
ルミネッセントナノ結晶の調製方法、得られたナノ結晶およびそれらの使用
【課題】従来よりも高く、特に周囲温度において50%を超える蛍光量子収率を有し、光酸化に対して高い安定性を有するInP/ZnS コア/シェルナノ結晶を合成する新規な方法を提供すること。
【解決手段】本発明は、(i)+III酸化状態での金属または非金属を表すAおよび−III酸化状態での元素を表すBを含む半導体を含むコアを有し、このコアが、(ii)外側部分が式ZnS1−xExを有する半導体を含むシェルでコーティングされているナノ結晶を調製するための方法を含む。ここで、Eは−II酸化状態の元素を表し、xは0≦x<1となるように少数である。
【解決手段】本発明は、(i)+III酸化状態での金属または非金属を表すAおよび−III酸化状態での元素を表すBを含む半導体を含むコアを有し、このコアが、(ii)外側部分が式ZnS1−xExを有する半導体を含むシェルでコーティングされているナノ結晶を調製するための方法を含む。ここで、Eは−II酸化状態の元素を表し、xは0≦x<1となるように少数である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、可視スペクトルの大部分をカバーしながら、既存のシステムに対して高い色純度および向上した蛍光量子収率を有する半導体に基づく「コア/シェル」ナノ結晶に基づく新規なルミネッセント材料を製造する方法に関する。
本発明はまた、得られたルミネッセント材料およびそれらの種々の用途に関する。実際、これらのナノ結晶の用途分野は、例えば発光ダイオード、太陽電池、および化学または生物学的分子の蛍光標識を含む。
【背景技術】
【0002】
半導体結晶は、数十年にわたって知られているルミネッセント材料である。1980年代から1990年代において、それらの発光スペクトルは、その結晶サイズが十分に小さくなる場合にはそのサイズに依存することが明示された。「ナノ結晶」または「量子ドット」と呼ばれる1〜10nmの範囲にほぼ位置するサイズの結晶に関して、こうした依存性は極めて強く現れる。事実、可視および近赤外および紫外範囲の色パレット全体は、半導体ナノ結晶を用いて、その結晶サイズおよび組成を適切に選択することによって得ることができる。ライトおよびディスプレイ(フラットスクリーン)の分野において非常に重要な可視スペクトルをカバーするために、最も研究された材料は、カドミウムカルコゲニド(CdS、CdSe、CdTe)である。しかし、欧州RoHS(「危険物質に関する制限」) の指令は、2006年7月1日から欧州にて販売される電気および電子機器(EEE)において、次の物質を除去することを目標としている:鉛、水銀、カドミウム、六価クロム、ポリブロモビフェニル(PBB)、ポリブロモジフェニルエーテル(PBDE)。故に、有利なことには、所望の光学特性を保持しながら、ナノ結晶の製造用にこれらの物質を含有しない代替材料を見出すことが重要である。
【0003】
通則として、ナノ結晶からなるルミネッセント材料の光学品質は複数のパラメータに依存し、そのうち最も重要なものは:
−発光波長を支配するナノ結晶のサイズ;
−発光バンド幅を制御するナノ結晶のサイズ分布;
−蛍光量子収率および経時的な安定性に関与するナノ結晶の表面不活性化
である。
リン化インジウム(InP)は、カドミウム系ナノ結晶を置換するために最も有利な材料の1つである。1.35eVの禁制バンド幅のために、そのサイズを変更することによって、可視スペクトルおよび近赤外スペクトルにおけるInPナノ結晶の発光波長を変更できる。
InPナノ結晶を調製するためには複数の方法があり、その方法のうち、現時点では、有機溶媒中、高温での迅速な前駆体注入方法が最も狭いサイズ分散に近づくことができる。ナノ結晶のサイズ分散が狭いと、狭い発光スペクトル(純粋な発光色)を生じる;すなわちこれは、技術的な用途に有利である。2つの主要な合成プロセスが利用可能である。
【0004】
いわゆる「従来の」有機金属合成プロセスである第1のプロセスは、高温(270〜300℃)において、反応性媒体として作用するトリオクチルホスフィンオキシド(TOPO)中、インジウム前駆体(例えば塩化インジウムまたはシュウ酸クロロインジウム)をリン前駆体(トリス(トリメエチルシリル)ホスフィン、P(TMS)3、P(Si(CH3)3)3)とを反応させることからなる(非特許文献1〜3)。TOPOは、ナノ結晶の表面での結合能力を強め、コロイド形態にてナノ結晶を安定化させるために「配位溶媒」と呼ばれる。この方法の主な欠点は、調製されるナノ結晶のサイズ分散が広く、反応時間が長いことである(3〜7日間)。
第2の合成プロセスは、2002年に公開された「従来の」合成プロセスにおける変更である。ここで、反応性媒体は、TOPOの代わりに、「非配位溶媒」として分類される単なるアルケンの1−オクタデセンである(非特許文献4)。さらに、ラウリン酸、ミリスチン酸またはパルミチン酸のような脂肪酸が、安定化配位子として使用された。反応は、インジウム前駆体(酢酸インジウム)および安定化剤を含有する300℃に加熱された溶媒中で、リン前駆体(P(TMS)3)の迅速な注入から開始される。この著者らは、約10%の範囲にあるサイズ分散の改善を報告している。さらに、反応時間は3〜4時間に短縮される。
しかし、上述の調製方法は、蛍光量子収率が依然として低いままである(通常1%未満)という問題を解決できない。種々の材料(例えばCdSe、CdSなど)のナノ結晶の量子収率を増大させるために広く使用されている方法の1つは、「コア」の周りに、より大きな禁制バンド幅を有する半導体シェルを成長させることによってそれらの表面を不活性化することからなる。これは、科学文献において「コア/シェル」システムと呼ばれる。シェルを堆積させるために使用される方法は、本質的にコアを調製するために使用されるものと同じである。
InPナノ結晶の光学特性を改善するために、種々のシェル材料が提案されている。Micicらは、コアとシェル材料との間の格子不整合を最小限にするために、InP表面上での三元化合物CdZnSe2の合成を記載している(非特許文献5)。顕著な格子不整合は、シェル中にてまたはコア/シェル界面にて結晶上の欠陥を誘導し、それにより蛍光効率を低下させ得る。それでもなお、このコア/シェルシステムで達成される量子収率(10%)は、カドミウム系ナノ結晶で得られる値(50〜85%)に比べて大きく劣っている。
【0005】
Hauboldらは、InPをZnSでコーティングし、3日後の15%から3週間後の23%までの間の量子収率を得た(非特許文献6)。これらのコア/シェルナノ結晶のこうした合成に使用される方法は:
a1)上述した第1の合成プロセスに従うInPコア結晶の合成;
a2)沈殿およびメタノールでの洗浄によるInP結晶の精製;
a3)ZnSシェルの成長。この工程に関して、InPナノ結晶は、200℃にてトリオクチルホスフィン(TOP)中に分散され、ジエチル亜鉛およびビス(トリメチルシリル)スルフィドの混合物が注入される。次いで温度を一時的に260℃まで上昇させた後、1時間で100℃に低下させる。続いて、反応混合物を不活性雰囲気(窒素)中に数日間または数週間保持する;
a4)ナノ結晶の沈殿およびメタノールでの洗浄による精製;
a5)選択的沈殿によるサイズ画分へのサンプル分離
を含む。
この方法に関連する問題は、シェルの成長のための自然発火性の前駆体(ジエチル亜鉛およびビス(トリメチルシリル)スルフィド)の使用、非常に長期間(数週間)での光学特性の進行、および最終的にII−VI半導体に基づくコア/シェルシステムに関連する相対的に低い量子収率が原因である。著者らは、InPナノ結晶のZnSシェルによるコーティングが完全でなく、そのことが蛍光量子収率を制限すると結論している。
【0006】
より最近では、Pengらが、InP/ZnS コア/シェルナノ結晶合成のための新しいプロトコルを提案した(非特許文献7)。この場合、コア結晶の合成は、上述されたような第2の合成プロセスと同様であるが、リン前駆体と同時に注入されるアミン(1−オクチルアミン)の添加により、270〜300℃の代わりに180〜190℃での反応にすることができる。次いでシェルは次のように成長される:
b1)150℃にて、亜鉛ジステアレート(亜鉛前駆体)の特定量を、InPナノ結晶を含有するフラスコに注入する。10分後、オクタデセン中に溶解した同量の元素硫黄(硫黄前駆体)を添加する;
b2)温度を30分間220℃に上昇させて、ZnSシェルを成長させる;
b3)温度を再び150℃に低下させて、より多量にて、上記と同じ順番で亜鉛前駆体、その後の硫黄前駆体の添加を繰り返す;
b4)温度を30分間で220℃に上昇させる;
b5)ナノ結晶を、ヘキサンを添加し、メタノールで抽出することによって精製する。
【0007】
この方法により、シェルを成長させるための自然発火性の前駆体を排除でき、コア結晶の合成とシェルの成長との間での中間精製を排除できるが、この方法は、実施が複雑なままである。それらを産業規模へ適合させることに関する問題は、特に、InP結晶の合成中の自然発火性リン前駆体の迅速な注入および複数のZnS前駆体注入中での温度制御に関連している。この方法で得られたナノ結晶の蛍光量子収率は40%までである。この値はInP系ナノ結晶に関して公開された最良のものであるが、依然としてカルシウム−カルゴゲニド系ナノ結晶よりも劣る。
要約すれば、これまで報告されたInP系ナノ結晶を製造するための方法は、50%を超える蛍光量子収率を達成できず、その技術的用途が制限される。低いサイズ分散を有するInPナノ結晶の合成について記載されている方法は、高温にて、反応媒体中への自然発火性前駆体の迅速な注入を含み、それは大規模で行うには困難である。
最後に、InP/ZnS コア/シェルナノ結晶の合成は、2工程の方法を必要とし、InPコア結晶の合成と、続くZnSシェルの成長を必要とし、その間に、正確に温度を制御しながら、再び前駆体を反応媒体に注入する必要がある。
【先行技術文献】
【非特許文献】
【0008】
【非特許文献1】Micic, O. I.;Curtis, C. J.; Jones, K. M.; Sprague, J. R.; Nozik, A. J., SYNTHESIS ANDCHARACTERIZATION OF INP QUANTUM DOTS. Journal of Physical Chemistry 1994, 98,(19), 4966-4969
【非特許文献2】Guzelian, A. A.;Katari, J. E. B.; Kadavanich, A. V.; Banin, U.; Hamad, K.; Juban, E.;Alivisatos, A. P.; Wolters, R. H.; Arnold, C. C.; Heath, J. R., Synthesis ofsize-selected, surface-passivated InP nanocrystals. Journal of PhysicalChemistry 1996, 100, (17), 7212-7219
【非特許文献3】Micic, O. I.;Sprague, J. R.; Curtis, C. J.; Jones, K. M.; Machol, J. L.; Nozik, A. J.;Giessen, H.; Fluegel, B.; Mohs, G.; Peyghambarian, N., SYNTHESIS ANDCHARACTERIZATION OF INP, GAP, AND GAINP2 QUANTUM DOTS. Journal of PhysicalChemistry 1995, 99, (19), 7754-7759
【非特許文献4】Battaglia, D.;Peng, X. G., Formation of high quality InP and InAs nanocrystals in anoncoordinating solvent. Nano Letters 2002, 2, (9), 1027-1030
【非特許文献5】Micic, O. I.; Smith, B. B.; Nozik, A. J., Core-shell quantum dots of lattice-matched ZnCdSe2 shellson InP cores: Experiment and theory. Journal of Physical Chemistry B 2000, 104,(51), 12149-12156
【非特許文献6】Haubold, S.;Haase, M.; Kornowski, A.; Weller, H., Strongly luminescent InP/ZnS core-shellnanoparticles. Chemphyschem 2001, 2, (5), 331-334
【非特許文献7】Xie, R.;Battaglia, D.; Peng, X., Colloidal InP nanocrystals as efficient emitterscovering blue to near-infrared. Journal of the American Chemical Society 2007,129, (50), 15432-15433
【発明の概要】
【発明が解決しようとする課題】
【0009】
本発明は、上記で列挙された技術的問題および欠点を解決する。
実際、本発明は、現在までに報告されている値よりも高く、特に周囲温度において50%を超える蛍光量子収率を有し、光酸化に対して高い安定性を有するInP/ZnS コア/シェルナノ結晶を合成する新規な方法を提案する。
本発明に従う方法は、InP/ZnS コア/シェルナノ結晶合成が単一工程にて行われるので、先行技術に記載されるすべての方法よりも非常に簡便である。本発明に従う方法は、中間精製工程を必要としない。さらに、サイズ分類工程(例えば選択的沈殿による)は、合成直後に得られたサンプルのサイズ分散が狭いので必要でない。
さらに、方法は、前駆体の注入、特に高温にて反応媒体中への自然発火性リン前駆体の迅速な注入がないことを特徴とする。前駆体は、周囲温度にて反応媒体に単に添加するだけでよい。さらに、反応の開始後および温度の上昇後にいかなる前駆体の添加も必要としない。
最後に、本発明に従う方法は、InP/ZnS コア/シェルナノ結晶の調製を可能にすることだけでなく、一般化として式ABを有する半導体を含むコアおよび式CDを有する少なくとも1つの半導体を含むシェルを有するコア/シェルナノ結晶の調製にも好適であることを特徴とし、ここでAは+III酸化状態における金属または非金属を表し、Bは−III酸化状態の元素を表し、Cは+II酸化状態の金属または非金属を表し、Dは−II酸化状態の元素を表す。
