
リチウム−多孔性金属酸化物組成物、及びリチウム試薬−多孔性金属組成物
本発明はリチウム金属/多孔性金属酸化物組成物に関する。これらのリチウム金属組成物は、液体リチウム金属を多孔性金属酸化物孔に吸収させるのに十分な発熱条件下で、不活性雰囲気において、液体リチウム金属と多孔性金属酸化物とを混合することにより調製される。本発明のリチウム金属/多孔性金属酸化物組成物は、最高約40重量%で、リチウム金属を担持しているのが好ましく、約20重量%〜40重量%の担持が最も好ましい。本発明はまた、多孔性酸化物に吸収されたRLiを含有するリチウム試薬−多孔性金属酸化物組成物に関する。RLiの式中、Rはアルキル基、アルケニル基、アルキニル基、アリール基、アルカリール基又はNR1R2基であり、R1はアルキル基、アルケニル基、アルキニル基、アリール基、アルカリール基であり、R2は水素、アルキル基、アルケニル基、アルキニル基、アリール基及びアルカリール基である。本発明はまた、これらの組成物の調製方法及び使用にも関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は多孔性金属酸化物粉(例えばアルミナゲル)と金属リチウムとの相互作用によって調製されるリチウム金属−多孔性酸化物組成物、並びに、有機リチウム及びリチウムアミド試薬の調製へのそれらの、in situでの使用、及び固体状で自由に流動する、取り扱いや貯蔵が容易な材料としての使用に関する。これらの組成物は貯蔵、出荷及び使用の際、可燃性溶媒及び/又は冷却条件を必要とせず、にもかかわらず、吸収されたリチウム及びリチウム試薬は、それらの反応性及び合成における有用性を保持している。
【背景技術】
【0002】
有機リチウム及びリチウムアミド化合物は、合成化学における変換の際に一般的に使用される重要な試薬である。有機リチウム試薬は伝統的に、微細に分散されたリチウム金属と有機ハロゲン化合物の溶液との低温での混合、又は金属−ハロゲン交換反応により調製される。リチウムアミドは最も一般的には、有機リチウム試薬を使用したアミンの脱プロトン化により調製される。しかしながら、有機リチウム化合物の安定性、貯蔵及び取扱いには課題が残されたままであり、そのことがしばしば有機合成へのそれらの使用(リチウムアミドの調製へのそれらの使用を含む)を困難にしている。
【0003】
本明細書で用いられ、また当技術分野で一般的に用いられる有機リチウムとは、炭素中心を有するアニオンのリチウム化合物のことを指す。有機リチウム試薬は、強い塩基であり、有効な求核種であり、またラジカル重合及びアニオン重合における有効な触媒であるため、それらは合成反応において有用である。しかしながらかかる試薬は非常に反応性が高く、しばしば空気の存在下で自然発火する。これらの危険をコントロールするため、それらは、炭化水素又はエーテル溶媒中の溶液としてのみ市販されている。これらの溶媒は有機リチウムの自然発火性を緩和できるが、それ自身揮発性かつ可燃性であるため、有機リチウム試薬の使用に関連する危険性を助長しうる。
【0004】
本発明で使用するリチウムアミドとは、第一級および第二級アミンのリチウム塩を意味する。リチウムアミド試薬は強塩基であり、一般の有機溶媒に自由に溶解し、非常に用途が広いため、合成的に有用である。しかしながらこれらの試薬は、非常に反応性が高く、取り扱いが困難である。若干の例外はあるが、それらは市販されておらず、それらの使用の直前に、第一級又は第二級アミンを有機リチウム試薬(例えばブチルリチウム)に添加することによって合成しなければならない。
【0005】
リチウム金属は有機リチウム試薬の生成に一般に使用されるものであり、その試薬は炭素原子とリチウム原子との間の直接的な結合を有する有機金属化合物である。リチウムの陽電気を帯びた性質により、結合中の大部分の電荷密度が炭素原子に配置されるため、カルボアニオン種が生じる。これにより、有機リチウム試薬が極めて強力な塩基及び求核試薬として作用しうる。有機リチウム試薬は典型的には、
R−X+2Li→R−Li+LiX
で表される、リチウム金属と有機ハロゲン化物との反応によって商業的に合成される(Weissその他、特許文献1:米国特許第5523447号、及びEmmelその他、特許文献2:米国特許出願公開第2006/0049379号を参照)。この合成の間に生じる副反応としては、特にヨウ化アルキルによるウルツ反応が挙げられる(Rがそれ自体でカップリングする)。この副反応は、低温、又はハロゲンとしての塩素若しくは臭素の使用によりほとんど排除できる。例えば、他の有機リチウム試薬の調製方法としては以下のものが挙げられる:
(i)有機ハロゲン化物とラジカルアニオンリチウム塩との反応、
(ii)有機ハロゲン化合物と有機リチウム種との間での金属−ハロゲン交換の実施(例えば、非特許文献1:Gilman,H.ら,J.Am.Chem.Soc.1932;54,1957)、
(iii)有機リチウム種と他の有機金属化合物との間での交換、
(iv)有機リチウム試薬による有機化合物の脱プロトン化、
(v)炭素−ヘテロ原子(例えば硫黄、酸素、リン又はケイ素)結合の還元的切断(例えば非特許文献2:Gilman,H.,ら,Org.Chem.1958;23,2044)、又は
(vi)DMSO中でのLiOH及びトルエンからのリチウム−水素交換によるベンジルリチウムの調製(特許文献3:Everettら、米国特許出願公開第2006/0170118号)。
【0006】
有機リチウム試薬、特にブチルリチウム(BuLi)、メチルリチウム(MeLi)、フェニルリチウム(PhLi)などは、製薬産業及び製造業における化学的成分及び強塩基として広く用いられている。リチウムアミドには関連用途も発見されている。例えばリチウムジイソプロピルアミド(LDA)及びリチウムヘキサメチルジシラジド(LiHMDS)は、両方とも、リチウムの強い配位能力によるエナンチオ選択的アルキル化を実施することもできる強塩基である(非特許文献3:Hilpert,H.Tetrahedron,2001,57,7675)。炭素中心を有する有機リチウム(例えばnBuLi)は、同時に強力な求核種及び強塩基であると考えられる(非特許文献4:Askin,D.;Wallace,M.A.;Vacca,J.P.;Reamer,R,A.;Volante,R.P.;Shinkai,I.J.Org.Chem.1992,57,2771)。これらの特徴により、アニオン重合の開始剤としての使用が可能となる(特許文献4:Hungenberg,Klaus−Dieter;Loth,Wolfgang;Knoll,Konrad;Janko,Lutz;Bandermann,Friedhelm.Method for producing statistical styrene−butadiene copolymers.国際公開第9936451A1号)。
【0007】
通常の有機リチウム及びリチウムアミド試薬の反応性又は選択性を変更することはほとんど不可能であるが、反応性または選択性の変更は、多くの化学工業、特に医薬調製および重合プロセス、において需要が高まっている。かかる化合物の両方を用いた従来の撹拌−バッチモードでの合成では、廃棄溶媒が顕著な量で生じるため、化学プロセスにおいては望ましくない。大規模な商業的合成においては、より環境に優しいプロセス(溶媒を含まない有機リチウム、リチウムアミド類又は充填層流通式反応器の設置)が理想的である。なぜなら、それにより廃棄溶媒の問題が減らされ、また長時間にわたる精製又はワークアップ(work−up)工程が省略できるからである。1つの解決法は、流動式の化学反応において使用でき、プロセスにおける試薬の効率及び効果をコントロールする、有機リチウム試薬の固体ソースの開発であろう。このアイデアを元に、近年、結晶質のグリニャール試薬(非特許文献5:Marcus,V.,ら,Angew Chem,Int.Ed.2000,39,3435)及び他の固体カルボアニオン源(非特許文献6:Davies,S.G.ら,J.Am.Chem.Soc.1977,4,135及び非特許文献7:Eaborn,C.,ら,J.Am.Chem.Soc.1994,116,12071)の開発がなされてきた。しかしながら、これらの結晶質の試薬の調製方法は通常時間がかかり、また特定のカルボアニオン系のみに限定されるため、それらの大規模スケールでの応用、またそれらの一般のカルボアニオン源としての応用が限定されたものとなる。グリニャール試薬(カルボアニオンドナーの最も一般的な種類)は更に、2当量のアルキル又はアリールマグネシウムハロゲン化物(2RMgX)と、各々1当量のジアルキル−又はジアリールマグネシウム化合物(RMgR)及びマグネシウムハライド塩(MgX2)との間におけるSchlenk平衡を受ける。この不均化反応は反応種を増加させ、この不均化反応は、場合によっては、カルボアニオン源としての使用に問題のある複雑化である。
【0008】
幾つかの公知の有機リチウム化合物(例えばブチルリチウム、メチルリチウム及びフェニルリチウム)は市販されている。市販されていない多くの有機リチウムは、金属−ハロゲン交換反応から調製する必要があり(総説として、非特許文献8:Wakefield,B.Organolithium Methods,Academic Press:London,1988を参照)交換反応においては1つの有機リチウム試薬と有機ハロゲン化物とを使用する。あるいは、純粋なリチウム金属と有機ハロゲン化物とを反応させて、有機リチウムを形成させてもよい。これらの変換は、有機ハロゲン化物、リチウム金属、リチウムハロゲン化物及び有機リチウム化合物との間での平衡反応を示す。純度の高い有機リチウム生成物を合成することは、未反応の有機ハロゲン化物がしばしば混入するため、困難であり、それにより大規模プロセスへの応用も困難となる。リチオ化反応は、脱プロトン反応によって、又はエーテル及びチオエーテルの還元的切断(非特許文献9:Schlosser,M.Organometallics in Syntheses;Wiley and Sons:Chichester,1994,47)、シャピロ方法(非特許文献10:Shapiro,R.H.Org.React.1976,23,405)、又はアレーンにより触媒されるリチオ化反応(非特許文献11:Yus,M.;Ramon,D.J.;J.Chem.Soc.,Chem.Commun.1991,398)により実施してもよい。これらの方法は有機リチウム自体を利用し、塩基として機能させることで開始するため、それらは合成の観点からは原子効率的ではない。また多くの工業において、微粉末化されたLi金属の高い反応性及び自然発火しやすい性質のため、ハロゲン化形態の標的有機基とリチウム金属とを直接反応させるアプローチは強く避けられる。還流炭化水素中につくられる分散リチウムも更に大きなスケールアップを困難にする(非特許文献12:Joshi,D.K.;Sutton,J.W.;Carver,S.;Blanchard,J.P.Org.Process Res.Dev.;2005;9(6);997−1002)。別法として、水銀−リチウム法(非特許文献13:Schollkopf,U.;Gerhart,F.Angew.Chem.Int.Ed.Engl.1981,20,795)、テルル−リチウム法(非特許文献14:Shiner,C.S.;Berks,A.H.;Fisher,A.M.J.Am.Chem.Soc.1988,110,957、非特許文献15:Hiiro,T.;Mogami,T.;Kambe,N.;Fujiwara,S−I;Sonoda,N.Synth.Commun.1990,20,703)、及びスズ−リチウム法(非特許文献16:Hoffmann,R.W.;Breitfelder,S.;Schlapbach,A.Helv.Chim.Acta 1996,79,346)による金属交換反応を実施してもよいが、水銀、テルル及びスズ化合物は通常有毒であり、これらの試薬を大規模な工業的プロセスに使用することは不適当である。
【0009】
本発明と異なるストラテジーにおいては、アルキルリチウム(MeLi、EtLi)を、無孔性の無機支持体(例えばSiO2、CaO又はAl2O3)の表面上へ吸着させることによって安定化させ、次にパラフィンワックスでコーティングする(特許文献5:Deberitzその他、米国特許第5149889号)。しかしながらこの場合、事前調製したアルキルリチウム試薬を使用しなければならない。更に、反応性を高めるため、ユーザはオイル、ワックス又は炭化水素を除去しなければならず、このことは望ましくない別の分離ステップを追加する。本発明との更なる大きな相違は、アルキルリチウムが無機支持体の表面上へ吸着され、支持体中には吸収されないという事実である。このストラテジーは、安定化され、かつ容易に使用きるアルキルリチウム試薬が、化学工業において要望されていることを物語るものともいえる。
【0010】
有機リチウム化合物と同様、リチウムアミドも商業的な入手可能性が限られている。リチウムと第一級若しくは第二級アミンからそれらを調製するのは困難であるが、最も便利には、第一級若しくは第二級アミンを有機リチウム試薬で処理することにより調製される。すなわち、リチウムアミドの使用は、それらが典型的に由来する有機リチウム試薬と同様の多数の限界に直面する。リチウムアミドの生成に使用する3つの最も一般的な有機リチウム試薬は、メチルリチウム、ブチルリチウム及びフェニルリチウムであり、それらは反応の間、それぞれメタン、ブタン及びベンゼンを発生させる。これらの全ての副産物は全て揮発性、可燃性の材料であるため、製造環境の悪化を生じさせる。メタン及びブタンは室温で可燃性の気体であり、大規模な製造における副産物としてのそれらの化学量論的な発生は、多くの問題を生じさせ、またコストを高めることにもなる。またベンゼンは有毒であり、発癌物質であることが周知である。
【先行技術文献】
【特許文献】
【0011】
【特許文献1】米国特許第5523447号
【特許文献2】米国特許出願公開第2006/0049379号
【特許文献3】米国特許出願公開第2006/0170118号
【特許文献4】国際公開第9936451A1号
【特許文献5】米国特許第5149889号
【非特許文献】
【0012】
【非特許文献1】Gilman,H.ら,J.Am.Chem.Soc.1932;54,1957
【非特許文献2】Gilman,H.,ら,Org.Chem.1958;23,2044
【非特許文献3】Hilpert,H.Tetrahedron,2001,57,7675
【非特許文献4】Askin,D.;Wallace,M.A.;Vacca,J.P.;Reamer,R,A.;Volante,R.P.;Shinkai,I.J.Org.Chem.1992,57,2771
【非特許文献5】Marcus,V.,ら,Angew Chem,Int.Ed.2000,39,3435
【非特許文献6】Davies,S.G.ら,J.Am.Chem.Soc.1977,4,135
【非特許文献7】Eaborn,C.,ら,J.Am.Chem.Soc.1994,116,12071
【非特許文献8】Wakefield,B.Organolithium Methods,Academic Press:London,1988
【非特許文献9】Schlosser,M.Organometallics in Syntheses;Wiley and Sons:Chichester,1994,47
【非特許文献10】Shapiro,R.H.Org.React.1976,23,405
【非特許文献11】Yus,M.;Ramon,D.J.;J.Chem.Soc.,Chem.Commun.1991,398
【非特許文献12】Joshi,D.K.;Sutton,J.W.;Carver,S.;Blanchard,J.P.Org.Process Res.Dev.;2005;9(6);997−1002
【非特許文献13】Schollkopf,U.;Gerhart,F.Angew.Chem.Int.Ed.Engl.1981,20,795
【非特許文献14】Shiner,C.S.;Berks,A.H.;Fisher,A.M.J.Am.Chem.Soc.1988,110,957
【非特許文献15】Hiiro,T.;Mogami,T.;Kambe,N.;Fujiwara,S−I;Sonoda,N.Synth.Commun.1990,20,703
【非特許文献16】Hoffmann,R.W.;Breitfelder,S.;Schlapbach,A.Helv.Chim.Acta 1996,79,346
【発明の概要】
