
一本鎖DNAライブラリーの調製方法
本発明は、増幅反応および配列決定反応に用いる一本鎖DNAライブラリーの作製方法に関する。さまざまな局面において、開示の方法は、DNAを断片化する段階; 断片の末端をポリッシングする段階; 断片をユニバーサルアダプターに連結する段階; ニックの入った断片の鎖置換および鎖伸長を行う段階; 二本鎖の連結産物を精製する段階; 二本鎖の連結産物を固相支持体に捕捉する段階; ならびに一本鎖DNAライブラリー断片を単離する段階、およびこれらの断片を別の固相支持体に結合させる段階を含む。

【発明の詳細な説明】
【技術分野】
【0001】
発明の分野
本発明は、タンパク質化学、分子生物学、および配列解析用の一本鎖ライブラリーの調製方法に関する。より具体的には、本発明は、増幅反応および配列決定反応に用いるDNAの処理方法を含む。
【0002】
関連出願
本出願は、以下の出願に対する優先権の恩典を主張するものである: 2003年1月29日付で出願した米国特許出願第60/443,471号、2003年4月23日付で出願した米国特許出願第60/465,071号; 2003年6月6日付で出願した米国特許出願第60/476,504号、2003年6月6日付で出願した米国特許出願第60/476,313号、2003年6月6日付で出願した米国特許出願第60/476,592号、2003年6月6日付で出願した米国特許出願第60/476,602号、2003年6月6日付で出願した米国特許出願第60/476,592号、および2003年8月25日付で出願した米国特許出願第60/497,985号。本段落の全ての特許および特許出願は、その全体が参照により本明細書に組み入れられる。
【0003】
本出願には同様に、以下の同時係属中の米国特許出願の2004年1月29日付で出願した「Bead Emulsion Nucleic Acid Amplification」、2004年1月29日付で出願した「Bead Emulsion Nucleic Acid Amplification with Continuous Flow」、2004年1月29日付で出願した「Double Ended Sequencing」、および2004年1月29日付で出願した「Methods Of Amplifying And Sequencing Nucleic Acids」が参照により組み入れられる。
【背景技術】
【0004】
発明の背景
ポリメラーゼ連鎖反応(PCR)による増幅では、二つのプライマーが、ある数のヌクレオチドによりDNA鋳型分子上で分離されるそれぞれのプライマーに相補的な位置で、鋳型DNAにハイブリダイズするように設計される。プライマー間のおよびプライマーを含む鋳型DNAの塩基配列が反復的な相補鎖伸長反応により増幅され、それにより、多数の標的DNA断片のコピーが数百倍分だけ増幅される。増幅は2nのように指数関数的である(式中nは増幅サイクルの数に等しい)。PCR後、増幅されたDNAは、従来の配列決定法により配列決定することができる(米国特許第6,274,320号を参照されたい)。
【0005】
大きい鋳型DNAを含む試料または長いヌクレオチド配列を含む全ゲノムDNAは、PCRによる効率的な増幅に寄与しない。これらの長い分子にはもともと、プライマーのハイブリダイゼーションに有用な配列がない。さらに、プライマーのハイブリダイゼーション配列を二本鎖DNA分子に付加する場合、増幅されたDNA分子の方向性を確定するのは困難であり、これが配列決定の取り組みの妨げになる。
【0006】
