
リグノセルロース系バイオマスからのエタノール醗酵微生物
【目的】 キシロースの醗酵能が向上した微生物の提供。
【解決手段】 本発明はグリシン合成系タンパク質の遺伝子及び/又はメチオニン合成系タンパク質の遺伝子の発現機能を喪失させ、かつキシロース代謝酵素遺伝子を導入した微生物、前記微生物の製造方法及び前記微生物を用いたエタノールの製造方法を提供する。
【解決手段】 本発明はグリシン合成系タンパク質の遺伝子及び/又はメチオニン合成系タンパク質の遺伝子の発現機能を喪失させ、かつキシロース代謝酵素遺伝子を導入した微生物、前記微生物の製造方法及び前記微生物を用いたエタノールの製造方法を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、グリシン合成系タンパク質の遺伝子及び/又はメチオニン合成系タンパク質の遺伝子の発現機能を喪失させたエタノール醗酵微生物に関する。また、本発明は、グリシン合成系タンパク質の遺伝子及び/又はメチオニン合成系タンパク質の遺伝子の発現機能を喪失させることにより、キシロース醗酵能力の増加した醗酵微生物を作製する方法に関する。さらに、本発明は、前記微生物と、キシロース含有原料とを接触させる工程を含む、エタノールの製造方法に関する。
【背景技術】
【0002】
近年、化石燃料の枯渇や、CO2ガス削減の必要といった観点から、従来廃棄物であった、コーンコブ、稲ワラ、スイッチグラス、エリアンサス、廃材などといった、バイオマスから燃料としてエタノールを生成する研究が進められている。人類は何千年も前より、デンプンから酵母サッカロミセス・セレビシエ(Saccharomyces cerevisiae)による醗酵でエタノールにする技術を持っていた。デンプンはグルコースがα-1,4結合した多糖であり、様々な生物が持つ加水分解酵素によって容易に分解することができる。また、グルコースは酵母サッカロミセス・セレビシエの最も好ましいC源であり、1分子のグルコースから2分子のエタノールが醗酵により生成される。これに対し、バイオマスは多糖として、セルロース、あるいはヘミセルロースを含んでいる。
【0003】
このうち、セルロースは、グルコースがβ-1,4結合した多糖であり、結晶構造をとっている。結晶構造を壊すための前処理が必要であることや、セルロースを分解するために必要なセロビオハイドロラーゼI、セロビオハイドロラーゼII、エンドグルカナーゼ等の活性が十分でないこと等の問題はあるが、分解後に生じる糖はグルコースであるため、酵母サッカロミセス・セレビシエで醗酵するには何の問題もない。
【0004】
これに対し、ヘミセルロースはグルコースの他に、キシロース、アラビノースと言った五炭糖を含んでいるが、従来酵母サッカロミセス・セレビジエはこれらの五炭糖を醗酵することができない。そこで、キシロースからエタノールを醗酵するために、キシロースを醗酵する能力のある、酵母ピキア・スティピティス(Pichia stipitis)のキシロース還元酵素遺伝子、キシリトール脱水素酵素遺伝子を酵母サッカロミセス・セレビシエにて高発現させる方法が、よく用いられてきた(非特許文献1〜4)。ピキア・スティピティスのキシロース還元酵素は補酵素として主にNADPHを用いる酵素であり、反応後、一分子のNADPが生成する。一方、キシリトール脱水素酵素は、補酵素として主にNADを用いる酵素であり、反応後一分子のNADHを生じる。従って、グルコースを醗酵する際には、NADP/NADPHあるいはNAD/NADHのバランスは変化しないが、キシロースからエタノールを醗酵する場合には、NADPあるいは、NADHが増加する方向に傾いてしまう。このことが、キシロースからのエタノール生成における生産量の低下の原因であると考えられている。これに対し、これまで補酵素特異性を改変した酵素や補酵素特異性が異なる酵素を発現させる試みがなされてきたが、十分な効果は得られていない(非特許文献2〜4)。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Petschacher B, Nidetzky B. Microb Cell Fact. 2008 Mar 17; 7: 9.
【非特許文献2】Watanabe S, Saleh AA, Pack SP, Annaluru N, Kodaki T, Makino K. J Biotechnol. 2007 Jun 30; 130(3): 316-9. Epub 2007 Apr 29.
【非特許文献3】Verho R, Londesborough J, Penttile M, Richard P. Appl Environ Microbiol. 2003 Oct; 69(10): 5892-7.
【非特許文献4】Mikael Anderlund et.al. Appl Environ Microbiol. 1999: June 65(6):2333-2340
【発明の概要】
【発明が解決しようとする課題】
【0006】
このような状況下、キシロースの醗酵能が向上した微生物の開発が必要とされる。
【課題を解決するための手段】
【0007】
本発明者らは、酵母ピキア・スティピティスのキシロース還元酵素、キシリトール脱水素酵素遺伝子および、酵母サッカロミセス・セレビジエのキシルロースリン酸化酵素遺伝子の発現カセットを導入することによって、キシロース醗酵性を付与した複数種類の酵母を用いて醗酵試験を行い、キシロースからのエタノール醗酵における細胞内代謝物のプロファイル分析を行った。得られたデータと、醗酵パフォーマンスを表す数値として、(1)エタノール生成量・速度・収率と、(2)キシロース資化量・速度・収率と、(3)副生成物の生成量・速度・収率等とを比較し、データと(1)〜(3)との相関関係を解析することにより、キシロースからエタノールへの代謝経路に属する物質、すなわち、キシリトール、キシルロース、キシルロース5リン酸、およびペントースリン酸回路と解糖系に属する中間代謝物以外の新たな物質と、エタノール醗酵パフォーマンスとの関連を見出して、育種ターゲットとなる遺伝子を決定し、その遺伝子に関する育種を行った。その結果、グリシン生合成経路の遺伝子GLY1を破壊することにより、キシロースの資化速度が上昇することが判明した。また、メチオニン生合成経路の遺伝子、MET6あるいはMET32を破壊することにより、副産物である、キシリトールの生成量が減少することが判明した。
本願発明は、上記知見に基づくものである。
【0008】
すなわち、本発明は以下に関する。
[1] グリシン合成系タンパク質の遺伝子及び/又はメチオニン合成系タンパク質の遺伝子の発現機能を喪失させ、かつキシロース代謝酵素遺伝子を導入した微生物。
[2] 前記グリシン合成系タンパク質の遺伝子がGLY1遺伝子である、前記[1]に記載の微生物。
[3] 前記メチオニン合成系タンパク質の遺伝子がMET6又はMET32遺伝子である、前記[1]又は[2]に記載の微生物。
[4] 酵母である、前記[1]〜[3]のいずれか1つに記載の微生物。
[5] サッカロミセス・セレビシエである、前記[1]〜[4]のいずれか1つに記載の微生物。
[6] 前記キシロース代謝酵素遺伝子が、キシロース還元酵素遺伝子、キシリトール脱水素酵素遺伝子及びキシルロースリン酸化酵素遺伝子からなる群より選択される少なくとも1つのものである、前記[1]〜[5]のいずれか1つに記載の微生物。
[7] グリシン合成系タンパク質の遺伝子及び/又はメチオニン合成系タンパク質の遺伝子の発現機能を喪失させることにより、キシロース醗酵能力の増加した微生物を作製する方法。
[8] 前記グリシン合成系タンパク質の遺伝子がGLY1遺伝子である、前記[7]に記載の方法。
[9] 前記メチオニン合成系タンパク質の遺伝子がMET6又はMET32遺伝子である、前記[7]又は[8]に記載の方法。
[10] 前記微生物が酵母である、前記[7]〜[9]のいずれか1つに記載の方法。
[11] 前記微生物がサッカロミセス・セレビシエである、前記[7]〜[10]のいずれか1つに記載の微生物。
[12] 前記微生物がキシロース代謝酵素遺伝子が導入されたものである、前記[7]〜[11]のいずれか1つに記載の方法。
[13] 前記キシロース代謝酵素遺伝子が、キシロース還元酵素遺伝子、キシリトール脱水素酵素遺伝子及びキシルロースリン酸化酵素遺伝子からなる群より選択される少なくとも1つのものである、前記[12]に記載の方法。
[14] 前記[1]〜[6]のいずれか1つに記載の微生物と、キシロース含有原料とを接触させる工程を含む、エタノールの製造方法。
【発明の効果】
【0009】
本発明で育種した酵母を使用することによって、キシロースからのエタノール醗酵能、あるいは醗酵速度を改善することができる。どのような醸造用酵母、実験室酵母においても、同様の育種をすることができる。また、キシロースを含むものであれば、どのようなバイオマスから得られたもろみであっても、本育種酵母で同様の効果が見込める。
本発明により、優れたキシロース醗酵能を有する微生物が提供される。また、本発明の微生物の作製方法を用いることにより、広範囲な微生物を宿主として、同様に高いキシロース醗酵能を有する育種を作成することができる。本発明の微生物を用いることにより、キシロース含有原料から効率よくエタノールを製造することができる。また、本発明の微生物を用いると、例えばアミノ酸含有量の低いバイオマスを原料とした場合でも、効率よくエタノールを製造することができる。
【図面の簡単な説明】
【0010】
【図1】実施例で用いたプラスミドpIUX1X2XKOの略図である。
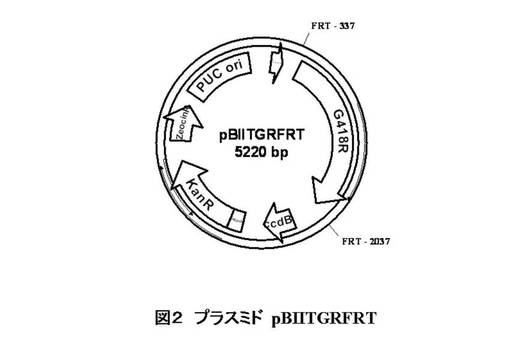
【図2】実施例で用いたプラスミドpBIITGRFRTの略図である。
【図3】実施例で用いたプラスミドpUGRFRTの略図である。
【図4】実施例で用いたプラスミドYRNFLPの略図である。
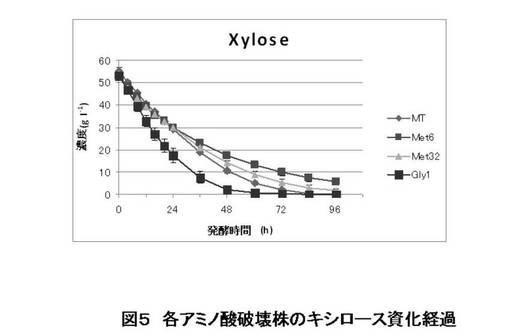
【図5】本発明の微生物を用いたキシロースからのエタノール醗酵試験の結果を示す(キシロース資化量)。
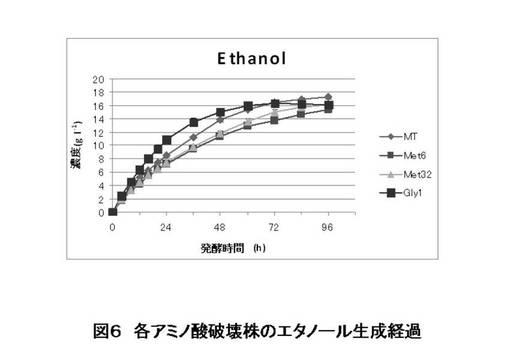
【図6】本発明の微生物を用いたキシロースからのエタノール醗酵試験の結果を示す(エタノール生産量)。
【図7】本発明の微生物を用いたキシロースからのエタノール醗酵試験の結果を示す(キシリトール生成量)。
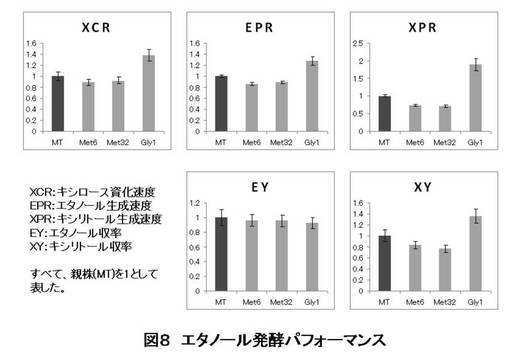
【図8】本発明の微生物を用いたキシロースからのエタノール醗酵試験の結果を示す(XCR:キシロース資化速度、EPRエタノール生成速度、XPR:キシリトール生成速度、EY:エタノール収率、XY:キシリトール収率、Met6:MET6遺伝子破壊株、Met32:MET32遺伝子破壊株、Gly1:GLY1遺伝子破壊株、MT:親株)。
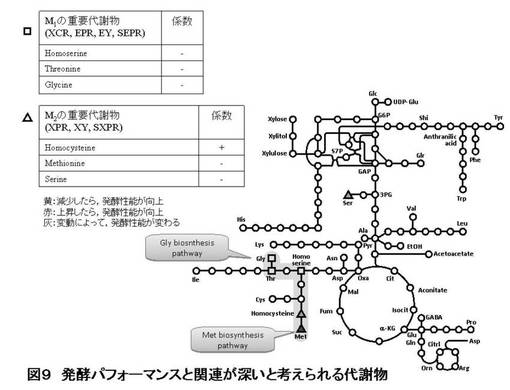
【図9】実施例1のキシロース醗酵試験及び酵母細胞の細胞内成分の網羅的分析の結果を示す図である。
【発明を実施するための形態】
【0011】
本発明者らは、グリシン合成系タンパク質の遺伝子の発現を抑制した微生物株を用いた場合、野生型同種株と比べて、キシロースの資化速度が上昇することを見出し、さらに、メチオニン合成系タンパク質の遺伝子の発現を抑制した微生物株では、キシロースを原料とするエタノール生成において副産物であるキシリトールの生産量が減少することを見出し、本発明を完成させた。以下において、本願発明の各実施態様を記載する。
【0012】
1. グリシン合成系タンパク質遺伝子及び/又はメチオニン合成系タンパク質遺伝子機能喪失微生物
本発明は、ある実施態様において、グリシン合成系タンパク質遺伝子及び/又はメチオニン合成系タンパク質遺伝子の発現機能を喪失させた、微生物を提供する。
【0013】
本発明において「微生物」とは、肉眼でその存在が判別できず、顕微鏡などによって観察できる程度以下の大きさの生物を意味する。微生物の例としては、細菌、藍色細菌、古細菌等の原核生物、並びに糸状菌、酵母、変形菌、担子菌、単細胞性の藻類及び原生動物等の真核生物が挙げられる。
好ましくは、微生物は、糖に対する醗酵能を有する種であり、より好ましくは、酵母である。酵母は、出芽酵母又は分裂酵母のいずれであってもよい。ある実施態様では、酵母は、サッカロミセス・セレビシエNBRC1951、NBRC1952、NBRC1953、NBRC1954、X2180-1A (ATCC26786)、CB11 (Berkley Stock Center)、W303-1A (BY4848)、MT8-1(BY2685)、NBRC1440、NBRC1445等の出芽酵母であってもよい。別の実施態様では、酵母は、シゾサッカロミセス・ジャポニクス(Schizosaccharomyces japonicus (Hasegawaea japonicus))、シゾサッカロミセス・オクトスポルス(Schizosaccharomyces octosporus (Octosporomyces octosporus))及びシゾサッカロミセス・ポンベ(Schizosaccharomyces pombe)等の分裂酵母であってもよい。
【0014】
本発明において「グリシン合成系タンパク質の遺伝子」又は「グリシン合成系タンパク質遺伝子」とは、グリシン合成に関わる酵素タンパク質や転写調節因子をコードする遺伝子を意味し、DNA又はRNAのいずれであってもよい。例えば、酵母由来のものでは、Saccharomyces genome database (http://www.yeastgenome.org/)に記載のsuperpathway of serine and glycine biosynthesis (http://www.yeastgenome.org/cgi-bin/search/ textSearch.pl?query=glycine&type=pathway) に関与する遺伝子、SHM2 、SHM1、GLY1 、SER3、AGX1 、SER1、 SER2 、SER33などが挙げられる。 グリシン合成系タンパク質とは、グリシン合成に関わる酵素タンパク質及びその転写調節因子であり、例えば、酵母由来のものでは、上記superpathwayのSHM2 、SHM1、GLY1 、SER3、AGX1 、SER1、 SER2 、SER33にコードされるタンパク質がある。好ましくは、L-トレオニンを加水分解してグリシンを生成する活性(以下、「L-トレオニン・アルドラーゼ活性」)を有するタンパク質(以下、「L-トレオニン・アルドラーゼ」)であり、酵母の場合はGLY1にコードされる。
【0015】
本明細書中で使用される場合、「グリシン合成系タンパク質遺伝子」は、サッカロミセス・セレビシエ由来のL-トレオニン・アルドラーゼ「GLY1」(配列番号2)をコードする遺伝子(配列番号1、Saccharomyces Genome Database Accession No:YEL046C)に限定されず、L-トレオニン・アルドラーゼ活性を示す、L-トレオニン・アルドラーゼと同じファミリーに属するホモログタンパク質をコードする遺伝子でもよい。L-トレオニン・アルドラーゼと同じファミリーに属するタンパク質は多くの植物において見出されており、ファミリー内全体で高度に保存されている。L-トレオニン・アルドラーゼは、グリシン合成に必須のタンパク質であるため、その遺伝子はファミリー内全体で高度に保存されており、複数の微生物のL-トレオニン・アルドラーゼ又はそのホモログタンパク質の遺伝子については既に塩基配列が解析され、その配列情報がデータベースに登録されている。微生物由来のL-トレオニン・アルドラーゼ及びそのホモログをコードする遺伝子を表1に示すが、これらに限定されるものではない。
【表1】

サッカロミセス・セレビシエを用いて本発明の微生物を作製する場合、グリシン合成系タンパク質遺伝子は、好ましくはGLY1遺伝子である。
【0016】
一実施形態において、グリシン合成系タンパク質遺伝子は、L-トレオニン・アルドラーゼ活性を有するポリペプチドをコードするポリヌクレオチドの塩基配列において1または複数個の塩基が欠失、挿入、置換、または付加された変異体であってもよい。変異体は、コードもしくは非コード領域、またはその両方において変異され得る。コード領域における変異は、保存的または非保存的なアミノ酸の欠失、挿入、置換および/または付加を生成し得る。本明細書中で使用される場合、配列番号2のアミノ酸配列をコードする遺伝子、または当該アミノ酸配列において1もしくは複数個(例えば、1〜40個、1〜20個、1〜15個、1〜10個、1〜9個、1〜8個、1〜7個、1〜6個、1〜5個、1〜4個、1〜3個、1〜2個、1個など)または1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列をコードする遺伝子もまた、「グリシン合成系タンパク質の遺伝子」に包含される。
【0017】
