
ルテニウムを主成分とした触媒錯体、およびオレフィンメタセシスのための前記錯体の使用
本発明は、式(I)または(II)[式中、Lは中性配位子であり、X、X’は陰イオン配位子であり、R1およびR2はそれぞれ独立して、水素、C1〜C6アルキル、C1〜C6ペルハロゲン化アルキル、アルデヒド、ケトン、エステル、ニトリル、アリール、場合により置換されるC5〜C6アルキルピリジニウム、ペルハロゲン化アルキルピリジニウムもしくはシクロヘキシル、CnH2nYもしくはCnF2nY基10[式中、nは1〜6からなり、Yはイオンマーカーである]、または次式:の基であり、化合物が式(I)である場合は、R1が式(Ibis)の基であり得、あるいは化合物が式(II)である場合は、R1が式(IIbis)の基であり得、R3は、C1〜C6アルキルまたはC5〜C6シクロアルキルまたはC5〜C6アリールであり、R0、R4、R5、R6、R7、R8、R9、R10、R11はそれぞれ独立して、水素、C1〜C6アルキル、C1〜C6ペルハロゲン化アルキル、またはC5〜C6アリールであり、R9、R10、R11は複素環であり得、X1は陰イオンであり、R1およびR2は、それぞれが結合するNおよびCと一緒になって、複素環を形成し得る]のすべての化合物に関する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、活性ルテニウムおよび再利用可能ルテニウムを主成分とした新規触媒錯体、ならびに前記錯体の合成方法を対象とする。
【0002】
本発明はまた、オレフィンメタセシスのための前記触媒錯体の使用にも関する。
【0003】
再利用可能ルテニウムまたは活性ルテニウムを主成分とした触媒錯体の開発は、第2世代グラブス触媒と呼ばれる、米国カリフォルニア工科大学ロバート・グラブス教授のルテニウム錯体2a(予備触媒2b)に関する研究に基づいている。
【0004】
【化1】
【0005】
例えば、スチレニルエーテル配位子(「ブーメラン」と呼ばれる配位子)を有する最初の再利用可能錯体3a(予備触媒3b)については、米国ボストン大学ホベイダ教授が記載している。
【0006】
【化2】
【0007】
本化合物については、特に国際特許第0214376号に記載されている。
【0008】
この錯体の第1の利点は、反応終了時に再利用のために回収される予備触媒の再利用を可能にすることである。
【0009】
しかし、本触媒には、1サイクル毎に10%程度の大きな損失をもたらす欠点がある。
【0010】
本錯体の第2の利点は、反応生成物中に含まれる有毒金属残滓(ルテニウム)を最小限にすることである。
【0011】
しかし、本錯体は、上述のグラブス触媒2bよりも活性が低いことが明らかにされている。
【0012】
最初の活性錯体4は、上述のホベイダのスチレニルエーテル配位子を有する錯体にニトロ基(NO2)が存在することにより誘発される電子効果を基に2002年に記載されている。
【0013】
【化3】
【0014】
本活性錯体については、国際特許出願第2004035596号に記載されている。
【0015】
本予備触媒の活性化は、触媒サイクルの迅速な開始とそれによる反応速度の大幅な向上をもたらすスチレニルエーテル配位子の大幅に加速した離脱に基づいている。その際、反応は、より柔軟な条件で(実際には室温で)より少ない触媒充填量で行われ得る。
【0016】
しかし、本錯体は簡単には再利用されず、しかも反応生成物中の有毒金属残滓(ルテニウム)による重大な汚染をもたらす。前記欠点は、特に医薬分子のような高付加価値を有する特定の生成物を合成するに当たって厄介となる。
【0017】
従って、従来技術によれば、実際には再利用を損ねて活性の増大が図られ、逆に触媒種の反応性を損ねて再利用の増大が図られることから、前記ルテニウム錯体の反応性と再利用は2つの矛盾する特性であることが明らかになる。
【0018】
本発明の目的は、これらの矛盾する特性の中間を最適に調製し得る活性ルテニウム錯体と再利用可能ルテニウム錯体、すなわち適切な再利用を維持しつつ優れた活性を兼ね備え得る錯体について記載することである。
【0019】
従って、本発明の目的は、使用することで触媒充填量の削減を可能にし得る前記錯体を提案することである。前記目的は、これらの触媒の高い費用を考慮すると重要なことである。
【0020】
従って、本発明の目的は、再利用可能性の度合いにより最終生成物中の有毒金属廃棄物が大幅に削減されるような錯体を提案することでもある。
【0021】
最善の場合には、本発明による触媒によって、ルテニウム含有量が非常に少ない(実際には10〜20ppm未満の)生成物を得ることが可能となるであろう。
【0022】
本発明により達成されるこれらの目的は、式(I)または(II)
【化4】
[式中、
Lは中性配位子であり、
X、X’は陰イオン配位子であり、
R1およびR2はそれぞれ独立して、水素、C1〜C6アルキル、C1〜C6ペルハロゲン化アルキル、アルデヒド、ケトン、エステル、アミド、ニトリル、場合により置換されるアリール、場合により置換されるC5〜C6アルキルピリジニウム、ペルハロゲン化アルキルピリジニウムもしくはシクロヘキシル、CnH2nYもしくはCnF2nY基[式中、nは1〜6からなり、Yはイオンマーカーである]、または次式:
【化5】
の基であり、
化合物が式(I)である場合は、R1が式(Ibis)
【化6】
の基であり得、
あるいは化合物が式(II)である場合は、R1が式(IIbis)
【化7】
の基であり得、
R3は、C1〜C6アルキルまたはC5〜C6シクロアルキルまたはC5〜C6アリールであり、
R0、R4、R5、R6、R7、R8、R9、R10、R11はそれぞれ独立して、水素、C1〜C6アルキル、C1〜C6ペルハロゲン化アルキル、またはC5〜C6アリールであり、R9、R10、R11は複素環を形成し得、
X1は陰イオン、すなわちハロゲン、テトラフルオロホウ酸塩([BF4]-)、[テトラキス−(3,5−ビス−(トリフルオロメチル)−フェニル)ホウ酸塩]([BARF]-)、ヘキサフルオロリン酸塩([PF6]-)、ヘキサフルオロアンチモン([SbF6]-)、ヘキサフルオロ砒酸塩([AsF6])、トリフルオロメチルスルホン酸塩([CF3]2N)-)であり、
R1およびR2は、それぞれが結合するNおよびCと一緒になって、次式
【化8】
[式中、ハルはハロゲンであり、R12は、水素、C1〜C6アルキルもしくはC5〜C6シクロアルキル、またはC5〜C6アリールである]
の複素環を形成し得る]
のすべての化合物に関する。
【0023】
好ましくは、LはP(R13)3[式中、R13は、C1〜C6アルキルもしくはアリールまたはC5〜C6シクロアルキルである]である。
【0024】
また、好ましくは、Lは、式7a、7b、7c、7dまたは7e
【化9】
[式中、
n1は0、1、2、3であり、
R14、R15、R16、R17、R18、R19、R20、R21、R22、R23、R24、R25、R26、R27、R28はそれぞれ独立して、C1〜C6アルキル、C3〜C20シクロアルキル、C2〜C20アルケニル、ナフチル、アントラセンまたはフェニルであり、前記フェニルは、C1〜C6アルキル、C1〜C6アルコキシおよびハロゲンの中から選択される最高5つの群から置換され得、R16およびR17ならびにR26およびR27は、3員、4員、5員、6員、7員の環を形成し得、R28は独立して、連結した6員の芳香族環を形成し得る]
の配位子である。
【0025】
好都合には、LはPCy3であり、Cyはシクロヘキシルであるか、あるいはLは式7aまたは7bの配位子であり、
Xは塩素であり、
X’は塩素であり、
イオンマーカーYは、好ましくは
【化10】
からなる群から選択される。
【0026】
一変形例によれば、本発明による化合物は、式(I)[式中、R1はCH3、CF3,C6F5、pNO3C6H4からなる群から選択される]に対応する。
【0027】
一変形例によれば、R1はCF3である。
【0028】
一変形例によれば、前記化合物は、式1a
【化11】
に対応する。
【0029】
別の変形例によれば、前記化合物は、式1b
【化12】
に対応する。
【0030】
別の変形例によれば、前記化合物は、式1c
【化13】
に対応する。
【0031】
別の変形例によれば、前記化合物は、式1d
【化14】
に対応する。
【0032】
別の変形例によれば、前記化合物は、式1e
【化15】
に対応する。
【0033】
別の変形例によれば、前記化合物は、式1f
【化16】
に対応する。
【0034】
別の変形例によれば、前記化合物は、式1g
【化17】
に対応する。
【0035】
別の変形例によれば、前記化合物は、式1h
【化18】
に対応する。
【0036】
別の変形例によれば、前記化合物は、式1i
【化19】
に対応する。
【0037】
別の変形例によれば、前記化合物は、式1j
【化20】
に対応する。
【0038】
別の変形例によれば、前記化合物は、式1k
【化21】
に対応する。
【0039】
別の変形例によれば、前記化合物は、式11
【化22】
に対応する。
