
リンカーを有するアゾ化合物及び当該アゾ化合物を用いた金属イオン回収方法
【課題】金属イオンの捕捉・脱離を制御し得るアゾベンゼン誘導体を提供するとともに、当該アゾベンゼン誘導体を用いて金属イオンの捕捉・脱離を制御する方法を提供する。
【解決手段】下記一般式(1)で表されるアゾ化合物、

(式中、R1a、R1b、R1c、R2a及びR2bは直鎖状のアルキレン基を示し、Xはリンカー部位を示し、Yは固相基板に対して結合性を有する固相基板結合部位を示す。)並びに、アゾ化合物に結合した金属イオンを脱離させる工程、を含む金属イオン回収方法。
【解決手段】下記一般式(1)で表されるアゾ化合物、
(式中、R1a、R1b、R1c、R2a及びR2bは直鎖状のアルキレン基を示し、Xはリンカー部位を示し、Yは固相基板に対して結合性を有する固相基板結合部位を示す。)並びに、アゾ化合物に結合した金属イオンを脱離させる工程、を含む金属イオン回収方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、アゾ化合物に関し、より詳細にはリンカーを有するアゾ化合物に関する。本発明はまた、当該アゾ化合物を用いた金属イオン回収方法に関する。
【背景技術】
【0002】
金属元素は現在の化学産業を支える必要不可欠な元素であり、我々生物が生きていくためにも重要な役割を果たしている。しかし、重金属資源は有限であり、生物にとっても重金属イオンの過剰摂取は毒となりうる。そのため、重金属イオンの検出、捕捉に関する研究が盛んに行われてきている。現行の重金属イオンの処理は、例えば、カルボン酸のような配位性官能基を有する高分子材料への重金属イオンの吸着により固体表面へ固定化(捕捉)し、廃棄することにより実施されている。しかし、重金属資源の枯渇が懸念される現状から、捕捉した金属の再利用が望まれており、金属の捕捉・再利用のために、溶液などから金属イオンを取り出す時には強く結合し回収する時には容易に金属イオンを脱離できるような材料の開発が切望されている。
【0003】
そこで、金属イオンの結合を制御する方法に利用できる可能性があるとして、例えばアゾベンゼン(及びその誘導体)のような光応答性分子に注目が集まっている。アゾベンゼンは熱力学的に安定なtrans-体に紫外光を照射することでcis-体へと異性化し、cis-体への可視光照射もしくは加熱によりtrans-体を再度生成する。
【0004】
【化1】
【0005】
これまでに、アゾベンゼンにキレート環を含む複数の配位性官能基を導入したアゾベンゼン誘導体を用い、上述の光応答性を利用することで、金属イオンの捕捉・脱離を制御する試みがなされている(例えば非特許文献1)。すなわち、当該アゾベンゼン誘導体がtrans-体のときには配位性官能基が立体的に離れた位置に存在するために金属イオンを捕捉できないが、cis-体のときには配位性官能基が立体的に近い位置に存在し、当該配位性官能基が配位子として金属イオンと結合して、金属イオンを捕捉できると予想されたため、当該予想を裏付けるための検討がなされてきたのである。
【0006】
しかしながら、例えば非特許文献1に示されるように、溶液中のアゾベンゼン誘導体はtrans-体・cis-体のいずれであっても金属イオンと強く結合してしまい、期待された金属イオンの捕捉・脱離を制御する効果は得られていない。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Shinkai, S.; Nakamura, S.; Nakashima, M.; Manabe, O.; Iwamoto, M. Bull. Chem. Soc. Jpn. 1985, 58, 2340-2347.
【発明の概要】
【発明が解決しようとする課題】
【0008】
本発明は、金属イオンの捕捉・脱離を制御し得るアゾ化合物を提供するとともに、当該アゾ化合物を用いて金属イオンの捕捉・脱離を制御する方法を提供することを課題とする。
【課題を解決するための手段】
【0009】
本発明者らは、驚くべき事に、下記一般式(1)で表されるアゾ化合物を固相基板に結合させ、当該アゾ化合物結合基板を用いて金属イオンの捕捉・脱離を制御できることを見出し、さらに改良を重ねて本発明を完成させるに至った。
【0010】
すなわち、本発明は例えば以下の項1〜7のアゾ化合物、アゾ化合物結合固相基板及び金属イオン回収方法に係るものである。
項1.
下記一般式(1)で表されるアゾ化合物。
【0011】
【化2】
【0012】
(式中、
R1aは、直鎖状のC1〜C10アルキレン基を示し、
R1b及びR1cは、同一又は異なって、直鎖状のC1〜C6アルキレン基を示す。
R2aは、直鎖状のC1〜C10アルキレン基を示し、
R2bは、直鎖状のC1〜C6アルキレン基を示す。
Xはリンカー部位を示し、Yは固相基板に対して結合性を有する固相基板結合部位を示す。)
項2.
前記リンカー部位がアルキレン鎖中に−NHCO−を有するアルキレン基であり、
前記構造基板結合部位がチオール基、ジスルフィド基、スルフィド基、ジチオラン基又はシロキシ基である、項1に記載のアゾ化合物。
項3.
項1又は2に記載のアゾ化合物を前記固相基板結合部位により固相基板に結合させたアゾ化合物結合固相基板。
項4.
前記固相基板が、表面を金でコーティングされた固相基板である、項3に記載のアゾ化合物結合固相基板。
項5.
(i)金属イオン含有溶液に、項3又は4に記載のアゾ化合物結合固相基板を浸積してアゾ化合物に金属イオンを結合させる工程、
(ii)当該アゾ化合物結合固相基板のアゾ化合物に可視光を照射する工程、
(iii)当該アゾ化合物結合固相基板を作用電極として金属イオンの酸化還元反応を繰り返して、アゾ化合物に結合した金属イオンを脱離させる工程、
を含む金属イオン回収方法。
項6.
前記酸化還元反応が、前記金属イオンの酸化還元電位より300mV低い電位から、当該酸化還元電位より300mV高い電位の幅で30回以上繰り返される、
項5に記載の金属イオン回収方法。
項7.
項5又は6に記載の工程(i)〜(iii)を含む工程を繰り返し行うことにより、金属イオンを回収する方法。
【発明の効果】
【0013】
本発明によって、金属イオンの捕捉・脱離に利用できる新規なアゾ化合物が提供される。また、当該アゾ化合物が結合した固相基板が提供される。さらに、当該アゾ化合物結合固相基板を用いた金属イオン回収方法が提供される。
【0014】
当該アゾ化合物結合固相基板のアゾ化合物は、非常に強い金属イオン結合能を有するため、これまで金属イオン回収が不可能であった金属イオン含有溶液(例えば、金属イオン濃度が薄い溶液、金属イオン以外の夾雑物が多量に溶解した溶液(廃液)等)からも金属イオン回収が可能である。
【0015】
さらに、当該アゾ化合物は、cis体・trans体のいずれであっても溶液中で金属イオンとほぼ同様の強さで結合するが、これを固相基板に結合させた当該アゾ化合物結合固相基板を用いると、trans体に比べcis体の方が金属イオンとより強く結合する。従って、当該アゾ化合物結合固相基板においては、cis体に結合した金属イオンよりtrans体に結合した金属イオンの方が脱離しやすく、当該アゾ化合物のcis体・trans体を紫外光及び可視光照射等によって制御することにより、当該アゾ化合物に結合した金属イオンの脱離を制御することができる。
【図面の簡単な説明】
【0016】
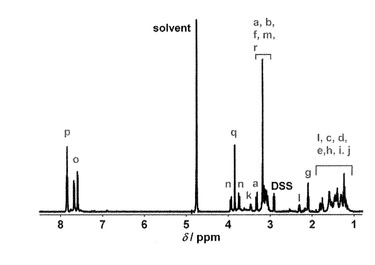
【図1】trans-L1の1H-NMRスペクトル(in D2O)を示す。
【図2】キセノンランプ照射に伴うtrans-L1 (a)およびtrans-L2 (b)の吸収スペクトル変化を示す。(1 mM in H2O; 黒線: trans-L, 青線: 紫外光照射時の光定常状態, 赤線: 可視光照射時の光定常状態)
【図3】キセノンランプ照射に伴うtrans-L1の1H-NMRスペクトル変化を示す。(1 mM in D2O; a: trans-L1, b: 紫外光照射時の光定常状態, c: 可視光照射時の光定常状態)
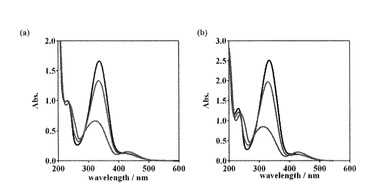
【図4】キセノンランプ照射に伴う[ZnII(trans-L1)] (a)および[CuII(trans-L1)] (b)の吸収スペクトル変化を示す。(1 mM in H2O; 灰線: trans-L1, 黒線: [MII(trans-L1)], 青線: 紫外光照射時の光定常状態, 赤線: 可視光照射時の光定常状態)
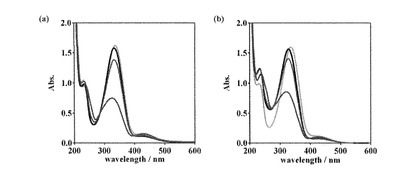
【図5】キセノンランプ照射に伴う[ZnII(trans-L2)] (a)および[CuII(trans-L2)] (b)の吸収スペクトル変化を示す。(1 mM in H2O; 灰線: trans-L1, 黒線: [MII(trans-L2)], 青線: 紫外光照射時の光定常状態, 赤線: 可視光照射時の光定常状態)
【図6】CuII(ClO4)2, L1, [CuII(L1)]のサイクリックボルタモグラムを示す。(1 mM in 0.1 M NaClO4水溶液, 掃引速度: 50 mV s-1; 黒線: CuII(ClO4)2, 赤点線: trans-L1, 赤実線: [CuII(trans-L1)], 青点線: cis-L1, 青実線: [CuII(cis-L1)])
【図7】各修飾電極及びその作製過程の模式図を示す。
【図8】(trans-H4L1)/C6-Au, (trans-L1)/C6-Au, [CuII(trans-L1)]/C6-Auのサイクリックボルタモグラムを示す。(電解質溶液: 0.1 M NaClO4水溶液, 掃引速度: 50 mV s-1: 黒点線: (trans-H4L1)/C6-Au, 黒実線: (trans-L1)/C6-Au, 赤線: [CuII(trans-L1)]/C6-Au)
【図9】[CuII(trans-L1)]/C6-Auのサイクリックボルタモグラムを示す。(電解質溶液: 0.1 M NaClO4水溶液, 掃引速度: 50 mV s-1; 赤線: 1サイクル目, 灰線: 5, 15, 40サイクル目, 緑線: 60サイクル目)
【図10】(cis-H4L1)/C6-Au, (cis-L1)/C6-Au, [CuII(cis-L1)]/C6-Auのサイクリックボルタモグラムを示す。(電解質溶液: 0.1 M NaClO4水溶液, 掃引速度: 50 mV s-1 : 黒点線: (cis-H4L1)/C6-Au, 黒実線: (cis-L1)/C6-Au, 青線: [CuII(cis-L1)]/C6-Au)
【図11】[CuII(cis-L1)]/C6-Auのサイクリックボルタモグラム(電解質溶液: 0.1 M NaClO4水溶液, 掃引速度: 50 mV s-1; 青線: 1サイクル目, 灰線: 5, 15, 40サイクル目, 緑線: 60サイクル目)
【図12】[CuII(trans-L1)]/C6-Au (赤)および[CuII(cis-L1)]/C6-Au (青)の還元ピーク電気量(1回目の電気量で規格化)と掃引回数との関係を表したグラフを示す。
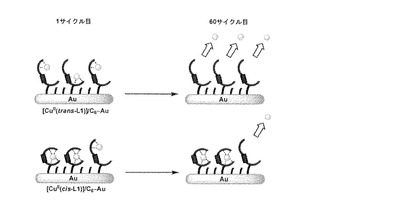
【図13】[CuII(L1)]/C6-Auの1サイクル目と60サイクル目におけるCuイオン結合状態の模式図を示す。
【図14】[CuII(cis-L1)]/C6-Auへの可視光照射前後のサイクリックボルタモグラムを示す。(電解質溶液: 0.1 M NaClO4水溶液, 掃引速度: 50 mV s-1; 緑線: 照射前, 黒線: 90分照射後, 灰線: 180分照射後, 赤線: 270分照射後)
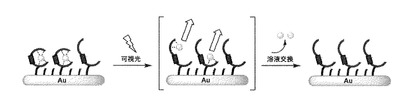
【図15】[CuII(cis-L1)]/C6-Au への可視光照射に伴うCuIIイオン脱離過程の模式図を示す。
【発明を実施するための形態】
【0017】
以下、本発明について、さらに詳細に説明する。
アゾ化合物
【0018】
【化3】
【0019】
本発明は、上記一般式(1)で表されるアゾ化合物に係る。
【0020】
一般式(1)中、R1aは、直鎖又は分岐鎖状のC1〜C10アルキレン基、好ましくはC1〜C6アルキレン基、より好ましくはC1〜C3アルキレン基を示す。また、直鎖状であることが好ましい。具体的には、メチレン基、エチレン基、トリメチレン基、プロピレン基等が例示でき、特にメチレン基が好ましい。
【0021】
また、R1b及びR1cは、同一又は異なって、直鎖又は分岐鎖状のC1〜C6アルキレン基、好ましくはC1〜C4アルキレン基、より好ましくはC1〜C3アルキレン基を示す。また、直鎖状であることが好ましい。具体的には、メチレン基、エチレン基、トリメチレン基、プロピレン基等が例示でき、特にメチレン基が好ましい。なお、R1b及びR1cのアルキレン基は、同一であるものがより好ましい。
【0022】
また、一般式(1)中、R2aは、直鎖又は分岐鎖状のC1〜C10アルキレン基、好ましくはC1〜C6アルキレン基、より好ましくはC1〜C3アルキレン基を示す。また、直鎖状であることが好ましい。具体的には、メチレン基、エチレン基、トリメチレン基、プロピレン基等が例示でき、特にメチレン基が好ましい。
【0023】
また、R2bは、直鎖又は分岐鎖状のC1〜C6アルキレン基、好ましくはC1〜C4アルキレン基、より好ましくはC1〜C3アルキレン基を示す。また、直鎖状であることが好ましい。具体的には、メチレン基、エチレン基、トリメチレン基、プロピレン基等が例示でき、特にメチレン基が好ましい。
【0024】
なお、R1a及びR2aのアルキレン基は同一であるものが好ましい。
【0025】
また、Xはリンカー部位を示し、Yは固相基板に対して結合性を有する固相基板結合部位を示す。なお、以下、一般式(1)で表されるアゾ化合物において、−X−Y部位を除いた部分を「金属イオン捕捉部位」と表記することがある。
【0026】
前記固相基板結合部位は、固相基板に対して結合性を有し、好ましくは固相基板表面上に高密度かつ高配向性の自己組織化単分子膜(Self-Assembled Monolayer, SAM)を構築することが可能な官能基から形成される。該官能基は固相基板の種類によって選択され、例えば、固相基板表面が金の場合は、チオール基、ジスルフィド基、スルフィド基、ジチオラン基を、ガラスの場合はシロキシ基を用いると、SAM膜が好適に形成されることが知られているが、これらに限定されるものではない。
【0027】
前記リンカー部位は、一般式(1)で表されるアゾ化合物を固相基板に結合させた際、金属イオン捕捉部位を当該固相基板から一定間隔離して存在せしめるための部位である。従って、当該機能を有すればリンカー部位の構造は特に制限されないが、例えば、直鎖もしくは無置換のC1〜C20アルキレン基をリンカー部位として用いることができる。当該アルキレン基は直鎖又は分岐鎖状であってよいが、直鎖であることが好ましい。また、当該アルキレン鎖の炭素数は1〜20が好ましく、3〜14がより好ましく、3〜8がさらに好ましい。中でも、アルキレン鎖中に−NHCO−を有していてもよいアルキレン基が好ましく、−NHCO−を有するアルキレン基がより好ましい。
【0028】
なお、リンカー部位がアルキレン鎖中に−NHCO−を有する直鎖状アルキレン基である場合は、−NHCO−と−Yとの間に存在する炭素数が1〜10、好ましくは1〜6、より好ましくは1〜4であり、−NHCO−と金属イオン捕捉部位との間に存在する炭素数が1〜4、好ましくは2〜4である。また、当該−NHCO−のC末端側が固相基板結合部位側であることが好ましい。
【0029】
また、上述のアルキレン鎖中に−NHCO−を有するアルキレン基において、当該−NHCO−に、荷電を有さないアミノ酸が1以上ペプチド結合により連続して結合したペプチドが連結され、当該アルキレン鎖中に含まれるものも、本発明のアゾ化合物のリンカー部位として好ましく用いることができる。
【0030】
すなわち、アルキレン鎖中に−NHCO−を有し、当該−NHCO−のC末端側が固相基板結合部位側であるアルキレン基において、金属イオン捕捉部位から−NHCO−までをアルキレン基(α)、−NHCO−から固相基板結合部位までをアルキレン基(β)と表記した場合、−X−Yは、
−アルキレン基(α)−NHCO−アルキレン基(β)−Y
であってよいし、また、
−アルキレン基(α)−NHCO−(電荷を有さないアミノ酸が1以上ペプチド結合により連続して結合したペプチド)−NHCO−アルキレン基(β)−Y
であってもよい。(なお下線を引いたCO及びNHは当該ペプチドに由来する。)
ここで、アルキレン基(α)は、炭素数2〜4の直鎖状アルキレン基であることが好ましく、アルキレン基(β)は、炭素数1〜10(好ましくは1〜6、より好ましくは1〜4)の直鎖又は分岐鎖上のアルキレン基であることが好ましい。
【0031】
また、(荷電を有さないアミノ酸が1以上ペプチド結合により連続して結合したペプチド)における「荷電を有さないアミノ酸」としては、グリシン、アラニン、バリン、ロイシン、イソロイシン、フェニルアラニン、トリプトファン、メチオニン、プロリン、セリン、トレオニン、チロシン、システイン等が挙げられ、特にグリシン、アラニン、バリン、ロイシン、イソロイシンが好ましい。
【0032】
ここでの「1以上」は好ましくは1〜20、より好ましくは1〜15、さらに好ましくは1〜10、さらにより好ましくは1〜5、特に好ましくは1〜3である。
【0033】
なお、当該ペプチドの固相基板結合部位側側末端がシステインである場合は、当該システインの−SH基が固相基板結合部位として働き得るため、−X−Yは、
−アルキレン基(α)−NHCO−(荷電を有さないアミノ酸が1以上ペプチド結合により連続して結合したペプチド)
であり得る。
【0034】
上記一般式(1)で表されるアゾ化合物は、種々の方法により製造され得る。その一例として、下記式で表されるアゾ化合物(以下「化合物(A)」と表記することがある)の合成方法を以下に示す。当該化合物(A)の製造において、用いる出発物質を変えれば、上記一般式(1)で表される種々のアゾ化合物を合成できることが、当業者は容易に理解できる。
【0035】
【化4】
【0036】
〔式中、lは1〜10の整数を示し、m及びnは同一又は異なって1〜6の整数を示し、kは1〜6の整数を示し、jは2〜4の整数を示し、pは1〜10の整数を示す。〕
なお、一般式(1)における−X−は、化合物(A)においては、
【0037】
【化5】
【0038】
〔j及びpは前記に同じ〕
に該当し、一般式(1)における−Yは、化合物(A)においては
【0039】
【化6】
【0040】
(すなわちジチオラン基)に該当する。
【0041】
化合物(A)は、下記化合物(1A)をTFAに溶解させ、室温にて1〜24時間程度撹拌して反応させることにより、得ることができる。当該反応終了後、溶液を減圧乾固し、得られた残渣を例えばジエチルエーテルで洗浄してもよい。なお、これ以降の化合物の構造式においてButは第三ブチル基を示す。
【0042】
【化7】
【0043】
〔式中、l、m、n、k、j及びpは前記に同じ〕
化合物(A1)は、例えば、下記反応式−1Aに示される方法により製造される。なお、これ以降の反応式において、Butは第三ブチル基を示し、DIEAはジイソプロピルエチルアミンを示す。
[反応式−1A]
【0044】
【化8】
【0045】
〔式中、l、m、n、k、j及びpは前記に同じ〕
化合物(A1)は、例えば、化合物(B1)及び化合物(C1)を溶解させたDMF溶液に、DIEA、NaIを添加し、室温にて1〜24時間撹拌して反応させることにより得ることができる。また、反応後、酢酸エチル等で抽出し、シリカゲルカラムクロマトグラフィー等の常法により精製することができる。
【0046】
上記反応式−1Aにおいて出発原料として用いられている化合物(B1)は、公知の化合物であるか、又は公知の方法により容易に製造される。例えば、下記反応式−1Bに示す方法により製造できる。
[反応式−1B]
【0047】
【化9】
【0048】
〔式中、l、m及びnは前記に同じ〕
化合物(B1)は、例えば、化合物(B2)及び化合物(B3)を溶解したDMF溶液に、DIEA及びNaIを添加し、室温にて1〜12時間撹拌して反応させて得ることができる。また、反応後、酢酸エチル等で抽出し、シリカゲルカラムクロマトグラフィー等の常法により精製することができる。
【0049】
上記反応式−1Bにおいて出発原料として用いられている化合物(B2)は、公知の化合物であるか、又は公知の方法により容易に製造される。例えば、下記反応式−2Bに示す方法により製造できる。
[反応式−2B]
【0050】
【化10】
【0051】
〔式中、lは前記に同じ〕
化合物(B2)は、例えば、次のようにして得ることができる。まず、熱EtOHにNaOH、p−ニトロベンジルアルコール、亜鉛粉末をこの順に添加し、70〜90℃にて1〜6時間撹拌した後、亜鉛粉末をろ去し、当該ろ液を半分以下の容量にまで減圧濃縮する。そして、これを1NHCl溶液によりpHを約5に調整した後、酢酸エチル等を用いて抽出すればよい。これを水・飽和食塩水等で洗浄し、減圧乾固させる。さらに、このようにして得られた化合物をDMFに溶解し、SOCl2を加えて室温で撹拌し、減圧乾固し、シリカゲルカラムクロマトグラフィー等の常法により精製して化合物(B2)を得ることができる。
