
ラパマイシン炭酸エステル類似体、薬剤組成物、製造方法及び用途
本発明は、構造式Iに示すラパマイシン炭酸エステル類似体、その薬学的に受容可能な塩、薬剤組成物、その製造方法及び用途に関する。前述ラパマイシン炭酸エステル類似体は構造式Iに示す構造を有し、抗腫瘍薬として適用できる。ラパマイシンと比較すると、構造式Iに示すラパマイシン炭酸エステル類似体は、水溶性、薬理学的な性質及び薬物代謝学的な性質などの特性が改善されている。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、薬物化学領域に属し、具体的に、新規な構造を有するラパマイシン炭酸エステル類似体(Rapalogs)又はその薬学的に受容可能な塩、及び該化合物を含む薬剤組成物に関する。また、本発明は、該化合物の製造方法、該化合物の抗腫瘍薬及び/又は抗癌薬或いは免疫抑制薬の研究における用途にも関する。
【背景技術】
【0002】
癌は、細胞の異常増殖及び転移を特徴とする一種の疾病であり、すでに人間の健康を脅かす疾病の一つとなっている。世界保健機関(WHO)の統計によれば、世界において癌の新規患者が毎年六百万人に達している。中国において、癌はすでに脳血管疾患に次ぐ致死性病因となっている。
【0003】
現在、臨床に良く使用されている抗腫瘍薬が主に細胞毒性薬剤であり、選択性が悪く、毒性副作用が強く、耐薬性を生じやすいなどの欠点が存在する。近年のゲノム工学技術、分子腫瘍学及び分子薬理学に関する研究の著しい発展に伴い、細胞癌化の本質が細胞シグナル伝達の失調にあり、大部分の腫瘍のシグナル伝達経路の活性が高すぎて、細胞が無限に増殖することが認知されてきている。細胞シグナル伝達分子は、新型抗腫瘍薬を探すための重要な切り口であり、一部の腫瘍細胞の分化増殖に相関する細胞シグナル伝達経路の重要な酵素を薬品に作用させて標的のスクリーニングを行い、選択的に標的に作用する効果的、且つ毒性の低く、特異性の強い新型抗腫瘍薬を発見することがすでに現在の抗腫瘍薬の研究開発の新しい方向とされている。
【0004】
PI3K−mTORシグナル経路は、最も重要なチロシンキナーゼシグナル伝達の手段の一つである。ホスホイノシチド3−キナーゼ(PI3K)がリン酸化によりプロティンキナーゼB(PKB)を活性化させ、後者が哺乳類動物のラパマイシン標的(mTOR)をリン酸化させて活性化させる。mTORは、直接的又は間接的に多数の細胞増殖及び成長に相関するプロセスの調節に関与し、細胞増殖の中心的な役割を果たすと認識されている。多くの研究結果によると、腫瘍細胞において、PI3K− mTORシグナル経路が異常な伝達を示し、腫瘍の発生及び発展において重要な役割を果たしている。従って、該経路を遮断し、特にmTORの活性を抑制することにより、特異的に腫瘍細胞の成長を抑制できる。PI3K− mTORシグナル伝達経路は、すでに有望な抗腫瘍治療の標的となっている。
【0005】
ラパマイシン(Rapamycin)は、一種のトリエンマクロライド抗生物質であり、シロリムス(Sirolimus)とも呼ばれる。1975年にWyeth Ayerst実験室によりRapanui島で分離したストレプトマイセス・ハイグロスコピカスを発酵させて初めて得られ、抗菌活性を有するとともに、効果的な免疫抑制薬としてすでに臨床治療に用いられている。最近の研究により、ラパマイシンがmTORの特異性抑制剤として明らかな抗腫瘍活性を有することが証明された。in vitroでは、約1ng/mlのラパマイシンだけで、明らかに横紋筋肉腫細胞の成長を抑制できる。世界中の多数の研究室による研究においても、Rapamycinが良い腫瘍治療候補薬品であると支持されている。横紋筋肉腫、神経芽細胞腫、膠芽腫、髄芽腫、小細胞肺癌などの腫瘍細胞に対して、強い抑制作用を有し、また腫瘍細胞の成長に対する抑制がmTORと結合する結果であることが確認されている。ラパマイシンが前臨床段階において良い抗癌活性を示しているが、そのマクロライドの低水溶性と化学安定性の理由で、臨床開発が制限されている。
【0006】
近年、各大企業により数種のmTORの腫瘍標的治療に用いるラパマイシン類似体を開発されている。その中、代表的なものとして、Wyeth社により開発されたCCI−779(TemRapamycin)、Novarti社により開発されたRAD−001(Everolimus)、及びAriad社のAP23576である。これらの類似物はラパマイシンと類似な抗腫瘍作用を有し、かつ薬理学的な性質が改善され、明らかな毒性副作用も有しない。CCI−779が静脈注射に適用でき、すでに末期腎臓癌患者の臨床治療に用いられている。RAD−001は経口用として、小細胞肺癌などに対する治療が第二相臨床試験段階まできている。現在AP23576の血液癌及び固形癌に対する治療は第三相臨床試験段階であり、良い製薬の可能性を示している。従って、ラパマイシンを母体として構造の改造を行うことで、高い応用価値を有し、効果的、低毒性、且つ特異性の強い抗腫瘍薬を発見することが期待されている。
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明は、ラパマイシンの31位と42位のヒドロキシ基に対して構造修飾又は改造を行うことにより、一連の体内外の抗腫瘍及び/又は抗癌活性或いは免疫抑制活性を有し、かつ新規な構造を備えるラパマイシン炭酸エステル類似体を合成した。水溶性、体内外の薬剤効果、経口バイオアベイラビリティ、薬物代謝などの方面の考察によると、更に研究を進める価値があり、抗腫瘍薬に用いられ、又は免疫抑制薬の候補薬として研究開発する価値があることが判明された。従って、本発明の一つの目的は、一種の新規構造を有するラパマイシン炭酸エステル類似物又は薬学的に受容可能な塩を提供することである。
【0008】
本発明のもう一つの目的は、前述ラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩を活性成分とする薬剤組成物を提供することである。
【0009】
本発明のもう一つの目的は、抗腫瘍及び/又は抗癌薬或いは免疫抑制薬の製造における前述ラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩の用途を提供することである。
【課題を解決するための手段】
【0010】
本発明で提供されたラパマイシン炭酸エステル類似体は構造式Iに示す構造を有しており、
【化1】
ここで、R1とR2は、各々独立に、H又は、
【化2】
であり、且つR1、R2同時にHではなく、その中nは1〜6の整数であり、R3は、
【化3】
又は
【化4】
であり、R4、R5とR6は、各々独立にH、炭素数1〜6のヒドロキシアルキル基、炭素数1〜6のアルキル基又は炭素数2〜6のアルケニル基であり、R7とR8が、各々独立にH又は炭素数1〜6のアルキル基である。
【0011】
本発明の好ましい実施形態において、前述R1とR2は、各々独立に、H又は、
【化5】
であり、且つR1、R2は同時にHではなく、その中、nは1〜4の整数であり、R3は、
【化6】
又は
【化7】
であり、R4、R5とR6が、各々独立に、H、炭素数1〜4のヒドロキシアルキル基であり、R7とR8が、各々独立に、炭素数1〜4のアルキル基である。
【0012】
本発明の好ましい実施形態において、前述R1とR2は、各々独立に、H又は、
【化8】
であり、且つR1、R2は同時にHではなく、その中、nは1〜2の整数であり、宜しくはR3が、
【化9】
又は
【化10】
であり、R7とR8が炭素数1〜4のアルキル基である。
【0013】
更に、本発明の代表的な化合物が以下の化合物の何れかである:
【化11】
【化12】
【化13】
【化14】
【0014】
本発明のラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩において、前述R3の中にキラル中心を含有する場合、前述ラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩は、各種の光学異性体又は光学異性体の混合物であっても良い。
【0015】
本発明は、構造式Iに示すラパマイシン炭酸エステル類似体の製造方法を提供する。即ち、R1、R2が同じく
【化15】
であり、R3が
【化16】
又は
【化17】
である場合、以下の方法により製造できる。
【0016】
構造式1に示すアルコールは、p−トルエンスルホン酸の触媒作用で、DMSO、DMFなどの溶媒中において、カルボニル化合物R7COR8又はそのアセタール化合物(アセトン、2,2−ジメトキシプロパンなど)と反応し、構造式2に示すアルコールが得られる。アルカリにより、トリホスゲンが構造式2に示したアルコールと反応し、アシルクロリド3を形成する。また、アルカリにより、該アシルクロリド3がラパマイシンと反応し、R3が
【化18】
である、31位、42位の二置換ラパマイシン炭酸エステル類似体を得る。更に、該ラパマイシン炭酸エステル類似体を加水分解することにより、R3が
【化19】
であるラパマイシン炭酸エステル類似体が得られる。
【0017】
反応式は以下のようである:
【化20】
式中、化合物1は市販で入手でき、例えば、国薬集団化学試材有限公司(Sinopharm Chemical Reagent Co., Ltd.)、Aldrich社等から購入できる。
【0018】
更に具体的に、アルコール1はオルト位の二つのヒドロキシ基の保護により、アルコール2を得る。DMA、DMF、アセトニトリル、ジクロロメタン又はテトラヒドロフラン溶媒において、ピリジン、トリエチルアミン又はジイソプロピルアミンなどのアルカリ性条件で、アルコール2がトリホスゲンと反応し、アシルクロリド3を得る。次に、DMA、DMF、アセトニトリル、ジクロロメタン又はテトラヒドロフラン溶媒において、ピリジン、トリエチルアミン、DMAP又はジイソプロピルアミンなどのアルカリ性条件で、ラパマイシンがアシルクロリド3と反応し、31位及び42位をエステル化された、ラパマイシン炭酸エステル類似体を得る。その中、ラパマイシンは福建科瑞薬業公司(Fujian Kerui Pharmaceutical Co., Ltd.)から購入できる。
【0019】
一方、本発明のラパマイシン炭酸エステル類似体において、R1とR2が異なり、各々独立に、H又は、
【化21】
であり、R3が
【化22】
又は
【化23】
である場合、ラパマイシンの31位、42位のヒドロキシ基の選択的保護により、異なる一置換又は二置換の炭酸エステル類似体が得られても良い。しかし、ラパマイシンが31位及び42位において二つの第2級アルコール基を有するため、選択的にラパマイシンの42位又は31位でのモノエステル化を実現することが以前から困難であった。米国特許第6,277,983号に二段階で42位のモノエステル化合物の製造方法を記載したが、実際の操作性が悪く、且つ長時間及び低温条件が必要である。本発明者は、米国特許第6,277,983号の方法を実施したとき、反応過程においてラパマイシンがすぐラパマイシンの31位、42位の二置換生成物に変化することを発見した。また、本発明者は、時間の延長とともに、前述二置換生成物が反応条件で、更にラパマイシン31位の一置換生成物及び一部のラパマイシン生成物と変化することを発見した。TLCにより反応過程を追跡し、反応時間を制御することにより、ラパマイシンの31位のみ置換された生成物の製造が可能となる。
【0020】
本発明者は、適切な比例のイミダゾールとクロロトリメチルシランを用い、ジクロロメタン、ジクロロエタン、酢酸エチル、テトラヒドロフラン、アセトニトリル又はDMFなどを溶媒として使用し、ラパマイシンにより室温条件で高速的、且つ効率的に31位のみ保護された生成物Rapamycin−31−OTMSを得た。それに基づき、42位のヒドロキシ基をTBSにより保護してから、安定性の悪い31位でのシリル系保護基を脱離させる方法により、42位のみ保護するRapamycin−42−OTBSを得ることが可能である。
【0021】
反応式が以下のようである。
【化24】
【0022】
式中、Rapamycin−31−OTMSの製造において、該反応の溶媒がジクロロメタン、ジクロロエタン、酢酸エチル、テトラヒドロフラン、アセトニトリル、N,N−ジメチルホルムアミドであり、反応温度が0℃〜40℃であり、反応時間は2〜48時間である。反応中のラパマイシン:イミダゾール:クロロトリメチルシランの適切な当量比は1:5〜30:2〜6であり、最も宜しい当量比は、1:10〜15:2〜4である。
【0023】
続いて、Rapamycin−31−OTMSが直接アシルクロリド3、
【化25】
と反応し、42位をエステル化する生成物が得られ、31位の保護基を脱離させた後、対応する42位をモノエステル化したラパマイシン炭酸エステル類似体が得られる。
【0024】
42位のみ保護する生成物Rapamycin−42−OTBSとアシルクロリド3、
【化26】
と反応させ、31位をエステルした生成物を得て、42位の保護基を脱離させた後、対応する31位のみモノエステル化したラパマイシン炭酸エステル類似体を得る。
【0025】
本発明のラパマイシン炭酸エステル類似体の薬学的に受容可能な塩は、本発明のラパマイシン炭酸エステル類似体から通常の方法により製造できる。
【0026】
本発明の薬剤組成物は、治療的有効量の活性成分としての一種又は数種の本発明に記載のラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩と、一種又は数種の薬学的に受容可能なキャリアとを含む。
【発明の効果】
【0027】
一方、本発明者は実験により、本発明のラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩が、ラパマイシンより明らかに高い抗腫瘍又は抗癌活性を有し、薬理学及び薬物代謝学の性質が良く、ヒト横紋筋肉腫、前立腺癌、非小細胞肺癌、乳癌、結腸癌、腎臓癌、肺腺癌、子宮頸癌又は白血病を治療するための薬剤の製造に応用できることを発見した。また、本発明のラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩は、ラパマイシンの水溶性を改善したほか、ラパマイシンに相当又はそれより優れた免疫抑制活性を維持している。
【0028】
本発明で提供された抗腫瘍化合物は、各種の腫瘍細胞及び/又は癌細胞に有効に対抗でき、且つラパマイシンに比べ、ヒドロキシ基などの水溶性、極性基を導入することにより、水溶性が増強され、薬学的な性質が改善されている。in vitro実験中に、異なる腫瘍株における実験により、抗腫瘍活性が明らかにラパマイシンより優れていることを示された(表4−8及び図3−9を参照)。細胞レベルの研究により、Y50がRh30、PC−3、MCF−7、CAKI−1及びHL−60腫瘍細胞の成長に対して、ラパマイシンに相当する抑制作用を有しており(図1に示す)、Y50が濃度依存的にRh30、PC−3、MCF−7及びCAKI−1細胞中のmTORの下流因子のリン酸化促進能力を抑制でき、同濃度でその抑制能力がラパマイシンに相当している(図2に示す)ことが判明された。SPR(表面プラズモン共鳴)の結果によると(表4及び図9に示す) 、1)ラパマイシン、CCI−779、Y50及びY31はいずれも濃度依存的にFKBP12と結合できる。ラパマイシンと比べると、同濃度でY50及びY31がラパマイシンより高いRU(Response Unit)を有し、同濃度のY50及びY31とFKBP−12の結合がラパマイシンより強い、 2)Y50及びY31とFKBP12の結合が飽和に達するまで必要な濃度がラパマイシンより低い、3)Y50とY31のFKBP12との解離速度がラパマイシンより低く、同様にCCI−779よりも低く、Y50及びY31の解離係数がラパマイシン及びCCI−779より低い。動物実験の研究により、Y50の経口投与がヒト横紋筋肉腫RH−30異種移植マウスの移植腫に対して、明らかにラパマイシンより優れた成長抑制作用を有する(表5及び図3から図4)。Y50の経口投与によりヒト前立腺癌PC−3異種移植マウスの移植腫に対する成長抑制作用も、明らかにラパマイシンより優れる(表6及び図5から図6に示す)。そのT/Cがそれぞれ10.0%と40.2%であり、陽性対照としてのラパマイシンの同用量群に対応するT/Cがそれぞれ30.9%と46.5%であった。Y31の経口投与は、ヒト骨肉腫U2SO異種移植マウスの移植腫の成長に対して、明らかな成長抑制作用を有する。低用量群(2.5mg/kg)の場合、CCI−779及びラパマイシンは、ヒト骨肉癌U2SO異種移植マウスの移植腫の成長に対して、明らかな抑制作用を有さず、そのT/C値がそれぞれ69.0%、60.0%であった。なお、低用量(2.5mg/kg)の場合、化合物Y31が該腫瘍の成長に対する抑制作用が明らかにラパマイシン及びCCI−779より優れている(表8及び図8に示す)。
【0029】
本発明で提供された化合物を更に研究するための抗腫瘍実験中に、該化合物及びラパマイシンは、市販しているラパマイシン類似体CCI−779と比べると、Y31がより良い薬物動態パラメータを示している(表9〜12及び図10〜12に示す)。これは、ヒドロキシ基などの水溶性、極性基を導入することと関係する可能性がある。特に、Y31を投与した後、薬物が腫瘍組織での選択的吸収が一番良かった(表12及び図12に示す)。実験によると、マウスにY31を投与した後、体内で迅速に代謝物ラパマイシン(Rapamycin)と変化し、血漿及び組織中のプロトタイプ薬の濃度が低く、最大濃度が20ng/ml(又はng/g)未満で、薬剤を投与してから5時間でプロトタイプ薬を検出できなかった。Y31を投与した後、血漿、肝臓、及び腫瘍中に、Rapamycinの暴露量とRapamycin投与群の比率がそれぞれ1.22、1.32、1.93であった。
【0030】
本発明のラパマイシン炭酸エステル類似体の薬学的に受容可能な塩は、前述の抗腫瘍活性及び優れた薬物動態パラメータを示すだけではなく、ラパマイシンに相当する又はそれより優れた免疫抑制活性も保持している。ラパマイシンを対照として、化合物Y31 を例として系統的免疫抑制活性実験を行った(結果を表13〜15及び図13〜16に示す)。
【0031】
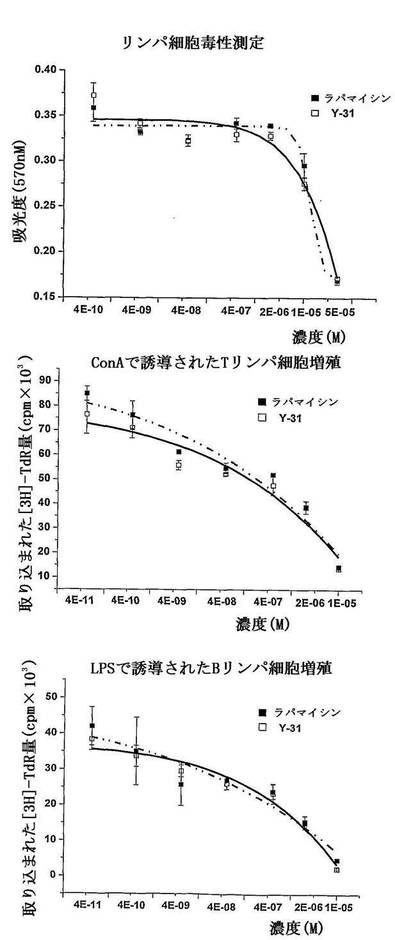
(1)マイトジェン/同種異体抗原で誘導された正常マウス脾臓リンパ細胞の増殖活性に対するRapamycin及びY31の影響:
結果に示すように、Rapamycin及びその誘導体Y31がマイトジェン/同種異体抗原で誘導されたリンパ細胞の増殖反応を明かに抑制し、強いin vitro免疫抑制活性を有する(表14及び図13に示す)。
