
リポソーム抗新生物薬剤およびその使用
【課題】新規の臨床的有用性を有する、脂質処方された抗新生物剤の提供。
【解決手段】本発明は、リポソーム抗新生物薬剤組成物に関する。本発明に用いられるリポソーム抗新生物薬剤としては、例えば、カンプトセシンが挙げられる。本発明は、さらに、新生物を処置するために、このような組成物を用いる方法に関する。本発明は、リポソーム組成物、および新生物を処置するためおよび新脈管形成を阻害するためにこのような組成物を使用する法に関する。本発明は、活性薬剤(例えば、トポテカン)の血漿循環半減期を調節するために有用な組成物および方法を提供する。リポソーム処方物は、臨床的効力を増加し、そして付帯的な毒性を減少する。さらに、本発明は、新生物の処置および新脈管形成の阻害のための方法およびリポソーム組成物を提供する。
【解決手段】本発明は、リポソーム抗新生物薬剤組成物に関する。本発明に用いられるリポソーム抗新生物薬剤としては、例えば、カンプトセシンが挙げられる。本発明は、さらに、新生物を処置するために、このような組成物を用いる方法に関する。本発明は、リポソーム組成物、および新生物を処置するためおよび新脈管形成を阻害するためにこのような組成物を使用する法に関する。本発明は、活性薬剤(例えば、トポテカン)の血漿循環半減期を調節するために有用な組成物および方法を提供する。リポソーム処方物は、臨床的効力を増加し、そして付帯的な毒性を減少する。さらに、本発明は、新生物の処置および新脈管形成の阻害のための方法およびリポソーム組成物を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
(関連出願の相互参照)
本出願は、「Liposomal Camptothecins and U
ses Thereof」と表題された、2000年6月30日出願の米国仮出
願第60/215,556号、および「Liposomal Antineop
lastic Drugs and Uses Thereof」と表題された
、2001年1月26日出願の米国仮出願第60/264,616号に関し、こ
れらの両方が、全ての目的について本明細書中にこれらの全体が参照として援用
される。「Liposomal Camptothecins and Use
s Thereof」と表題された、2001年6月29日出願の米国特許出願
第_号(代理人整理番号016303−008020を有する)は、全ての目的
について本明細書中に参照として援用される。
【背景技術】
【0002】
(発明の背景)
本発明は、リポソーム組成物、および新生物を処置するためおよび新脈管形成
を阻害するためにこのような組成物を使用する方法に関する。
【0003】
多くの抗癌剤または抗新生物薬物が、リポソーム中にカプセル化されている。
これらとしては、アルキル化剤、ニトロソウレア、シスプラチン、代謝拮抗剤、
およびアントラサイクリンが挙げられる。アントラサイクリン抗生物質を含むリ
ポソームを用いる研究は、遊離薬物を与えられるコントロールと比較して、心毒
性および皮膚毒性の減少、ならびに腫瘍を保有する動物の長期の生存を明らかに
示した。
【0004】
リポソーム抗癌剤は、それらの遊離薬物対照物と比較して薬物動態学を改変す
る。リポソーム薬物処方について、薬物動態学は、キャリアが血液から取り除か
れる速度、およびその薬物がキャリアから放出される速度によってほぼ決定され
る。血液からの遅いクリアランスを示すリポソームキャリア組成物を同定するた
めに、かなりの努力がなされており、そして、長循環(long−circul
ating)キャリアが、多くの科学論文および特許に記載されている。例えば
、コントロール放出に対する膜ポテンシャルを使用する、リポソームキャリアか
らの薬物漏出速度を制御するための努力もまた、なされた。
【0005】
治療カンプトセシン(例えば、トポテカン(9−ジメチルアミノメチル−10
−ヒドロキシ−カンプトセシン;HycamtinTM)、およびイリノテカン
は、カンプトセシンの半合成の水可溶性誘導体であり、シナ蝋Camptoth
eca acuminataの幹から抽出されるアルカロイドである(Wall
ら、J.Am.Chem.Soc.88:3888−3890(1966))。
カンプトセシンは、トポイソメラーゼインヒビタークラスの抗新生物剤に属し、
DNA複製に関連する核酵素トポイソメラーゼIの作用を特異的に阻害する(H
siangら、Cancer Res.48:1722−1726(1988)
)。このように、トポテカンは、細胞周期特異的作用機構を示し、S期(DNA
複製)の間に作用して、G2細胞周期阻止およびアポトーシスを最終的に導く、
DNA中の不可逆的な二本鎖崩壊を生じる。遊離形態において、この薬剤は、腫
瘍細胞株およびマウス同種移植片腫瘍モデルおよびヒト異種移植片腫瘍モデルに
わたって広範囲の活性を有する(McCabe,F.L.ら、Cancer I
nvest 12:308−313(1994);Emersonら、Canc
er Res.55:603−609(1995);Thompson,Bio
chim.Biophys.Acta 1400:301−319(1998)
;Ormrodら、Drugs 58:533−551(1999);Hard
manら、Anticancer Res.19:2269−2274(199
9))。より最近には、トポテカンが、抗腫瘍作用機構に寄与し得る強力な抗血
管形成性特性を有するということが示された(O’Learyら、Clin.C
ancer Res.5:181−187(1999);Clementsら、
Cancer Chemother.Pharmacol.44:411−41
6(1999))。これらの処置全てが、用量制限毒性(例えば、貧血(ana
emia)、好中球減少症および血小板減少症を導く非蓄積性の骨髄抑制(my
elosuppression))、ならびに粘膜症(musositis)お
よび下痢を含む胃腸関連毒性に関連する。臨床的には、トポテカンは、卵巣癌お
よび小細胞肺癌(SCLC)における第二期治療(second−line t
herapy)について認可されており、そして現在広範な臨床的評価の焦点で
ある。
【0006】
カンプトセシンの脂質処方物は、治療剤として提唱されてきた(米国特許第5
,552,156号およびPCT公開WO95/08986を参照のこと)。し
かし、脂質処方物の全てが、薬物送達の目的について等しいというのではなく、
広範な研究が、薬物の充填および貯蔵、薬物投与、薬学的動態、生物学的分布、
漏出速度、腫瘍蓄積、毒性プロファイルなどについて好ましい特性を示す処方物
に関して継続中である。カンプトセシンに対して、この分野は、さらに複雑であ
る。なぜなら、ヒトにおける用量制限毒性は、マウスにおけるものより1/10
の低さであり得るからである(Erickson−Millerら、Cance
r Chemother.Pharmacol.39:467−472(199
7))。
【発明の概要】
【発明が解決しようとする課題】
【0007】
抗新生物剤の改善されたリポソーム処方物は、非常に有用であることを証明し
得る。新規の臨床的有用性を有する、脂質処方された抗新生物剤を提供すること
が、本発明の目的である。
【課題を解決するための手段】
【0008】
(発明の要旨)
本発明は、活性薬剤(例えば、トポテカン)の血漿循環半減期を調節するため
に有用な組成物および方法を提供する。リポソーム処方物は、臨床的効力を増加
し、そして付帯的な毒性を減少する。さらに、本発明は、新生物の処置および新
脈管形成の阻害のための方法およびリポソーム組成物を提供する。
【0009】
このように、1つの実施形態において、本発明は、活性薬剤の血漿循環半減期
を調節するための方法を提供し、本方法は、以下の工程を包含する:(a)リポ
ソームを提供する工程であって、該リポソーム中に、遊離活性薬剤と沈殿した活
性薬剤がカプセル化されている、工程;ならびに(b)このリポソーム中に沈殿
した活性薬剤の量を変化させる工程。驚くことに、リポソーム中に沈殿した活性
薬剤の量を変化させることによって、血漿中への活性薬剤の放出動態を調節する
ことが可能である。好ましい活性薬剤は、抗新生物薬物(例えば、カンプトセシ
ン(例えば、トポテカン))である。
【0010】
別の実施形態において、本発明は、a)抗新生物薬物とb)遊離抗新生物薬物
および沈殿された抗新生物薬物を有するリポソーム、とを含むリポソーム処方物
を提供し、ここで、このリポソーム中に沈殿した抗新生物薬物は、抗新生物薬物
全体の少なくとも50%である。リポソーム中に沈殿した抗新生物薬物の量を調
整することによって、この薬物の放出をインビトロおよびインビボの両方で調節
し得る。特定の好ましい実施形態において、高いリポソーム内濃度の活性薬剤(
例えば、トポテカン)は、多量の沈殿形態を生じる。この局面において、その後
のインビボでの薬物の放出速度は、遅い。特定の局面において、遅い放出速度は
、速い放出速度と比べて好ましくかつより効果的である。
【0011】
なお別の実施形態において、本発明は、a)活性薬剤;b)その中にカプセル
化された、遊離活性薬剤と沈殿した活性薬剤とを有するリポソーム;ならびに、
c)空のリポソーム、を含む、リポソーム処方物を提供する。
【0012】
この局面において、リポソームの血清半減期は、空のリポソームを処方物中に
含むことによって長くされる。種々の脂質のいずれかを使用して本発明のリポソ
ーム組成物を形成し得ることは、当業者には容易に明らかである。現在好ましい
実施形態において、この脂質は、スフィンゴミエリンおよびコレステロールの混
合物を含み、好ましくは、約30:70〜約60:40のスフィンゴミエリン:
コレステロール比(モル比)である。1つの好ましい実施形態において、このリ
ポソームは、55:45比でスフィンゴミエリンおよびコレステロールを含む。
【0013】
さらに別の局面において、本発明は、固形腫瘍を、それを罹患するヒトにおい
て処置するための方法を提供し、本方法は、薬学的に受容可能なキャリア中にあ
る有効量の本発明のリポソーム処方物を、このヒトに投与する工程を包含する。
種々の固形腫瘍は、本発明の処方物を使用して処置され得る。好ましい実施形態
において、処置される固形腫瘍は、肺の固形腫瘍、乳房の固形腫瘍、結腸の固形
腫瘍および前立腺の固形腫瘍からなる群より選択される。別の好ましい実施形態
において、本発明はさらに、好中球減少症および血小板欠損を処置するために適
切な処置または活性薬剤の同時投与を包含する。
【0014】
好ましい実施形態において、リポソームトポテカンを使用して、固形腫瘍を処
置する。さらに、種々の脂質のいずれを使用しても本発明のリポソーム組成物を
形成し得ることは、当業者には容易に明らかである。
【0015】
本発明の他の特性、目的および利点、ならびにその好ましい実施形態は、以下
の詳細な説明から明らかになる。
例えば本願発明は以下の項目を提供する。
(項目1) 活性薬剤の血漿循環半減期を調節するための方法であって、
該方法は、以下:
(a) リポソームを提供する工程であって、該リポソーム中に、遊離の活性
薬剤および沈殿した活性薬剤がカプセル化されている、工程;および
(b) 該リポソーム中の沈殿した該活性薬剤の量を変化させる工程、
を包含する、方法。
(項目2) 前記工程(b)が、前記活性薬剤 対 脂質の割合を変化さ
せる工程を包含する、項目1に記載の方法。
(項目3) 前記活性薬剤 対 脂質の割合が、空のリポソームの添加に
より変化する、項目2に記載の方法。
(項目4) 前記工程(b)が、前記リポソームのサイズを変化させる工
程を包含する、項目1に記載の方法。
(項目5) 前記工程(b)が、前記活性薬剤の沈殿を増強する成分を添
加する工程を包含する、項目1に記載の方法。
(項目6) 前記成分が、モノアニオン、ジアニオン、トリアニオン、ま
たは多価アニオンである、項目5に記載の方法。
(項目7) 前記工程(b)が、前記活性薬剤 対 脂質の割合、および
前記リポソームのサイズの両方を変化させる工程を包含する、項目1に記載の
方法。
(項目8) 前記活性薬剤が、抗新生物薬物である、項目1に記載の方
法。
(項目9) 前記抗新生物薬物が、カンプトセシンである、項目8に記
載の方法。
(項目10) 前記カンプトセシンが、イリノテカン、トポテカン、9−
アミノカンプトセシン、10,11−メチレンジオキシカンプトセシン、9−ニ
トロカンプトセシン、TAS103、7−(4−メチル−ピペラジノ−メチレン
)−10,11−エチレンジオキシ−20(S)−カンプトセシン、および7−
(2−N−イソプロピルアミノ)エチル)−20(S)−カンプトセシンからな
る群より選択されるメンバーである、項目9に記載の方法。
(項目11) 前記カンプトセシンが、トポテカンである、項目10に
記載の方法。
(項目12) 前記活性抗新生物薬物が、ビンカアルカロイドである、請
求項1に記載の方法。
(項目13) 前記ビンカアルカロイドが、ビンクリスチン、ビンブラス
チン、ビンオレルビン、およびビンデシンからなる群より選択されるメンバーで
ある、項目12に記載の方法。
(項目14) 前記リポソーム中にカプセル化される沈殿した活性薬剤が
、該活性薬剤の総量の少なくとも50%である、項目1に記載の方法。
(項目15) 前記リポソーム中にカプセル化される沈殿した活性薬剤が
、該活性薬剤の総量の少なくとも60%である、項目14に記載の方法。
(項目16) 前記リポソーム中にカプセル化される沈殿した活性薬剤が
、該活性薬剤の総量の少なくとも70%である、項目15に記載の方法。
(項目17) 前記リポソームが、スフィンゴミエリンおよびコレステロ
ールを含む、項目1に記載の方法。
(項目18) 前記リポソームが、55:45の割合でスフィンゴミエリ
ンとコレステロールとを含む、項目17に記載の方法。
(項目19) 前記活性薬剤の血漿循環半減期が、最適効力に調節される
、項目1に記載の方法。
(項目20) 前記活性薬剤 対 脂質の割合が、約0.005〜1:1
(w/w)である、項目1に記載の方法。
(項目21) 前記活性薬剤 対 脂質の割合が、約0.05〜0.9:
1(w/w)である、項目20に記載の方法。
(項目22) 前記活性薬剤 対 脂質の割合が、約0.1〜0.5:1
(w/w)である、項目21に記載の方法。
(項目23) 活性薬剤の血漿循環半減期を調節するための方法であって
、該方法は、以下:
(a) リポソームを提供する工程であって、該リポソーム中に、遊離の活性
薬剤および沈殿した活性薬剤がカプセル化されている、工程;および
(b) 該リポソームに非カプセル化活性薬剤を添加する工程、
を包含する、方法。
(項目24) 活性薬剤を含むリポソーム 対 カプセル化された薬剤を
有さないリポソームの割合が、約1:0.5〜1:1000である、項目23
に記載の方法。
(項目25) 活性薬剤を含むリポソーム 対 カプセル化された薬剤を
有さないリポソームの割合が、約1:1〜1:100である、項目24に記載
の方法。
(項目26) 活性薬剤を含むリポソーム 対 カプセル化された薬剤を
有さないリポソームの割合が、約1:2〜1:10である、項目25に記載の
方法。
(項目27) 活性薬剤を含むリポソーム 対 カプセル化された薬剤を
有さないリポソームの割合が、約1:3〜1:5である、項目26に記載の方
法。
(項目28) 前記活性薬剤が、抗新生物薬物である、項目23に記載
の方法。
(項目29) 前記抗新生物薬物が、カンプトセシンである、項目28
に記載の方法。
(項目30) 前記カンプトセシンが、イリノテカン、トポテカン、9−
アミノカンプトセシン、10,11−メチレンジオキシカンプトセシン、9−ニ
トロカンプトセシン、TAS103、7−(4−メチル−ピペラジノ−メチレン
)−10,11−エチレンジオキシ−20(S)−カンプトセシン、および7−
(2−N−イソプロピルアミノ)エチル)−20(S)−カンプトセシンからな
る群より選択されるメンバーである、項目29に記載の方法。
(項目31) 前記カンプトセシンが、トポテカンである、項目30に
記載の方法。
(項目32) リポソーム処方物であって、該リポソーム処方物は、以下
:
a) 抗新生物薬物;および
b) 遊離の抗新生物薬物および沈殿した抗新生物薬物を有するリポソーム、
を含み、ここで、該リポソーム中の沈殿した抗新生物薬物は、抗新生物薬物の総
量の少なくとも50%である、処方物。
(項目33) 前記抗新生物薬物が、カンプトセシンである、項目32
に記載のリポソーム処方物。
(項目34) 前記カンプトセシンが、イリノテカン、トポテカン、9−
アミノカンプトセシン、10,11−メチレンジオキシカンプトセシン、9−ニ
トロカンプトセシン、TAS103、7−(4−メチル−ピペラジノ−メチレン
)−10,11−エチレンジオキシ−20(S)−カンプトセシン、および7−
(2−N−イソプロピルアミノ)エチル)−20(S)−カンプトセシンからな
る群より選択されるメンバーである、項目33に記載のリポソーム処方物。
(項目35) 前記カンプトセシンが、トポテカンである、項目34に
記載のリポソーム処方物。
(項目36) 前記活性抗新生物薬物が、ビンカアルカロイドである、請
求項33に記載のリポソーム処方物。
(項目37) 前記遊離の抗新生物薬物と前記沈殿した抗新生物薬物とが
異なる、項目32に記載のリポソーム処方物。
(項目38) 前記ビンカアルカロイドが、ビンクリスチン、ビンブラス
チン、ビンオレルビン、およびビンデシンからなる群より選択されるメンバーで
ある、項目36に記載のリポソーム処方物。
(項目39) 前記抗新生物薬物 対 脂質の割合が、約0.005〜1
:1(w/w)である、項目32に記載のリポソーム処方物。
(項目40) 前記抗新生物薬物 対 脂質の割合が、約0.05〜0.
