
ラパマイシンの39−デスメトキシ誘導体
本発明は新規39−デスメトキシラパマイシン誘導体、その産生方法およびその用途に関する。別の観点において本発明はこれら39−デスメトキシラパマイシン誘導体をガンおよび/またはB細胞系悪性腫瘍の治療、免疫抑制の誘導または維持、臓器移植拒絶反応、移植片対宿主拒絶病、自己免疫疾患、炎症疾患、血管疾患および線維症の治療、神経再生の刺激または感染症の治療に使用することについての用途を提供する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は新規な39−デスメトキシラパマイシン誘導体、その製造方法およびその用途に関する。別の観点において、本発明は39−デスメトキシラパマイシン誘導体を、ガンおよび/またはB細胞系悪性腫瘍の治療、免疫抑制の誘導または維持、臓器移植拒絶反応、移植片対宿主拒絶病、自己免疫疾患、炎症疾患、血管疾患および線維症の治療、神経再生の刺激または感染症の治療に使用することについての用途に関する。
【背景技術】
【0002】
ラパマイシン(rapamycin、シロリムス)(図1)は、ピペコリン酸ラクトン(Paivaら、1991年)に連結された1,2,3−トリカルボニル部分を有する、ストレプトマイセス・ヒグロスコピカス(Streptomyces hygroscopicus)NRRL5491により産生される親油性マクロライド(Sehgalら、1975;Vezinaら、1975;米国特許第3,929,992号;米国特許第3,993,749号)。本発明の目的のため、ラパマイシンは、Findlayら(1980年)または化情報索(第11版インデックス蓄積本、1982〜1986年、p60719CS)の番号付け慣例よりは、McAlpineら(1991年)の番号付け慣例により記述される。
【0003】
ラパマイシンは、その化合物によって示される広い活性スペクトルの故に顕著な薬理的価値を持つ。ラパマイシンは主としてカンジタ(Candida)属に対して、しかしまた糸状菌に対しても穏やかな抗菌活性を示す(Bakerら、1978年;Sehgalら、1975年;Vezinaら、1975年;米国特許3,929,992;米国特許3,993,749)。ラパマイシンは、多くの細胞の種類において信号伝達経路を標的にすることにより、たとえば細胞サイクルのG1からS相への成長を許容するシグナリング経路を示すことにより細胞増殖を示す(Kuoら、1992年)。T細胞内でラパマイシンはIL−2受容体およびその後の免疫抑制をもたらすT細胞自己増殖からのシグナリングを示す。ラパマイシンは多くの哺乳類細胞種の増殖を抑制する(Brunnら、1996年)から、ラパマイシンのこの抑制効果はT細胞に限らない。ラパマイシンは、したがって、臓器移植拒絶反応の予防や自己免疫疾患の治療において確立されたまたは予測される治療的適用を持った有力な免疫抑制剤である(Kahanら、1991年)。40−O−(2−ヒドロキシ)エチル−ラパマイシン(SDZ RAD、RAD001、Certican、エベロリムスeverolimus)は免疫抑制的薬理効果を示すラパマイシンの半合成相同物であり、抗ガン剤としての研究も進められている(Sedrani,Rら、1998年;Kirchnerら、2000年;米国特許5,665,772、Boulayら、2004年)。この薬剤は欧州で2003年に免疫抑制剤として承認された。ラパマイシンエステル誘導体CCI−779(Wyeth−Ayerst)は生体外で細胞成長を示し、生体内で腫瘍成長を示す(Yuら、2001年)。CCI−779は潜在的抗ガン剤として現在第2相臨床試験が行われている。慢性尋常性乾癬の治療(KirbyおよびGriffiths、2001年)、PC12細胞内神経突起伸長刺激などの潜在的効用(Lyonsら、1994年)、機械的損傷後の血管および平滑筋細胞によるサイトカインに対する増殖反応の抑制(Gregoryら、1993年)におけるラパマイシンの価値、および同種移植性線維症(allograft fibrosis)の予防におけるその役割(WallerおよびNicholson、2001年)が熱心な研究領域となっている(KahanおよびCamardo、2001年)。最近の報告によれば、ラパマイシンは臓器移植患者に対する長期免疫抑制治療において、他の免疫抑制レジメより低い発ガン率を示し、この発ガン率低下は血管新生(angiogenesis)の抑制によるとされている(Gubaら、2002年)。イムノフィリンリガンドの神経向性作用はそれらの免疫抑制活性とは無関係であり(Steinerら、1997年)、親出願WO01/03692に要約される成熟ステロイド受容体複合体の分裂によって神経細胞刺激が増進されると報じられた。催奇的影響と共に高脂血症や血小板減少症などの副作用が報告されている(Hentgesら、2001年;KahanおよびCamardo、2001年)。
【0004】
ラパマイシンのポリケチドバックボーンは、I型ポリケチド合成酵素を含む非常に大きな多機能タンパク質(rapPKS、Paivaら、1991年)により、全部で7個のプロピオン酸エステルおよび7個の酢酸エステルのユニットをシキマート由来シクロヘキサンカルボン酸スターターユニットにhead−to−tail縮合することにより合成される。L−リシン由来アミノ酸、ピペコリン酸がアミド架橋を経由してポリケチドバックボーンの最終酢酸エステルに縮合され(Paivaら、1993年)、次いでラクトン化によりマクロ環を形成する。
【0005】
3つのラパマイシンPKS遺伝子、NRPSエンコード遺伝子およびフランキング後期遺伝子配列および対応ポリペプチドの各々についてのヌクレオチド配列がAparicioら、1996およびSchweckeら、1995年によって同定され、受入番号X86780の下にNCBIに寄託され、この配列に対する訂正が最近WO04/007709に発表された。
【0006】
ラパマイシン生合成クラスタの最初の無酵素製品がプレラパマイシンである(WO04/007709、Gregoryら、2004年)。この完全にプロセス化されたラパマイシンの製造は、ラパマイシン後期遺伝子RapJ、RapN、RapO、RapM、RapQおよびRapIにエンコードされた酵素によるポリペプチド/NRPSコアの追加的処理を必要とする。
【0007】
今日までに特性付けられているラパマイシンの薬理作用は、FKBPと名付けられたサイトソル受容体との相互作用によるメディエートされるものと考えられている。真核T細胞の主要な細胞内ラパマイシン受容体はFKBP12であり(DiLellaおよびCraig、1991年)、産生される複合体が標的タンパク質と特異的に相互作用して細胞の信号伝達カスケードを示す。
【0008】
ラパマイシン−FKBP12複合体の標的は酵母内においてTOR(ラパマイシン標的タンパク質)として同定され(Alarconら、1999年)、哺乳類タンパク質はFRAP(FKBP−ラパマイシン付随タンパク質)ないしmTOR(ラパマイシンの哺乳類標的タンパク質)として知られている(Brownら、1994年)。
【0009】
mTOR伝達経路と神経単位に局所化されるタンパク質向性との関連、翻訳調整に包含されるタンパク質のリン酸化状態に対するその作用、転写および翻訳レベルにおける翻訳メカニズムの化合物存在性、アミノ酸ペルミアーゼ活性、および代謝経路に含まれる多くの酵素の転写調整が記述されている(Raughtら、2001年)。ラパマイシン感応性シグナル経路はまた、胎児の脳発達、学習および記憶形成に重要な役割を果たすものと考えられている(Tangら、2002年)。酵母内のTORタンパク質についての研究は、栄養感受性シグナル経路を調節する上でも役割を果たしていることを示した(Hardwickら、1999年)。同様に、mTORはタンパク質キナーゼB(akt)の活性に対する直接標的物質であり、インシュリンの信号伝達において主要な役割を果たすものとして確認されている(Shepherdら、1998年;Naveら、1999年)。哺乳類のTORはアクチン細胞骨格の分極化および翻訳開始の制御に関係していた(Alarconら、1999年)。mTORなどのホスファチジルイノストール−3−キナーゼは、細胞周期進行、付着、細胞生存および血管新生のような幾つかの腫瘍病原の見地で機能的である(RoymansおよびSiegers、2001年)。
【0010】
ラパマイシンおよびラパマイシン相同物の薬物動態学的研究により、溶液中でより安定的であり、代謝攻撃に対してより抵抗性を持ち、および/または細胞膜透過性が向上され且つエフラックスが減少されていて、より優れた経口バイオアベイラビリティを示すような新規ラパマイシン化合物の開発に対する要求が実証された。
【0011】
分子の化学的に利用可能な部位を用いて合成された一群のラパマイシン相同物が報告された。下記化合物に対する記述には、図1に記述されたラパマイシン分子の付番方式を適用させた。相同化または置換のための分子上の化学的に利用可能な部位には、C40およびC28ヒドロキシル基(例えば、米国特許第5,665,772号、米国特許第5,362,718号)、C39およびC16メトキシ基(例えば、WO96/41807号、米国特許第5,728,710号)、C32、C26およびC9ケト基(例えば、米国特許第5,378,836号、米国特許第5,138,051号、米国特許第5,665,772号)が含まれる。トリエンを標的にしたC17、C19および/またはC21での水添は、抗真菌活性は維持するが、免疫抑制特性の相対的損失をもたらした(例えば、米国特許第5,391,730号、米国特許第5,023,262号)。分子の安全性に対する相当な改善(例えば、C32、C40および/またはC28でのオキシムの形成、米国特許第5,563,145号、米国特許第5,446,048号)、代謝作用攻撃に対する耐性(例えば、米国特許第5,912,253号)、生体利用可能性(例えば、米国特許第5,221,670号、米国特許第5,955,457号、WO98/04279号)および前駆薬物の製造(例えば、米国特許第6,015,815号、米国特許第5,432,183号)が、相同化を通じて達成された。
【0012】
しかしながら、より優れた代謝安定性、より優れた細胞膜透過性および/または減少されたエフラックス率を備えたより広範な群のラパマイシン誘導体のための要求がなお存在している。そのようなラパマイシン誘導体は広範な病状の治療により大きな有用性を持つであろう。本発明は、より優れた代謝安定性、より優れた細胞膜透過性および/または減少されたエフラックス率を備え、および/またはラパマイシンとは異なる細胞抑制プロファイルを備えた一群の39−デスメトキシラパマイシン誘導体を提供する。この化合物は医療において、特にガンおよび/またはB細胞系悪性腫瘍の治療、免疫抑制の誘導または維持、臓器移植拒絶反応、移植片対宿主拒絶病、自己免疫疾患、炎症疾患、血管疾患および線維症の治療、神経再生の刺激または感染症の治療に有効である。
【発明の開示】
【0013】
本発明はラパマイシンの39−デスメトキシ誘導体、該化合物の調製方法、その中間産生物および該化合物の医療的用途のための方法に関する。
【0014】
その最も広い観点において、本発明は、40−ヒドロキシ位置がカルボン酸エステル、エーテル、リン酸エステル、ホスフィン酸エステル、アセタールまたはグリコシルとして誘導化されたことを特徴とするラパマイシンの39−デスメトキシ誘導体を提供する。
【0015】
本発明化合物の代謝安定性、細胞膜透過性、エフラックスおよびバイオアベイラビリティは下記のようにして試験することができる。
【0016】
39−デスメトキシラパマイシンがカルボン酸エステル、エーテルまたはアセタールとして誘導化される場合、誘導基は12個を越えない炭素原子を含むことが好ましい(特に7個以下、さらには5個以下の炭素原子数が好ましい)。好ましくは、それは−CF2PO(OH)2、−PO(OH)2、−COOH、−OHおよび−NH2から、好ましくは−COOHおよび−OH、より好ましくは−OHから選ばれる一以上の官能基(特には2以上の官能基)を含む。
【0017】
39−デスメトキシラパマイシンがグリコシル基由来のアセタールとして誘導化される場合、各グリコシルは好ましくは12個を越えない炭素原子を含む(特に7個以下、さらには6個以下の炭素原子数が好ましい)糖またはグリコシドから形成することが好ましい。好ましくは、それは−COOH、−OHおよび−NH2から、好ましくは−NH2および−OH、より好ましくは−OHから選ばれる一以上の官能基(特には2以上の官能基)を含む。
【0018】
39−デスメトキシラパマイシンがリン酸エステルとして誘導化される場合、好ましくはアルキル基は4個を越えない炭素原子を含む。
【0019】
39−デスメトキシラパマイシンがホスフィン酸エステルとして誘導化される場合、好ましくはアルキル基は4個を越えない炭素原子を含むことが好ましく、一例はホスフィン酸で形成されるエステルである。
【0020】
誘導部分の具体例を下記に示す。
【0021】
より具体的な観点において、本発明は、化1による39−デスメトキシラパマイシン誘導体またはその薬学的に許容可能な塩を提供する。
【0022】
【化1】
ここで、
Xは結合またはCH2を表し、
R1はケト基または(H,H)を表し、
R2はOHまたはOMeを表し、
R3はH、OHまたはOMeを表し、
R4およびR5は各々独立してHまたはOHを表し、
R6は−R7、−C(O)R7、−(CH2)2−O−[CR21R22−O]a−C(O)−R23;−CR21R22−O−C(O)−R23;POR19R20、−PO(OR19)(OR20)またはY−R15を表し、
R7は−(CR8R9)m(CR10R11)pCR12R13R14を表し、
R8およびR9は各々独立してC1−C4アルキル、C2−C4アルケニルまたはC2−C4アルキニルを表し、これらのいずれの基においても−PO(OH)2、CF2PO(OH)2、−OH、−COOHまたは−NH2で任意に置換可能であり、あるいはR8およびR9は各々独立してH、トリフルオロメチルまたはFを表し、
R10、R11、R12、R13およびR14は各々独立してC1−C4アルキル、C2−C4アルケニルまたはC2−C4アルキニルを表し、これらのいずれの基においても−PO(OH)2、CF2PO(OH)2、−OH、−COOHまたは−NH2で任意に置換可能であり、あるいはR10、R11、R12、R13およびR14は各々独立してH、−(CR8R9)qNH2、−(CR8R9)qOH、CF3、F、COOHから選択することができ、あるいはR10とR11またはR12とR13またはR13とR14は一緒になってそれらが結合している炭素と共にC3−C6シクロアルキルまたはN、OおよびSから選ばれる一以上のヘテロ原子を含み5個までの−(CR8R9)qOH、−(CR8R9)qNH2または−COOH基で任意に置換可能である3〜6員ヘテロアルキル環を形成しており、
ここでYは結合、−C(O)−O−または−(CH2)2−O−C(O)−O−であり、
R15は化2、化3または化4のいずれかであり、
【化2】
【化3】
【化4】
R16は各々独立してHまたはOHであり、
R17はH、OHおよびNH2から独立して選ばれ、
R18はH、−CH3、−CH2OHおよび−COOHから選ばれ、
ただしR16、R17およびR18から選ばれる2以上の基はHまたはCH3を表し、
R19およびR20は各々独立してHまたはC1−C4アルキルをあらわし、あるいはR19およびR20の両方で=CH2を示し、
R21はH、CH3から独立して選ばれ、
R22はH、−CH3、−CH=CH2、−CH2Cl、−CHCl2、−CCl3、−CH(OH)Me、−CH2OH、−CH2CH3、−CH(Cl)Meから独立して選ばれ、
R23は独立してR7、Y−R15または5または6員アリルまたはヘテロアリル環であって任意にOH、F、Cl、Br、NO2およびNH2から選ばれる1〜3個の基で置換されるもの、
aは0または1を表し、
m、pおよびqは各々独立して0〜4の整数を表し、
ただしR7部分は12個を超える炭素原子を含有せず、-PO(OH)2、-CF2PO(OH)2、-COOH、OHまたはNH2から選ばれる1以上の官能基を有し、あるいはそれらの薬学的に許容可能な塩である。
【0023】
上記構造は代表的な互変異体を示しており、本発明は化1の化合物のあらゆる互変異体、たとえばエノール化合物が示される場合のケト化合物およびその逆を包含する。
【0024】
具体的な立体異性体が特定的に示されない限り(たとえば、構造式中の立体中心における太字または点線の結合により、構造式中にEまたはZ配置を有する二重結合が示されることにより、または立体科学を示す術後を用いることにより)、すべての立体異性体は純粋化合物としてであれその混合物としてであれ、本発明の範囲内に含まれる。そうでないように示されない限り、個々のエナンチオマー、ジアステレオマー、幾何異性体、およびこれらの組み合わせおよび混合物はすべて本発明に包含される。多形結晶体、溶媒和物および水和物もまた本発明に包含される。
【0025】
さらに別の観点において、本発明は化1で示される39-デスメトキシラパマイシン誘導体またはその薬学的に許容可能な塩を薬剤として用いる用途を提供する。
【0026】
定義
冠詞“a”または“an”はここでは一または一以上(すなわち少なくとも一)の文法上の対象物を指すために用いられる。一例として、“an analogue”は一つの相同物または一以上の相同物を意味する。
【0027】
ここで用いる「相同物」の語は、ある化合物と構造的に近似しているが組成において(たとえば1個の原子が他に置換され、あるいは特定の1個の官能基が存在または不存在)若干異なる化合物を意味する。
【0028】
特に、「39-デスメトキシラパマイシン相同物」の語はWO2004/007709の方法によって製造される、および/または化1で示される39−デスメトキシラパマイシン化合物である。この化合物はまた「親化合物」として参照され、これらの語を本出願において互換的に使用する。本出願において「39−デスメトキシラパマイシン相同物」の語は39−デスメトキシラパマイシンそれ自体に対する参照を含む。
【0029】
ここで用いる「誘導体」の語は、半合成有機化学により親化合物から変性された化合物を指す。
【0030】
特に、「39−デスメトキシラパマイシン誘導体」の語は、化1で示される39−デスメトキシラパマイシン誘導体、またはその薬学的に許容され得る塩を意味し、39−デスメトキシラパマイシン相同物の半合成変性によって製造される。該化合物はまた「本発明化合物」または「ラパマイシンの39−デスメトキシ誘導体」として参照され、本出願においてこれらの語を互換的に使用する。
【0031】
ここで用いる「自己免疫疾患」の語は、限定的ではないが、全身性エリテマトーデス(SLE)、関節リウマチ、重症筋無力症および多発性硬化症を含む。
【0032】
ここで用いる「炎症疾患」の語は、限定的ではないが、乾癬、皮膚炎、湿疹、脂漏、炎症性腸疾患(限定的ではないが潰瘍性大腸炎およびクローン病を含む)、肺炎(限定できではないが喘息、慢性閉塞性肺疾患、肺気腫、急性呼吸窮迫症候群および気管支炎を含む)、リウマチ性関節炎および眼ブドウ膜炎を含む。
【0033】
ここで用いる「ガン」の語は、限定的ではないが、皮膚や限定的ではないがたとえば胸腺、前立腺、肺、腎臓、膵臓、胃または腸などの体器官中の悪性の細胞成長を意味する。ガンは隣接組織に侵入して、離れた器官、たとえば非限定的例示として骨や肝臓、肺または脳などに拡がっていく(転移)傾向がある。ここで用いる「ガン」の語は、非限定的な例示としてメラノーマ、リンフォーマ、白血病、繊維肉腫、横紋筋肉腫および悪性肥満細胞腫などの転移性腫瘍細胞タイプと、非限定的な例示として結腸大腸ガン、前立腺ガン、小細胞肺ガンおよび非小細胞肺ガン、乳ガン、膵臓ガン、膀胱ガン、腎臓ガン、胃ガン、グリオブラストーマ、肝ガン、卵巣ガンなどの細胞ガンタイプの両方を含む。
【0034】
ここで用いる「B細胞系悪性腫瘍」の語は、慢性リンパ性白血病(CLL)、多発性骨髄腫、非ホジキンリンパ腫(NHL)などの疾患群を含む。これは血液または造血器官の新生物疾患である。これは骨髄および免疫システムの機能不全を生じさせ、宿主を感染や出血しやすいものにする。
【0035】
ここで用いる「血液疾患」の語は、限定的ではないが、超増殖性血管障害(レステノシスおよび血管閉塞など)、アテローム性動脈硬化症、心血管疾患、脳血管障害および末梢性血液疾患(冠状動脈疾患、動脈硬化症、アテローム性動脈硬化症、非アテローム性動脈硬化症または血管壁損傷など)を含む。
【0036】
ここで用いる「神経再生」の語は神経細胞成長刺激を意味し、神経突起伸長および神経細胞の機能的回復を含む。神経再生がその治療に顕著に有効であろうとされる疾患および疾病は、限定的ではないが、アルツハイマー病、パーキンソン病、ハンチントン舞踏、筋萎縮性側索硬化症、三叉神経痛、舌咽神経痛、ベル麻痺、筋ジストロフィー、脳卒中、進行性筋萎縮症、進行性球脊髄性筋萎縮症、変形性頸椎症、ギラン・バレー症候群、痴呆症、末梢性神経障害および末梢神経損傷を含む、これらは身体的損傷(脊髄損傷またはトラウマ、座骨神経や顔面神経の外傷や損傷など)によって生ずるか、あるいは病状(糖尿病など)によって生ずるかを問わない。
【0037】
ここで用いる「線維症」の語は、細胞外マトリックスの過剰産生に関連する疾患を意味し、(限定的ではないが)サルコイドーシス、ケロイド、糸球体腎炎、末期腎臓疾患、肝繊維化症(非限定的例示として肝硬変、アルコール性肝障害および脂肪肝を含む)、慢性腎障害、手術創癒着、動脈硬化症、心臓繊維化症、肺線維症(非限定的例示として突発性肺線維症および突発性繊維性肺胞炎を含む)、黄斑変性症、新生児および糖尿病網膜症、化学療法または放射線誘発線維症を含む。
【0038】
ここで用いる「移植片対宿主拒絶病」の語は、同種幹細胞/骨髄移植後に見られる合併症を意味する。それは、ドナーから与えられた感染と闘う細胞が患者の体を異なるまたは異質であると認識したときに発症する。これらの感染攻撃細胞は、感染を攻撃するかの如く、患者の体内組織を攻撃する。移植片対宿主拒絶病は、それが移植後最初の100日以内に発症すれば急性とされ、移植後100日経過後に発症すれば慢性とされる。これに関連する主な組織は肝臓、消化管および皮膚である。慢性移植片対宿主拒絶病は幹細胞/骨髄移植患者の約10〜40%に発生する。
【0039】
ここで用いる「バイオアベイラビィリティ」の語は、投与後の薬剤または他の物質が生物学的活性領域で吸収されまたは有効に働く程度ないし割合を意味する。この性質は、化合物の可溶性、消化管への吸収率、タンパク質結合および代謝の程度などの数多くの要因に依存する。ここに当業界において当業者に良く知られている様々なバイオアベイラビィリティ試験を記述する(さらにTrepanierら、1998年、Gallant−Haidnerら、2000年も参照されたい)。
【0040】
ここで用いる「水溶性」の語は、水性媒体、たとえばpH7.4におけるリン酸緩衝サリン(PBS)中の溶解性を意味する。
【0041】
化1の化合物のような本発明化合物の薬学的に許容可能な塩は、薬学的に許容可能な無機または有機の酸または基から形成される慣習的な塩を含み、さらに第四アンモニウム酸添加塩をも含む。好適な酸塩についてより詳しく例示すれば、塩化水素、臭化水素、硫酸、リン酸、硝酸、過塩素酸、フマル酸、酢酸、プロピオン酸、コハク酸、グリコール酸、蟻酸、乳酸、マレイン酸、酒石酸、クエン酸、パルモイン酸、マロン酸、ヒドロキシマレイン酸、フェニル酢酸、グルタミン酸、安息香酸、サリチル酸、フマル酸、トルエンスルホン酸、メタンスルホン酸、ナフタリン−2−スルホン酸、ベンゼンスルホン酸、ヒドロキシナフタリン酸、よう化水素酸、リンゴ酸、ステロイン酸、タンニン酸などである。シュウ酸などの他の酸はそれ自身は薬学的に許容可能なものではないが、塩として調製することにより本発明化合物およびその薬学的許容可能塩を得るための中間生成物として有用である。より詳しく好適な基酸を例示すれば、ナトリウム、リチウム、カリウム、マグネシウム、アルミニウム、カルシウム、亜鉛、N−N’−ジベンジルエチレンジアミン、クロロプロカイン、コリン、ジエタノールアミン、エチレンジアミン、N−メチルグルカミンおよびプロカインの塩である。以降において本発明による化合物を参照するときは化1の化合物とその薬学的許容可能な塩の両方を含む。
【0042】
アルキル、アルケニルおよびアルキニル基は直鎖または分枝である。
【0043】
C1−C4アルキル基の例はメチル、エチル、n−プロピル、i−プロピルおよびn−ブチルを含む。
【0044】
C2−C4アルケニル基の例はエテニルおよび2−プロペニルを含む。
【0045】
C2−4アルキニル基の例はエチニルを含む。
【0046】
C3−C6シクロアルキル基は3〜6個の炭素原子を含むシクロアルキル環を意味し、それらは任意に分枝していても良い。例示すればシクロプロピル、シクロブチル、メチル−シクロブチル、シクロペンチルおよびシクロヘキシルである。
【0047】
N、OおよびSから選ばれる1以上のヘテロ原子を含む3〜6員ヘテロアルキル環は、1個または2個のヘテロ原子を含み、特に1個のヘテロ原子を含む。例示すればフラン、ピラン、オキセタン、オキシラン、ピペリジン、ピロリジン、アゼチジン、アジリジン、チイラン、チエタン、チオフェン、チオピランおよびモルフォリンを含む。
【0048】
3〜6員ヘテロアルキル環についての任意置換の例は、−OH、−CH2OH、NH2、CH2NH2およびCOOHを含む。特に3〜6員ヘテロアルキル環は置換されていないか、あるいは1個または2個、たとえば1個の置換基で置換されている。
【0049】
発明の記述
本発明は上述の39−デスメトキシラパマイシン誘導体、該化合物の調製方法、その中間産生物および該化合物の医療的用途を提供する。
【0050】
好ましくはR7は7個以下、特に5個以下の炭素原子を含む。
【0051】
R7は好ましくは−PO(OH)2、−OH、−COOHおよび−NH2から、より好ましくは−OH、−COOHまたは−NH2から、特に−OHおよび−COOHから、とりわけOHから選ばれる1以上の官能基を含む。好ましくはR7は2以上の置換基、たとえば2つのOH基を含む。
【0052】
好ましくはXはCH2を表す。
【0053】
好ましくはaは0を表す。
【0054】
好ましくはpは0または1を表す。
【0055】
好ましくはmは0または1を表す。
【0056】
好ましくはqは0、1または2を表す。
【0057】
好ましくはR11はHを表す。好ましくはR12はHを表す。
【0058】
好ましくはR13はHまたはOHを表す。
【0059】
pが1を表すとき、好ましくはR10はMe、OHまたはCH2OHを表す。
【0060】
pが1を表すとき、好ましくはR11はMe、HまたはCH2OHを表す。
【0061】
mとpが共に0を表すとき、好ましくはR12とR13は共にHを表し、R14は−(CR8R9)q−OHを表し、ここでqは0または1を表し、R8とR9は共にHを表す。
【0062】
pが1を表しmが0を表すとき、好ましくはR10とR11は共にHを表し、R12はHを表し、R13はH、OHまたはNH2を表し、R14は−(CR8R9)q−OHを表し、ここでqは0または1を表し、R8とR9は共にHを表す。
【0063】
R6が−POR15R16を表すとき、好ましくはR15とR16は共にCH3を表すかまたは共にCH2CH3を表す。
【0064】
好ましくはR6は、ヒドロキシ酢酸、3−ヒドロキシ−2,2−ジメチルプロピオン酸、2,3−ジヒドロキシプロピオン酸、3−ヒドロキシ−2−ヒドロキシメチルプロピオン酸または2,2−ビス(ヒドロキシメチル)プロピオン酸によるエステル形成由来の残留物を表す。
【0065】
化合物の一例セットにおいてR6はC(O) R7を表す。
【0066】
好ましくはR7は、ヒドロキシ酢酸、3−ヒドロキシ−2,2−ジメチルプロピオン酸、2,3−ジヒドロキシプロピオン酸、3−ヒドロキシ−2−ジヒドロキシメチルプロピオン酸および2,2−ビス(ヒドロキシメチル)プロピオン酸からなる群から選ばれる酸、特に2,2−ビス(ヒドロキシメチル)プロピオン酸で大環状アルコールを縮合することにより形成される部分である。
【0067】
R15が化2を表すとき、この部分の例は、(1)グルコース(すなわちR18がCH2OHを表し、各R16およびR17がOHを表す)、たとえばD−グルコース、(2)グルコサミン(すなわちR18がCH2OHを表し、各R16がOHを表し、R17がNH2を表す)、たとえばD−グルコサミン、(3)グルクロン酸(すなわちR18がCOOHを表し、各R16およびR17がOHを表す)、たとえばD−グルクロン酸、および(4)アラビノース(すなわちR18がHを表し、各R16およびR17がOHを表す)、たとえばD−アラビノースでアセタールを形成することにより形成される部分を含む。
【化2】
【0068】
R15が化3を示すとき、この部分の例は、フルクトース(すなわちR16が各々OHを表す)、たとえばD−フルクトースでアセタールを形成することにより形成される部分を含む。
【化3】
【0069】
R15が化4を示すとき、この部分の例は、グルクロン酸(すなわち各R16がOHを表す)、たとえばD−グルクロン酸でエステルを形成することにより形成される部分を含む。
【化4】
【0070】
概して言えば、本発明化合物は化5の39−デスメトキシラパマイシン相同物の半合成誘導により産生される。
【化5】
【0071】
すなわち、化1の化合物またはその薬学的に許容可能な塩の調製方法は、
(a)化5の39−デスメトキシラパマイシン相同物(RAはHまたは(CH2)2−OH)またはその保護された誘導体を、HO−R6またはR6の活性化された誘導体と反応させる工程、
(b)化1の化合物またはその塩を他の化1の化合物またはその薬学的に許容され得る塩に形質転換する工程、または、
(c)保護された化1の化合物を脱保護する工程を含む。
【0072】
ここで用いる「活性化された誘導体」の語は、非限定的例示として、エステルの場合であればカルボン酸、ハロゲン化アシル、混合酸無水物、対称酸無水物またはカルボン酸エステル、エーテルの場合であればハロゲン化アルキル、アルキルメシレート、アルキルトリフレート、アルキルトシレートまたは他の適当な活性化アルキル誘導体、リン酸エステルまたはホスホン酸エステルの場合であればクロロホスフェート、ジアルキルシアノホスフェート、ジアルキルジアルキルホスホラミデートまたはクロロホスファイト、グリコシル基由来アセタールの場合であればハロゲン化グリコシル、チオグリコシド、1−O−アシルグリコシド、オルトエステル、1−Oまたは1−S炭酸塩、トリクロロイミデート、4−ペンテニルグリコシド、グリコシルリン酸エステル、1−O−スルホニルまたは1−O−シリル化グリコシドなどのグリコシルドナーを用いることを意味する。
【0073】
工程(a)において、化5の39−デスメトキシラパマイシン相同物はWO2004/007709に記述され、さらには後述実施例に述べるようにして調製することができる。
【0074】
ここに記述される具体的方法および参照に加えて、当業者は、非限定的例示としてVogelの実際有機化学の教本(Furnissら、1989年)やMarchの高等有機化学(SmithおよびMarch、2001年)にょうな合成法のための標準的教本参照を顧慮することができる。
【0075】
さらに、当業者に知られている多くの標準的ヒドロキシ保護ストラテジーの一つによってヒドロキシル基を保護することができる。ヒドロキシル基は、限定的ではないがたとえば置換アルキルエーテル、置換ベンゼンエーテル、シリルエーテルなどのエーテルを生成させることによって保護することができる。好適には、シラン(限定的ではないが塩化シリル、シリルトリフラートを含む)の活性形態を適当な塩基の存在の下で39−デスメトキシラパマイシンと反応させることにより、限定的ではないがたとえばトリメチルシリル、トリエチルシリル、t−ブチルジメチルシリル、t−ブチルジフェニルシリルなどのシリルエーテルを生成させる。次いで酸加水分解またはフッ素酸アシスト開裂のいずれかにより保護基を除去することができる。アセトン誘導体の縮合に基づいて、1,2−ジオールがアセトニドとして保護され得る。これは酸触媒によって除去され得る。
【0076】
化5の39−デスメトキシラパマイシン相同物はさらなる半合成(すなわち工程(a))のためのテンプレートとしても用いることができる。C−40におけるペンダントヒドロキシル基は、当業者に知られている多くの合成的形質転換を介して、たとえばアシル化、アルキル化、グリコシル化またはリン酸化などによって感応化され得る。
【0077】
工程(a)において、R6が−C(O)R7またはY−R15であって、ここでR15が化4を表し、Yが結合であるとき、ヒドロキシエステルの形成ないしO−アシル化は、化5の化合物のヒドロキシルが好ましくは活性化状態である対応カルボキシル酸、たとえば化6または化7の化合物と反応することによって、あるいは化8の化合物と反応することによって、メディエートされ得る。
【化4】
【化6】
【化7】
【化8】
【0078】
ここで、Wはカルボキシル酸を求核攻撃に対して活性化する基である。カルボキシル酸は非限定的例示としてハロゲン化アキル(たとえばW=Cl)、混合酸無水物(すなわちW=OC(O)R’)、対称酸無水物(W=OC(O)R7)またはカルボキシルエステル(すなわちW=OR’)の形成により活性化され得る。
【0079】
化6、化7または化8の化合物はそれらの入手可能なカルボキシル酸から当業者に公知の標準的方法を用いて調製することができ、一つの特定の観点において、R7が−(CR8R9)m(CR10R11)pCR12R13R14を表す化6の化合物は米国特許5,362,718、米国特許「5,665,772または欧州特許0663916に記述されるような方法を用いて調製することができる。
【0080】
好ましくは39−デスメトキシラパマイシン相同物を有機媒体中で塩基の存在下で酸塩化物または混合酸無水物と反応させる。塩基の非限定的例示は、ピリジン、4,4−ジメチルアミノピリジン(DMAP)、2,6−ルチジン、2,6−ジ−タート−ブチルピリジン、トリエチルアミン、ジイソプロピルエチルアミン、他のトリアルキルアミン、1,8−ジアザビシクロ[5.4.0]ウンデク−7―エン(DBU)または1,5−ジアザビシクロ[4.3.0]ノン−5―エン(DBN)を含む。
【0081】
工程(a)において、R6が−C(O)R7またはY−R15であって、ここでR15が化4を表し、Yが−C(O)O−または−(CH2)2−OC(O)O−を表すとき、該ヒドロキシエステルの形成は、化5の化合物または40−O−(ヒドロキシエチル)−化5化合物のヒドロキシル基が化9の化合物のような活性化炭酸エステルを形成する試薬と反応させることを要求する。
【化4】
【化9】
【0082】
化9においてTは結合または−O(CH2)2−であり、R24はアルキルまたはアリル基、好ましくはアリル基、特にパラ−ニトロフェニル基である。
【0083】
化9の化合物は次いで化6、化7または化8の化合物と反応して、炭酸塩リンカー(carbonate linker)を介して、R6が40−ヒドロキシル基または40−O−(ヒドロキシルエチル)基に付着した化合物を生成する(WO2004/101583)。
【0084】
同様に、39−デスメトキシラパマイシン相同物を適当に選択した活性化されたアルキル誘導体と反応させることにより、39−デスメトキシラパマイシン相同物をC−40において異なるヒドロキシエステルで誘導することができ、これによって40−O−アルキル−39−デスメトキシラパマイシン誘導体を生成する。活性化されたアルキル基は、非限定的例示としてハロゲン化アキル(RCl、Rl、RBr)、アルキルメシレート(ROS(O)2CH3)、アルキルトリフレート(ROS(O)2CF3)、アルキルトシレート(ROS(O)2PhMe)の形成を含む多くの方法の一つにより活性化されたアルキル基を指す。活性化されたアルキル基は次いで有機溶媒中で適当な塩基の存在下において39−デスメトキシラパマイシン相同物と反応する。反応条件を最適化する標準的方法が当業者によって採用されることができ、これにより他の反応位置におけるアルキル化を防止する。
【0085】
