
ロキソプロフェン誘導体及びそれを含有する医薬
【課題】胃腸障害等の副作用を回避すると共に、優れた抗炎症・鎮痛作用を有する新規なロキソプロフェン誘導体を提供する。
【解決手段】次式(I)又は(II):

(式中、R1及びR2はハロゲン原子又は置換若しくは非置換フェニル基を表す)で示されるロキソプロフェン誘導体又はその薬理学的に許容される塩、並びに上記において、ハロゲン原子が塩素原子、臭素原子、フッ素原子、ヨウ素原子から選択されるものであり、置換フェニル基における置換基が、ハロゲン原子、水酸基、置換若しくは非置換低級アルキル基、低級アルキルチオ基、低級アルコキシ基、ニトロ基、アミノ基又はカルボキシル基であるロキソプロフェン誘導体又はその薬理学的に許容される塩。
【解決手段】次式(I)又は(II):
(式中、R1及びR2はハロゲン原子又は置換若しくは非置換フェニル基を表す)で示されるロキソプロフェン誘導体又はその薬理学的に許容される塩、並びに上記において、ハロゲン原子が塩素原子、臭素原子、フッ素原子、ヨウ素原子から選択されるものであり、置換フェニル基における置換基が、ハロゲン原子、水酸基、置換若しくは非置換低級アルキル基、低級アルキルチオ基、低級アルコキシ基、ニトロ基、アミノ基又はカルボキシル基であるロキソプロフェン誘導体又はその薬理学的に許容される塩。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、優れた消炎作用を有し且つ安全性の高い新規なロキソプロフェン誘導体に関する。詳細には、特に胃腸障害などの副作用を示さない、医薬品として有用なロキソプロフェン誘導体に関する。
【背景技術】
【0002】
ロキソプロフェンは、そのナトリウム塩の水和物(一般名:ロキソプロフェンナトリウム水和物)が優れた鎮痛、抗炎症、解熱作用を有する医薬品として臨床的に広く使用されている。
これまでに、ロキソプロフェンの優れた鎮痛、抗炎症、解熱作用を維持した各種誘導体の提案が種々行われてきており、例えば、次式A、B及びCで示される誘導体が知られている(特許文献1〜3)。
【0003】
【化1】
【0004】
具体的には、特許文献1には上記式(A)で示される化合物が開示されており、抗炎症、鎮痛及び解熱作用を有することが報告されている。また、特許文献2には、上記式(B)で形式的に示される誘導体が広範囲に開示されているが、本願発明が提供するロキソプロフェン誘導体とは異なるものである。
さらに、特許文献3には上記式(C)で形式的に示される誘導体が広範囲に開示されており、これらはアスピリン或いはインドメタシンに代表される既存の非ステロイド性酸性消炎剤(酸性NSAIDs)よりも更に強力な抗炎症作用及び鎮痛作用を有し、胃腸管障害等の副作用が極めて少ないものであると報告されている。
【0005】
しかしながら、具体的な胃腸障害等の副作用の程度は、個々の医薬品により異なるものであり、消化管、特に胃粘膜における潰瘍発生の重傷度については大きく異なっている。
したがって、これまでに、抗消炎・鎮痛作用を示す化合物について、その薬理作用と副作用の分離が種々検討されてきているが、これといった成果が上げられていないのが現状である。
本発明者は、かかる現状下、ロキソプロフェン誘導体について検討を行った結果、胃腸障害等の副作用を回避し、優れた抗炎症、鎮痛効果を有する化合物を合成することに成功し、本願発明を完成させるに至った。
【特許文献1】特開昭58−4699号公報
【特許文献2】特開昭54−103852号公報
【特許文献3】国際公開WO93/02999号
【発明の開示】
【発明が解決しようとする課題】
【0006】
したがって本願発明は胃腸障害等の副作用を回避すると共に、優れた抗炎症・鎮痛作用を有する新規なロキソプロフェン誘導体を提供することを課題とする。
【課題を解決するための手段】
【0007】
かかる課題を解決する本発明は、次式(I)又は(II):
【0008】
【化2】
【0009】
(式中、R1及びR2はハロゲン原子又は置換若しくは非置換フェニル基を表す)
で示されるロキソプロフェン誘導体又はその薬理学的に許容される塩である。
【0010】
具体的には、本発明は、上記式(I)及び(II)中、R1及びR2のハロゲン原子が塩素原子、臭素原子、フッ素原子、ヨウ素原子から選択されるものであるロキソプロフェン誘導体又はその薬理学的に許容される塩である。
【0011】
より具体的には、本発明は、上記式(I)中、R1の置換フェニル基における置換基が、ハロゲン原子、水酸基、置換若しくは非置換低級アルキル基、低級アルキルチオ基、低級アルコキシ基、ニトロ基、アミノ基又はカルボキシル基であるロキソプロフェン誘導体又はその薬理学的に許容される塩であり、また上記式(II)中、R2の置換フェニル基における置換基が、ハロゲン原子又は置換低級アルキル基であるロキソプロフェン誘導体又はその薬理学的に許容される塩である。
【0012】
また本発明は別の態様として、上記の式(I)又は(II)で示されるロキソプロフェン誘導体又はその薬理学的に許容される塩を有効成分として含有する医薬である。
【0013】
そのなかでも、特に好ましい本発明は、上記式(I)において、置換基R1がフッ素原子、臭素原子、p−ヒドロキシフェニル基又はp−アミノフェニル基であるロキソプロフェン誘導体又はその薬理学的に許容される塩であり、これら化合物を有効成分として含有する医薬である。
【発明の効果】
【0014】
本発明が提供するロキソプロフェン誘導体は、これまで知られていない新規な化合物であると共に、従来の酸性NSAIDsにみられた胃腸障害等の副作用がなく、その上、臨床的に使用されているロキソプロフェンより抗炎症、鎮痛作用が強いものである。
したがって、その安全域が大きいことから、ヒトに対して安全に使用できる点で、極めて有効なものといえる。
【発明を実施するための最良の形態】
【0015】
本発明は、上記したようにその基本は、下記式(I)又は(II):
【0016】
【化3】
【0017】
(式中、R1及びR2はハロゲン原子又は置換若しくは非置換フェニル基を表す)
で示されるロキソプロフェン誘導体又はその薬理学的に許容される塩である。
【0018】
本明細書において、置換基「R1」又は「R2」におけるハロゲン原子とは、塩素原子、臭素原子、フッ素原子又はヨウ素原子から選択されるハロゲン原子をいう。
【0019】
また、置換基「R1」又は「R2」で示される置換フェニル基における置換基である低級アルキル基とは炭素原子数1〜6程度の置換若しくは非置換アルキル基をいい、具体的には、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、sec-ブチル基、tert-ブチル基、ペンチル基、イソペンチル基、ヘキシル基等を意味する。
これらの低級アルキル基の置換基としては水酸基、アミノ基、ニトロ基等を意味する。
【0020】
低級アルコキシ基としては、炭素原子数1〜6程度の低級アルキルオキシ基であり、具体的には、メトキシ基、エトキシ基、プロポキシ基、ブトキシ基、イソブトキシ基、sec-ブチルオキシ基、tert-ブチルオキシ基、ペンチルオキシ基、イソペンチルオキシ基、ヘキシルオキシ基等を意味する。
【0021】
置換フェニル基における置換基の位置、またその数は特に限定されないが、好ましくはモノ置換フェニル基であってその置換位置がメタ位或いはパラ位であることが好ましい。
【0022】
したがって、本願発明が提供する新規なロキソプロフェン誘導体としては、具体的には以下の化合物を列記することができる。