これらの欠点のいくつかを克服するため、さまざまな方法が設計されている。例えば、米国特許第5,508,169号は、核酸断片のサブセットを同一でない5'突出付着末端または3'突出付着末端に含まれる情報に基づいて指標付け(すなわち、選択または標的化)できることについて記載している。これには、II型制限酵素および断続性の回文構造を認識するII型制限酵素によるDNAの切断により露呈されるもののような、3塩基、4塩基または5塩基の付着末端を有する断片が含まれる。その特許は、(認識配列ではなく)制限酵素による切断部位の付着末端に相補的な突出一本鎖を含むアダプターに似た核酸分子(指標付けリンカー(indexing linker)といわれる)について記載している。さまざまな官能基または特定の用途のために設計した特異的な核酸配列を上記の断片のサブセットに選択的に付着させてもよい。周知の塩基配列を付着末端に有する指標付けリンカーの、その相補的な付着末端を持つ断片のサブセットとの選択的付着は、断片のサブセットの検出、同定、単離、増幅、および操作に使用することができる。
【0007】
米国特許第6,468,748号は、いくつかの段階を含む遺伝子および/または遺伝子断片の選別方法について記載している。第一に、ds cDNA分子を逆転写により、任意でポリ-T配列の上流に一般的プライマー-鋳型配列を有するポリ-Tプライマーを用いてmRNA分子から調製し、任意で一般的プライマー-鋳型配列を有する、ポリ-T配列を持つds cDNA分子を得る。第二に、ds cDNA分子を、一定数の任意のヌクレオチドのssDNA突出配列が入った付着末端を有する消化cDNA分子を生成する制限酵素で消化する。第三に、消化cDNA分子をdsDNAオリゴヌクレオチドアダプターのセットであって、そのアダプターのそれぞれが、消化cDNAの考えられるssDNA突出配列の一つに相補的な付着末端性ssDNAアダプター配列をその末端の一端に、ssDNAアダプター相補配列に特有の特異的プライマー-鋳型配列を他端に、およびそのセットの異なるアダプターの全てで同じである一定配列をその両端の間に有するdsDNAオリゴヌクレオチドアダプターのセットに連結させる。第四に、連結cDNA分子を、各個別のポリメラーゼ連鎖反応を目的に、任意でcDNAの一般的プライマー-鋳型(配列)を有するcDNAのポリ-T配列にアニーリングするプライマーと、cDNAの特異的プライマー-鋳型配列にアニーリングする異なる特異的プライマーのセットのあるプライマーとを使用して、個別のポリメラーゼ連鎖反応により増幅させる。第五に、増幅cDNA分子を各個別のポリメラーゼ連鎖反応後の増幅産物を回収することにより重複のない群(特異的プライマー-鋳型配列にアニーリングして、ポリメラーゼ連鎖反応を開始する特異的プライマーにより決定される増幅cDNA分子の各群)に選別する。
【0008】
米国特許第5,863,722号は、オリゴヌクレオチドタグを有するポリヌクレオチドを選別するための方法および材料について記載している。オリゴヌクレオチドタグは、天然型オリゴヌクレオチドと比べて結合強度および特異性の増加したサブユニットからなる相補的なオリゴマー化合物にハイブリダイズすることができる。そのような相補的なオリゴマー化合物は「タグ相補体」といわれる。タグ相補体のサブユニットは、「アンチセンス単量体」といわれる、非天然型ヌクレオチド類似体の単量体からなってもよく、またはそれらには、アンチセンス単量体を含む、3〜6個の範囲内の長さのヌクレオチドを有するオリゴマーもしくはその類似体であって、最小限に交差ハイブリダイズするセットから選択されるオリゴマーが含まれてもよい。そのようなセットの場合、セットのオリゴマーとセットの他のオリゴマーの相補体とからなる二重鎖には、少なくとも2つのミスマッチが含まれる。言い換えれば、最小限に交差ハイブリダイズするセットのオリゴマーは、せいぜい、同じセットの他のオリゴマーの相補体と少なくとも2つのミスマッチを有する二重鎖を形成するだけである。固相支持体に付着されるタグ相補体を使用して、ポリヌクレオチドの混合物からそれぞれタグを含んだポリヌクレオチドを選別する。各支持体の表面は、特定の配列を有するただ一種類のタグ相補体により誘導体化される。同様に、選別されるポリヌクレオチドはそれぞれ、同じポリヌクレオチドが同じタグを有し、異なるポリヌクレオチドが異なるタグを有するように、オリゴヌクレオチドタグをレパートリーとして含む。従って、支持体およびポリヌクレオチドの集団が、オリゴヌクレオチドタグのその各相補体との特異的ハイブリダイゼーションを可能とする条件の下で混合される場合、同じポリヌクレオチドの亜集団が特定のビーズまたは領域に選別される。その後、ポリヌクレオチドの亜集団をマイクロ生化学的技術により固相支持体上で操作することができる。
