐色の澄明な溶液が形成され、それが次第に微細なスラリーに変るので、それを室温で30分間攪拌した。ジクロロメタン(50mL)中の(6)(14.4gアッセイ、102mmol、>99%ee)及びDMAP(1.25g、10.2mmol、0.10当量)からなる溶液を一度で加えた。非常に穏やかな発熱が観察され、温度が23℃で最大に達した。反応混合物を室温で2.5時間攪拌した。水(10mL)を加え、反応混合物を室温で2.5時間攪拌した。この時点でのHPLC分析は、過剰の無水ナフトエ酸が完全に加水分解されていることを示した。反応混合物をMTBE(500mL)と併合し、飽和のNaHCO3水(2×500mL)、水(500mL)、1M−HCl水(500mL)及び最後に水(4×500mL)で洗浄した。暗褐色の濁りのある有機相をソルカフロックの短いパッドで濾過し、濾液中に91%のアッセイ収率を得た。この溶液を200mL容量まで濃縮し、ヘプタン(200mL)を加え、この混合物を〜10gのシリカゲルで濾過し、ヘプタン−MTBE(2:1)100mLで溶出した。濾液を200mL容量まで濃縮し、種結晶を加えた。さらに50mLの溶媒を40℃で除去し、残りの懸濁液は室温にもどしながら2時間攪拌した。室温で15時間後、懸濁液を濾過し、濾過ケーキを50mLのヘプタンで洗い(母液損失4%)、23.2gの(7)を微細白色粉末として、77%収率及び99%LCAPで得た。
【0164】
工程4−Pd−触媒によるエーテル化反応:
【0165】
【化52】
【0166】
手法:
250mL容量の丸底フラスコに(8)(23.3g、90.3mmol、1.2当量)を容れ、排気してから窒素を充填した。THF(75mL)を加え、得られる溶液を氷浴で+5℃に冷やした。1.0M−Et2Zn/ヘプタン(45mL、45mmol)を加えると、緩やかに発熱し、15℃となった。冷却浴を除き、得られる澄明な溶液を室温で1時間攪拌した。
【0167】
別個の500mL丸底フラスコに、(7)(22.3g、75.3mmol)、Pd(OAc)2(676mg、3.01mmol、4mol%)、1,3−ビス(ジフェニルホスフィノ)プロパン(1.86g、4.51mmol、6mol%)及びL−トリプトファン(1.54g、7.54mmol、10mol%)を装入した。次いで、フラスコを排気してから窒素で充満させ、氷浴にて冷却した。第一のフラスコからのアルコキシド溶液はカニューレを介して第二のフラスコに移した。冷浴を除去し、得られる緑褐色懸濁液を室温で16時間攪拌した。この時点でのHPLC分析は、ナフトエ酸エステルの完全な変換を示していた。淡褐色懸濁液を氷浴で冷却し、1M−HCl水(100mL)及びMTBE(100mL)と併合すると発熱して15℃となった。この懸濁液を5℃で15分間攪拌し、次いでソルカフロックで濾過してMTBE(200mL)で溶出した。淡黄色濾液を水(3×200mL)、5%NaHCO3水(2×300mL)、及び水(2×200mL)で洗浄した。有機相をHPLCで分析すると、生成物を78%のアッセイ収率で得た。ダルコ(Darco)KB−B(15g)を有機相に加え、懸濁液を室温で3時間攪拌し、次いで、ソルカフロックで濾過した。ほぼ無色の濾液をMeOHと溶媒交換し、最終容量120mLとした。水(5mL)を加え、この溶液に種結晶を加え、さらに水(70mL)を15分間で滴下した。スラリーを室温で1時間攪拌し、次いで濾過した。得られる白色結晶を真空乾燥し、20.4gの(9)を71%の収率及び98%のLCAPで得た。
【0168】
工程5−Rh−触媒による共役付加/加水分解/酸単離:
【0169】
【化53】
【0170】
手法:
磁気攪拌機、熱電対、及び窒素送入口を備えた250mL容量三頚丸底フラスコに、(9)(19.0g、0.0497mol)、NaHCO3(1.94g、0.023mol)、フルオロホウ酸塩(16.1g、0.080mol)、及び[CODRh(OH)]2(646mg、0.0015mol)を加えた。フラスコを密封し、1時間窒素を送入した。1時間窒素で脱ガスしてあったエタノール(150mL)を、固形物を容れた反応容器にカニューレを介して供給し、その反応混合物を90℃に加熱した。HPLC分析によると、反応は3時間以内に完結した。混合物を50℃に冷却し、11mLの25wt%NaOMe/MeOHを供給した。この混合物を50℃で4時間熟成した;このものは95:5のジアステレオマー比を示し、完全な平衡にあった。混合物を40℃に冷やし、3N−KOH(40mL)を供給し、40℃で1時間熟成すると、HPLC分析で完全な加水分解を示した。混合物を室温に冷やし、水270mLとヘプタン270mLで希釈した。ヘプタン層を除去し、水層をヘプタン300mLと混合し、50mLの濃HClで中和した。層分離し、ヘプタン層を水(200mL)で2回洗浄した。ヘプタン溶液をアッセイすると、(10)が18.4g(83%)であることを示した。ヘプタン溶液を共沸蒸留させてKf<500とし、次いで、150mLの容量に濃縮した。この溶液をMTBE(15mL)で希釈し、45℃に加温し、NEt3(3.0mL、0.0215mol)を加えた。このバッチを15分間熟成した後、この混合物に種結晶(10mg)を加え、バッチを2時間かけて室温までゆっくりと冷やした。バッチを1時間熟成し、上清をアッセイし(10.8mg/mL)、溶融硝子漏斗にて濾過した。得られる結晶性固体をヘプタン:MTBE(10:1)50mLで洗い、19.4gの(2)(76%)を得た。母液への損失=1.95g(7.6%)。単離した固体を分析すると、98.0wt%の純度、98.2LCAPを示し、最大の不純物は(10a)由来のジアステレオマーであった(1.3面積%)。ベンジル性ジアステレオマーも0.3面積%で存在した。
【0171】
工程6−メチルエステル化/クロロメチル化/ヨード化:
【0172】
【化54】
【0173】
手法:
DMF(480mL)中、K2CO3(63.5g、460mmol)及び(2b)(160g、98.6w%、306mmol)の懸濁液に、ヨウ化メチル(38.2mL、613mmol)を23℃で30分かけて加えた。混合物を室温で20時間攪拌した。この反応混合物に、MTBE(1280mL)、10%食塩水(800ml)、及びH2O(800ml)を加えた。有機層を分離し、0.5Nリン酸バッファー(800ml、pH6.8)及び10%食塩水(800ml)で洗った。有機層(1240ml)を40℃でのMTBEとの共沸処理により乾燥し、次いで、溶媒を40℃でTHFと置き換えた。THF溶液中のこの粗製の(2c)(480ml、136gアッセイ、283mmol、92.5%、KF<400ppm)は、さらに生成することなしに次工程で使用した。
【0174】
1.9M−n−BuMgCl/THF(6当量、1700mmol、895mL)の溶液に、ジイソプロピルアミン(6.6当量、1870mmol、262mL)を25〜30℃で加え、得られるスラリーを2時間20〜25℃で攪拌した。THF(678mL)中、(2c)(136g、238mmol)及びクロロ酢酸(3当量、850mmol、80.3g)の混合物をこのスラリーに1時間かけて、15℃以下で滴下した。反応混合物を20〜25℃で16時間攪拌し、次いで、6N−HCl水(15当量、4.25mol、709mL)及びMTBE(1360mL)からなる混合物に15℃以下で、30分かけて移行した。有機層を分離し、0.5Nリン酸バッファー(800ml、pH6.8)により、水層のpHが>6.0となるまで洗い、次いで、23%食塩水で洗った。有機層を溶液のKFが<500ppmとなるまで、MTBEとの共沸処理により乾燥し、次いで、全容量を1440mLに調整した。(4c)のHPLCによるアッセイ収率は125g(251mmol、88.7%)であった。
【0175】
