
レボミルナシプランの安定した剤形
本発明は、レボミルナシプランとその薬学的に許容される塩の安定した剤形に関する。これらの剤形の調製方法およびこれらの剤形の使用方法も同様に記載されている。
【発明の詳細な説明】
【技術分野】
【0001】
本出願は、2010年1月14日出願の米国仮特許出願第61/294,898号に対する35U.S.C.119(e)に基づく優先権;およびそれ自体2009年11月6日出願の米国仮特許出願第61/258,652号に対する35U.S.C.119(e)に基づく優先権を主張している2010年11月8日出願の米国特許出願第12/941,293号に対する35U.S.C.120に基づく優先権を主張するものである。
【0002】
本発明は、レボミルナシプランまたはその薬学的に許容される塩の安定した投薬調合物に関する。これらの剤形の調製用プロセスおよびこれらの剤形の使用方法も同様に記載されている。
【背景技術】
【0003】
レボミルナシプランは、(1S,2R)−2−(アミノメチル)−N,N−ジエチル−1−フェニルシクロプロパンカルボキサミドの国際一般名である。これは、5−HT再取り込み阻害よりもNE再取り込み阻害に対して大きな選択性を有する非常に強力な選択的ノルエピネフリン(NE)およびセロトニン(5−HT)再取り込み阻害薬である。詳細には、レボミルナシプランは、NE:5−HTについておよそ1.5:1という阻害選択性比を有する。したがって、レボミルナシプランは、ノルエピネフリン−セロトニン再取り込み阻害薬(NSRI)とみなされており、これはNEに対してよりも5−HTに対して同等以上の阻害選択性を有するセロトニン−ノルエピネフリン再取り込み阻害薬(SNRI)とは薬理学的に全く異なる。
【0004】
レボミルナシプランの調合物については先行技術において一般的に論述されているものの、レボミルナシプランの安定した剤形を調製する上で問題点に直面してきた。これらの問題点は、少なくとも一部には、ある種の反応条件に対するレボミルナシプランの感応性および一般的に使用される一部の賦形剤とのその反応性に起因して発生していた。
【発明の概要】
【発明が解決しようとする課題】
【0005】
したがって、改善された純度と安定性を有するレボミルナシプランの改善された調合物に対するニーズが現在ひき続き存在している。さらに、安定性が改善した調合物は、患者における望ましくない不利な事象(例えば吐き気、嘔吐および胃出血)の発生率低下と結びつけられる望ましい薬物動態プロファイルを達成しなければならない。
【課題を解決するための手段】
【0006】
現在、使用環境に入った時点で所望されるレボミルナシプラン放出を達成しかつ意外なほど高い安定性を有する改良型レボミルナシプラン調合物が、発見されている。これらの改良型レボミルナシプラン調合物が本明細書中で記述されている。
【0007】
本発明は新規のレボミルナシプラン剤形、ならびにこれらの剤形の調製方法および剤形の使用方法に関する。
【0008】
一部の実施形態において、本発明は、レボミルナシプランまたはその薬学的に許容される塩を含む安定した剤形に関する。
【0009】
一部の実施形態において、本発明は、実質的に純粋なレボミルナシプランまたはその薬学的に許容される塩を含む活性成分を含むまたは本質的にこの活性成分からなる安定した剤形に関する。
【0010】
一部の実施形態において、本発明は、少なくとも約98重量%(例えば少なくとも98重量%)のレボミルナシプランまたはその薬学的に許容される塩を含む活性成分を含むまたは本質的にこの活性成分からなる安定した剤形に関する。
【0011】
一部の実施形態において、本発明は、剤形が12.0、20.1および22.5±0.2度2θに特徴的ピークを含むX線粉末回折(XRD)パターンを含む、レボミルナシプランまたはその薬学的に許容される塩を含む安定した剤形に関する。一部の実施形態において、XRDパターンは同様に32.7±0.2度2θに特徴的ピークを含む。一部の実施形態において、XRDパターンは同様に32.7±0.2度2θに特徴的ピークを含む。一部の実施形態において、XRDパターンは同様に6.0±0.2度2θに特徴的ピークを含む。
【0012】
一部の実施形態において、本発明は、レボミルナシプランまたはその薬学的に許容される塩を含む安定した剤形において、6.0、12.0および20.1±0.2度2θに特徴的ピークを含むX線粉末回折(XRD)パターンを含む安定した剤形に関する。一部の実施形態において、XRDパターンは同様に22.5±0.2度θに特徴的ピークを含む。
【0013】
一部の実施形態において、本発明は、レボミルナシプランまたはその薬学的に許容される塩および約0.001重量%〜約0.5重量%の(1S,5R)1−フェニル−3−アザビシクロ[3−1−0]ヘキサン−2−オンを含む安定した剤形に関する。
【0014】
一部の実施形態において、本発明は、レボミルナシプランまたはその薬学的に許容される塩および約0.001%重量%〜約0.2重量%の(1S,5R)1−フェニル−3−アザビシクロ[3−1−0]ヘキサン−2−オンを含む安定した剤形に関する。
【0015】
一部の実施形態において、安定した剤形は、実質的に純粋なレボミルナシプランまたはその薬学的に許容される塩を含む約45重量%〜約60重量%の活性成分を含む。
【0016】
一部の実施形態において、安定した剤形は、実質的に純粋なレボミルナシプランまたはその薬学的に許容される塩を含む約45重量%〜約60重量%の活性成分と、約4重量%〜約10重量%の結合剤を含む。
【0017】
一部の実施形態において、安定した剤形は、実質的に純粋なレボミルナシプランまたはその薬学的に許容される塩を含む約45重量%〜約60重量%の活性成分と、約30重量%〜約45重量%の不活性基質または充填剤と、約4重量%〜約10重量%の結合剤とを含む。
【0018】
一部の実施形態において、安定した剤形は、実質的に純粋なレボミルナシプランまたはその薬学的に許容される塩を含む約45重量%〜約60重量%の活性成分と、約30重量%〜約45重量%の不活性基質または充填剤と、約4重量%〜約10重量%の結合剤と、約1重量%〜約5重量%の付着防止剤または潤滑剤とを含む。
【0019】
一部の実施形態において、安定した剤形は、約50重量%〜約60重量%のレボミルナシプランまたはその薬学的に許容される塩と、約30重量%〜約40重量%の不活性基質または充填剤と、約4重量%〜約8重量%の結合剤と、約1重量%〜約5重量%の付着防止剤または潤滑剤とを含む。
【0020】
一部の実施形態において、安定した剤形は、約40重量%〜約55重量%のレボミルナシプランまたはその薬学的に許容される塩と、約5重量%〜約15重量%の放出制御剤と、約25重量%〜約40重量%の不活性基質と、約3重量%〜約10重量%の結合剤と、約3重量%〜約10重量%の付着防止剤と、約0.1重量%〜約5重量%の可塑剤とを含む。
【0021】
一部の実施形態において、安定した剤形は、使用環境内に入ってから約6時間〜約16時間後に少なくとも約80%の溶出率を提供する。
【0022】
一部の実施形態において、本発明は、約10mg〜約200mgのレボミルナシプランを含む活性成分を含む安定した経口剤形において、12.0、20.1および22.5±0.2度2θに特徴的ピークを含むX線粉末回折(XRD)パターンを含む、安定した経口剤形に関する。
【0023】
一部の実施形態において、本発明は、約10mg〜約200mgのレボミルナシプランと約0.0001重量%〜約0.5重量%(例えば約0.0001重量%〜約0.2重量%さらには約0.0001重量%〜約0.1重量%)の(1S,5R)1−フェニル−3−アザビシクロ[3−1−0]ヘキサン−2−オンを含む経口剤形に関する。
【0024】
一部の実施形態において、本発明は、約10mg〜約200mgのレボミルナシプランと約0.0001重量%〜約0.5重量%(例えば約0.0001重量%〜約0.2重量%さらには約0.0001重量%〜約0.1重量%)の(1S,5R)1−フェニル−3−アザビシクロ[3−1−0]ヘキサン−2−オンを含む経口剤形において、剤形が、12.0、20.1および22.5±0.2度2θに特徴的ピークを含むX線粉末回折(XRD)パターンを含む経口剤形に関する。
【0025】
一部の実施形態において、剤形は、使用環境内に入ってから約6時間〜約16時間後に少なくとも約80%の溶出率を提供する。
【0026】
一部の実施形態において、本発明は、レボミルナシプランまたはその薬学的に許容される塩と放出制御剤を含む安定した剤形において、使用環境内に入った後レボミルナシプランまたはその薬学的に許容される塩の放出を持続する安定した剤形に関する。
【0027】
一部の実施形態において、本発明は、約20mg、約40mg、約80mgまたは約120mgのレボミルナシプランまたはその薬学的に許容される塩を含む安定した剤形に関する。
【0028】
一部の実施形態において、本発明は、治療を要する患者に対してレボミルナシプランまたはその薬学的に許容される塩の安定した剤形を投与するステップを含む、大うつ病障害の治療方法に関する。
【0029】
一部の実施形態において、本発明は、治療を要する患者に対してレボミルナシプランまたはその薬学的に許容される塩の安定した剤形を投与するステップを含む、倦怠感を同時に伴う大うつ病障害の治療方法に関する。
【0030】
一部の実施形態において、本発明は、不活性基質をレボミルナシプランまたはその薬学的に許容される塩および脱水アルコールと接触させるステップを含む、安定した剤形の調製方法に関する。
【図面の簡単な説明】
【0031】
【図1】本発明の一実施形態に係るレボミルナシプランの安定した剤形についての溶出率を示す。
【図2】40℃および相対湿度(RH)75%で1カ月の保管後の本発明の一実施形態に係るレボミルナシプランの安定した剤形についての溶出率を示す。
【図3】40℃および75%RHで2カ月の保管後の本発明の一実施形態に係るレボミルナシプランの安定した剤形についての溶出率を示す。
【図4】40℃および75%RHで3カ月の保管後の本発明の一実施形態に係るレボミルナシプランの安定した剤形についての溶出率を示す。
【図5】40℃および75%RHで3カ月の保管後の、1グラムの乾燥剤を含む本発明の一実施形態に係るレボミルナシプランの安定した剤形についての溶出率を示す。
【図6】実質的に純粋なレボミルナシプランを含む活性薬剤成分についてのX線粉末回折パターン(XRD)を示す。
【図7】本発明の一実施形態に係るレボミルナシプランの安定した即時放出型剤形のXRDを示す。
【図8】本発明の一実施形態に係るレボミルナシプランの安定した持続放出型剤形のXRDを示す。
【図9】ヒトの患者に対するレボミルナシプランの安定した剤形の単回投与を介して達成され得る時間に対するレボミルナシプランの平均血漿濃度を示す。
【発明を実施するための形態】
【0032】
レボミルナシプランの新規の安定した剤形、これらの剤形を使用した治療方法およびこれらの剤形の調製方法が、本明細書中で提供されている。レボミルナシプランの剤形は、使用環境に入った時点で所望の溶出プロファイルを達成し、驚くほど高い安定性を有することが発見された。
【0033】
定義
本明細書中で使用される「レボミルナシプラン」という用語は、(1S,2R)−2−(アミノメチル)−N,N−ジエチル−1−フェニルシクロプロパンカルボキサミドおよびその薬学的に許容される塩に関する。この用語は2−(アミノメチル)−N,N−ジエチル−1−フェニルシクロプロパンカルボキサミドの他の異性体(例えば(1R,2S)−2−(アミノメチル)−N,N−ジエチル−1−フェニルシクロプロパンカルボキサミド)または(1S,2R)−2−(アミノメチル)−N,N−ジエチル−1−フェニルシクロプロパンカルボキサミドの分解生成物(例えば(1S,5R)−1−フェニル−3−アザビシクロ[3−1−0]ヘキサン−2−オン)を含まない。「薬学的に許容される塩」という用語は、患者が生理学的に耐容するレボミルナシプランのあらゆる塩(例えば塩酸レボミルナシプラン)を意味する。レボミルナシプランの構造式を以下に示す:
【化1】

【0034】
「実質的に純粋なレボミルナシプラン」という用語は、本明細書中で、少なくとも98重量%のレボミルナシプランを意味するように使用されている。例えば、実質的に純粋なレボミルナシプランを含む活性薬剤成分(すなわち活性成分)には、少なくとも98重量%(例えば約98.5重量%)のレボミルナシプランおよび多くとも2重量%の全ての組合わされた他の構成成分(例えば2−(アミノメチル)−N,N−ジエチル−1−フェニルシクロプロパンカルボキサミドの他の異性体および/または(1S,2R)−2−(アミノメチル)−N,N−ジエチル−1−フェニルシクロプロパンカルボキサミドの分解生成物)が含まれる。
【0035】
「安定した」という用語は、本明細書中で使用される場合、剤形が実質的に純粋なレボミルナシプランを含む活性成分を含んでいることを意味する。
【0036】
「脱水アルコール」および「脱水溶媒」という用語は、本明細書において米国薬局方中のその定義の通り、99.5体積%以下の水に対応する0.8重量%以下の水を含むアルコールまたは溶媒を意味するものと定義される。「脱水溶媒(dehydrated solvent)」という用語は、本明細書において、「実質的に純粋な溶媒」、「無水溶媒(anhydrous solvent)」および「純溶媒(absolute solvent)」という用語と同義的に使用されている。同様にして、「脱水アルコール(dehydrated alcohol)」という用語は、「実質的に純粋なアルコール」、「無水アルコール(anhydrous alcohol)」および「純アルコール(absolute alcohol)」と同義的に使用されている。
【0037】
本明細書中で使用される「治療する」という用語は、ノルエピネフリン(NE)およびセロトニン(5−HT)の再取り込み阻害により治療してよい患者の疾病、障害または身体条件の少なくとも1つの疾候を緩和、軽減、遅延、削減、逆転、改善または予防することを意味する。「治療する」という用語は、ノルエピネフリン(NE)およびセロトニン(5−HT)の再取り込み阻害により治療してよい患者の疾病、疾病または身体条件の開始(すなわち疾病、障害または身体条件の臨床的徴候に先立つ期間)を阻むこと、遅延させることかつ/またはそれらの発達または悪化の危険性を低減させることも意味し得る。
【0038】
「剤形」という用語は、別段の指摘のないかぎり、本明細書において、ヒトの患者に対する経口投与に適したあらゆるレボミルナシプラン調合物を意味する。例えば、「剤形」という用語は、あらゆる経口剤形またはあらゆる固形経口剤形、例えばカプセル内に装填するのに適した組成物(例えば、ビーズ、顆粒、微小顆粒など)、錠剤、ジェルキャップ、カプレット、トローチ剤または粉剤を包含する。一部の実施形態において、剤形は、カプセル内への装填に適した剤形(例えばビーズ、顆粒、微小顆粒など)である。一部の実施形態において、剤形は、即時放出型剤形(例えば即時放出型固形剤形、即時放出型経口剤形または即時放出型固形経口剤形)である。
【0039】
一部の実施形態において、剤形は、放出調節型(例えば持続放出、遅延放出および/または徐放型)組成物でコーティングされた即時放出型組成物である。一部の実施形態において、剤形は、持続放出型剤形(例えば持続放出型固形剤形、持続放出型経口剤形、または持続放出型固形経口剤形)である。一部の実施形態において、剤形はカプセル(例えばビーズ、顆粒または微小顆粒が充填されたカプセル)である。一部の実施形態において、剤形は錠剤である。一部の実施形態において、剤形は一日一回の固形経口剤形である。一部の実施形態において、剤形は一日一回のカプセルである。
【0040】
「持続放出」という用語は、本明細書では、別段の指摘のないかぎり、投与直後以外の時点で、例えばレボミルナシプランの従来の瞬間放出および即時放出型剤形からの薬物放出時間を上回る長時間にわたりレボミルナシプラン(そして任意には中に含まれている追加の活性作用物質)を放出する剤形を意味するように使用されている。
【0041】
「使用環境内に入る」という用語は、本明細書では、別段の指摘のないかぎり、レボミルナシプランの安定した剤形と、それが投与される患者の胃液または腸液、胃液または腸液をシミュレートするように意図された流体または温度が約37℃でかつ75rpmのUSP装置IIに付された脱イオン水(例えば温度が約37℃でかつ75rpmのUSP装置IIに付された脱イオン水1000mL)との接触を意味するように使用されている。
【0042】
本明細書中で使用されるように、別段の指摘のないかぎり、溶出率は、当初安定した剤形内に入っていたレボミルナシプランの剤形が、使用環境内に入ってから、特定の時間内に剤形から放出される分率を定義する。
【0043】
別段の指摘のないかぎり、本明細書中で使用される「有効量」および「治療上有効な量」という用語は、患者に投与された場合に相応の生物学的応答を惹起するのに充分であるレボミルナシプランの量または数量を意味する。例えば、「有効量」および「治療上有効な量」という用語は、ノルエピネフリン(NE)およびセロトニン(5−HT)の再取り込み阻害により治療してよい疾病、身体条件または障害を治療するために患者(例えばヒトまたは他の哺乳動物)に投与された場合に、その疾病、傷害または身体条件の1つ以上の症候のこのような治療をもたらすのに充分であるレボミルナシプラン(またはその剤形中に入っている追加の活性作用物質)の量、または、患者の体内でのNEおよび5−HTの再取り込み阻害に充分であるレボミルナシプラン(またはその剤形内に入っている追加の活性作用物質)の量を意味する。精確な治療的用量は、患者の年令、身体条件、体重など、および治療対象の身体条件の性質によって左右され、究極的には主治医の判断に基づく。
【0044】
例えば、一部の実施形態において、うつ病(例えば大うつ病障害)を治療するための安定した剤形内のレボミルナシプランの治療上有効な投薬量は、実質的に純粋なレボミルナシプランを含む活性成分約10mg〜約150mg(例えば活性成分約20mg〜約120mg)であることがわかった。一部の実施形態において、剤形は実質的に純粋なレボミルナシプランを含む活性成分を約15mg〜約25mg(例えば約20mg)含む。一部の実施形態において、剤形は、実質的に純粋なレボミルナシプランを含む活性成分を約35mg〜45mg(例えば約40mg)含む。一部の実施形態において、剤形は、実質的に純粋なレボミルナシプランを含む活性成分を約70mg〜約90mg(例えば約80mg)含む。一部の実施形態において、実質的に純粋なレボミルナシプランを含む活性成分を約100mg〜約140mg(例えば約120mg)含む。
【0045】
本明細書で使用される通り、別段の指摘のないかぎり、安定した剤形に言及して使用される「純度」という用語は、その剤形が特定の望ましくない構成成分または不純物(例えば分解生成物など)を含まない(またはそれらが欠如している)度合いを意味する。
【0046】
「本質的に〜からなる」という用語は、剤形に関連して使用される場合、その剤形が追加の活性薬剤成分を全く含まないものの、追加の不活性構成成分または賦形剤は含んでよいということを意味する。
【0047】
別段の指摘のないかぎり、本明細書中で使用される「約」および「およそ(〜程度)」という用語は、特定の値について、その値の測定方法または決定方法すなわち測定システムの限界により一部左右されるものである当業者によって決定される許容誤差範囲内を意味するものと理解されるべきである。例えば、「約」は、当該技術分野における実践方法によると、1以内または1超の標準偏差を意味することができる。あるいは、「約」とは、所与の値の最高20%、好ましくは最高10%、より好ましくは最高5%、そしてより好ましくはさらに最高1%の範囲を意味し得る。
【0048】
経口剤形のX線粉末回折パターン
一部の好ましい実施形態において、安定した剤形または剤形内に入っている活性成分は結晶構造を有する。一部の好ましい実施形態においては、安定した剤形および安定した剤形内に入っている活性成分が結晶構造を有する。一部の実施形態において、安定した剤形は表1に提供されている1つ以上の特徴的ピークを含むX線粉末回折パターン(XRD)を有する。一部の実施形態において、安定した剤形は、レボミルナシプランを含む活性成分を含み、ここでこの活性成分は表1に提供されている1つ以上の特徴的ピークを含むX線粉末回折パターン(XRD)を有する。
【0049】
別段の指摘のないかぎり、本明細書中で使用される「1つ以上のピーク」という語句は、(i)この語句の後に記された全てのピーク値においてXRDピークを有する安定した剤形、(ii)この語句の後に記されたピーク値の少なくとも1つにおいてXRDピークを有する安定した剤形、ならびに(iii)この語句の後に記されたピーク値の2つ以上(例えば3つ以上、4つ以上、5つ以上、6つ以上さらには7つ以上)においてXRDピークを有する安定した剤形を包含するものとして理解されるべきである。
【0050】
【表1】
【0051】
一部の実施形態において、レボミルナシプランの安定した剤形は、表2に提供されている1つ以上の特徴的ピークを含むX線粉末回折パターン(XRD)を有する。
【0052】
【表2】
【0053】
一部の実施形態において、レボミルナシプランの安定した剤形は、放出調節型剤形(例えば持続放出型剤形)であり、表3に提供されている1つ以上の特徴的ピークを含むX線粉末回折パターン(XRD)を有する。
【0054】
【表3】
【0055】
一部の実施形態において、本発明は、レボミルナシプランまたはその薬学的に許容される塩を含む安定した剤形において、12.0、20.1および22.5±0.2度2θに特徴的ピークを含むX線粉末回折(XRD)パターンを含む安定した剤形に関する。一部の実施形態において、XRDパターンは同様に32.7±0.2度2θに特徴的ピークを含む。一部の実施形態において、XRDパターンは同様に、32.7±0.2度2θに特徴的ピークを含む。一部の実施形態において、XRDパターンは6.0±0.2度2θにも特徴的ピークを含む。
【0056】
一部の実施形態において、レボミルナシプランの安定した剤形は、約6.0、約12.0および約20.1±0.2度2θに特徴的ピークを含むXRDを有する。一部の実施形態において、安定した剤形は、約6.0、約12.0および約22.4±0.2度2θに特徴的ピークを含むXRDを有する。一部の実施形態において、安定した剤形は、約6.0、約12.0、約20.1および約22.4±0.2度2θに特徴的ピークを含むXRDを有する。一部の実施形態において、安定した剤形は、約6.0、約20.1および約22.4±0.2度2θに特徴的ピークを含むXRDを有する。一部の実施形態において、安定した剤形は、約12.0、約20.1および約22.4±0.2度2θに特徴的ピークを含むXRDを有する。一部の実施形態において、安定した剤形は、約6.0および約12.0±0.2度2θに特徴的ピークを含むXRDを有する。一部の実施形態において、安定した剤形は、約6.0および約20.1±0.2度2θに特徴的ピークを含むXRDを有する。一部の実施形態において、安定した剤形は約6.0および約22.4±0.2度2θに特徴的ピークを含むXRDを有する。
【0057】
一部の実施形態において、安定した剤形は、約12.0および約20.1±0.2度2θに特徴的ピークを含むXRDを有する。一部の実施形態において、安定した剤形は、約12.0および約22.4±0.2度2θに特徴的ピークを含むXRDを有する。一部の実施形態において、安定した剤形は、約20.1±0.2度2θおよび約22.4±0.2度2θに特徴的ピークを含むXRDを有する。一部の実施形態において、安定した剤形は、約6.0±0.2度2θ、約12.0±0.2度2θ、約20.1±0.2度2θおよび約22.4±0.2度2θの1つ以上に特徴的ピークを含むXRDを有するレボミルナシプランの結晶形態を含む。
【0058】
一部の実施形態において、安定した剤形は、実質的に純粋なレボミルナシプランを含む活性成分を含み、ここでこの活性成分は表1に示された特徴的ピークの1つ以上を含んでいる。一部の実施形態において、安定した剤形は、実質的に純粋なレボミルナシプランを含む活性成分を含み、ここでこの安定した剤形は表1または表2に示された特徴的ピークの1つ以上を含む。
【0059】
安定した剤形の純度
安定した剤形および剤形中の活性成分は、
【化2】
という式(II)により表わされる(1S,5R)1−フェニル−3−アザビシクロ[3−1−0]ヘキサン−2−オンを驚くほど低い濃度で含むことが発見されている。