【0032】
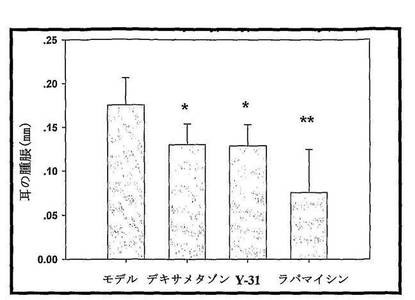
(2)マウス遅延型過敏反応に対するRapamycin及びY31の影響:
DNFBで誘導されたDTH反応は、Th1細胞を介した、T細胞の活性化及び数種の細胞因子の発現を含むアレルギー反応である。我々は、BALB/cマウスのDTH応答中での化合物の役割を測定し、実験結果を図14に示した。DNFBで誘導されて遅延型過敏反応を生じたマウスを対照群として使用し、その両耳の耳の腫脹の平均増加が0.175mmである。陽性対照薬Dex 2mg/kg投与群のマウスの耳の腫脹の平均増加が0.13mmであり、対照群と比較すると、明らかな差異を有する。Rapamycin投与群のマウスの耳の腫脹の平均増加が0.076mmであり、対照群と比較すると、差異が明らかである。Y31投与群のマウスの耳の腫脹の平均増加が0.129mmであり、対照群と比較すると、差異が明らかである。
実験結果によれば、Rapamycin及びY31が明かにDNFBで誘導されたマウス遅延型過敏反応を抑制できる(図14に示す)。
【0033】
(3)SRBCで誘導されたマウス脾臓リンパ細胞中の特異的抗体産生細胞に対するRapamycin及びY31の影響:
Rapamycin(1.5mg/kg)及びその誘導体Y31(1.5mg/kg)を腹腔内投与すると、マウス脾臓中の特異的な抗SRBC抗体産生細胞の生成量を明かに抑制でき、その抑制作用が陽性対照薬CsAより強く、マウスの体液性免疫機能に対して、明らかな抑制作用を有していた(表15に示す)。
【0034】
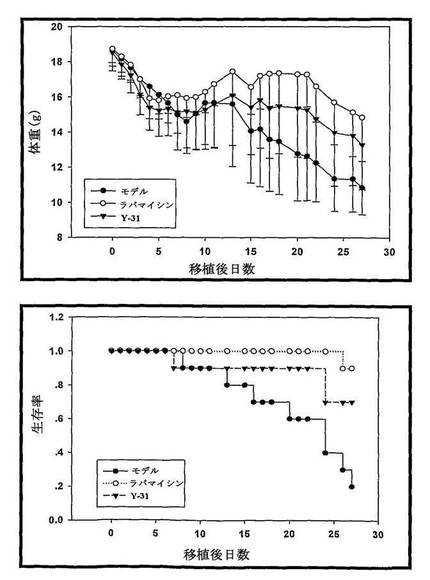
(4)マウス急性移植片対宿主病(aGVHD)に対するRapamycin及びY31の薬効学研究:
本実験の結果により、急性移植片対宿主病(aGVHD)の動物モデルに対して、Rapamycin及び誘導体Y31が良い治療効果を有することが証明された(図15に示す)。
【0035】
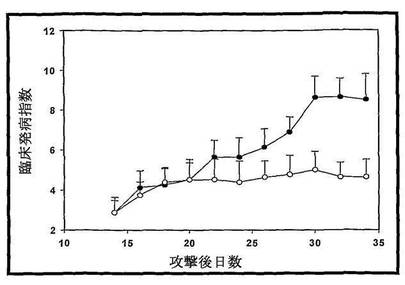
(5)DBA/1マウスでの牛II型コラーゲン誘導関節炎に対するY31の治療効果
牛II型コラーゲンを用いて皮下注射を二回行うことで、DBA/1マウス関節炎を誘導できる。刺激してから四日後関節腫脹が現れ、一週間後100%のマウスが関節炎を発症した。且つ、関節腫脹が進行的に悪化し、第14日から薬を投与開始した。Y31を投与してから、明かにCIAの発症程度が低減し、マウスの四肢(足、爪)の腫脹が明らかに減少された。Y31の経口投与は、DBA/1マウスでのコラーゲン誘導関節炎の発病を抑制できる(図16に示す)。
【0036】
本発明で提供されたラパマイシン炭酸エステル類似体は、優れた腫瘍抑制活性及び免疫抑制活性、比較的に良い薬物動態パラメータを有し、製造方法が簡単で、操作性に優れたほか、高い生産率、及び良好な薬剤開発可能性を有している。
【図面の簡単な説明】
【0037】
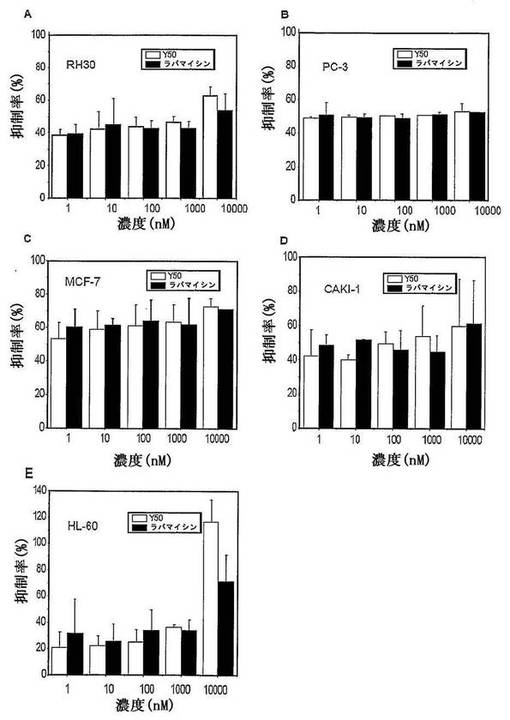
【図1】異なる濃度の化合物Y50のRh30(ヒト横紋筋肉腫、A)、PC−3(ヒト前立腺癌、B)、MCF(ヒト乳癌、C)、CAK−1(ヒト腎臓細胞癌、D)及びHL−60(ヒト白血病、E)細胞の成長に対する抑制を示す図である。
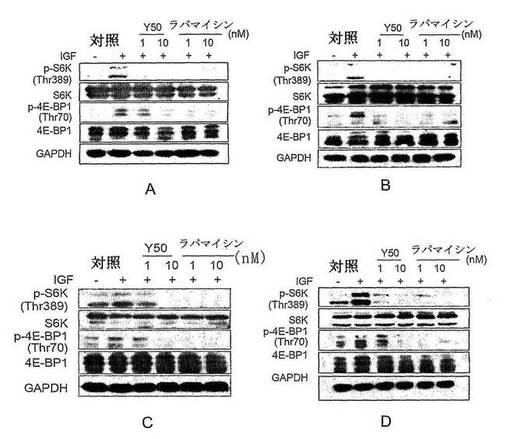
【図2】Y50のRh30、PC−3、MCF−7.CAK−1細胞中のp70S6K及び4E−BP1に対するリン酸化レベルの評価を示す図である。
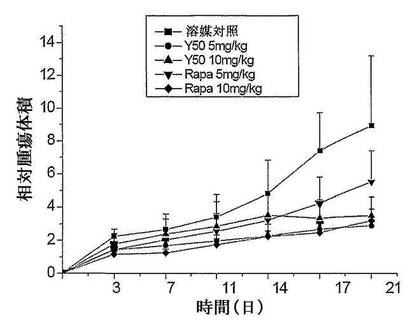
【図3】化合物Y50のヒト横紋筋肉腫Rh30異種移植マウスの移植腫の成長に対す抑制曲線を示す図である。
【図4】化合物Y50のヒト横紋筋肉腫Rh30異種移植マウスの移植腫の成長に対する抑制を示す図である。
【図5】化合物Y50のヒト前立腺癌PC−3異種移植マウスの移植腫の成長に対する抑制曲線を示す図である。
【図6】化合物Y50のヒト前立腺癌PC−3異種移植マウスの移植腫の成長に対する抑制を示す図である。
【図7】化合物Y50、Y31、Y31−1、Rapamycin(ラパマイシン)のヒト横紋筋肉腫Rh30異種移植マウスの移植腫の成長に対する抑制曲線を示す図である。
【図8】化合物Y31、CCI−779、 Rapamycin(ラパマイシン)のヒト骨肉癌U2SO異種移植マウスの移植腫に対する実験治療効果を示す図である。
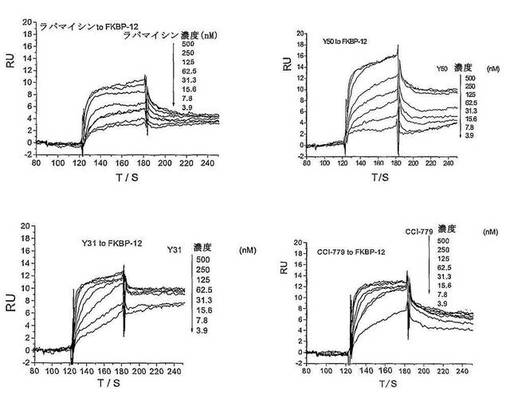
【図9】SPR(表面プラズモン共鳴)の方法により測定した小分子化合物とタンパク質FKBP−12との結合活性を示す図である。
【図10】マウスにそれぞれY31、CCI−779及びRapamycin(ラパマイシン)を投与した後、血漿中のRapamycin(ラパマイシン)の濃度−時間曲線を示す図である。
【図11】マウスにそれぞれY31、CCI−779及びRapamycin(ラパマイシン)を投与した後、肝臓中のRapamycin(ラパマイシン)の濃度−時間曲線を示す図である。
【図12】マウスにそれぞれY31、CCI−779及びRapamycin(ラパマイシン)を投与した後、腫瘍中のRapamycin(ラパマイシン)の濃度−時間曲線を示す図である。
【図13】マイトジェン/同種異体抗原で誘導された正常マウス脾臓リンパ細胞の増殖活性に関するRapamycin(ラパマイシン)及びY31の影響を示す図である。
【図14】マウス遅延型過敏反応に関するRapamycin(ラパマイシン)及びY31の影響を示す図である。
【図15】マウス急性移植片対宿主病(aGVHD)に関するRapamycin(ラパマイシン)及びY31の薬効学研究を示す図である。
【図16】DBA/1マウスでの牛II型コラーゲン誘導関節炎に関するY31の治療効果を示す図である。
【発明を実施するための形態】
【0038】
具体的な実施例に基づき、更に詳しく本発明を説明する。しかし、本発明は実施例に限定されない。
【0039】
<ラパマイシン炭酸エステル類似体の製造に関する実施例>
後述実施例中に、通常の後処理方法は、反応を終えた後、反応液に適量の水を加え、有機相と水相に分離し、水相が有機溶媒により充分に抽出された後、有機相を合併する。必要に応じて、順次に5%HCl溶液及び/又は飽和NaHCO3溶液、水及び飽和食塩水を用いて洗浄する。無水Na2SO4又は無水MgSO4により有機相を乾燥させ、濾過してから減圧スピン乾燥を行い、粗生成物を得て、更にカラムクロマトグラフィーにより純化した後、最終生成物を得た。
【0040】
後述製造の実施例中に、Varianにより製造されたMercury−Vx600M装置を用い、NMRを測定した。δH/C 7.26/77.0ppm(CDCl3)をNMRキャリブレーションに使用した。試薬が主に上海化学試剤公司(Shanghai Chemical Reagent Co., Ltd.)により提供され、生成物の純化が主にカラムクロマトグラフィーを使用した。シリカゲル(200−300目)について、カラムクロマトグラフィーに用いたシリカゲルのタイプはMacro−pored silica Gel (ZLX−II)であり、青島海洋化工場分場(Qingdao Haiyang Chemical Co., Ltd.)により製造されたものである。
【0041】
<製造の実施例1:化合物Y230、Y72及びY50の製造>
【化27】
276mg(3mmol)のグリセロールを取り、2mlのDMSO中に溶解させ、窒素雰囲気で、0.44mlの2,2−ジメトキシプロパンを注入し、触媒量のp−トルエンスルホン酸を加えた。室温で数時間攪拌し、TLCにより反応終了まで追跡し、通常の後処理を経て合計173mgの液状の生成物2iを得た。
【0042】
50mlのナスフラスコに173mg(1.31mmol)の化合物2i、130mg(0.44mmol)のトリホスゲンを加え、窒素雰囲気で25mlの二重蒸留CH2Cl2溶液を注入し、氷水浴で170μl(1.31mmol)の乾燥ピリジンを一滴ずつ滴加し、滴加を終えたら、自然に室温まで昇温して2時間反応させ、200mg(0.22mmol)のラパマイシンを加え、更に0.2mlのピリジンを追加し、TLCにより反応終了まで追跡した。次に、ナスフラスコに1NのHClを加え弱酸性に中和させ、ジクロロメタンで抽出し、ジクロロメタンの抽出液を水洗い、飽和食塩水により洗浄し、無水硫酸マグネシウムにより乾燥して濃縮し、石油エーテル/アセトン(体積比5/1)を展開液としてカラムクロマトグラフィーにより分離させて240mgの化合物Y230を得て、副生成物として20mgのY72を得た。
【0043】
240mgの化合物Y230を取り、3mlのTHFに溶解させ、0〜5℃で1.7mlの2NのH2SO4を滴加し、TLCにより反応終了まで追跡した。5%NaHCO3を加え弱アルカリ性まで中和させ、酢酸エチルで抽出し、酢酸エチルの抽出液を飽和食塩水により洗浄し、無水硫酸マグネシウムにより乾燥し、減圧濃縮し、石油エーテル/アセトン(体積比1/1)を展開液としてフラッシュカラムクロマトグラフィーにより分離させて120mgの化合物Y50を得た(全収率:38%)。
【0044】
【表1】
【0045】
<製造の実施例2:化合物Y31の製造>
【化28】
【0046】
400mg(0.44mmol)のラパマイシン、449mg(6.6mmol)のピリジンを20mlの二重蒸留CH2Cl2に溶解させ、0.22ml(1.76mmol) のクロロトリメチルシラン(TMSCI)を滴加した。滴加を終えた後、TLCにより追跡し、6時間ほど反応させ、減圧濃縮し、石油エーテル/アセトン(体積比4/1) を展開液としてカラムクロマトグラフィーにより合計277mgの化合物Rapamycin−31−OTMSを得た。
【0047】
50mlのナスフラスコに573mg(4.34mmol)の化合物3i、453mg(1.53mmol)のトリホスゲンを加え、窒素雰囲気で30mlの二重蒸留CH2Cl2溶液を注入し、氷水浴で377μl(4.67mmol)の乾燥ピリジンを滴加し、滴加を終えてから、自然に室温まで昇温して2時間反応させ、277mg(0.28mmol) のRapamycin−31−OTMSを加えた。4時間後TLCにより反応終了を追跡したら、弱酸性になるように1NのHClを加え、ジクロロメタンで抽出し、水洗い、飽和食塩水により洗浄し、無水硫酸マグネシウムにより乾燥し、濃縮し、石油エーテル/アセトン(体積比4/1) を展開液としてカラムクロマトグラフィーにより分離させて合計240mgの化合物Y44を得た。
【0048】
240mgの化合物Y40を4mlのTHFに溶解させ、0〜5℃で1.7mlの2NのH2SO4を滴加し、TLCにより反応終了まで追跡した。弱アルカリ性になるように、5%のNaHCO3を加え、酢酸エチルで抽出し、飽和食塩水により洗浄し、無水硫酸マグネシウムにより乾燥し、減圧濃縮し、石油エーテル/アセトン(体積比1.5/1) を展開液としてカラムクロマトグラフィーにより分離させて合計120mgの化合物Y31を得た。
【0049】
【表2】
【0050】
<製造の実施例3:化合物Y31−1の製造>
化合物Rapamycin−31−OTMSの製造に関して、製造の実施例2を参照する。
【0051】
25mlのナスフラスコに200mg(0.2mmol) の化合物Rapamycin−31−OTMS、206mg(3mmol) のピリジンを加え、7mlのDMFを注入し、更に184mg(1.22mmol) のtert−ブチルジメチルクロロシラン(TBSCI)を滴加した。TLCにより追跡し、室温で48時間反応させてから水を加えて希釈し、酢酸エチルで抽出し、水洗い、飽和食塩水により洗浄し、硫酸マグネシウムにより乾燥させ、石油エーテル/アセトン(体積比3/1) を展開液としてカラムクロマトグラフィーにより分離し、合計120mgの生成物Y028を得た。
【0052】
50mlのナスフラスコに173mg(1.31mmol)の化合物3i、130mg(0.44mmol)のトリホスゲンを加え、窒素雰囲気で25mlの二重蒸留CH2Cl2溶液を注入し、氷水浴で170μl(1.31mmol)の乾燥ピリジンを滴加し、滴加を終えてから、自然に室温まで昇温して2時間反応させ、120mg(0.12mmol) のY028を加え、更に0.2mlのピリジンを追加し、TLCにより反応終了まで追跡した。次に、弱酸性になるように、ナスフラスコに1NのHClを加え、ジクロロメタンで抽出し、水洗い、飽和食塩水により洗浄し、無水硫酸マグネシウムにより乾燥し、濃縮し、石油エーテル/アセトン(体積比3/1) を展開液としてカラムクロマトグラフィーにより分離させて合計100mgの化合物Y86を得た。
【0053】
100mgの化合物Y86を1.5mlのTHFに溶解させ、0〜5℃で0.8mlの2NのH2SO4を滴加し、TLCにより反応終了まで追跡した。弱アルカリ性になるように5%のNaHCO3を加え、酢酸エチルで抽出し、飽和食塩水により洗浄し、無水硫酸マグネシウムにより乾燥し、減圧濃縮させ、石油エーテル/アセトン(体積比1/1) を展開液としてフラッシュカラムクロマトグラフィーにより分離させて合計80mgの化合物Y31−1を得た。
【0054】
【化29】
【0055】
【表3】
【0056】
<生物実験の実施例>
<実施例1:細胞レベルの抗腫瘍活性測定実験>
I.Y50のRh30、PC−3、MCF−7、CAKI−1及びHL−60細胞の成長に対する抑制。
【0057】
異なる濃度の化合物により、Rh30細胞を72時間処理し、SRB(Sulforhodamine B)法により細胞の生存率を測定した。
【0058】
指数増殖期に属する前記の各種の腫瘍細胞を90μl/穴で96穴プレートに接種し、24時間接着培養し、更に10μl/穴で薬を加えた。各濃度に関して、同条件の穴を三つ設けた。また、相応な濃度の溶媒対照(生理食塩水)及アポトーシスを発生しない穴を設けた。37℃、5%CO2の条件で腫瘍細胞を72時間培養した後、培養液を取り除き、10%冷TCA(トリクロロ酢酸)を用いて細胞を固定させ、4℃で1時間放置してから、蒸留水により5回洗浄し、空気中で自然乾燥させた。1%氷酢酸から調製した4mg/mlのSRB(Sigma)溶液を100μl/穴で加え、室温で15分間染色し、上澄みを取り除き、1%酢酸により5回洗浄し、空気中で乾燥させた。最後に150μl/穴でTris溶液を加え、ELISAにより波長520nmでA値を測定した。以下の公式により、腫瘍細胞成長に対する抑制率を計算した。
【0059】
抑制率=(A520対照群−A520投与群)/ A520対照群×100%
【0060】
結果から図1に示すように、前記の各種の腫瘍細胞成長に対して、Y50がラパマイシンに相当する抑制作用を有することが証明された。
【0061】
II.Y50のRh30、PC−3、MCF−7及びCAKI−1細胞中のp70S6K及び4E−BP1のリン酸化レベルに対する抑制。
【0062】
一定の密度で細胞を12穴プレートに接種し、細胞が壁と接着する状態で一晩放置し、新鮮な無血清培養液に変え、飢餓状態で24時間保持した後、相応な濃度の薬を加えて1時間反応させ、IGFにより10分間刺激させ、サンプルを採取し、Western blotにより細胞中のp70S6K及び4E−BP1のリン酸化レベルを測定した。結果を図2に示し、実験結果からY50が濃度依存的に各種の腫瘍細胞中のmTORの下流因子のリン酸化促進能力を抑制できることが示された。同濃度で、その抑制能力がラパマイシンと相当する。
【0063】
III.化合物Y50、Y31と標的タンパク質FKBP−12の結合活性の測定実験。
【0064】
1.材料と装置:
(1)FKBP−12タンパク質はSigma社から購入した。
(2)HBS−EP緩衝液(10mM Hepes,150mM NaCl,3.4mM EDTA,0.005%(v/v) Surfactant P20,pH7.4)。
(3)活性化試薬のEDC、NHS及びブロッキング試薬(Blocking reagent)のEthanolamineなどは、BLACORE AB社(Uppsala,Sweden)から購入した。
(4)Biacore3000、CM5チップは、BLACORE AB社(Uppsala,Sweden)から購入した。
【0065】
2.実験方法:
(1)FKBP−12タンパク質のカップリング:
Biacore3000プログラム中のアミンカップリングのWizardを用いてTubulinタンパク質を、CM5チップのFC2通路にカップリングさせた。pH4.6、10mM NaACで、3.3g/LのFKBP−12タンパク質を最終濃度の66μg/mlまで希釈した。0.1Mの1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩(EDCI)と0.1MのN−ヒドロキシスクシンイミド(NHS)を1:1混合させて、チップ表面において20μL/minの流速で7分間試料を導入し、タンパク質溶液を注射し、活性化チップの表面をブロッキングさせるように、pH8.5、1Mのエタノールアミンを7分間導入した。
【0066】
化合物の初期スクリーニング(Preliminary screening)及びダイナミクス測定:
SPR(表面プラズモン共鳴)の方法を用いて小分子化合物とFKBP−12タンパク質の結合活性を測定した。化合物の原液濃度が10mMであり、HBS−EP緩衝液を一定の比例で希釈し、Biacore3000プログラム中のダイナミクス解析Wizardによりダイナミクス実験(Dynamic experiment)を行った。得られた実験データに関して、Biacore3000解析ソフト中の1:1 Langmuir結合モデル又は定常状態モデルに基づき近似を行い、確実な動的定数及び熱力学定数を得た。
【0067】
SPR実験結果(表4及び図8に示す):
1.Rapamycin(ラパマイシン)、CCI−779、Y50及びY31の何れもが濃度依存的にFKBP12と結合できる。Y50及びY31は、Rapamycinと比較すると、同濃度でのRU(Response Unit)がRapamycinより高く、同濃度のY50及びY31とFKBP−12の結合がRapamycinより強いことを意味する。
2.Y50及びY31とFKBP12の結合の飽和に達するまでに必要な濃度がRapamycinより低い。
3.Y50及びY31とFKBP12の解離速度がRapamycinより低く、かつCCI−779よりも低い。Y50及びY31の解離定数がRapamycin及びCCI−779より低い。
