
レボフロキサシンおよびその形態の調製
【課題】キラルなフッ素化カルボキシキノロンであり、化学名が(S)−9−フルオロ−2,3−ジヒドロ−3−メチル−10−(4−メチル−1−ピペラジニル)−7−オキソ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸であるレボフロキサシンの新規な調製方法の提供。
【解決手段】(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸をN−メチルピペラジンと極性溶媒中、又はニート混合物中で反応させ、新規な6種類の形態のレボフロキサシン(A〜C,F〜H)を調製する(反応温度80℃:半水和物、形態A,B,G,H,85℃:形態C,還流温度:形態F)。レボフロキサシンを更に極性溶媒中に溶解し、その後貧溶媒を添加して冷却下に沈殿処理することにより新規なレボフロキサシン形態が生成する。
【解決手段】(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸をN−メチルピペラジンと極性溶媒中、又はニート混合物中で反応させ、新規な6種類の形態のレボフロキサシン(A〜C,F〜H)を調製する(反応温度80℃:半水和物、形態A,B,G,H,85℃:形態C,還流温度:形態F)。レボフロキサシンを更に極性溶媒中に溶解し、その後貧溶媒を添加して冷却下に沈殿処理することにより新規なレボフロキサシン形態が生成する。
【発明の詳細な説明】
【技術分野】
【0001】
本願は2001年10月3日に提出された仮出願整理番号第60/326,958号、2001年11月29日に提出された同第60/334,316号および2002年2月11日に提出された同第60/354,939号ならびに本願と同時に提出された非仮出願整理番号(未定)[代理人整理番号1662/58004号]の優先権を主張する。これらの各出願の全内容が参照により本願明細書に取り込まれる。
【0002】
本発明はレボフロキサシンおよびその新規な形態の調製方法に関する。
【背景技術】
【0003】
レボフロキサシンは広スペクトルの合成抗生物質である。レボフロキサシンは、フルオロキノロン抗微生物剤であるオフロキサシン・ラセミ体のS−エナンチオマーである。オフロキサシンの抗菌活性は主にS−エナンチオマーにある。レボフロキサシンおよび他のフルオロキノロン抗微生物剤の作用機構は、DNA複製、転写修復および組換えに必要な酵素であるDNAジャイレース(細菌型トポイソメラーゼII)の阻害を含む。レボフロキサシンは、経口投与または静脈内投与し得るLEVAQUIN(登録商標)として利用可能である。
【0004】
レボフロキサシンはキラルなフッ素化カルボキシキノロンである。その化学名は(S)−9−フルオロ−2,3−ジヒドロ−3−メチル−10−(4−メチル−1−ピペラジニル)−7−オキソ−7H−ピリド[1,2,3−de]−1,4−ベンズオキサジン−6−カルボン酸(CAS登録番号第100986−85−4号)である。レボフロキサシンの化学構造を次式Iに示す。
【化1】

【0005】
米国特許第4,382,892号はピリド[1,2,3−de][1,4]ベンズオキサジン誘導体およびその調製方法に関する。
【0006】
米国特許第5,053,407号は光学活性ピリドベンズオキサジン誘導体、該誘導体の調製方法および該誘導体の調製に有用な中間体に関する。
【0007】
米国特許第5,051,505号はピペラジニルキノロン誘導体の調製方法に関する。この方法はアセトニトリル、ジメチルホルムアミド、ピリジン、スルホランおよびジメチルスルホキシドのような極性溶媒の存在下でジハロキノロンをピペラジン誘導体およびハロゲン化テトラアルキルアンモニウムと反応させることを含む。
【0008】
米国特許第5,155,223号はキノリンカルボン酸の調製に関する。
【0009】
米国特許第5,545,737号は、結晶化時にレボフロキサシンが溶解される水性溶媒の含水量を調節することによりレボフロキサシン半水和物または一水和物を選択的に生成することを開示している。
【0010】
レボフロキサシン形態
レボフロキサシンの3種の多形形態(polymorphic form)(無水α、β、γ)および2種の偽多形形態(pseudepolymorphic form)(半水和物および一水和物)が文献に言及されている。半水和物および一水和物形態は EP 0444 678 B1および米国特許第5,545,737号で言及されている。これら2件の特許は、一水和物を含まない半水和物の調製方法および半水和物を含まない一水和物の調製方法に関する。
【0011】
"Effect of dehydration on the formation of Lovofloxacine Pseudepolymorphs (レボフロキサシン偽多形の生成に対する脱水の効果)" と題された論文Chem. Pharm. Bull. 43(4) 649-653 (1995) はレボフロキサシンの水和物形態の物性を検討している。この論文によれば、半水和物形態を加熱することによって、水和水が除去されて無水形態γが得られる。更に加熱することによって、無水形態βが生成し、次に無水形態αが生成する。一水和物形態を加熱することによって、水和水が除去されて無水形態αが得られる。形態γおよび形態αは通常の相対湿度の下で急速に水蒸気を吸収し、それぞれ半水和物および一水和物に変換される。
【0012】
本発明はレボフロキサシンの固体物性に関する。固体物性としては、例えば粉砕された固体の流動性がある。流動性は、医薬品の製造時に該物質を取り扱う容易さに影響する。パウダー状化合物の粒子が相互に容易に流動しない場合に、配合専門家はタブレットまたはカプセル配合物の開発を考慮しなければならず、コロイド状二酸化ケイ素、タルク、スターチまたは三塩基性リン酸カルシウム等の流動促進剤(glidant)の使用を必要とする場合がある。
【0013】
医薬化合物の他の重要な固体物性は、水性流体中でのその溶解速度である。患者の胃液中での活性成分の溶解速度は、経口投与される活性成分が患者の血流に到達することができる速度に対する上限となるために、治療効果を有することができる。溶解速度はまた、シロップ剤、エリキシル剤および他の液状医薬品の配合における考慮事項でもある。化合物の固体形態は、圧蜜化とその貯蔵安定性に対する化合物の挙動にも影響する。
【0014】
これらの実際の物理特性は、物質の特定の多形形態を規定する単位胞中の分子の立体配座と配向によって影響を受ける。多形形態は、非晶質物質や他の多形形態の熱的挙動とは異なる熱的挙動を示し得る。熱的挙動は研究室においてキャピラリー融点、熱重量分析(TGA)、示差走査熱量測定法(DSC)等のような技法により測定され、これを用いてある多形形態を他の多形形態と区別することができる。特定の多形形態は、粉末X線結晶学、固体状態C−NMR分光測定および赤外分光測定によって検出可能な明確な分光的性質も示し得る。
【発明の開示】
【0015】
1つの態様において、本発明は、(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸(「化合物I」)をN−メチルピペラジンと反応させて、レボフロキサシンを形成すること、および該レボフロキサシンを回収することを含んでなる、レボフロキサシンの調製方法を提供する。化合物IはN−メチルピペラジンと、極性溶媒中で又はニート(neat:そのままの)混合物として反応させてよい。
【0016】
本発明の他の態様において、新規な結晶形態A、B、C、F、GおよびH、ならびにそれらの調製方法が記載される。1つの実施態様において、レボフロキサシンの調製方法は、レボフロキサシンを上昇した温度で保持すること、極性溶媒を添加すること、およびレボフロキサシン形態を回収することを含んでなる。好ましくは、この方法は、レボフロキサシン−溶媒混合物を周囲温度よりも低い温度に冷却および保持することを更に含む。
【発明を実施するための最良の形態】
【0017】
本発明の1つの態様は、レボフロキサシンの調製方法を提供する。幾つかの実施態様において、約70%〜約85%またはそれ以上の精製レボフロキサシンの収率が達成される。本願明細書で用いられる「粗」および「精製」とは、それぞれ純度の低いまたは純度の高いことを意味する相対的用語である。従来技術の方法に対し、高度に濃縮された混合物の使用に一部起因して、本発明によって高い収率が得られる。
【0018】
特に明示しない限り、「レボフロキサシン」および「レボフロキサシン形態」という用語は、レボフロキサシンの塩、水和物、および溶媒和物を包含する。この用語は、レボフロキサシンの多形形態がレボフロキサシンの塩、水和物、または溶媒和物でないと考えられる範囲でレボフロキサシンの全ての多形形態も包含する。
【0019】
本発明の1つの態様は、(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸(「化合物I」)をN−メチルピペラジンと極性溶媒中で、好ましくは上昇した温度で、反応させて、レボフロキサシンを生成させることを含んでなる、レボフロキサシンの調製方法を提供する。前記上昇した温度は、好ましくは約70〜120℃である。該レボフロキサシンは、次に、当該技術分野でよく知られた技法を使用して沈殿および回収することができる。本願明細書で用いられる「沈殿」という用語は、溶液中の固体の形成またはスラリー中の固体量の増大を包含する。化合物Iの調製については、例えば、米国特許第4,382,892号に記載されており、参照により本願明細書に取り込まれる。
【0020】
適した極性溶媒は、レボフロキサシンを溶解可能ないずれの極性溶媒でもよい。好ましくは、極性溶媒はジメチルスルホキシド(DMSO)、アルコール(好ましくはイソブタノール)、ケトン、プロピレン・グリコール・モノメチル・エーテル(PGME)またはジメチルアセトアミド(DMA)である。本願明細書で用いられる「極性溶媒」という用語は、他の溶媒よりも比較的に極性の高いことを意味する相対的用語であることが意図される。
【0021】
1つの実施態様において、溶媒の体積は化合物Iのグラム当たり約14ml〜約4mlである。この実施態様において、溶媒は好ましくはイソブタノールおよびPGMEからなる群より選択される。別の実施態様において、溶媒の体積は化合物Iのグラム当たり約3mlよりも少ない。この後者の実施態様において、溶媒は好ましくはDMSOおよびDMAからなる群より選択される。有利なのは、DMSOを用いる場合に高収率を得るために必要とされる反応時間が短いことである。
【0022】
溶媒としてDMSOを用いる場合に、好ましい溶媒の体積は約3ml溶媒/g化合物までであり、好ましくは約0.5〜約3ml溶媒/g化合物であるが、それより多くの量も使用し得る。溶媒としてPGMEを用いる場合に。好ましい溶媒の体積は約4〜約14ml溶媒/g化合物であるが、それより多くの量も使用し得る。
【0023】
