
ラサギリンの持続放出性製剤およびその使用
本発明は、特にパーキンソン病および神経系の損傷の治療に有用な活性化合物の持続放出のために製剤化された、特に経口投与のための各種医薬組成物を提供する。この組成物に含まれる活性化合物は、好ましくはN−プロパルギル−1−アミノインダン、その鏡像異性体またはその薬学的に許容される塩、より好ましくはラサギリンまたはその薬学的に許容される塩から選択される。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、神経変性疾患、特にパーキンソン病および神経系の損傷の治療に有用な活性化合物の持続放出のために製剤化された医薬組成物に関する。
【背景技術】
【0002】
いくつかのプロパルギルアミン誘導体は、ドーパミンのようなモノアミン作動性の神経伝達物質を不活性化する、モノアミンオキシダーゼ(MAO)−Bおよび/またはMAO−Aの活性を選択的に阻害するため、ドーパミンレベルが低下しているパーキンソン病(PD)およびアルツハイマー病(AD)のような神経変性疾患の治療に適しているということが示されている。さらに、これらの化合物がアポトーシスを防ぐことにより神経変性から保護するということが示されている。
【0003】
MAO−Bを選択的に阻害することがわかった最初の化合物は、L−(−)−デプレニル、R−(−)−デプレニルまたはセレギリンとしても知られる、R−(−)−N−メチル−N−(プロプ−2−イニル)−2−アミノフェニルプロパンであった。PDに加え、セレギリンが有効であることが明らかとなった他の疾患および状態としては、離脱症状(精神刺激薬、オピエート、麻薬およびバルビツール酸塩の離脱症状が含まれる、特許文献1);うつ病(特許文献2);AD;黄斑変性症(特許文献3);腎機能、および空間学習能力により証明されている認知機能および認知機能を含めた、加齢による退化(特許文献4);ヒトおよび非ヒトにおける下垂体性クッシング病(特許文献5);ヒト(特許文献6)および動物(特許文献7)の両方における免疫系機能不全;哺乳動物での加齢による体重減少(特許文献8);統合失調症(特許文献9);ならびに乳癌および下垂体癌のような癌を含めた各種腫瘍状態が挙げられる。特許文献10では、神経筋および神経変性疾患の治療、および低酸素症、低血糖、虚血性脳卒中または外傷によるCNS損傷の治療でのセレギリンの使用が開示されている。さらに、神経細胞に対するセレギリンの生化学的作用が広く研究されている(例えば、非特許文献1;ならびに非特許文献2を参照されたい)。特許文献11には、セレギリン応答性の疾患および状態に対するメチルセレギリンの使用が開示されている。
【0004】
ラサギリン、すなわちR(+)−N−プロパルギル−1−アミノインダンは非常に強力な選択的不可逆的MAO−B阻害剤であり、欧州、イスラエルおよび米国ではAZILECT(登録商標)またはAGILECT(登録商標)(Teva Pharmaceutical Industries Ltd.、Petach Tikvah、Israel)という名称でPDの治療に認可されている。ラサギリンは、各種障害に対して神経保護活性および抗アポトーシス作用を示すことが細胞培養およびin vivoで示されている(非特許文献3)。培養されたドーパミン作動性のSH−SY5YおよびPC12細胞において、ラサギリンによる神経保護の基礎となる機序が、N−メチル(R)サルソリノール、ペルオキシ亜硝酸供与体であるN−モルホリノ−シドノンイミン(SIN−1)、6−ヒドロキシドーパミンおよび血清と神経成長因子の除去により誘発されるアポトーシスに対して研究されている(非特許文献4;非特許文献5、非特許文献6、非特許文献7;非特許文献8、非特許文献9、非特許文献10)。
【0005】
ラサギリンおよびその薬学的に許容される塩は、PD、記憶障害、アルツハイマー型認知症、うつ病および多動性症候群の治療に有用であるとして、特許文献12、特許文献13、特許文献14、特許文献15、特許文献16、特許文献17、特許文献18および特許文献19で最初に開示された。4−フルオロ−、5−フルオロ−および6−フルオロ−N−プロパルギル−1−アミノインダン誘導体が同じ目的で特許文献20に開示された。特許文献21、特許文献22、特許文献23、特許文献24、特許文献25、特許文献26、特許文献15、特許文献16、特許文献17、特許文献18および特許文献19には、ラサギリンおよびその薬学的に許容される塩が、さらなる適応症、特に情緒疾患、神経の低酸素症または無酸素症、神経変性疾患、神経毒損傷、脳卒中、脳虚血、頭部外傷、脊髄外傷、統合失調症、注意欠陥障害、多発性硬化症および離脱症状の治療に有用であるとして開示されている。
【0006】
特許文献27にN−プロパルギル−フェニルエチルアミン化合物が、また特許文献28、特許文献29および特許文献30にN−プロパルギル−1−アミノインダンおよびN−プロパルギル−1−アミノテトラリン化合物が、うつ病、注意欠陥障害、注意欠陥/多動性障害、トゥレット症候群、AD、ならびにその他の認知症、例えば老人性認知症、パーキンソン型認知症、血管性認知症およびレビー小体型認知症などの治療に有用なものとして記載されている。
【0007】
これまでの研究で、ラサギリンおよび関連するプロパルギルアミン誘導体が、透過性遷移およびカスパーゼ3の活性化に起因するアポトーシス前のミトコンドリア膜電位(ΔΨm)の低下、グリセルアルデヒド−3−リン酸デヒドロゲナーゼの核移行、ならびにヌクレオソームDNA断片化のアポトーシス過程を防ぐことにより、ミトコンドリアから始まるアポトーシス死のカスケードを抑制することが示唆されている(YoudimおよびWeinstock,2002b)。対照単独療法において、L−ドーパの補助剤としてラサギリンが抗パーキンソン活性を示している。
【0008】
ラサギリンのMAO阻害特性および神経保護特性と、AD患者での有効性が証明されている薬物であるリバスチグミンのコリンエステラーゼ(ChE)阻害活性とを組み合わせる試みとして、カルバミン酸部分を含む2種類のラサギリン類似体が合成されている。これらの類似体は、ChE阻害活性とMAO−AおよびMAO−B阻害活性をともに有する(N−プロパルギル−(3R)アミノインダン−5イル)−エチルメチルカルバマート(TV3326)、ならびにChEを阻害するがMAOは阻害しないそのS−異性体(TV3279)である(非特許文献11;GrossbergおよびDesai,2001)。TV3326およびTV3279は、ラサギリンと同様に各種損傷に対する神経保護特性を有するが、この特性はChEおよびMAO阻害活性に依存するのではなく、プロパルギルアミン部分に内在する何らかの薬理活性に由来する可能性がある(非特許文献3)。さらにこれらの化合物は、タンパク質キナーゼCおよびマイトジェン活性化プロテインキナーゼ経路の活性化を介して、アミロイドを形成しない神経栄養性/神経保護性の可溶性アミロイド前駆体(sAPPβ)の放出を刺激する(非特許文献12)。したがってこれらの薬物は、潜在的にアミロイド形成性の誘導体の形成に影響を及ぼす可能性があり、またADの治療において臨床的に重要となる可能性がある。
【0009】
特許文献31、特許文献32および特許文献33には、選択的MAO−B阻害剤、神経保護剤および細胞レスキュー剤として脂肪族プロパルギルアミンが開示されている。リード化合物の(R)−N−(2−ヘプチル)メチル−プロパルギルアミンは、強力なMAO−B阻害剤および抗アポトーシス剤であることが示されている(非特許文献13)。
【0010】
プロパルギルアミンが作用機序に基づいた銅含有ウシ血漿アミンオキシダーゼ(BPAO)の阻害剤であることが何年も前に報告されたが、その効力は大きくはなかった。特許文献34には、弱いグリシン切断系阻害剤としてプロパルギルアミンが開示されている。
【先行技術文献】
【特許文献】
【0011】
【特許文献1】国際公開第92/21333号パンフレット
【特許文献2】米国特許第4,861,800号明細書
【特許文献3】米国特許第5,242,950号明細書
【特許文献4】米国特許第5,151,449号明細書
【特許文献5】米国特許第5,192,808号明細書
【特許文献6】米国特許第5,387,615号明細書
【特許文献7】米国特許第5,276,057号明細書
【特許文献8】米国特許第5,225,446号明細書
【特許文献9】米国特許第5,151,419号明細書
【特許文献10】国際公開第92/17169号パンフレット
【特許文献11】米国特許第6,562,365号明細書
【特許文献12】米国特許第5,387,612号明細書
【特許文献13】米国特許第5,453,446号明細書
【特許文献14】米国特許第5,457,133号明細書
【特許文献15】米国特許第5,576,353号明細書
【特許文献16】米国特許第5,668,181号明細書
【特許文献17】米国特許第5,786,390号明細書
【特許文献18】米国特許第5,891,923号明細書
【特許文献19】米国特許第6,630,514号明細書
【特許文献20】米国特許第5,486,541号明細書
【特許文献21】米国特許第5,519,061号明細書
【特許文献22】米国特許第5,532,415号明細書
【特許文献23】米国特許第5,599,991号明細書
【特許文献24】米国特許第5,744,500号明細書
【特許文献25】米国特許第6,277,886号明細書
【特許文献26】米国特許第6,316,504号明細書
【特許文献27】米国特許第6,251,938号明細書
【特許文献28】米国特許第6,303,650号明細書
【特許文献29】米国特許第6,462,222号明細書
【特許文献30】米国特許第6,538,025号明細書
【特許文献31】米国特許第5,169,868号明細書
【特許文献32】米国特許第5,840,979号明細書
【特許文献33】米国特許第6,251,950号明細書
【特許文献34】米国特許第6,395,780号明細書
【非特許文献】
【0012】
【非特許文献1】Tatton W.G.、「Selegiline can mediate neuronal rescue rather than neuronal protection」、Movement Disorders、1993年、第8巻(Supp.I)、S20−S30頁
【非特許文献2】Tatton W.G.、Greenwood C.E.、「Rescue of dying neurons:a new action for deprenyl in MPTP parkinsonism」、J Neurosci Res.、1991年、第30巻、666−672頁
【非特許文献3】Youdim M.B.H.、Weinstock M.、「ovel neuroprotective anti−Alzheimer drugs with antidepressant activity derived from the anti−Parkinson drug,rasagiline、「Mechanisms of Ageing & Developments」、2002年、第123巻、1081−1086頁
【非特許文献4】Youdim M.B.H.、Wadia A.、Tatton N.A.、Weinstock M.、「The anti−Parkinson drug rasagiline and its cholinesterase inhibitor derivatives exert neuroprotection unrelated to MAO inhibition in cell culture and in vivo」、Ann N Y Acad Sci、2001年、第939巻、450−458頁
【非特許文献5】Akao Y.、Nakagawa Y.、Maruyama W.、Takahashi T.、Naoi M.、「Apoptosis induced by an endogenous neurotoxin,N−methyl(R)salsolinol,is mediated by activation of caspase−3」、Neurosci.Lett.、1999年、第267巻、153−156頁
【非特許文献6】Akao Y.、Maruyama W.、Shimizu S.、Yi H.、Nakagawa Y.、Shamoto−Nagai M.、Youdim M.B.H.、Tsujimoto Y.、Naoi M.、「Mitochondrial permeability transition mediates apoptosis induced by N−methyl(R)salsolinol,an endogenous neurotoxin,and is inhibited by Bcl−2 and Rasagiline,N−Propargyl−1(R)−aminoindan」、J.Neurochem.、2002年、第82巻、913−923頁
【非特許文献7】Akao Y.、Maruyama W.、Yi H.,Shamoto−Nagai M.、Youdim M.B.H.、Naoi M.、「An anti−Parkinson’s disease drug,N−propargyl−1(R)−aminoindan(rasagiline),enhances expression of anti−apoptotic Bcl−2 in human dopaminergic SH−SY5Y cells」、Neurosci.Lett.、2002年、第326巻、105−108頁
【非特許文献8】Maruyama W.、Boulton A.A.、Davis B.A.、Dostert P.、Naoi M.、「Enantio−specific induction of apoptosis by an endogenous neurotoxin,N−methyl(R)salsolinol,in dopaminergic SH−SY5Y cells:suppression of apoptosis by N−(2−heptyl)−N−methylpropargylamine」、J.Neural Transm.、2001年、第108巻、11−24頁
【非特許文献9】Maruyama W.、Akao Y.、Youdim M.B.H.、Boulton A.A.、Davis B.A.、Naoi M.、「Transfection−enforced Bcl−2 overexpression and an anti−Parkinson drug,rasagiline,prevent nuclear accumulation of glyceraldehyde−3 phosphate dehydrogenase induced by an endogenous dopaminergic neurotoxin,N−methyl(R)salsolinol」、J.Neurochem.、2001年、第78巻、727−735頁
【非特許文献10】Maruyama W.、Takahashi T.、Youdim,M.B.H.、Naoi M.、「The anti−Parkinson drug,rasagiline,prevents apoptotic DNA damage induced by peroxynitrite in human dopaminergic neuroblastoma SH−SY5Y cells」、J.Neural Transm.、2002年、第109巻、467−481頁
【非特許文献11】Weinstock M.、「Selectivity of cholinesterase inhibition:Clinical implications for the treatment of Alzheimer’s disease」、CNS Drugs、1999年、第12巻、307−323頁
【非特許文献12】Yogev−Falach M.、Amit T.、Bar−Am O.、Sagi Y.、Weinstock M.、Youdim M.B.H.、「The involvement of mitogen−activated protein(MAP)kinase in the regulation of amyloid precursor protein processing by novel cholinesterase inhibitors derived from rasagiline」、FASEB J.、2002年、第16巻、1674−1676頁
【非特許文献13】Durden D.A.、Dyck L.E.、Davis B.A.、Liu Y.D.、Boulton A.A.、「Metabolism and pharmacokinetics,in the rat,of(R)−N−(2−heptyl)methyl−propargylamine(R−2HMP),a new potent monoamine oxidase inhibitor and antiapoptotic agent」、Drug Metab Dispos.、2000年、第28巻、147−154頁
【発明の概要】
【課題を解決するための手段】
【0013】
本発明では、薬物への曝露が急性投与によるものよりも大幅に持続される徐放方式でのラサギリンの投与が、CNSへの様々な傷害からの最適な神経保護を得るために重要であり得るということがわかった。より具体的には、N−メチル−4−フェニル−1,2,3,6−テトラヒドロピリジン(MPTP)−パーキンソン病(PD)マウスモデルにおいて、用量を漸増させたラサギリン(0.1、0.12または0.15mg/kg)の急性投与は、マウスのドーパミンレベルに対して実質的に同様の効果を示し、未処置マウスに比べてドーパミン量が約60%増加したのに対し、同じ3種類の用量の薬物を徐放方式で24時間にわたって投与したところ、ドーパミンレベルが未処置マウスに比べて、それぞれ57%、74%および88%という有意な用量反応性が見られた。このことは、MPTP処置したマウス脳において、徐放投与が即放投与に比べてドーパミンレベルに対して極めて有益な効果を有することを示唆している。興味深いことに、ラサギリン代謝産物である1−アミノインダンの徐放投与でも同様の結果が得られ、同じ薬物用量を1日1回、同じ期間だけ投与したマウスと比較して、ドーパミンレベルの有意な回復が見られた。
【0014】
さらに、6−ヒドロキシドーパミン(6−OHDA)ラットPDモデルを用いて、徐放方式の投与でラサギリン処置したラットでは、同じ薬物を毎日の注射により処置したラットと比べて、アンフェタミンに誘発された正味の回旋数において有意向上した効果が観察されるということがわかった。
【0015】
したがって一態様では、本発明は、上記活性薬剤の持続放出のために製剤化された、薬学的に許容される担体と、プロパルギルアミン部分、アミノインダン部分もしくはプロパルギルアミン部分とアミノインダン部分の両方を含む活性薬剤またはその薬学的に許容される塩とを含む、医薬組成物を提供する。好適な実施形態では、医薬組成物に含まれる活性薬剤は、R(+)−N−プロパルギル−1−アミノインダン(ラサギリン)またはその薬学的に許容される塩である。
【0016】
別の態様では、本発明は、
(i)不活性なペレットコアと、
(ii)前記ペレットコアをコーティングしている薬物層であって、プロパルギルアミン部分、アミノインダン部分もしくはプロパルギルアミン部分とアミノインダン部分の両方を含む活性薬剤またはその薬学的に許容される塩を含み、任意に結合剤および/またはフィルム形成ポリマーと適切に混合され、かつ任意に滑剤とさらに混合されている薬物層と、
(iii)任意に、前記薬物層をコーティングしている隔離/保護サブコーティング層と、
(iv)もし存在すれば前記サブコーティング層を、または前記薬物層をコーティングしている持続放出性コーティング層と
を含む、持続放出性ペレットを提供する。
【0017】
さらに別の態様では、本発明は、上で定義される持続放出性ペレットを含む経口医薬組成物を提供する。
【0018】
本発明の各種医薬組成物は、神経変性疾患、好ましくはパーキンソン病および神経系の損傷の治療に有用である。
【0019】
したがって、さらなる態様では、本発明は、治療を必要とする個人における神経変性疾患または神経系の損傷の治療方法であって、上で定義される医薬組成物を前記個人に投与することを含む治療方法に関する。
【0020】
さらなる態様では、本発明は、プロパルギルアミン部分、アミノインダン部分もしくはプロパルギルアミン部分とアミノインダン部分の両方を含む活性薬剤またはその薬学的に許容される塩の持続放出性製剤の調製方法であって、
(i)前記活性薬剤を、任意に結合剤および/または滑剤と適切に混合し、適当な溶媒に溶解させて、均一な懸濁液を調製する段階と、
(ii)(i)で得られた懸濁液の被覆を不活性なノンパレルシードのような不活性なペレットに塗布する段階と、
(iii)任意に、(ii)で得られた活性薬剤を充填したペレットを、隔離/保護サブコーティング層でコーティングする段階と、
(iv)(ii)または(iii)で得られたペレットを前記活性薬剤の持続放出を可能にする持続放出性コーティング層でコーティングすることにより前記持続放出性製剤を得る段階と、
(v)任意に、(iv)で得られたコーティング済みのペレットを適当な添加剤と混合する段階と
を含む調製方法に関する。
【図面の簡単な説明】
【0021】
【図1】プラミペキソール用量を一定(0.5mg/kg)にしてラサギリン用量を変化させた(0.1、0.12または0.15mg/kg)ラサギリン−プラミペキソール配合剤(それぞれComb1、2および3で表す)の脳内ドーパミン(DA)レベルに対する効果を示す図である。具体的には、薬物処置を行わない(生理食塩水IPおよび生理食塩水SR)MPTP投与により、未処置マウス(未処置マウスIPおよび未処置マウスSR)に比べて80%を超えるドーパミンレベルの欠乏が生じたことが示されている。ラサギリン−プラミペキソール配合剤での処置(IP投与)によりドーパミンレベルが未処置マウスの約60%まで回復したが、この効果は3種類の配合剤すべてにおいて同様であった。しかし、ALZETポンプを用いて同じ3種類の配合剤を徐放(SR)により投与した場合、ラサギリン用量の増加に伴って、57%、74%および88%というドーパミンレベルの有意な用量反応性の増加が見られた。
【図2】SR投与を用いた、脳内ドーパミン(DA)レベルに対するラサギリン代謝産物であるアミノインダンの効果を示す図である。具体的には、MPTP処置により未処置マウスに比べて90%超えるドーパミンレベルの欠乏が生じた。徐放(SR)投与によるアミノインダン処置により、ドーパミンレベルが溶媒処置マウスまたは毎日のIP注射によりアミノインダンを投与したマウスに比べて有意に回復した。
【図3】アンフェタミンに誘発された正味の回旋数を示す図である。正味の回旋数は、実施例3に記載されているようにラサギリン処置ラットで測定した、反時計回りの回旋数を差し引いた後の時計回りの回旋数(CW−CCW)である。ALZETポンプを用いた徐放(SR)ラサギリンで処置したラットでは、毎日のIP注射により即放(IR)ラサギリンで処置したラットに比べて、正味の回旋数において有意に向上した効果が見られる。
【図4】実施例4〜6のメシル酸ラサギリン(1.0mg)持続放出(ER)コーティングペレット(それぞれ15%ER、22%ERおよび28%ER)のIFS緩衝液中でのin vitro溶出データを示す図である。
【図5】実施例7〜8のサブコーティングを有するメシル酸ラサギリン(1.0mg)持続放出(ER)コーティングペレット(それぞれ、15%ERおよび16%ER)のIFS緩衝液中でのin vitro溶出データを示す図である。
【図6】(i)腸管内の条件を模したIFS緩衝液(pH6.8);(ii)空腹条件を模したGFS緩衝液(pH1.2);(iii)GFS緩衝液で2時間、次いでIFS緩衝液でさらに20時間;(iv)満腹条件を模した酢酸緩衝液(pH4.5);および(v)蒸留水(DI)における、実施例7のサブコーティングを有するメシル酸ラサギリン(1.0mg)持続放出(ER)コーティングペレット(15%ER)のin vitro溶出データを示す図である。
【図7】IFS緩衝剤における、実施例7のサブコーティングを有するメシル酸ラサギリン(1.0mg)持続放出(ER)コーティングペレット(15%ER)のゼロ時間(作製直後)、40℃、湿度75%で1ヵ月後(1M Acc.)、ならびに40℃、湿度75%で2か月後および3か月後(それぞれ、2M Acc.および3M Acc)でのin vitro安定性データを示す図である。
【図8】実施例7のサブコーティングを有するメシル酸ラサギリン持続放出(ER)コーティングペレット(27%ER)のIFS緩衝液中でのin vitro溶出データを示す図である。
【図9】静脈内ボーラス、十二指腸ボーラスまたは結腸ボーラスにより投与したラサギリンの血漿中濃度(ng/ml)対時間プロットを示す図である。
【発明を実施するための形態】
【0022】
パーキンソン病におけるモノアミンオキシダーゼB(MAO−B)阻害作用の主な理論的根拠は、症状による運動への効果を生じる線条体のドーパミン活性の増強にある。MAO−Bが特にドーパミン加水分解に関与するため、MAO−B阻害作用はドーパミンレベルを増加させる。記載されている作用機序によれば、ラサギリンによるMAO−B阻害作用が不可逆的であり、したがって新たなMAO−Bが産生されるまで(すなわち、約2〜3週間)、その阻害作用から得られる効果が存続するということにより、ラサギリンの活性がその薬物動態から切り離される。したがって、徐放方式でのラサギリン投与による効果は全くないと推測され得る。しかし、ラサギリンがアポトーシスまたはその他の経路の阻害を介した別の機序で神経保護作用を有し得ることを示唆する証拠が、最近得られている。さらに、ラサギリンは著しく代謝され、その主要な代謝産物である1−アミノインダンがMAO−B阻害作用と関係のない神経保護活性を有するということも知られている(Bar−Amら,2007;Weinrebら,2010)。
【0023】
ラサギリン、セレギリンおよびその他の構造的に関連するプロパルギルアミン誘導体は、一部はアポトーシスを減少させることにより、MAO−B阻害作用とは関係なくニューロンの生存を増加させる(Tattonら,2002)。この作用は、ミトコンドリアの膜透過性に影響を与えたり、酸化性ラジカルを除去したり、特定のアポトーシスシグナル伝達経路に関与したりするタンパク質のレベルまたは細胞内局在の変化により調節されている可能性が最も高い。ラサギリンおよびセレギリンはともに、他のプロパルギルアミン誘導体と同様に、各種傷害により誘発される細胞死からニューロンを保護するということが、パーキンソン病およびアルツハイマー病のような神経変性障害の細胞モデルおよび動物モデルで確認されている。プロパルギルアミン鎖により、多数の実験モデルにおいて神経保護と関連付けられてきた用量依存性の抗酸化作用および抗アポトーシス作用がもたらされる。最近の刊行物によれば、ラサギリンの神経保護作用はラサリギン(rasaligine)およびその代謝産物である1−アミノインダンの組合せに関連している可能性がある(Tazikら,2009;Bar−Am,2010)。
【0024】
一態様では、本発明は、薬学的に許容される担体と、プロパルギルアミン部分、アミノインダン部分もしくはプロパルギルアミン部分とアミノインダン部分の両方を含む活性薬剤またはその薬学的に許容される塩とを含む、前記活性薬剤の持続放出のために製剤化された医薬組成物を提供する。
【0025】
本発明の基礎となる概念は、後の実施例の節で示される知見に基づくものである。実施例1では、PDのMPTPマウスモデルにおいて、用量を漸増させたラサギリン(0.1、0.12または0.15mg/kg)の急性投与は、マウスのドーパミンレベルに対して実質的に同様の効果を示し、未処置(MPTP処置)マウスに比べてドーパミン量が約60%増加したのに対し、同じ3種類の用量の薬物を徐放方式で24時間にわたって投与したところ、ドーパミンレベルが未処置マウスに比べて、それぞれ57%、74%および88%という有意な用量反応性が見られたことを示す。このことは、MPTP処置したマウス脳において、徐放投与が即放投与に比べてドーパミンレベルに対して極めて有益な効果を有することを示唆している。実施例2では、同じマウスPDモデルを用いて、ラサギリン代謝産物である1−アミノインダンでマウスを処置した実験が記載されており、徐放方式で投与する1−アミノインダンによる処置により、溶媒(生理食塩水)処置マウスまたは同じ薬物を毎日の注射により投与したマウスに比べて、ドーパミンレベルが有意に回復したことを示す。これらの知見は、実施例3に記載されている実験によりさらに裏付けられる。実施例3では、6−OHDAラットPDモデルにおいて、徐放方式で投与するラサギリンで処置したラットでは、毎日の注射による同じ薬物で処置したラットに比べて、アンフェタミンに誘発された正味の回旋数(CW−CCW)において有意に向上した作用が観察されることを示す。
【0026】
