
ラッカーゼによる天然フレーバーの製造
【課題】従来技術の欠点を有しておらず、ヌートカトンの商業的に利用できる製造方法を提供する。
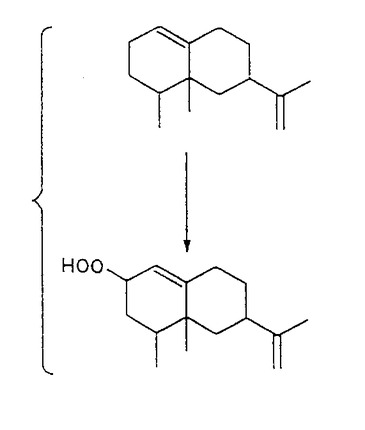
【解決手段】本発明は、バレンセンのラッカーゼ触媒酸化によるヌートカトンの製造方法を提供する。本発明による方法は、バレンセンとラッカーゼ活性を有する組成物とを、酸素供給源の存在下に、0.1%よりも大きいバレンセン濃度で反応させ、バレンセンヒドロペルオキシドを生成させる。所望により、ラッカーゼ活性を維持する濃度で、溶剤および/またはメディエーターをまた、包含させることができる。このバレンセンヒドロペルオキシドを分解させ、ヌートカトンを生成し、次いでヌートカトンを回収する。この方法は、ヌートカトンを商業的に実施可能な収率で生成させる。
【解決手段】本発明は、バレンセンのラッカーゼ触媒酸化によるヌートカトンの製造方法を提供する。本発明による方法は、バレンセンとラッカーゼ活性を有する組成物とを、酸素供給源の存在下に、0.1%よりも大きいバレンセン濃度で反応させ、バレンセンヒドロペルオキシドを生成させる。所望により、ラッカーゼ活性を維持する濃度で、溶剤および/またはメディエーターをまた、包含させることができる。このバレンセンヒドロペルオキシドを分解させ、ヌートカトンを生成し、次いでヌートカトンを回収する。この方法は、ヌートカトンを商業的に実施可能な収率で生成させる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は一般に、天然フレーバーの製造に関し、詳細には、ラッカーゼ触媒作用による天然フレーバーの製造に関する。
【背景技術】
【0002】
食品および飲料品工業界、ならびにその他の工業界、例えば化粧品業界および健康管理製品業界においては、それら製品の魅力を高めるために、フレーバーが常用される。健康、適合、安全、美学などの理由で、天然フレーバーが消費者に特に望まれている。従って、全世界的な、消費者による天然フレーバーの需要は、連続して増加している。天然フレーバーもしくは天然風味料(flavorings)は、21 C.F.R.§101.22に、スパイス、果実若しくは果汁、野菜若しくは野菜汁、可食性酵母、ハーブ、樹皮、芽、根、葉若しくは類似の植物材料、肉、海産物、家禽、卵、乳製品又はそれらの発酵生成物に由来する風味付与成分であって、その重要な機能が栄養よりも、風味付与にある上記風味付与成分を含有する、精油(essential oil)、オレオレジン(oleoresin)、エッセンス若しくはエキス(extractive)、タンパク質加水分解生成物、蒸留物、又は焙煎、加熱または酵素分解の生成物の全部であると定義される。
【0003】
このようなフレーバーの1種にヌートカトン(nootkatone)(4,4a,5,6,7,8−ヘキサヒドロ−6−イソプロペニル−4,4a−ジメチル−2(3II)−ナフタレノン)がある。ヌートカトンは、グレープフルーツの重要なフレーバー成分であり、ソフトドリンクおよびその他の飲料製品の風味付与に商業的に使用されており、また香料分野でも使用されている。ヌートカトンの慣用の調製方法は、バレンセン(valencene)の酸化による方法である。しかしながら、標準的化学酸化法によって調製されたヌートカトンは、天然の風味付与剤と見做すことはできない。従って、商業的にあまり望ましいとはいえない。さらに、出発物質であるバレンセンは、高価であり、従って、バレンセンを消費する方法は、商業的により受け入れ難い。これらの欠点から、天然ヌートカトンを調製するための商業的に望ましい方法に対するニーズが依然存在する。
【0004】
数種のヌートカトン製造方法が存在しているが、これらの方法はそれぞれ、制限を有する。例えば、米国特許No.5,847,226には、不飽和脂肪酸ヒドロペルオキシドの存在下で、バレンセンをヌートカトン、ヌートカトール(nootkatol)またはヌートカトンとヌートカトールとの混合物に酸化することによるヌートカトンの製造が開示されている。しかしながら、この特許においては、脂肪酸ヒドロペルオキシドを、自動酸化、光酸化、化学的触媒酸化またはリポオキシゲナーゼを用いる酵素酸化により生成し、次いでこの脂肪酸ヒドロペルオキシドを、バレンセンの自動酸化の触媒として用いている。このよう自動酸化は一般に、選択的方法ではない。微生物または酵素による変換を用いてヌートカトンを調製する従来の試みは一般に、低収率に終わった。例えば、Drawert 等(1984)には、細胞懸濁培地中でのバレンセンのヌートカトンへの生物転換(biotransformation)が報告されているが、バレンセンの出発レベルが実用のためには低すぎた。del Rio 等(1991)は、シトラス(Citrus)種のカルス(癒傷組織)培養によるヌートカトンおよびバレンセンの蓄積を研究している。9ヶ月経過したカルス培養物中のヌートカトンレベルは、16〜160g/100g生重量であった。さらに最近になって、日本国特許は、選択的微生物発酵を用いるバレンセンのヌートカトンへの生物転換を開示しているが、その総合収率は、低かった。50mlの操作容積を有する振盪フラスコ実験において、出発バレンセン500mgからヌートカトン2.5mgが得られたのみであった(Okuda等、1994)。
【0005】
従って、ラッカーゼを用いるヌートカトンの商業的に利用できる製造方法が、望ましい。
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明の課題は、従来技術の欠点を有しておらず、ヌートカトンの商業的に利用できる製造方法を提供することにある。
【課題を解決するための手段】
【0007】
本発明は、0.1%よりも大きいバレンセン濃度を用いて、バレンセンを、酸素供給源の存在下で、ラッカーゼ活性を有する組成物と反応させて、バレンセンヒドロペルオキシドを生成させることによりヌートカトン(nootkatone)を製造する方法に向けられている。このヒドロペルオキシドを分解してヌートカトンを生成し、次いでこのヌートカトンを回収する。ヒドロペルオキシドは、加熱により、および/または触媒との接触により分解させることができる。ラッカーゼは、微生物源、例えば菌類、ボトリチス シネレア(Botrytis cinerea)61〜34および/またはトラメテス ベルシコロール(Trametes versicolor)からのラッカーゼ、又は組換えDNA源からのラッカーゼであることができる。メディエーターおよび/または溶剤を、バレンセンおよびラッカーゼ活性を有する組成物に添加することができる。溶剤は、ラッカーゼ活性を維持する濃度で添加する。
【0008】
本発明はまた、ヌートカトンの製造方法であって、0.1%よりも大きいバレンセン濃度を用いて、バレンセンを、酸素供給源の存在下でラッカーゼ活性を有する組成物と反応させて、バレンセンヒドロペルオキシドを生成させ、次いでこのヒドロペルオキシドを分解させてヌートカトンを生成し、次いでこのヌートカトンを回収する上記方法に向けられている。ラッカーゼ活性を有する組成物は、約64重量%までのバレンセン濃度を有する溶液であることができる。別法として、ラッカーゼは、不動化させることができ、溶剤の不存在下に、バレンセンの酸化に使用することができる。
本発明はさらに、0.1%よりも大きいバレンセン濃度を用いて、バレンセンを、メディエーターおよび酸素供給源の存在下にpH約3〜7において、ボトリチス シネレア(Botrytis cinerea)61〜34またはトラメテス ベルシコロール(Trametes versicolor)のどちらか、または両方からのラッカーゼを含有する組成物と反応させ、バレンセンヒドロペルオキシドを生成させることによる、ヌートカトンの製造方法に向けられている。このヒドロペルオキシドを分解させてヌートカトンを生成し、次いでこのヌートカトンを回収する。
【0009】
本発明はさらにまた、バレンセンヒドロペルオキシドの製造方法に向けられている。バレンセンとラッカーゼ活性を有する組成物とを、酸素供給源の存在下で、バレンセンヒドロペルオキシドを生成させる条件下で反応させる。バレンセンの出発濃度は、0.1%よりも大きい。ラッカーゼ活性を維持する濃度のメディエーターおよび/または溶剤を添加することができる。
本発明はまた、バレンセンと不動化されたラッカーゼとを、酸素供給源の存在下に反応させ、バレンセンヒドロペルオキシドを生成させ、このバレンセンヒドロペルオキシドを加熱分解し、ヌートカトンを生成させ、次いでこのヌートカトンを回収することによる、ヌートカトンの製造方法に向けられている。この場合、反応中のバレンセンの濃度は、ラッカーゼ活性を阻害することなく、100%近くにすることができる。ラッカーゼ活性を維持する濃度のメディエーターおよび/または溶剤を、バレンセンおよび不動化されたラッカーゼに添加することができる。
【0010】
本発明はさらにまた、増加されたヌートカトンを含有する精油の製造方法に関する。バレンセンを含有する精油とラッカーゼ含有組成物とを、酸素供給源の存在下に反応させ、ヌートカトン含有精油の混合物を生成させる。この混合物を加熱し、ヌートカトンの量を増加させる。増加されたヌートカトンを含有する精油を回収する。この精油の出発濃度は、約0.5%よりも大きくする。この精油は、オレンジ油、ビターオレンジ油、グレープフルーツ油、レモン油および/またはタンジェリン油であることができる。精油は、分留することができ、例えばこれにより抽出物または蒸留物を得ることができる。
天然ヌートカトンを商業的に利用できる収量で、かつ比較的に副生成物をほとんど伴わずに、うまく製造することができる酵素的方法を開示する。ラッカーゼは、バレンセンの酸化を触媒するために使用された。
【0011】
ボトリチス シネレア(Botrytis cinerea)、トラメテス ベルシコロール(Trametes versicolor)又はその他の微生物源由来のラッカーゼ、ならびに商業的販売源から購入することができるラッカーゼおよび/または組換え技術を用いて生成されたラッカーゼを反応組成物中で、または不動化された形態で使用することができた。pH範囲は、pH3〜pH7であり、pH3.5が最適である。1種または2種以上のメディエーターおよび/または溶剤を、ラッカーゼ活性を維持する濃度で添加することができる。この方法は、当該酵素の幅広い基質特異性により、一連の天然フレーバー化学物質の転換に使用することができることがまた、見出された。