9:1(w/w)である、項目39に記載のリポソーム処方物。
(項目41) 前記抗新生物薬物 対 脂質の割合が、約0.1〜0.5
:1(w/w)である、項目40に記載のリポソーム処方物。
(項目42) 前記リポソームが、スフィンゴミエリンおよびコレステロ
ールを含む、項目32に記載のリポソーム処方物。
(項目43) 前記リポソームが、55:45の割合でスフィンゴミエリ
ンとコレステロールとを含む、項目42に記載のリポソーム処方物。
(項目44) カプセル化された活性薬剤を含まないリポソームをさらに
含む、項目32に記載のリポソーム処方物。
(項目45) 活性薬剤を含むリポソーム 対 カプセル化された薬剤を
有さないリポソームの割合が、約1:0.5〜1:1000である、項目44
に記載のリポソーム処方物。
(項目46) 活性薬剤を含むリポソーム 対 カプセル化された薬剤を
有さないリポソームの割合が、約1:1〜1:100である、項目45に記載
のリポソーム処方物。
(項目47) 活性薬剤を含むリポソーム 対 カプセル化された薬剤を
有さないリポソームの割合が、約1:2〜1:10である、項目46に記載の
リポソーム処方物。
(項目48) 活性薬剤を含むリポソーム 対 カプセル化された薬剤を
有さないリポソームの割合が、約1:3〜1:5である、項目47に記載のリ
ポソーム処方物。
(項目49) リポソーム処方物であって、該リポソーム処方物は、以下
:
a) 活性薬剤;
b) リポソームであって、該リポソーム中に、遊離の活性薬剤および沈殿し
た活性薬剤がカプセル化されている、リポソーム;および
c) 空のリポソーム、
を含む、リポソーム処方物。
(項目50) 前記活性薬剤を含むリポソーム 対 前記空のリポソーム
の割合が、約1:0.5〜1:1000である、項目49に記載のリポソーム
処方物。
(項目51) 前記活性薬剤を含むリポソーム 対 前記空のリポソーム
の割合が、約1:1〜1:100である、項目50に記載のリポソーム処方物
。
(項目52) 前記活性薬剤を含むリポソーム 対 前記空のリポソーム
の割合が、約1:2〜1:10である、項目51に記載のリポソーム処方物。
(項目53) 前記活性薬剤を含むリポソーム 対 前記空のリポソーム
の割合が、約1:3〜1:5である、項目52に記載のリポソーム処方物。
(項目54) 前記活性薬剤が、抗新生物薬物である、項目49に記載
のリポソーム処方物。
(項目55) 前記抗新生物薬物が、カンプトセシンである、項目54
に記載のリポソーム処方物。
(項目56) 前記カンプトセシンが、イリノテカン、トポテカン、9−
アミノカンプトセシン、10,11−メチレンジオキシカンプトセシン、9−ニ
トロカンプトセシン、TAS103、7−(4−メチル−ピペラジノ−メチレン
)−10,11−エチレンジオキシ−20(S)−カンプトセシン、および7−
(2−N−イソプロピルアミノ)エチル)−20(S)−カンプトセシンからな
る群より選択されるメンバーである、項目55に記載のリポソーム処方物。
(項目57) 前記カンプトセシンが、トポテカンである、項目56に
記載のリポソーム処方物。
(項目58) 前記活性抗新生物薬物が、ビンカアルカロイドである、請
求項57に記載のリポソーム処方物。
(項目59) 前記ビンカアルカロイドが、ビンクリスチン、ビンブラス
チン、ビンオレルビン、およびビンデシンからなる群より選択されるメンバーで
ある、項目58に記載のリポソーム処方物。
(項目60) 前記活性薬剤 対 脂質の割合が、約0.005〜1:1
(w/w)である、項目49に記載のリポソーム処方物。
(項目61) 前記活性薬剤 対 脂質の割合が、約0.05〜0.9:
1(w/w)である、項目60に記載のリポソーム処方物。
(項目62) 前記活性薬剤 対 脂質の割合が、約0.1〜0.5:1
(w/w)である、項目61に記載のリポソーム処方物。
(項目63) 前記リポソームが、スフィンゴミエリンおよびコレステロ
ールを含む、項目49に記載のリポソーム処方物。
【図面の簡単な説明】
【0016】
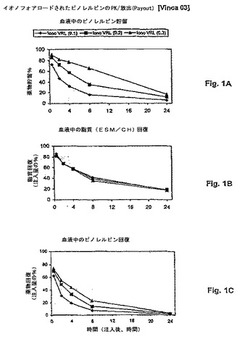
【図1】図1は、ビノレルビンのリポソーム処方物の薬物動態学的挙動を示す。パネルAは、異なる薬物:脂質比(0.1:1、0.2:1、0.3:1)の3つの処方物について、循環するキャリアからの薬物漏出速度を示す。薬物放出は、薬物:脂質比に依存し、最小の放出速度が最高の比(0.3:1)について見られる。パネルBは、血液中での脂質回復を示す。パネルCは、キャリアからの薬物放出速度の調節がビノレルビンについての血液クリアランス半減期に対する変化を生じることを示す。
【図2】図2は、血漿薬物レベルが薬物動態に従って使用される場合の、対応する挙動を示す。パネルAは、薬物貯留 対 時間を示す。パネルBは、脂質回復 対 時間を示す。パネルCは、薬物回復 対 時間を示す。
【図3】図3は、薬物:脂質比の関数として、リポソームビンブラスチンの処方物の薬物動態学的挙動(血液PK)を示す。リポソームキャリアからの薬物漏出は、最初の薬物:脂質比によって決定され、より高い薬物比の処方物について、より遅い放出を伴う。パネルAは、薬物貯留 対 時間を示す。パネルBは、脂質回復 対 時間を示す。パネルCは、薬物放出速度が血液からの薬物クリアランス半減期に対する変化と相関することを示す。
【図4】図4は、薬物:脂質比の関数として、リポソームビンブラスチンの処方物の薬物動態学的挙動(血漿PK)を示す。パネルAは、薬物貯留 対 時間を示す。パネルBは、脂質回復 対 時間を示す。パネルCは、薬物放出速度が血漿からの薬物クリアランス半減期に対する変化と相関することを示す。
【図5】図5は、脂質用量の、PK挙動(血液PK)に対する影響を示す。ここに例示されるように、類似する薬物放出速度(A)、脂質クリアランス速度(B)および薬物クリアランス速度(C)が、16.6mg/kg〜50mg/kgの脂質用量範囲にわたって薬物:脂質比が0.3:1であるリポソームビンブラスチン処方物について見られる。
【図6】図6は、脂質用量の、PKの動きに対する影響を示す(血漿PK)。ここに例示されるように、類似する薬物放出速度(A)、脂質クリアランス速度(B)および薬物クリアランス速度(C)が、16.6mg/kg〜50mg/kgの用量範囲にわたって薬物:脂質比が0.3:1であるリポソームビンブラスチン処方物について見られる。
【図7】図7は、異なる薬物:脂質比のリポソームトポテカンの2つの処方物の薬物動態的挙動を示す。トポテカンが薬物:脂質比0.11:1でロードされる場合、より低い薬物:脂質比0.02:1の処方物を有するパネルBと比較して、より長い血漿クリアランス速度を生じる、よりかなり遅い薬物放出速度が見られること、パネルAは示す。
【発明を実施するための形態】
【0017】
(発明の詳細な説明および好ましい実施形態)
多くの抗癌剤の活性は、その薬物動態的挙動に依存する。この薬物動態的挙動
は、薬物濃度、および癌細胞が薬物に暴露される期間を規定する。ほとんどの抗
癌剤の場合、より長い暴露時間は、これが癌細胞の増加した死滅を生じるので好
ましい。一般に、いくつかのパラメーターを使用して、薬物の薬物動態を記載す
る。血漿クリアランス半減期および曲線下面積(area under the
curve)(AUC)が、例である。この血漿クリアランス半減期は、投与
された薬物の半分が血漿から除去されるために必要とする時間である。AUCは
、経時的な血漿薬物レベルの尺度であり、そして全薬物暴露の指標を提供する。
一般に、抗癌剤に関する、増加した血漿クリアランス半減期および血漿AUCは
、増加した治療効果に相関する。
【0018】
(I.活性薬剤放出を調節すること)
本発明は、リポソームからの薬物放出を調節するための方法および処方物を提
供する。1つの実施形態において、本発明は、活性薬剤の血漿循環半減期を調節
するための方法を提供し、本方法は、以下の工程を包含する:(a)リポソーム
を提供する工程であって、このリポソーム中に、遊離活性薬剤と沈殿した活性薬
剤とがカプセル化されている、工程;ならびに(b)このリポソーム中に沈殿し
た活性薬剤の量を変化させる工程。好ましくは、「遊離活性薬剤」および「沈殿
した活性薬剤」は、同一の活性薬剤であるが、本発明は、そのように限定しない
。本明細書中で使用される場合、用語「調節すること(工程)」は、リポソーム
キャリアから活性薬剤を放出する速度を増加することかまたは減少することかの
いずれかを意味し得る。抗新生物活性薬剤について、調節することは、好ましく
は、活性薬剤の放出速度を減少することかまたは遅くすることかである。
【0019】
好ましい局面において、本発明のリポソームは、ともにカプセル化された、遊
離活性薬剤および沈殿した活性薬剤を含む。リポソーム中に沈殿した活性薬剤の
量は、種々の機構を使用して変化され得る。例えば、活性薬剤対脂質の比を変化
させることによって、沈殿した活性薬剤の量は、増加または減少され得る。低い
薬物:脂質比での薬物ローディングは、低濃度の活性薬剤(例えば、トポテカン
)をリポソーム内部に生じ、従って、薬物全体のうちのほとんど(すべてではな
いにしても)は溶液中にある(すなわち、沈殿されておらず、すなわち遊離して
いる)。少量の沈殿は、リポソームからの薬物の速い放出速度を生じる。逆に、
高い薬物:脂質比は、高いリポソーム内部濃度および高沈殿量を生じる。薬物が
沈殿形態の場合、その後の、インビボまたはインビトロでの放出速度は、遅い。
抗新生物薬物(例えば、トポテカン)について、遅い放出速度が好ましい。
【0020】
いかなる特定の理論によって束縛されることもないが、本発明のリポソームは
、薬物放出を指示する「沈殿−溶解機構」(PDM)を受けると考えられている
。本発明のPDM機構において、リポソーム内部の沈殿した活性薬剤(例えば、
トポテカン)の、リポソームの内部溶液への溶解速度は、リポソーム外部の活性
薬剤の、外部への放出速度に比べて遅く、従って律速である。すなわち、リポソ
ーム内部での沈殿した薬物から遊離薬物への溶解速度は、その薬物が血漿中にい
かに速く放出されるかを決定する。
【0021】
特定の実施形態において、活性薬剤対脂質比は、空のリポソームの添加によっ
て変えられ得る。一般に、空のリポソームまたはその中に活性薬剤が含まれてい
るリポソームのいずれもが、細網内皮系(RES)の細胞によってクリアランス
される。代表的に、RESは、一定用量の注射されたリポソームの80〜95%
を1時間以内に排除し、リポソームの取り込みのための選択された標的部位を効
果的に競合的除外(outcompeting)する。リポソームのRES取り
込みの速度に影響を与える種々の因子が、報告されており、これらには、リポソ
ームのサイズ、電荷、脂質飽和度、および表面成分が挙げられる。空のリポソー
ム小胞を含めることによって、RESから活性薬剤を含むリポソームを保護する
ことが可能である。このように、空のリポソーム小胞は、実際に、「おとり」と
して作用することによってリポソームの血液循環寿命を延長する。延長された循
環時間は、しばしば、リポソームが注射部位から標的領域、標的細胞または標的
部位に到達するために必要とされる。空のリポソーム小胞は、RESを活発に維
持し、結果として、その中に活性薬剤が含まれるリポソームの血清半減期が増加
される。
【0022】
特定の他の局面において、活性薬剤の沈降を増強する成分が、リポソームに添
加される。この局面において、種々の荷電イオンを使用して、小胞内部の沈降さ
れた活性薬剤の量を増加し得る。好ましい局面において、二価、三価または多価
アニオンが使用される。適切なアニオンとしては、カルボキシレート(−CO2
−)、スルホネート(SO3−)、スルフェート(SO4−2)、ヒドロキシド
(−OH)、アルコキシド、ホスフェート(−PO4−2)およびホスホネート
(−PO3−2)が挙げられるが、これらに限定されない。リポソーム内部の沈
降された活性薬剤の量を増強する他の成分は、当業者に公知である。
【0023】
さらに、薬物:脂質比は、リポソームのサイズを使用して変えられ得る。使用
するリポソーム小胞が大きいほど、薬物:脂質比はより小さい。特定の局面にお
いて、活性薬剤対脂質比およびリポソームのサイズの両方を変えて、活性薬剤の
効力を最適化する。
小胞中に沈降されるカプセル化された活性薬剤の量は、変化し、そしてこれは
、活性薬剤自体に幾分依存する。特定の実施形態において、沈降された活性薬剤
の量は、総活性薬剤の少なくとも約25%〜約95%(例えば、約25%、30
%、35%、40%、45%、50%、55%、60%、65%、70%、75
%、80%、85%、90%および95%)である。トポテカンについて、リポ
ソーム中にカプセル化される沈降された活性薬剤の量は、総活性薬剤の少なくと
も50%である。
【0024】
好ましい局面において、活性薬剤が抗腫瘍薬物である場合、高い薬物:脂質比
を使用することが、より多くの量のカプセル化され沈降された薬物を生じる。結
果として、インビボでのリポソームからの薬物放出は、より低い薬物:脂質比で
調整された同様の組成物よりも、よりゆっくりである。これらのより高い薬物:
脂質比のリポソームは、延長された血漿半減期および増加された血漿AUC値を
示す。有利には、これらの処方物は、改善された抗腫瘍効力を示す。
【0025】
特定の実施形態において、活性薬剤:脂質の比は、約0.005〜1:1(w
/w)である。
【0026】
好ましくは、活性薬剤:脂質の比は、約0.05〜0.9:1(w/w)であ
り、そしてより好ましくは、活性薬剤:脂質の比は、約0.1〜0.5:1(w
/w)である。活性薬剤の血漿循環半減期を調節することによって、このように
して、活性薬剤の効力を最大化または最適化することが可能である。
【0027】
(II.リポソーム処方物を作製する組成物および方法)
リポソーム、小胞およびリポソーム小胞は、水性の内部を包囲する脂質含有膜
を有する構造を示すことが理解される。これらの構造は、他に示されない限り、
1以上の脂質膜を有し得るが、一般に、リポソームは、たった1つの膜を有する
。このような単一層のリポソームは、本明細書中で「単層」と呼ばれる。多重層
のリポソームは、本明細書中で「多層」と呼ばれる。
【0028】
本発明において使用されるリポソームは、好ましくは、合わせられた場合に比
較的安定な小胞を形成する脂質から形成される。このようなリポソームを生成す
るために使用され得る、多くの種々の脂質が、当該分野で公知である。好ましい
脂質としては、中性または負に荷電したリン脂質またはスフィンゴ脂質およびス
テロール(例えば、コレステロール)が挙げられるが、これらに限定されない。
脂質の選択は、一般に、例えば、リポソームサイズおよび血流中のリポソームの
安定性を考慮して導かれる。
【0029】
本発明の使用のために好ましいリポソーム組成物としては、スフィンゴミエリ
ンおよびコレステロールを含有するリポソームが挙げられる。このリポソーム組
成物中のスフィンゴミエリン対コレステロールの比は、変化し得るが、一般に、
約75/25mol%/mol%のスフィンゴミエリン/コレステロール〜約3
0/50mol%/mol%のスフィンゴミエリン/コレステロールの範囲にあ
り、より好ましくは、約70/30mol%/mol%のスフィンゴミエリン/
コレステロール〜約40/45mol%/mol%のスフィンゴミエリン/コレ
ステロール、そしてさらにより好ましくは、約55/45mol%/mol%の
スフィンゴミエリン/コレステロールである。他の脂質は、必要であり得る場合
(例えば、脂質酸化を妨げるために、またはリポソーム表面上にリガンドを付着
するために)、本発明のリポソーム組成物中に含まれ得る。一般に、脂質が含ま
れる場合、このような脂質の他の含有は、スフィンゴミエリン/コレステロール
比の減少を生じる。この型のリポソームは、スフィンゴソームとして公知であり
、そして米国特許第5,814,335号(この教示は、本明細書中で参考とし
て援用される)において、より十分に記載される。
【0030】
例えば、以下に記載されるような、種々の方法が、リポソームを調製するため
に利用可能である:Szokaら、Ann.Rev.Biophys.Bioe
ng.9:467(1980);米国特許第4,235,871号;同第4,5
01,728号;同第4,837,028号、テキストLiposome,Ma
rc J.Ostro編、Marcel Dekker,Inc.,New Y
ork,1983,第1章;およびHopeら、Chem.Phys.Lip.
40:89(1986)(これら全てが、本明細書中で参考として援用される)
。リポソームを生成するためのプロトコルは、一般に以下の工程を包含し、これ
らの全ては、当該分野で周知である:有機溶媒中で脂質成分を混合する工程;乾
燥しそして水性溶媒中でリポソームを再構成する工程;およびリポソームをサイ
ズ化する工程(例えば、押出しによって)。
【0031】
リポソームを調製する代替的方法もまた、利用可能である。例えば、界面活性
剤透析に基づく、脂質粒子の自己アセンブリを含む方法が、Wheelerらに
対して発行された米国特許第5,976,567号において開示および権利化さ
れ、これは、時間のかかる難しい、比例性の乾燥および再構成工程を回避する。
連続フロー水和を使用してリポソームを調製するさらなる方法が、開発中であり
、そしてしばしば、最も効果的な大規模製造プロセスを提供し得る。
【0032】
活性薬剤(例えば、カンプトセシン)を有するリポソーム処方物の調製は、リ
ポソームへの薬物の充填を必要とする。充填は、受動的または能動的であり得る
。受動充填は、一般に、再構成工程の時点での緩衝液への薬物の添加を必要とす
る。これは、薬物がリポソーム内部に捕捉されるのを可能にし、ここでは、薬物
が脂溶性でない場合および小胞がインタクトなままである場合に、薬物が維持さ
れる(このような方法は、例えば、PCT公開番号WO95/08986(この
教示は、本明細書中で参考として援用される)において使用される)。
【0033】
能動充填は、多くの点で好ましく、そして広範に種々の治療薬剤が、膜のpH
勾配またはイオン勾配を使用することによって、ほぼ100%のカプセル化効率
でリポソーム中に充填され得る(Mayerら、Biochim.Biophy
s.Acta 1025:143−151(1990)およびMaddenら、
Chem.Phys.Lipids 53:37−46(1990)を参照のこ
と)。能動充填の多くの方法が、当業者に公知である。このような全ての方法は
、脂質性化合物をリポソームの内部に引き込む、いくつかの形態の勾配の確立を
包含し、ここでは、それらが、勾配が維持される限り存在し得る。非常に高い量
の所望の薬物が、内部に得られ得、薬物が、内部で沈降し、そして継続した取り
込みの勾配を生じる。
【0034】
イオノフォア媒介充填は、本発明と共に使用するために特に好ましく、これは
、米国特許第5,837,282号(この教示は、参考として本明細書中に援用
される)に開示および権利化されている。イオノフォア媒介充填は、電気的中性
のプロセスであり、これは、膜電位の形成を生じない。小胞への水素イオン輸送
によって、小胞からの並存するマグネシウムイオン輸送が、2:1比で存在する
(すなわち、正味電荷の輸送はない)。トポテカンの場合、その薬剤は、中性状
態(電荷なし)で膜を通過すると考えられる。小胞内への進入時に、トポテカン
は、正に荷電される。イオノフォア媒介充填は、電気的中性のプロセスであるの
で、膜電位は生成されない。
【0035】
薬学的目的のリポソームカンプトセシンの重要な特徴は、最終処方物の薬物対
脂質比である。先に議論したように、薬物:脂質比は、以下の2つの様式:1)
同じ薬物:脂質比を各々含有する均質なリポソームを使用するか;または2)空
のリポソームを高い薬物:脂質比を有するリポソームと混合することによって、
適切な平均薬物:脂質比を提供することによって確立され得る。異なる適用のた
めに、異なる薬物:脂質比が、所望され得る。特定の薬物:脂質比を生成するた
めの技術は、当該分野で周知である。薬物:脂質比は、重量に基づいて、モル濃
度に基づいてまたは任意の他の指定された基準に基づいて測定され得る。好まし
い薬物:脂質比は、約0.005:1の薬物:脂質(重量)〜約0.2:1の薬
物:脂質(重量)、そしてより好ましくは、約0.1:1の薬物:脂質(重量)
〜約0.3:1の薬物:脂質(重量)の範囲にある。
【0036】
さらなる重要な特徴は、リポソーム粒子のサイズである。本発明における使用
のために、約0.05ミクロン〜約0.15ミクロンのサイズを有するリポソー
ムが、好ましい。
【0037】
本発明はまた、キット形態でリポソーム組成物(例えば、カンプトセシン)を
提供する。キットは、既製の処方物または投与前に医薬の混合を必要とする処方
物を備え得る。キットは、代表的に、キットの種々のエレメントを保持するため
の仕切られたコンテナを備える。キットは、可能には水和形態で、本発明の組成
物またはその成分を、それらの再水和および投与のための指示書と共に備える。
【0038】
例えば、本明細書中に記載される方法によって調製されるリポソーム組成物は
、単独でか、または投与経路および標準的な薬務に従って選択された生理学的に
受容可能なキャリア(例えば、生理学的食塩水またはリン酸緩衝液)との混合物
のいずれかで投与され得る。一般に、通常の生理食塩水が、薬理学的に受容可能
なキャリアとして使用される。他の適切なキャリアとしては、例えば、水、緩衝
化水、0.4% 生理食塩水、0.3% グリシンなどが挙げられ、これらは、
安定性を増強するための糖タンパク質(例えば、アルブミン)、リポタンパク質
、グロブリンなどを含む。これらの組成物は、従来の周知の滅菌技術によって滅
菌され得る。得られた水溶液は、使用のためにパッケージされ得るか、または無
菌条件下でろ過され、そして凍結乾燥され得、この凍結乾燥調製物が、投与前に
滅菌水溶液と合わせられる。これらの組成物はまた、生理学的条件を近似するた
めに必要とされる、薬学的に受容可能な補助物質(例えば、pH調整剤および緩
衝化剤、張度調整剤など(例えば、酢酸ナトリウム、乳酸ナトリウム、塩化ナト
リウム、塩化カリウム、塩化カルシウムなど))を含み得る。さらに、この組成
物は、脂質保護剤を含み得、これは、保存中のフリーラジカルおよび脂質過酸化
損傷に対して脂質を保護する。親油性のフリーラジカル失活剤(例えば、α−ト
コフェロールおよび水溶性のイオン特異的キレート剤(例えば、フェリオキサミ
ン(ferrioxamine))が適切である。
【0039】
広範に種々の活性薬剤が、本発明のリポソーム組成物および方法に適切である
。好ましい局面において、活性薬剤は、抗腫瘍薬物である。現在、約20の認め
られたクラスの承認された抗腫瘍薬物が存在する。この分類は、特定の薬物によ
って共有される共通の構造に基づいてか、または薬物による作用の共通の機構に
基づく総括である。分類した抗腫瘍薬剤の一般に公知の商業的に認証された(ま
た開発中の)いくつかの部分的リストは、以下のとおりである:
構造に基づく分類:
1.フルオロピリミジン−−5−FU、フルオロデオキシウリジン、フトラフ
ァー(Ftorafur)、5’−デオキシフルオロウリジン、UFT、S−1
カペシタビン;
2.ピリミジンヌクレオシド−−デオキシシチジン、シトシンアラビノシド、
5−アザシトシン、ゲンシタビン、5−アザシトシン−アラビノシド;
3.プリン−−6−メルカプトプリン、チオグアニン、アザチオプリン、アロ
プリノール、クラドリビン(Cladribine)、フルダラビン、ペントス
タチン、2−クロロアデノシン;
4.白金アナログ−−シスプラチン、カルボプラチン、オキサリプラチン(O
xaliplatin)、テトラプラチン、白金−DACH、オルマプラチン(
Ormaplatin)、CI−973、JM−216;
5.アントラサイクリン/アントラセンジオン−−ドキソルビシン、ダウノル
ビシン、エピルビシン、イダルビシン、ミトキサントロン;
6.エピポドフィロトキシン−−エトポシド、テニポシド;
7.カンプトセシン−−イリノテカン、トポテカン、9−アミノカンプトセシ
ン、10,11−メチレンジオキシカンプトセシン、9−ニトロカンプトセシン
、TAS 103、7−(4−メチル−ピペラジノ−メチレン)−10,11−
エチレンジオキシ−20(S)−カンプトセシン、7−(2−N−イソプロピル
アミノ)エチル)−20(S)−カンプトセシン;
8.ホルモンおよびホルモンアナログ−−ジエチルスチルベストロール、タモ
キシフェン、トレメフィン、トルムデックス(Tolmudex)、チミタック
(Thymitaq)、フルタミド、ビカルタミド、フィナステリド、エストラ
ジオール、トリオキシフェン、ドロキシフェン(Droloxifene)、メ
ドロキシプロゲステロンアセテート、メゲステロールアセテート、アミノグルテ
チミド、テストラクトンなど;
9.酵素、タンパク質および抗体−−アスパラギナーゼ、インターロイキン、
インターフェロン、ロイプロリド、ペガスパラガーゼ(Pegaspargas
e)など;
10.ビンカアルカロイド−−ビンクリスチン、ビンブラスチン、ビノレルビ
ン、ビンデシン;
11.タキサン−−パクリタキセル、ドセタキセル。
【0040】
機構ベースのクラス:
1.抗ホルモン−ホルモンおよびホルモンアナログの分類を参照のこと、アナ
ストロゾール(Anastrozole);
2.抗葉酸(Antiforate)−−メトトレキサート、アミノプテリン
、トリメトレキサート、トリメトプリム、ピリトレキシム(Pyritrexi
m)、ピリメタミン、エダトレキサート(Edatrexate)、MDAM;
3.抗微小管剤−−タキサンおよびビンカアルカロイド;
4.アルキル化剤(古典的および非古典的)−−ナイトロジェンマスタード(
メクロレタミン、クロランブシル、メルファラン、ウラシルマスタード)、オキ
ザホスホリン(イフォスファミド、シクロホスファミド、パーホスファミド(P
erfosfamide)、トロホスファミド(Trophosphamide
))、アルキルスルフォネート(ブスルファン)、ニトロソ尿素(カルムスチン
、ロムスシン、ストレプトゾシン)、チオテパ、ダカルバジンなど;
5.代謝拮抗物質−−プリン、ピリミジンおよびヌクレオシド(上記);
6.抗生物質−−アントラサイクリン/アントラセンジオン、ブレオマイシン
、ダクチノマイシン、マイトマイシン、プリカマイシン(Plucamycin
)、ペントスタチン、ストレプトゾシン;
7.トポイソメラーゼインヒビター−−カンプトセシン(Topo I)、エ
ピポドフィロトキシン、m−AMSA,エリプチシン(Topo II);
8.抗ウイルス剤−−AZT、ザルシタビン(Zalcitabine)、ゲ
ンシタビン(Gemcitabine)、ジダノシンなど;
9.種々雑多な細胞傷害剤−−ヒドロキシ尿素、ミトーテン、融合毒素、PZ
A、ブリオスタチン(Bryostatin)、レチノイド、ブチル酸および誘
導体、ペントサン、フマギリンなど。
【0041】
全ての抗腫瘍薬剤の目的は、癌細胞の排除(治療)または癌細胞の増殖および
伝播の遅延(寛解)である。上記に列挙した抗腫瘍薬剤の大半は、主要な細胞傷
害活性を有し、癌細胞に対して直接的な殺傷をもたらすことによって、この目的
を追求する。他の抗腫瘍薬物は、身体の自然の免疫を刺激して、癌細胞の殺傷を
もたらす。文献は、上記の薬物の全ての活性および機構についての考察などで充
たされる。
【0042】
リポソームカンプトセシン、および特に、リポソームトポテカンの特的の処方
物を作製する例示的方法を、以下の実施例において示す。
【0043】
(III.リポソームカンプトセシンを使用する方法)
本発明のリポソーム組成物(例えば、カンプトセシン)は、動物(例えば、ヒ
ト)における固形腫瘍の処置において使用される。以下の実施例は、薬物:脂質
比、投与される活性薬剤および脂質の投薬量、および異なる腫瘍型を処置するた
めの好ましい投与スケジュールの重要なパラメーターを示す。
【0044】
好ましくは、薬学的組成物は、非経口的(すなわち、関節内、静脈内、腹腔内
、皮下または筋内)に投与される。より好ましくは、薬学的組成物は、静脈内点
滴によって投与されるか、またはボーラス注射によって腹腔内投与される。薬学
的処方物中のリポソームの濃度は、広範に、すなわち、約0.05重量%未満か
ら、通常、少なくとも約2〜5重量%〜多くて10〜30重量%で変化し得、そ
して選択される特定の投与様式に従って、流体容量、粘性などによって主に選択
される。例えば、濃度を増加して、処置に関連する流体負荷を低下させ得る。あ
るいは、刺激性脂質から構成されるリポソームを低い濃度に希釈して、投与部位
での炎症を小さくし得る。投与されるリポソームの量は、使用される特定のカン
プトセシン、処置される疾患状態および主治医の判断に依存するが、一般に、ヒ
トにおいて、約0.01mg/kg体重と約50mg/kg体重との間、好まし
くは、約5mg/kg体重と約40mg/kg体重との間である。より高い脂質
用量(例えば、50〜120mg/kg)が、マウスに適切である。
【0045】
活性薬剤(例えば、カンプトテシン)の投薬量は、患者の年齢、体重および状
態、ならびに処置計画に基づいて、投与する医師の意見に依存する。小細胞肺癌
における遊離トポテカンの推奨用量は、1用量あたり1.5mg/M2(5日間
毎日)であり、3週間ごとに繰り返される。以下の実施例で示された処置におけ
る改善が原因で、ヒトにおける活性薬剤(例えば、トポテカン)の用量は、0.