【0023】
【化4】
【0024】
【化5】
【0025】
【化6】
【0026】
なお、上記式(I−a)、(I−b)及び(II−a)において、フェニルプロピオン酸部分のメチル基は、その立体配置がα−位或いはβ−位を取り得るが、本発明においてはメチル基の配位は、その両者並びにその混合物であってもよい。
更に、式(II−a)におけるシクロペンタン環の水酸基(1位)とフェニル基(2位)の配位は、シス−及びトランス−配位を取り得るが、本発明においては、1,2−シス体であっても、または1,2−トランス体であっても、或いはそのジアステレオマーの混合物であってもよい。
【0027】
本発明が提供する新規なロキソプロフェン誘導体は、具体的には以下のようにして製造することができる。
なお、以下に説明する製造方法は、具体的な一製造方法であり、これに限定されるものでなく、一般的な化学教科書を参照し、製造し得ることはいうまでもない。
【0028】
本発明の式(I)で示されるロキソプロフェン誘導体のなかで、置換基「R1」がハロゲン原子である場合のロキソプロフェン誘導体は、例えば、下記化学反応式で示す製造スキーム1に従って合成することができる。
[製造スキーム1]
【0029】
【化7】
【0030】
式中、算用数字は具体的化合物番号を表し、ローマ数字は製造スキーム1における工程番号を表す(以下の製造スキームにおいて同じ)。
また、以下の各製造スキームに基づく各製造工程における反応条件(反応時間、反応温度等)、反応試薬、溶媒、触媒等は、好ましい製造例として例示するものであり、これらに限定されるものではない。
【0031】
まず、市販されている種々のハロゲンを含む3置換芳香族化合物(1a−d)を出発原料として、アミノ基末端をホルミル化反応によってアルデヒド化合物(2a−d)へと変換した。
かかる変換は、例えば、第i工程として、塩酸/硝酸ナトリウム/硫酸銅/亜硫酸ナトリウム/酢酸ナトリウムによるジアゾ化反応の後、第ii工程としてパラホルムアルデヒドの存在下、ヒドロキシムアミン塩酸塩との処理の後、第iii工程として塩酸による分解で目的とするアルデヒド化合物を得ることができる。
【0032】
次いで得られたアルデヒド化合物(2a−d)に対して、例えば、トルエン中MeOCH2P(Ph3)Cl、C6H18KNSi2によるウィテッヒ(Wittig)反応(第iv工程)、さらに例えばアセトン中塩酸等の酸処理(第v工程)による炭素鎖を伸長したフェニルアルデヒド化合物(3a−d)とし、2モル%のPFC存在下、過ヨウ素酸による酸化反応(第vi工程)によりフェニル酢酸化合物(4a−d)とした後、酸−アルコールによるエステル化反応(第vii工程)を行い、フェニル酢酸エステル化合物(5a−d)へと変換する。
【0033】
さらに、得られたフェニル酢酸エステル化合物(5a−d)を、乾燥テトラヒドロフラン中、2モル程度のリチウムジイソプロピルアミド(LDA)の存在下、ヨウ化メチルによりα−メチル化反応(第viii工程)を行い、プロピン酸エステル化合物(6a−d)へと順次変換した。当該反応は、低温で行うのが好ましく、例えば、−78℃から徐々に−40℃程度の温度下に実施するのが好ましい。
【0034】
次いで、例えば、四塩化炭素中N−ブロモスクシンイミド(NBS)、アゾイソブチロニトリル(AIBN)による還流処理により、メチル基末端をブロモメチレンへと変換(第ix工程)した化合物(7a−d)を経て、溶媒(例えば、乾燥アセトン)中炭酸カリウムの存在下、2−オキソシクロペンタンカルボン酸メチルとの反応(第x工程)により、シクロペンタン環を含むジエステル化合物(8a−d)を中間体として得る。
最後に、これらの中間体化合物(8a−d)を、例えば、酢酸/塩酸による酸加水分解・脱炭酸することによって、目的とする本発明の式(I)で示されるロキソプロフェン誘導体のなかで、置換基「R1」がハロゲン原子である場合のロキソプロフェン誘導体(9a−d)が製造される。
【0035】
また、本発明の式(I)で示されるロキソプロフェン誘導体のなかで、置換基「R1」が置換若しくは非置換フェニル基である場合のロキソプロフェン誘導体は、例えば下記化学反応式で示す製造スキーム2及び製造スキーム3に従って合成することができる。
[製造スキーム2]
【0036】
【化8】
【0037】
[製造スキーム3]
【0038】
【化9】
【0039】
すなわち、製造スキーム2及び製造スキーム3は、本発明のロキソプロフェン誘導体のなかで、置換基「R1」が置換若しくは非置換フェニル基である場合のロキソプロフェン誘導体(19−25、31−35)の合成工程を示した。
まず、製造スキーム1で得られた芳香環が臭素で置換されたロキソプロフェン誘導体(9c)のカルボン酸末端を、1,2−ジクロロエタン中、4−アミノジメチルピリジン(4−DMAP)の存在下、メタノールまたはベンジルアルコール(BnOH)によりカルボキシル基をエステル化(第i工程)でエステル保護した化合物(10c、11c)を合成し、これらを基質とした種々のボロン化合物との鈴木−宮浦クロスカップリング反応(第ii工程)を行うことにより、エステルで保護されたビフェニル型のロキソプロフェン誘導体(12−17)を得た。
【0040】
当該鈴木−宮浦クロスカップリング反応は、例えば、R2として上記に示した置換基を有するボロン化合物[R2−PhB(OH)2]を用い、2モル程度の炭酸ナトリウムの存在下、含水テトラヒドロフラン中、Pd(PPh3)4と還流処理をすることによって行うことができる。
【0041】
また、ビフェニル末端がニトロ基で置換された化合物(17)については、パラジウム−炭素を用いた還元反応によりアミノ基へと変換し(化合物18)、最終的にこれらの化合物(12−18)のエステル基末端を接触還元または加水分解することによって、目的とするビフェニル型のロキソプロフェン誘導体(19−25)を得た。
接触還元は、例えば、10%パラジウム−炭素を触媒とし、メタノール或いはエタノール等のアルコール溶媒中水素ガスを吸収させることにより、また加水分解は、通常のアルカリ加水分解(例えば、アルコール溶媒中、アルカリ金属水酸化物による加水分解)で実施することができる。
【0042】
一方、反応スキーム3に示した別のビフェニル型ロキソプロフェン誘導体(31−35)については、反応スキーム1で得られたシクロペンタノン環を含むジエステル化合物(8c)を基質として、種々のボロン酸化合物との鈴木−宮浦クロスカップリング反応により、目的化合物の前駆体としてのジエステル化合物(26-30)を合成し、最終的にこれらを加水分解・脱炭酸することによって、目的とする別のロキソプロフェン誘導体(31−35)を得た。
【0043】
なお、ボロン酸化合物との鈴木−宮浦クロスカップリング反応(第i工程)、及び加水分解・脱炭酸反応(第ii工程)は、上記の反応スキーム2及び反応スキーム1で行ったと同様の反応で実施することができる。
【0044】
さらに、本発明の目的化合物である式(III)で示されるシクロペンタノール環を有する還元型ロキソプロフェン誘導体の製造は、例えば下記化学反応式で示す製造スキーム4に従って合成することができる。
[製造スキーム4]
【0045】
【化10】
【0046】
まず、製造スキーム1で示した方法により製造された芳香環がフッ素原子または臭素原子で置換されたハロゲン型のロキソプロフェン誘導体(9a、9c)、及び製造スキーム2に示した方法により製造されたビフェニル末端基が水酸基(パラ位)で置換されたビフェニル型のロキソプロフェン誘導体(19)のカルボン酸末端を、例えば1,2−ジクロロエタン中、4−アミノジメチルピリジン(4−DMAP)の存在下、メタノールまたはベンジルアルコール(BnOH)によりカルボキシル基をエステル化(第i工程)でエステル保護した化合物(10c、11c、12)を合成した。