MTBE(1440mL)中の(4c)(125g、251mmol)の溶液をアセトンと溶媒置換し、全容量を1120mLに調整した。この溶液にNaI(56.5g、377mmol、1.5当量)を加え、さらにアセトン(125ml)を室温で加えた。混合物を外気温で16時間攪拌した。反応混合物を(4a)の種結晶(125mg)と水(2120mL)の懸濁液に1時間かけて滴下し、反応容器をアセトン(125ml)ですすいだ。2時間の熟成の後、(4a)の結晶を濾取し、濾液がpH>4.5となるまで水洗した。乾燥後、(4a)を黄色結晶(153g、93.8wt%、99.2%収率)として得た。
【0176】
工程7−オキサゾリジノンのカップリング結合:
【0177】
【化55】
【0178】
手法:
トルエン(100mL)中、DMPU(15.0mL)及びDMI(15.0mL)の混合物に、1.0M−LHMDS/ヘキサン(40.8mL、40.8mmol、2.4当量)を20〜25℃で加えた。得られる混合物を5分間以上攪拌し、次いで、−75℃以下に冷却した。この溶液に、トルエン(100mL)中のオキサゾリジノン(3a)(13.2g、42.5mmol、2.5当量)を−70℃以下で1時間かけてゆっくりと加え、得られる溶液を−70℃以下で30分間攪拌した。次いで、上記溶液に、トルエン(50.0mL)中の(4a)(10.0g、17.0mmol)の溶液を−70℃以下で1時間かけてゆっくりと加えた。−70℃以下で30分間攪拌した後、反応混合物に10%クエン酸水(100mL)を加えて反応停止し、外気温まで加温した。有機層を分離し、10%NaHCO3水(50mL)で洗い、次いで、外気温で18時間、1N−LiOH水(100mL、100mmol、5.9当量)で処理した。水層を分離した後、有機層を1N−NaHSO3水(100mL、100mmol、5.9当量)及び10%食塩水(50.0mL)で洗浄した。生成物(2a)をトルエン溶液(9.44g、72%、254mL)として得、さらに精製することなく次工程で使用した。
【0179】
工程8−エナミン形成:
【0180】
【化56】
【0181】
手法:
(2a)(2.02gA、2.62mmol)のトルエン溶液を40℃でTHF(約23.0mL)と溶媒交換し、次いで、外気温で24時間、25%NH3水(20.6当量、4.04mL、54.0mmol)で処理した。反応混合物に5%食塩水(10.1mL)及びAcOEt(10.1mL)を加えた。有機層を分離し、10%NaHSO3水(10.1mL×2)、10%K2HPO4水(10.1mL)、及び20%食塩水(10.1mL)で洗った。アミナール(14a,b)をAcOEt溶液(1.23g、1.80mmol、70.8%、31.8mL)として得た。
【0182】
(14a,b)(4.04g、KF4.6%)のAcOEt溶液をAcOEtにより共沸乾燥し、40℃で20.2mL(KF<0.3%)まで濃縮した。次いで、この混合物に0〜5℃でMsOH(0.31mL、1当量)を加え、反応混合物を0〜5℃で1時間攪拌した。反応混合物に水(20.2mL)を加え、有機層を分離し、0〜10℃で10%K2HPO4水(20.2mL)により、15〜25℃で20%NaCl水(20.2mL)により順次洗い、次いで、24〜27℃で活性炭(シラサギP:312mg及びダルコKB−B:312mg)により30分間処理した。混合物を濾過し、AcOEt(12.1mL)ですすいだ。濾液と洗液を併合し、CH3CNと溶媒交換し、15.9mLに濃縮した。CH3CN中、エナミン(15a,b)を位置異性体の混合物(3.19g、100%、異性体比76:24)として得た。
【0183】
工程9−最終段階(シラン還元/脱保護/結晶化):
【0184】
【化57】
【0185】
手法:
MsOH(5.85mL、80.3mmol、8.7当量)及びEt3SiH(1.93mL、12.1mmol、1.3当量)を、0℃以下で連続して、CH3CN中のエナミン(15a,b)の溶液(30mL、6.15g、9.25mmol、1当量)に加え、次いで、得られる混合物を−5〜0℃で4時間攪拌した。(15a,b)が完全に消費された後、30%HBr/AcOH(3.20mL、16.1mmol、3.5当量)を5℃以下で滴下した。この混合物を38〜42℃に加温し、同温度で一夜攪拌し、次いで、0℃に冷却した。この反応混合物に、水(30.8mL)及び活性炭(シラサギP、1.23g)を連続して加えた。得られた混合物を1時間攪拌した後、濾過し、CH3CN/H2O=1/1(18.5mL)ですすいだ。濾液と洗液を併合し、ヘプタン(61.5mL×3)で洗浄した。水溶液を20℃以下で5N−NaOH水(17mL)によりpH3〜4に調整した。この反応混合物にMTBE/t−BuOH=2/1(30.8mL)を加え、得られた混合物を20℃以下で5N−NaOH水(19.5mL)によりpH=9〜10の塩基性とした。有機層を分離した後、水層をMTBE/t−BuOH=2/1(30.8mL)で抽出した。容器をMTBE/t−BuOH=2/1(12.3mL)ですすいだ。有機層を併合し、12%リン酸バッファー(pH=6.5;30.8mL)及び23%食塩水(30.8mL)により順次洗浄した。有機溶液はIPAcと溶媒交換(KF=444ppm)すると、不均一系となった。IPAc懸濁液を濾過し、濾過固体をIPAc(12.3mL)で洗った。併合した濾液を35mLに濃縮した。IPAc中、有機塩基としての化合物(1)を褐色溶液として得た(4.93g、100%)。
【0186】
IPAc中、(1)遊離塩基の溶液(221mg/mL、5.0mL、1.11g、2.08mmol、1当量)をIPAc(9.43mL)で希釈した。この溶液に、PhSO3H・H2O/IPA(1.5M、1.38mL、2.08mmol、1.0当量)を2時間かけて40℃で滴下した。得られたスラリーを40℃で一夜攪拌し、このスラリーにヘプタン(16.7mL)を1時間で添加した。40℃で21時間攪拌した後、このスラリーを外気温まで冷やした。生成物を濾集し、IPAc/ヘプタン(1/1:8.3mL×2)で洗い、減圧下、40℃で一夜乾燥した。化合物(1)はベンゼンスルホン酸塩の無色固体(1.21g、99wt%、84.6%、I型)として得た。
【0187】
実験手法−第2部:
工程1−シクロペンテノンの臭素化:
【0188】
【化58】
【0189】
手法:
磁気攪拌バー、窒素送入口、滴下漏斗及び内部温度測定用電極を備えた3L容量の丸底フラスコに、1.25LのCH2Cl2中の2−シクロペンテノン(100g、1225mmol)を装入し、−20℃に冷却した。HBr(27.3mL、245mmol)を加え、その淡黄色溶液を5分間攪拌した。滴下漏斗に臭素(61.9mL、1225mmol)を容れ、内部温度を−24ないし−20℃に維持しながら、1時間かけてそれを滴下した。添加の間に、臭素は急速に脱色した。この黄色溶液を、TLCが出発エノンの消失を示すまで、30分間、−20℃で攪拌した。内部温度を−20℃に維持しながら、ピリジン(149mL、1837mmol)を滴下した。滴下終了後、この溶液を0℃で1時間攪拌した。1M−Na2S2O3(1L)を加えて反応を停止し、MTBE(2L)で希釈した。有機相を1M−HCl(2×1L)で洗い、次いで、H2O(1L)で洗った。暗色の有機相をNa2SO4で乾燥し、濾過した。CH2Cl2の残部をMTBEと溶媒交換し、MTBE:CH2Cl2の最終の比が8:1になるようにした。この暗色溶液を30%(60g)ダルコKB−Bと一夜攪拌した。ダルコはソルカフロックのショートパッドで濾去し、無色の溶液を得た。減圧下で溶媒を除去し、170gの(15)を白色結晶性固体(95.83wt%;87%単離収率)として得た。
【0190】
工程2−シアノ化/脱離:
【0191】
【化59】
【0192】
手法:
MeOH(400mL)中、(15)(62.5gアッセイ;0.388mol)とAcOH(22mL、0.384mol)の溶液に、15℃(吸熱溶解による)で、固形のNaCN(取り扱い注意;猛毒性;28.5g、0.582mol、1.5当量)を加えた。温度は5分間で15℃から30℃に上昇し、その時点でフラスコを氷水浴で冷却した。内部温度が18℃まで下がったところで冷却浴を除いた。1.5時間室温で攪拌した(TLCによると変換は不完全)。次いで、追加の固形NaCN(9.5g、0.194mol)を加え、室温で2時間攪拌した(TLCによると完全に変換)。
【0193】
フラスコの底に溶けずに残っていた約5gの固形NaCNから傾斜して、褐色の反応混合物を3Lの分液漏斗に移した。この溶液を水(1L)と併合し、CH2Cl2(1L+400mL+400mL)で抽出した。3つの有機相中の生成物をアッセイ(HPLC)すると、それぞれ、76%、4%、及び0.6%であった。3度目の抽出後の水相の生成物の損失は0.3%であった。
【0194】
第一及び第二の有機相(合計アッセイ収率80%)を組み合わせ、シリカ(約100gのシリカ)の短いプラグで濾過し、濾液を47.1gの重量(HPLCにより66wt%純度)に濃縮した。暗褐色油の20.6gアリコートを1mmHg、60〜70℃で蒸留し、12.7gの(16)を淡黄色液体として得た(94wt%純度、このアリコートにもとづく収率66%)。
【0195】
工程3−酵素還元:
【0196】
【化60】
【0197】
カリウム二塩基性バッファー(0.1M、pH7.0、1L)に、グルコース(100g)及びニコチンアミド・アデニン・ジヌクレオチド(NAD、4g)を加え、バッファーのpHを6.7に低下させた。このバッファーに酵素、アルコール・デヒドロゲナーゼRE(1g、37KU)、グルコース・デヒドロゲナーゼ103(1g、67KU)を加えた。35℃の温度及び400rpmの攪拌による1Lの反応のために、2つの500mL反応機を使用した。基質(16)(20g、0.19mol)を各反応機に10gずつ直接加えた。2.5M炭酸カリウムによりpHを6.5に調整した。基質(16)はpH>8.0で不安定であることが知られており、そのため、上記の表面添加により、また通常使用される2N−NaOHの替わりに弱い塩基(2.5M炭酸カリウム)を用いて、塩基との接触を最小とした。反応を20時間熟成させ、その時点での変換率は95%より高かった。この変換はグルコン酸の形成から生じるpH変化を調節する塩基の消費量により容易にモニターすることができる。補助因子リサイクルからのグルコン酸の形成は、形成されるアリルアルコール生成物の量に正比例した。反応液を酢酸エチル又はイソプロピルアルコールで抽出(2倍容量の抽出)し、次いで、減圧濃縮した。(17)の通算収率は92%であり、抽出による損失は1%、残余エノンは<2%であった。5%の質量バランスの損失は、反応条件下での(16)の分解によるものであった。
【0198】
工程4−アシル化:
【0199】
【化61】
【0200】
手法:
磁気攪拌バー及び窒素送入口を備えた1L容量の一頚丸底フラスコに、2−ナフトエ酸(20.98g、122mmol)及び塩化2−ナフトイル(23.49g、122mmol)及びジクロロメタン(135mL)を装入した。フラスコを内部温度0℃に冷却した。ジイソプロピルエチルアミン(76mL、436mmol)を加え、その間、内部温度を<5℃に維持した。得られた濁りのある褐色溶液を室温に加温し、30分間攪拌した。(S)−1−シアノ−1−シクロペンテン−3−オール(17、9.50g、87.1mmol)及びDMAP(1.07g、8.71mmol)をジクロロメタン(50mL)に溶かした。この溶液を反応混合物に一度に加え、室温で3時間攪拌した。水(8.50mL、472mmol)を加え、反応液を室温で90分間攪拌した。反応液をMTBE(400mL)で希釈し、飽和NaHCO3(2×400mL)で洗浄した。次いで、有機層をH2O(400mL)、1M−HCl(400mL)及びH2O(4×400mL)で洗った。合計水相損失は1.5%であった。暗色有機層をダルコKB−B(5.7g)とともに3時間攪拌した。この溶液をソルカフロックのパッドで濾過し、減圧下、40℃で濃縮し、淡黄色固体を得た。この固体をMTBE(300mL)に溶かし、40℃で減圧下で一定容量のヘプタンと溶媒交換した。この固体を濾過し、ヘプタンで洗い、19.24gの(18)を淡黄色固体として得た(100.0wt%、84%単離収率)。3.4%の損失が母液で起こった。
【0201】
工程5−Pd−触媒によるエーテル化:
【0202】
【化62】
【0203】
手法:
熱電対及び窒素送入口を備えた500mL三頚丸底フラスコを排気してから、窒素を充満させた。次いで、フラスコに、空気との接触を最小としながら、注意深く、アルコール(8)(31.0g、120mmol、1.0当量)を装入し、隔壁で密封し、THF(150mL)をシリンジで装入した。この溶液を氷浴にて+5℃に冷却した。1.0M−Et2Zn/ヘキサン(63mL、63mmol)を加えると、おだやかに発熱し、13℃となった。この溶液を氷浴中で30分間攪拌し、この反応混合物に、ニトリル(18)(31.6g、120mmol)、Pd(OAc)2(1.35g、6.01mmol、5mol%)、1,3−ビス(ジフェニルホスフィノ)プロパン(3.71g、9.00mmol、7.5mol%)、及びL−トリプトファン(2.45g、12.0mmol、10mol%)を、空気との接触を最小としながら、注意深く、固形物として加えた。15分後、氷浴を除去し、反応混合物を放置して室温とした。1時間後、緩和な発熱反応が始まり、内部温度が27℃に達した(外部冷却は適用されず)。さらに2時間後、HPLCにて分析すると、(18)の完全な変換を示した。薄い懸濁液を1Lのフラスコに移し、15gのソルカフロックを加え、次いで、激しく攪拌しながらMTBE(300mL)を加えた。懸濁液を30分間攪拌し、ソルカフロックで濾過し、濾液をジクロロメタン(200mL)と併合し、1M−HCl水(2×500mL)、水(500mL)、5%Na2CO3水(3×500mL)、及び水(2×500mL)で洗った。有機相をHPLC分析すると、(19)のアッセイ収率は84%であることを示した。有機相にダルコKB−B(15g)を加え、その懸濁液を室温で18時間攪拌し、ソルカフロックで濾過した。ほぼ無色の濾液を油状まで濃縮し、81wt%の(19)(77%単離収率)、14wt%のアルコール(8)、及び0.7%のエチルエーテル(30)を含む淡黄褐色の油39.78gを得た。
【0204】
工程6−メチルケトン形成:
【0205】
【化63】
【0206】
手法:
窒素送入口、熱電対及び磁気攪拌機を備えた1Lの三頚丸底フラスコに、MTBE(530mL)を加えた。この溶液を0℃に冷却し、MeLi溶液(1.6M/Et2O、123mL、0.196mol)を加えた。MTBE(160mL)中、(19)(41.33g、83wt%、34.3gアッセイ量、0.098mol)の溶液を、バッチ温度を5℃以下(tmax=4℃)に維持しながら、滴下漏斗経由で添加した。添加終了後、バッチを0℃で1時間熟成し、−70℃に冷却し、次いで、トリフルオロ酢酸(24mL、0.32mol)を一度で装入すると、温度が−40℃に上昇した。10%H3PO4溶液(250mL)を加え、得られる混合物を室温まで上昇させ、30分間熟成した。2相混合物を200mLのMTBEと200mLの10%H3PO4に加えた。有機相を水(500mL)で、次いで、1M−Na2CO3(500mL)、次いで、水(500mL)で、次いで、水(250mL)で洗った。有機相を硫酸ナトリウムで乾燥し、アッセイすると、34.5gの(20)を示した(96%アッセイ収率、87.9LCAP、Pd−エーテル化からのベンジルアルコール(8)10.6%)。
