
リグノフェノール誘導体の分子量制御方法

【課題】バイオマスとフェノール化合物を反応させて、リグノフェノール誘導体を製造する方法において、生成するリグノフェノール誘導体の分子量を制御する方法を提供する。
【解決手段】リグノセルロース材料とフェノール化合物とを反応させてリグノフェノール誘導体を製造する方法において、リグノセルロース材料にフェノール化合物と酸とを添加して得られた反応液から水層を相分離して反応系から取り除き、フェノール溶液中で該フェノール溶液に残存した酸と高分子量ポリマーを含むリグノフェノール誘導体とを反応させることを特徴とする分子量を制御する方法である。
【解決手段】リグノセルロース材料とフェノール化合物とを反応させてリグノフェノール誘導体を製造する方法において、リグノセルロース材料にフェノール化合物と酸とを添加して得られた反応液から水層を相分離して反応系から取り除き、フェノール溶液中で該フェノール溶液に残存した酸と高分子量ポリマーを含むリグノフェノール誘導体とを反応させることを特徴とする分子量を制御する方法である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、リグノセルロース材料にフェノール化合物と酸とを添加して得られるリグノフェノール誘導体の分子量制御方法に関する。
【背景技術】
【0002】
これまで廃棄又は焼却して処分していた木材や草、廃木材、建築廃材等のバイオマス由来のリグニンを有効に活用するための研究開発が盛んに行われている。リグニンを利用する技術として、例えば、特許文献1には、植物由来のリグニンをフェノール化合物及び酸と反応させてリグノフェノール誘導体に変換する方法が開示されている。
【0003】
このリグノフェノール誘導体の製造方法は、以下の(1)〜(4)の工程からなる。
(1)フェノール化合物(具体的にはクレゾール)で溶媒和したリグノセルロース材料に酸を加え、リグノフェノール誘導体を生成する工程(フェノールのグラフティング工程)。
(2)フェノール化合物層と酸層とを相分離してフェノール化合物層を得る工程(相分離工程)。
(3)得られたフェノール化合物層をリグノフェノール誘導体の貧溶媒(具体的にはジエチルエーテルやジイソプロピルエーテル)に滴下して、リグノフェノール誘導体をフェノール化合物と分離して不溶区分として回収する工程(1次精製工程)。
(4)得られた不溶区分をアセトンに溶解してさらにリグノフェノール誘導体の貧溶媒に滴下することにより、不溶区分に含まれるリグノフェノール誘導体を分離して回収する工程(2次精製工程)。
リグニンは、シキミ酸経路を介して生産されるフェニルアラニンやチロシンから誘導されるフェニルプロパノイドp-hydroxycinnamic alcohol類(例えば、p−クマリルアルコール、コニフェリルアルコール、シナピルアルコール)が、ペルオキシダーゼにより脱水素重合して分子量約3,000程度のサブユニットを形成していると考えられている。さらに脱水素重合後に生成するキノンメチドへH2O、リグニン単位、炭水化物の求核付加が生じ、より複雑な3次元網目構造を持つ巨大ポリマーが形成される。
【0004】
リグノフェノール誘導体は、酸の存在下、リグニンの反応活性の高いベンジルエーテル結合が開裂され、フェニルプロパン骨格のベンジル位にフェノール化合物が結合した1,1−ビス(アリール)プロパンを構成ユニットとするポリマーである。反応時間が短いと未反応のベンジルエーテル結合が残ってしまうため、生成するリグノフェノールは高分子量のものが多く生成される。また反応時間を長くした場合には、ベンジル位以外にフェノール化合物が結合することもあり、分子量分布の幅が大きくなってしまうことがある。特許文献1に記載された方法では、リグノフェノール誘導体の分子量は3,000〜100,000の幅広い分子量分布を有している。
【0005】
幅広い分子量分布を有することが望ましい場合もあるが、一般的に、樹脂原料とする場合には、分子量分布の幅が狭く高分子量サイドに平均分子量がシフトしているリグノフェノール誘導体が望まれ、反応性や溶解性の良いものを望む場合には平均分子量が低分子量側にシフトしたリグノフェノール誘導体が望まれている。
【0006】
非特許文献1には、精製したリグノフェノール誘導体をアルカリ処理することによって高分子リグノフェノール誘導体を低分子化する方法が報告されている。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】特開平02−233701号公報
【非特許文献】
【0008】
【非特許文献1】Polymer International, 47, 277-290 (1998)
【発明の開示】
【発明が解決しようとする課題】
【0009】
特許文献1に記載されたフェノール化合物とリグニンの反応時間を長くすれば、ベンジルエーテル結合の開裂反応が継続的に起こり、リグノフェノール誘導体の低分子化が進むことが考えられる。しかし、ベンジル位以外の結合も開裂され、長時間処理により、リグニン主要単位間結合であるβ−アリールエーテルの開裂が生じる可能性があり、リグノフェノールの特性であるβ−アリールエーテルを中心としたリニア型ポリマー特性が損なわれる可能性がある。また、リグニンのスルホン化や単糖のフルフラール類への分解などの副反応が起きるため、リグノフェノールの収率低下あるいはリグノセルロース材料の有効活用の観点からも好ましくない。
【0010】
リグノフェノール誘導体のアルカリ処理による分子量制御の場合、主要単位間結合であるβ−アリールエーテルの開裂が生じることにより低分子化し、高分子のリグノフェノール誘導体の特性であるβ−アリールエーテルを中心としたリニア型ポリマーの特性が損なわれる可能性がある。
【0011】
本発明は、リグニンのスルホン化や単糖のフルフラール類への分解を抑制し、目的とする特性を維持させてリグノフェノール誘導体の分子量をコントロールする方法を提供することを目的とする。
【課題を解決するための手段】
【0012】
本発明のリグノフェノール誘導体の分子量制御方法は、リグノセルロース材料とフェノール化合物とを反応させてリグノフェノール誘導体を製造する方法において、リグノセルロース材料にフェノール化合物と酸とを添加して得られた反応液から水層を相分離して反応系から取り除き、フェノール溶液中で該フェノール溶液に残存した酸とリグノフェノール誘導体とを反応させることを特徴とする。
【0013】
また、本発明のリグノフェノール誘導体の分子量制御方法は、リグノセルロース材料とフェノール化合物とを反応させてリグノフェノール誘導体を製造する方法において、リグノセルロース材料にフェノール化合物と酸とを添加して得られた反応液の水層の酸濃度を低下させる工程と、フェノール溶液中で該フェノール溶液に残存した酸とリグノフェノール誘導体とを反応させる工程とを含む。
【発明の効果】
【0014】
本発明により、リグノセルロース材料とフェノール化合物とを反応させて得られる高分子量のリグノフェノール誘導体を、目的とする平均分子量のリグノフェノール誘導体に変換制御することが可能となった。リグノフェノール誘導体の収率も低下することもほとんどない。