【0060】
一部の実施形態において、安定した剤形は、例えば40℃および相対湿度75%で1カ月、2カ月または3カ月の保管期間中保管した後、調和国際会議(ICH)指針により定義されている約0.0001〜約0.2重量%の(1S,5R)1−フェニル−3−アザビシクロ[3−1−0]ヘキサン−2−オンを含む。一部の実施形態において、安定した剤形は、例えば40℃、相対湿度75%で1カ月、2カ月、または3カ月の保管期間中保管した後、約0.2重量%未満の(1S,5R)1−フェニル−3−アザビシクロ[3−1−0]ヘキサン−2−オンを含む。一部の実施形態において、安定した剤形は、例えば40℃、相対湿度75%で1カ月、2カ月または3カ月の保管期間中保管した後、約0.0001〜約0.1重量%の(1S,5R)1−フェニル−3−アザビシクロ[3−1−0]ヘキサン−2−オンを含む。
【0061】
一部の好ましい実施形態において、安定した剤形は、例えば40℃、相対湿度75%で1カ月、2カ月、3カ月、4カ月、5カ月または6カ月の保管期間中保管した後、約0.001〜約0.2重量%、約0.01〜約0.2重量%、約0.0001〜約0.15重量%、約0.001〜約0.15重量%、約0.01〜約0.15重量%、約0.001〜約0.1重量%、約0.01〜約0.1重量%、約0.01〜約0.08重量%、さらには約0.001〜約0.08重量%の(1S,5R)1−フェニル−3−アザビシクロ[3−1−0]ヘキサン−2−オンを含む。一部の実施形態において、安定した剤形は、例えば40℃、相対湿度75%で1カ月、2カ月、3カ月、4カ月、5カ月または6カ月の保管期間中保管した後、約0.01〜約0.08重量%(例えば約0.001〜約0.08重量%)の(1S,5R)1−フェニル−3−アザビシクロ[3−1−0]ヘキサン−2−オンを含む。
【0062】
安定した剤形の調製
レボミルナシプランの安定した剤形は、適切な任意の方法によって調製可能である。一部の好ましい実施形態において、安定した剤形は、レボミルナシプラン、溶媒(例えば5重量%未満、4重量%未満、3重量%未満、2重量%未満、1重量%未満、0.8重量%未満、0.5重量%未満、さらには0.1重量%未満の水を含む溶媒)(例えば200プルーフエタノールなどの脱水アルコール)および任意には結合剤および付着防止剤または潤滑剤を含む(または本質的にこれからなるまたはこれからなる)溶液と不活性基質または充填剤とを接触させてレボミルナシプラン組成物を形成するステップを含む方法によって調製される。一部の実施形態において、レボミルナシプランの剤形は次に、放出制御剤、溶媒(例えば5重量%未満、4重量%未満、3重量%未満、2重量%未満、1重量%未満、0.8重量%未満、0.5重量%未満さらには0.1重量%未満の水を含む溶媒)(例えば200プルーフエタノールなどの脱水アルコール)そして任意には可塑剤、付着防止剤または流動促進剤を含む溶液とレボミルナシプラン組成物とを接触させることによってコーティングされて、持続放出型剤形となる。
【0063】
一部の好ましい実施形態において、安定した剤形は、レボミルナシプラン、溶媒(例えば5重量%未満、4重量%未満、3重量%未満、2重量%未満、1重量%未満、0.8重量%未満、0.5重量%未満、さらには0.1重量%未満の水を含む溶媒)結合剤および付着防止剤または潤滑剤を含む溶液と不活性基質または充填剤とを接触させてレボミルナシプラン組成物を形成するステップを含む方法によって調製される。一部の実施形態において、レボミルナシプランの剤形は次に、放出制御剤、可塑剤、付着防止剤または流動促進剤および溶媒(例えば5重量%未満、4重量%未満、3重量%未満、2重量%未満、1重量%未満、0.8重量%未満、0.5重量%未満さらには0.1重量%未満の水を含む溶媒)を含む溶液とレボミルナシプラン組成物とを接触させることによってコーティングされて、持続放出型剤形となる。
【0064】
一部の実施形態において、安定した剤形は、レボミルナシプラン、そして任意には結合剤、付着防止剤または潤滑剤および/または溶媒(例えば5重量%未満、4重量%未満、3重量%未満、2重量%未満、1重量%未満、0.8重量%未満、0.5重量%未満、さらには0.1重量%未満の水を含む溶媒)と不活性基質または充填剤とを接触させてレボミルナシプラン組成物(レボミルナシプランコアまたはレボミルナシプランビーズまたは顆粒)を形成するステップを含む方法によって調製される。一部の実施形態において、レボミルナシプランの剤形は次に、放出制御剤、そして任意には可塑剤、付着防止剤または潤滑剤および/または溶媒(例えば5重量%未満、4重量%未満、3重量%未満、2重量%未満、1重量%未満、0.8重量%未満、0.5重量%未満さらには0.1重量%未満の水を含む溶媒)とレボミルナシプラン組成物とを接触させることによってコーティングされて、持続放出型剤形となる。
【0065】
一部の実施形態において、安定した剤形は、レボミルナシプラン、溶媒(例えば5重量%未満、4重量%未満、3重量%未満、2重量%未満、1重量%未満、0.8重量%未満、0.5重量%未満、さらには0.1重量%未満の水を含む溶媒)および任意には結合剤および付着防止剤(または潤滑剤)を含む溶液と不活性基質または充填剤とを接触させてレボミルナシプラン組成物(例えばレボミルナシプランを含むコア、ビーズまたは顆粒)を形成するステップを含む方法によって調製される。一部の実施形態において、この方法はさらに、放出制御剤、そして任意には可塑剤、付着防止剤または潤滑剤および/または溶媒(例えば5重量%未満、4重量%未満、3重量%未満、2重量%未満、1重量%未満、0.8重量%未満、0.5重量%未満さらには0.1重量%未満の水を含む溶媒)を含むまたは本質的にこれらからなるまたはこれらからなる溶液とレボミルナシプラン組成物とを接触させるステップを含む。
【0066】
一部の実施形態において、安定した剤形は、レボミルナシプラン、結合剤および付着防止剤または潤滑剤および溶媒(例えば5重量%未満、4重量%未満、3重量%未満、2重量%未満、1重量%未満、0.8重量%未満、0.5重量%未満、さらには0.1重量%未満の水を含む溶媒)を含む溶液と不活性基質または充填剤とを接触させるステップを含む方法によって調製される。一部の実施形態において、接触ステップは本質的にレボミルナシプラン、結合剤、付着防止剤または潤滑剤および溶媒(例えば5重量%未満、4重量%未満、3重量%未満、2重量%未満、1重量%未満、0.8重量%未満、0.5重量%未満さらには0.1重量%未満の水を含む溶媒)からなる薬物層状化溶液で不活性基質を層状化するステップを含む。
【0067】
一部の好ましい実施形態において、接触ステップ(例えば薬物層状化ステップ)は、ウルスター法(Wurster process)(例えばウルスター装置内部で)などによって実施される。一部の好ましい実施形態において、コーティングステップは、ウルスター法(例えばウルスター装置内で)などにより実施される。一部の好ましい実施形態において、組合せステップと接触ステップの両方とも、ウルスター法などによって実施される。
【0068】
活性薬剤成分中のレボミルナシプランおよび2−(アミノメチル)−N,N−ジエチル−1−フェニルシクロプロパンカルボキサミドの他の異性体(例えば(1R,2S)−2−(アミノメチル)−N,N−ジエチル−1−フェニルシクロプロパンカルボキサミド))の相対的百分率を決定するためのプロセスは、任意の適切な方法を用いて、好ましくは逆相高性能液体クロマトグラフ(RP HPLC)(例えば220nmでのUV検出を用いる)によって実施可能である。
【0069】
安定した剤形の構成成分
安定した剤形は、任意の治療上有効な量のレボミルナシプランを含み得る。一部の実施形態において、安定した剤形は約5〜約200mgのレボミルナシプランを含む。一部の実施形態において、安定した剤形は約10〜約180mgのレボミルナシプランを含む。一部の実施形態において、安定した剤形は約20〜約150mgのレボミルナシプランを含む。一部の実施形態において、安定した剤形は約20〜約120mgのレボミルナシプランを含む。例えば、安定した剤形は約20mg、約40mg、約50mg、約60mg、約80mg、約100mg、120mgまたは約240mgのレボミルナシプランを含むことができる。この点に関して、安定した剤形は、剤形の他の構成成分との関係において任意の適切な重量百分率のレボミルナシプランを含むことができる。例えば、安定した剤形は、約35〜約65重量%(例えば約35〜約60重量%、約35〜約55重量%、約40〜約55重量%または約40〜約50重量%)のレボミルナシプランを含むことができる。
【0070】
レボミルナシプランの安定した剤形は同様に、不活性基質または充填剤も含んでいる。一部の好ましい実施形態において、安定した剤形は、糖例えばスクロース(例えば糖球)を含む不活性基質を含む。他の適切な不活性基質または充填剤としては、例えばイソマルト、リン酸ジカルシウム二水和物、硫酸カルシウム、ラクトース、マンニトール、ソルビトール、セルロース、微晶質セルロース、カオリン、塩化ナトリウム、乾燥デンプン、加水分解デンプン類、アルファー化デンプン、二酸化シリコーン、酸化チタン、ケイ酸アルミニウムマグネシウム、またはその混合物が含まれる。
【0071】
安定した剤形は、任意の適切な量の不活性基質または充填剤(例えば糖球)を含み得る。一部の実施形態において、安定した剤形は約15重量%〜約45重量%の不活性基質または充填剤を含む。一部の実施形態において、安定した剤形は、約20〜約40重量%の不活性基質または充填剤を含む。一部の実施形態において、安定した剤形は約25〜約40重量%の不活性基質または充填剤を含む。一部の実施形態において、安定した剤形は約30〜約40重量%の不活性基質または充填剤を含む。一部の実施形態において、安定した剤形は約35〜約40重量%の不活性基質または充填剤を含む。
【0072】
安定した剤形は、任意の適切なサイズで糖球を含むことができる。一部の実施形態において、安定した剤形は、約20〜約50メッシュのサイズを有する糖球を含む。一部の実施形態において、安定した剤形は、約25〜約45メッシュ程度のサイズを有する糖球を含む。一部の実施形態において、安定した剤形は約25〜約40メッシュのサイズを有する糖球を含む。一部の好ましい実施形態において、安定した剤形は、約30〜約40メッシュ程度(例えば約30〜約35メッシュ)のサイズを有する糖球を含む。例えば、安定した剤形は約30〜約40メッシュのサイズを有する糖球を約30〜約40重量%(例えば約35〜約40重量%)含んでいてよい。
【0073】
安定した剤形は同様に、例えばポリビニルピロリドン(例えばPovidone K30)などの結合剤を一部の好ましい実施形態において含んでいる。他の適切な結合剤としては、例えばデンプン、ポリビニルアルコール、アルファー化デンプン、ゼラチン、スクロース、グルコース、デキストロース、ラクトース、ソルビトール、ポリエチレングリコール、ワックス類、天然および合成ゴム類、例えばアカシア、トラガカント、アルギン酸ナトリウム、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース、エチルセルロース、veegumおよび合成ポリマー類、例えばアクリル酸およびメタクリル酸コポリマー類、メタクリル酸コポリマー類、メタクリル酸メチル、コポリマー類、メタクリル酸アミノアルキルコポリマー類、ポリアクリル酸/ポリメタクリル酸またはその混合物が含まれる。
【0074】
安定した剤形は、任意の適切な量の結合剤(例えばPVP)を含むことができる。一部の実施形態において、安定した剤形は約0.1〜約15重量%の結合剤を含む。一部の実施形態において、安定した剤形は、約1〜約12重量%の結合剤を含む。一部の実施形態において、安定した剤形は、約1〜約10重量%の結合剤を含む。一部の実施形態において、安定した剤形は、約2〜約10重量%の結合剤を含む。
【0075】
一部の好ましい実施形態において、安定した剤形は約3〜約10重量%の結合剤(例えばPVP)を含む。一部の実施形態において、安定した剤形は約4〜約10重量%の結合剤を含む。一部の実施形態において、安定した剤形は約2〜約8重量%の結合剤を含む。一部の実施形態において、安定した剤形は約4〜約8重量%の結合剤を含む。一部の実施形態において、安定した剤形は約5〜約7重量%の結合剤を含む。
【0076】
安定した剤形は同様に、一部の好ましい実施形態において、例えばタルクなどの付着防止剤または潤滑剤も含んでいる。他の適切な付着防止剤または潤滑剤としては、例えばステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸、ベヘン酸グリセロール、ポリエチレングリコール、タルク、鉱油、フマル酸ステアリルナトリウムまたはその混合物が含まれる。
【0077】
安定した剤形は、任意の適切な量の潤滑剤または付着防止剤(例えばタルク)を含むことができる。一部の実施形態において、安定した剤形は、約0.1〜約15重量%の潤滑剤または付着防止剤を含む。一部の実施形態において、安定した剤形は、約1〜約12重量%の潤滑剤または付着防止剤を含む。一部の実施形態において、安定した剤形は、約2〜約10重量%の潤滑剤または付着防止剤を含む。一部の実施形態において、安定した剤形は約3〜約10重量%の潤滑剤または付着防止剤を含む。一部の実施形態において、安定した剤形は約4重量%〜約10重量%の潤滑剤または付着防止剤を含む。一部の実施形態において、安定した剤形は約4〜約8重量%の潤滑剤または付着防止剤を含む。一部の実施形態において、安定した剤形は約5〜約8重量%の潤滑剤または付着防止剤を含む。
【0078】
一部の好ましい実施形態において、安定した剤形は約4〜約7.5重量%の潤滑剤または付着防止剤を含む。一部の好ましい実施形態において、安定した剤形は約5〜約7重量%の潤滑剤または付着防止剤を含む。
【0079】
一部の実施形態において、安定した剤形は、持続放出(SR)剤形であり、放出制御剤、ポリマー剤またはコーティングポリマー(例えばエチルセルロース)を含み、これは実質的に剤形からのレボミルナシプランの放出を持続させることに寄与する。他の適切な放出制御剤としては、例えばセルロースおよびセルロース誘導体、ワックス、カルボマー、ポリアルキレンポリオール、ポリカルボフィル、メタクリル酸誘導体、ゼラチン、ゴム、ポリ酸化エチレンおよびポリビニルピロリドン、またはその混合物が含まれる。一部の実施形態において、放出制御剤、ポリマー添加剤またはコーティングポリマーは、エチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシエチルセルロース、ヒドロキシメチルセルロース、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルメチルセルロースアセテートスクシネート、ヒドロキシプロピルメチルセルロースフタレート、メチルセルロース、セルロースアセテート、セルロースアセテートフタレート、セルロースアセテートトリメリテートおよびカルボキシメチルセルロースナトリウム;アクリル酸ポリマー類およびコポリマー類(好ましくはアクリル酸、メタクリル酸、アクリル酸メチル、アクリル酸エチル、メタクリル酸メチルおよび/またはメタクリル酸エチルから形成されたもの)およびEudragitTML30D−55およびL100−55、EudragitTM、EudragitTMおよびEudragitTMNE、RLおよびRSを含む、EudragitTM(Rohm Pharma;Westerstadt、Germany)の商標名の下で市販されている他のメタクリル樹脂;ビニルポリマー類およびコポリマー類、例えばポリビニルピロリドン、酢酸ビニル、酢酸ビニルフタレート、酢酸ビニルクロトン酸コポリマー、およびエチレン−酢酸ビニルコポリマー;酵素分解性ポリマー類、例えばアゾポリマー類、ペクチン、キトサン、アミロースおよびグアーゴム;ゼインおよびシェラックまたはその混合物から選択される。
【0080】
安定した剤形は、任意の適切な量の放出制御剤、ポリマー剤またはコーティングポリマー(例えばエチルセルロース)を含むことができる。安定した剤形は好ましくは約5〜約15重量%の放出制御剤、ポリマー剤またはコーティングポリマーを含む。しかしながら、一部の実施形態において、安定した剤形は約2〜約20重量%の放出制御剤、ポリマー剤またはコーティングポリマーを含む。一部の実施形態において、安定した剤形は、約5〜約12重量%の放出制御剤、ポリマー剤またはコーティングポリマーを含む。一部の実施形態において、安定した剤形は約8〜約12重量%の放出制御剤、ポリマー剤またはコーティングポリマーを含む。一部の実施形態において、安定した剤形は約8〜約11重量%の放出制御剤、ポリマー剤またはコーティングポリマーを含む。一部の実施形態において、安定した剤形は、約8〜約10重量%の放出制御剤、ポリマー剤またはコーティングポリマーを含む。
【0081】
安定した剤形は同様に、一部の好ましい実施形態において、例えばクエン酸トリエチルなどの可塑剤も含んでいる。他の適切な可塑剤としては、ポリエチレングリコール、プロピレングリコール、トリアセチン、フタル酸ジメチル、フタル酸ジエチル、フタル酸ジブチル、セバシン酸ジブチル、クエン酸トリブチル、クエン酸トリエチルアセチル、モノステアリン酸グリセロール、ヒマシ油、アセチル化モノグリセリド類またはその混合物が含まれる。
【0082】
安定した剤形は、任意の適切な量の可塑剤(例えばクエン酸トリエチル)を含むことができる。一部の実施形態において、安定した剤形は約0.1〜約10重量%の可塑剤を含む。一部の実施形態において、安定した剤形は約0.5〜約8重量%の可塑剤を含む。一部の実施形態において、安定した剤形は、約0.5〜約5重量%の可塑剤を含む。一部の実施形態において、安定した剤形は約1〜約5重量%の可塑剤を含む。一部の実施形態において、安定した剤形は約1〜約3重量%の可塑剤を含む。
【0083】
さらに、レボミルナシプランの安定した剤形は、一部の実施形態において、可塑剤、顔料、着色剤、安定剤、流動促進剤などの任意の追加の賦形剤または添加剤を含むことができる。
【0084】
一部の好ましい実施形態において、剤形は、約30重量%〜約65重量%(例えば約40重量%〜約60重量%、約45重量%〜約60重量%さらには約50重量%〜約60重量%)のレボミルナシプラン(または少なくとも98重量%さらには実質的に純粋なレボミルナシプランを含む活性成分);約25重量%〜約55重量%(例えば約30重量%〜約45重量%さらには約30重量%〜約40重量%)の不活性基質または充填剤;約2重量%〜約12重量%(例えば約4重量%〜約10重量%さらには約4重量%〜約8重量%)の結合剤;そして約0.5重量%〜約10重量%(例えば約1重量%〜約8重量%、約1重量%〜約5重量%さらには約2重量%〜約5重量%)の付着防止剤または潤滑剤を含む。
【0085】
一部の好ましい実施形態において、剤形は約45重量%〜約60重量%のレボミルナシプラン(または少なくとも98重量%さらには実質的に純粋なレボミルナシプランを含む活性成分);約30重量%〜約45重量%の不活性基質または充填剤;約4重量%〜約10重量%の結合剤;および約1重量%〜約5重量%の付着防止剤または潤滑剤を含む。
【0086】
一部の好ましい実施形態において、剤形は約50重量%〜約60重量%のレボミルナシプラン(または少なくとも98重量%さらには実質的に純粋なレボミルナシプランを含む活性成分);約30重量%〜約40重量%の不活性基質または充填剤;約4重量%〜約8重量%の結合剤;および約1重量%〜約5重量%の付着防止剤または潤滑剤を含む。
【0087】
一部の好ましい実施形態において、安定した剤形は40〜約55重量%(例えば約40〜約50重量%)のレボミルナシプラン;約3〜約10重量%(例えば約4〜約8重量%)の結合剤(例えばPVP);および約0.1〜約4重量%(例えば約1〜約3重量%)の可塑剤(例えばクエン酸トリエチル)を含む。
【0088】
一部の好ましい実施形態において、安定した剤形は40〜約55重量%(例えば約40〜約50重量%)のレボミルナシプラン;約3〜約7重量%(例えば約4〜約7重量%)の付着防止剤または潤滑剤(例えばタルク);および約0.1〜約4重量%(例えば約1〜約3重量%)の可塑剤(例えばクエン酸トリエチル)を含む。
【0089】
一部の好ましい実施形態において、安定した剤形は40〜約55重量%(例えば約40〜約50重量%)のレボミルナシプラン;約3〜約10重量%(例えば約4〜約8重量%)の結合剤(例えばPVP);約3〜約7重量%(例えば約4〜約7重量%)の付着防止剤または潤滑剤(例えばタルク);および約0.1〜約4重量%(例えば約1〜約3重量%)の可塑剤(例えばクエン酸トリエチル)を含む。
【0090】
一部の好ましい実施形態において、安定した剤形は40〜約55重量%(例えば約40〜約50重量%)のレボミルナシプラン;約3〜約10重量%(例えば約4〜約8重量%)の結合剤(例えばPVP);および約5〜約10.5重量%(例えば約6〜約10重量%)の放出制御剤(例えばエチルセルロース)を含む。
【0091】
一部の好ましい実施形態において、安定した剤形は40〜約55重量%(例えば約40〜約50重量%)のレボミルナシプラン;約3〜約7重量%(例えば約4〜約7重量%)の付着防止剤または潤滑剤(例えばタルク);および約5〜約10.5重量%(例えば約6〜約10重量%)の放出制御剤(例えばエチルセルロース)を含む。
【0092】
一部の好ましい実施形態において、安定した剤形は40〜約55重量%(例えば約40〜約50重量%)のレボミルナシプラン;約3〜約10重量%(例えば約4〜約8重量%)の結合剤(例えばPVP);約5〜約10.5重量%(例えば約6〜約10重量%)の放出制御剤(例えばエチルセルロース);および約5〜約10.5重量%(例えば約6〜約10重量%)の放出制御剤(例えばエチルセルロース)を含む。
【0093】
一部の好ましい実施形態において、安定した剤形は40〜約55重量%(例えば約40〜約50重量%)のレボミルナシプラン;約3〜約7重量%(例えば約4〜約7重量%)の付着防止剤または潤滑剤(例えばタルク);約5〜約10.5重量%(例えば約6〜約10重量%)の放出制御剤(例えばエチルセルロース);および約5〜約10.5重量%(例えば約6〜約10重量%)の放出制御剤(例えばエチルセルロース)を含む。
【0094】
一部の好ましい実施形態において、安定した剤形は、約35〜約60重量%(例えば約35〜約55重量%または約40〜約55重量%)のレボミルナシプラン;約20〜約45重量%(例えば約20〜約40重量%、約25〜約40重量%または約25〜35重量%)の不活性基質または充填剤(例えば糖球);約1〜約15重量%(例えば約2〜約12重量%、約3〜約10重量%、約3〜約9重量%または約4〜約8重量%)の結合剤(例えばPVP);約1〜約15重量%(例えば約2〜約12重量%、約3〜約10重量%、約3〜約9重量%、約4〜約8重量%または約4〜約7重量%)の付着防止剤または潤滑剤(例えばタルク);約1〜20重量%(例えば約5〜約15重量%または約8〜約12重量%)の放出制御剤(例えばエチルセルロース);および約0.1〜約10重量%(例えば約0.1〜約5重量%または約1〜約5重量%または約1〜約3重量%)の可塑剤(例えばクエン酸トリエチル)を含む。
【0095】
一部の好ましい実施形態において、安定した剤形は、約35〜約60重量%(例えば約35〜約55重量%または約40〜約55重量%)のレボミルナシプラン;約20〜約45重量%(例えば約20〜約40重量%、約25〜約40重量%または約25〜35重量%)の不活性基質(例えば糖球)(例えば約30〜35メッシュの糖球);約1〜約15重量%(例えば約2〜約12重量%、約3〜約10重量%、約3〜約9重量%または約4〜約8重量%)のPVP(例えばPovidone K30);約1〜約15重量%(例えば約2〜約12重量%、約3〜約10重量%、約3〜約9重量%、約3〜約8重量%、約4〜約8重量%または約4〜約7重量%)のタルク;約1〜20重量%(例えば約5〜約15重量%または約8〜約12重量%)のエチルセルロース;および約0.1〜約10重量%(例えば約0.1〜約5重量%または約1〜約5重量%または約1〜約3重量%)のクエン酸トリエチルを含む。
【0096】