同様に、39−デスメトキシラパマイシン相同物をリン酸化することができ、これによりリン酸エステルの脱保護の後に40−O−ホスフォ−39−デスメトキシラパマイシン誘導体または40−O−ジアルキルホスフォ−39−デスメトキシラパマイシン誘導体を産生する。これらの塩は当業者に知られている方法で産生することができる。リン酸エステルは、3価ホスフェート(亜リン酸)が5価ホスフェートに酸化(好ましくは非限定的例示としてmCPBAのような過酸活性によって)されているO−ホスファイトを介して、直接または間接に生成し得る。直接リン酸化方法は、非限定的例示として、好ましくは有機溶媒中でDMPA存在下において39−デスメトキシラパマイシン相同物を、保護されたクロロホスフェート(たとえば(BnO)2P(O)Cl、(アルキルO)2P(O)Cl)と反応させ、または、トリエチルアミンなどの塩基の存在下において39−デスメトキシラパマイシン相同物をオキシ塩化リン(POCl3)と反応させ、次いで得られたO−ジクロロホスフェート(すなわちROP(O)Cl2)の酸加水分解またはジアルキルシアノホスフェートへの結合(WO01/81355)を行うことを含む。ジアルキルまたはジアリルクロロホスフェートは、塩基の存在下でジアルキルまたはジアリルホスファイト(すなわち(RO)2P(O)H)を四塩化炭素と反応させることにより自然位で(in situ)生成させることができる。O−ホスフェートの生成(O−ホスフェートへの酸化のために)方法は、非限定的例示として、塩基(好ましくはテトラゾル)の存在下で39−デスメトキシラパマイシン相同物ジアルキル−ジアルキルホスフォラミデート(dialkylphosphoramidate)に結合させること、塩基の存在下でクロロホスファイトを用いて結合すること(Evansら、1992年)を含む。保護基の選択は重要であり、ホスフェートのエチルおよびメチルエステルは酸性や塩基条件では容易にはリン酸化しない。好ましい保護基は、非限定的例示として、ベンジルエステル(ヨウ化ナトリウム/クロロトリメチルシラン促進加水分解を介して開裂、WO01/81355)または2−シアノエチルエステル(緩やかな塩基触媒開裂を介して開裂)を含む。同様に、40−O−ジアルキルホスフォノ−39−デスメトキシラパマイシン誘導体は39−デスメトキシラパマイシン相同物を適当な活性化(上述)ジアルキルホスフォネートまたはジアルキルホスファイトと反応させることにより生成することができる。
【0086】
工程(a)において、R15が化2または化3の部分を示すとき、グリコシド結合の形成すなわちO−グリコシル化は、ヒドロキシル基が好ましくは活性化形態にある対応グリコシルドナー、たとえば化10の化合物または化11の化合物と反応することによってメディエートされ得る(ToshimaおよびTatsuta(1993年)参照)。
【化10】
【化11】
【0087】
非限定的例示として、ハロゲン化グリコシル(Z=F、Cl、Br)、チオグリコシド(Z=SMえ、Set、SPh、SPy、SCN)、1−O−アシルグリコシド(Z=OC(O)R)、オルトエステル(Z=OC(Me)(R)(O−化10/化11のC2)、1−Oまたは1−S炭酸塩(Z=OC(S)SMe、Z=OC(O)イミダゾール、Z=OC(S)イミダゾール、Z=SC(S)OEt)、トリクロロイミデート(Z=OC(=NH)CCl3)、4−ペンテニルグリコシド(Z=OCH2CH2CH2CH=CH2)、リン酸エステル(たとえばZ=OP(O)(OPh)2)、1−O−スルホニル(Z=トシル)、または1−O−シリル化グリコシド(Z=OTMSまたはOTBS)を含むグリコシルドナーの使用に際し、39−デスメトキシラパマイシン相同物を有機溶媒中で、好ましくは活性剤(たとえばルイス酸または重金属塩など、ToshimaおよびTatsuta、1993年、参照)の存在下で、グリコシル化させることができる。用いる特定のグリコシルドナーおよび反応条件によって、αまたはβグリコシドのいずれが生成されるかが決定される。アシル化についての以前と同様、親化合物に存在するあらゆるヒドロキシル基は、グリコシルドナーの1当量を用いることが40−O−アシル化をもたらすように保護され遮蔽され得る。グリコシルドナーに残るヒドロキシル基はたとえばO−酢酸エステル、O−ベンゾエート、1,2−アセトナイドなどで保護され、したがってさらに脱保護が必要となる。さらに、グリカールのような2−デオキシグリコシルを用いて(還元工程がまた要求される)、2’−デオキシ−39−デスメトキシラパマイシングリコシドを生成させることができ、2,6−アンヒドロ−2−チオ糖のような2,6−ジデオキシグリコシルドナーを用いて2’−6’−ジデオキシ−39−デスメトキシラパマイシングリコシドを生成させることができる。
【0088】
工程(b)において、塩の生成および交換は当業者に知られている従来法により行うことができる。化1の化合物の分子変換は既知の方法で行うことができ、たとえば本出願のどこかに記述されているように酸化/還元によりヒドロキシおよびケト基の変換を行うことができる。R6が−PO(OH)2を表す化1の化合物は、R6がOHを表す化1の対応化合物をリン酸化することで調製可能である。適当な条件は本出願のどこかで与えられる。
【0089】
工程(a)および(c)において、保護基の例およびその除去方法をT W Greene「Protective Groups in Organic Synthesis」(J WileyおよびSons、1991年)に見ることができる。適当なヒドロキシル保護基は、加水分解で除去することができるものとしてアルキル(たとえばメチル)、アセタール(たとえばアセトニド)およびアシル(たとえばアセチルまたはベンゾイル)、触媒加水分解で除去することができるものとしてアリルアルキル(たとえばベンジル)、または酸加水分解ないしフッ化イオンアシスト開裂により除去することができるものとしてシリルエーテルを含む。
【0090】
工程(a)に加えて、R6がR7を示す化1の39−デスメトキシラパマイシン相同物はリパーゼ触媒トランスエステル化によって合成することができる。非限定的例示として、化5の39−デスメトキシラパマイシン相同物をリパーゼPS−C「Amano」IIの存在下で化12のエステルと反応させることができる。この時の反応条件はGuら(2005年)および本実施例に記述されている。この方法論はビニルエステルの使用に限定されず、トランスエステル化を他のリパーゼやエステラーゼで触媒作用を発揮させても良い。
【化12】
【0091】
本発明の他の化合物はそれ自体既知である方法または上述の方法と相同的な方法によって調製することができる。
【0092】
新規39−デスメトキシラパマイシン誘導体は、免疫抑制剤、抗真菌剤、抗ガン剤、抗炎症剤、神経再生剤、または臓器移植拒絶反応、移植片対宿主拒絶病、自己免疫疾患、炎症疾患、血管疾患および/または線維症の治療薬として有効な化合物を製剤するために直接的に有用であり、また、さらに別の半合成または生物学的変換のためのテンプレートとして有用である。ラパマイシンおよびその相同物の半合成的誘導法は当業界において良く知られており、非限定的例示として、たとえば米国特許5,665,772、米国特許5,362,718、WO96/41807、米国特許5,728,710、米国特許5,378,836、米国特許5,138,051、米国特許5,665,772、米国特許5,955,457、WO98/04279、米国特許6,015,815および米国特許5,432,183に記述される改良を含む。
【0093】
中間産生物(たとえば化5の化合物)の上記構造は互変異性化可能であり、代表的な互変異性体が示されている場合、すべての互変異性体、たとえばエノール化合物が示されている場合のケト化合物およびその逆も包含することが意図されていることを理解すべきである。
【0094】
さらに別の観点において本発明は39−デスメトキシラパマイシン誘導体の医療用途を提供する。さらに別の観点において本発明は39−デスメトキシラパマイシン誘導体を免疫抑制の誘発または維持、神経再生の刺激、またはガン、B細胞系悪性腫瘍、真菌感染、臓器移植拒絶反応、移植片対宿主拒絶病、自己免疫疾患、炎症疾患、血管疾患および線維症の治療のための薬剤、または創傷治癒制御に用いる薬剤を製剤することの用途を提供する。
【0095】
多剤耐性(MDR)はガンおよびB細胞系悪性腫瘍の治療において重要な問題である。それは多くのガンにおいて薬剤耐性の開発を遅れさせる主要な原因となっている(Persidis A、1999年)。MDRはアデノシン三リン酸結合カセットトランスポーター(ABCトランスポーター)の増大レベル、特にP糖タンパク質(P−gp)のためにエンコードするMDR1遺伝子またはMRP1をエンコードするMRP1遺伝子の発現の増加に関連する。MDR1遺伝子発現レベルは異なるガン由来細胞ラインに亘って大きく変化し、幾つかの細胞ラインでは検出不能であるが、他の細胞ラインでは標準的対照と比較すると最大10ないし100倍の発現増加を示すことがある。
【0096】
したがって、本発明のさらなる態様は、39−デスメトキシラパマイシン誘導体をMDRガンまたはB細胞系悪性腫瘍の治療に用いる用途を提供する。一つの特定の態様において、本発明は39−デスメトキシラパマイシン誘導体をP−gp発現ガンまたはB細胞系悪性腫瘍の治療に用いる用途を提供する。さらにより好適な実施形態において、本発明は39−デスメトキシラパマイシン誘導体を高P−gp発現ガンまたはB細胞系悪性腫瘍の治療に用いる用途を提供する。特に、高P−gp発現ガンまたはB細胞系悪性腫瘍は比較対照に比べて2倍、5倍、10倍、20倍、25倍、50倍または100倍増大した発現を有する。適当な対照はP−gpを発現しないか、P−gp発現レベルが低いか、あるいはMDR機能が低い細胞であり、当業者はそのような細胞ラインを知っているかあるいは認知することができる。適当な細胞ラインを非限定的に例示すれば、MDA435/LCC6、SBC−3/CDDP、MCF7、NCI−H23、NCI−H522、A549/ATCC、EKVX、NCI−H226、NCI−H322M、NCI−H460、HOP−18、HOP−92、LXFL529、DMS114、DMS273、HT29、HCC−2998、HCT−116、COLO205、KM12、KM20L2、MDA−MB−231/ATCC、MDA−MB−435、MDA−N、BT−549、T−47D、OVCAR−3、OVCAR−4、OVCAR−5、OVCAR−8、IGROV1、SK−OV−3、K−562、MOLT−4、HL−60(TB)、RPMI−8226、SR、SN12C、RXF−631、786−0、TK−10、LOX IMVI、MALME−3M、SK−MEL−2、SK−MEL−5、SK−MEL−28、M14、UACC−62、UACC−257、PC−3、DU−145、SNB−19、SNB−75、SNB−78、U251、SF−268、SF−539、XF498。
【0097】
別の観点において本発明は39−デスメトキシラパマイシン誘導体をMDRガンまたはB細胞系悪性腫瘍の治療用薬剤の調製に用いる用途を提供する。特定の態様において、本発明は39−デスメトキシラパマイシン誘導体をP−gp発現ガンまたはB細胞系悪性腫瘍の治療用薬剤の調製に用いる用途を提供する。さらにより好適な実施形態において、本発明は39−デスメトキシラパマイシン誘導体を高P−gp発現ガンまたはB細胞系悪性腫瘍の治療用薬剤の調製に用いる用途を提供する。特に、高P−gp発現ガンまたはB細胞系悪性腫瘍は比較対照に比べて2倍、5倍、10倍、20倍、25倍、50倍または100倍増大した発現を有する。適当な対照は上記のものである。
【0098】
サンプル中のP−gp発現レベルを決定する方法は後述する。
【0099】
したがって、別の観点において本発明は、39−デスメトキシラパマイシン誘導体の治療的有効量を投与することからなるP−gp発現ガンまたはB細胞系悪性腫瘍の治療法を提供する。特定のガン種におけるP糖タンパク質(P−gp)発現レベルは、限定的ではないがたとえばリアルタイムRT−PCT(Szakacsら、2004年;Steinら2002年;Langmannら、2003年;Alvarezら、1995年;Boydら、1995年)を含む技術を用いて、免疫組織化学によって(Steinら、2002年)、またはマイクロアレイを用いて(Leeら、2003年)、当業者により決定することができる。これらの方法は単に例示として挙げたにすぎず、他の適当な方法も当業者に与えられるであろう。
【0100】
当業者はルーチン的実験によって当該化合物の真菌成長抑制能力を決定することができるであろう(たとえばBaker,Hら、1978年;NCCLS Refernce method for broth dilution antifungal susceptibility testing for yeasts:Approved standard M27−A、17(9)、1997年)。さらに、当業者はルーチン的実験によって当該化合物の腫瘍細胞成長抑制能力を決定することができるであろう(Dudkin,Lら、2001年;Yuら、2001年)。さらに別の観点において本発明化合物は免疫抑制を誘発するのに有用であり、この分野における化合物の効能を決定するためのアッセイは当業者に良く知られている。非限定的例示として、免疫抑制活性−Warner,L.M.ら、1992年;Kahanら(1991年)およびKahan&Camardo、2001年);同種移植−Fishbein,T.M.ら、2002年;Kirchnerら、2000年;自己免疫/炎症性/喘息−Calson,R.P.ら、1993年;Powell,N.ら、2001年;糖尿病I−Rabinovitch,Aら、2002年;乾癬−Reitamo,S.ら、2001年;慢性関節リウマチ−Foey,A.ら、2002年;線維症−Zhu,J.ら、1999年;Jain,S.ら、2001年;Gregoryら、1993年。
【0101】
本発明の39−デスメトキシラパマイシン誘導体の免疫抑制誘発能力はこの目的のための標準的試験において実証することができる。さらに別の観点において本発明の39−デスメトキシラパマイシン誘導体は抗繊維化、神経再生および抗血管形成のメカニズムに関連して有用であり、当業者はルーチン的実験によって当該化合物の血管形成阻止能力を決定することができるであろう(たとえばGuba,M.ら、2002年)。当業者はルーチン的実験によって、たとえば薬剤溶出ステント中で、当該化合物を血管超増殖の治療に用いることの有用性を決定することができるであろう(たとえばMorice,M.C.ら、2002年)。加えて、当業者はルーチン的実験によって当該化合物の神経再生能力を決定することができるであろう(たとえばMyckatyn,T.M.ら、2002年、Steinerら、1997年)。
【0102】
本発明はまた、39−デスメトキシラパマイシン誘導体を薬学的に許容可能なキャリアと共に用いてなる薬剤化合物を提供する。
【0103】
臨床試験で用いられているCCI−779およびRAD001などのラパマイシンおよび関連化合物は薬理プロファイルが不十分であり、水溶性が不十分であり、バイオアベイラビリティが不十分である。本発明は安定性および/または細胞膜透過性などの物性が向上された39−デスメトキシラパマイシン誘導体を提供する。当業者は標準的方法を用いて本発明の与えられた化合物の可溶性を容易に決定することができるであろう。代表的な方法が本実施例に示されている。
【0104】
さらに、当業者であれば、本発明化合物の薬物動態プロファイルおよびバイオアベイラビリティを、当業者に知られている生体内および生体外の方法、たとえば非限定的な例示として、後述や実施例記載の方法を用いて決定することができるであろうし、他の代替的アッセイも、非限定的例示としてGallant−Haidnerら、2000年およびTrepanierら、1998年に記述されたものをここで参照して、これらに記述されているように当業者に良く知られている。化合物のバイオアベイラビリティは多くの要因(水溶性、消化管吸収率、タンパク質結合および代謝の程度など)によって決定され、これらは後述するような生体外試験によって決定することができる。当業者には理解されていることであるが、これら要因の一以上の向上は化合物のバイオアベイラビリティを向上させることに繋がる。あるいは、化合物のバイオアベイラビリティは、下記に詳述するような生体内方法を用いて測定することも可能である。
【0105】
Caco−2透過アッセイ
In Vitro Technologies Inc.(IVT Inc.、Baltimore、Maryland、米国)によって提供されるような、24個のウェルCorning Costar Transwellフォーマット内の合流型Caco−2細胞(Li,A.P.、1992年;Grass,G.M.ら、1992年;Volpe,D.A.ら、2001年)を用いることができる。尖室(apical chamber)は0.15mLのハンクス平衡緩衝溶液(HBBS)pH7.4、1%DMSO、0.1mMルシファー・イエローを含む。底室(basal chamber)は0.6mLのHBBS pH7.4、1%DMSOを含む。次いで対照および試験体を湿潤インキュベータ内で37℃でインキュベートし、130rpmで1時間シェークした。ルシファー・イエローは細胞間(密着結合間)経路を介してのみ透過し、ルシファー・イエローについての高い見かけ透過係数(Papp)はアッセイ中の細胞損傷を示し、全てのそのようなウェルは拒絶された。プロプラノロール(トランスポーター効果は知られていないが良好な受動的透過性を持つ)およびアセブタロール(P糖タンパク質による活性的エフラックスにより減衰されて受動的透過性が低い)を対照化合物として用いる。化合物を尖室または底室に適用する(0.01mMで)ことにより一方向および二方向フォーマットで試験することができる。尖室または底室中の化合物をHPLC−MSで分析する。結果を見かけ透過係数Papp(nm/s)およびフラックス率(「A対B」対「B対A」)として示す。
【0106】
Papp(nm/s)=[ボリュームアクセプター/(面積×ドナー)]×(△アクセプター/△時間)
ボリュームアクセプター:0.6mL(A>B)および0.15mL(B>A)
単分子層の面積:0.33cm2
△時間:60分
フラックス率についての正の値は細胞の尖端面からの活性的エフラックスを示す。
【0107】
ヒト肝ミクロソーム(HLM)安定性アッセイ
肝ホモジェネートが、CYP450s(たとえばCYP2C8、CYP2D6、CYP1A、CYP3A4、CYP2E1)エステラーゼ、アミダーゼおよびモノオキシゼナーゼを含む対フェースI(酸化的)酵素群固有脆弱性化合物の測定を与える。
【0108】
試験化合物をヒト肝ホモジェネートに露出し、LC−MSでその消失を観測することにより半減期(T1/2)を決定した。0.001mMで化合物を40分間、37℃で、0.1M Tris−HCl、pH7.4でインキュベートした。0.25mg/mLタンパク質、NADPHの飽和レベルのヒト肝ミクロソーム細胞下分画を補因子として用いた。時間インターバルで試験サンプルにアセトニトリルを添加してタンパク質を沈殿させ、代謝を終了させた。サンプルを遠心処理し、HPLC−MSにより親化合物を分析した。
【0109】
生体内バイオアベイラビリティアッセイ
生体内アッセイは化合物のバイオアベイラビリティを測定するためにも使用することができる(たとえばCroweら、1999年を参照)。一般的に言えば、化合物を腹腔内(ip)または静脈(iv)および経口(po)の両方で試験動物に投与し、血液サンプルを所定インターバルで取り出して薬剤のプラズマ濃度の経時的変化を調べる。プラズマ濃度の経時的変化は、標準モデルを用いたパーセンテージとして該化合物の絶対的バイオアベイラビリティを算定するために用いることができる。主なプロトコルの例を下記に挙げる。
【0110】
本発明化合物またはその親化合物をivで3mg/kgまたは本発明化合物または園生や化合物をpoで10mg/kgマウスに投薬する。血液サンプルを5分、15分、1時間、4時間および24時間のインターバルで取り出し、サンプル中の本発明化合物またはその親化合物の濃度をHPLCで同定する。次いで、プラズマ濃度の経時的変化を用いて、プラズマ濃度−時間曲線下面積(AUC、全身循環に到達する不変薬剤の全量に正比例する)、最大(ピーク)プラズマまたは血中薬剤濃度、最大(ピーク)プラズマ薬剤濃度が発生する時間(ピーク時間)、などの主要なパラメータを得るために用いることができる。バイオアベイラビリティの正確な決定に用いられるその他の要因には、該化合物の消失半減期、全身クリアランス、定常状態分布容積およびF%が含まれる。次いでこれらのパラメータを非境界要素法または境界要素法で分析して、算定されたパーセンテージのバイオアベイラビリティを得る。この種の方法の一例がGallant−Haidnerら、2000年およびTrepanierら、1998年に記載されており、それらをここで参照する。
【0111】
上述の39−デスメトキシラパマイシン誘導体またはその処方は従来のいかなる方法によっても投与可能であり、限定的ではないがたとえば、非経口、経口、局所(口内、舌下、経皮を含む)的に投与することができ、ステントなどの医療器具を用いて投与することができ、吸入法や注射(皮下または筋肉)によっても投与可能である。治療は一度の投薬または一定期間に亘る複数回の投薬で行うことができる。
【0112】
本発明化合物を単独で投与することも可能であるが、薬剤処方として一以上の許容されるキャリアと共に存在させることが好ましい。キャリアは本発明化合物とコンパチブルであり且つそのレシピエントに対して有害でないという意味において「許容される」ものでなければならない。好適なキャリアの数例を下記に詳述する。
【0113】
本発明の39−デスメトキシラパマイシン誘導体は単独または他の治療薬と組み合わせて投与することができ、2つ(またはそれ以上)の薬剤の併用投与は各々を単独で投与する場合に比べて格段に少ない投薬量で済み、したがってそれらの副作用を軽減させる。
【0114】
一つの実施形態において、免疫抑制の誘導または維持、臓器移植拒絶反応、移植片対宿主拒絶病、自己免疫疾患、神経変性症状、血管炎症疾患の治療のために、39−デスメトキシラパマイシン誘導体を他の治療薬と併用投与する。好適な治療薬の非限定的例示として、アザチオプリン、コルチコステロイド、シクロフォスファミド、シクロスポリンA、FK506、ミコフェノール酸モフェチル、OKT−3およびATGなどの免疫調節剤を含む。
【0115】
別の実施形態において、39−デスメトキシラパマイシン誘導体はガンまたはB細胞系悪性腫瘍の治療のために他の治療薬と併用投与される。好適な治療薬の非限定的例示として、メトトレキセート、リューコボリン、アドリアマイシン、プレニゾン、ブレオマイシン、シクロフォスファミド、5−フルオロウラシル、パクリタキセル、ドセタキセル、ビンクリスチン、ビンブラスチン、ビノレルビン、ドキソルビシン、タモキシフェン、トレミフェン、酢酸メゲストロール、アナストロゾール、ゴセレリン、抗HER2単クローン抗体(Herceptin(商標名)など)、カペシタビン、塩酸ラロキシフェン、EGFR抑制剤(Iressa(登録商標)、Tarceva(商標名)、Erbitux(商標名)など)、VEGF抑制剤(Avastin(商標名)など)、プロテアソーム抑制剤(Velcade(商標名)など)、Glivec(登録商標)、hsp90抑制剤(17−AAGなど)を含む。加えて、39−デスメトキシラパマイシン誘導体は非限定的例示として放射線療法や手術を含む他の療法と組み合わせて投与しても良い。
【0116】
一実施形態において、39−デスメトキシラパマイシン誘導体は血管系疾患の治療のために他の治療薬と併用投与される。好適な治療薬の非限定的例示として、ACE抑制剤、アンギオテンシンII受容体拮抗薬、フィブリン酸誘導体、HMG−CoAリダクターゼ抑制剤、βアドレナリン遮断剤、カルシウムチャンネル遮断剤、抗酸化剤、抗凝集剤、血小板抑制剤(Plavix(商標名)など)を含む。
【0117】
一実施形態において、39−デスメトキシラパマイシン誘導体は神経再生の刺激のための他の治療薬と併用投与される。好適な治療薬の非限定的例示として、神経成長因子、グリア細胞由来成長因子、脳由来成長因子、毛様体神経栄養因子、ニューロトロフィン−3などの神経栄養因子を含む。
【0118】
一実施形態において、39−デスメトキシラパマイシン誘導体は感染症の治療のための他の治療薬と併用投与される。好適な治療薬の非限定的例示として、アンフォテリシンB、フルサイトシン、エキノカンジン(カスポファンギン、アニデュラファンギンまたはミカファンギンなど)、グリセオフルビン、イミダゾール系またはトリアゾール系抗真菌剤(クロトリマゾール、ミコナゾール、ケトコナゾール、エコナゾール、ブトコナゾール、オキシコナゾール、テルコナゾール、イトラコナゾール、フルコナゾオール、ボリコナゾールなど)を含む。
【0119】
当業者には明らかなことであろうが、併用投与には、同一の治療レジメの一部として2またはそれ以上の治療薬を患者に輸送するいかなる手段をも含む。2またはそれ以上の薬剤を単一の処方において同時に投与することも可能であるが、これは必須ではない。これらの薬剤は異なる処方で異なる時期にも投与可能である。
【0120】
処方は従来のように単位投薬形態として存在しても要旨、調剤分野で公知のいかなる方法によって調製しても良い。このような方法は、活性成分(本発明化合物)を一以上のアクセサリ成分を構成するキャリアに関連付けるステップを含む。一般的に言えば、活性成分を液体キャリアまたは微細に分割した固体キャリアまたはその両方に均一且つ周到に関連付けた後、必要であれば製品に仕上げることによって処方を調製する。
【0121】
本発明の39−デスメトキシラパマイシン誘導体は通常の場合、活性成分からなる薬剤処方の形態で、任意的に非毒性の有機または無機、酸または基、添加塩、薬学的に許容し得る投薬形態で、経口または何らかの非経口ルートによって投与される。治療すべき疾患や患者および投薬ルートに応じて異なる投薬量で化合物を投与することができる。
【0122】
一例として、本発明化合物は、即時的または時間を置いたあるいはリリースコントロールされた投与のために、香料剤や着色剤を含み得る錠剤、カプセル、胚珠、エリキシル、溶液または懸濁液の形態にして、経口、口内または舌下に投与することができる。
【0123】
経口投与に適した39−デスメトキシラパマイシン誘導体の溶液または懸濁液はまた、N,N−ジメチルアセトアミドなどの賦形剤、ポリソルベート80などの分散剤、表面活性剤、Phosal50PG(フォスファチジルコリン、大豆脂肪酸、エタノール、モノ/ジグリセリド、プロピレングリコールおよびアスコルビン酸パルミチン酸エステルからなる)などの溶解剤を含むことができる。
【0124】
錠剤には、微結晶セルロース、ラクトース(ラクトース一水和物、ラクトース無水物など)、クエン酸ナトリウム、炭酸カルシウム、リン酸水素カルシウムおよびグリシンなどの賦形剤、デンプン(好ましくはコーン、ポテトまたはタピオカのデンプン)、デンプングリコール酸ナトリウム、クロスカルメロースナトリウムなどの崩壊剤、ある種の錯体ケイ酸塩、ポリビニルピロリドン、ヒドロキシプロピルメチルセルロース(HPMC)、ヒドロキシプロピルセルロース(HPC)、マクロゴル8000、サクロース、ゼラチン、アカシアなどの顆粒結合剤を含むことができる。さらに、ステアリン酸マグネシウム、ステアリン酸、ベヘン酸グリセリル、タルクなどの潤滑剤を含有しても良い。
【0125】
類似タイプの固形化合物はゼラチンカプセル内の充填物としても使用可能である。この態様における好適な賦形剤は、ラクトース、デンプン、セルロース、乳糖または高分子ポリエチレングリコールなどである。液体懸濁物および/またはエリキシルの場合、本発明化合物は様々な甘味剤または香料剤、着色剤または顔料、乳化剤および/または懸濁剤、水やエタノール、プロピレングリコール、グリセリンおよびこれらの混合物と組み合わせて用いることができる。
【0126】
錠剤は任意の一以上の副成分と共に圧縮または成型によって調製することができる。圧縮法による錠剤は、粉体または粒体のような自由流動形態にある有効成分を適当な機械の中で圧縮することによって調製することができる。該有効成分は、結合剤(ポビドン、ゼラチン、ヒドロキシプロピルメチルセルロースなど)、潤滑剤、不活性希釈剤、防腐剤、崩壊剤(グリコールデンプンナトリウム、架橋ポビドン、架橋カルボキシメチルセルロースナトリウムなど)、表面活性剤または分散剤と任意に混合して用いることができる。成型法による錠剤は、不活性希釈剤で湿潤された粉状の化合物を適当な機械の中で成型することによって調製することができる。錠剤には任意にコーティングまたはスコアリングが施され、たとえばヒドロキシプロピルメチルセルロースを様々な配合で用いて所望のリリースプロファイルを与えることにより、その中の有効成分のリリースを遅らせ、あるいは制御するように処方することができる。
【0127】
経口投与に適した本発明の処方は、各々が所定量の有効成分を含むカプセル剤、カシュ剤、錠剤などの個別ユニットとして、粉状物または粒状物として、水性液または非水性液中の溶液または懸濁液として、あるいは水中油型液体エマルジョンまたは油中水型液体エマルジョンとして、提供することができる。有効成分はボーラス(急速静注)投与、練り薬またはペーストとして提供することもできる。
【0128】
口内局所投与に適した処方は、香り付けされた通常はサクロースまたはアカシアまたはトラガントなどの基質内の活性成分からなるロゼンジ、ゼラチン、グリセリン、サクロース、アカシアなどの不活性基質内の活性成分からなる芳香製剤、適当な液体キャリア内の活性成分からなるマウスウォッシュを含む。
【0129】
上述の成分に加えて、本発明の処方は、問題となる処方のタイプを考慮して、当業界に公知である他の薬剤を含み得ることを理解しなければならない。たとえば、経口投与に適したものは香料剤を含むことができる。
【0130】
局所投与に適合した薬剤化合物は、軟膏、クリーム、ローション、パウダー、溶液、ペースト、ジェル、含浸包帯、スプレー、エアロゾル、オイル、経皮デバイス、ダスティングパウダーなどとして設計することができる。これらの化合物は活性剤を含む従来法で調剤可能である。これらは従来公知の親水性キャリアおよび添加剤、たとえば防腐剤、薬剤の浸透をアシストする溶媒、クリームまたは軟膏内のエモリエント、ローションのためのエタノールまたはオレイルアルコールを含むことができる。このようなキャリアは化合物の約1%〜約98%存在することができる。より一般的にはこれらは化合物の約80%を上限として存在する。例示的に述べるにすぎないが、クリームまたは軟膏は、十分な量の親水性材料と化合物の約5〜10重量%を含む水とを混合して所望のコンシステンシーを有するクリームまたは軟膏として製造することができる。
【0131】
経皮投与に適合した薬剤化合物は、レシピエントの表皮と長時間密接し続けるように意図された不連続パッチとして提供することができる。たとえば、活性剤をパッチからイオントフォレーシスして供給することができる。
【0132】
口や皮膚などの外部組織への投与のために、化合物は好ましくは局所用の軟膏またはクリームとして提供される。軟膏として設計される場合は、活性剤をパラフィン系または水溶性の軟膏ベースと共に用いることができる。
【0133】
あるいは、活性剤を水中油クリームベースまたは油中水ベースと共にクリームに処方しても良い。
【0134】
静脈投与の場合、流体ユニットの単位投薬量フォームは、活性剤と、非限定的例示として水やアルコール、ポリオール、グリセリン、植物性油など、特に水が好ましいが、これらの無菌ベヒクルを用いて調製される。活性成分は、使用されるベヒクルと濃度に応じて、ベヒクルに懸濁または溶解され得る。溶液を調製する場合、活性成分は注射のために水に溶解し、適当なバイアルまたはアンプルに注入する前にフィルタ殺菌してシールする。
【0135】
好ましくは、局所麻酔薬、防腐剤、緩衝剤などの添加剤がベヒクルに溶解される。安定性を高めるために、バイアルに注入する前に化合物を凍結して真空下で水を除去することができる。凍結乾燥したパウダーは次いでバイアル内でシールされ、使用前に注射用の水バイアルが供給されて液体を再構成する。
【0136】
非経口投与懸濁液は、活性成分が溶解される代わりにベヒクル内に懸濁され、また、ろ過を伴わずに殺菌されることを除いて、溶液の場合とほぼ同様に調製される。活性成分は無菌ベヒクルに懸濁される前にエチレン酸化物に晒されることによって殺菌される。好ましくは、活性成分の均一な分散を促進させるために、界面活性剤または湿潤剤が化合物に含まれる。
【0137】
本発明化合物は、また、当業界において公知の医療機器を用いて投与することができる。たとえば、一実施例によれば、本発明の薬剤化合物は無針(needleless)皮下注射器を用いて投与することができる。このような注射器は、米国特許第5399163号、米国特許第5383851号、米国特許第5312335号、米国特許第5064413号、米国特許第4941880号、米国特許第4790824号または米国特許第4596556号に開示されている。本発明に有用な公知のインプラントおよびモジュールの例を挙げれば、制御された速度で投薬するためのインプラント可能なマイクロインフュージョンポンプ(micro-infusion pump)を開示する米国特許第4487603号、外皮を通して投薬する医療器具を開示する米国特許第4486194号、厳密な注入速度で薬剤をデリバーするための薬剤インフュージョンポンプを開示する米国特許第4447233号、連続薬剤デリバリーのための可変速度インプラント可能インフュージョン装置を開示する米国特許第4447224号、マルチチャンバーのコンパートメントを有する浸透性薬剤デリバリーシステムを開示する米国特許第4439196号、および、浸透性薬剤デリバリーシステムを開示する米国特許第4475196号である。一つの特定の実施形態において、39−デスメトキシラパマイシン誘導体は、WO01/87263に記述されるものや関連する刊行物あるいはPerin(Perin、EC、2005年)によって記述されるものに対応するような薬剤溶出ステントを用いて投与することができる。その他多くのインプラント、デリバリーシステムおよびモジュールが当業者に知られている。
【0138】
本発明の39−デスメトキシラパマイシン誘導体の1回投薬量は特定の化合物、関係する疾患、サブジェクト、疾患の性質および深刻度、サブジェクトの物質的な条件、および選択された投薬経路によって変わる。適切な1回投薬量は当業者であれば容易に決定することができる。
【0139】
薬剤化合物には、投薬方法に依存して、本発明化合物が0.1重量%以上、好ましくは5〜60重量%、より好ましくは10〜30%含まれる。
【0140】
当業者には認識し得るように、本発明化合物の1回投薬量の適量と投薬間隔は、治療する症状の性質および程度、投与の形態、経路および部位、治療患者の年齢および状態などによって決定され、最終的には医師が適切な投薬量を決定する。この投薬量は適切である限り頻繁に繰り返され得る。副作用が発症した場合は、通常の臨床プラクティスに基づいて投薬の量および/または頻度を変更または減少させる。
【実施例】
【0141】
一般的方法および材料
【0142】
材料
すべての試薬は市販品から入手したものであり、特記しない限りさらに精製すること無しに使用した。
【0143】
培養
S.ヒグロスコピカス MG2−10[IJMNOQLhis](WO04/007709;Gregoryら、2004年)を培地1の寒天プレート(下記)上で28℃に維持した。芽胞株を培地1で培養した後に調製した。それは、20%w/vグリセリンと10%w/vラクトースを含む蒸留水に保存して−80℃で保管した。凍結株0.1mlを250mlフラスコ中の50ml培地2(下記)に接種することによって植物培養菌を調製した。この培養菌を36〜48時間、28℃、300rpmでインキュベートした。
【0144】
製造法
植物培養菌を2.5〜5%v/vで培地3に接種した。6〜7日間にわたり26℃、300rpmで培養した。
【0145】
培養法
