
レトロウイルスベクターによる遺伝子導入鳥類での遺伝子発現法およびそれによって得られる遺伝子導入鳥類
【課題】抗体を産生するG0トランスジェニックキメラ鳥類を提供する。
【解決手段】複製能欠失型レトロウイルスベクターによって抗体遺伝子が導入され、導入遺伝子を発現することにより抗体を産生するG0トランスジェニックキメラ鳥類、及び作製したG0トランスジェニックキメラ鳥類が発現する抗体を回収することよりなる抗体生産法、及び鳥類受精卵を孵卵し、孵卵開始直後を除くそれ以降の受精卵にレトロウイルスベクターをインジェクションすることよりなるG0トランスジェニックキメラ鳥類の作製法。
【解決手段】複製能欠失型レトロウイルスベクターによって抗体遺伝子が導入され、導入遺伝子を発現することにより抗体を産生するG0トランスジェニックキメラ鳥類、及び作製したG0トランスジェニックキメラ鳥類が発現する抗体を回収することよりなる抗体生産法、及び鳥類受精卵を孵卵し、孵卵開始直後を除くそれ以降の受精卵にレトロウイルスベクターをインジェクションすることよりなるG0トランスジェニックキメラ鳥類の作製法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、血液中ならびに卵中に、抗体、例えばscFv−Fc抗体を産生するG0トランスジェニックキメラ鳥類に関する。また本発明は、複製能欠失型レトロウイルスベクターによって外来性抗体遺伝子が導入されたG0トランスジェニックキメラ鳥類を作製し、血中、卵白中あるいは卵黄中に産生した抗体を回収することからなる抗体産生法に関する。更に本発明は、効率的に導入遺伝子を発現するG0トランスジェニックキメラ鳥類の作製法ならびに該作製法より得られるG0トランスジェニックキメラ鳥類に関する。
【背景技術】
【0002】
遺伝子機能の研究手段として、外来遺伝子を宿主に組み込んだ遺伝子導入(トランスジェニック)動物の研究が盛んに行われているが、このトランスジェニック動物は、基礎研究だけでなく、品種改良や物質生産、移植用臓器のドナー等、産業応用にも有効である。ウシ、ヤギ、ヒツジ等の乳汁に生理活性物質を産生させる試みが実用に近づいており、その代表例としてα1−アンチトリプシンやアンチスロンビンは、医薬品への応用を目的として現在臨床段階にある。
【0003】
ウズラやニワトリに代表される家禽鳥類は、食肉用、採卵用家畜としての飼育実績が長く、トランスジェニック研究の目的としても耐病性や肉質向上等、品種改良に係るさまざまなアプローチが考えられる。また、鳥類は性成熟までの期間が短く、小さなスペースで飼育可能なことから、安価なコストで実施可能なタンパク質発現系と考えられ、抗体医薬品や希少タンパク質の産生手段としてのトランスジェニック作製に期待が持たれている。鳥類の卵は多量のタンパク質を含有し、毎日連続的に生産されることから、導入遺伝子産物を組換えタンパク質として意図的に卵中に産生させることができれば、効率的な生産システムとなると考えられる。
【0004】
近年数多くの品目が上市され、医薬品として注目されているモノクローナル抗体は、大腸菌などによる安価な発現システムでは生産できず、単価が高いことが普及の妨げとなっている。鳥類細胞は抗体タンパク質を構成する機能を備えており、トランスジェニック鳥類は、従来大量生産が困難であった医療用抗体等の生産手段として期待できる。更にこれらのタンパク質生産物には、鳥類細胞によって糖鎖が付与されることにより血中での安定性が向上する等、医薬、検査薬としての応用上有利な性質を備える可能性が高いと考えられる。
【0005】
このように、有用タンパク質の生産手段としての応用が待たれるトランスジェニック鳥類であるが、一方で現在までに様々な試みがあるにも係らず、鳥類の卵に実用レベルで目的とする組換えタンパク質を蓄積させた例はない。また、抗体のような複数のユニットからなる高次構造をもったタンパク質を、高濃度に発現するトランスジェニック鳥類を作製した例も、未だ報告されていない。
【0006】
Harveyら(非特許文献1)はエビアン・ロイコシス・ウイルス由来のベクターを用い、β−ラクタマーゼ遺伝子をニワトリに導入し、G0トランスジェニックキメラニワトリを作製したが、血清あるいは卵への酵素発現量は50〜250ng/ml程度であった。このG0トランスジェニックキメラニワトリの交配により作製されたG1〜G3トランスジェニックニワトリは、全身の体細胞に遺伝子が導入されることにより、酵素の発現量が増加したが、それでも数μg/mlのレベルに留まり、実用には遠い値であった。
【0007】
一般的に遺伝子導入動物を作製するには、受精卵の前核へDNAをマイクロインジェクションする手法がとられるが、鳥類にはこの方法は応用できない。鳥類では1細胞期の胚を取得するのが困難なこと、また取得できたとしても、卵内の核を見分ける手法がないことがその理由である。1細胞期の胚を取得するには、雌性鳥類の輸卵管内から受精直後の卵を取得し、正常に発生させなければならない。近年Perryによって鶏の卵割前細胞を取得し、体外で培養するシステムが確立された(非特許文献2)が、この手法によっても卵内の核を見分け、核内へ目的遺伝子を導入することは不可能である。
【0008】
従って、鳥類受精卵への遺伝子導入は、細胞質へのDNA注入に限られ、DNA封入脂質二重膜(リポソーム)の利用やリン酸カルシウム法、電気導入法(エレクトロポレーション)の利用が試みられてきた。しかし、これらの手法では遺伝子の導入効率が悪く、また導入されたプラスミドDNAは、染色体に取り込まれる確率が低い。マイクロインジェクションによる遺伝子導入法は、効率的に目的DNAを受精卵に送り込むことができるが、導入されたプラスミドDNAは宿主染色体に組み込まれないため、宿主の体細胞分裂に伴って導入遺伝子プラスミドは脱落してしまい、安定な遺伝子導入効果は望めない。
【0009】
1986年に初めてレトロウイルスベクターによるトランスジェニックニワトリの作製例が報告されている(非特許文献3)。レトロウイルスベクターをマイクロインジェクション法で受精卵に注入する手法は、遺伝子の導入効率の高い手法であり、核内へ直接DNAを注入できない鳥類において、染色体へ目的遺伝子が挿入された安定なトランスジェニックを作製する唯一の実用的方法である。
【0010】
本発明者らは鋭意研究を重ね、遺伝子治療にも応用されている安全な自己複製能欠失型ウイルスベクターを使用し、効率的に目的遺伝子を導入するトランスジェニック鳥類の作製法を見出した(特許文献1)。これにより、安全かつ効率的に複数の導入遺伝子コピー数をもつトランスジェニック鳥類の作製が可能となった。またこの手法によれば、導入遺伝子は高い効率で次世代に伝達されることもわかり、物質生産システムとしての遺伝子導入鳥類の利用が実用に近づいた。
【先行技術文献】
【特許文献】
【0011】
【特許文献1】特開2002−176880号公報
【非特許文献】
【0012】
【非特許文献1】Harvey,A.Jら(2002)NatureBiotechnology.19,396
【非特許文献2】Perry、M.M(1988)Nature,331
【非特許文献3】Salter,D.Wら(1986)Poultry Sci.,65
【発明の概要】
【発明が解決しようとする課題】
【0013】
遺伝子治療にも応用されている安全な自己複製能欠失型ウイルスベクターを使用すれば、効率的に目的遺伝子を導入してトランスジェニック鳥類を作製することが可能であるが、このとき導入された遺伝子の大部分は、発生初期に宿主によって不活性化されるため(遺伝子のサイレンシングと呼ばれる)、遺伝子発現の結果としてのタンパク質産生はごく微量であった。導入遺伝子が不活性化される生物学的メカニズムは未だ解明されていないが、この不活性化を回避し、目的遺伝子を効率的に発現させる技術が、トランスジェニック鳥類の応用開発に必須である。
【課題を解決するための手段】
【0014】
発明の要約
レトロウイルスベクターによって受精卵に導入された遺伝子が、発生後どの時期に不活性化されるのかを特定するため、本発明者らはβ−ガラクトシダーゼ発現遺伝子を指標とし、受精後の様々な時期にベクターを導入して発現量がどう変化するかを検討した。その結果、導入遺伝子の発現量は該遺伝子が胚発生のどの時期に導入されたかによって大きく変化してくることを発見し、本発明に至った。すなわち、導入遺伝子の不活性化は、放卵直後に導入された遺伝子に対して顕著であり、放卵から一定時間経過後に導入された遺伝子は、発現頻度が高いというものである。
【0015】
本発明者らはこの知見を利用し、鳥の種類によって決まる特定の時間が孵卵開始時より経過した後に、その初期胚へ外来遺伝子を導入すれば、宿主による不活性化の影響を受けず、目的遺伝子を効率的に発現させることが可能となることを見出した。
【0016】
さらに、医薬品として有用なキメラ抗体、例えばscFv−Fc(1本鎖抗体)をコードした遺伝子を組み込んだベクターを作り、この方法によりウズラに導入して、G0トランスジェニックキメラ鳥類を作製したところ、血中、卵白、卵黄中に導入遺伝子に由来する抗体が高濃度に発現することが見出された。
【0017】
すなわち第一の本発明は、鳥類受精卵を孵卵し、放卵直後の胚盤葉期(ステージX)を除くそれ以降の初期胚へ複製能欠失型レトロウイルスベクターを感染させ、その胚を孵化させることからなるG0トランスジェニックキメラ鳥類の作製法、及び該方法により作製されたG0トランスジェニックキメラ鳥類に関し、第二の本発明は、複製能欠失型レトロウイルスベクターによって外来性抗体遺伝子が導入されたG0トランスジェニックキメラ鳥類であって、導入遺伝子に由来する抗体を血中、卵白中及び卵黄中の少なくとも1つに産生することを特徴とするG0トランスジェニックキメラ鳥類に関し、第三の本発明は、上記G0トランスジェニックキメラ鳥類を作製し、該G0トランスジェニックキメラ鳥類の血中及び/又は卵中から抗体を回収することよりなる抗体の生産法に関する。
【0018】
本発明のG0トランスジェニックキメラ鳥類による導入遺伝子の発現は、誕生したG0トランスジェニックキメラ鳥類が成烏になっても維持されることが確認された。したがって、本発明により特定の遺伝子を鳥類に導入し、目的タンパク質を成長後も産生する、実用的な生産システムを構築することができる。また、本発明によって作製されたG0トランスジェニックキメラ鳥類の導入遺伝子は、交配により高い伝播効率で次世代に受け継がれる。導入遺伝子は、染色体に組み込まれたかたちで次世代に受け継がれるため、このトランスジェニック鳥類による生産システムは、安定的な物質生産が可能である。
【0019】
本発明により、トランスジェニック鳥類がその体細胞中に産生させた目的タンパク質は、血液中に分泌され、血清から分離することで利用可能となる。
【0020】
更に本発明者らは、ヒトIgGクラスの定常領域をもつ抗体や、ウズラIgG、ニワトリIgG又はマウスIgGの定常領域をもつ抗体が、ウズラ、ニワトリにおいて血中から卵中に効率的に移行することを見出した。これらの定常領域をもつ抗体を本発明の方法により作製されたG0トランスジェニックキメラ鳥類に発現させた場合、血中に分泌された抗体が卵中に高濃度に蓄積される。
【0021】
トランスジェニック動物に遺伝子を導入して目的タンパク質を取得する場合、哺乳類では乳汁中に分泌させて回収するのが一般的だが、鳥類のトランスジェニックでは卵に目的物を蓄積させ、その卵白、卵黄から回収、精製するのが実際的である。
【0022】
鶏卵などでは全成分の30%がタンパク質であり、その主成分となるタンパク質はオボアルブミンである。従来トランスジェニック鳥類の卵中に組換えタンパク質を発現させる方法として、このオボアルブミン等の発現プロモーターを利用し、その下流に目的タンパク質をコードする遺伝子を組み込んで、卵白中にオボアルブミンに代えて目的物を発現させるという考え方がとられていた。
【0023】
しかし、本発明の方法において、構成的なプロモーター、例えばニワトリβ−アクチンプロモーター等の制御のもとに、大量に発現した抗体、例えば前記IgGクラス抗体等を血中に分泌させ、卵中に蓄積させることが可能となる。なお本明細書において、構成的なプロモーターとは、全身性に発現するプロモーターのことを意味する。
【0024】
更に抗体の構造のなかで、卵中への移行に必須な部分を特定するため、抗体をFab、Fcフラグメントとしたものを、ウズラ、ニワトリ血中に接種して卵への移行性を調べた。その結果、卵内にはFcフラグメントが蓄積され、抗体の卵内への移行はFcレセプターを介して行われることが示唆された。
【0025】
このことからトランスジェニック鳥類による抗体の卵内での生産法を、タンパク質全般の生産に汎用化する方法として、ヒトIgGの定常領域(Fc)が融合した構造をもつタンパク質を生産するベクターを設計し、トランスジェニック鳥類を作製して卵から目的物を含むタンパク質を回収し、Fc部を切断して目的物を精製する生産法が考えられる。
【0026】
また、従来哺乳類のトランスジェニック動物を使って抗体を生産した場合、産生動物がもつ自己抗体と目的抗体を分離精製するのが困難との問題が指摘されていた。産生動物に鳥類を使う抗体生産法の利点として、鳥類の自己抗体がプロテインA、プロテインGカラムに吸着されないため、目的とする組換え抗体との分離が容易な点も挙げられる。
【0027】
前述のように、ヒトモノクローナル抗体は、医薬品として有用であるにも係らず、その生産には動物細胞培養やマウス腹水といった高価な生産手段しかなく、単価が高いことが普及に際して妨げとなっている。
【0028】
近年遺伝子工学的手法を用いて、抗体のH鎖、L鎖のV領域のみをリンカー配列でつなげたscFv(1本鎖抗体)が作製されているが、これは大腸菌でも生産されるという利点があり、コストの点から注目されている。しかし、低分子抗体と呼ばれるこれらのタンパク質は、血中での安定性が低いため、治療用、試験用としての実用性に難があった。
【0029】
scFvにFc領域をつなげたscFv−Fcは、血中でも安定であり、より実用的と考えられるが、大腸菌では生産できず、動物細胞を使ったバイオリアクターでしか供給できない。ニワトリなど他の動物に作らせた結合領域をもつscFvにヒトのFcを結合させたヒト化scFv−Fcは治療用としても有望であり、このタンパク質を本発明によるG0トランスジェニックキメラ鳥類により大量に生産できれば、その有用性は大きい。
【0030】
本発明のG0トランスジェニックキメラ鳥類は、scFv−Fcのような組換え抗体、キメラ抗体、ヒトモノクローナル抗体など、従来の方法では少量しか生産できなかった抗体タンパク質を安価に大量に生産し、実用的に回収、精製して利用することに応用できる。
【0031】
このように本発明ではレトロウイルスベクターによるG0トランスジェニックキメラ鳥類において、導入遺伝子を効率的に発現させる作製法を開示する。また本発明では、特定遺伝子を導入することにより、鳥類体細胞に有用な目的タンパクを産生させるG0トランスジェニックキメラ鳥類の作製法を開示する。更に従来大腸菌等で産生することが不可能だったモノクローナルヒト型抗体や、キメラ抗体、scFv−Fc抗体、複雑な高次構造をもった機能性タンパク等、医薬、検査薬として有用な物質を鳥類細胞に産生させ、血清や卵中から回収して利用する、生産コストの低いタンパク生産システムを開示する。
【0032】
また本発明のG0トランスジェニックキメラ鳥類の作製法を応用して、家禽鳥類を好ましい方向へ改質する手法および改質された鳥類を提供することも考えられる。家禽鳥類の好ましい性質としては、肉質改善、耐病性の向上、生育速度の向上等が挙げられる。鳥類はまた愛玩用途としても多種の需要をもっているが、本発明の作製法は、羽毛色の改善、攻撃性の低下等、愛玩用として好ましい形質へ短期間で品種改良するための手段としても応用しうる。
【0033】
更に、現在哺乳類、両生類、魚類等のトランスジェニック作製は、核移植によって行われるのが通常であるが、本発明はそれに代わる新たなトランスジェニック動物作製の効率的手法としても応用できる。
【0034】
即ち、本発明は、複製能欠失型レトロウイルスベクターによって外来性抗体遺伝子が導入されたG0トランスジェニックキメラ鳥類であって、導入遺伝子に由来する抗体を、血中、卵白中及び卵黄中の少なくとも1つに産生するG0トランスジェニックキメラ鳥類である。上記抗体の定常領域は、クラスがヒトIgGであるか、サブクラスがヒトIgG1であるか、ウズラIgG、ニワトリIgG、あるいはマウスIgGであることが好ましい。上記抗体遺伝子は、構成的なプロモーターにより制御されていることが好ましく、上記構成的なプロモーターは、ニワトリβ−アクチンプロモーターであることが好ましい。上記レトロウイルスベクターは、モロニー・ミューリン・ロイケミア・ウイルス由来ベクターであることが好ましく、VSV−Gシュードタイプであることが好ましい。本発明のG0トランスジェニックキメラ鳥類は、ニワトリあるいはウズラであることが好ましい。上記抗体は、キメラ抗体であることが好ましく、上記キメラ抗体の産生量は、血液中には、0.5μg/ml以上であることが好ましく、より好ましくは5μg/ml以上であり、卵白中には、0.1μg/ml以上であることが好ましく、より好ましくは1μg/ml以上であり、卵黄中には、0.1μg/ml以上であることが好ましく、より好ましくは1μg/ml以上である。また、上記抗体は、scFv−Fc抗体であることが好ましく、上記scFv−Fc抗体の産生量は、血液中には、20μg/ml以上であることが好ましく、より好ましくは2000μg/ml以上であり、卵白中には、5μg/ml以上であることが好ましく、より好ましくは500μg/ml以上であり、卵黄中には、5μg/ml以上であることが好ましく、より好ましくは500μg/ml以上である。本発明のG0トランスジェニックキメラ鳥類を作製し、上記G0トランスジェニックキメラ鳥類の血中及び/又は卵中から抗体を回収する抗体の生産法もまた、本発明の1つである。
【0035】
鳥類受精卵を孵卵し、放卵直後の胚盤葉期を除くそれ以降の初期胚へ複製能欠失型レトロウイルスベクターを感染させ、その胚を孵化させることからなるG0トランスジェニックキメラ鳥類の作製法もまた、本発明の1つである。本発明のG0トランスジェニックキメラ鳥類の作製法において、複製能欠失型レトロウイルスベクターを感染させる時期は、孵卵開始から24時間以降であることが好ましい。上記複製能欠失型レトロウイルスベクターを感染させる方法は、初期胚に形成される心臓ないしは血管内へマイクロインジェクションすることであるのが好ましい。上記心臓ないしは血管は、孵卵開始から24時間以降の初期胚に形成されるものであることが好ましい。マイクロインジェクションする複製能欠失型レトロウイルスベクターの活性は、1×107cfu/ml以上のタイターであることが好ましく、1×108cfu/ml以上のタイターであることがより好ましく、1×109cfu/ml以上のタイターであることが更に好ましい。上記レトロウイルスベクターは、モロニー・ミューリン・ロイケミア・ウイルス由来ベクターであることが好ましく、VSV−Gシュードタイプであることが好ましい。本発明で作製するG0トランスジェニックキメラ鳥類は、ニワトリまたはウズラであることが好ましい。上記複製能欠失型レトロウイルスベクターに組み込まれる導入遺伝子は、レトロウイルスに由来しない遺伝子配列を含有することが好ましい。上記レトロウイルスに由来しない遺伝子配列は、ニワトリβ−アクチンプロモーターにより制御されている遺伝子配列であることが好ましく、抗体遺伝子又は融合タンパク遺伝子をコードする遺伝子配列であることが好ましい。上記抗体遺伝子は、キメラ抗体遺伝子であることが好ましく、scFv−Fc抗体遺伝子であることが好ましい。
【0036】
本発明のG0トランスジェニックキメラ鳥類の作製法で作製されたG0トランスジェニックキメラ鳥類もまた、本発明の1つである。
【発明の効果】
【0037】
本発明の方法によれば、鳥の種類によって決まる特定の時間が孵卵開始時より経過した後に、その初期胚へ外来遺伝子を導入するので、宿主による不活性化の影響を受けず、目的遺伝子を効率的に発現させることが可能となる。本発明の方法でG0トランスジェニックキメラ鳥類を作製したところ、血中、卵白、卵黄中に導入遺伝子に由来するタンパク質が高濃度に発現する。
【図面の簡単な説明】
【0038】
【図1】図1は、複製能欠失型レトロウイルスベクターのベクターコンストラクトpMSCVNΔAβの構造を示す。Neorはネオマイシン耐性遺伝子を、Amprはアンピシリン耐性遺伝子を示す。PΔActはβ−アクチンプロモーター遺伝子を示す。β−Galはβ−ガラクトシダーゼ発現遺伝子を示す。Ψ+はパッケージングシグナル配列を示す。5’LTR及び3’LTRはそれぞれMoMLVのロングターミナルリピート配列を示す。
【図2】図2は、G0トランスジェニックキメラウズラにおける、遺伝子の導入時期とβ−ガラクトシダーゼ活性発現との関係を示す。横軸は孵卵時間(hr)を、縦軸はmUnit/mgで表されるβ−ガラクトシダーゼ活性を示す。
【図3】図3は、G0トランスジェニックキメラニワトリにおける、遺伝子の導入時期とβ−ガラクトシダーゼ活性発現との関係を示す。横軸は孵卵時間(hr)を、縦軸はmUnit/mgで表されるβ−ガラクトシダーゼ活性を示す。
【図4】図4は、導入したレトロウイルスベクターのタイターとG0トランスジェニックキメラウズラでのβ−ガラクトシダーゼ活性発現との関係を示す。横軸はcfu/mlで表されるウイルスタイターを、縦軸はmUnit/mgで表されるβ−ガラクトシダーゼ活性を示す。
【図5】図5は、導入したレトロウイルスベクターのタイターとG0トランスジェニックキメラニワトリでのβ−ガラクトシダーゼ活性発現との関係を示す。横軸はcfu/mlで表されるウイルスタイターを、縦軸はmUnit/mgで表されるβ−ガラクトシダーゼ活性を示す。
