
レルカニジピンの塩
本発明はレルカニジピンならびに(i)無機酸、(ii)スルホン酸、(iii)モノカルボン酸、(iv)ジカルボン酸、(v)トリカルボン酸および(vi)芳香族スルホンイミドからなる群より選択される対イオンを含む新しい付加塩に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明はレルカニジピンの新しい酸塩に関するものであり、特に、非晶質および結晶のレルカニジピンの塩とその製造方法に関する。本発明は、また、水和および溶媒和した形での新しい非晶質および結晶のレルカニジピンの塩に関する。さらに、本発明は、ここに開示する新しいレルカニジピンの塩を含む医薬品組成物に関する。
【背景技術】
【0002】
下記に示す、メチル1,1,N‐トリメチル‐N‐(3,3‐ジフェニルプロピル)‐2‐アミノエチル1,4‐ジヒドロ‐2,6‐ジメチル‐4‐(3‐ニトロフェニル)‐ピリジン‐3,5‐ジカルボキシレート(INN(医薬品の国際一般名):レルカニジピン)は、長時間作用性で血管選択性の高い、高脂肪親和性のジヒドロピリジンカルシウム拮抗薬である。
【化1】

【0003】
レルカニジピンの塩酸塩(塩酸レルカニジピン)は、レコルダッチ社(Recordati S.p.A.)(イタリア、ミラノ)から市販されている。レルカニジピンの遊離塩基およびその塩酸塩の製法は、特許文献1〜5において、レルカニジピンを個々の光学異性体に分割する方法とともに、以前より述べられている。
【特許文献1】米国特許第4,705,797号明細書
【特許文献2】米国特許第5,767,136号明細書
【特許文献3】米国特許第4,968,832号明細書
【特許文献4】米国特許第5,912,351号明細書
【特許文献5】米国特許第5,696,139号明細書
【特許文献6】米国特許出願公開第2003/0069285号明細書
【特許文献7】米国特許出願公開第2003/0083355号明細書
【発明の開示】
【発明が解決しようとする課題】
【0004】
レルカニジピンを調製する工程での主に不便な点は、特許文献1で述べられているように、開示されている環化反応では様々な副生成物を生じ、目的とする生成物の収率が低下してしまうことである。特許文献4は塩酸レルカニジピンの調製のための、より簡素化された工程について述べている。その工程では非水和で非吸湿性の結晶構造を有する塩酸レルカニジピンを生じ、不要な副生成物の生成を防ぐと同時に、それに続いて行うクロマトグラフィーカラムによる精製工程を省いている。
【0005】
しかしながら、結晶構造を持つ塩酸レルカニジピンを単離することもまた、非常に手間がかかる。さらに、塩酸レルカニジピンは、それぞれが相異なる物理的性質を持つ少なくとも4種類の多形体のいずれかの形で存在している可能性がある(特許文献7および特許文献8参照)。したがって、当業者にとって、レルカニジピンの塩、特に結晶のレルカニジピンの塩を生成する、より簡素な製造工程が必要とされる。限定はしないが、患者によるばらつきが少ない、食物の影響が少ない、および多形が少ないか存在しない、塩酸レルカニジピンの以前に単離された構造とは異なる、好ましくは、より望ましい溶解性および/または他の物理的性質を持つ、レルカニジピンの塩もまた、必要とされている。
【課題を解決するための手段】
【0006】
定義および本明細書中で用いられる略語
「DSC」:示差走査熱量測定法
「非晶質」は、固体の状態の特性である、実質的な結晶格子構造を持たない化合物を表現するのに用いられる。これらの化合物では、結晶質の化合物の特徴である鋭い発熱ピ‐クの代わりに、ガラス転移と定義される、独特な幅の広い発熱を伴う変化を示すDSCのプロットが見られる。
【0007】
「結晶」は、融点ならびに結晶構造のX線スペクトル特性を持つ化合物を表現するのに用いられる。これらの化合物では、独特の鋭い発熱ピークを有するDSCのプロットが見られる。
【0008】
「ベシル酸レルカニジピン(lercanidipine besylate)」:レルカニジピンの酸塩の一種であり、ベンゼンスルホン酸をモル比1:1の対イオンとしている。
【0009】
「ナパジシル酸レルカニジピン(lercanidipine napadisylate)」:レルカニジピンの酸塩の一種であり、ナフタレン‐1,5‐ジスルホン酸をモル比2:1の対イオンとしている。
【0010】
「多形の」または「多形」:化合物中に少なくとも2種以上の異なる構造が存在する特性。異なる結晶構造は結晶学的技術によって直接的に、あるいは、それぞれ特有の多形に関連する物理的および/または化学的性質の差異の評価によって非直接的に検出される。
【0011】
ある態様において、本発明は、酸の対イオンが
(i)塩酸以外の、臭化水素酸、リン酸、硫酸などの無機酸、
(ii)メタンスルホン酸、ベンゼンスルホン酸、トルエンスルホン酸、およびナフタレン‐1,5‐ジスルホン酸などのスルホン酸、
(iii)酢酸、(+)‐L‐乳酸、DL‐乳酸、DL‐マンデル酸、グルコン酸、ケイヒ酸、サリチル酸、およびゲンチシン酸などのモノカルボン酸、
(iv)シュウ酸、2‐オキソ‐グルタル酸、マロン酸、(‐)‐L‐リンゴ酸、粘液酸、(+)‐L‐酒石酸、フマル酸、マレイン酸、およびテレフタル酸などのジカルボン酸、
(v)クエン酸などのトリカルボン酸、
(vi)サッカリンなどの芳香族スルホンイミド、
からなる群より選択されることを特徴とする、レルカニジピンの酸付加塩を提供する。
【0012】
本発明に従ったレルカニジピンの塩としては、L‐乳酸塩、ケイヒ酸塩、サリチル酸塩、マレイン酸塩、およびサッカリン酸塩が好ましい。本発明に従ったレルカニジピンの塩として最も好ましいのは、ベシル酸レルカニジピンおよびナパジシル酸レルカニジピンである。これらの好ましい、および最も好ましい塩は、すべて非晶質の形態で生成することができ、ベシル酸レルカニジピンおよびナパジシル酸レルカニジピンは、結晶の形態で生成することも可能である。
【0013】
本発明にかかる結晶のレルカニジピンの塩は、多形であって差し支えない。
【0014】
本発明にかかるレルカニジピンの塩は、溶媒和および水和した形態で存在して差し支えない。これらの溶媒和または水和した形態は、配位数が1または2の溶媒和物または水和物であって構わない。レルカニジピンの塩を生成するときに溶媒を用いた結果として、固体の格子構造中に溶媒和物および水和物を形成してもよい。レルカニジピンの塩が生成する間に、溶媒和物および水和物が形成されることから、特定の溶媒和または水和の構造の形成は、塩の生成に用いる条件および方法によって大きく左右される。溶媒は、医薬品として容認可能なものであるべきである。
【0015】
結晶のベシル酸レルカニジピンは黄白色で、良い安定性を示す。22℃での0.1モル塩酸への溶解度は約25〜35mg/lであり、さらに具体的には約30mg/lである。これと比較して、同じ溶媒への塩酸レルカニジピンの結晶の溶解度は約10mg/lである。ベシル酸レルカニジピンの結晶の融点(DSCピーク)は、約170℃〜約175℃の範囲であり、さらに具体的には、約172℃である。下記に示す実施例では、ベシル酸レルカニジピンの結晶は水和または溶媒和のどちらの構造も得られていないが、これらの構造は本発明に包含され、不定量の水を含む極性溶媒から再結晶化することによって得ることが可能である。下記の実施例での生成のように、ベシル酸レルカニジピンの結晶はゆっくりと生じ、結晶の種を加えて初めて高収率の結晶が生成され、多形は見られない。
【0016】
結晶のナパジシル酸レルカニジピンの結晶もまた黄白色で、安定性が良い。結晶の大きさは、ベシル酸レルカニジピンの結晶よりも大きい。ナパジシル酸レルカニジピンの結晶の0.1モル塩酸への溶解度は約3mg/l〜4mg/lであり、さらに具体的には約3.5mg/lである。これは、結晶の塩酸レルカニジピンや結晶のベシル酸レルカニジピンよりも低い値である。結晶のナパジシル酸レルカニジピンの融点(DSCピーク)は、約145℃〜約155℃の範囲であり、さらに具体的には約150℃である。ナパジシル酸レルカニジピンの結晶は、溶媒和した水和物、特にジメタノラート水和物(dimethanolate hydrate)として、または非水和の形で生成してもよい。下記の実施例に示すとおり、結晶のナパジシル酸レルカニジピンは、自然発生的に生成する。
【0017】
本発明にかかる結晶のレルカニジピンの塩は、実質的に純粋な形態で生成し、溶媒はほとんど残らない。特に、結晶のベシル酸レルカニジピンは、残留溶媒の含量が約0.1〜最大約0.5重量%、さらに詳しく言えば最大約0.2重量%以下で生成する可能性がある。結晶のナパジシル酸レルカニジピンは、残留溶媒の含量が約2.5〜最大約5重量%、さらに詳しく言えば最大約4重量%以下で生成する可能性がある。これらの結晶のレルカニジピンの塩の生成物は、純度99.5%、残留溶媒含量0.3重量%(3000ppm)以下で単離することが可能であるが、より純度の低い(および/または溶媒または水分含量がもっと高い)生成物であれば、従来法にて生成することも可能である。医薬品として容認可能な各不純物のレベルは、一般に0.1%以下であり、有機溶媒では、各溶媒の毒性に応じて、0.5%(5000ppm)〜0.0002%(2ppm)の範囲である。本発明にかかるレルカニジピンの塩は、異なる溶媒から結晶化することによって精製することができ、制御された条件下での乾燥、または共沸によって除去することにより、溶媒の含有量を減じることが可能である。
【0018】
本発明にかかる非晶質のレルカニジピンの塩の物性は、ベシル酸塩、ナパジシル酸塩および塩酸塩を含む結晶形態の物性とは異なる。非晶質のレルカニジピンの塩は、多様な溶媒と多様な結晶化の条件の下で結晶化を繰り返し行ってもなお、結晶物質を生じないという特徴がある。結晶物質が存在しないことは、偏光顕微鏡検査、FTラマン分光法およびDSCによって確認した。ある試料は、交差偏光器で複屈折性が見られず、広範なFTラマンスペクトル、またはガラス転移温度を有するDSC曲線を示し、特有の融解ピークが見られないことから、非晶質であると特定された。FTラマン分光法を用いた結晶物質の検出限界は、一般に、約5〜約10%(試料に対する重量比)であり、DSCを用いた検出限界は、一般に、約5〜約10%(試料に対する重量比)である。
【0019】
結晶構造のレルカニジピンの塩と比較して、非晶質のレルカニジピンの塩は、0.1モル塩酸への溶解性が大きく、明確な相転移よりは、むしろガラス転移温度(例えば広範なDSC曲線)を有する。本発明にかかる非晶質のレルカニジピンの塩は、また、本発明にかかる2種類の新しい結晶の塩とは異なる、また、塩酸レルカニジピンの結晶とも異なるFTラマンスペクトルを有する。
【0020】
本発明は、さらに、本発明にかかる新しいレルカニジピン酸塩の生成方法を、適した溶媒に溶解した酸の対イオンの溶液を、適した溶媒に溶解したレルカニジピンの遊離塩基の溶液へ加え、次に溶媒を除去することにより、提供する。レルカニジピンの結晶塩は、
(a)有機溶媒中でレルカニジピンを塩酸以外の酸の対イオンと反応させて、レルカニジピンの塩を生成し、
(b)有機溶媒を除去し、それによって生成したレルカニジピンの塩を単離し、