シクロヘキサンカルボン酸の供給/添加を接種24〜48時間後に行い、他に述べられていない限り1〜2mMの最終濃度で供給した。
【0146】
培地1:
成分 ソース カタログ# L当たり
コーンスターチパウダー Sigma C−8160 2.5g
酵母エキス Difco 0127−17 3g
炭酸カルシウム Sigma C5929 3g
硫酸鉄 Sigma F8633 0.3g
BACTO寒天 Difco 2140−10 20g
小麦デンプン Sigma S2760 10g
水 1L
その後培地を121℃で20分間オートクレーブ殺菌
【0147】
培地2:RapV7菌種培地
成分 L当たり
トーストしたニュートリソイ(ADM Ingredients Ltd) 5g
Avedex W80 デキストリン(Deymer Ingredients Ltd) 35g
コーンスチープ固形物(Sigma) 4g
グルコース 10g
(NH4)2SO4 2g
ラクチン酸(80%) 1.6mL
CaCO3(Caltec) 7g
1MのNaOHでpHを7.5に調整
その後培地を121℃で20分間オートクレーブ殺菌
殺菌後16mLの40%グルコースを各7mLの培地に添加
【0148】
培地3:MD6培地(発酵培地)
成分 L当たり
トーストしたニュートリソイ(ADM Ingredients Ltd) 30g
コーンスターチ(Sigma) 30g
Avedex W80 デキストリン(Deymer Ingredients Ltd) 19g
酵母(Allinson) 3g
コーンスチープ固形物(Sigma) 1g
KH2PO4 2.5g
K2HPO4 2.5g
(NH4)2SO4 10g
NaCl 5g
CaCO3(Caltec) 10g
MnCl2・4H2O 10mg
MgSO4・7H2O 2.5mg
FeSO4・7H2O 120mg
ZnSO4・7H2O 50mg
MES(2−モルホリノエタンスルホン酸一水和物) 21.2g
1MのNaOHでpHを6.0に調整
殺菌前にSigmaのα−アミラーゼ(BAN250)を1L培地に添加
培地を121℃で20分間オートクレーブ殺菌
殺菌後0.35mLの滅菌40%フルクトースと0.10mLのL−リシン(水中に140mg/mL、フィルター殺菌済)を各7mLに添加
【0149】
培地4:RapV7a菌種培地
成分 L当たり
トーストしたニュートリソイ(ADM Ingredients Ltd) 5g
Avedex W80 デキストリン(Deymer Ingredients Ltd) 35g
コーンスチープ固形物(Sigma) 4g
(NH4)2SO4 2g
ラクチン酸(80%) 1.6mL
CaCO3(Caltec) 7g
1MのNaOHでpHを7.5に調整
その後培地を121℃で20分間オートクレーブ殺菌
【0150】
培地5:MD6/5−1培地(発酵培地)
成分 L当たり
トーストしたニュートリソイ(ADM Ingredients Ltd) 15g
Avedex W80 デキストリン(Deymer Ingredients Ltd) 50g
酵母(Allinson) 3g
コーンスチープ固形物(Sigma) 1g
KH2PO4 2.5g
K2HPO4 2.5g
(NH4)2SO4 10g
NaCl 13g
CaCO3(Caltec) 10g
MnCl2・4H2O 3.5mg
MgSO4・7H2O 15mg
FeSO4・7H2O 150mg
ZnSO4・7H2O 60mg
SAG471 0.1mL
培地を121℃で30分間オートクレーブ殺菌
殺菌後L当たり15gフルクトースを添加
48時間後0.5g/LのL−リシンを添加
【0151】
分析法
方法A
注入容量:0.005〜0.1mL(感受性に応じた要求に従う)。HPLCをアジレント「Spherisorb」「Rapid Resolution」カートリッジSB C8上に、3ミクロン、30mm×2.1mmで、下記移動相で作動させた。
【0152】
移動相A:純水中に0.01%蟻酸
移動相B:アセトニトリル中に0.01%蟻酸
流速:1mL/分
0分時の5%Bから2.5分時の95%Bまでリニアグラジエントを用い、4分間95%Bに維持し、次のサイクルまでに5%Bに戻した。検出は、254nmでのUV吸収および/またはMicromasss Quattro−Micro装置を用いたエレクトロスプレーイオン化質量分析(ポジティブまたはネガティブ)によって行った。
【0153】
方法B
注入容量:0.02mL。HPLCを3ミクロンBDS C18 Hypersil(ThermoHypersil−Keystone Ltd)カラム上に、150mm×4.6mmで、50℃に維持し、下記移動相で作動させた。
移動相A:アセトニトリル(100mL)、トリフルオラセト酸(1mL)、1M酢酸アンモニウム(10mL)、脱イオン水と共に1Lまで調製
移動相B:脱イオン水(100mL)、トリフルオラセト酸(1mL)、1M酢酸アンモニウム(10mL)、アセトニトリルと共に1Lまで調製
流速:1mL/分
10分間で55%Bから95%Bまでリニアグラジエントを用った後、95%Bで2分間、55%Bで0.5分間、55%Bでさらに2.5分間維持した。化合物の検出は280nmでのUV吸収によって行った。
【0154】
方法C
HPLCシステムとしてAgilentHP1100を用い、3ミクロンBDS C18 Hypersil(ThermoHypersil−Keystone Ltd)カラム上に、150mm×4.6mmで、40℃に維持し、下記移動相で作動させた。
移動相A:脱イオン水
移動相B:アセトニトリル
流速:1mL/分
このシステムをBruker Daltonics Esquire3000エレクトロスプレー質量分析装置に接続した。500〜1000Daltonの走査領域に亘ってポジティブ/ネガティブ切替を用いた。10分間で55%Bから95%Bまでリニアグラジエントを用いた後、95%Bで2分間、55%Bで0.5分間、55%Bでさらに2.5分間維持した。
【0155】
合成法
特記しない限り全ての反応は市場入手可能な乾燥溶媒を用いて無水条件下で行った。エレクトロスプレー源を備えたBruker Daltonics Esquire3000+質量分析装置に連結したAgilent1100HPLC上で、LC−UV−MSにより反応をモニターした。水:アセトニトリルv:v30:70〜100%アセトニトリルの直線勾配で10分間、次いで100%アセトニトリルで5分間の無勾配期の処理により、Phenomenex Hyperclone カラム、BDS C18 3u(150×4.6mm)において1mL/分で分解が実行された。
【0156】
NMRスペクトルがCDCl3中に記録され、δHおよびδC化学シフトが溶媒(7.26ppmおよび77.0ppm)参照付けられる。39−デスメトキシラパマイシンおよびその誘導体はコンホマーの混合物として存在するので、すべてのアサインメントは主要コンホマーにのみ対応する。
【0157】
抗ガン作用のための生体外バイオアッセイ
化合物の抗ガン作用を、単分子層増殖アッセイ中の12個のヒト腫瘍細胞ラインのパネルにおいて、FreiburgのExperimental OncologymOncotest GmbHの研究機関であるOncostest試験場で生体外評価を行った。選択された12個の細胞ラインの特性を表1に要約して示す。
【0158】
【表1】
【0159】
Rothら、1999年に記述されるように、ヒト腫瘍異種移植片からOncotest細胞ラインが確立された。ドナー異種移植片の起源はFiebigら、1999年に記述された。他の細胞ラインが、NCl(H460、SF−268、OVCAR−3、DU145、MDA−MB−231、MDA−MB−468)から得られ、またはドイツ国BraunshweigのDSMZから購入することができる(LNCAP)。
【0160】
他に特記しない限り、すべての細胞サインは、RPMI1640培地と、10%ウシ胎児血清と0.1mg/mLゲンタミシン(PAA、ドイツ国コルベ)を含む「すぐに混合できる」培地にて、加湿雰囲気(95%空気、5%二酸化炭素)中で37℃で培養した。
【0161】
単分子層アッセイ−プロトコル1の要約:
変性ヨウ化プロピジウムアッセイを用いて、12個のヒト腫瘍細胞ラインの成長に対する試験化合物の影響を評価した(Denglerら、1995年)。
【0162】
要約すれば、トリプシン化によって指数関数的位相培養(exponential phase cultures)から細胞を取り出し、細胞ラインに応じた細胞密度(5〜10,000の範囲で変動する細胞数/ウェル)にて96ウェル底面平滑マイクロタイタープレートに接種してカウントした。24時間後に回収して指数関数的な増殖を再開させた後、培養培地0.01ml(プレート当たり6個の対照ウェル)またはマクベシン(macbecin)含有培地をウェルに添加した。各濃度を3つ培養した。39−デスメトキシラパマイシンを2つの濃度(0.001mMおよび0.01mM)で適用した。4日間の連続的インキュベーションの後、39−デスメトキシラパマイシンを用いまたは用いずに細胞培養培地を0.2mlの水性死細胞染色用蛍光色素PI溶液(7mg/l)で置換した。生存細胞の割合を測定するため、プレート凍結によって細胞を透過させた。プレートを解凍した後、Cytofluor4000マイクロプレートリーダー(励起530nm、放出620nm)を用いて蛍光発光を測定して、生存可能な細胞の総数との直接関係を得た。
【0163】
増殖抑制が(被処理/対照)×100(%T/C)として表された。活性化合物についてIC50およびIC70の値を細胞生存性に対する化合物濃度をプロットすることによって評価した。
【0164】
実施例1:39−デスメトキシラパマイシンの発酵と単離
後述のようにS.ヒグロスコピカス MG2−10[IJMNOQLhis]の培養菌を成長させ、シクロヘキサンカルボン酸(CHCA)で培養することにより39−デスメトキシラパマイシンを産生した。
【0165】
液体培養
S.ヒグロスコピカス MG2−10[IJMNOQLhis]の栄養体培養菌を「Materials & Methods」に記述されるようにして培養した。産生培養を栄養体培養菌により0.5mLで50mLチューブ内の7mL培地3に接種した。7日間に亘り26℃、300rpmで培養した。1:1アセトニトリルで30分間シェイクして1mLのサンプルを抽出し、13,000回転で10分間延伸処理し、分析法Bに基づいて分析および定量化を行った(「Materials & Methods」参照)。産生物の確認は分析法C(「Materials & Methods」参照)を用いて質量分析により決定した。
【0166】
下記特性に記述した分析データを基に、観察されたラパマイシン相同物が所望の39−デスメトキシラパマイシンであると認められた。
【0167】
発酵
S.ヒグロスコピカス MG2−10[IJMNOQLhis]の培地4内の一次的栄養体培養菌を実質的に「Materials & Methods」に記載されるところにより培養した。培地4内の二次的栄養体培養菌を10%v/v、28℃、250rpmで24時間接種した。栄養体培養菌を5%v/vで20L発酵槽中の培地5(「Materials & Methods」参照)に接種した。6日間、26℃で培養し、インペラー先端速度を最小速度1.18ms−1から最大速度2.75ms−1の範囲で変化させることにより0.5vvm.>30%溶解酸素を維持した。シクロヘキサンカルボン酸の供給を接種の24時間後および48時間後に行って、最終濃度2mMを得た。
【0168】
抽出と精製
発酵汁(30L)を等量メタノールで2時間撹拌した後、遠心処理(10分間、3500rpm)して細胞をペレット状にした。上清をDiaion(登録商標)HP20樹脂(43g/L)で1.5時間撹拌した後にろ過した。アセトン(合計量7.5L)で樹脂をバッチ洗浄してラパマイシン相同物を剥ぎ取り、溶剤を減圧下で除去した。得られた水性抽出物を水で2Lに希釈し、酢酸エチル(3×2L)で溶離した。溶剤を減圧下で除去して褐色油を得た(20.5g)。
【0169】
この抽出物をアセトンに溶解し、シリカに乾燥させ、シリカカラム(6×6.5cm径)に適用し、徐々に比率を変えたアセトン/ヘキサン(20%−40%)で抽出した。ラパマイシン相同物含有フラクションをプールし、溶剤を減圧下で除去した。残余物(2.6g)をSephadex LH20でクロマトグラフィ処理し(3回バッチ)、10:10:1クロロホルム/ヘプタン/エタノールで抽出した。半精製されたラパマイシン相同物(1.7g)をGilson HPLCを用いて逆位相(C18)プレパラティブHPLCにより精製し、21mL/分の65%アセトニトリル/水でPhenomenex21.2×250mm Luna5μmC18BDSカラムを溶離した。最も純粋なフラクション(HPLC分析、方法Bにより同定)を結合し、溶媒を減圧下で除去して、39−デスメトキシラパマイシン(563mg)を得た。
【0170】
特性
39−デスメトキシラパマイシンの1H NMRスペクトルは標準(P.Lowden、物理学博士、学位論文、ケンブリッジ大学、1997年)のそれと同等であった。13C-NMR (125 MHz), δc (ppm): 215.75, 208.27, 169.19, 166.71 , 140.13, 135.94, 133.61, 130.10, 129.62, 126.80, 126.33, 98.42, 84.77, 84.37, 75.85, 70.91, 67.10, 59.44, 55.82, 51.21 , 46.50, 44.17, 41.39, 40.70, 40.16, 38.74, 38.37, 35.44, 35.26, 35.08, 33.78, 33.64, 33.04, 32.37, 31.22, 30.41 , 27.24, 27.02, 25.27, 21.48, 20.58, 16.24, 15.95, 15.78, 13.74, 13.00, 10.12
【0171】
培養抽出物のLCMSおよびLCMSn分析は、新規ラパマイシン相同物のm/z比がラパマイシンのそれよりも30原子質量単位だけ小さく、メトキシ基の欠乏と一致することを示した。観測されたイオンは、[M−H]−882.3、[M+NH4]+901.4、[M+Na]+906.2、[M+K]+922.2。ナトリウム付加化合物のフラグメンテーションは、規定の解離経路(図2)(J.A.Reather、理学博士、学位論文、ケンブリッジ大学、2000年)に従い、39−デスメトキシラパマイシンとして予期されたイオンを与えた。この質量分析解離データは、シクロヘキシル成分を含むC28−C42フラグメントにメトキシ欠損が生じている新規ラパマイシン相同物の領域を限定している。
【0172】
この質量分析解離データは39−デスメトキシラパマイシンに完全に合致している。
【0173】
実施例2:39−デスメトキシ−40−O−[2,2−ビス(ヒドロキシメチル)プロピオニル]ラパマイシン
下記工程により39−デスメトキシラパマイシンから39−デスメトキシ−40−O−[2,2−ビス(ヒドロキシメチル)プロピオニル]ラパマイシンを合成した。
【0174】
2.1 39−デスメトキシ−28−O−トリメチルシリルラパマイシンの合成
39−デスメトキシラパマイシン(170mg、0.17mmol)およびイミダゾール(51mg、0.75mmol)を5mLの酢酸エチルに0℃で溶解した。この冷たい溶液にクロロトリメチルシラン(77mg、0.09mL、0.71mmol)を10分間に亘り滴状で添加した。さらに60分間撹拌を連続して行って、28,39−ビス−O−トリメチルシリルエーテルの生成を完了した。この期間後、0.4mLの0.5N硫酸水溶液を添加し、混合物を0℃で2.5時間撹拌した。20mLの酢酸エチルを添加し、塩水、飽和炭酸水素ナトリウム溶液および水で有機層を洗浄した。硫酸ナトリウムでの乾燥および減圧下での濃縮により、無色の固形物として28−O−トリメチルシリルエーテルが産生され、これをさらに精製すること無しに次工程の反応に用いた。1H-NMR (400 MHz, CDCI3), δ (ppm): 4.07 (d, 1H, J=6.5 Hz, 0(28J-H)1 0.00 (s, 9H, 28-O-TMS).
【0175】
2.2 2,4,6−トリクロロ安息香酸2’,2’,5’−トリメチル−1’,3’−ジオキサン−5’カルボキシル無水物の合成
2,2−ジメトキシプロパン(13.5g、130mmol)およびρ−トルエンスルホン酸一水和物(100mg、0.53mmol、0.4mol%)を、アセトン(100mL)中の2,2−ビス(ヒドロキシメチル)プロピオン酸(13.5g、100mmol)溶液に添加した。反応混合物を室温で2時間撹拌した。この時間の後、炭酸水素ナトリウム溶液を添加し、その混合物をさらに5分間撹拌した。上清を除去し、減圧下で濃縮した。得られた固体物をジエチルエーテル(3×50mL)で処理し、組み合わせられた有機抽出物を減圧下で濃縮して白色の固形物16.2gを産生した。1H-NMR (400 MHz, CDCI3), δ (ppm): 4.19 (d, 1H, J=12.0Hz) 3.68 (d, 1 H, J=12.0Hz) 1.45 (s, 1 H) 1.41 (s, 1H) 1.20 (s, 1 H).
【0176】
この物質を米国特許5,362,718の方法で活性化混合無水物に形質転換した。すなわち、アセトニド(1.04g、5.98mmol)を0℃に冷却したTHF(20mL)に溶解し、トリエチルアミン(0.83mL、5.98mmol)および2,4,6−トリクロロ安息香酸塩化物(0.93mL、5.98mmol)の滴下添加で処理した。次いで反応物を室温で5時間撹拌した。得られた沈殿物をろ過し、THF(10mL)で洗浄した。組み合わされたろ過物質を真空内で還元して白色のアモルファスを産生し、これをさらに精製すること無しに以下において用いた。
【0177】
2.3 39−デスメトキシラパマイシン 28−O−トリメチルシリルエーテル、2,2,5−トリメチル[1,3−ジオキサン]−5−カルボキシル酸の40−エステルの合成
実施例2.1からの粗製28−O−トリメチルシリル−39−デスメトキシラパマイシン(200mg、0.17mmolの39−デスメトキシラパマイシンから)を2mLのジクロロメタンに溶解した。溶液を0℃に冷却してDMAP(102mg、0.84mmol)を添加した。次いで、1mLジクロロメタン中の2,4,6−トリクロロ安息香酸2’,2’,5’−トリメチル−1’,3’−ジオキサン−5’カルボキシル無水物(159mg、0.42mmol)溶液を10分間に亘って添加した。反応混合物を0℃で5時間撹拌し、LC/MSで形質転換をモニターした。反応混合物を7mLのジクロロメタンで希釈し、5mLの加水により冷却した。有機層を0.5N硫酸、炭酸水素ナトリウム溶液および水で順次に除去および洗浄した。硫酸ナトリウムでの乾燥および減圧下での濃縮により、無色の泡状物として表題の化合物が産生され、これをさらに精製すること無しに直ちに用いた。MS (ESI) m/z 1111 [M-H]"
【0178】
2.4 39−デスメトキシラパマイシン−40−O−[2,2−ビス(ヒドロキシメチル)プロピオニル]ラパマイシン
実施例2.3からの粗製39−デスメトキシラパマイシン−28−O−トリメチルシリルエーテル 2,2,5−トリメチル[1,3−ジオキサン]−5−カルボキシル酸の40−エステルを2mLアセトンに溶解し、0.5mLの0.5N硫酸を添加した。反応混合物を室温で5時間撹拌し、次いで5mLの飽和炭酸水素ナトリウム溶液と5mLの水を添加して中和した。混合液を酢酸エチルで抽出し、組み合わされた有機抽出物を硫酸ナトリウムで乾燥させた。減圧下での濃縮により無色の固形物が得られ、これをSephadex LH20にてサイズ排除クロマトグラフィにより精製した。溶離剤にはクロロホルム/ヘプタン/エタノール(v:v:v=10:10:1)を用いた。1H-NMR (500 MHz, CDCI3), δ (ppm): 4.72 (m, 1 H, C(40)-H), 3.87 (m, 2H), 3.69 (m, 2H), 1.03 (s, 3H); 13C-NMR (125 MHz, CDCI3), δ (ppm): 175.52, 74.04 (C(40)), 68.73 (2C), 48.90, 17.09. MS (ESI) m/z 1023 [M+Na]+
【0179】
実施例3:39−デスメトキシ−40−O−(2−ヒドロキシ)エチルラパマイシン
3.1 2−(タート−ブチルジメチルシリル)オキシエチルトリフレート
6mLのジクロロメタン中の2−(タート−ブチルジメチルシリル)−エチレングリコール(125mg、0.71mmol)および2,6−ルチデンlutidene(0.08mL、0.69mmol)溶液を−78℃に冷却した。トリフルオロメタンスルホン酸無水物(0.11mL、0.65mmol)を5分間に亘って添加し、さらに15分間−78℃で撹拌を続けてトリフレートの生成を完了した。このトリフレートを下記3.2に記述の反応に自然位で(in situ)用いた。
【0180】
3.2 40−O−[2−(タート−ブチルジメチルシリル)]エチル−39−デスメトキシラパマイシン
39−デスメトキシラパマイシン(300mg、0.34mmol)および2,6−ジ−タート−ブチルピリジン(1.5mL、6.68mmol)を室温にて2−(タート−ブチルジメチルシリル)オキシエチルトリフレート(6mLジクロロメタン中に0.65mmol)で処理した。この溶液を緩やかな窒素流でその当初容積の1/3に濃縮し、得られた懸濁液をさらに72時間室温で撹拌した。この期間の後、飽和炭酸水素ナトリウム溶液(5mL)および水(5mL)を添加し、この混合物を30分間撹拌した。有機層を分離し、溶液相を酢酸エチル(2×5mL)で抽出した。組み合わされた有機抽出物を硫酸ナトリウムで乾燥させ、減圧下でのUShくして無色の油分を得た。ヘキサンからヘキサン/アセトン(v:v=1:1)の勾配を用いたシリカカラムクロマトグラフィによる精製により無色の固形物としての製品を得た。1H-NMR (500 MHz, CDCI3), δ (ppm): 4.16 (d, 1H, J = 6.5 Hz, C(28)-H), 3.73 (t, 2H, J = 5.7 Hz), 3.52 (t, 2H, J = 5.7 Hz), 0.89 (s, 9H), 0.06 (s, 6H); 13C-NMR (125 MHz, CDCI3), δ (ppm): 76.61 (C-40), 69.31 (CH2), 63.03 (CH2), 25.92 (3C), 18.36, -5.23 (2C). MS (ESI) m/z 1065 [M+Na]+
【0181】
3.3 39−デスメトキシ−40−O−(2−ヒドロキシ)エチルラパマイシン
2mLアセトン中の40−O−[2−(タート−ブチルジメチルシリル)]エチル−39−デスメトキシラパマイシン(160mg、0.15mmol)の溶液を室温にて0.3mLの0.5N硫酸で処理した。この溶液を室温で3時間放置し、その後5mLの飽和炭酸水素ナトリウム溶液と10mLの水を添加して冷却した。この混合液を酢酸エチル(3×10mL)で抽出し、組み合わされた有機抽出物を硫酸ナトリウムで乾燥した。減圧下での濃縮により無色の固形物が得られ、これをHPLC(水/アセトンvv:v=20/80)でさらに精製した。1H-NMR (500 MHz, CDCI3), δ (ppm): 4.16 (d, 1H, J = 6 Hz), 3.70 (m, 2H), 3.57 (m, 2H), 3.20 (m,1H, C(40)-H); 13C-NMR (125 MHz, CDCI3), δ (ppm): 78.65 (C-40), 77.20 (C-28), 68.93 (CH2O),62.10 (CH2O).MS (ESI) m/z 951 [M+Na]+
【0182】
実施例4:39−デスメトキシラパマイシンのリパーゼ触媒エステル化を通じた39−デスメトキシ−40−O−[2,2−ビス(ヒドロキシメチル)プロピオニル]ラパマイシン
無水タート−ブチルメチルエーテル(3.5mL)中の39−デスメトキシラパマイシン(720mg、0.83mmol)、ビニル2,2,5−トリメチル[1,3−ジオキサン]−5−カルボキシル酸(244mg、1.22mmol)、リパーゼPS−C「アマノ」II(720mg)および分子篩0.5nm(250mg)の混合物をアルゴン雰囲気中で43℃に加熱した。48時間後LC/MSモニターは出発物質の完全な形質転換を示した。THF(10mL)を添加し、混合物をセライトのパッドでろ過した。この酵素をTHF(2×10mL)で洗浄し、組み合わされた有機抽出物を減圧下で濃縮した。残留物をTHF(50mL)に溶解し、H2SO4(15mL、0.5N)を添加した。この溶液を室温で5時間放置した後、NaHCO3(50mL、5%)および塩水(50mL)の添加により反応物を冷却した。この混合液をEtOAc(3×100mL)で抽出し、組み合わされた有機抽出物をMgSO4で乾燥した。溶媒を除去して半固形物としての製品を得た。フラッシュクロマトグラフィ(ヘキサン/アセトン 1:1)による精製により無色固形物としての製品を得た。NMRデータは実施例2.4と同じ。ナトリウム付加物のMS (ESI) m/z 1022 [M+Na]+ 断片化は図3に示す断片化パターンによりm/z 850, 728, 693, 614, 560, 545, 441および431でイオンを与えた。
【0183】
実施例5:39−デスメトキシ−40−O−[2−ヒドロキシエチル 3−ヒドロキシ−2−(ヒドロキシメチル)−2−メチルプロパノエート]ラパマイシン
無水タート−ブチルメチルエーテル(2mL)中の39−デスメトキシ−40−O−(2−ヒドロキシ)エチルラパマイシン(40mg、0.04mmol)、ビニル2,2,5−トリメチル[1,3−ジオキサン]−5−カルボキシル酸(25mg、0.13mmol)、リパーゼPS−C「アマノ」II(40mg)および分子篩0.5nm(40mg)の混合物をアルゴン雰囲気中で43℃に加熱した。72時間後LC/MSモニターは出発物質の完全な形質転換を示した。THF(10mL)を添加し、混合物をセライトのパッドでろ過した。この酵素をTHF(2×10mL)で洗浄し、組み合わされた有機抽出物を減圧下で濃縮した。残留物をアセトン(7.5mL)に溶解し、H2SO4(2.5mL、0.5N)を添加した。この溶液を室温で2時間放置した後、飽和NaHCO3(10mL)および水(10mL)の添加により反応物を冷却した。この混合液をEtOAc(3×10mL)で抽出し、組み合わされた有機抽出物をMgSO4で乾燥した。溶媒を除去して黄色っぽい固形物としての製品を得た。70:30 MeCN/水から100%MeCNへの勾配を用いてPhenomenex21.2×50mm Luna5μmC18BDSカラム上でプレパラティブHPLCにより15分間精製して、無色固形物としての製品を得た。ナトリウム付加物のMS (ESI) m/z 1067 [M+Na]+ 断片化は図4に示す断片化パターンによりm/z 894, 772, 738, 614, 604, 589, 475および441でイオンを与えた。.
【0184】
実施例6 27−O−デスメチル−39−デスメトキシ−40−O−[2,2−ビス(ヒドロキシメチル)プロピオニル]ラパマイシン
6.1 27−O−デスメチル−39−デスメトキシラパマイシン、2,2,5−トリメチル[1,3−ジオキサン]−5−カルボキシル酸の40−エステル
無水タート−ブチルメチルエーテル(2mL)中の27−O−デスメチル−39−デスメトキシラパマイシン(30mg、0.034mmol)、ビニル2,2,5−トリメチル[1,3−ジオキサン]−5−カルボキシル酸(34mg、0.17mmol)、リパーゼPS−C「アマノ」II(30mg)および分子篩0.5nm(30mg)の混合物をアルゴン雰囲気中で43℃に72時間加熱した。THF(10mL)を添加し、混合物をセライトのパッドでろ過した。この酵素をTHF(2×10mL)で洗浄し、組み合わされた有機抽出物を減圧下で濃縮して、黄色っぽい半固形物としての製品を得た。ヘキサン:アセトン(v:v=2:1)を用いたフラッシュクロマトグラフィによる精製で薄黄色の固形物としての製品を得た。MS (ESI) m/z 1049 [M+Na]+
【0185】
6.2 27−O−デスメチル−39−デスメトキシ−40−O−[2,2−ビス(ヒドロキシメチル)プロピオニル]ラパマイシン
6.1からの材料をアセトン(6mL)に溶解し、H2SO4(2mL、0.5N)を添加した。この溶液を室温で2時間放置した後、飽和NaHCO3(10mL)および水(10mL)の添加により反応物を冷却した。この混合液をEtOAc(3×10mL)で抽出し、組み合わされた有機抽出物をMgSO4で乾燥した。溶媒を除去して黄色っぽい固形物としての製品を得た。70:30 MeCN/水から100%MeCNへの勾配を用いてPhenomenex21.2×50mm Luna5μmC18BDSカラム上でプレパラティブHPLCにより15分間精製して、無色固形物としての製品を得た。ナトリウム付加物のMS (ESI) m/z 1009 [M+Na]+ 断片化は図5に示す断片化パターンによりm/z 836, 679, 600, 560, 531, 431, 427でイオンを与えた。1 H NMR (500 MHz1 CDCI3J δ ppm 4.73 (m, 1 H, C(40)-H), 4.32 (d, J=4.5 Hz, 1 H, 0(27J-H)1 4.19 (d, J=4.5 Hz, 1 H, C(28)-H), 3.89 (m, 2 H), 3.70 (m, 2 H) 1.03 (s, 3 H).
【0186】
実施例7:39−デスメトキシ−40−O−(2−ヒドロキシ)エチルラパマイシンおよび39−デスメトキシ−40−O−[2,2−ビス(ヒドロキシメチル)プロピオニル]ラパマイシンの抗ガン活性の生体外評価
単分子相増殖アッセイ中の12個のヒト腫瘍細胞ラインのパネルにおいて、39−デスメトキシ−40−O−(2−ヒドロキシ)エチルラパマイシンおよび39−デスメトキシ−40−O−[2,2−ビス(ヒドロキシメチル)プロピオニル]ラパマイシンの抗ガン活性の生体外評価を、上記一般的方法におけるプロトコル1に記述したところにより変性ヨウ化プロピジウムアッセイを用いて行った。
【0187】
結果を下の表2に示す。各結果は2回の実験の平均値を示す。表3は試験細胞ラインに対するこれら化合物についての平均IC50およびIC70を示し、ラパマイシンが対照として示されている。
【0188】
【表2】
【0189】
【表3】
【0190】
実施例8:生体外結合アッセイ
FKBP12
FKBP12は化学的変性剤である塩酸グアニジン(GdnHCl)中で可逆的にアンフォールドし、このアンフォールドをタンパク質の内在蛍光における変化によって観測した(Mainら、1998年)。FKBP12の天然状態を特異的に結合し安定させるリガンドは、タンパク質がより高い化学的変性剤濃度でアンフォールドするように変性曲線をシフトする(Mainら、1999年)。安定性の相違から、下記式を用いてリガンド結合定数を求めることができる。
【0191】
【数1】
ここで△Gappは遊離およびリガンド結合形態の間のアンフォールドの自由エネルギーにおける見掛け上の相違、△GH2OD−Nは遊離タンパク質水中アンフォールド自由エネルギー、[L]はリガンド濃度、Kdはタンパク質−リガンド複合体の解離定数である(Meieringら、1992年)。アンフォールドの自由エネルギーは下記式を用いてアンフォールド変位中間点に関連付けることができる。
【0192】
【数2】
ここでmD−Nは与えられたタンパク質と与えられた変性剤についての定数であって残余物のアンフォールドへの露出度変化に比例する(Tanford1968年およびTanford1970年)。[D]50%はアンフォールド中間点に対応する変性剤濃度である。我々は△GLD−NをFKBP12の安定性におけるラパマイシンと未知リガンド(同一リガンド濃度)の相違として下記のように定義する。
【0193】
【数3】
ここで<mD−N>はアンフォールド変位の平均m値であり、△[D]50%はラパマイシン−FKBP12アンフォールド変位と未知リガンド−FKBP12複合体アンフォールド変位の中間点の相違である。[L]>Kdの条件では△△△GD−Nを下記式から2つの化合物の相対的Kdsに関連付けることができる。
【0194】
【数4】
ここでKrapdはラパマイシンの解離定数、Kxdは未知リガンドXの解離定数である。したがって下記式が得られる。
【0195】
【数5】
各変性曲線のフィッティングはmD−Nのための値を生成し、この値を用いてm平均値<mD−N>および△[D]50%、したがってKxdを算出することができる。0.2nMのKrapdの文献値が用いられる。
【0196】
幾つかの場合においては、試験化合物の低い溶解性の故に、ラパマイシン対照実験におけるよりも低い試験化合物濃度が用いられた。これらの場合において、試験化合物濃度とラパマイシン対照濃度との相違を下記式を用いて考慮した。
【数6】
【0197】
【表4】
【0198】
mTOR
mTORの抑制は、mTOR経路およびp70S6キナーゼおよびS6の代用マーカーのリン酸化レベルの測定によって間接的に確立されている(Brunnら、1997年;Mothe−Satneyら、2000年;TeeおよびProud、2002年;HuangおよびHoughton、2002年)。
【0199】
HEK293細胞をFLAGがタグされたmTORおよびmycがタグされた受容体と共感染させ、24時間培養後、一晩血清飢餓させた。細胞を100nMインシュリンで刺激した後、収穫し、3回の凍結/融解サイクルで溶解した。溶解物をプールし、当量をmTOR/受容体複合物のためのFLAG抗体と免疫沈降させた。免疫沈降体を次のように処理した:化合物(0.00001〜0.003mM)で処理したサンプルを30分間30℃でFKBP12/ラパマイシン、FKBP12/39−デスメトキシラパマイシン誘導体またはベヒクル(DMSO)でプレインキュベートし、非処理サンプルをキナーゼ緩衝液中でインキュベートした。免疫沈降体を次いで3mMのATP、10mMのMn2+およびGST−4E−BP1の基質存在下でキナーゼアッセイに生体外適用した。4倍サンプルバッファで反応を停止させ、15%SDS−PAGE処理し、PVDF膜に移して、Phospho−4E−BP1(T37/46)のためにプルーブした。ウエスタンブロットバンドをJ画像を用いた画像分析(http://rsb.info.nih.gov/ij/)により計量した。図6Aはラパマイシン(黒塗り四角)および39−デスメトキシ−40−O−[2,2−ビス(ヒドロキシメチル)プロピオニル]ラパマイシン(黒塗り三角)についての用量反応曲線を示す。図6Bはラパマイシン(黒塗り四角)および39−デスメトキシ−40−O−(2−ヒドロキシ)エチルラパマイシン(黒塗り三角)についての用量反応曲線を示す。
【0200】
代替的に、HEK293細胞を6つのウェルプレートに接種して24時間プレインキュベートし、一晩中血清飢餓させた。次いで細胞をベヒクルまたは化合物で30分間30℃で予備処理し、100nMインシュリンで30分間30℃で刺激し、3回の凍結/融解サイクルで溶解し、タンパク質濃度を分析した。等量タンパク質を供給してSDS−PAGEジェル上で分離した。タンパク質をPVDF膜に湿潤転写し、Phospho−S6(S235/36)またはPhospho−p70 S6K(T389)のためにプルーブした。ウエスタンブロットバンドをJ画像を用いた画像分析(http://rsb.info.nih.gov/ij/)により計量した。
【0201】
参考文献
Alarcon, CM., Heitman, J., and Cardenas, M. E. (1999) Protein kinase activity and identification of a toxic effector domain of the target of rapamycin TOR proteins in yeast. Molecular Biology of the Cell 10: 2531-2546.