場合により、本発明に係る方法は、反応工程後に前記溶媒に貧溶媒を添加して収率を増大させることを更に含んでよい。本願明細書で用いられる「貧溶媒」(anti-solvent)という用語は、その中でレボフロキサシンが低溶解性である液体であって貧溶媒を溶媒に添加することによってレボフロキサシンの溶解度を低下させるようにするものである。好ましくは、貧溶媒は以下の1種以上のものである:n−ヘプタン、ヘキサン、イソプロピルアルコール、水中のイソプロピルアルコール(約5%以上のイソプロピルアルコール)、ブタノール、アセトニトリル、メチルエチルケトン、またはDMSO/水。溶媒がプロピレン・グリコール・モノメチル・エーテルまたはイソブタノールである場合に、好ましい貧溶媒はヘプタンまたはヘキサンである。溶媒がDMSOである場合には、好ましい貧溶媒はイソプロパノールである。
【0024】
他の実施態様において、化合物IはN−メチルピペラジンとニート混合物として反応する。この実施態様において、化合物Iは好ましくはN−メチルピペラジンの懸濁液に溶解される。
【0025】
1つの実施態様において、N−メチルピペラジンは化合物Iよりもモル過剰である。好ましくは、モル過剰は約2倍〜約4倍である。より好ましくは、モル過剰は約2倍〜約2.5倍である。
【0026】
好ましい反応工程の時間は、効率の最大化ならびに/あるいは副反応および/または分解の最小化を図りながら、反応を完了させるという要望を釣り合わせること、つまり、反応条件、特に溶媒の選択と温度に依存することになる。例えば、反応工程は、溶媒が用いられる場合に、約1時間〜約24時間の時間で一般に行われる。反応がニート混合物として行われる場合に、反応の時間は1時間よりも短くてよい。
【0027】
反応工程は、約110℃〜約120℃程の温度またはそれよりも高い温度で行ってよい。反応がニート混合物として行われる場合に、反応工程はほぼ還流温度で行われるのが好ましい。
【0028】
レボフロキサシン形態の調製
本発明の他の実施態様において、レボフロキサシン形態の調製方法は、レボフロキサシンを、好ましくは第1溶媒中で、第1の上昇した温度で保持すること;極性溶媒を添加すること;およびレボフロキサシン形態を回収することを含む。好ましくは、この方法は、レボフロキサシン−溶媒混合物を周囲温度よりも低い温度に冷却および保持することを更に含む。
【0029】
前記第1溶媒は、レボフロキサシンを溶解可能であって、好ましくは比較的高い沸点を有する極性溶媒である。例としては、PGME、DMAおよびDMSOが挙げられる。レボフロキサシンは、第1の上昇した温度に加熱してよい。しかしながら、上述のように、化合物IをN−メチルピペラジンと反応させる等により、最初にレボフロキサシンを合成し、その反応混合物を次に、極性溶媒を添加するために適した第2の上昇した温度に直接もって行くことが好ましい。第1の上昇した温度は、特定の溶媒に依存するが、一般には約70℃〜約120℃の範囲、好ましくは約80℃〜約85℃にある。第2の上昇した温度は、特定の極性溶媒に依存するが、一般には約60℃〜約80℃の範囲、好ましくは約75℃〜約79℃にある。
【0030】
極性溶媒は、第2の上昇した温度で、レボフロキサシンに、好ましくはゆっくりと添加される。極性溶媒は、好ましくは、約2時間にわたり添加される。場合により、更なる時間、好ましくは攪拌または他の掻き混ぜを行いながら、混合物が保持される。
【0031】
レボフロキサシンを回収することは、一般に、混合物を冷却してレボフロキサシンを沈殿させた後に濾過を行うことを包含する。
【0032】
レボフロキサシン半水和物の調製のためには、極性溶媒は、水を含み、好ましくはイソプロパノールと水、より好ましくは約3%〜約4%(v/v)の水、の混合物を含む。この実施態様において、第1の上昇した温度は約80℃であることが好ましく、第2の上昇した温度は約75℃であることが好ましい。好ましくは、極性溶媒が約2時間にわたり滴下により添加される。添加工程の後に、レボフロキサシン−極性溶媒混合物は周囲温度よりも低い温度、好ましくは約0℃〜約20℃の範囲、より好ましくは約5℃にゆっくりと冷却される。好ましくは、冷却工程は約1〜約10時間、より好ましくは約4時間にわたり行われる。好ましくは、前記周囲温度よりも低い温度が更に約2時間保持される。
【0033】
形態Cの調製のためには、極性溶媒は好ましくはイソプロパノールを含む。この実施態様において、第1の上昇した温度は約85℃であることが好ましく、第2の上昇した温度は約79℃であることが好ましい。好ましくは、極性溶媒が約2時間にわたり滴下により添加された後に、この温度で更に約2時間保持される。
【0034】
形態Aの調製のためには、極性溶媒は好ましくはイソプロパノールを含む。この実施態様において、第1の上昇した温度は約80℃であることが好ましく、第2の上昇した温度は約75℃であることが好ましい。添加工程の後に、レボフロキサシン−極性溶媒混合物は周囲温度よりも低い温度、好ましくは約0℃〜約20℃の範囲、より好ましくは約5℃にゆっくりと冷却される。好ましくは、冷却工程は約1〜約10時間、より好ましくは約4時間にわたり行われる。好ましくは、前記周囲温度よりも低い温度が更に約2時間保持される。
【0035】
形態Gおよび形態Bの調製のためには、極性溶媒は好ましくはイソプロパノールを含む。この実施態様において、第1の上昇した温度は約80℃であることが好ましく、第2の上昇した温度は約75℃であることが好ましい。添加工程の後に、レボフロキサシン−極性溶媒混合物は周囲温度よりも低い温度、好ましくは約0℃〜約20℃の範囲、より好ましくは約5℃にゆっくりと冷却される。好ましくは、冷却工程は約1〜約10時間、より好ましくは約4時間にわたり行われる。好ましくは、前記周囲温度よりも低い温度が更に約11時間保持される。好ましくは、沈殿が濾過され、次に乾燥される。形態Gを得るためには、第3の上昇した温度で約3〜約6時間および第4の上昇した温度で約3時間乾燥が行われる。第3の上昇した温度は約40℃であることが好ましく、第4の上昇した温度は約60℃であることが好ましい。形態Bを得るためには、第3の上昇した温度で少なくとも約20時間、好ましくは約21時間、および第4の上昇した温度で少なくとも約6時間、乾燥が行われる。第3の上昇した温度は約40℃であることが好ましく、第4の上昇した温度は約60℃であることが好ましい。
【0036】
形態Hの調製のためには、極性溶媒は、イソプロパノールと水、より好ましくは約0.3%〜約0.4%(v/v)の水、の混合物を含む。この実施態様において、第1の上昇した温度は約80℃であることが好ましく、第2の上昇した温度は約75℃であることが好ましい。好ましくは、極性溶媒が約1時間にわたり滴下により添加される。添加工程の後に、レボフロキサシン−極性溶媒混合物は、好ましくは、第2の上昇した温度で約2時間保持される。次に、その混合物が周囲温度よりも低い温度、好ましくは約0℃〜約20℃の範囲、より好ましくは約5℃にゆっくりと冷却される。好ましくは、冷却工程は約1〜約10時間、より好ましくは約4時間にわたり行われる。好ましくは、前記周囲温度よりも低い温度が更に約12時間保持される。
【0037】
本発明の他の実施態様において、レボフロキサシン形態の調製方法は、レボフロキサシンと極性溶媒の第1混合物を第1の上昇した温度で約4時間以上、好ましくは少なくとも4.5時間、保持すること、該第1混合物を第2の上昇した温度に冷却すること、および該冷却された第1混合物に追加の極性溶媒を添加して、第2混合物を形成すること、該第2混合物をレボフロキサシンが完全に溶解されるまで第3の上昇した温度で保持すること、場合により、前記保持工程中に該第2混合物に追加の極性溶媒を添加すること、該第2混合物を冷却して、レボフロキサシン形態を形成すること、および該レボフロキサシン形態を回収することを含んでなる。
【0038】
形態Fの調製のためには、極性溶媒はイソブチルアルコールを含む。この実施態様において、第1および第3の上昇した温度は還流温度であることが好ましく、第2の上昇した温度は約80℃であることが好ましい。好ましくは、レボフロキサシンを完全に溶解させるために十分な最少量の極性溶媒が第2混合物に添加される。第2混合物は周囲温度よりも低い温度、好ましくは約−5℃〜約20℃の範囲、より好ましくは約5℃にゆっくりと冷却される。好ましくは、冷却工程は約1〜約10時間、より好ましくは約1.5時間にわたり行われる。
【0039】
本発明の他の実施態様は、治療的に有効量の形態A、形態B、形態C、形態F、形態G、形態Hまたはこれらの組合せ、および場合により、薬学的に許容される担体を含んでなる。
【0040】
本発明の他の実施態様は、形態A、形態B、形態C、形態F、形態Gおよび形態Hからなる群より選択される1種以上の形態を、該1種以上の形態が大気中の水分(atomospheric water)の吸収によりレボフロキサシン半水和物に転化するために十分な時間の間、貯蔵することを含んでなる、レボフロキサシン半水和物の調製方法である。例えば、形態A、形態B、形態C、形態F、形態Gおよび形態Hは、室温(RT)で閉鎖された瓶内で7〜29日の貯蔵後に半水和物に転化する。該サンプルを開放された瓶内で保持する場合に、半水和物への転化は比較的速い(約24時間)。
【0041】
本発明の他の実施態様は、レボフロキサシン形態の調製方法である。(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸がN−メチルピペラジンと上昇した温度で反応されて、レボフロキサシンを生成する。該レボフロキサシンは次に、第1溶媒中で、第1の上昇した温度で、沈殿および保持される。極性溶媒が添加されて、レボフロキサシン形態C、形態A、形態G、形態B、形態H、形態Fまたは半水和物形態が沈殿し、これを次に濾過等の公知の手段により回収することができる。
【0042】
レボフロキサシン形態の物理特性
【表1】
【0043】
DTGサーモグラムを、Shimadzu DTG−50 ,加熱速度:10℃/分で行った。融点は、DTA曲線中の吸熱ピークにより、全ての議論される結晶形態について約225〜230℃であることが決定された。DTA曲線とTGA曲線における大きな差が160℃までの温度範囲で観測された。
【0044】
レボフロキサシン形態の熱分析
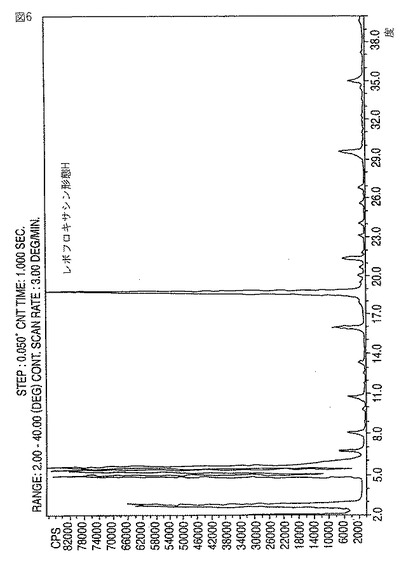
レボフロキサシン新規形態Aは、約100℃の吸熱ピークにより特徴付けられる。結晶からの溶媒の除去に起因して、この温度範囲で約18〜22%の重量損失段(weight loss step)が観測される。図7参照。
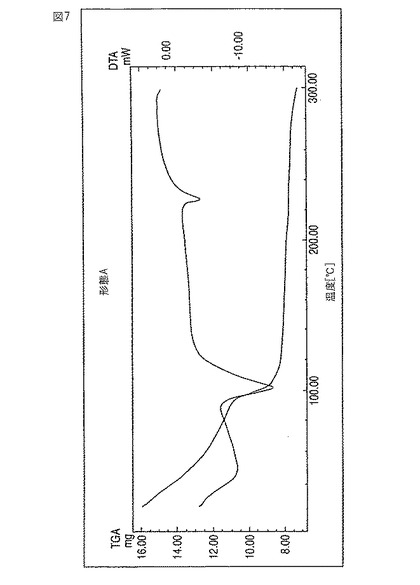
レボフロキサシン新規形態Cは、高DMSO含有量(30〜50%)およびKFによる約3.5%の含水量により特徴付けられる。図8参照。
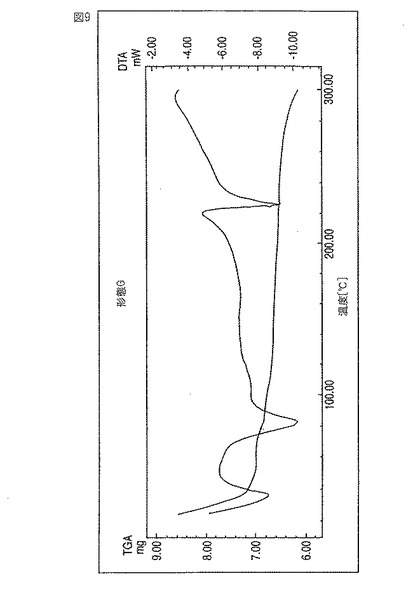
レボフロキサシン新規形態Gは、約82℃および約103℃の吸熱ピークにより特徴付けられる。この温度範囲で3〜6%の重量損失段が観測される。この重量損失値は該サンプル中のDMSO含有量と一致する。図9参照。
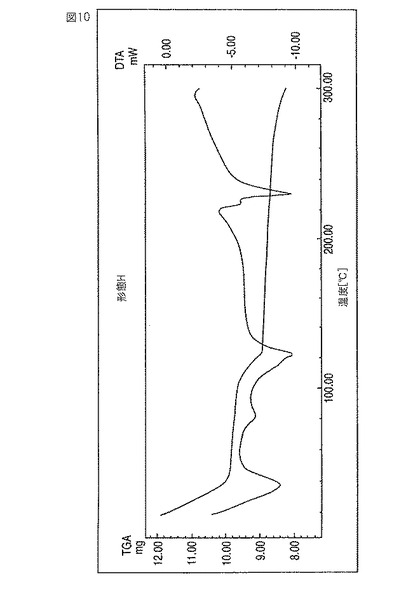
レボフロキサシン新規形態Hは、約122℃の吸熱ピークにより特徴付けられる。60〜150℃の温度範囲で約8%の重量損失段が観測される。この重量損失率は、1:0.5の比のレボフロキサシン:IPA溶媒和物に相当する期待値(これは7.7%である)に等しい。図10参照。
【0045】