【図面の簡単な説明】
【0015】
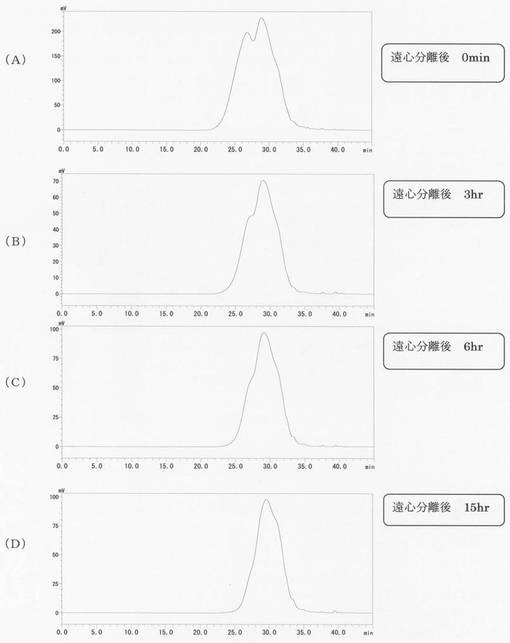
【図1】本発明に係る実施例1のリグノフェノール誘導体のGPC(ゲル浸透クロマト)分析結果を示す図で、(A)〜(D)は各々、遠心分離0分後、3時間後、6時間後、15時間後の結果である。
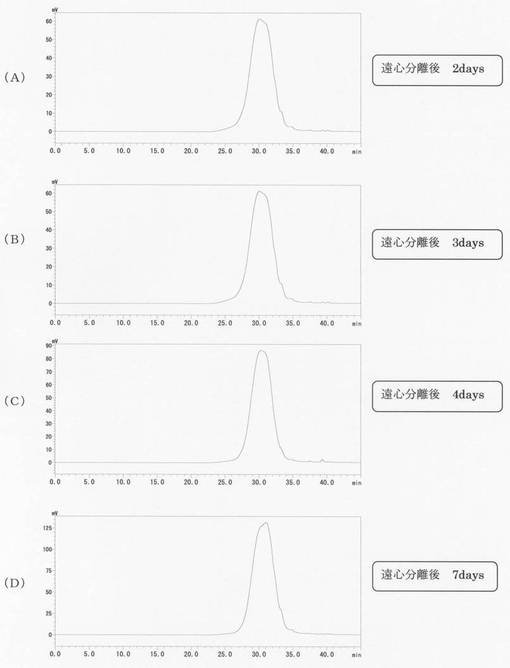
【図2】実施例1のリグノフェノール誘導体のGPC(ゲル浸透クロマト)分析結果を示す図であり、(A)〜(D)は各々、遠心分離2日後、3日後、4日後、7日後の結果である。
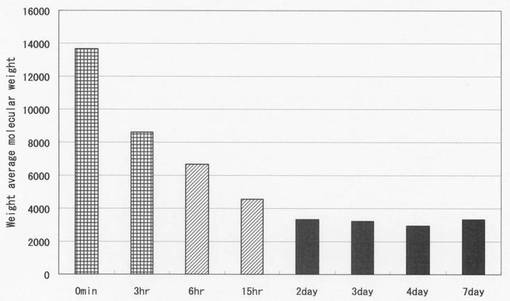
【図3】リグノフェノール誘導体の平均分子量の経時変化を示す図である。
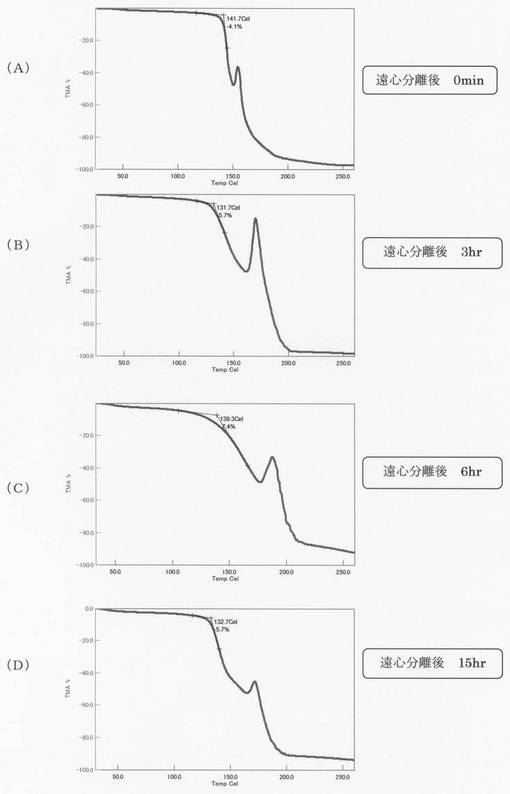
【図4】実施例1の熱機械分析(TMA)の結果を示す図で、(A)〜(D)は各々、遠心分離0分後、3時間後、6時間後、15時間後の結果である。
【図5】実施例1の精製リグノフェノール誘導体収率を示す図である。
【図6】実施例1のリグノフェノール誘導体製造に使用したクレゾール量(モル濃度)を示す図である。
【図7】実施例1の各リグノフェノール誘導体を0.5NのNaOHで170℃、1時間処理し、GPC(ゲル浸透クロマト)分析した結果を示す図で、(A)〜(D)は各々、遠心分離0分後、3時間後、6時間後、15時間後の結果である。
【図8】実施例2の遠心分離後すぐに水処理をしたときのフェノール層から回収されたリグノフェノール誘導体のGPC(ゲル浸透クロマト)分析結果を示す図である。
【図9】実施例2の水処理後約15時間後にフェノール層から回収されたリグノフェノール誘導体のGPC(ゲル浸透クロマト)分析結果(遠心分離2日後)を示す図である。
【発明を実施するための形態】
【0016】
以下、本発明について幾つかの実施形態に基づいて詳細に説明する。
【0017】
[実施形態1]
本実施形態1は、リグノセルロース材料とフェノール化合物とを反応させてリグノフェノール誘導体を製造する方法において、リグノセルロース材料にフェノール化合物と酸とを添加して得られたリグノフェノール誘導体を含む反応液を相分離して水層を反応系から取り除き、フェノール溶液中でこのフェノール溶液に残存した酸と高分子量ポリマーのリグノフェノール誘導体とを反応させるリグノフェノール誘導体の分子量制御方法である。具体的には、以下の工程からなる、高分子量ポリマーのリグノフェノール誘導体の低分子化方法である。
(1)フェノール化合物で溶媒和したリグノセルロース材料に酸を加え、リグノフェノール誘導体を生成する工程(フェノールのグラフティング工程)。
(2)処理液のフェノール層と酸層とを相分離してフェノール溶液を得る工程(相分離工程)。
(3)フェノール溶液を室温に放置する工程(低分子化工程)。
【0018】
得られた低分子量リグノフェノール誘導体は、以下(イ)及び/又は(ロ)の工程により精製することができる。
(イ)フェノール溶液をリグノフェノール誘導体の貧溶媒、具体的にはジエチルエーテルやジイソプロピルエーテルに滴下して、リグノフェノール誘導体をフェノール化合物と分離して不溶区分として回収する工程(1次精製工程)。
(ロ)得られた不溶区分をアセトンに溶解してさらにリグノフェノール誘導体の貧溶媒に滴下することにより、不溶区分に含まれるリグノフェノール誘導体を分離して回収する工程(2次精製工程)。なお、本明細書中で「フェノール溶液」とは、酸と高分子リグノフェノール誘導体及び/又は低分子リグノフェノール誘導体とを含むフェノール化合物溶液をいう。
【0019】
フェノールのグラフティング工程では、リグノセルロース材料、フェノール化合物及び酸水溶液を攪拌する。この段階で、フェノール化合物で親和され膨潤化した木片等のリグニンのフェニルプロパンのベンジル位が開裂され、フェノール化合物と結合する反応が始まるが、この工程では、反応材料を攪拌して、硫酸とフェノール化合物を十分に混合することが重要である。攪拌により、酸がフェノール化合物の溶液に混じる。
【0020】
フェノール化合物は、親油性のフェノール化合物の溶液、例えば、p−クレゾール、m−クレゾール、o−クレゾール、アニソール、2,4−ジメトキシフェノール、2,6−ジメトキシフェノール、2,4−ジメチルフェノール、2,6−ジメチルフェノール、プロピルフェノール、i−プロピルフェノール、tert−ブチルフェノール、カテコール、レゾルシノール、ピロガロール、フロログルシノール、ビスフェノール、バニリン、シリンゴール、グアイアゴール、フェルラ酸、クマル酸の何れか、又は、これらの組み合わせからなる溶液を使用することができる。酸は、バイオマスの加水分解に用いられる酸であればよく、塩酸、硫酸等の無機酸であり、硫酸が望ましい。
【0021】
反応時間は、リグノセルロース材料、反応温度によって異なるが、長時間の反応は、リグニンのスルホニル化や加水分解された糖のフルフラールなどへの分解が進行するため好ましくない。この段階で生ずるリグノフェノール誘導体は、平均分子量を9,000〜14,000程度の高分子量ポリマーを含む。反応時間は、長くなるより短いほうが好ましく、一般的には最長でも1時間程度である。
【0022】
生成したリグノフェノール誘導体をフェノール化合物溶液に溶解する。次いで、反応溶液を相分離により、リグノフェノール誘導体を含む溶液の層(本明細書中では「フェノール溶液」という。)と酸液層とに分離する。相分離は自然比重分離又は遠心分離処理を行えばよい。フェノール化合物は、わずかではあるが親水性を有するので、フェノール溶液にはわずかに酸が残存することになる。
【0023】
相分離され反応系から酸層が取り除かれたフェノール溶液は、室温で放置又は緩やかな攪拌が行われ、フェノール溶液に残存する酸とリグノフェノール誘導体とを反応させる。この反応系は、わずかに残存する酸とフェノール化合物に溶解したリグノフェノール誘導体との反応であるため、フェニルプロパンが穏やかな条件下にあり、先の工程で未反応であったベンジル位の選択的な開裂反応が、プロパンの他の炭素に優先して進むものと考えられる。したがって、ベンジル位の開裂に伴ったフェノール化合物の結合とリグノフェノール誘導体の低分子化が優先的に進むことになる。
【0024】
反応時間は、目的とするリグノフェノール誘導体の平均分子量によって異なるので、適宜決定すればよい。相分離後2日で平均分子量は約3,000となり、これ以上の低分子化は惹起されない。これはリグニンが分子量約3,000のサブユニットにまで分解されて、この反応条件下ではこれ以上の反応が起こらないためと考えられる。
【0025】
低分子量リグノフェノール誘導体も高分子量リグノフェノールが有している基本特性である熱軟化特性を有している。このことは、ベンジル位以外の炭素での開裂やフェノール化合物との結合が起きていないためと考えられる。このことは、リグニンの主要単位間結合であるβ−アリールエーテル結合が保持され、リニア型ポリマー特性を有していることを意味する。
【0026】