【0207】
炭酸ナトリウムでの洗浄は次工程の共役付加の成功にとって最重要である。この洗浄がない場合、〜20%の変換が観察されたのみであった。水洗が何回必要であるかは未知である。
【0208】
工程7−共役付加/異性化:
【0209】
【化64】
【0210】
手法:
窒素送入口、熱電対及び磁気攪拌機を備えた1Lの三頚丸底フラスコに、CuI(8.8g、0.046mol)を加えた。フラスコに窒素を1時間送入した。フラスコにTHF(255ml)を装入し、スラリーを0℃に冷却した。グリニヤール溶液(2.0M/Et2O、68.2mL、0.136mol)を0℃で加え、その温度を10℃以下に保持した。30分間熟成した後、その混合物を−70℃に冷却し、TMSCl(32.0mL、0.252mol)を加え、次いで、THF(180mL+70mL洗浄)中(20)(41.4g、79wt%、32.7gアッセイ量、0.089mol)の溶液を0℃で加えた。この添加の間、反応温度は−40℃を超えないようにした。1時間の間に、反応液は−40℃から−20℃に上昇させ、HPLC分析すると、変換率98.4%を示した。次いで、反応液を0℃に加温し、1時間熟成させると、完全な変換を示した。この混合物に0℃で1M−HCl(255mL)を加えて反応停止させると、27℃まで発熱した。この混合物を室温で1時間熟成させ、MTBE(500mL)に移行させ、層分離した。有機層を1M−HCl(500mL)と、次いで水(500mL)で洗った。この時点で、溶液から固形物が沈殿した。混合物をソルカフロック床(MTBEで湿潤)で濾過し、床を250mLのMTBEで洗った。層分離し、有機層を250mLの水で洗った。有機層を硫酸ナトリウムで乾燥し、〜600mLに濃縮し、アッセイすると(21)のアッセイ収量は39.52gであった(ジアステレオマーの15:85混合物)。
【0211】
MTBE中、生成物の粗製溶液を濃縮して油とし、窒素送入口、熱電対及び磁気攪拌機を備えた1Lの三頚丸底フラスコ中、MeOH400mLで希釈した。フラスコを20℃の水浴に浸漬し、NaOMe(25wt%/MeOH、10mL、0.044mol)を反応液にゆっくり加え、温度を25℃未満に維持した。1時間だけ熟成した後、ジアステレオマーの比は15:85から98:2に上昇した。反応液を0℃に冷やし、600mLのヘプタンで希釈し、1M−HCl(500mL)で反応停止させた。二相性混合物を静置し、有機層を分離した(水層への損失2%)。有機層を2回水洗(各250mL)し、硫酸ナトリウムで乾燥し、濃縮して、(21)を油として得た(47.9g、80.8wt%の(21)、98%アッセイ収率、79.8LCAP、10.5面積%ベンジルアルコール(8))。
【0212】
工程8−ヨードケトン形成:
【0213】
【化65】
【0214】
手法:
窒素送入口、熱電対及び磁気攪拌機を備えた500mLの三頚丸底フラスコに、メタノール(230mL)中の(21)(24.6g、80.8wt%、19.8gアッセイ量、0.0429mol)を加えた。この溶液を20℃の水浴に浸漬し、ICl(1.0M/CH2Cl2、77.5mL、0.0775mol)を20分間で滴下し、その温度を25℃以下に保持した。反応液を室温で2時間熟成させ、その時点で完全な変換がHPLC分析により観察された。この混合物をMTBE(250mL)と10%Na2S2O3/5%NaHCO3(250mL)で−10℃にて反応停止させた。この混合物をさらにMTBE(200mL)と10%Na2S2O3/5%NaHCO3(200mL)で希釈し、層分離した。有機層を2回水洗し(各300mL)、硫酸ナトリウムで乾燥し、アッセイすると、22.0gのヨードケトン(3)(87%収率)を示した。有機溶液を濃縮して固体とし、メタノール(170mL)で希釈した。容器に50mgのヨードケトンを播種し、この反応混合物に2時間かけて水(37.5mL)を滴下した。この混合物を一夜熟成させ、その上清(5.9mg/mL)についてアッセイし、濾過し、70mLのMeOH:H2O(70:30)で洗浄し、22.4gの(3)を白色固体として得た(93wt%ヨードケトン、20.8アッセイ収量、83%;97.3LCAP、0.9%Cl−ケトン、1.4%ケトン異性体)。
【0215】
塩:
式(Ia)で示される化合物の種々結晶性塩を調製し、評価した。
【0216】
【化66】
【0217】
これらの塩についての物理化学的データを以下の表に示す。
【0218】
【表9】
【0219】
ベシル酸塩の無水結晶形I型は物理的及び化学的共に安定であり、熱力学的にII型よりも安定であり、また通常非吸湿性であった。
【0220】
X線粉末回折での検討は、分子構造、結晶化度、及び多形を特性化するために広く使用される。ベシル酸塩の無水結晶形I型のX線粉末回折パターンは、PW3040/60コンソールを有するフィリップス分析用X′パートPRO−X線回折システムにて生成させた。PW3373/00セラミックCu LEF X線チューブK−アルファ放射を線源として用いた。
【0221】
図1はベシル酸塩の無水結晶形I型のX線回折パターンを示す。当該無水I型は、21.2、9.1、及び8.5オングストロームのd−間隔に相当する特徴的な回折ピークを示した。当該無水I型はさらに、13,5、10.9及び5.5オングストロームのd−間隔より特徴づけられた。当該無水I型はなおさらに、4.5、4.3、及び4.2オングストロームのd−間隔により特徴づけられた。
【0222】
DSCデータはTAインストルメンツDSC2910又は同等の装置を使用して、閉鎖したパン中、窒素気流下に、10℃/分の加熱速度で取得した。
【0223】
図2はベシル酸塩の無水結晶形I型の示差熱量走査を示す。当該無水結晶形I型は、273.2℃の開始温度で融解するために吸熱性を示し、ピーク温度は274.4℃であり、エンタルピー変化は61.3J/gである。
【特許請求の範囲】
【請求項1】
式(I):
【化1】
[式中、
R2は以下からなる群より選択され:
(1)水素、及び
(2)C1−6アルキル;
Rは以下からなる群より選択され:
(1)未置換であるか、又はR11、R12及びR13の1個以上により置換されたフェニル;
(2)未置換であるか、又は以下から選択される1個以上の置換基で置換されたC1−8アルキル:
(a)ヒドロキシ、
(b)オキソ、
(c)C1−6アルコキシ、
(d)フェニル−C1−3アルコキシ、
(e)フェニル、
(f)−CN、
(g)ハロ、
(h)−NR9R10(ここで、R9及びR10は、独立して、
(1)水素、
(2)C1−6アルキル、
(3)ヒドロキシ−C1−6アルキル、及び
(4)フェニル、から選択される)、
(i)−NR9COR10(ここで、R9及びR10は上記定義のとおり)、
(j)−NR9CO2R10(ここで、R9及びR10は上記定義のとおり)、
(k)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(l)−COR9(ここで、R9は上記定義のとおり)、及び
(m)−CO2R9(ここで、R9は上記定義のとおり);
(3)未置換であるか、又は以下から選択される1個以上の置換基で置換されたC2−6アルケニル:
(a)ヒドロキシ、
(b)オキソ、
(c)C1−6アルコキシ、
(d)フェニル−C1−3アルコキシ、
(e)フェニル、
(f)−CN、
(g)ハロ、
(h)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(i)−COR9(ここで、R9は上記定義のとおり)、及び
(j)−CO2R9(ここで、R9は上記定義のとおり);
(4)へテロ環(ここで、該ヘテロ環は、
(A)ベンゾイミダゾリル、
