
ワクチン組成物
本発明は、HIVポリペプチドをコードするオリゴヌクレオチドを含むウイルスベクターに関し、特にこのウイルスベクターはアデノウイルスである。さらに特定すると、このようなアデノウイルスは、非ヒト霊長類アデノウイルス、例えばサルのアデノウイルス、特にチンパンジーのアデノウイルスである。本発明は特に、複数の異なるHIV抗原、例えば2種もしくは3種またはそれ以上のHIV抗原をコードするHIVポリヌクレオチド配列を含有するアデノウイルスベクターに関する。本発明はさらに、上記ウイルスベクターの作製方法、この方法で作製されたウイルスベクター、ならびに医療、特に予防上または治療上のワクチン接種における上記ベクターの使用に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、HIVポリペプチドをコードするオリゴヌクレオチドを含むウイルスベクター、特にこのウイルスベクターがアデノウイルスであるウイルスベクターに関する。特にこのようなアデノウイルスは、非ヒト霊長類のアデノウイルス、例えばサルのアデノウイルス、特にチンパンジーのアデノウイルスである。特に本発明は、複数の異なるHIV抗原、例えば2種もしくは3種またはそれ以上のHIV抗原をコードするHIVポリヌクレオチド配列を含有するアデノウイルスベクターに関する。本発明はさらに、上記ウイルスベクターの作製方法、この方法で作製されたウイルスベクター、および医療、特に予防用または治療用ワクチン接種におけるこれらのベクターの使用に関する。
【背景技術】
【0002】
HIV-1は、世界の主要な健康問題の1つであると見なされている後天性免疫不全症候群(AIDS)の主な原因である。世界中で広範囲の研究が行われているが、ワクチンを製造しようとする取り組みは今のところ成功していない。
【0003】
HIV-1は、レトロウイルス科のRNAウイルスである。HIVゲノムは少なくとも9種類のタンパク質をコードし、これらは以下の3つのクラスに分けられる:主要な構造タンパク質Gag、PolおよびEnv、調節タンパク質TatおよびRev、ならびにアクセサリータンパク質Vpu、Vpr、VifおよびNef。このHIVゲノムは、全てのレトロウイルスに共通な5'LTR-gag-pol-env-LTR3'の構成を示す。
【0004】
アデノウイルスは、約36 kbのゲノムサイズを有する二本鎖DNAウイルスであり、その様々な標的組織において高効率の遺伝子導入を達成する能力と、大きなトランスジーンの収容力のために、遺伝子導入の用途に広く使用されている。従来、アデノウイルスのE1遺伝子は、欠失されるか、または好適なプロモーター、目的遺伝子のcDNA配列およびポリAシグナルからなる導入遺伝子カセットで置換され、その結果として複製欠損型の組換えウイルスをもたらす。
【0005】
アデノウイルスは、3つの主要タンパク質、ヘキソン(II)、ペントンベース(III)およびこぶ状のファイバー(IV)と、いくつかの他のマイナーなタンパク質VI、VIII、IX、IIIaおよびIVa2とからなる正二十面体のキャプシドをもつ特徴的形態を有する(Russell W.C. 2000, Gen Virol, 81:2573-2604)。このウイルスゲノムは、逆方向末端反復配列(ITR)を有する5'末端に末端タンパク質が共有結合で結合している、線状の二本鎖DNAである。このウイルスDNAは、高塩基性タンパク質VIIおよびmuと呼ばれる小型のペプチドと密接に会合している。もう1つのタンパク質Vは、このDNA-タンパク質複合体と共にパッケージングされ、タンパク質VIを介してキャプシドとの構造的連結を与える。このウイルスはまた、構造タンパク質の一部をプロセシングして、成熟した感染性ウイルスを生成するのに必要とされる、ウイルスにコードされたプロテアーゼを含む。
【0006】
様々な哺乳動物種に感染する、100を越える異なった血清型のアデノウイルスが分離されているが、そのうち51の血清型はヒト起源である。このようなヒト起源からのアデノウイルスの例は、Ad1、Ad2、Ad4、Ad5、Ad6、Ad11、Ad24、Ad34、Ad35である。上記のヒト血清型は、いくつかの生物学的、化学的、免疫学的および構造的基準に基づいて、6つの亜属(A〜F)に分類されている[WO 04018627、第1頁]。
【0007】
Ad5ベースのベクターが多くの遺伝子治療試験で広く用いられているが、一般集団には自然感染による既存の免疫が存在するため、Ad5と他のグループCのアデノウイルスベクターの使用には限界があるだろう。Ad5と他のグループCのメンバーは、どちらかといえば、最も優勢な血清型に入る。存在するベクターに対する免疫は、治療中に該ベクターにさらされた結果として発生し得る。優勢血清型のベクターに対する、こうした種類の既存のまたは発生した免疫は、遺伝子治療またはワクチン接種の努力の成果を制限する可能性がある。したがって、これらに代わるアデノウイルス血清型は、宿主の免疫応答を回避し得る遺伝子送達系の研究において極めて重要な目標となる。
【0008】
このような代替血清型の1分野は、非ヒト霊長類、特にチンパンジーのアデノウイルスである。2種類のチンパンジーアデノウイルスのゲノムについて記載している米国特許第6,083,716号を参照されたい。
【0009】
チンパンジー(「Pan」または「C」)アデノウイルスベクターは、ヒトアデノウイルスベクターと同程度に効率的に、トランスジーン産物に対して強い免疫応答を惹起することが示されている(Fitzgeraldら J. Immunol. 170:1416)。
【0010】
HIVのTatおよびNefタンパク質は初期タンパク質である。すなわち、これらのタンパク質は、感染の初期に、構造タンパク質の非存在下で発現する。
【0011】
Nef遺伝子は、幾つかの活性を有することが示されている初期アクセサリーHIVタンパク質をコードする。例えば、Nefタンパク質は、HIV受容体であるCD4の細胞表面からの除去を引き起こすことが知られているが、この機能の生物学的重要性については論議がある。さらに、NefはT細胞のシグナル伝達経路と相互作用して活性状態を誘導し、続いてより効率的な遺伝子発現を促進し得る。一部のHIV分離株はこの領域に変異を有し、そのためこれらの分離株は機能性タンパク質をコードすることができず、in vivoでのこれらの複製および病原性が大いに損なわれる。
【0012】
Gag遺伝子は、完全長RNAから翻訳されて前駆体ポリタンパク質を生成し、その後この前駆体タンパク質は、3〜5個のキャプシドタンパク質、すなわち、マトリックスタンパク質、キャプシドタンパク質および核酸結合タンパク質、ならびにプロテアーゼに切断される (Fundamental Virology, Fields BN, Knipe DMおよびHowley M 1996 2. Fields Virology vol 2 1996)。
【0013】
Gag遺伝子は、p55とも呼ばれる55キロダルトン(kD)のGag前駆体タンパク質を生じさせるが、このタンパク質は、スプライシングされていないウイルスmRNAから発現される。翻訳の間、p55のN末端はミリストイル化されて、細胞膜の細胞質側に結合するようになる。膜結合したGagポリタンパク質は、ウイルスゲノムRNAの2つのコピーを動員し、他のウイルス性および細胞性タンパク質と一緒になって、感染細胞の表面からの該ウイルス粒子の出芽を引き起こす。出芽後、p55はウイルスにコードされたプロテアーゼ(Pol遺伝子の産物)によってMA(マトリックス[p17])、CA(キャプシド[p24])、NC(ヌクレオキャプシド[p9])、およびp6(4)と呼ばれる4つのより小さなタンパク質へとウイルスの成熟過程で切断される。
【0014】
3つの主要なGagタンパク質(p17、p24およびp9)に加えて、全てのGag前駆体は幾つかの他の領域を含み、これらは切り出されて、様々なサイズのペプチドとしてビリオン(ウイルス粒子)中に残る。これらのタンパク質は多様な役割を有し、例えばp2タンパク質は、プロテアーゼ活性の調節において提案された役割を果たし、タンパク質分解プロセシングの正確なタイミングに寄与する。
【0015】
MAポリペプチドはp55のミリストイル化されたN末端に由来する。大部分のMA分子は、ビリオンの脂質二重層の内表面に結合した状態で存在し、この粒子を安定化させる。MAのサブセットは、ビリオンのより深い層の内部に動員されて、そこでウイルスDNAを核につなげる複合体の一部となる。これらのMA分子は、MA上の核親和性シグナルが細胞性核内輸送機構によって認識されるため、ウイルスゲノムの核輸送を容易にする。この現象は、HIVが非分裂細胞に感染することを可能とするが、レトロウイルスには珍しい特性である。
【0016】
p24(CA)タンパク質は、ウイルス粒子の円錐形のコア部を形成する。シクロフィリンAは、p55のp24領域と相互作用し、HIV粒子へのその取込みをもたらすことが実証されている。GagとシクロフィリンAとの相互作用は不可欠である。なぜなら、シクロスポリンAによる上記の相互作用の破壊がウイルス複製を阻害するからである。
【0017】
GagのNC領域は、いわゆるHIVのパッケージングシグナルを特異的に認識することに関与する。このパッケージングシグナルは、ウイルスRNAの5'末端の近くに位置する4つのステムループ構造からなり、HIV-1ウイルス粒子への異種RNAの取込みを媒介するのに十分である。NCは、2つのジンクフィンガーモチーフにより媒介される相互作用を通じてパッケージングシグナルと結合する。NCは逆転写も促進する。
【0018】
p6ポリペプチド領域は、p55 Gagとアクセサリータンパク質Vprとの相互作用を媒介し、組み立てつつあるビリオンへのVprの取込みをもたらす。p6領域はまた、感染細胞からの出芽ビリオンの効率的な放出に必要とされる、いわゆる後期ドメインを含む。
【0019】
Pol遺伝子は、初期感染においてウイルスが必要とする活性を有する3種類のタンパク質、逆転写酵素RT、プロテアーゼ、および細胞DNAへのウイルスDNAの組込みに必要とされるインテグラーゼタンパク質をコードする。Polの一次産物は、ビリオンのプロテアーゼによって切断されて、DNA合成に必要な活性を含むアミノ末端RTペプチド(RNAおよびDNA特異的DNAポリメラーゼ、リボヌクレアーゼH)およびカルボキシ末端インテグラーゼタンパク質をもたらす。HIVのRTは完全長RT(p66)のヘテロ二量体であり、分解産物(p51)はカルボキシ末端のRNアーゼインテグラーゼドメインを欠いている。
【0020】
RTは、レトロウイルスゲノムによりコードされるタンパク質のうち最も高度に保存されたタンパク質の1つである。RTの2つの主要な活性は、DNAポリメラーゼとリボヌクレアーゼHである。RTのDNAポリメラーゼ活性は、RNAとDNAを鋳型として交互に使用し、公知の全てのDNAポリメラーゼと同様に、新生DNA合成を開始することはできず、プライマーとしての役割を果たす既存の分子(RNA)を必要とする。
【0021】
全てのRTタンパク質に固有のRNアーゼH活性は、複製の初期に、DNA合成が進むにつれてRNAゲノムを除去するという重要な役割を果たす。RNアーゼHは、全てのRNA-DNAハイブリッド分子からRNAを選択的に分解する。構造的に、DNAポリメラーゼとリボヌクレアーゼHは、Pol内で別個の重複しないドメインを占めており、Polのアミノ末端3分の2の範囲に及ぶ。
【0022】
p66触媒サブユニットは、5つの異なるサブドメインへと折り畳まれる。これらのアミノ末端23は、RT活性を有する部分を含む。これらのカルボキシ末端は、RNアーゼ Hドメインである。
【0023】
宿主細胞への感染後、レトロウイルスRNAゲノムは、感染粒子中に存在する逆転写酵素により線状二本鎖DNAへとコピーされる。インテグラーゼ(Skalka AM'99 Adv in Virus Res 52 271-273に概説)はウイルスDNAの両端を認識し、これらを切り取って、宿主の染色体部位にウイルスDNAを連れて行き、組込みを触媒する。宿主DNAの多くの部位が組込みの標的部位となり得る。上記のインテグラーゼはin vitro組込みを触媒するのに十分であるが、これは、in vivoではウイルスDNAと関連した唯一のタンパク質ではない。感染細胞から単離される大型のタンパク質-ウイルスDNA複合体は、プレインテグレーション複合体(preintegration complex)を表している。この複合体は、子孫ウイルスゲノムによる宿主細胞遺伝子の獲得を促進する。
【0024】
上記のインテグラーゼは、N末端ドメイン、触媒コアおよびC末端ドメインの3つの異なるドメインで構成されている。触媒コアドメインは、ポリヌクレオチド移行の化学作用の必要条件を全て備えている。
【0025】
複数の外来遺伝子を含むウイルスベクター、特にアデノウイルスベクターは、常に簡単に製造できるわけではない。ベクターの安定性の問題、および挿入された遺伝子の効率のよい発現を得ることの困難性が存在しうる。特に、ワクチンにおいて使用し得る2つ以上または3つ以上のHIVポリヌクレオチドを含むアデノウイルスの製造は、これまで成功を収めていない。
【0026】
非ヒト霊長類アデノウイルスは、チンパンジーの腸間膜リンパ節から単離することができる。チンパンジーアデノウイルスはヒトC亜型アデノウイルスと十分に類似しており、HEK293細胞でのE1欠失ウイルスの複製が可能である。しかしチンパンジーアデノウイルスは、より一般的なヒト血清型(Ad2およびAd5)とは系統学的に区別される。Pan 6は、Pan5、7および9とはそれほど近縁でなく、血清学的に区別される。
【0027】
異種DNAをアデノウイルスに挿入することに関連して、一定のサイズ制限がある。ヒトアデノウイルスは、野生型ゲノム長の105%までパッケージする能力を有する(Bettら 1993, J Virol 67 (10), 5911-21)。ヒトアデノウイルスのパッケージングの下限値は、野生型ゲノム長の75%であることが示されている(Parksら 1995, J Virol 71(4), 3293-8)。
【0028】
HIVに対して有効なワクチンを見出す必要性が依然として存在する。
【発明の開示】
【0029】
本発明は、1つ以上の領域が欠失されたアデノウイルスベクターであって、該ベクターが少なくとも3つのHIV抗原またはこれらの免疫原性誘導体もしくは免疫原性断片をコードするポリヌクレオチド(1つまたは複数)を含み、ここで該ベクターは哺乳動物の宿主において上記の抗原または断片もしくは誘導体を発現することが可能であり、欠失のサイズおよびHIVポリヌクレオチド(1つまたは複数)のサイズは、ベクターゲノムの全長が野生型ウイルスのゲノム長の85〜105%となるようなサイズである、上記アデノウイルスベクターを提供する。
【0030】
本発明の1つの実施形態において、ポリヌクレオチド(1つまたは複数)によりコードされるHIV抗原は、Gag、NefおよびPolであり得る。他の実施形態において、Polは、RT部分のみを含むものでよい。本発明のさらに別の実施形態において、HIV抗原をコードするポリヌクレオチド(1つまたは複数)は、Gag、RT、Nefの順に転写されるように、すなわちGag部分が、結果として生じる融合タンパク質のN末端に位置するように、配置することができる。
【0031】
全ベクターゲノムのサイズは、例えば野生型ウイルスゲノムのサイズの90〜100%、または野生型ゲノムサイズの95〜100%であってよい。1つの実施形態において、ベクターの全サイズは、野生型ウイルスゲノムのサイズの約96%でありうる。
【0032】
本発明のアデノウイルスベクターに挿入するための特定のHIV抗原は、Pol、NefおよびGagまたはこれらの免疫原性誘導体もしくは免疫原性断片である。
【0033】
このようなアデノウイルスベクターを、製薬上許容される賦形剤、担体、希釈剤またはアジュバントと共に製剤化し、HIV感染およびAIDSの治療および/または予防に適した医薬組成物またはワクチン組成物を含む免疫原性組成物を製造することができる。
【0034】
本発明において用いられるのは、ヒト集団中で一般的な天然に存在する血清型(例えばAd2およびAd5)とは区別されるアデノウイルスである。このウイルスは、該ベクターに対する強い免疫応答の誘導を回避するが、かかる免疫応答は、中和抗体を介してベクターの取込みを阻害し、毒性を与えることにより、その後の同じ血清型の投与の有効性を制限する。
【0035】
従って、本発明のアデノウイルスは、一般的な天然に存在するヒトウイルス血清型ではないアデノウイルスとすることができる。動物から分離されるアデノウイルスは、免疫学的に区別されるキャプシド、ヘキソン、ペントンおよびファイバー成分を有するが、系統学的には密接な関係がある。特に本発明のウイルスは、非ヒトアデノウイルス、例えばサルアデノウイルス、特にPan 5、6、7または9などのチンパンジーアデノウイルスであってよい。このようなウイルス株の例はWO 03/000283に記載されており、American Type Culture Collection (10801 University Boulevard, Manassas, Virginia 20110-2209)、および他の供給元から入手可能である。望ましいチンパンジーアデノウイルス株は、Pan 5 [ATCC VR-591]、Pan 6 [ATCC VR-592]、およびPan 7 [ATCC VR-593]である。他の好適なアデノウイルスとしては、限定するものではないが、米国特許第6,083,716号に記載されるチンパンジーアデノウイルスC1とC68 (Pan9)、サルアデノウイルス、例えば、限定するものではないが、SV1 [VR-195]、SV25 [SV-201]、SV35、SV15、SV-34、SV-36、SV-37、およびヒヒアデノウイルス[VR-275]が挙げられる。Pan 5 (C5とも呼ばれる)、Pan 6 (C6とも呼ばれる)、Pan 7 (C7とも呼ばれる)、SV1、SV25、およびSV39の配列は文献に記載されている[WO 03/046124、2003年6月5日公開]。ハイブリッドアデノウイルスベクターおよびサルアデノウイルスSA18から構築されたベクターが記載されている国際公開番号WO 04/16614も参照されたい。