フェノール溶液内での酸とリグノフェノール誘導体との反応を停止するためには、フェノール溶液をリグノフェノール誘導体の貧溶媒に滴下して、リグノフェノール誘導体をフェノール化合物と分離すればよい。
【0027】
リグノフェノール誘導体の貧溶媒は、非対称エーテル類、対称アルキルエーテル、環状炭化水素類、直鎖状炭化水素、エステル類を使用することができる。具体的には、ジイソプロピルエーテル、ジエチルエーテル、n−ヘキサン、ペンタン、ベンゼン、トルエン、酢酸エチルを挙げることができ、ジイソプロピルエーテルが好ましい。なお、上述した貧溶媒を混合して、極性を変化させて使用することは当分野において常套手段であり、極性の異なる溶媒を組み合わせたものを貧溶媒として使用してもよい。
【0028】
反応開始から反応停止までの時間を1時間〜2日の間で選択することで、リグノフェノール誘導体の平均分子量を、例えば、約14,000〜約3,500、好ましくは約10,000〜約3,500の範囲で制御することができる。平均分子量を約10,000とするのであれば、反応開始から3時間以内、平均分子量約5,000とする場合は15時間程度、5,000以下にする場合は、2日間程度の範囲にコントロールすることが好ましい。
【0029】
得られた貧溶媒の不溶区分は、さらに、アセトンに溶解して再度リグノフェノール誘導体の貧溶媒に滴下することにより、不溶区分に含まれるリグノフェノール誘導体を分離(2次精製)してもよい。
【0030】
[実施形態2]
本実施形態2は、フェノール溶液の酸濃度をさらに低下させることでフェノール溶液内での酸とリグノフェノール誘導体との反応を止める点で実施形態1と異なる。
【0031】
本実施形態では、酸をフェノール溶液から効率的に取り除くため、フェノール溶液を水と接触させる処理を行う。水との接触処理として、具体的には以下の操作を挙げることができる。
(1)フェノール溶液を水に滴下し、懸濁させて水分散液とする、又は、
(2)フェノール溶液を水で洗浄する。
【0032】
フェノール溶液を水分散液とするためには、フェノール溶液より多い水を必要とするが、水洗浄の場合には少ない水で繰り返し洗浄することで水の量を減らすことが可能である。また、フェノール溶液と接触させる水をアルカリ性水溶液とし、酸の中和を行ってもよい。
【0033】
フェノール層と酸層を分離した後、分離して得られたフェノール溶液を水と接触させる、またはアルカリ処理する時間により反応時間をコントロールすることができる。実施形態1と同様、反応開始から反応停止までの時間を1時間〜2日の間で選択することで、リグノフェノール誘導体の平均分子量を、例えば、約14,000〜約3,500の範囲で制御することができる。平均分子量を約10,000とするのであれば、反応開始から3時間以内、平均分子量約5,000とする場合は15時間程度、5,000以下にする場合は、2日間程度の範囲にコントロールすることが好ましい。
【0034】
フェノール溶液と水との接触処理の後、処理液から水を除去する操作を行ってリグノフェノール誘導体を水と分離する。水の除去操作は、例えば、処理液を液層分離して、酸を含む水層をフェノール溶液から取り除けばよい。
【0035】
水層と分離されたリグノフェノール誘導体を含む溶液をさらに、リグノフェノール誘導体の貧溶媒に添加して、リグノフェノール誘導体を沈殿物として得ることができる。
【0036】
得られたリグノフェノール誘導体は、精製度合いを高めるために、アセトンやメチルエチルケトンに溶解し、さらにジイソプロピルエーテル又はメチルエチルケトンと液体状の直鎖状炭化水素との混合溶媒に滴下して沈殿させてもよい。
【0037】
また、得られたリグノフェノール誘導体は、精製度を高めるために、アセトンやメチルエチルケトンに溶解し、さらにリグノフェノール誘導体の貧溶媒に滴下して再沈殿させてもよい。
【0038】
本発明者らにより、リグノフェノール誘導体の溶媒溶解性は、次の序列になることがわかっている。
【0039】
強アルカリ溶液>アセトン、メチルエチルケトンなどのアルキルケトン類>ジメチルフォルムアミド(DMF)、N-メチルピロリドン(NMP)のような非プロトン極性溶媒>テトラヒドロフラン(THF)、ジオキサンのような環状エーテル類>ピリジンのようなヘテロ環状化合物>エタノール、セロソルブのようなアルコール類>tert−ブチルメチルエーテル(TBME)、シクロペンチルメチルエーテル(CPME)などの非対称エーテル類>ジエチルエーテルやジプロピルエーテル、ジイソプロピルエーテルなどの対称アルキルエーテル>トルエン又はキシレンとヘキサンとの混合溶媒>ベンゼン、トルエン、ヘキサンなどの芳香族及び環状炭化水素類
リグノフェノール誘導体の貧溶媒は、非対称エーテル類、対称アルキルエーテル、環状炭化水素類、直鎖状炭化水素、エステル類を使用することができる。具体的には、ジイソプロピルエーテル、ジエチルエーテル、n−ヘキサン、ペンタン、ベンゼン、トルエン、酢酸エチルを挙げることができ、ジイソプロピルエーテルが好ましい。なお、上述した貧溶媒を混合して、極性を変化させて使用することは当分野において常套手段であり、極性の異なる溶媒を組み合わせたものを貧溶媒として使用してもよい。
【0040】
[実施形態3]
本実施形態3は、フェノール溶液を水と接触させた後の処理液から水を除去する操作が実施形態2と異なる。
【0041】
すなわち、水との接触処理の後、処理液に非水溶性極性溶媒を加える。非水溶性極性溶媒は、リグノフェノール誘導体を溶解でき、水分離しやすい溶媒である。このような溶媒として、メチルエチルケトン、クロロホルム、ジクロロメタン、酢酸エチル、エーテル類、トルエン、ベンゼンを挙げることができる。リグノフェノール誘導体の溶解性と疎水性とを考慮するとメチルエチルケトンが好ましい。
【0042】
この操作により、処理液は水層と非水溶性極性溶媒層とに分離する。非水溶性極性溶媒層には、リグノフェノール誘導体が含まれるが、酸はほとんど含まれない。したがって、リグノフェノール誘導体は酸の影響を受けることがない。
【0043】
得られた非水溶性極性溶媒層をさらに、リグノフェノール誘導体の貧溶媒に添加して、リグノフェノール誘導体を沈殿物として得ることができる。
【0044】
得られたリグノフェノール誘導体は、精製度を高めるために、アセトンやメチルエチルケトンに溶解し、さらにリグノフェノール誘導体の貧溶媒に滴下して再沈殿させてもよい。
【0045】
なお、メチルエチルケトン、貧溶媒、フェノール化合物の分離は蒸留にてほぼ分離可能である。
【実施例1】
【0046】
3mol倍のp−クレゾールを収着したヒノキ木粉1.5g(木粉として1g)に72%の硫酸を10mリットル添加し、30℃で1時間攪拌した。遠心分離の10分前にm,p−クレゾールを5mリットル加え、攪拌した。反応終了後、遠心分離し、クレゾール層と硫酸層に分離した。得られたクレゾール層を室温で0時間〜7日間放置し、クレゾール層をジイソプロピルエーテルに滴下し、リグノフェノール誘導体を精製して、平均分子量の変化を調べた。精製溶媒はジイソプロピルエーテルを使用した。
【0047】