【0047】
次に、これらの化合物のカルボニル基を、例えば溶媒としてジクロロメタンを用い、水素化ホウ素ナトリウムによる還元反応(第ii工程)によりアルコール化合物(36a,c;37a,c;38;39)と変換し、最終的にエステル基末端を接触還元または加水分解(第iii又はiv工程)することによって目的とする還元型ロキソプロフェン誘導体(40a,c;41a,c;42;43)を得た。
この場合の接触還元または加水分解反応は、製造スキーム2における第iv又はv工程の方法と同様の手段採用される。
【0048】
なお、これらの化合物とその前駆体は、化学構造中におけるシクロペンタン環の2箇所(1位及び2位)において不斉炭素で区別されるシスまたはトランスの幾何異性体をシリカゲルクロマトグラフィーによって分離し、それぞれのα位不斉炭素で区別される立体異性体を含めた、4つのジアステレオアイソマーの混合物として単離することができる。
【0049】
以上に記載した製造方法により、本願発明の目的化合物であるロキソプロフェン誘導体を得ることができるが、得られた化合物の物理化学的性質を下記表1〜3にまとめて示した。
【0050】
【表1】
【0051】
【表2】
【0052】
【表3】
【0053】
本発明が提供する上記のロキソプロフェン誘導体は、遊離カルボン酸のまま、或いはその薬理学的に許容される塩として使用することができる。
薬理学的に許容される塩としては、ナトリウム塩、カリウム塩などのアルカリ金属塩或いはアンモニウム塩を挙げることができる。
【0054】
本発明が提供するロキソプロフェン誘導体またはその薬理学的に許容される塩を医薬組成物として投与する場合、例えば、当該誘導体またはその薬学的に許容される塩である有効成分を単独、または慣用の賦形剤と共にカプセル剤、錠剤、注射剤等の適宜な剤形として、経口的または非経口的に投与することができる。具体的には、例えば、カプセル剤は、ロキソプロフェン誘導体またはその塩を乳糖、澱粉またはその誘導体、セルロース誘導体等の賦形剤と混合してゼラチンカプセルに充填し調製することができる。
【0055】
また、錠剤は、上記賦形剤の他に、カルボキシメチルセルロースナトリウム、アルギン酸、アラビアゴム等の結合剤と水を加えて練合し、必要により顆粒とした後、さらにタルク、ステアリン酸等の潤滑剤を添加して、通常の圧縮打錠機を用いて錠剤に調製することができる。
【0056】
さらに、注射による非経口投与に際しては、ロキソプロフェン誘導体またはその塩を溶解補助剤と共に滅菌蒸留水または滅菌生理食塩水に溶解し、アンプルに封入して注射用製剤とする。必要により安定化剤、緩衝物質等を含有させてもよい。これらの非経口投与製剤は、静脈内投与、あるいは点滴静注により投与することができる。
【0057】
本発明が提供するロキソプロフェン誘導体の投与量は、種々の要因、例えば治療すべき患者の症状、重症度、年齢、合併症の有無等によって一概には限定できない。
また、投与経路、剤形、投与回数等によっても異なるものであるが、一般的には、経口投与の場合は、有効成分として、通常、0.1〜1000mg/日/ヒト、好ましくは1〜500mg/日/ヒトの範囲内、また、非経口投与の場合は、経口投与の場合における投与量の約1/100〜1/2量程度の範囲内で適宜選別し、投与することができる。なお、これらの投与量は、患者の年齢、症状等により適宜増減することが可能であることは勿論である。
【実施例】
【0058】
以下、実施例に代わる試験例に基づいて、本発明を更に具体的に説明するが、本発明の範囲をこれらの例に限定するものではないことはいうまでもない。
【0059】
インビトロ(in vitro)及びインビボ(in vivo)試験で用いた本発明のロキソプロフェン誘導体について、その化学構造を以下に示した。
【0060】
【化11】
【0061】
【表4】
【0062】
試験例1:ヒト全血アッセイ(インビトロ)
試験は、Inflamm. Res., 45: 68-74 (1996) に記載される方法に準じて行った。
A:インビトロにおけるCOX−1アッセイ
採血対象者は、少なくとも1週間以上にNSAIDsを服用しておらず、採取日に健康である人を選択した。
血液は、血液凝固抑制剤無添加で採取し、直後にアッセイに用いた。採取した血液を500μLずつチューブ(Protein Lobingdin tube, Eppenduf Co. LTD., Tokyo, Janan)に分注し、適切な溶媒(DMSO又はMilliQ water)に溶解させた試験化合物2μL(最終濃度:0.1μM−1000μM)を添加し、37℃/24時間血液凝固が認められるまでインキュベーションした。
インキュベーション後、サンプルを12,000×g/5分間の遠心分離を行い、血清を分離した。血中タンパクを除外するため、得られた血清100μLをエタノール400μLに添加し、再び12,000×g/5分間の遠心分離を行った。上清中のTXB2を酵素免疫測定法(EIA)kit[Cayman (Ann, Arbor, MI,USA)#519031]を用いて定量した。プロトコルは、付属のプロトコルに従った。
【0063】
B:インビトロにおけるCOX−2アッセイ
採血対象者は、少なくとも1週間以上にNSAIDsを服用しておらず、採取日に健康である人を選択した。
血液は、ヘパリン処理が施された試験管(Venojectll blood collection tubes、テルモ社製)に採取し、炎症性刺激物質であるリポポリサッカロイド(LPS)[Sigma-Aldrich Japan Inc,#L2880 from E. coli055:B5、最終濃度が100μg/mLとなるように燐酸緩衝生理食塩水(PBS)で希釈した]を添加した。500μLずつチューブに分注し、適切な溶媒(DMSO又はMilliQ water)に溶解させた試験化合物2μL(最終濃度:0.1μM−1000μM)を添加し、COX−2を誘導するために37℃/24時間インキュベーションした。
インキュベーション後、サンプルを12,000×g/5分間の遠心分離を行い、血清を分離した。血中タンパクを除外するため、得られた血清100μLをエタノール400μLに添加し、再び12,000×g/5分間の遠心分離を行った。上清中のPGE2を酵素免疫測定法(EIA)kit[Cayman (Ann, Arbor, MI,USA)#514040]を用いて定量した。プロトコルは、付属のプロトコルに従った。
【0064】
試験例2:腎障害アッセイ(インビボ)
試験は、Biochem. Biophysical. Res. Com., 323; 1032-39 (2004) に記載の方法に従った。
A:リポソーム膜調製法
卵黄ホスファチジルコリン(10μM、7.7mg:関東化学社製)をクロロホルム/メタノール(1:2、v/v)に溶解し、乾燥後、1.5mLのジエチルエーテルに溶解した。100mM calcein-NaOH(pH7.4)を1mL加え、1分間超音波処理した後、ジエチルエーテルをconventional rotary evaporator(25℃)で除去し、リン酸緩衝液に懸濁したものをリポソーム膜溶液とした(reversed-phase evaporation法)。
【0065】
B:膜傷害測定法
(1)水を溶剤として用いる測定方法