【0052】
上記反応式−2Bにおいて出発原料として用いられている化合物(B4)は、公知の化合物であるか、又は公知の方法により容易に製造される。
【0053】
なお、反応式−2Bにおいて、出発原料として化合物(B4)だけでなく、化合物(B4)とはlの値が異なる化合物を混合して出発物質とすることにより、下記の化合物(B2’)を得ることもできる。化合物(B2’)を反応式−1Bの出発原料とすることもできる。
【0054】
【化11】
【0055】
〔式中l及びl’はそれぞれ異なって1〜10の整数を示す。〕
また、上記反応式−1Bにおいて出発原料として用いられている化合物(B3)は、公知の化合物であるか、又は公知の方法により容易に製造される。例えば、下記反応式−3Bに示す方法により製造できる。
[反応式−3B]
【0056】
【化12】
【0057】
〔式中、m及びnは前記に同じ〕
化合物(B3)は、例えば、化合物(B5)を酢酸第三ブチルに溶解させ、これに過塩素酸水溶液を添加し、室温にて10〜120分撹拌して、K2CO3等でpHを約9に調整後、例えば酢酸エチル等で抽出して得ることができる。さらに、例えば、これを水又は飽和食塩水等で洗浄した後、無水硫酸ナトリウムを加えて数時間室温で放置した後、硫酸ナトリウムをろ去し、ろ液を減圧乾固して固体として得ることができる。
【0058】
上記反応式−3Bにおいて出発原料として用いられている化合物(B5)は、公知の化合物であるか、又は公知の方法により容易に製造される。
【0059】
上記反応式−1Aにおいて用いられている化合物(C1)は、公知の化合物であるか、又は公知の方法により容易に製造される。例えば、下記反応式−1Cに示す方法により製造できる。
[反応式−1C]
【0060】
【化13】
【0061】
〔式中、k、j及びpは前記に同じ。〕
化合物(C1)は、例えば、化合物(D1)及びトリエチルアミンをDMSOに溶解させ、当該溶液に化合物(E1)のDMSO溶液を滴下し、室温にて1〜12時間撹拌して反応させることによって得ることができる。反応終了後、例えば酢酸エチル等で抽出し、常法に従って減圧乾固することもできる。
【0062】
上記反応式−1Cにおいて出発原料として用いられている化合物(D1)は、公知の化合物であるか、又は公知の方法により容易に製造される。例えば、下記反応式−1Dに示す方法により製造できる。
[反応式−1D]
【0063】
【化14】
【0064】
〔式中、k及びjは前記に同じ。k’はkより1小さい整数を示す。〕
化合物(D1)は、例えば、次のようにして得ることができる。化合物(D2)(塩酸塩)をギ酸にヨウ化し、当該溶液に化合物(D3)(水和物)を添加して、20〜60℃にて1〜12時間撹拌し、溶液を減圧乾固する。得られた残渣を1NHCl水溶液に溶解し、2〜12時間撹拌し、さらに当該溶液を減圧乾固する。得られた残渣を酢酸第三ブチルに溶解し、過塩素酸水溶液を添加した後、室温にて6〜24時間撹拌して得ることができる。これを常法に従って精製することができる。
【0065】
上記反応式−1Dにおいて出発原料として用いられている化合物(D2)及び(D3)は、それぞれ、公知の化合物であるか、又は公知の方法により容易に製造される。
【0066】
なお、化合物(D2)のかわりに、以下の化合物(D2’)を出発原料として用いることもできる。
【0067】
【化15】
【0068】
(式中、jは前記に同じ。−RNは“−NHCO−(荷電を有さないアミノ酸が1以上ペプチド結合により連続して結合したペプチド)”を示す。)
また、上記反応式−1Cにおいて出発原料として用いられている化合物(E1)は、公知の化合物であるか、又は公知の方法により容易に製造される。例えば、下記反応式−1Eに示す方法により製造できる。
[反応式−1E]
【0069】
【化16】
【0070】
〔式中、pは前記に同じ。〕
化合物(E1)は、例えば、化合物(E2)及びN−ヒドロキシスクシンイミド(NHS)をTHFに溶解し、低温(約0〜10℃)で6〜24時間程度撹拌した後、室温でさらに12〜48時間撹拌して反応させ、得ることができる。さらに常法に従い、精製することができる。
【0071】
上記反応式−1Eにおいて出発原料として用いられている化合物(E2)は、公知の化合物であるか、又は公知の方法により容易に製造される。
【0072】
なお、化合物(E2)において、基
【0073】
【化17】
【0074】
(すなわちジチオラン基)が、例えばチオール基、ジスルフィド基、スルフィド基又はシロキシ基等である化合物を、上記反応式−1Eにおいて化合物(E2)のかわりに出発原料として用いることも可能である。このような化合物は、公知の化合物であるか、又は公知の方法により容易に製造される。
【0075】
アゾ化合物結合固相基板
上述した一般式(1)で表されるアゾ化合物を、固相基板に結合させアゾ化合物結合固相基板を得ることができる。本発明はこのようなアゾ化合物結合固相基板に係る。
【0076】
本発明において用いる固相基板とは、その表面においてアゾ化合物の固相基板結合部位の官能基が好適にSAM膜を形成する限り、特に限定されない。当該固相基板の材料および厚さの選択は、アゾ化合物の固相基板結合部位の種類等に依存して、当業者が最適な条件を適宜選択することが可能である。基板材料に好適な例として、ガラス基板、金属基板(例えば金、銀、銅、アルミニウム、白金、酸化アルミニウム、SrTiO3、LaAlO3、NdGaO3、ZrO2等)、シリコン基板(例えば酸化珪素)、ポリマー樹脂基板(例えばポリエチレンテレフタレート、ポリカーボネート)、炭素(グラファイト)、マイカ等が例示できる。
【0077】
本発明において用いる固相基板は、前記材料のうち単一の材料からなる基板であってもよいし、1つの基板材料(第1の基板)の表面に別種類の少なくとも1つの材料からなる薄膜(第1の層)を形成していてもよいし、更に、前記第1の基板と前記第1の層との間に少なくとも1つの他の介在層(第2の層、第3の層、等)が存在していてもよい。好適な固相基板の具体例を挙げると、前記第1の基板としてマイカ基板を用い、前記第1の層として表面に金属膜(好適には金薄膜、銀薄膜、銅薄膜、白金薄膜)を有する前記マイカ基板等が挙げられる。なお、前記ガラス基板と前記金属膜との間には、他の材料からなる介在層が施されていてもよい。例えば、マイカ基板に金がコーティングされた固相基板を、本発明のアゾ化合物結合固相基板の作製のために好ましく用いることができる。
【0078】
前記第1の層を初めとする各金属膜の形成(コーティング)は、公知の方法により可能である。例えば電気めっき法、無電解めっき法、スパッタ法、蒸着法、イオンプレーティング法、等により形成することができる。なお、前記金属膜表面を有機溶剤で洗浄し、更に必要に応じて強酸で洗浄することによる分解除去、紫外線により発生するオゾン等による分解除去、等の方法を用いて汚染を除去することもできる。
【0079】
本発明において用いる固相基板の厚さは特に限定されないが、通常、前記第1の基板であれば、0.1mm〜30mm程度であり、好適には、0.1mm〜2mm程度である。
【0080】
一般式(1)で表されるアゾ化合物を固相基板に固定化する方法としては、固相基板結合部位及び使用する固相基板の種類によって、適宜適当な方法を選択することができる。例えば、当該アゾ化合物を含む溶液と固相基板とを接触させてインキュベートすることにより、当該アゾ化合物を当該固相基板上に吸着させる方法を用いることができる。
【0081】
さらに、例えば上述のようにしてアゾ化合物を固相基板に結合させた後、当該化合物が結合していない固相基板表面をブロッキングする工程を行ってもよい。このようなブロッキング操作は、適宜適当なブロッキング試薬及びブロッキング方法を選択して行うことができる。例えば、固相基板表面が金属である場合は、ヘキサンチオール溶液に浸積することにより、アゾ化合物が結合していない固相基板表面部分にヘキサンチオールがチオール基を介して結合するため、このように処理をしてブロッキング処理を行うことができる。
【0082】
このようにして得られるアゾ化合物結合固相基板は、金属イオンの回収のため、好ましく用いることができる。
【0083】
金属イオン回収方法
上述のようにして得られるアゾ化合物結合固相基板を用いて、効率よく金属イオンを回収することができる。本発明はこのような金属イオン回収方法に係る。
【0084】
当該固相基板に結合したアゾ化合物は、trans-体に紫外光を照射するとcis-体へと異性化し、cis-体へ可視光照射もしくは加熱を行うことによりtrans-体を再度生成する。trans-体、cis-体のいずれであっても、金属イオンを結合することができるが、その結合力はcis-体の方が強い。特に、金属イオンを捕捉(結合)したアゾ化合物が結合した、アゾ化合物結合固相基板を作用電極として金属イオンの酸化還元反応を繰り返すと、trans-体に結合した金属イオンはほぼ全て脱離してしまうが、cis-体に結合した金属イオンはほとんど脱離せず、結合したままとなる。本発明のアゾ化合物結合固相基板が有する当該性質を用いることにより、金属イオン含有廃液等から金属イオンを回収することが可能となる。
【0085】
具体的には、例えば以下の工程(i)〜(iii)を含む金属イオン回収方法により、金属イオンを簡便に回収することができる。
(i)金属イオン含有溶液に上述のアゾ化合物結合固相基板を浸積してアゾ化合物に金属イオンを結合させる工程
(ii)当該アゾ化合物結合固相基板のアゾ化合物に可視光を照射する工程
(iii)当該アゾ化合物結合固相基板を作用電極として金属イオンの酸化還元反応を繰り返して、アゾ化合物に結合した金属イオンを脱離させる工程
工程(i)において、金属イオン含有溶液は、金属イオンを含有する溶液であれば特に制限されないが、例えば産業廃液など、有効利用することができない溶液であると、これまで手間をかけて廃棄しなければならなかったものを有効利用することができるため、好ましい。また、ここでの金属イオンは、当該固相基板に結合したアゾ化合物の金属捕捉部位が捕捉できる金属イオンであれば特に制限されるものではなく、例えば銅イオン、亜鉛イオン、ニッケルイオン、コバルトイオン、クロムイオン、鉄イオン、マンガンイオン等が挙げられ、なかでも銅イオン、亜鉛イオンが好ましい。
【0086】
なお、工程(i)を行うにあたっては、当該固相基板に結合したアゾ加工物はtrans-体、cis-体のいずれであってもよいが、上述のようにcis-体の方がより強く金属イオンを結合することができるため、trans-体では金属イオンを結合することができない溶液(例えば、金属イオン濃度が薄い溶液、金属イオン以外の夾雑物が多量に溶解した溶液〔廃液〕等)から回収を行う場合は、cis-体であることが好ましい。当該固相基板に結合したアゾ化合物に紫外光を照射することにより、当該アゾ化合物をcis-体とすることができる。よって、当該金属イオン回収方法は、工程(i)の前に、当該アゾ化合物結合固相基板のアゾ化合物に紫外光を照射する工程(工程0)を含んでもよい。
【0087】
工程(ii)において、当該アゾ化合物結合固相基板のアゾ化合物に可視光を照射する。可視光照射時間は特に制限されるものではないが、通常0.5〜3時間、好ましくは1〜2時間程度である。これにより、当該アゾ化合物がtrans-体となる。ただし、当該固相基板に結合したアゾ化合物の全てをtrans-体とする必要はなく、一定量(好ましくは5割以上、より好ましくは8割以上)のアゾ化合物がtrans-体となればよい。
【0088】
工程(iii)において、当該アゾ化合物結合固相基板(結合したアゾ化合物の多くはtrans-体で、金属イオンを結合している)を作用電極として用い、例えばサイクリックボルタンメトリー(CV)を行って、結合している金属イオンの酸化還元反応を繰り返すことにより、当該金属イオンをアゾ化合物から脱離させる。当該酸化還元反応は、結合している金属イオンの種類、当該金属イオンの酸化電位、還元電位、酸化還元電位等により、適宜その条件を設定することができる。また、当該酸化還元反応については、例えばCV(サイクリックボルタンメトリー)測定により確認することができる。
【0089】
当該酸化還元反応の電位幅は、特に制限されるものではないが、通常、酸化電位及び還元電位が含まれる幅で行われる。アゾ化合物に結合した金属イオンの酸化還元電位±(200〜300 mV)で行うことが好ましい。酸化還元電位については、予備実験等により簡単に計測することができる。
【0090】
当該酸化還元反応に用いる電解質溶液は、特に制限されないが、通常NaClO4溶液、KClO4溶液、NaNO3溶液、KNO3溶液等を用いることができる。なお、アゾ化合物から脱離した金属イオンは、カウンターアニオンの選択により望みの金属塩へと変換(リサイクル)できるため、このことを考慮したうえで電解質溶液を選択することが好ましい。
【0091】
掃引スピードも特に制限されないが、通常20〜80mVs−1、好ましくは40〜60mVs−1である。また、掃引サイクル数も、結合した金属イオンが脱離する(CVの場合、CV測定により酸化還元応答が実質的に消失する(すなわち、サイクリックボルタモグラムのピークが消失する))まで行えばよく、通常30回以上、好ましくは30〜100回、より好ましくは30〜80回である。
【0092】
サイクリックボルタンメトリーは例えば三電極系で行うことができる。この場合、作用電極には本発明に係るアゾ化合物結合固相基板を用いるが、対極及び参照電極は特に制限されず、例えば対極にPt線、参照電極にAg/AgClを用いることができる。
【0093】
なお、工程(i)及び(ii)の間あるいは工程(ii)及び(iii)の間に、当該アゾ化合物結合固相基板を金属イオン含有溶液から取り出し、異なる溶液(例えば1M NaClO4溶液等)中に移動させてもよい。このようにすれば、移動先の溶液中へ金属イオンが脱離することとなる(移動先の溶液を回収溶液とする)。すなわち、本発明の金属イオン回収方法は、工程(i)と(ii)の間に、当該アゾ化合物結合固相基板を回収溶液中に移動させる工程(工程(i-2))を含んでもよいし、工程(ii)と(iii)の間に、当該アゾ化合物結合固相基板を回収溶液中に移動させる工程(工程(ii-2))を含んでもよい。工程(ii)を経るとアゾ化合物がtrans体となり、金属イオンとの結合力がcis体より弱くなるため、工程(ii-2)よりも工程(i-2)を含むことが好ましい。
【0094】
さらに、本発明の金属イオン回収方法は、同一のアゾ化合物結合固相基板を用いて繰り返し行うことが可能である。すなわち、工程(iii)の後、固相基板上のアゾ化合物の多くはtrans-体であって金属イオンが脱離した状態にあるので、これを再度工程(i)に供することができる。また、上述したように、工程(i)に供する前に、固相基板に結合したアゾ化合物に紫外光を照射することにより(すなわち工程(0)を行うことにより)、これをcis-体としてもよい。紫外光照射時間は特に制限されず、通常0.5〜3時間、好ましくは0.5〜2時間程度である。
【0095】
なお、前述の工程(i-2)及び/又は工程(ii-2)を経た場合は、工程(i)に供する前に、アゾ化合物結合固相基板を回収溶液から工程(i)の金属含有溶液へと移動させる工程(工程iv)を経る必要がある。
【0096】
このように、本発明の金属イオン回収方法は、上述した工程(i)〜(iii)を含む工程を繰り返し行うことによって実施することもできる。また、当該繰り返し行われる工程には、工程(i)〜(iii)以外の工程が含まれてもよく、例えば上述した工程(0)、工程(i-2)、工程(ii-2)等を好ましく含むことができる。
【0097】
例えば、本発明は以下の工程(0)、(i)、(i-2)、(ii)、(iii)及び(iv)を含む金属イオン回収方法を含む。
(0)アゾ化合物結合固相基板のアゾ化合物に紫外光を照射する工程
(i)金属イオン含有溶液に上述のアゾ化合物結合固相基板を浸積してアゾ化合物に金属イオンを結合させる工程
(i-2)当該アゾ化合物結合固相基板を回収溶液中に移動させる工程
(ii)当該アゾ化合物結合固相基板のアゾ化合物に可視光を照射する工程
(iii)当該アゾ化合物結合固相基板を作用電極として金属イオンの酸化還元反応を繰り返して、アゾ化合物に結合した金属イオンを脱離させる工程
(iv)当該アゾ化合物結合固相基板を回収溶液から金属イオン含有溶液へと移動させる工程
またさらに本発明は、例えば、上記の工程(0)、(i)、(i-2)、(ii)、(iii)及び(iv)を含む工程を繰り返し行うことにより、金属イオンを回収する方法を含む。
【実施例】
【0098】
以下、本発明を具体的に説明するが、本発明は下記の例に限定されるものではない。
【0099】
なお、以下の例において、全ての試薬及び溶媒は、ナカライテスク、和光純薬、東京化成、石津製薬から購入し、そのまま使用した。ミリQ水はMillipore Milli-Q biocel Aにより得た。
【0100】
また、以下の例においては、1H-NMRスペクトル、MALDI-TOF Massスペクトル、UV-visスペクトル、CV、及びEQCMの測定は下記のようにして行った。
<1H-NMRスペクトル>
測定装置はJEOL社製JNM-ECP600分光計を使用し、δ= -0.1-10.1 ppmについて測定した。内径4 mmのサンプルチューブを使用し、溶媒はCDCl3またはD2Oを用いた。ケミカルシフトは内部標準のテトラメチルシランまたはDSSを基準として表記した。
<MALDI-TOF Massスペクトル>
測定装置はApplied Biosystems社製DE-STR Voyagerを使用し、マトリックスはa-シアノ-4-ヒドロキシケイ皮酸を用いた。キャリブレーションはポリエチレングリコール(分子量: 1000)を用いて行った。
<UV-visスペクトル>
測定装置は島津製作所製UV-3100PCを使用し、200-900 nmについて測定した。セルは光路長1 cmまたは0.1 cmの石英セルを使用した。サンプルは1 mM, 5 mMのアゾベンゼン化合物溶液(水, DMF)を用いた。
<CV>
測定装置はBAS社製CV-50Wを使用した。測定は三電極系で行い、作用電極に本発明に係るアゾ化合物結合固相基板(以下「修飾電極」と表記することがある;図7を参照)、対極にPt線、参照電極にAg/AgCl (3 M NaCl)を使用した。測定には0.1 M NaClO4水溶液または0.5 M KOH水溶液を用い、測定前に約15分間Arバブリングすることで溶存酸素を除去した。なお、実施例において酸化還元反応は全てCVによって行った。
【0101】
<合成例1:アゾ化合物の合成>
以下のようにして、下記の化学式で表されるアゾ化合物(trans-体)を合成した。なお、以下、下記式で表されるアゾ化合物をH4L1と表記することがある。(H4L1のH4は分子中に存在するCOOH基のHが4つあることを表している。従って、当該基がCOONaとなっている場合、Na4L1と表記される。)
【0102】
【化18】
【0103】
まず、H4L1の合成スキームを次に示す。なお、当該スキーム中に番号1〜5をふった化合物はそれぞれ以下「アゾベンゼン誘導体1」、「リジン誘導体2」、「リポ酸誘導体3」、「リジン誘導体4」、「アゾベンゼン誘導体5」と表記することがある。
【0104】
【化19】
【0105】
4,4’-ビスクロロメチルアゾベンゼンの合成
4,4’-ビスクロロメチルアゾベンゼンの合成は既報の方法((a) Mori, Y.; Niwa, T.; Toyoshi, K. Chem. Pharm. Bull. 1981, 29, 1439-1442, (b) Maie, K.; Nakamura, M.; Yamana, K. Nucleos. Nucleot. Nucl. 2006, 25, 453-462.)を改良して行った。 熱EtOH 120 mLにNaOH 9.6 g (240 mmol)、p-ニトロベンジルアルコール 6.0 g (39 mmol)、亜鉛粉末 5.0 g (77 mmol)をこの順に添加した。80℃にて3時間攪拌した後、亜鉛粉末をろ去した。ろ液を50 mLまで減圧濃縮し、水50 mLを加えた。1 N HCl水溶液によりpH 5に調整した後、酢酸エチルにて抽出した。得られた有機層を水、飽和食塩水で洗浄した後、ろ液を減圧乾固することで4,4’-ビスヒドロキシメチルアゾベンゼンの粗製物(1.9 g, 純度約60 %)を得た。
【0106】
得られた粗製物1.9 g (4.7 mmol)をDMF 20 mLに溶解し、SOCl20.86 mL (12 mmol)を加え、室温にて1時間攪拌した。反応溶液を減圧乾固し、残渣を少量のクロロホルムに溶解してシリカゲルカラムクロマトグラフィー(溶離液; クロロホルム:ヘキサン= 1 : 2)を行った。目的物はTLC (Rf= 0.95 (展開溶媒; クロロホルム))により確認し、得られた分画を減圧乾固した。得られた橙色粉末を真空乾燥し、1H-NMRスペクトルから目的物であることを確認した。
【0107】
【化20】
【0108】
収量 510 mg (収率 5 %); 1H-NMR (600 MHz, CDCl3) δ7.92 (d, J = 8.4 Hz, 4H, φ-Hc), 7.55 (d, J = 8.4 Hz, 4H, φ-Hb), 4.66 (s, 4H, -CHa2-).