【0009】
米国特許第5,728,524号は、核酸配列の分類の方法であって、それぞれが所定のヌクレオチド塩基を含む配列に対する結合特異性を示すアダプター分子の集団に、その核酸配列を結合させる方法について記載している。得られる連結配列をその後、特定塩基に対する選択に基づいて分類する。
【0010】
しかしながら、当技術分野では、それぞれが他方とは異なる二つの既知配列を、それぞれが(未知断片配列の)各末端に隣接するようにしてさらに含んだ未知断片配列のライブラリーを作製するための方法については記載されていない。このように、先行技術の欠点を克服する方法に対する必要性が存在する。従って、本発明は、試料中の複数のDNA配列の操作を容易とするのに必要な方法、材料、およびキットを説明するものである。
【発明の開示】
【0011】
発明の簡単な概要
本発明は、ライブラリーがさらなる定量分析および比較分析に適するような、特に複数の核酸配列が未知であって、大きい鋳型DNAまたは全ての(もしくは一部の)ゲノムDNAに由来するような、試料から複数の核酸配列のライブラリーを調製するための新規な方法について記載する。本発明のある種の態様において、一本鎖DNA(ssDNA)の配列は、大きい鋳型DNAまたは全てのもしくは一部のゲノムDNAの試料から断片化、ポリッシング(polishing)、アダプター連結、ニック修復、およびssDNAの単離を経て調製される。
【0012】
従って、ある局面において、本発明により、複数のssDNAを含んだライブラリーをクローニングにより単離するための方法であって、その際に各ssDNAは第一の一本鎖ユニバーサルアダプターと第二の一本鎖ユニバーサルアダプターとを含み、以下の段階を含む方法が提供される:
(a) 大きい鋳型DNA分子を断片化して複数の断片化したDNA分子を作製する段階;
(b)第一または第二の二本鎖ユニバーサルアダプターを断片化した各DNA分子の第一の末端におよび第一または第二のユニバーサルアダプターを断片化した各DNA分子の第二の末端に付着させて、アダプター連結DNA分子の混合物を形成させる段階;
(c) それぞれが第一の一本鎖ユニバーサルアダプターと第二の一本鎖ユニバーサルアダプターとを含む複数の一本鎖DNA分子を単離する段階; および
(d) 複数のリアクターに一つのDNA分子が含まれるように一本鎖DNA分子をリアクターに送達し、それによってライブラリーをクローニングにより単離する段階。
【0013】
ある種の局面において、一本鎖DNA分子は、油中水型乳濁液中の液滴(すなわち、マイクロリアクター)内に、またはマルチウェル表面(例えば、PicoTiterプレート)上に送達される。一本鎖DNA分子は、固相支持体(例えば、ビーズ)への付着により送達されてもよい。
【0014】
他の局面において、第一の二本鎖ユニバーサルアダプターと第二の二本鎖ユニバーサルアダプターとを含むアダプター連結DNA分子は、二本鎖ユニバーサルアダプターの片鎖を介して(第一または第二のユニバーサルアダプターを介して)固相支持体に付着される。固相支持体に付着されていないアダプター連結DNA分子は洗い流されて、アダプター連結DNA分子の片鎖が遊離される。これにより、第一および第二のユニバーサルアダプター対を有する一本鎖分子の集団を含んだ複数のssDNAを含有する混合物が得られ、それによってライブラリーが得られる。
【0015】
断片化したDNAの配列は、既知であっても未知であってもよい。好ましい態様において、断片化したDNAの配列、特に断片化したDNAの末端の配列は未知である。
【0016】
