
ラクダ科動物抗体の重鎖可変ドメイン(VHH)の生産方法
【課題】生産性が向上したVHHの生産方法に用いるためのDNAを提供する。
【解決手段】以下の(a)〜(d)のいずれかのDNA及びラクダ科動物抗体の重鎖可変ドメイン(VHH)をコードするDNAを融合したDNA:(a)特定の塩基配列からなるDNA(b)特定の塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、且つグルコアミラーゼ活性を有するタンパク質をコードするDNA(c)特定の塩基配列からなる別のDNA(d)特定の塩基配列からなる別のDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、且つエンドグルカナーゼ活性を有するタンパク質をコードするDNA。
【解決手段】以下の(a)〜(d)のいずれかのDNA及びラクダ科動物抗体の重鎖可変ドメイン(VHH)をコードするDNAを融合したDNA:(a)特定の塩基配列からなるDNA(b)特定の塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、且つグルコアミラーゼ活性を有するタンパク質をコードするDNA(c)特定の塩基配列からなる別のDNA(d)特定の塩基配列からなる別のDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、且つエンドグルカナーゼ活性を有するタンパク質をコードするDNA。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ラクダ科動物抗体の重鎖可変ドメイン(以下、VHHと呼ぶこともある)の生産方法、並びに当該方法の実施に有用なDNA、糸状菌用発現ベクター及び形質転換体に関する。更に、本発明は、当該方法により得られたVHHを含むタンパク質に関する。
【背景技術】
【0002】
抗体の標準的な構成は2本の軽鎖と2本の重鎖からなる。軽鎖と重鎖は免疫グロブリンドメインにより構成され、軽鎖は2つ(VL、CL)、重鎖は4つ(VH, CH1, CH2, CH3)のドメインからできている。抗体の多様性には軽鎖と重鎖の可変領域の組み合わせが重要であるため、軽鎖、重鎖からなる標準的な抗体フォーマットは普遍的なものとして考えられてきた。しかし、1993年にHamers-Castermanらにより、ラクダ科動物の血液中に重鎖のみからなる抗体(重鎖抗体(Heavy-chain antibody))が多量に存在することが発見された。当該重鎖抗体はCH1ドメインを有していないことも特徴的である。ラクダ抗体のVHドメインはVariable domain of a heavy-chain antibody (VHH)と呼ばれることがある。
【0003】
通常、抗体を利用した最小の抗原認識分子は、VH、VLの2つのドメインをリンカーで結合させた一本鎖抗体(scFv)である。scFvには長いリンカーが必要なことやVHドメインの可溶性が低いことなどの理由から、リフォールディングが難しい分子として知られている。それに対して、ラクダ抗体では1個の免疫グロブリンドメイン(VHドメイン)のみで抗原を認識することができるため、たった1個のドメインからなる抗原認識分子を作製することが可能である。更に、VHHは可溶性が高く高温でも安定であることが証明され、VHHの物性研究により、当該分子がリフォールディング効率の高い分子であることが分かって来ている。
【0004】
このように、VHHは抗体の最も小さい認識ユニットであり、フォールディングが簡便であり、リフォールディング効率が高いという優れた特徴を有している。また、VHHは抗原認識部位(パラトープ)の形状が通常の抗体とは異なり、酵素の活性部位の溝に突き刺さることができるため、酵素の活性を阻害する抗体(中和抗体)として機能できるという特徴も有している。このような利点を活かしたVHHの利用法として、抗体融合酵素、多価抗体、イントラボディ、不活化環境での利用などが提案されている(非特許文献1、特許文献1〜3)。
【0005】
また、VHHに更に優れた性質を付与するために、特許文献4では、ラクダ科抗体VHHドメインに2個のシステイン残基を導入し、ジスルフィド結合を形成させることにより熱安定性を高めたことが報告されている。
【0006】
このような優れた特徴を有するVHHが広く利用されるためには、安価且つ大量にVHHを製造できる方法が開発される必要がある。微生物を用いたVHHの生産系の中でも、酵素の分泌能に優れているという点から麹菌(糸状菌)が特にVHHの製造に適していると考えられる。非特許文献2及び3では、そのような麹菌を用いたVHHの製造が報告されている。
【0007】
非特許文献2では、アスペルギルス・アワモリ(Aspergillus awamori)によりVHHを発現、生産させたことが報告されており、その生産量は培地中に約1.5 mg/lであり、タンパク質の分解を防ぐために培地中にBSAを添加した場合は7.5 mg/lであったことが報告されている。
【0008】
非特許文献3では、ペロオキシダーゼ遺伝子とVHHをコードする遺伝子の融合遺伝子を含む発現カセットを有するアスペルギルス・アワモリにより、当該融合タンパク質を培地中に30 mg/l生産させたことが報告されている。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】特許第3444885号公報
【特許文献2】特許第3660270号公報
【特許文献3】特開2005-170953号公報
【特許文献4】特開2008-212125号公報
【非特許文献】
【0010】
【非特許文献1】萩原義久, 次世代抗体:ラクダ抗体の分子論, BIO INDUSTROY、Vol.25, No.5, 15-22(2008)
【非特許文献2】Joosten V, Gouka RJ, van den Hodel CAMJJ, Verrips CT, Lokmann BC, Appl Microbiol Biotechnol, 66, 384-392(2005)
【非特許文献3】Joosten V, Roelofs MS, van den Dries N, Goosen T, Verrips CT, van den Hondel CAMJJ, Lokman BC, J Biotechnol, 120, 347-359(2005)
【発明の概要】
【発明が解決しようとする課題】
【0011】
しかしながら、非特許文献2及び3におけるVHHの生産量は最大でも培地中に30 mg/lしかなく、工業的なレベルでVHHを大量生産するためには更に生産量を上げる必要がある。
【0012】
そこで、本発明は、生産性が向上したVHHの生産方法を提供することを目的とする。また、本発明は、当該生産方法に用いるための材料、すなわちDNA、糸状菌用発現ベクター及び形質転換体を提供することを目的とする。更に、本発明は、当該方法により得られたVHHを含むタンパク質を提供する。
【課題を解決するための手段】
【0013】
本発明者らは、上記課題を解決するために研究を重ねた結果、VHHを特定のリーダータンパク質と融合させてアスペルギルス・オリゼ(Aspergillus oryzae)により生産させることで、VHHの生産性を向上させることができるという知見を得た。本発明は、これら知見に基づき、更に検討を重ねて完成されたものであり、次のDNA、糸状菌用発現ベクター、形質転換体、VHHの生産方法及びタンパク質を提供するものである。
【0014】
(I) DNA
(I-1) 以下の(a)〜(d)のいずれかのDNA及びVHHをコードするDNAを融合したDNA:
(a)配列番号1に記載される塩基配列からなるDNA
(b)配列番号1に記載される塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、且つグルコアミラーゼ活性を有するタンパク質をコードするDNA
(c)配列番号2に記載される塩基配列からなるDNA
(d)配列番号2に記載される塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、且つエンドグルカナーゼ活性を有するタンパク質をコードするDNA。
(I-2) (I-1)に記載のDNAに更に精製用のアミノ酸配列をコードするDNAを融合したDNA。
(I-3) 前記精製用のアミノ酸配列がHisタグである、(I-2)に記載のDNA。
【0015】
(II) 糸状菌用発現ベクター
(II-1) 糸状菌体内で機能するプロモーター遺伝子及び(I-1)〜(I-3)のいずれかに記載のDNAを含む遺伝子発現カセットを有する糸状菌用発現ベクター。
【0016】
(III) 形質転換体
(III-1) (II-1)に記載の糸状菌用発現ベクターで形質転換されてなるアスペルギルス・オリゼ。
【0017】
(IV) VHHの生産方法
(IV-1) (III-1)に記載のアスペルギルス・オリゼを培地で培養し、当該培地からVHHを含むタンパク質を回収する工程を含む、VHHを含むタンパク質の生産方法。
(IV-2) 前記アスペルギルス・オリゼを液体培地で培養して、前記VHHを含むタンパク質を菌体外に分泌させることにより生成蓄積させることを特徴とする、(IV-1)に記載の方法。
(IV-3) (IV-1)又は(IV-2)に記載の方法において、更に前記VHHを含むタンパク質からVHHに結合したペプチドを切断する工程を有する、VHHの生産方法。
【0018】
(V) タンパク質
(V-1) (IV-1)又は(IV-2)に記載の方法により得られたVHHを含むタンパク質。
【発明の効果】
【0019】
本発明の生産方法によれば、アスペルギルス・オリゼを宿主としてVHHの生産性を大幅に向上させることが出来る。そのため、本発明によれば、優れた特徴を有するVHHの量産化が可能となり、VHHの生産コストを低減することも可能となる。
【図面の簡単な説明】
【0020】
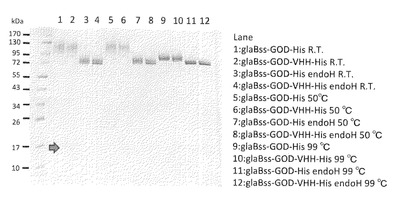
【図1】試験例1のSDS-PAGEの結果の写真である。レーン1−4:室温でインキュベーション、レーン5−8:50℃でインキュベーション、レーン9−12:99℃でインキュベーション、レーン3、4、7、8、11、12:endoH処理
【図2】試験例2のSDS-PAGEの結果の写真である。
【図3】試験例3のSDS-PAGEの結果の写真である。
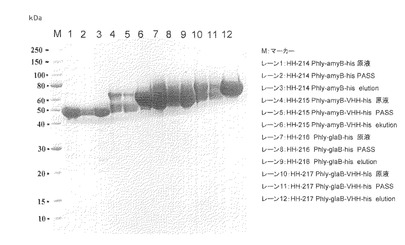
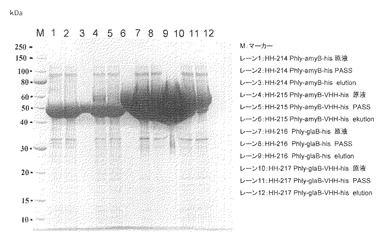
【図4】試験例4のSDS-PAGEの結果の写真である。レーン1、4、7、10:原液、レーン2、5、8、11:素通り画分、レーン3、6、9、12:溶出画分
【図5】試験例5のSDS-PAGEの結果の写真である。レーン1、4、7、10:原液、レーン2、5、8、11:素通り画分、レーン3、6、9、12:溶出画分
【図6】試験例6のSDS-PAGEの結果の写真である。レーン1、3、5、7:EndoH処理(37℃、24時間)、レーン2、4、6、8:EndoH処理せずに4℃で24時間静置
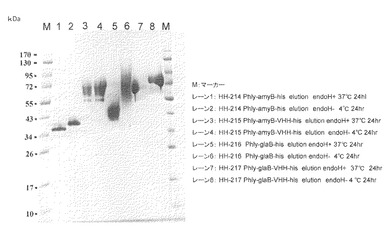
【図7】試験例7のSDS-PAGEの結果の写真である。レーン1、3、5、7:EndoH処理(37℃、24時間)、レーン2、4、6、8:EndoH処理せずに4℃で24時間静置
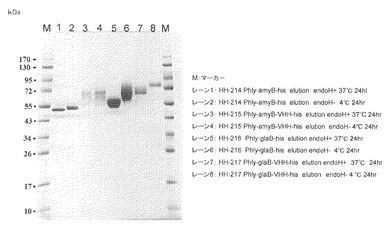
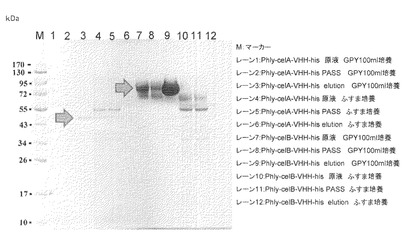
【図8】試験例8のSDS-PAGEの結果の写真である。
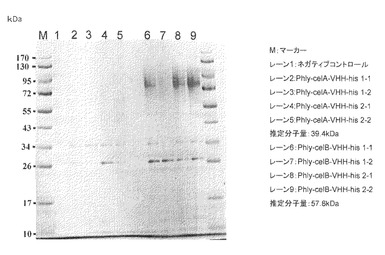
【図9】試験例9のSDS-PAGEの結果の写真である。レーン1、4、7、10:原液、レーン2、5、8、11:素通り画分、レーン3、6、9、12:溶出画分
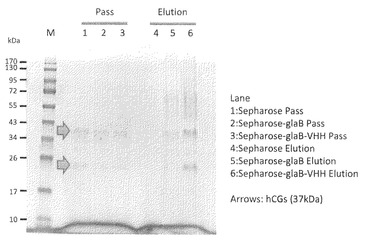
【図10】試験例10のSDS-PAGEの結果の写真である。レーン1−3:素通り画分、レーン34−6:溶出画分
【発明を実施するための形態】
【0021】
以下、本発明の実施の形態を詳細に説明する。
【0022】
本発明が対象とする糸状菌としては、長年清酒、醤油、味噌、みりんなどの醸造食品に利用されてきた安全な微生物であり、わが国の産業において利用頻度の高い宿主である点で、麹菌アスペルギルス・オリゼが好ましい。
【0023】
(1)DNA
本発明のDNAは、以下の(a)〜(d)のいずれかのDNA及びVHHをコードするDNAを融合したDNAであることを特徴とする:
(a)配列番号1に記載される塩基配列からなるDNA
(b)配列番号1に記載される塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、且つグルコアミラーゼ活性を有するタンパク質をコードするDNA
(c)配列番号2に記載される塩基配列からなるDNA
(d)配列番号2に記載される塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、且つエンドグルカナーゼ活性を有するタンパク質をコードするDNA。
【0024】
本明細書において、上記(b)又は(d)のDNAによりコードされるタンパク質を改変体と呼ぶことがある。
【0025】
(1-1)リーダータンパク質
本発明が対象とするVHHと融合させて発現させるためのリーダータンパク質は、配列番号3に記載されるアミノ酸配列を有するグルコアミラーゼ(glaB)若しくはその改変体、又は配列番号4に記載されるアミノ酸配列を有するエンドグルカナーゼ(CelB)若しくはその改変体である。リーダータンパク質としては、配列番号4に記載されるアミノ酸配列を有するエンドグルカナーゼ(CelB)又はその改変体がより好ましい。なお、配列番号3で表されるアミノ酸配列をコードする遺伝子の塩基配列は配列番号1に表され、配列番号4で表されるアミノ酸配列をコードする遺伝子の塩基配列は配列番号2に表される。
【0026】
VHHにこれらのリーダータンパク質を融合させて糸状菌で発現させることで、VHHの生産性を大幅に向上させることが可能となる。
【0027】
上記のストリンジェントな条件とは、例えば、65℃で5×SSC溶液(1倍濃度のSSC溶液の組成は、150 mM塩化ナトリウム、15 mMクエン酸ナトリウム)中でハイブリダイズさせ、更に0.1%のSDSを含有する0.5×SSC溶液で65℃で洗浄する条件を意味する。ストリンジェントな条件下でのハイブリダイゼーションの各操作は、「Molecular Cloning (Third Edition)」(J. Sambrook & D. W. Russell, Cold Spring Harbor Laboratory Press, 2001)に記載されている方法等、従来公知の方法で行うことができる。通常、温度が高いほど、塩濃度が低いほどストリンジェンシーは高くなる。
【0028】
ストリンジェントな条件下でハイブリダイズするDNAは、通常、プローブとして使用するDNAの塩基配列と一定以上の同一性を有し、その同一性は、例えば70%以上、好ましくは80%以上、更に好ましくは90%以上、特に好ましくは95%以上が挙げられる。塩基配列の同一性は、市販の又は電気通信回線(インターネット)を通じて利用可能な解析ツールを用いて算出することができる。塩基配列の同一性(%)は、当該分野で慣用のプログラム(例えば、BLAST、FASTA等)を初期設定で用いて決定することができる。
【0029】
グルコアミラーゼ活性は今井ら(1993)(生物工学会誌: seibutsu-kogaku kaishi 71(2), 93-97, 1993-03-25)の報告に記載の方法により、エンドグルカナーゼ活性は特開2003-164284号公報の段落[0017]に記載の方法により測定することができる。
【0030】
このようなリーダータンパク質の改変体の製造は常法に従って行うことができる。
【0031】
(1-2)ラクダ科動物抗体の重鎖可変ドメイン(VHH)
本発明で使用するのは、ラクダ科動物由来抗体のフラグメントである重鎖の可変ドメイン(VHH)である。ラクダ科動物としては、特に限定されないが、例えば、フタコブラクダ、ヒトコブラクダ、ラマ、アルパカ、ビクーナ、グアナコ等が挙げられる。抗体としては、軽鎖を有さず、CH1ドメインを有さない重鎖のみからなるラクダ科動物由来の抗体であれば特に限定されない。また、VHHの抗原の種類も特に限定されず、どのような抗原を対象とするかはVHHの使用用途に応じて適宜決定される。
【0032】
VHHとしては、例えば、配列番号5、配列番号6、配列番号7で表されるアミノ酸配列からなるVHHが挙げられる。
【0033】
本発明におけるVHHは、天然に存在するものに加えて、天然のVHHのアミノ酸配列に1個以上のアミン酸を置換、欠失及び/又は付加したものも含む。
【0034】
特定の抗原に対するVHHの取得方法としては、ラクダの免疫、ファージディスプレイによる選択、B細胞ディスプレイによる選択、酵母細胞表層ディスプレイによる選択など複数あるが、抗体ファージライブラリーからの特異的な抗体の単離法であるバイオパニングが有効である。バイオパニングは、ラムダgt11などの抗体を提示できるファージライブラリーを作成し、目的の抗原を固定化したものと複数回接触させ、特異的な抗体を有するファージを濃縮選択する方法である(Phage Display, A Laboratory Manual, Cold Spring Harbor Laboratory Press 2001)。