(c)単離したレルカニジピンの塩を、該レルカニジピンの塩の、
(i)非プロトン性溶媒、および
(ii)プロトン性溶媒、
の溶液から、順不同で、二連続工程のうちの少なくとも一方において再結晶化し、
それによって、実質的に純粋な結晶塩であるレルカニジピンを単離すること、
により生成して差し支えない。続いて行われる精製方法には、異なる温度において異なる溶媒で洗浄すること、あるいは、さらに、異なる溶媒または混合溶媒によって再結晶化を行うことを包含してもよい。
【0021】
医薬用組成物
本発明は、さらに、医薬品として容認可能な賦形剤および/または担体と混合した、本発明に従った酸付加塩を含む医薬用組成物を提供する。賦形剤としては、希釈剤、香料添加剤、甘味料、保存料、染料、結合剤、懸濁剤、分散剤、着色剤、錠剤分解物質、被膜剤、滑剤、可塑剤、食用油、または前述の物質の2種類以上の組合せを含めてよい。
【0022】
本発明に従った医薬用組成物には、さらに、塩酸レルカニジピンを含めてもよく、結晶の塩酸レルカニジピンであることが好ましい。さらに追加して、または代用として、他の活性成分またはアンギオテンシンII受容体遮断薬および/またはアンギオテンシン変換酵素阻害薬および/または利尿薬なども含んで差し支えない。
【0023】
結晶および非晶質のレルカニジピンの塩は、どちらも従来法のいずれかを利用して、微粉末化することが可能である。ある実施の形態では、微粉末化はマイクロネットM300(Nuova Guseo社(イタリア、ピアツェンツァ、ヴィッラノーヴァ・スッラルダ)から市販されている)を用いたジェット・ミル処理によって行ってよい。パラメーターを次に示す:射出圧力 約490kPa(5kg/cmq)、微粉末化圧力 約882kPa(9kg/cmq)、およびサイクロン圧力 約245kPa(2.5kg/cmq)。微粉末化の受容能力は16kg/時である。粒径は、ガライCis1レーザー機器(ガライ社(Galai)(イスラエル、ハイファ)製)を用いたレーザー光散乱法により検出する。微粉末化は、平均粒径が、90%径<15μm、好ましくは90%径<15μm、50%径が2〜8μmになるように行う。
【0024】
適切な、医薬品として容認可能な担体または希釈剤には、エタノール、水、グリセロール、プロピレングリコール、アロエ・ジェル、アラントイン、グリセリン、ビタミンA油、ビタミンE油、ミネラルオイル、プロピオン酸PPG2ミリスチル、炭酸マグネシウム、リン酸カリウム、植物油、動物油、およびソルケタールが含まれる。
【0025】
適切な結合剤には、デンプン、ゼラチン、ブドウ糖、しょ糖、乳糖などの天然糖、コーン甘味料、アカシア、トラガカント、植物ガム、アルギン酸ナトリウムなどの天然ゴムおよび合成ゴム、カルボキシメチルセルロース、ヒドロキシプロピルメチルセルロース、ポリエチレングリコール、ポビドン、ワックス類などが含まれる。好ましい結合剤は、乳糖、ヒドロキシプロピルメチルセルロースおよびポビドンである。
【0026】
適切な錠剤分解物質には、デンプン(例えば、コーンスタ‐チまたは加工デンプン)、メチルセルロース、寒天、ベントナイト、キサンタンガム、デンプングリコール酸ナトリウム、クロスポビドンなどが含まれる。好ましい結合剤は、デンプングリコール酸ナトリウムである。
【0027】
適切な滑剤としては、オレイン酸ナトリウム、ステアリン酸ナトリウム、ステアリルフマル酸ナトリウム、ステアリン酸マグネシウム、安息香酸ナトリウム、酢酸ナトリウム、塩化ナトリウムなどが含まれる。好ましい滑剤は、ステアリン酸マグネシウムである。
【0028】
好ましい懸濁剤としては、ベントナイト、エトキシ化イソステアリルアルコール、ポリオキシエチレンソルビトールエステルおよびポリオキシエチレンソルビタンエステル、微結晶性セルロース、水酸化酸化アルミニウム(aluminum metahydroxide)、寒天、トラガカント、またはこれらの物質の2種類以上の組合せ、などが含まれる。好ましい懸濁剤は、微結晶性セルロースである。
【0029】
適切な分散剤および懸濁剤には、植物ガム、トラガカント、アカシア、アルギン酸塩、デキストラン、カルボキシメチルセルロースナトリウム、メチルセルロース、ポリビニルピロリドンおよびゼラチンなどの合成および天然ゴムが含まれる。
【0030】
適切な被膜剤には、ヒドロキシプロピルメチルセルロース、エチルセルロース、ポリメタクリル酸が含まれる。
【0031】
適切な可塑剤には、異なる分子量のポリエチレングリコール(例えば200〜8000ダルトン(Da))およびプロピレングリコールが含まれる。好ましいのはポリエチレングリコール6000である。
【0032】
適切な着色料には、酸化鉄、二酸化チタン、天然および合成漆が含まれる。好ましいのは、酸化鉄および二酸化チタンである。
【0033】
好ましい食用油には、綿実油、ゴマ油、ココナツ油、ピ‐ナツ油が含まれる。
【0034】
追加する添加物の例には、ソルビトール、タルク、ステアリン酸、第二リン酸カルシウム、ポリデキストロースが含まれる。
【0035】
医薬用組成物は、錠剤、丸薬、カプセル、カプレット、ボーラス(大丸薬)、粉末、顆粒、滅菌非経口溶液、滅菌非経口懸濁液、滅菌非経口乳濁液、エリキシル剤、チンキ剤、定量制の噴霧器または液体スプレー、点滴薬、アンプル、自動注入装置および座剤などの単位投薬形態を形成しうる。単位投薬形態は、経口、非経口、経鼻、耳下または直腸投与、または吸入あるいは吹送、経皮貼付、凍結乾燥組織の態様で用いて構わない。一般に、全身に効くような、活性物質のどのような投与を利用しても差し支えない。単位投薬形態として好ましいのは、経口投薬の形態であり、固体の経口投薬形態が最も好ましい。したがって、投薬の形態は、錠剤、丸薬、カプレットおよびカプセルが好ましい。しかしながら、特に、経口投与が難しい、あるいは不可能な状況下では、非経口の製剤もまた好ましい。
【0036】
固体の単位投薬形態は、本発明にかかる活性物質を、前述のように、医薬品的に容認可能な担体および他の所望の添加物とともに混合することによって調製して差し支えない。一般的には、本発明にかかる活性物質と担体と他の所望の添加物が均一な混合物になるまで、言い換えれば、活性物質が組成物の中に均一に分散されるまで、その混合物を混ぜ合わせる。この場合、組成物は乾燥した、または湿った顆粒状になる。
【0037】
錠剤または丸薬は、表面をコーティングすることが可能であり、または、調合によって、好ましくは放出を制御する特徴を有する、単位投薬形態を形成することが可能である。例えば、錠剤または丸薬は、内側の投薬成分と外側の投薬成分とを含む構成にすることが可能であり、後者は層状または前者を包み込む形態をしている。放出を調節する層で2つの成分を隔てることにより、長時間にわたって中核となる成分から活性物質を溶解させることができる。あるいは、放出調節物質は、ゆっくりと分解する基質である。付加的な放出調節の形成に関しては、当業者にとっては明らかであろう。
【0038】
活性物質の放出を調節するための生分解性ポリマーには、ポリ乳酸、ポリイプシロンカプロラクトン、ポリヒドロキシ酪酸、ポリオルトエステル、ポリアセタール、ポリジヒドロピラン、ポリシアノアクリレート、架橋されているか、あるいは両親媒性のブロック共重合体となっているハイドロゲルが含まれるが、これらに限定されない。
【0039】
液体投薬形態として用いるため、活性物質またはそれらの生理的に容認可能な塩を、追加に、可溶化剤、乳化剤または他の助剤などの通常用いられる物質と共に、溶液、懸濁液または乳濁液にする。活性の組合せおよびこれらに対応する生理的に容認可能な塩に用いる溶媒には、水、生理的食塩水、および、エタノ‐ル、プロパンジオール、またはグリセロ‐ルといったアルコール類が含まれる。追加に、ブドウ糖溶液またはマンニトール溶液などの糖溶液も利用して差し支えない。さらには言及した様々な溶媒の混合物も本発明に利用してよい。
【0040】
経皮的な投薬形態もまた、本発明の範囲に含まれる。経皮の形態は、液タンク(fluid reservoir)または粘着製剤のマトリクスシステムのどちらかを用いた拡散促進経皮システム(経皮パッチ)であってよい。他の経皮投薬形態としては、局所用のゲル、水薬、軟膏、経粘膜的方法および用具、およびイオン化導入(iontohoretic)(電気拡散)デリバリーシステムが含まれるが、これらに限定されない。経皮的な投薬形態は、本発明にかかる活性物質の徐放および持続放出に利用してよい。
【0041】
非経口投与、特に注入による投与のための本発明にかかる医薬用組成物および単位投薬形態には、前述したように、医薬品として容認可能な担体が一般に含まれる。好ましい液体担体は植物油である。注入は、例えば、静脈注射、髄膜注射、筋肉注射、胃内注入、気管内注入および皮下注射であってよい。
【0042】
活性物質は、さらに、小さな単層ベシクル、大きな単層ベシクルおよび多層膜ベシクルなどのリポソームデリバリーシステムによる投与が可能である。リポソームは、コレステロール、ステアリルアミンまたはフォスファチジルコリンなどの様々なリン脂質から生成することが可能である。
【0043】
本発明にかかる結晶化合物は、さらに、目標設定が可能な薬物担体である水溶性ポリマーと共に用いてもよい。これらポリマーには、ポリビニルピロリドン、ピラン共重合体、ポリヒドロキシプロピルメタクリル‐アミドフェノール、ポリヒドロキシ‐エチルアスパルトアミドフェノール、およびパルミトイル残基で置換されたポリエチル‐エネオキシドポリリシンが含まれるが、これらに限定されない。
【実施例】
【0044】
本発明を以下の実施例および図面によって例証する。
【0045】
実施例1:非晶質のベシル酸レルカニジピンの生成
0.2mlのテトラヒドロフラン(THF)に181.7mgのレルカニジピン遊離塩基(レコルダッチ社(Recordati S.p.A.)(イタリア、ミラノ)製)を溶解し、ベシル酸レルカニジピン遊離塩基の原液を調製した。0.1mlのTHFに65.3mgのベンゼンスルホン酸を溶解し、酸の原液を調製した。レルカニジピン原液(0.2ml)と酸原液(0.072ml)の等モル混合物を調製した。溶媒をすべて真空除去した。溶媒を除去する際に、非晶質物質の特徴であるガラス状の膜が観察された。その非晶質物質をメタノール(MeOH)に溶かし、結晶の種となる結晶性の固体を加え、黄白色の結晶性物質を得た。
【0046】
最初の試みは、非晶質のベシル酸レルカニジピンを結晶化させるため、様々な溶媒の組合せを用いて行われた。これらの試みはすべて、結晶のベシル酸レルカニジピンの形成には功を奏さなかった(ベシル酸レルカニジピンの結晶化に成功したのはわずか2例であり、ひとつは下記に示す実施例2であり、2例目は1例目の方法を用いて生成した生成物を結晶の種として利用している)。非晶質のベシル酸レルカニジピンを結晶化させる試みのための一般的な実験スキームには、(1)35mgの非晶質のベシル酸レルカニジピン(前述の方法にて調製)を、MeOH、アセトニトリル(MeCN)、エタノール(EtOH)またはジクロロメタン(CH2Cl2)から選択する有機溶媒0.5mlに溶かし、(2)その溶媒を周囲条件下で少なくとも20日間以上かけてゆっくりと蒸発させ、(3)その試料を真空下で完全に乾燥させ、(4)約0.025mlのEtOHに試料を溶かし、(5)得られた試料を−18℃で5日間、閉所にて保管する、各工程が含まれる。