30μLのリポソーム膜溶液を、5mLのリン酸緩衝液に懸濁させ、遠心(10,500×g/20分間)し、リン酸緩衝液で2回洗浄した。ペレットを1mLのリン酸緩衝液に懸濁し、その懸濁液を膜傷害実験に用いた。
1.5mLチューブに6μLずつ分注し、そのリポソーム懸濁液に、濃度が異なるテストサンプルのリン酸緩衝液6μLを加え、30℃/10分間インキュベーションした。氷上で冷却後、384穴wellに混合溶液を10μL分注した後、漏出したcalceinの蛍光強度を測定した(励起波長:490nm)。
【0066】
(2)DMSOを溶剤として用いる測定方法
リポソーム懸濁液を遠心(10,500×g/20分間)し、リン酸緩衝液で2回洗浄した。ペレットを5mLのリン酸緩衝液に懸濁し、その懸濁液の0.75mLを50mLのリン酸緩衝液で希釈したものを膜傷害実験に用いた。
1.5mLチューブに400μLずつ分注し、そのリポソーム懸濁液に、濃度が異なるテストサンプルのDMSO溶液を加え、30℃/10分間インキュベーションした。氷上で冷却後、96穴wellに混合溶液を200μL分注した後に、漏出したcalceinの蛍光強度を測定した(励起波長:490nm)。
【0067】
C:膜傷害性の評価
100%膜傷害のコントロールとして、triton X-100の25%溶液を用い、各テストサンプルの蛍光強度を測定後、100%膜傷害のコントロールの値に対する割合を百分率で表したものを膜傷害(calcein release)とした。
【0068】
これらの結果をまとめて下記表に示した。
【0069】
【表5】
【0070】
試験例3:動物アッセイ(インビボ試験)
A:ロキソプロフェン誘導体による胃潰瘍の形成
試験は、Biochem. Pharmacol., 67; 575-85 (2004) に記載の方法に従った。
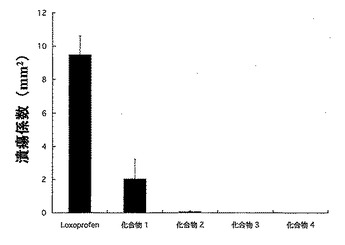
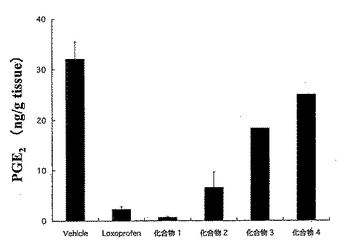
Wister系雄性ラット(体重:180-200g)を18時間絶食させ、試験化合物を経口投与した(投与量は、ロキソプロフェンと等しい物質量となるようにした)。8時間後、胃を摘出し、胃内部に生じた潰瘍の面積を測定した。全ての潰瘍の面積を合計し、潰瘍係数とした。
また、胃粘膜のPGE2量は、ELISA法により測定した。
なお、プロトコルは、キット付属のプロトコルに従った。
【0071】
それらの結果を図1〜図3に示した。
図1にロキソプロフェン40mg/kg量に相当する試験化合物を投与した場合の結果を示し、図2にロキソプロフェン50mg/kg量に相当する試験化合物を投与した場合の結果を示し、図3に胃粘膜PGE2量の変化を示した。
図中に示した結果からも判明するように、本発明のロキソプロフェン誘導体(化合物1〜4)は、ロキソプロフェンに比較して顕著に潰瘍係数が低く、化合物3及び4では殆ど潰瘍の形成が認められないものであった。
また、胃粘膜のPGE2量は、化合物2、3及び4において、ロキソプロフェンより高いものであった。
【0072】
B:カラゲニン惹起性浮腫に対するロキソプロフェン誘導体の効果
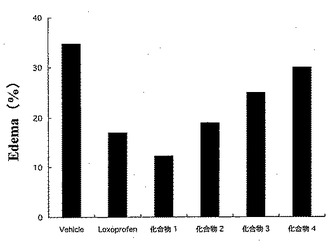
試験は、Br. J. Pharmacol., 151; 285-91 (2007) に記載の方法に従った。
Wister系雄性ラット(体重:180-200g)を18時間絶食させ、試験化合物を経口投与した(投与量は、ロキソプロフェンと等しい物質量となるようにした)。1時間後、左足蹠皮下に1%カラゲニン(生理食塩水に溶解)100μLを注射し、浮腫を惹起させた。
カラゲニン投与前、投与後3時間及び6時間後の足容積を、Plethysmometerを用いて測定した。
また、足浮腫PGE2量は、ELISA法により測定した。なお、プロトコルは、キット付属のプロトコルに従った。
【0073】
それらの結果を図4〜図6に示した。
図4にロキソプロフェン10mg/kg量に相当する試験化合物を投与した6時間後におけるカラゲニン浮腫(%)を示し、図5に浮腫抑制率(%)を示した。
なお、浮腫抑制率は、以下の計算による。
抑制率(%)=100−(化合物投与時の浮腫容積/Vehicle投与時の浮腫容積)×100
また、図6に、投与後3時間及び6時間後の足容積の変化を示した。
【0074】
これらの結果からも判明するように、本発明のロキソプロフェン誘導体(化合物1〜4)は、良好な消炎作用を示すものであると共に、副作用である潰瘍形成を生じないものであり、薬理作用と副作用の分離が良好に行われた化合物であることが判明する。
【0075】
製剤例1:錠剤
化合物1 50mg
乳糖 100mg
ヒドロキシプロピルセルロース 150mg
ステアリン酸マグネシウム 50mg
上記処方を基本とし、顆粒を調製後、打錠し重量350mgの錠剤を、常法により調製した。
【0076】
製剤例2:顆粒剤
化合物2 50mg
乳糖 100mg
トウモロコシデンプンルロース 150mg
上記処方を基本とし、200mg顆粒中有効成分50mg含有の顆粒を常法により調製した。
【産業上の利用可能性】
【0077】
以上記載のように、本発明が提供するロキソプロフェン誘導体は、これまで知られていない新規な化合物であると共に、従来のNSAIDsにみられた胃腸障害等の副作用がなく、その上、臨床的に使用されているロキソプロフェンより抗炎症、鎮痛作用が強いものである。
したがって、その安全域が大きいことから、ヒトに対して安全に使用できる点で、極めて有効なものであり、産業上の貢献度は多大なものである。
【図面の簡単な説明】
【0078】
【図1】試験例3における、ロキソプロフェン40mg/kg相当量の化合物量を投与した場合の潰瘍係数の結果を示した図である。
【図2】試験例3における、ロキソプロフェン50mg/kg相当量の化合物量を投与した場合の潰瘍係数の結果を示した図である。
【図3】試験例3における、ロキソプロフェン40mg/kg相当量の化合物量を投与した場合の胃粘膜PGE2量の変化を示した図である。
【図4】試験例3における、ロキソプロフェン10mg/kg相当量の試験化合物を投与した6時間後におけるカラゲニン浮腫(%)を示した図である。
【図5】試験例3における、ロキソプロフェン10mg/kg相当量の試験化合物を投与した6時間後におけるカラゲニン浮腫抑制率(%)を示した図である。
【図6】試験例3における、ロキソプロフェン10mg/kg相当量の試験化合物を投与した場合の、投与後3時間及び6時間後の足容積の変化を示した図である。
【技術分野】
【0001】
本発明は、優れた消炎作用を有し且つ安全性の高い新規なロキソプロフェン誘導体に関する。詳細には、特に胃腸障害などの副作用を示さない、医薬品として有用なロキソプロフェン誘導体に関する。
【背景技術】
【0002】
ロキソプロフェンは、そのナトリウム塩の水和物(一般名:ロキソプロフェンナトリウム水和物)が優れた鎮痛、抗炎症、解熱作用を有する医薬品として臨床的に広く使用されている。
これまでに、ロキソプロフェンの優れた鎮痛、抗炎症、解熱作用を維持した各種誘導体の提案が種々行われてきており、例えば、次式A、B及びCで示される誘導体が知られている(特許文献1〜3)。
【0003】
【化1】
【0004】
具体的には、特許文献1には上記式(A)で示される化合物が開示されており、抗炎症、鎮痛及び解熱作用を有することが報告されている。また、特許文献2には、上記式(B)で形式的に示される誘導体が広範囲に開示されているが、本願発明が提供するロキソプロフェン誘導体とは異なるものである。