【発明が解決しようとする課題】
【0013】
したがって、反応性の顕著な損失がなく、取扱、貯蔵、使用が容易であり、乾燥形態において入手可能な、有機リチウム及びリチウムアミド試薬に対するニーズが存在する。本発明はそのニーズに応えるものである。
【課題を解決するための手段】
【0014】
一実施形態では、本発明はリチウム金属/多孔性金属酸化物組成物に関する。これらのリチウム金属組成物は、液体リチウム金属を多孔性金属酸化物孔に吸収させるのに十分な発熱条件下で、不活性雰囲気において、液体リチウム金属と多孔性金属酸化物とを混合することにより調製される。本発明のリチウム金属/多孔性金属酸化物組成物は、約40重量%以下のリチウム金属を担持するのが好ましく、最も好ましくは約20重量%〜40重量%で担持する。
【0015】
他の実施形態では、本発明はまた、多孔性酸化物に吸収されたRLiを含有する、リチウム試薬−多孔性金属酸化物組成物に関する。上記の式RLiにおいて、Rはアルキル基、アルケニル基、アルキニル基、アリール基、アルカリール基又はNR1R2基であり、R1はアルキル基、アルケニル基、アルキニル基、アリール基、アルカリール基であり、R2は水素、アルキル基、アルケニル基、アルキニル基、アリール基、アルカリール基である。
【0016】
したがって、本発明はまた、多孔性金属酸化物(アルミナ(Al2O3、アルミナゲル)など)中に形成され、貯蔵されたリチウム、有機リチウム及びリチウムアミド種の新規な組成物の調製方法に関する。多孔性Al2O3に吸収されたリチウムを用いることにより、孔内部において有機リチウム及びリチウムアミド化合物を調製及び形成することができ、並びに、求核付加反応及び重合反応のためのカルボアニオンのin situ生成を可能とする。予備調製された有機リチウム化合物を、固体状の無機支持体(例えば二酸化ケイ素又はアルミナゲル)の孔に吸収させることができ、それにより、合成変換での使用のための溶媒フリーの貯蔵及び配送が可能となる。これらの新規な組成物は、バッチ反応器及び流通式反応器の両方における使用に適する、固体状の流動性の粉末である。
【図面の簡単な説明】
【0017】
【図1】図1A〜1Cは、実施例3の結果の代表的なスペクトルを示す。
【図2】図2は、実施例4の結果の代表的な1H NMRスペクトルの結果を示す。
【図3】図3A〜3Cは、実施例7の結果の、代表的な1H NMR、13C NMR及びESI−MSスペクトルを示す。
【図4】図4は、実施例8の結果の代表的な1H NMRスペクトルを示す。
【図5】図5A〜5Cは、実施例9の結果の代表的な1H NMR、GC−MS及び13C NMRスペクトルを示す。
【発明を実施するための形態】
【0018】
アルカリ金属、それらの同等物、並びにそれらの誘導体を簡便に利用する技術の開発は、学界、化学工業および水素生産業に求められ続けてきた。そのニーズに応えて、本発明は、リチウム金属を多孔性金属酸化物に吸収させることに関する。本発明の組成物で利用される多孔性金属酸化物は還元不可能な多孔性金属酸化物であり、特に多孔性アルミナが好ましい多孔性金属酸化物である。本発明はまた、リチウム試薬−多孔性金属酸化物組成物、多孔性金属酸化物に吸収された有機リチウム試薬及びリチウムアミドに関する。これらのリチウム試薬−多孔性金属酸化物組成物は、より多くの種類の利用しやすい求核試薬を提供し、取扱い及び貯蔵において現在知られているリチウム試薬を上回る大きな優位を有する。
【0019】
リチウム金属−多孔性金属酸化物組成物:
リチウム(Liの元素記号)は、周期表の1族に属するアルカリ金属である。それは全ての金属の中で最も軽く、水の半分の密度である。リチウムは軟らかい銀色の金属であり、一個の価電子を有するが、それは容易に失われて陽イオンになる。このため、リチウムは酸素又は窒素、特に水に曝露したときに燃焼し、激しく反応する。したがってこの金属は、非反応性の雰囲気中、又は非反応性の液体(例えば炭化水素又はナフサ)中に貯蔵する必要がある。リチウムはまた、1族の中でも、2族のアルカリ土類金属の特性を若干示すことが知られている。
【0020】
リチウム−多孔性金属酸化物組成物の調製においては、リチウム金属を多孔性金属酸化物(例えば多孔性アルミナゲル)と混合し、次にリチウム金属が溶解して金属酸化物の孔に吸収されるまで加熱するのが好ましい。これを行うための1つの方法としては、不活性雰囲気(例えばアルゴン又はヘリウム)下で、リチウム金属を、多孔性金属酸化物と混合する前に加熱することが挙げられる。あるいは、リチウム金属を固体として多孔性アルミナと混合し、その混合物を加熱してリチウム金属を溶解させてもよい。リチウム金属を吸収させる熱処理は、160〜325℃、好ましくは160〜225℃の間の温度で調節できる。リチウム金属を多孔性アルミナに取り込ませるための他の利用可能な方法としては、ゼオライトを用いた気相における反応が挙げられる(A.S.Ichimura,J.L.Dye,M.A.Camblor and L.A.Villaescusa,J.Am.Chem.Soc.,124,1170−1171(2002)、及びD.P.Wernette,A.S.Ichimura,S.A.Urbin and J.L.Dye,Chem.Mater.15,1441−1448,(2003)を参照のこと)。他の利用可能な方法として、リチウム金属を、金属−アンモニア溶液から多孔性アルミナ内に堆積させることが挙げられる(M.Makesya and K.Grala,Syn.Lett.1997,pp.267−268を参照のこと)。
【0021】
リチウム−多孔性金属酸化物組成物(例えば好適なリチウム−アルミナゲル(Li−AG))は、バルク若しくはシェービングされた(shaved)リチウム金属と、焼成多孔性金属酸化物(例えばアルミナ)との混合物を加熱して直接調製し、親金属の還元能力を大部分保持する、脆い黒色の粉末(loose black powders)を形成させてもよい。この反応は、好ましくは不活性雰囲気(例えばアルゴン又はヘリウム)下で実施する。リチウム−多孔性金属酸化物組成物はおそらく、吸収された中性のリチウム金属の小さいクラスタを多孔性金属酸化物孔に有し、並びに、電子が原子から非局在化した、イオン化リチウム金属(Li+)を孔の壁に有すると考えられる。上記材料は自然発火性であるが、空気中においては、微細粉末化された純粋なリチウム金属よりも反応性が低い。リチウム金属と多孔性アルミナゲルの加熱は、約160℃〜約325℃、好ましくは約160℃〜約225℃の温度範囲内で実施する。上記加熱は、リチウム金属の全てが吸収されるまで、あるいは一晩実施してもよい。
【0022】
リチウム金属は、アルミナゲル又は他の多孔性金属酸化物の内部で、非常に微細に分散すると考えられている。リチウム−アルミナゲルは、ほぼ完全に窒素(N2)ガスと反応できる。窒素ガスとリチウム−アルミナゲルとの反応により、アルミナの孔内部で、窒化リチウム(Li3N)の微粉末が分散される。このLi3Nが水と穏やかに反応すると、必要に応じて以下の反応(式中、AGはアルミナゲルを示す)によって高純度のアンモニアガスが発生する:
【数1】

従って、この材料はアンモニアを、その輸送、転送及び貯蔵における危険が回避される方法で発生させる。この純粋なアンモニアは、限定されないが化石燃料燃焼プロセスから発生する、窒素酸化物(NOx)を含有する煙道ガスの処理などの多くの用途に使用可能である。したがって、本発明のリチウム−多孔性金属酸化物組成物は不活性雰囲気からの窒素ガスの洗浄に使用できる。
【0023】
本発明のリチウム金属−多孔性金属酸化物組成は、約40重量%以下でリチウム金属を担持するのが好ましく、約10重量%〜20重量%で担持するのが最も好ましい。
【0024】
使用できる多孔性金属酸化物は、リチウム金属により還元されない、いかなる金属酸化物でもあってもよい。本発明において使用する、最も好ましい多孔性金属酸化物粉末は、多孔性アルミナ(また、アルミナゲル(特にγ−アルミナゲル)と呼ばれる)である。本発明に使用できる他の多孔性金属酸化物粉としては、リチウム金属により還元されないあらゆる遷移金属酸化物であり、例えば、多孔性酸化チタン(すなわちTiO、TiO2、Ti2O3、Ti3O5)、多孔性酸化カルシウム(CaO)、多孔性酸化ジルコニウム(すなわちZrO2)、多孔性酸化鉄(すなわちFe2O3又はFe3O4)、多孔性Co3O4、多孔性金属ホスフェート(MPO)、多孔性ハイブリッドホスホシリケート、多孔性アルミネート、多孔性アルミノシリケート、多孔性バナデート、モリブデート、などが挙げられる。シリカゲルは、二酸化ケイ素と純粋なリチウム金属との間の高い反応性ゆえに、予備調製された溶媒和有機リチウム試薬の吸収に使用できるだけである。多孔性を有することにより、これらの多孔性金属酸化物は、大量の物質を吸収して取り込むことができる。本発明の組成物は、生成したカルボアニオン又はアミド種に加えて、酸化物の孔に吸収されたリチウム金属を有する。多孔性アルミナは、多くの企業(例えばW.R.Grace&Co.社、又はAlmatis AC社)から購入できる。
【0025】
本発明の多孔性金属酸化物組成物において使用する多孔性金属酸化物は、好ましくは30Å〜500Åの平均孔径を有する。より好ましくは、上記平均孔径は60Å〜190Åである。
【0026】
多孔性金属酸化物は、購入したときは流動性の粉末であるが、それらは典型的にはガス状の物質(例えば水及び空気)を含有する。ゆえに、これらは、多孔性酸化物粉末とリチウム金属とを混合して本発明の組成物を調製する前に除去するのが好ましい。多孔性金属酸化物は、当業者に既知の方法を使用してガス除去することができる。例えば、ガス状物質を除去するため、多孔性金属酸化物を、真空の下で、排気可能なフラスコ中で(最初は温風ドライヤーで、次にトーチで)加熱してもよい。かかる加熱は、約300℃の温度で実施する。空気中で多孔性金属酸化物を600℃若しくはより高い温度(〜900℃)で加熱することにより、より容易にガスを除去し、活性部位を不動態化すること(焼成)も可能であり、実際上好ましい。多孔性金属酸化物は典型的には、本発明の組成物を調製する前に、乾燥した(また好ましくは不活性)雰囲気下で、室温に冷却される。
【0027】
リチウム試薬−多孔性金属酸化物組成物:
【0028】
本発明はまた、多孔性の金属酸化物又は非金属酸化物に吸収されたRLiを有する、リチウム試薬−多孔性金属酸化物組成物の提供に関する。上記の式RLiにおいて、Rはアルキル基、アルケニル基、アルキニル基、アリール基、アルカリール基又はNR1R2基であり、R1はアルキル基、アルケニル基、アルキニル基、アリール基、アルカリール基、XRn基(Xはヘテロ原子(例えばSi、S、Sn、Ge、P)であってもよく、nは整数である)、又はSi(R3)3基であり、R2は水素、アルキル基、アルケニル基、アルキニル基、アリール基、アルカリール基又はSi(R3)3基であり、R3はアルキル基、アルケニル基、アルキニル基、アリール基又はアルカリール基である。リチウム試薬−多孔性金属酸化物組成物は、リチウム金属−多孔性金属酸化物組成物と有機ハロゲン化物(RX)とを反応させることによって調製しても、リチウム金属と多孔性金属酸化物と有機ハロゲン化物(RX)とのin situでの混合によって調製しても、リチウム試薬(RLi)の多孔性金属酸化物への吸収によって調製しても、リチウム金属−多孔性酸化物組成物を第1級若しくは第2級アミン(HNR1R2)と反応させることによって調製してもよい。R、R1、R2及びR3基は、官能基によって更に置換されても、ヘテロ原子(例えばN、O、S、など)を更に含んでもよく、これらは、本発明のリチウム試薬−多孔性金属酸化物組成の形成を防止するような方法ではリチウムと反応しない。次に、R、R1、R2及びR3の好適かつ典型的な実施形態について説明する。
【0029】
本発明のリチウム試薬−多孔性金属酸化物組成物を調製する際、Li−多孔性金属酸化物組成物(上記の通りに調製)を、反応容器中で約0℃〜約−78℃に冷却してもよい。次に、有機ハロゲン化合物(又はR=R1R2Nのときは第一級又は第二級アミン(両方ともRX))を、Li−多孔性金属酸化物(純粋であるか又は溶媒中の物質)に徐々に添加する。有機ハロゲン化合物又はアミンが孔へ移動し、金属酸化物の孔内部で、リチウム金属と反応し、有機リチウム化合物が生成する。室温になるまで反応液を加熱した後、いかなる過剰の有機ハロゲン化合物、アミン、及び/又は溶媒も蒸発除去し、乾燥した、流動性の粉末状のLi試薬−多孔性金属酸化物組成物の形成を完了する。下記の実施例8に示すように、有機リチウム−多孔性金属酸化物組成物を、炭素−ヘテロ原子結合の還元的切断(例えばエチルビニルエーテルからのビニルリチウムの調製)により調製してもよい。
【0030】
リチウム試薬が有機リチウム化合物であるとき、使用できる有機ハロゲン化合物(RX)としては、限定されないがハロゲン化アルキル、ハロゲン化アルケニル、ハロゲン化アリール及びハロゲン化アルカリールなどが挙げられる。好ましいハロゲンは、塩化物、臭化物及びヨウ化物である。上記アルキル基、アルケニル基及びアルキニル基は、直鎖状若しくは分岐鎖状であってもよく、環状及び複素環式のアルキル基、アルケニル基又はアルキニル基であってもよい。上記アリール基及びアルカリール基は、ヘテロアリール基及びalk−ヘテロアリール基であってもよい。上記アルキル基は、好ましくはC1−C12アルキル基又はC5−C12シクロアルキル基であってもよい。上記アルケニル基は、好ましくはC2−C12アルケニル基又はC3−C12シクロアルケニル基であってもよい。上記アルキニル基は、好ましくはC2−C12アルキニル基又はC3−C12シクロアルキニル基であってもよい。上記アリール基及びアルカリール基は、ヘテロアリール基及びalk−ヘテロアリール基であってもよい。アルカリール基の「alk」部分は、本願明細書に記載のアルキル基、アルケニル基又はアルキニル基のいずれでもよい。
【0031】
好ましいアルキル基は、例えばメチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、t−ブチル基、イソブチル基、sec−ブチル基、シクロプロピル基、シクロブチル基、シクロヘキシル基、(シクロヘキシル)メチル基、シクロプロピルメチル基、並びに、n−ペンチル基、n−ヘキシル基、n−ヘプチル基、n−オクチル基の同族体及び異性体、などである。好ましいアルケニル基としては、エテニル基、アリル基、並びにブテニル基、ペンテニル基、ヘキセニル基、ヘプテニル基、オクテニル基、ノネニル基、デセニル基、シクロプロペニル基、シクロブテニル基、シクロペンテニル基、シクロペンタジエニル基、シクロヘキセニル基、シクロヘプテニル基、シクロヘプタジエニル基、シクロオクテニル基、シクロオクタジエニル基などの異性体が挙げられる。好ましいアルケニル基としては、アセチレニル基、並びにブチニル基、ペンチニル基、ヘキシニル基、ヘプチニル基、オクチニル基、ノニニル基、デシニル基の異性体が挙げられる。好ましいアリール基及びアルカリール基としては、フェニル基、ベンジル基、並びにトリル基、アニシル基、アナリニル基、ナフチル基、インデニル基、アントリル基、ピロリル基、ピリジル基、ピリミジル基、イミダゾリル基、ピラジニル基、オキサゾリル基、イソオキサゾリル基、チアゾリル基、フリル基、チエニル基、イミダゾリル基、ピラゾリル基、チオフェン基、N−アルキル化ピロール基、α−アルキル化ピリジン基、インドリル基、キノリニル基、イソキノリニル基などの異性体が挙げられる。
【0032】
RLiがR1R2NLiであるときに、Liアミド−アルミナゲル組成物の形成に用いる第一級若しくは第二級アミンとしては、限定されないがアルキルアミン、ジアルキルアミン、アルキルアリールアミン及びジアリールアミン並びに環状の第二級アミンなどが挙げられる。これらのアミン中の窒素と結合する有機基、又はシリル基Si(R3)3の場合にはR3は、Rに関して上記したものが挙げられる。好ましいアルキル基は、好ましくはメチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、トリメチルシリル基、トリエチルシリル基、t−ブチル基、イソブチル基、sec−ブチル基、並びに、例えばn−ペンチル基、n−ヘキシル基、n−ヘプチル基、n−オクチル基などの同族体及び異性体である。好ましいアリール基及びアルカリール基は、フェニル基、ベンジル基、並びにトリル基、アニシル基、アナリニル基、ナフチル基、インデニル基、アントリル基の異性体である。好ましい環状の第二級アミンは、ピロリジン、ピペリジン及び2,2,5,5−テトラメチルピペリジンである。