得られたリグノフェノール誘導体のGPC(ゲル浸透クロマト)分析の結果を図1及び図2に示す。図1(A)〜(D)は各々、遠心分離0分後、3時間後、6時間後、15時間後、図2(A)〜(D)は各々、遠心分離2日後、3日後、4日後、7日後の結果である。時間が経つにつれ溶出時間が長くなり分子量が小さくなり、2日後〜7日後はほとんど変化していないのが分かる。
【0048】
図3に平均分子量を示す。すぐに精製したリグノフェノール誘導体の重量平均分子量は約14,000であったが、3時間室温放置で約8,500まで低下した。15時間経過すると分子量は約4,200まで低下した。7日目まで経時変化を調べたが、2日目以降は、分子量約3,500で更なる低分子化は見られなかった。
また、熱機械分析(TMA)の結果を図4に示す。TMA分析は、SEIKO EXSTAR 6000 TMA/SSにより行った。試料約2mgを5mmΦ,深さ2mmのアルミニウムパンに入れ、ハンドプレスで表面が平滑になるように圧縮し、その上にアルミニウム板を載せ、その上からTMA測定機の石英測定針でプレスした。測定は鉛直方向に4.9 Nの荷重をかけながら温度を変化させ、測定針の変化を測定するペネトレート法で分析した。なお、温度上昇: 2.0度/min、窒素流量:150cm3/min、測定温度:50−250℃の条件で行った。(A)〜(D)は各々、遠心分離0分後、3時間後、6時間後、15時間後の結果を示す。遠心分離3時間後、6時間後、15時間後の低分子量のリグノフェノール誘導体であっても、熱軟化特性を有していることがわかる。
【0049】
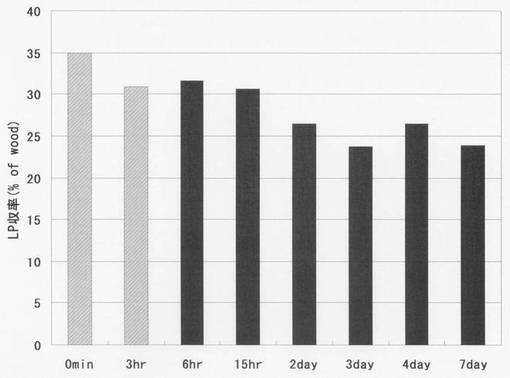
図5に、精製リグノフェノール誘導体の収率を示す。低分子化後2日以降のリグノフェノール誘導体の収率は多少の変化があるものの、25%前後で安定している。
【0050】
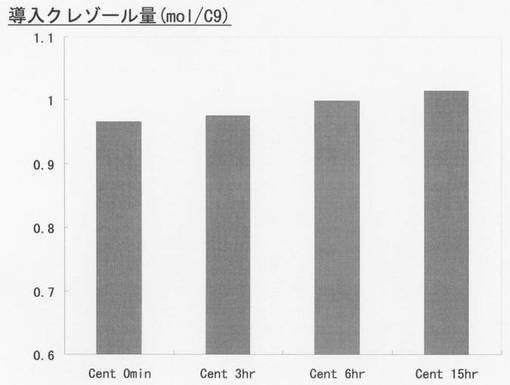
1H−NMR分析を行い、得られたリグノフェノールに含まれる結合フェノール量を測定した。遠心0分後、3時間後、6時間後及び15時間後のリグノフェノール誘導体の導入フェノール量を図6に示す。縦軸の「導入フェノール量mol/C9」はリグニン1ユニットであるフェニルプロパン単位あたり何モルフェノールが導入されたかを示す指標である。遠心後の時間経過が長いほど導入フェノール量mol/C9が少し増えており、ベンジルエーテル結合が開裂され、そこにクレゾールが導入されたことを示している。
【0051】
精製した各リグノフェノール誘導体を0.5NのNaOHで170℃、1時間処理し、前述と同じ条件でGPC分析を行った。結果を図7に示す。(A)〜(D)は各々、遠心分離0分後、3時間後、6時間後、15時間後の結果を示す。低分子化したリグノフェノール誘導体のアルカリ処理結果も高分子リグノフェノール誘導体のアルカリ処理の結果とほぼ同様のアルカリ分解挙動が見られた。これらの結果から、低分子化リグノフェノールにおいてもリグニン主要単位間結合であるβ−アリールエーテル結合が保持されていることが示された。
【実施例2】
【0052】
3mol倍のp−クレゾールを収着したヒノキ木粉15g(木粉として10g)に72%硫酸100mlを加え、30℃で60分攪拌し、終了10分前にm,p−クレゾール80mリットルを加えた。これを遠心分離し、フェノール層を回収した。フェノール層に固形分も少なく、分離性は良好であった。界面洗浄として10mリットルのm,p−クレゾールを加え、界面を洗浄した。クレゾール層100mリットルが回収された。このクレゾール層を600mリットルの水に滴下した。滴下後は淡桃白色の分散液となった。これに200mリットルのメチルエチルケトンを加え、分液ロートに入れ、一晩放置した。翌日、メチルエチルケトン層の濁りは少なくなっていた。水層を抜き、メチルエチルケトン層160mリットルを回収した。
【0053】
上記操作において、水処理−メチルエチルケトン抽出処理後のメチルエチルケトン層中でのリグノフェノール誘導体の安定性を確認するため、メチルエチルケトン層の放置時間を変えた実験を行った。
遠心分離後のフェノール層にすぐに水を加えてメチルエチルケトンで抽出した後、メチルエチルケトン層10mリットルをすぐに120mリットルのジイソプロピルエーテルに滴下し、1次精製リグノフェノール誘導体を得た。1次精製リグノフェノール誘導体をアセトンに溶解後、ジイソプロピルエーテルに滴下し2次精製を行い、0.125gのリグノフェノール誘導体を得た。GPC分析の結果を図8に示す。GPC分析条件は実施例1と同じである。
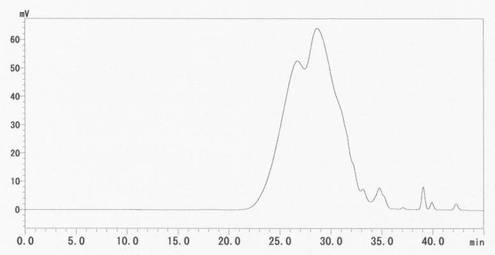
遠心分離後のフェノール層にすぐに水を加え、メチルエチルケトンで抽出し15時間放置した後、メチルエチルケトン層10mリットルを120mリットルのジイソプロピルエーテルに滴下し、1次精製リグノフェノール誘導体を得た。1次精製リグノフェノール誘導体をアセトンに溶解後、ジイソプロピルエーテルに滴下し2次精製を行い、0.136gのリグノフェノール誘導体を得た。GPC分析の結果を図9に示す。GPC分析条件は実施例1と同じである。
GPCを見るとクレゾール分離1日後のリグノフェノール誘導体は、若干、重量平均分子量にばらつきがあり、高分子区分が少なくなっているが、ほとんど差は見られなかった。この結果から、フェノール層と水層を分離してから目的の分子量となる時間が経過したところで水処理するとその分子量で止めることができることがわかる。
【技術分野】
【0001】
本発明は、リグノセルロース材料にフェノール化合物と酸とを添加して得られるリグノフェノール誘導体の分子量制御方法に関する。
【背景技術】
【0002】
これまで廃棄又は焼却して処分していた木材や草、廃木材、建築廃材等のバイオマス由来のリグニンを有効に活用するための研究開発が盛んに行われている。リグニンを利用する技術として、例えば、特許文献1には、植物由来のリグニンをフェノール化合物及び酸と反応させてリグノフェノール誘導体に変換する方法が開示されている。
【0003】
このリグノフェノール誘導体の製造方法は、以下の(1)〜(4)の工程からなる。
(1)フェノール化合物(具体的にはクレゾール)で溶媒和したリグノセルロース材料に酸を加え、リグノフェノール誘導体を生成する工程(フェノールのグラフティング工程)。
(2)フェノール化合物層と酸層とを相分離してフェノール化合物層を得る工程(相分離工程)。
(3)得られたフェノール化合物層をリグノフェノール誘導体の貧溶媒(具体的にはジエチルエーテルやジイソプロピルエーテル)に滴下して、リグノフェノール誘導体をフェノール化合物と分離して不溶区分として回収する工程(1次精製工程)。
(4)得られた不溶区分をアセトンに溶解してさらにリグノフェノール誘導体の貧溶媒に滴下することにより、不溶区分に含まれるリグノフェノール誘導体を分離して回収する工程(2次精製工程)。