【0040】
別の変形例によれば、前記化合物は、式12
【化23】
に対応する。
【0041】
別の変形例によれば、前記化合物は、式13
【化24】
に対応する。
【0042】
別の変形例によれば、前記化合物は、式14
【化25】
に対応する。
【0043】
本発明はまた、式(I)の化合物のすべての合成方法であって、4−イソプロポキシ−3−ビニルアニリンとアシル基を有する化合物とを反応させてアミド配位子を得る第1段階と、前記アミド配位子を式(III)
【化26】
の化合物と反応させる第2段階とからなることを特徴とする方法にも関する。
【0044】
好ましくは、式(III)の前記化合物は、グラブス予備触媒(2b)またはノラン予備触媒(2c)である。
【0045】
【化27】
【0046】
本発明に基づくスチレニルエーテル配位子へのアミド官能基の導入は、触媒の活性化を促進する特殊性を有する。
【0047】
特に、アミド官能基がペルフッ素化メチル(トリフルオロメチル)を有する場合は、きわめて短い間に比較的高い転化率を特徴とする触媒の高活性化が認められる。こうした条件では、効率を損なうことなくメタセシス反応における触媒充填量を大幅に削減することで、経済的な効果が考えられる。
【0048】
さらに、前記アミド官能基は、水相および/またはイオン相ならびに固体支持体上への固定化のためのイオンマーカー(「タグ」)の導入においてスペーサーの役割を果たし得る。
【0049】
このようなイオンマーカーにより、水−イオン混合溶媒中または固体支持体上(連続した流れ反応)における触媒錯体のより良好な再利用を可能にし、特に医薬分子の合成方法の範囲内において、高付加価値生成物による汚染を回避し、反応費用の大幅な削減を図ることが可能となる。
【0050】
図面を参照して示した本発明の種々の実施例に関する以下の説明により、本発明ならびに本発明が有する種々の利点がより容易に理解されるであろう。
【図面の簡単な説明】
【0051】
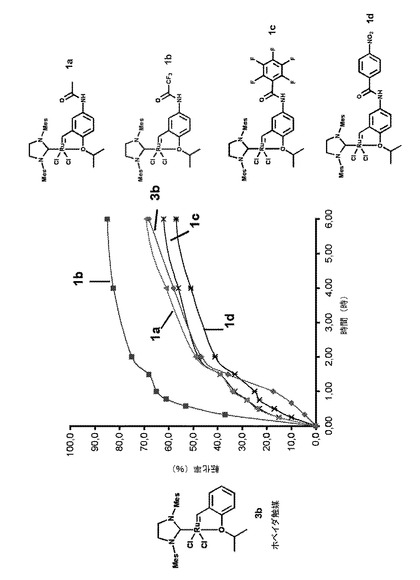
【図1】1モル%のホベイダ錯体3bならびに本発明による触媒錯体1a、1b、1c、1dの存在下において室温で環化したメタセシス反応における、メタリル−アリルマロン酸ジエチル化合物の反応時間に対する転化率を示すグラフである。
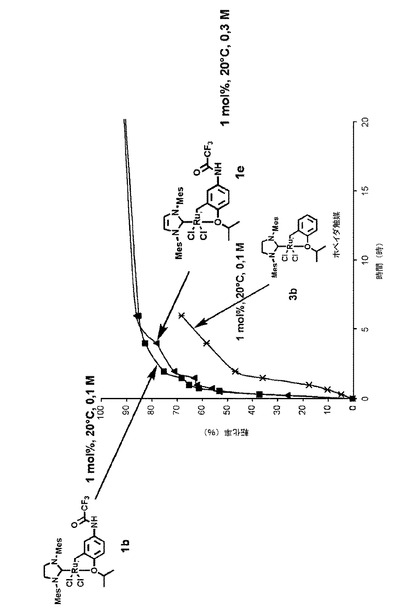
【図2】1モル%のホベイダ錯体3bならびに本発明による触媒錯体1b、1eの存在下において室温で環化するメタセシス反応における、メタリル−アリルマロン酸ジエチル化合物の反応時間に対する転化率を示すグラフである。
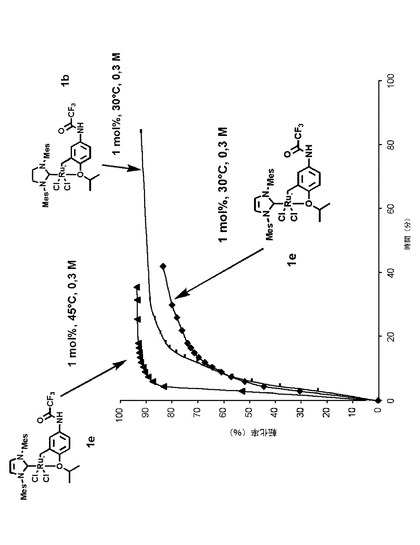
【図3】1モル%の本発明による触媒錯体1eの存在下において45℃で環化するメタセシス反応における、メタリル−アリルマロン酸ジエチル化合物の反応時間に対する転化率を示すグラフである。
【図4】1モル%の本発明による触媒錯体1bと0.3モル%の本発明による触媒錯体1bの存在下において30℃で環化するメタセシス反応における、メタリル−アリルマロン酸ジエチル化合物の反応時間に対する転化率を示すグラフである。
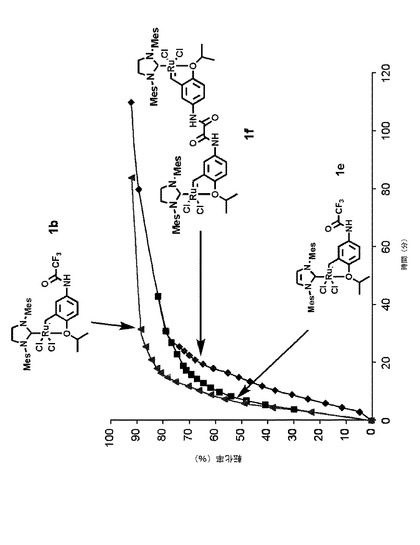
【図5】1モル%の本発明による触媒錯体1b、1eおよび1fの存在下において30℃で環化するメタセシス反応における、メタリル−アリルマロン酸ジエチル化合物の反応時間に対する転化率を示すグラフである。
【図6】ルテニウム錯体1aの実施例のNMRスペクトルを示す。
【図7】ルテニウム錯体1bの実施例のNMRスペクトルを示す。
【図8】ルテニウム錯体1cの実施例のNMRスペクトルを示す。
【図9】ルテニウム錯体1dの実施例のNMRスペクトルを示す。
【図10】ルテニウム錯体1eの実施例のNMRスペクトルを示す。
【図11】ルテニウム錯体1fの実施例のNMRスペクトルを示す。
【0052】
最初に、本発明による種々の錯体例の合成について以下に説明する。
【0053】
本発明による錯体1a、1b、1c、1d、1eおよび1fは、官能化したアニリン5から2段階で得られる。
【0054】
この官能化したアニリン5をパラニトロフェノールから合成する4段階の方法については、論文"Activated pyridinium−tagged ruthenium complex as efficient catalyst for Ring−Closing Metathesis."D.Rix,H.Clavier,Y.Coutard,L.Gulajski,K.Grela*,M.Mauduit*,J.Organomet.Chem.,2006,691,5397−5405に記載されている。
【0055】
次のスキームは、この2段階合成を端的に示している。
【0056】
【化28】
【0057】
第1段階:4−イソプロポキシ−3−ビニルアニリン5からのアミド6a、6b、6c、6d、6f、9a、9b、10aおよび10bの合成
一般的な方法に従って、4−イソプロポキシ−3−ビニルアニリン5(1当量、約0.2mmol)を、窒素雰囲気にした丸底フラスコに投入し、無水ジクロロメタン(2〜3mL)に溶解した。ピリジン(1.5当量)を溶液に添加し、0℃に冷却した。次いで、塩化アシルまたは無水物(1.2当量)を緩徐に添加した後、反応媒質を窒素雰囲気下にて2時間室温で攪拌した。
【0058】
次いで、原料をジクロロメタン(10mL)で希釈し、塩酸水溶液1N(2mL)、次に炭酸水素ナトリウム飽和溶液(2×2mL)、最後に塩化ナトリウム飽和溶液(3×2mL)で洗浄した。有機相を回収し、硫酸マグネシウムで乾燥させ、真空濃縮した。
【0059】
残渣物をクロマトグラフィーによりシリカゲル上で浄化した。
【0060】
N−(4−イソプロポキシ−3−ビニルフェニル)アセトアミド化合物6aの合成
【化29】
【0061】
塩化アセチル(15μL)を用いて4−イソプロポキシ−3−ビニルアニリン5(50mg、0.3mmol)からアミドを得る一般的な方法により、クロマトグラフィー後にはシリカゲル(溶離用:CH2Cl2/AcOEt(4:1))上に淡紅色の固体(49mg、78%)状のアセトアミドが得られた。
【0062】
【0063】
N−(4−イソプロポキシ−3−ビニルフェニル)トリフルオロアセトアミド化合物6bの合成
【化30】
【0064】
無水トリフルオロ酢酸(25μL)を用いて4−イソプロポキシ−3−ビニルアニリン5(26mg、0.14mmol)からアミドを得る一般的な方法により、クロマトグラフィー後にはシリカゲル(溶離用:CH2Cl2/EP(9:1))上に黄色がかった固体(23mg、59%)状のトリフルオロアセトアミドが得られた。
【0065】
【0066】
N−(4−イソプロポキシ−3−ビニルフェニル)ペンタフルオロベンズアミド化合物6cの合成
【化31】
【0067】
塩化ペンタフルオロベンゾイル(38μL)を用いて4−イソプロポキシ−3−ビニルアニリン5(39mg、0.22mmol)からアミドを得る一般的な方法により、クロマトグラフィー後にはシリカゲル(溶離用:CH2Cl2/EP(9:1))上に淡紅色の固体(75mg、92%)状のペンタフルオロベンズアミドが得られた。
【0068】
【0069】
N−(4−イソプロポキシ−3−ビニルフェニル)パラニトロベンズアミド化合物6dの合成
【化32】
【0070】
塩化パラニトロベンゾイル(48mg)を用いて4−イソプロポキシ−3−ビニルアニリン5(38mg、0.22mmol)からアミドを得る一般的な方法により、クロマトグラフィー後にはシリカゲル(溶離用:CH2Cl2)上に黄色のオイル(67mg、96%)状のパラニトロベンズアミドが得られた。
【0071】
【0072】
N,N’−ビス(4−イソプロポキシ−3−ビニルフェニル)オキサミド化合物6fの合成
【化33】
【0073】
4−イソプロポキシ−3−ビニルアニリン5(30mg、1当量、0.2mmol)を、窒素雰囲気にした丸底フラスコに投入し、無水ジクロロメタン(3mL)に溶解した。ピリジン(21μL、1.5当量)を溶液に添加し、0℃に冷却した。次いで、塩化オキサリル(8.8μL、1.2当量)を緩徐に添加した後、反応媒質を窒素雰囲気下にて2時間室温で攪拌した。
【0074】
次いで、原料をジクロロメタン(10mL)で希釈し、塩酸水溶液1N(2mL)、次に炭酸水素ナトリウム飽和溶液(2×2mL)、最後に塩化ナトリウム飽和溶液(3×2mL)で洗浄した。有機相を回収し、硫酸マグネシウムで乾燥させ、真空濃縮した。
【0075】
残渣物をクロマトグラフィーによりシリカゲル(溶離用:CH2Cl2/EP(9:1))上で浄化し、白色の固体(14mg、20%)状の化合物6fを生じた。
【0076】
【0077】
N−(4−イソプロポキシ−3−ビニルフェニル)ジフルオロクロロアセトアミド化合物9aの合成
【化34】
【0078】