【図6】図6は、ウズラ卵中に蓄積したヒトIgG抗体を示す。抗体値は3羽のウズラで行った同一の実験結果の平均値を示している。
【図7】図7は、ニワトリ卵中に蓄積したヒトIgG抗体を示す。抗体値は3羽のニワトリで行った同一の実験結果の平均値を示している。
【図8】図8は、ウズラ卵中に蓄積したFabフラグメントを示す。抗体値は3羽のウズラで行った同一の実験結果の平均値を示している。
【図9】図9は、ニワトリ卵中に蓄積したFabフラグメントを示す。抗体値は3羽のニワトリで行った同一の実験結果の平均値を示している。
【図10】図10は、ウズラ卵中に蓄積したFcフラグメントを示す。抗体値は3羽のウズラで行った同一の実験結果の平均値を示している。
【図11】図11は、ニワトリ卵中に蓄積したFcフラグメントを示す。抗体値は3羽のニワトリで行った同一の実験結果の平均値を示している。
【図12】図12は、抗CD2抗体発現ベクターコンストラクトpMSCV/GΔAL(12(A))、pMSCV/GΔAH(12(B))及びpMSCV/GΔALIH(12(C))の構造を示す。Amprはアンピシリン耐性遺伝子を示す。PΔActはβ−アクチンプロモーター遺伝子を示す。Ψ+はパッケージングシグナル配列を示す。GFPはグリーン・フルオレッセント・プロテイン遺伝子を示す。Lは抗CD2抗体軽鎖遺伝子を示す。Hは抗CD2抗体重鎖遺伝子を示す。5’LTR及び3’LTRはそれぞれMoMLVのロングターミナルリピート配列を示す。
【図13】図13は、scFv−Fc抗体発現ベクターコンストラクトpMSCV/GΔAscFv−Fcの構造を示す。Amprはアンピシリン耐性遺伝子を示す。PΔActはβ−アクチンプロモーター遺伝子を示す。Ψ+はパッケージングシグナル配列を示す。GFPはグリーン・フルオレッセント・プロテイン遺伝子を示す。scFv−FcはscFv−Fc抗体遺伝子を示す。5’LTR及び3’LTRはそれぞれMoMLVのロングターミナルリピート配列を示す。
【図14】図14は、G0トランスジェニックキメラウズラ血清中に発現したscFv−Fc量を示す。横軸は個体番号、縦軸はscFv−Fc抗体濃度(μg/ml)を示す。
【図15】図15は、G0トランスジェニックウズラ卵中に発現したscFv−Fc量をしめす。横軸は産卵開始からの採卵日、縦軸はscFv−Fc抗体濃度(μg/ml)を示す。
【図16】図16は、精製したscFv−FcのSDS−PAGEによる解析結果を示す。レーン1は低分子量マーカー(LMW)を、レーン4は高分子量マーカー(HMW)を示す。レーン2は還元処理したscFv−Fcを、レーン3は還元処理していないscFv−Fcを電気泳動した結果を示す。
【発明を実施するための形態】
【0039】
以下に本発明を詳述する。
本発明のG0トランスジェニックキメラ鳥類は、複製能欠失型レトロウイルスベクターによって外来性抗体遺伝子が導入された鳥類であって、導入遺伝子に由来する抗体を、血中、卵白中あるいは卵黄中に産生することを特徴とする。
【0040】
本発明で使用する鳥類としては、特に限定されず、例えばニワトリ、七面鳥、カモ、ダチョウ、ウズラなど、食肉、採卵目的で家畜化されている家禽鳥類や愛玩用鳥類を挙げることができる。なかでもニワトリやウズラは入手が容易で、産卵種としても多産である点が好ましい。
【0041】
本発明で使用されるレトロウイルスベクターとしては、モロニー・ミューリン・ロイケミア・ウイルス(MoMLV)、エビアン・ロイコシス・ウイルス(ALV)等に由来するベクターが挙げられる。なかでもMoMLVに由来するものが好ましいが、本発明はこれに限定されるものではない。
【0042】
安全性を考慮し、遺伝子導入ベクターとして用いられるウイルスは、通常ウイルス粒子の複製に必要な3種の遺伝子gag、pol、envのうちのいずれか又は全てを欠くことにより、自己複製能を欠失したウイルスが用いられる。鳥類細胞にこのウイルスベクターを効率的に感染させるため、外皮タンパク質を人工的にVSV−G(水疱性口内炎ウイルス由来)シュードタイプとしたウイルスベクターが好ましいが、本発明はこのウイルスタイプに限定されるものではない。
【0043】
パッケージング細胞またはヘルパーウイルス等を利用することにより調製されたシュードタイプのウイルスベクターは、通常のマイクロインジェクション法(Bosselman,R.Aら(1989)Science 243、533)により、初期胚、血管内、心臓内へ導入される。遺伝子導入法としては、この他にもリポフェクションやエレクトロポレーション法等が考えられる。
【0044】
本発明により鳥類に導入される遺伝子は特に限定されないが、マーカー遺伝子や目的タンパク質を発現するための構造遺伝子、これらの遺伝子発現をコントロールするプロモーター遺伝子、分泌シグナル遺伝子等により構成される。
【0045】
上記マーカー遺伝子としては、ネオマイシン耐性遺伝子、β−ガラクトシターゼ遺伝子、LacZ遺伝子、蛍光タンパク質、例えばGFP(グリーン・フロオロレッセント・プロテイン)等をコードした遺伝子が挙げられる。
【0046】
上記目的タンパク質を発現するための構造遺伝子としては特に制限されず、ヒトモノクローナル抗体など遺伝子産業上有用な抗体、酵素等をコードした遺伝子などが挙げられる。また、その他の有用生理活性物質の遺伝子を用いることもできる。特に、卵中での蓄積がよいことから、ヒトIgGクラスの定常領域をもつ抗体の遺伝子、ヒトIgG1のサブクラスの定常領域をもつ抗体の遺伝子、ウズラIgG、ニワトリIgGやマウスIgGの定常領域をもつ抗体の遺伝子など外来性抗体の構造遺伝子が好ましい。
【0047】
また好ましい上記構造遺伝子としては、キメラ抗体の構造遺伝子が挙げられる。
キメラ抗体とは、2種以上の異なる遺伝形質から構成される抗体のことをいう。
【0048】
従来、マウスハイブリドーマによって作製された医療用抗体はマウス由来であるため、ヒト体内に投与されると免疫系による拒絶反応が引き起こされるという問題があった。上記キメラ抗体としては、例えば、抗ヒトCD2抗体、抗CD20受容体抗体、抗TNF抗体など、当該欠点を改良し、マウス抗体のうち抗原タンパク質と結合する領域以外をヒト抗体で置き換え、拒絶反応をなくしたキメラ抗体が挙げられ、すでに医薬品として上市されているものもある。
【0049】
さらに好ましい上記構造遺伝子としては、scFv−Fc抗体の構造遺伝子が挙げられる。
医療用の組換え抗体には、低分子抗体と称される一群がある。免疫グロブリンIgGには直接抗原と結合する可変領域(Fv:Fragment of variable regeon)と呼ばれるVH、VLのヘテロ二量体からなるドメインがあり、このFvドメインはIgGの約5分の1の分子量でありながら、単独で充分な抗原結合能を持つ。VH、VLドメイン間を人工的にペプチドリンカーで結合したものが1本鎖抗体(scFv:single chain Fv)と呼ばれる低分子抗体で、VH、VL単独よりも安定性が向上することが知られていた。
【0050】
Powersら(Powers,D.Bら(2000)J Immunol Method.251,123)は、このscFvにヒトIgG1に由来するFc部を融合させることで、血中での安定性が増すことを見出した。このscFv−Fc抗体は医療用として有用と考えられるが、安価な大量生産システムである大腸菌では生産されない。
【0051】
他の好ましい上記遺伝子配列として、融合タンパクの構造遺伝子が挙げられる。
遺伝子組み換えにより2種あるいはそれ以上のタンパクの一部を融合させた人工的な一群のタンパクは融合タンパクと称され、医薬品として実用化されているものに、TNF受容体に免疫グロブリンのFcを融合させたTNFR−FcやLFA3にFcを融合させたLFA3−Fcなどがある。これらはFcを融合させることで可溶化され、より強力な生理活性をもつように設計された人工タンパクである。
【0052】
本発明のG0トランスジェニックキメラ鳥類において、鳥類に導入される遺伝子として、これらヒトモノクローナル抗体遺伝子、キメラ抗体遺伝子、scFv−Fc抗体遺伝子を使用することにより、従来生産が困難だった抗体医薬品を安価に大量生産できる。
【0053】
例えば、キメラ抗体遺伝子を導入したG0トランスジェニックキメラ鳥類の場合、血液中の抗体含有量は、好ましくは0.5μg/ml以上、より好ましくは5μg/ml以上である。また、卵白中の抗体含有量は、好ましくは0.1μg/ml以上、より好ましくは1μg/ml以上であり、卵黄中の抗体含有量は、好ましくは0.1μg/ml以上、より好ましくは1μg/ml以上である。
【0054】
また、scFv−Fc抗体遺伝子を導入したG0トランスジェニックキメラ鳥類の場合、血液中の抗体含有量は、好ましくは20μg/ml以上、より好ましくは2000μg/ml以上である。また、卵白中の抗体含有量は、好ましくは5μg/ml以上、より好ましくは500μg/ml以上である。卵黄中の抗体含有量は、好ましくは5μg/ml以上、より好ましくは500μg/ml以上である。
【0055】
上記プロモーター遺伝子としては、構成的なプロモーターが挙げられる。抗体遺伝子が構成的なプロモーターにより制御されている場合、抗体遺伝子発現が安定してよいので好ましい。より好ましい構成的なプロモーターとして、ニワトリβ−アクチンプロモーターが挙げられる。
【0056】
本発明の抗体の生産法は、本発明のG0トランスジェニックキメラ鳥類を作成し、上記G0トランスジェニックキメラ鳥類の血中及び/又は卵中から抗体を回収することを特徴とする。
【0057】
次に本発明のG0トランスジェニックキメラ鳥類の作製法について説明する。
当該作製法の1つとして、鳥類受精卵を孵卵し、放卵直後の胚盤葉期を除くそれ以降の初期胚へ複製能欠失型レトロウイルスベクターを感染させ、その胚を孵化させる作製法が挙げられる。また、鳥類受精卵を孵卵し、孵卵開始から24時間以降の初期胚へ複製能欠失型レトロウイルスベクターを感染させ、その胚を孵化させる作製法も、本発明の作製法の1つとして挙げられる。
【0058】
より好ましくは、上記初期胚に形成される心臓ないしは血管内へ、複製能欠失型レトロウイルスベクターをマイクロインジェクションする方法である。
【0059】
つまり、本発明のG0トランスジェニックキメラ鳥類の作製法は、放卵後特定の時間経過した受精卵に複製能欠失型レトロウイルスベクターをマイクロインジェクションすることよりなる。放卵後の受精卵の初期発生についてニワトリを例にとると、まず輸卵管内で受精した卵は、受精後約1.5時間で卵割を開始する。細胞質がつながったまま盤割が始まった卵は、1日かけて体外に放出される間に分裂し、約6万個の細胞からなる胚盤葉と呼ばれる胚になる(胚盤葉期)。この胚盤葉は、卵黄中央部の直径3〜4mmの白いリングとして観察される。この胚は、上層と下層に分裂し、割腔を形成する。放卵は胚盤葉下層が形成されるころに起こり、原条が形成され、胚盤葉は上、中、下の三重構造を取り、三胚葉が形成される。その後、胚の形成、成長を経て、排卵から22日目に孵化する。胚盤葉期は、ステージXとも呼ばれ、この時期の細胞の一部から生殖細胞が生じることから、従来は遺伝子導入の対象としてこの時期の受精卵を使用している。
【0060】
本発明においては、放卵直後、胚盤葉期の受精卵を孵化条件、例えばニワトリならば37.7〜37.8℃、湿度50〜70%程度の孵化に適した環境条件においた時間を0時間とし、これを孵卵開始時として、経時的に各種の処置を行った。ウズラでは孵卵開始から36時間後、ニワトリでは孵卵開始後50時間頃から、卵黄上に血管系の形成が観察され、心臓に分化する器官の脈動が観察できた。
【0061】
上述の遺伝子を導入した受精卵の孵化には、本発明者らが開発した人工卵殻による方法(Kamihira,M.ら(1998)Develop.Growth Differ.,40,449)等が応用できる。
【0062】
本発明の作製法において使用する複製能欠失型レトロウイルスベクター、導入する遺伝子、遺伝子導入鳥類としては、上述したG0トランスジェニックキメラ鳥類と同様のものが挙げられる。
【0063】
上記複製能欠失型レトロウイルスベクターに組み込まれる導入遺伝子は、レトロウイルスに由来しない遺伝子配列を含有することが好ましい。なお本発明の作製法において、「レトロウイルスに由来しない遺伝子」としては、上述した構造遺伝子、プロモーター遺伝子、分泌シグナル遺伝子等が挙げられる。上記レトロウイルスに由来しない遺伝子配列は、ニワトリβ−アクチンプロモーターにより制御されている遺伝子配列であることが好ましく、抗体遺伝子又は融合タンパク遺伝子をコードする遺伝子配列であることが好ましい。
【0064】
本発明の作製法では、1×107cfu/ml以上、好ましくは1×108cfu/ml以上、より好ましくは1×109cfu/ml以上のタイターを持つ複製能欠失型レトロウイルスベクターをマイクロインジェクションすることが、遺伝子を効率よく導入できる点で好ましい。
【0065】
本発明の作製法で受精卵に遺伝子を導入された鳥類は、その体細胞にモザイク状に導入遺伝子をもったトランスジェニック鳥類として成長する。この一世代目の遺伝子導入鳥類をG0トランスジェニックキメラ鳥類と呼ぶ。
【0066】
このような本発明の作製法によって得られるG0トランスジェニックキメラ鳥類も、本発明の1つである。
【0067】
G0トランスジェニックキメラ鳥類と非トランスジェニック鳥類、あるいはG0トランスジェニックキメラ鳥類同士を交配させて誕生する二世代目、三世代目が導入遺伝子を染色体にもつ生殖細胞から発生した場合、全身の体細胞に導入遺伝子を含有する個体として成長する。G0トランスジェニックキメラ鳥類個体から導入遺伝子を受け継ぐ子孫を、代々G1〜G2〜G3トランスジェニック鳥類と称する。
【0068】
本発明によるG0トランスジェニックキメラ鳥類を同種の非トランスジェニック鳥類あるいは配偶型G0トランスジェニックキメラ鳥類と交配させることにより、導入遺伝子を子孫に伝播させることができるとともに、全身の体細胞に導入遺伝子をもつ完全なトランスジェニック鳥類を作製できる。完全なトランスジェニック鳥類は、導入遺伝子をもつ体細胞の割合が多いことから、G0トランスジェニックキメラ鳥類に比べ、導入遺伝子に由来する組換えタンパク質の産生量が増加することが期待できる。更に、安定的に導入遺伝子を伝播するトランスジェニック鳥類の系統を確立することで、タンパク質産生システムとしての品質の安定化が可能である。
【実施例】
【0069】
以下、実施例により本発明を詳述するが、本発明はこれらの実施例により限定されるものではない。
【0070】
(実施例1)β−ガラクトシダーゼ発現ベクターコンストラクトの調製
β−ガラクトシダーゼ発現ベクターコンストラクトpMSCVNΔAβは、以下のように作製した。
【0071】
1.プラスミドpLXRN(クロンテック社製)からラウス・ザルコーマ・ウイルス(RSV)プロモーター断片を制限酵素XhoI及びHindIIIによって切り出し、プラスミドpBluescriptIISK(+)(ストラタジーン社製)のXhoI、HindIIIサイトへ挿入し、プラスミドpBlue/RSVを作製した。
【0072】
2.プラスミドpCMVβ(クロンテック社製)からβ−ガラクトシダーゼ(β−Gal)遺伝子断片を制限酵素NotIによって切り出し、プラスミドpZeoSV2(+)(インビトロジェン社製)のNotIサイトへ挿入した。T7プロモーターと同方向にβ−Gal遺伝子が挿入された構造のプラスミドをpZeo/lacZとした。
【0073】
3.pBlue/RSVからRSVプロモーター断片を制限酵素XhoI及びPstIによって切り出した。pZeo/lacZからβ−Gal遺伝子断片を制限酵素PstI及びXhoIによって切り出した。制限酵素XhoIによって処理したプラスミドpLNHX(クロンテック社製)のベクター断片に上記切り出した2断片を連結し、プラスミドpLNRβを作製した。
【0074】
4.pLNHXから一連のモロニー・ミューリン・ザルコーマ・ウイルス(MoMuSV)5’−ロング・ターミナル・リピート(LTR)、ウイルス・パッケージング・シグナル及びネオマイシン耐性(Neor)遺伝子を含む断片を、制限酵素SacII及びXhoIによって切り出し、制限酵素SacII及びXhoIによって処理したpLXRNのベクター断片に連結し、プラスミドpLXLを作製した。
【0075】
5.pZeo/lacZからβ−Gal遺伝子断片を制限酵素HindIII及びXhoIによって切り出し、制限酵素HindIII及びXhoIによって処理したpLXLのベクター断片に連結し、プラスミドpLZLを作製した。
【0076】
6.2つの化学合成オリゴヌクレオチド5’−cggtctagaggaattcagtggttcg−3’(配列番号1)及び5’−ccaggatccgacgttgtaaaacgacg−3’(配列番号2;下線部はBamHI制限酵素サイト)をプライマーとするPCR(94℃/15秒→55℃/30秒→72℃/1分30秒:35サイクル;KOD−Plus−DNAポリメラーゼ(東洋紡社製))により、プラスミドpMiwZ(Suemori et al.,1990,Cell Diff.Dev.29:181−185)からRSVプロモーターとニワトリβアクチン(Act)プロモーターのハイブリッドプロモーター(Miwプロモーター)の5’領域断片を増幅後、制限酵素BamHI及びMunIによって切り出し、プラスミドpGREEN LANTERN−1(ギブコBRL社製)のBamHI、MunIサイトへ挿入し、プラスミドpGmiw5’を作製した。
【0077】
7.pMiwZからMiwプロモーター5’側中央領域断片を制限酵素MunI及びClaIによって切り出し、pGmiw5’のMunI、ClaIサイトへ挿入し、プラスミドpGmiw5’−2を作製した。
【0078】
8.pGmiw5’−2からMiwプロモーター5’領域及び5’側中央領域を含む断片を制限酵素BamHI及びEcoRIによって切り出し、pBluescriptIISK(+)のBamHI、EcoRIサイトへ挿入し、プラスミドpBlue/Miw5’を作製した。
【0079】
9.2つの化学合成オリゴヌクレオチド5’−ccaaagcttgccgcagccattgcctttt−3’(配列番号3;下線部はHindIII制限酵素サイト)及び5’−atacctaggggctggctgcggaggaac−3’(配列番号4;下線部はBlnI制限酵素サイト)をプライマーとするPCR(98℃/15秒→60℃/30秒→72℃/30秒:35サイクル)によりpMiwZからMiwプロモーター3’領域断片を増幅後、制限酵素HindIII及びBlnIによって切り出し、制限酵素HindIII及びBlnIによって処理したpLXLのベクター断片に連結し、プラスミドpLMiw3’を作製した。
【0080】
10.pMiwZからMiwプロモーター3’側中央領域断片を制限酵素EcoRI及びMboIIによって切り出した。pLMiw3’からMiwプロモーター3’領域断片を制限酵素MboII及びKpnIによって切り出した。pBlue/Miw5’のEcoRI、KpnIサイトへ上記切り出した2断片を挿入し、プラスミドpBlue/Miwを作製した。
【0081】
11.pBlue/MiwからMiwプロモーター全長を含む断片を制限酵素BamHI及びBlnIによって切り出し、制限酵素BamHI及びBlnIによって処理したpLXLのベクター断片に連結し、プラスミドpLMLを作製した。
【0082】
12.pLMLからActプロモーター断片を制限酵素SmaI及びXbaIで切り出し、pBluescriptIISK(+)のEcoRV、XbaIサイトへ挿入し、プラスミドpBlue/Actを作製した。
【0083】
13.pLMLからMiwプロモーター断片を制限酵素HindIII及びBglIIによって切り出し、制限酵素HindIII及びBamHIによって処理したpLZLのベクター断片に連結し、プラスミドpLMβLを作製した。
【0084】
14.pBlue/ActからActプロモーター断片を制限酵素SalI及びBlnIによって切り出した。pLMβLからβ−Gal遺伝子断片を制限酵素BlnI及びBglIIによって切り出した。制限酵素XhoI及びBglIIによって処理したpLNRβのベクター断片に上記切り出した2断片を連結し、プラスミドpLNAβを作製した。
【0085】
15.2つの化学合成オリゴヌクレオチド5’−tttagctagctgcagctcagtgcatgcac−3’(配列番号5;下線部はNheI制限酵素サイト)及び5’−ataatctagaaacgcagcgactcccgc−3’(配列番号6;下線部はXbaI制限酵素サイト)をプライマーとするPCR(98℃/15秒→60℃/30秒→68℃/2分:30サイクル)によりpMiwZからイントロン欠失アクチン(ΔAct)プロモーター断片を増幅後、制限酵素XhoI(XhoI制限酵素サイトは増幅断片中に存在)及びXbaIによってΔActプロモーターの一部を含む断片を切り出した。ΔActプロモーターの残りの部分とβ−Gal遺伝子を含む断片をpLNAβから制限酵素BlnI及びBglIIによって切り出した。制限酵素XbaI及びBglIIによって処理したpLNAβのベクター断片に上記切り出した2断片を連結し、プラスミドpLNΔAβを作製した。
【0086】
16.pLNΔAβから一連のNeor遺伝子、ΔActプロモーター、及びβ−Gal遺伝子を含む断片を制限酵素BlnI及びBglIIによって切り出し、制限酵素BlnI及びBglIIによって処理したpLXLのベクター断片に連結し、プラスミドpLNΔAβ−2を作製した。
【0087】