【0012】
本発明の目的およびその他の利点は、下記の図面、詳細な説明および例を参照してさらに理解されるはずである。
【図面の簡単な説明】
【0013】
【図1】バレンセンヒドロペルオキシドの生成反応を示す図である。
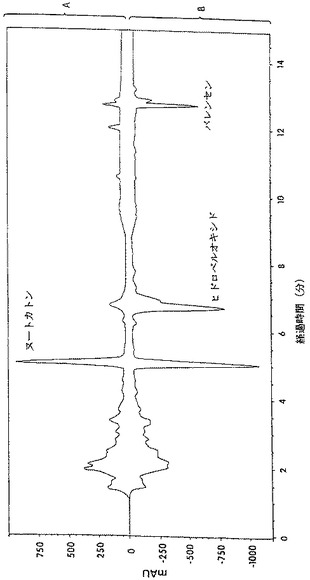
【図2】酵素反応の生成混合物の高速液体クロマトグラムである。
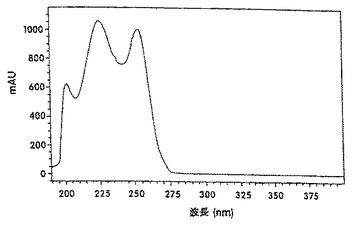
【図3A】ヌートカトンの紫外線スペクトルを示す図である。
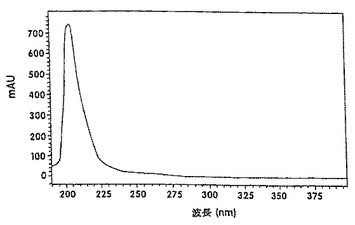
【図3B】バレンセンのヒドロペルオキシドの紫外線スペクトルを示す図である。
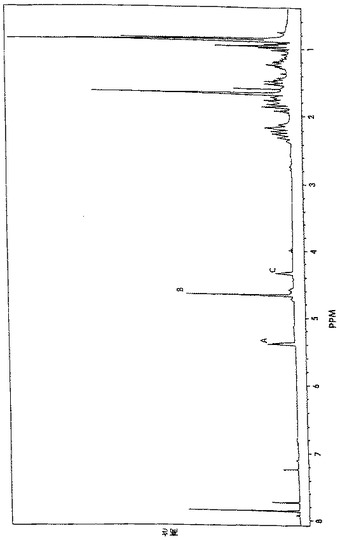
【図4A】酵素酸化後のバレンセンヒドロペルオキシドを表示する1H核磁気共鳴スペクトルを示す図である。
【図4B】図4Aにプロフィールされる化合物を示す図である。
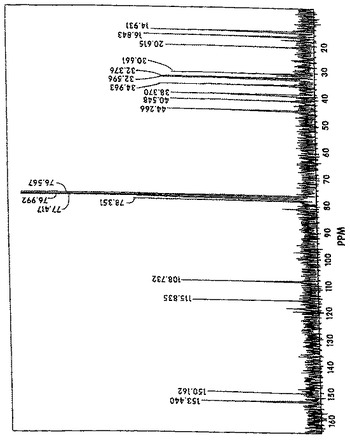
【図5A】酵素酸化後のバレンセンヒドロペルオキシドを表示する13C核磁気共鳴スペクトルを示す図である。
【図5B】図5Aにプロフィールされる化合物を示す図である。
【図6】酵素反応生成物のガスクロマトグラフィープロフィールを示す図である。
【図7】計算および試験(カッコ内)による13C化学シフトを示すバレンセンヒドロペルオキシドの化学構造を示す図である。
【発明を実施するための形態】
【0014】
本発明は、ラッカーゼを用いる天然ヌートカトンの商業的に利用することができる製造方法に関する。驚くべきことに、本発明により、酵素ラッカーゼ(ベンゼンジオール:オキシドレダクターゼ:EC1.10.3.2)が、商業的に利用できる収率でのバレンセンのヌートカトンへの酸化を触媒するために用いることができることが見出された。モノマー状または多量体状のいずれかの銅−含有糖タンパク質であるラッカーゼは、リグニンを合成する樹木植物中に、およびリグニンを分解する菌種中に見出される天然産出リグニン分解酵素である。
【0015】
材料
ラッカーゼは、微生物源、商業的販売業者[例えば、シグマ(Sigma)]から得ることができ、又は当業者に公知の方法を用いる組換えDNA技術により製造することができる。ラッカーゼの微生物源の例には、ボトリチス シネレア(Botrytis cinerea)またはトラメテス ベルシコロール(Trametes versicolor)などの白色腐朽菌(white rot fungi)がある。ボトリチス シネレア61−34およびトラメテス ベルシコロールの培養液からのラッカーゼは、College of Environmental Science and Forestryat the State University of New York in SyracuseのDr.James P.Nakasからの寄贈品であった。そのラッカーゼ活性レベルは、630〜1130nkat/mlであった。この酵素は、冷蔵下(約4℃)で少なくとも6ヶ月間、安定である。
【0016】
バレンセンは、GivaudanRoure Flavors,Lakeland,FLから、84〜85%の純度で得た。バレンセンはまた、その他の製造業者から、また精油の分留、例えば抽出または蒸留技術を用いることによる分別によって得ることができる。精油は通常、これらが得られる植物の特徴的匂いまたは香りを有する揮発性油であり、香料および風味付与剤に使用される。精油の例には、オレンジ油、ビターオレンジ油、グレープフルーツ油、レモン油、タンジェリン油、柑橘類油などが包含される。
【0017】
本明細書で使用されるメディエーター(mediator)の用語は、酸化性酵素により活性化され、また酵素上の活性部位から感受性構造に拡散する、拡散性分子であると定義される。ラッカーゼは、独立して、触媒として機能することができるものの、或る種のメディエーターの存在によりラッカーゼ触媒反応が強化され得ることが知られている。下記化学物質は、メディエーターとして活性であることが見出された:
1−ヒドロキシベンゾトリアゾール(HBT)、2,2´−アジノ−ビス(3−エチルベンズチアゾリン−6−スルホン酸)(ABTS)、フェルラ酸(ferulic aced)、ジメトキシベンジルアルコール、ジメチルアミノ安息香酸、カテキン、エピカテキン、p−ヒドロキシフェニル酢酸、ケルセチン(quercetin)、クロロプロマジン、フェノチアジン、ナリンジン(naringin)、プロマジン(promazine)、ホモバニリン酸、4−アミノ−サリチル酸、シリンガ酸(syringic acid)、4−ヒドロキシケイ皮酸、4−アミノ−3−ヒドロキシ安息香酸、バニリン酸、イソバニリン酸、カフェー酸、α−レゾルシル酸、β−レゾルシル酸、γ−レゾルシル酸、2,3−ジヒドロキシ安息香酸、3,4−ジヒドロキシ安息香酸、4−ヒドロキシ安息香酸、3−ヒドロキシ安息香酸、2,4,6−トリヒドロキシ安息香酸、安息香酸、ケイ皮酸、安息香酸ナトリウムおよびサリチル酸。使用するメディエーターは、Sigma Chemical Co.またはAldrich ChemicalCo.から購入した。その他の関連化学物質もまた、メディエーターとして有用であることは、当業者により認識されることである。
【0018】
酵素的転換方法
バレンセンとラッカーゼ活性を有する組成物とを、酸素供給源の存在下で反応させた。商業的に実施可能であるためには、少なくとも約0.1%のバレンセン出発濃度が必要である。ラッカーゼ活性を有する組成物が、溶液形態である場合、そのバレンセン濃度は、約64重量%までであることができる。
別法として、ラッカーゼなどの酵素は、当業者に公知であるように、固形支持体上で不動化または結合させることができる。この不動化技術の例は、J.Mol.Cat.B:Enzymatic 6(1999),29〜39; Chibata 等、Biocatalysis:Immobilized cells and enzymes,J.Mol.Cat.,37(1986),1〜24;Sharma 等、Immobilized Biomaterials Techniques and Applications,Angew.Chem.Int.,Ed.Engl.,21(1982),837〜854;Laskin(Ed.),Enzymes and Immobilized Cells in Biotechnologyに見出され、これらの記載の全体を引用して、ここに組み入れる。不動化された酵素が、しばしば未結合状態の酵素に比較して、有機溶剤に対して増大した耐性を示すことは、一般に公知である。すなわち、ラッカーゼを一度、不動化するか、または架橋させた酵素結晶(Altus Biologics, Inc.)などのように、別の方法により安定化すると、さらに高濃度のバレンセンを使用することができるものと見做すことができる。固体支持体、例えばカラム上に不動化させた場合、反応混合物中のバレンセンの濃度は、100%に近づけることができる。従って、本発明の精神または範囲を逸脱することなく、ラッカーゼまたは反応条件に対する種々の変化、修正または変更をなすことができる。
【0019】
酸素供給源は、純粋な酸素または酸素含有気体の混合物、例えば空気またはその他の気体混合物であることができる。場合により、1種または2種以上のメディエーターを、約50mMまでの濃度、好ましくは0.1mMよりは大きく、約5mMまでの濃度で包含させることができ、これにより反応を強化することができる。好適メディエーターは、HBT、フェルラ酸、4−ヒドロキシケイ皮酸、p−ヒドロキシフェニル酢酸、β−レゾルシル酸であり、HBTは、特に好適である。また、1種または2種以上の溶剤、例えば水、ヘキサンまたはその他の炭化水素を、ラッカーゼが当該反応を触媒することができる条件として定義される、ラッカーゼ活性を維持する濃度で包含させることができる。これらの条件には、適当なpHが包含され、このpHは、ラッカーゼについて約pH3〜7の範囲、最適には、pH3.5である。その他の反応条件には、酵素を変性させない溶剤の使用および酵素を触媒作用を得るのに適する立体配置にする一定の水分レベルが包含される。別段の記載がないかぎり、反応は、0.1Mシトラート緩衝液(pH3.5)中で行った。
【0020】
バレンセンを先ず、反応容器中でpH3〜7において、シトラート緩衝液と混合した。使用する場合、メディエーターおよび/または乳化剤を、この混合物にまた添加した。酵素を添加し、次いで反応を室温で継続させた。この反応は、いずれかの長さの期間、例えば1週間まで、またはそれ以上の期間にわたり進行させることができるが、少なくも約24時間の反応により、商業的に実施可能な量で生産される。この混合物を、約700rpmまでの速度で連続撹拌した。一態様において、反応は、約25℃〜30℃およびpH3〜7の範囲のpHにおいて、約48時間、生じさせた。試料を、定期的に採取し、バレンセンのヌートカトンへの変換を追跡した。炭酸ナトリウムによりpHをpH9.0に高め、また少なくとも約55℃に加熱することによって、反応を停止させた。当該ヒドロペルオキシドの分解を促進するために、加熱中、または加熱の代わりに、触媒を添加することができる。一態様において、触媒は、生理学的化合物、例えば鉄またはアスコルビン酸などである。その他の触媒の例には、コバルトおよび銅がある。
【0021】
被験酵素レベルは、約10nkat/ml〜620nkat/mlであり、またバレンセンの出発濃度は、約0.1%〜約64%であった。他の態様では、バレンセンを、約1%〜約50%、または約1%〜5%の濃度で含有させた。大部分の実験において、この出発濃度は、約0.5%〜約1.5%の範囲内であった。
ツイーン(Tween)−80またはトリトン(Triton)X−100などの乳化剤をまた、0.5%の濃度で使用することができる。
【0022】
粗生成物の回収
