
リガンド親和性複合タンパク質の取得方法、抗体スクリーニング方法、抗体スクリーニング用キット、医療方法、抗体医薬、非免疫反応性抗体医薬、投薬方法

【課題】 極めて多種類のリガンド親和性タンパク質の中から特定された1種又は2種以上のタンパク質を迅速・大量・高純度に取得する技術の提供。
【解決手段】 1)ヒト又は非ヒト動物たる検体から、多種類の複合タンパク質Xpの各専用の生産手段Xmを備えた抗体産生細胞を単細胞分離によって厳密に分離し、2)生産手段Xmを逆転写PCR法により複製手段Xcへ一旦転換させて、3)この複製手段XcをPCR法で大量複製し、4)転写用配列を付加したもとで、5)in vitro転写/翻訳共役系へ投入して複合タンパク質Xpを1種類ごとに大量生産し、必要に応じて6)複合タンパク質Xpのキャラクタリゼーションを行う、リガンド親和性複合タンパク質の取得方法。
【解決手段】 1)ヒト又は非ヒト動物たる検体から、多種類の複合タンパク質Xpの各専用の生産手段Xmを備えた抗体産生細胞を単細胞分離によって厳密に分離し、2)生産手段Xmを逆転写PCR法により複製手段Xcへ一旦転換させて、3)この複製手段XcをPCR法で大量複製し、4)転写用配列を付加したもとで、5)in vitro転写/翻訳共役系へ投入して複合タンパク質Xpを1種類ごとに大量生産し、必要に応じて6)複合タンパク質Xpのキャラクタリゼーションを行う、リガンド親和性複合タンパク質の取得方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、リガンド親和性複合タンパク質の取得方法、抗体スクリーニング方法、抗体スクリーニング用キット、医療方法、抗体医薬、非免疫反応性抗体医薬、及び投薬方法に関する。
【0002】
更に詳しくは、本発明は、特定されたリガンドに対して選択的な親和性を示す極めて多種類の複合タンパク質Xpの中から選ばれる1種又は2種以上の複合タンパク質を迅速かつ大量に取得することを可能とした、リガンド親和性複合タンパク質の取得方法に関する。このリガンド親和性複合タンパク質の取得方法は、ヒトに対して適用することができ、更に、少なくとも免疫反応による抗体の生成が認められる非ヒト動物に対しても適用することができる。
【0003】
又、本発明は、上記のリガンド親和性複合タンパク質の取得方法に基づいた、ハイ・スループット(High-throughput )なスクリーニングを迅速に実行可能とする抗体スクリーニング方法と、この抗体スクリーニング方法を行うための抗体スクリーニング用キットに関する。
【0004】
又、本発明は、上記の抗体スクリーニング方法によって得られた抗体を用いる、ヒト又は上記の非ヒト動物に対する医療方法と、当該抗体を有効成分とする抗体医薬とに関する。
【0005】
更に、本発明は、ヒトを適用対象とする上記のリガンド親和性複合タンパク質の取得方法によって得られた抗体を有効成分とする医薬であって、適用対象たる当該ヒト個体に投与することを目的とする非免疫反応性抗体医薬と、この非免疫反応性抗体医薬を用いる投薬方法とに関する。
【0006】
なお、本発明において、「リガンド」とは、任意の無機化合物や有機化合物、及び抗原たり得る各種の無機化合物や有機化合物、タンパク質、ウイルス、微生物等を制限なく含む概念である。リガンドが抗原である場合において、本発明のリガンド親和性複合タンパク質は抗体あるいはモノクローナル抗体(以下、単に「Mab」とも言う)であり得る。
【背景技術】
【0007】
任意のリガンドに対して選択的な親和性を示すタンパク質は、一般的な技術分野においては、当該リガンドの検出手段、分離手段あるいは精製手段として重要である。
【0008】
一方、医学あるいは生理学の分野においては、このような親和性タンパク質として、ヒト又は少なくとも免疫反応による抗体の産生が認められる非ヒト動物において生成される極めて多種類の抗体を代表的に例示することができる。抗体は免疫学における重要な生体分子であり、ヒト又は非ヒト動物の疾患の予防、診断あるいは治療等に広範囲に応用することができる。非ヒト動物を用いた病理研究等にも広範囲に応用することができる。抗体の内でも、特に、いわゆるハイブリドーマ技術によって大量生産が可能となったMabは極めて重要である。
【0009】
一般的に、リガンド親和性タンパク質を取得する場合において、例えば任意の抗原即ちリガンドで免疫した非ヒト動物、ヒトにおける各種の感染症患者、あるいは、ガン細胞がリガンドとなって抗体を生成しているガン患者等に典型的に見られるように、リガンドに対する親和性又は結合力等に差異のある極めて多種類の類似タンパク質が存在する場合が多い。
【0010】
例えばポリクローナル抗体を使用する場合のように、このような類似タンパク質群を混ぜ物としてそのまま利用することも、場合によって可能ではある。しかし、リガンドの検出手段、分離手段あるいは精製手段としても、疾患の予防、診断あるいは治療等の手段としても、多種類の類似タンパク質群の内から特定された1種類ごとの優れたリガンド親和性タンパク質のみを選択的に大量取得することが、圧倒的に有利である。
【0011】
ハイブリドーマ技術は、1種類のリガンド親和性タンパク質のみを、Mabとして選択的に大量取得する可能性を切り開いた点で重要な意味を持つ。しかし、この技術では、動物の免疫反応を利用して抗原親和性タンパク質たる各種Mabの生産源である多数のB細胞を調製する点は良いとしても、その後、例えば安定したハイブリドーマの作成(タンパク質の生産手段の調製)に困難があったり、ハイブリドーマの培養と増殖(タンパク質の生産手段の大規模化)に非常に手間がかかる等の問題がある。従って、実用技術としては、迅速な大量生産及び高速多検体スクリーニングと言う要求に十分に応えることができない。
【0012】
近年、ハイブリドーマ技術の上記問題点に鑑み、B細胞の生成と組織培養を不要化するため、例えば、キメラモノクローナル抗体(Vaquelo et al., 1999)、遺伝子導入マウス(Green et al., 1994)、ファージディスプレー(MacCafferty
et al., 1990)、リボソームディスプレー(Hanes & Plucthun, 1997)のような種々の遺伝子工学的技術の開発が図られている。しかしこれらの技術は、いわば要素技術に過ぎず、多様なリガンド親和性タンパク質を大量生産し高速多検体スクリーニングを行うと言う要求に全体的に応えることはできないし、より具体的なレベルの問題として、抗体のH鎖とL鎖とのオリジナルで正確なペアリングは得られていない。
【0013】
【非特許文献1】Coronella, J.A., Telleman, P., Truong, T.D., Ylera, F., Junghans, R.P., 2000. "Amplification of IgG VH and VL(Fab) fromsingle cell human plasma cells and B cells" Nucleic Acids Res. 28,e85 又、B細胞を迅速・正確に単離する技術、キット化された実験試薬、シークエンシングとその解析の自動化、単細胞逆転写PCR技術等が進歩し、例えば上記の非特許文献1で記載されているように、種々の細胞由来の抗体遺伝子の増幅が報告されて来た。即ち、単細胞逆転写PCRの利点により、単一の細胞から得た抗体のL鎖遺伝子とH鎖遺伝子とをそれぞれ増幅し、組換え大腸菌を用いた発現システムを利用すれば、抗体のH鎖とL鎖とのペアリングは実現できる。
【0014】
しかし、逆転写PCR産物たる抗体遺伝子を、発現のためにベクターにクローニングする必要があり、そのクローニングと微生物での発現には非常に面倒な作業と長時間を要する。又、細胞毒性を有する抗体を扱うことが困難であるとか、極めて多数のクローン(極めて多数の組換え大腸菌)を扱うことが困難であるとかの問題がある。
【0015】
以上の点は、特定されたリガンド親和性タンパク質に対する迅速な大量生産と高速多検体スクリーニングと言う一般的な技術手段としての要求に関する問題の指摘であるが、この問題を医学又は生理学の立場から見ると、優れた抗体スクリーニング方法、医療方法、抗体医薬等の提供の問題に直結するものである。更に、ヒトを対象とする場合には、免疫反応が関連する疾患等において、拒絶反応を伴わない非経口的投与を可能とする非免疫反応性抗体医薬の提供の問題にも直結するものである。
【発明の開示】
【発明が解決しようとする課題】
【0016】
本願発明者は、極めて多種類のリガンド親和性複合タンパク質の中から特定された1種又は2種以上のタンパク質を迅速・大量に取得するための技術として、感染症患者等からヒトの抗体産生細胞を非侵襲的手段によって採取し、又は免疫反応を利用して非ヒト動物の抗体産生細胞を調製し、これをハイブリドーマ技術に替わるものとしての単細胞逆転写PCR技術と組み合わせ、更に、クローン化された遺伝子からのタンパク質製造技術として近年注目されている無細胞タンパク質合成系を組み合わせることを着想した。
【0017】
しかし、このような生産技術体系を構想する場合、とりわけ、極めて多種類のリガンド親和性複合タンパク質の中から優れた特性を有する1種又は2種以上のタンパク質を選択的に迅速・大量取得しようとする場合、下記イ)、ロ)、ハ)に起因する実質的に無限とも言える多様性の制約の中で、その目的を果たさねばならない。
イ)抗体として得られる目的タンパク質には実質的に無限とも言える極めて多種のバリエーションがある。
ロ)タンパク質の各分子が複数のフラグメントからなる複合タンパク質であって、各フラグメントが個別の構造遺伝子でコードされている。
ハ)この複合タンパク質においてIgG、IgA、IgE等の各クラスと、L鎖におけるκ鎖やλ鎖等の各アイソタイプがある。
【0018】
本願発明者の知る限りにおいて、このような多様性の制約の中で特定の複合タンパク質を選択的に迅速・大量取得しようとする課題に取り組んだリガンド親和性複合タンパク質の取得方法は、未だ提案されたことがない。本願発明者は、このような前例のない課題の解決に取り組み、本願発明に係るリガンド親和性複合タンパク質の取得方法を完成した。更に、このリガンド親和性複合タンパク質の取得方法を基礎として、抗体スクリーニング方法、抗体スクリーニング用キット、医療方法、抗体医薬、非免疫反応性抗体医薬、及び投薬方法の各発明をも完成した。
【課題を解決するための手段】
【0019】
(第1発明の構成)
上記課題を解決するための本願第1発明の構成は、任意に特定されたリガンドXに対して選択的な親和性を示す極めて多種類の複合タンパク質Xpの中から、無作為にあるいは特性評価を通じて選ばれる1種又は2種以上の複合タンパク質を取得する方法であって、少なくとも下記の1)生産手段分離工程、2)生産手段転換工程、3)大量生産準備工程、4)転写用配列付加工程及び5)単一種大量生産工程を含み、必要に応じて更に下記の6)キャラクタリゼーション工程を含む、リガンド親和性複合タンパク質の取得方法である。
【0020】
1)極めて多種類にわたる前記複合タンパク質Xpのそれぞれ専用の生産手段(mRNA)であって、容易に大量複製できる複製手段Xc(cDNA)への転換が可能な生産手段Xmを備えた抗体産生細胞の1個又は複数個の単細胞を、ヒト又は非ヒト動物たる検体の適宜な組織又は生体構成部分から個別に分離することにより、目的タンパク質の種類ごとに生産手段Xmを厳密に分離して、以下の各工程における生産手段Xm、複製手段Xc及び複合タンパク質Xpの異種間混在を予め防止する生産手段分離工程。
【0021】
2)互いに分離された1種又は複数種の前記生産手段Xmについて、それぞれの生産手段Xmを構成するmRNAの全部又は一部を逆転写PCR法により複製手段Xcへ転換させる生産手段転換工程。
【0022】
3)逆転写PCR法により得られた前記複製手段XcをPCR法で大量複製してタンパク質の大量生産能力を準備する大量生産準備工程。
【0023】
4)大量複製された前記複製手段Xc(cDNA)に対して、その無細胞系での生産手段Xm(mRNA)への再転換に必要な転写用配列を付加する転写用配列付加工程。
【0024】
5)転写用配列を付加された前記複製手段Xcを、少なくとも複製手段Xcの生産手段Xmへの再転換を行う要素と、生産手段Xmに基づきタンパク質の生産を行う要素とを備えた無細胞系へ投入することにより、前記複合タンパク質Xpを、又は前記生産手段転換工程で規定した一部のmRNAに対応する複合タンパク質Xpを、1種類ごとに大量生産する単一種大量生産工程。
【0025】
6)以上の各工程により生産された複合タンパク質Xpについて、特性評価を行い、優れた特性を持つ複合タンパク質Xpをスクリーニングし、あるいはこれらの複合タンパク質Xpの配列決定を行うキャラクタリゼーション工程。
【0026】
(第2発明の構成)
上記課題を解決するための本願第2発明の構成は、前記第1発明に係る検体がヒトである場合において、抗原たるリガンドXに対する抗体を既に体内に生成している生理状態にあると推定される被検者を選択したもとで、当該被検者における適宜な組織又は生体構成部分を社会通念上許容される非侵襲的手段によって採取し、この組織又は生体構成部分から前記1)の1個又は複数個の単細胞を分離する、リガンド親和性複合タンパク質の取得方法である。
【0027】
(第3発明の構成)
上記課題を解決するための本願第3発明の構成は、前記第2発明に係る被検者から、前記組織又は生体構成部分として末梢血を採取し、この末梢血から抗体産生細胞たるB細胞及び/又は形質細胞の単細胞を分離する、リガンド親和性複合タンパク質の取得方法である。
【0028】
(第4発明の構成)
上記課題を解決するための本願第4発明の構成は、前記第1発明に係る検体が非ヒト動物である場合において、この動物をリガンドXで免疫したもとで、当該動物の適宜な組織又は生体構成部分から前記1)の1個又は複数個の単細胞を分離する、リガンド親和性複合タンパク質の取得方法である。
【0029】
(第5発明の構成)
上記課題を解決するための本願第5発明の構成は、前記第1発明〜第4発明のいずれかに係る複合タンパク質Xpが各1対のH鎖及びL鎖からなる免疫グロブリン(抗体)、そのFab(抗原結合性フラグメント)、又はこれらに準ずるタンパク質フラグメントである、リガンド親和性複合タンパク質の取得方法である。
【0030】
(第6発明の構成)
上記課題を解決するための本願第6発明の構成は、前記第1発明〜第5発明のいずれかに係る生産手段分離工程において、前記検体の組織又は生体構成部分を液体中に投入して抗体産生細胞を分散・懸濁させ、一定の分離用抗体と結合させた磁性微粒子と共にインキュベートした後、分離用抗体を介して抗体産生細胞に結合した磁性微粒子をマグネットで回収することにより、前記懸濁液から目的とする抗体産生細胞の単細胞を分離する、リガンド親和性複合タンパク質の取得方法である。
【0031】
(第7発明の構成)
上記課題を解決するための本願第7発明の構成は、前記第1発明〜第6発明のいずれかに係る生産手段転換工程において、逆転写PCRに用いるプライマーの設計により、特定の分類カテゴリーに属する複合タンパク質XpをコードするmRNAのみを複製手段Xcへ転換させ、及び/又は複合タンパク質Xpを構成する特定のフラグメントをコードするmRNAのみを前記複製手段Xcへ転換させる、リガンド親和性複合タンパク質の取得方法である。
【0032】
(第8発明の構成)
上記課題を解決するための本願第8発明の構成は、前記第1発明〜第7発明のいずれかに係る大量生産準備工程において、複製手段XcのPCR法による大量複製を、次の2段階PCRによって行う、リガンド親和性複合タンパク質の取得方法である。
【0033】
第1段階PCR:複製手段Xcを構成する全ての種類のcDNAに対応する各種類のプライマーにおいて、それらの5’末端側に同一の付加配列を設けておいて増幅を行う。
【0034】
第2段階PCR:第1段階PCRで増幅された全ての種類のDNAフラグメントに対して、前記付加配列に対して相補的な配列部分を備えた単一種類のプライマーを用いて増幅を行う。
【0035】
(第9発明の構成)
上記課題を解決するための本願第9発明の構成は、前記第8発明に係る第1段階PCRで用いる各種類のプライマーの付加配列中に、前記5)の無細胞系の由来生物を考慮した適宜な種類の翻訳開始促進配列部分を含めておく、リガンド親和性複合タンパク質の取得方法である。
【0036】
(第10発明の構成)
上記課題を解決するための本願第10発明の構成は、前記第1発明〜第9発明のいずれかに係る転写用配列付加工程において、大量生産準備工程で増幅されたDNAフラグメントに対して、少なくともプロモーター配列を付加するPCRを行う、リガンド親和性複合タンパク質の取得方法である。
【0037】
(第11発明の構成)
上記課題を解決するための本願第11発明の構成は、前記第1発明〜第10発明のいずれかに係る単一種大量生産工程において、前記無細胞系を還元化しないままの条件で用いる、リガンド親和性複合タンパク質の取得方法である。
【0038】
(第12発明の構成)
上記課題を解決するための本願第12発明の構成は、前記第1発明〜第11発明のいずれかに係るキャラクタリゼーション工程が、複合タンパク質XpのELISA法によるリガンド親和性評価と、複合タンパク質Xp又はその遺伝子の配列決定と、形質転換体を用いて生産させた複合タンパク質XpのKd値の測定との内の少なくとも1項目を含む、リガンド親和性複合タンパク質の取得方法である。
【0039】
(第13発明の構成)
上記課題を解決するための本願第13発明の構成は、前記第1発明〜第12発明のいずれかに係るリガンド親和性複合タンパク質の取得方法を、ヒト又は非ヒト動物におけるリガンドXに対する抗体のスクリーニングに用いる、抗体スクリーニング方法である。
【0040】
(第14発明の構成)
上記課題を解決するための本願第14発明の構成は、第13発明に係る抗体スクリーニング方法を実施するために用いられるキットであって、少なくとも以下の構成要素を含んで構成される、抗体スクリーニング用キットである。
【0041】
11)第6発明に係る分散・懸濁用の液体と、磁性微粒子とのセット。
【0042】
12)第7発明に係る逆転写PCR用のプライマーセット。
【0043】
13)第8発明に係る2段階のPCR用のプライマーセット。
【0044】
14)第1発明の5)に係る無細胞系。
【0045】
(第15発明の構成)
上記課題を解決するための本願第15発明の構成は、第1発明及び第4発明〜第12発明のいずれかに係るリガンド親和性複合タンパク質の取得方法によって得られた抗体を用いて、非ヒト動物の疾患の予防、診断又は治療を行う、医療方法である。
【0046】
(第16発明の構成)
上記課題を解決するための本願第16発明の構成は、第1発明〜第3発明及び第5発明〜第12発明のいずれかに係るリガンド親和性複合タンパク質の取得方法によって得られた抗体を用いて、ヒトの疾患の予防、診断又は治療を行う、医療方法である。
【0047】
(第17発明の構成)
上記課題を解決するための本願第17発明の構成は、第1発明〜第12発明のいずれかに係るリガンド親和性複合タンパク質の取得方法によって得られた抗体を有効成分とし、その由来に応じてヒト又は非ヒト動物に適用されるものである、抗体医薬である。
【0048】
(第18発明の構成)
上記課題を解決するための本願第18発明の構成は、第1発明〜第3発明及び第5発明〜第12発明のいずれかに係るリガンド親和性複合タンパク質の取得方法によって得られた抗体を有効成分とし、その由来するヒト個体に対して適用されるものである、非免疫反応性抗体医薬である。
【0049】
(第19発明の構成)
上記課題を解決するための本願第19発明の構成は、第18発明に係る非免疫反応性抗体医薬を、非経口的な投与方法によって、その由来する前記ヒト個体の体内へ投与する、投薬方法である。
【発明の効果】
【0050】
(第1発明の効果)
第1発明においては、生産手段分離工程において、ヒト又は非ヒト動物たる検体を利用して、複合タンパク質Xpのそれぞれ専用の生産手段Xmを備えた抗体産生細胞の1個又は複数個の単細胞を調製するので、極めて多種類の複合タンパク質Xpの生産手段を簡易かつ迅速に一括して調製できる。
【0051】
上記により一括調製した生産手段Xmたる各mRNAは、各細胞単位で個別に生成し分離されているので、生産手段Xmを複合タンパク質Xpの種類ごとに厳密に分離して絞り込むことができ、後工程における生産手段Xm、複製手段Xc及び複合タンパク質Xpの異種間混在を完全に予防できる。しかも、これらの生産手段Xmは各複合タンパク質Xpの専用の生産手段として機能するので、タンパク質生産手段ライブラリーとして極めて好適である。更に、前記したハイブリドーマ技術のような、安定した生産手段の調製(ハイブリドーマ作成)上の困難がない。
【0052】
しかし、上記のタンパク質生産手段ライブラリーにおいては、各mRNAが極めて微量であるために生産規模が微少であって、大量生産の要求に応えることができない。そこで次の生産手段転換工程において、直接に生産手段としては機能しないが容易に大量複製することができる複製手段Xc(cDNA)へ一旦転換させるのである。そして、逆転写PCR法を利用する生産手段転換工程では、生産手段Xmを構成するmRNAの全部を転換させても良いが、mRNAの一部を選択的に転換させることにより、前記した生産手段分離工程において絞り込まれたタンパク質の種類ごとのmRNA(mRNAセット)の内から、更にmRNAの種別の絞り込みを行うことができる。
【0053】
こうして絞り込まれた種類のmRNAについて得た複製手段Xc(cDNA)を大量生産準備工程において大量複製する。この工程はPCR法を利用するので、前記したハイブリドーマ技術や非特許文献1に記載の技術のように、大量生産規模を整える(ハイブリドーマや形質転換体を培養・増殖する)上で面倒な作業と長時間を要すると言う困難がない。しかも前記の生産手段転換工程でmRNAの種別の絞り込みを行う場合には、使用するプライマーの種類を著しく低減させることができるため、副産物が非常に少なく、特異性の高いPCR産物を得ることができる。
【0054】
こうして大量複製された複製手段Xc(cDNA)に対して、転写用配列付加工程において、その無細胞系での生産手段Xm(mRNA)への再転換に必要な転写用配列を付加する。
【0055】
次に単一種大量生産工程において、上記のように転写用配列を付加された複製手段Xc(cDNA)を無細胞系へ投入することにより、cDNAに対応する複合タンパク質Xpの種類ごとに、タンパク質の大量生産を行うことができる。
【0056】
前記のように、生産手段分離工程において生産手段Xmが複合タンパク質Xpの種類ごとに厳密に分離され、必要に応じて生産手段転換工程ではmRNAの種別の絞り込みが行われる。従って、転写用配列を有効に機能し得る状態で付加することができると共に、大量生産準備工程では特異性の高いPCR産物が得られる。
【0057】
そのため、単一種大量生産工程においては、目的とする単一種の複合タンパク質に対して分子量、立体構造、リガンド親和性等の面で類似する複合タンパク質の副産物は殆ど生産されず、一方では目的とする単一種の複合タンパク質が有効に大量生産される。その結果、工程終了後の無細胞系からの目的タンパク質Xpの高純度な分離・回収を容易かつ迅速に行うことができる。
【0058】
従って、キャラクタリゼーション工程を行う場合においても、特性評価、スクリーニング、配列決定を迅速かつ正確に行うことが可能である。
【0059】
以上のように、本願第1発明によれば、実質的に無限とも言える多様性の制約の中で、互いに類似した極めて多種類のリガンド親和性タンパク質の中から特定された1種又は2種以上のタンパク質を迅速・大量にクローニングして高純度で取得することができる。こうして取得したリガンド親和性複合タンパク質においては、H鎖とL鎖が正しくペアリングしていることが確認されている。
【0060】
第1発明のリガンド親和性複合タンパク質の取得方法を極めて迅速に行い得る点に関しては、例えば次のような評価が可能である。即ち、このような極めて多種類のリガンド親和性複合タンパク質の中から機能的に特に優れた1種又は2種以上の複合タンパク質を取得する場合(例えばMabを取得する場合)、通常は3ケ月又はそれ以上の長期間を要する、と言われている。これに対して第1発明では、約5時間の逆転写PCRその他の工程と、約1時間の単一種大量生産工程(即ち、in vitro転写/翻訳)とを含め、クローニングから抗体遺伝子のキャラクタリゼーションまでを実質的に1日で完了することが可能である。
【0061】
(第2発明の効果)
ヒトを検体として第1発明の生産手段分離工程を行う場合、一定の病理症状からリガンドXに対する抗体を生成している患者(被検者)に協力を求め、なるべく非侵襲的な手段で抗体産生細胞たる前記単細胞を分離することができる。この場合、得られるリガンド親和性タンパク質はモノクローナル抗体であり、しかも当該被検者にとっては自身に由来する非免疫反応性抗体であるから、その患者の体内に投与しても免疫上の拒絶反応を起こさない。従って、患者に対する最適のオーダーメード医療として抗体投与治療を行うことができる。
【0062】
従来の技術に基づいても、このような抗体投与治療が可能ではあった。しかし、患者の容体等の問題から極めて迅速なワクチンの提供を要求される場合が多く、従来の技術では常に迅速性がネックとなって来た。第2発明によれば、このような迅速性の問題を解消できるので、医療上の価値は極めて大きい。
【0063】
(第3発明の効果)
ヒトを検体とする場合、なるべく非侵襲的で被検者に負担を与えない手段により抗体産生細胞を採取するため、好ましい一例として、被検者の末梢血を少量採取し、その末梢血からB細胞及び/又は形質細胞の単細胞を分離する方法を挙げることができる。
【0064】
(第4発明の効果)
非ヒト動物を検体として第1発明の生産手段分離工程を行う場合、その動物をリガンドXで免疫したもとで、その適宜な組織又は生体構成部分から抗体産生細胞の単細胞を分離すると言う方法が一般的であり、かつ便宜である。
【0065】
(第5発明の効果)
前記した第1発明におけるリガンド親和性複合タンパク質の好適な例として、各1対のH鎖及びL鎖からなる免疫グロブリン(抗体)、そのFab(抗原結合性フラグメント)、又はこれらに準ずる有効なリガンド親和性を示すタンパク質フラグメントを挙げることができる。
【0066】
(第6発明の効果)
生産手段分離工程においては、第6発明のように一定の分離用抗体と結合させた磁性微粒子を利用し、その分離用抗体と特異的に結合する抗体産生細胞を細胞懸濁液から分離することにより、抗体産生細胞を選択的かつ効率的に回収できると言うメリットがある。
【0067】
(第7発明の効果)
第1発明で述べたように、生産手段転換工程において、生産手段Xmを構成するmRNAの一部を複製手段Xcへ転換させることができる。
【0068】
即ち、生産手段転換工程における生産手段Xmは実際には複合タンパク質Xpの各フラグメントをコードしているmRNAの1セットである。そして、例えば抗体におけるFab(抗原結合性断片)のように、リガンド親和性複合タンパク質の本来の構成フラグメントの一部を欠くが、リガンド親和性は維持しているようなタンパク質があり得る。しかも、例えば抗体の場合には複合タンパク質Xpの各フラグメントにおいて、各クラスや各アイソタイプ別の相違がある。
【0069】
そこで、mRNAの一部を複製手段Xcへ転換させる場合の一つの有利な実施形態として、逆転写PCRに用いるプライマーの設計により、特定のクラスやアイソタイプの分類カテゴリーに属する複合タンパク質XpをコードするmRNAのみを複製手段Xcへ転換させると言う実施形態が例示される。あるいはFabのように、複合タンパク質Xpを構成する特定のフラグメントをコードするmRNAのみを複製手段Xcへ転換させると言う実施形態が例示される。
【0070】
第7発明によって、逆転写PCRに用いるプライマーの種類を著しく低減させることができ、従って生成されるcDNAの種類も著しく低減させることができるので、その後のPCR法による大量生産準備工程を、特異性の高い有利な条件で行うことができる。
【0071】
(第8発明の効果)
第8発明も、技術的な意味合いの大きな改良点の一つである。即ち、一般論として、PCRにおいては基本的にプライマーダイマーや非特異的アニーリング等による副産物の生成は避けられないため、1段階のPCRによって複製手段XcたるcDNAを増幅することは、実質的に困難である。
【0072】
例えば、一旦生成した副産物たるDNAは、標的cDNAよりも鎖長が短い場合には標的cDNAよりも増幅し易い、と言う問題がある。しかも極めて微量な鋳型DNAから出発した場合には、このような負の効果が顕著である。
【0073】
この困難に対する通常の対処策として、入れ子構造になった2組のPCRプライマーセットを用いる、ネスティッドPCRと呼ばれる公知の2段階PCR法がある。しかし、この方法では、増幅しようとするcDNAの種類ごとに各2組のPCRプライマーセットを用いる必要があるため、抗体遺伝子のように多様性のある場合に応用するのは容易ではない。
【0074】
例えばヒトのB細胞の抗体遺伝子におけるL鎖の可変領域を増幅させる場合、10本の縮重が見られるプライマーを1段階用い、更に2段階目でもそれと同数のプライマーを用いなければならない。即ち、プライマーの数が多いだけでなく、多くの組み合わせに由来する副産物の生成が避けられないため、目的のDNA以外にも多数の増幅バンドが検出され、その後のタンパク質生産工程等に大きな困難をもたらす。即ち、例えば無細胞系における目的タンパク質の合成量が顕著に低下するし、あるいはタンパク質の配列決定のための大腸菌等へのクローニングが、著しく手間と時間のかかるものとなる。
【0075】
第8発明によれば、第1段階PCRでは複製手段Xcを構成する各cDNAに対応する各種類のプライマーを用いるものの、それらのプライマーに設けた共通の付加配列を利用して、第2段階PCRでは単一種類のプライマーによって全ての複製手段Xcを構成する各cDNAのみを一括して増幅することができる。従って、複製手段Xcが抗体遺伝子のように多様性のある場合にも、容易に応用することができる。
【0076】
第2段階PCRのように単一種類のプライマーを用いた場合でも、一定量のプライマーダイマーは生成する。しかし、この場合のプライマーダイマーは、異種プライマーの会合によるダイマーとは異なり、熱で解離させて冷却すると、プライマーの5’末端と3’末端が会合する構造(パンハンドル構造)を取って、新たに会合できなくなる。そのため、増幅能力が実質的に失われる。
【0077】
従って、第8発明によれば、無細胞系における目的タンパク質の合成量の低下を有効に防止でき、あるいはタンパク質の配列決定のための大腸菌等へのクローニングが著しく簡易化される。
【0078】
(第9発明の効果)
上記した第8発明の第1段階PCRにおいて各プライマーに付加配列を設ける点を利用し、その付加配列中に適宜な(第1発明の単一種大量生産工程で用いる無細胞系の由来生物を考慮した)翻訳開始促進配列部分を含めておくことができる。その場合、後の無細胞系での単一種大量生産工程において目的タンパク質の翻訳効率を向上させることができる。
【0079】
(第10発明の効果)
転写用配列付加工程において、大量生産準備工程で増幅されたDNAフラグメントに対して、PCRにより少なくともプロモーター配列を付加しておくことができる。そのことにより、無細胞系におけるDNAからmRNAへの再転換を準備できる。
【0080】
通常、抗体遺伝子のように多様性のある鋳型遺伝子に対して、PCRでこのような各種配列を付加しても、それらの付加配列の効果を十分に発揮させることは困難である。しかし、前記した第1発明、第7発明、第8発明等により、このようなPCRに供する鋳型遺伝子が十分に絞り込まれているため、これらの付加配列の効果が十分に発揮される。