【0035】
(1-3)精製用タグ
本発明のDNAには、リーダータンパク質をコードするDNA及びVHHをコードするDNAに更に精製用のアミノ酸配列をコードするDNAが融合されていても良い。精製用のアミノ酸配列としては、例えばHisタグ、GST、MBP(マルトース結合タンパク質)などが挙げられ、好ましくはHisタグである。このような精製用のアミノ酸配列がVHHと結合していることで、アフィニティークロマトグラフによるVHHの精製が可能になる。
【0036】
上記の各DNAの順序は、本発明の効果が得られるものであればどのような順序であってもよいが、好ましくは5'末端からリーダータンパク質→VHH、又はリーダータンパク質→VHH→精製用アミノ酸配列の順である。
【0037】
また、発現後に目的とするVHHを容易に回収できるように、ペプチド結合が切断可能な認識部位を介在させて、上記の各DNAを融合させても良い。該認識部位としては、プロセッシング酵素、消化酵素などのプロテアーゼで切断可能なアミノ酸配列(2以上のアミノ酸からなる)、システインやメチオニンなどの化学修飾剤、超音波、レーザー、熱などの物理的開裂により切断可能なアミノ酸などが挙げられる。認識部位を切断可能な消化酵素(プロセッシング酵素)としては、好ましくは第Xa因子、トロンビン、レニン、トリプシン、V8プロテアーゼ、Pseudomonasエンドプロテアーゼ、Arthrobacterリシルエンドペプチダーゼ等が挙げられ、認識部位としては、例えば第Xa因子認識配列であるIle-Glu-Gly-Argが挙げられる。化学修飾剤としては、Metを認識するCNBr (Cyanogen bromide)、Asn、Asp、Gluを認識する希塩酸、及びCysを認識するDMAP-CNが挙げられる。
【0038】
(2)遺伝子発現カセット
本発明における遺伝子発現カセットは、糸状菌体内で機能するプロモーター遺伝子と上記DNAを含むことを特徴とする。
【0039】
(2-1)プロモーター
プロモーターは、糸状菌体内、特にアスペルギルス・オリゼの菌体内でプロモーターとして機能するものであればよい。かかるプロモーターとして具体的には、例えばα−アミラーゼ遺伝子プロモーターamyB(Biosci Biotechnol Biochem, 56, 1849-1853(1992))、グルコアミラーゼ遺伝子プロモーターglaA(Gene, 108, 145-150(1992))、α−グルコシダーゼ遺伝子プロモーターagdA(Curr Genet, 30, 432-438(1996))、マンガンSOD遺伝子プロモーターsodM(特開2000-224381号公報)、チロシナーゼ遺伝子プロモーターmelO(特開2001-046078号公報)、グルコアミラーゼ遺伝子プロモーターglaB(特開2000-245465号公報、特開平11-243965号公報)、hlyプロモーター(特開2009-296958号公報)等が挙げられる。中でも好ましいのは、hlyプロモーターである。
【0040】
遺伝子発現カセットにおけるプロモーターの位置は、糸状菌体内でプロモーターが機能し、所望のVHHが発現し産生できる位置であれば、特に限定されないが、通常、VHH遺伝子配列の上流域に配置される。
【0041】
(2-2)ターミネーター
ターミネーターは、糸状菌体内、特にアスペルギルス・オリゼの菌体内でターミネーターとして機能するものであれば良い。ターミネーターとしては、例えばα−アミラーゼ遺伝子のターミネーター、又はグルコアミラーゼ(glaB)ターミネーター(Gene. 207, 127-134(1998))等を挙げることができる。
【0042】
本発明の糸状菌用遺伝子発現カセットは、これらのプロモーター、リーダータンパク質をコードするDNA、VHHをコードするDNA、及びターミネーターが、直接連結されてなるものであってもよいが、それらの間に1〜2000塩基程度のヌクレオチドが挟まれていてもよい。例えば、目的とするVHHをコードするDNAのオープンリーディングフレームの上流に糸状菌、好ましくはアスペルギルス・オリゼの既知分泌タンパク質の遺伝子を配置しても良く、そうすることにより、目的とするVHHは、上記分泌タンパク質との融合タンパク質として発現産生され、斯くして産生された融合タンパク質は菌体外に分泌される。また、その他のヌクレオチドとして、通常ベクターが備える制限酵素切断部位などを備えていてもよい。
【0043】
なお、そのような分泌タンパク質のアスペルギルス糸状菌由来のものとしては、α−アミラーゼ、グルコアミラーゼ、α−グルコシダーゼ、エンドグルカナーゼ、セロビオハイドロラーゼ、β−グルコシダーゼ、酸性プロテアーゼ、中性プロテアーゼ、アルカリプロテアーゼ、カルボキシペプチダーゼ、リパーゼ、ホスホリパーゼ、キシラナーゼ、ガラクトシダーゼ、フラクトフラノシダーゼ、キシロシダーゼ、ペクチンリアーゼ、フィターゼなどが知られている。
【0044】
(3)糸状菌用発現ベクター
本発明の糸状菌用発現ベクターは、上記遺伝子発現カセットを有することを特徴とする。
【0045】
本発明の糸状菌用発現ベクターには、選択マーカー遺伝子が含まれていても良く、コトランスフォーメーションを行う場合は、当該糸状菌用発現ベクターとは別のベクターに選択マーカー遺伝子が存在していれば良い。
【0046】
本発明の糸状菌用発現ベクターは、これらの選択マーカー遺伝子及び遺伝子発現カセットが、直接連結されてなるものであってもよいが、それらの間に1〜2000塩基程度のヌクレオチドが挟まれていてもよい。
【0047】
また、本発明には、直鎖状の糸状菌用発現ベクター及び環状の糸状菌用発現ベクターの両者が含まれるが、(1)構築したプラスミドから1ステップのPCRで取得できるなど、環状の発現ベクターと比べて少ないステップで調製することができること、(2)宿主としてロイシン要求性変異株を使用する場合に、当該変異を直接相補することができること、及び(3)形質転換効率が環状の発現ベクターと比べて高いという点から、好ましくは直鎖状の糸状菌用発現ベクターである。
【0048】
(3-1)選択マーカー遺伝子
選択マーカー遺伝子は、糸状菌体内、特にアスペルギルス・オリゼの菌体内で選択マーカーとして機能するもの、すなわち発現できるものであれば良い。かかる選択マーカー遺伝子として、例えば、niaD(Biosci. Biotechnol. Biochem., 59, 1795-1797(1995))、argB(Enzyme Microbiol Technol, 6, 386-389(1984))、sC(Gene, 84, 329-334(1989))、ptrA(Biosci Biotechnol Biochem, 64, 1416-1421(2000))、pyrG(Biochem Biophys Res Commun, 112, 284-289(1983))、amdS(Gene, 26, 205-221(1983))、オーレオバシジン耐性遺伝子(Mol Gen Genet, 261, 290-296(1999))、ベノミル耐性遺伝子(Proc Natl Acad Sci USA, 83, 4869-4873(1986))、及びハイグロマイシン耐性遺伝子(Gene, 57, 21-26(1987))から選ばれるマーカー遺伝子を挙げることができる。
【0049】
これらの選択マーカー遺伝子は、上記文献に記載された配列に基づいて設計されたプライマーを用いて、アスペルギルス属糸状菌の染色体DNAを鋳型としてPCRを行う方法、上記文献に記載された配列に基づいて設計されたプローブを用いてアスペルギルス属糸状菌の染色体DNAライブラリーからハイブリダイゼーションにより取得する方法などにより容易に入手することができる。また、宿主の糸状菌として、例えばロイシン要求性などの栄養要求性変異株を用いる場合、選択マーカー遺伝子として当該栄養要求性を相補する野生型遺伝子を用いることもできる。例えば、宿主がロイシン要求性変異株である場合、そのロイシン要求性を相補できる選択マーカー遺伝子を用いることができ、かかる遺伝子としてはβ−イソプロピルリンゴ酸デヒドロゲナーゼをコードするアスペルギルス・ニジュランス由来の遺伝子ANleu2及びアスペルギルス・オリゼ由来の遺伝子leu2を挙げることができる。なお、これらの場合、宿主として用いる糸状菌は、選定された選択マーカーについての機能的遺伝子を有していない株を用いる必要がある。
【0050】
(4)形質転換体
本発明の形質転換体は、上記糸状菌用発現ベクターでアスペルギルス・オリゼを形質転換したものである。
【0051】
アスペルギルス・オリゼを宿主とすることは、高いタンパク質生産能及び醸造微生物としての安全性の点で好ましい。なお、宿主として用いるアスペルギルス・オリゼは、野生株に限らず、変異株であってもよい。かかる変異株としては、前述する選択マーカーに関する機能的遺伝子を有さないように変異してなる株を挙げることができ、具体的には正常にロイシンの生合成ができないようにロイシン生合成を担う遺伝子が変異又は欠失しているロイシン要求性変異株:正常にアルギニンの生合成ができないようにアルギニン生合成を担う遺伝子が変異又は欠失しているアルギニン要求性変異株:正常にメチオニンの生合成ができないようにメチオニン生合成を担う遺伝子が変異又は欠失しているメチオニン要求性変異株:正常にヒスチジンの生合成ができないようにヒスチジン生合成を担う遺伝子が変異又は欠失しているヒスチジン要求性変異株:正常に硝酸が資化できないように硝酸資化性遺伝子が変異又は欠失しているniaD変異株:正常に硝酸イオンが資化できないように硝酸イオン資化性遺伝子が欠失しているsC変異株などを挙げることができる。
【0052】
宿主の形質転換方法は、特に限定されず、従来公知の方法を用いて行うことができる。例えば、Cohenらの方法(塩化カルシウム法)(Proc. Natl. Acad. Sci. USA, 69:2110 (1972))、プロトプラスト法(Mol. Gen. Genet., 168:111 (1979))、コンピテント法(J. Mol. Biol., 56:209 (1971))、エレクトロポレーション法等を挙げることができる。
【0053】
(5)VHHの生産方法
本発明のVHHを含むタンパク質の生産方法は、上記のアスペルギルス・オリゼを培地で培養し、当該培地からVHHを含むタンパク質を回収する工程を含むことを特徴とする。
【0054】
本発明の方法によれば、VHH(又はVHHを含むタンパク質)を培地中に1000 mg/l以上、好ましくは1500 mg/l以上生産することが可能となる。
【0055】
本発明において、「VHHを含むタンパク質」とはVHHにリーダータンパク質、精製用アミノ酸配列等が融合したタンパク質のことを意味している。
【0056】
培養は、固体培地を用いる固体培養、又は液体培地を用いる液体培養のいずれでも行うことができるが、好ましくは液体培地を用いた液体培養である。
【0057】
固体培地としては、アスペルギルス・オリゼの培養に使用される公知の固体培地を制限なく使用することができる。固体培地とはその固形の支持担体が栄養源を含むか、又は固形の支持担体に栄養源が添加されものであり、そこにアスペルギルス・オリゼが生育できる固形培地を指し示す。このような固体培地として主に、フスマ(小麦などの穀物の殻)、デンプン粉末、米・小麦・大豆の生あるいは蒸したもの、更にはメンブレンや多孔質の人工物(例えば園芸に使われるバーミキュライト)等に栄養源を添加したもの等が挙げられる。特にフスマ及び蒸した米が好ましい。
【0058】
また、液体培地は、アスペルギルス・オリゼの培養に使用される公知の液体培地を制限なく使用できる。例えば、用いられる培地としては、炭素源としてグルコース、マルトース、フルクトース、グリセロール、スターチなどの炭水化物を含有するものが挙げられる。また、無機もしくは有機窒素源(例えば、硫酸アンモニウム、塩化アンモニウム、カゼインの加水分解物、酵母抽出物、ポリペプトン、バクトトリプトン、ビーフ抽出物等)を含有するものも挙げられる。これらの炭素源及び窒素源は、純粋な形で使用する必要はなく、純度の低いものも微量の生育因子や無機栄養素を豊富に含んでいるので有利である。さらに所望により、他の栄養源[例えば、無機塩(例えば、二リン酸ナトリウム、二リン酸カリウム、リン酸水素二カリウム、塩化マグネシウム、硫酸マグネシウム、塩化カルシウム)、ビタミン類(例えば、ビタミンB1)、抗生物質(例えば、アンピシリン、カナマイシン)など]を培地中に添加してもよい。具体的な液体培地としては、例えば、ポテトデキストロース培地(ニッスイ社)、最少培地(2%グルコース(又はスターチ)、0.3% NaNO3、0.2% KCl、0.1% KH2PO4、0.05% MgSO4、0.002% FeSO4、pH6.0)、GPY培地(グルコース2%、ペプトン1%、酵母抽出物0.5%)等を挙げることができる。なお、これらは1.5%程度の寒天を添加することで、固体培地として調製することもできる。
【0059】
本発明のアスペルギルス・オリゼの培養は、通常pH5.5〜8.5、好ましくはpH6〜8のpH条件;通常25〜42℃、好適には30〜37℃の温度条件で行うことができる。培養時間は、その他の条件によって異なるが、通常2〜7日間程度、好ましくは3〜5日間程度を挙げることができる。
【0060】
本発明の方法によれば、目的とするVHHを含むタンパク質を菌体外に生成することもできる。その場合、VHHを含むタンパク質の取得に際しては、培養上清を回収すればよい。また、寒天培地などのように固体培地を使用する場合も培養上清としての寒天培地を回収すれば良い。さらに、これらの上清を、公知のタンパク質精製方法、例えばイオン交換、疎水、ゲルろ過などの各種クロマトグラフィーに供することにより目的とするVHHを含むタンパク質を単離し回収することができる。VHHを含むタンパク質が精製用のアミノ酸配列と結合している場合は、アフィニティークロマトグラフにより精製を行うことができる。
【0061】
本発明の方法で得られたVHHを含むタンパク質は、リーダータンパク質を含む形であっても抗原結合活性を有している。しかしながら、本発明の方法では、前記の2工程に加えて、培地から回収したVHHを含むタンパク質からリーダータンパク質(及び精製用のアミノ酸配列)を切断する工程を行うことによって、VHHのみを取得しても良い。
【実施例】
【0062】
以下、実施例を示して本発明をより詳細に説明するが、本発明はこれらに限定されるものではない。なお、一般的な実験方法は、実験書(サムブルックら(Sambrook, J.)のMolecular Cloning:A Laboratory Manual 第3版)に従った。
【0063】
試験例で使用した遺伝子の塩基配列及びアミノ酸配列
・選択マーカー
硝酸還元酵素遺伝子niaD<配列番号8>
・プロモーター
hlyプロモーター<配列番号9>
・ターミネーター
glaBターミネーター<配列番号10>
・リーダータンパク質
glaBss-GOD<配列番号11、12>、amyB<配列番号13、14>、glaB<配列番号1、3>、celA<配列番号15、16>、celB<配列番号2、4>(すべてストップコドンなし)
・VHH
試験例1〜10で使用したVHH<配列番号17、5>、試験例11で使用した抗EGFRのVHH<配列番号18、6>、試験例12で使用した抗R66のVHH<配列番号19、7>(すべてストップコドンなし)
【0064】
糸状菌用発現ベクターの構築
<niaD遺伝子の大腸菌ベクターへの組み込み>
麹菌アスペルギルス・オリゼ由来の硝酸還元酵素遺伝子niaDをPstI-HindIII断片となるようにプライマーA(5’-aactgcagaacaggccccaaattcaattaattgca-3’:配列番号20)とプライマーB(5’-cccaagctttggatttcctacgtcttcaatacaaacc-3’:配列番号21)を用いて麹菌ゲノムDNAを鋳型としてLA-Taq(宝ホールディングス)を用いて以下の条件のPCRにより増幅した。
PCR条件
・ 96℃ (5分間)を1サイクル
・ 96℃ (20秒間)、60℃ (30秒間)、72℃ (5分間) を30サイクル
・ 72℃ (7分間)を1サイクル
得られたPCR増幅産物を制限酵素PstI-HindIIIで37℃で処理後、アガロースゲル電気泳動で切り出した。切り出しは、QIAquick Gel Extraction kit (QIAGEN)を用いた。大腸菌プラスミドpUC119(宝ホールディングス)にDNA Ligation kit ver.1(宝ホールディングス)を用いてライゲーションし、大腸菌(E. coli)JM109株に形質転換した。その結果、niaDマーカーがサブクローニングされたプラスミドpNIA2を得た。プラスミドpNIA2は、PstI、SalI部位の両方に遺伝子をunique siteとして導入できる。
【0065】
<glaBターミネーターの組み込み>