015mg/M2/用量程度の低さの範囲で有効であり、投与計画に依存して、
15〜75mg/M2/用量程度の高さでなお寛容可能である。用量は、単一用
量であり得、この用量は、4時間毎、6時間毎、または12時間毎、あるいは、
1日毎、2日毎、3日毎、4日毎、5日毎、6日毎、7日毎、8日毎、9日毎、
10日毎またはそれらの組み合わせで繰り返して投与され得る。好ましい計画は
、1週間毎、2週間毎、3週間毎、4週間毎、5週間毎、もしくは6週間毎また
はそれらの組み合わせで繰り返される処置サイクルを用い得る。現在好ましい実
施形態において、処置は、1週間に1回行われ、この用量は、代表的には、1.
5mg/M2未満である。
【0046】
特に好ましいトポテカン投薬量および計画は、以下のとおりである:
【0047】
【表1A】

本発明は、具体的実施例により、より詳細に記載される。以下の実施例は、例
示目的で提供され、如何様にも本発明を限定することを意図しない。当業者は、
重要ではない種々のパラメーターを容易に認識し、これらのパラメーターは、本
質的に同じ結果を得るために変更または改変され得る。
【0048】
(IV.実施例)
(A.材料および方法)
1.材料 トポテカン(HycamtinTM,SmithKline Be
echam)を、British Columbia Cancer Agen
cyの薬局から購入した。スフィンゴミエリン(SM)を、Avanti Po
lar Lipidsから購入した。Northern Lipidsのスフィ
ンゴミエリンを初期の研究において用いたが、Avantiバージョンのものよ
り、あまりエタノールに可溶性ではなかった。コレステロール(CH)および二
価の陽イオンイオノフォアA23187をSigmaから購入した。[3H]−
コレステリルヘキサデシルエーテル(Dupont)を脂質マーカーとして用い
た。
【0049】
2.マウス。雌性のICR、BDF−1または無胸腺nu/nu(6−8週齢
)を、Harlan−Sprague Dawley(Indianapoli
s,IN)から購入した。すべての動物を、使用する前に1週間にわたり隔離し
た。すべての研究を、Canadian Council on Animal
Care(CCAC)およびInstitutional Animal C
are and User Committee(IACUC)により制定され
たガイドラインに沿って行った。
【0050】
3.Mg−A23187法によるトポテカンの処方。トポテカンを、米国特許
第5,837,282号に従うMg−A23187イオノフォア法を用いて、S
M:CH(55:45,mol/mol)リポソーム中にカプセル化した。最初
の薬物 対 脂質比は、0.10(w/w)であり、薬物負荷は、代表的には、
95−100%であった。外用緩衝液(external buffer)は、
10mM PBS,pH7.5および300mM スクロースからなった。すべ
ての処方物を粒子サイズ、薬物負荷効率、pH、および薬物と脂質の濃度に関し
て分析した。
【0051】
4.薬物調製および投薬。トポテカン(HycamtinTM)の各バイアル
を、1.0mlの滅菌水で水和し、4.0mg/mlのトポテカン濃度にした。
引き続いて、薬物のラクトン種について必要な低pHを維持するために、0.9
%滅菌生理食塩水中で希釈した。水保存溶液中の非使用薬物(4.0mg/ml
)を、遮光して4℃で保存した。リポソームにカプセル化したトポテカンを、0
.9%生理食塩水で、投与に必要な濃度に希釈した。すべての薬物投与は、側方
尾静脈を介した10ml/kg(200μl/20gマウス)であった。
【0052】
5.薬物動態およびインビボ漏出研究。遊離トポテカンおよびリポソームカプ
セル化トポテカンの薬物動態および薬物漏出を、側方尾静脈を介したi.v.投
与後24時間にわたり、ICRマウスにおいて評価した。2つの異なる薬物 対
脂質比(すなわち、0.10(w/w)および0.02(w/w))を用いて
、薬物漏出およびPK挙動に対する薬物 対 脂質比および脂質用量の影響を試
験した。カプセル化されたトポテカンを、1mg/kg(10または50mg/
kg脂質)および5mg/kgトポテカン(50mg/kg脂質)で投与した。
対応して、遊離のトポテカンのPK挙動を、1mg/kgおよび5mg/kgで
評価した。血中総トポテカンを、血漿タンパク質の沈降後の蛍光アッセイにより
決定した。トポテカンを、それぞれ、380nmおよび518nmの励起波長(
2.5nmスリット幅)および発光波長(2.5nmスリット幅)で分光蛍光器
により定量した。血漿中の脂質レベルを、[3H]−CHE標識の液体シンチレ
ーション計測により決定した。
【0053】
6.MTD研究。MTD研究を、各腫瘍モデルに対応する宿主マウス系統にお
いて行った。単一用量および複数用量のMTDを、経時的な体重減少をモニター
することにより決定した。MTDを、20%体重減少を生じる用量と規定した。
【0054】
7.骨髄抑制および好中球減少症研究。トポテカン投与の結果としての末梢血
細胞レベルの変化を、4〜6週間にわたりICRマウスにおいて評価した。10
mg/kgの遊離トポテカンまたはリポソームカプセル化トポテカンをi.v.
投与して1日、3日、5日、7日、14日、および21日目に、血液を、EDT
A微量チューブ(microtainer tube)に採取した。空の小胞を
、コントロールとして投与した。CBCおよび差示的分析をCentral L
abs for Veterinarians(Langley,BC)で行っ
て、細胞レベル、比および形態を定量した。
【0055】
8.腫瘍モデル。標準的プロトコルで用いられるように、L1210マウス白
血病モデルおよびCT−26マウス結腸転移モデルを用いた。ヒトMX−1およ
びLX−1細胞株をDCTD Tumor Repository in Fr
ederick,MDから得た。これらの細胞株を、腫瘍フラグメントとして回
収し、3mm×3mmフラグメントの連続的移植によりNCrヌードマウスにお
いて増殖させた。細胞株がヌードマウスにおいて3継代を過ぎ、腫瘍株が継代数
が10に達したときに再開始するまで、実験を開始しなかった。
【0056】
9.効力研究。遊離トポテカンおよびリポソームトポテカンの投薬すべてを、
側方尾静脈を介した10ml/kgの静脈内経路により投与した。L1210お
よびCT−26モデルにおいて、投薬を1日目に行った(腫瘍細胞注入=0日目
)。MX−1およびLX−1腫瘍モデルについて、腫瘍容積を、腫瘍寸法の垂直
測定の繰り返しおよび以下の式を用いることにより決定した:
容積(mm3)=(L×W2)/2
腫瘍が明らかに増殖を示し、100−300mm3の範囲にある場合に、MX
−1モデルおよびLX−1モデルで投薬を開始した。
【0057】
大部分の薬物が生物学的効果と毒性との間で平衡を示すので、これらの属性の
両方を組み込むパラメーターを試験することは有用である。最も一般に用いられ
るパラメーターは、治療指数(therapeutic index)(TI)
である。伝統的には、治療指数は、以下のように規定されている:
TI=LD50/ED50
しかし、LD50研究を行うことはもはや許されないので、これらの研究につ
いての治療指数を、以下のように規定した:
TI=MTD/MED
上記の式において、MTDは最大寛容用量であり、動物の群において20%の
平均体重減少を引き起こす用量として規定される;そしてMEDは、最小有効用
量であり、固形腫瘍モデルにおいて40以下の最適%T/C値を生じる用量また
は生存モデルにおいて50±10%の%ILSを生じる用量として規定される。
【0058】
(B.結果)
1.薬物動態および薬物漏出。トポテカンの血漿薬物動態および薬物漏出に対
するリポソームカプセル化および薬物 対 脂質比の影響を、ICRマウスにお
いて24時間にわたり試験した。トポテカンのリポソームカプセル化(薬物 対
脂質比、0.11、wt/wt)は、この薬物の薬物動態パラメーターに対し
て劇的な影響を与えた(図1、上部;および表1を参照のこと)。5mg/kg
用量のトポテカンでは、遊離薬物に対するリポソーム薬物について、血漿AUC
において164倍の増加、Cmaxにおいて24倍の増加および血漿α半減期に
おいて24倍の増加が観察された(表1を参照のこと)。歴史的には、リポソー
ム薬物のAUCおよび血清半減期における大きな改善により、疾患部位(例えば
、腫瘍)への薬物の送達(「疾患部位標的化」として公知のプロセス)の増強が
得られた。
【0059】
この研究において用いた処方物を、Mg−A23187イオノフォア法により
調製した。iv投与して最初の10〜30分後に、最初に薬物が急激に放出され
(図1、下部を参照のこと)、続いてより緩やかな放出相が生じた。Mn−A2
3187処方物およびMg−A23187処方物についてのtl/2放出は、そ
れぞれ、約3時間および約5〜7時間であった;しかし、24時間では、いずれ
の処方物においても薬物はほとんど存在しなかった。
【0060】
大部分のリポソーム薬物処方物について、カプセル化薬物の薬物動態特性を、
脂質組成および用量により制御する。リポソームトポテカンは、非常に低い薬物
用量ですら(0.5mg/kg;薬物 対 脂質比、0.10、wt/wt)、
別格の抗腫瘍活性を示すことが示された。これらの薬物用量および薬物 対 脂
質比において、血漿からのリポソーム排除は、迅速であることが期待される。従
って、低用量でのトポテカンの薬物動態が改善され得るか否かを決定するために
、トポテカンの低い薬物 対 脂質比(0.02,wt/wt)処方物を調査し
た。興味深いことに、この研究において、低い薬物 対 脂質比の処方物が、よ
り高い薬物 対 脂質比(0.11,wt/wt)の処方物よりはるかに速く薬
物を放出した。この結果は予測外であった。
【0061】
【表1】
すべてのパラメーターは、WINNONLIN PKモデリングソフトウェアを
用いて、1または2区画モデルから導出した。
【0062】
2.最大寛容用量。単一および複数用量のMTD研究を、腫瘍保有Balb/
cマウス、BDF−1マウスおよびNCr nu/nuマウスにおいて行った。
個々のマウスの体重を、各研究の間中モニターして、遊離トポテカンおよびリポ
ソームトポテカンの全身的な寛容性を評価し、可能ならば、MTDを確立した(
図2を参照のこと)。リポソームトポテカンの最大寛容用量は、単一投与に関し
て10mg/kg、q7dx3スケジュールに関して7.5mg/kgおよびq
3dx4スケジュールに関して5mg/mlであった。マウスにおける1回の静
脈内注入後の遊離トポテカンの報告されたLD50は、75mg/M2(約25
mg/kg)であった[HycamtinTM製品小論];しかし、40mg/
kgまでの用量で体重減少はほとんど観察されなかったが、これは、急性応答に
起因したMTDと考えられた。薬物量は、制限されていたので、40mg/kg
より高い用量(5〜10分にわたり投与された)については追求しなかった。q
dx5スケジュールに対する遊離トポテカンのLD10は、14mg/M2/用
量(約4.7mg/kg/用量)であることが以前に示されている(Groch
ow,et al.,Drug Metab.Dispos.20:706−7
13(1992))。
【0063】
3.毒性。ヒトにおいて5日間連続(dx5)にわたり1.5mg/M2/用
量で毎日投与される遊離トポテカンの主要な用量制限毒性(MTD)は、非累積
性の骨髄抑制である。先に言及されたように、ヒトは、マウスよりも、骨髄抑制
に対してより感受性であり、マウスにおけるMTDのわずか11%を寛容し得る
にすぎない(14mg/M2に対して1.5mg/M2)。この点において、イ
ヌは、ヒトにおけるトポテカンの骨髄抑制のはるかに良好な予測者であることが
示された(Burris,et al.,J.Natl.Cancer Ins
t.84:1816−1820 (1992))。しかし、マウスは、遊離トポ
テカンおよびリポソームカプセル化トポテカンの相対的骨髄抑制効果を比較する
に適切であるはずである。
【0064】
1つの研究において、末梢WBC数の最大減少が、リポソームトポテカンを投
与して3日目に生じた。次いで、末梢血球レベルおよび形態の比較を、遊離トポ
テカンもしくはリポソームカプセル化トポテカンまたは空の小胞を投与して3日
目に行った(表2を参照のこと)。この比較に用いた用量は、リポソームカプセ
ル化トポテカンのMTDであった(10mg/kg)。リポソームトポテカンに
ついて遊離トポテカン(約10倍)、空の小胞(約10倍)、またはコントロー
ル動物(約20倍)と比べて、循環好中球の有意な減少が観察された。総WBC
レベルおよびリンパ球亜集団は、コントロール動物に対してリポソームトポテカ
ンについて約2倍減少した。同用量では、遊離トポテカンについてのこれらのパ
ラメーターにおいて、有意差はまったくなかった。注射して21日目に、リポソ
ームトポテカンについての総WBCレベルは、正常動物より約2.5倍低いまま
であった;しかし、好中球レベルは、正常マウスと比較して、20倍〜3倍の減
少まで回復した。リンパ球レベルは、正常マウスより約2倍低いままであった。
他の有意差は観察されなかった。
【0065】
注射して3日後の血清化学パラメーターの分析により、未処置動物に対してわ
ずかな変化が明らかになった(表3を参照のこと)。注意すべきわずかな変化は
、リポソームトポテカンで処置した動物に関するグロブリンレベルの統計的に有
意な増加(約2倍)およびアルブミン/グロブリン比の付随した減少であった。
他の有意な変化は観察されなかった。
【0066】
【表2】
(表3.遊離トポテカンまたはリポソームカプセル化トポテカンの10mg/
kg静脈内投薬量で処理したICRマウスの血清化学パネル−注射後3日目−)
【0067】
【表3】
(C.マウス腫瘍モデルおよびヒト腫瘍モデルにおける効力の研究:単回用量
研究)
(1.L1210マウス白血病) 静脈内L1210マウス白血病モデルが、
遊離の化学療法剤とリポソームカプセル化化学療法剤との間の差次的活性を評価
するために広範に使用されており、そしてこのモデルは、新規な化学療法剤のイ
ンビボNCIスクリーニングにおける元々の(1955〜1975)モデルのう
りの1つであった(Plowmanら、Human tumor xenogr
aft models in NCI drug development、「
Anticancer Drug Development Guide:Pr
eclinical Screening,Clinical Trials,
and Approval」(B.Teicher編)、Humana Pre
ss Inc.,Totowa(1997);Waud、Murine L12
10 and P388 leukemisas、「Anticancer D
rug Development Guide」Preclinical Sc
reening,Clinical Trials,and Approval
」(B.Teicher編)、Humana Press Inc.,Toto
wa(1997))。このモデルは、迅速であり−未処置動物の平均生存は代表
的には約7〜8日である−、そして投与された腫瘍細胞は、肝臓および骨髄に播
種される(seed)。
【0068】
遊離のトポテカンを単回静脈内用量として投与すると、L1210モデルにお
ける生存に対して、最小の効果しか有さなかった(図3Aを参照のこと)。最高
用量の遊離のトポテカンでは、生存中央値13日(44% ILS)が観察され
た。この群において、1つの長期生存体(60日)が存在した。対照的に、5m
g/kgまたは10mg/kgのいずれかのリポソームトポテカンの単回静脈内
(i.e.)投与は、60日目で100%の生存を生じた(図3Bを参照のこと
)。1mg/kg用量についての生存中央値は13日(44% ILS)であり
、そして生存曲線は、30mg/kgで投与された遊離トポテカンの生存曲線と
ほぼ同一であった−30倍の効力増強−。より高用量(30mg/kg)のリポ
ソームトポテカンでは、毒性死が観察された。リポソームトポテカンについての
MTDは、単回静脈内投与の後、BDF−1マウスにおいて20mg/kgであ
った。
【0069】
(2.CT−26マウス結腸癌) マウスCT−26結腸細胞株は、薬物スク
リーニングのために有用である。なぜなら、この細胞株は、皮下固形腫瘍として
容易に増殖するか、またはこの細胞株を静脈投与して生存モデルとして使用し得
るからである。さらに、この腫瘍細胞を、脾内(i.s.)注射により投与し、
その後、脾摘出術を行って、この細胞を、肝臓へと播種して実験的転移モデルを
生じる。このモデルは、結腸直腸癌の臨床経過によく似ている。このモデルは、
広範に使用されており、そして、例えば、他の箇所に詳細に記載される。
【0070】
このCT−26モデルにおいて、トポテカンの単回用量の投与は、生存に対し
て穏やかな影響を有し、用量範囲5〜40mg/kgにわたって23〜60%の
ILS%を生じた(図4を参照のこと)。しかし、リポソームカプセル化トポテ
カンは、5mg/kgより大きい用量では非常に活性であり、90日目で100
%の生存(8/8)を生じた。10mg/kgでは、87.5%の生存(7/8
)が90日目に観察されたが、死んだ動物の腫瘍負荷は非常に低く、このことは
、この動物が、他の要因(例えば、骨髄抑制に関連する感染)に起因して死んだ
かもしれないことを示唆する。リポソームトポテカンに関する用量応答が観察さ
れ、2mg/kg用量は、54%のILS%を生じた。これは、MEDであるこ
とが決定され、そしてこれは、40mg/kgの遊離トポテカンを使用して達成
されるILS%(58%)に匹敵した−20倍の効力増強−。
【0071】
(3.MX−1ヒト乳癌腫) MX−1は、ヒト乳癌の実験モデルであり、そ
して倍化時間3.9日を有することが報告されている。この研究において、倍化
時間中央値は、一貫して3.6〜3.7日であった。この腫瘍細胞株は、29年
齢の雌の原発性腫瘍に由来し、化学療法を受けた既往歴はなかった。そしてこの
腫瘍細胞株は、ヌードマウスにおいて連続継代される腫瘍フラグメントとしてD
CTD(NCI)腫瘍貯蔵所(tumor repository)により提供
される。組織学的に、MX−1は、ほどんど分化していない乳癌腫であり、腺形
成またはムチン産生の形跡はない。MX−1は、NCIインビボ腫瘍パネルおよ
びプレスクリーニング物(1976〜1986)を含んだ、新規な化学療法剤を
評価するための3つの異種移植片モデル(MX−1、LX−1、CX−1)のう
ちの1つであった(Plowmanら、Human tumor xenogr
aft models in NCI drug development.「
Anticancer Drug Development Guide:Pr
eclinical Screening,Clinical Trials,
and Approval」(B.Teicher編)、Humana Pre
ss Inc.,Totowa(1997))。その後、「化合物指向的」発見
から「疾患指向的」発見へのNCIの戦略のシフトを反映して、MX−1はより
大きな乳癌モデル群(計12)へと組み込まれた。
【0072】
進行した(staged)(100〜300mm3)MX−1腫瘍において、
遊離のトポテカンは、腫瘍増殖の用量依存性阻害を示した(図5;表1を参照の
こと)。最高用量(40mg/kg)において、最適T/C% 24%が得られ
たが、一方、10mg/kgおよび5mg/kgについての最適T/C%は、そ
れぞれ、66%および78%であった。薬物関連死は観察されず、そしてすべて
の動物は、この研究を通じて体重を増した。トポテカンのリポソームカプセル化
は、T/C%に対して顕著な影響を有し、2mg/kg、5mg/kgまたは1
0mg/kgの薬物の単回投与の後、それぞれ、最適T/C%が8%、−49%
および−62%であった。負のT/C%値は、もとの進行した(staged)
腫瘍サイズ(100〜300mm3)からの腫瘍体積の後退を示す。NCIのガ
イドラインによると、最適T/C%が<10%であることは、有意な活性と見な
され、一方、<42%の値は、薬物を開発へとさらに前進させるために許容可能
な最低限度である(Corbett,T.編、In vivo mehods
for screening and preclinical testin
g.「Anticancer Drug Development Guide
:Preclinincal Screening,Clinical Tri
als,and Approval」(B.Teicher編)Humana
Press Inc.,Totowa(1997))。リポソームカプセル化は
、トポテカンの毒性を増加し、遊離トポテカンについてのMIDを、>40mg
/kgから10mg/kgへと減少させた。
【0073】
(4.LX−1ヒト肺癌) LX−1は、ヒト小細胞癌(SCLC)の実験モ
デルである。この腫瘍細胞株は、48歳の男性において見出された転移病変の外