一部の好ましい実施形態において、安定した剤形は40〜約55重量%(例えば約40〜約50重量%)のレボミルナシプラン;約25〜約40重量%の糖球(例えば約30〜35メッシュの糖球);約2〜約10重量%(例えば約4〜約8重量%)のPVP(例えばPovidone K30);約2〜約10重量%(例えば約4〜約8重量%)のタルク;約5〜約15重量%のエチルセルロース;約0.1〜約5重量%(例えば約1〜約5重量%)のクエン酸トリエチルを含む。
【0097】
安定した剤形は、所望のpKプロファイルを達成するため任意の適切な厚みで放出制御剤によりコーティングされているレボミルナシプランのビーズまたは顆粒(例えば微小顆粒または他の同様のコア)を含むことができる。一部の実施形態において、例えば安定した剤形は、ビーズまたは顆粒(または類似のコア)が入ったカプセルであり、ここでビーズまたは顆粒(または類似のコア)は、任意の所望の平均厚みを有する放出制御剤(および任意には可塑剤、付着防止剤または潤滑剤および/または溶媒)を含むコーティング組成物でコーティングされている。例えば、コーティング組成物は、約1〜約100ミクロン(例えば約5〜約75ミクロン、約5〜約60ミクロン、約5〜約50ミクロン、約5〜約40ミクロン、約5〜約30ミクロン、約10〜約30ミクロン、約15〜約30ミクロン、約20〜約30ミクロン、約25〜約35ミクロンさらには約25〜約35ミクロン)の平均厚みで、ビーズまたは顆粒(または類似のコア)に対して塗布可能である。
【0098】
一部の好ましい実施形態において、例えば安定した剤形は、レボミルナシプランのコーティングされたビーズまたは顆粒(または類似のコア)を含み、ここでコーティングは1つ以上の放出制御剤(例えばエチルセルロース)を含み、ビーズまたは顆粒(または類似のコア)上のコーティングの平均厚みは約20〜約35ミクロン(例えば約20〜約30ミクロン)である。例えば、剤形はビーズ、顆粒または微小顆粒が充填されたカプセルであり得、ここでビーズ、顆粒または微小顆粒(または類似のコア)は、約20〜約30ミクロン(例えばおよそ25ミクロン)の平均厚みでコーティング組成物(例えばエチルセルロースを含む)によりコーティングされている。
【0099】
一部の実施形態において、安定した剤形は、約400〜約900ミクロンの平均直径を有するビーズまたは顆粒(例えばコーティングされたビーズまたは顆粒)の形をしている。一部の実施形態において、安定した剤形は、約500〜約800ミクロンの平均直径を有するビーズまたは顆粒(例えばコーティングされたビーズまたは顆粒)の形をしている。一部の実施形態において、安定した剤形は、約600〜約800ミクロンの平均直径を有するビーズまたは顆粒(例えばコーティングされたビーズまたは顆粒)の形をしている。一部の実施形態において、安定した剤形は、約600〜約750ミクロンの平均直径を有するビーズまたは顆粒(例えばコーティングされたビーズまたは顆粒)の形をしている。一部の実施形態において、安定した剤形は、約650〜約850ミクロンの平均直径を有するビーズまたは顆粒(例えばコーティングされたビーズまたは顆粒)の形をしている。一部の実施形態において、安定した剤形は、約1000ミクロン未満の平均直径を有するビーズまたは顆粒(例えばコーティングされたビーズまたは顆粒)の形をしている。一部の実施形態において、安定した剤形は、約900ミクロン未満の平均直径を有するビーズまたは顆粒(例えばコーティングされたビーズまたは顆粒)の形をしている。
【0100】
本発明は同様に、5−HTおよびNEの再取り込み阻害によって管理可能である障害、例えば不安障害またはうつ病(例えば大うつ病障害)の治療のための医薬品の製造におけるレボミルナシプランの安定した剤形の使用方法も提供する。
【0101】
一部の実施形態において、安定した剤形はカプセル(例えばHPMCまたはゼラチンカプセル)内に装填される。例えば一部の好ましい実施形態において、安定した剤形はHPMCカプセル内に装填される。このようなHPMCカプセルは次に、乾燥剤(例えば約0.01〜約2グラム、約0.01〜約1グラムさらには約0.01〜約0.8グラムの乾燥剤)を伴ってまたは伴わずにボトルまたはキャニスタ内に包装され得る。一部の好ましい実施形態において、安定した剤形はHPMCカプセル内に収納され、乾燥剤無しで包装される。一部の好ましい実施形態において、安定した剤形はHPMCカプセル中に収納され、乾燥剤を伴って包装される。一部の実施形態において、安定した剤形は、ゼラチンカプセル内に収納され、乾燥剤無しで包装される。一部の実施形態において、安定した剤形はゼラチンカプセル内に収納され、乾燥剤を伴って包装される。
【0102】
安定した剤形の溶出率
レボミルナシプランの安定した剤形は、使用環境内に入った後所望の溶出率を提供することがわかっている。一部の実施形態において、安定した剤形は、使用環境内に入ってから約6時間〜約16時間後に少なくとも約80%(例えば少なくとも80%)の溶出率(例えば単相溶出率(single phase dissolution rate)を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約6時間〜約12時間後に少なくとも約80%の溶出率を提供する。
【0103】
一部の実施形態において、安定した剤形は、使用環境内に入ってから約2時間後に約60%未満の溶出率(例えば単相溶出率)を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約2時間後に約55%未満の溶出率(例えば単相溶出率)を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約2時間後に約50%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約2時間後に約45%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約2時間後に約40%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約2時間後に約20%〜約60%の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約2時間後に約25%〜約55%の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約2時間後に約30%〜約50%の溶出率を提供する。
【0104】
一部の実施形態において、安定した剤形は、使用環境内に入ってから約4時間後に約90%未満の溶出率(例えば単相溶出率)を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約4時間後に約80%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約4時間後に約70%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約4時間後に約65%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約4時間後に約60%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約4時間後に約40%〜約80%の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約4時間後に約45%〜約75%の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約4時間後に約40%〜約70%の溶出率を提供する。
【0105】
一部の実施形態において、安定した剤形は、使用環境内に入ってから約6時間後に約90%未満の溶出率(例えば単相溶出率)を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約6時間後に約85%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約6時間後に約80%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約6時間後に約75%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約6時間後に約70%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約6時間後に約40%〜約95%の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約6時間後に約50%〜約90%の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約6時間後に約60%〜約85%の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約6時間後に約60%〜約80%の溶出率を提供する。
【0106】
一部の実施形態において、安定した剤形は、使用環境内に入ってから約8時間後に約95%未満の溶出率(例えば単相溶出率)を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約8時間後に約90%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約8時間後に約85%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約8時間後に約65%〜約95%の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約8時間後に約65%〜約90%の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約8時間後に約65%〜約85%の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約8時間後に約70%〜約85%の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約8時間後に約70%〜約80%の溶出率を提供する。
【0107】
一部の実施形態において、安定した剤形は、使用環境内に入ってから約12時間後に約95%未満の溶出率(例えば単相溶出率)を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約12時間後に約90%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約12時間後に約75%〜約95%の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約12時間後に約80%〜約95%の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約12時間後に約80%〜約90%の溶出率を提供する。
【0108】
一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約6時間〜約16時間後に、少なくとも約80%(例えば少なくとも80%)の溶出率(例えば単相溶出率)を提供する(例えば温度が37℃で75rpmのUSP装置IIに付された1000mLの脱イオン水中に入った後であり、ここでレボミルナシプランは220nmの波長でUV検出器を伴うHPLCを用いて定量化されている)。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約6時間〜約12時間後に、少なくとも約80%の溶出率を提供する。
【0109】
一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約2時間後に約60%未満の溶出率(例えば単相溶出率)を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約2時間後に約55%未満の溶出率(例えば単相溶出率)を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約2時間後に約50%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約2時間後に約45%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約2時間後に約40%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約2時間後に約20%〜約60%の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約2時間後に約25%〜約55%の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約2時間後に約30%〜約50%の溶出率を提供する。
【0110】
一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約4時間後に約90%未満の溶出率(例えば単相溶出率)を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約4時間後に約80%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約4時間後に約70%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約4時間後に約65%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約4時間後に約60%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約4時間後に約40%〜約80%の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約4時間後に約45%〜約75%の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約4時間後に約40%〜約70%の溶出率を提供する。
【0111】
一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約6時間後に約90%未満の溶出率(例えば単相溶出率)を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約6時間後に約85%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約6時間後に約80%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約6時間後に約75%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約6時間後に約70%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約6時間後に約40%〜約95%の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約6時間後に約50%〜約90%の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約6時間後に約60%〜約85%の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約6時間後に約60%〜約80%の溶出率を提供する。
【0112】
一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約8時間後に約95%未満の溶出率(例えば単相溶出率)を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約8時間後に約90%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約8時間後に約85%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約8時間後に約65%〜約95%の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約8時間後に約65%〜約90%の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約8時間後に約65%〜約85%の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約8時間後に約70%〜約85%の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約8時間後に約70%〜約80%の溶出率を提供する。
【0113】
一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約12時間後に約95%未満の溶出率(例えば単相溶出率)を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約12時間後に約90%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約12時間後に約75%〜約95%の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約12時間後に約80%〜約95%の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約12時間後に約80%〜約90%の溶出率を提供する。
【0114】
一部の実施形態において、レボミルナシプランの安定した剤形は、40℃、相対湿度(RH)75%で1カ月、2カ月さらには3カ月間剤形を保管した後、論述された溶出率を達成する。
【0115】
安定した剤形の薬物動態(pK)性能
安定した剤形は、使用環境内に入った場合に長時間にわたるレボミルナシプランの持続放出を提供し、ヒトの患者に対して投与した場合に所望のpKプロファイルを達成するものと予期されている。一部の実施形態において、安定した剤形は、例えば単回投与の後およそ24時間にわたり、治療的血漿レベルのレボミルナシプランを提供し得る(すなわちこれを達成するものと予期される)。例えば、一部の実施形態において、安定した剤形は使用環境内に剤形が入ってから約4時間〜約24時間(例えば約5時間〜約24時間、さらには約6時間〜約24時間)の間レボミルナシプランを放出し得る。
【0116】
一部の実施形態において、安定した剤形は、患者に投与(例えば単回投与)した後、少なくとも1時間の平均Tmax(最大血漿濃度までの平均時間)を提供し得る。一部の実施形態において、安定した剤形は、患者に投与(例えば単回投与)した後、少なくとも2時間の平均Tmax(最大血漿濃度までの平均時間)を提供し得る。一部の実施形態において、安定した剤形は、患者に投与(例えば単回投与)した後、少なくとも3時間の平均Tmax(最大血漿濃度までの平均時間)を提供し得る。一部の実施形態において、安定した剤形は、少なくとも3.5時間の平均Tmaxを提供し得る。好ましくは、本発明の安定した剤形は、少なくとも4時間の平均Tmaxを提供し得る。例えば安定した剤形は、少なくとも4.5時間の平均Tmaxを提供し得る。
【0117】
一部の実施形態において、剤形は少なくとも5時間の平均Tmaxを提供し得る。一部の実施形態において、剤形は少なくとも5.5時間の平均Tmaxを提供し得る。一部の実施形態において、剤形は少なくとも6時間の平均Tmaxを提供し得る。安定した剤形は同様に、約4時間〜約12時間の平均Tmaxを提供できる。例えば、安定した剤形は約4時間〜約10時間の平均Tmaxを提供できる。一部の実施形態において、安定した剤形は約4.5時間〜約12時間の平均Tmaxを提供し得る。一部の実施形態において、剤形は約4.5時間〜約10時間の平均Tmaxを提供し得る。一部の実施形態において、剤形は約5時間〜約12時間の平均Tmaxを提供し得る。一部の実施形態において、剤形は約5時間〜約10時間の平均Tmaxを提供し得る。一部の実施形態において、剤形は約4時間〜約8時間の平均Tmaxを提供し得る。一部の実施形態において、剤形は約4.5時間〜約8.5時間の平均Tmaxを提供し得る。一部の実施形態において、剤形は約5時間〜約8時間の平均Tmaxを提供し得る。一部の実施形態において、剤形は約4時間〜約9時間の平均Tmaxを提供し得る。
【0118】
一部の実施形態において、安定した剤形は約500〜約20,000ng hr/mLの平均AUC0−∞(経時的レボミルナシプラン血漿濃度)を提供し得る。一部の実施形態において、安定した剤形は約500〜約15,000ng hr/mLの平均AUC0−∞(経時的レボミルナシプラン血漿濃度)を提供し得る。一部の実施形態において、安定した剤形は約500〜約10,000ng hr/mLの平均AUC0−∞(経時的レボミルナシプラン血漿濃度)を提供し得る。一部の実施形態において、安定した剤形は約1000〜約9000ngの平均AUC0−∞(経時的レボミルナシプラン血漿濃度)を提供し得る。一部の実施形態において、安定した剤形は約500〜約5,000ng hr/mLの平均AUC0−∞(経時的レボミルナシプラン血漿濃度)を提供し得る。一部の実施形態において、安定した剤形は約500〜約2500ng hr/mLの平均AUC0−∞(経時的レボミルナシプラン血漿濃度)を提供し得る。一部の実施形態において、剤形は、約500〜約2200ng hr/mLの平均AUC0−∞を提供し得る。一部の実施形態において、剤形は、約700〜約2500ng hr/mLの平均AUC0−∞を提供し得る。
【0119】
一部の実施形態において、剤形は約700〜約2200ng hr/mLの平均AUC0−∞を提供し得る。一部の実施形態において、剤形は約800〜約2200ng hr/mLの平均AUC0−∞を提供し得る。一部の実施形態において、剤形は約700〜約2300ng hr/mLの平均AUC0−∞を提供し得る。一部の実施形態において、剤形は約1000〜約2000ng hr/mLの平均AUC0−∞を提供し得る。一部の実施形態において、剤形は、約1000〜約1800ng hr/mLの平均AUC0−∞を提供し得る。一部の実施形態において、剤形は、約1100〜約1800ng hr/mLの平均AUC0−∞を提供し得る。一部の実施形態において、剤形は、約1200〜約1700ng hr/mLの平均AUC0−∞を提供し得る。一部の実施形態において、剤形は、約1300〜約1700ng hr/mLの平均AUC0−∞を提供し得る。一部の実施形態において、剤形は、約1300〜約1650ng hr/mLの平均AUC0−∞を提供し得る。
【0120】
安定した剤形により提供される平均最大血漿濃度(Cmax)は、剤形の強度を変更することによって(例えば剤形のTmaxに実質的な影響を及ぼすことなく)修正可能である。一部の実施形態において、剤形は、患者に対する投与(例えば単回投与)の後約200ng/ml未満の平均Cmaxを提供し得る。一部の実施形態において、剤形は、約180ng/ml未満の平均Cmaxを提供し得る。一部の実施形態において、剤形は、約170ng/ml未満の平均Cmaxを提供し得る。一部の実施形態において、剤形は約160ng/ml未満の平均Cmaxを提供し得る。一部の実施形態において、剤形は約150ng/ml未満の平均Cmaxを提供し得る。一部の実施形態において、剤形は約140ng/ml未満の平均Cmaxを提供し得る。一部の実施形態において、剤形は、約130ng/ml未満の平均Cmaxを提供し得る。
【0121】
一部の実施形態において、剤形は、約120ng/ml未満の平均Cmaxを提供し得る。一部の実施形態において、剤形は約110ng/ml未満の平均Cmaxを提供し得る。一部の実施形態において、剤形は、約100ng/ml未満の平均Cmaxを提供し得る。一部の実施形態において、剤形は約20〜約250ng/mLの平均Cmaxを提供し得る。一部の実施形態において、剤形は約20〜約200ng/mLの平均Cmaxを提供し得る。一部の実施形態において、剤形は約20〜約180ng/mLの平均Cmaxを提供し得る。一部の実施形態において、剤形は約30〜約140ng/mLの平均Cmaxを提供し得る。一部の実施形態において、剤形は約40〜約140ng/mLの平均Cmaxを提供し得る。一部の実施形態において、剤形は約20〜約150ng/mLの平均Cmaxを提供し得る。