麹菌由来His-tag付きglaBターミネーターの遺伝子のpNIA2への挿入を試みた。プライマーC(5'-acgcgtcgacCACCATCACCACCACCACTAAATGTACTTTCCAGTGCGTGTAGTCTACTCTG-3'配列番号22)とプライマーD(5'-acgcctcgagCTGCAGATCGGCTGAAGTTAGGAGCGGCCATTGTC-3'配列番号23)を用いて麹菌ゲノムDNAを鋳型としてLA-Taq(宝ホールディングス)を用いて以下の条件のPCRにより増幅した。
PCR条件
・ 96℃ (5分間)を1サイクル
・ 96℃ (20秒間)、60℃ (30秒間)、72℃ (5分間) を30サイクル
・ 72℃ (7分間)を1サイクル
得られたPCR増幅産物を制限酵素SalI-XhoIで37℃で処理後、アガロースゲル電気泳動で切り出した。切り出しは、QIAquick Gel Extraction kit (QIAGEN)を用いた。大腸菌プラスミドpNIA2のSalI部位にDNA Ligation kit ver.1(宝ホールディングス)を用いてライゲーションし、大腸菌(E. coli)JM109株に形質転換した。その結果、His-tag付きglaBターミネーターがサブクローニングされたプラスミドpNIATを得た。プラスミドpNIATは、PstI、SalI部位の両方に遺伝子をunique siteとして導入できる。
【0066】
<hlyプロモーターの組み込み>
麹菌由来hlyプロモーターの遺伝子のpNIATへの挿入を試みた。プライマーE(5'-aaCTGCAGGCAGATGTAGCCGTGGCACCACAA-3'配列番号24)とプライマーF(5'-acgcgtcgacGGTGTTGTGGTGTGAAGGGTGATTGATGTGAGACCG-3'配列番号25)を用いて麹菌ゲノムDNAを鋳型としてLA-Taq(宝ホールディングス)を用いて以下の条件のPCRにより増幅した。
PCR条件
・ 96℃ (5分間)を1サイクル
・ 96℃ (20秒間)、60℃ (30秒間)、72℃ (5分間) を30サイクル
・ 72℃ (7分間)を1サイクル
得られたPCR増幅産物を制限酵素SalI-PstIで37℃で処理後、アガロースゲル電気泳動で切り出した。切り出しは、QIAquick Gel Extraction kit (QIAGEN)を用いた。大腸菌プラスミドpNIATのPstI-SalI部位にDNA Ligation kit ver.1(宝ホールディングス)を用いてライゲーションし、大腸菌(E. coli)JM109株に形質転換した。その結果、hlyプロモーターがサブクローニングされたプラスミドpNMBを得た。プラスミドpNMBは、SalI部位に麹菌で発現させたいタンパク質をコードする遺伝子をunique siteとして導入できる。
【0067】
<目的遺伝子の組み込み>
(1)ベクターの調製
pNMBを制限酵素SalIで37℃処理後、dNTPを最終10 mMとなるように添加してT4 DNAポリメラーゼ(宝ホールディングス)で37℃1時間処理した。さらに、バクテリア由来アルカリホスファターゼ(宝ホールディングス)で50℃、30分反応させた。得られた反応物をPCRクリーンアップカラム(プロメガ社製)で処理溶出させ、ベクターとした。
【0068】
(2)インサートの調製
麹菌で発現させたいタンパク質をコードする遺伝子を目的遺伝子として、上記ベクターにサブクローニングするインサートとした。
【0069】
目的遺伝子の開始コドンから下流30bpのセンス鎖のプライマー(5'末端をリン酸化)と、目的遺伝子の終止コドンから上流30bpのアンチセンス鎖のプライマー(5'末端をリン酸化)を用いて、目的遺伝子の起源生物由来のゲノムDNAを鋳型として、pfu Taqポリメラーゼ(東洋紡績株式会社)を用いて下記条件でPCRを行った。
PCR条件
・ 96℃ (5分間)を1サイクル
・ 96℃ (20秒間)、60℃ (30秒間)、 72℃ (5分間) を30サイクル
・ 72℃ (7分間)を1サイクル
得られたPCR断片をPCRクリーンアップカラム(プロメガ社製)で処理溶出させ、インサートとした。
【0070】
(3)ベクターとインサートのライゲーション
モル数で、ベクター:インサート=1:4となるように添加して、DNA Ligation kit ver.1(宝ホールディングス)を用いてライゲーションし、大腸菌(E. coli)JM109株に形質転換した。形質転換体からプラスミドを調製し、目的遺伝子の読み枠がhlyプロモーターに対して正方向にサブクローニングされ、なお且つ翻訳時の読み枠がフレームシフトしておらず、更にHis-tagの読み枠と合致しているプラスミドを目的遺伝子発現プラスミドとして調製した。
【0071】
目的遺伝子発現株の取得
定法であるPEG-カルシウム法(Mol Gen Genet, 218, 99-104, (1989))により、上記目的遺伝子発現プラスミドを用いて、アスペルギルス・オリゼのniaD変異株(独立行政法人産業技術総合研究所特許生物寄託センターにFERM P-17707として寄託されている)を形質転換した。硝酸を単一窒素源とするツアペクドックス(Czapek-Dox)培地(2%グルコース、0.1%リン酸1水素2カリウム、0.05%塩化カリウム、0.05%硫酸マグネシウム、0.001%硫酸鉄、0.3%硝酸ナトリウム)で生育できる株を選択することにより、目的遺伝子発現プラスミドを保持する形質転換体を複数得た。
【0072】
目的遺伝子産物の取得
上記形質転換体をポテトデキストロース培地で胞子形成させ、滅菌水で胞子を回収した。500 ml容三角フラスコに入った100 ml GPY液体培地(2%グルコース、1%ポリペプトン、0.5%イーストエキストラクト、0.1%リン酸1水素2カリウム、0.05%塩化カリウム、0.05%硫酸マグネシウム、0.001%硫酸鉄、0.3%硝酸ナトリウム)に最終胞子濃度1×106/mlとなるように植菌した。30℃3日間の液体培養で目的遺伝子産物が培地中に分泌発現し、当該培養液をサンプルとした。
【0073】
サンプルの更なる精製は、His GraviTrapカラム(GEヘルスケア社)を用い、所定のマニュアル通りに5-15mLのサンプルをカラムにアプライし自然落下により吸着(20 mMリン酸ナトリウム, 500 mM NaCl, 45 mMイミダゾール, pH 7.4)させ、カラムに吸着せずに出てきた画分を素通り画分とした。10mLで洗浄(20 mMリン酸ナトリウム, 500 mM NaCl, 45 mMイミダゾール, pH 7.4)した後、イミダゾール溶液(20 mMリン酸ナトリウム, 500 mM NaCl, 500 mMイミダゾール, pH 7.4)を同様にアプライし、出てきた画分を溶出画分とした。
【0074】
試験例1:アスペルギルス・ニガー(Aspergillus niger)由来グルコースオキシダーゼタンパク質とのVHHの融合タンパク質の培地中への分泌生産の結果
目的:モデルとして麹菌には類縁体が存在しないアスペルギルス・ニガー由来グルコースオキシダーゼタンパク質(GOD)をリーダータンパク質としてVHHを融合させて、麹菌でGOD-VHH融合タンパク質を培地中への分泌生産させることを試みる。
【0075】
方法:
1.ベクターの構築方法
ベクターはhlyプロモーターが組み込まれた発現プラスミドから「ベクターの調製」に従って調製し、インサートはglaBss-GOD遺伝子を合成遺伝子配列(北海道システムサイエンス社製)を鋳型として、「インサートの調製」に従って調製した。「ベクターとインサートのライゲーション」に従い、Phly-glaBss-GOD-his分泌発現ベクターを構築した。融合タンパク質の分泌発現ベクターPhly-glaBss-GOD-VHH-hisは、インサート1:glaBss-GOD遺伝子を鋳型としたものと、インサート2:VHH遺伝子を合成遺伝子配列(北海道システムサイエンス社製)を鋳型としたものを、「インサートの調製」に従ってそれぞれ調製した。ベクターとインサート1及び2を同時に混和して、「ベクターとインサートのライゲーション」に従い、Phly-glaBss-GOD-VHH-hisを構築した。
【0076】
2.タンパク質の発現分泌回収
「目的遺伝子発現株の取得」に従い、麹菌に1で作成した分泌発現ベクターを麹菌に形質転換し、「目的遺伝子産物の取得」によりサンプルを回収した。
【0077】
3.SDS-PAGE解析
2で得られたサンプルをVIVA SPIN 500 (Sartorius社製)により濃縮し、5μgタンパク質相当をSDS-PAGEにて解析を行った。EndoH処理は、Endoglycosidase H (Streptomyces griseus由来、生化学バイオビジネス株式会社製)により行った。各サンプルはSDS電気泳動用のサンプルバッファー(0.1 M Tris-HCl (pH 6.8)、2% SDS、20% glycerol、0.2% BPB、0.2M DTT)と1対1で混和し、室温、50℃又は99℃で5分間インキュベートした後にSDS-PAGE解析に供した。結果を図1に示した。
【0078】
結果及び考察:
インキュベートの温度は、高温になるほどバンドがシャープになり、99℃が最適であると考えられた。また、EndoH処理を行ったサンプルは全て72kDa近傍にバンドが検出されたことから、おおよそglaBss-GOD-hisをコードする部分は生産できたと考えられる。しかしならが、glaBss-GOD-VHH-hisもほぼ同じ位置にバンドが検出されたことから、VHHの部分だけ取り除かれている可能性がある。glaBss-GOD-VHH-hisを泳動したレーン全てにVHH単独の分子量に近い17kDa近傍にバンドが検出されていることから(矢印)、glaBss-GOD-VHH-hisはglaBss-GODとVHH-hisに分解されて生産されている或いは生産できる可能性が示唆された。
【0079】
試験例2:アスペルギルス・オリゼ由来α-アミラーゼ(amyB)とのVHHの融合タンパク質の培地中への分泌生産の結果
目的:アスペルギルス・オリゼ由来α-アミラーゼ(amyB)をリーダータンパク質としてVHHを融合させて、麹菌でamyB-VHH融合タンパク質を培地中への分泌生産させることを試みる。
【0080】
方法:
1.ベクターの構築方法
ベクターはhlyプロモーターが組み込まれた発現プラスミドから「ベクターの調製」に従って調製し、インサートはamyB遺伝子をアスペルギルス・オリゼ由来amyB(cDNA型)遺伝子を鋳型として、「インサートの調製」に従って調製した。「ベクターとインサートのライゲーション」に従い、Phly-amyB-his分泌発現ベクターを構築した。融合タンパク質の分泌発現ベクターPhly-amyB-VHH-hisは、インサート1:amyB(cDNA型)遺伝子を鋳型としたものと、インサート2:VHH遺伝子を合成遺伝子配列(北海道システムサイエンス社製)を鋳型としたものを、「インサートの調製」に従ってそれぞれ調製した。ベクターとインサート1及び2を同時に混和して、「ベクターとインサートのライゲーション」に従い、Phly-amyB-VHH-hisを構築した。
【0081】
2.タンパク質の発現分泌回収
「目的遺伝子発現株の取得」に従い、麹菌に1で作成した分泌発現ベクターを麹菌に形質転換し、「目的遺伝子産物の取得」によりサンプルを回収した。
【0082】
3.SDS-PAGE解析
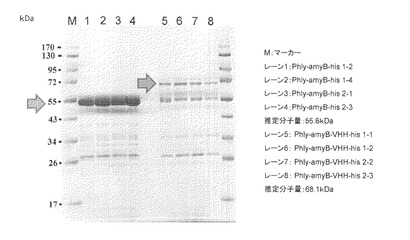
2で得られたサンプル100μLをVIVA SPIN 500 (Sartorius社製)により濃縮し、SDS-PAGEにて解析を行った。結果を図2に示した。
【0083】
結果及び考察:
amyB-his遺伝子導入した独立の4株のサンプル(レーン1−4)は良好にamyBが生産されていることが観察された。amyB-VHH-his遺伝子導入した独立の4株のサンプル(レーン5−8)にはVHHの分子量増加に相当する72kDaの位置にバンドが観察され(中央の矢印)、融合タンパク質の形でVHHが生産できたことを示唆する結果を得た。しかし、amyB-hisと比較してamyB-VHH-hisのバンドのシグナル強度は弱く、生産性は低かった。
【0084】
試験例3:アスペルギルス・オリゼ由来グルコアミラーゼ(glaB)とのVHHの融合タンパク質の培地中への分泌生産の結果
目的:アスペルギルス・オリゼ由来グルコアミラーゼ(glaB)をリーダータンパク質としてVHHを融合させて、麹菌でglaB-VHH融合タンパク質を培地中への分泌生産させることを試みる。
【0085】
方法:
1.ベクターの構築方法
ベクターはhlyプロモーターが組み込まれた発現プラスミドから「ベクターの調製」に従って調製し、インサートはglaB遺伝子をアスペルギルス・オリゼ由来glaB(cDNA型)遺伝子を鋳型として、「インサートの調製」に従って調製した。「ベクターとインサートのライゲーション」に従い、Phly-glaB-his分泌発現ベクターを構築した。融合タンパク質の分泌発現ベクターPhly-glaB-VHH-hisは、インサート1:glaB(cDNA型)遺伝子を鋳型としたものと、インサート2:VHH遺伝子を合成遺伝子配列(北海道システムサイエンス社製)を鋳型としたものを、「インサートの調製」に従ってそれぞれ調製した。ベクターとインサート1及び2を同時に混和して、「ベクターとインサートのライゲーション」に従い、Phly-glaB-VHH-hisを構築した。
【0086】
2.タンパク質の発現分泌回収
「目的遺伝子発現株の取得」に従い、麹菌に1で作成した分泌発現ベクターを麹菌に形質転換し、「目的遺伝子産物の取得」によりサンプルを回収した。
【0087】
3.SDS-PAGE解析
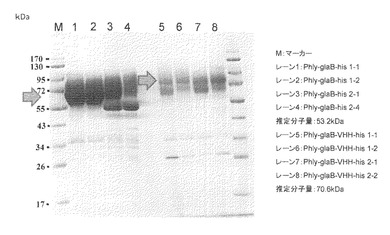
2で得られたサンプル100μLをVIVA SPIN 500 (Sartorius社製)により濃縮し、SDS-PAGEにて解析を行った。結果を図3に示した。
【0088】
結果及び考察:
glaB-his遺伝子導入した独立の4株のサンプル(レーン1−4)は良好にglaBが生産されていることが観察された。しかしながら、3と4レーンのバンドはスプリットしており、一部hisタグが切断された可能性が示唆された。glaB-VHH-his遺伝子導入した独立の4株のサンプル(レーン5−8)にはVHHの分子量増加に相当する80kDaの位置にバンドが観察され(中央の矢印)、融合タンパク質の形でVHHが生産できたことを示唆する結果を得た。5−8の全てのレーンもスプリットしており、一部hisタグが切断された可能性が示唆された。しかし、glaB-hisと比較してglaB-VHH-hisのバンドのシグナル強度は弱かったが、全体としての生産量は高かった。
【0089】
試験例4:α-アミラーゼ及びグルコアミラーゼと融合させたVHHタンパク質のHis-tagカラムでの精製結果
目的:amyB-VHH-his及びglaB-VHH-hisタンパク質がhis-tagまで完全に作られているかをHis-tagカラム精製により検証する。
【0090】
方法:
試験例2と3で用いたamyB-his、amyB-VHH-his、glaB-his及びglaB-VHH-hisサンプルをHis-tagカラムであるHis GraviTrap (GEヘルスケア社製)にて精製を行い、各サンプル100μLをVIVA SPIN 500 (Sartorius社製)により濃縮し、SDS-PAGEにて解析を行った。結果を図4に示した。
【0091】
結果及び考察:
amyB-his、amyB-VHH-his、glaB-his及びglaB-VHH-hisサンプルをHis-tagカラムであるHis GraviTrap (GEヘルスケア社製)にて精製を行った。amyB-hisはサンプル(レーン1)と比較して素通り画分(レーン2)のバンドの強度が若干弱くなっていること、及び溶出画分(レーン3)にはっきりとしたバンドが確認できることからamyB-hisの一部はhis-tagまでが分解されずに培地に分泌されていることが確認できた。同様にamyB-VHH-hisのサンプル(レーン4)、素通り画分(レーン5)及び溶出画分(レーン6)のバンドパターンからamyB-VHH-hisの一部はhis-tagまでが分解されずに培地中に生産できることが明らかとなった。また、スプリットしたバンドは溶出画分には見られなかったことから、70kDaのバンドがhis-tagまでの完全長産物であり、50kDaのバンドはVHH及びHis-tag部分分解産物であることが考えられる。glaB-hisについてもamyB-hisと同様に精製ができ、傾向も同じであった(レーン7−9)。glaB-VHH-hisについてはamyB-VHH-hisと同様に60kDaのバンドはカラム生成できなかったことから、70kDaのバンドがhis-tagまでの完全長産物であることが明らかとなった。完全長のバンドのシグナル強度はamyB-VHH-hisよりもglaB-VHH-hisが強く、glaBをリーダータンパク質とすることで分解を受けていない完全長型のVHHをより高生産できることを明らかにした。
【0092】
試験例5:固体培養におけるα-アミラーゼ及びグルコアミラーゼと融合させたVHHタンパク質の生産結果
目的:amyB-VHH-his及びglaB-VHH-hisタンパク質がフスマ固体培養で液体培養より効率よくhis-tagまで完全に作られているかを検証する。
【0093】
方法:
試験例2及び3で得られたamyB-VHH-his及びglaB-VHH-hisタンパク質生産麹菌を、フスマ固体培養で培養を行った。100 mL容のガラス製三角フラスコ(AGC社製)に乾燥した小麦フスマ5 gを入れ、オートクレーブで滅菌した後、各麹菌の胞子を滅菌水に懸濁した胞子懸濁液(106胞子/mL)を加えて良く混和し、30℃で3日間静置培養を行った。培養物に50 mMリン酸ナトリウム緩衝液(pH 7.0)を100 mL加えて、タンパク質の抽出を行い、0.45μmのフィルターでろ過後、His GraviTrap(GEヘルスケア社製)にて精製を行い、各サンプルを100μLをVIVA SPIN 500 (Sartorius社製)により濃縮し、SDS-PAGEにて解析を行った。結果を図5に示した。
【0094】
結果及び考察:
amyB-hisはGPY液体培養同様に精製できたが、素通り画分のバンドの強度が原液画分とほぼ同じであり、かつ溶出画分の強度が弱かったことから大半がhis-tagを欠損していることが明らかとなった(レーン1−3)。amyB-VHH-hisに関してはVHHに相当するバンドの分子量増加が見られず、また溶出画分に何も存在しなかったことから、his-tagのみならずVHHの部分もプロテアーゼなどにより分解されている可能性が示唆された(レーン4−6)。glaB-hisについてもamyB-his同様に素通り画分のバンドの強度が原液画分とほぼ同じであり、かつ溶出画分の強度が弱かったことから大半がhis-tagを欠損していることが明らかとなった(レーン7−9)。glaB-VHH-hisについてもamyB-VHH-hisと同様にVHHに相当するバンドの分子量増加が見られず、また溶出画分に何も存在しなかったことから、his-tagのみならずVHHの部分もプロテアーゼなどにより分解されている可能性が示唆された(レーン10−12)。フスマ固体培養においてはHis-tagのみならずVHHも分解されることからGPY液体培養と比較してVHH生産には不向きである事が明らかとなった。
【0095】
試験例6:amyB-VHH-his及びglaB-VHH-hisタンパク質のN型糖鎖除去
目的:amyB-VHH-his及びglaB-VHH-hisタンパク質が目的分子量を保持しているかを確認する。
【0096】
方法:
試験例4で得られたHis-tagカラム精製タンパク質amyB-his、amyB-VHH-his、glaB-his、及びglaB-VHH-hisをVIVA SPIN 500 (Sartorius社製)により濃縮し、5μgタンパク質相当をSDS-PAGEにて解析を行った。EndoH処理は、Endoglycosidase H (Streptomyces griseus由来、生化学バイオビジネス株式会社製)により行った。各サンプルはSDS電気泳動用のサンプルバッファー(0.1 M Tris-HCl (pH 6.8)、2% SDS、20%グリセロール、0.2% BPB、0.2M DTT)と1対1で混和し、室温、5分間インキュベートしたのちにSDS-PAGE解析に供した。結果を図6に示した。
【0097】
結果及び考察:
各サンプルともに、Endo H処理をすることにより、低分子化し糖鎖が付加していることが明らかとなった。しかしながらバンドの位置及びその大きさから、幾つかのサンプルについては試験例1と同様に高温でインキュベートする必要が考えられた。
【0098】
試験例7:amyB-VHH-his及びglaB-VHH-hisタンパク質のN型糖鎖除去(高温処理)
目的:amyB-VHH-his及びglaB-VHH-hisタンパク質が目的分子量を保持しているかを確認する。
【0099】
方法:
試験例4で得られたHis-tagカラム精製タンパク質amyB-his、amyB-VHH-his、glaB-his及びglaB-VHH-hisをVIVA SPIN 500 (Sartorius社製)により濃縮し、5μgタンパク質相当をSDS-PAGEにて解析を行った。EndoH処理は、Endoglycosidase H (Streptomyces griseus由来、生化学バイオビジネス株式会社製)により行った。各サンプルはSDS電気泳動用のサンプルバッファー(0.1 M Tris-HCl (pH 6.8)、2% SDS、20% グリセロール、0.2% BPB、0.2M DTT)と1対1で混和し、99℃、5分間インキュベートしたのちにSDS-PAGE解析に供した。結果を図7に示した。
【0100】
結果及び考察:
各サンプルともに、Endo H処理をすることにより、低分子化し糖鎖が付加している事が明らかとなった。amyBをリーダーとしたときはN型糖鎖の付加か少なく、glaBをリーダーとしたときは糖鎖付加により10kDa近い分子量の増加がみられた。またそれぞれの分子量の位置が理論値と一致しており、99℃でインキュベートすることによりVHHの付加による分子量増大が明らかとなった(amyB-VHH-hisはレーン1と3の比較、glaB-VHH-hisはレーン5と7の比較)。
【0101】
試験例8:他のリーダータンパク質の検討、エンドグルカナーゼcelA及びcelB
目的:glaBを超えるVHH生産に最適なリーダータンパク質を見出す。
【0102】
方法:
1.ベクターの構築方法
ベクターはhlyプロモーターが組み込まれた発現プラスミドから「ベクターの調製」に従って調製し、インサートはcelA遺伝子をアスペルギルス・オリゼ由来celA(cDNA型)及びcelB(cDNA型)遺伝子を鋳型として、「インサートの調製」に従ってそれぞれ調製した。「ベクターとインサートのライゲーション」に従い、Phly-celA-his及びPhly-celA-his分泌発現ベクターをそれぞれ構築した。融合タンパク質の分泌発現ベクターPhly-celA-VHH-hisは、インサート1:celA(cDNA型)、インサート2:celB(cDNA型)遺伝子、インサート3:VHH遺伝子(合成遺伝子配列、北海道システムサイエンス社製)を鋳型としたものを、「インサートの調製」に従ってそれぞれ調製した。ベクターとインサート1及び3及びベクターとインサート2及び3同時に混和して、「ベクターとインサートのライゲーション」に従い、Phly-celA-VHH-his及びPhly-celB-VHH-hisを構築した。
【0103】
2.タンパク質の発現分泌回収
「目的遺伝子発現株の取得」に従い、麹菌に1で作成した分泌発現ベクターを麹菌に形質転換し、「目的遺伝子産物の取得」によりサンプルを回収した。
【0104】
3.SDS-PAGE解析
2で得られたサンプル100μLをVIVA SPIN 500 (Sartorius社製)により濃縮し、SDS-PAGEにて解析を行った。結果を図8に示した。
【0105】
結果及び考察:
celB-VHH-his遺伝子導入した独立の4株のサンプル(レーン6−9)は目的産物が良好に生産されている事が観察された。celA-VHH-his遺伝子導入した独立の4株のサンプル(レーン2−5)には目的産物がほとんど生産されていなかった。celBはリーダータンパク質として有望な可能性がある。
【0106】
試験例9:エンドグルカナーゼcelA及びcelBと融合させたVHHタンパク質のHis-tagカラムでの精製結果
目的:
celA-VHH-his及びcelB-VHH-hisタンパク質がhis-tagまで完全に作られているかをHis-tagカラム精製により検証する。
【0107】
方法:
GPY液体培養(試験例8)及びフスマ固体培養(試験例5に従って実施)から得られたcelA-VHH-his及びcelB-VHH-hisサンプルをHis-tagカラムであるHis GraviTrap (GEヘルスケア社製)にて精製を行った。得られたサンプル100μLをVIVA SPIN 500 (Sartorius社製)により濃縮し、SDS-PAGEにて解析を行った。結果を図9に示した。
【0108】
結果及び考察:
フスマ固体培養では目的物タンパク質自体を検出することができなかった。他方GPY液体培養では、溶出画分にのみcelA-VHH-his産物を検出することができた。また、celB-VHH-hisサンプルからはglaB-VHH-hisと同程度の溶出画分サンプル量を得ることができ、glaBと並んでcelBは有望なリーダータンパク質であることが明らかとなった。
【0109】
試験例10:glaB-VHH-hisと抗原hCGとの結合性試験
目的:glaB-VHH-hisの抗原結合能を検証する。
【0110】
方法:
glaB-his、glaB-VHH-hisを臭化シアン活性化セファロース(アマシャムバイオサイエンス社)に付属のプロトコールにより固相化した。glaB-his、glaB-VHH-hisを結合した20μLのセファロース樹脂(10μgのVHHを結合)と10μgのhCG (Human Chorionic Gonadotropin(ヒト絨毛性ゴナドトロピン)、シグマアルドリッチ社)をバッファー(20 mM Tris-HCl 、0.15 M NaCl、pH 8)中、室温で90分間インキュベートした後、3回同じバッファーで樹脂を洗浄した。洗浄した樹脂に10μLのSDS電気泳動用のサンプルバッファー(78 mM Tris-HCl、pH6.8、2.5% SDS、2%グリセロール、1.25 mg/mLブロモフェノールブルー、120 mM β-メルカプトエタノール)を加えて、95℃で15分間インキュベートし、VHHに結合したhCGを溶出した。樹脂を遠心で沈澱させ、上清をSDS電気泳動にかけ、クーマジーブリリアントブルーで染色を行なった。結果を図10に示した。
【0111】
結果及び考察:
素通り画分(レーン1−3)では、活性化セファロースのみ、glaB-hisを結合させたもの、glaB-VHH-hisを結合させたものに大きな差異はなく、hCGは大過剰量添加されていると言える。溶出画分ではglaB-VHH-hisのみに明瞭なバンド(レーン4−6、矢印)が検出されたことから、glaB-VHH-hisはhCGに対して強い結合能をもつことが明らかとなった。
【0112】
試験例11.抗EGFR抗体の麹菌での生産
上皮成長因子受容体(Epidermal Growth Factor Receptor; EGFR)は、細胞の増殖や成長を制御する上皮成長因子(EGF)を認識し、シグナル伝達を行う受容体である。このEGFRに遺伝子増幅や遺伝子変異、構造変化が起きると、発癌、および癌の増殖、浸潤、転移などに関与するようになる。
【0113】
hlyプロモーター支配下で、Lama glama由来の抗EGFRのVHH抗体(GenBank Accession Number: ABX79398.1)のcDNAを麹菌で発現させた。N末端にリーダータンパク質としてamyB, glaB, celA, celBからなる群の1つの遺伝子とLama glama由来の抗EGFRのVHH抗体のcDNAとHisタグをコードする遺伝子の順からなる融合遺伝子をpNMBベクターにサブクローニングし、得られたベクターを、定法であるPEG-カルシウム法(Mol Gen Genet, 218, 99-104, (1989))により、アスペルギルス・オリゼのniaD変異株(独立行政法人産業技術総合研究所特許生物寄託センターにFERM P-17707として寄託済み)を形質転換した。
【0114】
得られた形質転換体をポテトデキストロース培地で胞子形成させ、滅菌水で胞子を回収した。500 ml容三角フラスコに入った100 ml GPY液体培地(2%グルコース、1%ポリペプトン、0.5%イーストエキストラクト、0.1%リン酸1水素2カリウム、0.05%塩化カリウム、0.05%硫酸マグネシウム、0.001%硫酸鉄、0.3%硝酸ナトリウム)に最終胞子濃度1×106/mlとなるように植菌した。30℃3日間の液体培養で目的遺伝子産物が培地中に分泌発現させ、分泌タンパク質をSDS-PAGEで解析した。
【0115】
分泌された目的融合遺伝子産物の培地あたりのタンパク質生産量を決定するために、菌体外のタンパク質総量をBradfordプロテインアッセイ試薬(バイオラッドラボラトリーズ)で測定し、菌体外のタンパク質に対する目的融合遺伝子産物の量比と純度をAgilent Technologies社製の商品名2100 Bioanalyzer(Protein chip 80を使用)で測定した。その結果の生産量と全分泌タンパク質に対する目的融合遺伝子産物の純度を下記表1に示す。対照は、ベクターpNMBのみを形質転換した麹菌形質転換体である。その結果、当該抗EGFRのVHH抗体についても、glaBとcelBをリーダータンパク質に選択することで、良好に分泌発現でき、1528 mg/Lの著量の分泌発現に成功し、その純度は94.2%となった。
【0116】
【表1】

【0117】
次に、celB+Lama glama由来の抗EGFRのVHH抗体+Hisタグ融合タンパク質をNiカラムにより公知の方法で精製し、表面プラズモン共鳴シグナルによる抗体活性を測定した。前記精製タンパク質を、金薄膜がコートされたセンサーチップ表面に固定化した。なお、固定化は、予め金薄膜の表面にカルボキシメチル基を導入しておき、融合タンパク質中のアミノ基と結合させることにより行った。得られたセンサーチップを、表面プラズモン共鳴シグナル解析装置(ビアコア社製、BIACORE X)にセットし、リガンドサンプルとして10〜100 nMのEGFR (ミズーリ州セントルイス、シグマ社)溶液をフローセルに流してリガンド物質を結合させた後、pH2.5のグリシン溶液によって解離させた。この結合・解離に伴う質量変化をリアルタイムで測定し、解析ソフトウウェアBIAevaluation9.1 (ビアコア社)によって解離定数(Kd)を算出したところ14.7 nMであった。
【0118】
なお、コントロール実験として、celB+Hisタグ融合タンパク質のみをセンサーチップに固定化して同様の実験を行ったところ、EGFR(ミズーリ州セントルイス、シグマ社)との結合は全く検出されなかった。よって、Lama glama由来の抗EGFRのVHH抗体は、融合タンパク質の状態で抗原結合活性を有することが判明した。
【0119】
試験例12.抗RR6抗体の麹菌での生産
RR6は、Reactive Red 6という色素である。Lama glama由来の抗EGFRのVHH抗体に代えてLama glama由来の抗RR6のVHH抗体(GenBank Accession Number: AJ236100.1)を使用した以外は試験例11と同様の実験を行った。
【0120】
その結果の生産量と全分泌タンパク質に対する目的融合遺伝子産物の純度を表2に示す。対照は、ベクターpNMBのみを形質転換した麹菌形質転換体である。その結果、当該抗RR6のVHH抗体についても、glaBとcelBをリーダータンパク質に選択することで、良好に分泌発現でき、1624 mg/Lの著量の分泌発現に成功し、その純度は92.6%となった。
【0121】
【表2】
【0122】
次に、celB+Lama glama由来の抗RR6のVHH抗体+Hisタグ融合タンパク質をNiカラムにより公知の方法で精製し、表面プラズモン共鳴シグナルによる抗体活性を測定した。前記精製タンパク質を、金薄膜がコートされたセンサーチップ表面に固定化した。なお、固定化は、予め金薄膜の表面にカルボキシメチル基を導入しておき、融合タンパク質中のアミノ基と結合させることにより行った。得られたセンサーチップを、表面プラズモン共鳴シグナル解析装置(ビアコア社製、BIACORE X)にセットし、リガンドサンプルとして10〜100 nMのRR6 (Reactive Red 6, Procion Rubine MX-B, ICI)溶液をフローセルに流してリガンド物質を結合させた後、pH2.5のグリシン溶液によって解離させた。この結合・解離に伴う質量変化をリアルタイムで測定し、解析ソフトウウェアBIAevaluation9.1(ビアコア社)によって解離定数(Kd)を算出したところ14.7 nMであった。
【0123】
なお、コントロール実験として、celB+Hisタグ融合タンパク質のみをセンサーチップに固定化して同様の実験を行ったところ、RR6との結合は全く検出されなかった。よって、Lama glama由来の抗RR6のVHH抗体は、融合タンパク質の状態で抗原結合活性を有することが判明した。
【0124】
以上すべての実施例を総合すると、glaB又はcelBをリーダータンパク質に選択すれば、すべての抗原に対するVHH抗体を、融合タンパク質として活性型で、著量高純度で分泌発現させることが可能なことが明らかとなった。
【技術分野】
【0001】
本発明は、ラクダ科動物抗体の重鎖可変ドメイン(以下、VHHと呼ぶこともある)の生産方法、並びに当該方法の実施に有用なDNA、糸状菌用発現ベクター及び形質転換体に関する。更に、本発明は、当該方法により得られたVHHを含むタンパク質に関する。
【背景技術】
【0002】
抗体の標準的な構成は2本の軽鎖と2本の重鎖からなる。軽鎖と重鎖は免疫グロブリンドメインにより構成され、軽鎖は2つ(VL、CL)、重鎖は4つ(VH, CH1, CH2, CH3)のドメインからできている。抗体の多様性には軽鎖と重鎖の可変領域の組み合わせが重要であるため、軽鎖、重鎖からなる標準的な抗体フォーマットは普遍的なものとして考えられてきた。しかし、1993年にHamers-Castermanらにより、ラクダ科動物の血液中に重鎖のみからなる抗体(重鎖抗体(Heavy-chain antibody))が多量に存在することが発見された。当該重鎖抗体はCH1ドメインを有していないことも特徴的である。ラクダ抗体のVHドメインはVariable domain of a heavy-chain antibody (VHH)と呼ばれることがある。