実際に本明細書で初めて示されるように、ラサギリンを持続放出方式で送達した場合、薬物またはその活性代謝物である1−アミノインダンへの暴露が著しく延長されることにより、患者の状態を著しく向上させ得るさらに効果的な神経保護が可能になる。この概念によれば、パーキンソン病の治療に適応されるMAO−B阻害剤のラサギリンおよびセレギリンはともに、その他のプロパルギルアミン誘導体と同様に、活性薬剤を連続的に放出する「プロドラッグ」またはプロパギルアミン(propagylamine)/アミノインダン「送達媒体」であると考えることができる。このようなプロドラッグまたは送達媒体はそのMAO阻害活性に関係なく、様々な段階のアポトーシス過程の間に、上で定義される活性薬剤、すなわち、プロパルギルアミン部分、アミノインダン部分もしくはプロパルギルアミン部分とアミノインダン部分の両方を含む活性薬剤またはその薬学的に許容される塩への慢性的な持続的曝露によりニューロン細胞を保護する。
【0027】
本発明では、活性薬剤の任意の薬学的に許容される塩を使用することができる。薬学的に許容される塩の例としては、特に限定されるわけではないが、メシル酸塩、エシル酸塩、トシル酸塩、硫酸塩、スルホン酸塩、リン酸塩、カルボン酸塩、マレイン酸塩、フマル酸塩、酒石酸塩、安息香酸塩、酢酸塩、塩酸塩および臭化水素酸塩が挙げられる。
【0028】
特定の実施形態では、本発明の医薬組成物に含まれる活性薬剤は、N−プロパルギル−1−アミノインダン、その鏡像異性体、その代謝産物、その類似体または上記のうちのいずれかの薬学的に許容される塩である。
【0029】
具体的な実施形態では、活性薬剤は、例えば米国特許第6,630,514号に記載されているようなラセミ体のN−プロパルギル−1−アミノインダン、またはその薬学的に許容される塩である。
【0030】
他の具体的な一実施形態では、活性薬剤は、R(+)−N−プロパルギル−1−アミノインダン(ラサギリン)、そのS−鏡像異性体であるS−(−)−N−プロパルギル−1−アミノインダンまたはその薬学的に許容される塩である。より具体的な実施形態では、活性薬剤は、ラサギリンまたはS−(−)−N−プロパルギル−1−アミノインダンのいずれかのメシル酸塩、エシル酸塩、トシル酸塩、硫酸塩、スルホン酸塩、リン酸塩、カルボン酸塩、マレイン酸塩、フマル酸塩、酒石酸塩、安息香酸塩、酢酸塩、塩酸塩または臭化水素酸塩である。好適な実施形態では、活性薬剤は、例えば米国特許第5,532,415号に記載されているメシル酸ラサギリン;例えば米国特許第5,599,991号に記載されているラサギリンエシル酸塩もしくはラサギリン硫酸塩;または例えば米国特許第6,630,514号に記載されているラサギリン塩酸塩、より好ましくはメシル酸ラサギリンである。
【0031】
さらなる具体的な実施形態では、活性薬剤はラサギリン代謝産物である1−アミノインダンまたはその薬学的に許容される塩である。
【0032】
さらに他の具体的な実施形態では、活性薬剤は、N−プロパルギル−1−アミノインダンの類似体、その鏡像異性体またはその薬学的に許容される塩である。このような類似体の例としては、米国特許第5,486,541号に記載されている化合物、例えば特に限定されないが、4−フルオロ−N−プロパルギル−1−アミノインダン、5−フルオロ−N−プロパルギル−1−アミノインダンおよび6−フルオロ−N−プロパルギル−1−アミノインダンなど;米国特許第6,251,938号に記載されている化合物、例えば特に限定されないが、3−(N−メチル,N−プロピル−カルバミルオキシ)−α−メチル−N’−プロパルギルフェネチルアミン;3−(N,N−ジメチル−カルバミルオキシ)−α−メチル−N’−メチル,N’−プロパルギルフェネチルアミン;3−(N−メチル,N−ヘキシル−カルバミルオキシ)−α−メチル−N’−メチル,N’−プロパルギルフェネチルアミン;3−(N−メチル,N−シクロヘキシル−カルバミルオキシ)−α−メチル−N’−メチル,N’−プロパルギルフェネチルアミンなど;および3−(N−メチル,N−ヘキシル−カルバミルオキシ)−α−メチル−N’−メチル,N’−プロパルギルフェネチルアミンなど;米国特許第6,303,650号に記載されている化合物、例えば特に限定されないが、6−(N−メチル,N−エチル−カルバミルオキシ)−N’−プロパルギル−1−アミノインダン;6−(N,N−ジメチル−カルバミルオキシ)−N’−メチル−N’−プロパルギル−1−アミノインダン;6−(N−メチル,N−エチル−カルバミルオキシ−N’−プロパルギル−1−アミノテトラリン;6−(N,N−ジメチル−チオカルバミルオキシ)−1−アミノインダン;6−(N−プロピル−カルバミルオキシ)−N’−プロパルギル−1−アミノインダン;5−クロロ−6−(N−メチル,N−プロピル−カルバミルオキシ)−N’−プロパルギル−1−アミノインダン;および6−(N−メチル),N−プロピル−カルバミルオキシ)−N’−プロパルギル−1−アミノインダンなど;ならびに米国特許第6,462,222号に記載されている化合物、例えば特に限定されないが、6−(N−メチル,N−エチル−カルバミルオキシ)−N’−メチル,N’−プロパルギル−1−アミノインダンなどが挙げられる。
【0033】
他の特定の実施形態では、本発明の医薬組成物に含まれる活性薬剤は、プロパルギルアミン、脂肪族プロパルギルアミンまたはその薬学的に許容される塩である。
【0034】
具体的な一実施形態では、活性薬剤はプロパルギルアミンまたはその薬学的に許容される塩である。
【0035】
他の具体的な実施形態では、活性薬剤は、米国特許第5,169,868号、米国特許第5,840,979号または米国特許第6,251,950号に記載されている脂肪族プロパルギルアミン、例えば特に限定されないが、N−(1−ヘプチル)プロパルギルアミン;N−(1−オクチル)プロパルギルアミン;N−(1−ノニル)プロパルギルアミン;N−(1−デシル)プロパルギルアミン;N−(1−ウンデシル)プロパルギルアミン;N−(1−ドデシル)プロパルギルアミン;N−(2−ブチル)プロパルギルアミン;N−(2−ペンチル)プロパルギルアミン;N−(2−ヘキシル)プロパルギルアミン;N−(2−ヘプチル)プロパルギルアミン;N−(2−オクチル)プロパルギルアミン;N−(2−ノニル)プロパルギルアミン;N−(2−デシル)プロパルギルアミン;N−(2−ウンデシル)プロパルギルアミン;N−(2−ドデシル)プロパルギルアミン;N−(1−ブチル)−N−メチルプロパルギルアミン;N−(2−ブチル)−N−メチルプロパルギルアミン;N−(2−ペンチル)−N−メチルプロパルギルアミン;(1−ペンチル)−N−メチルプロパルギルアミン;N−(2−ヘキシル)−N−メチルプロパルギルアミン;(2−ヘプチル)−N−メチルプロパルギルアミン;N−(2−デシル)−N−メチルプロパルギルアミン;(2−ドデシル)−N−メチルプロパルギルアミンなど;その鏡像異性体;またはその薬学的に許容される塩である。
さらなる特定の実施形態では、本発明の医薬組成物に含まれる活性薬剤は、セレギリン、デスメチルセレギリン、パルギリンまたはクロルギリンである。
【0036】
さらなる特定の実施形態では、本発明の医薬組成物に含まれる活性薬剤は、CGP3466としても知られ、Zimmermannら(1999)に記載されている、(N−メチル−N−プロパルギル)−10−アミノメチル−ジベンゾ[b,f]オキセピンである。
【0037】
上で引用した米国特許およびその他の刊行物はすべて、本明細書において完全に開示された場合と同様に、その内容全体が参照により本明細書に組み込まれる。
【0038】
本明細書で互換的に使用される、「持続放出」、「制御放出」または「徐放」という用語は、ある期間にわたって身体に吸収されるように活性薬剤をその製剤から放出する様式を表す。活性薬剤の持続放出性の製剤化は、例えば、有効成分がコーティングから徐々に規則的に浸出するように、身体の中で徐々に溶解する入り組んだ物質中に活性薬剤を埋め込むことにより、または薬物が半透性の層から徐々に出て行くように、活性薬剤で膨潤させて表面がほぼ不透性のゲルを形成することにより達成し得る。
【0039】
制御放出性製剤の開発における主な原則は、意図する部位(標的化)において、一定速度で、必要とされる治療域内で物質を放出することである。血中の一定濃度の活性物質を長期間維持する、膨潤システムおよび浸透システムのような溶媒制御システムの原理に基づく機序により、少ない副作用でより効果的な薬物レベルが達成される。言い換えれば、治療域とは、効果が得られる量(有効量)と所望の効果よりも副作用の方が大きい量の間の投薬量のことである。その範囲になるまで、各化合物の個々のバイオアベイラビリティー、作用部位および吸収特性に従って、各薬物の溶出プロファイルを設計するべきである。
【0040】
本発明の医薬組成物は、薬物、すなわち活性薬剤の制御放出を提供するであろう。特定の実施形態では、薬物がゼロ次、一次、二次またはその他の放出プロファイル(N次)の制御放出方式で医薬組成物から放出される。薬物の制御放出は、好ましくは緩徐であるべきであり、特定の実施形態では、連続的な薬物徐放、パルス状の薬物放出、多相性の薬物放出またはそれらの組合せが得られるように医薬組成物を製剤化する。
【0041】
本発明の医薬組成物は、例えば、Remington:The Science and Practice of Pharmacy,第19版,1995に記載されている従来の技術により調製してもよく、任意の従来の形態であってもよく、また各種の用量で提供してもよい。
【0042】
組成物を任意の適当な投与経路、例えば、静脈内、動脈内、筋肉内、皮下または腹腔内投与用に製剤化することができるが、好ましくは、組成物を経口投与用に製剤化する。
【0043】
用量は患者の状態によって異なり、実施者が適切であると判断する量で決定される。具体的な実施形態では、用量は、60kgの成人では1日当たり0.1〜2.0mg、好ましくは0.2〜1.5mg、より好ましくは0.5〜1.0mgである。本発明の組成物は、種々の継続期間、例えば週、月、年または10年で、例えば継続的に、毎日、1日2回、1日3回または1日4回、投与してよい。
【0044】
本発明の医薬組成物は、例えば、既知の技術により適当な分散剤、湿潤剤または懸濁化剤を用いて製剤化し得る、無菌の水性または油性懸濁注射液の形態であってよい。また無菌注射用製剤は、無毒の非経口的に許容される希釈剤または溶剤での無菌の注射溶液または懸濁液注射であってもよい。使用し得る許容される溶媒および溶剤としては、水、リンガー溶液および等張塩化ナトリウム溶液が非限定的に挙げられる。
【0045】
本発明の医薬組成物は、経口投与用に製剤化する場合、錠剤、トローチ剤(troches、lozenges)、水性もしくは油性懸濁剤、分散性の粉末剤もしくは顆粒剤、乳剤、硬もしくは軟カプセル剤、またはシロップ剤もしくはエリキシル剤の形態であってよい。経口使用のための医薬組成物は、当業者に公知の医薬組成物製造のための任意の方法に従って調製してよく、また医薬品として洗練された味の良い製剤を作製するために、甘味剤、着香剤、着色剤および保存剤から選択される1つ以上の薬剤をさらに含んでもよい。錠剤は、錠剤の製造に適した無毒の薬学的に許容される添加剤と混合された活性薬剤を含有する。このような添加剤は、例えば、不活性希釈剤、例えば炭酸カルシウム、炭酸ナトリウム、ラクトース、リン酸カルシウムまたはリン酸ナトリウムなど;造粒剤および崩壊剤、例えばコーンスターチまたはアルギン酸;結合剤;ならびに滑沢剤であり得る。好ましくは、消化管での分解および吸収を遅延させるために既知の技術を用いて錠剤をコーティングして、長期間にわたる薬物の持続放出をもたらす。例えば、モノステアリン酸グリセリルまたはジステアリン酸グリセリルのような時間遅延物質を使用し得る。また米国特許第4,256,108号、同第4,166,452号および同第4,265,874号に記載されている技術を用いて錠剤をコーティングし、制御放出用の浸透圧性の治療錠剤を形成してもよい。また本発明の医薬組成物は、2つ以上の異なる顆粒層が、それぞれ異なる様式の薬物放出が生じるように製剤化され互いに重なり合って圧縮されている、二層テーブルの形態であってもよい。また本発明の経口医薬組成物は、水中油型乳剤の形態であってもよい。
【0046】
また本発明の医薬組成物を制御放出マトリックスとして、例えば、溶出させる液体(in vitro)または胃腸管液(in vivo)と接触する親水性ポリマーを膨潤させて形成されたゲルを介した能動拡散を有することにより可溶性の活性薬剤の放出が制御される、制御放出マトリックス錠剤として製剤化してもよい。このようなゲルを形成することができるものとして、例えば、セルロース誘導体、特にセルロースエーテル、例えばヒドロキシプロピルセルロース、ヒドロキシメチルセルロース、メチルセルロースまたはヒドロキシプロピルメチルセルロースなどのポリマーが数多く記載されており、様々な市販グレードの上記エーテルの中には、かなり高い粘性を示すものがある。他の形状では、組成物は制御放出用にマイクロカプセル化剤形で製剤化された活性薬剤を含む。マイクロカプセル化剤形では、活性薬剤の小滴がコーティングまたは膜に取り囲まれて数マイクロメートルから数ミリメートルの範囲の粒子を形成している。
【0047】
考慮される別の製剤は生分解性ポリマーに基づくデポーシステムである。このシステムでは、ポリマーが分解されるときに活性薬剤が徐々に放出される。最も一般的な生分解性ポリマーのクラスは、乳酸、グリコール酸またはこれらの組合せから調製される加水分解に不安定なポリエステルである。
【0048】
本発明の医薬組成物は1つ以上の薬学的に許容される添加剤を含み得る。例えば、錠剤は、少なくとも1つの賦形剤、例えばラクトース、エチルセルロース、微結晶性セルロース、ケイ化微結晶性セルロース;少なくとも1つの崩壊剤、例えば架橋ポリビニルピロリジノン;少なくとも1つの結合剤、例えばポリビニルピリドン、ヒドロキシプロピルメチルセルロース;少なくとも1つの界面活性剤、例えばラウリル硫酸ナトリウム;少なくとも1つの滑剤、例えばコロイド状二酸化ケイ素;および少なくとも1つの滑沢剤、例えばステアリン酸マグネシウムを含み得る。
【0049】
後の実施例4〜6では、不活性なペレットをコーティングしている薬物層と、薬物層をコーティングしている持続放出性の、すなわち機能性の層とを含む、3種類のメシル酸ラサギリン持続放出(ER)コーティングペレット(ER層が15%、22%および28%)の調製に関して記載する。薬物層の調製では、ポビドンUSP(PVP K29/32)を蒸留水とエタノールの混合物に溶解させ、形成された溶液に薬物を溶解させ、次いで、この溶液に超微末タルクを分散させて加え、均一な懸濁液を形成し、600〜710μm(直径)の糖球状顆粒にコーティングした。各種厚さのERコーティングフィルムの調製では、1つの溶液を調製し、様々な層の厚さを生じる様々な量のスプレーされた溶液に対応する、コーティング工程中の様々な時点での試料をそこから採取した。溶液は、アセトンとエタノールの混合物に溶解したEthocel 45cps(エチルセルロース;放出制御ポリマー);および蒸留水に溶解したポリエチレングリコール(PEG)4000で構成され、次いでこの2つを混合して均質な溶液を形成した。この機能性の溶液を上記の薬物充填ペレットにコーティングして、各種厚さのERフィルムを形成した。各種ERコーティングペレットの溶出プロファイルを、USP(米国薬局方)装置1(バスケット)により、腸内条件を模した腸液溶液(IFS、pH6.8)を用いて軸回転速度100rpmおよび温度37℃で評価した。示されるように、放出速度はフィルムの厚さに影響され、その放出パターンは機能層が厚くなるほど遅かった。
【0050】
実施例7〜8では、不活性なペレットをコーティングしている薬物層と、前記薬物層をコーティングしているサブコーティング層と、サブコーティング層をコーティングしている機能層とを含む、2種類のメシル酸ラサギリン持続放出(ER)コーティングペレット(ER層が15%および18%)の調製に関して記載する。薬物層の調製では、ポビドン(PVP K25)を蒸留水とエタノールの混合物に溶解させ、形成された溶液に薬物を溶解させ、次いで、この溶液に超微末タルクを分散させて加え、均一な懸濁液を形成し、次いでこれを600〜710μmの糖球状顆粒にコーティングした。PVP K25を蒸留水とエタノールの混合物に溶解させてサブコーティング溶液を調製し、次いで、これを薬物充填ペレットにコーティングした。各種厚さのERコーティングフィルムの調製では、1つの溶液を調製し、様々な層の厚さを生じる様々な量のスプレーされた溶液に対応する、コーティング工程中の様々な時点での試料をそこから採取した。溶液は、アセトンとエタノールの混合物に溶解したEthocel 45cps;および蒸留水に溶解したPEG3000で構成され、次いでこの2つを混合して均質な溶液を形成した。この溶液に超微末タルクを蒸留水中に分散させて加え、均一な懸濁液を形成し、次いで、これをサブコーティングしたペレットにコーティングして、各種厚さのERフィルムを形成した。次いで、得られたペレットをAerosil200と乾燥混合した。上記2種類のERコーティングペレットの溶出プロファイルを、USP装置1により、(i)腸内条件を模した腸液溶液(IFS、pH6.8);(ii)空腹条件を模した胃液溶液(GFS、pH1.2)で2時間、次いで、IFSでさらに20時間;および(iii)満腹条件を模した酢酸緩衝液(pH4.5)を用いて、100rpmおよび温度37℃で評価した。示されるように、pH1.2〜6.8の範囲では、ER層中のpH依存性ポリマーにより放出速度が一定に維持され(溶出試験で±10%の許容範囲内)、また安定性促進条件への曝露にもかかわらず3か月間安定であった。機能層の厚さの違いにより、15%のERコーティングペレットからの放出速度は18%のERコーティングペレットよりも速かった。
【0051】
実施例9では、不活性なペレットをコーティングしている薬物層と、サブコーティング層と、外側機能層とを含む、高いパーセンテージ(27%)のERコーティング層を有する第三の種類のメシル酸ラサギリンERコーティングペレットの調製について記載する。本実施例では、ペレットを、実施例7〜8で使用したAerosil200の代わりに二酸化ケイ素と乾燥混合した。上記ペレットの溶出プロファイルを、USP装置1により、IFS(pH6.8)を用いて、100rpmおよび温度37℃で評価した。示されるように、この場合の放出パターンは、機能層の厚さの違いにより、実施例7〜8のペレットで観察されたパターンよりも遅いものであった。
【0052】
様々な薬物放出プロファイルのために設計された、サブコーティング層を有するまたは有さないメシル酸ラサギリンERペレットに関して実施例10でさらに記載する。
【0053】
経口投与用の24時間型持続放出性製剤を設計する場合、全放出時間を通して薬物が吸収される、すなわち、十二指腸および結腸の両方を含めた胃腸管のすべての部分から吸収される必要がある。実施例11では、薬物動態実験に関して記載する。この実験では、ラットの結腸、十二指腸または頸静脈に水溶液としてラサギリンの単回ボーラス投与を行い、投与の5分前、ならびに投与の5、15、30、50、90、150および200分後にラットから血液試料を採取し、ラサギリンとその代謝産物の血漿レベルの両方を測定した。示されるように、結腸および十二指腸投与群の親化合物のT1/2は、IV投与後のT1/2よりも長かった。さらに、IVと十二指腸投与で同様の血中濃度曲線下面積(AUC)値が算出され、このことは完全な経口吸収を示唆するものであった。また、結腸投与後のAUC値はIV投与のAUC値の約28%であり、このことは結腸吸収の実現可能性を示すものであった。
【0054】
上記各種メシル酸ラサギリンERコーティングペレットに関して得られた溶出プロファイルおよび胃腸管の各種部分からのラサギリン吸収を示す上記実験を考慮して、特定の実施形態では、本発明の医薬組成物を経口投与用に製剤化する。具体的な実施形態では、医薬組成物は、実施例10に記載のいくつかのメシル酸ラサギリンERペレットに関して示されているように、当該技術分野で公知の任意の従来の方法により、混合してカプセルもしくは小袋に充填される、または圧縮して錠剤にされる、顆粒、粒、ビーズまたはペレットの形態の固体であってよい。例えば、活性薬剤が少なくとも2つの分離した層に存在する錠剤、すなわち、二層または多層錠が提供され、このような錠剤では、各層が任意に、中間の不活性な層、例えば1つ以上の崩壊剤を含む層により分離されている。また医薬組成物は半固体または液体系であってもよい。
【0055】
特定の実施形態では、本発明の医薬組成物は、経口投与用に製剤化される場合、モノリシックなマトリックス、すなわち、個別の大きさと形状を有する三次元的に安定なマトリックス材料を含む構造;錠剤、例えば二層もしくは多層錠、マトリックス錠、崩壊錠、溶解錠またはチュアブル錠など;または顆粒、粒、ビーズもしくはペレットを充填したカプセルもしくは小袋の形態である。他の特定の実施形態では、本発明の医薬組成物は、経口投与用に製剤化される場合、ポリマーが分解されるときに活性薬剤が徐々に放出される、生分解性ポリマーに基づくデポーシステムの形態である。最も一般的な生分解性ポリマーのクラスは、乳酸、グリコール酸またはこれらの組合せから調製される加水分解に不安定なポリエステルである。上記特定のモノマーから調製される生分解性ポリマーの例としては、ポリ(D,L−ラクチド)(PLA)、ポリグリコリド(ポリグリコール酸;PGA)およびコポリマーポリ(D,L−ラクチド−co−グリコリド)(PLGA)が非限定的に挙げられる。
【0056】
特定の具体的な実施形態では、本発明は、上で定義される医薬組成物、すなわち、プロパルギルアミン部分、アミノインダン部分もしくはプロパルギルアミン部分とアミノインダン部分の両方を含む活性薬剤またはその薬学的に許容される塩を含む医薬組成物を提供し、前記医薬組成物は、USP装置1(バスケット)において、50〜150rpm、好ましくは100rpmで、7.4以下のpH値、好ましくは1.2〜6.8のpH値、37℃で、以下のような溶出プロファイルを有する。
【0057】
【表1】

【0058】
好適な医薬組成物は、USP装置1(バスケット)において、50〜150rpm、好ましくは100rpmで、7.4以下のpH値、好ましくは1.2〜6.8のpH値、37℃で、以下のような溶出プロファイルを有する。
【0059】
【表2】
【0060】
より具体的な実施形態では、この組成物に含まれる活性薬剤は、N−プロパルギル−1−アミノインダン;その鏡像異性体、すなわち、ラサギリンまたはS−(−)−N−プロパルギル−1−アミノインダン;その代謝産物、より具体的には1−アミノインダン;その類似体または上記のもののいずれかの薬学的に許容される塩である。最も具体的な実施形態では、この組成物に含まれる活性薬剤はラサギリンまたはその薬学的に許容される塩である。
【0061】
特定の実施形態では、本発明の医薬組成物は、即放性剤形に比べて投与の際のCmaxおよび変動指数が小さく、望ましくない副作用が少ない。本明細書で使用される「Cmax」という用語は治療薬の最大血漿中濃度を表し、また本明細書で使用される「変動指数」という用語は、薬物投与後の時間の関数としての治療薬の血清中濃度の変動を表す。ラサギリンの望ましくない副作用としては、重篤なアレルギー反応(発疹;じん麻疹;かゆみ;呼吸困難;胸部逼迫;口腔、顔面、口唇または舌の腫脹);黒便または血便;血尿;霧視;性的能力または性欲の変化;胸痛;錯乱;抑うつ;瞳孔散大;速いまたは不規則な心拍;発熱;幻覚;じっとしていられない;手足の痺れまたは刺痛;片側脱力;発作;光敏感性;重度の頭痛;皮膚の変化;頸部の痛みまたは硬直;振戦;思考または歩行の困難;原因不明の嘔気または嘔吐;異常発汗;視覚または発話の障害;下痢;めまい;傾眠;口渇;インフルエンザ様の症状;頭痛;関節痛;意識もうろう;不眠;胃の不調;および鼻詰まりが挙げられるが、これらに限定されない。
【0062】
別の態様では、本発明は、
(i)不活性なペレットコアと、
(ii)上記ペレットコアをコーティングしている薬物層であって、プロパルギルアミン部分、アミノインダン部分もしくはプロパルギルアミン部分とアミノインダン部分の両方を含み、任意に結合剤および/またはフィルム形成ポリマーと適切に混合され、さらに任意に滑剤と混合されている活性薬剤またはその薬学的に許容される塩を含む薬物層と、
(iii)任意に、前記薬物層をコーティングしている隔離/保護サブコーティング層と、
(iv)もし存在すれば前記サブコーティング層を、または前記薬物層をコーティングしている持続放出性コーティング層と
を含む、持続放出性(ER)ペレットを提供する。
【0063】
本発明のERペレットは、上記薬物層をコーティングしている隔離/保護サブコーティング層を任意に含み得る。このサブコーティング層の役割は、活性物質層を外側のERコーティングから分離して、その安定性に影響を与え、活性成分(API)の分解産物を生じ得る可能性のある活性薬剤との相互作用から保護することである。特定の実施形態では、サブコーティング層は、フィルム形成ポリマーおよび任意に滑剤を含む。
【0064】
本発明のERペレットは、存在すればサブコーティング層を、または薬物層をコーティングしている、本明細書では「機能層」とも呼ばれる外側ERコーティング層を含む。
【0065】
特定の実施形態では、ERコーティング層は、少なくとも1つのpH非依存性ポリマー、すなわち水膨潤性/不水溶性/疎水性ポリマー、および任意に細孔形成剤を含み、この場合、持続放出性ペレットは、pH非依存性のin vitro放出特性を有する。他の実施形態では、機能層は、pH非依存性ポリマー、細孔形成剤として働く親水性放出調節ポリマー、および任意に疎水性または親水性の可塑剤、および/または滑剤を含む。さらなる特定の実施形態では、ERコーティング層は、pH依存性の腸溶性コーティングポリマーとpH非依存性ポリマーの混合物を含み、この場合、持続放出性ペレットは、酸性pHまたは生理的pH(すなわち、7.4以下のpH値)においてゼロ次に近いin vitro放出特性を有する。
【0066】
製薬学的用途のための結合剤は、その粘着性および凝集性により固形剤形の製造で使用される、糖類および天然または合成起源のポリマーのような親水性物質である。結合剤の役割は、粉末の凝集性を増加させることにより賦形を補助し、必要な結合力を顆粒剤および錠剤に与えることである。結合剤は、これらの製剤の外観、硬さ、および破砕性を向上させるが、活性物質の崩壊および溶出速度に影響を与えることを目的としていない。これまで一般に使用されてきた天然起源の結合剤としては、アラビアゴム、ゼラチン、デンプンおよび加水分解デンプンが挙げられる。これらの物質は、合成起源の結合剤に取って代わられており、その中で最も重要なのはポビドンおよび各種セルロース誘導体である。本発明のERペレットの薬物層コーティングの活性薬剤と混合することができる結合剤の例としては、ポリビニルピロリドン(PVP)、ヒドロキシプロピルメチルセルロース(HPMC)、ヒドロキシプロピルセルロース(HPC)、微結晶性セルロースおよびこれらの組合せが非限定的に挙げられる。結合剤は、ペレットの総重量の約0.5%〜約20%、好ましくは約0.5%〜約10%の量で存在していてよい。
【0067】
本明細書で使用される「フィルム形成ポリマー」という用語は、硬化して密着性のフィルムを形成することができるポリマーを表す。さらに、コーティングに不可欠なこのポリマーの物理的特性は、フィルムを形成する能力またはコーティングする材料へのある種の粘着性である。フィルム形成ポリマーの例としては、PVP、HPMC、HPC、微結晶性セルロースおよびこれらの組合せが非限定的に挙げられる。フィルム形成ポリマーは、薬物層に含まれる場合、薬物層の総重量の90%以下、好ましくはペレットの総重量の約0.5%〜約20%の量で存在していてよい。サブコーティング層中のフィルム形成ポリマーの量は、サブコーティング層の総重量の100%以下、好ましくはペレットの総重量の約0.5%〜約10%である。
【0068】
滑剤は通常、顆粒および粉末の摩擦および表面電荷を低減して流動性を増強するために、医薬組成物に添加される。さらに滑剤は、抗粘着剤としてコーティング工程で使用される。タルクおよびモノステアリン酸グリセリルのような特定の滑剤が低い製品温度での粘着傾向を低減する抗粘着剤として、コーティング製剤に一般的に用いられる。二酸化ケイ素コロイドのような他の滑剤は、その小さい粒子サイズおよび特有の大きい表面積により、錠剤化およびカプセル化のような数多くの工程において乾燥粉末の流動性を向上させるために利用される、望ましい流動特性を与える。滑剤の非限定的な例としては、タルク、特に超微末タルク、コロイド状二酸化ケイ素、モノステアリン酸グリセリルおよびこれらの組合せが挙げられる。
【0069】
滑剤は、薬物層に含まれる場合、薬物層の総重量の30%以下、好ましくはペレットの総重量の約0.5%〜約5%の量で存在していてよい。滑剤がサブコーティング層に含まれる場合、その量はサブコーティング層の総重量の約10%以下、好ましくはペレットの総重量の約0.5%〜約5%であってよい。
【0070】
本発明のERペレットに含まれ得るpH非依存性ポリマーの例としては、エチルセルロース、Surelease(登録商標)、アクリル酸エステルとメタクリル酸エステルのコポリマー、例えばEudragit(登録商標)RL(ポリ(アクリル酸エチル、メタクリル酸メチル、メタクリル酸トリメチルアンモニオエチルクロリド)、1:2:0.2)、Eudragit(登録商標)RS(ポリ(アクリル酸エチル、メタクリル酸メチル、メタクリル酸トリメチルアンモニオエチルクロリド)、1:2:0.1)、Eudragit(登録商標)NE(ポリ(アクリル酸エチル、メタクリル酸メチル)、2:1)など、およびこれらの組合せが非限定的に挙げられる。pH非依存性ポリマーは、ペレットの総重量の約10%〜約50%、好ましくは約10%〜約30%の量で存在していてよい。