【0068】
【表4】
【0069】
<実施例2:動物レベルの抗腫瘍活性測定実験>
実験目的:ヒト横紋筋肉腫RH−30異種移植マウスの移植腫の成長に対する抑制作用を観察する。
【0070】
実験物:5%ツイン(Tween)80、5%PEG400及びDDWを溶媒として適用し、Y50を経口投与製剤に調製した。
【0071】
陽性対照薬:5%ツイン80、5%PEG400及びDDWを溶媒として適用し、ラパマイシンを経口投与製剤に調製した。
【0072】
投与量の設置:Y50の経口投与量を5mg/kg、10mg/kgの二つの用量群と設け、毎日一回の経口投与を行った。ラパマイシンについて、同様な投与量で経口投与を行った。
【0073】
動物:BALB/cAマウス、オス、40〜45日齢、体重21±2g、中国科学院上海薬物研究所により提供された。生産ライセンス:SCXK(濾) 2004−0002。各群の動物数:陰性対照群6匹、投与群6匹。
【0074】
移植腫:ヒト横紋筋肉腫RH−30異種移植マウスの移植腫。ヒト横紋筋肉腫RH−30細胞株をマウスの皮下に接種して作成した。5×106の細胞接種量で接種して移植腫を形成した後、マウス体内に遺伝して3世代目以降を使用した。
【0075】
実験方法:成長増殖の活発な時期の腫瘍組織を約1.5mm3にカットし、無菌条件で、マウスの右側の脇の下の皮下に接種した。マウスの移植腫に対して、ノギスにより移植腫の直径を測定し、腫瘍が100〜200mm3まで成長してから、ランダムに動物を組み分けした。実験群に経口投与し、毎日一回投与を行い、三週間連続行った。陽性対照薬のラパマイシンについても、同様な方法で、同用量を投与し、合計三週間行った。陰性対照群に対して、0.2ml/匹で溶媒を経口投与した。毎週腫瘍の直径を二回測定し、同時にマウスの体重も測定した。腫瘍体積(Tumor volume, TV)の計算式は、TV = 1/2×a×b2であり、その中、a、bがそれぞれ長さ、厚さを示す。測定した結果により、相対腫瘍体積(Relative tumor volume,RTV)を計算した。その計算式は、RTV = Vt/V0であり、その中、V0は投与開始のとき(すなわち、d0のとき)の測定した腫瘍体積で、Vtが毎回測定するときの腫瘍体積である。抗腫瘍活性の評価指標は、相対腫瘍増殖率T/C(%)である。
【0076】
計算式:T/C(%)=(TRTV/CRTV)×100。その中、TRTVは治療群RTVで、CRTVは陰性対照群RTVである。
【0077】
治療の評価指標:T/C(%)>60%であれば、無効である。T/C(%)≦60%、且つ統計学処理によりp<0.05であれば、有効である。
【0078】
結果:Y50のヒト横紋筋肉腫RH−30異種移植マウスの移植腫の成長に対する抑制作用を表5及び図3、図4に示す。実験結果により、三週間連続毎日一回5mg/kg、10mg/kg のY50を経口投与した、二つの用量群がヒト横紋筋肉腫RH−30異種移植マウスの移植腫の成長に対して明らかな抑制作用を有し、そのT/Cがそれぞれ32.5%、32.9%であり、陽性対照群のラパマイシンの高用量群(10mg/kg)に相当した。一方、ラパマイシンの低用量群(5mg/kg)が、ヒト横紋筋肉腫RH−30異種移植マウスの移植腫の成長に対して、明らかな抑制作用がなく、そのT/C値が61.8%であった。実験群のマウスの死亡例が見られなかった。
【0079】
結論:ヒト横紋筋肉腫RH−30異種移植マウスの移植腫に対して、Y50経口投与がラパマイシンより明らかに優れた成長抑制作用を有する。下の表5、図3及び図4に示す。
【0080】
【表5】
【0081】
同様な実験により、Y50のヒト前立腺癌PC−3異種移植マウスの移植癌の成長に対する抑制作用を観察した。結果に示すように、異なる用量群において、ヒト前立腺癌PC−3異種移植マウスの移植癌に対する成長抑制作用について、Y50経口投与がラパマイシンより明らかに優れる。そのT/Cがそれぞれ10.0%、40.2%であった。一方、陽性対照のラパマイシンの同用量群に対応するT/Cがそれぞれ30.9%、46.5%であった。下の表6、図5及び図6に示す。
【0082】
【表6】
【0083】
【表7】
【0084】
前記と同様の実験により、Y50、Y31、Y31−1、Rapamycinのヒト横紋筋肉腫RH−30異種移植マウスの移植腫の成長に対する抑制作用を観察した。ヒト横紋筋肉腫RH−30異種移植マウスの移植腫の成長に対して、Y31、Y50、Y31−1の経口投与が明らかな成長抑制作用を有した。Y31、Y50の該腫瘍の成長に対する抑制作用がラパマイシンより優れ、その中、ラパマイシンが低用量群(2.5mg/kg)のとき、ヒト横紋筋肉腫RH−30異種移植マウスの移植腫に対して、明らかな抑制作用を有していない。そのT/C値が63.7%であった。一方、Y31は低使用量(2.5mg/kg)のときでも、比較的に高い抑制作用を達成でき、Y50及びラパマイシンの高用量群(5mg/kg)より明かに優れる。表7及び図7に示す。
【0085】
【表8】
【0086】
同実験により、Y31、CCI−779、Rapamycinのヒト骨肉癌U2SO異種移植マウスの移植腫の成長に対する抑制作用を観察した。結果により、ヒト骨肉癌U2SO異種移植マウスの移植腫の成長に対して、Y31経口投与が明らかな成長抑制作用を有することが示された。低用量群(2.5mg/kg)のとき、ヒト骨肉癌U2SO異種移植マウスの移植腫に対して、CCI−779及びラパマイシンが明らかな抑制作用を有しない。そのT/C値はそれぞれ69.0%、60.0%であった。一方、低用量(2.5mg/kg)のとき、化合物Y31の該腫瘍の成長に対する抑制作用がラパマイシン及びCCI−779より明かに優れる。
【0087】
<実施例3:Y31、Rapamycin及びCCI−779を投与した後のマウス体内におけるRapamycinの分布に関する研究実験>
本実験において、マウスにY31、Rapamycin及びCCI−779を投与した後のマウス体内におけるRapamycinの分布特徴に関して、検討を行った。マウスに薬剤を投与した後、異なる時間での血漿、肝臓及び腫瘍組織を採集し、液体クロマトグラフィー・タンデム質量分析法(LC−MS/MS)により血漿及び組織中のプロトンタイプ薬物及びRapamycinの濃度を測定した。マウスにY31、Rapamycin及びCCI−779を投与した後、血漿及び組織中のプロトンタイプ薬物及びRapamycinの濃度を表9〜11に示し、主な薬物動態パラメータを表12に示す。
【0088】
【表9】
【0089】
【表10】
【0090】
【表11】
【0091】
【表12】
【0092】
結果によると、ヒト横紋筋肉腫RH−30異種移植マウスの移植腫を移植した動物にY31、Rapamycin及びCCI−779をそれぞれ投与してからマウスにY31を投与した後、体内に迅速に代謝物Rapamycinと変化し、血漿及び組織中のプロトンタイプ薬の濃度が低く、最大濃度が20ng/ml又はng/gより低かった。投薬してから5時間でプロトンタイプ薬を検出できなくなった。Y31を投与した後、血漿、肝臓及び腫瘍中のRapamycinの暴露量とRapamycin投与群の比例がそれぞれ1.22、1.32、1.93であった。
【0093】
マウスにCCI−779を投与した後、血漿及び組織中にプロトンタイプ薬CCI−779及び代謝物Rapamycinを同時に検出でき、血漿、肝臓及び腫瘍組織中の代謝物Rapamycinとプロトンタイプ薬の暴露量の比例がそれぞれ10.4、0.94と1.89であった。CCI−779を投与した後、血漿、肝臓及び腫瘍中のRapamycinの暴露量とRapamycin投与群の比例がそれぞれ0.75、0.54と0.63であった。
【0094】
マウスにRapamycinを投与した後、血漿中にRapamycinが迅速に吸収され、ピークに達する時間が0.5〜2時間であった。肝臓中にピークに達する時間が0.5時間であり、血漿中の場合に近かった。腫瘍中にピークに達する時間が比較的に遅く、2〜5時間であった。Rapamycinは血漿及び肝臓でより速く除去される。
【0095】
三つの投与群において、投薬してから48時間、血漿及び肝臓中の濃度が、ピーク濃度の1.96%〜6.42%となった。Rapamycinは腫瘍中で比較的に遅く除去され、投与してから48時間、腫瘍中の濃度がピーク濃度の40%〜51%となった。三つの投薬群において、肝臓中のRapamycinの暴露量がそれぞれ血漿中の暴露量の2.26、2.01、1.51倍であり、腫瘍中のRapamycinの暴露量がそれぞれ血漿中の暴露量の1.66、1.05と0.87倍であった。
【0096】
Y31を陽性対照薬Rapamycin及びCCI−779と比較すると、共有の有効成分Rapamycinの肝臓中の暴露量がそれぞれ血漿中の暴露量の2.26、2.01と1.51倍であった。腫瘍中の暴露量がそれぞれ血漿中の暴露量の1.66、1.05と0.87倍であった。Y31が明らかな腫瘍組織特異性を有することが示された。
【0097】
<実施例4:細胞レベルの免疫抑制活性測定実験>
【0098】
【表13】
【0099】
化合物Y50、Y31、Y230の免疫抑制活性実験の結果により、化合物Y31のIC50値が50nMに達し、親化合物のラパマイシン及び化合物Y50より明かに優れ、化合物Y31が比較的に高い安全指数を有し、化合物Y50、Y230の免疫抑制活性がラパマイシンに相当することが示された。
【0100】
<実施例5:Y31の免疫抑制活性測定の系統的実験>
I.マイトジェン/同種異体抗原で誘導された正常マウス脾臓リンパ細胞の増殖活性に関するRapamycin及びY31の影響:
実験目的:
3H−チミジン(3H−thymidine)取り込みにより、マイトジェン/同種異型マウス脾臓リンパ細胞の混合培養による正常マウス脾臓リンパ細胞の増殖機能に対する、化合物のin vitro投与の影響を測定し、そのin vitro免疫抑制活性を評価する。
3−(4,5−ジメチル−2−チアゾリル)−2,5−ジフェニルテトラゾリウムブロミド (MTT)法により、正常マウス脾臓リンパ細胞活性に対して、化合物のin vitro投与の影響を測定し、その細胞毒性効果を評価した。
【0101】
測定薬物:
名称:Rapamycin、Y31。性状、含有量:白い粉末。
【0102】
調製方法:使用する前4℃で保存した。実験前にDMSOに溶解させ、原液を調製した。使用時、培養液で必要な濃度まで希釈した。細胞を培養するとき、DMSO中の最終濃度が<0.02%であった。該濃度のDMSOが細胞成長に影響しない。
【0103】
実験動物:由来、品種、系統:BALB/c純系マウス、メス、18〜20g、中国科学院上海実験動物中心から購入。動物生産ライセンス:SCXK(濾)2002−0010号。動物は上海薬物研究所SPF動物飼育施設で飼育。実験動物使用ライセンス:SYXK(濾)2003−0029号、少なくとも一週間飼育してから使用した。温度が22±1℃であり、湿度が55±5%であり、12時間交代の明暗サイクルで飼育した。餌と水が全部消毒してから動物が自由に摂取した。全部の実験について、完全に実験動物に関する条例に基づいて行った。
【0104】
実験方法及び評価:
MTT法によりマウス脾臓リンパ細胞の活性に対する化合物の影響を測定した。
100μl(4×105/孔)のマウス脾臓リンパ細胞の懸濁液を96穴プレートに接種するとともに、異なる濃度の化合物を加え、相応な溶媒対照及び培養液のバックグランド対照を設け、全体積が200μlであった。37℃で5%のCO2のインキュベータで48時間培養した。培養を終了する6−7時間前に、20μl(5mg/ml)のMTT溶液を加えた。各孔から100μlの上澄みを取り出し、更に100μlのMTT溶解液を加え、インキュベータで6〜7時間放置してから、ELISAにより570nMでOD値を測定した。
【0105】
3H−TdR取り込みにより、マイトジェンで誘導された正常マウス脾臓リンパ細胞の増殖機能に対する化合物の影響を測定した。100μl(4×105/孔)のマウス脾臓リンパ細胞の懸濁液を96穴プレートに接種し、50μlのConA(最終濃度5μg/ml)又は50μlのLPS(最終濃度10μg/ml)を加え、異なる濃度の化合物50μlを注入し、全体積が200μlとなった。各濃度に関して、三つの穴を設け、相応なConA/LPS非投与対照穴及び薬物非投与対照穴を設けた。37℃で5%のCO2のインキュベータで48時間培養した。培養を終了する8時間前に、各孔に25μl(10μCi/ml)の3H−チミジンを加えた。実験終了まで、培養を継続した。細胞収集器(Cell harvester)により細胞をガラス繊維膜に収集し、シンチレーション液を加えてからBetaカウンタ(MicroBeta Trilux, PerkinElmer)により細胞DNAに取り込まれた3H−TdR量を測定し、cpm値を用いて細胞増殖の状況を示す。
【0106】
3H−TdR取り込みにより、同種異体抗原で誘導された正常マウス脾臓リンパ細胞の増殖機能に対する化合物の影響を測定した。
刺激細胞の準備:BALB/cマウス脾臓リンパ細胞の懸濁液、Gamma線照射装置(Gammacell 3000)を用いてセシウム137放射線照射(3000Rads)を行ってその増殖能力を無くしてから、RPMI1640で2回洗浄し、細胞濃度を5×106/mlまで調節した。
【0107】
反応細胞の準備:C57BL/6マウス脾臓リンパ細胞を反応細胞として使用し、細胞濃度は5×106/mlであった。
【0108】
リンパ細胞混合培養:50μl のC57BL/6マウス脾臓リンパ細胞の懸濁液を96穴プレートに接種し、50μl のセシウム137放射線で処理した後のBALB/Cマウス脾臓リンパ細胞の懸濁液を加え、全体積を200μlになるように異なる濃度の化合物50μlを加えた。全体積が200μlより少ない場合、RMPI−1640培養液で補足した。実験において、BALB/cマウス群、C57BL/6マウス群、及びBALB/CとC57BL/6の混合培養群の合計3群を設けた。各濃度に対して、三つの穴を設け、又は相応な非投与対照穴、刺激細胞対照穴及び反応細胞対照穴を設けた。37℃で5%のCO2のインキュベータで3〜5日培養した。培養を終了する8時間前に、各孔に25μl(10μCi/ml)の3H−チミジンを加えた。実験終了まで、培養を継続した。細胞収集器により細胞をガラス繊維膜に収集し、シンチレーション液を加えてからBetaカウンタ(MicroBeta Trilux, PerkinElmer)により細胞DNAに取り込まれた3H−TdR量を測定し、cpm値を用いて細胞増殖の状況を示した。
【0109】
結果処理及び統計方法:全データを平均値±標準偏差で示し、各指標の検査結果をExcel 2000及びSPSS 11.0統計ソフトで処理を行った。
【0110】
実験結果により用量反応曲線(Dose−response curve)を描き、CC50値(50% cytotoxic concentration)、すなわち50%細胞の致死濃度を計算した。また、IC50(50% inhibitory concentration)、すなわち50%抑制効果を有するときの化合物濃度を計算した。
【0111】
実験結果:実験結果を図13及び表14に示す。
【0112】
正常マウス脾臓リンパ細胞に対するRapamycinの細胞毒性は、CC50=45.51μMであった。濃度依存的にConA/LPSで誘導された正常マウス脾臓T/Bリンパ細胞増殖反応を抑制し、その中、IC50がそれぞれ196.4nMと48.8nMであった。正常マウス脾臓リンパ細胞に対するY31の細胞毒性は、CC50=36.9μMであった。濃度依存的にConA/LPSで誘導された正常マウス脾臓T/Bリンパ細胞の増殖反応を抑制し、そのIC50がそれぞれ330.7nMと97.8nMであった。Rapamycin及びY31が、同種異体抗原で誘導された正常マウス脾臓リンパ細胞の増殖反応に対して、抑制活性を示しており、Y31の免疫抑制活性がより強かった。
【0113】
【表14】
【0114】
結果により、Rapamycin及びその誘導体Y31がマイトジェン/同種異体抗原で誘導されたリンパ細胞の増殖反応を明かに抑制し、強いin vitro免疫抑制活性を有することが示された。
【0115】
II.マウス遅延型過敏反応に対するRapamycin及びY31の影響:
実験目的:DNFBで誘導されたマウス遅延型過敏反応(DTH)を用い、DTHに対する化合物in vivo作用の抑制作用を観察した。誘導によりマウスが遅延型過敏反応を生じ、すなわち、マウスがDNFB感作後にDNFBで攻撃されると、マウスが耳介腫脹反応を生じる。マウスの耳介腫脹反応の進行状況に対する化合物の影響を観察することで、マウスDTHに対する影響を評価し、生体細胞の免疫反応に対する影響を検討した。
【0116】
実験薬剤:
名称:Rapamycin、Y31。
性状、含有量:白い粉末。
調製方法:無水アルコールに溶解させ、50mg/mlの保存液を調製した。使用時、5%PEG400+5%Tween−80無菌水を溶媒として必要な濃度まで保存液を希釈した。
【0117】
実験動物:
由来、品種、系統:BALB/c純系マウス、メス、18〜20g、中国科学院上海実験動物中心から購入された。動物生産ライセンス:SCXK(濾)2002−0010号。動物が上海薬物研究所SPF動物飼育施設で飼育された。実験動物使用ライセンス:SYXK(濾)2003−0029号、少なくとも一週間飼育してから使用した。温度が22±1℃であり、湿度が55±5%であり、12時間交代の明暗サイクルで飼育した。餌と水を全部消毒してから動物が自由に摂取した。全部の実験に関して、完全に実験動物に関する条例に基づいて行った。
【0118】
実験方法:
2,4−ジニトロフルオロベンゼン (DNFB)は一種のハプテン(Incomplete antigen)であり、DNFBでマウスの足部を敏感にさせた後、皮膚タンパク質と結合して完全抗原となり、更に一週間後DNFBでマウスの耳を攻撃し、部分的に遅延型アレルギー反応を生じさせ、耳の腫脹の遅延型過敏反応を起こさせた。一方、攻撃されなかった耳の方では耳の腫脹の遅延型過敏反応を生じなかった。耳の腫脹の増加により、ジニトロフルオロベンゼンで誘導されたマウスの遅延型過敏反応に対する進行状況を表した。
(1)20μlの、アセトン:オリーブオイル(4:1)を溶媒とした0.5%DNFB溶液をマウスの各後足に塗布し、敏感にさせた。