【0122】
安定した剤形は同様に、一部の実施形態において、少なくとも約6時間の平均半減期(T1/2)を提供することが発見されている。一部の実施形態において、剤形は少なくとも約7時間の平均T1/2を提供し得る。一部の実施形態において、剤形は少なくとも約8時間の平均T1/2を提供し得る。一部の実施形態において、剤形は少なくとも約9時間の平均T1/2を提供し得る。一部の実施形態において、剤形は、少なくとも約10時間の平均T1/2を提供し得る。一部の実施形態において、剤形は少なくとも約11時間の平均T1/2を提供し得る。
【0123】
一部の実施形態において、剤形は、少なくとも約12時間の平均T1/2を提供し得る。一部の実施形態において、投薬量は、約6時間〜約24時間の平均T1/2を提供し得る。一部の実施形態において、投薬量は、約6時間〜約18時間の平均T1/2を提供し得る。一部の実施形態において、投薬量は約7時間〜約18時間の平均T1/2を提供し得る。一部の実施形態において、投薬量は約8時間〜約24時間の平均T1/2を提供し得る。一部の実施形態において、投薬量は約8時間〜約18時間の平均T1/2を提供し得る。
【0124】
一部の好ましい実施形態において、安定した剤形は放出調節型剤形であり、約1000〜約9000ng hr/mLの平均AUC0−∞を提供する。
【0125】
一部の好ましい実施形態において、安定した剤形は放出調節型剤形であり、約50〜約350ng/mlの平均Cmaxを提供する。
【0126】
一部の好ましい実施形態において、安定した剤形は放出調節型剤形であり、約5〜12時間の平均Tmaxを提供する。
【0127】
一部の好ましい実施形態において、安定した剤形は放出調節型剤形であり、約9時間〜約20時間の平均T1/2を提供する。
【0128】
一部の好ましい実施形態において、安定した剤形は放出調節型剤形であり、約1000〜約9000ng hr/mLの平均AUC0−∞、約50〜約350ng/mlの平均Cmax、約5〜12時間の平均Tmaxおよび約9時間〜約20時間の平均T1/2を提供する。
【0129】
一部の実施形態において、安定した剤形は約200ng/ml未満の平均Cmax、約2500ng hr/mL未満の平均AUC0−∞および少なくとも約4時間の平均Tmaxを提供し得る。
【0130】
一部の実施形態において、安定した剤形は、約180ng/ml未満の平均Cmax、約2500ng hr/mL未満の平均AUC0−∞および少なくとも約4時間の平均Tmaxを提供し得る。
【0131】
一部の実施形態において、安定した剤形は、約160ng/ml未満の平均Cmax、約2500ng hr/mL未満の平均AUC0−∞および少なくとも約4時間の平均Tmaxを提供し得る。
【0132】
一部の実施形態において、安定した剤形は、約150ng/ml未満の平均Cmax、約2500ng hr/mL未満の平均AUC0−∞および少なくとも約4時間の平均Tmaxを提供し得る。
【0133】
一部の実施形態において、安定した剤形は、約140ng/ml未満の平均Cmax、約2500ng hr/mL未満の平均AUC0−∞および少なくとも約4時間の平均Tmaxを提供し得る。
【0134】
一部の実施形態において、安定した剤形は、約140ng/mlの平均Cmax、約2200ng hr/mL未満の平均AUC0−∞および少なくとも約4時間の平均Tmaxを提供し得る。
【0135】
一部の実施形態において、安定した剤形は、約180ng/ml未満の平均Cmax、約2200ng hr/mL未満の平均AUC0−∞および少なくとも約5時間の平均Tmaxを提供し得る。
【0136】
一部の実施形態において、安定した剤形は、約180ng/ml未満の平均Cmax、約2200ng hr/mL未満の平均AUC0−∞および少なくとも約6時間の平均Tmaxを提供し得る。
【0137】
一部の実施形態において、安定した剤形は、約10〜約200ng/mlの平均Cmax、約500〜約2500ng hr/mLの平均AUC0−∞および約4〜約10時間の平均Tmaxを提供し得る。
【0138】
一部の実施形態において、安定した剤形は、約25〜約175ng/mlの平均Cmax、約500〜約2500ng hr/mLの平均AUC0−∞および約4時間〜約10時間の平均Tmaxを提供し得る。
【0139】
一部の実施形態において、安定した剤形は、約30〜約150ng/mlの平均Cmax、約500〜約2500ng hr/mLの平均AUC0−∞および約4〜約10時間の平均Tmaxを提供し得る。
【0140】
一部の実施形態において、安定した剤形は、約30〜約120ng/mlの平均Cmax、約500〜約2500ng hr/mLの平均AUC0−∞および約4〜約10時間の平均Tmaxを提供し得る。
【0141】
一部の実施形態において、安定した剤形は、約10〜約200ng/mlの平均Cmax、約600〜約2200ng hr/mLの平均AUC0−∞および約4〜約10時間の平均Tmaxを提供し得る。
【0142】
一部の実施形態において、安定した剤形は、約10〜約200ng/mlの平均Cmax、約800〜約2100ng hr/mLの平均AUC0−∞および約4〜約10時間の平均Tmaxを提供し得る。
【0143】
一部の実施形態において、安定した剤形は、約10〜約200ng/mlの平均Cmax、約900〜約2100ng hr/mLの平均AUC0−∞および約4〜約10時間の平均Tmaxを提供し得る。
【0144】
一部の実施形態において、安定した剤形は、約30〜約150ng/mlの平均Cmax、約600〜約2200ng hr/mLの平均AUC0−∞および約4〜約10時間の平均Tmaxを提供し得る。
【0145】
一部の実施形態において、安定した剤形は、約30〜約120ng/mlの平均Cmax、約600〜約2200ng hr/mLの平均AUC0−∞および約4〜約10時間の平均Tmaxを提供し得る。
【0146】
一部の実施形態において、安定した剤形は、約10〜約200ng/mlの平均Cmax、約500〜約2500ng hr/mLの平均AUC0−∞および約4〜約8時間の平均Tmaxを伴う生体内血漿プロファイルを提供し得る。
【0147】
一部の実施形態において、安定した剤形は、約10〜約200ng/mlの平均Cmax、約500〜約2500ng hr/mLの平均AUC0−∞および約5〜約8時間の平均Tmaxを伴う生体内血漿プロファイルを提供し得る。
【0148】
一部の実施形態において、安定した剤形は、約25〜約175ng/mlの平均Cmax、約600〜約2200ng hr/mLの平均AUC0−∞および約4〜約9時間の平均Tmaxを伴う生体内血漿プロファイルを提供し得る。
【0149】
一部の実施形態において、安定した剤形は、約30〜約150ng/mlの平均Cmax、約800〜約2100ng hr/mLの平均AUC0−∞および約4〜約9時間の平均Tmaxを伴う生体内血漿プロファイルを提供し得る。
【0150】
一部の実施形態において、約125ng/ml未満の平均Cmax、約1000〜約2200ng hr/mLの平均AUC0−∞および少なくとも約4時間の平均Tmaxを伴う生体内血漿プロファイルを提供し得る剤形が提供されている。
【0151】
安定した剤形を使用する治療方法
本発明は同様に、治療を必要としている患者に対して安定した剤形を投与することによって哺乳動物(例えばヒト)における不安障害またはうつ病(例えば大うつ病性障害(MDD))などの5−HTおよびNEの再取り込み阻害(例えば2重阻害および/または選択的阻害)により管理可能な疾病、障害または身体条件を治療するための方法も提供する。
【0152】
一部の実施形態においては、治療を必要としている患者におけるうつ病(例えば非定型うつ病またはMDD)、不安神経症(例えば全般的不安障害)またはうつ病や不安神経症に付随する倦怠感を、前記患者に対してレボミルナシプランの安定した剤形を投与することによって治療または予防するための方法が提供されている。一部の実施形態において、治療を必要とする患者における大うつ病性障害(MDD)(例えば急性MDDまたは非定型MDD)を、前記患者に対してレボミルナシプランの安定した剤形を投与することによって治療または予防するための方法が提供されている。一部の実施形態において、治療を必要とする患者における未解決の、併発するまたは随伴する倦怠感を伴うMDDを、前記患者に対してレボミルナシプランの安定した剤形を投与することによって治療または予防するための方法が提供されている。
【0153】
一部の実施形態において、患者に剤形を投与することによって治療を必要とする患者におけるMDDの再発を治療または予防するために、レボミルナシプランの安定した剤形が使用される。一部の実施形態において、安定した剤形は、患者に剤形を投与することによって治療を必要とする患者における倦怠感(例えばMDDまたは他の形態のうつ病に付随する倦怠感)を治療または予防するために使用される。一部の実施形態において、安定した剤形は、患者に剤形を投与することによって治療を必要とする患者における性的機能不全(例えば勃起不全)を治療または予防するために使用される。一部の実施形態において、安定した剤形は、患者に剤形を投与することによって治療を必要とする患者におけるうつ病(例えばMDD)に付随する疼痛を治療または予防するために使用される。
【0154】
一部の実施形態において、本発明の安定した剤形は、患者に安定した剤形を投与することによって治療を必要とする患者におけるうつ病(例えばMDD)に付随する(またはそれと併発する)うつ状態、気分変調、傾眠、認識機能障害、睡眠障害および/または高脂血症を治療または予防するために使用される。
【0155】
一部の実施形態においては、治療を必要とする患者において、神経因性疼痛(例えば糖尿病性多発神経障害性疼痛(DPNP))を治療または予防するための方法において、患者に対してレボミルナシプランの安定した剤形を有効量投与するステップを含む方法が提供されている。
【0156】
レボミルナシプランの安定した剤形を投与することを通して、薬物血漿濃度対時間の平坦化されたプロファイルを得、それにより一日多数回の投薬で得ることのできるものよりも密な血漿療法範囲の制御を提供するための方法が提供されている。換言すると、レボミルナシプランの従来の即時放出型調合物での一日多数回の投薬により誘発される血漿薬物レベルにおける急なピークおよびトラフを除去するための方法が提供されている。本質的には、レボミルナシプランの血漿レベルは、本発明の安定した剤形を投与した後数時間にわたり上昇し、その後下降し始め、およそ24時間の残りの時間中ピーク血漿レベルからの実質的に線形の持続的な減少を長々と続け、全期間中少なくともレボミルナシプランの閾値治療レベルを維持する。
【0157】
以下の実施例は単に本発明を例示しているにすぎず、当業者には本開示を読んだ上で本発明により包含される多くの変形形態および等価物が明白となるため、いかなる形であれ本発明の範囲を限定するものとしてみなされてはならない。
【実施例】
【0158】
実施例1:レボミルナシプラン剤形の調製
糖球(およそ30〜35メッシュ)を予熱しウルスタープロセスを介して約3.5時間にわたり薬物層溶液で予熱済み糖球を層状化して薬物装填済みビーズを形成することにより、レボミルナシプランビーズを調製した。薬物層溶液は、レボミルナシプラン、Povidone K30、タルクおよび脱水アルコールを含んでいた。流動床内で約30分間薬物装填済みビーズを乾燥させ、篩過して、表4に示されている構成成分の濃度をおおよそ含む即時放出型レボミルナシプランビーズ(およそ540mg/g)を生成した。
【0159】
【表4】
【0160】
次に、レボミルナシプランビーズを予熱し、ウルスタープロセスを介して分散溶液でコーティングした。分散溶液は、エチルセルロースN22、クエン酸トリエチル、タルクおよび脱水アルコールを含んでいた。コーティングされたビーズを流動床内で入念に硬化させ、篩分けしてレボミルナシプランの持続放出型ビーズ(およそ460mg/g)を生成してからカプセル内に充填した。持続放出型レボミルナシプランビーズは、おおよそ表5に示された構成成分濃度を含んでいた。
【0161】
【表5】
【0162】
実施例2:実施例1で調製された剤形の安定性
2つの異なるタイプのカプセル(すなわち硬質ゼラチンカプセルおよびHPMCカプセル)内の剤形を40℃、相対湿度(RH)75%で3カ月間保管した後、実施例1で調製したレボミルナシプランの剤形の安定性を査定した。
【0163】
表6は、3カ月の保管後にレボミルナシプランの安定した剤形の硬質ゼラチンカプセル内に見出された不純物のおおよその濃度を示す。
【0164】
【表6】
【0165】
表7は、レボミルナシプランの安定した剤形のHPMCカプセル内に見出された不純物のおおよその濃度を示す。
【0166】
【表7】
【0167】
実施例3:実施例1で調製された剤形の溶出率
実施例1で調製されたレボミルナシプランの安定した剤形の溶出率を、75rpmのUSP装置IIを用いて37℃の脱イオン水1000mLで評価した。レボミルナシプランの定量化は、220nmの波長でUV検出器を伴うHPLCを用いて判定した。
【0168】
実施例1で調製した(乾燥剤を全く含まないHPMCカプセル内に装填された)安定した剤形の溶出プロファイルを、図1および表8に示す。
【0169】
実施例1で調製した安定した剤形の溶出プロファイルを同様に、40℃および相対湿度(RH)75%で1、2および3カ月間剤形を保管した後にも判定し、この場合剤形は表8および図1〜5で示す通り、(1グラムの乾燥剤を含むかまたは乾燥剤を全く含まない)HPMCカプセル内に装填した。
【0170】
詳細には、図1は、保管を全くしていないレボミルナシプランの安定した剤形についての溶出率を示しており、ここで剤形は乾燥剤を全く含まないHPMCカプセル内に装填されている。図2は、40℃、75%RHで1カ月保管した後のレボミルナシプランの安定した剤形についての溶出率を示しており、ここでも剤形は、乾燥剤を全く含まないHPMCカプセル内に装填されている。図3は、40℃、75%RHで2カ月保管した後のレボミルナシプランの安定した剤形についての溶出率を示しており、ここでも剤形は乾燥剤を全く含まないHPMCカプセル内に装填されている。図4は、40℃、75%RHで3カ月保管した後のレボミルナシプランの安定した剤形についての溶出率を示しており、ここでも剤形は、乾燥剤を全く含まないHPMCカプセル内に装填されている。図5は、40℃、75%RHで3カ月保管した後のレボミルナシプランの安定した剤形についての溶出率を示しており、ここで剤形は乾燥剤を1グラム含むHPMCカプセル内に装填されている。
【0171】
【表8】
【0172】
実施例4− 実質的に純粋なレボミルナシプランを含む活性成分のX線粉末回折(XRD)分析
塩酸レボミルナシプランを少なくとも98重量%含む活性成分の試料を深いアルミニウムホルダー上に装填し、広角ベンチトップX線回折計(D8型、Bruker AXS Inc.,Madison WI)内でCuKα放射(40kV×40mA)に曝露した。計器を0.05°2θの増分でステップ走査モードで作動させた。角度範囲は5〜40°2θであり、走査速度は0.15°2θ/分であった。市販のソフトウェアを用いてデータ収集および分析を実施した。
【0173】
図6のXRDパターンについてのピーク位置を表1に示す。
【0174】
【表9】
【0175】
実施例5− 実施例1で調製した即時放出型レボミルナシプランビーズのX線粉末回折(XRD)分析
実施例1で調製したIR(即時放出型)レボミルナシプランビーズを深いアルミニウムホルダー上に装填し、広角ベンチトップX線回折計(D8型、Bruker AXS Inc.,Madison WI)内でCuKα放射(40kV×40mA)に曝露した。計器を0.05°2θの増分でステップ走査モードで作動させた。角度範囲は5〜40°2θであり、走査速度は0.15°2θ/分であった。市販のソフトウェアを用いてデータ収集および分析を実施した。
【0176】
図7のXRDパターンについてのピーク位置を表2に示す。
【0177】
【表10】
【0178】
実施例6− 実施例1で調製した持続放出型剤形のX線粉末回折(XRD)分析
実施例1で調製した少量の持続放出型剤形を深いアルミニウムホルダー上に装填し、広角ベンチトップX線回折計(D8型、Bruker AXS Inc.,Madison WI)内でCuKα放射(40kV×40mA)に曝露した。計器を0.05°2θの増分でステップ走査モードで作動させた。角度範囲は5〜40°2θであり、走査速度は0.15°2θ/分であった。市販のソフトウェア(具体的には、DIFFRACplus XRD Commander,Bruker−AXS GmbH;およびJADE,Materials Data,Inc.)を用いてデータ収集および分析を実施した。
【0179】
レボミルナシプランの安定した剤形についてのXRDパターンを図7に示す。図8のXRDパターンについてのピーク位置は表3に提供されている。
【0180】
【表11】
【0181】
実施例7− ヒトの患者に対するレボミルナシプランの安定した剤形の投与(理論上の予測)
50mgのレボミルナシプランが入ったカプセルの形でヒトの患者に対して本発明のレボミルナシプランの安定した剤形を投与することができる。カプセルには、約7.5重量%のエチルセルロース(EC)(「剤形1」)、約10重量%のEC(「剤形2」)および約12.5重量%EC(「剤形3」)でコーティングされた微小顆粒が入り得る。
【0182】
剤形1〜3は、少なくとも約10時間の絶食期間後に患者に投与することができる。投与前および投与後複数の時点(例えば30分、1時間、2時間、3時間、4時間、5時間、6時間、7時間、8時間、10時間、12時間、24時間、48時間および72時間後)に各患者から血液試料を収集することができる。各患者からの血漿試料を、LC/MS−MS検出と連動させた有効オンライン抽出方法(乱流クロマトグラフィ)を用いて、レボミルナシプランの定量化について各患者を査定することができる。
【0183】
安定した剤形を経口投与することにより、表9に剤形1〜3のいずれかについて実質的に示されている通りの薬物動態(pK)パラメータが生成されることが予期される。一部の好ましい実施形態において、安定した剤形は、実質的に剤形2について示されている通りのpKパラメータを生成する。これらのpKパラメータは、幾何平均(幾何学的CV%)および[範囲]としても表現されている。
【0184】
【表12】
【0185】
単回経口投与後(投薬から最高24時間後)に得られうる平均レボミルナシプラン血漿濃度対時間プロファイルは、図9(幾何平均として表わされた対数−線形スケール)に示されている。
【0186】
本発明について、その例示的実施形態を参考にして描写し記述してきたが、このような参照は本発明に対する限定を意味せず、このような限定が暗示されることは一切ない。本発明は、本開示の利益を受ける関連技術分野の当業者が思いつく形態および機能面の多数の修正、改変および等価物の可能性を含むものである。
【0187】
描写したおよび記述した本発明の実施形態は、単なる一例にすぎず、本発明の範囲を完全に網羅するものではない。したがって、本発明は添付クレームの精神および範囲によってのみ制限され、あらゆる面で等価物を認知するものである。
【技術分野】
【0001】
本出願は、2010年1月14日出願の米国仮特許出願第61/294,898号に対する35U.S.C.119(e)に基づく優先権;およびそれ自体2009年11月6日出願の米国仮特許出願第61/258,652号に対する35U.S.C.119(e)に基づく優先権を主張している2010年11月8日出願の米国特許出願第12/941,293号に対する35U.S.C.120に基づく優先権を主張するものである。
【0002】
本発明は、レボミルナシプランまたはその薬学的に許容される塩の安定した投薬調合物に関する。これらの剤形の調製用プロセスおよびこれらの剤形の使用方法も同様に記載されている。
【背景技術】
【0003】
レボミルナシプランは、(1S,2R)−2−(アミノメチル)−N,N−ジエチル−1−フェニルシクロプロパンカルボキサミドの国際一般名である。これは、5−HT再取り込み阻害よりもNE再取り込み阻害に対して大きな選択性を有する非常に強力な選択的ノルエピネフリン(NE)およびセロトニン(5−HT)再取り込み阻害薬である。詳細には、レボミルナシプランは、NE:5−HTについておよそ1.5:1という阻害選択性比を有する。したがって、レボミルナシプランは、ノルエピネフリン−セロトニン再取り込み阻害薬(NSRI)とみなされており、これはNEに対してよりも5−HTに対して同等以上の阻害選択性を有するセロトニン−ノルエピネフリン再取り込み阻害薬(SNRI)とは薬理学的に全く異なる。
【0004】
レボミルナシプランの調合物については先行技術において一般的に論述されているものの、レボミルナシプランの安定した剤形を調製する上で問題点に直面してきた。これらの問題点は、少なくとも一部には、ある種の反応条件に対するレボミルナシプランの感応性および一般的に使用される一部の賦形剤とのその反応性に起因して発生していた。
【発明の概要】
【発明が解決しようとする課題】
【0005】
したがって、改善された純度と安定性を有するレボミルナシプランの改善された調合物に対するニーズが現在ひき続き存在している。さらに、安定性が改善した調合物は、患者における望ましくない不利な事象(例えば吐き気、嘔吐および胃出血)の発生率低下と結びつけられる望ましい薬物動態プロファイルを達成しなければならない。
【課題を解決するための手段】
【0006】
現在、使用環境に入った時点で所望されるレボミルナシプラン放出を達成しかつ意外なほど高い安定性を有する改良型レボミルナシプラン調合物が、発見されている。これらの改良型レボミルナシプラン調合物が本明細書中で記述されている。
【0007】
本発明は新規のレボミルナシプラン剤形、ならびにこれらの剤形の調製方法および剤形の使用方法に関する。
【0008】
一部の実施形態において、本発明は、レボミルナシプランまたはその薬学的に許容される塩を含む安定した剤形に関する。
【0009】
一部の実施形態において、本発明は、実質的に純粋なレボミルナシプランまたはその薬学的に許容される塩を含む活性成分を含むまたは本質的にこの活性成分からなる安定した剤形に関する。
【0010】
一部の実施形態において、本発明は、少なくとも約98重量%(例えば少なくとも98重量%)のレボミルナシプランまたはその薬学的に許容される塩を含む活性成分を含むまたは本質的にこの活性成分からなる安定した剤形に関する。
【0011】
一部の実施形態において、本発明は、剤形が12.0、20.1および22.5±0.2度2θに特徴的ピークを含むX線粉末回折(XRD)パターンを含む、レボミルナシプランまたはその薬学的に許容される塩を含む安定した剤形に関する。一部の実施形態において、XRDパターンは同様に32.7±0.2度2θに特徴的ピークを含む。一部の実施形態において、XRDパターンは同様に32.7±0.2度2θに特徴的ピークを含む。一部の実施形態において、XRDパターンは同様に6.0±0.2度2θに特徴的ピークを含む。
【0012】
一部の実施形態において、本発明は、レボミルナシプランまたはその薬学的に許容される塩を含む安定した剤形において、6.0、12.0および20.1±0.2度2θに特徴的ピークを含むX線粉末回折(XRD)パターンを含む安定した剤形に関する。一部の実施形態において、XRDパターンは同様に22.5±0.2度θに特徴的ピークを含む。
【0013】
一部の実施形態において、本発明は、レボミルナシプランまたはその薬学的に許容される塩および約0.001重量%〜約0.5重量%の(1S,5R)1−フェニル−3−アザビシクロ[3−1−0]ヘキサン−2−オンを含む安定した剤形に関する。
【0014】
一部の実施形態において、本発明は、レボミルナシプランまたはその薬学的に許容される塩および約0.001%重量%〜約0.2重量%の(1S,5R)1−フェニル−3−アザビシクロ[3−1−0]ヘキサン−2−オンを含む安定した剤形に関する。
【0015】
一部の実施形態において、安定した剤形は、実質的に純粋なレボミルナシプランまたはその薬学的に許容される塩を含む約45重量%〜約60重量%の活性成分を含む。
【0016】