17.pLNΔAβ−2から一連のNeor遺伝子、ΔActプロモーター、及びβ−Gal遺伝子を含む断片を制限酵素BamHI及びBglIIによって切り出し、制限酵素BamHI及びBglIIによって処理したプラスミドpMSCVneo(クロンテック社製)のベクター断片に連結した。BamHI及びBglIIサイトが消失したプラスミドをpMSCVNΔAβとした。
このように作製した複製能欠失型レトロウイルスベクターのベクターコンストラクトpMSCVNΔAβの構造を図1に示した。
【0088】
(実施例2)β−ガラクトシダーゼ発現レトロウイルスベクターの調製
実施例1で作製したベクターコンストラクトpMSCVNΔAβよりレトロウイルスベクターを調製するため、パッケージング細胞GP293(クロンテック社製)を直径100mmの培養ディッシュに5×106細胞植え、培養した。培地を新鮮なDMEM(ダルベッコ変法イーグル培地)に交換し、p−VSV−Gベクター(クロンテック社製)8μgとpMSCVNΔAβ8μgをリポフェクション法により前記GP293細胞に導入した。48時間後、ウイルス粒子を含む培養上清を回収し、0.45μm酢酸セルロースフィルター(アドバンテック社製)を通して、夾雑物を除去した。得られた溶液にポリブレン(シグマ社製)を10μg/mlとなるように加えウイルス液とした。
【0089】
調製したウイルス液を別に培養したGP293細胞に加え、48時間培養後、600μg/mlのG418(GIBCO BRL社製)を含む培養液で植え継ぎ、G418安定形質転換GP293株を取得した。
【0090】
得られた安定形質転換株を80%コンフルエントとなるよう直径100mmディッシュに培養し、16μgのpVSV−Gベクターをリポフェクション法で導入した。48時間後ウイルス粒子を含む培養上清12mlを回収した。
【0091】
この培養上清を50,000×g、4℃で1.5時間遠心を行い、ウイルスを沈殿させた。上清を除き、ウイルス粒子を含む沈殿物に50μlの50mM Tris−HCl(pH7.8)、130mM NaCl、1mM EDTA溶液を加え、4℃で一晩放置後、よく懸濁してウイルス溶液を回収した。このようにして得られた高タイターウイルスベクターは、108〜109cfu/mlであった。
【0092】
ウイルスタイターの測定は、以下のように行った。測定の前日にNIH3T3細胞(アメリカン・タイプ・カルチャー・コレクションより入手)を直径35mmのディッシュに7×104細胞植え、培養した。102〜106倍に希釈したウイルス溶液を各ディッシュに1ml加え、48時間後に蛍光顕微鏡によりGFP(グリーン・フルオレッセント・プロテイン)を発現している細胞の割合を測定し、以下の計算式によりタイターを決定した。
ウイルスタイター=細胞数×希釈率×発現割合(cfu/ml)
【0093】
(実施例3)ウズラ胚へのレトロウイルスベクターのインジェクション
WE系統のウズラ受精卵(日本生物科学研究所)を使用した。この受精卵を自動転卵装置が内蔵された孵卵器(昭和フランキ社製P−008型)内で37.9℃、湿度65%環境に置いた時刻を孵卵開始時刻(0時間)とし、以後15分毎に90度転卵しながら孵卵を行った。
【0094】
孵卵開始時、受精卵の卵殻を70%エタノールで消毒し、鋭端部を直径2cmの円形にダイヤモンドカッター(MINIMO7C710、ミニター社製)で切り取り、胚を露出させた。胚盤葉を実体顕微鏡で観察しながら、ガラス管(CD−1、オリンパス社製)をマイクロピペット製作器(PC−10、オリンパス社製)で加工し、外径約20μmとなるよう先端を折って作製した針を刺し、マイクロインジェクター(Transjector5246、エッペンドルフ社製)を用いて、胚盤下腔の中央に実施例2で調製したウイルス溶液約2μlを微量注入した。この卵殻の切り口まで卵白を満たした後、卵白を糊としてテフロン(登録商標)膜(ミリラップ、ミリポア社製)とポリ塩化ビニリデンラップ(サランラップ、旭化成社製)とで蓋をし、15分毎に90度転卵しながら孵卵を行った。
【0095】
孵卵開始から12時間後、24時間後の受精卵に同様の方法でウイルスを注入した。孵卵後約36時間目から卵黄表面に血管の発生が認められ、その一部が脈動して心臓の原基となることが実体顕微鏡で観察できる。孵卵後36時間、48時間、55時間の心臓内へ、実施例2で調製したウイルス溶液2μlをマイクロインジェクターにより微量注入した。
【0096】
(実施例4)ニワトリ胚へのレトロウイルスベクターのインジェクション
ニワトリ受精卵(日本生物科学研究所)を使用した。この受精卵を自動転卵装置が内蔵された孵卵器(昭和フランキ社製P−008型)内で37.9℃、湿度65%環境に置いた時刻を孵卵開始時刻(0時間)とし、以後15分毎に90度転卵しながら孵卵を行った。
【0097】
孵卵開始時、受精卵の卵殻を70%エタノールで消毒し、鋭端部を直径3.5cmの円形にダイヤモンドカッター(MINIMO7C710、ミニター社製)で切り取り、胚を露出させた。胚盤葉を実体顕微鏡で観察しながら、ガラス管(CD−1、オリンパス社製)をマイクロピペット製作器(PC−10、オリンパス社製)で加工し、外径約20μmとなるよう先端を折って作製した針を刺し、マイクロインジェクター(Transjector5246、エッペンドルフ社製)を用いて胚盤下腔の中央に、実施例2で調製したウイルス溶液約2μlを微量注入した。この卵殻の切り口まで卵白を満たした後、卵白を糊としてテフロン(登録商標)膜(ミリラップ、ミリポア社製)とポリ塩化ビニリデンラップ(サランラップ、旭化成社製)とで蓋をし、15分毎に90度転卵しながら孵卵を行った。
【0098】
孵卵開始から12時間後、24時間後の受精卵を同様に処理した。孵卵後約50時間目から卵黄表面に血管の発生が認められ、その一部が脈動して心臓の原基となることが実体顕微鏡で観察できる。孵卵後50時間、55時間、60時間の心臓内へ、実施例2で調製したウイルス溶液2μlをマイクロインジェクターにより微量注入した。
【0099】
(実施例5)β−ガラクトシダーゼ活性測定
孵卵開始から115時間後、胚を卵殻より取り出しPBS(リン酸緩衝液)にて洗浄し、胚をとり囲む膜を取り除いた。摘出した胚を細かくせん断し、0.8mlの反応バッファー(10mM KCl、1mM MgCl2、0.1% Triton X−100(和光純薬工業製)、5mM 2−メルカプトエタノール(和光純薬工業製)、2mM リン酸バッファー pH7.5)を加えて超音波破砕し、細胞液を得た。
【0100】
0.6mlの細胞液を、37℃で10分間インキュベートし、あらかじめ暖めておいた4mg/mlのo−ニトロフェニル−β−D−ガラクトピラノシド(ONPG)(シグマ社製)を0.1Mリン酸バッファー(pH7.5)に溶解した液0.1mlを添加した。反応後0.3mlの1M Na2CO3(和光純薬工業製)を加え、吸光度計にて420nmの波長強度を計測した。
【0101】
β−ガラクトシダーゼの活性はONPGunitで表した(1unit:1μmolのo−ニトロフェノールが1分間に生成されるときの活性)。3回実験を行い、その平均値をβ−ガラクトシダーゼ活性とした。
【0102】
ニワトリおよびウズラの、遺伝子導入時期とβ−ガラクトシダーゼ活性測定結果との関係を図2、図3に示した。ウズラでは孵卵後48時間後、ニワトリでは孵卵後55時間後に導入したβ−ガラクトシダーゼ遺伝子は、それ以前の時期に導入されたものに比べ、強く発現した。これは鳥類受精卵が、レトロウイルスによる外来遺伝子を不活性化する(サイレンシング)メカニズムが受精直後に顕著であり、時間を経るごとに弱くなっていくことを示唆する。このことから、鳥類の種類によって決まる特定の時間経過した受精卵に目的遺伝子を導入することで、不活性化されることなく効率的に導入遺伝子を発現するG0トランスジェニックキメラ鳥類の作製が可能になる。
【0103】
(実施例6)ウイルスタイターによる遺伝子発現の効率
実施例2で調製した1×108cfu/mlのウイルス液を、希釈溶媒(50mM Tris−HCl(pH7.8)、130mM NaCl、1mM EDTA溶液)で10倍、100倍、1000倍に3段階希釈し、1×107、1×106、1×105cfu/mlのタイター・ウイルス溶液とした。ウズラ受精卵を孵卵し、48時間後の発生初期心臓に、調製したウイルス液2μlをマイクロインジェクションした。同様にして48時間後の発生初期心臓に希釈溶媒のみを2μlインジェクションしたものを対照とした。
【0104】
孵卵開始から115時間後、実施例5に準じてβ−ガラクトシダーゼ活性を測定した。ウズラでのウイルスタイターと遺伝子発現結果との関係を図4に示した。
【0105】
同様にニワトリ受精卵を孵卵し、55時間後の発生初期心臓に、調製したウイルス液2μlをマイクロインジェクションした。55時間後の発生初期心臓に希釈溶媒のみを2μlインジェクションしたものを対照とした。
【0106】
孵卵開始から115時間後、実施例5に準じてβ−ガラクトシダーゼ活性を測定した。ニワトリでのウイルスタイターと遺伝子発現結果との関係を図5に示した。
【0107】
1×108cfu/mlのウイルスが注入されたウズラおよびニワトリで顕著なβ−ガラクトシダーゼ活性が確認され、それ以下の濃度では発現量は低かった。導入遺伝子の発現にはウイルスタイターが大きく影響すること、すなわち本発明によるG0トランスジェニックキメラ鳥類で、導入遺伝子を効率的に発現させるためには高タイターの複製能欠失型ベクターを使用するのが有効であるということが示唆された。
【0108】
(実施例7)ヒト抗体のウズラ、ニワトリ卵内への移行性
3種のサブクラスをもつヒト抗体(IgG1、2、3)(コスモバイオ社製)を混合したもの、および対応する3種の抗体フラグメント(Fab−1、Fab−2、Fab−3、Fc−1、Fc−2、Fc−3)(コスモバイオ社製)を100μg/mlとなるようPBSで希釈し、希釈液100μlをウズラ成鳥(3羽)またはニワトリ成鳥(3羽)の翼下静脈に注射した。
【0109】
抗体を静脈に注射した翌日から20日目まで採卵を行い、卵中に移行した抗体を定量した。卵黄は50%(W/V)、卵白は10%(V/V)となるようPBSを用いて希釈し、凍結保存して測定用サンプルとした。
【0110】
(実施例8)ELISA法による卵黄中抗体の定量
PBSで希釈した抗ヒトIgG抗体(コスモバイオ社製)をELISAプレートに100μg/well入れて、4℃で一晩静置した。PBS−0.05%Tween20溶液を200μlで各wellを3回洗浄した後、PBS−0.05%Tween20溶液−2%スキムミルクを150μl/well入れた。
【0111】
室温で2時間静置後、wellを200μlのPBS−0.05%Tween20溶液で3回洗浄し、採取した血液、卵白、卵黄サンプルを120μl入れ、4℃で一晩静置した。このELISAプレートを室温に戻した後、PBS−Tween20溶液で3回各wellを洗浄し、PBS−0.05%Tween20溶液で希釈したPeroxide(POD)標識抗ヒトIgG抗体(コスモバイオ社製)を100μl/well入れて、室温で1時間静置した。
【0112】
PBS−0.05%Tween20溶液でwellを4回洗浄し、発色液(10mgのo−フェニレンジアミン(片山化学工業製)を1mlのメタノールに溶解し、蒸留水で100mlとしたものに、10μlの過酸化水素水(和光純薬工業製)を加えて調製した)100μlをwellに加えた。8M硫酸を50μl添加して反応を止め、490nmの蛍光強度をプレートリーダーで測定して、標準検量線から濃度を計算した。ウズラ、ニワトリ各々3羽から得られたサンプルの抗体濃度を平均したものを結果とした。
【0113】
標準検量線作製のための標準抗体(コスモバイオ社製)は、50%卵黄−PBS(W/V)で希釈した。
【0114】
ウズラおよびニワトリの卵中に蓄積された抗体濃度を図6、7に示した。ウズラおよびニワトリの卵中に蓄積されたFab,Fcフラグメント濃度を図8、9、10、11に示した。
【0115】
ウズラ、ニワトリにおいて、ヒトIgG抗体、サブクラスではヒトIgG2、ヒトIgG1が効率よく卵中に蓄積されることが示された。またFcフラグメントが卵内への高い移行性を示したことから、ヒトIgGの移行はFcレセプターを介したものであることが示唆された。
【0116】
(実施例9)抗CD2抗体発現ベクターコンストラクトの作製
抗CD2抗体発現用ベクターコンストラクトpMSCV/GΔAH、pMSCV/GΔAL及びpMSCV/GΔALIHは、以下のように作製した。
【0117】
1.ヒト抗体(IgM)産生ハイブリドーマ細胞D253−15−6(アメリカン・タイプ・カルチャー・コレクションHB−8789)からQuick Prep Micro mRNA Purification Kit(ファルマシア社製)を用いてmRNAを取得し、得られたmRNAからFirst−Strand cDNA Synthesis Kit(ファルマシア社製)を用いてcDNAライブラリを調製した。2つの化学合成オリゴヌクレオチド5’−atcctcgagaggccaaagtacagtg−3’(配列番号7;下線部はXhoI制限酵素サイト)及び5’−cccggatccctaacactctcccctgttgaagct−3’(配列番号8;下線部はBamHI制限酵素サイト)をプライマーとするPCR(94℃/1分→50℃/1分→72℃/1分30秒:25サイクル;TaqDNAポリメラーゼ(パーキンエルマー社製))により上記cDNAライブラリからヒト抗体L鎖κ定常領域(hCκ)の遺伝子断片を増幅後、制限酵素XhoI及びBamHIによって切り出し、プラスミドpBluescriptIIKS(−)(ストラタジーン社製)のXhoI、BamHIサイトへ挿入し、プラスミドpBlue/hCκを作製した。
【0118】
2.同様にして、2つの化学合成オリゴヌクレオチド5’−agcggccgctacaggtgtccactccgacatcgtgatgacccagtctcc−3’(配列番号9;下線部はNotI制限酵素サイト)及び5’−cctctcgaggatagaagttattcagcaggcacac−3’(配列番号10;下線部はXhoI制限酵素サイト)をプライマーとするPCRにより上記cDNAライブラリからヒト抗体L鎖可変領域(hVL)の遺伝子断片を増幅後、制限酵素NotI及びXhoIによって切り出し、pBluescriptIIKS(−)のNotI、XhoIサイトへ挿入し、プラスミドpBlue/hVLを作製した。
【0119】
3.同様にして、2つの化学合成オリゴヌクレオチド5’−acctcgagcgtggccgttggctgcctcgcaca−3’(配列番号11;下線部はXhoI制限酵素サイト)及び5’−actaagcttacgttgtacagggtgggtttacc−3’(配列番号12;下線部はHindIII制限酵素サイト)をプライマーとするPCRにより上記cDNAライブラリからヒト抗体H鎖μ定常領域(hCμ)の遺伝子断片を増幅後、制限酵素XhoI及びHindIIIによって切り出し、pBluescriptIIKS(−)のXhoI、HindIIIサイトへ挿入し、プラスミドpBlue/hCμを作製した。
【0120】
4.同様にして、2つの化学合成オリゴヌクレオチド5’−agcggccgctacaggtgtccactccgaggtgcagctggtggagtctgg−3’(配列番号13;下線部はNotI制限酵素サイト)及び5’−cacgctcgaggtatccgacggggaattctcacagga−3’(配列番号14;下線部はXhoI制限酵素サイト)をプライマーとするPCRにより上記cDNAライブラリからヒト抗体H鎖可変領域(hVH)の遺伝子断片を増幅後、制限酵素NotI及びXhoIによって切り出し、pBluescriptIIKS(−)のNotI、XhoIサイトへ挿入し、プラスミドpBlue/hVHを作製した。
【0121】
5.pBlue/hCκからhCκ遺伝子断片を制限酵素XhoI及びBamHIによって切り出し、プラスミドpCEP4(インビトロジェン社製)のXhoI、BamHIサイトへ挿入し、プラスミドpCEP4/hCκを作製した。
【0122】
6.2つの化学合成オリゴヌクレオチド5’−cccaagcttgatctccactgggatggtgggggccctcctcttgctgctg−3’(配列番号15;下線部はHindIII制限酵素サイト)及び5’−cccggatcctcagtcaaggcgccttcgcatgaagaggccgatccccagggccaccaccagcagcaagaggagggcccc−3’(配列番号16;下線部はBamHI制限酵素サイト)を21bpsにわたって相補的な3’末端でアニールさせ、T4DNAポリメラーゼ(宝酒造社製)を用いたDNA2重鎖合成反応によって上皮増殖因子受容体膜貫通領域(TM)の遺伝子断片を調製した。得られたTM遺伝子断片を制限酵素HindIII及びBamHIによって処理後、pBluescriptIIKS(−)のHindIII、BamHIサイトに挿入し、プラスミドpBlue/TMを作製した。
【0123】
7.pBlue/hCμからhCμ遺伝子断片を制限酵素XhoI及びHindIIIによって切り出し、pBlue/TMのXhoI、HindIIIサイトへ挿入し、プラスミドpBlue/hCμTMを作製した。
【0124】
8.pBlue/hCμTMから一連のhCμ遺伝子及びTM遺伝子を含む断片を制限酵素XhoI及びBamHIによって切り出し、pCEP4のXhoI、BamHIサイトへ挿入し、プラスミドpCEP4/hCμTMを作製した。
【0125】
9.pCEP4/hCμTMを制限酵素BamHIによって切断し、末端をT4DNAポリメラーゼによって平滑処理後、セルフライゲーションによりプラスミドpCEP4/hCμTMΔBを作製した。
【0126】
10.化学合成オリゴヌクレオチド5’−tgaagacagatggcgccgccacagttcgttt−3’(配列番号17;下線部はNarI制限酵素サイト)を用いた部位特異的変異導入によりpBlue/hVLが保有するhVLの3’末端にアミノ酸暗号の変更を伴わずに制限酵素NarIサイトを導入し、プラスミドpBlue/hVLNを作製した。
【0127】
11.化学合成オリゴヌクレオチド5’−tggggcggatgcggatcctgaggagacggt−3’(配列番号18;下線部はBamHI制限酵素サイト)を用いた部位特異的変異導入によりpBlue/hVHが保有するhVHの3’末端にアミノ酸暗号の変更を伴わずに制限酵素BamHIサイトを導入し、プラスミドpBlue/hVHBを作製した。
【0128】
12.抗ヒトCD2マウス抗体産生ハイブリドーマ細胞TS2/18.1.1(アメリカン・タイプ・カルチャー・コレクションHB−195)からQuick Prep Micro mRNA Purification Kitを用いてmRNAを取得し、得られたmRNAからFirst−Strand cDNA Synthesis Kitを用いてcDNAライブラリを調製した。2つの化学合成オリゴヌクレオチド5’−cgcggccgcctcagggaaagtttgaagatg−3’(配列番号19;下線部はNotI制限酵素サイト)及び5’−cggcgccgccacagtccgttttatttccagcttggt−3’(配列番号20;下線部はNarI制限酵素サイト)をプライマーとするPCRにより上記cDNAライブラリからマウス抗体L鎖可変領域(mVL)の遺伝子断片を増幅後、制限酵素NotI及びNarIによって切り出し、pBlue/hVLNのNotI、NarIサイトへ挿入し、プラスミドpBlue/mVLを作製した。
【0129】
13.同様にして、2つの化学合成オリゴヌクレオチド5’−cgcggccgcgaacacggamccctcaccatg−3’(配列番号21;下線部はNotI制限酵素サイト)及び5’−cggatcctgcagagacagtgaccagagt−3’(配列番号22;下線部はBamHI制限酵素サイト)をプライマーとするPCRにより上記cDNAライブラリからマウス抗体H鎖可変領域(mVH)の遺伝子断片を増幅後、制限酵素NotI及びBamHIによって切り出し、pBluescriptIIKS(−)のNotI、BamHIサイトへ挿入し、プラスミドpBlue/mVHを作製した。
【0130】
14.pBlue/mVLからmVL遺伝子断片を制限酵素NotI及びXhoIによって切り出し、pCEP4/hCκのNotI、XhoIサイトへ挿入し、プラスミドpCEP4/IgLκを作製した。
【0131】
15.pBlue/hVHBからhVH遺伝子断片を制限酵素NotI及びXhoIによって切り出し、pCEP4/hCμTMΔBのNotI、XhoIサイトへ挿入し、プラスミドpCEP4/hIgHμTMを作製した。
【0132】
16.pBlue/mVHからmVH遺伝子断片を制限酵素NotI及びBamHIによって切り出し、制限酵素NotI及びBamHIによって処理したpCEP4/hIgHμTMのベクター断片に連結し、プラスミドpCEP4/IgHμTMを作製した。
【0133】
17.プラスミドpMSCVneo(クロンテック社製)から一連のミューリン・ホスホグリセレート・キナーゼ(PGK)プロモーター及びNeor遺伝子を含む断片を制限酵素BglII及びBamHIによって除去し、残ったベクター断片のセルフライゲーションによりプラスミドpMSCVを作製した。
【0134】
18.プラスミドpGREEN LANTERN−1(ギブコBRL社製)からGFP遺伝子断片を制限酵素NotIによって切り出し、pZeoSV2(+)のNotIサイトに挿入した。T7プロモーターと同方向にGFP遺伝子が挿入された構造のプラスミドをpZeo/GFPとした。
【0135】
19.pZeo/GFPからGFP遺伝子断片を制限酵素EcoRI及びXhoIによって切り出し、制限酵素EcoRI及びXhoIによって処理したpMSCVのベクター断片に連結し、プラスミドpMSCV/Gを作製した。