リグニンは、シキミ酸経路を介して生産されるフェニルアラニンやチロシンから誘導されるフェニルプロパノイドp-hydroxycinnamic alcohol類(例えば、p−クマリルアルコール、コニフェリルアルコール、シナピルアルコール)が、ペルオキシダーゼにより脱水素重合して分子量約3,000程度のサブユニットを形成していると考えられている。さらに脱水素重合後に生成するキノンメチドへH2O、リグニン単位、炭水化物の求核付加が生じ、より複雑な3次元網目構造を持つ巨大ポリマーが形成される。
【0004】
リグノフェノール誘導体は、酸の存在下、リグニンの反応活性の高いベンジルエーテル結合が開裂され、フェニルプロパン骨格のベンジル位にフェノール化合物が結合した1,1−ビス(アリール)プロパンを構成ユニットとするポリマーである。反応時間が短いと未反応のベンジルエーテル結合が残ってしまうため、生成するリグノフェノールは高分子量のものが多く生成される。また反応時間を長くした場合には、ベンジル位以外にフェノール化合物が結合することもあり、分子量分布の幅が大きくなってしまうことがある。特許文献1に記載された方法では、リグノフェノール誘導体の分子量は3,000〜100,000の幅広い分子量分布を有している。
【0005】
幅広い分子量分布を有することが望ましい場合もあるが、一般的に、樹脂原料とする場合には、分子量分布の幅が狭く高分子量サイドに平均分子量がシフトしているリグノフェノール誘導体が望まれ、反応性や溶解性の良いものを望む場合には平均分子量が低分子量側にシフトしたリグノフェノール誘導体が望まれている。
【0006】
非特許文献1には、精製したリグノフェノール誘導体をアルカリ処理することによって高分子リグノフェノール誘導体を低分子化する方法が報告されている。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】特開平02−233701号公報
【非特許文献】
【0008】
【非特許文献1】Polymer International, 47, 277-290 (1998)
【発明の開示】
【発明が解決しようとする課題】
【0009】
特許文献1に記載されたフェノール化合物とリグニンの反応時間を長くすれば、ベンジルエーテル結合の開裂反応が継続的に起こり、リグノフェノール誘導体の低分子化が進むことが考えられる。しかし、ベンジル位以外の結合も開裂され、長時間処理により、リグニン主要単位間結合であるβ−アリールエーテルの開裂が生じる可能性があり、リグノフェノールの特性であるβ−アリールエーテルを中心としたリニア型ポリマー特性が損なわれる可能性がある。また、リグニンのスルホン化や単糖のフルフラール類への分解などの副反応が起きるため、リグノフェノールの収率低下あるいはリグノセルロース材料の有効活用の観点からも好ましくない。
【0010】
リグノフェノール誘導体のアルカリ処理による分子量制御の場合、主要単位間結合であるβ−アリールエーテルの開裂が生じることにより低分子化し、高分子のリグノフェノール誘導体の特性であるβ−アリールエーテルを中心としたリニア型ポリマーの特性が損なわれる可能性がある。
【0011】
本発明は、リグニンのスルホン化や単糖のフルフラール類への分解を抑制し、目的とする特性を維持させてリグノフェノール誘導体の分子量をコントロールする方法を提供することを目的とする。
【課題を解決するための手段】
【0012】
本発明のリグノフェノール誘導体の分子量制御方法は、リグノセルロース材料とフェノール化合物とを反応させてリグノフェノール誘導体を製造する方法において、リグノセルロース材料にフェノール化合物と酸とを添加して得られた反応液から水層を相分離して反応系から取り除き、フェノール溶液中で該フェノール溶液に残存した酸とリグノフェノール誘導体とを反応させることを特徴とする。
【0013】
また、本発明のリグノフェノール誘導体の分子量制御方法は、リグノセルロース材料とフェノール化合物とを反応させてリグノフェノール誘導体を製造する方法において、リグノセルロース材料にフェノール化合物と酸とを添加して得られた反応液の水層の酸濃度を低下させる工程と、フェノール溶液中で該フェノール溶液に残存した酸とリグノフェノール誘導体とを反応させる工程とを含む。
【発明の効果】
【0014】
本発明により、リグノセルロース材料とフェノール化合物とを反応させて得られる高分子量のリグノフェノール誘導体を、目的とする平均分子量のリグノフェノール誘導体に変換制御することが可能となった。リグノフェノール誘導体の収率も低下することもほとんどない。
【図面の簡単な説明】
【0015】
【図1】本発明に係る実施例1のリグノフェノール誘導体のGPC(ゲル浸透クロマト)分析結果を示す図で、(A)〜(D)は各々、遠心分離0分後、3時間後、6時間後、15時間後の結果である。
【図2】実施例1のリグノフェノール誘導体のGPC(ゲル浸透クロマト)分析結果を示す図であり、(A)〜(D)は各々、遠心分離2日後、3日後、4日後、7日後の結果である。
【図3】リグノフェノール誘導体の平均分子量の経時変化を示す図である。
【図4】実施例1の熱機械分析(TMA)の結果を示す図で、(A)〜(D)は各々、遠心分離0分後、3時間後、6時間後、15時間後の結果である。
【図5】実施例1の精製リグノフェノール誘導体収率を示す図である。
【図6】実施例1のリグノフェノール誘導体製造に使用したクレゾール量(モル濃度)を示す図である。
【図7】実施例1の各リグノフェノール誘導体を0.5NのNaOHで170℃、1時間処理し、GPC(ゲル浸透クロマト)分析した結果を示す図で、(A)〜(D)は各々、遠心分離0分後、3時間後、6時間後、15時間後の結果である。
【図8】実施例2の遠心分離後すぐに水処理をしたときのフェノール層から回収されたリグノフェノール誘導体のGPC(ゲル浸透クロマト)分析結果を示す図である。
【図9】実施例2の水処理後約15時間後にフェノール層から回収されたリグノフェノール誘導体のGPC(ゲル浸透クロマト)分析結果(遠心分離2日後)を示す図である。
【発明を実施するための形態】
【0016】
以下、本発明について幾つかの実施形態に基づいて詳細に説明する。
【0017】
[実施形態1]
本実施形態1は、リグノセルロース材料とフェノール化合物とを反応させてリグノフェノール誘導体を製造する方法において、リグノセルロース材料にフェノール化合物と酸とを添加して得られたリグノフェノール誘導体を含む反応液を相分離して水層を反応系から取り除き、フェノール溶液中でこのフェノール溶液に残存した酸と高分子量ポリマーのリグノフェノール誘導体とを反応させるリグノフェノール誘導体の分子量制御方法である。具体的には、以下の工程からなる、高分子量ポリマーのリグノフェノール誘導体の低分子化方法である。
(1)フェノール化合物で溶媒和したリグノセルロース材料に酸を加え、リグノフェノール誘導体を生成する工程(フェノールのグラフティング工程)。
(2)処理液のフェノール層と酸層とを相分離してフェノール溶液を得る工程(相分離工程)。
(3)フェノール溶液を室温に放置する工程(低分子化工程)。
【0018】
得られた低分子量リグノフェノール誘導体は、以下(イ)及び/又は(ロ)の工程により精製することができる。
(イ)フェノール溶液をリグノフェノール誘導体の貧溶媒、具体的にはジエチルエーテルやジイソプロピルエーテルに滴下して、リグノフェノール誘導体をフェノール化合物と分離して不溶区分として回収する工程(1次精製工程)。