【0033】
リチウムと反応しないあらゆる不活性溶媒を、本発明のリチウム試薬組成物の調製に使用できる。例えば、ヘキサン又はヘプタンなどの非反応性の炭化水素、又はTHFなどの非反応性のエーテルが使用できる。通常THFが好適である。リチウム試薬−多孔性金属酸化物組成物を形成するとき、極性の高い溶媒は試薬RXと競合するため好ましくない。
【0034】
本発明の有機リチウム−多孔性金属酸化物組成物を生成する他の方法は、炭素−水素活性化を介する方法であり、すなわちそれは、有機リチウム−アルミナゲルが添加された試薬の炭素原子の脱プロトン化を行い、新規なカルボアニオン種を生成することを意味する。この有機リチウムの交換反応は、典型的には低い温度(例えば約23℃〜約−80℃)で実施する。新規な有機リチウム試薬が形成させた後、それは、利用できる基質との次の反応における、求核種又は塩基として使用できる。この反応は、バッチ反応器又は流通式反応器において実施できる。
【0035】
本発明の有機リチウム−多孔性金属酸化物組成物を生成する更に別の方法としては、in situで金属−ハロゲン(金属−元素)交換反応を行わせて、第1の有機リチウム−多孔性金属酸化物を、第2の有機リチウム−多孔性金属酸化物に変換することが挙げられる。1つの反応容器(バッチ反応容器又は流通式反応容器)に、有機ハロゲン化合物(RX)、又はC−Z結合を有するあらゆる活性化された分子種を添加し(Zはいかなるハロゲン化物又は酸性水素などであってもよい)、最初に、低い温度でTHF中で分散させ、次に、当該容器に有機リチウム−多孔性金属酸化物を添加することにより実施できる。冷却(すなわち約23℃〜約−80℃)したスラリーを一定時間十分に混合した後、上記の有機リチウムは、従来の方法で次に添加される試薬と反応可能になる。
【0036】
有機リチウム−アルミナゲルはまた、リチウムの切片、多孔性アルミナゲル及びハロゲン化アルキル溶液(好ましくは、例えば炭化水素又はエーテル中)を使用して、in situで反応させ、溶媒を蒸発させることにより調製できる。
【0037】
本発明の有機リチウム試薬−多孔性金属酸化物組成物はまた、低温条件下でシリカゲル又はアルミナゲルの孔に、溶媒中で予備調製した有機リチウム種を吸収させることにより調製できる。過剰の溶媒を次に留去し、粉末を乾燥真空する。生成物は自然発火性を有さず、それらは好ましくは有機リチウム−シリカゲル又は有機リチウム−アルミナゲルと呼ばれる。上記の方法では、低温条件(例えば約23℃〜約−80℃)下で、多孔性材料に有機リチウム種を接触させて有機リチウム種を多孔性金属酸化物材の孔に吸収させ、あらゆる過剰の溶媒を留去し、得られる有機リチウム材料を乾燥させる。
【0038】
得られる流動性の粉末、すなわちRLiが多孔性金属酸化物に吸収した有機リチウム−多孔性金属酸化物組成物は、特に、従来の有機リチウム化合物の全ての反応(例えば求電子物質に対する求核付加反応、重合反応の開始、及び塩基により触媒される反応)を行う。材料中にリチウム金属が残留しないとき、上記粉末は窒素雰囲気下でも安定である。残留リチウムの有無にかかわらず、この材料は、市販され、従来公知の、有機リチウム化合物の溶液(それらが貯蔵される溶媒中で凝集し、反応し、分解する傾向を有する)と比較し、室温における長期の貯蔵寿命を示す。
【0039】
本発明に係るリチウムアミド−多孔性金属酸化物を生成する他の方法は、有機リチウム−多孔性金属酸化物を用いて、第一級若しくは第二級アミンを非プロトン化することである。1つの反応容器(バッチ反応器又は流通式反応器)中に、第一級若しくは第二級アミン(好ましくは溶媒中に溶解)を、有機リチウム−多孔性金属酸化物の撹拌バッチに投入する。スラリーをよく混合し、反応させた後、あらゆる過剰のアミン及び/又は溶媒をデカントし、真空除去し、又は低温で蒸発させ、乾燥した、流動性を有するリチウムアミド−多孔性金属酸化物の粉末の形成を完成させる。
【0040】
リチウムアミド−多孔性金属酸化物組成物(R=R1R2N)(自由に流動する粉末)は、従来のリチウムアミド化合物に関して既知の反応をする。これらの反応としては、限定はされないが、アルコール及びカルボニル化合物(アルデヒド及びケトン)などの大部分の一般的な炭素酸の脱プロトンが挙げられる。リチウム金属が材料中に残留しないとき、上記粉末は窒素下でも安定である。残留リチウムの有無にかかわらず、この材料は、凝集し、徐々に分解する傾向がある市販若しくは従来公知のリチウムアミド化合物と比較し、室温で長期にわたる貯蔵寿命を示す。これらのリチウムアミドを調製するための一般的な方法は、アルキルリチウム種の、対応するアミンとの反応(例えばDetlefら、国際公開第03033505号)、金属の融点より高い温度で、リチウムと、アミンとの混合物の加熱(Chiuら、米国特許第5420322号)、又は少量の電子担体としてのイソプレンとの混合物の加熱(Corellaら、米国特許第6169203号)を含む。
【0041】
上記のように、リチウム−多孔性金属酸化物組成物(例えばリチウム−アルミナゲル組成物)は、本発明の対応するリチウム試薬組成物(例えば対応する有機リチウム−アルミナゲル又はリチウムアミド−アルミナゲル組成物)の生成の際、開始成分として使用できる。かかる対応する組成物の調製に関する更なる具体例は、以下のとおりである:リチウム−アルミナゲルを約が−80℃〜約0℃の温度範囲で冷却し、数分間撹拌する。撹拌しながら、リチウム−アルミナゲルに、有機ハロゲン化合物(純粋な形態又は非反応性の炭化水素又はエーテル溶媒(例えばテトラヒドロフラン(THF))中の溶液)を徐々に添加する。反応混合物を、低温で数時間撹拌する。あらゆる過剰な未反応有機ハロゲン化合物又は溶媒を、真空下で蒸留させ、一方で系の温度を室温まで上昇させる。最終的な有機リチウム−アルミナゲル組成物の強度(1グラムあたりの反応当量)を、別の反応系において、求電子種への求核付加反応を実施させることにより測定する。臭化ブチルの場合、上記反応は以下のとおりと考えられる:
【数2】
LiBr(3.464g/cm3)及びBuLi(0.67g/cm3)の純粋な化合物密度を使用すると、生成物のmolあたりの体積は、LiBrの場合は25.1cm3で、BuLiの場合は95.5cm3で、合計120.6cm3/molである。アルミナゲルの空隙の体積は、水銀ポロシメーターにおいて1.56cm3/gである。生成物が、体積を増加させることなく孔にフィットしたと仮定した場合、8.0gのアルミナゲルのサンプルでは、空隙部は最高12.5cm3である。すなわち、8.0gのAGにより、12.5/120.6=0.110molの反応(1.53gのLiを必要とする)が可能となる。これは、(1.53/9.53)×100=16.0重量%のLiの担持に対応する。ゆえに、BuLiの担持は、0.110/9.53=0.0115mol/(リチウム−アルミナゲル1g)又は11.5mmol/(リチウム−アルミナゲル1g)である。
【0042】
2つの例示的な系における有機リチウム−アルミナゲル、ブチルリチウム−アルミナゲル及びメチルリチウム−アルミナゲルの最大の担持量を、使用するリチウム−アルミナゲルの合計量を10gとして、下記の表1に示す:
【0043】
【表1】
【0044】
リチウム金属の担持量は、実際に使用する多孔性金属酸化物の孔径及び孔密度に依る。典型的には、リチウム金属は最高約40重量%で、本発明の組成物に存在してもよい。上記の材料を典型的な還元反応及びin situでのリチオ化反応に用いる場合、好ましくは、上記金属の量は20重量%〜40重量%の範囲である。有機リチウム−多孔性金属酸化物及びリチウムアミド−アルミナゲル組成物を生成させる際の好ましい担持量は、10重量%〜20重量%の範囲のリチウムである。本発明のリチウム−多孔性金属酸化物組成物において、約40重量%超で担持すると、多孔性金属酸化物の表面上に若干のフリーの金属が残留することがありえる。
【0045】
有機金属リチウム試薬を安定させることが知られている、様々な添加剤を、リチウム試薬−多孔性金属酸化物と共に使用してもよい。例えば限定されないが、テトラメチルエチレンジアミン(TMEDA)、ジアザビシクロウンデカン(DBU)、スパルテイン(Tsumakiら,米国特許第6024897号を参照)などの安定化剤を更に添加して、これらの固体状カルボアニオンの安定性を増加させてもよい。例えば、安定化された有機リチウム種を、合成反応において添加剤としてテトラメチルエチレンジアミン(TMEDA)を用いて調製できる。典型的には、リチウム2molごとに、有機ハロゲン化合物を1mol及びTMEDAを1mol用いて、有機リチウム−TMEDA−多孔性金属酸化物を調製する。TMEDAの分子構造は、1,2−ジアミノエタン1分子中の、2つの各々のアミン窒素原子上に、2つのメチル基が結合してアルキル化された構造に対応する。TMEDA又は他の第三級アミン安定化剤は、リチウムカチオンと第三級窒素との配位結合によってこれらの試薬の安定化を助ける添加剤として使用できる。潜在的に、このイオン錯体形成はリチウムカチオンの有効半径を拡大し、求電子性を低下させ、それをカルボアニオン部から分離させ、それによりアルキルカルボアニオンからのβヒドリドを脱離させる能力が制限される。この安定化態様はまた、外部から添加された試薬との反応に対するカルボアニオンの有効性を強化するとも考えられる。
【0046】
リチウム−及びリチウム試薬−多孔性金属酸化物組成物の使用:
【0047】
本発明のリチウム−多孔性金属酸化物組成物及びリチウム試薬−多孔性金属酸化物組成物は、リチウム金属、有機リチウム試薬及びリチウムアミド試薬と同様の反応に使用できる。かかる反応としては、限定されないが求核付加反応、重合反応及び塩基により触媒される反応などが挙げられる。表2は、かかる反応の典型的なリストを示す。しかしながら本発明の組成物はフリーの流動性の粉末であり、貯蔵寿命が長いという効果を発揮する。
【0048】
【表2−1】
【0049】
【表2−2】
【0050】
更に、固体試薬としての、本発明のリチウム−多孔性金属酸化物組成物及びリチウム試薬−多孔性金属酸化物組成物は、幾つかの方法による反応に使用できる。最も単純な様式は、固体試薬を、1つ以上の基質を含有する溶液と共にスラリー中で撹拌する、従来公知のバッチ反応である。ここで、市販の固体状のリチウムの調製、又は有機リチウム若しくはリチウムアミド試薬の溶液の調製に勝る根本的な利点としては、
(a)固体の取扱の容易さ、及び
(b)反応、クエンチング及びワークアップ手順における、有機溶媒の使用の最小化などが挙げられる。好ましいケースでは、固体状の酸がスラリー中に含有されるとき、中性化生成物のみを含有する有機溶液の直接単離が、固体酸化物ゲル(例えばアルミナ又は二酸化ケイ素)のイオン副産物に対する親和性によって促進され、別の有機/水性分配や、洗浄工程により除去しなくてもよい。
【0051】
第2の使用態様は、流通式反応器(例えばカラム流通式反応器)におけるプロセスである。そのプロセスでは、反応基質の溶液を、固体状のリチウムアルミナゲル、有機リチウム−固体状酸化物又はリチウムアミド−固体状酸化物試薬の充填層にパーコレートさせる。流通式反応器又は連続流通式反応器を用い、新しい反応溶液を反応器中の固体状試薬に連続的に添加する。反応生成物を連続的に取り除き、一方で、廃棄物を支持試薬に結合させたままにするか、又は所望の生成物と共に溶出させる。流通式反応器を使用することによる利点は非常に大きい。例えば、当該反応器の使用により、全ての試薬の効率的な利用が確保され、その反応能力が完全に消耗されたときにのみ、反応が終了する。その結果、従来のバッチ反応と比較し、ゲル中及び溶媒中においても、生産性が高く、効率的な方法となる。流通式反応器の例としては、固定床流通式反応器が挙げられる。当該反応器では、反応基質の溶液を、固体試薬(例えば有機リチウム−アルミナゲル)のカラムを通してパーコレートし、カラムの出口で生成物溶液を直接回収する。実質的にいかなるタイプの反応プロセス及び反応器も、本願明細書に記載の反応に使用できるが、流通式反応器(例えば固定床流通式カラム反応器)が、本発明の反応に好ましい反応器のタイプである。
【0052】
溶媒の選択及び有機部分の性質に応じて、リチウム−アルミナゲルと、有機ハロゲン化合物若しくは有機アミンの反応を経て形成される有機リチウム又はリチウムアミド種は、抽出されるのに十分な可溶性を有し得るため、純粋な有機リチウム又はリチウムアミド試薬の新しく調製した溶液を容易に入手できる。あるいは、目的の有機リチウム種が特に不安定若しくは短寿命であると考えられるとき、その求電子パートナー(例えばプロトン源、ケトンなど)を充填カラムの層内に固体状態で直接含有させ、カラム出口で再び、クエンチされた純粋な形態の中性生成物として調製してもよい。
【0053】
あるいは、経時的な反応方式を採用してもよい。当該反応では、in situでリチウム−アルミナゲルから形成された、固体酸化物に結合した有機リチウム又はリチウムアミドを更に、同じ若しくは異なる溶媒中の基質溶液と反応させ、生成物を形成され、更にカラムから溶出させる。すなわちこの方法は、以下の点で有利である:
(a)リチウムの反応性を効率的に使用できること、
(b)溶出された生成物がリチウム金属を含有しないため、安全であること、
(c)不安定な有機リチウム種の中間体が必要となる用途に利用できること、及び
(d)支持された反応物質の連続的な形成及びそれによる反応、更に極性/イオン性副産物及び未反応リチウム金属からそれらの溶出が可能であること。
【実施例】
【0054】
実施例1:LiAG(10%(w/w))の調製
【0055】
Heグローブボックス中で、3.16gのリチウムリボン(1.425g/フィート)を計量し、小片にカットし、28.4gのアルミナゲル(AG)で完全にコーティングし、スチール製の反応器に添加した。スチール製の反応器を、100℃で6時間、165℃で一晩、182℃で8時間、及び190℃で一晩加熱した。スチール製の反応器を冷却し、グローブボックスに入れた。76.7mgの代表的な小さいサンプルを用いて、水によるH2発生に関して試験した。回収されたH2の量は、Liの9.2%当量であった。多孔性アルミナゲルに対するリチウム金属の化学量論比率を変化させて、リチウムの担持量が5重量%〜40重量%の範囲になるように調製した。
【0056】
実施例2:リチウム−アルミナゲル中で安定化された、有機リチウム種の調製:
【数3】
式中、RXは脂肪族(メチル基、ブチル基など)、芳香族(フェニル基)、ベンジル基、アリル基、ホモアリル基、プロパルギル基、sec−アルキル基、tert−アルキル基又はネオペンチル基のハロゲン化物(塩化物、臭化物又はヨウ化物)であってもよい。
【0057】
γ−Al2O3中の10% Li 1g(14mmolのLi)を丸底フラスコに添加し、ドライアイス−アセトン浴で−78℃で冷却し、次にスラリーを撹拌しながら、ヨウ化メチル(2mL)(純粋)を滴状添加した。冷却しながら、反応液を2時間撹拌した。添加が完了し、混合物が室温に達した後、過剰なヨウ化メチルを蒸発させた。この調製の間、少量のエタン及びI2が生じたが、このウルツカップリングによる生成物は、低温を長時間維持することにより最小化できる。アルミナ中のMeLiの強度は、過剰なベンゾフェノンによる求核付加反応により測定した。幾つかの反応について測定した結果、アルミナ中のMeLi(10〜25%のLi/γ−Al2O3から調製)の担持量が3〜7mmol/gの間であった。有機リチウム及びベンゾフェノンの化学量論反応の場合、求核付加反応の生成物の収率は75%〜90%の範囲であり、またオルトリチオ化反応生成物が微量で存在した。
【0058】
PhCH2Li、n−BuLi、アリルリチウム及びホモアリルリチウムを、同じ手順を用いて調製し、ベンゾフェノンとの反応に関して試験した。
【化1】
【0059】
実施例3:TMEDA安定化剤を用いた有機リチウム−アルミナゲルの調製:
【0060】
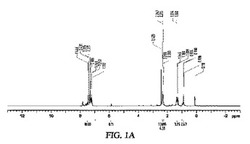
2gのLi−AG(25%重量のLi)を含むフラスコ中に、20mLのペンタン中に2gのTMEDA、3gのn−ブチルブロミドを含有する混合液を精製した後添加し、−80℃で3時間撹拌し、ドライアイス入りの−40℃浴に移し、更に反応液が室温に達するまで一晩撹拌した。溶媒を真空下で除去し、自由に流動する灰色の粉末を得た。この材料を200mg用い、1.1mmol(200mg)のPhCOPhと反応させ、60%の収率でブチルジフェニルアルコールを得た。図1Aは、ブチルジフェニルアルコール生成物の1H NMRを示す。