科的外植片に由来し、そしてヌードマウスにおいて連続継代される腫瘍フラグメ
ントとして、DCTD(NCI)腫瘍貯蔵所により提供される。このLX−1モ
デルは、1976〜1986のNCIインビボ腫瘍群の一部であった(Plow
man,J.ら、Human tumor xenograft models
in NCI drug development.「Anticancer
Drug Development Guide:Preclinical
Screening,Clinical Trials,and Approv
al」(B.Teicher編)、Humana Press Inc.,To
towa(1997))。現在は頻度を減らしてしか使用されないが、このLX
−1モデルは、その迅速な増殖速度が理由で、依然として、遊離薬物とリポソー
ム薬物との間の比較活性研究についての有用な異種移植片である。
【0074】
一般に、このLX−1モデルは、遊離薬物およびリポソームカプセル化薬物の
両方について、MX−1モデルよりもトポテカンの効果に対して感受性が低い(
図6;表1を参照のこと)。用量30mg/kg、10mg/kg、または5m
g/kgの遊離トポテカンについての最適T/C%は、それぞれ、43%、55
%、および67%であった。抗腫瘍活性が、カプセル化を介して改善され、用量
30mg/kg、10mg/kg、または5mg/kgについてのT/C%が、
それぞれ、8%、11%、および13%となった。興味深いことに、このリポソ
ームトポテカン用量のすべてが、類似する活性を示した。これは、初期の研究で
あり、他のモデルにおけるその後の研究(図4〜6を参照のこと)は、用量<5
mg/kgで始まる用量応答を示す。このことは、カンプトテシンクラス化合物
(およびおそらく他の抗腫瘍性薬剤)が「自己制限的」効力を示し得、それによ
り臨界閾値用量を超える用量では、さらなる活性の利点が全く観察されないとい
う知見(Thompson、Biochim.Biophys.Acta 14
00:301〜319(1998))と、一致する。この状況は、おそらく、そ
の薬物が制限された腫瘍細胞への接近を有する場合かまたはその薬物が腫瘍血管
に対して作用して破壊する(すなわち、抗脈管形成活性を有する)場合に、生じ
得る。両方の場合において、より高用量の薬物は、ごく小量の利点しか有さない
と予期される。
【0075】
L1210研究において観察されるように、トポテカンのカプセル化は、その
薬物の毒性を増強し、そしてそのMTDを減少した。腫瘍を保有するヌードマウ
スにおけるMTDは、10mg/kg(約16%の体重損失)であった。30m
g/kgでは、4/6の薬物関連毒性死が観察され、そして最大体重損失は、約
29%に達した(27〜34%範囲)。
【0076】
(D.マウス腫瘍モデルおよびヒト腫瘍モデルにおける効力研究:多数回用量
研究)
(1.MX−1ヒト乳癌腫) 複数回投与および薬物に対する腫瘍の長期曝露
の効果を調べるために、2つの複数用量プロトコル(q3dx4スケジュールお
よびq7dx3スケジュール)を、MX−1異種移植片において調べた。q4d
x3スケジュールにおいて、遊離トポテカンは、2.5mg/kg/用量および
10mg/kg/用量では中程度の活性を示し、1.25mg/kg/用量では
、最小の活性を示した(図7;表IIを参照のこと)。この投与スケジュールに
関する遊離トポテカンの最適T/C%値は、1.25mg/kg/用量、2.5
mg/kg/用量および10mg/kg/用量について、それぞれ、55%、3
0%、および27%であった。同じ投与スケジュールで投与されたカプセル化ト
ポテカンについて、最適T/C%値は、0.5mg/kg/用量、1.25mg
/kg/用量、2.5mg/kg/用量および5mg/kg/用量について、そ
れぞれ、15%、100%、100%、および100%であった。後退したすべ
ての腫瘍を、60日間モニターした。>1.25mg/kg/用量のリポソーム
トポテカンで処理したすべての動物は、この期間の終わりに、腫瘍を有さないと
見なされた。
【0077】
q7dx3投与スケジュールにおいて、5mg/kg/用量または10mg/
kg/用量のいずれの遊離トポテカンを用いても、ほとんど活性は観察されなか
った(図8;表IIを参照のこと)。同じ用量で、リポソームトポテカンは、進
行した(staged)腫瘍の完全な後退を誘導した。しかし、この投与スケジ
ュールにおいて、10mg/kg/用量は毒性があり過ぎた。そして、6/6毒
性死(または安楽死)が24日目に観察されたので、この研究のこの部分は停止
した。
【0078】
(2.LX−1ヒト肺癌) LX−1モデルにおける最初の研究(単回用量)
は、遊離トポテカンが評価用量<30mg/kgでは不活性であり、そしてリポ
ソームトポテカンは腫瘍増殖を阻害するが後退は誘導しないことを示した。この
活性を改善するために、複数回(q7dx3)スケジュールを、遊離トポテカン
およびリポソームトポテカンの両方について試験した。この場合、単回用量研究
と比較して、かなり大きな活性が遊離トポテカンについて観察され、30mg/
kg/用量および10mg/kg/用量について、それぞれ、最適T/C%値5
および40が得られた。リポソームトポテカンもまた、有意に改善した活性を示
し、5mg/kg/用量で完全な後退(およびその後の再増殖)を生じた。この
モデルにおけるリポソームトポテカンについての最適T/C%値は、5mg/k
g/日、2.5mg/kg/日、1.25mg/kg/日について、それぞれ、
55、3および16であった。
【0079】
(3.治療指数(TI)の比較) 遊離トポテカンおよびリポソームトポテカ
ンの治療指数を、いくつかの異なる投与スケジュールで4つの異なる腫瘍モデル
において評価した(表4を参照のこと)。これらの数を作製するために使用した
仮定および定義が、表IIIに見出される。いくつかの場合、真のMEDも真の
MTDも観察されなかった。従って、真のMEDも真のMTDを、用量応答トレ
ンドに基づいて数学的評価した。例えば、急性MTD 40mg/kgが、単回
ボーラス注射として投与された遊離トポテカンについて観察されたが、真のMT
D(重量損失に基づく)は、その薬物が5〜10分間にわたって注入された場合
には60mg/kgにより近づくようである。また、この分析をいくらか複雑に
したのは、リポソーム処方物の効力のレベルであった。低薬物用量にて、有意な
抗腫瘍活性が達成され、MEDが、特定の研究において評価されなければならな
かった。これらの場合、表4において注釈をつけた。
【0080】
一般に、リポソームトポテカンについての治療指数の増加は、単回用量投与に
ついて比較的大きく(モデルに依存して、5倍、10倍、15倍および18倍)
、そして投薬頻度が増加するにつれて減少した。このことが表4に示され、q7
dx3スケジュールおよびq3dx4スケジュールについて、それぞれ、TIT
CS/TIFree比は、4.7〜7.5および3.3であった。投与がより頻
繁になるにつれてこのTITCS/TIFree比が減少することは、遊離トポ
テカンの効力および毒性がスケジュール依存性であることを示す前臨床研究およ
び臨床研究と一致する。
【0081】
(表4.マウス腫瘍モデルおよびヒト腫瘍モデルにおける遊離トポテカンおよ
びリポソームトポテカンの相対的治療指数a)
【0082】
【表4】
a 表IIおよび表IIIのデータ;表IVにおける式および定義に基づく
b 急性MTD 40mg/kgを使用して得た;第2の値は、推定MTD(
体重)に基づく
c 約2倍大きくあり得る控えめな推定値;低用量での高い活性に起因して、
このMEDを評価することは困難である。
【0083】
(E.考察)
トポテカンは、リポソームカプセル化のための優れた候補である。簡単に述べ
ると、トポテカンは、細胞周期(S期)特異的であり、そして長期の曝露ととも
に活性が大いに増強され、トポテカンは、迅速な血漿薬物動態を示し、そしてこ
の薬物は、生物学的活性を保持するためにpH6.0より下で維持される必要が
ある。これは、酸性水性コアを有する比較的非漏出性のリポソーム処方物(例え
ば、SM:CH、55:45)を使用するための理想的シナリオである。必要な
酸性内部は、例えば、pHローディング法またはイオノフォアローディング法に
よって、作製され得る。ここで、Mg−A23187法によるSM/CHリポソ
ーム中へのトポテカンのカプセル化は、抗腫瘍効力の劇的な増強を生じることを
示した。毒性の中程度の増強もまた、リポソームトポテカンについて観察される
が、これは、遊離薬物に対して匹敵する効力(および多くの場合はより優れた効
力)を達成する、実質的用量の減少によって、大きく相殺された。
【0084】
治療指数(TI)は、薬物活性の有用なパラメーターである。なぜなら、それ
は、生物学的活性(ユーザーが規定する指標、すなわち、MED、ED50また
はED80)に対する毒性(MTD)の比の尺度であるからである。一般に、T
Iが低い程、毒性の危険が高い。なぜなら、生物学的効果を惹起するために必要
な薬物の容量は、MTDに近づくからである。治療指数は、リポソーム薬物の評
価のために特に有用である。なぜなら、TIの相対的変化は、カプセル化の利点
(またはその欠如)を規定するために使用され得るからである。本明細書中に示
されるように、TIは、3倍から18倍に改善され、それは、使用されるモデル
および投与スケジュールに依存する。従って、トポテカンのリポソームカプセル
化後に観察される生物学的活性の改善は、いかなる毒性の増加も補償するにとど
まらない。
【0085】
いかなる理論によっても拘束されることを意図しないが、抗腫瘍活性の有意な
改善およびリポソーム形態の薬物の毒性増加は、薬物動態の改善および活性ラク
トン形態での薬物の維持から生じると考えられる。これらの研究において、24
時間後には、84%のトポテカンが、血漿中にラクトン種として存在したのに対
して、たった5分後には遊離トポテカンについて48%のラクトンが存在した。
さらに、同じ用量(10mg/kg)の遊離トポテカンおよびリポソームトポテ
カンを、マウスに静脈投与した場合、ラクトン濃度は、<1時間の時点で、約4
0倍高かった。24時間にて、リポソーム薬物についてのラクトン血漿濃度は、
5.4μg/mlであったのに対して、遊離の薬物について5分で1.5μg/
mgであり、遊離トポテカンについてのピークラクトン濃度よりなお3.5倍高
かった。
【0086】
(表I)
(単回用量抗腫瘍活性および毒性パラメーターの要旨)
【0087】
【表5】
a 最終処理後の最適T/C%。負の値は、腫瘍後退を示す。
b 腫瘍増殖遅延(処理した腫瘍およびコントロール腫瘍が500mm3に達す
るための時間の差異)
c 未処理腫瘍に対する寿命の増加(%として表す)
d log(細胞殺傷)(量)
e 研究終了時に腫瘍を有さない動物(すなわち、可視的腫瘍がないか、または
長期生存体)
f 薬物関連死
g 処理群当たりの最大平均重量損失
h 正の重量変化(すなわち、処理前の重量を下回る重量低下が、どの時点でも
なかった)
** 長期生存体
(表II)
(複数回用量抗腫瘍活性および毒性パラメーターの要旨)
【0088】
【表6】
a 最終処理後の最適T/C%。負の値は、腫瘍後退を示す。
b 腫瘍増殖遅延(処理した腫瘍およびコントロール腫瘍が500mm3に達す
るための時間の差異)
c 未処理腫瘍に対する寿命の増加(%として表す)
d log(細胞殺傷)(量)
e 研究終了時に腫瘍を有さない動物(すなわち、可視的腫瘍がないか、または
長期生存体)
f 薬物関連死
g 処理群当たりの最大平均重量損失
h 正の重量変化(すなわち、処理前の重量を下回る重量低下が、どの時点でも
なかった)
i 測定せず;リポソームカプセル化群における毒性死
** 長期生存体;60日目まで可視的腫瘍が存在しない。
【0089】
表III.
毒性および抗腫瘍活性パラメーターについての規定および式
DRD 薬物関連死亡。動物が死亡するか、または薬物ANDでの最終処置後
15日以内に安楽死させたか、その腫瘍重量がコントロールマウスに対する致死
的負荷より小さいか、またはその体重減少が、コントロール動物の体重減少より
20%を超えた場合に、死亡が薬物に関連するとみなした。
【0090】
GI50 インビトロでの細胞の集団において50%増殖阻害を引き起こす薬
物の濃度。NCIは、時間0の時点で細胞数についての補正を強調するためにI
C50パラメーターを改名した。従って、式は、以下のとおりである:
GI50=(T−T0)/(C−T0)×100=50
TおよびT0は、それぞれ、48時間および0時間での光学密度である;Cは0
時間でのコントロール(細胞数)光学密度である。
【0091】
%ILS 寿命の増加(%)。生存モデルについて、処置動物(T処置)およ
び未処置動物(Tコントロール)についてメジアン生存時間を用いて、以下に従
って、これを計算した:
(T処置−Tコントロール)/Tコントロール×100
固形腫瘍モデルについては、腫瘍が2000mm3(体重の約10%)に達する
までの時間を、メジアン生存の代わりに倫理的カットオフとして用いた。
【0092】
LCK 細胞殺傷の対数(総数)。このパラメーターは、処置の最後に殺傷さ
れた細胞のlog10単位の数を以下の式に従って推定する:
(T−C)×0.301/メジアン倍化時間
正味の細胞殺傷の対数は、以下のように、腫瘍増殖遅延(T−C)パラメーター
から処置の持続時間を差し引きすることにより計算され得る:
[(T−C)−処置の持続時間]×0.301/メジアン倍化時間
0の細胞殺傷の対数は、処置の最後における細胞集団が、処置の開始における細
胞集団と同じであることを示す。しかし、例えば、4の細胞殺傷の対数は、開始
細胞集団における99.99%減少を示す。
【0093】
MBWL 最大体重減少(%)。動物を、最初の薬物投与の前(Wi)および
研究の間の種々の日(Wd)に体重測定した。体重の%変化は、以下により計算
される:
MBWL=(Wd−Wi)/Wi×100
MED 最小有効用量。これは、いくらか任意のパラメーターである。これら
の研究について、本発明者らは、40以下の最適%T/C(固形腫瘍モデルにつ
いて)または40〜60%の%ILS(生存モデルについて)を達成する最低用
量としてMEDを規定した。
【0094】
MTD 最大寛容用量。20%以下のMBWLを生じる薬物用量。
【0095】
%T/C 処置の最初の過程後に得られたコントロール腫瘍に対する処置腫瘍
の最適比。これらの値は、以下の式に従って、各観察日での腫瘍重量から、処置
の初日でのメジアン腫瘍重量(TiまたはCi)を差し引きすることにより得ら
れる:
%T/C=(ΔT/ΔC)×100(ここで、ΔT>0)または
%T/C=(ΔT/Ti)×100(ここで、ΔT<0)
NCI活性基準に従って、以下のスコア付けシステム(Plowman,et
al,Human tumor xenograft models in N
CI drug development、「Anticancer Drug
Development Guide:Preclinical Scree
ning,Clinical Trials,and Approval」(B
. Teicher,Ed.),Humana Press Inc.,Tot
owa(1997)[22])を適用する:
0=不活性、%T/C>40
1=腫瘍阻害、%T/C範囲は1〜40
2=腫瘍静止、%T/C範囲は0〜−40
3=腫瘍退行、%T/C範囲は−50〜−100
4=%T/C範囲は−50〜−100および>30%(腫瘍なしマウス)
。
【0096】
TGD 腫瘍増殖遅延(T−Cともあらわされる)。このパラメーターは、任
意のサイズ(代表的には、500または1000mm3)を得るために処置腫瘍
および未処置腫瘍についての時間差(日数で)をあらわす。
【0097】
TI 治療指数。治療指数は、毒性パラメーター(すなわち、LD50、LD
10、MTD)および生物学的活性パラメーター(すなわち、ED50−処置群
の50%において、規定された生物学的応答を生じる用量)の比である。一般に
、TIは、薬物についての安全性の限界を示す。動物モデル研究について、これ
は、伝統的には、以下の式によって記載される:
TI=LD50/ED50
しかし、LD50研究を行うことは、もはや倫理的に許されないので、本発明者
らは、これらの研究のために、以下のように治療指数を規定した:
TI=MTD/MED。
【0098】
上記の説明は、例示であって、制限するものでないと意図することが理解され
るべきである。多くの実施形態は、上記の説明を読めば当業者に明らかである。
従って、本発明の範囲は、上記の説明を参照して決定されるのではなく、添付の
特許請求の範囲を参照してこのような特許請求の範囲により権利が与えられる等
価物の全範囲とともに決定されるべきである。全ての学術文献および参考文献(
特許出願および刊行物を含む)の開示は、全ての目的のために、本明細書中に参
考として援用される。
【技術分野】
【0001】
(関連出願の相互参照)
本出願は、「Liposomal Camptothecins and U
ses Thereof」と表題された、2000年6月30日出願の米国仮出
願第60/215,556号、および「Liposomal Antineop
lastic Drugs and Uses Thereof」と表題された
、2001年1月26日出願の米国仮出願第60/264,616号に関し、こ
れらの両方が、全ての目的について本明細書中にこれらの全体が参照として援用
される。「Liposomal Camptothecins and Use
s Thereof」と表題された、2001年6月29日出願の米国特許出願
第_号(代理人整理番号016303−008020を有する)は、全ての目的
について本明細書中に参照として援用される。
【背景技術】
【0002】
(発明の背景)
本発明は、リポソーム組成物、および新生物を処置するためおよび新脈管形成
を阻害するためにこのような組成物を使用する方法に関する。
【0003】
多くの抗癌剤または抗新生物薬物が、リポソーム中にカプセル化されている。
これらとしては、アルキル化剤、ニトロソウレア、シスプラチン、代謝拮抗剤、
およびアントラサイクリンが挙げられる。アントラサイクリン抗生物質を含むリ
ポソームを用いる研究は、遊離薬物を与えられるコントロールと比較して、心毒
性および皮膚毒性の減少、ならびに腫瘍を保有する動物の長期の生存を明らかに
示した。
【0004】
リポソーム抗癌剤は、それらの遊離薬物対照物と比較して薬物動態学を改変す
る。リポソーム薬物処方について、薬物動態学は、キャリアが血液から取り除か
れる速度、およびその薬物がキャリアから放出される速度によってほぼ決定され
る。血液からの遅いクリアランスを示すリポソームキャリア組成物を同定するた
めに、かなりの努力がなされており、そして、長循環(long−circul
ating)キャリアが、多くの科学論文および特許に記載されている。例えば
、コントロール放出に対する膜ポテンシャルを使用する、リポソームキャリアか
らの薬物漏出速度を制御するための努力もまた、なされた。
【0005】
治療カンプトセシン(例えば、トポテカン(9−ジメチルアミノメチル−10
−ヒドロキシ−カンプトセシン;HycamtinTM)、およびイリノテカン
は、カンプトセシンの半合成の水可溶性誘導体であり、シナ蝋Camptoth
eca acuminataの幹から抽出されるアルカロイドである(Wall
ら、J.Am.Chem.Soc.88:3888−3890(1966))。
カンプトセシンは、トポイソメラーゼインヒビタークラスの抗新生物剤に属し、
DNA複製に関連する核酵素トポイソメラーゼIの作用を特異的に阻害する(H
siangら、Cancer Res.48:1722−1726(1988)
)。このように、トポテカンは、細胞周期特異的作用機構を示し、S期(DNA
複製)の間に作用して、G2細胞周期阻止およびアポトーシスを最終的に導く、
DNA中の不可逆的な二本鎖崩壊を生じる。遊離形態において、この薬剤は、腫
瘍細胞株およびマウス同種移植片腫瘍モデルおよびヒト異種移植片腫瘍モデルに
わたって広範囲の活性を有する(McCabe,F.L.ら、Cancer I
nvest 12:308−313(1994);Emersonら、Canc
er Res.55:603−609(1995);Thompson,Bio
chim.Biophys.Acta 1400:301−319(1998)
;Ormrodら、Drugs 58:533−551(1999);Hard
manら、Anticancer Res.19:2269−2274(199
9))。より最近には、トポテカンが、抗腫瘍作用機構に寄与し得る強力な抗血
管形成性特性を有するということが示された(O’Learyら、Clin.C
ancer Res.5:181−187(1999);Clementsら、
Cancer Chemother.Pharmacol.44:411−41
6(1999))。これらの処置全てが、用量制限毒性(例えば、貧血(ana