無水2−クロロ−2,2−ジフルオロエタン(63μL)を用いて4−イソプロポキシ−3−ビニルアニリン5(50mg、0.3mmol)からアミドを得る一般的な方法により、クロマトグラフィー後にはシリカゲル(溶離用:CH2Cl2/AcOEt(4:1))上に淡紅色の固体(65mg、75%)状のアセトアミドが得られた。
【0079】
【0080】
3−{1,1−ジフルオロ−2−[4−イソプロポキシ−3−ビニルフェニルアミノ]−2−オキソエチル}−1−メチル−1H−イミダゾール−3−イウム化合物9cの合成
【化35】
【0081】
塩化アミド9a(20mg、0.07mmol)を無水トルエン(2.5mL)に溶解した。N−メチルイミダゾール(1mL、20当量)を溶液に添加した後、一晩還流した。次いで、揮発相を減圧除去し、濃橙色のオイル状の標識化合物を回収した。
【0082】
【0083】
3−クロロ−2,2,3,3−テトラフルオロ−N−(4−イソプロポキシ−3−ビニルフェニル)プロパンアミド化合物9bの合成
【化36】
【0084】
塩化3−クロロ−2,2,3,3−テトラフルオロプロパノイル(81mg)を用いて4−イソプロポキシ−3−ビニルアニリン5(50mg、0.3mmol)からアミドを得る一般的な方法により、クロマトグラフィー後にはシリカゲル(溶離用:CH2Cl2/AcOEt(4:1))上に白色の固体(65mg、57%)状のアセトアミドが得られた。
【0085】
【0086】
第2段階:アミド6a、6b、6c、6d、6f、10bからのルテニウム錯体1a、1b、1c、1d、1e、1f、11、12の合成
一般的な方法に従って、アミド配位子(1当量)、塩化銅(I)(1当量)およびインデニリデン予備触媒(1当量)をアルゴン雰囲気下にて丸底フラスコに投入し、そこに無水ジクロロメタン(2〜3mL)を添加した。次いで、アルゴン雰囲気下にて、30〜33℃にした反応媒質から3回脱気を行い、攪拌しながら約5時間保持した。
【0087】
次いで、反応原料を真空濃縮した。残渣物をアセトン(1〜2mL)に戻して、セライトで濾過した。濾過液を真空濃縮して、残渣物をクロマトグラフィーによりシリカゲル上で浄化した。
ルテニウム錯体1aの合成
【化37】
【0088】
N−(4−イソプロポキシ−3−ビニルフェニル)アセトアミド6a(24mg、0.011mmol)を用いてルテニウム錯体を得る一般的な方法により、クロマトグラフィー後にはシリカゲル(溶離用:EP/アセトン(1:1))上に緑色の固体(73mg、98%)状の錯体1aが得られた。
【0089】
【0090】
ルテニウム錯体1bの合成
【化38】
【0091】
N−(4−イソプロポキシ−3−ビニルフェニル)トリフルオロアセトアミド6b(11.7mg、0.04mmol)を用いてルテニウム錯体を得る一般的な方法により、クロマトグラフィー後にはシリカゲル(溶離用:EP/アセトン(7:3))上に緑色の固体(26.1mg、88%)状の錯体1bが得られた。
【0092】
【0093】
ルテニウム錯体1cの合成
【化39】
【0094】
N−(4−イソプロポキシ−3−ビニルフェニル)ペンタフルオロベンズアミド6c(9mg、0.02mmol)を用いてルテニウム錯体を得る一般的な方法により、クロマトグラフィー後にはシリカゲル(溶離用:EP/アセトン(7:3))上に緑色の固体(10mg、50%)状の錯体1cが得られた。
【0095】
【0096】
ルテニウム錯体1dの合成
【化40】
【0097】
N−(4−イソプロポキシ−3−ビニルフェニル)パラニトロベンズアミド6d(8mg、0.02mmol)を用いてルテニウム錯体を得る一般的な方法により、クロマトグラフィー後にはシリカゲル(溶離用:EP/アセトン(6:4))上に緑色の固体(18mg、95%)状の錯体1dが得られた。
【0098】
【0099】
ルテニウム錯体1eの合成
【化41】
【0100】
N−(4−イソプロポキシ−3−ビニルフェニル)トリフルオロアセトアミド配位子6b(22mg、0.08mmol、1当量)、塩化銅(I)(8mg、1当量)、および式で表される第2世代ノラン予備触媒2c(68mg、1当量)をアルゴン雰囲気下にて丸底フラスコに投入し、そこに無水ジクロロメタン(3mL)を添加した。次いで、アルゴン雰囲気下にて30〜33℃にした反応媒質から3回脱気を行い、攪拌しながら約5時間保持した。
【0101】
次いで、反応原料を真空濃縮した。残渣物をアセトン(1〜2mL)に戻して、セライトで濾過した。濾過液を真空濃縮して、残渣物をクロマトグラフィーによりシリカゲル上で浄化した。
【0102】
クロマトグラフィー後にはシリカゲル(溶離用:EP/アセトン(4:1))上に緑色の固体(52mg、88%)状の錯体1eが得られた。
【0103】
【0104】
ルテニウム錯体1fの合成
【化42】
【0105】
N,N’−ビス(4−イソプロポキシ−3−ビニルフェニル)オキサミド配位子6f(8mg、0.02mmol、1当量)、塩化銅(I)(4mg、2.1当量)およびインデニリデン予備触媒(37mg、2.1当量)をアルゴン雰囲気下にて丸底フラスコに投入し、そこに無水ジクロロメタン(5mL)を添加した。次いで、アルゴン雰囲気下にて30〜33℃にした反応媒質から3回脱気を行い、攪拌しながら約5時間保持した。
【0106】
次いで、反応原料を真空濃縮した。残渣物をアセトン(2mL)に戻して、焼結濾過を行った。このようにして緑色の固体(15mg、59%)状の錯体1fを単離した。
【0107】
【0108】
標識ルテニウム錯体11の合成
トリフルロアセトアミド官能基を予備触媒の活性化により適した基として明確に特定されれば、イオンモチーフ(イオンタグ)の導入が可能となる。
【0109】
このため、本発明では、化合物10a中の塩素原子を第3アミン(イミダゾール、ピリジンなど)に置換することを提案する。
【0110】
従って、本発明者等は、所期のイオン配位子10bを容易に生じるために、4−クロロ−N−(4−イソプロポキシ−3−ビニルフェニル)ブタンアミド10a上でピリジンの置換を行った。第2世代グラブス触媒を用いたピリジンの錯体化により、錯体11を生じた。
【0111】
【化43】
【0112】
4−クロロ−N−(4−イソプロポキシ−3−ビニルフェニル)ブタンアミド化合物10aの合成
【化44】
【0113】
塩化3−クロロプロパノイル(15μL)を用いて4−イソプロポキシ−3−ビニルアニリン5(50mg、0.3mmol)からアミドを得る一般的な方法により、クロマトグラフィー後にはシリカゲル(溶離用:CH2Cl2)上に淡紅色の固体(52mg、65%)状のアセトアミドが得られた。
【0114】
【0115】
1−(4−(4−イソプロポキシ−3−ビニルフェニルアミノ)−4−オキソブチル) ヘキサフルオロリン酸ピリジニウム化合物(V)10bの合成
【化45】
【0116】
無水トルエン中のアセトアミド溶液10a(52mg、0.19mmol)にピリジン(1mL)を添加した後、混合物を攪拌しながら2日間還流した。溶媒を気化させた後、残渣物を水に溶かして、KPF6(38mg)を添加した。室温で2時間攪拌した後、ジクロロメタンから水相を抽出し、次いでNaCl飽和溶液を用いて有機相を洗浄し、硫酸マグネシウムで乾燥させた。溶媒を気化させた後、クロマトグラフィーによりシリカゲル(溶離用:CH2Cl2/MeOH(4:1))上で無定形の固体(38mg、44%)状のピリジニウム塩を浄化した。
【0117】
【0118】
ルテニウム錯体11の合成
【化46】
【0119】
1−(4−(4−イソプロポキシ−3−ビニルフェニルアミノ)−4−オキソブチル)ヘキサフルオロリン酸ピリジニウム配位子(V)10b(5mg、0.011mmol、1当量)、塩化銅(I)(2mg、1当量)およびインデニリデン予備触媒(9.6mg、1当量)をアルゴン雰囲気下にて丸底フラスコに投入し、そこに無水ジクロロメタン(3mL)を添加した。次いで、アルゴン雰囲気下にて30〜33℃にした反応媒質から3回脱気を行い、攪拌しながら約5時間保持した。
【0120】
次いで、反応原料を真空濃縮した。残渣物をアセトン(2mL)に戻して、焼結濾過を行った。このようにして深緑色無定形の固体状の錯体11を単離した。
【0121】
【0122】
次に、活性ルテニウム錯体1a、1b、1c、1d、1eおよび1fの活性化について検討した。
【0123】
以下の反応スキームに従い、1モル%の錯体の存在下においてジクロロメタン中でメタリル−アリルマロン酸ジエチルと室温で環化するオレフィンメタセシス反応にて、本発明による錯体1a、1b、1c、1dとホベイダの従来技術による錯体3bを検討した。
【0124】
【化47】
【0125】
これらの化合物を用いて得られた転化率の結果は、図1のグラフに示す。
【0126】
これらの結果は、アセトアミド官能基の活性化効果を明確に示している。
【0127】
特に、このアセトアミド官能基がトリフルオロメチル基(錯体1b)を有する場合は、ホベイダ錯体3bの場合に5%であったのに対して、反応開始からわずか15分後に37%以上の転化率が得られた。
【0128】
化合物1e(ノラン触媒2cを用いた錯体化から生成)と化合物1b(第2世代グラブス触媒2bを用いた錯体化から生成)の活性、ならびにホベイダの従来技術による錯体3bの活性を、同じ反応と同じ反応条件で検討した。