【0136】
20.ヒト抗体(IgG1)産生ミエローマ細胞IM−9(ジャパニーズ・コレクション・オブ・リサーチ・バイオリソーシズ 0024)からmRNA isolation Kit(ロッシュ社製)を用いてmRNAを取得し、得られたmRNAからReverTra Ace(東洋紡社製)を用いてcDNAライブラリを調製した。2つの化学合成オリゴヌクレオチド5’−caagcttcaagggcccat−3’(配列番号23)及び5’−atttacccggagacaggga−3’(配列番号24)をプライマーとするPCR(95℃/2分→52℃/30秒→74℃/3分:30サイクル;Pfu DNAポリメラーゼ(プロメガ社製))により上記cDNAライブラリからヒト抗体H鎖γ1定常領域(hCγ1)の遺伝子断片を増幅した。さらに、2つの化学合成オリゴヌクレオチド5’−ataggatccgctagcttcaagggcccatcg−3’(配列番号25;下線部はBamHI制限酵素サイト)及び5’−agcaagctttcatttacccggagacaggga−3’(配列番号26;下線部はHindIII制限酵素サイト)をプライマーとするPCR(94℃/15秒→58℃/30秒→68℃/1分:30サイクル;KOD−plus−DNAポリメラーゼ)により上記PCR産物からhCγ1遺伝子断片を増幅後、制限酵素BamHI及びHindIIIによって切り出し、pBluescriptIISK(+)のBamHI、HindIIIサイトへ挿入し、プラスミドpBlue/hCγ1を作製した。
【0137】
21.pCEP4/IgHμTMからmVH遺伝子断片を制限酵素HindIII及びBamHIによって切り出した。pBlue/hCγ1からhCγ1遺伝子断片を制限酵素BamHI及びHindIIIによって切り出した。制限酵素HindIIIによって処理したプラスミドpETBlue−2(ノバジェン社製)のベクター断片に上記切り出した2断片を連結し、プラスミドpETBlue/IgHγ1を作製した。
【0138】
22.pETBlue/IgHγ1から抗体H鎖γ1(IgHγ1)の遺伝子断片を制限酵素HindIIIによって切り出し、pMSCV/GのHindIIIサイトへ挿入した。GFP遺伝子と同方向にIgHγ1遺伝子が挿入された構造のプラスミドをpMSCV/GHとした。
【0139】
23.2つの化学合成オリゴヌクレオチド5’−acgcgtcgacgtgcatgcacgctcattg−3’(配列番号27;下線部はSalI制限酵素サイト)及び5’−acgcgtcgacaacgcagcgactcccg−3’(配列番号28;下線部はSalI制限酵素サイト)をプライマーとするPCR(94℃/15秒→50℃/30秒→68℃/1分:10サイクル;94℃/15秒→62℃/30秒→68℃/1分:30サイクル)によりpMiwZからΔActプロモーター断片を増幅後、制限酵素SalIによってΔActプロモーター断片を切り出し、pETBlue−2のSalIサイトへ挿入し、プラスミドpETBlue/ΔActを作製した。
【0140】
24.pETBlue/ΔActからΔActプロモーター断片を制限酵素SalIによって切り出し、pMSCV/GHのXhoIサイトへ挿入した。IgHγ1遺伝子と同方向にΔActプロモーターが挿入された構造のプラスミドをpMSCV/GΔAHとした。
【0141】
25.2つの化学合成オリゴヌクレオチド5’−aatgtcgacatggtgtccacttctcagctc−3’(配列番号29;下線部はSalI制限酵素サイト)及び5’−ttcgtcgacctaacactctcccctgttgaa−3’(配列番号30;下線部はSalI制限酵素サイト)をプライマーとするPCR(95℃/30秒→50℃/30秒→74℃/2分:10サイクル;95℃/30秒→60℃/30秒→74℃/2分:30サイクル;Pfu DNAポリメラーゼ)によりpCEP4/IgLκから抗体L鎖κ(IgLκ)の遺伝子断片を増幅後、制限酵素SalIによって切り出し、pETBlue−2のSalIサイトへ挿入し、プラスミドpETBlue/IgLκを作製した。
【0142】
26.pETBlue/ΔActからΔActプロモーター断片を制限酵素SalIによって切り出し、pMSCV/GのXhoIサイトへ挿入した。GFP遺伝子と同方向にΔActプロモーターが挿入された構造のプラスミドをpMSCV/GΔAとした。
【0143】
27.pETBlue/IgLκからIgLκ遺伝子断片を制限酵素SalIによって切り出し、pMSCV/GΔAのSalIサイトへ挿入した。ΔActプロモーターと同方向にIgLκ遺伝子断片が挿入された構造のプラスミドをpMSCV/GΔALとした。
【0144】
28.2つの化学合成オリゴヌクレオチド5’−acgcgtcgaccgcccctctccctccccc−3’(配列番号31;下線部はSalI制限酵素サイト)及び5’−ccgctcgagattatcatcgtgtttttcaaaggaaaaccacgtc−3’(配列番号32;下線部はXhoI制限酵素サイト)をプライマーとするPCR(94℃/15秒→60℃/30秒→68℃/1分:30サイクル)によりプラスミドpLXIN(クロンテック社製)からIRES断片を増幅後、制限酵素SalI及びXhoIによって切り出し、pETBlue−2のSalI、XhoIサイトへ挿入し、プラスミドpETBlue/IRESを作製した。
【0145】
29.pETBlue/IRESからIRES断片を制限酵素SalI及びXhoIによって切り出し、pMSCV/GΔAHのSalIサイトへ挿入した。IgHγ1遺伝子と同方向にIRESが挿入された構造のプラスミドをpMSCV/GΔAIHとした。
【0146】
30.pETBlue/IgLκからIgLκ遺伝子断片を制限酵素SalIによって切り出し、pMSCV/GΔAIHのSalIサイトへ挿入した。ΔActプロモーターと同方向にIgLκ遺伝子断片が挿入された構造のプラスミドをpMSCV/GΔALIHとした。
【0147】
このように作製した複製能欠失型レトロウイルスベクターのベクターコンストラクトpMSCV/GΔAH、pMSCV/GΔAL及びpMSCV/GΔALIHの構造を図12に示した。
【0148】
(実施例10)抗CD2抗体発現G0トランスジェニックキメラウズラの作製
実施例2に準じて、ベクターコンストラクトpMSCV/GΔAH、pMSCV/GΔAL及びpMSCV/GΔALIHより3種類のレトロウイルスベクターを調製した。このレトロウイルスベクターのタイターを測定したところ、108cfu/ml〜109cfu/mlだった。
【0149】
得られたレトロウイルスベクターを、実施例3に準じて孵卵後36時間のウズラ受精卵心臓にマイクロインジェクションし、37.9℃,湿度65%で15分毎に90度転卵させながら孵卵した。
【0150】
軽鎖(Light Chain)、重鎖(Heavy Chain)からなる抗体をトランスジェニック動物で発現させるには、軽鎖、重鎖それぞれを発現させるベクターを各々単独で導入する方法と、軽鎖を発現する遺伝子と重鎖を発現する遺伝子をIRESのような配列で区切り、同一のベクターとして導入する方法が考えられる。そこでインジェクションは、pMSCV/GΔAH、pMSCV/GΔALを同時に感染させた場合(実施例11、実験例1)と、pMSCV/GΔALIHより調製したベクターを単独で導入した場合(実施例11、実験例2)に分けて行った。
【0151】
孵卵48時間後、発生が正常に進行していることを確認し、ニワトリのSサイズの卵殻の鈍端部に直径4cmの穴を開けたものに、このウイルス導入胚を移した。胚を上にして空気に触れるようにし、濃度50mg/mlで卵白に懸濁した乳酸カルシウム(シグマ社製)溶液を0.5ml添加後、卵白を糊としてラップで密閉した。再度孵卵器に入れ、37.9℃、湿度65%で1時間毎に60度転卵しながら13日間培養した。転卵を止めて静置状態にし、胚が肺呼吸に移行したら、ラップに針で小さな穴を開け、呼吸を助けた。漿尿膜の血が引いたら培養器から雛を出し、孵化させた。
【0152】
(実施例11)血清中、卵中の抗CD2抗体濃度の測定
実施例10により孵化させたG0トランスジェニックキメラウズラを1ヶ月間飼育して雛を成長させた。30日後および60日後、成長したG0トランスジェニックウズラの翼下静脈より採血を行い、血液サンプルを得た。得られた血液を15,000rpmで10分間遠心し、上清として得られる血清から抗CD2抗体量の測定を行った。
孵化から1.5ヵ月後、産卵を始めた雌性トランスジェニックウズラより採卵を行い、実施例7に準じて調製した卵白、卵黄中の抗CD2抗体量を、実施例8に準じてELISA法により定量した。
実験例1、実験例2の定量結果を示す。
【0153】
(実験例1)
pMSCV/GΔAH、pMSCV/GΔALから調製したベクター(3〜4×108cfu/ml)を同時に感染させたG0トランスジェニックキメラウズラ(個体識別番号#1113)は、卵黄中に0.6μg/ml、卵白中に0.5μg/mlの抗CD2抗体を発現した。
【0154】
(実験例2)
pMSCV/GΔALIHより調製したベクター(5×108cfu/ml)を単独で導入したG0トランスジェニックキメラウズラ(#4202)は、血清中に5.2μg/mlの抗CD2抗体を発現した。
【0155】
(実施例12)scFv−Fc抗体発現ベクターコンストラクトの作製
ScFv−Fc抗体発現ベクターコンストラクトpMSCV/scFv−Fcは、以下のように作製した。
【0156】
1.5’末端をリン酸化した2つの化学合成オリゴヌクレオチド5’−ctagaccatgaggtctttgctaatcttggtgctttgcttcctgcccctggctgctctgggg−3’(配列番号33;ctagaはXbaI認識部位末端、ggはHaeIII認識部位末端)及び5’−ccccagagcagccaggggcaggaagcaaagcaccaagattagcaaagacctcatggt−3’(配列番号34;ccはXbaI認識部位末端、tはHaeIII認識部位末端)をアニールさせ、リゾチーム分泌シグナルの遺伝子断片を調製した。2つの化学合成オリゴヌクレオチド5’−gcgtttaaagtgacgttggacgtccg−3’(配列番号35;tttaaaはDraI制限酵素サイト)及び5’−attaggatccgcgcttaaggacggtcagg−3’(配列番号36;ggatccはBamHI制限酵素サイト)をプライマーとするPCR(94℃/15秒→58℃/30秒→68℃/1分:30サイクル;KOD−Plus−DNAポリメラーゼ)により、HUC2−13細胞のニワトリ抗体可変領域遺伝子から調製された一本鎖抗体(scFv)の遺伝子を含んだプラスミドpPDS/scFv(Nakamura et al.,2000,Cytotechnology32:191−198)からscFv遺伝子断片を増幅後、制限酵素DraI及びBamHIによって切り出した。pBluescriptIISK(+)のXbaI、BamHIサイトに上記調製した2断片を挿入し、プラスミドpBlue/scFvを作製した。
【0157】
2.pBlue/scFvからscFv遺伝子断片を制限酵素NotI及びBamHIによって切り出し、pCEP4のNotI、BamHIサイトへ挿入し、プラスミドpCEP4/scFvを作製した。
【0158】
3.ヒトIgG1産生ミエローマ細胞IM−9からmRNA isolation Kitを用いてmRNAを取得し、得られたmRNAからReverTra Aceを用いてcDNAライブラリを調製した。2つの化学合成オリゴヌクレオチド5’−caagcttcaagggcccat−3’(配列番号23)及び5’−atttacccggagacaggga−3’(配列番号24)をプライマーとするPCR(95℃/2分→52℃/30秒→74℃/3分:30サイクル;Pfu DNAポリメラーゼ)により上記cDNAライブラリからhCγ1遺伝子断片を増幅した。さらに、2つの化学合成オリゴヌクレオチド5’−attaggatccgagcccaaatcttgtgacaaaactc−3’(配列番号37;ggatccはBamHI制限酵素サイト)及び5’−agcaagctttcatttacccggagacaggga−3’(配列番号26;aagcttはHindIII制限酵素サイト)をプライマーとするPCR(94℃/15秒→58℃/30秒→68℃/1分:30サイクル;KOD−plus−DNAポリメラーゼ)により上記PCR産物からヒト抗体H鎖γ1のFc領域(Fc)の遺伝子断片を増幅後、制限酵素BamHI及びHindIIIによって切り出し、pBluescriptIISK(+)のBamHI、HindIIIサイトへ挿入し、プラスミドpBlue/Fcを作製した。
【0159】
4.pCEP4/scFvからscFv遺伝子断片を制限酵素HindIII及びBamHIによって切り出した。pBlue/FcからFc遺伝子断片を制限酵素BamHI及びHindIIIによって切り出した。pBluescriptIISK(+)のHindIIIサイトへ上記切り出した2断片を挿入し、プラスミドpBlue/scFv−Fcを作製した。
【0160】
5.pBlue/scFv−Fcからニワトリ一本鎖抗体可変領域にヒト抗体H鎖γ1・Fcが連結した構造(scFv−Fc)の遺伝子断片を制限酵素HindIIIによって切り出し、制限酵素HindIIIによって処理したpMSCV/GΔAHのベクター断片に連結した。ΔActプロモーターと同方向にscFv−Fc遺伝子が連結された構造のプラスミドをpMSCV/GΔAscFv−Fcとした。
【0161】
このように作製した複製能欠失型レトロウイルスベクターのベクターコンストラクトpMSCV/GΔAscFv−Fcの構造を図13に示した。
【0162】
(実施例13)scFv−Fc抗体発現G0トランスジェニックキメラウズラの作製
実施例2に準じて、ベクターコンストラクトpMSCV/GΔAscFv−Fcよりレトロウイルスベクターを調製した。このレトロウイルスベクターのタイターを測定したところ、108cfu/ml〜109cfu/mlだった。
【0163】
得られたウイルスベクター溶液を、実施例3に準じて孵卵後36時間のウズラ受精卵心臓にマイクロインジェクションし、実施例10に準じて孵化させることによりG0トランスジェスジェニックキメラウズラを誕生させた。
【0164】
(実施例14)血清中、卵中のscFv−Fc濃度の測定
実施例13により誕生したG0トランスジェニックキメラウズラを1ヶ月間飼育して雛を成長させた。30日後および60日後、成長したG0トランスジェニックキメラウズラ(個体識別番号#3303、#3306、#3310、#3311、#3313)の翼下静脈より採血を行い、血液サンプルを得た。得られた血液を15,000rpmで10分間遠心し、上清として得られる血清からscFv−Fc抗体量の測定を行った。
孵化から1.5ヵ月後、産卵を始めた雌性トランスジェニックウズラ(個体識別番号#3310)より採卵を行い、実施例7に準じて調製した卵白、卵黄中のscFv−Fc抗体量を、実施例8に準じてELISA法により定量した。
【0165】
標準検量線は精製したscFv−Fcを用いて作製した。実施例12で作製したベクターコンストラクトpMSCV/scFv−Fcをリポフェクション法によりGP293細胞に導入し、その培養上清を4℃、10分間、3000rpmで遠心して固形物を除去した。この上清を冷却しながら攪拌し、50%飽和となるよう細かく砕いた硫酸アンモニウムを徐々に加え(313g硫安/1000ml水)、タンパク質を沈殿させた。これを4℃で一晩静置した後、4℃で10分間、15,000rpmで遠心して沈殿を完全に沈降させ、少量のPBSで溶解した。2LのPBSで3回透析して硫安を除去した。
【0166】
精製用のプロテインGカラム(パーゼプティブバイオシステムズ社製)の初期洗浄をBinding Buffer(NaHPO4・2H2O 1.56g/l、NaHPO4・12H2O 7.16g/l)10mL、Wash Buffer(酢酸20%、蒸留水80%)10ml、Binding Buffer10mlの順で行った(流速2ml/分)。PBSに溶解したタンパク質液を1ml/分で流し、scFv−Fcをカラムに吸着させた。Binding Buffer20mlを1.7ml/分で流して、不要なタンパク質を除去し、Elution Buffer(グリシン7.507g/l、2N HClにてpH2.5〜3.0に調製)を1.5ml/分で流して、scFv−Fcを溶出した。
【0167】
溶出分画をPBS(2L)で3回透析し、精製scFv−Fcとし、波長280nmの吸光度よりタンパク質濃度を定量した。
【0168】
実施例14で作製したG0トランスジェニックキメラウズラの30日後、60日後採血血清中のscFv−Fc量を図14に示した。scFv−Fc抗体の発現遺伝子を導入されたG0トランスジェニックキメラウズラは、30日目に約2mg/ml〜4mg/mlの抗体を血清中に発現し、5羽のうち3羽が60日後も同程度の発現量を示した。
【0169】
G0トランスジェニックキメラウズラ(#3310)が産卵を始めた日から、卵黄、卵白中のscFv−Fc量を図15に示した。抗体は卵白、卵黄中に約500μg/ml〜1mg/ml発現し、産卵開始から17日目まで若干の変動はあるものの安定した発現量を維持した。
【0170】
(実施例15)scFv−Fc構造の確認
実施例13で作製したG0トランスジェニックキメラウズラ血清1mlから実施例10に準じて、硫安沈殿とプロテインGカラムによりscFv−Fcを精製した。精製したscFv−FcをSDS−PAGEで解析し、その結果を図16に示した。未処理のレーンより、scFv−Fcの分子量約120kDaが示された。還元処理したscFv−Fcの分子量は未処理の場合のほぼ半分(約60kDa)になっていることから、G0トランスジェニックキメラウズラにより産生されたscFv−FcはS−S結合による2量体を形成していることがわかった。これらは、S−S結合に関与するシステイン残基をFc部に有するscFv−Fcの構造的特徴に符合し、G0トランスジェニックキメラウズラにより産生されたscFv−Fcは正しい構造を保持していることが示唆された。
【0171】
(実施例16)TNFR−Fc融合タンパク発現G0トランスジェニックキメラニワトリの作製
実施例9に準じてTNFR−Fc発現ベクターコンストラクトを作製し、実施例2に準じてレトロウイルスベクターを作製した。このレトロウイルスベクターのタイターは、1.7×107cfu/mlだった。
【0172】
得られたウイルスベクター溶液を、実施例4に準じて孵卵後55時間のニワトリ受精卵心臓にマイクロインジェクションし、実施例10に準じて孵化させることによりG0トランスジェニックキメラニワトリを誕生させた。
【0173】
8個の受精卵にインジェクションしたところ4羽が孵化し、実施例14に準じてその血清中のTNFR−Fcを定量したところ、最高で50μg/mlのTNFR−Fcが発現していた。
【産業上の利用可能性】
【0174】
本発明のG0トランスジェニックキメラ鳥類は、複製能欠失型レトロウイルスベクターで導入された遺伝子を不活性化することなく効率的に発現することができる。また、本発明のG0トランスジェニックキメラ鳥類の作製法は、キメラ抗体、例えばscFv−Fc抗体の遺伝子を導入し、血中、卵中に抗体を効率的に発現することができる鳥類の作製を可能にする。更に、本発明の抗体生産法は、キメラ抗体、例えばscFv−Fc抗体を産生するG0トランスジェニックキメラ鳥類を作製し、抗体を鳥類の血清、卵中から回収、精製することからなるので、効率よい抗体生産を可能にする。
【技術分野】
【0001】
本発明は、血液中ならびに卵中に、抗体、例えばscFv−Fc抗体を産生するG0トランスジェニックキメラ鳥類に関する。また本発明は、複製能欠失型レトロウイルスベクターによって外来性抗体遺伝子が導入されたG0トランスジェニックキメラ鳥類を作製し、血中、卵白中あるいは卵黄中に産生した抗体を回収することからなる抗体産生法に関する。更に本発明は、効率的に導入遺伝子を発現するG0トランスジェニックキメラ鳥類の作製法ならびに該作製法より得られるG0トランスジェニックキメラ鳥類に関する。
【背景技術】
【0002】
遺伝子機能の研究手段として、外来遺伝子を宿主に組み込んだ遺伝子導入(トランスジェニック)動物の研究が盛んに行われているが、このトランスジェニック動物は、基礎研究だけでなく、品種改良や物質生産、移植用臓器のドナー等、産業応用にも有効である。ウシ、ヤギ、ヒツジ等の乳汁に生理活性物質を産生させる試みが実用に近づいており、その代表例としてα1−アンチトリプシンやアンチスロンビンは、医薬品への応用を目的として現在臨床段階にある。
【0003】
ウズラやニワトリに代表される家禽鳥類は、食肉用、採卵用家畜としての飼育実績が長く、トランスジェニック研究の目的としても耐病性や肉質向上等、品種改良に係るさまざまなアプローチが考えられる。また、鳥類は性成熟までの期間が短く、小さなスペースで飼育可能なことから、安価なコストで実施可能なタンパク質発現系と考えられ、抗体医薬品や希少タンパク質の産生手段としてのトランスジェニック作製に期待が持たれている。鳥類の卵は多量のタンパク質を含有し、毎日連続的に生産されることから、導入遺伝子産物を組換えタンパク質として意図的に卵中に産生させることができれば、効率的な生産システムとなると考えられる。
【0004】
近年数多くの品目が上市され、医薬品として注目されているモノクローナル抗体は、大腸菌などによる安価な発現システムでは生産できず、単価が高いことが普及の妨げとなっている。鳥類細胞は抗体タンパク質を構成する機能を備えており、トランスジェニック鳥類は、従来大量生産が困難であった医療用抗体等の生産手段として期待できる。更にこれらのタンパク質生産物には、鳥類細胞によって糖鎖が付与されることにより血中での安定性が向上する等、医薬、検査薬としての応用上有利な性質を備える可能性が高いと考えられる。