【0036】
チンパンジーアデノウイルスは、標的集団中にアデノウイルスに対する既存の免疫(特に交差中和抗体)が存在しないため、ヒトアデノウイルス血清型より有利であると考えられる。チンパンジーアデノウイルスと既存の中和抗体応答との交差反応は、標的集団の2%に存在するにすぎないのに対して、ある種の候補ヒトアデノウイルスベクターの場合には35%に存在する。チンパンジーアデノウイルスは、より一般的なヒトアデノウイルスのサブタイプAd2およびAd5とは明確に区別されるが、一般的なサブタイプではないヒトアデノウイルスのサブグループEのAd4とはより密接な関係がある。Pan 6はPan 5、7および9とそれほど密接な関係はない。
【0037】
本発明のアデノウイルスは複製欠損型であってよい。これは、該ウイルスが野生型ウイルスと比較して、非補完細胞(non-complementing cell)における複製能力が低下していることを意味する。この複製能の低下は、ウイルスを変異させることにより、例えば複製に関与する遺伝子を欠失させること(例えば、E1a、E1b、E3またはE4遺伝子の欠失)により、もたらされ得る。
【0038】
本発明によるアデノウイルスベクターは、機能的E1欠失を含む複製欠損型アデノウイルスであってよい。このように、本発明によるアデノウイルスベクターは、アデノウイルスのE1aとE1bを発現する能力の欠如のため、すなわちE1aとE1bが機能的に欠失されているため、複製欠損型でありうる。上記の組換えアデノウイルスはまた、他の遺伝子に機能的欠失を有していてもよく(WO 03/000283を参照)、例えば、E3またはE4遺伝子に欠失を有する。アデノウイルス遅延型初期遺伝子E3は、組換えウイルスの一部を形成するサルアデノウイルス配列から除去することができる。E3の機能は、組換えアデノウイルス粒子の生産に必要ではない。したがって、本発明において有用な組換えサルアデノウイルスをパッケージングするために、この遺伝子産物の機能をもとに戻すことは不必要である。1つの特定の実施形態において、上記の組換え(サル)アデノウイルスは、E1およびE3遺伝子が機能的に欠失している。このようなベクターの構築は、Royら, Human Gene Therapy 15:519-530, 2004に記載されている。
【0039】
組換えアデノウイルスはまた、E4遺伝子の機能的欠失を有するように構築することもできるが、E4のORF6機能を保持していることが望ましい。本発明によるアデノウイルスベクターは、遅延型初期遺伝子E2aに欠失を含んでいてもよい。欠失は、サルアデノウイルスゲノムの後期遺伝子L1〜L5のいずれかに生じさせることもできる。同様に、中期遺伝子IXおよびIVaの欠失も有用であり得る。
【0040】
他の欠失は、アデノウイルスの他の構造遺伝子または非構造遺伝子に生じさせることができる。こうした欠失は個々に用いることができる。すなわち、本発明において用いるアデノウイルス配列は、E1欠失のみを含み得る。あるいは、遺伝子全体の欠失または生物学的活性を破壊するのに有効な遺伝子の一部の欠失を任意の組み合わせで用いてもよい。例えば、ある代表的なベクターにおいて、アデノウイルス配列は、E1遺伝子とE4遺伝子の欠失、またはE1、E2aおよびE3遺伝子の欠失、またはE1およびE3遺伝子の欠失(例えば、E1aおよびE1bの機能的欠失、ならびにE3の少なくとも一部の欠失)、またはE1、E2aおよびE4遺伝子の欠失(E3遺伝子などの欠失を伴うまたは伴わない)を有し得る。このような欠失は、これらの遺伝子の一部の欠失または完全な欠失であってよく、所望の結果を達成するために、温度感受性変異などの他の変異と組み合わせて用いることができる。
【0041】
本発明のアデノウイルスベクターは、ウイルスが複製し得る好適な細胞株で製造することができる。特に、ウイルスベクターから失われて複製特性の欠陥をもたらす因子を供給する、補完性細胞株を使用することができる。例えば、補完性細胞株は、E1、またはE1およびE3、またはE1、E3およびE4を発現することができる。限定するものではないが、このような細胞株は、とりわけHeLa [ATCC受託番号CCL 2]、A549 [ATCC受託番号CCL 185]、HEK 293、KB [CCL 17]、Detroit [例えば、Detroit 510, CCL 72]およびWI-38 [CCL 75]細胞であってよい。これらの細胞株は全て、American Type Culture Collection (10801 University Boulevard, Manassas, Virginia 20110-2209)から入手することができる。他の好適な親細胞株は他の供給元から入手することが可能であり、例えばPER.C6(登録商標)細胞のような、Centre for Applied Microbiology and Research (CAMR, UK)所在のEuropean Collection of Animal Cell Cultures (ECACC)にECACC番号96022940として寄託された細胞に代表される。
【0042】
別の態様において、本発明は、少なくともHIV抗原RT、NefおよびGagまたはこれらの免疫原性誘導体もしくは免疫原性断片を、Gag、RT、Nefの順にコードするポリヌクレオチド(1つまたは複数)を含むアデノウイルスベクター、すなわち、Gag、RT、Nefの順に転写されるように配置された少なくともHIV抗原RT、NefおよびGagまたはこれらの免疫原性誘導体もしくは免疫原性断片をコードするポリヌクレオチド(1つまたは複数)を含むアデノウイルスベクターを提供する。
【0043】
例えば、本発明のアデノウイルスベクターは、Gagまたはその免疫原性誘導体もしくは免疫原性断片がRTまたはその免疫原性誘導体もしくは免疫原性断片をコードするポリヌクレオチド配列に融合し、これがNefまたはその免疫原性誘導体もしくは免疫原性断片をコードするポリヌクレオチド配列に融合しているポリヌクレオチドを、単一の異種プロモーターの制御下に含有することができ、その際、該遺伝子のGag部分は該ポリヌクレオチドの5'末端に存在する。
【0044】
本発明の別の実施形態において、3つの抗原は各々、それ自身のプロモーターを通じて発現され、該プロモーターは各々同じでも異なっていてもよい。本発明のさらに別の実施形態において、3つの抗原のうち2つは、単一のプロモーターに連結された融合体を形成し、第3の抗原は、第1のプロモーターと同一でも異なっていてもよい第2のプロモーターに連結される。例えば、GagとRTが第1のプロモーターに連結され、Nefが第2のプロモーターに連結されていてもよい。
【0045】
少なくとも3つのHIV抗原またはこれらの免疫原性誘導体もしくは免疫原性断片をコードするポリヌクレオチド(1つまたは複数)を、アデノ(Adeno)欠失領域のいずれか、例えばE1欠失領域に挿入することができる。
【0046】
抗原をコードする2つ以上のポリヌクレオチドを1つの融合体として連結することができるが、結果として生じるタンパク質は、融合タンパク質として発現させてもよいし、または別個のタンパク質産物として発現させてもよいし、あるいは融合タンパク質として発現させた後、より小さなサブユニットに分解してもよい。
【0047】
1つの態様において、本発明は、本発明のベクターによって発現される融合タンパク質、例えば、ヒト体内で産生される融合タンパク質を提供する。
【0048】
本発明によるベクターに含まれる、例えばNef、GagまたはRTをコードする、1つ以上のHIV配列は、例えば、高発現されたヒト遺伝子にコドン使用が似るように、哺乳動物細胞用にコドンを最適化することができる。これらのHIV配列のコドン最適化は、WO 03/025003にさらに記載されている。
【0049】
例えば、本発明によるアデノウイルスベクター中でGagおよび/またはRTをコードするポリヌクレオチドは、上記のようにコドン最適化することができる。
【0050】
本発明によるアデノウイルスベクター中のGag配列は、Gag p6ポリペプチドをコードする配列を除外し得る。本発明において使用するGag配列の特定の例には、p17および/またはp24コード配列が含まれる。
【0051】
本発明のRT配列は、いずれかの逆転写酵素活性を実質的に不活化する変異をコードし得る。1つの特定の不活化変異は、W(トリプトファン)229によるK(リシン)の置換を含む。WO 03/025003を参照されたい。
【0052】
RT遺伝子は、上記のとおり、HIVゲノム中の大きいPol遺伝子の成分である。本発明によるアデノウイルスベクターに含まれるRTコード配列は、Pol、または少なくともRTをコードするPol断片の状態で存在しうることが理解されるだろう。このようなPol断片は、Polの主要なCTLエピトープを保有する。1つの具体例において、RTは、RTのp51断片のみまたはp66断片のみとして含まれる。
【0053】
場合により、本発明において用いるNef配列は、N末端領域をコードする配列を除去するため、すなわち30〜85アミノ酸、例えば60〜85アミノ酸、特にN末端の65アミノ酸を除去するため、末端切断(トランケーション)されている(本明細書では、後者の切断型をtrNefと呼ぶ)。あるいは、またはさらに、Nefは1つ以上のミリストイル化部位を除去するように改変されていてもよい。例えば、Gly 2ミリストイル化部位を欠失または置換によって除去する。あるいは、またはさらに、Nefは、ロイシン174とロイシン175のジロイシンモチーフを、1個もしくは両方のロイシンの欠失または置換により変更するように改変することができる。CD4下方制御におけるジロイシンモチーフの重要性は、例えば、Bresnahan P.A.ら(1998) Current Biology, 8(22): 1235-8に記載されている。
【0054】
本発明による構築物は、Gag、PolおよびNefを含むことが可能であり、その際、これら天然抗原のCTLエピトープの少なくとも75%、少なくとも90%、または少なくとも95%、例えば96%が存在する。
【0055】
上記のp17/p24 Gag、p66 RT、および末端切断型Nefを含む本発明の構築物においては、天然Gag PolおよびNef抗原のCTLエピトープの96%が存在する。
【0056】
本発明の1つの実施形態は、p17、p24(コドン最適化)Gag、p66 RT(コドン最適化)、末端切断型Nef(末端アミノ酸1-85をコードするヌクレオチドを欠く-「trNef」)をGag、RT、Nefの順にコードするポリヌクレオチド(1つまたは複数)を含むアデノウイルスベクターを提供する。
【0057】
本発明の構築物は、
1. p17、p24(コドン最適化)Gag - p66 RT(コドン最適化) - 末端切断型Nef;
2. 末端切断型Nef - p66 RT(コドン最適化) - p17、p24(コドン最適化)Gag;
3. 末端切断型Nef - p17、p24(コドン最適化)Gag - p66 RT(コドン最適化);
4. p66 RT(コドン最適化) - p17、p24(コドン最適化)Gag - 末端切断型Nef;
5. p66 RT(コドン最適化) - 末端切断型Nef - p17、p24(コドン最適化)Gag;
6. p17、p24(コドン最適化)Gag - 末端切断型Nef - p66 RT(コドン最適化)
を含む。
【0058】
本発明のポリヌクレオチド(1つまたは複数)は、Gag、RTおよびNefをコードする配列の間に存在するリンカー配列を有してもよい。このようなリンカー配列の長さは、例えば、最大20アミノ酸までである。特定の例において、この長さは1〜10アミノ酸、または1〜6アミノ酸、例えば2〜4アミノ酸であってよい。
【0059】
本発明のポリヌクレオチドは、さらなるHIV配列を含んでいてもよい。特に、これらのポリヌクレオチドはHIVのenvタンパク質またはその免疫原性誘導体もしくは免疫原性断片を含み得る。envの好適な形態は、gp120、gp140およびgp160である。他の好適なHIV配列としては、限定するものではないが、Tat、Rev、Vpu、VprおよびVifが挙げられる。このように本発明は、HIVのenvタンパク質またはその免疫原性誘導体もしくは免疫原性断片と共に、HIV抗原RT、NefおよびGagまたはこれらの免疫原性誘導体もしくは免疫原性断片を、Gag、RT、Nefの順にコードするポリヌクレオチド(1つまたは複数)を含むアデノウイルスベクターをさらに提供する。
【0060】
本発明はさらに、本発明のアデノウイルスベクターを、1つ以上のHIV抗原をコードするポリヌクレオチド(1つまたは複数)を含む第2のアデノウイルスベクターと組み合わせて含有する免疫原性組成物を含む。
【0061】
本発明に含まれる全てのHIV配列について、これらの配列は、必ずしも完全長つまり天然のタンパク質をコードする配列に相当しないことが理解されるだろう。末端切断された、あるいは変更された(例えば変異した)タンパク質のような免疫原性誘導体も意図され、少なくとも1つのHIVエピトープ、例えばCTLエピトープ、典型的には少なくとも8アミノ酸のペプチドをコードする断片などの免疫原性誘導体も意図される。長さが少なくとも8アミノ酸、例えば8〜10アミノ酸、または20、50、60、70、100、150もしくは200アミノ酸までの断片をコードするポリヌクレオチドは、コードされるオリゴペプチドまたはポリペプチドがHIV抗原性を示す限り、すなわち主要なCTLエピトープが上記のオリゴペプチドまたはポリペプチドに保持される限り、本発明の範囲に含まれると考えられる。本明細書において、主要なCTLエピトープは、in vivoで免疫応答を誘発することができるエピトープとして定義される。本発明のポリヌクレオチド配列によってコードされるHIVポリペプチド分子は、天然タンパク質の長さの少なくとも50%の断片であってよく、該断片は複数の変異を含み得るが、少なくとも1つのHIVエピトープを保持してHIV抗原性を示すものである。このようなHIV抗原性は、例えば抗体または細胞性応答を測定することにより確認することができる。同様に、本発明の免疫原性誘導体はHIV抗原性を示さなければならない。免疫原性誘導体は、天然タンパク質に比して幾つかの潜在的な利点(例えば、酵素活性(RT)またはCD4下方制御(Nef)などの、ワクチン抗原には好ましくない天然タンパク質の機能の低下または除去)を提供し得る。本発明のポリヌクレオチド配列は、本明細書に記載される本発明のコドン最適化態様に従って、哺乳動物細胞用にコドン最適化することができる。
【0062】
本発明はさらに、以下のステップ:
a) アデノウイルスベクターを提供するステップ、
b) 好適なプロモーターに機能的に連結されたHIV抗原配列を含有するプラスミドを提供するステップ、
c) プラスミドとベクターの両方を用いて細胞をトランスフェクトするステップ、
d) 組換えが起こるのに十分な時間を与えるステップ、および
e) HIV抗原配列を含有する組換えウイルスベクターを回収するステップ、
を含んでなる、本発明のベクターを作製する方法を提供する。
【0063】
別の態様において、本発明は、哺乳動物に好適な量の本発明の免疫原性組成物を投与することを含んでなる、哺乳動物に免疫応答を引き起こす方法を提供する。
【0064】
本発明は特に、HIV-1に関係し得る。本明細書に記載される構築物は、任意のHIVクレード、例えばクレードBまたはクレードC、特にクレードBに由来し得る。
【0065】
本発明のアデノウイルスベクターに用いるプロモーターは、HCMV IE遺伝子に由来するプロモーターとすることができ、例えば、WO 02/36792に記載されるような、エキソン1を含むHCMV IE遺伝子の5'非翻訳領域が含まれる。
【0066】
本発明の医薬組成物は、標的細胞への形質導入を可能にするのに十分な量で、また、十分なレベルの遺伝子導入と発現をもたらして、過度の副作用を伴うことなく、あるいは医学的に許容可能な生理的効果を伴って、治療効果を与えるのに十分な量で、投与することが可能であり、こうした投与量は、医療技術分野の当業者が決定し得る。通常の製薬上許容される投与経路としては、限定するものではないが、網膜への直接送達および他の眼内送達法、肝臓への直接送達、吸入、鼻腔内、静脈内、筋肉内、気管内、皮下、皮内、直腸、口腔および他の非経口投与経路が挙げられる。
【0067】
投与経路は、必要に応じて組み合わせて、または投与される遺伝子産物もしくは症状に応じて調整することができる。投与経路は、主に治療対象の症状の性質によって決まるだろう。
【0068】