本発明のこれらの及び他の実施態様の機能及び利点は、下の実施例からより良く理解されるであろう。以下の実施例は、本発明の全範囲を例証するのではなく、本発明の利益を例示することを意図するものである。
【実施例】
【0046】
例1:DMSO中でのレボフロキサシンの合成
5g(17.8ミリモル)の(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸を2.5mLのDMSOおよび4.2mL(37.9ミリモル)のN−メチルピペラジン中に懸濁させた。その反応混合物を120℃に加熱し、懸濁液が溶解するようになった。2.5時間後に反応が完了した。次に、その混合物を70℃に冷却し、次に、この温度でイソプロピルアルコール(25mL)を添加した。その反応混合物を周囲温度で1時間スラリー化し、濾過し、終夜乾燥して、5.86g(91.3%)のレボフロキサシンを得た。
【0047】
例2:PGME中でのレボフロキサシンの合成
3g(10.67ミリモル)の(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸を30mLのPGMEおよび4.75mL(43ミリモル)のN−メチルピペラジン中に懸濁させた。その反応混合物を加熱して反応完了まで23時間還流した。その間に反応混合物が溶解するようになった。次に、その混合物を90℃に冷却し、この温度でn−ヘプタン(10mL)を添加した。次に、その反応混合物を0℃に冷却し、沈殿が65℃付近で生じた。その反応混合物を0℃で3時間置き、減圧下で濾過し、終夜乾燥して、2.98g(77.3%)のレボフロキサシンを得た。
【0048】
例3:イソブタノール中でのレボフロキサシンの合成
3g(10.67ミリモル)の(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸を21mLのイソブタノールおよび4.75mL(43ミリモル)のN−メチルピペラジン中に懸濁させた。その反応混合物を加熱して6時間還流し、次に周囲温度で60時間スラリー化し、再び加熱して反応完了まで7時間還流した。その間に反応混合物が還流温度で溶解するようになった。次に、その混合物を0℃に冷却し、減圧下で濾過し、7mLのイソブタノールおよび10mLのn−ヘプタンで洗浄し、終夜乾燥して、2.83g(77.3%)のレボフロキサシンを得た。
【0049】
例4:レボフロキサシンの合成(ニート)
5g(17.79ミリモル)の(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸を6.8mL(0.06モル)のN−メチルピペラジン中に懸濁させた。その反応混合物を加熱して反応完了まで40分間還流した。次に、その混合物を80℃に冷却した。この温度でIPA(10mL)およびn−ヘプタン(10mL)を添加した。その固体を減圧下で濾過し、n−ヘプタンで濯いだ。n−ヘプタンの添加後に、母液が更に沈殿を与えた。両方の沈殿を減圧下で濾過し、終夜乾燥して、4.9g(76%)のレボフロキサシンを得た。
【0050】
例5:DMA中でのレボフロキサシンの合成
10g(35.6ミリモル)の(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸を5mLのDMA(ジメチルアセトアミド)および8.3mL(75ミリモル)のN−メチルピペラジン中に懸濁させた。その反応混合物を出発物質が完全に転化されるまで約1.5時間110℃に加熱した。次に、その反応混合物を80℃に冷却し、60mlのイソプロピルアルコールを添加した。次に、その反応混合物を周囲温度で3時間スラリー化し、減圧下で濾過し、40mlのIPAで洗浄し、真空オーブン内で終夜乾燥して、11.48g(89.3%)のレボフロキサシンを得た。
【0051】
例6:半水和物の合成
メカニカルスターラー、凝縮器および温度計を備え、80℃に加熱された1リットルの反応器に、87.5g(0.31モル)の(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸、61.3mLのDMSOおよび86.3mL(0.77モル)のN−メチルピペラジンを入れた。そのスラリーを80℃で窒素雰囲気の下で250rpmの速度で(HPLCによりモニタリングして)反応完了まで攪拌した。次に、そのスラリーを75℃に冷却し、この温度でイソプロパノール(675mL)と水(25mL)の混合物を2時間にわたり滴下により添加した。次に、そのスラリーを4時間にわたり5℃に冷却し、この温度で2時間保持し、この温度で減圧下に濾過した。次に、その固体を175mLのイソプロパノールで洗浄(2回リンス)し、減圧下で乾燥して、レボフロキサシン半水和物を得た。
【0052】
例7:形態Cの合成
調製
メカニカルスターラー、凝縮器および温度計を備え、85℃に加熱された1リットルの反応器に、(S)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸(87.5g)、DMSO(61.3ml)およびN−メチルピペラジン(86.3ml)を入れた。そのスラリーを窒素雰囲気の下で250rpmの速度で攪拌した。加熱を(HPLCによりモニタリングして)反応完了まで4.5時間続けた。次に、そのスラリーを79℃に冷却し、この温度でイソプロパノール(700ml)を2時間にわたり滴下により添加し、この温度で更に2時間攪拌した。その終了時に、サンプルを減圧下で濾過し、イソプロパノールで洗浄した。
【0053】
例8:形態Aの合成
調製
メカニカルスターラー、凝縮器および温度計を備え、80℃に加熱された1リットルの反応器に、(S)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸(87.5g)、DMSO(61.3ml)およびN−メチルピペラジン(86.3ml)を入れた。そのスラリーを空気雰囲気の下で250rpmの速度で攪拌した。加熱を(HPLCによりモニタリングして)反応完了まで4.5時間続けた。次に、そのスラリーを75℃に冷却し、この温度でイソプロパノール(700ml)を2時間にわたり滴下により添加した。サンプルをXRD分析にかけた。混合物をこの温度で攪拌しながら2時間保持した。次に、その反応混合物を5℃まで4時間にわたり冷却し、この温度で攪拌しながら2時間保持した。その終了時に、反応混合物を減圧下で濾過し、イソプロパノール(175ml)で洗浄して、171gの湿った物質(106.8gの乾燥物質、92.7%)を得た。
【0054】
例9:形態Gおよび形態Bの合成
調製
メカニカルスターラー、凝縮器および温度計を備え、80℃に加熱された1リットルの反応器に、(S)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸(87.5g)、DMSO(61.3ml)およびN−メチルピペラジン(86.3ml)を入れた。そのスラリーを窒素雰囲気の下で250rpmの速度で攪拌した。加熱を反応完了まで4.5時間続けた。次に、そのスラリーを75℃に冷却し、この温度でイソプロパノール(700ml)を2時間にわたり滴下により添加した。
添加終了時に、攪拌を75℃で2時間保持し、次に、5℃まで4時間にわたり冷却し、この温度で攪拌しながら11時間保持した。そのスラリーを減圧下で濾過し、イソプロパノール(175ml)で洗浄して、149gの湿った物質を得た。
その湿った物質を乾燥のために2つの部分に分けた。第1部分は減圧下で攪拌しながら40℃で21時間乾燥し、第2画分は減圧下で攪拌しながら60℃で21時間乾燥した。40℃で3時間または6時間乾燥後そして60℃で3時間乾燥後にレボフロキサシン形態Gが生じた。40℃で21時間乾燥後そして60℃で6時間、9時間および21時間乾燥後にレボフロキサシン形態Bが生じた。
【0055】
例10:形態Hの合成
調製
メカニカルスターラー、凝縮器および温度計を備え、80℃に加熱された1リットルの反応器に、(S)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸(87.5g)、DMSO(61.3ml)およびN−メチルピペラジン(86.3ml)を入れた。そのスラリーを窒素雰囲気の下で250rpmの速度で攪拌した。加熱を(HPLCによりモニタリングして)反応完了まで4.5時間続けた。次に、そのスラリー混合物を75℃に冷却し、この温度で H2O(2.5ml)と混合されたイソプロパノール(697.5ml)を1時間にわたり滴下により添加した。添加終了時に、攪拌を2時間保持し、次に、5℃まで4時間にわたり冷却した。この温度で攪拌を12時間保持した。次に、その反応混合物を減圧下で濾過し、イソプロパノール(175ml)で洗浄して、150gの湿った物質を得た。
【0056】
例11:形態Fの合成
調製
凝縮器を備えたフラスコに、4.0gのレボフロキサシンを入れた。イソブチルアルコール(8mL)を添加し、その混合物を還流温度に加熱した。15分後、その混合物を80℃に冷却し、4mLのイソブチルアルコールを添加した。次に、その混合物を再び還流温度に加熱した。イソブチルアルコール(6mL)を完全に溶解するまで添加した。溶液が透明になり、その混合物を1.5時間で0℃に冷却した。次に、その沈殿を減圧下で濾過し、イソブチルアルコール(4mL)で洗浄し、減圧下で60℃において乾燥した。
【0057】
例12:形態の時間により誘導される転化
メカニカルスターラー、凝縮器および温度計を備えた1リットルの反応器に、(S)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸(87.5g)、DMSO(61.3ml)、N−メチルピペラジン(86.3ml)および H2O(0.44ml)を入れた。次に、そのスラリーを80℃に加熱し、窒素雰囲気の下で250rpmの速度で攪拌した。加熱を反応完了まで4.5時間続けた。次に、そのスラリー混合物を75℃に冷却し、この温度でイソプロパノール(700ml)を2時間にわたり滴下により添加した。
次に、その混合物を攪拌しながら75℃で2時間保持した。次に、その混合物を5℃まで4時間にわたり冷却し、この温度で混合物を攪拌しながら9時間保持した。そのスラリーを減圧下で濾過し、イソプロパノール(175ml)で洗浄して、147gの湿った多形形態Hを得た。
その湿った物質を(保護のため)紙で覆って室温(RT)で空気に曝した。3時間、6時間、9時間後、その湿ったサンプルの多形は形態Gであった。24時間後、その湿ったサンプルは半水和物の多形形態であった。
【0058】
例13:スラリー中の形態Hの転化
メカニカルスターラー、凝縮器および温度計を備えた1リットルの反応器を80℃に加熱し、(S)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸(87.5g)、DMSO(61.3ml)およびN−メチルピペラジン(86.3ml)を入れた。そのスラリーを窒素雰囲気の下で250rpmの速度で攪拌した。加熱を(HPLCによりモニタリングして)反応完了まで4.5時間続けた。次に、そのスラリー混合物を75℃に冷却し、この温度で H2O(2.5ml)と混合されたイソプロパノール(697.5ml)を1時間にわたり滴下により添加した。