【0005】
このように、有用タンパク質の生産手段としての応用が待たれるトランスジェニック鳥類であるが、一方で現在までに様々な試みがあるにも係らず、鳥類の卵に実用レベルで目的とする組換えタンパク質を蓄積させた例はない。また、抗体のような複数のユニットからなる高次構造をもったタンパク質を、高濃度に発現するトランスジェニック鳥類を作製した例も、未だ報告されていない。
【0006】
Harveyら(非特許文献1)はエビアン・ロイコシス・ウイルス由来のベクターを用い、β−ラクタマーゼ遺伝子をニワトリに導入し、G0トランスジェニックキメラニワトリを作製したが、血清あるいは卵への酵素発現量は50〜250ng/ml程度であった。このG0トランスジェニックキメラニワトリの交配により作製されたG1〜G3トランスジェニックニワトリは、全身の体細胞に遺伝子が導入されることにより、酵素の発現量が増加したが、それでも数μg/mlのレベルに留まり、実用には遠い値であった。
【0007】
一般的に遺伝子導入動物を作製するには、受精卵の前核へDNAをマイクロインジェクションする手法がとられるが、鳥類にはこの方法は応用できない。鳥類では1細胞期の胚を取得するのが困難なこと、また取得できたとしても、卵内の核を見分ける手法がないことがその理由である。1細胞期の胚を取得するには、雌性鳥類の輸卵管内から受精直後の卵を取得し、正常に発生させなければならない。近年Perryによって鶏の卵割前細胞を取得し、体外で培養するシステムが確立された(非特許文献2)が、この手法によっても卵内の核を見分け、核内へ目的遺伝子を導入することは不可能である。
【0008】
従って、鳥類受精卵への遺伝子導入は、細胞質へのDNA注入に限られ、DNA封入脂質二重膜(リポソーム)の利用やリン酸カルシウム法、電気導入法(エレクトロポレーション)の利用が試みられてきた。しかし、これらの手法では遺伝子の導入効率が悪く、また導入されたプラスミドDNAは、染色体に取り込まれる確率が低い。マイクロインジェクションによる遺伝子導入法は、効率的に目的DNAを受精卵に送り込むことができるが、導入されたプラスミドDNAは宿主染色体に組み込まれないため、宿主の体細胞分裂に伴って導入遺伝子プラスミドは脱落してしまい、安定な遺伝子導入効果は望めない。
【0009】
1986年に初めてレトロウイルスベクターによるトランスジェニックニワトリの作製例が報告されている(非特許文献3)。レトロウイルスベクターをマイクロインジェクション法で受精卵に注入する手法は、遺伝子の導入効率の高い手法であり、核内へ直接DNAを注入できない鳥類において、染色体へ目的遺伝子が挿入された安定なトランスジェニックを作製する唯一の実用的方法である。
【0010】
本発明者らは鋭意研究を重ね、遺伝子治療にも応用されている安全な自己複製能欠失型ウイルスベクターを使用し、効率的に目的遺伝子を導入するトランスジェニック鳥類の作製法を見出した(特許文献1)。これにより、安全かつ効率的に複数の導入遺伝子コピー数をもつトランスジェニック鳥類の作製が可能となった。またこの手法によれば、導入遺伝子は高い効率で次世代に伝達されることもわかり、物質生産システムとしての遺伝子導入鳥類の利用が実用に近づいた。
【先行技術文献】
【特許文献】
【0011】
【特許文献1】特開2002−176880号公報
【非特許文献】
【0012】
【非特許文献1】Harvey,A.Jら(2002)NatureBiotechnology.19,396
【非特許文献2】Perry、M.M(1988)Nature,331
【非特許文献3】Salter,D.Wら(1986)Poultry Sci.,65
【発明の概要】
【発明が解決しようとする課題】
【0013】
遺伝子治療にも応用されている安全な自己複製能欠失型ウイルスベクターを使用すれば、効率的に目的遺伝子を導入してトランスジェニック鳥類を作製することが可能であるが、このとき導入された遺伝子の大部分は、発生初期に宿主によって不活性化されるため(遺伝子のサイレンシングと呼ばれる)、遺伝子発現の結果としてのタンパク質産生はごく微量であった。導入遺伝子が不活性化される生物学的メカニズムは未だ解明されていないが、この不活性化を回避し、目的遺伝子を効率的に発現させる技術が、トランスジェニック鳥類の応用開発に必須である。
【課題を解決するための手段】
【0014】
発明の要約
レトロウイルスベクターによって受精卵に導入された遺伝子が、発生後どの時期に不活性化されるのかを特定するため、本発明者らはβ−ガラクトシダーゼ発現遺伝子を指標とし、受精後の様々な時期にベクターを導入して発現量がどう変化するかを検討した。その結果、導入遺伝子の発現量は該遺伝子が胚発生のどの時期に導入されたかによって大きく変化してくることを発見し、本発明に至った。すなわち、導入遺伝子の不活性化は、放卵直後に導入された遺伝子に対して顕著であり、放卵から一定時間経過後に導入された遺伝子は、発現頻度が高いというものである。
【0015】
本発明者らはこの知見を利用し、鳥の種類によって決まる特定の時間が孵卵開始時より経過した後に、その初期胚へ外来遺伝子を導入すれば、宿主による不活性化の影響を受けず、目的遺伝子を効率的に発現させることが可能となることを見出した。
【0016】
さらに、医薬品として有用なキメラ抗体、例えばscFv−Fc(1本鎖抗体)をコードした遺伝子を組み込んだベクターを作り、この方法によりウズラに導入して、G0トランスジェニックキメラ鳥類を作製したところ、血中、卵白、卵黄中に導入遺伝子に由来する抗体が高濃度に発現することが見出された。
【0017】
すなわち第一の本発明は、鳥類受精卵を孵卵し、放卵直後の胚盤葉期(ステージX)を除くそれ以降の初期胚へ複製能欠失型レトロウイルスベクターを感染させ、その胚を孵化させることからなるG0トランスジェニックキメラ鳥類の作製法、及び該方法により作製されたG0トランスジェニックキメラ鳥類に関し、第二の本発明は、複製能欠失型レトロウイルスベクターによって外来性抗体遺伝子が導入されたG0トランスジェニックキメラ鳥類であって、導入遺伝子に由来する抗体を血中、卵白中及び卵黄中の少なくとも1つに産生することを特徴とするG0トランスジェニックキメラ鳥類に関し、第三の本発明は、上記G0トランスジェニックキメラ鳥類を作製し、該G0トランスジェニックキメラ鳥類の血中及び/又は卵中から抗体を回収することよりなる抗体の生産法に関する。
【0018】
本発明のG0トランスジェニックキメラ鳥類による導入遺伝子の発現は、誕生したG0トランスジェニックキメラ鳥類が成烏になっても維持されることが確認された。したがって、本発明により特定の遺伝子を鳥類に導入し、目的タンパク質を成長後も産生する、実用的な生産システムを構築することができる。また、本発明によって作製されたG0トランスジェニックキメラ鳥類の導入遺伝子は、交配により高い伝播効率で次世代に受け継がれる。導入遺伝子は、染色体に組み込まれたかたちで次世代に受け継がれるため、このトランスジェニック鳥類による生産システムは、安定的な物質生産が可能である。
【0019】
本発明により、トランスジェニック鳥類がその体細胞中に産生させた目的タンパク質は、血液中に分泌され、血清から分離することで利用可能となる。
【0020】
更に本発明者らは、ヒトIgGクラスの定常領域をもつ抗体や、ウズラIgG、ニワトリIgG又はマウスIgGの定常領域をもつ抗体が、ウズラ、ニワトリにおいて血中から卵中に効率的に移行することを見出した。これらの定常領域をもつ抗体を本発明の方法により作製されたG0トランスジェニックキメラ鳥類に発現させた場合、血中に分泌された抗体が卵中に高濃度に蓄積される。
【0021】
トランスジェニック動物に遺伝子を導入して目的タンパク質を取得する場合、哺乳類では乳汁中に分泌させて回収するのが一般的だが、鳥類のトランスジェニックでは卵に目的物を蓄積させ、その卵白、卵黄から回収、精製するのが実際的である。
【0022】
鶏卵などでは全成分の30%がタンパク質であり、その主成分となるタンパク質はオボアルブミンである。従来トランスジェニック鳥類の卵中に組換えタンパク質を発現させる方法として、このオボアルブミン等の発現プロモーターを利用し、その下流に目的タンパク質をコードする遺伝子を組み込んで、卵白中にオボアルブミンに代えて目的物を発現させるという考え方がとられていた。
【0023】
しかし、本発明の方法において、構成的なプロモーター、例えばニワトリβ−アクチンプロモーター等の制御のもとに、大量に発現した抗体、例えば前記IgGクラス抗体等を血中に分泌させ、卵中に蓄積させることが可能となる。なお本明細書において、構成的なプロモーターとは、全身性に発現するプロモーターのことを意味する。
【0024】
更に抗体の構造のなかで、卵中への移行に必須な部分を特定するため、抗体をFab、Fcフラグメントとしたものを、ウズラ、ニワトリ血中に接種して卵への移行性を調べた。その結果、卵内にはFcフラグメントが蓄積され、抗体の卵内への移行はFcレセプターを介して行われることが示唆された。
【0025】
このことからトランスジェニック鳥類による抗体の卵内での生産法を、タンパク質全般の生産に汎用化する方法として、ヒトIgGの定常領域(Fc)が融合した構造をもつタンパク質を生産するベクターを設計し、トランスジェニック鳥類を作製して卵から目的物を含むタンパク質を回収し、Fc部を切断して目的物を精製する生産法が考えられる。
【0026】
また、従来哺乳類のトランスジェニック動物を使って抗体を生産した場合、産生動物がもつ自己抗体と目的抗体を分離精製するのが困難との問題が指摘されていた。産生動物に鳥類を使う抗体生産法の利点として、鳥類の自己抗体がプロテインA、プロテインGカラムに吸着されないため、目的とする組換え抗体との分離が容易な点も挙げられる。
【0027】
前述のように、ヒトモノクローナル抗体は、医薬品として有用であるにも係らず、その生産には動物細胞培養やマウス腹水といった高価な生産手段しかなく、単価が高いことが普及に際して妨げとなっている。
【0028】
近年遺伝子工学的手法を用いて、抗体のH鎖、L鎖のV領域のみをリンカー配列でつなげたscFv(1本鎖抗体)が作製されているが、これは大腸菌でも生産されるという利点があり、コストの点から注目されている。しかし、低分子抗体と呼ばれるこれらのタンパク質は、血中での安定性が低いため、治療用、試験用としての実用性に難があった。
【0029】
scFvにFc領域をつなげたscFv−Fcは、血中でも安定であり、より実用的と考えられるが、大腸菌では生産できず、動物細胞を使ったバイオリアクターでしか供給できない。ニワトリなど他の動物に作らせた結合領域をもつscFvにヒトのFcを結合させたヒト化scFv−Fcは治療用としても有望であり、このタンパク質を本発明によるG0トランスジェニックキメラ鳥類により大量に生産できれば、その有用性は大きい。
【0030】
本発明のG0トランスジェニックキメラ鳥類は、scFv−Fcのような組換え抗体、キメラ抗体、ヒトモノクローナル抗体など、従来の方法では少量しか生産できなかった抗体タンパク質を安価に大量に生産し、実用的に回収、精製して利用することに応用できる。
【0031】
このように本発明ではレトロウイルスベクターによるG0トランスジェニックキメラ鳥類において、導入遺伝子を効率的に発現させる作製法を開示する。また本発明では、特定遺伝子を導入することにより、鳥類体細胞に有用な目的タンパクを産生させるG0トランスジェニックキメラ鳥類の作製法を開示する。更に従来大腸菌等で産生することが不可能だったモノクローナルヒト型抗体や、キメラ抗体、scFv−Fc抗体、複雑な高次構造をもった機能性タンパク等、医薬、検査薬として有用な物質を鳥類細胞に産生させ、血清や卵中から回収して利用する、生産コストの低いタンパク生産システムを開示する。
【0032】
また本発明のG0トランスジェニックキメラ鳥類の作製法を応用して、家禽鳥類を好ましい方向へ改質する手法および改質された鳥類を提供することも考えられる。家禽鳥類の好ましい性質としては、肉質改善、耐病性の向上、生育速度の向上等が挙げられる。鳥類はまた愛玩用途としても多種の需要をもっているが、本発明の作製法は、羽毛色の改善、攻撃性の低下等、愛玩用として好ましい形質へ短期間で品種改良するための手段としても応用しうる。
【0033】
更に、現在哺乳類、両生類、魚類等のトランスジェニック作製は、核移植によって行われるのが通常であるが、本発明はそれに代わる新たなトランスジェニック動物作製の効率的手法としても応用できる。
【0034】
即ち、本発明は、複製能欠失型レトロウイルスベクターによって外来性抗体遺伝子が導入されたG0トランスジェニックキメラ鳥類であって、導入遺伝子に由来する抗体を、血中、卵白中及び卵黄中の少なくとも1つに産生するG0トランスジェニックキメラ鳥類である。上記抗体の定常領域は、クラスがヒトIgGであるか、サブクラスがヒトIgG1であるか、ウズラIgG、ニワトリIgG、あるいはマウスIgGであることが好ましい。上記抗体遺伝子は、構成的なプロモーターにより制御されていることが好ましく、上記構成的なプロモーターは、ニワトリβ−アクチンプロモーターであることが好ましい。上記レトロウイルスベクターは、モロニー・ミューリン・ロイケミア・ウイルス由来ベクターであることが好ましく、VSV−Gシュードタイプであることが好ましい。本発明のG0トランスジェニックキメラ鳥類は、ニワトリあるいはウズラであることが好ましい。上記抗体は、キメラ抗体であることが好ましく、上記キメラ抗体の産生量は、血液中には、0.5μg/ml以上であることが好ましく、より好ましくは5μg/ml以上であり、卵白中には、0.1μg/ml以上であることが好ましく、より好ましくは1μg/ml以上であり、卵黄中には、0.1μg/ml以上であることが好ましく、より好ましくは1μg/ml以上である。また、上記抗体は、scFv−Fc抗体であることが好ましく、上記scFv−Fc抗体の産生量は、血液中には、20μg/ml以上であることが好ましく、より好ましくは2000μg/ml以上であり、卵白中には、5μg/ml以上であることが好ましく、より好ましくは500μg/ml以上であり、卵黄中には、5μg/ml以上であることが好ましく、より好ましくは500μg/ml以上である。本発明のG0トランスジェニックキメラ鳥類を作製し、上記G0トランスジェニックキメラ鳥類の血中及び/又は卵中から抗体を回収する抗体の生産法もまた、本発明の1つである。
【0035】
鳥類受精卵を孵卵し、放卵直後の胚盤葉期を除くそれ以降の初期胚へ複製能欠失型レトロウイルスベクターを感染させ、その胚を孵化させることからなるG0トランスジェニックキメラ鳥類の作製法もまた、本発明の1つである。本発明のG0トランスジェニックキメラ鳥類の作製法において、複製能欠失型レトロウイルスベクターを感染させる時期は、孵卵開始から24時間以降であることが好ましい。上記複製能欠失型レトロウイルスベクターを感染させる方法は、初期胚に形成される心臓ないしは血管内へマイクロインジェクションすることであるのが好ましい。上記心臓ないしは血管は、孵卵開始から24時間以降の初期胚に形成されるものであることが好ましい。マイクロインジェクションする複製能欠失型レトロウイルスベクターの活性は、1×107cfu/ml以上のタイターであることが好ましく、1×108cfu/ml以上のタイターであることがより好ましく、1×109cfu/ml以上のタイターであることが更に好ましい。上記レトロウイルスベクターは、モロニー・ミューリン・ロイケミア・ウイルス由来ベクターであることが好ましく、VSV−Gシュードタイプであることが好ましい。本発明で作製するG0トランスジェニックキメラ鳥類は、ニワトリまたはウズラであることが好ましい。上記複製能欠失型レトロウイルスベクターに組み込まれる導入遺伝子は、レトロウイルスに由来しない遺伝子配列を含有することが好ましい。上記レトロウイルスに由来しない遺伝子配列は、ニワトリβ−アクチンプロモーターにより制御されている遺伝子配列であることが好ましく、抗体遺伝子又は融合タンパク遺伝子をコードする遺伝子配列であることが好ましい。上記抗体遺伝子は、キメラ抗体遺伝子であることが好ましく、scFv−Fc抗体遺伝子であることが好ましい。
【0036】
本発明のG0トランスジェニックキメラ鳥類の作製法で作製されたG0トランスジェニックキメラ鳥類もまた、本発明の1つである。
【発明の効果】
【0037】
本発明の方法によれば、鳥の種類によって決まる特定の時間が孵卵開始時より経過した後に、その初期胚へ外来遺伝子を導入するので、宿主による不活性化の影響を受けず、目的遺伝子を効率的に発現させることが可能となる。本発明の方法でG0トランスジェニックキメラ鳥類を作製したところ、血中、卵白、卵黄中に導入遺伝子に由来するタンパク質が高濃度に発現する。
【図面の簡単な説明】
【0038】
【図1】図1は、複製能欠失型レトロウイルスベクターのベクターコンストラクトpMSCVNΔAβの構造を示す。Neorはネオマイシン耐性遺伝子を、Amprはアンピシリン耐性遺伝子を示す。PΔActはβ−アクチンプロモーター遺伝子を示す。β−Galはβ−ガラクトシダーゼ発現遺伝子を示す。Ψ+はパッケージングシグナル配列を示す。5’LTR及び3’LTRはそれぞれMoMLVのロングターミナルリピート配列を示す。
【図2】図2は、G0トランスジェニックキメラウズラにおける、遺伝子の導入時期とβ−ガラクトシダーゼ活性発現との関係を示す。横軸は孵卵時間(hr)を、縦軸はmUnit/mgで表されるβ−ガラクトシダーゼ活性を示す。
【図3】図3は、G0トランスジェニックキメラニワトリにおける、遺伝子の導入時期とβ−ガラクトシダーゼ活性発現との関係を示す。横軸は孵卵時間(hr)を、縦軸はmUnit/mgで表されるβ−ガラクトシダーゼ活性を示す。
【図4】図4は、導入したレトロウイルスベクターのタイターとG0トランスジェニックキメラウズラでのβ−ガラクトシダーゼ活性発現との関係を示す。横軸はcfu/mlで表されるウイルスタイターを、縦軸はmUnit/mgで表されるβ−ガラクトシダーゼ活性を示す。
【図5】図5は、導入したレトロウイルスベクターのタイターとG0トランスジェニックキメラニワトリでのβ−ガラクトシダーゼ活性発現との関係を示す。横軸はcfu/mlで表されるウイルスタイターを、縦軸はmUnit/mgで表されるβ−ガラクトシダーゼ活性を示す。
【図6】図6は、ウズラ卵中に蓄積したヒトIgG抗体を示す。抗体値は3羽のウズラで行った同一の実験結果の平均値を示している。
【図7】図7は、ニワトリ卵中に蓄積したヒトIgG抗体を示す。抗体値は3羽のニワトリで行った同一の実験結果の平均値を示している。
【図8】図8は、ウズラ卵中に蓄積したFabフラグメントを示す。抗体値は3羽のウズラで行った同一の実験結果の平均値を示している。
【図9】図9は、ニワトリ卵中に蓄積したFabフラグメントを示す。抗体値は3羽のニワトリで行った同一の実験結果の平均値を示している。
【図10】図10は、ウズラ卵中に蓄積したFcフラグメントを示す。抗体値は3羽のウズラで行った同一の実験結果の平均値を示している。
【図11】図11は、ニワトリ卵中に蓄積したFcフラグメントを示す。抗体値は3羽のニワトリで行った同一の実験結果の平均値を示している。
【図12】図12は、抗CD2抗体発現ベクターコンストラクトpMSCV/GΔAL(12(A))、pMSCV/GΔAH(12(B))及びpMSCV/GΔALIH(12(C))の構造を示す。Amprはアンピシリン耐性遺伝子を示す。PΔActはβ−アクチンプロモーター遺伝子を示す。Ψ+はパッケージングシグナル配列を示す。GFPはグリーン・フルオレッセント・プロテイン遺伝子を示す。Lは抗CD2抗体軽鎖遺伝子を示す。Hは抗CD2抗体重鎖遺伝子を示す。5’LTR及び3’LTRはそれぞれMoMLVのロングターミナルリピート配列を示す。
【図13】図13は、scFv−Fc抗体発現ベクターコンストラクトpMSCV/GΔAscFv−Fcの構造を示す。Amprはアンピシリン耐性遺伝子を示す。PΔActはβ−アクチンプロモーター遺伝子を示す。Ψ+はパッケージングシグナル配列を示す。GFPはグリーン・フルオレッセント・プロテイン遺伝子を示す。scFv−FcはscFv−Fc抗体遺伝子を示す。5’LTR及び3’LTRはそれぞれMoMLVのロングターミナルリピート配列を示す。
【図14】図14は、G0トランスジェニックキメラウズラ血清中に発現したscFv−Fc量を示す。横軸は個体番号、縦軸はscFv−Fc抗体濃度(μg/ml)を示す。
【図15】図15は、G0トランスジェニックウズラ卵中に発現したscFv−Fc量をしめす。横軸は産卵開始からの採卵日、縦軸はscFv−Fc抗体濃度(μg/ml)を示す。
【図16】図16は、精製したscFv−FcのSDS−PAGEによる解析結果を示す。レーン1は低分子量マーカー(LMW)を、レーン4は高分子量マーカー(HMW)を示す。レーン2は還元処理したscFv−Fcを、レーン3は還元処理していないscFv−Fcを電気泳動した結果を示す。
【発明を実施するための形態】
【0039】
以下に本発明を詳述する。
本発明のG0トランスジェニックキメラ鳥類は、複製能欠失型レトロウイルスベクターによって外来性抗体遺伝子が導入された鳥類であって、導入遺伝子に由来する抗体を、血中、卵白中あるいは卵黄中に産生することを特徴とする。
【0040】
本発明で使用する鳥類としては、特に限定されず、例えばニワトリ、七面鳥、カモ、ダチョウ、ウズラなど、食肉、採卵目的で家畜化されている家禽鳥類や愛玩用鳥類を挙げることができる。なかでもニワトリやウズラは入手が容易で、産卵種としても多産である点が好ましい。
【0041】