さらに、特許文献3には上記式(C)で形式的に示される誘導体が広範囲に開示されており、これらはアスピリン或いはインドメタシンに代表される既存の非ステロイド性酸性消炎剤(酸性NSAIDs)よりも更に強力な抗炎症作用及び鎮痛作用を有し、胃腸管障害等の副作用が極めて少ないものであると報告されている。
【0005】
しかしながら、具体的な胃腸障害等の副作用の程度は、個々の医薬品により異なるものであり、消化管、特に胃粘膜における潰瘍発生の重傷度については大きく異なっている。
したがって、これまでに、抗消炎・鎮痛作用を示す化合物について、その薬理作用と副作用の分離が種々検討されてきているが、これといった成果が上げられていないのが現状である。
本発明者は、かかる現状下、ロキソプロフェン誘導体について検討を行った結果、胃腸障害等の副作用を回避し、優れた抗炎症、鎮痛効果を有する化合物を合成することに成功し、本願発明を完成させるに至った。
【特許文献1】特開昭58−4699号公報
【特許文献2】特開昭54−103852号公報
【特許文献3】国際公開WO93/02999号
【発明の開示】
【発明が解決しようとする課題】
【0006】
したがって本願発明は胃腸障害等の副作用を回避すると共に、優れた抗炎症・鎮痛作用を有する新規なロキソプロフェン誘導体を提供することを課題とする。
【課題を解決するための手段】
【0007】
かかる課題を解決する本発明は、次式(I)又は(II):
【0008】
【化2】
【0009】
(式中、R1及びR2はハロゲン原子又は置換若しくは非置換フェニル基を表す)
で示されるロキソプロフェン誘導体又はその薬理学的に許容される塩である。
【0010】
具体的には、本発明は、上記式(I)及び(II)中、R1及びR2のハロゲン原子が塩素原子、臭素原子、フッ素原子、ヨウ素原子から選択されるものであるロキソプロフェン誘導体又はその薬理学的に許容される塩である。
【0011】
より具体的には、本発明は、上記式(I)中、R1の置換フェニル基における置換基が、ハロゲン原子、水酸基、置換若しくは非置換低級アルキル基、低級アルキルチオ基、低級アルコキシ基、ニトロ基、アミノ基又はカルボキシル基であるロキソプロフェン誘導体又はその薬理学的に許容される塩であり、また上記式(II)中、R2の置換フェニル基における置換基が、ハロゲン原子又は置換低級アルキル基であるロキソプロフェン誘導体又はその薬理学的に許容される塩である。
【0012】
また本発明は別の態様として、上記の式(I)又は(II)で示されるロキソプロフェン誘導体又はその薬理学的に許容される塩を有効成分として含有する医薬である。
【0013】
そのなかでも、特に好ましい本発明は、上記式(I)において、置換基R1がフッ素原子、臭素原子、p−ヒドロキシフェニル基又はp−アミノフェニル基であるロキソプロフェン誘導体又はその薬理学的に許容される塩であり、これら化合物を有効成分として含有する医薬である。
【発明の効果】
【0014】
本発明が提供するロキソプロフェン誘導体は、これまで知られていない新規な化合物であると共に、従来の酸性NSAIDsにみられた胃腸障害等の副作用がなく、その上、臨床的に使用されているロキソプロフェンより抗炎症、鎮痛作用が強いものである。
したがって、その安全域が大きいことから、ヒトに対して安全に使用できる点で、極めて有効なものといえる。
【発明を実施するための最良の形態】
【0015】
本発明は、上記したようにその基本は、下記式(I)又は(II):
【0016】
【化3】
【0017】
(式中、R1及びR2はハロゲン原子又は置換若しくは非置換フェニル基を表す)
で示されるロキソプロフェン誘導体又はその薬理学的に許容される塩である。
【0018】
本明細書において、置換基「R1」又は「R2」におけるハロゲン原子とは、塩素原子、臭素原子、フッ素原子又はヨウ素原子から選択されるハロゲン原子をいう。
【0019】
また、置換基「R1」又は「R2」で示される置換フェニル基における置換基である低級アルキル基とは炭素原子数1〜6程度の置換若しくは非置換アルキル基をいい、具体的には、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、イソブチル基、sec-ブチル基、tert-ブチル基、ペンチル基、イソペンチル基、ヘキシル基等を意味する。
これらの低級アルキル基の置換基としては水酸基、アミノ基、ニトロ基等を意味する。
【0020】
低級アルコキシ基としては、炭素原子数1〜6程度の低級アルキルオキシ基であり、具体的には、メトキシ基、エトキシ基、プロポキシ基、ブトキシ基、イソブトキシ基、sec-ブチルオキシ基、tert-ブチルオキシ基、ペンチルオキシ基、イソペンチルオキシ基、ヘキシルオキシ基等を意味する。
【0021】
置換フェニル基における置換基の位置、またその数は特に限定されないが、好ましくはモノ置換フェニル基であってその置換位置がメタ位或いはパラ位であることが好ましい。
【0022】
したがって、本願発明が提供する新規なロキソプロフェン誘導体としては、具体的には以下の化合物を列記することができる。
【0023】
【化4】
【0024】
【化5】
【0025】
【化6】
【0026】
なお、上記式(I−a)、(I−b)及び(II−a)において、フェニルプロピオン酸部分のメチル基は、その立体配置がα−位或いはβ−位を取り得るが、本発明においてはメチル基の配位は、その両者並びにその混合物であってもよい。
更に、式(II−a)におけるシクロペンタン環の水酸基(1位)とフェニル基(2位)の配位は、シス−及びトランス−配位を取り得るが、本発明においては、1,2−シス体であっても、または1,2−トランス体であっても、或いはそのジアステレオマーの混合物であってもよい。
【0027】
本発明が提供する新規なロキソプロフェン誘導体は、具体的には以下のようにして製造することができる。
なお、以下に説明する製造方法は、具体的な一製造方法であり、これに限定されるものでなく、一般的な化学教科書を参照し、製造し得ることはいうまでもない。
【0028】
本発明の式(I)で示されるロキソプロフェン誘導体のなかで、置換基「R1」がハロゲン原子である場合のロキソプロフェン誘導体は、例えば、下記化学反応式で示す製造スキーム1に従って合成することができる。
[製造スキーム1]
【0029】
【化7】
【0030】
式中、算用数字は具体的化合物番号を表し、ローマ数字は製造スキーム1における工程番号を表す(以下の製造スキームにおいて同じ)。
また、以下の各製造スキームに基づく各製造工程における反応条件(反応時間、反応温度等)、反応試薬、溶媒、触媒等は、好ましい製造例として例示するものであり、これらに限定されるものではない。
【0031】
まず、市販されている種々のハロゲンを含む3置換芳香族化合物(1a−d)を出発原料として、アミノ基末端をホルミル化反応によってアルデヒド化合物(2a−d)へと変換した。
かかる変換は、例えば、第i工程として、塩酸/硝酸ナトリウム/硫酸銅/亜硫酸ナトリウム/酢酸ナトリウムによるジアゾ化反応の後、第ii工程としてパラホルムアルデヒドの存在下、ヒドロキシムアミン塩酸塩との処理の後、第iii工程として塩酸による分解で目的とするアルデヒド化合物を得ることができる。
【0032】