本発明のウイルスベクターの投与量は、主に患者の治療される症状、年齢、体重および健康状態などの要因に応じて決まり、このため患者間で異なり得る。例えば、上記ウイルスベクターの、治療上有効な成人または動物に対する投与量は、通常、約1 x 106〜約1 x 1015個、約1 x 1011〜1 x 1013個、または約1 x 109〜1 x 1012個のウイルス粒子の濃度を有する担体約100μL〜約100 mLの範囲内である。投与量は、投与される動物の大きさおよび投与経路に応じて変動するだろう。例えば、ヒトまたは動物への筋肉注射用の好適な投与量(約80 kgの動物に対する)は、単一の部位について、1 mL当たり約1 x 109〜約5 x 1012個の粒子の範囲である。場合により、複数の投与部位へ送達することができる。別の例において、ヒトまたは動物への好適な投与量は、口腔製剤について、約1 x 1011〜約1 x 1015個の粒子の範囲である。当業者であれば、投与経路および上記の組換えベクターを用いる治療用途またはワクチン用途に応じて、これらの投与量を調整することができる。治療用産物の発現レベル、または免疫原については循環抗体のレベルをモニターして、投与量の投与頻度を決定することができる。しかし、当業者であれば、投与スケジュールを決定するための他の方法を容易に理解するであろう。
【0069】
医薬組成物の投与は、1つまたは2つ以上の個別用量の形式をとることが可能であり、例えば、同一のポリヌクレオチドを含むアデノウイルスの反復投与として、または異種「プライム・ブースト」ワクチン接種計画で投与することができる。異種プライム・ブースト計画は、初回免疫と追加免疫とで異なる形態のワクチンを投与するもので、各免疫は、それぞれ2回以上の投与を含み得る。初回免疫組成物と追加免疫組成物とは少なくとも1種類の抗原を共通して含むと考えられるが、それは必ずしも同一の形態の抗原である必要はなく、同一の抗原の異なる形態であってよい。
【0070】
本発明のベクターを用いるプライム・ブースト計画は、異種DNAとアデノウイルスベクターのプライム・ブースト(例えば、裸のDNAの初回量とその後のアデノウイルスベクターのブースト、あるいは、例えばアデノウイルスベクターのプライムとその後の1回以上の裸のDNAのブースト)の形式をとり得る。このようなDNAブーストは、DNAの筋肉内投与または皮内投与により、または粒子加速法により送達することができる。あるいはまた、このようなプライム・ブースト計画は、例えば、本発明のタンパク質およびアデノウイルスベクターを含むことが可能であり、この場合、初回量がタンパク質を含み、追加量がアデノウイルスベクターを含んでいてもよいし、または初回量がアデノウイルスベクターを含み、追加量がタンパク質を含んでいてもよい。
【実施例】
【0071】
実施例1:E1/E3が欠失されたPan 6およびPan 7アデノウイルスの構築
1. 組換えE1欠失SV-25ベクターの作製
遺伝子操作でE1を欠失させたことを除いて完全なSV-25ゲノムを含むプラスミドを構築した。このE1欠失部位に、制限酵素I-CeuIとPI-SceIの認識部位を挿入した。これらの認識部位は、これら2つの酵素認識部位にトランスジーン発現カセットが挟まれているシャトルプラスミドからのトランスジーンの挿入を可能にすると考えられる。
【0072】
制限部位SwaI-SnaBI-SpeI-AflII-EcoRV-SwaIを含む合成リンカーを、EcoRIとNdeIで切断したpBR322中にクローン化した。この操作は、2種類の合成オリゴマーSV25T (5'-AAT TTA AAT ACG TAG CGC ACT AGT CGC GCT AAG CGC GGA TAT CAT TTA AA-3')とSV25B (5'-TAT TTA AAT GAT ATC CGC GCT TAA GCG CGA CTA GTG CGC TAC GTA TTT A-3')を一緒にアニーリングし、これをEcoRIとNdeIで消化したpBR322に挿入することによって行った。アデノウイルス(Ad) SV25の左端(bp 1〜1057)を、上記リンカーのSnaBI部位とSpeI部位の間にクローン化した。Ad SV25の右端(bp 28059〜31042)は、該リンカーのAflII部位とEcoRV部位の間にクローン化した。その後アデノウイルスE1を、クローン化した左端からEcoRI部位(bp 547)とXhoI部位(bp 2031)の間で以下のとおり切り出した。PCRで生成した、pShuttle (Clontech)に由来するI-CeuI-PI-SceIカセットを、EcoRI部位とSpeI部位の間に挿入した。その後、Ad SV-25の10154bpのXhoI断片(bp2031〜12185)をSpeI部位に挿入した。結果として生じたプラスミドをHindIIIで消化し、Ad SV-25の18344bpのHindIII断片(bp 11984〜30328)を挿入することにより構築物(pSV25)を完成させ、組換えアデノウイルスの作製に適したE1欠失アデノウイルスSV25の完全な分子クローンを生成した。場合により、新たに作出したpSV25ベクタープラスミドのI-CeuI部位とPI-SceI部位に所望のトランスジーンを挿入してもよい。
【0073】
マーカー遺伝子を含むAd SV25を作製するため、プラスミドpShuttle(Clontech)中にあらかじめクローン化されたGFP(緑色蛍光タンパク質)発現カセットを制限酵素I-CeuIとPI-SceIで切除し、同じ酵素で消化したpSV25(または本明細書中に記載される別のAdチンパンジープラスミド)中に連結した。結果として生じたプラスミド(pSV25GFP)をSwaIで消化して細菌プラスミド骨格を分離し、E1補完性細胞株HEK293にトランスフェクトした。約10日後、複製ウイルスの存在を示す細胞変性効果が観察された。GFPを発現する、Ad SV25に基づくアデノウイルスベクターが首尾よく生成されたことは、トランスフェクトされた培養物からの上清を新鮮な細胞培養物に加えることにより確認した。二次的な感染細胞の存在は、これらの細胞集団における緑色蛍光の観察により確認した。
【0074】
2. E3欠失Pan-6およびPan-7ベクターの構築
本発明のアデノウイルスベクターのクローニング能力を高めるため、E3領域を欠失させることができる。なぜならこの領域は、培養中のウイルスの増殖に必要のない遺伝子をコードするからである。そのために、Pan-5、Pan-6、Pan-7、およびC68のE3欠失型が作製された(E31-9を含む3.5kbのNru-AvrII断片が欠失される)。
【0075】
Pan6に基づくベクターにおけるE3欠失
E1欠失pPan6-pkGFP分子クローンをSbf IとNot Iで消化し、19.3kbの断片を単離してSbf I部位に連結し直した。結果として生じた構築物pPan6-Sbf I-E3をEco 47 IIIとSwa Iで処理し、pPan6-E3を生成した。最終的に、pPan6-pkGFPのSbf I消化から得られた21kbのSbf I断片をpPan6-E3にサブクローン化し、E3に4kbの欠失を有するpPan6-E3-pkGFPを作出した。
【0076】
E3欠失Pan7ベクター
Pan 7でE3欠失を達成するために、上記と同じ方法を用いた。最初に、E3領域にわたる5.8kbのAvr II断片をpSL-1180にサブクローン化し、その後Nru I消化によりE3を欠失させた。結果として生じたプラスミドをSpe IとAvr IIで処理して4.4kbの断片を取得し、pPan7-pkGFPのAvr II部位にクローン化することにより、もとのE3を含むAvr II断片をそれぞれ置換した。最終的なpPan7-E3-pkGFP構築物は、3.5kbのE3欠失を有していた。
【0077】
これらのおよび他のPanアデノウイルス血清型におけるE1、E3およびE4欠失型の構築についての十分な説明は、WO 03/0046124に記載されている。さらなる情報は、Human Gene Therapy 15:519-530 (WO 03/046124)からも入手可能である。
【0078】
実施例2
Gag、RT、Nef配列の構築
この構築は、WO 03/025003に十分に記載されている。
【0079】
プラスミドp73i-Tgrn
1. プラスミド:p73i-GRN2クローン #19(p17/p24(コドン最適化)/RT(コドン最適化)trNef) - 修復型
目的遺伝子
HIV-1クレードBのHXB2株由来の、lowa長さのHCMVプロモーター + エキソン1の下流で、かつウサギβグロビンポリアデニル化シグナルの上流の、コドン最適化Gagのp17/p24部分、コドン最適化RTおよび末端切断型Nef遺伝子。
【0080】
プラスミドp17/24trNef1に由来するtrNef遺伝子を含むプラスミドはPCRエラーを含み、このエラーは、Nefの末端から19アミノ酸の位置にアルギニン(R)からヒスチジン(H)へのアミノ酸変化をもたらす。このエラーをPCR突然変異誘発によって修正し、修正したNefをp7077-RT3に由来するコドン最適化RTにPCRで継ぎ合わせ、継ぎ合わせた断片をApaIとBamHIで切断して、ApaI/BamHIで切断したp73i-GRNにクローン化した。
【0081】
プライマー
プライマーを用いたp7077-RT3からのコドン最適化RTのPCR増幅:
(PCR全体を通して、ポリメラーゼ = PWO (Roche))
センス:U1
GAATTCGCGGCCGCGATGGGCCCCATCAGTCCCATCGAGACCGTGCCGGTGAAGCTGAAACCCGGGAT
アンチセンス:AScoRT-Nef
GGTGTGACTGGAAAACCCACCATCAGCACCTTTCTAATCCCCGC
サイクル:95℃(30秒)、その後95℃(30秒)、55℃(30秒)、72℃(180秒)を20サイクル、その後72℃(120秒)にして4℃に保持
1.7kbのPCR産物をゲル精製した。
【0082】
プライマーを用いたp17/24trNef1からの5' NefのPCR増幅:
センス:S-Nef
ATGGTGGGTTTTCCAGTCACACC
アンチセンス:ASNef-G:
GATGAAATGCTAGGCGGCTGTCAAACCTC
サイクル:95℃(30秒)、その後95℃(30秒)、55℃(30秒)、72℃(60秒)を15サイクル、その後72℃(120秒)にして4℃に保持。
【0083】
プライマーを用いたp17/24trNef1からの3' NefのPCR増幅:
センス:SNEF-G
GAGGTTTGACAGCCGCCTAGCATTTCATC
アンチセンス:AStrNef
CGCGGATCCTCAGCAGTTCTTGAAGTACTCC
サイクル:95℃(30秒)、その後95℃(30秒)、55℃(30秒)、72℃(60秒)を15サイクル、その後72℃(120秒)にして4℃に保持。
【0084】
上記のPCR産物をゲル精製した。最初に、5'(S-Nef)と3'(AstrNef)のプライマーを用いて2つのNef産物を継ぎ合わせた。
サイクル:95℃(30秒)、その後95℃(30秒)、55℃(30秒)、72℃(60秒)を15サイクル、その後72℃(180秒)にして4℃に保持。
【0085】
このPCR産物をPCR洗浄し、U1とAstrNefのプライマーを用いて上記のRT産物に継ぎ合わせた。
サイクル:95℃(30秒)、その後95℃(30秒)、55℃(30秒)、72℃(180秒)を20サイクル、その後72℃(180秒)にして4℃に保持。
【0086】
上記の2.1kbの産物をゲル精製し、ApaIとBamHIで切断した。プラスミドp73I-GRNもApaIとBamHIで切断してゲル精製し、ApaI-Bam RT3trNefと連結してp17/p24(コドン最適化)/RT(コドン最適化)trNef遺伝子を再生成した。
【0087】
2. プラスミド:p73I-RT w229k (不活性化RT)
目的遺伝子
lowa長さのHCMVプロモーター + エキソン1の下流で、かつウサギβグロビンポリアデニル化シグナルの上流の不活性化RT遺伝子の生成。
【0088】
治療用ワクチンに活性型HIV RT種を使用することの懸念のため、この遺伝子は不活性化させることが望ましい。不活性化は、上記RT(P73I-GRN2由来)の229位のアミノ酸をトリプトファン(Trp)からリシン(Lys)にPCR突然変異誘発させることにより達成した(R7271 p1-28)。
【0089】
プライマー
プライマーを用いた5'RT + 変異のPCR増幅:
(PCR全体を通して、ポリメラーゼ = PWO (Roche))
センス:RT3-u:1
GAATTCGCGGCCGCGATGGGCCCCATCAGTCCCATCGAGACCGTGCCGGTGAAGCTGAAACCCGGGAT
アンチセンス:AScoRT-Trp229Lys
GGAGCTCGTAGCCCATCTTCAGGAATGGCGGCTCCTTCT
サイクル:
1 x [94℃ (30秒)]
15 x [94℃ (30秒)/55℃ (30秒)/72℃ (60秒)]
1 x [72℃ (180秒)]
PCR ゲル精製。
【0090】
プライマーを用いた3'RT + 変異のPCR増幅:
アンチセンス:RT3-l:1
GAATTCGGATCCTTACAGCACCTTTCTAATCCCCGCACTCACCAGCTTGTCGACCTGCTCGTTGCCGC
センス:ScoRT-Trp229Lys
CCTGAAGATGGGCTACGAGCTCCATG
サイクル:
1 x [94℃ (30秒)]
15 x [94℃ (30秒)/55℃ (30秒)/72℃ (60秒)]
1 x [72℃ (180秒)]
PCR ゲル精製。
【0091】
上記のPCR産物をゲル精製し、RTの5'末端と3'末端を、5'(RT3-U1)および3'(RT3-L1)プライマーを用いて継ぎ合わせた。
サイクル:
1 x [94℃ (30秒)]
15 x [94℃ (30秒)/55℃ (30秒)/72℃ (120秒)]
1 x [72℃ (180秒)]
【0092】
上記のPCR産物をゲル精製し、NotIとBamHIの制限部位を利用してp7313ieにクローン化し、p73I-RT w229kを生成した(図13参照)。
【0093】
3. プラスミド:p73i-Tgrn
目的遺伝子
HIV-1クレードBのHXB2株由来の、lowa長さのHCMVプロモーター + エキソン1の下流で、かつウサギβグロビンポリアデニル化シグナルの上流のコドン最適化Gagのp17/p24部分、コドン最適化RTおよび末端切断型Nef遺伝子。
【0094】
活性型RTを含む3種融合構築物は、規制当局がヒトへの使用を認めていないため、p73i-RT w229kからNheIとApaIで切断した断片をNheI/ApaI切断したp73i-GRN2#19に挿入することにより、RTの不活性化を達成した(図14)。この操作により、RTの229位にトリプトファン(W)→リシン(K)変更が生じる。
【0095】
Tgrnプラスミド挿入物の完全配列を図7に示す。この配列は、p17 p24(コドン最適化)Gag、p66 RT(コドン最適化且つ不活性型)および末端切断型Nefを含む。
【0096】
Gag、RTおよびNefの代替構築物は以下のとおりである:
trNef - p66 RT(コドン最適化) - p17、p24(コドン最適化)Gag、
trNef - p17、p24(コドン最適化)Gag - p66 RT(コドン最適化)、
p66 RT(コドン最適化) - p17、p24(コドン最適化)Gag - trNef、
p66 RT(コドン最適化) - trNef - p17、p24(コドン最適化)Gag、
p17、p24(コドン最適化)Gag - trNef - p66 RT(コドン最適化)
これらの構築物の完全配列を、それぞれ図8〜12に示す。
【0097】
実施例3
Gag、RT、Nef配列のアデノウイルスへの挿入
pShuttleプラスミドへのGRN発現カセットのサブクローニング
プロモーター、cDNAおよびポリアデニル化シグナルからなる発現カセット全体を、Sph IとEcoR Iの二重消化によってpT-GRN構築物から単離した。Sph I/EcoR I断片のSph I末端をKlenowで埋めて、pShuttleプラスミドのEcoR I部位とMlu I部位(Mlu I末端を平滑化した)の間にクローン化した。
【0098】
クローニング工程の間に、付加的なフランキング配列がHIV発現カセットに付随するようになった。この配列はCer配列として知られており、その機能は不明である。
【0099】
Pan6 およびPan7ベクターのE1/E3欠失分子クローンへのGRN発現カセットの導入
発現カセットをI-Ceu IとPI-Sce Iとの消化によりpShuttleから回収し、Pan6およびPan7ベクターの分子クローンの同じ部位にクローン化した。組換えクローンは青白選択を通じて同定し、広範囲の制限酵素解析で確認した。
【0100】
組換えウイルスのレスキューと増殖
C6およびC7ベクターの分子クローンを適切な制限エンドヌクレアーゼ(それぞれPmeIとPacI)で処理してインタクトな線状ベクターゲノムを放出させ、リン酸カルシウム法を用いて293細胞にトランスフェクトした。トランスフェクトされた細胞中で十分な細胞変性効果が観察された場合、粗ウイルス溶解物を回収し、293細胞中でラージスケールの感染(1x109細胞)へと徐々に増やした。ラージスケール感染物からのウイルスを標準的なCsCl沈降法で精製した。
【0101】
また、pShuttleプラスミドは、EcoRIとXmnIで切断することにより3'リンカー配列を除去してさらに切り取り、プラスミドのサイズを縮小してpShuttleGRNcを生成することができる。この改変プラスミドは、上記の方法を用いてさらなるPan7ウイルス(C7-GRNc)を生成するために使用し得る。