本発明で使用されるレトロウイルスベクターとしては、モロニー・ミューリン・ロイケミア・ウイルス(MoMLV)、エビアン・ロイコシス・ウイルス(ALV)等に由来するベクターが挙げられる。なかでもMoMLVに由来するものが好ましいが、本発明はこれに限定されるものではない。
【0042】
安全性を考慮し、遺伝子導入ベクターとして用いられるウイルスは、通常ウイルス粒子の複製に必要な3種の遺伝子gag、pol、envのうちのいずれか又は全てを欠くことにより、自己複製能を欠失したウイルスが用いられる。鳥類細胞にこのウイルスベクターを効率的に感染させるため、外皮タンパク質を人工的にVSV−G(水疱性口内炎ウイルス由来)シュードタイプとしたウイルスベクターが好ましいが、本発明はこのウイルスタイプに限定されるものではない。
【0043】
パッケージング細胞またはヘルパーウイルス等を利用することにより調製されたシュードタイプのウイルスベクターは、通常のマイクロインジェクション法(Bosselman,R.Aら(1989)Science 243、533)により、初期胚、血管内、心臓内へ導入される。遺伝子導入法としては、この他にもリポフェクションやエレクトロポレーション法等が考えられる。
【0044】
本発明により鳥類に導入される遺伝子は特に限定されないが、マーカー遺伝子や目的タンパク質を発現するための構造遺伝子、これらの遺伝子発現をコントロールするプロモーター遺伝子、分泌シグナル遺伝子等により構成される。
【0045】
上記マーカー遺伝子としては、ネオマイシン耐性遺伝子、β−ガラクトシターゼ遺伝子、LacZ遺伝子、蛍光タンパク質、例えばGFP(グリーン・フロオロレッセント・プロテイン)等をコードした遺伝子が挙げられる。
【0046】
上記目的タンパク質を発現するための構造遺伝子としては特に制限されず、ヒトモノクローナル抗体など遺伝子産業上有用な抗体、酵素等をコードした遺伝子などが挙げられる。また、その他の有用生理活性物質の遺伝子を用いることもできる。特に、卵中での蓄積がよいことから、ヒトIgGクラスの定常領域をもつ抗体の遺伝子、ヒトIgG1のサブクラスの定常領域をもつ抗体の遺伝子、ウズラIgG、ニワトリIgGやマウスIgGの定常領域をもつ抗体の遺伝子など外来性抗体の構造遺伝子が好ましい。
【0047】
また好ましい上記構造遺伝子としては、キメラ抗体の構造遺伝子が挙げられる。
キメラ抗体とは、2種以上の異なる遺伝形質から構成される抗体のことをいう。
【0048】
従来、マウスハイブリドーマによって作製された医療用抗体はマウス由来であるため、ヒト体内に投与されると免疫系による拒絶反応が引き起こされるという問題があった。上記キメラ抗体としては、例えば、抗ヒトCD2抗体、抗CD20受容体抗体、抗TNF抗体など、当該欠点を改良し、マウス抗体のうち抗原タンパク質と結合する領域以外をヒト抗体で置き換え、拒絶反応をなくしたキメラ抗体が挙げられ、すでに医薬品として上市されているものもある。
【0049】
さらに好ましい上記構造遺伝子としては、scFv−Fc抗体の構造遺伝子が挙げられる。
医療用の組換え抗体には、低分子抗体と称される一群がある。免疫グロブリンIgGには直接抗原と結合する可変領域(Fv:Fragment of variable regeon)と呼ばれるVH、VLのヘテロ二量体からなるドメインがあり、このFvドメインはIgGの約5分の1の分子量でありながら、単独で充分な抗原結合能を持つ。VH、VLドメイン間を人工的にペプチドリンカーで結合したものが1本鎖抗体(scFv:single chain Fv)と呼ばれる低分子抗体で、VH、VL単独よりも安定性が向上することが知られていた。
【0050】
Powersら(Powers,D.Bら(2000)J Immunol Method.251,123)は、このscFvにヒトIgG1に由来するFc部を融合させることで、血中での安定性が増すことを見出した。このscFv−Fc抗体は医療用として有用と考えられるが、安価な大量生産システムである大腸菌では生産されない。
【0051】
他の好ましい上記遺伝子配列として、融合タンパクの構造遺伝子が挙げられる。
遺伝子組み換えにより2種あるいはそれ以上のタンパクの一部を融合させた人工的な一群のタンパクは融合タンパクと称され、医薬品として実用化されているものに、TNF受容体に免疫グロブリンのFcを融合させたTNFR−FcやLFA3にFcを融合させたLFA3−Fcなどがある。これらはFcを融合させることで可溶化され、より強力な生理活性をもつように設計された人工タンパクである。
【0052】
本発明のG0トランスジェニックキメラ鳥類において、鳥類に導入される遺伝子として、これらヒトモノクローナル抗体遺伝子、キメラ抗体遺伝子、scFv−Fc抗体遺伝子を使用することにより、従来生産が困難だった抗体医薬品を安価に大量生産できる。
【0053】
例えば、キメラ抗体遺伝子を導入したG0トランスジェニックキメラ鳥類の場合、血液中の抗体含有量は、好ましくは0.5μg/ml以上、より好ましくは5μg/ml以上である。また、卵白中の抗体含有量は、好ましくは0.1μg/ml以上、より好ましくは1μg/ml以上であり、卵黄中の抗体含有量は、好ましくは0.1μg/ml以上、より好ましくは1μg/ml以上である。
【0054】
また、scFv−Fc抗体遺伝子を導入したG0トランスジェニックキメラ鳥類の場合、血液中の抗体含有量は、好ましくは20μg/ml以上、より好ましくは2000μg/ml以上である。また、卵白中の抗体含有量は、好ましくは5μg/ml以上、より好ましくは500μg/ml以上である。卵黄中の抗体含有量は、好ましくは5μg/ml以上、より好ましくは500μg/ml以上である。
【0055】
上記プロモーター遺伝子としては、構成的なプロモーターが挙げられる。抗体遺伝子が構成的なプロモーターにより制御されている場合、抗体遺伝子発現が安定してよいので好ましい。より好ましい構成的なプロモーターとして、ニワトリβ−アクチンプロモーターが挙げられる。
【0056】
本発明の抗体の生産法は、本発明のG0トランスジェニックキメラ鳥類を作成し、上記G0トランスジェニックキメラ鳥類の血中及び/又は卵中から抗体を回収することを特徴とする。
【0057】
次に本発明のG0トランスジェニックキメラ鳥類の作製法について説明する。
当該作製法の1つとして、鳥類受精卵を孵卵し、放卵直後の胚盤葉期を除くそれ以降の初期胚へ複製能欠失型レトロウイルスベクターを感染させ、その胚を孵化させる作製法が挙げられる。また、鳥類受精卵を孵卵し、孵卵開始から24時間以降の初期胚へ複製能欠失型レトロウイルスベクターを感染させ、その胚を孵化させる作製法も、本発明の作製法の1つとして挙げられる。
【0058】
より好ましくは、上記初期胚に形成される心臓ないしは血管内へ、複製能欠失型レトロウイルスベクターをマイクロインジェクションする方法である。
【0059】
つまり、本発明のG0トランスジェニックキメラ鳥類の作製法は、放卵後特定の時間経過した受精卵に複製能欠失型レトロウイルスベクターをマイクロインジェクションすることよりなる。放卵後の受精卵の初期発生についてニワトリを例にとると、まず輸卵管内で受精した卵は、受精後約1.5時間で卵割を開始する。細胞質がつながったまま盤割が始まった卵は、1日かけて体外に放出される間に分裂し、約6万個の細胞からなる胚盤葉と呼ばれる胚になる(胚盤葉期)。この胚盤葉は、卵黄中央部の直径3〜4mmの白いリングとして観察される。この胚は、上層と下層に分裂し、割腔を形成する。放卵は胚盤葉下層が形成されるころに起こり、原条が形成され、胚盤葉は上、中、下の三重構造を取り、三胚葉が形成される。その後、胚の形成、成長を経て、排卵から22日目に孵化する。胚盤葉期は、ステージXとも呼ばれ、この時期の細胞の一部から生殖細胞が生じることから、従来は遺伝子導入の対象としてこの時期の受精卵を使用している。
【0060】
本発明においては、放卵直後、胚盤葉期の受精卵を孵化条件、例えばニワトリならば37.7〜37.8℃、湿度50〜70%程度の孵化に適した環境条件においた時間を0時間とし、これを孵卵開始時として、経時的に各種の処置を行った。ウズラでは孵卵開始から36時間後、ニワトリでは孵卵開始後50時間頃から、卵黄上に血管系の形成が観察され、心臓に分化する器官の脈動が観察できた。
【0061】
上述の遺伝子を導入した受精卵の孵化には、本発明者らが開発した人工卵殻による方法(Kamihira,M.ら(1998)Develop.Growth Differ.,40,449)等が応用できる。
【0062】
本発明の作製法において使用する複製能欠失型レトロウイルスベクター、導入する遺伝子、遺伝子導入鳥類としては、上述したG0トランスジェニックキメラ鳥類と同様のものが挙げられる。
【0063】
上記複製能欠失型レトロウイルスベクターに組み込まれる導入遺伝子は、レトロウイルスに由来しない遺伝子配列を含有することが好ましい。なお本発明の作製法において、「レトロウイルスに由来しない遺伝子」としては、上述した構造遺伝子、プロモーター遺伝子、分泌シグナル遺伝子等が挙げられる。上記レトロウイルスに由来しない遺伝子配列は、ニワトリβ−アクチンプロモーターにより制御されている遺伝子配列であることが好ましく、抗体遺伝子又は融合タンパク遺伝子をコードする遺伝子配列であることが好ましい。
【0064】
本発明の作製法では、1×107cfu/ml以上、好ましくは1×108cfu/ml以上、より好ましくは1×109cfu/ml以上のタイターを持つ複製能欠失型レトロウイルスベクターをマイクロインジェクションすることが、遺伝子を効率よく導入できる点で好ましい。
【0065】
本発明の作製法で受精卵に遺伝子を導入された鳥類は、その体細胞にモザイク状に導入遺伝子をもったトランスジェニック鳥類として成長する。この一世代目の遺伝子導入鳥類をG0トランスジェニックキメラ鳥類と呼ぶ。
【0066】
このような本発明の作製法によって得られるG0トランスジェニックキメラ鳥類も、本発明の1つである。
【0067】
G0トランスジェニックキメラ鳥類と非トランスジェニック鳥類、あるいはG0トランスジェニックキメラ鳥類同士を交配させて誕生する二世代目、三世代目が導入遺伝子を染色体にもつ生殖細胞から発生した場合、全身の体細胞に導入遺伝子を含有する個体として成長する。G0トランスジェニックキメラ鳥類個体から導入遺伝子を受け継ぐ子孫を、代々G1〜G2〜G3トランスジェニック鳥類と称する。
【0068】
本発明によるG0トランスジェニックキメラ鳥類を同種の非トランスジェニック鳥類あるいは配偶型G0トランスジェニックキメラ鳥類と交配させることにより、導入遺伝子を子孫に伝播させることができるとともに、全身の体細胞に導入遺伝子をもつ完全なトランスジェニック鳥類を作製できる。完全なトランスジェニック鳥類は、導入遺伝子をもつ体細胞の割合が多いことから、G0トランスジェニックキメラ鳥類に比べ、導入遺伝子に由来する組換えタンパク質の産生量が増加することが期待できる。更に、安定的に導入遺伝子を伝播するトランスジェニック鳥類の系統を確立することで、タンパク質産生システムとしての品質の安定化が可能である。
【実施例】
【0069】
以下、実施例により本発明を詳述するが、本発明はこれらの実施例により限定されるものではない。
【0070】
(実施例1)β−ガラクトシダーゼ発現ベクターコンストラクトの調製
β−ガラクトシダーゼ発現ベクターコンストラクトpMSCVNΔAβは、以下のように作製した。
【0071】
1.プラスミドpLXRN(クロンテック社製)からラウス・ザルコーマ・ウイルス(RSV)プロモーター断片を制限酵素XhoI及びHindIIIによって切り出し、プラスミドpBluescriptIISK(+)(ストラタジーン社製)のXhoI、HindIIIサイトへ挿入し、プラスミドpBlue/RSVを作製した。
【0072】
2.プラスミドpCMVβ(クロンテック社製)からβ−ガラクトシダーゼ(β−Gal)遺伝子断片を制限酵素NotIによって切り出し、プラスミドpZeoSV2(+)(インビトロジェン社製)のNotIサイトへ挿入した。T7プロモーターと同方向にβ−Gal遺伝子が挿入された構造のプラスミドをpZeo/lacZとした。
【0073】
3.pBlue/RSVからRSVプロモーター断片を制限酵素XhoI及びPstIによって切り出した。pZeo/lacZからβ−Gal遺伝子断片を制限酵素PstI及びXhoIによって切り出した。制限酵素XhoIによって処理したプラスミドpLNHX(クロンテック社製)のベクター断片に上記切り出した2断片を連結し、プラスミドpLNRβを作製した。
【0074】
4.pLNHXから一連のモロニー・ミューリン・ザルコーマ・ウイルス(MoMuSV)5’−ロング・ターミナル・リピート(LTR)、ウイルス・パッケージング・シグナル及びネオマイシン耐性(Neor)遺伝子を含む断片を、制限酵素SacII及びXhoIによって切り出し、制限酵素SacII及びXhoIによって処理したpLXRNのベクター断片に連結し、プラスミドpLXLを作製した。
【0075】
5.pZeo/lacZからβ−Gal遺伝子断片を制限酵素HindIII及びXhoIによって切り出し、制限酵素HindIII及びXhoIによって処理したpLXLのベクター断片に連結し、プラスミドpLZLを作製した。
【0076】
6.2つの化学合成オリゴヌクレオチド5’−cggtctagaggaattcagtggttcg−3’(配列番号1)及び5’−ccaggatccgacgttgtaaaacgacg−3’(配列番号2;下線部はBamHI制限酵素サイト)をプライマーとするPCR(94℃/15秒→55℃/30秒→72℃/1分30秒:35サイクル;KOD−Plus−DNAポリメラーゼ(東洋紡社製))により、プラスミドpMiwZ(Suemori et al.,1990,Cell Diff.Dev.29:181−185)からRSVプロモーターとニワトリβアクチン(Act)プロモーターのハイブリッドプロモーター(Miwプロモーター)の5’領域断片を増幅後、制限酵素BamHI及びMunIによって切り出し、プラスミドpGREEN LANTERN−1(ギブコBRL社製)のBamHI、MunIサイトへ挿入し、プラスミドpGmiw5’を作製した。
【0077】
7.pMiwZからMiwプロモーター5’側中央領域断片を制限酵素MunI及びClaIによって切り出し、pGmiw5’のMunI、ClaIサイトへ挿入し、プラスミドpGmiw5’−2を作製した。
【0078】
8.pGmiw5’−2からMiwプロモーター5’領域及び5’側中央領域を含む断片を制限酵素BamHI及びEcoRIによって切り出し、pBluescriptIISK(+)のBamHI、EcoRIサイトへ挿入し、プラスミドpBlue/Miw5’を作製した。
【0079】
9.2つの化学合成オリゴヌクレオチド5’−ccaaagcttgccgcagccattgcctttt−3’(配列番号3;下線部はHindIII制限酵素サイト)及び5’−atacctaggggctggctgcggaggaac−3’(配列番号4;下線部はBlnI制限酵素サイト)をプライマーとするPCR(98℃/15秒→60℃/30秒→72℃/30秒:35サイクル)によりpMiwZからMiwプロモーター3’領域断片を増幅後、制限酵素HindIII及びBlnIによって切り出し、制限酵素HindIII及びBlnIによって処理したpLXLのベクター断片に連結し、プラスミドpLMiw3’を作製した。
【0080】
10.pMiwZからMiwプロモーター3’側中央領域断片を制限酵素EcoRI及びMboIIによって切り出した。pLMiw3’からMiwプロモーター3’領域断片を制限酵素MboII及びKpnIによって切り出した。pBlue/Miw5’のEcoRI、KpnIサイトへ上記切り出した2断片を挿入し、プラスミドpBlue/Miwを作製した。
【0081】
11.pBlue/MiwからMiwプロモーター全長を含む断片を制限酵素BamHI及びBlnIによって切り出し、制限酵素BamHI及びBlnIによって処理したpLXLのベクター断片に連結し、プラスミドpLMLを作製した。
【0082】
12.pLMLからActプロモーター断片を制限酵素SmaI及びXbaIで切り出し、pBluescriptIISK(+)のEcoRV、XbaIサイトへ挿入し、プラスミドpBlue/Actを作製した。
【0083】
13.pLMLからMiwプロモーター断片を制限酵素HindIII及びBglIIによって切り出し、制限酵素HindIII及びBamHIによって処理したpLZLのベクター断片に連結し、プラスミドpLMβLを作製した。
【0084】
14.pBlue/ActからActプロモーター断片を制限酵素SalI及びBlnIによって切り出した。pLMβLからβ−Gal遺伝子断片を制限酵素BlnI及びBglIIによって切り出した。制限酵素XhoI及びBglIIによって処理したpLNRβのベクター断片に上記切り出した2断片を連結し、プラスミドpLNAβを作製した。
【0085】
15.2つの化学合成オリゴヌクレオチド5’−tttagctagctgcagctcagtgcatgcac−3’(配列番号5;下線部はNheI制限酵素サイト)及び5’−ataatctagaaacgcagcgactcccgc−3’(配列番号6;下線部はXbaI制限酵素サイト)をプライマーとするPCR(98℃/15秒→60℃/30秒→68℃/2分:30サイクル)によりpMiwZからイントロン欠失アクチン(ΔAct)プロモーター断片を増幅後、制限酵素XhoI(XhoI制限酵素サイトは増幅断片中に存在)及びXbaIによってΔActプロモーターの一部を含む断片を切り出した。ΔActプロモーターの残りの部分とβ−Gal遺伝子を含む断片をpLNAβから制限酵素BlnI及びBglIIによって切り出した。制限酵素XbaI及びBglIIによって処理したpLNAβのベクター断片に上記切り出した2断片を連結し、プラスミドpLNΔAβを作製した。
【0086】
16.pLNΔAβから一連のNeor遺伝子、ΔActプロモーター、及びβ−Gal遺伝子を含む断片を制限酵素BlnI及びBglIIによって切り出し、制限酵素BlnI及びBglIIによって処理したpLXLのベクター断片に連結し、プラスミドpLNΔAβ−2を作製した。
【0087】
17.pLNΔAβ−2から一連のNeor遺伝子、ΔActプロモーター、及びβ−Gal遺伝子を含む断片を制限酵素BamHI及びBglIIによって切り出し、制限酵素BamHI及びBglIIによって処理したプラスミドpMSCVneo(クロンテック社製)のベクター断片に連結した。BamHI及びBglIIサイトが消失したプラスミドをpMSCVNΔAβとした。
このように作製した複製能欠失型レトロウイルスベクターのベクターコンストラクトpMSCVNΔAβの構造を図1に示した。
【0088】
(実施例2)β−ガラクトシダーゼ発現レトロウイルスベクターの調製
実施例1で作製したベクターコンストラクトpMSCVNΔAβよりレトロウイルスベクターを調製するため、パッケージング細胞GP293(クロンテック社製)を直径100mmの培養ディッシュに5×106細胞植え、培養した。培地を新鮮なDMEM(ダルベッコ変法イーグル培地)に交換し、p−VSV−Gベクター(クロンテック社製)8μgとpMSCVNΔAβ8μgをリポフェクション法により前記GP293細胞に導入した。48時間後、ウイルス粒子を含む培養上清を回収し、0.45μm酢酸セルロースフィルター(アドバンテック社製)を通して、夾雑物を除去した。得られた溶液にポリブレン(シグマ社製)を10μg/mlとなるように加えウイルス液とした。
【0089】
調製したウイルス液を別に培養したGP293細胞に加え、48時間培養後、600μg/mlのG418(GIBCO BRL社製)を含む培養液で植え継ぎ、G418安定形質転換GP293株を取得した。
【0090】
得られた安定形質転換株を80%コンフルエントとなるよう直径100mmディッシュに培養し、16μgのpVSV−Gベクターをリポフェクション法で導入した。48時間後ウイルス粒子を含む培養上清12mlを回収した。
【0091】
この培養上清を50,000×g、4℃で1.5時間遠心を行い、ウイルスを沈殿させた。上清を除き、ウイルス粒子を含む沈殿物に50μlの50mM Tris−HCl(pH7.8)、130mM NaCl、1mM EDTA溶液を加え、4℃で一晩放置後、よく懸濁してウイルス溶液を回収した。このようにして得られた高タイターウイルスベクターは、108〜109cfu/mlであった。
【0092】
ウイルスタイターの測定は、以下のように行った。測定の前日にNIH3T3細胞(アメリカン・タイプ・カルチャー・コレクションより入手)を直径35mmのディッシュに7×104細胞植え、培養した。102〜106倍に希釈したウイルス溶液を各ディッシュに1ml加え、48時間後に蛍光顕微鏡によりGFP(グリーン・フルオレッセント・プロテイン)を発現している細胞の割合を測定し、以下の計算式によりタイターを決定した。
ウイルスタイター=細胞数×希釈率×発現割合(cfu/ml)
【0093】
(実施例3)ウズラ胚へのレトロウイルスベクターのインジェクション
WE系統のウズラ受精卵(日本生物科学研究所)を使用した。この受精卵を自動転卵装置が内蔵された孵卵器(昭和フランキ社製P−008型)内で37.9℃、湿度65%環境に置いた時刻を孵卵開始時刻(0時間)とし、以後15分毎に90度転卵しながら孵卵を行った。
【0094】