次に、その混合物を攪拌しながら2時間保持した。次に、その混合物を5℃まで4時間にわたり冷却し、この温度で混合物を12時間保持した。次に、その反応混合物を減圧下で濾過し、イソプロパノール(175ml)で洗浄して、150gの湿った多形形態Hを得た。
その湿った物質の第1部分を5vのアセトニトリル中で75℃で2時間攪拌し、次に減圧下で濾過した。その湿ったサンプルの多形は半水和物であった。その湿った物質の第2部分を(保護のため)紙で覆って室温(RT)で空気に曝した。数時間後、その湿ったサンプルの多形は半水和物であった。
【0059】
例14:スラリー中の形態Gの転化
メカニカルスターラー、凝縮器および温度計を備えた1リットルの反応器に、(S)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸(87.5g)、DMSO(61.3ml)およびN−メチルピペラジン(86.3ml)を周囲温度で入れた。次に、そのスラリーを80℃に加熱し、窒素雰囲気の下で250rpmの速度で攪拌した。加熱を反応完了まで4時間続けた。次に、そのスラリー混合物を75℃に冷却し、この温度で H2O(21ml)と混合されたイソプロパノール(679ml)を2時間にわたり滴下により添加した。
次に、その混合物を攪拌しながら75℃で2時間保持した。次に、その混合物を5℃まで4時間にわたり冷却し、この温度で混合物を攪拌しながら10時間保持した。そのスラリーを減圧下で濾過し、イソプロパノール(175ml)で洗浄して、166.5gの湿った多形形態Gを得た。
その湿った物質(162g)をアセトニトリル(486ml)と混合した。そのスラリーを250rpmの速度で27℃において1時間攪拌した。次に、そのスラリーを減圧下で濾過し、アセトニトリル(162ml)で洗浄した。その湿ったサンプルは半水和物の多形形態であった。その固体を真空オーブン内で40℃において6時間乾燥した。その乾燥した粗レボフロキサシンは半水和物の多形形態であった。
【0060】
例15:乾燥
メカニカルスターラー、凝縮器および温度計を備えた1リットルの反応器に、(S)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸(87.5g)、DMSO(61.3ml)およびN−メチルピペラジン(86.3ml)を周囲温度で入れた。次に、そのスラリーを80℃に加熱し、窒素雰囲気の下で250rpmの速度で攪拌した。加熱を反応完了まで4.5時間続けた。次に、そのスラリー混合物を75℃に冷却し、イソプロパノール(700ml)を一度に添加した。添加終了時に、その混合物を300rpmの速度で攪拌した。その反応混合物を7℃まで2時間にわたり冷却した。この温度において350rpmの速度で2時間攪拌を保持した。次に、その反応混合物を減圧下で濾過し、イソプロパノール(175ml)で洗浄した。
その湿った物質(162g)をイソプロパノール(180ml)と混合した。そのスラリーを250rpmの速度で40℃において1時間攪拌した。次に、そのスラリーを減圧下で濾過し、イソプロパノール(60ml)で洗浄した。その湿ったサンプルの多形形態はHであった。
その固体を真空オーブン内で60℃において14時間乾燥して、106g(92%)の乾燥レボフロキサシン形態Bを得た。
【0061】
例16:乾燥
メカニカルスターラー、凝縮器および温度計を備えた1リットルの反応器に、(S)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸(87.5g)、DMSO(61.3ml)およびN−メチルピペラジン(86.3ml)を周囲温度で入れた。次に、そのスラリーを80℃に加熱し、窒素雰囲気の下で250rpmの速度で攪拌した。加熱を反応完了まで4.5時間続けた。次に、そのスラリー混合物を75℃に冷却し、イソプロパノール(700ml)を一度に添加した。添加終了時に、その混合物を300rpmの速度で攪拌した。その反応混合物を7℃まで2時間にわたり冷却した。この温度において350rpmの速度で2時間攪拌を保持した。次に、その反応混合物を減圧下で濾過し、イソプロパノール(175ml)で洗浄した。
その湿った物質(162g)をイソプロパノール(180ml)と混合した。そのスラリーを250rpmの速度で40℃において1時間攪拌した。次に、そのスラリーを減圧下で濾過し、イソプロパノール(60ml)で洗浄した。その湿ったサンプルの多形形態はGであった。
その固体を真空オーブン内で60℃において14時間乾燥して、106g(92%)の乾燥レボフロキサシン形態Bを得た。
【0062】
例17:n−BuOHを用いた半水和物の調製
1gの粗レボフロキサシンを7mlのn−BuOHに懸濁させた。その混合物をその物質が完全に溶解するまで還流温度に加熱した。次に、その溶液を2.5時間の時間にわたり室温(RT)に冷却した。その沈殿を減圧下で濾過し、n−BuOHで洗浄し、真空オーブン内で60℃において乾燥して、810mg(81%)の精製レボフロキサシン半水和物を得た。
【0063】
例18:ACNを用いた半水和物の調製
1.5gの粗レボフロキサシンを10.5mlのACNに懸濁させた。その混合物をその物質が完全に溶解するまで還流温度に加熱した。次に、その溶液を20分の時間にわたり0℃に冷却した。その沈殿を減圧下で濾過し、ACN(1.5ml)で洗浄し、真空オーブン内で30℃において乾燥して、1.15g(77%)の精製レボフロキサシン(半水和物/一水和物混合物)を得た。精製レボフロキサシンは粗サンプル中の量のほぼ半分の量のデスメチル・レボフロキサシンを含有した。
【0064】
例19:DMSO/水を用いた半水和物の調製
1gの粗レボフロキサシンを1.5mlのDMSOに懸濁させた。その混合物をその物質が完全に溶解するまで108℃に加熱した。次に、 H2O(7.5ml)を10分間にわたり添加し、その混合物を室温(RT)に冷却した。その沈殿を減圧下で濾過し、1mlのDMSO:H2O 1:5混合物で洗浄し、気流オーブン内で60℃において乾燥して、840mg(84%)の精製レボフロキサシン半水和物を得た。
【0065】
例20:MEKを用いた半水和物の調製
1.5gの粗レボフロキサシンを15mlのMEKに懸濁させた。その混合物をその物質が完全に溶解するまで還流温度に加熱した。次に、その溶液を3時間の時間にわたり−5℃に冷却した。その沈殿を減圧下で濾過し、1.5mlのMEKで洗浄し、真空オーブン内で30℃において乾燥して、840mg(84%)の精製レボフロキサシン半水和物を得た。
【図面の簡単な説明】
【0066】
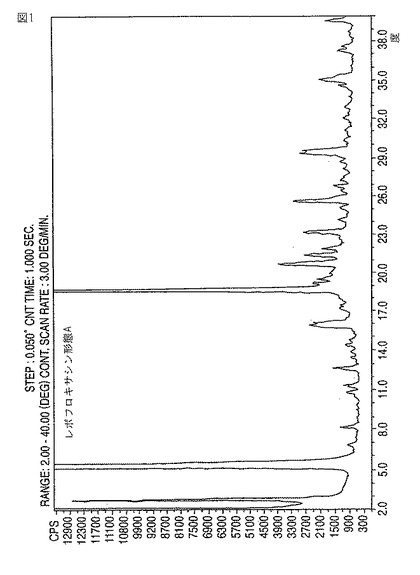
【図1】新規レボフロキサシン結晶形態AのXRDディフラクトグラムである。
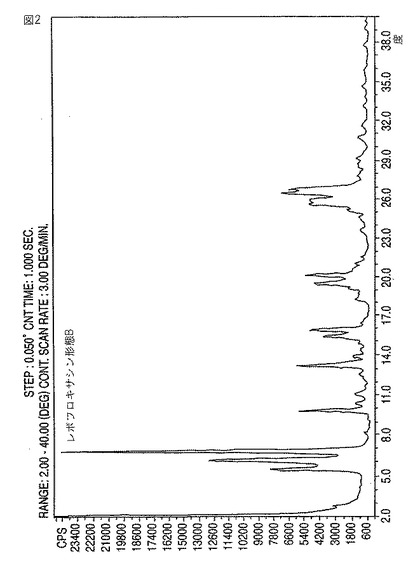
【図2】新規レボフロキサシン結晶形態BのXRDディフラクトグラムである。
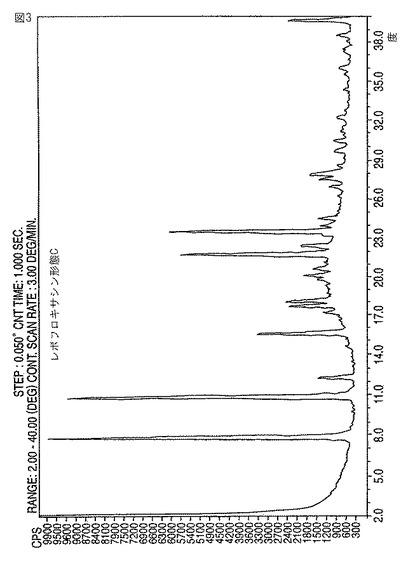
【図3】新規レボフロキサシン結晶形態CのXRDディフラクトグラムである。
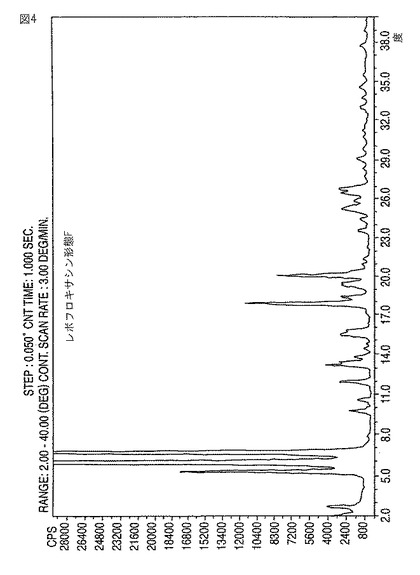
【図4】新規レボフロキサシン結晶形態FのXRDディフラクトグラムである。
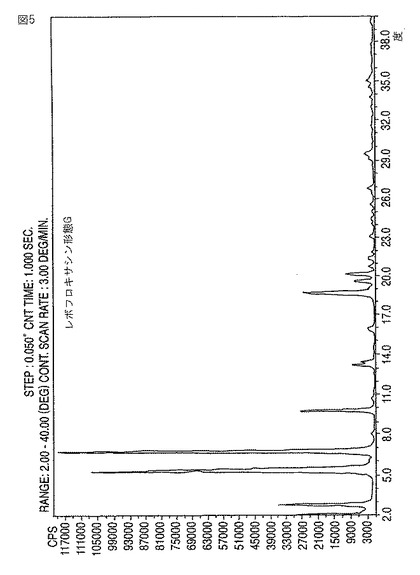
【図5】新規レボフロキサシン結晶形態GのXRDディフラクトグラムである。
【図6】新規レボフロキサシン結晶形態HのXRDディフラクトグラムである。
【図7】新規レボフロキサシン結晶形態AのDTGサーモグラムである。
【図8】新規レボフロキサシン結晶形態CのDTGサーモグラムである。
【図9】新規レボフロキサシン結晶形態GのDTGサーモグラムである。
【図10】新規レボフロキサシン結晶形態HのDTGサーモグラムである。
【技術分野】
【0001】
本願は2001年10月3日に提出された仮出願整理番号第60/326,958号、2001年11月29日に提出された同第60/334,316号および2002年2月11日に提出された同第60/354,939号ならびに本願と同時に提出された非仮出願整理番号(未定)[代理人整理番号1662/58004号]の優先権を主張する。これらの各出願の全内容が参照により本願明細書に取り込まれる。
【0002】
本発明はレボフロキサシンおよびその新規な形態の調製方法に関する。
【背景技術】
【0003】
レボフロキサシンは広スペクトルの合成抗生物質である。レボフロキサシンは、フルオロキノロン抗微生物剤であるオフロキサシン・ラセミ体のS−エナンチオマーである。オフロキサシンの抗菌活性は主にS−エナンチオマーにある。レボフロキサシンおよび他のフルオロキノロン抗微生物剤の作用機構は、DNA複製、転写修復および組換えに必要な酵素であるDNAジャイレース(細菌型トポイソメラーゼII)の阻害を含む。レボフロキサシンは、経口投与または静脈内投与し得るLEVAQUIN(登録商標)として利用可能である。
【0004】
レボフロキサシンはキラルなフッ素化カルボキシキノロンである。その化学名は(S)−9−フルオロ−2,3−ジヒドロ−3−メチル−10−(4−メチル−1−ピペラジニル)−7−オキソ−7H−ピリド[1,2,3−de]−1,4−ベンズオキサジン−6−カルボン酸(CAS登録番号第100986−85−4号)である。レボフロキサシンの化学構造を次式Iに示す。
【化1】
【0005】
米国特許第4,382,892号はピリド[1,2,3−de][1,4]ベンズオキサジン誘導体およびその調製方法に関する。
【0006】
米国特許第5,053,407号は光学活性ピリドベンズオキサジン誘導体、該誘導体の調製方法および該誘導体の調製に有用な中間体に関する。
【0007】
米国特許第5,051,505号はピペラジニルキノロン誘導体の調製方法に関する。この方法はアセトニトリル、ジメチルホルムアミド、ピリジン、スルホランおよびジメチルスルホキシドのような極性溶媒の存在下でジハロキノロンをピペラジン誘導体およびハロゲン化テトラアルキルアンモニウムと反応させることを含む。
【0008】
米国特許第5,155,223号はキノリンカルボン酸の調製に関する。
【0009】
米国特許第5,545,737号は、結晶化時にレボフロキサシンが溶解される水性溶媒の含水量を調節することによりレボフロキサシン半水和物または一水和物を選択的に生成することを開示している。
【0010】
レボフロキサシン形態
レボフロキサシンの3種の多形形態(polymorphic form)(無水α、β、γ)および2種の偽多形形態(pseudepolymorphic form)(半水和物および一水和物)が文献に言及されている。半水和物および一水和物形態は EP 0444 678 B1および米国特許第5,545,737号で言及されている。これら2件の特許は、一水和物を含まない半水和物の調製方法および半水和物を含まない一水和物の調製方法に関する。
【0011】
"Effect of dehydration on the formation of Lovofloxacine Pseudepolymorphs (レボフロキサシン偽多形の生成に対する脱水の効果)" と題された論文Chem. Pharm. Bull. 43(4) 649-653 (1995) はレボフロキサシンの水和物形態の物性を検討している。この論文によれば、半水和物形態を加熱することによって、水和水が除去されて無水形態γが得られる。更に加熱することによって、無水形態βが生成し、次に無水形態αが生成する。一水和物形態を加熱することによって、水和水が除去されて無水形態αが得られる。形態γおよび形態αは通常の相対湿度の下で急速に水蒸気を吸収し、それぞれ半水和物および一水和物に変換される。