【0047】
実施例2:結晶のベシル酸レルカニジピンの生成
212.3mgのレルカニジピン遊離塩基(レコルダッチ社製)を1mlのテトラヒドロフラン(THF)に溶かし、レルカニジピン遊離塩基の原液を調製した。0.1mlのTHFに21.7mgのベンゼンスルホン酸を溶解し、酸の原液を調製した。レルカニジピンの原液(0.236ml)と酸原液(0.06ml)の混合物を調製し、25℃で24時間、閉所にて保管した。続いてその混合物を−18℃で24時間、および25℃でさらに24時間、保管した。この時点では沈殿物は観察されなかった。
【0048】
溶媒は周囲条件下で蒸発させた。ガラス状の膜を0.15mlの酢酸エチル(EtAc)に溶かした。8日後、沈殿物は観察されなかった。再度、溶媒を蒸発させ、ガラス状の膜を生じさせた。そのガラス状の膜を0.05mlのt‐ブチルメチルエーテル(TBME)に溶かし、その試料を、室温で6日間、閉所に置いた。この時点で沈殿は観察されなかった。
【0049】
さらに0.05mlのTBMEを加え、温度サイクル(20‐40‐20℃を5サイクル、立ち上がり時間2時間、立ち下がり時間2時間、20℃および40℃における等温時間10分間)に晒した。沈殿は観察されず、溶媒を穏やかな窒素流下で蒸発させた。
【0050】
溶媒を蒸発させた後、粘着性の試料が観察された。試料を真空下で24時間保管した。試料を0.05mlのメタノール(MeOH)に懸濁し、20℃の閉所にて保管した。試料から、いくつかの微粒子を含んだ粘稠液が生じた。さらに3日間、20℃で保管した後、試料は完全な固体となり、交差偏光器で複屈折性が実証され、結晶性物質の存在が確認された。
【0051】
結晶性の物質は、結晶のベシル酸レルカニジピンの収率を増大させるための第2の実験に、結晶の種として用いられた。再度、レルカニジピン遊離塩基の原液(1.10gの遊離塩基を2.2mlのMeOHに溶解)と酸(0.2844gのベンゼンスルホン酸を0.5mlのMeOHに溶解)を調製し、共に混ぜ合わせた。溶媒を穏やかな窒素流下で溶液から蒸発させた。溶液量が2mlまで減少したとき、上述のベシル酸レルカニジピンの結晶を溶液に加えた。再び、穏やかな窒素流下で溶液量が1mlに減少するまで溶媒を除去した。試料をガラスフィルターで濾過し、真空下で乾燥させた。
【0052】
最終収量1.21gの黄白色で結晶性のベシル酸レルカニジピンが得られた。元素分析により、結晶塩の組成はC42H47N2O9S(分子量755.9、溶媒和なし)であることが明らかとなり、このことは酸と遊離塩基の比が1:1(モル/モル)であることと一致する。塩は溶媒和を持たず、非吸湿性であり、TG‐FTIRによる検出では0.1%の質量放出が観察された。
【0053】
以下の通り、さらに大規模な生成を行った。96mlのメタノールに48gのレルカニジピン遊離塩基を溶かした溶液に、室温で22mlのメタノールに溶かした12.7gのベンゼンスルホン酸を加えた。得られた懸濁液を濾過し、最終的な液量が50mlになるまで、55℃で蒸発させた。その溶液に、結晶の種となるベシル酸レルカニジピンの結晶を加え、室温で24時間放置し、続いて5℃で6日間保管した。得られた結晶の集まりを吸引回収し、40mlのメタノールで2回洗浄し、P2O5の存在下で、真空下乾燥を行った。51.1gのベシル酸レルカニジピンを得た。
【0054】
実施例3: 結晶(および非晶質)のナパジシル酸レルカニジピンの生成
169.6mgのレルカニジピン遊離塩基(レコルダッチ社製)を0.82mlのメタノール(MeOH)に溶かし、レルカニジピン遊離塩基の原液を調製した。0.2mlのレルカニジピン遊離塩基の原液と0.195mlのナフタレン‐1,5‐ジスルホン酸水溶液(50mg/ml)を混合し、溶液を調製した。混合時に沈殿が観察された。その沈殿に1.0mlのMeOHを加え、溶解させた。試料を−18℃で4日間保管したが、観察のため、毎日、室温に晒された。次に、溶媒を蒸発させるため、試料を室温で7日間、開放系で保管した。溶媒の蒸発の後、結晶が確認された。その結晶を0.25mlのH2Oと0.01mlのMeOHで懸濁した。懸濁に続いて、遠心分離フィルタ(10,000RPM、0.22μmフィルタ)で結晶を回収し、真空乾燥した。
【0055】
結晶物質は、さらに多量のナパジシル酸レルカニジピンの結晶を得るため、第2の実験において、種結晶として用いられた。1.1gのレルカニジピン遊離塩基を4.4mlのMeOHに溶かし、レルカニジピン遊離塩基の溶液を調製した。その遊離塩基の溶液に5.184mlのナフタレンジスルホン酸水溶液(50mg/ml)を加え、直ちに沈殿が生成した。その沈殿に23mlのMeOHを加えて溶解させた。溶液に、上述の結晶物質を種として加え、4℃で4日間、続いてさらに3日間、−18℃で保管した。結晶を遠心分離フィルタで回収し、真空乾燥した。最終収量0.905g、黄白色のナパジシル酸レルカニジピンの結晶を得た。元素分析により、結晶塩の組成は、C82H90N6O18S2(分子量1511.76、溶媒和なし)であることが明らかになり、このことは、酸と遊離塩基の比が1:2(モル/モル)であることと一致する。その塩は、ジメトキシドの塩水和物として存在し、TG‐FTIRによる検出では4.1%(H2O放出0.4%、MeOH放出3.7%)の質量放出が観察された。
【0056】
非晶質のナパジシル酸レルカニジピンは、本実施例3で得られた溶液を真空で急速に蒸発させて生成する。
【0057】
実施例4: 非晶質塩の生成およびさらに別の結晶のレルカニジピンの塩の生成の試み
結晶のレルカニジピンの塩を生成することが可能な対イオンを判別するため、多くの対イオンとレルカニジピン遊離塩基を用いて塩のスクリーニングを行った。スクリーニング実験では、11種の対イオン、すなわち酢酸塩、ケイヒ酸塩、フマル酸塩、L‐乳酸塩、DL‐乳酸塩、L‐マレイン酸塩、DL‐マンデル酸塩、メシル酸塩、硫酸塩およびトシル酸塩の研究を行った。各対イオンについて、数回、結晶化を試みた。数回の試行後も、対イオンのうち、結晶のレルカニジピンを生成する能力のあるものは見つからなかった。11種の各対イオンのスクリーニングに用いられた一般的な実験スキームの詳細を下記に記す。結晶のレルカニジピンを得ることが難しいことが周知であることから、結晶化技術には十分な保管時間とゆっくりとした工程が選択された。
【0058】
塩スクリーニング実験のそれぞれから得た最終的なレルカニジピンの塩を完全に乾燥させ、化学分析および物理分析を行った。各塩の化学組成を元素分析、赤外分光法と連結した熱重量分析、および含水量分析によって決定した。さらに、FT‐ラマン分析法を用いた物理試験および溶解度試験も行った。
【0059】
実施例4a: テトラヒドロフランに溶解した対イオン
下記に示す通り、テトラヒドロフラン(THF)に溶かしたレルカニジピン遊離塩基(レコルダッチ社製)とTHFに溶かしたそれぞれの酸を溶解させ、レルカニジピン遊離塩基と酸それぞれの原液を調製した。
【0060】
酸と塩基のモル比と同時に、酸の立体的配置に配慮して、レルカニジピン遊離塩基と酸の原液を等量ずつ混合した。原液の混合物には沈殿は見られなかった。試料を25℃で24時間閉所に保管し、続けてさらに4℃で24時間、および−18℃で24時間保管した。保管後、固体は観察されなかった。
【0061】
試料から溶媒を周囲条件下で蒸発させて除去した。4日後、ガラス状の膜が見られた。そのガラス状の膜をアセトンに溶かし、室温で2日間放置した。沈殿は観察されなかった。試料をさらに6日間放置したが、沈殿は見られなかった。
【0062】
溶媒を穏やかな窒素流下で蒸発させ、ガラス状の膜を形成した。ガラス状の膜をt‐ブチルメチルエーテル(TBME)に溶かし、その試料を室温で6日間放置した。さらにTBMEを加え、その試料を
温度サイクル(20‐40‐20℃、5サイクル、立ち上がり時間2時間、立ち下がり時間2時間、20℃および40℃における等温時間10分間)に晒した。沈殿は観察されず、再度、溶媒を穏やかな窒素流下で蒸発させた。
【0063】
溶媒を蒸発させた後、粘着性の試料が観察された。試料を真空下で24時間保管した。その試料をメタノール(MeOH)に懸濁し、20℃の閉所に保管した。試料は粘着性の塊が生じたが、結晶の微粒子は見られなかった。試料をさらに24時間、真空下で保管し、溶媒を除去した。溶媒除去後も結晶物質は観察されなかった。
【0064】
本試料から、2種類の非晶質のレルカニジピンの塩、すなわち、ケイヒ酸レルカニジピンおよびマレイン酸レルカニジピンを生じた。本試料の各塩の化学組成を元素分析、赤外分光法と連結した熱重量分析、および含水量分析によって決定した。各非晶質について、さらに、FT‐ラマン分析法を用いた物理試験および溶解度試験も行った。結果を下記に示す。
【0065】
実施例4b: メタノールに溶解させた対イオン
レルカニジピン遊離塩基および酸のそれぞれの原液を混合して、さらに塩スクリーニング実験を行った。原液は、下記の通り、TBMEに溶かしたレルカニジピン遊離塩基(レコルダッチ社製)およびMeOHに酸をそれぞれ溶解して調製した。
【0066】
酸と塩基のモル比と同時に、酸の立体化学を説明するため、レルカニジピン遊離塩基と酸の原液を等量ずつ混合した。原液の混合物には沈殿は見られなかった。試料を25℃で24時間、閉所に保管し、続けてさらに60℃で8時間、および4℃で6日間保管した。保管後も固体は観察されなかった。
【0067】
試料へ水を加え、続いて周囲条件下で溶媒を蒸発させた。4日後、ガラス状の膜が観察された。試料を真空下で24時間保管した。ガラス状の膜をMeOHに溶かし、20℃で24時間、閉所に保管した。24時間後、粘着性の塊が観察されたが、固体の粒子は見られなかった。試料をさらに2日間保管したが、それでも固体の粒子は見られなかった。真空下で24時間保管し、サンプルから溶媒を除去した。溶媒を除去した後も、結晶物質は見られなかった。
【0068】
本実施例によって、L‐乳酸レルカニジピンが生成した。本実施例にかかる塩の化学組成を元素分析、赤外分光法と連結した熱重量分析、および含水量分析によって決定した。本非晶質塩について、さらに、FT‐ラマン分析法を用いた物理試験および溶解度試験も行った。物性試験の結果を下記に簡潔にまとめる。
【0069】
実施例5: 非晶質の塩の調製および結晶のレルカニジピンの塩の生成の試み
実施例4で述べた塩スクリーニング実験に加えて、結晶のレルカニジピンの塩を生成するための試みをさらに12種類の酸、すなわちクエン酸塩、粘液酸塩、ゲンチシン酸塩、グルコン酸塩、2‐オキソグルタル酸塩、リン酸塩、サッカリン酸塩、サリチル酸塩、L‐酒石酸塩、テレフタル酸塩、マロン酸塩、シュウ酸塩について行った。12種類の対イオンのそれぞれについて、結晶のレルカニジピンの塩を生成する試みを1回ずつ行った。12種類の対イオンのいずれも、結晶のレルカニジピンの塩を生成させることができず、特徴的な非晶質塩を生成したのもほんのわずかであった。対イオンのスクリーニングに用いられた一般的な実験スキームの詳細を下記に記す。結晶のレルカニジピンを得ることが難しいことが周知であることから、結晶化技術には十分な保管時間とゆっくりとした工程が選択された。