【0061】
また、ブチルアニオンの存在を、この材料と水とのガス発生反応を行わせ、GC−MSでガス分析することにより確認した。以下の反応に従うブタン(MW=58)の形成が示された。図1Aから1Bを参照。
【数4】
【0062】
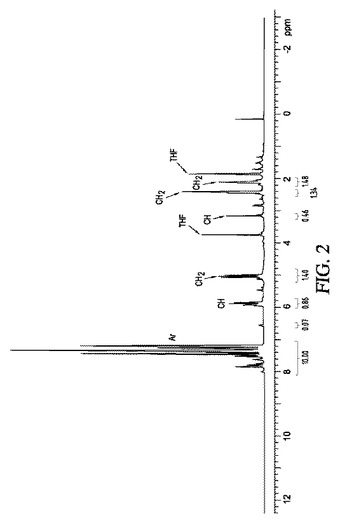
実施例4:有機リチウム−多孔性金属酸化物組成物の生成及びケトンに対するOne−Potでの求核付加反応
【化2】
【0063】
1.5mmolのホモアリルブロミド(195mg)を、5mLのドライ蒸留THFに溶解させ、この溶液を−78℃に冷却した。この冷却溶液に、0.5gのγ−Al2O3中のLi(8〜12%w/w、7〜10mmolのLi)を、15分間にわたり、固体添加用のチューブを用いて添加した。同じ低温条件下でスラリーを30分間撹拌した。低温スラリーを撹拌しながら、3mLのTHF中に溶解させた1mmolのベンゾフェノン(182mg)を、15分間にわたり添加した。反応液を徐々に(6時間)室温に加温し、冷水でクエンチし、更にEtOAcで抽出した。有機層を窒素雰囲気下で蒸発させ、油状の残渣200mg(85%)を得た。
【0064】
図2は、以下のピークを有する、生成物である1H ホモアリルジフェニルメチルケトンの1H NMR(δ、ppm)を示す:7.2−7.8(m,10H),5.8−6.0(m,1H),5.0−5.2(m,2H),2.4−2.6(m,2H),2.05−2.15(m,2H)。
【0065】
実施例5:アルミナゲル中のリチウムと、第二級アミンとの反応による、有機リチウム(リチウムアミド)の生成:
【0066】
1gのリチウム−アルミナゲル(25重量%のLi担持量)を、−60℃でジメチルアミン(〜4mL)と反応させ、濃青色の溶液を得た。徐々に室温に加温すると、この溶液混合物は、水素ガスの形成に起因すると思われる泡を立て始めた。更に室温で1時間静置したところ、溶液の青色が完全に消失した。過剰な未反応のジメチルアミンを、真空下で蒸発させ、得られる固体を秤量した。1.35gのMe2NLi−アルミナゲルを得た(使用したLiの量に対して〜40%の収率)。
【数5】
【0067】
比較例1:リチウム金属と第二級アミンとの反応から、リチウムジメチルアミドを生成させる試み:
【0068】
調製直後の微粉末化したリチウム金属30mgを、真空ストッパーを備えた丸底フラスコに添加し、そこでMe2NHを濃縮した。反応容器を最初の数分、−60℃に維持し、徐々に室温に加温し、その温度で圧力計を用いて一晩維持した。Me2NHの呈色もH2ガス発生も観察されなかった。
【0069】
比較例2:アルミナゲルの存在下での、リチウム金属と第二級アミンとの反応からリチウムジメチルアミドを生成させる試み:
【0070】
30mgの多孔性アルミナゲルを、真空ストッパーを備えているフラスコ中で、20mgの微粉末化したリチウム金属と混合し、この混合物にMe2NHを濃縮した。反応容器を最初に低温に維持し、次に徐々に室温に加温し、その状態で2時間維持した。Me2NHの呈色もH2ガス発生も観察されなかった。
【0071】
比較例1及び2により、リチウム金属は、単独でもアルミナの存在下でも、ジメチルアミンからのリチウムアミド形成を行わないことが証明された。
【0072】
実施例6:カラム中での、有機リチウムの連続的流動反応:
【化3】
【0073】
5mLのTHF中に溶解させた1mmol(182mg)のベンゾフェノンを、1時間にわたり、MeLi/アルミナ(5mM、2g)を充填し、冷却ジャケットを装備したカラム(−40℃)中を通過させた。カラムを過剰量のTHFで洗浄し、回収した生成物を冷水中でクエンチし、更にEtOAcで抽出し、有機層を蒸発させ、92%の収率の粗製(オルトリチオ化反応生成物を若干含む)の油状の液体(185mg)を得た。MeOHで溶出させることによる逆相(C18)カラムクロマトグラフィによって、純粋なアルコールを単離した(73%)。
【0074】
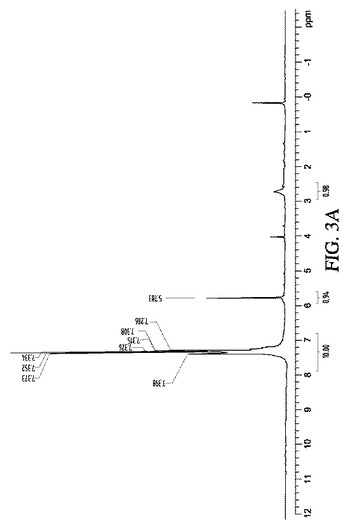
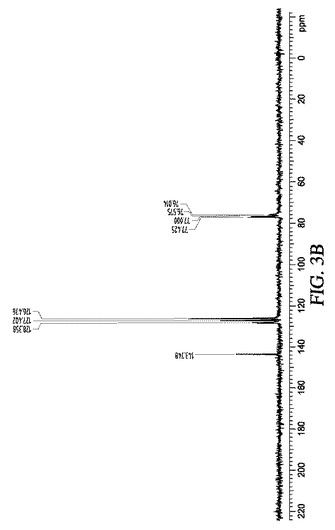
実施例7:リチウム−アルミナゲルを使用するケトンの還元:
【化4】
【0075】
5mLのTHF中に溶解させた1mmol(182mg)のベンゾフェノンを、0.5gの12% Li−AG及び固体状のプロトン源(例えばNH4Cl、(NH4)2HPO4)と共に、又は室温で濃青色の溶液となった後に氷酢酸を滴状添加しながら撹拌した。反応容器の着色が徐々に消失し、最終的には灰色になった。すなわちリチウムの完全な消費を示した。反応混合物を濾過し、固体をヘキサンで洗浄し、溶媒を真空下で混合有機溶液から除去し、1H、13C NMR及びESI−MSで確認し、ジフェニルメタノールに対応する白色固体を得た。図3Aから3Cを参照のこと。
【0076】
実施例8:RX、リチウム金属及びアルミナゲルの3つの成分による反応からの、RLi−AGの調製:
【0077】
2gの焼成アルミナゲル、202mgのリチウムリボン(5つの小片にカットした)をフラスコに添加し、ガラスコートしたマグネチックスターラーバー及びゴム隔壁を装着した。このフラスコを、クライオ浴(cryo bath)中で−10℃で10分間冷却した。このフラスコに、40mLのドライヘキサン、更にn−BuBrを200μL添加した。反応液を30分間撹拌し、それにより、リチウム片が輝くようになった。更に次の1時間でn−BuBrを500μL添加し、次の20分間でn−BuBrを更に800μL添加した。反応混合物を、冷却しながら、全てのリチウム及び有機誘導体がアルミナゲルに取り込まれるまで、更に3時間かけて撹拌した。この時点で反応を停止させ、低温に維持し、ヘキサン層を、n−BuLiに関して分析した。
【0078】
THF中に212.5mgのジフェニル酢酸を含有する溶液を、丸底フラスコに添加し、上記の反応混合物のヘキサン層中のn−BuLiを添加した。ジフェニル酢酸の色を変化させるのに、15.5mLのヘキサン溶液が必要であった(2mmolのn−BuLiに対応)。以上より、全ヘキサン層はn−BuLiを5.16mmolで含有し、固体アルミナゲルに吸収されたn−BuLiが8.84mmolであることになる。
【0079】
609mgのBuLiAGを1mmolのPhCOPh(182mg)と反応させたとき、1H NMRで解析した結果、所望の求核付加物が約90%で転換されていた。δ(ppm):7.2−7.5(m,10H),2.3(t,2H),1.4(m,4H),0.95(t,3H)(図4を参照)。
【化5】
【0080】
実施例9:エチルビニルエーテルの還元的切断による、ビニルリチウムの調製:
【化6】
【0081】
500mgのLi−AG(12%w/w)を、マグネチックスターラーバー及びゴム隔壁を装備した丸底フラスコ中で秤量した。このフラスコに、シリンジを介して、−40℃で、2mLのエチルビニルエーテルを添加した。−40℃で20分間、混合物を撹拌した後、3mLのエチルビニルエーテル中に1mmolのベンゾフェノンを含有する溶液を、反応混合物に滴状添加した。ベンゾケトイルラジカルアニオンの青色が維持されないように、添加速度を維持した(各添加の後、青色が消えるまで待つ)。全て添加するのに約1時間かかり、この時点の後、反応液の色が赤色に変化し始めた。反応液を、低温で更に1時間撹拌し、次に室温にし、その後、水でクエンチした。有機層をエーテルで抽出(複数回)し、乾燥させ、粗生成物として200mg(57%)を得た。
【0082】
粗製混合物の1H NMR(図5A)においても、54%の転換を示した。1H NMR(δ、ppm)のピークは以下のとおりである:2.27(br,1H),5.32(d,1H),5.33(d,1H),6.51(dd,1H),7.2−7.5(m,10H)。
【0083】
GC−MS(図5B)では、210の位置でのm/zピークに対応する求核付加物の形成が示された。
【0084】
CDCl3中の13C NMR(δ、ppm)(図5C)から、145.7、143.5、128.6、127.9、126.9、114、79.4の位置においてピークが示された。
【図1A】
【図1B】
【図1C】
【技術分野】
【0001】
本発明は多孔性金属酸化物粉(例えばアルミナゲル)と金属リチウムとの相互作用によって調製されるリチウム金属−多孔性酸化物組成物、並びに、有機リチウム及びリチウムアミド試薬の調製へのそれらの、in situでの使用、及び固体状で自由に流動する、取り扱いや貯蔵が容易な材料としての使用に関する。これらの組成物は貯蔵、出荷及び使用の際、可燃性溶媒及び/又は冷却条件を必要とせず、にもかかわらず、吸収されたリチウム及びリチウム試薬は、それらの反応性及び合成における有用性を保持している。
【背景技術】
【0002】
有機リチウム及びリチウムアミド化合物は、合成化学における変換の際に一般的に使用される重要な試薬である。有機リチウム試薬は伝統的に、微細に分散されたリチウム金属と有機ハロゲン化合物の溶液との低温での混合、又は金属−ハロゲン交換反応により調製される。リチウムアミドは最も一般的には、有機リチウム試薬を使用したアミンの脱プロトン化により調製される。しかしながら、有機リチウム化合物の安定性、貯蔵及び取扱いには課題が残されたままであり、そのことがしばしば有機合成へのそれらの使用(リチウムアミドの調製へのそれらの使用を含む)を困難にしている。
【0003】
本明細書で用いられ、また当技術分野で一般的に用いられる有機リチウムとは、炭素中心を有するアニオンのリチウム化合物のことを指す。有機リチウム試薬は、強い塩基であり、有効な求核種であり、またラジカル重合及びアニオン重合における有効な触媒であるため、それらは合成反応において有用である。しかしながらかかる試薬は非常に反応性が高く、しばしば空気の存在下で自然発火する。これらの危険をコントロールするため、それらは、炭化水素又はエーテル溶媒中の溶液としてのみ市販されている。これらの溶媒は有機リチウムの自然発火性を緩和できるが、それ自身揮発性かつ可燃性であるため、有機リチウム試薬の使用に関連する危険性を助長しうる。
【0004】
本発明で使用するリチウムアミドとは、第一級および第二級アミンのリチウム塩を意味する。リチウムアミド試薬は強塩基であり、一般の有機溶媒に自由に溶解し、非常に用途が広いため、合成的に有用である。しかしながらこれらの試薬は、非常に反応性が高く、取り扱いが困難である。若干の例外はあるが、それらは市販されておらず、それらの使用の直前に、第一級又は第二級アミンを有機リチウム試薬(例えばブチルリチウム)に添加することによって合成しなければならない。
【0005】
リチウム金属は有機リチウム試薬の生成に一般に使用されるものであり、その試薬は炭素原子とリチウム原子との間の直接的な結合を有する有機金属化合物である。リチウムの陽電気を帯びた性質により、結合中の大部分の電荷密度が炭素原子に配置されるため、カルボアニオン種が生じる。これにより、有機リチウム試薬が極めて強力な塩基及び求核試薬として作用しうる。有機リチウム試薬は典型的には、
R−X+2Li→R−Li+LiX
で表される、リチウム金属と有機ハロゲン化物との反応によって商業的に合成される(Weissその他、特許文献1:米国特許第5523447号、及びEmmelその他、特許文献2:米国特許出願公開第2006/0049379号を参照)。この合成の間に生じる副反応としては、特にヨウ化アルキルによるウルツ反応が挙げられる(Rがそれ自体でカップリングする)。この副反応は、低温、又はハロゲンとしての塩素若しくは臭素の使用によりほとんど排除できる。例えば、他の有機リチウム試薬の調製方法としては以下のものが挙げられる:
(i)有機ハロゲン化物とラジカルアニオンリチウム塩との反応、
(ii)有機ハロゲン化合物と有機リチウム種との間での金属−ハロゲン交換の実施(例えば、非特許文献1:Gilman,H.ら,J.Am.Chem.Soc.1932;54,1957)、
(iii)有機リチウム種と他の有機金属化合物との間での交換、
(iv)有機リチウム試薬による有機化合物の脱プロトン化、
(v)炭素−ヘテロ原子(例えば硫黄、酸素、リン又はケイ素)結合の還元的切断(例えば非特許文献2:Gilman,H.,ら,Org.Chem.1958;23,2044)、又は
(vi)DMSO中でのLiOH及びトルエンからのリチウム−水素交換によるベンジルリチウムの調製(特許文献3:Everettら、米国特許出願公開第2006/0170118号)。
【0006】
有機リチウム試薬、特にブチルリチウム(BuLi)、メチルリチウム(MeLi)、フェニルリチウム(PhLi)などは、製薬産業及び製造業における化学的成分及び強塩基として広く用いられている。リチウムアミドには関連用途も発見されている。例えばリチウムジイソプロピルアミド(LDA)及びリチウムヘキサメチルジシラジド(LiHMDS)は、両方とも、リチウムの強い配位能力によるエナンチオ選択的アルキル化を実施することもできる強塩基である(非特許文献3:Hilpert,H.Tetrahedron,2001,57,7675)。炭素中心を有する有機リチウム(例えばnBuLi)は、同時に強力な求核種及び強塩基であると考えられる(非特許文献4:Askin,D.;Wallace,M.A.;Vacca,J.P.;Reamer,R,A.;Volante,R.P.;Shinkai,I.J.Org.Chem.1992,57,2771)。これらの特徴により、アニオン重合の開始剤としての使用が可能となる(特許文献4:Hungenberg,Klaus−Dieter;Loth,Wolfgang;Knoll,Konrad;Janko,Lutz;Bandermann,Friedhelm.Method for producing statistical styrene−butadiene copolymers.国際公開第9936451A1号)。
【0007】
通常の有機リチウム及びリチウムアミド試薬の反応性又は選択性を変更することはほとんど不可能であるが、反応性または選択性の変更は、多くの化学工業、特に医薬調製および重合プロセス、において需要が高まっている。かかる化合物の両方を用いた従来の撹拌−バッチモードでの合成では、廃棄溶媒が顕著な量で生じるため、化学プロセスにおいては望ましくない。大規模な商業的合成においては、より環境に優しいプロセス(溶媒を含まない有機リチウム、リチウムアミド類又は充填層流通式反応器の設置)が理想的である。なぜなら、それにより廃棄溶媒の問題が減らされ、また長時間にわたる精製又はワークアップ(work−up)工程が省略できるからである。1つの解決法は、流動式の化学反応において使用でき、プロセスにおける試薬の効率及び効果をコントロールする、有機リチウム試薬の固体ソースの開発であろう。このアイデアを元に、近年、結晶質のグリニャール試薬(非特許文献5:Marcus,V.,ら,Angew Chem,Int.Ed.2000,39,3435)及び他の固体カルボアニオン源(非特許文献6:Davies,S.G.ら,J.Am.Chem.Soc.1977,4,135及び非特許文献7:Eaborn,C.,ら,J.Am.Chem.Soc.1994,116,12071)の開発がなされてきた。しかしながら、これらの結晶質の試薬の調製方法は通常時間がかかり、また特定のカルボアニオン系のみに限定されるため、それらの大規模スケールでの応用、またそれらの一般のカルボアニオン源としての応用が限定されたものとなる。グリニャール試薬(カルボアニオンドナーの最も一般的な種類)は更に、2当量のアルキル又はアリールマグネシウムハロゲン化物(2RMgX)と、各々1当量のジアルキル−又はジアリールマグネシウム化合物(RMgR)及びマグネシウムハライド塩(MgX2)との間におけるSchlenk平衡を受ける。この不均化反応は反応種を増加させ、この不均化反応は、場合によっては、カルボアニオン源としての使用に問題のある複雑化である。
【0008】