【0071】
本発明のERペレットに含まれ得るpH依存性の腸溶性コーティングポリマーの例としては、Eudragit(登録商標)S(ポリ(メタクリル酸、メチルメタクリル酸)、1:2)、Eudragit(登録商標)L55(ポリ(メタクリル酸、アクリル酸エチル)、1:1)、Kollicoat(登録商標)(ポリ(メタクリル酸、アクリル酸エチル)、1:1)、ヒドロキシプロピルメチルセルロースフタラート(HPMCP)、アルギン酸、カルボキシメチルセルロースおよびこれらの組合せが非限定的に挙げられる。pH依存性の腸溶性コーティングポリマーは、ペレットの総重量の約10%〜約50%、好ましくは約10%〜約30%の量で存在していてよい。
【0072】
本明細書で使用される「細孔形成剤」という用語は、体内環境で溶解して、コーティング層を通過する活性薬剤の拡散速度を増加させる開口細孔をマトリックス中に形成する物質を表す。形成される細孔の大きさは、使用する固体粒子材料の大きさによりある程度まで制御することができる。細孔を均一にするために、粒子材料を次第に微細になるメッシュ篩で選別して、所望の粒子の大きさにする。本発明のERペレットに含まれ得る細孔形成剤は無機物質または有機物質のいずれかであり、このような物質としては、例えば、ポリビニルピロリドン(PVP)、ポリエチレングリコール(PEG)、HPMC、HPC、メチルセルロース、1,2−プロピレングリコール、ラクトース、スクロース、タルク、特に超微末タルクおよびこれらの組合せが挙げられる。細孔形成剤は、ペレットの総重量の約0.1%〜約20%、好ましくは約0.1%〜約10%の量で存在していてよい。
【0073】
本明細書で使用される「親水性放出調節ポリマー」という用語は、水溶性であり、かつ活性薬剤の放出を制御するポリマーを表す。しかし、特定の実施形態では、本発明のERペレットのERコーティング層に含まれる親水性放出調節ポリマーは、実際には細孔形成剤として働く。親水性放出調節ポリマーの例としては、PVP、PEG、HPMC、HPCおよびこれらの組合せが非限定的に挙げられる。親水性放出調節ポリマーは、ペレットの総重量の約0.1%〜約20%、好ましくは約0.1%〜約10%の量で存在していてよい。
【0074】
本明細書で使用される「可塑剤」という用語は、本発明のERペレットで使用するポリマーを可塑化または軟化することができる、任意の化合物または化合物の組合せを包含する。ERコーティング層の製造において、可塑剤は、使用するポリマーまたはポリマーの組合せの融解温度またはガラス転移温度(軟化点温度)を下げること、前記ポリマーまたはポリマーの組合せの平均分子量の幅を広げること、およびコーティング溶液の簡便な加工のために前記ポリマーまたはポリマーの組合せの粘度をさらに減少させることができる。可塑剤の非限定的な例としては、セバシン酸ジブチル;フタル酸ジブチル;クエン酸トリエチルのようなクエン酸エステルおよびトリアセチン;プロピレングリコール;PEG、ポリ(プロピレングリコール)およびポリ(エチレン/プロピレングリコール)のような低分子量ポリ(アルキレンオキシド);およびこれらの組合せが挙げられる。可塑剤は、ペレットの総重量の約0.1%〜約20%、好ましくは約0.1%〜約10%の量で存在していてよい。
【0075】
本発明のERペレットの薬物層コーティングは、上で定義される任意の活性薬剤、すなわち、プロパルギルアミン部分、アミノインダン部分もしくはプロパルギルアミン部分とアミノインダン部分の両方を含む任意の活性薬剤またはその薬学的に許容される塩を含み得る。具体的な実施形態では、活性薬剤は、N−プロパルギル−1−アミノインダン、その鏡像異性体、その代謝産物、その類似体またはその薬学的に許容される塩から選択される。より具体的な実施形態では、活性薬剤はラサギリンまたはその薬学的に許容される塩である。
本発明のERペレットは浸透圧/等張化剤のような不活性成分をさらに含み得る。このような薬剤は、パルス状の薬物送達が必要な場合に崩壊時間を制御するために一般的に使用される。ERペレットの調製で使用し得る適当な浸透圧/等張化添加剤の例としては、塩化ナトリウムおよびマンニトールが非限定的に挙げられる。浸透圧/等張化剤は、ERペレットに含まれる場合、ペレットの総重量の20%以下、好ましくは約0.5%〜約10%の量で存在していてよい。
【0076】
本明細書で例示される特定の実施形態では、本明細書で例示されるERペレットは、不活性なペレットコア;フィルム形成ポリマー/結合剤としてのPVPおよび滑剤としての超微末タルクと混合した活性薬剤を含む薬物層;ならびにpH非依存性ポリマーとしてエチルセルロースおよび細孔形成剤としてPEGを含むERコーティング層を含み、前記フィルム形成ポリマー/結合剤の量は、薬物層の総重量の90%以下、またはペレットの総重量の約0.5%〜約20%であり;前記滑剤の量は、薬物層の総重量の30%以下、またはペレットの総重量の約0.1%〜約10%であり;前記pH非依存性ポリマーの量は、ERコーティング層の総重量の約50%〜約90%、またはペレットの総重量の約10%〜約30%であり;かつ前記細孔形成剤の量は、ERコーティング層の総重量の約1%〜約20%、またはペレットの総重量の約0.1%〜約10%である。
【0077】
本明細書で例示されている他の特定の実施形態では、本発明のERペレットは、不活性なペレットコア;フィルム形成ポリマー/結合剤としてのPVPおよび滑剤としての超微末タルクと混合した前記活性薬剤を含む薬物層;フィルム形成ポリマーとしてPVPを含む隔離/保護サブコーティング層;ならびにpH非依存性ポリマーとしてエチルセルロース、細孔形成剤としてPEGおよび滑剤として超微末タルクを含むERコーティング層を含み、前記薬物層中の前記フィルム形成ポリマー/結合剤の量は、薬物層の総重量の90%以下、またはペレットの総重量の約0.5%〜約20%であり;前記薬物層中の前記滑剤の量は、薬物層の総重量の30%以下、またはペレットの総重量の約0.1%〜約10%であり;前記サブコーティング層中の前記フィルム形成ポリマーの量は、サブコーティング層の総重量の100%以下、またはペレットの総重量の約0.5%〜約20%であり;前記pH非依存性ポリマーの量は、ERコーティング層の総重量の約50%〜約90%、またはペレットの総重量の約10%〜約30%であり;前記細孔形成剤の量は、ERコーティング層の総重量の約1%〜約20%またはペレットの総重量の約0.1%〜約10%であり;かつ前記ERコーティング層中の前記滑剤の量は、ERコーティング層の総重量の約0.1%〜約20%、またはペレットの総重量の約0.1%〜約10%である。
【0078】
さらに別の態様では、本発明は、上で定義されるERペレットを含む経口医薬組成物を提供する。特定の実施形態では、この組成物に含まれるERペレットは、1つ以上の適当な添加剤と混合されて、カプセルに充填されているか、または錠剤に圧縮されている。このようなカプセル剤または錠剤の調製は、当該技術分野で公知の任意の適当な技術を用いて行い得る。
【0079】
経口医薬組成物の調製で使用し得る適当な添加剤の例としては、二酸化ケイ素および上で定義される当該技術分野で公知の他の滑剤が非限定的に挙げられる。
【0080】
錠剤賦形剤は錠剤またはカプセルの大きさを膨らませて、作製を実用的なものにし、また消費者が使用しやすいものにする。賦形剤でかさを増加させすることにより、患者が取り扱うのに適した体積を最終製品にもたせることが可能となる。好ましい賦形剤は、不活性で、製剤の他の成分と適合性があり、非吸湿性であり、比較的安価で、圧縮可能であり、好ましくは無味であるか味が良くなければならない。植物性セルロース(純植物性賦形剤)は、錠剤または硬ゼラチンカプセル剤で一般的な賦形剤である。二塩基性リン酸カルシウムがもう1つの一般的な錠剤賦形剤である。様々な植物油脂を軟ゼラチンカプセル剤に使用することができる。錠剤賦形剤としては、例えば、ラクトース、マンニトール/Parteck(登録商標)、ソルビトール、デンプンおよびこれらの組合せが挙げられる。
【0081】
崩壊剤は、消化管内で錠剤が水分により崩れるときに膨張して溶解し、吸収されるべき有効成分を放出する。崩壊剤の種類には、水分吸収促進剤および錠剤破壊促進剤がある。これらは、錠剤が水分と接触したときに錠剤を迅速に小片に崩壊させて、溶解を促進する。崩壊剤の非限定的な例としては、架橋ポリビニルピロリドン(クロスポビドン)、ナトリウム/カルシウムカルボキシメチルセルロース(CMC)、低置換度クロスカルメロースナトリウムヒドロキシプロピルセルロース、炭酸水素ナトリウム、デンプン、デンプングリコール酸ナトリウムおよびこれらの組合せが挙げられる。
【0082】
特定の加工特性を向上させるために、錠剤およびカプセル製剤に滑沢剤を少量添加する。より具体的には、このような薬剤は、成分が凝集したり、打錠機やカプセル充填機に付着したりするのを防ぐ。また滑沢剤は、固形物とダイ壁間の摩擦が少ない状態での錠剤の成形および排出を可能にする。滑沢剤の例としては、グリセリルベヘナート、ステアリン酸、タルク、ステアリン酸亜鉛、ステアリン酸カルシウムおよびこれらの組合せが非限定的に挙げられる。
【0083】
後の実施例の節で、具体的にラサギリンおよびその代謝産物である1−アミノインダンに関して示されるように、本発明の医薬組成物は、パーキンソン病、さらにはこの組成物に含まれる活性薬剤が有効であることが開示されている、他の任意の神経変性疾患または状態、および神経系の損傷の治療に有用である。上記神経変性疾患または状態の例としては、アルツハイマー病;精神刺激薬、オピエート、麻薬およびバルビツール酸塩の離脱症状を含めた離脱症状;うつ病;腎機能、および空間学習能力により証明されている認知機能を含めた、加齢による退化加齢;ヒトおよび非ヒトにおける下垂体性クッシング病;ヒトおよび動物の両方における免疫系機能不全;哺乳動物での加齢による体重減少;統合失調症;乳癌および下垂体癌のような癌を含めた各種腫瘍状態;神経筋および神経変性疾患;老人性認知症のような認知症、例えば、パーキンソン型認知症、アルツハイマー型認知症、血管性認知症およびレビー小体型認知症;多動性症候群;情緒疾患;注意欠陥障害;多動性障害;多発性硬化症;ならびにトゥレット症候群が非限定的に挙げられる。本発明の医薬組成物により治療され得る具体的な神経系損傷としては、頭部外傷、低酸素症、無酸素症、低血糖症、神経毒損傷、虚血性脳卒中および外傷によるCNS損傷、ならびにアポトーシス過程が生じる他の神経傷害が非限定的に挙げられる。
【0084】
したがってさらなる態様では、本発明は、治療を必要とする個人における神経変性疾患または神経系の損傷の治療方法に関し、この方法は、上で定義される医薬組成物を前記個人に投与することを含む。
【0085】
特定の実施形態では、本発明の方法で使用する医薬組成物を経口投与用に製剤化する。具体的な実施形態では、本発明の方法は、上で定義される活性薬剤の持続放出のために製剤化された経口医薬組成物を投与することを含み、より具体的には、活性薬剤はN−プロパルギル−1−アミノインダン、その鏡像異性体、その代謝産物、その類似体またはその薬学的に許容される塩から選択され、好ましくは、活性薬剤はラサギリンまたはその薬学的に許容される塩である。
【0086】
本発明の方法は、上で定義される任意の神経変性疾患または神経系の損傷の治療に使用することができる。具体的な実施形態では、神経変性疾患はパーキンソン病またはアルツハイマー病であり、神経系の損傷は、脳卒中のような急性脳損傷、または外傷性脳損傷である。
【0087】
さらなる態様では、本発明は、上で定義される活性薬剤、すなわち、プロパルギルアミン部分、アミノインダン部分もしくはプロパルギルアミン部分とアミノインダン部分の両方を含む活性薬剤またはその薬学的に許容される塩の持続放出性製剤の調製方法であって、
(i)任意に結合剤および/または滑剤と適切に混合した前記活性薬剤を適切な溶媒系に溶解させて、均一な懸濁液に調製する段階と、
(ii)不活性ノンパレルシードのような不活性なペレットに、(i)で得られた懸濁液の被覆を塗布する段階と、
(iii)(ii)で得られた活性薬剤充填ペレットを、任意に隔離/保護サブコーティング層でコーティングする段階と、
(iv)(ii)または(iii)で得られたペレットを、持続放出性コーティング層、すなわち、前記活性薬剤の持続放出を可能にするポリマー層でコーティングして前記持続放出性製剤を得る段階と、
(v)(iv)で得られたコーティングペレットを、任意に適当な添加剤と混合する段階と
を含む方法に関する。
【0088】
上で定義される活性薬剤の持続放出性製剤の調製のための、本発明に記載されている方法は、当該技術分野で公知の任意の適当な技術、例えば、後の実施例の節で詳細に記載されているような技術を用いて行うことができる。特定の実施形態では、この方法の段階(ii)および(iv)の1つ以上、ならびに段階(iii)および(v)(もし行った場合)を流動床加工装置を用いて行う。
【0089】
特定の実施形態では、この方法に従って調製した持続放出性製剤をさらに、カプセルに充填させるか、または圧縮して錠剤にする。
【0090】
これより、以下の非限定的な実施例により本発明を説明する。
【実施例】
【0091】
実験
パーキンソン病モデル
パーキンソン病(PD)の実験モデルは、この疾患の考え得る病理学的機序に関する洞察を得るために必要であり、さらに、薬理学的またはその他の新たな治療戦略の開発および試験に不可欠である。
【0092】
MPTPマウスモデル
ヒト脳の剖検研究および動物モデルの生化学的データの重要な主要部は、ドーパミン作動性神経変性を惹起する可能性のある、黒質での進行中の酸化ストレスの過程を示している。酸化ストレスが一次事象であるか二次事象であるかは不明であるが、抗酸化神経保護薬を開発する目的で、神経毒MPTP(N−メチル−4−フェニル−1,2,3,6−テトラヒドロピリジン)により誘発される酸化ストレスを動物モデルに用いて、神経変性の過程が調べられてきた。
【0093】
MPTPは、脳内で酵素モノアミンオキシダーゼ(MAO)−Bにより正荷電分子MPP+(1−メチル−4−フェニルピリジニウム)に変換され、黒質内にある特定のドーパミン産生ニューロンを殺すことにより、霊長類のパーキンソニズムを引き起こす。MPP+はミトコンドリアでの酸化的リン酸化を妨げることにより作用して、ATPの枯渇と細胞死を引き起こす。またMPP+は、カテコールアミンの合成を阻害し、ドーパミンおよび心臓ノルエピネフリンのレベルを低下させ、チロシンヒドロキシラーゼを不活性化する。
【0094】
6−OHDAラットモデル
PDのモデル化は、カテコールアミン神経毒である6−ヒドロキシドーパミン(6−OHDA)により進歩した。この分子は、ドーパミン作動性およびノルアドレナリン作動性の両方のニューロンの細胞体および線維内に輸送されて、神経終末の変性を引き起こし、また特に細胞体領域に投与した場合、細胞体にも影響を及ぼし得る。6−OHDAの神経毒性は、ミトコンドリアの呼吸酵素(呼吸鎖複合体IおよびIV)に対するその強力な阻害作用に基づくものである。これらの酵素のブロックによる代謝欠陥により、ニューロンは正常な生理的機能を発揮することができなくなり、その結果死に至る。PDでは、変性の対象となるのは主としてドーパミン作動性黒質線条体経路であるため、黒質線条体束の主要な遠心性投射に毒素を片側注射により直接注射してドーパミン作動性系の6−OHDA損傷を生じさせた動物モデルが開発されてきた。
【0095】
ドーパミンおよび代謝産物のHPLC解析用の試料調製
500μlのホモジナイゼーション緩衝液(0.1M過塩素酸、0.02%EDTAおよび1%ETOH)中の線条体組織試料を、OMNI International社製のOMNI Tipホモジナイジングキットを用いて(中速、5秒のインターバルで3×10秒)氷中でホモジナイズした。ホモジネートを5分間、超音波処理し、次いで4℃で15分間、15,000rpmで遠心分離した。上清を新たなチューブに移し、HPLCによりドーパミン含有量を解析した。
【0096】
実施例1.PDのMPTPマウスモデルにおけるラサギリン−プラミペキソール配合剤に関するin vivo実験
この実験にはそれぞれ約7〜9匹のマウスからなる10個の群が含まれ、それぞれ表1のように処置した。具体的には、マウスにMPTPを腹腔内(IP)投与してパーキンソン病(PD)モデルを誘発し、一定用量のプラミペキソール(初期段階のPDの治療に適応される非エルゴリン系ドーパミンアゴニスト)と種々の用量のラサギリンとを含有する配合剤で処置した。MPTPを最初の5日間(0〜4日目)、毎日注射し、配合剤を、IPまたは徐放を模擬するALZETポンプ(ALZET微小浸透圧ポンプ、1002モデル、速度0.25μl/時、DURECT社、Cupertino、USA)を用いて、0〜11日目に投与した。群5〜7には、最初の5日間(0〜4日目)は毎日、MPTP投与の30分前に配合剤を投与し、残りの日(5〜11日目)は、各投与日のほぼ同じ時間に薬物を投与した。ALZETポンプを最初のMPTP投与の15〜17時間前に腹腔内に埋め込んだ(群8〜10)。投与期間中にポンプにより投与した薬物の総量は、IP注射群に投与した総量と同じであった。対照は、生理食塩水を注射した未処置マウスおよび生理食塩水を注射したMPTP投与マウスであった。
【0097】
投与前および投与期間中の毎日に体重を測定し、各個体の体重変化を計算した。全実験期間を通して、臨床兆候を1週間に2回記録した。12日目の実験終了時に、全個体をC02窒息により安楽死させた。脳を素早く摘出し、冷却したプレート上に置いて解剖した。左右の線条体を摘出して重量を測定し、液体窒素で急速凍結してから、次の処置まで−70℃で保管した。線条体試料を実験の章に記載されている通りにHPLC用に調製した。
【0098】
【表3】
【0099】
図1に示されるように、3種類のラサギリン+プラミペキソール配合剤をIPで投与した場合、マウスのドーパミンレベルの対するその効果はほぼ同じであり、ドーパミン含有量が未処置マウスに比べて約60%増加した。しかし、24時間にわたって同じ量を投与するALZETポンプを用いた徐放(SR)方式で3種類の配合剤を投与した場合、有意な用量反応性が見られ、ドーパミンレベルはラサギリン用量の増加に従って増加した。全配合剤のプラミペキソールの量は同じであったことから、観察された効果はラサギリンの用量増加によるものに違いない。このことは、MPTP投与マウス脳のドーパミンレベルに対して、徐放投与が即放投与に比べて極めて有用な効果を有することを示唆している。
【0100】
実施例2.PDのMPTPマウスモデルにおけるラサギリン代謝産物アミノインダンに関するin vivo実験
すべての実験で体重20±1gのC57B1/6雄性マウスを使用した(1群当たり10匹のマウス)。MPTPを1日当たり40mg/kgの用量で5日間、IP注射により投与した。対照は、生理食塩水を注射した未処置マウスおよび生理食塩水を注射したMPTP投与マウスであった。アミノインダンを、毎日のIP注射により、または腹腔内に埋め込まれたALZETポンプを用いた徐放により12日間投与した。実験終了時にマウスから採取した左右の線条体中のドーパミンおよびその代謝産物(ジヒドロキシフェニル酢酸およびホモバニリン酸)を測定して処置効果を評価した。線条体組織試料を実験の章に記載されている通りにHPLC用に調製した。
【0101】
図2に示されるように、MPTP処置により、対照未処置マウスに比べて90%を超えるドーパミンレベルの欠乏が生じた。ALZETポンプを用いた徐放(SR)方式で投与したラサギリン代謝産物アミノインダンによる処置により、溶媒(生理食塩水)処置マウスまたは同じ薬物を毎日のIP注射で投与したマウスと比べて、ドーパミンレベルが有意に回復した。
【0102】
実施例3.PDの6−OHDAラットモデルにおけるラサギリンに関するin vivo実験
6−OHDAによる内側前脳束(MFB)の片側損傷は、片側での黒質線条体経路のドーパミン作動性ニューロン破壊を生じ、ラットの非対称な運動行動を引き起こす。損傷を受けたラットにドーパミン系に作用する薬物を与えると、ラットは活発な回旋行動を示す。より具体的には、DA放出薬剤であるD−アンフェタミンの投与により、損傷を受けていない黒質線条体投射に好発するドーパミンの不均衡が生じ、これにより時計回りの回旋が生じる。処置効果により、より多くのDAが利用可能になり、時計回りの回旋が増えると予想される。薬物誘発性の回旋は、回旋ボウルとラットの胴体に取り付ける回り継手とからなる自動ロタメータを用いて測定される。
【0103】
PDのMPTPマウスモデルドーパミン含有量の生化学的エンドポイントに対するラサギリンまたはその代謝産物アミノインダンによる徐放(SR)処置の明らかな利点を示唆する実施例1〜2で示される結果に加え、この実験では、PDの6−OHDAラットモデルを用いて、行動エンドポイントに対するラサギリンのSR投与の処置効果を試験した。
【0104】
体重250〜300gの成体Sprague−Dawley雄性ラットの中央前脳束(MFB)に6−OHDAで損傷を与えた。ラットをケタミン−キシラジン(85:15)0.1ml/100gで麻酔し、脳定位固定装置に載せた。以下の脳定位座標:ブレグマからAP−2.8mm、ML−2mm、および硬膜からDV−9に従って、片側のMFB内に6−OHDAを注入した。注入速度は、注入ポンプおよびHamiltonマイクロシリンジを用いて1μl/分であった。注入後、マイクロシリンジを注入部位に5分間静置し、骨ロウで穴を閉じた。
【0105】
同じ用量のラサギリンを、IP注射により毎日、28日間(6−OHDA投与の7日前から6−OHDA投与の21日後まで)投与するか、または6−OHDA投与の7日前から、埋め込んだALZETポンプを用いて毎日24時間にわたって一定速度で投与し、6−OHDA投与後さらに21日間投与を続け、合計28日間投与した。どちらの場合も、処置後に10日間の休薬を行ってからラットを屠殺した。実験終了時、すなわち6−OHDA投与後32日目に、誘発剤としてアンフェタミンを用いて、運動の非対称性をロタメータにより評価した。アンフェタミンはドーパミン放出剤であるため、6−OHDA投与後にも生存して機能するドーパミン作動性(DAegic)ニューロンの数に左右される。アンフェタミンを1.5mg/kgの単回用量でIP注射した後、ロタメータで60分間の回旋記録を行った。
【0106】
図3に示されるように、徐放を示すALZETポンプを用いてラサギリンで処置したラットでは、即放を示す毎日のIP注射によりラサギリンで処置したラットに比べて、正味の回旋数、すなわち、反時計回り回旋数を差し引いた後の時計回り回旋数(CW−CCW)において有意に向上した効果が観察された。
【0107】
実施例4〜6.サブコーティングを有さないラサギリン持続放出コーティングペレット
サブコーティングを有さない表2に示される組成のメシル酸ラサギリン持続放出(ER)ペレットを調製した。具体的には、薬物層の調製では、ポビドンUSP(PVP K29/32)を蒸留水と96%エタノールの混合物に溶解させ、次いで、形成された溶液にメシル酸ラサギリンを溶解させた。超微末タルクを分散させて形成された溶液に加え均一な懸濁液を形成し、次いで、流動床コーティング機を用いて600〜710μm(直径)の糖球状顆粒にコーティングした。アセトンと96%エタノールの混合物中にEthocel 45cps(エチルセルロース;放出制御ポリマー)を溶解させて機能性のコーティング懸濁液を調製し、次いでポリエチレングリコール(PEG)4000を蒸留水中に溶解させ、これを形成された溶液に加えた。得られた懸濁液を、流動床コーティング機を用いて薬物充填ペレットにコーティングした。
【0108】
【表4】
【0109】
各種ERコーティングペレットの溶出プロファイルを次の条件下で評価した:USP(米国薬局方)装置1を用いて、溶出液(900mlの腸液溶液、IFS、pH6.8)を100rpmの主軸回転速度および37℃の温度で攪拌した。溶出プロファイルを表3および図4に示す。
【0110】
【表5】
【0111】
実施例7〜8.サブコーティングを有するラサギリン持続放出コーティングペレット
サブコーティングを有する表4に示される組成のメシル酸ラサギリンERペレットを調製した。具体的には、薬物層の調製では、ポビドン(PVP K25)を蒸留水と96%エタノールの混合物に溶解させ、次いで、形成された溶液にメシル酸ラサギリンを溶解させた。形成された溶液に超微末タルクを分散させて加え、均一な懸濁液を形成し、次いでこれを600〜710μmの糖球状顆粒に流動床コーティング機を用いてコーティングした。PVP K25を蒸留水と96%エタノールの混合物に溶解させてサブコーティング溶液を調製し、次いで、得られた溶液を薬物充填ペレットに流動床コーティング機を用いてコーティングした。アセトンと96%エタノールの混合物中にEthocel 45cpsを溶解させて機能性のコーティング懸濁液を調製し、次いでPEG3000を蒸留水中に溶解させ、これを形成された溶液に加えた。得られた懸濁液を、流動床コーティング機を用いて薬物充填ペレットにコーティングした。形成された溶液に超微末タルクを分散させて加え、均一な懸濁液を形成し、次いで、これをサブコーティングされたペレットに流動床コーティング機を用いてコーティングした。Tumbler Bin Blenderを用いて、ラサギリンERペレットとAerosil200の乾燥混合物を調製した。
【0112】
各種ERコーティングペレットの溶出プロファイルを実施例4〜6で用いた条件下で評価し、これを表5および図5に示す。(i)腸管内の条件を模したIFS緩衝液(pH6.8);(ii)空腹条件を模したGFS(胃液溶液)緩衝液(pH1.2);(iii)GFS緩衝液で2時間、次いでIFS緩衝液でさらに20時間;および(iv)満腹条件を模した酢酸緩衝液(pH4.5)における実施例7(15%ER)のメシル酸ラサギリンERコーティングペレットのin vitro溶出データを図6に示す。同じERコーティングペレットのゼロ時間(作製直後)、安定性促進条件下(40℃、湿度75%)での1か月後、ならびに同じ安定性促進条件下での2か月後および3か月後におけるIFS緩衝液中でのin vitro安定性のデータを図7に示す。
【0113】
【表6】
【0114】
【表7】
【0115】
実施例9.サブコーティングを有するラサギリン持続放出カプセル剤
サブコーティングを有する表6に示される組成物のメシル酸ラサギリンERペレットを、実施例7〜8に記載されている通りに調製した。ただし、乾燥混合物の調製には、Aerosil200の代わりにコロイド状二酸化ケイ素を使用した。調製したこれらのERコーティングペレットの溶出プロファイルを、実施例4〜8で用いた条件下で評価し、これを表7および図8に示す。
【0116】
【表8】
【0117】
【表9】
【0118】
実施例10.サブコーティングを有する/有さないラサギリン持続放出コーティングペレット
サブコーティングを有するまたは有さない、表8〜12に示される組成のさらなるメシル酸ラサギリンERペレットを、実施例4〜9に記載されている手順に従って調製することができる。本実施例に含まれる各表で言及されている脚注は、表16の下に記載されている。
【0119】
【表10】
【0120】
【表11】
【0121】
【表12】
【0122】
【表13】
*2つのビーズ集団の代わりに、均一な集団として多相放出粒子を調製することもできる:糖球状顆粒またはその他の不活性なコア上の薬物層(0.25〜4.75mg)(第1相)→サブコーティング(第2相)→ERコーティング(第3相)→追加の薬物層(0.25〜4.75mg)→上塗りコーティング(第2相として)→カプセル殻。
【0123】
【表14】
【0124】
上の表2、4および6に記載されているメシル酸ラサギリン製剤、ならびに表8〜12に記載されているメシル酸ラサギリン製剤を錠剤に圧縮して、ラサギリンERコーティング錠を形成してもよい。この目的のために、ラサギリンERコーティングペレットを追加の添加剤と乾燥混合して均質な混合物を作製し、次いでこれを圧縮して、上塗り/装飾/非機能性のコーティング層でコーティングされた錠剤にする(例えば、表13を参照されたい)。
【0125】
【表15】
【0126】
表14は、湿式造粒で調製し、次いで乾燥、粉砕、乾燥混合、錠剤化を行い、最後にERコーティングを施した、ラサギリン0.2mgのERコーティング錠製剤を示したものである。
【0127】
【表16】
【0128】
表15は、放出制御ポリマーを含む湿式造粒で調製し、次いで乾燥、粉砕、乾燥混合、錠剤化を行い、最後に上塗りコーティングを施した、ラサギリン0.2mgのERコーティング錠製剤を示したものである。
【0129】
【表17】
【0130】
表16は、湿式造粒で調製し、次いで乾燥、粉砕、乾燥混合、二層錠剤化を行い、最後に上塗りコーティングを施した、ラサギリン5mgのERコーティング錠製剤を示したものである。
【0131】
【表18】
1製剤は0.2〜5mgのメシル酸ラサギリンを含み得る。
2浸透圧剤の代わりに、またはそれに加えて追加のpH依存性ポリマーを含ませて、パルス状放出性製剤を作製し得る。
3別の結合剤としては、例えば、ヒドロキシプロピルメチルセルロース(HPMC)、ポビドン(PVP)、微結晶性セルロースおよび前記結合剤の組合せが挙げられる。
4別の滑剤としては、例えば、コロイド状二酸化ケイ素、モノステアリン酸グリセリル、ステアリン酸マグネシウムおよび前記滑剤の組合せが挙げられる。