孵卵開始時、受精卵の卵殻を70%エタノールで消毒し、鋭端部を直径2cmの円形にダイヤモンドカッター(MINIMO7C710、ミニター社製)で切り取り、胚を露出させた。胚盤葉を実体顕微鏡で観察しながら、ガラス管(CD−1、オリンパス社製)をマイクロピペット製作器(PC−10、オリンパス社製)で加工し、外径約20μmとなるよう先端を折って作製した針を刺し、マイクロインジェクター(Transjector5246、エッペンドルフ社製)を用いて、胚盤下腔の中央に実施例2で調製したウイルス溶液約2μlを微量注入した。この卵殻の切り口まで卵白を満たした後、卵白を糊としてテフロン(登録商標)膜(ミリラップ、ミリポア社製)とポリ塩化ビニリデンラップ(サランラップ、旭化成社製)とで蓋をし、15分毎に90度転卵しながら孵卵を行った。
【0095】
孵卵開始から12時間後、24時間後の受精卵に同様の方法でウイルスを注入した。孵卵後約36時間目から卵黄表面に血管の発生が認められ、その一部が脈動して心臓の原基となることが実体顕微鏡で観察できる。孵卵後36時間、48時間、55時間の心臓内へ、実施例2で調製したウイルス溶液2μlをマイクロインジェクターにより微量注入した。
【0096】
(実施例4)ニワトリ胚へのレトロウイルスベクターのインジェクション
ニワトリ受精卵(日本生物科学研究所)を使用した。この受精卵を自動転卵装置が内蔵された孵卵器(昭和フランキ社製P−008型)内で37.9℃、湿度65%環境に置いた時刻を孵卵開始時刻(0時間)とし、以後15分毎に90度転卵しながら孵卵を行った。
【0097】
孵卵開始時、受精卵の卵殻を70%エタノールで消毒し、鋭端部を直径3.5cmの円形にダイヤモンドカッター(MINIMO7C710、ミニター社製)で切り取り、胚を露出させた。胚盤葉を実体顕微鏡で観察しながら、ガラス管(CD−1、オリンパス社製)をマイクロピペット製作器(PC−10、オリンパス社製)で加工し、外径約20μmとなるよう先端を折って作製した針を刺し、マイクロインジェクター(Transjector5246、エッペンドルフ社製)を用いて胚盤下腔の中央に、実施例2で調製したウイルス溶液約2μlを微量注入した。この卵殻の切り口まで卵白を満たした後、卵白を糊としてテフロン(登録商標)膜(ミリラップ、ミリポア社製)とポリ塩化ビニリデンラップ(サランラップ、旭化成社製)とで蓋をし、15分毎に90度転卵しながら孵卵を行った。
【0098】
孵卵開始から12時間後、24時間後の受精卵を同様に処理した。孵卵後約50時間目から卵黄表面に血管の発生が認められ、その一部が脈動して心臓の原基となることが実体顕微鏡で観察できる。孵卵後50時間、55時間、60時間の心臓内へ、実施例2で調製したウイルス溶液2μlをマイクロインジェクターにより微量注入した。
【0099】
(実施例5)β−ガラクトシダーゼ活性測定
孵卵開始から115時間後、胚を卵殻より取り出しPBS(リン酸緩衝液)にて洗浄し、胚をとり囲む膜を取り除いた。摘出した胚を細かくせん断し、0.8mlの反応バッファー(10mM KCl、1mM MgCl2、0.1% Triton X−100(和光純薬工業製)、5mM 2−メルカプトエタノール(和光純薬工業製)、2mM リン酸バッファー pH7.5)を加えて超音波破砕し、細胞液を得た。
【0100】
0.6mlの細胞液を、37℃で10分間インキュベートし、あらかじめ暖めておいた4mg/mlのo−ニトロフェニル−β−D−ガラクトピラノシド(ONPG)(シグマ社製)を0.1Mリン酸バッファー(pH7.5)に溶解した液0.1mlを添加した。反応後0.3mlの1M Na2CO3(和光純薬工業製)を加え、吸光度計にて420nmの波長強度を計測した。
【0101】
β−ガラクトシダーゼの活性はONPGunitで表した(1unit:1μmolのo−ニトロフェノールが1分間に生成されるときの活性)。3回実験を行い、その平均値をβ−ガラクトシダーゼ活性とした。
【0102】
ニワトリおよびウズラの、遺伝子導入時期とβ−ガラクトシダーゼ活性測定結果との関係を図2、図3に示した。ウズラでは孵卵後48時間後、ニワトリでは孵卵後55時間後に導入したβ−ガラクトシダーゼ遺伝子は、それ以前の時期に導入されたものに比べ、強く発現した。これは鳥類受精卵が、レトロウイルスによる外来遺伝子を不活性化する(サイレンシング)メカニズムが受精直後に顕著であり、時間を経るごとに弱くなっていくことを示唆する。このことから、鳥類の種類によって決まる特定の時間経過した受精卵に目的遺伝子を導入することで、不活性化されることなく効率的に導入遺伝子を発現するG0トランスジェニックキメラ鳥類の作製が可能になる。
【0103】
(実施例6)ウイルスタイターによる遺伝子発現の効率
実施例2で調製した1×108cfu/mlのウイルス液を、希釈溶媒(50mM Tris−HCl(pH7.8)、130mM NaCl、1mM EDTA溶液)で10倍、100倍、1000倍に3段階希釈し、1×107、1×106、1×105cfu/mlのタイター・ウイルス溶液とした。ウズラ受精卵を孵卵し、48時間後の発生初期心臓に、調製したウイルス液2μlをマイクロインジェクションした。同様にして48時間後の発生初期心臓に希釈溶媒のみを2μlインジェクションしたものを対照とした。
【0104】
孵卵開始から115時間後、実施例5に準じてβ−ガラクトシダーゼ活性を測定した。ウズラでのウイルスタイターと遺伝子発現結果との関係を図4に示した。
【0105】
同様にニワトリ受精卵を孵卵し、55時間後の発生初期心臓に、調製したウイルス液2μlをマイクロインジェクションした。55時間後の発生初期心臓に希釈溶媒のみを2μlインジェクションしたものを対照とした。
【0106】
孵卵開始から115時間後、実施例5に準じてβ−ガラクトシダーゼ活性を測定した。ニワトリでのウイルスタイターと遺伝子発現結果との関係を図5に示した。
【0107】
1×108cfu/mlのウイルスが注入されたウズラおよびニワトリで顕著なβ−ガラクトシダーゼ活性が確認され、それ以下の濃度では発現量は低かった。導入遺伝子の発現にはウイルスタイターが大きく影響すること、すなわち本発明によるG0トランスジェニックキメラ鳥類で、導入遺伝子を効率的に発現させるためには高タイターの複製能欠失型ベクターを使用するのが有効であるということが示唆された。
【0108】
(実施例7)ヒト抗体のウズラ、ニワトリ卵内への移行性
3種のサブクラスをもつヒト抗体(IgG1、2、3)(コスモバイオ社製)を混合したもの、および対応する3種の抗体フラグメント(Fab−1、Fab−2、Fab−3、Fc−1、Fc−2、Fc−3)(コスモバイオ社製)を100μg/mlとなるようPBSで希釈し、希釈液100μlをウズラ成鳥(3羽)またはニワトリ成鳥(3羽)の翼下静脈に注射した。
【0109】
抗体を静脈に注射した翌日から20日目まで採卵を行い、卵中に移行した抗体を定量した。卵黄は50%(W/V)、卵白は10%(V/V)となるようPBSを用いて希釈し、凍結保存して測定用サンプルとした。
【0110】
(実施例8)ELISA法による卵黄中抗体の定量
PBSで希釈した抗ヒトIgG抗体(コスモバイオ社製)をELISAプレートに100μg/well入れて、4℃で一晩静置した。PBS−0.05%Tween20溶液を200μlで各wellを3回洗浄した後、PBS−0.05%Tween20溶液−2%スキムミルクを150μl/well入れた。
【0111】
室温で2時間静置後、wellを200μlのPBS−0.05%Tween20溶液で3回洗浄し、採取した血液、卵白、卵黄サンプルを120μl入れ、4℃で一晩静置した。このELISAプレートを室温に戻した後、PBS−Tween20溶液で3回各wellを洗浄し、PBS−0.05%Tween20溶液で希釈したPeroxide(POD)標識抗ヒトIgG抗体(コスモバイオ社製)を100μl/well入れて、室温で1時間静置した。
【0112】
PBS−0.05%Tween20溶液でwellを4回洗浄し、発色液(10mgのo−フェニレンジアミン(片山化学工業製)を1mlのメタノールに溶解し、蒸留水で100mlとしたものに、10μlの過酸化水素水(和光純薬工業製)を加えて調製した)100μlをwellに加えた。8M硫酸を50μl添加して反応を止め、490nmの蛍光強度をプレートリーダーで測定して、標準検量線から濃度を計算した。ウズラ、ニワトリ各々3羽から得られたサンプルの抗体濃度を平均したものを結果とした。
【0113】
標準検量線作製のための標準抗体(コスモバイオ社製)は、50%卵黄−PBS(W/V)で希釈した。
【0114】
ウズラおよびニワトリの卵中に蓄積された抗体濃度を図6、7に示した。ウズラおよびニワトリの卵中に蓄積されたFab,Fcフラグメント濃度を図8、9、10、11に示した。
【0115】
ウズラ、ニワトリにおいて、ヒトIgG抗体、サブクラスではヒトIgG2、ヒトIgG1が効率よく卵中に蓄積されることが示された。またFcフラグメントが卵内への高い移行性を示したことから、ヒトIgGの移行はFcレセプターを介したものであることが示唆された。
【0116】
(実施例9)抗CD2抗体発現ベクターコンストラクトの作製
抗CD2抗体発現用ベクターコンストラクトpMSCV/GΔAH、pMSCV/GΔAL及びpMSCV/GΔALIHは、以下のように作製した。
【0117】
1.ヒト抗体(IgM)産生ハイブリドーマ細胞D253−15−6(アメリカン・タイプ・カルチャー・コレクションHB−8789)からQuick Prep Micro mRNA Purification Kit(ファルマシア社製)を用いてmRNAを取得し、得られたmRNAからFirst−Strand cDNA Synthesis Kit(ファルマシア社製)を用いてcDNAライブラリを調製した。2つの化学合成オリゴヌクレオチド5’−atcctcgagaggccaaagtacagtg−3’(配列番号7;下線部はXhoI制限酵素サイト)及び5’−cccggatccctaacactctcccctgttgaagct−3’(配列番号8;下線部はBamHI制限酵素サイト)をプライマーとするPCR(94℃/1分→50℃/1分→72℃/1分30秒:25サイクル;TaqDNAポリメラーゼ(パーキンエルマー社製))により上記cDNAライブラリからヒト抗体L鎖κ定常領域(hCκ)の遺伝子断片を増幅後、制限酵素XhoI及びBamHIによって切り出し、プラスミドpBluescriptIIKS(−)(ストラタジーン社製)のXhoI、BamHIサイトへ挿入し、プラスミドpBlue/hCκを作製した。
【0118】
2.同様にして、2つの化学合成オリゴヌクレオチド5’−agcggccgctacaggtgtccactccgacatcgtgatgacccagtctcc−3’(配列番号9;下線部はNotI制限酵素サイト)及び5’−cctctcgaggatagaagttattcagcaggcacac−3’(配列番号10;下線部はXhoI制限酵素サイト)をプライマーとするPCRにより上記cDNAライブラリからヒト抗体L鎖可変領域(hVL)の遺伝子断片を増幅後、制限酵素NotI及びXhoIによって切り出し、pBluescriptIIKS(−)のNotI、XhoIサイトへ挿入し、プラスミドpBlue/hVLを作製した。
【0119】
3.同様にして、2つの化学合成オリゴヌクレオチド5’−acctcgagcgtggccgttggctgcctcgcaca−3’(配列番号11;下線部はXhoI制限酵素サイト)及び5’−actaagcttacgttgtacagggtgggtttacc−3’(配列番号12;下線部はHindIII制限酵素サイト)をプライマーとするPCRにより上記cDNAライブラリからヒト抗体H鎖μ定常領域(hCμ)の遺伝子断片を増幅後、制限酵素XhoI及びHindIIIによって切り出し、pBluescriptIIKS(−)のXhoI、HindIIIサイトへ挿入し、プラスミドpBlue/hCμを作製した。
【0120】
4.同様にして、2つの化学合成オリゴヌクレオチド5’−agcggccgctacaggtgtccactccgaggtgcagctggtggagtctgg−3’(配列番号13;下線部はNotI制限酵素サイト)及び5’−cacgctcgaggtatccgacggggaattctcacagga−3’(配列番号14;下線部はXhoI制限酵素サイト)をプライマーとするPCRにより上記cDNAライブラリからヒト抗体H鎖可変領域(hVH)の遺伝子断片を増幅後、制限酵素NotI及びXhoIによって切り出し、pBluescriptIIKS(−)のNotI、XhoIサイトへ挿入し、プラスミドpBlue/hVHを作製した。
【0121】
5.pBlue/hCκからhCκ遺伝子断片を制限酵素XhoI及びBamHIによって切り出し、プラスミドpCEP4(インビトロジェン社製)のXhoI、BamHIサイトへ挿入し、プラスミドpCEP4/hCκを作製した。
【0122】
6.2つの化学合成オリゴヌクレオチド5’−cccaagcttgatctccactgggatggtgggggccctcctcttgctgctg−3’(配列番号15;下線部はHindIII制限酵素サイト)及び5’−cccggatcctcagtcaaggcgccttcgcatgaagaggccgatccccagggccaccaccagcagcaagaggagggcccc−3’(配列番号16;下線部はBamHI制限酵素サイト)を21bpsにわたって相補的な3’末端でアニールさせ、T4DNAポリメラーゼ(宝酒造社製)を用いたDNA2重鎖合成反応によって上皮増殖因子受容体膜貫通領域(TM)の遺伝子断片を調製した。得られたTM遺伝子断片を制限酵素HindIII及びBamHIによって処理後、pBluescriptIIKS(−)のHindIII、BamHIサイトに挿入し、プラスミドpBlue/TMを作製した。
【0123】
7.pBlue/hCμからhCμ遺伝子断片を制限酵素XhoI及びHindIIIによって切り出し、pBlue/TMのXhoI、HindIIIサイトへ挿入し、プラスミドpBlue/hCμTMを作製した。
【0124】
8.pBlue/hCμTMから一連のhCμ遺伝子及びTM遺伝子を含む断片を制限酵素XhoI及びBamHIによって切り出し、pCEP4のXhoI、BamHIサイトへ挿入し、プラスミドpCEP4/hCμTMを作製した。
【0125】
9.pCEP4/hCμTMを制限酵素BamHIによって切断し、末端をT4DNAポリメラーゼによって平滑処理後、セルフライゲーションによりプラスミドpCEP4/hCμTMΔBを作製した。
【0126】
10.化学合成オリゴヌクレオチド5’−tgaagacagatggcgccgccacagttcgttt−3’(配列番号17;下線部はNarI制限酵素サイト)を用いた部位特異的変異導入によりpBlue/hVLが保有するhVLの3’末端にアミノ酸暗号の変更を伴わずに制限酵素NarIサイトを導入し、プラスミドpBlue/hVLNを作製した。
【0127】
11.化学合成オリゴヌクレオチド5’−tggggcggatgcggatcctgaggagacggt−3’(配列番号18;下線部はBamHI制限酵素サイト)を用いた部位特異的変異導入によりpBlue/hVHが保有するhVHの3’末端にアミノ酸暗号の変更を伴わずに制限酵素BamHIサイトを導入し、プラスミドpBlue/hVHBを作製した。
【0128】
12.抗ヒトCD2マウス抗体産生ハイブリドーマ細胞TS2/18.1.1(アメリカン・タイプ・カルチャー・コレクションHB−195)からQuick Prep Micro mRNA Purification Kitを用いてmRNAを取得し、得られたmRNAからFirst−Strand cDNA Synthesis Kitを用いてcDNAライブラリを調製した。2つの化学合成オリゴヌクレオチド5’−cgcggccgcctcagggaaagtttgaagatg−3’(配列番号19;下線部はNotI制限酵素サイト)及び5’−cggcgccgccacagtccgttttatttccagcttggt−3’(配列番号20;下線部はNarI制限酵素サイト)をプライマーとするPCRにより上記cDNAライブラリからマウス抗体L鎖可変領域(mVL)の遺伝子断片を増幅後、制限酵素NotI及びNarIによって切り出し、pBlue/hVLNのNotI、NarIサイトへ挿入し、プラスミドpBlue/mVLを作製した。
【0129】
13.同様にして、2つの化学合成オリゴヌクレオチド5’−cgcggccgcgaacacggamccctcaccatg−3’(配列番号21;下線部はNotI制限酵素サイト)及び5’−cggatcctgcagagacagtgaccagagt−3’(配列番号22;下線部はBamHI制限酵素サイト)をプライマーとするPCRにより上記cDNAライブラリからマウス抗体H鎖可変領域(mVH)の遺伝子断片を増幅後、制限酵素NotI及びBamHIによって切り出し、pBluescriptIIKS(−)のNotI、BamHIサイトへ挿入し、プラスミドpBlue/mVHを作製した。
【0130】
14.pBlue/mVLからmVL遺伝子断片を制限酵素NotI及びXhoIによって切り出し、pCEP4/hCκのNotI、XhoIサイトへ挿入し、プラスミドpCEP4/IgLκを作製した。
【0131】
15.pBlue/hVHBからhVH遺伝子断片を制限酵素NotI及びXhoIによって切り出し、pCEP4/hCμTMΔBのNotI、XhoIサイトへ挿入し、プラスミドpCEP4/hIgHμTMを作製した。
【0132】
16.pBlue/mVHからmVH遺伝子断片を制限酵素NotI及びBamHIによって切り出し、制限酵素NotI及びBamHIによって処理したpCEP4/hIgHμTMのベクター断片に連結し、プラスミドpCEP4/IgHμTMを作製した。
【0133】
17.プラスミドpMSCVneo(クロンテック社製)から一連のミューリン・ホスホグリセレート・キナーゼ(PGK)プロモーター及びNeor遺伝子を含む断片を制限酵素BglII及びBamHIによって除去し、残ったベクター断片のセルフライゲーションによりプラスミドpMSCVを作製した。
【0134】
18.プラスミドpGREEN LANTERN−1(ギブコBRL社製)からGFP遺伝子断片を制限酵素NotIによって切り出し、pZeoSV2(+)のNotIサイトに挿入した。T7プロモーターと同方向にGFP遺伝子が挿入された構造のプラスミドをpZeo/GFPとした。
【0135】
19.pZeo/GFPからGFP遺伝子断片を制限酵素EcoRI及びXhoIによって切り出し、制限酵素EcoRI及びXhoIによって処理したpMSCVのベクター断片に連結し、プラスミドpMSCV/Gを作製した。
【0136】
20.ヒト抗体(IgG1)産生ミエローマ細胞IM−9(ジャパニーズ・コレクション・オブ・リサーチ・バイオリソーシズ 0024)からmRNA isolation Kit(ロッシュ社製)を用いてmRNAを取得し、得られたmRNAからReverTra Ace(東洋紡社製)を用いてcDNAライブラリを調製した。2つの化学合成オリゴヌクレオチド5’−caagcttcaagggcccat−3’(配列番号23)及び5’−atttacccggagacaggga−3’(配列番号24)をプライマーとするPCR(95℃/2分→52℃/30秒→74℃/3分:30サイクル;Pfu DNAポリメラーゼ(プロメガ社製))により上記cDNAライブラリからヒト抗体H鎖γ1定常領域(hCγ1)の遺伝子断片を増幅した。さらに、2つの化学合成オリゴヌクレオチド5’−ataggatccgctagcttcaagggcccatcg−3’(配列番号25;下線部はBamHI制限酵素サイト)及び5’−agcaagctttcatttacccggagacaggga−3’(配列番号26;下線部はHindIII制限酵素サイト)をプライマーとするPCR(94℃/15秒→58℃/30秒→68℃/1分:30サイクル;KOD−plus−DNAポリメラーゼ)により上記PCR産物からhCγ1遺伝子断片を増幅後、制限酵素BamHI及びHindIIIによって切り出し、pBluescriptIISK(+)のBamHI、HindIIIサイトへ挿入し、プラスミドpBlue/hCγ1を作製した。
【0137】
21.pCEP4/IgHμTMからmVH遺伝子断片を制限酵素HindIII及びBamHIによって切り出した。pBlue/hCγ1からhCγ1遺伝子断片を制限酵素BamHI及びHindIIIによって切り出した。制限酵素HindIIIによって処理したプラスミドpETBlue−2(ノバジェン社製)のベクター断片に上記切り出した2断片を連結し、プラスミドpETBlue/IgHγ1を作製した。
【0138】
22.pETBlue/IgHγ1から抗体H鎖γ1(IgHγ1)の遺伝子断片を制限酵素HindIIIによって切り出し、pMSCV/GのHindIIIサイトへ挿入した。GFP遺伝子と同方向にIgHγ1遺伝子が挿入された構造のプラスミドをpMSCV/GHとした。
【0139】
23.2つの化学合成オリゴヌクレオチド5’−acgcgtcgacgtgcatgcacgctcattg−3’(配列番号27;下線部はSalI制限酵素サイト)及び5’−acgcgtcgacaacgcagcgactcccg−3’(配列番号28;下線部はSalI制限酵素サイト)をプライマーとするPCR(94℃/15秒→50℃/30秒→68℃/1分:10サイクル;94℃/15秒→62℃/30秒→68℃/1分:30サイクル)によりpMiwZからΔActプロモーター断片を増幅後、制限酵素SalIによってΔActプロモーター断片を切り出し、pETBlue−2のSalIサイトへ挿入し、プラスミドpETBlue/ΔActを作製した。
【0140】
24.pETBlue/ΔActからΔActプロモーター断片を制限酵素SalIによって切り出し、pMSCV/GHのXhoIサイトへ挿入した。IgHγ1遺伝子と同方向にΔActプロモーターが挿入された構造のプラスミドをpMSCV/GΔAHとした。
【0141】