【0003】
通常、抗体を利用した最小の抗原認識分子は、VH、VLの2つのドメインをリンカーで結合させた一本鎖抗体(scFv)である。scFvには長いリンカーが必要なことやVHドメインの可溶性が低いことなどの理由から、リフォールディングが難しい分子として知られている。それに対して、ラクダ抗体では1個の免疫グロブリンドメイン(VHドメイン)のみで抗原を認識することができるため、たった1個のドメインからなる抗原認識分子を作製することが可能である。更に、VHHは可溶性が高く高温でも安定であることが証明され、VHHの物性研究により、当該分子がリフォールディング効率の高い分子であることが分かって来ている。
【0004】
このように、VHHは抗体の最も小さい認識ユニットであり、フォールディングが簡便であり、リフォールディング効率が高いという優れた特徴を有している。また、VHHは抗原認識部位(パラトープ)の形状が通常の抗体とは異なり、酵素の活性部位の溝に突き刺さることができるため、酵素の活性を阻害する抗体(中和抗体)として機能できるという特徴も有している。このような利点を活かしたVHHの利用法として、抗体融合酵素、多価抗体、イントラボディ、不活化環境での利用などが提案されている(非特許文献1、特許文献1〜3)。
【0005】
また、VHHに更に優れた性質を付与するために、特許文献4では、ラクダ科抗体VHHドメインに2個のシステイン残基を導入し、ジスルフィド結合を形成させることにより熱安定性を高めたことが報告されている。
【0006】
このような優れた特徴を有するVHHが広く利用されるためには、安価且つ大量にVHHを製造できる方法が開発される必要がある。微生物を用いたVHHの生産系の中でも、酵素の分泌能に優れているという点から麹菌(糸状菌)が特にVHHの製造に適していると考えられる。非特許文献2及び3では、そのような麹菌を用いたVHHの製造が報告されている。
【0007】
非特許文献2では、アスペルギルス・アワモリ(Aspergillus awamori)によりVHHを発現、生産させたことが報告されており、その生産量は培地中に約1.5 mg/lであり、タンパク質の分解を防ぐために培地中にBSAを添加した場合は7.5 mg/lであったことが報告されている。
【0008】
非特許文献3では、ペロオキシダーゼ遺伝子とVHHをコードする遺伝子の融合遺伝子を含む発現カセットを有するアスペルギルス・アワモリにより、当該融合タンパク質を培地中に30 mg/l生産させたことが報告されている。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】特許第3444885号公報
【特許文献2】特許第3660270号公報
【特許文献3】特開2005-170953号公報
【特許文献4】特開2008-212125号公報
【非特許文献】
【0010】
【非特許文献1】萩原義久, 次世代抗体:ラクダ抗体の分子論, BIO INDUSTROY、Vol.25, No.5, 15-22(2008)
【非特許文献2】Joosten V, Gouka RJ, van den Hodel CAMJJ, Verrips CT, Lokmann BC, Appl Microbiol Biotechnol, 66, 384-392(2005)
【非特許文献3】Joosten V, Roelofs MS, van den Dries N, Goosen T, Verrips CT, van den Hondel CAMJJ, Lokman BC, J Biotechnol, 120, 347-359(2005)
【発明の概要】
【発明が解決しようとする課題】
【0011】
しかしながら、非特許文献2及び3におけるVHHの生産量は最大でも培地中に30 mg/lしかなく、工業的なレベルでVHHを大量生産するためには更に生産量を上げる必要がある。
【0012】
そこで、本発明は、生産性が向上したVHHの生産方法を提供することを目的とする。また、本発明は、当該生産方法に用いるための材料、すなわちDNA、糸状菌用発現ベクター及び形質転換体を提供することを目的とする。更に、本発明は、当該方法により得られたVHHを含むタンパク質を提供する。
【課題を解決するための手段】
【0013】
本発明者らは、上記課題を解決するために研究を重ねた結果、VHHを特定のリーダータンパク質と融合させてアスペルギルス・オリゼ(Aspergillus oryzae)により生産させることで、VHHの生産性を向上させることができるという知見を得た。本発明は、これら知見に基づき、更に検討を重ねて完成されたものであり、次のDNA、糸状菌用発現ベクター、形質転換体、VHHの生産方法及びタンパク質を提供するものである。
【0014】
(I) DNA
(I-1) 以下の(a)〜(d)のいずれかのDNA及びVHHをコードするDNAを融合したDNA:
(a)配列番号1に記載される塩基配列からなるDNA
(b)配列番号1に記載される塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、且つグルコアミラーゼ活性を有するタンパク質をコードするDNA
(c)配列番号2に記載される塩基配列からなるDNA
(d)配列番号2に記載される塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、且つエンドグルカナーゼ活性を有するタンパク質をコードするDNA。
(I-2) (I-1)に記載のDNAに更に精製用のアミノ酸配列をコードするDNAを融合したDNA。
(I-3) 前記精製用のアミノ酸配列がHisタグである、(I-2)に記載のDNA。
【0015】
(II) 糸状菌用発現ベクター
(II-1) 糸状菌体内で機能するプロモーター遺伝子及び(I-1)〜(I-3)のいずれかに記載のDNAを含む遺伝子発現カセットを有する糸状菌用発現ベクター。
【0016】
(III) 形質転換体
(III-1) (II-1)に記載の糸状菌用発現ベクターで形質転換されてなるアスペルギルス・オリゼ。
【0017】
(IV) VHHの生産方法
(IV-1) (III-1)に記載のアスペルギルス・オリゼを培地で培養し、当該培地からVHHを含むタンパク質を回収する工程を含む、VHHを含むタンパク質の生産方法。
(IV-2) 前記アスペルギルス・オリゼを液体培地で培養して、前記VHHを含むタンパク質を菌体外に分泌させることにより生成蓄積させることを特徴とする、(IV-1)に記載の方法。
(IV-3) (IV-1)又は(IV-2)に記載の方法において、更に前記VHHを含むタンパク質からVHHに結合したペプチドを切断する工程を有する、VHHの生産方法。
【0018】
(V) タンパク質
(V-1) (IV-1)又は(IV-2)に記載の方法により得られたVHHを含むタンパク質。
【発明の効果】
【0019】
本発明の生産方法によれば、アスペルギルス・オリゼを宿主としてVHHの生産性を大幅に向上させることが出来る。そのため、本発明によれば、優れた特徴を有するVHHの量産化が可能となり、VHHの生産コストを低減することも可能となる。
【図面の簡単な説明】
【0020】
【図1】試験例1のSDS-PAGEの結果の写真である。レーン1−4:室温でインキュベーション、レーン5−8:50℃でインキュベーション、レーン9−12:99℃でインキュベーション、レーン3、4、7、8、11、12:endoH処理
【図2】試験例2のSDS-PAGEの結果の写真である。
【図3】試験例3のSDS-PAGEの結果の写真である。
【図4】試験例4のSDS-PAGEの結果の写真である。レーン1、4、7、10:原液、レーン2、5、8、11:素通り画分、レーン3、6、9、12:溶出画分
【図5】試験例5のSDS-PAGEの結果の写真である。レーン1、4、7、10:原液、レーン2、5、8、11:素通り画分、レーン3、6、9、12:溶出画分
【図6】試験例6のSDS-PAGEの結果の写真である。レーン1、3、5、7:EndoH処理(37℃、24時間)、レーン2、4、6、8:EndoH処理せずに4℃で24時間静置
【図7】試験例7のSDS-PAGEの結果の写真である。レーン1、3、5、7:EndoH処理(37℃、24時間)、レーン2、4、6、8:EndoH処理せずに4℃で24時間静置
【図8】試験例8のSDS-PAGEの結果の写真である。
【図9】試験例9のSDS-PAGEの結果の写真である。レーン1、4、7、10:原液、レーン2、5、8、11:素通り画分、レーン3、6、9、12:溶出画分
【図10】試験例10のSDS-PAGEの結果の写真である。レーン1−3:素通り画分、レーン34−6:溶出画分
【発明を実施するための形態】
【0021】
以下、本発明の実施の形態を詳細に説明する。
【0022】
本発明が対象とする糸状菌としては、長年清酒、醤油、味噌、みりんなどの醸造食品に利用されてきた安全な微生物であり、わが国の産業において利用頻度の高い宿主である点で、麹菌アスペルギルス・オリゼが好ましい。
【0023】
(1)DNA
本発明のDNAは、以下の(a)〜(d)のいずれかのDNA及びVHHをコードするDNAを融合したDNAであることを特徴とする:
(a)配列番号1に記載される塩基配列からなるDNA
(b)配列番号1に記載される塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、且つグルコアミラーゼ活性を有するタンパク質をコードするDNA
(c)配列番号2に記載される塩基配列からなるDNA
(d)配列番号2に記載される塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、且つエンドグルカナーゼ活性を有するタンパク質をコードするDNA。
【0024】
本明細書において、上記(b)又は(d)のDNAによりコードされるタンパク質を改変体と呼ぶことがある。
【0025】
(1-1)リーダータンパク質
本発明が対象とするVHHと融合させて発現させるためのリーダータンパク質は、配列番号3に記載されるアミノ酸配列を有するグルコアミラーゼ(glaB)若しくはその改変体、又は配列番号4に記載されるアミノ酸配列を有するエンドグルカナーゼ(CelB)若しくはその改変体である。リーダータンパク質としては、配列番号4に記載されるアミノ酸配列を有するエンドグルカナーゼ(CelB)又はその改変体がより好ましい。なお、配列番号3で表されるアミノ酸配列をコードする遺伝子の塩基配列は配列番号1に表され、配列番号4で表されるアミノ酸配列をコードする遺伝子の塩基配列は配列番号2に表される。
【0026】
VHHにこれらのリーダータンパク質を融合させて糸状菌で発現させることで、VHHの生産性を大幅に向上させることが可能となる。
【0027】
上記のストリンジェントな条件とは、例えば、65℃で5×SSC溶液(1倍濃度のSSC溶液の組成は、150 mM塩化ナトリウム、15 mMクエン酸ナトリウム)中でハイブリダイズさせ、更に0.1%のSDSを含有する0.5×SSC溶液で65℃で洗浄する条件を意味する。ストリンジェントな条件下でのハイブリダイゼーションの各操作は、「Molecular Cloning (Third Edition)」(J. Sambrook & D. W. Russell, Cold Spring Harbor Laboratory Press, 2001)に記載されている方法等、従来公知の方法で行うことができる。通常、温度が高いほど、塩濃度が低いほどストリンジェンシーは高くなる。
【0028】
ストリンジェントな条件下でハイブリダイズするDNAは、通常、プローブとして使用するDNAの塩基配列と一定以上の同一性を有し、その同一性は、例えば70%以上、好ましくは80%以上、更に好ましくは90%以上、特に好ましくは95%以上が挙げられる。塩基配列の同一性は、市販の又は電気通信回線(インターネット)を通じて利用可能な解析ツールを用いて算出することができる。塩基配列の同一性(%)は、当該分野で慣用のプログラム(例えば、BLAST、FASTA等)を初期設定で用いて決定することができる。
【0029】
グルコアミラーゼ活性は今井ら(1993)(生物工学会誌: seibutsu-kogaku kaishi 71(2), 93-97, 1993-03-25)の報告に記載の方法により、エンドグルカナーゼ活性は特開2003-164284号公報の段落[0017]に記載の方法により測定することができる。
【0030】
このようなリーダータンパク質の改変体の製造は常法に従って行うことができる。
【0031】
(1-2)ラクダ科動物抗体の重鎖可変ドメイン(VHH)
本発明で使用するのは、ラクダ科動物由来抗体のフラグメントである重鎖の可変ドメイン(VHH)である。ラクダ科動物としては、特に限定されないが、例えば、フタコブラクダ、ヒトコブラクダ、ラマ、アルパカ、ビクーナ、グアナコ等が挙げられる。抗体としては、軽鎖を有さず、CH1ドメインを有さない重鎖のみからなるラクダ科動物由来の抗体であれば特に限定されない。また、VHHの抗原の種類も特に限定されず、どのような抗原を対象とするかはVHHの使用用途に応じて適宜決定される。
【0032】
VHHとしては、例えば、配列番号5、配列番号6、配列番号7で表されるアミノ酸配列からなるVHHが挙げられる。
【0033】
本発明におけるVHHは、天然に存在するものに加えて、天然のVHHのアミノ酸配列に1個以上のアミン酸を置換、欠失及び/又は付加したものも含む。
【0034】
特定の抗原に対するVHHの取得方法としては、ラクダの免疫、ファージディスプレイによる選択、B細胞ディスプレイによる選択、酵母細胞表層ディスプレイによる選択など複数あるが、抗体ファージライブラリーからの特異的な抗体の単離法であるバイオパニングが有効である。バイオパニングは、ラムダgt11などの抗体を提示できるファージライブラリーを作成し、目的の抗原を固定化したものと複数回接触させ、特異的な抗体を有するファージを濃縮選択する方法である(Phage Display, A Laboratory Manual, Cold Spring Harbor Laboratory Press 2001)。
【0035】
(1-3)精製用タグ
本発明のDNAには、リーダータンパク質をコードするDNA及びVHHをコードするDNAに更に精製用のアミノ酸配列をコードするDNAが融合されていても良い。精製用のアミノ酸配列としては、例えばHisタグ、GST、MBP(マルトース結合タンパク質)などが挙げられ、好ましくはHisタグである。このような精製用のアミノ酸配列がVHHと結合していることで、アフィニティークロマトグラフによるVHHの精製が可能になる。
【0036】
上記の各DNAの順序は、本発明の効果が得られるものであればどのような順序であってもよいが、好ましくは5'末端からリーダータンパク質→VHH、又はリーダータンパク質→VHH→精製用アミノ酸配列の順である。
【0037】
また、発現後に目的とするVHHを容易に回収できるように、ペプチド結合が切断可能な認識部位を介在させて、上記の各DNAを融合させても良い。該認識部位としては、プロセッシング酵素、消化酵素などのプロテアーゼで切断可能なアミノ酸配列(2以上のアミノ酸からなる)、システインやメチオニンなどの化学修飾剤、超音波、レーザー、熱などの物理的開裂により切断可能なアミノ酸などが挙げられる。認識部位を切断可能な消化酵素(プロセッシング酵素)としては、好ましくは第Xa因子、トロンビン、レニン、トリプシン、V8プロテアーゼ、Pseudomonasエンドプロテアーゼ、Arthrobacterリシルエンドペプチダーゼ等が挙げられ、認識部位としては、例えば第Xa因子認識配列であるIle-Glu-Gly-Argが挙げられる。化学修飾剤としては、Metを認識するCNBr (Cyanogen bromide)、Asn、Asp、Gluを認識する希塩酸、及びCysを認識するDMAP-CNが挙げられる。
【0038】
(2)遺伝子発現カセット
本発明における遺伝子発現カセットは、糸状菌体内で機能するプロモーター遺伝子と上記DNAを含むことを特徴とする。
【0039】
(2-1)プロモーター
プロモーターは、糸状菌体内、特にアスペルギルス・オリゼの菌体内でプロモーターとして機能するものであればよい。かかるプロモーターとして具体的には、例えばα−アミラーゼ遺伝子プロモーターamyB(Biosci Biotechnol Biochem, 56, 1849-1853(1992))、グルコアミラーゼ遺伝子プロモーターglaA(Gene, 108, 145-150(1992))、α−グルコシダーゼ遺伝子プロモーターagdA(Curr Genet, 30, 432-438(1996))、マンガンSOD遺伝子プロモーターsodM(特開2000-224381号公報)、チロシナーゼ遺伝子プロモーターmelO(特開2001-046078号公報)、グルコアミラーゼ遺伝子プロモーターglaB(特開2000-245465号公報、特開平11-243965号公報)、hlyプロモーター(特開2009-296958号公報)等が挙げられる。中でも好ましいのは、hlyプロモーターである。