【0109】
イミノ二酢酸ジ第三ブチルエステルの合成
イミノ二酢酸ジ第三ブチルエステルの合成は既報の方法(Chen, H.; Feng, Y.; Xu, Z.; Ye, T. Tetrahedron 2005, 61, 11132-11140.)を参考にして行った。イミノ二酢酸 1.0 g (7.5 mmol) を酢酸第三ブチル200 mL (1.5 mol) に溶解した。その溶液に60 %過塩素酸水溶液2.6 mL (40 mmol)を添加し、室温にて20時間撹拌した。その後、10 % K2CO3水溶液によりpH 9に調整し、酢酸エチルにて抽出した。得られた有機層を水、飽和食塩水にて洗浄した後、無水硫酸ナトリウムを加えて1時間放置した。硫酸ナトリウムをろ去し、ろ液を減圧乾固することで白色固体を得た。得られた白色固体を真空乾燥し、1H-NMRスペクトルから目的物であることを確認した。
【0110】
【化21】
【0111】
収量1.2 g (収率 66 %); 1H-NMR (600 MHz, CDCl3) δ 3.34 (s, 4H, -CHa2-), 1.47 (s, 18H, -CHb3).
【0112】
アゾベンゼン誘導体1の合成
4,4’-ビスクロロメチルアゾベンゼン 300 mg (1.08 mmol)、イミノ二酢酸ジ第三ブチルエステル 264 mg (1.08 mmol)のDMF 溶液(20 mL)にジイソプロピルエチルアミン(DIEA) 360μL (2.16 mmol)、NaI 726 mg (3.24 mmol)を添加し、室温にて3時間攪拌した。反応終了後、水100 mLを加え、酢酸エチルにて抽出した。得られた有機層を1 N HCl水溶液、水、飽和食塩水にて洗浄した後、ろ液を減圧乾固することで橙色固体を得た。得られた残渣を少量のクロロホルムに溶解してシリカゲルカラムクロマトグラフィー(溶離液; クロロホルム)を行った。目的物はTLC (Rf = 0.20 (展開溶媒; クロロホルム))により確認し、得られた分画を減圧乾固した。得られた橙色固体を真空乾燥し、1H-NMRスペクトルから目的物であることを確認した。
【0113】
【化22】
【0114】
収量150 mg (収率29 %); 1H-NMR (600 MHz, CDCl3) δ 7.91 (d, J = 8.4 Hz, 2H, φ-Hc), 7.88 (d, J = 8.4 Hz, 2H, φ-Hd), 7.57 (d, J = 8.4 Hz, 2H, φ-He), 7.54 (d, J = 8.4 Hz, 2H, φ-Hb), 4.66 (s, 2H, -CHa2-), 3.98 (s, 2H, -CHf2-), 3.45 (s, 4H, -CHg2-), 1.48 (s, 18H, -CHh3).
【0115】
リジン誘導体2の合成
リジン誘導体2の合成は既報の方法(Kihlberg, J.; Bergman, R.; Wickberg, B. Acta Chem. Scand. B 1983, 37, 911-916.)を参考にして行った。Lys・HCl 546 mg (3.0 mmol) をギ酸 30 mL に溶解し、その溶液にグリオキシル酸一水和物600 mg (6.6 mmol)を添加した。40℃にて4時間攪拌し、溶液を減圧乾固した。得られた残渣を1 N HCl水溶液2 mLに溶解し、5時間攪拌した後、溶液を減圧乾固した。得られたオイル状残渣550 mgを酢酸第三ブチル60 mLに溶解し、60 %過塩素酸水溶液900μL (13 mmol)を添加した後、室温にて14時間撹拌した。反応終了後、10 % K2CO3 水溶液によりpH 9に調整し、酢酸エチルにて抽出した。得られた有機層を水、飽和食塩水にて洗浄した後、無水硫酸ナトリウムを加えて1時間放置した。硫酸ナトリウムをろ去し、ろ液を減圧乾固することで黄色油状物を得た。得られた黄色油状物を真空乾燥し、1H-NMRスペクトルから目的物であることを確認した。
【0116】
【化23】
【0117】
収量400 mg (収率 42 %); 1H-NMR (600 MHz, CDCl3) . δ 3.31 (d, J = 16.8 Hz, 1H, -CHa2-), 3.23 (d, J = 16.8 Hz, 1H, -CHa2-), 3.14 (t, J = 6.6 Hz, 1H, -CHb-), 2.69 (t, J = 6.6 Hz, 2H, -CHf2-), 1.63 (m, 2H, -CHc2-), 1.51-1.39 (m, 22H, -CHd2-, -CHe2-, -CHg3, -CHh3).
【0118】
リポ酸誘導体3の合成
リポ酸誘導体3の合成は既報の方法(Ha, T. H.; Jung, S. O.; Lee, K. Y.; Lee, Y.; Park, J. S.; Chung, B. H. Anal. Chem. 2007, 79, 546-556.)を改良して行った。DL-a-リポ酸1.0 g (5.0 mmol)、N-ヒドロキシスクシンイミド 0.58 g (5.0 mmol)をTHF 5 mLに溶解し、氷浴上で撹拌しながらジシクロヘキシルカルボジイミド1.1 g (5.1 mmol)をゆっくりと添加した。氷浴上にて10時間撹拌した後、反応温度を室温に変えて24時間撹拌した。析出した固体をろ去し、ろ液を減圧乾固した。得られた残渣を酢酸エチル-ヘキサンから再結晶することで黄色針状結晶を得た。得られた黄色針状結晶を真空乾燥し、1H-NMRスペクトルから目的物であることを確認した。
【0119】
【化24】
【0120】
収量1.1 g (収率 75 %); 1H-NMR (600 MHz, CDCl3) δ 3.59 (m, 1H, -CHc-), 3.19 (m, 1H, -CHa2-), 3.12 (m, 1H, -CHa2-), 2.84 (br, 4H, -CHh2-), 2.63 (t, J = 7.2 Hz, 2H, -CHg2-), 2.47 (m, 1H, -CHb2-), 1.93 (m, 1H, -CHb2-), 1.81-1.56 (m, 6H, -CHd2-, -CHe2-, -CHf2-).
【0121】
リジン誘導体4の合成
リジン誘導体2を200 mg (0.63 mmol)、トリエチルアミン 170μL (1.3 mmol)とともにDMSO 4 mLに溶解した。その溶液にリポ酸誘導体3を 230 mg (0.76 mmol)溶解したDMSO溶液(4 mL)を30分かけて滴下し、室温にて4時間撹拌した。反応終了後、水30 mLを加えて室温にて12時間撹拌した。酢酸エチルにて抽出を行った後、得られた有機層を水、飽和食塩水にて洗浄し、無水硫酸ナトリウムを加えて1時間放置した。硫酸ナトリウムをろ去し、ろ液を減圧乾固することで黄色油状物を得た。得られた黄色油状物を真空乾燥し、1H-NMRスペクトルから目的物であることを確認した。
【0122】
【化25】
【0123】
収量220 mg (収率 69 %); 1H-NMR (600 MHz, CDCl3) δ 5.68 (br, 1H, -NHi-), 3.57 (m, 1H, -CHn-), 3.32 (d, J = 16.8 Hz, 1H, -CHa2-), 3.25 (t, J = 6.6 Hz, 2H, -CHf2-), 3.21 (d, J = 16.8 Hz, 1H, -CHa2-), 3.18 (m, 1H, -CHp2-), 3.12 (t, J = 6.6 Hz, 1H, -CHb-), 3.11 (m, 1H, -CHp2-), 2.46 (m, 1H, -CHo2-), 2.17 (d, J = 7.2 Hz, 1H, -CHj2-), 2.16 (d, J = 7.2 Hz, 1H, -CHj2-), 1.91 (m, 1H, -CHo2-), 1.71-1.41 (m, 30H, -CHc2-, -CHd2-, -CHe2-, -CHk2-, -CHl2-, -CHm2-, -CHg3, -CHh3).
【0124】
アゾベンゼン誘導体5の合成
アゾベンゼン誘導体1を137 mg (0.27 mmol)及びリジン誘導体4を 86 mg (0.18 mmol)溶解させたDMF溶液(4 mL)にDIEA 30μL (0.30 mmol)、NaI 66 mg (0.30 mmol)を添加し、室温にて15時間攪拌した。反応終了後、溶液を減圧乾固した。得られた残渣を水20 mLで溶解し、酢酸エチルにて抽出した。得られた有機層を水、飽和食塩水にて洗浄した後、ろ液を減圧乾固した。得られた残渣を少量の酢酸エチルに溶解してシリカゲルカラムクロマトグラフィー(溶離液; 酢酸エチル:ヘキサン= 1 : 5)を行った。目的物はTLC (Rf= 0.1 (展開溶媒; 酢酸エチル:ヘキサン= 1 : 5))により確認し、得られた分画を減圧乾固した。得られた橙色固体を真空乾燥し、1H-NMRスペクトルから目的物であることを確認した。
【0125】
【化26】
【0126】
収量60 mg (収率35 %); 1H-NMR (600 MHz, CDCl3) δ 7.87 (d, J = 8.4 Hz, 2H, φ-Hs), 7.86 (d, J = 8.4 Hz, 2H, φ-Hs), 7.56 (d, J = 8.4 Hz, 2H, φ-Hu), 7.55 (d, J = 8.4 Hz, 2H, φ-Hu), 5.77 (t, J = 5.4 Hz, 1H, -NHi-), 3.98 (s, 2H, -CHt2-), 3.98 (d, J = 14.4 Hz, 1H, -CHq2-), 3.80 (d, J = 14.4 Hz, 1H, -CHq2-), 3.54 (m, 1H, -CHn-), 3.45 (s, 4H, -CHu2-), 3.44 (d, J = 16.8 Hz, 1H, -CHa2-), 3.33 (d, J = 16.8 Hz, 1H, -CHa2-), 3.25 (m, 1H, -CHf2-), 3.22 (dd, J = 7.2, 9.0 Hz 1H, -CHb-) 3.17-3.12 (m, 2H, -CHf2-, -CHp2-), 3.08 (m, 1H, -CHp2-), 2.42 (m, 1H, -CHo2-), 2.15 (t, J = 7.2 Hz, 2H, -CHj2-), 1.87 (m, 1H, -CHo2-), 1.65 (m, 2H, -CHc2-), 1.51-1.37 (m, 46H, -CHd2-, -CHe2-, -CHk2-, -CHl2-, -CHm2-, -CHg3, -CHh3, -CHv3).
【0127】
trans-H4L1の合成
アゾベンゼン誘導体5を60 mg (0.063 mmol)、TFA 18 mL に溶解し、室温にて13時間撹拌した。反応終了後、溶液を減圧乾固して得られた残渣をジエチルエーテルにて洗浄した。得られた黄色固体を真空乾燥し、MALDI-TOF Massスペクトルから目的物であることを確認した。また、30 % NaOD水溶液を加えてtrans-L1とした場合の1H-NMRスペクトルから目的物(trans-Na4L1)であることを確認した(図1)。
【0128】
【化27】
【0129】
収量42 mg (収率 91 %); 1H-NMR (600 MHz, D2O) δ 7.85 (d, J = 8.4 Hz, 2H, φ-Hp), 7.84 (d, J = 8.4 Hz, 2H, φ-Hp), 7.67 (d, J = 8.4 Hz, 2H, φ-Ho), 7.59 (d, J = 8.4 Hz, 2H, φ-Ho), 3.93 (d, J = 13.2 Hz, 1H, -CHn2-), 3.84 (s, 2H, -CHq2-), 3.72 (d, J = 13.2 Hz, 1H, -CHn2-), 3.47 (m, 1H, -CHk-), 3.33 (d, J = 16.8 Hz, 1H, -CHa2-), 3.18 (s, 4H, -CHr2-), 3.16-3.09 (m, 8H, -CHa2-, -CHb-, -CHf2-, -CHm2-), 2.31 (m, 1H, -CHl2-), 2.10 (t, J = 7.2 Hz, 2H, -CHg2-), 1.81 (m, 1H, -CHl2-), 1.59-1.22 (m, 12H, -CHc2-, -CHd2-, -CHe2-, -CHh2-, -CHi2-, -CHj2-); MS (MALDI-TOF) m/z: 732.33 [H4L1 + H+]+, 754.14 [H4L1 + Na+]+.
【0130】
<参考例1:アゾ化合物の合成>
以下のようにして、下記の化学式で表されるアゾ化合物(trans-体)を合成した。なお、以下、下記式で表されるアゾ化合物をH4L2と呼ぶ。(H4L2のH4は分子中に存在するCOOH基のHが4つあることを表している。従って、当該基がCOONaとなっている場合、Na4L2と表記される。)
【0131】
【化28】
【0132】
trans-H4L2の合成
4,4’-ビスクロロメチルアゾベンゼン 0.187 g (0.67 mmol)、イミノ二酢酸ジ第三ブチルエステル 0.82 g (3.35 mmol)のDMF溶液(28 ml)にDIEA 224μL (2.2 mmol)、NaI 0.40 g (2.68 mmol)を添加し、室温にて一晩撹拌した。反応終了後、溶液を減圧乾固した。得られた残渣をクロロホルムに溶解し、1 N HCl水溶液にて洗浄した。有機層を5 % NaHCO3水溶液、飽和食塩水で洗浄した後、無水硫酸ナトリウムを加えて静置した。硫酸ナトリウムをろ去し、ろ液を減圧乾固した。得られた残渣をTFA 30 mLに溶解し、室温にて一晩撹拌した後、溶液を減圧乾固した。得られた黄色固体をジエチルエーテルにて洗浄し、真空乾燥した。30 % NaOD水溶液を加えて1H-NMRスペクトルから目的物(Na4L2)であることを確認した。なお、当該アゾ化合物は、既に上記非特許文献1において報告されている。
【0133】
【化29】
【0134】
収量 285 mg (収率 90 %); 1H-NMR (600 MHz, D2O) δ 7.85 (d, J = 8.4 Hz, 4H, φ-Ha), 7.61 (d, J = 8.4 Hz, 4H, φ-Hb), 3.88 (s, 4H, -CHc2-), 3.22 (s, 8H, -CHd2-).