別の局面において、本発明には、以下の段階を含む、固相支持体に結合されたssDNAライブラリーを作製するための方法が含まれる: (a) 鋳型ssDNAのライブラリーを作製する段階; (b) 鋳型ssDNAを固相支持体に付着させる段階; および(c) 一つの鋳型ssDNAが付着された固相支持体を単離する段階。別の局面において、本発明には、本明細書に記載の方法により作製される可動性の固相支持体のライブラリーが含まれる。
【0017】
発明の詳細な説明
本発明は、増幅反応および配列決定反応のための試料DNAの調製に関する。本発明には、以下の段階からなる試料DNAの調製方法が含まれる: (a) 大きい鋳型DNAまたは全ゲノムDNAの試料を断片化して複数のDNA消化断片を作製させる段階; (b) 複数のDNA消化試料に適合末端を作製する段階; (c) 断片化したDNA分子の末端にユニバーサルアダプター配列のセットを連結して複数のアダプター連結DNA分子を作製する段階であって、その際に各ユニバーサルアダプター配列はPCR用プライマー配列と、配列決定用プライマー配列と識別可能なキー配列とを含み、およびその際に一方のアダプターにはビオチンが付着している段階; (d) 複数の連結DNA断片を分離かつ単離する段階; (e) 複数の連結DNA断片の任意部分を取り出す段階; (f) 複数の連結DNA断片をニック修復および鎖伸長する段階; (g) それぞれの連結DNA断片を固相支持体に付着させる段階; および(h) 各末端に特有のアダプターが存在する(すなわち、方向性を与える)一本鎖のアダプター連結DNA断片を含んだ集団を単離する段階。
【0018】
特別の定めのない限り、本明細書で使用される全ての技術用語および科学用語は、当業者が通常理解しているのと同じ意味を有する。本発明の実施に際して、本明細書に記載のものに類似であるかまたは等価である方法および材料を使用することができるが、例示として適当な方法および材料を以下に記載する。例えば、三つまたはそれ以上の段階を含む方法が記載される場合がある。そのような方法では、全ての段階が規定の目標を達成するのに必要とされるとは限らない可能性があり、本発明はこれら(段階)の個別の目標を達成するのに、孤立的段階の使用を想定する。全ての刊行物、特許出願、特許、および他の参照文献の開示は、その全体が参照により本明細書に組み入れられる。さらに、材料、方法、および例は、単なる例示に過ぎず限定を意図するものではない。
【0019】
本明細書で用いられる場合の「ユニバーサルアダプター」という用語は、PCRのプライミングのためのヌクレオチド配列と配列決定のプライミングのためのヌクレオチド配列とを含むように設計される二本の相補的な且つアニーリングするオリゴヌクレオチドを指す。任意で、ユニバーサルアダプターは、非反復ヌクレオチド配列からなる特有の識別可能なキー配列(すなわち、ACGT、CAGTなど)をさらに含むことができる。ユニバーサルアダプターのセットには、二本鎖DNAの末端に連結され得る二本の特有であり且つ異なる二本鎖配列が含まれる。従って、同じユニバーサルアダプターまたは異なるユニバーサルアダプターをDNA分子の両端に連結することができる。一本鎖である大きいDNA分子に含まれる場合またはオリゴヌクレオチドとして存在する場合、ユニバーサルアダプターは、一本鎖のユニバーサルアダプターということができる。
【0020】
本明細書で用いられる場合の「識別可能なキー配列」という用語は、4種類のデオキシリボヌクレオチド(すなわち、A、C、G、T)の組み合わせを含む配列を指す。DNA断片の丸ごとのライブラリーには、識別可能な同一配列を使用することができる。または、異なる生物に由来するDNA断片のライブラリーを追跡するには、識別可能な異なるキー配列を使用することができる。二つまたはそれ以上のライブラリーの混合には、識別可能なより長いキー配列を使用することができる。
【0021】