【0081】
(第11発明の効果)
従来、無細胞系(in vitro転写/翻訳共役系)は還元化した状態下で利用する必要があるとの見解があり、そのためDTT(ジチオスレイトール)等の高価な還元化剤を添加することが多かった。しかし、本願発明者は、酸化条件下の in
vitro 転写/翻訳共役系においても遜色なく目的タンパク質を生産できることを既に確認している。従って、無細胞系を還元化しないままの条件で用いることにより、コストダウンを実現することができる。
【0082】
(第12発明の効果)
前記した第1発明におけるキャラクタリゼーション工程の内容は必ずしも限定されない。例えば、複合タンパク質XpのELISA法によるリガンド親和性評価、複合タンパク質Xp又はその遺伝子の配列決定、あるいは複合タンパク質XpのKd値の測定等を好ましく行うことができる。Kd値を測定する場合には、複合タンパク質Xpの遺伝子を導入した形質転換体を用いて複合タンパク質Xpを生産させ、この複合タンパク質Xpについて測定することが好ましい。
【0083】
そして、単一種大量生産工程では、複合タンパク質Xpが非常に副産物の少ない状態で大量に取得されているため、これらの評価や配列決定、あるいはKd値の測定を迅速かつ正確に行うことができる。
【0084】
(第13発明の効果)
リガンド親和性複合タンパク質の取得方法は各種の実験目的、研究目的、医療目的等に利用することができるが、重要な具体的用途の一つとして、ヒト又は非ヒト動物におけるリガンドXに対する抗体のスクリーニングに用いることが例示される。
【0085】
(第14発明の効果)
上記の第13発明に係る抗体スクリーニング方法を行うに当たり、第14発明に係る抗体スクリーニング用キットを好ましく利用することができる。この抗体スクリーニング用キットの各構成要素は、一部のプライマーの構成を除き、個々には特段に目新しいものではない。しかし、第14発明における11)〜14)の各構成要素を必須のコンポーネントとするキットの全体は、上記のリガンド親和性複合タンパク質の取得方法が着想されない限り成り立ち得ない内容であり、今までに、提案されたことも、市場に提供されたこともない。
【0086】
(第15発明及び第16発明の効果)
ヒトの医療あるいは非ヒト動物(例えば、ペット動物や家畜)の医療において、Mabを用いる点で第15発明又は第16発明の医療方法と同様の医療方法は、例えばハイブリドーマ技術等の従来技術に基づいても成立可能である。しかしながら、従来技術に基づく場合は、極めて多種類のMabの内から最適のMabを迅速・大量にスクリーニング及び製造することが困難である。
【0087】
第15発明及び第16発明に係る医療方法によれば、感染症における抗原や各種のガンにおけるガン細胞等に対して最適のMabを、迅速・大量にスクリーニング及び製造することができる。従って、医療現場における実用性がある。この点は、特にヒトの医療現場における一刻を争う救命医療等において、極めて重要な意義を持つ。
【0088】
(第17発明の効果)
第17発明に係る抗体医薬と機能的に同等な抗体医薬は、従来技術に基づいても供給可能である。しかし、従来技術に基づいて供給される抗体医薬は、迅速に供給することが困難であり、又、製造コストの面から安価に供給することも困難である。第17発明に係る抗体医薬は、迅速・大量にかつ安価に供給することができる。
【0089】
(第18発明及び第19発明の効果)
第18発明に係る非免疫反応性抗体医薬は、上記した第17発明の効果に加え、その由来するヒト個体に非経口的に投与しても、副作用としての免疫上の拒絶反応を誘発しない。従って、第19発明のような非経口的な投薬方法を安全に行うことが可能となる。更にその投薬の際、免疫抑制剤の併用を不要とすることができる。従って、患者の免疫能力低下に起因する治療上のリスクや患者の負担あるいは苦痛を軽減でき、更に、免疫抑制剤の節約により治療コストも低減させることができる。
【発明を実施するための最良の形態】
【0090】
次に、本願の第1発明〜第19発明の実施形態を、その最良の形態を含めて説明する。以下において単に「本発明」と言うときは、上記の各発明の全部又は一部を一括して指している。
【0091】
〔リガンド親和性複合タンパク質の取得方法〕
本発明に係るリガンド親和性複合タンパク質の取得方法は、任意に特定されたリガンドXに対して選択的な親和性を示す極めて多種類の複合タンパク質Xpの中から、無作為にあるいは特性評価を通じて選ばれる1種又は2種以上の複合タンパク質を取得する方法である。ここにおいて、「2種以上」とは、数種類であり得るし、数十種類であり得るし、更に数百種類又はそれ以上であり得る。
【0092】
複合タンパク質Xpの種類は限定されないが、例えば、各1対のH鎖及びL鎖からなる免疫グロブリン(抗体)であり得るし、その特定部分のフラグメントたるFabであり得るし、又は免疫グロブリンやFabに準ずるリガンド親和性を示す任意のタンパク質フラグメントであり得る。一方、リガンドXの種類は限定されず、抗原たり得る各種の無機化合物や有機化合物、タンパク質、ウイルス、微生物等を制限なく含む。
【0093】
リガンド親和性複合タンパク質の取得方法は、少なくとも後述する1)生産手段分離工程、2)生産手段転換工程、3)大量生産準備工程、4)転写用配列付加工程及び5)単一種大量生産工程を含む。更に必要に応じて、後述する6)キャラクタリゼーション工程を含むことができる。更に、上記1)工程に対する任意の前工程を含むことができ、上記6)工程に対する任意の後工程を含むことができ、又は1)〜6)工程中の任意の工程間に任意の中間工程を介在させることもできる。
【0094】
〔生産手段分離工程〕
生産手段分離工程とは、極めて多種類にわたる前記複合タンパク質Xpのそれぞれ専用の生産手段(mRNA)であって、容易に大量複製できる複製手段Xc(cDNA)への転換が可能な生産手段Xmを備えた抗体産生細胞の1個又は複数個の単細胞を、ヒト又は非ヒト動物たる検体の適宜な組織又は生体構成部分から個別に分離することにより、目的タンパク質の種類ごとに生産手段Xmを厳密に分離して、以下の各工程における生産手段Xm、複製手段Xc及び複合タンパク質Xpの異種間混在を予め防止する工程である。この工程により生産手段Xmが複合タンパク質Xpの種類ごとに厳密に分離される。
【0095】
ここにおいて、生産手段Xmとは、具体的にはmRNAであり、より具体的には、複合タンパク質Xpを構成する各タンパク質フラグメントをコードする各mRNAの1セットである。
【0096】
この生産手段分離工程において、検体がヒトである場合、その体内に人為的にリガンドXに対する抗体を生成させることは許されない。従って一定の疾患状態にある患者、即ち抗原たるリガンドXに対する抗体を既に体内に生成している生理状態にあると推定される被検者を選択し、その被検者の承諾のもとに生産手段分離工程を行う必要がある。具体的には、当該被検者における適宜な組織又は生体構成部分を社会通念上許容される非侵襲的手段によって採取し、この組織又は生体構成部分から前記の1個又は複数個の単細胞を分離する。
【0097】
上記のような患者の種類は必ずしも限定されないが、例えばリガンドXに感染した感染症患者、リガンドとしてのガン細胞に対する抗体を生成している各種のガン患者等を例示することができる。患者からは、医療倫理上及び関連法規上から採取することを許される組織又は生体構成部分に限り、しかも社会通念上許容される非侵襲的な手段によってのみ、抗体産生細胞を含む組織又は生体構成部分を採取することができる。「組織又は生体構成部分」には、血液その他の体液も含まれる。
【0098】
このような組織又は生体構成部分の採取と、そこからの抗体産生細胞の分離の具体的な一事例として、被検者から末梢血を採取し、この末梢血から抗体産生細胞たるB細胞及び/又は形質細胞の単細胞を分離する場合が挙げられる。
【0099】
又、他の具体的な一事例として、別途の治療上の目的から抗体産生細胞を含む組織又は生体構成部分の摘出手術や分離処置等を受けた患者において、その者の了解を得て、当該組織又は生体構成部分(例えば、脾臓、体液、骨髄等)を利用する場合が挙げられる。
【0100】
この生産手段分離工程において検体が非ヒト動物である場合、この動物をリガンドXで免疫したもとで、当該動物の適宜な組織又は生体構成部分から抗体産生細胞である1個又は複数個の単細胞を分離することができる。なお、既にリガンドXに対する抗体を生成している非ヒト動物においては、リガンドXで人為的に免疫する必要はない。
【0101】
非ヒト動物の種類は、少なくとも免疫反応による抗体の生成が認められる非ヒト動物である限りにおいて、限定されない。具体的には、入手及び利用が容易な各種の実験動物や、抗体医療の必要性が考えられる家畜又はペット動物等を好ましく例示することができる。より具体的には、例えばマウス、ラット、ウサギ、サル、牛、馬、豚、犬、猫等の哺乳動物が考えられるが、哺乳動物以外の非ヒト動物も対象となり得る。
【0102】
非ヒト動物を対象とする生産手段分離工程においては、動物愛護関連の法規又は倫理を遵守すると言う条件のもとで、抗体産生細胞、これを含む組織又は生体構成部分の種類、及びその採取方法は限定されない。抗体産生細胞と、これを含む組織又は生体構成部分としては、検体がヒトである場合においては、抗体産生細胞としてのB細胞、形質細胞を含む骨髄組織等を好ましく例示することができる。
【0103】
生産手段分離工程において、採取した組織又は生体構成部分から抗体産生細胞の1個又は複数個の単細胞を分離して取得する方法は限定されない。好ましくは、これらの組織又は生体構成部分を液体(好ましくは、適当な組成の液体培地)中に投入して抗体産生細胞を分散・懸濁させ、一定の分離用抗体と結合させた磁性微粒子と共にインキュベートした後、分離用抗体を介して抗体産生細胞に結合した磁性微粒子をマグネットで回収することにより、前記懸濁液から目的とする抗体産生細胞の単細胞を分離する、と言う方法がある。
【0104】
上記の分離用抗体としては、例えばマウスの場合における抗マウス CD19 抗体等、任意の抗体を利用することができる。
【0105】
〔生産手段転換工程〕
生産手段転換工程とは、単離したそれぞれの抗体産生細胞に含まれる生産手段Xm、即ち、複合タンパク質Xpの各タンパク質フラグメントをコードするmRNAのセットを細胞外へ溶出させ、逆転写PCR法により、これらのmRNAの全部又は一部を複製手段Xc(cDNA)へ転換させる工程を言う。
【0106】
ここにおいて、mRNAの一部をcDNAへ転換させる場合の一形態として、特定の分類カテゴリーに属する複合タンパク質XpをコードするmRNAのみをcDNAへ転換させる形態が挙げられる。例えば抗体である複合タンパク質Xpの内、特定のクラスやアイソタイプに属するタンパク質をコードするmRNAのみをcDNAへ転換させる場合が例示される。
【0107】
mRNAの一部をcDNAへ転換させる場合の他の一形態として、複合タンパク質Xpを構成する特定のフラグメント(例えばFab)をコードするmRNAのみをcDNAへ転換させる形態が挙げられる。
【0108】
生産手段Xmを構成するmRNAのセットの全部をcDNAへ転換させたり、上記のような各種の形態でその一部をcDNAへ転換させたりする逆転写PCRは、使用するプライマーの選択により、容易に選択して行うことができる。
【0109】
なお、抗体産生細胞に含まれる生産手段Xmを細胞外へ溶出させる手段は限定されないが、公知の適当な手段、例えばB細胞を界面活性剤や低浸透圧の液等で破壊する方法等を任意に採用することができる。
【0110】
〔大量生産準備工程〕
大量生産準備工程とは、上記した逆転写PCR法により得られた複製手段Xc(cDNA)をPCR法で大量複製して、タンパク質の大量生産能力を準備する工程を言う。
【0111】
この工程におけるPCR法の内容、実施条件あるいは使用するプライマーの種類は特段に限定されないが、2段階のPCRによって行うことが好ましく、特に次の内容の2段階PCRによって行うことが、前記「第8発明の作用・効果」欄で記載した理由から、好ましい。
【0112】
(第1段階PCR)
複製手段Xcを構成する全ての種類のcDNAに対応する各種類のプライマーにおいて、それらの5’末端側に同一の付加配列を設けて増幅を行う。更に、この付加配列中には、次の単一種大量生産工程において有利に働く任意の塩基配列、例えば後述する無細胞系の由来生物を考慮した翻訳開始促進配列配列を含めておくことができる。
【0113】
翻訳開始促進配列配列としては、例えばrbs(リボソーム結合サイト)や、無細胞系の由来生物が大腸菌である場合のSD(シャイン・ダルガーノ)配列、無細胞系の由来生物が真核生物である場合のKozak配列、ウイルスに由来する5’UTR配列等が挙げられる。
【0114】
この第1段階PCRの実施条件は限定されないが、例えば以下のプログラムで行うことが好ましい。
94°C−3分;
94°C−30秒/50°C−45秒/72°C−45秒を30サイクル;
72°C−7分;
(第2段階PCR)
上記の第1段階PCRで増幅された全ての種類のDNAフラグメントに対して、前記付加配列に相補的な配列部分を備えた単一種類のプライマーを用いて増幅を行う。
【0115】
この第2段階PCRの実施条件は限定されないが、例えば以下のプログラムで行うことが好ましい。
94°C−3分;
96°C−5秒/45°C−10秒/72°C−45秒を65サイクル;
72°C−7分
〔転写用配列付加工程〕
転写用配列付加工程とは、大量複製された前記複製手段Xc(cDNA)に対して、その無細胞系での生産手段Xm(mRNA)への再転換に必要な転写用配列を付加する工程である。転写用配列としては、限定はされないが、少なくともプロモーター配列を含むことが好ましい。プロモーター配列に加えてターミネーター配列も含むことが、更に好ましい。
【0116】
この工程を行う方法は必ずしも限定されないが、好ましくは、大量生産準備工程で増幅されたDNAフラグメントに対し、PCR、例えばオーバーラッピングPCRによって、前記の転写用配列を付加することにより行われる。
【0117】
〔単一種大量生産工程〕
単一種大量生産工程とは、上記の工程により転写用配列を付加された複製手段Xc(DNA)を、少なくとも複製手段Xcの生産手段Xm(mRNA)への再転換を行う要素と、生産手段Xmに基づきタンパク質の生産を行う要素とを備えた無細胞系へ投入することにより、複合タンパク質Xpを、又は前記生産手段転換工程で規定した一部のmRNAに対応する複合タンパク質Xpを、1種類ごとに大量生産する工程を言う。
【0118】
この無細胞系において、DNAをmRNAへ転換させる要素としては転写酵素が挙げられる。転写酵素としては、T7RNAポリメラーゼ、T3RNAポリメラーゼ、 SP6RNAポリメラーゼ、大腸菌等の微生物由来のRNAポリメラーゼ等の利用が可能である。mRNAに基づきタンパク質の生産を行う要素としてはリボソーム、各種のdNTP等が例示される。このような無細胞系は in vitro 転写/翻訳共役系として公知であって、例えば大腸菌由来、小麦胚芽由来、ウサギ網状赤血球由来等のキットが市販されているので、わざわざ調製しなくても、適宜にこれらを利用することができる。
【0119】
in vitro 転写/翻訳共役系としては、高価なDTT等を添加して還元状態としたものを使用することもできるが、これを添加しない酸化状態の in vitro 転写/翻訳共役系も遜色のない機能を発揮するので、これを使用する方が、安価で有利である。
【0120】
〔キャラクタリゼーション工程〕
キャラクタリゼーション工程とは、上記の各工程により生産された複合タンパク質Xpについて、特性評価を行ったり、優れた特性を持つ複合タンパク質Xpをスクリーニングしたり、あるいは、これらの複合タンパク質Xpの配列決定を行ったりする工程を言う。
【0121】
この工程は、単なるリガンド親和性複合タンパク質の取得方法としての観点から見れば、本来的に不可欠の工程ではない。しかし、リガンド親和性複合タンパク質の取得方法が後述の抗体のスクリーニング、抗体医療、抗体医薬等の発明の前提であると言う観点からは、不可欠の工程あるいは重要な工程である。
【0122】
キャラクタリゼーション工程においては、好ましくは、複合タンパク質XpのELISA法によるリガンド親和性評価と、複合タンパク質Xp又はその遺伝子の配列決定と、形質転換体を用いて生産させた複合タンパク質XpのKd値の測定との内の少なくとも1項目を含む。
【0123】
〔抗体スクリーニング方法〕
本発明に係る抗体スクリーニング方法とは、上記したリガンド親和性複合タンパク質の取得方法を、ヒト又は非ヒト動物におけるリガンドXに対する抗体のスクリーニングに用いる方法である。
【0124】
即ち、リガンド親和性複合タンパク質の取得方法における前記1)の生産手段分離工程〜前記5)の単一種大量生産工程によって得られた2種以上のリガンド親和性複合タンパク質の中から、前記6)のキャラクタリゼーション工程を通じて、特に優れた抗リガンドX抗体をスクリーニングする方法である。
【0125】
従って、従来の各種の抗体スクリーニング方法に比較して、最適の抗リガンドX抗体を容易、迅速かつ大量に得ることができる。
【0126】
〔抗体スクリーニング用キット〕
本発明に係る抗体スクリーニング用キットは、上記の抗体スクリーニング方法を実施するために用いられるキットであって、少なくとも以下の構成要素を含んで構成されるものである。
11)前記第6発明に係る分散・懸濁用の液体と、磁性微粒子とのセット。
12)前記第7発明に係る逆転写PCR用のプライマーセット。
13)前記第8発明に係る2段階のPCR用のプライマーセット。
14)前記第1発明の5)に規定する無細胞系。
【0127】
上記の11)〜14)の各構成要素は、この種の商品として通常に採用される剤型あるいは包装形態において提供することができる。例えば12)又は13)のプライマーセットは、保存性や使用上の便宜等を考慮して、乾燥品等の形態で提供することができる。
【0128】
又、抗体スクリーニング用キットにおいては、上記の11)〜14)の各構成要素の他に、例えば転写用配列付加工程においてプロモーター配列やターミネーター配列の付加のために利用するベクター、任意の目的で使用する緩衝液やpH調整液等の任意の有益な付加的構成要素を含むことができる。
【0129】
〔医療方法〕
本発明に係る医療方法には、非ヒト動物を検体として得られたリガンド親和性複合タンパク質(抗体)を用いて非ヒト動物の疾患の予防、診断又は治療を行う医療方法と、ヒトを検体として得られたリガンド親和性複合タンパク質(抗体)を用いてヒトの疾患の予防、診断又は治療を行う医療方法とを含む。
【0130】
これらの医療方法において、非ヒト動物又はヒトの疾患の種類は限定されないが、リガンドXによる感染症、ガン細胞がリガンドXとなっている各種のガン等を代表的に例示することができる。
【0131】
〔抗体医薬〕
本発明に係る抗体医薬は、前記したリガンド親和性複合タンパク質の取得方法によって得られた抗体を有効成分とするものであって、その抗体の由来(検体とされたヒト又は非ヒト動物)に応じて、ヒト又は非ヒト動物に適用されるものである。この抗体医薬の投与方法は限定されないが、非経口的な方法によることが好ましい。
【0132】
抗体医薬の剤型は限定されず、例えば凍結乾燥剤、粉末製剤、pHを調整された緩衝液による溶液製剤、注射用マイクロカプセル剤等として提供できる。これらの抗体医薬は、この種の抗体医薬において含有されることがある任意の有益な成分(例えば、増量剤、pH調整剤、保存安定剤等)を含むことができる。
【0133】
〔非免疫反応性抗体医薬及び投薬方法〕
本発明に係る非免疫反応性抗体医薬は、ヒトを検体として得られたリガンド親和性複合タンパク質(抗体)を有効成分とし、その由来するヒト個体に対して投薬されるものである。この非免疫反応性抗体医薬の剤型や含有成分については、上記の抗体医薬の場合と同様の構成が可能である。
【0134】
非免疫反応性抗体医薬の投薬方法は非経口的な投与方法による。非経口的な投与方法の種類は限定されないが、例えば静脈注射、皮下注射、筋肉注射等を例示できる。他にも、近年注目されている各種のドラッグデリバリー方法、例えば、磁性微粒子に抗体分子を結合させて磁力により効果的にガン細胞周辺に集め、最終的に抗体によりターゲット細胞をとらえると言う方法等の投与方法も採用可能である。
【実施例】
【0135】
以下に本発明の実施例を説明する。本発明の技術的範囲がこれらの実施例によって限定されないことは、もち論である。
【0136】
〔マウスを検体とする実施例1〕
(実施例1の概要)
実施例1の概略を述べる。まず、抗原である CBP40(他の任意の抗原を用いても良い)で免疫したマウスの脾臓から、抗マウス CD19 抗体と結合した磁性マイクロビースを用いて、B細胞を単離した。単離したB細胞のL鎖(Lc)遺伝子とH鎖のFd部(VHドメインとCH1ドメイン)遺伝子とを、別々に逆転写PCR(RT−PCR)によって増幅した。
【0137】
次いで、 in vitro 合成に適したDNA断片を得るため、これら2種類の遺伝子に対して、オーバーラッピングPCRによって T7 プロモーター、リボソーム結合部位( rbs)及び T7 ターミネーターの各配列を付加した。このH鎖遺伝子とL鎖遺伝子の全長DNA断片の対を同じ反応用試験管に投入し、 in vitro で転写及び翻訳を行わせてFab断片を得た後,ELISA法によるスクリーニングを行った。このような一連のプロセスにより、種々の細胞と組織から抗原特異的なモノクローナル抗体を得ることができる。
【0138】
以上の全体的な実験操作(工程)のフローを図1に概念化して示す。
【0139】
(実施例1−1:B細胞の単離)
BALA/c 系マウスを50μgの CBP40(Calcium Binding Protein 40)で免疫した。5週間後にその脾臓を摘出し、F-10 HAM栄養培地(英国 Sigma-Aldrich社製)中で細胞単位に分散させた。次に、4°Cで1000 r.p.m. の遠心分離を3分間行って脾細胞を集め、 PBS(phosphate buffered saline )で一回洗浄した。
【0140】
脾細胞の懸濁液を、抗マウス CD19 抗体と結合した磁性粒子である CD19MACS
マイクロビーズ(Miltenyi Biotec 社)と共にインキュベートし、 CD19 を発現しているB細胞をマイクロビーズに吸着させた。次いでマグネットにより細胞を集めて上澄みを取り除いた。更に PBS(2.5% BSA)で洗浄し、次いでマグネットにより細胞を集め、マイクロビーズに結合したB細胞を回収した。
【0141】
単離したB細胞を顕微鏡(日本国オリンパス社製 LX70 )下でカウントし、希釈してPCRチューブ1本当たり1個の細胞を収容させ、直ちにcDNA合成に供した。
【0142】
(実施例1−2:単細胞逆転写PCR−cDNAの合成)
各細胞のmRNAを鋳型にして、逆転写用キットを用いてcDNA合成を行った。ここでは Invitrogen 社の逆転写PCR用 SUPERSCRIPT First Strand 合成システムを用いたが、他のメーカーのものでも構わない。
【0143】
まず PBSバッファーで平均2細胞/μLになるようにB細胞を調製し、1.5μLの DEPC 処理( RNase失活)滅菌水を加え、そこに、プロコールに従い全量5μLの反応液〔0.25μL dNTP,0.50μL 10×RTバッファー:200mM トリス−塩酸(pH8.4),500mM KCl,1.0μL 25mM MgCl2 ,0.5μL 0.1M DTT,0.25μLRnaseインヒビター, 0.25μL Reverse trriptase:SUPERSCRIPT II RT ,0.25μL 2μM 各プライマー〕になるように液を加え、42°Cで90分処理した後、70°Cで15分処理した。
【0144】
この合成反応は、目的とする全てのcDNA、即ちB細胞のL鎖遺伝子(κ鎖)とH鎖(γ鎖)のFd部(VHドメインとCH1ドメイン)遺伝子とに対応する全てのcDNAを得るために必要な下記1)〜5)の各プライマーをそれぞれ2μM含む混合液を用いて行った。又、コンタミネーションをチェックするため、B細胞が存在しない系でのコントロールの合成反応を別途に行った。
1)プライマー CK−J1(表1及び配列表の配列番号1に示す)
2)プライマー CH−IgG1-1(表1及び配列表の配列番号2に示す)
3)プライマー CH−IgG2A-1 (表1及び配列表の配列番号3に示す)
4)プライマー CH−IgG2B-1 (表1及び配列表の配列番号4に示す)
5)プライマー CH−IgG3-1(表1及び配列表の配列番号5に示す)
なお、上記1)〜5)の各プライマー及び後述の全てのプライマーの塩基配列を、表/図と配列表とにそれぞれ示すが、万が一、表/図に示す塩基配列と配列表に示す塩基配列とに不一致が存在した場合には、表/図に示す塩基配列が正しい配列である。
【0145】
【表1】

(実施例1−3:DNAの増幅−第1段階のPCR)
上記により得られたcDNAの混合物を第1段階のPCR反応のテンプレートとして使用した。その際、発現されるマウスIg遺伝子の95%はL鎖がκアイソタイプである(Honjo and Alt, 1995 )ため、λアイソタイプのL鎖は無視することにし、抗体L鎖のκアイソタイププライマーのみを用いた。又、高い抗原結合能を持つ抗体を得ると言う目的から、H鎖についてはIgG(イムノグロブリンG)のみを標的とした。第1段階のPCRで用いた下記の縮重プライマーは Kabatのデータベース( http://immuno.bme.nwu.edu)に基づいて設計した。
【0146】
このような設計によりプライマー数を低減させると、好ましくない増幅を減少させるのに有益であろうと言うことを考慮している。なお、これらの縮重プライマーは鋳型として十分に理想的とは言えないので、増幅反応液には、下記のように、3'エクソヌクレアーゼ活性を欠き、かつ他のDNAポリメラーゼに比較して縮重プライマーと鋳型とのミスマッチに対して大きな耐性をもつ TaqDNAポリメラーゼ(Wang et al., 2000 )を用いている。
【0147】
L鎖遺伝子は、いずれも縮重プライマーである6)プライマー VK− M2 (表1及び配列表の配列番号6に示す)、及び7)プライマー CK−His6M2(表1及び配列表の配列番号7に示す)を用いて増幅した。
【0148】
H鎖遺伝子は、縮重プライマーである8)プライマー VH−M2(表1及び配列表の配列番号8に示す)と9)プライマーV'H−M2(表1及び配列表の配列番号9に示す)との混合物と、定常領域プライマーである10)プライマー CH−IgG1-M2
(表1及び配列表の配列番号10に示す)、11)プライマー CH−IgG2A-M2(表1及び配列表の配列番号11に示す)、12)プライマー CH−IgG2B-M2(表1及び配列表の配列番号12に示す)及び13)プライマー CH−Ig G3-M2(表1及び配列表の配列番号13に示す)の混合物とを用いて増幅した。
【0149】
これらのL鎖遺伝子とH鎖遺伝子との増幅反応は、0.25単位の TaqDNAポリメラーゼ(日本国 宝酒造社製)、0.2mMの各dNTP、0.05μMの各プライマー、0.5μLの前記cDNA混合物及びリアクションバッファーからなる5μLの反応液中で行った。PCRは以下のプログラムで行った。
94°C−3分;
94°C−30秒/50°C−45秒/72°C−45秒を30サイクル;
72°C−7分;
(実施例1−4:DNAの増幅−第2段階のPCR)
第1段階のPCR産物の10分の1量を、単一種の14)プライマー SP2(表1及び配列表の配列番号14に示す)を用いた第2段階のPCRにおいて増幅した。
【0150】
この増幅反応は、0.25単位の Pfu Turbo(商標)DNAポリメラーゼ(Stratagene社製)、0.125mMの各dNTP、0.5μMのプライマー SP2及びリアクションバッファーからなる5μLの反応液中で行った。PCRは以下のプログラムで行った。
94°C−3分;
96°C−5秒/45°C−10秒/72°C−45秒を65サイクル;
72°C−7分
図2に、H鎖遺伝子(「Hc」で示す)及びκタイプL鎖遺伝子(「 Lc 」で示す)の増幅結果を示す。これらの結果は、PCR産物2μLを1.0%アガロースゲル上で解析したものである。 EcoT14Iで消化したλ−DNAマーカーを図の左側に示している。
【0151】
図2に示すように、これらの反応条件によって単一のB細胞に由来するFab断片のL鎖とH鎖をコードする遺伝子が別々に増幅され、しかも副産物の生成は認められない。L鎖PCR産物は、単一B細胞の72検体中、39検体から得られた(39/72=54%)。H鎖遺伝子に関しては、特異的なPCR産物は、単一B細胞の72検体中、28検体から得られた(28/72=39%)。全体として、L鎖とH鎖がペアリングしたFabは72検体中の20検体から得られ、回収率は28%であった。
【0152】
(実施例1−5:PCRの全体的な説明)
上記の全てのPCR反応は GeneAmp(登録商標)PCRシステム(PE Applied Biosystems 社製)を用いて行った。PCR産物のシークエンスを確認するため、幾つかのL鎖遺伝子とH鎖遺伝子を EcoRVで消化した pBluescriptSKII+ ベクター(Stratagene社製)にクローニングした。クローニング領域のシークエンシングにはThermo SequenaseII(商標)dye terminator cycle sequencing premix kit(Amersham Pharmacia Biotech社製)と、 ABIPRISM (商標)310 Genetic
Analyzerとを、メーカー推奨のプロトコールに従って用いた。
【0153】
(実施例1−6:オーバーラッピングPCR)
実施例6において、下記のように、 T7 プロモーター、 rbs及び T7 ターミネーターを含む2つの断片をオーバーラッピングPCRによりそれぞれ増幅されたL鎖遺伝子及びH鎖遺伝子と結合させた。オーバーラッピングPCR産物の解析結果を図3に示す。図3は、オーバーラッピングPCR産物2μLを1.0%アガロースゲル上で解析した結果であり、L鎖遺伝子がレーン1〜3に、H鎖遺伝子がレーン4〜6に、それぞれ示されている。
【0154】
T7 プロモーターおよびSD配列を増幅するためのプライマーセットとして、配列表の配列番号15に示す15)プライマーIn-F(5'-CCAATACGCAAACCGCCTCTCC-3')と、配列表の配列番号16に示す16)プライマー T7PR-SD(5'-ATAATCTCCTTCTTATCTAATAACAAAATTATTTCTAGAGGGAAACCG-3')とを用い、pRSET-B ベクター(Invitrogen社製)から T7 プロモーターとリボソーム結合部位を含む断片を増幅した。
【0155】
又、 T7 ターミネーターを増幅するためのプライマーセットとして、配列表の配列番号17に示す17)プライマーT7T-NFM2(5'-TAATCAATAATCTCCTTCTTATCTAATTCCGGCTGCTAACAAAGCCCG-3 )と、配列表の配列番号18に示す18)プライマーIn-R( 5'-TACAGGGCGCGTCCCATTCG-3' )とを用い、pRSET-B ベクターから転写ターミネーター断片を増幅した。
【0156】
オーバーラッピングPCRによって、上記2種類のDNA断片の間に第2ラウンドのPCRに係るL鎖あるいはH鎖を挿入し、in vitroタンパク質合成に適した全長DNA断片を生成させた。オーバーラッピングPCRは、0.75単位の Ex taq DNAポリメラーゼ(宝酒造社製)、0.2mMの各dNTP、それぞれ0.15μLの各DNA断片、及び反応バッファーからなる15μLの反応液中で次のPCRプログラムに従って行った。