Alvarez, M., Paull, K., Monks, A., Hose, C, Lee, J. S., Weinstein, J., Grever, M., Bates, S., Fojo, T., 1995. Generation of a drug resistance profile by quantitation of mdr-1/P-glycoprotein in the cell lines of the National Cancer Institute Anticancer Drug Screen. Journal of Clinical Investigation, 95, 2205-2214.
Aparicio, J. F., Molnar, I., Schwecke, T., Kδnig, A., Haydock, S. F., Khaw, L.E., Staunton, J., and Leadlay, P. F. (1996) Organization of the biosynthetic gene cluster for rapamycin in Streptomyces hygroscopicus: analysis of the enzymatic domains in the modular polyketide synthase. Gene 169: 9-16.
Baker, H., Sidorowicz, A., Sehgal, S. N., and Vezina, C. (1978) Rapamycin (AY-22,989), a new antifungal antibiotic. 111. In vitro and in vivo evaluation. Journal of Antibiotics 31: 539-545.
Boulay, A., Zumstein-Mecker, S., Stephan, C, Beuvink, I., Zilbermann, F., Haller, R., Tobler, S., Heusser, C, O'Reilly, T., Stolz, B., Marti, A., Thomas, G., Lane, H.A.,. 2004, Antitumor efficacy of intermittent treatment schedules with the rapamycin derivative RADO01 correlates with prolonged inactivation of ribosomal protein S6 kinase 1 in peripheral blood mononuclear cells. Cancer Res. 64(1), 252-61.
Boyd, M.R. and Paull, K.D., 1995. Some Practical Considerations and Applications of the National Cancer Institute In Vitro Anticancer Drug Discovery Screen. Drug Development Research 34, 91-109,
Brown, E.J. , Albers, M.W., Shin, T.B., lchikawa, K., Keith, C.T., Lane, W.S., and Schreiber, S. L. (1994) A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 369: 756-758.
Brunn, GJ. , Fadden, P., Haystead, TA, Lawrence, J. C. Jr.1997 The mammalian target of rapamycin phosphorylates sites having a (Ser/Thr)-Pro motif and is activated by antibodies to a region near its COOH terminus. J Biol. Chem^ 272(51), 32547-32550.
Brunn, GJ. , Williams, J., Sabers, C1 Wiederrecht, G., Lawrence, J. C, and Abraham, RT. (1996) Direct inhibition of the signaling functions of the mammalian target of rapamycin by the phosphoinositide 3-kinase inhibitors, wortmannin and LY294002. EMBO Journal 15: 5256-5267.
Carlson, R.P., Hartman, D.A., Tomchek, LA, Walter, T.L., Lugay, J. R., Calhoun, W., Sehgal, , S. N., Chang, J. Y. (1993). Rapamycin, a potential disease-modifying antiarthritic drug. J. Pharmacol. Exp. Ther. 266(2):1125-38.
Crowe A, Bruelisauer A, Duerr L, Guntz P1 Lemaire M. (1999) Absorption and intestinal metabolism of SDZ-RAD and rapamycin in rats. Drug Metab Dispos, 27(5), 627-32
Dengler W.A., Schulte J., Berger D. P., Mertelsmann R. and Fiebig HH. (.1995) Development of a propidium iodide fluorescence assay for proliferation and cytotoxicity assay. Anti-Cancer Drugs, 6:522-532.
DiLeIIa, A.G., and Craig, RJ. (1991 ) Exon organization of the human FKBP-12 gene: correlation with structural and functional protein domains. Biochemistry 30: 8512-8517.
Dudkin, L, Dilling, M. B., Cheshire, P.J., Harwood, F.C., Hollingshead, M., Arbuck, S. G., Travis, R., Sausville, E.A., Houghton, PJ. (2001). Biochemical correlates of mTCJR inhibition by the rapamycin ester CCI-779 and tumor growth inhibition. Clin. Cancer Res. 7(6): 1758-64
Evans D.A., Gage J. R. and Leighton J. L. (1992) Assymetric synthesis of calyculin A. 3. Assemblage of the calyculin skeleton and the introduction of a new phosphate monoester synthesis. J. Org. Chem., 57:1964-1966
Fiebig H. H., Dengler W.A. and Roth T. (1999) Human tumor xenografts: Predictivity, characterization, and discovery of new anticancer agents. In: Fiebig HH, Burger AM (eds). Relevance of Tumor Models for Anticancer Drug Development. Contrib. Oncol., 54: 29 - 50.
Findlay J.A, and Radios, L (1980) Canadian Journal of Chemistry 58:579.
Fishbein, T.M., Florman, S., Gondolesi, G., Schiano, T., LeLeiko, N., Tschernia, A., Kaufman, S. (2002). Intestinal transplantation before and after the introduction of sirolimus. Transplantation. 73(10): 1538-42.
Foey, A., Green, P., Foxwell, B., Feldmann, M., Brennan, F. (2002). Cytokine-stimulated T cells induce macrophage IL-10 production dependent on phosphatidylinositol 3-kinase and p70S6K: implications for rheumatoid arthritis. Arthritis Res. 4(1):64-70. Epub 2001 Oct 10.
Furniss B.S., Hannaford AJ., Smith P.W.G. and Tatchell A.R. (1989) Vogel's textbook of practical organic chemistry, 5th Ed, Pearson, Prentice Hall, Harlow, UK.
Gallant-Haidner HL, Trepanier DJ, Freitag DG, Yatscoff RW. 2000, "Pharmacokinetics and metabolism of sirolimus". Ther Drug Monit 22(1), 31-5.
Grass, G. M., Rubas, W., Jezyk, N., (1992) Evaluation of CACO-2 monolayers as a predictor of drug permeability in colonic tissues. FASEB Journal, 6, A1002.
Gregory, C.R., Huie, P., Billingham, M. E. and Morris, R.E. (1993). Rapamycin inhibits arterial intimal thickening caused by both alloimmune and mechanical injury. Its effect on cellular, growth factor and cytokine response in injured vessels. Transplantation 55(6): 1409-1418.
Gregory MA, Gaisser S, LiII RE, Hong H, Sheridan RM, Wilkinson B, Petkovic H, Weston AJ, Carletti I, Lee HL, Staunton J, Leadlay PF. (2004) "Isolation and characterization of pre- rapamycin, the first macrocyclic intermediate in the biosynthesis of the immunosuppressant rapamycin by S. hygroscopicus". Angew Chem lnt Ed Engl. 43(19), 2551-3
Gu, J, Ruppen ME, Cai P. (2005), "Lipase-Catalyzed Regioselective Esterification of Rapamycin: Synthesis of Temsirolimus (CCI-779). Org. Lett. 7(18): 3945-3948.
Guba, M., von Breitenbuch, P., Steinbauer, M., Koehl, G., Flegel, S., Hornung, M., Bruns, C. J., Zuelke, C, Farkas, S., Anthuber, M., Jauch, K.W., and Geissler, E.K. (2002) Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nature Medicine 8: 128-135.
Hardwick, J. S., Kuruvilla, F.G., Tong, J. K., Shamji, A.F., and Schreiber, S. L (1999) Rapamycin- modulated transcription defines the subset of nutrient-sensitive signaling pathways directly controlled by the Tor proteins. Proceedings of the National Academy of Sciences of the United States of America 96: 14866-14870.
Hentges, K.E., Sirry, B., Gingeras, A.C., Sarbassov, D., Sonenberg, N., Sabatini, D., and Peterson, A.S. (2001) FRAP/mTOR is required for proliferation and patterning during embryonic development in the mouse. Proceedings of the National Academy of Sciences of the United States of America 98: 13796-13801.
Huang, S. and Houghton, P. J., 2002. Mechanisms of resistance to rapamycins. Drug Resist. Update, 4(6), 378-391.
Jain, S., Bicknell, G. R., Whiting, P. H., Nicholson, M. L. (2001 ). Rapamycin reduces expression of fibrosis-associated genes in an experimental model of renal ischaemia reperfusion injury. Transplant Proc. 33(1-2):556-8.
Kahan, B. D., and Camardo, J. S. (2001) Rapamycin: Clinical results and future opportunities. Transplantation 72:1181-1193.
Kahan, B. D., Chang, J. Y., and Sehgal, S.N. (1991 ) Preclinical evaluation of a new potent immunosuppressive agent, rapamycin. Transplantation 52: 185-191.
Kirby, B., and Griffiths, C. E. M. (2001 ) Psoriasis: the future. British Journal of Dermatology 144:37-43.
Kirchner, G. I., Winkler, M., Mueller L, Vidal, C, Jacobsen, W., Franzke, A., Wagner, S., Blick, S., Manns M. P., and Sewing K.-F.(2000) Pharmacokinetics of SDZ RAD and cyclosporin including their metabolites in seven kidney graft patients after the first dose of SDZ RAD. British Journal of Clinical Pharmacology 50:449-454.
Kuo, CJ. , Chung, J. K., Fiorentino, D. F., Flanagan, W.M., Blenis, J., and Crabtree, G. R. (1992) Rapamycin selectively inhibits interleukin-2 activation of p70 S6 kinase. Nature 358: 70-73.
Langmann T1 Mauerer R, Zahn A, Moehle C, Probst M, Stremmel W, Schmitz G. (2003) "Realtime reverse transcription-PCR expression profiling of the complete human ATP-binding cassette transporter superfamily in various tissues". Clin Chem. 49(2), 230-8.
Lee, J-S, Paul!, K., Alvarez, M., Hose, C1 Monks, A., Grever, M., Fojo, AT., Bates, S. E., 1994. Rhodamine efflux patterns predict P-glycoprotein substrates in the National Cancer Institute drug screen. Molecular Pharmacology 46,627-638.
Li, AP. (1992) Screening for human ADME/Tox drug properties in drug discovery. Drug Discovery Today, 6, 357-366.
Lowden, P. A. S., (1997) Ph.D. Dissertation, University of Cambridge. "Studies on the biosynthesis of rapamycin".
Lyons, W.E., George, E.B., Dawson, T.M., Steiner, J. P., and Snyder, S. H. (1994) Immunosuppressant FK506 promotes neurite outgrowth in cultures of PC12 cells and sensory ganglia. Proceedings of the National Academy of Sciences of the United States of America 91:3191-3195.
Main, E. R. G., Fulton, K. F. & Jackson, S. E. (1998). The Context-Dependent Nature of Destabilising Mutations on The Stability of FKBP12. Biochemistry 37 , 6145-6153.
Main, E. R. G., Fulton, K. F. & Jackson, S. E. (1999a). Folding of FKBP12: Pathway of Folding and Characterisation of the Transition State. J. MoI. Biol. 291 , 429-444.
McAlpine, J. B,.Swanson S. J., Jackson, M., Whittem, D.N. (1991). Revised NMR assignments for rapamycin. Journal of Antibiotics 44: 688-690.
Meiering, E. M., Serrano, L. & Fersht, A. R. (1992). Effect of Active Site Residues in Bamase on Activity and Stability. J. MoI. Biol. 225, 585-589.
Morice, M. C, Serruys, P.W., Sousa, J. E., Fajadet, J., Ban Hayashi, E., Perin, M., Colombo, A., Schuler, G., Barragan, P., Guagliumi, G., Molnar, F., Falotico, R. (2002). RAVEL Study Group. Randomized Study with the Sirolimus-Coated Bx Velocity Balloon-Expandable Stent in the Treatment of Patients with de Novo Native Coronary Artery Lesions. A randomized comparison of a sirolimus-eluting stent with a standard stent for coronary revascularization. N. Eng.l J. Med. 346(23):1773-80.
Mothe-Satney, I., Brunn, G.J., McMahon, L. P., Capaldo, C. T., Abraham, R.T., Lawrence, J. C. Jr-. 2000 Mammalian target of rapamycin-dependent phosphorylation of PHAS-I in four (SfT)P sites detected by phospho-specific antibodies. J Biol Chem. 275(43), 33836-33843.
Myckatyn, T.M., Ellis, R.A., Grand, A.G., Sen, S. K., Lowe, J. B, 3rd, Hunter, D.A., Mackinnon, S. E. (2002). The effects of rapamycin in murine peripheral nerve isografts and allografts. Plast Reconstr. Surg. 109(7):2405-17.
Nave, B.T., Ouwens, D. M., Withers, DJ. , Alessi, D. R., and Sheperd, P.R. (1999) Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochemical Journal 344:427-431.
NCCLS Reference Method for Broth Dilution Antifungal Susceptibility Testing for Yeasts: Approved Standard M27-A, vol. 17 No. 9. (1997).
Paiva, N. L, Demain, A.L., and Roberts, M. F. (1991 ) Incorporation of acetate, propionate, and methionine into rapamycin By Streptomyces hygroscopicus. Journal of Natural Products 54: 167-177.
Paiva, N. L., Demain, A.L., and Roberts, M. F. (1993) The immediate precursor of the nitrogen- containing ring of rapamycin is free pipecolic acid. Enzyme and Microbial Technology 15: 581-585.
Perin, E C, (2005), "Choosing a Drug-Eluting Stent: A Comparison Between CYPHER and TAXUS", Reviews in Cardiovascular Medicine, 6 (suppl 1 ), ppS13-S21.
Persidis A. (1999), "Cancer multidrug resistance" Nat Biotechnol. 17: 94-5
Powell, N., Till, S., Bungre, J., Corrigan, C. (2001 ). The immunomodulatory drugs cyclosporin A, mycophenolate mofetil, and sirolimus (rapamycin) inhibit allergen-induced proliferation and IL-5 production by PBMCs from atopic asthmatic patients. J. Allergy Clin. Immunol. 108(6):915-7
Rabinovitch, A., Suarez-Pinzon, W.L., Shapiro, A.M., Rajotte, R. V., Power, R. (2002). Combination therapy with sirolimus and interleukin-2 prevents spontaneous and recurrent autoimmune diabetes in NOD mice. Diabetes. 51(3):638-45.
Raught, B., Gingras, A.C., and Sonenberg, N. (2001) The target of rapamycin (TOR) proteins. Proceedings of the National Academy of Sciences of the United States of America 98: 7037-7044.
Reather, J. A., (2000), Ph.D. Dissertation, University of Cambridge. "Late steps in the biosynthesis of macrocyclic lactones".
Reitamo, S., Spuls, P., Sassolas, B., Lahfa, M., Claudy, A., Griffiths, C. E.; Sirolimus European Psoriasis Study Group. (2001). Efficacy of sirolimus (rapamycin) administered concomitantly with a subtherapeutic dose of cyclosporin in the treatment of severe psoriasis: a randomized controlled trial. Br. J. Dermatol. 145(3):438-45.
Roth T., Burger A.M., Dengler W., Willmann H. and Fiebig H. H. (1999) Human tumor cell lines demonstrating the characteristics of patient tumors as useful models for anticancer drug screening. In: Fiebig HH, Burger AM (eds). Relevance of Tumor Models for Anticancer Drug Development. Contrib. Oncol., 54: 145 - 156.
Royrnans, D., and Siegers, H. (2001) Phosphaditidylinositol 3-kinases in tumor progression. European Journal of Biochemistry 268:487-498.
Schwecke, T., Aparicio, J. F., Molnar, I., Kδnig, A., Khaw, L.E., Haydock, S. F., Oliynyk, M., Caffrey, P., Cortes, J., Lester, J. B., Bδhm, GA, Staunton, J., and Leadlay, P.F. (1995) The biosynthetic gene cluster for the polyketide immunosuppressant rapamycin. Proceedings of the National Academy of Sciences of the United States of America 92: 7839-7843.
Sedrani, R., Cottens, S., Kalien, J., and Schuler, W. (1998) Chemical modifications of rapamycin: the discovery of SDZ RAD. Transplantation Proceedings 30: 2192-2194.
Sehgal, S. N., Baker, H., and Vezina, C. (1975) Rapamycin (AY-22,989), a new antifungal antibiotic II. Fermentation, isolation and characterization. The Journal of Antibiotics 28: 727-733.
Shepherd, P.R , Withers, DJ. , and Siddle K. (1998) Phosphoinositide 3-kinase: the key switch mechanism in insulin signalling. Biochemical Journal 333: 471-490.
Smith M. B. and March J. (2001) March's advanced organic chemistry, 5th Ed, John Wiley and Sons Inc., UK
Steiπer, J. P., Hamilton, G.S., Ross, D. T., Valentine, H. L1 Guo, H., Connolly, M.A., Liang, S., Ramsey, C, Li, J.-H.J., Huang, W., Howorth, P., Soni, R., Fuller, M., Sauer, H., Nowotnik, A.C., and Suzdak, P. D. (1997) Neutrophic immunophilin ligands stimulate structural and functional recovery in neurodegenerative animal models. Proceedings of the National Academy of Sciences of the United States of America 94:2019-2024.
Stein U, Jurchott K, Schlafke M, Hohenberger P. (2002) "Expression of multidrug resistance genes MVP, MDR1 , and MRP1 determined sequentially before, during, and after hyperthermic isolated limb perfusion of soft tissue sarcoma and melanoma patients". J Clin Oncol. 20(15):3282-92.
Szakacs G, Annereau JP, Lababidi S, Shankavaram U, Arciello A, Bussey KJ, Reinhold W, Guo Y, Kruh GD, Reimers M, Weinstein JN, Gottesman MM. 2004, "Predicting drug sensitivity and resistance: profiling ABC transporter genes in cancer cells". Cancer Cell. 6(2): 129-37.
Tanford, C. (1968). Protein Denaturation. Adv. Prot. Chem. 23, 121-282.
Tanford, C. (1970). Protein Denaturation. Part C. Theoretical models for the mechanism of denaturation. /Advances in Protein Chemistry 24, 1-95
Tang, S.J., Reis, G., Kang, H., Gingras, A.-C, Sonenberg, N., and Schuman, E.M. (2002) A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proceedings of the National Academy of Sciences of the United States of America 1:467-472.
Tee, A.R. and Proud, CG. 2002 Caspase cleavage of initiation factor 4E-binding protein 1 yields a dominant inhibitor of Cap-dependent translation and reveals a novel regulatory motif. MoI. Cell. Biol. 22, 1674-1683
Toshima K. and Tatsuta K. (1993) Recent progress in O-glycosylation methods and its application to natural product synthesis. Chem. Rev., 93:1503-1531.
Trepanier DJ, Gallant H, Legatt DF, Yatscoff RW. (1998), "Rapamycin: distribution, pharmacokinetics and therapeutic range investigations: an update". CHn Biochem. 31(5):345-51.
Vezina, C1 Kudelski, A., and Sehgal, S.N. (1975) Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. The Journal of Antibiotics 28: 721-726.
Volpe, D.A., Faustino, PJ., Yu, L.X., (2001) Towards standardisation of an in vitro method of drug absorption. Pharmacopeia! Forum, 27, 2916-2922.
Waller, J. R., and Nicholson, M. L (2001 ) Molecular mechanisms of renal allograft fibrosis. British Journal of Surgery 88: 1429-1441.
Warner, LM., Adams, LM., Chang, J.Y., Sehgal, S.N. (1992). A modification of the in vivo mixed lymphocyte reaction and rapamycin's effect in this model. Clin. Immunol. Immunopathol. 64(3):242-7.
Yu, K., Toral-Barza, L1 Discafani, C1 Zhang, W.G., Skotnicki, J., Frost, P., Gibbons, JJ. (2001 ) mTOR, a novel target in breast cancer: the effect of CCI-779, an mTOR inhibitor, in preclinical models of breast cancer. Endocrine-Related Cancer 8:249-258.
Zhu, J., Wu J., Frizell, E., Liu, S.L, Bashey, R., Rubin, R., Norton, P., Zern, M.A. (1999). Rapamycin inhibits hepatic stellate cell proliferation in vitro and limits fibrogenesis in an in vivo model of liver fibrosis. Gastroenterology. 117(5):1198-204.
【図面の簡単な説明】
【0202】
【図1】ラパマイシンの構造を示す。
【図2】39−デスメトキシラパマイシンの断裂経路を示す。
【図3】39−デスメトキシ−40−O−[2,2−ビス(ヒドロキシメチル)プロピオニル]ラパマイシンの断裂経路を示す。
【図4】39−デスメトキシ−40−O−[2−ヒドロキシエチル 3−ヒドロキシ−2−(ヒドロキシメチル)−2−メチルプロパノアート]ラパマイシンの断裂経路を示す。
【図5】27−O−デスメチル−39−デスメトキシ−40−O−[2,2−ビス(ヒドロキシメチル)プロピオニル]ラパマイシンの断裂経路を示す。
【図6】39−デスメトキシ−40−O−[2,2−ビス(ヒドロキシメチル)プロピオニル]ラパマイシン(A−黒塗三角)および39−デスメトキシ−40−O−(2−ヒドロキシ)エチルラパマイシン(B−黒塗り三角)のmTOR抑制活性をラパマイシン(黒塗り四角)と比較して示す。
【技術分野】
【0001】
本発明は新規な39−デスメトキシラパマイシン誘導体、その製造方法およびその用途に関する。別の観点において、本発明は39−デスメトキシラパマイシン誘導体を、ガンおよび/またはB細胞系悪性腫瘍の治療、免疫抑制の誘導または維持、臓器移植拒絶反応、移植片対宿主拒絶病、自己免疫疾患、炎症疾患、血管疾患および線維症の治療、神経再生の刺激または感染症の治療に使用することについての用途に関する。
【背景技術】
【0002】
ラパマイシン(rapamycin、シロリムス)(図1)は、ピペコリン酸ラクトン(Paivaら、1991年)に連結された1,2,3−トリカルボニル部分を有する、ストレプトマイセス・ヒグロスコピカス(Streptomyces hygroscopicus)NRRL5491により産生される親油性マクロライド(Sehgalら、1975;Vezinaら、1975;米国特許第3,929,992号;米国特許第3,993,749号)。本発明の目的のため、ラパマイシンは、Findlayら(1980年)または化情報索(第11版インデックス蓄積本、1982〜1986年、p60719CS)の番号付け慣例よりは、McAlpineら(1991年)の番号付け慣例により記述される。
【0003】
ラパマイシンは、その化合物によって示される広い活性スペクトルの故に顕著な薬理的価値を持つ。ラパマイシンは主としてカンジタ(Candida)属に対して、しかしまた糸状菌に対しても穏やかな抗菌活性を示す(Bakerら、1978年;Sehgalら、1975年;Vezinaら、1975年;米国特許3,929,992;米国特許3,993,749)。ラパマイシンは、多くの細胞の種類において信号伝達経路を標的にすることにより、たとえば細胞サイクルのG1からS相への成長を許容するシグナリング経路を示すことにより細胞増殖を示す(Kuoら、1992年)。T細胞内でラパマイシンはIL−2受容体およびその後の免疫抑制をもたらすT細胞自己増殖からのシグナリングを示す。ラパマイシンは多くの哺乳類細胞種の増殖を抑制する(Brunnら、1996年)から、ラパマイシンのこの抑制効果はT細胞に限らない。ラパマイシンは、したがって、臓器移植拒絶反応の予防や自己免疫疾患の治療において確立されたまたは予測される治療的適用を持った有力な免疫抑制剤である(Kahanら、1991年)。40−O−(2−ヒドロキシ)エチル−ラパマイシン(SDZ RAD、RAD001、Certican、エベロリムスeverolimus)は免疫抑制的薬理効果を示すラパマイシンの半合成相同物であり、抗ガン剤としての研究も進められている(Sedrani,Rら、1998年;Kirchnerら、2000年;米国特許5,665,772、Boulayら、2004年)。この薬剤は欧州で2003年に免疫抑制剤として承認された。ラパマイシンエステル誘導体CCI−779(Wyeth−Ayerst)は生体外で細胞成長を示し、生体内で腫瘍成長を示す(Yuら、2001年)。CCI−779は潜在的抗ガン剤として現在第2相臨床試験が行われている。慢性尋常性乾癬の治療(KirbyおよびGriffiths、2001年)、PC12細胞内神経突起伸長刺激などの潜在的効用(Lyonsら、1994年)、機械的損傷後の血管および平滑筋細胞によるサイトカインに対する増殖反応の抑制(Gregoryら、1993年)におけるラパマイシンの価値、および同種移植性線維症(allograft fibrosis)の予防におけるその役割(WallerおよびNicholson、2001年)が熱心な研究領域となっている(KahanおよびCamardo、2001年)。最近の報告によれば、ラパマイシンは臓器移植患者に対する長期免疫抑制治療において、他の免疫抑制レジメより低い発ガン率を示し、この発ガン率低下は血管新生(angiogenesis)の抑制によるとされている(Gubaら、2002年)。イムノフィリンリガンドの神経向性作用はそれらの免疫抑制活性とは無関係であり(Steinerら、1997年)、親出願WO01/03692に要約される成熟ステロイド受容体複合体の分裂によって神経細胞刺激が増進されると報じられた。催奇的影響と共に高脂血症や血小板減少症などの副作用が報告されている(Hentgesら、2001年;KahanおよびCamardo、2001年)。