(ロ)得られた不溶区分をアセトンに溶解してさらにリグノフェノール誘導体の貧溶媒に滴下することにより、不溶区分に含まれるリグノフェノール誘導体を分離して回収する工程(2次精製工程)。なお、本明細書中で「フェノール溶液」とは、酸と高分子リグノフェノール誘導体及び/又は低分子リグノフェノール誘導体とを含むフェノール化合物溶液をいう。
【0019】
フェノールのグラフティング工程では、リグノセルロース材料、フェノール化合物及び酸水溶液を攪拌する。この段階で、フェノール化合物で親和され膨潤化した木片等のリグニンのフェニルプロパンのベンジル位が開裂され、フェノール化合物と結合する反応が始まるが、この工程では、反応材料を攪拌して、硫酸とフェノール化合物を十分に混合することが重要である。攪拌により、酸がフェノール化合物の溶液に混じる。
【0020】
フェノール化合物は、親油性のフェノール化合物の溶液、例えば、p−クレゾール、m−クレゾール、o−クレゾール、アニソール、2,4−ジメトキシフェノール、2,6−ジメトキシフェノール、2,4−ジメチルフェノール、2,6−ジメチルフェノール、プロピルフェノール、i−プロピルフェノール、tert−ブチルフェノール、カテコール、レゾルシノール、ピロガロール、フロログルシノール、ビスフェノール、バニリン、シリンゴール、グアイアゴール、フェルラ酸、クマル酸の何れか、又は、これらの組み合わせからなる溶液を使用することができる。酸は、バイオマスの加水分解に用いられる酸であればよく、塩酸、硫酸等の無機酸であり、硫酸が望ましい。
【0021】
反応時間は、リグノセルロース材料、反応温度によって異なるが、長時間の反応は、リグニンのスルホニル化や加水分解された糖のフルフラールなどへの分解が進行するため好ましくない。この段階で生ずるリグノフェノール誘導体は、平均分子量を9,000〜14,000程度の高分子量ポリマーを含む。反応時間は、長くなるより短いほうが好ましく、一般的には最長でも1時間程度である。
【0022】
生成したリグノフェノール誘導体をフェノール化合物溶液に溶解する。次いで、反応溶液を相分離により、リグノフェノール誘導体を含む溶液の層(本明細書中では「フェノール溶液」という。)と酸液層とに分離する。相分離は自然比重分離又は遠心分離処理を行えばよい。フェノール化合物は、わずかではあるが親水性を有するので、フェノール溶液にはわずかに酸が残存することになる。
【0023】
相分離され反応系から酸層が取り除かれたフェノール溶液は、室温で放置又は緩やかな攪拌が行われ、フェノール溶液に残存する酸とリグノフェノール誘導体とを反応させる。この反応系は、わずかに残存する酸とフェノール化合物に溶解したリグノフェノール誘導体との反応であるため、フェニルプロパンが穏やかな条件下にあり、先の工程で未反応であったベンジル位の選択的な開裂反応が、プロパンの他の炭素に優先して進むものと考えられる。したがって、ベンジル位の開裂に伴ったフェノール化合物の結合とリグノフェノール誘導体の低分子化が優先的に進むことになる。
【0024】
反応時間は、目的とするリグノフェノール誘導体の平均分子量によって異なるので、適宜決定すればよい。相分離後2日で平均分子量は約3,000となり、これ以上の低分子化は惹起されない。これはリグニンが分子量約3,000のサブユニットにまで分解されて、この反応条件下ではこれ以上の反応が起こらないためと考えられる。
【0025】
低分子量リグノフェノール誘導体も高分子量リグノフェノールが有している基本特性である熱軟化特性を有している。このことは、ベンジル位以外の炭素での開裂やフェノール化合物との結合が起きていないためと考えられる。このことは、リグニンの主要単位間結合であるβ−アリールエーテル結合が保持され、リニア型ポリマー特性を有していることを意味する。
【0026】
フェノール溶液内での酸とリグノフェノール誘導体との反応を停止するためには、フェノール溶液をリグノフェノール誘導体の貧溶媒に滴下して、リグノフェノール誘導体をフェノール化合物と分離すればよい。
【0027】
リグノフェノール誘導体の貧溶媒は、非対称エーテル類、対称アルキルエーテル、環状炭化水素類、直鎖状炭化水素、エステル類を使用することができる。具体的には、ジイソプロピルエーテル、ジエチルエーテル、n−ヘキサン、ペンタン、ベンゼン、トルエン、酢酸エチルを挙げることができ、ジイソプロピルエーテルが好ましい。なお、上述した貧溶媒を混合して、極性を変化させて使用することは当分野において常套手段であり、極性の異なる溶媒を組み合わせたものを貧溶媒として使用してもよい。
【0028】
反応開始から反応停止までの時間を1時間〜2日の間で選択することで、リグノフェノール誘導体の平均分子量を、例えば、約14,000〜約3,500、好ましくは約10,000〜約3,500の範囲で制御することができる。平均分子量を約10,000とするのであれば、反応開始から3時間以内、平均分子量約5,000とする場合は15時間程度、5,000以下にする場合は、2日間程度の範囲にコントロールすることが好ましい。
【0029】
得られた貧溶媒の不溶区分は、さらに、アセトンに溶解して再度リグノフェノール誘導体の貧溶媒に滴下することにより、不溶区分に含まれるリグノフェノール誘導体を分離(2次精製)してもよい。
【0030】
[実施形態2]
本実施形態2は、フェノール溶液の酸濃度をさらに低下させることでフェノール溶液内での酸とリグノフェノール誘導体との反応を止める点で実施形態1と異なる。
【0031】
本実施形態では、酸をフェノール溶液から効率的に取り除くため、フェノール溶液を水と接触させる処理を行う。水との接触処理として、具体的には以下の操作を挙げることができる。
(1)フェノール溶液を水に滴下し、懸濁させて水分散液とする、又は、
(2)フェノール溶液を水で洗浄する。
【0032】
フェノール溶液を水分散液とするためには、フェノール溶液より多い水を必要とするが、水洗浄の場合には少ない水で繰り返し洗浄することで水の量を減らすことが可能である。また、フェノール溶液と接触させる水をアルカリ性水溶液とし、酸の中和を行ってもよい。
【0033】
フェノール層と酸層を分離した後、分離して得られたフェノール溶液を水と接触させる、またはアルカリ処理する時間により反応時間をコントロールすることができる。実施形態1と同様、反応開始から反応停止までの時間を1時間〜2日の間で選択することで、リグノフェノール誘導体の平均分子量を、例えば、約14,000〜約3,500の範囲で制御することができる。平均分子量を約10,000とするのであれば、反応開始から3時間以内、平均分子量約5,000とする場合は15時間程度、5,000以下にする場合は、2日間程度の範囲にコントロールすることが好ましい。
【0034】
フェノール溶液と水との接触処理の後、処理液から水を除去する操作を行ってリグノフェノール誘導体を水と分離する。水の除去操作は、例えば、処理液を液層分離して、酸を含む水層をフェノール溶液から取り除けばよい。
【0035】