5別のフィルム形成ポリマーとしては、例えば、HPMC、PVP、微結晶性セルロース、ポリエチレングリコール(PEG)および前記フィルム形成ポリマーの組合せが挙げられる。
6別のpH非依存性ポリマーとしては、例えば、Surelease(登録商標)、Eudragit(登録商標)RL、Eudragit(登録商標)RS、Eudragit(登録商標)NEおよび前記ポリマーの組合せが挙げられる。
7別の細孔形成剤としては、例えば、HPMC、PVP、PEGおよび前記細孔形成剤の組合せが挙げられる。
8別の可塑剤としては、例えば、セバシン酸ジブチル/フタル酸ジブチル、トリアセチン、クエン酸トリエチルおよび前記可塑剤の組合せが挙げられる。
9別のpH依存性腸溶性コーティングポリマーとしては、例えば、Eudragit(登録商標)S、Kollicoat(登録商標)、ヒドロキシプロピルメチルセルロースフタラート(HPMCP)および前記薬剤の組合せが挙げられる。
10別の錠剤賦形剤としては、例えば、ラクトース、マンニトール/Parteck(登録商標)、ソルビトール、デンプンおよび前記錠剤賦形剤の組合せが挙げられる。
11別の崩壊剤としては、例えば、ナトリウムCMC/カルシウムCMC、クロスポビドン、低置換度クロスカルメロースナトリウムヒドロキシプロピルセルロース、炭酸水素ナトリウム、デンプン、デンプングリコール酸ナトリウムおよび前記崩壊剤の組合せが挙げられる。
12別の滑沢剤としては、例えば、グリセリルベヘナート、ステアリン酸、タルク、ステアリン酸亜鉛、ステアリン酸カルシウムおよび前記滑沢剤の組合せが挙げられる。
【0132】
実施例11.胃腸管の各種部分からのラサギリン吸収
薬物は胃腸管の各種部分から異なって吸収される。経口投与用の24時間型徐放性製剤を設計するためには、薬物が全時間にわたって、すなわち、胃腸管のすべての部分から吸収される必要である。薬物の大部分は十二指腸からよく吸収されるが、多くの薬物は結腸からはあまり吸収されないということが知られている。薬物は体内から排泄されるまでに、かなりの長時間にわたって結腸内に留まるため、放出プロファイルを効率的に設計するためには、結腸からの薬物吸収を評価することが重要である。
【0133】
この実験では、ラサギリン(1.5mg/kg)を0.5mg/mlの水溶液として、薬物動態実験の1日前に埋め込まれたポリエチレンカニューレにより自由行動Wistar雄性ラットに投与した。結腸、十二指腸および静脈内のボーラス投与のために、カニューレをそれぞれ結腸、十二指腸および頸静脈内に留置した。単回ボーラス投与を各区画に対して行った。さらに、全身の血液採取のために、各個体の右側静脈に第二の留置カニューレを留置した。投与5分前、投与の5、15、30、50、90、150および200分後に血液試料(0.5ml)を採取した。脱水症を予防するために、血液試料の採取後に毎回、等体積の生理溶液をラットに投与した。遠心分離により血漿を分離した後、LC−MS−MSトリプル四重極を用いて、ラサギリンおよびその主要代謝産物である1−アミノインダンの分析的定量を行った。Excelソフトウェアを用いてノンコンパートメント薬物動態解を行った。最終的な測定可能な試料に対するノンコンパートメント解析により、対数線形台形法を用いて血中濃度曲線下面積(AUC)を算出した。ラサギリンの経口バイオアベイラビリティ(F)を、AUC(十二指腸)/AU(IV)またはAUC(結腸)/AUC(IV)のパーセント比として算出した。
【0134】
表17および図9は、最大(またはピーク)血漿中濃度(Cmax)およびAUCの十二指腸投与群と結腸投与群間での違いを示している(データは平均±SEで表されており、n=4〜5である)。具体的には、結腸および十二指腸投与群の親化合物のT1/2は、IV投与後のT1/2よりも長かった。IV投与と十二指腸投与で同様のAUC値が算出され、このことは完全な経口吸収を示唆するものであった。結腸投与後のAUCはIV投与のAUCの約28%であり、このことは結腸吸収の実現可能性を示すものであった。これらの結果から、ラサギリンの制御放出送達システムの設計は実現可能かつ実用的である。
【0135】
【表19】
*Cmax−最大血漿中濃度;Tmax−Cmaxが生じた時間;Vss−定常状態での分布容積;CI−1kg当たりのクリアランス;F−ラサギリンの経口バイオアベイラビリティー。データは平均±SEとして表されている(n=4〜5)。
【0136】
参考文献
Akao Y.,Nakagawa Y.,Maruyama W.,Takahashi T.,Naoi M.,Apoptosis induced by an endogenous neurotoxin,N−methyl(R)salsolinol,is mediated by activation of caspase−3,Neurosci.Lett.,1999,267,153−156
Akao Y.,Maruyama W.,Shimizu S.,Yi H.,Nakagawa Y.,Shamoto−Nagai M.,Youdim M.B.H.,Tsujimoto Y.,Naoi M.,Mitochondrial permeability transition mediates apoptosis induced by N−methyl(R)salsolinol,an endogenous neurotoxin,and is inhibited by Bcl−2 and Rasagiline,N−Propargyl−1(R)−aminoindan,J.Neurochem.,2002a,82,913−923
Akao Y.,Maruyama W.,Yi H.,Shamoto−Nagai M.,Youdim M.B.H.,Naoi M.,An anti−Parkinson’s disease drug,N−propargyl−1(R)−aminoindan(rasagiline),enhances expression of anti−apoptotic Bcl−2 in human dopaminergic SH−SY5Y cells,Neurosci.Lett.,2002b,326,105−108
Bar−Am O.,Amit T.,Youdim M.B.,Aminoindan and hydroxyaminoindan,metabolites of rasagiline and ladostigil,respectively,exert neuroprotective properties in vitro,J.Neurochem.,2007,103(2),500−508
Bar−Am O.,Weinreb O.,Amit T.,Youdim M.B.,The neuroprotective mechanism of 1−(R)−aminoindan,the major metabolite of the anti−parkinsonian drug rasagiline,J.Neurochem.,2010,112,1131−1137
Durden D.A.,Dyck L.E.,Davis B.A.,Liu Y.D.,Boulton A.A.,Metabolism and pharmacokinetics,in the rat,of(R)−N−(2−heptyl)methyl−propargylamine(R−2HMP),a new potent monoamine oxidase inhibitor and antiapoptotic agent,Drug Metab Dispos.,2000,28,147−154
Grossberg G.,Desai A.,Review of rivastigmine and its clinical applications in Alzheimer’s disease and related disorders,Expert Opin.Pharmacother.,2000,2,653−666
Maruyama W.,Boulton A.A.,Davis B.A.,Dostert P.,Naoi M.,Enantio−specific induction of apoptosis by an endogenous neurotoxin,N−methyl(R)salsolinol,in dopaminergic SH−SY5Y cells:suppression of apoptosis by N−(2−heptyl)−N−methylpropargylamine,J.Neural Transm.,2001a,108,11−24
Maruyama W.,Akao Y.,Youdim M.B.H.,Boulton A.A.,Davis B.A.,Naoi M.,Transfection−enforced Bcl−2 overexpression and an anti−Parkinson drug,rasagiline,prevent nuclear accumulation of glyceraldehyde−3 phosphate dehydrogenase induced by an endogenous dopaminergic neurotoxin,N−methyl(R)salsolinol,J.Neurochem.,2001b,78,727−735
Maruyama W.,Takahashi T.,Youdim,M.B.H.,Naoi M.,The anti−Parkinson drug,rasagiline,prevents apoptotic DNA damage induced by peroxynitrite in human dopaminergic neuroblastoma SH−SY5Y cells,J.Neural Transm.,2002,109,467−481
Tazik S.,Johnson S.,Lu D.,Johnson C,Youdim M.B.,Stockmeier C.A.,Ou X.M.,Comparative neuroprotective effects of rasagiline and aminoindan with selegiline on dexamethasone−induced brain cell apoptosis,Neurotoxicity Research,2009,15,284−290
Tatton W.G.,Chalmers−Redman R.M.,Ju W.J.,Mammen M.,Carlile G.W.,Pong A.W.,Tatton N.A.,Propargylamines induce antiapoptotic new protein synthesis in serum−and nerve growth factor(NGF)−withdrawn,NGF−differentiated PC−12 cells,J Pharmacol Exp Then,2002,301,753−764
Tatton W.G.,Greenwood C.E.,Rescue of dying neurons:a new action for deprenyl in MPTP parkinsonism,J Neurosci Res.,1991,30,666−672
Tatton W.G.,Selegiline can mediate neuronal rescue rather than neuronal protection,Movement Disorders 8(Supp.I),1993,S20−S30
Weinreb O.,Amit T.,Bar−Am O.,Yousim M.B.,Rasagiline:a novel antiparkinsonian monoamine oxidase−B inhibitor with neuroprotective activity,Prog Neurobiol,2010,92(3),330−344
Weinstock M.,Selectivity of cholinesterase inhibition:Clinical implications for the treatment of Alzheimer’s disease,CNS Drugs,1999,12,307−323
Yogev−Falach M.,Amit T.,Bar−Am O.,Sagi Y.,Weinstock M.,Youdim M.B.H.,The involvement of mitogen−activated protein(MAP)kinase in the regulation of amyloid precursor protein processing by novel cholinesterase inhibitors derived from rasagiline,FASEB J.,2002,16,1674−1676
Youdim M.B.H.,Weinstock M.,ovel neuroprotective anti−Alzheimer drugs with antidepressant activity derived from the anti−Parkinson drug,rasagiline,Mechanisms of Ageing & Developments,2002a,123,1081−1086
Youdim M.B.H.,Gross A.,Finberg J.P.M.,Rasagiline[N−Propargyl−1R(+)−aminoindan],a selective and potent inhibitor of mitochondrial monoamine oxidase B,Br.J.Pharmacol.,2001a,132,500−506
Youdim M.B.H.,Wadia A.,Tatton N.A.,Weinstock M.,The anti−Parkinson drug rasagiline and its cholinesterase inhibitor derivatives exert neuroprotection unrelated to MAO inhibition in cell culture and in vivo,Ann N Y Acad Sci,2001b,939,450−458
Zimmermann K,Waldmeier P.C.,Tatton W.G.,Dibenzoxepines as treatments for neurodegenerative diseases,Pure Appl Chem,1999,71,2039−2046
【技術分野】
【0001】
本発明は、神経変性疾患、特にパーキンソン病および神経系の損傷の治療に有用な活性化合物の持続放出のために製剤化された医薬組成物に関する。
【背景技術】
【0002】
いくつかのプロパルギルアミン誘導体は、ドーパミンのようなモノアミン作動性の神経伝達物質を不活性化する、モノアミンオキシダーゼ(MAO)−Bおよび/またはMAO−Aの活性を選択的に阻害するため、ドーパミンレベルが低下しているパーキンソン病(PD)およびアルツハイマー病(AD)のような神経変性疾患の治療に適しているということが示されている。さらに、これらの化合物がアポトーシスを防ぐことにより神経変性から保護するということが示されている。
【0003】
MAO−Bを選択的に阻害することがわかった最初の化合物は、L−(−)−デプレニル、R−(−)−デプレニルまたはセレギリンとしても知られる、R−(−)−N−メチル−N−(プロプ−2−イニル)−2−アミノフェニルプロパンであった。PDに加え、セレギリンが有効であることが明らかとなった他の疾患および状態としては、離脱症状(精神刺激薬、オピエート、麻薬およびバルビツール酸塩の離脱症状が含まれる、特許文献1);うつ病(特許文献2);AD;黄斑変性症(特許文献3);腎機能、および空間学習能力により証明されている認知機能および認知機能を含めた、加齢による退化(特許文献4);ヒトおよび非ヒトにおける下垂体性クッシング病(特許文献5);ヒト(特許文献6)および動物(特許文献7)の両方における免疫系機能不全;哺乳動物での加齢による体重減少(特許文献8);統合失調症(特許文献9);ならびに乳癌および下垂体癌のような癌を含めた各種腫瘍状態が挙げられる。特許文献10では、神経筋および神経変性疾患の治療、および低酸素症、低血糖、虚血性脳卒中または外傷によるCNS損傷の治療でのセレギリンの使用が開示されている。さらに、神経細胞に対するセレギリンの生化学的作用が広く研究されている(例えば、非特許文献1;ならびに非特許文献2を参照されたい)。特許文献11には、セレギリン応答性の疾患および状態に対するメチルセレギリンの使用が開示されている。
【0004】
ラサギリン、すなわちR(+)−N−プロパルギル−1−アミノインダンは非常に強力な選択的不可逆的MAO−B阻害剤であり、欧州、イスラエルおよび米国ではAZILECT(登録商標)またはAGILECT(登録商標)(Teva Pharmaceutical Industries Ltd.、Petach Tikvah、Israel)という名称でPDの治療に認可されている。ラサギリンは、各種障害に対して神経保護活性および抗アポトーシス作用を示すことが細胞培養およびin vivoで示されている(非特許文献3)。培養されたドーパミン作動性のSH−SY5YおよびPC12細胞において、ラサギリンによる神経保護の基礎となる機序が、N−メチル(R)サルソリノール、ペルオキシ亜硝酸供与体であるN−モルホリノ−シドノンイミン(SIN−1)、6−ヒドロキシドーパミンおよび血清と神経成長因子の除去により誘発されるアポトーシスに対して研究されている(非特許文献4;非特許文献5、非特許文献6、非特許文献7;非特許文献8、非特許文献9、非特許文献10)。
【0005】
ラサギリンおよびその薬学的に許容される塩は、PD、記憶障害、アルツハイマー型認知症、うつ病および多動性症候群の治療に有用であるとして、特許文献12、特許文献13、特許文献14、特許文献15、特許文献16、特許文献17、特許文献18および特許文献19で最初に開示された。4−フルオロ−、5−フルオロ−および6−フルオロ−N−プロパルギル−1−アミノインダン誘導体が同じ目的で特許文献20に開示された。特許文献21、特許文献22、特許文献23、特許文献24、特許文献25、特許文献26、特許文献15、特許文献16、特許文献17、特許文献18および特許文献19には、ラサギリンおよびその薬学的に許容される塩が、さらなる適応症、特に情緒疾患、神経の低酸素症または無酸素症、神経変性疾患、神経毒損傷、脳卒中、脳虚血、頭部外傷、脊髄外傷、統合失調症、注意欠陥障害、多発性硬化症および離脱症状の治療に有用であるとして開示されている。
【0006】
特許文献27にN−プロパルギル−フェニルエチルアミン化合物が、また特許文献28、特許文献29および特許文献30にN−プロパルギル−1−アミノインダンおよびN−プロパルギル−1−アミノテトラリン化合物が、うつ病、注意欠陥障害、注意欠陥/多動性障害、トゥレット症候群、AD、ならびにその他の認知症、例えば老人性認知症、パーキンソン型認知症、血管性認知症およびレビー小体型認知症などの治療に有用なものとして記載されている。
【0007】
これまでの研究で、ラサギリンおよび関連するプロパルギルアミン誘導体が、透過性遷移およびカスパーゼ3の活性化に起因するアポトーシス前のミトコンドリア膜電位(ΔΨm)の低下、グリセルアルデヒド−3−リン酸デヒドロゲナーゼの核移行、ならびにヌクレオソームDNA断片化のアポトーシス過程を防ぐことにより、ミトコンドリアから始まるアポトーシス死のカスケードを抑制することが示唆されている(YoudimおよびWeinstock,2002b)。対照単独療法において、L−ドーパの補助剤としてラサギリンが抗パーキンソン活性を示している。
【0008】
ラサギリンのMAO阻害特性および神経保護特性と、AD患者での有効性が証明されている薬物であるリバスチグミンのコリンエステラーゼ(ChE)阻害活性とを組み合わせる試みとして、カルバミン酸部分を含む2種類のラサギリン類似体が合成されている。これらの類似体は、ChE阻害活性とMAO−AおよびMAO−B阻害活性をともに有する(N−プロパルギル−(3R)アミノインダン−5イル)−エチルメチルカルバマート(TV3326)、ならびにChEを阻害するがMAOは阻害しないそのS−異性体(TV3279)である(非特許文献11;GrossbergおよびDesai,2001)。TV3326およびTV3279は、ラサギリンと同様に各種損傷に対する神経保護特性を有するが、この特性はChEおよびMAO阻害活性に依存するのではなく、プロパルギルアミン部分に内在する何らかの薬理活性に由来する可能性がある(非特許文献3)。さらにこれらの化合物は、タンパク質キナーゼCおよびマイトジェン活性化プロテインキナーゼ経路の活性化を介して、アミロイドを形成しない神経栄養性/神経保護性の可溶性アミロイド前駆体(sAPPβ)の放出を刺激する(非特許文献12)。したがってこれらの薬物は、潜在的にアミロイド形成性の誘導体の形成に影響を及ぼす可能性があり、またADの治療において臨床的に重要となる可能性がある。
【0009】
特許文献31、特許文献32および特許文献33には、選択的MAO−B阻害剤、神経保護剤および細胞レスキュー剤として脂肪族プロパルギルアミンが開示されている。リード化合物の(R)−N−(2−ヘプチル)メチル−プロパルギルアミンは、強力なMAO−B阻害剤および抗アポトーシス剤であることが示されている(非特許文献13)。
【0010】
プロパルギルアミンが作用機序に基づいた銅含有ウシ血漿アミンオキシダーゼ(BPAO)の阻害剤であることが何年も前に報告されたが、その効力は大きくはなかった。特許文献34には、弱いグリシン切断系阻害剤としてプロパルギルアミンが開示されている。
【先行技術文献】
【特許文献】
【0011】
【特許文献1】国際公開第92/21333号パンフレット
【特許文献2】米国特許第4,861,800号明細書
【特許文献3】米国特許第5,242,950号明細書
【特許文献4】米国特許第5,151,449号明細書
【特許文献5】米国特許第5,192,808号明細書
【特許文献6】米国特許第5,387,615号明細書
【特許文献7】米国特許第5,276,057号明細書
【特許文献8】米国特許第5,225,446号明細書
【特許文献9】米国特許第5,151,419号明細書
【特許文献10】国際公開第92/17169号パンフレット
【特許文献11】米国特許第6,562,365号明細書
【特許文献12】米国特許第5,387,612号明細書
【特許文献13】米国特許第5,453,446号明細書
【特許文献14】米国特許第5,457,133号明細書
【特許文献15】米国特許第5,576,353号明細書
【特許文献16】米国特許第5,668,181号明細書
【特許文献17】米国特許第5,786,390号明細書
【特許文献18】米国特許第5,891,923号明細書
【特許文献19】米国特許第6,630,514号明細書
【特許文献20】米国特許第5,486,541号明細書
【特許文献21】米国特許第5,519,061号明細書
【特許文献22】米国特許第5,532,415号明細書
【特許文献23】米国特許第5,599,991号明細書
【特許文献24】米国特許第5,744,500号明細書
【特許文献25】米国特許第6,277,886号明細書
【特許文献26】米国特許第6,316,504号明細書
【特許文献27】米国特許第6,251,938号明細書
【特許文献28】米国特許第6,303,650号明細書
【特許文献29】米国特許第6,462,222号明細書
【特許文献30】米国特許第6,538,025号明細書
【特許文献31】米国特許第5,169,868号明細書
【特許文献32】米国特許第5,840,979号明細書
【特許文献33】米国特許第6,251,950号明細書
【特許文献34】米国特許第6,395,780号明細書
【非特許文献】
【0012】
【非特許文献1】Tatton W.G.、「Selegiline can mediate neuronal rescue rather than neuronal protection」、Movement Disorders、1993年、第8巻(Supp.I)、S20−S30頁
【非特許文献2】Tatton W.G.、Greenwood C.E.、「Rescue of dying neurons:a new action for deprenyl in MPTP parkinsonism」、J Neurosci Res.、1991年、第30巻、666−672頁
【非特許文献3】Youdim M.B.H.、Weinstock M.、「ovel neuroprotective anti−Alzheimer drugs with antidepressant activity derived from the anti−Parkinson drug,rasagiline、「Mechanisms of Ageing & Developments」、2002年、第123巻、1081−1086頁
【非特許文献4】Youdim M.B.H.、Wadia A.、Tatton N.A.、Weinstock M.、「The anti−Parkinson drug rasagiline and its cholinesterase inhibitor derivatives exert neuroprotection unrelated to MAO inhibition in cell culture and in vivo」、Ann N Y Acad Sci、2001年、第939巻、450−458頁
【非特許文献5】Akao Y.、Nakagawa Y.、Maruyama W.、Takahashi T.、Naoi M.、「Apoptosis induced by an endogenous neurotoxin,N−methyl(R)salsolinol,is mediated by activation of caspase−3」、Neurosci.Lett.、1999年、第267巻、153−156頁
【非特許文献6】Akao Y.、Maruyama W.、Shimizu S.、Yi H.、Nakagawa Y.、Shamoto−Nagai M.、Youdim M.B.H.、Tsujimoto Y.、Naoi M.、「Mitochondrial permeability transition mediates apoptosis induced by N−methyl(R)salsolinol,an endogenous neurotoxin,and is inhibited by Bcl−2 and Rasagiline,N−Propargyl−1(R)−aminoindan」、J.Neurochem.、2002年、第82巻、913−923頁
【非特許文献7】Akao Y.、Maruyama W.、Yi H.,Shamoto−Nagai M.、Youdim M.B.H.、Naoi M.、「An anti−Parkinson’s disease drug,N−propargyl−1(R)−aminoindan(rasagiline),enhances expression of anti−apoptotic Bcl−2 in human dopaminergic SH−SY5Y cells」、Neurosci.Lett.、2002年、第326巻、105−108頁
【非特許文献8】Maruyama W.、Boulton A.A.、Davis B.A.、Dostert P.、Naoi M.、「Enantio−specific induction of apoptosis by an endogenous neurotoxin,N−methyl(R)salsolinol,in dopaminergic SH−SY5Y cells:suppression of apoptosis by N−(2−heptyl)−N−methylpropargylamine」、J.Neural Transm.、2001年、第108巻、11−24頁
【非特許文献9】Maruyama W.、Akao Y.、Youdim M.B.H.、Boulton A.A.、Davis B.A.、Naoi M.、「Transfection−enforced Bcl−2 overexpression and an anti−Parkinson drug,rasagiline,prevent nuclear accumulation of glyceraldehyde−3 phosphate dehydrogenase induced by an endogenous dopaminergic neurotoxin,N−methyl(R)salsolinol」、J.Neurochem.、2001年、第78巻、727−735頁
【非特許文献10】Maruyama W.、Takahashi T.、Youdim,M.B.H.、Naoi M.、「The anti−Parkinson drug,rasagiline,prevents apoptotic DNA damage induced by peroxynitrite in human dopaminergic neuroblastoma SH−SY5Y cells」、J.Neural Transm.、2002年、第109巻、467−481頁