【0004】
ラパマイシンのポリケチドバックボーンは、I型ポリケチド合成酵素を含む非常に大きな多機能タンパク質(rapPKS、Paivaら、1991年)により、全部で7個のプロピオン酸エステルおよび7個の酢酸エステルのユニットをシキマート由来シクロヘキサンカルボン酸スターターユニットにhead−to−tail縮合することにより合成される。L−リシン由来アミノ酸、ピペコリン酸がアミド架橋を経由してポリケチドバックボーンの最終酢酸エステルに縮合され(Paivaら、1993年)、次いでラクトン化によりマクロ環を形成する。
【0005】
3つのラパマイシンPKS遺伝子、NRPSエンコード遺伝子およびフランキング後期遺伝子配列および対応ポリペプチドの各々についてのヌクレオチド配列がAparicioら、1996およびSchweckeら、1995年によって同定され、受入番号X86780の下にNCBIに寄託され、この配列に対する訂正が最近WO04/007709に発表された。
【0006】
ラパマイシン生合成クラスタの最初の無酵素製品がプレラパマイシンである(WO04/007709、Gregoryら、2004年)。この完全にプロセス化されたラパマイシンの製造は、ラパマイシン後期遺伝子RapJ、RapN、RapO、RapM、RapQおよびRapIにエンコードされた酵素によるポリペプチド/NRPSコアの追加的処理を必要とする。
【0007】
今日までに特性付けられているラパマイシンの薬理作用は、FKBPと名付けられたサイトソル受容体との相互作用によるメディエートされるものと考えられている。真核T細胞の主要な細胞内ラパマイシン受容体はFKBP12であり(DiLellaおよびCraig、1991年)、産生される複合体が標的タンパク質と特異的に相互作用して細胞の信号伝達カスケードを示す。
【0008】
ラパマイシン−FKBP12複合体の標的は酵母内においてTOR(ラパマイシン標的タンパク質)として同定され(Alarconら、1999年)、哺乳類タンパク質はFRAP(FKBP−ラパマイシン付随タンパク質)ないしmTOR(ラパマイシンの哺乳類標的タンパク質)として知られている(Brownら、1994年)。
【0009】
mTOR伝達経路と神経単位に局所化されるタンパク質向性との関連、翻訳調整に包含されるタンパク質のリン酸化状態に対するその作用、転写および翻訳レベルにおける翻訳メカニズムの化合物存在性、アミノ酸ペルミアーゼ活性、および代謝経路に含まれる多くの酵素の転写調整が記述されている(Raughtら、2001年)。ラパマイシン感応性シグナル経路はまた、胎児の脳発達、学習および記憶形成に重要な役割を果たすものと考えられている(Tangら、2002年)。酵母内のTORタンパク質についての研究は、栄養感受性シグナル経路を調節する上でも役割を果たしていることを示した(Hardwickら、1999年)。同様に、mTORはタンパク質キナーゼB(akt)の活性に対する直接標的物質であり、インシュリンの信号伝達において主要な役割を果たすものとして確認されている(Shepherdら、1998年;Naveら、1999年)。哺乳類のTORはアクチン細胞骨格の分極化および翻訳開始の制御に関係していた(Alarconら、1999年)。mTORなどのホスファチジルイノストール−3−キナーゼは、細胞周期進行、付着、細胞生存および血管新生のような幾つかの腫瘍病原の見地で機能的である(RoymansおよびSiegers、2001年)。
【0010】
ラパマイシンおよびラパマイシン相同物の薬物動態学的研究により、溶液中でより安定的であり、代謝攻撃に対してより抵抗性を持ち、および/または細胞膜透過性が向上され且つエフラックスが減少されていて、より優れた経口バイオアベイラビリティを示すような新規ラパマイシン化合物の開発に対する要求が実証された。
【0011】
分子の化学的に利用可能な部位を用いて合成された一群のラパマイシン相同物が報告された。下記化合物に対する記述には、図1に記述されたラパマイシン分子の付番方式を適用させた。相同化または置換のための分子上の化学的に利用可能な部位には、C40およびC28ヒドロキシル基(例えば、米国特許第5,665,772号、米国特許第5,362,718号)、C39およびC16メトキシ基(例えば、WO96/41807号、米国特許第5,728,710号)、C32、C26およびC9ケト基(例えば、米国特許第5,378,836号、米国特許第5,138,051号、米国特許第5,665,772号)が含まれる。トリエンを標的にしたC17、C19および/またはC21での水添は、抗真菌活性は維持するが、免疫抑制特性の相対的損失をもたらした(例えば、米国特許第5,391,730号、米国特許第5,023,262号)。分子の安全性に対する相当な改善(例えば、C32、C40および/またはC28でのオキシムの形成、米国特許第5,563,145号、米国特許第5,446,048号)、代謝作用攻撃に対する耐性(例えば、米国特許第5,912,253号)、生体利用可能性(例えば、米国特許第5,221,670号、米国特許第5,955,457号、WO98/04279号)および前駆薬物の製造(例えば、米国特許第6,015,815号、米国特許第5,432,183号)が、相同化を通じて達成された。
【0012】
しかしながら、より優れた代謝安定性、より優れた細胞膜透過性および/または減少されたエフラックス率を備えたより広範な群のラパマイシン誘導体のための要求がなお存在している。そのようなラパマイシン誘導体は広範な病状の治療により大きな有用性を持つであろう。本発明は、より優れた代謝安定性、より優れた細胞膜透過性および/または減少されたエフラックス率を備え、および/またはラパマイシンとは異なる細胞抑制プロファイルを備えた一群の39−デスメトキシラパマイシン誘導体を提供する。この化合物は医療において、特にガンおよび/またはB細胞系悪性腫瘍の治療、免疫抑制の誘導または維持、臓器移植拒絶反応、移植片対宿主拒絶病、自己免疫疾患、炎症疾患、血管疾患および線維症の治療、神経再生の刺激または感染症の治療に有効である。
【発明の開示】
【0013】
本発明はラパマイシンの39−デスメトキシ誘導体、該化合物の調製方法、その中間産生物および該化合物の医療的用途のための方法に関する。
【0014】
その最も広い観点において、本発明は、40−ヒドロキシ位置がカルボン酸エステル、エーテル、リン酸エステル、ホスフィン酸エステル、アセタールまたはグリコシルとして誘導化されたことを特徴とするラパマイシンの39−デスメトキシ誘導体を提供する。
【0015】
本発明化合物の代謝安定性、細胞膜透過性、エフラックスおよびバイオアベイラビリティは下記のようにして試験することができる。
【0016】
39−デスメトキシラパマイシンがカルボン酸エステル、エーテルまたはアセタールとして誘導化される場合、誘導基は12個を越えない炭素原子を含むことが好ましい(特に7個以下、さらには5個以下の炭素原子数が好ましい)。好ましくは、それは−CF2PO(OH)2、−PO(OH)2、−COOH、−OHおよび−NH2から、好ましくは−COOHおよび−OH、より好ましくは−OHから選ばれる一以上の官能基(特には2以上の官能基)を含む。
【0017】
39−デスメトキシラパマイシンがグリコシル基由来のアセタールとして誘導化される場合、各グリコシルは好ましくは12個を越えない炭素原子を含む(特に7個以下、さらには6個以下の炭素原子数が好ましい)糖またはグリコシドから形成することが好ましい。好ましくは、それは−COOH、−OHおよび−NH2から、好ましくは−NH2および−OH、より好ましくは−OHから選ばれる一以上の官能基(特には2以上の官能基)を含む。
【0018】
39−デスメトキシラパマイシンがリン酸エステルとして誘導化される場合、好ましくはアルキル基は4個を越えない炭素原子を含む。
【0019】
39−デスメトキシラパマイシンがホスフィン酸エステルとして誘導化される場合、好ましくはアルキル基は4個を越えない炭素原子を含むことが好ましく、一例はホスフィン酸で形成されるエステルである。
【0020】
誘導部分の具体例を下記に示す。
【0021】
より具体的な観点において、本発明は、化1による39−デスメトキシラパマイシン誘導体またはその薬学的に許容可能な塩を提供する。
【0022】
【化1】
ここで、
Xは結合またはCH2を表し、
R1はケト基または(H,H)を表し、
R2はOHまたはOMeを表し、
R3はH、OHまたはOMeを表し、
R4およびR5は各々独立してHまたはOHを表し、
R6は−R7、−C(O)R7、−(CH2)2−O−[CR21R22−O]a−C(O)−R23;−CR21R22−O−C(O)−R23;POR19R20、−PO(OR19)(OR20)またはY−R15を表し、
R7は−(CR8R9)m(CR10R11)pCR12R13R14を表し、
R8およびR9は各々独立してC1−C4アルキル、C2−C4アルケニルまたはC2−C4アルキニルを表し、これらのいずれの基においても−PO(OH)2、CF2PO(OH)2、−OH、−COOHまたは−NH2で任意に置換可能であり、あるいはR8およびR9は各々独立してH、トリフルオロメチルまたはFを表し、
R10、R11、R12、R13およびR14は各々独立してC1−C4アルキル、C2−C4アルケニルまたはC2−C4アルキニルを表し、これらのいずれの基においても−PO(OH)2、CF2PO(OH)2、−OH、−COOHまたは−NH2で任意に置換可能であり、あるいはR10、R11、R12、R13およびR14は各々独立してH、−(CR8R9)qNH2、−(CR8R9)qOH、CF3、F、COOHから選択することができ、あるいはR10とR11またはR12とR13またはR13とR14は一緒になってそれらが結合している炭素と共にC3−C6シクロアルキルまたはN、OおよびSから選ばれる一以上のヘテロ原子を含み5個までの−(CR8R9)qOH、−(CR8R9)qNH2または−COOH基で任意に置換可能である3〜6員ヘテロアルキル環を形成しており、
ここでYは結合、−C(O)−O−または−(CH2)2−O−C(O)−O−であり、
R15は化2、化3または化4のいずれかであり、
【化2】
【化3】
【化4】
R16は各々独立してHまたはOHであり、
R17はH、OHおよびNH2から独立して選ばれ、
R18はH、−CH3、−CH2OHおよび−COOHから選ばれ、
ただしR16、R17およびR18から選ばれる2以上の基はHまたはCH3を表し、
R19およびR20は各々独立してHまたはC1−C4アルキルをあらわし、あるいはR19およびR20の両方で=CH2を示し、
R21はH、CH3から独立して選ばれ、
R22はH、−CH3、−CH=CH2、−CH2Cl、−CHCl2、−CCl3、−CH(OH)Me、−CH2OH、−CH2CH3、−CH(Cl)Meから独立して選ばれ、
R23は独立してR7、Y−R15または5または6員アリルまたはヘテロアリル環であって任意にOH、F、Cl、Br、NO2およびNH2から選ばれる1〜3個の基で置換されるもの、
aは0または1を表し、
m、pおよびqは各々独立して0〜4の整数を表し、
ただしR7部分は12個を超える炭素原子を含有せず、-PO(OH)2、-CF2PO(OH)2、-COOH、OHまたはNH2から選ばれる1以上の官能基を有し、あるいはそれらの薬学的に許容可能な塩である。
【0023】
上記構造は代表的な互変異体を示しており、本発明は化1の化合物のあらゆる互変異体、たとえばエノール化合物が示される場合のケト化合物およびその逆を包含する。
【0024】
具体的な立体異性体が特定的に示されない限り(たとえば、構造式中の立体中心における太字または点線の結合により、構造式中にEまたはZ配置を有する二重結合が示されることにより、または立体科学を示す術後を用いることにより)、すべての立体異性体は純粋化合物としてであれその混合物としてであれ、本発明の範囲内に含まれる。そうでないように示されない限り、個々のエナンチオマー、ジアステレオマー、幾何異性体、およびこれらの組み合わせおよび混合物はすべて本発明に包含される。多形結晶体、溶媒和物および水和物もまた本発明に包含される。
【0025】
さらに別の観点において、本発明は化1で示される39-デスメトキシラパマイシン誘導体またはその薬学的に許容可能な塩を薬剤として用いる用途を提供する。
【0026】
定義
冠詞“a”または“an”はここでは一または一以上(すなわち少なくとも一)の文法上の対象物を指すために用いられる。一例として、“an analogue”は一つの相同物または一以上の相同物を意味する。
【0027】
ここで用いる「相同物」の語は、ある化合物と構造的に近似しているが組成において(たとえば1個の原子が他に置換され、あるいは特定の1個の官能基が存在または不存在)若干異なる化合物を意味する。
【0028】
特に、「39-デスメトキシラパマイシン相同物」の語はWO2004/007709の方法によって製造される、および/または化1で示される39−デスメトキシラパマイシン化合物である。この化合物はまた「親化合物」として参照され、これらの語を本出願において互換的に使用する。本出願において「39−デスメトキシラパマイシン相同物」の語は39−デスメトキシラパマイシンそれ自体に対する参照を含む。
【0029】
ここで用いる「誘導体」の語は、半合成有機化学により親化合物から変性された化合物を指す。
【0030】
特に、「39−デスメトキシラパマイシン誘導体」の語は、化1で示される39−デスメトキシラパマイシン誘導体、またはその薬学的に許容され得る塩を意味し、39−デスメトキシラパマイシン相同物の半合成変性によって製造される。該化合物はまた「本発明化合物」または「ラパマイシンの39−デスメトキシ誘導体」として参照され、本出願においてこれらの語を互換的に使用する。
【0031】
ここで用いる「自己免疫疾患」の語は、限定的ではないが、全身性エリテマトーデス(SLE)、関節リウマチ、重症筋無力症および多発性硬化症を含む。
【0032】
ここで用いる「炎症疾患」の語は、限定的ではないが、乾癬、皮膚炎、湿疹、脂漏、炎症性腸疾患(限定的ではないが潰瘍性大腸炎およびクローン病を含む)、肺炎(限定できではないが喘息、慢性閉塞性肺疾患、肺気腫、急性呼吸窮迫症候群および気管支炎を含む)、リウマチ性関節炎および眼ブドウ膜炎を含む。
【0033】
ここで用いる「ガン」の語は、限定的ではないが、皮膚や限定的ではないがたとえば胸腺、前立腺、肺、腎臓、膵臓、胃または腸などの体器官中の悪性の細胞成長を意味する。ガンは隣接組織に侵入して、離れた器官、たとえば非限定的例示として骨や肝臓、肺または脳などに拡がっていく(転移)傾向がある。ここで用いる「ガン」の語は、非限定的な例示としてメラノーマ、リンフォーマ、白血病、繊維肉腫、横紋筋肉腫および悪性肥満細胞腫などの転移性腫瘍細胞タイプと、非限定的な例示として結腸大腸ガン、前立腺ガン、小細胞肺ガンおよび非小細胞肺ガン、乳ガン、膵臓ガン、膀胱ガン、腎臓ガン、胃ガン、グリオブラストーマ、肝ガン、卵巣ガンなどの細胞ガンタイプの両方を含む。
【0034】
ここで用いる「B細胞系悪性腫瘍」の語は、慢性リンパ性白血病(CLL)、多発性骨髄腫、非ホジキンリンパ腫(NHL)などの疾患群を含む。これは血液または造血器官の新生物疾患である。これは骨髄および免疫システムの機能不全を生じさせ、宿主を感染や出血しやすいものにする。
【0035】
ここで用いる「血液疾患」の語は、限定的ではないが、超増殖性血管障害(レステノシスおよび血管閉塞など)、アテローム性動脈硬化症、心血管疾患、脳血管障害および末梢性血液疾患(冠状動脈疾患、動脈硬化症、アテローム性動脈硬化症、非アテローム性動脈硬化症または血管壁損傷など)を含む。
【0036】
ここで用いる「神経再生」の語は神経細胞成長刺激を意味し、神経突起伸長および神経細胞の機能的回復を含む。神経再生がその治療に顕著に有効であろうとされる疾患および疾病は、限定的ではないが、アルツハイマー病、パーキンソン病、ハンチントン舞踏、筋萎縮性側索硬化症、三叉神経痛、舌咽神経痛、ベル麻痺、筋ジストロフィー、脳卒中、進行性筋萎縮症、進行性球脊髄性筋萎縮症、変形性頸椎症、ギラン・バレー症候群、痴呆症、末梢性神経障害および末梢神経損傷を含む、これらは身体的損傷(脊髄損傷またはトラウマ、座骨神経や顔面神経の外傷や損傷など)によって生ずるか、あるいは病状(糖尿病など)によって生ずるかを問わない。
【0037】
ここで用いる「線維症」の語は、細胞外マトリックスの過剰産生に関連する疾患を意味し、(限定的ではないが)サルコイドーシス、ケロイド、糸球体腎炎、末期腎臓疾患、肝繊維化症(非限定的例示として肝硬変、アルコール性肝障害および脂肪肝を含む)、慢性腎障害、手術創癒着、動脈硬化症、心臓繊維化症、肺線維症(非限定的例示として突発性肺線維症および突発性繊維性肺胞炎を含む)、黄斑変性症、新生児および糖尿病網膜症、化学療法または放射線誘発線維症を含む。
【0038】
ここで用いる「移植片対宿主拒絶病」の語は、同種幹細胞/骨髄移植後に見られる合併症を意味する。それは、ドナーから与えられた感染と闘う細胞が患者の体を異なるまたは異質であると認識したときに発症する。これらの感染攻撃細胞は、感染を攻撃するかの如く、患者の体内組織を攻撃する。移植片対宿主拒絶病は、それが移植後最初の100日以内に発症すれば急性とされ、移植後100日経過後に発症すれば慢性とされる。これに関連する主な組織は肝臓、消化管および皮膚である。慢性移植片対宿主拒絶病は幹細胞/骨髄移植患者の約10〜40%に発生する。
【0039】
ここで用いる「バイオアベイラビィリティ」の語は、投与後の薬剤または他の物質が生物学的活性領域で吸収されまたは有効に働く程度ないし割合を意味する。この性質は、化合物の可溶性、消化管への吸収率、タンパク質結合および代謝の程度などの数多くの要因に依存する。ここに当業界において当業者に良く知られている様々なバイオアベイラビィリティ試験を記述する(さらにTrepanierら、1998年、Gallant−Haidnerら、2000年も参照されたい)。
【0040】
ここで用いる「水溶性」の語は、水性媒体、たとえばpH7.4におけるリン酸緩衝サリン(PBS)中の溶解性を意味する。
【0041】
化1の化合物のような本発明化合物の薬学的に許容可能な塩は、薬学的に許容可能な無機または有機の酸または基から形成される慣習的な塩を含み、さらに第四アンモニウム酸添加塩をも含む。好適な酸塩についてより詳しく例示すれば、塩化水素、臭化水素、硫酸、リン酸、硝酸、過塩素酸、フマル酸、酢酸、プロピオン酸、コハク酸、グリコール酸、蟻酸、乳酸、マレイン酸、酒石酸、クエン酸、パルモイン酸、マロン酸、ヒドロキシマレイン酸、フェニル酢酸、グルタミン酸、安息香酸、サリチル酸、フマル酸、トルエンスルホン酸、メタンスルホン酸、ナフタリン−2−スルホン酸、ベンゼンスルホン酸、ヒドロキシナフタリン酸、よう化水素酸、リンゴ酸、ステロイン酸、タンニン酸などである。シュウ酸などの他の酸はそれ自身は薬学的に許容可能なものではないが、塩として調製することにより本発明化合物およびその薬学的許容可能塩を得るための中間生成物として有用である。より詳しく好適な基酸を例示すれば、ナトリウム、リチウム、カリウム、マグネシウム、アルミニウム、カルシウム、亜鉛、N−N’−ジベンジルエチレンジアミン、クロロプロカイン、コリン、ジエタノールアミン、エチレンジアミン、N−メチルグルカミンおよびプロカインの塩である。以降において本発明による化合物を参照するときは化1の化合物とその薬学的許容可能な塩の両方を含む。
【0042】
アルキル、アルケニルおよびアルキニル基は直鎖または分枝である。
【0043】
C1−C4アルキル基の例はメチル、エチル、n−プロピル、i−プロピルおよびn−ブチルを含む。
【0044】
C2−C4アルケニル基の例はエテニルおよび2−プロペニルを含む。
【0045】
C2−4アルキニル基の例はエチニルを含む。
【0046】
C3−C6シクロアルキル基は3〜6個の炭素原子を含むシクロアルキル環を意味し、それらは任意に分枝していても良い。例示すればシクロプロピル、シクロブチル、メチル−シクロブチル、シクロペンチルおよびシクロヘキシルである。
【0047】
N、OおよびSから選ばれる1以上のヘテロ原子を含む3〜6員ヘテロアルキル環は、1個または2個のヘテロ原子を含み、特に1個のヘテロ原子を含む。例示すればフラン、ピラン、オキセタン、オキシラン、ピペリジン、ピロリジン、アゼチジン、アジリジン、チイラン、チエタン、チオフェン、チオピランおよびモルフォリンを含む。
【0048】
3〜6員ヘテロアルキル環についての任意置換の例は、−OH、−CH2OH、NH2、CH2NH2およびCOOHを含む。特に3〜6員ヘテロアルキル環は置換されていないか、あるいは1個または2個、たとえば1個の置換基で置換されている。
【0049】
発明の記述
本発明は上述の39−デスメトキシラパマイシン誘導体、該化合物の調製方法、その中間産生物および該化合物の医療的用途を提供する。
【0050】
好ましくはR7は7個以下、特に5個以下の炭素原子を含む。
【0051】
R7は好ましくは−PO(OH)2、−OH、−COOHおよび−NH2から、より好ましくは−OH、−COOHまたは−NH2から、特に−OHおよび−COOHから、とりわけOHから選ばれる1以上の官能基を含む。好ましくはR7は2以上の置換基、たとえば2つのOH基を含む。
【0052】
好ましくはXはCH2を表す。
【0053】
好ましくはaは0を表す。
【0054】
好ましくはpは0または1を表す。
【0055】
好ましくはmは0または1を表す。
【0056】
好ましくはqは0、1または2を表す。
【0057】
好ましくはR11はHを表す。好ましくはR12はHを表す。
【0058】
好ましくはR13はHまたはOHを表す。
【0059】
pが1を表すとき、好ましくはR10はMe、OHまたはCH2OHを表す。
【0060】
pが1を表すとき、好ましくはR11はMe、HまたはCH2OHを表す。
【0061】
mとpが共に0を表すとき、好ましくはR12とR13は共にHを表し、R14は−(CR8R9)q−OHを表し、ここでqは0または1を表し、R8とR9は共にHを表す。
【0062】
pが1を表しmが0を表すとき、好ましくはR10とR11は共にHを表し、R12はHを表し、R13はH、OHまたはNH2を表し、R14は−(CR8R9)q−OHを表し、ここでqは0または1を表し、R8とR9は共にHを表す。
【0063】
R6が−POR15R16を表すとき、好ましくはR15とR16は共にCH3を表すかまたは共にCH2CH3を表す。
【0064】
好ましくはR6は、ヒドロキシ酢酸、3−ヒドロキシ−2,2−ジメチルプロピオン酸、2,3−ジヒドロキシプロピオン酸、3−ヒドロキシ−2−ヒドロキシメチルプロピオン酸または2,2−ビス(ヒドロキシメチル)プロピオン酸によるエステル形成由来の残留物を表す。
【0065】
化合物の一例セットにおいてR6はC(O) R7を表す。
【0066】
好ましくはR7は、ヒドロキシ酢酸、3−ヒドロキシ−2,2−ジメチルプロピオン酸、2,3−ジヒドロキシプロピオン酸、3−ヒドロキシ−2−ジヒドロキシメチルプロピオン酸および2,2−ビス(ヒドロキシメチル)プロピオン酸からなる群から選ばれる酸、特に2,2−ビス(ヒドロキシメチル)プロピオン酸で大環状アルコールを縮合することにより形成される部分である。
【0067】
R15が化2を表すとき、この部分の例は、(1)グルコース(すなわちR18がCH2OHを表し、各R16およびR17がOHを表す)、たとえばD−グルコース、(2)グルコサミン(すなわちR18がCH2OHを表し、各R16がOHを表し、R17がNH2を表す)、たとえばD−グルコサミン、(3)グルクロン酸(すなわちR18がCOOHを表し、各R16およびR17がOHを表す)、たとえばD−グルクロン酸、および(4)アラビノース(すなわちR18がHを表し、各R16およびR17がOHを表す)、たとえばD−アラビノースでアセタールを形成することにより形成される部分を含む。
【化2】
【0068】
R15が化3を示すとき、この部分の例は、フルクトース(すなわちR16が各々OHを表す)、たとえばD−フルクトースでアセタールを形成することにより形成される部分を含む。
【化3】
【0069】
R15が化4を示すとき、この部分の例は、グルクロン酸(すなわち各R16がOHを表す)、たとえばD−グルクロン酸でエステルを形成することにより形成される部分を含む。
【化4】
【0070】
概して言えば、本発明化合物は化5の39−デスメトキシラパマイシン相同物の半合成誘導により産生される。
【化5】
【0071】
すなわち、化1の化合物またはその薬学的に許容可能な塩の調製方法は、
(a)化5の39−デスメトキシラパマイシン相同物(RAはHまたは(CH2)2−OH)またはその保護された誘導体を、HO−R6またはR6の活性化された誘導体と反応させる工程、
(b)化1の化合物またはその塩を他の化1の化合物またはその薬学的に許容され得る塩に形質転換する工程、または、
(c)保護された化1の化合物を脱保護する工程を含む。
【0072】
ここで用いる「活性化された誘導体」の語は、非限定的例示として、エステルの場合であればカルボン酸、ハロゲン化アシル、混合酸無水物、対称酸無水物またはカルボン酸エステル、エーテルの場合であればハロゲン化アルキル、アルキルメシレート、アルキルトリフレート、アルキルトシレートまたは他の適当な活性化アルキル誘導体、リン酸エステルまたはホスホン酸エステルの場合であればクロロホスフェート、ジアルキルシアノホスフェート、ジアルキルジアルキルホスホラミデートまたはクロロホスファイト、グリコシル基由来アセタールの場合であればハロゲン化グリコシル、チオグリコシド、1−O−アシルグリコシド、オルトエステル、1−Oまたは1−S炭酸塩、トリクロロイミデート、4−ペンテニルグリコシド、グリコシルリン酸エステル、1−O−スルホニルまたは1−O−シリル化グリコシドなどのグリコシルドナーを用いることを意味する。
【0073】
工程(a)において、化5の39−デスメトキシラパマイシン相同物はWO2004/007709に記述され、さらには後述実施例に述べるようにして調製することができる。
【0074】
ここに記述される具体的方法および参照に加えて、当業者は、非限定的例示としてVogelの実際有機化学の教本(Furnissら、1989年)やMarchの高等有機化学(SmithおよびMarch、2001年)にょうな合成法のための標準的教本参照を顧慮することができる。
【0075】
さらに、当業者に知られている多くの標準的ヒドロキシ保護ストラテジーの一つによってヒドロキシル基を保護することができる。ヒドロキシル基は、限定的ではないがたとえば置換アルキルエーテル、置換ベンゼンエーテル、シリルエーテルなどのエーテルを生成させることによって保護することができる。好適には、シラン(限定的ではないが塩化シリル、シリルトリフラートを含む)の活性形態を適当な塩基の存在の下で39−デスメトキシラパマイシンと反応させることにより、限定的ではないがたとえばトリメチルシリル、トリエチルシリル、t−ブチルジメチルシリル、t−ブチルジフェニルシリルなどのシリルエーテルを生成させる。次いで酸加水分解またはフッ素酸アシスト開裂のいずれかにより保護基を除去することができる。アセトン誘導体の縮合に基づいて、1,2−ジオールがアセトニドとして保護され得る。これは酸触媒によって除去され得る。
【0076】
化5の39−デスメトキシラパマイシン相同物はさらなる半合成(すなわち工程(a))のためのテンプレートとしても用いることができる。C−40におけるペンダントヒドロキシル基は、当業者に知られている多くの合成的形質転換を介して、たとえばアシル化、アルキル化、グリコシル化またはリン酸化などによって感応化され得る。
【0077】
工程(a)において、R6が−C(O)R7またはY−R15であって、ここでR15が化4を表し、Yが結合であるとき、ヒドロキシエステルの形成ないしO−アシル化は、化5の化合物のヒドロキシルが好ましくは活性化状態である対応カルボキシル酸、たとえば化6または化7の化合物と反応することによって、あるいは化8の化合物と反応することによって、メディエートされ得る。
【化4】
【化6】
【化7】
【化8】
【0078】
ここで、Wはカルボキシル酸を求核攻撃に対して活性化する基である。カルボキシル酸は非限定的例示としてハロゲン化アキル(たとえばW=Cl)、混合酸無水物(すなわちW=OC(O)R’)、対称酸無水物(W=OC(O)R7)またはカルボキシルエステル(すなわちW=OR’)の形成により活性化され得る。
【0079】
化6、化7または化8の化合物はそれらの入手可能なカルボキシル酸から当業者に公知の標準的方法を用いて調製することができ、一つの特定の観点において、R7が−(CR8R9)m(CR10R11)pCR12R13R14を表す化6の化合物は米国特許5,362,718、米国特許「5,665,772または欧州特許0663916に記述されるような方法を用いて調製することができる。
【0080】
好ましくは39−デスメトキシラパマイシン相同物を有機媒体中で塩基の存在下で酸塩化物または混合酸無水物と反応させる。塩基の非限定的例示は、ピリジン、4,4−ジメチルアミノピリジン(DMAP)、2,6−ルチジン、2,6−ジ−タート−ブチルピリジン、トリエチルアミン、ジイソプロピルエチルアミン、他のトリアルキルアミン、1,8−ジアザビシクロ[5.4.0]ウンデク−7―エン(DBU)または1,5−ジアザビシクロ[4.3.0]ノン−5―エン(DBN)を含む。
【0081】
工程(a)において、R6が−C(O)R7またはY−R15であって、ここでR15が化4を表し、Yが−C(O)O−または−(CH2)2−OC(O)O−を表すとき、該ヒドロキシエステルの形成は、化5の化合物または40−O−(ヒドロキシエチル)−化5化合物のヒドロキシル基が化9の化合物のような活性化炭酸エステルを形成する試薬と反応させることを要求する。
【化4】
【化9】
【0082】
化9においてTは結合または−O(CH2)2−であり、R24はアルキルまたはアリル基、好ましくはアリル基、特にパラ−ニトロフェニル基である。
【0083】
化9の化合物は次いで化6、化7または化8の化合物と反応して、炭酸塩リンカー(carbonate linker)を介して、R6が40−ヒドロキシル基または40−O−(ヒドロキシルエチル)基に付着した化合物を生成する(WO2004/101583)。
【0084】
同様に、39−デスメトキシラパマイシン相同物を適当に選択した活性化されたアルキル誘導体と反応させることにより、39−デスメトキシラパマイシン相同物をC−40において異なるヒドロキシエステルで誘導することができ、これによって40−O−アルキル−39−デスメトキシラパマイシン誘導体を生成する。活性化されたアルキル基は、非限定的例示としてハロゲン化アキル(RCl、Rl、RBr)、アルキルメシレート(ROS(O)2CH3)、アルキルトリフレート(ROS(O)2CF3)、アルキルトシレート(ROS(O)2PhMe)の形成を含む多くの方法の一つにより活性化されたアルキル基を指す。活性化されたアルキル基は次いで有機溶媒中で適当な塩基の存在下において39−デスメトキシラパマイシン相同物と反応する。反応条件を最適化する標準的方法が当業者によって採用されることができ、これにより他の反応位置におけるアルキル化を防止する。
【0085】
同様に、39−デスメトキシラパマイシン相同物をリン酸化することができ、これによりリン酸エステルの脱保護の後に40−O−ホスフォ−39−デスメトキシラパマイシン誘導体または40−O−ジアルキルホスフォ−39−デスメトキシラパマイシン誘導体を産生する。これらの塩は当業者に知られている方法で産生することができる。リン酸エステルは、3価ホスフェート(亜リン酸)が5価ホスフェートに酸化(好ましくは非限定的例示としてmCPBAのような過酸活性によって)されているO−ホスファイトを介して、直接または間接に生成し得る。直接リン酸化方法は、非限定的例示として、好ましくは有機溶媒中でDMPA存在下において39−デスメトキシラパマイシン相同物を、保護されたクロロホスフェート(たとえば(BnO)2P(O)Cl、(アルキルO)2P(O)Cl)と反応させ、または、トリエチルアミンなどの塩基の存在下において39−デスメトキシラパマイシン相同物をオキシ塩化リン(POCl3)と反応させ、次いで得られたO−ジクロロホスフェート(すなわちROP(O)Cl2)の酸加水分解またはジアルキルシアノホスフェートへの結合(WO01/81355)を行うことを含む。ジアルキルまたはジアリルクロロホスフェートは、塩基の存在下でジアルキルまたはジアリルホスファイト(すなわち(RO)2P(O)H)を四塩化炭素と反応させることにより自然位で(in situ)生成させることができる。O−ホスフェートの生成(O−ホスフェートへの酸化のために)方法は、非限定的例示として、塩基(好ましくはテトラゾル)の存在下で39−デスメトキシラパマイシン相同物ジアルキル−ジアルキルホスフォラミデート(dialkylphosphoramidate)に結合させること、塩基の存在下でクロロホスファイトを用いて結合すること(Evansら、1992年)を含む。保護基の選択は重要であり、ホスフェートのエチルおよびメチルエステルは酸性や塩基条件では容易にはリン酸化しない。好ましい保護基は、非限定的例示として、ベンジルエステル(ヨウ化ナトリウム/クロロトリメチルシラン促進加水分解を介して開裂、WO01/81355)または2−シアノエチルエステル(緩やかな塩基触媒開裂を介して開裂)を含む。同様に、40−O−ジアルキルホスフォノ−39−デスメトキシラパマイシン誘導体は39−デスメトキシラパマイシン相同物を適当な活性化(上述)ジアルキルホスフォネートまたはジアルキルホスファイトと反応させることにより生成することができる。
【0086】
工程(a)において、R15が化2または化3の部分を示すとき、グリコシド結合の形成すなわちO−グリコシル化は、ヒドロキシル基が好ましくは活性化形態にある対応グリコシルドナー、たとえば化10の化合物または化11の化合物と反応することによってメディエートされ得る(ToshimaおよびTatsuta(1993年)参照)。
【化10】
【化11】
【0087】
非限定的例示として、ハロゲン化グリコシル(Z=F、Cl、Br)、チオグリコシド(Z=SMえ、Set、SPh、SPy、SCN)、1−O−アシルグリコシド(Z=OC(O)R)、オルトエステル(Z=OC(Me)(R)(O−化10/化11のC2)、1−Oまたは1−S炭酸塩(Z=OC(S)SMe、Z=OC(O)イミダゾール、Z=OC(S)イミダゾール、Z=SC(S)OEt)、トリクロロイミデート(Z=OC(=NH)CCl3)、4−ペンテニルグリコシド(Z=OCH2CH2CH2CH=CH2)、リン酸エステル(たとえばZ=OP(O)(OPh)2)、1−O−スルホニル(Z=トシル)、または1−O−シリル化グリコシド(Z=OTMSまたはOTBS)を含むグリコシルドナーの使用に際し、39−デスメトキシラパマイシン相同物を有機溶媒中で、好ましくは活性剤(たとえばルイス酸または重金属塩など、ToshimaおよびTatsuta、1993年、参照)の存在下で、グリコシル化させることができる。用いる特定のグリコシルドナーおよび反応条件によって、αまたはβグリコシドのいずれが生成されるかが決定される。アシル化についての以前と同様、親化合物に存在するあらゆるヒドロキシル基は、グリコシルドナーの1当量を用いることが40−O−アシル化をもたらすように保護され遮蔽され得る。グリコシルドナーに残るヒドロキシル基はたとえばO−酢酸エステル、O−ベンゾエート、1,2−アセトナイドなどで保護され、したがってさらに脱保護が必要となる。さらに、グリカールのような2−デオキシグリコシルを用いて(還元工程がまた要求される)、2’−デオキシ−39−デスメトキシラパマイシングリコシドを生成させることができ、2,6−アンヒドロ−2−チオ糖のような2,6−ジデオキシグリコシルドナーを用いて2’−6’−ジデオキシ−39−デスメトキシラパマイシングリコシドを生成させることができる。
【0088】
工程(b)において、塩の生成および交換は当業者に知られている従来法により行うことができる。化1の化合物の分子変換は既知の方法で行うことができ、たとえば本出願のどこかに記述されているように酸化/還元によりヒドロキシおよびケト基の変換を行うことができる。R6が−PO(OH)2を表す化1の化合物は、R6がOHを表す化1の対応化合物をリン酸化することで調製可能である。適当な条件は本出願のどこかで与えられる。
【0089】
工程(a)および(c)において、保護基の例およびその除去方法をT W Greene「Protective Groups in Organic Synthesis」(J WileyおよびSons、1991年)に見ることができる。適当なヒドロキシル保護基は、加水分解で除去することができるものとしてアルキル(たとえばメチル)、アセタール(たとえばアセトニド)およびアシル(たとえばアセチルまたはベンゾイル)、触媒加水分解で除去することができるものとしてアリルアルキル(たとえばベンジル)、または酸加水分解ないしフッ化イオンアシスト開裂により除去することができるものとしてシリルエーテルを含む。
【0090】
工程(a)に加えて、R6がR7を示す化1の39−デスメトキシラパマイシン相同物はリパーゼ触媒トランスエステル化によって合成することができる。非限定的例示として、化5の39−デスメトキシラパマイシン相同物をリパーゼPS−C「Amano」IIの存在下で化12のエステルと反応させることができる。この時の反応条件はGuら(2005年)および本実施例に記述されている。この方法論はビニルエステルの使用に限定されず、トランスエステル化を他のリパーゼやエステラーゼで触媒作用を発揮させても良い。
【化12】
【0091】
本発明の他の化合物はそれ自体既知である方法または上述の方法と相同的な方法によって調製することができる。
【0092】
新規39−デスメトキシラパマイシン誘導体は、免疫抑制剤、抗真菌剤、抗ガン剤、抗炎症剤、神経再生剤、または臓器移植拒絶反応、移植片対宿主拒絶病、自己免疫疾患、炎症疾患、血管疾患および/または線維症の治療薬として有効な化合物を製剤するために直接的に有用であり、また、さらに別の半合成または生物学的変換のためのテンプレートとして有用である。ラパマイシンおよびその相同物の半合成的誘導法は当業界において良く知られており、非限定的例示として、たとえば米国特許5,665,772、米国特許5,362,718、WO96/41807、米国特許5,728,710、米国特許5,378,836、米国特許5,138,051、米国特許5,665,772、米国特許5,955,457、WO98/04279、米国特許6,015,815および米国特許5,432,183に記述される改良を含む。
【0093】
中間産生物(たとえば化5の化合物)の上記構造は互変異性化可能であり、代表的な互変異性体が示されている場合、すべての互変異性体、たとえばエノール化合物が示されている場合のケト化合物およびその逆も包含することが意図されていることを理解すべきである。
【0094】
さらに別の観点において本発明は39−デスメトキシラパマイシン誘導体の医療用途を提供する。さらに別の観点において本発明は39−デスメトキシラパマイシン誘導体を免疫抑制の誘発または維持、神経再生の刺激、またはガン、B細胞系悪性腫瘍、真菌感染、臓器移植拒絶反応、移植片対宿主拒絶病、自己免疫疾患、炎症疾患、血管疾患および線維症の治療のための薬剤、または創傷治癒制御に用いる薬剤を製剤することの用途を提供する。
【0095】
多剤耐性(MDR)はガンおよびB細胞系悪性腫瘍の治療において重要な問題である。それは多くのガンにおいて薬剤耐性の開発を遅れさせる主要な原因となっている(Persidis A、1999年)。MDRはアデノシン三リン酸結合カセットトランスポーター(ABCトランスポーター)の増大レベル、特にP糖タンパク質(P−gp)のためにエンコードするMDR1遺伝子またはMRP1をエンコードするMRP1遺伝子の発現の増加に関連する。MDR1遺伝子発現レベルは異なるガン由来細胞ラインに亘って大きく変化し、幾つかの細胞ラインでは検出不能であるが、他の細胞ラインでは標準的対照と比較すると最大10ないし100倍の発現増加を示すことがある。
【0096】
したがって、本発明のさらなる態様は、39−デスメトキシラパマイシン誘導体をMDRガンまたはB細胞系悪性腫瘍の治療に用いる用途を提供する。一つの特定の態様において、本発明は39−デスメトキシラパマイシン誘導体をP−gp発現ガンまたはB細胞系悪性腫瘍の治療に用いる用途を提供する。さらにより好適な実施形態において、本発明は39−デスメトキシラパマイシン誘導体を高P−gp発現ガンまたはB細胞系悪性腫瘍の治療に用いる用途を提供する。特に、高P−gp発現ガンまたはB細胞系悪性腫瘍は比較対照に比べて2倍、5倍、10倍、20倍、25倍、50倍または100倍増大した発現を有する。適当な対照はP−gpを発現しないか、P−gp発現レベルが低いか、あるいはMDR機能が低い細胞であり、当業者はそのような細胞ラインを知っているかあるいは認知することができる。適当な細胞ラインを非限定的に例示すれば、MDA435/LCC6、SBC−3/CDDP、MCF7、NCI−H23、NCI−H522、A549/ATCC、EKVX、NCI−H226、NCI−H322M、NCI−H460、HOP−18、HOP−92、LXFL529、DMS114、DMS273、HT29、HCC−2998、HCT−116、COLO205、KM12、KM20L2、MDA−MB−231/ATCC、MDA−MB−435、MDA−N、BT−549、T−47D、OVCAR−3、OVCAR−4、OVCAR−5、OVCAR−8、IGROV1、SK−OV−3、K−562、MOLT−4、HL−60(TB)、RPMI−8226、SR、SN12C、RXF−631、786−0、TK−10、LOX IMVI、MALME−3M、SK−MEL−2、SK−MEL−5、SK−MEL−28、M14、UACC−62、UACC−257、PC−3、DU−145、SNB−19、SNB−75、SNB−78、U251、SF−268、SF−539、XF498。