本発明において「メチオニン合成系タンパク質の遺伝子」又は「メチオニン合成系タンパク質遺伝子」とは、メチオニン合成に関わる酵素タンパク質や転写調節因子をコードする遺伝子を意味し、DNA又はRNAのいずれであってもよい。例えば、酵母由来のものでは、Saccharomyces genome databaseに記載のsuperpathway of threonine and methionine biosynthesis(http://pathway.yeastgenome.org/YEAST/new-image?type=PATHWAY&object=P4-PWY&detail-level=2) に関与する遺伝子、MET2、HOM3、FOL3、HOM2、MET7、MET17、THR1、THR4、HOM6、MET6 や、superpathway of methionine salvage pathway(http://pathway. yeastgenome.org/YEAST/new-image?type=PATHWAY&object=PWY3O-351&detail-level=2)に関与する遺伝子、MRI1 、 BAT1 、ARO9 、 MDE1 、SAM2、 ARO8 、SPE2 、SPE3、SPE4、UTR4、MEU1、ADI1、BAT2、SAM1、および、これら遺伝子の転写調節に関わる遺伝子、MET4、MET28、MET31、MET32が挙げられる。メチオニン合成系タンパク質とは、メチオニン合成に関わる酵素タンパク質及びその転写調節因子であり、例えば、酵母由来のものでは、上記superpathwayのMET2、HOM3、FOL3、HOM2、MET7、MET17、THR1、THR4、HOM6、MET6、MRI1 、 BAT1 、ARO9 、 MDE1 、SAM2、 ARO8 、SPE2 、SPE3、SPE4、UTR4、MEU1、ADI1、BAT2、SAM1、MET4、MET28、MET31、MET32にコードされるタンパク質がある。好ましくは、ホモシステインの硫黄をメチル化してメチオニンを生成する活性(以下、「メチオニン・シンターゼ活性」)を有するタンパク質(以下、「メチオニン・シンターゼ」)及びメチオニン・シンターゼの転写制御因子である。酵母の場合、メチオニン・シンターゼの遺伝子の例としてはMET6が挙げられ、その転写制御因子をコードする遺伝子としてはMET32が挙げられる。
【0018】
本明細書中で使用される場合、「メチオニン合成系タンパク質遺伝子」は、サッカロミセス・セレビシエ由来のメチオニン・シンターゼ「MET6」(配列番号4)をコードする遺伝子(配列番号3、Saccharomyces Genome Database Accession No:YER091C)及びその転写因子である「MET32」(配列番号6)をコードする遺伝子(配列番号5、Saccharomyces Genome Database Accession No:YDR253C)に限定されず、メチオニン・シンターゼ活性を示す、MET6と同じファミリーに属するホモログタンパク質をコードする遺伝子(即ち、MET6のホモログ遺伝子)でもよく、前記MET6ホモログ遺伝子の転写因子をコードする遺伝子(即ち、MET32のホモログ遺伝子)であってもよい。MET6及びMET32と同じファミリーに属するタンパク質は多くの植物において見出されており、ファミリー内全体で高度に保存されている。微生物由来のMET6及びそのホモログをコードする遺伝子を表2に示し、微生物由来のMET32及びそのホモログをコードする遺伝子を表3に示すが、これらに限定されるものではない。
【表2】
【表3】
サッカロミセス・セレビシエを用いて本発明の微生物を作製する場合、メチオニン合成系タンパク質遺伝子は、好ましくはMET6又はMET32遺伝子である。
【0019】
一実施形態において、メチオニン合成系タンパク質遺伝子は、メチオニン・シンターゼ活性を有するポリペプチド又はその転写因子をコードするポリヌクレオチドの塩基配列において1または複数個の塩基が欠失、挿入、置換、または付加された変異体であってもよい。変異体は、コードもしくは非コード領域、またはその両方において変異され得る。コード領域における変異は、保存的または非保存的なアミノ酸の欠失、挿入、置換および/または付加を生成し得る。本明細書中で使用される場合、配列番号4又は6のアミノ酸配列をコードする遺伝子、または当該アミノ酸配列において1もしくは複数個(例えば、1〜40個、1〜20個、1〜15個、1〜10個、1〜9個、1〜8個、1〜7個、1〜6個、1〜5個、1〜4個、1〜3個、1〜2個、1個など)または1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列をコードする遺伝子もまた、「メチオニン合成系タンパク質の遺伝子」に包含される。
【0020】
本発明において、「グリシン合成系タンパク質の遺伝子及び/又はメチオニン合成系タンパク質の遺伝子の発現機能を喪失させた」とは、グリシン合成系タンパク質遺伝子及び/又はメチオニン合成系タンパク質遺伝子(以下、「対象タンパク質遺伝子」と総称)から本来の生物学的機能を有する対象タンパク質が発現されないことを意味する。このような状態の例としては、対象タンパク質遺伝子の発現産物が全く生成されない状態だけでなく、当該遺伝子の発現産物(例えば、hnRNA、mRNA又はタンパク質)は発現されるが、それら発現産物が本来の正常な機能を有しない状態が挙げられる。このような対象タンパク質遺伝子の機能喪失は、対象タンパク質遺伝子又はその転写調節領域若しくはプロモーター領域を含む発現制御領域上における1又は複数のヌクレオチドの欠失、置換、及び/又は挿入等によって生じさせることができる。なお、前記欠失、置換、及び/又は挿入を行う部位や、欠失、置換、及び/又は挿入される配列は、対象タンパク質遺伝子の正常な機能が喪失しうる限り、特に限定されないが、好ましくは、対象タンパク質の活性部位をコードする遺伝子配列の少なくとも1つが欠失していることが好ましい。
【0021】
本発明の微生物は、特に真核生物の場合、前記対象タンパク質の発現機能が、染色体上の少なくとも一方のアレル(ヘテロ接合型)で喪失していれば効果がある場合もあるが、好ましくは、両アレルで喪失していること(ホモ接合型)が好ましい。
【0022】
2. キシロース醗酵能力の増加した醗酵微生物を作製する方法
本発明は、宿主微生物のグリシン合成系タンパク質の遺伝子及び/又はメチオニン合成系タンパク質の遺伝子の発現機能を喪失させることにより、キシロース醗酵能力の増加した醗酵微生物を作製する方法を提供する。
グリシン合成系タンパク質遺伝子及び/又はメチオニン合成系タンパク質遺伝子の発現機能を喪失させる方法としては、ジーンターゲティング又はRNAi等のノックアウト又はノックダウン技術が挙げられる
【0023】
(1)ジーンターゲティング
ジーンターゲティングは、相同組換えを利用して染色体上の特定遺伝子に変異を導入する手法である(Capeccchi, M.R. Science, 244, 1288-1292, 1989)。
【0024】
まず、対象タンパク質遺伝子機能を喪失させるためのターゲティングベクターを構築する。ターゲティングベクターを構築するに当たり、対象微生物のゲノムDNAライブラリーを調製する。このゲノムDNAライブラリーは、多型等により組換え頻度が低下しないよう、ジーンターゲティングの対象となる微生物種と同系統の微生物、好ましくは、同種の微生物に由来するゲノムDNAから作製したライブラリーを用いることが好ましい。そのようなライブラリーとしては、市販のものを用いてもよい。あるいは、対象タンパク質のcDNA又はその部分配列をプローブとしてスクリーニングを行い、対象タンパク質のゲノム遺伝子のDNA配列をクローニングしてもよく、あるいは、対象タンパク質のcDNA又はその部分配列に基づいてプライマーを作成し、ゲノムの対象タンパク質遺伝子をPCR増幅することにより調製することができる。ゲノムDNAライブラリーの調製方法の詳細については、以下を参照できる。"Sambrook & Russell, Molecular Cloning: A Laboratory Manual Vol. 3, Cold Spring Harbor Laboratory Press 2001"、"Ausubel, Current Protocols in Molecular Biology, John Wiley &Sons 1987-1997"
【0025】
次に、クローニングされたゲノムDNAを、シークエンシング、サザンブロッティング、制限酵素消化等によって解析してエクソン(真核生物の場合)の位置及び制限酵素サイトを同定する。このような解析により得られた配列情報を基に変異導入部位等を決定する。
【0026】
本発明において、染色体上に導入する変異(欠失、置換、及び/又は挿入)は対象タンパク質遺伝子の正常な機能が損なわれる限り特に限定されず、これらの変異は、対象タンパク質遺伝子のイントロン領域、エクソン領域又は対象タンパク質遺伝子の発現制御領域に存在していてよい。前記変異は、対象タンパク質遺伝子のエクソン領域に存在することが好ましく、さらに好ましくは、対象タンパク質遺伝子の少なくとも1つのエクソンが欠失した変異、最も好ましくは全てのエクソンが欠失した変異である。このような変異であれば、確実に対象タンパク質遺伝子の機能を欠損させることができるからである。
【0027】
ターゲティングベクターは、変異導入部位の3’及び5’側の相同領域(それぞれ3’アーム及び5’アーム)に加え、組み換え体のセレクション用の選択マーカーを含んでいてもよい。選択マーカーの例としては、Nourseothricin dihydrogen sulfate(NAT)耐性遺伝子、ネオマイシン耐性遺伝子、ハイグロマイシンBホスホトランスフェラーゼ遺伝子、カナマイシン耐性遺伝子等のポジティブセレクションマーカー、LacZ、GFP(Green Fluorescence Protein)及びルシフェラーゼ遺伝子などの破壊対象遺伝子の発現レポーター、単純ヘルペスウイルスチミジンキナーゼ遺伝子(HSV-TK)、ジフテリア毒素Aフラグメント(DTA)等のネガティブセレクションマーカー等が挙げられる。特に、本発明は、微生物を宿主とするため、栄養要求性を相補する遺伝子、例えばURA3を用いれば、ウラシルを含まない寒天培地でセレクションが可能であり、宿主の栄養要求性によっては、使用が可能である。また、シクロヘキシミドに対する薬剤耐性遺伝子(YAP1)を用いれば、シクロヘキシミドを含んだ培地でセレクションを行うことができる。これらのセレクションマーカーを3’アーム及び5’アームの間に挿入した配列を用いることにより、ターゲット遺伝子を破壊することもできる。ベクターは相同領域の外側に、ベクターを直鎖化するための適当な制限酵素切断部位を含んでいてもよい。
このようなターゲティングベクターは、市販のプラスミドベクター(例えば、pENTR/D-TOPO(登録商標:Invitrogen)やpBluescriptII (Stratagene)等)をベースにして構築することができる。
【0028】
本発明の微生物は、対象タンパク質遺伝子の破壊後に、相同組換に用いた選択マーカーが、細胞から脱落していることが好ましい。例えば、上記ターゲティングベクターがコードする選択マーカー遺伝子の両端に、組換え配列を挿入し、当該組換え配列間で組換えを生じさせて、選択マーカー遺伝子をループアウトさせることにより、選択マーカーを宿主細胞から脱落させることができる。このようにして作製される本発明の微生物は、選択マーカーを有しないため、複数の対象タンパク質遺伝子を破壊する際に、選択マーカーが制限されないという優れた特徴を有する。
【0029】
選択マーカーの脱落に使用可能な組換え配列としては、Flippase Recognition Target 配列(FRT配列)、バクテリオファージP1由来のloxP配列、醤油酵母(Zygosaccharomyces rouxii)由来のRS配列、バクテリオファージMu由来のgix配列等が挙げられる。
これらの組換え配列間に組換えを生じさせるためには、適切なリコンビナーゼを発現させる必要がある。上記の組換え配列間に組換えを生じさせるリコンビナーゼは、それぞれ、Flippase(FLP:FLP-FRTリコンビネーション)、Cre(Cre-loxPリコンビネーション)、R Recombinase (R:RS リコンビネーション)、GIX recombinase (R:RS リコンビネーション)等が挙げられる。
さらに、リコンビナーゼをコードするプラスミドも宿主細胞から脱落していることが好ましい。宿主細胞の複数の対象タンパク質遺伝子の破壊において、選択マーカーが制限されないからである。
したがって、リコンビナーゼをコードするプラスミドは、宿主細胞内で不安定なものが好ましく、特に、当該プラスミドの選択マーカーに対する選択圧をかけない状態において、宿主細胞から脱落するものが好ましい。
そのようなプラスミドの例としては、YRpタイプのプラスミドが挙げられるが、これに限定されるものではない。
【0030】
選択マーカーは、任意の薬剤耐性(NAT耐性、ネオマイシン耐性、ハイグロマイシンBホスホトランスフェラーゼ耐性、カナマイシン耐性、ハイグロマイシン耐性、ゼオシン耐性、ジェネティシン耐性)、栄養要求性マーカー(ura3、ura5、niaD)、銅耐性マーカー(CUP1)(Marin et al., Proc. Natl. Acad. Sci. USA, 81, 337 1984)、セルレニン耐性マーカー(fas2m, PDR4)等であってもよいが、これらに限定されるものではない。
【0031】
図2及び3に、実施例で用いたサッカロミセス・セレビシエの対象タンパク質遺伝子のノックアウトに用いた断片を得る為のベクターの例を示す。このコンストラクトは、その上流と下流にFRT配列を持つG418耐性遺伝子、FRT-G418R-FRT断片を含む。G418Rは、ジェネティシン耐性遺伝子であり、対象タンパク質遺伝子が破壊された株のポジティブセレクションマーカーとして用いられる。これらプラスミドを鋳型として、対象タンパク質遺伝子の各ORFの開始コドンから下流80塩基の配列にFRT-G418R-FRT断片のすぐ上流20塩基が並んだプライマーと、終始コドンから上流80塩基の配列にFRT-G418R-FRT断片のすぐ下流20塩基が並んだプライマーを用いてPCRを行う。得られたPCR増幅断片は、対象タンパク質遺伝子の各ORFの開始コドンから下流80塩基の配列、あるいは終始コドンから上流80塩基の配列がそれぞれ両端に付加された、FRT-G418R-FRT断片を含む。この断片を用いて、宿主細胞を形質転換し、相同組換えによりジェネティシン耐性を獲得した株を取得する。
次に、取得した株にFLPをコードするプラスミド(図4に示すpYRNFLP)を導入してFRT配列間に組換えを生じさせ、ジェネティシン耐性遺伝子を脱落させる。
さらに、pYRNFLPも不安定であるため、非選択圧下において宿主細胞から脱落している。このようにして作製された本発明の微生物は、選択マーカーを有しないため、複数の遺伝子の破壊を行う際に、先の遺伝子の破壊に使用した選択マーカーと、同一の選択マーカーを繰り返し用いることが可能である。
【0032】
(2)RNA干渉
上記以外にも、対象タンパク質遺伝子の発現機能を欠損させる方法として、siRNA(small interfering RNA)等を用いたRNA干渉(RNA interference: RNAi)法が挙げられる。RNAi は、複数の段階を経て行われるマルチステッププロセスである。最初に、RNAi発現ベクターから発現した二本鎖RNA(Double Stranded RNA: dsRNA)又はヘアピン状のshRNA(Small Hairpin RNA)が Dicerによって認識され、21〜23 ヌクレオチドの siRNAs に分解される。次に、siRNAs は RNA 誘導型サイレンシング複合体 (RNA-Induced Silencing Complex: RISC) と呼ばれる RNAi 標的複合体に組み込まれ、RISC とsiRNAsとの複合体がsiRNAの配列と相補的な配列を含む標的mRNAに結合し、mRNAを分解する。標的mRNAは、siRNAに相補的な領域の中央で切断され、最終的に標的mRNAが速やかに分解されてタンパク発現量が低下する。最も効力の高い siRNA 二重鎖は、19bpの二重鎖の各3’末端にウリジン残基2個の突出部分を持つ 21 ヌクレオチド長の配列であることが知られている(Elbashir S.M. et al., Genes and Dev, 15, 188-200 (2001))。
【0033】
従って、対象タンパク質遺伝子ノックダウン微生物を得るには、まず、表1〜3に示されるような対象タンパク質遺伝子配列(これらに限定されない)の一部と相補的な配列を含むヌクレオチドを、適切なRNAi発現ベクターにdsRNA又はshRNAとして発現可能な状態で挿入してRNAiベクターを作製する。次に、前記ベクターを宿主細胞に導入し、形質転換体をセレクションすることにより対象タンパク質遺伝子ノックダウン微生物を得ることができる。
【0034】
dsRNA又はshRNAの設計及び合成は、市販のDNA/RNAシンセサイザー、例えば、Applied Biosystems394型で行うことも可能であり、あるいは第三者機関(例えば、TAKARA Bio)に委託することもできる。
【0035】
対象タンパク質遺伝子の発現機能の喪失は、正常な対象タンパク質遺伝子の発現量の低下として確認することができる。対象タンパク質遺伝子の発現量は、本発明の微生物の抽出物を用いたRT-PCR法及びアガロースゲル電気泳動、Real-Time PCR法、ノーザンブロッティング法、マイクロアレイ解析並びに質量分析法等によって測定することができる。これらの測定方法に用いるプライマー又はプローブは、当該対象タンパク質遺伝子の配列に基づいて設計及び合成することができる。対象タンパク質遺伝子の発現産物が正常であるかどうかは、当該発現産物の配列解析を行うことにより確認することができる。
【0036】
本発明の微生物は、対象タンパク質遺伝子の発現機能が喪失していることを特徴とするが、さらに、キシロース代謝酵素遺伝子が発現可能な状態で導入されていてもよい。キシロース代謝酵素遺伝子は、キシロースからエタノールを生成する一連の化学反応のうち、いずれかの反応を触媒する活性を有するタンパク質(以下、「キシロース代謝酵素」)をコードする遺伝子を意味する。キシロース代謝酵素遺伝子の例としては、キシロース還元酵素遺伝子、キシリトール脱水素酵素遺伝子、キシルロースリン酸化酵素遺伝子が挙げられる。
このようなキシロース代謝酵素遺伝子は、例えば、ピキア・スティピティス、サッカロミセス・セレビシエ、大腸菌、ラクトバシラス・カゼイ、ラクトバシラス・アシドフィラス、アスペルギルス・フミガツス、スタフィロコッカス・アウレウス、ピキア・パストリス、シゾサッカロミセス・ポンベ等に由来するものであってもよい。