本明細書で用いられる場合の「複数の分子」という用語は、同じ供給源から単離されたDNAを指し、したがって、異なる生物は同じ方法により別々に調製することができる。ある態様において、複数のDNA試料は、大きいDNA断片、例えば、ゲノムDNA、cDNA、ウイルスDNA、プラスミドDNA、コスミドDNA、人工染色体DNA(例えば、BAC、YAC、MAC、PAC)、合成DNA、ファージミドDNA、ファセミド(phasemid)DNAから、またはウイルスRNAの逆転写産物から得られる。このDNAは、任意の哺乳類(すなわち、ヒト、ヒト以外の霊長類、齧歯類、またはイヌ)、植物、鳥類、爬虫類、魚類、真菌、細菌、またはウイルスを含む、任意の供給源から得ることができる。
【0022】
本明細書で用いられる場合の「ライブラリー」という用語は、大きい鋳型DNA、例えば、断片化したゲノムまたは全ゲノムから作製された小さなサイズのDNA種のサブセットを指す。
【0023】
本明細書で用いられる場合の「特有のPCRプライミング領域」などで、「特有の」という用語は、増幅されるまたは配列決定されるDNA分子内に存在しないまたは極端に低いコピーレベルで存在する配列を指す。
【0024】
本明細書で用いられる場合の「適合する」という用語は、アダプター分子が付着できる二本鎖DNAの末端(すなわち、平滑末端または付着末端)を指す。
【0025】
本明細書で用いられる場合の「断片化する」という用語は、大きいDNA分子を小さなDNA断片に変換する工程を指す。
【0026】
本明細書で用いられる場合の「大きい鋳型DNA」とは、5 kb、10 kb、または25 kbを超える、好ましくは500 kbを超える、より好ましくは1 MBを超える、および最も好ましくは5 MBまたはそれ以上のDNAとすることができる。
【0027】
本明細書で用いられる場合の「ストリンジェントなハイブリダイゼーション条件」という用語は、完全に相補的な配列のみが互いにハイブリダイズできる条件を指す。
【0028】
以下の説明は、本発明の方法に含まれる基本的段階を要約している。各段階は特定の順序で列挙されているが、しかしながら、当業者に公知のように、各工程の順序は同じ結果を達成するように操作されてもよい。そのような操作は、本発明者らによって予想されている。さらに、一部の段階は、当業者に同様にして公知であるように最小化されてもよい。
【0029】
断片化
本発明の方法の実施に際して、DNA試料の断片化は、当業者に周知の任意の手段により行われることができる。断片化は酵素的手段、化学的手段、または機械的手段により行われることが好ましい。機械的手段には、超音波処理、フレンチプレス、HPLC、HydroShear(GeneMachines, San Carlos, CA)および噴霧を含むことができる。酵素的手段は、デオキシリボヌクレアーゼI(DNaseI)、非特異的ヌクレアーゼ、または単一のもしくは複合の制限エンドヌクレアーゼを用いた消化により行われることができる。好ましい態様において、断片化により、末端に隣接する配列が未知の末端が生ずる。末端に隣接する配列は、少なくとも5塩基、10塩基、20塩基、30塩基、または50塩基とすることができる。
【0030】
酵素による断片化
好ましい態様において、酵素的手段はDNaseIである。DNaseIは、二本鎖DNA(dsDNA)を非特異的に切断して5'-リン酸化オリゴヌクレオチド産物を放出する汎用酵素である。DNaseIは、Mn2+、Mg2+およびCa2+を含有する緩衝液中で至適活性を有する。DNaseIによる消化段階の目的は、大きいゲノムDNAを、ライブラリーを構成するより小さな種に断片化することである。DNaseIの切断特性により、鋳型DNAの無作為消化(すなわち、最小配列の偏り)をもたらし、マンガンに基づく緩衝液の存在下で使用する場合には平滑末端dsDNA断片が圧倒的に多い量になる(Melgar, E. and D.A. Goldthwait.1968. Deoxyribonucleic acid nucleases. II. The effects of metal on the mechanism of action of Deoxyribonuclease I. J. Biol. Chem. 243: 4409)。鋳型ゲノムのDNase I処理後に生ずる消化産物の範囲は、次の三つの要因に依存する: i) 使用する酵素の量(単位); ii) 消化の温度(℃); およびiii) インキュベーション時間(分)。以下に概略を説明するDNase I消化条件は、50〜700塩基対(bp)に及ぶサイズのゲノムライブラリーを得るために最適化された。