【非特許文献11】Weinstock M.、「Selectivity of cholinesterase inhibition:Clinical implications for the treatment of Alzheimer’s disease」、CNS Drugs、1999年、第12巻、307−323頁
【非特許文献12】Yogev−Falach M.、Amit T.、Bar−Am O.、Sagi Y.、Weinstock M.、Youdim M.B.H.、「The involvement of mitogen−activated protein(MAP)kinase in the regulation of amyloid precursor protein processing by novel cholinesterase inhibitors derived from rasagiline」、FASEB J.、2002年、第16巻、1674−1676頁
【非特許文献13】Durden D.A.、Dyck L.E.、Davis B.A.、Liu Y.D.、Boulton A.A.、「Metabolism and pharmacokinetics,in the rat,of(R)−N−(2−heptyl)methyl−propargylamine(R−2HMP),a new potent monoamine oxidase inhibitor and antiapoptotic agent」、Drug Metab Dispos.、2000年、第28巻、147−154頁
【発明の概要】
【課題を解決するための手段】
【0013】
本発明では、薬物への曝露が急性投与によるものよりも大幅に持続される徐放方式でのラサギリンの投与が、CNSへの様々な傷害からの最適な神経保護を得るために重要であり得るということがわかった。より具体的には、N−メチル−4−フェニル−1,2,3,6−テトラヒドロピリジン(MPTP)−パーキンソン病(PD)マウスモデルにおいて、用量を漸増させたラサギリン(0.1、0.12または0.15mg/kg)の急性投与は、マウスのドーパミンレベルに対して実質的に同様の効果を示し、未処置マウスに比べてドーパミン量が約60%増加したのに対し、同じ3種類の用量の薬物を徐放方式で24時間にわたって投与したところ、ドーパミンレベルが未処置マウスに比べて、それぞれ57%、74%および88%という有意な用量反応性が見られた。このことは、MPTP処置したマウス脳において、徐放投与が即放投与に比べてドーパミンレベルに対して極めて有益な効果を有することを示唆している。興味深いことに、ラサギリン代謝産物である1−アミノインダンの徐放投与でも同様の結果が得られ、同じ薬物用量を1日1回、同じ期間だけ投与したマウスと比較して、ドーパミンレベルの有意な回復が見られた。
【0014】
さらに、6−ヒドロキシドーパミン(6−OHDA)ラットPDモデルを用いて、徐放方式の投与でラサギリン処置したラットでは、同じ薬物を毎日の注射により処置したラットと比べて、アンフェタミンに誘発された正味の回旋数において有意向上した効果が観察されるということがわかった。
【0015】
したがって一態様では、本発明は、上記活性薬剤の持続放出のために製剤化された、薬学的に許容される担体と、プロパルギルアミン部分、アミノインダン部分もしくはプロパルギルアミン部分とアミノインダン部分の両方を含む活性薬剤またはその薬学的に許容される塩とを含む、医薬組成物を提供する。好適な実施形態では、医薬組成物に含まれる活性薬剤は、R(+)−N−プロパルギル−1−アミノインダン(ラサギリン)またはその薬学的に許容される塩である。
【0016】
別の態様では、本発明は、
(i)不活性なペレットコアと、
(ii)前記ペレットコアをコーティングしている薬物層であって、プロパルギルアミン部分、アミノインダン部分もしくはプロパルギルアミン部分とアミノインダン部分の両方を含む活性薬剤またはその薬学的に許容される塩を含み、任意に結合剤および/またはフィルム形成ポリマーと適切に混合され、かつ任意に滑剤とさらに混合されている薬物層と、
(iii)任意に、前記薬物層をコーティングしている隔離/保護サブコーティング層と、
(iv)もし存在すれば前記サブコーティング層を、または前記薬物層をコーティングしている持続放出性コーティング層と
を含む、持続放出性ペレットを提供する。
【0017】
さらに別の態様では、本発明は、上で定義される持続放出性ペレットを含む経口医薬組成物を提供する。
【0018】
本発明の各種医薬組成物は、神経変性疾患、好ましくはパーキンソン病および神経系の損傷の治療に有用である。
【0019】
したがって、さらなる態様では、本発明は、治療を必要とする個人における神経変性疾患または神経系の損傷の治療方法であって、上で定義される医薬組成物を前記個人に投与することを含む治療方法に関する。
【0020】
さらなる態様では、本発明は、プロパルギルアミン部分、アミノインダン部分もしくはプロパルギルアミン部分とアミノインダン部分の両方を含む活性薬剤またはその薬学的に許容される塩の持続放出性製剤の調製方法であって、
(i)前記活性薬剤を、任意に結合剤および/または滑剤と適切に混合し、適当な溶媒に溶解させて、均一な懸濁液を調製する段階と、
(ii)(i)で得られた懸濁液の被覆を不活性なノンパレルシードのような不活性なペレットに塗布する段階と、
(iii)任意に、(ii)で得られた活性薬剤を充填したペレットを、隔離/保護サブコーティング層でコーティングする段階と、
(iv)(ii)または(iii)で得られたペレットを前記活性薬剤の持続放出を可能にする持続放出性コーティング層でコーティングすることにより前記持続放出性製剤を得る段階と、
(v)任意に、(iv)で得られたコーティング済みのペレットを適当な添加剤と混合する段階と
を含む調製方法に関する。
【図面の簡単な説明】
【0021】
【図1】プラミペキソール用量を一定(0.5mg/kg)にしてラサギリン用量を変化させた(0.1、0.12または0.15mg/kg)ラサギリン−プラミペキソール配合剤(それぞれComb1、2および3で表す)の脳内ドーパミン(DA)レベルに対する効果を示す図である。具体的には、薬物処置を行わない(生理食塩水IPおよび生理食塩水SR)MPTP投与により、未処置マウス(未処置マウスIPおよび未処置マウスSR)に比べて80%を超えるドーパミンレベルの欠乏が生じたことが示されている。ラサギリン−プラミペキソール配合剤での処置(IP投与)によりドーパミンレベルが未処置マウスの約60%まで回復したが、この効果は3種類の配合剤すべてにおいて同様であった。しかし、ALZETポンプを用いて同じ3種類の配合剤を徐放(SR)により投与した場合、ラサギリン用量の増加に伴って、57%、74%および88%というドーパミンレベルの有意な用量反応性の増加が見られた。
【図2】SR投与を用いた、脳内ドーパミン(DA)レベルに対するラサギリン代謝産物であるアミノインダンの効果を示す図である。具体的には、MPTP処置により未処置マウスに比べて90%超えるドーパミンレベルの欠乏が生じた。徐放(SR)投与によるアミノインダン処置により、ドーパミンレベルが溶媒処置マウスまたは毎日のIP注射によりアミノインダンを投与したマウスに比べて有意に回復した。
【図3】アンフェタミンに誘発された正味の回旋数を示す図である。正味の回旋数は、実施例3に記載されているようにラサギリン処置ラットで測定した、反時計回りの回旋数を差し引いた後の時計回りの回旋数(CW−CCW)である。ALZETポンプを用いた徐放(SR)ラサギリンで処置したラットでは、毎日のIP注射により即放(IR)ラサギリンで処置したラットに比べて、正味の回旋数において有意に向上した効果が見られる。
【図4】実施例4〜6のメシル酸ラサギリン(1.0mg)持続放出(ER)コーティングペレット(それぞれ15%ER、22%ERおよび28%ER)のIFS緩衝液中でのin vitro溶出データを示す図である。
【図5】実施例7〜8のサブコーティングを有するメシル酸ラサギリン(1.0mg)持続放出(ER)コーティングペレット(それぞれ、15%ERおよび16%ER)のIFS緩衝液中でのin vitro溶出データを示す図である。
【図6】(i)腸管内の条件を模したIFS緩衝液(pH6.8);(ii)空腹条件を模したGFS緩衝液(pH1.2);(iii)GFS緩衝液で2時間、次いでIFS緩衝液でさらに20時間;(iv)満腹条件を模した酢酸緩衝液(pH4.5);および(v)蒸留水(DI)における、実施例7のサブコーティングを有するメシル酸ラサギリン(1.0mg)持続放出(ER)コーティングペレット(15%ER)のin vitro溶出データを示す図である。
【図7】IFS緩衝剤における、実施例7のサブコーティングを有するメシル酸ラサギリン(1.0mg)持続放出(ER)コーティングペレット(15%ER)のゼロ時間(作製直後)、40℃、湿度75%で1ヵ月後(1M Acc.)、ならびに40℃、湿度75%で2か月後および3か月後(それぞれ、2M Acc.および3M Acc)でのin vitro安定性データを示す図である。
【図8】実施例7のサブコーティングを有するメシル酸ラサギリン持続放出(ER)コーティングペレット(27%ER)のIFS緩衝液中でのin vitro溶出データを示す図である。
【図9】静脈内ボーラス、十二指腸ボーラスまたは結腸ボーラスにより投与したラサギリンの血漿中濃度(ng/ml)対時間プロットを示す図である。
【発明を実施するための形態】
【0022】
パーキンソン病におけるモノアミンオキシダーゼB(MAO−B)阻害作用の主な理論的根拠は、症状による運動への効果を生じる線条体のドーパミン活性の増強にある。MAO−Bが特にドーパミン加水分解に関与するため、MAO−B阻害作用はドーパミンレベルを増加させる。記載されている作用機序によれば、ラサギリンによるMAO−B阻害作用が不可逆的であり、したがって新たなMAO−Bが産生されるまで(すなわち、約2〜3週間)、その阻害作用から得られる効果が存続するということにより、ラサギリンの活性がその薬物動態から切り離される。したがって、徐放方式でのラサギリン投与による効果は全くないと推測され得る。しかし、ラサギリンがアポトーシスまたはその他の経路の阻害を介した別の機序で神経保護作用を有し得ることを示唆する証拠が、最近得られている。さらに、ラサギリンは著しく代謝され、その主要な代謝産物である1−アミノインダンがMAO−B阻害作用と関係のない神経保護活性を有するということも知られている(Bar−Amら,2007;Weinrebら,2010)。
【0023】
ラサギリン、セレギリンおよびその他の構造的に関連するプロパルギルアミン誘導体は、一部はアポトーシスを減少させることにより、MAO−B阻害作用とは関係なくニューロンの生存を増加させる(Tattonら,2002)。この作用は、ミトコンドリアの膜透過性に影響を与えたり、酸化性ラジカルを除去したり、特定のアポトーシスシグナル伝達経路に関与したりするタンパク質のレベルまたは細胞内局在の変化により調節されている可能性が最も高い。ラサギリンおよびセレギリンはともに、他のプロパルギルアミン誘導体と同様に、各種傷害により誘発される細胞死からニューロンを保護するということが、パーキンソン病およびアルツハイマー病のような神経変性障害の細胞モデルおよび動物モデルで確認されている。プロパルギルアミン鎖により、多数の実験モデルにおいて神経保護と関連付けられてきた用量依存性の抗酸化作用および抗アポトーシス作用がもたらされる。最近の刊行物によれば、ラサギリンの神経保護作用はラサリギン(rasaligine)およびその代謝産物である1−アミノインダンの組合せに関連している可能性がある(Tazikら,2009;Bar−Am,2010)。
【0024】
一態様では、本発明は、薬学的に許容される担体と、プロパルギルアミン部分、アミノインダン部分もしくはプロパルギルアミン部分とアミノインダン部分の両方を含む活性薬剤またはその薬学的に許容される塩とを含む、前記活性薬剤の持続放出のために製剤化された医薬組成物を提供する。
【0025】
本発明の基礎となる概念は、後の実施例の節で示される知見に基づくものである。実施例1では、PDのMPTPマウスモデルにおいて、用量を漸増させたラサギリン(0.1、0.12または0.15mg/kg)の急性投与は、マウスのドーパミンレベルに対して実質的に同様の効果を示し、未処置(MPTP処置)マウスに比べてドーパミン量が約60%増加したのに対し、同じ3種類の用量の薬物を徐放方式で24時間にわたって投与したところ、ドーパミンレベルが未処置マウスに比べて、それぞれ57%、74%および88%という有意な用量反応性が見られたことを示す。このことは、MPTP処置したマウス脳において、徐放投与が即放投与に比べてドーパミンレベルに対して極めて有益な効果を有することを示唆している。実施例2では、同じマウスPDモデルを用いて、ラサギリン代謝産物である1−アミノインダンでマウスを処置した実験が記載されており、徐放方式で投与する1−アミノインダンによる処置により、溶媒(生理食塩水)処置マウスまたは同じ薬物を毎日の注射により投与したマウスに比べて、ドーパミンレベルが有意に回復したことを示す。これらの知見は、実施例3に記載されている実験によりさらに裏付けられる。実施例3では、6−OHDAラットPDモデルにおいて、徐放方式で投与するラサギリンで処置したラットでは、毎日の注射による同じ薬物で処置したラットに比べて、アンフェタミンに誘発された正味の回旋数(CW−CCW)において有意に向上した作用が観察されることを示す。
【0026】
実際に本明細書で初めて示されるように、ラサギリンを持続放出方式で送達した場合、薬物またはその活性代謝物である1−アミノインダンへの暴露が著しく延長されることにより、患者の状態を著しく向上させ得るさらに効果的な神経保護が可能になる。この概念によれば、パーキンソン病の治療に適応されるMAO−B阻害剤のラサギリンおよびセレギリンはともに、その他のプロパルギルアミン誘導体と同様に、活性薬剤を連続的に放出する「プロドラッグ」またはプロパギルアミン(propagylamine)/アミノインダン「送達媒体」であると考えることができる。このようなプロドラッグまたは送達媒体はそのMAO阻害活性に関係なく、様々な段階のアポトーシス過程の間に、上で定義される活性薬剤、すなわち、プロパルギルアミン部分、アミノインダン部分もしくはプロパルギルアミン部分とアミノインダン部分の両方を含む活性薬剤またはその薬学的に許容される塩への慢性的な持続的曝露によりニューロン細胞を保護する。
【0027】
本発明では、活性薬剤の任意の薬学的に許容される塩を使用することができる。薬学的に許容される塩の例としては、特に限定されるわけではないが、メシル酸塩、エシル酸塩、トシル酸塩、硫酸塩、スルホン酸塩、リン酸塩、カルボン酸塩、マレイン酸塩、フマル酸塩、酒石酸塩、安息香酸塩、酢酸塩、塩酸塩および臭化水素酸塩が挙げられる。
【0028】
特定の実施形態では、本発明の医薬組成物に含まれる活性薬剤は、N−プロパルギル−1−アミノインダン、その鏡像異性体、その代謝産物、その類似体または上記のうちのいずれかの薬学的に許容される塩である。
【0029】
具体的な実施形態では、活性薬剤は、例えば米国特許第6,630,514号に記載されているようなラセミ体のN−プロパルギル−1−アミノインダン、またはその薬学的に許容される塩である。
【0030】
他の具体的な一実施形態では、活性薬剤は、R(+)−N−プロパルギル−1−アミノインダン(ラサギリン)、そのS−鏡像異性体であるS−(−)−N−プロパルギル−1−アミノインダンまたはその薬学的に許容される塩である。より具体的な実施形態では、活性薬剤は、ラサギリンまたはS−(−)−N−プロパルギル−1−アミノインダンのいずれかのメシル酸塩、エシル酸塩、トシル酸塩、硫酸塩、スルホン酸塩、リン酸塩、カルボン酸塩、マレイン酸塩、フマル酸塩、酒石酸塩、安息香酸塩、酢酸塩、塩酸塩または臭化水素酸塩である。好適な実施形態では、活性薬剤は、例えば米国特許第5,532,415号に記載されているメシル酸ラサギリン;例えば米国特許第5,599,991号に記載されているラサギリンエシル酸塩もしくはラサギリン硫酸塩;または例えば米国特許第6,630,514号に記載されているラサギリン塩酸塩、より好ましくはメシル酸ラサギリンである。
【0031】
さらなる具体的な実施形態では、活性薬剤はラサギリン代謝産物である1−アミノインダンまたはその薬学的に許容される塩である。
【0032】
さらに他の具体的な実施形態では、活性薬剤は、N−プロパルギル−1−アミノインダンの類似体、その鏡像異性体またはその薬学的に許容される塩である。このような類似体の例としては、米国特許第5,486,541号に記載されている化合物、例えば特に限定されないが、4−フルオロ−N−プロパルギル−1−アミノインダン、5−フルオロ−N−プロパルギル−1−アミノインダンおよび6−フルオロ−N−プロパルギル−1−アミノインダンなど;米国特許第6,251,938号に記載されている化合物、例えば特に限定されないが、3−(N−メチル,N−プロピル−カルバミルオキシ)−α−メチル−N’−プロパルギルフェネチルアミン;3−(N,N−ジメチル−カルバミルオキシ)−α−メチル−N’−メチル,N’−プロパルギルフェネチルアミン;3−(N−メチル,N−ヘキシル−カルバミルオキシ)−α−メチル−N’−メチル,N’−プロパルギルフェネチルアミン;3−(N−メチル,N−シクロヘキシル−カルバミルオキシ)−α−メチル−N’−メチル,N’−プロパルギルフェネチルアミンなど;および3−(N−メチル,N−ヘキシル−カルバミルオキシ)−α−メチル−N’−メチル,N’−プロパルギルフェネチルアミンなど;米国特許第6,303,650号に記載されている化合物、例えば特に限定されないが、6−(N−メチル,N−エチル−カルバミルオキシ)−N’−プロパルギル−1−アミノインダン;6−(N,N−ジメチル−カルバミルオキシ)−N’−メチル−N’−プロパルギル−1−アミノインダン;6−(N−メチル,N−エチル−カルバミルオキシ−N’−プロパルギル−1−アミノテトラリン;6−(N,N−ジメチル−チオカルバミルオキシ)−1−アミノインダン;6−(N−プロピル−カルバミルオキシ)−N’−プロパルギル−1−アミノインダン;5−クロロ−6−(N−メチル,N−プロピル−カルバミルオキシ)−N’−プロパルギル−1−アミノインダン;および6−(N−メチル),N−プロピル−カルバミルオキシ)−N’−プロパルギル−1−アミノインダンなど;ならびに米国特許第6,462,222号に記載されている化合物、例えば特に限定されないが、6−(N−メチル,N−エチル−カルバミルオキシ)−N’−メチル,N’−プロパルギル−1−アミノインダンなどが挙げられる。
【0033】
他の特定の実施形態では、本発明の医薬組成物に含まれる活性薬剤は、プロパルギルアミン、脂肪族プロパルギルアミンまたはその薬学的に許容される塩である。
【0034】
具体的な一実施形態では、活性薬剤はプロパルギルアミンまたはその薬学的に許容される塩である。
【0035】
他の具体的な実施形態では、活性薬剤は、米国特許第5,169,868号、米国特許第5,840,979号または米国特許第6,251,950号に記載されている脂肪族プロパルギルアミン、例えば特に限定されないが、N−(1−ヘプチル)プロパルギルアミン;N−(1−オクチル)プロパルギルアミン;N−(1−ノニル)プロパルギルアミン;N−(1−デシル)プロパルギルアミン;N−(1−ウンデシル)プロパルギルアミン;N−(1−ドデシル)プロパルギルアミン;N−(2−ブチル)プロパルギルアミン;N−(2−ペンチル)プロパルギルアミン;N−(2−ヘキシル)プロパルギルアミン;N−(2−ヘプチル)プロパルギルアミン;N−(2−オクチル)プロパルギルアミン;N−(2−ノニル)プロパルギルアミン;N−(2−デシル)プロパルギルアミン;N−(2−ウンデシル)プロパルギルアミン;N−(2−ドデシル)プロパルギルアミン;N−(1−ブチル)−N−メチルプロパルギルアミン;N−(2−ブチル)−N−メチルプロパルギルアミン;N−(2−ペンチル)−N−メチルプロパルギルアミン;(1−ペンチル)−N−メチルプロパルギルアミン;N−(2−ヘキシル)−N−メチルプロパルギルアミン;(2−ヘプチル)−N−メチルプロパルギルアミン;N−(2−デシル)−N−メチルプロパルギルアミン;(2−ドデシル)−N−メチルプロパルギルアミンなど;その鏡像異性体;またはその薬学的に許容される塩である。
さらなる特定の実施形態では、本発明の医薬組成物に含まれる活性薬剤は、セレギリン、デスメチルセレギリン、パルギリンまたはクロルギリンである。
【0036】
さらなる特定の実施形態では、本発明の医薬組成物に含まれる活性薬剤は、CGP3466としても知られ、Zimmermannら(1999)に記載されている、(N−メチル−N−プロパルギル)−10−アミノメチル−ジベンゾ[b,f]オキセピンである。
【0037】
上で引用した米国特許およびその他の刊行物はすべて、本明細書において完全に開示された場合と同様に、その内容全体が参照により本明細書に組み込まれる。
【0038】
本明細書で互換的に使用される、「持続放出」、「制御放出」または「徐放」という用語は、ある期間にわたって身体に吸収されるように活性薬剤をその製剤から放出する様式を表す。活性薬剤の持続放出性の製剤化は、例えば、有効成分がコーティングから徐々に規則的に浸出するように、身体の中で徐々に溶解する入り組んだ物質中に活性薬剤を埋め込むことにより、または薬物が半透性の層から徐々に出て行くように、活性薬剤で膨潤させて表面がほぼ不透性のゲルを形成することにより達成し得る。
【0039】
制御放出性製剤の開発における主な原則は、意図する部位(標的化)において、一定速度で、必要とされる治療域内で物質を放出することである。血中の一定濃度の活性物質を長期間維持する、膨潤システムおよび浸透システムのような溶媒制御システムの原理に基づく機序により、少ない副作用でより効果的な薬物レベルが達成される。言い換えれば、治療域とは、効果が得られる量(有効量)と所望の効果よりも副作用の方が大きい量の間の投薬量のことである。その範囲になるまで、各化合物の個々のバイオアベイラビリティー、作用部位および吸収特性に従って、各薬物の溶出プロファイルを設計するべきである。
【0040】
本発明の医薬組成物は、薬物、すなわち活性薬剤の制御放出を提供するであろう。特定の実施形態では、薬物がゼロ次、一次、二次またはその他の放出プロファイル(N次)の制御放出方式で医薬組成物から放出される。薬物の制御放出は、好ましくは緩徐であるべきであり、特定の実施形態では、連続的な薬物徐放、パルス状の薬物放出、多相性の薬物放出またはそれらの組合せが得られるように医薬組成物を製剤化する。
【0041】
本発明の医薬組成物は、例えば、Remington:The Science and Practice of Pharmacy,第19版,1995に記載されている従来の技術により調製してもよく、任意の従来の形態であってもよく、また各種の用量で提供してもよい。
【0042】
組成物を任意の適当な投与経路、例えば、静脈内、動脈内、筋肉内、皮下または腹腔内投与用に製剤化することができるが、好ましくは、組成物を経口投与用に製剤化する。
【0043】
用量は患者の状態によって異なり、実施者が適切であると判断する量で決定される。具体的な実施形態では、用量は、60kgの成人では1日当たり0.1〜2.0mg、好ましくは0.2〜1.5mg、より好ましくは0.5〜1.0mgである。本発明の組成物は、種々の継続期間、例えば週、月、年または10年で、例えば継続的に、毎日、1日2回、1日3回または1日4回、投与してよい。
【0044】
本発明の医薬組成物は、例えば、既知の技術により適当な分散剤、湿潤剤または懸濁化剤を用いて製剤化し得る、無菌の水性または油性懸濁注射液の形態であってよい。また無菌注射用製剤は、無毒の非経口的に許容される希釈剤または溶剤での無菌の注射溶液または懸濁液注射であってもよい。使用し得る許容される溶媒および溶剤としては、水、リンガー溶液および等張塩化ナトリウム溶液が非限定的に挙げられる。
【0045】
本発明の医薬組成物は、経口投与用に製剤化する場合、錠剤、トローチ剤(troches、lozenges)、水性もしくは油性懸濁剤、分散性の粉末剤もしくは顆粒剤、乳剤、硬もしくは軟カプセル剤、またはシロップ剤もしくはエリキシル剤の形態であってよい。経口使用のための医薬組成物は、当業者に公知の医薬組成物製造のための任意の方法に従って調製してよく、また医薬品として洗練された味の良い製剤を作製するために、甘味剤、着香剤、着色剤および保存剤から選択される1つ以上の薬剤をさらに含んでもよい。錠剤は、錠剤の製造に適した無毒の薬学的に許容される添加剤と混合された活性薬剤を含有する。このような添加剤は、例えば、不活性希釈剤、例えば炭酸カルシウム、炭酸ナトリウム、ラクトース、リン酸カルシウムまたはリン酸ナトリウムなど;造粒剤および崩壊剤、例えばコーンスターチまたはアルギン酸;結合剤;ならびに滑沢剤であり得る。好ましくは、消化管での分解および吸収を遅延させるために既知の技術を用いて錠剤をコーティングして、長期間にわたる薬物の持続放出をもたらす。例えば、モノステアリン酸グリセリルまたはジステアリン酸グリセリルのような時間遅延物質を使用し得る。また米国特許第4,256,108号、同第4,166,452号および同第4,265,874号に記載されている技術を用いて錠剤をコーティングし、制御放出用の浸透圧性の治療錠剤を形成してもよい。また本発明の医薬組成物は、2つ以上の異なる顆粒層が、それぞれ異なる様式の薬物放出が生じるように製剤化され互いに重なり合って圧縮されている、二層テーブルの形態であってもよい。また本発明の経口医薬組成物は、水中油型乳剤の形態であってもよい。
【0046】
また本発明の医薬組成物を制御放出マトリックスとして、例えば、溶出させる液体(in vitro)または胃腸管液(in vivo)と接触する親水性ポリマーを膨潤させて形成されたゲルを介した能動拡散を有することにより可溶性の活性薬剤の放出が制御される、制御放出マトリックス錠剤として製剤化してもよい。このようなゲルを形成することができるものとして、例えば、セルロース誘導体、特にセルロースエーテル、例えばヒドロキシプロピルセルロース、ヒドロキシメチルセルロース、メチルセルロースまたはヒドロキシプロピルメチルセルロースなどのポリマーが数多く記載されており、様々な市販グレードの上記エーテルの中には、かなり高い粘性を示すものがある。他の形状では、組成物は制御放出用にマイクロカプセル化剤形で製剤化された活性薬剤を含む。マイクロカプセル化剤形では、活性薬剤の小滴がコーティングまたは膜に取り囲まれて数マイクロメートルから数ミリメートルの範囲の粒子を形成している。
【0047】
考慮される別の製剤は生分解性ポリマーに基づくデポーシステムである。このシステムでは、ポリマーが分解されるときに活性薬剤が徐々に放出される。最も一般的な生分解性ポリマーのクラスは、乳酸、グリコール酸またはこれらの組合せから調製される加水分解に不安定なポリエステルである。
【0048】
本発明の医薬組成物は1つ以上の薬学的に許容される添加剤を含み得る。例えば、錠剤は、少なくとも1つの賦形剤、例えばラクトース、エチルセルロース、微結晶性セルロース、ケイ化微結晶性セルロース;少なくとも1つの崩壊剤、例えば架橋ポリビニルピロリジノン;少なくとも1つの結合剤、例えばポリビニルピリドン、ヒドロキシプロピルメチルセルロース;少なくとも1つの界面活性剤、例えばラウリル硫酸ナトリウム;少なくとも1つの滑剤、例えばコロイド状二酸化ケイ素;および少なくとも1つの滑沢剤、例えばステアリン酸マグネシウムを含み得る。
【0049】
後の実施例4〜6では、不活性なペレットをコーティングしている薬物層と、薬物層をコーティングしている持続放出性の、すなわち機能性の層とを含む、3種類のメシル酸ラサギリン持続放出(ER)コーティングペレット(ER層が15%、22%および28%)の調製に関して記載する。薬物層の調製では、ポビドンUSP(PVP K29/32)を蒸留水とエタノールの混合物に溶解させ、形成された溶液に薬物を溶解させ、次いで、この溶液に超微末タルクを分散させて加え、均一な懸濁液を形成し、600〜710μm(直径)の糖球状顆粒にコーティングした。各種厚さのERコーティングフィルムの調製では、1つの溶液を調製し、様々な層の厚さを生じる様々な量のスプレーされた溶液に対応する、コーティング工程中の様々な時点での試料をそこから採取した。溶液は、アセトンとエタノールの混合物に溶解したEthocel 45cps(エチルセルロース;放出制御ポリマー);および蒸留水に溶解したポリエチレングリコール(PEG)4000で構成され、次いでこの2つを混合して均質な溶液を形成した。この機能性の溶液を上記の薬物充填ペレットにコーティングして、各種厚さのERフィルムを形成した。各種ERコーティングペレットの溶出プロファイルを、USP(米国薬局方)装置1(バスケット)により、腸内条件を模した腸液溶液(IFS、pH6.8)を用いて軸回転速度100rpmおよび温度37℃で評価した。示されるように、放出速度はフィルムの厚さに影響され、その放出パターンは機能層が厚くなるほど遅かった。
【0050】