【0102】
他の構築物も同様にPan 6およびPan 7アデノウイルスの両方に挿入した。しかし、p66 RT(コドン最適化) - trNef - p17、p24(コドン最適化)Gag挿入物を有するPan 6の作製は成功しなかった。
【0103】
実施例4
マウス免疫原性モデル
HIV抗原RT、NefおよびGagの再配列挿入物を含有する一連のPan6およびPan7ベクター(RGN、NRG、NGR、GRN、およびGNR)をin vivoでの一次免疫反応について試験した。Pan6ウイルスを試験するため3つの実験を、Pan7ウイルスを試験するため2つの実験を行った。各アデノウイルスを1x108粒子の用量でBalb/c(K2d)マウスの片方の後肢に50μl量で筋肉内投与した。この用量を選択したのは、優れたレベルの細胞性免疫応答を誘導することが以前にわかっていた(未公開)ためである。表1は、これらの実験で比較したアデノウイルスをまとめたものである。
【表1】

【0104】
Gag、NefおよびRTの特異的エピトープに対するペプチドまたはタンパク質を用いたin vitro刺激の後、CD8応答とCD4応答の発生をELIspotアッセイにより初回免疫の14日後と28日後に測定した。これらの結果は、全ての変異体が、空ベクターの対照と比較して強い一次免疫応答(IFNγとIL-2の両方の生成により測定される)を生じる能力があるという強力な証拠を提供する(データ示されず)。
【0105】
これらの研究から得られたデータは、SASプログラム(version 9.1.3 Service Pack 2)による混合モデル(Proc Mixed)の分散分析(ANOVA)を使用して統計的に解析し、Pan6およびPan7におけるRNG変異体のランクを別々の時点で決定した。IFNγ産生に関しは、CD8ペプチドに対する応答の合計をGagとRTについて定量し、一方、IL-2のELIspotデータは、CD4ペプチドに対する応答の合計でGag、NefおよびRTについて評価した。
【0106】
前記パネルの変異体のランク付けは、Bayesianモデルを用いて計算し(100,000個の事後サンプルをもたらす平坦事前分布(flat prior)を有するProc MixedのPriorステートメントを使用して実施した;Tierney, L. (1994), "Markov Chains for Exploring Posterior Distributions"(考察を含む)、ならびにAnnals of Statistics, 22, 1701-1762; Gelfand, A.E., Hills, S.E., Racine-Poon, A.およびSmith, A.F.M. (1990), "Illustration of Bayesian Inference in Normal Data Models Using Gibbs Sampling," Journal of the American Statistical Association, 85, 972-985)を参照)、各変異体の「最良」としての確率を、研究された実験条件から得られたデータに基づいて推測した。
【0107】
図1は、Bayesian法で推測された、14日目と28日目の、各ペプチドによるIFNγとIL-2に関するPan6のCD4およびCD8応答の合計を表す。
【0108】
図2は、Bayesian法で推測された、14日目と28日目の、各ペプチドによるIFNγとIL-2に関するPan7のCD4およびCD8応答の合計を表す。
【0109】
挿入物は全て、空ベクターの対照と比較して免疫応答の著しい増加を示す。統計分析は、異なるウイルス間に有意差がないことを示す。
【0110】
実施例5
ブタ免疫原性モデル
幾つかの研究から得られた結果は、ブタが候補ワクチンの免疫原性を試験するための優れたモデルであることを示していた。4つの候補NHPアデノウイルスの免疫原性をミニブタで検証するための研究を設定した。1グループ5匹のミニブタをPAN6GRN、PAN6NGR、PAN7GRNまたはPAN7NGRで初回免疫した(使用したバッチの詳細については表2を参照)。各動物に合計3 x 1010個のアデノウイルス粒子を筋肉内経路により(1.0ml量を各内側大腿筋間で均等に分割して用いて)投与した。
【表2】
【0111】
免疫前、および免疫後に間隔をあけて各動物から血液サンプルを回収した。末梢血単核細胞を単離し、RT、NefおよびGagペプチドライブラリーのプールとタンパク質を用いてin vitroで再刺激した。上記のペプチドライブラリーのプールは、RT、NefおよびGagの配列全体にわたり11アミノ酸だけ重複している15-merのペプチドからなり、in vivoマウス実験で使用したペプチドと同一であった。
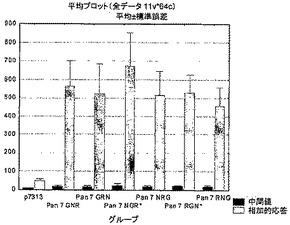
【0112】
これらのブタ細胞によるインターフェロンγの産生を、ELIspotアッセイを用いて測定した。図3は、4回のサンプリング時点におけるRT、NefおよびGagペプチドライブラリーのプールに対する応答を示す。
【0113】
4種のウイルス全てに対する応答を免疫の7日後に検出した。4種のNHPウイルス全てに対する細胞性応答は、初回免疫後少なくとも5週間まで維持される。PAN6-GRNは、初回免疫の7日後に、IFN-γ ELIspotにより最も強い応答をもたらす。
【0114】
実施例6
霊長類免疫原性モデル
霊長類の試験的研究から得られた結果は、RT、NefおよびGagを発現するNHPアデノウイルスの筋肉内注射が、カニクイザルにおいて細胞性免疫応答を誘発したことを示した。
【0115】
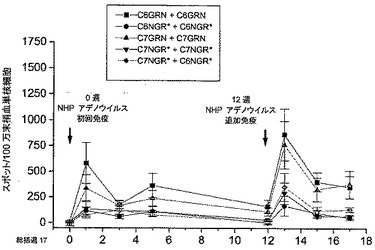
4種類の候補NHPアデノウイルスの免疫原性をカニクイザルで検証するための研究を設定した。動物グループをPAN6-GRN、PAN6-NGR、PAN7-GRNまたはPAN7-NGRで一次免疫した(使用したウイルスバッチの詳細については表3を参照)。各動物に合計1011個のアデノウイルス粒子を筋肉内経路により(1.0ml量を各内側大腿筋間で均等に分割して使用して)投与した。
【表3】
【0116】
血液サンプルを免疫前と、免疫後に1週間間隔で回収した。末梢血単核細胞を単離し、RT、NefおよびGagペプチドライブラリーのプールを用いてin vitroで再刺激した。これらの霊長類細胞によるインターフェロンγの産生を、ELIspotアッセイを用いて測定した。図4は、3回のサンプリング時点における各グループの応答を示す。
【0117】
結果は、全グループが初回免疫の1週間後に強く応答し、この応答が免疫の少なくとも7週間後まで維持されたことを示す。この結果は、霊長類においてこの用量(すなわち1011個の粒子)で使用した場合、ベクター間に大差がないことを示唆する。
【0118】
実施例7
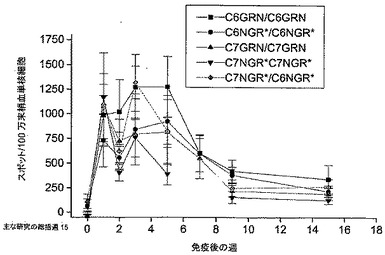
筋肉内投与(i.m.)で送達された、HIV GRN抗原をコードするNHPアデノウイルスの一定用量範囲に対する一次免疫後の応答
一次免疫で用いたアデノウイルスの用量の影響を評価するために、1グループ5匹のマウスを、漸増用量のNHPアデノウイルス(107〜1010個の粒子)で筋肉内(i.m.)に免疫した。陽性対照として、1グループの動物を、粒子媒介表皮送達を用いてDNA(2μg)で免疫した(ND5)。免疫後6日目と19日目に、動物を動物(科学的処置)法付則1に基づいて安楽死させ、脾臓を取り出した。免疫応答は、脾細胞を一晩刺激するために各抗原(GAGおよびRT)のペプチドライブラリーのプールを用いて、IFN-γ ELISPOTアッセイによりモニターした。図5は、2回のサンプリング時点における各グループの応答を示す。
【0119】
実施例8
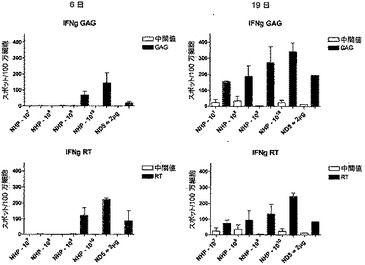
皮内投与(i.d.)で送達された、HIV GRN抗原をコードするNHPアデノウイルスの一定用量範囲に対する一次免疫後の応答
一次免疫で用いたアデノウイルスの用量の影響を評価するために、1グループ5匹のマウスを、漸増用量のNHPアデノウイルス(107〜1010個の粒子)で皮内(i.d.)に免疫した。陽性対照として、1グループの動物を、粒子媒介表皮送達(PMED)を使用してDNA(1μg)で免疫した。免疫後7日目と14日目に、動物を動物(科学的処置)法付則1に基づいて安楽死させ、脾臓を取り出した。免疫応答はIFN-γ ELISPOTアッセイによりモニターした。脾細胞を、CD4またはCD8 T細胞を特異的に刺激する各抗原(GAGおよびRT)の確かなペプチドを用いて一晩刺激した。図6は、2回のサンプリング時点における各グループの応答を示す。
【0120】
これらの結果は、筋肉内免疫と皮内免疫の両方が、本発明の組成物の有効な投与経路であることを示唆する。
【表4】
【図面の簡単な説明】
【0121】
【図1】Pan6 HIVアデノウイルスのランク付け。これは、Bayesian法で推測された、14日目と28日目の、各ペプチドによるIFNγとIL-2に関するPan6のCD4およびCD8応答の合計を表す。y軸は、脾細胞100万個当たりのスポット形成細胞を表す。
【図2】Pan7 HIVアデノウイルスのランク付け。これは、Bayesian法で推測された、14日目と28日目の、各ペプチドによるIFNγとIL-2に関するPan7のCD4およびCD8応答の合計を表す。y軸は、脾細胞100万個当たりのスポット形成細胞を表す。
【図3】初回免疫の0、1、3および5週間後のRT、NefおよびGagペプチドライブラリーのプールに対するミニブタの応答。結果は、各動物についての、各ペプチドライブラリーのプールに対する応答の合計の平均±標準誤差である。データは、ペンシルバニア大学(University of Pennsylvania)から入手した。
【図4】初回免疫の0、1および2週間後のRT、NefおよびGagペプチドライブラリーのプールに対する霊長類の応答。結果は、各動物についての、各ペプチドライブラリーのプールに対する応答の合計の平均±標準誤差である。
【図5】筋肉内(i.m.)に送達された、HIV GRN抗原をコードするNHPアデノウイルスの一定用量範囲に対する一次免疫後の応答。マウスのグループ(n=5)を様々な用量のNHPアデノウイルス(107〜1010個の粒子)で免疫し、各抗原のペプチドライブラリーのプールに対する細胞性免疫応答を、IFN-γ ELISPOTアッセイを用いてモニターした(6日目および19日目)。
【図6】皮内(i.d.)に送達された、HIV GRN抗原をコードするNHPアデノウイルスの一定用量範囲に対する一次免疫後の応答。マウスのグループ(n=3)を様々な用量のNHPアデノウイルス(107〜1010個の粒子)で免疫し、特異的ペプチドに対する細胞性免疫応答を、IFN-γ ELISPOTアッセイを用いてモニターした(7日目および14日目)。
【図7−1】実施例2に記載される構築物p73i-Tgrnのポリヌクレオチド配列、アミノ酸配列および制限地図を示す。
【図7−2】実施例2に記載される構築物p73i-Tgrnのポリヌクレオチド配列、アミノ酸配列および制限地図を示す。
【図8−1】実施例2に記載される構築物p73i-Tnrgのポリヌクレオチド配列、アミノ酸配列および制限地図を示す。
【図8−2】実施例2に記載される構築物p73i-Tnrgのポリヌクレオチド配列、アミノ酸配列および制限地図を示す。
【図9−1】実施例2に記載される構築物p73i-Tngrのポリヌクレオチド配列、アミノ酸配列および制限地図を示す。
【図9−2】実施例2に記載される構築物p73i-Tngrのポリヌクレオチド配列、アミノ酸配列および制限地図を示す。
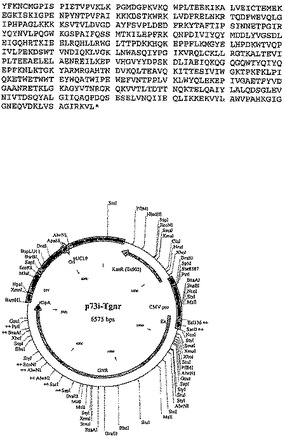
【図10−1】実施例2に記載される構築物p73i-Trgnのポリヌクレオチド配列、アミノ酸配列および制限地図を示す。
【図10−2】実施例2に記載される構築物p73i-Trgnのポリヌクレオチド配列、アミノ酸配列および制限地図を示す。
【図11−1】実施例2に記載される構築物p73i-Trngのポリヌクレオチド配列、アミノ酸配列および制限地図を示す。
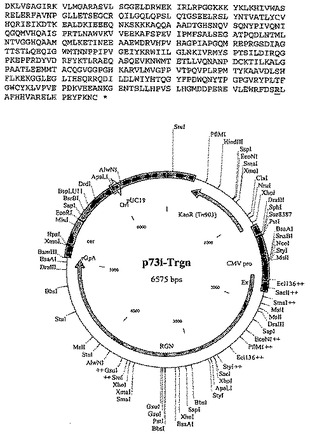
【図11−2】実施例2に記載される構築物p73i-Trngのポリヌクレオチド配列、アミノ酸配列および制限地図を示す。
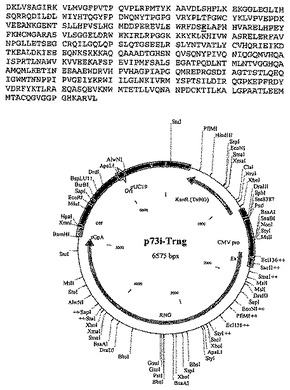
【図12−1】実施例2に記載される構築物p73i-Tgnrのポリヌクレオチド配列、アミノ酸配列および制限地図を示す。
【図12−2】実施例2に記載される構築物p73i-Tgnrのポリヌクレオチド配列、アミノ酸配列および制限地図を示す。
【技術分野】
【0001】
本発明は、HIVポリペプチドをコードするオリゴヌクレオチドを含むウイルスベクター、特にこのウイルスベクターがアデノウイルスであるウイルスベクターに関する。特にこのようなアデノウイルスは、非ヒト霊長類のアデノウイルス、例えばサルのアデノウイルス、特にチンパンジーのアデノウイルスである。特に本発明は、複数の異なるHIV抗原、例えば2種もしくは3種またはそれ以上のHIV抗原をコードするHIVポリヌクレオチド配列を含有するアデノウイルスベクターに関する。本発明はさらに、上記ウイルスベクターの作製方法、この方法で作製されたウイルスベクター、および医療、特に予防用または治療用ワクチン接種におけるこれらのベクターの使用に関する。
【背景技術】
【0002】
HIV-1は、世界の主要な健康問題の1つであると見なされている後天性免疫不全症候群(AIDS)の主な原因である。世界中で広範囲の研究が行われているが、ワクチンを製造しようとする取り組みは今のところ成功していない。
【0003】
HIV-1は、レトロウイルス科のRNAウイルスである。HIVゲノムは少なくとも9種類のタンパク質をコードし、これらは以下の3つのクラスに分けられる:主要な構造タンパク質Gag、PolおよびEnv、調節タンパク質TatおよびRev、ならびにアクセサリータンパク質Vpu、Vpr、VifおよびNef。このHIVゲノムは、全てのレトロウイルスに共通な5'LTR-gag-pol-env-LTR3'の構成を示す。
【0004】
アデノウイルスは、約36 kbのゲノムサイズを有する二本鎖DNAウイルスであり、その様々な標的組織において高効率の遺伝子導入を達成する能力と、大きなトランスジーンの収容力のために、遺伝子導入の用途に広く使用されている。従来、アデノウイルスのE1遺伝子は、欠失されるか、または好適なプロモーター、目的遺伝子のcDNA配列およびポリAシグナルからなる導入遺伝子カセットで置換され、その結果として複製欠損型の組換えウイルスをもたらす。
【0005】
アデノウイルスは、3つの主要タンパク質、ヘキソン(II)、ペントンベース(III)およびこぶ状のファイバー(IV)と、いくつかの他のマイナーなタンパク質VI、VIII、IX、IIIaおよびIVa2とからなる正二十面体のキャプシドをもつ特徴的形態を有する(Russell W.C. 2000, Gen Virol, 81:2573-2604)。このウイルスゲノムは、逆方向末端反復配列(ITR)を有する5'末端に末端タンパク質が共有結合で結合している、線状の二本鎖DNAである。このウイルスDNAは、高塩基性タンパク質VIIおよびmuと呼ばれる小型のペプチドと密接に会合している。もう1つのタンパク質Vは、このDNA-タンパク質複合体と共にパッケージングされ、タンパク質VIを介してキャプシドとの構造的連結を与える。このウイルスはまた、構造タンパク質の一部をプロセシングして、成熟した感染性ウイルスを生成するのに必要とされる、ウイルスにコードされたプロテアーゼを含む。
【0006】
様々な哺乳動物種に感染する、100を越える異なった血清型のアデノウイルスが分離されているが、そのうち51の血清型はヒト起源である。このようなヒト起源からのアデノウイルスの例は、Ad1、Ad2、Ad4、Ad5、Ad6、Ad11、Ad24、Ad34、Ad35である。上記のヒト血清型は、いくつかの生物学的、化学的、免疫学的および構造的基準に基づいて、6つの亜属(A〜F)に分類されている[WO 04018627、第1頁]。
【0007】
Ad5ベースのベクターが多くの遺伝子治療試験で広く用いられているが、一般集団には自然感染による既存の免疫が存在するため、Ad5と他のグループCのアデノウイルスベクターの使用には限界があるだろう。Ad5と他のグループCのメンバーは、どちらかといえば、最も優勢な血清型に入る。存在するベクターに対する免疫は、治療中に該ベクターにさらされた結果として発生し得る。優勢血清型のベクターに対する、こうした種類の既存のまたは発生した免疫は、遺伝子治療またはワクチン接種の努力の成果を制限する可能性がある。したがって、これらに代わるアデノウイルス血清型は、宿主の免疫応答を回避し得る遺伝子送達系の研究において極めて重要な目標となる。
【0008】
このような代替血清型の1分野は、非ヒト霊長類、特にチンパンジーのアデノウイルスである。2種類のチンパンジーアデノウイルスのゲノムについて記載している米国特許第6,083,716号を参照されたい。
【0009】
チンパンジー(「Pan」または「C」)アデノウイルスベクターは、ヒトアデノウイルスベクターと同程度に効率的に、トランスジーン産物に対して強い免疫応答を惹起することが示されている(Fitzgeraldら J. Immunol. 170:1416)。