(2)第1回敏感にしてから5日、0.2%DNFB溶液を用いてマウスの左耳の両側を塗布して免疫攻撃を行った。対照として、アセトン:オリーブオイル(4:1)溶媒だけで右耳を塗布した。
(3)マウスをモデル群、Dex群(2mg/kg/d、経口投与)、Rapamycin群(1.5mg/kg/d、腹腔内注射投与)、Y31群(1.5mg/kg/d、腹腔内注射投与)と、ランダムに4群と分けた。
(4)スパイラルマイクロメータ(Spiral micrometer)で各マウスの左、右耳の厚さをそれぞれ測定し、右耳の厚さから左耳の厚さを引き、その差を腫脹の増加として記録した。
【0119】
実験結果を図14に示す。
【0120】
DNFBで誘導されたDTH反応は、Th1細胞を介した、T細胞の活性化及び多種の細胞因子の生成を含むアレルギー反応である。我々はBALB/cマウスDTH応答中における化合物の効果を測定し、その結果を以下の図に示した。DNFBで誘導されて遅延型過敏反応を生じたマウスをモデル対照群として用い、その両耳の耳の腫脹の平均増加は0.175mmであった。陽性対照薬Dex 2mg/kg群のマウスの耳の腫脹の平均増加は0.13mmであり、モデル群と比較すると、明らかな差異を有していた。Rapamycin群のマウスの耳の腫脹の平均増加が0.076mmであり、モデル群と比較すると、明らかな差異を有した。Y31群のマウスの耳の腫脹の平均増加が0.129mmであり、モデル群と比較すると、明らかな差異を有していた。
【0121】
実験結果により、Rapamycin及びY31がDNFBで誘導されたマウス遅延型過敏反応に対して、明らかに抑制できることが示された。
【0122】
III.SRBCで誘導されたマウス脾臓リンパ細胞中の特異的抗体産生細胞に対するRapamycin及びY31の影響:
実験目的:
マウスがヒツジ赤血細胞で免疫されると、マウスの脾臓リンパ細胞中に特異的抗体産生細胞が現れる。Rapamycin及びその誘導体を投与した後のマウスの脾臓リンパ細胞の特性的抗体産生細胞の生成量の変化を測定することで、Rapamycin及びその誘導体のマウスの体液性免疫への影響を観察した。
【0123】
実験薬剤:
名称:Rapamycin、Y31。
性状、含有量:白い粉末。
調製方法:無水アルコールに溶解させ、50mg/mlの保存液を調製した。使用時、5%PEG400+5%Tween−80無菌水を溶媒として必要な濃度まで保存液を希釈した。
【0124】
実験動物:
由来、品種、系統:BALB/c純系マウス、メス、18〜20g、中国科学院上海実験動物中心から購入された。動物生産ライセンス:SCXK(濾)2002−0010号。動物が上海薬物研究所SPF動物飼育施設で飼育された。実験動物使用ライセンス:SYXK(濾)2003−0029号、少なくとも一週間飼育してから使用した。温度が22±1℃であり、湿度が55±5%であり、12時間交代の明暗サイクルで飼育した。餌と水を全部消毒してから動物が自由に摂取した。全部の実験に関して、完全に実験動物に関する条例に基づいて行った。
【0125】
マウスは中国科学院上海実験動物中心から購入され、血清(補体)を取るために使用した。
【0126】
その他の実験材料:ヒツジ赤血細胞(SRBC)は上海江南生物技術工程有限公司から購入した。
【0127】
実験原理:
SRBCで誘導されたマウス体液性免疫反応のモデルの中に、SRBCの溶血反応で抗原特性的抗体の生産状況を測定することは、典型的な実験方法である。ヒツジ赤血細胞定量溶血反応検定(Quantitative hemolysis of sheep red blood cells, QHS)は、Bリンパ細胞(プラズマ細胞)で特異的な抗SRBC抗体を生じてヒツジ赤血細胞を破砕し、ヘモグロビンを解放して抗体の生成状況を表す一種の実験方法である。ヒツジ赤血細胞(SRBC)でマウスを敏感にさせた後、マウス脾臓リンパ細胞中に特異的抗体産生細胞が現れる。この細胞の産生する抗体とSRBCが補体の協同作用で溶血反応を生じ、分光光度法により溶血程度を測定し、特異性抗体産生細胞数の生成量を表した。
【0128】
実験方法:
1.BALB/cマウスをランダムに正常対照群、モデル対照群、陽性対照群(CsA10mg/kg)、Rapamycin群(1.5mg/kg/d、腹腔内注射投与)、Y31群(1.5mg/kg/d、腹腔内注射投与)の5群と分け、各群に6匹のマウスを設けた。
免疫にする場合、各群のマウスに腹腔内投与し始め、免疫後の5日まで毎日一回投与した。モデル対照群に対して、毎日溶媒対照を行った。
【0129】
2.PBSで新鮮なヒツジ赤血細胞(SRBC)を3回洗浄した後、1:5(v/v)に希釈し、各マウスに0.2ml希釈されたSRBCを腹腔内注射して、敏感にさせた。
【0130】
3.免疫後の5日でQHS測定を行い、マウス脾臓を取り、脾臓リンパ細胞を調製し。
分光光度法により溶血程度の測定:リンパ細胞5×106、0.2%SRBC、最適希釈比率のマウスの血清補体を均一に混合させ、37℃で1時間定温放置してから、3000rpm、10分間遠心分離し、上澄みを取り出した。540nmの所でOD値を測定し、特異的抗体産生細胞の生成量を表した。
【0131】
実験結果(表15に示す):
ヒツジ赤血細胞定量溶血反応検定(QHS)で、体内におけるB細胞の分泌した抗体でヒツジ赤血細胞を破砕して解放されたヘモグロビン(OD値)を用いて表した。その結果で、生体の体液性免疫機能を表す。
【0132】
特異的抗体産生細胞抑制%=(モデル対照群OD−投与群OD)/(モデル対照群OD−正常対照群OD)
【0133】
【表15】
【0134】
実験結論:
Rapamycin(1.5mg/kg)及びその誘導物Y31(1.5mg/kg)の腹腔内投与が、マウス脾臓中の特異的な抗SRBC抗体産生細胞の生成量を明らかに抑制でき、その抑制作用が陽性対照薬CsAより強く、マウスの体液性免疫機能に対して、明らかな抑制作用を有した。
【0135】
IV.マウス急性移植片対宿主病(aGVHD)に対するRapamycin及びY31の薬効学研究:
実験目的:
BABL/Cマウスをドナーとして、C57B6/6マウスを受容体として用い、BABL/Cマウス骨髄細胞及びリンパ細胞を致死量γ射線で照射したC57B/6マウス体内に移植し、急性移植片対宿主病(aGVHD)モデルを作成し、マウスのaGVHDに対するRapamycin及び誘導体の薬効学効果を考察した。
【0136】
実験薬剤:
名称:Rapamycin、Y31。
性状、含有量:白い粉末。
調製方法:無水アルコールに溶解させ、50mg/mlの保存液を調製した。使用時、5%PEG400+5%Tween−80無菌水を溶媒として必要な濃度まで保存液を希釈した。
【0137】
実験動物:
由来、品種、系統:BALB/c純系マウス、メス、4週齢、C57B/6マウス、メス、7〜8週齢、中国科学院上海実験動物中心から購入された。動物生産ライセンス:SCXK(濾)2002−0010号。動物が上海薬物研究所SPF動物飼育施設で飼育された。実験動物使用ライセンス:SYXK(濾)2003−0029号、少なくとも一週間飼育してから使用した。温度が22±1℃であり、湿度が55±5%であり、12時間交代の明暗サイクルで飼育した。餌と水を全部消毒してから動物が自由に摂取した。全部の実験に関して、完全に実験動物に関する条例に基づいて行った。
【0138】
実験方法:
1.全身照射(TBI):
C57B/6(H−2b)、メス、7週齢のマウスを受容体マウスとして使用した。
Gammacellで8.5Gy全身照射した。
【0139】
2.骨髄移植:
受容体マウスに4〜6時間照射した後、異種骨髄移植を行った。
BABL/C(H−2d)、メス、4週齢のマウスをドナーマウスとして使用した。マウスの四肢長骨骨髄細胞及び脾臓リンパ細胞を採取した。二種類の細胞濃度を1×108まで調製し、PBS緩衝液中に懸濁させた。
等比例で二種類の細胞を混合させ、各受容体マウスに混合細胞の懸濁液を0.5mlずつ静脈注射した。
【0140】
3.群分け及び投与:
ランダムにモデル群(溶媒対照)、Rapamycin群(1.5mg/kg/d、腹腔内注射投与)、Y−31群(1.5mg/kg/d、腹腔内注射投与)と3群に分け、各群のサンプル数はn=10であり、骨髄移植の当日から投与開始し、毎日1回行った。
【0141】
4.測定指標:
(1)体重:毎日1回計測した。
(2)BMT後のマウスの生存時間を記録した。
【0142】
実験結果:
本実験において、急性移植片対宿主病(aGVHD)のマウスモデルを作成し、C57BL/6マウスが半数致死量(Sub−fatal dose)8.5Gyの全身照射を受けた後、BALB/Cマウス骨髄細胞及び脾臓細胞を注入し、aGVHDモデルを複製し、aGVHDマウスの生存及び体重変化に対する化合物の影響を観察した。
実験結果を図15示した。骨髄移植した後、aGVHDモデルマウスの体重が明らかに低減し、且つ死亡例が見られた。aGVHD異体移植によるマウス体重の低減に対して、Rapamycin及び誘導体Y31が明らかな緩衝作用を有し、aGVHDマウスの生存率を上昇させ、治療効果が明らかであった。
実験結果により、急性移植片対宿主病(aGVHD)動物モデルに対して、Rapamycin及び誘導体Y31が良好な治療効果を有することが証明された。
【0143】
V.DBA/1マウスでの牛II型コラーゲン誘導関節炎に関するY31の治療効果:
実験目的:本実験において牛型コラーゲンでDBA/1マウス関節炎を誘導し、Y31を体内へ投与し、マウス関節炎の発病指数(Disease index)の変化を観察することで、マウス関節炎に対するY31の治療効果を観察・評価した。
【0144】
実験薬剤:
名称:Y31。
性状、含有量:白い粉末。
調製方法:無水アルコールに溶解させ、50mg/mlの保存液を調製した。使用時、5%PEG400+5%Tween−80無菌水を溶媒として必要な濃度まで保存液を希釈した。
【0145】
実験動物及び材料:
DBA/1マウス、7〜8週齢、体重20〜22g、日本大阪大学医学部Hiromi Fujiwara教授より提供された。動物は上海薬物研究所SPF動物飼育施設で飼育され、少なくとも一週間飼育してから使用した。温度が22±1℃であり、湿度が55±5%であり、12時間交代の明暗サイクルで飼育した。餌と水を全部消毒してから動物が自由に摂取した。全部の実験に関して、完全に実験動物に関する条例に基づいて行った。
Mycobacterium tuberculosisstrain H37Rvを含むフロイント完全アジュバント(Freund’s complete adjuvant)がWako Pure Chemical Industries Ltd(Osaka, Japan)から購入。
【0146】
実験方法:
関節炎モデル:牛II型コラーゲンに0.1Mの酢酸を加え、濃度を20mg/mlに調製し、4℃の冷蔵庫で一晩放置して溶解させた。コラーゲンが同体積のフロイント完全アジュバント(結核菌を含むH37Rv株)と充分乳化し、オスのDBA/1マウスを麻酔した後、25μl/匹(すなわち、250μg/匹)でその尾部を敏感にさせた。3週間後、同用量で攻撃した。肉眼でマウスの四肢を観察し、関節炎の程度に関して、0〜4段階評価を行った:0=正常、1=1個又は数個の趾関節が発赤又は腫脹、2=膝関節以下が中等度発赤腫脹、3=膝関節を含む部分が重度発赤腫脹、4=膝関節を含む部分が完全に発赤腫脹、関節変形、硬直、機能障害。各マウスの最高評価点数が、16点であった。
【0147】
薬物治療:
ランダムにマウスをモデル対照群、Y31(1mg/kg)投与群(攻撃した後14日から投与開始し、三週連続投与した)と、二群に分けた。
【0148】
実験結果:
牛II型コラーゲンを皮下に二回注射することでDBA/1マウス関節炎を誘導できた。関節腫脹が攻撃された後の4日から現れ、一週間後100%のマウスが関節炎を発症し、且つ関節腫脹が進行性に悪化し、14日から投与開始した。Y31を投与した後、CIAの発症程度が明らかに低減され、マウスの四肢(足、爪)の腫脹が明らかに軽減された(図16、p<0.05)。Y31経口投与がコラーゲンで誘導されたDBA/1マウス関節炎の発症を抑制できることが示された。
【技術分野】
【0001】
本発明は、薬物化学領域に属し、具体的に、新規な構造を有するラパマイシン炭酸エステル類似体(Rapalogs)又はその薬学的に受容可能な塩、及び該化合物を含む薬剤組成物に関する。また、本発明は、該化合物の製造方法、該化合物の抗腫瘍薬及び/又は抗癌薬或いは免疫抑制薬の研究における用途にも関する。
【背景技術】
【0002】
癌は、細胞の異常増殖及び転移を特徴とする一種の疾病であり、すでに人間の健康を脅かす疾病の一つとなっている。世界保健機関(WHO)の統計によれば、世界において癌の新規患者が毎年六百万人に達している。中国において、癌はすでに脳血管疾患に次ぐ致死性病因となっている。
【0003】
現在、臨床に良く使用されている抗腫瘍薬が主に細胞毒性薬剤であり、選択性が悪く、毒性副作用が強く、耐薬性を生じやすいなどの欠点が存在する。近年のゲノム工学技術、分子腫瘍学及び分子薬理学に関する研究の著しい発展に伴い、細胞癌化の本質が細胞シグナル伝達の失調にあり、大部分の腫瘍のシグナル伝達経路の活性が高すぎて、細胞が無限に増殖することが認知されてきている。細胞シグナル伝達分子は、新型抗腫瘍薬を探すための重要な切り口であり、一部の腫瘍細胞の分化増殖に相関する細胞シグナル伝達経路の重要な酵素を薬品に作用させて標的のスクリーニングを行い、選択的に標的に作用する効果的、且つ毒性の低く、特異性の強い新型抗腫瘍薬を発見することがすでに現在の抗腫瘍薬の研究開発の新しい方向とされている。
【0004】
PI3K−mTORシグナル経路は、最も重要なチロシンキナーゼシグナル伝達の手段の一つである。ホスホイノシチド3−キナーゼ(PI3K)がリン酸化によりプロティンキナーゼB(PKB)を活性化させ、後者が哺乳類動物のラパマイシン標的(mTOR)をリン酸化させて活性化させる。mTORは、直接的又は間接的に多数の細胞増殖及び成長に相関するプロセスの調節に関与し、細胞増殖の中心的な役割を果たすと認識されている。多くの研究結果によると、腫瘍細胞において、PI3K− mTORシグナル経路が異常な伝達を示し、腫瘍の発生及び発展において重要な役割を果たしている。従って、該経路を遮断し、特にmTORの活性を抑制することにより、特異的に腫瘍細胞の成長を抑制できる。PI3K− mTORシグナル伝達経路は、すでに有望な抗腫瘍治療の標的となっている。
【0005】
ラパマイシン(Rapamycin)は、一種のトリエンマクロライド抗生物質であり、シロリムス(Sirolimus)とも呼ばれる。1975年にWyeth Ayerst実験室によりRapanui島で分離したストレプトマイセス・ハイグロスコピカスを発酵させて初めて得られ、抗菌活性を有するとともに、効果的な免疫抑制薬としてすでに臨床治療に用いられている。最近の研究により、ラパマイシンがmTORの特異性抑制剤として明らかな抗腫瘍活性を有することが証明された。in vitroでは、約1ng/mlのラパマイシンだけで、明らかに横紋筋肉腫細胞の成長を抑制できる。世界中の多数の研究室による研究においても、Rapamycinが良い腫瘍治療候補薬品であると支持されている。横紋筋肉腫、神経芽細胞腫、膠芽腫、髄芽腫、小細胞肺癌などの腫瘍細胞に対して、強い抑制作用を有し、また腫瘍細胞の成長に対する抑制がmTORと結合する結果であることが確認されている。ラパマイシンが前臨床段階において良い抗癌活性を示しているが、そのマクロライドの低水溶性と化学安定性の理由で、臨床開発が制限されている。
【0006】
近年、各大企業により数種のmTORの腫瘍標的治療に用いるラパマイシン類似体を開発されている。その中、代表的なものとして、Wyeth社により開発されたCCI−779(TemRapamycin)、Novarti社により開発されたRAD−001(Everolimus)、及びAriad社のAP23576である。これらの類似物はラパマイシンと類似な抗腫瘍作用を有し、かつ薬理学的な性質が改善され、明らかな毒性副作用も有しない。CCI−779が静脈注射に適用でき、すでに末期腎臓癌患者の臨床治療に用いられている。RAD−001は経口用として、小細胞肺癌などに対する治療が第二相臨床試験段階まできている。現在AP23576の血液癌及び固形癌に対する治療は第三相臨床試験段階であり、良い製薬の可能性を示している。従って、ラパマイシンを母体として構造の改造を行うことで、高い応用価値を有し、効果的、低毒性、且つ特異性の強い抗腫瘍薬を発見することが期待されている。
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明は、ラパマイシンの31位と42位のヒドロキシ基に対して構造修飾又は改造を行うことにより、一連の体内外の抗腫瘍及び/又は抗癌活性或いは免疫抑制活性を有し、かつ新規な構造を備えるラパマイシン炭酸エステル類似体を合成した。水溶性、体内外の薬剤効果、経口バイオアベイラビリティ、薬物代謝などの方面の考察によると、更に研究を進める価値があり、抗腫瘍薬に用いられ、又は免疫抑制薬の候補薬として研究開発する価値があることが判明された。従って、本発明の一つの目的は、一種の新規構造を有するラパマイシン炭酸エステル類似物又は薬学的に受容可能な塩を提供することである。
【0008】
本発明のもう一つの目的は、前述ラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩を活性成分とする薬剤組成物を提供することである。
【0009】
本発明のもう一つの目的は、抗腫瘍及び/又は抗癌薬或いは免疫抑制薬の製造における前述ラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩の用途を提供することである。
【課題を解決するための手段】
【0010】
本発明で提供されたラパマイシン炭酸エステル類似体は構造式Iに示す構造を有しており、
【化1】
ここで、R1とR2は、各々独立に、H又は、
【化2】
であり、且つR1、R2同時にHではなく、その中nは1〜6の整数であり、R3は、
【化3】
又は
【化4】
であり、R4、R5とR6は、各々独立にH、炭素数1〜6のヒドロキシアルキル基、炭素数1〜6のアルキル基又は炭素数2〜6のアルケニル基であり、R7とR8が、各々独立にH又は炭素数1〜6のアルキル基である。
【0011】
本発明の好ましい実施形態において、前述R1とR2は、各々独立に、H又は、
【化5】
であり、且つR1、R2は同時にHではなく、その中、nは1〜4の整数であり、R3は、