94°C−5分;
94°C−30秒/48°C−20秒/72°C−90秒を10サイクル;
その後、15μLのPCR溶液を、メーカー推奨の反応バッファー( Ex taq
DNAポリメラーゼ1.5単位、dNTP各0.2mM、プライマー In-F 及び In-R 各0.5mM)を用いて30μLに調製し、以下のPCRプログラムにより増幅した。
94°C−5分;
94°C−30秒/55°C−30秒/72°C−90秒を25サイクル;
72°C−7分
(実施例1−7:in vitro転写/翻訳共役系)
上記の30μLの反応液(同一の細胞に由来するH鎖とL鎖遺伝子とのオーバーラッピングPCR混合物)のうち、3μLを用いて in vitro 転写/翻訳共役反応を行った。この反応は Jiangらの報告(Jiang et al., 2001)に従い、次の要素を含む in vitro 転写/翻訳系を用いた。
トリス酢酸(pH7.4) 56.4mM;
ATP 1.2mM;
GTP、CTP及びUTP 各1.0mM;
クレアチンリン酸 40mM;
全20種の非ラベルアミノ酸 各0.32mM;
ポリエチレングリコール6000 4%(W/V);
フォリン酸 34.6mg/mL;
大腸菌( E. coli)tRNAs 0.17mg/mL;
酢酸アンモニウム 36mM;
Mg(OAc)2 8mM;
KOAc 100mM;
リファンピシン 10mg/mL;
クレアチンキナーゼ 0.15mg/mL;
T7 RNAポリメラーゼ 7.7mg/mL;
大腸菌 S30抽出液 28.3%(V/V);
又、 in vitro 転写/翻訳共役反応は次のように行った。即ち、上記オーバーラッピングPCR産物の3μLずつを30°Cで60分間転写・翻訳反応させ、続いて氷の上で冷やして反応を停止させた。同時に、DNAのテンプレートを含まない点のみが異なる同様の系でコントロールの反応を行った。反応系には、オートラジオグラフィーのため、14Cでラベルしたロイシンを0.016mM添加した。
【0157】
(実施例1−8:SDS−PAGEとオートラジオグラフィー)
5μLのサンプルを、非酸化条件とするための同量のローディングバッファー(pH6.8の50mMトリス塩酸、10%グリセロール、2%SDS、0.1%ブロモフェノールブルー)と煮沸した。煮沸後のサンプルをSDS−12.5%−ポリアクリルアミドゲル上で電気泳動させた後、ゲルをワットマン3Mのろ紙にのせ、ゲルドライヤーで乾燥した。次いで、イメージングプレート(富士フィルム社)に感光させた後、BAS-2000にて解析した。その結果を図4に示すが、ゲル上にFab断片のバンド(46.0 kDa)が観察される。
【0158】
(実施例1−9:ELISA解析)
本実施例では、上記 in vitro 系で合成されたいずれのFab断片が抗原親和性を持つかについて、下記のように抗原に対するELISA解析を行った。
【0159】
96ウエルのプレート(デンマーク Nunc 社製)を、5μg/mLの CBP40で50μL/ウエルとなるようにコーティングした。これらのウエルを4°Cで一晩インキュベートした後、蒸留水に溶かした 1/4×Block Ace (大日本製薬社製)を用いて37°C、30分でブロックし、次いで洗浄液(1/10×Block Ace 、0.05% Tween20)で2回洗浄した。
【0160】
無細胞反応混合物30μLを PBS(pH7.4)で5倍に希釈した。この希釈液50μLを、コーティングしたプレートに加え、25°Cで2時間インキュベートした後、上記と同じ洗浄液で2回洗浄した。同時に、DNAのテンプレートを含まない反応液もコントロールとして同様に処理した。
【0161】
150μLの抗マウスIgG(5μg/蒸留水1mL)を各ウエルに加え、25°Cで1時間インキュベートした。そして2回洗浄した後、酵素免疫反応(ELISA)を行った。パーオキシダーゼ反応の基質として、o−フェニレンジアミンと過酸化水素を含む溶液を用いた。発色反応は2M硫酸を用いて停止させた。そして492nmの吸光度を測定した。なお、ELISAの発色はVectastain ABCキット( Vector Laboratories社製)を用いた。
【0162】
ELISA解析の結果を示す図5によれば、 No.9のクローンのELISAシグナルが他のクローンより著しく強く、合成Fab中、このクローンが免疫抗原を結合できることが強く示唆される。
【0163】
(実施例1−10:遺伝子の配列決定)
そこで、上記した No.9のクローンのL鎖及びH鎖(Fd部)の配列決定を行った。L鎖のアミノ酸配列及び遺伝子塩基配列を図6及び配列表の配列番号19に、H鎖(Fd部)のアミノ酸配列及び遺伝子塩基配列を図7及び配列表の配列番号20に、それぞれ示す。
【0164】
上記で決定した配列を、 National Center for Biotechnology Informationの IgBLASTデータベース(http://www.ncbi,nlm, nih.gov/igblast)によって解析した。その結果、L鎖の配列は、 ce9 germline 遺伝子(GenBank accession Nos. AJ239197 )と98%の同一性を持っていた。H鎖の配列は V130 germline遺伝子(GenBank accession Nos. M11700 )と91%の同一性を持っていた。これらの結果から、新規なマウスL鎖遺伝子及びH鎖遺伝子を得たことを確認し、これらの2種遺伝子から形成されたFab断片を「 JM9-Fab」と名付けた。
【0165】
(実施例1−11:宿主での発現、リフォールディング、Fab断片精製)
オーバーラッピングPCR産物中の陽性Fab断片のL鎖とH鎖を XbaI-NaeIで消化し、pRSET-B ベクター(Invitrogen社製)の XbaI-NaeIサイトに挿入した。こうして得た、L鎖を含むプラスミド( pRSETB-Lcと命名)及びH鎖を含むプラスミド( pRSETB-Hcと命名)を、それぞれL鎖とH鎖の発現に用いた。
【0166】
プラスミド pRSETB-Lcとプラスミド pRSETB-Hcとを別々に、コンピテント大腸菌 BL21(DE3)plysS 細胞(Stratagene社製)に導入した。Fab断片の大腸菌での発現とリフォールディングとは、実質的に Wibbenmeyerらの方法と同様に行った。
【0167】
即ち、各ペプチド断片を封入体として発現させた後、遠心分離で回収・洗浄して精製した。その後可溶化バッファー(6M 塩酸グアニジン、0.4M L−アルギニン、pH8.0の100M トリス塩酸、2.5mM EDTA)により可溶化し、100mM DTT(ジチオスレイトール)を添加した後、L鎖とK鎖を等モルになるよう混合した。そしてDTTを含まない可溶化バッファーにて50倍に希釈した後、4mMの酸化型グルタチオンを添加し、透析により徐々に塩酸グアニジン、EDTA等を取り除き、リフォールドさせた。
【0168】
Fab断片の精製は次のプロセスで行った。即ち、0.1M NiSO4 溶液1.0mLを HiTrap キレートカラム(1mL)(Pharmacia Biotech 社製)に流し、10mLの純水で洗浄した後、結合バッファー(PBSに0.5M NaCl、5mMイミダゾールを添加したもの)でカラムを平衡化したものに、上記のFab溶液を流し、結合させた。合計20mLの結合バッファーで洗浄した後、溶出バッファー(PBSに0.5MNaCl、500mM イミダゾールを添加したもの)で溶出させた。
【0169】
(実施例1−12:発現されたFab断片の Kd 値)
JM9-Fab の親和性評価のため、L鎖遺伝子とH鎖遺伝子のシークエンスをそれぞれ pRSET-Bベクターにクローニングして、 pRSETB-JM9Lc と pRSETB-JM9Hc とを得た。これらのクローニングベクター pRSETB-JM9Lc と、 pRSETB-JM9Hc を、別々にコンピテント大腸菌 E. coli BL21(DE3)plysS 細胞(Stratagene社製)に導入した。そして JM9-Fabを発現させ、リフォールディングさせ、精製した。
【0170】
Stevevs の方法による JM9-FabのKd値は1.7(±0.3)×10−8であった。この結果は、このFab断片が従来のハイブリドーマ法による抗体と同等の高い抗原結合活性を有することを示している。
【0171】
〔ヒトを検体とする実施例2〕
この実施例2では、健常成人女性を検体とし、その人の承諾を得て、その末梢血2mLを採血させてもらい、この末梢血から分離した抗体産生細胞(B細胞)を用いて、実施例1と同様の手順に従う各工程を行った。
【0172】
実施例2の工程及び内容中、実施例1と異なる点は、以下の(a)〜(d)である。従って、以下において、まず実施例2の概要を図8に基づいて説明すると共に、続いて(a)〜(d)について順次説明する。これらの点以外は実施例1と同様に行っている。
(a)B細胞の単離工程
(b)モノクローナル抗体の構成
(c)各工程のPCRで使用したプライマー
(d)ELISA解析、配列決定、抗原に対する結合活性
(実施例2の概要)
図8に概念化して示すように、まず、末梢血からB細胞を単離した。単離したB細胞のL鎖(Lc)遺伝子とH鎖(Hc)遺伝子とを、別々に逆転写PCR(RT−PCR)によって増幅した。次いで in vitro 合成に適したDNA断片を得るため、これらの遺伝子に対してオーバーラッピングPCRによって T7 プロモーター、リボソーム結合部位( rbs)及び T7 ターミネーターの配列を付加した。このH鎖遺伝子とL鎖遺伝子の全長DNA断片の対を同じ反応用試験管に投入し、in vitroで転写及び翻訳を行わせてMabを得た後、ELISA法によるスクリーニングを行った。
【0173】
(B細胞の単離工程)
健常成人女性を検体とし、その承諾を得て、その末梢血2mLを採血させてもらった。採血は、血液凝固を防ぐため、ヘパリン存在下で行った。次に Ficllo-Plaque Plus (Amersham Pharmacia Biotech Inc. )を用いた遠心密度勾配(400×g、40分、18°C)により末梢血単球を分離した。
【0174】
回収した細胞をPBS緩衝液で2度洗浄し、ナイロン(商標)ネット(41μmメッシュ)により凝固体を取り除き、CD19+ 細胞をMACS磁性ビーズ(Miltenyi Biotech社, ドイツ)により単離した。磁性ビーズを取り除いた後、抗ヒトCD19抗体PEコンジュゲート(Santa Cruz Biotechnology, Inc., 米国)により染色した。PBSにより2度洗浄した後、分取用フローサイトメトリー(EPICA ELITE, Beckman Coulter社, 日本)により、蛍光標識された細胞だけを分取した。
【0175】
単離したB細胞を顕微鏡(日本国オリンパス社製 LX70 )下でカウントし、希釈してPCRチューブ1本当たり1個の細胞を収容させ、直ちに次工程たるcDNA合成に供した。
【0176】
(モノクローナル抗体の構成)
実施例1では、単離したマウスB細胞のL鎖(Lc)遺伝子とH鎖のFd部遺伝子とを別々に逆転写PCRによって増幅したが、実施例2においては、単離したヒトB細胞のH鎖遺伝子(Hc)、κタイプL鎖遺伝子( Lc(kappa))及びλタイプL鎖遺伝子( Lc(lamda))を別々に逆転写PCRによって増幅し、最終的に、これらのフラグメントからなるMabを取得した。
【0177】
実施例1の場合と同様に行った第2段階のPCRによるこれらの遺伝子の増幅結果を図9に示す。図9中の「Hc」、「 Lc(kappa)」、「 Lc(lamda)」の各図において、 EcoT14Iで消化したλ−DNAマーカーを図の左側に示している。
【0178】
図9に示すように、単一のB細胞に由来するFab断片のH鎖遺伝子、κタイプL鎖遺伝子及びλタイプL鎖遺伝子が別々に増幅され、しかも副産物の生成は認められない。
【0179】
(各工程のPCRで使用したプライマー)
実施例2においては、各工程のPCRで使用したプライマーが実施例1の場合と異なるので、その一覧を表2に示すと共に、それらの塩基配列を順次配列表にも記載した。表2に示した各プライマーについて、以下に簡単に述べる。
【0180】
【表2】
(逆転写PCR用プライマー)
前記実施例1−2において使用した1)〜5)の各プライマーに相当するものが、次の21)〜23)のプライマーである。
【0181】
21)プライマー HuCH-1 :H鎖遺伝子合成用のプライマーであり、配列表の配列番号21に塩基配列を示す。
【0182】
22)プライマー HuCκ-1:κタイプL鎖遺伝子合成用のプライマーであり、配列表の配列番号22に塩基配列を示す。
【0183】
23)プライマー HuCλ-2:λタイプL鎖遺伝子合成用のプライマーであり、配列表の配列番号23に塩基配列を示す。
【0184】
(第1段階PCR用プライマー)
前記実施例1−3で使用した6)〜13)の各プライマーに相当するものが、次の24)〜37)のプライマーである。
【0185】
24)プライマー HuVH-1 :H鎖可変領域用のプライマーであり、配列表の配列番号24に塩基配列を示す。
【0186】
25)プライマー HuVH-2 :H鎖可変領域用のプライマーであり、配列表の配列番号25に塩基配列を示す。
【0187】
26)プライマー HuVH-3 :H鎖可変領域用のプライマーであり、配列表の配列番号26に塩基配列を示す。
【0188】
27)プライマー HuVH-4 :H鎖可変領域用のプライマーであり、配列表の配列番号27に塩基配列を示す。
【0189】
28)プライマー HuVκ-1:L鎖κ遺伝子可変領域用のプライマーであり、配列表の配列番号28に塩基配列を示す。
【0190】
29)プライマー HuVκ-2:L鎖κ遺伝子可変領域用のプライマーであり、配列表の配列番号29に塩基配列を示す。
【0191】
30)プライマー HuVκ-3:L鎖κ遺伝子可変領域用のプライマーであり、配列表の配列番号30に塩基配列を示す。
【0192】
31)プライマー HuVκ-4:L鎖κ遺伝子可変領域用のプライマーであり、配列表の配列番号31に塩基配列を示す。
【0193】
32)プライマー HuVλ-1:L鎖λ遺伝子可変領域用のプライマーであり、配列表の配列番号32に塩基配列を示す。
【0194】
33)プライマー HuVλ-2:L鎖λ遺伝子可変領域用のプライマーであり、配列表の配列番号33に塩基配列を示す。
【0195】
34)プライマー HuVλ-3:L鎖λ遺伝子可変領域用のプライマーであり、配列表の配列番号34に塩基配列を示す。
【0196】
35)プライマー HuCH-2 :H鎖定常領域用のプライマーであり、配列表の配列番号35に塩基配列を示す。
【0197】
36)プライマー HuCκ-2:L鎖κ遺伝子定常領域用のプライマーであり、配列表の配列番号36に塩基配列を示す。
【0198】
37)プライマー HuCλ-2:L鎖λ遺伝子定常領域用のプライマーであり、配列表の配列番号37に塩基配列を示す。
【0199】
(第2段階PCR用プライマー)
前記実施例1−4で使用した14)プライマーに相当するものが次の38)のプライマーである。
【0200】
38)プライマー HuSP1:第2段階PCR用の単一種のプライマーであり、配列表の配列番号38に塩基配列を示す。
【0201】
(ELISA解析、配列決定、抗原に対する結合活性)
実施例1と同様にして増幅された抗体遺伝子より活性体の抗体分子(Fab)が合成されていることを証明するため、血液型抗原B型に対するELISA反応により検討した。検体成人はA型の血液型であるため、B型抗原に対する血液型抗体を当然有すると推測される。このことは、ある感染症に感染した患者の血液中に、その感染症を引き起こす病原体に対する抗体が存在すると言う状況と同じである。
【0202】
B型抗原 BSAを抗原としてELISAを行った結果を図10に示す。実験方法は実施例1の場合とほぼ同様に行った。実施例1の場合と異なる点は、抗原としてCBP40 の代わりにBlood Group B Trisaccharide-BSA 10-atom spacer(Destra laboratoriesm Ltd, )を用いた点である。
【0203】
この結果得られた抗体群は、B型抗原に対しそれぞれ異なる結合活性を有していることがわかった。人体は様々な種類の抗体を産生し、体内に有していることを考えると、十分に予想される結果であり、本実験が予想通り進行したことを示唆している。
【0204】
これらのうち、ELISAシグナルが高かった No.2及び No.10のクローンのL鎖及びH鎖の配列を決定した。 No.2のクローンのL鎖のアミノ酸配列及び遺伝子塩基配列を図11及び配列表の配列番号39に、 No.2のクローンのH鎖のアミノ酸配列及び遺伝子塩基配列を図12及び配列表の配列番号40に、 No.10のクローンのL鎖のアミノ酸配列及び遺伝子塩基配列を図13及び配列表の配列番号41に、 No.10のクローンのH鎖のアミノ酸配列及び遺伝子塩基配列を図14及び配列表の配列番号42に、それぞれ示す。
【0205】
これらの配列について、ヒト抗体のデータベースIgBlast に対してホモロジー検索を行った。その結果、 No.10のクローンのL鎖は、 V1-18germline遺伝子(GeneBank accession Nos.D187018)と95%のホモロジーが検出された。又、 No.10のクローンのH鎖は VH2-70germiline遺伝子 GeneBank accession Nos.AB019437) と91%のホモロジーが検出された。 No.2のクローンについても同様に検索し、L鎖は V1-5germline (GeneBank accession Nos. D87015)と100%の相同性が、H鎖は VH4-4(GeneBank accession Nos. AB019441)と90%の相同性が、それぞれ認められた。
【0206】
これらの結果より、本発明の方法を用いることで、目的とする抗原に対して結合能を有するヒトMabと、そのcDNAとを取得できることを証明した。
【産業上の利用可能性】
【0207】
本発明によって、特定のリガンドに対して親和性を示す極めて多種類のリガンド親和性複合タンパク質の中から、1種又は2種以上のタンパク質を迅速、大量かつ高純度に取得することが可能となる。
[配列表]
SEQUENCE LISTING
<110> National University Corporation Nagoya University
<120> リガンド親和性複合タンパク質の取得方法、抗体スクリーニング方法、抗体スクリーニング用キット、医療方法、抗体医薬、非免疫反応性抗体医薬、投薬方法
<130> POK-04-029
<160> 42
<210> 1
<211> 21
<212> DNA
<213> Artificial Sequence
<400> 1
ttaacactca ttcctgttga a 21
<210> 2
<211> 21
<212> DNA
<213> Artificial Sequence
<400> 2
aattttcttg tccaccttgg t 21
<210> 3
<211> 21
<212> DNA
<213> Artificial Sequence
<400> 3
aattttcttg tccaccttgg t 21
<210> 4
<211> 21
<212> DNA
<213> Artificial Sequence
<400> 4
aagttttttg tccaccgtgg t 21
<210> 5
<211> 21
<212> DNA
<213> Artificial Sequence
<400> 5
gattctcttg atcaactcag t 21
<210> 6
<211> 51
<212> DNA
<213> Artificial Sequence
<400> 6
attagataag aaggagatta ttgaatgga(c/t) attgtg(a/c)t(c/g)a 40
c(a/c)ca(a/g)(a/t)ct(a/c)c a 51
<210> 7
<211> 66
<212> DNA
<213> Artificial Sequence
<400> 7
attagataag aaggagatta ttgattagtg gtggtggtgg tggtgacact cattcctgtt 60
gaagct 66
<210> 8
<211> 47
<212> DNA
<213> Artificial Sequence
<400> 8
attagataag aaggagatta ttgaatg(c/g)a(a/g) gt(a/c/g/t)(a/c)agctg(c/g) 40
ag(c/g)agtc 47
<210> 9
<211> 50
<212> DNA
<213> Artificial Sequence
<400> 9
attagataag aaggagatta ttgaatg(c/g)a(a/g) gt(a/c/g/t)(a/c)agctg(c/g) 40
ag(c/g)agtc(a/t)gg 50
<210> 10
<211> 54
<212> DNA
<213> Artificial Sequence
<400> 10
attagataag aaggagatta ttgattaaat tttcttgtcc accttggtgc tgct 54
<210> 11
<211> 52
<212> DNA
<213> Artificial Sequence
<400> 11
attagataag aaggagatta ttgattaaat tttcttgtcc accttggtgc tg 52
<210> 12
<211> 52
<212> DNA
<213> Artificial Sequence
<400> 12
attagataag aaggagatta ttgattaaag ttttttgtcc accgtggtgc tg 52
<210> 13
<211> 54
<212> DNA
<213> Artificial Sequence
<400> 13
attagataag aaggagatta ttgattagat tctcttgatc aactcagtct tgct 54
<210> 14
<211> 24
<212> DNA
<213> Artificial Sequence
<400> 14
attagataag aaggagatta ttga 24
<210> 15
<211> 22
<212> DNA
<213> Artificial Sequence
<400> 15
ccaatacgca aaccgcctct cc 22
<210> 16
<211> 48
<212> DNA
<213> Artificial Sequence
<400> 16
ataatctcct tcttatctaa taacaaaatt atttctagag ggaaaccg 48
<210> 17
<211> 48
<212> DNA
<213> Artificial Sequence
<400> 17
taatcaataa tctccttctt atctaattcc ggctgctaac aaagcccg 48
<210> 18
<211> 20
<212> DNA
<213> Artificial Sequence
<400> 18
tacagggcgc gtcccattcg 20
<210> 19
<211> 219
<212> PRT
<213> Mus musculus
<400> 19
Asp Ile Val Met Thr Gln Ser Pro Ser Ser Leu Ser Ala Ser Leu Gly 16
GAC ATT GTG ATG ACC CAG TCT CCA TCC TCC CTG TCT GCC TCT CTG GGA 48
Asp Arg Val Thr Ile Ser Cys Arg Ala Ser Gln Asp Ile Ser Asn Tyr 32
GAC AGA GTC ACC ATC AGT TGC AGG GCA AGT CAG GAC ATT AGC AAT TAT 96
Leu Asn Trp Tyr Gln Gln Lys Pro Asp Gly Thr Val Lys Leu Leu Ile 48
TTA AAC TGG TAT CAG CAG AAA CCA GAT GGA ACT GTT AAA CTC CTG ATC 144
Tyr Tyr Thr Ser Arg Leu His Ser Gly Val Pro Ser Arg Phe Ser Gly 64
TAC TAC ACA TCA AGA TTA CAC TCA GGA GTC CCA TCA AGG TTC AGT GGC 192
Ser Gly Ser Gly Thr Asp Tyr Ser Leu Thr Ile Ser Asn Leu Glu Gln 80
AGT GGG TCT GGA ACA GAT TAT TCT CTC ACC ATT AGC AAC CTG GAG CAA 240
Glu Asp Ile Ala Thr Tyr Phe Cys Gln Gln Gly Asn Thr Leu Tyr Thr 96
GAA GAT ATT GCC ACT TAC TTT TGC CAA CAG GGT AAT ACG CTG TAC ACG 288
Phe Gly Gly Gly Thr Lys Leu Glu Ile Lys Arg Ala Asp Ala Ala Pro 112
TTC GGA GGG GGG ACC AAG CTG GAA ATA AAA CGG GCT GAT GCT GCA CCA 336
Thr Val Ser Ile Phe Pro Pro Ser Ser Glu Gln Leu Thr Ser Gly Gly 128
ACT GTA TCC ATC TTC CCA CCA TCC AGT GAG CAG TTA ACA TCT GGA GGT 384
Ala Ser Val Val Cys Phe Leu Asn Asn Phe Tyr Pro Lys Asp Ile Asn 144
GCC TCA GTC GTG TGC TTC TTG AAC AAC TTC TAC CCC AAA GAC ATC AAT 432
Val Lys Trp Lys Ile Asp Gly Ser Glu Arg Gln Asn Gly Val Leu Asn 160
GTC AAG TGG AAG ATT GAT GGC AGT GAA CGA CAA AAT GGC GTC CTG AAC 480
Ser Trp Thr Asp Gln Asp Ser Lys Asp Ser Thr Tyr Ser Met Ser Ser 176
AGT TGG ACT GAT CAG GAC AGC AAA GAC AGC ACC TAC AGC ATG AGC AGC 528
Thr Leu Thr Leu Thr Lys Asp Glu Tyr Glu Arg His Asn Ser Tyr Thr 192
ACC CTC ACG TTG ACC AAG GAC GAG TAT GAA CGA CAT AAC AGC TAT ACC 576
Cys Glu Ala Thr His Lys Thr Ser Thr Ser Pro Ile Val Lys Ser Phe 208
TGT GAG GCC ACT CAC AAG ACA TCA ACT TCA CCC ATT GTC AAG AGC TTC 624
Asn Arg Asn Glu Cys His His His His His His 219
AAC AGG AAT GAG TGT CAC CAC CAC CAC CAC CAC 657
<210> 20
<211> 213
<212> PRT
<213> Mus musculus
<400> 20
Glu Val Lys Leu Gln Gln Ser Gly Ala Glu Phe Val Arg Pro Gly Ala 16
GAA GTA AAG CTG CAG CAG TCT GGG GCT GAG TTT GTA AGG CCA GGG GCC 45
Leu Val Lys Leu Ser Cys Lys Thr Ser Gly Phe Asn Ile Lys Asp Tyr 32
TTA GTC AAG TTG TCC TGC AAA ACT TCT GGC TTC AAC ATT AAA GAC TAC 90
Tyr Met His Trp Val Lys Gln Arg Pro Glu Gln Gly Leu Glu Trp Ile 48
TAT ATG CAC TGG GTG AAG CAG AGG CCT GAA CAG GGC CTG GAG TGG ATT 135
Gly Trp Ile Asp Pro Glu Asn Asp Asp Thr Ile Tyr Asp Pro Lys Phe 64
GGA TGG ATT GAT CCT GAG AAT GAT GAT ACT ATA TAT GAC CCG AAA TTC 180
Gln Gly Lys Ala Ser Ile Thr Ala Asp Thr Ser Ser Asn Thr Ala Tyr 80
CAG GGC AAG GCC AGT ATA ACA GCT GAC ACA TCC TCC AAC ACA GCC TAC 225
Leu Gln Leu Ser Ser Leu Thr Ser Glu Asp Thr Ala Val Tyr Phe Cys 96
CTG CAG CTC AGC AGC CTG ACA TCT GAG GAC ACT GCC GTC TAT TTC TGT 270
Ala Gly Asp Gly Tyr Ser Gly Asn Tyr Trp Gly Gln Gly Thr Ser Val 112
GCT GGT GAT GGT TAC TCC GGG AAC TAC TGG GGT CAA GGA ACC TCA GTC 315
Thr Val Ser Ser Ala Lys Thr Thr Pro Pro Ser Val Tyr Pro Leu Ala 128
ACC GTC TCC TCA GCC AAA ACG ACA CCC CCA TCT GTC TAT CCA CTG GCC 360
Pro Gly Ser Ala Ala Gln Thr Asn Ser Met Val Thr Leu Gly Cys Leu 144
CCT GGA TCT GCT GCC CAA ACT AAC TCC ATG GTG ACC CTG GGA TGC CTG 405
Val Lys Gly Tyr Phe Pro Glu Pro Val Thr Val Thr Trp Asn Ser Gly 160
GTC AAG GGC TAT TTC CCT GAG CCA GTG ACA GTG ACC TGG AAC TCT GGA 450
Ser Leu Ser Ser Gly Val His Thr Phe Pro Ala Val Leu Gln Ser Asp 176
TCC CTG TCC AGC GGT GTG CAC ACC TTC CCA GCT GTC CTG CAG TCT GAC 495
Leu Tyr Thr Leu Ser Ser Ser Val Thr Val Pro Ser Ser Thr Trp Pro 192
CTC TAC ACT CTG AGC AGC TCA GTG ACT GTC CCC TCC AGC ACC TGG CCC 540
Ser Glu Thr Val Thr Cys Asn Val Ala His Pro Ala Ser Ser Thr Lys 208