emia)、好中球減少症および血小板減少症を導く非蓄積性の骨髄抑制(my
elosuppression))、ならびに粘膜症(musositis)お
よび下痢を含む胃腸関連毒性に関連する。臨床的には、トポテカンは、卵巣癌お
よび小細胞肺癌(SCLC)における第二期治療(second−line t
herapy)について認可されており、そして現在広範な臨床的評価の焦点で
ある。
【0006】
カンプトセシンの脂質処方物は、治療剤として提唱されてきた(米国特許第5
,552,156号およびPCT公開WO95/08986を参照のこと)。し
かし、脂質処方物の全てが、薬物送達の目的について等しいというのではなく、
広範な研究が、薬物の充填および貯蔵、薬物投与、薬学的動態、生物学的分布、
漏出速度、腫瘍蓄積、毒性プロファイルなどについて好ましい特性を示す処方物
に関して継続中である。カンプトセシンに対して、この分野は、さらに複雑であ
る。なぜなら、ヒトにおける用量制限毒性は、マウスにおけるものより1/10
の低さであり得るからである(Erickson−Millerら、Cance
r Chemother.Pharmacol.39:467−472(199
7))。
【発明の概要】
【発明が解決しようとする課題】
【0007】
抗新生物剤の改善されたリポソーム処方物は、非常に有用であることを証明し
得る。新規の臨床的有用性を有する、脂質処方された抗新生物剤を提供すること
が、本発明の目的である。
【課題を解決するための手段】
【0008】
(発明の要旨)
本発明は、活性薬剤(例えば、トポテカン)の血漿循環半減期を調節するため
に有用な組成物および方法を提供する。リポソーム処方物は、臨床的効力を増加
し、そして付帯的な毒性を減少する。さらに、本発明は、新生物の処置および新
脈管形成の阻害のための方法およびリポソーム組成物を提供する。
【0009】
このように、1つの実施形態において、本発明は、活性薬剤の血漿循環半減期
を調節するための方法を提供し、本方法は、以下の工程を包含する:(a)リポ
ソームを提供する工程であって、該リポソーム中に、遊離活性薬剤と沈殿した活
性薬剤がカプセル化されている、工程;ならびに(b)このリポソーム中に沈殿
した活性薬剤の量を変化させる工程。驚くことに、リポソーム中に沈殿した活性
薬剤の量を変化させることによって、血漿中への活性薬剤の放出動態を調節する
ことが可能である。好ましい活性薬剤は、抗新生物薬物(例えば、カンプトセシ
ン(例えば、トポテカン))である。
【0010】
別の実施形態において、本発明は、a)抗新生物薬物とb)遊離抗新生物薬物
および沈殿された抗新生物薬物を有するリポソーム、とを含むリポソーム処方物
を提供し、ここで、このリポソーム中に沈殿した抗新生物薬物は、抗新生物薬物
全体の少なくとも50%である。リポソーム中に沈殿した抗新生物薬物の量を調
整することによって、この薬物の放出をインビトロおよびインビボの両方で調節
し得る。特定の好ましい実施形態において、高いリポソーム内濃度の活性薬剤(
例えば、トポテカン)は、多量の沈殿形態を生じる。この局面において、その後
のインビボでの薬物の放出速度は、遅い。特定の局面において、遅い放出速度は
、速い放出速度と比べて好ましくかつより効果的である。
【0011】
なお別の実施形態において、本発明は、a)活性薬剤;b)その中にカプセル
化された、遊離活性薬剤と沈殿した活性薬剤とを有するリポソーム;ならびに、
c)空のリポソーム、を含む、リポソーム処方物を提供する。
【0012】
この局面において、リポソームの血清半減期は、空のリポソームを処方物中に
含むことによって長くされる。種々の脂質のいずれかを使用して本発明のリポソ
ーム組成物を形成し得ることは、当業者には容易に明らかである。現在好ましい
実施形態において、この脂質は、スフィンゴミエリンおよびコレステロールの混
合物を含み、好ましくは、約30:70〜約60:40のスフィンゴミエリン:
コレステロール比(モル比)である。1つの好ましい実施形態において、このリ
ポソームは、55:45比でスフィンゴミエリンおよびコレステロールを含む。
【0013】
さらに別の局面において、本発明は、固形腫瘍を、それを罹患するヒトにおい
て処置するための方法を提供し、本方法は、薬学的に受容可能なキャリア中にあ
る有効量の本発明のリポソーム処方物を、このヒトに投与する工程を包含する。
種々の固形腫瘍は、本発明の処方物を使用して処置され得る。好ましい実施形態
において、処置される固形腫瘍は、肺の固形腫瘍、乳房の固形腫瘍、結腸の固形
腫瘍および前立腺の固形腫瘍からなる群より選択される。別の好ましい実施形態
において、本発明はさらに、好中球減少症および血小板欠損を処置するために適
切な処置または活性薬剤の同時投与を包含する。
【0014】
好ましい実施形態において、リポソームトポテカンを使用して、固形腫瘍を処
置する。さらに、種々の脂質のいずれを使用しても本発明のリポソーム組成物を
形成し得ることは、当業者には容易に明らかである。
【0015】
本発明の他の特性、目的および利点、ならびにその好ましい実施形態は、以下
の詳細な説明から明らかになる。
例えば本願発明は以下の項目を提供する。
(項目1) 活性薬剤の血漿循環半減期を調節するための方法であって、
該方法は、以下:
(a) リポソームを提供する工程であって、該リポソーム中に、遊離の活性
薬剤および沈殿した活性薬剤がカプセル化されている、工程;および
(b) 該リポソーム中の沈殿した該活性薬剤の量を変化させる工程、
を包含する、方法。
(項目2) 前記工程(b)が、前記活性薬剤 対 脂質の割合を変化さ
せる工程を包含する、項目1に記載の方法。
(項目3) 前記活性薬剤 対 脂質の割合が、空のリポソームの添加に
より変化する、項目2に記載の方法。
(項目4) 前記工程(b)が、前記リポソームのサイズを変化させる工
程を包含する、項目1に記載の方法。
(項目5) 前記工程(b)が、前記活性薬剤の沈殿を増強する成分を添
加する工程を包含する、項目1に記載の方法。
(項目6) 前記成分が、モノアニオン、ジアニオン、トリアニオン、ま
たは多価アニオンである、項目5に記載の方法。
(項目7) 前記工程(b)が、前記活性薬剤 対 脂質の割合、および
前記リポソームのサイズの両方を変化させる工程を包含する、項目1に記載の
方法。
(項目8) 前記活性薬剤が、抗新生物薬物である、項目1に記載の方
法。
(項目9) 前記抗新生物薬物が、カンプトセシンである、項目8に記
載の方法。
(項目10) 前記カンプトセシンが、イリノテカン、トポテカン、9−
アミノカンプトセシン、10,11−メチレンジオキシカンプトセシン、9−ニ
トロカンプトセシン、TAS103、7−(4−メチル−ピペラジノ−メチレン
)−10,11−エチレンジオキシ−20(S)−カンプトセシン、および7−
(2−N−イソプロピルアミノ)エチル)−20(S)−カンプトセシンからな
る群より選択されるメンバーである、項目9に記載の方法。
(項目11) 前記カンプトセシンが、トポテカンである、項目10に
記載の方法。
(項目12) 前記活性抗新生物薬物が、ビンカアルカロイドである、請
求項1に記載の方法。
(項目13) 前記ビンカアルカロイドが、ビンクリスチン、ビンブラス
チン、ビンオレルビン、およびビンデシンからなる群より選択されるメンバーで
ある、項目12に記載の方法。
(項目14) 前記リポソーム中にカプセル化される沈殿した活性薬剤が
、該活性薬剤の総量の少なくとも50%である、項目1に記載の方法。
(項目15) 前記リポソーム中にカプセル化される沈殿した活性薬剤が
、該活性薬剤の総量の少なくとも60%である、項目14に記載の方法。
(項目16) 前記リポソーム中にカプセル化される沈殿した活性薬剤が
、該活性薬剤の総量の少なくとも70%である、項目15に記載の方法。
(項目17) 前記リポソームが、スフィンゴミエリンおよびコレステロ
ールを含む、項目1に記載の方法。
(項目18) 前記リポソームが、55:45の割合でスフィンゴミエリ
ンとコレステロールとを含む、項目17に記載の方法。
(項目19) 前記活性薬剤の血漿循環半減期が、最適効力に調節される
、項目1に記載の方法。
(項目20) 前記活性薬剤 対 脂質の割合が、約0.005〜1:1
(w/w)である、項目1に記載の方法。
(項目21) 前記活性薬剤 対 脂質の割合が、約0.05〜0.9:
1(w/w)である、項目20に記載の方法。
(項目22) 前記活性薬剤 対 脂質の割合が、約0.1〜0.5:1
(w/w)である、項目21に記載の方法。
(項目23) 活性薬剤の血漿循環半減期を調節するための方法であって
、該方法は、以下:
(a) リポソームを提供する工程であって、該リポソーム中に、遊離の活性
薬剤および沈殿した活性薬剤がカプセル化されている、工程;および
(b) 該リポソームに非カプセル化活性薬剤を添加する工程、
を包含する、方法。
(項目24) 活性薬剤を含むリポソーム 対 カプセル化された薬剤を
有さないリポソームの割合が、約1:0.5〜1:1000である、項目23
に記載の方法。
(項目25) 活性薬剤を含むリポソーム 対 カプセル化された薬剤を
有さないリポソームの割合が、約1:1〜1:100である、項目24に記載
の方法。
(項目26) 活性薬剤を含むリポソーム 対 カプセル化された薬剤を
有さないリポソームの割合が、約1:2〜1:10である、項目25に記載の
方法。
(項目27) 活性薬剤を含むリポソーム 対 カプセル化された薬剤を
有さないリポソームの割合が、約1:3〜1:5である、項目26に記載の方
法。
(項目28) 前記活性薬剤が、抗新生物薬物である、項目23に記載
の方法。
(項目29) 前記抗新生物薬物が、カンプトセシンである、項目28
に記載の方法。
(項目30) 前記カンプトセシンが、イリノテカン、トポテカン、9−
アミノカンプトセシン、10,11−メチレンジオキシカンプトセシン、9−ニ
トロカンプトセシン、TAS103、7−(4−メチル−ピペラジノ−メチレン
)−10,11−エチレンジオキシ−20(S)−カンプトセシン、および7−
(2−N−イソプロピルアミノ)エチル)−20(S)−カンプトセシンからな
る群より選択されるメンバーである、項目29に記載の方法。
(項目31) 前記カンプトセシンが、トポテカンである、項目30に
記載の方法。
(項目32) リポソーム処方物であって、該リポソーム処方物は、以下
:
a) 抗新生物薬物;および
b) 遊離の抗新生物薬物および沈殿した抗新生物薬物を有するリポソーム、
を含み、ここで、該リポソーム中の沈殿した抗新生物薬物は、抗新生物薬物の総
量の少なくとも50%である、処方物。
(項目33) 前記抗新生物薬物が、カンプトセシンである、項目32
に記載のリポソーム処方物。
(項目34) 前記カンプトセシンが、イリノテカン、トポテカン、9−
アミノカンプトセシン、10,11−メチレンジオキシカンプトセシン、9−ニ
トロカンプトセシン、TAS103、7−(4−メチル−ピペラジノ−メチレン
)−10,11−エチレンジオキシ−20(S)−カンプトセシン、および7−
(2−N−イソプロピルアミノ)エチル)−20(S)−カンプトセシンからな
る群より選択されるメンバーである、項目33に記載のリポソーム処方物。
(項目35) 前記カンプトセシンが、トポテカンである、項目34に
記載のリポソーム処方物。
(項目36) 前記活性抗新生物薬物が、ビンカアルカロイドである、請
求項33に記載のリポソーム処方物。
(項目37) 前記遊離の抗新生物薬物と前記沈殿した抗新生物薬物とが
異なる、項目32に記載のリポソーム処方物。
(項目38) 前記ビンカアルカロイドが、ビンクリスチン、ビンブラス
チン、ビンオレルビン、およびビンデシンからなる群より選択されるメンバーで
ある、項目36に記載のリポソーム処方物。
(項目39) 前記抗新生物薬物 対 脂質の割合が、約0.005〜1
:1(w/w)である、項目32に記載のリポソーム処方物。
(項目40) 前記抗新生物薬物 対 脂質の割合が、約0.05〜0.
9:1(w/w)である、項目39に記載のリポソーム処方物。
(項目41) 前記抗新生物薬物 対 脂質の割合が、約0.1〜0.5
:1(w/w)である、項目40に記載のリポソーム処方物。
(項目42) 前記リポソームが、スフィンゴミエリンおよびコレステロ
ールを含む、項目32に記載のリポソーム処方物。
(項目43) 前記リポソームが、55:45の割合でスフィンゴミエリ
ンとコレステロールとを含む、項目42に記載のリポソーム処方物。
(項目44) カプセル化された活性薬剤を含まないリポソームをさらに
含む、項目32に記載のリポソーム処方物。
(項目45) 活性薬剤を含むリポソーム 対 カプセル化された薬剤を
有さないリポソームの割合が、約1:0.5〜1:1000である、項目44
に記載のリポソーム処方物。
(項目46) 活性薬剤を含むリポソーム 対 カプセル化された薬剤を
有さないリポソームの割合が、約1:1〜1:100である、項目45に記載
のリポソーム処方物。
(項目47) 活性薬剤を含むリポソーム 対 カプセル化された薬剤を
有さないリポソームの割合が、約1:2〜1:10である、項目46に記載の
リポソーム処方物。
(項目48) 活性薬剤を含むリポソーム 対 カプセル化された薬剤を
有さないリポソームの割合が、約1:3〜1:5である、項目47に記載のリ
ポソーム処方物。
(項目49) リポソーム処方物であって、該リポソーム処方物は、以下
:
a) 活性薬剤;
b) リポソームであって、該リポソーム中に、遊離の活性薬剤および沈殿し
た活性薬剤がカプセル化されている、リポソーム;および
c) 空のリポソーム、
を含む、リポソーム処方物。
(項目50) 前記活性薬剤を含むリポソーム 対 前記空のリポソーム
の割合が、約1:0.5〜1:1000である、項目49に記載のリポソーム
処方物。
(項目51) 前記活性薬剤を含むリポソーム 対 前記空のリポソーム
の割合が、約1:1〜1:100である、項目50に記載のリポソーム処方物
。
(項目52) 前記活性薬剤を含むリポソーム 対 前記空のリポソーム
の割合が、約1:2〜1:10である、項目51に記載のリポソーム処方物。
(項目53) 前記活性薬剤を含むリポソーム 対 前記空のリポソーム
の割合が、約1:3〜1:5である、項目52に記載のリポソーム処方物。
(項目54) 前記活性薬剤が、抗新生物薬物である、項目49に記載
のリポソーム処方物。
(項目55) 前記抗新生物薬物が、カンプトセシンである、項目54
に記載のリポソーム処方物。
(項目56) 前記カンプトセシンが、イリノテカン、トポテカン、9−
アミノカンプトセシン、10,11−メチレンジオキシカンプトセシン、9−ニ
トロカンプトセシン、TAS103、7−(4−メチル−ピペラジノ−メチレン
)−10,11−エチレンジオキシ−20(S)−カンプトセシン、および7−
(2−N−イソプロピルアミノ)エチル)−20(S)−カンプトセシンからな
る群より選択されるメンバーである、項目55に記載のリポソーム処方物。
(項目57) 前記カンプトセシンが、トポテカンである、項目56に
記載のリポソーム処方物。
(項目58) 前記活性抗新生物薬物が、ビンカアルカロイドである、請
求項57に記載のリポソーム処方物。
(項目59) 前記ビンカアルカロイドが、ビンクリスチン、ビンブラス
チン、ビンオレルビン、およびビンデシンからなる群より選択されるメンバーで
ある、項目58に記載のリポソーム処方物。
(項目60) 前記活性薬剤 対 脂質の割合が、約0.005〜1:1
(w/w)である、項目49に記載のリポソーム処方物。
(項目61) 前記活性薬剤 対 脂質の割合が、約0.05〜0.9:
1(w/w)である、項目60に記載のリポソーム処方物。
(項目62) 前記活性薬剤 対 脂質の割合が、約0.1〜0.5:1
(w/w)である、項目61に記載のリポソーム処方物。
(項目63) 前記リポソームが、スフィンゴミエリンおよびコレステロ
ールを含む、項目49に記載のリポソーム処方物。
【図面の簡単な説明】
【0016】
【図1】図1は、ビノレルビンのリポソーム処方物の薬物動態学的挙動を示す。パネルAは、異なる薬物:脂質比(0.1:1、0.2:1、0.3:1)の3つの処方物について、循環するキャリアからの薬物漏出速度を示す。薬物放出は、薬物:脂質比に依存し、最小の放出速度が最高の比(0.3:1)について見られる。パネルBは、血液中での脂質回復を示す。パネルCは、キャリアからの薬物放出速度の調節がビノレルビンについての血液クリアランス半減期に対する変化を生じることを示す。
【図2】図2は、血漿薬物レベルが薬物動態に従って使用される場合の、対応する挙動を示す。パネルAは、薬物貯留 対 時間を示す。パネルBは、脂質回復 対 時間を示す。パネルCは、薬物回復 対 時間を示す。
【図3】図3は、薬物:脂質比の関数として、リポソームビンブラスチンの処方物の薬物動態学的挙動(血液PK)を示す。リポソームキャリアからの薬物漏出は、最初の薬物:脂質比によって決定され、より高い薬物比の処方物について、より遅い放出を伴う。パネルAは、薬物貯留 対 時間を示す。パネルBは、脂質回復 対 時間を示す。パネルCは、薬物放出速度が血液からの薬物クリアランス半減期に対する変化と相関することを示す。
【図4】図4は、薬物:脂質比の関数として、リポソームビンブラスチンの処方物の薬物動態学的挙動(血漿PK)を示す。パネルAは、薬物貯留 対 時間を示す。パネルBは、脂質回復 対 時間を示す。パネルCは、薬物放出速度が血漿からの薬物クリアランス半減期に対する変化と相関することを示す。
【図5】図5は、脂質用量の、PK挙動(血液PK)に対する影響を示す。ここに例示されるように、類似する薬物放出速度(A)、脂質クリアランス速度(B)および薬物クリアランス速度(C)が、16.6mg/kg〜50mg/kgの脂質用量範囲にわたって薬物:脂質比が0.3:1であるリポソームビンブラスチン処方物について見られる。
【図6】図6は、脂質用量の、PKの動きに対する影響を示す(血漿PK)。ここに例示されるように、類似する薬物放出速度(A)、脂質クリアランス速度(B)および薬物クリアランス速度(C)が、16.6mg/kg〜50mg/kgの用量範囲にわたって薬物:脂質比が0.3:1であるリポソームビンブラスチン処方物について見られる。
【図7】図7は、異なる薬物:脂質比のリポソームトポテカンの2つの処方物の薬物動態的挙動を示す。トポテカンが薬物:脂質比0.11:1でロードされる場合、より低い薬物:脂質比0.02:1の処方物を有するパネルBと比較して、より長い血漿クリアランス速度を生じる、よりかなり遅い薬物放出速度が見られること、パネルAは示す。
【発明を実施するための形態】
【0017】
(発明の詳細な説明および好ましい実施形態)
多くの抗癌剤の活性は、その薬物動態的挙動に依存する。この薬物動態的挙動
は、薬物濃度、および癌細胞が薬物に暴露される期間を規定する。ほとんどの抗
癌剤の場合、より長い暴露時間は、これが癌細胞の増加した死滅を生じるので好
ましい。一般に、いくつかのパラメーターを使用して、薬物の薬物動態を記載す
る。血漿クリアランス半減期および曲線下面積(area under the
curve)(AUC)が、例である。この血漿クリアランス半減期は、投与
された薬物の半分が血漿から除去されるために必要とする時間である。AUCは
、経時的な血漿薬物レベルの尺度であり、そして全薬物暴露の指標を提供する。
一般に、抗癌剤に関する、増加した血漿クリアランス半減期および血漿AUCは
、増加した治療効果に相関する。
【0018】
(I.活性薬剤放出を調節すること)
本発明は、リポソームからの薬物放出を調節するための方法および処方物を提
供する。1つの実施形態において、本発明は、活性薬剤の血漿循環半減期を調節
するための方法を提供し、本方法は、以下の工程を包含する:(a)リポソーム
を提供する工程であって、このリポソーム中に、遊離活性薬剤と沈殿した活性薬
剤とがカプセル化されている、工程;ならびに(b)このリポソーム中に沈殿し
た活性薬剤の量を変化させる工程。好ましくは、「遊離活性薬剤」および「沈殿
した活性薬剤」は、同一の活性薬剤であるが、本発明は、そのように限定しない
。本明細書中で使用される場合、用語「調節すること(工程)」は、リポソーム
キャリアから活性薬剤を放出する速度を増加することかまたは減少することかの
いずれかを意味し得る。抗新生物活性薬剤について、調節することは、好ましく
は、活性薬剤の放出速度を減少することかまたは遅くすることかである。
【0019】
好ましい局面において、本発明のリポソームは、ともにカプセル化された、遊
離活性薬剤および沈殿した活性薬剤を含む。リポソーム中に沈殿した活性薬剤の
量は、種々の機構を使用して変化され得る。例えば、活性薬剤対脂質の比を変化
させることによって、沈殿した活性薬剤の量は、増加または減少され得る。低い
薬物:脂質比での薬物ローディングは、低濃度の活性薬剤(例えば、トポテカン
)をリポソーム内部に生じ、従って、薬物全体のうちのほとんど(すべてではな