(B)ベンゾフラニル、
(C)ベンゾチオフェニル、
(D)ベンゾオキサゾリル、
(E)フラニル、
(F)イミダゾリル、
(G)インドリル、
(H)イソオキサゾリル、
(I)イソチアゾリル、
(J)オキサジアゾリル、
(K)オキサゾリル、
(L)ピラジニル、
(M)ピラゾリル、
(N)ピリジル、
(O)ピリミジル、
(P)ピロリル、
(Q)キノリル、
(R)テトラゾリル、
(S)チアジアゾリル、
(T)チアゾリル、
(U)チエニル、
(V)トリアゾリル、
(W)アゼチジニル、
(X)1,4−ジオキサニル、
(Y)ヘキサヒドロアゼピニル、
(Z)ピペラジニル、
(AA)ピペリジニル、
(AB)ピロリジニル、
(AC)テトラヒドロフラニル、及び
(AD)テトラヒドロチエニル、
からなる群より選択され、また、該へテロ環は未置換であるか、又は
(i)未置換であるか、又はハロ、−CF3、−OCH3、若しくはフェニルで置換されたC1−6アルキル、
(ii)C1−6アルコキシ、
(iii)オキソ、
(iv)ヒドロキシ、
(v)チオキソ、
(vi)−SR9(ここで、R9は上記定義のとおり)、
(vii)ハロ、
(viii)シアノ、
(ix)フェニル、
(x)トリフルオロメチル、
(xi)−(CH2)m−NR9R10(ここで、mは0、1又は2であり;R9及びR10は上記定義のとおり)、
(xii)−NR9COR10(ここで、R9及びR10は上記定義のとおり)、
(xiii)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(xiv)−CO2R9(ここで、R9は上記定義のとおり)、及び
(xv)−(CH2)m−OR9(ここで、m及びR9は上記定義のとおり)
から選択される1個以上の置換基で置換されている);
R1は以下からなる群より選択され:
(1)
【化2】
(2)−C1−8アルキル(ここで、アルキルは未置換であるか、又は以下から選択される1個以上の置換基で置換されている:
(a)ヒドロキシ、
(b)オキソ、
(c)C1−6アルコキシ、
(d)フェニル−C1−3アルコキシ、
(e)フェニル、
(f)−CN、
(g)ハロ、
(h)−NR9R10(ここで、R9及びR10は上記定義のとおり)、
(i)−NR9COR10(ここで、R9及びR10は上記定義のとおり)、
(j)−NR9CO2R10(ここで、R9及びR10は上記定義のとおり)、
(k)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(l)−COR9(ここで、R9は上記定義のとおり)、及び
(m)−CO2R9(ここで、R9は上記定義のとおり));
(3)−C2−6アルケニル(ここで、アルケニルは未置換であるか、又は以下から選択される1個以上の置換基で置換されている:
(a)ヒドロキシ、
(b)オキソ、
(c)C1−6アルコキシ、
(d)フェニル−C1−3アルコキシ、
(e)フェニル、
(f)−CN、
(g)ハロ、
(h)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(i)−COR9(ここで、R9は上記定義のとおり)、
(j)−CO2R9(ここで、R9は上記定義のとおり));
(4)−(CO)−フェニル(ここで、該フェニルは未置換であるか、又はR6、R7及びR8の1個以上により置換されている);
R6、R7及びR8は、独立して、以下からなる群より選択され:
(1)水素;
(2)未置換であるか、又は以下から選択される1個以上の置換基で置換されたC1−6アルキル:
(a)ヒドロキシ、
(b)オキソ、
(c)C1−6アルコキシ、
(d)フェニル−C1−3アルコキシ、
(e)フェニル、
(f)−CN、
(g)ハロ、
(h)−NR9R10(ここで、R9及びR10は上記定義のとおり)、
(i)−NR9COR10(ここで、R9及びR10は上記定義のとおり)、
(j)−NR9CO2R10(ここで、R9及びR10は上記定義のとおり)、
(k)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(l)−COR9(ここで、R9は上記定義のとおり)、及び
(m)−CO2R9(ここで、R9は上記定義のとおり);
(3)未置換であるか、又は以下から選択される1個以上の置換基で置換されたC2−6アルケニル:
(a)ヒドロキシ、
(b)オキソ、
(c)C1−6アルコキシ、
(d)フェニル−C1−3アルコキシ、
(e)フェニル、
(f)−CN、
(g)ハロ、
(h)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(i)−COR9(ここで、R9は上記定義のとおり)、
(j)−CO2R9(ここで、R9は上記定義のとおり);
(4)C2−6アルキニル;
(5)未置換であるか、又は以下から選択される1個以上の置換基で置換されたフェニル:
(a)ヒドロキシ、
(b)C1−6アルコキシ、
(c)C1−6アルキル、
(d)C2−5アルケニル、
(e)ハロ、
(f)−CN、
(g)−NO2、
(h)−CF3、
(i)−(CH2)m−NR9R10(ここで、m、R9及びR10は上記定義のとおり)、
(j)−NR9COR10(ここで、R9及びR10は上記定義のとおり)
(k)−NR9CO2R10(ここで、R9及びR10は上記定義のとおり)、
(l)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(m)−CO2NR9R10(ここで、R9及びR10は上記定義のとおり)、
(n)−COR9(ここで、R9は上記定義のとおり)、
(o)−CO2R9(ここで、R9は上記定義のとおり);
(6)ハロ、
(7)−CN、
(8)−CF3、
(9)−NO2、
(10)−SR14(ここで、R14は水素又はC1−5アルキルである)、
(11)−SOR14(ここで、R14は上記定義のとおり)、
(12)−SO2R14(ここで、R14は上記定義のとおり)、
(13)NR9COR10(ここで、R9及びR10は上記定義のとおり)、
(14)CONR9COR10(ここで、R9及びR10は上記定義のとおり)、
(15)NR9R10(ここで、R9及びR10は上記定義のとおり)、
(16)NR9CO2R10(ここで、R9及びR10は上記定義のとおり)、
(17)ヒドロキシ、
(18)C1−6アルコキシ、
(19)COR9(ここで、R9は上記定義のとおり)、
(20)CO2R9(ここで、R9は上記定義のとおり)、
(21)2−ピリジル、
(22)3−ピリジル、
(23)4−ピリジル、
(24)5−テトラゾリル、
(25)2−オキサゾリル、及び
(26)2−チアゾリル;
R11、R12及びR13は、独立して、R6、R7及びR8の定義から選択され;そして
Zは、
(1)水素、
(2)C1−6アルキル、及び
(3)ヒドロキシル
から選択される]で示される化合物又はその薬学的に許容され得る塩の製造法であって、
式(A):
【化3】
[式中、R14はR6から選択される]
で示される化合物と還元剤とを酸性条件下で反応させて、式(B):
【化4】
で示される化合物とし、次いで、式(B)の化合物と強酸とを反応させて式(I)で示される化合物とすることを含んでなり、次いで、所望により、式(I)で示される化合物の薬学的に許容され得る塩を形成するために、式(I)で示される化合物と対応する塩の酸とを反応させて、式(I)で示される化合物の薬学的に許容され得る塩を形成することを含んでなる製造法。
【請求項2】
さらに、式(A)の化合物を調製するために、式(C):
【化5】
で示される化合物と酸とを反応させることにより、式(A)で示される化合物を調製することを含んでなる請求項1記載の製造法。
【請求項3】
さらに、式(C)の化合物を得るために、式(D):