次いで得られたアルデヒド化合物(2a−d)に対して、例えば、トルエン中MeOCH2P(Ph3)Cl、C6H18KNSi2によるウィテッヒ(Wittig)反応(第iv工程)、さらに例えばアセトン中塩酸等の酸処理(第v工程)による炭素鎖を伸長したフェニルアルデヒド化合物(3a−d)とし、2モル%のPFC存在下、過ヨウ素酸による酸化反応(第vi工程)によりフェニル酢酸化合物(4a−d)とした後、酸−アルコールによるエステル化反応(第vii工程)を行い、フェニル酢酸エステル化合物(5a−d)へと変換する。
【0033】
さらに、得られたフェニル酢酸エステル化合物(5a−d)を、乾燥テトラヒドロフラン中、2モル程度のリチウムジイソプロピルアミド(LDA)の存在下、ヨウ化メチルによりα−メチル化反応(第viii工程)を行い、プロピン酸エステル化合物(6a−d)へと順次変換した。当該反応は、低温で行うのが好ましく、例えば、−78℃から徐々に−40℃程度の温度下に実施するのが好ましい。
【0034】
次いで、例えば、四塩化炭素中N−ブロモスクシンイミド(NBS)、アゾイソブチロニトリル(AIBN)による還流処理により、メチル基末端をブロモメチレンへと変換(第ix工程)した化合物(7a−d)を経て、溶媒(例えば、乾燥アセトン)中炭酸カリウムの存在下、2−オキソシクロペンタンカルボン酸メチルとの反応(第x工程)により、シクロペンタン環を含むジエステル化合物(8a−d)を中間体として得る。
最後に、これらの中間体化合物(8a−d)を、例えば、酢酸/塩酸による酸加水分解・脱炭酸することによって、目的とする本発明の式(I)で示されるロキソプロフェン誘導体のなかで、置換基「R1」がハロゲン原子である場合のロキソプロフェン誘導体(9a−d)が製造される。
【0035】
また、本発明の式(I)で示されるロキソプロフェン誘導体のなかで、置換基「R1」が置換若しくは非置換フェニル基である場合のロキソプロフェン誘導体は、例えば下記化学反応式で示す製造スキーム2及び製造スキーム3に従って合成することができる。
[製造スキーム2]
【0036】
【化8】
【0037】
[製造スキーム3]
【0038】
【化9】
【0039】
すなわち、製造スキーム2及び製造スキーム3は、本発明のロキソプロフェン誘導体のなかで、置換基「R1」が置換若しくは非置換フェニル基である場合のロキソプロフェン誘導体(19−25、31−35)の合成工程を示した。
まず、製造スキーム1で得られた芳香環が臭素で置換されたロキソプロフェン誘導体(9c)のカルボン酸末端を、1,2−ジクロロエタン中、4−アミノジメチルピリジン(4−DMAP)の存在下、メタノールまたはベンジルアルコール(BnOH)によりカルボキシル基をエステル化(第i工程)でエステル保護した化合物(10c、11c)を合成し、これらを基質とした種々のボロン化合物との鈴木−宮浦クロスカップリング反応(第ii工程)を行うことにより、エステルで保護されたビフェニル型のロキソプロフェン誘導体(12−17)を得た。
【0040】
当該鈴木−宮浦クロスカップリング反応は、例えば、R2として上記に示した置換基を有するボロン化合物[R2−PhB(OH)2]を用い、2モル程度の炭酸ナトリウムの存在下、含水テトラヒドロフラン中、Pd(PPh3)4と還流処理をすることによって行うことができる。
【0041】
また、ビフェニル末端がニトロ基で置換された化合物(17)については、パラジウム−炭素を用いた還元反応によりアミノ基へと変換し(化合物18)、最終的にこれらの化合物(12−18)のエステル基末端を接触還元または加水分解することによって、目的とするビフェニル型のロキソプロフェン誘導体(19−25)を得た。
接触還元は、例えば、10%パラジウム−炭素を触媒とし、メタノール或いはエタノール等のアルコール溶媒中水素ガスを吸収させることにより、また加水分解は、通常のアルカリ加水分解(例えば、アルコール溶媒中、アルカリ金属水酸化物による加水分解)で実施することができる。
【0042】
一方、反応スキーム3に示した別のビフェニル型ロキソプロフェン誘導体(31−35)については、反応スキーム1で得られたシクロペンタノン環を含むジエステル化合物(8c)を基質として、種々のボロン酸化合物との鈴木−宮浦クロスカップリング反応により、目的化合物の前駆体としてのジエステル化合物(26-30)を合成し、最終的にこれらを加水分解・脱炭酸することによって、目的とする別のロキソプロフェン誘導体(31−35)を得た。
【0043】
なお、ボロン酸化合物との鈴木−宮浦クロスカップリング反応(第i工程)、及び加水分解・脱炭酸反応(第ii工程)は、上記の反応スキーム2及び反応スキーム1で行ったと同様の反応で実施することができる。
【0044】
さらに、本発明の目的化合物である式(III)で示されるシクロペンタノール環を有する還元型ロキソプロフェン誘導体の製造は、例えば下記化学反応式で示す製造スキーム4に従って合成することができる。
[製造スキーム4]
【0045】
【化10】
【0046】
まず、製造スキーム1で示した方法により製造された芳香環がフッ素原子または臭素原子で置換されたハロゲン型のロキソプロフェン誘導体(9a、9c)、及び製造スキーム2に示した方法により製造されたビフェニル末端基が水酸基(パラ位)で置換されたビフェニル型のロキソプロフェン誘導体(19)のカルボン酸末端を、例えば1,2−ジクロロエタン中、4−アミノジメチルピリジン(4−DMAP)の存在下、メタノールまたはベンジルアルコール(BnOH)によりカルボキシル基をエステル化(第i工程)でエステル保護した化合物(10c、11c、12)を合成した。
【0047】
次に、これらの化合物のカルボニル基を、例えば溶媒としてジクロロメタンを用い、水素化ホウ素ナトリウムによる還元反応(第ii工程)によりアルコール化合物(36a,c;37a,c;38;39)と変換し、最終的にエステル基末端を接触還元または加水分解(第iii又はiv工程)することによって目的とする還元型ロキソプロフェン誘導体(40a,c;41a,c;42;43)を得た。
この場合の接触還元または加水分解反応は、製造スキーム2における第iv又はv工程の方法と同様の手段採用される。
【0048】
なお、これらの化合物とその前駆体は、化学構造中におけるシクロペンタン環の2箇所(1位及び2位)において不斉炭素で区別されるシスまたはトランスの幾何異性体をシリカゲルクロマトグラフィーによって分離し、それぞれのα位不斉炭素で区別される立体異性体を含めた、4つのジアステレオアイソマーの混合物として単離することができる。
【0049】
以上に記載した製造方法により、本願発明の目的化合物であるロキソプロフェン誘導体を得ることができるが、得られた化合物の物理化学的性質を下記表1〜3にまとめて示した。
【0050】
【表1】
【0051】
【表2】
【0052】
【表3】
【0053】
本発明が提供する上記のロキソプロフェン誘導体は、遊離カルボン酸のまま、或いはその薬理学的に許容される塩として使用することができる。
薬理学的に許容される塩としては、ナトリウム塩、カリウム塩などのアルカリ金属塩或いはアンモニウム塩を挙げることができる。
【0054】
本発明が提供するロキソプロフェン誘導体またはその薬理学的に許容される塩を医薬組成物として投与する場合、例えば、当該誘導体またはその薬学的に許容される塩である有効成分を単独、または慣用の賦形剤と共にカプセル剤、錠剤、注射剤等の適宜な剤形として、経口的または非経口的に投与することができる。具体的には、例えば、カプセル剤は、ロキソプロフェン誘導体またはその塩を乳糖、澱粉またはその誘導体、セルロース誘導体等の賦形剤と混合してゼラチンカプセルに充填し調製することができる。
【0055】