【0012】
本発明はレボフロキサシンの固体物性に関する。固体物性としては、例えば粉砕された固体の流動性がある。流動性は、医薬品の製造時に該物質を取り扱う容易さに影響する。パウダー状化合物の粒子が相互に容易に流動しない場合に、配合専門家はタブレットまたはカプセル配合物の開発を考慮しなければならず、コロイド状二酸化ケイ素、タルク、スターチまたは三塩基性リン酸カルシウム等の流動促進剤(glidant)の使用を必要とする場合がある。
【0013】
医薬化合物の他の重要な固体物性は、水性流体中でのその溶解速度である。患者の胃液中での活性成分の溶解速度は、経口投与される活性成分が患者の血流に到達することができる速度に対する上限となるために、治療効果を有することができる。溶解速度はまた、シロップ剤、エリキシル剤および他の液状医薬品の配合における考慮事項でもある。化合物の固体形態は、圧蜜化とその貯蔵安定性に対する化合物の挙動にも影響する。
【0014】
これらの実際の物理特性は、物質の特定の多形形態を規定する単位胞中の分子の立体配座と配向によって影響を受ける。多形形態は、非晶質物質や他の多形形態の熱的挙動とは異なる熱的挙動を示し得る。熱的挙動は研究室においてキャピラリー融点、熱重量分析(TGA)、示差走査熱量測定法(DSC)等のような技法により測定され、これを用いてある多形形態を他の多形形態と区別することができる。特定の多形形態は、粉末X線結晶学、固体状態C−NMR分光測定および赤外分光測定によって検出可能な明確な分光的性質も示し得る。
【発明の開示】
【0015】
1つの態様において、本発明は、(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸(「化合物I」)をN−メチルピペラジンと反応させて、レボフロキサシンを形成すること、および該レボフロキサシンを回収することを含んでなる、レボフロキサシンの調製方法を提供する。化合物IはN−メチルピペラジンと、極性溶媒中で又はニート(neat:そのままの)混合物として反応させてよい。
【0016】
本発明の他の態様において、新規な結晶形態A、B、C、F、GおよびH、ならびにそれらの調製方法が記載される。1つの実施態様において、レボフロキサシンの調製方法は、レボフロキサシンを上昇した温度で保持すること、極性溶媒を添加すること、およびレボフロキサシン形態を回収することを含んでなる。好ましくは、この方法は、レボフロキサシン−溶媒混合物を周囲温度よりも低い温度に冷却および保持することを更に含む。
【発明を実施するための最良の形態】
【0017】
本発明の1つの態様は、レボフロキサシンの調製方法を提供する。幾つかの実施態様において、約70%〜約85%またはそれ以上の精製レボフロキサシンの収率が達成される。本願明細書で用いられる「粗」および「精製」とは、それぞれ純度の低いまたは純度の高いことを意味する相対的用語である。従来技術の方法に対し、高度に濃縮された混合物の使用に一部起因して、本発明によって高い収率が得られる。
【0018】
特に明示しない限り、「レボフロキサシン」および「レボフロキサシン形態」という用語は、レボフロキサシンの塩、水和物、および溶媒和物を包含する。この用語は、レボフロキサシンの多形形態がレボフロキサシンの塩、水和物、または溶媒和物でないと考えられる範囲でレボフロキサシンの全ての多形形態も包含する。
【0019】
本発明の1つの態様は、(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸(「化合物I」)をN−メチルピペラジンと極性溶媒中で、好ましくは上昇した温度で、反応させて、レボフロキサシンを生成させることを含んでなる、レボフロキサシンの調製方法を提供する。前記上昇した温度は、好ましくは約70〜120℃である。該レボフロキサシンは、次に、当該技術分野でよく知られた技法を使用して沈殿および回収することができる。本願明細書で用いられる「沈殿」という用語は、溶液中の固体の形成またはスラリー中の固体量の増大を包含する。化合物Iの調製については、例えば、米国特許第4,382,892号に記載されており、参照により本願明細書に取り込まれる。
【0020】
適した極性溶媒は、レボフロキサシンを溶解可能ないずれの極性溶媒でもよい。好ましくは、極性溶媒はジメチルスルホキシド(DMSO)、アルコール(好ましくはイソブタノール)、ケトン、プロピレン・グリコール・モノメチル・エーテル(PGME)またはジメチルアセトアミド(DMA)である。本願明細書で用いられる「極性溶媒」という用語は、他の溶媒よりも比較的に極性の高いことを意味する相対的用語であることが意図される。
【0021】
1つの実施態様において、溶媒の体積は化合物Iのグラム当たり約14ml〜約4mlである。この実施態様において、溶媒は好ましくはイソブタノールおよびPGMEからなる群より選択される。別の実施態様において、溶媒の体積は化合物Iのグラム当たり約3mlよりも少ない。この後者の実施態様において、溶媒は好ましくはDMSOおよびDMAからなる群より選択される。有利なのは、DMSOを用いる場合に高収率を得るために必要とされる反応時間が短いことである。
【0022】
溶媒としてDMSOを用いる場合に、好ましい溶媒の体積は約3ml溶媒/g化合物までであり、好ましくは約0.5〜約3ml溶媒/g化合物であるが、それより多くの量も使用し得る。溶媒としてPGMEを用いる場合に。好ましい溶媒の体積は約4〜約14ml溶媒/g化合物であるが、それより多くの量も使用し得る。
【0023】
場合により、本発明に係る方法は、反応工程後に前記溶媒に貧溶媒を添加して収率を増大させることを更に含んでよい。本願明細書で用いられる「貧溶媒」(anti-solvent)という用語は、その中でレボフロキサシンが低溶解性である液体であって貧溶媒を溶媒に添加することによってレボフロキサシンの溶解度を低下させるようにするものである。好ましくは、貧溶媒は以下の1種以上のものである:n−ヘプタン、ヘキサン、イソプロピルアルコール、水中のイソプロピルアルコール(約5%以上のイソプロピルアルコール)、ブタノール、アセトニトリル、メチルエチルケトン、またはDMSO/水。溶媒がプロピレン・グリコール・モノメチル・エーテルまたはイソブタノールである場合に、好ましい貧溶媒はヘプタンまたはヘキサンである。溶媒がDMSOである場合には、好ましい貧溶媒はイソプロパノールである。
【0024】
他の実施態様において、化合物IはN−メチルピペラジンとニート混合物として反応する。この実施態様において、化合物Iは好ましくはN−メチルピペラジンの懸濁液に溶解される。
【0025】
1つの実施態様において、N−メチルピペラジンは化合物Iよりもモル過剰である。好ましくは、モル過剰は約2倍〜約4倍である。より好ましくは、モル過剰は約2倍〜約2.5倍である。
【0026】
好ましい反応工程の時間は、効率の最大化ならびに/あるいは副反応および/または分解の最小化を図りながら、反応を完了させるという要望を釣り合わせること、つまり、反応条件、特に溶媒の選択と温度に依存することになる。例えば、反応工程は、溶媒が用いられる場合に、約1時間〜約24時間の時間で一般に行われる。反応がニート混合物として行われる場合に、反応の時間は1時間よりも短くてよい。
【0027】
反応工程は、約110℃〜約120℃程の温度またはそれよりも高い温度で行ってよい。反応がニート混合物として行われる場合に、反応工程はほぼ還流温度で行われるのが好ましい。
【0028】
レボフロキサシン形態の調製
本発明の他の実施態様において、レボフロキサシン形態の調製方法は、レボフロキサシンを、好ましくは第1溶媒中で、第1の上昇した温度で保持すること;極性溶媒を添加すること;およびレボフロキサシン形態を回収することを含む。好ましくは、この方法は、レボフロキサシン−溶媒混合物を周囲温度よりも低い温度に冷却および保持することを更に含む。
【0029】
前記第1溶媒は、レボフロキサシンを溶解可能であって、好ましくは比較的高い沸点を有する極性溶媒である。例としては、PGME、DMAおよびDMSOが挙げられる。レボフロキサシンは、第1の上昇した温度に加熱してよい。しかしながら、上述のように、化合物IをN−メチルピペラジンと反応させる等により、最初にレボフロキサシンを合成し、その反応混合物を次に、極性溶媒を添加するために適した第2の上昇した温度に直接もって行くことが好ましい。第1の上昇した温度は、特定の溶媒に依存するが、一般には約70℃〜約120℃の範囲、好ましくは約80℃〜約85℃にある。第2の上昇した温度は、特定の極性溶媒に依存するが、一般には約60℃〜約80℃の範囲、好ましくは約75℃〜約79℃にある。
【0030】
極性溶媒は、第2の上昇した温度で、レボフロキサシンに、好ましくはゆっくりと添加される。極性溶媒は、好ましくは、約2時間にわたり添加される。場合により、更なる時間、好ましくは攪拌または他の掻き混ぜを行いながら、混合物が保持される。
【0031】
レボフロキサシンを回収することは、一般に、混合物を冷却してレボフロキサシンを沈殿させた後に濾過を行うことを包含する。
【0032】
レボフロキサシン半水和物の調製のためには、極性溶媒は、水を含み、好ましくはイソプロパノールと水、より好ましくは約3%〜約4%(v/v)の水、の混合物を含む。この実施態様において、第1の上昇した温度は約80℃であることが好ましく、第2の上昇した温度は約75℃であることが好ましい。好ましくは、極性溶媒が約2時間にわたり滴下により添加される。添加工程の後に、レボフロキサシン−極性溶媒混合物は周囲温度よりも低い温度、好ましくは約0℃〜約20℃の範囲、より好ましくは約5℃にゆっくりと冷却される。好ましくは、冷却工程は約1〜約10時間、より好ましくは約4時間にわたり行われる。好ましくは、前記周囲温度よりも低い温度が更に約2時間保持される。
【0033】
形態Cの調製のためには、極性溶媒は好ましくはイソプロパノールを含む。この実施態様において、第1の上昇した温度は約85℃であることが好ましく、第2の上昇した温度は約79℃であることが好ましい。好ましくは、極性溶媒が約2時間にわたり滴下により添加された後に、この温度で更に約2時間保持される。
【0034】
形態Aの調製のためには、極性溶媒は好ましくはイソプロパノールを含む。この実施態様において、第1の上昇した温度は約80℃であることが好ましく、第2の上昇した温度は約75℃であることが好ましい。添加工程の後に、レボフロキサシン−極性溶媒混合物は周囲温度よりも低い温度、好ましくは約0℃〜約20℃の範囲、より好ましくは約5℃にゆっくりと冷却される。好ましくは、冷却工程は約1〜約10時間、より好ましくは約4時間にわたり行われる。好ましくは、前記周囲温度よりも低い温度が更に約2時間保持される。
【0035】