実施例7〜8では、不活性なペレットをコーティングしている薬物層と、前記薬物層をコーティングしているサブコーティング層と、サブコーティング層をコーティングしている機能層とを含む、2種類のメシル酸ラサギリン持続放出(ER)コーティングペレット(ER層が15%および18%)の調製に関して記載する。薬物層の調製では、ポビドン(PVP K25)を蒸留水とエタノールの混合物に溶解させ、形成された溶液に薬物を溶解させ、次いで、この溶液に超微末タルクを分散させて加え、均一な懸濁液を形成し、次いでこれを600〜710μmの糖球状顆粒にコーティングした。PVP K25を蒸留水とエタノールの混合物に溶解させてサブコーティング溶液を調製し、次いで、これを薬物充填ペレットにコーティングした。各種厚さのERコーティングフィルムの調製では、1つの溶液を調製し、様々な層の厚さを生じる様々な量のスプレーされた溶液に対応する、コーティング工程中の様々な時点での試料をそこから採取した。溶液は、アセトンとエタノールの混合物に溶解したEthocel 45cps;および蒸留水に溶解したPEG3000で構成され、次いでこの2つを混合して均質な溶液を形成した。この溶液に超微末タルクを蒸留水中に分散させて加え、均一な懸濁液を形成し、次いで、これをサブコーティングしたペレットにコーティングして、各種厚さのERフィルムを形成した。次いで、得られたペレットをAerosil200と乾燥混合した。上記2種類のERコーティングペレットの溶出プロファイルを、USP装置1により、(i)腸内条件を模した腸液溶液(IFS、pH6.8);(ii)空腹条件を模した胃液溶液(GFS、pH1.2)で2時間、次いで、IFSでさらに20時間;および(iii)満腹条件を模した酢酸緩衝液(pH4.5)を用いて、100rpmおよび温度37℃で評価した。示されるように、pH1.2〜6.8の範囲では、ER層中のpH依存性ポリマーにより放出速度が一定に維持され(溶出試験で±10%の許容範囲内)、また安定性促進条件への曝露にもかかわらず3か月間安定であった。機能層の厚さの違いにより、15%のERコーティングペレットからの放出速度は18%のERコーティングペレットよりも速かった。
【0051】
実施例9では、不活性なペレットをコーティングしている薬物層と、サブコーティング層と、外側機能層とを含む、高いパーセンテージ(27%)のERコーティング層を有する第三の種類のメシル酸ラサギリンERコーティングペレットの調製について記載する。本実施例では、ペレットを、実施例7〜8で使用したAerosil200の代わりに二酸化ケイ素と乾燥混合した。上記ペレットの溶出プロファイルを、USP装置1により、IFS(pH6.8)を用いて、100rpmおよび温度37℃で評価した。示されるように、この場合の放出パターンは、機能層の厚さの違いにより、実施例7〜8のペレットで観察されたパターンよりも遅いものであった。
【0052】
様々な薬物放出プロファイルのために設計された、サブコーティング層を有するまたは有さないメシル酸ラサギリンERペレットに関して実施例10でさらに記載する。
【0053】
経口投与用の24時間型持続放出性製剤を設計する場合、全放出時間を通して薬物が吸収される、すなわち、十二指腸および結腸の両方を含めた胃腸管のすべての部分から吸収される必要がある。実施例11では、薬物動態実験に関して記載する。この実験では、ラットの結腸、十二指腸または頸静脈に水溶液としてラサギリンの単回ボーラス投与を行い、投与の5分前、ならびに投与の5、15、30、50、90、150および200分後にラットから血液試料を採取し、ラサギリンとその代謝産物の血漿レベルの両方を測定した。示されるように、結腸および十二指腸投与群の親化合物のT1/2は、IV投与後のT1/2よりも長かった。さらに、IVと十二指腸投与で同様の血中濃度曲線下面積(AUC)値が算出され、このことは完全な経口吸収を示唆するものであった。また、結腸投与後のAUC値はIV投与のAUC値の約28%であり、このことは結腸吸収の実現可能性を示すものであった。
【0054】
上記各種メシル酸ラサギリンERコーティングペレットに関して得られた溶出プロファイルおよび胃腸管の各種部分からのラサギリン吸収を示す上記実験を考慮して、特定の実施形態では、本発明の医薬組成物を経口投与用に製剤化する。具体的な実施形態では、医薬組成物は、実施例10に記載のいくつかのメシル酸ラサギリンERペレットに関して示されているように、当該技術分野で公知の任意の従来の方法により、混合してカプセルもしくは小袋に充填される、または圧縮して錠剤にされる、顆粒、粒、ビーズまたはペレットの形態の固体であってよい。例えば、活性薬剤が少なくとも2つの分離した層に存在する錠剤、すなわち、二層または多層錠が提供され、このような錠剤では、各層が任意に、中間の不活性な層、例えば1つ以上の崩壊剤を含む層により分離されている。また医薬組成物は半固体または液体系であってもよい。
【0055】
特定の実施形態では、本発明の医薬組成物は、経口投与用に製剤化される場合、モノリシックなマトリックス、すなわち、個別の大きさと形状を有する三次元的に安定なマトリックス材料を含む構造;錠剤、例えば二層もしくは多層錠、マトリックス錠、崩壊錠、溶解錠またはチュアブル錠など;または顆粒、粒、ビーズもしくはペレットを充填したカプセルもしくは小袋の形態である。他の特定の実施形態では、本発明の医薬組成物は、経口投与用に製剤化される場合、ポリマーが分解されるときに活性薬剤が徐々に放出される、生分解性ポリマーに基づくデポーシステムの形態である。最も一般的な生分解性ポリマーのクラスは、乳酸、グリコール酸またはこれらの組合せから調製される加水分解に不安定なポリエステルである。上記特定のモノマーから調製される生分解性ポリマーの例としては、ポリ(D,L−ラクチド)(PLA)、ポリグリコリド(ポリグリコール酸;PGA)およびコポリマーポリ(D,L−ラクチド−co−グリコリド)(PLGA)が非限定的に挙げられる。
【0056】
特定の具体的な実施形態では、本発明は、上で定義される医薬組成物、すなわち、プロパルギルアミン部分、アミノインダン部分もしくはプロパルギルアミン部分とアミノインダン部分の両方を含む活性薬剤またはその薬学的に許容される塩を含む医薬組成物を提供し、前記医薬組成物は、USP装置1(バスケット)において、50〜150rpm、好ましくは100rpmで、7.4以下のpH値、好ましくは1.2〜6.8のpH値、37℃で、以下のような溶出プロファイルを有する。
【0057】
【表1】
【0058】
好適な医薬組成物は、USP装置1(バスケット)において、50〜150rpm、好ましくは100rpmで、7.4以下のpH値、好ましくは1.2〜6.8のpH値、37℃で、以下のような溶出プロファイルを有する。
【0059】
【表2】
【0060】
より具体的な実施形態では、この組成物に含まれる活性薬剤は、N−プロパルギル−1−アミノインダン;その鏡像異性体、すなわち、ラサギリンまたはS−(−)−N−プロパルギル−1−アミノインダン;その代謝産物、より具体的には1−アミノインダン;その類似体または上記のもののいずれかの薬学的に許容される塩である。最も具体的な実施形態では、この組成物に含まれる活性薬剤はラサギリンまたはその薬学的に許容される塩である。
【0061】
特定の実施形態では、本発明の医薬組成物は、即放性剤形に比べて投与の際のCmaxおよび変動指数が小さく、望ましくない副作用が少ない。本明細書で使用される「Cmax」という用語は治療薬の最大血漿中濃度を表し、また本明細書で使用される「変動指数」という用語は、薬物投与後の時間の関数としての治療薬の血清中濃度の変動を表す。ラサギリンの望ましくない副作用としては、重篤なアレルギー反応(発疹;じん麻疹;かゆみ;呼吸困難;胸部逼迫;口腔、顔面、口唇または舌の腫脹);黒便または血便;血尿;霧視;性的能力または性欲の変化;胸痛;錯乱;抑うつ;瞳孔散大;速いまたは不規則な心拍;発熱;幻覚;じっとしていられない;手足の痺れまたは刺痛;片側脱力;発作;光敏感性;重度の頭痛;皮膚の変化;頸部の痛みまたは硬直;振戦;思考または歩行の困難;原因不明の嘔気または嘔吐;異常発汗;視覚または発話の障害;下痢;めまい;傾眠;口渇;インフルエンザ様の症状;頭痛;関節痛;意識もうろう;不眠;胃の不調;および鼻詰まりが挙げられるが、これらに限定されない。
【0062】
別の態様では、本発明は、
(i)不活性なペレットコアと、
(ii)上記ペレットコアをコーティングしている薬物層であって、プロパルギルアミン部分、アミノインダン部分もしくはプロパルギルアミン部分とアミノインダン部分の両方を含み、任意に結合剤および/またはフィルム形成ポリマーと適切に混合され、さらに任意に滑剤と混合されている活性薬剤またはその薬学的に許容される塩を含む薬物層と、
(iii)任意に、前記薬物層をコーティングしている隔離/保護サブコーティング層と、
(iv)もし存在すれば前記サブコーティング層を、または前記薬物層をコーティングしている持続放出性コーティング層と
を含む、持続放出性(ER)ペレットを提供する。
【0063】
本発明のERペレットは、上記薬物層をコーティングしている隔離/保護サブコーティング層を任意に含み得る。このサブコーティング層の役割は、活性物質層を外側のERコーティングから分離して、その安定性に影響を与え、活性成分(API)の分解産物を生じ得る可能性のある活性薬剤との相互作用から保護することである。特定の実施形態では、サブコーティング層は、フィルム形成ポリマーおよび任意に滑剤を含む。
【0064】
本発明のERペレットは、存在すればサブコーティング層を、または薬物層をコーティングしている、本明細書では「機能層」とも呼ばれる外側ERコーティング層を含む。
【0065】
特定の実施形態では、ERコーティング層は、少なくとも1つのpH非依存性ポリマー、すなわち水膨潤性/不水溶性/疎水性ポリマー、および任意に細孔形成剤を含み、この場合、持続放出性ペレットは、pH非依存性のin vitro放出特性を有する。他の実施形態では、機能層は、pH非依存性ポリマー、細孔形成剤として働く親水性放出調節ポリマー、および任意に疎水性または親水性の可塑剤、および/または滑剤を含む。さらなる特定の実施形態では、ERコーティング層は、pH依存性の腸溶性コーティングポリマーとpH非依存性ポリマーの混合物を含み、この場合、持続放出性ペレットは、酸性pHまたは生理的pH(すなわち、7.4以下のpH値)においてゼロ次に近いin vitro放出特性を有する。
【0066】
製薬学的用途のための結合剤は、その粘着性および凝集性により固形剤形の製造で使用される、糖類および天然または合成起源のポリマーのような親水性物質である。結合剤の役割は、粉末の凝集性を増加させることにより賦形を補助し、必要な結合力を顆粒剤および錠剤に与えることである。結合剤は、これらの製剤の外観、硬さ、および破砕性を向上させるが、活性物質の崩壊および溶出速度に影響を与えることを目的としていない。これまで一般に使用されてきた天然起源の結合剤としては、アラビアゴム、ゼラチン、デンプンおよび加水分解デンプンが挙げられる。これらの物質は、合成起源の結合剤に取って代わられており、その中で最も重要なのはポビドンおよび各種セルロース誘導体である。本発明のERペレットの薬物層コーティングの活性薬剤と混合することができる結合剤の例としては、ポリビニルピロリドン(PVP)、ヒドロキシプロピルメチルセルロース(HPMC)、ヒドロキシプロピルセルロース(HPC)、微結晶性セルロースおよびこれらの組合せが非限定的に挙げられる。結合剤は、ペレットの総重量の約0.5%〜約20%、好ましくは約0.5%〜約10%の量で存在していてよい。
【0067】
本明細書で使用される「フィルム形成ポリマー」という用語は、硬化して密着性のフィルムを形成することができるポリマーを表す。さらに、コーティングに不可欠なこのポリマーの物理的特性は、フィルムを形成する能力またはコーティングする材料へのある種の粘着性である。フィルム形成ポリマーの例としては、PVP、HPMC、HPC、微結晶性セルロースおよびこれらの組合せが非限定的に挙げられる。フィルム形成ポリマーは、薬物層に含まれる場合、薬物層の総重量の90%以下、好ましくはペレットの総重量の約0.5%〜約20%の量で存在していてよい。サブコーティング層中のフィルム形成ポリマーの量は、サブコーティング層の総重量の100%以下、好ましくはペレットの総重量の約0.5%〜約10%である。
【0068】
滑剤は通常、顆粒および粉末の摩擦および表面電荷を低減して流動性を増強するために、医薬組成物に添加される。さらに滑剤は、抗粘着剤としてコーティング工程で使用される。タルクおよびモノステアリン酸グリセリルのような特定の滑剤が低い製品温度での粘着傾向を低減する抗粘着剤として、コーティング製剤に一般的に用いられる。二酸化ケイ素コロイドのような他の滑剤は、その小さい粒子サイズおよび特有の大きい表面積により、錠剤化およびカプセル化のような数多くの工程において乾燥粉末の流動性を向上させるために利用される、望ましい流動特性を与える。滑剤の非限定的な例としては、タルク、特に超微末タルク、コロイド状二酸化ケイ素、モノステアリン酸グリセリルおよびこれらの組合せが挙げられる。
【0069】
滑剤は、薬物層に含まれる場合、薬物層の総重量の30%以下、好ましくはペレットの総重量の約0.5%〜約5%の量で存在していてよい。滑剤がサブコーティング層に含まれる場合、その量はサブコーティング層の総重量の約10%以下、好ましくはペレットの総重量の約0.5%〜約5%であってよい。
【0070】
本発明のERペレットに含まれ得るpH非依存性ポリマーの例としては、エチルセルロース、Surelease(登録商標)、アクリル酸エステルとメタクリル酸エステルのコポリマー、例えばEudragit(登録商標)RL(ポリ(アクリル酸エチル、メタクリル酸メチル、メタクリル酸トリメチルアンモニオエチルクロリド)、1:2:0.2)、Eudragit(登録商標)RS(ポリ(アクリル酸エチル、メタクリル酸メチル、メタクリル酸トリメチルアンモニオエチルクロリド)、1:2:0.1)、Eudragit(登録商標)NE(ポリ(アクリル酸エチル、メタクリル酸メチル)、2:1)など、およびこれらの組合せが非限定的に挙げられる。pH非依存性ポリマーは、ペレットの総重量の約10%〜約50%、好ましくは約10%〜約30%の量で存在していてよい。
【0071】
本発明のERペレットに含まれ得るpH依存性の腸溶性コーティングポリマーの例としては、Eudragit(登録商標)S(ポリ(メタクリル酸、メチルメタクリル酸)、1:2)、Eudragit(登録商標)L55(ポリ(メタクリル酸、アクリル酸エチル)、1:1)、Kollicoat(登録商標)(ポリ(メタクリル酸、アクリル酸エチル)、1:1)、ヒドロキシプロピルメチルセルロースフタラート(HPMCP)、アルギン酸、カルボキシメチルセルロースおよびこれらの組合せが非限定的に挙げられる。pH依存性の腸溶性コーティングポリマーは、ペレットの総重量の約10%〜約50%、好ましくは約10%〜約30%の量で存在していてよい。
【0072】
本明細書で使用される「細孔形成剤」という用語は、体内環境で溶解して、コーティング層を通過する活性薬剤の拡散速度を増加させる開口細孔をマトリックス中に形成する物質を表す。形成される細孔の大きさは、使用する固体粒子材料の大きさによりある程度まで制御することができる。細孔を均一にするために、粒子材料を次第に微細になるメッシュ篩で選別して、所望の粒子の大きさにする。本発明のERペレットに含まれ得る細孔形成剤は無機物質または有機物質のいずれかであり、このような物質としては、例えば、ポリビニルピロリドン(PVP)、ポリエチレングリコール(PEG)、HPMC、HPC、メチルセルロース、1,2−プロピレングリコール、ラクトース、スクロース、タルク、特に超微末タルクおよびこれらの組合せが挙げられる。細孔形成剤は、ペレットの総重量の約0.1%〜約20%、好ましくは約0.1%〜約10%の量で存在していてよい。
【0073】
本明細書で使用される「親水性放出調節ポリマー」という用語は、水溶性であり、かつ活性薬剤の放出を制御するポリマーを表す。しかし、特定の実施形態では、本発明のERペレットのERコーティング層に含まれる親水性放出調節ポリマーは、実際には細孔形成剤として働く。親水性放出調節ポリマーの例としては、PVP、PEG、HPMC、HPCおよびこれらの組合せが非限定的に挙げられる。親水性放出調節ポリマーは、ペレットの総重量の約0.1%〜約20%、好ましくは約0.1%〜約10%の量で存在していてよい。
【0074】
本明細書で使用される「可塑剤」という用語は、本発明のERペレットで使用するポリマーを可塑化または軟化することができる、任意の化合物または化合物の組合せを包含する。ERコーティング層の製造において、可塑剤は、使用するポリマーまたはポリマーの組合せの融解温度またはガラス転移温度(軟化点温度)を下げること、前記ポリマーまたはポリマーの組合せの平均分子量の幅を広げること、およびコーティング溶液の簡便な加工のために前記ポリマーまたはポリマーの組合せの粘度をさらに減少させることができる。可塑剤の非限定的な例としては、セバシン酸ジブチル;フタル酸ジブチル;クエン酸トリエチルのようなクエン酸エステルおよびトリアセチン;プロピレングリコール;PEG、ポリ(プロピレングリコール)およびポリ(エチレン/プロピレングリコール)のような低分子量ポリ(アルキレンオキシド);およびこれらの組合せが挙げられる。可塑剤は、ペレットの総重量の約0.1%〜約20%、好ましくは約0.1%〜約10%の量で存在していてよい。
【0075】
本発明のERペレットの薬物層コーティングは、上で定義される任意の活性薬剤、すなわち、プロパルギルアミン部分、アミノインダン部分もしくはプロパルギルアミン部分とアミノインダン部分の両方を含む任意の活性薬剤またはその薬学的に許容される塩を含み得る。具体的な実施形態では、活性薬剤は、N−プロパルギル−1−アミノインダン、その鏡像異性体、その代謝産物、その類似体またはその薬学的に許容される塩から選択される。より具体的な実施形態では、活性薬剤はラサギリンまたはその薬学的に許容される塩である。
本発明のERペレットは浸透圧/等張化剤のような不活性成分をさらに含み得る。このような薬剤は、パルス状の薬物送達が必要な場合に崩壊時間を制御するために一般的に使用される。ERペレットの調製で使用し得る適当な浸透圧/等張化添加剤の例としては、塩化ナトリウムおよびマンニトールが非限定的に挙げられる。浸透圧/等張化剤は、ERペレットに含まれる場合、ペレットの総重量の20%以下、好ましくは約0.5%〜約10%の量で存在していてよい。
【0076】
本明細書で例示される特定の実施形態では、本明細書で例示されるERペレットは、不活性なペレットコア;フィルム形成ポリマー/結合剤としてのPVPおよび滑剤としての超微末タルクと混合した活性薬剤を含む薬物層;ならびにpH非依存性ポリマーとしてエチルセルロースおよび細孔形成剤としてPEGを含むERコーティング層を含み、前記フィルム形成ポリマー/結合剤の量は、薬物層の総重量の90%以下、またはペレットの総重量の約0.5%〜約20%であり;前記滑剤の量は、薬物層の総重量の30%以下、またはペレットの総重量の約0.1%〜約10%であり;前記pH非依存性ポリマーの量は、ERコーティング層の総重量の約50%〜約90%、またはペレットの総重量の約10%〜約30%であり;かつ前記細孔形成剤の量は、ERコーティング層の総重量の約1%〜約20%、またはペレットの総重量の約0.1%〜約10%である。
【0077】
本明細書で例示されている他の特定の実施形態では、本発明のERペレットは、不活性なペレットコア;フィルム形成ポリマー/結合剤としてのPVPおよび滑剤としての超微末タルクと混合した前記活性薬剤を含む薬物層;フィルム形成ポリマーとしてPVPを含む隔離/保護サブコーティング層;ならびにpH非依存性ポリマーとしてエチルセルロース、細孔形成剤としてPEGおよび滑剤として超微末タルクを含むERコーティング層を含み、前記薬物層中の前記フィルム形成ポリマー/結合剤の量は、薬物層の総重量の90%以下、またはペレットの総重量の約0.5%〜約20%であり;前記薬物層中の前記滑剤の量は、薬物層の総重量の30%以下、またはペレットの総重量の約0.1%〜約10%であり;前記サブコーティング層中の前記フィルム形成ポリマーの量は、サブコーティング層の総重量の100%以下、またはペレットの総重量の約0.5%〜約20%であり;前記pH非依存性ポリマーの量は、ERコーティング層の総重量の約50%〜約90%、またはペレットの総重量の約10%〜約30%であり;前記細孔形成剤の量は、ERコーティング層の総重量の約1%〜約20%またはペレットの総重量の約0.1%〜約10%であり;かつ前記ERコーティング層中の前記滑剤の量は、ERコーティング層の総重量の約0.1%〜約20%、またはペレットの総重量の約0.1%〜約10%である。
【0078】
さらに別の態様では、本発明は、上で定義されるERペレットを含む経口医薬組成物を提供する。特定の実施形態では、この組成物に含まれるERペレットは、1つ以上の適当な添加剤と混合されて、カプセルに充填されているか、または錠剤に圧縮されている。このようなカプセル剤または錠剤の調製は、当該技術分野で公知の任意の適当な技術を用いて行い得る。
【0079】
経口医薬組成物の調製で使用し得る適当な添加剤の例としては、二酸化ケイ素および上で定義される当該技術分野で公知の他の滑剤が非限定的に挙げられる。
【0080】
錠剤賦形剤は錠剤またはカプセルの大きさを膨らませて、作製を実用的なものにし、また消費者が使用しやすいものにする。賦形剤でかさを増加させすることにより、患者が取り扱うのに適した体積を最終製品にもたせることが可能となる。好ましい賦形剤は、不活性で、製剤の他の成分と適合性があり、非吸湿性であり、比較的安価で、圧縮可能であり、好ましくは無味であるか味が良くなければならない。植物性セルロース(純植物性賦形剤)は、錠剤または硬ゼラチンカプセル剤で一般的な賦形剤である。二塩基性リン酸カルシウムがもう1つの一般的な錠剤賦形剤である。様々な植物油脂を軟ゼラチンカプセル剤に使用することができる。錠剤賦形剤としては、例えば、ラクトース、マンニトール/Parteck(登録商標)、ソルビトール、デンプンおよびこれらの組合せが挙げられる。
【0081】
崩壊剤は、消化管内で錠剤が水分により崩れるときに膨張して溶解し、吸収されるべき有効成分を放出する。崩壊剤の種類には、水分吸収促進剤および錠剤破壊促進剤がある。これらは、錠剤が水分と接触したときに錠剤を迅速に小片に崩壊させて、溶解を促進する。崩壊剤の非限定的な例としては、架橋ポリビニルピロリドン(クロスポビドン)、ナトリウム/カルシウムカルボキシメチルセルロース(CMC)、低置換度クロスカルメロースナトリウムヒドロキシプロピルセルロース、炭酸水素ナトリウム、デンプン、デンプングリコール酸ナトリウムおよびこれらの組合せが挙げられる。
【0082】
特定の加工特性を向上させるために、錠剤およびカプセル製剤に滑沢剤を少量添加する。より具体的には、このような薬剤は、成分が凝集したり、打錠機やカプセル充填機に付着したりするのを防ぐ。また滑沢剤は、固形物とダイ壁間の摩擦が少ない状態での錠剤の成形および排出を可能にする。滑沢剤の例としては、グリセリルベヘナート、ステアリン酸、タルク、ステアリン酸亜鉛、ステアリン酸カルシウムおよびこれらの組合せが非限定的に挙げられる。
【0083】
後の実施例の節で、具体的にラサギリンおよびその代謝産物である1−アミノインダンに関して示されるように、本発明の医薬組成物は、パーキンソン病、さらにはこの組成物に含まれる活性薬剤が有効であることが開示されている、他の任意の神経変性疾患または状態、および神経系の損傷の治療に有用である。上記神経変性疾患または状態の例としては、アルツハイマー病;精神刺激薬、オピエート、麻薬およびバルビツール酸塩の離脱症状を含めた離脱症状;うつ病;腎機能、および空間学習能力により証明されている認知機能を含めた、加齢による退化加齢;ヒトおよび非ヒトにおける下垂体性クッシング病;ヒトおよび動物の両方における免疫系機能不全;哺乳動物での加齢による体重減少;統合失調症;乳癌および下垂体癌のような癌を含めた各種腫瘍状態;神経筋および神経変性疾患;老人性認知症のような認知症、例えば、パーキンソン型認知症、アルツハイマー型認知症、血管性認知症およびレビー小体型認知症;多動性症候群;情緒疾患;注意欠陥障害;多動性障害;多発性硬化症;ならびにトゥレット症候群が非限定的に挙げられる。本発明の医薬組成物により治療され得る具体的な神経系損傷としては、頭部外傷、低酸素症、無酸素症、低血糖症、神経毒損傷、虚血性脳卒中および外傷によるCNS損傷、ならびにアポトーシス過程が生じる他の神経傷害が非限定的に挙げられる。
【0084】
したがってさらなる態様では、本発明は、治療を必要とする個人における神経変性疾患または神経系の損傷の治療方法に関し、この方法は、上で定義される医薬組成物を前記個人に投与することを含む。
【0085】
特定の実施形態では、本発明の方法で使用する医薬組成物を経口投与用に製剤化する。具体的な実施形態では、本発明の方法は、上で定義される活性薬剤の持続放出のために製剤化された経口医薬組成物を投与することを含み、より具体的には、活性薬剤はN−プロパルギル−1−アミノインダン、その鏡像異性体、その代謝産物、その類似体またはその薬学的に許容される塩から選択され、好ましくは、活性薬剤はラサギリンまたはその薬学的に許容される塩である。
【0086】
本発明の方法は、上で定義される任意の神経変性疾患または神経系の損傷の治療に使用することができる。具体的な実施形態では、神経変性疾患はパーキンソン病またはアルツハイマー病であり、神経系の損傷は、脳卒中のような急性脳損傷、または外傷性脳損傷である。
【0087】
さらなる態様では、本発明は、上で定義される活性薬剤、すなわち、プロパルギルアミン部分、アミノインダン部分もしくはプロパルギルアミン部分とアミノインダン部分の両方を含む活性薬剤またはその薬学的に許容される塩の持続放出性製剤の調製方法であって、
(i)任意に結合剤および/または滑剤と適切に混合した前記活性薬剤を適切な溶媒系に溶解させて、均一な懸濁液に調製する段階と、
(ii)不活性ノンパレルシードのような不活性なペレットに、(i)で得られた懸濁液の被覆を塗布する段階と、
(iii)(ii)で得られた活性薬剤充填ペレットを、任意に隔離/保護サブコーティング層でコーティングする段階と、
(iv)(ii)または(iii)で得られたペレットを、持続放出性コーティング層、すなわち、前記活性薬剤の持続放出を可能にするポリマー層でコーティングして前記持続放出性製剤を得る段階と、
(v)(iv)で得られたコーティングペレットを、任意に適当な添加剤と混合する段階と
を含む方法に関する。
【0088】
上で定義される活性薬剤の持続放出性製剤の調製のための、本発明に記載されている方法は、当該技術分野で公知の任意の適当な技術、例えば、後の実施例の節で詳細に記載されているような技術を用いて行うことができる。特定の実施形態では、この方法の段階(ii)および(iv)の1つ以上、ならびに段階(iii)および(v)(もし行った場合)を流動床加工装置を用いて行う。
【0089】
特定の実施形態では、この方法に従って調製した持続放出性製剤をさらに、カプセルに充填させるか、または圧縮して錠剤にする。
【0090】
これより、以下の非限定的な実施例により本発明を説明する。
【実施例】
【0091】
実験
パーキンソン病モデル
パーキンソン病(PD)の実験モデルは、この疾患の考え得る病理学的機序に関する洞察を得るために必要であり、さらに、薬理学的またはその他の新たな治療戦略の開発および試験に不可欠である。
【0092】
MPTPマウスモデル
ヒト脳の剖検研究および動物モデルの生化学的データの重要な主要部は、ドーパミン作動性神経変性を惹起する可能性のある、黒質での進行中の酸化ストレスの過程を示している。酸化ストレスが一次事象であるか二次事象であるかは不明であるが、抗酸化神経保護薬を開発する目的で、神経毒MPTP(N−メチル−4−フェニル−1,2,3,6−テトラヒドロピリジン)により誘発される酸化ストレスを動物モデルに用いて、神経変性の過程が調べられてきた。
【0093】
MPTPは、脳内で酵素モノアミンオキシダーゼ(MAO)−Bにより正荷電分子MPP+(1−メチル−4−フェニルピリジニウム)に変換され、黒質内にある特定のドーパミン産生ニューロンを殺すことにより、霊長類のパーキンソニズムを引き起こす。MPP+はミトコンドリアでの酸化的リン酸化を妨げることにより作用して、ATPの枯渇と細胞死を引き起こす。またMPP+は、カテコールアミンの合成を阻害し、ドーパミンおよび心臓ノルエピネフリンのレベルを低下させ、チロシンヒドロキシラーゼを不活性化する。
【0094】
6−OHDAラットモデル
PDのモデル化は、カテコールアミン神経毒である6−ヒドロキシドーパミン(6−OHDA)により進歩した。この分子は、ドーパミン作動性およびノルアドレナリン作動性の両方のニューロンの細胞体および線維内に輸送されて、神経終末の変性を引き起こし、また特に細胞体領域に投与した場合、細胞体にも影響を及ぼし得る。6−OHDAの神経毒性は、ミトコンドリアの呼吸酵素(呼吸鎖複合体IおよびIV)に対するその強力な阻害作用に基づくものである。これらの酵素のブロックによる代謝欠陥により、ニューロンは正常な生理的機能を発揮することができなくなり、その結果死に至る。PDでは、変性の対象となるのは主としてドーパミン作動性黒質線条体経路であるため、黒質線条体束の主要な遠心性投射に毒素を片側注射により直接注射してドーパミン作動性系の6−OHDA損傷を生じさせた動物モデルが開発されてきた。
