
リコンビナーゼポリメラーゼ増幅
【課題】リコンビナーゼ−ポリメラーゼ増幅(RPA)と称するDNAの増幅方法を提供すること。
【解決手段】本開示において、リコンビナーゼおよび関連するタンパク質の特性を利用し、DNAポリメラーゼ反応の配列特異的なプライミングを可能にする単鎖相同性DNAを有する二本鎖DNA侵入する、標的DNAのRPAに関連する新規な方法を記載する。開示する方法は、熱サイクリングまたは好熱性酵素を必要とせず、したがって、他の増幅方法よりも容易で手ごろな実施および携帯性を提供するという利点を有する。さらに、RPA反応のリアルタイムモニタリングを可能にする条件、光を用いてRPA反応を調節する方法、あるいは、増幅種の性質をゲル電気泳動の必要なく測定する方法、RPA反応におけるシグナル対ノイズ比を改善および最適化する方法、ならびにオリゴヌクレオチドプライマー機能を最適化する方法等を開示する。
【解決手段】本開示において、リコンビナーゼおよび関連するタンパク質の特性を利用し、DNAポリメラーゼ反応の配列特異的なプライミングを可能にする単鎖相同性DNAを有する二本鎖DNA侵入する、標的DNAのRPAに関連する新規な方法を記載する。開示する方法は、熱サイクリングまたは好熱性酵素を必要とせず、したがって、他の増幅方法よりも容易で手ごろな実施および携帯性を提供するという利点を有する。