反応は、反応ブロスのpHを、炭酸ナトリウムによりpH9.0に調節することによって停止させた。この混合物を次いで、少なくとも約55℃に加熱することによって、ヒドロペルオキシドを分解させた。この加熱時間は、選択する温度に依存する。例えば、80℃の温度で2時間、加熱する。この混合物を、各反応ブロス1000mlに対して、それぞれメチレンクロライド(250ml×3回)またはヘプタン(250ml×6回)により抽出した。この抽出液を、硫酸ナトリウム上で乾燥させ、次いで溶剤を、減圧下で除去した。
【0023】
生成物の精製
この粗生成物を、クーゲルロアー(Kugelrohr) 装置で高減圧下、例えば5mmHgで蒸留した。クーゲルロアー蒸留からの精製された生成物の重量および外部標定曲線から測定するものとして、この生成物中のヌートカトン濃度を使用し、バレンセンからの全体的なヌートカトン収率を計算した。ヌートカトンを採取するための別法、例えば水蒸気蒸留、クロマトグラフィーおよび結晶化を、当業者に知られているように、使用することもできる。
未反応のバレンセンはまた、回収し、次いで当業者に公知の方法、例えばシリカゲルクロマトグラフィー、蒸留または結晶化によって、後続の反応において基質として使用するためにリサイクルすることができる。再使用するための未反応バレンセンの回収は、バレンセンが高価であることから、本発明の商業的利用価値を高める。
ラッカーゼ活性の測定は、2,6−ジメトキシフェノール(DMOP)の酸化的二量化に基づいており、この方法は、Slomczynski等(1995)により開示されており、その記載全体を引用して、ここに組み入れる。簡単に言えば、5mMの2,6−DMOP500リットルに、0.1Mクエン酸塩NaOH(pH3.5)を添加し、次いで酵素稀釈液各10リットルを添加した。反応混合物を、477nmにおける分光光度測定により追跡した。
【0024】
分析方法
反応の間に、試料(1ml)を、定期的に採取し、次いでメチレンクロライド0.8mlで抽出した。分離したメチレンクロライド層を、ガスクロマトグラフィーによる分析に付した。反応の停止後、クーゲルロアー蒸留後に回収された生成物を、例えば95%エタノール、メチルtert−ブチルエーテル(MTBE)またはメチレンクロライドにより、2.5%濃度に稀釈し、次いでガスクロマトグラフィー(GC)により分析した。バレンセンおよびヌートカトンを分析するためには、スペルコ(Supelco)SPB−1カラム(30m、内径(ID)0.25mm、フィルム0.25ミクロン)を、250℃のインジェクター温度および10℃/分で120℃〜270℃のプログラム温度、および20分間の保持時間をもって使用した。
【0025】
図1に示されているように、バレンセンの酸化は、ヒドロペルオキシドをもたらす。この酵素方法で生成されたヒドロペルオキシドは、ガスクロマトグラフィーのインジェクター口で分解させる。当該酵素反応中のヒドロペルオキシドの生成を特性決定するために、ヒドロペルオキシドを、AOAC公定分析法(Official Method Analysis)(1997)または薄層クロマトグラフィー(TLC)法(Johnson and Nidy,1975)のどちらかを用いて測定した。ヒドロペルオキシドはまた、ヘルドリッチ(Herderich)等の方法(1997)およびシュナイダー(Schneider)等の方法(1997)を用いる液体クロマトグラフィー/質量分析法(LC/MS)により検出した。
バレンセンヒドロペルオキシドが、酵素による酸化後に反応混合物中に存在していたことが高速液体クロマトグラフィー(HPLC)により単離された試料の核磁気共鳴(NMR)分析により確認された。
【0026】
バレンセンヒドロペルオキシドの分析
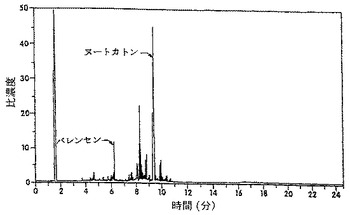
酵素反応の生成混合物中に、高いペルオキシド値が見出され、この数値は、還流後にほぼゼロに減少した。しかしながら、ガスクロマトグラフィー(GC)分析によれば、還流前と還流後との組成上の差異は、有意ではなかった。これは、ヒドロペルオキシドの熱不安定性によるものと見做された。図2に示されているように、反応混合物のHPLC分析から、6.7分の保持時間にある一つのピークが、反応混合物の還流後に消失したことがわかった。酵素反応の生成混合物のHPLCクロマトグラムを、還流後(図2A)および還流前(図2B)に得た。ルナ(Luna)C18(2)、150×2mmの3個のカラムを使用した。溶出液は0.25ml/分の流速で、メタノール:水(80:20)による4分間から100%メタノールで1分間までの勾配であった。検出は、210nmで行った。ピークに相当するフラクションを採取した。これらのフラクションは、ヨウ化カリウム/デンプンおよびFeSO4/NH4SCN溶液(AOAC,1990; Johnson and Nidy,1975)を用いる試験に基づき、ヒドロペルオキシド活性を明確に示した。ヌートカトンの紫外線(UV)スペクトル分析(図3A)を、仮定の(hypothetical)ヒドロペルオキシドピーク(図3B)のUVスペクトルと比較した。スペクトルは、バレンセンのスペクトルとは類似しているが、ヌートカトンのスペクトルとは相違しており、230nmおよび255nmにおけるエノン吸光に欠けていた。
【0027】
バレンセンヒドロペルオキシドの単離
反応混合物の粗製メチレンクロライド抽出液を、フラッシュクロマトグラフィーにより分別した[フラッシュ(Flash)40M、KP−Sil,31−63m,90g)。溶剤系は、薄層クロマトグラフィー(TLC)試験に基づき選択した。試料を、メチレンクロライドで、次いでMTBEで溶出した。全部で70のフラクションを集め、可視化試薬としてチオシアネート硫酸鉄(II)を用いるTLCにより分析した。ヒドロペルオキシドは、Fe(II)を、チオシアネートと反応するFe(III)に酸化し、血赤色のFe(SCN)52-を生成する(JohnsonおよびNidy、1975)。TLC試験は、フラクション7−12がヒドロペルオキシド活性であることを示した。集めたフラクションについてHPLC分析を行った。フラクション7−12中の主要成分は、仮定ヒドロペルオキシドのHPLCピークに相当した。フラクション7−12から、溶剤を蒸発させ、得られた生成物は、バレンセンヒドロペルオキシドとして同定された。
【0028】
NMR試験
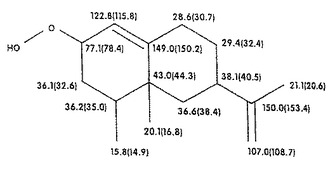
1H−、13C−、DEPT、1H1H−関連性および1H13C−関連性試験を、フラクション7−12からの生成物について行った。図4Aは、図4Bに示されている化合物のプロトン(1H)核磁気共鳴(NMR)スペクトルを示している。図5Aは、図5Bに示されている化合物の13C NMRスペクトルを示している。13Cシグナルの多重度および化学シフトから、その炭素骨格がバレンセンの炭素骨格に非常に類似していることが判る。図5Aを参照すると、1個の炭素共鳴ラインが、78.4ppmに見出され、これは酸素原子に結合している炭素に典型的である。しかしながら、この炭素ラインは、対応するアルコールに比較して、さらに13.9ppm下部にある。炭素(C−O)に結合したプロトンの化学シフトは、4.33ppmにあるのに対して、アルコールの相当するプロトンは、4.04ppmの化学シフトを有する。図7は、フラクション7−12からの生成物を用いて得られたバレンセンヒドロペルオキシドの計算された13C化学シフトに相当する(試験結果は、カッコ内に示されている)。バレンセンヒドロペルオキシドの計算された13C化学シフトは、図5Aに示されているフラクション群7−12中の主要成分のものと非常に近接している。これらの試験結果から、酵素反応混合物中のヒドロペルオキシドは、図7に示されている化学構造を有するものと結論することができる。
【実施例】
【0029】
本発明は、下記例によって、さらに理解される。
例1
バレンセンの酵素酸化を、1.5リットル反応容器(New BrurswickScientific)中で行った。この反応混合物は、0.1Mシトラート緩衝液(pH3.5)450ml、0.1M HBT 2.5ml、ツイーン(Tween)−80 2.5ml、バレンセン2.5mlおよびボトリチス(Botrytis)発酵ブロス(ラッカーゼ活性レベル:110nkat/ml)50mlからなるものであった。酸素供給源として空気を使用し、高速撹拌(約500〜700rpm)を用いて供給した。この反応は、室温(約25℃〜30℃)で行った。2日後、混合物のpHを炭酸ナトリウムにより9.0に変え、沸騰水浴中で2時間、加熱することによって、反応を停止させた。
室温まで冷却させた後、この混合物を、メチレンクロライドで抽出した(3×各150ml)。有機相を集め、Na2SO4上で乾燥させ、次いで減圧下で濃縮した。この粗生成物(3.5ml)を、クーゲルロアー(Kugelrohr)装置を用いて蒸留し、次いでガスクロマトグラフィー(GS)により分析した。図6を参照すると、蒸留後の生成物の2.5%溶液の典型的GCプロフィールは、ヌートカトンが最大ピークであり、外部曲線から測定して、43.3%w/wを占めたことが示されている。この反応からのヌートカトンの総合収率は、28.6%w/wであった。未反応バレンセンは、33.4%w/wであった。
【0030】
例2
バレンセンの酵素酸化を、2リットル反応容器中で行った。この反応混合物は、0.1Mシトラート緩衝液(pH3.5)1405ml、0.1M HBT 30ml、ツイーン(Tween)−80 30mlおよびバレンセン30mlからなるものであった。トラメテス ベルシコローア(Trametes versicolor)からの発酵ブロス(ラッカーゼ活性レベル:11nkat/ml)5mlの添加によって、反応を開始させた。空気を酸素供給源として使用し、350rpmの撹拌により供給した。この反応は、30℃の温度で行った。9日間後、例1と同様に、反応を停止させ、生成物を回収した。この反応からのヌートカトンの総合収率は、22.38%w/wであった。未反応バレンセンは、37.34%w/wであった。
【0031】
例3
振盪フラスコにおいて、0.1Mシトラート緩衝液(pH3.5)、HBTメディエーター(最終濃度0.5〜16mM)、ツイーン−80(0.5%)、バレンセン(0.5〜64%)およびラッカーゼ(活性レベル:60nkat/ml)からなる反応混合物(20ml)を、225rpmで振盪させながら48時間、30℃でインキュベートした。反応を停止させた時点で、各フラスコから、試料2mlを採取し、次いでメチレンクロライド1.2mlで抽出した。分離した溶剤層を、GCにより分析した。バレンセンおよびヌートカトンに相当する面積パーセンテージを、下表に示す:
【0032】
【表1】

【0033】
例4
ラッカーゼおよび/またはメディエーターの存在および不存在における、バレンセンのヌートカトンへの変換を評価した。各フラスコ中の総反応容積は、10mlであった。ラッカーゼは、60nkat/mlの活性レベルを有するボトリチス シネレア(Botrytis cinerea)由来であった。メディエーターは、0.5mMの最終濃度のHBTであった。出発基質(バレンセン)は、0.5%であった。4日後、これらの反応混合物を、メチレンクロライドで抽出し、次いで分離した有機相を、GCにより分析した。結果を、下表に示す。
【表2】
【0034】
例5
活性レベル620nkat/mlのラッカーゼ、基質としてバレンセン0.5%およびツイーン−80 0.5%からなる総反応容積10mlにおいて、下記化合物を、0.5mMの濃度でメディエーターとして試験した:1−ヒドロキシベンゾトリアゾール(HBT)、2,2´−アジノ−ビス(3−エチルベンズチアゾリン−6−スルホン酸(ABTS)、フェルラ酸(FA)、ジメトキシベンジルアルコール(DBA)およびジメチルアミノ安息香酸(DBAD)。各種メディエーターの存在下に4日間の反応後のバレンセンおよびヌートカトンの相対量を下表に示す。
【0035】
【表3】
【0036】
例6
活性レベル60nkat/mlのラッカーゼ、基質としてバレンセン1%およびツイーン−80 1%からなる反応容積20mlにおいて、下記化合物を、5mMの濃度でメディエーターとして試験した:カテキン、エピカテキン、p−ヒドロキシフェニル酢酸、ケルセチン、クロロプロマジン、フェノチアジン、ナリンジン、プロマジン、ホモバニリン酸、4−アミノ−サリチル酸、シリンガ酸、4−アミノ−3−ヒドロキシ安息香酸、バニリン酸、カフェー酸、プロトカテキュ酸(protocatechuic acid)、α−レゾルシル酸、β−レゾルシル酸、γ−レゾルシル酸、2,3−ジヒドロキシ安息香酸、4−ヒドロキシ安息香酸、3−ヒドロキシ安息香酸、2,4,6−トリヒドロキシ安息香酸、安息香酸、ケイ皮酸、イソバニリン酸、安息香酸ナトリウムおよびサリチル酸。各場合、変換率は、メディエーターの不存在下に得られた結果に比較して2〜8倍大きかった。この結果は、これらの化合物が、本方法においてメディエーターとして機能することを示している。
【0037】
例7
ボトリチス シネレア(Botrytis cinerea)からの60nkat/mlレベルのラッカーゼ、基質としてバレンセン0.5%、ツイーン(Tween)−80 0.5%、メディエーターとして0.5mMの最終濃度のHBTおよびpHを調整するための緩衝液(0.1M)からなる総量10mlの反応混合物を、225rpmで撹拌しながら30℃でインキュベートした。6日間の反応後、試料2mlを、各フラスコから採取し、次いでメチレンクロライドで抽出し、次いでGC分析に付す。異なるpH条件下での結果を、下表に示す。
【表4】
【0038】
例8
バレンセンを含有するオレンジ油の酵素酸化を、1.5リットル容器中で行った。0.1Mシトラート緩衝液(pH3.5)875ml、ツイーン−80 5ml、0.1M HBT 5ml、オレンジ油5mlおよびボトリチス シネレア(Botrytis cinerea)からのラッカーゼブロス100ml(活性レベル:63nkat/ml)からなる反応ブロスを、500rpmで撹拌しながら30℃でインキュベートした。4日間の反応後、クーゲルロアー(Kigelrohr) 蒸留後の生成物を、例1に記載の方法に従い回収した。この生成物中のヌートカトンの存在は、GC/MSによる分析により確認した。
酸化前および酸化後の両方のオレンジ油の感覚器官試験を、下記組成物を用いて、9人の熟練のフレーバーリスト(flavorists)により行った。
【表5】
【0039】
これらのフレーバーリストの全員が、混合物Aを混合物Bと区別することができた。これらのフレーバーリストは、混合物Bのフレーバーを、フルーティ、ヌートカトン様フレーバーおよび強力なウッディノートを有し、長い持続性を有すると評価した。
明細書中に示され、説明されている本発明の態様は、当業者である発明者の態様を示すのみであって、いかなる点でも制限するものではない。例えば、固体支持体に結合させた酵素は、酵素の安定性および有機溶剤に対するその耐性を高めることができる。もう一つの例として、多くの種々の化学物質を、メディエーターとして使用することができる。さらにもう一つの例として、フレーバーを生成させるために、カリオフィレン、フェニルピルビン酸、ガイエン(guaiene)、アネトール、ファメゾールおよびホップ油などの別種の基質の酸化を包含することができる。従って、本発明の精神および特許請求の範囲の範囲から逸脱することなく、これらの態様の種々の変化、修正または変更をなすことができ、または頼ることができる。
【0040】
ヌートカトンはフレーバーとして有用である。
本発明では、バレンセンのラッカーゼ触媒酸化によるヌートカトンの製造方法が提供される。本発明の方法によれば、ヌートカトンが商業的に実用性のある収率で製造される。
【技術分野】
【0001】
本発明は一般に、天然フレーバーの製造に関し、詳細には、ラッカーゼ触媒作用による天然フレーバーの製造に関する。
【背景技術】
【0002】
食品および飲料品工業界、ならびにその他の工業界、例えば化粧品業界および健康管理製品業界においては、それら製品の魅力を高めるために、フレーバーが常用される。健康、適合、安全、美学などの理由で、天然フレーバーが消費者に特に望まれている。従って、全世界的な、消費者による天然フレーバーの需要は、連続して増加している。天然フレーバーもしくは天然風味料(flavorings)は、21 C.F.R.§101.22に、スパイス、果実若しくは果汁、野菜若しくは野菜汁、可食性酵母、ハーブ、樹皮、芽、根、葉若しくは類似の植物材料、肉、海産物、家禽、卵、乳製品又はそれらの発酵生成物に由来する風味付与成分であって、その重要な機能が栄養よりも、風味付与にある上記風味付与成分を含有する、精油(essential oil)、オレオレジン(oleoresin)、エッセンス若しくはエキス(extractive)、タンパク質加水分解生成物、蒸留物、又は焙煎、加熱または酵素分解の生成物の全部であると定義される。
【0003】
このようなフレーバーの1種にヌートカトン(nootkatone)(4,4a,5,6,7,8−ヘキサヒドロ−6−イソプロペニル−4,4a−ジメチル−2(3II)−ナフタレノン)がある。ヌートカトンは、グレープフルーツの重要なフレーバー成分であり、ソフトドリンクおよびその他の飲料製品の風味付与に商業的に使用されており、また香料分野でも使用されている。ヌートカトンの慣用の調製方法は、バレンセン(valencene)の酸化による方法である。しかしながら、標準的化学酸化法によって調製されたヌートカトンは、天然の風味付与剤と見做すことはできない。従って、商業的にあまり望ましいとはいえない。さらに、出発物質であるバレンセンは、高価であり、従って、バレンセンを消費する方法は、商業的により受け入れ難い。これらの欠点から、天然ヌートカトンを調製するための商業的に望ましい方法に対するニーズが依然存在する。
【0004】
数種のヌートカトン製造方法が存在しているが、これらの方法はそれぞれ、制限を有する。例えば、米国特許No.5,847,226には、不飽和脂肪酸ヒドロペルオキシドの存在下で、バレンセンをヌートカトン、ヌートカトール(nootkatol)またはヌートカトンとヌートカトールとの混合物に酸化することによるヌートカトンの製造が開示されている。しかしながら、この特許においては、脂肪酸ヒドロペルオキシドを、自動酸化、光酸化、化学的触媒酸化またはリポオキシゲナーゼを用いる酵素酸化により生成し、次いでこの脂肪酸ヒドロペルオキシドを、バレンセンの自動酸化の触媒として用いている。このよう自動酸化は一般に、選択的方法ではない。微生物または酵素による変換を用いてヌートカトンを調製する従来の試みは一般に、低収率に終わった。例えば、Drawert 等(1984)には、細胞懸濁培地中でのバレンセンのヌートカトンへの生物転換(biotransformation)が報告されているが、バレンセンの出発レベルが実用のためには低すぎた。del Rio 等(1991)は、シトラス(Citrus)種のカルス(癒傷組織)培養によるヌートカトンおよびバレンセンの蓄積を研究している。9ヶ月経過したカルス培養物中のヌートカトンレベルは、16〜160g/100g生重量であった。さらに最近になって、日本国特許は、選択的微生物発酵を用いるバレンセンのヌートカトンへの生物転換を開示しているが、その総合収率は、低かった。50mlの操作容積を有する振盪フラスコ実験において、出発バレンセン500mgからヌートカトン2.5mgが得られたのみであった(Okuda等、1994)。
【0005】
従って、ラッカーゼを用いるヌートカトンの商業的に利用できる製造方法が、望ましい。
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明の課題は、従来技術の欠点を有しておらず、ヌートカトンの商業的に利用できる製造方法を提供することにある。
【課題を解決するための手段】
【0007】
本発明は、0.1%よりも大きいバレンセン濃度を用いて、バレンセンを、酸素供給源の存在下で、ラッカーゼ活性を有する組成物と反応させて、バレンセンヒドロペルオキシドを生成させることによりヌートカトン(nootkatone)を製造する方法に向けられている。このヒドロペルオキシドを分解してヌートカトンを生成し、次いでこのヌートカトンを回収する。ヒドロペルオキシドは、加熱により、および/または触媒との接触により分解させることができる。ラッカーゼは、微生物源、例えば菌類、ボトリチス シネレア(Botrytis cinerea)61〜34および/またはトラメテス ベルシコロール(Trametes versicolor)からのラッカーゼ、又は組換えDNA源からのラッカーゼであることができる。メディエーターおよび/または溶剤を、バレンセンおよびラッカーゼ活性を有する組成物に添加することができる。溶剤は、ラッカーゼ活性を維持する濃度で添加する。