本発明の微生物には、前記キシロース代謝酵素のうち、キシロース還元酵素遺伝子、キシリトール脱水素酵素遺伝子又はキシルロースリン酸化酵素遺伝子からなる群より選択される少なくとも1つが導入されていることが好ましく、これら遺伝子の2つ以上、あるいは、3つ全ての遺伝子が導入されていてもよい。
【0037】
キシロース還元酵素遺伝子、キシリトール脱水素酵素遺伝子又はキシルロースリン酸化酵素遺伝子の例としては、以下が挙げられるが、これに限定されるものではない。
【表4】
【表5】
【表6】
【0038】
キシロース還元酵素は、キシロース代謝において、以下の反応を触媒する酵素である。
Xylose+NADPH→Xylitol+NADP
【0039】
キシリトール脱水素酵素は、キシロース代謝において、以下の反応を触媒する酵素である。
Xylitol+NAD→Xylulose+NADH
【0040】
キシルロースリン酸化酵素は、キシロース代謝において、以下の反応を触媒する酵素である。
Xylulose+ATP→Xylulose-5P+ADP
【0041】
一実施形態において、キシロース代謝酵素遺伝子(キシロース還元酵素、キシリトール脱水素酵素又はキシルロースリン酸化酵素等)は、キシロース代謝活性を有するタンパク質をコードするものである限り、上記キシロース代謝酵素遺伝子の変異体であってもよい。変異体の例としては、上記キシロース代謝酵素タンパク質をコードするポリヌクレオチドの塩基配列において1または複数個の塩基が欠失、挿入、置換、または付加された変異体であってもよい。変異体は、コードもしくは非コード領域、またはその両方において変異され得る。コード領域における変異は、保存的または非保存的なアミノ酸の欠失、挿入、置換および/または付加を生成し得る。本明細書中で使用される場合、配列番号8、10又は12のアミノ酸配列をコードする遺伝子(配列番号7、9又は11)、または当該アミノ酸配列において1もしくは複数個(例えば、1〜40個、1〜20個、1〜15個、1〜10個、1〜9個、1〜8個、1〜7個、1〜6個、1〜5個、1〜4個、1〜3個、1〜2個、1個など)または1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列をコードする遺伝子もまた、「キシロース代謝酵素遺伝子」に包含される。
【0042】
ポリペプチドのアミノ酸配列中のいくつかのアミノ酸が、このポリペプチドの構造または機能に有意に影響することなく容易に改変され得ることは、当該分野において周知である。さらに、人為的に改変させるだけではく、天然のタンパク質において、当該タンパク質の構造または機能を有意に変化させない変異体が存在することもまた周知である。
【0043】
当業者は、周知技術を使用してポリペプチドのアミノ酸配列において1または複数個のアミノ酸を容易に改変させることができる。例えば、公知の点変異導入法に従えば、ポリペプチドをコードするポリヌクレオチドの任意の塩基を改変させることができる。また、ポリペプチドをコードするポリヌクレオチドの任意の部位に対応するプライマーを設計して欠失変異体または付加変異体を作製することができる。さらに、本明細書中に記載される方法を用いれば、作製した改変体が所望の活性を有するか否かを容易に決定し得る。
【0044】
好ましい改変体は、保存性もしくは非保存性アミノ酸置換、欠失、または添加を有する。好ましくは、サイレント置換、添加、および欠失であり、特に好ましくは、保存性置換である。これらは、本発明に係るポリペプチド活性を変化させない。
【0045】
代表的に保存性置換と見られるのは、脂肪族アミノ酸Ala、Val、Leu、およびIleの中での1つのアミノ酸の別のアミノ酸への置換;ヒドロキシル残基SerおよびThrの交換、酸性残基AspおよびGluの交換、アミド残基AsnおよびGlnの間の置換、塩基性残基LysおよびArgの交換、ならびに芳香族残基Phe、Tyrの間の置換である。
【0046】
上記に詳細に示されるように、どのアミノ酸の変化が表現型的にサイレントでありそうか(すなわち、機能に対して有意に有害な効果を有しそうにないか)に関するさらなるガイダンスは、Bowie, J.U.ら「Deciphering the Message in Protein Sequences: Tolerance to Amino Acid Substitutions」,Science 247:1306-1310(1990)(本明細書中に参考として援用される)に見出され得る。
【0047】
上記に挙げられる本発明に係るポリヌクレオチドを使用すれば、形質転換体または細胞においてキシロース代謝酵素活性を有するポリペプチドを合成することができる。
【0048】
キシロース代謝酵素遺伝子は、宿主細胞において発現可能な発現ベクターに組み込まれた状態で、導入されることが好ましい。発現ベクターは、以下の(i)〜(iii)の要素を含みうる。
(i)宿主細胞内で転写可能なプロモーター;
(ii)該プロモーターに結合した、キシロース代謝酵素遺伝子;及び
(iii)RNA分子の転写終結及びポリアデニル化に関し、宿主細胞内で機能するシグナルを構成要素として含む発現カセット
を含むように構成される。
【0049】
このようなベクターとしては、例えば、pYE22m(酵母用:Biosci. Biotech. Biochem., 59, 1221-1228, 1995)、YCp50 (酵母用:X70276)、YIp1 (酵母用:X70480)が挙げられる。
【0050】
宿主細胞中での遺伝子発現を調節するためのプロモーター/ターミネーターとしては、宿主細胞中で機能する限り、任意の組み合わせでよい。例えば、酵母で利用する場合はTDH3、PYK1、PGK 、GAP 、TPI、GAL1、GAL10 、ADH2、PHO5、CUP1、MFα1等のプロモーターを用いることができるが、これに限定されるものではない。構成的高発現プロモーターとしては、TDH3、TPI1、PGK1、PGI1、PYK1、ADH1等のプロモーターを使用することができる。
形質転換の際に用いる選択マーカーとしては、栄養要求性マーカー(ura3、ura5、niaD)、薬剤耐性マーカー(ハイグロマイシン耐性、ゼオシン耐性、ジェネチシン耐性)、銅耐性遺伝子(CUP1)(Marin et al., Proc. Natl. Acad. Sci. USA, 81, 337 1984)、セルレニン耐性遺伝子(fas2m, PDR4)(それぞれ猪腰淳嗣ら, 生化学, 64, 660, 1992; Hussain et al., gene, 101, 149, 1991)等が利用可能である。
【0051】
宿主細胞の形質転換方法としては、一般に用いられる公知の方法が利用できる。例えば、原核生物の場合、エレクトロポレーション法(Mackenxie D. A. et al. Appl. Environ. Microbiol., 66, 4655-4661, 2000)やヒートショック法が利用できる。また、酵母の場合は、エレクトロポレーション法、スフェロプラスト法(Proc. Natl. Acad. Sci. USA, 75 p1929(1978))、酢酸リチウム法(J. Bacteriology, 153, p163(1983))、Proc. Natl. Acad. Sci. USA, 75 p1929 (1978)、Methods in yeast genetics, 2000 Edition: A Cold Spring Harbor Laboratory Course Manual等に記載の方法で実施可能であるが、これらに限定されない。
【0052】
その他、一般的なクローニング技術に関しては、"Sambrook & Russell, Molecular Cloning: A Laboratory Manual Vol. 3, Cold Spring Harbor Laboratory Press 2001"、"Methods in Yeast Genetics、A laboratory manual (Cold Spring Harbor Laboratory Press、Cold Spring Harbor, NY)"等を参照することができる。
【0053】
3.エタノールの製造方法
上記のようにして作製された本発明の微生物は、同種の野生型微生物と比べて、キシロース醗酵能が増加している。ここで、キシロース醗酵とは、キシロースを資化することにより、エタノールを生成することを意味する。したがって、本発明の微生物をキシロース含有原料の存在下で培養した場合、同種の野生型微生物を同一条件下で培養した場合よりも、早くキシロースを資化することが出来、副生成物であるキシリトールの生成量が少なく、キシロースからのエタノール醗酵能が上昇する。
このことから、本発明は、別の実施態様において、本発明の微生物と、キシロース含有原料とを接触させる工程を含む、エタノールの製造方法を提供する。
【0054】
本発明の微生物と、キシロース含有材料とを接触させる工程は、本発明の微生物と、キシロース含有材料とを混合することにより達成できる。好ましくは、本発明の微生物は、活性な状態でキシロース含有材料と接触させる。
本発明の微生物を活性な状態でキシロース含有材料と接触させる方法の例としては、本発明の微生物が増殖可能な培養条件において、かつ、キシロース含有原料の存在下で、本発明の微生物を培養する方法が挙げられる。このような、培養条件は、微生物種によって異なるが、例えば、酵母であれば、キシロース含有原料を添加したYPD培地(イーストエキストラクト 10 g/L、ポリペプトン 20 g/L、グルコース 20 g/L)、SX培地(キシロース50 g/l、6.7 g/l yeast nitrogen baseアミノ酸抜き、アデニン20 mg/l、ヒスチジン 20 mg/l、ロイシン 100 mg/l、トリプトファン 20mg/l)等の適切な培地中で30℃にて培養することができる。
あるいは、本発明の微生物は、キシロース含有原料の存在下で培養を行う前に、本発明の微生物が増殖可能な培養条件において培養して菌体を増殖させ、その後、キシロース含有原料の存在下で、本発明の微生物を用いて醗酵を行なってもよい。
但し、培養条件は、上記に限定されるものではない。当業者であれば、適宜、微生物種に応じた適切な培養条件を選択し、実際に培養を行うことができる。
【0055】
キシロースからのエタノール醗酵能は、本発明の微生物とキシロース含有原料とを接触させ、一定時間(例えば、72時間、48時間、24時間、12時間、10時間、8時間、6時間、4時間、2時間、1時間)が経過した後に系のエタノール生成量を測定することにより確認することができる。
【0056】
キシロース資化量及びエタノール生成量は、本発明の微生物とキシロース含有原料とを含む醗酵系の成分を、高速液体クロマトグラフィー等の公知の分析手法によって分析することにより、容易に測定することができる。
【0057】
「キシロース含有原料」は、キシロースを含む原料であれば、特に限定されないが、好ましくは、キシロースを含む生物由来の原料、即ち、「キシロース含有バイオマス」である。バイオマスは、日本国農林水産省が発行した「バイオマス・ニッポン総合戦略」において、「再生可能な、生物由来の有機性資源で化石資源を除いたもの」と定義されている。
よって、この定義に基づき、「キシロース含有バイオマス」は、「再生可能な、生物由来の有機性資源で化石資源を除いたものであって、キシロースを含むもの」と定義することができる。
キシロース含有バイオマスの具体例としては、サトウキビ、トウモロコシ、テンサイ、ジャガイモ、サツマイモ、麦、モロコシ(=こうりゃん)、ソルガムなどの非可食部や、廃木材、パルプ廃液、バガス、もみ殻、コーンコブ、稲ワラ、スイッチグラス、エリアンサス、ネピアグラスが挙げられるが、これらに限定されるものではない。
【0058】
以下、実施例を用いて本発明を詳細に説明するが、本発明は実施例に記載された態様に限定されるものではない。
【実施例】
【0059】
試験方法:
本実施例で用いた試験項目および試験方法を以下に示す。特に断りのない限り、本実施例における試験方法はこれに準じた。
【0060】
<酵母株>
本発明において用いた株は、以下の(a)〜(e)のとおりである。いずれの株も、P.stipitisのキシロース還元酵素遺伝子(XYL1:配列番号7)とキシリトール脱水素酵素遺伝子(XYL2:配列番号9)とS.cerevisiaeのキシルロースリン酸化酵素遺伝子(XKS1:配列番号11)とをS.cerevisiaeのTDH3遺伝子のプロモーター制御下に連結させた発現ユニットを持つプラスミド・pIUX1X2XK(図1)が、染色体上のURA3遺伝子領域に相同組換えで導入されている。株については、これに限定されるものではなく、キシロース醗酵能を持つ酵母であれば、何でも良い。
(a) MT8-1/pIUX1X2XK(Mata ade his3 leu2 trp1/pIUX1X2XK)
(b) MT8-1/pIUX1X2XK/Ad(Mata ade his3 leu2 trp1, キシロース培地に順化した株)
(c) MT8-1/pIUX1X2XK/pGK404-ScTal(Mata ade his3 leu2 /pIUX1X2XK/pGK404-ScTal)
(d) NBRC1440/pIUX1X2XK(Matα・his3 trp1/pIUX1X2XK)
(e) NBRC1445/pIUX1X2XK(Mata his3 trp1/pIUX1X2XK)
MT8-1は、ナショナルバイオリソースプロジェクトにNBRP No. BY2685として登録されている。
【0061】
<遺伝子破壊方法>
以下の各々のプライマー(1)〜(16)を用いて、プラスミドpBIITGRFRT(図2)((1)+(2))、あるいはpUGRFRT(図3)((3)+(4)、(5)+(6))を鋳型としてPCRを行った。これらにより、MET6((1) +(2))、MET32((3)+(4))、GLY1 ((5)+(6))の各ORFの開始コドンから下流80塩基の配列、あるいは終始コドンから上流80塩基の配列がそれぞれ両端に付加された、FRT-G418R-FRT断片を増幅される。FRTとは酵母2μプラスミド上にある、リコンビナーゼによって組換えが起こる、組換え部位であり、その配列は、5’-GAAGTTCCTATTCTCTAGAAAGTATAGGAACTTC-3’(配列番号13)である。G418Rとは、G418耐性マーカー遺伝子( PGK1p::KanMX)であり、pBIITGRFRTとpUGRFRTは両者とも、FRT-G418R-FRT断片を持つプラスミドである。増幅した各ORFの両末端配列を持つFRT-G418R-FRT断片でMT8-1/pIUX1X2XKを形質転換し、形質転換後の菌体をジェネティシン 300 μg/mlを含むYPD (イーストエキストラクト 10 g/L、ポリペプトン 20 g/L、グルコース*20 g/L(ろ過滅菌添加*))に塗布した。生育した株において、FRT-G418R-FRT断片が、それぞれMET6、MET32、GLY1のORFと置き換わったことを確認するために、各々、数個のコロニーからゲノミックDNAを調製した。これを鋳型として、MET6については、(7)と(10)、MET32については(8)と(10)、GLY1については(9)と(10)、のプライマーを用いてPCRを行った。プライマー(7)、(8)、(9)は、各ORFの5’上流の配列、(10)はG418耐性遺伝子に相補的な配列である。PCRで適切な大きさのDNAが増幅された株については、さらにプラスミドpYRNFLP (図4)を形質転換し、Nourseothricin dihydrogen sulfate 50 μg/mlを含むYPDに塗布した。
(1) MET6DF1: 5’-ATGGTTCAATCTGCTGTCTTAGGGTTCCCAAGAATCGGTCCAAACAGAGAATTAAAGAAGGCCACTGAAGGTTACTGGAAACTCAAGCTATGCATCAAGC-3’ (配列番号14)
(2) MET6DR1: 5’-TTAATTCTTGTATTGTTCACGGAAGTACTTGGCGGCTTCGACCATATGAGTCAAAGACAATCT
AGTTTCTTCCCAGCCTCAATACGACTCACTATAGGGC-3’ (配列番号15)
(3) MET32DF1:5’-ATGGAGGATCAGGATGCTGCATTTATCAAACAGGCTACAGAAGCAATAGTGGATGTATCATTA
AATATAGATAACATAGAGTAAAACGACGGCCAGTGCC-3’ (配列番号16)
(4) MET32DR2: 5’-TCAGCCATTACTGCTACCATTGTGGTTGTTATCTTGACGATGAACGATGTTGGATTGCTTG
ACTTTTTTCAGTAATTCATCCGGCTCGTATGTTGTGTGG-3’ (配列番号17)
(5) GLY1DF1: 5’-ATGACTGAATTCGAATTGCCTCCAAAATATATCACCGCTGCTAACGACTTGCGGTCAGACACA
TTCACCACTCCAACTGCGTAAAACGACGGCCAGTGCC-3’ (配列番号18)
(6) GLY1DR1: 5’-TCAGTATTTGTAGGTTTTTATTTCGCGGATAGCGTTGCCATCAACGTCGACCTCGGTGGATTC
ACTACGGTAAATCTGGGCCGGCTCGTATGTTGTGTGG-3’ (配列番号19)
(7) MET6-5UF1: 5’-AAACTGTGGTAGTCATAGCTC-3’ (配列番号20)
(8) MET32-5UF1: 5’-TATACTAGTCAAAGATAGTATCCACC-3’ (配列番号21)
(9) GLY1-UF1: 5’-ATGTCACGCGAAACGGACC-3’ (配列番号22)
(10) G418MER: 5’-AAATGCTTGATGGTCGGAAG-3’ (配列番号23)
(11) MET6NF1: 5’-ATGGTTCAATCTGCTGTCTTAGGGT-3’ (配列番号24)