水層と分離されたリグノフェノール誘導体を含む溶液をさらに、リグノフェノール誘導体の貧溶媒に添加して、リグノフェノール誘導体を沈殿物として得ることができる。
【0036】
得られたリグノフェノール誘導体は、精製度合いを高めるために、アセトンやメチルエチルケトンに溶解し、さらにジイソプロピルエーテル又はメチルエチルケトンと液体状の直鎖状炭化水素との混合溶媒に滴下して沈殿させてもよい。
【0037】
また、得られたリグノフェノール誘導体は、精製度を高めるために、アセトンやメチルエチルケトンに溶解し、さらにリグノフェノール誘導体の貧溶媒に滴下して再沈殿させてもよい。
【0038】
本発明者らにより、リグノフェノール誘導体の溶媒溶解性は、次の序列になることがわかっている。
【0039】
強アルカリ溶液>アセトン、メチルエチルケトンなどのアルキルケトン類>ジメチルフォルムアミド(DMF)、N-メチルピロリドン(NMP)のような非プロトン極性溶媒>テトラヒドロフラン(THF)、ジオキサンのような環状エーテル類>ピリジンのようなヘテロ環状化合物>エタノール、セロソルブのようなアルコール類>tert−ブチルメチルエーテル(TBME)、シクロペンチルメチルエーテル(CPME)などの非対称エーテル類>ジエチルエーテルやジプロピルエーテル、ジイソプロピルエーテルなどの対称アルキルエーテル>トルエン又はキシレンとヘキサンとの混合溶媒>ベンゼン、トルエン、ヘキサンなどの芳香族及び環状炭化水素類
リグノフェノール誘導体の貧溶媒は、非対称エーテル類、対称アルキルエーテル、環状炭化水素類、直鎖状炭化水素、エステル類を使用することができる。具体的には、ジイソプロピルエーテル、ジエチルエーテル、n−ヘキサン、ペンタン、ベンゼン、トルエン、酢酸エチルを挙げることができ、ジイソプロピルエーテルが好ましい。なお、上述した貧溶媒を混合して、極性を変化させて使用することは当分野において常套手段であり、極性の異なる溶媒を組み合わせたものを貧溶媒として使用してもよい。
【0040】
[実施形態3]
本実施形態3は、フェノール溶液を水と接触させた後の処理液から水を除去する操作が実施形態2と異なる。
【0041】
すなわち、水との接触処理の後、処理液に非水溶性極性溶媒を加える。非水溶性極性溶媒は、リグノフェノール誘導体を溶解でき、水分離しやすい溶媒である。このような溶媒として、メチルエチルケトン、クロロホルム、ジクロロメタン、酢酸エチル、エーテル類、トルエン、ベンゼンを挙げることができる。リグノフェノール誘導体の溶解性と疎水性とを考慮するとメチルエチルケトンが好ましい。
【0042】
この操作により、処理液は水層と非水溶性極性溶媒層とに分離する。非水溶性極性溶媒層には、リグノフェノール誘導体が含まれるが、酸はほとんど含まれない。したがって、リグノフェノール誘導体は酸の影響を受けることがない。
【0043】
得られた非水溶性極性溶媒層をさらに、リグノフェノール誘導体の貧溶媒に添加して、リグノフェノール誘導体を沈殿物として得ることができる。
【0044】
得られたリグノフェノール誘導体は、精製度を高めるために、アセトンやメチルエチルケトンに溶解し、さらにリグノフェノール誘導体の貧溶媒に滴下して再沈殿させてもよい。
【0045】
なお、メチルエチルケトン、貧溶媒、フェノール化合物の分離は蒸留にてほぼ分離可能である。
【実施例1】
【0046】
3mol倍のp−クレゾールを収着したヒノキ木粉1.5g(木粉として1g)に72%の硫酸を10mリットル添加し、30℃で1時間攪拌した。遠心分離の10分前にm,p−クレゾールを5mリットル加え、攪拌した。反応終了後、遠心分離し、クレゾール層と硫酸層に分離した。得られたクレゾール層を室温で0時間〜7日間放置し、クレゾール層をジイソプロピルエーテルに滴下し、リグノフェノール誘導体を精製して、平均分子量の変化を調べた。精製溶媒はジイソプロピルエーテルを使用した。
【0047】
得られたリグノフェノール誘導体のGPC(ゲル浸透クロマト)分析の結果を図1及び図2に示す。図1(A)〜(D)は各々、遠心分離0分後、3時間後、6時間後、15時間後、図2(A)〜(D)は各々、遠心分離2日後、3日後、4日後、7日後の結果である。時間が経つにつれ溶出時間が長くなり分子量が小さくなり、2日後〜7日後はほとんど変化していないのが分かる。
【0048】
図3に平均分子量を示す。すぐに精製したリグノフェノール誘導体の重量平均分子量は約14,000であったが、3時間室温放置で約8,500まで低下した。15時間経過すると分子量は約4,200まで低下した。7日目まで経時変化を調べたが、2日目以降は、分子量約3,500で更なる低分子化は見られなかった。
また、熱機械分析(TMA)の結果を図4に示す。TMA分析は、SEIKO EXSTAR 6000 TMA/SSにより行った。試料約2mgを5mmΦ,深さ2mmのアルミニウムパンに入れ、ハンドプレスで表面が平滑になるように圧縮し、その上にアルミニウム板を載せ、その上からTMA測定機の石英測定針でプレスした。測定は鉛直方向に4.9 Nの荷重をかけながら温度を変化させ、測定針の変化を測定するペネトレート法で分析した。なお、温度上昇: 2.0度/min、窒素流量:150cm3/min、測定温度:50−250℃の条件で行った。(A)〜(D)は各々、遠心分離0分後、3時間後、6時間後、15時間後の結果を示す。遠心分離3時間後、6時間後、15時間後の低分子量のリグノフェノール誘導体であっても、熱軟化特性を有していることがわかる。
【0049】
図5に、精製リグノフェノール誘導体の収率を示す。低分子化後2日以降のリグノフェノール誘導体の収率は多少の変化があるものの、25%前後で安定している。
【0050】
1H−NMR分析を行い、得られたリグノフェノールに含まれる結合フェノール量を測定した。遠心0分後、3時間後、6時間後及び15時間後のリグノフェノール誘導体の導入フェノール量を図6に示す。縦軸の「導入フェノール量mol/C9」はリグニン1ユニットであるフェニルプロパン単位あたり何モルフェノールが導入されたかを示す指標である。遠心後の時間経過が長いほど導入フェノール量mol/C9が少し増えており、ベンジルエーテル結合が開裂され、そこにクレゾールが導入されたことを示している。
【0051】
精製した各リグノフェノール誘導体を0.5NのNaOHで170℃、1時間処理し、前述と同じ条件でGPC分析を行った。結果を図7に示す。(A)〜(D)は各々、遠心分離0分後、3時間後、6時間後、15時間後の結果を示す。低分子化したリグノフェノール誘導体のアルカリ処理結果も高分子リグノフェノール誘導体のアルカリ処理の結果とほぼ同様のアルカリ分解挙動が見られた。これらの結果から、低分子化リグノフェノールにおいてもリグニン主要単位間結合であるβ−アリールエーテル結合が保持されていることが示された。
【実施例2】
【0052】