AGC GAG ACC GTC ACC TGC AAC GTT GCC CAC CCG GCC AGC AGC ACC AAG 585
Val Asp Lys Lys Ile 213
GTG GAC AAG AAA ATT 639
<210> 21
<211> 18
<212> DNA
<213> Artificial Sequence
<400> 21
gctggcctct caccaact 18
<210> 22
<211> 18
<212> DNA
<213> Artificial Sequence
<400> 22
acactctccc ctgttgaa 18
<210> 23
<211> 18
<212> DNA
<213> Artificial Sequence
<400> 23
tgaacattcy gyaggggc 18
<210> 24
<211> 50
<212> DNA
<213> Artificial Sequence
<400> 24
gaaacaagaa tagaaggaga tattgtaatg caggtgcagc tggtgcagtc 50
<210> 25
<211> 53
<212> DNA
<213> Artificial Sequence
<400> 25
gaaacaagaa tagaaggaga tattgtaatg caggtcaact taagggagtc tgg 53
<210> 26
<211> 50
<212> DNA
<213> Artificial Sequence
<400> 26
gaaacaagaa tagaaggaga tattgtaatg gaggtgcagc tgstgsagtc 50
<210> 27
<211> 50
<212> DNA
<213> Artificial Sequence
<400> 27
gaaacaagaa tagaaggaga tattgtaatg caggtrcagc tgcagsagtc 50
<210> 28
<211> 51
<212> DNA
<213> Artificial Sequence
<400> 28
gaaacaagaa tagaaggaga tattgtaatg gacatcswga tgacccagtc t 51
<210> 29
<211> 53
<212> DNA
<213> Artificial Sequence
<400> 29
gaaacaagaa tagaaggaga tattgtaatg gatattgtgm tgactcagtc tcc 53
<210> 30
<211> 53
<212> DNA
<213> Artificial Sequence
<400> 30
gaaacaagaa tagaaggaga tattgtaatg gaaattgtgt tgacgcagtc tcc 53
<210> 31
<211> 52
<212> DNA
<213> Artificial Sequence
<400> 31
gaaacaagaa tagaaggaga tattgtaatg gaaacgacac tcacgcagtc tc 52
<210> 32
<211> 50
<212> DNA
<213> Artificial Sequence
<400> 32
gaaacaagaa tagaaggaga tattgtaatg cagtctgtgc tgactcagcc 50
<210> 33
<211> 50
<212> DNA
<213> Artificial Sequence
<400> 33
gaaacaagaa tagaaggaga tattgtaatg cagtctgccc tgactcagcc 50
<210> 34
<211> 50
<212> DNA
<213> Artificial Sequence
<400> 34
gaaacaagaa tagaaggaga tattgtaatg tcttatgagc tgacwcagcc 50
<210> 35
<211> 73
<212> DNA
<213> Artificial Sequence
<400> 35
gaaacaagaa tagaaggaga tattgtatta gtggtggtgg tggtggtgaa ctbtcttgtc 60
caccttggtg ttg 73
<210> 36
<211> 52
<212> DNA
<213> Artificial Sequence
<400> 36
gaaacaagaa tagaaggaga tattgtatta acactctccc ctgttgaagc tc 52
<210> 37
<211> 52
<212> DNA
<213> Artificial Sequence
<400> 37
gaaacaagaa tagaaggaga tattgtatta tgaacattcy gyaggggcma ct 52
<210> 38
<211> 27
<212> DNA
<213> Artificial Sequence
<400> 38
gaaacaagaa tagaaggaga tattgta 27
<210> 39
<211> 213
<212> PRT
<213> Homo sapiens
<400> 39
Gln Ser Ala Leu Thr Gln Pro Pro Lys Val Leu Gly Leu Gln Ala Arg 16
CAG TCT GCC CTG ACT CAG CCT CCC AAA GTG TTG GGA TTA CAG GCG CGA 48
Ala Thr Thr Pro Gly Tyr Leu Phe His Phe Ile Leu Met Thr Thr Leu 32
GCC ACC ACG CCT GGT TAT TTA TTT CAT TTT ATA CTT ATG ACA ACC CTG 96
Gly Ser Pro Ser Val Ile Leu Cys Gln Pro Leu Thr Leu Ser Phe Leu 48
GGA AGT CCC TCT GTC ATT CTA TGC CAG CCG TTG ACA TTG TCA TTC TTG 144
Pro Phe Tyr Thr Gly Arg Asn Lys Leu Arg Glu Lys Val Met Ser Ile 64
CCA TTT TAT ACA GGA CGA AAT AAA CTC AGA GAG AAG GTA ATG AGC ATA 192
Ile Cys Leu Arg Ser Gln Asp Thr Trp Gln Ser Gly Asn Ser Asn Pro 80
ATC TGT CTG AGG TCA CAG GAT ACG TGG CAG AGC GGA AAT TCG AAT CCT 240
Ser Ile Leu Thr Ser Glu Pro Val Ser Trp Thr Leu Trp Leu Leu Ser 96
AGC ATT CTG ACT TCA GAA CCT GTT TCC TGG ACA CTC TGG CTT CTC TCG 288
Met Trp Ser Pro Ala Arg Gln His Lys Thr Leu Val Gln Pro Lys Ala 112
ATG TGG TCC CCG GCC CGG CAG CAT AAG ACA CTA GTC CAG CCC AAG GCT 336
Ala Pro Ser Val Thr Leu Phe Pro Pro Ser Ser Glu Glu Leu Gln Ala 128
GCC CCC TCG GTC ACT CTG TTC CCG CCC TCC TCT GAG GAG CTT CAA GCC 384
Asn Lys Ala Thr Leu Val Cys Leu Ile Ser Asp Phe Tyr Pro Gly Ala 144
AAC AAG GCC ACA CTG GTG TGT CTC ATA AGT GAC TTC TAC CCG GGA GCC 432
Val Thr Val Ala Trp Lys Ala Asp Ser Ser Pro Val Lys Ala Gly Val 160
GTG ACA GTG GCT TGG AAA GCA GAT AGC AGC CCC GTC AAG GCG GGA GTG 480
Glu Thr Thr Thr Pro Ser Lys Gln Ser Asn Asn Lys Tyr Ala Ala Ser 176
GAG ACC ACC ACA CCC TCC AAA CAA AGC AAC AAC AAG TAC GCG GCC AGC 528
Ser Tyr Leu Ser Leu Thr Pro Glu Gln Trp Lys Ser His Arg Ser Tyr 192
AGC TAT CTG AGC CTG ACG CCT GAG CAG TGG AAG TCC CAC AGA AGC TAC 576
Ser Cys Gln Val Thr His Glu Gly Ser Thr Val Glu Lys Thr Val Ala 208
AGC TGC CAG GTC ACG CAT GAA GGG AGC ACC GTG GAG AAG ACA GTG GCC 624
Pro Thr Glu Cys Ser 213
CCT ACA GAA TGT TCA 639
<210> 40
<211> 219
<212> PRT
<213> Homo sapiens
<400> 40
Gln Val Gln Leu Gln Gln Ser Gly Tyr Pro Ser His Glu His Pro Ile 16
CAG GTA CAG CTG CAG CAG TCC GGA TAT CCC TCC CAT GAG CAT CCT ATC 48
Leu Trp Thr Ser Asp Val Leu Tyr Lys Glu Ala Pro Ser Asn Phe Leu 32
CTC TGG ACT TCT GAT GTT CTA TAT AAA GAG GCA CCC TCA AAT TTT CTT 96
Phe Ser Glu Ala Lys Pro Trp Arg Glu Trp Glu Tyr Pro Tyr Glu Glu 48
TTC TCC GAA GCT AAG CCT TGG AGA GAA TGG GAG TAC CCC TAC GAA GAG 144
Phe Leu Leu Ser Val Ser Arg Leu Ser Glu Cys Asn Tyr Gln Ser His 64
TTT CTA CTC TCA GTG TCC AGG TTA TCT GAG TGC AAT TAC CAG TCC CAT 192
Asn Ile Pro Ile Pro Glu Ser Tyr Val Arg Leu Phe Pro Glu Glu His 80
AAC ATA CCA ATT CCA GAA TCA TAT GTG CGT CTC TTT CCA GAA GAA CAT 240
Arg Ser Tyr Leu Lys Gly Cys Cys Asp Thr Asn Ser Tyr Asp Met Leu 96
AGA AGT TAT CTC AAA GGG TGT TGT GAC ACT AAC AGC TAC GAT ATG TTA 288
Arg Tyr Leu Ala Glu Tyr Gln Val Glu Gly Glu Thr Leu Tyr Asn Arg 112
AGG TAT TTG GCA GAA TAT CAG GTA GAA GGT GAA ACA CTT TAT AAC AGG 336
Leu Leu Ser Val Ser Thr Lys Gly Pro Ser Val Phe Pro Leu Ala Pro 128
CTA TTA TCA GTA TCC ACC AAG GGC CCA TCC GTC TTC CCC CTG GCG CCC 384
Cys Ser Arg Ser Thr Ser Glu Ser Thr Ala Ala Leu Gly Cys Leu Val 144
TGC TCC AGG AGC ACC TCC GAG AGC ACA GCC GCC CTG GGC TGC CTG GTC 432
Lys Asp Tyr Phe Pro Glu Pro Val Thr Val Ser Trp Asn Ser Gly Ala 160
AAG GAC TAC TTC CCC GAA CCG GTG ACG GTG TCG TGG AAC TCA GGC GCC 480
Leu Thr Ser Gly Val His Thr Phe Pro Ala Val Leu Gln Ser Ser Gly 176
CTG ACC AGC GGC GTG CAC ACC TTC CCG GCT GTC CTA CAG TCC TCA GGA 528
Leu Tyr Ser Leu Ser Ser Val Val Thr Val Pro Ser Ser Ser Leu Gly 192
CTC TAC TCC CTC AGC AGC GTG GTG ACC GTG CCC TCC AGC AGC TTG GGC 576
Thr Lys Thr Tyr Thr Cys Asn Val Asp His Lys Pro Ser Asn Thr Lys 208
ACG AAG ACC TAC ACC TGC AAC GTA GAT CAC AAG CCC AGC AAC ACC AAG 624
Val Asp Lys Arg Val His His His His His His 219
GTG GAC AAG AGA GTT CAC CAC CAC CAC CAC CAC 657
<210> 41
<211> 213
<212> PRT
<213> Homo sapiens
<400> 41
Gln Ser Val Leu Thr Gln Pro Tyr Pro Gly Glu Gly Phe Leu Ile Tyr 16
CAG TCT GTG CTG ACT CAG CCT TAT CCA GGT GAA GGC TTT CTG ATA TAT 48
Gln Ser Ala Tyr Leu Ala Pro Phe Cys Leu Ser Tyr Tyr Thr Val Lys 32
CAA TCT GCT TAT TTG GCT CCA TTC TGT CTC TCA TAC TAT ACT GTG AAG 96
Arg Arg Pro Cys Leu Ile His Val Asp Thr Thr His Ser Arg Ile Met 48
CGA AGA CCA TGT CTC ATT CAT GTA GAC ACT ACA CAC AGT AGG ATA ATG 144
His Val Glu Leu Asn Trp Leu Met His Glu Tyr Ala Ser Tyr Gln Arg 64
CAT GTT GAG TTA AAT TGG CTT ATG CAT GAA TAT GCT TCC TAC CAG CGA 192
Gly Val Asn Gly Leu Ser Lys Asp Glu His His Leu Phe Leu Tyr Ile 80
GGA GTC AAT GGT CTA AGT AAG GAT GAA CAT CAT CTG TTT CTG TAC ATT 240
Tyr Phe Gln Thr Val Val Arg Leu Gly Val Pro Arg Ser Lys Asp Tyr 96
TAC TTT CAA ACA GTA GTG AGG CTG GGA GTA CCT AGA AGC AAG GAC TAT 288
Val Leu Thr Gln Val Ser Lys Phe Ile Phe Tyr Ser Phe Pro Lys Ala 112
GTT TTA ACC CAA GTT TCA AAA TTT ATC TTT TAT TCG TTT CCC AAG GCC 336
Asn Pro Thr Val Thr Leu Phe Pro Pro Ser Ser Glu Glu Leu Gln Ala 128
AAC CCC ACG GTC ACT CTG TTC CCG CCC TCC TCT GAG GAG CTC CAA GCC 384
Asn Lys Ala Thr Leu Val Cys Leu Ile Ser Asp Phe Tyr Pro Gly Ala 144
AAC AAG GCC ACA CTA GTG TGT CTG ATC AGT GAC TTC TAC CCG GGA GCT 432
Val Thr Val Ala Trp Lys Ala Asp Gly Ser Pro Val Lys Ala Gly Val 160
GTG ACA GTG GCT TGG AAG GCA GAT GGC AGC CCC GTC AAG GCG GGA GTG 480
Glu Thr Thr Lys Pro Ser Lys Gln Ser Asn Asn Lys Tyr Ala Ala Ser 176
GAG ACG ACC AAA CCC TCC AAA CAG AGC AAC AAC AAG TAC GCG GCC AGC 528
Ser Tyr Leu Ser Leu Thr Pro Glu Gln Trp Lys Ser His Arg Ser Tyr 192
AGC TAC CTG AGC CTG ACG CCC GAG CAG TGG AAG TCC CAC AGA AGC TAC 576
Ser Cys Gln Val Thr His Glu Gly Ser Thr Val Glu Lys Thr Val Ala 208
AGC TGC CAG GTC ACG CAT GAA GGG AGC ACC GTG GAG AAG ACA GTG GCC 624
Pro Thr Glu Cys Ser 213
CCT ACA GAA TGT TCA 639
<210> 42
<211> 219
<212> PRT
<213> Homo sapiens
<400> 42
Gln Val Asn Leu Arg Glu Ser Gly Gln Leu Met Pro Ala Ser Ser Pro 16
CAG GTC AAC TTA AGG GAG TCT GGA CAA TTA ATG CCT GCC TCC TCT CCC 48
Tyr Pro Thr Asn Val His Ile Leu Ile Leu Arg Thr Cys Glu Tyr Gly 32
TAC CCC ACA AAT GTT CAC ATC CTA ATT CTT AGA ACC TGT GAA TAT GGT 96
Glu Tyr Val Glu Tyr Val Thr Tyr His Gly Gln Glu Val Leu Tyr Arg 48
GAA TAT GTT GAA TAT GTT ACC TAC CAT GGT CAA GAG GTA CTT TAC AGA 144
Tyr Glu Gln Leu Arg Thr Leu Arg Glu Ala Gln Ser Ser Arg Leu Leu 64
TAT GAG CAA TTA AGG ACC CTG AGA GAG GCA CAA TCA TCT AGA TTA CTC 192
Arg Gln Ala Gln Ser Gly His Gln Gly Pro Cys Lys Arg Lys Val Gly 80
AGG CAA GCC CAA AGT GGC CAC CAG GGT CCC TGT AAG AGG AAG GTA GGG 240
Arg Val Lys Arg Gln Gln Ile Lys Ser Arg Asp Glu Ser Asp Ala Leu 96
AGG GTT AAG AGA CAG CAG ATA AAG AGC AGA GAC GAG AGT GAT GCA CTT 288
Gly Lys Met Lys Glu Gly His Glu Val Lys Glu Cys Trp Trp Pro Leu 112
GGA AAG ATG AAA GAA GGT CAT GAG GTT AAG GAA TGC TGG TGG CCT CTA 336
Glu Ala Arg Lys Ser Thr Lys Gly Pro Ser Val Phe Pro Leu Ala Pro 128
GAA GCT AGA AAG TCC ACC AAG GGC CCA TCG GTC TTC CCC CTG GCA CCC 384
Ser Ser Lys Ser Thr Ser Gly Gly Thr Ala Ala Leu Gly Cys Leu Val 144
TCC TCC AAG AGC ACC TCT GGG GGC ACA GCG GCC CTG GGC TGC CTG GTC 432
Lys Asp Tyr Phe Pro Glu Pro Val Thr Val Ser Trp Asn Ser Gly Ala 160
AAG GAC TAC TTC CCC GAA CCG GTG ACG GTG TCG TGG AAC TCA GGC GCC 480
Leu Thr Ser Gly Val His Thr Phe Pro Ala Val Leu Gln Ser Ser Gly 176
CTG ACC AGC GGC GTG CAC ACC TTC CCG GCT GTC CTA CAG TCC TCA GGA 528
Leu Tyr Ser Leu Ser Ser Val Val Thr Val Pro Ser Ser Ser Leu Gly 192
CTC TAC TCC CTC AGC AGC GTG GTG ACC GTG CCC TCC AGC AGC TTG GGC 576
Thr Gln Thr Tyr Ile Cys Asn Val Asn His Lys Pro Ser Asn Thr Lys 208
ACC CAG ACC TAC ATC TGC AAC GTG AAT CAC AAG CCC AGC AAC ACC AAG 624
Val Asp Lys Arg Val His His His His His His 219
GTG GAC AAG AGA GTT CAC CAC CAC CAC CAC CAC 657
【図面の簡単な説明】
【0208】
【図1】実施例1の全体的なフローを概念化して示す図である。
【0209】
【図2】実施例1におけるH鎖遺伝子とL鎖遺伝子の増幅結果を示す図である。
【0210】
【図3】実施例1のオーバーラッピングPCR産物の解析結果を示す図である。
【0211】
【図4】実施例1におけるオートラジオグラフィーの結果を示す図である。
【0212】
【図5】実施例1におけるELISA解析の結果を示す図である。
【0213】
【図6】実施例1におけるL鎖の配列決定の結果を示す図である。
【0214】
【図7】実施例1におけるH鎖の配列決定の結果を示す図である。
【0215】
【図8】実施例2の全体的なフローを概念化して示す図である。
【0216】
【図9】実施例2におけるH鎖遺伝子とL鎖遺伝子の増幅結果を示す図である。
【0217】
【図10】実施例2におけるELISA解析の結果を示す図である。
【0218】
【図11】実施例2における No.2クローンのL鎖配列決定の結果を示す図である。
【0219】
【図12】実施例2における No.2クローンのH鎖配列決定の結果を示す図である。
【0220】
【図13】実施例2における No.10クローンのL鎖配列決定の結果を示す図である。
【0221】
【図14】実施例2における No.10クローンのH鎖配列決定の結果を示す図である。
【技術分野】
【0001】
本発明は、リガンド親和性複合タンパク質の取得方法、抗体スクリーニング方法、抗体スクリーニング用キット、医療方法、抗体医薬、非免疫反応性抗体医薬、及び投薬方法に関する。
【0002】
更に詳しくは、本発明は、特定されたリガンドに対して選択的な親和性を示す極めて多種類の複合タンパク質Xpの中から選ばれる1種又は2種以上の複合タンパク質を迅速かつ大量に取得することを可能とした、リガンド親和性複合タンパク質の取得方法に関する。このリガンド親和性複合タンパク質の取得方法は、ヒトに対して適用することができ、更に、少なくとも免疫反応による抗体の生成が認められる非ヒト動物に対しても適用することができる。
【0003】
又、本発明は、上記のリガンド親和性複合タンパク質の取得方法に基づいた、ハイ・スループット(High-throughput )なスクリーニングを迅速に実行可能とする抗体スクリーニング方法と、この抗体スクリーニング方法を行うための抗体スクリーニング用キットに関する。
【0004】
又、本発明は、上記の抗体スクリーニング方法によって得られた抗体を用いる、ヒト又は上記の非ヒト動物に対する医療方法と、当該抗体を有効成分とする抗体医薬とに関する。
【0005】
更に、本発明は、ヒトを適用対象とする上記のリガンド親和性複合タンパク質の取得方法によって得られた抗体を有効成分とする医薬であって、適用対象たる当該ヒト個体に投与することを目的とする非免疫反応性抗体医薬と、この非免疫反応性抗体医薬を用いる投薬方法とに関する。
【0006】
なお、本発明において、「リガンド」とは、任意の無機化合物や有機化合物、及び抗原たり得る各種の無機化合物や有機化合物、タンパク質、ウイルス、微生物等を制限なく含む概念である。リガンドが抗原である場合において、本発明のリガンド親和性複合タンパク質は抗体あるいはモノクローナル抗体(以下、単に「Mab」とも言う)であり得る。
【背景技術】
【0007】
任意のリガンドに対して選択的な親和性を示すタンパク質は、一般的な技術分野においては、当該リガンドの検出手段、分離手段あるいは精製手段として重要である。
【0008】
一方、医学あるいは生理学の分野においては、このような親和性タンパク質として、ヒト又は少なくとも免疫反応による抗体の産生が認められる非ヒト動物において生成される極めて多種類の抗体を代表的に例示することができる。抗体は免疫学における重要な生体分子であり、ヒト又は非ヒト動物の疾患の予防、診断あるいは治療等に広範囲に応用することができる。非ヒト動物を用いた病理研究等にも広範囲に応用することができる。抗体の内でも、特に、いわゆるハイブリドーマ技術によって大量生産が可能となったMabは極めて重要である。
【0009】
一般的に、リガンド親和性タンパク質を取得する場合において、例えば任意の抗原即ちリガンドで免疫した非ヒト動物、ヒトにおける各種の感染症患者、あるいは、ガン細胞がリガンドとなって抗体を生成しているガン患者等に典型的に見られるように、リガンドに対する親和性又は結合力等に差異のある極めて多種類の類似タンパク質が存在する場合が多い。
【0010】
例えばポリクローナル抗体を使用する場合のように、このような類似タンパク質群を混ぜ物としてそのまま利用することも、場合によって可能ではある。しかし、リガンドの検出手段、分離手段あるいは精製手段としても、疾患の予防、診断あるいは治療等の手段としても、多種類の類似タンパク質群の内から特定された1種類ごとの優れたリガンド親和性タンパク質のみを選択的に大量取得することが、圧倒的に有利である。
【0011】
ハイブリドーマ技術は、1種類のリガンド親和性タンパク質のみを、Mabとして選択的に大量取得する可能性を切り開いた点で重要な意味を持つ。しかし、この技術では、動物の免疫反応を利用して抗原親和性タンパク質たる各種Mabの生産源である多数のB細胞を調製する点は良いとしても、その後、例えば安定したハイブリドーマの作成(タンパク質の生産手段の調製)に困難があったり、ハイブリドーマの培養と増殖(タンパク質の生産手段の大規模化)に非常に手間がかかる等の問題がある。従って、実用技術としては、迅速な大量生産及び高速多検体スクリーニングと言う要求に十分に応えることができない。
【0012】
近年、ハイブリドーマ技術の上記問題点に鑑み、B細胞の生成と組織培養を不要化するため、例えば、キメラモノクローナル抗体(Vaquelo et al., 1999)、遺伝子導入マウス(Green et al., 1994)、ファージディスプレー(MacCafferty
et al., 1990)、リボソームディスプレー(Hanes & Plucthun, 1997)のような種々の遺伝子工学的技術の開発が図られている。しかしこれらの技術は、いわば要素技術に過ぎず、多様なリガンド親和性タンパク質を大量生産し高速多検体スクリーニングを行うと言う要求に全体的に応えることはできないし、より具体的なレベルの問題として、抗体のH鎖とL鎖とのオリジナルで正確なペアリングは得られていない。
【0013】
【非特許文献1】Coronella, J.A., Telleman, P., Truong, T.D., Ylera, F., Junghans, R.P., 2000. "Amplification of IgG VH and VL(Fab) fromsingle cell human plasma cells and B cells" Nucleic Acids Res. 28,e85 又、B細胞を迅速・正確に単離する技術、キット化された実験試薬、シークエンシングとその解析の自動化、単細胞逆転写PCR技術等が進歩し、例えば上記の非特許文献1で記載されているように、種々の細胞由来の抗体遺伝子の増幅が報告されて来た。即ち、単細胞逆転写PCRの利点により、単一の細胞から得た抗体のL鎖遺伝子とH鎖遺伝子とをそれぞれ増幅し、組換え大腸菌を用いた発現システムを利用すれば、抗体のH鎖とL鎖とのペアリングは実現できる。
【0014】
しかし、逆転写PCR産物たる抗体遺伝子を、発現のためにベクターにクローニングする必要があり、そのクローニングと微生物での発現には非常に面倒な作業と長時間を要する。又、細胞毒性を有する抗体を扱うことが困難であるとか、極めて多数のクローン(極めて多数の組換え大腸菌)を扱うことが困難であるとかの問題がある。
【0015】
以上の点は、特定されたリガンド親和性タンパク質に対する迅速な大量生産と高速多検体スクリーニングと言う一般的な技術手段としての要求に関する問題の指摘であるが、この問題を医学又は生理学の立場から見ると、優れた抗体スクリーニング方法、医療方法、抗体医薬等の提供の問題に直結するものである。更に、ヒトを対象とする場合には、免疫反応が関連する疾患等において、拒絶反応を伴わない非経口的投与を可能とする非免疫反応性抗体医薬の提供の問題にも直結するものである。
【発明の開示】
【発明が解決しようとする課題】
【0016】
本願発明者は、極めて多種類のリガンド親和性複合タンパク質の中から特定された1種又は2種以上のタンパク質を迅速・大量に取得するための技術として、感染症患者等からヒトの抗体産生細胞を非侵襲的手段によって採取し、又は免疫反応を利用して非ヒト動物の抗体産生細胞を調製し、これをハイブリドーマ技術に替わるものとしての単細胞逆転写PCR技術と組み合わせ、更に、クローン化された遺伝子からのタンパク質製造技術として近年注目されている無細胞タンパク質合成系を組み合わせることを着想した。
【0017】
しかし、このような生産技術体系を構想する場合、とりわけ、極めて多種類のリガンド親和性複合タンパク質の中から優れた特性を有する1種又は2種以上のタンパク質を選択的に迅速・大量取得しようとする場合、下記イ)、ロ)、ハ)に起因する実質的に無限とも言える多様性の制約の中で、その目的を果たさねばならない。
イ)抗体として得られる目的タンパク質には実質的に無限とも言える極めて多種のバリエーションがある。
ロ)タンパク質の各分子が複数のフラグメントからなる複合タンパク質であって、各フラグメントが個別の構造遺伝子でコードされている。
ハ)この複合タンパク質においてIgG、IgA、IgE等の各クラスと、L鎖におけるκ鎖やλ鎖等の各アイソタイプがある。