(12) MET6CR: 5’-TTAATTCTTGTATTGTTCACGGAAGTAC-3’ (配列番号25)
(13) MET32NF1: 5’-ATGGAGGATCAGGATGCTGCATTT-3’ (配列番号26)
(14) MET32CR2: 5’-TCAGCCATTACTGCTACCATTGTG-3’ (配列番号27)
(15) GLY1NF1: 5’-TGACTGAATTCGAATTGCCTC-3’ (配列番号28)
(16) GLY1CR1: 5’-ATAGCGTTGCCATCAACGTC-3’ (配列番号29)
【0062】
pYRNFLPは、酵母2μプラスミド上にあるFLPリコンビナーゼをTDH3p制御下で発現させるYRpタイプのプラスミドである。生育した株のゲノミックDNAを鋳型として、MET6については、(11)と(12)、MET32については(13)と(14)、GLY1については(15)と(16)のプライマーを用いてPCRを行った。(11)〜(16)のプライマーは全長の各ORFを増幅できるプライマーであり、これらを用いてPCRを行うと、FRT領域で組換えが起こりG418耐性遺伝子がループアウトした株では、ORFの長さに関係なく、約350bpの短い断片が増幅される。取得した各遺伝子破壊株は、G418耐性遺伝子がなくなっている。また、YRpタイプのプラスミドは安定性が悪く、選択圧をかけない状態では、細胞からすぐに脱落してしまうので、Nourseothricin dihydrogen sulfate(NAT)を含まない培地で生育させることにより、pYRNFLPも脱落させることができる。従って本方法で遺伝子を破壊した株は、選択マーカー(薬剤耐性)を持たないため、同じ方法で、さらに複数の遺伝子を破壊することができる。
【0063】
<PCRのための鋳型DNA調製方法>
酵母細胞を50 μlのLysis buffer(0.125 mg/ml ザイモリアーゼ100T、1 M ソルビトール、40 mM リン酸カリウムバッファー pH6.8、1 mMジチオスレイトール)に懸濁する。30℃で1時間インキュベートした後、プロテアーゼE (1 mg/ml) を5μl加え、さらに55℃で20分、99℃で10分インキュベートする。15,000 rpmで10分遠心を行い、上清を鋳型DNAとした。
【0064】
<キシロースからのエタノール醗酵試験>
SD培地(20 g/lグルコース、6.7 g/l yeast nitrogen baseアミノ酸抜き、アデニン20 mg/l、ヒスチジン 20 mg/l、ロイシン 100 mg/l、トリプトファン 20 mg/l)5 mlで前培養した酵母を初期OD600が0.03になるように500 mlのSD培地(casamino acid 20 g/lを含有)に移し、48 h、30℃で振とう培養機で好気培養した。細胞を遠心分離機で回収し、滅菌水で洗浄した後、初期OD600が20になるように100 mlのSX培地(キシロース50 g/l、6.7 g/l yeast nitrogen baseアミノ酸抜き、アデニン20 mg/l、ヒスチジン 20 mg/l、ロイシン 100 mg/l、トリプトファン 20 mg/l)に入れ、醗酵を開始した。醗酵は、生成した二酸化炭素ガスを逃すための出口がついた100 mlメディウム瓶で行い、100-150 rpmのスターラーで培養液を撹拌した。醗酵の進行を監視するために、培地を1 ml回収し、1000 gで遠心分離した後、上澄みを回収し、HPLCで分析した。
【0065】
<メタボローム解析用サンプルの調製法>
醗酵液から経時的にサンプリングした酵母細胞は遠心分離(1000 g、 3分)によって収穫した。細胞のペレットは、MilliQ水(Millipore、 Tokyo、Japan)で2回洗浄した後、液体窒素で凍結した。細胞は凍結乾燥し、使用時まで-80°Cで保存した。
【0066】
<醗酵もろみのHPLC分析>
醗酵培地中のキシロース、キシリトール、エタノール濃度はShimPack SPR-Pbカラム(Shimadzu、Kyoto、Japan)を用いたHPLC法(GL-7400システム、GL Sciences)によって定量した。カラムは80℃に保温し、MilliQ水を移動相として用い、流量は0.6 ml/minと設定した。各化合物はGL-7454の示差屈折率検出器(Shimadzu)によって検出した。
【0067】
細胞内代謝物分析方法
<GC-MS分析>
・ 10 mgの酵母の乾燥細胞を2 mlチューブに入れる
・ 混合溶媒1000 ml (MeOH : CHCl3 : H2O = 2.5 : 1 : 1)と内部標準溶液60 ml (2-isopropylmalic acid 1 mg/ml )を加える
・ シェーカーで1200 rpm、 37℃ 、 30分混合する
・ 遠心分離機で15000 x g、 4℃、 3分遠心分離する
・ 上澄み900 mlを1.5 mlチューブに移し、MilliQ水を400 ml加え、vortexで混合する
・ 遠心分離機で15000 x g、 4℃、 3分で遠心分離する
・ 上澄み200 mlを1.5 mlチューブに移し、遠心濃縮機及び凍結乾燥機で乾燥させる
・ メトキシアミン(ピリミジンで溶解)20 mg/mlを100 ml加える
・ シェーカーで1200 rpm、 30℃、 90分混合する
・ MSTFA 1 mg/mlを50 ml加え、シェーカーで1200 rpm、 37℃、 30分混合する
・ バイアルに移し、1 mlをGC-TOF/MSに注入し、分析する
分析条件
GCシステム : 6890N (Agilent)
カラム : CP-Sil 8 CB-Low Bleed 30 m x 0.25 mm ID (DF = 0.25 mm)
オーブン温度 : 80℃ (2分保持)、 15℃/分で330℃まで温度上昇 後、6分保持
インジェクター温度 : 230℃
イオン源温度 : 200℃
MS システム : PegasusIII TOF-MS (Leco)
マスレンジ : 85-500 m/z
スキャン速度 : 20 スキャン/秒
【0068】
<CE-MS分析>
・ 10 mgの酵母の乾燥細胞を2 mlチューブに入れる
・ MeOH 500ml、 CHCl3 500ml、 MilliQ水180ml、と内部標準溶液20ml (100mM ribitol、 100mM PIPES)を加え、vortexで混合する
・ 遠心分離機で15000 x g、 4℃、 3分遠心分離する
・ 上澄み350 mlを1.5 mlチューブに移し、MilliQ水を100ml加え、vortexで混合する
・ 沈殿物にMilliQ水100mlを加え、vortexで混合する
・ 両サンプルを遠心分離機で15000 x g、 4℃、 3分遠心分離する
・ 上澄みを2本の減外ろ過チューブ(Millpore Ultrafree-MC 5kDa MWCO)にチューブに移す
・ 遠心濃縮機でオーバーナイト乾燥させる
・ MilliQ水50mlで溶解する
・ バイアルに移し、分析する
分析条件
CEシステム : P/ACE MDQ (Beckman Coulter)
キャピラリー : Fused silica capillary 50μm i.d. x 80 cm
シーズ液 : 5 mM HCOONH3 in 50% MeOH
電解質 : 50 mM CH3COONH3 (pH 9.0)
電圧 : 30.0 kV
MSシステム : 4000 QTRAP (Applied Biosystems)
イオン化法 : ESI
送液ポンプ : MP-711 (GL Sciences)
【0069】
実施例1:複数のキシロース醗酵性酵母による、エタノール醗酵試験1
7種類のキシロース醗酵性酵母 ((1) MT8-1/pIUX1X2XK (2) MT8-1/pIUX1X2XK/Ad (3) MT8-1/pIUX1X2XK/pGK404-ScTal (4) NBRC1440/pIUX1X2XK (5) NBRC1445/pIUX1X2XK)を、実験方法に示した方法にてSD培地にて前培養し、細胞を集菌した後、滅菌水で洗浄した。酵母細胞をOD600=20となるように100 ml のSX培地に植菌して、キシロースからのエタノール醗酵試験を行った。適時醗酵もろみを1mlサンプリングし、HPLCでキシロース、キシリトール、エタノール濃度を測定した。また、同時に酵母細胞をサンプリングし、実験方法に示した方法で、GC-MSおよびCE-MS分析を行い、細胞内成分の網羅的分析を行った。得られた代謝物プロファイルを説明変数、諸性能評価値を応答変数としたOPLS回帰モデルを構築して、モデル構築に重要な説明変数を解析した結果、下記の化合物がキシロースからのエタノール醗酵性能と深く関与することがわかった。
Homoserine
Homocysteine
Methionine
Threonine
Glycine
Serine
【0070】
Homoserine、Threonine、GlycineはXCR(キシロース資化速度 g-comsumed xylose/h)、 EPR(エタノール生成速度 g-ethanol/h)、 EY(エタノール収率 g-ethanol/ g-comsumed xylose)、 SEPR (比エタノール生産速度 g-ethanol/g-cell-dried weight/h)の各醗酵性能の予測モデルに重要な意味を持つ代謝物と考えられた。これらの代謝物はグリシン生合成経路の中間体であるため、上記の醗酵性能はグリシン生合成経路と関連すると予測される。予測モデルから説明変数の係数値を解析したところ、これらの代謝物の細胞内蓄積量を減少させることによって上記の醗酵性能が向上されると予想された。そこで、これらの物質の細胞内蓄積量の減少を実現するために、グリシン生合成経路にある遺伝子の1つ、GLY1を破壊するターゲット遺伝子として選択した。
【0071】
一方、Homocysteine、Methionine、Serineは、 XPR(キシリトール生成速度 g-xylitol/h)、 XY(キシリトール収率 g-xylitol/g-comsumed xylose)、 SXPR(比キシリトール生産速度 g-xylitol/g-cell-dried weight/h)の各醗酵性能の予測モデルに重要な意味を持つ代謝物と考えられた。これらの代謝物はメチオニン生合成経路の中間体であり、上記の醗酵性能はこの経路と関連すると予測される。予測モデルから説明変数の係数値を解析したところ、Homocysteineの細胞内蓄積量を減少することによってキシリトール関連性能(XPR、XY、SXPR)が減少すると予想された。そこで、メチオニン合成経路関連遺伝子を破壊、することによって、Homocysteineの蓄積量を減少させることができれば、キシリトール生産を抑えることができると予測した。破壊するターゲット遺伝子として、MET6とMET32を選択した。図9に醗酵パフォーマンスと関連が深いと考えられる代謝物とそのマップ、係数等をまとめた。
【0072】
実施例2:MET6、MET32、GLY1の破壊株の作製
MT8-1/pIUX1X2XK株のMET6、MET32、GLY1遺伝子を実験方法に示したように、破壊した。すなわち、各遺伝子の開始コドンから下流80 bpと、終始コドンから上流80 bpの配列を両端に持ち、その間にFRT-G418R-FRTを持つDNA断片をPCRにて調製し、MT8-1/pIUX1X2XK株に形質転換して、ジェネティシン耐性を持つ株を選択した。複数の株よりPCRでMET6、MET32、GLY1各遺伝子に、FRT-G418R-FRTが挿入されたものを選択し、続いてpYRNFLPを形質転換した。Nourseothricin dihydrogen sulfate耐性を持つ株を選択し、pYRNFLP から発現される、リコンビナーゼの活性により、FRT配列間での組換えが起こり、G418耐性遺伝子がループアウトした株を、PCRにて選択した。
【0073】
実施例3:MET6、MET32、GLY1の破壊株のキシロース培地でのキシロースからのエタノール醗酵
MT8-1/pIUX1X2XK株(親株)とMET6、MET32、GLY1の破壊株を用いて、実験方法に示した方法にてSD培地にて前培養し、細胞を集菌した後、滅菌水で洗浄した。酵母細胞をOD=20となるように100 ml のSX培地に植菌して、エタノール醗酵試験を行った(n=3)。適時醗酵もろみを1 mlサンプリングし、HPLCでキシロース、キシリトール、エタノール濃度を測定した。キシロース資化経過を図5に、エタノール生成経過を図6に、キシリトール生成経過を図7に示す。また、醗酵パフォーマンスを表すいくつかの指標を図8に示した。
【0074】
この結果より、グリシン生合成経路の遺伝子GLY1を破壊することにより、キシロースの資化速度を早めることができた。また、メチオニン生合成経路の遺伝子、MET6あるいはMET32を破壊することにより、副産物の一つである、キシリトール生成量を減少させることができた。これらの育種を組み合わせる、あるいは他の育種と組み合わせることにより、エタノール収率も向上させることができると考えられる。
【0075】
<まとめ>
以上記載したように、解糖系や、ペントースリン酸経路、あるいは、導入したキシロース還元酵素、キシリトール脱水素酵素のような、キシロースからのエタノールへの代謝経路にあたる酵素ではなく、直接には関係のない、グリシン生合成経路、メチオニン生合成経路に関与する遺伝子を破壊することで、キシロースからのエタノール生成パフォーマンスを向上させることができた。
【産業上の利用可能性】
【0076】
バイオマスから生じた、5炭糖を炭素源として、エタノール醗酵を行うにあたり、エタノール生成収率を上げることができる。特に、ヘミセルロース含量が多い場合には、有効である。
本発明で育種した酵母を使用することによって、キシロースからのエタノール醗酵能、あるいは醗酵速度を改善することができる。どのような醸造用酵母、実験室酵母においても、同様の育種をすることができる。また、キシロースを含むものであれば、どのようなバイオマスから得られたもろみであっても、本育種酵母で同様の効果が見込める。
【配列表フリーテキスト】
【0077】
配列番号13:合成DNA
配列番号14:合成DNA
配列番号15:合成DNA
配列番号16:合成DNA
配列番号17:合成DNA
配列番号18:合成DNA
配列番号19:合成DNA
配列番号20:合成DNA
配列番号21:合成DNA
配列番号22:合成DNA
配列番号23:合成DNA
配列番号24:合成DNA
配列番号25:合成DNA
配列番号26:合成DNA
配列番号27:合成DNA
配列番号28:合成DNA
配列番号29:合成DNA
【技術分野】
【0001】
本発明は、グリシン合成系タンパク質の遺伝子及び/又はメチオニン合成系タンパク質の遺伝子の発現機能を喪失させたエタノール醗酵微生物に関する。また、本発明は、グリシン合成系タンパク質の遺伝子及び/又はメチオニン合成系タンパク質の遺伝子の発現機能を喪失させることにより、キシロース醗酵能力の増加した醗酵微生物を作製する方法に関する。さらに、本発明は、前記微生物と、キシロース含有原料とを接触させる工程を含む、エタノールの製造方法に関する。
【背景技術】
【0002】
近年、化石燃料の枯渇や、CO2ガス削減の必要といった観点から、従来廃棄物であった、コーンコブ、稲ワラ、スイッチグラス、エリアンサス、廃材などといった、バイオマスから燃料としてエタノールを生成する研究が進められている。人類は何千年も前より、デンプンから酵母サッカロミセス・セレビシエ(Saccharomyces cerevisiae)による醗酵でエタノールにする技術を持っていた。デンプンはグルコースがα-1,4結合した多糖であり、様々な生物が持つ加水分解酵素によって容易に分解することができる。また、グルコースは酵母サッカロミセス・セレビシエの最も好ましいC源であり、1分子のグルコースから2分子のエタノールが醗酵により生成される。これに対し、バイオマスは多糖として、セルロース、あるいはヘミセルロースを含んでいる。
【0003】
このうち、セルロースは、グルコースがβ-1,4結合した多糖であり、結晶構造をとっている。結晶構造を壊すための前処理が必要であることや、セルロースを分解するために必要なセロビオハイドロラーゼI、セロビオハイドロラーゼII、エンドグルカナーゼ等の活性が十分でないこと等の問題はあるが、分解後に生じる糖はグルコースであるため、酵母サッカロミセス・セレビシエで醗酵するには何の問題もない。
【0004】
これに対し、ヘミセルロースはグルコースの他に、キシロース、アラビノースと言った五炭糖を含んでいるが、従来酵母サッカロミセス・セレビジエはこれらの五炭糖を醗酵することができない。そこで、キシロースからエタノールを醗酵するために、キシロースを醗酵する能力のある、酵母ピキア・スティピティス(Pichia stipitis)のキシロース還元酵素遺伝子、キシリトール脱水素酵素遺伝子を酵母サッカロミセス・セレビシエにて高発現させる方法が、よく用いられてきた(非特許文献1〜4)。ピキア・スティピティスのキシロース還元酵素は補酵素として主にNADPHを用いる酵素であり、反応後、一分子のNADPが生成する。一方、キシリトール脱水素酵素は、補酵素として主にNADを用いる酵素であり、反応後一分子のNADHを生じる。従って、グルコースを醗酵する際には、NADP/NADPHあるいはNAD/NADHのバランスは変化しないが、キシロースからエタノールを醗酵する場合には、NADPあるいは、NADHが増加する方向に傾いてしまう。このことが、キシロースからのエタノール生成における生産量の低下の原因であると考えられている。これに対し、これまで補酵素特異性を改変した酵素や補酵素特異性が異なる酵素を発現させる試みがなされてきたが、十分な効果は得られていない(非特許文献2〜4)。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Petschacher B, Nidetzky B. Microb Cell Fact. 2008 Mar 17; 7: 9.