【課題を解決するための手段】
【0010】
本発明は、(i)+III酸化状態での金属または非金属を表すAおよび−III酸化状態での元素を表すBを含む半導体を含むコアを有し、このコアが、(ii)外側部分が式ZnS1−xExを有する半導体を含むシェルでコーティングされているナノ結晶を調製するための方法に関し、ここでEは−II酸化状態の元素を表し、xは0≦x<1となるように少数であり、この方法は、Aの少なくとも1つの前駆体、Bの少なくとも1つの前駆体、亜鉛の少なくとも1つの前駆体、硫黄の少なくとも1つの前駆体、および任意にEの少なくとも1つの前駆体の混合物を、まずコアが形成され、次いでシェルが形成されるように上昇する様式で温度T1から、T1を超える温度T2まで加熱することからなる工程を含む。
【0011】
より詳細には、本発明に従う方法は次の工程を含む:
a)Aの少なくとも1つの前駆体、Bの少なくとも1つの前駆体、少なくとも1つの亜鉛前駆体、少なくとも1つの硫黄前駆体、および任意にEの少なくとも1つの前駆体の混合物を温度T1で調製する工程、
b)工程(a)にて得られた混合物を温度T2に加熱する工程、
c)工程(b)にて得られた、式ABを有する半導体を含むコアを有し、外側層が式ZnS1−xExを有する半導体を含むシェルによって囲まれているナノ結晶を精製する工程。
本発明に従う方法に従って調製されたナノ結晶のコアは、+III酸化状態での金属または非金属を表すAおよび−III酸化状態での元素を表すBを含む半導体を含み、このタイプの半導体はIII−Vと称される。
+III酸化状態での金属または非金属であって、Bとともに、本発明に従って調製されるナノ結晶のコアに含まれる半導体を形成するAは、ガリウム(Ga)、インジウム(In)、アルミニウム(Al)、およびこれらの混合物から選択される。
【0012】
−III酸化状態での元素であって、Aと共に、本発明に従って調製されるナノ結晶のコアに含まれる半導体を形成するBは、アンチモン(Sb)、ヒ素(As)、リン(P)、窒素(N)およびこれらの混合物から選択される。
本発明に従う方法に従って調製されたナノ結晶のコアに含まれる式ABを有する半導体の例は、GaAs、GaSb、GaN、GaP、InAs、InSb、InN、InP、AlAs、AlSb、AlN、AlP、InGaAs、InGaSb、InGaN、InGaP、InAlAs、InAlSb、InAlN、InAlP、GaAlAs、GaAlSb、GaAlN、GaAlPおよびこれらの混合物である。特に、ナノ結晶のコアは、InP、InAs、InGaPおよびこれらの混合物から選択される式ABを有する半導体を含み、より詳細にはこの半導体はInPである。
有利なことには、本発明に従う方法に従って調製されるナノ結晶のコアは、上記で定義された半導体だけからなる。
【0013】
本発明の方法に従って調製されたナノ結晶はシェルを含み、このシェルの外側部分は、式ZnS1−xExを有する半導体を含み、ここでEは−II酸化状態の元素を表し、xは0≦x<1となるような少数である。
Eは、特に酸素(O)、セレン(Se)、テルル(Te)およびこれらの混合物から選択される−II酸化状態の元素である。
本発明に従う方法に従って調製されるナノ結晶のシェルは、単層または複数の層(すなわち、多層シェル)からなってもよい。シェルが単層のみからなる場合、シェルの外側部分はこの層に対応する。
シェルが複数の層からなる場合、シェルの外側部分は、シェルの外側層に対応する。「外側層」という用語は、本発明の範囲内において、ナノ結晶のコアから最も遠く、ナノ結晶が置かれた媒体または環境と直接接触するシェルの層を指す。多層シェルは、2〜10、特に2〜5個の異なる半導体層を含んでいてもよい。このようにして、種々の代替実施形態が、本発明に従う方法を用いて調製されるナノ結晶のシェルについて想定できる。
【0014】
第1の代替実施形態において、式ZnS1−xExのxは0に等しく、シェルは、1つの層、すなわちZnS層でのみ形成されている。
第2の代替実施形態において、式ZnS1−xExのxは、0<x<1となるようなものであり、シェルは1つの層でのみ形成される。
第3の代替実施形態において式ZnS1−xExのxは0に等しく、シェルは少なくとも2つの異なる層を含み、その外側層はZnS層である。
第4の代替実施形態において、式ZnS1−xExのxは、0<x<1となるようなものであり、シェルは少なくとも2つの異なる層を含む。
さらに、本発明に従う方法に従って調製されるナノ結晶のシェルの層は、均一な化学組成を有していてもよく、または同じ層内において、異なる化学組成を有していてもよく、特に化学組成が勾配の形態であってもよい。この場合、シェルの外側部分は、1つの層を有するシェルの外側区域および多層シェルの外側層の外側区域からなる。
多層シェルの場合、ナノ結晶のコアと、上記で定義された式ZnS1−xExを有する外側層との間に含まれる層は、上記で定義された式ABを有する半導体および/または式CDを有する半導体を含んでいてもよく、ここでCは+II酸化状態での金属または非金属を表し、Dは−II酸化状態での元素を表す。式CDを有する半導体はまたII−VIと称される。
【0015】
+II酸化状態の金属または非金属Cは、特に、マグネシウム(Mg)、カルシウム(Ca)、ストロンチウム(Sr)、バリウム(Ba)、亜鉛(Zn)、カドミウム(Cd)、水銀(Hg)、スズ(Sn)、鉛(Pb)、およびこれらの混合物から選択される。−II酸化状態の元素Dは、特に、酸素(O)、硫黄(S)、セレン(Se)、テルル(Te)、およびこれらの混合物から選択される。ナノ結晶のコアとシェルの外側層との間に含まれる層に存在してもよい半導体の例は、例えば、MgS、MgSe、MgTe、CaS、CaSe、CaTe、SrS、SrSe、SrTe、BaS、BaSe、BaTe、ZnO、ZnS、ZnSe、ZnTe、CdS、CdSe、CdTe、HgS、HgSe、HgTe、SnS、SnSe、SnTe、PbS、PbSe、PbTeおよびこれらの混合物である。より詳細には、ナノ結晶のコアとシェルの外側層との間に含まれる層に存在してもよい半導体の例は、MgS、MgSe、MgTe、CaS、CaSe、CaTe、SrS、SrSe、SrTe、BaS、BaTe、ZnO、ZnS、ZnSe、ZnTe、Sns、SnSe、SnTeおよびこれらの混合物からなる群から選択される。
故に、多層シェルの場合、本発明の範囲内、特に工程(a)にて使用される反応混合物は、シェルの外側層以外の層を形成する元素の前駆体をさらに含有することは明らかである。
【0016】
本発明の方法に従って調製されるナノ結晶は、15nm未満、特に12nm未満、特に10nm未満の直径を有する。
本発明に従う方法を用いて調製されたナノ結晶のコアは、1〜10nmの直径、特に1.5〜8nm、特に2〜6nmの直径を有する。
本発明に従う方法を用いて調製されるナノ結晶のシェルは、0.3〜6nm、特に0.5〜4nm、特に1〜2nmの厚さを有する。
本発明の範囲内において、使用されるAの前駆体は、インジウム前駆体、ガリウム前駆体、アルミニウム前駆体およびこれらの混合物からなる群から選択される。当業者に既知のインジウム、アルミニウムおよびガリウム前駆体の全て、特に液体または固体形態の前駆体は、本発明に使用するのに好適である。
有利なことに、Aの前駆体は、Aの塩、Aのハロゲン化物、Aの酸化物およびAの有機金属化合物から選択される。「Aの有機金属化合物」という用語は、より詳細には、Aの三置換化合物、Aのカルボン酸塩、またはAのホスホン酸塩を指す。
【0017】
「Aの三置換化合物」という用語は、本発明の範囲内において、式(R1)3Aを有する化合物を指し、ここで各R1は、同一または異なって、1〜20個の炭素原子を有する炭化水素基、例えばアルキルラジカル、アルケニルラジカル、アルコキシラジカル、アリールラジカルまたはアリールオキシラジカルを表す。
「Aのカルボン酸塩」という用語は、本発明の範囲内において、式(R2COO)3Aを有する化合物を指し、ここで、各R2は、同一または異なって、1〜20個の炭素原子を有する炭化水素基、例えばアルキルラジカル、アルケニルラジカル、アルコキシラジカル、アリールラジカルまたはアリールオキシラジカルを表す。
「Aのホスホン酸塩」という用語は、本発明の範囲内において、式[R3−P(OR4)(OR5)O]3Aを有する化合物を指し、ここで
−各R3は、同一または異なって、1〜20個の炭素原子を有する炭化水素基、例えばアルキルラジカル、アルケニルラジカル、アルコキシラジカル、アリールラジカルまたはアリールオキシラジカルを表し;
−各R4は、同一または異なって、水素原子または1〜20個の炭素原子を有する炭化水素基、例えばアルキルラジカル、アルケニルラジカル、アルコキシラジカル、アリールラジカルまたはアリールオキシラジカルを表し;
−各R5は、同一または異なって、水素原子または1〜20個の炭素原子を有する炭化水素基、例えばアルキルラジカル、アルケニルラジカル、アルコキシラジカル、アリールラジカルまたはアリールオキシラジカルを表す。
【0018】
本発明の範囲内において、特に指示がない限り、「アルキル基」という用語は、1〜20個の炭素原子、特に1〜15個の炭素原子、とりわけ1〜10個の炭素原子を有する直鎖、分枝鎖または環状の、任意に置換されたアルキル基を指す。
本発明の範囲内において、「アルケニル基」という用語は、2〜20個の炭素原子、特に2〜15個の炭素原子、とりわけ2〜10個の炭素原子を有する直鎖、分枝鎖または環状の、任意に置換されたアルケニル基を指す。
【0019】
本発明の範囲内において、「アルコキシ基」という用語は、上記で定義されたアルキルによって置換された酸素原子を指す。
本発明の範囲内において、「アリール基」という用語は、6〜20個の炭素原子、特に6〜14個の炭素原子、とりわけ6〜8個の炭素原子を有する単環式または多環式の、任意に置換された芳香族基を指す。
本発明の範囲内において、「アリールオキシ基」という用語は、上記で定義されたアリールによって置換された酸素原子を指す。
本発明の範囲内において、「任意に置換された」という用語は、アルキル基、アルコキシ基、ハロゲン、ヒドロキシル、シアノ、トリフルオロメチルまたはニトロから選択される1つまたは複数の基によって置換されたラジカルを指す。
本発明の範囲内において、「ハロゲン」という用語は、フッ素、塩素、臭素またはヨウ素を指す。
【0020】
本発明の範囲内にて使用するのが好適なAの前駆体の例は、Aがインジウムである場合、インジウムトリクロライド、トリエチル−インジウム、インジウムトリアセテート、インジウムトリ(アセチル−アセトネート)、インジウムトリオクトネート、インジウムトリステアレート、インジウムトリラウレート、インジウムトリパルミテート、インジウムトリミリステート、インジウムトリオレエート、およびこれらの混合物が挙げられる。
【0021】
本発明の範囲内において、使用されるBの前駆体は、アンチモン前駆体、ヒ素前駆体、リン前駆体、窒素前駆体およびこれらの混合物から成る群から選択される。当業者に既知のアンチモン、ヒ素、リンおよび窒素前駆体のすべて、特に液体または固体形態の前駆体は、本発明に使用するのに好適である。
有利なことには、本発明に使用されるBの前駆体は、式B(F(R6)3)3または式B(R7)3を有する化合物であり、ここで
−各Fは、シリカ(Si)、ゲルマニウム(Ge)およびスズ(Sn)から成る群から選択され;
−各R6は、同一または異なって、1〜10個の炭素原子、特に1〜6個の炭素原子、とりわけ1〜3個の炭素原子を有する直鎖、分枝鎖または環状の、任意に置換されたアルキル基であり;
−各R7は、同一または異なって、水素原子、ハロゲン、例えば塩素(Cl)、臭素(Br)、ヨウ素(I)もしくはフッ素(F)、または1〜10個の炭素原子、特に1〜6個の炭素原子、とりわけ1〜3個の炭素原子を有する直鎖、分枝鎖または環状の、任意に置換されたアルキル基である。
特に、Bの前駆体、特に好ましいリン前駆体は、式B(Si(CH3)3)3またはB(TMS)3を有するBのトリス(トリメチルシリル)化合物である。
【0022】
本発明の範囲内において、使用される亜鉛前駆体は、亜鉛塩、ハロゲン化亜鉛、酸化亜鉛および有機金属亜鉛化合物から成る群から選択される。「有機金属亜鉛化合物」という用語は、より詳細には、二置換された亜鉛化合物、カルボン酸亜鉛、またはホスホン酸亜鉛を指す。
本発明の範囲内において、「二置換された亜鉛化合物」という用語は、式(R8)2Znを有する化合物を指し、各R8は、同一または異なって、1〜20個の炭素原子を有する炭化水素基、例えばアルキルラジカル、アルケニルラジカル、アルコキシラジカル、アリールラジカルまたはアリールオキシラジカルを表す。
本発明の範囲内において、「カルボン酸亜鉛」という用語は、式(R9COO)2Znを有する化合物を指し、ここで各R9は、
同一または異なって、1〜20個の炭素原子を有する炭化水素基、例えばアルキルラジカル、アルケニルラジカル、アルコキシラジカル、アリールラジカルまたはアリールオキシラジカルを表す。
本発明の範囲内において、「ホスホン酸亜鉛」という用語は、式[R10−P(OR11)(OR12)O]2Znを有する化合物を指し、ここで
−各R10は、同一または異なって、1〜20個の炭素原子を有する炭化水素基、例えばアルキルラジカル、アルケニルラジカル、アルコキシラジカル、アリールラジカルまたはアリールオキシラジカルを表し;
−各R11は、同一または異なって、1〜20個の炭素原子を有する炭化水素基、例えばアルキルラジカル、アルケニルラジカル、アルコキシラジカル、アリールラジカルまたはアリールオキシラジカルを表し;
−各R12は、同一または異なって、1〜20個の炭素原子を有する炭化水素基、例えばアルキルラジカル、アルケニルラジカル、アルコキシラジカル、アリールラジカルまたはアリールオキシラジカルを表す。
アルキル、アルケニル、アルコキシ、アリールおよびアリールオキシラジカルは、Aの前駆体に関して定義された通りである。
【0023】
本発明の範囲内において、使用される硫黄前駆体は、脂肪族チオール、有機溶媒に溶解した元素硫黄、および式S(Si(R13)3)2を有する化合物から成る群から選択され、ここで、各R13は、同一または異なって、1〜10個の炭素原子、特に1〜6個の炭素原子、とりわけ1〜3個の炭素原子を有する直鎖、分枝鎖または環状の、任意に置換されたアルキル基である。