一部の実施形態において、安定した剤形は、実質的に純粋なレボミルナシプランまたはその薬学的に許容される塩を含む約45重量%〜約60重量%の活性成分と、約4重量%〜約10重量%の結合剤を含む。
【0017】
一部の実施形態において、安定した剤形は、実質的に純粋なレボミルナシプランまたはその薬学的に許容される塩を含む約45重量%〜約60重量%の活性成分と、約30重量%〜約45重量%の不活性基質または充填剤と、約4重量%〜約10重量%の結合剤とを含む。
【0018】
一部の実施形態において、安定した剤形は、実質的に純粋なレボミルナシプランまたはその薬学的に許容される塩を含む約45重量%〜約60重量%の活性成分と、約30重量%〜約45重量%の不活性基質または充填剤と、約4重量%〜約10重量%の結合剤と、約1重量%〜約5重量%の付着防止剤または潤滑剤とを含む。
【0019】
一部の実施形態において、安定した剤形は、約50重量%〜約60重量%のレボミルナシプランまたはその薬学的に許容される塩と、約30重量%〜約40重量%の不活性基質または充填剤と、約4重量%〜約8重量%の結合剤と、約1重量%〜約5重量%の付着防止剤または潤滑剤とを含む。
【0020】
一部の実施形態において、安定した剤形は、約40重量%〜約55重量%のレボミルナシプランまたはその薬学的に許容される塩と、約5重量%〜約15重量%の放出制御剤と、約25重量%〜約40重量%の不活性基質と、約3重量%〜約10重量%の結合剤と、約3重量%〜約10重量%の付着防止剤と、約0.1重量%〜約5重量%の可塑剤とを含む。
【0021】
一部の実施形態において、安定した剤形は、使用環境内に入ってから約6時間〜約16時間後に少なくとも約80%の溶出率を提供する。
【0022】
一部の実施形態において、本発明は、約10mg〜約200mgのレボミルナシプランを含む活性成分を含む安定した経口剤形において、12.0、20.1および22.5±0.2度2θに特徴的ピークを含むX線粉末回折(XRD)パターンを含む、安定した経口剤形に関する。
【0023】
一部の実施形態において、本発明は、約10mg〜約200mgのレボミルナシプランと約0.0001重量%〜約0.5重量%(例えば約0.0001重量%〜約0.2重量%さらには約0.0001重量%〜約0.1重量%)の(1S,5R)1−フェニル−3−アザビシクロ[3−1−0]ヘキサン−2−オンを含む経口剤形に関する。
【0024】
一部の実施形態において、本発明は、約10mg〜約200mgのレボミルナシプランと約0.0001重量%〜約0.5重量%(例えば約0.0001重量%〜約0.2重量%さらには約0.0001重量%〜約0.1重量%)の(1S,5R)1−フェニル−3−アザビシクロ[3−1−0]ヘキサン−2−オンを含む経口剤形において、剤形が、12.0、20.1および22.5±0.2度2θに特徴的ピークを含むX線粉末回折(XRD)パターンを含む経口剤形に関する。
【0025】
一部の実施形態において、剤形は、使用環境内に入ってから約6時間〜約16時間後に少なくとも約80%の溶出率を提供する。
【0026】
一部の実施形態において、本発明は、レボミルナシプランまたはその薬学的に許容される塩と放出制御剤を含む安定した剤形において、使用環境内に入った後レボミルナシプランまたはその薬学的に許容される塩の放出を持続する安定した剤形に関する。
【0027】
一部の実施形態において、本発明は、約20mg、約40mg、約80mgまたは約120mgのレボミルナシプランまたはその薬学的に許容される塩を含む安定した剤形に関する。
【0028】
一部の実施形態において、本発明は、治療を要する患者に対してレボミルナシプランまたはその薬学的に許容される塩の安定した剤形を投与するステップを含む、大うつ病障害の治療方法に関する。
【0029】
一部の実施形態において、本発明は、治療を要する患者に対してレボミルナシプランまたはその薬学的に許容される塩の安定した剤形を投与するステップを含む、倦怠感を同時に伴う大うつ病障害の治療方法に関する。
【0030】
一部の実施形態において、本発明は、不活性基質をレボミルナシプランまたはその薬学的に許容される塩および脱水アルコールと接触させるステップを含む、安定した剤形の調製方法に関する。
【図面の簡単な説明】
【0031】
【図1】本発明の一実施形態に係るレボミルナシプランの安定した剤形についての溶出率を示す。
【図2】40℃および相対湿度(RH)75%で1カ月の保管後の本発明の一実施形態に係るレボミルナシプランの安定した剤形についての溶出率を示す。
【図3】40℃および75%RHで2カ月の保管後の本発明の一実施形態に係るレボミルナシプランの安定した剤形についての溶出率を示す。
【図4】40℃および75%RHで3カ月の保管後の本発明の一実施形態に係るレボミルナシプランの安定した剤形についての溶出率を示す。
【図5】40℃および75%RHで3カ月の保管後の、1グラムの乾燥剤を含む本発明の一実施形態に係るレボミルナシプランの安定した剤形についての溶出率を示す。
【図6】実質的に純粋なレボミルナシプランを含む活性薬剤成分についてのX線粉末回折パターン(XRD)を示す。
【図7】本発明の一実施形態に係るレボミルナシプランの安定した即時放出型剤形のXRDを示す。
【図8】本発明の一実施形態に係るレボミルナシプランの安定した持続放出型剤形のXRDを示す。
【図9】ヒトの患者に対するレボミルナシプランの安定した剤形の単回投与を介して達成され得る時間に対するレボミルナシプランの平均血漿濃度を示す。
【発明を実施するための形態】
【0032】
レボミルナシプランの新規の安定した剤形、これらの剤形を使用した治療方法およびこれらの剤形の調製方法が、本明細書中で提供されている。レボミルナシプランの剤形は、使用環境に入った時点で所望の溶出プロファイルを達成し、驚くほど高い安定性を有することが発見された。
【0033】
定義
本明細書中で使用される「レボミルナシプラン」という用語は、(1S,2R)−2−(アミノメチル)−N,N−ジエチル−1−フェニルシクロプロパンカルボキサミドおよびその薬学的に許容される塩に関する。この用語は2−(アミノメチル)−N,N−ジエチル−1−フェニルシクロプロパンカルボキサミドの他の異性体(例えば(1R,2S)−2−(アミノメチル)−N,N−ジエチル−1−フェニルシクロプロパンカルボキサミド)または(1S,2R)−2−(アミノメチル)−N,N−ジエチル−1−フェニルシクロプロパンカルボキサミドの分解生成物(例えば(1S,5R)−1−フェニル−3−アザビシクロ[3−1−0]ヘキサン−2−オン)を含まない。「薬学的に許容される塩」という用語は、患者が生理学的に耐容するレボミルナシプランのあらゆる塩(例えば塩酸レボミルナシプラン)を意味する。レボミルナシプランの構造式を以下に示す:
【化1】
【0034】
「実質的に純粋なレボミルナシプラン」という用語は、本明細書中で、少なくとも98重量%のレボミルナシプランを意味するように使用されている。例えば、実質的に純粋なレボミルナシプランを含む活性薬剤成分(すなわち活性成分)には、少なくとも98重量%(例えば約98.5重量%)のレボミルナシプランおよび多くとも2重量%の全ての組合わされた他の構成成分(例えば2−(アミノメチル)−N,N−ジエチル−1−フェニルシクロプロパンカルボキサミドの他の異性体および/または(1S,2R)−2−(アミノメチル)−N,N−ジエチル−1−フェニルシクロプロパンカルボキサミドの分解生成物)が含まれる。
【0035】
「安定した」という用語は、本明細書中で使用される場合、剤形が実質的に純粋なレボミルナシプランを含む活性成分を含んでいることを意味する。
【0036】
「脱水アルコール」および「脱水溶媒」という用語は、本明細書において米国薬局方中のその定義の通り、99.5体積%以下の水に対応する0.8重量%以下の水を含むアルコールまたは溶媒を意味するものと定義される。「脱水溶媒(dehydrated solvent)」という用語は、本明細書において、「実質的に純粋な溶媒」、「無水溶媒(anhydrous solvent)」および「純溶媒(absolute solvent)」という用語と同義的に使用されている。同様にして、「脱水アルコール(dehydrated alcohol)」という用語は、「実質的に純粋なアルコール」、「無水アルコール(anhydrous alcohol)」および「純アルコール(absolute alcohol)」と同義的に使用されている。
【0037】
本明細書中で使用される「治療する」という用語は、ノルエピネフリン(NE)およびセロトニン(5−HT)の再取り込み阻害により治療してよい患者の疾病、障害または身体条件の少なくとも1つの疾候を緩和、軽減、遅延、削減、逆転、改善または予防することを意味する。「治療する」という用語は、ノルエピネフリン(NE)およびセロトニン(5−HT)の再取り込み阻害により治療してよい患者の疾病、疾病または身体条件の開始(すなわち疾病、障害または身体条件の臨床的徴候に先立つ期間)を阻むこと、遅延させることかつ/またはそれらの発達または悪化の危険性を低減させることも意味し得る。
【0038】
「剤形」という用語は、別段の指摘のないかぎり、本明細書において、ヒトの患者に対する経口投与に適したあらゆるレボミルナシプラン調合物を意味する。例えば、「剤形」という用語は、あらゆる経口剤形またはあらゆる固形経口剤形、例えばカプセル内に装填するのに適した組成物(例えば、ビーズ、顆粒、微小顆粒など)、錠剤、ジェルキャップ、カプレット、トローチ剤または粉剤を包含する。一部の実施形態において、剤形は、カプセル内への装填に適した剤形(例えばビーズ、顆粒、微小顆粒など)である。一部の実施形態において、剤形は、即時放出型剤形(例えば即時放出型固形剤形、即時放出型経口剤形または即時放出型固形経口剤形)である。
【0039】
一部の実施形態において、剤形は、放出調節型(例えば持続放出、遅延放出および/または徐放型)組成物でコーティングされた即時放出型組成物である。一部の実施形態において、剤形は、持続放出型剤形(例えば持続放出型固形剤形、持続放出型経口剤形、または持続放出型固形経口剤形)である。一部の実施形態において、剤形はカプセル(例えばビーズ、顆粒または微小顆粒が充填されたカプセル)である。一部の実施形態において、剤形は錠剤である。一部の実施形態において、剤形は一日一回の固形経口剤形である。一部の実施形態において、剤形は一日一回のカプセルである。
【0040】
「持続放出」という用語は、本明細書では、別段の指摘のないかぎり、投与直後以外の時点で、例えばレボミルナシプランの従来の瞬間放出および即時放出型剤形からの薬物放出時間を上回る長時間にわたりレボミルナシプラン(そして任意には中に含まれている追加の活性作用物質)を放出する剤形を意味するように使用されている。
【0041】
「使用環境内に入る」という用語は、本明細書では、別段の指摘のないかぎり、レボミルナシプランの安定した剤形と、それが投与される患者の胃液または腸液、胃液または腸液をシミュレートするように意図された流体または温度が約37℃でかつ75rpmのUSP装置IIに付された脱イオン水(例えば温度が約37℃でかつ75rpmのUSP装置IIに付された脱イオン水1000mL)との接触を意味するように使用されている。
【0042】
本明細書中で使用されるように、別段の指摘のないかぎり、溶出率は、当初安定した剤形内に入っていたレボミルナシプランの剤形が、使用環境内に入ってから、特定の時間内に剤形から放出される分率を定義する。
【0043】
別段の指摘のないかぎり、本明細書中で使用される「有効量」および「治療上有効な量」という用語は、患者に投与された場合に相応の生物学的応答を惹起するのに充分であるレボミルナシプランの量または数量を意味する。例えば、「有効量」および「治療上有効な量」という用語は、ノルエピネフリン(NE)およびセロトニン(5−HT)の再取り込み阻害により治療してよい疾病、身体条件または障害を治療するために患者(例えばヒトまたは他の哺乳動物)に投与された場合に、その疾病、傷害または身体条件の1つ以上の症候のこのような治療をもたらすのに充分であるレボミルナシプラン(またはその剤形中に入っている追加の活性作用物質)の量、または、患者の体内でのNEおよび5−HTの再取り込み阻害に充分であるレボミルナシプラン(またはその剤形内に入っている追加の活性作用物質)の量を意味する。精確な治療的用量は、患者の年令、身体条件、体重など、および治療対象の身体条件の性質によって左右され、究極的には主治医の判断に基づく。
【0044】
例えば、一部の実施形態において、うつ病(例えば大うつ病障害)を治療するための安定した剤形内のレボミルナシプランの治療上有効な投薬量は、実質的に純粋なレボミルナシプランを含む活性成分約10mg〜約150mg(例えば活性成分約20mg〜約120mg)であることがわかった。一部の実施形態において、剤形は実質的に純粋なレボミルナシプランを含む活性成分を約15mg〜約25mg(例えば約20mg)含む。一部の実施形態において、剤形は、実質的に純粋なレボミルナシプランを含む活性成分を約35mg〜45mg(例えば約40mg)含む。一部の実施形態において、剤形は、実質的に純粋なレボミルナシプランを含む活性成分を約70mg〜約90mg(例えば約80mg)含む。一部の実施形態において、実質的に純粋なレボミルナシプランを含む活性成分を約100mg〜約140mg(例えば約120mg)含む。
【0045】
本明細書で使用される通り、別段の指摘のないかぎり、安定した剤形に言及して使用される「純度」という用語は、その剤形が特定の望ましくない構成成分または不純物(例えば分解生成物など)を含まない(またはそれらが欠如している)度合いを意味する。
【0046】
「本質的に〜からなる」という用語は、剤形に関連して使用される場合、その剤形が追加の活性薬剤成分を全く含まないものの、追加の不活性構成成分または賦形剤は含んでよいということを意味する。
【0047】
別段の指摘のないかぎり、本明細書中で使用される「約」および「およそ(〜程度)」という用語は、特定の値について、その値の測定方法または決定方法すなわち測定システムの限界により一部左右されるものである当業者によって決定される許容誤差範囲内を意味するものと理解されるべきである。例えば、「約」は、当該技術分野における実践方法によると、1以内または1超の標準偏差を意味することができる。あるいは、「約」とは、所与の値の最高20%、好ましくは最高10%、より好ましくは最高5%、そしてより好ましくはさらに最高1%の範囲を意味し得る。
【0048】
経口剤形のX線粉末回折パターン
一部の好ましい実施形態において、安定した剤形または剤形内に入っている活性成分は結晶構造を有する。一部の好ましい実施形態においては、安定した剤形および安定した剤形内に入っている活性成分が結晶構造を有する。一部の実施形態において、安定した剤形は表1に提供されている1つ以上の特徴的ピークを含むX線粉末回折パターン(XRD)を有する。一部の実施形態において、安定した剤形は、レボミルナシプランを含む活性成分を含み、ここでこの活性成分は表1に提供されている1つ以上の特徴的ピークを含むX線粉末回折パターン(XRD)を有する。
【0049】
別段の指摘のないかぎり、本明細書中で使用される「1つ以上のピーク」という語句は、(i)この語句の後に記された全てのピーク値においてXRDピークを有する安定した剤形、(ii)この語句の後に記されたピーク値の少なくとも1つにおいてXRDピークを有する安定した剤形、ならびに(iii)この語句の後に記されたピーク値の2つ以上(例えば3つ以上、4つ以上、5つ以上、6つ以上さらには7つ以上)においてXRDピークを有する安定した剤形を包含するものとして理解されるべきである。
【0050】
【表1】
【0051】
一部の実施形態において、レボミルナシプランの安定した剤形は、表2に提供されている1つ以上の特徴的ピークを含むX線粉末回折パターン(XRD)を有する。
【0052】
【表2】
【0053】
一部の実施形態において、レボミルナシプランの安定した剤形は、放出調節型剤形(例えば持続放出型剤形)であり、表3に提供されている1つ以上の特徴的ピークを含むX線粉末回折パターン(XRD)を有する。
【0054】
【表3】
【0055】
一部の実施形態において、本発明は、レボミルナシプランまたはその薬学的に許容される塩を含む安定した剤形において、12.0、20.1および22.5±0.2度2θに特徴的ピークを含むX線粉末回折(XRD)パターンを含む安定した剤形に関する。一部の実施形態において、XRDパターンは同様に32.7±0.2度2θに特徴的ピークを含む。一部の実施形態において、XRDパターンは同様に、32.7±0.2度2θに特徴的ピークを含む。一部の実施形態において、XRDパターンは6.0±0.2度2θにも特徴的ピークを含む。
【0056】
一部の実施形態において、レボミルナシプランの安定した剤形は、約6.0、約12.0および約20.1±0.2度2θに特徴的ピークを含むXRDを有する。一部の実施形態において、安定した剤形は、約6.0、約12.0および約22.4±0.2度2θに特徴的ピークを含むXRDを有する。一部の実施形態において、安定した剤形は、約6.0、約12.0、約20.1および約22.4±0.2度2θに特徴的ピークを含むXRDを有する。一部の実施形態において、安定した剤形は、約6.0、約20.1および約22.4±0.2度2θに特徴的ピークを含むXRDを有する。一部の実施形態において、安定した剤形は、約12.0、約20.1および約22.4±0.2度2θに特徴的ピークを含むXRDを有する。一部の実施形態において、安定した剤形は、約6.0および約12.0±0.2度2θに特徴的ピークを含むXRDを有する。一部の実施形態において、安定した剤形は、約6.0および約20.1±0.2度2θに特徴的ピークを含むXRDを有する。一部の実施形態において、安定した剤形は約6.0および約22.4±0.2度2θに特徴的ピークを含むXRDを有する。
【0057】
一部の実施形態において、安定した剤形は、約12.0および約20.1±0.2度2θに特徴的ピークを含むXRDを有する。一部の実施形態において、安定した剤形は、約12.0および約22.4±0.2度2θに特徴的ピークを含むXRDを有する。一部の実施形態において、安定した剤形は、約20.1±0.2度2θおよび約22.4±0.2度2θに特徴的ピークを含むXRDを有する。一部の実施形態において、安定した剤形は、約6.0±0.2度2θ、約12.0±0.2度2θ、約20.1±0.2度2θおよび約22.4±0.2度2θの1つ以上に特徴的ピークを含むXRDを有するレボミルナシプランの結晶形態を含む。
【0058】
一部の実施形態において、安定した剤形は、実質的に純粋なレボミルナシプランを含む活性成分を含み、ここでこの活性成分は表1に示された特徴的ピークの1つ以上を含んでいる。一部の実施形態において、安定した剤形は、実質的に純粋なレボミルナシプランを含む活性成分を含み、ここでこの安定した剤形は表1または表2に示された特徴的ピークの1つ以上を含む。
【0059】
安定した剤形の純度
安定した剤形および剤形中の活性成分は、
【化2】
という式(II)により表わされる(1S,5R)1−フェニル−3−アザビシクロ[3−1−0]ヘキサン−2−オンを驚くほど低い濃度で含むことが発見されている。
【0060】
一部の実施形態において、安定した剤形は、例えば40℃および相対湿度75%で1カ月、2カ月または3カ月の保管期間中保管した後、調和国際会議(ICH)指針により定義されている約0.0001〜約0.2重量%の(1S,5R)1−フェニル−3−アザビシクロ[3−1−0]ヘキサン−2−オンを含む。一部の実施形態において、安定した剤形は、例えば40℃、相対湿度75%で1カ月、2カ月、または3カ月の保管期間中保管した後、約0.2重量%未満の(1S,5R)1−フェニル−3−アザビシクロ[3−1−0]ヘキサン−2−オンを含む。一部の実施形態において、安定した剤形は、例えば40℃、相対湿度75%で1カ月、2カ月または3カ月の保管期間中保管した後、約0.0001〜約0.1重量%の(1S,5R)1−フェニル−3−アザビシクロ[3−1−0]ヘキサン−2−オンを含む。
【0061】
一部の好ましい実施形態において、安定した剤形は、例えば40℃、相対湿度75%で1カ月、2カ月、3カ月、4カ月、5カ月または6カ月の保管期間中保管した後、約0.001〜約0.2重量%、約0.01〜約0.2重量%、約0.0001〜約0.15重量%、約0.001〜約0.15重量%、約0.01〜約0.15重量%、約0.001〜約0.1重量%、約0.01〜約0.1重量%、約0.01〜約0.08重量%、さらには約0.001〜約0.08重量%の(1S,5R)1−フェニル−3−アザビシクロ[3−1−0]ヘキサン−2−オンを含む。一部の実施形態において、安定した剤形は、例えば40℃、相対湿度75%で1カ月、2カ月、3カ月、4カ月、5カ月または6カ月の保管期間中保管した後、約0.01〜約0.08重量%(例えば約0.001〜約0.08重量%)の(1S,5R)1−フェニル−3−アザビシクロ[3−1−0]ヘキサン−2−オンを含む。
【0062】
安定した剤形の調製
レボミルナシプランの安定した剤形は、適切な任意の方法によって調製可能である。一部の好ましい実施形態において、安定した剤形は、レボミルナシプラン、溶媒(例えば5重量%未満、4重量%未満、3重量%未満、2重量%未満、1重量%未満、0.8重量%未満、0.5重量%未満、さらには0.1重量%未満の水を含む溶媒)(例えば200プルーフエタノールなどの脱水アルコール)および任意には結合剤および付着防止剤または潤滑剤を含む(または本質的にこれからなるまたはこれからなる)溶液と不活性基質または充填剤とを接触させてレボミルナシプラン組成物を形成するステップを含む方法によって調製される。一部の実施形態において、レボミルナシプランの剤形は次に、放出制御剤、溶媒(例えば5重量%未満、4重量%未満、3重量%未満、2重量%未満、1重量%未満、0.8重量%未満、0.5重量%未満さらには0.1重量%未満の水を含む溶媒)(例えば200プルーフエタノールなどの脱水アルコール)そして任意には可塑剤、付着防止剤または流動促進剤を含む溶液とレボミルナシプラン組成物とを接触させることによってコーティングされて、持続放出型剤形となる。
【0063】
一部の好ましい実施形態において、安定した剤形は、レボミルナシプラン、溶媒(例えば5重量%未満、4重量%未満、3重量%未満、2重量%未満、1重量%未満、0.8重量%未満、0.5重量%未満、さらには0.1重量%未満の水を含む溶媒)結合剤および付着防止剤または潤滑剤を含む溶液と不活性基質または充填剤とを接触させてレボミルナシプラン組成物を形成するステップを含む方法によって調製される。一部の実施形態において、レボミルナシプランの剤形は次に、放出制御剤、可塑剤、付着防止剤または流動促進剤および溶媒(例えば5重量%未満、4重量%未満、3重量%未満、2重量%未満、1重量%未満、0.8重量%未満、0.5重量%未満さらには0.1重量%未満の水を含む溶媒)を含む溶液とレボミルナシプラン組成物とを接触させることによってコーティングされて、持続放出型剤形となる。
【0064】
一部の実施形態において、安定した剤形は、レボミルナシプラン、そして任意には結合剤、付着防止剤または潤滑剤および/または溶媒(例えば5重量%未満、4重量%未満、3重量%未満、2重量%未満、1重量%未満、0.8重量%未満、0.5重量%未満、さらには0.1重量%未満の水を含む溶媒)と不活性基質または充填剤とを接触させてレボミルナシプラン組成物(レボミルナシプランコアまたはレボミルナシプランビーズまたは顆粒)を形成するステップを含む方法によって調製される。一部の実施形態において、レボミルナシプランの剤形は次に、放出制御剤、そして任意には可塑剤、付着防止剤または潤滑剤および/または溶媒(例えば5重量%未満、4重量%未満、3重量%未満、2重量%未満、1重量%未満、0.8重量%未満、0.5重量%未満さらには0.1重量%未満の水を含む溶媒)とレボミルナシプラン組成物とを接触させることによってコーティングされて、持続放出型剤形となる。
【0065】