【0097】
別の観点において本発明は39−デスメトキシラパマイシン誘導体をMDRガンまたはB細胞系悪性腫瘍の治療用薬剤の調製に用いる用途を提供する。特定の態様において、本発明は39−デスメトキシラパマイシン誘導体をP−gp発現ガンまたはB細胞系悪性腫瘍の治療用薬剤の調製に用いる用途を提供する。さらにより好適な実施形態において、本発明は39−デスメトキシラパマイシン誘導体を高P−gp発現ガンまたはB細胞系悪性腫瘍の治療用薬剤の調製に用いる用途を提供する。特に、高P−gp発現ガンまたはB細胞系悪性腫瘍は比較対照に比べて2倍、5倍、10倍、20倍、25倍、50倍または100倍増大した発現を有する。適当な対照は上記のものである。
【0098】
サンプル中のP−gp発現レベルを決定する方法は後述する。
【0099】
したがって、別の観点において本発明は、39−デスメトキシラパマイシン誘導体の治療的有効量を投与することからなるP−gp発現ガンまたはB細胞系悪性腫瘍の治療法を提供する。特定のガン種におけるP糖タンパク質(P−gp)発現レベルは、限定的ではないがたとえばリアルタイムRT−PCT(Szakacsら、2004年;Steinら2002年;Langmannら、2003年;Alvarezら、1995年;Boydら、1995年)を含む技術を用いて、免疫組織化学によって(Steinら、2002年)、またはマイクロアレイを用いて(Leeら、2003年)、当業者により決定することができる。これらの方法は単に例示として挙げたにすぎず、他の適当な方法も当業者に与えられるであろう。
【0100】
当業者はルーチン的実験によって当該化合物の真菌成長抑制能力を決定することができるであろう(たとえばBaker,Hら、1978年;NCCLS Refernce method for broth dilution antifungal susceptibility testing for yeasts:Approved standard M27−A、17(9)、1997年)。さらに、当業者はルーチン的実験によって当該化合物の腫瘍細胞成長抑制能力を決定することができるであろう(Dudkin,Lら、2001年;Yuら、2001年)。さらに別の観点において本発明化合物は免疫抑制を誘発するのに有用であり、この分野における化合物の効能を決定するためのアッセイは当業者に良く知られている。非限定的例示として、免疫抑制活性−Warner,L.M.ら、1992年;Kahanら(1991年)およびKahan&Camardo、2001年);同種移植−Fishbein,T.M.ら、2002年;Kirchnerら、2000年;自己免疫/炎症性/喘息−Calson,R.P.ら、1993年;Powell,N.ら、2001年;糖尿病I−Rabinovitch,Aら、2002年;乾癬−Reitamo,S.ら、2001年;慢性関節リウマチ−Foey,A.ら、2002年;線維症−Zhu,J.ら、1999年;Jain,S.ら、2001年;Gregoryら、1993年。
【0101】
本発明の39−デスメトキシラパマイシン誘導体の免疫抑制誘発能力はこの目的のための標準的試験において実証することができる。さらに別の観点において本発明の39−デスメトキシラパマイシン誘導体は抗繊維化、神経再生および抗血管形成のメカニズムに関連して有用であり、当業者はルーチン的実験によって当該化合物の血管形成阻止能力を決定することができるであろう(たとえばGuba,M.ら、2002年)。当業者はルーチン的実験によって、たとえば薬剤溶出ステント中で、当該化合物を血管超増殖の治療に用いることの有用性を決定することができるであろう(たとえばMorice,M.C.ら、2002年)。加えて、当業者はルーチン的実験によって当該化合物の神経再生能力を決定することができるであろう(たとえばMyckatyn,T.M.ら、2002年、Steinerら、1997年)。
【0102】
本発明はまた、39−デスメトキシラパマイシン誘導体を薬学的に許容可能なキャリアと共に用いてなる薬剤化合物を提供する。
【0103】
臨床試験で用いられているCCI−779およびRAD001などのラパマイシンおよび関連化合物は薬理プロファイルが不十分であり、水溶性が不十分であり、バイオアベイラビリティが不十分である。本発明は安定性および/または細胞膜透過性などの物性が向上された39−デスメトキシラパマイシン誘導体を提供する。当業者は標準的方法を用いて本発明の与えられた化合物の可溶性を容易に決定することができるであろう。代表的な方法が本実施例に示されている。
【0104】
さらに、当業者であれば、本発明化合物の薬物動態プロファイルおよびバイオアベイラビリティを、当業者に知られている生体内および生体外の方法、たとえば非限定的な例示として、後述や実施例記載の方法を用いて決定することができるであろうし、他の代替的アッセイも、非限定的例示としてGallant−Haidnerら、2000年およびTrepanierら、1998年に記述されたものをここで参照して、これらに記述されているように当業者に良く知られている。化合物のバイオアベイラビリティは多くの要因(水溶性、消化管吸収率、タンパク質結合および代謝の程度など)によって決定され、これらは後述するような生体外試験によって決定することができる。当業者には理解されていることであるが、これら要因の一以上の向上は化合物のバイオアベイラビリティを向上させることに繋がる。あるいは、化合物のバイオアベイラビリティは、下記に詳述するような生体内方法を用いて測定することも可能である。
【0105】
Caco−2透過アッセイ
In Vitro Technologies Inc.(IVT Inc.、Baltimore、Maryland、米国)によって提供されるような、24個のウェルCorning Costar Transwellフォーマット内の合流型Caco−2細胞(Li,A.P.、1992年;Grass,G.M.ら、1992年;Volpe,D.A.ら、2001年)を用いることができる。尖室(apical chamber)は0.15mLのハンクス平衡緩衝溶液(HBBS)pH7.4、1%DMSO、0.1mMルシファー・イエローを含む。底室(basal chamber)は0.6mLのHBBS pH7.4、1%DMSOを含む。次いで対照および試験体を湿潤インキュベータ内で37℃でインキュベートし、130rpmで1時間シェークした。ルシファー・イエローは細胞間(密着結合間)経路を介してのみ透過し、ルシファー・イエローについての高い見かけ透過係数(Papp)はアッセイ中の細胞損傷を示し、全てのそのようなウェルは拒絶された。プロプラノロール(トランスポーター効果は知られていないが良好な受動的透過性を持つ)およびアセブタロール(P糖タンパク質による活性的エフラックスにより減衰されて受動的透過性が低い)を対照化合物として用いる。化合物を尖室または底室に適用する(0.01mMで)ことにより一方向および二方向フォーマットで試験することができる。尖室または底室中の化合物をHPLC−MSで分析する。結果を見かけ透過係数Papp(nm/s)およびフラックス率(「A対B」対「B対A」)として示す。
【0106】
Papp(nm/s)=[ボリュームアクセプター/(面積×ドナー)]×(△アクセプター/△時間)
ボリュームアクセプター:0.6mL(A>B)および0.15mL(B>A)
単分子層の面積:0.33cm2
△時間:60分
フラックス率についての正の値は細胞の尖端面からの活性的エフラックスを示す。
【0107】
ヒト肝ミクロソーム(HLM)安定性アッセイ
肝ホモジェネートが、CYP450s(たとえばCYP2C8、CYP2D6、CYP1A、CYP3A4、CYP2E1)エステラーゼ、アミダーゼおよびモノオキシゼナーゼを含む対フェースI(酸化的)酵素群固有脆弱性化合物の測定を与える。
【0108】
試験化合物をヒト肝ホモジェネートに露出し、LC−MSでその消失を観測することにより半減期(T1/2)を決定した。0.001mMで化合物を40分間、37℃で、0.1M Tris−HCl、pH7.4でインキュベートした。0.25mg/mLタンパク質、NADPHの飽和レベルのヒト肝ミクロソーム細胞下分画を補因子として用いた。時間インターバルで試験サンプルにアセトニトリルを添加してタンパク質を沈殿させ、代謝を終了させた。サンプルを遠心処理し、HPLC−MSにより親化合物を分析した。
【0109】
生体内バイオアベイラビリティアッセイ
生体内アッセイは化合物のバイオアベイラビリティを測定するためにも使用することができる(たとえばCroweら、1999年を参照)。一般的に言えば、化合物を腹腔内(ip)または静脈(iv)および経口(po)の両方で試験動物に投与し、血液サンプルを所定インターバルで取り出して薬剤のプラズマ濃度の経時的変化を調べる。プラズマ濃度の経時的変化は、標準モデルを用いたパーセンテージとして該化合物の絶対的バイオアベイラビリティを算定するために用いることができる。主なプロトコルの例を下記に挙げる。
【0110】
本発明化合物またはその親化合物をivで3mg/kgまたは本発明化合物または園生や化合物をpoで10mg/kgマウスに投薬する。血液サンプルを5分、15分、1時間、4時間および24時間のインターバルで取り出し、サンプル中の本発明化合物またはその親化合物の濃度をHPLCで同定する。次いで、プラズマ濃度の経時的変化を用いて、プラズマ濃度−時間曲線下面積(AUC、全身循環に到達する不変薬剤の全量に正比例する)、最大(ピーク)プラズマまたは血中薬剤濃度、最大(ピーク)プラズマ薬剤濃度が発生する時間(ピーク時間)、などの主要なパラメータを得るために用いることができる。バイオアベイラビリティの正確な決定に用いられるその他の要因には、該化合物の消失半減期、全身クリアランス、定常状態分布容積およびF%が含まれる。次いでこれらのパラメータを非境界要素法または境界要素法で分析して、算定されたパーセンテージのバイオアベイラビリティを得る。この種の方法の一例がGallant−Haidnerら、2000年およびTrepanierら、1998年に記載されており、それらをここで参照する。
【0111】
上述の39−デスメトキシラパマイシン誘導体またはその処方は従来のいかなる方法によっても投与可能であり、限定的ではないがたとえば、非経口、経口、局所(口内、舌下、経皮を含む)的に投与することができ、ステントなどの医療器具を用いて投与することができ、吸入法や注射(皮下または筋肉)によっても投与可能である。治療は一度の投薬または一定期間に亘る複数回の投薬で行うことができる。
【0112】
本発明化合物を単独で投与することも可能であるが、薬剤処方として一以上の許容されるキャリアと共に存在させることが好ましい。キャリアは本発明化合物とコンパチブルであり且つそのレシピエントに対して有害でないという意味において「許容される」ものでなければならない。好適なキャリアの数例を下記に詳述する。
【0113】
本発明の39−デスメトキシラパマイシン誘導体は単独または他の治療薬と組み合わせて投与することができ、2つ(またはそれ以上)の薬剤の併用投与は各々を単独で投与する場合に比べて格段に少ない投薬量で済み、したがってそれらの副作用を軽減させる。
【0114】
一つの実施形態において、免疫抑制の誘導または維持、臓器移植拒絶反応、移植片対宿主拒絶病、自己免疫疾患、神経変性症状、血管炎症疾患の治療のために、39−デスメトキシラパマイシン誘導体を他の治療薬と併用投与する。好適な治療薬の非限定的例示として、アザチオプリン、コルチコステロイド、シクロフォスファミド、シクロスポリンA、FK506、ミコフェノール酸モフェチル、OKT−3およびATGなどの免疫調節剤を含む。
【0115】
別の実施形態において、39−デスメトキシラパマイシン誘導体はガンまたはB細胞系悪性腫瘍の治療のために他の治療薬と併用投与される。好適な治療薬の非限定的例示として、メトトレキセート、リューコボリン、アドリアマイシン、プレニゾン、ブレオマイシン、シクロフォスファミド、5−フルオロウラシル、パクリタキセル、ドセタキセル、ビンクリスチン、ビンブラスチン、ビノレルビン、ドキソルビシン、タモキシフェン、トレミフェン、酢酸メゲストロール、アナストロゾール、ゴセレリン、抗HER2単クローン抗体(Herceptin(商標名)など)、カペシタビン、塩酸ラロキシフェン、EGFR抑制剤(Iressa(登録商標)、Tarceva(商標名)、Erbitux(商標名)など)、VEGF抑制剤(Avastin(商標名)など)、プロテアソーム抑制剤(Velcade(商標名)など)、Glivec(登録商標)、hsp90抑制剤(17−AAGなど)を含む。加えて、39−デスメトキシラパマイシン誘導体は非限定的例示として放射線療法や手術を含む他の療法と組み合わせて投与しても良い。
【0116】
一実施形態において、39−デスメトキシラパマイシン誘導体は血管系疾患の治療のために他の治療薬と併用投与される。好適な治療薬の非限定的例示として、ACE抑制剤、アンギオテンシンII受容体拮抗薬、フィブリン酸誘導体、HMG−CoAリダクターゼ抑制剤、βアドレナリン遮断剤、カルシウムチャンネル遮断剤、抗酸化剤、抗凝集剤、血小板抑制剤(Plavix(商標名)など)を含む。
【0117】
一実施形態において、39−デスメトキシラパマイシン誘導体は神経再生の刺激のための他の治療薬と併用投与される。好適な治療薬の非限定的例示として、神経成長因子、グリア細胞由来成長因子、脳由来成長因子、毛様体神経栄養因子、ニューロトロフィン−3などの神経栄養因子を含む。
【0118】
一実施形態において、39−デスメトキシラパマイシン誘導体は感染症の治療のための他の治療薬と併用投与される。好適な治療薬の非限定的例示として、アンフォテリシンB、フルサイトシン、エキノカンジン(カスポファンギン、アニデュラファンギンまたはミカファンギンなど)、グリセオフルビン、イミダゾール系またはトリアゾール系抗真菌剤(クロトリマゾール、ミコナゾール、ケトコナゾール、エコナゾール、ブトコナゾール、オキシコナゾール、テルコナゾール、イトラコナゾール、フルコナゾオール、ボリコナゾールなど)を含む。
【0119】
当業者には明らかなことであろうが、併用投与には、同一の治療レジメの一部として2またはそれ以上の治療薬を患者に輸送するいかなる手段をも含む。2またはそれ以上の薬剤を単一の処方において同時に投与することも可能であるが、これは必須ではない。これらの薬剤は異なる処方で異なる時期にも投与可能である。
【0120】
処方は従来のように単位投薬形態として存在しても要旨、調剤分野で公知のいかなる方法によって調製しても良い。このような方法は、活性成分(本発明化合物)を一以上のアクセサリ成分を構成するキャリアに関連付けるステップを含む。一般的に言えば、活性成分を液体キャリアまたは微細に分割した固体キャリアまたはその両方に均一且つ周到に関連付けた後、必要であれば製品に仕上げることによって処方を調製する。
【0121】
本発明の39−デスメトキシラパマイシン誘導体は通常の場合、活性成分からなる薬剤処方の形態で、任意的に非毒性の有機または無機、酸または基、添加塩、薬学的に許容し得る投薬形態で、経口または何らかの非経口ルートによって投与される。治療すべき疾患や患者および投薬ルートに応じて異なる投薬量で化合物を投与することができる。
【0122】
一例として、本発明化合物は、即時的または時間を置いたあるいはリリースコントロールされた投与のために、香料剤や着色剤を含み得る錠剤、カプセル、胚珠、エリキシル、溶液または懸濁液の形態にして、経口、口内または舌下に投与することができる。
【0123】
経口投与に適した39−デスメトキシラパマイシン誘導体の溶液または懸濁液はまた、N,N−ジメチルアセトアミドなどの賦形剤、ポリソルベート80などの分散剤、表面活性剤、Phosal50PG(フォスファチジルコリン、大豆脂肪酸、エタノール、モノ/ジグリセリド、プロピレングリコールおよびアスコルビン酸パルミチン酸エステルからなる)などの溶解剤を含むことができる。
【0124】
錠剤には、微結晶セルロース、ラクトース(ラクトース一水和物、ラクトース無水物など)、クエン酸ナトリウム、炭酸カルシウム、リン酸水素カルシウムおよびグリシンなどの賦形剤、デンプン(好ましくはコーン、ポテトまたはタピオカのデンプン)、デンプングリコール酸ナトリウム、クロスカルメロースナトリウムなどの崩壊剤、ある種の錯体ケイ酸塩、ポリビニルピロリドン、ヒドロキシプロピルメチルセルロース(HPMC)、ヒドロキシプロピルセルロース(HPC)、マクロゴル8000、サクロース、ゼラチン、アカシアなどの顆粒結合剤を含むことができる。さらに、ステアリン酸マグネシウム、ステアリン酸、ベヘン酸グリセリル、タルクなどの潤滑剤を含有しても良い。
【0125】
類似タイプの固形化合物はゼラチンカプセル内の充填物としても使用可能である。この態様における好適な賦形剤は、ラクトース、デンプン、セルロース、乳糖または高分子ポリエチレングリコールなどである。液体懸濁物および/またはエリキシルの場合、本発明化合物は様々な甘味剤または香料剤、着色剤または顔料、乳化剤および/または懸濁剤、水やエタノール、プロピレングリコール、グリセリンおよびこれらの混合物と組み合わせて用いることができる。
【0126】
錠剤は任意の一以上の副成分と共に圧縮または成型によって調製することができる。圧縮法による錠剤は、粉体または粒体のような自由流動形態にある有効成分を適当な機械の中で圧縮することによって調製することができる。該有効成分は、結合剤(ポビドン、ゼラチン、ヒドロキシプロピルメチルセルロースなど)、潤滑剤、不活性希釈剤、防腐剤、崩壊剤(グリコールデンプンナトリウム、架橋ポビドン、架橋カルボキシメチルセルロースナトリウムなど)、表面活性剤または分散剤と任意に混合して用いることができる。成型法による錠剤は、不活性希釈剤で湿潤された粉状の化合物を適当な機械の中で成型することによって調製することができる。錠剤には任意にコーティングまたはスコアリングが施され、たとえばヒドロキシプロピルメチルセルロースを様々な配合で用いて所望のリリースプロファイルを与えることにより、その中の有効成分のリリースを遅らせ、あるいは制御するように処方することができる。
【0127】
経口投与に適した本発明の処方は、各々が所定量の有効成分を含むカプセル剤、カシュ剤、錠剤などの個別ユニットとして、粉状物または粒状物として、水性液または非水性液中の溶液または懸濁液として、あるいは水中油型液体エマルジョンまたは油中水型液体エマルジョンとして、提供することができる。有効成分はボーラス(急速静注)投与、練り薬またはペーストとして提供することもできる。
【0128】
口内局所投与に適した処方は、香り付けされた通常はサクロースまたはアカシアまたはトラガントなどの基質内の活性成分からなるロゼンジ、ゼラチン、グリセリン、サクロース、アカシアなどの不活性基質内の活性成分からなる芳香製剤、適当な液体キャリア内の活性成分からなるマウスウォッシュを含む。
【0129】
上述の成分に加えて、本発明の処方は、問題となる処方のタイプを考慮して、当業界に公知である他の薬剤を含み得ることを理解しなければならない。たとえば、経口投与に適したものは香料剤を含むことができる。
【0130】
局所投与に適合した薬剤化合物は、軟膏、クリーム、ローション、パウダー、溶液、ペースト、ジェル、含浸包帯、スプレー、エアロゾル、オイル、経皮デバイス、ダスティングパウダーなどとして設計することができる。これらの化合物は活性剤を含む従来法で調剤可能である。これらは従来公知の親水性キャリアおよび添加剤、たとえば防腐剤、薬剤の浸透をアシストする溶媒、クリームまたは軟膏内のエモリエント、ローションのためのエタノールまたはオレイルアルコールを含むことができる。このようなキャリアは化合物の約1%〜約98%存在することができる。より一般的にはこれらは化合物の約80%を上限として存在する。例示的に述べるにすぎないが、クリームまたは軟膏は、十分な量の親水性材料と化合物の約5〜10重量%を含む水とを混合して所望のコンシステンシーを有するクリームまたは軟膏として製造することができる。
【0131】
経皮投与に適合した薬剤化合物は、レシピエントの表皮と長時間密接し続けるように意図された不連続パッチとして提供することができる。たとえば、活性剤をパッチからイオントフォレーシスして供給することができる。
【0132】
口や皮膚などの外部組織への投与のために、化合物は好ましくは局所用の軟膏またはクリームとして提供される。軟膏として設計される場合は、活性剤をパラフィン系または水溶性の軟膏ベースと共に用いることができる。
【0133】
あるいは、活性剤を水中油クリームベースまたは油中水ベースと共にクリームに処方しても良い。
【0134】
静脈投与の場合、流体ユニットの単位投薬量フォームは、活性剤と、非限定的例示として水やアルコール、ポリオール、グリセリン、植物性油など、特に水が好ましいが、これらの無菌ベヒクルを用いて調製される。活性成分は、使用されるベヒクルと濃度に応じて、ベヒクルに懸濁または溶解され得る。溶液を調製する場合、活性成分は注射のために水に溶解し、適当なバイアルまたはアンプルに注入する前にフィルタ殺菌してシールする。
【0135】
好ましくは、局所麻酔薬、防腐剤、緩衝剤などの添加剤がベヒクルに溶解される。安定性を高めるために、バイアルに注入する前に化合物を凍結して真空下で水を除去することができる。凍結乾燥したパウダーは次いでバイアル内でシールされ、使用前に注射用の水バイアルが供給されて液体を再構成する。
【0136】
非経口投与懸濁液は、活性成分が溶解される代わりにベヒクル内に懸濁され、また、ろ過を伴わずに殺菌されることを除いて、溶液の場合とほぼ同様に調製される。活性成分は無菌ベヒクルに懸濁される前にエチレン酸化物に晒されることによって殺菌される。好ましくは、活性成分の均一な分散を促進させるために、界面活性剤または湿潤剤が化合物に含まれる。
【0137】
本発明化合物は、また、当業界において公知の医療機器を用いて投与することができる。たとえば、一実施例によれば、本発明の薬剤化合物は無針(needleless)皮下注射器を用いて投与することができる。このような注射器は、米国特許第5399163号、米国特許第5383851号、米国特許第5312335号、米国特許第5064413号、米国特許第4941880号、米国特許第4790824号または米国特許第4596556号に開示されている。本発明に有用な公知のインプラントおよびモジュールの例を挙げれば、制御された速度で投薬するためのインプラント可能なマイクロインフュージョンポンプ(micro-infusion pump)を開示する米国特許第4487603号、外皮を通して投薬する医療器具を開示する米国特許第4486194号、厳密な注入速度で薬剤をデリバーするための薬剤インフュージョンポンプを開示する米国特許第4447233号、連続薬剤デリバリーのための可変速度インプラント可能インフュージョン装置を開示する米国特許第4447224号、マルチチャンバーのコンパートメントを有する浸透性薬剤デリバリーシステムを開示する米国特許第4439196号、および、浸透性薬剤デリバリーシステムを開示する米国特許第4475196号である。一つの特定の実施形態において、39−デスメトキシラパマイシン誘導体は、WO01/87263に記述されるものや関連する刊行物あるいはPerin(Perin、EC、2005年)によって記述されるものに対応するような薬剤溶出ステントを用いて投与することができる。その他多くのインプラント、デリバリーシステムおよびモジュールが当業者に知られている。
【0138】
本発明の39−デスメトキシラパマイシン誘導体の1回投薬量は特定の化合物、関係する疾患、サブジェクト、疾患の性質および深刻度、サブジェクトの物質的な条件、および選択された投薬経路によって変わる。適切な1回投薬量は当業者であれば容易に決定することができる。
【0139】
薬剤化合物には、投薬方法に依存して、本発明化合物が0.1重量%以上、好ましくは5〜60重量%、より好ましくは10〜30%含まれる。
【0140】
当業者には認識し得るように、本発明化合物の1回投薬量の適量と投薬間隔は、治療する症状の性質および程度、投与の形態、経路および部位、治療患者の年齢および状態などによって決定され、最終的には医師が適切な投薬量を決定する。この投薬量は適切である限り頻繁に繰り返され得る。副作用が発症した場合は、通常の臨床プラクティスに基づいて投薬の量および/または頻度を変更または減少させる。
【実施例】
【0141】
一般的方法および材料
【0142】
材料
すべての試薬は市販品から入手したものであり、特記しない限りさらに精製すること無しに使用した。
【0143】
培養
S.ヒグロスコピカス MG2−10[IJMNOQLhis](WO04/007709;Gregoryら、2004年)を培地1の寒天プレート(下記)上で28℃に維持した。芽胞株を培地1で培養した後に調製した。それは、20%w/vグリセリンと10%w/vラクトースを含む蒸留水に保存して−80℃で保管した。凍結株0.1mlを250mlフラスコ中の50ml培地2(下記)に接種することによって植物培養菌を調製した。この培養菌を36〜48時間、28℃、300rpmでインキュベートした。
【0144】
製造法
植物培養菌を2.5〜5%v/vで培地3に接種した。6〜7日間にわたり26℃、300rpmで培養した。
【0145】
培養法
シクロヘキサンカルボン酸の供給/添加を接種24〜48時間後に行い、他に述べられていない限り1〜2mMの最終濃度で供給した。
【0146】
培地1:
成分 ソース カタログ# L当たり
コーンスターチパウダー Sigma C−8160 2.5g
酵母エキス Difco 0127−17 3g
炭酸カルシウム Sigma C5929 3g
硫酸鉄 Sigma F8633 0.3g
BACTO寒天 Difco 2140−10 20g
小麦デンプン Sigma S2760 10g
水 1L
その後培地を121℃で20分間オートクレーブ殺菌
【0147】
培地2:RapV7菌種培地
成分 L当たり
トーストしたニュートリソイ(ADM Ingredients Ltd) 5g
Avedex W80 デキストリン(Deymer Ingredients Ltd) 35g
コーンスチープ固形物(Sigma) 4g
グルコース 10g
(NH4)2SO4 2g
ラクチン酸(80%) 1.6mL
CaCO3(Caltec) 7g
1MのNaOHでpHを7.5に調整
その後培地を121℃で20分間オートクレーブ殺菌
殺菌後16mLの40%グルコースを各7mLの培地に添加
【0148】
培地3:MD6培地(発酵培地)
成分 L当たり
トーストしたニュートリソイ(ADM Ingredients Ltd) 30g
コーンスターチ(Sigma) 30g
Avedex W80 デキストリン(Deymer Ingredients Ltd) 19g
酵母(Allinson) 3g
コーンスチープ固形物(Sigma) 1g
KH2PO4 2.5g
K2HPO4 2.5g
(NH4)2SO4 10g
NaCl 5g
CaCO3(Caltec) 10g
MnCl2・4H2O 10mg
MgSO4・7H2O 2.5mg
FeSO4・7H2O 120mg
ZnSO4・7H2O 50mg
MES(2−モルホリノエタンスルホン酸一水和物) 21.2g
1MのNaOHでpHを6.0に調整
殺菌前にSigmaのα−アミラーゼ(BAN250)を1L培地に添加
培地を121℃で20分間オートクレーブ殺菌
殺菌後0.35mLの滅菌40%フルクトースと0.10mLのL−リシン(水中に140mg/mL、フィルター殺菌済)を各7mLに添加
【0149】
培地4:RapV7a菌種培地
成分 L当たり
トーストしたニュートリソイ(ADM Ingredients Ltd) 5g
Avedex W80 デキストリン(Deymer Ingredients Ltd) 35g
コーンスチープ固形物(Sigma) 4g
(NH4)2SO4 2g
ラクチン酸(80%) 1.6mL
CaCO3(Caltec) 7g
1MのNaOHでpHを7.5に調整
その後培地を121℃で20分間オートクレーブ殺菌
【0150】
培地5:MD6/5−1培地(発酵培地)
成分 L当たり
トーストしたニュートリソイ(ADM Ingredients Ltd) 15g
Avedex W80 デキストリン(Deymer Ingredients Ltd) 50g
酵母(Allinson) 3g
コーンスチープ固形物(Sigma) 1g
KH2PO4 2.5g
K2HPO4 2.5g
(NH4)2SO4 10g
NaCl 13g
CaCO3(Caltec) 10g
MnCl2・4H2O 3.5mg
MgSO4・7H2O 15mg
FeSO4・7H2O 150mg
ZnSO4・7H2O 60mg
SAG471 0.1mL
培地を121℃で30分間オートクレーブ殺菌
殺菌後L当たり15gフルクトースを添加
48時間後0.5g/LのL−リシンを添加
【0151】
分析法
方法A
注入容量:0.005〜0.1mL(感受性に応じた要求に従う)。HPLCをアジレント「Spherisorb」「Rapid Resolution」カートリッジSB C8上に、3ミクロン、30mm×2.1mmで、下記移動相で作動させた。
【0152】
移動相A:純水中に0.01%蟻酸
移動相B:アセトニトリル中に0.01%蟻酸
流速:1mL/分
0分時の5%Bから2.5分時の95%Bまでリニアグラジエントを用い、4分間95%Bに維持し、次のサイクルまでに5%Bに戻した。検出は、254nmでのUV吸収および/またはMicromasss Quattro−Micro装置を用いたエレクトロスプレーイオン化質量分析(ポジティブまたはネガティブ)によって行った。
【0153】
方法B
注入容量:0.02mL。HPLCを3ミクロンBDS C18 Hypersil(ThermoHypersil−Keystone Ltd)カラム上に、150mm×4.6mmで、50℃に維持し、下記移動相で作動させた。
移動相A:アセトニトリル(100mL)、トリフルオラセト酸(1mL)、1M酢酸アンモニウム(10mL)、脱イオン水と共に1Lまで調製
移動相B:脱イオン水(100mL)、トリフルオラセト酸(1mL)、1M酢酸アンモニウム(10mL)、アセトニトリルと共に1Lまで調製
流速:1mL/分
10分間で55%Bから95%Bまでリニアグラジエントを用った後、95%Bで2分間、55%Bで0.5分間、55%Bでさらに2.5分間維持した。化合物の検出は280nmでのUV吸収によって行った。
【0154】
方法C
HPLCシステムとしてAgilentHP1100を用い、3ミクロンBDS C18 Hypersil(ThermoHypersil−Keystone Ltd)カラム上に、150mm×4.6mmで、40℃に維持し、下記移動相で作動させた。
移動相A:脱イオン水
移動相B:アセトニトリル
流速:1mL/分
このシステムをBruker Daltonics Esquire3000エレクトロスプレー質量分析装置に接続した。500〜1000Daltonの走査領域に亘ってポジティブ/ネガティブ切替を用いた。10分間で55%Bから95%Bまでリニアグラジエントを用いた後、95%Bで2分間、55%Bで0.5分間、55%Bでさらに2.5分間維持した。
【0155】
合成法
特記しない限り全ての反応は市場入手可能な乾燥溶媒を用いて無水条件下で行った。エレクトロスプレー源を備えたBruker Daltonics Esquire3000+質量分析装置に連結したAgilent1100HPLC上で、LC−UV−MSにより反応をモニターした。水:アセトニトリルv:v30:70〜100%アセトニトリルの直線勾配で10分間、次いで100%アセトニトリルで5分間の無勾配期の処理により、Phenomenex Hyperclone カラム、BDS C18 3u(150×4.6mm)において1mL/分で分解が実行された。
【0156】
NMRスペクトルがCDCl3中に記録され、δHおよびδC化学シフトが溶媒(7.26ppmおよび77.0ppm)参照付けられる。39−デスメトキシラパマイシンおよびその誘導体はコンホマーの混合物として存在するので、すべてのアサインメントは主要コンホマーにのみ対応する。
【0157】
抗ガン作用のための生体外バイオアッセイ
化合物の抗ガン作用を、単分子層増殖アッセイ中の12個のヒト腫瘍細胞ラインのパネルにおいて、FreiburgのExperimental OncologymOncotest GmbHの研究機関であるOncostest試験場で生体外評価を行った。選択された12個の細胞ラインの特性を表1に要約して示す。
【0158】
【表1】
【0159】
Rothら、1999年に記述されるように、ヒト腫瘍異種移植片からOncotest細胞ラインが確立された。ドナー異種移植片の起源はFiebigら、1999年に記述された。他の細胞ラインが、NCl(H460、SF−268、OVCAR−3、DU145、MDA−MB−231、MDA−MB−468)から得られ、またはドイツ国BraunshweigのDSMZから購入することができる(LNCAP)。
【0160】
他に特記しない限り、すべての細胞サインは、RPMI1640培地と、10%ウシ胎児血清と0.1mg/mLゲンタミシン(PAA、ドイツ国コルベ)を含む「すぐに混合できる」培地にて、加湿雰囲気(95%空気、5%二酸化炭素)中で37℃で培養した。
【0161】
単分子層アッセイ−プロトコル1の要約:
変性ヨウ化プロピジウムアッセイを用いて、12個のヒト腫瘍細胞ラインの成長に対する試験化合物の影響を評価した(Denglerら、1995年)。
【0162】
要約すれば、トリプシン化によって指数関数的位相培養(exponential phase cultures)から細胞を取り出し、細胞ラインに応じた細胞密度(5〜10,000の範囲で変動する細胞数/ウェル)にて96ウェル底面平滑マイクロタイタープレートに接種してカウントした。24時間後に回収して指数関数的な増殖を再開させた後、培養培地0.01ml(プレート当たり6個の対照ウェル)またはマクベシン(macbecin)含有培地をウェルに添加した。各濃度を3つ培養した。39−デスメトキシラパマイシンを2つの濃度(0.001mMおよび0.01mM)で適用した。4日間の連続的インキュベーションの後、39−デスメトキシラパマイシンを用いまたは用いずに細胞培養培地を0.2mlの水性死細胞染色用蛍光色素PI溶液(7mg/l)で置換した。生存細胞の割合を測定するため、プレート凍結によって細胞を透過させた。プレートを解凍した後、Cytofluor4000マイクロプレートリーダー(励起530nm、放出620nm)を用いて蛍光発光を測定して、生存可能な細胞の総数との直接関係を得た。
【0163】
増殖抑制が(被処理/対照)×100(%T/C)として表された。活性化合物についてIC50およびIC70の値を細胞生存性に対する化合物濃度をプロットすることによって評価した。
【0164】
実施例1:39−デスメトキシラパマイシンの発酵と単離
後述のようにS.ヒグロスコピカス MG2−10[IJMNOQLhis]の培養菌を成長させ、シクロヘキサンカルボン酸(CHCA)で培養することにより39−デスメトキシラパマイシンを産生した。
【0165】
液体培養
S.ヒグロスコピカス MG2−10[IJMNOQLhis]の栄養体培養菌を「Materials & Methods」に記述されるようにして培養した。産生培養を栄養体培養菌により0.5mLで50mLチューブ内の7mL培地3に接種した。7日間に亘り26℃、300rpmで培養した。1:1アセトニトリルで30分間シェイクして1mLのサンプルを抽出し、13,000回転で10分間延伸処理し、分析法Bに基づいて分析および定量化を行った(「Materials & Methods」参照)。産生物の確認は分析法C(「Materials & Methods」参照)を用いて質量分析により決定した。
【0166】
下記特性に記述した分析データを基に、観察されたラパマイシン相同物が所望の39−デスメトキシラパマイシンであると認められた。
【0167】
発酵
S.ヒグロスコピカス MG2−10[IJMNOQLhis]の培地4内の一次的栄養体培養菌を実質的に「Materials & Methods」に記載されるところにより培養した。培地4内の二次的栄養体培養菌を10%v/v、28℃、250rpmで24時間接種した。栄養体培養菌を5%v/vで20L発酵槽中の培地5(「Materials & Methods」参照)に接種した。6日間、26℃で培養し、インペラー先端速度を最小速度1.18ms−1から最大速度2.75ms−1の範囲で変化させることにより0.5vvm.>30%溶解酸素を維持した。シクロヘキサンカルボン酸の供給を接種の24時間後および48時間後に行って、最終濃度2mMを得た。
【0168】
抽出と精製
発酵汁(30L)を等量メタノールで2時間撹拌した後、遠心処理(10分間、3500rpm)して細胞をペレット状にした。上清をDiaion(登録商標)HP20樹脂(43g/L)で1.5時間撹拌した後にろ過した。アセトン(合計量7.5L)で樹脂をバッチ洗浄してラパマイシン相同物を剥ぎ取り、溶剤を減圧下で除去した。得られた水性抽出物を水で2Lに希釈し、酢酸エチル(3×2L)で溶離した。溶剤を減圧下で除去して褐色油を得た(20.5g)。
【0169】
この抽出物をアセトンに溶解し、シリカに乾燥させ、シリカカラム(6×6.5cm径)に適用し、徐々に比率を変えたアセトン/ヘキサン(20%−40%)で抽出した。ラパマイシン相同物含有フラクションをプールし、溶剤を減圧下で除去した。残余物(2.6g)をSephadex LH20でクロマトグラフィ処理し(3回バッチ)、10:10:1クロロホルム/ヘプタン/エタノールで抽出した。半精製されたラパマイシン相同物(1.7g)をGilson HPLCを用いて逆位相(C18)プレパラティブHPLCにより精製し、21mL/分の65%アセトニトリル/水でPhenomenex21.2×250mm Luna5μmC18BDSカラムを溶離した。最も純粋なフラクション(HPLC分析、方法Bにより同定)を結合し、溶媒を減圧下で除去して、39−デスメトキシラパマイシン(563mg)を得た。
【0170】
特性
39−デスメトキシラパマイシンの1H NMRスペクトルは標準(P.Lowden、物理学博士、学位論文、ケンブリッジ大学、1997年)のそれと同等であった。13C-NMR (125 MHz), δc (ppm): 215.75, 208.27, 169.19, 166.71 , 140.13, 135.94, 133.61, 130.10, 129.62, 126.80, 126.33, 98.42, 84.77, 84.37, 75.85, 70.91, 67.10, 59.44, 55.82, 51.21 , 46.50, 44.17, 41.39, 40.70, 40.16, 38.74, 38.37, 35.44, 35.26, 35.08, 33.78, 33.64, 33.04, 32.37, 31.22, 30.41 , 27.24, 27.02, 25.27, 21.48, 20.58, 16.24, 15.95, 15.78, 13.74, 13.00, 10.12
【0171】
培養抽出物のLCMSおよびLCMSn分析は、新規ラパマイシン相同物のm/z比がラパマイシンのそれよりも30原子質量単位だけ小さく、メトキシ基の欠乏と一致することを示した。観測されたイオンは、[M−H]−882.3、[M+NH4]+901.4、[M+Na]+906.2、[M+K]+922.2。ナトリウム付加化合物のフラグメンテーションは、規定の解離経路(図2)(J.A.Reather、理学博士、学位論文、ケンブリッジ大学、2000年)に従い、39−デスメトキシラパマイシンとして予期されたイオンを与えた。この質量分析解離データは、シクロヘキシル成分を含むC28−C42フラグメントにメトキシ欠損が生じている新規ラパマイシン相同物の領域を限定している。
【0172】
この質量分析解離データは39−デスメトキシラパマイシンに完全に合致している。
【0173】
実施例2:39−デスメトキシ−40−O−[2,2−ビス(ヒドロキシメチル)プロピオニル]ラパマイシン
下記工程により39−デスメトキシラパマイシンから39−デスメトキシ−40−O−[2,2−ビス(ヒドロキシメチル)プロピオニル]ラパマイシンを合成した。
【0174】
2.1 39−デスメトキシ−28−O−トリメチルシリルラパマイシンの合成
39−デスメトキシラパマイシン(170mg、0.17mmol)およびイミダゾール(51mg、0.75mmol)を5mLの酢酸エチルに0℃で溶解した。この冷たい溶液にクロロトリメチルシラン(77mg、0.09mL、0.71mmol)を10分間に亘り滴状で添加した。さらに60分間撹拌を連続して行って、28,39−ビス−O−トリメチルシリルエーテルの生成を完了した。この期間後、0.4mLの0.5N硫酸水溶液を添加し、混合物を0℃で2.5時間撹拌した。20mLの酢酸エチルを添加し、塩水、飽和炭酸水素ナトリウム溶液および水で有機層を洗浄した。硫酸ナトリウムでの乾燥および減圧下での濃縮により、無色の固形物として28−O−トリメチルシリルエーテルが産生され、これをさらに精製すること無しに次工程の反応に用いた。1H-NMR (400 MHz, CDCI3), δ (ppm): 4.07 (d, 1H, J=6.5 Hz, 0(28J-H)1 0.00 (s, 9H, 28-O-TMS).