形態Gおよび形態Bの調製のためには、極性溶媒は好ましくはイソプロパノールを含む。この実施態様において、第1の上昇した温度は約80℃であることが好ましく、第2の上昇した温度は約75℃であることが好ましい。添加工程の後に、レボフロキサシン−極性溶媒混合物は周囲温度よりも低い温度、好ましくは約0℃〜約20℃の範囲、より好ましくは約5℃にゆっくりと冷却される。好ましくは、冷却工程は約1〜約10時間、より好ましくは約4時間にわたり行われる。好ましくは、前記周囲温度よりも低い温度が更に約11時間保持される。好ましくは、沈殿が濾過され、次に乾燥される。形態Gを得るためには、第3の上昇した温度で約3〜約6時間および第4の上昇した温度で約3時間乾燥が行われる。第3の上昇した温度は約40℃であることが好ましく、第4の上昇した温度は約60℃であることが好ましい。形態Bを得るためには、第3の上昇した温度で少なくとも約20時間、好ましくは約21時間、および第4の上昇した温度で少なくとも約6時間、乾燥が行われる。第3の上昇した温度は約40℃であることが好ましく、第4の上昇した温度は約60℃であることが好ましい。
【0036】
形態Hの調製のためには、極性溶媒は、イソプロパノールと水、より好ましくは約0.3%〜約0.4%(v/v)の水、の混合物を含む。この実施態様において、第1の上昇した温度は約80℃であることが好ましく、第2の上昇した温度は約75℃であることが好ましい。好ましくは、極性溶媒が約1時間にわたり滴下により添加される。添加工程の後に、レボフロキサシン−極性溶媒混合物は、好ましくは、第2の上昇した温度で約2時間保持される。次に、その混合物が周囲温度よりも低い温度、好ましくは約0℃〜約20℃の範囲、より好ましくは約5℃にゆっくりと冷却される。好ましくは、冷却工程は約1〜約10時間、より好ましくは約4時間にわたり行われる。好ましくは、前記周囲温度よりも低い温度が更に約12時間保持される。
【0037】
本発明の他の実施態様において、レボフロキサシン形態の調製方法は、レボフロキサシンと極性溶媒の第1混合物を第1の上昇した温度で約4時間以上、好ましくは少なくとも4.5時間、保持すること、該第1混合物を第2の上昇した温度に冷却すること、および該冷却された第1混合物に追加の極性溶媒を添加して、第2混合物を形成すること、該第2混合物をレボフロキサシンが完全に溶解されるまで第3の上昇した温度で保持すること、場合により、前記保持工程中に該第2混合物に追加の極性溶媒を添加すること、該第2混合物を冷却して、レボフロキサシン形態を形成すること、および該レボフロキサシン形態を回収することを含んでなる。
【0038】
形態Fの調製のためには、極性溶媒はイソブチルアルコールを含む。この実施態様において、第1および第3の上昇した温度は還流温度であることが好ましく、第2の上昇した温度は約80℃であることが好ましい。好ましくは、レボフロキサシンを完全に溶解させるために十分な最少量の極性溶媒が第2混合物に添加される。第2混合物は周囲温度よりも低い温度、好ましくは約−5℃〜約20℃の範囲、より好ましくは約5℃にゆっくりと冷却される。好ましくは、冷却工程は約1〜約10時間、より好ましくは約1.5時間にわたり行われる。
【0039】
本発明の他の実施態様は、治療的に有効量の形態A、形態B、形態C、形態F、形態G、形態Hまたはこれらの組合せ、および場合により、薬学的に許容される担体を含んでなる。
【0040】
本発明の他の実施態様は、形態A、形態B、形態C、形態F、形態Gおよび形態Hからなる群より選択される1種以上の形態を、該1種以上の形態が大気中の水分(atomospheric water)の吸収によりレボフロキサシン半水和物に転化するために十分な時間の間、貯蔵することを含んでなる、レボフロキサシン半水和物の調製方法である。例えば、形態A、形態B、形態C、形態F、形態Gおよび形態Hは、室温(RT)で閉鎖された瓶内で7〜29日の貯蔵後に半水和物に転化する。該サンプルを開放された瓶内で保持する場合に、半水和物への転化は比較的速い(約24時間)。
【0041】
本発明の他の実施態様は、レボフロキサシン形態の調製方法である。(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸がN−メチルピペラジンと上昇した温度で反応されて、レボフロキサシンを生成する。該レボフロキサシンは次に、第1溶媒中で、第1の上昇した温度で、沈殿および保持される。極性溶媒が添加されて、レボフロキサシン形態C、形態A、形態G、形態B、形態H、形態Fまたは半水和物形態が沈殿し、これを次に濾過等の公知の手段により回収することができる。
【0042】
レボフロキサシン形態の物理特性
【表1】
【0043】
DTGサーモグラムを、Shimadzu DTG−50 ,加熱速度:10℃/分で行った。融点は、DTA曲線中の吸熱ピークにより、全ての議論される結晶形態について約225〜230℃であることが決定された。DTA曲線とTGA曲線における大きな差が160℃までの温度範囲で観測された。
【0044】
レボフロキサシン形態の熱分析
レボフロキサシン新規形態Aは、約100℃の吸熱ピークにより特徴付けられる。結晶からの溶媒の除去に起因して、この温度範囲で約18〜22%の重量損失段(weight loss step)が観測される。図7参照。
レボフロキサシン新規形態Cは、高DMSO含有量(30〜50%)およびKFによる約3.5%の含水量により特徴付けられる。図8参照。
レボフロキサシン新規形態Gは、約82℃および約103℃の吸熱ピークにより特徴付けられる。この温度範囲で3〜6%の重量損失段が観測される。この重量損失値は該サンプル中のDMSO含有量と一致する。図9参照。
レボフロキサシン新規形態Hは、約122℃の吸熱ピークにより特徴付けられる。60〜150℃の温度範囲で約8%の重量損失段が観測される。この重量損失率は、1:0.5の比のレボフロキサシン:IPA溶媒和物に相当する期待値(これは7.7%である)に等しい。図10参照。
【0045】
本発明のこれらの及び他の実施態様の機能及び利点は、下の実施例からより良く理解されるであろう。以下の実施例は、本発明の全範囲を例証するのではなく、本発明の利益を例示することを意図するものである。
【実施例】
【0046】
例1:DMSO中でのレボフロキサシンの合成
5g(17.8ミリモル)の(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸を2.5mLのDMSOおよび4.2mL(37.9ミリモル)のN−メチルピペラジン中に懸濁させた。その反応混合物を120℃に加熱し、懸濁液が溶解するようになった。2.5時間後に反応が完了した。次に、その混合物を70℃に冷却し、次に、この温度でイソプロピルアルコール(25mL)を添加した。その反応混合物を周囲温度で1時間スラリー化し、濾過し、終夜乾燥して、5.86g(91.3%)のレボフロキサシンを得た。
【0047】
例2:PGME中でのレボフロキサシンの合成
3g(10.67ミリモル)の(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸を30mLのPGMEおよび4.75mL(43ミリモル)のN−メチルピペラジン中に懸濁させた。その反応混合物を加熱して反応完了まで23時間還流した。その間に反応混合物が溶解するようになった。次に、その混合物を90℃に冷却し、この温度でn−ヘプタン(10mL)を添加した。次に、その反応混合物を0℃に冷却し、沈殿が65℃付近で生じた。その反応混合物を0℃で3時間置き、減圧下で濾過し、終夜乾燥して、2.98g(77.3%)のレボフロキサシンを得た。
【0048】
例3:イソブタノール中でのレボフロキサシンの合成
3g(10.67ミリモル)の(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸を21mLのイソブタノールおよび4.75mL(43ミリモル)のN−メチルピペラジン中に懸濁させた。その反応混合物を加熱して6時間還流し、次に周囲温度で60時間スラリー化し、再び加熱して反応完了まで7時間還流した。その間に反応混合物が還流温度で溶解するようになった。次に、その混合物を0℃に冷却し、減圧下で濾過し、7mLのイソブタノールおよび10mLのn−ヘプタンで洗浄し、終夜乾燥して、2.83g(77.3%)のレボフロキサシンを得た。
【0049】
例4:レボフロキサシンの合成(ニート)
5g(17.79ミリモル)の(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸を6.8mL(0.06モル)のN−メチルピペラジン中に懸濁させた。その反応混合物を加熱して反応完了まで40分間還流した。次に、その混合物を80℃に冷却した。この温度でIPA(10mL)およびn−ヘプタン(10mL)を添加した。その固体を減圧下で濾過し、n−ヘプタンで濯いだ。n−ヘプタンの添加後に、母液が更に沈殿を与えた。両方の沈殿を減圧下で濾過し、終夜乾燥して、4.9g(76%)のレボフロキサシンを得た。
【0050】
例5:DMA中でのレボフロキサシンの合成
10g(35.6ミリモル)の(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸を5mLのDMA(ジメチルアセトアミド)および8.3mL(75ミリモル)のN−メチルピペラジン中に懸濁させた。その反応混合物を出発物質が完全に転化されるまで約1.5時間110℃に加熱した。次に、その反応混合物を80℃に冷却し、60mlのイソプロピルアルコールを添加した。次に、その反応混合物を周囲温度で3時間スラリー化し、減圧下で濾過し、40mlのIPAで洗浄し、真空オーブン内で終夜乾燥して、11.48g(89.3%)のレボフロキサシンを得た。
【0051】
例6:半水和物の合成
メカニカルスターラー、凝縮器および温度計を備え、80℃に加熱された1リットルの反応器に、87.5g(0.31モル)の(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸、61.3mLのDMSOおよび86.3mL(0.77モル)のN−メチルピペラジンを入れた。そのスラリーを80℃で窒素雰囲気の下で250rpmの速度で(HPLCによりモニタリングして)反応完了まで攪拌した。次に、そのスラリーを75℃に冷却し、この温度でイソプロパノール(675mL)と水(25mL)の混合物を2時間にわたり滴下により添加した。次に、そのスラリーを4時間にわたり5℃に冷却し、この温度で2時間保持し、この温度で減圧下に濾過した。次に、その固体を175mLのイソプロパノールで洗浄(2回リンス)し、減圧下で乾燥して、レボフロキサシン半水和物を得た。
【0052】
例7:形態Cの合成
調製
メカニカルスターラー、凝縮器および温度計を備え、85℃に加熱された1リットルの反応器に、(S)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸(87.5g)、DMSO(61.3ml)およびN−メチルピペラジン(86.3ml)を入れた。そのスラリーを窒素雰囲気の下で250rpmの速度で攪拌した。加熱を(HPLCによりモニタリングして)反応完了まで4.5時間続けた。次に、そのスラリーを79℃に冷却し、この温度でイソプロパノール(700ml)を2時間にわたり滴下により添加し、この温度で更に2時間攪拌した。その終了時に、サンプルを減圧下で濾過し、イソプロパノールで洗浄した。
【0053】