幾つかの公知の有機リチウム化合物(例えばブチルリチウム、メチルリチウム及びフェニルリチウム)は市販されている。市販されていない多くの有機リチウムは、金属−ハロゲン交換反応から調製する必要があり(総説として、非特許文献8:Wakefield,B.Organolithium Methods,Academic Press:London,1988を参照)交換反応においては1つの有機リチウム試薬と有機ハロゲン化物とを使用する。あるいは、純粋なリチウム金属と有機ハロゲン化物とを反応させて、有機リチウムを形成させてもよい。これらの変換は、有機ハロゲン化物、リチウム金属、リチウムハロゲン化物及び有機リチウム化合物との間での平衡反応を示す。純度の高い有機リチウム生成物を合成することは、未反応の有機ハロゲン化物がしばしば混入するため、困難であり、それにより大規模プロセスへの応用も困難となる。リチオ化反応は、脱プロトン反応によって、又はエーテル及びチオエーテルの還元的切断(非特許文献9:Schlosser,M.Organometallics in Syntheses;Wiley and Sons:Chichester,1994,47)、シャピロ方法(非特許文献10:Shapiro,R.H.Org.React.1976,23,405)、又はアレーンにより触媒されるリチオ化反応(非特許文献11:Yus,M.;Ramon,D.J.;J.Chem.Soc.,Chem.Commun.1991,398)により実施してもよい。これらの方法は有機リチウム自体を利用し、塩基として機能させることで開始するため、それらは合成の観点からは原子効率的ではない。また多くの工業において、微粉末化されたLi金属の高い反応性及び自然発火しやすい性質のため、ハロゲン化形態の標的有機基とリチウム金属とを直接反応させるアプローチは強く避けられる。還流炭化水素中につくられる分散リチウムも更に大きなスケールアップを困難にする(非特許文献12:Joshi,D.K.;Sutton,J.W.;Carver,S.;Blanchard,J.P.Org.Process Res.Dev.;2005;9(6);997−1002)。別法として、水銀−リチウム法(非特許文献13:Schollkopf,U.;Gerhart,F.Angew.Chem.Int.Ed.Engl.1981,20,795)、テルル−リチウム法(非特許文献14:Shiner,C.S.;Berks,A.H.;Fisher,A.M.J.Am.Chem.Soc.1988,110,957、非特許文献15:Hiiro,T.;Mogami,T.;Kambe,N.;Fujiwara,S−I;Sonoda,N.Synth.Commun.1990,20,703)、及びスズ−リチウム法(非特許文献16:Hoffmann,R.W.;Breitfelder,S.;Schlapbach,A.Helv.Chim.Acta 1996,79,346)による金属交換反応を実施してもよいが、水銀、テルル及びスズ化合物は通常有毒であり、これらの試薬を大規模な工業的プロセスに使用することは不適当である。
【0009】
本発明と異なるストラテジーにおいては、アルキルリチウム(MeLi、EtLi)を、無孔性の無機支持体(例えばSiO2、CaO又はAl2O3)の表面上へ吸着させることによって安定化させ、次にパラフィンワックスでコーティングする(特許文献5:Deberitzその他、米国特許第5149889号)。しかしながらこの場合、事前調製したアルキルリチウム試薬を使用しなければならない。更に、反応性を高めるため、ユーザはオイル、ワックス又は炭化水素を除去しなければならず、このことは望ましくない別の分離ステップを追加する。本発明との更なる大きな相違は、アルキルリチウムが無機支持体の表面上へ吸着され、支持体中には吸収されないという事実である。このストラテジーは、安定化され、かつ容易に使用きるアルキルリチウム試薬が、化学工業において要望されていることを物語るものともいえる。
【0010】
有機リチウム化合物と同様、リチウムアミドも商業的な入手可能性が限られている。リチウムと第一級若しくは第二級アミンからそれらを調製するのは困難であるが、最も便利には、第一級若しくは第二級アミンを有機リチウム試薬で処理することにより調製される。すなわち、リチウムアミドの使用は、それらが典型的に由来する有機リチウム試薬と同様の多数の限界に直面する。リチウムアミドの生成に使用する3つの最も一般的な有機リチウム試薬は、メチルリチウム、ブチルリチウム及びフェニルリチウムであり、それらは反応の間、それぞれメタン、ブタン及びベンゼンを発生させる。これらの全ての副産物は全て揮発性、可燃性の材料であるため、製造環境の悪化を生じさせる。メタン及びブタンは室温で可燃性の気体であり、大規模な製造における副産物としてのそれらの化学量論的な発生は、多くの問題を生じさせ、またコストを高めることにもなる。またベンゼンは有毒であり、発癌物質であることが周知である。
【先行技術文献】
【特許文献】
【0011】
【特許文献1】米国特許第5523447号
【特許文献2】米国特許出願公開第2006/0049379号
【特許文献3】米国特許出願公開第2006/0170118号
【特許文献4】国際公開第9936451A1号
【特許文献5】米国特許第5149889号
【非特許文献】
【0012】
【非特許文献1】Gilman,H.ら,J.Am.Chem.Soc.1932;54,1957
【非特許文献2】Gilman,H.,ら,Org.Chem.1958;23,2044
【非特許文献3】Hilpert,H.Tetrahedron,2001,57,7675
【非特許文献4】Askin,D.;Wallace,M.A.;Vacca,J.P.;Reamer,R,A.;Volante,R.P.;Shinkai,I.J.Org.Chem.1992,57,2771
【非特許文献5】Marcus,V.,ら,Angew Chem,Int.Ed.2000,39,3435
【非特許文献6】Davies,S.G.ら,J.Am.Chem.Soc.1977,4,135
【非特許文献7】Eaborn,C.,ら,J.Am.Chem.Soc.1994,116,12071
【非特許文献8】Wakefield,B.Organolithium Methods,Academic Press:London,1988
【非特許文献9】Schlosser,M.Organometallics in Syntheses;Wiley and Sons:Chichester,1994,47
【非特許文献10】Shapiro,R.H.Org.React.1976,23,405
【非特許文献11】Yus,M.;Ramon,D.J.;J.Chem.Soc.,Chem.Commun.1991,398
【非特許文献12】Joshi,D.K.;Sutton,J.W.;Carver,S.;Blanchard,J.P.Org.Process Res.Dev.;2005;9(6);997−1002
【非特許文献13】Schollkopf,U.;Gerhart,F.Angew.Chem.Int.Ed.Engl.1981,20,795
【非特許文献14】Shiner,C.S.;Berks,A.H.;Fisher,A.M.J.Am.Chem.Soc.1988,110,957
【非特許文献15】Hiiro,T.;Mogami,T.;Kambe,N.;Fujiwara,S−I;Sonoda,N.Synth.Commun.1990,20,703
【非特許文献16】Hoffmann,R.W.;Breitfelder,S.;Schlapbach,A.Helv.Chim.Acta 1996,79,346
【発明の概要】
【発明が解決しようとする課題】
【0013】
したがって、反応性の顕著な損失がなく、取扱、貯蔵、使用が容易であり、乾燥形態において入手可能な、有機リチウム及びリチウムアミド試薬に対するニーズが存在する。本発明はそのニーズに応えるものである。
【課題を解決するための手段】
【0014】
一実施形態では、本発明はリチウム金属/多孔性金属酸化物組成物に関する。これらのリチウム金属組成物は、液体リチウム金属を多孔性金属酸化物孔に吸収させるのに十分な発熱条件下で、不活性雰囲気において、液体リチウム金属と多孔性金属酸化物とを混合することにより調製される。本発明のリチウム金属/多孔性金属酸化物組成物は、約40重量%以下のリチウム金属を担持するのが好ましく、最も好ましくは約20重量%〜40重量%で担持する。
【0015】
他の実施形態では、本発明はまた、多孔性酸化物に吸収されたRLiを含有する、リチウム試薬−多孔性金属酸化物組成物に関する。上記の式RLiにおいて、Rはアルキル基、アルケニル基、アルキニル基、アリール基、アルカリール基又はNR1R2基であり、R1はアルキル基、アルケニル基、アルキニル基、アリール基、アルカリール基であり、R2は水素、アルキル基、アルケニル基、アルキニル基、アリール基、アルカリール基である。
【0016】
したがって、本発明はまた、多孔性金属酸化物(アルミナ(Al2O3、アルミナゲル)など)中に形成され、貯蔵されたリチウム、有機リチウム及びリチウムアミド種の新規な組成物の調製方法に関する。多孔性Al2O3に吸収されたリチウムを用いることにより、孔内部において有機リチウム及びリチウムアミド化合物を調製及び形成することができ、並びに、求核付加反応及び重合反応のためのカルボアニオンのin situ生成を可能とする。予備調製された有機リチウム化合物を、固体状の無機支持体(例えば二酸化ケイ素又はアルミナゲル)の孔に吸収させることができ、それにより、合成変換での使用のための溶媒フリーの貯蔵及び配送が可能となる。これらの新規な組成物は、バッチ反応器及び流通式反応器の両方における使用に適する、固体状の流動性の粉末である。
【図面の簡単な説明】
【0017】
【図1】図1A〜1Cは、実施例3の結果の代表的なスペクトルを示す。
【図2】図2は、実施例4の結果の代表的な1H NMRスペクトルの結果を示す。
【図3】図3A〜3Cは、実施例7の結果の、代表的な1H NMR、13C NMR及びESI−MSスペクトルを示す。
【図4】図4は、実施例8の結果の代表的な1H NMRスペクトルを示す。
【図5】図5A〜5Cは、実施例9の結果の代表的な1H NMR、GC−MS及び13C NMRスペクトルを示す。
【発明を実施するための形態】
【0018】
アルカリ金属、それらの同等物、並びにそれらの誘導体を簡便に利用する技術の開発は、学界、化学工業および水素生産業に求められ続けてきた。そのニーズに応えて、本発明は、リチウム金属を多孔性金属酸化物に吸収させることに関する。本発明の組成物で利用される多孔性金属酸化物は還元不可能な多孔性金属酸化物であり、特に多孔性アルミナが好ましい多孔性金属酸化物である。本発明はまた、リチウム試薬−多孔性金属酸化物組成物、多孔性金属酸化物に吸収された有機リチウム試薬及びリチウムアミドに関する。これらのリチウム試薬−多孔性金属酸化物組成物は、より多くの種類の利用しやすい求核試薬を提供し、取扱い及び貯蔵において現在知られているリチウム試薬を上回る大きな優位を有する。
【0019】
リチウム金属−多孔性金属酸化物組成物:
リチウム(Liの元素記号)は、周期表の1族に属するアルカリ金属である。それは全ての金属の中で最も軽く、水の半分の密度である。リチウムは軟らかい銀色の金属であり、一個の価電子を有するが、それは容易に失われて陽イオンになる。このため、リチウムは酸素又は窒素、特に水に曝露したときに燃焼し、激しく反応する。したがってこの金属は、非反応性の雰囲気中、又は非反応性の液体(例えば炭化水素又はナフサ)中に貯蔵する必要がある。リチウムはまた、1族の中でも、2族のアルカリ土類金属の特性を若干示すことが知られている。
【0020】
リチウム−多孔性金属酸化物組成物の調製においては、リチウム金属を多孔性金属酸化物(例えば多孔性アルミナゲル)と混合し、次にリチウム金属が溶解して金属酸化物の孔に吸収されるまで加熱するのが好ましい。これを行うための1つの方法としては、不活性雰囲気(例えばアルゴン又はヘリウム)下で、リチウム金属を、多孔性金属酸化物と混合する前に加熱することが挙げられる。あるいは、リチウム金属を固体として多孔性アルミナと混合し、その混合物を加熱してリチウム金属を溶解させてもよい。リチウム金属を吸収させる熱処理は、160〜325℃、好ましくは160〜225℃の間の温度で調節できる。リチウム金属を多孔性アルミナに取り込ませるための他の利用可能な方法としては、ゼオライトを用いた気相における反応が挙げられる(A.S.Ichimura,J.L.Dye,M.A.Camblor and L.A.Villaescusa,J.Am.Chem.Soc.,124,1170−1171(2002)、及びD.P.Wernette,A.S.Ichimura,S.A.Urbin and J.L.Dye,Chem.Mater.15,1441−1448,(2003)を参照のこと)。他の利用可能な方法として、リチウム金属を、金属−アンモニア溶液から多孔性アルミナ内に堆積させることが挙げられる(M.Makesya and K.Grala,Syn.Lett.1997,pp.267−268を参照のこと)。
【0021】
リチウム−多孔性金属酸化物組成物(例えば好適なリチウム−アルミナゲル(Li−AG))は、バルク若しくはシェービングされた(shaved)リチウム金属と、焼成多孔性金属酸化物(例えばアルミナ)との混合物を加熱して直接調製し、親金属の還元能力を大部分保持する、脆い黒色の粉末(loose black powders)を形成させてもよい。この反応は、好ましくは不活性雰囲気(例えばアルゴン又はヘリウム)下で実施する。リチウム−多孔性金属酸化物組成物はおそらく、吸収された中性のリチウム金属の小さいクラスタを多孔性金属酸化物孔に有し、並びに、電子が原子から非局在化した、イオン化リチウム金属(Li+)を孔の壁に有すると考えられる。上記材料は自然発火性であるが、空気中においては、微細粉末化された純粋なリチウム金属よりも反応性が低い。リチウム金属と多孔性アルミナゲルの加熱は、約160℃〜約325℃、好ましくは約160℃〜約225℃の温度範囲内で実施する。上記加熱は、リチウム金属の全てが吸収されるまで、あるいは一晩実施してもよい。
【0022】
リチウム金属は、アルミナゲル又は他の多孔性金属酸化物の内部で、非常に微細に分散すると考えられている。リチウム−アルミナゲルは、ほぼ完全に窒素(N2)ガスと反応できる。窒素ガスとリチウム−アルミナゲルとの反応により、アルミナの孔内部で、窒化リチウム(Li3N)の微粉末が分散される。このLi3Nが水と穏やかに反応すると、必要に応じて以下の反応(式中、AGはアルミナゲルを示す)によって高純度のアンモニアガスが発生する:
【数1】
従って、この材料はアンモニアを、その輸送、転送及び貯蔵における危険が回避される方法で発生させる。この純粋なアンモニアは、限定されないが化石燃料燃焼プロセスから発生する、窒素酸化物(NOx)を含有する煙道ガスの処理などの多くの用途に使用可能である。したがって、本発明のリチウム−多孔性金属酸化物組成物は不活性雰囲気からの窒素ガスの洗浄に使用できる。
【0023】
本発明のリチウム金属−多孔性金属酸化物組成は、約40重量%以下でリチウム金属を担持するのが好ましく、約10重量%〜20重量%で担持するのが最も好ましい。
【0024】
使用できる多孔性金属酸化物は、リチウム金属により還元されない、いかなる金属酸化物でもあってもよい。本発明において使用する、最も好ましい多孔性金属酸化物粉末は、多孔性アルミナ(また、アルミナゲル(特にγ−アルミナゲル)と呼ばれる)である。本発明に使用できる他の多孔性金属酸化物粉としては、リチウム金属により還元されないあらゆる遷移金属酸化物であり、例えば、多孔性酸化チタン(すなわちTiO、TiO2、Ti2O3、Ti3O5)、多孔性酸化カルシウム(CaO)、多孔性酸化ジルコニウム(すなわちZrO2)、多孔性酸化鉄(すなわちFe2O3又はFe3O4)、多孔性Co3O4、多孔性金属ホスフェート(MPO)、多孔性ハイブリッドホスホシリケート、多孔性アルミネート、多孔性アルミノシリケート、多孔性バナデート、モリブデート、などが挙げられる。シリカゲルは、二酸化ケイ素と純粋なリチウム金属との間の高い反応性ゆえに、予備調製された溶媒和有機リチウム試薬の吸収に使用できるだけである。多孔性を有することにより、これらの多孔性金属酸化物は、大量の物質を吸収して取り込むことができる。本発明の組成物は、生成したカルボアニオン又はアミド種に加えて、酸化物の孔に吸収されたリチウム金属を有する。多孔性アルミナは、多くの企業(例えばW.R.Grace&Co.社、又はAlmatis AC社)から購入できる。
【0025】
本発明の多孔性金属酸化物組成物において使用する多孔性金属酸化物は、好ましくは30Å〜500Åの平均孔径を有する。より好ましくは、上記平均孔径は60Å〜190Åである。
【0026】