【0135】
実施例1:L1の光異性化挙動
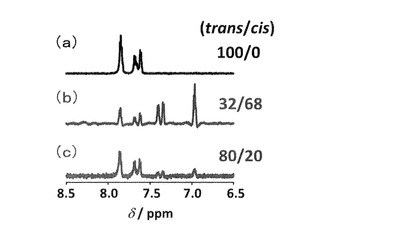
合成したtrans-H4L1は水に不溶であるため、4当量のNaOHの添加により1 mM trans-L1水溶液を調製した。trans-L1水溶液のUV-visスペクトルは、229 (1.1 × 104), 334 nm (1.9 × 104 M-1 cm-1)にtrans-型アゾベンゼンに特徴的なπ-π*遷移を、420 nm付近(ca. 1.3 × 103M-1cm-1)にn-π*遷移を示した(図2(a))。この溶液の1H-NMRスペクトルを測定したところ、芳香族領域にはtrans-型アゾベンゼンに由来する4種類のダブレットのみが観測された(図3(a))。これらの結果から、合成終了段階では熱力学的に安定なtrans-体のみが存在すると考えられた(trans/cis = 100/0)。
【0136】
次に、L1を用いた光異性化挙動について検討した。キセノンランプを光源とし、カットフィルターを用いて紫外光(320-400 nm)、可視光(420-1800 nm)をそれぞれ調整した。1 mM trans-L1水溶液に紫外光を照射すると、そのUV-visスペクトル(紫外可視吸収スペクトル)は徐々に変化し、80 分間照射すると光定常状態(photostationary state; PSS)となった(図2(a))。光定常状態(紫外光)のUV-visスペクトルは、trans-L1における334 nmのπ-π*遷移の吸光度が大きく減少して324 nmに短波長シフトし、420 nmのn-π*遷移の吸光度がわずかに増大していた。これはcis-型アゾベンゼンに特徴的なスペクトルであるため、光異性化が進行しているものと考えられた。
【0137】
上述の非特許文献1に開示されるように、一連のアゾベンゼン誘導体の光定常状態における異性体比をtrans-型アゾベンゼンのπ-π*遷移(334 nm)の吸光度を用いて下記式(I)から算出することができる。
【0138】
【数1】
【0139】
ここで、APSSは光定常状態におけるπ-π*遷移の吸光度、Atransはtrans-型アゾベンゼンにおけるπ-π*遷移の吸光度である。式(1)により算出した紫外光照射時の光定常状態におけるL1の異性体比はtrans/cis = 38/62であった。
【0140】
実施例2:金属イオン添加時の光異性化挙動および錯形成挙動
上述の非特許文献1には、cis-L2に1当量の金属イオン(ZnII, CuII)を添加した場合、cis-L2と金属イオンとの錯形成により熱力学的な安定性が増大し、cis-体からtrans-体への熱異性化速度が激減することが記載されている。そこで、L1, L2に1当量の金属イオンを添加した場合の光異性化挙動およびその錯形成挙動についての検討を行った。ここで、金属錯体[MII(trans-L1)]はtrans-L1水溶液に1当量のMII(ClO4)2(MII = ZnII, CuII)を添加することで調製した。
【0141】
1 mM [ZnII(trans-L1)]水溶液のUV-visスペクトルは、trans-L1における334 nmのπ-π*遷移が332 nmに短波長シフトし、420 nmのn-π*遷移の吸収がわずかに減少していた(図4a)。この結果は、添加したZnIIイオンがtrans-L1に配位したことを示している。[ZnII(trans-L1)]に紫外光を照射すると、UV-visスペクトルは徐々に変化し、90分間照射することで光定常状態に達した(図4a)。光定常状態(紫外光)のUV-visスペクトルは、332 nm付近のπ-π*遷移が減少するとともに、420 nm付近のn-π*遷移の吸収がわずかに増大した。これはtrans-体([ZnII(trans-L1)])からcis-体([ZnII(cis-L1)])への異性化を示しており、吸光度から算出した異性体比はtrans/cis = 47/53であった。さらにこの溶液に可視光を照射した場合、UV-visスペクトルが変化し、10分間照射することで光定常状態へと達した(図4a)。光定常状態(可視光)のUV-visスペクトルは、[ZnII(trans-L1)]のスペクトルに非常に類似したスペクトルとなり、吸光度から算出した異性体比はtrans/cis = 87/13であった。これらの結果から、1当量のZnIIイオンを添加した場合においても光による異性化が可能であることを確認した。
【0142】
同様の光異性化実験を1 mM [CuII(trans-L1)], [ZnII(trans-L2)], [CuII(trans-L2)]水溶液に対して行い、それらのUV-visスペクトル変化および吸光度から得られた異性体比を図4、図5、表1にまとめた。いずれの場合も金属イオンの添加によってπ-π*遷移の低波長シフトが観測され、金属イオンの結合が示唆された。また、光異性化挙動に関しても上述の[ZnII(trans-L1)]の場合と同様であったため、金属イオン添加時にも光による異性化が可能であることを確認した。
【0143】
異性体比に着目すると、紫外光照射時(trans-体からcis-体への異性化)では金属添加時の方が残存するtrans-体の割合が大きかった。一方、可視光照射時(cis-体からtrans-体への異性化)では逆に金属添加時の方が残存するcis-体の割合が少なかった。したがって、金属イオンの添加はtrans-体からcis-体への異性化には不利に働くが、cis-体からtrans-体への異性化に対しては有利に働くことがわかった。
【0144】
【表1】
【0145】
次に、[CuII(trans-L1)], [CuII(cis-L1)]の電気化学的な性質を調べるためにCV測定を行った(図6)。ここで、[CuII(cis-L1)]は前述のようにtrans-L1への紫外光照射後にCuIIイオンを添加することで調製し、参照のためにtrans-L1, cis-L1, CuII(ClO4)2についても測定を行った。trans-L1およびcis-L1はいずれも-800 mV付近に還元波を示し、対応する酸化波は観測されなかった。この不可逆な還元波は、アゾ基からヒドロアゾ基への還元反応(2e-, 2H+)に由来するものと考えられ、両異性体間での違いはほとんどなかった。このような還元波は[CuII(trans-L1)], [CuII(cis-L1)]についても同様に観測された。また、CuII(ClO4)2は-60 mV付近にCuI/IIの還元に由来すると考えられる応答を示したが、この還元波は[CuII(trans-L1)], [CuII(cis-L1)]の場合においては全く観測されなかった。したがって、いずれも遊離しているCuIIイオンは存在せず、全てのCuIIイオンは配位子(−COO−)と結合していると考えられた。
【0146】
これらの結果から、L1に1当量のCuIIイオンを添加した場合、全てのCuIIイオンは配位子と結合しており、少なくとも溶液中では遊離しているCuIIイオンは存在しないことが示された。また、溶液中では、L1はcis体でもtrans体でも金属イオンを結合する強さにほとんどかわりがないと考えられた。
【0147】
実施例3:修飾電極と銅イオンとの結合強度の検討
<L1修飾電極の作製>
以下のようにして金薄膜がコーティングされたマイカ基板を作製し、これにL1分子を修飾してアゾ化合物結合固相基板(修飾電極として使用)を作製した。
【0148】
蒸着金の作製および評価
蒸着金の作製はシンク社製高真空抵抗加熱装置JIS-300Ak型を使用した。金原料には田中貴金属製のφ1.0 mm金線(99.999 %)を用いた。ニラコ社製の天然マイカ上を14 × 14 mm四方の大きさにカットし、装置のチャンバー内部に固定した。また蒸着用バスケットに清浄な金線を適当量静置した。ロータリーポンプ、ターボ分子ポンプを順に起動し、チャンバー内圧を10-5 Pa以下となるまで減圧した。ランプヒーターによりマイカを300℃で3時間加熱した後、金線を一定温度で加熱することで金蒸着を行った。蒸着速度は1.0Å s-1に制御し、厚さ1000 Åまで蒸着することで目的の蒸着金を得た。
【0149】
蒸着金の表面積は、水素炎でアニール処理後の蒸着金を用いた0.1 M H2SO4水溶液中でのCV測定を行い、酸化被膜の還元反応時の電気量(理論値: 444μC cm-2)から算出した(A = 1.1 cm2, roughness factor: 2.1)。
【0150】
修飾電極の作製
金表面への分子修飾は上述のように作製した蒸着金を水素炎でアニール処理した後、各溶液に浸漬することで作製した。各修飾電極及びその作製過程の模式図を図7に示す。
【0151】
修飾電極H4L1-Au(H4L1結合固相基板(金蒸着あり))の作製(図7)
(trans-H4L1)-Au は、アニール処理した蒸着金を1 mM trans-H4L1溶液(DMF)に15時間浸漬することで作製した。(cis-H4L1)-Au は、1 mM trans-H4L溶液(DMF)に紫外光を80分間照射した光定常状態の溶液に蒸着金を浸漬し、紫外光を照射しながら15時間浸漬することで作製した。いずれもDMF、水の順にリンスした後に電気化学測定に使用した。なお、(cis-H4L1)-Auでは全てのH4L1がcis体となるわけではなく、一部trans体も含まれるが、(cis-H4L1)-Auと表記する(図7)。以下も同様とする。
【0152】
修飾電極H4L1/C6-Au(ヘキサンチオールでブロッキングされたH4L1結合固相基板(金蒸着あり))の作製(図7)
(trans-H4L1)/C6-Au, (cis-H4L1)/C6-Au は、上述のようにして作製した(trans-H4L1)-Au, (cis-H4L1)-Auを0.1 mM ヘキサンチオール(C6)溶液(MeOH)に5分間浸漬することでそれぞれ作製した。いずれもMeOH、水の順にてリンスした後に電気化学測定に用いた。
【0153】
なお、ヘキサンチオールは、修飾電極として用いた際バックグラウンド電流を小さくするため、及びCuIIイオンのAu-S結合への影響を抑えるためのブロッキングのために用いた。
【0154】
修飾電極L1/C6-Au(ヘキサンチオールでブロッキングされたL1結合固相基板(金蒸着あり))の作製(図7)
(trans-L1)/C6-Au, (cis-L1)/C6-Auは、上述のようにして作製した(trans-H4L1)/C6-Au, (cis-H4L1)/C6-Auを0.5 M NaOH水溶液に90分間浸漬することでそれぞれ作製した。いずれも水でリンスした後に電気化学測定に用いた。
【0155】
修飾電極[CuII(L1)]/C6-Au(ヘキサンチオールでブロッキングされたL1結合固相基板であって、L1が銅イオンを結合している基板(金蒸着あり))の作製(図7)
[CuII(trans-L1)]/C6-Au, [CuII(cis-L1)]/C6-Auは、上述のようにして作製した(trans-L1)/C6-Au, (cis-L1)/C6-Auを1 mM CuII(ClO4)2水溶液に30分間浸漬することでそれぞれ作製した。いずれも水でリンスした後に電気化学測定に用いた。
【0156】
実施例4 trans-L1修飾電極と銅イオンとの結合の検討
上述のようにして作製したアゾ化合物結合固相基板((trans-H4L)/C6-Au)を用い、固体表面上に配列された配位子L1へのCuIIイオンの結合を電気化学的に評価した。作製した(trans-H4L1)/C6-Auの0.1 M NaClO4水溶液中でのCV測定を行ったところ、測定電位範囲(-100 mVから+400 mV)において酸化還元応答は観測されなかった(図8)。また、(trans-H4L1)/C6-Auを0.5 M NaOH水溶液に浸漬することでH4L1部位を脱プロトン化した(trans-L1)/C6-Auもまた測定電位範囲では酸化還元応答を示さず、プロトン化体((trans-H4L1)/C6-Au)に比べてバックグラウンド電流がわずかに増加していた(図8)。水溶液中においてL1は-800 mV付近に還元波を示すが、(trans-H4L1)-Auは-900 mV付近に脱離波を示すためこれ以上負側への掃引は行わなかった。
【0157】
また、上述のようにして作製した[CuII(trans-L1)]/C6-Auを用いた0.1 M NaClO4水溶液中(CuIIイオン非存在下)でのボルタモグラムは、+308, +136 mVに一対の酸化還元応答を示した(E1/2 = +222 mV,図8)。(trans-H4L1)/C6-Au, (trans-L1)/C6-Auではこの測定電位範囲において酸化還元応答を示さなかったことから、この酸化還元波は表面上に固定化されたL1のカルボン酸部位と結合したCuI/IIの1電子酸化還元反応に由来するものと考えられる。還元波のピーク電気量(Qpc)は18μC cm-2であった。
【0158】
この酸化還元応答は掃引を繰り返すことでピークが減少していき、60サイクル目には酸化還元応答がほぼ消失した(図9)。これはCuイオンの固体表面上からの脱離を意味しており、酸化還元反応を繰り返すことでCuイオンが遊離していくことがわかった。また、このことから、(trans-L1)/C6-AuとCuイオンとの結合は比較的弱いことがわかった。なお、この場合、表面上のtrans-L1はCuイオン結合型からNa+イオン結合型(CuIIイオンを浸漬する前の状態)へと変換され、遊離したCuイオンは溶液中で過塩素酸銅へと変換されたと考えられた。
【0159】
以上の結果より、表面上のtrans-L1とCuイオンとの結合は比較的弱く、酸化還元反応を繰り返すことでCuイオンを溶液中に放出できることがわかった。またこのことから、trans-L1部位の金属イオン結合能の再活性化とともに、カウンターアニオンの選択により望みの金属塩へと変換(リサイクル)できることがわかった。
【0160】
実施例5 cis-L1修飾電極と銅イオンとの結合の検討
上述のようにして作製した (cis-L1)/C6-Au)を作用電極としたボルタモグラムは、trans-体の場合と同様に測定電位範囲において酸化還元応答を示さなかった(図10)。
【0161】
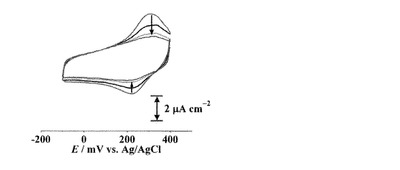
(cis-L1)/C6-AuをCuII(ClO4)2水溶液中に浸漬することで作製した[CuII(cis-L1)]/C6-Auのボルタモグラムは、+313, +247 mVに酸化還元応答を示した(E1/2 = +280 mV)。これはL1のカルボン酸部位に結合したCuI/IIの酸化還元に由来するものと考えられる(図10)。還元波のピーク電気量(Qpc)は12μC cm-2であった。
【0162】
[CuII(cis-L1)]/C6-Auの酸化還元応答もまた掃引を繰り返すことでピークが減少していくが、trans-体の場合とは異なり15サイクル目付近からピーク強度がほぼ一定になった(図11)。そこで、[CuII(trans-L1)]/C6-Au, [CuII(cis-L1)]/C6-Auの還元波のピーク電気量を1サイクル目の電気量を基準として規格化し、それぞれサイクル数に対してプロットした(図12)。trans-体の場合ではサイクル数の増加に伴いピーク電気量は減少していき、最終的にはほぼピークが消失した。一方、cis-体の場合では10サイクル目付近まではtrans-体と同様にピーク電気量が減少したが、それ以降ではピーク電気量がほぼ一定になった(約6.6μC cm-2, 図12)。[CuII(cis-L1)]/C6-Auはcis-, trans-体の両異性体が修飾されており、前述のようにtrans-体は酸化還元を繰り返すことでCuイオンが脱離する。したがって、10サイクル目あたりまでのピーク電気量の減少はtrans-L1部位からのCuイオンの脱離と考えられ、それ以降の一定のピーク電気量はcis-L1部位からCuイオンが脱離しないことを示している(図13)。すなわち、溶液中とは異なり、固相基板に結合したcis-L1及びtrans-L1のCuIIイオンとの結合能力には明確な差が存在し、cis-体の場合は掃引によってほとんど脱離しないことがわかった。
【0163】
次に、固相基板表面上でのcis-体からtrans-体への光異性化を試みた。[CuII(cis-L1)]/C6-Auを75サイクル掃引した後、すぐに溶液を交換することでcis-L1のみにCuIIイオンが結合した電極を作製した。この電極を0.1 M NaClO4水溶液に浸し、金表面に可視光を90分間照射した。照射後すぐに溶液を新しい溶液に交換してCV測定を行ったところ、可視光照射前のものに比べてCuI/II由来の酸化還元応答が減少していた(図14)。同様に可視光180分照射後、270分照射後のボルタモグラムにおいてもピークの減少が観測され、270分照射後にはCuI/II由来の酸化還元応答はほぼ消失した (図14)。ここで、全てのボルタモグラムについて掃引に伴うピークの減少は観測されなかったことから、得られたピークはCuII(cis-L1)に由来するものと考えられる。したがって、比較的結合が強いCuII(cis-L1)部位は、可視光照射によってCuIIイオンとの結合が弱いCuII(trans-L1)へと異性化し、酸化還元反応を繰り返したことによりCuIIイオンは溶液中へと遊離し、溶液交換により完全に系外に除去されたと考えられる(図15)。この結果から、固相基板に結合した状態においてもアゾ化合物がcis-体からtrans-体へと光異性化できることがわかった。またさらに、当該光異性化により、金属イオンとの結合強度を制御できることもわかった。
【技術分野】
【0001】
本発明は、アゾ化合物に関し、より詳細にはリンカーを有するアゾ化合物に関する。本発明はまた、当該アゾ化合物を用いた金属イオン回収方法に関する。
【背景技術】
【0002】
金属元素は現在の化学産業を支える必要不可欠な元素であり、我々生物が生きていくためにも重要な役割を果たしている。しかし、重金属資源は有限であり、生物にとっても重金属イオンの過剰摂取は毒となりうる。そのため、重金属イオンの検出、捕捉に関する研究が盛んに行われてきている。現行の重金属イオンの処理は、例えば、カルボン酸のような配位性官能基を有する高分子材料への重金属イオンの吸着により固体表面へ固定化(捕捉)し、廃棄することにより実施されている。しかし、重金属資源の枯渇が懸念される現状から、捕捉した金属の再利用が望まれており、金属の捕捉・再利用のために、溶液などから金属イオンを取り出す時には強く結合し回収する時には容易に金属イオンを脱離できるような材料の開発が切望されている。
【0003】
そこで、金属イオンの結合を制御する方法に利用できる可能性があるとして、例えばアゾベンゼン(及びその誘導体)のような光応答性分子に注目が集まっている。アゾベンゼンは熱力学的に安定なtrans-体に紫外光を照射することでcis-体へと異性化し、cis-体への可視光照射もしくは加熱によりtrans-体を再度生成する。
【0004】
【化1】
【0005】
これまでに、アゾベンゼンにキレート環を含む複数の配位性官能基を導入したアゾベンゼン誘導体を用い、上述の光応答性を利用することで、金属イオンの捕捉・脱離を制御する試みがなされている(例えば非特許文献1)。すなわち、当該アゾベンゼン誘導体がtrans-体のときには配位性官能基が立体的に離れた位置に存在するために金属イオンを捕捉できないが、cis-体のときには配位性官能基が立体的に近い位置に存在し、当該配位性官能基が配位子として金属イオンと結合して、金属イオンを捕捉できると予想されたため、当該予想を裏付けるための検討がなされてきたのである。
【0006】
しかしながら、例えば非特許文献1に示されるように、溶液中のアゾベンゼン誘導体はtrans-体・cis-体のいずれであっても金属イオンと強く結合してしまい、期待された金属イオンの捕捉・脱離を制御する効果は得られていない。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】Shinkai, S.; Nakamura, S.; Nakashima, M.; Manabe, O.; Iwamoto, M. Bull. Chem. Soc. Jpn. 1985, 58, 2340-2347.