例8:形態Aの合成
調製
メカニカルスターラー、凝縮器および温度計を備え、80℃に加熱された1リットルの反応器に、(S)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸(87.5g)、DMSO(61.3ml)およびN−メチルピペラジン(86.3ml)を入れた。そのスラリーを空気雰囲気の下で250rpmの速度で攪拌した。加熱を(HPLCによりモニタリングして)反応完了まで4.5時間続けた。次に、そのスラリーを75℃に冷却し、この温度でイソプロパノール(700ml)を2時間にわたり滴下により添加した。サンプルをXRD分析にかけた。混合物をこの温度で攪拌しながら2時間保持した。次に、その反応混合物を5℃まで4時間にわたり冷却し、この温度で攪拌しながら2時間保持した。その終了時に、反応混合物を減圧下で濾過し、イソプロパノール(175ml)で洗浄して、171gの湿った物質(106.8gの乾燥物質、92.7%)を得た。
【0054】
例9:形態Gおよび形態Bの合成
調製
メカニカルスターラー、凝縮器および温度計を備え、80℃に加熱された1リットルの反応器に、(S)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸(87.5g)、DMSO(61.3ml)およびN−メチルピペラジン(86.3ml)を入れた。そのスラリーを窒素雰囲気の下で250rpmの速度で攪拌した。加熱を反応完了まで4.5時間続けた。次に、そのスラリーを75℃に冷却し、この温度でイソプロパノール(700ml)を2時間にわたり滴下により添加した。
添加終了時に、攪拌を75℃で2時間保持し、次に、5℃まで4時間にわたり冷却し、この温度で攪拌しながら11時間保持した。そのスラリーを減圧下で濾過し、イソプロパノール(175ml)で洗浄して、149gの湿った物質を得た。
その湿った物質を乾燥のために2つの部分に分けた。第1部分は減圧下で攪拌しながら40℃で21時間乾燥し、第2画分は減圧下で攪拌しながら60℃で21時間乾燥した。40℃で3時間または6時間乾燥後そして60℃で3時間乾燥後にレボフロキサシン形態Gが生じた。40℃で21時間乾燥後そして60℃で6時間、9時間および21時間乾燥後にレボフロキサシン形態Bが生じた。
【0055】
例10:形態Hの合成
調製
メカニカルスターラー、凝縮器および温度計を備え、80℃に加熱された1リットルの反応器に、(S)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸(87.5g)、DMSO(61.3ml)およびN−メチルピペラジン(86.3ml)を入れた。そのスラリーを窒素雰囲気の下で250rpmの速度で攪拌した。加熱を(HPLCによりモニタリングして)反応完了まで4.5時間続けた。次に、そのスラリー混合物を75℃に冷却し、この温度で H2O(2.5ml)と混合されたイソプロパノール(697.5ml)を1時間にわたり滴下により添加した。添加終了時に、攪拌を2時間保持し、次に、5℃まで4時間にわたり冷却した。この温度で攪拌を12時間保持した。次に、その反応混合物を減圧下で濾過し、イソプロパノール(175ml)で洗浄して、150gの湿った物質を得た。
【0056】
例11:形態Fの合成
調製
凝縮器を備えたフラスコに、4.0gのレボフロキサシンを入れた。イソブチルアルコール(8mL)を添加し、その混合物を還流温度に加熱した。15分後、その混合物を80℃に冷却し、4mLのイソブチルアルコールを添加した。次に、その混合物を再び還流温度に加熱した。イソブチルアルコール(6mL)を完全に溶解するまで添加した。溶液が透明になり、その混合物を1.5時間で0℃に冷却した。次に、その沈殿を減圧下で濾過し、イソブチルアルコール(4mL)で洗浄し、減圧下で60℃において乾燥した。
【0057】
例12:形態の時間により誘導される転化
メカニカルスターラー、凝縮器および温度計を備えた1リットルの反応器に、(S)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸(87.5g)、DMSO(61.3ml)、N−メチルピペラジン(86.3ml)および H2O(0.44ml)を入れた。次に、そのスラリーを80℃に加熱し、窒素雰囲気の下で250rpmの速度で攪拌した。加熱を反応完了まで4.5時間続けた。次に、そのスラリー混合物を75℃に冷却し、この温度でイソプロパノール(700ml)を2時間にわたり滴下により添加した。
次に、その混合物を攪拌しながら75℃で2時間保持した。次に、その混合物を5℃まで4時間にわたり冷却し、この温度で混合物を攪拌しながら9時間保持した。そのスラリーを減圧下で濾過し、イソプロパノール(175ml)で洗浄して、147gの湿った多形形態Hを得た。
その湿った物質を(保護のため)紙で覆って室温(RT)で空気に曝した。3時間、6時間、9時間後、その湿ったサンプルの多形は形態Gであった。24時間後、その湿ったサンプルは半水和物の多形形態であった。
【0058】
例13:スラリー中の形態Hの転化
メカニカルスターラー、凝縮器および温度計を備えた1リットルの反応器を80℃に加熱し、(S)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸(87.5g)、DMSO(61.3ml)およびN−メチルピペラジン(86.3ml)を入れた。そのスラリーを窒素雰囲気の下で250rpmの速度で攪拌した。加熱を(HPLCによりモニタリングして)反応完了まで4.5時間続けた。次に、そのスラリー混合物を75℃に冷却し、この温度で H2O(2.5ml)と混合されたイソプロパノール(697.5ml)を1時間にわたり滴下により添加した。
次に、その混合物を攪拌しながら2時間保持した。次に、その混合物を5℃まで4時間にわたり冷却し、この温度で混合物を12時間保持した。次に、その反応混合物を減圧下で濾過し、イソプロパノール(175ml)で洗浄して、150gの湿った多形形態Hを得た。
その湿った物質の第1部分を5vのアセトニトリル中で75℃で2時間攪拌し、次に減圧下で濾過した。その湿ったサンプルの多形は半水和物であった。その湿った物質の第2部分を(保護のため)紙で覆って室温(RT)で空気に曝した。数時間後、その湿ったサンプルの多形は半水和物であった。
【0059】
例14:スラリー中の形態Gの転化
メカニカルスターラー、凝縮器および温度計を備えた1リットルの反応器に、(S)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸(87.5g)、DMSO(61.3ml)およびN−メチルピペラジン(86.3ml)を周囲温度で入れた。次に、そのスラリーを80℃に加熱し、窒素雰囲気の下で250rpmの速度で攪拌した。加熱を反応完了まで4時間続けた。次に、そのスラリー混合物を75℃に冷却し、この温度で H2O(21ml)と混合されたイソプロパノール(679ml)を2時間にわたり滴下により添加した。
次に、その混合物を攪拌しながら75℃で2時間保持した。次に、その混合物を5℃まで4時間にわたり冷却し、この温度で混合物を攪拌しながら10時間保持した。そのスラリーを減圧下で濾過し、イソプロパノール(175ml)で洗浄して、166.5gの湿った多形形態Gを得た。
その湿った物質(162g)をアセトニトリル(486ml)と混合した。そのスラリーを250rpmの速度で27℃において1時間攪拌した。次に、そのスラリーを減圧下で濾過し、アセトニトリル(162ml)で洗浄した。その湿ったサンプルは半水和物の多形形態であった。その固体を真空オーブン内で40℃において6時間乾燥した。その乾燥した粗レボフロキサシンは半水和物の多形形態であった。
【0060】
例15:乾燥
メカニカルスターラー、凝縮器および温度計を備えた1リットルの反応器に、(S)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸(87.5g)、DMSO(61.3ml)およびN−メチルピペラジン(86.3ml)を周囲温度で入れた。次に、そのスラリーを80℃に加熱し、窒素雰囲気の下で250rpmの速度で攪拌した。加熱を反応完了まで4.5時間続けた。次に、そのスラリー混合物を75℃に冷却し、イソプロパノール(700ml)を一度に添加した。添加終了時に、その混合物を300rpmの速度で攪拌した。その反応混合物を7℃まで2時間にわたり冷却した。この温度において350rpmの速度で2時間攪拌を保持した。次に、その反応混合物を減圧下で濾過し、イソプロパノール(175ml)で洗浄した。
その湿った物質(162g)をイソプロパノール(180ml)と混合した。そのスラリーを250rpmの速度で40℃において1時間攪拌した。次に、そのスラリーを減圧下で濾過し、イソプロパノール(60ml)で洗浄した。その湿ったサンプルの多形形態はHであった。
その固体を真空オーブン内で60℃において14時間乾燥して、106g(92%)の乾燥レボフロキサシン形態Bを得た。
【0061】
例16:乾燥
メカニカルスターラー、凝縮器および温度計を備えた1リットルの反応器に、(S)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸(87.5g)、DMSO(61.3ml)およびN−メチルピペラジン(86.3ml)を周囲温度で入れた。次に、そのスラリーを80℃に加熱し、窒素雰囲気の下で250rpmの速度で攪拌した。加熱を反応完了まで4.5時間続けた。次に、そのスラリー混合物を75℃に冷却し、イソプロパノール(700ml)を一度に添加した。添加終了時に、その混合物を300rpmの速度で攪拌した。その反応混合物を7℃まで2時間にわたり冷却した。この温度において350rpmの速度で2時間攪拌を保持した。次に、その反応混合物を減圧下で濾過し、イソプロパノール(175ml)で洗浄した。
その湿った物質(162g)をイソプロパノール(180ml)と混合した。そのスラリーを250rpmの速度で40℃において1時間攪拌した。次に、そのスラリーを減圧下で濾過し、イソプロパノール(60ml)で洗浄した。その湿ったサンプルの多形形態はGであった。
その固体を真空オーブン内で60℃において14時間乾燥して、106g(92%)の乾燥レボフロキサシン形態Bを得た。
【0062】
例17:n−BuOHを用いた半水和物の調製
1gの粗レボフロキサシンを7mlのn−BuOHに懸濁させた。その混合物をその物質が完全に溶解するまで還流温度に加熱した。次に、その溶液を2.5時間の時間にわたり室温(RT)に冷却した。その沈殿を減圧下で濾過し、n−BuOHで洗浄し、真空オーブン内で60℃において乾燥して、810mg(81%)の精製レボフロキサシン半水和物を得た。
【0063】
例18:ACNを用いた半水和物の調製
1.5gの粗レボフロキサシンを10.5mlのACNに懸濁させた。その混合物をその物質が完全に溶解するまで還流温度に加熱した。次に、その溶液を20分の時間にわたり0℃に冷却した。その沈殿を減圧下で濾過し、ACN(1.5ml)で洗浄し、真空オーブン内で30℃において乾燥して、1.15g(77%)の精製レボフロキサシン(半水和物/一水和物混合物)を得た。精製レボフロキサシンは粗サンプル中の量のほぼ半分の量のデスメチル・レボフロキサシンを含有した。