さらに、RPA反応のリアルタイムモニタリングを可能にする条件、光を用いてRPA反応を調節する方法、あるいは、増幅種の性質をゲル電気泳動の必要なく測定する方法、RPA反応におけるシグナル対ノイズ比を改善および最適化する方法、ならびにオリゴヌクレオチドプライマー機能を最適化する方法等を開示する。
Notice: Undefined index: DEJ in /mnt/www/gzt_disp.php on line 298
【特許請求の範囲】
【請求項1】
明細書中に記載の発明。
【請求項1】
明細書中に記載の発明。
【図1】



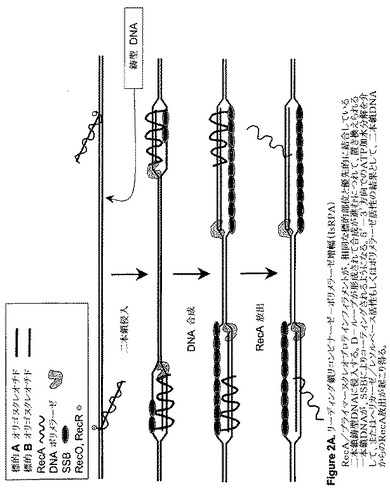
【図2A】
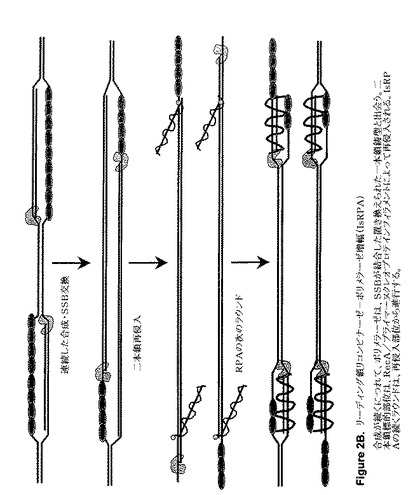
【図2B】
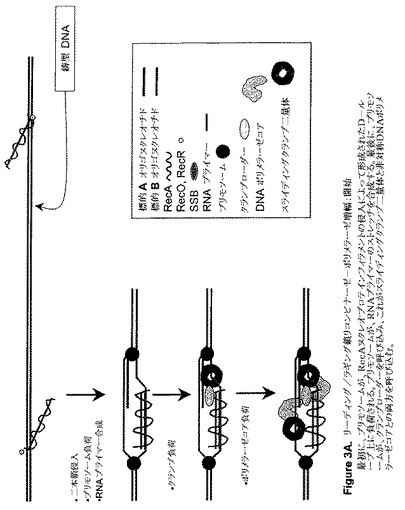
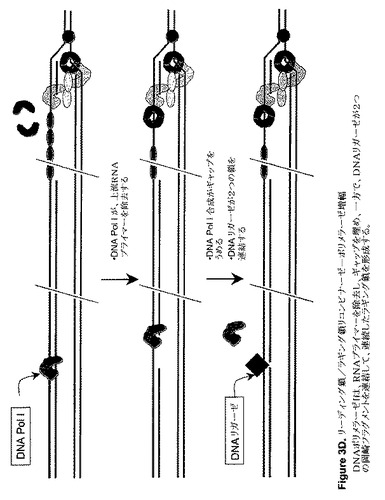
【図3A】
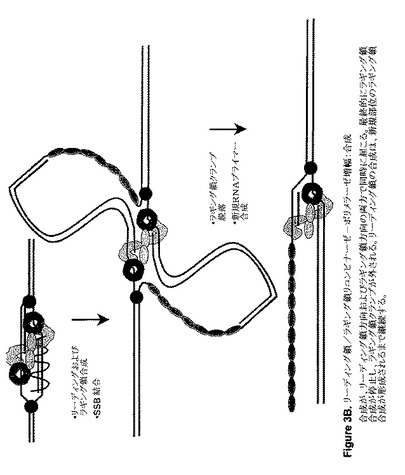
【図3B】
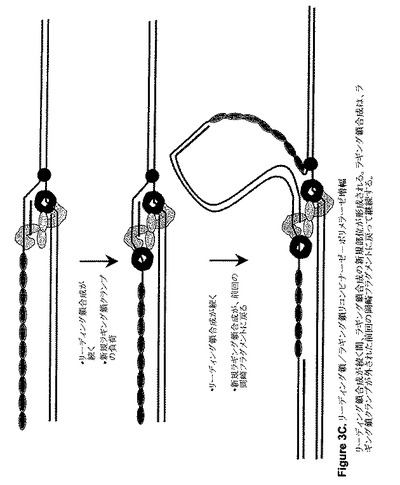
【図3C】
【図3D】
【図4】

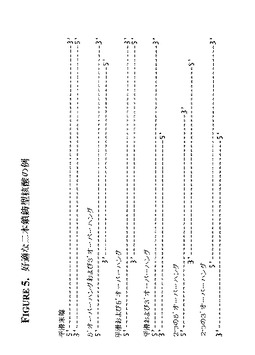
【図5】
【図6】

【図7】

【図8】
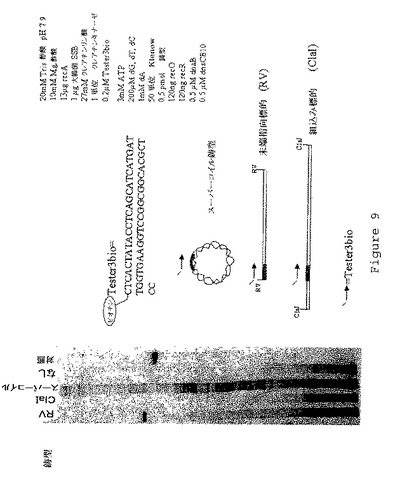
【図9】