【0070】
塩スクリーニング実験のそれぞれから得た最終的なレルカニジピンの塩を完全に乾燥させ、化学分析および物理分析を行った。各塩の化学組成を元素分析、赤外分光法と連結した熱重量分析、および含水量分析によって決定した。さらに、FT‐ラマン分析法を用いた物理試験および溶解度試験も行った。
【0071】
レルカニジピンの原液は、2.67mlのメタノール(MeOH)に530mgのレルカニジピン遊離塩基(レコルダッチ社製)溶解して調製した。結晶をふるいにかけるため、レルカニジピン原液の等量(0.1ml)を下記のように酸と混合した。
【0072】
本実施例の各試料は、完全に同じように取り扱われた。レルカニジピン遊離塩基と対応する対イオンを混ぜ合わせた後、試料を−18℃で2日間、閉所に置いた。2日後、試料のいずれにも沈殿は見られなかった。次に、試料を周囲条件下で8時間、開放系に置き、続いて−18℃でさらに5日間、保管した。保管期間の終了後も、また、沈殿は見られなかった。次に、試料を周囲条件下で15時間、開放系で保管し、−18℃でさらに2日間、置いた。保管後、どの試料にも沈殿は見られなかった。溶媒を真空下でそれぞれの試料から除去し、残った固体を閉所に保管した。
【0073】
レルカニジピン遊離塩基と本実施例にかかる対イオンから結晶のレルカニジピンの塩を生成するという試みはすべて失敗し、結晶物質は生成しなかった。本実施例は、実施例4で述べたと同様、レルカニジピンの結晶塩の生成の困難さ、および予測し難さを示すものである。
【0074】
本実施例では、2種類のレルカニジピンの非晶質塩、すなわちサッカリン酸塩およびサリチル酸塩を生成した。本実施例にかかる各塩の化学組成を元素分析、赤外分光法と連結した熱重量分析、および含水量分析によって決定した。各非晶質塩のFT‐ラマン分析法を用いた物理試験および溶解度試験も行った。物性試験の結果を下記に簡潔にまとめる。
【0075】
実施例6:非晶質および結晶のレルカニジピンの塩の化学組成
乾式燃焼/熱伝導率および非分散型IR検出を用いて、非晶質および結晶のレルカニジピンの塩の双方の元素組成を決定した。元素分析の結果を表1に要約する。
【0076】
非晶質および結晶のレルカニジピンの塩の双方の残留溶媒の内容物を、赤外分光計と一体となった重量分析を用いて決定した。分析には、ブルカー社(Bruker)(スイス、フェランデン)製のFTIR分光計Vector 22と共に、ネッチ社(Netzsch)(ドイツ、ゼルプ)製のThermobalance TG209を用いた。分析は、以下の条件にしたがって行われた。すなわち、2〜5mgの試料を窒素環境下でアルミニウムのるつぼ内で、加熱速度10℃/分で25℃から250℃まで熱した。重量分析の結果を表1に示す。
【0077】
非晶質および結晶のレルカニジピンの塩の双方の吸湿性を、水分吸着測定装置(Surface Measurement System Ltd社(イギリス、バッキンガムシャー、マリオン)製)を用いた動的な蒸気吸着測定(DVS analysis)によって決定した。測定は以下の方法に従って行った。すなわち、10〜15mgの試料を石英またはプラチナの容器に入れ、その容器をマイクロバランスの上に内転させて置き、試料を25℃で湿度サイクルが相対湿度(RH)で0〜95%(50‐95‐0‐50%、速度5%RH/hr)に晒した。吸湿性の測定結果を下記の表1に要約する。
【表1】
【0078】
実施例7:非晶質および結晶のレルカニジピンの塩の溶解度
結晶のベシル酸レルカニジピン、ナパジシル酸レルカニジピンおよび塩酸レルカニジピン、ならびに非晶質のベシル酸レルカニジピンの溶解度は、22℃、0.1モルHCl水溶液(pH1)中でUV‐可視分光法によって測定した。0.1モル塩酸水溶液を用いて、それぞれの化合物のおよそ0.3mg/mlの懸濁液を調製し、24時間振動させて液を平衡にした。平衡にした後、試料を濾過し(0.1μmフィルタ‐)、濃縮物をパーキンエルマー社(Perkin Elmer)(ドイツ、ユーベリンゲン)製のLambda16を用いて、光度測定を行った。基準測定は、共溶媒として20%アセトニトリルを用いて行った。
【表2】
【0079】
結晶のベシル酸塩および結晶のナパシジル酸塩の両方とも、非晶質の塩に比べて溶解度が低いことが表2から見て取れる。結晶塩の溶解度は変化が大きく、結晶のベシル酸塩は、実質、結晶の塩酸塩およびナパジシル酸塩よりも溶解度が大きいことも、表2から読み取れる。
【0080】
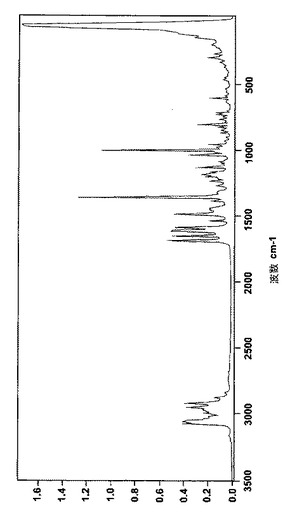
実施例8: 新しいレルカニジピンの塩のラマンスペクトル
FTラマン分光法を用いて、新しいレルカニジピンの塩を分析した。ブルカー社製のFTラマンRFS100分光光度計を次の一般的な方法で利用した:試料(前処理なし)約10mg、走査64、分解能2cm‐1、レーザー出力100mW、Ge検出。
【0081】
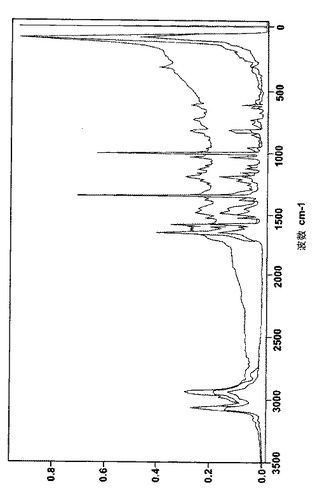
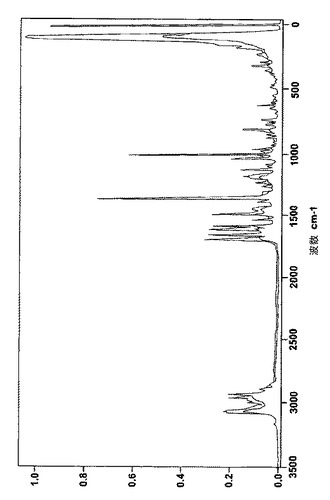
次の表3、表4、表5は、非晶質のベシル酸レルカニジピンと共に、結晶のベシル酸レルカニジピンおよびナパジシル酸レルカニジピンそれぞれのラマンスペクトルの最も特徴的なピークを示している。
【表3】
【表4】
【表5】
【0082】
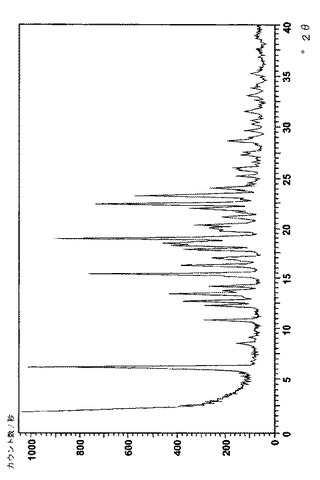
実施例9: 新しい結晶のレルカニジピンの塩のX線回折パタ‐ン
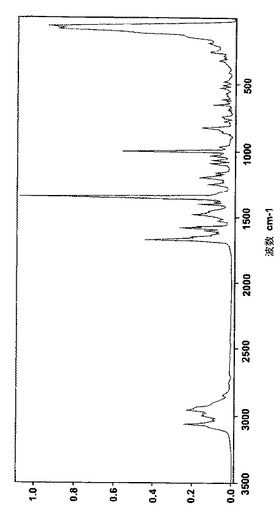
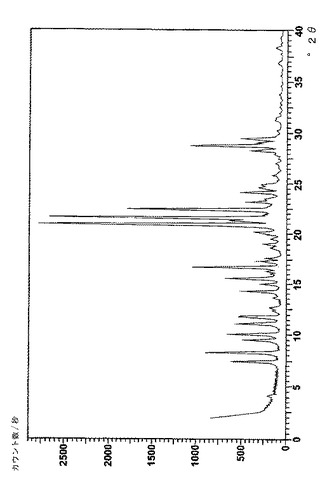
フィリップス社(オランダ、アイントホーフェン)製のX‐pert PW3040またはPW1710粉末回折計を用いて、次の一般的な条件下で、結晶のベシル酸レルカニジピンおよびナパジシル酸レルカニジピンのX線回折パターンを得た:試料5〜70mg(前処理なし)をわずかな圧力を用いて平らな試料とし、周囲空気雰囲気、Cu Kα線、2θ 0.02°、ステップサイズ 2s/step、測定範囲2θ 2〜50°。得られたスペクトルを図4(ベシル酸塩)および図6(ナパジシル酸塩)に、それに対応する主なピークを表6(ナパジシル酸塩)および表7(ベシル酸塩)に示す。2θの値は約±0.10から約±0.20°の範囲で再現可能であり、一方で個々のピークの相対強度は試料同士の比によって変化しうるということは、当業者にとって理解されよう。United States Pharmacopiea XXV (2002), p. 2088‐2089を参照のこと。
【表6】
【表7】
【0083】
実施例10: 結晶のベシル酸レルカニジピンおよびナパジシル酸レルカニジピンのDSC分析
示差走査熱量測定法(DSC)を用いて、本発明にかかる結晶のレルカニジピンの塩および結晶の塩酸レルカニジピンの融点を測定した。DSC分析は、加熱によって供与試料に起こる変化を測定するものであり、その変化によって、転移する相を特定する。相転移のときに起こるエンタルピーの変化を、曲線の下の領域に基づいて計算する。最も一般的な転移の相としては、融解および昇華が挙げられる。転移が始まる温度、すなわち開始温度は、曲線からベースラインが逸れ始めた点(変曲点)で表される。
【0084】
結晶のベシル酸レルカニジピンのDSC:4.040mgの結晶のベシル酸レルカニジピンをPerkin Elmer社製の装置DSC7の金製の皿(pan)に入れた。試験中の加熱速度は10℃/分であった。
【0085】
結晶のナパジシル酸レルカニジピンのDSC:3.697mgの結晶のナパジシル酸レルカニジピンをパーキンエルマー社製装置DSC7の金製の皿(pan)に入れた。試験中の加熱速度は10℃/分であった。
【0086】
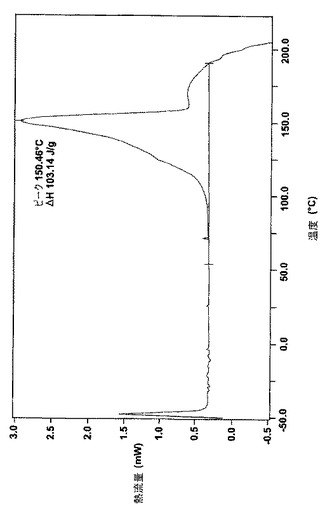
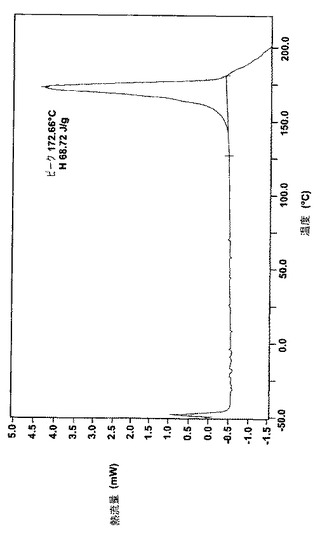
データを図1(ナパジシル酸塩)および図2(ベシル酸塩)に示し、図の特性点を表8に簡潔にまとめる。
【表8】
【図面の簡単な説明】
【0087】
【図1】図1は結晶のナパジシル酸レルカニジピンの走査熱量の特徴の差異を示す。
【図2】図2は結晶のベシル酸レルカニジピンの走査熱量の特徴の差異を示す。
【図3】図3は結晶のベシル酸レルカニジピンのFTラマンパターンを表す。
【図4】図2は結晶のベシル酸レルカニジピンのX線回折像を表す。
【図5】図5は結晶のナパジシル酸レルカニジピンのFTラマンパターンを表す。
【図6】図6は結晶のナパジシル酸レルカニジピンのX線回折像を表す。
【図7】図7は非晶質のベシル酸レルカニジピンを、等モルのレルカニジピン遊離塩基とベンゼンスルホン酸の固体の状態の混合物とを比較したFTラマンパターンを表す。
【図8】図8は非晶質と結晶のベシル酸レルカニジピンを比較したFTラマンパターンを表す。
【技術分野】
【0001】
本発明はレルカニジピンの新しい酸塩に関するものであり、特に、非晶質および結晶のレルカニジピンの塩とその製造方法に関する。本発明は、また、水和および溶媒和した形での新しい非晶質および結晶のレルカニジピンの塩に関する。さらに、本発明は、ここに開示する新しいレルカニジピンの塩を含む医薬品組成物に関する。
【背景技術】
【0002】
下記に示す、メチル1,1,N‐トリメチル‐N‐(3,3‐ジフェニルプロピル)‐2‐アミノエチル1,4‐ジヒドロ‐2,6‐ジメチル‐4‐(3‐ニトロフェニル)‐ピリジン‐3,5‐ジカルボキシレート(INN(医薬品の国際一般名):レルカニジピン)は、長時間作用性で血管選択性の高い、高脂肪親和性のジヒドロピリジンカルシウム拮抗薬である。
【化1】
【0003】
レルカニジピンの塩酸塩(塩酸レルカニジピン)は、レコルダッチ社(Recordati S.p.A.)(イタリア、ミラノ)から市販されている。レルカニジピンの遊離塩基およびその塩酸塩の製法は、特許文献1〜5において、レルカニジピンを個々の光学異性体に分割する方法とともに、以前より述べられている。
【特許文献1】米国特許第4,705,797号明細書
【特許文献2】米国特許第5,767,136号明細書
【特許文献3】米国特許第4,968,832号明細書
【特許文献4】米国特許第5,912,351号明細書
【特許文献5】米国特許第5,696,139号明細書
【特許文献6】米国特許出願公開第2003/0069285号明細書
【特許文献7】米国特許出願公開第2003/0083355号明細書
【発明の開示】
【発明が解決しようとする課題】
【0004】
レルカニジピンを調製する工程での主に不便な点は、特許文献1で述べられているように、開示されている環化反応では様々な副生成物を生じ、目的とする生成物の収率が低下してしまうことである。特許文献4は塩酸レルカニジピンの調製のための、より簡素化された工程について述べている。その工程では非水和で非吸湿性の結晶構造を有する塩酸レルカニジピンを生じ、不要な副生成物の生成を防ぐと同時に、それに続いて行うクロマトグラフィーカラムによる精製工程を省いている。
【0005】