また、錠剤は、上記賦形剤の他に、カルボキシメチルセルロースナトリウム、アルギン酸、アラビアゴム等の結合剤と水を加えて練合し、必要により顆粒とした後、さらにタルク、ステアリン酸等の潤滑剤を添加して、通常の圧縮打錠機を用いて錠剤に調製することができる。
【0056】
さらに、注射による非経口投与に際しては、ロキソプロフェン誘導体またはその塩を溶解補助剤と共に滅菌蒸留水または滅菌生理食塩水に溶解し、アンプルに封入して注射用製剤とする。必要により安定化剤、緩衝物質等を含有させてもよい。これらの非経口投与製剤は、静脈内投与、あるいは点滴静注により投与することができる。
【0057】
本発明が提供するロキソプロフェン誘導体の投与量は、種々の要因、例えば治療すべき患者の症状、重症度、年齢、合併症の有無等によって一概には限定できない。
また、投与経路、剤形、投与回数等によっても異なるものであるが、一般的には、経口投与の場合は、有効成分として、通常、0.1〜1000mg/日/ヒト、好ましくは1〜500mg/日/ヒトの範囲内、また、非経口投与の場合は、経口投与の場合における投与量の約1/100〜1/2量程度の範囲内で適宜選別し、投与することができる。なお、これらの投与量は、患者の年齢、症状等により適宜増減することが可能であることは勿論である。
【実施例】
【0058】
以下、実施例に代わる試験例に基づいて、本発明を更に具体的に説明するが、本発明の範囲をこれらの例に限定するものではないことはいうまでもない。
【0059】
インビトロ(in vitro)及びインビボ(in vivo)試験で用いた本発明のロキソプロフェン誘導体について、その化学構造を以下に示した。
【0060】
【化11】
【0061】
【表4】
【0062】
試験例1:ヒト全血アッセイ(インビトロ)
試験は、Inflamm. Res., 45: 68-74 (1996) に記載される方法に準じて行った。
A:インビトロにおけるCOX−1アッセイ
採血対象者は、少なくとも1週間以上にNSAIDsを服用しておらず、採取日に健康である人を選択した。
血液は、血液凝固抑制剤無添加で採取し、直後にアッセイに用いた。採取した血液を500μLずつチューブ(Protein Lobingdin tube, Eppenduf Co. LTD., Tokyo, Janan)に分注し、適切な溶媒(DMSO又はMilliQ water)に溶解させた試験化合物2μL(最終濃度:0.1μM−1000μM)を添加し、37℃/24時間血液凝固が認められるまでインキュベーションした。
インキュベーション後、サンプルを12,000×g/5分間の遠心分離を行い、血清を分離した。血中タンパクを除外するため、得られた血清100μLをエタノール400μLに添加し、再び12,000×g/5分間の遠心分離を行った。上清中のTXB2を酵素免疫測定法(EIA)kit[Cayman (Ann, Arbor, MI,USA)#519031]を用いて定量した。プロトコルは、付属のプロトコルに従った。
【0063】
B:インビトロにおけるCOX−2アッセイ
採血対象者は、少なくとも1週間以上にNSAIDsを服用しておらず、採取日に健康である人を選択した。
血液は、ヘパリン処理が施された試験管(Venojectll blood collection tubes、テルモ社製)に採取し、炎症性刺激物質であるリポポリサッカロイド(LPS)[Sigma-Aldrich Japan Inc,#L2880 from E. coli055:B5、最終濃度が100μg/mLとなるように燐酸緩衝生理食塩水(PBS)で希釈した]を添加した。500μLずつチューブに分注し、適切な溶媒(DMSO又はMilliQ water)に溶解させた試験化合物2μL(最終濃度:0.1μM−1000μM)を添加し、COX−2を誘導するために37℃/24時間インキュベーションした。
インキュベーション後、サンプルを12,000×g/5分間の遠心分離を行い、血清を分離した。血中タンパクを除外するため、得られた血清100μLをエタノール400μLに添加し、再び12,000×g/5分間の遠心分離を行った。上清中のPGE2を酵素免疫測定法(EIA)kit[Cayman (Ann, Arbor, MI,USA)#514040]を用いて定量した。プロトコルは、付属のプロトコルに従った。
【0064】
試験例2:腎障害アッセイ(インビボ)
試験は、Biochem. Biophysical. Res. Com., 323; 1032-39 (2004) に記載の方法に従った。
A:リポソーム膜調製法
卵黄ホスファチジルコリン(10μM、7.7mg:関東化学社製)をクロロホルム/メタノール(1:2、v/v)に溶解し、乾燥後、1.5mLのジエチルエーテルに溶解した。100mM calcein-NaOH(pH7.4)を1mL加え、1分間超音波処理した後、ジエチルエーテルをconventional rotary evaporator(25℃)で除去し、リン酸緩衝液に懸濁したものをリポソーム膜溶液とした(reversed-phase evaporation法)。
【0065】
B:膜傷害測定法
(1)水を溶剤として用いる測定方法
30μLのリポソーム膜溶液を、5mLのリン酸緩衝液に懸濁させ、遠心(10,500×g/20分間)し、リン酸緩衝液で2回洗浄した。ペレットを1mLのリン酸緩衝液に懸濁し、その懸濁液を膜傷害実験に用いた。
1.5mLチューブに6μLずつ分注し、そのリポソーム懸濁液に、濃度が異なるテストサンプルのリン酸緩衝液6μLを加え、30℃/10分間インキュベーションした。氷上で冷却後、384穴wellに混合溶液を10μL分注した後、漏出したcalceinの蛍光強度を測定した(励起波長:490nm)。
【0066】
(2)DMSOを溶剤として用いる測定方法
リポソーム懸濁液を遠心(10,500×g/20分間)し、リン酸緩衝液で2回洗浄した。ペレットを5mLのリン酸緩衝液に懸濁し、その懸濁液の0.75mLを50mLのリン酸緩衝液で希釈したものを膜傷害実験に用いた。
1.5mLチューブに400μLずつ分注し、そのリポソーム懸濁液に、濃度が異なるテストサンプルのDMSO溶液を加え、30℃/10分間インキュベーションした。氷上で冷却後、96穴wellに混合溶液を200μL分注した後に、漏出したcalceinの蛍光強度を測定した(励起波長:490nm)。
【0067】
C:膜傷害性の評価
100%膜傷害のコントロールとして、triton X-100の25%溶液を用い、各テストサンプルの蛍光強度を測定後、100%膜傷害のコントロールの値に対する割合を百分率で表したものを膜傷害(calcein release)とした。
【0068】
これらの結果をまとめて下記表に示した。
【0069】
【表5】
【0070】
試験例3:動物アッセイ(インビボ試験)
A:ロキソプロフェン誘導体による胃潰瘍の形成
試験は、Biochem. Pharmacol., 67; 575-85 (2004) に記載の方法に従った。
Wister系雄性ラット(体重:180-200g)を18時間絶食させ、試験化合物を経口投与した(投与量は、ロキソプロフェンと等しい物質量となるようにした)。8時間後、胃を摘出し、胃内部に生じた潰瘍の面積を測定した。全ての潰瘍の面積を合計し、潰瘍係数とした。
また、胃粘膜のPGE2量は、ELISA法により測定した。
なお、プロトコルは、キット付属のプロトコルに従った。
【0071】
それらの結果を図1〜図3に示した。