【非特許文献2】Watanabe S, Saleh AA, Pack SP, Annaluru N, Kodaki T, Makino K. J Biotechnol. 2007 Jun 30; 130(3): 316-9. Epub 2007 Apr 29.
【非特許文献3】Verho R, Londesborough J, Penttile M, Richard P. Appl Environ Microbiol. 2003 Oct; 69(10): 5892-7.
【非特許文献4】Mikael Anderlund et.al. Appl Environ Microbiol. 1999: June 65(6):2333-2340
【発明の概要】
【発明が解決しようとする課題】
【0006】
このような状況下、キシロースの醗酵能が向上した微生物の開発が必要とされる。
【課題を解決するための手段】
【0007】
本発明者らは、酵母ピキア・スティピティスのキシロース還元酵素、キシリトール脱水素酵素遺伝子および、酵母サッカロミセス・セレビジエのキシルロースリン酸化酵素遺伝子の発現カセットを導入することによって、キシロース醗酵性を付与した複数種類の酵母を用いて醗酵試験を行い、キシロースからのエタノール醗酵における細胞内代謝物のプロファイル分析を行った。得られたデータと、醗酵パフォーマンスを表す数値として、(1)エタノール生成量・速度・収率と、(2)キシロース資化量・速度・収率と、(3)副生成物の生成量・速度・収率等とを比較し、データと(1)〜(3)との相関関係を解析することにより、キシロースからエタノールへの代謝経路に属する物質、すなわち、キシリトール、キシルロース、キシルロース5リン酸、およびペントースリン酸回路と解糖系に属する中間代謝物以外の新たな物質と、エタノール醗酵パフォーマンスとの関連を見出して、育種ターゲットとなる遺伝子を決定し、その遺伝子に関する育種を行った。その結果、グリシン生合成経路の遺伝子GLY1を破壊することにより、キシロースの資化速度が上昇することが判明した。また、メチオニン生合成経路の遺伝子、MET6あるいはMET32を破壊することにより、副産物である、キシリトールの生成量が減少することが判明した。
本願発明は、上記知見に基づくものである。
【0008】
すなわち、本発明は以下に関する。
[1] グリシン合成系タンパク質の遺伝子及び/又はメチオニン合成系タンパク質の遺伝子の発現機能を喪失させ、かつキシロース代謝酵素遺伝子を導入した微生物。
[2] 前記グリシン合成系タンパク質の遺伝子がGLY1遺伝子である、前記[1]に記載の微生物。
[3] 前記メチオニン合成系タンパク質の遺伝子がMET6又はMET32遺伝子である、前記[1]又は[2]に記載の微生物。
[4] 酵母である、前記[1]〜[3]のいずれか1つに記載の微生物。
[5] サッカロミセス・セレビシエである、前記[1]〜[4]のいずれか1つに記載の微生物。
[6] 前記キシロース代謝酵素遺伝子が、キシロース還元酵素遺伝子、キシリトール脱水素酵素遺伝子及びキシルロースリン酸化酵素遺伝子からなる群より選択される少なくとも1つのものである、前記[1]〜[5]のいずれか1つに記載の微生物。
[7] グリシン合成系タンパク質の遺伝子及び/又はメチオニン合成系タンパク質の遺伝子の発現機能を喪失させることにより、キシロース醗酵能力の増加した微生物を作製する方法。
[8] 前記グリシン合成系タンパク質の遺伝子がGLY1遺伝子である、前記[7]に記載の方法。
[9] 前記メチオニン合成系タンパク質の遺伝子がMET6又はMET32遺伝子である、前記[7]又は[8]に記載の方法。
[10] 前記微生物が酵母である、前記[7]〜[9]のいずれか1つに記載の方法。
[11] 前記微生物がサッカロミセス・セレビシエである、前記[7]〜[10]のいずれか1つに記載の微生物。
[12] 前記微生物がキシロース代謝酵素遺伝子が導入されたものである、前記[7]〜[11]のいずれか1つに記載の方法。
[13] 前記キシロース代謝酵素遺伝子が、キシロース還元酵素遺伝子、キシリトール脱水素酵素遺伝子及びキシルロースリン酸化酵素遺伝子からなる群より選択される少なくとも1つのものである、前記[12]に記載の方法。
[14] 前記[1]〜[6]のいずれか1つに記載の微生物と、キシロース含有原料とを接触させる工程を含む、エタノールの製造方法。
【発明の効果】
【0009】
本発明で育種した酵母を使用することによって、キシロースからのエタノール醗酵能、あるいは醗酵速度を改善することができる。どのような醸造用酵母、実験室酵母においても、同様の育種をすることができる。また、キシロースを含むものであれば、どのようなバイオマスから得られたもろみであっても、本育種酵母で同様の効果が見込める。
本発明により、優れたキシロース醗酵能を有する微生物が提供される。また、本発明の微生物の作製方法を用いることにより、広範囲な微生物を宿主として、同様に高いキシロース醗酵能を有する育種を作成することができる。本発明の微生物を用いることにより、キシロース含有原料から効率よくエタノールを製造することができる。また、本発明の微生物を用いると、例えばアミノ酸含有量の低いバイオマスを原料とした場合でも、効率よくエタノールを製造することができる。
【図面の簡単な説明】
【0010】
【図1】実施例で用いたプラスミドpIUX1X2XKOの略図である。
【図2】実施例で用いたプラスミドpBIITGRFRTの略図である。
【図3】実施例で用いたプラスミドpUGRFRTの略図である。
【図4】実施例で用いたプラスミドYRNFLPの略図である。
【図5】本発明の微生物を用いたキシロースからのエタノール醗酵試験の結果を示す(キシロース資化量)。
【図6】本発明の微生物を用いたキシロースからのエタノール醗酵試験の結果を示す(エタノール生産量)。
【図7】本発明の微生物を用いたキシロースからのエタノール醗酵試験の結果を示す(キシリトール生成量)。
【図8】本発明の微生物を用いたキシロースからのエタノール醗酵試験の結果を示す(XCR:キシロース資化速度、EPRエタノール生成速度、XPR:キシリトール生成速度、EY:エタノール収率、XY:キシリトール収率、Met6:MET6遺伝子破壊株、Met32:MET32遺伝子破壊株、Gly1:GLY1遺伝子破壊株、MT:親株)。
【図9】実施例1のキシロース醗酵試験及び酵母細胞の細胞内成分の網羅的分析の結果を示す図である。
【発明を実施するための形態】
【0011】
本発明者らは、グリシン合成系タンパク質の遺伝子の発現を抑制した微生物株を用いた場合、野生型同種株と比べて、キシロースの資化速度が上昇することを見出し、さらに、メチオニン合成系タンパク質の遺伝子の発現を抑制した微生物株では、キシロースを原料とするエタノール生成において副産物であるキシリトールの生産量が減少することを見出し、本発明を完成させた。以下において、本願発明の各実施態様を記載する。
【0012】
1. グリシン合成系タンパク質遺伝子及び/又はメチオニン合成系タンパク質遺伝子機能喪失微生物
本発明は、ある実施態様において、グリシン合成系タンパク質遺伝子及び/又はメチオニン合成系タンパク質遺伝子の発現機能を喪失させた、微生物を提供する。
【0013】
本発明において「微生物」とは、肉眼でその存在が判別できず、顕微鏡などによって観察できる程度以下の大きさの生物を意味する。微生物の例としては、細菌、藍色細菌、古細菌等の原核生物、並びに糸状菌、酵母、変形菌、担子菌、単細胞性の藻類及び原生動物等の真核生物が挙げられる。
好ましくは、微生物は、糖に対する醗酵能を有する種であり、より好ましくは、酵母である。酵母は、出芽酵母又は分裂酵母のいずれであってもよい。ある実施態様では、酵母は、サッカロミセス・セレビシエNBRC1951、NBRC1952、NBRC1953、NBRC1954、X2180-1A (ATCC26786)、CB11 (Berkley Stock Center)、W303-1A (BY4848)、MT8-1(BY2685)、NBRC1440、NBRC1445等の出芽酵母であってもよい。別の実施態様では、酵母は、シゾサッカロミセス・ジャポニクス(Schizosaccharomyces japonicus (Hasegawaea japonicus))、シゾサッカロミセス・オクトスポルス(Schizosaccharomyces octosporus (Octosporomyces octosporus))及びシゾサッカロミセス・ポンベ(Schizosaccharomyces pombe)等の分裂酵母であってもよい。
【0014】
本発明において「グリシン合成系タンパク質の遺伝子」又は「グリシン合成系タンパク質遺伝子」とは、グリシン合成に関わる酵素タンパク質や転写調節因子をコードする遺伝子を意味し、DNA又はRNAのいずれであってもよい。例えば、酵母由来のものでは、Saccharomyces genome database (http://www.yeastgenome.org/)に記載のsuperpathway of serine and glycine biosynthesis (http://www.yeastgenome.org/cgi-bin/search/ textSearch.pl?query=glycine&type=pathway) に関与する遺伝子、SHM2 、SHM1、GLY1 、SER3、AGX1 、SER1、 SER2 、SER33などが挙げられる。 グリシン合成系タンパク質とは、グリシン合成に関わる酵素タンパク質及びその転写調節因子であり、例えば、酵母由来のものでは、上記superpathwayのSHM2 、SHM1、GLY1 、SER3、AGX1 、SER1、 SER2 、SER33にコードされるタンパク質がある。好ましくは、L-トレオニンを加水分解してグリシンを生成する活性(以下、「L-トレオニン・アルドラーゼ活性」)を有するタンパク質(以下、「L-トレオニン・アルドラーゼ」)であり、酵母の場合はGLY1にコードされる。
【0015】
本明細書中で使用される場合、「グリシン合成系タンパク質遺伝子」は、サッカロミセス・セレビシエ由来のL-トレオニン・アルドラーゼ「GLY1」(配列番号2)をコードする遺伝子(配列番号1、Saccharomyces Genome Database Accession No:YEL046C)に限定されず、L-トレオニン・アルドラーゼ活性を示す、L-トレオニン・アルドラーゼと同じファミリーに属するホモログタンパク質をコードする遺伝子でもよい。L-トレオニン・アルドラーゼと同じファミリーに属するタンパク質は多くの植物において見出されており、ファミリー内全体で高度に保存されている。L-トレオニン・アルドラーゼは、グリシン合成に必須のタンパク質であるため、その遺伝子はファミリー内全体で高度に保存されており、複数の微生物のL-トレオニン・アルドラーゼ又はそのホモログタンパク質の遺伝子については既に塩基配列が解析され、その配列情報がデータベースに登録されている。微生物由来のL-トレオニン・アルドラーゼ及びそのホモログをコードする遺伝子を表1に示すが、これらに限定されるものではない。
【表1】
サッカロミセス・セレビシエを用いて本発明の微生物を作製する場合、グリシン合成系タンパク質遺伝子は、好ましくはGLY1遺伝子である。
【0016】
一実施形態において、グリシン合成系タンパク質遺伝子は、L-トレオニン・アルドラーゼ活性を有するポリペプチドをコードするポリヌクレオチドの塩基配列において1または複数個の塩基が欠失、挿入、置換、または付加された変異体であってもよい。変異体は、コードもしくは非コード領域、またはその両方において変異され得る。コード領域における変異は、保存的または非保存的なアミノ酸の欠失、挿入、置換および/または付加を生成し得る。本明細書中で使用される場合、配列番号2のアミノ酸配列をコードする遺伝子、または当該アミノ酸配列において1もしくは複数個(例えば、1〜40個、1〜20個、1〜15個、1〜10個、1〜9個、1〜8個、1〜7個、1〜6個、1〜5個、1〜4個、1〜3個、1〜2個、1個など)または1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列をコードする遺伝子もまた、「グリシン合成系タンパク質の遺伝子」に包含される。
【0017】
本発明において「メチオニン合成系タンパク質の遺伝子」又は「メチオニン合成系タンパク質遺伝子」とは、メチオニン合成に関わる酵素タンパク質や転写調節因子をコードする遺伝子を意味し、DNA又はRNAのいずれであってもよい。例えば、酵母由来のものでは、Saccharomyces genome databaseに記載のsuperpathway of threonine and methionine biosynthesis(http://pathway.yeastgenome.org/YEAST/new-image?type=PATHWAY&object=P4-PWY&detail-level=2) に関与する遺伝子、MET2、HOM3、FOL3、HOM2、MET7、MET17、THR1、THR4、HOM6、MET6 や、superpathway of methionine salvage pathway(http://pathway. yeastgenome.org/YEAST/new-image?type=PATHWAY&object=PWY3O-351&detail-level=2)に関与する遺伝子、MRI1 、 BAT1 、ARO9 、 MDE1 、SAM2、 ARO8 、SPE2 、SPE3、SPE4、UTR4、MEU1、ADI1、BAT2、SAM1、および、これら遺伝子の転写調節に関わる遺伝子、MET4、MET28、MET31、MET32が挙げられる。メチオニン合成系タンパク質とは、メチオニン合成に関わる酵素タンパク質及びその転写調節因子であり、例えば、酵母由来のものでは、上記superpathwayのMET2、HOM3、FOL3、HOM2、MET7、MET17、THR1、THR4、HOM6、MET6、MRI1 、 BAT1 、ARO9 、 MDE1 、SAM2、 ARO8 、SPE2 、SPE3、SPE4、UTR4、MEU1、ADI1、BAT2、SAM1、MET4、MET28、MET31、MET32にコードされるタンパク質がある。好ましくは、ホモシステインの硫黄をメチル化してメチオニンを生成する活性(以下、「メチオニン・シンターゼ活性」)を有するタンパク質(以下、「メチオニン・シンターゼ」)及びメチオニン・シンターゼの転写制御因子である。酵母の場合、メチオニン・シンターゼの遺伝子の例としてはMET6が挙げられ、その転写制御因子をコードする遺伝子としてはMET32が挙げられる。
【0018】
本明細書中で使用される場合、「メチオニン合成系タンパク質遺伝子」は、サッカロミセス・セレビシエ由来のメチオニン・シンターゼ「MET6」(配列番号4)をコードする遺伝子(配列番号3、Saccharomyces Genome Database Accession No:YER091C)及びその転写因子である「MET32」(配列番号6)をコードする遺伝子(配列番号5、Saccharomyces Genome Database Accession No:YDR253C)に限定されず、メチオニン・シンターゼ活性を示す、MET6と同じファミリーに属するホモログタンパク質をコードする遺伝子(即ち、MET6のホモログ遺伝子)でもよく、前記MET6ホモログ遺伝子の転写因子をコードする遺伝子(即ち、MET32のホモログ遺伝子)であってもよい。MET6及びMET32と同じファミリーに属するタンパク質は多くの植物において見出されており、ファミリー内全体で高度に保存されている。微生物由来のMET6及びそのホモログをコードする遺伝子を表2に示し、微生物由来のMET32及びそのホモログをコードする遺伝子を表3に示すが、これらに限定されるものではない。
【表2】
【表3】
サッカロミセス・セレビシエを用いて本発明の微生物を作製する場合、メチオニン合成系タンパク質遺伝子は、好ましくはMET6又はMET32遺伝子である。
【0019】
一実施形態において、メチオニン合成系タンパク質遺伝子は、メチオニン・シンターゼ活性を有するポリペプチド又はその転写因子をコードするポリヌクレオチドの塩基配列において1または複数個の塩基が欠失、挿入、置換、または付加された変異体であってもよい。変異体は、コードもしくは非コード領域、またはその両方において変異され得る。コード領域における変異は、保存的または非保存的なアミノ酸の欠失、挿入、置換および/または付加を生成し得る。本明細書中で使用される場合、配列番号4又は6のアミノ酸配列をコードする遺伝子、または当該アミノ酸配列において1もしくは複数個(例えば、1〜40個、1〜20個、1〜15個、1〜10個、1〜9個、1〜8個、1〜7個、1〜6個、1〜5個、1〜4個、1〜3個、1〜2個、1個など)または1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列をコードする遺伝子もまた、「メチオニン合成系タンパク質の遺伝子」に包含される。
【0020】