いにしても)は溶液中にある(すなわち、沈殿されておらず、すなわち遊離して
いる)。少量の沈殿は、リポソームからの薬物の速い放出速度を生じる。逆に、
高い薬物:脂質比は、高いリポソーム内部濃度および高沈殿量を生じる。薬物が
沈殿形態の場合、その後の、インビボまたはインビトロでの放出速度は、遅い。
抗新生物薬物(例えば、トポテカン)について、遅い放出速度が好ましい。
【0020】
いかなる特定の理論によって束縛されることもないが、本発明のリポソームは
、薬物放出を指示する「沈殿−溶解機構」(PDM)を受けると考えられている
。本発明のPDM機構において、リポソーム内部の沈殿した活性薬剤(例えば、
トポテカン)の、リポソームの内部溶液への溶解速度は、リポソーム外部の活性
薬剤の、外部への放出速度に比べて遅く、従って律速である。すなわち、リポソ
ーム内部での沈殿した薬物から遊離薬物への溶解速度は、その薬物が血漿中にい
かに速く放出されるかを決定する。
【0021】
特定の実施形態において、活性薬剤対脂質比は、空のリポソームの添加によっ
て変えられ得る。一般に、空のリポソームまたはその中に活性薬剤が含まれてい
るリポソームのいずれもが、細網内皮系(RES)の細胞によってクリアランス
される。代表的に、RESは、一定用量の注射されたリポソームの80〜95%
を1時間以内に排除し、リポソームの取り込みのための選択された標的部位を効
果的に競合的除外(outcompeting)する。リポソームのRES取り
込みの速度に影響を与える種々の因子が、報告されており、これらには、リポソ
ームのサイズ、電荷、脂質飽和度、および表面成分が挙げられる。空のリポソー
ム小胞を含めることによって、RESから活性薬剤を含むリポソームを保護する
ことが可能である。このように、空のリポソーム小胞は、実際に、「おとり」と
して作用することによってリポソームの血液循環寿命を延長する。延長された循
環時間は、しばしば、リポソームが注射部位から標的領域、標的細胞または標的
部位に到達するために必要とされる。空のリポソーム小胞は、RESを活発に維
持し、結果として、その中に活性薬剤が含まれるリポソームの血清半減期が増加
される。
【0022】
特定の他の局面において、活性薬剤の沈降を増強する成分が、リポソームに添
加される。この局面において、種々の荷電イオンを使用して、小胞内部の沈降さ
れた活性薬剤の量を増加し得る。好ましい局面において、二価、三価または多価
アニオンが使用される。適切なアニオンとしては、カルボキシレート(−CO2
−)、スルホネート(SO3−)、スルフェート(SO4−2)、ヒドロキシド
(−OH)、アルコキシド、ホスフェート(−PO4−2)およびホスホネート
(−PO3−2)が挙げられるが、これらに限定されない。リポソーム内部の沈
降された活性薬剤の量を増強する他の成分は、当業者に公知である。
【0023】
さらに、薬物:脂質比は、リポソームのサイズを使用して変えられ得る。使用
するリポソーム小胞が大きいほど、薬物:脂質比はより小さい。特定の局面にお
いて、活性薬剤対脂質比およびリポソームのサイズの両方を変えて、活性薬剤の
効力を最適化する。
小胞中に沈降されるカプセル化された活性薬剤の量は、変化し、そしてこれは
、活性薬剤自体に幾分依存する。特定の実施形態において、沈降された活性薬剤
の量は、総活性薬剤の少なくとも約25%〜約95%(例えば、約25%、30
%、35%、40%、45%、50%、55%、60%、65%、70%、75
%、80%、85%、90%および95%)である。トポテカンについて、リポ
ソーム中にカプセル化される沈降された活性薬剤の量は、総活性薬剤の少なくと
も50%である。
【0024】
好ましい局面において、活性薬剤が抗腫瘍薬物である場合、高い薬物:脂質比
を使用することが、より多くの量のカプセル化され沈降された薬物を生じる。結
果として、インビボでのリポソームからの薬物放出は、より低い薬物:脂質比で
調整された同様の組成物よりも、よりゆっくりである。これらのより高い薬物:
脂質比のリポソームは、延長された血漿半減期および増加された血漿AUC値を
示す。有利には、これらの処方物は、改善された抗腫瘍効力を示す。
【0025】
特定の実施形態において、活性薬剤:脂質の比は、約0.005〜1:1(w
/w)である。
【0026】
好ましくは、活性薬剤:脂質の比は、約0.05〜0.9:1(w/w)であ
り、そしてより好ましくは、活性薬剤:脂質の比は、約0.1〜0.5:1(w
/w)である。活性薬剤の血漿循環半減期を調節することによって、このように
して、活性薬剤の効力を最大化または最適化することが可能である。
【0027】
(II.リポソーム処方物を作製する組成物および方法)
リポソーム、小胞およびリポソーム小胞は、水性の内部を包囲する脂質含有膜
を有する構造を示すことが理解される。これらの構造は、他に示されない限り、
1以上の脂質膜を有し得るが、一般に、リポソームは、たった1つの膜を有する
。このような単一層のリポソームは、本明細書中で「単層」と呼ばれる。多重層
のリポソームは、本明細書中で「多層」と呼ばれる。
【0028】
本発明において使用されるリポソームは、好ましくは、合わせられた場合に比
較的安定な小胞を形成する脂質から形成される。このようなリポソームを生成す
るために使用され得る、多くの種々の脂質が、当該分野で公知である。好ましい
脂質としては、中性または負に荷電したリン脂質またはスフィンゴ脂質およびス
テロール(例えば、コレステロール)が挙げられるが、これらに限定されない。
脂質の選択は、一般に、例えば、リポソームサイズおよび血流中のリポソームの
安定性を考慮して導かれる。
【0029】
本発明の使用のために好ましいリポソーム組成物としては、スフィンゴミエリ
ンおよびコレステロールを含有するリポソームが挙げられる。このリポソーム組
成物中のスフィンゴミエリン対コレステロールの比は、変化し得るが、一般に、
約75/25mol%/mol%のスフィンゴミエリン/コレステロール〜約3
0/50mol%/mol%のスフィンゴミエリン/コレステロールの範囲にあ
り、より好ましくは、約70/30mol%/mol%のスフィンゴミエリン/
コレステロール〜約40/45mol%/mol%のスフィンゴミエリン/コレ
ステロール、そしてさらにより好ましくは、約55/45mol%/mol%の
スフィンゴミエリン/コレステロールである。他の脂質は、必要であり得る場合
(例えば、脂質酸化を妨げるために、またはリポソーム表面上にリガンドを付着
するために)、本発明のリポソーム組成物中に含まれ得る。一般に、脂質が含ま
れる場合、このような脂質の他の含有は、スフィンゴミエリン/コレステロール
比の減少を生じる。この型のリポソームは、スフィンゴソームとして公知であり
、そして米国特許第5,814,335号(この教示は、本明細書中で参考とし
て援用される)において、より十分に記載される。
【0030】
例えば、以下に記載されるような、種々の方法が、リポソームを調製するため
に利用可能である:Szokaら、Ann.Rev.Biophys.Bioe
ng.9:467(1980);米国特許第4,235,871号;同第4,5
01,728号;同第4,837,028号、テキストLiposome,Ma
rc J.Ostro編、Marcel Dekker,Inc.,New Y
ork,1983,第1章;およびHopeら、Chem.Phys.Lip.
40:89(1986)(これら全てが、本明細書中で参考として援用される)
。リポソームを生成するためのプロトコルは、一般に以下の工程を包含し、これ
らの全ては、当該分野で周知である:有機溶媒中で脂質成分を混合する工程;乾
燥しそして水性溶媒中でリポソームを再構成する工程;およびリポソームをサイ
ズ化する工程(例えば、押出しによって)。
【0031】
リポソームを調製する代替的方法もまた、利用可能である。例えば、界面活性
剤透析に基づく、脂質粒子の自己アセンブリを含む方法が、Wheelerらに
対して発行された米国特許第5,976,567号において開示および権利化さ
れ、これは、時間のかかる難しい、比例性の乾燥および再構成工程を回避する。
連続フロー水和を使用してリポソームを調製するさらなる方法が、開発中であり
、そしてしばしば、最も効果的な大規模製造プロセスを提供し得る。
【0032】
活性薬剤(例えば、カンプトセシン)を有するリポソーム処方物の調製は、リ
ポソームへの薬物の充填を必要とする。充填は、受動的または能動的であり得る
。受動充填は、一般に、再構成工程の時点での緩衝液への薬物の添加を必要とす
る。これは、薬物がリポソーム内部に捕捉されるのを可能にし、ここでは、薬物
が脂溶性でない場合および小胞がインタクトなままである場合に、薬物が維持さ
れる(このような方法は、例えば、PCT公開番号WO95/08986(この
教示は、本明細書中で参考として援用される)において使用される)。
【0033】
能動充填は、多くの点で好ましく、そして広範に種々の治療薬剤が、膜のpH
勾配またはイオン勾配を使用することによって、ほぼ100%のカプセル化効率
でリポソーム中に充填され得る(Mayerら、Biochim.Biophy
s.Acta 1025:143−151(1990)およびMaddenら、
Chem.Phys.Lipids 53:37−46(1990)を参照のこ
と)。能動充填の多くの方法が、当業者に公知である。このような全ての方法は
、脂質性化合物をリポソームの内部に引き込む、いくつかの形態の勾配の確立を
包含し、ここでは、それらが、勾配が維持される限り存在し得る。非常に高い量
の所望の薬物が、内部に得られ得、薬物が、内部で沈降し、そして継続した取り
込みの勾配を生じる。
【0034】
イオノフォア媒介充填は、本発明と共に使用するために特に好ましく、これは
、米国特許第5,837,282号(この教示は、参考として本明細書中に援用
される)に開示および権利化されている。イオノフォア媒介充填は、電気的中性
のプロセスであり、これは、膜電位の形成を生じない。小胞への水素イオン輸送
によって、小胞からの並存するマグネシウムイオン輸送が、2:1比で存在する
(すなわち、正味電荷の輸送はない)。トポテカンの場合、その薬剤は、中性状
態(電荷なし)で膜を通過すると考えられる。小胞内への進入時に、トポテカン
は、正に荷電される。イオノフォア媒介充填は、電気的中性のプロセスであるの
で、膜電位は生成されない。
【0035】
薬学的目的のリポソームカンプトセシンの重要な特徴は、最終処方物の薬物対
脂質比である。先に議論したように、薬物:脂質比は、以下の2つの様式:1)
同じ薬物:脂質比を各々含有する均質なリポソームを使用するか;または2)空
のリポソームを高い薬物:脂質比を有するリポソームと混合することによって、
適切な平均薬物:脂質比を提供することによって確立され得る。異なる適用のた
めに、異なる薬物:脂質比が、所望され得る。特定の薬物:脂質比を生成するた
めの技術は、当該分野で周知である。薬物:脂質比は、重量に基づいて、モル濃
度に基づいてまたは任意の他の指定された基準に基づいて測定され得る。好まし
い薬物:脂質比は、約0.005:1の薬物:脂質(重量)〜約0.2:1の薬
物:脂質(重量)、そしてより好ましくは、約0.1:1の薬物:脂質(重量)
〜約0.3:1の薬物:脂質(重量)の範囲にある。
【0036】
さらなる重要な特徴は、リポソーム粒子のサイズである。本発明における使用
のために、約0.05ミクロン〜約0.15ミクロンのサイズを有するリポソー
ムが、好ましい。
【0037】
本発明はまた、キット形態でリポソーム組成物(例えば、カンプトセシン)を
提供する。キットは、既製の処方物または投与前に医薬の混合を必要とする処方
物を備え得る。キットは、代表的に、キットの種々のエレメントを保持するため
の仕切られたコンテナを備える。キットは、可能には水和形態で、本発明の組成
物またはその成分を、それらの再水和および投与のための指示書と共に備える。
【0038】
例えば、本明細書中に記載される方法によって調製されるリポソーム組成物は
、単独でか、または投与経路および標準的な薬務に従って選択された生理学的に
受容可能なキャリア(例えば、生理学的食塩水またはリン酸緩衝液)との混合物
のいずれかで投与され得る。一般に、通常の生理食塩水が、薬理学的に受容可能
なキャリアとして使用される。他の適切なキャリアとしては、例えば、水、緩衝
化水、0.4% 生理食塩水、0.3% グリシンなどが挙げられ、これらは、
安定性を増強するための糖タンパク質(例えば、アルブミン)、リポタンパク質
、グロブリンなどを含む。これらの組成物は、従来の周知の滅菌技術によって滅
菌され得る。得られた水溶液は、使用のためにパッケージされ得るか、または無
菌条件下でろ過され、そして凍結乾燥され得、この凍結乾燥調製物が、投与前に
滅菌水溶液と合わせられる。これらの組成物はまた、生理学的条件を近似するた
めに必要とされる、薬学的に受容可能な補助物質(例えば、pH調整剤および緩
衝化剤、張度調整剤など(例えば、酢酸ナトリウム、乳酸ナトリウム、塩化ナト
リウム、塩化カリウム、塩化カルシウムなど))を含み得る。さらに、この組成
物は、脂質保護剤を含み得、これは、保存中のフリーラジカルおよび脂質過酸化
損傷に対して脂質を保護する。親油性のフリーラジカル失活剤(例えば、α−ト
コフェロールおよび水溶性のイオン特異的キレート剤(例えば、フェリオキサミ
ン(ferrioxamine))が適切である。
【0039】
広範に種々の活性薬剤が、本発明のリポソーム組成物および方法に適切である
。好ましい局面において、活性薬剤は、抗腫瘍薬物である。現在、約20の認め
られたクラスの承認された抗腫瘍薬物が存在する。この分類は、特定の薬物によ
って共有される共通の構造に基づいてか、または薬物による作用の共通の機構に
基づく総括である。分類した抗腫瘍薬剤の一般に公知の商業的に認証された(ま
た開発中の)いくつかの部分的リストは、以下のとおりである:
構造に基づく分類:
1.フルオロピリミジン−−5−FU、フルオロデオキシウリジン、フトラフ
ァー(Ftorafur)、5’−デオキシフルオロウリジン、UFT、S−1
カペシタビン;
2.ピリミジンヌクレオシド−−デオキシシチジン、シトシンアラビノシド、
5−アザシトシン、ゲンシタビン、5−アザシトシン−アラビノシド;
3.プリン−−6−メルカプトプリン、チオグアニン、アザチオプリン、アロ
プリノール、クラドリビン(Cladribine)、フルダラビン、ペントス
タチン、2−クロロアデノシン;
4.白金アナログ−−シスプラチン、カルボプラチン、オキサリプラチン(O
xaliplatin)、テトラプラチン、白金−DACH、オルマプラチン(
Ormaplatin)、CI−973、JM−216;
5.アントラサイクリン/アントラセンジオン−−ドキソルビシン、ダウノル
ビシン、エピルビシン、イダルビシン、ミトキサントロン;
6.エピポドフィロトキシン−−エトポシド、テニポシド;
7.カンプトセシン−−イリノテカン、トポテカン、9−アミノカンプトセシ
ン、10,11−メチレンジオキシカンプトセシン、9−ニトロカンプトセシン
、TAS 103、7−(4−メチル−ピペラジノ−メチレン)−10,11−
エチレンジオキシ−20(S)−カンプトセシン、7−(2−N−イソプロピル
アミノ)エチル)−20(S)−カンプトセシン;
8.ホルモンおよびホルモンアナログ−−ジエチルスチルベストロール、タモ
キシフェン、トレメフィン、トルムデックス(Tolmudex)、チミタック
(Thymitaq)、フルタミド、ビカルタミド、フィナステリド、エストラ
ジオール、トリオキシフェン、ドロキシフェン(Droloxifene)、メ
ドロキシプロゲステロンアセテート、メゲステロールアセテート、アミノグルテ
チミド、テストラクトンなど;
9.酵素、タンパク質および抗体−−アスパラギナーゼ、インターロイキン、
インターフェロン、ロイプロリド、ペガスパラガーゼ(Pegaspargas
e)など;
10.ビンカアルカロイド−−ビンクリスチン、ビンブラスチン、ビノレルビ
ン、ビンデシン;
11.タキサン−−パクリタキセル、ドセタキセル。
【0040】
機構ベースのクラス:
1.抗ホルモン−ホルモンおよびホルモンアナログの分類を参照のこと、アナ
ストロゾール(Anastrozole);
2.抗葉酸(Antiforate)−−メトトレキサート、アミノプテリン
、トリメトレキサート、トリメトプリム、ピリトレキシム(Pyritrexi
m)、ピリメタミン、エダトレキサート(Edatrexate)、MDAM;
3.抗微小管剤−−タキサンおよびビンカアルカロイド;
4.アルキル化剤(古典的および非古典的)−−ナイトロジェンマスタード(
メクロレタミン、クロランブシル、メルファラン、ウラシルマスタード)、オキ
ザホスホリン(イフォスファミド、シクロホスファミド、パーホスファミド(P
erfosfamide)、トロホスファミド(Trophosphamide
))、アルキルスルフォネート(ブスルファン)、ニトロソ尿素(カルムスチン
、ロムスシン、ストレプトゾシン)、チオテパ、ダカルバジンなど;
5.代謝拮抗物質−−プリン、ピリミジンおよびヌクレオシド(上記);
6.抗生物質−−アントラサイクリン/アントラセンジオン、ブレオマイシン
、ダクチノマイシン、マイトマイシン、プリカマイシン(Plucamycin
)、ペントスタチン、ストレプトゾシン;
7.トポイソメラーゼインヒビター−−カンプトセシン(Topo I)、エ
ピポドフィロトキシン、m−AMSA,エリプチシン(Topo II);
8.抗ウイルス剤−−AZT、ザルシタビン(Zalcitabine)、ゲ
ンシタビン(Gemcitabine)、ジダノシンなど;
9.種々雑多な細胞傷害剤−−ヒドロキシ尿素、ミトーテン、融合毒素、PZ
A、ブリオスタチン(Bryostatin)、レチノイド、ブチル酸および誘
導体、ペントサン、フマギリンなど。
【0041】
全ての抗腫瘍薬剤の目的は、癌細胞の排除(治療)または癌細胞の増殖および
伝播の遅延(寛解)である。上記に列挙した抗腫瘍薬剤の大半は、主要な細胞傷
害活性を有し、癌細胞に対して直接的な殺傷をもたらすことによって、この目的
を追求する。他の抗腫瘍薬物は、身体の自然の免疫を刺激して、癌細胞の殺傷を
もたらす。文献は、上記の薬物の全ての活性および機構についての考察などで充
たされる。
【0042】
リポソームカンプトセシン、および特に、リポソームトポテカンの特的の処方
物を作製する例示的方法を、以下の実施例において示す。
【0043】
(III.リポソームカンプトセシンを使用する方法)
本発明のリポソーム組成物(例えば、カンプトセシン)は、動物(例えば、ヒ
ト)における固形腫瘍の処置において使用される。以下の実施例は、薬物:脂質
比、投与される活性薬剤および脂質の投薬量、および異なる腫瘍型を処置するた
めの好ましい投与スケジュールの重要なパラメーターを示す。
【0044】
好ましくは、薬学的組成物は、非経口的(すなわち、関節内、静脈内、腹腔内
、皮下または筋内)に投与される。より好ましくは、薬学的組成物は、静脈内点
滴によって投与されるか、またはボーラス注射によって腹腔内投与される。薬学
的処方物中のリポソームの濃度は、広範に、すなわち、約0.05重量%未満か
ら、通常、少なくとも約2〜5重量%〜多くて10〜30重量%で変化し得、そ
して選択される特定の投与様式に従って、流体容量、粘性などによって主に選択
される。例えば、濃度を増加して、処置に関連する流体負荷を低下させ得る。あ
るいは、刺激性脂質から構成されるリポソームを低い濃度に希釈して、投与部位
での炎症を小さくし得る。投与されるリポソームの量は、使用される特定のカン
プトセシン、処置される疾患状態および主治医の判断に依存するが、一般に、ヒ
トにおいて、約0.01mg/kg体重と約50mg/kg体重との間、好まし
くは、約5mg/kg体重と約40mg/kg体重との間である。より高い脂質
用量(例えば、50〜120mg/kg)が、マウスに適切である。
【0045】
活性薬剤(例えば、カンプトテシン)の投薬量は、患者の年齢、体重および状
態、ならびに処置計画に基づいて、投与する医師の意見に依存する。小細胞肺癌
における遊離トポテカンの推奨用量は、1用量あたり1.5mg/M2(5日間
毎日)であり、3週間ごとに繰り返される。以下の実施例で示された処置におけ
る改善が原因で、ヒトにおける活性薬剤(例えば、トポテカン)の用量は、0.
015mg/M2/用量程度の低さの範囲で有効であり、投与計画に依存して、
15〜75mg/M2/用量程度の高さでなお寛容可能である。用量は、単一用
量であり得、この用量は、4時間毎、6時間毎、または12時間毎、あるいは、
1日毎、2日毎、3日毎、4日毎、5日毎、6日毎、7日毎、8日毎、9日毎、
10日毎またはそれらの組み合わせで繰り返して投与され得る。好ましい計画は
、1週間毎、2週間毎、3週間毎、4週間毎、5週間毎、もしくは6週間毎また
はそれらの組み合わせで繰り返される処置サイクルを用い得る。現在好ましい実
施形態において、処置は、1週間に1回行われ、この用量は、代表的には、1.