【0010】
HIVのTatおよびNefタンパク質は初期タンパク質である。すなわち、これらのタンパク質は、感染の初期に、構造タンパク質の非存在下で発現する。
【0011】
Nef遺伝子は、幾つかの活性を有することが示されている初期アクセサリーHIVタンパク質をコードする。例えば、Nefタンパク質は、HIV受容体であるCD4の細胞表面からの除去を引き起こすことが知られているが、この機能の生物学的重要性については論議がある。さらに、NefはT細胞のシグナル伝達経路と相互作用して活性状態を誘導し、続いてより効率的な遺伝子発現を促進し得る。一部のHIV分離株はこの領域に変異を有し、そのためこれらの分離株は機能性タンパク質をコードすることができず、in vivoでのこれらの複製および病原性が大いに損なわれる。
【0012】
Gag遺伝子は、完全長RNAから翻訳されて前駆体ポリタンパク質を生成し、その後この前駆体タンパク質は、3〜5個のキャプシドタンパク質、すなわち、マトリックスタンパク質、キャプシドタンパク質および核酸結合タンパク質、ならびにプロテアーゼに切断される (Fundamental Virology, Fields BN, Knipe DMおよびHowley M 1996 2. Fields Virology vol 2 1996)。
【0013】
Gag遺伝子は、p55とも呼ばれる55キロダルトン(kD)のGag前駆体タンパク質を生じさせるが、このタンパク質は、スプライシングされていないウイルスmRNAから発現される。翻訳の間、p55のN末端はミリストイル化されて、細胞膜の細胞質側に結合するようになる。膜結合したGagポリタンパク質は、ウイルスゲノムRNAの2つのコピーを動員し、他のウイルス性および細胞性タンパク質と一緒になって、感染細胞の表面からの該ウイルス粒子の出芽を引き起こす。出芽後、p55はウイルスにコードされたプロテアーゼ(Pol遺伝子の産物)によってMA(マトリックス[p17])、CA(キャプシド[p24])、NC(ヌクレオキャプシド[p9])、およびp6(4)と呼ばれる4つのより小さなタンパク質へとウイルスの成熟過程で切断される。
【0014】
3つの主要なGagタンパク質(p17、p24およびp9)に加えて、全てのGag前駆体は幾つかの他の領域を含み、これらは切り出されて、様々なサイズのペプチドとしてビリオン(ウイルス粒子)中に残る。これらのタンパク質は多様な役割を有し、例えばp2タンパク質は、プロテアーゼ活性の調節において提案された役割を果たし、タンパク質分解プロセシングの正確なタイミングに寄与する。
【0015】
MAポリペプチドはp55のミリストイル化されたN末端に由来する。大部分のMA分子は、ビリオンの脂質二重層の内表面に結合した状態で存在し、この粒子を安定化させる。MAのサブセットは、ビリオンのより深い層の内部に動員されて、そこでウイルスDNAを核につなげる複合体の一部となる。これらのMA分子は、MA上の核親和性シグナルが細胞性核内輸送機構によって認識されるため、ウイルスゲノムの核輸送を容易にする。この現象は、HIVが非分裂細胞に感染することを可能とするが、レトロウイルスには珍しい特性である。
【0016】
p24(CA)タンパク質は、ウイルス粒子の円錐形のコア部を形成する。シクロフィリンAは、p55のp24領域と相互作用し、HIV粒子へのその取込みをもたらすことが実証されている。GagとシクロフィリンAとの相互作用は不可欠である。なぜなら、シクロスポリンAによる上記の相互作用の破壊がウイルス複製を阻害するからである。
【0017】
GagのNC領域は、いわゆるHIVのパッケージングシグナルを特異的に認識することに関与する。このパッケージングシグナルは、ウイルスRNAの5'末端の近くに位置する4つのステムループ構造からなり、HIV-1ウイルス粒子への異種RNAの取込みを媒介するのに十分である。NCは、2つのジンクフィンガーモチーフにより媒介される相互作用を通じてパッケージングシグナルと結合する。NCは逆転写も促進する。
【0018】
p6ポリペプチド領域は、p55 Gagとアクセサリータンパク質Vprとの相互作用を媒介し、組み立てつつあるビリオンへのVprの取込みをもたらす。p6領域はまた、感染細胞からの出芽ビリオンの効率的な放出に必要とされる、いわゆる後期ドメインを含む。
【0019】
Pol遺伝子は、初期感染においてウイルスが必要とする活性を有する3種類のタンパク質、逆転写酵素RT、プロテアーゼ、および細胞DNAへのウイルスDNAの組込みに必要とされるインテグラーゼタンパク質をコードする。Polの一次産物は、ビリオンのプロテアーゼによって切断されて、DNA合成に必要な活性を含むアミノ末端RTペプチド(RNAおよびDNA特異的DNAポリメラーゼ、リボヌクレアーゼH)およびカルボキシ末端インテグラーゼタンパク質をもたらす。HIVのRTは完全長RT(p66)のヘテロ二量体であり、分解産物(p51)はカルボキシ末端のRNアーゼインテグラーゼドメインを欠いている。
【0020】
RTは、レトロウイルスゲノムによりコードされるタンパク質のうち最も高度に保存されたタンパク質の1つである。RTの2つの主要な活性は、DNAポリメラーゼとリボヌクレアーゼHである。RTのDNAポリメラーゼ活性は、RNAとDNAを鋳型として交互に使用し、公知の全てのDNAポリメラーゼと同様に、新生DNA合成を開始することはできず、プライマーとしての役割を果たす既存の分子(RNA)を必要とする。
【0021】
全てのRTタンパク質に固有のRNアーゼH活性は、複製の初期に、DNA合成が進むにつれてRNAゲノムを除去するという重要な役割を果たす。RNアーゼHは、全てのRNA-DNAハイブリッド分子からRNAを選択的に分解する。構造的に、DNAポリメラーゼとリボヌクレアーゼHは、Pol内で別個の重複しないドメインを占めており、Polのアミノ末端3分の2の範囲に及ぶ。
【0022】
p66触媒サブユニットは、5つの異なるサブドメインへと折り畳まれる。これらのアミノ末端23は、RT活性を有する部分を含む。これらのカルボキシ末端は、RNアーゼ Hドメインである。
【0023】
宿主細胞への感染後、レトロウイルスRNAゲノムは、感染粒子中に存在する逆転写酵素により線状二本鎖DNAへとコピーされる。インテグラーゼ(Skalka AM'99 Adv in Virus Res 52 271-273に概説)はウイルスDNAの両端を認識し、これらを切り取って、宿主の染色体部位にウイルスDNAを連れて行き、組込みを触媒する。宿主DNAの多くの部位が組込みの標的部位となり得る。上記のインテグラーゼはin vitro組込みを触媒するのに十分であるが、これは、in vivoではウイルスDNAと関連した唯一のタンパク質ではない。感染細胞から単離される大型のタンパク質-ウイルスDNA複合体は、プレインテグレーション複合体(preintegration complex)を表している。この複合体は、子孫ウイルスゲノムによる宿主細胞遺伝子の獲得を促進する。
【0024】
上記のインテグラーゼは、N末端ドメイン、触媒コアおよびC末端ドメインの3つの異なるドメインで構成されている。触媒コアドメインは、ポリヌクレオチド移行の化学作用の必要条件を全て備えている。
【0025】
複数の外来遺伝子を含むウイルスベクター、特にアデノウイルスベクターは、常に簡単に製造できるわけではない。ベクターの安定性の問題、および挿入された遺伝子の効率のよい発現を得ることの困難性が存在しうる。特に、ワクチンにおいて使用し得る2つ以上または3つ以上のHIVポリヌクレオチドを含むアデノウイルスの製造は、これまで成功を収めていない。
【0026】
非ヒト霊長類アデノウイルスは、チンパンジーの腸間膜リンパ節から単離することができる。チンパンジーアデノウイルスはヒトC亜型アデノウイルスと十分に類似しており、HEK293細胞でのE1欠失ウイルスの複製が可能である。しかしチンパンジーアデノウイルスは、より一般的なヒト血清型(Ad2およびAd5)とは系統学的に区別される。Pan 6は、Pan5、7および9とはそれほど近縁でなく、血清学的に区別される。
【0027】
異種DNAをアデノウイルスに挿入することに関連して、一定のサイズ制限がある。ヒトアデノウイルスは、野生型ゲノム長の105%までパッケージする能力を有する(Bettら 1993, J Virol 67 (10), 5911-21)。ヒトアデノウイルスのパッケージングの下限値は、野生型ゲノム長の75%であることが示されている(Parksら 1995, J Virol 71(4), 3293-8)。
【0028】
HIVに対して有効なワクチンを見出す必要性が依然として存在する。
【発明の開示】
【0029】
本発明は、1つ以上の領域が欠失されたアデノウイルスベクターであって、該ベクターが少なくとも3つのHIV抗原またはこれらの免疫原性誘導体もしくは免疫原性断片をコードするポリヌクレオチド(1つまたは複数)を含み、ここで該ベクターは哺乳動物の宿主において上記の抗原または断片もしくは誘導体を発現することが可能であり、欠失のサイズおよびHIVポリヌクレオチド(1つまたは複数)のサイズは、ベクターゲノムの全長が野生型ウイルスのゲノム長の85〜105%となるようなサイズである、上記アデノウイルスベクターを提供する。
【0030】
本発明の1つの実施形態において、ポリヌクレオチド(1つまたは複数)によりコードされるHIV抗原は、Gag、NefおよびPolであり得る。他の実施形態において、Polは、RT部分のみを含むものでよい。本発明のさらに別の実施形態において、HIV抗原をコードするポリヌクレオチド(1つまたは複数)は、Gag、RT、Nefの順に転写されるように、すなわちGag部分が、結果として生じる融合タンパク質のN末端に位置するように、配置することができる。
【0031】
全ベクターゲノムのサイズは、例えば野生型ウイルスゲノムのサイズの90〜100%、または野生型ゲノムサイズの95〜100%であってよい。1つの実施形態において、ベクターの全サイズは、野生型ウイルスゲノムのサイズの約96%でありうる。
【0032】
本発明のアデノウイルスベクターに挿入するための特定のHIV抗原は、Pol、NefおよびGagまたはこれらの免疫原性誘導体もしくは免疫原性断片である。
【0033】
このようなアデノウイルスベクターを、製薬上許容される賦形剤、担体、希釈剤またはアジュバントと共に製剤化し、HIV感染およびAIDSの治療および/または予防に適した医薬組成物またはワクチン組成物を含む免疫原性組成物を製造することができる。
【0034】
本発明において用いられるのは、ヒト集団中で一般的な天然に存在する血清型(例えばAd2およびAd5)とは区別されるアデノウイルスである。このウイルスは、該ベクターに対する強い免疫応答の誘導を回避するが、かかる免疫応答は、中和抗体を介してベクターの取込みを阻害し、毒性を与えることにより、その後の同じ血清型の投与の有効性を制限する。
【0035】
従って、本発明のアデノウイルスは、一般的な天然に存在するヒトウイルス血清型ではないアデノウイルスとすることができる。動物から分離されるアデノウイルスは、免疫学的に区別されるキャプシド、ヘキソン、ペントンおよびファイバー成分を有するが、系統学的には密接な関係がある。特に本発明のウイルスは、非ヒトアデノウイルス、例えばサルアデノウイルス、特にPan 5、6、7または9などのチンパンジーアデノウイルスであってよい。このようなウイルス株の例はWO 03/000283に記載されており、American Type Culture Collection (10801 University Boulevard, Manassas, Virginia 20110-2209)、および他の供給元から入手可能である。望ましいチンパンジーアデノウイルス株は、Pan 5 [ATCC VR-591]、Pan 6 [ATCC VR-592]、およびPan 7 [ATCC VR-593]である。他の好適なアデノウイルスとしては、限定するものではないが、米国特許第6,083,716号に記載されるチンパンジーアデノウイルスC1とC68 (Pan9)、サルアデノウイルス、例えば、限定するものではないが、SV1 [VR-195]、SV25 [SV-201]、SV35、SV15、SV-34、SV-36、SV-37、およびヒヒアデノウイルス[VR-275]が挙げられる。Pan 5 (C5とも呼ばれる)、Pan 6 (C6とも呼ばれる)、Pan 7 (C7とも呼ばれる)、SV1、SV25、およびSV39の配列は文献に記載されている[WO 03/046124、2003年6月5日公開]。ハイブリッドアデノウイルスベクターおよびサルアデノウイルスSA18から構築されたベクターが記載されている国際公開番号WO 04/16614も参照されたい。
【0036】
チンパンジーアデノウイルスは、標的集団中にアデノウイルスに対する既存の免疫(特に交差中和抗体)が存在しないため、ヒトアデノウイルス血清型より有利であると考えられる。チンパンジーアデノウイルスと既存の中和抗体応答との交差反応は、標的集団の2%に存在するにすぎないのに対して、ある種の候補ヒトアデノウイルスベクターの場合には35%に存在する。チンパンジーアデノウイルスは、より一般的なヒトアデノウイルスのサブタイプAd2およびAd5とは明確に区別されるが、一般的なサブタイプではないヒトアデノウイルスのサブグループEのAd4とはより密接な関係がある。Pan 6はPan 5、7および9とそれほど密接な関係はない。
【0037】
本発明のアデノウイルスは複製欠損型であってよい。これは、該ウイルスが野生型ウイルスと比較して、非補完細胞(non-complementing cell)における複製能力が低下していることを意味する。この複製能の低下は、ウイルスを変異させることにより、例えば複製に関与する遺伝子を欠失させること(例えば、E1a、E1b、E3またはE4遺伝子の欠失)により、もたらされ得る。
【0038】
本発明によるアデノウイルスベクターは、機能的E1欠失を含む複製欠損型アデノウイルスであってよい。このように、本発明によるアデノウイルスベクターは、アデノウイルスのE1aとE1bを発現する能力の欠如のため、すなわちE1aとE1bが機能的に欠失されているため、複製欠損型でありうる。上記の組換えアデノウイルスはまた、他の遺伝子に機能的欠失を有していてもよく(WO 03/000283を参照)、例えば、E3またはE4遺伝子に欠失を有する。アデノウイルス遅延型初期遺伝子E3は、組換えウイルスの一部を形成するサルアデノウイルス配列から除去することができる。E3の機能は、組換えアデノウイルス粒子の生産に必要ではない。したがって、本発明において有用な組換えサルアデノウイルスをパッケージングするために、この遺伝子産物の機能をもとに戻すことは不必要である。1つの特定の実施形態において、上記の組換え(サル)アデノウイルスは、E1およびE3遺伝子が機能的に欠失している。このようなベクターの構築は、Royら, Human Gene Therapy 15:519-530, 2004に記載されている。
【0039】
組換えアデノウイルスはまた、E4遺伝子の機能的欠失を有するように構築することもできるが、E4のORF6機能を保持していることが望ましい。本発明によるアデノウイルスベクターは、遅延型初期遺伝子E2aに欠失を含んでいてもよい。欠失は、サルアデノウイルスゲノムの後期遺伝子L1〜L5のいずれかに生じさせることもできる。同様に、中期遺伝子IXおよびIVaの欠失も有用であり得る。
【0040】
他の欠失は、アデノウイルスの他の構造遺伝子または非構造遺伝子に生じさせることができる。こうした欠失は個々に用いることができる。すなわち、本発明において用いるアデノウイルス配列は、E1欠失のみを含み得る。あるいは、遺伝子全体の欠失または生物学的活性を破壊するのに有効な遺伝子の一部の欠失を任意の組み合わせで用いてもよい。例えば、ある代表的なベクターにおいて、アデノウイルス配列は、E1遺伝子とE4遺伝子の欠失、またはE1、E2aおよびE3遺伝子の欠失、またはE1およびE3遺伝子の欠失(例えば、E1aおよびE1bの機能的欠失、ならびにE3の少なくとも一部の欠失)、またはE1、E2aおよびE4遺伝子の欠失(E3遺伝子などの欠失を伴うまたは伴わない)を有し得る。このような欠失は、これらの遺伝子の一部の欠失または完全な欠失であってよく、所望の結果を達成するために、温度感受性変異などの他の変異と組み合わせて用いることができる。
【0041】
本発明のアデノウイルスベクターは、ウイルスが複製し得る好適な細胞株で製造することができる。特に、ウイルスベクターから失われて複製特性の欠陥をもたらす因子を供給する、補完性細胞株を使用することができる。例えば、補完性細胞株は、E1、またはE1およびE3、またはE1、E3およびE4を発現することができる。限定するものではないが、このような細胞株は、とりわけHeLa [ATCC受託番号CCL 2]、A549 [ATCC受託番号CCL 185]、HEK 293、KB [CCL 17]、Detroit [例えば、Detroit 510, CCL 72]およびWI-38 [CCL 75]細胞であってよい。これらの細胞株は全て、American Type Culture Collection (10801 University Boulevard, Manassas, Virginia 20110-2209)から入手することができる。他の好適な親細胞株は他の供給元から入手することが可能であり、例えばPER.C6(登録商標)細胞のような、Centre for Applied Microbiology and Research (CAMR, UK)所在のEuropean Collection of Animal Cell Cultures (ECACC)にECACC番号96022940として寄託された細胞に代表される。