【0018】
本願発明者の知る限りにおいて、このような多様性の制約の中で特定の複合タンパク質を選択的に迅速・大量取得しようとする課題に取り組んだリガンド親和性複合タンパク質の取得方法は、未だ提案されたことがない。本願発明者は、このような前例のない課題の解決に取り組み、本願発明に係るリガンド親和性複合タンパク質の取得方法を完成した。更に、このリガンド親和性複合タンパク質の取得方法を基礎として、抗体スクリーニング方法、抗体スクリーニング用キット、医療方法、抗体医薬、非免疫反応性抗体医薬、及び投薬方法の各発明をも完成した。
【課題を解決するための手段】
【0019】
(第1発明の構成)
上記課題を解決するための本願第1発明の構成は、任意に特定されたリガンドXに対して選択的な親和性を示す極めて多種類の複合タンパク質Xpの中から、無作為にあるいは特性評価を通じて選ばれる1種又は2種以上の複合タンパク質を取得する方法であって、少なくとも下記の1)生産手段分離工程、2)生産手段転換工程、3)大量生産準備工程、4)転写用配列付加工程及び5)単一種大量生産工程を含み、必要に応じて更に下記の6)キャラクタリゼーション工程を含む、リガンド親和性複合タンパク質の取得方法である。
【0020】
1)極めて多種類にわたる前記複合タンパク質Xpのそれぞれ専用の生産手段(mRNA)であって、容易に大量複製できる複製手段Xc(cDNA)への転換が可能な生産手段Xmを備えた抗体産生細胞の1個又は複数個の単細胞を、ヒト又は非ヒト動物たる検体の適宜な組織又は生体構成部分から個別に分離することにより、目的タンパク質の種類ごとに生産手段Xmを厳密に分離して、以下の各工程における生産手段Xm、複製手段Xc及び複合タンパク質Xpの異種間混在を予め防止する生産手段分離工程。
【0021】
2)互いに分離された1種又は複数種の前記生産手段Xmについて、それぞれの生産手段Xmを構成するmRNAの全部又は一部を逆転写PCR法により複製手段Xcへ転換させる生産手段転換工程。
【0022】
3)逆転写PCR法により得られた前記複製手段XcをPCR法で大量複製してタンパク質の大量生産能力を準備する大量生産準備工程。
【0023】
4)大量複製された前記複製手段Xc(cDNA)に対して、その無細胞系での生産手段Xm(mRNA)への再転換に必要な転写用配列を付加する転写用配列付加工程。
【0024】
5)転写用配列を付加された前記複製手段Xcを、少なくとも複製手段Xcの生産手段Xmへの再転換を行う要素と、生産手段Xmに基づきタンパク質の生産を行う要素とを備えた無細胞系へ投入することにより、前記複合タンパク質Xpを、又は前記生産手段転換工程で規定した一部のmRNAに対応する複合タンパク質Xpを、1種類ごとに大量生産する単一種大量生産工程。
【0025】
6)以上の各工程により生産された複合タンパク質Xpについて、特性評価を行い、優れた特性を持つ複合タンパク質Xpをスクリーニングし、あるいはこれらの複合タンパク質Xpの配列決定を行うキャラクタリゼーション工程。
【0026】
(第2発明の構成)
上記課題を解決するための本願第2発明の構成は、前記第1発明に係る検体がヒトである場合において、抗原たるリガンドXに対する抗体を既に体内に生成している生理状態にあると推定される被検者を選択したもとで、当該被検者における適宜な組織又は生体構成部分を社会通念上許容される非侵襲的手段によって採取し、この組織又は生体構成部分から前記1)の1個又は複数個の単細胞を分離する、リガンド親和性複合タンパク質の取得方法である。
【0027】
(第3発明の構成)
上記課題を解決するための本願第3発明の構成は、前記第2発明に係る被検者から、前記組織又は生体構成部分として末梢血を採取し、この末梢血から抗体産生細胞たるB細胞及び/又は形質細胞の単細胞を分離する、リガンド親和性複合タンパク質の取得方法である。
【0028】
(第4発明の構成)
上記課題を解決するための本願第4発明の構成は、前記第1発明に係る検体が非ヒト動物である場合において、この動物をリガンドXで免疫したもとで、当該動物の適宜な組織又は生体構成部分から前記1)の1個又は複数個の単細胞を分離する、リガンド親和性複合タンパク質の取得方法である。
【0029】
(第5発明の構成)
上記課題を解決するための本願第5発明の構成は、前記第1発明〜第4発明のいずれかに係る複合タンパク質Xpが各1対のH鎖及びL鎖からなる免疫グロブリン(抗体)、そのFab(抗原結合性フラグメント)、又はこれらに準ずるタンパク質フラグメントである、リガンド親和性複合タンパク質の取得方法である。
【0030】
(第6発明の構成)
上記課題を解決するための本願第6発明の構成は、前記第1発明〜第5発明のいずれかに係る生産手段分離工程において、前記検体の組織又は生体構成部分を液体中に投入して抗体産生細胞を分散・懸濁させ、一定の分離用抗体と結合させた磁性微粒子と共にインキュベートした後、分離用抗体を介して抗体産生細胞に結合した磁性微粒子をマグネットで回収することにより、前記懸濁液から目的とする抗体産生細胞の単細胞を分離する、リガンド親和性複合タンパク質の取得方法である。
【0031】
(第7発明の構成)
上記課題を解決するための本願第7発明の構成は、前記第1発明〜第6発明のいずれかに係る生産手段転換工程において、逆転写PCRに用いるプライマーの設計により、特定の分類カテゴリーに属する複合タンパク質XpをコードするmRNAのみを複製手段Xcへ転換させ、及び/又は複合タンパク質Xpを構成する特定のフラグメントをコードするmRNAのみを前記複製手段Xcへ転換させる、リガンド親和性複合タンパク質の取得方法である。
【0032】
(第8発明の構成)
上記課題を解決するための本願第8発明の構成は、前記第1発明〜第7発明のいずれかに係る大量生産準備工程において、複製手段XcのPCR法による大量複製を、次の2段階PCRによって行う、リガンド親和性複合タンパク質の取得方法である。
【0033】
第1段階PCR:複製手段Xcを構成する全ての種類のcDNAに対応する各種類のプライマーにおいて、それらの5’末端側に同一の付加配列を設けておいて増幅を行う。
【0034】
第2段階PCR:第1段階PCRで増幅された全ての種類のDNAフラグメントに対して、前記付加配列に対して相補的な配列部分を備えた単一種類のプライマーを用いて増幅を行う。
【0035】
(第9発明の構成)
上記課題を解決するための本願第9発明の構成は、前記第8発明に係る第1段階PCRで用いる各種類のプライマーの付加配列中に、前記5)の無細胞系の由来生物を考慮した適宜な種類の翻訳開始促進配列部分を含めておく、リガンド親和性複合タンパク質の取得方法である。
【0036】
(第10発明の構成)
上記課題を解決するための本願第10発明の構成は、前記第1発明〜第9発明のいずれかに係る転写用配列付加工程において、大量生産準備工程で増幅されたDNAフラグメントに対して、少なくともプロモーター配列を付加するPCRを行う、リガンド親和性複合タンパク質の取得方法である。
【0037】
(第11発明の構成)
上記課題を解決するための本願第11発明の構成は、前記第1発明〜第10発明のいずれかに係る単一種大量生産工程において、前記無細胞系を還元化しないままの条件で用いる、リガンド親和性複合タンパク質の取得方法である。
【0038】
(第12発明の構成)
上記課題を解決するための本願第12発明の構成は、前記第1発明〜第11発明のいずれかに係るキャラクタリゼーション工程が、複合タンパク質XpのELISA法によるリガンド親和性評価と、複合タンパク質Xp又はその遺伝子の配列決定と、形質転換体を用いて生産させた複合タンパク質XpのKd値の測定との内の少なくとも1項目を含む、リガンド親和性複合タンパク質の取得方法である。
【0039】
(第13発明の構成)
上記課題を解決するための本願第13発明の構成は、前記第1発明〜第12発明のいずれかに係るリガンド親和性複合タンパク質の取得方法を、ヒト又は非ヒト動物におけるリガンドXに対する抗体のスクリーニングに用いる、抗体スクリーニング方法である。
【0040】
(第14発明の構成)
上記課題を解決するための本願第14発明の構成は、第13発明に係る抗体スクリーニング方法を実施するために用いられるキットであって、少なくとも以下の構成要素を含んで構成される、抗体スクリーニング用キットである。
【0041】
11)第6発明に係る分散・懸濁用の液体と、磁性微粒子とのセット。
【0042】
12)第7発明に係る逆転写PCR用のプライマーセット。
【0043】
13)第8発明に係る2段階のPCR用のプライマーセット。
【0044】
14)第1発明の5)に係る無細胞系。
【0045】
(第15発明の構成)
上記課題を解決するための本願第15発明の構成は、第1発明及び第4発明〜第12発明のいずれかに係るリガンド親和性複合タンパク質の取得方法によって得られた抗体を用いて、非ヒト動物の疾患の予防、診断又は治療を行う、医療方法である。
【0046】
(第16発明の構成)
上記課題を解決するための本願第16発明の構成は、第1発明〜第3発明及び第5発明〜第12発明のいずれかに係るリガンド親和性複合タンパク質の取得方法によって得られた抗体を用いて、ヒトの疾患の予防、診断又は治療を行う、医療方法である。
【0047】
(第17発明の構成)
上記課題を解決するための本願第17発明の構成は、第1発明〜第12発明のいずれかに係るリガンド親和性複合タンパク質の取得方法によって得られた抗体を有効成分とし、その由来に応じてヒト又は非ヒト動物に適用されるものである、抗体医薬である。
【0048】
(第18発明の構成)
上記課題を解決するための本願第18発明の構成は、第1発明〜第3発明及び第5発明〜第12発明のいずれかに係るリガンド親和性複合タンパク質の取得方法によって得られた抗体を有効成分とし、その由来するヒト個体に対して適用されるものである、非免疫反応性抗体医薬である。
【0049】
(第19発明の構成)
上記課題を解決するための本願第19発明の構成は、第18発明に係る非免疫反応性抗体医薬を、非経口的な投与方法によって、その由来する前記ヒト個体の体内へ投与する、投薬方法である。
【発明の効果】
【0050】
(第1発明の効果)
第1発明においては、生産手段分離工程において、ヒト又は非ヒト動物たる検体を利用して、複合タンパク質Xpのそれぞれ専用の生産手段Xmを備えた抗体産生細胞の1個又は複数個の単細胞を調製するので、極めて多種類の複合タンパク質Xpの生産手段を簡易かつ迅速に一括して調製できる。
【0051】
上記により一括調製した生産手段Xmたる各mRNAは、各細胞単位で個別に生成し分離されているので、生産手段Xmを複合タンパク質Xpの種類ごとに厳密に分離して絞り込むことができ、後工程における生産手段Xm、複製手段Xc及び複合タンパク質Xpの異種間混在を完全に予防できる。しかも、これらの生産手段Xmは各複合タンパク質Xpの専用の生産手段として機能するので、タンパク質生産手段ライブラリーとして極めて好適である。更に、前記したハイブリドーマ技術のような、安定した生産手段の調製(ハイブリドーマ作成)上の困難がない。
【0052】
しかし、上記のタンパク質生産手段ライブラリーにおいては、各mRNAが極めて微量であるために生産規模が微少であって、大量生産の要求に応えることができない。そこで次の生産手段転換工程において、直接に生産手段としては機能しないが容易に大量複製することができる複製手段Xc(cDNA)へ一旦転換させるのである。そして、逆転写PCR法を利用する生産手段転換工程では、生産手段Xmを構成するmRNAの全部を転換させても良いが、mRNAの一部を選択的に転換させることにより、前記した生産手段分離工程において絞り込まれたタンパク質の種類ごとのmRNA(mRNAセット)の内から、更にmRNAの種別の絞り込みを行うことができる。
【0053】
こうして絞り込まれた種類のmRNAについて得た複製手段Xc(cDNA)を大量生産準備工程において大量複製する。この工程はPCR法を利用するので、前記したハイブリドーマ技術や非特許文献1に記載の技術のように、大量生産規模を整える(ハイブリドーマや形質転換体を培養・増殖する)上で面倒な作業と長時間を要すると言う困難がない。しかも前記の生産手段転換工程でmRNAの種別の絞り込みを行う場合には、使用するプライマーの種類を著しく低減させることができるため、副産物が非常に少なく、特異性の高いPCR産物を得ることができる。
【0054】
こうして大量複製された複製手段Xc(cDNA)に対して、転写用配列付加工程において、その無細胞系での生産手段Xm(mRNA)への再転換に必要な転写用配列を付加する。
【0055】
次に単一種大量生産工程において、上記のように転写用配列を付加された複製手段Xc(cDNA)を無細胞系へ投入することにより、cDNAに対応する複合タンパク質Xpの種類ごとに、タンパク質の大量生産を行うことができる。
【0056】
前記のように、生産手段分離工程において生産手段Xmが複合タンパク質Xpの種類ごとに厳密に分離され、必要に応じて生産手段転換工程ではmRNAの種別の絞り込みが行われる。従って、転写用配列を有効に機能し得る状態で付加することができると共に、大量生産準備工程では特異性の高いPCR産物が得られる。
【0057】
そのため、単一種大量生産工程においては、目的とする単一種の複合タンパク質に対して分子量、立体構造、リガンド親和性等の面で類似する複合タンパク質の副産物は殆ど生産されず、一方では目的とする単一種の複合タンパク質が有効に大量生産される。その結果、工程終了後の無細胞系からの目的タンパク質Xpの高純度な分離・回収を容易かつ迅速に行うことができる。
【0058】
従って、キャラクタリゼーション工程を行う場合においても、特性評価、スクリーニング、配列決定を迅速かつ正確に行うことが可能である。
【0059】
以上のように、本願第1発明によれば、実質的に無限とも言える多様性の制約の中で、互いに類似した極めて多種類のリガンド親和性タンパク質の中から特定された1種又は2種以上のタンパク質を迅速・大量にクローニングして高純度で取得することができる。こうして取得したリガンド親和性複合タンパク質においては、H鎖とL鎖が正しくペアリングしていることが確認されている。
【0060】
第1発明のリガンド親和性複合タンパク質の取得方法を極めて迅速に行い得る点に関しては、例えば次のような評価が可能である。即ち、このような極めて多種類のリガンド親和性複合タンパク質の中から機能的に特に優れた1種又は2種以上の複合タンパク質を取得する場合(例えばMabを取得する場合)、通常は3ケ月又はそれ以上の長期間を要する、と言われている。これに対して第1発明では、約5時間の逆転写PCRその他の工程と、約1時間の単一種大量生産工程(即ち、in vitro転写/翻訳)とを含め、クローニングから抗体遺伝子のキャラクタリゼーションまでを実質的に1日で完了することが可能である。
【0061】
(第2発明の効果)
ヒトを検体として第1発明の生産手段分離工程を行う場合、一定の病理症状からリガンドXに対する抗体を生成している患者(被検者)に協力を求め、なるべく非侵襲的な手段で抗体産生細胞たる前記単細胞を分離することができる。この場合、得られるリガンド親和性タンパク質はモノクローナル抗体であり、しかも当該被検者にとっては自身に由来する非免疫反応性抗体であるから、その患者の体内に投与しても免疫上の拒絶反応を起こさない。従って、患者に対する最適のオーダーメード医療として抗体投与治療を行うことができる。
【0062】
従来の技術に基づいても、このような抗体投与治療が可能ではあった。しかし、患者の容体等の問題から極めて迅速なワクチンの提供を要求される場合が多く、従来の技術では常に迅速性がネックとなって来た。第2発明によれば、このような迅速性の問題を解消できるので、医療上の価値は極めて大きい。
【0063】
(第3発明の効果)
ヒトを検体とする場合、なるべく非侵襲的で被検者に負担を与えない手段により抗体産生細胞を採取するため、好ましい一例として、被検者の末梢血を少量採取し、その末梢血からB細胞及び/又は形質細胞の単細胞を分離する方法を挙げることができる。
【0064】
(第4発明の効果)
非ヒト動物を検体として第1発明の生産手段分離工程を行う場合、その動物をリガンドXで免疫したもとで、その適宜な組織又は生体構成部分から抗体産生細胞の単細胞を分離すると言う方法が一般的であり、かつ便宜である。
【0065】
(第5発明の効果)
前記した第1発明におけるリガンド親和性複合タンパク質の好適な例として、各1対のH鎖及びL鎖からなる免疫グロブリン(抗体)、そのFab(抗原結合性フラグメント)、又はこれらに準ずる有効なリガンド親和性を示すタンパク質フラグメントを挙げることができる。
【0066】
(第6発明の効果)
生産手段分離工程においては、第6発明のように一定の分離用抗体と結合させた磁性微粒子を利用し、その分離用抗体と特異的に結合する抗体産生細胞を細胞懸濁液から分離することにより、抗体産生細胞を選択的かつ効率的に回収できると言うメリットがある。
【0067】
(第7発明の効果)
第1発明で述べたように、生産手段転換工程において、生産手段Xmを構成するmRNAの一部を複製手段Xcへ転換させることができる。
【0068】
即ち、生産手段転換工程における生産手段Xmは実際には複合タンパク質Xpの各フラグメントをコードしているmRNAの1セットである。そして、例えば抗体におけるFab(抗原結合性断片)のように、リガンド親和性複合タンパク質の本来の構成フラグメントの一部を欠くが、リガンド親和性は維持しているようなタンパク質があり得る。しかも、例えば抗体の場合には複合タンパク質Xpの各フラグメントにおいて、各クラスや各アイソタイプ別の相違がある。
【0069】
そこで、mRNAの一部を複製手段Xcへ転換させる場合の一つの有利な実施形態として、逆転写PCRに用いるプライマーの設計により、特定のクラスやアイソタイプの分類カテゴリーに属する複合タンパク質XpをコードするmRNAのみを複製手段Xcへ転換させると言う実施形態が例示される。あるいはFabのように、複合タンパク質Xpを構成する特定のフラグメントをコードするmRNAのみを複製手段Xcへ転換させると言う実施形態が例示される。
【0070】
第7発明によって、逆転写PCRに用いるプライマーの種類を著しく低減させることができ、従って生成されるcDNAの種類も著しく低減させることができるので、その後のPCR法による大量生産準備工程を、特異性の高い有利な条件で行うことができる。
【0071】
(第8発明の効果)
第8発明も、技術的な意味合いの大きな改良点の一つである。即ち、一般論として、PCRにおいては基本的にプライマーダイマーや非特異的アニーリング等による副産物の生成は避けられないため、1段階のPCRによって複製手段XcたるcDNAを増幅することは、実質的に困難である。
【0072】
例えば、一旦生成した副産物たるDNAは、標的cDNAよりも鎖長が短い場合には標的cDNAよりも増幅し易い、と言う問題がある。しかも極めて微量な鋳型DNAから出発した場合には、このような負の効果が顕著である。
【0073】
この困難に対する通常の対処策として、入れ子構造になった2組のPCRプライマーセットを用いる、ネスティッドPCRと呼ばれる公知の2段階PCR法がある。しかし、この方法では、増幅しようとするcDNAの種類ごとに各2組のPCRプライマーセットを用いる必要があるため、抗体遺伝子のように多様性のある場合に応用するのは容易ではない。
【0074】
例えばヒトのB細胞の抗体遺伝子におけるL鎖の可変領域を増幅させる場合、10本の縮重が見られるプライマーを1段階用い、更に2段階目でもそれと同数のプライマーを用いなければならない。即ち、プライマーの数が多いだけでなく、多くの組み合わせに由来する副産物の生成が避けられないため、目的のDNA以外にも多数の増幅バンドが検出され、その後のタンパク質生産工程等に大きな困難をもたらす。即ち、例えば無細胞系における目的タンパク質の合成量が顕著に低下するし、あるいはタンパク質の配列決定のための大腸菌等へのクローニングが、著しく手間と時間のかかるものとなる。
【0075】
第8発明によれば、第1段階PCRでは複製手段Xcを構成する各cDNAに対応する各種類のプライマーを用いるものの、それらのプライマーに設けた共通の付加配列を利用して、第2段階PCRでは単一種類のプライマーによって全ての複製手段Xcを構成する各cDNAのみを一括して増幅することができる。従って、複製手段Xcが抗体遺伝子のように多様性のある場合にも、容易に応用することができる。
【0076】
第2段階PCRのように単一種類のプライマーを用いた場合でも、一定量のプライマーダイマーは生成する。しかし、この場合のプライマーダイマーは、異種プライマーの会合によるダイマーとは異なり、熱で解離させて冷却すると、プライマーの5’末端と3’末端が会合する構造(パンハンドル構造)を取って、新たに会合できなくなる。そのため、増幅能力が実質的に失われる。
【0077】
従って、第8発明によれば、無細胞系における目的タンパク質の合成量の低下を有効に防止でき、あるいはタンパク質の配列決定のための大腸菌等へのクローニングが著しく簡易化される。
【0078】
(第9発明の効果)
上記した第8発明の第1段階PCRにおいて各プライマーに付加配列を設ける点を利用し、その付加配列中に適宜な(第1発明の単一種大量生産工程で用いる無細胞系の由来生物を考慮した)翻訳開始促進配列部分を含めておくことができる。その場合、後の無細胞系での単一種大量生産工程において目的タンパク質の翻訳効率を向上させることができる。
【0079】
(第10発明の効果)
転写用配列付加工程において、大量生産準備工程で増幅されたDNAフラグメントに対して、PCRにより少なくともプロモーター配列を付加しておくことができる。そのことにより、無細胞系におけるDNAからmRNAへの再転換を準備できる。
【0080】
通常、抗体遺伝子のように多様性のある鋳型遺伝子に対して、PCRでこのような各種配列を付加しても、それらの付加配列の効果を十分に発揮させることは困難である。しかし、前記した第1発明、第7発明、第8発明等により、このようなPCRに供する鋳型遺伝子が十分に絞り込まれているため、これらの付加配列の効果が十分に発揮される。
【0081】
(第11発明の効果)
従来、無細胞系(in vitro転写/翻訳共役系)は還元化した状態下で利用する必要があるとの見解があり、そのためDTT(ジチオスレイトール)等の高価な還元化剤を添加することが多かった。しかし、本願発明者は、酸化条件下の in
vitro 転写/翻訳共役系においても遜色なく目的タンパク質を生産できることを既に確認している。従って、無細胞系を還元化しないままの条件で用いることにより、コストダウンを実現することができる。
【0082】
(第12発明の効果)
前記した第1発明におけるキャラクタリゼーション工程の内容は必ずしも限定されない。例えば、複合タンパク質XpのELISA法によるリガンド親和性評価、複合タンパク質Xp又はその遺伝子の配列決定、あるいは複合タンパク質XpのKd値の測定等を好ましく行うことができる。Kd値を測定する場合には、複合タンパク質Xpの遺伝子を導入した形質転換体を用いて複合タンパク質Xpを生産させ、この複合タンパク質Xpについて測定することが好ましい。
【0083】
そして、単一種大量生産工程では、複合タンパク質Xpが非常に副産物の少ない状態で大量に取得されているため、これらの評価や配列決定、あるいはKd値の測定を迅速かつ正確に行うことができる。
【0084】
(第13発明の効果)
リガンド親和性複合タンパク質の取得方法は各種の実験目的、研究目的、医療目的等に利用することができるが、重要な具体的用途の一つとして、ヒト又は非ヒト動物におけるリガンドXに対する抗体のスクリーニングに用いることが例示される。
【0085】
(第14発明の効果)
上記の第13発明に係る抗体スクリーニング方法を行うに当たり、第14発明に係る抗体スクリーニング用キットを好ましく利用することができる。この抗体スクリーニング用キットの各構成要素は、一部のプライマーの構成を除き、個々には特段に目新しいものではない。しかし、第14発明における11)〜14)の各構成要素を必須のコンポーネントとするキットの全体は、上記のリガンド親和性複合タンパク質の取得方法が着想されない限り成り立ち得ない内容であり、今までに、提案されたことも、市場に提供されたこともない。
【0086】
(第15発明及び第16発明の効果)
ヒトの医療あるいは非ヒト動物(例えば、ペット動物や家畜)の医療において、Mabを用いる点で第15発明又は第16発明の医療方法と同様の医療方法は、例えばハイブリドーマ技術等の従来技術に基づいても成立可能である。しかしながら、従来技術に基づく場合は、極めて多種類のMabの内から最適のMabを迅速・大量にスクリーニング及び製造することが困難である。
【0087】
第15発明及び第16発明に係る医療方法によれば、感染症における抗原や各種のガンにおけるガン細胞等に対して最適のMabを、迅速・大量にスクリーニング及び製造することができる。従って、医療現場における実用性がある。この点は、特にヒトの医療現場における一刻を争う救命医療等において、極めて重要な意義を持つ。
【0088】
(第17発明の効果)
第17発明に係る抗体医薬と機能的に同等な抗体医薬は、従来技術に基づいても供給可能である。しかし、従来技術に基づいて供給される抗体医薬は、迅速に供給することが困難であり、又、製造コストの面から安価に供給することも困難である。第17発明に係る抗体医薬は、迅速・大量にかつ安価に供給することができる。
【0089】
(第18発明及び第19発明の効果)
第18発明に係る非免疫反応性抗体医薬は、上記した第17発明の効果に加え、その由来するヒト個体に非経口的に投与しても、副作用としての免疫上の拒絶反応を誘発しない。従って、第19発明のような非経口的な投薬方法を安全に行うことが可能となる。更にその投薬の際、免疫抑制剤の併用を不要とすることができる。従って、患者の免疫能力低下に起因する治療上のリスクや患者の負担あるいは苦痛を軽減でき、更に、免疫抑制剤の節約により治療コストも低減させることができる。
【発明を実施するための最良の形態】
【0090】
次に、本願の第1発明〜第19発明の実施形態を、その最良の形態を含めて説明する。以下において単に「本発明」と言うときは、上記の各発明の全部又は一部を一括して指している。
【0091】
〔リガンド親和性複合タンパク質の取得方法〕
本発明に係るリガンド親和性複合タンパク質の取得方法は、任意に特定されたリガンドXに対して選択的な親和性を示す極めて多種類の複合タンパク質Xpの中から、無作為にあるいは特性評価を通じて選ばれる1種又は2種以上の複合タンパク質を取得する方法である。ここにおいて、「2種以上」とは、数種類であり得るし、数十種類であり得るし、更に数百種類又はそれ以上であり得る。
【0092】
複合タンパク質Xpの種類は限定されないが、例えば、各1対のH鎖及びL鎖からなる免疫グロブリン(抗体)であり得るし、その特定部分のフラグメントたるFabであり得るし、又は免疫グロブリンやFabに準ずるリガンド親和性を示す任意のタンパク質フラグメントであり得る。一方、リガンドXの種類は限定されず、抗原たり得る各種の無機化合物や有機化合物、タンパク質、ウイルス、微生物等を制限なく含む。
【0093】
リガンド親和性複合タンパク質の取得方法は、少なくとも後述する1)生産手段分離工程、2)生産手段転換工程、3)大量生産準備工程、4)転写用配列付加工程及び5)単一種大量生産工程を含む。更に必要に応じて、後述する6)キャラクタリゼーション工程を含むことができる。更に、上記1)工程に対する任意の前工程を含むことができ、上記6)工程に対する任意の後工程を含むことができ、又は1)〜6)工程中の任意の工程間に任意の中間工程を介在させることもできる。
【0094】
〔生産手段分離工程〕
生産手段分離工程とは、極めて多種類にわたる前記複合タンパク質Xpのそれぞれ専用の生産手段(mRNA)であって、容易に大量複製できる複製手段Xc(cDNA)への転換が可能な生産手段Xmを備えた抗体産生細胞の1個又は複数個の単細胞を、ヒト又は非ヒト動物たる検体の適宜な組織又は生体構成部分から個別に分離することにより、目的タンパク質の種類ごとに生産手段Xmを厳密に分離して、以下の各工程における生産手段Xm、複製手段Xc及び複合タンパク質Xpの異種間混在を予め防止する工程である。この工程により生産手段Xmが複合タンパク質Xpの種類ごとに厳密に分離される。
【0095】
ここにおいて、生産手段Xmとは、具体的にはmRNAであり、より具体的には、複合タンパク質Xpを構成する各タンパク質フラグメントをコードする各mRNAの1セットである。
【0096】
この生産手段分離工程において、検体がヒトである場合、その体内に人為的にリガンドXに対する抗体を生成させることは許されない。従って一定の疾患状態にある患者、即ち抗原たるリガンドXに対する抗体を既に体内に生成している生理状態にあると推定される被検者を選択し、その被検者の承諾のもとに生産手段分離工程を行う必要がある。具体的には、当該被検者における適宜な組織又は生体構成部分を社会通念上許容される非侵襲的手段によって採取し、この組織又は生体構成部分から前記の1個又は複数個の単細胞を分離する。
【0097】
上記のような患者の種類は必ずしも限定されないが、例えばリガンドXに感染した感染症患者、リガンドとしてのガン細胞に対する抗体を生成している各種のガン患者等を例示することができる。患者からは、医療倫理上及び関連法規上から採取することを許される組織又は生体構成部分に限り、しかも社会通念上許容される非侵襲的な手段によってのみ、抗体産生細胞を含む組織又は生体構成部分を採取することができる。「組織又は生体構成部分」には、血液その他の体液も含まれる。
【0098】
このような組織又は生体構成部分の採取と、そこからの抗体産生細胞の分離の具体的な一事例として、被検者から末梢血を採取し、この末梢血から抗体産生細胞たるB細胞及び/又は形質細胞の単細胞を分離する場合が挙げられる。
【0099】
又、他の具体的な一事例として、別途の治療上の目的から抗体産生細胞を含む組織又は生体構成部分の摘出手術や分離処置等を受けた患者において、その者の了解を得て、当該組織又は生体構成部分(例えば、脾臓、体液、骨髄等)を利用する場合が挙げられる。
【0100】
この生産手段分離工程において検体が非ヒト動物である場合、この動物をリガンドXで免疫したもとで、当該動物の適宜な組織又は生体構成部分から抗体産生細胞である1個又は複数個の単細胞を分離することができる。なお、既にリガンドXに対する抗体を生成している非ヒト動物においては、リガンドXで人為的に免疫する必要はない。
【0101】
非ヒト動物の種類は、少なくとも免疫反応による抗体の生成が認められる非ヒト動物である限りにおいて、限定されない。具体的には、入手及び利用が容易な各種の実験動物や、抗体医療の必要性が考えられる家畜又はペット動物等を好ましく例示することができる。より具体的には、例えばマウス、ラット、ウサギ、サル、牛、馬、豚、犬、猫等の哺乳動物が考えられるが、哺乳動物以外の非ヒト動物も対象となり得る。
【0102】
非ヒト動物を対象とする生産手段分離工程においては、動物愛護関連の法規又は倫理を遵守すると言う条件のもとで、抗体産生細胞、これを含む組織又は生体構成部分の種類、及びその採取方法は限定されない。抗体産生細胞と、これを含む組織又は生体構成部分としては、検体がヒトである場合においては、抗体産生細胞としてのB細胞、形質細胞を含む骨髄組織等を好ましく例示することができる。
【0103】