【0129】
これらの化合物を用いて得られた転化率の結果は、図2のグラフに示す。
【0130】
非常に驚くことに、これらの結果では、触媒1eおよび1bについては同様の活性が示されているが、第2世代グラブス錯体2b(SIMes配位子を有する)はノラン錯体2c(IMes配位子を有する)よりも活性がきわめて高かった。この結果は非常に興味深い。なぜなら、IMes配位子を有する触媒種(ノラン錯体2cから生成)は、SIMes配位子を有する触媒種(第2世代グラブス錯体2bから生成)よりも熱安定性がきわめ高いためである。
【0131】
このため、本発明は、基質が非常に密集している場合(例えば、テトラ置換オレフィン)に、より厳しい条件(より高温の暖房)下で活性錯体1eを用いてオレフィンメタセシス反応を実現する可能性をもたらす。従って、メタリル−アリルマロン酸ジエチル化合物を環化するメタセシス反応は、1モル%の触媒錯体1eの存在下において45℃で、ならびに1モル%の触媒錯体1bおよび1eの存在下において30℃で実現される。これらの化合物を用いて得られた転化率の結果は、図3のグラフに示す。予想通り、IMes活性触媒1eは、反応開始からわずか6分後に87%の転化率を達成した注目すべき活性を示している。
【0132】
メタリル−アリルマロン酸ジエチル化合物を環化するメタセシス反応では、触媒充填量を削減する活性錯体1bの活性についても評価した。図4は、1モル%の本発明による触媒錯体1bと0.3モル%の本発明による触媒錯体1bの存在下において30℃で環化するメタセシス反応における、メタリル−アリルマロン酸ジエチル化合物の反応時間に対する転化率を示すグラフである。このグラフでは反応性のわずかな低下が示されているが、反応開始からわずか40分後に75%の転化率が認められていることから、この低下も依然として注目に値する。
【0133】
最後に、二量体活性錯体1fについても評価し、その活性を活性錯体1bおよび1eと比較した。図5は、1モル%の本発明による触媒錯体1b、1eおよび1fの存在下において30℃で環化するメタセシス反応における、メタリル−アリルマロン酸ジエチル化合物の反応時間に対する転化率を示すグラフである。
【技術分野】
【0001】
本発明は、活性ルテニウムおよび再利用可能ルテニウムを主成分とした新規触媒錯体、ならびに前記錯体の合成方法を対象とする。
【0002】
本発明はまた、オレフィンメタセシスのための前記触媒錯体の使用にも関する。
【0003】
再利用可能ルテニウムまたは活性ルテニウムを主成分とした触媒錯体の開発は、第2世代グラブス触媒と呼ばれる、米国カリフォルニア工科大学ロバート・グラブス教授のルテニウム錯体2a(予備触媒2b)に関する研究に基づいている。
【0004】
【化1】
【0005】
例えば、スチレニルエーテル配位子(「ブーメラン」と呼ばれる配位子)を有する最初の再利用可能錯体3a(予備触媒3b)については、米国ボストン大学ホベイダ教授が記載している。
【0006】
【化2】
【0007】
本化合物については、特に国際特許第0214376号に記載されている。
【0008】
この錯体の第1の利点は、反応終了時に再利用のために回収される予備触媒の再利用を可能にすることである。
【0009】
しかし、本触媒には、1サイクル毎に10%程度の大きな損失をもたらす欠点がある。
【0010】
本錯体の第2の利点は、反応生成物中に含まれる有毒金属残滓(ルテニウム)を最小限にすることである。
【0011】
しかし、本錯体は、上述のグラブス触媒2bよりも活性が低いことが明らかにされている。
【0012】
最初の活性錯体4は、上述のホベイダのスチレニルエーテル配位子を有する錯体にニトロ基(NO2)が存在することにより誘発される電子効果を基に2002年に記載されている。
【0013】
【化3】
【0014】
本活性錯体については、国際特許出願第2004035596号に記載されている。
【0015】
本予備触媒の活性化は、触媒サイクルの迅速な開始とそれによる反応速度の大幅な向上をもたらすスチレニルエーテル配位子の大幅に加速した離脱に基づいている。その際、反応は、より柔軟な条件で(実際には室温で)より少ない触媒充填量で行われ得る。
【0016】
しかし、本錯体は簡単には再利用されず、しかも反応生成物中の有毒金属残滓(ルテニウム)による重大な汚染をもたらす。前記欠点は、特に医薬分子のような高付加価値を有する特定の生成物を合成するに当たって厄介となる。
【0017】
従って、従来技術によれば、実際には再利用を損ねて活性の増大が図られ、逆に触媒種の反応性を損ねて再利用の増大が図られることから、前記ルテニウム錯体の反応性と再利用は2つの矛盾する特性であることが明らかになる。
【0018】
本発明の目的は、これらの矛盾する特性の中間を最適に調製し得る活性ルテニウム錯体と再利用可能ルテニウム錯体、すなわち適切な再利用を維持しつつ優れた活性を兼ね備え得る錯体について記載することである。
【0019】
従って、本発明の目的は、使用することで触媒充填量の削減を可能にし得る前記錯体を提案することである。前記目的は、これらの触媒の高い費用を考慮すると重要なことである。
【0020】
従って、本発明の目的は、再利用可能性の度合いにより最終生成物中の有毒金属廃棄物が大幅に削減されるような錯体を提案することでもある。
【0021】
最善の場合には、本発明による触媒によって、ルテニウム含有量が非常に少ない(実際には10〜20ppm未満の)生成物を得ることが可能となるであろう。
【0022】
本発明により達成されるこれらの目的は、式(I)または(II)
【化4】
[式中、
Lは中性配位子であり、
X、X’は陰イオン配位子であり、
R1およびR2はそれぞれ独立して、水素、C1〜C6アルキル、C1〜C6ペルハロゲン化アルキル、アルデヒド、ケトン、エステル、アミド、ニトリル、場合により置換されるアリール、場合により置換されるC5〜C6アルキルピリジニウム、ペルハロゲン化アルキルピリジニウムもしくはシクロヘキシル、CnH2nYもしくはCnF2nY基[式中、nは1〜6からなり、Yはイオンマーカーである]、または次式:
【化5】
の基であり、
化合物が式(I)である場合は、R1が式(Ibis)
【化6】
の基であり得、
あるいは化合物が式(II)である場合は、R1が式(IIbis)
【化7】
の基であり得、
R3は、C1〜C6アルキルまたはC5〜C6シクロアルキルまたはC5〜C6アリールであり、
R0、R4、R5、R6、R7、R8、R9、R10、R11はそれぞれ独立して、水素、C1〜C6アルキル、C1〜C6ペルハロゲン化アルキル、またはC5〜C6アリールであり、R9、R10、R11は複素環を形成し得、
X1は陰イオン、すなわちハロゲン、テトラフルオロホウ酸塩([BF4]-)、[テトラキス−(3,5−ビス−(トリフルオロメチル)−フェニル)ホウ酸塩]([BARF]-)、ヘキサフルオロリン酸塩([PF6]-)、ヘキサフルオロアンチモン([SbF6]-)、ヘキサフルオロ砒酸塩([AsF6])、トリフルオロメチルスルホン酸塩([CF3]2N)-)であり、
R1およびR2は、それぞれが結合するNおよびCと一緒になって、次式
【化8】
[式中、ハルはハロゲンであり、R12は、水素、C1〜C6アルキルもしくはC5〜C6シクロアルキル、またはC5〜C6アリールである]
の複素環を形成し得る]
のすべての化合物に関する。
【0023】
好ましくは、LはP(R13)3[式中、R13は、C1〜C6アルキルもしくはアリールまたはC5〜C6シクロアルキルである]である。
【0024】
また、好ましくは、Lは、式7a、7b、7c、7dまたは7e
【化9】
[式中、
n1は0、1、2、3であり、
R14、R15、R16、R17、R18、R19、R20、R21、R22、R23、R24、R25、R26、R27、R28はそれぞれ独立して、C1〜C6アルキル、C3〜C20シクロアルキル、C2〜C20アルケニル、ナフチル、アントラセンまたはフェニルであり、前記フェニルは、C1〜C6アルキル、C1〜C6アルコキシおよびハロゲンの中から選択される最高5つの群から置換され得、R16およびR17ならびにR26およびR27は、3員、4員、5員、6員、7員の環を形成し得、R28は独立して、連結した6員の芳香族環を形成し得る]
の配位子である。
【0025】
好都合には、LはPCy3であり、Cyはシクロヘキシルであるか、あるいはLは式7aまたは7bの配位子であり、
Xは塩素であり、
X’は塩素であり、
イオンマーカーYは、好ましくは
【化10】
からなる群から選択される。
【0026】
一変形例によれば、本発明による化合物は、式(I)[式中、R1はCH3、CF3,C6F5、pNO3C6H4からなる群から選択される]に対応する。
【0027】
一変形例によれば、R1はCF3である。
【0028】
一変形例によれば、前記化合物は、式1a
【化11】
に対応する。
【0029】
別の変形例によれば、前記化合物は、式1b
【化12】
に対応する。