【0040】
遺伝子発現カセットにおけるプロモーターの位置は、糸状菌体内でプロモーターが機能し、所望のVHHが発現し産生できる位置であれば、特に限定されないが、通常、VHH遺伝子配列の上流域に配置される。
【0041】
(2-2)ターミネーター
ターミネーターは、糸状菌体内、特にアスペルギルス・オリゼの菌体内でターミネーターとして機能するものであれば良い。ターミネーターとしては、例えばα−アミラーゼ遺伝子のターミネーター、又はグルコアミラーゼ(glaB)ターミネーター(Gene. 207, 127-134(1998))等を挙げることができる。
【0042】
本発明の糸状菌用遺伝子発現カセットは、これらのプロモーター、リーダータンパク質をコードするDNA、VHHをコードするDNA、及びターミネーターが、直接連結されてなるものであってもよいが、それらの間に1〜2000塩基程度のヌクレオチドが挟まれていてもよい。例えば、目的とするVHHをコードするDNAのオープンリーディングフレームの上流に糸状菌、好ましくはアスペルギルス・オリゼの既知分泌タンパク質の遺伝子を配置しても良く、そうすることにより、目的とするVHHは、上記分泌タンパク質との融合タンパク質として発現産生され、斯くして産生された融合タンパク質は菌体外に分泌される。また、その他のヌクレオチドとして、通常ベクターが備える制限酵素切断部位などを備えていてもよい。
【0043】
なお、そのような分泌タンパク質のアスペルギルス糸状菌由来のものとしては、α−アミラーゼ、グルコアミラーゼ、α−グルコシダーゼ、エンドグルカナーゼ、セロビオハイドロラーゼ、β−グルコシダーゼ、酸性プロテアーゼ、中性プロテアーゼ、アルカリプロテアーゼ、カルボキシペプチダーゼ、リパーゼ、ホスホリパーゼ、キシラナーゼ、ガラクトシダーゼ、フラクトフラノシダーゼ、キシロシダーゼ、ペクチンリアーゼ、フィターゼなどが知られている。
【0044】
(3)糸状菌用発現ベクター
本発明の糸状菌用発現ベクターは、上記遺伝子発現カセットを有することを特徴とする。
【0045】
本発明の糸状菌用発現ベクターには、選択マーカー遺伝子が含まれていても良く、コトランスフォーメーションを行う場合は、当該糸状菌用発現ベクターとは別のベクターに選択マーカー遺伝子が存在していれば良い。
【0046】
本発明の糸状菌用発現ベクターは、これらの選択マーカー遺伝子及び遺伝子発現カセットが、直接連結されてなるものであってもよいが、それらの間に1〜2000塩基程度のヌクレオチドが挟まれていてもよい。
【0047】
また、本発明には、直鎖状の糸状菌用発現ベクター及び環状の糸状菌用発現ベクターの両者が含まれるが、(1)構築したプラスミドから1ステップのPCRで取得できるなど、環状の発現ベクターと比べて少ないステップで調製することができること、(2)宿主としてロイシン要求性変異株を使用する場合に、当該変異を直接相補することができること、及び(3)形質転換効率が環状の発現ベクターと比べて高いという点から、好ましくは直鎖状の糸状菌用発現ベクターである。
【0048】
(3-1)選択マーカー遺伝子
選択マーカー遺伝子は、糸状菌体内、特にアスペルギルス・オリゼの菌体内で選択マーカーとして機能するもの、すなわち発現できるものであれば良い。かかる選択マーカー遺伝子として、例えば、niaD(Biosci. Biotechnol. Biochem., 59, 1795-1797(1995))、argB(Enzyme Microbiol Technol, 6, 386-389(1984))、sC(Gene, 84, 329-334(1989))、ptrA(Biosci Biotechnol Biochem, 64, 1416-1421(2000))、pyrG(Biochem Biophys Res Commun, 112, 284-289(1983))、amdS(Gene, 26, 205-221(1983))、オーレオバシジン耐性遺伝子(Mol Gen Genet, 261, 290-296(1999))、ベノミル耐性遺伝子(Proc Natl Acad Sci USA, 83, 4869-4873(1986))、及びハイグロマイシン耐性遺伝子(Gene, 57, 21-26(1987))から選ばれるマーカー遺伝子を挙げることができる。
【0049】
これらの選択マーカー遺伝子は、上記文献に記載された配列に基づいて設計されたプライマーを用いて、アスペルギルス属糸状菌の染色体DNAを鋳型としてPCRを行う方法、上記文献に記載された配列に基づいて設計されたプローブを用いてアスペルギルス属糸状菌の染色体DNAライブラリーからハイブリダイゼーションにより取得する方法などにより容易に入手することができる。また、宿主の糸状菌として、例えばロイシン要求性などの栄養要求性変異株を用いる場合、選択マーカー遺伝子として当該栄養要求性を相補する野生型遺伝子を用いることもできる。例えば、宿主がロイシン要求性変異株である場合、そのロイシン要求性を相補できる選択マーカー遺伝子を用いることができ、かかる遺伝子としてはβ−イソプロピルリンゴ酸デヒドロゲナーゼをコードするアスペルギルス・ニジュランス由来の遺伝子ANleu2及びアスペルギルス・オリゼ由来の遺伝子leu2を挙げることができる。なお、これらの場合、宿主として用いる糸状菌は、選定された選択マーカーについての機能的遺伝子を有していない株を用いる必要がある。
【0050】
(4)形質転換体
本発明の形質転換体は、上記糸状菌用発現ベクターでアスペルギルス・オリゼを形質転換したものである。
【0051】
アスペルギルス・オリゼを宿主とすることは、高いタンパク質生産能及び醸造微生物としての安全性の点で好ましい。なお、宿主として用いるアスペルギルス・オリゼは、野生株に限らず、変異株であってもよい。かかる変異株としては、前述する選択マーカーに関する機能的遺伝子を有さないように変異してなる株を挙げることができ、具体的には正常にロイシンの生合成ができないようにロイシン生合成を担う遺伝子が変異又は欠失しているロイシン要求性変異株:正常にアルギニンの生合成ができないようにアルギニン生合成を担う遺伝子が変異又は欠失しているアルギニン要求性変異株:正常にメチオニンの生合成ができないようにメチオニン生合成を担う遺伝子が変異又は欠失しているメチオニン要求性変異株:正常にヒスチジンの生合成ができないようにヒスチジン生合成を担う遺伝子が変異又は欠失しているヒスチジン要求性変異株:正常に硝酸が資化できないように硝酸資化性遺伝子が変異又は欠失しているniaD変異株:正常に硝酸イオンが資化できないように硝酸イオン資化性遺伝子が欠失しているsC変異株などを挙げることができる。
【0052】
宿主の形質転換方法は、特に限定されず、従来公知の方法を用いて行うことができる。例えば、Cohenらの方法(塩化カルシウム法)(Proc. Natl. Acad. Sci. USA, 69:2110 (1972))、プロトプラスト法(Mol. Gen. Genet., 168:111 (1979))、コンピテント法(J. Mol. Biol., 56:209 (1971))、エレクトロポレーション法等を挙げることができる。
【0053】
(5)VHHの生産方法
本発明のVHHを含むタンパク質の生産方法は、上記のアスペルギルス・オリゼを培地で培養し、当該培地からVHHを含むタンパク質を回収する工程を含むことを特徴とする。
【0054】
本発明の方法によれば、VHH(又はVHHを含むタンパク質)を培地中に1000 mg/l以上、好ましくは1500 mg/l以上生産することが可能となる。
【0055】
本発明において、「VHHを含むタンパク質」とはVHHにリーダータンパク質、精製用アミノ酸配列等が融合したタンパク質のことを意味している。
【0056】
培養は、固体培地を用いる固体培養、又は液体培地を用いる液体培養のいずれでも行うことができるが、好ましくは液体培地を用いた液体培養である。
【0057】
固体培地としては、アスペルギルス・オリゼの培養に使用される公知の固体培地を制限なく使用することができる。固体培地とはその固形の支持担体が栄養源を含むか、又は固形の支持担体に栄養源が添加されものであり、そこにアスペルギルス・オリゼが生育できる固形培地を指し示す。このような固体培地として主に、フスマ(小麦などの穀物の殻)、デンプン粉末、米・小麦・大豆の生あるいは蒸したもの、更にはメンブレンや多孔質の人工物(例えば園芸に使われるバーミキュライト)等に栄養源を添加したもの等が挙げられる。特にフスマ及び蒸した米が好ましい。
【0058】
また、液体培地は、アスペルギルス・オリゼの培養に使用される公知の液体培地を制限なく使用できる。例えば、用いられる培地としては、炭素源としてグルコース、マルトース、フルクトース、グリセロール、スターチなどの炭水化物を含有するものが挙げられる。また、無機もしくは有機窒素源(例えば、硫酸アンモニウム、塩化アンモニウム、カゼインの加水分解物、酵母抽出物、ポリペプトン、バクトトリプトン、ビーフ抽出物等)を含有するものも挙げられる。これらの炭素源及び窒素源は、純粋な形で使用する必要はなく、純度の低いものも微量の生育因子や無機栄養素を豊富に含んでいるので有利である。さらに所望により、他の栄養源[例えば、無機塩(例えば、二リン酸ナトリウム、二リン酸カリウム、リン酸水素二カリウム、塩化マグネシウム、硫酸マグネシウム、塩化カルシウム)、ビタミン類(例えば、ビタミンB1)、抗生物質(例えば、アンピシリン、カナマイシン)など]を培地中に添加してもよい。具体的な液体培地としては、例えば、ポテトデキストロース培地(ニッスイ社)、最少培地(2%グルコース(又はスターチ)、0.3% NaNO3、0.2% KCl、0.1% KH2PO4、0.05% MgSO4、0.002% FeSO4、pH6.0)、GPY培地(グルコース2%、ペプトン1%、酵母抽出物0.5%)等を挙げることができる。なお、これらは1.5%程度の寒天を添加することで、固体培地として調製することもできる。
【0059】
本発明のアスペルギルス・オリゼの培養は、通常pH5.5〜8.5、好ましくはpH6〜8のpH条件;通常25〜42℃、好適には30〜37℃の温度条件で行うことができる。培養時間は、その他の条件によって異なるが、通常2〜7日間程度、好ましくは3〜5日間程度を挙げることができる。
【0060】
本発明の方法によれば、目的とするVHHを含むタンパク質を菌体外に生成することもできる。その場合、VHHを含むタンパク質の取得に際しては、培養上清を回収すればよい。また、寒天培地などのように固体培地を使用する場合も培養上清としての寒天培地を回収すれば良い。さらに、これらの上清を、公知のタンパク質精製方法、例えばイオン交換、疎水、ゲルろ過などの各種クロマトグラフィーに供することにより目的とするVHHを含むタンパク質を単離し回収することができる。VHHを含むタンパク質が精製用のアミノ酸配列と結合している場合は、アフィニティークロマトグラフにより精製を行うことができる。
【0061】
本発明の方法で得られたVHHを含むタンパク質は、リーダータンパク質を含む形であっても抗原結合活性を有している。しかしながら、本発明の方法では、前記の2工程に加えて、培地から回収したVHHを含むタンパク質からリーダータンパク質(及び精製用のアミノ酸配列)を切断する工程を行うことによって、VHHのみを取得しても良い。
【実施例】
【0062】
以下、実施例を示して本発明をより詳細に説明するが、本発明はこれらに限定されるものではない。なお、一般的な実験方法は、実験書(サムブルックら(Sambrook, J.)のMolecular Cloning:A Laboratory Manual 第3版)に従った。
【0063】
試験例で使用した遺伝子の塩基配列及びアミノ酸配列
・選択マーカー
硝酸還元酵素遺伝子niaD<配列番号8>
・プロモーター
hlyプロモーター<配列番号9>
・ターミネーター
glaBターミネーター<配列番号10>
・リーダータンパク質
glaBss-GOD<配列番号11、12>、amyB<配列番号13、14>、glaB<配列番号1、3>、celA<配列番号15、16>、celB<配列番号2、4>(すべてストップコドンなし)
・VHH
試験例1〜10で使用したVHH<配列番号17、5>、試験例11で使用した抗EGFRのVHH<配列番号18、6>、試験例12で使用した抗R66のVHH<配列番号19、7>(すべてストップコドンなし)
【0064】
糸状菌用発現ベクターの構築
<niaD遺伝子の大腸菌ベクターへの組み込み>
麹菌アスペルギルス・オリゼ由来の硝酸還元酵素遺伝子niaDをPstI-HindIII断片となるようにプライマーA(5’-aactgcagaacaggccccaaattcaattaattgca-3’:配列番号20)とプライマーB(5’-cccaagctttggatttcctacgtcttcaatacaaacc-3’:配列番号21)を用いて麹菌ゲノムDNAを鋳型としてLA-Taq(宝ホールディングス)を用いて以下の条件のPCRにより増幅した。
PCR条件
・ 96℃ (5分間)を1サイクル
・ 96℃ (20秒間)、60℃ (30秒間)、72℃ (5分間) を30サイクル
・ 72℃ (7分間)を1サイクル
得られたPCR増幅産物を制限酵素PstI-HindIIIで37℃で処理後、アガロースゲル電気泳動で切り出した。切り出しは、QIAquick Gel Extraction kit (QIAGEN)を用いた。大腸菌プラスミドpUC119(宝ホールディングス)にDNA Ligation kit ver.1(宝ホールディングス)を用いてライゲーションし、大腸菌(E. coli)JM109株に形質転換した。その結果、niaDマーカーがサブクローニングされたプラスミドpNIA2を得た。プラスミドpNIA2は、PstI、SalI部位の両方に遺伝子をunique siteとして導入できる。
【0065】
<glaBターミネーターの組み込み>
麹菌由来His-tag付きglaBターミネーターの遺伝子のpNIA2への挿入を試みた。プライマーC(5'-acgcgtcgacCACCATCACCACCACCACTAAATGTACTTTCCAGTGCGTGTAGTCTACTCTG-3'配列番号22)とプライマーD(5'-acgcctcgagCTGCAGATCGGCTGAAGTTAGGAGCGGCCATTGTC-3'配列番号23)を用いて麹菌ゲノムDNAを鋳型としてLA-Taq(宝ホールディングス)を用いて以下の条件のPCRにより増幅した。
PCR条件
・ 96℃ (5分間)を1サイクル
・ 96℃ (20秒間)、60℃ (30秒間)、72℃ (5分間) を30サイクル
・ 72℃ (7分間)を1サイクル
得られたPCR増幅産物を制限酵素SalI-XhoIで37℃で処理後、アガロースゲル電気泳動で切り出した。切り出しは、QIAquick Gel Extraction kit (QIAGEN)を用いた。大腸菌プラスミドpNIA2のSalI部位にDNA Ligation kit ver.1(宝ホールディングス)を用いてライゲーションし、大腸菌(E. coli)JM109株に形質転換した。その結果、His-tag付きglaBターミネーターがサブクローニングされたプラスミドpNIATを得た。プラスミドpNIATは、PstI、SalI部位の両方に遺伝子をunique siteとして導入できる。
【0066】
<hlyプロモーターの組み込み>
麹菌由来hlyプロモーターの遺伝子のpNIATへの挿入を試みた。プライマーE(5'-aaCTGCAGGCAGATGTAGCCGTGGCACCACAA-3'配列番号24)とプライマーF(5'-acgcgtcgacGGTGTTGTGGTGTGAAGGGTGATTGATGTGAGACCG-3'配列番号25)を用いて麹菌ゲノムDNAを鋳型としてLA-Taq(宝ホールディングス)を用いて以下の条件のPCRにより増幅した。
PCR条件
・ 96℃ (5分間)を1サイクル
・ 96℃ (20秒間)、60℃ (30秒間)、72℃ (5分間) を30サイクル
・ 72℃ (7分間)を1サイクル
得られたPCR増幅産物を制限酵素SalI-PstIで37℃で処理後、アガロースゲル電気泳動で切り出した。切り出しは、QIAquick Gel Extraction kit (QIAGEN)を用いた。大腸菌プラスミドpNIATのPstI-SalI部位にDNA Ligation kit ver.1(宝ホールディングス)を用いてライゲーションし、大腸菌(E. coli)JM109株に形質転換した。その結果、hlyプロモーターがサブクローニングされたプラスミドpNMBを得た。プラスミドpNMBは、SalI部位に麹菌で発現させたいタンパク質をコードする遺伝子をunique siteとして導入できる。
【0067】
<目的遺伝子の組み込み>
(1)ベクターの調製
pNMBを制限酵素SalIで37℃処理後、dNTPを最終10 mMとなるように添加してT4 DNAポリメラーゼ(宝ホールディングス)で37℃1時間処理した。さらに、バクテリア由来アルカリホスファターゼ(宝ホールディングス)で50℃、30分反応させた。得られた反応物をPCRクリーンアップカラム(プロメガ社製)で処理溶出させ、ベクターとした。
【0068】
(2)インサートの調製
麹菌で発現させたいタンパク質をコードする遺伝子を目的遺伝子として、上記ベクターにサブクローニングするインサートとした。