一部の実施形態において、安定した剤形は、レボミルナシプラン、溶媒(例えば5重量%未満、4重量%未満、3重量%未満、2重量%未満、1重量%未満、0.8重量%未満、0.5重量%未満、さらには0.1重量%未満の水を含む溶媒)および任意には結合剤および付着防止剤(または潤滑剤)を含む溶液と不活性基質または充填剤とを接触させてレボミルナシプラン組成物(例えばレボミルナシプランを含むコア、ビーズまたは顆粒)を形成するステップを含む方法によって調製される。一部の実施形態において、この方法はさらに、放出制御剤、そして任意には可塑剤、付着防止剤または潤滑剤および/または溶媒(例えば5重量%未満、4重量%未満、3重量%未満、2重量%未満、1重量%未満、0.8重量%未満、0.5重量%未満さらには0.1重量%未満の水を含む溶媒)を含むまたは本質的にこれらからなるまたはこれらからなる溶液とレボミルナシプラン組成物とを接触させるステップを含む。
【0066】
一部の実施形態において、安定した剤形は、レボミルナシプラン、結合剤および付着防止剤または潤滑剤および溶媒(例えば5重量%未満、4重量%未満、3重量%未満、2重量%未満、1重量%未満、0.8重量%未満、0.5重量%未満、さらには0.1重量%未満の水を含む溶媒)を含む溶液と不活性基質または充填剤とを接触させるステップを含む方法によって調製される。一部の実施形態において、接触ステップは本質的にレボミルナシプラン、結合剤、付着防止剤または潤滑剤および溶媒(例えば5重量%未満、4重量%未満、3重量%未満、2重量%未満、1重量%未満、0.8重量%未満、0.5重量%未満さらには0.1重量%未満の水を含む溶媒)からなる薬物層状化溶液で不活性基質を層状化するステップを含む。
【0067】
一部の好ましい実施形態において、接触ステップ(例えば薬物層状化ステップ)は、ウルスター法(Wurster process)(例えばウルスター装置内部で)などによって実施される。一部の好ましい実施形態において、コーティングステップは、ウルスター法(例えばウルスター装置内で)などにより実施される。一部の好ましい実施形態において、組合せステップと接触ステップの両方とも、ウルスター法などによって実施される。
【0068】
活性薬剤成分中のレボミルナシプランおよび2−(アミノメチル)−N,N−ジエチル−1−フェニルシクロプロパンカルボキサミドの他の異性体(例えば(1R,2S)−2−(アミノメチル)−N,N−ジエチル−1−フェニルシクロプロパンカルボキサミド))の相対的百分率を決定するためのプロセスは、任意の適切な方法を用いて、好ましくは逆相高性能液体クロマトグラフ(RP HPLC)(例えば220nmでのUV検出を用いる)によって実施可能である。
【0069】
安定した剤形の構成成分
安定した剤形は、任意の治療上有効な量のレボミルナシプランを含み得る。一部の実施形態において、安定した剤形は約5〜約200mgのレボミルナシプランを含む。一部の実施形態において、安定した剤形は約10〜約180mgのレボミルナシプランを含む。一部の実施形態において、安定した剤形は約20〜約150mgのレボミルナシプランを含む。一部の実施形態において、安定した剤形は約20〜約120mgのレボミルナシプランを含む。例えば、安定した剤形は約20mg、約40mg、約50mg、約60mg、約80mg、約100mg、120mgまたは約240mgのレボミルナシプランを含むことができる。この点に関して、安定した剤形は、剤形の他の構成成分との関係において任意の適切な重量百分率のレボミルナシプランを含むことができる。例えば、安定した剤形は、約35〜約65重量%(例えば約35〜約60重量%、約35〜約55重量%、約40〜約55重量%または約40〜約50重量%)のレボミルナシプランを含むことができる。
【0070】
レボミルナシプランの安定した剤形は同様に、不活性基質または充填剤も含んでいる。一部の好ましい実施形態において、安定した剤形は、糖例えばスクロース(例えば糖球)を含む不活性基質を含む。他の適切な不活性基質または充填剤としては、例えばイソマルト、リン酸ジカルシウム二水和物、硫酸カルシウム、ラクトース、マンニトール、ソルビトール、セルロース、微晶質セルロース、カオリン、塩化ナトリウム、乾燥デンプン、加水分解デンプン類、アルファー化デンプン、二酸化シリコーン、酸化チタン、ケイ酸アルミニウムマグネシウム、またはその混合物が含まれる。
【0071】
安定した剤形は、任意の適切な量の不活性基質または充填剤(例えば糖球)を含み得る。一部の実施形態において、安定した剤形は約15重量%〜約45重量%の不活性基質または充填剤を含む。一部の実施形態において、安定した剤形は、約20〜約40重量%の不活性基質または充填剤を含む。一部の実施形態において、安定した剤形は約25〜約40重量%の不活性基質または充填剤を含む。一部の実施形態において、安定した剤形は約30〜約40重量%の不活性基質または充填剤を含む。一部の実施形態において、安定した剤形は約35〜約40重量%の不活性基質または充填剤を含む。
【0072】
安定した剤形は、任意の適切なサイズで糖球を含むことができる。一部の実施形態において、安定した剤形は、約20〜約50メッシュのサイズを有する糖球を含む。一部の実施形態において、安定した剤形は、約25〜約45メッシュ程度のサイズを有する糖球を含む。一部の実施形態において、安定した剤形は約25〜約40メッシュのサイズを有する糖球を含む。一部の好ましい実施形態において、安定した剤形は、約30〜約40メッシュ程度(例えば約30〜約35メッシュ)のサイズを有する糖球を含む。例えば、安定した剤形は約30〜約40メッシュのサイズを有する糖球を約30〜約40重量%(例えば約35〜約40重量%)含んでいてよい。
【0073】
安定した剤形は同様に、例えばポリビニルピロリドン(例えばPovidone K30)などの結合剤を一部の好ましい実施形態において含んでいる。他の適切な結合剤としては、例えばデンプン、ポリビニルアルコール、アルファー化デンプン、ゼラチン、スクロース、グルコース、デキストロース、ラクトース、ソルビトール、ポリエチレングリコール、ワックス類、天然および合成ゴム類、例えばアカシア、トラガカント、アルギン酸ナトリウム、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース、エチルセルロース、veegumおよび合成ポリマー類、例えばアクリル酸およびメタクリル酸コポリマー類、メタクリル酸コポリマー類、メタクリル酸メチル、コポリマー類、メタクリル酸アミノアルキルコポリマー類、ポリアクリル酸/ポリメタクリル酸またはその混合物が含まれる。
【0074】
安定した剤形は、任意の適切な量の結合剤(例えばPVP)を含むことができる。一部の実施形態において、安定した剤形は約0.1〜約15重量%の結合剤を含む。一部の実施形態において、安定した剤形は、約1〜約12重量%の結合剤を含む。一部の実施形態において、安定した剤形は、約1〜約10重量%の結合剤を含む。一部の実施形態において、安定した剤形は、約2〜約10重量%の結合剤を含む。
【0075】
一部の好ましい実施形態において、安定した剤形は約3〜約10重量%の結合剤(例えばPVP)を含む。一部の実施形態において、安定した剤形は約4〜約10重量%の結合剤を含む。一部の実施形態において、安定した剤形は約2〜約8重量%の結合剤を含む。一部の実施形態において、安定した剤形は約4〜約8重量%の結合剤を含む。一部の実施形態において、安定した剤形は約5〜約7重量%の結合剤を含む。
【0076】
安定した剤形は同様に、一部の好ましい実施形態において、例えばタルクなどの付着防止剤または潤滑剤も含んでいる。他の適切な付着防止剤または潤滑剤としては、例えばステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸、ベヘン酸グリセロール、ポリエチレングリコール、タルク、鉱油、フマル酸ステアリルナトリウムまたはその混合物が含まれる。
【0077】
安定した剤形は、任意の適切な量の潤滑剤または付着防止剤(例えばタルク)を含むことができる。一部の実施形態において、安定した剤形は、約0.1〜約15重量%の潤滑剤または付着防止剤を含む。一部の実施形態において、安定した剤形は、約1〜約12重量%の潤滑剤または付着防止剤を含む。一部の実施形態において、安定した剤形は、約2〜約10重量%の潤滑剤または付着防止剤を含む。一部の実施形態において、安定した剤形は約3〜約10重量%の潤滑剤または付着防止剤を含む。一部の実施形態において、安定した剤形は約4重量%〜約10重量%の潤滑剤または付着防止剤を含む。一部の実施形態において、安定した剤形は約4〜約8重量%の潤滑剤または付着防止剤を含む。一部の実施形態において、安定した剤形は約5〜約8重量%の潤滑剤または付着防止剤を含む。
【0078】
一部の好ましい実施形態において、安定した剤形は約4〜約7.5重量%の潤滑剤または付着防止剤を含む。一部の好ましい実施形態において、安定した剤形は約5〜約7重量%の潤滑剤または付着防止剤を含む。
【0079】
一部の実施形態において、安定した剤形は、持続放出(SR)剤形であり、放出制御剤、ポリマー剤またはコーティングポリマー(例えばエチルセルロース)を含み、これは実質的に剤形からのレボミルナシプランの放出を持続させることに寄与する。他の適切な放出制御剤としては、例えばセルロースおよびセルロース誘導体、ワックス、カルボマー、ポリアルキレンポリオール、ポリカルボフィル、メタクリル酸誘導体、ゼラチン、ゴム、ポリ酸化エチレンおよびポリビニルピロリドン、またはその混合物が含まれる。一部の実施形態において、放出制御剤、ポリマー添加剤またはコーティングポリマーは、エチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシエチルセルロース、ヒドロキシメチルセルロース、ヒドロキシプロピルメチルセルロース、ヒドロキシプロピルメチルセルロースアセテートスクシネート、ヒドロキシプロピルメチルセルロースフタレート、メチルセルロース、セルロースアセテート、セルロースアセテートフタレート、セルロースアセテートトリメリテートおよびカルボキシメチルセルロースナトリウム;アクリル酸ポリマー類およびコポリマー類(好ましくはアクリル酸、メタクリル酸、アクリル酸メチル、アクリル酸エチル、メタクリル酸メチルおよび/またはメタクリル酸エチルから形成されたもの)およびEudragitTML30D−55およびL100−55、EudragitTM、EudragitTMおよびEudragitTMNE、RLおよびRSを含む、EudragitTM(Rohm Pharma;Westerstadt、Germany)の商標名の下で市販されている他のメタクリル樹脂;ビニルポリマー類およびコポリマー類、例えばポリビニルピロリドン、酢酸ビニル、酢酸ビニルフタレート、酢酸ビニルクロトン酸コポリマー、およびエチレン−酢酸ビニルコポリマー;酵素分解性ポリマー類、例えばアゾポリマー類、ペクチン、キトサン、アミロースおよびグアーゴム;ゼインおよびシェラックまたはその混合物から選択される。
【0080】
安定した剤形は、任意の適切な量の放出制御剤、ポリマー剤またはコーティングポリマー(例えばエチルセルロース)を含むことができる。安定した剤形は好ましくは約5〜約15重量%の放出制御剤、ポリマー剤またはコーティングポリマーを含む。しかしながら、一部の実施形態において、安定した剤形は約2〜約20重量%の放出制御剤、ポリマー剤またはコーティングポリマーを含む。一部の実施形態において、安定した剤形は、約5〜約12重量%の放出制御剤、ポリマー剤またはコーティングポリマーを含む。一部の実施形態において、安定した剤形は約8〜約12重量%の放出制御剤、ポリマー剤またはコーティングポリマーを含む。一部の実施形態において、安定した剤形は約8〜約11重量%の放出制御剤、ポリマー剤またはコーティングポリマーを含む。一部の実施形態において、安定した剤形は、約8〜約10重量%の放出制御剤、ポリマー剤またはコーティングポリマーを含む。
【0081】
安定した剤形は同様に、一部の好ましい実施形態において、例えばクエン酸トリエチルなどの可塑剤も含んでいる。他の適切な可塑剤としては、ポリエチレングリコール、プロピレングリコール、トリアセチン、フタル酸ジメチル、フタル酸ジエチル、フタル酸ジブチル、セバシン酸ジブチル、クエン酸トリブチル、クエン酸トリエチルアセチル、モノステアリン酸グリセロール、ヒマシ油、アセチル化モノグリセリド類またはその混合物が含まれる。
【0082】
安定した剤形は、任意の適切な量の可塑剤(例えばクエン酸トリエチル)を含むことができる。一部の実施形態において、安定した剤形は約0.1〜約10重量%の可塑剤を含む。一部の実施形態において、安定した剤形は約0.5〜約8重量%の可塑剤を含む。一部の実施形態において、安定した剤形は、約0.5〜約5重量%の可塑剤を含む。一部の実施形態において、安定した剤形は約1〜約5重量%の可塑剤を含む。一部の実施形態において、安定した剤形は約1〜約3重量%の可塑剤を含む。
【0083】
さらに、レボミルナシプランの安定した剤形は、一部の実施形態において、可塑剤、顔料、着色剤、安定剤、流動促進剤などの任意の追加の賦形剤または添加剤を含むことができる。
【0084】
一部の好ましい実施形態において、剤形は、約30重量%〜約65重量%(例えば約40重量%〜約60重量%、約45重量%〜約60重量%さらには約50重量%〜約60重量%)のレボミルナシプラン(または少なくとも98重量%さらには実質的に純粋なレボミルナシプランを含む活性成分);約25重量%〜約55重量%(例えば約30重量%〜約45重量%さらには約30重量%〜約40重量%)の不活性基質または充填剤;約2重量%〜約12重量%(例えば約4重量%〜約10重量%さらには約4重量%〜約8重量%)の結合剤;そして約0.5重量%〜約10重量%(例えば約1重量%〜約8重量%、約1重量%〜約5重量%さらには約2重量%〜約5重量%)の付着防止剤または潤滑剤を含む。
【0085】
一部の好ましい実施形態において、剤形は約45重量%〜約60重量%のレボミルナシプラン(または少なくとも98重量%さらには実質的に純粋なレボミルナシプランを含む活性成分);約30重量%〜約45重量%の不活性基質または充填剤;約4重量%〜約10重量%の結合剤;および約1重量%〜約5重量%の付着防止剤または潤滑剤を含む。
【0086】
一部の好ましい実施形態において、剤形は約50重量%〜約60重量%のレボミルナシプラン(または少なくとも98重量%さらには実質的に純粋なレボミルナシプランを含む活性成分);約30重量%〜約40重量%の不活性基質または充填剤;約4重量%〜約8重量%の結合剤;および約1重量%〜約5重量%の付着防止剤または潤滑剤を含む。
【0087】
一部の好ましい実施形態において、安定した剤形は40〜約55重量%(例えば約40〜約50重量%)のレボミルナシプラン;約3〜約10重量%(例えば約4〜約8重量%)の結合剤(例えばPVP);および約0.1〜約4重量%(例えば約1〜約3重量%)の可塑剤(例えばクエン酸トリエチル)を含む。
【0088】
一部の好ましい実施形態において、安定した剤形は40〜約55重量%(例えば約40〜約50重量%)のレボミルナシプラン;約3〜約7重量%(例えば約4〜約7重量%)の付着防止剤または潤滑剤(例えばタルク);および約0.1〜約4重量%(例えば約1〜約3重量%)の可塑剤(例えばクエン酸トリエチル)を含む。
【0089】
一部の好ましい実施形態において、安定した剤形は40〜約55重量%(例えば約40〜約50重量%)のレボミルナシプラン;約3〜約10重量%(例えば約4〜約8重量%)の結合剤(例えばPVP);約3〜約7重量%(例えば約4〜約7重量%)の付着防止剤または潤滑剤(例えばタルク);および約0.1〜約4重量%(例えば約1〜約3重量%)の可塑剤(例えばクエン酸トリエチル)を含む。
【0090】
一部の好ましい実施形態において、安定した剤形は40〜約55重量%(例えば約40〜約50重量%)のレボミルナシプラン;約3〜約10重量%(例えば約4〜約8重量%)の結合剤(例えばPVP);および約5〜約10.5重量%(例えば約6〜約10重量%)の放出制御剤(例えばエチルセルロース)を含む。
【0091】
一部の好ましい実施形態において、安定した剤形は40〜約55重量%(例えば約40〜約50重量%)のレボミルナシプラン;約3〜約7重量%(例えば約4〜約7重量%)の付着防止剤または潤滑剤(例えばタルク);および約5〜約10.5重量%(例えば約6〜約10重量%)の放出制御剤(例えばエチルセルロース)を含む。
【0092】
一部の好ましい実施形態において、安定した剤形は40〜約55重量%(例えば約40〜約50重量%)のレボミルナシプラン;約3〜約10重量%(例えば約4〜約8重量%)の結合剤(例えばPVP);約5〜約10.5重量%(例えば約6〜約10重量%)の放出制御剤(例えばエチルセルロース);および約5〜約10.5重量%(例えば約6〜約10重量%)の放出制御剤(例えばエチルセルロース)を含む。
【0093】
一部の好ましい実施形態において、安定した剤形は40〜約55重量%(例えば約40〜約50重量%)のレボミルナシプラン;約3〜約7重量%(例えば約4〜約7重量%)の付着防止剤または潤滑剤(例えばタルク);約5〜約10.5重量%(例えば約6〜約10重量%)の放出制御剤(例えばエチルセルロース);および約5〜約10.5重量%(例えば約6〜約10重量%)の放出制御剤(例えばエチルセルロース)を含む。
【0094】
一部の好ましい実施形態において、安定した剤形は、約35〜約60重量%(例えば約35〜約55重量%または約40〜約55重量%)のレボミルナシプラン;約20〜約45重量%(例えば約20〜約40重量%、約25〜約40重量%または約25〜35重量%)の不活性基質または充填剤(例えば糖球);約1〜約15重量%(例えば約2〜約12重量%、約3〜約10重量%、約3〜約9重量%または約4〜約8重量%)の結合剤(例えばPVP);約1〜約15重量%(例えば約2〜約12重量%、約3〜約10重量%、約3〜約9重量%、約4〜約8重量%または約4〜約7重量%)の付着防止剤または潤滑剤(例えばタルク);約1〜20重量%(例えば約5〜約15重量%または約8〜約12重量%)の放出制御剤(例えばエチルセルロース);および約0.1〜約10重量%(例えば約0.1〜約5重量%または約1〜約5重量%または約1〜約3重量%)の可塑剤(例えばクエン酸トリエチル)を含む。
【0095】
一部の好ましい実施形態において、安定した剤形は、約35〜約60重量%(例えば約35〜約55重量%または約40〜約55重量%)のレボミルナシプラン;約20〜約45重量%(例えば約20〜約40重量%、約25〜約40重量%または約25〜35重量%)の不活性基質(例えば糖球)(例えば約30〜35メッシュの糖球);約1〜約15重量%(例えば約2〜約12重量%、約3〜約10重量%、約3〜約9重量%または約4〜約8重量%)のPVP(例えばPovidone K30);約1〜約15重量%(例えば約2〜約12重量%、約3〜約10重量%、約3〜約9重量%、約3〜約8重量%、約4〜約8重量%または約4〜約7重量%)のタルク;約1〜20重量%(例えば約5〜約15重量%または約8〜約12重量%)のエチルセルロース;および約0.1〜約10重量%(例えば約0.1〜約5重量%または約1〜約5重量%または約1〜約3重量%)のクエン酸トリエチルを含む。
【0096】
一部の好ましい実施形態において、安定した剤形は40〜約55重量%(例えば約40〜約50重量%)のレボミルナシプラン;約25〜約40重量%の糖球(例えば約30〜35メッシュの糖球);約2〜約10重量%(例えば約4〜約8重量%)のPVP(例えばPovidone K30);約2〜約10重量%(例えば約4〜約8重量%)のタルク;約5〜約15重量%のエチルセルロース;約0.1〜約5重量%(例えば約1〜約5重量%)のクエン酸トリエチルを含む。
【0097】
安定した剤形は、所望のpKプロファイルを達成するため任意の適切な厚みで放出制御剤によりコーティングされているレボミルナシプランのビーズまたは顆粒(例えば微小顆粒または他の同様のコア)を含むことができる。一部の実施形態において、例えば安定した剤形は、ビーズまたは顆粒(または類似のコア)が入ったカプセルであり、ここでビーズまたは顆粒(または類似のコア)は、任意の所望の平均厚みを有する放出制御剤(および任意には可塑剤、付着防止剤または潤滑剤および/または溶媒)を含むコーティング組成物でコーティングされている。例えば、コーティング組成物は、約1〜約100ミクロン(例えば約5〜約75ミクロン、約5〜約60ミクロン、約5〜約50ミクロン、約5〜約40ミクロン、約5〜約30ミクロン、約10〜約30ミクロン、約15〜約30ミクロン、約20〜約30ミクロン、約25〜約35ミクロンさらには約25〜約35ミクロン)の平均厚みで、ビーズまたは顆粒(または類似のコア)に対して塗布可能である。
【0098】
一部の好ましい実施形態において、例えば安定した剤形は、レボミルナシプランのコーティングされたビーズまたは顆粒(または類似のコア)を含み、ここでコーティングは1つ以上の放出制御剤(例えばエチルセルロース)を含み、ビーズまたは顆粒(または類似のコア)上のコーティングの平均厚みは約20〜約35ミクロン(例えば約20〜約30ミクロン)である。例えば、剤形はビーズ、顆粒または微小顆粒が充填されたカプセルであり得、ここでビーズ、顆粒または微小顆粒(または類似のコア)は、約20〜約30ミクロン(例えばおよそ25ミクロン)の平均厚みでコーティング組成物(例えばエチルセルロースを含む)によりコーティングされている。
【0099】
一部の実施形態において、安定した剤形は、約400〜約900ミクロンの平均直径を有するビーズまたは顆粒(例えばコーティングされたビーズまたは顆粒)の形をしている。一部の実施形態において、安定した剤形は、約500〜約800ミクロンの平均直径を有するビーズまたは顆粒(例えばコーティングされたビーズまたは顆粒)の形をしている。一部の実施形態において、安定した剤形は、約600〜約800ミクロンの平均直径を有するビーズまたは顆粒(例えばコーティングされたビーズまたは顆粒)の形をしている。一部の実施形態において、安定した剤形は、約600〜約750ミクロンの平均直径を有するビーズまたは顆粒(例えばコーティングされたビーズまたは顆粒)の形をしている。一部の実施形態において、安定した剤形は、約650〜約850ミクロンの平均直径を有するビーズまたは顆粒(例えばコーティングされたビーズまたは顆粒)の形をしている。一部の実施形態において、安定した剤形は、約1000ミクロン未満の平均直径を有するビーズまたは顆粒(例えばコーティングされたビーズまたは顆粒)の形をしている。一部の実施形態において、安定した剤形は、約900ミクロン未満の平均直径を有するビーズまたは顆粒(例えばコーティングされたビーズまたは顆粒)の形をしている。
【0100】
本発明は同様に、5−HTおよびNEの再取り込み阻害によって管理可能である障害、例えば不安障害またはうつ病(例えば大うつ病障害)の治療のための医薬品の製造におけるレボミルナシプランの安定した剤形の使用方法も提供する。
【0101】
一部の実施形態において、安定した剤形はカプセル(例えばHPMCまたはゼラチンカプセル)内に装填される。例えば一部の好ましい実施形態において、安定した剤形はHPMCカプセル内に装填される。このようなHPMCカプセルは次に、乾燥剤(例えば約0.01〜約2グラム、約0.01〜約1グラムさらには約0.01〜約0.8グラムの乾燥剤)を伴ってまたは伴わずにボトルまたはキャニスタ内に包装され得る。一部の好ましい実施形態において、安定した剤形はHPMCカプセル内に収納され、乾燥剤無しで包装される。一部の好ましい実施形態において、安定した剤形はHPMCカプセル中に収納され、乾燥剤を伴って包装される。一部の実施形態において、安定した剤形は、ゼラチンカプセル内に収納され、乾燥剤無しで包装される。一部の実施形態において、安定した剤形はゼラチンカプセル内に収納され、乾燥剤を伴って包装される。
【0102】
安定した剤形の溶出率
レボミルナシプランの安定した剤形は、使用環境内に入った後所望の溶出率を提供することがわかっている。一部の実施形態において、安定した剤形は、使用環境内に入ってから約6時間〜約16時間後に少なくとも約80%(例えば少なくとも80%)の溶出率(例えば単相溶出率(single phase dissolution rate)を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約6時間〜約12時間後に少なくとも約80%の溶出率を提供する。
【0103】