【化6】
又は
【化7】
であり、R4、R5とR6が、各々独立に、H、炭素数1〜4のヒドロキシアルキル基であり、R7とR8が、各々独立に、炭素数1〜4のアルキル基である。
【0012】
本発明の好ましい実施形態において、前述R1とR2は、各々独立に、H又は、
【化8】
であり、且つR1、R2は同時にHではなく、その中、nは1〜2の整数であり、宜しくはR3が、
【化9】
又は
【化10】
であり、R7とR8が炭素数1〜4のアルキル基である。
【0013】
更に、本発明の代表的な化合物が以下の化合物の何れかである:
【化11】
【化12】
【化13】
【化14】
【0014】
本発明のラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩において、前述R3の中にキラル中心を含有する場合、前述ラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩は、各種の光学異性体又は光学異性体の混合物であっても良い。
【0015】
本発明は、構造式Iに示すラパマイシン炭酸エステル類似体の製造方法を提供する。即ち、R1、R2が同じく
【化15】
であり、R3が
【化16】
又は
【化17】
である場合、以下の方法により製造できる。
【0016】
構造式1に示すアルコールは、p−トルエンスルホン酸の触媒作用で、DMSO、DMFなどの溶媒中において、カルボニル化合物R7COR8又はそのアセタール化合物(アセトン、2,2−ジメトキシプロパンなど)と反応し、構造式2に示すアルコールが得られる。アルカリにより、トリホスゲンが構造式2に示したアルコールと反応し、アシルクロリド3を形成する。また、アルカリにより、該アシルクロリド3がラパマイシンと反応し、R3が
【化18】
である、31位、42位の二置換ラパマイシン炭酸エステル類似体を得る。更に、該ラパマイシン炭酸エステル類似体を加水分解することにより、R3が
【化19】
であるラパマイシン炭酸エステル類似体が得られる。
【0017】
反応式は以下のようである:
【化20】
式中、化合物1は市販で入手でき、例えば、国薬集団化学試材有限公司(Sinopharm Chemical Reagent Co., Ltd.)、Aldrich社等から購入できる。
【0018】
更に具体的に、アルコール1はオルト位の二つのヒドロキシ基の保護により、アルコール2を得る。DMA、DMF、アセトニトリル、ジクロロメタン又はテトラヒドロフラン溶媒において、ピリジン、トリエチルアミン又はジイソプロピルアミンなどのアルカリ性条件で、アルコール2がトリホスゲンと反応し、アシルクロリド3を得る。次に、DMA、DMF、アセトニトリル、ジクロロメタン又はテトラヒドロフラン溶媒において、ピリジン、トリエチルアミン、DMAP又はジイソプロピルアミンなどのアルカリ性条件で、ラパマイシンがアシルクロリド3と反応し、31位及び42位をエステル化された、ラパマイシン炭酸エステル類似体を得る。その中、ラパマイシンは福建科瑞薬業公司(Fujian Kerui Pharmaceutical Co., Ltd.)から購入できる。
【0019】
一方、本発明のラパマイシン炭酸エステル類似体において、R1とR2が異なり、各々独立に、H又は、
【化21】
であり、R3が
【化22】
又は
【化23】
である場合、ラパマイシンの31位、42位のヒドロキシ基の選択的保護により、異なる一置換又は二置換の炭酸エステル類似体が得られても良い。しかし、ラパマイシンが31位及び42位において二つの第2級アルコール基を有するため、選択的にラパマイシンの42位又は31位でのモノエステル化を実現することが以前から困難であった。米国特許第6,277,983号に二段階で42位のモノエステル化合物の製造方法を記載したが、実際の操作性が悪く、且つ長時間及び低温条件が必要である。本発明者は、米国特許第6,277,983号の方法を実施したとき、反応過程においてラパマイシンがすぐラパマイシンの31位、42位の二置換生成物に変化することを発見した。また、本発明者は、時間の延長とともに、前述二置換生成物が反応条件で、更にラパマイシン31位の一置換生成物及び一部のラパマイシン生成物と変化することを発見した。TLCにより反応過程を追跡し、反応時間を制御することにより、ラパマイシンの31位のみ置換された生成物の製造が可能となる。
【0020】
本発明者は、適切な比例のイミダゾールとクロロトリメチルシランを用い、ジクロロメタン、ジクロロエタン、酢酸エチル、テトラヒドロフラン、アセトニトリル又はDMFなどを溶媒として使用し、ラパマイシンにより室温条件で高速的、且つ効率的に31位のみ保護された生成物Rapamycin−31−OTMSを得た。それに基づき、42位のヒドロキシ基をTBSにより保護してから、安定性の悪い31位でのシリル系保護基を脱離させる方法により、42位のみ保護するRapamycin−42−OTBSを得ることが可能である。
【0021】
反応式が以下のようである。
【化24】
【0022】
式中、Rapamycin−31−OTMSの製造において、該反応の溶媒がジクロロメタン、ジクロロエタン、酢酸エチル、テトラヒドロフラン、アセトニトリル、N,N−ジメチルホルムアミドであり、反応温度が0℃〜40℃であり、反応時間は2〜48時間である。反応中のラパマイシン:イミダゾール:クロロトリメチルシランの適切な当量比は1:5〜30:2〜6であり、最も宜しい当量比は、1:10〜15:2〜4である。
【0023】
続いて、Rapamycin−31−OTMSが直接アシルクロリド3、
【化25】
と反応し、42位をエステル化する生成物が得られ、31位の保護基を脱離させた後、対応する42位をモノエステル化したラパマイシン炭酸エステル類似体が得られる。
【0024】
42位のみ保護する生成物Rapamycin−42−OTBSとアシルクロリド3、
【化26】
と反応させ、31位をエステルした生成物を得て、42位の保護基を脱離させた後、対応する31位のみモノエステル化したラパマイシン炭酸エステル類似体を得る。
【0025】
本発明のラパマイシン炭酸エステル類似体の薬学的に受容可能な塩は、本発明のラパマイシン炭酸エステル類似体から通常の方法により製造できる。
【0026】
本発明の薬剤組成物は、治療的有効量の活性成分としての一種又は数種の本発明に記載のラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩と、一種又は数種の薬学的に受容可能なキャリアとを含む。
【発明の効果】
【0027】
一方、本発明者は実験により、本発明のラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩が、ラパマイシンより明らかに高い抗腫瘍又は抗癌活性を有し、薬理学及び薬物代謝学の性質が良く、ヒト横紋筋肉腫、前立腺癌、非小細胞肺癌、乳癌、結腸癌、腎臓癌、肺腺癌、子宮頸癌又は白血病を治療するための薬剤の製造に応用できることを発見した。また、本発明のラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩は、ラパマイシンの水溶性を改善したほか、ラパマイシンに相当又はそれより優れた免疫抑制活性を維持している。
【0028】
本発明で提供された抗腫瘍化合物は、各種の腫瘍細胞及び/又は癌細胞に有効に対抗でき、且つラパマイシンに比べ、ヒドロキシ基などの水溶性、極性基を導入することにより、水溶性が増強され、薬学的な性質が改善されている。in vitro実験中に、異なる腫瘍株における実験により、抗腫瘍活性が明らかにラパマイシンより優れていることを示された(表4−8及び図3−9を参照)。細胞レベルの研究により、Y50がRh30、PC−3、MCF−7、CAKI−1及びHL−60腫瘍細胞の成長に対して、ラパマイシンに相当する抑制作用を有しており(図1に示す)、Y50が濃度依存的にRh30、PC−3、MCF−7及びCAKI−1細胞中のmTORの下流因子のリン酸化促進能力を抑制でき、同濃度でその抑制能力がラパマイシンに相当している(図2に示す)ことが判明された。SPR(表面プラズモン共鳴)の結果によると(表4及び図9に示す) 、1)ラパマイシン、CCI−779、Y50及びY31はいずれも濃度依存的にFKBP12と結合できる。ラパマイシンと比べると、同濃度でY50及びY31がラパマイシンより高いRU(Response Unit)を有し、同濃度のY50及びY31とFKBP−12の結合がラパマイシンより強い、 2)Y50及びY31とFKBP12の結合が飽和に達するまで必要な濃度がラパマイシンより低い、3)Y50とY31のFKBP12との解離速度がラパマイシンより低く、同様にCCI−779よりも低く、Y50及びY31の解離係数がラパマイシン及びCCI−779より低い。動物実験の研究により、Y50の経口投与がヒト横紋筋肉腫RH−30異種移植マウスの移植腫に対して、明らかにラパマイシンより優れた成長抑制作用を有する(表5及び図3から図4)。Y50の経口投与によりヒト前立腺癌PC−3異種移植マウスの移植腫に対する成長抑制作用も、明らかにラパマイシンより優れる(表6及び図5から図6に示す)。そのT/Cがそれぞれ10.0%と40.2%であり、陽性対照としてのラパマイシンの同用量群に対応するT/Cがそれぞれ30.9%と46.5%であった。Y31の経口投与は、ヒト骨肉腫U2SO異種移植マウスの移植腫の成長に対して、明らかな成長抑制作用を有する。低用量群(2.5mg/kg)の場合、CCI−779及びラパマイシンは、ヒト骨肉癌U2SO異種移植マウスの移植腫の成長に対して、明らかな抑制作用を有さず、そのT/C値がそれぞれ69.0%、60.0%であった。なお、低用量(2.5mg/kg)の場合、化合物Y31が該腫瘍の成長に対する抑制作用が明らかにラパマイシン及びCCI−779より優れている(表8及び図8に示す)。
【0029】
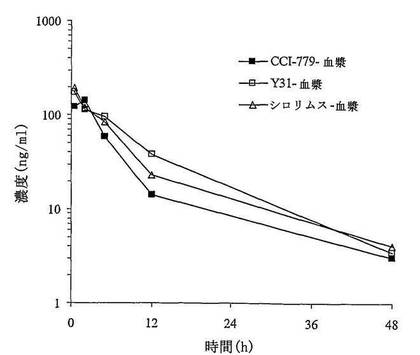
本発明で提供された化合物を更に研究するための抗腫瘍実験中に、該化合物及びラパマイシンは、市販しているラパマイシン類似体CCI−779と比べると、Y31がより良い薬物動態パラメータを示している(表9〜12及び図10〜12に示す)。これは、ヒドロキシ基などの水溶性、極性基を導入することと関係する可能性がある。特に、Y31を投与した後、薬物が腫瘍組織での選択的吸収が一番良かった(表12及び図12に示す)。実験によると、マウスにY31を投与した後、体内で迅速に代謝物ラパマイシン(Rapamycin)と変化し、血漿及び組織中のプロトタイプ薬の濃度が低く、最大濃度が20ng/ml(又はng/g)未満で、薬剤を投与してから5時間でプロトタイプ薬を検出できなかった。Y31を投与した後、血漿、肝臓、及び腫瘍中に、Rapamycinの暴露量とRapamycin投与群の比率がそれぞれ1.22、1.32、1.93であった。
【0030】
本発明のラパマイシン炭酸エステル類似体の薬学的に受容可能な塩は、前述の抗腫瘍活性及び優れた薬物動態パラメータを示すだけではなく、ラパマイシンに相当する又はそれより優れた免疫抑制活性も保持している。ラパマイシンを対照として、化合物Y31 を例として系統的免疫抑制活性実験を行った(結果を表13〜15及び図13〜16に示す)。
【0031】
(1)マイトジェン/同種異体抗原で誘導された正常マウス脾臓リンパ細胞の増殖活性に対するRapamycin及びY31の影響:
結果に示すように、Rapamycin及びその誘導体Y31がマイトジェン/同種異体抗原で誘導されたリンパ細胞の増殖反応を明かに抑制し、強いin vitro免疫抑制活性を有する(表14及び図13に示す)。
【0032】
(2)マウス遅延型過敏反応に対するRapamycin及びY31の影響:
DNFBで誘導されたDTH反応は、Th1細胞を介した、T細胞の活性化及び数種の細胞因子の発現を含むアレルギー反応である。我々は、BALB/cマウスのDTH応答中での化合物の役割を測定し、実験結果を図14に示した。DNFBで誘導されて遅延型過敏反応を生じたマウスを対照群として使用し、その両耳の耳の腫脹の平均増加が0.175mmである。陽性対照薬Dex 2mg/kg投与群のマウスの耳の腫脹の平均増加が0.13mmであり、対照群と比較すると、明らかな差異を有する。Rapamycin投与群のマウスの耳の腫脹の平均増加が0.076mmであり、対照群と比較すると、差異が明らかである。Y31投与群のマウスの耳の腫脹の平均増加が0.129mmであり、対照群と比較すると、差異が明らかである。
実験結果によれば、Rapamycin及びY31が明かにDNFBで誘導されたマウス遅延型過敏反応を抑制できる(図14に示す)。
【0033】
(3)SRBCで誘導されたマウス脾臓リンパ細胞中の特異的抗体産生細胞に対するRapamycin及びY31の影響:
Rapamycin(1.5mg/kg)及びその誘導体Y31(1.5mg/kg)を腹腔内投与すると、マウス脾臓中の特異的な抗SRBC抗体産生細胞の生成量を明かに抑制でき、その抑制作用が陽性対照薬CsAより強く、マウスの体液性免疫機能に対して、明らかな抑制作用を有していた(表15に示す)。
【0034】
(4)マウス急性移植片対宿主病(aGVHD)に対するRapamycin及びY31の薬効学研究:
本実験の結果により、急性移植片対宿主病(aGVHD)の動物モデルに対して、Rapamycin及び誘導体Y31が良い治療効果を有することが証明された(図15に示す)。
【0035】
(5)DBA/1マウスでの牛II型コラーゲン誘導関節炎に対するY31の治療効果
牛II型コラーゲンを用いて皮下注射を二回行うことで、DBA/1マウス関節炎を誘導できる。刺激してから四日後関節腫脹が現れ、一週間後100%のマウスが関節炎を発症した。且つ、関節腫脹が進行的に悪化し、第14日から薬を投与開始した。Y31を投与してから、明かにCIAの発症程度が低減し、マウスの四肢(足、爪)の腫脹が明らかに減少された。Y31の経口投与は、DBA/1マウスでのコラーゲン誘導関節炎の発病を抑制できる(図16に示す)。
【0036】
本発明で提供されたラパマイシン炭酸エステル類似体は、優れた腫瘍抑制活性及び免疫抑制活性、比較的に良い薬物動態パラメータを有し、製造方法が簡単で、操作性に優れたほか、高い生産率、及び良好な薬剤開発可能性を有している。
【図面の簡単な説明】
【0037】
【図1】異なる濃度の化合物Y50のRh30(ヒト横紋筋肉腫、A)、PC−3(ヒト前立腺癌、B)、MCF(ヒト乳癌、C)、CAK−1(ヒト腎臓細胞癌、D)及びHL−60(ヒト白血病、E)細胞の成長に対する抑制を示す図である。
【図2】Y50のRh30、PC−3、MCF−7.CAK−1細胞中のp70S6K及び4E−BP1に対するリン酸化レベルの評価を示す図である。
【図3】化合物Y50のヒト横紋筋肉腫Rh30異種移植マウスの移植腫の成長に対す抑制曲線を示す図である。
【図4】化合物Y50のヒト横紋筋肉腫Rh30異種移植マウスの移植腫の成長に対する抑制を示す図である。
【図5】化合物Y50のヒト前立腺癌PC−3異種移植マウスの移植腫の成長に対する抑制曲線を示す図である。
【図6】化合物Y50のヒト前立腺癌PC−3異種移植マウスの移植腫の成長に対する抑制を示す図である。
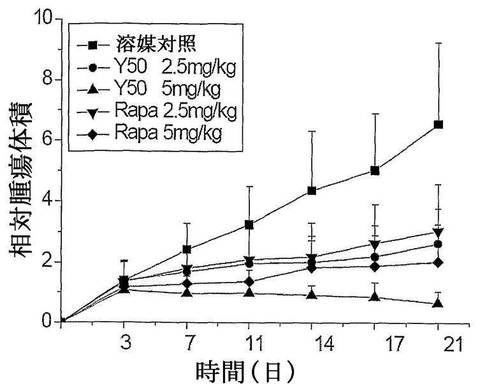
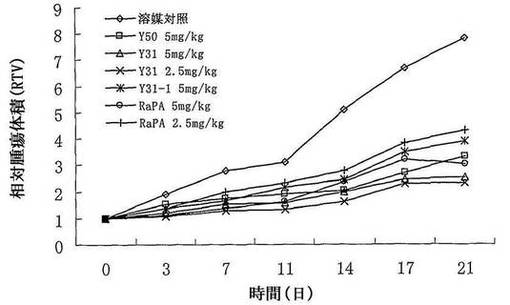
【図7】化合物Y50、Y31、Y31−1、Rapamycin(ラパマイシン)のヒト横紋筋肉腫Rh30異種移植マウスの移植腫の成長に対する抑制曲線を示す図である。
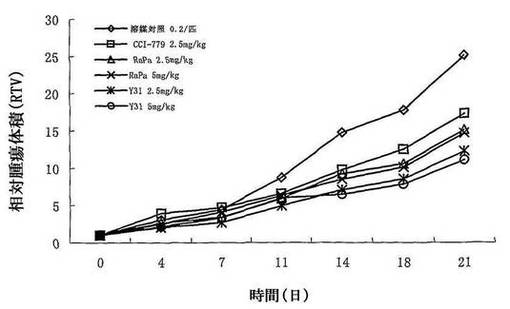
【図8】化合物Y31、CCI−779、 Rapamycin(ラパマイシン)のヒト骨肉癌U2SO異種移植マウスの移植腫に対する実験治療効果を示す図である。
【図9】SPR(表面プラズモン共鳴)の方法により測定した小分子化合物とタンパク質FKBP−12との結合活性を示す図である。
【図10】マウスにそれぞれY31、CCI−779及びRapamycin(ラパマイシン)を投与した後、血漿中のRapamycin(ラパマイシン)の濃度−時間曲線を示す図である。
【図11】マウスにそれぞれY31、CCI−779及びRapamycin(ラパマイシン)を投与した後、肝臓中のRapamycin(ラパマイシン)の濃度−時間曲線を示す図である。
【図12】マウスにそれぞれY31、CCI−779及びRapamycin(ラパマイシン)を投与した後、腫瘍中のRapamycin(ラパマイシン)の濃度−時間曲線を示す図である。
【図13】マイトジェン/同種異体抗原で誘導された正常マウス脾臓リンパ細胞の増殖活性に関するRapamycin(ラパマイシン)及びY31の影響を示す図である。
【図14】マウス遅延型過敏反応に関するRapamycin(ラパマイシン)及びY31の影響を示す図である。
【図15】マウス急性移植片対宿主病(aGVHD)に関するRapamycin(ラパマイシン)及びY31の薬効学研究を示す図である。
【図16】DBA/1マウスでの牛II型コラーゲン誘導関節炎に関するY31の治療効果を示す図である。
【発明を実施するための形態】
【0038】
具体的な実施例に基づき、更に詳しく本発明を説明する。しかし、本発明は実施例に限定されない。
【0039】
<ラパマイシン炭酸エステル類似体の製造に関する実施例>
後述実施例中に、通常の後処理方法は、反応を終えた後、反応液に適量の水を加え、有機相と水相に分離し、水相が有機溶媒により充分に抽出された後、有機相を合併する。必要に応じて、順次に5%HCl溶液及び/又は飽和NaHCO3溶液、水及び飽和食塩水を用いて洗浄する。無水Na2SO4又は無水MgSO4により有機相を乾燥させ、濾過してから減圧スピン乾燥を行い、粗生成物を得て、更にカラムクロマトグラフィーにより純化した後、最終生成物を得た。
【0040】
後述製造の実施例中に、Varianにより製造されたMercury−Vx600M装置を用い、NMRを測定した。δH/C 7.26/77.0ppm(CDCl3)をNMRキャリブレーションに使用した。試薬が主に上海化学試剤公司(Shanghai Chemical Reagent Co., Ltd.)により提供され、生成物の純化が主にカラムクロマトグラフィーを使用した。シリカゲル(200−300目)について、カラムクロマトグラフィーに用いたシリカゲルのタイプはMacro−pored silica Gel (ZLX−II)であり、青島海洋化工場分場(Qingdao Haiyang Chemical Co., Ltd.)により製造されたものである。