しかしながら、結晶構造を持つ塩酸レルカニジピンを単離することもまた、非常に手間がかかる。さらに、塩酸レルカニジピンは、それぞれが相異なる物理的性質を持つ少なくとも4種類の多形体のいずれかの形で存在している可能性がある(特許文献7および特許文献8参照)。したがって、当業者にとって、レルカニジピンの塩、特に結晶のレルカニジピンの塩を生成する、より簡素な製造工程が必要とされる。限定はしないが、患者によるばらつきが少ない、食物の影響が少ない、および多形が少ないか存在しない、塩酸レルカニジピンの以前に単離された構造とは異なる、好ましくは、より望ましい溶解性および/または他の物理的性質を持つ、レルカニジピンの塩もまた、必要とされている。
【課題を解決するための手段】
【0006】
定義および本明細書中で用いられる略語
「DSC」:示差走査熱量測定法
「非晶質」は、固体の状態の特性である、実質的な結晶格子構造を持たない化合物を表現するのに用いられる。これらの化合物では、結晶質の化合物の特徴である鋭い発熱ピ‐クの代わりに、ガラス転移と定義される、独特な幅の広い発熱を伴う変化を示すDSCのプロットが見られる。
【0007】
「結晶」は、融点ならびに結晶構造のX線スペクトル特性を持つ化合物を表現するのに用いられる。これらの化合物では、独特の鋭い発熱ピークを有するDSCのプロットが見られる。
【0008】
「ベシル酸レルカニジピン(lercanidipine besylate)」:レルカニジピンの酸塩の一種であり、ベンゼンスルホン酸をモル比1:1の対イオンとしている。
【0009】
「ナパジシル酸レルカニジピン(lercanidipine napadisylate)」:レルカニジピンの酸塩の一種であり、ナフタレン‐1,5‐ジスルホン酸をモル比2:1の対イオンとしている。
【0010】
「多形の」または「多形」:化合物中に少なくとも2種以上の異なる構造が存在する特性。異なる結晶構造は結晶学的技術によって直接的に、あるいは、それぞれ特有の多形に関連する物理的および/または化学的性質の差異の評価によって非直接的に検出される。
【0011】
ある態様において、本発明は、酸の対イオンが
(i)塩酸以外の、臭化水素酸、リン酸、硫酸などの無機酸、
(ii)メタンスルホン酸、ベンゼンスルホン酸、トルエンスルホン酸、およびナフタレン‐1,5‐ジスルホン酸などのスルホン酸、
(iii)酢酸、(+)‐L‐乳酸、DL‐乳酸、DL‐マンデル酸、グルコン酸、ケイヒ酸、サリチル酸、およびゲンチシン酸などのモノカルボン酸、
(iv)シュウ酸、2‐オキソ‐グルタル酸、マロン酸、(‐)‐L‐リンゴ酸、粘液酸、(+)‐L‐酒石酸、フマル酸、マレイン酸、およびテレフタル酸などのジカルボン酸、
(v)クエン酸などのトリカルボン酸、
(vi)サッカリンなどの芳香族スルホンイミド、
からなる群より選択されることを特徴とする、レルカニジピンの酸付加塩を提供する。
【0012】
本発明に従ったレルカニジピンの塩としては、L‐乳酸塩、ケイヒ酸塩、サリチル酸塩、マレイン酸塩、およびサッカリン酸塩が好ましい。本発明に従ったレルカニジピンの塩として最も好ましいのは、ベシル酸レルカニジピンおよびナパジシル酸レルカニジピンである。これらの好ましい、および最も好ましい塩は、すべて非晶質の形態で生成することができ、ベシル酸レルカニジピンおよびナパジシル酸レルカニジピンは、結晶の形態で生成することも可能である。
【0013】
本発明にかかる結晶のレルカニジピンの塩は、多形であって差し支えない。
【0014】
本発明にかかるレルカニジピンの塩は、溶媒和および水和した形態で存在して差し支えない。これらの溶媒和または水和した形態は、配位数が1または2の溶媒和物または水和物であって構わない。レルカニジピンの塩を生成するときに溶媒を用いた結果として、固体の格子構造中に溶媒和物および水和物を形成してもよい。レルカニジピンの塩が生成する間に、溶媒和物および水和物が形成されることから、特定の溶媒和または水和の構造の形成は、塩の生成に用いる条件および方法によって大きく左右される。溶媒は、医薬品として容認可能なものであるべきである。
【0015】
結晶のベシル酸レルカニジピンは黄白色で、良い安定性を示す。22℃での0.1モル塩酸への溶解度は約25〜35mg/lであり、さらに具体的には約30mg/lである。これと比較して、同じ溶媒への塩酸レルカニジピンの結晶の溶解度は約10mg/lである。ベシル酸レルカニジピンの結晶の融点(DSCピーク)は、約170℃〜約175℃の範囲であり、さらに具体的には、約172℃である。下記に示す実施例では、ベシル酸レルカニジピンの結晶は水和または溶媒和のどちらの構造も得られていないが、これらの構造は本発明に包含され、不定量の水を含む極性溶媒から再結晶化することによって得ることが可能である。下記の実施例での生成のように、ベシル酸レルカニジピンの結晶はゆっくりと生じ、結晶の種を加えて初めて高収率の結晶が生成され、多形は見られない。
【0016】
結晶のナパジシル酸レルカニジピンの結晶もまた黄白色で、安定性が良い。結晶の大きさは、ベシル酸レルカニジピンの結晶よりも大きい。ナパジシル酸レルカニジピンの結晶の0.1モル塩酸への溶解度は約3mg/l〜4mg/lであり、さらに具体的には約3.5mg/lである。これは、結晶の塩酸レルカニジピンや結晶のベシル酸レルカニジピンよりも低い値である。結晶のナパジシル酸レルカニジピンの融点(DSCピーク)は、約145℃〜約155℃の範囲であり、さらに具体的には約150℃である。ナパジシル酸レルカニジピンの結晶は、溶媒和した水和物、特にジメタノラート水和物(dimethanolate hydrate)として、または非水和の形で生成してもよい。下記の実施例に示すとおり、結晶のナパジシル酸レルカニジピンは、自然発生的に生成する。
【0017】
本発明にかかる結晶のレルカニジピンの塩は、実質的に純粋な形態で生成し、溶媒はほとんど残らない。特に、結晶のベシル酸レルカニジピンは、残留溶媒の含量が約0.1〜最大約0.5重量%、さらに詳しく言えば最大約0.2重量%以下で生成する可能性がある。結晶のナパジシル酸レルカニジピンは、残留溶媒の含量が約2.5〜最大約5重量%、さらに詳しく言えば最大約4重量%以下で生成する可能性がある。これらの結晶のレルカニジピンの塩の生成物は、純度99.5%、残留溶媒含量0.3重量%(3000ppm)以下で単離することが可能であるが、より純度の低い(および/または溶媒または水分含量がもっと高い)生成物であれば、従来法にて生成することも可能である。医薬品として容認可能な各不純物のレベルは、一般に0.1%以下であり、有機溶媒では、各溶媒の毒性に応じて、0.5%(5000ppm)〜0.0002%(2ppm)の範囲である。本発明にかかるレルカニジピンの塩は、異なる溶媒から結晶化することによって精製することができ、制御された条件下での乾燥、または共沸によって除去することにより、溶媒の含有量を減じることが可能である。
【0018】
本発明にかかる非晶質のレルカニジピンの塩の物性は、ベシル酸塩、ナパジシル酸塩および塩酸塩を含む結晶形態の物性とは異なる。非晶質のレルカニジピンの塩は、多様な溶媒と多様な結晶化の条件の下で結晶化を繰り返し行ってもなお、結晶物質を生じないという特徴がある。結晶物質が存在しないことは、偏光顕微鏡検査、FTラマン分光法およびDSCによって確認した。ある試料は、交差偏光器で複屈折性が見られず、広範なFTラマンスペクトル、またはガラス転移温度を有するDSC曲線を示し、特有の融解ピークが見られないことから、非晶質であると特定された。FTラマン分光法を用いた結晶物質の検出限界は、一般に、約5〜約10%(試料に対する重量比)であり、DSCを用いた検出限界は、一般に、約5〜約10%(試料に対する重量比)である。
【0019】
結晶構造のレルカニジピンの塩と比較して、非晶質のレルカニジピンの塩は、0.1モル塩酸への溶解性が大きく、明確な相転移よりは、むしろガラス転移温度(例えば広範なDSC曲線)を有する。本発明にかかる非晶質のレルカニジピンの塩は、また、本発明にかかる2種類の新しい結晶の塩とは異なる、また、塩酸レルカニジピンの結晶とも異なるFTラマンスペクトルを有する。
【0020】
本発明は、さらに、本発明にかかる新しいレルカニジピン酸塩の生成方法を、適した溶媒に溶解した酸の対イオンの溶液を、適した溶媒に溶解したレルカニジピンの遊離塩基の溶液へ加え、次に溶媒を除去することにより、提供する。レルカニジピンの結晶塩は、
(a)有機溶媒中でレルカニジピンを塩酸以外の酸の対イオンと反応させて、レルカニジピンの塩を生成し、
(b)有機溶媒を除去し、それによって生成したレルカニジピンの塩を単離し、
(c)単離したレルカニジピンの塩を、該レルカニジピンの塩の、
(i)非プロトン性溶媒、および
(ii)プロトン性溶媒、
の溶液から、順不同で、二連続工程のうちの少なくとも一方において再結晶化し、
それによって、実質的に純粋な結晶塩であるレルカニジピンを単離すること、
により生成して差し支えない。続いて行われる精製方法には、異なる温度において異なる溶媒で洗浄すること、あるいは、さらに、異なる溶媒または混合溶媒によって再結晶化を行うことを包含してもよい。
【0021】
医薬用組成物
本発明は、さらに、医薬品として容認可能な賦形剤および/または担体と混合した、本発明に従った酸付加塩を含む医薬用組成物を提供する。賦形剤としては、希釈剤、香料添加剤、甘味料、保存料、染料、結合剤、懸濁剤、分散剤、着色剤、錠剤分解物質、被膜剤、滑剤、可塑剤、食用油、または前述の物質の2種類以上の組合せを含めてよい。
【0022】
本発明に従った医薬用組成物には、さらに、塩酸レルカニジピンを含めてもよく、結晶の塩酸レルカニジピンであることが好ましい。さらに追加して、または代用として、他の活性成分またはアンギオテンシンII受容体遮断薬および/またはアンギオテンシン変換酵素阻害薬および/または利尿薬なども含んで差し支えない。
【0023】
結晶および非晶質のレルカニジピンの塩は、どちらも従来法のいずれかを利用して、微粉末化することが可能である。ある実施の形態では、微粉末化はマイクロネットM300(Nuova Guseo社(イタリア、ピアツェンツァ、ヴィッラノーヴァ・スッラルダ)から市販されている)を用いたジェット・ミル処理によって行ってよい。パラメーターを次に示す:射出圧力 約490kPa(5kg/cmq)、微粉末化圧力 約882kPa(9kg/cmq)、およびサイクロン圧力 約245kPa(2.5kg/cmq)。微粉末化の受容能力は16kg/時である。粒径は、ガライCis1レーザー機器(ガライ社(Galai)(イスラエル、ハイファ)製)を用いたレーザー光散乱法により検出する。微粉末化は、平均粒径が、90%径<15μm、好ましくは90%径<15μm、50%径が2〜8μmになるように行う。
【0024】
適切な、医薬品として容認可能な担体または希釈剤には、エタノール、水、グリセロール、プロピレングリコール、アロエ・ジェル、アラントイン、グリセリン、ビタミンA油、ビタミンE油、ミネラルオイル、プロピオン酸PPG2ミリスチル、炭酸マグネシウム、リン酸カリウム、植物油、動物油、およびソルケタールが含まれる。
【0025】