【0030】
別の変形例によれば、前記化合物は、式1c
【化13】
に対応する。
【0031】
別の変形例によれば、前記化合物は、式1d
【化14】
に対応する。
【0032】
別の変形例によれば、前記化合物は、式1e
【化15】
に対応する。
【0033】
別の変形例によれば、前記化合物は、式1f
【化16】
に対応する。
【0034】
別の変形例によれば、前記化合物は、式1g
【化17】
に対応する。
【0035】
別の変形例によれば、前記化合物は、式1h
【化18】
に対応する。
【0036】
別の変形例によれば、前記化合物は、式1i
【化19】
に対応する。
【0037】
別の変形例によれば、前記化合物は、式1j
【化20】
に対応する。
【0038】
別の変形例によれば、前記化合物は、式1k
【化21】
に対応する。
【0039】
別の変形例によれば、前記化合物は、式11
【化22】
に対応する。
【0040】
別の変形例によれば、前記化合物は、式12
【化23】
に対応する。
【0041】
別の変形例によれば、前記化合物は、式13
【化24】
に対応する。
【0042】
別の変形例によれば、前記化合物は、式14
【化25】
に対応する。
【0043】
本発明はまた、式(I)の化合物のすべての合成方法であって、4−イソプロポキシ−3−ビニルアニリンとアシル基を有する化合物とを反応させてアミド配位子を得る第1段階と、前記アミド配位子を式(III)
【化26】
の化合物と反応させる第2段階とからなることを特徴とする方法にも関する。
【0044】
好ましくは、式(III)の前記化合物は、グラブス予備触媒(2b)またはノラン予備触媒(2c)である。
【0045】
【化27】
【0046】
本発明に基づくスチレニルエーテル配位子へのアミド官能基の導入は、触媒の活性化を促進する特殊性を有する。
【0047】
特に、アミド官能基がペルフッ素化メチル(トリフルオロメチル)を有する場合は、きわめて短い間に比較的高い転化率を特徴とする触媒の高活性化が認められる。こうした条件では、効率を損なうことなくメタセシス反応における触媒充填量を大幅に削減することで、経済的な効果が考えられる。
【0048】
さらに、前記アミド官能基は、水相および/またはイオン相ならびに固体支持体上への固定化のためのイオンマーカー(「タグ」)の導入においてスペーサーの役割を果たし得る。
【0049】
このようなイオンマーカーにより、水−イオン混合溶媒中または固体支持体上(連続した流れ反応)における触媒錯体のより良好な再利用を可能にし、特に医薬分子の合成方法の範囲内において、高付加価値生成物による汚染を回避し、反応費用の大幅な削減を図ることが可能となる。
【0050】
図面を参照して示した本発明の種々の実施例に関する以下の説明により、本発明ならびに本発明が有する種々の利点がより容易に理解されるであろう。
【図面の簡単な説明】
【0051】
【図1】1モル%のホベイダ錯体3bならびに本発明による触媒錯体1a、1b、1c、1dの存在下において室温で環化したメタセシス反応における、メタリル−アリルマロン酸ジエチル化合物の反応時間に対する転化率を示すグラフである。
【図2】1モル%のホベイダ錯体3bならびに本発明による触媒錯体1b、1eの存在下において室温で環化するメタセシス反応における、メタリル−アリルマロン酸ジエチル化合物の反応時間に対する転化率を示すグラフである。
【図3】1モル%の本発明による触媒錯体1eの存在下において45℃で環化するメタセシス反応における、メタリル−アリルマロン酸ジエチル化合物の反応時間に対する転化率を示すグラフである。
【図4】1モル%の本発明による触媒錯体1bと0.3モル%の本発明による触媒錯体1bの存在下において30℃で環化するメタセシス反応における、メタリル−アリルマロン酸ジエチル化合物の反応時間に対する転化率を示すグラフである。
【図5】1モル%の本発明による触媒錯体1b、1eおよび1fの存在下において30℃で環化するメタセシス反応における、メタリル−アリルマロン酸ジエチル化合物の反応時間に対する転化率を示すグラフである。
【図6】ルテニウム錯体1aの実施例のNMRスペクトルを示す。
【図7】ルテニウム錯体1bの実施例のNMRスペクトルを示す。
【図8】ルテニウム錯体1cの実施例のNMRスペクトルを示す。
【図9】ルテニウム錯体1dの実施例のNMRスペクトルを示す。
【図10】ルテニウム錯体1eの実施例のNMRスペクトルを示す。
【図11】ルテニウム錯体1fの実施例のNMRスペクトルを示す。
【0052】
最初に、本発明による種々の錯体例の合成について以下に説明する。
【0053】
本発明による錯体1a、1b、1c、1d、1eおよび1fは、官能化したアニリン5から2段階で得られる。
【0054】
この官能化したアニリン5をパラニトロフェノールから合成する4段階の方法については、論文"Activated pyridinium−tagged ruthenium complex as efficient catalyst for Ring−Closing Metathesis."D.Rix,H.Clavier,Y.Coutard,L.Gulajski,K.Grela*,M.Mauduit*,J.Organomet.Chem.,2006,691,5397−5405に記載されている。
【0055】
次のスキームは、この2段階合成を端的に示している。
【0056】
【化28】
【0057】
第1段階:4−イソプロポキシ−3−ビニルアニリン5からのアミド6a、6b、6c、6d、6f、9a、9b、10aおよび10bの合成
一般的な方法に従って、4−イソプロポキシ−3−ビニルアニリン5(1当量、約0.2mmol)を、窒素雰囲気にした丸底フラスコに投入し、無水ジクロロメタン(2〜3mL)に溶解した。ピリジン(1.5当量)を溶液に添加し、0℃に冷却した。次いで、塩化アシルまたは無水物(1.2当量)を緩徐に添加した後、反応媒質を窒素雰囲気下にて2時間室温で攪拌した。
【0058】
次いで、原料をジクロロメタン(10mL)で希釈し、塩酸水溶液1N(2mL)、次に炭酸水素ナトリウム飽和溶液(2×2mL)、最後に塩化ナトリウム飽和溶液(3×2mL)で洗浄した。有機相を回収し、硫酸マグネシウムで乾燥させ、真空濃縮した。
【0059】
残渣物をクロマトグラフィーによりシリカゲル上で浄化した。
【0060】
N−(4−イソプロポキシ−3−ビニルフェニル)アセトアミド化合物6aの合成
【化29】
【0061】
塩化アセチル(15μL)を用いて4−イソプロポキシ−3−ビニルアニリン5(50mg、0.3mmol)からアミドを得る一般的な方法により、クロマトグラフィー後にはシリカゲル(溶離用:CH2Cl2/AcOEt(4:1))上に淡紅色の固体(49mg、78%)状のアセトアミドが得られた。
【0062】
【0063】
N−(4−イソプロポキシ−3−ビニルフェニル)トリフルオロアセトアミド化合物6bの合成
【化30】
【0064】
無水トリフルオロ酢酸(25μL)を用いて4−イソプロポキシ−3−ビニルアニリン5(26mg、0.14mmol)からアミドを得る一般的な方法により、クロマトグラフィー後にはシリカゲル(溶離用:CH2Cl2/EP(9:1))上に黄色がかった固体(23mg、59%)状のトリフルオロアセトアミドが得られた。
【0065】
【0066】
N−(4−イソプロポキシ−3−ビニルフェニル)ペンタフルオロベンズアミド化合物6cの合成
【化31】
【0067】
塩化ペンタフルオロベンゾイル(38μL)を用いて4−イソプロポキシ−3−ビニルアニリン5(39mg、0.22mmol)からアミドを得る一般的な方法により、クロマトグラフィー後にはシリカゲル(溶離用:CH2Cl2/EP(9:1))上に淡紅色の固体(75mg、92%)状のペンタフルオロベンズアミドが得られた。
【0068】
【0069】
N−(4−イソプロポキシ−3−ビニルフェニル)パラニトロベンズアミド化合物6dの合成
【化32】
【0070】
塩化パラニトロベンゾイル(48mg)を用いて4−イソプロポキシ−3−ビニルアニリン5(38mg、0.22mmol)からアミドを得る一般的な方法により、クロマトグラフィー後にはシリカゲル(溶離用:CH2Cl2)上に黄色のオイル(67mg、96%)状のパラニトロベンズアミドが得られた。
【0071】
【0072】
N,N’−ビス(4−イソプロポキシ−3−ビニルフェニル)オキサミド化合物6fの合成
【化33】
【0073】
4−イソプロポキシ−3−ビニルアニリン5(30mg、1当量、0.2mmol)を、窒素雰囲気にした丸底フラスコに投入し、無水ジクロロメタン(3mL)に溶解した。ピリジン(21μL、1.5当量)を溶液に添加し、0℃に冷却した。次いで、塩化オキサリル(8.8μL、1.2当量)を緩徐に添加した後、反応媒質を窒素雰囲気下にて2時間室温で攪拌した。
【0074】
次いで、原料をジクロロメタン(10mL)で希釈し、塩酸水溶液1N(2mL)、次に炭酸水素ナトリウム飽和溶液(2×2mL)、最後に塩化ナトリウム飽和溶液(3×2mL)で洗浄した。有機相を回収し、硫酸マグネシウムで乾燥させ、真空濃縮した。