有利なことには、脂肪族チオールは、式CnH2n+1−SHを有し、ここでnは1〜25、特に5〜20、とりわけ8〜18の整数を表す。本発明の範囲内にて使用するのに好適な脂肪族チオールの例としては、オクタンチオール(n=8)、オクトデカンチオール(n=18)、ドデカンチオール(n=12)およびこれらの混合物が挙げられる。
有利なことには、元素硫黄が溶解する有機溶媒は、トリアルキルホスフィン(ここでアルキル基は4〜12個の炭素原子を含む)およびアルケンから選択される。使用するのに好適な有機溶媒の例としては、1−オクタデセン、トリブチルホスフィンおよびトリオクチルホスフィンが挙げられる。
【0024】
本発明の範囲内において、任意に使用されるEの前駆体は、酸素前駆体、セレン前駆体、テルル前駆体およびこれらの混合物からなる群から選択される。
有利なことには、Eの前駆体は、有機溶媒に溶解した元素セレン;有機溶媒に溶解した元素テルル;酢酸亜鉛;ホスフィンセレニド;酸化ホスフィン;式E’(Si(R14)3)2(式中、E’はSeまたはTeを表し、各R14は、同一または異なって、1〜10個の炭素原子、特に1〜6個の炭素原子、とりわけ1〜3個の炭素原子を有する直鎖、分岐鎖または環状の、任意に置換されたアルキル基を表す)を有する化合物、およびこれらの混合物から選択される。
有利なことには、元素セレンおよび/またはテルル元素が溶解する有機溶媒は、トリアルキルホスフィン(ここでアルキル基は4〜12個の炭素原子を含む)およびアルケンから選択される。使用するのに好適な有機溶媒の例としては、1−オクタデセン、トリブチルホスフィンおよびトリオクチルホスフィンが挙げられる。
より詳細には、本発明の範囲内にて使用するのに好適なホスフィンセレニドおよび酸化ホスフィンは、それぞれ、トリアルキルホスフィンセレニドおよびトリアルキルホスフィンオキシドから選択され、ここでアルキル基は4〜12個の炭素原子を含む。
【0025】
Aの少なくとも1つの前駆体、Bの少なくとも1つの前駆体、少なくとも1つの亜鉛前駆体、少なくとも1つの硫黄前駆体、および任意にEの少なくとも1つの前駆体の混合物、特に本発明に従う方法の工程(a)にて調製された混合物は、有機溶媒中にて製造される。
有利なことには、有機溶媒は、T2より高い、すなわち本発明に従う方法の工程(b)のために選択される温度よりも高い沸点を有する、アルカン、二級または三級アミン、またはアルケンである。
「アルカン」という用語は、本発明の範囲内において、1〜40個の炭素原子、特に10〜25個の炭素原子、特に14〜20個の炭素原子を有する直鎖、分岐鎖または環状の、任意に置換されたアルカンを指す。
「二級または三級アミン」という用語は、本発明の範囲内においてジアルキルアミンおよびトリアルキルアミンを指し、ここでアルキル基は、4〜24個の炭素原子、特に8〜20個の炭素原子を含む。例えば、本発明の範囲内において使用するのに好適な二級(三級)アミンは、ジオクチルアミン(トリオクチルアミン)であり、それはアルキル鎖あたり8個の炭素原子を含む。
【0026】
「アルケン」という用語は、本発明の範囲内において、2〜40個の炭素原子、特に10〜25個の炭素原子、とりわけ14〜20個の炭素原子を有する直鎖、分岐鎖または環状の、任意に置換されたアルケンを指す。
より詳細には本発明に従う方法において前駆体混合物を調製するために使用される溶媒の1つは1−オクタデセンである。
さらに、この溶媒中の前駆体混合物はさらに、一級アミンを含有してもよい。有利なことには、一級アミンはアルキルアミンであり、ここでアルキル基は4〜24個の炭素原子、特に8〜20個の炭素原子を含む。例えば、本発明の範囲内において使用するのに好適な一級アミンは、ヘキサデシルアミン(HAD、16個の炭素原子を有するアルキルアミンである)である。
方法の工程(a)中、混合物中のAの前駆体の濃度およびBの前駆体の濃度は、2.5〜150mmol/l、特に5〜100mmol/l、とりわけ10〜20mmol/lである。
亜鉛前駆体の濃度および硫黄前駆体の濃度、任意にEの前駆体の濃度は、5〜300mmol/l、特に10〜200mmol/l、とりわけ20〜40mmol/lである。
ナノ結晶のシェルが多層である場合、シェルの外側層以外の層を形成する種々の元素、金属および非金属の前駆体は、反応混合物中、5〜300mmol/l、特に10〜200mmol/l、とりわけ20〜40mmol/lの濃度で存在する。
【0027】
本発明は、Aの少なくとも1つの前駆体、Bの少なくとも1つの前駆体、少なくとも1つの亜鉛前駆体、少なくとも1つの硫黄前駆体、および任意にEの少なくとも1つの前駆体を含む反応混合物を漸進加熱する必須工程に基づく。この工程の間、混合物の初期温度、すなわち温度T1から高温T2への混合物の漸進加熱により、ナノ結晶のコアの形成が得られた後、続いてシェルの形成を連続的に得ることができる。多層シェルの場合、層の形成は、シェルの連続する層の一連の形成からなる。
【0028】
有利なことには、前駆体混合物の温度T1は、50℃未満、特に40℃未満、とりわけ30℃未満である。より詳細には、前駆体混合物は、周囲温度にある。「周囲温度」という用語は、20℃±5℃の温度を指す。
温度T2は、180℃を超える、特に210℃を超える、とりわけ210〜320℃、より詳細には230℃〜300℃である。有利なことには、温度T2は、270℃の領域にある。「270℃の領域」という用語は、270℃±20℃の温度、特に270℃±10℃の温度を指す。
本発明に従う方法の加熱工程、特に工程(b)の第1の実施形態において、温度T1から温度T2への移行は、線形上昇様式にて行われる。
有利なことには、温度の線形上昇は、毎秒1〜20℃の勾配、特に毎秒2.5〜15℃の勾配、より詳細には毎秒5〜10℃の勾配を用いて行われる。
本発明に従う方法の加熱工程、特に工程(b)の第2の実施形態において、温度T1から温度T2への移行は、少なくとも1つの段階を有する上昇様式で行われる。
「段階」という用語は、T1とT2との間にある温度Tを指し、それは5秒から2時間、特に15秒から1時間、とりわけ30秒から30分間、より詳細には1分から15分の時間一定に維持される。温度T1とT2との間にある温度の上昇は、1〜10段階、特に2〜5段階を含んでいてもよい。T1と第1の段階との間、2つの連続段階の間、および最終段階とT2との間において、温度は、加熱工程の第1の実施形態に関して定義された通りの条件下で線形様式で上昇する。
【0029】
本発明に従う方法の加熱工程中(すなわち、工程(b))、温度T2に到達したら、この温度は、5分から5時間、特に15分から3.5時間、とりわけ30分から2時間の期間一定に維持されてもよい。
当業者には、前駆体混合物を温度T1から温度T2まで漸進的に加熱するための種々の技術および種々の手段が公知である。例としては、前駆体混合物を含有するサーモスタット制御のプログラム可能なフラスコまたは反応容器の使用、または前駆体混合物を含有するフラスコもしくは反応容器が浸漬される1つの段階の温度もしくは温度T2であり得る必須温度に予め加熱された浴の使用が挙げられる。
【0030】
本発明の範囲内で使用されるすべての前駆体は、市販の製品または当業者が少なくとも1つの調製方法を知っている製品のいずれかである。
本発明に従う方法の工程(a)の第1の代替実施形態において、使用される種々の前駆体、すなわちAの前駆体、Bの前駆体、亜鉛前駆体、硫黄前駆体および任意にEの前駆体は、任意に予め調製された後、共に混合される。有利なことには、方法の工程(a)にて使用される少なくとも1つの前駆体が予め調製される場合、工程(a)中に使用される溶媒と同一または異なる、有利なことには同一の非プロトン性溶媒中で調製される。
混合物の調製中、種々の前駆体は、同時に、次々におよび/または少なくとも2つの前駆体の群として上記で定義されたような溶媒に添加されてもよい。使用される前駆体に従って、当業者は、混合物中に前駆体を導入するのに最良の方法を決定できる。
【0031】
以降の例は、本発明に従う方法の工程(a)の第1の代替実施形態の例である。この例において、Aの前駆体は予め調製された(工程(i))インジウムトリミリステートであり、Bの前駆体はトリス(トリメチルシリル)ホスフィン(P(TMS)3)であり、亜鉛前駆体は亜鉛ジステアレートであり、硫黄前駆体はドデカンチオール(DDT)である。
この例において、方法の工程(a)は、先行する工程(i)と共に工程(ii)および(iii)に対応し、これらの工程は:
i)約100℃にて低真空下で1時間加熱することによって、オクタデセン中、酢酸インジウムを3当量のミリスチン酸(CH3−(CH2)12−COOH)と反応させることによりインジウムトリミリステートを調製する工程。この混合物を次いで周囲温度まで冷却し、不活性雰囲気(窒素またはアルゴン)中に保存する;
ii)工程(i)で調製されたインジウム前駆体の必須量を、アルゴンまたは窒素フロー中のフラスコまたは反応容器中に入れ、撹拌し、および任意にオクタデセンで希釈する工程;
iii)この混合物に、周囲温度でP(TMS)3、亜鉛ジステアレートおよびドデカンチオールを添加する工程
からなる。
工程(iii)の後、得られた反応混合物を本発明に従う方法の工程(b)および(c)に供する。
本発明に従う方法の工程(a)の第2の代替実施形態において、使用される種々の前駆体の少なくとも1つ、すなわちAの前駆体、Bの前駆体、亜鉛前駆体、硫黄前駆体、および任意にEの前駆体から選択される少なくとも1つの前駆体は、工程(a)中にインサイチュで調製される。
この代替実施形態において、インサイチュで調製されない前駆体はそれぞれ、前駆体が調製される溶媒に、互いに独立して、この調製前、調製中および/または調製後に添加されてもよい。前駆体は、次々におよび/または少なくとも2つの前駆体の群にて添加されてもよい。使用される前駆体および1つ(またはそれ以上の)前駆体をインサイチュで調製するために使用される条件に従って、当業者は、インサイチュで調製されない前駆体を混合物中に導入するための最適な方法を決定できる。
【0032】
以降の例は、本発明に従う方法の工程(a)の第2の代替実施形態の例である。この例において、Aの前駆体は、インジウムトリアセテートおよびミリスチン酸を用いてインサイチュで調製されたインジウムトリミリステートであり、Bの前駆体はP(TMS)3であり、亜鉛前駆体は亜鉛ジステアレートであり、硫黄前駆体はDDTである。工程(a)は、次からなる連続工程(i’)、(ii’)および(iii’)を含む:
i’)インジウムトリアセテート、ミリスチン酸およびP(TMS)3をフラスコ中で混合する工程;
ii’)低真空中(特に10−2mBarの領域での真空)、工程(i’)からの混合物を50〜150℃の温度に(特に110℃に)30分〜3時間の期間(特に1時間)加熱して、インジウム前駆体を形成する工程;
iii’)工程(ii’)の混合物を不活性ガス(アルゴンまたは窒素)でパージし、P(TMS)3およびDDTを添加する工程。
工程iii’)の後、得られた反応混合物を本発明に従う方法の工程(b)および(c)に供する。
【0033】
この特定例において、リン前駆体および硫黄前駆体は、インジウム前駆体の調製後に添加されて、真空中、100℃での部分蒸発を防止することに注目すべきである。
有利なことには、本発明に従う方法の工程(a)および(b)は、撹拌下で行われる。当業者に既知の種々の手段が、本発明に従う方法の工程(a)および(b)に使用される混合物を撹拌するために使用できる。例えば、混合物は、撹拌器、磁性棒、超音波浴またはホモジナイザーを用いて撹拌されてもよい。
本発明に従う方法の工程(c)中、得られたナノ結晶は、反応混合物から精製される。当業者には、沈殿工程、希釈工程および/またはろ過工程を用いる精製のための種々の技術が公知である。ルミネッセントナノ結晶を精製するために先行技術において使用される技術は、本発明に従う方法の工程(c)の範囲内に使用するのに好適である。
有利なことには、方法の工程(c)は、温度T2未満の温度にて実施される。このようにして、方法の工程(b)の後に得られた反応混合物は、冷却される、または周囲温度まで放冷される。次いでナノ結晶は、溶媒または適切な溶媒の混合物を用いて沈殿により精製される。
例えば、工程(b)の後に得られた反応混合物を希釈して体積を2倍にし、次いで過剰のアセトンを用いて沈殿させるために、メタノールおよびクロロホルムのようなアルコールの混合物(有利なことには1:1vol:volの割合で)を使用できる。ナノ結晶は、遠心分離によって回収され、次いでヘキサン、トルエンまたはクロロホルムのような有機溶媒中に分散されてもよい。本発明に従う方法の精製工程(c)は、任意に1回以上繰り返されてもよい。
【0034】
本発明は、本発明に従う方法によって得られるのに好適な、+III酸化状態での金属または非金属を表すAおよび−III酸化状態での元素を表すBを含む半導体を含むコアを有し、そのコアが、外側部分が式ZnS1−xExを有する半導体を含むシェル(ii)でコーティングされたナノ結晶のいずれかを指し、ここでEは−II酸化状態での元素を表し、xは0≦x<1となるような少数である、任意のナノ結晶に関する。
より詳細には本発明は、本発明に従う方法によって得られるのに好適な、外側部分が亜鉛および硫黄を含む半導体を含むシェルでコーティングされたインジウムおよびリンを含む半導体を含むコアを有し、周囲温度にて45%を超える、特に50%を超える、より詳細には55%を超える蛍光量子収率を有するナノ結晶に関する。
当業者は、所与のナノ結晶に関して、周囲温度にてその蛍光量子収率を得ることができる種々の技術を知っている。例としては、励起波長Yにて光学密度Xを有するヘキサン中の本発明に従うナノ結晶分散物のスペクトルに関して積分された発光強度を、同じ励起波長にて同じ光学密度を有するエタノール中のローダミン6G溶液の発光強度と比較することからなる技術が挙げられる。
【0035】
本発明がカバーする、外側部分が亜鉛および硫黄を含む半導体を含むシェルでコーティングされたインジウムおよびリンを含む半導体を含むコアを有するナノ結晶は、80nm未満、有利なことには60nm未満、とりわけ50nm未満のフォトルミネッセンス線幅を有する。
本発明は、発光ダイオード、太陽電池において、および化学または生物学的分子の蛍光標識のための、本発明に従う方法に従って調製されるのに好適なナノ結晶または本発明に従うナノ結晶の使用に関する。
本発明のさらなる特徴および利点は、例示のために与えられ、限定を目的としない以降の実施例を添付の図面を参照して読む際に、当業者に明らかになる。
【図面の簡単な説明】