【化6】
[式中、X1はC1−6アルキル又はフェニルである]
で示される化合物と、アンモニア又はその塩とを反応させることにより、式(C)で示される化合物を調製することを含んでなる請求項2記載の製造法。
【請求項4】
さらに、式(D)の化合物を得るために、式(E):
【化7】
[式中、Yはハロゲンである]
で示される化合物と、式(F):
【化8】
で示される化合物、及び式M1N(R15)2又はM1N(Si(R15)3)2(ここで、M1はLi、Na、K又はMgであり、各R15は、独立して、C1−4アルキルから選択される)で示される金属アミドとを、第一の非プロトン性有機溶媒中で反応させることにより、式(D)で示される化合物を調製することを含んでなる請求項3記載の製造法。
【請求項5】
さらに、式(E)の化合物を得るために、式(G):
【化9】
で示される化合物と、ハロゲン化剤とを反応させることにより、式(E)で示される化合物を調製することを含んでなる請求項4記載の製造法。
【請求項6】
さらに、式(G)の化合物を得るために、式(H):
【化10】
で示される化合物と、R−M2(ここで、M2は金属である)とを、第一の遷移金属触媒及びルイス酸の存在下で反応させることにより、式(G)で示される化合物を調製することを含んでなる請求項5記載の製造法。
【請求項7】
さらに、式(H)の化合物を得るために、式(J):
【化11】
で示される化合物と、CH3Liとを、tert−ブチルメチルエーテル中で反応させることにより、式(H)で示される化合物を調製することを含んでなる請求項6記載の製造法。
【請求項8】
さらに、式(J)の化合物を得るために、式(K):
【化12】
[式中、X2はRから選択される]
で示される化合物と、R1−OHとを、第二の遷移金属触媒、リガンド及び亜鉛付加物の存在下で反応させることにより、式(J)で示される化合物を調製することを含んでなる請求項7記載の製造法。
【請求項9】
さらに、式(K)の化合物を得るために、式(L):
【化13】
で示される化合物を酵素的に還元し、次いで、その生成物と、X2−COClとを反応させることにより、式(K)で示される化合物を調製することを含んでなる請求項8記載の製造法。
【請求項10】
さらに、式(L)の化合物を得るために、式(M):
【化14】
で示される化合物とブロム化剤とを反応させ、次いでバッファーの存在下でシアノ化剤を反応させることにより、式(L)で示される化合物を調製することを含んでなる請求項9記載の製造法。
【請求項11】
YがIであり、さらに、式(E)の化合物を得るために、式(N):
【化15】
で示される化合物と、M3−I(ここで、M3はLi、Na又はKである)とを反応させることにより、式(E)で示される化合物を調製することを含んでなる請求項4記載の製造法。
【請求項12】
さらに、式(N)の化合物を得るために、式(O):
【化16】
で示される化合物と、ClCH2CO2H、ClCH2I、又はClCH2Br及び式M4N(R16)2又はM4N(Si(R16)3)2(ここで、M4はLi、Na、K、又はMgであり、各R16は、独立して、C1−4アルキルから選択される)で示される金属アミドとを、第二の非プロトン性有機溶媒中、約−20℃ないし約40℃の範囲の温度で反応させることにより、式(N)で示される化合物を調製することを含んでなる請求項11記載の製造法。
【請求項13】
さらに、式(O)の化合物を得るために、式(P):
【化17】
で示される化合物又はそのトリエチルアミン塩とメチル化剤とを反応させることにより、式(O)で示される化合物を調製することを含んでなる請求項12記載の製造法。
【請求項14】
さらに、式(P)の化合物又はそのトリエチルアミン塩を得るために、式(Q):
【化18】
で示される化合物と、R−M5(ここで、M5はM2同様の金属である)とを、第一の遷移金属触媒及びルイス酸の存在下で反応させ、次いで、所望により、トリエチルアミドを反応させて塩を形成することにより、式(P)で示される化合物を調製することを含んでなる請求項13記載の製造法。
【請求項15】
式(Q)の化合物を得るために、式(R):
【化19】
[式中、X3はRから選択される]
で示される化合物とR1−OHとを、第二の遷移金属触媒、リガンド及び亜鉛付加物の存在下で反応させることにより、式(Q)で示される化合物が製造されることを特徴とする請求項14記載の製造法。
【請求項16】
さらに、式(R)の化合物を得るために、式(S):
【化20】
で示される化合物を酵素的に還元し、次いで、その生成物とX3−COClとを反応させることにより、式(R)で示される化合物を調製することを含んでなる請求項15記載の製造法。
【請求項17】
さらに、式(S)の化合物を得るために、式(T):
【化21】
で示される化合物と酸化剤とを反応させることにより、式(S)で示される化合物を調製することを含んでなる請求項16記載の製造法。
【請求項18】
R1が
【化22】
である請求項1記載の製造法。
【請求項19】
Rが
【化23】
である請求項1記載の製造法。
【請求項20】
R2がメチルである請求項1記載の製造法。
【請求項21】
式(I)の化合物が式(Ia):
【化24】
の化合物である請求項1記載の製造法。
【請求項22】
式(Ia)の化合物が薬学的に許容され得る塩である請求項21記載の製造法。
【請求項23】
薬学的に許容され得る塩がベンゼンスルホン酸塩であり、該塩の相当する酸がベンゼンスルホン酸である請求項22記載の製造法。
【請求項24】
式(Ia):
【化25】
で示される化合物のベンゼンスルホン酸塩。
【請求項25】
I型で示され、約21.2、9.1、及び8.5オングストロームのd−間隔に相当する特徴的な回折ピークを示す請求項24記載のベンゼンスルホン酸塩の無水結晶形。
【請求項26】
さらに、13.5、10.9及び5.5オングストロームのd−間隔により特徴づけられる請求項25のI型で示される無水結晶形。
【請求項27】
さらに、4.5、4.3及び4.2オングストロームのd−間隔により特徴づけられる請求項26のI型で示される無水結晶形。
【請求項1】
式(I):
【化1】
[式中、
R2は以下からなる群より選択され:
(1)水素、及び
(2)C1−6アルキル;
Rは以下からなる群より選択され:
(1)未置換であるか、又はR11、R12及びR13の1個以上により置換されたフェニル;
(2)未置換であるか、又は以下から選択される1個以上の置換基で置換されたC1−8アルキル:
(a)ヒドロキシ、
(b)オキソ、
(c)C1−6アルコキシ、
(d)フェニル−C1−3アルコキシ、
(e)フェニル、
(f)−CN、
(g)ハロ、
(h)−NR9R10(ここで、R9及びR10は、独立して、
(1)水素、
(2)C1−6アルキル、
(3)ヒドロキシ−C1−6アルキル、及び
(4)フェニル、から選択される)、
(i)−NR9COR10(ここで、R9及びR10は上記定義のとおり)、
(j)−NR9CO2R10(ここで、R9及びR10は上記定義のとおり)、
(k)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(l)−COR9(ここで、R9は上記定義のとおり)、及び
(m)−CO2R9(ここで、R9は上記定義のとおり);
(3)未置換であるか、又は以下から選択される1個以上の置換基で置換されたC2−6アルケニル:
(a)ヒドロキシ、
(b)オキソ、
(c)C1−6アルコキシ、
(d)フェニル−C1−3アルコキシ、
(e)フェニル、
(f)−CN、
(g)ハロ、
(h)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(i)−COR9(ここで、R9は上記定義のとおり)、及び
(j)−CO2R9(ここで、R9は上記定義のとおり);