多孔性金属酸化物は、購入したときは流動性の粉末であるが、それらは典型的にはガス状の物質(例えば水及び空気)を含有する。ゆえに、これらは、多孔性酸化物粉末とリチウム金属とを混合して本発明の組成物を調製する前に除去するのが好ましい。多孔性金属酸化物は、当業者に既知の方法を使用してガス除去することができる。例えば、ガス状物質を除去するため、多孔性金属酸化物を、真空の下で、排気可能なフラスコ中で(最初は温風ドライヤーで、次にトーチで)加熱してもよい。かかる加熱は、約300℃の温度で実施する。空気中で多孔性金属酸化物を600℃若しくはより高い温度(〜900℃)で加熱することにより、より容易にガスを除去し、活性部位を不動態化すること(焼成)も可能であり、実際上好ましい。多孔性金属酸化物は典型的には、本発明の組成物を調製する前に、乾燥した(また好ましくは不活性)雰囲気下で、室温に冷却される。
【0027】
リチウム試薬−多孔性金属酸化物組成物:
【0028】
本発明はまた、多孔性の金属酸化物又は非金属酸化物に吸収されたRLiを有する、リチウム試薬−多孔性金属酸化物組成物の提供に関する。上記の式RLiにおいて、Rはアルキル基、アルケニル基、アルキニル基、アリール基、アルカリール基又はNR1R2基であり、R1はアルキル基、アルケニル基、アルキニル基、アリール基、アルカリール基、XRn基(Xはヘテロ原子(例えばSi、S、Sn、Ge、P)であってもよく、nは整数である)、又はSi(R3)3基であり、R2は水素、アルキル基、アルケニル基、アルキニル基、アリール基、アルカリール基又はSi(R3)3基であり、R3はアルキル基、アルケニル基、アルキニル基、アリール基又はアルカリール基である。リチウム試薬−多孔性金属酸化物組成物は、リチウム金属−多孔性金属酸化物組成物と有機ハロゲン化物(RX)とを反応させることによって調製しても、リチウム金属と多孔性金属酸化物と有機ハロゲン化物(RX)とのin situでの混合によって調製しても、リチウム試薬(RLi)の多孔性金属酸化物への吸収によって調製しても、リチウム金属−多孔性酸化物組成物を第1級若しくは第2級アミン(HNR1R2)と反応させることによって調製してもよい。R、R1、R2及びR3基は、官能基によって更に置換されても、ヘテロ原子(例えばN、O、S、など)を更に含んでもよく、これらは、本発明のリチウム試薬−多孔性金属酸化物組成の形成を防止するような方法ではリチウムと反応しない。次に、R、R1、R2及びR3の好適かつ典型的な実施形態について説明する。
【0029】
本発明のリチウム試薬−多孔性金属酸化物組成物を調製する際、Li−多孔性金属酸化物組成物(上記の通りに調製)を、反応容器中で約0℃〜約−78℃に冷却してもよい。次に、有機ハロゲン化合物(又はR=R1R2Nのときは第一級又は第二級アミン(両方ともRX))を、Li−多孔性金属酸化物(純粋であるか又は溶媒中の物質)に徐々に添加する。有機ハロゲン化合物又はアミンが孔へ移動し、金属酸化物の孔内部で、リチウム金属と反応し、有機リチウム化合物が生成する。室温になるまで反応液を加熱した後、いかなる過剰の有機ハロゲン化合物、アミン、及び/又は溶媒も蒸発除去し、乾燥した、流動性の粉末状のLi試薬−多孔性金属酸化物組成物の形成を完了する。下記の実施例8に示すように、有機リチウム−多孔性金属酸化物組成物を、炭素−ヘテロ原子結合の還元的切断(例えばエチルビニルエーテルからのビニルリチウムの調製)により調製してもよい。
【0030】
リチウム試薬が有機リチウム化合物であるとき、使用できる有機ハロゲン化合物(RX)としては、限定されないがハロゲン化アルキル、ハロゲン化アルケニル、ハロゲン化アリール及びハロゲン化アルカリールなどが挙げられる。好ましいハロゲンは、塩化物、臭化物及びヨウ化物である。上記アルキル基、アルケニル基及びアルキニル基は、直鎖状若しくは分岐鎖状であってもよく、環状及び複素環式のアルキル基、アルケニル基又はアルキニル基であってもよい。上記アリール基及びアルカリール基は、ヘテロアリール基及びalk−ヘテロアリール基であってもよい。上記アルキル基は、好ましくはC1−C12アルキル基又はC5−C12シクロアルキル基であってもよい。上記アルケニル基は、好ましくはC2−C12アルケニル基又はC3−C12シクロアルケニル基であってもよい。上記アルキニル基は、好ましくはC2−C12アルキニル基又はC3−C12シクロアルキニル基であってもよい。上記アリール基及びアルカリール基は、ヘテロアリール基及びalk−ヘテロアリール基であってもよい。アルカリール基の「alk」部分は、本願明細書に記載のアルキル基、アルケニル基又はアルキニル基のいずれでもよい。
【0031】
好ましいアルキル基は、例えばメチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、t−ブチル基、イソブチル基、sec−ブチル基、シクロプロピル基、シクロブチル基、シクロヘキシル基、(シクロヘキシル)メチル基、シクロプロピルメチル基、並びに、n−ペンチル基、n−ヘキシル基、n−ヘプチル基、n−オクチル基の同族体及び異性体、などである。好ましいアルケニル基としては、エテニル基、アリル基、並びにブテニル基、ペンテニル基、ヘキセニル基、ヘプテニル基、オクテニル基、ノネニル基、デセニル基、シクロプロペニル基、シクロブテニル基、シクロペンテニル基、シクロペンタジエニル基、シクロヘキセニル基、シクロヘプテニル基、シクロヘプタジエニル基、シクロオクテニル基、シクロオクタジエニル基などの異性体が挙げられる。好ましいアルケニル基としては、アセチレニル基、並びにブチニル基、ペンチニル基、ヘキシニル基、ヘプチニル基、オクチニル基、ノニニル基、デシニル基の異性体が挙げられる。好ましいアリール基及びアルカリール基としては、フェニル基、ベンジル基、並びにトリル基、アニシル基、アナリニル基、ナフチル基、インデニル基、アントリル基、ピロリル基、ピリジル基、ピリミジル基、イミダゾリル基、ピラジニル基、オキサゾリル基、イソオキサゾリル基、チアゾリル基、フリル基、チエニル基、イミダゾリル基、ピラゾリル基、チオフェン基、N−アルキル化ピロール基、α−アルキル化ピリジン基、インドリル基、キノリニル基、イソキノリニル基などの異性体が挙げられる。
【0032】
RLiがR1R2NLiであるときに、Liアミド−アルミナゲル組成物の形成に用いる第一級若しくは第二級アミンとしては、限定されないがアルキルアミン、ジアルキルアミン、アルキルアリールアミン及びジアリールアミン並びに環状の第二級アミンなどが挙げられる。これらのアミン中の窒素と結合する有機基、又はシリル基Si(R3)3の場合にはR3は、Rに関して上記したものが挙げられる。好ましいアルキル基は、好ましくはメチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、トリメチルシリル基、トリエチルシリル基、t−ブチル基、イソブチル基、sec−ブチル基、並びに、例えばn−ペンチル基、n−ヘキシル基、n−ヘプチル基、n−オクチル基などの同族体及び異性体である。好ましいアリール基及びアルカリール基は、フェニル基、ベンジル基、並びにトリル基、アニシル基、アナリニル基、ナフチル基、インデニル基、アントリル基の異性体である。好ましい環状の第二級アミンは、ピロリジン、ピペリジン及び2,2,5,5−テトラメチルピペリジンである。
【0033】
リチウムと反応しないあらゆる不活性溶媒を、本発明のリチウム試薬組成物の調製に使用できる。例えば、ヘキサン又はヘプタンなどの非反応性の炭化水素、又はTHFなどの非反応性のエーテルが使用できる。通常THFが好適である。リチウム試薬−多孔性金属酸化物組成物を形成するとき、極性の高い溶媒は試薬RXと競合するため好ましくない。
【0034】
本発明の有機リチウム−多孔性金属酸化物組成物を生成する他の方法は、炭素−水素活性化を介する方法であり、すなわちそれは、有機リチウム−アルミナゲルが添加された試薬の炭素原子の脱プロトン化を行い、新規なカルボアニオン種を生成することを意味する。この有機リチウムの交換反応は、典型的には低い温度(例えば約23℃〜約−80℃)で実施する。新規な有機リチウム試薬が形成させた後、それは、利用できる基質との次の反応における、求核種又は塩基として使用できる。この反応は、バッチ反応器又は流通式反応器において実施できる。
【0035】
本発明の有機リチウム−多孔性金属酸化物組成物を生成する更に別の方法としては、in situで金属−ハロゲン(金属−元素)交換反応を行わせて、第1の有機リチウム−多孔性金属酸化物を、第2の有機リチウム−多孔性金属酸化物に変換することが挙げられる。1つの反応容器(バッチ反応容器又は流通式反応容器)に、有機ハロゲン化合物(RX)、又はC−Z結合を有するあらゆる活性化された分子種を添加し(Zはいかなるハロゲン化物又は酸性水素などであってもよい)、最初に、低い温度でTHF中で分散させ、次に、当該容器に有機リチウム−多孔性金属酸化物を添加することにより実施できる。冷却(すなわち約23℃〜約−80℃)したスラリーを一定時間十分に混合した後、上記の有機リチウムは、従来の方法で次に添加される試薬と反応可能になる。
【0036】
有機リチウム−アルミナゲルはまた、リチウムの切片、多孔性アルミナゲル及びハロゲン化アルキル溶液(好ましくは、例えば炭化水素又はエーテル中)を使用して、in situで反応させ、溶媒を蒸発させることにより調製できる。
【0037】
本発明の有機リチウム試薬−多孔性金属酸化物組成物はまた、低温条件下でシリカゲル又はアルミナゲルの孔に、溶媒中で予備調製した有機リチウム種を吸収させることにより調製できる。過剰の溶媒を次に留去し、粉末を乾燥真空する。生成物は自然発火性を有さず、それらは好ましくは有機リチウム−シリカゲル又は有機リチウム−アルミナゲルと呼ばれる。上記の方法では、低温条件(例えば約23℃〜約−80℃)下で、多孔性材料に有機リチウム種を接触させて有機リチウム種を多孔性金属酸化物材の孔に吸収させ、あらゆる過剰の溶媒を留去し、得られる有機リチウム材料を乾燥させる。
【0038】
得られる流動性の粉末、すなわちRLiが多孔性金属酸化物に吸収した有機リチウム−多孔性金属酸化物組成物は、特に、従来の有機リチウム化合物の全ての反応(例えば求電子物質に対する求核付加反応、重合反応の開始、及び塩基により触媒される反応)を行う。材料中にリチウム金属が残留しないとき、上記粉末は窒素雰囲気下でも安定である。残留リチウムの有無にかかわらず、この材料は、市販され、従来公知の、有機リチウム化合物の溶液(それらが貯蔵される溶媒中で凝集し、反応し、分解する傾向を有する)と比較し、室温における長期の貯蔵寿命を示す。
【0039】
本発明に係るリチウムアミド−多孔性金属酸化物を生成する他の方法は、有機リチウム−多孔性金属酸化物を用いて、第一級若しくは第二級アミンを非プロトン化することである。1つの反応容器(バッチ反応器又は流通式反応器)中に、第一級若しくは第二級アミン(好ましくは溶媒中に溶解)を、有機リチウム−多孔性金属酸化物の撹拌バッチに投入する。スラリーをよく混合し、反応させた後、あらゆる過剰のアミン及び/又は溶媒をデカントし、真空除去し、又は低温で蒸発させ、乾燥した、流動性を有するリチウムアミド−多孔性金属酸化物の粉末の形成を完成させる。
【0040】
リチウムアミド−多孔性金属酸化物組成物(R=R1R2N)(自由に流動する粉末)は、従来のリチウムアミド化合物に関して既知の反応をする。これらの反応としては、限定はされないが、アルコール及びカルボニル化合物(アルデヒド及びケトン)などの大部分の一般的な炭素酸の脱プロトンが挙げられる。リチウム金属が材料中に残留しないとき、上記粉末は窒素下でも安定である。残留リチウムの有無にかかわらず、この材料は、凝集し、徐々に分解する傾向がある市販若しくは従来公知のリチウムアミド化合物と比較し、室温で長期にわたる貯蔵寿命を示す。これらのリチウムアミドを調製するための一般的な方法は、アルキルリチウム種の、対応するアミンとの反応(例えばDetlefら、国際公開第03033505号)、金属の融点より高い温度で、リチウムと、アミンとの混合物の加熱(Chiuら、米国特許第5420322号)、又は少量の電子担体としてのイソプレンとの混合物の加熱(Corellaら、米国特許第6169203号)を含む。
【0041】
上記のように、リチウム−多孔性金属酸化物組成物(例えばリチウム−アルミナゲル組成物)は、本発明の対応するリチウム試薬組成物(例えば対応する有機リチウム−アルミナゲル又はリチウムアミド−アルミナゲル組成物)の生成の際、開始成分として使用できる。かかる対応する組成物の調製に関する更なる具体例は、以下のとおりである:リチウム−アルミナゲルを約が−80℃〜約0℃の温度範囲で冷却し、数分間撹拌する。撹拌しながら、リチウム−アルミナゲルに、有機ハロゲン化合物(純粋な形態又は非反応性の炭化水素又はエーテル溶媒(例えばテトラヒドロフラン(THF))中の溶液)を徐々に添加する。反応混合物を、低温で数時間撹拌する。あらゆる過剰な未反応有機ハロゲン化合物又は溶媒を、真空下で蒸留させ、一方で系の温度を室温まで上昇させる。最終的な有機リチウム−アルミナゲル組成物の強度(1グラムあたりの反応当量)を、別の反応系において、求電子種への求核付加反応を実施させることにより測定する。臭化ブチルの場合、上記反応は以下のとおりと考えられる:
【数2】
LiBr(3.464g/cm3)及びBuLi(0.67g/cm3)の純粋な化合物密度を使用すると、生成物のmolあたりの体積は、LiBrの場合は25.1cm3で、BuLiの場合は95.5cm3で、合計120.6cm3/molである。アルミナゲルの空隙の体積は、水銀ポロシメーターにおいて1.56cm3/gである。生成物が、体積を増加させることなく孔にフィットしたと仮定した場合、8.0gのアルミナゲルのサンプルでは、空隙部は最高12.5cm3である。すなわち、8.0gのAGにより、12.5/120.6=0.110molの反応(1.53gのLiを必要とする)が可能となる。これは、(1.53/9.53)×100=16.0重量%のLiの担持に対応する。ゆえに、BuLiの担持は、0.110/9.53=0.0115mol/(リチウム−アルミナゲル1g)又は11.5mmol/(リチウム−アルミナゲル1g)である。
【0042】
2つの例示的な系における有機リチウム−アルミナゲル、ブチルリチウム−アルミナゲル及びメチルリチウム−アルミナゲルの最大の担持量を、使用するリチウム−アルミナゲルの合計量を10gとして、下記の表1に示す:
【0043】
【表1】
【0044】
リチウム金属の担持量は、実際に使用する多孔性金属酸化物の孔径及び孔密度に依る。典型的には、リチウム金属は最高約40重量%で、本発明の組成物に存在してもよい。上記の材料を典型的な還元反応及びin situでのリチオ化反応に用いる場合、好ましくは、上記金属の量は20重量%〜40重量%の範囲である。有機リチウム−多孔性金属酸化物及びリチウムアミド−アルミナゲル組成物を生成させる際の好ましい担持量は、10重量%〜20重量%の範囲のリチウムである。本発明のリチウム−多孔性金属酸化物組成物において、約40重量%超で担持すると、多孔性金属酸化物の表面上に若干のフリーの金属が残留することがありえる。
【0045】
有機金属リチウム試薬を安定させることが知られている、様々な添加剤を、リチウム試薬−多孔性金属酸化物と共に使用してもよい。例えば限定されないが、テトラメチルエチレンジアミン(TMEDA)、ジアザビシクロウンデカン(DBU)、スパルテイン(Tsumakiら,米国特許第6024897号を参照)などの安定化剤を更に添加して、これらの固体状カルボアニオンの安定性を増加させてもよい。例えば、安定化された有機リチウム種を、合成反応において添加剤としてテトラメチルエチレンジアミン(TMEDA)を用いて調製できる。典型的には、リチウム2molごとに、有機ハロゲン化合物を1mol及びTMEDAを1mol用いて、有機リチウム−TMEDA−多孔性金属酸化物を調製する。TMEDAの分子構造は、1,2−ジアミノエタン1分子中の、2つの各々のアミン窒素原子上に、2つのメチル基が結合してアルキル化された構造に対応する。TMEDA又は他の第三級アミン安定化剤は、リチウムカチオンと第三級窒素との配位結合によってこれらの試薬の安定化を助ける添加剤として使用できる。潜在的に、このイオン錯体形成はリチウムカチオンの有効半径を拡大し、求電子性を低下させ、それをカルボアニオン部から分離させ、それによりアルキルカルボアニオンからのβヒドリドを脱離させる能力が制限される。この安定化態様はまた、外部から添加された試薬との反応に対するカルボアニオンの有効性を強化するとも考えられる。