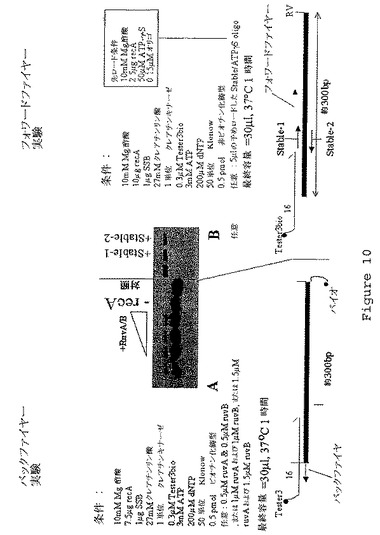
【図10】
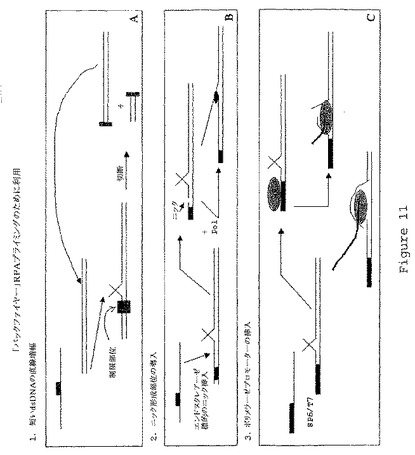
【図11】
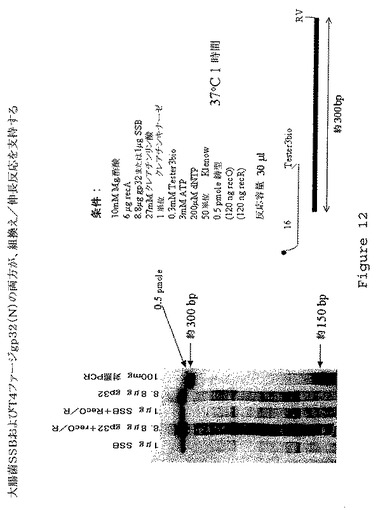
【図12】
【図13】
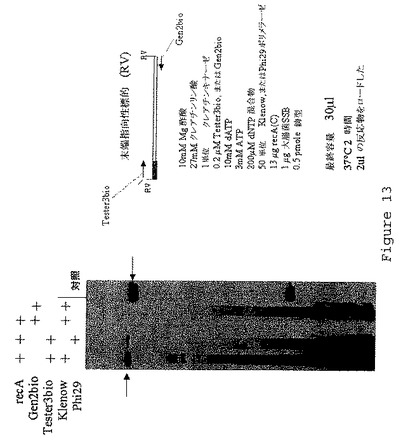
【図14】
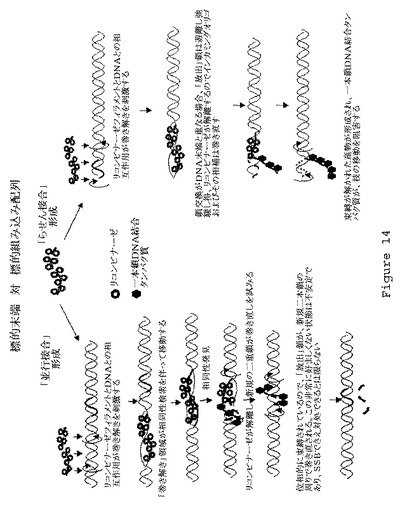
【図15】
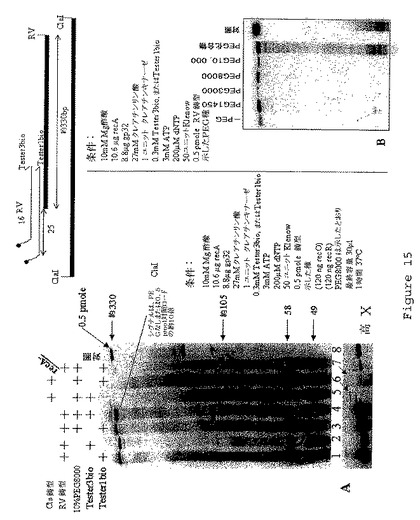
【図16】
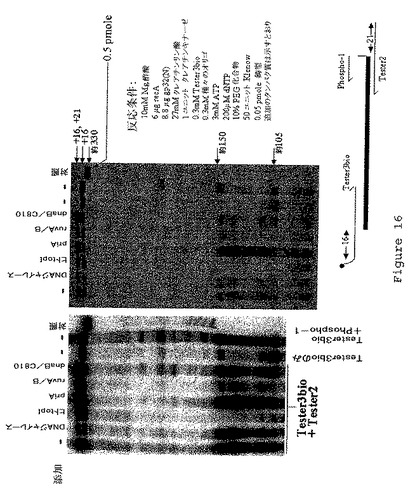
【図17】
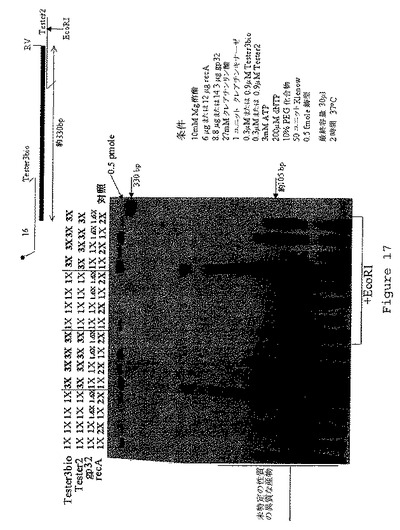
【図18】
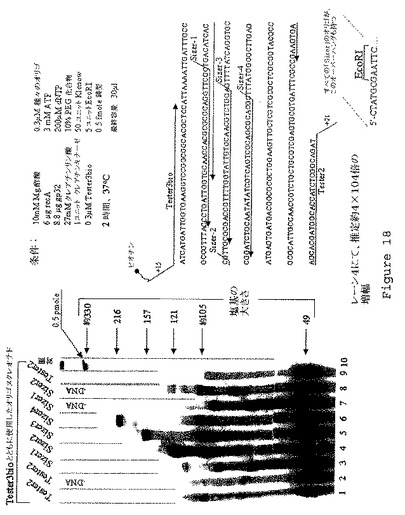
【図19】
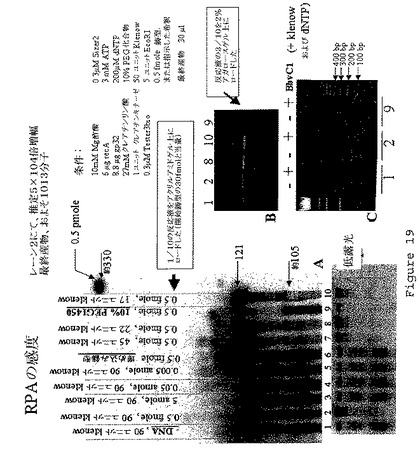
【図20】
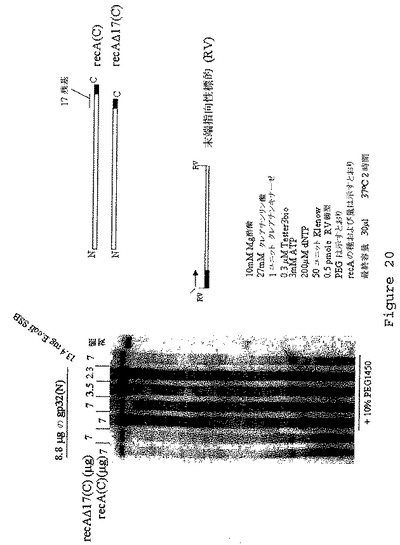
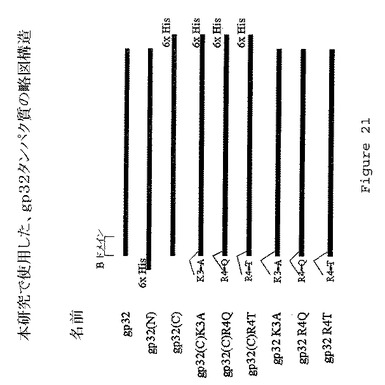
【図21】
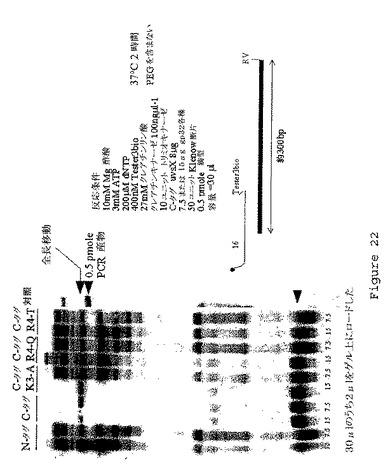
【図22】
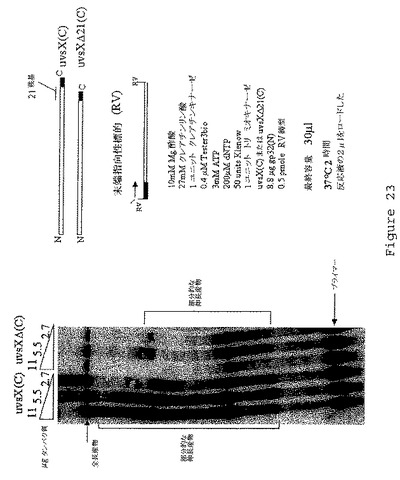