生産手段分離工程において、採取した組織又は生体構成部分から抗体産生細胞の1個又は複数個の単細胞を分離して取得する方法は限定されない。好ましくは、これらの組織又は生体構成部分を液体(好ましくは、適当な組成の液体培地)中に投入して抗体産生細胞を分散・懸濁させ、一定の分離用抗体と結合させた磁性微粒子と共にインキュベートした後、分離用抗体を介して抗体産生細胞に結合した磁性微粒子をマグネットで回収することにより、前記懸濁液から目的とする抗体産生細胞の単細胞を分離する、と言う方法がある。
【0104】
上記の分離用抗体としては、例えばマウスの場合における抗マウス CD19 抗体等、任意の抗体を利用することができる。
【0105】
〔生産手段転換工程〕
生産手段転換工程とは、単離したそれぞれの抗体産生細胞に含まれる生産手段Xm、即ち、複合タンパク質Xpの各タンパク質フラグメントをコードするmRNAのセットを細胞外へ溶出させ、逆転写PCR法により、これらのmRNAの全部又は一部を複製手段Xc(cDNA)へ転換させる工程を言う。
【0106】
ここにおいて、mRNAの一部をcDNAへ転換させる場合の一形態として、特定の分類カテゴリーに属する複合タンパク質XpをコードするmRNAのみをcDNAへ転換させる形態が挙げられる。例えば抗体である複合タンパク質Xpの内、特定のクラスやアイソタイプに属するタンパク質をコードするmRNAのみをcDNAへ転換させる場合が例示される。
【0107】
mRNAの一部をcDNAへ転換させる場合の他の一形態として、複合タンパク質Xpを構成する特定のフラグメント(例えばFab)をコードするmRNAのみをcDNAへ転換させる形態が挙げられる。
【0108】
生産手段Xmを構成するmRNAのセットの全部をcDNAへ転換させたり、上記のような各種の形態でその一部をcDNAへ転換させたりする逆転写PCRは、使用するプライマーの選択により、容易に選択して行うことができる。
【0109】
なお、抗体産生細胞に含まれる生産手段Xmを細胞外へ溶出させる手段は限定されないが、公知の適当な手段、例えばB細胞を界面活性剤や低浸透圧の液等で破壊する方法等を任意に採用することができる。
【0110】
〔大量生産準備工程〕
大量生産準備工程とは、上記した逆転写PCR法により得られた複製手段Xc(cDNA)をPCR法で大量複製して、タンパク質の大量生産能力を準備する工程を言う。
【0111】
この工程におけるPCR法の内容、実施条件あるいは使用するプライマーの種類は特段に限定されないが、2段階のPCRによって行うことが好ましく、特に次の内容の2段階PCRによって行うことが、前記「第8発明の作用・効果」欄で記載した理由から、好ましい。
【0112】
(第1段階PCR)
複製手段Xcを構成する全ての種類のcDNAに対応する各種類のプライマーにおいて、それらの5’末端側に同一の付加配列を設けて増幅を行う。更に、この付加配列中には、次の単一種大量生産工程において有利に働く任意の塩基配列、例えば後述する無細胞系の由来生物を考慮した翻訳開始促進配列配列を含めておくことができる。
【0113】
翻訳開始促進配列配列としては、例えばrbs(リボソーム結合サイト)や、無細胞系の由来生物が大腸菌である場合のSD(シャイン・ダルガーノ)配列、無細胞系の由来生物が真核生物である場合のKozak配列、ウイルスに由来する5’UTR配列等が挙げられる。
【0114】
この第1段階PCRの実施条件は限定されないが、例えば以下のプログラムで行うことが好ましい。
94°C−3分;
94°C−30秒/50°C−45秒/72°C−45秒を30サイクル;
72°C−7分;
(第2段階PCR)
上記の第1段階PCRで増幅された全ての種類のDNAフラグメントに対して、前記付加配列に相補的な配列部分を備えた単一種類のプライマーを用いて増幅を行う。
【0115】
この第2段階PCRの実施条件は限定されないが、例えば以下のプログラムで行うことが好ましい。
94°C−3分;
96°C−5秒/45°C−10秒/72°C−45秒を65サイクル;
72°C−7分
〔転写用配列付加工程〕
転写用配列付加工程とは、大量複製された前記複製手段Xc(cDNA)に対して、その無細胞系での生産手段Xm(mRNA)への再転換に必要な転写用配列を付加する工程である。転写用配列としては、限定はされないが、少なくともプロモーター配列を含むことが好ましい。プロモーター配列に加えてターミネーター配列も含むことが、更に好ましい。
【0116】
この工程を行う方法は必ずしも限定されないが、好ましくは、大量生産準備工程で増幅されたDNAフラグメントに対し、PCR、例えばオーバーラッピングPCRによって、前記の転写用配列を付加することにより行われる。
【0117】
〔単一種大量生産工程〕
単一種大量生産工程とは、上記の工程により転写用配列を付加された複製手段Xc(DNA)を、少なくとも複製手段Xcの生産手段Xm(mRNA)への再転換を行う要素と、生産手段Xmに基づきタンパク質の生産を行う要素とを備えた無細胞系へ投入することにより、複合タンパク質Xpを、又は前記生産手段転換工程で規定した一部のmRNAに対応する複合タンパク質Xpを、1種類ごとに大量生産する工程を言う。
【0118】
この無細胞系において、DNAをmRNAへ転換させる要素としては転写酵素が挙げられる。転写酵素としては、T7RNAポリメラーゼ、T3RNAポリメラーゼ、 SP6RNAポリメラーゼ、大腸菌等の微生物由来のRNAポリメラーゼ等の利用が可能である。mRNAに基づきタンパク質の生産を行う要素としてはリボソーム、各種のdNTP等が例示される。このような無細胞系は in vitro 転写/翻訳共役系として公知であって、例えば大腸菌由来、小麦胚芽由来、ウサギ網状赤血球由来等のキットが市販されているので、わざわざ調製しなくても、適宜にこれらを利用することができる。
【0119】
in vitro 転写/翻訳共役系としては、高価なDTT等を添加して還元状態としたものを使用することもできるが、これを添加しない酸化状態の in vitro 転写/翻訳共役系も遜色のない機能を発揮するので、これを使用する方が、安価で有利である。
【0120】
〔キャラクタリゼーション工程〕
キャラクタリゼーション工程とは、上記の各工程により生産された複合タンパク質Xpについて、特性評価を行ったり、優れた特性を持つ複合タンパク質Xpをスクリーニングしたり、あるいは、これらの複合タンパク質Xpの配列決定を行ったりする工程を言う。
【0121】
この工程は、単なるリガンド親和性複合タンパク質の取得方法としての観点から見れば、本来的に不可欠の工程ではない。しかし、リガンド親和性複合タンパク質の取得方法が後述の抗体のスクリーニング、抗体医療、抗体医薬等の発明の前提であると言う観点からは、不可欠の工程あるいは重要な工程である。
【0122】
キャラクタリゼーション工程においては、好ましくは、複合タンパク質XpのELISA法によるリガンド親和性評価と、複合タンパク質Xp又はその遺伝子の配列決定と、形質転換体を用いて生産させた複合タンパク質XpのKd値の測定との内の少なくとも1項目を含む。
【0123】
〔抗体スクリーニング方法〕
本発明に係る抗体スクリーニング方法とは、上記したリガンド親和性複合タンパク質の取得方法を、ヒト又は非ヒト動物におけるリガンドXに対する抗体のスクリーニングに用いる方法である。
【0124】
即ち、リガンド親和性複合タンパク質の取得方法における前記1)の生産手段分離工程〜前記5)の単一種大量生産工程によって得られた2種以上のリガンド親和性複合タンパク質の中から、前記6)のキャラクタリゼーション工程を通じて、特に優れた抗リガンドX抗体をスクリーニングする方法である。
【0125】
従って、従来の各種の抗体スクリーニング方法に比較して、最適の抗リガンドX抗体を容易、迅速かつ大量に得ることができる。
【0126】
〔抗体スクリーニング用キット〕
本発明に係る抗体スクリーニング用キットは、上記の抗体スクリーニング方法を実施するために用いられるキットであって、少なくとも以下の構成要素を含んで構成されるものである。
11)前記第6発明に係る分散・懸濁用の液体と、磁性微粒子とのセット。
12)前記第7発明に係る逆転写PCR用のプライマーセット。
13)前記第8発明に係る2段階のPCR用のプライマーセット。
14)前記第1発明の5)に規定する無細胞系。
【0127】
上記の11)〜14)の各構成要素は、この種の商品として通常に採用される剤型あるいは包装形態において提供することができる。例えば12)又は13)のプライマーセットは、保存性や使用上の便宜等を考慮して、乾燥品等の形態で提供することができる。
【0128】
又、抗体スクリーニング用キットにおいては、上記の11)〜14)の各構成要素の他に、例えば転写用配列付加工程においてプロモーター配列やターミネーター配列の付加のために利用するベクター、任意の目的で使用する緩衝液やpH調整液等の任意の有益な付加的構成要素を含むことができる。
【0129】
〔医療方法〕
本発明に係る医療方法には、非ヒト動物を検体として得られたリガンド親和性複合タンパク質(抗体)を用いて非ヒト動物の疾患の予防、診断又は治療を行う医療方法と、ヒトを検体として得られたリガンド親和性複合タンパク質(抗体)を用いてヒトの疾患の予防、診断又は治療を行う医療方法とを含む。
【0130】
これらの医療方法において、非ヒト動物又はヒトの疾患の種類は限定されないが、リガンドXによる感染症、ガン細胞がリガンドXとなっている各種のガン等を代表的に例示することができる。
【0131】
〔抗体医薬〕
本発明に係る抗体医薬は、前記したリガンド親和性複合タンパク質の取得方法によって得られた抗体を有効成分とするものであって、その抗体の由来(検体とされたヒト又は非ヒト動物)に応じて、ヒト又は非ヒト動物に適用されるものである。この抗体医薬の投与方法は限定されないが、非経口的な方法によることが好ましい。
【0132】
抗体医薬の剤型は限定されず、例えば凍結乾燥剤、粉末製剤、pHを調整された緩衝液による溶液製剤、注射用マイクロカプセル剤等として提供できる。これらの抗体医薬は、この種の抗体医薬において含有されることがある任意の有益な成分(例えば、増量剤、pH調整剤、保存安定剤等)を含むことができる。
【0133】
〔非免疫反応性抗体医薬及び投薬方法〕
本発明に係る非免疫反応性抗体医薬は、ヒトを検体として得られたリガンド親和性複合タンパク質(抗体)を有効成分とし、その由来するヒト個体に対して投薬されるものである。この非免疫反応性抗体医薬の剤型や含有成分については、上記の抗体医薬の場合と同様の構成が可能である。
【0134】
非免疫反応性抗体医薬の投薬方法は非経口的な投与方法による。非経口的な投与方法の種類は限定されないが、例えば静脈注射、皮下注射、筋肉注射等を例示できる。他にも、近年注目されている各種のドラッグデリバリー方法、例えば、磁性微粒子に抗体分子を結合させて磁力により効果的にガン細胞周辺に集め、最終的に抗体によりターゲット細胞をとらえると言う方法等の投与方法も採用可能である。
【実施例】
【0135】
以下に本発明の実施例を説明する。本発明の技術的範囲がこれらの実施例によって限定されないことは、もち論である。
【0136】
〔マウスを検体とする実施例1〕
(実施例1の概要)
実施例1の概略を述べる。まず、抗原である CBP40(他の任意の抗原を用いても良い)で免疫したマウスの脾臓から、抗マウス CD19 抗体と結合した磁性マイクロビースを用いて、B細胞を単離した。単離したB細胞のL鎖(Lc)遺伝子とH鎖のFd部(VHドメインとCH1ドメイン)遺伝子とを、別々に逆転写PCR(RT−PCR)によって増幅した。
【0137】
次いで、 in vitro 合成に適したDNA断片を得るため、これら2種類の遺伝子に対して、オーバーラッピングPCRによって T7 プロモーター、リボソーム結合部位( rbs)及び T7 ターミネーターの各配列を付加した。このH鎖遺伝子とL鎖遺伝子の全長DNA断片の対を同じ反応用試験管に投入し、 in vitro で転写及び翻訳を行わせてFab断片を得た後,ELISA法によるスクリーニングを行った。このような一連のプロセスにより、種々の細胞と組織から抗原特異的なモノクローナル抗体を得ることができる。
【0138】
以上の全体的な実験操作(工程)のフローを図1に概念化して示す。
【0139】
(実施例1−1:B細胞の単離)
BALA/c 系マウスを50μgの CBP40(Calcium Binding Protein 40)で免疫した。5週間後にその脾臓を摘出し、F-10 HAM栄養培地(英国 Sigma-Aldrich社製)中で細胞単位に分散させた。次に、4°Cで1000 r.p.m. の遠心分離を3分間行って脾細胞を集め、 PBS(phosphate buffered saline )で一回洗浄した。
【0140】
脾細胞の懸濁液を、抗マウス CD19 抗体と結合した磁性粒子である CD19MACS
マイクロビーズ(Miltenyi Biotec 社)と共にインキュベートし、 CD19 を発現しているB細胞をマイクロビーズに吸着させた。次いでマグネットにより細胞を集めて上澄みを取り除いた。更に PBS(2.5% BSA)で洗浄し、次いでマグネットにより細胞を集め、マイクロビーズに結合したB細胞を回収した。
【0141】
単離したB細胞を顕微鏡(日本国オリンパス社製 LX70 )下でカウントし、希釈してPCRチューブ1本当たり1個の細胞を収容させ、直ちにcDNA合成に供した。
【0142】
(実施例1−2:単細胞逆転写PCR−cDNAの合成)
各細胞のmRNAを鋳型にして、逆転写用キットを用いてcDNA合成を行った。ここでは Invitrogen 社の逆転写PCR用 SUPERSCRIPT First Strand 合成システムを用いたが、他のメーカーのものでも構わない。
【0143】
まず PBSバッファーで平均2細胞/μLになるようにB細胞を調製し、1.5μLの DEPC 処理( RNase失活)滅菌水を加え、そこに、プロコールに従い全量5μLの反応液〔0.25μL dNTP,0.50μL 10×RTバッファー:200mM トリス−塩酸(pH8.4),500mM KCl,1.0μL 25mM MgCl2 ,0.5μL 0.1M DTT,0.25μLRnaseインヒビター, 0.25μL Reverse trriptase:SUPERSCRIPT II RT ,0.25μL 2μM 各プライマー〕になるように液を加え、42°Cで90分処理した後、70°Cで15分処理した。
【0144】
この合成反応は、目的とする全てのcDNA、即ちB細胞のL鎖遺伝子(κ鎖)とH鎖(γ鎖)のFd部(VHドメインとCH1ドメイン)遺伝子とに対応する全てのcDNAを得るために必要な下記1)〜5)の各プライマーをそれぞれ2μM含む混合液を用いて行った。又、コンタミネーションをチェックするため、B細胞が存在しない系でのコントロールの合成反応を別途に行った。
1)プライマー CK−J1(表1及び配列表の配列番号1に示す)
2)プライマー CH−IgG1-1(表1及び配列表の配列番号2に示す)
3)プライマー CH−IgG2A-1 (表1及び配列表の配列番号3に示す)
4)プライマー CH−IgG2B-1 (表1及び配列表の配列番号4に示す)
5)プライマー CH−IgG3-1(表1及び配列表の配列番号5に示す)
なお、上記1)〜5)の各プライマー及び後述の全てのプライマーの塩基配列を、表/図と配列表とにそれぞれ示すが、万が一、表/図に示す塩基配列と配列表に示す塩基配列とに不一致が存在した場合には、表/図に示す塩基配列が正しい配列である。
【0145】
【表1】
(実施例1−3:DNAの増幅−第1段階のPCR)
上記により得られたcDNAの混合物を第1段階のPCR反応のテンプレートとして使用した。その際、発現されるマウスIg遺伝子の95%はL鎖がκアイソタイプである(Honjo and Alt, 1995 )ため、λアイソタイプのL鎖は無視することにし、抗体L鎖のκアイソタイププライマーのみを用いた。又、高い抗原結合能を持つ抗体を得ると言う目的から、H鎖についてはIgG(イムノグロブリンG)のみを標的とした。第1段階のPCRで用いた下記の縮重プライマーは Kabatのデータベース( http://immuno.bme.nwu.edu)に基づいて設計した。
【0146】
このような設計によりプライマー数を低減させると、好ましくない増幅を減少させるのに有益であろうと言うことを考慮している。なお、これらの縮重プライマーは鋳型として十分に理想的とは言えないので、増幅反応液には、下記のように、3'エクソヌクレアーゼ活性を欠き、かつ他のDNAポリメラーゼに比較して縮重プライマーと鋳型とのミスマッチに対して大きな耐性をもつ TaqDNAポリメラーゼ(Wang et al., 2000 )を用いている。
【0147】
L鎖遺伝子は、いずれも縮重プライマーである6)プライマー VK− M2 (表1及び配列表の配列番号6に示す)、及び7)プライマー CK−His6M2(表1及び配列表の配列番号7に示す)を用いて増幅した。
【0148】
H鎖遺伝子は、縮重プライマーである8)プライマー VH−M2(表1及び配列表の配列番号8に示す)と9)プライマーV'H−M2(表1及び配列表の配列番号9に示す)との混合物と、定常領域プライマーである10)プライマー CH−IgG1-M2
(表1及び配列表の配列番号10に示す)、11)プライマー CH−IgG2A-M2(表1及び配列表の配列番号11に示す)、12)プライマー CH−IgG2B-M2(表1及び配列表の配列番号12に示す)及び13)プライマー CH−Ig G3-M2(表1及び配列表の配列番号13に示す)の混合物とを用いて増幅した。
【0149】
これらのL鎖遺伝子とH鎖遺伝子との増幅反応は、0.25単位の TaqDNAポリメラーゼ(日本国 宝酒造社製)、0.2mMの各dNTP、0.05μMの各プライマー、0.5μLの前記cDNA混合物及びリアクションバッファーからなる5μLの反応液中で行った。PCRは以下のプログラムで行った。
94°C−3分;
94°C−30秒/50°C−45秒/72°C−45秒を30サイクル;
72°C−7分;
(実施例1−4:DNAの増幅−第2段階のPCR)
第1段階のPCR産物の10分の1量を、単一種の14)プライマー SP2(表1及び配列表の配列番号14に示す)を用いた第2段階のPCRにおいて増幅した。
【0150】
この増幅反応は、0.25単位の Pfu Turbo(商標)DNAポリメラーゼ(Stratagene社製)、0.125mMの各dNTP、0.5μMのプライマー SP2及びリアクションバッファーからなる5μLの反応液中で行った。PCRは以下のプログラムで行った。
94°C−3分;
96°C−5秒/45°C−10秒/72°C−45秒を65サイクル;
72°C−7分
図2に、H鎖遺伝子(「Hc」で示す)及びκタイプL鎖遺伝子(「 Lc 」で示す)の増幅結果を示す。これらの結果は、PCR産物2μLを1.0%アガロースゲル上で解析したものである。 EcoT14Iで消化したλ−DNAマーカーを図の左側に示している。
【0151】
図2に示すように、これらの反応条件によって単一のB細胞に由来するFab断片のL鎖とH鎖をコードする遺伝子が別々に増幅され、しかも副産物の生成は認められない。L鎖PCR産物は、単一B細胞の72検体中、39検体から得られた(39/72=54%)。H鎖遺伝子に関しては、特異的なPCR産物は、単一B細胞の72検体中、28検体から得られた(28/72=39%)。全体として、L鎖とH鎖がペアリングしたFabは72検体中の20検体から得られ、回収率は28%であった。
【0152】
(実施例1−5:PCRの全体的な説明)
上記の全てのPCR反応は GeneAmp(登録商標)PCRシステム(PE Applied Biosystems 社製)を用いて行った。PCR産物のシークエンスを確認するため、幾つかのL鎖遺伝子とH鎖遺伝子を EcoRVで消化した pBluescriptSKII+ ベクター(Stratagene社製)にクローニングした。クローニング領域のシークエンシングにはThermo SequenaseII(商標)dye terminator cycle sequencing premix kit(Amersham Pharmacia Biotech社製)と、 ABIPRISM (商標)310 Genetic
Analyzerとを、メーカー推奨のプロトコールに従って用いた。
【0153】
(実施例1−6:オーバーラッピングPCR)
実施例6において、下記のように、 T7 プロモーター、 rbs及び T7 ターミネーターを含む2つの断片をオーバーラッピングPCRによりそれぞれ増幅されたL鎖遺伝子及びH鎖遺伝子と結合させた。オーバーラッピングPCR産物の解析結果を図3に示す。図3は、オーバーラッピングPCR産物2μLを1.0%アガロースゲル上で解析した結果であり、L鎖遺伝子がレーン1〜3に、H鎖遺伝子がレーン4〜6に、それぞれ示されている。
【0154】
T7 プロモーターおよびSD配列を増幅するためのプライマーセットとして、配列表の配列番号15に示す15)プライマーIn-F(5'-CCAATACGCAAACCGCCTCTCC-3')と、配列表の配列番号16に示す16)プライマー T7PR-SD(5'-ATAATCTCCTTCTTATCTAATAACAAAATTATTTCTAGAGGGAAACCG-3')とを用い、pRSET-B ベクター(Invitrogen社製)から T7 プロモーターとリボソーム結合部位を含む断片を増幅した。
【0155】
又、 T7 ターミネーターを増幅するためのプライマーセットとして、配列表の配列番号17に示す17)プライマーT7T-NFM2(5'-TAATCAATAATCTCCTTCTTATCTAATTCCGGCTGCTAACAAAGCCCG-3 )と、配列表の配列番号18に示す18)プライマーIn-R( 5'-TACAGGGCGCGTCCCATTCG-3' )とを用い、pRSET-B ベクターから転写ターミネーター断片を増幅した。
【0156】
オーバーラッピングPCRによって、上記2種類のDNA断片の間に第2ラウンドのPCRに係るL鎖あるいはH鎖を挿入し、in vitroタンパク質合成に適した全長DNA断片を生成させた。オーバーラッピングPCRは、0.75単位の Ex taq DNAポリメラーゼ(宝酒造社製)、0.2mMの各dNTP、それぞれ0.15μLの各DNA断片、及び反応バッファーからなる15μLの反応液中で次のPCRプログラムに従って行った。
94°C−5分;
94°C−30秒/48°C−20秒/72°C−90秒を10サイクル;
その後、15μLのPCR溶液を、メーカー推奨の反応バッファー( Ex taq
DNAポリメラーゼ1.5単位、dNTP各0.2mM、プライマー In-F 及び In-R 各0.5mM)を用いて30μLに調製し、以下のPCRプログラムにより増幅した。
94°C−5分;
94°C−30秒/55°C−30秒/72°C−90秒を25サイクル;
72°C−7分
(実施例1−7:in vitro転写/翻訳共役系)
上記の30μLの反応液(同一の細胞に由来するH鎖とL鎖遺伝子とのオーバーラッピングPCR混合物)のうち、3μLを用いて in vitro 転写/翻訳共役反応を行った。この反応は Jiangらの報告(Jiang et al., 2001)に従い、次の要素を含む in vitro 転写/翻訳系を用いた。
トリス酢酸(pH7.4) 56.4mM;
ATP 1.2mM;
GTP、CTP及びUTP 各1.0mM;
クレアチンリン酸 40mM;
全20種の非ラベルアミノ酸 各0.32mM;
ポリエチレングリコール6000 4%(W/V);
フォリン酸 34.6mg/mL;
大腸菌( E. coli)tRNAs 0.17mg/mL;
酢酸アンモニウム 36mM;
Mg(OAc)2 8mM;
KOAc 100mM;
リファンピシン 10mg/mL;
クレアチンキナーゼ 0.15mg/mL;
T7 RNAポリメラーゼ 7.7mg/mL;
大腸菌 S30抽出液 28.3%(V/V);
又、 in vitro 転写/翻訳共役反応は次のように行った。即ち、上記オーバーラッピングPCR産物の3μLずつを30°Cで60分間転写・翻訳反応させ、続いて氷の上で冷やして反応を停止させた。同時に、DNAのテンプレートを含まない点のみが異なる同様の系でコントロールの反応を行った。反応系には、オートラジオグラフィーのため、14Cでラベルしたロイシンを0.016mM添加した。
【0157】
(実施例1−8:SDS−PAGEとオートラジオグラフィー)
5μLのサンプルを、非酸化条件とするための同量のローディングバッファー(pH6.8の50mMトリス塩酸、10%グリセロール、2%SDS、0.1%ブロモフェノールブルー)と煮沸した。煮沸後のサンプルをSDS−12.5%−ポリアクリルアミドゲル上で電気泳動させた後、ゲルをワットマン3Mのろ紙にのせ、ゲルドライヤーで乾燥した。次いで、イメージングプレート(富士フィルム社)に感光させた後、BAS-2000にて解析した。その結果を図4に示すが、ゲル上にFab断片のバンド(46.0 kDa)が観察される。
【0158】
(実施例1−9:ELISA解析)
本実施例では、上記 in vitro 系で合成されたいずれのFab断片が抗原親和性を持つかについて、下記のように抗原に対するELISA解析を行った。
【0159】
96ウエルのプレート(デンマーク Nunc 社製)を、5μg/mLの CBP40で50μL/ウエルとなるようにコーティングした。これらのウエルを4°Cで一晩インキュベートした後、蒸留水に溶かした 1/4×Block Ace (大日本製薬社製)を用いて37°C、30分でブロックし、次いで洗浄液(1/10×Block Ace 、0.05% Tween20)で2回洗浄した。
【0160】
無細胞反応混合物30μLを PBS(pH7.4)で5倍に希釈した。この希釈液50μLを、コーティングしたプレートに加え、25°Cで2時間インキュベートした後、上記と同じ洗浄液で2回洗浄した。同時に、DNAのテンプレートを含まない反応液もコントロールとして同様に処理した。
【0161】
150μLの抗マウスIgG(5μg/蒸留水1mL)を各ウエルに加え、25°Cで1時間インキュベートした。そして2回洗浄した後、酵素免疫反応(ELISA)を行った。パーオキシダーゼ反応の基質として、o−フェニレンジアミンと過酸化水素を含む溶液を用いた。発色反応は2M硫酸を用いて停止させた。そして492nmの吸光度を測定した。なお、ELISAの発色はVectastain ABCキット( Vector Laboratories社製)を用いた。
【0162】
ELISA解析の結果を示す図5によれば、 No.9のクローンのELISAシグナルが他のクローンより著しく強く、合成Fab中、このクローンが免疫抗原を結合できることが強く示唆される。
【0163】
(実施例1−10:遺伝子の配列決定)
そこで、上記した No.9のクローンのL鎖及びH鎖(Fd部)の配列決定を行った。L鎖のアミノ酸配列及び遺伝子塩基配列を図6及び配列表の配列番号19に、H鎖(Fd部)のアミノ酸配列及び遺伝子塩基配列を図7及び配列表の配列番号20に、それぞれ示す。
【0164】
上記で決定した配列を、 National Center for Biotechnology Informationの IgBLASTデータベース(http://www.ncbi,nlm, nih.gov/igblast)によって解析した。その結果、L鎖の配列は、 ce9 germline 遺伝子(GenBank accession Nos. AJ239197 )と98%の同一性を持っていた。H鎖の配列は V130 germline遺伝子(GenBank accession Nos. M11700 )と91%の同一性を持っていた。これらの結果から、新規なマウスL鎖遺伝子及びH鎖遺伝子を得たことを確認し、これらの2種遺伝子から形成されたFab断片を「 JM9-Fab」と名付けた。
【0165】
(実施例1−11:宿主での発現、リフォールディング、Fab断片精製)
オーバーラッピングPCR産物中の陽性Fab断片のL鎖とH鎖を XbaI-NaeIで消化し、pRSET-B ベクター(Invitrogen社製)の XbaI-NaeIサイトに挿入した。こうして得た、L鎖を含むプラスミド( pRSETB-Lcと命名)及びH鎖を含むプラスミド( pRSETB-Hcと命名)を、それぞれL鎖とH鎖の発現に用いた。
【0166】
プラスミド pRSETB-Lcとプラスミド pRSETB-Hcとを別々に、コンピテント大腸菌 BL21(DE3)plysS 細胞(Stratagene社製)に導入した。Fab断片の大腸菌での発現とリフォールディングとは、実質的に Wibbenmeyerらの方法と同様に行った。
【0167】
即ち、各ペプチド断片を封入体として発現させた後、遠心分離で回収・洗浄して精製した。その後可溶化バッファー(6M 塩酸グアニジン、0.4M L−アルギニン、pH8.0の100M トリス塩酸、2.5mM EDTA)により可溶化し、100mM DTT(ジチオスレイトール)を添加した後、L鎖とK鎖を等モルになるよう混合した。そしてDTTを含まない可溶化バッファーにて50倍に希釈した後、4mMの酸化型グルタチオンを添加し、透析により徐々に塩酸グアニジン、EDTA等を取り除き、リフォールドさせた。
【0168】
Fab断片の精製は次のプロセスで行った。即ち、0.1M NiSO4 溶液1.0mLを HiTrap キレートカラム(1mL)(Pharmacia Biotech 社製)に流し、10mLの純水で洗浄した後、結合バッファー(PBSに0.5M NaCl、5mMイミダゾールを添加したもの)でカラムを平衡化したものに、上記のFab溶液を流し、結合させた。合計20mLの結合バッファーで洗浄した後、溶出バッファー(PBSに0.5MNaCl、500mM イミダゾールを添加したもの)で溶出させた。
【0169】
(実施例1−12:発現されたFab断片の Kd 値)
JM9-Fab の親和性評価のため、L鎖遺伝子とH鎖遺伝子のシークエンスをそれぞれ pRSET-Bベクターにクローニングして、 pRSETB-JM9Lc と pRSETB-JM9Hc とを得た。これらのクローニングベクター pRSETB-JM9Lc と、 pRSETB-JM9Hc を、別々にコンピテント大腸菌 E. coli BL21(DE3)plysS 細胞(Stratagene社製)に導入した。そして JM9-Fabを発現させ、リフォールディングさせ、精製した。
【0170】
Stevevs の方法による JM9-FabのKd値は1.7(±0.3)×10−8であった。この結果は、このFab断片が従来のハイブリドーマ法による抗体と同等の高い抗原結合活性を有することを示している。
【0171】
〔ヒトを検体とする実施例2〕
この実施例2では、健常成人女性を検体とし、その人の承諾を得て、その末梢血2mLを採血させてもらい、この末梢血から分離した抗体産生細胞(B細胞)を用いて、実施例1と同様の手順に従う各工程を行った。
【0172】
実施例2の工程及び内容中、実施例1と異なる点は、以下の(a)〜(d)である。従って、以下において、まず実施例2の概要を図8に基づいて説明すると共に、続いて(a)〜(d)について順次説明する。これらの点以外は実施例1と同様に行っている。
(a)B細胞の単離工程
(b)モノクローナル抗体の構成
(c)各工程のPCRで使用したプライマー
(d)ELISA解析、配列決定、抗原に対する結合活性
(実施例2の概要)
図8に概念化して示すように、まず、末梢血からB細胞を単離した。単離したB細胞のL鎖(Lc)遺伝子とH鎖(Hc)遺伝子とを、別々に逆転写PCR(RT−PCR)によって増幅した。次いで in vitro 合成に適したDNA断片を得るため、これらの遺伝子に対してオーバーラッピングPCRによって T7 プロモーター、リボソーム結合部位( rbs)及び T7 ターミネーターの配列を付加した。このH鎖遺伝子とL鎖遺伝子の全長DNA断片の対を同じ反応用試験管に投入し、in vitroで転写及び翻訳を行わせてMabを得た後、ELISA法によるスクリーニングを行った。