(4)へテロ環(ここで、該ヘテロ環は、
(A)ベンゾイミダゾリル、
(B)ベンゾフラニル、
(C)ベンゾチオフェニル、
(D)ベンゾオキサゾリル、
(E)フラニル、
(F)イミダゾリル、
(G)インドリル、
(H)イソオキサゾリル、
(I)イソチアゾリル、
(J)オキサジアゾリル、
(K)オキサゾリル、
(L)ピラジニル、
(M)ピラゾリル、
(N)ピリジル、
(O)ピリミジル、
(P)ピロリル、
(Q)キノリル、
(R)テトラゾリル、
(S)チアジアゾリル、
(T)チアゾリル、
(U)チエニル、
(V)トリアゾリル、
(W)アゼチジニル、
(X)1,4−ジオキサニル、
(Y)ヘキサヒドロアゼピニル、
(Z)ピペラジニル、
(AA)ピペリジニル、
(AB)ピロリジニル、
(AC)テトラヒドロフラニル、及び
(AD)テトラヒドロチエニル、
からなる群より選択され、また、該へテロ環は未置換であるか、又は
(i)未置換であるか、又はハロ、−CF3、−OCH3、若しくはフェニルで置換されたC1−6アルキル、
(ii)C1−6アルコキシ、
(iii)オキソ、
(iv)ヒドロキシ、
(v)チオキソ、
(vi)−SR9(ここで、R9は上記定義のとおり)、
(vii)ハロ、
(viii)シアノ、
(ix)フェニル、
(x)トリフルオロメチル、
(xi)−(CH2)m−NR9R10(ここで、mは0、1又は2であり;R9及びR10は上記定義のとおり)、
(xii)−NR9COR10(ここで、R9及びR10は上記定義のとおり)、
(xiii)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(xiv)−CO2R9(ここで、R9は上記定義のとおり)、及び
(xv)−(CH2)m−OR9(ここで、m及びR9は上記定義のとおり)
から選択される1個以上の置換基で置換されている);
R1は以下からなる群より選択され:
(1)
【化2】
(2)−C1−8アルキル(ここで、アルキルは未置換であるか、又は以下から選択される1個以上の置換基で置換されている:
(a)ヒドロキシ、
(b)オキソ、
(c)C1−6アルコキシ、
(d)フェニル−C1−3アルコキシ、
(e)フェニル、
(f)−CN、
(g)ハロ、
(h)−NR9R10(ここで、R9及びR10は上記定義のとおり)、
(i)−NR9COR10(ここで、R9及びR10は上記定義のとおり)、
(j)−NR9CO2R10(ここで、R9及びR10は上記定義のとおり)、
(k)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(l)−COR9(ここで、R9は上記定義のとおり)、及び
(m)−CO2R9(ここで、R9は上記定義のとおり));
(3)−C2−6アルケニル(ここで、アルケニルは未置換であるか、又は以下から選択される1個以上の置換基で置換されている:
(a)ヒドロキシ、
(b)オキソ、
(c)C1−6アルコキシ、
(d)フェニル−C1−3アルコキシ、
(e)フェニル、
(f)−CN、
(g)ハロ、
(h)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(i)−COR9(ここで、R9は上記定義のとおり)、
(j)−CO2R9(ここで、R9は上記定義のとおり));
(4)−(CO)−フェニル(ここで、該フェニルは未置換であるか、又はR6、R7及びR8の1個以上により置換されている);
R6、R7及びR8は、独立して、以下からなる群より選択され:
(1)水素;
(2)未置換であるか、又は以下から選択される1個以上の置換基で置換されたC1−6アルキル:
(a)ヒドロキシ、
(b)オキソ、
(c)C1−6アルコキシ、
(d)フェニル−C1−3アルコキシ、
(e)フェニル、
(f)−CN、
(g)ハロ、
(h)−NR9R10(ここで、R9及びR10は上記定義のとおり)、
(i)−NR9COR10(ここで、R9及びR10は上記定義のとおり)、
(j)−NR9CO2R10(ここで、R9及びR10は上記定義のとおり)、
(k)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(l)−COR9(ここで、R9は上記定義のとおり)、及び
(m)−CO2R9(ここで、R9は上記定義のとおり);
(3)未置換であるか、又は以下から選択される1個以上の置換基で置換されたC2−6アルケニル:
(a)ヒドロキシ、
(b)オキソ、
(c)C1−6アルコキシ、
(d)フェニル−C1−3アルコキシ、
(e)フェニル、
(f)−CN、
(g)ハロ、
(h)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(i)−COR9(ここで、R9は上記定義のとおり)、
(j)−CO2R9(ここで、R9は上記定義のとおり);
(4)C2−6アルキニル;
(5)未置換であるか、又は以下から選択される1個以上の置換基で置換されたフェニル:
(a)ヒドロキシ、
(b)C1−6アルコキシ、
(c)C1−6アルキル、
(d)C2−5アルケニル、
(e)ハロ、
(f)−CN、
(g)−NO2、
(h)−CF3、
(i)−(CH2)m−NR9R10(ここで、m、R9及びR10は上記定義のとおり)、
(j)−NR9COR10(ここで、R9及びR10は上記定義のとおり)
(k)−NR9CO2R10(ここで、R9及びR10は上記定義のとおり)、
(l)−CONR9R10(ここで、R9及びR10は上記定義のとおり)、
(m)−CO2NR9R10(ここで、R9及びR10は上記定義のとおり)、
(n)−COR9(ここで、R9は上記定義のとおり)、
(o)−CO2R9(ここで、R9は上記定義のとおり);
(6)ハロ、
(7)−CN、
(8)−CF3、
(9)−NO2、
(10)−SR14(ここで、R14は水素又はC1−5アルキルである)、
(11)−SOR14(ここで、R14は上記定義のとおり)、
(12)−SO2R14(ここで、R14は上記定義のとおり)、
(13)NR9COR10(ここで、R9及びR10は上記定義のとおり)、
(14)CONR9COR10(ここで、R9及びR10は上記定義のとおり)、
(15)NR9R10(ここで、R9及びR10は上記定義のとおり)、
(16)NR9CO2R10(ここで、R9及びR10は上記定義のとおり)、
(17)ヒドロキシ、
(18)C1−6アルコキシ、
(19)COR9(ここで、R9は上記定義のとおり)、
(20)CO2R9(ここで、R9は上記定義のとおり)、
(21)2−ピリジル、
(22)3−ピリジル、
(23)4−ピリジル、
(24)5−テトラゾリル、
(25)2−オキサゾリル、及び
(26)2−チアゾリル;
R11、R12及びR13は、独立して、R6、R7及びR8の定義から選択され;そして
Zは、
(1)水素、
(2)C1−6アルキル、及び
(3)ヒドロキシル
から選択される]で示される化合物又はその薬学的に許容され得る塩の製造法であって、
式(A):
【化3】
[式中、R14はR6から選択される]
で示される化合物と還元剤とを酸性条件下で反応させて、式(B):
【化4】
で示される化合物とし、次いで、式(B)の化合物と強酸とを反応させて式(I)で示される化合物とすることを含んでなり、次いで、所望により、式(I)で示される化合物の薬学的に許容され得る塩を形成するために、式(I)で示される化合物と対応する塩の酸とを反応させて、式(I)で示される化合物の薬学的に許容され得る塩を形成することを含んでなる製造法。
【請求項2】
さらに、式(A)の化合物を調製するために、式(C):
【化5】
で示される化合物と酸とを反応させることにより、式(A)で示される化合物を調製することを含んでなる請求項1記載の製造法。