本発明において、「グリシン合成系タンパク質の遺伝子及び/又はメチオニン合成系タンパク質の遺伝子の発現機能を喪失させた」とは、グリシン合成系タンパク質遺伝子及び/又はメチオニン合成系タンパク質遺伝子(以下、「対象タンパク質遺伝子」と総称)から本来の生物学的機能を有する対象タンパク質が発現されないことを意味する。このような状態の例としては、対象タンパク質遺伝子の発現産物が全く生成されない状態だけでなく、当該遺伝子の発現産物(例えば、hnRNA、mRNA又はタンパク質)は発現されるが、それら発現産物が本来の正常な機能を有しない状態が挙げられる。このような対象タンパク質遺伝子の機能喪失は、対象タンパク質遺伝子又はその転写調節領域若しくはプロモーター領域を含む発現制御領域上における1又は複数のヌクレオチドの欠失、置換、及び/又は挿入等によって生じさせることができる。なお、前記欠失、置換、及び/又は挿入を行う部位や、欠失、置換、及び/又は挿入される配列は、対象タンパク質遺伝子の正常な機能が喪失しうる限り、特に限定されないが、好ましくは、対象タンパク質の活性部位をコードする遺伝子配列の少なくとも1つが欠失していることが好ましい。
【0021】
本発明の微生物は、特に真核生物の場合、前記対象タンパク質の発現機能が、染色体上の少なくとも一方のアレル(ヘテロ接合型)で喪失していれば効果がある場合もあるが、好ましくは、両アレルで喪失していること(ホモ接合型)が好ましい。
【0022】
2. キシロース醗酵能力の増加した醗酵微生物を作製する方法
本発明は、宿主微生物のグリシン合成系タンパク質の遺伝子及び/又はメチオニン合成系タンパク質の遺伝子の発現機能を喪失させることにより、キシロース醗酵能力の増加した醗酵微生物を作製する方法を提供する。
グリシン合成系タンパク質遺伝子及び/又はメチオニン合成系タンパク質遺伝子の発現機能を喪失させる方法としては、ジーンターゲティング又はRNAi等のノックアウト又はノックダウン技術が挙げられる
【0023】
(1)ジーンターゲティング
ジーンターゲティングは、相同組換えを利用して染色体上の特定遺伝子に変異を導入する手法である(Capeccchi, M.R. Science, 244, 1288-1292, 1989)。
【0024】
まず、対象タンパク質遺伝子機能を喪失させるためのターゲティングベクターを構築する。ターゲティングベクターを構築するに当たり、対象微生物のゲノムDNAライブラリーを調製する。このゲノムDNAライブラリーは、多型等により組換え頻度が低下しないよう、ジーンターゲティングの対象となる微生物種と同系統の微生物、好ましくは、同種の微生物に由来するゲノムDNAから作製したライブラリーを用いることが好ましい。そのようなライブラリーとしては、市販のものを用いてもよい。あるいは、対象タンパク質のcDNA又はその部分配列をプローブとしてスクリーニングを行い、対象タンパク質のゲノム遺伝子のDNA配列をクローニングしてもよく、あるいは、対象タンパク質のcDNA又はその部分配列に基づいてプライマーを作成し、ゲノムの対象タンパク質遺伝子をPCR増幅することにより調製することができる。ゲノムDNAライブラリーの調製方法の詳細については、以下を参照できる。"Sambrook & Russell, Molecular Cloning: A Laboratory Manual Vol. 3, Cold Spring Harbor Laboratory Press 2001"、"Ausubel, Current Protocols in Molecular Biology, John Wiley &Sons 1987-1997"
【0025】
次に、クローニングされたゲノムDNAを、シークエンシング、サザンブロッティング、制限酵素消化等によって解析してエクソン(真核生物の場合)の位置及び制限酵素サイトを同定する。このような解析により得られた配列情報を基に変異導入部位等を決定する。
【0026】
本発明において、染色体上に導入する変異(欠失、置換、及び/又は挿入)は対象タンパク質遺伝子の正常な機能が損なわれる限り特に限定されず、これらの変異は、対象タンパク質遺伝子のイントロン領域、エクソン領域又は対象タンパク質遺伝子の発現制御領域に存在していてよい。前記変異は、対象タンパク質遺伝子のエクソン領域に存在することが好ましく、さらに好ましくは、対象タンパク質遺伝子の少なくとも1つのエクソンが欠失した変異、最も好ましくは全てのエクソンが欠失した変異である。このような変異であれば、確実に対象タンパク質遺伝子の機能を欠損させることができるからである。
【0027】
ターゲティングベクターは、変異導入部位の3’及び5’側の相同領域(それぞれ3’アーム及び5’アーム)に加え、組み換え体のセレクション用の選択マーカーを含んでいてもよい。選択マーカーの例としては、Nourseothricin dihydrogen sulfate(NAT)耐性遺伝子、ネオマイシン耐性遺伝子、ハイグロマイシンBホスホトランスフェラーゼ遺伝子、カナマイシン耐性遺伝子等のポジティブセレクションマーカー、LacZ、GFP(Green Fluorescence Protein)及びルシフェラーゼ遺伝子などの破壊対象遺伝子の発現レポーター、単純ヘルペスウイルスチミジンキナーゼ遺伝子(HSV-TK)、ジフテリア毒素Aフラグメント(DTA)等のネガティブセレクションマーカー等が挙げられる。特に、本発明は、微生物を宿主とするため、栄養要求性を相補する遺伝子、例えばURA3を用いれば、ウラシルを含まない寒天培地でセレクションが可能であり、宿主の栄養要求性によっては、使用が可能である。また、シクロヘキシミドに対する薬剤耐性遺伝子(YAP1)を用いれば、シクロヘキシミドを含んだ培地でセレクションを行うことができる。これらのセレクションマーカーを3’アーム及び5’アームの間に挿入した配列を用いることにより、ターゲット遺伝子を破壊することもできる。ベクターは相同領域の外側に、ベクターを直鎖化するための適当な制限酵素切断部位を含んでいてもよい。
このようなターゲティングベクターは、市販のプラスミドベクター(例えば、pENTR/D-TOPO(登録商標:Invitrogen)やpBluescriptII (Stratagene)等)をベースにして構築することができる。
【0028】
本発明の微生物は、対象タンパク質遺伝子の破壊後に、相同組換に用いた選択マーカーが、細胞から脱落していることが好ましい。例えば、上記ターゲティングベクターがコードする選択マーカー遺伝子の両端に、組換え配列を挿入し、当該組換え配列間で組換えを生じさせて、選択マーカー遺伝子をループアウトさせることにより、選択マーカーを宿主細胞から脱落させることができる。このようにして作製される本発明の微生物は、選択マーカーを有しないため、複数の対象タンパク質遺伝子を破壊する際に、選択マーカーが制限されないという優れた特徴を有する。
【0029】
選択マーカーの脱落に使用可能な組換え配列としては、Flippase Recognition Target 配列(FRT配列)、バクテリオファージP1由来のloxP配列、醤油酵母(Zygosaccharomyces rouxii)由来のRS配列、バクテリオファージMu由来のgix配列等が挙げられる。
これらの組換え配列間に組換えを生じさせるためには、適切なリコンビナーゼを発現させる必要がある。上記の組換え配列間に組換えを生じさせるリコンビナーゼは、それぞれ、Flippase(FLP:FLP-FRTリコンビネーション)、Cre(Cre-loxPリコンビネーション)、R Recombinase (R:RS リコンビネーション)、GIX recombinase (R:RS リコンビネーション)等が挙げられる。
さらに、リコンビナーゼをコードするプラスミドも宿主細胞から脱落していることが好ましい。宿主細胞の複数の対象タンパク質遺伝子の破壊において、選択マーカーが制限されないからである。
したがって、リコンビナーゼをコードするプラスミドは、宿主細胞内で不安定なものが好ましく、特に、当該プラスミドの選択マーカーに対する選択圧をかけない状態において、宿主細胞から脱落するものが好ましい。
そのようなプラスミドの例としては、YRpタイプのプラスミドが挙げられるが、これに限定されるものではない。
【0030】
選択マーカーは、任意の薬剤耐性(NAT耐性、ネオマイシン耐性、ハイグロマイシンBホスホトランスフェラーゼ耐性、カナマイシン耐性、ハイグロマイシン耐性、ゼオシン耐性、ジェネティシン耐性)、栄養要求性マーカー(ura3、ura5、niaD)、銅耐性マーカー(CUP1)(Marin et al., Proc. Natl. Acad. Sci. USA, 81, 337 1984)、セルレニン耐性マーカー(fas2m, PDR4)等であってもよいが、これらに限定されるものではない。
【0031】
図2及び3に、実施例で用いたサッカロミセス・セレビシエの対象タンパク質遺伝子のノックアウトに用いた断片を得る為のベクターの例を示す。このコンストラクトは、その上流と下流にFRT配列を持つG418耐性遺伝子、FRT-G418R-FRT断片を含む。G418Rは、ジェネティシン耐性遺伝子であり、対象タンパク質遺伝子が破壊された株のポジティブセレクションマーカーとして用いられる。これらプラスミドを鋳型として、対象タンパク質遺伝子の各ORFの開始コドンから下流80塩基の配列にFRT-G418R-FRT断片のすぐ上流20塩基が並んだプライマーと、終始コドンから上流80塩基の配列にFRT-G418R-FRT断片のすぐ下流20塩基が並んだプライマーを用いてPCRを行う。得られたPCR増幅断片は、対象タンパク質遺伝子の各ORFの開始コドンから下流80塩基の配列、あるいは終始コドンから上流80塩基の配列がそれぞれ両端に付加された、FRT-G418R-FRT断片を含む。この断片を用いて、宿主細胞を形質転換し、相同組換えによりジェネティシン耐性を獲得した株を取得する。
次に、取得した株にFLPをコードするプラスミド(図4に示すpYRNFLP)を導入してFRT配列間に組換えを生じさせ、ジェネティシン耐性遺伝子を脱落させる。
さらに、pYRNFLPも不安定であるため、非選択圧下において宿主細胞から脱落している。このようにして作製された本発明の微生物は、選択マーカーを有しないため、複数の遺伝子の破壊を行う際に、先の遺伝子の破壊に使用した選択マーカーと、同一の選択マーカーを繰り返し用いることが可能である。
【0032】
(2)RNA干渉
上記以外にも、対象タンパク質遺伝子の発現機能を欠損させる方法として、siRNA(small interfering RNA)等を用いたRNA干渉(RNA interference: RNAi)法が挙げられる。RNAi は、複数の段階を経て行われるマルチステッププロセスである。最初に、RNAi発現ベクターから発現した二本鎖RNA(Double Stranded RNA: dsRNA)又はヘアピン状のshRNA(Small Hairpin RNA)が Dicerによって認識され、21〜23 ヌクレオチドの siRNAs に分解される。次に、siRNAs は RNA 誘導型サイレンシング複合体 (RNA-Induced Silencing Complex: RISC) と呼ばれる RNAi 標的複合体に組み込まれ、RISC とsiRNAsとの複合体がsiRNAの配列と相補的な配列を含む標的mRNAに結合し、mRNAを分解する。標的mRNAは、siRNAに相補的な領域の中央で切断され、最終的に標的mRNAが速やかに分解されてタンパク発現量が低下する。最も効力の高い siRNA 二重鎖は、19bpの二重鎖の各3’末端にウリジン残基2個の突出部分を持つ 21 ヌクレオチド長の配列であることが知られている(Elbashir S.M. et al., Genes and Dev, 15, 188-200 (2001))。
【0033】
従って、対象タンパク質遺伝子ノックダウン微生物を得るには、まず、表1〜3に示されるような対象タンパク質遺伝子配列(これらに限定されない)の一部と相補的な配列を含むヌクレオチドを、適切なRNAi発現ベクターにdsRNA又はshRNAとして発現可能な状態で挿入してRNAiベクターを作製する。次に、前記ベクターを宿主細胞に導入し、形質転換体をセレクションすることにより対象タンパク質遺伝子ノックダウン微生物を得ることができる。
【0034】
dsRNA又はshRNAの設計及び合成は、市販のDNA/RNAシンセサイザー、例えば、Applied Biosystems394型で行うことも可能であり、あるいは第三者機関(例えば、TAKARA Bio)に委託することもできる。
【0035】
対象タンパク質遺伝子の発現機能の喪失は、正常な対象タンパク質遺伝子の発現量の低下として確認することができる。対象タンパク質遺伝子の発現量は、本発明の微生物の抽出物を用いたRT-PCR法及びアガロースゲル電気泳動、Real-Time PCR法、ノーザンブロッティング法、マイクロアレイ解析並びに質量分析法等によって測定することができる。これらの測定方法に用いるプライマー又はプローブは、当該対象タンパク質遺伝子の配列に基づいて設計及び合成することができる。対象タンパク質遺伝子の発現産物が正常であるかどうかは、当該発現産物の配列解析を行うことにより確認することができる。
【0036】
本発明の微生物は、対象タンパク質遺伝子の発現機能が喪失していることを特徴とするが、さらに、キシロース代謝酵素遺伝子が発現可能な状態で導入されていてもよい。キシロース代謝酵素遺伝子は、キシロースからエタノールを生成する一連の化学反応のうち、いずれかの反応を触媒する活性を有するタンパク質(以下、「キシロース代謝酵素」)をコードする遺伝子を意味する。キシロース代謝酵素遺伝子の例としては、キシロース還元酵素遺伝子、キシリトール脱水素酵素遺伝子、キシルロースリン酸化酵素遺伝子が挙げられる。
このようなキシロース代謝酵素遺伝子は、例えば、ピキア・スティピティス、サッカロミセス・セレビシエ、大腸菌、ラクトバシラス・カゼイ、ラクトバシラス・アシドフィラス、アスペルギルス・フミガツス、スタフィロコッカス・アウレウス、ピキア・パストリス、シゾサッカロミセス・ポンベ等に由来するものであってもよい。
本発明の微生物には、前記キシロース代謝酵素のうち、キシロース還元酵素遺伝子、キシリトール脱水素酵素遺伝子又はキシルロースリン酸化酵素遺伝子からなる群より選択される少なくとも1つが導入されていることが好ましく、これら遺伝子の2つ以上、あるいは、3つ全ての遺伝子が導入されていてもよい。
【0037】
キシロース還元酵素遺伝子、キシリトール脱水素酵素遺伝子又はキシルロースリン酸化酵素遺伝子の例としては、以下が挙げられるが、これに限定されるものではない。
【表4】
【表5】
【表6】
【0038】
キシロース還元酵素は、キシロース代謝において、以下の反応を触媒する酵素である。
Xylose+NADPH→Xylitol+NADP
【0039】
キシリトール脱水素酵素は、キシロース代謝において、以下の反応を触媒する酵素である。
Xylitol+NAD→Xylulose+NADH
【0040】
キシルロースリン酸化酵素は、キシロース代謝において、以下の反応を触媒する酵素である。
Xylulose+ATP→Xylulose-5P+ADP
【0041】
一実施形態において、キシロース代謝酵素遺伝子(キシロース還元酵素、キシリトール脱水素酵素又はキシルロースリン酸化酵素等)は、キシロース代謝活性を有するタンパク質をコードするものである限り、上記キシロース代謝酵素遺伝子の変異体であってもよい。変異体の例としては、上記キシロース代謝酵素タンパク質をコードするポリヌクレオチドの塩基配列において1または複数個の塩基が欠失、挿入、置換、または付加された変異体であってもよい。変異体は、コードもしくは非コード領域、またはその両方において変異され得る。コード領域における変異は、保存的または非保存的なアミノ酸の欠失、挿入、置換および/または付加を生成し得る。本明細書中で使用される場合、配列番号8、10又は12のアミノ酸配列をコードする遺伝子(配列番号7、9又は11)、または当該アミノ酸配列において1もしくは複数個(例えば、1〜40個、1〜20個、1〜15個、1〜10個、1〜9個、1〜8個、1〜7個、1〜6個、1〜5個、1〜4個、1〜3個、1〜2個、1個など)または1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列をコードする遺伝子もまた、「キシロース代謝酵素遺伝子」に包含される。
【0042】
ポリペプチドのアミノ酸配列中のいくつかのアミノ酸が、このポリペプチドの構造または機能に有意に影響することなく容易に改変され得ることは、当該分野において周知である。さらに、人為的に改変させるだけではく、天然のタンパク質において、当該タンパク質の構造または機能を有意に変化させない変異体が存在することもまた周知である。
【0043】
当業者は、周知技術を使用してポリペプチドのアミノ酸配列において1または複数個のアミノ酸を容易に改変させることができる。例えば、公知の点変異導入法に従えば、ポリペプチドをコードするポリヌクレオチドの任意の塩基を改変させることができる。また、ポリペプチドをコードするポリヌクレオチドの任意の部位に対応するプライマーを設計して欠失変異体または付加変異体を作製することができる。さらに、本明細書中に記載される方法を用いれば、作製した改変体が所望の活性を有するか否かを容易に決定し得る。
【0044】
好ましい改変体は、保存性もしくは非保存性アミノ酸置換、欠失、または添加を有する。好ましくは、サイレント置換、添加、および欠失であり、特に好ましくは、保存性置換である。これらは、本発明に係るポリペプチド活性を変化させない。
【0045】