【0046】
リチウム−及びリチウム試薬−多孔性金属酸化物組成物の使用:
【0047】
本発明のリチウム−多孔性金属酸化物組成物及びリチウム試薬−多孔性金属酸化物組成物は、リチウム金属、有機リチウム試薬及びリチウムアミド試薬と同様の反応に使用できる。かかる反応としては、限定されないが求核付加反応、重合反応及び塩基により触媒される反応などが挙げられる。表2は、かかる反応の典型的なリストを示す。しかしながら本発明の組成物はフリーの流動性の粉末であり、貯蔵寿命が長いという効果を発揮する。
【0048】
【表2−1】
【0049】
【表2−2】
【0050】
更に、固体試薬としての、本発明のリチウム−多孔性金属酸化物組成物及びリチウム試薬−多孔性金属酸化物組成物は、幾つかの方法による反応に使用できる。最も単純な様式は、固体試薬を、1つ以上の基質を含有する溶液と共にスラリー中で撹拌する、従来公知のバッチ反応である。ここで、市販の固体状のリチウムの調製、又は有機リチウム若しくはリチウムアミド試薬の溶液の調製に勝る根本的な利点としては、
(a)固体の取扱の容易さ、及び
(b)反応、クエンチング及びワークアップ手順における、有機溶媒の使用の最小化などが挙げられる。好ましいケースでは、固体状の酸がスラリー中に含有されるとき、中性化生成物のみを含有する有機溶液の直接単離が、固体酸化物ゲル(例えばアルミナ又は二酸化ケイ素)のイオン副産物に対する親和性によって促進され、別の有機/水性分配や、洗浄工程により除去しなくてもよい。
【0051】
第2の使用態様は、流通式反応器(例えばカラム流通式反応器)におけるプロセスである。そのプロセスでは、反応基質の溶液を、固体状のリチウムアルミナゲル、有機リチウム−固体状酸化物又はリチウムアミド−固体状酸化物試薬の充填層にパーコレートさせる。流通式反応器又は連続流通式反応器を用い、新しい反応溶液を反応器中の固体状試薬に連続的に添加する。反応生成物を連続的に取り除き、一方で、廃棄物を支持試薬に結合させたままにするか、又は所望の生成物と共に溶出させる。流通式反応器を使用することによる利点は非常に大きい。例えば、当該反応器の使用により、全ての試薬の効率的な利用が確保され、その反応能力が完全に消耗されたときにのみ、反応が終了する。その結果、従来のバッチ反応と比較し、ゲル中及び溶媒中においても、生産性が高く、効率的な方法となる。流通式反応器の例としては、固定床流通式反応器が挙げられる。当該反応器では、反応基質の溶液を、固体試薬(例えば有機リチウム−アルミナゲル)のカラムを通してパーコレートし、カラムの出口で生成物溶液を直接回収する。実質的にいかなるタイプの反応プロセス及び反応器も、本願明細書に記載の反応に使用できるが、流通式反応器(例えば固定床流通式カラム反応器)が、本発明の反応に好ましい反応器のタイプである。
【0052】
溶媒の選択及び有機部分の性質に応じて、リチウム−アルミナゲルと、有機ハロゲン化合物若しくは有機アミンの反応を経て形成される有機リチウム又はリチウムアミド種は、抽出されるのに十分な可溶性を有し得るため、純粋な有機リチウム又はリチウムアミド試薬の新しく調製した溶液を容易に入手できる。あるいは、目的の有機リチウム種が特に不安定若しくは短寿命であると考えられるとき、その求電子パートナー(例えばプロトン源、ケトンなど)を充填カラムの層内に固体状態で直接含有させ、カラム出口で再び、クエンチされた純粋な形態の中性生成物として調製してもよい。
【0053】
あるいは、経時的な反応方式を採用してもよい。当該反応では、in situでリチウム−アルミナゲルから形成された、固体酸化物に結合した有機リチウム又はリチウムアミドを更に、同じ若しくは異なる溶媒中の基質溶液と反応させ、生成物を形成され、更にカラムから溶出させる。すなわちこの方法は、以下の点で有利である:
(a)リチウムの反応性を効率的に使用できること、
(b)溶出された生成物がリチウム金属を含有しないため、安全であること、
(c)不安定な有機リチウム種の中間体が必要となる用途に利用できること、及び
(d)支持された反応物質の連続的な形成及びそれによる反応、更に極性/イオン性副産物及び未反応リチウム金属からそれらの溶出が可能であること。
【実施例】
【0054】
実施例1:LiAG(10%(w/w))の調製
【0055】
Heグローブボックス中で、3.16gのリチウムリボン(1.425g/フィート)を計量し、小片にカットし、28.4gのアルミナゲル(AG)で完全にコーティングし、スチール製の反応器に添加した。スチール製の反応器を、100℃で6時間、165℃で一晩、182℃で8時間、及び190℃で一晩加熱した。スチール製の反応器を冷却し、グローブボックスに入れた。76.7mgの代表的な小さいサンプルを用いて、水によるH2発生に関して試験した。回収されたH2の量は、Liの9.2%当量であった。多孔性アルミナゲルに対するリチウム金属の化学量論比率を変化させて、リチウムの担持量が5重量%〜40重量%の範囲になるように調製した。
【0056】
実施例2:リチウム−アルミナゲル中で安定化された、有機リチウム種の調製:
【数3】
式中、RXは脂肪族(メチル基、ブチル基など)、芳香族(フェニル基)、ベンジル基、アリル基、ホモアリル基、プロパルギル基、sec−アルキル基、tert−アルキル基又はネオペンチル基のハロゲン化物(塩化物、臭化物又はヨウ化物)であってもよい。
【0057】
γ−Al2O3中の10% Li 1g(14mmolのLi)を丸底フラスコに添加し、ドライアイス−アセトン浴で−78℃で冷却し、次にスラリーを撹拌しながら、ヨウ化メチル(2mL)(純粋)を滴状添加した。冷却しながら、反応液を2時間撹拌した。添加が完了し、混合物が室温に達した後、過剰なヨウ化メチルを蒸発させた。この調製の間、少量のエタン及びI2が生じたが、このウルツカップリングによる生成物は、低温を長時間維持することにより最小化できる。アルミナ中のMeLiの強度は、過剰なベンゾフェノンによる求核付加反応により測定した。幾つかの反応について測定した結果、アルミナ中のMeLi(10〜25%のLi/γ−Al2O3から調製)の担持量が3〜7mmol/gの間であった。有機リチウム及びベンゾフェノンの化学量論反応の場合、求核付加反応の生成物の収率は75%〜90%の範囲であり、またオルトリチオ化反応生成物が微量で存在した。
【0058】
PhCH2Li、n−BuLi、アリルリチウム及びホモアリルリチウムを、同じ手順を用いて調製し、ベンゾフェノンとの反応に関して試験した。
【化1】
【0059】
実施例3:TMEDA安定化剤を用いた有機リチウム−アルミナゲルの調製:
【0060】
2gのLi−AG(25%重量のLi)を含むフラスコ中に、20mLのペンタン中に2gのTMEDA、3gのn−ブチルブロミドを含有する混合液を精製した後添加し、−80℃で3時間撹拌し、ドライアイス入りの−40℃浴に移し、更に反応液が室温に達するまで一晩撹拌した。溶媒を真空下で除去し、自由に流動する灰色の粉末を得た。この材料を200mg用い、1.1mmol(200mg)のPhCOPhと反応させ、60%の収率でブチルジフェニルアルコールを得た。図1Aは、ブチルジフェニルアルコール生成物の1H NMRを示す。
【0061】
また、ブチルアニオンの存在を、この材料と水とのガス発生反応を行わせ、GC−MSでガス分析することにより確認した。以下の反応に従うブタン(MW=58)の形成が示された。図1Aから1Bを参照。
【数4】
【0062】
実施例4:有機リチウム−多孔性金属酸化物組成物の生成及びケトンに対するOne−Potでの求核付加反応
【化2】
【0063】
1.5mmolのホモアリルブロミド(195mg)を、5mLのドライ蒸留THFに溶解させ、この溶液を−78℃に冷却した。この冷却溶液に、0.5gのγ−Al2O3中のLi(8〜12%w/w、7〜10mmolのLi)を、15分間にわたり、固体添加用のチューブを用いて添加した。同じ低温条件下でスラリーを30分間撹拌した。低温スラリーを撹拌しながら、3mLのTHF中に溶解させた1mmolのベンゾフェノン(182mg)を、15分間にわたり添加した。反応液を徐々に(6時間)室温に加温し、冷水でクエンチし、更にEtOAcで抽出した。有機層を窒素雰囲気下で蒸発させ、油状の残渣200mg(85%)を得た。
【0064】
図2は、以下のピークを有する、生成物である1H ホモアリルジフェニルメチルケトンの1H NMR(δ、ppm)を示す:7.2−7.8(m,10H),5.8−6.0(m,1H),5.0−5.2(m,2H),2.4−2.6(m,2H),2.05−2.15(m,2H)。
【0065】
実施例5:アルミナゲル中のリチウムと、第二級アミンとの反応による、有機リチウム(リチウムアミド)の生成:
【0066】
1gのリチウム−アルミナゲル(25重量%のLi担持量)を、−60℃でジメチルアミン(〜4mL)と反応させ、濃青色の溶液を得た。徐々に室温に加温すると、この溶液混合物は、水素ガスの形成に起因すると思われる泡を立て始めた。更に室温で1時間静置したところ、溶液の青色が完全に消失した。過剰な未反応のジメチルアミンを、真空下で蒸発させ、得られる固体を秤量した。1.35gのMe2NLi−アルミナゲルを得た(使用したLiの量に対して〜40%の収率)。
【数5】
【0067】
比較例1:リチウム金属と第二級アミンとの反応から、リチウムジメチルアミドを生成させる試み:
【0068】
調製直後の微粉末化したリチウム金属30mgを、真空ストッパーを備えた丸底フラスコに添加し、そこでMe2NHを濃縮した。反応容器を最初の数分、−60℃に維持し、徐々に室温に加温し、その温度で圧力計を用いて一晩維持した。Me2NHの呈色もH2ガス発生も観察されなかった。
【0069】
比較例2:アルミナゲルの存在下での、リチウム金属と第二級アミンとの反応からリチウムジメチルアミドを生成させる試み:
【0070】
30mgの多孔性アルミナゲルを、真空ストッパーを備えているフラスコ中で、20mgの微粉末化したリチウム金属と混合し、この混合物にMe2NHを濃縮した。反応容器を最初に低温に維持し、次に徐々に室温に加温し、その状態で2時間維持した。Me2NHの呈色もH2ガス発生も観察されなかった。
【0071】
比較例1及び2により、リチウム金属は、単独でもアルミナの存在下でも、ジメチルアミンからのリチウムアミド形成を行わないことが証明された。
【0072】
実施例6:カラム中での、有機リチウムの連続的流動反応:
【化3】
【0073】
5mLのTHF中に溶解させた1mmol(182mg)のベンゾフェノンを、1時間にわたり、MeLi/アルミナ(5mM、2g)を充填し、冷却ジャケットを装備したカラム(−40℃)中を通過させた。カラムを過剰量のTHFで洗浄し、回収した生成物を冷水中でクエンチし、更にEtOAcで抽出し、有機層を蒸発させ、92%の収率の粗製(オルトリチオ化反応生成物を若干含む)の油状の液体(185mg)を得た。MeOHで溶出させることによる逆相(C18)カラムクロマトグラフィによって、純粋なアルコールを単離した(73%)。
【0074】
実施例7:リチウム−アルミナゲルを使用するケトンの還元:
【化4】
【0075】
5mLのTHF中に溶解させた1mmol(182mg)のベンゾフェノンを、0.5gの12% Li−AG及び固体状のプロトン源(例えばNH4Cl、(NH4)2HPO4)と共に、又は室温で濃青色の溶液となった後に氷酢酸を滴状添加しながら撹拌した。反応容器の着色が徐々に消失し、最終的には灰色になった。すなわちリチウムの完全な消費を示した。反応混合物を濾過し、固体をヘキサンで洗浄し、溶媒を真空下で混合有機溶液から除去し、1H、13C NMR及びESI−MSで確認し、ジフェニルメタノールに対応する白色固体を得た。図3Aから3Cを参照のこと。
【0076】
実施例8:RX、リチウム金属及びアルミナゲルの3つの成分による反応からの、RLi−AGの調製:
【0077】
2gの焼成アルミナゲル、202mgのリチウムリボン(5つの小片にカットした)をフラスコに添加し、ガラスコートしたマグネチックスターラーバー及びゴム隔壁を装着した。このフラスコを、クライオ浴(cryo bath)中で−10℃で10分間冷却した。このフラスコに、40mLのドライヘキサン、更にn−BuBrを200μL添加した。反応液を30分間撹拌し、それにより、リチウム片が輝くようになった。更に次の1時間でn−BuBrを500μL添加し、次の20分間でn−BuBrを更に800μL添加した。反応混合物を、冷却しながら、全てのリチウム及び有機誘導体がアルミナゲルに取り込まれるまで、更に3時間かけて撹拌した。この時点で反応を停止させ、低温に維持し、ヘキサン層を、n−BuLiに関して分析した。
【0078】
THF中に212.5mgのジフェニル酢酸を含有する溶液を、丸底フラスコに添加し、上記の反応混合物のヘキサン層中のn−BuLiを添加した。ジフェニル酢酸の色を変化させるのに、15.5mLのヘキサン溶液が必要であった(2mmolのn−BuLiに対応)。以上より、全ヘキサン層はn−BuLiを5.16mmolで含有し、固体アルミナゲルに吸収されたn−BuLiが8.84mmolであることになる。
【0079】
609mgのBuLiAGを1mmolのPhCOPh(182mg)と反応させたとき、1H NMRで解析した結果、所望の求核付加物が約90%で転換されていた。δ(ppm):7.2−7.5(m,10H),2.3(t,2H),1.4(m,4H),0.95(t,3H)(図4を参照)。
【化5】
【0080】
実施例9:エチルビニルエーテルの還元的切断による、ビニルリチウムの調製:
【化6】
【0081】
500mgのLi−AG(12%w/w)を、マグネチックスターラーバー及びゴム隔壁を装備した丸底フラスコ中で秤量した。このフラスコに、シリンジを介して、−40℃で、2mLのエチルビニルエーテルを添加した。−40℃で20分間、混合物を撹拌した後、3mLのエチルビニルエーテル中に1mmolのベンゾフェノンを含有する溶液を、反応混合物に滴状添加した。ベンゾケトイルラジカルアニオンの青色が維持されないように、添加速度を維持した(各添加の後、青色が消えるまで待つ)。全て添加するのに約1時間かかり、この時点の後、反応液の色が赤色に変化し始めた。反応液を、低温で更に1時間撹拌し、次に室温にし、その後、水でクエンチした。有機層をエーテルで抽出(複数回)し、乾燥させ、粗生成物として200mg(57%)を得た。
【0082】
粗製混合物の1H NMR(図5A)においても、54%の転換を示した。1H NMR(δ、ppm)のピークは以下のとおりである:2.27(br,1H),5.32(d,1H),5.33(d,1H),6.51(dd,1H),7.2−7.5(m,10H)。
【0083】
GC−MS(図5B)では、210の位置でのm/zピークに対応する求核付加物の形成が示された。
【0084】
CDCl3中の13C NMR(δ、ppm)(図5C)から、145.7、143.5、128.6、127.9、126.9、114、79.4の位置においてピークが示された。
【図1A】
【図1B】
【図1C】
【特許請求の範囲】
【請求項1】
液体リチウム金属を多孔性金属酸化物孔に吸収させるのに十分な発熱条件下で、不活性雰囲気において、液体リチウム金属と多孔性金属酸化物とを混合するステップを包含する方法により調製される、リチウム金属/多孔性金属酸化物組成物。
【請求項2】
前記リチウム金属が、最高約40重量%で担持され、
前記多孔性金属酸化物の孔が、30Å〜500Åの平均孔径を有し、
前記多孔性金属酸化物が、多孔性アルミナ、多孔性酸化チタン、多孔性酸化カルシウム、多孔性酸化ジルコニウム、多孔性酸化鉄、多孔性Co3O4、多孔性金属ホスフェート、多孔性ハイブリッドホスホシリケート、多孔性アルミネート、多孔性バナデート及びモリブデートから選択される、請求項1記載のリチウム金属/多孔性金属酸化物組成物。
【請求項3】
前記リチウム金属が約20重量%〜40重量%で担持されている、請求項2記載のリチウム金属/多孔性金属酸化物組成物。
【請求項4】
前記多孔性金属酸化物がアルミナである、請求項2記載のリチウム金属/多孔性金属酸化物組成物。
【請求項5】
前記多孔性金属酸化物の孔が60Å〜190Åの平均孔径を有する、請求項2記載のリチウム金属/多孔性金属酸化物組成物。
【請求項6】
多孔性酸化物に吸収されたRLiを含有するリチウム試薬−多孔性金属酸化物組成物