【0008】
本発明はまた、ヌートカトンの製造方法であって、0.1%よりも大きいバレンセン濃度を用いて、バレンセンを、酸素供給源の存在下でラッカーゼ活性を有する組成物と反応させて、バレンセンヒドロペルオキシドを生成させ、次いでこのヒドロペルオキシドを分解させてヌートカトンを生成し、次いでこのヌートカトンを回収する上記方法に向けられている。ラッカーゼ活性を有する組成物は、約64重量%までのバレンセン濃度を有する溶液であることができる。別法として、ラッカーゼは、不動化させることができ、溶剤の不存在下に、バレンセンの酸化に使用することができる。
本発明はさらに、0.1%よりも大きいバレンセン濃度を用いて、バレンセンを、メディエーターおよび酸素供給源の存在下にpH約3〜7において、ボトリチス シネレア(Botrytis cinerea)61〜34またはトラメテス ベルシコロール(Trametes versicolor)のどちらか、または両方からのラッカーゼを含有する組成物と反応させ、バレンセンヒドロペルオキシドを生成させることによる、ヌートカトンの製造方法に向けられている。このヒドロペルオキシドを分解させてヌートカトンを生成し、次いでこのヌートカトンを回収する。
【0009】
本発明はさらにまた、バレンセンヒドロペルオキシドの製造方法に向けられている。バレンセンとラッカーゼ活性を有する組成物とを、酸素供給源の存在下で、バレンセンヒドロペルオキシドを生成させる条件下で反応させる。バレンセンの出発濃度は、0.1%よりも大きい。ラッカーゼ活性を維持する濃度のメディエーターおよび/または溶剤を添加することができる。
本発明はまた、バレンセンと不動化されたラッカーゼとを、酸素供給源の存在下に反応させ、バレンセンヒドロペルオキシドを生成させ、このバレンセンヒドロペルオキシドを加熱分解し、ヌートカトンを生成させ、次いでこのヌートカトンを回収することによる、ヌートカトンの製造方法に向けられている。この場合、反応中のバレンセンの濃度は、ラッカーゼ活性を阻害することなく、100%近くにすることができる。ラッカーゼ活性を維持する濃度のメディエーターおよび/または溶剤を、バレンセンおよび不動化されたラッカーゼに添加することができる。
【0010】
本発明はさらにまた、増加されたヌートカトンを含有する精油の製造方法に関する。バレンセンを含有する精油とラッカーゼ含有組成物とを、酸素供給源の存在下に反応させ、ヌートカトン含有精油の混合物を生成させる。この混合物を加熱し、ヌートカトンの量を増加させる。増加されたヌートカトンを含有する精油を回収する。この精油の出発濃度は、約0.5%よりも大きくする。この精油は、オレンジ油、ビターオレンジ油、グレープフルーツ油、レモン油および/またはタンジェリン油であることができる。精油は、分留することができ、例えばこれにより抽出物または蒸留物を得ることができる。
天然ヌートカトンを商業的に利用できる収量で、かつ比較的に副生成物をほとんど伴わずに、うまく製造することができる酵素的方法を開示する。ラッカーゼは、バレンセンの酸化を触媒するために使用された。
【0011】
ボトリチス シネレア(Botrytis cinerea)、トラメテス ベルシコロール(Trametes versicolor)又はその他の微生物源由来のラッカーゼ、ならびに商業的販売源から購入することができるラッカーゼおよび/または組換え技術を用いて生成されたラッカーゼを反応組成物中で、または不動化された形態で使用することができた。pH範囲は、pH3〜pH7であり、pH3.5が最適である。1種または2種以上のメディエーターおよび/または溶剤を、ラッカーゼ活性を維持する濃度で添加することができる。この方法は、当該酵素の幅広い基質特異性により、一連の天然フレーバー化学物質の転換に使用することができることがまた、見出された。
【0012】
本発明の目的およびその他の利点は、下記の図面、詳細な説明および例を参照してさらに理解されるはずである。
【図面の簡単な説明】
【0013】
【図1】バレンセンヒドロペルオキシドの生成反応を示す図である。
【図2】酵素反応の生成混合物の高速液体クロマトグラムである。
【図3A】ヌートカトンの紫外線スペクトルを示す図である。
【図3B】バレンセンのヒドロペルオキシドの紫外線スペクトルを示す図である。
【図4A】酵素酸化後のバレンセンヒドロペルオキシドを表示する1H核磁気共鳴スペクトルを示す図である。
【図4B】図4Aにプロフィールされる化合物を示す図である。
【図5A】酵素酸化後のバレンセンヒドロペルオキシドを表示する13C核磁気共鳴スペクトルを示す図である。
【図5B】図5Aにプロフィールされる化合物を示す図である。
【図6】酵素反応生成物のガスクロマトグラフィープロフィールを示す図である。
【図7】計算および試験(カッコ内)による13C化学シフトを示すバレンセンヒドロペルオキシドの化学構造を示す図である。
【発明を実施するための形態】
【0014】
本発明は、ラッカーゼを用いる天然ヌートカトンの商業的に利用することができる製造方法に関する。驚くべきことに、本発明により、酵素ラッカーゼ(ベンゼンジオール:オキシドレダクターゼ:EC1.10.3.2)が、商業的に利用できる収率でのバレンセンのヌートカトンへの酸化を触媒するために用いることができることが見出された。モノマー状または多量体状のいずれかの銅−含有糖タンパク質であるラッカーゼは、リグニンを合成する樹木植物中に、およびリグニンを分解する菌種中に見出される天然産出リグニン分解酵素である。
【0015】
材料
ラッカーゼは、微生物源、商業的販売業者[例えば、シグマ(Sigma)]から得ることができ、又は当業者に公知の方法を用いる組換えDNA技術により製造することができる。ラッカーゼの微生物源の例には、ボトリチス シネレア(Botrytis cinerea)またはトラメテス ベルシコロール(Trametes versicolor)などの白色腐朽菌(white rot fungi)がある。ボトリチス シネレア61−34およびトラメテス ベルシコロールの培養液からのラッカーゼは、College of Environmental Science and Forestryat the State University of New York in SyracuseのDr.James P.Nakasからの寄贈品であった。そのラッカーゼ活性レベルは、630〜1130nkat/mlであった。この酵素は、冷蔵下(約4℃)で少なくとも6ヶ月間、安定である。
【0016】
バレンセンは、GivaudanRoure Flavors,Lakeland,FLから、84〜85%の純度で得た。バレンセンはまた、その他の製造業者から、また精油の分留、例えば抽出または蒸留技術を用いることによる分別によって得ることができる。精油は通常、これらが得られる植物の特徴的匂いまたは香りを有する揮発性油であり、香料および風味付与剤に使用される。精油の例には、オレンジ油、ビターオレンジ油、グレープフルーツ油、レモン油、タンジェリン油、柑橘類油などが包含される。
【0017】
本明細書で使用されるメディエーター(mediator)の用語は、酸化性酵素により活性化され、また酵素上の活性部位から感受性構造に拡散する、拡散性分子であると定義される。ラッカーゼは、独立して、触媒として機能することができるものの、或る種のメディエーターの存在によりラッカーゼ触媒反応が強化され得ることが知られている。下記化学物質は、メディエーターとして活性であることが見出された:
1−ヒドロキシベンゾトリアゾール(HBT)、2,2´−アジノ−ビス(3−エチルベンズチアゾリン−6−スルホン酸)(ABTS)、フェルラ酸(ferulic aced)、ジメトキシベンジルアルコール、ジメチルアミノ安息香酸、カテキン、エピカテキン、p−ヒドロキシフェニル酢酸、ケルセチン(quercetin)、クロロプロマジン、フェノチアジン、ナリンジン(naringin)、プロマジン(promazine)、ホモバニリン酸、4−アミノ−サリチル酸、シリンガ酸(syringic acid)、4−ヒドロキシケイ皮酸、4−アミノ−3−ヒドロキシ安息香酸、バニリン酸、イソバニリン酸、カフェー酸、α−レゾルシル酸、β−レゾルシル酸、γ−レゾルシル酸、2,3−ジヒドロキシ安息香酸、3,4−ジヒドロキシ安息香酸、4−ヒドロキシ安息香酸、3−ヒドロキシ安息香酸、2,4,6−トリヒドロキシ安息香酸、安息香酸、ケイ皮酸、安息香酸ナトリウムおよびサリチル酸。使用するメディエーターは、Sigma Chemical Co.またはAldrich ChemicalCo.から購入した。その他の関連化学物質もまた、メディエーターとして有用であることは、当業者により認識されることである。
【0018】
酵素的転換方法
バレンセンとラッカーゼ活性を有する組成物とを、酸素供給源の存在下で反応させた。商業的に実施可能であるためには、少なくとも約0.1%のバレンセン出発濃度が必要である。ラッカーゼ活性を有する組成物が、溶液形態である場合、そのバレンセン濃度は、約64重量%までであることができる。
別法として、ラッカーゼなどの酵素は、当業者に公知であるように、固形支持体上で不動化または結合させることができる。この不動化技術の例は、J.Mol.Cat.B:Enzymatic 6(1999),29〜39; Chibata 等、Biocatalysis:Immobilized cells and enzymes,J.Mol.Cat.,37(1986),1〜24;Sharma 等、Immobilized Biomaterials Techniques and Applications,Angew.Chem.Int.,Ed.Engl.,21(1982),837〜854;Laskin(Ed.),Enzymes and Immobilized Cells in Biotechnologyに見出され、これらの記載の全体を引用して、ここに組み入れる。不動化された酵素が、しばしば未結合状態の酵素に比較して、有機溶剤に対して増大した耐性を示すことは、一般に公知である。すなわち、ラッカーゼを一度、不動化するか、または架橋させた酵素結晶(Altus Biologics, Inc.)などのように、別の方法により安定化すると、さらに高濃度のバレンセンを使用することができるものと見做すことができる。固体支持体、例えばカラム上に不動化させた場合、反応混合物中のバレンセンの濃度は、100%に近づけることができる。従って、本発明の精神または範囲を逸脱することなく、ラッカーゼまたは反応条件に対する種々の変化、修正または変更をなすことができる。