【発明の概要】
【発明が解決しようとする課題】
【0008】
本発明は、金属イオンの捕捉・脱離を制御し得るアゾ化合物を提供するとともに、当該アゾ化合物を用いて金属イオンの捕捉・脱離を制御する方法を提供することを課題とする。
【課題を解決するための手段】
【0009】
本発明者らは、驚くべき事に、下記一般式(1)で表されるアゾ化合物を固相基板に結合させ、当該アゾ化合物結合基板を用いて金属イオンの捕捉・脱離を制御できることを見出し、さらに改良を重ねて本発明を完成させるに至った。
【0010】
すなわち、本発明は例えば以下の項1〜7のアゾ化合物、アゾ化合物結合固相基板及び金属イオン回収方法に係るものである。
項1.
下記一般式(1)で表されるアゾ化合物。
【0011】
【化2】
【0012】
(式中、
R1aは、直鎖状のC1〜C10アルキレン基を示し、
R1b及びR1cは、同一又は異なって、直鎖状のC1〜C6アルキレン基を示す。
R2aは、直鎖状のC1〜C10アルキレン基を示し、
R2bは、直鎖状のC1〜C6アルキレン基を示す。
Xはリンカー部位を示し、Yは固相基板に対して結合性を有する固相基板結合部位を示す。)
項2.
前記リンカー部位がアルキレン鎖中に−NHCO−を有するアルキレン基であり、
前記構造基板結合部位がチオール基、ジスルフィド基、スルフィド基、ジチオラン基又はシロキシ基である、項1に記載のアゾ化合物。
項3.
項1又は2に記載のアゾ化合物を前記固相基板結合部位により固相基板に結合させたアゾ化合物結合固相基板。
項4.
前記固相基板が、表面を金でコーティングされた固相基板である、項3に記載のアゾ化合物結合固相基板。
項5.
(i)金属イオン含有溶液に、項3又は4に記載のアゾ化合物結合固相基板を浸積してアゾ化合物に金属イオンを結合させる工程、
(ii)当該アゾ化合物結合固相基板のアゾ化合物に可視光を照射する工程、
(iii)当該アゾ化合物結合固相基板を作用電極として金属イオンの酸化還元反応を繰り返して、アゾ化合物に結合した金属イオンを脱離させる工程、
を含む金属イオン回収方法。
項6.
前記酸化還元反応が、前記金属イオンの酸化還元電位より300mV低い電位から、当該酸化還元電位より300mV高い電位の幅で30回以上繰り返される、
項5に記載の金属イオン回収方法。
項7.
項5又は6に記載の工程(i)〜(iii)を含む工程を繰り返し行うことにより、金属イオンを回収する方法。
【発明の効果】
【0013】
本発明によって、金属イオンの捕捉・脱離に利用できる新規なアゾ化合物が提供される。また、当該アゾ化合物が結合した固相基板が提供される。さらに、当該アゾ化合物結合固相基板を用いた金属イオン回収方法が提供される。
【0014】
当該アゾ化合物結合固相基板のアゾ化合物は、非常に強い金属イオン結合能を有するため、これまで金属イオン回収が不可能であった金属イオン含有溶液(例えば、金属イオン濃度が薄い溶液、金属イオン以外の夾雑物が多量に溶解した溶液(廃液)等)からも金属イオン回収が可能である。
【0015】
さらに、当該アゾ化合物は、cis体・trans体のいずれであっても溶液中で金属イオンとほぼ同様の強さで結合するが、これを固相基板に結合させた当該アゾ化合物結合固相基板を用いると、trans体に比べcis体の方が金属イオンとより強く結合する。従って、当該アゾ化合物結合固相基板においては、cis体に結合した金属イオンよりtrans体に結合した金属イオンの方が脱離しやすく、当該アゾ化合物のcis体・trans体を紫外光及び可視光照射等によって制御することにより、当該アゾ化合物に結合した金属イオンの脱離を制御することができる。
【図面の簡単な説明】
【0016】
【図1】trans-L1の1H-NMRスペクトル(in D2O)を示す。
【図2】キセノンランプ照射に伴うtrans-L1 (a)およびtrans-L2 (b)の吸収スペクトル変化を示す。(1 mM in H2O; 黒線: trans-L, 青線: 紫外光照射時の光定常状態, 赤線: 可視光照射時の光定常状態)
【図3】キセノンランプ照射に伴うtrans-L1の1H-NMRスペクトル変化を示す。(1 mM in D2O; a: trans-L1, b: 紫外光照射時の光定常状態, c: 可視光照射時の光定常状態)
【図4】キセノンランプ照射に伴う[ZnII(trans-L1)] (a)および[CuII(trans-L1)] (b)の吸収スペクトル変化を示す。(1 mM in H2O; 灰線: trans-L1, 黒線: [MII(trans-L1)], 青線: 紫外光照射時の光定常状態, 赤線: 可視光照射時の光定常状態)
【図5】キセノンランプ照射に伴う[ZnII(trans-L2)] (a)および[CuII(trans-L2)] (b)の吸収スペクトル変化を示す。(1 mM in H2O; 灰線: trans-L1, 黒線: [MII(trans-L2)], 青線: 紫外光照射時の光定常状態, 赤線: 可視光照射時の光定常状態)
【図6】CuII(ClO4)2, L1, [CuII(L1)]のサイクリックボルタモグラムを示す。(1 mM in 0.1 M NaClO4水溶液, 掃引速度: 50 mV s-1; 黒線: CuII(ClO4)2, 赤点線: trans-L1, 赤実線: [CuII(trans-L1)], 青点線: cis-L1, 青実線: [CuII(cis-L1)])
【図7】各修飾電極及びその作製過程の模式図を示す。
【図8】(trans-H4L1)/C6-Au, (trans-L1)/C6-Au, [CuII(trans-L1)]/C6-Auのサイクリックボルタモグラムを示す。(電解質溶液: 0.1 M NaClO4水溶液, 掃引速度: 50 mV s-1: 黒点線: (trans-H4L1)/C6-Au, 黒実線: (trans-L1)/C6-Au, 赤線: [CuII(trans-L1)]/C6-Au)
【図9】[CuII(trans-L1)]/C6-Auのサイクリックボルタモグラムを示す。(電解質溶液: 0.1 M NaClO4水溶液, 掃引速度: 50 mV s-1; 赤線: 1サイクル目, 灰線: 5, 15, 40サイクル目, 緑線: 60サイクル目)
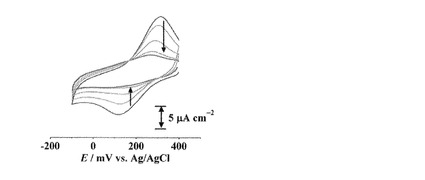
【図10】(cis-H4L1)/C6-Au, (cis-L1)/C6-Au, [CuII(cis-L1)]/C6-Auのサイクリックボルタモグラムを示す。(電解質溶液: 0.1 M NaClO4水溶液, 掃引速度: 50 mV s-1 : 黒点線: (cis-H4L1)/C6-Au, 黒実線: (cis-L1)/C6-Au, 青線: [CuII(cis-L1)]/C6-Au)
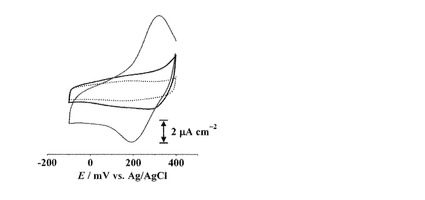
【図11】[CuII(cis-L1)]/C6-Auのサイクリックボルタモグラム(電解質溶液: 0.1 M NaClO4水溶液, 掃引速度: 50 mV s-1; 青線: 1サイクル目, 灰線: 5, 15, 40サイクル目, 緑線: 60サイクル目)
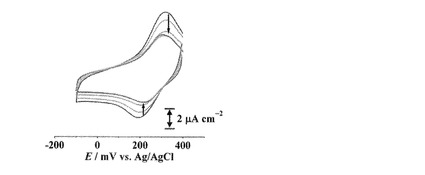
【図12】[CuII(trans-L1)]/C6-Au (赤)および[CuII(cis-L1)]/C6-Au (青)の還元ピーク電気量(1回目の電気量で規格化)と掃引回数との関係を表したグラフを示す。
【図13】[CuII(L1)]/C6-Auの1サイクル目と60サイクル目におけるCuイオン結合状態の模式図を示す。
【図14】[CuII(cis-L1)]/C6-Auへの可視光照射前後のサイクリックボルタモグラムを示す。(電解質溶液: 0.1 M NaClO4水溶液, 掃引速度: 50 mV s-1; 緑線: 照射前, 黒線: 90分照射後, 灰線: 180分照射後, 赤線: 270分照射後)
【図15】[CuII(cis-L1)]/C6-Au への可視光照射に伴うCuIIイオン脱離過程の模式図を示す。
【発明を実施するための形態】
【0017】
以下、本発明について、さらに詳細に説明する。
アゾ化合物
【0018】
【化3】
【0019】
本発明は、上記一般式(1)で表されるアゾ化合物に係る。
【0020】
一般式(1)中、R1aは、直鎖又は分岐鎖状のC1〜C10アルキレン基、好ましくはC1〜C6アルキレン基、より好ましくはC1〜C3アルキレン基を示す。また、直鎖状であることが好ましい。具体的には、メチレン基、エチレン基、トリメチレン基、プロピレン基等が例示でき、特にメチレン基が好ましい。
【0021】
また、R1b及びR1cは、同一又は異なって、直鎖又は分岐鎖状のC1〜C6アルキレン基、好ましくはC1〜C4アルキレン基、より好ましくはC1〜C3アルキレン基を示す。また、直鎖状であることが好ましい。具体的には、メチレン基、エチレン基、トリメチレン基、プロピレン基等が例示でき、特にメチレン基が好ましい。なお、R1b及びR1cのアルキレン基は、同一であるものがより好ましい。
【0022】
また、一般式(1)中、R2aは、直鎖又は分岐鎖状のC1〜C10アルキレン基、好ましくはC1〜C6アルキレン基、より好ましくはC1〜C3アルキレン基を示す。また、直鎖状であることが好ましい。具体的には、メチレン基、エチレン基、トリメチレン基、プロピレン基等が例示でき、特にメチレン基が好ましい。
【0023】
また、R2bは、直鎖又は分岐鎖状のC1〜C6アルキレン基、好ましくはC1〜C4アルキレン基、より好ましくはC1〜C3アルキレン基を示す。また、直鎖状であることが好ましい。具体的には、メチレン基、エチレン基、トリメチレン基、プロピレン基等が例示でき、特にメチレン基が好ましい。
【0024】
なお、R1a及びR2aのアルキレン基は同一であるものが好ましい。
【0025】
また、Xはリンカー部位を示し、Yは固相基板に対して結合性を有する固相基板結合部位を示す。なお、以下、一般式(1)で表されるアゾ化合物において、−X−Y部位を除いた部分を「金属イオン捕捉部位」と表記することがある。
【0026】
前記固相基板結合部位は、固相基板に対して結合性を有し、好ましくは固相基板表面上に高密度かつ高配向性の自己組織化単分子膜(Self-Assembled Monolayer, SAM)を構築することが可能な官能基から形成される。該官能基は固相基板の種類によって選択され、例えば、固相基板表面が金の場合は、チオール基、ジスルフィド基、スルフィド基、ジチオラン基を、ガラスの場合はシロキシ基を用いると、SAM膜が好適に形成されることが知られているが、これらに限定されるものではない。
【0027】
前記リンカー部位は、一般式(1)で表されるアゾ化合物を固相基板に結合させた際、金属イオン捕捉部位を当該固相基板から一定間隔離して存在せしめるための部位である。従って、当該機能を有すればリンカー部位の構造は特に制限されないが、例えば、直鎖もしくは無置換のC1〜C20アルキレン基をリンカー部位として用いることができる。当該アルキレン基は直鎖又は分岐鎖状であってよいが、直鎖であることが好ましい。また、当該アルキレン鎖の炭素数は1〜20が好ましく、3〜14がより好ましく、3〜8がさらに好ましい。中でも、アルキレン鎖中に−NHCO−を有していてもよいアルキレン基が好ましく、−NHCO−を有するアルキレン基がより好ましい。
【0028】
なお、リンカー部位がアルキレン鎖中に−NHCO−を有する直鎖状アルキレン基である場合は、−NHCO−と−Yとの間に存在する炭素数が1〜10、好ましくは1〜6、より好ましくは1〜4であり、−NHCO−と金属イオン捕捉部位との間に存在する炭素数が1〜4、好ましくは2〜4である。また、当該−NHCO−のC末端側が固相基板結合部位側であることが好ましい。
【0029】
また、上述のアルキレン鎖中に−NHCO−を有するアルキレン基において、当該−NHCO−に、荷電を有さないアミノ酸が1以上ペプチド結合により連続して結合したペプチドが連結され、当該アルキレン鎖中に含まれるものも、本発明のアゾ化合物のリンカー部位として好ましく用いることができる。
【0030】
すなわち、アルキレン鎖中に−NHCO−を有し、当該−NHCO−のC末端側が固相基板結合部位側であるアルキレン基において、金属イオン捕捉部位から−NHCO−までをアルキレン基(α)、−NHCO−から固相基板結合部位までをアルキレン基(β)と表記した場合、−X−Yは、
−アルキレン基(α)−NHCO−アルキレン基(β)−Y
であってよいし、また、
−アルキレン基(α)−NHCO−(電荷を有さないアミノ酸が1以上ペプチド結合により連続して結合したペプチド)−NHCO−アルキレン基(β)−Y
であってもよい。(なお下線を引いたCO及びNHは当該ペプチドに由来する。)
ここで、アルキレン基(α)は、炭素数2〜4の直鎖状アルキレン基であることが好ましく、アルキレン基(β)は、炭素数1〜10(好ましくは1〜6、より好ましくは1〜4)の直鎖又は分岐鎖上のアルキレン基であることが好ましい。
【0031】
また、(荷電を有さないアミノ酸が1以上ペプチド結合により連続して結合したペプチド)における「荷電を有さないアミノ酸」としては、グリシン、アラニン、バリン、ロイシン、イソロイシン、フェニルアラニン、トリプトファン、メチオニン、プロリン、セリン、トレオニン、チロシン、システイン等が挙げられ、特にグリシン、アラニン、バリン、ロイシン、イソロイシンが好ましい。
【0032】
ここでの「1以上」は好ましくは1〜20、より好ましくは1〜15、さらに好ましくは1〜10、さらにより好ましくは1〜5、特に好ましくは1〜3である。
【0033】
なお、当該ペプチドの固相基板結合部位側側末端がシステインである場合は、当該システインの−SH基が固相基板結合部位として働き得るため、−X−Yは、
−アルキレン基(α)−NHCO−(荷電を有さないアミノ酸が1以上ペプチド結合により連続して結合したペプチド)
であり得る。
【0034】
上記一般式(1)で表されるアゾ化合物は、種々の方法により製造され得る。その一例として、下記式で表されるアゾ化合物(以下「化合物(A)」と表記することがある)の合成方法を以下に示す。当該化合物(A)の製造において、用いる出発物質を変えれば、上記一般式(1)で表される種々のアゾ化合物を合成できることが、当業者は容易に理解できる。
【0035】
【化4】
【0036】
〔式中、lは1〜10の整数を示し、m及びnは同一又は異なって1〜6の整数を示し、kは1〜6の整数を示し、jは2〜4の整数を示し、pは1〜10の整数を示す。〕
なお、一般式(1)における−X−は、化合物(A)においては、
【0037】
【化5】
【0038】
〔j及びpは前記に同じ〕
に該当し、一般式(1)における−Yは、化合物(A)においては
【0039】
【化6】
【0040】
(すなわちジチオラン基)に該当する。
【0041】
化合物(A)は、下記化合物(1A)をTFAに溶解させ、室温にて1〜24時間程度撹拌して反応させることにより、得ることができる。当該反応終了後、溶液を減圧乾固し、得られた残渣を例えばジエチルエーテルで洗浄してもよい。なお、これ以降の化合物の構造式においてButは第三ブチル基を示す。
【0042】
【化7】
【0043】
〔式中、l、m、n、k、j及びpは前記に同じ〕
化合物(A1)は、例えば、下記反応式−1Aに示される方法により製造される。なお、これ以降の反応式において、Butは第三ブチル基を示し、DIEAはジイソプロピルエチルアミンを示す。
[反応式−1A]
【0044】
【化8】
【0045】
〔式中、l、m、n、k、j及びpは前記に同じ〕
化合物(A1)は、例えば、化合物(B1)及び化合物(C1)を溶解させたDMF溶液に、DIEA、NaIを添加し、室温にて1〜24時間撹拌して反応させることにより得ることができる。また、反応後、酢酸エチル等で抽出し、シリカゲルカラムクロマトグラフィー等の常法により精製することができる。
【0046】
上記反応式−1Aにおいて出発原料として用いられている化合物(B1)は、公知の化合物であるか、又は公知の方法により容易に製造される。例えば、下記反応式−1Bに示す方法により製造できる。
[反応式−1B]
【0047】
【化9】
【0048】
〔式中、l、m及びnは前記に同じ〕
化合物(B1)は、例えば、化合物(B2)及び化合物(B3)を溶解したDMF溶液に、DIEA及びNaIを添加し、室温にて1〜12時間撹拌して反応させて得ることができる。また、反応後、酢酸エチル等で抽出し、シリカゲルカラムクロマトグラフィー等の常法により精製することができる。
【0049】
上記反応式−1Bにおいて出発原料として用いられている化合物(B2)は、公知の化合物であるか、又は公知の方法により容易に製造される。例えば、下記反応式−2Bに示す方法により製造できる。
[反応式−2B]
【0050】
【化10】
【0051】
〔式中、lは前記に同じ〕
化合物(B2)は、例えば、次のようにして得ることができる。まず、熱EtOHにNaOH、p−ニトロベンジルアルコール、亜鉛粉末をこの順に添加し、70〜90℃にて1〜6時間撹拌した後、亜鉛粉末をろ去し、当該ろ液を半分以下の容量にまで減圧濃縮する。そして、これを1NHCl溶液によりpHを約5に調整した後、酢酸エチル等を用いて抽出すればよい。これを水・飽和食塩水等で洗浄し、減圧乾固させる。さらに、このようにして得られた化合物をDMFに溶解し、SOCl2を加えて室温で撹拌し、減圧乾固し、シリカゲルカラムクロマトグラフィー等の常法により精製して化合物(B2)を得ることができる。
【0052】
上記反応式−2Bにおいて出発原料として用いられている化合物(B4)は、公知の化合物であるか、又は公知の方法により容易に製造される。
【0053】
なお、反応式−2Bにおいて、出発原料として化合物(B4)だけでなく、化合物(B4)とはlの値が異なる化合物を混合して出発物質とすることにより、下記の化合物(B2’)を得ることもできる。化合物(B2’)を反応式−1Bの出発原料とすることもできる。
【0054】
【化11】
【0055】
〔式中l及びl’はそれぞれ異なって1〜10の整数を示す。〕
また、上記反応式−1Bにおいて出発原料として用いられている化合物(B3)は、公知の化合物であるか、又は公知の方法により容易に製造される。例えば、下記反応式−3Bに示す方法により製造できる。
[反応式−3B]
【0056】
【化12】
【0057】
〔式中、m及びnは前記に同じ〕
化合物(B3)は、例えば、化合物(B5)を酢酸第三ブチルに溶解させ、これに過塩素酸水溶液を添加し、室温にて10〜120分撹拌して、K2CO3等でpHを約9に調整後、例えば酢酸エチル等で抽出して得ることができる。さらに、例えば、これを水又は飽和食塩水等で洗浄した後、無水硫酸ナトリウムを加えて数時間室温で放置した後、硫酸ナトリウムをろ去し、ろ液を減圧乾固して固体として得ることができる。
【0058】
上記反応式−3Bにおいて出発原料として用いられている化合物(B5)は、公知の化合物であるか、又は公知の方法により容易に製造される。
【0059】
上記反応式−1Aにおいて用いられている化合物(C1)は、公知の化合物であるか、又は公知の方法により容易に製造される。例えば、下記反応式−1Cに示す方法により製造できる。
[反応式−1C]
【0060】
【化13】
【0061】
〔式中、k、j及びpは前記に同じ。〕