【0075】
残渣物をクロマトグラフィーによりシリカゲル(溶離用:CH2Cl2/EP(9:1))上で浄化し、白色の固体(14mg、20%)状の化合物6fを生じた。
【0076】
【0077】
N−(4−イソプロポキシ−3−ビニルフェニル)ジフルオロクロロアセトアミド化合物9aの合成
【化34】
【0078】
無水2−クロロ−2,2−ジフルオロエタン(63μL)を用いて4−イソプロポキシ−3−ビニルアニリン5(50mg、0.3mmol)からアミドを得る一般的な方法により、クロマトグラフィー後にはシリカゲル(溶離用:CH2Cl2/AcOEt(4:1))上に淡紅色の固体(65mg、75%)状のアセトアミドが得られた。
【0079】
【0080】
3−{1,1−ジフルオロ−2−[4−イソプロポキシ−3−ビニルフェニルアミノ]−2−オキソエチル}−1−メチル−1H−イミダゾール−3−イウム化合物9cの合成
【化35】
【0081】
塩化アミド9a(20mg、0.07mmol)を無水トルエン(2.5mL)に溶解した。N−メチルイミダゾール(1mL、20当量)を溶液に添加した後、一晩還流した。次いで、揮発相を減圧除去し、濃橙色のオイル状の標識化合物を回収した。
【0082】
【0083】
3−クロロ−2,2,3,3−テトラフルオロ−N−(4−イソプロポキシ−3−ビニルフェニル)プロパンアミド化合物9bの合成
【化36】
【0084】
塩化3−クロロ−2,2,3,3−テトラフルオロプロパノイル(81mg)を用いて4−イソプロポキシ−3−ビニルアニリン5(50mg、0.3mmol)からアミドを得る一般的な方法により、クロマトグラフィー後にはシリカゲル(溶離用:CH2Cl2/AcOEt(4:1))上に白色の固体(65mg、57%)状のアセトアミドが得られた。
【0085】
【0086】
第2段階:アミド6a、6b、6c、6d、6f、10bからのルテニウム錯体1a、1b、1c、1d、1e、1f、11、12の合成
一般的な方法に従って、アミド配位子(1当量)、塩化銅(I)(1当量)およびインデニリデン予備触媒(1当量)をアルゴン雰囲気下にて丸底フラスコに投入し、そこに無水ジクロロメタン(2〜3mL)を添加した。次いで、アルゴン雰囲気下にて、30〜33℃にした反応媒質から3回脱気を行い、攪拌しながら約5時間保持した。
【0087】
次いで、反応原料を真空濃縮した。残渣物をアセトン(1〜2mL)に戻して、セライトで濾過した。濾過液を真空濃縮して、残渣物をクロマトグラフィーによりシリカゲル上で浄化した。
ルテニウム錯体1aの合成
【化37】
【0088】
N−(4−イソプロポキシ−3−ビニルフェニル)アセトアミド6a(24mg、0.011mmol)を用いてルテニウム錯体を得る一般的な方法により、クロマトグラフィー後にはシリカゲル(溶離用:EP/アセトン(1:1))上に緑色の固体(73mg、98%)状の錯体1aが得られた。
【0089】
【0090】
ルテニウム錯体1bの合成
【化38】
【0091】
N−(4−イソプロポキシ−3−ビニルフェニル)トリフルオロアセトアミド6b(11.7mg、0.04mmol)を用いてルテニウム錯体を得る一般的な方法により、クロマトグラフィー後にはシリカゲル(溶離用:EP/アセトン(7:3))上に緑色の固体(26.1mg、88%)状の錯体1bが得られた。
【0092】
【0093】
ルテニウム錯体1cの合成
【化39】
【0094】
N−(4−イソプロポキシ−3−ビニルフェニル)ペンタフルオロベンズアミド6c(9mg、0.02mmol)を用いてルテニウム錯体を得る一般的な方法により、クロマトグラフィー後にはシリカゲル(溶離用:EP/アセトン(7:3))上に緑色の固体(10mg、50%)状の錯体1cが得られた。
【0095】
【0096】
ルテニウム錯体1dの合成
【化40】
【0097】
N−(4−イソプロポキシ−3−ビニルフェニル)パラニトロベンズアミド6d(8mg、0.02mmol)を用いてルテニウム錯体を得る一般的な方法により、クロマトグラフィー後にはシリカゲル(溶離用:EP/アセトン(6:4))上に緑色の固体(18mg、95%)状の錯体1dが得られた。
【0098】
【0099】
ルテニウム錯体1eの合成
【化41】
【0100】
N−(4−イソプロポキシ−3−ビニルフェニル)トリフルオロアセトアミド配位子6b(22mg、0.08mmol、1当量)、塩化銅(I)(8mg、1当量)、および式で表される第2世代ノラン予備触媒2c(68mg、1当量)をアルゴン雰囲気下にて丸底フラスコに投入し、そこに無水ジクロロメタン(3mL)を添加した。次いで、アルゴン雰囲気下にて30〜33℃にした反応媒質から3回脱気を行い、攪拌しながら約5時間保持した。
【0101】
次いで、反応原料を真空濃縮した。残渣物をアセトン(1〜2mL)に戻して、セライトで濾過した。濾過液を真空濃縮して、残渣物をクロマトグラフィーによりシリカゲル上で浄化した。
【0102】
クロマトグラフィー後にはシリカゲル(溶離用:EP/アセトン(4:1))上に緑色の固体(52mg、88%)状の錯体1eが得られた。
【0103】
【0104】
ルテニウム錯体1fの合成
【化42】
【0105】
N,N’−ビス(4−イソプロポキシ−3−ビニルフェニル)オキサミド配位子6f(8mg、0.02mmol、1当量)、塩化銅(I)(4mg、2.1当量)およびインデニリデン予備触媒(37mg、2.1当量)をアルゴン雰囲気下にて丸底フラスコに投入し、そこに無水ジクロロメタン(5mL)を添加した。次いで、アルゴン雰囲気下にて30〜33℃にした反応媒質から3回脱気を行い、攪拌しながら約5時間保持した。
【0106】
次いで、反応原料を真空濃縮した。残渣物をアセトン(2mL)に戻して、焼結濾過を行った。このようにして緑色の固体(15mg、59%)状の錯体1fを単離した。
【0107】
【0108】
標識ルテニウム錯体11の合成
トリフルロアセトアミド官能基を予備触媒の活性化により適した基として明確に特定されれば、イオンモチーフ(イオンタグ)の導入が可能となる。
【0109】
このため、本発明では、化合物10a中の塩素原子を第3アミン(イミダゾール、ピリジンなど)に置換することを提案する。
【0110】
従って、本発明者等は、所期のイオン配位子10bを容易に生じるために、4−クロロ−N−(4−イソプロポキシ−3−ビニルフェニル)ブタンアミド10a上でピリジンの置換を行った。第2世代グラブス触媒を用いたピリジンの錯体化により、錯体11を生じた。
【0111】
【化43】
【0112】
4−クロロ−N−(4−イソプロポキシ−3−ビニルフェニル)ブタンアミド化合物10aの合成
【化44】
【0113】
塩化3−クロロプロパノイル(15μL)を用いて4−イソプロポキシ−3−ビニルアニリン5(50mg、0.3mmol)からアミドを得る一般的な方法により、クロマトグラフィー後にはシリカゲル(溶離用:CH2Cl2)上に淡紅色の固体(52mg、65%)状のアセトアミドが得られた。
【0114】
【0115】
1−(4−(4−イソプロポキシ−3−ビニルフェニルアミノ)−4−オキソブチル) ヘキサフルオロリン酸ピリジニウム化合物(V)10bの合成
【化45】
【0116】
無水トルエン中のアセトアミド溶液10a(52mg、0.19mmol)にピリジン(1mL)を添加した後、混合物を攪拌しながら2日間還流した。溶媒を気化させた後、残渣物を水に溶かして、KPF6(38mg)を添加した。室温で2時間攪拌した後、ジクロロメタンから水相を抽出し、次いでNaCl飽和溶液を用いて有機相を洗浄し、硫酸マグネシウムで乾燥させた。溶媒を気化させた後、クロマトグラフィーによりシリカゲル(溶離用:CH2Cl2/MeOH(4:1))上で無定形の固体(38mg、44%)状のピリジニウム塩を浄化した。
【0117】
【0118】
ルテニウム錯体11の合成
【化46】
【0119】
1−(4−(4−イソプロポキシ−3−ビニルフェニルアミノ)−4−オキソブチル)ヘキサフルオロリン酸ピリジニウム配位子(V)10b(5mg、0.011mmol、1当量)、塩化銅(I)(2mg、1当量)およびインデニリデン予備触媒(9.6mg、1当量)をアルゴン雰囲気下にて丸底フラスコに投入し、そこに無水ジクロロメタン(3mL)を添加した。次いで、アルゴン雰囲気下にて30〜33℃にした反応媒質から3回脱気を行い、攪拌しながら約5時間保持した。
【0120】
次いで、反応原料を真空濃縮した。残渣物をアセトン(2mL)に戻して、焼結濾過を行った。このようにして深緑色無定形の固体状の錯体11を単離した。
【0121】
【0122】
次に、活性ルテニウム錯体1a、1b、1c、1d、1eおよび1fの活性化について検討した。
【0123】
以下の反応スキームに従い、1モル%の錯体の存在下においてジクロロメタン中でメタリル−アリルマロン酸ジエチルと室温で環化するオレフィンメタセシス反応にて、本発明による錯体1a、1b、1c、1dとホベイダの従来技術による錯体3bを検討した。
【0124】
【化47】