適切な結合剤には、デンプン、ゼラチン、ブドウ糖、しょ糖、乳糖などの天然糖、コーン甘味料、アカシア、トラガカント、植物ガム、アルギン酸ナトリウムなどの天然ゴムおよび合成ゴム、カルボキシメチルセルロース、ヒドロキシプロピルメチルセルロース、ポリエチレングリコール、ポビドン、ワックス類などが含まれる。好ましい結合剤は、乳糖、ヒドロキシプロピルメチルセルロースおよびポビドンである。
【0026】
適切な錠剤分解物質には、デンプン(例えば、コーンスタ‐チまたは加工デンプン)、メチルセルロース、寒天、ベントナイト、キサンタンガム、デンプングリコール酸ナトリウム、クロスポビドンなどが含まれる。好ましい結合剤は、デンプングリコール酸ナトリウムである。
【0027】
適切な滑剤としては、オレイン酸ナトリウム、ステアリン酸ナトリウム、ステアリルフマル酸ナトリウム、ステアリン酸マグネシウム、安息香酸ナトリウム、酢酸ナトリウム、塩化ナトリウムなどが含まれる。好ましい滑剤は、ステアリン酸マグネシウムである。
【0028】
好ましい懸濁剤としては、ベントナイト、エトキシ化イソステアリルアルコール、ポリオキシエチレンソルビトールエステルおよびポリオキシエチレンソルビタンエステル、微結晶性セルロース、水酸化酸化アルミニウム(aluminum metahydroxide)、寒天、トラガカント、またはこれらの物質の2種類以上の組合せ、などが含まれる。好ましい懸濁剤は、微結晶性セルロースである。
【0029】
適切な分散剤および懸濁剤には、植物ガム、トラガカント、アカシア、アルギン酸塩、デキストラン、カルボキシメチルセルロースナトリウム、メチルセルロース、ポリビニルピロリドンおよびゼラチンなどの合成および天然ゴムが含まれる。
【0030】
適切な被膜剤には、ヒドロキシプロピルメチルセルロース、エチルセルロース、ポリメタクリル酸が含まれる。
【0031】
適切な可塑剤には、異なる分子量のポリエチレングリコール(例えば200〜8000ダルトン(Da))およびプロピレングリコールが含まれる。好ましいのはポリエチレングリコール6000である。
【0032】
適切な着色料には、酸化鉄、二酸化チタン、天然および合成漆が含まれる。好ましいのは、酸化鉄および二酸化チタンである。
【0033】
好ましい食用油には、綿実油、ゴマ油、ココナツ油、ピ‐ナツ油が含まれる。
【0034】
追加する添加物の例には、ソルビトール、タルク、ステアリン酸、第二リン酸カルシウム、ポリデキストロースが含まれる。
【0035】
医薬用組成物は、錠剤、丸薬、カプセル、カプレット、ボーラス(大丸薬)、粉末、顆粒、滅菌非経口溶液、滅菌非経口懸濁液、滅菌非経口乳濁液、エリキシル剤、チンキ剤、定量制の噴霧器または液体スプレー、点滴薬、アンプル、自動注入装置および座剤などの単位投薬形態を形成しうる。単位投薬形態は、経口、非経口、経鼻、耳下または直腸投与、または吸入あるいは吹送、経皮貼付、凍結乾燥組織の態様で用いて構わない。一般に、全身に効くような、活性物質のどのような投与を利用しても差し支えない。単位投薬形態として好ましいのは、経口投薬の形態であり、固体の経口投薬形態が最も好ましい。したがって、投薬の形態は、錠剤、丸薬、カプレットおよびカプセルが好ましい。しかしながら、特に、経口投与が難しい、あるいは不可能な状況下では、非経口の製剤もまた好ましい。
【0036】
固体の単位投薬形態は、本発明にかかる活性物質を、前述のように、医薬品的に容認可能な担体および他の所望の添加物とともに混合することによって調製して差し支えない。一般的には、本発明にかかる活性物質と担体と他の所望の添加物が均一な混合物になるまで、言い換えれば、活性物質が組成物の中に均一に分散されるまで、その混合物を混ぜ合わせる。この場合、組成物は乾燥した、または湿った顆粒状になる。
【0037】
錠剤または丸薬は、表面をコーティングすることが可能であり、または、調合によって、好ましくは放出を制御する特徴を有する、単位投薬形態を形成することが可能である。例えば、錠剤または丸薬は、内側の投薬成分と外側の投薬成分とを含む構成にすることが可能であり、後者は層状または前者を包み込む形態をしている。放出を調節する層で2つの成分を隔てることにより、長時間にわたって中核となる成分から活性物質を溶解させることができる。あるいは、放出調節物質は、ゆっくりと分解する基質である。付加的な放出調節の形成に関しては、当業者にとっては明らかであろう。
【0038】
活性物質の放出を調節するための生分解性ポリマーには、ポリ乳酸、ポリイプシロンカプロラクトン、ポリヒドロキシ酪酸、ポリオルトエステル、ポリアセタール、ポリジヒドロピラン、ポリシアノアクリレート、架橋されているか、あるいは両親媒性のブロック共重合体となっているハイドロゲルが含まれるが、これらに限定されない。
【0039】
液体投薬形態として用いるため、活性物質またはそれらの生理的に容認可能な塩を、追加に、可溶化剤、乳化剤または他の助剤などの通常用いられる物質と共に、溶液、懸濁液または乳濁液にする。活性の組合せおよびこれらに対応する生理的に容認可能な塩に用いる溶媒には、水、生理的食塩水、および、エタノ‐ル、プロパンジオール、またはグリセロ‐ルといったアルコール類が含まれる。追加に、ブドウ糖溶液またはマンニトール溶液などの糖溶液も利用して差し支えない。さらには言及した様々な溶媒の混合物も本発明に利用してよい。
【0040】
経皮的な投薬形態もまた、本発明の範囲に含まれる。経皮の形態は、液タンク(fluid reservoir)または粘着製剤のマトリクスシステムのどちらかを用いた拡散促進経皮システム(経皮パッチ)であってよい。他の経皮投薬形態としては、局所用のゲル、水薬、軟膏、経粘膜的方法および用具、およびイオン化導入(iontohoretic)(電気拡散)デリバリーシステムが含まれるが、これらに限定されない。経皮的な投薬形態は、本発明にかかる活性物質の徐放および持続放出に利用してよい。
【0041】
非経口投与、特に注入による投与のための本発明にかかる医薬用組成物および単位投薬形態には、前述したように、医薬品として容認可能な担体が一般に含まれる。好ましい液体担体は植物油である。注入は、例えば、静脈注射、髄膜注射、筋肉注射、胃内注入、気管内注入および皮下注射であってよい。
【0042】
活性物質は、さらに、小さな単層ベシクル、大きな単層ベシクルおよび多層膜ベシクルなどのリポソームデリバリーシステムによる投与が可能である。リポソームは、コレステロール、ステアリルアミンまたはフォスファチジルコリンなどの様々なリン脂質から生成することが可能である。
【0043】
本発明にかかる結晶化合物は、さらに、目標設定が可能な薬物担体である水溶性ポリマーと共に用いてもよい。これらポリマーには、ポリビニルピロリドン、ピラン共重合体、ポリヒドロキシプロピルメタクリル‐アミドフェノール、ポリヒドロキシ‐エチルアスパルトアミドフェノール、およびパルミトイル残基で置換されたポリエチル‐エネオキシドポリリシンが含まれるが、これらに限定されない。
【実施例】
【0044】
本発明を以下の実施例および図面によって例証する。
【0045】
実施例1:非晶質のベシル酸レルカニジピンの生成
0.2mlのテトラヒドロフラン(THF)に181.7mgのレルカニジピン遊離塩基(レコルダッチ社(Recordati S.p.A.)(イタリア、ミラノ)製)を溶解し、ベシル酸レルカニジピン遊離塩基の原液を調製した。0.1mlのTHFに65.3mgのベンゼンスルホン酸を溶解し、酸の原液を調製した。レルカニジピン原液(0.2ml)と酸原液(0.072ml)の等モル混合物を調製した。溶媒をすべて真空除去した。溶媒を除去する際に、非晶質物質の特徴であるガラス状の膜が観察された。その非晶質物質をメタノール(MeOH)に溶かし、結晶の種となる結晶性の固体を加え、黄白色の結晶性物質を得た。
【0046】
最初の試みは、非晶質のベシル酸レルカニジピンを結晶化させるため、様々な溶媒の組合せを用いて行われた。これらの試みはすべて、結晶のベシル酸レルカニジピンの形成には功を奏さなかった(ベシル酸レルカニジピンの結晶化に成功したのはわずか2例であり、ひとつは下記に示す実施例2であり、2例目は1例目の方法を用いて生成した生成物を結晶の種として利用している)。非晶質のベシル酸レルカニジピンを結晶化させる試みのための一般的な実験スキームには、(1)35mgの非晶質のベシル酸レルカニジピン(前述の方法にて調製)を、MeOH、アセトニトリル(MeCN)、エタノール(EtOH)またはジクロロメタン(CH2Cl2)から選択する有機溶媒0.5mlに溶かし、(2)その溶媒を周囲条件下で少なくとも20日間以上かけてゆっくりと蒸発させ、(3)その試料を真空下で完全に乾燥させ、(4)約0.025mlのEtOHに試料を溶かし、(5)得られた試料を−18℃で5日間、閉所にて保管する、各工程が含まれる。
【0047】
実施例2:結晶のベシル酸レルカニジピンの生成
212.3mgのレルカニジピン遊離塩基(レコルダッチ社製)を1mlのテトラヒドロフラン(THF)に溶かし、レルカニジピン遊離塩基の原液を調製した。0.1mlのTHFに21.7mgのベンゼンスルホン酸を溶解し、酸の原液を調製した。レルカニジピンの原液(0.236ml)と酸原液(0.06ml)の混合物を調製し、25℃で24時間、閉所にて保管した。続いてその混合物を−18℃で24時間、および25℃でさらに24時間、保管した。この時点では沈殿物は観察されなかった。
【0048】
溶媒は周囲条件下で蒸発させた。ガラス状の膜を0.15mlの酢酸エチル(EtAc)に溶かした。8日後、沈殿物は観察されなかった。再度、溶媒を蒸発させ、ガラス状の膜を生じさせた。そのガラス状の膜を0.05mlのt‐ブチルメチルエーテル(TBME)に溶かし、その試料を、室温で6日間、閉所に置いた。この時点で沈殿は観察されなかった。
【0049】
さらに0.05mlのTBMEを加え、温度サイクル(20‐40‐20℃を5サイクル、立ち上がり時間2時間、立ち下がり時間2時間、20℃および40℃における等温時間10分間)に晒した。沈殿は観察されず、溶媒を穏やかな窒素流下で蒸発させた。
【0050】
溶媒を蒸発させた後、粘着性の試料が観察された。試料を真空下で24時間保管した。試料を0.05mlのメタノール(MeOH)に懸濁し、20℃の閉所にて保管した。試料から、いくつかの微粒子を含んだ粘稠液が生じた。さらに3日間、20℃で保管した後、試料は完全な固体となり、交差偏光器で複屈折性が実証され、結晶性物質の存在が確認された。
【0051】
結晶性の物質は、結晶のベシル酸レルカニジピンの収率を増大させるための第2の実験に、結晶の種として用いられた。再度、レルカニジピン遊離塩基の原液(1.10gの遊離塩基を2.2mlのMeOHに溶解)と酸(0.2844gのベンゼンスルホン酸を0.5mlのMeOHに溶解)を調製し、共に混ぜ合わせた。溶媒を穏やかな窒素流下で溶液から蒸発させた。溶液量が2mlまで減少したとき、上述のベシル酸レルカニジピンの結晶を溶液に加えた。再び、穏やかな窒素流下で溶液量が1mlに減少するまで溶媒を除去した。試料をガラスフィルターで濾過し、真空下で乾燥させた。
【0052】