化合物(C1)は、例えば、化合物(D1)及びトリエチルアミンをDMSOに溶解させ、当該溶液に化合物(E1)のDMSO溶液を滴下し、室温にて1〜12時間撹拌して反応させることによって得ることができる。反応終了後、例えば酢酸エチル等で抽出し、常法に従って減圧乾固することもできる。
【0062】
上記反応式−1Cにおいて出発原料として用いられている化合物(D1)は、公知の化合物であるか、又は公知の方法により容易に製造される。例えば、下記反応式−1Dに示す方法により製造できる。
[反応式−1D]
【0063】
【化14】
【0064】
〔式中、k及びjは前記に同じ。k’はkより1小さい整数を示す。〕
化合物(D1)は、例えば、次のようにして得ることができる。化合物(D2)(塩酸塩)をギ酸にヨウ化し、当該溶液に化合物(D3)(水和物)を添加して、20〜60℃にて1〜12時間撹拌し、溶液を減圧乾固する。得られた残渣を1NHCl水溶液に溶解し、2〜12時間撹拌し、さらに当該溶液を減圧乾固する。得られた残渣を酢酸第三ブチルに溶解し、過塩素酸水溶液を添加した後、室温にて6〜24時間撹拌して得ることができる。これを常法に従って精製することができる。
【0065】
上記反応式−1Dにおいて出発原料として用いられている化合物(D2)及び(D3)は、それぞれ、公知の化合物であるか、又は公知の方法により容易に製造される。
【0066】
なお、化合物(D2)のかわりに、以下の化合物(D2’)を出発原料として用いることもできる。
【0067】
【化15】
【0068】
(式中、jは前記に同じ。−RNは“−NHCO−(荷電を有さないアミノ酸が1以上ペプチド結合により連続して結合したペプチド)”を示す。)
また、上記反応式−1Cにおいて出発原料として用いられている化合物(E1)は、公知の化合物であるか、又は公知の方法により容易に製造される。例えば、下記反応式−1Eに示す方法により製造できる。
[反応式−1E]
【0069】
【化16】
【0070】
〔式中、pは前記に同じ。〕
化合物(E1)は、例えば、化合物(E2)及びN−ヒドロキシスクシンイミド(NHS)をTHFに溶解し、低温(約0〜10℃)で6〜24時間程度撹拌した後、室温でさらに12〜48時間撹拌して反応させ、得ることができる。さらに常法に従い、精製することができる。
【0071】
上記反応式−1Eにおいて出発原料として用いられている化合物(E2)は、公知の化合物であるか、又は公知の方法により容易に製造される。
【0072】
なお、化合物(E2)において、基
【0073】
【化17】
【0074】
(すなわちジチオラン基)が、例えばチオール基、ジスルフィド基、スルフィド基又はシロキシ基等である化合物を、上記反応式−1Eにおいて化合物(E2)のかわりに出発原料として用いることも可能である。このような化合物は、公知の化合物であるか、又は公知の方法により容易に製造される。
【0075】
アゾ化合物結合固相基板
上述した一般式(1)で表されるアゾ化合物を、固相基板に結合させアゾ化合物結合固相基板を得ることができる。本発明はこのようなアゾ化合物結合固相基板に係る。
【0076】
本発明において用いる固相基板とは、その表面においてアゾ化合物の固相基板結合部位の官能基が好適にSAM膜を形成する限り、特に限定されない。当該固相基板の材料および厚さの選択は、アゾ化合物の固相基板結合部位の種類等に依存して、当業者が最適な条件を適宜選択することが可能である。基板材料に好適な例として、ガラス基板、金属基板(例えば金、銀、銅、アルミニウム、白金、酸化アルミニウム、SrTiO3、LaAlO3、NdGaO3、ZrO2等)、シリコン基板(例えば酸化珪素)、ポリマー樹脂基板(例えばポリエチレンテレフタレート、ポリカーボネート)、炭素(グラファイト)、マイカ等が例示できる。
【0077】
本発明において用いる固相基板は、前記材料のうち単一の材料からなる基板であってもよいし、1つの基板材料(第1の基板)の表面に別種類の少なくとも1つの材料からなる薄膜(第1の層)を形成していてもよいし、更に、前記第1の基板と前記第1の層との間に少なくとも1つの他の介在層(第2の層、第3の層、等)が存在していてもよい。好適な固相基板の具体例を挙げると、前記第1の基板としてマイカ基板を用い、前記第1の層として表面に金属膜(好適には金薄膜、銀薄膜、銅薄膜、白金薄膜)を有する前記マイカ基板等が挙げられる。なお、前記ガラス基板と前記金属膜との間には、他の材料からなる介在層が施されていてもよい。例えば、マイカ基板に金がコーティングされた固相基板を、本発明のアゾ化合物結合固相基板の作製のために好ましく用いることができる。
【0078】
前記第1の層を初めとする各金属膜の形成(コーティング)は、公知の方法により可能である。例えば電気めっき法、無電解めっき法、スパッタ法、蒸着法、イオンプレーティング法、等により形成することができる。なお、前記金属膜表面を有機溶剤で洗浄し、更に必要に応じて強酸で洗浄することによる分解除去、紫外線により発生するオゾン等による分解除去、等の方法を用いて汚染を除去することもできる。
【0079】
本発明において用いる固相基板の厚さは特に限定されないが、通常、前記第1の基板であれば、0.1mm〜30mm程度であり、好適には、0.1mm〜2mm程度である。
【0080】
一般式(1)で表されるアゾ化合物を固相基板に固定化する方法としては、固相基板結合部位及び使用する固相基板の種類によって、適宜適当な方法を選択することができる。例えば、当該アゾ化合物を含む溶液と固相基板とを接触させてインキュベートすることにより、当該アゾ化合物を当該固相基板上に吸着させる方法を用いることができる。
【0081】
さらに、例えば上述のようにしてアゾ化合物を固相基板に結合させた後、当該化合物が結合していない固相基板表面をブロッキングする工程を行ってもよい。このようなブロッキング操作は、適宜適当なブロッキング試薬及びブロッキング方法を選択して行うことができる。例えば、固相基板表面が金属である場合は、ヘキサンチオール溶液に浸積することにより、アゾ化合物が結合していない固相基板表面部分にヘキサンチオールがチオール基を介して結合するため、このように処理をしてブロッキング処理を行うことができる。
【0082】
このようにして得られるアゾ化合物結合固相基板は、金属イオンの回収のため、好ましく用いることができる。
【0083】
金属イオン回収方法
上述のようにして得られるアゾ化合物結合固相基板を用いて、効率よく金属イオンを回収することができる。本発明はこのような金属イオン回収方法に係る。
【0084】
当該固相基板に結合したアゾ化合物は、trans-体に紫外光を照射するとcis-体へと異性化し、cis-体へ可視光照射もしくは加熱を行うことによりtrans-体を再度生成する。trans-体、cis-体のいずれであっても、金属イオンを結合することができるが、その結合力はcis-体の方が強い。特に、金属イオンを捕捉(結合)したアゾ化合物が結合した、アゾ化合物結合固相基板を作用電極として金属イオンの酸化還元反応を繰り返すと、trans-体に結合した金属イオンはほぼ全て脱離してしまうが、cis-体に結合した金属イオンはほとんど脱離せず、結合したままとなる。本発明のアゾ化合物結合固相基板が有する当該性質を用いることにより、金属イオン含有廃液等から金属イオンを回収することが可能となる。
【0085】
具体的には、例えば以下の工程(i)〜(iii)を含む金属イオン回収方法により、金属イオンを簡便に回収することができる。
(i)金属イオン含有溶液に上述のアゾ化合物結合固相基板を浸積してアゾ化合物に金属イオンを結合させる工程
(ii)当該アゾ化合物結合固相基板のアゾ化合物に可視光を照射する工程
(iii)当該アゾ化合物結合固相基板を作用電極として金属イオンの酸化還元反応を繰り返して、アゾ化合物に結合した金属イオンを脱離させる工程
工程(i)において、金属イオン含有溶液は、金属イオンを含有する溶液であれば特に制限されないが、例えば産業廃液など、有効利用することができない溶液であると、これまで手間をかけて廃棄しなければならなかったものを有効利用することができるため、好ましい。また、ここでの金属イオンは、当該固相基板に結合したアゾ化合物の金属捕捉部位が捕捉できる金属イオンであれば特に制限されるものではなく、例えば銅イオン、亜鉛イオン、ニッケルイオン、コバルトイオン、クロムイオン、鉄イオン、マンガンイオン等が挙げられ、なかでも銅イオン、亜鉛イオンが好ましい。
【0086】
なお、工程(i)を行うにあたっては、当該固相基板に結合したアゾ加工物はtrans-体、cis-体のいずれであってもよいが、上述のようにcis-体の方がより強く金属イオンを結合することができるため、trans-体では金属イオンを結合することができない溶液(例えば、金属イオン濃度が薄い溶液、金属イオン以外の夾雑物が多量に溶解した溶液〔廃液〕等)から回収を行う場合は、cis-体であることが好ましい。当該固相基板に結合したアゾ化合物に紫外光を照射することにより、当該アゾ化合物をcis-体とすることができる。よって、当該金属イオン回収方法は、工程(i)の前に、当該アゾ化合物結合固相基板のアゾ化合物に紫外光を照射する工程(工程0)を含んでもよい。
【0087】
工程(ii)において、当該アゾ化合物結合固相基板のアゾ化合物に可視光を照射する。可視光照射時間は特に制限されるものではないが、通常0.5〜3時間、好ましくは1〜2時間程度である。これにより、当該アゾ化合物がtrans-体となる。ただし、当該固相基板に結合したアゾ化合物の全てをtrans-体とする必要はなく、一定量(好ましくは5割以上、より好ましくは8割以上)のアゾ化合物がtrans-体となればよい。
【0088】
工程(iii)において、当該アゾ化合物結合固相基板(結合したアゾ化合物の多くはtrans-体で、金属イオンを結合している)を作用電極として用い、例えばサイクリックボルタンメトリー(CV)を行って、結合している金属イオンの酸化還元反応を繰り返すことにより、当該金属イオンをアゾ化合物から脱離させる。当該酸化還元反応は、結合している金属イオンの種類、当該金属イオンの酸化電位、還元電位、酸化還元電位等により、適宜その条件を設定することができる。また、当該酸化還元反応については、例えばCV(サイクリックボルタンメトリー)測定により確認することができる。
【0089】
当該酸化還元反応の電位幅は、特に制限されるものではないが、通常、酸化電位及び還元電位が含まれる幅で行われる。アゾ化合物に結合した金属イオンの酸化還元電位±(200〜300 mV)で行うことが好ましい。酸化還元電位については、予備実験等により簡単に計測することができる。
【0090】
当該酸化還元反応に用いる電解質溶液は、特に制限されないが、通常NaClO4溶液、KClO4溶液、NaNO3溶液、KNO3溶液等を用いることができる。なお、アゾ化合物から脱離した金属イオンは、カウンターアニオンの選択により望みの金属塩へと変換(リサイクル)できるため、このことを考慮したうえで電解質溶液を選択することが好ましい。
【0091】
掃引スピードも特に制限されないが、通常20〜80mVs−1、好ましくは40〜60mVs−1である。また、掃引サイクル数も、結合した金属イオンが脱離する(CVの場合、CV測定により酸化還元応答が実質的に消失する(すなわち、サイクリックボルタモグラムのピークが消失する))まで行えばよく、通常30回以上、好ましくは30〜100回、より好ましくは30〜80回である。
【0092】
サイクリックボルタンメトリーは例えば三電極系で行うことができる。この場合、作用電極には本発明に係るアゾ化合物結合固相基板を用いるが、対極及び参照電極は特に制限されず、例えば対極にPt線、参照電極にAg/AgClを用いることができる。
【0093】
なお、工程(i)及び(ii)の間あるいは工程(ii)及び(iii)の間に、当該アゾ化合物結合固相基板を金属イオン含有溶液から取り出し、異なる溶液(例えば1M NaClO4溶液等)中に移動させてもよい。このようにすれば、移動先の溶液中へ金属イオンが脱離することとなる(移動先の溶液を回収溶液とする)。すなわち、本発明の金属イオン回収方法は、工程(i)と(ii)の間に、当該アゾ化合物結合固相基板を回収溶液中に移動させる工程(工程(i-2))を含んでもよいし、工程(ii)と(iii)の間に、当該アゾ化合物結合固相基板を回収溶液中に移動させる工程(工程(ii-2))を含んでもよい。工程(ii)を経るとアゾ化合物がtrans体となり、金属イオンとの結合力がcis体より弱くなるため、工程(ii-2)よりも工程(i-2)を含むことが好ましい。
【0094】
さらに、本発明の金属イオン回収方法は、同一のアゾ化合物結合固相基板を用いて繰り返し行うことが可能である。すなわち、工程(iii)の後、固相基板上のアゾ化合物の多くはtrans-体であって金属イオンが脱離した状態にあるので、これを再度工程(i)に供することができる。また、上述したように、工程(i)に供する前に、固相基板に結合したアゾ化合物に紫外光を照射することにより(すなわち工程(0)を行うことにより)、これをcis-体としてもよい。紫外光照射時間は特に制限されず、通常0.5〜3時間、好ましくは0.5〜2時間程度である。
【0095】
なお、前述の工程(i-2)及び/又は工程(ii-2)を経た場合は、工程(i)に供する前に、アゾ化合物結合固相基板を回収溶液から工程(i)の金属含有溶液へと移動させる工程(工程iv)を経る必要がある。
【0096】
このように、本発明の金属イオン回収方法は、上述した工程(i)〜(iii)を含む工程を繰り返し行うことによって実施することもできる。また、当該繰り返し行われる工程には、工程(i)〜(iii)以外の工程が含まれてもよく、例えば上述した工程(0)、工程(i-2)、工程(ii-2)等を好ましく含むことができる。
【0097】
例えば、本発明は以下の工程(0)、(i)、(i-2)、(ii)、(iii)及び(iv)を含む金属イオン回収方法を含む。
(0)アゾ化合物結合固相基板のアゾ化合物に紫外光を照射する工程
(i)金属イオン含有溶液に上述のアゾ化合物結合固相基板を浸積してアゾ化合物に金属イオンを結合させる工程
(i-2)当該アゾ化合物結合固相基板を回収溶液中に移動させる工程
(ii)当該アゾ化合物結合固相基板のアゾ化合物に可視光を照射する工程
(iii)当該アゾ化合物結合固相基板を作用電極として金属イオンの酸化還元反応を繰り返して、アゾ化合物に結合した金属イオンを脱離させる工程
(iv)当該アゾ化合物結合固相基板を回収溶液から金属イオン含有溶液へと移動させる工程
またさらに本発明は、例えば、上記の工程(0)、(i)、(i-2)、(ii)、(iii)及び(iv)を含む工程を繰り返し行うことにより、金属イオンを回収する方法を含む。
【実施例】
【0098】
以下、本発明を具体的に説明するが、本発明は下記の例に限定されるものではない。
【0099】
なお、以下の例において、全ての試薬及び溶媒は、ナカライテスク、和光純薬、東京化成、石津製薬から購入し、そのまま使用した。ミリQ水はMillipore Milli-Q biocel Aにより得た。
【0100】
また、以下の例においては、1H-NMRスペクトル、MALDI-TOF Massスペクトル、UV-visスペクトル、CV、及びEQCMの測定は下記のようにして行った。
<1H-NMRスペクトル>
測定装置はJEOL社製JNM-ECP600分光計を使用し、δ= -0.1-10.1 ppmについて測定した。内径4 mmのサンプルチューブを使用し、溶媒はCDCl3またはD2Oを用いた。ケミカルシフトは内部標準のテトラメチルシランまたはDSSを基準として表記した。
<MALDI-TOF Massスペクトル>
測定装置はApplied Biosystems社製DE-STR Voyagerを使用し、マトリックスはa-シアノ-4-ヒドロキシケイ皮酸を用いた。キャリブレーションはポリエチレングリコール(分子量: 1000)を用いて行った。
<UV-visスペクトル>
測定装置は島津製作所製UV-3100PCを使用し、200-900 nmについて測定した。セルは光路長1 cmまたは0.1 cmの石英セルを使用した。サンプルは1 mM, 5 mMのアゾベンゼン化合物溶液(水, DMF)を用いた。
<CV>
測定装置はBAS社製CV-50Wを使用した。測定は三電極系で行い、作用電極に本発明に係るアゾ化合物結合固相基板(以下「修飾電極」と表記することがある;図7を参照)、対極にPt線、参照電極にAg/AgCl (3 M NaCl)を使用した。測定には0.1 M NaClO4水溶液または0.5 M KOH水溶液を用い、測定前に約15分間Arバブリングすることで溶存酸素を除去した。なお、実施例において酸化還元反応は全てCVによって行った。
【0101】
<合成例1:アゾ化合物の合成>
以下のようにして、下記の化学式で表されるアゾ化合物(trans-体)を合成した。なお、以下、下記式で表されるアゾ化合物をH4L1と表記することがある。(H4L1のH4は分子中に存在するCOOH基のHが4つあることを表している。従って、当該基がCOONaとなっている場合、Na4L1と表記される。)
【0102】
【化18】
【0103】
まず、H4L1の合成スキームを次に示す。なお、当該スキーム中に番号1〜5をふった化合物はそれぞれ以下「アゾベンゼン誘導体1」、「リジン誘導体2」、「リポ酸誘導体3」、「リジン誘導体4」、「アゾベンゼン誘導体5」と表記することがある。
【0104】
【化19】
【0105】
4,4’-ビスクロロメチルアゾベンゼンの合成
4,4’-ビスクロロメチルアゾベンゼンの合成は既報の方法((a) Mori, Y.; Niwa, T.; Toyoshi, K. Chem. Pharm. Bull. 1981, 29, 1439-1442, (b) Maie, K.; Nakamura, M.; Yamana, K. Nucleos. Nucleot. Nucl. 2006, 25, 453-462.)を改良して行った。 熱EtOH 120 mLにNaOH 9.6 g (240 mmol)、p-ニトロベンジルアルコール 6.0 g (39 mmol)、亜鉛粉末 5.0 g (77 mmol)をこの順に添加した。80℃にて3時間攪拌した後、亜鉛粉末をろ去した。ろ液を50 mLまで減圧濃縮し、水50 mLを加えた。1 N HCl水溶液によりpH 5に調整した後、酢酸エチルにて抽出した。得られた有機層を水、飽和食塩水で洗浄した後、ろ液を減圧乾固することで4,4’-ビスヒドロキシメチルアゾベンゼンの粗製物(1.9 g, 純度約60 %)を得た。
【0106】
得られた粗製物1.9 g (4.7 mmol)をDMF 20 mLに溶解し、SOCl20.86 mL (12 mmol)を加え、室温にて1時間攪拌した。反応溶液を減圧乾固し、残渣を少量のクロロホルムに溶解してシリカゲルカラムクロマトグラフィー(溶離液; クロロホルム:ヘキサン= 1 : 2)を行った。目的物はTLC (Rf= 0.95 (展開溶媒; クロロホルム))により確認し、得られた分画を減圧乾固した。得られた橙色粉末を真空乾燥し、1H-NMRスペクトルから目的物であることを確認した。
【0107】
【化20】
【0108】
収量 510 mg (収率 5 %); 1H-NMR (600 MHz, CDCl3) δ7.92 (d, J = 8.4 Hz, 4H, φ-Hc), 7.55 (d, J = 8.4 Hz, 4H, φ-Hb), 4.66 (s, 4H, -CHa2-).