一部の実施形態において、安定した剤形は、使用環境内に入ってから約2時間後に約60%未満の溶出率(例えば単相溶出率)を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約2時間後に約55%未満の溶出率(例えば単相溶出率)を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約2時間後に約50%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約2時間後に約45%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約2時間後に約40%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約2時間後に約20%〜約60%の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約2時間後に約25%〜約55%の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約2時間後に約30%〜約50%の溶出率を提供する。
【0104】
一部の実施形態において、安定した剤形は、使用環境内に入ってから約4時間後に約90%未満の溶出率(例えば単相溶出率)を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約4時間後に約80%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約4時間後に約70%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約4時間後に約65%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約4時間後に約60%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約4時間後に約40%〜約80%の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約4時間後に約45%〜約75%の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約4時間後に約40%〜約70%の溶出率を提供する。
【0105】
一部の実施形態において、安定した剤形は、使用環境内に入ってから約6時間後に約90%未満の溶出率(例えば単相溶出率)を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約6時間後に約85%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約6時間後に約80%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約6時間後に約75%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約6時間後に約70%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約6時間後に約40%〜約95%の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約6時間後に約50%〜約90%の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約6時間後に約60%〜約85%の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約6時間後に約60%〜約80%の溶出率を提供する。
【0106】
一部の実施形態において、安定した剤形は、使用環境内に入ってから約8時間後に約95%未満の溶出率(例えば単相溶出率)を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約8時間後に約90%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約8時間後に約85%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約8時間後に約65%〜約95%の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約8時間後に約65%〜約90%の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約8時間後に約65%〜約85%の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約8時間後に約70%〜約85%の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約8時間後に約70%〜約80%の溶出率を提供する。
【0107】
一部の実施形態において、安定した剤形は、使用環境内に入ってから約12時間後に約95%未満の溶出率(例えば単相溶出率)を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約12時間後に約90%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約12時間後に約75%〜約95%の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約12時間後に約80%〜約95%の溶出率を提供する。一部の実施形態において、安定した剤形は、使用環境内に入ってから約12時間後に約80%〜約90%の溶出率を提供する。
【0108】
一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約6時間〜約16時間後に、少なくとも約80%(例えば少なくとも80%)の溶出率(例えば単相溶出率)を提供する(例えば温度が37℃で75rpmのUSP装置IIに付された1000mLの脱イオン水中に入った後であり、ここでレボミルナシプランは220nmの波長でUV検出器を伴うHPLCを用いて定量化されている)。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約6時間〜約12時間後に、少なくとも約80%の溶出率を提供する。
【0109】
一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約2時間後に約60%未満の溶出率(例えば単相溶出率)を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約2時間後に約55%未満の溶出率(例えば単相溶出率)を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約2時間後に約50%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約2時間後に約45%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約2時間後に約40%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約2時間後に約20%〜約60%の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約2時間後に約25%〜約55%の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約2時間後に約30%〜約50%の溶出率を提供する。
【0110】
一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約4時間後に約90%未満の溶出率(例えば単相溶出率)を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約4時間後に約80%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約4時間後に約70%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約4時間後に約65%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約4時間後に約60%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約4時間後に約40%〜約80%の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約4時間後に約45%〜約75%の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約4時間後に約40%〜約70%の溶出率を提供する。
【0111】
一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約6時間後に約90%未満の溶出率(例えば単相溶出率)を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約6時間後に約85%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約6時間後に約80%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約6時間後に約75%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約6時間後に約70%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約6時間後に約40%〜約95%の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約6時間後に約50%〜約90%の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約6時間後に約60%〜約85%の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約6時間後に約60%〜約80%の溶出率を提供する。
【0112】
一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約8時間後に約95%未満の溶出率(例えば単相溶出率)を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約8時間後に約90%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約8時間後に約85%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約8時間後に約65%〜約95%の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約8時間後に約65%〜約90%の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約8時間後に約65%〜約85%の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約8時間後に約70%〜約85%の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約8時間後に約70%〜約80%の溶出率を提供する。
【0113】
一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約12時間後に約95%未満の溶出率(例えば単相溶出率)を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約12時間後に約90%未満の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約12時間後に約75%〜約95%の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水に入ってから約12時間後に約80%〜約95%の溶出率を提供する。一部の実施形態において、安定した剤形は、温度が約37℃で75rpmのUSP装置IIに付された脱イオン水中に入ってから約12時間後に約80%〜約90%の溶出率を提供する。
【0114】
一部の実施形態において、レボミルナシプランの安定した剤形は、40℃、相対湿度(RH)75%で1カ月、2カ月さらには3カ月間剤形を保管した後、論述された溶出率を達成する。
【0115】
安定した剤形の薬物動態(pK)性能
安定した剤形は、使用環境内に入った場合に長時間にわたるレボミルナシプランの持続放出を提供し、ヒトの患者に対して投与した場合に所望のpKプロファイルを達成するものと予期されている。一部の実施形態において、安定した剤形は、例えば単回投与の後およそ24時間にわたり、治療的血漿レベルのレボミルナシプランを提供し得る(すなわちこれを達成するものと予期される)。例えば、一部の実施形態において、安定した剤形は使用環境内に剤形が入ってから約4時間〜約24時間(例えば約5時間〜約24時間、さらには約6時間〜約24時間)の間レボミルナシプランを放出し得る。
【0116】
一部の実施形態において、安定した剤形は、患者に投与(例えば単回投与)した後、少なくとも1時間の平均Tmax(最大血漿濃度までの平均時間)を提供し得る。一部の実施形態において、安定した剤形は、患者に投与(例えば単回投与)した後、少なくとも2時間の平均Tmax(最大血漿濃度までの平均時間)を提供し得る。一部の実施形態において、安定した剤形は、患者に投与(例えば単回投与)した後、少なくとも3時間の平均Tmax(最大血漿濃度までの平均時間)を提供し得る。一部の実施形態において、安定した剤形は、少なくとも3.5時間の平均Tmaxを提供し得る。好ましくは、本発明の安定した剤形は、少なくとも4時間の平均Tmaxを提供し得る。例えば安定した剤形は、少なくとも4.5時間の平均Tmaxを提供し得る。
【0117】
一部の実施形態において、剤形は少なくとも5時間の平均Tmaxを提供し得る。一部の実施形態において、剤形は少なくとも5.5時間の平均Tmaxを提供し得る。一部の実施形態において、剤形は少なくとも6時間の平均Tmaxを提供し得る。安定した剤形は同様に、約4時間〜約12時間の平均Tmaxを提供できる。例えば、安定した剤形は約4時間〜約10時間の平均Tmaxを提供できる。一部の実施形態において、安定した剤形は約4.5時間〜約12時間の平均Tmaxを提供し得る。一部の実施形態において、剤形は約4.5時間〜約10時間の平均Tmaxを提供し得る。一部の実施形態において、剤形は約5時間〜約12時間の平均Tmaxを提供し得る。一部の実施形態において、剤形は約5時間〜約10時間の平均Tmaxを提供し得る。一部の実施形態において、剤形は約4時間〜約8時間の平均Tmaxを提供し得る。一部の実施形態において、剤形は約4.5時間〜約8.5時間の平均Tmaxを提供し得る。一部の実施形態において、剤形は約5時間〜約8時間の平均Tmaxを提供し得る。一部の実施形態において、剤形は約4時間〜約9時間の平均Tmaxを提供し得る。
【0118】
一部の実施形態において、安定した剤形は約500〜約20,000ng hr/mLの平均AUC0−∞(経時的レボミルナシプラン血漿濃度)を提供し得る。一部の実施形態において、安定した剤形は約500〜約15,000ng hr/mLの平均AUC0−∞(経時的レボミルナシプラン血漿濃度)を提供し得る。一部の実施形態において、安定した剤形は約500〜約10,000ng hr/mLの平均AUC0−∞(経時的レボミルナシプラン血漿濃度)を提供し得る。一部の実施形態において、安定した剤形は約1000〜約9000ngの平均AUC0−∞(経時的レボミルナシプラン血漿濃度)を提供し得る。一部の実施形態において、安定した剤形は約500〜約5,000ng hr/mLの平均AUC0−∞(経時的レボミルナシプラン血漿濃度)を提供し得る。一部の実施形態において、安定した剤形は約500〜約2500ng hr/mLの平均AUC0−∞(経時的レボミルナシプラン血漿濃度)を提供し得る。一部の実施形態において、剤形は、約500〜約2200ng hr/mLの平均AUC0−∞を提供し得る。一部の実施形態において、剤形は、約700〜約2500ng hr/mLの平均AUC0−∞を提供し得る。
【0119】
一部の実施形態において、剤形は約700〜約2200ng hr/mLの平均AUC0−∞を提供し得る。一部の実施形態において、剤形は約800〜約2200ng hr/mLの平均AUC0−∞を提供し得る。一部の実施形態において、剤形は約700〜約2300ng hr/mLの平均AUC0−∞を提供し得る。一部の実施形態において、剤形は約1000〜約2000ng hr/mLの平均AUC0−∞を提供し得る。一部の実施形態において、剤形は、約1000〜約1800ng hr/mLの平均AUC0−∞を提供し得る。一部の実施形態において、剤形は、約1100〜約1800ng hr/mLの平均AUC0−∞を提供し得る。一部の実施形態において、剤形は、約1200〜約1700ng hr/mLの平均AUC0−∞を提供し得る。一部の実施形態において、剤形は、約1300〜約1700ng hr/mLの平均AUC0−∞を提供し得る。一部の実施形態において、剤形は、約1300〜約1650ng hr/mLの平均AUC0−∞を提供し得る。
【0120】
安定した剤形により提供される平均最大血漿濃度(Cmax)は、剤形の強度を変更することによって(例えば剤形のTmaxに実質的な影響を及ぼすことなく)修正可能である。一部の実施形態において、剤形は、患者に対する投与(例えば単回投与)の後約200ng/ml未満の平均Cmaxを提供し得る。一部の実施形態において、剤形は、約180ng/ml未満の平均Cmaxを提供し得る。一部の実施形態において、剤形は、約170ng/ml未満の平均Cmaxを提供し得る。一部の実施形態において、剤形は約160ng/ml未満の平均Cmaxを提供し得る。一部の実施形態において、剤形は約150ng/ml未満の平均Cmaxを提供し得る。一部の実施形態において、剤形は約140ng/ml未満の平均Cmaxを提供し得る。一部の実施形態において、剤形は、約130ng/ml未満の平均Cmaxを提供し得る。
【0121】
一部の実施形態において、剤形は、約120ng/ml未満の平均Cmaxを提供し得る。一部の実施形態において、剤形は約110ng/ml未満の平均Cmaxを提供し得る。一部の実施形態において、剤形は、約100ng/ml未満の平均Cmaxを提供し得る。一部の実施形態において、剤形は約20〜約250ng/mLの平均Cmaxを提供し得る。一部の実施形態において、剤形は約20〜約200ng/mLの平均Cmaxを提供し得る。一部の実施形態において、剤形は約20〜約180ng/mLの平均Cmaxを提供し得る。一部の実施形態において、剤形は約30〜約140ng/mLの平均Cmaxを提供し得る。一部の実施形態において、剤形は約40〜約140ng/mLの平均Cmaxを提供し得る。一部の実施形態において、剤形は約20〜約150ng/mLの平均Cmaxを提供し得る。
【0122】
安定した剤形は同様に、一部の実施形態において、少なくとも約6時間の平均半減期(T1/2)を提供することが発見されている。一部の実施形態において、剤形は少なくとも約7時間の平均T1/2を提供し得る。一部の実施形態において、剤形は少なくとも約8時間の平均T1/2を提供し得る。一部の実施形態において、剤形は少なくとも約9時間の平均T1/2を提供し得る。一部の実施形態において、剤形は、少なくとも約10時間の平均T1/2を提供し得る。一部の実施形態において、剤形は少なくとも約11時間の平均T1/2を提供し得る。
【0123】
一部の実施形態において、剤形は、少なくとも約12時間の平均T1/2を提供し得る。一部の実施形態において、投薬量は、約6時間〜約24時間の平均T1/2を提供し得る。一部の実施形態において、投薬量は、約6時間〜約18時間の平均T1/2を提供し得る。一部の実施形態において、投薬量は約7時間〜約18時間の平均T1/2を提供し得る。一部の実施形態において、投薬量は約8時間〜約24時間の平均T1/2を提供し得る。一部の実施形態において、投薬量は約8時間〜約18時間の平均T1/2を提供し得る。
【0124】
一部の好ましい実施形態において、安定した剤形は放出調節型剤形であり、約1000〜約9000ng hr/mLの平均AUC0−∞を提供する。
【0125】
一部の好ましい実施形態において、安定した剤形は放出調節型剤形であり、約50〜約350ng/mlの平均Cmaxを提供する。
【0126】
一部の好ましい実施形態において、安定した剤形は放出調節型剤形であり、約5〜12時間の平均Tmaxを提供する。
【0127】
一部の好ましい実施形態において、安定した剤形は放出調節型剤形であり、約9時間〜約20時間の平均T1/2を提供する。
【0128】
一部の好ましい実施形態において、安定した剤形は放出調節型剤形であり、約1000〜約9000ng hr/mLの平均AUC0−∞、約50〜約350ng/mlの平均Cmax、約5〜12時間の平均Tmaxおよび約9時間〜約20時間の平均T1/2を提供する。
【0129】
一部の実施形態において、安定した剤形は約200ng/ml未満の平均Cmax、約2500ng hr/mL未満の平均AUC0−∞および少なくとも約4時間の平均Tmaxを提供し得る。
【0130】
一部の実施形態において、安定した剤形は、約180ng/ml未満の平均Cmax、約2500ng hr/mL未満の平均AUC0−∞および少なくとも約4時間の平均Tmaxを提供し得る。
【0131】
一部の実施形態において、安定した剤形は、約160ng/ml未満の平均Cmax、約2500ng hr/mL未満の平均AUC0−∞および少なくとも約4時間の平均Tmaxを提供し得る。
【0132】
一部の実施形態において、安定した剤形は、約150ng/ml未満の平均Cmax、約2500ng hr/mL未満の平均AUC0−∞および少なくとも約4時間の平均Tmaxを提供し得る。
【0133】
一部の実施形態において、安定した剤形は、約140ng/ml未満の平均Cmax、約2500ng hr/mL未満の平均AUC0−∞および少なくとも約4時間の平均Tmaxを提供し得る。
【0134】
一部の実施形態において、安定した剤形は、約140ng/mlの平均Cmax、約2200ng hr/mL未満の平均AUC0−∞および少なくとも約4時間の平均Tmaxを提供し得る。
【0135】
一部の実施形態において、安定した剤形は、約180ng/ml未満の平均Cmax、約2200ng hr/mL未満の平均AUC0−∞および少なくとも約5時間の平均Tmaxを提供し得る。
【0136】
一部の実施形態において、安定した剤形は、約180ng/ml未満の平均Cmax、約2200ng hr/mL未満の平均AUC0−∞および少なくとも約6時間の平均Tmaxを提供し得る。
【0137】
一部の実施形態において、安定した剤形は、約10〜約200ng/mlの平均Cmax、約500〜約2500ng hr/mLの平均AUC0−∞および約4〜約10時間の平均Tmaxを提供し得る。
【0138】
一部の実施形態において、安定した剤形は、約25〜約175ng/mlの平均Cmax、約500〜約2500ng hr/mLの平均AUC0−∞および約4時間〜約10時間の平均Tmaxを提供し得る。
【0139】
一部の実施形態において、安定した剤形は、約30〜約150ng/mlの平均Cmax、約500〜約2500ng hr/mLの平均AUC0−∞および約4〜約10時間の平均Tmaxを提供し得る。
【0140】
一部の実施形態において、安定した剤形は、約30〜約120ng/mlの平均Cmax、約500〜約2500ng hr/mLの平均AUC0−∞および約4〜約10時間の平均Tmaxを提供し得る。
【0141】
一部の実施形態において、安定した剤形は、約10〜約200ng/mlの平均Cmax、約600〜約2200ng hr/mLの平均AUC0−∞および約4〜約10時間の平均Tmaxを提供し得る。
【0142】
一部の実施形態において、安定した剤形は、約10〜約200ng/mlの平均Cmax、約800〜約2100ng hr/mLの平均AUC0−∞および約4〜約10時間の平均Tmaxを提供し得る。