25.2つの化学合成オリゴヌクレオチド5’−aatgtcgacatggtgtccacttctcagctc−3’(配列番号29;下線部はSalI制限酵素サイト)及び5’−ttcgtcgacctaacactctcccctgttgaa−3’(配列番号30;下線部はSalI制限酵素サイト)をプライマーとするPCR(95℃/30秒→50℃/30秒→74℃/2分:10サイクル;95℃/30秒→60℃/30秒→74℃/2分:30サイクル;Pfu DNAポリメラーゼ)によりpCEP4/IgLκから抗体L鎖κ(IgLκ)の遺伝子断片を増幅後、制限酵素SalIによって切り出し、pETBlue−2のSalIサイトへ挿入し、プラスミドpETBlue/IgLκを作製した。
【0142】
26.pETBlue/ΔActからΔActプロモーター断片を制限酵素SalIによって切り出し、pMSCV/GのXhoIサイトへ挿入した。GFP遺伝子と同方向にΔActプロモーターが挿入された構造のプラスミドをpMSCV/GΔAとした。
【0143】
27.pETBlue/IgLκからIgLκ遺伝子断片を制限酵素SalIによって切り出し、pMSCV/GΔAのSalIサイトへ挿入した。ΔActプロモーターと同方向にIgLκ遺伝子断片が挿入された構造のプラスミドをpMSCV/GΔALとした。
【0144】
28.2つの化学合成オリゴヌクレオチド5’−acgcgtcgaccgcccctctccctccccc−3’(配列番号31;下線部はSalI制限酵素サイト)及び5’−ccgctcgagattatcatcgtgtttttcaaaggaaaaccacgtc−3’(配列番号32;下線部はXhoI制限酵素サイト)をプライマーとするPCR(94℃/15秒→60℃/30秒→68℃/1分:30サイクル)によりプラスミドpLXIN(クロンテック社製)からIRES断片を増幅後、制限酵素SalI及びXhoIによって切り出し、pETBlue−2のSalI、XhoIサイトへ挿入し、プラスミドpETBlue/IRESを作製した。
【0145】
29.pETBlue/IRESからIRES断片を制限酵素SalI及びXhoIによって切り出し、pMSCV/GΔAHのSalIサイトへ挿入した。IgHγ1遺伝子と同方向にIRESが挿入された構造のプラスミドをpMSCV/GΔAIHとした。
【0146】
30.pETBlue/IgLκからIgLκ遺伝子断片を制限酵素SalIによって切り出し、pMSCV/GΔAIHのSalIサイトへ挿入した。ΔActプロモーターと同方向にIgLκ遺伝子断片が挿入された構造のプラスミドをpMSCV/GΔALIHとした。
【0147】
このように作製した複製能欠失型レトロウイルスベクターのベクターコンストラクトpMSCV/GΔAH、pMSCV/GΔAL及びpMSCV/GΔALIHの構造を図12に示した。
【0148】
(実施例10)抗CD2抗体発現G0トランスジェニックキメラウズラの作製
実施例2に準じて、ベクターコンストラクトpMSCV/GΔAH、pMSCV/GΔAL及びpMSCV/GΔALIHより3種類のレトロウイルスベクターを調製した。このレトロウイルスベクターのタイターを測定したところ、108cfu/ml〜109cfu/mlだった。
【0149】
得られたレトロウイルスベクターを、実施例3に準じて孵卵後36時間のウズラ受精卵心臓にマイクロインジェクションし、37.9℃,湿度65%で15分毎に90度転卵させながら孵卵した。
【0150】
軽鎖(Light Chain)、重鎖(Heavy Chain)からなる抗体をトランスジェニック動物で発現させるには、軽鎖、重鎖それぞれを発現させるベクターを各々単独で導入する方法と、軽鎖を発現する遺伝子と重鎖を発現する遺伝子をIRESのような配列で区切り、同一のベクターとして導入する方法が考えられる。そこでインジェクションは、pMSCV/GΔAH、pMSCV/GΔALを同時に感染させた場合(実施例11、実験例1)と、pMSCV/GΔALIHより調製したベクターを単独で導入した場合(実施例11、実験例2)に分けて行った。
【0151】
孵卵48時間後、発生が正常に進行していることを確認し、ニワトリのSサイズの卵殻の鈍端部に直径4cmの穴を開けたものに、このウイルス導入胚を移した。胚を上にして空気に触れるようにし、濃度50mg/mlで卵白に懸濁した乳酸カルシウム(シグマ社製)溶液を0.5ml添加後、卵白を糊としてラップで密閉した。再度孵卵器に入れ、37.9℃、湿度65%で1時間毎に60度転卵しながら13日間培養した。転卵を止めて静置状態にし、胚が肺呼吸に移行したら、ラップに針で小さな穴を開け、呼吸を助けた。漿尿膜の血が引いたら培養器から雛を出し、孵化させた。
【0152】
(実施例11)血清中、卵中の抗CD2抗体濃度の測定
実施例10により孵化させたG0トランスジェニックキメラウズラを1ヶ月間飼育して雛を成長させた。30日後および60日後、成長したG0トランスジェニックウズラの翼下静脈より採血を行い、血液サンプルを得た。得られた血液を15,000rpmで10分間遠心し、上清として得られる血清から抗CD2抗体量の測定を行った。
孵化から1.5ヵ月後、産卵を始めた雌性トランスジェニックウズラより採卵を行い、実施例7に準じて調製した卵白、卵黄中の抗CD2抗体量を、実施例8に準じてELISA法により定量した。
実験例1、実験例2の定量結果を示す。
【0153】
(実験例1)
pMSCV/GΔAH、pMSCV/GΔALから調製したベクター(3〜4×108cfu/ml)を同時に感染させたG0トランスジェニックキメラウズラ(個体識別番号#1113)は、卵黄中に0.6μg/ml、卵白中に0.5μg/mlの抗CD2抗体を発現した。
【0154】
(実験例2)
pMSCV/GΔALIHより調製したベクター(5×108cfu/ml)を単独で導入したG0トランスジェニックキメラウズラ(#4202)は、血清中に5.2μg/mlの抗CD2抗体を発現した。
【0155】
(実施例12)scFv−Fc抗体発現ベクターコンストラクトの作製
ScFv−Fc抗体発現ベクターコンストラクトpMSCV/scFv−Fcは、以下のように作製した。
【0156】
1.5’末端をリン酸化した2つの化学合成オリゴヌクレオチド5’−ctagaccatgaggtctttgctaatcttggtgctttgcttcctgcccctggctgctctgggg−3’(配列番号33;ctagaはXbaI認識部位末端、ggはHaeIII認識部位末端)及び5’−ccccagagcagccaggggcaggaagcaaagcaccaagattagcaaagacctcatggt−3’(配列番号34;ccはXbaI認識部位末端、tはHaeIII認識部位末端)をアニールさせ、リゾチーム分泌シグナルの遺伝子断片を調製した。2つの化学合成オリゴヌクレオチド5’−gcgtttaaagtgacgttggacgtccg−3’(配列番号35;tttaaaはDraI制限酵素サイト)及び5’−attaggatccgcgcttaaggacggtcagg−3’(配列番号36;ggatccはBamHI制限酵素サイト)をプライマーとするPCR(94℃/15秒→58℃/30秒→68℃/1分:30サイクル;KOD−Plus−DNAポリメラーゼ)により、HUC2−13細胞のニワトリ抗体可変領域遺伝子から調製された一本鎖抗体(scFv)の遺伝子を含んだプラスミドpPDS/scFv(Nakamura et al.,2000,Cytotechnology32:191−198)からscFv遺伝子断片を増幅後、制限酵素DraI及びBamHIによって切り出した。pBluescriptIISK(+)のXbaI、BamHIサイトに上記調製した2断片を挿入し、プラスミドpBlue/scFvを作製した。
【0157】
2.pBlue/scFvからscFv遺伝子断片を制限酵素NotI及びBamHIによって切り出し、pCEP4のNotI、BamHIサイトへ挿入し、プラスミドpCEP4/scFvを作製した。
【0158】
3.ヒトIgG1産生ミエローマ細胞IM−9からmRNA isolation Kitを用いてmRNAを取得し、得られたmRNAからReverTra Aceを用いてcDNAライブラリを調製した。2つの化学合成オリゴヌクレオチド5’−caagcttcaagggcccat−3’(配列番号23)及び5’−atttacccggagacaggga−3’(配列番号24)をプライマーとするPCR(95℃/2分→52℃/30秒→74℃/3分:30サイクル;Pfu DNAポリメラーゼ)により上記cDNAライブラリからhCγ1遺伝子断片を増幅した。さらに、2つの化学合成オリゴヌクレオチド5’−attaggatccgagcccaaatcttgtgacaaaactc−3’(配列番号37;ggatccはBamHI制限酵素サイト)及び5’−agcaagctttcatttacccggagacaggga−3’(配列番号26;aagcttはHindIII制限酵素サイト)をプライマーとするPCR(94℃/15秒→58℃/30秒→68℃/1分:30サイクル;KOD−plus−DNAポリメラーゼ)により上記PCR産物からヒト抗体H鎖γ1のFc領域(Fc)の遺伝子断片を増幅後、制限酵素BamHI及びHindIIIによって切り出し、pBluescriptIISK(+)のBamHI、HindIIIサイトへ挿入し、プラスミドpBlue/Fcを作製した。
【0159】
4.pCEP4/scFvからscFv遺伝子断片を制限酵素HindIII及びBamHIによって切り出した。pBlue/FcからFc遺伝子断片を制限酵素BamHI及びHindIIIによって切り出した。pBluescriptIISK(+)のHindIIIサイトへ上記切り出した2断片を挿入し、プラスミドpBlue/scFv−Fcを作製した。
【0160】
5.pBlue/scFv−Fcからニワトリ一本鎖抗体可変領域にヒト抗体H鎖γ1・Fcが連結した構造(scFv−Fc)の遺伝子断片を制限酵素HindIIIによって切り出し、制限酵素HindIIIによって処理したpMSCV/GΔAHのベクター断片に連結した。ΔActプロモーターと同方向にscFv−Fc遺伝子が連結された構造のプラスミドをpMSCV/GΔAscFv−Fcとした。
【0161】
このように作製した複製能欠失型レトロウイルスベクターのベクターコンストラクトpMSCV/GΔAscFv−Fcの構造を図13に示した。
【0162】
(実施例13)scFv−Fc抗体発現G0トランスジェニックキメラウズラの作製
実施例2に準じて、ベクターコンストラクトpMSCV/GΔAscFv−Fcよりレトロウイルスベクターを調製した。このレトロウイルスベクターのタイターを測定したところ、108cfu/ml〜109cfu/mlだった。
【0163】
得られたウイルスベクター溶液を、実施例3に準じて孵卵後36時間のウズラ受精卵心臓にマイクロインジェクションし、実施例10に準じて孵化させることによりG0トランスジェスジェニックキメラウズラを誕生させた。
【0164】
(実施例14)血清中、卵中のscFv−Fc濃度の測定
実施例13により誕生したG0トランスジェニックキメラウズラを1ヶ月間飼育して雛を成長させた。30日後および60日後、成長したG0トランスジェニックキメラウズラ(個体識別番号#3303、#3306、#3310、#3311、#3313)の翼下静脈より採血を行い、血液サンプルを得た。得られた血液を15,000rpmで10分間遠心し、上清として得られる血清からscFv−Fc抗体量の測定を行った。
孵化から1.5ヵ月後、産卵を始めた雌性トランスジェニックウズラ(個体識別番号#3310)より採卵を行い、実施例7に準じて調製した卵白、卵黄中のscFv−Fc抗体量を、実施例8に準じてELISA法により定量した。
【0165】
標準検量線は精製したscFv−Fcを用いて作製した。実施例12で作製したベクターコンストラクトpMSCV/scFv−Fcをリポフェクション法によりGP293細胞に導入し、その培養上清を4℃、10分間、3000rpmで遠心して固形物を除去した。この上清を冷却しながら攪拌し、50%飽和となるよう細かく砕いた硫酸アンモニウムを徐々に加え(313g硫安/1000ml水)、タンパク質を沈殿させた。これを4℃で一晩静置した後、4℃で10分間、15,000rpmで遠心して沈殿を完全に沈降させ、少量のPBSで溶解した。2LのPBSで3回透析して硫安を除去した。
【0166】
精製用のプロテインGカラム(パーゼプティブバイオシステムズ社製)の初期洗浄をBinding Buffer(NaHPO4・2H2O 1.56g/l、NaHPO4・12H2O 7.16g/l)10mL、Wash Buffer(酢酸20%、蒸留水80%)10ml、Binding Buffer10mlの順で行った(流速2ml/分)。PBSに溶解したタンパク質液を1ml/分で流し、scFv−Fcをカラムに吸着させた。Binding Buffer20mlを1.7ml/分で流して、不要なタンパク質を除去し、Elution Buffer(グリシン7.507g/l、2N HClにてpH2.5〜3.0に調製)を1.5ml/分で流して、scFv−Fcを溶出した。
【0167】
溶出分画をPBS(2L)で3回透析し、精製scFv−Fcとし、波長280nmの吸光度よりタンパク質濃度を定量した。
【0168】
実施例14で作製したG0トランスジェニックキメラウズラの30日後、60日後採血血清中のscFv−Fc量を図14に示した。scFv−Fc抗体の発現遺伝子を導入されたG0トランスジェニックキメラウズラは、30日目に約2mg/ml〜4mg/mlの抗体を血清中に発現し、5羽のうち3羽が60日後も同程度の発現量を示した。
【0169】
G0トランスジェニックキメラウズラ(#3310)が産卵を始めた日から、卵黄、卵白中のscFv−Fc量を図15に示した。抗体は卵白、卵黄中に約500μg/ml〜1mg/ml発現し、産卵開始から17日目まで若干の変動はあるものの安定した発現量を維持した。
【0170】
(実施例15)scFv−Fc構造の確認
実施例13で作製したG0トランスジェニックキメラウズラ血清1mlから実施例10に準じて、硫安沈殿とプロテインGカラムによりscFv−Fcを精製した。精製したscFv−FcをSDS−PAGEで解析し、その結果を図16に示した。未処理のレーンより、scFv−Fcの分子量約120kDaが示された。還元処理したscFv−Fcの分子量は未処理の場合のほぼ半分(約60kDa)になっていることから、G0トランスジェニックキメラウズラにより産生されたscFv−FcはS−S結合による2量体を形成していることがわかった。これらは、S−S結合に関与するシステイン残基をFc部に有するscFv−Fcの構造的特徴に符合し、G0トランスジェニックキメラウズラにより産生されたscFv−Fcは正しい構造を保持していることが示唆された。
【0171】
(実施例16)TNFR−Fc融合タンパク発現G0トランスジェニックキメラニワトリの作製
実施例9に準じてTNFR−Fc発現ベクターコンストラクトを作製し、実施例2に準じてレトロウイルスベクターを作製した。このレトロウイルスベクターのタイターは、1.7×107cfu/mlだった。
【0172】
得られたウイルスベクター溶液を、実施例4に準じて孵卵後55時間のニワトリ受精卵心臓にマイクロインジェクションし、実施例10に準じて孵化させることによりG0トランスジェニックキメラニワトリを誕生させた。
【0173】
8個の受精卵にインジェクションしたところ4羽が孵化し、実施例14に準じてその血清中のTNFR−Fcを定量したところ、最高で50μg/mlのTNFR−Fcが発現していた。
【産業上の利用可能性】
【0174】
本発明のG0トランスジェニックキメラ鳥類は、複製能欠失型レトロウイルスベクターで導入された遺伝子を不活性化することなく効率的に発現することができる。また、本発明のG0トランスジェニックキメラ鳥類の作製法は、キメラ抗体、例えばscFv−Fc抗体の遺伝子を導入し、血中、卵中に抗体を効率的に発現することができる鳥類の作製を可能にする。更に、本発明の抗体生産法は、キメラ抗体、例えばscFv−Fc抗体を産生するG0トランスジェニックキメラ鳥類を作製し、抗体を鳥類の血清、卵中から回収、精製することからなるので、効率よい抗体生産を可能にする。
【特許請求の範囲】
【請求項1】
鳥類受精卵を孵卵し、孵卵開始から36時間以降の初期胚に形成される心臓ないしは血管内へ複製能欠失型レトロウイルスベクターをマイクロインジェクションすることにより感染させ、その胚を孵化させることからなるG0トランスジェニックキメラ鳥類の作製法。
【請求項2】
鳥類がニワトリまたはウズラである請求項1に記載のG0トランスジェニックキメラ鳥類の作製法。
【請求項3】
レトロウイルスに由来しない遺伝子配列が、複製能欠失型レトロウイルスベクターに組み込まれる導入遺伝子に含有される請求項1−2のいずれか1項に記載のG0トランスジェニックキメラ鳥類の作製法。
【請求項4】
レトロウイルスに由来しない遺伝子配列は、抗体遺伝子をコードする遺伝子配列である請求項3に記載のG0トランスジェニックキメラ鳥類の作製法。
【請求項5】
抗体遺伝子がキメラ抗体遺伝子である請求項4に記載のG0トランスジェニックキメラ鳥類の作製法。
【請求項6】
抗体遺伝子がscFv−Fc抗体遺伝子である請求項5に記載のG0トランスジェニックキメラ鳥類の作製法。
【請求項7】
請求項1−6のいずれか1項に記載の方法で作製され、目的タンパク質を血液中に0.5μg/ml以上、卵白中に0.1μg/ml以上、又は卵黄中に0.1μg/ml以上含む、G0トランスジェニックキメラ鳥類。
【請求項8】
請求項1−7のいずれか1項に記載の方法で作製されたG0トランスジェニックキメラ鳥類を、配偶型の同種鳥類と交配させ、その卵を孵化することよりなるトランスジェニック鳥類の作製法。
【請求項1】
鳥類受精卵を孵卵し、孵卵開始から36時間以降の初期胚に形成される心臓ないしは血管内へ複製能欠失型レトロウイルスベクターをマイクロインジェクションすることにより感染させ、その胚を孵化させることからなるG0トランスジェニックキメラ鳥類の作製法。
【請求項2】
鳥類がニワトリまたはウズラである請求項1に記載のG0トランスジェニックキメラ鳥類の作製法。
【請求項3】
レトロウイルスに由来しない遺伝子配列が、複製能欠失型レトロウイルスベクターに組み込まれる導入遺伝子に含有される請求項1−2のいずれか1項に記載のG0トランスジェニックキメラ鳥類の作製法。
【請求項4】
レトロウイルスに由来しない遺伝子配列は、抗体遺伝子をコードする遺伝子配列である請求項3に記載のG0トランスジェニックキメラ鳥類の作製法。
【請求項5】
抗体遺伝子がキメラ抗体遺伝子である請求項4に記載のG0トランスジェニックキメラ鳥類の作製法。
【請求項6】
抗体遺伝子がscFv−Fc抗体遺伝子である請求項5に記載のG0トランスジェニックキメラ鳥類の作製法。
【請求項7】
請求項1−6のいずれか1項に記載の方法で作製され、目的タンパク質を血液中に0.5μg/ml以上、卵白中に0.1μg/ml以上、又は卵黄中に0.1μg/ml以上含む、G0トランスジェニックキメラ鳥類。
【請求項8】
請求項1−7のいずれか1項に記載の方法で作製されたG0トランスジェニックキメラ鳥類を、配偶型の同種鳥類と交配させ、その卵を孵化することよりなるトランスジェニック鳥類の作製法。
【図1】


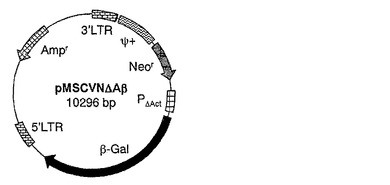
【図2】
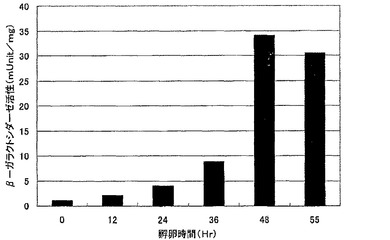
【図3】
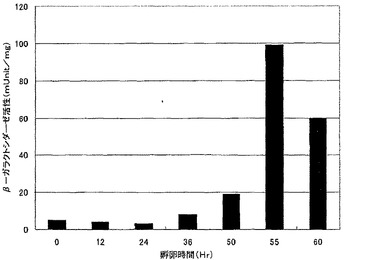
【図4】

【図5】

【図6】
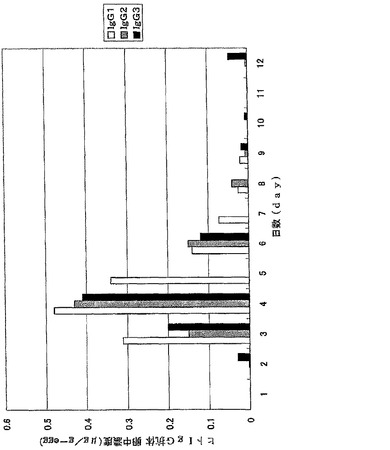
【図7】

【図8】

【図9】
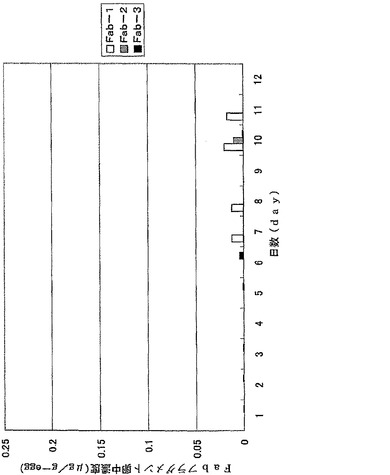
【図10】
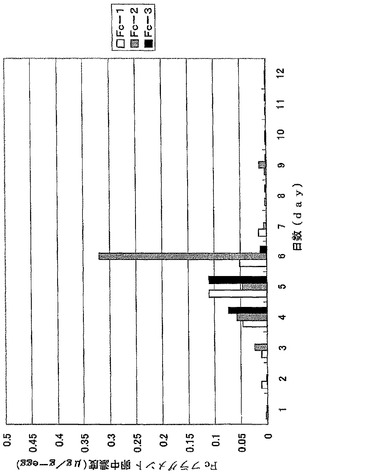
【図11】
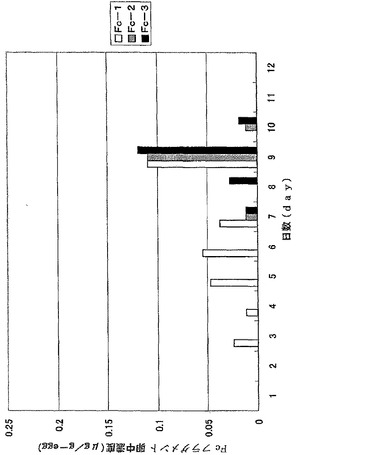
【図12】
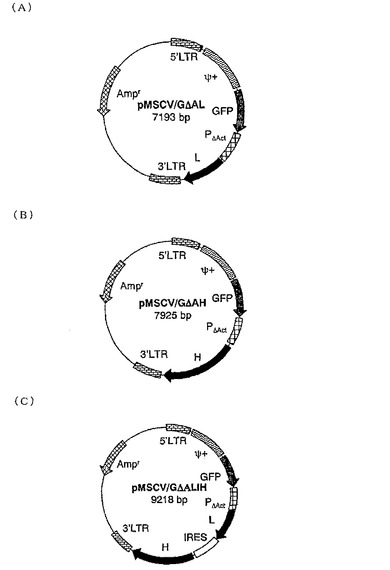
【図13】
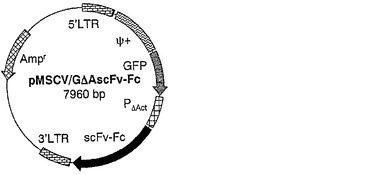
【図14】

【図15】
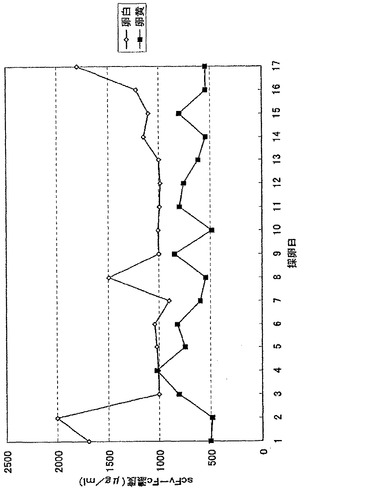
【図16】
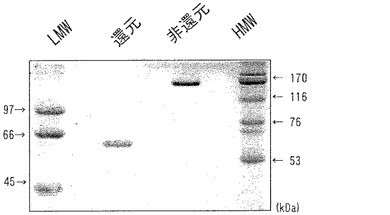
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【公開番号】特開2011−147447(P2011−147447A)
【公開日】平成23年8月4日(2011.8.4)
【国際特許分類】
【出願番号】特願2011−40380(P2011−40380)
【出願日】平成23年2月25日(2011.2.25)
【分割の表示】特願2004−528857(P2004−528857)の分割
【原出願日】平成15年8月11日(2003.8.11)
【出願人】(000000941)株式会社カネカ (3,932)
【出願人】(598091860)財団法人名古屋産業科学研究所 (23)
【Fターム(参考)】
【公開日】平成23年8月4日(2011.8.4)
【国際特許分類】
【出願日】平成23年2月25日(2011.2.25)
【分割の表示】特願2004−528857(P2004−528857)の分割
【原出願日】平成15年8月11日(2003.8.11)
【出願人】(000000941)株式会社カネカ (3,932)
【出願人】(598091860)財団法人名古屋産業科学研究所 (23)
【Fターム(参考)】
[ Back to top ]