【0041】
<製造の実施例1:化合物Y230、Y72及びY50の製造>
【化27】
276mg(3mmol)のグリセロールを取り、2mlのDMSO中に溶解させ、窒素雰囲気で、0.44mlの2,2−ジメトキシプロパンを注入し、触媒量のp−トルエンスルホン酸を加えた。室温で数時間攪拌し、TLCにより反応終了まで追跡し、通常の後処理を経て合計173mgの液状の生成物2iを得た。
【0042】
50mlのナスフラスコに173mg(1.31mmol)の化合物2i、130mg(0.44mmol)のトリホスゲンを加え、窒素雰囲気で25mlの二重蒸留CH2Cl2溶液を注入し、氷水浴で170μl(1.31mmol)の乾燥ピリジンを一滴ずつ滴加し、滴加を終えたら、自然に室温まで昇温して2時間反応させ、200mg(0.22mmol)のラパマイシンを加え、更に0.2mlのピリジンを追加し、TLCにより反応終了まで追跡した。次に、ナスフラスコに1NのHClを加え弱酸性に中和させ、ジクロロメタンで抽出し、ジクロロメタンの抽出液を水洗い、飽和食塩水により洗浄し、無水硫酸マグネシウムにより乾燥して濃縮し、石油エーテル/アセトン(体積比5/1)を展開液としてカラムクロマトグラフィーにより分離させて240mgの化合物Y230を得て、副生成物として20mgのY72を得た。
【0043】
240mgの化合物Y230を取り、3mlのTHFに溶解させ、0〜5℃で1.7mlの2NのH2SO4を滴加し、TLCにより反応終了まで追跡した。5%NaHCO3を加え弱アルカリ性まで中和させ、酢酸エチルで抽出し、酢酸エチルの抽出液を飽和食塩水により洗浄し、無水硫酸マグネシウムにより乾燥し、減圧濃縮し、石油エーテル/アセトン(体積比1/1)を展開液としてフラッシュカラムクロマトグラフィーにより分離させて120mgの化合物Y50を得た(全収率:38%)。
【0044】
【表1】
【0045】
<製造の実施例2:化合物Y31の製造>
【化28】
【0046】
400mg(0.44mmol)のラパマイシン、449mg(6.6mmol)のピリジンを20mlの二重蒸留CH2Cl2に溶解させ、0.22ml(1.76mmol) のクロロトリメチルシラン(TMSCI)を滴加した。滴加を終えた後、TLCにより追跡し、6時間ほど反応させ、減圧濃縮し、石油エーテル/アセトン(体積比4/1) を展開液としてカラムクロマトグラフィーにより合計277mgの化合物Rapamycin−31−OTMSを得た。
【0047】
50mlのナスフラスコに573mg(4.34mmol)の化合物3i、453mg(1.53mmol)のトリホスゲンを加え、窒素雰囲気で30mlの二重蒸留CH2Cl2溶液を注入し、氷水浴で377μl(4.67mmol)の乾燥ピリジンを滴加し、滴加を終えてから、自然に室温まで昇温して2時間反応させ、277mg(0.28mmol) のRapamycin−31−OTMSを加えた。4時間後TLCにより反応終了を追跡したら、弱酸性になるように1NのHClを加え、ジクロロメタンで抽出し、水洗い、飽和食塩水により洗浄し、無水硫酸マグネシウムにより乾燥し、濃縮し、石油エーテル/アセトン(体積比4/1) を展開液としてカラムクロマトグラフィーにより分離させて合計240mgの化合物Y44を得た。
【0048】
240mgの化合物Y40を4mlのTHFに溶解させ、0〜5℃で1.7mlの2NのH2SO4を滴加し、TLCにより反応終了まで追跡した。弱アルカリ性になるように、5%のNaHCO3を加え、酢酸エチルで抽出し、飽和食塩水により洗浄し、無水硫酸マグネシウムにより乾燥し、減圧濃縮し、石油エーテル/アセトン(体積比1.5/1) を展開液としてカラムクロマトグラフィーにより分離させて合計120mgの化合物Y31を得た。
【0049】
【表2】
【0050】
<製造の実施例3:化合物Y31−1の製造>
化合物Rapamycin−31−OTMSの製造に関して、製造の実施例2を参照する。
【0051】
25mlのナスフラスコに200mg(0.2mmol) の化合物Rapamycin−31−OTMS、206mg(3mmol) のピリジンを加え、7mlのDMFを注入し、更に184mg(1.22mmol) のtert−ブチルジメチルクロロシラン(TBSCI)を滴加した。TLCにより追跡し、室温で48時間反応させてから水を加えて希釈し、酢酸エチルで抽出し、水洗い、飽和食塩水により洗浄し、硫酸マグネシウムにより乾燥させ、石油エーテル/アセトン(体積比3/1) を展開液としてカラムクロマトグラフィーにより分離し、合計120mgの生成物Y028を得た。
【0052】
50mlのナスフラスコに173mg(1.31mmol)の化合物3i、130mg(0.44mmol)のトリホスゲンを加え、窒素雰囲気で25mlの二重蒸留CH2Cl2溶液を注入し、氷水浴で170μl(1.31mmol)の乾燥ピリジンを滴加し、滴加を終えてから、自然に室温まで昇温して2時間反応させ、120mg(0.12mmol) のY028を加え、更に0.2mlのピリジンを追加し、TLCにより反応終了まで追跡した。次に、弱酸性になるように、ナスフラスコに1NのHClを加え、ジクロロメタンで抽出し、水洗い、飽和食塩水により洗浄し、無水硫酸マグネシウムにより乾燥し、濃縮し、石油エーテル/アセトン(体積比3/1) を展開液としてカラムクロマトグラフィーにより分離させて合計100mgの化合物Y86を得た。
【0053】
100mgの化合物Y86を1.5mlのTHFに溶解させ、0〜5℃で0.8mlの2NのH2SO4を滴加し、TLCにより反応終了まで追跡した。弱アルカリ性になるように5%のNaHCO3を加え、酢酸エチルで抽出し、飽和食塩水により洗浄し、無水硫酸マグネシウムにより乾燥し、減圧濃縮させ、石油エーテル/アセトン(体積比1/1) を展開液としてフラッシュカラムクロマトグラフィーにより分離させて合計80mgの化合物Y31−1を得た。
【0054】
【化29】
【0055】
【表3】
【0056】
<生物実験の実施例>
<実施例1:細胞レベルの抗腫瘍活性測定実験>
I.Y50のRh30、PC−3、MCF−7、CAKI−1及びHL−60細胞の成長に対する抑制。
【0057】
異なる濃度の化合物により、Rh30細胞を72時間処理し、SRB(Sulforhodamine B)法により細胞の生存率を測定した。
【0058】
指数増殖期に属する前記の各種の腫瘍細胞を90μl/穴で96穴プレートに接種し、24時間接着培養し、更に10μl/穴で薬を加えた。各濃度に関して、同条件の穴を三つ設けた。また、相応な濃度の溶媒対照(生理食塩水)及アポトーシスを発生しない穴を設けた。37℃、5%CO2の条件で腫瘍細胞を72時間培養した後、培養液を取り除き、10%冷TCA(トリクロロ酢酸)を用いて細胞を固定させ、4℃で1時間放置してから、蒸留水により5回洗浄し、空気中で自然乾燥させた。1%氷酢酸から調製した4mg/mlのSRB(Sigma)溶液を100μl/穴で加え、室温で15分間染色し、上澄みを取り除き、1%酢酸により5回洗浄し、空気中で乾燥させた。最後に150μl/穴でTris溶液を加え、ELISAにより波長520nmでA値を測定した。以下の公式により、腫瘍細胞成長に対する抑制率を計算した。
【0059】
抑制率=(A520対照群−A520投与群)/ A520対照群×100%
【0060】
結果から図1に示すように、前記の各種の腫瘍細胞成長に対して、Y50がラパマイシンに相当する抑制作用を有することが証明された。
【0061】
II.Y50のRh30、PC−3、MCF−7及びCAKI−1細胞中のp70S6K及び4E−BP1のリン酸化レベルに対する抑制。
【0062】
一定の密度で細胞を12穴プレートに接種し、細胞が壁と接着する状態で一晩放置し、新鮮な無血清培養液に変え、飢餓状態で24時間保持した後、相応な濃度の薬を加えて1時間反応させ、IGFにより10分間刺激させ、サンプルを採取し、Western blotにより細胞中のp70S6K及び4E−BP1のリン酸化レベルを測定した。結果を図2に示し、実験結果からY50が濃度依存的に各種の腫瘍細胞中のmTORの下流因子のリン酸化促進能力を抑制できることが示された。同濃度で、その抑制能力がラパマイシンと相当する。
【0063】
III.化合物Y50、Y31と標的タンパク質FKBP−12の結合活性の測定実験。
【0064】
1.材料と装置:
(1)FKBP−12タンパク質はSigma社から購入した。
(2)HBS−EP緩衝液(10mM Hepes,150mM NaCl,3.4mM EDTA,0.005%(v/v) Surfactant P20,pH7.4)。
(3)活性化試薬のEDC、NHS及びブロッキング試薬(Blocking reagent)のEthanolamineなどは、BLACORE AB社(Uppsala,Sweden)から購入した。
(4)Biacore3000、CM5チップは、BLACORE AB社(Uppsala,Sweden)から購入した。
【0065】
2.実験方法:
(1)FKBP−12タンパク質のカップリング:
Biacore3000プログラム中のアミンカップリングのWizardを用いてTubulinタンパク質を、CM5チップのFC2通路にカップリングさせた。pH4.6、10mM NaACで、3.3g/LのFKBP−12タンパク質を最終濃度の66μg/mlまで希釈した。0.1Mの1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド塩酸塩(EDCI)と0.1MのN−ヒドロキシスクシンイミド(NHS)を1:1混合させて、チップ表面において20μL/minの流速で7分間試料を導入し、タンパク質溶液を注射し、活性化チップの表面をブロッキングさせるように、pH8.5、1Mのエタノールアミンを7分間導入した。
【0066】
化合物の初期スクリーニング(Preliminary screening)及びダイナミクス測定:
SPR(表面プラズモン共鳴)の方法を用いて小分子化合物とFKBP−12タンパク質の結合活性を測定した。化合物の原液濃度が10mMであり、HBS−EP緩衝液を一定の比例で希釈し、Biacore3000プログラム中のダイナミクス解析Wizardによりダイナミクス実験(Dynamic experiment)を行った。得られた実験データに関して、Biacore3000解析ソフト中の1:1 Langmuir結合モデル又は定常状態モデルに基づき近似を行い、確実な動的定数及び熱力学定数を得た。
【0067】
SPR実験結果(表4及び図8に示す):
1.Rapamycin(ラパマイシン)、CCI−779、Y50及びY31の何れもが濃度依存的にFKBP12と結合できる。Y50及びY31は、Rapamycinと比較すると、同濃度でのRU(Response Unit)がRapamycinより高く、同濃度のY50及びY31とFKBP−12の結合がRapamycinより強いことを意味する。
2.Y50及びY31とFKBP12の結合の飽和に達するまでに必要な濃度がRapamycinより低い。
3.Y50及びY31とFKBP12の解離速度がRapamycinより低く、かつCCI−779よりも低い。Y50及びY31の解離定数がRapamycin及びCCI−779より低い。
【0068】
【表4】
【0069】
<実施例2:動物レベルの抗腫瘍活性測定実験>
実験目的:ヒト横紋筋肉腫RH−30異種移植マウスの移植腫の成長に対する抑制作用を観察する。
【0070】
実験物:5%ツイン(Tween)80、5%PEG400及びDDWを溶媒として適用し、Y50を経口投与製剤に調製した。
【0071】
陽性対照薬:5%ツイン80、5%PEG400及びDDWを溶媒として適用し、ラパマイシンを経口投与製剤に調製した。
【0072】
投与量の設置:Y50の経口投与量を5mg/kg、10mg/kgの二つの用量群と設け、毎日一回の経口投与を行った。ラパマイシンについて、同様な投与量で経口投与を行った。
【0073】
動物:BALB/cAマウス、オス、40〜45日齢、体重21±2g、中国科学院上海薬物研究所により提供された。生産ライセンス:SCXK(濾) 2004−0002。各群の動物数:陰性対照群6匹、投与群6匹。
【0074】
移植腫:ヒト横紋筋肉腫RH−30異種移植マウスの移植腫。ヒト横紋筋肉腫RH−30細胞株をマウスの皮下に接種して作成した。5×106の細胞接種量で接種して移植腫を形成した後、マウス体内に遺伝して3世代目以降を使用した。
【0075】
実験方法:成長増殖の活発な時期の腫瘍組織を約1.5mm3にカットし、無菌条件で、マウスの右側の脇の下の皮下に接種した。マウスの移植腫に対して、ノギスにより移植腫の直径を測定し、腫瘍が100〜200mm3まで成長してから、ランダムに動物を組み分けした。実験群に経口投与し、毎日一回投与を行い、三週間連続行った。陽性対照薬のラパマイシンについても、同様な方法で、同用量を投与し、合計三週間行った。陰性対照群に対して、0.2ml/匹で溶媒を経口投与した。毎週腫瘍の直径を二回測定し、同時にマウスの体重も測定した。腫瘍体積(Tumor volume, TV)の計算式は、TV = 1/2×a×b2であり、その中、a、bがそれぞれ長さ、厚さを示す。測定した結果により、相対腫瘍体積(Relative tumor volume,RTV)を計算した。その計算式は、RTV = Vt/V0であり、その中、V0は投与開始のとき(すなわち、d0のとき)の測定した腫瘍体積で、Vtが毎回測定するときの腫瘍体積である。抗腫瘍活性の評価指標は、相対腫瘍増殖率T/C(%)である。
【0076】
計算式:T/C(%)=(TRTV/CRTV)×100。その中、TRTVは治療群RTVで、CRTVは陰性対照群RTVである。
【0077】
治療の評価指標:T/C(%)>60%であれば、無効である。T/C(%)≦60%、且つ統計学処理によりp<0.05であれば、有効である。
【0078】
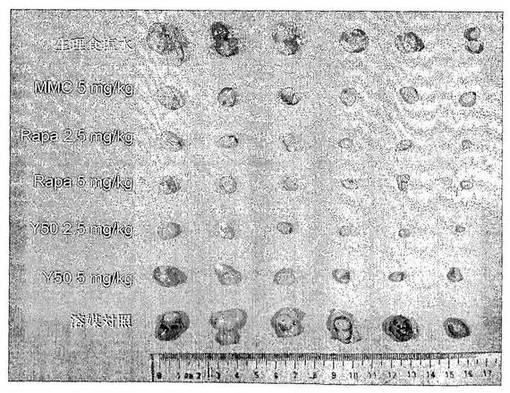
結果:Y50のヒト横紋筋肉腫RH−30異種移植マウスの移植腫の成長に対する抑制作用を表5及び図3、図4に示す。実験結果により、三週間連続毎日一回5mg/kg、10mg/kg のY50を経口投与した、二つの用量群がヒト横紋筋肉腫RH−30異種移植マウスの移植腫の成長に対して明らかな抑制作用を有し、そのT/Cがそれぞれ32.5%、32.9%であり、陽性対照群のラパマイシンの高用量群(10mg/kg)に相当した。一方、ラパマイシンの低用量群(5mg/kg)が、ヒト横紋筋肉腫RH−30異種移植マウスの移植腫の成長に対して、明らかな抑制作用がなく、そのT/C値が61.8%であった。実験群のマウスの死亡例が見られなかった。
【0079】
結論:ヒト横紋筋肉腫RH−30異種移植マウスの移植腫に対して、Y50経口投与がラパマイシンより明らかに優れた成長抑制作用を有する。下の表5、図3及び図4に示す。
【0080】
【表5】
【0081】
同様な実験により、Y50のヒト前立腺癌PC−3異種移植マウスの移植癌の成長に対する抑制作用を観察した。結果に示すように、異なる用量群において、ヒト前立腺癌PC−3異種移植マウスの移植癌に対する成長抑制作用について、Y50経口投与がラパマイシンより明らかに優れる。そのT/Cがそれぞれ10.0%、40.2%であった。一方、陽性対照のラパマイシンの同用量群に対応するT/Cがそれぞれ30.9%、46.5%であった。下の表6、図5及び図6に示す。
【0082】
【表6】
【0083】
【表7】
【0084】
前記と同様の実験により、Y50、Y31、Y31−1、Rapamycinのヒト横紋筋肉腫RH−30異種移植マウスの移植腫の成長に対する抑制作用を観察した。ヒト横紋筋肉腫RH−30異種移植マウスの移植腫の成長に対して、Y31、Y50、Y31−1の経口投与が明らかな成長抑制作用を有した。Y31、Y50の該腫瘍の成長に対する抑制作用がラパマイシンより優れ、その中、ラパマイシンが低用量群(2.5mg/kg)のとき、ヒト横紋筋肉腫RH−30異種移植マウスの移植腫に対して、明らかな抑制作用を有していない。そのT/C値が63.7%であった。一方、Y31は低使用量(2.5mg/kg)のときでも、比較的に高い抑制作用を達成でき、Y50及びラパマイシンの高用量群(5mg/kg)より明かに優れる。表7及び図7に示す。
【0085】
【表8】
【0086】
同実験により、Y31、CCI−779、Rapamycinのヒト骨肉癌U2SO異種移植マウスの移植腫の成長に対する抑制作用を観察した。結果により、ヒト骨肉癌U2SO異種移植マウスの移植腫の成長に対して、Y31経口投与が明らかな成長抑制作用を有することが示された。低用量群(2.5mg/kg)のとき、ヒト骨肉癌U2SO異種移植マウスの移植腫に対して、CCI−779及びラパマイシンが明らかな抑制作用を有しない。そのT/C値はそれぞれ69.0%、60.0%であった。一方、低用量(2.5mg/kg)のとき、化合物Y31の該腫瘍の成長に対する抑制作用がラパマイシン及びCCI−779より明かに優れる。
【0087】
<実施例3:Y31、Rapamycin及びCCI−779を投与した後のマウス体内におけるRapamycinの分布に関する研究実験>
本実験において、マウスにY31、Rapamycin及びCCI−779を投与した後のマウス体内におけるRapamycinの分布特徴に関して、検討を行った。マウスに薬剤を投与した後、異なる時間での血漿、肝臓及び腫瘍組織を採集し、液体クロマトグラフィー・タンデム質量分析法(LC−MS/MS)により血漿及び組織中のプロトンタイプ薬物及びRapamycinの濃度を測定した。マウスにY31、Rapamycin及びCCI−779を投与した後、血漿及び組織中のプロトンタイプ薬物及びRapamycinの濃度を表9〜11に示し、主な薬物動態パラメータを表12に示す。
【0088】
【表9】
【0089】
【表10】
【0090】
【表11】
【0091】
【表12】
【0092】
結果によると、ヒト横紋筋肉腫RH−30異種移植マウスの移植腫を移植した動物にY31、Rapamycin及びCCI−779をそれぞれ投与してからマウスにY31を投与した後、体内に迅速に代謝物Rapamycinと変化し、血漿及び組織中のプロトンタイプ薬の濃度が低く、最大濃度が20ng/ml又はng/gより低かった。投薬してから5時間でプロトンタイプ薬を検出できなくなった。Y31を投与した後、血漿、肝臓及び腫瘍中のRapamycinの暴露量とRapamycin投与群の比例がそれぞれ1.22、1.32、1.93であった。