【0036】
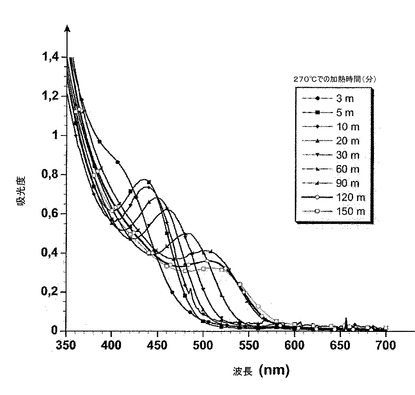
【図1】以降の項目IIにて記載される実験中にとったサンプルのUV−可視吸収スペクトルを示す。
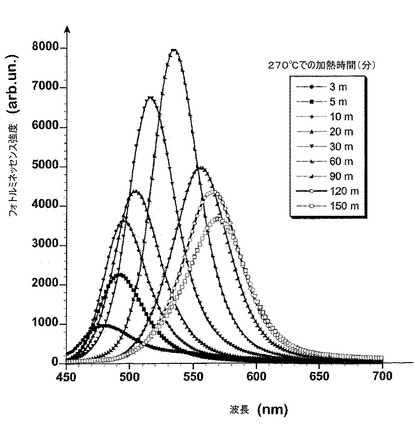
【図2】以降の項目IIに記載される実験中にとったサンプルのフォトルミネッセンススペクトルを示す。励起波長は420nmである。
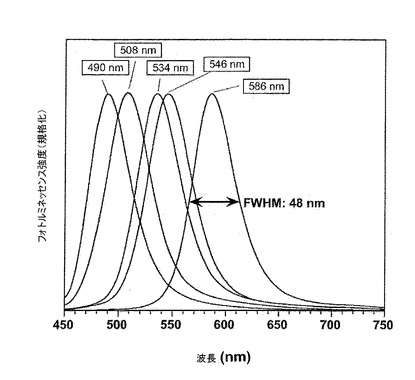
【図3】以降の項目IIIに記載されるプロトコルに従って調製されたサンプルのフォトルミネッセンススペクトルを示す。励起波長は420nmである。
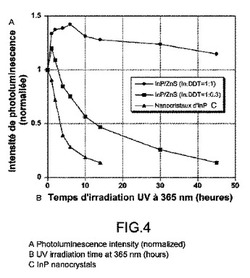
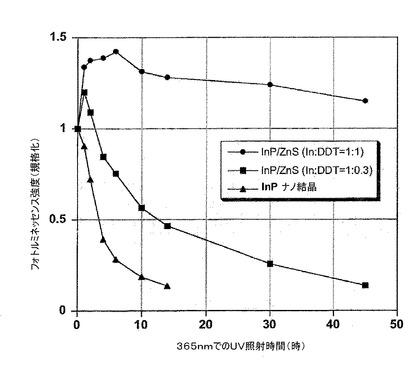
【図4】空気の存在下、UVランプ(365nm)での照射時間の関数としてのフォトルミネッセンス強度を表す。InPコアナノ結晶は、少量のドデカンチオールで調製されたナノ結晶(In:DDT=1:0.3)および以降の項目Iに記載されるプロトコルで合成されたInP/ZnS コア/シェルナノ結晶(In:DDT=1:1)に相当する。
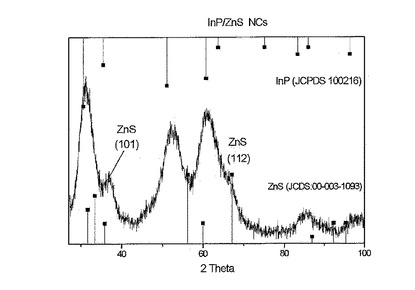
【図5】以降の項目IIに記載された方法に従って調製されるが、0.06mmolのP(TMS)3を用いて、240℃の温度にて1時間で調製されたサンプルのX線粉末回折図を示す。
【発明を実施するための形態】
【0037】
空気感応性材料の取扱はすべて、グローブボックスにてまたは真空装置を用いて行なわれる(シュレンク法)。
特徴付けのために、UV−可視吸収スペクトルは、Hewlett−Packard8425A分光計(波長スペクトル範囲:190〜820nm、解像度2nm)にて測定し、フォトルミネッセンススペクトルは、Hitachi F−4500分光計で得た。これらの分光測定に関して、ヘキサン中に希釈したコロイド状のナノ結晶溶液を、1cmの光学経路の石英セルに入れた。周囲温度での蛍光量子収率は、ヘキサン中のナノ結晶分散物のスペクトルに関して積分された発光強度を、エタノール中のローダミン6G溶液の発光強度と比較することによって得たが、両方の溶液は励起波長(490nm)にて同じ光学密度(<0.03)を有していた。X線回折図は、Coソースを用いて、50kVおよび35mAにてPhilips X’Pert装置にて得た。
亜鉛ジステアレート(Riedel de Haen)を除いてすべての製品をSigma−Aldrichから購入し、そのまま使用した:酢酸インジウム(99.99%純度)、ミリスチン酸(純度>99%)、亜鉛ジステアレート(90%純度)、ドデカンチオール(97%純度)、1−オクタデセン(90%純度)。
【0038】
I 標準プロトコル
I.A.インジウム前駆体(インジウムミリステート)の調製
2mmolのインジウムトリアセテート、6mmolのミリスチン酸、および20mlの1−オクタデセンを、50mlのフラスコ中に入れ、不活性雰囲気(アルゴンまたは窒素)中、磁性撹拌器を用いて混合する。温度を、110℃に上昇させ、フラスコを低真空ポンプを用いて1時間ポンプする(フラスコ中の圧力は10−2mBarの領域)。均質な無色の溶液が得られる。次いでフラスコを不活性ガス(アルゴンおよび窒素)で満たし、周囲温度に冷却する。前駆体溶液を不活性雰囲気中、例えばグローブボックス中に保存する。
I.B.InP/ZnS コア/シェルナノ結晶合成
不活性雰囲気中、項目I.A.にて記載されたように調製された1mlのインジウム前駆体、0.2mmolの亜鉛ジステアレート、0.1mmolのP(TMS)3、0.2mmolのドデカンチオールおよび6mlの1−オクタデセンを、冷却器を備えた50mlの3つ口フラスコに入れ、磁性撹拌器を用いて混合する。混合物を、約5℃/秒の勾配にて230〜300℃に加熱し、この温度を1〜3時間維持する。
周囲温度まで冷却した後、InP/ZnSナノ結晶は、クロロホルム/メタノール混合物(1:1 vol:vol)の1体積当量およびアセトンの10体積当量を添加し、続いて遠心分離することによって単離する。ナノ結晶を含有する得られた沈殿物は、ヘキサン、トルエンまたはクロロホルムのような有機溶媒中に分散させてもよい。
【0039】
II.インジウム前駆体溶液を予め調製しない合成プロトコル。
冷却器を備えた50mlのフラスコに、0.1mmolのインジウムトリアセテート、0.3mmolのミリスチン酸、0.1mmolの亜鉛ジステアレート、および8.6mlの1−オクタデセンを入れ、不活性雰囲気中にて磁性撹拌器を用いて混合する。
温度を110℃に上昇させ、フラスコを低真空ポンプを用いて1時間ポンプする(フラスコ中の圧力は10−2mBarの領域)。均質な無色の溶液が得られる。次いでフラスコを不活性ガス(アルゴンおよび窒素)で満たし、周囲温度に冷却する(任意)。0.1mmolのP(TMS)3および1mlの1−オクタデセン中に希釈された1mmolを、混合物に添加し、混合物を、約5℃/秒の勾配にて270℃に加熱し、この温度を150分間維持する。
サンプルを、周期的に採取し(1mlのクロロホルムで希釈された0.1mlの反応混合物)、UV−可視吸収およびフォトルミネッセンス分光法によって分析する。
周囲温度まで冷却した後、InP/ZnSナノ結晶は、クロロホルム/メタノール混合物(1:1 vol:vol)の1体積当量およびアセトンの10体積当量を添加し、続いて遠心分離することによって単離する。ナノ結晶を含有する得られた沈殿物は、ヘキサン、トルエンまたはクロロホルムのような有機溶媒中に分散させてもよい。
【0040】
III.項目IIのプロトコルの変形例
この実施例において、InP/ZnS コア/シェルナノ結晶の合成を、上記で記載されたプロトコルIIの変形例に従って行った。
プロトコルは、項目IIにて記載された方法を順守するが、以降の表に要約される反応パラメータを用いる:
【表1】

【0041】
IV.本発明に従う方法を用いて得られたナノ結晶の特徴付け
IV.A.光学特性
得られたナノ結晶のサイズ、すなわちそれらの発光色を制御するための2つの手段がある:1)反応時間を用いて2)反応パラメータを調節することによって。
項目IIに記載されたような方法の後で得られたサンプルのUV−可視吸収およびフォトルミネッセンススペクトルをそれぞれ図1および2に示す。項目IIIに記載されるような方法を用いて得られたサンプルのフォトルミネッセンススペクトルを図3に示す。
サンプルの励起子ピークが、特に反応時間の5分から120分間に得られるピークに関して明確に現れる(図1)。この特徴は狭いサイズ分散を示し、それはフォトルミネッセンススペクトル(図2)における線幅を分析することによって確認され、約40〜50nmにある(FWHM、半値全幅)。
【0042】
IV.B.光学特性の安定性
蛍光ナノ結晶の技術的用途に関して、経時的なそれらの光学特性の安定性、特に放射線誘導エージングに対して特に注意を払うできである。
得られたサンプルの安定性を試験するために、それらを365nmにて発光するUVランプでの連続照射に供した。
ランプは、空気の存在下で石英セル中にあるサンプルから4cm離して配置する。この距離における照射出力は約2〜3mWである。
図4は、3つの異なるサンプルのフォトルミネッセンス強度の進行を示す。
迅速な減衰は、ZnSシェルをもたないInPナノ結晶(本発明がカバーしていない比較の目的のために使用されるナノ結晶)に関して観察され、10〜15時間照射後にそれらの発光は実際すべて失われる。
同様に、項目Iに記載されるような方法に従って調製された薄いシェルを有するサンプル(In:DDT=1:0.3)は、ZnSシェルを持たないナノ結晶の安定性と比較して改善された光学特性の安定性を有する。このようにして、12時間の照射後、50%の初期フォトルミネッセンス強度が依然として検出されるが、結局発光は実際には40〜50時間後に停止する。
最後に、多量の硫黄前駆体(In:DDT=1:1)を用いて製造されたコア/シェルサンプルは、全く異なるように挙動する。照射開始時(0〜10時間)、フォトルミネッセンス強度において初期値の約140%までの増加が観察される。45時間(実験の停止)後であっても、初期強度の110%超過が測定され、これはサンプルの非常に満足のいく光安定性を裏付ける。
【0043】
IV.C.物理化学的なX線回折分析
図5に示されるコア/シェルサンプルについてのX線粉末回折図は、InP/ZnSヘテロ構造を確認する。本実施例において使用されるコア/シェルサンプルは、項目IIに記載されるような方法に従って調製した。
結晶がナノメートルサイズであることによる拡がったピークは、InP立方相(「Joint Committee on Powder Diffraction Standard」card,JCPDS,No.100216)およびZnSの亜鉛ブレンド構造(JCPDS card No. 003−1093)の両方の寄与を含む。
【技術分野】
【0001】
本発明は、可視スペクトルの大部分をカバーしながら、既存のシステムに対して高い色純度および向上した蛍光量子収率を有する半導体に基づく「コア/シェル」ナノ結晶に基づく新規なルミネッセント材料を製造する方法に関する。
本発明はまた、得られたルミネッセント材料およびそれらの種々の用途に関する。実際、これらのナノ結晶の用途分野は、例えば発光ダイオード、太陽電池、および化学または生物学的分子の蛍光標識を含む。
【背景技術】
【0002】
半導体結晶は、数十年にわたって知られているルミネッセント材料である。1980年代から1990年代において、それらの発光スペクトルは、その結晶サイズが十分に小さくなる場合にはそのサイズに依存することが明示された。「ナノ結晶」または「量子ドット」と呼ばれる1〜10nmの範囲にほぼ位置するサイズの結晶に関して、こうした依存性は極めて強く現れる。事実、可視および近赤外および紫外範囲の色パレット全体は、半導体ナノ結晶を用いて、その結晶サイズおよび組成を適切に選択することによって得ることができる。ライトおよびディスプレイ(フラットスクリーン)の分野において非常に重要な可視スペクトルをカバーするために、最も研究された材料は、カドミウムカルコゲニド(CdS、CdSe、CdTe)である。しかし、欧州RoHS(「危険物質に関する制限」) の指令は、2006年7月1日から欧州にて販売される電気および電子機器(EEE)において、次の物質を除去することを目標としている:鉛、水銀、カドミウム、六価クロム、ポリブロモビフェニル(PBB)、ポリブロモジフェニルエーテル(PBDE)。故に、有利なことには、所望の光学特性を保持しながら、ナノ結晶の製造用にこれらの物質を含有しない代替材料を見出すことが重要である。
【0003】
通則として、ナノ結晶からなるルミネッセント材料の光学品質は複数のパラメータに依存し、そのうち最も重要なものは:
−発光波長を支配するナノ結晶のサイズ;
−発光バンド幅を制御するナノ結晶のサイズ分布;
−蛍光量子収率および経時的な安定性に関与するナノ結晶の表面不活性化
である。
リン化インジウム(InP)は、カドミウム系ナノ結晶を置換するために最も有利な材料の1つである。1.35eVの禁制バンド幅のために、そのサイズを変更することによって、可視スペクトルおよび近赤外スペクトルにおけるInPナノ結晶の発光波長を変更できる。
InPナノ結晶を調製するためには複数の方法があり、その方法のうち、現時点では、有機溶媒中、高温での迅速な前駆体注入方法が最も狭いサイズ分散に近づくことができる。ナノ結晶のサイズ分散が狭いと、狭い発光スペクトル(純粋な発光色)を生じる;すなわちこれは、技術的な用途に有利である。2つの主要な合成プロセスが利用可能である。
【0004】
いわゆる「従来の」有機金属合成プロセスである第1のプロセスは、高温(270〜300℃)において、反応性媒体として作用するトリオクチルホスフィンオキシド(TOPO)中、インジウム前駆体(例えば塩化インジウムまたはシュウ酸クロロインジウム)をリン前駆体(トリス(トリメエチルシリル)ホスフィン、P(TMS)3、P(Si(CH3)3)3)とを反応させることからなる(非特許文献1〜3)。TOPOは、ナノ結晶の表面での結合能力を強め、コロイド形態にてナノ結晶を安定化させるために「配位溶媒」と呼ばれる。この方法の主な欠点は、調製されるナノ結晶のサイズ分散が広く、反応時間が長いことである(3〜7日間)。
第2の合成プロセスは、2002年に公開された「従来の」合成プロセスにおける変更である。ここで、反応性媒体は、TOPOの代わりに、「非配位溶媒」として分類される単なるアルケンの1−オクタデセンである(非特許文献4)。さらに、ラウリン酸、ミリスチン酸またはパルミチン酸のような脂肪酸が、安定化配位子として使用された。反応は、インジウム前駆体(酢酸インジウム)および安定化剤を含有する300℃に加熱された溶媒中で、リン前駆体(P(TMS)3)の迅速な注入から開始される。この著者らは、約10%の範囲にあるサイズ分散の改善を報告している。さらに、反応時間は3〜4時間に短縮される。
しかし、上述の調製方法は、蛍光量子収率が依然として低いままである(通常1%未満)という問題を解決できない。種々の材料(例えばCdSe、CdSなど)のナノ結晶の量子収率を増大させるために広く使用されている方法の1つは、「コア」の周りに、より大きな禁制バンド幅を有する半導体シェルを成長させることによってそれらの表面を不活性化することからなる。これは、科学文献において「コア/シェル」システムと呼ばれる。シェルを堆積させるために使用される方法は、本質的にコアを調製するために使用されるものと同じである。
InPナノ結晶の光学特性を改善するために、種々のシェル材料が提案されている。Micicらは、コアとシェル材料との間の格子不整合を最小限にするために、InP表面上での三元化合物CdZnSe2の合成を記載している(非特許文献5)。顕著な格子不整合は、シェル中にてまたはコア/シェル界面にて結晶上の欠陥を誘導し、それにより蛍光効率を低下させ得る。それでもなお、このコア/シェルシステムで達成される量子収率(10%)は、カドミウム系ナノ結晶で得られる値(50〜85%)に比べて大きく劣っている。
【0005】
Hauboldらは、InPをZnSでコーティングし、3日後の15%から3週間後の23%までの間の量子収率を得た(非特許文献6)。これらのコア/シェルナノ結晶のこうした合成に使用される方法は:
a1)上述した第1の合成プロセスに従うInPコア結晶の合成;