【0125】
これらの化合物を用いて得られた転化率の結果は、図1のグラフに示す。
【0126】
これらの結果は、アセトアミド官能基の活性化効果を明確に示している。
【0127】
特に、このアセトアミド官能基がトリフルオロメチル基(錯体1b)を有する場合は、ホベイダ錯体3bの場合に5%であったのに対して、反応開始からわずか15分後に37%以上の転化率が得られた。
【0128】
化合物1e(ノラン触媒2cを用いた錯体化から生成)と化合物1b(第2世代グラブス触媒2bを用いた錯体化から生成)の活性、ならびにホベイダの従来技術による錯体3bの活性を、同じ反応と同じ反応条件で検討した。
【0129】
これらの化合物を用いて得られた転化率の結果は、図2のグラフに示す。
【0130】
非常に驚くことに、これらの結果では、触媒1eおよび1bについては同様の活性が示されているが、第2世代グラブス錯体2b(SIMes配位子を有する)はノラン錯体2c(IMes配位子を有する)よりも活性がきわめて高かった。この結果は非常に興味深い。なぜなら、IMes配位子を有する触媒種(ノラン錯体2cから生成)は、SIMes配位子を有する触媒種(第2世代グラブス錯体2bから生成)よりも熱安定性がきわめ高いためである。
【0131】
このため、本発明は、基質が非常に密集している場合(例えば、テトラ置換オレフィン)に、より厳しい条件(より高温の暖房)下で活性錯体1eを用いてオレフィンメタセシス反応を実現する可能性をもたらす。従って、メタリル−アリルマロン酸ジエチル化合物を環化するメタセシス反応は、1モル%の触媒錯体1eの存在下において45℃で、ならびに1モル%の触媒錯体1bおよび1eの存在下において30℃で実現される。これらの化合物を用いて得られた転化率の結果は、図3のグラフに示す。予想通り、IMes活性触媒1eは、反応開始からわずか6分後に87%の転化率を達成した注目すべき活性を示している。
【0132】
メタリル−アリルマロン酸ジエチル化合物を環化するメタセシス反応では、触媒充填量を削減する活性錯体1bの活性についても評価した。図4は、1モル%の本発明による触媒錯体1bと0.3モル%の本発明による触媒錯体1bの存在下において30℃で環化するメタセシス反応における、メタリル−アリルマロン酸ジエチル化合物の反応時間に対する転化率を示すグラフである。このグラフでは反応性のわずかな低下が示されているが、反応開始からわずか40分後に75%の転化率が認められていることから、この低下も依然として注目に値する。
【0133】
最後に、二量体活性錯体1fについても評価し、その活性を活性錯体1bおよび1eと比較した。図5は、1モル%の本発明による触媒錯体1b、1eおよび1fの存在下において30℃で環化するメタセシス反応における、メタリル−アリルマロン酸ジエチル化合物の反応時間に対する転化率を示すグラフである。
【特許請求の範囲】
【請求項1】
式(I)または(II)
【化1】
[式中、
Lは中性配位子であり、
X、X’は陰イオン配位子であり、
R1およびR2はそれぞれ独立して、水素、C1〜C6アルキル、C1〜C6ペルハロゲン化アルキル、アルデヒド、ケトン、エステル、アミド、ニトリル、場合により置換されるアリール、場合により置換されるアルキルピリジニウム、ペルハロゲン化アルキルピリジニウムもしくはC5〜C6シクロヘキシル、CnH2nYもしくはCnF2nY基[式中、nは1〜6からなり、Yはイオンマーカーである]、または次式:
【化2】
の基であり、
化合物が式(I)である場合は、R1が式(Ibis)
【化3】
の基であり得、
あるいは化合物が式(II)である場合は、R1が式(IIbis)
【化4】
の基であり得、
R3は、C1〜C6アルキルまたはC5〜C6シクロアルキルまたはC5〜C6アリールであり、
R0、R4、R5、R6、R7、R8、R9、R10、R11はそれぞれ独立して、水素、C1〜C6アルキル、C1〜C6ペルハロゲン化アルキル、またはC5〜C6アリールであり、R9、R10、R11は複素環を形成し得、
X1は陰イオン、すなわちハロゲン、テトラフルオロホウ酸塩([BF4]-)、[テトラキス−(3,5−ビス−(トリフルオロメチル)−フェニル)ホウ酸塩]([BARF]-)、ヘキサフルオロリン酸塩([PF6]-)、ヘキサフルオロアンチモン([SbF6]-)、ヘキサフルオロ砒酸塩([AsF6])、トリフルオロメチルスルホン酸塩([CF3]2N)-)であり、
R1およびR2は、それぞれが結合するNおよびCと一緒になって、次式
【化5】
[式中、ハルはハロゲンであり、R12は、水素、C1〜C6アルキルもしくはC5〜C6シクロアルキル、またはC5〜C6アリールである]
の複素環を形成し得る]
の化合物。
【請求項2】
LがP(R13)3[式中、R13は、C1〜C6アルキルもしくはアリールまたはC5〜C6シクロアルキルである]であることを特徴とする、請求項1に記載の化合物。
【請求項3】
Lが、式7a、7b、7c、7dまたは7e
【化6】
[式中、
n1は0、1、2、3であり、
R14、R15、R16、R17、R18、R19、R20、R21、R22、R23、R24、R25、R26、R27、R28はそれぞれ独立して、C1〜C6アルキル、C3〜C20シクロアルキル、C2〜C20アルケニル、ナフチル、アントラセンまたはフェニルであり、前記フェニルは、C1〜C6アルキル、C1〜C6アルコキシおよびハロゲンの中から選択される最高5つの群から置換され得、R16およびR17ならびにR26およびR27は、3員、4員、5員、6員、7員の環を形成し得、R28は独立して、連結した6員の芳香族環を形成し得る]
の配位子であることを特徴とする、請求項1に記載の化合物。
【請求項4】
LがPCy3であり、Cyがシクロヘキシルであるか、あるいはLが式7aまたは7bの配位子であり、
Xが塩素であり、
X’が塩素である
ことを特徴とする、請求項2または3に記載の化合物。
【請求項5】
イオンマーカーYが、
【化7】
からなる群から選択されることを特徴とする、請求項2から4までのいずれか1項に記載の化合物。
【請求項6】
式(I)[式中、R1はCH3、CF3、C6F5、pNO3C6H4からなる群から選択される]に対応することを特徴とする、請求項4に記載の化合物。
【請求項7】
式(I)[式中、R1はCF3である]に対応することを特徴とする、請求項6に記載の化合物。
【請求項8】
式1a
【化8】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項9】
式1b
【化9】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項10】
式1c
【化10】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項11】
式1d
【化11】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項12】
式1e
【化12】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項13】
式1f
【化13】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項14】
式1g
【化14】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項15】
式1h
【化15】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項16】
式1i
【化16】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項17】
式1j
【化17】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項18】
式1k
【化18】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項19】
式11
【化19】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項20】
式12
【化20】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項21】
式13
【化21】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項22】
式14
【化22】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項23】
請求項1から22までのいずれか1項に記載の化合物の合成方法であって、4−イソプロポキシ−3−ビニルアニリンとアシル基を有する化合物とを反応させてアミド配位子を得る第1段階と、前記アミド配位子を式(III)
【化23】
の化合物と反応させる第2段階とからなることを特徴とする方法。
【請求項24】
式(III)の前記化合物が、グラブス予備触媒(2b)またはノラン予備触媒(2c)であることを特徴とする、請求項23に記載の方法。