代表的に保存性置換と見られるのは、脂肪族アミノ酸Ala、Val、Leu、およびIleの中での1つのアミノ酸の別のアミノ酸への置換;ヒドロキシル残基SerおよびThrの交換、酸性残基AspおよびGluの交換、アミド残基AsnおよびGlnの間の置換、塩基性残基LysおよびArgの交換、ならびに芳香族残基Phe、Tyrの間の置換である。
【0046】
上記に詳細に示されるように、どのアミノ酸の変化が表現型的にサイレントでありそうか(すなわち、機能に対して有意に有害な効果を有しそうにないか)に関するさらなるガイダンスは、Bowie, J.U.ら「Deciphering the Message in Protein Sequences: Tolerance to Amino Acid Substitutions」,Science 247:1306-1310(1990)(本明細書中に参考として援用される)に見出され得る。
【0047】
上記に挙げられる本発明に係るポリヌクレオチドを使用すれば、形質転換体または細胞においてキシロース代謝酵素活性を有するポリペプチドを合成することができる。
【0048】
キシロース代謝酵素遺伝子は、宿主細胞において発現可能な発現ベクターに組み込まれた状態で、導入されることが好ましい。発現ベクターは、以下の(i)〜(iii)の要素を含みうる。
(i)宿主細胞内で転写可能なプロモーター;
(ii)該プロモーターに結合した、キシロース代謝酵素遺伝子;及び
(iii)RNA分子の転写終結及びポリアデニル化に関し、宿主細胞内で機能するシグナルを構成要素として含む発現カセット
を含むように構成される。
【0049】
このようなベクターとしては、例えば、pYE22m(酵母用:Biosci. Biotech. Biochem., 59, 1221-1228, 1995)、YCp50 (酵母用:X70276)、YIp1 (酵母用:X70480)が挙げられる。
【0050】
宿主細胞中での遺伝子発現を調節するためのプロモーター/ターミネーターとしては、宿主細胞中で機能する限り、任意の組み合わせでよい。例えば、酵母で利用する場合はTDH3、PYK1、PGK 、GAP 、TPI、GAL1、GAL10 、ADH2、PHO5、CUP1、MFα1等のプロモーターを用いることができるが、これに限定されるものではない。構成的高発現プロモーターとしては、TDH3、TPI1、PGK1、PGI1、PYK1、ADH1等のプロモーターを使用することができる。
形質転換の際に用いる選択マーカーとしては、栄養要求性マーカー(ura3、ura5、niaD)、薬剤耐性マーカー(ハイグロマイシン耐性、ゼオシン耐性、ジェネチシン耐性)、銅耐性遺伝子(CUP1)(Marin et al., Proc. Natl. Acad. Sci. USA, 81, 337 1984)、セルレニン耐性遺伝子(fas2m, PDR4)(それぞれ猪腰淳嗣ら, 生化学, 64, 660, 1992; Hussain et al., gene, 101, 149, 1991)等が利用可能である。
【0051】
宿主細胞の形質転換方法としては、一般に用いられる公知の方法が利用できる。例えば、原核生物の場合、エレクトロポレーション法(Mackenxie D. A. et al. Appl. Environ. Microbiol., 66, 4655-4661, 2000)やヒートショック法が利用できる。また、酵母の場合は、エレクトロポレーション法、スフェロプラスト法(Proc. Natl. Acad. Sci. USA, 75 p1929(1978))、酢酸リチウム法(J. Bacteriology, 153, p163(1983))、Proc. Natl. Acad. Sci. USA, 75 p1929 (1978)、Methods in yeast genetics, 2000 Edition: A Cold Spring Harbor Laboratory Course Manual等に記載の方法で実施可能であるが、これらに限定されない。
【0052】
その他、一般的なクローニング技術に関しては、"Sambrook & Russell, Molecular Cloning: A Laboratory Manual Vol. 3, Cold Spring Harbor Laboratory Press 2001"、"Methods in Yeast Genetics、A laboratory manual (Cold Spring Harbor Laboratory Press、Cold Spring Harbor, NY)"等を参照することができる。
【0053】
3.エタノールの製造方法
上記のようにして作製された本発明の微生物は、同種の野生型微生物と比べて、キシロース醗酵能が増加している。ここで、キシロース醗酵とは、キシロースを資化することにより、エタノールを生成することを意味する。したがって、本発明の微生物をキシロース含有原料の存在下で培養した場合、同種の野生型微生物を同一条件下で培養した場合よりも、早くキシロースを資化することが出来、副生成物であるキシリトールの生成量が少なく、キシロースからのエタノール醗酵能が上昇する。
このことから、本発明は、別の実施態様において、本発明の微生物と、キシロース含有原料とを接触させる工程を含む、エタノールの製造方法を提供する。
【0054】
本発明の微生物と、キシロース含有材料とを接触させる工程は、本発明の微生物と、キシロース含有材料とを混合することにより達成できる。好ましくは、本発明の微生物は、活性な状態でキシロース含有材料と接触させる。
本発明の微生物を活性な状態でキシロース含有材料と接触させる方法の例としては、本発明の微生物が増殖可能な培養条件において、かつ、キシロース含有原料の存在下で、本発明の微生物を培養する方法が挙げられる。このような、培養条件は、微生物種によって異なるが、例えば、酵母であれば、キシロース含有原料を添加したYPD培地(イーストエキストラクト 10 g/L、ポリペプトン 20 g/L、グルコース 20 g/L)、SX培地(キシロース50 g/l、6.7 g/l yeast nitrogen baseアミノ酸抜き、アデニン20 mg/l、ヒスチジン 20 mg/l、ロイシン 100 mg/l、トリプトファン 20mg/l)等の適切な培地中で30℃にて培養することができる。
あるいは、本発明の微生物は、キシロース含有原料の存在下で培養を行う前に、本発明の微生物が増殖可能な培養条件において培養して菌体を増殖させ、その後、キシロース含有原料の存在下で、本発明の微生物を用いて醗酵を行なってもよい。
但し、培養条件は、上記に限定されるものではない。当業者であれば、適宜、微生物種に応じた適切な培養条件を選択し、実際に培養を行うことができる。
【0055】
キシロースからのエタノール醗酵能は、本発明の微生物とキシロース含有原料とを接触させ、一定時間(例えば、72時間、48時間、24時間、12時間、10時間、8時間、6時間、4時間、2時間、1時間)が経過した後に系のエタノール生成量を測定することにより確認することができる。
【0056】
キシロース資化量及びエタノール生成量は、本発明の微生物とキシロース含有原料とを含む醗酵系の成分を、高速液体クロマトグラフィー等の公知の分析手法によって分析することにより、容易に測定することができる。
【0057】
「キシロース含有原料」は、キシロースを含む原料であれば、特に限定されないが、好ましくは、キシロースを含む生物由来の原料、即ち、「キシロース含有バイオマス」である。バイオマスは、日本国農林水産省が発行した「バイオマス・ニッポン総合戦略」において、「再生可能な、生物由来の有機性資源で化石資源を除いたもの」と定義されている。
よって、この定義に基づき、「キシロース含有バイオマス」は、「再生可能な、生物由来の有機性資源で化石資源を除いたものであって、キシロースを含むもの」と定義することができる。
キシロース含有バイオマスの具体例としては、サトウキビ、トウモロコシ、テンサイ、ジャガイモ、サツマイモ、麦、モロコシ(=こうりゃん)、ソルガムなどの非可食部や、廃木材、パルプ廃液、バガス、もみ殻、コーンコブ、稲ワラ、スイッチグラス、エリアンサス、ネピアグラスが挙げられるが、これらに限定されるものではない。
【0058】
以下、実施例を用いて本発明を詳細に説明するが、本発明は実施例に記載された態様に限定されるものではない。
【実施例】
【0059】
試験方法:
本実施例で用いた試験項目および試験方法を以下に示す。特に断りのない限り、本実施例における試験方法はこれに準じた。
【0060】
<酵母株>
本発明において用いた株は、以下の(a)〜(e)のとおりである。いずれの株も、P.stipitisのキシロース還元酵素遺伝子(XYL1:配列番号7)とキシリトール脱水素酵素遺伝子(XYL2:配列番号9)とS.cerevisiaeのキシルロースリン酸化酵素遺伝子(XKS1:配列番号11)とをS.cerevisiaeのTDH3遺伝子のプロモーター制御下に連結させた発現ユニットを持つプラスミド・pIUX1X2XK(図1)が、染色体上のURA3遺伝子領域に相同組換えで導入されている。株については、これに限定されるものではなく、キシロース醗酵能を持つ酵母であれば、何でも良い。
(a) MT8-1/pIUX1X2XK(Mata ade his3 leu2 trp1/pIUX1X2XK)
(b) MT8-1/pIUX1X2XK/Ad(Mata ade his3 leu2 trp1, キシロース培地に順化した株)
(c) MT8-1/pIUX1X2XK/pGK404-ScTal(Mata ade his3 leu2 /pIUX1X2XK/pGK404-ScTal)
(d) NBRC1440/pIUX1X2XK(Matα・his3 trp1/pIUX1X2XK)
(e) NBRC1445/pIUX1X2XK(Mata his3 trp1/pIUX1X2XK)
MT8-1は、ナショナルバイオリソースプロジェクトにNBRP No. BY2685として登録されている。
【0061】
<遺伝子破壊方法>
以下の各々のプライマー(1)〜(16)を用いて、プラスミドpBIITGRFRT(図2)((1)+(2))、あるいはpUGRFRT(図3)((3)+(4)、(5)+(6))を鋳型としてPCRを行った。これらにより、MET6((1) +(2))、MET32((3)+(4))、GLY1 ((5)+(6))の各ORFの開始コドンから下流80塩基の配列、あるいは終始コドンから上流80塩基の配列がそれぞれ両端に付加された、FRT-G418R-FRT断片を増幅される。FRTとは酵母2μプラスミド上にある、リコンビナーゼによって組換えが起こる、組換え部位であり、その配列は、5’-GAAGTTCCTATTCTCTAGAAAGTATAGGAACTTC-3’(配列番号13)である。G418Rとは、G418耐性マーカー遺伝子( PGK1p::KanMX)であり、pBIITGRFRTとpUGRFRTは両者とも、FRT-G418R-FRT断片を持つプラスミドである。増幅した各ORFの両末端配列を持つFRT-G418R-FRT断片でMT8-1/pIUX1X2XKを形質転換し、形質転換後の菌体をジェネティシン 300 μg/mlを含むYPD (イーストエキストラクト 10 g/L、ポリペプトン 20 g/L、グルコース*20 g/L(ろ過滅菌添加*))に塗布した。生育した株において、FRT-G418R-FRT断片が、それぞれMET6、MET32、GLY1のORFと置き換わったことを確認するために、各々、数個のコロニーからゲノミックDNAを調製した。これを鋳型として、MET6については、(7)と(10)、MET32については(8)と(10)、GLY1については(9)と(10)、のプライマーを用いてPCRを行った。プライマー(7)、(8)、(9)は、各ORFの5’上流の配列、(10)はG418耐性遺伝子に相補的な配列である。PCRで適切な大きさのDNAが増幅された株については、さらにプラスミドpYRNFLP (図4)を形質転換し、Nourseothricin dihydrogen sulfate 50 μg/mlを含むYPDに塗布した。
(1) MET6DF1: 5’-ATGGTTCAATCTGCTGTCTTAGGGTTCCCAAGAATCGGTCCAAACAGAGAATTAAAGAAGGCCACTGAAGGTTACTGGAAACTCAAGCTATGCATCAAGC-3’ (配列番号14)
(2) MET6DR1: 5’-TTAATTCTTGTATTGTTCACGGAAGTACTTGGCGGCTTCGACCATATGAGTCAAAGACAATCT
AGTTTCTTCCCAGCCTCAATACGACTCACTATAGGGC-3’ (配列番号15)
(3) MET32DF1:5’-ATGGAGGATCAGGATGCTGCATTTATCAAACAGGCTACAGAAGCAATAGTGGATGTATCATTA
AATATAGATAACATAGAGTAAAACGACGGCCAGTGCC-3’ (配列番号16)
(4) MET32DR2: 5’-TCAGCCATTACTGCTACCATTGTGGTTGTTATCTTGACGATGAACGATGTTGGATTGCTTG
ACTTTTTTCAGTAATTCATCCGGCTCGTATGTTGTGTGG-3’ (配列番号17)
(5) GLY1DF1: 5’-ATGACTGAATTCGAATTGCCTCCAAAATATATCACCGCTGCTAACGACTTGCGGTCAGACACA
TTCACCACTCCAACTGCGTAAAACGACGGCCAGTGCC-3’ (配列番号18)
(6) GLY1DR1: 5’-TCAGTATTTGTAGGTTTTTATTTCGCGGATAGCGTTGCCATCAACGTCGACCTCGGTGGATTC
ACTACGGTAAATCTGGGCCGGCTCGTATGTTGTGTGG-3’ (配列番号19)
(7) MET6-5UF1: 5’-AAACTGTGGTAGTCATAGCTC-3’ (配列番号20)
(8) MET32-5UF1: 5’-TATACTAGTCAAAGATAGTATCCACC-3’ (配列番号21)
(9) GLY1-UF1: 5’-ATGTCACGCGAAACGGACC-3’ (配列番号22)
(10) G418MER: 5’-AAATGCTTGATGGTCGGAAG-3’ (配列番号23)
(11) MET6NF1: 5’-ATGGTTCAATCTGCTGTCTTAGGGT-3’ (配列番号24)
(12) MET6CR: 5’-TTAATTCTTGTATTGTTCACGGAAGTAC-3’ (配列番号25)
(13) MET32NF1: 5’-ATGGAGGATCAGGATGCTGCATTT-3’ (配列番号26)
(14) MET32CR2: 5’-TCAGCCATTACTGCTACCATTGTG-3’ (配列番号27)
(15) GLY1NF1: 5’-TGACTGAATTCGAATTGCCTC-3’ (配列番号28)
(16) GLY1CR1: 5’-ATAGCGTTGCCATCAACGTC-3’ (配列番号29)
【0062】
pYRNFLPは、酵母2μプラスミド上にあるFLPリコンビナーゼをTDH3p制御下で発現させるYRpタイプのプラスミドである。生育した株のゲノミックDNAを鋳型として、MET6については、(11)と(12)、MET32については(13)と(14)、GLY1については(15)と(16)のプライマーを用いてPCRを行った。(11)〜(16)のプライマーは全長の各ORFを増幅できるプライマーであり、これらを用いてPCRを行うと、FRT領域で組換えが起こりG418耐性遺伝子がループアウトした株では、ORFの長さに関係なく、約350bpの短い断片が増幅される。取得した各遺伝子破壊株は、G418耐性遺伝子がなくなっている。また、YRpタイプのプラスミドは安定性が悪く、選択圧をかけない状態では、細胞からすぐに脱落してしまうので、Nourseothricin dihydrogen sulfate(NAT)を含まない培地で生育させることにより、pYRNFLPも脱落させることができる。従って本方法で遺伝子を破壊した株は、選択マーカー(薬剤耐性)を持たないため、同じ方法で、さらに複数の遺伝子を破壊することができる。
【0063】
<PCRのための鋳型DNA調製方法>
酵母細胞を50 μlのLysis buffer(0.125 mg/ml ザイモリアーゼ100T、1 M ソルビトール、40 mM リン酸カリウムバッファー pH6.8、1 mMジチオスレイトール)に懸濁する。30℃で1時間インキュベートした後、プロテアーゼE (1 mg/ml) を5μl加え、さらに55℃で20分、99℃で10分インキュベートする。15,000 rpmで10分遠心を行い、上清を鋳型DNAとした。
【0064】
<キシロースからのエタノール醗酵試験>
SD培地(20 g/lグルコース、6.7 g/l yeast nitrogen baseアミノ酸抜き、アデニン20 mg/l、ヒスチジン 20 mg/l、ロイシン 100 mg/l、トリプトファン 20 mg/l)5 mlで前培養した酵母を初期OD600が0.03になるように500 mlのSD培地(casamino acid 20 g/lを含有)に移し、48 h、30℃で振とう培養機で好気培養した。細胞を遠心分離機で回収し、滅菌水で洗浄した後、初期OD600が20になるように100 mlのSX培地(キシロース50 g/l、6.7 g/l yeast nitrogen baseアミノ酸抜き、アデニン20 mg/l、ヒスチジン 20 mg/l、ロイシン 100 mg/l、トリプトファン 20 mg/l)に入れ、醗酵を開始した。醗酵は、生成した二酸化炭素ガスを逃すための出口がついた100 mlメディウム瓶で行い、100-150 rpmのスターラーで培養液を撹拌した。醗酵の進行を監視するために、培地を1 ml回収し、1000 gで遠心分離した後、上澄みを回収し、HPLCで分析した。