【図23】
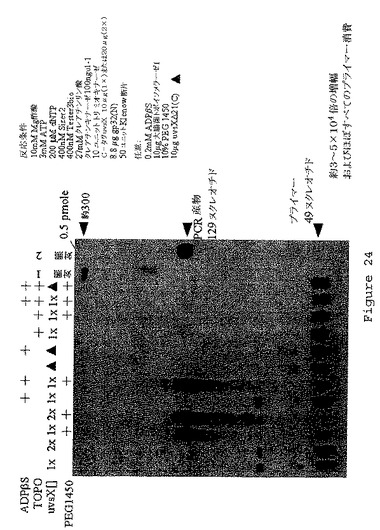
【図24】
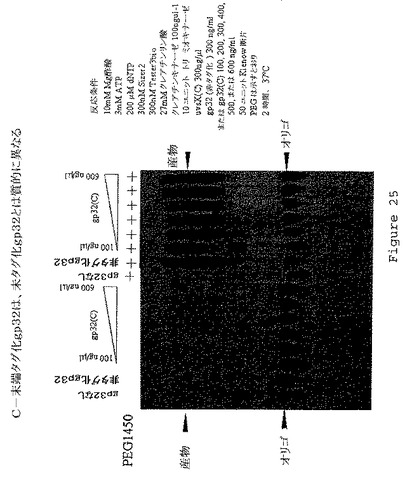
【図25】
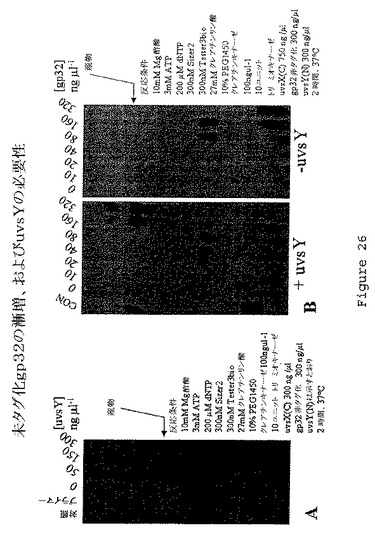
【図26】
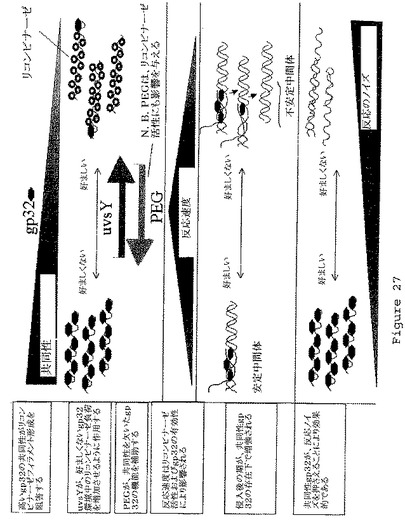
【図27】
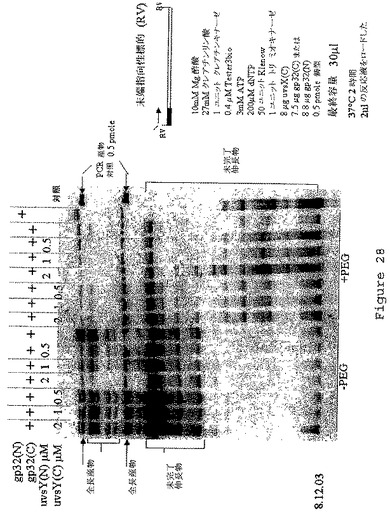
【図28】
【図29】
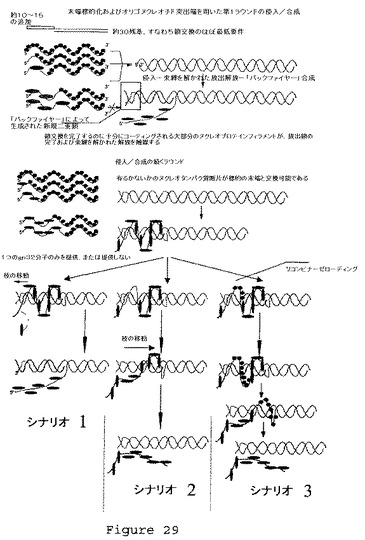
【図30】
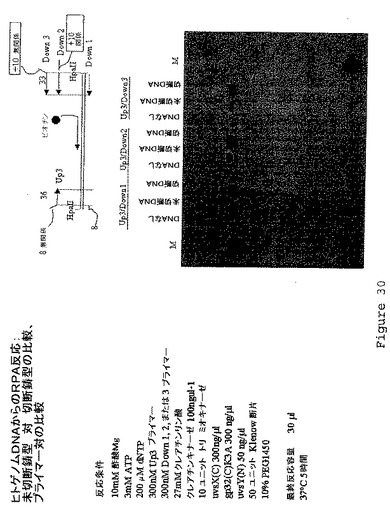
【図31】
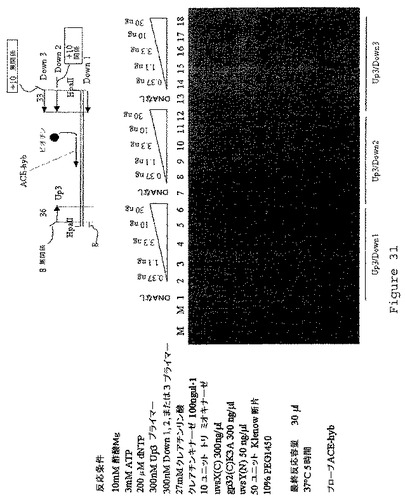
【図32】
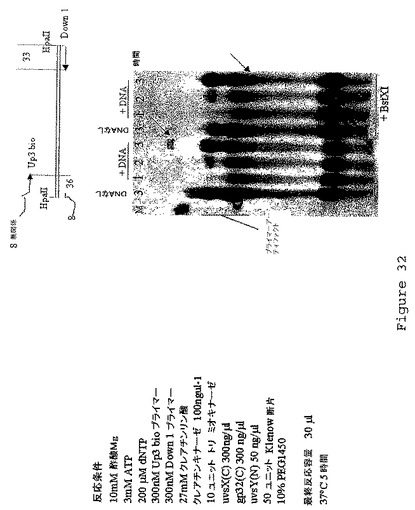
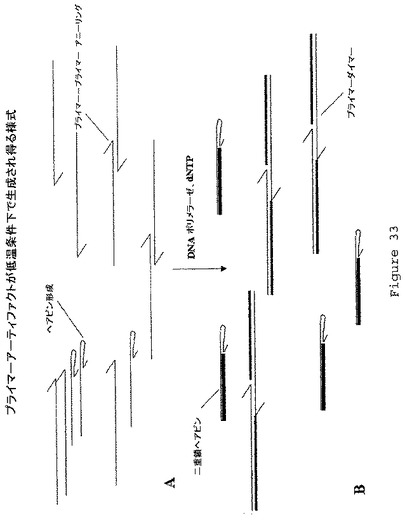
【図33】
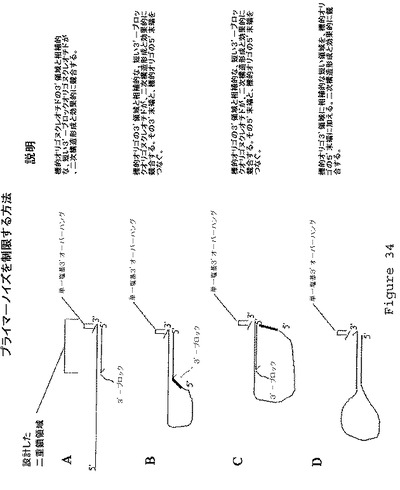
【図34】
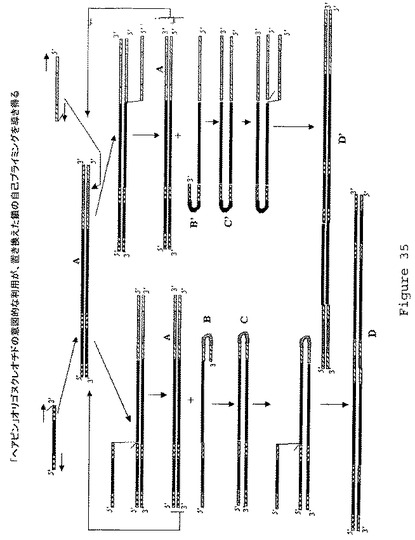
【図35】
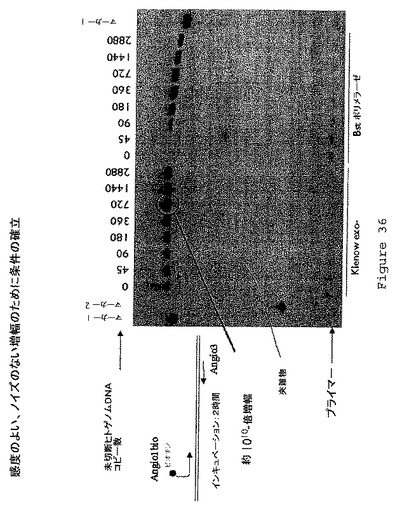
【図36】
【図37】