【0093】
マウスにCCI−779を投与した後、血漿及び組織中にプロトンタイプ薬CCI−779及び代謝物Rapamycinを同時に検出でき、血漿、肝臓及び腫瘍組織中の代謝物Rapamycinとプロトンタイプ薬の暴露量の比例がそれぞれ10.4、0.94と1.89であった。CCI−779を投与した後、血漿、肝臓及び腫瘍中のRapamycinの暴露量とRapamycin投与群の比例がそれぞれ0.75、0.54と0.63であった。
【0094】
マウスにRapamycinを投与した後、血漿中にRapamycinが迅速に吸収され、ピークに達する時間が0.5〜2時間であった。肝臓中にピークに達する時間が0.5時間であり、血漿中の場合に近かった。腫瘍中にピークに達する時間が比較的に遅く、2〜5時間であった。Rapamycinは血漿及び肝臓でより速く除去される。
【0095】
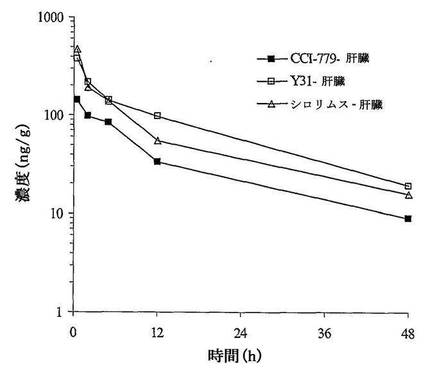
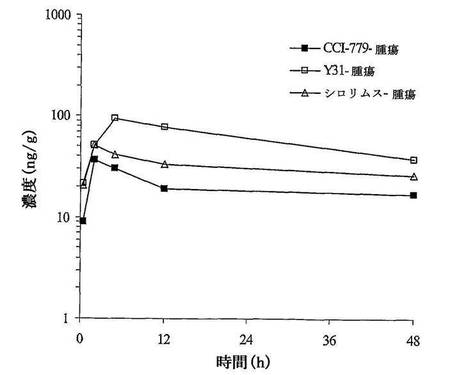
三つの投与群において、投薬してから48時間、血漿及び肝臓中の濃度が、ピーク濃度の1.96%〜6.42%となった。Rapamycinは腫瘍中で比較的に遅く除去され、投与してから48時間、腫瘍中の濃度がピーク濃度の40%〜51%となった。三つの投薬群において、肝臓中のRapamycinの暴露量がそれぞれ血漿中の暴露量の2.26、2.01、1.51倍であり、腫瘍中のRapamycinの暴露量がそれぞれ血漿中の暴露量の1.66、1.05と0.87倍であった。
【0096】
Y31を陽性対照薬Rapamycin及びCCI−779と比較すると、共有の有効成分Rapamycinの肝臓中の暴露量がそれぞれ血漿中の暴露量の2.26、2.01と1.51倍であった。腫瘍中の暴露量がそれぞれ血漿中の暴露量の1.66、1.05と0.87倍であった。Y31が明らかな腫瘍組織特異性を有することが示された。
【0097】
<実施例4:細胞レベルの免疫抑制活性測定実験>
【0098】
【表13】
【0099】
化合物Y50、Y31、Y230の免疫抑制活性実験の結果により、化合物Y31のIC50値が50nMに達し、親化合物のラパマイシン及び化合物Y50より明かに優れ、化合物Y31が比較的に高い安全指数を有し、化合物Y50、Y230の免疫抑制活性がラパマイシンに相当することが示された。
【0100】
<実施例5:Y31の免疫抑制活性測定の系統的実験>
I.マイトジェン/同種異体抗原で誘導された正常マウス脾臓リンパ細胞の増殖活性に関するRapamycin及びY31の影響:
実験目的:
3H−チミジン(3H−thymidine)取り込みにより、マイトジェン/同種異型マウス脾臓リンパ細胞の混合培養による正常マウス脾臓リンパ細胞の増殖機能に対する、化合物のin vitro投与の影響を測定し、そのin vitro免疫抑制活性を評価する。
3−(4,5−ジメチル−2−チアゾリル)−2,5−ジフェニルテトラゾリウムブロミド (MTT)法により、正常マウス脾臓リンパ細胞活性に対して、化合物のin vitro投与の影響を測定し、その細胞毒性効果を評価した。
【0101】
測定薬物:
名称:Rapamycin、Y31。性状、含有量:白い粉末。
【0102】
調製方法:使用する前4℃で保存した。実験前にDMSOに溶解させ、原液を調製した。使用時、培養液で必要な濃度まで希釈した。細胞を培養するとき、DMSO中の最終濃度が<0.02%であった。該濃度のDMSOが細胞成長に影響しない。
【0103】
実験動物:由来、品種、系統:BALB/c純系マウス、メス、18〜20g、中国科学院上海実験動物中心から購入。動物生産ライセンス:SCXK(濾)2002−0010号。動物は上海薬物研究所SPF動物飼育施設で飼育。実験動物使用ライセンス:SYXK(濾)2003−0029号、少なくとも一週間飼育してから使用した。温度が22±1℃であり、湿度が55±5%であり、12時間交代の明暗サイクルで飼育した。餌と水が全部消毒してから動物が自由に摂取した。全部の実験について、完全に実験動物に関する条例に基づいて行った。
【0104】
実験方法及び評価:
MTT法によりマウス脾臓リンパ細胞の活性に対する化合物の影響を測定した。
100μl(4×105/孔)のマウス脾臓リンパ細胞の懸濁液を96穴プレートに接種するとともに、異なる濃度の化合物を加え、相応な溶媒対照及び培養液のバックグランド対照を設け、全体積が200μlであった。37℃で5%のCO2のインキュベータで48時間培養した。培養を終了する6−7時間前に、20μl(5mg/ml)のMTT溶液を加えた。各孔から100μlの上澄みを取り出し、更に100μlのMTT溶解液を加え、インキュベータで6〜7時間放置してから、ELISAにより570nMでOD値を測定した。
【0105】
3H−TdR取り込みにより、マイトジェンで誘導された正常マウス脾臓リンパ細胞の増殖機能に対する化合物の影響を測定した。100μl(4×105/孔)のマウス脾臓リンパ細胞の懸濁液を96穴プレートに接種し、50μlのConA(最終濃度5μg/ml)又は50μlのLPS(最終濃度10μg/ml)を加え、異なる濃度の化合物50μlを注入し、全体積が200μlとなった。各濃度に関して、三つの穴を設け、相応なConA/LPS非投与対照穴及び薬物非投与対照穴を設けた。37℃で5%のCO2のインキュベータで48時間培養した。培養を終了する8時間前に、各孔に25μl(10μCi/ml)の3H−チミジンを加えた。実験終了まで、培養を継続した。細胞収集器(Cell harvester)により細胞をガラス繊維膜に収集し、シンチレーション液を加えてからBetaカウンタ(MicroBeta Trilux, PerkinElmer)により細胞DNAに取り込まれた3H−TdR量を測定し、cpm値を用いて細胞増殖の状況を示す。
【0106】
3H−TdR取り込みにより、同種異体抗原で誘導された正常マウス脾臓リンパ細胞の増殖機能に対する化合物の影響を測定した。
刺激細胞の準備:BALB/cマウス脾臓リンパ細胞の懸濁液、Gamma線照射装置(Gammacell 3000)を用いてセシウム137放射線照射(3000Rads)を行ってその増殖能力を無くしてから、RPMI1640で2回洗浄し、細胞濃度を5×106/mlまで調節した。
【0107】
反応細胞の準備:C57BL/6マウス脾臓リンパ細胞を反応細胞として使用し、細胞濃度は5×106/mlであった。
【0108】
リンパ細胞混合培養:50μl のC57BL/6マウス脾臓リンパ細胞の懸濁液を96穴プレートに接種し、50μl のセシウム137放射線で処理した後のBALB/Cマウス脾臓リンパ細胞の懸濁液を加え、全体積を200μlになるように異なる濃度の化合物50μlを加えた。全体積が200μlより少ない場合、RMPI−1640培養液で補足した。実験において、BALB/cマウス群、C57BL/6マウス群、及びBALB/CとC57BL/6の混合培養群の合計3群を設けた。各濃度に対して、三つの穴を設け、又は相応な非投与対照穴、刺激細胞対照穴及び反応細胞対照穴を設けた。37℃で5%のCO2のインキュベータで3〜5日培養した。培養を終了する8時間前に、各孔に25μl(10μCi/ml)の3H−チミジンを加えた。実験終了まで、培養を継続した。細胞収集器により細胞をガラス繊維膜に収集し、シンチレーション液を加えてからBetaカウンタ(MicroBeta Trilux, PerkinElmer)により細胞DNAに取り込まれた3H−TdR量を測定し、cpm値を用いて細胞増殖の状況を示した。
【0109】
結果処理及び統計方法:全データを平均値±標準偏差で示し、各指標の検査結果をExcel 2000及びSPSS 11.0統計ソフトで処理を行った。
【0110】
実験結果により用量反応曲線(Dose−response curve)を描き、CC50値(50% cytotoxic concentration)、すなわち50%細胞の致死濃度を計算した。また、IC50(50% inhibitory concentration)、すなわち50%抑制効果を有するときの化合物濃度を計算した。
【0111】
実験結果:実験結果を図13及び表14に示す。
【0112】
正常マウス脾臓リンパ細胞に対するRapamycinの細胞毒性は、CC50=45.51μMであった。濃度依存的にConA/LPSで誘導された正常マウス脾臓T/Bリンパ細胞増殖反応を抑制し、その中、IC50がそれぞれ196.4nMと48.8nMであった。正常マウス脾臓リンパ細胞に対するY31の細胞毒性は、CC50=36.9μMであった。濃度依存的にConA/LPSで誘導された正常マウス脾臓T/Bリンパ細胞の増殖反応を抑制し、そのIC50がそれぞれ330.7nMと97.8nMであった。Rapamycin及びY31が、同種異体抗原で誘導された正常マウス脾臓リンパ細胞の増殖反応に対して、抑制活性を示しており、Y31の免疫抑制活性がより強かった。
【0113】
【表14】
【0114】
結果により、Rapamycin及びその誘導体Y31がマイトジェン/同種異体抗原で誘導されたリンパ細胞の増殖反応を明かに抑制し、強いin vitro免疫抑制活性を有することが示された。
【0115】
II.マウス遅延型過敏反応に対するRapamycin及びY31の影響:
実験目的:DNFBで誘導されたマウス遅延型過敏反応(DTH)を用い、DTHに対する化合物in vivo作用の抑制作用を観察した。誘導によりマウスが遅延型過敏反応を生じ、すなわち、マウスがDNFB感作後にDNFBで攻撃されると、マウスが耳介腫脹反応を生じる。マウスの耳介腫脹反応の進行状況に対する化合物の影響を観察することで、マウスDTHに対する影響を評価し、生体細胞の免疫反応に対する影響を検討した。
【0116】
実験薬剤:
名称:Rapamycin、Y31。
性状、含有量:白い粉末。
調製方法:無水アルコールに溶解させ、50mg/mlの保存液を調製した。使用時、5%PEG400+5%Tween−80無菌水を溶媒として必要な濃度まで保存液を希釈した。
【0117】
実験動物:
由来、品種、系統:BALB/c純系マウス、メス、18〜20g、中国科学院上海実験動物中心から購入された。動物生産ライセンス:SCXK(濾)2002−0010号。動物が上海薬物研究所SPF動物飼育施設で飼育された。実験動物使用ライセンス:SYXK(濾)2003−0029号、少なくとも一週間飼育してから使用した。温度が22±1℃であり、湿度が55±5%であり、12時間交代の明暗サイクルで飼育した。餌と水を全部消毒してから動物が自由に摂取した。全部の実験に関して、完全に実験動物に関する条例に基づいて行った。
【0118】
実験方法:
2,4−ジニトロフルオロベンゼン (DNFB)は一種のハプテン(Incomplete antigen)であり、DNFBでマウスの足部を敏感にさせた後、皮膚タンパク質と結合して完全抗原となり、更に一週間後DNFBでマウスの耳を攻撃し、部分的に遅延型アレルギー反応を生じさせ、耳の腫脹の遅延型過敏反応を起こさせた。一方、攻撃されなかった耳の方では耳の腫脹の遅延型過敏反応を生じなかった。耳の腫脹の増加により、ジニトロフルオロベンゼンで誘導されたマウスの遅延型過敏反応に対する進行状況を表した。
(1)20μlの、アセトン:オリーブオイル(4:1)を溶媒とした0.5%DNFB溶液をマウスの各後足に塗布し、敏感にさせた。
(2)第1回敏感にしてから5日、0.2%DNFB溶液を用いてマウスの左耳の両側を塗布して免疫攻撃を行った。対照として、アセトン:オリーブオイル(4:1)溶媒だけで右耳を塗布した。
(3)マウスをモデル群、Dex群(2mg/kg/d、経口投与)、Rapamycin群(1.5mg/kg/d、腹腔内注射投与)、Y31群(1.5mg/kg/d、腹腔内注射投与)と、ランダムに4群と分けた。
(4)スパイラルマイクロメータ(Spiral micrometer)で各マウスの左、右耳の厚さをそれぞれ測定し、右耳の厚さから左耳の厚さを引き、その差を腫脹の増加として記録した。
【0119】
実験結果を図14に示す。
【0120】
DNFBで誘導されたDTH反応は、Th1細胞を介した、T細胞の活性化及び多種の細胞因子の生成を含むアレルギー反応である。我々はBALB/cマウスDTH応答中における化合物の効果を測定し、その結果を以下の図に示した。DNFBで誘導されて遅延型過敏反応を生じたマウスをモデル対照群として用い、その両耳の耳の腫脹の平均増加は0.175mmであった。陽性対照薬Dex 2mg/kg群のマウスの耳の腫脹の平均増加は0.13mmであり、モデル群と比較すると、明らかな差異を有していた。Rapamycin群のマウスの耳の腫脹の平均増加が0.076mmであり、モデル群と比較すると、明らかな差異を有した。Y31群のマウスの耳の腫脹の平均増加が0.129mmであり、モデル群と比較すると、明らかな差異を有していた。
【0121】
実験結果により、Rapamycin及びY31がDNFBで誘導されたマウス遅延型過敏反応に対して、明らかに抑制できることが示された。
【0122】
III.SRBCで誘導されたマウス脾臓リンパ細胞中の特異的抗体産生細胞に対するRapamycin及びY31の影響:
実験目的:
マウスがヒツジ赤血細胞で免疫されると、マウスの脾臓リンパ細胞中に特異的抗体産生細胞が現れる。Rapamycin及びその誘導体を投与した後のマウスの脾臓リンパ細胞の特性的抗体産生細胞の生成量の変化を測定することで、Rapamycin及びその誘導体のマウスの体液性免疫への影響を観察した。
【0123】
実験薬剤:
名称:Rapamycin、Y31。
性状、含有量:白い粉末。
調製方法:無水アルコールに溶解させ、50mg/mlの保存液を調製した。使用時、5%PEG400+5%Tween−80無菌水を溶媒として必要な濃度まで保存液を希釈した。
【0124】
実験動物:
由来、品種、系統:BALB/c純系マウス、メス、18〜20g、中国科学院上海実験動物中心から購入された。動物生産ライセンス:SCXK(濾)2002−0010号。動物が上海薬物研究所SPF動物飼育施設で飼育された。実験動物使用ライセンス:SYXK(濾)2003−0029号、少なくとも一週間飼育してから使用した。温度が22±1℃であり、湿度が55±5%であり、12時間交代の明暗サイクルで飼育した。餌と水を全部消毒してから動物が自由に摂取した。全部の実験に関して、完全に実験動物に関する条例に基づいて行った。
【0125】
マウスは中国科学院上海実験動物中心から購入され、血清(補体)を取るために使用した。
【0126】
その他の実験材料:ヒツジ赤血細胞(SRBC)は上海江南生物技術工程有限公司から購入した。
【0127】
実験原理:
SRBCで誘導されたマウス体液性免疫反応のモデルの中に、SRBCの溶血反応で抗原特性的抗体の生産状況を測定することは、典型的な実験方法である。ヒツジ赤血細胞定量溶血反応検定(Quantitative hemolysis of sheep red blood cells, QHS)は、Bリンパ細胞(プラズマ細胞)で特異的な抗SRBC抗体を生じてヒツジ赤血細胞を破砕し、ヘモグロビンを解放して抗体の生成状況を表す一種の実験方法である。ヒツジ赤血細胞(SRBC)でマウスを敏感にさせた後、マウス脾臓リンパ細胞中に特異的抗体産生細胞が現れる。この細胞の産生する抗体とSRBCが補体の協同作用で溶血反応を生じ、分光光度法により溶血程度を測定し、特異性抗体産生細胞数の生成量を表した。
【0128】
実験方法:
1.BALB/cマウスをランダムに正常対照群、モデル対照群、陽性対照群(CsA10mg/kg)、Rapamycin群(1.5mg/kg/d、腹腔内注射投与)、Y31群(1.5mg/kg/d、腹腔内注射投与)の5群と分け、各群に6匹のマウスを設けた。
免疫にする場合、各群のマウスに腹腔内投与し始め、免疫後の5日まで毎日一回投与した。モデル対照群に対して、毎日溶媒対照を行った。
【0129】
2.PBSで新鮮なヒツジ赤血細胞(SRBC)を3回洗浄した後、1:5(v/v)に希釈し、各マウスに0.2ml希釈されたSRBCを腹腔内注射して、敏感にさせた。
【0130】
3.免疫後の5日でQHS測定を行い、マウス脾臓を取り、脾臓リンパ細胞を調製し。
分光光度法により溶血程度の測定:リンパ細胞5×106、0.2%SRBC、最適希釈比率のマウスの血清補体を均一に混合させ、37℃で1時間定温放置してから、3000rpm、10分間遠心分離し、上澄みを取り出した。540nmの所でOD値を測定し、特異的抗体産生細胞の生成量を表した。
【0131】
実験結果(表15に示す):
ヒツジ赤血細胞定量溶血反応検定(QHS)で、体内におけるB細胞の分泌した抗体でヒツジ赤血細胞を破砕して解放されたヘモグロビン(OD値)を用いて表した。その結果で、生体の体液性免疫機能を表す。
【0132】
特異的抗体産生細胞抑制%=(モデル対照群OD−投与群OD)/(モデル対照群OD−正常対照群OD)
【0133】
【表15】
【0134】
実験結論:
Rapamycin(1.5mg/kg)及びその誘導物Y31(1.5mg/kg)の腹腔内投与が、マウス脾臓中の特異的な抗SRBC抗体産生細胞の生成量を明らかに抑制でき、その抑制作用が陽性対照薬CsAより強く、マウスの体液性免疫機能に対して、明らかな抑制作用を有した。
【0135】
IV.マウス急性移植片対宿主病(aGVHD)に対するRapamycin及びY31の薬効学研究:
実験目的:
BABL/Cマウスをドナーとして、C57B6/6マウスを受容体として用い、BABL/Cマウス骨髄細胞及びリンパ細胞を致死量γ射線で照射したC57B/6マウス体内に移植し、急性移植片対宿主病(aGVHD)モデルを作成し、マウスのaGVHDに対するRapamycin及び誘導体の薬効学効果を考察した。
【0136】
実験薬剤:
名称:Rapamycin、Y31。
性状、含有量:白い粉末。
調製方法:無水アルコールに溶解させ、50mg/mlの保存液を調製した。使用時、5%PEG400+5%Tween−80無菌水を溶媒として必要な濃度まで保存液を希釈した。
【0137】
実験動物:
由来、品種、系統:BALB/c純系マウス、メス、4週齢、C57B/6マウス、メス、7〜8週齢、中国科学院上海実験動物中心から購入された。動物生産ライセンス:SCXK(濾)2002−0010号。動物が上海薬物研究所SPF動物飼育施設で飼育された。実験動物使用ライセンス:SYXK(濾)2003−0029号、少なくとも一週間飼育してから使用した。温度が22±1℃であり、湿度が55±5%であり、12時間交代の明暗サイクルで飼育した。餌と水を全部消毒してから動物が自由に摂取した。全部の実験に関して、完全に実験動物に関する条例に基づいて行った。