最終収量1.21gの黄白色で結晶性のベシル酸レルカニジピンが得られた。元素分析により、結晶塩の組成はC42H47N2O9S(分子量755.9、溶媒和なし)であることが明らかとなり、このことは酸と遊離塩基の比が1:1(モル/モル)であることと一致する。塩は溶媒和を持たず、非吸湿性であり、TG‐FTIRによる検出では0.1%の質量放出が観察された。
【0053】
以下の通り、さらに大規模な生成を行った。96mlのメタノールに48gのレルカニジピン遊離塩基を溶かした溶液に、室温で22mlのメタノールに溶かした12.7gのベンゼンスルホン酸を加えた。得られた懸濁液を濾過し、最終的な液量が50mlになるまで、55℃で蒸発させた。その溶液に、結晶の種となるベシル酸レルカニジピンの結晶を加え、室温で24時間放置し、続いて5℃で6日間保管した。得られた結晶の集まりを吸引回収し、40mlのメタノールで2回洗浄し、P2O5の存在下で、真空下乾燥を行った。51.1gのベシル酸レルカニジピンを得た。
【0054】
実施例3: 結晶(および非晶質)のナパジシル酸レルカニジピンの生成
169.6mgのレルカニジピン遊離塩基(レコルダッチ社製)を0.82mlのメタノール(MeOH)に溶かし、レルカニジピン遊離塩基の原液を調製した。0.2mlのレルカニジピン遊離塩基の原液と0.195mlのナフタレン‐1,5‐ジスルホン酸水溶液(50mg/ml)を混合し、溶液を調製した。混合時に沈殿が観察された。その沈殿に1.0mlのMeOHを加え、溶解させた。試料を−18℃で4日間保管したが、観察のため、毎日、室温に晒された。次に、溶媒を蒸発させるため、試料を室温で7日間、開放系で保管した。溶媒の蒸発の後、結晶が確認された。その結晶を0.25mlのH2Oと0.01mlのMeOHで懸濁した。懸濁に続いて、遠心分離フィルタ(10,000RPM、0.22μmフィルタ)で結晶を回収し、真空乾燥した。
【0055】
結晶物質は、さらに多量のナパジシル酸レルカニジピンの結晶を得るため、第2の実験において、種結晶として用いられた。1.1gのレルカニジピン遊離塩基を4.4mlのMeOHに溶かし、レルカニジピン遊離塩基の溶液を調製した。その遊離塩基の溶液に5.184mlのナフタレンジスルホン酸水溶液(50mg/ml)を加え、直ちに沈殿が生成した。その沈殿に23mlのMeOHを加えて溶解させた。溶液に、上述の結晶物質を種として加え、4℃で4日間、続いてさらに3日間、−18℃で保管した。結晶を遠心分離フィルタで回収し、真空乾燥した。最終収量0.905g、黄白色のナパジシル酸レルカニジピンの結晶を得た。元素分析により、結晶塩の組成は、C82H90N6O18S2(分子量1511.76、溶媒和なし)であることが明らかになり、このことは、酸と遊離塩基の比が1:2(モル/モル)であることと一致する。その塩は、ジメトキシドの塩水和物として存在し、TG‐FTIRによる検出では4.1%(H2O放出0.4%、MeOH放出3.7%)の質量放出が観察された。
【0056】
非晶質のナパジシル酸レルカニジピンは、本実施例3で得られた溶液を真空で急速に蒸発させて生成する。
【0057】
実施例4: 非晶質塩の生成およびさらに別の結晶のレルカニジピンの塩の生成の試み
結晶のレルカニジピンの塩を生成することが可能な対イオンを判別するため、多くの対イオンとレルカニジピン遊離塩基を用いて塩のスクリーニングを行った。スクリーニング実験では、11種の対イオン、すなわち酢酸塩、ケイヒ酸塩、フマル酸塩、L‐乳酸塩、DL‐乳酸塩、L‐マレイン酸塩、DL‐マンデル酸塩、メシル酸塩、硫酸塩およびトシル酸塩の研究を行った。各対イオンについて、数回、結晶化を試みた。数回の試行後も、対イオンのうち、結晶のレルカニジピンを生成する能力のあるものは見つからなかった。11種の各対イオンのスクリーニングに用いられた一般的な実験スキームの詳細を下記に記す。結晶のレルカニジピンを得ることが難しいことが周知であることから、結晶化技術には十分な保管時間とゆっくりとした工程が選択された。
【0058】
塩スクリーニング実験のそれぞれから得た最終的なレルカニジピンの塩を完全に乾燥させ、化学分析および物理分析を行った。各塩の化学組成を元素分析、赤外分光法と連結した熱重量分析、および含水量分析によって決定した。さらに、FT‐ラマン分析法を用いた物理試験および溶解度試験も行った。
【0059】
実施例4a: テトラヒドロフランに溶解した対イオン
下記に示す通り、テトラヒドロフラン(THF)に溶かしたレルカニジピン遊離塩基(レコルダッチ社製)とTHFに溶かしたそれぞれの酸を溶解させ、レルカニジピン遊離塩基と酸それぞれの原液を調製した。
【0060】
酸と塩基のモル比と同時に、酸の立体的配置に配慮して、レルカニジピン遊離塩基と酸の原液を等量ずつ混合した。原液の混合物には沈殿は見られなかった。試料を25℃で24時間閉所に保管し、続けてさらに4℃で24時間、および−18℃で24時間保管した。保管後、固体は観察されなかった。
【0061】
試料から溶媒を周囲条件下で蒸発させて除去した。4日後、ガラス状の膜が見られた。そのガラス状の膜をアセトンに溶かし、室温で2日間放置した。沈殿は観察されなかった。試料をさらに6日間放置したが、沈殿は見られなかった。
【0062】
溶媒を穏やかな窒素流下で蒸発させ、ガラス状の膜を形成した。ガラス状の膜をt‐ブチルメチルエーテル(TBME)に溶かし、その試料を室温で6日間放置した。さらにTBMEを加え、その試料を
温度サイクル(20‐40‐20℃、5サイクル、立ち上がり時間2時間、立ち下がり時間2時間、20℃および40℃における等温時間10分間)に晒した。沈殿は観察されず、再度、溶媒を穏やかな窒素流下で蒸発させた。
【0063】
溶媒を蒸発させた後、粘着性の試料が観察された。試料を真空下で24時間保管した。その試料をメタノール(MeOH)に懸濁し、20℃の閉所に保管した。試料は粘着性の塊が生じたが、結晶の微粒子は見られなかった。試料をさらに24時間、真空下で保管し、溶媒を除去した。溶媒除去後も結晶物質は観察されなかった。
【0064】
本試料から、2種類の非晶質のレルカニジピンの塩、すなわち、ケイヒ酸レルカニジピンおよびマレイン酸レルカニジピンを生じた。本試料の各塩の化学組成を元素分析、赤外分光法と連結した熱重量分析、および含水量分析によって決定した。各非晶質について、さらに、FT‐ラマン分析法を用いた物理試験および溶解度試験も行った。結果を下記に示す。
【0065】
実施例4b: メタノールに溶解させた対イオン
レルカニジピン遊離塩基および酸のそれぞれの原液を混合して、さらに塩スクリーニング実験を行った。原液は、下記の通り、TBMEに溶かしたレルカニジピン遊離塩基(レコルダッチ社製)およびMeOHに酸をそれぞれ溶解して調製した。
【0066】
酸と塩基のモル比と同時に、酸の立体化学を説明するため、レルカニジピン遊離塩基と酸の原液を等量ずつ混合した。原液の混合物には沈殿は見られなかった。試料を25℃で24時間、閉所に保管し、続けてさらに60℃で8時間、および4℃で6日間保管した。保管後も固体は観察されなかった。
【0067】
試料へ水を加え、続いて周囲条件下で溶媒を蒸発させた。4日後、ガラス状の膜が観察された。試料を真空下で24時間保管した。ガラス状の膜をMeOHに溶かし、20℃で24時間、閉所に保管した。24時間後、粘着性の塊が観察されたが、固体の粒子は見られなかった。試料をさらに2日間保管したが、それでも固体の粒子は見られなかった。真空下で24時間保管し、サンプルから溶媒を除去した。溶媒を除去した後も、結晶物質は見られなかった。
【0068】
本実施例によって、L‐乳酸レルカニジピンが生成した。本実施例にかかる塩の化学組成を元素分析、赤外分光法と連結した熱重量分析、および含水量分析によって決定した。本非晶質塩について、さらに、FT‐ラマン分析法を用いた物理試験および溶解度試験も行った。物性試験の結果を下記に簡潔にまとめる。
【0069】
実施例5: 非晶質の塩の調製および結晶のレルカニジピンの塩の生成の試み
実施例4で述べた塩スクリーニング実験に加えて、結晶のレルカニジピンの塩を生成するための試みをさらに12種類の酸、すなわちクエン酸塩、粘液酸塩、ゲンチシン酸塩、グルコン酸塩、2‐オキソグルタル酸塩、リン酸塩、サッカリン酸塩、サリチル酸塩、L‐酒石酸塩、テレフタル酸塩、マロン酸塩、シュウ酸塩について行った。12種類の対イオンのそれぞれについて、結晶のレルカニジピンの塩を生成する試みを1回ずつ行った。12種類の対イオンのいずれも、結晶のレルカニジピンの塩を生成させることができず、特徴的な非晶質塩を生成したのもほんのわずかであった。対イオンのスクリーニングに用いられた一般的な実験スキームの詳細を下記に記す。結晶のレルカニジピンを得ることが難しいことが周知であることから、結晶化技術には十分な保管時間とゆっくりとした工程が選択された。
【0070】
塩スクリーニング実験のそれぞれから得た最終的なレルカニジピンの塩を完全に乾燥させ、化学分析および物理分析を行った。各塩の化学組成を元素分析、赤外分光法と連結した熱重量分析、および含水量分析によって決定した。さらに、FT‐ラマン分析法を用いた物理試験および溶解度試験も行った。
【0071】
レルカニジピンの原液は、2.67mlのメタノール(MeOH)に530mgのレルカニジピン遊離塩基(レコルダッチ社製)溶解して調製した。結晶をふるいにかけるため、レルカニジピン原液の等量(0.1ml)を下記のように酸と混合した。
【0072】
本実施例の各試料は、完全に同じように取り扱われた。レルカニジピン遊離塩基と対応する対イオンを混ぜ合わせた後、試料を−18℃で2日間、閉所に置いた。2日後、試料のいずれにも沈殿は見られなかった。次に、試料を周囲条件下で8時間、開放系に置き、続いて−18℃でさらに5日間、保管した。保管期間の終了後も、また、沈殿は見られなかった。次に、試料を周囲条件下で15時間、開放系で保管し、−18℃でさらに2日間、置いた。保管後、どの試料にも沈殿は見られなかった。溶媒を真空下でそれぞれの試料から除去し、残った固体を閉所に保管した。
【0073】
レルカニジピン遊離塩基と本実施例にかかる対イオンから結晶のレルカニジピンの塩を生成するという試みはすべて失敗し、結晶物質は生成しなかった。本実施例は、実施例4で述べたと同様、レルカニジピンの結晶塩の生成の困難さ、および予測し難さを示すものである。
【0074】
本実施例では、2種類のレルカニジピンの非晶質塩、すなわちサッカリン酸塩およびサリチル酸塩を生成した。本実施例にかかる各塩の化学組成を元素分析、赤外分光法と連結した熱重量分析、および含水量分析によって決定した。各非晶質塩のFT‐ラマン分析法を用いた物理試験および溶解度試験も行った。物性試験の結果を下記に簡潔にまとめる。
【0075】
実施例6:非晶質および結晶のレルカニジピンの塩の化学組成
乾式燃焼/熱伝導率および非分散型IR検出を用いて、非晶質および結晶のレルカニジピンの塩の双方の元素組成を決定した。元素分析の結果を表1に要約する。
【0076】
非晶質および結晶のレルカニジピンの塩の双方の残留溶媒の内容物を、赤外分光計と一体となった重量分析を用いて決定した。分析には、ブルカー社(Bruker)(スイス、フェランデン)製のFTIR分光計Vector 22と共に、ネッチ社(Netzsch)(ドイツ、ゼルプ)製のThermobalance TG209を用いた。分析は、以下の条件にしたがって行われた。すなわち、2〜5mgの試料を窒素環境下でアルミニウムのるつぼ内で、加熱速度10℃/分で25℃から250℃まで熱した。重量分析の結果を表1に示す。