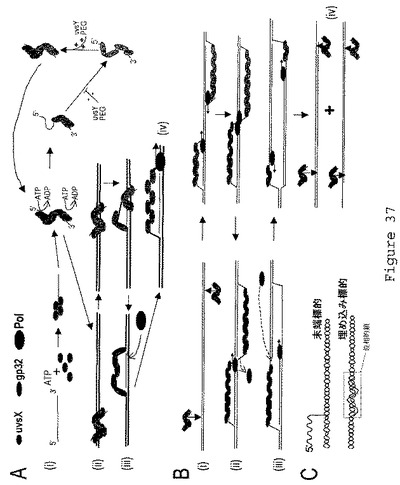
【図38】
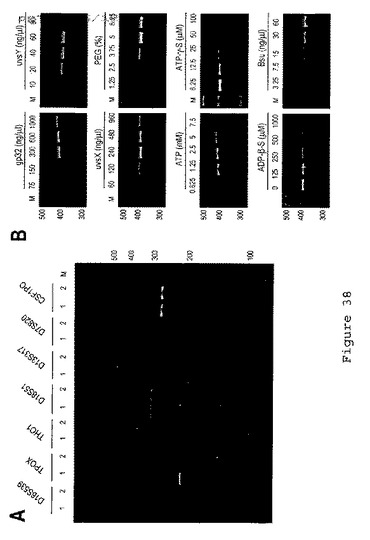
【図39】
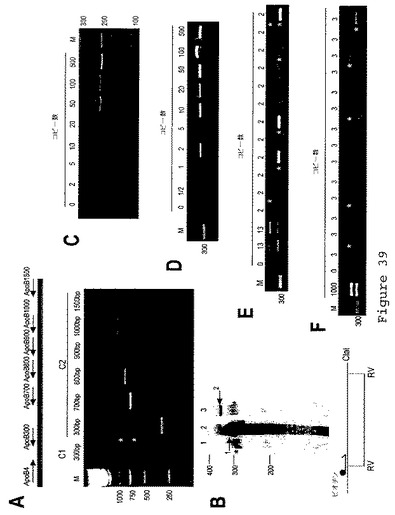
【図40】
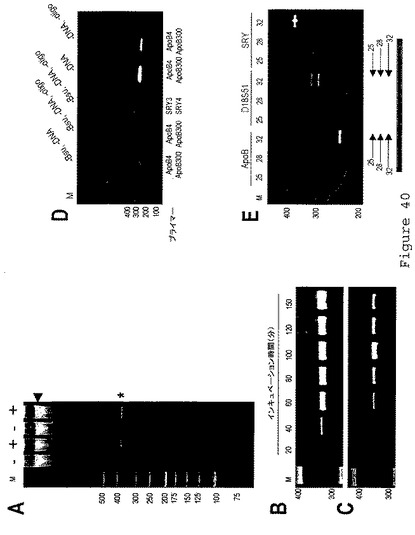
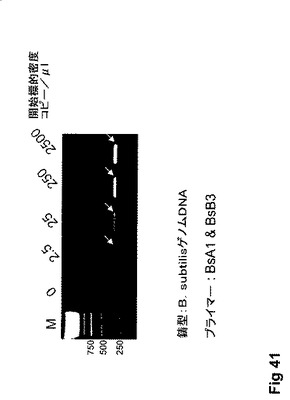
【図41】
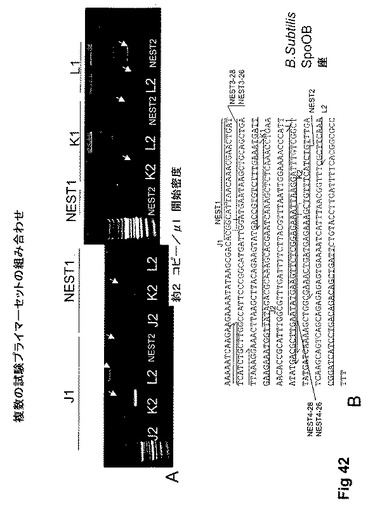
【図42】
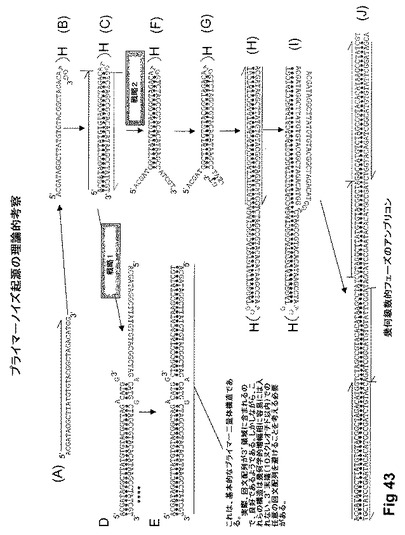
【図43】
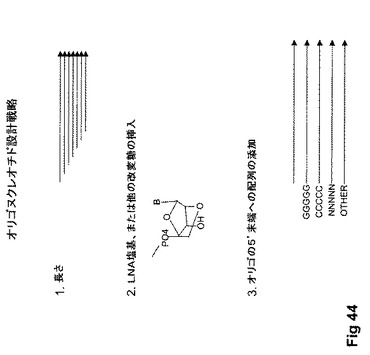
【図44】
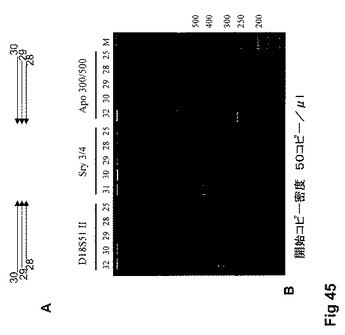
【図45】
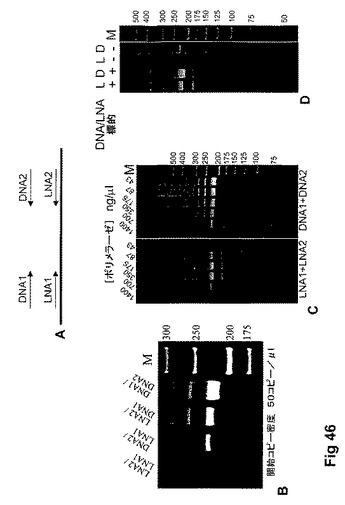
【図46】
【図47】

【図48】

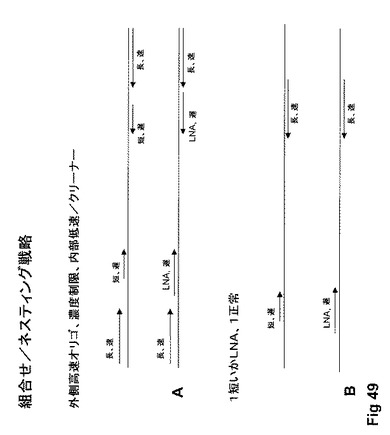
【図49】
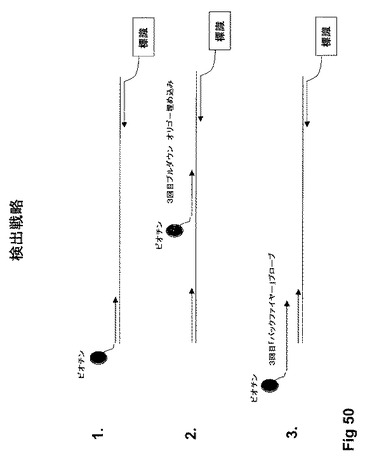
【図50】
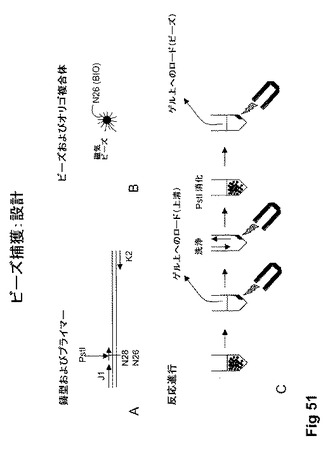
【図51】
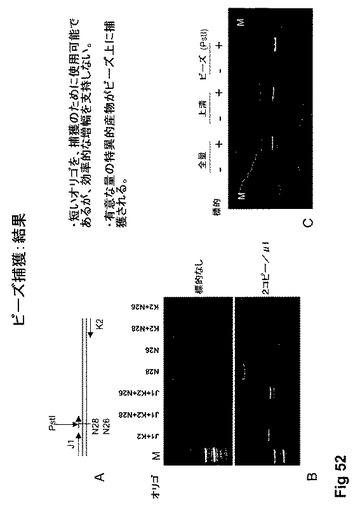