【0138】
実験方法:
1.全身照射(TBI):
C57B/6(H−2b)、メス、7週齢のマウスを受容体マウスとして使用した。
Gammacellで8.5Gy全身照射した。
【0139】
2.骨髄移植:
受容体マウスに4〜6時間照射した後、異種骨髄移植を行った。
BABL/C(H−2d)、メス、4週齢のマウスをドナーマウスとして使用した。マウスの四肢長骨骨髄細胞及び脾臓リンパ細胞を採取した。二種類の細胞濃度を1×108まで調製し、PBS緩衝液中に懸濁させた。
等比例で二種類の細胞を混合させ、各受容体マウスに混合細胞の懸濁液を0.5mlずつ静脈注射した。
【0140】
3.群分け及び投与:
ランダムにモデル群(溶媒対照)、Rapamycin群(1.5mg/kg/d、腹腔内注射投与)、Y−31群(1.5mg/kg/d、腹腔内注射投与)と3群に分け、各群のサンプル数はn=10であり、骨髄移植の当日から投与開始し、毎日1回行った。
【0141】
4.測定指標:
(1)体重:毎日1回計測した。
(2)BMT後のマウスの生存時間を記録した。
【0142】
実験結果:
本実験において、急性移植片対宿主病(aGVHD)のマウスモデルを作成し、C57BL/6マウスが半数致死量(Sub−fatal dose)8.5Gyの全身照射を受けた後、BALB/Cマウス骨髄細胞及び脾臓細胞を注入し、aGVHDモデルを複製し、aGVHDマウスの生存及び体重変化に対する化合物の影響を観察した。
実験結果を図15示した。骨髄移植した後、aGVHDモデルマウスの体重が明らかに低減し、且つ死亡例が見られた。aGVHD異体移植によるマウス体重の低減に対して、Rapamycin及び誘導体Y31が明らかな緩衝作用を有し、aGVHDマウスの生存率を上昇させ、治療効果が明らかであった。
実験結果により、急性移植片対宿主病(aGVHD)動物モデルに対して、Rapamycin及び誘導体Y31が良好な治療効果を有することが証明された。
【0143】
V.DBA/1マウスでの牛II型コラーゲン誘導関節炎に関するY31の治療効果:
実験目的:本実験において牛型コラーゲンでDBA/1マウス関節炎を誘導し、Y31を体内へ投与し、マウス関節炎の発病指数(Disease index)の変化を観察することで、マウス関節炎に対するY31の治療効果を観察・評価した。
【0144】
実験薬剤:
名称:Y31。
性状、含有量:白い粉末。
調製方法:無水アルコールに溶解させ、50mg/mlの保存液を調製した。使用時、5%PEG400+5%Tween−80無菌水を溶媒として必要な濃度まで保存液を希釈した。
【0145】
実験動物及び材料:
DBA/1マウス、7〜8週齢、体重20〜22g、日本大阪大学医学部Hiromi Fujiwara教授より提供された。動物は上海薬物研究所SPF動物飼育施設で飼育され、少なくとも一週間飼育してから使用した。温度が22±1℃であり、湿度が55±5%であり、12時間交代の明暗サイクルで飼育した。餌と水を全部消毒してから動物が自由に摂取した。全部の実験に関して、完全に実験動物に関する条例に基づいて行った。
Mycobacterium tuberculosisstrain H37Rvを含むフロイント完全アジュバント(Freund’s complete adjuvant)がWako Pure Chemical Industries Ltd(Osaka, Japan)から購入。
【0146】
実験方法:
関節炎モデル:牛II型コラーゲンに0.1Mの酢酸を加え、濃度を20mg/mlに調製し、4℃の冷蔵庫で一晩放置して溶解させた。コラーゲンが同体積のフロイント完全アジュバント(結核菌を含むH37Rv株)と充分乳化し、オスのDBA/1マウスを麻酔した後、25μl/匹(すなわち、250μg/匹)でその尾部を敏感にさせた。3週間後、同用量で攻撃した。肉眼でマウスの四肢を観察し、関節炎の程度に関して、0〜4段階評価を行った:0=正常、1=1個又は数個の趾関節が発赤又は腫脹、2=膝関節以下が中等度発赤腫脹、3=膝関節を含む部分が重度発赤腫脹、4=膝関節を含む部分が完全に発赤腫脹、関節変形、硬直、機能障害。各マウスの最高評価点数が、16点であった。
【0147】
薬物治療:
ランダムにマウスをモデル対照群、Y31(1mg/kg)投与群(攻撃した後14日から投与開始し、三週連続投与した)と、二群に分けた。
【0148】
実験結果:
牛II型コラーゲンを皮下に二回注射することでDBA/1マウス関節炎を誘導できた。関節腫脹が攻撃された後の4日から現れ、一週間後100%のマウスが関節炎を発症し、且つ関節腫脹が進行性に悪化し、14日から投与開始した。Y31を投与した後、CIAの発症程度が明らかに低減され、マウスの四肢(足、爪)の腫脹が明らかに軽減された(図16、p<0.05)。Y31経口投与がコラーゲンで誘導されたDBA/1マウス関節炎の発症を抑制できることが示された。
【特許請求の範囲】
【請求項1】
構造式Iに示すラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩において、
【化1】
式中、R1とR2は、各々独立に、H又は、
【化2】
であり、且つR1、R2同時にHではなく、
その中、nは1〜6の整数であり、R3は、
【化3】
又は
【化4】
であり、R4、R5とR6は、各々独立に、H、炭素数1〜6のヒドロキシアルキル基、炭素数1〜6のアルキル基又は炭素数2〜6のアルケニル基であり、R7とR8は、各々独立に、H又は炭素数1〜6のアルキル基である、
ラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩。
【請求項2】
請求項1に記載の構造式Iに示すラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩において、
nは1〜4の整数であり、R3は、
【化5】
又は
【化6】
であり、
その中、R4、R5とR6が、各々独立に、H又は炭素数1〜4のヒドロキシアルキル基であり、R7とR8が、各々独立に、炭素数1〜4のアルキル基である、
ことを特徴とするラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩。
【請求項3】
請求項2に記載の構造式Iに示すラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩において、
nは1〜2の整数であり、R3は、
【化7】
又は
【化8】
であり、
その中、R7とR8は、各々独立に、炭素数1〜4のアルキル基である、
ことを特徴とするラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩。
【請求項4】
請求項3に記載の構造式Iに示すラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩において、
前記化合物が以下のいずれかである、
【化9】
【化10】
【化11】
【化12】
ことを特徴とするラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩。
【請求項5】
請求項1に記載の構造式Iに示すラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩において、
前記R3の中にキラル中心を有する場合、前記ラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩が光学異性体又は光学異性体の混合物である、
ことを特徴とするラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩。
【請求項6】
請求項1から請求項5のいずれか一項に記載のラパマイシン炭酸エステル類似体の製造方法であって、
R1、R2が同じく、
【化13】
で、R3が、
【化14】
又は
【化15】
である場合、以下の方法により製造する:
構造式1に示すアルコールが、p−トルエンスルホン酸の触媒作用で、DMSO、DMFなどの溶媒中において、カルボニル化合物R7COR8又はそのアセタール化合物と反応し、構造式2に示すアルコールを得て、
アルカリにより、トリホスゲンが構造式2に示したアルコールと反応し、アシルクロリド3を形成し、
更に、アルカリにより該アシルクロリド3がラパマイシンと反応し、R3が、
【化16】
である31位、42位の二置換ラパマイシン炭酸エステル類似体を得て、
更に、該ラパマイシン炭酸エステル類似体を加水分解することにより、R3が、
【化17】
であるラパマイシン炭酸エステル類似体を得て、
その反応式が以下のようであり、
【化18】
式中、R4、R5、R6、R7及びR8の定義が引用した請求項の記載と同様であり、
或いは、
R1とR2が異なり、それぞれH又は、
【化19】
であり、R3が、
【化20】
又は
【化21】
である場合、以下の方法で製造する:
適切な比例のイミダゾールとクロロトリメチルシランの存在下で、ジクロロメタン、ジクロロエタン、テトラヒドロフラン、アセトニトリル又はDMFを溶媒として、ラパマイシンとクロロトリメチルシランとを反応させ、31位のみ保護する生成物Rapamycin−31−OTMSを得て、
Rapamycin−31−OTMSの42位のヒドロキシ基をTBSにより保護してから、安定性の悪い31位での保護基を脱離させることにより、42位のみ保護するRapamycin−42−OTBSを得て、
その反応式は以下のようであり、
【化22】
Rapamycin−31−OTMSが直接にアシルクロリド(式3)
【化23】
と反応し、42位をエステル化する生成物を得て、更に31位のシリル系保護基を脱離させた後、対応する42位をモノエステル化したラパマイシン炭酸エステル類似体を得て、
又は、42位のみ保護する生成物Rapamycin−42-OTBSとアシルクロリド(式3)
【化24】
とを反応させ、31位をエステルした化生成物が得られ、更に42位のシリル系保護基を脱離させ、対応する31位のみモノエステル化したラパマイシン炭酸エステル類似体を得る、
ことを特徴とするラパマイシン炭酸エステル類似体の製造方法。
【請求項7】
抗腫瘍活性及び抗癌活性又は免疫抑制活性を有する薬剤組成物であって、
活性成分として治療有効量の一種又は数種の請求項1に記載のラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩と、一種又は数種の薬学的に受容可能なキャリアとを含む、
ことを特徴とする薬剤組成物。
【請求項8】
請求項1から請求項5のいずれか一項に記載のラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩の、抗腫瘍薬及び/又は抗癌薬或いは免疫抑制薬 の製造における使用。
【請求項9】
請求項1から請求項5のいずれか一項に記載のラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩の、ヒト横紋筋肉腫、前立腺癌、非小細胞肺癌、乳癌、結腸癌、腎臓癌、肺腺癌、子宮頸癌又は白血病の治療薬の製造における使用。
【請求項1】
構造式Iに示すラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩において、
【化1】
式中、R1とR2は、各々独立に、H又は、
【化2】
であり、且つR1、R2同時にHではなく、
その中、nは1〜6の整数であり、R3は、
【化3】
又は
【化4】
であり、R4、R5とR6は、各々独立に、H、炭素数1〜6のヒドロキシアルキル基、炭素数1〜6のアルキル基又は炭素数2〜6のアルケニル基であり、R7とR8は、各々独立に、H又は炭素数1〜6のアルキル基である、
ラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩。
【請求項2】
請求項1に記載の構造式Iに示すラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩において、
nは1〜4の整数であり、R3は、
【化5】
又は
【化6】
であり、
その中、R4、R5とR6が、各々独立に、H又は炭素数1〜4のヒドロキシアルキル基であり、R7とR8が、各々独立に、炭素数1〜4のアルキル基である、
ことを特徴とするラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩。
【請求項3】
請求項2に記載の構造式Iに示すラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩において、
nは1〜2の整数であり、R3は、
【化7】
又は
【化8】
であり、
その中、R7とR8は、各々独立に、炭素数1〜4のアルキル基である、
ことを特徴とするラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩。
【請求項4】
請求項3に記載の構造式Iに示すラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩において、
前記化合物が以下のいずれかである、
【化9】
【化10】
【化11】
【化12】
ことを特徴とするラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩。
【請求項5】
請求項1に記載の構造式Iに示すラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩において、
前記R3の中にキラル中心を有する場合、前記ラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩が光学異性体又は光学異性体の混合物である、
ことを特徴とするラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩。
【請求項6】
請求項1から請求項5のいずれか一項に記載のラパマイシン炭酸エステル類似体の製造方法であって、
R1、R2が同じく、
【化13】
で、R3が、
【化14】
又は
【化15】
である場合、以下の方法により製造する:
構造式1に示すアルコールが、p−トルエンスルホン酸の触媒作用で、DMSO、DMFなどの溶媒中において、カルボニル化合物R7COR8又はそのアセタール化合物と反応し、構造式2に示すアルコールを得て、
アルカリにより、トリホスゲンが構造式2に示したアルコールと反応し、アシルクロリド3を形成し、
更に、アルカリにより該アシルクロリド3がラパマイシンと反応し、R3が、
【化16】
である31位、42位の二置換ラパマイシン炭酸エステル類似体を得て、
更に、該ラパマイシン炭酸エステル類似体を加水分解することにより、R3が、
【化17】
であるラパマイシン炭酸エステル類似体を得て、
その反応式が以下のようであり、
【化18】
式中、R4、R5、R6、R7及びR8の定義が引用した請求項の記載と同様であり、
或いは、
R1とR2が異なり、それぞれH又は、
【化19】
であり、R3が、
【化20】
又は
【化21】
である場合、以下の方法で製造する:
適切な比例のイミダゾールとクロロトリメチルシランの存在下で、ジクロロメタン、ジクロロエタン、テトラヒドロフラン、アセトニトリル又はDMFを溶媒として、ラパマイシンとクロロトリメチルシランとを反応させ、31位のみ保護する生成物Rapamycin−31−OTMSを得て、
Rapamycin−31−OTMSの42位のヒドロキシ基をTBSにより保護してから、安定性の悪い31位での保護基を脱離させることにより、42位のみ保護するRapamycin−42−OTBSを得て、
その反応式は以下のようであり、
【化22】
Rapamycin−31−OTMSが直接にアシルクロリド(式3)
【化23】
と反応し、42位をエステル化する生成物を得て、更に31位のシリル系保護基を脱離させた後、対応する42位をモノエステル化したラパマイシン炭酸エステル類似体を得て、
又は、42位のみ保護する生成物Rapamycin−42-OTBSとアシルクロリド(式3)
【化24】
とを反応させ、31位をエステルした化生成物が得られ、更に42位のシリル系保護基を脱離させ、対応する31位のみモノエステル化したラパマイシン炭酸エステル類似体を得る、
ことを特徴とするラパマイシン炭酸エステル類似体の製造方法。
【請求項7】
抗腫瘍活性及び抗癌活性又は免疫抑制活性を有する薬剤組成物であって、
活性成分として治療有効量の一種又は数種の請求項1に記載のラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩と、一種又は数種の薬学的に受容可能なキャリアとを含む、
ことを特徴とする薬剤組成物。
【請求項8】
請求項1から請求項5のいずれか一項に記載のラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩の、抗腫瘍薬及び/又は抗癌薬或いは免疫抑制薬 の製造における使用。
【請求項9】
請求項1から請求項5のいずれか一項に記載のラパマイシン炭酸エステル類似体又はその薬学的に受容可能な塩の、ヒト横紋筋肉腫、前立腺癌、非小細胞肺癌、乳癌、結腸癌、腎臓癌、肺腺癌、子宮頸癌又は白血病の治療薬の製造における使用。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【公表番号】特表2012−502930(P2012−502930A)
【公表日】平成24年2月2日(2012.2.2)
【国際特許分類】
【出願番号】特願2011−527184(P2011−527184)
【出願日】平成21年9月17日(2009.9.17)
【国際出願番号】PCT/CN2009/001042
【国際公開番号】WO2010/031251
【国際公開日】平成22年3月25日(2010.3.25)
【出願人】(507388845)中国科学院上海薬物研究所 (4)
【Fターム(参考)】
【公表日】平成24年2月2日(2012.2.2)
【国際特許分類】
【出願日】平成21年9月17日(2009.9.17)
【国際出願番号】PCT/CN2009/001042
【国際公開番号】WO2010/031251
【国際公開日】平成22年3月25日(2010.3.25)
【出願人】(507388845)中国科学院上海薬物研究所 (4)
【Fターム(参考)】
[ Back to top ]