(式中、Rは、アルキル基、アルケニル基、アルキニル基、アリール基、アルカリール基又はNR1R2基であり、
R1はアルキル基、アルケニル基、アルキニル基、アリール基、アルカリール基又はSi(R3)3基であり、
R2は水素、アルキル基、アルケニル基、アルキニル基、アリール基、アルカリール基又はSi(R3)3基であり、
R3はアルキル基、アルケニル基、アルキニル基、アリール基又はアルカリール基である)。
【請求項7】
Rがメチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、t−ブチル基、イソブチル基、sec−ブチル基、エテニル基、アリル基、シクロペンチル基、シクロヘキシル基、フェニル基及びベンジル基からなる群から選択される、請求項6記載のリチウム試薬−多孔性金属酸化物組成物。
【請求項8】
RがR1R2Nであり、
R1がメチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、t−ブチル基、イソブチル基、sec−ブチル基、エテニル基、アリル基、シクロペンチル基、シクロヘキシル基、フェニル基及びベンジル基からなる群から選択され、
R2が水素、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、t−ブチル基、イソブチル基、sec−ブチル基、エテニル基、アリル基、シクロペンチル基、シクロヘキシル基、フェニル基及びベンジル基からなる群から選択される、請求項6記載のリチウム試薬−多孔性金属酸化物組成物。
【請求項9】
液体リチウム金属を多孔性金属酸化物に吸収させるのに十分な発熱条件下で、不活性雰囲気において、液体リチウム金属と多孔性金属酸化物とを混合するステップと、
得られるリチウム金属/多孔性金属酸化物組成物を、約−80℃〜0℃の温度で化合物RXと反応させるステップと、を包含する、リチウム試薬−多孔性金属酸化物組成物の調製方法
(式中、Rはアルキル基、アルケニル基、アルキニル基、アリール基、アルカリール基又はNR1R2基であり、
R1はアルキル基、アルケニル基、アルキニル基、アリール基、アルカリール基又はSi(R3)3基であり、
R2は水素、アルキル基、アルケニル基、アルキニル基、アリール基、アルカリール基又はSi(R3)3基であり、
R3はアルキル基、アルケニル基、アルキニル基、アリール基又はアルカリール基であり、
Xはハロゲンである)。
【請求項10】
前記リチウム金属が、最高約40重量%で担持され、
前記多孔性金属酸化物の孔が、30Å〜500Åの平均孔径を有し、
前記多孔性金属酸化物が、多孔性アルミナ、多孔性酸化チタン、多孔性酸化カルシウム、多孔性酸化ジルコニウム、多孔性酸化鉄、多孔性Co3O4、多孔性金属ホスフェート、多孔性ハイブリッドホスホシリケート、多孔性アルミネート、多孔性バナデート及びモリブデートから選択される、請求項9記載のリチウム試薬−多孔性金属酸化物組成物の調製方法。
【請求項11】
前記リチウム金属が、約20重量%〜40重量%で担持されている、請求項9記載のリチウム試薬−多孔性金属酸化物組成物の調製方法。
【請求項12】
前記多孔性金属酸化物がアルミナである、請求項9記載のリチウム試薬−多孔性金属酸化物組成物の調製方法。
【請求項13】
前記多孔性金属酸化物の孔が、60Å〜190Åの平均孔径を有する、請求項9記載のリチウム試薬−多孔性金属酸化物組成物の調製方法。
【請求項14】
リチウムを多孔性金属酸化物の孔に吸収させるのに十分な条件下で、溶媒の存在下で、リチウム金属、多孔性金属酸化物及びハロゲン化アルキル溶液を混合するステップと、
いかなる過剰の溶媒も蒸発させるステップと、を包含する、有機リチウム−多孔性金属酸化物組成物の調製方法。
【請求項15】
前記リチウム金属が、最高約40重量%で担持され、
前記多孔性金属酸化物の孔が、30Å〜500Åの平均孔径を有し、
前記多孔性金属酸化物が、多孔性アルミナ、多孔性酸化チタン、多孔性酸化カルシウム、多孔性酸化ジルコニウム、多孔性酸化鉄、多孔性Co3O4、多孔性金属ホスフェート、多孔性ハイブリッドホスホシリケート、多孔性アルミネート、多孔性バナデート及びモリブデートから選択される、請求項14記載の有機リチウム−多孔性金属酸化物組成物の調製方法。
【請求項16】
前記リチウム金属が、約20重量%〜40重量%で担持されている、請求項14記載の有機リチウム−多孔性金属酸化物組成物の調製方法。
【請求項17】
前記多孔性金属酸化物がアルミナである、請求項14記載の有機リチウム−多孔性金属酸化物組成物の調製方法。
【請求項18】
前記多孔性金属酸化物の孔が、60Å〜190Åの平均孔径を有する、請求項14記載の有機リチウム−多孔性金属酸化物組成物の調製方法。
【請求項19】
低温条件下で多孔性材料と有機リチウム種とを接触させるステップであって、
前記多孔性材料が多孔性シリカゲル及び多孔性金属酸化物からなる群から選択されるステップと、
いかなる過剰の溶媒も蒸発させるステップと、得られる有機リチウム材を乾燥させるステップと、を包含する、有機リチウム種を多孔性材料の孔に吸収させる方法。
【請求項20】
請求項6記載のリチウム試薬−多孔性金属酸化物組成物の使用を包含する、有機リチウム試薬又はリチウムアミドを用いた化学反応。
【請求項21】
前記化学反応が、求核付加反応、重合反応及び塩基触媒反応からなる群から選択される、請求項20記載の化学反応。
【請求項22】
前記化学反応が、炭素中心求電子種に対する求核付加反応、有機クプレート又はギルマンの試薬の調製、適当な求電子物質からの有機リン化合物、有機硫黄化合物、有機ホウ素化合物、有機スズ化合物の調製、リチウムアミドの調製、配向的オルトリチオ化反応及びそれに続く求電子種によるクエンチング、エノラートの調製又は他の脱プロトン化、アニオン重合の開始、ウィッティヒ反応におけるイリドの生成、カービンの生成、アイソトープ標識、シャピロ反応、非求核塩基反応、ハロゲン−金属置換又はC−O、C−S又はC−Pの還元的切断による有機リチウム化合物の調製、カルボニル化合物又は芳香族化合物の還元(Birch還元)及び反応性中間体の生成、からなる群から選択される、請求項20記載の化学反応。
【請求項23】
化学反応が従来のバッチ反応において、又はプロセスフロー反応装置において生じる、請求項20記載の化学反応。
【請求項1】
液体リチウム金属を多孔性金属酸化物孔に吸収させるのに十分な発熱条件下で、不活性雰囲気において、液体リチウム金属と多孔性金属酸化物とを混合するステップを包含する方法により調製される、リチウム金属/多孔性金属酸化物組成物。
【請求項2】
前記リチウム金属が、最高約40重量%で担持され、
前記多孔性金属酸化物の孔が、30Å〜500Åの平均孔径を有し、
前記多孔性金属酸化物が、多孔性アルミナ、多孔性酸化チタン、多孔性酸化カルシウム、多孔性酸化ジルコニウム、多孔性酸化鉄、多孔性Co3O4、多孔性金属ホスフェート、多孔性ハイブリッドホスホシリケート、多孔性アルミネート、多孔性バナデート及びモリブデートから選択される、請求項1記載のリチウム金属/多孔性金属酸化物組成物。
【請求項3】
前記リチウム金属が約20重量%〜40重量%で担持されている、請求項2記載のリチウム金属/多孔性金属酸化物組成物。
【請求項4】
前記多孔性金属酸化物がアルミナである、請求項2記載のリチウム金属/多孔性金属酸化物組成物。
【請求項5】
前記多孔性金属酸化物の孔が60Å〜190Åの平均孔径を有する、請求項2記載のリチウム金属/多孔性金属酸化物組成物。
【請求項6】
多孔性酸化物に吸収されたRLiを含有するリチウム試薬−多孔性金属酸化物組成物
(式中、Rは、アルキル基、アルケニル基、アルキニル基、アリール基、アルカリール基又はNR1R2基であり、
R1はアルキル基、アルケニル基、アルキニル基、アリール基、アルカリール基又はSi(R3)3基であり、
R2は水素、アルキル基、アルケニル基、アルキニル基、アリール基、アルカリール基又はSi(R3)3基であり、
R3はアルキル基、アルケニル基、アルキニル基、アリール基又はアルカリール基である)。
【請求項7】
Rがメチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、t−ブチル基、イソブチル基、sec−ブチル基、エテニル基、アリル基、シクロペンチル基、シクロヘキシル基、フェニル基及びベンジル基からなる群から選択される、請求項6記載のリチウム試薬−多孔性金属酸化物組成物。
【請求項8】
RがR1R2Nであり、
R1がメチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、t−ブチル基、イソブチル基、sec−ブチル基、エテニル基、アリル基、シクロペンチル基、シクロヘキシル基、フェニル基及びベンジル基からなる群から選択され、
R2が水素、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、t−ブチル基、イソブチル基、sec−ブチル基、エテニル基、アリル基、シクロペンチル基、シクロヘキシル基、フェニル基及びベンジル基からなる群から選択される、請求項6記載のリチウム試薬−多孔性金属酸化物組成物。
【請求項9】
液体リチウム金属を多孔性金属酸化物に吸収させるのに十分な発熱条件下で、不活性雰囲気において、液体リチウム金属と多孔性金属酸化物とを混合するステップと、
得られるリチウム金属/多孔性金属酸化物組成物を、約−80℃〜0℃の温度で化合物RXと反応させるステップと、を包含する、リチウム試薬−多孔性金属酸化物組成物の調製方法
(式中、Rはアルキル基、アルケニル基、アルキニル基、アリール基、アルカリール基又はNR1R2基であり、
R1はアルキル基、アルケニル基、アルキニル基、アリール基、アルカリール基又はSi(R3)3基であり、
R2は水素、アルキル基、アルケニル基、アルキニル基、アリール基、アルカリール基又はSi(R3)3基であり、
R3はアルキル基、アルケニル基、アルキニル基、アリール基又はアルカリール基であり、
Xはハロゲンである)。
【請求項10】
前記リチウム金属が、最高約40重量%で担持され、
前記多孔性金属酸化物の孔が、30Å〜500Åの平均孔径を有し、
前記多孔性金属酸化物が、多孔性アルミナ、多孔性酸化チタン、多孔性酸化カルシウム、多孔性酸化ジルコニウム、多孔性酸化鉄、多孔性Co3O4、多孔性金属ホスフェート、多孔性ハイブリッドホスホシリケート、多孔性アルミネート、多孔性バナデート及びモリブデートから選択される、請求項9記載のリチウム試薬−多孔性金属酸化物組成物の調製方法。
【請求項11】
前記リチウム金属が、約20重量%〜40重量%で担持されている、請求項9記載のリチウム試薬−多孔性金属酸化物組成物の調製方法。
【請求項12】
前記多孔性金属酸化物がアルミナである、請求項9記載のリチウム試薬−多孔性金属酸化物組成物の調製方法。
【請求項13】
前記多孔性金属酸化物の孔が、60Å〜190Åの平均孔径を有する、請求項9記載のリチウム試薬−多孔性金属酸化物組成物の調製方法。
【請求項14】
リチウムを多孔性金属酸化物の孔に吸収させるのに十分な条件下で、溶媒の存在下で、リチウム金属、多孔性金属酸化物及びハロゲン化アルキル溶液を混合するステップと、
いかなる過剰の溶媒も蒸発させるステップと、を包含する、有機リチウム−多孔性金属酸化物組成物の調製方法。
【請求項15】
前記リチウム金属が、最高約40重量%で担持され、
前記多孔性金属酸化物の孔が、30Å〜500Åの平均孔径を有し、
前記多孔性金属酸化物が、多孔性アルミナ、多孔性酸化チタン、多孔性酸化カルシウム、多孔性酸化ジルコニウム、多孔性酸化鉄、多孔性Co3O4、多孔性金属ホスフェート、多孔性ハイブリッドホスホシリケート、多孔性アルミネート、多孔性バナデート及びモリブデートから選択される、請求項14記載の有機リチウム−多孔性金属酸化物組成物の調製方法。
【請求項16】
前記リチウム金属が、約20重量%〜40重量%で担持されている、請求項14記載の有機リチウム−多孔性金属酸化物組成物の調製方法。
【請求項17】
前記多孔性金属酸化物がアルミナである、請求項14記載の有機リチウム−多孔性金属酸化物組成物の調製方法。
【請求項18】
前記多孔性金属酸化物の孔が、60Å〜190Åの平均孔径を有する、請求項14記載の有機リチウム−多孔性金属酸化物組成物の調製方法。
【請求項19】
低温条件下で多孔性材料と有機リチウム種とを接触させるステップであって、
前記多孔性材料が多孔性シリカゲル及び多孔性金属酸化物からなる群から選択されるステップと、
いかなる過剰の溶媒も蒸発させるステップと、得られる有機リチウム材を乾燥させるステップと、を包含する、有機リチウム種を多孔性材料の孔に吸収させる方法。
【請求項20】
請求項6記載のリチウム試薬−多孔性金属酸化物組成物の使用を包含する、有機リチウム試薬又はリチウムアミドを用いた化学反応。
【請求項21】
前記化学反応が、求核付加反応、重合反応及び塩基触媒反応からなる群から選択される、請求項20記載の化学反応。
【請求項22】
前記化学反応が、炭素中心求電子種に対する求核付加反応、有機クプレート又はギルマンの試薬の調製、適当な求電子物質からの有機リン化合物、有機硫黄化合物、有機ホウ素化合物、有機スズ化合物の調製、リチウムアミドの調製、配向的オルトリチオ化反応及びそれに続く求電子種によるクエンチング、エノラートの調製又は他の脱プロトン化、アニオン重合の開始、ウィッティヒ反応におけるイリドの生成、カービンの生成、アイソトープ標識、シャピロ反応、非求核塩基反応、ハロゲン−金属置換又はC−O、C−S又はC−Pの還元的切断による有機リチウム化合物の調製、カルボニル化合物又は芳香族化合物の還元(Birch還元)及び反応性中間体の生成、からなる群から選択される、請求項20記載の化学反応。
【請求項23】
化学反応が従来のバッチ反応において、又はプロセスフロー反応装置において生じる、請求項20記載の化学反応。
【図2】

【図3A】
【図3B】
【図3C】
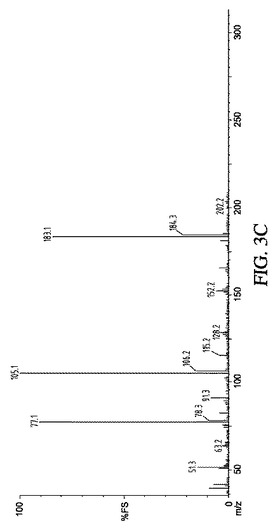
【図4】
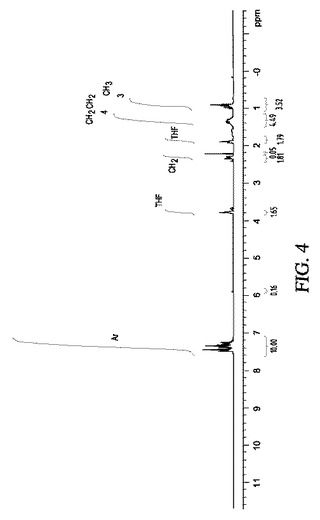
【図5A】

【図5B】
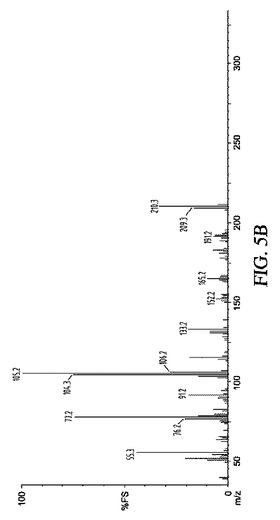
【図5C】
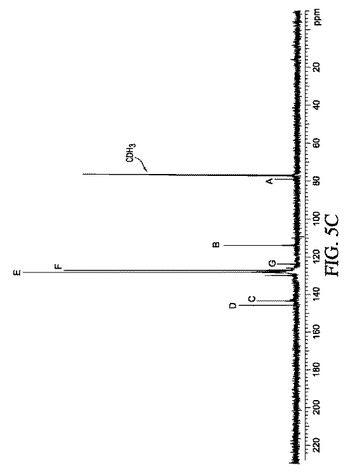
【図3A】
【図3B】
【図3C】
【図4】
【図5A】
【図5B】
【図5C】
【公表番号】特表2010−502843(P2010−502843A)
【公表日】平成22年1月28日(2010.1.28)
【国際特許分類】
【出願番号】特願2009−527613(P2009−527613)
【出願日】平成19年9月10日(2007.9.10)
【国際出願番号】PCT/US2007/078048
【国際公開番号】WO2008/031101
【国際公開日】平成20年3月13日(2008.3.13)
【出願人】(506414842)シグナ・ケミストリー・リミテッド・ライアビリティ・カンパニー (4)
【氏名又は名称原語表記】SIGNA CHEMISTRY LLC
【出願人】(509067935)
【氏名又は名称原語表記】Michael LEFENFELD
【出願人】(506249392)
【氏名又は名称原語表記】James L. DYE
【出願人】(509067728)
【氏名又は名称原語表記】Partha NANDI
【出願人】(509067739)
【氏名又は名称原語表記】James JACKSON
【Fターム(参考)】
【公表日】平成22年1月28日(2010.1.28)
【国際特許分類】
【出願日】平成19年9月10日(2007.9.10)
【国際出願番号】PCT/US2007/078048
【国際公開番号】WO2008/031101
【国際公開日】平成20年3月13日(2008.3.13)
【出願人】(506414842)シグナ・ケミストリー・リミテッド・ライアビリティ・カンパニー (4)
【氏名又は名称原語表記】SIGNA CHEMISTRY LLC
【出願人】(509067935)
【氏名又は名称原語表記】Michael LEFENFELD
【出願人】(506249392)
【氏名又は名称原語表記】James L. DYE
【出願人】(509067728)
【氏名又は名称原語表記】Partha NANDI
【出願人】(509067739)
【氏名又は名称原語表記】James JACKSON
【Fターム(参考)】
[ Back to top ]