【0031】
好ましい態様において、DNase Iを使用して大きい鋳型DNAまたは全ゲノムDNAを1〜2分間消化し、50〜500 bp、または50〜700 bpに及ぶオリゴヌクレオチドの集団を作製する。別の好ましい態様において、DNase I消化を10℃〜37℃の温度で行う。別の好ましい態様において、消化されたDNA断片は長さが50 bp〜700 bpである。
【0032】
機械的断片化
核酸の断片化のための別の好ましい方法は、機械的な断片化である。機械的な断片化方法には、超音波処理および噴霧、ならびにHydroShear、HPLC、およびフレンチプレスの使用が含まれる。超音波処理は、適当な緩衝液(すなわち、10 mM Tris、0.1 mM EDTA)に入れたDNAを含有しており、さまざまな回数の10秒バーストの間、最大出力と連続出力を使用して超音波処理する管により行われることができる。超音波破砕機は、例えば、Misonix社(Farmingdale, NY)から市販されており、本質的にはBankier and Barrell (Bankier, A.T., Weston, K.M., and Barrell, B.G.,「Random cloning and sequencing by the M13/dideoxynucleotide chain termination method」, Meth. Enzymol. 155, 51-93 (1987)により記載されているように使用することができる。超音波処理の場合、試料を氷上に保持することにより、核酸を均一な温度に維持することが好ましい。例えば、0℃の一定温度条件は、均一の断片分布を維持するのに好ましい。超音波処理に最適な条件は、調製用の超音波処理を行う前に、所定のDNA試料に対して実験的に決定することができる。例えば、DNAの一定分割量を超音波処理の下で異なる時間、処理することができ、DNAのサイズと質をPAGEにより分析することができる。最適な超音波処理条件が決定されると、残りのDNAをその予め決められた条件に従って超音波処理することができる。
【0033】
核酸の断片化のための別の好ましい方法は、噴霧装置による処理である(例えば、GeneMachines, San Carlos, Californiaから得られる手順、およびハードウェア。同様に米国特許第5,506,100号および米国特許第5,610,010号を参照されたい)。噴霧の場合、流体力学的剪断力を使用して、DNA鎖を断片化する。例えば、急縮小流路(abrupt contraction)を備えた管にDNA水溶液を通すことができる。溶液が縮小流路に接近するにつれ、流体は縮小流路の狭い領域を通過する体積流量を維持するように加速する。この加速の間、流体抵抗により、DNAは切れるまで引き延ばされる。任意で、DNA溶液は、断片をさらに剪断するにはあまりにも短いところまで、縮小流路に数回(例えば、15〜20サイクル)通すことができる。縮小流路および流体の流量を調整することにより、最終的なDNA断片のサイズを決定することができる。反応条件を制御するおよび監視するためのソフトウェアは、噴霧過程の自動化を可能とするのに使用することができる。別の利点として、噴霧化のための特別な緩衝液の必要性がない。例えば、DNAは、以下に限定されることはないが、水、Tris緩衝液、Tris-EDTA緩衝液、および0.5 MまでのNaClを有するTris-EDTAを含む、さまざまな溶液に懸濁させることができる。
【0034】
ポリッシング
Mn2+の存在下でDNase Iを用いて鋳型ゲノムDNA(gDNA)をポリッシング消化することで、平滑末端であるかまたは1塩基もしくは2塩基の長さの突出末端を有するDNAの断片が生成される。同様に、機械的手段によるDNAの断片化により、平滑末端または突出末端を有する断片の組み合わせが得られる。これらのDNA断片は、酵素的にまたは機械的に作製されたかにかかわらず、下記の手順を使用して「ポリッシングされる」ことができる。