【0173】
(B細胞の単離工程)
健常成人女性を検体とし、その承諾を得て、その末梢血2mLを採血させてもらった。採血は、血液凝固を防ぐため、ヘパリン存在下で行った。次に Ficllo-Plaque Plus (Amersham Pharmacia Biotech Inc. )を用いた遠心密度勾配(400×g、40分、18°C)により末梢血単球を分離した。
【0174】
回収した細胞をPBS緩衝液で2度洗浄し、ナイロン(商標)ネット(41μmメッシュ)により凝固体を取り除き、CD19+ 細胞をMACS磁性ビーズ(Miltenyi Biotech社, ドイツ)により単離した。磁性ビーズを取り除いた後、抗ヒトCD19抗体PEコンジュゲート(Santa Cruz Biotechnology, Inc., 米国)により染色した。PBSにより2度洗浄した後、分取用フローサイトメトリー(EPICA ELITE, Beckman Coulter社, 日本)により、蛍光標識された細胞だけを分取した。
【0175】
単離したB細胞を顕微鏡(日本国オリンパス社製 LX70 )下でカウントし、希釈してPCRチューブ1本当たり1個の細胞を収容させ、直ちに次工程たるcDNA合成に供した。
【0176】
(モノクローナル抗体の構成)
実施例1では、単離したマウスB細胞のL鎖(Lc)遺伝子とH鎖のFd部遺伝子とを別々に逆転写PCRによって増幅したが、実施例2においては、単離したヒトB細胞のH鎖遺伝子(Hc)、κタイプL鎖遺伝子( Lc(kappa))及びλタイプL鎖遺伝子( Lc(lamda))を別々に逆転写PCRによって増幅し、最終的に、これらのフラグメントからなるMabを取得した。
【0177】
実施例1の場合と同様に行った第2段階のPCRによるこれらの遺伝子の増幅結果を図9に示す。図9中の「Hc」、「 Lc(kappa)」、「 Lc(lamda)」の各図において、 EcoT14Iで消化したλ−DNAマーカーを図の左側に示している。
【0178】
図9に示すように、単一のB細胞に由来するFab断片のH鎖遺伝子、κタイプL鎖遺伝子及びλタイプL鎖遺伝子が別々に増幅され、しかも副産物の生成は認められない。
【0179】
(各工程のPCRで使用したプライマー)
実施例2においては、各工程のPCRで使用したプライマーが実施例1の場合と異なるので、その一覧を表2に示すと共に、それらの塩基配列を順次配列表にも記載した。表2に示した各プライマーについて、以下に簡単に述べる。
【0180】
【表2】
(逆転写PCR用プライマー)
前記実施例1−2において使用した1)〜5)の各プライマーに相当するものが、次の21)〜23)のプライマーである。
【0181】
21)プライマー HuCH-1 :H鎖遺伝子合成用のプライマーであり、配列表の配列番号21に塩基配列を示す。
【0182】
22)プライマー HuCκ-1:κタイプL鎖遺伝子合成用のプライマーであり、配列表の配列番号22に塩基配列を示す。
【0183】
23)プライマー HuCλ-2:λタイプL鎖遺伝子合成用のプライマーであり、配列表の配列番号23に塩基配列を示す。
【0184】
(第1段階PCR用プライマー)
前記実施例1−3で使用した6)〜13)の各プライマーに相当するものが、次の24)〜37)のプライマーである。
【0185】
24)プライマー HuVH-1 :H鎖可変領域用のプライマーであり、配列表の配列番号24に塩基配列を示す。
【0186】
25)プライマー HuVH-2 :H鎖可変領域用のプライマーであり、配列表の配列番号25に塩基配列を示す。
【0187】
26)プライマー HuVH-3 :H鎖可変領域用のプライマーであり、配列表の配列番号26に塩基配列を示す。
【0188】
27)プライマー HuVH-4 :H鎖可変領域用のプライマーであり、配列表の配列番号27に塩基配列を示す。
【0189】
28)プライマー HuVκ-1:L鎖κ遺伝子可変領域用のプライマーであり、配列表の配列番号28に塩基配列を示す。
【0190】
29)プライマー HuVκ-2:L鎖κ遺伝子可変領域用のプライマーであり、配列表の配列番号29に塩基配列を示す。
【0191】
30)プライマー HuVκ-3:L鎖κ遺伝子可変領域用のプライマーであり、配列表の配列番号30に塩基配列を示す。
【0192】
31)プライマー HuVκ-4:L鎖κ遺伝子可変領域用のプライマーであり、配列表の配列番号31に塩基配列を示す。
【0193】
32)プライマー HuVλ-1:L鎖λ遺伝子可変領域用のプライマーであり、配列表の配列番号32に塩基配列を示す。
【0194】
33)プライマー HuVλ-2:L鎖λ遺伝子可変領域用のプライマーであり、配列表の配列番号33に塩基配列を示す。
【0195】
34)プライマー HuVλ-3:L鎖λ遺伝子可変領域用のプライマーであり、配列表の配列番号34に塩基配列を示す。
【0196】
35)プライマー HuCH-2 :H鎖定常領域用のプライマーであり、配列表の配列番号35に塩基配列を示す。
【0197】
36)プライマー HuCκ-2:L鎖κ遺伝子定常領域用のプライマーであり、配列表の配列番号36に塩基配列を示す。
【0198】
37)プライマー HuCλ-2:L鎖λ遺伝子定常領域用のプライマーであり、配列表の配列番号37に塩基配列を示す。
【0199】
(第2段階PCR用プライマー)
前記実施例1−4で使用した14)プライマーに相当するものが次の38)のプライマーである。
【0200】
38)プライマー HuSP1:第2段階PCR用の単一種のプライマーであり、配列表の配列番号38に塩基配列を示す。
【0201】
(ELISA解析、配列決定、抗原に対する結合活性)
実施例1と同様にして増幅された抗体遺伝子より活性体の抗体分子(Fab)が合成されていることを証明するため、血液型抗原B型に対するELISA反応により検討した。検体成人はA型の血液型であるため、B型抗原に対する血液型抗体を当然有すると推測される。このことは、ある感染症に感染した患者の血液中に、その感染症を引き起こす病原体に対する抗体が存在すると言う状況と同じである。
【0202】
B型抗原 BSAを抗原としてELISAを行った結果を図10に示す。実験方法は実施例1の場合とほぼ同様に行った。実施例1の場合と異なる点は、抗原としてCBP40 の代わりにBlood Group B Trisaccharide-BSA 10-atom spacer(Destra laboratoriesm Ltd, )を用いた点である。
【0203】
この結果得られた抗体群は、B型抗原に対しそれぞれ異なる結合活性を有していることがわかった。人体は様々な種類の抗体を産生し、体内に有していることを考えると、十分に予想される結果であり、本実験が予想通り進行したことを示唆している。
【0204】
これらのうち、ELISAシグナルが高かった No.2及び No.10のクローンのL鎖及びH鎖の配列を決定した。 No.2のクローンのL鎖のアミノ酸配列及び遺伝子塩基配列を図11及び配列表の配列番号39に、 No.2のクローンのH鎖のアミノ酸配列及び遺伝子塩基配列を図12及び配列表の配列番号40に、 No.10のクローンのL鎖のアミノ酸配列及び遺伝子塩基配列を図13及び配列表の配列番号41に、 No.10のクローンのH鎖のアミノ酸配列及び遺伝子塩基配列を図14及び配列表の配列番号42に、それぞれ示す。
【0205】
これらの配列について、ヒト抗体のデータベースIgBlast に対してホモロジー検索を行った。その結果、 No.10のクローンのL鎖は、 V1-18germline遺伝子(GeneBank accession Nos.D187018)と95%のホモロジーが検出された。又、 No.10のクローンのH鎖は VH2-70germiline遺伝子 GeneBank accession Nos.AB019437) と91%のホモロジーが検出された。 No.2のクローンについても同様に検索し、L鎖は V1-5germline (GeneBank accession Nos. D87015)と100%の相同性が、H鎖は VH4-4(GeneBank accession Nos. AB019441)と90%の相同性が、それぞれ認められた。
【0206】
これらの結果より、本発明の方法を用いることで、目的とする抗原に対して結合能を有するヒトMabと、そのcDNAとを取得できることを証明した。
【産業上の利用可能性】
【0207】
本発明によって、特定のリガンドに対して親和性を示す極めて多種類のリガンド親和性複合タンパク質の中から、1種又は2種以上のタンパク質を迅速、大量かつ高純度に取得することが可能となる。
[配列表]
SEQUENCE LISTING
<110> National University Corporation Nagoya University
<120> リガンド親和性複合タンパク質の取得方法、抗体スクリーニング方法、抗体スクリーニング用キット、医療方法、抗体医薬、非免疫反応性抗体医薬、投薬方法
<130> POK-04-029
<160> 42
<210> 1
<211> 21
<212> DNA
<213> Artificial Sequence
<400> 1
ttaacactca ttcctgttga a 21
<210> 2
<211> 21
<212> DNA
<213> Artificial Sequence
<400> 2
aattttcttg tccaccttgg t 21
<210> 3
<211> 21
<212> DNA
<213> Artificial Sequence
<400> 3
aattttcttg tccaccttgg t 21
<210> 4
<211> 21
<212> DNA
<213> Artificial Sequence
<400> 4
aagttttttg tccaccgtgg t 21
<210> 5
<211> 21
<212> DNA
<213> Artificial Sequence
<400> 5
gattctcttg atcaactcag t 21
<210> 6
<211> 51
<212> DNA
<213> Artificial Sequence
<400> 6
attagataag aaggagatta ttgaatgga(c/t) attgtg(a/c)t(c/g)a 40
c(a/c)ca(a/g)(a/t)ct(a/c)c a 51
<210> 7
<211> 66
<212> DNA
<213> Artificial Sequence
<400> 7
attagataag aaggagatta ttgattagtg gtggtggtgg tggtgacact cattcctgtt 60
gaagct 66
<210> 8
<211> 47
<212> DNA
<213> Artificial Sequence
<400> 8
attagataag aaggagatta ttgaatg(c/g)a(a/g) gt(a/c/g/t)(a/c)agctg(c/g) 40
ag(c/g)agtc 47
<210> 9
<211> 50
<212> DNA
<213> Artificial Sequence
<400> 9
attagataag aaggagatta ttgaatg(c/g)a(a/g) gt(a/c/g/t)(a/c)agctg(c/g) 40
ag(c/g)agtc(a/t)gg 50
<210> 10
<211> 54
<212> DNA
<213> Artificial Sequence
<400> 10
attagataag aaggagatta ttgattaaat tttcttgtcc accttggtgc tgct 54
<210> 11
<211> 52
<212> DNA
<213> Artificial Sequence
<400> 11
attagataag aaggagatta ttgattaaat tttcttgtcc accttggtgc tg 52
<210> 12
<211> 52
<212> DNA
<213> Artificial Sequence
<400> 12
attagataag aaggagatta ttgattaaag ttttttgtcc accgtggtgc tg 52
<210> 13
<211> 54
<212> DNA
<213> Artificial Sequence
<400> 13
attagataag aaggagatta ttgattagat tctcttgatc aactcagtct tgct 54
<210> 14
<211> 24
<212> DNA
<213> Artificial Sequence
<400> 14
attagataag aaggagatta ttga 24
<210> 15
<211> 22
<212> DNA
<213> Artificial Sequence
<400> 15
ccaatacgca aaccgcctct cc 22
<210> 16
<211> 48
<212> DNA
<213> Artificial Sequence
<400> 16
ataatctcct tcttatctaa taacaaaatt atttctagag ggaaaccg 48
<210> 17
<211> 48
<212> DNA
<213> Artificial Sequence
<400> 17
taatcaataa tctccttctt atctaattcc ggctgctaac aaagcccg 48
<210> 18
<211> 20
<212> DNA
<213> Artificial Sequence
<400> 18
tacagggcgc gtcccattcg 20
<210> 19
<211> 219
<212> PRT
<213> Mus musculus
<400> 19
Asp Ile Val Met Thr Gln Ser Pro Ser Ser Leu Ser Ala Ser Leu Gly 16
GAC ATT GTG ATG ACC CAG TCT CCA TCC TCC CTG TCT GCC TCT CTG GGA 48
Asp Arg Val Thr Ile Ser Cys Arg Ala Ser Gln Asp Ile Ser Asn Tyr 32
GAC AGA GTC ACC ATC AGT TGC AGG GCA AGT CAG GAC ATT AGC AAT TAT 96
Leu Asn Trp Tyr Gln Gln Lys Pro Asp Gly Thr Val Lys Leu Leu Ile 48
TTA AAC TGG TAT CAG CAG AAA CCA GAT GGA ACT GTT AAA CTC CTG ATC 144
Tyr Tyr Thr Ser Arg Leu His Ser Gly Val Pro Ser Arg Phe Ser Gly 64
TAC TAC ACA TCA AGA TTA CAC TCA GGA GTC CCA TCA AGG TTC AGT GGC 192
Ser Gly Ser Gly Thr Asp Tyr Ser Leu Thr Ile Ser Asn Leu Glu Gln 80
AGT GGG TCT GGA ACA GAT TAT TCT CTC ACC ATT AGC AAC CTG GAG CAA 240
Glu Asp Ile Ala Thr Tyr Phe Cys Gln Gln Gly Asn Thr Leu Tyr Thr 96
GAA GAT ATT GCC ACT TAC TTT TGC CAA CAG GGT AAT ACG CTG TAC ACG 288
Phe Gly Gly Gly Thr Lys Leu Glu Ile Lys Arg Ala Asp Ala Ala Pro 112
TTC GGA GGG GGG ACC AAG CTG GAA ATA AAA CGG GCT GAT GCT GCA CCA 336
Thr Val Ser Ile Phe Pro Pro Ser Ser Glu Gln Leu Thr Ser Gly Gly 128
ACT GTA TCC ATC TTC CCA CCA TCC AGT GAG CAG TTA ACA TCT GGA GGT 384
Ala Ser Val Val Cys Phe Leu Asn Asn Phe Tyr Pro Lys Asp Ile Asn 144
GCC TCA GTC GTG TGC TTC TTG AAC AAC TTC TAC CCC AAA GAC ATC AAT 432
Val Lys Trp Lys Ile Asp Gly Ser Glu Arg Gln Asn Gly Val Leu Asn 160
GTC AAG TGG AAG ATT GAT GGC AGT GAA CGA CAA AAT GGC GTC CTG AAC 480
Ser Trp Thr Asp Gln Asp Ser Lys Asp Ser Thr Tyr Ser Met Ser Ser 176
AGT TGG ACT GAT CAG GAC AGC AAA GAC AGC ACC TAC AGC ATG AGC AGC 528
Thr Leu Thr Leu Thr Lys Asp Glu Tyr Glu Arg His Asn Ser Tyr Thr 192
ACC CTC ACG TTG ACC AAG GAC GAG TAT GAA CGA CAT AAC AGC TAT ACC 576
Cys Glu Ala Thr His Lys Thr Ser Thr Ser Pro Ile Val Lys Ser Phe 208
TGT GAG GCC ACT CAC AAG ACA TCA ACT TCA CCC ATT GTC AAG AGC TTC 624
Asn Arg Asn Glu Cys His His His His His His 219
AAC AGG AAT GAG TGT CAC CAC CAC CAC CAC CAC 657
<210> 20
<211> 213
<212> PRT
<213> Mus musculus
<400> 20
Glu Val Lys Leu Gln Gln Ser Gly Ala Glu Phe Val Arg Pro Gly Ala 16
GAA GTA AAG CTG CAG CAG TCT GGG GCT GAG TTT GTA AGG CCA GGG GCC 45
Leu Val Lys Leu Ser Cys Lys Thr Ser Gly Phe Asn Ile Lys Asp Tyr 32
TTA GTC AAG TTG TCC TGC AAA ACT TCT GGC TTC AAC ATT AAA GAC TAC 90
Tyr Met His Trp Val Lys Gln Arg Pro Glu Gln Gly Leu Glu Trp Ile 48
TAT ATG CAC TGG GTG AAG CAG AGG CCT GAA CAG GGC CTG GAG TGG ATT 135
Gly Trp Ile Asp Pro Glu Asn Asp Asp Thr Ile Tyr Asp Pro Lys Phe 64
GGA TGG ATT GAT CCT GAG AAT GAT GAT ACT ATA TAT GAC CCG AAA TTC 180
Gln Gly Lys Ala Ser Ile Thr Ala Asp Thr Ser Ser Asn Thr Ala Tyr 80
CAG GGC AAG GCC AGT ATA ACA GCT GAC ACA TCC TCC AAC ACA GCC TAC 225
Leu Gln Leu Ser Ser Leu Thr Ser Glu Asp Thr Ala Val Tyr Phe Cys 96
CTG CAG CTC AGC AGC CTG ACA TCT GAG GAC ACT GCC GTC TAT TTC TGT 270
Ala Gly Asp Gly Tyr Ser Gly Asn Tyr Trp Gly Gln Gly Thr Ser Val 112
GCT GGT GAT GGT TAC TCC GGG AAC TAC TGG GGT CAA GGA ACC TCA GTC 315
Thr Val Ser Ser Ala Lys Thr Thr Pro Pro Ser Val Tyr Pro Leu Ala 128
ACC GTC TCC TCA GCC AAA ACG ACA CCC CCA TCT GTC TAT CCA CTG GCC 360
Pro Gly Ser Ala Ala Gln Thr Asn Ser Met Val Thr Leu Gly Cys Leu 144
CCT GGA TCT GCT GCC CAA ACT AAC TCC ATG GTG ACC CTG GGA TGC CTG 405
Val Lys Gly Tyr Phe Pro Glu Pro Val Thr Val Thr Trp Asn Ser Gly 160
GTC AAG GGC TAT TTC CCT GAG CCA GTG ACA GTG ACC TGG AAC TCT GGA 450
Ser Leu Ser Ser Gly Val His Thr Phe Pro Ala Val Leu Gln Ser Asp 176
TCC CTG TCC AGC GGT GTG CAC ACC TTC CCA GCT GTC CTG CAG TCT GAC 495
Leu Tyr Thr Leu Ser Ser Ser Val Thr Val Pro Ser Ser Thr Trp Pro 192
CTC TAC ACT CTG AGC AGC TCA GTG ACT GTC CCC TCC AGC ACC TGG CCC 540
Ser Glu Thr Val Thr Cys Asn Val Ala His Pro Ala Ser Ser Thr Lys 208
AGC GAG ACC GTC ACC TGC AAC GTT GCC CAC CCG GCC AGC AGC ACC AAG 585
Val Asp Lys Lys Ile 213
GTG GAC AAG AAA ATT 639
<210> 21
<211> 18
<212> DNA
<213> Artificial Sequence
<400> 21
gctggcctct caccaact 18
<210> 22
<211> 18
<212> DNA
<213> Artificial Sequence
<400> 22
acactctccc ctgttgaa 18
<210> 23
<211> 18
<212> DNA
<213> Artificial Sequence
<400> 23
tgaacattcy gyaggggc 18
<210> 24
<211> 50
<212> DNA
<213> Artificial Sequence
<400> 24
gaaacaagaa tagaaggaga tattgtaatg caggtgcagc tggtgcagtc 50
<210> 25
<211> 53
<212> DNA
<213> Artificial Sequence
<400> 25
gaaacaagaa tagaaggaga tattgtaatg caggtcaact taagggagtc tgg 53
<210> 26
<211> 50
<212> DNA
<213> Artificial Sequence
<400> 26
gaaacaagaa tagaaggaga tattgtaatg gaggtgcagc tgstgsagtc 50
<210> 27
<211> 50
<212> DNA
<213> Artificial Sequence
<400> 27
gaaacaagaa tagaaggaga tattgtaatg caggtrcagc tgcagsagtc 50
<210> 28
<211> 51
<212> DNA
<213> Artificial Sequence
<400> 28
gaaacaagaa tagaaggaga tattgtaatg gacatcswga tgacccagtc t 51
<210> 29
<211> 53
<212> DNA
<213> Artificial Sequence
<400> 29
gaaacaagaa tagaaggaga tattgtaatg gatattgtgm tgactcagtc tcc 53
<210> 30
<211> 53
<212> DNA
<213> Artificial Sequence
<400> 30
gaaacaagaa tagaaggaga tattgtaatg gaaattgtgt tgacgcagtc tcc 53
<210> 31
<211> 52
<212> DNA
<213> Artificial Sequence
<400> 31
gaaacaagaa tagaaggaga tattgtaatg gaaacgacac tcacgcagtc tc 52
<210> 32
<211> 50
<212> DNA
<213> Artificial Sequence
<400> 32
gaaacaagaa tagaaggaga tattgtaatg cagtctgtgc tgactcagcc 50
<210> 33
<211> 50
<212> DNA
<213> Artificial Sequence
<400> 33
gaaacaagaa tagaaggaga tattgtaatg cagtctgccc tgactcagcc 50
<210> 34
<211> 50
<212> DNA
<213> Artificial Sequence
<400> 34
gaaacaagaa tagaaggaga tattgtaatg tcttatgagc tgacwcagcc 50
<210> 35
<211> 73
<212> DNA
<213> Artificial Sequence
<400> 35
gaaacaagaa tagaaggaga tattgtatta gtggtggtgg tggtggtgaa ctbtcttgtc 60
caccttggtg ttg 73
<210> 36
<211> 52
<212> DNA
<213> Artificial Sequence
<400> 36
gaaacaagaa tagaaggaga tattgtatta acactctccc ctgttgaagc tc 52
<210> 37
<211> 52
<212> DNA
<213> Artificial Sequence
<400> 37
gaaacaagaa tagaaggaga tattgtatta tgaacattcy gyaggggcma ct 52
<210> 38
<211> 27
<212> DNA
<213> Artificial Sequence
<400> 38
gaaacaagaa tagaaggaga tattgta 27
<210> 39
<211> 213
<212> PRT
<213> Homo sapiens
<400> 39
Gln Ser Ala Leu Thr Gln Pro Pro Lys Val Leu Gly Leu Gln Ala Arg 16
CAG TCT GCC CTG ACT CAG CCT CCC AAA GTG TTG GGA TTA CAG GCG CGA 48
Ala Thr Thr Pro Gly Tyr Leu Phe His Phe Ile Leu Met Thr Thr Leu 32
GCC ACC ACG CCT GGT TAT TTA TTT CAT TTT ATA CTT ATG ACA ACC CTG 96
Gly Ser Pro Ser Val Ile Leu Cys Gln Pro Leu Thr Leu Ser Phe Leu 48
GGA AGT CCC TCT GTC ATT CTA TGC CAG CCG TTG ACA TTG TCA TTC TTG 144
Pro Phe Tyr Thr Gly Arg Asn Lys Leu Arg Glu Lys Val Met Ser Ile 64
CCA TTT TAT ACA GGA CGA AAT AAA CTC AGA GAG AAG GTA ATG AGC ATA 192
Ile Cys Leu Arg Ser Gln Asp Thr Trp Gln Ser Gly Asn Ser Asn Pro 80
ATC TGT CTG AGG TCA CAG GAT ACG TGG CAG AGC GGA AAT TCG AAT CCT 240
Ser Ile Leu Thr Ser Glu Pro Val Ser Trp Thr Leu Trp Leu Leu Ser 96
AGC ATT CTG ACT TCA GAA CCT GTT TCC TGG ACA CTC TGG CTT CTC TCG 288
Met Trp Ser Pro Ala Arg Gln His Lys Thr Leu Val Gln Pro Lys Ala 112
ATG TGG TCC CCG GCC CGG CAG CAT AAG ACA CTA GTC CAG CCC AAG GCT 336
Ala Pro Ser Val Thr Leu Phe Pro Pro Ser Ser Glu Glu Leu Gln Ala 128
GCC CCC TCG GTC ACT CTG TTC CCG CCC TCC TCT GAG GAG CTT CAA GCC 384
Asn Lys Ala Thr Leu Val Cys Leu Ile Ser Asp Phe Tyr Pro Gly Ala 144
AAC AAG GCC ACA CTG GTG TGT CTC ATA AGT GAC TTC TAC CCG GGA GCC 432
Val Thr Val Ala Trp Lys Ala Asp Ser Ser Pro Val Lys Ala Gly Val 160
GTG ACA GTG GCT TGG AAA GCA GAT AGC AGC CCC GTC AAG GCG GGA GTG 480
Glu Thr Thr Thr Pro Ser Lys Gln Ser Asn Asn Lys Tyr Ala Ala Ser 176
GAG ACC ACC ACA CCC TCC AAA CAA AGC AAC AAC AAG TAC GCG GCC AGC 528
Ser Tyr Leu Ser Leu Thr Pro Glu Gln Trp Lys Ser His Arg Ser Tyr 192
AGC TAT CTG AGC CTG ACG CCT GAG CAG TGG AAG TCC CAC AGA AGC TAC 576
Ser Cys Gln Val Thr His Glu Gly Ser Thr Val Glu Lys Thr Val Ala 208
AGC TGC CAG GTC ACG CAT GAA GGG AGC ACC GTG GAG AAG ACA GTG GCC 624
Pro Thr Glu Cys Ser 213
CCT ACA GAA TGT TCA 639
<210> 40
<211> 219
<212> PRT
<213> Homo sapiens
<400> 40
Gln Val Gln Leu Gln Gln Ser Gly Tyr Pro Ser His Glu His Pro Ile 16