【0143】
一部の実施形態において、安定した剤形は、約10〜約200ng/mlの平均Cmax、約900〜約2100ng hr/mLの平均AUC0−∞および約4〜約10時間の平均Tmaxを提供し得る。
【0144】
一部の実施形態において、安定した剤形は、約30〜約150ng/mlの平均Cmax、約600〜約2200ng hr/mLの平均AUC0−∞および約4〜約10時間の平均Tmaxを提供し得る。
【0145】
一部の実施形態において、安定した剤形は、約30〜約120ng/mlの平均Cmax、約600〜約2200ng hr/mLの平均AUC0−∞および約4〜約10時間の平均Tmaxを提供し得る。
【0146】
一部の実施形態において、安定した剤形は、約10〜約200ng/mlの平均Cmax、約500〜約2500ng hr/mLの平均AUC0−∞および約4〜約8時間の平均Tmaxを伴う生体内血漿プロファイルを提供し得る。
【0147】
一部の実施形態において、安定した剤形は、約10〜約200ng/mlの平均Cmax、約500〜約2500ng hr/mLの平均AUC0−∞および約5〜約8時間の平均Tmaxを伴う生体内血漿プロファイルを提供し得る。
【0148】
一部の実施形態において、安定した剤形は、約25〜約175ng/mlの平均Cmax、約600〜約2200ng hr/mLの平均AUC0−∞および約4〜約9時間の平均Tmaxを伴う生体内血漿プロファイルを提供し得る。
【0149】
一部の実施形態において、安定した剤形は、約30〜約150ng/mlの平均Cmax、約800〜約2100ng hr/mLの平均AUC0−∞および約4〜約9時間の平均Tmaxを伴う生体内血漿プロファイルを提供し得る。
【0150】
一部の実施形態において、約125ng/ml未満の平均Cmax、約1000〜約2200ng hr/mLの平均AUC0−∞および少なくとも約4時間の平均Tmaxを伴う生体内血漿プロファイルを提供し得る剤形が提供されている。
【0151】
安定した剤形を使用する治療方法
本発明は同様に、治療を必要としている患者に対して安定した剤形を投与することによって哺乳動物(例えばヒト)における不安障害またはうつ病(例えば大うつ病性障害(MDD))などの5−HTおよびNEの再取り込み阻害(例えば2重阻害および/または選択的阻害)により管理可能な疾病、障害または身体条件を治療するための方法も提供する。
【0152】
一部の実施形態においては、治療を必要としている患者におけるうつ病(例えば非定型うつ病またはMDD)、不安神経症(例えば全般的不安障害)またはうつ病や不安神経症に付随する倦怠感を、前記患者に対してレボミルナシプランの安定した剤形を投与することによって治療または予防するための方法が提供されている。一部の実施形態において、治療を必要とする患者における大うつ病性障害(MDD)(例えば急性MDDまたは非定型MDD)を、前記患者に対してレボミルナシプランの安定した剤形を投与することによって治療または予防するための方法が提供されている。一部の実施形態において、治療を必要とする患者における未解決の、併発するまたは随伴する倦怠感を伴うMDDを、前記患者に対してレボミルナシプランの安定した剤形を投与することによって治療または予防するための方法が提供されている。
【0153】
一部の実施形態において、患者に剤形を投与することによって治療を必要とする患者におけるMDDの再発を治療または予防するために、レボミルナシプランの安定した剤形が使用される。一部の実施形態において、安定した剤形は、患者に剤形を投与することによって治療を必要とする患者における倦怠感(例えばMDDまたは他の形態のうつ病に付随する倦怠感)を治療または予防するために使用される。一部の実施形態において、安定した剤形は、患者に剤形を投与することによって治療を必要とする患者における性的機能不全(例えば勃起不全)を治療または予防するために使用される。一部の実施形態において、安定した剤形は、患者に剤形を投与することによって治療を必要とする患者におけるうつ病(例えばMDD)に付随する疼痛を治療または予防するために使用される。
【0154】
一部の実施形態において、本発明の安定した剤形は、患者に安定した剤形を投与することによって治療を必要とする患者におけるうつ病(例えばMDD)に付随する(またはそれと併発する)うつ状態、気分変調、傾眠、認識機能障害、睡眠障害および/または高脂血症を治療または予防するために使用される。
【0155】
一部の実施形態においては、治療を必要とする患者において、神経因性疼痛(例えば糖尿病性多発神経障害性疼痛(DPNP))を治療または予防するための方法において、患者に対してレボミルナシプランの安定した剤形を有効量投与するステップを含む方法が提供されている。
【0156】
レボミルナシプランの安定した剤形を投与することを通して、薬物血漿濃度対時間の平坦化されたプロファイルを得、それにより一日多数回の投薬で得ることのできるものよりも密な血漿療法範囲の制御を提供するための方法が提供されている。換言すると、レボミルナシプランの従来の即時放出型調合物での一日多数回の投薬により誘発される血漿薬物レベルにおける急なピークおよびトラフを除去するための方法が提供されている。本質的には、レボミルナシプランの血漿レベルは、本発明の安定した剤形を投与した後数時間にわたり上昇し、その後下降し始め、およそ24時間の残りの時間中ピーク血漿レベルからの実質的に線形の持続的な減少を長々と続け、全期間中少なくともレボミルナシプランの閾値治療レベルを維持する。
【0157】
以下の実施例は単に本発明を例示しているにすぎず、当業者には本開示を読んだ上で本発明により包含される多くの変形形態および等価物が明白となるため、いかなる形であれ本発明の範囲を限定するものとしてみなされてはならない。
【実施例】
【0158】
実施例1:レボミルナシプラン剤形の調製
糖球(およそ30〜35メッシュ)を予熱しウルスタープロセスを介して約3.5時間にわたり薬物層溶液で予熱済み糖球を層状化して薬物装填済みビーズを形成することにより、レボミルナシプランビーズを調製した。薬物層溶液は、レボミルナシプラン、Povidone K30、タルクおよび脱水アルコールを含んでいた。流動床内で約30分間薬物装填済みビーズを乾燥させ、篩過して、表4に示されている構成成分の濃度をおおよそ含む即時放出型レボミルナシプランビーズ(およそ540mg/g)を生成した。
【0159】
【表4】
【0160】
次に、レボミルナシプランビーズを予熱し、ウルスタープロセスを介して分散溶液でコーティングした。分散溶液は、エチルセルロースN22、クエン酸トリエチル、タルクおよび脱水アルコールを含んでいた。コーティングされたビーズを流動床内で入念に硬化させ、篩分けしてレボミルナシプランの持続放出型ビーズ(およそ460mg/g)を生成してからカプセル内に充填した。持続放出型レボミルナシプランビーズは、おおよそ表5に示された構成成分濃度を含んでいた。
【0161】
【表5】
【0162】
実施例2:実施例1で調製された剤形の安定性
2つの異なるタイプのカプセル(すなわち硬質ゼラチンカプセルおよびHPMCカプセル)内の剤形を40℃、相対湿度(RH)75%で3カ月間保管した後、実施例1で調製したレボミルナシプランの剤形の安定性を査定した。
【0163】
表6は、3カ月の保管後にレボミルナシプランの安定した剤形の硬質ゼラチンカプセル内に見出された不純物のおおよその濃度を示す。
【0164】
【表6】
【0165】
表7は、レボミルナシプランの安定した剤形のHPMCカプセル内に見出された不純物のおおよその濃度を示す。
【0166】
【表7】
【0167】
実施例3:実施例1で調製された剤形の溶出率
実施例1で調製されたレボミルナシプランの安定した剤形の溶出率を、75rpmのUSP装置IIを用いて37℃の脱イオン水1000mLで評価した。レボミルナシプランの定量化は、220nmの波長でUV検出器を伴うHPLCを用いて判定した。
【0168】
実施例1で調製した(乾燥剤を全く含まないHPMCカプセル内に装填された)安定した剤形の溶出プロファイルを、図1および表8に示す。
【0169】
実施例1で調製した安定した剤形の溶出プロファイルを同様に、40℃および相対湿度(RH)75%で1、2および3カ月間剤形を保管した後にも判定し、この場合剤形は表8および図1〜5で示す通り、(1グラムの乾燥剤を含むかまたは乾燥剤を全く含まない)HPMCカプセル内に装填した。
【0170】
詳細には、図1は、保管を全くしていないレボミルナシプランの安定した剤形についての溶出率を示しており、ここで剤形は乾燥剤を全く含まないHPMCカプセル内に装填されている。図2は、40℃、75%RHで1カ月保管した後のレボミルナシプランの安定した剤形についての溶出率を示しており、ここでも剤形は、乾燥剤を全く含まないHPMCカプセル内に装填されている。図3は、40℃、75%RHで2カ月保管した後のレボミルナシプランの安定した剤形についての溶出率を示しており、ここでも剤形は乾燥剤を全く含まないHPMCカプセル内に装填されている。図4は、40℃、75%RHで3カ月保管した後のレボミルナシプランの安定した剤形についての溶出率を示しており、ここでも剤形は、乾燥剤を全く含まないHPMCカプセル内に装填されている。図5は、40℃、75%RHで3カ月保管した後のレボミルナシプランの安定した剤形についての溶出率を示しており、ここで剤形は乾燥剤を1グラム含むHPMCカプセル内に装填されている。
【0171】
【表8】
【0172】
実施例4− 実質的に純粋なレボミルナシプランを含む活性成分のX線粉末回折(XRD)分析
塩酸レボミルナシプランを少なくとも98重量%含む活性成分の試料を深いアルミニウムホルダー上に装填し、広角ベンチトップX線回折計(D8型、Bruker AXS Inc.,Madison WI)内でCuKα放射(40kV×40mA)に曝露した。計器を0.05°2θの増分でステップ走査モードで作動させた。角度範囲は5〜40°2θであり、走査速度は0.15°2θ/分であった。市販のソフトウェアを用いてデータ収集および分析を実施した。
【0173】
図6のXRDパターンについてのピーク位置を表1に示す。
【0174】
【表9】
【0175】
実施例5− 実施例1で調製した即時放出型レボミルナシプランビーズのX線粉末回折(XRD)分析
実施例1で調製したIR(即時放出型)レボミルナシプランビーズを深いアルミニウムホルダー上に装填し、広角ベンチトップX線回折計(D8型、Bruker AXS Inc.,Madison WI)内でCuKα放射(40kV×40mA)に曝露した。計器を0.05°2θの増分でステップ走査モードで作動させた。角度範囲は5〜40°2θであり、走査速度は0.15°2θ/分であった。市販のソフトウェアを用いてデータ収集および分析を実施した。
【0176】
図7のXRDパターンについてのピーク位置を表2に示す。
【0177】
【表10】
【0178】
実施例6− 実施例1で調製した持続放出型剤形のX線粉末回折(XRD)分析
実施例1で調製した少量の持続放出型剤形を深いアルミニウムホルダー上に装填し、広角ベンチトップX線回折計(D8型、Bruker AXS Inc.,Madison WI)内でCuKα放射(40kV×40mA)に曝露した。計器を0.05°2θの増分でステップ走査モードで作動させた。角度範囲は5〜40°2θであり、走査速度は0.15°2θ/分であった。市販のソフトウェア(具体的には、DIFFRACplus XRD Commander,Bruker−AXS GmbH;およびJADE,Materials Data,Inc.)を用いてデータ収集および分析を実施した。
【0179】
レボミルナシプランの安定した剤形についてのXRDパターンを図7に示す。図8のXRDパターンについてのピーク位置は表3に提供されている。
【0180】
【表11】
【0181】
実施例7− ヒトの患者に対するレボミルナシプランの安定した剤形の投与(理論上の予測)
50mgのレボミルナシプランが入ったカプセルの形でヒトの患者に対して本発明のレボミルナシプランの安定した剤形を投与することができる。カプセルには、約7.5重量%のエチルセルロース(EC)(「剤形1」)、約10重量%のEC(「剤形2」)および約12.5重量%EC(「剤形3」)でコーティングされた微小顆粒が入り得る。
【0182】
剤形1〜3は、少なくとも約10時間の絶食期間後に患者に投与することができる。投与前および投与後複数の時点(例えば30分、1時間、2時間、3時間、4時間、5時間、6時間、7時間、8時間、10時間、12時間、24時間、48時間および72時間後)に各患者から血液試料を収集することができる。各患者からの血漿試料を、LC/MS−MS検出と連動させた有効オンライン抽出方法(乱流クロマトグラフィ)を用いて、レボミルナシプランの定量化について各患者を査定することができる。
【0183】
安定した剤形を経口投与することにより、表9に剤形1〜3のいずれかについて実質的に示されている通りの薬物動態(pK)パラメータが生成されることが予期される。一部の好ましい実施形態において、安定した剤形は、実質的に剤形2について示されている通りのpKパラメータを生成する。これらのpKパラメータは、幾何平均(幾何学的CV%)および[範囲]としても表現されている。
【0184】
【表12】
【0185】
単回経口投与後(投薬から最高24時間後)に得られうる平均レボミルナシプラン血漿濃度対時間プロファイルは、図9(幾何平均として表わされた対数−線形スケール)に示されている。
【0186】
本発明について、その例示的実施形態を参考にして描写し記述してきたが、このような参照は本発明に対する限定を意味せず、このような限定が暗示されることは一切ない。本発明は、本開示の利益を受ける関連技術分野の当業者が思いつく形態および機能面の多数の修正、改変および等価物の可能性を含むものである。
【0187】
描写したおよび記述した本発明の実施形態は、単なる一例にすぎず、本発明の範囲を完全に網羅するものではない。したがって、本発明は添付クレームの精神および範囲によってのみ制限され、あらゆる面で等価物を認知するものである。
【特許請求の範囲】
【請求項1】
レボミルナシプランまたはその薬学的に許容される塩を含む安定した剤形。
【請求項2】
少なくとも98重量%のレボミルナシプランまたはその薬学的に許容される塩を含む活性成分を含む安定した剤形。
【請求項3】
12.0、20.1および22.5±0.2度2θに特徴的ピークを含むX線粉末回折(XRD)パターンを含む、請求項2に記載の安定した剤形。
【請求項4】
XRDパターンがさらに32.7±0.2度2θに特徴的ピークを含む、請求項3に記載の安定した剤形。
【請求項5】
XRDパターンがさらに6.0±0.2度2θに特徴的ピークを含む、請求項3に記載の安定した剤形。
【請求項6】
6.0、12.0および20.1±0.2度2θに特徴的ピークを含むXRDパターンを含む、請求項2に記載の安定した剤形。
【請求項7】
剤形が、約0.001重量%〜約0.5重量%の(1S,5R)1−フェニル−3−アザビシクロ[3−1−0]ヘキサン−2−オンを含む、請求項2に記載の安定した剤形。
【請求項8】
剤形が、約0.001%重量%〜約0.2重量%の(1S,5R)1−フェニル−3−アザビシクロ[3−1−0]ヘキサン−2−オンを含む、請求項2に記載の安定した剤形。
【請求項9】
剤形が、約45重量%〜約60重量%の活性成分と、約30重量%〜約45重量%の不活性基質または充填剤とを含む、請求項2に記載の安定した剤形。
【請求項10】
剤形が、約45重量%〜約60重量%の活性成分と、約30重量%〜約45重量%の不活性基質または充填剤と、約4重量%〜約10重量%の結合剤と、約1重量%〜約5重量%の付着防止剤または潤滑剤とを含む、請求項2に記載の安定した剤形。
【請求項11】
剤形が、約50重量%〜約60重量%の活性成分と、約30重量%〜約40重量%の不活性基質または充填剤と、約4重量%〜約8重量%の結合剤と、約1重量%〜約5重量%の付着防止剤または潤滑剤とを含む、請求項2に記載の安定した剤形。
【請求項12】
剤形が、即時放出型経口剤形である、請求項9に記載の安定した剤形。
【請求項13】
剤形が、約40重量%〜約55重量%のレボミルナシプランまたはその薬学的に許容される塩と、約5重量%〜約15重量%の放出制御剤と、約25重量%〜約40重量%の不活性基質と、約3重量%〜約10重量%の結合剤と、約3重量%〜約10重量%の付着防止剤と、約0.1重量%〜約5重量%の可塑剤とを含む、請求項2に記載の安定した剤形。
【請求項14】
剤形が、放出調節型経口剤形である、請求項13に記載の安定した剤形。
【請求項15】
剤形が、使用環境内に入ってから約6時間〜約16時間後に少なくとも約80%の溶出率を提供する、請求項2に記載の剤形。
【請求項16】
実質的に純粋なレボミルナシプランまたはその薬学的に許容される塩を含む活性成分と、約0.01重量%〜約0.2重量%の(1S,5R)1−フェニル−3−アザビシクロ[3−1−0]ヘキサン−2−オンとを含む安定した剤形において、12.0、20.1および22.5±0.2度2θに特徴的ピークを含むX線粉末回折(XRD)パターンを含む、安定した剤形。
【請求項17】
約45重量%〜約60重量%の活性成分と、約4重量%〜約10重量%の結合剤を含む、請求項16に記載の安定した剤形。
【請求項18】
治療を要する患者に対して請求項1に記載の安定した剤形を投与するステップを含む、大うつ病性障害の治療方法。
【請求項19】
治療を要する患者に対して請求項2に記載の安定した剤形を投与するステップを含む、大うつ病性障害の治療方法。
【請求項20】
治療を要する患者に対して請求項16に記載の安定した剤形を投与するステップを含む、大うつ病性障害の治療方法。
【請求項21】
不活性基質をレボミルナシプランまたはその薬学的に許容される塩および脱水アルコールと接触させるステップを含む、請求項1に記載の安定した剤形の調製方法。
【請求項22】
不活性基質をレボミルナシプランまたはその薬学的に許容される塩および脱水アルコールと接触させるステップを含む、請求項2に記載の安定した剤形の調製方法。
【請求項23】
不活性基質をレボミルナシプランまたはその薬学的に許容される塩および脱水アルコールと接触させるステップを含む、請求項16に記載の安定した剤形の調製方法。
【請求項1】
レボミルナシプランまたはその薬学的に許容される塩を含む安定した剤形。
【請求項2】
少なくとも98重量%のレボミルナシプランまたはその薬学的に許容される塩を含む活性成分を含む安定した剤形。
【請求項3】
12.0、20.1および22.5±0.2度2θに特徴的ピークを含むX線粉末回折(XRD)パターンを含む、請求項2に記載の安定した剤形。
【請求項4】
XRDパターンがさらに32.7±0.2度2θに特徴的ピークを含む、請求項3に記載の安定した剤形。
【請求項5】
XRDパターンがさらに6.0±0.2度2θに特徴的ピークを含む、請求項3に記載の安定した剤形。
【請求項6】
6.0、12.0および20.1±0.2度2θに特徴的ピークを含むXRDパターンを含む、請求項2に記載の安定した剤形。
【請求項7】
剤形が、約0.001重量%〜約0.5重量%の(1S,5R)1−フェニル−3−アザビシクロ[3−1−0]ヘキサン−2−オンを含む、請求項2に記載の安定した剤形。
【請求項8】
剤形が、約0.001%重量%〜約0.2重量%の(1S,5R)1−フェニル−3−アザビシクロ[3−1−0]ヘキサン−2−オンを含む、請求項2に記載の安定した剤形。
【請求項9】
剤形が、約45重量%〜約60重量%の活性成分と、約30重量%〜約45重量%の不活性基質または充填剤とを含む、請求項2に記載の安定した剤形。
【請求項10】
剤形が、約45重量%〜約60重量%の活性成分と、約30重量%〜約45重量%の不活性基質または充填剤と、約4重量%〜約10重量%の結合剤と、約1重量%〜約5重量%の付着防止剤または潤滑剤とを含む、請求項2に記載の安定した剤形。
【請求項11】
剤形が、約50重量%〜約60重量%の活性成分と、約30重量%〜約40重量%の不活性基質または充填剤と、約4重量%〜約8重量%の結合剤と、約1重量%〜約5重量%の付着防止剤または潤滑剤とを含む、請求項2に記載の安定した剤形。
【請求項12】
剤形が、即時放出型経口剤形である、請求項9に記載の安定した剤形。
【請求項13】
剤形が、約40重量%〜約55重量%のレボミルナシプランまたはその薬学的に許容される塩と、約5重量%〜約15重量%の放出制御剤と、約25重量%〜約40重量%の不活性基質と、約3重量%〜約10重量%の結合剤と、約3重量%〜約10重量%の付着防止剤と、約0.1重量%〜約5重量%の可塑剤とを含む、請求項2に記載の安定した剤形。
【請求項14】
剤形が、放出調節型経口剤形である、請求項13に記載の安定した剤形。
【請求項15】
剤形が、使用環境内に入ってから約6時間〜約16時間後に少なくとも約80%の溶出率を提供する、請求項2に記載の剤形。
【請求項16】
実質的に純粋なレボミルナシプランまたはその薬学的に許容される塩を含む活性成分と、約0.01重量%〜約0.2重量%の(1S,5R)1−フェニル−3−アザビシクロ[3−1−0]ヘキサン−2−オンとを含む安定した剤形において、12.0、20.1および22.5±0.2度2θに特徴的ピークを含むX線粉末回折(XRD)パターンを含む、安定した剤形。
【請求項17】
約45重量%〜約60重量%の活性成分と、約4重量%〜約10重量%の結合剤を含む、請求項16に記載の安定した剤形。
【請求項18】
治療を要する患者に対して請求項1に記載の安定した剤形を投与するステップを含む、大うつ病性障害の治療方法。
【請求項19】
治療を要する患者に対して請求項2に記載の安定した剤形を投与するステップを含む、大うつ病性障害の治療方法。
【請求項20】
治療を要する患者に対して請求項16に記載の安定した剤形を投与するステップを含む、大うつ病性障害の治療方法。
【請求項21】
不活性基質をレボミルナシプランまたはその薬学的に許容される塩および脱水アルコールと接触させるステップを含む、請求項1に記載の安定した剤形の調製方法。
【請求項22】
不活性基質をレボミルナシプランまたはその薬学的に許容される塩および脱水アルコールと接触させるステップを含む、請求項2に記載の安定した剤形の調製方法。
【請求項23】
不活性基質をレボミルナシプランまたはその薬学的に許容される塩および脱水アルコールと接触させるステップを含む、請求項16に記載の安定した剤形の調製方法。
【図1】

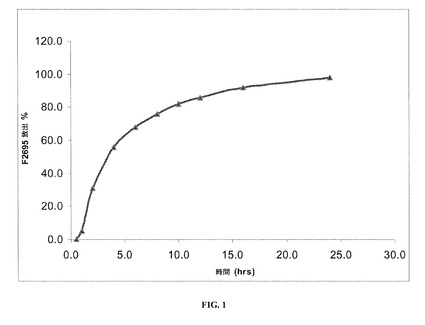
【図2】
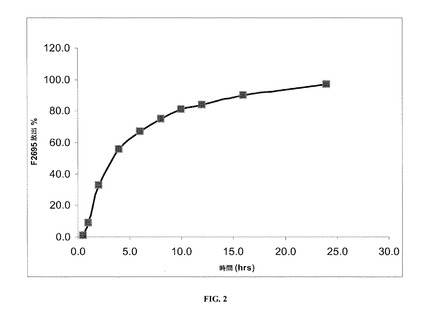
【図3】
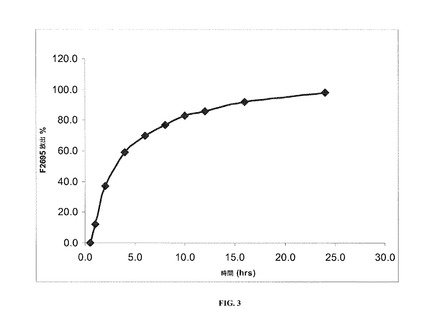
【図4】
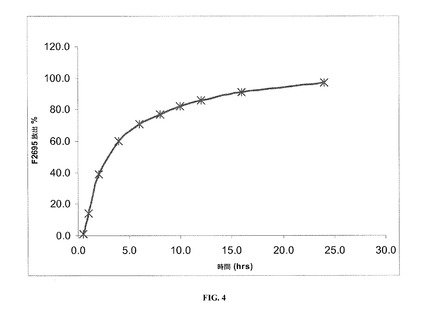
【図5】

【図6】
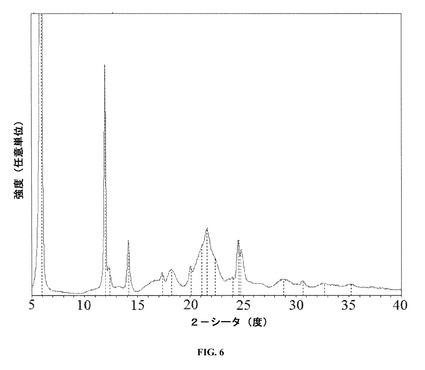
【図7】

【図8】
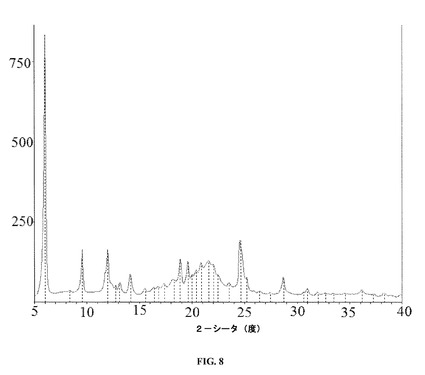
【図9】
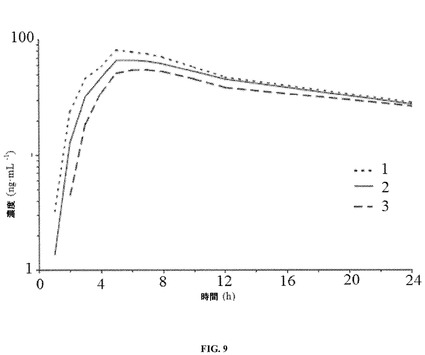
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公表番号】特表2013−517290(P2013−517290A)
【公表日】平成25年5月16日(2013.5.16)
【国際特許分類】
【出願番号】特願2012−549117(P2012−549117)
【出願日】平成23年1月14日(2011.1.14)
【国際出願番号】PCT/US2011/021315
【国際公開番号】WO2011/088331
【国際公開日】平成23年7月21日(2011.7.21)
【出願人】(504249145)
【氏名又は名称原語表記】PIERRE FABRE MEDICAMENT
【Fターム(参考)】
【公表日】平成25年5月16日(2013.5.16)
【国際特許分類】
【出願日】平成23年1月14日(2011.1.14)
【国際出願番号】PCT/US2011/021315
【国際公開番号】WO2011/088331
【国際公開日】平成23年7月21日(2011.7.21)
【出願人】(504249145)
【氏名又は名称原語表記】PIERRE FABRE MEDICAMENT
【Fターム(参考)】
[ Back to top ]