【0042】
別の態様において、本発明は、少なくともHIV抗原RT、NefおよびGagまたはこれらの免疫原性誘導体もしくは免疫原性断片を、Gag、RT、Nefの順にコードするポリヌクレオチド(1つまたは複数)を含むアデノウイルスベクター、すなわち、Gag、RT、Nefの順に転写されるように配置された少なくともHIV抗原RT、NefおよびGagまたはこれらの免疫原性誘導体もしくは免疫原性断片をコードするポリヌクレオチド(1つまたは複数)を含むアデノウイルスベクターを提供する。
【0043】
例えば、本発明のアデノウイルスベクターは、Gagまたはその免疫原性誘導体もしくは免疫原性断片がRTまたはその免疫原性誘導体もしくは免疫原性断片をコードするポリヌクレオチド配列に融合し、これがNefまたはその免疫原性誘導体もしくは免疫原性断片をコードするポリヌクレオチド配列に融合しているポリヌクレオチドを、単一の異種プロモーターの制御下に含有することができ、その際、該遺伝子のGag部分は該ポリヌクレオチドの5'末端に存在する。
【0044】
本発明の別の実施形態において、3つの抗原は各々、それ自身のプロモーターを通じて発現され、該プロモーターは各々同じでも異なっていてもよい。本発明のさらに別の実施形態において、3つの抗原のうち2つは、単一のプロモーターに連結された融合体を形成し、第3の抗原は、第1のプロモーターと同一でも異なっていてもよい第2のプロモーターに連結される。例えば、GagとRTが第1のプロモーターに連結され、Nefが第2のプロモーターに連結されていてもよい。
【0045】
少なくとも3つのHIV抗原またはこれらの免疫原性誘導体もしくは免疫原性断片をコードするポリヌクレオチド(1つまたは複数)を、アデノ(Adeno)欠失領域のいずれか、例えばE1欠失領域に挿入することができる。
【0046】
抗原をコードする2つ以上のポリヌクレオチドを1つの融合体として連結することができるが、結果として生じるタンパク質は、融合タンパク質として発現させてもよいし、または別個のタンパク質産物として発現させてもよいし、あるいは融合タンパク質として発現させた後、より小さなサブユニットに分解してもよい。
【0047】
1つの態様において、本発明は、本発明のベクターによって発現される融合タンパク質、例えば、ヒト体内で産生される融合タンパク質を提供する。
【0048】
本発明によるベクターに含まれる、例えばNef、GagまたはRTをコードする、1つ以上のHIV配列は、例えば、高発現されたヒト遺伝子にコドン使用が似るように、哺乳動物細胞用にコドンを最適化することができる。これらのHIV配列のコドン最適化は、WO 03/025003にさらに記載されている。
【0049】
例えば、本発明によるアデノウイルスベクター中でGagおよび/またはRTをコードするポリヌクレオチドは、上記のようにコドン最適化することができる。
【0050】
本発明によるアデノウイルスベクター中のGag配列は、Gag p6ポリペプチドをコードする配列を除外し得る。本発明において使用するGag配列の特定の例には、p17および/またはp24コード配列が含まれる。
【0051】
本発明のRT配列は、いずれかの逆転写酵素活性を実質的に不活化する変異をコードし得る。1つの特定の不活化変異は、W(トリプトファン)229によるK(リシン)の置換を含む。WO 03/025003を参照されたい。
【0052】
RT遺伝子は、上記のとおり、HIVゲノム中の大きいPol遺伝子の成分である。本発明によるアデノウイルスベクターに含まれるRTコード配列は、Pol、または少なくともRTをコードするPol断片の状態で存在しうることが理解されるだろう。このようなPol断片は、Polの主要なCTLエピトープを保有する。1つの具体例において、RTは、RTのp51断片のみまたはp66断片のみとして含まれる。
【0053】
場合により、本発明において用いるNef配列は、N末端領域をコードする配列を除去するため、すなわち30〜85アミノ酸、例えば60〜85アミノ酸、特にN末端の65アミノ酸を除去するため、末端切断(トランケーション)されている(本明細書では、後者の切断型をtrNefと呼ぶ)。あるいは、またはさらに、Nefは1つ以上のミリストイル化部位を除去するように改変されていてもよい。例えば、Gly 2ミリストイル化部位を欠失または置換によって除去する。あるいは、またはさらに、Nefは、ロイシン174とロイシン175のジロイシンモチーフを、1個もしくは両方のロイシンの欠失または置換により変更するように改変することができる。CD4下方制御におけるジロイシンモチーフの重要性は、例えば、Bresnahan P.A.ら(1998) Current Biology, 8(22): 1235-8に記載されている。
【0054】
本発明による構築物は、Gag、PolおよびNefを含むことが可能であり、その際、これら天然抗原のCTLエピトープの少なくとも75%、少なくとも90%、または少なくとも95%、例えば96%が存在する。
【0055】
上記のp17/p24 Gag、p66 RT、および末端切断型Nefを含む本発明の構築物においては、天然Gag PolおよびNef抗原のCTLエピトープの96%が存在する。
【0056】
本発明の1つの実施形態は、p17、p24(コドン最適化)Gag、p66 RT(コドン最適化)、末端切断型Nef(末端アミノ酸1-85をコードするヌクレオチドを欠く-「trNef」)をGag、RT、Nefの順にコードするポリヌクレオチド(1つまたは複数)を含むアデノウイルスベクターを提供する。
【0057】
本発明の構築物は、
1. p17、p24(コドン最適化)Gag - p66 RT(コドン最適化) - 末端切断型Nef;
2. 末端切断型Nef - p66 RT(コドン最適化) - p17、p24(コドン最適化)Gag;
3. 末端切断型Nef - p17、p24(コドン最適化)Gag - p66 RT(コドン最適化);
4. p66 RT(コドン最適化) - p17、p24(コドン最適化)Gag - 末端切断型Nef;
5. p66 RT(コドン最適化) - 末端切断型Nef - p17、p24(コドン最適化)Gag;
6. p17、p24(コドン最適化)Gag - 末端切断型Nef - p66 RT(コドン最適化)
を含む。
【0058】
本発明のポリヌクレオチド(1つまたは複数)は、Gag、RTおよびNefをコードする配列の間に存在するリンカー配列を有してもよい。このようなリンカー配列の長さは、例えば、最大20アミノ酸までである。特定の例において、この長さは1〜10アミノ酸、または1〜6アミノ酸、例えば2〜4アミノ酸であってよい。
【0059】
本発明のポリヌクレオチドは、さらなるHIV配列を含んでいてもよい。特に、これらのポリヌクレオチドはHIVのenvタンパク質またはその免疫原性誘導体もしくは免疫原性断片を含み得る。envの好適な形態は、gp120、gp140およびgp160である。他の好適なHIV配列としては、限定するものではないが、Tat、Rev、Vpu、VprおよびVifが挙げられる。このように本発明は、HIVのenvタンパク質またはその免疫原性誘導体もしくは免疫原性断片と共に、HIV抗原RT、NefおよびGagまたはこれらの免疫原性誘導体もしくは免疫原性断片を、Gag、RT、Nefの順にコードするポリヌクレオチド(1つまたは複数)を含むアデノウイルスベクターをさらに提供する。
【0060】
本発明はさらに、本発明のアデノウイルスベクターを、1つ以上のHIV抗原をコードするポリヌクレオチド(1つまたは複数)を含む第2のアデノウイルスベクターと組み合わせて含有する免疫原性組成物を含む。
【0061】
本発明に含まれる全てのHIV配列について、これらの配列は、必ずしも完全長つまり天然のタンパク質をコードする配列に相当しないことが理解されるだろう。末端切断された、あるいは変更された(例えば変異した)タンパク質のような免疫原性誘導体も意図され、少なくとも1つのHIVエピトープ、例えばCTLエピトープ、典型的には少なくとも8アミノ酸のペプチドをコードする断片などの免疫原性誘導体も意図される。長さが少なくとも8アミノ酸、例えば8〜10アミノ酸、または20、50、60、70、100、150もしくは200アミノ酸までの断片をコードするポリヌクレオチドは、コードされるオリゴペプチドまたはポリペプチドがHIV抗原性を示す限り、すなわち主要なCTLエピトープが上記のオリゴペプチドまたはポリペプチドに保持される限り、本発明の範囲に含まれると考えられる。本明細書において、主要なCTLエピトープは、in vivoで免疫応答を誘発することができるエピトープとして定義される。本発明のポリヌクレオチド配列によってコードされるHIVポリペプチド分子は、天然タンパク質の長さの少なくとも50%の断片であってよく、該断片は複数の変異を含み得るが、少なくとも1つのHIVエピトープを保持してHIV抗原性を示すものである。このようなHIV抗原性は、例えば抗体または細胞性応答を測定することにより確認することができる。同様に、本発明の免疫原性誘導体はHIV抗原性を示さなければならない。免疫原性誘導体は、天然タンパク質に比して幾つかの潜在的な利点(例えば、酵素活性(RT)またはCD4下方制御(Nef)などの、ワクチン抗原には好ましくない天然タンパク質の機能の低下または除去)を提供し得る。本発明のポリヌクレオチド配列は、本明細書に記載される本発明のコドン最適化態様に従って、哺乳動物細胞用にコドン最適化することができる。
【0062】
本発明はさらに、以下のステップ:
a) アデノウイルスベクターを提供するステップ、
b) 好適なプロモーターに機能的に連結されたHIV抗原配列を含有するプラスミドを提供するステップ、
c) プラスミドとベクターの両方を用いて細胞をトランスフェクトするステップ、
d) 組換えが起こるのに十分な時間を与えるステップ、および
e) HIV抗原配列を含有する組換えウイルスベクターを回収するステップ、
を含んでなる、本発明のベクターを作製する方法を提供する。
【0063】
別の態様において、本発明は、哺乳動物に好適な量の本発明の免疫原性組成物を投与することを含んでなる、哺乳動物に免疫応答を引き起こす方法を提供する。
【0064】
本発明は特に、HIV-1に関係し得る。本明細書に記載される構築物は、任意のHIVクレード、例えばクレードBまたはクレードC、特にクレードBに由来し得る。
【0065】
本発明のアデノウイルスベクターに用いるプロモーターは、HCMV IE遺伝子に由来するプロモーターとすることができ、例えば、WO 02/36792に記載されるような、エキソン1を含むHCMV IE遺伝子の5'非翻訳領域が含まれる。
【0066】
本発明の医薬組成物は、標的細胞への形質導入を可能にするのに十分な量で、また、十分なレベルの遺伝子導入と発現をもたらして、過度の副作用を伴うことなく、あるいは医学的に許容可能な生理的効果を伴って、治療効果を与えるのに十分な量で、投与することが可能であり、こうした投与量は、医療技術分野の当業者が決定し得る。通常の製薬上許容される投与経路としては、限定するものではないが、網膜への直接送達および他の眼内送達法、肝臓への直接送達、吸入、鼻腔内、静脈内、筋肉内、気管内、皮下、皮内、直腸、口腔および他の非経口投与経路が挙げられる。
【0067】
投与経路は、必要に応じて組み合わせて、または投与される遺伝子産物もしくは症状に応じて調整することができる。投与経路は、主に治療対象の症状の性質によって決まるだろう。
【0068】
本発明のウイルスベクターの投与量は、主に患者の治療される症状、年齢、体重および健康状態などの要因に応じて決まり、このため患者間で異なり得る。例えば、上記ウイルスベクターの、治療上有効な成人または動物に対する投与量は、通常、約1 x 106〜約1 x 1015個、約1 x 1011〜1 x 1013個、または約1 x 109〜1 x 1012個のウイルス粒子の濃度を有する担体約100μL〜約100 mLの範囲内である。投与量は、投与される動物の大きさおよび投与経路に応じて変動するだろう。例えば、ヒトまたは動物への筋肉注射用の好適な投与量(約80 kgの動物に対する)は、単一の部位について、1 mL当たり約1 x 109〜約5 x 1012個の粒子の範囲である。場合により、複数の投与部位へ送達することができる。別の例において、ヒトまたは動物への好適な投与量は、口腔製剤について、約1 x 1011〜約1 x 1015個の粒子の範囲である。当業者であれば、投与経路および上記の組換えベクターを用いる治療用途またはワクチン用途に応じて、これらの投与量を調整することができる。治療用産物の発現レベル、または免疫原については循環抗体のレベルをモニターして、投与量の投与頻度を決定することができる。しかし、当業者であれば、投与スケジュールを決定するための他の方法を容易に理解するであろう。
【0069】
医薬組成物の投与は、1つまたは2つ以上の個別用量の形式をとることが可能であり、例えば、同一のポリヌクレオチドを含むアデノウイルスの反復投与として、または異種「プライム・ブースト」ワクチン接種計画で投与することができる。異種プライム・ブースト計画は、初回免疫と追加免疫とで異なる形態のワクチンを投与するもので、各免疫は、それぞれ2回以上の投与を含み得る。初回免疫組成物と追加免疫組成物とは少なくとも1種類の抗原を共通して含むと考えられるが、それは必ずしも同一の形態の抗原である必要はなく、同一の抗原の異なる形態であってよい。
【0070】
本発明のベクターを用いるプライム・ブースト計画は、異種DNAとアデノウイルスベクターのプライム・ブースト(例えば、裸のDNAの初回量とその後のアデノウイルスベクターのブースト、あるいは、例えばアデノウイルスベクターのプライムとその後の1回以上の裸のDNAのブースト)の形式をとり得る。このようなDNAブーストは、DNAの筋肉内投与または皮内投与により、または粒子加速法により送達することができる。あるいはまた、このようなプライム・ブースト計画は、例えば、本発明のタンパク質およびアデノウイルスベクターを含むことが可能であり、この場合、初回量がタンパク質を含み、追加量がアデノウイルスベクターを含んでいてもよいし、または初回量がアデノウイルスベクターを含み、追加量がタンパク質を含んでいてもよい。
【実施例】
【0071】
実施例1:E1/E3が欠失されたPan 6およびPan 7アデノウイルスの構築
1. 組換えE1欠失SV-25ベクターの作製
遺伝子操作でE1を欠失させたことを除いて完全なSV-25ゲノムを含むプラスミドを構築した。このE1欠失部位に、制限酵素I-CeuIとPI-SceIの認識部位を挿入した。これらの認識部位は、これら2つの酵素認識部位にトランスジーン発現カセットが挟まれているシャトルプラスミドからのトランスジーンの挿入を可能にすると考えられる。
【0072】
制限部位SwaI-SnaBI-SpeI-AflII-EcoRV-SwaIを含む合成リンカーを、EcoRIとNdeIで切断したpBR322中にクローン化した。この操作は、2種類の合成オリゴマーSV25T (5'-AAT TTA AAT ACG TAG CGC ACT AGT CGC GCT AAG CGC GGA TAT CAT TTA AA-3')とSV25B (5'-TAT TTA AAT GAT ATC CGC GCT TAA GCG CGA CTA GTG CGC TAC GTA TTT A-3')を一緒にアニーリングし、これをEcoRIとNdeIで消化したpBR322に挿入することによって行った。アデノウイルス(Ad) SV25の左端(bp 1〜1057)を、上記リンカーのSnaBI部位とSpeI部位の間にクローン化した。Ad SV25の右端(bp 28059〜31042)は、該リンカーのAflII部位とEcoRV部位の間にクローン化した。その後アデノウイルスE1を、クローン化した左端からEcoRI部位(bp 547)とXhoI部位(bp 2031)の間で以下のとおり切り出した。PCRで生成した、pShuttle (Clontech)に由来するI-CeuI-PI-SceIカセットを、EcoRI部位とSpeI部位の間に挿入した。その後、Ad SV-25の10154bpのXhoI断片(bp2031〜12185)をSpeI部位に挿入した。結果として生じたプラスミドをHindIIIで消化し、Ad SV-25の18344bpのHindIII断片(bp 11984〜30328)を挿入することにより構築物(pSV25)を完成させ、組換えアデノウイルスの作製に適したE1欠失アデノウイルスSV25の完全な分子クローンを生成した。場合により、新たに作出したpSV25ベクタープラスミドのI-CeuI部位とPI-SceI部位に所望のトランスジーンを挿入してもよい。
【0073】
マーカー遺伝子を含むAd SV25を作製するため、プラスミドpShuttle(Clontech)中にあらかじめクローン化されたGFP(緑色蛍光タンパク質)発現カセットを制限酵素I-CeuIとPI-SceIで切除し、同じ酵素で消化したpSV25(または本明細書中に記載される別のAdチンパンジープラスミド)中に連結した。結果として生じたプラスミド(pSV25GFP)をSwaIで消化して細菌プラスミド骨格を分離し、E1補完性細胞株HEK293にトランスフェクトした。約10日後、複製ウイルスの存在を示す細胞変性効果が観察された。GFPを発現する、Ad SV25に基づくアデノウイルスベクターが首尾よく生成されたことは、トランスフェクトされた培養物からの上清を新鮮な細胞培養物に加えることにより確認した。二次的な感染細胞の存在は、これらの細胞集団における緑色蛍光の観察により確認した。