【0065】
<メタボローム解析用サンプルの調製法>
醗酵液から経時的にサンプリングした酵母細胞は遠心分離(1000 g、 3分)によって収穫した。細胞のペレットは、MilliQ水(Millipore、 Tokyo、Japan)で2回洗浄した後、液体窒素で凍結した。細胞は凍結乾燥し、使用時まで-80°Cで保存した。
【0066】
<醗酵もろみのHPLC分析>
醗酵培地中のキシロース、キシリトール、エタノール濃度はShimPack SPR-Pbカラム(Shimadzu、Kyoto、Japan)を用いたHPLC法(GL-7400システム、GL Sciences)によって定量した。カラムは80℃に保温し、MilliQ水を移動相として用い、流量は0.6 ml/minと設定した。各化合物はGL-7454の示差屈折率検出器(Shimadzu)によって検出した。
【0067】
細胞内代謝物分析方法
<GC-MS分析>
・ 10 mgの酵母の乾燥細胞を2 mlチューブに入れる
・ 混合溶媒1000 ml (MeOH : CHCl3 : H2O = 2.5 : 1 : 1)と内部標準溶液60 ml (2-isopropylmalic acid 1 mg/ml )を加える
・ シェーカーで1200 rpm、 37℃ 、 30分混合する
・ 遠心分離機で15000 x g、 4℃、 3分遠心分離する
・ 上澄み900 mlを1.5 mlチューブに移し、MilliQ水を400 ml加え、vortexで混合する
・ 遠心分離機で15000 x g、 4℃、 3分で遠心分離する
・ 上澄み200 mlを1.5 mlチューブに移し、遠心濃縮機及び凍結乾燥機で乾燥させる
・ メトキシアミン(ピリミジンで溶解)20 mg/mlを100 ml加える
・ シェーカーで1200 rpm、 30℃、 90分混合する
・ MSTFA 1 mg/mlを50 ml加え、シェーカーで1200 rpm、 37℃、 30分混合する
・ バイアルに移し、1 mlをGC-TOF/MSに注入し、分析する
分析条件
GCシステム : 6890N (Agilent)
カラム : CP-Sil 8 CB-Low Bleed 30 m x 0.25 mm ID (DF = 0.25 mm)
オーブン温度 : 80℃ (2分保持)、 15℃/分で330℃まで温度上昇 後、6分保持
インジェクター温度 : 230℃
イオン源温度 : 200℃
MS システム : PegasusIII TOF-MS (Leco)
マスレンジ : 85-500 m/z
スキャン速度 : 20 スキャン/秒
【0068】
<CE-MS分析>
・ 10 mgの酵母の乾燥細胞を2 mlチューブに入れる
・ MeOH 500ml、 CHCl3 500ml、 MilliQ水180ml、と内部標準溶液20ml (100mM ribitol、 100mM PIPES)を加え、vortexで混合する
・ 遠心分離機で15000 x g、 4℃、 3分遠心分離する
・ 上澄み350 mlを1.5 mlチューブに移し、MilliQ水を100ml加え、vortexで混合する
・ 沈殿物にMilliQ水100mlを加え、vortexで混合する
・ 両サンプルを遠心分離機で15000 x g、 4℃、 3分遠心分離する
・ 上澄みを2本の減外ろ過チューブ(Millpore Ultrafree-MC 5kDa MWCO)にチューブに移す
・ 遠心濃縮機でオーバーナイト乾燥させる
・ MilliQ水50mlで溶解する
・ バイアルに移し、分析する
分析条件
CEシステム : P/ACE MDQ (Beckman Coulter)
キャピラリー : Fused silica capillary 50μm i.d. x 80 cm
シーズ液 : 5 mM HCOONH3 in 50% MeOH
電解質 : 50 mM CH3COONH3 (pH 9.0)
電圧 : 30.0 kV
MSシステム : 4000 QTRAP (Applied Biosystems)
イオン化法 : ESI
送液ポンプ : MP-711 (GL Sciences)
【0069】
実施例1:複数のキシロース醗酵性酵母による、エタノール醗酵試験1
7種類のキシロース醗酵性酵母 ((1) MT8-1/pIUX1X2XK (2) MT8-1/pIUX1X2XK/Ad (3) MT8-1/pIUX1X2XK/pGK404-ScTal (4) NBRC1440/pIUX1X2XK (5) NBRC1445/pIUX1X2XK)を、実験方法に示した方法にてSD培地にて前培養し、細胞を集菌した後、滅菌水で洗浄した。酵母細胞をOD600=20となるように100 ml のSX培地に植菌して、キシロースからのエタノール醗酵試験を行った。適時醗酵もろみを1mlサンプリングし、HPLCでキシロース、キシリトール、エタノール濃度を測定した。また、同時に酵母細胞をサンプリングし、実験方法に示した方法で、GC-MSおよびCE-MS分析を行い、細胞内成分の網羅的分析を行った。得られた代謝物プロファイルを説明変数、諸性能評価値を応答変数としたOPLS回帰モデルを構築して、モデル構築に重要な説明変数を解析した結果、下記の化合物がキシロースからのエタノール醗酵性能と深く関与することがわかった。
Homoserine
Homocysteine
Methionine
Threonine
Glycine
Serine
【0070】
Homoserine、Threonine、GlycineはXCR(キシロース資化速度 g-comsumed xylose/h)、 EPR(エタノール生成速度 g-ethanol/h)、 EY(エタノール収率 g-ethanol/ g-comsumed xylose)、 SEPR (比エタノール生産速度 g-ethanol/g-cell-dried weight/h)の各醗酵性能の予測モデルに重要な意味を持つ代謝物と考えられた。これらの代謝物はグリシン生合成経路の中間体であるため、上記の醗酵性能はグリシン生合成経路と関連すると予測される。予測モデルから説明変数の係数値を解析したところ、これらの代謝物の細胞内蓄積量を減少させることによって上記の醗酵性能が向上されると予想された。そこで、これらの物質の細胞内蓄積量の減少を実現するために、グリシン生合成経路にある遺伝子の1つ、GLY1を破壊するターゲット遺伝子として選択した。
【0071】
一方、Homocysteine、Methionine、Serineは、 XPR(キシリトール生成速度 g-xylitol/h)、 XY(キシリトール収率 g-xylitol/g-comsumed xylose)、 SXPR(比キシリトール生産速度 g-xylitol/g-cell-dried weight/h)の各醗酵性能の予測モデルに重要な意味を持つ代謝物と考えられた。これらの代謝物はメチオニン生合成経路の中間体であり、上記の醗酵性能はこの経路と関連すると予測される。予測モデルから説明変数の係数値を解析したところ、Homocysteineの細胞内蓄積量を減少することによってキシリトール関連性能(XPR、XY、SXPR)が減少すると予想された。そこで、メチオニン合成経路関連遺伝子を破壊、することによって、Homocysteineの蓄積量を減少させることができれば、キシリトール生産を抑えることができると予測した。破壊するターゲット遺伝子として、MET6とMET32を選択した。図9に醗酵パフォーマンスと関連が深いと考えられる代謝物とそのマップ、係数等をまとめた。
【0072】
実施例2:MET6、MET32、GLY1の破壊株の作製
MT8-1/pIUX1X2XK株のMET6、MET32、GLY1遺伝子を実験方法に示したように、破壊した。すなわち、各遺伝子の開始コドンから下流80 bpと、終始コドンから上流80 bpの配列を両端に持ち、その間にFRT-G418R-FRTを持つDNA断片をPCRにて調製し、MT8-1/pIUX1X2XK株に形質転換して、ジェネティシン耐性を持つ株を選択した。複数の株よりPCRでMET6、MET32、GLY1各遺伝子に、FRT-G418R-FRTが挿入されたものを選択し、続いてpYRNFLPを形質転換した。Nourseothricin dihydrogen sulfate耐性を持つ株を選択し、pYRNFLP から発現される、リコンビナーゼの活性により、FRT配列間での組換えが起こり、G418耐性遺伝子がループアウトした株を、PCRにて選択した。
【0073】
実施例3:MET6、MET32、GLY1の破壊株のキシロース培地でのキシロースからのエタノール醗酵
MT8-1/pIUX1X2XK株(親株)とMET6、MET32、GLY1の破壊株を用いて、実験方法に示した方法にてSD培地にて前培養し、細胞を集菌した後、滅菌水で洗浄した。酵母細胞をOD=20となるように100 ml のSX培地に植菌して、エタノール醗酵試験を行った(n=3)。適時醗酵もろみを1 mlサンプリングし、HPLCでキシロース、キシリトール、エタノール濃度を測定した。キシロース資化経過を図5に、エタノール生成経過を図6に、キシリトール生成経過を図7に示す。また、醗酵パフォーマンスを表すいくつかの指標を図8に示した。
【0074】
この結果より、グリシン生合成経路の遺伝子GLY1を破壊することにより、キシロースの資化速度を早めることができた。また、メチオニン生合成経路の遺伝子、MET6あるいはMET32を破壊することにより、副産物の一つである、キシリトール生成量を減少させることができた。これらの育種を組み合わせる、あるいは他の育種と組み合わせることにより、エタノール収率も向上させることができると考えられる。
【0075】
<まとめ>
以上記載したように、解糖系や、ペントースリン酸経路、あるいは、導入したキシロース還元酵素、キシリトール脱水素酵素のような、キシロースからのエタノールへの代謝経路にあたる酵素ではなく、直接には関係のない、グリシン生合成経路、メチオニン生合成経路に関与する遺伝子を破壊することで、キシロースからのエタノール生成パフォーマンスを向上させることができた。
【産業上の利用可能性】
【0076】
バイオマスから生じた、5炭糖を炭素源として、エタノール醗酵を行うにあたり、エタノール生成収率を上げることができる。特に、ヘミセルロース含量が多い場合には、有効である。
本発明で育種した酵母を使用することによって、キシロースからのエタノール醗酵能、あるいは醗酵速度を改善することができる。どのような醸造用酵母、実験室酵母においても、同様の育種をすることができる。また、キシロースを含むものであれば、どのようなバイオマスから得られたもろみであっても、本育種酵母で同様の効果が見込める。
【配列表フリーテキスト】
【0077】
配列番号13:合成DNA
配列番号14:合成DNA
配列番号15:合成DNA
配列番号16:合成DNA
配列番号17:合成DNA
配列番号18:合成DNA
配列番号19:合成DNA
配列番号20:合成DNA
配列番号21:合成DNA
配列番号22:合成DNA
配列番号23:合成DNA
配列番号24:合成DNA
配列番号25:合成DNA
配列番号26:合成DNA
配列番号27:合成DNA
配列番号28:合成DNA
配列番号29:合成DNA
【特許請求の範囲】
【請求項1】
グリシン合成系タンパク質の遺伝子及び/又はメチオニン合成系タンパク質の遺伝子の発現機能を喪失させ、かつキシロース代謝酵素遺伝子を導入した微生物。
【請求項2】
前記グリシン合成系タンパク質の遺伝子がGLY1遺伝子である、請求項1に記載の微生物。
【請求項3】
前記メチオニン合成系タンパク質の遺伝子がMET6又はMET32遺伝子である、請求項1又は2に記載の微生物。
【請求項4】
酵母である、請求項1〜3のいずれか1項に記載の微生物。
【請求項5】
サッカロミセス・セレビシエである、請求項1〜4のいずれか1項に記載の微生物。
【請求項6】
前記キシロース代謝酵素遺伝子が、キシロース還元酵素遺伝子、キシリトール脱水素酵素遺伝子及びキシルロースリン酸化酵素遺伝子からなる群より選択される少なくとも1つのものである、請求項1〜5のいずれか1項に記載の微生物。
【請求項7】
グリシン合成系タンパク質の遺伝子及び/又はメチオニン合成系タンパク質の遺伝子の発現機能を喪失させることにより、キシロース醗酵能力の増加した微生物を作製する方法。
【請求項8】
前記グリシン合成系タンパク質の遺伝子がGLY1遺伝子である、請求項7に記載の方法。
【請求項9】
前記メチオニン合成系タンパク質の遺伝子がMET6又はMET32遺伝子である、請求項7又は8に記載の方法。
【請求項10】
前記微生物が酵母である、請求項7〜9のいずれか1項に記載の方法。
【請求項11】
前記微生物がサッカロミセス・セレビシエである、請求項7〜10のいずれか1項に記載の微生物。
【請求項12】
前記微生物がキシロース代謝酵素遺伝子が導入されたものである、請求項7〜11のいずれか1項に記載の方法。
【請求項13】
前記キシロース代謝酵素遺伝子が、キシロース還元酵素遺伝子、キシリトール脱水素酵素遺伝子及びキシルロースリン酸化酵素遺伝子からなる群より選択される少なくとも1つのものである、請求項12に記載の方法。
【請求項14】
請求項1〜6のいずれか1項に記載の微生物と、キシロース含有原料とを接触させる工程を含む、エタノールの製造方法。
【請求項1】
グリシン合成系タンパク質の遺伝子及び/又はメチオニン合成系タンパク質の遺伝子の発現機能を喪失させ、かつキシロース代謝酵素遺伝子を導入した微生物。
【請求項2】
前記グリシン合成系タンパク質の遺伝子がGLY1遺伝子である、請求項1に記載の微生物。
【請求項3】
前記メチオニン合成系タンパク質の遺伝子がMET6又はMET32遺伝子である、請求項1又は2に記載の微生物。
【請求項4】
酵母である、請求項1〜3のいずれか1項に記載の微生物。
【請求項5】
サッカロミセス・セレビシエである、請求項1〜4のいずれか1項に記載の微生物。
【請求項6】
前記キシロース代謝酵素遺伝子が、キシロース還元酵素遺伝子、キシリトール脱水素酵素遺伝子及びキシルロースリン酸化酵素遺伝子からなる群より選択される少なくとも1つのものである、請求項1〜5のいずれか1項に記載の微生物。
【請求項7】
グリシン合成系タンパク質の遺伝子及び/又はメチオニン合成系タンパク質の遺伝子の発現機能を喪失させることにより、キシロース醗酵能力の増加した微生物を作製する方法。
【請求項8】
前記グリシン合成系タンパク質の遺伝子がGLY1遺伝子である、請求項7に記載の方法。
【請求項9】
前記メチオニン合成系タンパク質の遺伝子がMET6又はMET32遺伝子である、請求項7又は8に記載の方法。
【請求項10】
前記微生物が酵母である、請求項7〜9のいずれか1項に記載の方法。
【請求項11】
前記微生物がサッカロミセス・セレビシエである、請求項7〜10のいずれか1項に記載の微生物。
【請求項12】
前記微生物がキシロース代謝酵素遺伝子が導入されたものである、請求項7〜11のいずれか1項に記載の方法。
【請求項13】
前記キシロース代謝酵素遺伝子が、キシロース還元酵素遺伝子、キシリトール脱水素酵素遺伝子及びキシルロースリン酸化酵素遺伝子からなる群より選択される少なくとも1つのものである、請求項12に記載の方法。
【請求項14】
請求項1〜6のいずれか1項に記載の微生物と、キシロース含有原料とを接触させる工程を含む、エタノールの製造方法。
【図1】


【図2】
【図3】

【図4】

【図5】
【図6】
【図7】

【図8】
【図9】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公開番号】特開2012−183013(P2012−183013A)
【公開日】平成24年9月27日(2012.9.27)
【国際特許分類】
【出願番号】特願2011−47628(P2011−47628)
【出願日】平成23年3月4日(2011.3.4)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成20年度独立行政法人新エネルギー・産業技術総合開発機構「新エネルギー技術研究開発/バイオマスエネルギー等高効率転換技術開発(先導技術開発)/セルロースエタノール高効率製造のための環境調和型統合プロセス開発」、産業技術力強化法19条の適用を受ける特許出願
【出願人】(309007911)サントリーホールディングス株式会社 (307)
【Fターム(参考)】
【公開日】平成24年9月27日(2012.9.27)
【国際特許分類】
【出願日】平成23年3月4日(2011.3.4)
【国等の委託研究の成果に係る記載事項】(出願人による申告)平成20年度独立行政法人新エネルギー・産業技術総合開発機構「新エネルギー技術研究開発/バイオマスエネルギー等高効率転換技術開発(先導技術開発)/セルロースエタノール高効率製造のための環境調和型統合プロセス開発」、産業技術力強化法19条の適用を受ける特許出願
【出願人】(309007911)サントリーホールディングス株式会社 (307)
【Fターム(参考)】
[ Back to top ]