【0175】
2.2 2,4,6−トリクロロ安息香酸2’,2’,5’−トリメチル−1’,3’−ジオキサン−5’カルボキシル無水物の合成
2,2−ジメトキシプロパン(13.5g、130mmol)およびρ−トルエンスルホン酸一水和物(100mg、0.53mmol、0.4mol%)を、アセトン(100mL)中の2,2−ビス(ヒドロキシメチル)プロピオン酸(13.5g、100mmol)溶液に添加した。反応混合物を室温で2時間撹拌した。この時間の後、炭酸水素ナトリウム溶液を添加し、その混合物をさらに5分間撹拌した。上清を除去し、減圧下で濃縮した。得られた固体物をジエチルエーテル(3×50mL)で処理し、組み合わせられた有機抽出物を減圧下で濃縮して白色の固形物16.2gを産生した。1H-NMR (400 MHz, CDCI3), δ (ppm): 4.19 (d, 1H, J=12.0Hz) 3.68 (d, 1 H, J=12.0Hz) 1.45 (s, 1 H) 1.41 (s, 1H) 1.20 (s, 1 H).
【0176】
この物質を米国特許5,362,718の方法で活性化混合無水物に形質転換した。すなわち、アセトニド(1.04g、5.98mmol)を0℃に冷却したTHF(20mL)に溶解し、トリエチルアミン(0.83mL、5.98mmol)および2,4,6−トリクロロ安息香酸塩化物(0.93mL、5.98mmol)の滴下添加で処理した。次いで反応物を室温で5時間撹拌した。得られた沈殿物をろ過し、THF(10mL)で洗浄した。組み合わされたろ過物質を真空内で還元して白色のアモルファスを産生し、これをさらに精製すること無しに以下において用いた。
【0177】
2.3 39−デスメトキシラパマイシン 28−O−トリメチルシリルエーテル、2,2,5−トリメチル[1,3−ジオキサン]−5−カルボキシル酸の40−エステルの合成
実施例2.1からの粗製28−O−トリメチルシリル−39−デスメトキシラパマイシン(200mg、0.17mmolの39−デスメトキシラパマイシンから)を2mLのジクロロメタンに溶解した。溶液を0℃に冷却してDMAP(102mg、0.84mmol)を添加した。次いで、1mLジクロロメタン中の2,4,6−トリクロロ安息香酸2’,2’,5’−トリメチル−1’,3’−ジオキサン−5’カルボキシル無水物(159mg、0.42mmol)溶液を10分間に亘って添加した。反応混合物を0℃で5時間撹拌し、LC/MSで形質転換をモニターした。反応混合物を7mLのジクロロメタンで希釈し、5mLの加水により冷却した。有機層を0.5N硫酸、炭酸水素ナトリウム溶液および水で順次に除去および洗浄した。硫酸ナトリウムでの乾燥および減圧下での濃縮により、無色の泡状物として表題の化合物が産生され、これをさらに精製すること無しに直ちに用いた。MS (ESI) m/z 1111 [M-H]"
【0178】
2.4 39−デスメトキシラパマイシン−40−O−[2,2−ビス(ヒドロキシメチル)プロピオニル]ラパマイシン
実施例2.3からの粗製39−デスメトキシラパマイシン−28−O−トリメチルシリルエーテル 2,2,5−トリメチル[1,3−ジオキサン]−5−カルボキシル酸の40−エステルを2mLアセトンに溶解し、0.5mLの0.5N硫酸を添加した。反応混合物を室温で5時間撹拌し、次いで5mLの飽和炭酸水素ナトリウム溶液と5mLの水を添加して中和した。混合液を酢酸エチルで抽出し、組み合わされた有機抽出物を硫酸ナトリウムで乾燥させた。減圧下での濃縮により無色の固形物が得られ、これをSephadex LH20にてサイズ排除クロマトグラフィにより精製した。溶離剤にはクロロホルム/ヘプタン/エタノール(v:v:v=10:10:1)を用いた。1H-NMR (500 MHz, CDCI3), δ (ppm): 4.72 (m, 1 H, C(40)-H), 3.87 (m, 2H), 3.69 (m, 2H), 1.03 (s, 3H); 13C-NMR (125 MHz, CDCI3), δ (ppm): 175.52, 74.04 (C(40)), 68.73 (2C), 48.90, 17.09. MS (ESI) m/z 1023 [M+Na]+
【0179】
実施例3:39−デスメトキシ−40−O−(2−ヒドロキシ)エチルラパマイシン
3.1 2−(タート−ブチルジメチルシリル)オキシエチルトリフレート
6mLのジクロロメタン中の2−(タート−ブチルジメチルシリル)−エチレングリコール(125mg、0.71mmol)および2,6−ルチデンlutidene(0.08mL、0.69mmol)溶液を−78℃に冷却した。トリフルオロメタンスルホン酸無水物(0.11mL、0.65mmol)を5分間に亘って添加し、さらに15分間−78℃で撹拌を続けてトリフレートの生成を完了した。このトリフレートを下記3.2に記述の反応に自然位で(in situ)用いた。
【0180】
3.2 40−O−[2−(タート−ブチルジメチルシリル)]エチル−39−デスメトキシラパマイシン
39−デスメトキシラパマイシン(300mg、0.34mmol)および2,6−ジ−タート−ブチルピリジン(1.5mL、6.68mmol)を室温にて2−(タート−ブチルジメチルシリル)オキシエチルトリフレート(6mLジクロロメタン中に0.65mmol)で処理した。この溶液を緩やかな窒素流でその当初容積の1/3に濃縮し、得られた懸濁液をさらに72時間室温で撹拌した。この期間の後、飽和炭酸水素ナトリウム溶液(5mL)および水(5mL)を添加し、この混合物を30分間撹拌した。有機層を分離し、溶液相を酢酸エチル(2×5mL)で抽出した。組み合わされた有機抽出物を硫酸ナトリウムで乾燥させ、減圧下でのUShくして無色の油分を得た。ヘキサンからヘキサン/アセトン(v:v=1:1)の勾配を用いたシリカカラムクロマトグラフィによる精製により無色の固形物としての製品を得た。1H-NMR (500 MHz, CDCI3), δ (ppm): 4.16 (d, 1H, J = 6.5 Hz, C(28)-H), 3.73 (t, 2H, J = 5.7 Hz), 3.52 (t, 2H, J = 5.7 Hz), 0.89 (s, 9H), 0.06 (s, 6H); 13C-NMR (125 MHz, CDCI3), δ (ppm): 76.61 (C-40), 69.31 (CH2), 63.03 (CH2), 25.92 (3C), 18.36, -5.23 (2C). MS (ESI) m/z 1065 [M+Na]+
【0181】
3.3 39−デスメトキシ−40−O−(2−ヒドロキシ)エチルラパマイシン
2mLアセトン中の40−O−[2−(タート−ブチルジメチルシリル)]エチル−39−デスメトキシラパマイシン(160mg、0.15mmol)の溶液を室温にて0.3mLの0.5N硫酸で処理した。この溶液を室温で3時間放置し、その後5mLの飽和炭酸水素ナトリウム溶液と10mLの水を添加して冷却した。この混合液を酢酸エチル(3×10mL)で抽出し、組み合わされた有機抽出物を硫酸ナトリウムで乾燥した。減圧下での濃縮により無色の固形物が得られ、これをHPLC(水/アセトンvv:v=20/80)でさらに精製した。1H-NMR (500 MHz, CDCI3), δ (ppm): 4.16 (d, 1H, J = 6 Hz), 3.70 (m, 2H), 3.57 (m, 2H), 3.20 (m,1H, C(40)-H); 13C-NMR (125 MHz, CDCI3), δ (ppm): 78.65 (C-40), 77.20 (C-28), 68.93 (CH2O),62.10 (CH2O).MS (ESI) m/z 951 [M+Na]+
【0182】
実施例4:39−デスメトキシラパマイシンのリパーゼ触媒エステル化を通じた39−デスメトキシ−40−O−[2,2−ビス(ヒドロキシメチル)プロピオニル]ラパマイシン
無水タート−ブチルメチルエーテル(3.5mL)中の39−デスメトキシラパマイシン(720mg、0.83mmol)、ビニル2,2,5−トリメチル[1,3−ジオキサン]−5−カルボキシル酸(244mg、1.22mmol)、リパーゼPS−C「アマノ」II(720mg)および分子篩0.5nm(250mg)の混合物をアルゴン雰囲気中で43℃に加熱した。48時間後LC/MSモニターは出発物質の完全な形質転換を示した。THF(10mL)を添加し、混合物をセライトのパッドでろ過した。この酵素をTHF(2×10mL)で洗浄し、組み合わされた有機抽出物を減圧下で濃縮した。残留物をTHF(50mL)に溶解し、H2SO4(15mL、0.5N)を添加した。この溶液を室温で5時間放置した後、NaHCO3(50mL、5%)および塩水(50mL)の添加により反応物を冷却した。この混合液をEtOAc(3×100mL)で抽出し、組み合わされた有機抽出物をMgSO4で乾燥した。溶媒を除去して半固形物としての製品を得た。フラッシュクロマトグラフィ(ヘキサン/アセトン 1:1)による精製により無色固形物としての製品を得た。NMRデータは実施例2.4と同じ。ナトリウム付加物のMS (ESI) m/z 1022 [M+Na]+ 断片化は図3に示す断片化パターンによりm/z 850, 728, 693, 614, 560, 545, 441および431でイオンを与えた。
【0183】
実施例5:39−デスメトキシ−40−O−[2−ヒドロキシエチル 3−ヒドロキシ−2−(ヒドロキシメチル)−2−メチルプロパノエート]ラパマイシン
無水タート−ブチルメチルエーテル(2mL)中の39−デスメトキシ−40−O−(2−ヒドロキシ)エチルラパマイシン(40mg、0.04mmol)、ビニル2,2,5−トリメチル[1,3−ジオキサン]−5−カルボキシル酸(25mg、0.13mmol)、リパーゼPS−C「アマノ」II(40mg)および分子篩0.5nm(40mg)の混合物をアルゴン雰囲気中で43℃に加熱した。72時間後LC/MSモニターは出発物質の完全な形質転換を示した。THF(10mL)を添加し、混合物をセライトのパッドでろ過した。この酵素をTHF(2×10mL)で洗浄し、組み合わされた有機抽出物を減圧下で濃縮した。残留物をアセトン(7.5mL)に溶解し、H2SO4(2.5mL、0.5N)を添加した。この溶液を室温で2時間放置した後、飽和NaHCO3(10mL)および水(10mL)の添加により反応物を冷却した。この混合液をEtOAc(3×10mL)で抽出し、組み合わされた有機抽出物をMgSO4で乾燥した。溶媒を除去して黄色っぽい固形物としての製品を得た。70:30 MeCN/水から100%MeCNへの勾配を用いてPhenomenex21.2×50mm Luna5μmC18BDSカラム上でプレパラティブHPLCにより15分間精製して、無色固形物としての製品を得た。ナトリウム付加物のMS (ESI) m/z 1067 [M+Na]+ 断片化は図4に示す断片化パターンによりm/z 894, 772, 738, 614, 604, 589, 475および441でイオンを与えた。.
【0184】
実施例6 27−O−デスメチル−39−デスメトキシ−40−O−[2,2−ビス(ヒドロキシメチル)プロピオニル]ラパマイシン
6.1 27−O−デスメチル−39−デスメトキシラパマイシン、2,2,5−トリメチル[1,3−ジオキサン]−5−カルボキシル酸の40−エステル
無水タート−ブチルメチルエーテル(2mL)中の27−O−デスメチル−39−デスメトキシラパマイシン(30mg、0.034mmol)、ビニル2,2,5−トリメチル[1,3−ジオキサン]−5−カルボキシル酸(34mg、0.17mmol)、リパーゼPS−C「アマノ」II(30mg)および分子篩0.5nm(30mg)の混合物をアルゴン雰囲気中で43℃に72時間加熱した。THF(10mL)を添加し、混合物をセライトのパッドでろ過した。この酵素をTHF(2×10mL)で洗浄し、組み合わされた有機抽出物を減圧下で濃縮して、黄色っぽい半固形物としての製品を得た。ヘキサン:アセトン(v:v=2:1)を用いたフラッシュクロマトグラフィによる精製で薄黄色の固形物としての製品を得た。MS (ESI) m/z 1049 [M+Na]+
【0185】
6.2 27−O−デスメチル−39−デスメトキシ−40−O−[2,2−ビス(ヒドロキシメチル)プロピオニル]ラパマイシン
6.1からの材料をアセトン(6mL)に溶解し、H2SO4(2mL、0.5N)を添加した。この溶液を室温で2時間放置した後、飽和NaHCO3(10mL)および水(10mL)の添加により反応物を冷却した。この混合液をEtOAc(3×10mL)で抽出し、組み合わされた有機抽出物をMgSO4で乾燥した。溶媒を除去して黄色っぽい固形物としての製品を得た。70:30 MeCN/水から100%MeCNへの勾配を用いてPhenomenex21.2×50mm Luna5μmC18BDSカラム上でプレパラティブHPLCにより15分間精製して、無色固形物としての製品を得た。ナトリウム付加物のMS (ESI) m/z 1009 [M+Na]+ 断片化は図5に示す断片化パターンによりm/z 836, 679, 600, 560, 531, 431, 427でイオンを与えた。1 H NMR (500 MHz1 CDCI3J δ ppm 4.73 (m, 1 H, C(40)-H), 4.32 (d, J=4.5 Hz, 1 H, 0(27J-H)1 4.19 (d, J=4.5 Hz, 1 H, C(28)-H), 3.89 (m, 2 H), 3.70 (m, 2 H) 1.03 (s, 3 H).
【0186】
実施例7:39−デスメトキシ−40−O−(2−ヒドロキシ)エチルラパマイシンおよび39−デスメトキシ−40−O−[2,2−ビス(ヒドロキシメチル)プロピオニル]ラパマイシンの抗ガン活性の生体外評価
単分子相増殖アッセイ中の12個のヒト腫瘍細胞ラインのパネルにおいて、39−デスメトキシ−40−O−(2−ヒドロキシ)エチルラパマイシンおよび39−デスメトキシ−40−O−[2,2−ビス(ヒドロキシメチル)プロピオニル]ラパマイシンの抗ガン活性の生体外評価を、上記一般的方法におけるプロトコル1に記述したところにより変性ヨウ化プロピジウムアッセイを用いて行った。
【0187】
結果を下の表2に示す。各結果は2回の実験の平均値を示す。表3は試験細胞ラインに対するこれら化合物についての平均IC50およびIC70を示し、ラパマイシンが対照として示されている。
【0188】
【表2】
【0189】
【表3】
【0190】
実施例8:生体外結合アッセイ
FKBP12
FKBP12は化学的変性剤である塩酸グアニジン(GdnHCl)中で可逆的にアンフォールドし、このアンフォールドをタンパク質の内在蛍光における変化によって観測した(Mainら、1998年)。FKBP12の天然状態を特異的に結合し安定させるリガンドは、タンパク質がより高い化学的変性剤濃度でアンフォールドするように変性曲線をシフトする(Mainら、1999年)。安定性の相違から、下記式を用いてリガンド結合定数を求めることができる。
【0191】
【数1】
ここで△Gappは遊離およびリガンド結合形態の間のアンフォールドの自由エネルギーにおける見掛け上の相違、△GH2OD−Nは遊離タンパク質水中アンフォールド自由エネルギー、[L]はリガンド濃度、Kdはタンパク質−リガンド複合体の解離定数である(Meieringら、1992年)。アンフォールドの自由エネルギーは下記式を用いてアンフォールド変位中間点に関連付けることができる。
【0192】
【数2】
ここでmD−Nは与えられたタンパク質と与えられた変性剤についての定数であって残余物のアンフォールドへの露出度変化に比例する(Tanford1968年およびTanford1970年)。[D]50%はアンフォールド中間点に対応する変性剤濃度である。我々は△GLD−NをFKBP12の安定性におけるラパマイシンと未知リガンド(同一リガンド濃度)の相違として下記のように定義する。
【0193】
【数3】
ここで<mD−N>はアンフォールド変位の平均m値であり、△[D]50%はラパマイシン−FKBP12アンフォールド変位と未知リガンド−FKBP12複合体アンフォールド変位の中間点の相違である。[L]>Kdの条件では△△△GD−Nを下記式から2つの化合物の相対的Kdsに関連付けることができる。
【0194】
【数4】
ここでKrapdはラパマイシンの解離定数、Kxdは未知リガンドXの解離定数である。したがって下記式が得られる。
【0195】
【数5】
各変性曲線のフィッティングはmD−Nのための値を生成し、この値を用いてm平均値<mD−N>および△[D]50%、したがってKxdを算出することができる。0.2nMのKrapdの文献値が用いられる。
【0196】
幾つかの場合においては、試験化合物の低い溶解性の故に、ラパマイシン対照実験におけるよりも低い試験化合物濃度が用いられた。これらの場合において、試験化合物濃度とラパマイシン対照濃度との相違を下記式を用いて考慮した。
【数6】
【0197】
【表4】
【0198】
mTOR
mTORの抑制は、mTOR経路およびp70S6キナーゼおよびS6の代用マーカーのリン酸化レベルの測定によって間接的に確立されている(Brunnら、1997年;Mothe−Satneyら、2000年;TeeおよびProud、2002年;HuangおよびHoughton、2002年)。
【0199】
HEK293細胞をFLAGがタグされたmTORおよびmycがタグされた受容体と共感染させ、24時間培養後、一晩血清飢餓させた。細胞を100nMインシュリンで刺激した後、収穫し、3回の凍結/融解サイクルで溶解した。溶解物をプールし、当量をmTOR/受容体複合物のためのFLAG抗体と免疫沈降させた。免疫沈降体を次のように処理した:化合物(0.00001〜0.003mM)で処理したサンプルを30分間30℃でFKBP12/ラパマイシン、FKBP12/39−デスメトキシラパマイシン誘導体またはベヒクル(DMSO)でプレインキュベートし、非処理サンプルをキナーゼ緩衝液中でインキュベートした。免疫沈降体を次いで3mMのATP、10mMのMn2+およびGST−4E−BP1の基質存在下でキナーゼアッセイに生体外適用した。4倍サンプルバッファで反応を停止させ、15%SDS−PAGE処理し、PVDF膜に移して、Phospho−4E−BP1(T37/46)のためにプルーブした。ウエスタンブロットバンドをJ画像を用いた画像分析(http://rsb.info.nih.gov/ij/)により計量した。図6Aはラパマイシン(黒塗り四角)および39−デスメトキシ−40−O−[2,2−ビス(ヒドロキシメチル)プロピオニル]ラパマイシン(黒塗り三角)についての用量反応曲線を示す。図6Bはラパマイシン(黒塗り四角)および39−デスメトキシ−40−O−(2−ヒドロキシ)エチルラパマイシン(黒塗り三角)についての用量反応曲線を示す。
【0200】
代替的に、HEK293細胞を6つのウェルプレートに接種して24時間プレインキュベートし、一晩中血清飢餓させた。次いで細胞をベヒクルまたは化合物で30分間30℃で予備処理し、100nMインシュリンで30分間30℃で刺激し、3回の凍結/融解サイクルで溶解し、タンパク質濃度を分析した。等量タンパク質を供給してSDS−PAGEジェル上で分離した。タンパク質をPVDF膜に湿潤転写し、Phospho−S6(S235/36)またはPhospho−p70 S6K(T389)のためにプルーブした。ウエスタンブロットバンドをJ画像を用いた画像分析(http://rsb.info.nih.gov/ij/)により計量した。
【0201】
参考文献
Alarcon, CM., Heitman, J., and Cardenas, M. E. (1999) Protein kinase activity and identification of a toxic effector domain of the target of rapamycin TOR proteins in yeast. Molecular Biology of the Cell 10: 2531-2546.
Alvarez, M., Paull, K., Monks, A., Hose, C, Lee, J. S., Weinstein, J., Grever, M., Bates, S., Fojo, T., 1995. Generation of a drug resistance profile by quantitation of mdr-1/P-glycoprotein in the cell lines of the National Cancer Institute Anticancer Drug Screen. Journal of Clinical Investigation, 95, 2205-2214.
Aparicio, J. F., Molnar, I., Schwecke, T., Kδnig, A., Haydock, S. F., Khaw, L.E., Staunton, J., and Leadlay, P. F. (1996) Organization of the biosynthetic gene cluster for rapamycin in Streptomyces hygroscopicus: analysis of the enzymatic domains in the modular polyketide synthase. Gene 169: 9-16.
Baker, H., Sidorowicz, A., Sehgal, S. N., and Vezina, C. (1978) Rapamycin (AY-22,989), a new antifungal antibiotic. 111. In vitro and in vivo evaluation. Journal of Antibiotics 31: 539-545.
Boulay, A., Zumstein-Mecker, S., Stephan, C, Beuvink, I., Zilbermann, F., Haller, R., Tobler, S., Heusser, C, O'Reilly, T., Stolz, B., Marti, A., Thomas, G., Lane, H.A.,. 2004, Antitumor efficacy of intermittent treatment schedules with the rapamycin derivative RADO01 correlates with prolonged inactivation of ribosomal protein S6 kinase 1 in peripheral blood mononuclear cells. Cancer Res. 64(1), 252-61.
Boyd, M.R. and Paull, K.D., 1995. Some Practical Considerations and Applications of the National Cancer Institute In Vitro Anticancer Drug Discovery Screen. Drug Development Research 34, 91-109,
Brown, E.J. , Albers, M.W., Shin, T.B., lchikawa, K., Keith, C.T., Lane, W.S., and Schreiber, S. L. (1994) A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 369: 756-758.
Brunn, GJ. , Fadden, P., Haystead, TA, Lawrence, J. C. Jr.1997 The mammalian target of rapamycin phosphorylates sites having a (Ser/Thr)-Pro motif and is activated by antibodies to a region near its COOH terminus. J Biol. Chem^ 272(51), 32547-32550.
Brunn, GJ. , Williams, J., Sabers, C1 Wiederrecht, G., Lawrence, J. C, and Abraham, RT. (1996) Direct inhibition of the signaling functions of the mammalian target of rapamycin by the phosphoinositide 3-kinase inhibitors, wortmannin and LY294002. EMBO Journal 15: 5256-5267.
Carlson, R.P., Hartman, D.A., Tomchek, LA, Walter, T.L., Lugay, J. R., Calhoun, W., Sehgal, , S. N., Chang, J. Y. (1993). Rapamycin, a potential disease-modifying antiarthritic drug. J. Pharmacol. Exp. Ther. 266(2):1125-38.
Crowe A, Bruelisauer A, Duerr L, Guntz P1 Lemaire M. (1999) Absorption and intestinal metabolism of SDZ-RAD and rapamycin in rats. Drug Metab Dispos, 27(5), 627-32
Dengler W.A., Schulte J., Berger D. P., Mertelsmann R. and Fiebig HH. (.1995) Development of a propidium iodide fluorescence assay for proliferation and cytotoxicity assay. Anti-Cancer Drugs, 6:522-532.
DiLeIIa, A.G., and Craig, RJ. (1991 ) Exon organization of the human FKBP-12 gene: correlation with structural and functional protein domains. Biochemistry 30: 8512-8517.
Dudkin, L, Dilling, M. B., Cheshire, P.J., Harwood, F.C., Hollingshead, M., Arbuck, S. G., Travis, R., Sausville, E.A., Houghton, PJ. (2001). Biochemical correlates of mTCJR inhibition by the rapamycin ester CCI-779 and tumor growth inhibition. Clin. Cancer Res. 7(6): 1758-64
Evans D.A., Gage J. R. and Leighton J. L. (1992) Assymetric synthesis of calyculin A. 3. Assemblage of the calyculin skeleton and the introduction of a new phosphate monoester synthesis. J. Org. Chem., 57:1964-1966
Fiebig H. H., Dengler W.A. and Roth T. (1999) Human tumor xenografts: Predictivity, characterization, and discovery of new anticancer agents. In: Fiebig HH, Burger AM (eds). Relevance of Tumor Models for Anticancer Drug Development. Contrib. Oncol., 54: 29 - 50.
Findlay J.A, and Radios, L (1980) Canadian Journal of Chemistry 58:579.
Fishbein, T.M., Florman, S., Gondolesi, G., Schiano, T., LeLeiko, N., Tschernia, A., Kaufman, S. (2002). Intestinal transplantation before and after the introduction of sirolimus. Transplantation. 73(10): 1538-42.
Foey, A., Green, P., Foxwell, B., Feldmann, M., Brennan, F. (2002). Cytokine-stimulated T cells induce macrophage IL-10 production dependent on phosphatidylinositol 3-kinase and p70S6K: implications for rheumatoid arthritis. Arthritis Res. 4(1):64-70. Epub 2001 Oct 10.
Furniss B.S., Hannaford AJ., Smith P.W.G. and Tatchell A.R. (1989) Vogel's textbook of practical organic chemistry, 5th Ed, Pearson, Prentice Hall, Harlow, UK.
Gallant-Haidner HL, Trepanier DJ, Freitag DG, Yatscoff RW. 2000, "Pharmacokinetics and metabolism of sirolimus". Ther Drug Monit 22(1), 31-5.
Grass, G. M., Rubas, W., Jezyk, N., (1992) Evaluation of CACO-2 monolayers as a predictor of drug permeability in colonic tissues. FASEB Journal, 6, A1002.
Gregory, C.R., Huie, P., Billingham, M. E. and Morris, R.E. (1993). Rapamycin inhibits arterial intimal thickening caused by both alloimmune and mechanical injury. Its effect on cellular, growth factor and cytokine response in injured vessels. Transplantation 55(6): 1409-1418.
Gregory MA, Gaisser S, LiII RE, Hong H, Sheridan RM, Wilkinson B, Petkovic H, Weston AJ, Carletti I, Lee HL, Staunton J, Leadlay PF. (2004) "Isolation and characterization of pre- rapamycin, the first macrocyclic intermediate in the biosynthesis of the immunosuppressant rapamycin by S. hygroscopicus". Angew Chem lnt Ed Engl. 43(19), 2551-3
Gu, J, Ruppen ME, Cai P. (2005), "Lipase-Catalyzed Regioselective Esterification of Rapamycin: Synthesis of Temsirolimus (CCI-779). Org. Lett. 7(18): 3945-3948.
Guba, M., von Breitenbuch, P., Steinbauer, M., Koehl, G., Flegel, S., Hornung, M., Bruns, C. J., Zuelke, C, Farkas, S., Anthuber, M., Jauch, K.W., and Geissler, E.K. (2002) Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nature Medicine 8: 128-135.
Hardwick, J. S., Kuruvilla, F.G., Tong, J. K., Shamji, A.F., and Schreiber, S. L (1999) Rapamycin- modulated transcription defines the subset of nutrient-sensitive signaling pathways directly controlled by the Tor proteins. Proceedings of the National Academy of Sciences of the United States of America 96: 14866-14870.
Hentges, K.E., Sirry, B., Gingeras, A.C., Sarbassov, D., Sonenberg, N., Sabatini, D., and Peterson, A.S. (2001) FRAP/mTOR is required for proliferation and patterning during embryonic development in the mouse. Proceedings of the National Academy of Sciences of the United States of America 98: 13796-13801.
Huang, S. and Houghton, P. J., 2002. Mechanisms of resistance to rapamycins. Drug Resist. Update, 4(6), 378-391.
Jain, S., Bicknell, G. R., Whiting, P. H., Nicholson, M. L. (2001 ). Rapamycin reduces expression of fibrosis-associated genes in an experimental model of renal ischaemia reperfusion injury. Transplant Proc. 33(1-2):556-8.
Kahan, B. D., and Camardo, J. S. (2001) Rapamycin: Clinical results and future opportunities. Transplantation 72:1181-1193.
Kahan, B. D., Chang, J. Y., and Sehgal, S.N. (1991 ) Preclinical evaluation of a new potent immunosuppressive agent, rapamycin. Transplantation 52: 185-191.
Kirby, B., and Griffiths, C. E. M. (2001 ) Psoriasis: the future. British Journal of Dermatology 144:37-43.
Kirchner, G. I., Winkler, M., Mueller L, Vidal, C, Jacobsen, W., Franzke, A., Wagner, S., Blick, S., Manns M. P., and Sewing K.-F.(2000) Pharmacokinetics of SDZ RAD and cyclosporin including their metabolites in seven kidney graft patients after the first dose of SDZ RAD. British Journal of Clinical Pharmacology 50:449-454.
Kuo, CJ. , Chung, J. K., Fiorentino, D. F., Flanagan, W.M., Blenis, J., and Crabtree, G. R. (1992) Rapamycin selectively inhibits interleukin-2 activation of p70 S6 kinase. Nature 358: 70-73.
Langmann T1 Mauerer R, Zahn A, Moehle C, Probst M, Stremmel W, Schmitz G. (2003) "Realtime reverse transcription-PCR expression profiling of the complete human ATP-binding cassette transporter superfamily in various tissues". Clin Chem. 49(2), 230-8.
Lee, J-S, Paul!, K., Alvarez, M., Hose, C1 Monks, A., Grever, M., Fojo, AT., Bates, S. E., 1994. Rhodamine efflux patterns predict P-glycoprotein substrates in the National Cancer Institute drug screen. Molecular Pharmacology 46,627-638.
Li, AP. (1992) Screening for human ADME/Tox drug properties in drug discovery. Drug Discovery Today, 6, 357-366.
Lowden, P. A. S., (1997) Ph.D. Dissertation, University of Cambridge. "Studies on the biosynthesis of rapamycin".
Lyons, W.E., George, E.B., Dawson, T.M., Steiner, J. P., and Snyder, S. H. (1994) Immunosuppressant FK506 promotes neurite outgrowth in cultures of PC12 cells and sensory ganglia. Proceedings of the National Academy of Sciences of the United States of America 91:3191-3195.
Main, E. R. G., Fulton, K. F. & Jackson, S. E. (1998). The Context-Dependent Nature of Destabilising Mutations on The Stability of FKBP12. Biochemistry 37 , 6145-6153.
Main, E. R. G., Fulton, K. F. & Jackson, S. E. (1999a). Folding of FKBP12: Pathway of Folding and Characterisation of the Transition State. J. MoI. Biol. 291 , 429-444.
McAlpine, J. B,.Swanson S. J., Jackson, M., Whittem, D.N. (1991). Revised NMR assignments for rapamycin. Journal of Antibiotics 44: 688-690.
Meiering, E. M., Serrano, L. & Fersht, A. R. (1992). Effect of Active Site Residues in Bamase on Activity and Stability. J. MoI. Biol. 225, 585-589.
Morice, M. C, Serruys, P.W., Sousa, J. E., Fajadet, J., Ban Hayashi, E., Perin, M., Colombo, A., Schuler, G., Barragan, P., Guagliumi, G., Molnar, F., Falotico, R. (2002). RAVEL Study Group. Randomized Study with the Sirolimus-Coated Bx Velocity Balloon-Expandable Stent in the Treatment of Patients with de Novo Native Coronary Artery Lesions. A randomized comparison of a sirolimus-eluting stent with a standard stent for coronary revascularization. N. Eng.l J. Med. 346(23):1773-80.
Mothe-Satney, I., Brunn, G.J., McMahon, L. P., Capaldo, C. T., Abraham, R.T., Lawrence, J. C. Jr-. 2000 Mammalian target of rapamycin-dependent phosphorylation of PHAS-I in four (SfT)P sites detected by phospho-specific antibodies. J Biol Chem. 275(43), 33836-33843.
Myckatyn, T.M., Ellis, R.A., Grand, A.G., Sen, S. K., Lowe, J. B, 3rd, Hunter, D.A., Mackinnon, S. E. (2002). The effects of rapamycin in murine peripheral nerve isografts and allografts. Plast Reconstr. Surg. 109(7):2405-17.
Nave, B.T., Ouwens, D. M., Withers, DJ. , Alessi, D. R., and Sheperd, P.R. (1999) Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochemical Journal 344:427-431.
NCCLS Reference Method for Broth Dilution Antifungal Susceptibility Testing for Yeasts: Approved Standard M27-A, vol. 17 No. 9. (1997).
Paiva, N. L, Demain, A.L., and Roberts, M. F. (1991 ) Incorporation of acetate, propionate, and methionine into rapamycin By Streptomyces hygroscopicus. Journal of Natural Products 54: 167-177.
Paiva, N. L., Demain, A.L., and Roberts, M. F. (1993) The immediate precursor of the nitrogen- containing ring of rapamycin is free pipecolic acid. Enzyme and Microbial Technology 15: 581-585.
Perin, E C, (2005), "Choosing a Drug-Eluting Stent: A Comparison Between CYPHER and TAXUS", Reviews in Cardiovascular Medicine, 6 (suppl 1 ), ppS13-S21.
Persidis A. (1999), "Cancer multidrug resistance" Nat Biotechnol. 17: 94-5
Powell, N., Till, S., Bungre, J., Corrigan, C. (2001 ). The immunomodulatory drugs cyclosporin A, mycophenolate mofetil, and sirolimus (rapamycin) inhibit allergen-induced proliferation and IL-5 production by PBMCs from atopic asthmatic patients. J. Allergy Clin. Immunol. 108(6):915-7
Rabinovitch, A., Suarez-Pinzon, W.L., Shapiro, A.M., Rajotte, R. V., Power, R. (2002). Combination therapy with sirolimus and interleukin-2 prevents spontaneous and recurrent autoimmune diabetes in NOD mice. Diabetes. 51(3):638-45.
Raught, B., Gingras, A.C., and Sonenberg, N. (2001) The target of rapamycin (TOR) proteins. Proceedings of the National Academy of Sciences of the United States of America 98: 7037-7044.
Reather, J. A., (2000), Ph.D. Dissertation, University of Cambridge. "Late steps in the biosynthesis of macrocyclic lactones".
Reitamo, S., Spuls, P., Sassolas, B., Lahfa, M., Claudy, A., Griffiths, C. E.; Sirolimus European Psoriasis Study Group. (2001). Efficacy of sirolimus (rapamycin) administered concomitantly with a subtherapeutic dose of cyclosporin in the treatment of severe psoriasis: a randomized controlled trial. Br. J. Dermatol. 145(3):438-45.
Roth T., Burger A.M., Dengler W., Willmann H. and Fiebig H. H. (1999) Human tumor cell lines demonstrating the characteristics of patient tumors as useful models for anticancer drug screening. In: Fiebig HH, Burger AM (eds). Relevance of Tumor Models for Anticancer Drug Development. Contrib. Oncol., 54: 145 - 156.
Royrnans, D., and Siegers, H. (2001) Phosphaditidylinositol 3-kinases in tumor progression. European Journal of Biochemistry 268:487-498.