5mg/M2未満である。
【0046】
特に好ましいトポテカン投薬量および計画は、以下のとおりである:
【0047】
【表1A】
本発明は、具体的実施例により、より詳細に記載される。以下の実施例は、例
示目的で提供され、如何様にも本発明を限定することを意図しない。当業者は、
重要ではない種々のパラメーターを容易に認識し、これらのパラメーターは、本
質的に同じ結果を得るために変更または改変され得る。
【0048】
(IV.実施例)
(A.材料および方法)
1.材料 トポテカン(HycamtinTM,SmithKline Be
echam)を、British Columbia Cancer Agen
cyの薬局から購入した。スフィンゴミエリン(SM)を、Avanti Po
lar Lipidsから購入した。Northern Lipidsのスフィ
ンゴミエリンを初期の研究において用いたが、Avantiバージョンのものよ
り、あまりエタノールに可溶性ではなかった。コレステロール(CH)および二
価の陽イオンイオノフォアA23187をSigmaから購入した。[3H]−
コレステリルヘキサデシルエーテル(Dupont)を脂質マーカーとして用い
た。
【0049】
2.マウス。雌性のICR、BDF−1または無胸腺nu/nu(6−8週齢
)を、Harlan−Sprague Dawley(Indianapoli
s,IN)から購入した。すべての動物を、使用する前に1週間にわたり隔離し
た。すべての研究を、Canadian Council on Animal
Care(CCAC)およびInstitutional Animal C
are and User Committee(IACUC)により制定され
たガイドラインに沿って行った。
【0050】
3.Mg−A23187法によるトポテカンの処方。トポテカンを、米国特許
第5,837,282号に従うMg−A23187イオノフォア法を用いて、S
M:CH(55:45,mol/mol)リポソーム中にカプセル化した。最初
の薬物 対 脂質比は、0.10(w/w)であり、薬物負荷は、代表的には、
95−100%であった。外用緩衝液(external buffer)は、
10mM PBS,pH7.5および300mM スクロースからなった。すべ
ての処方物を粒子サイズ、薬物負荷効率、pH、および薬物と脂質の濃度に関し
て分析した。
【0051】
4.薬物調製および投薬。トポテカン(HycamtinTM)の各バイアル
を、1.0mlの滅菌水で水和し、4.0mg/mlのトポテカン濃度にした。
引き続いて、薬物のラクトン種について必要な低pHを維持するために、0.9
%滅菌生理食塩水中で希釈した。水保存溶液中の非使用薬物(4.0mg/ml
)を、遮光して4℃で保存した。リポソームにカプセル化したトポテカンを、0
.9%生理食塩水で、投与に必要な濃度に希釈した。すべての薬物投与は、側方
尾静脈を介した10ml/kg(200μl/20gマウス)であった。
【0052】
5.薬物動態およびインビボ漏出研究。遊離トポテカンおよびリポソームカプ
セル化トポテカンの薬物動態および薬物漏出を、側方尾静脈を介したi.v.投
与後24時間にわたり、ICRマウスにおいて評価した。2つの異なる薬物 対
脂質比(すなわち、0.10(w/w)および0.02(w/w))を用いて
、薬物漏出およびPK挙動に対する薬物 対 脂質比および脂質用量の影響を試
験した。カプセル化されたトポテカンを、1mg/kg(10または50mg/
kg脂質)および5mg/kgトポテカン(50mg/kg脂質)で投与した。
対応して、遊離のトポテカンのPK挙動を、1mg/kgおよび5mg/kgで
評価した。血中総トポテカンを、血漿タンパク質の沈降後の蛍光アッセイにより
決定した。トポテカンを、それぞれ、380nmおよび518nmの励起波長(
2.5nmスリット幅)および発光波長(2.5nmスリット幅)で分光蛍光器
により定量した。血漿中の脂質レベルを、[3H]−CHE標識の液体シンチレ
ーション計測により決定した。
【0053】
6.MTD研究。MTD研究を、各腫瘍モデルに対応する宿主マウス系統にお
いて行った。単一用量および複数用量のMTDを、経時的な体重減少をモニター
することにより決定した。MTDを、20%体重減少を生じる用量と規定した。
【0054】
7.骨髄抑制および好中球減少症研究。トポテカン投与の結果としての末梢血
細胞レベルの変化を、4〜6週間にわたりICRマウスにおいて評価した。10
mg/kgの遊離トポテカンまたはリポソームカプセル化トポテカンをi.v.
投与して1日、3日、5日、7日、14日、および21日目に、血液を、EDT
A微量チューブ(microtainer tube)に採取した。空の小胞を
、コントロールとして投与した。CBCおよび差示的分析をCentral L
abs for Veterinarians(Langley,BC)で行っ
て、細胞レベル、比および形態を定量した。
【0055】
8.腫瘍モデル。標準的プロトコルで用いられるように、L1210マウス白
血病モデルおよびCT−26マウス結腸転移モデルを用いた。ヒトMX−1およ
びLX−1細胞株をDCTD Tumor Repository in Fr
ederick,MDから得た。これらの細胞株を、腫瘍フラグメントとして回
収し、3mm×3mmフラグメントの連続的移植によりNCrヌードマウスにお
いて増殖させた。細胞株がヌードマウスにおいて3継代を過ぎ、腫瘍株が継代数
が10に達したときに再開始するまで、実験を開始しなかった。
【0056】
9.効力研究。遊離トポテカンおよびリポソームトポテカンの投薬すべてを、
側方尾静脈を介した10ml/kgの静脈内経路により投与した。L1210お
よびCT−26モデルにおいて、投薬を1日目に行った(腫瘍細胞注入=0日目
)。MX−1およびLX−1腫瘍モデルについて、腫瘍容積を、腫瘍寸法の垂直
測定の繰り返しおよび以下の式を用いることにより決定した:
容積(mm3)=(L×W2)/2
腫瘍が明らかに増殖を示し、100−300mm3の範囲にある場合に、MX
−1モデルおよびLX−1モデルで投薬を開始した。
【0057】
大部分の薬物が生物学的効果と毒性との間で平衡を示すので、これらの属性の
両方を組み込むパラメーターを試験することは有用である。最も一般に用いられ
るパラメーターは、治療指数(therapeutic index)(TI)
である。伝統的には、治療指数は、以下のように規定されている:
TI=LD50/ED50
しかし、LD50研究を行うことはもはや許されないので、これらの研究につ
いての治療指数を、以下のように規定した:
TI=MTD/MED
上記の式において、MTDは最大寛容用量であり、動物の群において20%の
平均体重減少を引き起こす用量として規定される;そしてMEDは、最小有効用
量であり、固形腫瘍モデルにおいて40以下の最適%T/C値を生じる用量また
は生存モデルにおいて50±10%の%ILSを生じる用量として規定される。
【0058】
(B.結果)
1.薬物動態および薬物漏出。トポテカンの血漿薬物動態および薬物漏出に対
するリポソームカプセル化および薬物 対 脂質比の影響を、ICRマウスにお
いて24時間にわたり試験した。トポテカンのリポソームカプセル化(薬物 対
脂質比、0.11、wt/wt)は、この薬物の薬物動態パラメーターに対し
て劇的な影響を与えた(図1、上部;および表1を参照のこと)。5mg/kg
用量のトポテカンでは、遊離薬物に対するリポソーム薬物について、血漿AUC
において164倍の増加、Cmaxにおいて24倍の増加および血漿α半減期に
おいて24倍の増加が観察された(表1を参照のこと)。歴史的には、リポソー
ム薬物のAUCおよび血清半減期における大きな改善により、疾患部位(例えば
、腫瘍)への薬物の送達(「疾患部位標的化」として公知のプロセス)の増強が
得られた。
【0059】
この研究において用いた処方物を、Mg−A23187イオノフォア法により
調製した。iv投与して最初の10〜30分後に、最初に薬物が急激に放出され
(図1、下部を参照のこと)、続いてより緩やかな放出相が生じた。Mn−A2
3187処方物およびMg−A23187処方物についてのtl/2放出は、そ
れぞれ、約3時間および約5〜7時間であった;しかし、24時間では、いずれ
の処方物においても薬物はほとんど存在しなかった。
【0060】
大部分のリポソーム薬物処方物について、カプセル化薬物の薬物動態特性を、
脂質組成および用量により制御する。リポソームトポテカンは、非常に低い薬物
用量ですら(0.5mg/kg;薬物 対 脂質比、0.10、wt/wt)、
別格の抗腫瘍活性を示すことが示された。これらの薬物用量および薬物 対 脂
質比において、血漿からのリポソーム排除は、迅速であることが期待される。従
って、低用量でのトポテカンの薬物動態が改善され得るか否かを決定するために
、トポテカンの低い薬物 対 脂質比(0.02,wt/wt)処方物を調査し
た。興味深いことに、この研究において、低い薬物 対 脂質比の処方物が、よ
り高い薬物 対 脂質比(0.11,wt/wt)の処方物よりはるかに速く薬
物を放出した。この結果は予測外であった。
【0061】
【表1】
すべてのパラメーターは、WINNONLIN PKモデリングソフトウェアを
用いて、1または2区画モデルから導出した。
【0062】
2.最大寛容用量。単一および複数用量のMTD研究を、腫瘍保有Balb/
cマウス、BDF−1マウスおよびNCr nu/nuマウスにおいて行った。
個々のマウスの体重を、各研究の間中モニターして、遊離トポテカンおよびリポ
ソームトポテカンの全身的な寛容性を評価し、可能ならば、MTDを確立した(
図2を参照のこと)。リポソームトポテカンの最大寛容用量は、単一投与に関し
て10mg/kg、q7dx3スケジュールに関して7.5mg/kgおよびq
3dx4スケジュールに関して5mg/mlであった。マウスにおける1回の静
脈内注入後の遊離トポテカンの報告されたLD50は、75mg/M2(約25
mg/kg)であった[HycamtinTM製品小論];しかし、40mg/
kgまでの用量で体重減少はほとんど観察されなかったが、これは、急性応答に
起因したMTDと考えられた。薬物量は、制限されていたので、40mg/kg
より高い用量(5〜10分にわたり投与された)については追求しなかった。q
dx5スケジュールに対する遊離トポテカンのLD10は、14mg/M2/用
量(約4.7mg/kg/用量)であることが以前に示されている(Groch
ow,et al.,Drug Metab.Dispos.20:706−7
13(1992))。
【0063】
3.毒性。ヒトにおいて5日間連続(dx5)にわたり1.5mg/M2/用
量で毎日投与される遊離トポテカンの主要な用量制限毒性(MTD)は、非累積
性の骨髄抑制である。先に言及されたように、ヒトは、マウスよりも、骨髄抑制
に対してより感受性であり、マウスにおけるMTDのわずか11%を寛容し得る
にすぎない(14mg/M2に対して1.5mg/M2)。この点において、イ
ヌは、ヒトにおけるトポテカンの骨髄抑制のはるかに良好な予測者であることが
示された(Burris,et al.,J.Natl.Cancer Ins
t.84:1816−1820 (1992))。しかし、マウスは、遊離トポ
テカンおよびリポソームカプセル化トポテカンの相対的骨髄抑制効果を比較する
に適切であるはずである。
【0064】
1つの研究において、末梢WBC数の最大減少が、リポソームトポテカンを投
与して3日目に生じた。次いで、末梢血球レベルおよび形態の比較を、遊離トポ
テカンもしくはリポソームカプセル化トポテカンまたは空の小胞を投与して3日
目に行った(表2を参照のこと)。この比較に用いた用量は、リポソームカプセ
ル化トポテカンのMTDであった(10mg/kg)。リポソームトポテカンに
ついて遊離トポテカン(約10倍)、空の小胞(約10倍)、またはコントロー
ル動物(約20倍)と比べて、循環好中球の有意な減少が観察された。総WBC
レベルおよびリンパ球亜集団は、コントロール動物に対してリポソームトポテカ
ンについて約2倍減少した。同用量では、遊離トポテカンについてのこれらのパ
ラメーターにおいて、有意差はまったくなかった。注射して21日目に、リポソ
ームトポテカンについての総WBCレベルは、正常動物より約2.5倍低いまま
であった;しかし、好中球レベルは、正常マウスと比較して、20倍〜3倍の減
少まで回復した。リンパ球レベルは、正常マウスより約2倍低いままであった。
他の有意差は観察されなかった。
【0065】
注射して3日後の血清化学パラメーターの分析により、未処置動物に対してわ
ずかな変化が明らかになった(表3を参照のこと)。注意すべきわずかな変化は
、リポソームトポテカンで処置した動物に関するグロブリンレベルの統計的に有
意な増加(約2倍)およびアルブミン/グロブリン比の付随した減少であった。
他の有意な変化は観察されなかった。
【0066】
【表2】
(表3.遊離トポテカンまたはリポソームカプセル化トポテカンの10mg/
kg静脈内投薬量で処理したICRマウスの血清化学パネル−注射後3日目−)
【0067】
【表3】
(C.マウス腫瘍モデルおよびヒト腫瘍モデルにおける効力の研究:単回用量
研究)
(1.L1210マウス白血病) 静脈内L1210マウス白血病モデルが、
遊離の化学療法剤とリポソームカプセル化化学療法剤との間の差次的活性を評価
するために広範に使用されており、そしてこのモデルは、新規な化学療法剤のイ
ンビボNCIスクリーニングにおける元々の(1955〜1975)モデルのう
りの1つであった(Plowmanら、Human tumor xenogr
aft models in NCI drug development、「
Anticancer Drug Development Guide:Pr
eclinical Screening,Clinical Trials,
and Approval」(B.Teicher編)、Humana Pre
ss Inc.,Totowa(1997);Waud、Murine L12
10 and P388 leukemisas、「Anticancer D
rug Development Guide」Preclinical Sc
reening,Clinical Trials,and Approval
」(B.Teicher編)、Humana Press Inc.,Toto
wa(1997))。このモデルは、迅速であり−未処置動物の平均生存は代表
的には約7〜8日である−、そして投与された腫瘍細胞は、肝臓および骨髄に播
種される(seed)。
【0068】
遊離のトポテカンを単回静脈内用量として投与すると、L1210モデルにお
ける生存に対して、最小の効果しか有さなかった(図3Aを参照のこと)。最高
用量の遊離のトポテカンでは、生存中央値13日(44% ILS)が観察され
た。この群において、1つの長期生存体(60日)が存在した。対照的に、5m
g/kgまたは10mg/kgのいずれかのリポソームトポテカンの単回静脈内
(i.e.)投与は、60日目で100%の生存を生じた(図3Bを参照のこと
)。1mg/kg用量についての生存中央値は13日(44% ILS)であり
、そして生存曲線は、30mg/kgで投与された遊離トポテカンの生存曲線と
ほぼ同一であった−30倍の効力増強−。より高用量(30mg/kg)のリポ
ソームトポテカンでは、毒性死が観察された。リポソームトポテカンについての
MTDは、単回静脈内投与の後、BDF−1マウスにおいて20mg/kgであ
った。
【0069】
(2.CT−26マウス結腸癌) マウスCT−26結腸細胞株は、薬物スク
リーニングのために有用である。なぜなら、この細胞株は、皮下固形腫瘍として
容易に増殖するか、またはこの細胞株を静脈投与して生存モデルとして使用し得
るからである。さらに、この腫瘍細胞を、脾内(i.s.)注射により投与し、
その後、脾摘出術を行って、この細胞を、肝臓へと播種して実験的転移モデルを
生じる。このモデルは、結腸直腸癌の臨床経過によく似ている。このモデルは、
広範に使用されており、そして、例えば、他の箇所に詳細に記載される。
【0070】
このCT−26モデルにおいて、トポテカンの単回用量の投与は、生存に対し
て穏やかな影響を有し、用量範囲5〜40mg/kgにわたって23〜60%の
ILS%を生じた(図4を参照のこと)。しかし、リポソームカプセル化トポテ
カンは、5mg/kgより大きい用量では非常に活性であり、90日目で100
%の生存(8/8)を生じた。10mg/kgでは、87.5%の生存(7/8
)が90日目に観察されたが、死んだ動物の腫瘍負荷は非常に低く、このことは
、この動物が、他の要因(例えば、骨髄抑制に関連する感染)に起因して死んだ
かもしれないことを示唆する。リポソームトポテカンに関する用量応答が観察さ
れ、2mg/kg用量は、54%のILS%を生じた。これは、MEDであるこ
とが決定され、そしてこれは、40mg/kgの遊離トポテカンを使用して達成
されるILS%(58%)に匹敵した−20倍の効力増強−。
【0071】
(3.MX−1ヒト乳癌腫) MX−1は、ヒト乳癌の実験モデルであり、そ
して倍化時間3.9日を有することが報告されている。この研究において、倍化
時間中央値は、一貫して3.6〜3.7日であった。この腫瘍細胞株は、29年
齢の雌の原発性腫瘍に由来し、化学療法を受けた既往歴はなかった。そしてこの
腫瘍細胞株は、ヌードマウスにおいて連続継代される腫瘍フラグメントとしてD
CTD(NCI)腫瘍貯蔵所(tumor repository)により提供
される。組織学的に、MX−1は、ほどんど分化していない乳癌腫であり、腺形
成またはムチン産生の形跡はない。MX−1は、NCIインビボ腫瘍パネルおよ
びプレスクリーニング物(1976〜1986)を含んだ、新規な化学療法剤を
評価するための3つの異種移植片モデル(MX−1、LX−1、CX−1)のう
ちの1つであった(Plowmanら、Human tumor xenogr
aft models in NCI drug development.「
Anticancer Drug Development Guide:Pr
eclinical Screening,Clinical Trials,
and Approval」(B.Teicher編)、Humana Pre
ss Inc.,Totowa(1997))。その後、「化合物指向的」発見
から「疾患指向的」発見へのNCIの戦略のシフトを反映して、MX−1はより
大きな乳癌モデル群(計12)へと組み込まれた。
【0072】
進行した(staged)(100〜300mm3)MX−1腫瘍において、
遊離のトポテカンは、腫瘍増殖の用量依存性阻害を示した(図5;表1を参照の
こと)。最高用量(40mg/kg)において、最適T/C% 24%が得られ
たが、一方、10mg/kgおよび5mg/kgについての最適T/C%は、そ
れぞれ、66%および78%であった。薬物関連死は観察されず、そしてすべて
の動物は、この研究を通じて体重を増した。トポテカンのリポソームカプセル化
は、T/C%に対して顕著な影響を有し、2mg/kg、5mg/kgまたは1
0mg/kgの薬物の単回投与の後、それぞれ、最適T/C%が8%、−49%
および−62%であった。負のT/C%値は、もとの進行した(staged)
腫瘍サイズ(100〜300mm3)からの腫瘍体積の後退を示す。NCIのガ
イドラインによると、最適T/C%が<10%であることは、有意な活性と見な
され、一方、<42%の値は、薬物を開発へとさらに前進させるために許容可能
な最低限度である(Corbett,T.編、In vivo mehods
for screening and preclinical testin
g.「Anticancer Drug Development Guide
:Preclinincal Screening,Clinical Tri
als,and Approval」(B.Teicher編)Humana
Press Inc.,Totowa(1997))。リポソームカプセル化は
、トポテカンの毒性を増加し、遊離トポテカンについてのMIDを、>40mg
/kgから10mg/kgへと減少させた。
【0073】
(4.LX−1ヒト肺癌) LX−1は、ヒト小細胞癌(SCLC)の実験モ
デルである。この腫瘍細胞株は、48歳の男性において見出された転移病変の外
科的外植片に由来し、そしてヌードマウスにおいて連続継代される腫瘍フラグメ
ントとして、DCTD(NCI)腫瘍貯蔵所により提供される。このLX−1モ
デルは、1976〜1986のNCIインビボ腫瘍群の一部であった(Plow
man,J.ら、Human tumor xenograft models
in NCI drug development.「Anticancer
Drug Development Guide:Preclinical
Screening,Clinical Trials,and Approv
al」(B.Teicher編)、Humana Press Inc.,To
towa(1997))。現在は頻度を減らしてしか使用されないが、このLX
−1モデルは、その迅速な増殖速度が理由で、依然として、遊離薬物とリポソー