【0077】
非晶質および結晶のレルカニジピンの塩の双方の吸湿性を、水分吸着測定装置(Surface Measurement System Ltd社(イギリス、バッキンガムシャー、マリオン)製)を用いた動的な蒸気吸着測定(DVS analysis)によって決定した。測定は以下の方法に従って行った。すなわち、10〜15mgの試料を石英またはプラチナの容器に入れ、その容器をマイクロバランスの上に内転させて置き、試料を25℃で湿度サイクルが相対湿度(RH)で0〜95%(50‐95‐0‐50%、速度5%RH/hr)に晒した。吸湿性の測定結果を下記の表1に要約する。
【表1】
【0078】
実施例7:非晶質および結晶のレルカニジピンの塩の溶解度
結晶のベシル酸レルカニジピン、ナパジシル酸レルカニジピンおよび塩酸レルカニジピン、ならびに非晶質のベシル酸レルカニジピンの溶解度は、22℃、0.1モルHCl水溶液(pH1)中でUV‐可視分光法によって測定した。0.1モル塩酸水溶液を用いて、それぞれの化合物のおよそ0.3mg/mlの懸濁液を調製し、24時間振動させて液を平衡にした。平衡にした後、試料を濾過し(0.1μmフィルタ‐)、濃縮物をパーキンエルマー社(Perkin Elmer)(ドイツ、ユーベリンゲン)製のLambda16を用いて、光度測定を行った。基準測定は、共溶媒として20%アセトニトリルを用いて行った。
【表2】
【0079】
結晶のベシル酸塩および結晶のナパシジル酸塩の両方とも、非晶質の塩に比べて溶解度が低いことが表2から見て取れる。結晶塩の溶解度は変化が大きく、結晶のベシル酸塩は、実質、結晶の塩酸塩およびナパジシル酸塩よりも溶解度が大きいことも、表2から読み取れる。
【0080】
実施例8: 新しいレルカニジピンの塩のラマンスペクトル
FTラマン分光法を用いて、新しいレルカニジピンの塩を分析した。ブルカー社製のFTラマンRFS100分光光度計を次の一般的な方法で利用した:試料(前処理なし)約10mg、走査64、分解能2cm‐1、レーザー出力100mW、Ge検出。
【0081】
次の表3、表4、表5は、非晶質のベシル酸レルカニジピンと共に、結晶のベシル酸レルカニジピンおよびナパジシル酸レルカニジピンそれぞれのラマンスペクトルの最も特徴的なピークを示している。
【表3】
【表4】
【表5】
【0082】
実施例9: 新しい結晶のレルカニジピンの塩のX線回折パタ‐ン
フィリップス社(オランダ、アイントホーフェン)製のX‐pert PW3040またはPW1710粉末回折計を用いて、次の一般的な条件下で、結晶のベシル酸レルカニジピンおよびナパジシル酸レルカニジピンのX線回折パターンを得た:試料5〜70mg(前処理なし)をわずかな圧力を用いて平らな試料とし、周囲空気雰囲気、Cu Kα線、2θ 0.02°、ステップサイズ 2s/step、測定範囲2θ 2〜50°。得られたスペクトルを図4(ベシル酸塩)および図6(ナパジシル酸塩)に、それに対応する主なピークを表6(ナパジシル酸塩)および表7(ベシル酸塩)に示す。2θの値は約±0.10から約±0.20°の範囲で再現可能であり、一方で個々のピークの相対強度は試料同士の比によって変化しうるということは、当業者にとって理解されよう。United States Pharmacopiea XXV (2002), p. 2088‐2089を参照のこと。
【表6】
【表7】
【0083】
実施例10: 結晶のベシル酸レルカニジピンおよびナパジシル酸レルカニジピンのDSC分析
示差走査熱量測定法(DSC)を用いて、本発明にかかる結晶のレルカニジピンの塩および結晶の塩酸レルカニジピンの融点を測定した。DSC分析は、加熱によって供与試料に起こる変化を測定するものであり、その変化によって、転移する相を特定する。相転移のときに起こるエンタルピーの変化を、曲線の下の領域に基づいて計算する。最も一般的な転移の相としては、融解および昇華が挙げられる。転移が始まる温度、すなわち開始温度は、曲線からベースラインが逸れ始めた点(変曲点)で表される。
【0084】
結晶のベシル酸レルカニジピンのDSC:4.040mgの結晶のベシル酸レルカニジピンをPerkin Elmer社製の装置DSC7の金製の皿(pan)に入れた。試験中の加熱速度は10℃/分であった。
【0085】
結晶のナパジシル酸レルカニジピンのDSC:3.697mgの結晶のナパジシル酸レルカニジピンをパーキンエルマー社製装置DSC7の金製の皿(pan)に入れた。試験中の加熱速度は10℃/分であった。
【0086】
データを図1(ナパジシル酸塩)および図2(ベシル酸塩)に示し、図の特性点を表8に簡潔にまとめる。
【表8】
【図面の簡単な説明】
【0087】
【図1】図1は結晶のナパジシル酸レルカニジピンの走査熱量の特徴の差異を示す。
【図2】図2は結晶のベシル酸レルカニジピンの走査熱量の特徴の差異を示す。
【図3】図3は結晶のベシル酸レルカニジピンのFTラマンパターンを表す。
【図4】図2は結晶のベシル酸レルカニジピンのX線回折像を表す。
【図5】図5は結晶のナパジシル酸レルカニジピンのFTラマンパターンを表す。
【図6】図6は結晶のナパジシル酸レルカニジピンのX線回折像を表す。
【図7】図7は非晶質のベシル酸レルカニジピンを、等モルのレルカニジピン遊離塩基とベンゼンスルホン酸の固体の状態の混合物とを比較したFTラマンパターンを表す。
【図8】図8は非晶質と結晶のベシル酸レルカニジピンを比較したFTラマンパターンを表す。
【特許請求の範囲】
【請求項1】
(a)レルカニジピンと、
(b)(i)無機酸、(ii)スルホン酸、(iii)モノカルボン酸、(iv)ジカルボン酸、(v)トリカルボン酸、および(vi)芳香族スルホンイミドからなる群より選択される酸の対イオンとの、
酸の対イオンが塩酸ではないことを条件とする、酸付加塩。
【請求項2】
非晶質のレルカニジピンのL‐乳酸塩、ケイヒ酸塩、サリチル酸塩、マレイン酸塩またはサッカリン酸塩であることを特徴とする請求項1に記載の酸付加塩。
【請求項3】
塩が非晶質であって、モル比1:1のレルカニジピンのベンゼンスルホン酸塩であることを特徴とする請求項1に記載の酸付加塩。
【請求項4】
塩が結晶であって、モル比1:1のレルカニジピンのベンゼンスルホン酸塩であることを特徴とする請求項1に記載の酸付加塩。
【請求項5】
塩が非晶質であって、モル比1:2のレルカニジピンのナフタレン‐1,5‐ジスルホン酸塩であることを特徴とする請求項1に記載の酸付加塩。
【請求項6】
塩が結晶であって、モル比1:2のレルカニジピンのナフタレン‐1,5‐ジスルホン酸塩であることを特徴とする請求項1に記載の酸付加塩。
【請求項7】
水和または溶媒和されていることを特徴とする請求項1〜4いずれか1項に記載の酸付加塩。
【請求項8】
医薬品として容認可能な賦形剤および/または担体を用いた混合調剤中に、請求項1〜7いずれか1項に記載の酸付加塩を含む医薬用組成物。
【請求項9】
さらに塩酸レルカニジピンを含むことを特徴とする請求項8記載の医薬用組成物。
【請求項10】
(a)有機溶媒中でレルカニジピンを塩酸以外の酸の対イオンと反応させて、レルカニジピンの塩を生成し、
(b)有機溶媒を除去し、それによって生成したレルカニジピンの塩を単離し、
(c)単離したレルカニジピンの塩を、該レルカニジピンの塩の、
(i)非プロトン性溶媒、および
(ii)プロトン性溶媒、
の溶液から、順不同で、二連続工程のうちの少なくとも一方において再結晶化し、それによって、実質的に純粋な結晶塩であるレルカニジピンを単離する、
各工程を有してなるレルカニジピンの結晶塩の調製方法。
【請求項1】
(a)レルカニジピンと、
(b)(i)無機酸、(ii)スルホン酸、(iii)モノカルボン酸、(iv)ジカルボン酸、(v)トリカルボン酸、および(vi)芳香族スルホンイミドからなる群より選択される酸の対イオンとの、
酸の対イオンが塩酸ではないことを条件とする、酸付加塩。
【請求項2】
非晶質のレルカニジピンのL‐乳酸塩、ケイヒ酸塩、サリチル酸塩、マレイン酸塩またはサッカリン酸塩であることを特徴とする請求項1に記載の酸付加塩。
【請求項3】
塩が非晶質であって、モル比1:1のレルカニジピンのベンゼンスルホン酸塩であることを特徴とする請求項1に記載の酸付加塩。
【請求項4】
塩が結晶であって、モル比1:1のレルカニジピンのベンゼンスルホン酸塩であることを特徴とする請求項1に記載の酸付加塩。
【請求項5】
塩が非晶質であって、モル比1:2のレルカニジピンのナフタレン‐1,5‐ジスルホン酸塩であることを特徴とする請求項1に記載の酸付加塩。
【請求項6】
塩が結晶であって、モル比1:2のレルカニジピンのナフタレン‐1,5‐ジスルホン酸塩であることを特徴とする請求項1に記載の酸付加塩。
【請求項7】
水和または溶媒和されていることを特徴とする請求項1〜4いずれか1項に記載の酸付加塩。
【請求項8】
医薬品として容認可能な賦形剤および/または担体を用いた混合調剤中に、請求項1〜7いずれか1項に記載の酸付加塩を含む医薬用組成物。
【請求項9】
さらに塩酸レルカニジピンを含むことを特徴とする請求項8記載の医薬用組成物。
【請求項10】
(a)有機溶媒中でレルカニジピンを塩酸以外の酸の対イオンと反応させて、レルカニジピンの塩を生成し、
(b)有機溶媒を除去し、それによって生成したレルカニジピンの塩を単離し、
(c)単離したレルカニジピンの塩を、該レルカニジピンの塩の、
(i)非プロトン性溶媒、および
(ii)プロトン性溶媒、
の溶液から、順不同で、二連続工程のうちの少なくとも一方において再結晶化し、それによって、実質的に純粋な結晶塩であるレルカニジピンを単離する、
各工程を有してなるレルカニジピンの結晶塩の調製方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公表番号】特表2008−510754(P2008−510754A)
【公表日】平成20年4月10日(2008.4.10)
【国際特許分類】
【出願番号】特願2007−528715(P2007−528715)
【出願日】平成17年8月22日(2005.8.22)
【国際出願番号】PCT/EP2005/009043
【国際公開番号】WO2006/021397
【国際公開日】平成18年3月2日(2006.3.2)
【出願人】(507061708)レコルダーティ アイルランド リミテッド (7)
【氏名又は名称原語表記】RECORDATI IRELAND LIMITED
【Fターム(参考)】
【公表日】平成20年4月10日(2008.4.10)
【国際特許分類】
【出願日】平成17年8月22日(2005.8.22)
【国際出願番号】PCT/EP2005/009043
【国際公開番号】WO2006/021397
【国際公開日】平成18年3月2日(2006.3.2)
【出願人】(507061708)レコルダーティ アイルランド リミテッド (7)
【氏名又は名称原語表記】RECORDATI IRELAND LIMITED
【Fターム(参考)】
[ Back to top ]