a2)沈殿およびメタノールでの洗浄によるInP結晶の精製;
a3)ZnSシェルの成長。この工程に関して、InPナノ結晶は、200℃にてトリオクチルホスフィン(TOP)中に分散され、ジエチル亜鉛およびビス(トリメチルシリル)スルフィドの混合物が注入される。次いで温度を一時的に260℃まで上昇させた後、1時間で100℃に低下させる。続いて、反応混合物を不活性雰囲気(窒素)中に数日間または数週間保持する;
a4)ナノ結晶の沈殿およびメタノールでの洗浄による精製;
a5)選択的沈殿によるサイズ画分へのサンプル分離
を含む。
この方法に関連する問題は、シェルの成長のための自然発火性の前駆体(ジエチル亜鉛およびビス(トリメチルシリル)スルフィド)の使用、非常に長期間(数週間)での光学特性の進行、および最終的にII−VI半導体に基づくコア/シェルシステムに関連する相対的に低い量子収率が原因である。著者らは、InPナノ結晶のZnSシェルによるコーティングが完全でなく、そのことが蛍光量子収率を制限すると結論している。
【0006】
より最近では、Pengらが、InP/ZnS コア/シェルナノ結晶合成のための新しいプロトコルを提案した(非特許文献7)。この場合、コア結晶の合成は、上述されたような第2の合成プロセスと同様であるが、リン前駆体と同時に注入されるアミン(1−オクチルアミン)の添加により、270〜300℃の代わりに180〜190℃での反応にすることができる。次いでシェルは次のように成長される:
b1)150℃にて、亜鉛ジステアレート(亜鉛前駆体)の特定量を、InPナノ結晶を含有するフラスコに注入する。10分後、オクタデセン中に溶解した同量の元素硫黄(硫黄前駆体)を添加する;
b2)温度を30分間220℃に上昇させて、ZnSシェルを成長させる;
b3)温度を再び150℃に低下させて、より多量にて、上記と同じ順番で亜鉛前駆体、その後の硫黄前駆体の添加を繰り返す;
b4)温度を30分間で220℃に上昇させる;
b5)ナノ結晶を、ヘキサンを添加し、メタノールで抽出することによって精製する。
【0007】
この方法により、シェルを成長させるための自然発火性の前駆体を排除でき、コア結晶の合成とシェルの成長との間での中間精製を排除できるが、この方法は、実施が複雑なままである。それらを産業規模へ適合させることに関する問題は、特に、InP結晶の合成中の自然発火性リン前駆体の迅速な注入および複数のZnS前駆体注入中での温度制御に関連している。この方法で得られたナノ結晶の蛍光量子収率は40%までである。この値はInP系ナノ結晶に関して公開された最良のものであるが、依然としてカルシウム−カルゴゲニド系ナノ結晶よりも劣る。
要約すれば、これまで報告されたInP系ナノ結晶を製造するための方法は、50%を超える蛍光量子収率を達成できず、その技術的用途が制限される。低いサイズ分散を有するInPナノ結晶の合成について記載されている方法は、高温にて、反応媒体中への自然発火性前駆体の迅速な注入を含み、それは大規模で行うには困難である。
最後に、InP/ZnS コア/シェルナノ結晶の合成は、2工程の方法を必要とし、InPコア結晶の合成と、続くZnSシェルの成長を必要とし、その間に、正確に温度を制御しながら、再び前駆体を反応媒体に注入する必要がある。
【先行技術文献】
【非特許文献】
【0008】
【非特許文献1】Micic, O. I.;Curtis, C. J.; Jones, K. M.; Sprague, J. R.; Nozik, A. J., SYNTHESIS ANDCHARACTERIZATION OF INP QUANTUM DOTS. Journal of Physical Chemistry 1994, 98,(19), 4966-4969
【非特許文献2】Guzelian, A. A.;Katari, J. E. B.; Kadavanich, A. V.; Banin, U.; Hamad, K.; Juban, E.;Alivisatos, A. P.; Wolters, R. H.; Arnold, C. C.; Heath, J. R., Synthesis ofsize-selected, surface-passivated InP nanocrystals. Journal of PhysicalChemistry 1996, 100, (17), 7212-7219
【非特許文献3】Micic, O. I.;Sprague, J. R.; Curtis, C. J.; Jones, K. M.; Machol, J. L.; Nozik, A. J.;Giessen, H.; Fluegel, B.; Mohs, G.; Peyghambarian, N., SYNTHESIS ANDCHARACTERIZATION OF INP, GAP, AND GAINP2 QUANTUM DOTS. Journal of PhysicalChemistry 1995, 99, (19), 7754-7759
【非特許文献4】Battaglia, D.;Peng, X. G., Formation of high quality InP and InAs nanocrystals in anoncoordinating solvent. Nano Letters 2002, 2, (9), 1027-1030
【非特許文献5】Micic, O. I.; Smith, B. B.; Nozik, A. J., Core-shell quantum dots of lattice-matched ZnCdSe2 shellson InP cores: Experiment and theory. Journal of Physical Chemistry B 2000, 104,(51), 12149-12156
【非特許文献6】Haubold, S.;Haase, M.; Kornowski, A.; Weller, H., Strongly luminescent InP/ZnS core-shellnanoparticles. Chemphyschem 2001, 2, (5), 331-334
【非特許文献7】Xie, R.;Battaglia, D.; Peng, X., Colloidal InP nanocrystals as efficient emitterscovering blue to near-infrared. Journal of the American Chemical Society 2007,129, (50), 15432-15433
【発明の概要】
【発明が解決しようとする課題】
【0009】
本発明は、上記で列挙された技術的問題および欠点を解決する。
実際、本発明は、現在までに報告されている値よりも高く、特に周囲温度において50%を超える蛍光量子収率を有し、光酸化に対して高い安定性を有するInP/ZnS コア/シェルナノ結晶を合成する新規な方法を提案する。
本発明に従う方法は、InP/ZnS コア/シェルナノ結晶合成が単一工程にて行われるので、先行技術に記載されるすべての方法よりも非常に簡便である。本発明に従う方法は、中間精製工程を必要としない。さらに、サイズ分類工程(例えば選択的沈殿による)は、合成直後に得られたサンプルのサイズ分散が狭いので必要でない。
さらに、方法は、前駆体の注入、特に高温にて反応媒体中への自然発火性リン前駆体の迅速な注入がないことを特徴とする。前駆体は、周囲温度にて反応媒体に単に添加するだけでよい。さらに、反応の開始後および温度の上昇後にいかなる前駆体の添加も必要としない。
最後に、本発明に従う方法は、InP/ZnS コア/シェルナノ結晶の調製を可能にすることだけでなく、一般化として式ABを有する半導体を含むコアおよび式CDを有する少なくとも1つの半導体を含むシェルを有するコア/シェルナノ結晶の調製にも好適であることを特徴とし、ここでAは+III酸化状態における金属または非金属を表し、Bは−III酸化状態の元素を表し、Cは+II酸化状態の金属または非金属を表し、Dは−II酸化状態の元素を表す。
【課題を解決するための手段】
【0010】
本発明は、(i)+III酸化状態での金属または非金属を表すAおよび−III酸化状態での元素を表すBを含む半導体を含むコアを有し、このコアが、(ii)外側部分が式ZnS1−xExを有する半導体を含むシェルでコーティングされているナノ結晶を調製するための方法に関し、ここでEは−II酸化状態の元素を表し、xは0≦x<1となるように少数であり、この方法は、Aの少なくとも1つの前駆体、Bの少なくとも1つの前駆体、亜鉛の少なくとも1つの前駆体、硫黄の少なくとも1つの前駆体、および任意にEの少なくとも1つの前駆体の混合物を、まずコアが形成され、次いでシェルが形成されるように上昇する様式で温度T1から、T1を超える温度T2まで加熱することからなる工程を含む。
【0011】
より詳細には、本発明に従う方法は次の工程を含む:
a)Aの少なくとも1つの前駆体、Bの少なくとも1つの前駆体、少なくとも1つの亜鉛前駆体、少なくとも1つの硫黄前駆体、および任意にEの少なくとも1つの前駆体の混合物を温度T1で調製する工程、
b)工程(a)にて得られた混合物を温度T2に加熱する工程、
c)工程(b)にて得られた、式ABを有する半導体を含むコアを有し、外側層が式ZnS1−xExを有する半導体を含むシェルによって囲まれているナノ結晶を精製する工程。
本発明に従う方法に従って調製されたナノ結晶のコアは、+III酸化状態での金属または非金属を表すAおよび−III酸化状態での元素を表すBを含む半導体を含み、このタイプの半導体はIII−Vと称される。
+III酸化状態での金属または非金属であって、Bとともに、本発明に従って調製されるナノ結晶のコアに含まれる半導体を形成するAは、ガリウム(Ga)、インジウム(In)、アルミニウム(Al)、およびこれらの混合物から選択される。
【0012】
−III酸化状態での元素であって、Aと共に、本発明に従って調製されるナノ結晶のコアに含まれる半導体を形成するBは、アンチモン(Sb)、ヒ素(As)、リン(P)、窒素(N)およびこれらの混合物から選択される。
本発明に従う方法に従って調製されたナノ結晶のコアに含まれる式ABを有する半導体の例は、GaAs、GaSb、GaN、GaP、InAs、InSb、InN、InP、AlAs、AlSb、AlN、AlP、InGaAs、InGaSb、InGaN、InGaP、InAlAs、InAlSb、InAlN、InAlP、GaAlAs、GaAlSb、GaAlN、GaAlPおよびこれらの混合物である。特に、ナノ結晶のコアは、InP、InAs、InGaPおよびこれらの混合物から選択される式ABを有する半導体を含み、より詳細にはこの半導体はInPである。
有利なことには、本発明に従う方法に従って調製されるナノ結晶のコアは、上記で定義された半導体だけからなる。
【0013】
本発明の方法に従って調製されたナノ結晶はシェルを含み、このシェルの外側部分は、式ZnS1−xExを有する半導体を含み、ここでEは−II酸化状態の元素を表し、xは0≦x<1となるような少数である。
Eは、特に酸素(O)、セレン(Se)、テルル(Te)およびこれらの混合物から選択される−II酸化状態の元素である。
本発明に従う方法に従って調製されるナノ結晶のシェルは、単層または複数の層(すなわち、多層シェル)からなってもよい。シェルが単層のみからなる場合、シェルの外側部分はこの層に対応する。
シェルが複数の層からなる場合、シェルの外側部分は、シェルの外側層に対応する。「外側層」という用語は、本発明の範囲内において、ナノ結晶のコアから最も遠く、ナノ結晶が置かれた媒体または環境と直接接触するシェルの層を指す。多層シェルは、2〜10、特に2〜5個の異なる半導体層を含んでいてもよい。このようにして、種々の代替実施形態が、本発明に従う方法を用いて調製されるナノ結晶のシェルについて想定できる。
【0014】
第1の代替実施形態において、式ZnS1−xExのxは0に等しく、シェルは、1つの層、すなわちZnS層でのみ形成されている。
第2の代替実施形態において、式ZnS1−xExのxは、0<x<1となるようなものであり、シェルは1つの層でのみ形成される。
第3の代替実施形態において式ZnS1−xExのxは0に等しく、シェルは少なくとも2つの異なる層を含み、その外側層はZnS層である。
第4の代替実施形態において、式ZnS1−xExのxは、0<x<1となるようなものであり、シェルは少なくとも2つの異なる層を含む。
さらに、本発明に従う方法に従って調製されるナノ結晶のシェルの層は、均一な化学組成を有していてもよく、または同じ層内において、異なる化学組成を有していてもよく、特に化学組成が勾配の形態であってもよい。この場合、シェルの外側部分は、1つの層を有するシェルの外側区域および多層シェルの外側層の外側区域からなる。
多層シェルの場合、ナノ結晶のコアと、上記で定義された式ZnS1−xExを有する外側層との間に含まれる層は、上記で定義された式ABを有する半導体および/または式CDを有する半導体を含んでいてもよく、ここでCは+II酸化状態での金属または非金属を表し、Dは−II酸化状態での元素を表す。式CDを有する半導体はまたII−VIと称される。
【0015】
+II酸化状態の金属または非金属Cは、特に、マグネシウム(Mg)、カルシウム(Ca)、ストロンチウム(Sr)、バリウム(Ba)、亜鉛(Zn)、カドミウム(Cd)、水銀(Hg)、スズ(Sn)、鉛(Pb)、およびこれらの混合物から選択される。−II酸化状態の元素Dは、特に、酸素(O)、硫黄(S)、セレン(Se)、テルル(Te)、およびこれらの混合物から選択される。ナノ結晶のコアとシェルの外側層との間に含まれる層に存在してもよい半導体の例は、例えば、MgS、MgSe、MgTe、CaS、CaSe、CaTe、SrS、SrSe、SrTe、BaS、BaSe、BaTe、ZnO、ZnS、ZnSe、ZnTe、CdS、CdSe、CdTe、HgS、HgSe、HgTe、SnS、SnSe、SnTe、PbS、PbSe、PbTeおよびこれらの混合物である。より詳細には、ナノ結晶のコアとシェルの外側層との間に含まれる層に存在してもよい半導体の例は、MgS、MgSe、MgTe、CaS、CaSe、CaTe、SrS、SrSe、SrTe、BaS、BaTe、ZnO、ZnS、ZnSe、ZnTe、Sns、SnSe、SnTeおよびこれらの混合物からなる群から選択される。
故に、多層シェルの場合、本発明の範囲内、特に工程(a)にて使用される反応混合物は、シェルの外側層以外の層を形成する元素の前駆体をさらに含有することは明らかである。
【0016】