【図52】
【図53】

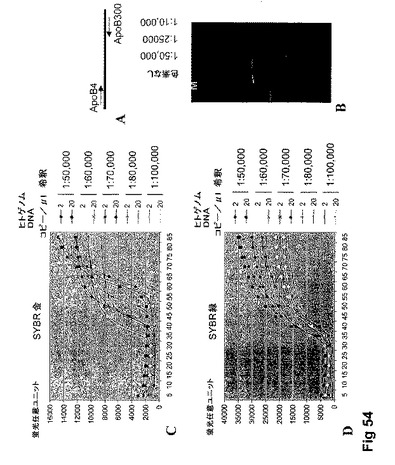
【図54】
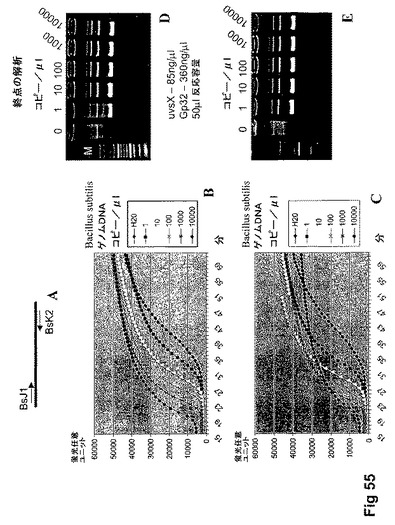
【図55】
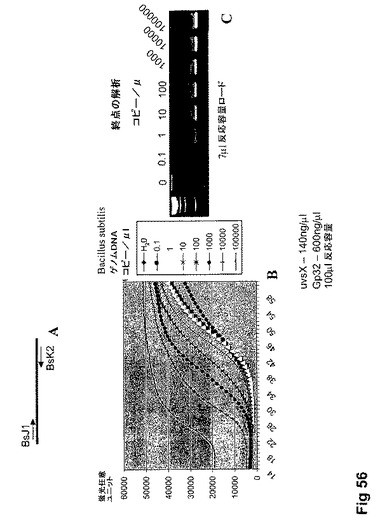
【図56】
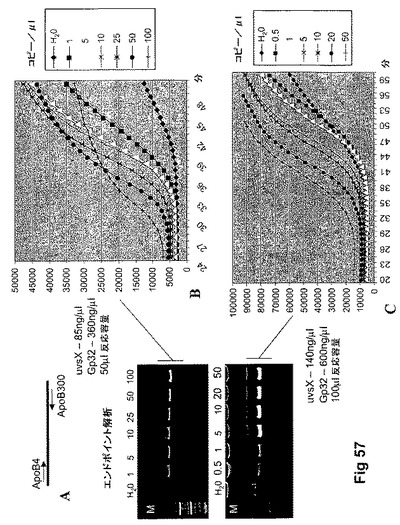
【図57】
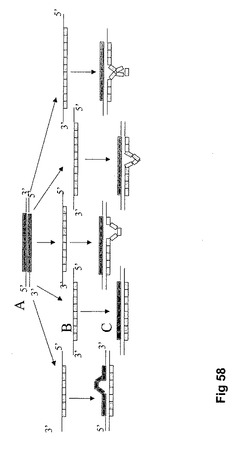
【図58】
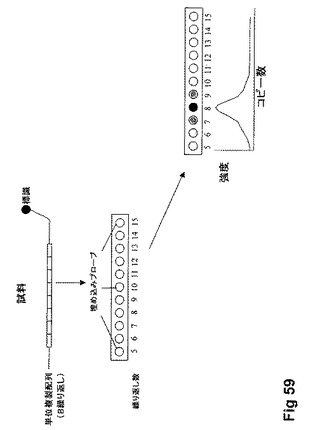
【図59】
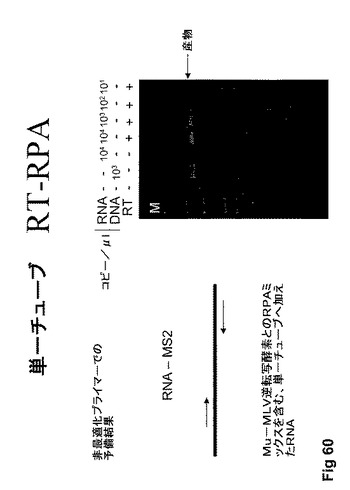
【図60】
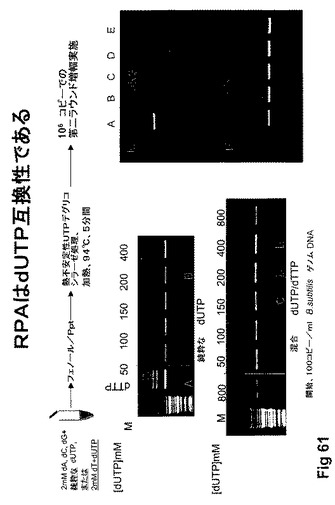
【図61】
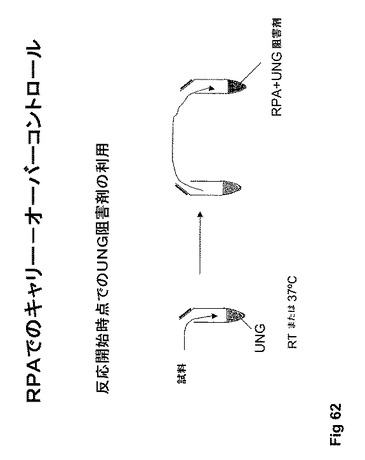
【図62】
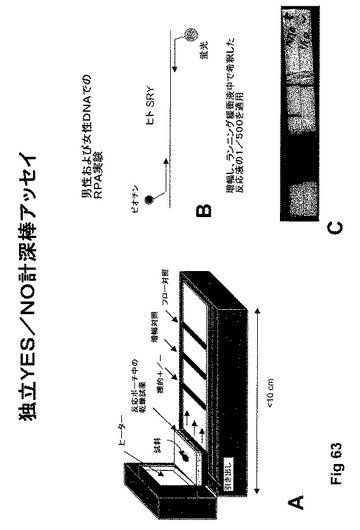
【図63】
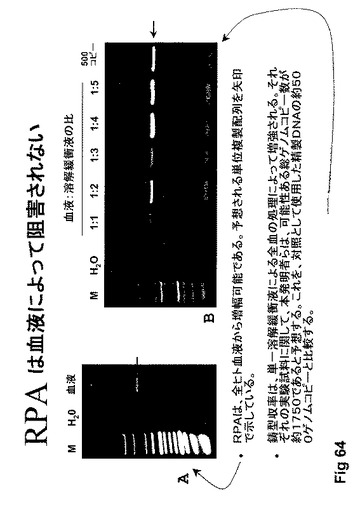
【図64】
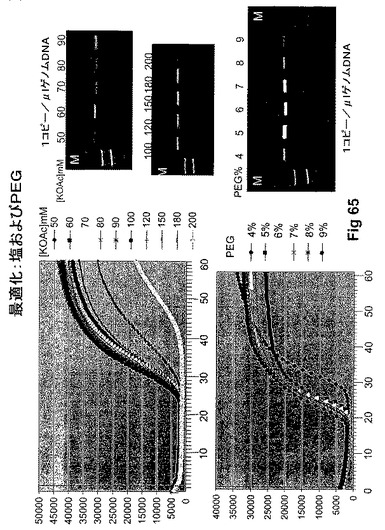
【図65】
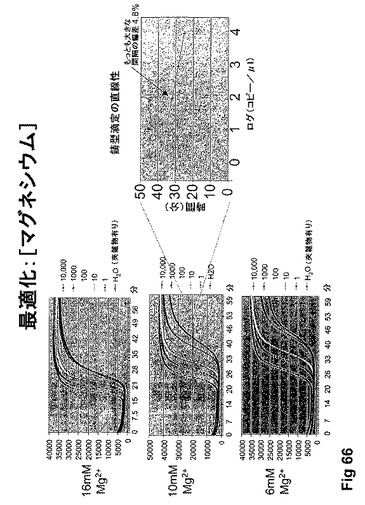
【図66】
【図67】