【0019】
酸素供給源は、純粋な酸素または酸素含有気体の混合物、例えば空気またはその他の気体混合物であることができる。場合により、1種または2種以上のメディエーターを、約50mMまでの濃度、好ましくは0.1mMよりは大きく、約5mMまでの濃度で包含させることができ、これにより反応を強化することができる。好適メディエーターは、HBT、フェルラ酸、4−ヒドロキシケイ皮酸、p−ヒドロキシフェニル酢酸、β−レゾルシル酸であり、HBTは、特に好適である。また、1種または2種以上の溶剤、例えば水、ヘキサンまたはその他の炭化水素を、ラッカーゼが当該反応を触媒することができる条件として定義される、ラッカーゼ活性を維持する濃度で包含させることができる。これらの条件には、適当なpHが包含され、このpHは、ラッカーゼについて約pH3〜7の範囲、最適には、pH3.5である。その他の反応条件には、酵素を変性させない溶剤の使用および酵素を触媒作用を得るのに適する立体配置にする一定の水分レベルが包含される。別段の記載がないかぎり、反応は、0.1Mシトラート緩衝液(pH3.5)中で行った。
【0020】
バレンセンを先ず、反応容器中でpH3〜7において、シトラート緩衝液と混合した。使用する場合、メディエーターおよび/または乳化剤を、この混合物にまた添加した。酵素を添加し、次いで反応を室温で継続させた。この反応は、いずれかの長さの期間、例えば1週間まで、またはそれ以上の期間にわたり進行させることができるが、少なくも約24時間の反応により、商業的に実施可能な量で生産される。この混合物を、約700rpmまでの速度で連続撹拌した。一態様において、反応は、約25℃〜30℃およびpH3〜7の範囲のpHにおいて、約48時間、生じさせた。試料を、定期的に採取し、バレンセンのヌートカトンへの変換を追跡した。炭酸ナトリウムによりpHをpH9.0に高め、また少なくとも約55℃に加熱することによって、反応を停止させた。当該ヒドロペルオキシドの分解を促進するために、加熱中、または加熱の代わりに、触媒を添加することができる。一態様において、触媒は、生理学的化合物、例えば鉄またはアスコルビン酸などである。その他の触媒の例には、コバルトおよび銅がある。
【0021】
被験酵素レベルは、約10nkat/ml〜620nkat/mlであり、またバレンセンの出発濃度は、約0.1%〜約64%であった。他の態様では、バレンセンを、約1%〜約50%、または約1%〜5%の濃度で含有させた。大部分の実験において、この出発濃度は、約0.5%〜約1.5%の範囲内であった。
ツイーン(Tween)−80またはトリトン(Triton)X−100などの乳化剤をまた、0.5%の濃度で使用することができる。
【0022】
粗生成物の回収
反応は、反応ブロスのpHを、炭酸ナトリウムによりpH9.0に調節することによって停止させた。この混合物を次いで、少なくとも約55℃に加熱することによって、ヒドロペルオキシドを分解させた。この加熱時間は、選択する温度に依存する。例えば、80℃の温度で2時間、加熱する。この混合物を、各反応ブロス1000mlに対して、それぞれメチレンクロライド(250ml×3回)またはヘプタン(250ml×6回)により抽出した。この抽出液を、硫酸ナトリウム上で乾燥させ、次いで溶剤を、減圧下で除去した。
【0023】
生成物の精製
この粗生成物を、クーゲルロアー(Kugelrohr) 装置で高減圧下、例えば5mmHgで蒸留した。クーゲルロアー蒸留からの精製された生成物の重量および外部標定曲線から測定するものとして、この生成物中のヌートカトン濃度を使用し、バレンセンからの全体的なヌートカトン収率を計算した。ヌートカトンを採取するための別法、例えば水蒸気蒸留、クロマトグラフィーおよび結晶化を、当業者に知られているように、使用することもできる。
未反応のバレンセンはまた、回収し、次いで当業者に公知の方法、例えばシリカゲルクロマトグラフィー、蒸留または結晶化によって、後続の反応において基質として使用するためにリサイクルすることができる。再使用するための未反応バレンセンの回収は、バレンセンが高価であることから、本発明の商業的利用価値を高める。
ラッカーゼ活性の測定は、2,6−ジメトキシフェノール(DMOP)の酸化的二量化に基づいており、この方法は、Slomczynski等(1995)により開示されており、その記載全体を引用して、ここに組み入れる。簡単に言えば、5mMの2,6−DMOP500リットルに、0.1Mクエン酸塩NaOH(pH3.5)を添加し、次いで酵素稀釈液各10リットルを添加した。反応混合物を、477nmにおける分光光度測定により追跡した。
【0024】
分析方法
反応の間に、試料(1ml)を、定期的に採取し、次いでメチレンクロライド0.8mlで抽出した。分離したメチレンクロライド層を、ガスクロマトグラフィーによる分析に付した。反応の停止後、クーゲルロアー蒸留後に回収された生成物を、例えば95%エタノール、メチルtert−ブチルエーテル(MTBE)またはメチレンクロライドにより、2.5%濃度に稀釈し、次いでガスクロマトグラフィー(GC)により分析した。バレンセンおよびヌートカトンを分析するためには、スペルコ(Supelco)SPB−1カラム(30m、内径(ID)0.25mm、フィルム0.25ミクロン)を、250℃のインジェクター温度および10℃/分で120℃〜270℃のプログラム温度、および20分間の保持時間をもって使用した。
【0025】
図1に示されているように、バレンセンの酸化は、ヒドロペルオキシドをもたらす。この酵素方法で生成されたヒドロペルオキシドは、ガスクロマトグラフィーのインジェクター口で分解させる。当該酵素反応中のヒドロペルオキシドの生成を特性決定するために、ヒドロペルオキシドを、AOAC公定分析法(Official Method Analysis)(1997)または薄層クロマトグラフィー(TLC)法(Johnson and Nidy,1975)のどちらかを用いて測定した。ヒドロペルオキシドはまた、ヘルドリッチ(Herderich)等の方法(1997)およびシュナイダー(Schneider)等の方法(1997)を用いる液体クロマトグラフィー/質量分析法(LC/MS)により検出した。
バレンセンヒドロペルオキシドが、酵素による酸化後に反応混合物中に存在していたことが高速液体クロマトグラフィー(HPLC)により単離された試料の核磁気共鳴(NMR)分析により確認された。
【0026】
バレンセンヒドロペルオキシドの分析
酵素反応の生成混合物中に、高いペルオキシド値が見出され、この数値は、還流後にほぼゼロに減少した。しかしながら、ガスクロマトグラフィー(GC)分析によれば、還流前と還流後との組成上の差異は、有意ではなかった。これは、ヒドロペルオキシドの熱不安定性によるものと見做された。図2に示されているように、反応混合物のHPLC分析から、6.7分の保持時間にある一つのピークが、反応混合物の還流後に消失したことがわかった。酵素反応の生成混合物のHPLCクロマトグラムを、還流後(図2A)および還流前(図2B)に得た。ルナ(Luna)C18(2)、150×2mmの3個のカラムを使用した。溶出液は0.25ml/分の流速で、メタノール:水(80:20)による4分間から100%メタノールで1分間までの勾配であった。検出は、210nmで行った。ピークに相当するフラクションを採取した。これらのフラクションは、ヨウ化カリウム/デンプンおよびFeSO4/NH4SCN溶液(AOAC,1990; Johnson and Nidy,1975)を用いる試験に基づき、ヒドロペルオキシド活性を明確に示した。ヌートカトンの紫外線(UV)スペクトル分析(図3A)を、仮定の(hypothetical)ヒドロペルオキシドピーク(図3B)のUVスペクトルと比較した。スペクトルは、バレンセンのスペクトルとは類似しているが、ヌートカトンのスペクトルとは相違しており、230nmおよび255nmにおけるエノン吸光に欠けていた。
【0027】
バレンセンヒドロペルオキシドの単離
反応混合物の粗製メチレンクロライド抽出液を、フラッシュクロマトグラフィーにより分別した[フラッシュ(Flash)40M、KP−Sil,31−63m,90g)。溶剤系は、薄層クロマトグラフィー(TLC)試験に基づき選択した。試料を、メチレンクロライドで、次いでMTBEで溶出した。全部で70のフラクションを集め、可視化試薬としてチオシアネート硫酸鉄(II)を用いるTLCにより分析した。ヒドロペルオキシドは、Fe(II)を、チオシアネートと反応するFe(III)に酸化し、血赤色のFe(SCN)52-を生成する(JohnsonおよびNidy、1975)。TLC試験は、フラクション7−12がヒドロペルオキシド活性であることを示した。集めたフラクションについてHPLC分析を行った。フラクション7−12中の主要成分は、仮定ヒドロペルオキシドのHPLCピークに相当した。フラクション7−12から、溶剤を蒸発させ、得られた生成物は、バレンセンヒドロペルオキシドとして同定された。
【0028】
NMR試験
1H−、13C−、DEPT、1H1H−関連性および1H13C−関連性試験を、フラクション7−12からの生成物について行った。図4Aは、図4Bに示されている化合物のプロトン(1H)核磁気共鳴(NMR)スペクトルを示している。図5Aは、図5Bに示されている化合物の13C NMRスペクトルを示している。13Cシグナルの多重度および化学シフトから、その炭素骨格がバレンセンの炭素骨格に非常に類似していることが判る。図5Aを参照すると、1個の炭素共鳴ラインが、78.4ppmに見出され、これは酸素原子に結合している炭素に典型的である。しかしながら、この炭素ラインは、対応するアルコールに比較して、さらに13.9ppm下部にある。炭素(C−O)に結合したプロトンの化学シフトは、4.33ppmにあるのに対して、アルコールの相当するプロトンは、4.04ppmの化学シフトを有する。図7は、フラクション7−12からの生成物を用いて得られたバレンセンヒドロペルオキシドの計算された13C化学シフトに相当する(試験結果は、カッコ内に示されている)。バレンセンヒドロペルオキシドの計算された13C化学シフトは、図5Aに示されているフラクション群7−12中の主要成分のものと非常に近接している。これらの試験結果から、酵素反応混合物中のヒドロペルオキシドは、図7に示されている化学構造を有するものと結論することができる。