【0064】
例19:DMSO/水を用いた半水和物の調製
1gの粗レボフロキサシンを1.5mlのDMSOに懸濁させた。その混合物をその物質が完全に溶解するまで108℃に加熱した。次に、 H2O(7.5ml)を10分間にわたり添加し、その混合物を室温(RT)に冷却した。その沈殿を減圧下で濾過し、1mlのDMSO:H2O 1:5混合物で洗浄し、気流オーブン内で60℃において乾燥して、840mg(84%)の精製レボフロキサシン半水和物を得た。
【0065】
例20:MEKを用いた半水和物の調製
1.5gの粗レボフロキサシンを15mlのMEKに懸濁させた。その混合物をその物質が完全に溶解するまで還流温度に加熱した。次に、その溶液を3時間の時間にわたり−5℃に冷却した。その沈殿を減圧下で濾過し、1.5mlのMEKで洗浄し、真空オーブン内で30℃において乾燥して、840mg(84%)の精製レボフロキサシン半水和物を得た。
【図面の簡単な説明】
【0066】
【図1】新規レボフロキサシン結晶形態AのXRDディフラクトグラムである。
【図2】新規レボフロキサシン結晶形態BのXRDディフラクトグラムである。
【図3】新規レボフロキサシン結晶形態CのXRDディフラクトグラムである。
【図4】新規レボフロキサシン結晶形態FのXRDディフラクトグラムである。
【図5】新規レボフロキサシン結晶形態GのXRDディフラクトグラムである。
【図6】新規レボフロキサシン結晶形態HのXRDディフラクトグラムである。
【図7】新規レボフロキサシン結晶形態AのDTGサーモグラムである。
【図8】新規レボフロキサシン結晶形態CのDTGサーモグラムである。
【図9】新規レボフロキサシン結晶形態GのDTGサーモグラムである。
【図10】新規レボフロキサシン結晶形態HのDTGサーモグラムである。
【特許請求の範囲】
【請求項1】
(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸をN−メチルピペラジンと、極性溶媒中において、70〜120℃の温度で反応させて、レボフロキサシンを形成すること、
該レボフロキサシンを沈殿させること、および、
該レボフロキサシンを回収すること
を含んでなる、レボフロキサシンの調製方法であって、
ここで、上記極性溶媒の体積が(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸のグラム当たり14ml〜4mlであり、そして上記極性溶媒がジメチルスルホキシド(DMSO)、イソブタノール、プロピレン・グリコール・モノメチル・エーテル(PGME)、ジメチルアセトアミド(DMA)およびこれらの混合物からなる群より選択される、方法。
【請求項2】
(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸をN−メチルピペラジンと、極性溶媒中において、70〜120℃の温度で反応させて、レボフロキサシンを形成すること、
該レボフロキサシンを沈殿させること、および、
該レボフロキサシンを回収すること
を含んでなる、レボフロキサシンの調製方法であって、
ここで、上記溶媒の体積が(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸のグラム当たり3mlよりも少ない、方法。
【請求項3】
(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸をN−メチルピペラジンと、70〜120℃の温度で反応させて、レボフロキサシンを形成すること、
該レボフロキサシンを沈殿させること、および、
該レボフロキサシンを回収すること
を含んでなる、レボフロキサシンの調製方法であって、
ここで上記反応工程が、そのままの混合物中で行われる、方法。
【請求項4】
収率が75%以上である、請求項1〜3のいずれか一項に記載の方法。
【請求項5】
収率が85%以上である、請求項4に記載の方法。
【請求項6】
溶媒がイソブタノールおよびPGMEからなる群より選択される、請求項1に記載の方法。
【請求項7】
溶媒がDMSOおよびDMAからなる群より選択される、請求項2に記載の方法。
【請求項8】
N−メチルピペラジンが(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸よりもモル過剰である、請求項1〜3のいずれか一項に記載の方法。
【請求項9】
モル過剰が2倍〜4倍である、請求項8に記載の方法。
【請求項10】
モル過剰が2倍〜2.5倍である、請求項8に記載の方法。
【請求項11】
前記沈殿工程において貧溶媒が添加されることを更に含んで成る、請求項1又は2のいずれか一項に記載の方法。
【請求項12】
貧溶媒がn−ヘプタン、ヘキサン、イソプロピルアルコール、イソプロピルアルコール、水中の少なくとも5%のイソプロピルアルコール、ブタノール、アセトニトリル、メチルエチルケトンおよび、DMSOと水の混合物からなる群より選択される、請求項11に記載の方法。
【請求項13】
前記反応工程が還流温度で行われる、請求項3に記載の方法。
【請求項14】
(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸がN−メチルピペラジン中に懸濁される、請求項3に記載の方法。
【請求項15】
回収されたレボフロキサシンが、
12.2、17.6、18.0、21.7、22.4、及び23.4±0.3度2θ;
5.5、11.3、12.6、及び18.8±0.3度2θ;
5.3、6.7、13.1、13.4、26.4、及び26.7±0.3度2θ;
15.2、15.8、25.5、及び25.8±0.3度2θ;
4.9、5.2、5.5、及び18.7±0.3度2θ;又は、
11.9、17.8、及び18.4;
におけるXRDピークにより特徴付けられる結晶形態、並びに半水和物形態から成る群から選択される形態である、請求項1又は2のいずれか一項に記載の方法。
【請求項1】
(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸をN−メチルピペラジンと、極性溶媒中において、70〜120℃の温度で反応させて、レボフロキサシンを形成すること、
該レボフロキサシンを沈殿させること、および、
該レボフロキサシンを回収すること
を含んでなる、レボフロキサシンの調製方法であって、
ここで、上記極性溶媒の体積が(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸のグラム当たり14ml〜4mlであり、そして上記極性溶媒がジメチルスルホキシド(DMSO)、イソブタノール、プロピレン・グリコール・モノメチル・エーテル(PGME)、ジメチルアセトアミド(DMA)およびこれらの混合物からなる群より選択される、方法。
【請求項2】
(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸をN−メチルピペラジンと、極性溶媒中において、70〜120℃の温度で反応させて、レボフロキサシンを形成すること、
該レボフロキサシンを沈殿させること、および、
該レボフロキサシンを回収すること
を含んでなる、レボフロキサシンの調製方法であって、
ここで、上記溶媒の体積が(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸のグラム当たり3mlよりも少ない、方法。
【請求項3】
(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸をN−メチルピペラジンと、70〜120℃の温度で反応させて、レボフロキサシンを形成すること、
該レボフロキサシンを沈殿させること、および、
該レボフロキサシンを回収すること
を含んでなる、レボフロキサシンの調製方法であって、
ここで上記反応工程が、そのままの混合物中で行われる、方法。
【請求項4】
収率が75%以上である、請求項1〜3のいずれか一項に記載の方法。
【請求項5】
収率が85%以上である、請求項4に記載の方法。
【請求項6】
溶媒がイソブタノールおよびPGMEからなる群より選択される、請求項1に記載の方法。
【請求項7】
溶媒がDMSOおよびDMAからなる群より選択される、請求項2に記載の方法。
【請求項8】
N−メチルピペラジンが(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸よりもモル過剰である、請求項1〜3のいずれか一項に記載の方法。
【請求項9】
モル過剰が2倍〜4倍である、請求項8に記載の方法。
【請求項10】
モル過剰が2倍〜2.5倍である、請求項8に記載の方法。
【請求項11】
前記沈殿工程において貧溶媒が添加されることを更に含んで成る、請求項1又は2のいずれか一項に記載の方法。
【請求項12】
貧溶媒がn−ヘプタン、ヘキサン、イソプロピルアルコール、イソプロピルアルコール、水中の少なくとも5%のイソプロピルアルコール、ブタノール、アセトニトリル、メチルエチルケトンおよび、DMSOと水の混合物からなる群より選択される、請求項11に記載の方法。
【請求項13】
前記反応工程が還流温度で行われる、請求項3に記載の方法。
【請求項14】
(S)−(−)−9,10−ジフルオロ−3−メチル−7−オキソ−2,3−ジヒドロ−7H−ピリド[1,2,3−de][1,4]ベンズオキサジン−6−カルボン酸がN−メチルピペラジン中に懸濁される、請求項3に記載の方法。
【請求項15】
回収されたレボフロキサシンが、
12.2、17.6、18.0、21.7、22.4、及び23.4±0.3度2θ;
5.5、11.3、12.6、及び18.8±0.3度2θ;
5.3、6.7、13.1、13.4、26.4、及び26.7±0.3度2θ;
15.2、15.8、25.5、及び25.8±0.3度2θ;
4.9、5.2、5.5、及び18.7±0.3度2θ;又は、
11.9、17.8、及び18.4;
におけるXRDピークにより特徴付けられる結晶形態、並びに半水和物形態から成る群から選択される形態である、請求項1又は2のいずれか一項に記載の方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【公開番号】特開2007−131628(P2007−131628A)
【公開日】平成19年5月31日(2007.5.31)
【国際特許分類】
【出願番号】特願2006−311904(P2006−311904)
【出願日】平成18年11月17日(2006.11.17)
【分割の表示】特願2003−532000(P2003−532000)の分割
【原出願日】平成14年10月3日(2002.10.3)
【出願人】(501079705)テバ ファーマシューティカル インダストリーズ リミティド (283)
【Fターム(参考)】
【公開日】平成19年5月31日(2007.5.31)
【国際特許分類】
【出願日】平成18年11月17日(2006.11.17)
【分割の表示】特願2003−532000(P2003−532000)の分割
【原出願日】平成14年10月3日(2002.10.3)
【出願人】(501079705)テバ ファーマシューティカル インダストリーズ リミティド (283)
【Fターム(参考)】
[ Back to top ]