CAG GTA CAG CTG CAG CAG TCC GGA TAT CCC TCC CAT GAG CAT CCT ATC 48
Leu Trp Thr Ser Asp Val Leu Tyr Lys Glu Ala Pro Ser Asn Phe Leu 32
CTC TGG ACT TCT GAT GTT CTA TAT AAA GAG GCA CCC TCA AAT TTT CTT 96
Phe Ser Glu Ala Lys Pro Trp Arg Glu Trp Glu Tyr Pro Tyr Glu Glu 48
TTC TCC GAA GCT AAG CCT TGG AGA GAA TGG GAG TAC CCC TAC GAA GAG 144
Phe Leu Leu Ser Val Ser Arg Leu Ser Glu Cys Asn Tyr Gln Ser His 64
TTT CTA CTC TCA GTG TCC AGG TTA TCT GAG TGC AAT TAC CAG TCC CAT 192
Asn Ile Pro Ile Pro Glu Ser Tyr Val Arg Leu Phe Pro Glu Glu His 80
AAC ATA CCA ATT CCA GAA TCA TAT GTG CGT CTC TTT CCA GAA GAA CAT 240
Arg Ser Tyr Leu Lys Gly Cys Cys Asp Thr Asn Ser Tyr Asp Met Leu 96
AGA AGT TAT CTC AAA GGG TGT TGT GAC ACT AAC AGC TAC GAT ATG TTA 288
Arg Tyr Leu Ala Glu Tyr Gln Val Glu Gly Glu Thr Leu Tyr Asn Arg 112
AGG TAT TTG GCA GAA TAT CAG GTA GAA GGT GAA ACA CTT TAT AAC AGG 336
Leu Leu Ser Val Ser Thr Lys Gly Pro Ser Val Phe Pro Leu Ala Pro 128
CTA TTA TCA GTA TCC ACC AAG GGC CCA TCC GTC TTC CCC CTG GCG CCC 384
Cys Ser Arg Ser Thr Ser Glu Ser Thr Ala Ala Leu Gly Cys Leu Val 144
TGC TCC AGG AGC ACC TCC GAG AGC ACA GCC GCC CTG GGC TGC CTG GTC 432
Lys Asp Tyr Phe Pro Glu Pro Val Thr Val Ser Trp Asn Ser Gly Ala 160
AAG GAC TAC TTC CCC GAA CCG GTG ACG GTG TCG TGG AAC TCA GGC GCC 480
Leu Thr Ser Gly Val His Thr Phe Pro Ala Val Leu Gln Ser Ser Gly 176
CTG ACC AGC GGC GTG CAC ACC TTC CCG GCT GTC CTA CAG TCC TCA GGA 528
Leu Tyr Ser Leu Ser Ser Val Val Thr Val Pro Ser Ser Ser Leu Gly 192
CTC TAC TCC CTC AGC AGC GTG GTG ACC GTG CCC TCC AGC AGC TTG GGC 576
Thr Lys Thr Tyr Thr Cys Asn Val Asp His Lys Pro Ser Asn Thr Lys 208
ACG AAG ACC TAC ACC TGC AAC GTA GAT CAC AAG CCC AGC AAC ACC AAG 624
Val Asp Lys Arg Val His His His His His His 219
GTG GAC AAG AGA GTT CAC CAC CAC CAC CAC CAC 657
<210> 41
<211> 213
<212> PRT
<213> Homo sapiens
<400> 41
Gln Ser Val Leu Thr Gln Pro Tyr Pro Gly Glu Gly Phe Leu Ile Tyr 16
CAG TCT GTG CTG ACT CAG CCT TAT CCA GGT GAA GGC TTT CTG ATA TAT 48
Gln Ser Ala Tyr Leu Ala Pro Phe Cys Leu Ser Tyr Tyr Thr Val Lys 32
CAA TCT GCT TAT TTG GCT CCA TTC TGT CTC TCA TAC TAT ACT GTG AAG 96
Arg Arg Pro Cys Leu Ile His Val Asp Thr Thr His Ser Arg Ile Met 48
CGA AGA CCA TGT CTC ATT CAT GTA GAC ACT ACA CAC AGT AGG ATA ATG 144
His Val Glu Leu Asn Trp Leu Met His Glu Tyr Ala Ser Tyr Gln Arg 64
CAT GTT GAG TTA AAT TGG CTT ATG CAT GAA TAT GCT TCC TAC CAG CGA 192
Gly Val Asn Gly Leu Ser Lys Asp Glu His His Leu Phe Leu Tyr Ile 80
GGA GTC AAT GGT CTA AGT AAG GAT GAA CAT CAT CTG TTT CTG TAC ATT 240
Tyr Phe Gln Thr Val Val Arg Leu Gly Val Pro Arg Ser Lys Asp Tyr 96
TAC TTT CAA ACA GTA GTG AGG CTG GGA GTA CCT AGA AGC AAG GAC TAT 288
Val Leu Thr Gln Val Ser Lys Phe Ile Phe Tyr Ser Phe Pro Lys Ala 112
GTT TTA ACC CAA GTT TCA AAA TTT ATC TTT TAT TCG TTT CCC AAG GCC 336
Asn Pro Thr Val Thr Leu Phe Pro Pro Ser Ser Glu Glu Leu Gln Ala 128
AAC CCC ACG GTC ACT CTG TTC CCG CCC TCC TCT GAG GAG CTC CAA GCC 384
Asn Lys Ala Thr Leu Val Cys Leu Ile Ser Asp Phe Tyr Pro Gly Ala 144
AAC AAG GCC ACA CTA GTG TGT CTG ATC AGT GAC TTC TAC CCG GGA GCT 432
Val Thr Val Ala Trp Lys Ala Asp Gly Ser Pro Val Lys Ala Gly Val 160
GTG ACA GTG GCT TGG AAG GCA GAT GGC AGC CCC GTC AAG GCG GGA GTG 480
Glu Thr Thr Lys Pro Ser Lys Gln Ser Asn Asn Lys Tyr Ala Ala Ser 176
GAG ACG ACC AAA CCC TCC AAA CAG AGC AAC AAC AAG TAC GCG GCC AGC 528
Ser Tyr Leu Ser Leu Thr Pro Glu Gln Trp Lys Ser His Arg Ser Tyr 192
AGC TAC CTG AGC CTG ACG CCC GAG CAG TGG AAG TCC CAC AGA AGC TAC 576
Ser Cys Gln Val Thr His Glu Gly Ser Thr Val Glu Lys Thr Val Ala 208
AGC TGC CAG GTC ACG CAT GAA GGG AGC ACC GTG GAG AAG ACA GTG GCC 624
Pro Thr Glu Cys Ser 213
CCT ACA GAA TGT TCA 639
<210> 42
<211> 219
<212> PRT
<213> Homo sapiens
<400> 42
Gln Val Asn Leu Arg Glu Ser Gly Gln Leu Met Pro Ala Ser Ser Pro 16
CAG GTC AAC TTA AGG GAG TCT GGA CAA TTA ATG CCT GCC TCC TCT CCC 48
Tyr Pro Thr Asn Val His Ile Leu Ile Leu Arg Thr Cys Glu Tyr Gly 32
TAC CCC ACA AAT GTT CAC ATC CTA ATT CTT AGA ACC TGT GAA TAT GGT 96
Glu Tyr Val Glu Tyr Val Thr Tyr His Gly Gln Glu Val Leu Tyr Arg 48
GAA TAT GTT GAA TAT GTT ACC TAC CAT GGT CAA GAG GTA CTT TAC AGA 144
Tyr Glu Gln Leu Arg Thr Leu Arg Glu Ala Gln Ser Ser Arg Leu Leu 64
TAT GAG CAA TTA AGG ACC CTG AGA GAG GCA CAA TCA TCT AGA TTA CTC 192
Arg Gln Ala Gln Ser Gly His Gln Gly Pro Cys Lys Arg Lys Val Gly 80
AGG CAA GCC CAA AGT GGC CAC CAG GGT CCC TGT AAG AGG AAG GTA GGG 240
Arg Val Lys Arg Gln Gln Ile Lys Ser Arg Asp Glu Ser Asp Ala Leu 96
AGG GTT AAG AGA CAG CAG ATA AAG AGC AGA GAC GAG AGT GAT GCA CTT 288
Gly Lys Met Lys Glu Gly His Glu Val Lys Glu Cys Trp Trp Pro Leu 112
GGA AAG ATG AAA GAA GGT CAT GAG GTT AAG GAA TGC TGG TGG CCT CTA 336
Glu Ala Arg Lys Ser Thr Lys Gly Pro Ser Val Phe Pro Leu Ala Pro 128
GAA GCT AGA AAG TCC ACC AAG GGC CCA TCG GTC TTC CCC CTG GCA CCC 384
Ser Ser Lys Ser Thr Ser Gly Gly Thr Ala Ala Leu Gly Cys Leu Val 144
TCC TCC AAG AGC ACC TCT GGG GGC ACA GCG GCC CTG GGC TGC CTG GTC 432
Lys Asp Tyr Phe Pro Glu Pro Val Thr Val Ser Trp Asn Ser Gly Ala 160
AAG GAC TAC TTC CCC GAA CCG GTG ACG GTG TCG TGG AAC TCA GGC GCC 480
Leu Thr Ser Gly Val His Thr Phe Pro Ala Val Leu Gln Ser Ser Gly 176
CTG ACC AGC GGC GTG CAC ACC TTC CCG GCT GTC CTA CAG TCC TCA GGA 528
Leu Tyr Ser Leu Ser Ser Val Val Thr Val Pro Ser Ser Ser Leu Gly 192
CTC TAC TCC CTC AGC AGC GTG GTG ACC GTG CCC TCC AGC AGC TTG GGC 576
Thr Gln Thr Tyr Ile Cys Asn Val Asn His Lys Pro Ser Asn Thr Lys 208
ACC CAG ACC TAC ATC TGC AAC GTG AAT CAC AAG CCC AGC AAC ACC AAG 624
Val Asp Lys Arg Val His His His His His His 219
GTG GAC AAG AGA GTT CAC CAC CAC CAC CAC CAC 657
【図面の簡単な説明】
【0208】
【図1】実施例1の全体的なフローを概念化して示す図である。
【0209】
【図2】実施例1におけるH鎖遺伝子とL鎖遺伝子の増幅結果を示す図である。
【0210】
【図3】実施例1のオーバーラッピングPCR産物の解析結果を示す図である。
【0211】
【図4】実施例1におけるオートラジオグラフィーの結果を示す図である。
【0212】
【図5】実施例1におけるELISA解析の結果を示す図である。
【0213】
【図6】実施例1におけるL鎖の配列決定の結果を示す図である。
【0214】
【図7】実施例1におけるH鎖の配列決定の結果を示す図である。
【0215】
【図8】実施例2の全体的なフローを概念化して示す図である。
【0216】
【図9】実施例2におけるH鎖遺伝子とL鎖遺伝子の増幅結果を示す図である。
【0217】
【図10】実施例2におけるELISA解析の結果を示す図である。
【0218】
【図11】実施例2における No.2クローンのL鎖配列決定の結果を示す図である。
【0219】
【図12】実施例2における No.2クローンのH鎖配列決定の結果を示す図である。
【0220】
【図13】実施例2における No.10クローンのL鎖配列決定の結果を示す図である。
【0221】
【図14】実施例2における No.10クローンのH鎖配列決定の結果を示す図である。
【特許請求の範囲】
【請求項1】
任意に特定されたリガンドXに対して選択的な親和性を示す極めて多種類の複合タンパク質Xpの中から、無作為にあるいは特性評価を通じて選ばれる1種又は2種以上の複合タンパク質を取得する方法であって、少なくとも下記の1)生産手段分離工程、2)生産手段転換工程、3)大量生産準備工程、4)転写用配列付加工程及び5)単一種大量生産工程を含み、必要に応じて更に下記の6)キャラクタリゼーション工程を含むことを特徴とするリガンド親和性複合タンパク質の取得方法。
1)極めて多種類にわたる前記複合タンパク質Xpのそれぞれ専用の生産手段(mRNA)であって、容易に大量複製できる複製手段Xc(cDNA)への転換が可能な生産手段Xmを備えた抗体産生細胞の1個又は複数個の単細胞を、ヒト又は非ヒト動物たる検体の適宜な組織又は生体構成部分から個別に分離することにより、目的タンパク質の種類ごとに生産手段Xmを厳密に分離して、以下の各工程における生産手段Xm、複製手段Xc及び複合タンパク質Xpの異種間混在を予め防止する生産手段分離工程。
2)互いに分離された1種又は複数種の前記生産手段Xmについて、それぞれの生産手段Xmを構成するmRNAの全部又は一部を逆転写PCR法により複製手段Xcへ転換させる生産手段転換工程。
3)逆転写PCR法により得られた前記複製手段XcをPCR法で大量複製してタンパク質の大量生産能力を準備する大量生産準備工程。
4)大量複製された前記複製手段Xc(cDNA)に対して、その無細胞系での生産手段Xm(mRNA)への再転換に必要な転写用配列を付加する転写用配列付加工程。
5)転写用配列を付加された前記複製手段Xcを、少なくとも複製手段Xcの生産手段Xmへの再転換を行う要素と、生産手段Xmに基づきタンパク質の生産を行う要素とを備えた無細胞系へ投入することにより、前記複合タンパク質Xpを、又は前記生産手段転換工程で規定した一部のmRNAに対応する複合タンパク質Xpを、1種類ごとに大量生産する単一種大量生産工程。
6)以上の各工程により生産された複合タンパク質Xpについて、特性評価を行い、優れた特性を持つ複合タンパク質Xpをスクリーニングし、あるいはこれらの複合タンパク質Xpの配列決定を行うキャラクタリゼーション工程。
【請求項2】
前記検体がヒトである場合において、抗原たるリガンドXに対する抗体を既に体内に生成している生理状態にあると推定される被検者を選択したもとで、当該被検者における適宜な組織又は生体構成部分を社会通念上許容される非侵襲的手段によって採取し、この組織又は生体構成部分から前記1)の1個又は複数個の単細胞を分離することを特徴とする請求項1に記載のリガンド親和性複合タンパク質の取得方法。
【請求項3】
前記被検者から、前記組織又は生体構成部分として末梢血を採取し、この末梢血から抗体産生細胞たるB細胞及び/又は形質細胞の単細胞を分離することを特徴とする請求項2に記載のリガンド親和性複合タンパク質の取得方法。
【請求項4】
前記検体が非ヒト動物である場合において、この動物をリガンドXで免疫したもとで、当該動物の適宜な組織又は生体構成部分から前記1)の1個又は複数個の単細胞を分離することを特徴とする請求項1に記載のリガンド親和性複合タンパク質の取得方法。
【請求項5】
前記複合タンパク質Xpが各1対のH鎖及びL鎖からなる免疫グロブリン(抗体)、そのFab(抗原結合性フラグメント)、又はこれらに準ずるタンパク質フラグメントであることを特徴とする請求項1〜請求項4のいずれかに記載のリガンド親和性複合タンパク質の取得方法。
【請求項6】
前記生産手段分離工程において、前記検体の組織又は生体構成部分を液体中に投入して抗体産生細胞を分散・懸濁させ、一定の分離用抗体と結合させた磁性微粒子と共にインキュベートした後、分離用抗体を介して抗体産生細胞に結合した磁性微粒子をマグネットで回収することにより、前記懸濁液から目的とする抗体産生細胞の単細胞を分離することを特徴とする請求項1〜請求項5のいずれかに記載のリガンド親和性複合タンパク質の取得方法。
【請求項7】
前記生産手段転換工程において、逆転写PCRに用いるプライマーの設計により、特定の分類カテゴリーに属する複合タンパク質XpをコードするmRNAのみを複製手段Xcへ転換させ、及び/又は複合タンパク質Xpを構成する特定のフラグメントをコードするmRNAのみを前記複製手段Xcへ転換させることを特徴とする請求項1〜請求項6のいずれかに記載のリガンド親和性複合タンパク質の取得方法。
【請求項8】
前記大量生産準備工程において、複製手段XcのPCR法による大量複製を、次の2段階PCRによって行うことを特徴とする請求項1〜請求項7のいずれかに記載のリガンド親和性複合タンパク質の取得方法。
第1段階PCR:複製手段Xcを構成する全ての種類のcDNAに対応する各種類のプライマーにおいて、それらの5’末端側に同一の付加配列を設けておいて増幅を行う。
第2段階PCR:第1段階PCRで増幅された全ての種類のDNAフラグメントに対して、前記付加配列に対して相補的な配列部分を備えた単一種類のプライマーを用いて増幅を行う。
【請求項9】
前記第1段階PCRで用いる各種類のプライマーの付加配列中に、前記5)の無細胞系の由来生物を考慮した適宜な種類の翻訳開始促進配列部分を含めておくことを特徴とする請求項8に記載のリガンド親和性複合タンパク質の取得方法。
【請求項10】
前記転写用配列付加工程において、大量生産準備工程で増幅されたDNAフラグメントに対して、少なくともプロモーター配列を付加するPCRを行うことを特徴とする請求項1〜請求項9のいずれかに記載のリガンド親和性複合タンパク質の取得方法。
【請求項11】
前記単一種大量生産工程において、前記無細胞系を還元化しないままの条件で用いることを特徴とする請求項1〜請求項10のいずれかに記載のリガンド親和性複合タンパク質の取得方法。
【請求項12】
前記キャラクタリゼーション工程が、複合タンパク質XpのELISA法によるリガンド親和性評価と、複合タンパク質Xp又はその遺伝子の配列決定と、形質転換体を用いて生産させた複合タンパク質XpのKd値の測定との内の少なくとも1項目を含むことを特徴とする請求項1〜請求項11のいずれかに記載のリガンド親和性複合タンパク質の取得方法。
【請求項13】
請求項1〜請求項12のいずれかに記載のリガンド親和性複合タンパク質の取得方法を、ヒト又は非ヒト動物におけるリガンドXに対する抗体のスクリーニングに用いることを特徴とする抗体スクリーニング方法。
【請求項14】
請求項13に記載の抗体スクリーニング方法を実施するために用いられるキットであって、少なくとも以下の構成要素を含んで構成されることを特徴とする抗体スクリーニング用キット。
11)請求項6に規定する分散・懸濁用の液体と、磁性微粒子とのセット。
12)請求項7に規定する逆転写PCR用のプライマーセット。
13)請求項8に規定する2段階のPCR用のプライマーセット。
14)請求項1の5)に規定する無細胞系。
【請求項15】
請求項1及び請求項4〜請求項12のいずれかに記載のリガンド親和性複合タンパク質の取得方法によって得られた抗体を用いて、非ヒト動物の疾患の予防、診断又は治療を行うことを特徴とする医療方法。
【請求項16】
請求項1〜請求項3及び請求項5〜請求項12のいずれかに記載のリガンド親和性複合タンパク質の取得方法によって得られた抗体を用いて、ヒトの疾患の予防、診断又は治療を行うことを特徴とする医療方法。
【請求項17】
請求項1〜請求項12のいずれかに記載のリガンド親和性複合タンパク質の取得方法によって得られた抗体を有効成分とし、その由来に応じてヒト又は非ヒト動物に適用されるものであることを特徴とする抗体医薬。
【請求項18】
請求項1〜請求項3及び請求項5〜請求項12のいずれかに記載のリガンド親和性複合タンパク質の取得方法によって得られた抗体を有効成分とし、その由来するヒト個体に対して適用されるものであることを特徴とする非免疫反応性抗体医薬。
【請求項19】
請求項18に記載の非免疫反応性抗体医薬を、非経口的な投与方法によって、その由来する前記ヒト個体の体内へ投与することを特徴とする投薬方法。
【請求項1】
任意に特定されたリガンドXに対して選択的な親和性を示す極めて多種類の複合タンパク質Xpの中から、無作為にあるいは特性評価を通じて選ばれる1種又は2種以上の複合タンパク質を取得する方法であって、少なくとも下記の1)生産手段分離工程、2)生産手段転換工程、3)大量生産準備工程、4)転写用配列付加工程及び5)単一種大量生産工程を含み、必要に応じて更に下記の6)キャラクタリゼーション工程を含むことを特徴とするリガンド親和性複合タンパク質の取得方法。
1)極めて多種類にわたる前記複合タンパク質Xpのそれぞれ専用の生産手段(mRNA)であって、容易に大量複製できる複製手段Xc(cDNA)への転換が可能な生産手段Xmを備えた抗体産生細胞の1個又は複数個の単細胞を、ヒト又は非ヒト動物たる検体の適宜な組織又は生体構成部分から個別に分離することにより、目的タンパク質の種類ごとに生産手段Xmを厳密に分離して、以下の各工程における生産手段Xm、複製手段Xc及び複合タンパク質Xpの異種間混在を予め防止する生産手段分離工程。
2)互いに分離された1種又は複数種の前記生産手段Xmについて、それぞれの生産手段Xmを構成するmRNAの全部又は一部を逆転写PCR法により複製手段Xcへ転換させる生産手段転換工程。
3)逆転写PCR法により得られた前記複製手段XcをPCR法で大量複製してタンパク質の大量生産能力を準備する大量生産準備工程。
4)大量複製された前記複製手段Xc(cDNA)に対して、その無細胞系での生産手段Xm(mRNA)への再転換に必要な転写用配列を付加する転写用配列付加工程。
5)転写用配列を付加された前記複製手段Xcを、少なくとも複製手段Xcの生産手段Xmへの再転換を行う要素と、生産手段Xmに基づきタンパク質の生産を行う要素とを備えた無細胞系へ投入することにより、前記複合タンパク質Xpを、又は前記生産手段転換工程で規定した一部のmRNAに対応する複合タンパク質Xpを、1種類ごとに大量生産する単一種大量生産工程。
6)以上の各工程により生産された複合タンパク質Xpについて、特性評価を行い、優れた特性を持つ複合タンパク質Xpをスクリーニングし、あるいはこれらの複合タンパク質Xpの配列決定を行うキャラクタリゼーション工程。
【請求項2】
前記検体がヒトである場合において、抗原たるリガンドXに対する抗体を既に体内に生成している生理状態にあると推定される被検者を選択したもとで、当該被検者における適宜な組織又は生体構成部分を社会通念上許容される非侵襲的手段によって採取し、この組織又は生体構成部分から前記1)の1個又は複数個の単細胞を分離することを特徴とする請求項1に記載のリガンド親和性複合タンパク質の取得方法。
【請求項3】
前記被検者から、前記組織又は生体構成部分として末梢血を採取し、この末梢血から抗体産生細胞たるB細胞及び/又は形質細胞の単細胞を分離することを特徴とする請求項2に記載のリガンド親和性複合タンパク質の取得方法。
【請求項4】
前記検体が非ヒト動物である場合において、この動物をリガンドXで免疫したもとで、当該動物の適宜な組織又は生体構成部分から前記1)の1個又は複数個の単細胞を分離することを特徴とする請求項1に記載のリガンド親和性複合タンパク質の取得方法。
【請求項5】
前記複合タンパク質Xpが各1対のH鎖及びL鎖からなる免疫グロブリン(抗体)、そのFab(抗原結合性フラグメント)、又はこれらに準ずるタンパク質フラグメントであることを特徴とする請求項1〜請求項4のいずれかに記載のリガンド親和性複合タンパク質の取得方法。
【請求項6】
前記生産手段分離工程において、前記検体の組織又は生体構成部分を液体中に投入して抗体産生細胞を分散・懸濁させ、一定の分離用抗体と結合させた磁性微粒子と共にインキュベートした後、分離用抗体を介して抗体産生細胞に結合した磁性微粒子をマグネットで回収することにより、前記懸濁液から目的とする抗体産生細胞の単細胞を分離することを特徴とする請求項1〜請求項5のいずれかに記載のリガンド親和性複合タンパク質の取得方法。
【請求項7】
前記生産手段転換工程において、逆転写PCRに用いるプライマーの設計により、特定の分類カテゴリーに属する複合タンパク質XpをコードするmRNAのみを複製手段Xcへ転換させ、及び/又は複合タンパク質Xpを構成する特定のフラグメントをコードするmRNAのみを前記複製手段Xcへ転換させることを特徴とする請求項1〜請求項6のいずれかに記載のリガンド親和性複合タンパク質の取得方法。
【請求項8】
前記大量生産準備工程において、複製手段XcのPCR法による大量複製を、次の2段階PCRによって行うことを特徴とする請求項1〜請求項7のいずれかに記載のリガンド親和性複合タンパク質の取得方法。
第1段階PCR:複製手段Xcを構成する全ての種類のcDNAに対応する各種類のプライマーにおいて、それらの5’末端側に同一の付加配列を設けておいて増幅を行う。
第2段階PCR:第1段階PCRで増幅された全ての種類のDNAフラグメントに対して、前記付加配列に対して相補的な配列部分を備えた単一種類のプライマーを用いて増幅を行う。
【請求項9】
前記第1段階PCRで用いる各種類のプライマーの付加配列中に、前記5)の無細胞系の由来生物を考慮した適宜な種類の翻訳開始促進配列部分を含めておくことを特徴とする請求項8に記載のリガンド親和性複合タンパク質の取得方法。
【請求項10】
前記転写用配列付加工程において、大量生産準備工程で増幅されたDNAフラグメントに対して、少なくともプロモーター配列を付加するPCRを行うことを特徴とする請求項1〜請求項9のいずれかに記載のリガンド親和性複合タンパク質の取得方法。
【請求項11】
前記単一種大量生産工程において、前記無細胞系を還元化しないままの条件で用いることを特徴とする請求項1〜請求項10のいずれかに記載のリガンド親和性複合タンパク質の取得方法。
【請求項12】
前記キャラクタリゼーション工程が、複合タンパク質XpのELISA法によるリガンド親和性評価と、複合タンパク質Xp又はその遺伝子の配列決定と、形質転換体を用いて生産させた複合タンパク質XpのKd値の測定との内の少なくとも1項目を含むことを特徴とする請求項1〜請求項11のいずれかに記載のリガンド親和性複合タンパク質の取得方法。
【請求項13】
請求項1〜請求項12のいずれかに記載のリガンド親和性複合タンパク質の取得方法を、ヒト又は非ヒト動物におけるリガンドXに対する抗体のスクリーニングに用いることを特徴とする抗体スクリーニング方法。
【請求項14】
請求項13に記載の抗体スクリーニング方法を実施するために用いられるキットであって、少なくとも以下の構成要素を含んで構成されることを特徴とする抗体スクリーニング用キット。
11)請求項6に規定する分散・懸濁用の液体と、磁性微粒子とのセット。
12)請求項7に規定する逆転写PCR用のプライマーセット。
13)請求項8に規定する2段階のPCR用のプライマーセット。
14)請求項1の5)に規定する無細胞系。
【請求項15】
請求項1及び請求項4〜請求項12のいずれかに記載のリガンド親和性複合タンパク質の取得方法によって得られた抗体を用いて、非ヒト動物の疾患の予防、診断又は治療を行うことを特徴とする医療方法。
【請求項16】
請求項1〜請求項3及び請求項5〜請求項12のいずれかに記載のリガンド親和性複合タンパク質の取得方法によって得られた抗体を用いて、ヒトの疾患の予防、診断又は治療を行うことを特徴とする医療方法。
【請求項17】
請求項1〜請求項12のいずれかに記載のリガンド親和性複合タンパク質の取得方法によって得られた抗体を有効成分とし、その由来に応じてヒト又は非ヒト動物に適用されるものであることを特徴とする抗体医薬。
【請求項18】
請求項1〜請求項3及び請求項5〜請求項12のいずれかに記載のリガンド親和性複合タンパク質の取得方法によって得られた抗体を有効成分とし、その由来するヒト個体に対して適用されるものであることを特徴とする非免疫反応性抗体医薬。
【請求項19】
請求項18に記載の非免疫反応性抗体医薬を、非経口的な投与方法によって、その由来する前記ヒト個体の体内へ投与することを特徴とする投薬方法。
【図1】

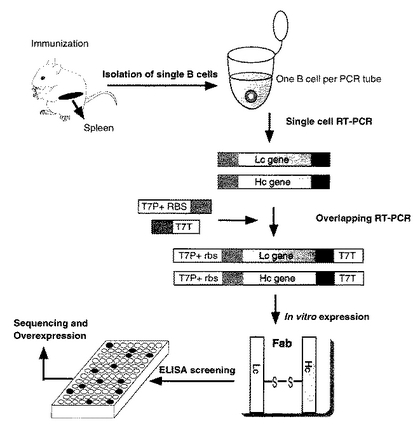
【図2】
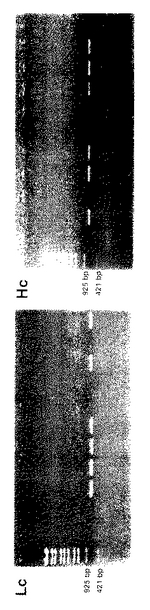
【図3】
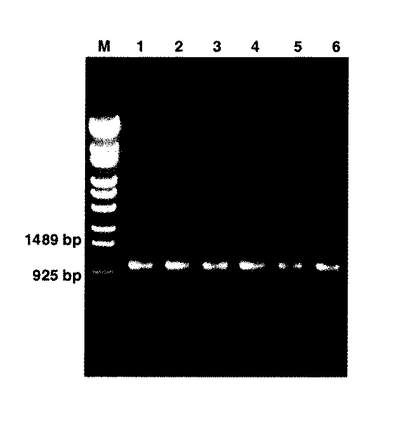
【図4】
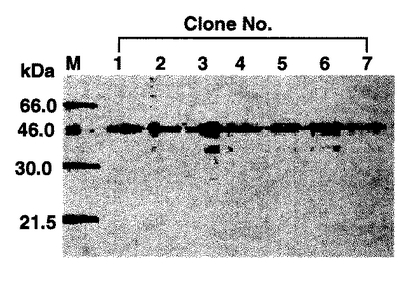
【図5】
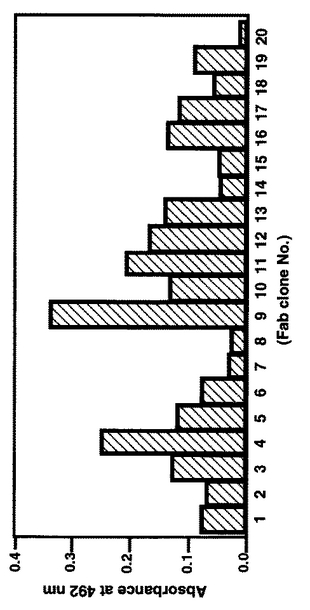
【図6】

【図7】

【図8】
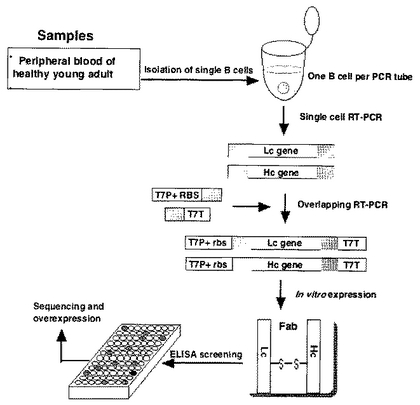
【図9】
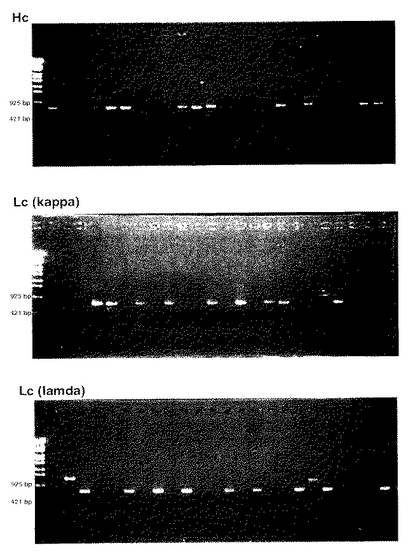
【図10】
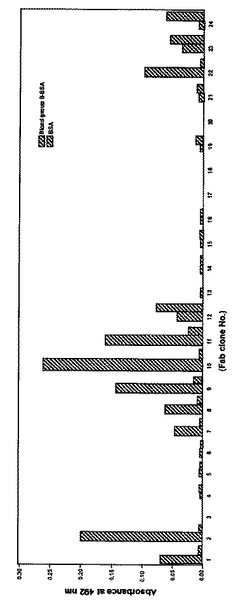
【図11】

【図12】

【図13】

【図14】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【公開番号】特開2006−158203(P2006−158203A)
【公開日】平成18年6月22日(2006.6.22)
【国際特許分類】
【出願番号】特願2004−349309(P2004−349309)
【出願日】平成16年12月2日(2004.12.2)
【出願人】(504139662)国立大学法人名古屋大学 (996)
【Fターム(参考)】
【公開日】平成18年6月22日(2006.6.22)
【国際特許分類】
【出願日】平成16年12月2日(2004.12.2)
【出願人】(504139662)国立大学法人名古屋大学 (996)
【Fターム(参考)】
[ Back to top ]