【0035】
ポリッシング(末端修復ともいわれる)とは、平滑末端DNAへの非平滑末端DNAの変換を指す。一つの方法として、ポリッシングは、BAL32ヌクレアーゼまたはマングビーン・ヌクレアーゼのような、一本鎖特異的エキソヌクレアーゼを用いた処理により行われることができる。一般に、ヌクレアーゼは使用前に正確に測定されるべきである。
【0036】
ある態様において、平滑末端は、Pfu DNAポリメラーゼによりもたらされる。他の態様において、平滑末端は、T4 DNAポリメラーゼまたはクレノーDNAポリメラーゼのような他のDNAポリメラーゼによりもたらされる。Pfu「ポリッシング」または平滑末端化を使用して、DNaseIによる鋳型ゲノムの消化後に作製される平滑末端種の量を増加させることができる。Pfu DNAポリメラーゼは、5'突出部を埋める。さらに、Pfu DNAポリメラーゼは、3'から5'方向のエキソヌクレアーゼ活性を示す。従って、この酵素を使用して一つのおよび二つのヌクレオチド伸長部分を除去し、アダプターの連結に使用可能な平滑末端DNA断片の量をさらに増加させることができる。
【0037】
アダプターの連結
DNAライブラリーの断片化および平滑末端化に続いて、ユニバーサルアダプター配列を各DNA断片に付加することができる。本発明の種々の態様において、ユニバーサルアダプターは、以下を含むように設計される: 1) 次の2)に隣接して位置する長さが通常10〜20 bpである(任意の適当なサイズを使用することができる)特有のPCRプライミング領域のセット; 2) 任意で次の3)に先行する、長さが通常10〜20 bpである(任意の適当なサイズを使用することができる)特有の配列決定プライミング領域のセット; 3) 4種のデオキシリボヌクレオチド(すなわち、A、C、G、T)のそれぞれの少なくとも1つの組み合わせを含む特有の識別可能なキー配列(例えば、長さが1〜12 bp)。好ましい態様において、識別可能なキー配列は4塩基長である。別の態様において、識別可能なキー配列は1〜4塩基の組み合わせとすることができる。別の態様において、キー配列は4種のヌクレオチドのそれぞれを1つ含む。ある種の態様において、キー配列は1つまたは複数のリボヌクレオチド、例えば、Uを含む。
【0038】
本発明のある態様において、それぞれの特有のユニバーサルアダプターは長さが44 bpであるが、任意の適当なサイズを使用することができる。好ましい態様において、ユニバーサルアダプターは、T4 DNAリガーゼによりDNA断片の各末端に連結されて、各DNA断片に対し全体で88 bpのヌクレオチド付加をもたらすが、任意の適当なサイズを使用することができる。異なるユニバーサルアダプターを各DNAライブラリーの調製専用に設計することができ、その結果、各生物に特有の識別子(identifier)を与えることができる。例えば、異なるライブラリーの調製に、異なるキー配列を採用することができる。当業者には明らかであるように、ユニバーサルアダプターのサイズおよび配列は変化させることができるものと理解される。従って、本発明で用いるアダプターは、本明細書に記載のサイズおよび配列に限定されることはない。
【0039】
例えば、二つの異なる(すなわち、「第一」および「第二」)ユニバーサルアダプターを調製するのに、一本鎖オリゴヌクレオチドを商業者(例えば、Integrated DNA Technologies, IAまたはOperon Technologies, CA)に注文することができる。本発明のある種の態様において、ライブラリーの第一アダプターの全てがPCRプライミング配列、配列決定用プライマー配列、および識別可能なキー配列を含む、あるヌクレオチド配列を共有し、その一方、第二アダプターの全てが別のヌクレオチド配列を共有する。