【0069】
目的遺伝子の開始コドンから下流30bpのセンス鎖のプライマー(5'末端をリン酸化)と、目的遺伝子の終止コドンから上流30bpのアンチセンス鎖のプライマー(5'末端をリン酸化)を用いて、目的遺伝子の起源生物由来のゲノムDNAを鋳型として、pfu Taqポリメラーゼ(東洋紡績株式会社)を用いて下記条件でPCRを行った。
PCR条件
・ 96℃ (5分間)を1サイクル
・ 96℃ (20秒間)、60℃ (30秒間)、 72℃ (5分間) を30サイクル
・ 72℃ (7分間)を1サイクル
得られたPCR断片をPCRクリーンアップカラム(プロメガ社製)で処理溶出させ、インサートとした。
【0070】
(3)ベクターとインサートのライゲーション
モル数で、ベクター:インサート=1:4となるように添加して、DNA Ligation kit ver.1(宝ホールディングス)を用いてライゲーションし、大腸菌(E. coli)JM109株に形質転換した。形質転換体からプラスミドを調製し、目的遺伝子の読み枠がhlyプロモーターに対して正方向にサブクローニングされ、なお且つ翻訳時の読み枠がフレームシフトしておらず、更にHis-tagの読み枠と合致しているプラスミドを目的遺伝子発現プラスミドとして調製した。
【0071】
目的遺伝子発現株の取得
定法であるPEG-カルシウム法(Mol Gen Genet, 218, 99-104, (1989))により、上記目的遺伝子発現プラスミドを用いて、アスペルギルス・オリゼのniaD変異株(独立行政法人産業技術総合研究所特許生物寄託センターにFERM P-17707として寄託されている)を形質転換した。硝酸を単一窒素源とするツアペクドックス(Czapek-Dox)培地(2%グルコース、0.1%リン酸1水素2カリウム、0.05%塩化カリウム、0.05%硫酸マグネシウム、0.001%硫酸鉄、0.3%硝酸ナトリウム)で生育できる株を選択することにより、目的遺伝子発現プラスミドを保持する形質転換体を複数得た。
【0072】
目的遺伝子産物の取得
上記形質転換体をポテトデキストロース培地で胞子形成させ、滅菌水で胞子を回収した。500 ml容三角フラスコに入った100 ml GPY液体培地(2%グルコース、1%ポリペプトン、0.5%イーストエキストラクト、0.1%リン酸1水素2カリウム、0.05%塩化カリウム、0.05%硫酸マグネシウム、0.001%硫酸鉄、0.3%硝酸ナトリウム)に最終胞子濃度1×106/mlとなるように植菌した。30℃3日間の液体培養で目的遺伝子産物が培地中に分泌発現し、当該培養液をサンプルとした。
【0073】
サンプルの更なる精製は、His GraviTrapカラム(GEヘルスケア社)を用い、所定のマニュアル通りに5-15mLのサンプルをカラムにアプライし自然落下により吸着(20 mMリン酸ナトリウム, 500 mM NaCl, 45 mMイミダゾール, pH 7.4)させ、カラムに吸着せずに出てきた画分を素通り画分とした。10mLで洗浄(20 mMリン酸ナトリウム, 500 mM NaCl, 45 mMイミダゾール, pH 7.4)した後、イミダゾール溶液(20 mMリン酸ナトリウム, 500 mM NaCl, 500 mMイミダゾール, pH 7.4)を同様にアプライし、出てきた画分を溶出画分とした。
【0074】
試験例1:アスペルギルス・ニガー(Aspergillus niger)由来グルコースオキシダーゼタンパク質とのVHHの融合タンパク質の培地中への分泌生産の結果
目的:モデルとして麹菌には類縁体が存在しないアスペルギルス・ニガー由来グルコースオキシダーゼタンパク質(GOD)をリーダータンパク質としてVHHを融合させて、麹菌でGOD-VHH融合タンパク質を培地中への分泌生産させることを試みる。
【0075】
方法:
1.ベクターの構築方法
ベクターはhlyプロモーターが組み込まれた発現プラスミドから「ベクターの調製」に従って調製し、インサートはglaBss-GOD遺伝子を合成遺伝子配列(北海道システムサイエンス社製)を鋳型として、「インサートの調製」に従って調製した。「ベクターとインサートのライゲーション」に従い、Phly-glaBss-GOD-his分泌発現ベクターを構築した。融合タンパク質の分泌発現ベクターPhly-glaBss-GOD-VHH-hisは、インサート1:glaBss-GOD遺伝子を鋳型としたものと、インサート2:VHH遺伝子を合成遺伝子配列(北海道システムサイエンス社製)を鋳型としたものを、「インサートの調製」に従ってそれぞれ調製した。ベクターとインサート1及び2を同時に混和して、「ベクターとインサートのライゲーション」に従い、Phly-glaBss-GOD-VHH-hisを構築した。
【0076】
2.タンパク質の発現分泌回収
「目的遺伝子発現株の取得」に従い、麹菌に1で作成した分泌発現ベクターを麹菌に形質転換し、「目的遺伝子産物の取得」によりサンプルを回収した。
【0077】
3.SDS-PAGE解析
2で得られたサンプルをVIVA SPIN 500 (Sartorius社製)により濃縮し、5μgタンパク質相当をSDS-PAGEにて解析を行った。EndoH処理は、Endoglycosidase H (Streptomyces griseus由来、生化学バイオビジネス株式会社製)により行った。各サンプルはSDS電気泳動用のサンプルバッファー(0.1 M Tris-HCl (pH 6.8)、2% SDS、20% glycerol、0.2% BPB、0.2M DTT)と1対1で混和し、室温、50℃又は99℃で5分間インキュベートした後にSDS-PAGE解析に供した。結果を図1に示した。
【0078】
結果及び考察:
インキュベートの温度は、高温になるほどバンドがシャープになり、99℃が最適であると考えられた。また、EndoH処理を行ったサンプルは全て72kDa近傍にバンドが検出されたことから、おおよそglaBss-GOD-hisをコードする部分は生産できたと考えられる。しかしならが、glaBss-GOD-VHH-hisもほぼ同じ位置にバンドが検出されたことから、VHHの部分だけ取り除かれている可能性がある。glaBss-GOD-VHH-hisを泳動したレーン全てにVHH単独の分子量に近い17kDa近傍にバンドが検出されていることから(矢印)、glaBss-GOD-VHH-hisはglaBss-GODとVHH-hisに分解されて生産されている或いは生産できる可能性が示唆された。
【0079】
試験例2:アスペルギルス・オリゼ由来α-アミラーゼ(amyB)とのVHHの融合タンパク質の培地中への分泌生産の結果
目的:アスペルギルス・オリゼ由来α-アミラーゼ(amyB)をリーダータンパク質としてVHHを融合させて、麹菌でamyB-VHH融合タンパク質を培地中への分泌生産させることを試みる。
【0080】
方法:
1.ベクターの構築方法
ベクターはhlyプロモーターが組み込まれた発現プラスミドから「ベクターの調製」に従って調製し、インサートはamyB遺伝子をアスペルギルス・オリゼ由来amyB(cDNA型)遺伝子を鋳型として、「インサートの調製」に従って調製した。「ベクターとインサートのライゲーション」に従い、Phly-amyB-his分泌発現ベクターを構築した。融合タンパク質の分泌発現ベクターPhly-amyB-VHH-hisは、インサート1:amyB(cDNA型)遺伝子を鋳型としたものと、インサート2:VHH遺伝子を合成遺伝子配列(北海道システムサイエンス社製)を鋳型としたものを、「インサートの調製」に従ってそれぞれ調製した。ベクターとインサート1及び2を同時に混和して、「ベクターとインサートのライゲーション」に従い、Phly-amyB-VHH-hisを構築した。
【0081】
2.タンパク質の発現分泌回収
「目的遺伝子発現株の取得」に従い、麹菌に1で作成した分泌発現ベクターを麹菌に形質転換し、「目的遺伝子産物の取得」によりサンプルを回収した。
【0082】
3.SDS-PAGE解析
2で得られたサンプル100μLをVIVA SPIN 500 (Sartorius社製)により濃縮し、SDS-PAGEにて解析を行った。結果を図2に示した。
【0083】
結果及び考察:
amyB-his遺伝子導入した独立の4株のサンプル(レーン1−4)は良好にamyBが生産されていることが観察された。amyB-VHH-his遺伝子導入した独立の4株のサンプル(レーン5−8)にはVHHの分子量増加に相当する72kDaの位置にバンドが観察され(中央の矢印)、融合タンパク質の形でVHHが生産できたことを示唆する結果を得た。しかし、amyB-hisと比較してamyB-VHH-hisのバンドのシグナル強度は弱く、生産性は低かった。
【0084】
試験例3:アスペルギルス・オリゼ由来グルコアミラーゼ(glaB)とのVHHの融合タンパク質の培地中への分泌生産の結果
目的:アスペルギルス・オリゼ由来グルコアミラーゼ(glaB)をリーダータンパク質としてVHHを融合させて、麹菌でglaB-VHH融合タンパク質を培地中への分泌生産させることを試みる。
【0085】
方法:
1.ベクターの構築方法
ベクターはhlyプロモーターが組み込まれた発現プラスミドから「ベクターの調製」に従って調製し、インサートはglaB遺伝子をアスペルギルス・オリゼ由来glaB(cDNA型)遺伝子を鋳型として、「インサートの調製」に従って調製した。「ベクターとインサートのライゲーション」に従い、Phly-glaB-his分泌発現ベクターを構築した。融合タンパク質の分泌発現ベクターPhly-glaB-VHH-hisは、インサート1:glaB(cDNA型)遺伝子を鋳型としたものと、インサート2:VHH遺伝子を合成遺伝子配列(北海道システムサイエンス社製)を鋳型としたものを、「インサートの調製」に従ってそれぞれ調製した。ベクターとインサート1及び2を同時に混和して、「ベクターとインサートのライゲーション」に従い、Phly-glaB-VHH-hisを構築した。
【0086】
2.タンパク質の発現分泌回収
「目的遺伝子発現株の取得」に従い、麹菌に1で作成した分泌発現ベクターを麹菌に形質転換し、「目的遺伝子産物の取得」によりサンプルを回収した。
【0087】
3.SDS-PAGE解析
2で得られたサンプル100μLをVIVA SPIN 500 (Sartorius社製)により濃縮し、SDS-PAGEにて解析を行った。結果を図3に示した。
【0088】
結果及び考察:
glaB-his遺伝子導入した独立の4株のサンプル(レーン1−4)は良好にglaBが生産されていることが観察された。しかしながら、3と4レーンのバンドはスプリットしており、一部hisタグが切断された可能性が示唆された。glaB-VHH-his遺伝子導入した独立の4株のサンプル(レーン5−8)にはVHHの分子量増加に相当する80kDaの位置にバンドが観察され(中央の矢印)、融合タンパク質の形でVHHが生産できたことを示唆する結果を得た。5−8の全てのレーンもスプリットしており、一部hisタグが切断された可能性が示唆された。しかし、glaB-hisと比較してglaB-VHH-hisのバンドのシグナル強度は弱かったが、全体としての生産量は高かった。
【0089】
試験例4:α-アミラーゼ及びグルコアミラーゼと融合させたVHHタンパク質のHis-tagカラムでの精製結果
目的:amyB-VHH-his及びglaB-VHH-hisタンパク質がhis-tagまで完全に作られているかをHis-tagカラム精製により検証する。
【0090】
方法:
試験例2と3で用いたamyB-his、amyB-VHH-his、glaB-his及びglaB-VHH-hisサンプルをHis-tagカラムであるHis GraviTrap (GEヘルスケア社製)にて精製を行い、各サンプル100μLをVIVA SPIN 500 (Sartorius社製)により濃縮し、SDS-PAGEにて解析を行った。結果を図4に示した。
【0091】
結果及び考察:
amyB-his、amyB-VHH-his、glaB-his及びglaB-VHH-hisサンプルをHis-tagカラムであるHis GraviTrap (GEヘルスケア社製)にて精製を行った。amyB-hisはサンプル(レーン1)と比較して素通り画分(レーン2)のバンドの強度が若干弱くなっていること、及び溶出画分(レーン3)にはっきりとしたバンドが確認できることからamyB-hisの一部はhis-tagまでが分解されずに培地に分泌されていることが確認できた。同様にamyB-VHH-hisのサンプル(レーン4)、素通り画分(レーン5)及び溶出画分(レーン6)のバンドパターンからamyB-VHH-hisの一部はhis-tagまでが分解されずに培地中に生産できることが明らかとなった。また、スプリットしたバンドは溶出画分には見られなかったことから、70kDaのバンドがhis-tagまでの完全長産物であり、50kDaのバンドはVHH及びHis-tag部分分解産物であることが考えられる。glaB-hisについてもamyB-hisと同様に精製ができ、傾向も同じであった(レーン7−9)。glaB-VHH-hisについてはamyB-VHH-hisと同様に60kDaのバンドはカラム生成できなかったことから、70kDaのバンドがhis-tagまでの完全長産物であることが明らかとなった。完全長のバンドのシグナル強度はamyB-VHH-hisよりもglaB-VHH-hisが強く、glaBをリーダータンパク質とすることで分解を受けていない完全長型のVHHをより高生産できることを明らかにした。
【0092】
試験例5:固体培養におけるα-アミラーゼ及びグルコアミラーゼと融合させたVHHタンパク質の生産結果
目的:amyB-VHH-his及びglaB-VHH-hisタンパク質がフスマ固体培養で液体培養より効率よくhis-tagまで完全に作られているかを検証する。
【0093】
方法:
試験例2及び3で得られたamyB-VHH-his及びglaB-VHH-hisタンパク質生産麹菌を、フスマ固体培養で培養を行った。100 mL容のガラス製三角フラスコ(AGC社製)に乾燥した小麦フスマ5 gを入れ、オートクレーブで滅菌した後、各麹菌の胞子を滅菌水に懸濁した胞子懸濁液(106胞子/mL)を加えて良く混和し、30℃で3日間静置培養を行った。培養物に50 mMリン酸ナトリウム緩衝液(pH 7.0)を100 mL加えて、タンパク質の抽出を行い、0.45μmのフィルターでろ過後、His GraviTrap(GEヘルスケア社製)にて精製を行い、各サンプルを100μLをVIVA SPIN 500 (Sartorius社製)により濃縮し、SDS-PAGEにて解析を行った。結果を図5に示した。
【0094】
結果及び考察:
amyB-hisはGPY液体培養同様に精製できたが、素通り画分のバンドの強度が原液画分とほぼ同じであり、かつ溶出画分の強度が弱かったことから大半がhis-tagを欠損していることが明らかとなった(レーン1−3)。amyB-VHH-hisに関してはVHHに相当するバンドの分子量増加が見られず、また溶出画分に何も存在しなかったことから、his-tagのみならずVHHの部分もプロテアーゼなどにより分解されている可能性が示唆された(レーン4−6)。glaB-hisについてもamyB-his同様に素通り画分のバンドの強度が原液画分とほぼ同じであり、かつ溶出画分の強度が弱かったことから大半がhis-tagを欠損していることが明らかとなった(レーン7−9)。glaB-VHH-hisについてもamyB-VHH-hisと同様にVHHに相当するバンドの分子量増加が見られず、また溶出画分に何も存在しなかったことから、his-tagのみならずVHHの部分もプロテアーゼなどにより分解されている可能性が示唆された(レーン10−12)。フスマ固体培養においてはHis-tagのみならずVHHも分解されることからGPY液体培養と比較してVHH生産には不向きである事が明らかとなった。
【0095】
試験例6:amyB-VHH-his及びglaB-VHH-hisタンパク質のN型糖鎖除去
目的:amyB-VHH-his及びglaB-VHH-hisタンパク質が目的分子量を保持しているかを確認する。
【0096】
方法:
試験例4で得られたHis-tagカラム精製タンパク質amyB-his、amyB-VHH-his、glaB-his、及びglaB-VHH-hisをVIVA SPIN 500 (Sartorius社製)により濃縮し、5μgタンパク質相当をSDS-PAGEにて解析を行った。EndoH処理は、Endoglycosidase H (Streptomyces griseus由来、生化学バイオビジネス株式会社製)により行った。各サンプルはSDS電気泳動用のサンプルバッファー(0.1 M Tris-HCl (pH 6.8)、2% SDS、20%グリセロール、0.2% BPB、0.2M DTT)と1対1で混和し、室温、5分間インキュベートしたのちにSDS-PAGE解析に供した。結果を図6に示した。
【0097】
結果及び考察:
各サンプルともに、Endo H処理をすることにより、低分子化し糖鎖が付加していることが明らかとなった。しかしながらバンドの位置及びその大きさから、幾つかのサンプルについては試験例1と同様に高温でインキュベートする必要が考えられた。
【0098】
試験例7:amyB-VHH-his及びglaB-VHH-hisタンパク質のN型糖鎖除去(高温処理)