ム薬物との間の比較活性研究についての有用な異種移植片である。
【0074】
一般に、このLX−1モデルは、遊離薬物およびリポソームカプセル化薬物の
両方について、MX−1モデルよりもトポテカンの効果に対して感受性が低い(
図6;表1を参照のこと)。用量30mg/kg、10mg/kg、または5m
g/kgの遊離トポテカンについての最適T/C%は、それぞれ、43%、55
%、および67%であった。抗腫瘍活性が、カプセル化を介して改善され、用量
30mg/kg、10mg/kg、または5mg/kgについてのT/C%が、
それぞれ、8%、11%、および13%となった。興味深いことに、このリポソ
ームトポテカン用量のすべてが、類似する活性を示した。これは、初期の研究で
あり、他のモデルにおけるその後の研究(図4〜6を参照のこと)は、用量<5
mg/kgで始まる用量応答を示す。このことは、カンプトテシンクラス化合物
(およびおそらく他の抗腫瘍性薬剤)が「自己制限的」効力を示し得、それによ
り臨界閾値用量を超える用量では、さらなる活性の利点が全く観察されないとい
う知見(Thompson、Biochim.Biophys.Acta 14
00:301〜319(1998))と、一致する。この状況は、おそらく、そ
の薬物が制限された腫瘍細胞への接近を有する場合かまたはその薬物が腫瘍血管
に対して作用して破壊する(すなわち、抗脈管形成活性を有する)場合に、生じ
得る。両方の場合において、より高用量の薬物は、ごく小量の利点しか有さない
と予期される。
【0075】
L1210研究において観察されるように、トポテカンのカプセル化は、その
薬物の毒性を増強し、そしてそのMTDを減少した。腫瘍を保有するヌードマウ
スにおけるMTDは、10mg/kg(約16%の体重損失)であった。30m
g/kgでは、4/6の薬物関連毒性死が観察され、そして最大体重損失は、約
29%に達した(27〜34%範囲)。
【0076】
(D.マウス腫瘍モデルおよびヒト腫瘍モデルにおける効力研究:多数回用量
研究)
(1.MX−1ヒト乳癌腫) 複数回投与および薬物に対する腫瘍の長期曝露
の効果を調べるために、2つの複数用量プロトコル(q3dx4スケジュールお
よびq7dx3スケジュール)を、MX−1異種移植片において調べた。q4d
x3スケジュールにおいて、遊離トポテカンは、2.5mg/kg/用量および
10mg/kg/用量では中程度の活性を示し、1.25mg/kg/用量では
、最小の活性を示した(図7;表IIを参照のこと)。この投与スケジュールに
関する遊離トポテカンの最適T/C%値は、1.25mg/kg/用量、2.5
mg/kg/用量および10mg/kg/用量について、それぞれ、55%、3
0%、および27%であった。同じ投与スケジュールで投与されたカプセル化ト
ポテカンについて、最適T/C%値は、0.5mg/kg/用量、1.25mg
/kg/用量、2.5mg/kg/用量および5mg/kg/用量について、そ
れぞれ、15%、100%、100%、および100%であった。後退したすべ
ての腫瘍を、60日間モニターした。>1.25mg/kg/用量のリポソーム
トポテカンで処理したすべての動物は、この期間の終わりに、腫瘍を有さないと
見なされた。
【0077】
q7dx3投与スケジュールにおいて、5mg/kg/用量または10mg/
kg/用量のいずれの遊離トポテカンを用いても、ほとんど活性は観察されなか
った(図8;表IIを参照のこと)。同じ用量で、リポソームトポテカンは、進
行した(staged)腫瘍の完全な後退を誘導した。しかし、この投与スケジ
ュールにおいて、10mg/kg/用量は毒性があり過ぎた。そして、6/6毒
性死(または安楽死)が24日目に観察されたので、この研究のこの部分は停止
した。
【0078】
(2.LX−1ヒト肺癌) LX−1モデルにおける最初の研究(単回用量)
は、遊離トポテカンが評価用量<30mg/kgでは不活性であり、そしてリポ
ソームトポテカンは腫瘍増殖を阻害するが後退は誘導しないことを示した。この
活性を改善するために、複数回(q7dx3)スケジュールを、遊離トポテカン
およびリポソームトポテカンの両方について試験した。この場合、単回用量研究
と比較して、かなり大きな活性が遊離トポテカンについて観察され、30mg/
kg/用量および10mg/kg/用量について、それぞれ、最適T/C%値5
および40が得られた。リポソームトポテカンもまた、有意に改善した活性を示
し、5mg/kg/用量で完全な後退(およびその後の再増殖)を生じた。この
モデルにおけるリポソームトポテカンについての最適T/C%値は、5mg/k
g/日、2.5mg/kg/日、1.25mg/kg/日について、それぞれ、
55、3および16であった。
【0079】
(3.治療指数(TI)の比較) 遊離トポテカンおよびリポソームトポテカ
ンの治療指数を、いくつかの異なる投与スケジュールで4つの異なる腫瘍モデル
において評価した(表4を参照のこと)。これらの数を作製するために使用した
仮定および定義が、表IIIに見出される。いくつかの場合、真のMEDも真の
MTDも観察されなかった。従って、真のMEDも真のMTDを、用量応答トレ
ンドに基づいて数学的評価した。例えば、急性MTD 40mg/kgが、単回
ボーラス注射として投与された遊離トポテカンについて観察されたが、真のMT
D(重量損失に基づく)は、その薬物が5〜10分間にわたって注入された場合
には60mg/kgにより近づくようである。また、この分析をいくらか複雑に
したのは、リポソーム処方物の効力のレベルであった。低薬物用量にて、有意な
抗腫瘍活性が達成され、MEDが、特定の研究において評価されなければならな
かった。これらの場合、表4において注釈をつけた。
【0080】
一般に、リポソームトポテカンについての治療指数の増加は、単回用量投与に
ついて比較的大きく(モデルに依存して、5倍、10倍、15倍および18倍)
、そして投薬頻度が増加するにつれて減少した。このことが表4に示され、q7
dx3スケジュールおよびq3dx4スケジュールについて、それぞれ、TIT
CS/TIFree比は、4.7〜7.5および3.3であった。投与がより頻
繁になるにつれてこのTITCS/TIFree比が減少することは、遊離トポ
テカンの効力および毒性がスケジュール依存性であることを示す前臨床研究およ
び臨床研究と一致する。
【0081】
(表4.マウス腫瘍モデルおよびヒト腫瘍モデルにおける遊離トポテカンおよ
びリポソームトポテカンの相対的治療指数a)
【0082】
【表4】
a 表IIおよび表IIIのデータ;表IVにおける式および定義に基づく
b 急性MTD 40mg/kgを使用して得た;第2の値は、推定MTD(
体重)に基づく
c 約2倍大きくあり得る控えめな推定値;低用量での高い活性に起因して、
このMEDを評価することは困難である。
【0083】
(E.考察)
トポテカンは、リポソームカプセル化のための優れた候補である。簡単に述べ
ると、トポテカンは、細胞周期(S期)特異的であり、そして長期の曝露ととも
に活性が大いに増強され、トポテカンは、迅速な血漿薬物動態を示し、そしてこ
の薬物は、生物学的活性を保持するためにpH6.0より下で維持される必要が
ある。これは、酸性水性コアを有する比較的非漏出性のリポソーム処方物(例え
ば、SM:CH、55:45)を使用するための理想的シナリオである。必要な
酸性内部は、例えば、pHローディング法またはイオノフォアローディング法に
よって、作製され得る。ここで、Mg−A23187法によるSM/CHリポソ
ーム中へのトポテカンのカプセル化は、抗腫瘍効力の劇的な増強を生じることを
示した。毒性の中程度の増強もまた、リポソームトポテカンについて観察される
が、これは、遊離薬物に対して匹敵する効力(および多くの場合はより優れた効
力)を達成する、実質的用量の減少によって、大きく相殺された。
【0084】
治療指数(TI)は、薬物活性の有用なパラメーターである。なぜなら、それ
は、生物学的活性(ユーザーが規定する指標、すなわち、MED、ED50また
はED80)に対する毒性(MTD)の比の尺度であるからである。一般に、T
Iが低い程、毒性の危険が高い。なぜなら、生物学的効果を惹起するために必要
な薬物の容量は、MTDに近づくからである。治療指数は、リポソーム薬物の評
価のために特に有用である。なぜなら、TIの相対的変化は、カプセル化の利点
(またはその欠如)を規定するために使用され得るからである。本明細書中に示
されるように、TIは、3倍から18倍に改善され、それは、使用されるモデル
および投与スケジュールに依存する。従って、トポテカンのリポソームカプセル
化後に観察される生物学的活性の改善は、いかなる毒性の増加も補償するにとど
まらない。
【0085】
いかなる理論によっても拘束されることを意図しないが、抗腫瘍活性の有意な
改善およびリポソーム形態の薬物の毒性増加は、薬物動態の改善および活性ラク
トン形態での薬物の維持から生じると考えられる。これらの研究において、24
時間後には、84%のトポテカンが、血漿中にラクトン種として存在したのに対
して、たった5分後には遊離トポテカンについて48%のラクトンが存在した。
さらに、同じ用量(10mg/kg)の遊離トポテカンおよびリポソームトポテ
カンを、マウスに静脈投与した場合、ラクトン濃度は、<1時間の時点で、約4
0倍高かった。24時間にて、リポソーム薬物についてのラクトン血漿濃度は、
5.4μg/mlであったのに対して、遊離の薬物について5分で1.5μg/
mgであり、遊離トポテカンについてのピークラクトン濃度よりなお3.5倍高
かった。
【0086】
(表I)
(単回用量抗腫瘍活性および毒性パラメーターの要旨)
【0087】
【表5】
a 最終処理後の最適T/C%。負の値は、腫瘍後退を示す。
b 腫瘍増殖遅延(処理した腫瘍およびコントロール腫瘍が500mm3に達す
るための時間の差異)
c 未処理腫瘍に対する寿命の増加(%として表す)
d log(細胞殺傷)(量)
e 研究終了時に腫瘍を有さない動物(すなわち、可視的腫瘍がないか、または
長期生存体)
f 薬物関連死
g 処理群当たりの最大平均重量損失
h 正の重量変化(すなわち、処理前の重量を下回る重量低下が、どの時点でも
なかった)
** 長期生存体
(表II)
(複数回用量抗腫瘍活性および毒性パラメーターの要旨)
【0088】
【表6】
a 最終処理後の最適T/C%。負の値は、腫瘍後退を示す。
b 腫瘍増殖遅延(処理した腫瘍およびコントロール腫瘍が500mm3に達す
るための時間の差異)
c 未処理腫瘍に対する寿命の増加(%として表す)
d log(細胞殺傷)(量)
e 研究終了時に腫瘍を有さない動物(すなわち、可視的腫瘍がないか、または
長期生存体)
f 薬物関連死
g 処理群当たりの最大平均重量損失
h 正の重量変化(すなわち、処理前の重量を下回る重量低下が、どの時点でも
なかった)
i 測定せず;リポソームカプセル化群における毒性死
** 長期生存体;60日目まで可視的腫瘍が存在しない。
【0089】
表III.
毒性および抗腫瘍活性パラメーターについての規定および式
DRD 薬物関連死亡。動物が死亡するか、または薬物ANDでの最終処置後
15日以内に安楽死させたか、その腫瘍重量がコントロールマウスに対する致死
的負荷より小さいか、またはその体重減少が、コントロール動物の体重減少より
20%を超えた場合に、死亡が薬物に関連するとみなした。
【0090】
GI50 インビトロでの細胞の集団において50%増殖阻害を引き起こす薬
物の濃度。NCIは、時間0の時点で細胞数についての補正を強調するためにI
C50パラメーターを改名した。従って、式は、以下のとおりである:
GI50=(T−T0)/(C−T0)×100=50
TおよびT0は、それぞれ、48時間および0時間での光学密度である;Cは0
時間でのコントロール(細胞数)光学密度である。
【0091】
%ILS 寿命の増加(%)。生存モデルについて、処置動物(T処置)およ
び未処置動物(Tコントロール)についてメジアン生存時間を用いて、以下に従
って、これを計算した:
(T処置−Tコントロール)/Tコントロール×100
固形腫瘍モデルについては、腫瘍が2000mm3(体重の約10%)に達する
までの時間を、メジアン生存の代わりに倫理的カットオフとして用いた。
【0092】
LCK 細胞殺傷の対数(総数)。このパラメーターは、処置の最後に殺傷さ
れた細胞のlog10単位の数を以下の式に従って推定する:
(T−C)×0.301/メジアン倍化時間
正味の細胞殺傷の対数は、以下のように、腫瘍増殖遅延(T−C)パラメーター
から処置の持続時間を差し引きすることにより計算され得る:
[(T−C)−処置の持続時間]×0.301/メジアン倍化時間
0の細胞殺傷の対数は、処置の最後における細胞集団が、処置の開始における細
胞集団と同じであることを示す。しかし、例えば、4の細胞殺傷の対数は、開始
細胞集団における99.99%減少を示す。
【0093】
MBWL 最大体重減少(%)。動物を、最初の薬物投与の前(Wi)および
研究の間の種々の日(Wd)に体重測定した。体重の%変化は、以下により計算
される:
MBWL=(Wd−Wi)/Wi×100
MED 最小有効用量。これは、いくらか任意のパラメーターである。これら
の研究について、本発明者らは、40以下の最適%T/C(固形腫瘍モデルにつ
いて)または40〜60%の%ILS(生存モデルについて)を達成する最低用
量としてMEDを規定した。
【0094】
MTD 最大寛容用量。20%以下のMBWLを生じる薬物用量。
【0095】
%T/C 処置の最初の過程後に得られたコントロール腫瘍に対する処置腫瘍
の最適比。これらの値は、以下の式に従って、各観察日での腫瘍重量から、処置
の初日でのメジアン腫瘍重量(TiまたはCi)を差し引きすることにより得ら
れる:
%T/C=(ΔT/ΔC)×100(ここで、ΔT>0)または
%T/C=(ΔT/Ti)×100(ここで、ΔT<0)
NCI活性基準に従って、以下のスコア付けシステム(Plowman,et
al,Human tumor xenograft models in N
CI drug development、「Anticancer Drug
Development Guide:Preclinical Scree
ning,Clinical Trials,and Approval」(B
. Teicher,Ed.),Humana Press Inc.,Tot
owa(1997)[22])を適用する:
0=不活性、%T/C>40
1=腫瘍阻害、%T/C範囲は1〜40
2=腫瘍静止、%T/C範囲は0〜−40
3=腫瘍退行、%T/C範囲は−50〜−100
4=%T/C範囲は−50〜−100および>30%(腫瘍なしマウス)
。
【0096】
TGD 腫瘍増殖遅延(T−Cともあらわされる)。このパラメーターは、任
意のサイズ(代表的には、500または1000mm3)を得るために処置腫瘍
および未処置腫瘍についての時間差(日数で)をあらわす。
【0097】
TI 治療指数。治療指数は、毒性パラメーター(すなわち、LD50、LD
10、MTD)および生物学的活性パラメーター(すなわち、ED50−処置群
の50%において、規定された生物学的応答を生じる用量)の比である。一般に
、TIは、薬物についての安全性の限界を示す。動物モデル研究について、これ
は、伝統的には、以下の式によって記載される:
TI=LD50/ED50
しかし、LD50研究を行うことは、もはや倫理的に許されないので、本発明者
らは、これらの研究のために、以下のように治療指数を規定した:
TI=MTD/MED。
【0098】
上記の説明は、例示であって、制限するものでないと意図することが理解され
るべきである。多くの実施形態は、上記の説明を読めば当業者に明らかである。
従って、本発明の範囲は、上記の説明を参照して決定されるのではなく、添付の
特許請求の範囲を参照してこのような特許請求の範囲により権利が与えられる等
価物の全範囲とともに決定されるべきである。全ての学術文献および参考文献(
特許出願および刊行物を含む)の開示は、全ての目的のために、本明細書中に参
考として援用される。
【特許請求の範囲】
【請求項1】
活性薬剤の血漿循環半減期を調節するための組成物であって、
リポソームであって、該リポソーム中に、遊離の活性薬剤および沈殿した活性薬剤がカプセル化されている、リポソーム
を包含し、該リポソーム中の沈殿している該活性薬剤の量は変化し、該活性薬剤 対 脂質の割合は、0.005〜1(重量)対1(重量)である、組成物。
【請求項2】
活性薬剤の血漿循環半減期を調節するための組成物であって、
リポソームであって、該リポソーム中に、遊離の活性薬剤および沈殿した活性薬剤がカプセル化されており、該活性薬剤 対 脂質の割合は、0.005〜1(重量)対1(重量)である、リポソーム;および
活性薬剤がカプセル化されていないリポソーム
を含む、組成物。
【請求項3】
リポソーム処方物であって、該リポソーム処方物は、
a)抗新生物薬物;および
b)遊離の抗新生物薬物および沈殿した抗新生物薬物を有するリポソーム
を含み、ここで、該リポソーム中の沈殿した抗新生物薬物は、抗新生物薬物の総量の少なくとも50%であり、該抗新生物薬物 対 脂質の割合は、0.005〜1(重量)対1(重量)である、リポソーム処方物。
【請求項4】
リポソーム処方物であって、該リポソーム処方物は、
a)活性薬剤;
b)リポソームであって、該リポソーム中に、遊離の活性薬剤および沈殿した活性薬剤がカプセル化されており、該活性薬剤 対 脂質の割合は、0.005〜1(重量)対1(重量)である、リポソーム;および
c)空のリポソーム、
を含む、リポソーム処方物。
【請求項1】
活性薬剤の血漿循環半減期を調節するための組成物であって、
リポソームであって、該リポソーム中に、遊離の活性薬剤および沈殿した活性薬剤がカプセル化されている、リポソーム
を包含し、該リポソーム中の沈殿している該活性薬剤の量は変化し、該活性薬剤 対 脂質の割合は、0.005〜1(重量)対1(重量)である、組成物。
【請求項2】
活性薬剤の血漿循環半減期を調節するための組成物であって、
リポソームであって、該リポソーム中に、遊離の活性薬剤および沈殿した活性薬剤がカプセル化されており、該活性薬剤 対 脂質の割合は、0.005〜1(重量)対1(重量)である、リポソーム;および
活性薬剤がカプセル化されていないリポソーム
を含む、組成物。
【請求項3】
リポソーム処方物であって、該リポソーム処方物は、
a)抗新生物薬物;および
b)遊離の抗新生物薬物および沈殿した抗新生物薬物を有するリポソーム
を含み、ここで、該リポソーム中の沈殿した抗新生物薬物は、抗新生物薬物の総量の少なくとも50%であり、該抗新生物薬物 対 脂質の割合は、0.005〜1(重量)対1(重量)である、リポソーム処方物。
【請求項4】
リポソーム処方物であって、該リポソーム処方物は、
a)活性薬剤;
b)リポソームであって、該リポソーム中に、遊離の活性薬剤および沈殿した活性薬剤がカプセル化されており、該活性薬剤 対 脂質の割合は、0.005〜1(重量)対1(重量)である、リポソーム;および
c)空のリポソーム、
を含む、リポソーム処方物。
【図1】

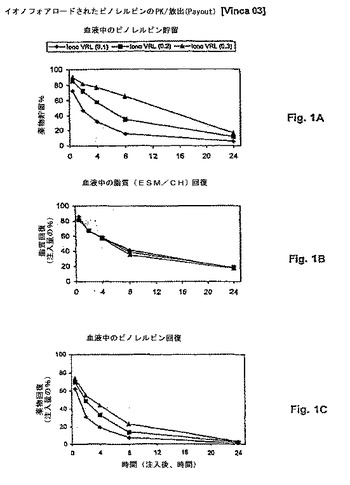
【図2】
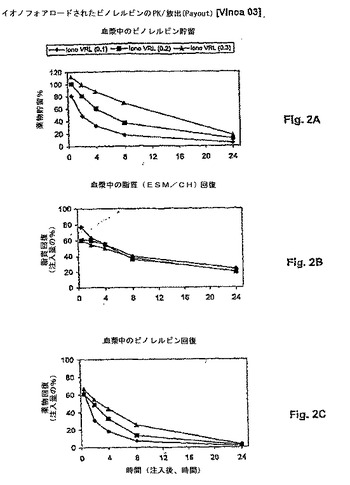
【図3】
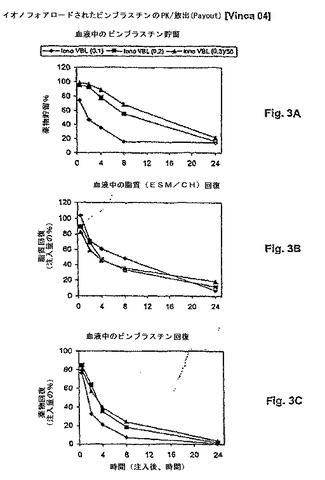
【図4】
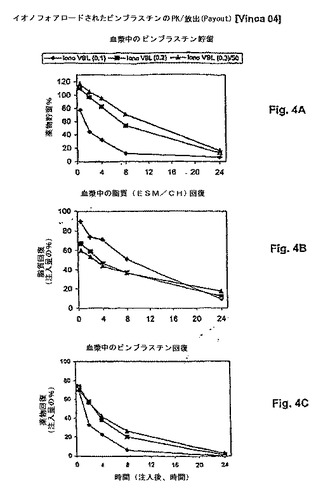
【図5】
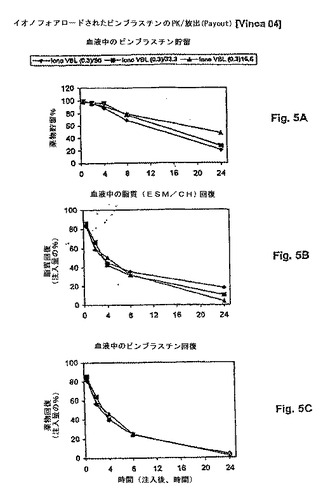
【図6】
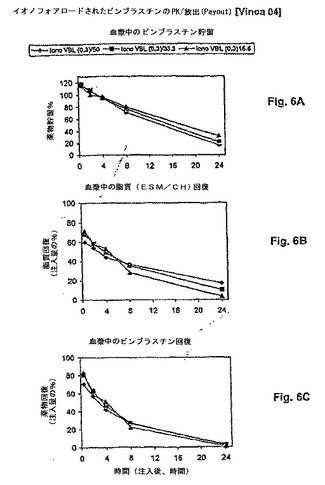
【図7】
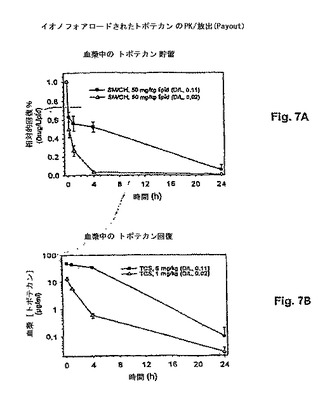
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2012−92148(P2012−92148A)
【公開日】平成24年5月17日(2012.5.17)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−25785(P2012−25785)
【出願日】平成24年2月9日(2012.2.9)
【分割の表示】特願2002−506699(P2002−506699)の分割
【原出願日】平成13年6月29日(2001.6.29)
【出願人】(510184830)テクミラ ファーマシューティカルズ コーポレイション (3)
【Fターム(参考)】
【公開日】平成24年5月17日(2012.5.17)
【国際特許分類】
【出願番号】特願2012−25785(P2012−25785)
【出願日】平成24年2月9日(2012.2.9)
【分割の表示】特願2002−506699(P2002−506699)の分割
【原出願日】平成13年6月29日(2001.6.29)
【出願人】(510184830)テクミラ ファーマシューティカルズ コーポレイション (3)
【Fターム(参考)】
[ Back to top ]