3mol倍のp−クレゾールを収着したヒノキ木粉15g(木粉として10g)に72%硫酸100mlを加え、30℃で60分攪拌し、終了10分前にm,p−クレゾール80mリットルを加えた。これを遠心分離し、フェノール層を回収した。フェノール層に固形分も少なく、分離性は良好であった。界面洗浄として10mリットルのm,p−クレゾールを加え、界面を洗浄した。クレゾール層100mリットルが回収された。このクレゾール層を600mリットルの水に滴下した。滴下後は淡桃白色の分散液となった。これに200mリットルのメチルエチルケトンを加え、分液ロートに入れ、一晩放置した。翌日、メチルエチルケトン層の濁りは少なくなっていた。水層を抜き、メチルエチルケトン層160mリットルを回収した。
【0053】
上記操作において、水処理−メチルエチルケトン抽出処理後のメチルエチルケトン層中でのリグノフェノール誘導体の安定性を確認するため、メチルエチルケトン層の放置時間を変えた実験を行った。
遠心分離後のフェノール層にすぐに水を加えてメチルエチルケトンで抽出した後、メチルエチルケトン層10mリットルをすぐに120mリットルのジイソプロピルエーテルに滴下し、1次精製リグノフェノール誘導体を得た。1次精製リグノフェノール誘導体をアセトンに溶解後、ジイソプロピルエーテルに滴下し2次精製を行い、0.125gのリグノフェノール誘導体を得た。GPC分析の結果を図8に示す。GPC分析条件は実施例1と同じである。
遠心分離後のフェノール層にすぐに水を加え、メチルエチルケトンで抽出し15時間放置した後、メチルエチルケトン層10mリットルを120mリットルのジイソプロピルエーテルに滴下し、1次精製リグノフェノール誘導体を得た。1次精製リグノフェノール誘導体をアセトンに溶解後、ジイソプロピルエーテルに滴下し2次精製を行い、0.136gのリグノフェノール誘導体を得た。GPC分析の結果を図9に示す。GPC分析条件は実施例1と同じである。
GPCを見るとクレゾール分離1日後のリグノフェノール誘導体は、若干、重量平均分子量にばらつきがあり、高分子区分が少なくなっているが、ほとんど差は見られなかった。この結果から、フェノール層と水層を分離してから目的の分子量となる時間が経過したところで水処理するとその分子量で止めることができることがわかる。
【特許請求の範囲】
【請求項1】
リグノセルロース材料とフェノール化合物とを反応させてリグノフェノール誘導体を製造する方法において、リグノセルロース材料にフェノール化合物と酸とを添加して得られた反応液から水層を相分離して反応系から取り除き、フェノール溶液中で該フェノール溶液に残存した酸とリグノフェノール誘導体とを反応させることを特徴とする、リグノフェノール誘導体の分子量制御方法。
【請求項2】
前記フェノール溶液中での反応は、前記リグノフェノール誘導体ベンジルエーテル基の開裂と前記フェノール化合物の結合である、請求項1記載のリグノフェノール誘導体の分子量制御方法。
【請求項3】
前記フェノール溶液中での反応は室温下で行われる、請求項1記載のリグノフェノール誘導体の分子量制御方法。
【請求項4】
前記リグノフェノールが、1,1−ビス(アリール)プロパンを構成ユニットとするポリマーである、請求項3記載のリグノフェノール誘導体の分子量制御方法。
【請求項5】
前記リグノフェノール誘導体の平均分子量が約14,000〜3,500である、請求項4記載のリグノフェノール誘導体の分子量制御方法。
【請求項6】
前記相分離が比重分離である、請求項5に記載のリグノフェノール誘導体の分子量制御方法。
【請求項7】
リグノセルロース材料とフェノール化合物とを反応させてリグノフェノール誘導体を製造する方法において、リグノセルロース材料にフェノール化合物と酸とを添加して得られた反応液の水層の酸濃度を低下させる工程と、フェノール溶液中で該フェノール溶液に残存した酸とリグノフェノール誘導体とを反応させる工程とを含む、リグノフェノール誘導体の分子量制御方法。
【請求項8】
前記フェノール溶液中での反応は、前記リグノフェノール誘導体ベンジルエーテル基の開裂と前記フェノール化合物の結合である、請求項7記載のリグノフェノール誘導体の分子量制御方法。
【請求項9】
前記リグノフェノールが、1,1−ビス(アリール)プロパンを構成ユニットとするポリマーである、請求項8記載のリグノフェノール誘導体の分子量制御方法。
【請求項1】
リグノセルロース材料とフェノール化合物とを反応させてリグノフェノール誘導体を製造する方法において、リグノセルロース材料にフェノール化合物と酸とを添加して得られた反応液から水層を相分離して反応系から取り除き、フェノール溶液中で該フェノール溶液に残存した酸とリグノフェノール誘導体とを反応させることを特徴とする、リグノフェノール誘導体の分子量制御方法。
【請求項2】
前記フェノール溶液中での反応は、前記リグノフェノール誘導体ベンジルエーテル基の開裂と前記フェノール化合物の結合である、請求項1記載のリグノフェノール誘導体の分子量制御方法。
【請求項3】
前記フェノール溶液中での反応は室温下で行われる、請求項1記載のリグノフェノール誘導体の分子量制御方法。
【請求項4】
前記リグノフェノールが、1,1−ビス(アリール)プロパンを構成ユニットとするポリマーである、請求項3記載のリグノフェノール誘導体の分子量制御方法。
【請求項5】
前記リグノフェノール誘導体の平均分子量が約14,000〜3,500である、請求項4記載のリグノフェノール誘導体の分子量制御方法。
【請求項6】
前記相分離が比重分離である、請求項5に記載のリグノフェノール誘導体の分子量制御方法。
【請求項7】
リグノセルロース材料とフェノール化合物とを反応させてリグノフェノール誘導体を製造する方法において、リグノセルロース材料にフェノール化合物と酸とを添加して得られた反応液の水層の酸濃度を低下させる工程と、フェノール溶液中で該フェノール溶液に残存した酸とリグノフェノール誘導体とを反応させる工程とを含む、リグノフェノール誘導体の分子量制御方法。
【請求項8】
前記フェノール溶液中での反応は、前記リグノフェノール誘導体ベンジルエーテル基の開裂と前記フェノール化合物の結合である、請求項7記載のリグノフェノール誘導体の分子量制御方法。
【請求項9】
前記リグノフェノールが、1,1−ビス(アリール)プロパンを構成ユニットとするポリマーである、請求項8記載のリグノフェノール誘導体の分子量制御方法。
【図1】


【図2】
【図3】
【図4】
【図5】
【図6】
【図7】

【図8】
【図9】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公開番号】特開2011−42640(P2011−42640A)
【公開日】平成23年3月3日(2011.3.3)
【国際特許分類】
【出願番号】特願2009−193438(P2009−193438)
【出願日】平成21年8月24日(2009.8.24)
【出願人】(503360115)独立行政法人科学技術振興機構 (1,734)
【出願人】(501269122)株式会社ケー・ワイ・ビー (2)
【Fターム(参考)】
【公開日】平成23年3月3日(2011.3.3)
【国際特許分類】
【出願日】平成21年8月24日(2009.8.24)
【出願人】(503360115)独立行政法人科学技術振興機構 (1,734)
【出願人】(501269122)株式会社ケー・ワイ・ビー (2)
【Fターム(参考)】
[ Back to top ]