本発明の方法に従って調製されるナノ結晶は、15nm未満、特に12nm未満、特に10nm未満の直径を有する。
本発明に従う方法を用いて調製されたナノ結晶のコアは、1〜10nmの直径、特に1.5〜8nm、特に2〜6nmの直径を有する。
本発明に従う方法を用いて調製されるナノ結晶のシェルは、0.3〜6nm、特に0.5〜4nm、特に1〜2nmの厚さを有する。
本発明の範囲内において、使用されるAの前駆体は、インジウム前駆体、ガリウム前駆体、アルミニウム前駆体およびこれらの混合物からなる群から選択される。当業者に既知のインジウム、アルミニウムおよびガリウム前駆体の全て、特に液体または固体形態の前駆体は、本発明に使用するのに好適である。
有利なことに、Aの前駆体は、Aの塩、Aのハロゲン化物、Aの酸化物およびAの有機金属化合物から選択される。「Aの有機金属化合物」という用語は、より詳細には、Aの三置換化合物、Aのカルボン酸塩、またはAのホスホン酸塩を指す。
【0017】
「Aの三置換化合物」という用語は、本発明の範囲内において、式(R1)3Aを有する化合物を指し、ここで各R1は、同一または異なって、1〜20個の炭素原子を有する炭化水素基、例えばアルキルラジカル、アルケニルラジカル、アルコキシラジカル、アリールラジカルまたはアリールオキシラジカルを表す。
「Aのカルボン酸塩」という用語は、本発明の範囲内において、式(R2COO)3Aを有する化合物を指し、ここで、各R2は、同一または異なって、1〜20個の炭素原子を有する炭化水素基、例えばアルキルラジカル、アルケニルラジカル、アルコキシラジカル、アリールラジカルまたはアリールオキシラジカルを表す。
「Aのホスホン酸塩」という用語は、本発明の範囲内において、式[R3−P(OR4)(OR5)O]3Aを有する化合物を指し、ここで
−各R3は、同一または異なって、1〜20個の炭素原子を有する炭化水素基、例えばアルキルラジカル、アルケニルラジカル、アルコキシラジカル、アリールラジカルまたはアリールオキシラジカルを表し;
−各R4は、同一または異なって、水素原子または1〜20個の炭素原子を有する炭化水素基、例えばアルキルラジカル、アルケニルラジカル、アルコキシラジカル、アリールラジカルまたはアリールオキシラジカルを表し;
−各R5は、同一または異なって、水素原子または1〜20個の炭素原子を有する炭化水素基、例えばアルキルラジカル、アルケニルラジカル、アルコキシラジカル、アリールラジカルまたはアリールオキシラジカルを表す。
【0018】
本発明の範囲内において、特に指示がない限り、「アルキル基」という用語は、1〜20個の炭素原子、特に1〜15個の炭素原子、とりわけ1〜10個の炭素原子を有する直鎖、分枝鎖または環状の、任意に置換されたアルキル基を指す。
本発明の範囲内において、「アルケニル基」という用語は、2〜20個の炭素原子、特に2〜15個の炭素原子、とりわけ2〜10個の炭素原子を有する直鎖、分枝鎖または環状の、任意に置換されたアルケニル基を指す。
【0019】
本発明の範囲内において、「アルコキシ基」という用語は、上記で定義されたアルキルによって置換された酸素原子を指す。
本発明の範囲内において、「アリール基」という用語は、6〜20個の炭素原子、特に6〜14個の炭素原子、とりわけ6〜8個の炭素原子を有する単環式または多環式の、任意に置換された芳香族基を指す。
本発明の範囲内において、「アリールオキシ基」という用語は、上記で定義されたアリールによって置換された酸素原子を指す。
本発明の範囲内において、「任意に置換された」という用語は、アルキル基、アルコキシ基、ハロゲン、ヒドロキシル、シアノ、トリフルオロメチルまたはニトロから選択される1つまたは複数の基によって置換されたラジカルを指す。
本発明の範囲内において、「ハロゲン」という用語は、フッ素、塩素、臭素またはヨウ素を指す。
【0020】
本発明の範囲内にて使用するのが好適なAの前駆体の例は、Aがインジウムである場合、インジウムトリクロライド、トリエチル−インジウム、インジウムトリアセテート、インジウムトリ(アセチル−アセトネート)、インジウムトリオクトネート、インジウムトリステアレート、インジウムトリラウレート、インジウムトリパルミテート、インジウムトリミリステート、インジウムトリオレエート、およびこれらの混合物が挙げられる。
【0021】
本発明の範囲内において、使用されるBの前駆体は、アンチモン前駆体、ヒ素前駆体、リン前駆体、窒素前駆体およびこれらの混合物から成る群から選択される。当業者に既知のアンチモン、ヒ素、リンおよび窒素前駆体のすべて、特に液体または固体形態の前駆体は、本発明に使用するのに好適である。
有利なことには、本発明に使用されるBの前駆体は、式B(F(R6)3)3または式B(R7)3を有する化合物であり、ここで
−各Fは、シリカ(Si)、ゲルマニウム(Ge)およびスズ(Sn)から成る群から選択され;
−各R6は、同一または異なって、1〜10個の炭素原子、特に1〜6個の炭素原子、とりわけ1〜3個の炭素原子を有する直鎖、分枝鎖または環状の、任意に置換されたアルキル基であり;
−各R7は、同一または異なって、水素原子、ハロゲン、例えば塩素(Cl)、臭素(Br)、ヨウ素(I)もしくはフッ素(F)、または1〜10個の炭素原子、特に1〜6個の炭素原子、とりわけ1〜3個の炭素原子を有する直鎖、分枝鎖または環状の、任意に置換されたアルキル基である。
特に、Bの前駆体、特に好ましいリン前駆体は、式B(Si(CH3)3)3またはB(TMS)3を有するBのトリス(トリメチルシリル)化合物である。
【0022】
本発明の範囲内において、使用される亜鉛前駆体は、亜鉛塩、ハロゲン化亜鉛、酸化亜鉛および有機金属亜鉛化合物から成る群から選択される。「有機金属亜鉛化合物」という用語は、より詳細には、二置換された亜鉛化合物、カルボン酸亜鉛、またはホスホン酸亜鉛を指す。
本発明の範囲内において、「二置換された亜鉛化合物」という用語は、式(R8)2Znを有する化合物を指し、各R8は、同一または異なって、1〜20個の炭素原子を有する炭化水素基、例えばアルキルラジカル、アルケニルラジカル、アルコキシラジカル、アリールラジカルまたはアリールオキシラジカルを表す。
本発明の範囲内において、「カルボン酸亜鉛」という用語は、式(R9COO)2Znを有する化合物を指し、ここで各R9は、
同一または異なって、1〜20個の炭素原子を有する炭化水素基、例えばアルキルラジカル、アルケニルラジカル、アルコキシラジカル、アリールラジカルまたはアリールオキシラジカルを表す。
本発明の範囲内において、「ホスホン酸亜鉛」という用語は、式[R10−P(OR11)(OR12)O]2Znを有する化合物を指し、ここで
−各R10は、同一または異なって、1〜20個の炭素原子を有する炭化水素基、例えばアルキルラジカル、アルケニルラジカル、アルコキシラジカル、アリールラジカルまたはアリールオキシラジカルを表し;
−各R11は、同一または異なって、1〜20個の炭素原子を有する炭化水素基、例えばアルキルラジカル、アルケニルラジカル、アルコキシラジカル、アリールラジカルまたはアリールオキシラジカルを表し;
−各R12は、同一または異なって、1〜20個の炭素原子を有する炭化水素基、例えばアルキルラジカル、アルケニルラジカル、アルコキシラジカル、アリールラジカルまたはアリールオキシラジカルを表す。
アルキル、アルケニル、アルコキシ、アリールおよびアリールオキシラジカルは、Aの前駆体に関して定義された通りである。
【0023】
本発明の範囲内において、使用される硫黄前駆体は、脂肪族チオール、有機溶媒に溶解した元素硫黄、および式S(Si(R13)3)2を有する化合物から成る群から選択され、ここで、各R13は、同一または異なって、1〜10個の炭素原子、特に1〜6個の炭素原子、とりわけ1〜3個の炭素原子を有する直鎖、分枝鎖または環状の、任意に置換されたアルキル基である。
有利なことには、脂肪族チオールは、式CnH2n+1−SHを有し、ここでnは1〜25、特に5〜20、とりわけ8〜18の整数を表す。本発明の範囲内にて使用するのに好適な脂肪族チオールの例としては、オクタンチオール(n=8)、オクトデカンチオール(n=18)、ドデカンチオール(n=12)およびこれらの混合物が挙げられる。
有利なことには、元素硫黄が溶解する有機溶媒は、トリアルキルホスフィン(ここでアルキル基は4〜12個の炭素原子を含む)およびアルケンから選択される。使用するのに好適な有機溶媒の例としては、1−オクタデセン、トリブチルホスフィンおよびトリオクチルホスフィンが挙げられる。
【0024】
本発明の範囲内において、任意に使用されるEの前駆体は、酸素前駆体、セレン前駆体、テルル前駆体およびこれらの混合物からなる群から選択される。
有利なことには、Eの前駆体は、有機溶媒に溶解した元素セレン;有機溶媒に溶解した元素テルル;酢酸亜鉛;ホスフィンセレニド;酸化ホスフィン;式E’(Si(R14)3)2(式中、E’はSeまたはTeを表し、各R14は、同一または異なって、1〜10個の炭素原子、特に1〜6個の炭素原子、とりわけ1〜3個の炭素原子を有する直鎖、分岐鎖または環状の、任意に置換されたアルキル基を表す)を有する化合物、およびこれらの混合物から選択される。
有利なことには、元素セレンおよび/またはテルル元素が溶解する有機溶媒は、トリアルキルホスフィン(ここでアルキル基は4〜12個の炭素原子を含む)およびアルケンから選択される。使用するのに好適な有機溶媒の例としては、1−オクタデセン、トリブチルホスフィンおよびトリオクチルホスフィンが挙げられる。
より詳細には、本発明の範囲内にて使用するのに好適なホスフィンセレニドおよび酸化ホスフィンは、それぞれ、トリアルキルホスフィンセレニドおよびトリアルキルホスフィンオキシドから選択され、ここでアルキル基は4〜12個の炭素原子を含む。
【0025】
Aの少なくとも1つの前駆体、Bの少なくとも1つの前駆体、少なくとも1つの亜鉛前駆体、少なくとも1つの硫黄前駆体、および任意にEの少なくとも1つの前駆体の混合物、特に本発明に従う方法の工程(a)にて調製された混合物は、有機溶媒中にて製造される。
有利なことには、有機溶媒は、T2より高い、すなわち本発明に従う方法の工程(b)のために選択される温度よりも高い沸点を有する、アルカン、二級または三級アミン、またはアルケンである。
「アルカン」という用語は、本発明の範囲内において、1〜40個の炭素原子、特に10〜25個の炭素原子、特に14〜20個の炭素原子を有する直鎖、分岐鎖または環状の、任意に置換されたアルカンを指す。
「二級または三級アミン」という用語は、本発明の範囲内においてジアルキルアミンおよびトリアルキルアミンを指し、ここでアルキル基は、4〜24個の炭素原子、特に8〜20個の炭素原子を含む。例えば、本発明の範囲内において使用するのに好適な二級(三級)アミンは、ジオクチルアミン(トリオクチルアミン)であり、それはアルキル鎖あたり8個の炭素原子を含む。
【0026】
「アルケン」という用語は、本発明の範囲内において、2〜40個の炭素原子、特に10〜25個の炭素原子、とりわけ14〜20個の炭素原子を有する直鎖、分岐鎖または環状の、任意に置換されたアルケンを指す。
より詳細には本発明に従う方法において前駆体混合物を調製するために使用される溶媒の1つは1−オクタデセンである。
さらに、この溶媒中の前駆体混合物はさらに、一級アミンを含有してもよい。有利なことには、一級アミンはアルキルアミンであり、ここでアルキル基は4〜24個の炭素原子、特に8〜20個の炭素原子を含む。例えば、本発明の範囲内において使用するのに好適な一級アミンは、ヘキサデシルアミン(HAD、16個の炭素原子を有するアルキルアミンである)である。
方法の工程(a)中、混合物中のAの前駆体の濃度およびBの前駆体の濃度は、2.5〜150mmol/l、特に5〜100mmol/l、とりわけ10〜20mmol/lである。
亜鉛前駆体の濃度および硫黄前駆体の濃度、任意にEの前駆体の濃度は、5〜300mmol/l、特に10〜200mmol/l、とりわけ20〜40mmol/lである。
ナノ結晶のシェルが多層である場合、シェルの外側層以外の層を形成する種々の元素、金属および非金属の前駆体は、反応混合物中、5〜300mmol/l、特に10〜200mmol/l、とりわけ20〜40mmol/lの濃度で存在する。
【0027】
本発明は、Aの少なくとも1つの前駆体、Bの少なくとも1つの前駆体、少なくとも1つの亜鉛前駆体、少なくとも1つの硫黄前駆体、および任意にEの少なくとも1つの前駆体を含む反応混合物を漸進加熱する必須工程に基づく。この工程の間、混合物の初期温度、すなわち温度T1から高温T2への混合物の漸進加熱により、ナノ結晶のコアの形成が得られた後、続いてシェルの形成を連続的に得ることができる。多層シェルの場合、層の形成は、シェルの連続する層の一連の形成からなる。
【0028】
有利なことには、前駆体混合物の温度T1は、50℃未満、特に40℃未満、とりわけ30℃未満である。より詳細には、前駆体混合物は、周囲温度にある。「周囲温度」という用語は、20℃±5℃の温度を指す。
温度T2は、180℃を超える、特に210℃を超える、とりわけ210〜320℃、より詳細には230℃〜300℃である。有利なことには、温度T2は、270℃の領域にある。「270℃の領域」という用語は、270℃±20℃の温度、特に270℃±10℃の温度を指す。
本発明に従う方法の加熱工程、特に工程(b)の第1の実施形態において、温度T1から温度T2への移行は、線形上昇様式にて行われる。
有利なことには、温度の線形上昇は、毎秒1〜20℃の勾配、特に毎秒2.5〜15℃の勾配、より詳細には毎秒5〜10℃の勾配を用いて行われる。
本発明に従う方法の加熱工程、特に工程(b)の第2の実施形態において、温度T1から温度T2への移行は、少なくとも1つの段階を有する上昇様式で行われる。
「段階」という用語は、T1とT2との間にある温度Tを指し、それは5秒から2時間、特に15秒から1時間、とりわけ30秒から30分間、より詳細には1分から15分の時間一定に維持される。温度T1とT2との間にある温度の上昇は、1〜10段階、特に2〜5段階を含んでいてもよい。T1と第1の段階との間、2つの連続段階の間、および最終段階とT2との間において、温度は、加熱工程の第1の実施形態に関して定義された通りの条件下で線形様式で上昇する。
【0029】
本発明に従う方法の加熱工程中(すなわち、工程(b))、温度T2に到達したら、この温度は、5分から5時間、特に15分から3.5時間、とりわけ30分から2時間の期間一定に維持されてもよい。