図1にロキソプロフェン40mg/kg量に相当する試験化合物を投与した場合の結果を示し、図2にロキソプロフェン50mg/kg量に相当する試験化合物を投与した場合の結果を示し、図3に胃粘膜PGE2量の変化を示した。
図中に示した結果からも判明するように、本発明のロキソプロフェン誘導体(化合物1〜4)は、ロキソプロフェンに比較して顕著に潰瘍係数が低く、化合物3及び4では殆ど潰瘍の形成が認められないものであった。
また、胃粘膜のPGE2量は、化合物2、3及び4において、ロキソプロフェンより高いものであった。
【0072】
B:カラゲニン惹起性浮腫に対するロキソプロフェン誘導体の効果
試験は、Br. J. Pharmacol., 151; 285-91 (2007) に記載の方法に従った。
Wister系雄性ラット(体重:180-200g)を18時間絶食させ、試験化合物を経口投与した(投与量は、ロキソプロフェンと等しい物質量となるようにした)。1時間後、左足蹠皮下に1%カラゲニン(生理食塩水に溶解)100μLを注射し、浮腫を惹起させた。
カラゲニン投与前、投与後3時間及び6時間後の足容積を、Plethysmometerを用いて測定した。
また、足浮腫PGE2量は、ELISA法により測定した。なお、プロトコルは、キット付属のプロトコルに従った。
【0073】
それらの結果を図4〜図6に示した。
図4にロキソプロフェン10mg/kg量に相当する試験化合物を投与した6時間後におけるカラゲニン浮腫(%)を示し、図5に浮腫抑制率(%)を示した。
なお、浮腫抑制率は、以下の計算による。
抑制率(%)=100−(化合物投与時の浮腫容積/Vehicle投与時の浮腫容積)×100
また、図6に、投与後3時間及び6時間後の足容積の変化を示した。
【0074】
これらの結果からも判明するように、本発明のロキソプロフェン誘導体(化合物1〜4)は、良好な消炎作用を示すものであると共に、副作用である潰瘍形成を生じないものであり、薬理作用と副作用の分離が良好に行われた化合物であることが判明する。
【0075】
製剤例1:錠剤
化合物1 50mg
乳糖 100mg
ヒドロキシプロピルセルロース 150mg
ステアリン酸マグネシウム 50mg
上記処方を基本とし、顆粒を調製後、打錠し重量350mgの錠剤を、常法により調製した。
【0076】
製剤例2:顆粒剤
化合物2 50mg
乳糖 100mg
トウモロコシデンプンルロース 150mg
上記処方を基本とし、200mg顆粒中有効成分50mg含有の顆粒を常法により調製した。
【産業上の利用可能性】
【0077】
以上記載のように、本発明が提供するロキソプロフェン誘導体は、これまで知られていない新規な化合物であると共に、従来のNSAIDsにみられた胃腸障害等の副作用がなく、その上、臨床的に使用されているロキソプロフェンより抗炎症、鎮痛作用が強いものである。
したがって、その安全域が大きいことから、ヒトに対して安全に使用できる点で、極めて有効なものであり、産業上の貢献度は多大なものである。
【図面の簡単な説明】
【0078】
【図1】試験例3における、ロキソプロフェン40mg/kg相当量の化合物量を投与した場合の潰瘍係数の結果を示した図である。
【図2】試験例3における、ロキソプロフェン50mg/kg相当量の化合物量を投与した場合の潰瘍係数の結果を示した図である。
【図3】試験例3における、ロキソプロフェン40mg/kg相当量の化合物量を投与した場合の胃粘膜PGE2量の変化を示した図である。
【図4】試験例3における、ロキソプロフェン10mg/kg相当量の試験化合物を投与した6時間後におけるカラゲニン浮腫(%)を示した図である。
【図5】試験例3における、ロキソプロフェン10mg/kg相当量の試験化合物を投与した6時間後におけるカラゲニン浮腫抑制率(%)を示した図である。
【図6】試験例3における、ロキソプロフェン10mg/kg相当量の試験化合物を投与した場合の、投与後3時間及び6時間後の足容積の変化を示した図である。
【特許請求の範囲】
【請求項1】
次式(I)又は(II):
【化1】
(式中、R1及びR2はハロゲン原子又は置換若しくは非置換フェニル基を表す)
で示されるロキソプロフェン誘導体又はその薬理学的に許容される塩。
【請求項2】
式(I)及び(II)中、R1及びR2のハロゲン原子が塩素原子、臭素原子、フッ素原子、ヨウ素原子から選択されるものである請求項1に記載のロキソプロフェン誘導体又はその薬理学的に許容される塩。
【請求項3】
式(I)中、R1の置換フェニル基における置換基が、ハロゲン原子、水酸基、置換若しくは非置換低級アルキル基、低級アルキルチオ基、低級アルコキシ基、ニトロ基、アミノ基又はカルボキシル基である請求項1に記載のロキソプロフェン誘導体又はその薬理学的に許容される塩。
【請求項4】
式(II)中、R2の置換フェニル基における置換基が、ハロゲン原子又は置換低級アルキル基である請求項1に記載のロキソプロフェン誘導体又はその薬理学的に許容される塩。
【請求項5】
請求項1に記載の式(I)又は(II)で示されるロキソプロフェン誘導体又はその薬理学的に許容される塩を有効成分として含有する医薬。
【請求項6】
請求項2〜4のいずれかに記載のロキソプロフェン誘導体又はその薬理学的に許容される塩を有効成分として含有する医薬。
【請求項1】
次式(I)又は(II):
【化1】
(式中、R1及びR2はハロゲン原子又は置換若しくは非置換フェニル基を表す)
で示されるロキソプロフェン誘導体又はその薬理学的に許容される塩。
【請求項2】
式(I)及び(II)中、R1及びR2のハロゲン原子が塩素原子、臭素原子、フッ素原子、ヨウ素原子から選択されるものである請求項1に記載のロキソプロフェン誘導体又はその薬理学的に許容される塩。
【請求項3】
式(I)中、R1の置換フェニル基における置換基が、ハロゲン原子、水酸基、置換若しくは非置換低級アルキル基、低級アルキルチオ基、低級アルコキシ基、ニトロ基、アミノ基又はカルボキシル基である請求項1に記載のロキソプロフェン誘導体又はその薬理学的に許容される塩。
【請求項4】
式(II)中、R2の置換フェニル基における置換基が、ハロゲン原子又は置換低級アルキル基である請求項1に記載のロキソプロフェン誘導体又はその薬理学的に許容される塩。
【請求項5】
請求項1に記載の式(I)又は(II)で示されるロキソプロフェン誘導体又はその薬理学的に許容される塩を有効成分として含有する医薬。
【請求項6】
請求項2〜4のいずれかに記載のロキソプロフェン誘導体又はその薬理学的に許容される塩を有効成分として含有する医薬。
【図1】

【図2】

【図3】
【図4】
【図5】

【図6】

【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2010−195727(P2010−195727A)
【公開日】平成22年9月9日(2010.9.9)
【国際特許分類】
【出願番号】特願2009−43801(P2009−43801)
【出願日】平成21年2月26日(2009.2.26)
【出願人】(504159235)国立大学法人 熊本大学 (314)
【出願人】(303010452)株式会社LTTバイオファーマ (27)
【Fターム(参考)】
【公開日】平成22年9月9日(2010.9.9)
【国際特許分類】
【出願日】平成21年2月26日(2009.2.26)
【出願人】(504159235)国立大学法人 熊本大学 (314)
【出願人】(303010452)株式会社LTTバイオファーマ (27)
【Fターム(参考)】
[ Back to top ]