【0095】
ドーパミンおよび代謝産物のHPLC解析用の試料調製
500μlのホモジナイゼーション緩衝液(0.1M過塩素酸、0.02%EDTAおよび1%ETOH)中の線条体組織試料を、OMNI International社製のOMNI Tipホモジナイジングキットを用いて(中速、5秒のインターバルで3×10秒)氷中でホモジナイズした。ホモジネートを5分間、超音波処理し、次いで4℃で15分間、15,000rpmで遠心分離した。上清を新たなチューブに移し、HPLCによりドーパミン含有量を解析した。
【0096】
実施例1.PDのMPTPマウスモデルにおけるラサギリン−プラミペキソール配合剤に関するin vivo実験
この実験にはそれぞれ約7〜9匹のマウスからなる10個の群が含まれ、それぞれ表1のように処置した。具体的には、マウスにMPTPを腹腔内(IP)投与してパーキンソン病(PD)モデルを誘発し、一定用量のプラミペキソール(初期段階のPDの治療に適応される非エルゴリン系ドーパミンアゴニスト)と種々の用量のラサギリンとを含有する配合剤で処置した。MPTPを最初の5日間(0〜4日目)、毎日注射し、配合剤を、IPまたは徐放を模擬するALZETポンプ(ALZET微小浸透圧ポンプ、1002モデル、速度0.25μl/時、DURECT社、Cupertino、USA)を用いて、0〜11日目に投与した。群5〜7には、最初の5日間(0〜4日目)は毎日、MPTP投与の30分前に配合剤を投与し、残りの日(5〜11日目)は、各投与日のほぼ同じ時間に薬物を投与した。ALZETポンプを最初のMPTP投与の15〜17時間前に腹腔内に埋め込んだ(群8〜10)。投与期間中にポンプにより投与した薬物の総量は、IP注射群に投与した総量と同じであった。対照は、生理食塩水を注射した未処置マウスおよび生理食塩水を注射したMPTP投与マウスであった。
【0097】
投与前および投与期間中の毎日に体重を測定し、各個体の体重変化を計算した。全実験期間を通して、臨床兆候を1週間に2回記録した。12日目の実験終了時に、全個体をC02窒息により安楽死させた。脳を素早く摘出し、冷却したプレート上に置いて解剖した。左右の線条体を摘出して重量を測定し、液体窒素で急速凍結してから、次の処置まで−70℃で保管した。線条体試料を実験の章に記載されている通りにHPLC用に調製した。
【0098】
【表3】
【0099】
図1に示されるように、3種類のラサギリン+プラミペキソール配合剤をIPで投与した場合、マウスのドーパミンレベルの対するその効果はほぼ同じであり、ドーパミン含有量が未処置マウスに比べて約60%増加した。しかし、24時間にわたって同じ量を投与するALZETポンプを用いた徐放(SR)方式で3種類の配合剤を投与した場合、有意な用量反応性が見られ、ドーパミンレベルはラサギリン用量の増加に従って増加した。全配合剤のプラミペキソールの量は同じであったことから、観察された効果はラサギリンの用量増加によるものに違いない。このことは、MPTP投与マウス脳のドーパミンレベルに対して、徐放投与が即放投与に比べて極めて有用な効果を有することを示唆している。
【0100】
実施例2.PDのMPTPマウスモデルにおけるラサギリン代謝産物アミノインダンに関するin vivo実験
すべての実験で体重20±1gのC57B1/6雄性マウスを使用した(1群当たり10匹のマウス)。MPTPを1日当たり40mg/kgの用量で5日間、IP注射により投与した。対照は、生理食塩水を注射した未処置マウスおよび生理食塩水を注射したMPTP投与マウスであった。アミノインダンを、毎日のIP注射により、または腹腔内に埋め込まれたALZETポンプを用いた徐放により12日間投与した。実験終了時にマウスから採取した左右の線条体中のドーパミンおよびその代謝産物(ジヒドロキシフェニル酢酸およびホモバニリン酸)を測定して処置効果を評価した。線条体組織試料を実験の章に記載されている通りにHPLC用に調製した。
【0101】
図2に示されるように、MPTP処置により、対照未処置マウスに比べて90%を超えるドーパミンレベルの欠乏が生じた。ALZETポンプを用いた徐放(SR)方式で投与したラサギリン代謝産物アミノインダンによる処置により、溶媒(生理食塩水)処置マウスまたは同じ薬物を毎日のIP注射で投与したマウスと比べて、ドーパミンレベルが有意に回復した。
【0102】
実施例3.PDの6−OHDAラットモデルにおけるラサギリンに関するin vivo実験
6−OHDAによる内側前脳束(MFB)の片側損傷は、片側での黒質線条体経路のドーパミン作動性ニューロン破壊を生じ、ラットの非対称な運動行動を引き起こす。損傷を受けたラットにドーパミン系に作用する薬物を与えると、ラットは活発な回旋行動を示す。より具体的には、DA放出薬剤であるD−アンフェタミンの投与により、損傷を受けていない黒質線条体投射に好発するドーパミンの不均衡が生じ、これにより時計回りの回旋が生じる。処置効果により、より多くのDAが利用可能になり、時計回りの回旋が増えると予想される。薬物誘発性の回旋は、回旋ボウルとラットの胴体に取り付ける回り継手とからなる自動ロタメータを用いて測定される。
【0103】
PDのMPTPマウスモデルドーパミン含有量の生化学的エンドポイントに対するラサギリンまたはその代謝産物アミノインダンによる徐放(SR)処置の明らかな利点を示唆する実施例1〜2で示される結果に加え、この実験では、PDの6−OHDAラットモデルを用いて、行動エンドポイントに対するラサギリンのSR投与の処置効果を試験した。
【0104】
体重250〜300gの成体Sprague−Dawley雄性ラットの中央前脳束(MFB)に6−OHDAで損傷を与えた。ラットをケタミン−キシラジン(85:15)0.1ml/100gで麻酔し、脳定位固定装置に載せた。以下の脳定位座標:ブレグマからAP−2.8mm、ML−2mm、および硬膜からDV−9に従って、片側のMFB内に6−OHDAを注入した。注入速度は、注入ポンプおよびHamiltonマイクロシリンジを用いて1μl/分であった。注入後、マイクロシリンジを注入部位に5分間静置し、骨ロウで穴を閉じた。
【0105】
同じ用量のラサギリンを、IP注射により毎日、28日間(6−OHDA投与の7日前から6−OHDA投与の21日後まで)投与するか、または6−OHDA投与の7日前から、埋め込んだALZETポンプを用いて毎日24時間にわたって一定速度で投与し、6−OHDA投与後さらに21日間投与を続け、合計28日間投与した。どちらの場合も、処置後に10日間の休薬を行ってからラットを屠殺した。実験終了時、すなわち6−OHDA投与後32日目に、誘発剤としてアンフェタミンを用いて、運動の非対称性をロタメータにより評価した。アンフェタミンはドーパミン放出剤であるため、6−OHDA投与後にも生存して機能するドーパミン作動性(DAegic)ニューロンの数に左右される。アンフェタミンを1.5mg/kgの単回用量でIP注射した後、ロタメータで60分間の回旋記録を行った。
【0106】
図3に示されるように、徐放を示すALZETポンプを用いてラサギリンで処置したラットでは、即放を示す毎日のIP注射によりラサギリンで処置したラットに比べて、正味の回旋数、すなわち、反時計回り回旋数を差し引いた後の時計回り回旋数(CW−CCW)において有意に向上した効果が観察された。
【0107】
実施例4〜6.サブコーティングを有さないラサギリン持続放出コーティングペレット
サブコーティングを有さない表2に示される組成のメシル酸ラサギリン持続放出(ER)ペレットを調製した。具体的には、薬物層の調製では、ポビドンUSP(PVP K29/32)を蒸留水と96%エタノールの混合物に溶解させ、次いで、形成された溶液にメシル酸ラサギリンを溶解させた。超微末タルクを分散させて形成された溶液に加え均一な懸濁液を形成し、次いで、流動床コーティング機を用いて600〜710μm(直径)の糖球状顆粒にコーティングした。アセトンと96%エタノールの混合物中にEthocel 45cps(エチルセルロース;放出制御ポリマー)を溶解させて機能性のコーティング懸濁液を調製し、次いでポリエチレングリコール(PEG)4000を蒸留水中に溶解させ、これを形成された溶液に加えた。得られた懸濁液を、流動床コーティング機を用いて薬物充填ペレットにコーティングした。
【0108】
【表4】
【0109】
各種ERコーティングペレットの溶出プロファイルを次の条件下で評価した:USP(米国薬局方)装置1を用いて、溶出液(900mlの腸液溶液、IFS、pH6.8)を100rpmの主軸回転速度および37℃の温度で攪拌した。溶出プロファイルを表3および図4に示す。
【0110】
【表5】
【0111】
実施例7〜8.サブコーティングを有するラサギリン持続放出コーティングペレット
サブコーティングを有する表4に示される組成のメシル酸ラサギリンERペレットを調製した。具体的には、薬物層の調製では、ポビドン(PVP K25)を蒸留水と96%エタノールの混合物に溶解させ、次いで、形成された溶液にメシル酸ラサギリンを溶解させた。形成された溶液に超微末タルクを分散させて加え、均一な懸濁液を形成し、次いでこれを600〜710μmの糖球状顆粒に流動床コーティング機を用いてコーティングした。PVP K25を蒸留水と96%エタノールの混合物に溶解させてサブコーティング溶液を調製し、次いで、得られた溶液を薬物充填ペレットに流動床コーティング機を用いてコーティングした。アセトンと96%エタノールの混合物中にEthocel 45cpsを溶解させて機能性のコーティング懸濁液を調製し、次いでPEG3000を蒸留水中に溶解させ、これを形成された溶液に加えた。得られた懸濁液を、流動床コーティング機を用いて薬物充填ペレットにコーティングした。形成された溶液に超微末タルクを分散させて加え、均一な懸濁液を形成し、次いで、これをサブコーティングされたペレットに流動床コーティング機を用いてコーティングした。Tumbler Bin Blenderを用いて、ラサギリンERペレットとAerosil200の乾燥混合物を調製した。
【0112】
各種ERコーティングペレットの溶出プロファイルを実施例4〜6で用いた条件下で評価し、これを表5および図5に示す。(i)腸管内の条件を模したIFS緩衝液(pH6.8);(ii)空腹条件を模したGFS(胃液溶液)緩衝液(pH1.2);(iii)GFS緩衝液で2時間、次いでIFS緩衝液でさらに20時間;および(iv)満腹条件を模した酢酸緩衝液(pH4.5)における実施例7(15%ER)のメシル酸ラサギリンERコーティングペレットのin vitro溶出データを図6に示す。同じERコーティングペレットのゼロ時間(作製直後)、安定性促進条件下(40℃、湿度75%)での1か月後、ならびに同じ安定性促進条件下での2か月後および3か月後におけるIFS緩衝液中でのin vitro安定性のデータを図7に示す。
【0113】
【表6】
【0114】
【表7】
【0115】
実施例9.サブコーティングを有するラサギリン持続放出カプセル剤
サブコーティングを有する表6に示される組成物のメシル酸ラサギリンERペレットを、実施例7〜8に記載されている通りに調製した。ただし、乾燥混合物の調製には、Aerosil200の代わりにコロイド状二酸化ケイ素を使用した。調製したこれらのERコーティングペレットの溶出プロファイルを、実施例4〜8で用いた条件下で評価し、これを表7および図8に示す。
【0116】
【表8】
【0117】
【表9】
【0118】
実施例10.サブコーティングを有する/有さないラサギリン持続放出コーティングペレット
サブコーティングを有するまたは有さない、表8〜12に示される組成のさらなるメシル酸ラサギリンERペレットを、実施例4〜9に記載されている手順に従って調製することができる。本実施例に含まれる各表で言及されている脚注は、表16の下に記載されている。
【0119】
【表10】
【0120】
【表11】
【0121】
【表12】
【0122】
【表13】
*2つのビーズ集団の代わりに、均一な集団として多相放出粒子を調製することもできる:糖球状顆粒またはその他の不活性なコア上の薬物層(0.25〜4.75mg)(第1相)→サブコーティング(第2相)→ERコーティング(第3相)→追加の薬物層(0.25〜4.75mg)→上塗りコーティング(第2相として)→カプセル殻。
【0123】
【表14】
【0124】
上の表2、4および6に記載されているメシル酸ラサギリン製剤、ならびに表8〜12に記載されているメシル酸ラサギリン製剤を錠剤に圧縮して、ラサギリンERコーティング錠を形成してもよい。この目的のために、ラサギリンERコーティングペレットを追加の添加剤と乾燥混合して均質な混合物を作製し、次いでこれを圧縮して、上塗り/装飾/非機能性のコーティング層でコーティングされた錠剤にする(例えば、表13を参照されたい)。
【0125】
【表15】
【0126】
表14は、湿式造粒で調製し、次いで乾燥、粉砕、乾燥混合、錠剤化を行い、最後にERコーティングを施した、ラサギリン0.2mgのERコーティング錠製剤を示したものである。
【0127】
【表16】
【0128】
表15は、放出制御ポリマーを含む湿式造粒で調製し、次いで乾燥、粉砕、乾燥混合、錠剤化を行い、最後に上塗りコーティングを施した、ラサギリン0.2mgのERコーティング錠製剤を示したものである。
【0129】
【表17】
【0130】
表16は、湿式造粒で調製し、次いで乾燥、粉砕、乾燥混合、二層錠剤化を行い、最後に上塗りコーティングを施した、ラサギリン5mgのERコーティング錠製剤を示したものである。
【0131】
【表18】
1製剤は0.2〜5mgのメシル酸ラサギリンを含み得る。
2浸透圧剤の代わりに、またはそれに加えて追加のpH依存性ポリマーを含ませて、パルス状放出性製剤を作製し得る。
3別の結合剤としては、例えば、ヒドロキシプロピルメチルセルロース(HPMC)、ポビドン(PVP)、微結晶性セルロースおよび前記結合剤の組合せが挙げられる。
4別の滑剤としては、例えば、コロイド状二酸化ケイ素、モノステアリン酸グリセリル、ステアリン酸マグネシウムおよび前記滑剤の組合せが挙げられる。
5別のフィルム形成ポリマーとしては、例えば、HPMC、PVP、微結晶性セルロース、ポリエチレングリコール(PEG)および前記フィルム形成ポリマーの組合せが挙げられる。
6別のpH非依存性ポリマーとしては、例えば、Surelease(登録商標)、Eudragit(登録商標)RL、Eudragit(登録商標)RS、Eudragit(登録商標)NEおよび前記ポリマーの組合せが挙げられる。
7別の細孔形成剤としては、例えば、HPMC、PVP、PEGおよび前記細孔形成剤の組合せが挙げられる。
8別の可塑剤としては、例えば、セバシン酸ジブチル/フタル酸ジブチル、トリアセチン、クエン酸トリエチルおよび前記可塑剤の組合せが挙げられる。
9別のpH依存性腸溶性コーティングポリマーとしては、例えば、Eudragit(登録商標)S、Kollicoat(登録商標)、ヒドロキシプロピルメチルセルロースフタラート(HPMCP)および前記薬剤の組合せが挙げられる。
10別の錠剤賦形剤としては、例えば、ラクトース、マンニトール/Parteck(登録商標)、ソルビトール、デンプンおよび前記錠剤賦形剤の組合せが挙げられる。
11別の崩壊剤としては、例えば、ナトリウムCMC/カルシウムCMC、クロスポビドン、低置換度クロスカルメロースナトリウムヒドロキシプロピルセルロース、炭酸水素ナトリウム、デンプン、デンプングリコール酸ナトリウムおよび前記崩壊剤の組合せが挙げられる。
12別の滑沢剤としては、例えば、グリセリルベヘナート、ステアリン酸、タルク、ステアリン酸亜鉛、ステアリン酸カルシウムおよび前記滑沢剤の組合せが挙げられる。
【0132】
実施例11.胃腸管の各種部分からのラサギリン吸収
薬物は胃腸管の各種部分から異なって吸収される。経口投与用の24時間型徐放性製剤を設計するためには、薬物が全時間にわたって、すなわち、胃腸管のすべての部分から吸収される必要である。薬物の大部分は十二指腸からよく吸収されるが、多くの薬物は結腸からはあまり吸収されないということが知られている。薬物は体内から排泄されるまでに、かなりの長時間にわたって結腸内に留まるため、放出プロファイルを効率的に設計するためには、結腸からの薬物吸収を評価することが重要である。
【0133】
この実験では、ラサギリン(1.5mg/kg)を0.5mg/mlの水溶液として、薬物動態実験の1日前に埋め込まれたポリエチレンカニューレにより自由行動Wistar雄性ラットに投与した。結腸、十二指腸および静脈内のボーラス投与のために、カニューレをそれぞれ結腸、十二指腸および頸静脈内に留置した。単回ボーラス投与を各区画に対して行った。さらに、全身の血液採取のために、各個体の右側静脈に第二の留置カニューレを留置した。投与5分前、投与の5、15、30、50、90、150および200分後に血液試料(0.5ml)を採取した。脱水症を予防するために、血液試料の採取後に毎回、等体積の生理溶液をラットに投与した。遠心分離により血漿を分離した後、LC−MS−MSトリプル四重極を用いて、ラサギリンおよびその主要代謝産物である1−アミノインダンの分析的定量を行った。Excelソフトウェアを用いてノンコンパートメント薬物動態解を行った。最終的な測定可能な試料に対するノンコンパートメント解析により、対数線形台形法を用いて血中濃度曲線下面積(AUC)を算出した。ラサギリンの経口バイオアベイラビリティ(F)を、AUC(十二指腸)/AU(IV)またはAUC(結腸)/AUC(IV)のパーセント比として算出した。
【0134】
表17および図9は、最大(またはピーク)血漿中濃度(Cmax)およびAUCの十二指腸投与群と結腸投与群間での違いを示している(データは平均±SEで表されており、n=4〜5である)。具体的には、結腸および十二指腸投与群の親化合物のT1/2は、IV投与後のT1/2よりも長かった。IV投与と十二指腸投与で同様のAUC値が算出され、このことは完全な経口吸収を示唆するものであった。結腸投与後のAUCはIV投与のAUCの約28%であり、このことは結腸吸収の実現可能性を示すものであった。これらの結果から、ラサギリンの制御放出送達システムの設計は実現可能かつ実用的である。
【0135】
【表19】
*Cmax−最大血漿中濃度;Tmax−Cmaxが生じた時間;Vss−定常状態での分布容積;CI−1kg当たりのクリアランス;F−ラサギリンの経口バイオアベイラビリティー。データは平均±SEとして表されている(n=4〜5)。
【0136】
参考文献
Akao Y.,Nakagawa Y.,Maruyama W.,Takahashi T.,Naoi M.,Apoptosis induced by an endogenous neurotoxin,N−methyl(R)salsolinol,is mediated by activation of caspase−3,Neurosci.Lett.,1999,267,153−156
Akao Y.,Maruyama W.,Shimizu S.,Yi H.,Nakagawa Y.,Shamoto−Nagai M.,Youdim M.B.H.,Tsujimoto Y.,Naoi M.,Mitochondrial permeability transition mediates apoptosis induced by N−methyl(R)salsolinol,an endogenous neurotoxin,and is inhibited by Bcl−2 and Rasagiline,N−Propargyl−1(R)−aminoindan,J.Neurochem.,2002a,82,913−923
Akao Y.,Maruyama W.,Yi H.,Shamoto−Nagai M.,Youdim M.B.H.,Naoi M.,An anti−Parkinson’s disease drug,N−propargyl−1(R)−aminoindan(rasagiline),enhances expression of anti−apoptotic Bcl−2 in human dopaminergic SH−SY5Y cells,Neurosci.Lett.,2002b,326,105−108
Bar−Am O.,Amit T.,Youdim M.B.,Aminoindan and hydroxyaminoindan,metabolites of rasagiline and ladostigil,respectively,exert neuroprotective properties in vitro,J.Neurochem.,2007,103(2),500−508
Bar−Am O.,Weinreb O.,Amit T.,Youdim M.B.,The neuroprotective mechanism of 1−(R)−aminoindan,the major metabolite of the anti−parkinsonian drug rasagiline,J.Neurochem.,2010,112,1131−1137
Durden D.A.,Dyck L.E.,Davis B.A.,Liu Y.D.,Boulton A.A.,Metabolism and pharmacokinetics,in the rat,of(R)−N−(2−heptyl)methyl−propargylamine(R−2HMP),a new potent monoamine oxidase inhibitor and antiapoptotic agent,Drug Metab Dispos.,2000,28,147−154
Grossberg G.,Desai A.,Review of rivastigmine and its clinical applications in Alzheimer’s disease and related disorders,Expert Opin.Pharmacother.,2000,2,653−666
Maruyama W.,Boulton A.A.,Davis B.A.,Dostert P.,Naoi M.,Enantio−specific induction of apoptosis by an endogenous neurotoxin,N−methyl(R)salsolinol,in dopaminergic SH−SY5Y cells:suppression of apoptosis by N−(2−heptyl)−N−methylpropargylamine,J.Neural Transm.,2001a,108,11−24
Maruyama W.,Akao Y.,Youdim M.B.H.,Boulton A.A.,Davis B.A.,Naoi M.,Transfection−enforced Bcl−2 overexpression and an anti−Parkinson drug,rasagiline,prevent nuclear accumulation of glyceraldehyde−3 phosphate dehydrogenase induced by an endogenous dopaminergic neurotoxin,N−methyl(R)salsolinol,J.Neurochem.,2001b,78,727−735
Maruyama W.,Takahashi T.,Youdim,M.B.H.,Naoi M.,The anti−Parkinson drug,rasagiline,prevents apoptotic DNA damage induced by peroxynitrite in human dopaminergic neuroblastoma SH−SY5Y cells,J.Neural Transm.,2002,109,467−481
Tazik S.,Johnson S.,Lu D.,Johnson C,Youdim M.B.,Stockmeier C.A.,Ou X.M.,Comparative neuroprotective effects of rasagiline and aminoindan with selegiline on dexamethasone−induced brain cell apoptosis,Neurotoxicity Research,2009,15,284−290
Tatton W.G.,Chalmers−Redman R.M.,Ju W.J.,Mammen M.,Carlile G.W.,Pong A.W.,Tatton N.A.,Propargylamines induce antiapoptotic new protein synthesis in serum−and nerve growth factor(NGF)−withdrawn,NGF−differentiated PC−12 cells,J Pharmacol Exp Then,2002,301,753−764
Tatton W.G.,Greenwood C.E.,Rescue of dying neurons:a new action for deprenyl in MPTP parkinsonism,J Neurosci Res.,1991,30,666−672
Tatton W.G.,Selegiline can mediate neuronal rescue rather than neuronal protection,Movement Disorders 8(Supp.I),1993,S20−S30
Weinreb O.,Amit T.,Bar−Am O.,Yousim M.B.,Rasagiline:a novel antiparkinsonian monoamine oxidase−B inhibitor with neuroprotective activity,Prog Neurobiol,2010,92(3),330−344
Weinstock M.,Selectivity of cholinesterase inhibition:Clinical implications for the treatment of Alzheimer’s disease,CNS Drugs,1999,12,307−323
Yogev−Falach M.,Amit T.,Bar−Am O.,Sagi Y.,Weinstock M.,Youdim M.B.H.,The involvement of mitogen−activated protein(MAP)kinase in the regulation of amyloid precursor protein processing by novel cholinesterase inhibitors derived from rasagiline,FASEB J.,2002,16,1674−1676
Youdim M.B.H.,Weinstock M.,ovel neuroprotective anti−Alzheimer drugs with antidepressant activity derived from the anti−Parkinson drug,rasagiline,Mechanisms of Ageing & Developments,2002a,123,1081−1086
Youdim M.B.H.,Gross A.,Finberg J.P.M.,Rasagiline[N−Propargyl−1R(+)−aminoindan],a selective and potent inhibitor of mitochondrial monoamine oxidase B,Br.J.Pharmacol.,2001a,132,500−506
Youdim M.B.H.,Wadia A.,Tatton N.A.,Weinstock M.,The anti−Parkinson drug rasagiline and its cholinesterase inhibitor derivatives exert neuroprotection unrelated to MAO inhibition in cell culture and in vivo,Ann N Y Acad Sci,2001b,939,450−458
Zimmermann K,Waldmeier P.C.,Tatton W.G.,Dibenzoxepines as treatments for neurodegenerative diseases,Pure Appl Chem,1999,71,2039−2046
【特許請求の範囲】
【請求項1】
薬学的に許容される担体と、プロパルギルアミン部分、アミノインダン部分もしくはプロパルギルアミン部分とアミノインダン部分の両方を含む活性薬剤またはその薬学的に許容される塩とを含む医薬組成物であって、前記活性薬剤の持続放出のために製剤化された医薬組成物。
【請求項2】
前記活性薬剤が、N−プロパルギル−1−アミノインダン、その鏡像異性体、その代謝産物、その類似体またはその薬学的に許容される塩である、請求項1に記載の医薬組成物。
【請求項3】
前記活性薬剤が、R(+)−N−プロパルギル−1−アミノインダン(ラサギリン)、S−(−)−N−プロパルギル−1−アミノインダンまたはその薬学的に許容される塩である、請求項2に記載の医薬組成物。
【請求項4】