当業者には、前駆体混合物を温度T1から温度T2まで漸進的に加熱するための種々の技術および種々の手段が公知である。例としては、前駆体混合物を含有するサーモスタット制御のプログラム可能なフラスコまたは反応容器の使用、または前駆体混合物を含有するフラスコもしくは反応容器が浸漬される1つの段階の温度もしくは温度T2であり得る必須温度に予め加熱された浴の使用が挙げられる。
【0030】
本発明の範囲内で使用されるすべての前駆体は、市販の製品または当業者が少なくとも1つの調製方法を知っている製品のいずれかである。
本発明に従う方法の工程(a)の第1の代替実施形態において、使用される種々の前駆体、すなわちAの前駆体、Bの前駆体、亜鉛前駆体、硫黄前駆体および任意にEの前駆体は、任意に予め調製された後、共に混合される。有利なことには、方法の工程(a)にて使用される少なくとも1つの前駆体が予め調製される場合、工程(a)中に使用される溶媒と同一または異なる、有利なことには同一の非プロトン性溶媒中で調製される。
混合物の調製中、種々の前駆体は、同時に、次々におよび/または少なくとも2つの前駆体の群として上記で定義されたような溶媒に添加されてもよい。使用される前駆体に従って、当業者は、混合物中に前駆体を導入するのに最良の方法を決定できる。
【0031】
以降の例は、本発明に従う方法の工程(a)の第1の代替実施形態の例である。この例において、Aの前駆体は予め調製された(工程(i))インジウムトリミリステートであり、Bの前駆体はトリス(トリメチルシリル)ホスフィン(P(TMS)3)であり、亜鉛前駆体は亜鉛ジステアレートであり、硫黄前駆体はドデカンチオール(DDT)である。
この例において、方法の工程(a)は、先行する工程(i)と共に工程(ii)および(iii)に対応し、これらの工程は:
i)約100℃にて低真空下で1時間加熱することによって、オクタデセン中、酢酸インジウムを3当量のミリスチン酸(CH3−(CH2)12−COOH)と反応させることによりインジウムトリミリステートを調製する工程。この混合物を次いで周囲温度まで冷却し、不活性雰囲気(窒素またはアルゴン)中に保存する;
ii)工程(i)で調製されたインジウム前駆体の必須量を、アルゴンまたは窒素フロー中のフラスコまたは反応容器中に入れ、撹拌し、および任意にオクタデセンで希釈する工程;
iii)この混合物に、周囲温度でP(TMS)3、亜鉛ジステアレートおよびドデカンチオールを添加する工程
からなる。
工程(iii)の後、得られた反応混合物を本発明に従う方法の工程(b)および(c)に供する。
本発明に従う方法の工程(a)の第2の代替実施形態において、使用される種々の前駆体の少なくとも1つ、すなわちAの前駆体、Bの前駆体、亜鉛前駆体、硫黄前駆体、および任意にEの前駆体から選択される少なくとも1つの前駆体は、工程(a)中にインサイチュで調製される。
この代替実施形態において、インサイチュで調製されない前駆体はそれぞれ、前駆体が調製される溶媒に、互いに独立して、この調製前、調製中および/または調製後に添加されてもよい。前駆体は、次々におよび/または少なくとも2つの前駆体の群にて添加されてもよい。使用される前駆体および1つ(またはそれ以上の)前駆体をインサイチュで調製するために使用される条件に従って、当業者は、インサイチュで調製されない前駆体を混合物中に導入するための最適な方法を決定できる。
【0032】
以降の例は、本発明に従う方法の工程(a)の第2の代替実施形態の例である。この例において、Aの前駆体は、インジウムトリアセテートおよびミリスチン酸を用いてインサイチュで調製されたインジウムトリミリステートであり、Bの前駆体はP(TMS)3であり、亜鉛前駆体は亜鉛ジステアレートであり、硫黄前駆体はDDTである。工程(a)は、次からなる連続工程(i’)、(ii’)および(iii’)を含む:
i’)インジウムトリアセテート、ミリスチン酸およびP(TMS)3をフラスコ中で混合する工程;
ii’)低真空中(特に10−2mBarの領域での真空)、工程(i’)からの混合物を50〜150℃の温度に(特に110℃に)30分〜3時間の期間(特に1時間)加熱して、インジウム前駆体を形成する工程;
iii’)工程(ii’)の混合物を不活性ガス(アルゴンまたは窒素)でパージし、P(TMS)3およびDDTを添加する工程。
工程iii’)の後、得られた反応混合物を本発明に従う方法の工程(b)および(c)に供する。
【0033】
この特定例において、リン前駆体および硫黄前駆体は、インジウム前駆体の調製後に添加されて、真空中、100℃での部分蒸発を防止することに注目すべきである。
有利なことには、本発明に従う方法の工程(a)および(b)は、撹拌下で行われる。当業者に既知の種々の手段が、本発明に従う方法の工程(a)および(b)に使用される混合物を撹拌するために使用できる。例えば、混合物は、撹拌器、磁性棒、超音波浴またはホモジナイザーを用いて撹拌されてもよい。
本発明に従う方法の工程(c)中、得られたナノ結晶は、反応混合物から精製される。当業者には、沈殿工程、希釈工程および/またはろ過工程を用いる精製のための種々の技術が公知である。ルミネッセントナノ結晶を精製するために先行技術において使用される技術は、本発明に従う方法の工程(c)の範囲内に使用するのに好適である。
有利なことには、方法の工程(c)は、温度T2未満の温度にて実施される。このようにして、方法の工程(b)の後に得られた反応混合物は、冷却される、または周囲温度まで放冷される。次いでナノ結晶は、溶媒または適切な溶媒の混合物を用いて沈殿により精製される。
例えば、工程(b)の後に得られた反応混合物を希釈して体積を2倍にし、次いで過剰のアセトンを用いて沈殿させるために、メタノールおよびクロロホルムのようなアルコールの混合物(有利なことには1:1vol:volの割合で)を使用できる。ナノ結晶は、遠心分離によって回収され、次いでヘキサン、トルエンまたはクロロホルムのような有機溶媒中に分散されてもよい。本発明に従う方法の精製工程(c)は、任意に1回以上繰り返されてもよい。
【0034】
本発明は、本発明に従う方法によって得られるのに好適な、+III酸化状態での金属または非金属を表すAおよび−III酸化状態での元素を表すBを含む半導体を含むコアを有し、そのコアが、外側部分が式ZnS1−xExを有する半導体を含むシェル(ii)でコーティングされたナノ結晶のいずれかを指し、ここでEは−II酸化状態での元素を表し、xは0≦x<1となるような少数である、任意のナノ結晶に関する。
より詳細には本発明は、本発明に従う方法によって得られるのに好適な、外側部分が亜鉛および硫黄を含む半導体を含むシェルでコーティングされたインジウムおよびリンを含む半導体を含むコアを有し、周囲温度にて45%を超える、特に50%を超える、より詳細には55%を超える蛍光量子収率を有するナノ結晶に関する。
当業者は、所与のナノ結晶に関して、周囲温度にてその蛍光量子収率を得ることができる種々の技術を知っている。例としては、励起波長Yにて光学密度Xを有するヘキサン中の本発明に従うナノ結晶分散物のスペクトルに関して積分された発光強度を、同じ励起波長にて同じ光学密度を有するエタノール中のローダミン6G溶液の発光強度と比較することからなる技術が挙げられる。
【0035】
本発明がカバーする、外側部分が亜鉛および硫黄を含む半導体を含むシェルでコーティングされたインジウムおよびリンを含む半導体を含むコアを有するナノ結晶は、80nm未満、有利なことには60nm未満、とりわけ50nm未満のフォトルミネッセンス線幅を有する。
本発明は、発光ダイオード、太陽電池において、および化学または生物学的分子の蛍光標識のための、本発明に従う方法に従って調製されるのに好適なナノ結晶または本発明に従うナノ結晶の使用に関する。
本発明のさらなる特徴および利点は、例示のために与えられ、限定を目的としない以降の実施例を添付の図面を参照して読む際に、当業者に明らかになる。
【図面の簡単な説明】
【0036】
【図1】以降の項目IIにて記載される実験中にとったサンプルのUV−可視吸収スペクトルを示す。
【図2】以降の項目IIに記載される実験中にとったサンプルのフォトルミネッセンススペクトルを示す。励起波長は420nmである。
【図3】以降の項目IIIに記載されるプロトコルに従って調製されたサンプルのフォトルミネッセンススペクトルを示す。励起波長は420nmである。
【図4】空気の存在下、UVランプ(365nm)での照射時間の関数としてのフォトルミネッセンス強度を表す。InPコアナノ結晶は、少量のドデカンチオールで調製されたナノ結晶(In:DDT=1:0.3)および以降の項目Iに記載されるプロトコルで合成されたInP/ZnS コア/シェルナノ結晶(In:DDT=1:1)に相当する。
【図5】以降の項目IIに記載された方法に従って調製されるが、0.06mmolのP(TMS)3を用いて、240℃の温度にて1時間で調製されたサンプルのX線粉末回折図を示す。
【発明を実施するための形態】
【0037】
空気感応性材料の取扱はすべて、グローブボックスにてまたは真空装置を用いて行なわれる(シュレンク法)。
特徴付けのために、UV−可視吸収スペクトルは、Hewlett−Packard8425A分光計(波長スペクトル範囲:190〜820nm、解像度2nm)にて測定し、フォトルミネッセンススペクトルは、Hitachi F−4500分光計で得た。これらの分光測定に関して、ヘキサン中に希釈したコロイド状のナノ結晶溶液を、1cmの光学経路の石英セルに入れた。周囲温度での蛍光量子収率は、ヘキサン中のナノ結晶分散物のスペクトルに関して積分された発光強度を、エタノール中のローダミン6G溶液の発光強度と比較することによって得たが、両方の溶液は励起波長(490nm)にて同じ光学密度(<0.03)を有していた。X線回折図は、Coソースを用いて、50kVおよび35mAにてPhilips X’Pert装置にて得た。
亜鉛ジステアレート(Riedel de Haen)を除いてすべての製品をSigma−Aldrichから購入し、そのまま使用した:酢酸インジウム(99.99%純度)、ミリスチン酸(純度>99%)、亜鉛ジステアレート(90%純度)、ドデカンチオール(97%純度)、1−オクタデセン(90%純度)。
【0038】
I 標準プロトコル
I.A.インジウム前駆体(インジウムミリステート)の調製
2mmolのインジウムトリアセテート、6mmolのミリスチン酸、および20mlの1−オクタデセンを、50mlのフラスコ中に入れ、不活性雰囲気(アルゴンまたは窒素)中、磁性撹拌器を用いて混合する。温度を、110℃に上昇させ、フラスコを低真空ポンプを用いて1時間ポンプする(フラスコ中の圧力は10−2mBarの領域)。均質な無色の溶液が得られる。次いでフラスコを不活性ガス(アルゴンおよび窒素)で満たし、周囲温度に冷却する。前駆体溶液を不活性雰囲気中、例えばグローブボックス中に保存する。
I.B.InP/ZnS コア/シェルナノ結晶合成
不活性雰囲気中、項目I.A.にて記載されたように調製された1mlのインジウム前駆体、0.2mmolの亜鉛ジステアレート、0.1mmolのP(TMS)3、0.2mmolのドデカンチオールおよび6mlの1−オクタデセンを、冷却器を備えた50mlの3つ口フラスコに入れ、磁性撹拌器を用いて混合する。混合物を、約5℃/秒の勾配にて230〜300℃に加熱し、この温度を1〜3時間維持する。
周囲温度まで冷却した後、InP/ZnSナノ結晶は、クロロホルム/メタノール混合物(1:1 vol:vol)の1体積当量およびアセトンの10体積当量を添加し、続いて遠心分離することによって単離する。ナノ結晶を含有する得られた沈殿物は、ヘキサン、トルエンまたはクロロホルムのような有機溶媒中に分散させてもよい。
【0039】
II.インジウム前駆体溶液を予め調製しない合成プロトコル。
冷却器を備えた50mlのフラスコに、0.1mmolのインジウムトリアセテート、0.3mmolのミリスチン酸、0.1mmolの亜鉛ジステアレート、および8.6mlの1−オクタデセンを入れ、不活性雰囲気中にて磁性撹拌器を用いて混合する。
温度を110℃に上昇させ、フラスコを低真空ポンプを用いて1時間ポンプする(フラスコ中の圧力は10−2mBarの領域)。均質な無色の溶液が得られる。次いでフラスコを不活性ガス(アルゴンおよび窒素)で満たし、周囲温度に冷却する(任意)。0.1mmolのP(TMS)3および1mlの1−オクタデセン中に希釈された1mmolを、混合物に添加し、混合物を、約5℃/秒の勾配にて270℃に加熱し、この温度を150分間維持する。
サンプルを、周期的に採取し(1mlのクロロホルムで希釈された0.1mlの反応混合物)、UV−可視吸収およびフォトルミネッセンス分光法によって分析する。
周囲温度まで冷却した後、InP/ZnSナノ結晶は、クロロホルム/メタノール混合物(1:1 vol:vol)の1体積当量およびアセトンの10体積当量を添加し、続いて遠心分離することによって単離する。ナノ結晶を含有する得られた沈殿物は、ヘキサン、トルエンまたはクロロホルムのような有機溶媒中に分散させてもよい。
【0040】
III.項目IIのプロトコルの変形例
この実施例において、InP/ZnS コア/シェルナノ結晶の合成を、上記で記載されたプロトコルIIの変形例に従って行った。
プロトコルは、項目IIにて記載された方法を順守するが、以降の表に要約される反応パラメータを用いる:
【表1】
【0041】
IV.本発明に従う方法を用いて得られたナノ結晶の特徴付け
IV.A.光学特性
得られたナノ結晶のサイズ、すなわちそれらの発光色を制御するための2つの手段がある:1)反応時間を用いて2)反応パラメータを調節することによって。
項目IIに記載されたような方法の後で得られたサンプルのUV−可視吸収およびフォトルミネッセンススペクトルをそれぞれ図1および2に示す。項目IIIに記載されるような方法を用いて得られたサンプルのフォトルミネッセンススペクトルを図3に示す。
サンプルの励起子ピークが、特に反応時間の5分から120分間に得られるピークに関して明確に現れる(図1)。この特徴は狭いサイズ分散を示し、それはフォトルミネッセンススペクトル(図2)における線幅を分析することによって確認され、約40〜50nmにある(FWHM、半値全幅)。
【0042】
IV.B.光学特性の安定性
蛍光ナノ結晶の技術的用途に関して、経時的なそれらの光学特性の安定性、特に放射線誘導エージングに対して特に注意を払うできである。
得られたサンプルの安定性を試験するために、それらを365nmにて発光するUVランプでの連続照射に供した。
ランプは、空気の存在下で石英セル中にあるサンプルから4cm離して配置する。この距離における照射出力は約2〜3mWである。