【請求項1】
式(I)または(II)
【化1】
[式中、
Lは中性配位子であり、
X、X’は陰イオン配位子であり、
R1およびR2はそれぞれ独立して、水素、C1〜C6アルキル、C1〜C6ペルハロゲン化アルキル、アルデヒド、ケトン、エステル、アミド、ニトリル、場合により置換されるアリール、場合により置換されるアルキルピリジニウム、ペルハロゲン化アルキルピリジニウムもしくはC5〜C6シクロヘキシル、CnH2nYもしくはCnF2nY基[式中、nは1〜6からなり、Yはイオンマーカーである]、または次式:
【化2】
の基であり、
化合物が式(I)である場合は、R1が式(Ibis)
【化3】
の基であり得、
あるいは化合物が式(II)である場合は、R1が式(IIbis)
【化4】
の基であり得、
R3は、C1〜C6アルキルまたはC5〜C6シクロアルキルまたはC5〜C6アリールであり、
R0、R4、R5、R6、R7、R8、R9、R10、R11はそれぞれ独立して、水素、C1〜C6アルキル、C1〜C6ペルハロゲン化アルキル、またはC5〜C6アリールであり、R9、R10、R11は複素環を形成し得、
X1は陰イオン、すなわちハロゲン、テトラフルオロホウ酸塩([BF4]-)、[テトラキス−(3,5−ビス−(トリフルオロメチル)−フェニル)ホウ酸塩]([BARF]-)、ヘキサフルオロリン酸塩([PF6]-)、ヘキサフルオロアンチモン([SbF6]-)、ヘキサフルオロ砒酸塩([AsF6])、トリフルオロメチルスルホン酸塩([CF3]2N)-)であり、
R1およびR2は、それぞれが結合するNおよびCと一緒になって、次式
【化5】
[式中、ハルはハロゲンであり、R12は、水素、C1〜C6アルキルもしくはC5〜C6シクロアルキル、またはC5〜C6アリールである]
の複素環を形成し得る]
の化合物。
【請求項2】
LがP(R13)3[式中、R13は、C1〜C6アルキルもしくはアリールまたはC5〜C6シクロアルキルである]であることを特徴とする、請求項1に記載の化合物。
【請求項3】
Lが、式7a、7b、7c、7dまたは7e
【化6】
[式中、
n1は0、1、2、3であり、
R14、R15、R16、R17、R18、R19、R20、R21、R22、R23、R24、R25、R26、R27、R28はそれぞれ独立して、C1〜C6アルキル、C3〜C20シクロアルキル、C2〜C20アルケニル、ナフチル、アントラセンまたはフェニルであり、前記フェニルは、C1〜C6アルキル、C1〜C6アルコキシおよびハロゲンの中から選択される最高5つの群から置換され得、R16およびR17ならびにR26およびR27は、3員、4員、5員、6員、7員の環を形成し得、R28は独立して、連結した6員の芳香族環を形成し得る]
の配位子であることを特徴とする、請求項1に記載の化合物。
【請求項4】
LがPCy3であり、Cyがシクロヘキシルであるか、あるいはLが式7aまたは7bの配位子であり、
Xが塩素であり、
X’が塩素である
ことを特徴とする、請求項2または3に記載の化合物。
【請求項5】
イオンマーカーYが、
【化7】
からなる群から選択されることを特徴とする、請求項2から4までのいずれか1項に記載の化合物。
【請求項6】
式(I)[式中、R1はCH3、CF3、C6F5、pNO3C6H4からなる群から選択される]に対応することを特徴とする、請求項4に記載の化合物。
【請求項7】
式(I)[式中、R1はCF3である]に対応することを特徴とする、請求項6に記載の化合物。
【請求項8】
式1a
【化8】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項9】
式1b
【化9】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項10】
式1c
【化10】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項11】
式1d
【化11】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項12】
式1e
【化12】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項13】
式1f
【化13】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項14】
式1g
【化14】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項15】
式1h
【化15】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項16】
式1i
【化16】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項17】
式1j
【化17】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項18】
式1k
【化18】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項19】
式11
【化19】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項20】
式12
【化20】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項21】
式13
【化21】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項22】
式14
【化22】
に対応することを特徴とする、請求項4に記載の化合物。
【請求項23】
請求項1から22までのいずれか1項に記載の化合物の合成方法であって、4−イソプロポキシ−3−ビニルアニリンとアシル基を有する化合物とを反応させてアミド配位子を得る第1段階と、前記アミド配位子を式(III)
【化23】
の化合物と反応させる第2段階とからなることを特徴とする方法。
【請求項24】
式(III)の前記化合物が、グラブス予備触媒(2b)またはノラン予備触媒(2c)であることを特徴とする、請求項23に記載の方法。
【図1】

【図2】
【図3】
【図4】

【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
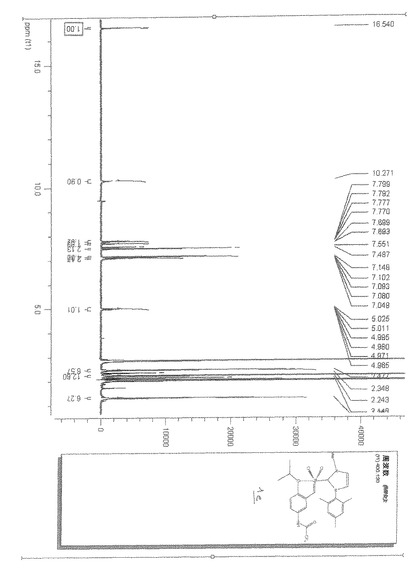
【図11】
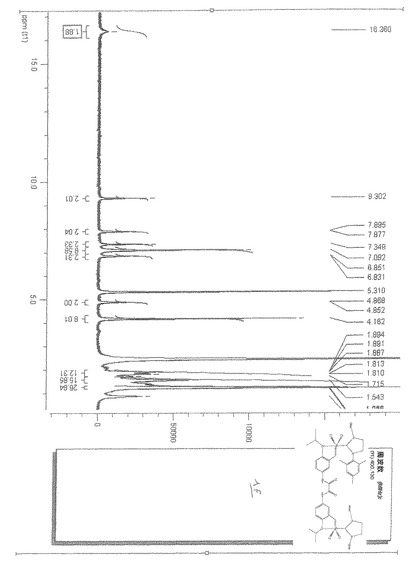
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【公表番号】特表2010−511017(P2010−511017A)
【公表日】平成22年4月8日(2010.4.8)
【国際特許分類】
【出願番号】特願2009−538722(P2009−538722)
【出願日】平成19年11月30日(2007.11.30)
【国際出願番号】PCT/EP2007/063062
【国際公開番号】WO2008/065187
【国際公開日】平成20年6月5日(2008.6.5)
【出願人】(509153962)
【氏名又は名称原語表記】Ecole Nationale Superieure de Chimie de Rennes
【住所又は居所原語表記】263 avenue du General Leclerc, F−35700 Rennes Cedex, France
【出願人】(500379381)サントル ナショナル ドゥ ラ ルシャルシュ シアンティフィク (17)
【氏名又は名称原語表記】Centre National de la Recherche Scientifique
【住所又は居所原語表記】3 rue Michel Ange, FR−75016 Paris, France
【Fターム(参考)】
【公表日】平成22年4月8日(2010.4.8)
【国際特許分類】
【出願日】平成19年11月30日(2007.11.30)
【国際出願番号】PCT/EP2007/063062
【国際公開番号】WO2008/065187
【国際公開日】平成20年6月5日(2008.6.5)
【出願人】(509153962)
【氏名又は名称原語表記】Ecole Nationale Superieure de Chimie de Rennes
【住所又は居所原語表記】263 avenue du General Leclerc, F−35700 Rennes Cedex, France
【出願人】(500379381)サントル ナショナル ドゥ ラ ルシャルシュ シアンティフィク (17)
【氏名又は名称原語表記】Centre National de la Recherche Scientifique
【住所又は居所原語表記】3 rue Michel Ange, FR−75016 Paris, France
【Fターム(参考)】
[ Back to top ]