前記薬学的に許容される塩が、R(+)−N−プロパルギル−1−アミノインダンまたはS(−)−N−プロパルギル−1−アミノインダンのメシル酸塩、エシル酸塩、トシル酸塩、硫酸塩、スルホン酸塩、リン酸塩、カルボン酸塩、マレイン酸塩、フマル酸塩、酒石酸塩、安息香酸塩、酢酸塩、塩酸塩または臭化水素酸塩、好ましくはR(+)−N−プロパルギル−1−アミノインダンのメシル酸塩から選択される、請求項3に記載の医薬組成物。
【請求項5】
前記N−プロパルギル−1−アミノインダンの代謝産物が、1−アミノインダンまたはその薬学的に許容される塩である、請求項2に記載の医薬組成物。
【請求項6】
前記N−プロパルギル−1−アミノインダンの類似体が、4−フルオロ−N−プロパルギル−1−アミノインダン、5−フルオロ−N−プロパルギル−1−アミノインダン、6−フルオロ−N−プロパルギル−1−アミノインダン、その鏡像異性体またはその薬学的に許容される塩である、請求項2に記載の医薬組成物。
【請求項7】
前記N−プロパルギル−1−アミノインダンの類似体が、3−(N−メチル,N−プロピル−カルバミルオキシ)−α−メチル−N’−プロパルギルフェネチルアミン;3−(N,N−ジメチル−カルバミルオキシ)−α−メチル−N’−メチル,N’−プロパルギルフェネチルアミン;3−(N−メチル,N−ヘキシル−カルバミルオキシ)−α−メチル−N’−メチル,N’−プロパルギルフェネチルアミン;3−(N−メチル,N−シクロヘキシル−カルバミルオキシ)−α−メチル−N’−メチル,N’−プロパルギルフェネチルアミン;3−(N−メチル,N−ヘキシル−カルバミルオキシ)−α−メチル−N’−メチル,N’−プロパルギルフェネチルアミン;6−(N−メチル,N−エチル−カルバミルオキシ)−N’−プロパルギル−1−アミノインダン;6−(N,N−ジメチル−カルバミルオキシ)−N’−メチル−N’−プロパルギル−1−アミノインダン;6−(N−メチル,N−エチル−カルバミルオキシ−N’−プロパルギル−1−アミノテトラリン;6−(N,N−ジメチル−チオカルバミルオキシ)−1−アミノインダン;6−(N−プロピル−カルバミルオキシ)−N’−プロパルギル−1−アミノインダン;5−クロロ−6−(N−メチル,N−プロピル−カルバミルオキシ)−N’−プロパルギル−1−アミノインダン;6−(N−メチル),N−プロピル−カルバミルオキシ)−N’−プロパルギル−1−アミノインダン;および6−(N−メチル,N−エチル−カルバミルオキシ)−N’−メチル,N’−プロパルギル−1−アミノインダン;その鏡像異性体;またはその薬学的に許容される塩である、請求項2に記載の医薬組成物。
【請求項8】
前記活性薬剤が、プロパルギルアミン、脂肪族プロパルギルアミンまたはその薬学的に許容される塩である、請求項1に記載の医薬組成物。
【請求項9】
前記脂肪族プロパルギルアミンが、N−(1−ヘプチル)プロパルギルアミン;N−(1−オクチル)プロパルギルアミン;N−(1−ノニル)プロパルギルアミン;N−(1−デシル)プロパルギルアミン;N−(1−ウンデシル)プロパルギルアミン;N−(1−ドデシル)プロパルギルアミン;N−(2−ブチル)プロパルギルアミン;N−(2−ペンチル)プロパルギルアミン;N−(2−ヘキシル)プロパルギルアミン;N−(2−ヘプチル)プロパルギルアミン;N−(2−オクチル)プロパルギルアミン;N−(2−ノニル)プロパルギルアミン;N−(2−デシル)プロパルギルアミン;N−(2−ウンデシル)プロパルギルアミン;N−(2−ドデシル)プロパルギルアミン;N−(1−ブチル)−N−メチルプロパルギルアミン;N−(2−ブチル)−N−メチルプロパルギルアミン;N−(2−ペンチル)−N−メチルプロパルギルアミン;(1−ペンチル)−N−メチルプロパルギルアミン;N−(2−ヘキシル)−N−メチルプロパルギルアミン;(2−ヘプチル)−N−メチルプロパルギルアミン;N−(2−デシル)−N−メチルプロパルギルアミン;(2−ドデシル)−N−メチルプロパルギルアミン;その鏡像異性体;またはその薬学的に許容される塩である、請求項8に記載の医薬組成物。
【請求項10】
前記活性薬剤が、セレギリン、デスメチルセレギリン、パルギリン、クロルギリンまたは(N−メチル−N−プロパルギル)−10−アミノメチル−ジベンゾ[b,f]オキセピンである、請求項1に記載の医薬組成物。
【請求項11】
前記活性薬剤の連続的徐放、パルス状放出または多相放出をもたらすように製剤化された、請求項1に記載の医薬組成物。
【請求項12】
経口投与のための請求項11に記載の医薬組成物。
【請求項13】
モノリシックなマトリックス;錠剤、好ましくは二層もしくは多層錠剤、マトリックス錠剤、崩壊錠剤、溶解錠またはチュアブル錠;好ましくは顆粒、粒、ビーズまたはペレットが充填された、カプセルまたは小袋;あるいはポリ(D,L−ラクチド)(PLA)、ポリグリコリド(PGA)およびポリ(D,L−ラクチド−co−グリコリド)(PLGA)のような生分解性ポリマーに基づくデポーシステムの形態である、請求項12に記載の医薬組成物。
【請求項14】
USP装置1(バスケット)37℃、7.4以下のpH値、50〜150rpmにおいて、以下の溶出プロファイル:
【表1】
を有する、請求項1〜13のいずれか1項に記載の医薬組成物。
【請求項15】
前記活性薬剤が、ラサギリンまたはその薬学的に許容される塩である、請求項14に記載の医薬組成物。
【請求項16】
(i)不活性なペレットコアと、
(ii)前記ペレットコアをコーティングしている薬物層であって、プロパルギルアミン部分、アミノインダン部分もしくはプロパルギルアミン部分とアミノインダン部分の両方を含み、任意に結合剤および/またはフィルム形成ポリマーと適切に混合され、さらに任意に滑剤と混合されている活性薬剤またはその薬学的に許容される塩を含む薬物層と、
(iii)任意に、前記薬物層をコーティングしている隔離/保護サブコーティング層と、
(iv)存在すれば前記サブコーティング層を、または前記薬物層をコーティングしている持続放出性コーティング層と
を含む、持続放出性ペレット。
【請求項17】
前記サブコーティング層が、フィルム形成ポリマーと、任意に滑剤とを含む、請求項16に記載の持続放出性ペレット。
【請求項18】
前記持続放出性コーティング層が、
(i)前記持続放出性ペレットがpH非依存性のin vitro放出特性を有する場合、少なくとも1つのpH非依存性ポリマーおよび任意に細孔形成剤を含む;または
(ii)pH非依存性ポリマー、親水性放出調節ポリマーおよび任意に疎水性または親水性の可塑剤および/または滑剤を含む、または
(iii)前記持続放出性ペレットがpH7.4の以下pH値でほぼゼロ次のin vitro放出特性を有する場合、pH依存性腸溶性コーティングポリマーとpH非依存性ポリマーの混合物を含む、
請求項16に記載の持続放出性ペレット。
【請求項19】
請求項16〜18のいずれか1項に記載の持続放出性ペレットであって、
(i)前記結合剤が、ポリビニルピロリドン(PVP)、ヒドロキシプロピルメチルセルロース(HPMC)、ヒドロキシプロピルセルロース(HPC)、微結晶性セルロースまたはそれらの組合せであり、
(ii)前記フィルム形成ポリマーが、PVP、HPMC、HPC、微結晶性セルロースまたはそれらの組合せであり、
(iii)前記滑剤が、タルク、コロイド状二酸化ケイ素、モノステアリン酸グリセリルまたはそれらの組合せであり、
(iv)前記pH非依存性ポリマーが、エチルセルロース、Surelease(登録商標)、Eudragit(登録商標)RL、Eudragit(登録商標)RS、Eudragit(登録商標)NEまたはそれらの組合せであり、
(v)前記pH依存性腸溶性コーティングポリマーが、Eudragit(登録商標)S、Eudragit(登録商標)L55、Kollicoat(登録商標)、フタル酸ヒドロキシプロピルメチルセルロース(HPMCP)、アルギン酸、カルボキシメチルセルロースまたはそれらの組合せであり、
(vi)前記細孔形成剤が、PVP、PEG、HPMC、HPC、メチルセルロース、1,2−プロピレングリコール、ラクトース、スクロース、タルクまたはそれらの組合せであり、
(vii)前記親水性放出調節ポリマーが、HPMC、HPC、PVP、PEGまたはそれらの組合せであり、かつ
(viii)前記可塑剤が、セバシン酸ジブチル;フタル酸ジブチル;クエン酸トリエチルのようなクエン酸エステルおよびトリアセチン;プロピレングリコール;PEG、ポリ(プロピレングリコール)およびポリ(エチレン/プロピレングリコール)のような低分子量ポリ(アルキレンオキシド);またはそれらの組合せである、
持続放出性ペレット。
【請求項20】
請求項16〜19のいずれか1項に記載の持続放出性ペレットであって、
(i)不活性なペレットコアと、フィルム形成ポリマー/結合剤としてのPVPおよび滑剤としての超微末タルクと混合した前記活性薬剤を含む薬物層と、pH非依存性ポリマーとしてエチルセルロースおよび細孔形成剤としてPEGを含む持続放出性(ER)コーティング層とを含み、
前記フィルム形成ポリマー/結合剤の量が前記薬物層の総重量の90%以下、または前記ペレットの総重量の約0.5%〜約20%であり、前記滑剤の量が前記薬物層の総重量の30%以下、または前記ペレットの総重量の約0.1%〜約10%であり、前記pH非依存性ポリマーの量が前記ERコーティング層の総重量の約50%〜約90%、または前記ペレットの総重量の約10%〜約30%であり、かつ前記細孔形成剤の量が前記ERコーティング層の総重量の約1%〜約20%、または前記ペレットの総重量の約0.1%〜約10%である、あるいは
(ii)不活性なペレットコアと、フィルム形成ポリマー/結合剤としてのPVPおよび滑剤としての超微末タルクと混合した前記活性薬剤を含む薬物層と、フィルム形成ポリマーとしてPVPを含む隔離/保護サブコーティング層と、pH非依存性ポリマーとしてエチルセルロース、細孔形成剤としてPEGおよび滑剤として超微末タルクを含むERコーティング層とを含み、
前記薬物層中の前記フィルム形成ポリマー/結合剤の量が前記薬物層の総重量の90%以下、または前記ペレットの総重量の約0.5%〜約20%であり、前記薬物層中の前記滑剤の量が前記薬物層の総重量の30%以下、または前記ペレットの総重量の約0.1%〜約10%であり、前記サブコーティング層中の前記フィルム形成ポリマーの量が前記サブコーティング層の総重量の100%以下、または前記ペレットの総重量の約0.5%〜約20%であり、前記pH非依存性ポリマーの量が前記ERコーティング層の総重量の約50%〜約90%、または前記ペレットの総重量の約10%〜約30%であり、前記細孔形成剤の量が前記ERコーティング層の総重量の約1%〜約20%、または前記ペレットの総重量の約0.1%〜約10%であり、かつ前記ERコーティング層中の前記滑剤の量が前記ERコーティング層の総重量の約0.1%〜約20%、または前記ペレットの総重量の約0.1%〜約10%である、
持続放出性ペレット。
【請求項21】
前記活性薬剤が、ラサギリンまたはその薬学的に許容される塩である、請求項16〜20のいずれか1項に記載の持続放出性ペレット。
【請求項22】
請求項16〜21のいずれか1項に記載の持続放出性ペレットを含む、経口医薬組成物。
【請求項23】
前記持続放出性ペレットが、1つ以上の適当な添加剤と混合されて、カプセルに充填されているか、または錠剤に圧縮されている、請求項22に記載の経口医薬組成物。
【請求項24】
神経変性疾患または神経系の損傷の治療のための、請求項1〜15、22または23のいずれか1項に記載の医薬組成物。
【請求項25】
前記神経変性疾患がパーキンソン病またはアルツハイマー病であり、かつ前記神経系の損傷が脳卒中のような急性脳損傷または外傷性脳損傷である、請求項24に記載の医薬組成物。
【請求項26】
治療を必要とする個人における神経変性疾患または神経系の損傷の治療方法であって、請求項1〜15、22または23のいずれか1項に記載の医薬組成物を前記個人に投与することを含む方法。
【請求項27】
前記神経変性疾患がパーキンソン病またはアルツハイマー病であり、かつ前記神経系の損傷が脳卒中のような急性脳損傷または外傷性脳損傷である、請求項26に記載の方法。
【請求項28】
前記医薬組成物が、ラサギリンまたはその薬学的に許容される塩を含む、請求項26または27に記載の方法。
【請求項29】
プロパルギルアミン部分、アミノインダン部分もしくはプロパルギルアミン部分とアミノインダン部分の両方を含む活性薬剤またはその薬学的に許容される塩の持続放出性製剤を調製する方法であって、
(i)任意に結合剤および/または滑剤と適切に混合した前記活性薬剤を適切な溶媒系に溶解させて、均一な懸濁液に調製する段階と、
(ii)不活性ノンパレルシードのような不活性なペレットに、(i)で得られた懸濁液の被覆を塗布する段階と、
(iii)(ii)で得られた活性薬剤充填ペレットを、任意に隔離/保護サブコーティング層でコーティングする段階と、
(iv)(ii)または(iii)で得られたペレットを、前記活性薬剤の持続放出を可能にする持続放出性コーティング層でコーティングして、前記持続放出性製剤を得る段階と、
(v)(iv)で得られたコーティングペレットを、任意に適当な添加剤と混合する段階と
を含む方法。
【請求項30】
前記持続放出性製剤を、カプセルに充填するか、または錠剤に圧縮する、請求項29に記載の方法。
【請求項1】
薬学的に許容される担体と、プロパルギルアミン部分、アミノインダン部分もしくはプロパルギルアミン部分とアミノインダン部分の両方を含む活性薬剤またはその薬学的に許容される塩とを含む医薬組成物であって、前記活性薬剤の持続放出のために製剤化された医薬組成物。
【請求項2】
前記活性薬剤が、N−プロパルギル−1−アミノインダン、その鏡像異性体、その代謝産物、その類似体またはその薬学的に許容される塩である、請求項1に記載の医薬組成物。
【請求項3】
前記活性薬剤が、R(+)−N−プロパルギル−1−アミノインダン(ラサギリン)、S−(−)−N−プロパルギル−1−アミノインダンまたはその薬学的に許容される塩である、請求項2に記載の医薬組成物。
【請求項4】
前記薬学的に許容される塩が、R(+)−N−プロパルギル−1−アミノインダンまたはS(−)−N−プロパルギル−1−アミノインダンのメシル酸塩、エシル酸塩、トシル酸塩、硫酸塩、スルホン酸塩、リン酸塩、カルボン酸塩、マレイン酸塩、フマル酸塩、酒石酸塩、安息香酸塩、酢酸塩、塩酸塩または臭化水素酸塩、好ましくはR(+)−N−プロパルギル−1−アミノインダンのメシル酸塩から選択される、請求項3に記載の医薬組成物。
【請求項5】
前記N−プロパルギル−1−アミノインダンの代謝産物が、1−アミノインダンまたはその薬学的に許容される塩である、請求項2に記載の医薬組成物。
【請求項6】
前記N−プロパルギル−1−アミノインダンの類似体が、4−フルオロ−N−プロパルギル−1−アミノインダン、5−フルオロ−N−プロパルギル−1−アミノインダン、6−フルオロ−N−プロパルギル−1−アミノインダン、その鏡像異性体またはその薬学的に許容される塩である、請求項2に記載の医薬組成物。
【請求項7】
前記N−プロパルギル−1−アミノインダンの類似体が、3−(N−メチル,N−プロピル−カルバミルオキシ)−α−メチル−N’−プロパルギルフェネチルアミン;3−(N,N−ジメチル−カルバミルオキシ)−α−メチル−N’−メチル,N’−プロパルギルフェネチルアミン;3−(N−メチル,N−ヘキシル−カルバミルオキシ)−α−メチル−N’−メチル,N’−プロパルギルフェネチルアミン;3−(N−メチル,N−シクロヘキシル−カルバミルオキシ)−α−メチル−N’−メチル,N’−プロパルギルフェネチルアミン;3−(N−メチル,N−ヘキシル−カルバミルオキシ)−α−メチル−N’−メチル,N’−プロパルギルフェネチルアミン;6−(N−メチル,N−エチル−カルバミルオキシ)−N’−プロパルギル−1−アミノインダン;6−(N,N−ジメチル−カルバミルオキシ)−N’−メチル−N’−プロパルギル−1−アミノインダン;6−(N−メチル,N−エチル−カルバミルオキシ−N’−プロパルギル−1−アミノテトラリン;6−(N,N−ジメチル−チオカルバミルオキシ)−1−アミノインダン;6−(N−プロピル−カルバミルオキシ)−N’−プロパルギル−1−アミノインダン;5−クロロ−6−(N−メチル,N−プロピル−カルバミルオキシ)−N’−プロパルギル−1−アミノインダン;6−(N−メチル),N−プロピル−カルバミルオキシ)−N’−プロパルギル−1−アミノインダン;および6−(N−メチル,N−エチル−カルバミルオキシ)−N’−メチル,N’−プロパルギル−1−アミノインダン;その鏡像異性体;またはその薬学的に許容される塩である、請求項2に記載の医薬組成物。
【請求項8】
前記活性薬剤が、プロパルギルアミン、脂肪族プロパルギルアミンまたはその薬学的に許容される塩である、請求項1に記載の医薬組成物。
【請求項9】
前記脂肪族プロパルギルアミンが、N−(1−ヘプチル)プロパルギルアミン;N−(1−オクチル)プロパルギルアミン;N−(1−ノニル)プロパルギルアミン;N−(1−デシル)プロパルギルアミン;N−(1−ウンデシル)プロパルギルアミン;N−(1−ドデシル)プロパルギルアミン;N−(2−ブチル)プロパルギルアミン;N−(2−ペンチル)プロパルギルアミン;N−(2−ヘキシル)プロパルギルアミン;N−(2−ヘプチル)プロパルギルアミン;N−(2−オクチル)プロパルギルアミン;N−(2−ノニル)プロパルギルアミン;N−(2−デシル)プロパルギルアミン;N−(2−ウンデシル)プロパルギルアミン;N−(2−ドデシル)プロパルギルアミン;N−(1−ブチル)−N−メチルプロパルギルアミン;N−(2−ブチル)−N−メチルプロパルギルアミン;N−(2−ペンチル)−N−メチルプロパルギルアミン;(1−ペンチル)−N−メチルプロパルギルアミン;N−(2−ヘキシル)−N−メチルプロパルギルアミン;(2−ヘプチル)−N−メチルプロパルギルアミン;N−(2−デシル)−N−メチルプロパルギルアミン;(2−ドデシル)−N−メチルプロパルギルアミン;その鏡像異性体;またはその薬学的に許容される塩である、請求項8に記載の医薬組成物。
【請求項10】
前記活性薬剤が、セレギリン、デスメチルセレギリン、パルギリン、クロルギリンまたは(N−メチル−N−プロパルギル)−10−アミノメチル−ジベンゾ[b,f]オキセピンである、請求項1に記載の医薬組成物。
【請求項11】
前記活性薬剤の連続的徐放、パルス状放出または多相放出をもたらすように製剤化された、請求項1に記載の医薬組成物。
【請求項12】
経口投与のための請求項11に記載の医薬組成物。
【請求項13】
モノリシックなマトリックス;錠剤、好ましくは二層もしくは多層錠剤、マトリックス錠剤、崩壊錠剤、溶解錠またはチュアブル錠;好ましくは顆粒、粒、ビーズまたはペレットが充填された、カプセルまたは小袋;あるいはポリ(D,L−ラクチド)(PLA)、ポリグリコリド(PGA)およびポリ(D,L−ラクチド−co−グリコリド)(PLGA)のような生分解性ポリマーに基づくデポーシステムの形態である、請求項12に記載の医薬組成物。
【請求項14】
USP装置1(バスケット)37℃、7.4以下のpH値、50〜150rpmにおいて、以下の溶出プロファイル:
【表1】
を有する、請求項1〜13のいずれか1項に記載の医薬組成物。
【請求項15】
前記活性薬剤が、ラサギリンまたはその薬学的に許容される塩である、請求項14に記載の医薬組成物。
【請求項16】
(i)不活性なペレットコアと、
(ii)前記ペレットコアをコーティングしている薬物層であって、プロパルギルアミン部分、アミノインダン部分もしくはプロパルギルアミン部分とアミノインダン部分の両方を含み、任意に結合剤および/またはフィルム形成ポリマーと適切に混合され、さらに任意に滑剤と混合されている活性薬剤またはその薬学的に許容される塩を含む薬物層と、
(iii)任意に、前記薬物層をコーティングしている隔離/保護サブコーティング層と、
(iv)存在すれば前記サブコーティング層を、または前記薬物層をコーティングしている持続放出性コーティング層と
を含む、持続放出性ペレット。
【請求項17】
前記サブコーティング層が、フィルム形成ポリマーと、任意に滑剤とを含む、請求項16に記載の持続放出性ペレット。
【請求項18】
前記持続放出性コーティング層が、
(i)前記持続放出性ペレットがpH非依存性のin vitro放出特性を有する場合、少なくとも1つのpH非依存性ポリマーおよび任意に細孔形成剤を含む;または
(ii)pH非依存性ポリマー、親水性放出調節ポリマーおよび任意に疎水性または親水性の可塑剤および/または滑剤を含む、または
(iii)前記持続放出性ペレットがpH7.4の以下pH値でほぼゼロ次のin vitro放出特性を有する場合、pH依存性腸溶性コーティングポリマーとpH非依存性ポリマーの混合物を含む、
請求項16に記載の持続放出性ペレット。
【請求項19】
請求項16〜18のいずれか1項に記載の持続放出性ペレットであって、
(i)前記結合剤が、ポリビニルピロリドン(PVP)、ヒドロキシプロピルメチルセルロース(HPMC)、ヒドロキシプロピルセルロース(HPC)、微結晶性セルロースまたはそれらの組合せであり、
(ii)前記フィルム形成ポリマーが、PVP、HPMC、HPC、微結晶性セルロースまたはそれらの組合せであり、
(iii)前記滑剤が、タルク、コロイド状二酸化ケイ素、モノステアリン酸グリセリルまたはそれらの組合せであり、
(iv)前記pH非依存性ポリマーが、エチルセルロース、Surelease(登録商標)、Eudragit(登録商標)RL、Eudragit(登録商標)RS、Eudragit(登録商標)NEまたはそれらの組合せであり、
(v)前記pH依存性腸溶性コーティングポリマーが、Eudragit(登録商標)S、Eudragit(登録商標)L55、Kollicoat(登録商標)、フタル酸ヒドロキシプロピルメチルセルロース(HPMCP)、アルギン酸、カルボキシメチルセルロースまたはそれらの組合せであり、
(vi)前記細孔形成剤が、PVP、PEG、HPMC、HPC、メチルセルロース、1,2−プロピレングリコール、ラクトース、スクロース、タルクまたはそれらの組合せであり、
(vii)前記親水性放出調節ポリマーが、HPMC、HPC、PVP、PEGまたはそれらの組合せであり、かつ
(viii)前記可塑剤が、セバシン酸ジブチル;フタル酸ジブチル;クエン酸トリエチルのようなクエン酸エステルおよびトリアセチン;プロピレングリコール;PEG、ポリ(プロピレングリコール)およびポリ(エチレン/プロピレングリコール)のような低分子量ポリ(アルキレンオキシド);またはそれらの組合せである、
持続放出性ペレット。
【請求項20】
請求項16〜19のいずれか1項に記載の持続放出性ペレットであって、
(i)不活性なペレットコアと、フィルム形成ポリマー/結合剤としてのPVPおよび滑剤としての超微末タルクと混合した前記活性薬剤を含む薬物層と、pH非依存性ポリマーとしてエチルセルロースおよび細孔形成剤としてPEGを含む持続放出性(ER)コーティング層とを含み、
前記フィルム形成ポリマー/結合剤の量が前記薬物層の総重量の90%以下、または前記ペレットの総重量の約0.5%〜約20%であり、前記滑剤の量が前記薬物層の総重量の30%以下、または前記ペレットの総重量の約0.1%〜約10%であり、前記pH非依存性ポリマーの量が前記ERコーティング層の総重量の約50%〜約90%、または前記ペレットの総重量の約10%〜約30%であり、かつ前記細孔形成剤の量が前記ERコーティング層の総重量の約1%〜約20%、または前記ペレットの総重量の約0.1%〜約10%である、あるいは
(ii)不活性なペレットコアと、フィルム形成ポリマー/結合剤としてのPVPおよび滑剤としての超微末タルクと混合した前記活性薬剤を含む薬物層と、フィルム形成ポリマーとしてPVPを含む隔離/保護サブコーティング層と、pH非依存性ポリマーとしてエチルセルロース、細孔形成剤としてPEGおよび滑剤として超微末タルクを含むERコーティング層とを含み、
前記薬物層中の前記フィルム形成ポリマー/結合剤の量が前記薬物層の総重量の90%以下、または前記ペレットの総重量の約0.5%〜約20%であり、前記薬物層中の前記滑剤の量が前記薬物層の総重量の30%以下、または前記ペレットの総重量の約0.1%〜約10%であり、前記サブコーティング層中の前記フィルム形成ポリマーの量が前記サブコーティング層の総重量の100%以下、または前記ペレットの総重量の約0.5%〜約20%であり、前記pH非依存性ポリマーの量が前記ERコーティング層の総重量の約50%〜約90%、または前記ペレットの総重量の約10%〜約30%であり、前記細孔形成剤の量が前記ERコーティング層の総重量の約1%〜約20%、または前記ペレットの総重量の約0.1%〜約10%であり、かつ前記ERコーティング層中の前記滑剤の量が前記ERコーティング層の総重量の約0.1%〜約20%、または前記ペレットの総重量の約0.1%〜約10%である、
持続放出性ペレット。
【請求項21】
前記活性薬剤が、ラサギリンまたはその薬学的に許容される塩である、請求項16〜20のいずれか1項に記載の持続放出性ペレット。
【請求項22】
請求項16〜21のいずれか1項に記載の持続放出性ペレットを含む、経口医薬組成物。
【請求項23】
前記持続放出性ペレットが、1つ以上の適当な添加剤と混合されて、カプセルに充填されているか、または錠剤に圧縮されている、請求項22に記載の経口医薬組成物。
【請求項24】
神経変性疾患または神経系の損傷の治療のための、請求項1〜15、22または23のいずれか1項に記載の医薬組成物。
【請求項25】
前記神経変性疾患がパーキンソン病またはアルツハイマー病であり、かつ前記神経系の損傷が脳卒中のような急性脳損傷または外傷性脳損傷である、請求項24に記載の医薬組成物。
【請求項26】
治療を必要とする個人における神経変性疾患または神経系の損傷の治療方法であって、請求項1〜15、22または23のいずれか1項に記載の医薬組成物を前記個人に投与することを含む方法。
【請求項27】
前記神経変性疾患がパーキンソン病またはアルツハイマー病であり、かつ前記神経系の損傷が脳卒中のような急性脳損傷または外傷性脳損傷である、請求項26に記載の方法。
【請求項28】
前記医薬組成物が、ラサギリンまたはその薬学的に許容される塩を含む、請求項26または27に記載の方法。
【請求項29】
プロパルギルアミン部分、アミノインダン部分もしくはプロパルギルアミン部分とアミノインダン部分の両方を含む活性薬剤またはその薬学的に許容される塩の持続放出性製剤を調製する方法であって、
(i)任意に結合剤および/または滑剤と適切に混合した前記活性薬剤を適切な溶媒系に溶解させて、均一な懸濁液に調製する段階と、
(ii)不活性ノンパレルシードのような不活性なペレットに、(i)で得られた懸濁液の被覆を塗布する段階と、
(iii)(ii)で得られた活性薬剤充填ペレットを、任意に隔離/保護サブコーティング層でコーティングする段階と、
(iv)(ii)または(iii)で得られたペレットを、前記活性薬剤の持続放出を可能にする持続放出性コーティング層でコーティングして、前記持続放出性製剤を得る段階と、
(v)(iv)で得られたコーティングペレットを、任意に適当な添加剤と混合する段階と
を含む方法。
【請求項30】
前記持続放出性製剤を、カプセルに充填するか、または錠剤に圧縮する、請求項29に記載の方法。
【図1】

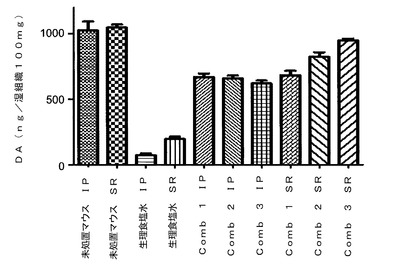
【図2】
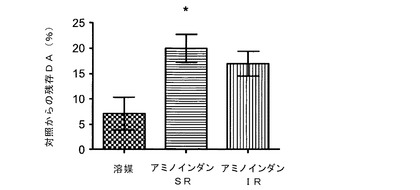
【図3】
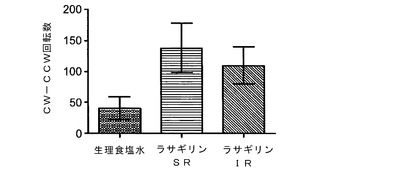
【図4】
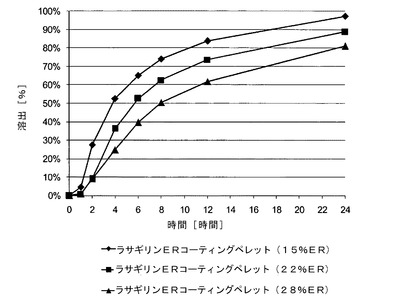
【図5】
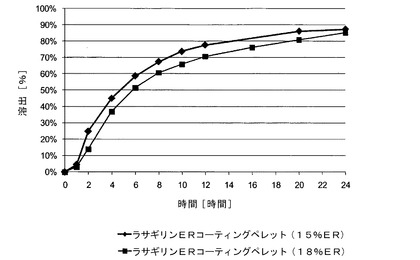
【図6】
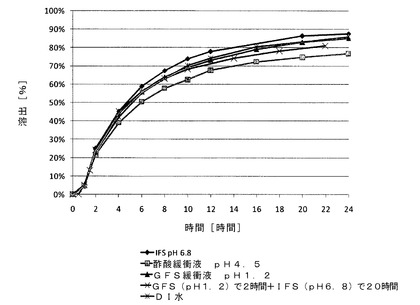
【図7】
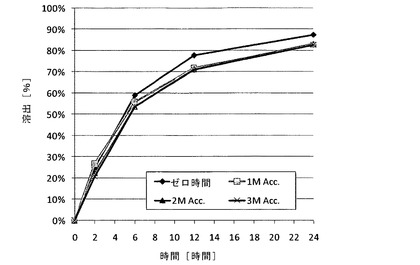
【図8】
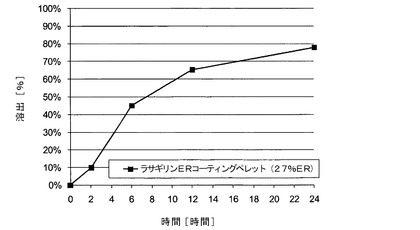
【図9】
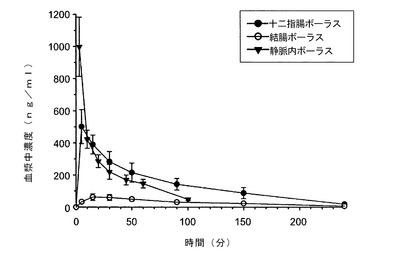
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公表番号】特表2013−518870(P2013−518870A)
【公表日】平成25年5月23日(2013.5.23)
【国際特許分類】
【出願番号】特願2012−551732(P2012−551732)
【出願日】平成23年2月3日(2011.2.3)
【国際出願番号】PCT/IL2011/000126
【国際公開番号】WO2011/095973
【国際公開日】平成23年8月11日(2011.8.11)
【出願人】(512202750)ファーマ ツー ビー リミテッド (1)
【Fターム(参考)】
【公表日】平成25年5月23日(2013.5.23)
【国際特許分類】
【出願日】平成23年2月3日(2011.2.3)
【国際出願番号】PCT/IL2011/000126
【国際公開番号】WO2011/095973
【国際公開日】平成23年8月11日(2011.8.11)
【出願人】(512202750)ファーマ ツー ビー リミテッド (1)
【Fターム(参考)】
[ Back to top ]