【0109】
イミノ二酢酸ジ第三ブチルエステルの合成
イミノ二酢酸ジ第三ブチルエステルの合成は既報の方法(Chen, H.; Feng, Y.; Xu, Z.; Ye, T. Tetrahedron 2005, 61, 11132-11140.)を参考にして行った。イミノ二酢酸 1.0 g (7.5 mmol) を酢酸第三ブチル200 mL (1.5 mol) に溶解した。その溶液に60 %過塩素酸水溶液2.6 mL (40 mmol)を添加し、室温にて20時間撹拌した。その後、10 % K2CO3水溶液によりpH 9に調整し、酢酸エチルにて抽出した。得られた有機層を水、飽和食塩水にて洗浄した後、無水硫酸ナトリウムを加えて1時間放置した。硫酸ナトリウムをろ去し、ろ液を減圧乾固することで白色固体を得た。得られた白色固体を真空乾燥し、1H-NMRスペクトルから目的物であることを確認した。
【0110】
【化21】
【0111】
収量1.2 g (収率 66 %); 1H-NMR (600 MHz, CDCl3) δ 3.34 (s, 4H, -CHa2-), 1.47 (s, 18H, -CHb3).
【0112】
アゾベンゼン誘導体1の合成
4,4’-ビスクロロメチルアゾベンゼン 300 mg (1.08 mmol)、イミノ二酢酸ジ第三ブチルエステル 264 mg (1.08 mmol)のDMF 溶液(20 mL)にジイソプロピルエチルアミン(DIEA) 360μL (2.16 mmol)、NaI 726 mg (3.24 mmol)を添加し、室温にて3時間攪拌した。反応終了後、水100 mLを加え、酢酸エチルにて抽出した。得られた有機層を1 N HCl水溶液、水、飽和食塩水にて洗浄した後、ろ液を減圧乾固することで橙色固体を得た。得られた残渣を少量のクロロホルムに溶解してシリカゲルカラムクロマトグラフィー(溶離液; クロロホルム)を行った。目的物はTLC (Rf = 0.20 (展開溶媒; クロロホルム))により確認し、得られた分画を減圧乾固した。得られた橙色固体を真空乾燥し、1H-NMRスペクトルから目的物であることを確認した。
【0113】
【化22】
【0114】
収量150 mg (収率29 %); 1H-NMR (600 MHz, CDCl3) δ 7.91 (d, J = 8.4 Hz, 2H, φ-Hc), 7.88 (d, J = 8.4 Hz, 2H, φ-Hd), 7.57 (d, J = 8.4 Hz, 2H, φ-He), 7.54 (d, J = 8.4 Hz, 2H, φ-Hb), 4.66 (s, 2H, -CHa2-), 3.98 (s, 2H, -CHf2-), 3.45 (s, 4H, -CHg2-), 1.48 (s, 18H, -CHh3).
【0115】
リジン誘導体2の合成
リジン誘導体2の合成は既報の方法(Kihlberg, J.; Bergman, R.; Wickberg, B. Acta Chem. Scand. B 1983, 37, 911-916.)を参考にして行った。Lys・HCl 546 mg (3.0 mmol) をギ酸 30 mL に溶解し、その溶液にグリオキシル酸一水和物600 mg (6.6 mmol)を添加した。40℃にて4時間攪拌し、溶液を減圧乾固した。得られた残渣を1 N HCl水溶液2 mLに溶解し、5時間攪拌した後、溶液を減圧乾固した。得られたオイル状残渣550 mgを酢酸第三ブチル60 mLに溶解し、60 %過塩素酸水溶液900μL (13 mmol)を添加した後、室温にて14時間撹拌した。反応終了後、10 % K2CO3 水溶液によりpH 9に調整し、酢酸エチルにて抽出した。得られた有機層を水、飽和食塩水にて洗浄した後、無水硫酸ナトリウムを加えて1時間放置した。硫酸ナトリウムをろ去し、ろ液を減圧乾固することで黄色油状物を得た。得られた黄色油状物を真空乾燥し、1H-NMRスペクトルから目的物であることを確認した。
【0116】
【化23】
【0117】
収量400 mg (収率 42 %); 1H-NMR (600 MHz, CDCl3) . δ 3.31 (d, J = 16.8 Hz, 1H, -CHa2-), 3.23 (d, J = 16.8 Hz, 1H, -CHa2-), 3.14 (t, J = 6.6 Hz, 1H, -CHb-), 2.69 (t, J = 6.6 Hz, 2H, -CHf2-), 1.63 (m, 2H, -CHc2-), 1.51-1.39 (m, 22H, -CHd2-, -CHe2-, -CHg3, -CHh3).
【0118】
リポ酸誘導体3の合成
リポ酸誘導体3の合成は既報の方法(Ha, T. H.; Jung, S. O.; Lee, K. Y.; Lee, Y.; Park, J. S.; Chung, B. H. Anal. Chem. 2007, 79, 546-556.)を改良して行った。DL-a-リポ酸1.0 g (5.0 mmol)、N-ヒドロキシスクシンイミド 0.58 g (5.0 mmol)をTHF 5 mLに溶解し、氷浴上で撹拌しながらジシクロヘキシルカルボジイミド1.1 g (5.1 mmol)をゆっくりと添加した。氷浴上にて10時間撹拌した後、反応温度を室温に変えて24時間撹拌した。析出した固体をろ去し、ろ液を減圧乾固した。得られた残渣を酢酸エチル-ヘキサンから再結晶することで黄色針状結晶を得た。得られた黄色針状結晶を真空乾燥し、1H-NMRスペクトルから目的物であることを確認した。
【0119】
【化24】
【0120】
収量1.1 g (収率 75 %); 1H-NMR (600 MHz, CDCl3) δ 3.59 (m, 1H, -CHc-), 3.19 (m, 1H, -CHa2-), 3.12 (m, 1H, -CHa2-), 2.84 (br, 4H, -CHh2-), 2.63 (t, J = 7.2 Hz, 2H, -CHg2-), 2.47 (m, 1H, -CHb2-), 1.93 (m, 1H, -CHb2-), 1.81-1.56 (m, 6H, -CHd2-, -CHe2-, -CHf2-).
【0121】
リジン誘導体4の合成
リジン誘導体2を200 mg (0.63 mmol)、トリエチルアミン 170μL (1.3 mmol)とともにDMSO 4 mLに溶解した。その溶液にリポ酸誘導体3を 230 mg (0.76 mmol)溶解したDMSO溶液(4 mL)を30分かけて滴下し、室温にて4時間撹拌した。反応終了後、水30 mLを加えて室温にて12時間撹拌した。酢酸エチルにて抽出を行った後、得られた有機層を水、飽和食塩水にて洗浄し、無水硫酸ナトリウムを加えて1時間放置した。硫酸ナトリウムをろ去し、ろ液を減圧乾固することで黄色油状物を得た。得られた黄色油状物を真空乾燥し、1H-NMRスペクトルから目的物であることを確認した。
【0122】
【化25】
【0123】
収量220 mg (収率 69 %); 1H-NMR (600 MHz, CDCl3) δ 5.68 (br, 1H, -NHi-), 3.57 (m, 1H, -CHn-), 3.32 (d, J = 16.8 Hz, 1H, -CHa2-), 3.25 (t, J = 6.6 Hz, 2H, -CHf2-), 3.21 (d, J = 16.8 Hz, 1H, -CHa2-), 3.18 (m, 1H, -CHp2-), 3.12 (t, J = 6.6 Hz, 1H, -CHb-), 3.11 (m, 1H, -CHp2-), 2.46 (m, 1H, -CHo2-), 2.17 (d, J = 7.2 Hz, 1H, -CHj2-), 2.16 (d, J = 7.2 Hz, 1H, -CHj2-), 1.91 (m, 1H, -CHo2-), 1.71-1.41 (m, 30H, -CHc2-, -CHd2-, -CHe2-, -CHk2-, -CHl2-, -CHm2-, -CHg3, -CHh3).
【0124】
アゾベンゼン誘導体5の合成
アゾベンゼン誘導体1を137 mg (0.27 mmol)及びリジン誘導体4を 86 mg (0.18 mmol)溶解させたDMF溶液(4 mL)にDIEA 30μL (0.30 mmol)、NaI 66 mg (0.30 mmol)を添加し、室温にて15時間攪拌した。反応終了後、溶液を減圧乾固した。得られた残渣を水20 mLで溶解し、酢酸エチルにて抽出した。得られた有機層を水、飽和食塩水にて洗浄した後、ろ液を減圧乾固した。得られた残渣を少量の酢酸エチルに溶解してシリカゲルカラムクロマトグラフィー(溶離液; 酢酸エチル:ヘキサン= 1 : 5)を行った。目的物はTLC (Rf= 0.1 (展開溶媒; 酢酸エチル:ヘキサン= 1 : 5))により確認し、得られた分画を減圧乾固した。得られた橙色固体を真空乾燥し、1H-NMRスペクトルから目的物であることを確認した。
【0125】
【化26】
【0126】
収量60 mg (収率35 %); 1H-NMR (600 MHz, CDCl3) δ 7.87 (d, J = 8.4 Hz, 2H, φ-Hs), 7.86 (d, J = 8.4 Hz, 2H, φ-Hs), 7.56 (d, J = 8.4 Hz, 2H, φ-Hu), 7.55 (d, J = 8.4 Hz, 2H, φ-Hu), 5.77 (t, J = 5.4 Hz, 1H, -NHi-), 3.98 (s, 2H, -CHt2-), 3.98 (d, J = 14.4 Hz, 1H, -CHq2-), 3.80 (d, J = 14.4 Hz, 1H, -CHq2-), 3.54 (m, 1H, -CHn-), 3.45 (s, 4H, -CHu2-), 3.44 (d, J = 16.8 Hz, 1H, -CHa2-), 3.33 (d, J = 16.8 Hz, 1H, -CHa2-), 3.25 (m, 1H, -CHf2-), 3.22 (dd, J = 7.2, 9.0 Hz 1H, -CHb-) 3.17-3.12 (m, 2H, -CHf2-, -CHp2-), 3.08 (m, 1H, -CHp2-), 2.42 (m, 1H, -CHo2-), 2.15 (t, J = 7.2 Hz, 2H, -CHj2-), 1.87 (m, 1H, -CHo2-), 1.65 (m, 2H, -CHc2-), 1.51-1.37 (m, 46H, -CHd2-, -CHe2-, -CHk2-, -CHl2-, -CHm2-, -CHg3, -CHh3, -CHv3).
【0127】
trans-H4L1の合成
アゾベンゼン誘導体5を60 mg (0.063 mmol)、TFA 18 mL に溶解し、室温にて13時間撹拌した。反応終了後、溶液を減圧乾固して得られた残渣をジエチルエーテルにて洗浄した。得られた黄色固体を真空乾燥し、MALDI-TOF Massスペクトルから目的物であることを確認した。また、30 % NaOD水溶液を加えてtrans-L1とした場合の1H-NMRスペクトルから目的物(trans-Na4L1)であることを確認した(図1)。
【0128】
【化27】
【0129】
収量42 mg (収率 91 %); 1H-NMR (600 MHz, D2O) δ 7.85 (d, J = 8.4 Hz, 2H, φ-Hp), 7.84 (d, J = 8.4 Hz, 2H, φ-Hp), 7.67 (d, J = 8.4 Hz, 2H, φ-Ho), 7.59 (d, J = 8.4 Hz, 2H, φ-Ho), 3.93 (d, J = 13.2 Hz, 1H, -CHn2-), 3.84 (s, 2H, -CHq2-), 3.72 (d, J = 13.2 Hz, 1H, -CHn2-), 3.47 (m, 1H, -CHk-), 3.33 (d, J = 16.8 Hz, 1H, -CHa2-), 3.18 (s, 4H, -CHr2-), 3.16-3.09 (m, 8H, -CHa2-, -CHb-, -CHf2-, -CHm2-), 2.31 (m, 1H, -CHl2-), 2.10 (t, J = 7.2 Hz, 2H, -CHg2-), 1.81 (m, 1H, -CHl2-), 1.59-1.22 (m, 12H, -CHc2-, -CHd2-, -CHe2-, -CHh2-, -CHi2-, -CHj2-); MS (MALDI-TOF) m/z: 732.33 [H4L1 + H+]+, 754.14 [H4L1 + Na+]+.
【0130】
<参考例1:アゾ化合物の合成>
以下のようにして、下記の化学式で表されるアゾ化合物(trans-体)を合成した。なお、以下、下記式で表されるアゾ化合物をH4L2と呼ぶ。(H4L2のH4は分子中に存在するCOOH基のHが4つあることを表している。従って、当該基がCOONaとなっている場合、Na4L2と表記される。)
【0131】
【化28】
【0132】
trans-H4L2の合成
4,4’-ビスクロロメチルアゾベンゼン 0.187 g (0.67 mmol)、イミノ二酢酸ジ第三ブチルエステル 0.82 g (3.35 mmol)のDMF溶液(28 ml)にDIEA 224μL (2.2 mmol)、NaI 0.40 g (2.68 mmol)を添加し、室温にて一晩撹拌した。反応終了後、溶液を減圧乾固した。得られた残渣をクロロホルムに溶解し、1 N HCl水溶液にて洗浄した。有機層を5 % NaHCO3水溶液、飽和食塩水で洗浄した後、無水硫酸ナトリウムを加えて静置した。硫酸ナトリウムをろ去し、ろ液を減圧乾固した。得られた残渣をTFA 30 mLに溶解し、室温にて一晩撹拌した後、溶液を減圧乾固した。得られた黄色固体をジエチルエーテルにて洗浄し、真空乾燥した。30 % NaOD水溶液を加えて1H-NMRスペクトルから目的物(Na4L2)であることを確認した。なお、当該アゾ化合物は、既に上記非特許文献1において報告されている。
【0133】
【化29】
【0134】
収量 285 mg (収率 90 %); 1H-NMR (600 MHz, D2O) δ 7.85 (d, J = 8.4 Hz, 4H, φ-Ha), 7.61 (d, J = 8.4 Hz, 4H, φ-Hb), 3.88 (s, 4H, -CHc2-), 3.22 (s, 8H, -CHd2-).