【0074】
2. E3欠失Pan-6およびPan-7ベクターの構築
本発明のアデノウイルスベクターのクローニング能力を高めるため、E3領域を欠失させることができる。なぜならこの領域は、培養中のウイルスの増殖に必要のない遺伝子をコードするからである。そのために、Pan-5、Pan-6、Pan-7、およびC68のE3欠失型が作製された(E31-9を含む3.5kbのNru-AvrII断片が欠失される)。
【0075】
Pan6に基づくベクターにおけるE3欠失
E1欠失pPan6-pkGFP分子クローンをSbf IとNot Iで消化し、19.3kbの断片を単離してSbf I部位に連結し直した。結果として生じた構築物pPan6-Sbf I-E3をEco 47 IIIとSwa Iで処理し、pPan6-E3を生成した。最終的に、pPan6-pkGFPのSbf I消化から得られた21kbのSbf I断片をpPan6-E3にサブクローン化し、E3に4kbの欠失を有するpPan6-E3-pkGFPを作出した。
【0076】
E3欠失Pan7ベクター
Pan 7でE3欠失を達成するために、上記と同じ方法を用いた。最初に、E3領域にわたる5.8kbのAvr II断片をpSL-1180にサブクローン化し、その後Nru I消化によりE3を欠失させた。結果として生じたプラスミドをSpe IとAvr IIで処理して4.4kbの断片を取得し、pPan7-pkGFPのAvr II部位にクローン化することにより、もとのE3を含むAvr II断片をそれぞれ置換した。最終的なpPan7-E3-pkGFP構築物は、3.5kbのE3欠失を有していた。
【0077】
これらのおよび他のPanアデノウイルス血清型におけるE1、E3およびE4欠失型の構築についての十分な説明は、WO 03/0046124に記載されている。さらなる情報は、Human Gene Therapy 15:519-530 (WO 03/046124)からも入手可能である。
【0078】
実施例2
Gag、RT、Nef配列の構築
この構築は、WO 03/025003に十分に記載されている。
【0079】
プラスミドp73i-Tgrn
1. プラスミド:p73i-GRN2クローン #19(p17/p24(コドン最適化)/RT(コドン最適化)trNef) - 修復型
目的遺伝子
HIV-1クレードBのHXB2株由来の、lowa長さのHCMVプロモーター + エキソン1の下流で、かつウサギβグロビンポリアデニル化シグナルの上流の、コドン最適化Gagのp17/p24部分、コドン最適化RTおよび末端切断型Nef遺伝子。
【0080】
プラスミドp17/24trNef1に由来するtrNef遺伝子を含むプラスミドはPCRエラーを含み、このエラーは、Nefの末端から19アミノ酸の位置にアルギニン(R)からヒスチジン(H)へのアミノ酸変化をもたらす。このエラーをPCR突然変異誘発によって修正し、修正したNefをp7077-RT3に由来するコドン最適化RTにPCRで継ぎ合わせ、継ぎ合わせた断片をApaIとBamHIで切断して、ApaI/BamHIで切断したp73i-GRNにクローン化した。
【0081】
プライマー
プライマーを用いたp7077-RT3からのコドン最適化RTのPCR増幅:
(PCR全体を通して、ポリメラーゼ = PWO (Roche))
センス:U1
GAATTCGCGGCCGCGATGGGCCCCATCAGTCCCATCGAGACCGTGCCGGTGAAGCTGAAACCCGGGAT
アンチセンス:AScoRT-Nef
GGTGTGACTGGAAAACCCACCATCAGCACCTTTCTAATCCCCGC
サイクル:95℃(30秒)、その後95℃(30秒)、55℃(30秒)、72℃(180秒)を20サイクル、その後72℃(120秒)にして4℃に保持
1.7kbのPCR産物をゲル精製した。
【0082】
プライマーを用いたp17/24trNef1からの5' NefのPCR増幅:
センス:S-Nef
ATGGTGGGTTTTCCAGTCACACC
アンチセンス:ASNef-G:
GATGAAATGCTAGGCGGCTGTCAAACCTC
サイクル:95℃(30秒)、その後95℃(30秒)、55℃(30秒)、72℃(60秒)を15サイクル、その後72℃(120秒)にして4℃に保持。
【0083】
プライマーを用いたp17/24trNef1からの3' NefのPCR増幅:
センス:SNEF-G
GAGGTTTGACAGCCGCCTAGCATTTCATC
アンチセンス:AStrNef
CGCGGATCCTCAGCAGTTCTTGAAGTACTCC
サイクル:95℃(30秒)、その後95℃(30秒)、55℃(30秒)、72℃(60秒)を15サイクル、その後72℃(120秒)にして4℃に保持。
【0084】
上記のPCR産物をゲル精製した。最初に、5'(S-Nef)と3'(AstrNef)のプライマーを用いて2つのNef産物を継ぎ合わせた。
サイクル:95℃(30秒)、その後95℃(30秒)、55℃(30秒)、72℃(60秒)を15サイクル、その後72℃(180秒)にして4℃に保持。
【0085】
このPCR産物をPCR洗浄し、U1とAstrNefのプライマーを用いて上記のRT産物に継ぎ合わせた。
サイクル:95℃(30秒)、その後95℃(30秒)、55℃(30秒)、72℃(180秒)を20サイクル、その後72℃(180秒)にして4℃に保持。
【0086】
上記の2.1kbの産物をゲル精製し、ApaIとBamHIで切断した。プラスミドp73I-GRNもApaIとBamHIで切断してゲル精製し、ApaI-Bam RT3trNefと連結してp17/p24(コドン最適化)/RT(コドン最適化)trNef遺伝子を再生成した。
【0087】
2. プラスミド:p73I-RT w229k (不活性化RT)
目的遺伝子
lowa長さのHCMVプロモーター + エキソン1の下流で、かつウサギβグロビンポリアデニル化シグナルの上流の不活性化RT遺伝子の生成。
【0088】
治療用ワクチンに活性型HIV RT種を使用することの懸念のため、この遺伝子は不活性化させることが望ましい。不活性化は、上記RT(P73I-GRN2由来)の229位のアミノ酸をトリプトファン(Trp)からリシン(Lys)にPCR突然変異誘発させることにより達成した(R7271 p1-28)。
【0089】
プライマー
プライマーを用いた5'RT + 変異のPCR増幅:
(PCR全体を通して、ポリメラーゼ = PWO (Roche))
センス:RT3-u:1
GAATTCGCGGCCGCGATGGGCCCCATCAGTCCCATCGAGACCGTGCCGGTGAAGCTGAAACCCGGGAT
アンチセンス:AScoRT-Trp229Lys
GGAGCTCGTAGCCCATCTTCAGGAATGGCGGCTCCTTCT
サイクル:
1 x [94℃ (30秒)]
15 x [94℃ (30秒)/55℃ (30秒)/72℃ (60秒)]
1 x [72℃ (180秒)]
PCR ゲル精製。
【0090】
プライマーを用いた3'RT + 変異のPCR増幅:
アンチセンス:RT3-l:1
GAATTCGGATCCTTACAGCACCTTTCTAATCCCCGCACTCACCAGCTTGTCGACCTGCTCGTTGCCGC
センス:ScoRT-Trp229Lys
CCTGAAGATGGGCTACGAGCTCCATG
サイクル:
1 x [94℃ (30秒)]
15 x [94℃ (30秒)/55℃ (30秒)/72℃ (60秒)]
1 x [72℃ (180秒)]
PCR ゲル精製。
【0091】
上記のPCR産物をゲル精製し、RTの5'末端と3'末端を、5'(RT3-U1)および3'(RT3-L1)プライマーを用いて継ぎ合わせた。
サイクル:
1 x [94℃ (30秒)]
15 x [94℃ (30秒)/55℃ (30秒)/72℃ (120秒)]
1 x [72℃ (180秒)]
【0092】
上記のPCR産物をゲル精製し、NotIとBamHIの制限部位を利用してp7313ieにクローン化し、p73I-RT w229kを生成した(図13参照)。
【0093】
3. プラスミド:p73i-Tgrn
目的遺伝子
HIV-1クレードBのHXB2株由来の、lowa長さのHCMVプロモーター + エキソン1の下流で、かつウサギβグロビンポリアデニル化シグナルの上流のコドン最適化Gagのp17/p24部分、コドン最適化RTおよび末端切断型Nef遺伝子。
【0094】
活性型RTを含む3種融合構築物は、規制当局がヒトへの使用を認めていないため、p73i-RT w229kからNheIとApaIで切断した断片をNheI/ApaI切断したp73i-GRN2#19に挿入することにより、RTの不活性化を達成した(図14)。この操作により、RTの229位にトリプトファン(W)→リシン(K)変更が生じる。
【0095】
Tgrnプラスミド挿入物の完全配列を図7に示す。この配列は、p17 p24(コドン最適化)Gag、p66 RT(コドン最適化且つ不活性型)および末端切断型Nefを含む。
【0096】
Gag、RTおよびNefの代替構築物は以下のとおりである:
trNef - p66 RT(コドン最適化) - p17、p24(コドン最適化)Gag、
trNef - p17、p24(コドン最適化)Gag - p66 RT(コドン最適化)、
p66 RT(コドン最適化) - p17、p24(コドン最適化)Gag - trNef、
p66 RT(コドン最適化) - trNef - p17、p24(コドン最適化)Gag、
p17、p24(コドン最適化)Gag - trNef - p66 RT(コドン最適化)
これらの構築物の完全配列を、それぞれ図8〜12に示す。
【0097】
実施例3
Gag、RT、Nef配列のアデノウイルスへの挿入
pShuttleプラスミドへのGRN発現カセットのサブクローニング
プロモーター、cDNAおよびポリアデニル化シグナルからなる発現カセット全体を、Sph IとEcoR Iの二重消化によってpT-GRN構築物から単離した。Sph I/EcoR I断片のSph I末端をKlenowで埋めて、pShuttleプラスミドのEcoR I部位とMlu I部位(Mlu I末端を平滑化した)の間にクローン化した。
【0098】
クローニング工程の間に、付加的なフランキング配列がHIV発現カセットに付随するようになった。この配列はCer配列として知られており、その機能は不明である。
【0099】
Pan6 およびPan7ベクターのE1/E3欠失分子クローンへのGRN発現カセットの導入
発現カセットをI-Ceu IとPI-Sce Iとの消化によりpShuttleから回収し、Pan6およびPan7ベクターの分子クローンの同じ部位にクローン化した。組換えクローンは青白選択を通じて同定し、広範囲の制限酵素解析で確認した。
【0100】
組換えウイルスのレスキューと増殖
C6およびC7ベクターの分子クローンを適切な制限エンドヌクレアーゼ(それぞれPmeIとPacI)で処理してインタクトな線状ベクターゲノムを放出させ、リン酸カルシウム法を用いて293細胞にトランスフェクトした。トランスフェクトされた細胞中で十分な細胞変性効果が観察された場合、粗ウイルス溶解物を回収し、293細胞中でラージスケールの感染(1x109細胞)へと徐々に増やした。ラージスケール感染物からのウイルスを標準的なCsCl沈降法で精製した。
【0101】
また、pShuttleプラスミドは、EcoRIとXmnIで切断することにより3'リンカー配列を除去してさらに切り取り、プラスミドのサイズを縮小してpShuttleGRNcを生成することができる。この改変プラスミドは、上記の方法を用いてさらなるPan7ウイルス(C7-GRNc)を生成するために使用し得る。
【0102】
他の構築物も同様にPan 6およびPan 7アデノウイルスの両方に挿入した。しかし、p66 RT(コドン最適化) - trNef - p17、p24(コドン最適化)Gag挿入物を有するPan 6の作製は成功しなかった。
【0103】
実施例4
マウス免疫原性モデル
HIV抗原RT、NefおよびGagの再配列挿入物を含有する一連のPan6およびPan7ベクター(RGN、NRG、NGR、GRN、およびGNR)をin vivoでの一次免疫反応について試験した。Pan6ウイルスを試験するため3つの実験を、Pan7ウイルスを試験するため2つの実験を行った。各アデノウイルスを1x108粒子の用量でBalb/c(K2d)マウスの片方の後肢に50μl量で筋肉内投与した。この用量を選択したのは、優れたレベルの細胞性免疫応答を誘導することが以前にわかっていた(未公開)ためである。表1は、これらの実験で比較したアデノウイルスをまとめたものである。
【表1】
【0104】
Gag、NefおよびRTの特異的エピトープに対するペプチドまたはタンパク質を用いたin vitro刺激の後、CD8応答とCD4応答の発生をELIspotアッセイにより初回免疫の14日後と28日後に測定した。これらの結果は、全ての変異体が、空ベクターの対照と比較して強い一次免疫応答(IFNγとIL-2の両方の生成により測定される)を生じる能力があるという強力な証拠を提供する(データ示されず)。
【0105】
これらの研究から得られたデータは、SASプログラム(version 9.1.3 Service Pack 2)による混合モデル(Proc Mixed)の分散分析(ANOVA)を使用して統計的に解析し、Pan6およびPan7におけるRNG変異体のランクを別々の時点で決定した。IFNγ産生に関しは、CD8ペプチドに対する応答の合計をGagとRTについて定量し、一方、IL-2のELIspotデータは、CD4ペプチドに対する応答の合計でGag、NefおよびRTについて評価した。
【0106】
前記パネルの変異体のランク付けは、Bayesianモデルを用いて計算し(100,000個の事後サンプルをもたらす平坦事前分布(flat prior)を有するProc MixedのPriorステートメントを使用して実施した;Tierney, L. (1994), "Markov Chains for Exploring Posterior Distributions"(考察を含む)、ならびにAnnals of Statistics, 22, 1701-1762; Gelfand, A.E., Hills, S.E., Racine-Poon, A.およびSmith, A.F.M. (1990), "Illustration of Bayesian Inference in Normal Data Models Using Gibbs Sampling," Journal of the American Statistical Association, 85, 972-985)を参照)、各変異体の「最良」としての確率を、研究された実験条件から得られたデータに基づいて推測した。
【0107】
図1は、Bayesian法で推測された、14日目と28日目の、各ペプチドによるIFNγとIL-2に関するPan6のCD4およびCD8応答の合計を表す。
【0108】
図2は、Bayesian法で推測された、14日目と28日目の、各ペプチドによるIFNγとIL-2に関するPan7のCD4およびCD8応答の合計を表す。
【0109】
挿入物は全て、空ベクターの対照と比較して免疫応答の著しい増加を示す。統計分析は、異なるウイルス間に有意差がないことを示す。
【0110】
実施例5
ブタ免疫原性モデル
幾つかの研究から得られた結果は、ブタが候補ワクチンの免疫原性を試験するための優れたモデルであることを示していた。4つの候補NHPアデノウイルスの免疫原性をミニブタで検証するための研究を設定した。1グループ5匹のミニブタをPAN6GRN、PAN6NGR、PAN7GRNまたはPAN7NGRで初回免疫した(使用したバッチの詳細については表2を参照)。各動物に合計3 x 1010個のアデノウイルス粒子を筋肉内経路により(1.0ml量を各内側大腿筋間で均等に分割して用いて)投与した。
【表2】
【0111】
免疫前、および免疫後に間隔をあけて各動物から血液サンプルを回収した。末梢血単核細胞を単離し、RT、NefおよびGagペプチドライブラリーのプールとタンパク質を用いてin vitroで再刺激した。上記のペプチドライブラリーのプールは、RT、NefおよびGagの配列全体にわたり11アミノ酸だけ重複している15-merのペプチドからなり、in vivoマウス実験で使用したペプチドと同一であった。
【0112】
これらのブタ細胞によるインターフェロンγの産生を、ELIspotアッセイを用いて測定した。図3は、4回のサンプリング時点におけるRT、NefおよびGagペプチドライブラリーのプールに対する応答を示す。
【0113】
4種のウイルス全てに対する応答を免疫の7日後に検出した。4種のNHPウイルス全てに対する細胞性応答は、初回免疫後少なくとも5週間まで維持される。PAN6-GRNは、初回免疫の7日後に、IFN-γ ELIspotにより最も強い応答をもたらす。
【0114】
実施例6
霊長類免疫原性モデル