【実施例】
【0029】
本発明は、下記例によって、さらに理解される。
例1
バレンセンの酵素酸化を、1.5リットル反応容器(New BrurswickScientific)中で行った。この反応混合物は、0.1Mシトラート緩衝液(pH3.5)450ml、0.1M HBT 2.5ml、ツイーン(Tween)−80 2.5ml、バレンセン2.5mlおよびボトリチス(Botrytis)発酵ブロス(ラッカーゼ活性レベル:110nkat/ml)50mlからなるものであった。酸素供給源として空気を使用し、高速撹拌(約500〜700rpm)を用いて供給した。この反応は、室温(約25℃〜30℃)で行った。2日後、混合物のpHを炭酸ナトリウムにより9.0に変え、沸騰水浴中で2時間、加熱することによって、反応を停止させた。
室温まで冷却させた後、この混合物を、メチレンクロライドで抽出した(3×各150ml)。有機相を集め、Na2SO4上で乾燥させ、次いで減圧下で濃縮した。この粗生成物(3.5ml)を、クーゲルロアー(Kugelrohr)装置を用いて蒸留し、次いでガスクロマトグラフィー(GS)により分析した。図6を参照すると、蒸留後の生成物の2.5%溶液の典型的GCプロフィールは、ヌートカトンが最大ピークであり、外部曲線から測定して、43.3%w/wを占めたことが示されている。この反応からのヌートカトンの総合収率は、28.6%w/wであった。未反応バレンセンは、33.4%w/wであった。
【0030】
例2
バレンセンの酵素酸化を、2リットル反応容器中で行った。この反応混合物は、0.1Mシトラート緩衝液(pH3.5)1405ml、0.1M HBT 30ml、ツイーン(Tween)−80 30mlおよびバレンセン30mlからなるものであった。トラメテス ベルシコローア(Trametes versicolor)からの発酵ブロス(ラッカーゼ活性レベル:11nkat/ml)5mlの添加によって、反応を開始させた。空気を酸素供給源として使用し、350rpmの撹拌により供給した。この反応は、30℃の温度で行った。9日間後、例1と同様に、反応を停止させ、生成物を回収した。この反応からのヌートカトンの総合収率は、22.38%w/wであった。未反応バレンセンは、37.34%w/wであった。
【0031】
例3
振盪フラスコにおいて、0.1Mシトラート緩衝液(pH3.5)、HBTメディエーター(最終濃度0.5〜16mM)、ツイーン−80(0.5%)、バレンセン(0.5〜64%)およびラッカーゼ(活性レベル:60nkat/ml)からなる反応混合物(20ml)を、225rpmで振盪させながら48時間、30℃でインキュベートした。反応を停止させた時点で、各フラスコから、試料2mlを採取し、次いでメチレンクロライド1.2mlで抽出した。分離した溶剤層を、GCにより分析した。バレンセンおよびヌートカトンに相当する面積パーセンテージを、下表に示す:
【0032】
【表1】
【0033】
例4
ラッカーゼおよび/またはメディエーターの存在および不存在における、バレンセンのヌートカトンへの変換を評価した。各フラスコ中の総反応容積は、10mlであった。ラッカーゼは、60nkat/mlの活性レベルを有するボトリチス シネレア(Botrytis cinerea)由来であった。メディエーターは、0.5mMの最終濃度のHBTであった。出発基質(バレンセン)は、0.5%であった。4日後、これらの反応混合物を、メチレンクロライドで抽出し、次いで分離した有機相を、GCにより分析した。結果を、下表に示す。
【表2】
【0034】
例5
活性レベル620nkat/mlのラッカーゼ、基質としてバレンセン0.5%およびツイーン−80 0.5%からなる総反応容積10mlにおいて、下記化合物を、0.5mMの濃度でメディエーターとして試験した:1−ヒドロキシベンゾトリアゾール(HBT)、2,2´−アジノ−ビス(3−エチルベンズチアゾリン−6−スルホン酸(ABTS)、フェルラ酸(FA)、ジメトキシベンジルアルコール(DBA)およびジメチルアミノ安息香酸(DBAD)。各種メディエーターの存在下に4日間の反応後のバレンセンおよびヌートカトンの相対量を下表に示す。
【0035】
【表3】
【0036】
例6
活性レベル60nkat/mlのラッカーゼ、基質としてバレンセン1%およびツイーン−80 1%からなる反応容積20mlにおいて、下記化合物を、5mMの濃度でメディエーターとして試験した:カテキン、エピカテキン、p−ヒドロキシフェニル酢酸、ケルセチン、クロロプロマジン、フェノチアジン、ナリンジン、プロマジン、ホモバニリン酸、4−アミノ−サリチル酸、シリンガ酸、4−アミノ−3−ヒドロキシ安息香酸、バニリン酸、カフェー酸、プロトカテキュ酸(protocatechuic acid)、α−レゾルシル酸、β−レゾルシル酸、γ−レゾルシル酸、2,3−ジヒドロキシ安息香酸、4−ヒドロキシ安息香酸、3−ヒドロキシ安息香酸、2,4,6−トリヒドロキシ安息香酸、安息香酸、ケイ皮酸、イソバニリン酸、安息香酸ナトリウムおよびサリチル酸。各場合、変換率は、メディエーターの不存在下に得られた結果に比較して2〜8倍大きかった。この結果は、これらの化合物が、本方法においてメディエーターとして機能することを示している。
【0037】
例7
ボトリチス シネレア(Botrytis cinerea)からの60nkat/mlレベルのラッカーゼ、基質としてバレンセン0.5%、ツイーン(Tween)−80 0.5%、メディエーターとして0.5mMの最終濃度のHBTおよびpHを調整するための緩衝液(0.1M)からなる総量10mlの反応混合物を、225rpmで撹拌しながら30℃でインキュベートした。6日間の反応後、試料2mlを、各フラスコから採取し、次いでメチレンクロライドで抽出し、次いでGC分析に付す。異なるpH条件下での結果を、下表に示す。
【表4】
【0038】
例8
バレンセンを含有するオレンジ油の酵素酸化を、1.5リットル容器中で行った。0.1Mシトラート緩衝液(pH3.5)875ml、ツイーン−80 5ml、0.1M HBT 5ml、オレンジ油5mlおよびボトリチス シネレア(Botrytis cinerea)からのラッカーゼブロス100ml(活性レベル:63nkat/ml)からなる反応ブロスを、500rpmで撹拌しながら30℃でインキュベートした。4日間の反応後、クーゲルロアー(Kigelrohr) 蒸留後の生成物を、例1に記載の方法に従い回収した。この生成物中のヌートカトンの存在は、GC/MSによる分析により確認した。
酸化前および酸化後の両方のオレンジ油の感覚器官試験を、下記組成物を用いて、9人の熟練のフレーバーリスト(flavorists)により行った。
【表5】
【0039】
これらのフレーバーリストの全員が、混合物Aを混合物Bと区別することができた。これらのフレーバーリストは、混合物Bのフレーバーを、フルーティ、ヌートカトン様フレーバーおよび強力なウッディノートを有し、長い持続性を有すると評価した。
明細書中に示され、説明されている本発明の態様は、当業者である発明者の態様を示すのみであって、いかなる点でも制限するものではない。例えば、固体支持体に結合させた酵素は、酵素の安定性および有機溶剤に対するその耐性を高めることができる。もう一つの例として、多くの種々の化学物質を、メディエーターとして使用することができる。さらにもう一つの例として、フレーバーを生成させるために、カリオフィレン、フェニルピルビン酸、ガイエン(guaiene)、アネトール、ファメゾールおよびホップ油などの別種の基質の酸化を包含することができる。従って、本発明の精神および特許請求の範囲の範囲から逸脱することなく、これらの態様の種々の変化、修正または変更をなすことができ、または頼ることができる。
【0040】
ヌートカトンはフレーバーとして有用である。
本発明では、バレンセンのラッカーゼ触媒酸化によるヌートカトンの製造方法が提供される。本発明の方法によれば、ヌートカトンが商業的に実用性のある収率で製造される。
【特許請求の範囲】
【請求項1】
ヌートカトンの製造方法であって、
a)酸素供給源の存在下に、バレンセンをラッカーゼを含む組成物と反応させて、バレンセンヒドロペルオキシドを生成させる工程であって、バレンセンの濃度が0.1%よりも大きい工程
b)このヒドロペルオキシドを分解させ、ヌートカトンを生成する工程、及び
c)ヌートカトンを回収する工程、
を包含する上記方法。
【請求項2】
ヒドロペルオキシドを加熱により分解させる、請求項1に記載の方法。
【請求項3】
ヒドロペルオキシドを触媒により分解させる、請求項1に記載の方法。
【請求項4】
触媒が、鉄、アスコルビン酸、コバルト、銅およびそれらの組合わせからなる群から選択される、請求項3に記載の方法。
【請求項5】
ラッカーゼが、微生物源由来である、請求項1〜4のいずれか一項に記載の方法。
【請求項6】
微生物源が、樹木を腐朽させる菌類である、請求項5に記載の方法。
【請求項7】
ラッカーゼが、組換えDNA由来である、請求項1〜4のいずれか一項に記載の方法。
【請求項8】
バレンセンおよびラッカーゼを含む組成物に、メディエーターを添加することをさらに包含する、請求項1〜7のいずれか一項に記載の方法。
【請求項9】
メディエーターが、1−ヒドロキシベンゾトリアゾール、2,2´−アジノ−ビス(3−エチルベンズチアゾリン−6−スルホン酸)、フェルラ酸、ジメトキシベンジルアルコール、ジメチルアミノ安息香酸、カテキン、エピカテキン、p−ヒドロキシフェニル酢酸、ケルセチン、クロロプロマジン、フェノチアジン、ナリンジン、プロマジン、ホモバニリン酸、4−アミノ−サリチル酸、シリンガ酸、4−ヒドロキシケイ皮酸、4−アミノ−3−ヒドロキシ安息香酸、バニリン酸、イソバニリン酸、カフェー酸、α−レゾルシル酸、β−レゾルシル酸、γ−レゾルシル酸、2,3−ジヒドロキシ安息香酸、3,4−ジヒドロキシ安息香酸、4−ヒドロキシ安息香酸、3−ヒドロキシ安息香酸、2,4,6−トリヒドロキシ安息香酸、安息香酸、ケイ皮酸、安息香酸ナトリウムおよびサリチル酸、ならびにそれらの組合わせからなる群から選択される、請求項8に記載の方法。