【0135】
実施例1:L1の光異性化挙動
合成したtrans-H4L1は水に不溶であるため、4当量のNaOHの添加により1 mM trans-L1水溶液を調製した。trans-L1水溶液のUV-visスペクトルは、229 (1.1 × 104), 334 nm (1.9 × 104 M-1 cm-1)にtrans-型アゾベンゼンに特徴的なπ-π*遷移を、420 nm付近(ca. 1.3 × 103M-1cm-1)にn-π*遷移を示した(図2(a))。この溶液の1H-NMRスペクトルを測定したところ、芳香族領域にはtrans-型アゾベンゼンに由来する4種類のダブレットのみが観測された(図3(a))。これらの結果から、合成終了段階では熱力学的に安定なtrans-体のみが存在すると考えられた(trans/cis = 100/0)。
【0136】
次に、L1を用いた光異性化挙動について検討した。キセノンランプを光源とし、カットフィルターを用いて紫外光(320-400 nm)、可視光(420-1800 nm)をそれぞれ調整した。1 mM trans-L1水溶液に紫外光を照射すると、そのUV-visスペクトル(紫外可視吸収スペクトル)は徐々に変化し、80 分間照射すると光定常状態(photostationary state; PSS)となった(図2(a))。光定常状態(紫外光)のUV-visスペクトルは、trans-L1における334 nmのπ-π*遷移の吸光度が大きく減少して324 nmに短波長シフトし、420 nmのn-π*遷移の吸光度がわずかに増大していた。これはcis-型アゾベンゼンに特徴的なスペクトルであるため、光異性化が進行しているものと考えられた。
【0137】
上述の非特許文献1に開示されるように、一連のアゾベンゼン誘導体の光定常状態における異性体比をtrans-型アゾベンゼンのπ-π*遷移(334 nm)の吸光度を用いて下記式(I)から算出することができる。
【0138】
【数1】
【0139】
ここで、APSSは光定常状態におけるπ-π*遷移の吸光度、Atransはtrans-型アゾベンゼンにおけるπ-π*遷移の吸光度である。式(1)により算出した紫外光照射時の光定常状態におけるL1の異性体比はtrans/cis = 38/62であった。
【0140】
実施例2:金属イオン添加時の光異性化挙動および錯形成挙動
上述の非特許文献1には、cis-L2に1当量の金属イオン(ZnII, CuII)を添加した場合、cis-L2と金属イオンとの錯形成により熱力学的な安定性が増大し、cis-体からtrans-体への熱異性化速度が激減することが記載されている。そこで、L1, L2に1当量の金属イオンを添加した場合の光異性化挙動およびその錯形成挙動についての検討を行った。ここで、金属錯体[MII(trans-L1)]はtrans-L1水溶液に1当量のMII(ClO4)2(MII = ZnII, CuII)を添加することで調製した。
【0141】
1 mM [ZnII(trans-L1)]水溶液のUV-visスペクトルは、trans-L1における334 nmのπ-π*遷移が332 nmに短波長シフトし、420 nmのn-π*遷移の吸収がわずかに減少していた(図4a)。この結果は、添加したZnIIイオンがtrans-L1に配位したことを示している。[ZnII(trans-L1)]に紫外光を照射すると、UV-visスペクトルは徐々に変化し、90分間照射することで光定常状態に達した(図4a)。光定常状態(紫外光)のUV-visスペクトルは、332 nm付近のπ-π*遷移が減少するとともに、420 nm付近のn-π*遷移の吸収がわずかに増大した。これはtrans-体([ZnII(trans-L1)])からcis-体([ZnII(cis-L1)])への異性化を示しており、吸光度から算出した異性体比はtrans/cis = 47/53であった。さらにこの溶液に可視光を照射した場合、UV-visスペクトルが変化し、10分間照射することで光定常状態へと達した(図4a)。光定常状態(可視光)のUV-visスペクトルは、[ZnII(trans-L1)]のスペクトルに非常に類似したスペクトルとなり、吸光度から算出した異性体比はtrans/cis = 87/13であった。これらの結果から、1当量のZnIIイオンを添加した場合においても光による異性化が可能であることを確認した。
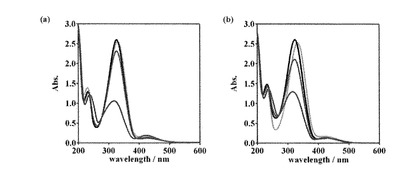
【0142】
同様の光異性化実験を1 mM [CuII(trans-L1)], [ZnII(trans-L2)], [CuII(trans-L2)]水溶液に対して行い、それらのUV-visスペクトル変化および吸光度から得られた異性体比を図4、図5、表1にまとめた。いずれの場合も金属イオンの添加によってπ-π*遷移の低波長シフトが観測され、金属イオンの結合が示唆された。また、光異性化挙動に関しても上述の[ZnII(trans-L1)]の場合と同様であったため、金属イオン添加時にも光による異性化が可能であることを確認した。
【0143】
異性体比に着目すると、紫外光照射時(trans-体からcis-体への異性化)では金属添加時の方が残存するtrans-体の割合が大きかった。一方、可視光照射時(cis-体からtrans-体への異性化)では逆に金属添加時の方が残存するcis-体の割合が少なかった。したがって、金属イオンの添加はtrans-体からcis-体への異性化には不利に働くが、cis-体からtrans-体への異性化に対しては有利に働くことがわかった。
【0144】
【表1】
【0145】
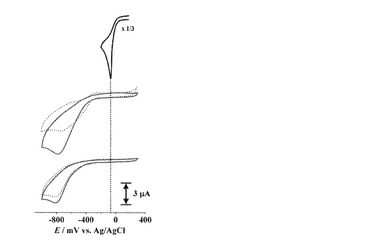
次に、[CuII(trans-L1)], [CuII(cis-L1)]の電気化学的な性質を調べるためにCV測定を行った(図6)。ここで、[CuII(cis-L1)]は前述のようにtrans-L1への紫外光照射後にCuIIイオンを添加することで調製し、参照のためにtrans-L1, cis-L1, CuII(ClO4)2についても測定を行った。trans-L1およびcis-L1はいずれも-800 mV付近に還元波を示し、対応する酸化波は観測されなかった。この不可逆な還元波は、アゾ基からヒドロアゾ基への還元反応(2e-, 2H+)に由来するものと考えられ、両異性体間での違いはほとんどなかった。このような還元波は[CuII(trans-L1)], [CuII(cis-L1)]についても同様に観測された。また、CuII(ClO4)2は-60 mV付近にCuI/IIの還元に由来すると考えられる応答を示したが、この還元波は[CuII(trans-L1)], [CuII(cis-L1)]の場合においては全く観測されなかった。したがって、いずれも遊離しているCuIIイオンは存在せず、全てのCuIIイオンは配位子(−COO−)と結合していると考えられた。
【0146】
これらの結果から、L1に1当量のCuIIイオンを添加した場合、全てのCuIIイオンは配位子と結合しており、少なくとも溶液中では遊離しているCuIIイオンは存在しないことが示された。また、溶液中では、L1はcis体でもtrans体でも金属イオンを結合する強さにほとんどかわりがないと考えられた。
【0147】
実施例3:修飾電極と銅イオンとの結合強度の検討
<L1修飾電極の作製>
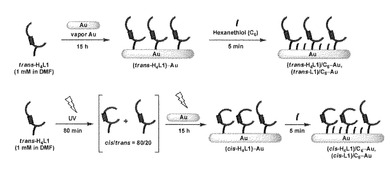
以下のようにして金薄膜がコーティングされたマイカ基板を作製し、これにL1分子を修飾してアゾ化合物結合固相基板(修飾電極として使用)を作製した。
【0148】
蒸着金の作製および評価
蒸着金の作製はシンク社製高真空抵抗加熱装置JIS-300Ak型を使用した。金原料には田中貴金属製のφ1.0 mm金線(99.999 %)を用いた。ニラコ社製の天然マイカ上を14 × 14 mm四方の大きさにカットし、装置のチャンバー内部に固定した。また蒸着用バスケットに清浄な金線を適当量静置した。ロータリーポンプ、ターボ分子ポンプを順に起動し、チャンバー内圧を10-5 Pa以下となるまで減圧した。ランプヒーターによりマイカを300℃で3時間加熱した後、金線を一定温度で加熱することで金蒸着を行った。蒸着速度は1.0Å s-1に制御し、厚さ1000 Åまで蒸着することで目的の蒸着金を得た。
【0149】
蒸着金の表面積は、水素炎でアニール処理後の蒸着金を用いた0.1 M H2SO4水溶液中でのCV測定を行い、酸化被膜の還元反応時の電気量(理論値: 444μC cm-2)から算出した(A = 1.1 cm2, roughness factor: 2.1)。
【0150】
修飾電極の作製
金表面への分子修飾は上述のように作製した蒸着金を水素炎でアニール処理した後、各溶液に浸漬することで作製した。各修飾電極及びその作製過程の模式図を図7に示す。
【0151】
修飾電極H4L1-Au(H4L1結合固相基板(金蒸着あり))の作製(図7)
(trans-H4L1)-Au は、アニール処理した蒸着金を1 mM trans-H4L1溶液(DMF)に15時間浸漬することで作製した。(cis-H4L1)-Au は、1 mM trans-H4L溶液(DMF)に紫外光を80分間照射した光定常状態の溶液に蒸着金を浸漬し、紫外光を照射しながら15時間浸漬することで作製した。いずれもDMF、水の順にリンスした後に電気化学測定に使用した。なお、(cis-H4L1)-Auでは全てのH4L1がcis体となるわけではなく、一部trans体も含まれるが、(cis-H4L1)-Auと表記する(図7)。以下も同様とする。
【0152】
修飾電極H4L1/C6-Au(ヘキサンチオールでブロッキングされたH4L1結合固相基板(金蒸着あり))の作製(図7)
(trans-H4L1)/C6-Au, (cis-H4L1)/C6-Au は、上述のようにして作製した(trans-H4L1)-Au, (cis-H4L1)-Auを0.1 mM ヘキサンチオール(C6)溶液(MeOH)に5分間浸漬することでそれぞれ作製した。いずれもMeOH、水の順にてリンスした後に電気化学測定に用いた。
【0153】
なお、ヘキサンチオールは、修飾電極として用いた際バックグラウンド電流を小さくするため、及びCuIIイオンのAu-S結合への影響を抑えるためのブロッキングのために用いた。
【0154】
修飾電極L1/C6-Au(ヘキサンチオールでブロッキングされたL1結合固相基板(金蒸着あり))の作製(図7)
(trans-L1)/C6-Au, (cis-L1)/C6-Auは、上述のようにして作製した(trans-H4L1)/C6-Au, (cis-H4L1)/C6-Auを0.5 M NaOH水溶液に90分間浸漬することでそれぞれ作製した。いずれも水でリンスした後に電気化学測定に用いた。
【0155】
修飾電極[CuII(L1)]/C6-Au(ヘキサンチオールでブロッキングされたL1結合固相基板であって、L1が銅イオンを結合している基板(金蒸着あり))の作製(図7)
[CuII(trans-L1)]/C6-Au, [CuII(cis-L1)]/C6-Auは、上述のようにして作製した(trans-L1)/C6-Au, (cis-L1)/C6-Auを1 mM CuII(ClO4)2水溶液に30分間浸漬することでそれぞれ作製した。いずれも水でリンスした後に電気化学測定に用いた。
【0156】
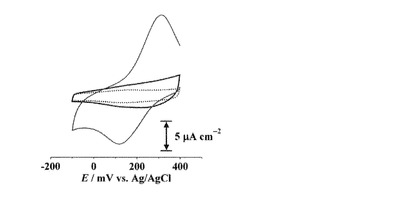
実施例4 trans-L1修飾電極と銅イオンとの結合の検討
上述のようにして作製したアゾ化合物結合固相基板((trans-H4L)/C6-Au)を用い、固体表面上に配列された配位子L1へのCuIIイオンの結合を電気化学的に評価した。作製した(trans-H4L1)/C6-Auの0.1 M NaClO4水溶液中でのCV測定を行ったところ、測定電位範囲(-100 mVから+400 mV)において酸化還元応答は観測されなかった(図8)。また、(trans-H4L1)/C6-Auを0.5 M NaOH水溶液に浸漬することでH4L1部位を脱プロトン化した(trans-L1)/C6-Auもまた測定電位範囲では酸化還元応答を示さず、プロトン化体((trans-H4L1)/C6-Au)に比べてバックグラウンド電流がわずかに増加していた(図8)。水溶液中においてL1は-800 mV付近に還元波を示すが、(trans-H4L1)-Auは-900 mV付近に脱離波を示すためこれ以上負側への掃引は行わなかった。
【0157】
また、上述のようにして作製した[CuII(trans-L1)]/C6-Auを用いた0.1 M NaClO4水溶液中(CuIIイオン非存在下)でのボルタモグラムは、+308, +136 mVに一対の酸化還元応答を示した(E1/2 = +222 mV,図8)。(trans-H4L1)/C6-Au, (trans-L1)/C6-Auではこの測定電位範囲において酸化還元応答を示さなかったことから、この酸化還元波は表面上に固定化されたL1のカルボン酸部位と結合したCuI/IIの1電子酸化還元反応に由来するものと考えられる。還元波のピーク電気量(Qpc)は18μC cm-2であった。
【0158】
この酸化還元応答は掃引を繰り返すことでピークが減少していき、60サイクル目には酸化還元応答がほぼ消失した(図9)。これはCuイオンの固体表面上からの脱離を意味しており、酸化還元反応を繰り返すことでCuイオンが遊離していくことがわかった。また、このことから、(trans-L1)/C6-AuとCuイオンとの結合は比較的弱いことがわかった。なお、この場合、表面上のtrans-L1はCuイオン結合型からNa+イオン結合型(CuIIイオンを浸漬する前の状態)へと変換され、遊離したCuイオンは溶液中で過塩素酸銅へと変換されたと考えられた。
【0159】
以上の結果より、表面上のtrans-L1とCuイオンとの結合は比較的弱く、酸化還元反応を繰り返すことでCuイオンを溶液中に放出できることがわかった。またこのことから、trans-L1部位の金属イオン結合能の再活性化とともに、カウンターアニオンの選択により望みの金属塩へと変換(リサイクル)できることがわかった。
【0160】
実施例5 cis-L1修飾電極と銅イオンとの結合の検討
上述のようにして作製した (cis-L1)/C6-Au)を作用電極としたボルタモグラムは、trans-体の場合と同様に測定電位範囲において酸化還元応答を示さなかった(図10)。
【0161】
(cis-L1)/C6-AuをCuII(ClO4)2水溶液中に浸漬することで作製した[CuII(cis-L1)]/C6-Auのボルタモグラムは、+313, +247 mVに酸化還元応答を示した(E1/2 = +280 mV)。これはL1のカルボン酸部位に結合したCuI/IIの酸化還元に由来するものと考えられる(図10)。還元波のピーク電気量(Qpc)は12μC cm-2であった。
【0162】
[CuII(cis-L1)]/C6-Auの酸化還元応答もまた掃引を繰り返すことでピークが減少していくが、trans-体の場合とは異なり15サイクル目付近からピーク強度がほぼ一定になった(図11)。そこで、[CuII(trans-L1)]/C6-Au, [CuII(cis-L1)]/C6-Auの還元波のピーク電気量を1サイクル目の電気量を基準として規格化し、それぞれサイクル数に対してプロットした(図12)。trans-体の場合ではサイクル数の増加に伴いピーク電気量は減少していき、最終的にはほぼピークが消失した。一方、cis-体の場合では10サイクル目付近まではtrans-体と同様にピーク電気量が減少したが、それ以降ではピーク電気量がほぼ一定になった(約6.6μC cm-2, 図12)。[CuII(cis-L1)]/C6-Auはcis-, trans-体の両異性体が修飾されており、前述のようにtrans-体は酸化還元を繰り返すことでCuイオンが脱離する。したがって、10サイクル目あたりまでのピーク電気量の減少はtrans-L1部位からのCuイオンの脱離と考えられ、それ以降の一定のピーク電気量はcis-L1部位からCuイオンが脱離しないことを示している(図13)。すなわち、溶液中とは異なり、固相基板に結合したcis-L1及びtrans-L1のCuIIイオンとの結合能力には明確な差が存在し、cis-体の場合は掃引によってほとんど脱離しないことがわかった。
【0163】
次に、固相基板表面上でのcis-体からtrans-体への光異性化を試みた。[CuII(cis-L1)]/C6-Auを75サイクル掃引した後、すぐに溶液を交換することでcis-L1のみにCuIIイオンが結合した電極を作製した。この電極を0.1 M NaClO4水溶液に浸し、金表面に可視光を90分間照射した。照射後すぐに溶液を新しい溶液に交換してCV測定を行ったところ、可視光照射前のものに比べてCuI/II由来の酸化還元応答が減少していた(図14)。同様に可視光180分照射後、270分照射後のボルタモグラムにおいてもピークの減少が観測され、270分照射後にはCuI/II由来の酸化還元応答はほぼ消失した (図14)。ここで、全てのボルタモグラムについて掃引に伴うピークの減少は観測されなかったことから、得られたピークはCuII(cis-L1)に由来するものと考えられる。したがって、比較的結合が強いCuII(cis-L1)部位は、可視光照射によってCuIIイオンとの結合が弱いCuII(trans-L1)へと異性化し、酸化還元反応を繰り返したことによりCuIIイオンは溶液中へと遊離し、溶液交換により完全に系外に除去されたと考えられる(図15)。この結果から、固相基板に結合した状態においてもアゾ化合物がcis-体からtrans-体へと光異性化できることがわかった。またさらに、当該光異性化により、金属イオンとの結合強度を制御できることもわかった。
【特許請求の範囲】
【請求項1】
下記一般式(1)で表されるアゾ化合物。
【化1】
(式中、
R1aは、直鎖状のC1〜C10アルキレン基を示し、
R1b及びR1cは、同一又は異なって、直鎖状のC1〜C6アルキレン基を示す。
R2aは、直鎖状のC1〜C10アルキレン基を示し、
R2bは、直鎖状のC1〜C6アルキレン基を示す。
Xはリンカー部位を示し、Yは固相基板に対して結合性を有する固相基板結合部位を示す。)
【請求項2】
前記リンカー部位がアルキレン鎖中に−NHCO−を有するアルキレン基であり、
前記構造基板結合部位がチオール基、ジスルフィド基、スルフィド基、ジチオラン基又はシロキシ基である、請求項1に記載のアゾ化合物。
【請求項3】
請求項1又は2に記載のアゾ化合物を前記固相基板結合部位により固相基板に結合させたアゾ化合物結合固相基板。
【請求項4】
前記固相基板が、表面を金でコーティングされた固相基板である、請求項3に記載のアゾ化合物結合固相基板。
【請求項5】
(i)金属イオン含有溶液に、請求項3又は4に記載のアゾ化合物結合固相基板を浸積してアゾ化合物に金属イオンを結合させる工程、
(ii)当該アゾ化合物結合固相基板のアゾ化合物に可視光を照射する工程、
(iii)当該アゾ化合物結合固相基板を作用電極として金属イオンの酸化還元反応を繰り返して、アゾ化合物に結合した金属イオンを脱離させる工程、
を含む金属イオン回収方法。
【請求項6】
前記酸化還元反応が、前記金属イオンの酸化還元電位より300mV低い電位から、当該酸化還元電位より300mV高い電位の幅で30回以上繰り返される、
請求項5に記載の金属イオン回収方法。
【請求項7】
請求項5又は6に記載の工程(i)〜(iii)を含む工程を繰り返し行うことにより、金属イオンを回収する方法。
【請求項1】
下記一般式(1)で表されるアゾ化合物。
【化1】
(式中、
R1aは、直鎖状のC1〜C10アルキレン基を示し、
R1b及びR1cは、同一又は異なって、直鎖状のC1〜C6アルキレン基を示す。
R2aは、直鎖状のC1〜C10アルキレン基を示し、
R2bは、直鎖状のC1〜C6アルキレン基を示す。
Xはリンカー部位を示し、Yは固相基板に対して結合性を有する固相基板結合部位を示す。)
【請求項2】
前記リンカー部位がアルキレン鎖中に−NHCO−を有するアルキレン基であり、
前記構造基板結合部位がチオール基、ジスルフィド基、スルフィド基、ジチオラン基又はシロキシ基である、請求項1に記載のアゾ化合物。
【請求項3】
請求項1又は2に記載のアゾ化合物を前記固相基板結合部位により固相基板に結合させたアゾ化合物結合固相基板。
【請求項4】
前記固相基板が、表面を金でコーティングされた固相基板である、請求項3に記載のアゾ化合物結合固相基板。
【請求項5】
(i)金属イオン含有溶液に、請求項3又は4に記載のアゾ化合物結合固相基板を浸積してアゾ化合物に金属イオンを結合させる工程、
(ii)当該アゾ化合物結合固相基板のアゾ化合物に可視光を照射する工程、
(iii)当該アゾ化合物結合固相基板を作用電極として金属イオンの酸化還元反応を繰り返して、アゾ化合物に結合した金属イオンを脱離させる工程、
を含む金属イオン回収方法。
【請求項6】
前記酸化還元反応が、前記金属イオンの酸化還元電位より300mV低い電位から、当該酸化還元電位より300mV高い電位の幅で30回以上繰り返される、
請求項5に記載の金属イオン回収方法。
【請求項7】
請求項5又は6に記載の工程(i)〜(iii)を含む工程を繰り返し行うことにより、金属イオンを回収する方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【公開番号】特開2010−202830(P2010−202830A)
【公開日】平成22年9月16日(2010.9.16)
【国際特許分類】
【出願番号】特願2009−52285(P2009−52285)
【出願日】平成21年3月5日(2009.3.5)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 1.発行者名 錯体化学会 刊行物名 第58回錯体化学討論会要旨集 発行年月日 平成20年9月5日 2.発行者名 国立大学法人奈良先端科学技術大学院大学 物質創成科学研究科 刊行物名 平成20年度修士論文要旨集 発行年月日 平成21年2月17日
【出願人】(504143441)国立大学法人 奈良先端科学技術大学院大学 (226)
【公開日】平成22年9月16日(2010.9.16)
【国際特許分類】
【出願日】平成21年3月5日(2009.3.5)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 1.発行者名 錯体化学会 刊行物名 第58回錯体化学討論会要旨集 発行年月日 平成20年9月5日 2.発行者名 国立大学法人奈良先端科学技術大学院大学 物質創成科学研究科 刊行物名 平成20年度修士論文要旨集 発行年月日 平成21年2月17日
【出願人】(504143441)国立大学法人 奈良先端科学技術大学院大学 (226)
[ Back to top ]