【請求項3】
さらに、式(C)の化合物を得るために、式(D):
【化6】
[式中、X1はC1−6アルキル又はフェニルである]
で示される化合物と、アンモニア又はその塩とを反応させることにより、式(C)で示される化合物を調製することを含んでなる請求項2記載の製造法。
【請求項4】
さらに、式(D)の化合物を得るために、式(E):
【化7】
[式中、Yはハロゲンである]
で示される化合物と、式(F):
【化8】
で示される化合物、及び式M1N(R15)2又はM1N(Si(R15)3)2(ここで、M1はLi、Na、K又はMgであり、各R15は、独立して、C1−4アルキルから選択される)で示される金属アミドとを、第一の非プロトン性有機溶媒中で反応させることにより、式(D)で示される化合物を調製することを含んでなる請求項3記載の製造法。
【請求項5】
さらに、式(E)の化合物を得るために、式(G):
【化9】
で示される化合物と、ハロゲン化剤とを反応させることにより、式(E)で示される化合物を調製することを含んでなる請求項4記載の製造法。
【請求項6】
さらに、式(G)の化合物を得るために、式(H):
【化10】
で示される化合物と、R−M2(ここで、M2は金属である)とを、第一の遷移金属触媒及びルイス酸の存在下で反応させることにより、式(G)で示される化合物を調製することを含んでなる請求項5記載の製造法。
【請求項7】
さらに、式(H)の化合物を得るために、式(J):
【化11】
で示される化合物と、CH3Liとを、tert−ブチルメチルエーテル中で反応させることにより、式(H)で示される化合物を調製することを含んでなる請求項6記載の製造法。
【請求項8】
さらに、式(J)の化合物を得るために、式(K):
【化12】
[式中、X2はRから選択される]
で示される化合物と、R1−OHとを、第二の遷移金属触媒、リガンド及び亜鉛付加物の存在下で反応させることにより、式(J)で示される化合物を調製することを含んでなる請求項7記載の製造法。
【請求項9】
さらに、式(K)の化合物を得るために、式(L):
【化13】
で示される化合物を酵素的に還元し、次いで、その生成物と、X2−COClとを反応させることにより、式(K)で示される化合物を調製することを含んでなる請求項8記載の製造法。
【請求項10】
さらに、式(L)の化合物を得るために、式(M):
【化14】
で示される化合物とブロム化剤とを反応させ、次いでバッファーの存在下でシアノ化剤を反応させることにより、式(L)で示される化合物を調製することを含んでなる請求項9記載の製造法。
【請求項11】
YがIであり、さらに、式(E)の化合物を得るために、式(N):
【化15】
で示される化合物と、M3−I(ここで、M3はLi、Na又はKである)とを反応させることにより、式(E)で示される化合物を調製することを含んでなる請求項4記載の製造法。
【請求項12】
さらに、式(N)の化合物を得るために、式(O):
【化16】
で示される化合物と、ClCH2CO2H、ClCH2I、又はClCH2Br及び式M4N(R16)2又はM4N(Si(R16)3)2(ここで、M4はLi、Na、K、又はMgであり、各R16は、独立して、C1−4アルキルから選択される)で示される金属アミドとを、第二の非プロトン性有機溶媒中、約−20℃ないし約40℃の範囲の温度で反応させることにより、式(N)で示される化合物を調製することを含んでなる請求項11記載の製造法。
【請求項13】
さらに、式(O)の化合物を得るために、式(P):
【化17】
で示される化合物又はそのトリエチルアミン塩とメチル化剤とを反応させることにより、式(O)で示される化合物を調製することを含んでなる請求項12記載の製造法。
【請求項14】
さらに、式(P)の化合物又はそのトリエチルアミン塩を得るために、式(Q):
【化18】
で示される化合物と、R−M5(ここで、M5はM2同様の金属である)とを、第一の遷移金属触媒及びルイス酸の存在下で反応させ、次いで、所望により、トリエチルアミドを反応させて塩を形成することにより、式(P)で示される化合物を調製することを含んでなる請求項13記載の製造法。
【請求項15】
式(Q)の化合物を得るために、式(R):
【化19】
[式中、X3はRから選択される]
で示される化合物とR1−OHとを、第二の遷移金属触媒、リガンド及び亜鉛付加物の存在下で反応させることにより、式(Q)で示される化合物が製造されることを特徴とする請求項14記載の製造法。
【請求項16】
さらに、式(R)の化合物を得るために、式(S):
【化20】
で示される化合物を酵素的に還元し、次いで、その生成物とX3−COClとを反応させることにより、式(R)で示される化合物を調製することを含んでなる請求項15記載の製造法。
【請求項17】
さらに、式(S)の化合物を得るために、式(T):
【化21】
で示される化合物と酸化剤とを反応させることにより、式(S)で示される化合物を調製することを含んでなる請求項16記載の製造法。
【請求項18】
R1が
【化22】
である請求項1記載の製造法。
【請求項19】
Rが
【化23】
である請求項1記載の製造法。
【請求項20】
R2がメチルである請求項1記載の製造法。
【請求項21】
式(I)の化合物が式(Ia):
【化24】
の化合物である請求項1記載の製造法。
【請求項22】
式(Ia)の化合物が薬学的に許容され得る塩である請求項21記載の製造法。
【請求項23】
薬学的に許容され得る塩がベンゼンスルホン酸塩であり、該塩の相当する酸がベンゼンスルホン酸である請求項22記載の製造法。
【請求項24】
式(Ia):
【化25】
で示される化合物のベンゼンスルホン酸塩。
【請求項25】
I型で示され、約21.2、9.1、及び8.5オングストロームのd−間隔に相当する特徴的な回折ピークを示す請求項24記載のベンゼンスルホン酸塩の無水結晶形。
【請求項26】
さらに、13.5、10.9及び5.5オングストロームのd−間隔により特徴づけられる請求項25のI型で示される無水結晶形。
【請求項27】
さらに、4.5、4.3及び4.2オングストロームのd−間隔により特徴づけられる請求項26のI型で示される無水結晶形。
【図1】

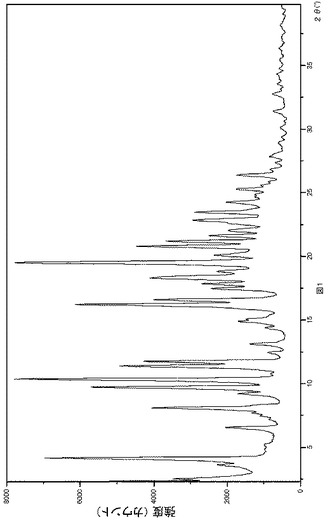
【図2】
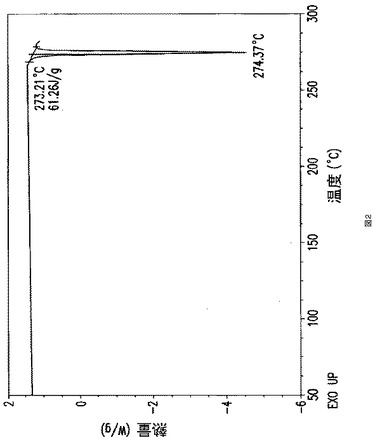
【図2】
【公表番号】特表2010−500351(P2010−500351A)
【公表日】平成22年1月7日(2010.1.7)
【国際特許分類】
【出願番号】特願2009−523797(P2009−523797)
【出願日】平成19年8月3日(2007.8.3)
【国際出願番号】PCT/US2007/017406
【国際公開番号】WO2008/021029
【国際公開日】平成20年2月21日(2008.2.21)
【出願人】(390023526)メルク エンド カムパニー インコーポレーテッド (924)
【氏名又は名称原語表記】MERCK & COMPANY INCOPORATED
【出願人】(000005072)萬有製薬株式会社 (51)
【Fターム(参考)】
【公表日】平成22年1月7日(2010.1.7)
【国際特許分類】
【出願日】平成19年8月3日(2007.8.3)
【国際出願番号】PCT/US2007/017406
【国際公開番号】WO2008/021029
【国際公開日】平成20年2月21日(2008.2.21)
【出願人】(390023526)メルク エンド カムパニー インコーポレーテッド (924)
【氏名又は名称原語表記】MERCK & COMPANY INCOPORATED
【出願人】(000005072)萬有製薬株式会社 (51)
【Fターム(参考)】
[ Back to top ]