【請求項10】
メディエーターの濃度が、0.1mMよりも大きく、50mMまでである、請求項8又は9に記載の方法。
【請求項11】
酸素供給源が、酸素含有気体混合物および純粋酸素からなる群から選択される、請求項1〜10のいずれか一項に記載の方法。
【請求項12】
バレンセンとラッカーゼを含む組成物とを、加熱に先立ち、少なくとも24時間反応させ、バレンセンヒドロペルオキシドを生成させる、請求項1又は2に記載の方法。
【請求項13】
少なくとも55℃の温度に加熱する、請求項1又は2に記載の方法。
【請求項14】
バレンセンおよびラッカーゼを含む組成物に、ラッカーゼ活性を維持する濃度で、水、ヘキサン又はそれらの混合物から選択される溶剤を添加することをさらに包含する、請求項1〜13のいずれか一項に記載の方法。
【請求項15】
組成物が溶液であり、およびバレンセンの濃度が64重量%までである、請求項1〜14のいずれか一項に記載の方法。
【請求項16】
ラッカーゼが、固定化されている、請求項1〜14のいずれか一項に記載の方法。
【請求項17】
バレンセンヒドロペルオキシドの製造方法であって、バレンセン、ラッカーゼを含む組成物および酸素供給源を、該バレンセンヒドロペルオキシドを生成させる条件下で反応させることを包含する上記方法。
【請求項18】
バレンセンの出発濃度が0.1%よりも大きい、請求項17に記載の方法。
【請求項19】
メディエーターを添加することをさらに包含する、請求項17又は18に記載の方法。
【請求項20】
ラッカーゼ活性を維持する濃度で、水、ヘキサン又はそれらの混合物から選択される溶剤を添加することをさらに包含する、請求項17〜19に記載の方法。
【請求項21】
ヌートカトンの製造方法であって、
a)バレンセンおよび固定化したラッカーゼを、酸素供給源の存在下に反応させ、バレンセンヒドロペルオキシドを生成させる工程、
b)加熱して、このバレンセンヒドロペルオキシドを分解させ、ヌートカトンを生成する工程、そして
c)ヌートカトンを回収する工程、
を包含する方法。
【請求項22】
バレンセンの濃度が、86%である、請求項21に記載の方法。
【請求項23】
バレンセンおよび固定化したラッカーゼに、メディエーターを添加することをさらに包含する、請求項21又は22に記載の方法。
【請求項24】
バレンセンおよび固定化したラッカーゼに、ラッカーゼ活性を維持する濃度で水、ヘキサン又はそれらの混合物から選択される溶剤を添加することをさらに包含する、請求項21〜23に記載の方法。
【請求項25】
増加されたヌートカトン含有量を有する精油の製造方法であって、
a)バレンセンを含有する精油およびラッカーゼを含む組成物を、酸素供給源の存在下に反応させ、ヌートカトンを含有する精油の混合物を生成させる工程、
b)この混合物を加熱し、ヌートカトンの量を増加させる工程、及び
c)増加されたヌートカトンを含有する精油を回収する工程、
を包含する上記方法。
【請求項26】
精油の出発濃度が0.5%よりも大きい、請求項25に記載の方法。
【請求項27】
精油が、オレンジ油、ビターオレンジ油、グレープフルーツ油、レモン油、タンジェリン油およびそれらの組合せからなる群から選択される、請求項25又は26に記載の方法。
【請求項28】
精油のフラクションを反応させる、請求項25〜27のいずれか一項に記載の方法。
【請求項29】
精油のフラクションが、抽出物および蒸留物からなる群から選択される、請求項28に記載の方法。
【請求項1】
ヌートカトンの製造方法であって、
a)酸素供給源の存在下に、バレンセンをラッカーゼを含む組成物と反応させて、バレンセンヒドロペルオキシドを生成させる工程であって、バレンセンの濃度が0.1%よりも大きい工程
b)このヒドロペルオキシドを分解させ、ヌートカトンを生成する工程、及び
c)ヌートカトンを回収する工程、
を包含する上記方法。
【請求項2】
ヒドロペルオキシドを加熱により分解させる、請求項1に記載の方法。
【請求項3】
ヒドロペルオキシドを触媒により分解させる、請求項1に記載の方法。
【請求項4】
触媒が、鉄、アスコルビン酸、コバルト、銅およびそれらの組合わせからなる群から選択される、請求項3に記載の方法。
【請求項5】
ラッカーゼが、微生物源由来である、請求項1〜4のいずれか一項に記載の方法。
【請求項6】
微生物源が、樹木を腐朽させる菌類である、請求項5に記載の方法。
【請求項7】
ラッカーゼが、組換えDNA由来である、請求項1〜4のいずれか一項に記載の方法。
【請求項8】
バレンセンおよびラッカーゼを含む組成物に、メディエーターを添加することをさらに包含する、請求項1〜7のいずれか一項に記載の方法。
【請求項9】
メディエーターが、1−ヒドロキシベンゾトリアゾール、2,2´−アジノ−ビス(3−エチルベンズチアゾリン−6−スルホン酸)、フェルラ酸、ジメトキシベンジルアルコール、ジメチルアミノ安息香酸、カテキン、エピカテキン、p−ヒドロキシフェニル酢酸、ケルセチン、クロロプロマジン、フェノチアジン、ナリンジン、プロマジン、ホモバニリン酸、4−アミノ−サリチル酸、シリンガ酸、4−ヒドロキシケイ皮酸、4−アミノ−3−ヒドロキシ安息香酸、バニリン酸、イソバニリン酸、カフェー酸、α−レゾルシル酸、β−レゾルシル酸、γ−レゾルシル酸、2,3−ジヒドロキシ安息香酸、3,4−ジヒドロキシ安息香酸、4−ヒドロキシ安息香酸、3−ヒドロキシ安息香酸、2,4,6−トリヒドロキシ安息香酸、安息香酸、ケイ皮酸、安息香酸ナトリウムおよびサリチル酸、ならびにそれらの組合わせからなる群から選択される、請求項8に記載の方法。
【請求項10】
メディエーターの濃度が、0.1mMよりも大きく、50mMまでである、請求項8又は9に記載の方法。
【請求項11】
酸素供給源が、酸素含有気体混合物および純粋酸素からなる群から選択される、請求項1〜10のいずれか一項に記載の方法。
【請求項12】
バレンセンとラッカーゼを含む組成物とを、加熱に先立ち、少なくとも24時間反応させ、バレンセンヒドロペルオキシドを生成させる、請求項1又は2に記載の方法。
【請求項13】
少なくとも55℃の温度に加熱する、請求項1又は2に記載の方法。
【請求項14】
バレンセンおよびラッカーゼを含む組成物に、ラッカーゼ活性を維持する濃度で、水、ヘキサン又はそれらの混合物から選択される溶剤を添加することをさらに包含する、請求項1〜13のいずれか一項に記載の方法。
【請求項15】
組成物が溶液であり、およびバレンセンの濃度が64重量%までである、請求項1〜14のいずれか一項に記載の方法。
【請求項16】
ラッカーゼが、固定化されている、請求項1〜14のいずれか一項に記載の方法。
【請求項17】
バレンセンヒドロペルオキシドの製造方法であって、バレンセン、ラッカーゼを含む組成物および酸素供給源を、該バレンセンヒドロペルオキシドを生成させる条件下で反応させることを包含する上記方法。
【請求項18】
バレンセンの出発濃度が0.1%よりも大きい、請求項17に記載の方法。
【請求項19】
メディエーターを添加することをさらに包含する、請求項17又は18に記載の方法。
【請求項20】
ラッカーゼ活性を維持する濃度で、水、ヘキサン又はそれらの混合物から選択される溶剤を添加することをさらに包含する、請求項17〜19に記載の方法。
【請求項21】
ヌートカトンの製造方法であって、
a)バレンセンおよび固定化したラッカーゼを、酸素供給源の存在下に反応させ、バレンセンヒドロペルオキシドを生成させる工程、
b)加熱して、このバレンセンヒドロペルオキシドを分解させ、ヌートカトンを生成する工程、そして
c)ヌートカトンを回収する工程、
を包含する方法。
【請求項22】
バレンセンの濃度が、86%である、請求項21に記載の方法。
【請求項23】
バレンセンおよび固定化したラッカーゼに、メディエーターを添加することをさらに包含する、請求項21又は22に記載の方法。
【請求項24】
バレンセンおよび固定化したラッカーゼに、ラッカーゼ活性を維持する濃度で水、ヘキサン又はそれらの混合物から選択される溶剤を添加することをさらに包含する、請求項21〜23に記載の方法。
【請求項25】
増加されたヌートカトン含有量を有する精油の製造方法であって、
a)バレンセンを含有する精油およびラッカーゼを含む組成物を、酸素供給源の存在下に反応させ、ヌートカトンを含有する精油の混合物を生成させる工程、
b)この混合物を加熱し、ヌートカトンの量を増加させる工程、及び
c)増加されたヌートカトンを含有する精油を回収する工程、
を包含する上記方法。
【請求項26】
精油の出発濃度が0.5%よりも大きい、請求項25に記載の方法。
【請求項27】
精油が、オレンジ油、ビターオレンジ油、グレープフルーツ油、レモン油、タンジェリン油およびそれらの組合せからなる群から選択される、請求項25又は26に記載の方法。
【請求項28】
精油のフラクションを反応させる、請求項25〜27のいずれか一項に記載の方法。
【請求項29】
精油のフラクションが、抽出物および蒸留物からなる群から選択される、請求項28に記載の方法。
【図1】

【図2】
【図3A】
【図3B】
【図4A】
【図4B】
【図5A】
【図5B】
【図6】
【図7】
【図2】
【図3A】
【図3B】
【図4A】
【図4B】
【図5A】
【図5B】
【図6】
【図7】
【公開番号】特開2012−90638(P2012−90638A)
【公開日】平成24年5月17日(2012.5.17)
【国際特許分類】
【出願番号】特願2012−175(P2012−175)
【出願日】平成24年1月4日(2012.1.4)
【分割の表示】特願2000−270975(P2000−270975)の分割
【原出願日】平成12年9月7日(2000.9.7)
【出願人】(591040281)ジボーダン ソシエテ アノニム (3)
【Fターム(参考)】
【公開日】平成24年5月17日(2012.5.17)
【国際特許分類】
【出願日】平成24年1月4日(2012.1.4)
【分割の表示】特願2000−270975(P2000−270975)の分割
【原出願日】平成12年9月7日(2000.9.7)
【出願人】(591040281)ジボーダン ソシエテ アノニム (3)
【Fターム(参考)】
[ Back to top ]