Schwecke, T., Aparicio, J. F., Molnar, I., Kδnig, A., Khaw, L.E., Haydock, S. F., Oliynyk, M., Caffrey, P., Cortes, J., Lester, J. B., Bδhm, GA, Staunton, J., and Leadlay, P.F. (1995) The biosynthetic gene cluster for the polyketide immunosuppressant rapamycin. Proceedings of the National Academy of Sciences of the United States of America 92: 7839-7843.
Sedrani, R., Cottens, S., Kalien, J., and Schuler, W. (1998) Chemical modifications of rapamycin: the discovery of SDZ RAD. Transplantation Proceedings 30: 2192-2194.
Sehgal, S. N., Baker, H., and Vezina, C. (1975) Rapamycin (AY-22,989), a new antifungal antibiotic II. Fermentation, isolation and characterization. The Journal of Antibiotics 28: 727-733.
Shepherd, P.R , Withers, DJ. , and Siddle K. (1998) Phosphoinositide 3-kinase: the key switch mechanism in insulin signalling. Biochemical Journal 333: 471-490.
Smith M. B. and March J. (2001) March's advanced organic chemistry, 5th Ed, John Wiley and Sons Inc., UK
Steiπer, J. P., Hamilton, G.S., Ross, D. T., Valentine, H. L1 Guo, H., Connolly, M.A., Liang, S., Ramsey, C, Li, J.-H.J., Huang, W., Howorth, P., Soni, R., Fuller, M., Sauer, H., Nowotnik, A.C., and Suzdak, P. D. (1997) Neutrophic immunophilin ligands stimulate structural and functional recovery in neurodegenerative animal models. Proceedings of the National Academy of Sciences of the United States of America 94:2019-2024.
Stein U, Jurchott K, Schlafke M, Hohenberger P. (2002) "Expression of multidrug resistance genes MVP, MDR1 , and MRP1 determined sequentially before, during, and after hyperthermic isolated limb perfusion of soft tissue sarcoma and melanoma patients". J Clin Oncol. 20(15):3282-92.
Szakacs G, Annereau JP, Lababidi S, Shankavaram U, Arciello A, Bussey KJ, Reinhold W, Guo Y, Kruh GD, Reimers M, Weinstein JN, Gottesman MM. 2004, "Predicting drug sensitivity and resistance: profiling ABC transporter genes in cancer cells". Cancer Cell. 6(2): 129-37.
Tanford, C. (1968). Protein Denaturation. Adv. Prot. Chem. 23, 121-282.
Tanford, C. (1970). Protein Denaturation. Part C. Theoretical models for the mechanism of denaturation. /Advances in Protein Chemistry 24, 1-95
Tang, S.J., Reis, G., Kang, H., Gingras, A.-C, Sonenberg, N., and Schuman, E.M. (2002) A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proceedings of the National Academy of Sciences of the United States of America 1:467-472.
Tee, A.R. and Proud, CG. 2002 Caspase cleavage of initiation factor 4E-binding protein 1 yields a dominant inhibitor of Cap-dependent translation and reveals a novel regulatory motif. MoI. Cell. Biol. 22, 1674-1683
Toshima K. and Tatsuta K. (1993) Recent progress in O-glycosylation methods and its application to natural product synthesis. Chem. Rev., 93:1503-1531.
Trepanier DJ, Gallant H, Legatt DF, Yatscoff RW. (1998), "Rapamycin: distribution, pharmacokinetics and therapeutic range investigations: an update". CHn Biochem. 31(5):345-51.
Vezina, C1 Kudelski, A., and Sehgal, S.N. (1975) Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. The Journal of Antibiotics 28: 721-726.
Volpe, D.A., Faustino, PJ., Yu, L.X., (2001) Towards standardisation of an in vitro method of drug absorption. Pharmacopeia! Forum, 27, 2916-2922.
Waller, J. R., and Nicholson, M. L (2001 ) Molecular mechanisms of renal allograft fibrosis. British Journal of Surgery 88: 1429-1441.
Warner, LM., Adams, LM., Chang, J.Y., Sehgal, S.N. (1992). A modification of the in vivo mixed lymphocyte reaction and rapamycin's effect in this model. Clin. Immunol. Immunopathol. 64(3):242-7.
Yu, K., Toral-Barza, L1 Discafani, C1 Zhang, W.G., Skotnicki, J., Frost, P., Gibbons, JJ. (2001 ) mTOR, a novel target in breast cancer: the effect of CCI-779, an mTOR inhibitor, in preclinical models of breast cancer. Endocrine-Related Cancer 8:249-258.
Zhu, J., Wu J., Frizell, E., Liu, S.L, Bashey, R., Rubin, R., Norton, P., Zern, M.A. (1999). Rapamycin inhibits hepatic stellate cell proliferation in vitro and limits fibrogenesis in an in vivo model of liver fibrosis. Gastroenterology. 117(5):1198-204.
【図面の簡単な説明】
【0202】
【図1】ラパマイシンの構造を示す。
【図2】39−デスメトキシラパマイシンの断裂経路を示す。
【図3】39−デスメトキシ−40−O−[2,2−ビス(ヒドロキシメチル)プロピオニル]ラパマイシンの断裂経路を示す。
【図4】39−デスメトキシ−40−O−[2−ヒドロキシエチル 3−ヒドロキシ−2−(ヒドロキシメチル)−2−メチルプロパノアート]ラパマイシンの断裂経路を示す。
【図5】27−O−デスメチル−39−デスメトキシ−40−O−[2,2−ビス(ヒドロキシメチル)プロピオニル]ラパマイシンの断裂経路を示す。
【図6】39−デスメトキシ−40−O−[2,2−ビス(ヒドロキシメチル)プロピオニル]ラパマイシン(A−黒塗三角)および39−デスメトキシ−40−O−(2−ヒドロキシ)エチルラパマイシン(B−黒塗り三角)のmTOR抑制活性をラパマイシン(黒塗り四角)と比較して示す。
【特許請求の範囲】
【請求項1】
40−ヒドロキシ位置がカルボン酸エステル、エーテル、リン酸エステル、ホスフィン酸エステル、アセタールまたはグリコシルとして誘導化されたことを特徴とするラパマイシンの39−デスメトキシ誘導体。
【請求項2】
【化1】
ここで、Xは結合またはCH2を表し、
R1はケト基または(H,H)を表し、
R2はOHまたはOMeを表し、
R3はH、OHまたはOMeを表し、
R4およびR5は各々独立してHまたはOHを表し、
R6は−R7、−C(O)R7、−(CH2)2−O−[CR21R22−O]a−C(O)−R23;−CR21R22−O−C(O)−R23;POR19R20、−PO(OR19)(OR20)またはY−R15を表し、
R7は−(CR8R9)m(CR10R11)pCR12R13R14を表し、
R8およびR9は各々独立してC1−C4アルキル、C2−C4アルケニルまたはC2−C4アルキニルを表し、これらのいずれの基においても−PO(OH)2、CF2PO(OH)2、−OH、−COOHまたは−NH2で任意に置換可能であり、あるいはR8およびR9は各々独立してH、トリフルオロメチルまたはFを表し、
R10、R11、R12、R13およびR14は各々独立してC1−C4アルキル、C2−C4アルケニルまたはC2−C4アルキニルを表し、これらのいずれの基においても−PO(OH)2、CF2PO(OH)2、−OH、−COOHまたは−NH2で任意に置換可能であり、あるいはR10、R11、R12、R13およびR14は各々独立してH、−(CR8R9)qNH2、−(CR8R9)qOH、CF3、F、COOHから選択することができ、あるいはR10とR11またはR12とR13またはR13とR14は一緒になってそれらが結合している炭素と共にC3−C6シクロアルキルまたはN、OおよびSから選ばれる一以上のヘテロ原子を含み5個までの−(CR8R9)qOH、−(CR8R9)qNH2または−COOH基で任意に置換可能である3〜6員ヘテロアルキル環を形成しており、
ここでYは結合、−C(O)−O−または−(CH2)2−O−C(O)−O−であり、
R15は化2、化3または化4のいずれかであり、
【化2】
【化3】
【化4】
R16は各々独立してHまたはOHであり、
R17はH、OHおよびNH2から独立して選ばれ、
R18はH、−CH3、−CH2OHおよび−COOHから選ばれ、
ただしR16、R17およびR18から選ばれる2以上の基はHまたはCH3を表し、
R19およびR20は各々独立してHまたはC1−C4アルキルをあらわし、あるいはR19およびR20の両方で=CH2を示し、
R21はH、CH3から独立して選ばれ、
R22はH、−CH3、−CH=CH2、−CH2Cl、−CHCl2、−CCl3、−CH(OH)Me、−CH2OH、−CH2CH3、−CH(Cl)Meから独立して選ばれ、
R23は独立してR7、Y−R15または5または6員アリルまたはヘテロアリル環であって任意にOH、F、Cl、Br、NO2およびNH2から選ばれる1〜3個の基で置換されるもの、
aは0または1を表し、
m、pおよびqは各々独立して0〜4の整数を表し、
ただしR7部分は12個を超える炭素原子を含有せず、-PO(OH)2、-CF2PO(OH)2、-COOH、OHまたはNH2から選ばれる1以上の官能基を有するところの化1の化合物、またはそれらの薬学的に許容可能な塩。
【請求項3】
R6が−R7を表す請求項2の化合物。
【請求項4】
R6が−C(O)R7を表す請求項2の化合物。
【請求項5】
R6が−(CH2)2-O−[CR21R22−O]a−C(O)−R23を表す請求項2の化合物。
【請求項6】
R23がR7を表す請求項5の化合物。
【請求項7】
R23がY−R15を表す請求項5の化合物。
【請求項8】
R7が7個またはそれより少ない数の炭素原子を含む請求項2〜6のいずれかの化合物。
【請求項9】
R7が5個またはそれより少ない数の炭素原子を含む請求項8の化合物。
【請求項10】
R7が−PO(OH)2、−CF2PO(OH)2、−OH、−COOHおよび−NH2から選ばれる2つの基を含む請求項2〜6、8または9のいずれかの化合物。
【請求項11】
R7が−COOH、OH、およびNH2から選ばれる少なくとも1つの官能基を含む請求項2〜6または8〜10のいずれかの化合物。
【請求項12】
pが0または1を表す請求項2〜6または8〜11の化合物。
【請求項13】
mが0または1を表す請求項2〜6または8〜12の化合物。
【請求項14】
qが0、1または2を表す請求項2〜6または8〜13の化合物。
【請求項15】
R11がHを表す請求項2〜6または8〜14の化合物。
【請求項16】
R12がHを表す請求項2〜6または8〜15の化合物。
【請求項17】
R13がHまたはOHを表す請求項2〜6または8〜16の化合物。
【請求項18】
pが1を表し、R10がMe、OHまたはCH2OHを表す請求項2〜6または8〜17の化合物。
【請求項19】
pが1を表し、R11がMe、HまたはCH2OHを表す請求項2〜6または8〜17の化合物。
【請求項20】
mとpが共に0を表し、R12とR13が共にHを表し、R14は−(CR8R9)q−OHを表し、ここでqは0または1であり、R8とR9は共にHを表す請求項2〜6または8〜11の化合物。
【請求項21】
pが1を表し、mが0を表し、R10とR11が共にHを表し、R12がHを表し、R13がH、OHまたはNH2を表し、R14が−(CR8R9)q−OHを表し、ここでqは0または1であり、R8とR9は共にHを表す請求項2〜6または8〜11の化合物。
【請求項22】
R6が、ヒドロキシ酢酸、3−ヒドロキシ−2,2−ジメチルプロピオン酸、2,3−ジヒドロキシプロピオン酸、3−ヒドロキシ−2−ヒドロキシメチルプロピオン酸または2,2−ビス(ヒドロキシメチル)プロピオン酸によるエステル形成由来の残留物を表す請求項2〜6または8〜11の化合物。
【請求項23】
R6が、ヒドロキシ酢酸、3−ヒドロキシ−2,2−ジメチルプロピオン酸、2,3−ジヒドロキシプロピオン酸、3−ヒドロキシ−2−ヒドロキシメチルプロピオン酸または2,2−ビス(ヒドロキシメチル)プロピオン酸によるエーテル形成由来の残留物を表す請求項2〜6または8〜11の化合物。
【請求項24】
39−デスメトキシ−40−O−[2,2−ビス(ヒドロキシメチル)プロピオニル]ラパマイシンまたはその薬学的に供され得る塩である請求項2の化合物。
【請求項25】
39−デスメトキシ−40−O−(2−ヒドロキシ)エチルラパマイシンまたはその薬学的に供され得る塩である請求項2の化合物。
【請求項26】
39−デスメトキシ−40−O−[2−ヒドロキシエチル 3−ヒドロキシ−2−(ヒドロキシメチル)−2−メチルプロパノエート]ラパマイシンまたはその薬学的に供され得る塩である請求項2の化合物。
【請求項27】
27−O−デスメチル−39−デスメトキシ−40−O−[2,2−ビス(ヒドロキシメチル)プロピオニル]ラパマイシンまたはその薬学的に供され得る塩である請求項2の化合物。
【請求項28】
R6が−POR19R20を表す請求項2の化合物。
【請求項29】
R6が−PO(OR19)(OR20)を表す請求項2の化合物。
【請求項30】
R19とR20が共にCH3を表すか、または共にCH2CH3を表す請求項28または請求項29の化合物。
【請求項31】
R6がY−R15を表す請求項2の化合物。
【請求項32】
R15基が化2を表す請求項31の化合物。
【化2】
【請求項33】
R15がグルコース、グルコサミン、グルクロン酸またはアラビノースでアセタールを形成することにより形成される部分である請求項32の化合物。
【請求項34】
R15がD−グルコースでアセタールを形成することにより形成される部分である請求項32の化合物。
【請求項35】
R15がD−グルコサミンでアセタールを形成することにより形成される部分である請求項32の化合物。
【請求項36】
R15がD−グルクロン酸でアセタールを形成することにより形成される部分である請求項32の化合物。
【請求項37】
R15が化3を表す請求項31の化合物。
【化3】
【請求項38】
R15がフルクトースでアセタールを形成することにより形成される部分である請求項37の化合物。
【請求項39】
R15が化4を表す請求項31の化合物。
【化4】
【請求項40】
R15がグルクロン酸でエステルを形成することにより形成される部分である請求項39の化合物。
【請求項41】
Yが結合を示す請求項31〜40のいずれかの化合物。
【請求項42】
Yが−(CH2)2−O−C(O)−O−を示す請求項31〜40のいずれかの化合物。
【請求項43】
Yが−C(O)−O−を示す請求項31〜40のいずれかの化合物。
【請求項44】
薬剤として用いられる請求項1〜43のいずれかの化合物。
【請求項45】
ガンおよび/またはB細胞系悪性腫瘍の治療、免疫抑制の誘導または維持、臓器移植拒絶反応、移植片対宿主拒絶病、自己免疫疾患、炎症疾患、血管疾患および線維症の治療、神経再生の刺激または感染症の治療に用いられる請求項1〜43のいずれかの化合物。
【請求項46】
ガンまたはB細胞系悪性腫瘍の治療薬として用いられる請求項1〜43のいずれかの化合物。
【請求項47】
請求項1〜43のいずれかの化合物を一以上の薬学的に許容され得る希釈剤または媒体と共に用いてなる薬剤化合物。
【請求項48】
請求項1〜43のいずれかの化合物の有効量を患者に投与して、ガンおよび/またはB細胞系悪性腫瘍の治療、免疫抑制の誘導または維持、臓器移植拒絶反応、移植片対宿主拒絶病、自己免疫疾患、炎症疾患、血管疾患および線維症の治療、神経再生の刺激または感染症の治療を行う方法。
【請求項49】
請求項1〜43のいずれかの化合物の有効量を患者に投与することからなるガンまたはB細胞系悪性腫瘍の治療方法。
【請求項50】
請求項1〜43のいずれかの化合物を、ガンおよび/またはB細胞系悪性腫瘍の治療、免疫抑制の誘導または維持、臓器移植拒絶反応、移植片対宿主拒絶病、自己免疫疾患、炎症疾患、血管疾患および線維症の治療、神経再生の刺激または感染症の治療のための薬剤調製に使用する用途。
【請求項51】
薬剤がガンまたはB細胞系悪性腫瘍の治療用である請求項50の用途。
【請求項52】
請求項2〜43のいずれかによる化1の化合物の調製方法であって、
【化1】
(a)化5の39−デスメトキシラパマイシン相同物(RAはHまたは(CH2)2−OHを表す)またはその保護された誘導体をHO−R6化合物(R6は化1の化合物について定義したと同じ、またはその保護された誘導体)またはその活性化された誘導体と反応させる工程、または、
【化5】
(b)化1の化合物またはその塩を他の化1の化合物またはその薬学的に許容され得る塩に形質転換する工程、または、
(c)保護された化1の化合物を脱保護する工程とを含む方法。
【請求項53】
請求項1〜43のいずれかによる化合物と、一以上の有効治療剤とからなる化合物tまたはパーツキット。
【請求項54】
一以上の有効治療剤が、メトトレキセート、リューコボリン、アドリアマイシン、プレニゾン、ブレオマイシン、シクロフォスファミド、5−フルオロウラシル、パクリタキセル、ドセタキセル、ビンクリスチン、ビンブラスチン、ビノレルビン、ドキソルビシン、タモキシフェン、トレミフェン、酢酸メゲストロール、アナストロゾール、ゴセレリン、抗HER2単クローン抗体(Herceptin(商標名)など)、カペシタビン、塩酸ラロキシフェン、EGFR抑制剤、VEGF抑制剤、プロテアソーム抑制剤、hsp90抑制剤、アザチオプリン、コルチコステロイド、シクロフォスファミド、シクロスポリンA、FK506、ミコフェノール酸モフェチル、OKT−3、ATG、アンフォテリシンB、フルサイトシン、エキノカンジン、グリセオフルビン、イミダゾール系およびトリアゾール系抗真菌剤よりなる群から選ばれる、請求項53の化合物またはパーツキット。
【請求項1】
40−ヒドロキシ位置がカルボン酸エステル、エーテル、リン酸エステル、ホスフィン酸エステル、アセタールまたはグリコシルとして誘導化されたことを特徴とするラパマイシンの39−デスメトキシ誘導体。
【請求項2】
【化1】
ここで、Xは結合またはCH2を表し、
R1はケト基または(H,H)を表し、
R2はOHまたはOMeを表し、
R3はH、OHまたはOMeを表し、
R4およびR5は各々独立してHまたはOHを表し、
R6は−R7、−C(O)R7、−(CH2)2−O−[CR21R22−O]a−C(O)−R23;−CR21R22−O−C(O)−R23;POR19R20、−PO(OR19)(OR20)またはY−R15を表し、
R7は−(CR8R9)m(CR10R11)pCR12R13R14を表し、
R8およびR9は各々独立してC1−C4アルキル、C2−C4アルケニルまたはC2−C4アルキニルを表し、これらのいずれの基においても−PO(OH)2、CF2PO(OH)2、−OH、−COOHまたは−NH2で任意に置換可能であり、あるいはR8およびR9は各々独立してH、トリフルオロメチルまたはFを表し、
R10、R11、R12、R13およびR14は各々独立してC1−C4アルキル、C2−C4アルケニルまたはC2−C4アルキニルを表し、これらのいずれの基においても−PO(OH)2、CF2PO(OH)2、−OH、−COOHまたは−NH2で任意に置換可能であり、あるいはR10、R11、R12、R13およびR14は各々独立してH、−(CR8R9)qNH2、−(CR8R9)qOH、CF3、F、COOHから選択することができ、あるいはR10とR11またはR12とR13またはR13とR14は一緒になってそれらが結合している炭素と共にC3−C6シクロアルキルまたはN、OおよびSから選ばれる一以上のヘテロ原子を含み5個までの−(CR8R9)qOH、−(CR8R9)qNH2または−COOH基で任意に置換可能である3〜6員ヘテロアルキル環を形成しており、
ここでYは結合、−C(O)−O−または−(CH2)2−O−C(O)−O−であり、
R15は化2、化3または化4のいずれかであり、
【化2】
【化3】
【化4】
R16は各々独立してHまたはOHであり、
R17はH、OHおよびNH2から独立して選ばれ、
R18はH、−CH3、−CH2OHおよび−COOHから選ばれ、
ただしR16、R17およびR18から選ばれる2以上の基はHまたはCH3を表し、
R19およびR20は各々独立してHまたはC1−C4アルキルをあらわし、あるいはR19およびR20の両方で=CH2を示し、
R21はH、CH3から独立して選ばれ、
R22はH、−CH3、−CH=CH2、−CH2Cl、−CHCl2、−CCl3、−CH(OH)Me、−CH2OH、−CH2CH3、−CH(Cl)Meから独立して選ばれ、
R23は独立してR7、Y−R15または5または6員アリルまたはヘテロアリル環であって任意にOH、F、Cl、Br、NO2およびNH2から選ばれる1〜3個の基で置換されるもの、
aは0または1を表し、
m、pおよびqは各々独立して0〜4の整数を表し、
ただしR7部分は12個を超える炭素原子を含有せず、-PO(OH)2、-CF2PO(OH)2、-COOH、OHまたはNH2から選ばれる1以上の官能基を有するところの化1の化合物、またはそれらの薬学的に許容可能な塩。
【請求項3】
R6が−R7を表す請求項2の化合物。
【請求項4】
R6が−C(O)R7を表す請求項2の化合物。
【請求項5】
R6が−(CH2)2-O−[CR21R22−O]a−C(O)−R23を表す請求項2の化合物。
【請求項6】
R23がR7を表す請求項5の化合物。
【請求項7】
R23がY−R15を表す請求項5の化合物。
【請求項8】
R7が7個またはそれより少ない数の炭素原子を含む請求項2〜6のいずれかの化合物。
【請求項9】
R7が5個またはそれより少ない数の炭素原子を含む請求項8の化合物。
【請求項10】
R7が−PO(OH)2、−CF2PO(OH)2、−OH、−COOHおよび−NH2から選ばれる2つの基を含む請求項2〜6、8または9のいずれかの化合物。
【請求項11】
R7が−COOH、OH、およびNH2から選ばれる少なくとも1つの官能基を含む請求項2〜6または8〜10のいずれかの化合物。
【請求項12】
pが0または1を表す請求項2〜6または8〜11の化合物。
【請求項13】
mが0または1を表す請求項2〜6または8〜12の化合物。
【請求項14】
qが0、1または2を表す請求項2〜6または8〜13の化合物。
【請求項15】
R11がHを表す請求項2〜6または8〜14の化合物。
【請求項16】
R12がHを表す請求項2〜6または8〜15の化合物。
【請求項17】
R13がHまたはOHを表す請求項2〜6または8〜16の化合物。
【請求項18】
pが1を表し、R10がMe、OHまたはCH2OHを表す請求項2〜6または8〜17の化合物。
【請求項19】
pが1を表し、R11がMe、HまたはCH2OHを表す請求項2〜6または8〜17の化合物。
【請求項20】
mとpが共に0を表し、R12とR13が共にHを表し、R14は−(CR8R9)q−OHを表し、ここでqは0または1であり、R8とR9は共にHを表す請求項2〜6または8〜11の化合物。
【請求項21】
pが1を表し、mが0を表し、R10とR11が共にHを表し、R12がHを表し、R13がH、OHまたはNH2を表し、R14が−(CR8R9)q−OHを表し、ここでqは0または1であり、R8とR9は共にHを表す請求項2〜6または8〜11の化合物。
【請求項22】
R6が、ヒドロキシ酢酸、3−ヒドロキシ−2,2−ジメチルプロピオン酸、2,3−ジヒドロキシプロピオン酸、3−ヒドロキシ−2−ヒドロキシメチルプロピオン酸または2,2−ビス(ヒドロキシメチル)プロピオン酸によるエステル形成由来の残留物を表す請求項2〜6または8〜11の化合物。
【請求項23】
R6が、ヒドロキシ酢酸、3−ヒドロキシ−2,2−ジメチルプロピオン酸、2,3−ジヒドロキシプロピオン酸、3−ヒドロキシ−2−ヒドロキシメチルプロピオン酸または2,2−ビス(ヒドロキシメチル)プロピオン酸によるエーテル形成由来の残留物を表す請求項2〜6または8〜11の化合物。
【請求項24】
39−デスメトキシ−40−O−[2,2−ビス(ヒドロキシメチル)プロピオニル]ラパマイシンまたはその薬学的に供され得る塩である請求項2の化合物。
【請求項25】
39−デスメトキシ−40−O−(2−ヒドロキシ)エチルラパマイシンまたはその薬学的に供され得る塩である請求項2の化合物。
【請求項26】
39−デスメトキシ−40−O−[2−ヒドロキシエチル 3−ヒドロキシ−2−(ヒドロキシメチル)−2−メチルプロパノエート]ラパマイシンまたはその薬学的に供され得る塩である請求項2の化合物。
【請求項27】
27−O−デスメチル−39−デスメトキシ−40−O−[2,2−ビス(ヒドロキシメチル)プロピオニル]ラパマイシンまたはその薬学的に供され得る塩である請求項2の化合物。
【請求項28】
R6が−POR19R20を表す請求項2の化合物。
【請求項29】
R6が−PO(OR19)(OR20)を表す請求項2の化合物。
【請求項30】
R19とR20が共にCH3を表すか、または共にCH2CH3を表す請求項28または請求項29の化合物。
【請求項31】
R6がY−R15を表す請求項2の化合物。
【請求項32】
R15基が化2を表す請求項31の化合物。
【化2】
【請求項33】
R15がグルコース、グルコサミン、グルクロン酸またはアラビノースでアセタールを形成することにより形成される部分である請求項32の化合物。
【請求項34】
R15がD−グルコースでアセタールを形成することにより形成される部分である請求項32の化合物。
【請求項35】
R15がD−グルコサミンでアセタールを形成することにより形成される部分である請求項32の化合物。
【請求項36】
R15がD−グルクロン酸でアセタールを形成することにより形成される部分である請求項32の化合物。
【請求項37】
R15が化3を表す請求項31の化合物。
【化3】
【請求項38】
R15がフルクトースでアセタールを形成することにより形成される部分である請求項37の化合物。
【請求項39】
R15が化4を表す請求項31の化合物。
【化4】
【請求項40】
R15がグルクロン酸でエステルを形成することにより形成される部分である請求項39の化合物。
【請求項41】
Yが結合を示す請求項31〜40のいずれかの化合物。
【請求項42】
Yが−(CH2)2−O−C(O)−O−を示す請求項31〜40のいずれかの化合物。
【請求項43】
Yが−C(O)−O−を示す請求項31〜40のいずれかの化合物。
【請求項44】
薬剤として用いられる請求項1〜43のいずれかの化合物。
【請求項45】
ガンおよび/またはB細胞系悪性腫瘍の治療、免疫抑制の誘導または維持、臓器移植拒絶反応、移植片対宿主拒絶病、自己免疫疾患、炎症疾患、血管疾患および線維症の治療、神経再生の刺激または感染症の治療に用いられる請求項1〜43のいずれかの化合物。
【請求項46】
ガンまたはB細胞系悪性腫瘍の治療薬として用いられる請求項1〜43のいずれかの化合物。
【請求項47】
請求項1〜43のいずれかの化合物を一以上の薬学的に許容され得る希釈剤または媒体と共に用いてなる薬剤化合物。
【請求項48】
請求項1〜43のいずれかの化合物の有効量を患者に投与して、ガンおよび/またはB細胞系悪性腫瘍の治療、免疫抑制の誘導または維持、臓器移植拒絶反応、移植片対宿主拒絶病、自己免疫疾患、炎症疾患、血管疾患および線維症の治療、神経再生の刺激または感染症の治療を行う方法。
【請求項49】
請求項1〜43のいずれかの化合物の有効量を患者に投与することからなるガンまたはB細胞系悪性腫瘍の治療方法。
【請求項50】
請求項1〜43のいずれかの化合物を、ガンおよび/またはB細胞系悪性腫瘍の治療、免疫抑制の誘導または維持、臓器移植拒絶反応、移植片対宿主拒絶病、自己免疫疾患、炎症疾患、血管疾患および線維症の治療、神経再生の刺激または感染症の治療のための薬剤調製に使用する用途。
【請求項51】
薬剤がガンまたはB細胞系悪性腫瘍の治療用である請求項50の用途。
【請求項52】
請求項2〜43のいずれかによる化1の化合物の調製方法であって、
【化1】
(a)化5の39−デスメトキシラパマイシン相同物(RAはHまたは(CH2)2−OHを表す)またはその保護された誘導体をHO−R6化合物(R6は化1の化合物について定義したと同じ、またはその保護された誘導体)またはその活性化された誘導体と反応させる工程、または、
【化5】
(b)化1の化合物またはその塩を他の化1の化合物またはその薬学的に許容され得る塩に形質転換する工程、または、
(c)保護された化1の化合物を脱保護する工程とを含む方法。
【請求項53】
請求項1〜43のいずれかによる化合物と、一以上の有効治療剤とからなる化合物tまたはパーツキット。
【請求項54】
一以上の有効治療剤が、メトトレキセート、リューコボリン、アドリアマイシン、プレニゾン、ブレオマイシン、シクロフォスファミド、5−フルオロウラシル、パクリタキセル、ドセタキセル、ビンクリスチン、ビンブラスチン、ビノレルビン、ドキソルビシン、タモキシフェン、トレミフェン、酢酸メゲストロール、アナストロゾール、ゴセレリン、抗HER2単クローン抗体(Herceptin(商標名)など)、カペシタビン、塩酸ラロキシフェン、EGFR抑制剤、VEGF抑制剤、プロテアソーム抑制剤、hsp90抑制剤、アザチオプリン、コルチコステロイド、シクロフォスファミド、シクロスポリンA、FK506、ミコフェノール酸モフェチル、OKT−3、ATG、アンフォテリシンB、フルサイトシン、エキノカンジン、グリセオフルビン、イミダゾール系およびトリアゾール系抗真菌剤よりなる群から選ばれる、請求項53の化合物またはパーツキット。
【図1】

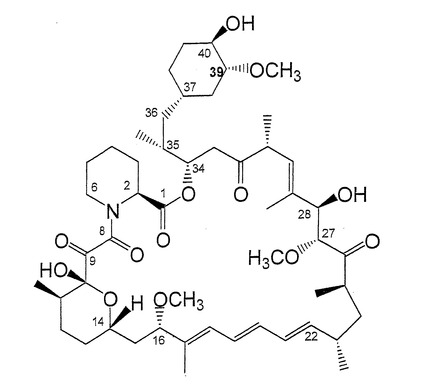
【図2】
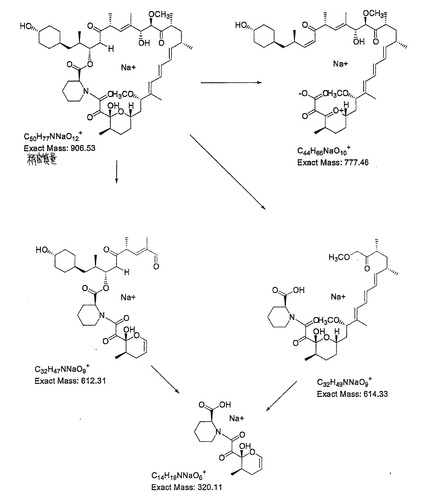
【図3】
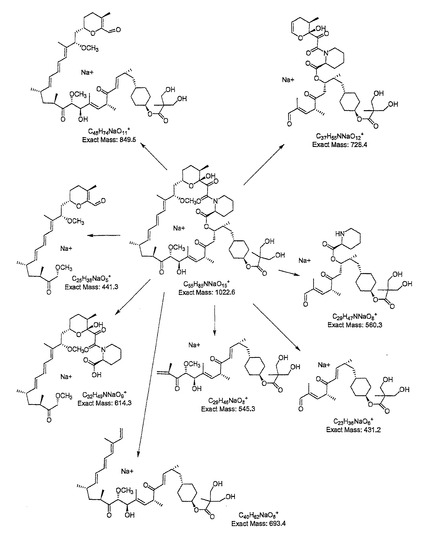
【図4】
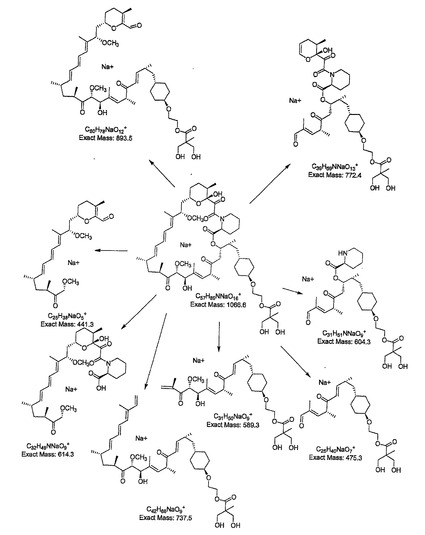
【図5】
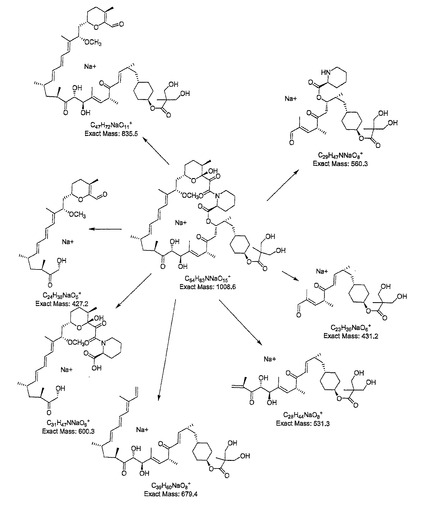
【図6】
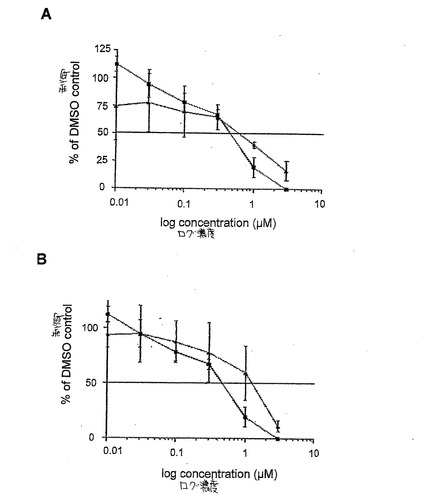
【図2】
【図3】
【図4】
【図5】
【図6】
【公表番号】特表2008−532991(P2008−532991A)
【公表日】平成20年8月21日(2008.8.21)
【国際特許分類】
【出願番号】特願2008−500271(P2008−500271)
【出願日】平成18年3月10日(2006.3.10)
【国際出願番号】PCT/GB2006/000853
【国際公開番号】WO2006/095185
【国際公開日】平成18年9月14日(2006.9.14)
【出願人】(507304591)バイオティカ テクノロジー リミテッド (2)
【氏名又は名称原語表記】BIOTICA TECHNOLOGY LIMITED
【住所又は居所原語表記】112 Hills Road, Cambridge CB2 1PH, Great Britain
【Fターム(参考)】
【公表日】平成20年8月21日(2008.8.21)
【国際特許分類】
【出願日】平成18年3月10日(2006.3.10)
【国際出願番号】PCT/GB2006/000853
【国際公開番号】WO2006/095185
【国際公開日】平成18年9月14日(2006.9.14)
【出願人】(507304591)バイオティカ テクノロジー リミテッド (2)
【氏名又は名称原語表記】BIOTICA TECHNOLOGY LIMITED
【住所又は居所原語表記】112 Hills Road, Cambridge CB2 1PH, Great Britain
【Fターム(参考)】
[ Back to top ]