目的:amyB-VHH-his及びglaB-VHH-hisタンパク質が目的分子量を保持しているかを確認する。
【0099】
方法:
試験例4で得られたHis-tagカラム精製タンパク質amyB-his、amyB-VHH-his、glaB-his及びglaB-VHH-hisをVIVA SPIN 500 (Sartorius社製)により濃縮し、5μgタンパク質相当をSDS-PAGEにて解析を行った。EndoH処理は、Endoglycosidase H (Streptomyces griseus由来、生化学バイオビジネス株式会社製)により行った。各サンプルはSDS電気泳動用のサンプルバッファー(0.1 M Tris-HCl (pH 6.8)、2% SDS、20% グリセロール、0.2% BPB、0.2M DTT)と1対1で混和し、99℃、5分間インキュベートしたのちにSDS-PAGE解析に供した。結果を図7に示した。
【0100】
結果及び考察:
各サンプルともに、Endo H処理をすることにより、低分子化し糖鎖が付加している事が明らかとなった。amyBをリーダーとしたときはN型糖鎖の付加か少なく、glaBをリーダーとしたときは糖鎖付加により10kDa近い分子量の増加がみられた。またそれぞれの分子量の位置が理論値と一致しており、99℃でインキュベートすることによりVHHの付加による分子量増大が明らかとなった(amyB-VHH-hisはレーン1と3の比較、glaB-VHH-hisはレーン5と7の比較)。
【0101】
試験例8:他のリーダータンパク質の検討、エンドグルカナーゼcelA及びcelB
目的:glaBを超えるVHH生産に最適なリーダータンパク質を見出す。
【0102】
方法:
1.ベクターの構築方法
ベクターはhlyプロモーターが組み込まれた発現プラスミドから「ベクターの調製」に従って調製し、インサートはcelA遺伝子をアスペルギルス・オリゼ由来celA(cDNA型)及びcelB(cDNA型)遺伝子を鋳型として、「インサートの調製」に従ってそれぞれ調製した。「ベクターとインサートのライゲーション」に従い、Phly-celA-his及びPhly-celA-his分泌発現ベクターをそれぞれ構築した。融合タンパク質の分泌発現ベクターPhly-celA-VHH-hisは、インサート1:celA(cDNA型)、インサート2:celB(cDNA型)遺伝子、インサート3:VHH遺伝子(合成遺伝子配列、北海道システムサイエンス社製)を鋳型としたものを、「インサートの調製」に従ってそれぞれ調製した。ベクターとインサート1及び3及びベクターとインサート2及び3同時に混和して、「ベクターとインサートのライゲーション」に従い、Phly-celA-VHH-his及びPhly-celB-VHH-hisを構築した。
【0103】
2.タンパク質の発現分泌回収
「目的遺伝子発現株の取得」に従い、麹菌に1で作成した分泌発現ベクターを麹菌に形質転換し、「目的遺伝子産物の取得」によりサンプルを回収した。
【0104】
3.SDS-PAGE解析
2で得られたサンプル100μLをVIVA SPIN 500 (Sartorius社製)により濃縮し、SDS-PAGEにて解析を行った。結果を図8に示した。
【0105】
結果及び考察:
celB-VHH-his遺伝子導入した独立の4株のサンプル(レーン6−9)は目的産物が良好に生産されている事が観察された。celA-VHH-his遺伝子導入した独立の4株のサンプル(レーン2−5)には目的産物がほとんど生産されていなかった。celBはリーダータンパク質として有望な可能性がある。
【0106】
試験例9:エンドグルカナーゼcelA及びcelBと融合させたVHHタンパク質のHis-tagカラムでの精製結果
目的:
celA-VHH-his及びcelB-VHH-hisタンパク質がhis-tagまで完全に作られているかをHis-tagカラム精製により検証する。
【0107】
方法:
GPY液体培養(試験例8)及びフスマ固体培養(試験例5に従って実施)から得られたcelA-VHH-his及びcelB-VHH-hisサンプルをHis-tagカラムであるHis GraviTrap (GEヘルスケア社製)にて精製を行った。得られたサンプル100μLをVIVA SPIN 500 (Sartorius社製)により濃縮し、SDS-PAGEにて解析を行った。結果を図9に示した。
【0108】
結果及び考察:
フスマ固体培養では目的物タンパク質自体を検出することができなかった。他方GPY液体培養では、溶出画分にのみcelA-VHH-his産物を検出することができた。また、celB-VHH-hisサンプルからはglaB-VHH-hisと同程度の溶出画分サンプル量を得ることができ、glaBと並んでcelBは有望なリーダータンパク質であることが明らかとなった。
【0109】
試験例10:glaB-VHH-hisと抗原hCGとの結合性試験
目的:glaB-VHH-hisの抗原結合能を検証する。
【0110】
方法:
glaB-his、glaB-VHH-hisを臭化シアン活性化セファロース(アマシャムバイオサイエンス社)に付属のプロトコールにより固相化した。glaB-his、glaB-VHH-hisを結合した20μLのセファロース樹脂(10μgのVHHを結合)と10μgのhCG (Human Chorionic Gonadotropin(ヒト絨毛性ゴナドトロピン)、シグマアルドリッチ社)をバッファー(20 mM Tris-HCl 、0.15 M NaCl、pH 8)中、室温で90分間インキュベートした後、3回同じバッファーで樹脂を洗浄した。洗浄した樹脂に10μLのSDS電気泳動用のサンプルバッファー(78 mM Tris-HCl、pH6.8、2.5% SDS、2%グリセロール、1.25 mg/mLブロモフェノールブルー、120 mM β-メルカプトエタノール)を加えて、95℃で15分間インキュベートし、VHHに結合したhCGを溶出した。樹脂を遠心で沈澱させ、上清をSDS電気泳動にかけ、クーマジーブリリアントブルーで染色を行なった。結果を図10に示した。
【0111】
結果及び考察:
素通り画分(レーン1−3)では、活性化セファロースのみ、glaB-hisを結合させたもの、glaB-VHH-hisを結合させたものに大きな差異はなく、hCGは大過剰量添加されていると言える。溶出画分ではglaB-VHH-hisのみに明瞭なバンド(レーン4−6、矢印)が検出されたことから、glaB-VHH-hisはhCGに対して強い結合能をもつことが明らかとなった。
【0112】
試験例11.抗EGFR抗体の麹菌での生産
上皮成長因子受容体(Epidermal Growth Factor Receptor; EGFR)は、細胞の増殖や成長を制御する上皮成長因子(EGF)を認識し、シグナル伝達を行う受容体である。このEGFRに遺伝子増幅や遺伝子変異、構造変化が起きると、発癌、および癌の増殖、浸潤、転移などに関与するようになる。
【0113】
hlyプロモーター支配下で、Lama glama由来の抗EGFRのVHH抗体(GenBank Accession Number: ABX79398.1)のcDNAを麹菌で発現させた。N末端にリーダータンパク質としてamyB, glaB, celA, celBからなる群の1つの遺伝子とLama glama由来の抗EGFRのVHH抗体のcDNAとHisタグをコードする遺伝子の順からなる融合遺伝子をpNMBベクターにサブクローニングし、得られたベクターを、定法であるPEG-カルシウム法(Mol Gen Genet, 218, 99-104, (1989))により、アスペルギルス・オリゼのniaD変異株(独立行政法人産業技術総合研究所特許生物寄託センターにFERM P-17707として寄託済み)を形質転換した。
【0114】
得られた形質転換体をポテトデキストロース培地で胞子形成させ、滅菌水で胞子を回収した。500 ml容三角フラスコに入った100 ml GPY液体培地(2%グルコース、1%ポリペプトン、0.5%イーストエキストラクト、0.1%リン酸1水素2カリウム、0.05%塩化カリウム、0.05%硫酸マグネシウム、0.001%硫酸鉄、0.3%硝酸ナトリウム)に最終胞子濃度1×106/mlとなるように植菌した。30℃3日間の液体培養で目的遺伝子産物が培地中に分泌発現させ、分泌タンパク質をSDS-PAGEで解析した。
【0115】
分泌された目的融合遺伝子産物の培地あたりのタンパク質生産量を決定するために、菌体外のタンパク質総量をBradfordプロテインアッセイ試薬(バイオラッドラボラトリーズ)で測定し、菌体外のタンパク質に対する目的融合遺伝子産物の量比と純度をAgilent Technologies社製の商品名2100 Bioanalyzer(Protein chip 80を使用)で測定した。その結果の生産量と全分泌タンパク質に対する目的融合遺伝子産物の純度を下記表1に示す。対照は、ベクターpNMBのみを形質転換した麹菌形質転換体である。その結果、当該抗EGFRのVHH抗体についても、glaBとcelBをリーダータンパク質に選択することで、良好に分泌発現でき、1528 mg/Lの著量の分泌発現に成功し、その純度は94.2%となった。
【0116】
【表1】
【0117】
次に、celB+Lama glama由来の抗EGFRのVHH抗体+Hisタグ融合タンパク質をNiカラムにより公知の方法で精製し、表面プラズモン共鳴シグナルによる抗体活性を測定した。前記精製タンパク質を、金薄膜がコートされたセンサーチップ表面に固定化した。なお、固定化は、予め金薄膜の表面にカルボキシメチル基を導入しておき、融合タンパク質中のアミノ基と結合させることにより行った。得られたセンサーチップを、表面プラズモン共鳴シグナル解析装置(ビアコア社製、BIACORE X)にセットし、リガンドサンプルとして10〜100 nMのEGFR (ミズーリ州セントルイス、シグマ社)溶液をフローセルに流してリガンド物質を結合させた後、pH2.5のグリシン溶液によって解離させた。この結合・解離に伴う質量変化をリアルタイムで測定し、解析ソフトウウェアBIAevaluation9.1 (ビアコア社)によって解離定数(Kd)を算出したところ14.7 nMであった。
【0118】
なお、コントロール実験として、celB+Hisタグ融合タンパク質のみをセンサーチップに固定化して同様の実験を行ったところ、EGFR(ミズーリ州セントルイス、シグマ社)との結合は全く検出されなかった。よって、Lama glama由来の抗EGFRのVHH抗体は、融合タンパク質の状態で抗原結合活性を有することが判明した。
【0119】
試験例12.抗RR6抗体の麹菌での生産
RR6は、Reactive Red 6という色素である。Lama glama由来の抗EGFRのVHH抗体に代えてLama glama由来の抗RR6のVHH抗体(GenBank Accession Number: AJ236100.1)を使用した以外は試験例11と同様の実験を行った。
【0120】
その結果の生産量と全分泌タンパク質に対する目的融合遺伝子産物の純度を表2に示す。対照は、ベクターpNMBのみを形質転換した麹菌形質転換体である。その結果、当該抗RR6のVHH抗体についても、glaBとcelBをリーダータンパク質に選択することで、良好に分泌発現でき、1624 mg/Lの著量の分泌発現に成功し、その純度は92.6%となった。
【0121】
【表2】
【0122】
次に、celB+Lama glama由来の抗RR6のVHH抗体+Hisタグ融合タンパク質をNiカラムにより公知の方法で精製し、表面プラズモン共鳴シグナルによる抗体活性を測定した。前記精製タンパク質を、金薄膜がコートされたセンサーチップ表面に固定化した。なお、固定化は、予め金薄膜の表面にカルボキシメチル基を導入しておき、融合タンパク質中のアミノ基と結合させることにより行った。得られたセンサーチップを、表面プラズモン共鳴シグナル解析装置(ビアコア社製、BIACORE X)にセットし、リガンドサンプルとして10〜100 nMのRR6 (Reactive Red 6, Procion Rubine MX-B, ICI)溶液をフローセルに流してリガンド物質を結合させた後、pH2.5のグリシン溶液によって解離させた。この結合・解離に伴う質量変化をリアルタイムで測定し、解析ソフトウウェアBIAevaluation9.1(ビアコア社)によって解離定数(Kd)を算出したところ14.7 nMであった。
【0123】
なお、コントロール実験として、celB+Hisタグ融合タンパク質のみをセンサーチップに固定化して同様の実験を行ったところ、RR6との結合は全く検出されなかった。よって、Lama glama由来の抗RR6のVHH抗体は、融合タンパク質の状態で抗原結合活性を有することが判明した。
【0124】
以上すべての実施例を総合すると、glaB又はcelBをリーダータンパク質に選択すれば、すべての抗原に対するVHH抗体を、融合タンパク質として活性型で、著量高純度で分泌発現させることが可能なことが明らかとなった。
【特許請求の範囲】
【請求項1】
以下の(a)〜(d)のいずれかのDNA及びラクダ科動物抗体の重鎖可変ドメイン(VHH)をコードするDNAを融合したDNA:
(a)配列番号1に記載される塩基配列からなるDNA
(b)配列番号1に記載される塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、且つグルコアミラーゼ活性を有するタンパク質をコードするDNA
(c)配列番号2に記載される塩基配列からなるDNA
(d)配列番号2に記載される塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、且つエンドグルカナーゼ活性を有するタンパク質をコードするDNA。
【請求項2】
請求項1に記載のDNAに更に精製用のアミノ酸配列をコードするDNAを融合したDNA。
【請求項3】
糸状菌体内で機能するプロモーター遺伝子及び請求項1又は2に記載のDNAを含む遺伝子発現カセットを有する糸状菌用発現ベクター。
【請求項4】
請求項3に記載の糸状菌用発現ベクターで形質転換されてなるアスペルギルス・オリゼ。
【請求項5】
請求項4に記載のアスペルギルス・オリゼを培地で培養し、当該培地からVHHを含むタンパク質を回収する工程を含む、VHHを含むタンパク質の生産方法。
【請求項6】
前記アスペルギルス・オリゼを液体培地で培養して、前記VHHを含むタンパク質を菌体外に分泌させることにより生成蓄積させることを特徴とする、請求項5に記載の方法。
【請求項7】
請求項5又は6に記載の方法において、更に前記VHHを含むタンパク質からVHHに結合したペプチドを切断する工程を有する、VHHの生産方法。
【請求項8】
請求項5又は6に記載の方法により得られたVHHを含むタンパク質。
【請求項1】
以下の(a)〜(d)のいずれかのDNA及びラクダ科動物抗体の重鎖可変ドメイン(VHH)をコードするDNAを融合したDNA:
(a)配列番号1に記載される塩基配列からなるDNA
(b)配列番号1に記載される塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、且つグルコアミラーゼ活性を有するタンパク質をコードするDNA
(c)配列番号2に記載される塩基配列からなるDNA
(d)配列番号2に記載される塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、且つエンドグルカナーゼ活性を有するタンパク質をコードするDNA。
【請求項2】
請求項1に記載のDNAに更に精製用のアミノ酸配列をコードするDNAを融合したDNA。
【請求項3】
糸状菌体内で機能するプロモーター遺伝子及び請求項1又は2に記載のDNAを含む遺伝子発現カセットを有する糸状菌用発現ベクター。
【請求項4】
請求項3に記載の糸状菌用発現ベクターで形質転換されてなるアスペルギルス・オリゼ。
【請求項5】
請求項4に記載のアスペルギルス・オリゼを培地で培養し、当該培地からVHHを含むタンパク質を回収する工程を含む、VHHを含むタンパク質の生産方法。
【請求項6】
前記アスペルギルス・オリゼを液体培地で培養して、前記VHHを含むタンパク質を菌体外に分泌させることにより生成蓄積させることを特徴とする、請求項5に記載の方法。
【請求項7】
請求項5又は6に記載の方法において、更に前記VHHを含むタンパク質からVHHに結合したペプチドを切断する工程を有する、VHHの生産方法。
【請求項8】
請求項5又は6に記載の方法により得られたVHHを含むタンパク質。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【公開番号】特開2012−157315(P2012−157315A)
【公開日】平成24年8月23日(2012.8.23)
【国際特許分類】
【出願番号】特願2011−20538(P2011−20538)
【出願日】平成23年2月2日(2011.2.2)
【出願人】(000165251)月桂冠株式会社 (88)
【Fターム(参考)】
【公開日】平成24年8月23日(2012.8.23)
【国際特許分類】
【出願日】平成23年2月2日(2011.2.2)
【出願人】(000165251)月桂冠株式会社 (88)
【Fターム(参考)】
[ Back to top ]