霊長類の試験的研究から得られた結果は、RT、NefおよびGagを発現するNHPアデノウイルスの筋肉内注射が、カニクイザルにおいて細胞性免疫応答を誘発したことを示した。
【0115】
4種類の候補NHPアデノウイルスの免疫原性をカニクイザルで検証するための研究を設定した。動物グループをPAN6-GRN、PAN6-NGR、PAN7-GRNまたはPAN7-NGRで一次免疫した(使用したウイルスバッチの詳細については表3を参照)。各動物に合計1011個のアデノウイルス粒子を筋肉内経路により(1.0ml量を各内側大腿筋間で均等に分割して使用して)投与した。
【表3】
【0116】
血液サンプルを免疫前と、免疫後に1週間間隔で回収した。末梢血単核細胞を単離し、RT、NefおよびGagペプチドライブラリーのプールを用いてin vitroで再刺激した。これらの霊長類細胞によるインターフェロンγの産生を、ELIspotアッセイを用いて測定した。図4は、3回のサンプリング時点における各グループの応答を示す。
【0117】
結果は、全グループが初回免疫の1週間後に強く応答し、この応答が免疫の少なくとも7週間後まで維持されたことを示す。この結果は、霊長類においてこの用量(すなわち1011個の粒子)で使用した場合、ベクター間に大差がないことを示唆する。
【0118】
実施例7
筋肉内投与(i.m.)で送達された、HIV GRN抗原をコードするNHPアデノウイルスの一定用量範囲に対する一次免疫後の応答
一次免疫で用いたアデノウイルスの用量の影響を評価するために、1グループ5匹のマウスを、漸増用量のNHPアデノウイルス(107〜1010個の粒子)で筋肉内(i.m.)に免疫した。陽性対照として、1グループの動物を、粒子媒介表皮送達を用いてDNA(2μg)で免疫した(ND5)。免疫後6日目と19日目に、動物を動物(科学的処置)法付則1に基づいて安楽死させ、脾臓を取り出した。免疫応答は、脾細胞を一晩刺激するために各抗原(GAGおよびRT)のペプチドライブラリーのプールを用いて、IFN-γ ELISPOTアッセイによりモニターした。図5は、2回のサンプリング時点における各グループの応答を示す。
【0119】
実施例8
皮内投与(i.d.)で送達された、HIV GRN抗原をコードするNHPアデノウイルスの一定用量範囲に対する一次免疫後の応答
一次免疫で用いたアデノウイルスの用量の影響を評価するために、1グループ5匹のマウスを、漸増用量のNHPアデノウイルス(107〜1010個の粒子)で皮内(i.d.)に免疫した。陽性対照として、1グループの動物を、粒子媒介表皮送達(PMED)を使用してDNA(1μg)で免疫した。免疫後7日目と14日目に、動物を動物(科学的処置)法付則1に基づいて安楽死させ、脾臓を取り出した。免疫応答はIFN-γ ELISPOTアッセイによりモニターした。脾細胞を、CD4またはCD8 T細胞を特異的に刺激する各抗原(GAGおよびRT)の確かなペプチドを用いて一晩刺激した。図6は、2回のサンプリング時点における各グループの応答を示す。
【0120】
これらの結果は、筋肉内免疫と皮内免疫の両方が、本発明の組成物の有効な投与経路であることを示唆する。
【表4】
【図面の簡単な説明】
【0121】
【図1】Pan6 HIVアデノウイルスのランク付け。これは、Bayesian法で推測された、14日目と28日目の、各ペプチドによるIFNγとIL-2に関するPan6のCD4およびCD8応答の合計を表す。y軸は、脾細胞100万個当たりのスポット形成細胞を表す。
【図2】Pan7 HIVアデノウイルスのランク付け。これは、Bayesian法で推測された、14日目と28日目の、各ペプチドによるIFNγとIL-2に関するPan7のCD4およびCD8応答の合計を表す。y軸は、脾細胞100万個当たりのスポット形成細胞を表す。
【図3】初回免疫の0、1、3および5週間後のRT、NefおよびGagペプチドライブラリーのプールに対するミニブタの応答。結果は、各動物についての、各ペプチドライブラリーのプールに対する応答の合計の平均±標準誤差である。データは、ペンシルバニア大学(University of Pennsylvania)から入手した。
【図4】初回免疫の0、1および2週間後のRT、NefおよびGagペプチドライブラリーのプールに対する霊長類の応答。結果は、各動物についての、各ペプチドライブラリーのプールに対する応答の合計の平均±標準誤差である。
【図5】筋肉内(i.m.)に送達された、HIV GRN抗原をコードするNHPアデノウイルスの一定用量範囲に対する一次免疫後の応答。マウスのグループ(n=5)を様々な用量のNHPアデノウイルス(107〜1010個の粒子)で免疫し、各抗原のペプチドライブラリーのプールに対する細胞性免疫応答を、IFN-γ ELISPOTアッセイを用いてモニターした(6日目および19日目)。
【図6】皮内(i.d.)に送達された、HIV GRN抗原をコードするNHPアデノウイルスの一定用量範囲に対する一次免疫後の応答。マウスのグループ(n=3)を様々な用量のNHPアデノウイルス(107〜1010個の粒子)で免疫し、特異的ペプチドに対する細胞性免疫応答を、IFN-γ ELISPOTアッセイを用いてモニターした(7日目および14日目)。
【図7−1】実施例2に記載される構築物p73i-Tgrnのポリヌクレオチド配列、アミノ酸配列および制限地図を示す。
【図7−2】実施例2に記載される構築物p73i-Tgrnのポリヌクレオチド配列、アミノ酸配列および制限地図を示す。
【図8−1】実施例2に記載される構築物p73i-Tnrgのポリヌクレオチド配列、アミノ酸配列および制限地図を示す。
【図8−2】実施例2に記載される構築物p73i-Tnrgのポリヌクレオチド配列、アミノ酸配列および制限地図を示す。
【図9−1】実施例2に記載される構築物p73i-Tngrのポリヌクレオチド配列、アミノ酸配列および制限地図を示す。
【図9−2】実施例2に記載される構築物p73i-Tngrのポリヌクレオチド配列、アミノ酸配列および制限地図を示す。
【図10−1】実施例2に記載される構築物p73i-Trgnのポリヌクレオチド配列、アミノ酸配列および制限地図を示す。
【図10−2】実施例2に記載される構築物p73i-Trgnのポリヌクレオチド配列、アミノ酸配列および制限地図を示す。
【図11−1】実施例2に記載される構築物p73i-Trngのポリヌクレオチド配列、アミノ酸配列および制限地図を示す。
【図11−2】実施例2に記載される構築物p73i-Trngのポリヌクレオチド配列、アミノ酸配列および制限地図を示す。
【図12−1】実施例2に記載される構築物p73i-Tgnrのポリヌクレオチド配列、アミノ酸配列および制限地図を示す。
【図12−2】実施例2に記載される構築物p73i-Tgnrのポリヌクレオチド配列、アミノ酸配列および制限地図を示す。
【特許請求の範囲】
【請求項1】
Gag、RT、Nefの順に転写されるように配列された、少なくともHIV抗原RT、NefおよびGagまたはこれらの免疫原性誘導体もしくは免疫原性断片をコードする1つまたは複数のポリヌクレオチドを含む、アデノウイルスベクター。
【請求項2】
RTが末端切断型である、請求項1に記載のアデノウイルスベクター。
【請求項3】
Nefが末端切断型である、請求項1または2に記載のアデノウイルスベクター。
【請求項4】
Gagがp17とp24のみである、請求項1〜3のいずれか1項に記載のアデノウイルスベクター。
【請求項5】
1つまたは複数のHIVポリヌクレオチドのサイズは、ベクター全体のサイズがウイルスのサイズの90〜100%となるようなサイズである、請求項1〜4のいずれか1項に記載のアデノウイルスベクター。
【請求項6】
ウイルスが非ヒト霊長類のアデノウイルスである、請求項1〜5のいずれか1項に記載のアデノウイルスベクター。
【請求項7】
ウイルスがチンパンジーのアデノウイルスである、請求項6に記載のアデノウイルスベクター。
【請求項8】
アデノウイルスがpan5、6、7および9から選択される、請求項7に記載のアデノウイルスベクター。
【請求項9】
アデノウイルスがpan6である、請求項8に記載のアデノウイルスベクター。
【請求項10】
アデノウイルスがpan7である、請求項8に記載のアデノウイルスベクター。
【請求項11】
ウイルスが複製欠損型である、請求項1〜10のいずれか1項に記載のアデノウイルスベクター。
【請求項12】
ウイルスのE1およびE3領域が欠失している、請求項1〜11のいずれか1項に記載のアデノウイルスベクター。
【請求項13】
HIV抗原をコードするポリヌクレオチド配列が融合体として配列されている、請求項1〜12のいずれか1項に記載のアデノウイルスベクター。
【請求項14】
以下のポリヌクレオチド構築物:
p17、p24(コドン最適化)Gag - p66 RT(コドン最適化) - 末端切断型Nef;
末端切断型Nef - p66 RT(コドン最適化) - p17、p24(コドン最適化)Gag;
末端切断型Nef - p17、p24(コドン最適化)Gag - p66 RT(コドン最適化);
p66 RT(コドン最適化) - p17、p24(コドン最適化)Gag - 末端切断型Nef;
p66 RT(コドン最適化) - 末端切断型Nef - p17、p24(コドン最適化)Gag;
p17、p24(コドン最適化)Gag - 末端切断型Nef - p66 RT(コドン最適化)
の1つを含む、チンパンジーアデノウイルスベクター。
【請求項15】
アデノウイルスがPan6またはPan7であり、アデノウイルスがPan6である場合は、前記構築物はp66 RT(コドン最適化) - 末端切断型Nef - p17、p24(コドン最適化)Gagではない、請求項14に記載のアデノウイルスベクター。
【請求項16】
請求項1〜15のいずれか1項に記載のウイルスベクターおよび製薬上許容される担体またはアジュバントを含む免疫原性組成物。
【請求項17】
HIV感染の治療または予防のための医薬の製造における、請求項1〜15のいずれか1項に記載のアデノウイルスベクターの使用。
【請求項18】
以下のステップ:
a) アデノウイルスベクターを供給するステップ;
b) 好適なプロモーターに機能的に連結されたHIV抗原配列を有するプラスミドを供給するステップ;
c) 該プラスミドと該ベクターの両方を用いて細胞をトランスフェクトするステップ;
d) 組換えを起こすのに十分な時間を与えるステップ;および
e) HIV抗原配列を有する組換えウイルスベクターを回収するステップ
を含んでなる、請求項1〜15のいずれか1項に記載のベクターの作製方法。
【請求項19】
哺乳動物に、好適な量の請求項16に記載の免疫原性組成物を投与することを含んでなる、哺乳動物において免疫応答を生じさせる方法。
【請求項20】
請求項1〜15のいずれか1項に記載のベクターにより発現される融合タンパク質。
【請求項21】
ヒト体内で産生される、請求項20に記載の融合タンパク質。
【請求項1】
Gag、RT、Nefの順に転写されるように配列された、少なくともHIV抗原RT、NefおよびGagまたはこれらの免疫原性誘導体もしくは免疫原性断片をコードする1つまたは複数のポリヌクレオチドを含む、アデノウイルスベクター。
【請求項2】
RTが末端切断型である、請求項1に記載のアデノウイルスベクター。
【請求項3】
Nefが末端切断型である、請求項1または2に記載のアデノウイルスベクター。
【請求項4】
Gagがp17とp24のみである、請求項1〜3のいずれか1項に記載のアデノウイルスベクター。
【請求項5】
1つまたは複数のHIVポリヌクレオチドのサイズは、ベクター全体のサイズがウイルスのサイズの90〜100%となるようなサイズである、請求項1〜4のいずれか1項に記載のアデノウイルスベクター。
【請求項6】
ウイルスが非ヒト霊長類のアデノウイルスである、請求項1〜5のいずれか1項に記載のアデノウイルスベクター。
【請求項7】
ウイルスがチンパンジーのアデノウイルスである、請求項6に記載のアデノウイルスベクター。
【請求項8】
アデノウイルスがpan5、6、7および9から選択される、請求項7に記載のアデノウイルスベクター。
【請求項9】
アデノウイルスがpan6である、請求項8に記載のアデノウイルスベクター。
【請求項10】
アデノウイルスがpan7である、請求項8に記載のアデノウイルスベクター。
【請求項11】
ウイルスが複製欠損型である、請求項1〜10のいずれか1項に記載のアデノウイルスベクター。
【請求項12】
ウイルスのE1およびE3領域が欠失している、請求項1〜11のいずれか1項に記載のアデノウイルスベクター。
【請求項13】
HIV抗原をコードするポリヌクレオチド配列が融合体として配列されている、請求項1〜12のいずれか1項に記載のアデノウイルスベクター。
【請求項14】
以下のポリヌクレオチド構築物:
p17、p24(コドン最適化)Gag - p66 RT(コドン最適化) - 末端切断型Nef;
末端切断型Nef - p66 RT(コドン最適化) - p17、p24(コドン最適化)Gag;
末端切断型Nef - p17、p24(コドン最適化)Gag - p66 RT(コドン最適化);
p66 RT(コドン最適化) - p17、p24(コドン最適化)Gag - 末端切断型Nef;
p66 RT(コドン最適化) - 末端切断型Nef - p17、p24(コドン最適化)Gag;
p17、p24(コドン最適化)Gag - 末端切断型Nef - p66 RT(コドン最適化)
の1つを含む、チンパンジーアデノウイルスベクター。
【請求項15】
アデノウイルスがPan6またはPan7であり、アデノウイルスがPan6である場合は、前記構築物はp66 RT(コドン最適化) - 末端切断型Nef - p17、p24(コドン最適化)Gagではない、請求項14に記載のアデノウイルスベクター。
【請求項16】
請求項1〜15のいずれか1項に記載のウイルスベクターおよび製薬上許容される担体またはアジュバントを含む免疫原性組成物。
【請求項17】
HIV感染の治療または予防のための医薬の製造における、請求項1〜15のいずれか1項に記載のアデノウイルスベクターの使用。
【請求項18】
以下のステップ:
a) アデノウイルスベクターを供給するステップ;
b) 好適なプロモーターに機能的に連結されたHIV抗原配列を有するプラスミドを供給するステップ;
c) 該プラスミドと該ベクターの両方を用いて細胞をトランスフェクトするステップ;
d) 組換えを起こすのに十分な時間を与えるステップ;および
e) HIV抗原配列を有する組換えウイルスベクターを回収するステップ
を含んでなる、請求項1〜15のいずれか1項に記載のベクターの作製方法。
【請求項19】
哺乳動物に、好適な量の請求項16に記載の免疫原性組成物を投与することを含んでなる、哺乳動物において免疫応答を生じさせる方法。
【請求項20】
請求項1〜15のいずれか1項に記載のベクターにより発現される融合タンパク質。
【請求項21】
ヒト体内で産生される、請求項20に記載の融合タンパク質。
【図1】


【図2】
【図3】
【図4】
【図5】
【図6】
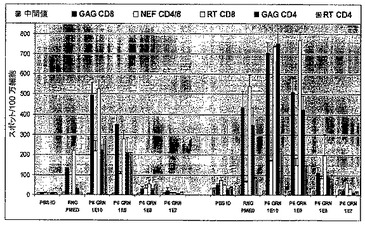
【図7−1】

【図7−2】
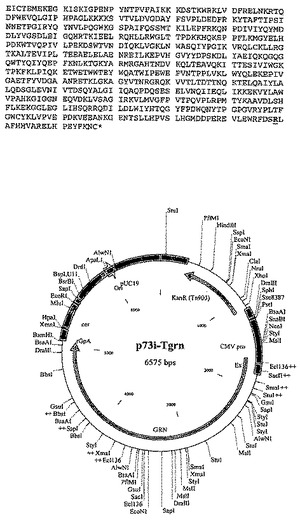
【図8−1】

【図8−2】
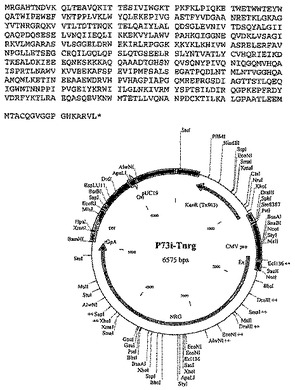
【図9−1】

【図9−2】
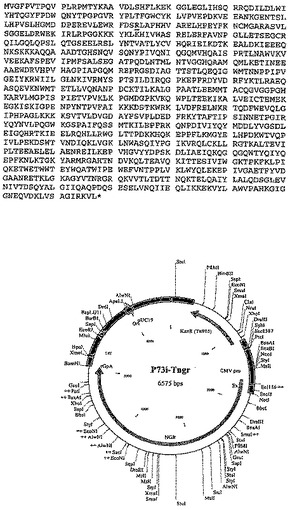
【図10−1】

【図10−2】
【図11−1】

【図11−2】
【図12−1】

【図12−2】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7−1】
【図7−2】
【図8−1】
【図8−2】
【図9−1】
【図9−2】
【図10−1】
【図10−2】
【図11−1】
【図11−2】
【図12−1】
【図12−2】
【公表番号】特表2008−539746(P2008−539746A)
【公表日】平成20年11月20日(2008.11.20)
【国際特許分類】
【出願番号】特願2008−510517(P2008−510517)
【出願日】平成18年5月10日(2006.5.10)
【国際出願番号】PCT/EP2006/004854
【国際公開番号】WO2006/120034
【国際公開日】平成18年11月16日(2006.11.16)
【出願人】(397009934)グラクソ グループ リミテッド (832)
【氏名又は名称原語表記】GLAXO GROUP LIMITED
【住所又は居所原語表記】Glaxo Wellcome House,Berkeley Avenue Greenford,Middlesex UB6 0NN,Great Britain
【Fターム(参考)】
【公表日】平成20年11月20日(2008.11.20)
【国際特許分類】
【出願日】平成18年5月10日(2006.5.10)
【国際出願番号】PCT/EP2006/004854
【国際公開番号】WO2006/120034
【国際公開日】平成18年11月16日(2006.11.16)
【出願人】(397009934)グラクソ グループ リミテッド (832)
【氏名又は名称原語表記】GLAXO GROUP LIMITED
【住所又は居所原語表記】Glaxo Wellcome House,Berkeley Avenue Greenford,Middlesex UB6 0NN,Great Britain
【Fターム(参考)】
[ Back to top ]