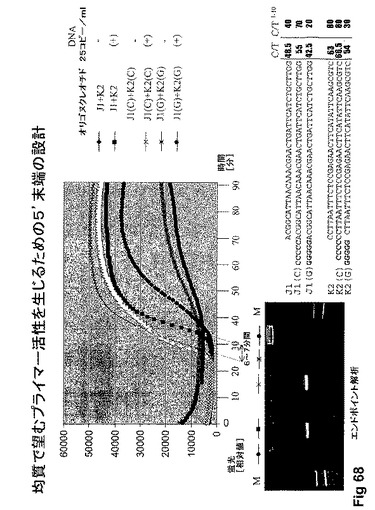
【図68】
【図69】
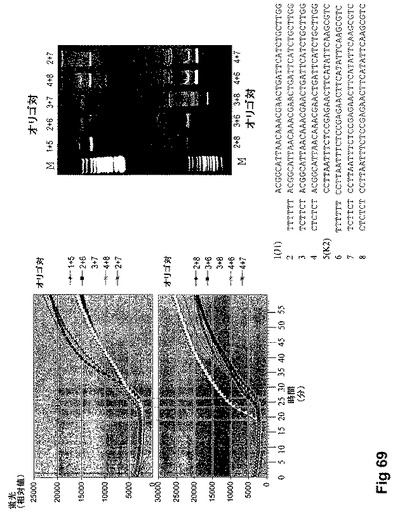
【図70】
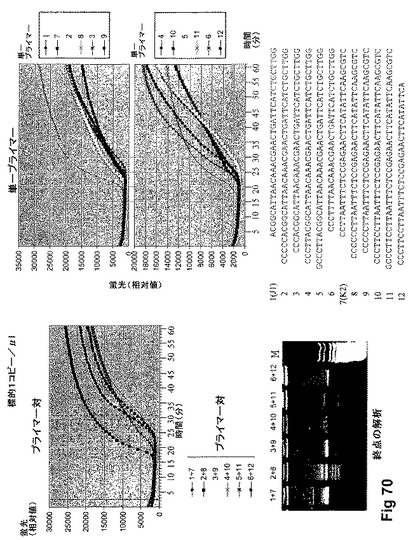
【図71】
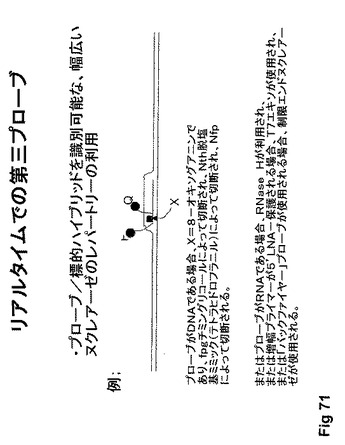
【図72】
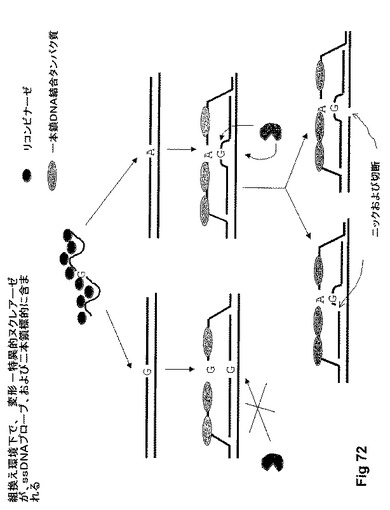
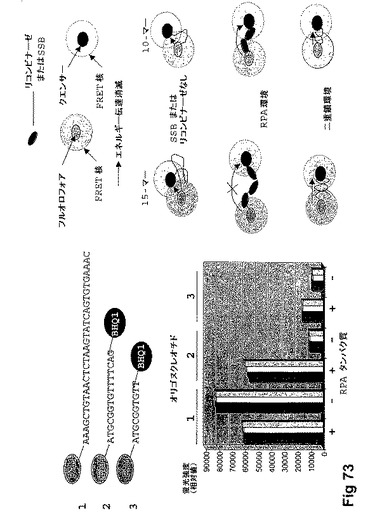
【図73】
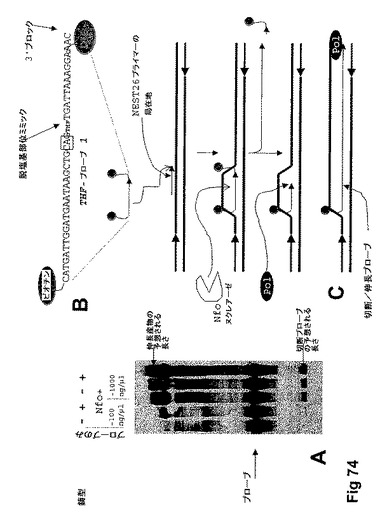
【図74】
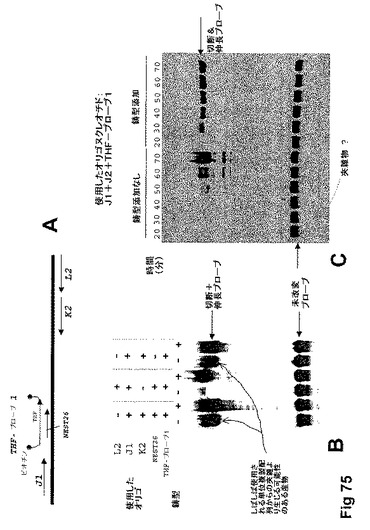
【図75】
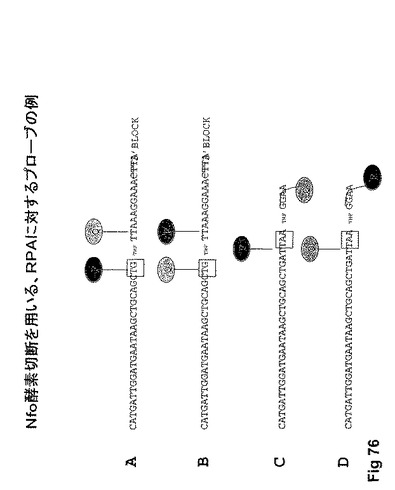
【図76】
【図2A】
【図2B】
【図3A】
【図3B】
【図3C】
【図3D】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【図31】
【図32】
【図33】
【図34】
【図35】
【図36】
【図37】
【図38】
【図39】
【図40】
【図41】
【図42】
【図43】
【図44】
【図45】
【図46】
【図47】
【図48】
【図49】
【図50】
【図51】
【図52】
【図53】
【図54】
【図55】
【図56】
【図57】
【図58】
【図59】
【図60】
【図61】
【図62】
【図63】
【図64】
【図65】
【図66】
【図67】
【図68】
【図69】
【図70】
【図71】
【図72】
【図73】
【図74】
【図75】
【図76】
【公開番号】特開2013−102769(P2013−102769A)
【公開日】平成25年5月30日(2013.5.30)
【国際特許分類】
【外国語出願】
【出願番号】特願2013−29664(P2013−29664)
【出願日】平成25年2月19日(2013.2.19)
【分割の表示】特願2011−44524(P2011−44524)の分割
【原出願日】平成17年4月11日(2005.4.11)
【出願人】(512051941)アリーア サン ディエゴ, インコーポレイテッド (7)
【Fターム(参考)】
【公開日】平成25年5月30日(2013.5.30)
【国際特許分類】
【出願番号】特願2013−29664(P2013−29664)
【出願日】平成25年2月19日(2013.2.19)
【分割の表示】特願2011−44524(P2011−44524)の分割
【原出願日】平成17年4月11日(2005.4.11)
【出願人】(512051941)アリーア サン ディエゴ, インコーポレイテッド (7)
【Fターム(参考)】
[ Back to top ]