図4は、3つの異なるサンプルのフォトルミネッセンス強度の進行を示す。
迅速な減衰は、ZnSシェルをもたないInPナノ結晶(本発明がカバーしていない比較の目的のために使用されるナノ結晶)に関して観察され、10〜15時間照射後にそれらの発光は実際すべて失われる。
同様に、項目Iに記載されるような方法に従って調製された薄いシェルを有するサンプル(In:DDT=1:0.3)は、ZnSシェルを持たないナノ結晶の安定性と比較して改善された光学特性の安定性を有する。このようにして、12時間の照射後、50%の初期フォトルミネッセンス強度が依然として検出されるが、結局発光は実際には40〜50時間後に停止する。
最後に、多量の硫黄前駆体(In:DDT=1:1)を用いて製造されたコア/シェルサンプルは、全く異なるように挙動する。照射開始時(0〜10時間)、フォトルミネッセンス強度において初期値の約140%までの増加が観察される。45時間(実験の停止)後であっても、初期強度の110%超過が測定され、これはサンプルの非常に満足のいく光安定性を裏付ける。
【0043】
IV.C.物理化学的なX線回折分析
図5に示されるコア/シェルサンプルについてのX線粉末回折図は、InP/ZnSヘテロ構造を確認する。本実施例において使用されるコア/シェルサンプルは、項目IIに記載されるような方法に従って調製した。
結晶がナノメートルサイズであることによる拡がったピークは、InP立方相(「Joint Committee on Powder Diffraction Standard」card,JCPDS,No.100216)およびZnSの亜鉛ブレンド構造(JCPDS card No. 003−1093)の両方の寄与を含む。
【特許請求の範囲】
【請求項1】
(i)+III酸化状態で金属または非金属を表すAおよび−III酸化状態での元素を表すBを含む半導体を含むコアを有し、このコアが、(ii)外側部分が式ZnS1−xEx有する半導体を含むシェルでコーティングされているナノ結晶を調製するための方法であって、ここでEは−II酸化状態の元素を表し、xは0≦x<1となるように少数であり、
この方法は、Aの少なくとも1つの前駆体、Bの少なくとも1つの前駆体、亜鉛の少なくとも1つの前駆体、硫黄の少なくとも1つの前駆体、および任意にEの少なくとも1つの前駆体の混合物を、まずコアが形成され、次いでシェルが形成されるように上昇させる様式で温度T1から、T1を超える温度T2まで加熱することを特徴とする、方法。
【請求項2】
前記方法が次の工程を含むことを特徴とする、請求項1に記載の調製方法:
a)Aの少なくとも1つの前駆体、Bの少なくとも1つの前駆体、少なくとも1つの亜鉛前駆体、少なくとも1つの硫黄前駆体、および任意にEの少なくとも1つの前駆体の混合物を温度T1で調製する工程、
b)工程(a)にて得られた混合物を温度T2に加熱する工程、
c)工程(b)にて得られた、式ABを有する半導体を含むコアを有し、外側が式ZnS1−xExを有する半導体を含むシェルによって囲まれているナノ結晶を精製する工程。
【請求項3】
前記ナノ結晶が、15nm未満、特に12nm未満、とりわけ10nm未満の直径を有することを特徴とする、請求項1または2のいずれか一項に記載の調製方法。
【請求項4】
前記Aの前駆体が、インジウム前駆体、ガリウム前駆体、アルミニウム前駆体およびこれらの混合物からなる群から選択されることを特徴とする、請求項1から3のいずれか一項に記載の調製方法。
【請求項5】
前記Aの前駆体が、Aの塩、Aのハロゲン化物、Aの酸化物およびAの有機金属化合物から選択されることを特徴とする、請求項1から4のいずれか一項に記載の調製方法。
【請求項6】
前記Bの前駆体が、アンチモン前駆体、ヒ素前駆体、リン前駆体、窒素前駆体およびこれらの混合物から成る群から選択されることを特徴とする、請求項1から5のいずれか一項に記載の調製方法。
【請求項7】
前記Bの前駆体が、式B(F(R6)3)3または式B(R7)3を有する化合物であり、ここで
−各Fは、シリカ(Si)、ゲルマニウム(Ge)およびスズ(Sn)から成る群から選択され;
−各R6は、同一または異なって、1〜10個の炭素原子を有する直鎖、分枝鎖または環状の、任意に置換されたアルキル基であり;
−各R7は、同一または異なって、水素原子、ハロゲン、または1〜10個の炭素原子を有する直鎖、分枝鎖または環状の、任意に置換されたアルキル基である
ことを特徴とする、請求項1から6のいずれか一項に記載の調製方法。
【請求項8】
前記亜鉛前駆体が、亜鉛塩、ハロゲン化亜鉛、酸化亜鉛および有機金属亜鉛化合物から成る群から選択されることを特徴とする、請求項1から7のいずれか一項に記載の調製方法。
【請求項9】
前記硫黄前駆体が、脂肪族チオール、有機溶媒に溶解した元素硫黄、および式S(Si(R13)3)2を有する化合物から成る群から選択され、ここで、各R13は、同一または異なって、1〜10個の炭素原子を有する直鎖、分枝鎖または環状の、任意に置換されたアルキル基であることを特徴とする、請求項1から8のいずれか一項に記載の調製方法。
【請求項10】
前記Eの前駆体が、酸素前駆体、セレン前駆体、テルル前駆体およびこれらの混合物からなる群から選択されることを特徴とする、請求項1から9のいずれか一項に記載の調製方法。
【請求項11】
前記Eの前駆体が、有機溶媒に溶解した元素セレン;有機溶媒に溶解した元素テルル;酢酸亜鉛;ホスフィンセレニド;酸化ホスフィン;式E’(Si(R14)3)2(式中、E’はSeまたはTeを表し、各R14は、同一または異なって、1〜10個の炭素原子を有する直鎖、分岐鎖または環状の、任意に置換されたアルキル基を表す)から選択されることを特徴とする、請求項1から10のいずれか一項に記載の調製方法。
【請求項12】
前記混合物が有機溶媒中に生成されることを特徴とする、請求項1から11のいずれか一項に記載の調製方法。
【請求項13】
前記有機溶媒が、T2より高い沸点を有する、アルカン、二級または三級アミン、またはアルケンであることを特徴とする、請求項12に記載の調製方法。
【請求項14】
前記混合物がさらに一級アミンを含有することを特徴とする、請求項1から13のいずれか一項に記載の調製方法。
【請求項15】
前駆体混合物の温度T1が50℃未満、特に40℃未満、とりわけ30℃未満であることを特徴とする、請求項1から14のいずれか一項に記載の調製方法。
【請求項16】
温度T2が180℃を超える、特に210℃を超える、とりわけ210℃〜320℃、より詳細には230℃〜300℃であることを特徴とする、請求項1から15のいずれか一項に記載の調製方法。
【請求項17】
温度T1から温度T2への移行は、線形上昇様式にて行われ、有利なことには毎秒1〜20℃の勾配で行われることを特徴とする、請求項1から16のいずれか一項に記載の調製方法。
【請求項18】
温度T1から温度T2への移行は、少なくとも1つの段階を有する上昇様式で行われることを特徴とする、請求項1から16のいずれか一項に記載の調製方法。
【請求項19】
温度T2に到達したら、この温度は、5分から5時間、特に15分から3.5時間、とりわけ30分から2時間の期間一定に維持されてもよいことを特徴とする、請求項1から18のいずれか一項に記載の調製方法。
【請求項20】
Aの前駆体、Bの前駆体、亜鉛前駆体、硫黄前駆体、および任意にEの前駆体から選択される少なくとも1つの前駆体が、工程(a)中にインサイチュで調製されることを特徴とする、請求項2から19のいずれか一項に記載の調製方法。
【請求項1】
(i)+III酸化状態で金属または非金属を表すAおよび−III酸化状態での元素を表すBを含む半導体を含むコアを有し、このコアが、(ii)外側部分が式ZnS1−xEx有する半導体を含むシェルでコーティングされているナノ結晶を調製するための方法であって、ここでEは−II酸化状態の元素を表し、xは0≦x<1となるように少数であり、
この方法は、Aの少なくとも1つの前駆体、Bの少なくとも1つの前駆体、亜鉛の少なくとも1つの前駆体、硫黄の少なくとも1つの前駆体、および任意にEの少なくとも1つの前駆体の混合物を、まずコアが形成され、次いでシェルが形成されるように上昇させる様式で温度T1から、T1を超える温度T2まで加熱することを特徴とする、方法。
【請求項2】
前記方法が次の工程を含むことを特徴とする、請求項1に記載の調製方法:
a)Aの少なくとも1つの前駆体、Bの少なくとも1つの前駆体、少なくとも1つの亜鉛前駆体、少なくとも1つの硫黄前駆体、および任意にEの少なくとも1つの前駆体の混合物を温度T1で調製する工程、
b)工程(a)にて得られた混合物を温度T2に加熱する工程、
c)工程(b)にて得られた、式ABを有する半導体を含むコアを有し、外側が式ZnS1−xExを有する半導体を含むシェルによって囲まれているナノ結晶を精製する工程。
【請求項3】
前記ナノ結晶が、15nm未満、特に12nm未満、とりわけ10nm未満の直径を有することを特徴とする、請求項1または2のいずれか一項に記載の調製方法。
【請求項4】
前記Aの前駆体が、インジウム前駆体、ガリウム前駆体、アルミニウム前駆体およびこれらの混合物からなる群から選択されることを特徴とする、請求項1から3のいずれか一項に記載の調製方法。
【請求項5】
前記Aの前駆体が、Aの塩、Aのハロゲン化物、Aの酸化物およびAの有機金属化合物から選択されることを特徴とする、請求項1から4のいずれか一項に記載の調製方法。
【請求項6】
前記Bの前駆体が、アンチモン前駆体、ヒ素前駆体、リン前駆体、窒素前駆体およびこれらの混合物から成る群から選択されることを特徴とする、請求項1から5のいずれか一項に記載の調製方法。
【請求項7】
前記Bの前駆体が、式B(F(R6)3)3または式B(R7)3を有する化合物であり、ここで
−各Fは、シリカ(Si)、ゲルマニウム(Ge)およびスズ(Sn)から成る群から選択され;
−各R6は、同一または異なって、1〜10個の炭素原子を有する直鎖、分枝鎖または環状の、任意に置換されたアルキル基であり;
−各R7は、同一または異なって、水素原子、ハロゲン、または1〜10個の炭素原子を有する直鎖、分枝鎖または環状の、任意に置換されたアルキル基である
ことを特徴とする、請求項1から6のいずれか一項に記載の調製方法。
【請求項8】
前記亜鉛前駆体が、亜鉛塩、ハロゲン化亜鉛、酸化亜鉛および有機金属亜鉛化合物から成る群から選択されることを特徴とする、請求項1から7のいずれか一項に記載の調製方法。
【請求項9】
前記硫黄前駆体が、脂肪族チオール、有機溶媒に溶解した元素硫黄、および式S(Si(R13)3)2を有する化合物から成る群から選択され、ここで、各R13は、同一または異なって、1〜10個の炭素原子を有する直鎖、分枝鎖または環状の、任意に置換されたアルキル基であることを特徴とする、請求項1から8のいずれか一項に記載の調製方法。
【請求項10】
前記Eの前駆体が、酸素前駆体、セレン前駆体、テルル前駆体およびこれらの混合物からなる群から選択されることを特徴とする、請求項1から9のいずれか一項に記載の調製方法。
【請求項11】
前記Eの前駆体が、有機溶媒に溶解した元素セレン;有機溶媒に溶解した元素テルル;酢酸亜鉛;ホスフィンセレニド;酸化ホスフィン;式E’(Si(R14)3)2(式中、E’はSeまたはTeを表し、各R14は、同一または異なって、1〜10個の炭素原子を有する直鎖、分岐鎖または環状の、任意に置換されたアルキル基を表す)から選択されることを特徴とする、請求項1から10のいずれか一項に記載の調製方法。
【請求項12】
前記混合物が有機溶媒中に生成されることを特徴とする、請求項1から11のいずれか一項に記載の調製方法。
【請求項13】
前記有機溶媒が、T2より高い沸点を有する、アルカン、二級または三級アミン、またはアルケンであることを特徴とする、請求項12に記載の調製方法。
【請求項14】
前記混合物がさらに一級アミンを含有することを特徴とする、請求項1から13のいずれか一項に記載の調製方法。
【請求項15】
前駆体混合物の温度T1が50℃未満、特に40℃未満、とりわけ30℃未満であることを特徴とする、請求項1から14のいずれか一項に記載の調製方法。
【請求項16】
温度T2が180℃を超える、特に210℃を超える、とりわけ210℃〜320℃、より詳細には230℃〜300℃であることを特徴とする、請求項1から15のいずれか一項に記載の調製方法。
【請求項17】
温度T1から温度T2への移行は、線形上昇様式にて行われ、有利なことには毎秒1〜20℃の勾配で行われることを特徴とする、請求項1から16のいずれか一項に記載の調製方法。
【請求項18】
温度T1から温度T2への移行は、少なくとも1つの段階を有する上昇様式で行われることを特徴とする、請求項1から16のいずれか一項に記載の調製方法。
【請求項19】
温度T2に到達したら、この温度は、5分から5時間、特に15分から3.5時間、とりわけ30分から2時間の期間一定に維持されてもよいことを特徴とする、請求項1から18のいずれか一項に記載の調製方法。
【請求項20】
Aの前駆体、Bの前駆体、亜鉛前駆体、硫黄前駆体、および任意にEの前駆体から選択される少なくとも1つの前駆体が、工程(a)中にインサイチュで調製されることを特徴とする、請求項2から19のいずれか一項に記載の調製方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公表番号】特表2011−520002(P2011−520002A)
【公表日】平成23年7月14日(2011.7.14)
【国際特許分類】
【出願番号】特願2011−507878(P2011−507878)
【出願日】平成21年4月29日(2009.4.29)
【国際出願番号】PCT/EP2009/055231
【国際公開番号】WO2009/135797
【国際公開日】平成21年11月12日(2009.11.12)
【出願人】(510097644)コミッサリア ア ロンネルジー アトミック エ オ ゾンネルジー ザルテルナティーフ (33)
【Fターム(参考)】
【公表日】平成23年7月14日(2011.7.14)
【国際特許分類】
【出願日】平成21年4月29日(2009.4.29)
【国際出願番号】PCT/EP2009/055231
【国際公開番号】WO2009/135797
【国際公開日】平成21年11月12日(2009.11.12)
【出願人】(510097644)コミッサリア ア ロンネルジー アトミック エ オ ゾンネルジー ザルテルナティーフ (33)
【Fターム(参考)】
[ Back to top ]