別の態様において、ユニバーサルアダプターのオリゴヌクレオチド配列は、ホスホジエステル結合に代えて一つまたは複数のホスホロチオエート結合を用いた合成の間に修飾される。例えば、アダプターのオリゴヌクレオチドは、二つまたは三つのホスホロチオエート結合を5'と3'双方の末端に、または一端に含むことができる。未修飾のオリゴヌクレオチドは通常、ヌクレオチド塩基間のホスホジエステル結合の加水分解を触媒する混入ヌクレアーゼによる急速な分解を受けやすい。オリゴヌクレオチドの用途で用いるのに使用可能な一つの単純かつ広く使用されるヌクレアーゼ抵抗性の化学的性質がホスホロチオエート修飾である。ホスホロチオエートでは、硫黄原子がオリゴヌクレオチド骨格中の非架橋酸素に取って代わることで、これを全ての形態のヌクレアーゼ消化に抵抗性(すなわち、エンドヌクレアーゼ消化およびエキソヌクレアーゼ消化の双方に抵抗性)とする。各オリゴヌクレオチドをHPLC精製して、合成オリゴヌクレオチド調製物に混入しないことまたは擬似オリゴヌクレオチド配列が存在しないことを確実にする。
【0040】
ユニバーサルアダプターは、断片化したDNAに対する定方向連結を可能とするように設計される。二本鎖ユニバーサルアダプターの各セットは、相互にまたは平滑末端DNA断片に連結できない非相補的な5'の4塩基突出部を含むPCRプライミング領域を考慮して設計される。従って、結合は、アダプターの3'末端とDNA断片の5'末端との間でまたはDNA断片の3'末端とアダプターの5'末端との間で起こる。二本鎖ユニバーサルアダプターの配列は、主に相補オリゴヌクレオチドをアニーリング可能として、二つの非相補オリゴヌクレオチド間の交差ハイブリダイゼーションを阻止可能とする配列を考慮して設計される一本鎖オリゴヌクレオチドを用いて作製される。
【0041】
ある態様において、ユニバーサルアダプターの95%が相補オリゴヌクレオチドのアニーリングから形成される。好ましい態様において、ユニバーサルアダプターの97%が相補オリゴヌクレオチドのアニーリングから形成される。より好ましい態様において、ユニバーサルアダプターの99%が相補オリゴヌクレオチドのアニーリングから形成される。最も好ましい態様において、ユニバーサルアダプターの100%が相補オリゴヌクレオチドのアニーリングから形成される。プライマーと擬似標的配列との間の交差ハイブリダイゼーションを最少化するためのプタイマー設計の典型的な方法が実施例2に示されている。
【0042】
本発明のある種の局面において、突出するヌクレオチド(例えば、T)を第一および第二アダプターの3'平滑末端に付加する。並行して、ポリメラーゼを使用して、鋳型DNAの5'平滑末端に突出するヌクレオチド(例えば、A)を付加する。アダプターおよび鋳型の突出ヌクレオチドは相補的であり、一層効率的なアダプターの連結を可能とする。
【0043】
他の局面において、プラスミド捕捉系を開示の方法に従って使用する。例えば、二本鎖ユニバーサルアダプターをプラスミドに挿入することができる。アダプター領域は、以下の配列を順番に含むことができる: 制限部位、PCRプライマー配列1、配列決定用プライマー配列1、キー配列1、制限部位、キー配列2、配列決定用プライマー配列2、PCRプライミング配列2、および制限部位。一つのアプローチでは、プラスミドをキー配列1とキー配列2との間を切断する一つまたは複数の制限酵素で消化する。断片化された鋳型DNAをキー配列1とキー配列2との間に連結する。連結されたコンストラクトを次に消化して、PCRプライミング配列2の後で切断する。PCRプライミング配列2に隣接する消化末端をビオチン化ヌクレオチドで埋める。ビオチン化コンストラクトを消化して、PCRプライマー配列1の前で切断する。アダプター-DNA断片-アダプター-ビオチン断片を切り出して、例えば、ストレプトアビジン磁気ビーズに結合させることで単離する。プラスミド捕捉系の他の態様が同様に、周知のクローニング技術の適用により可能である。これらの態様は同様に本発明に包含される。
【0044】
二つのアダプターのうちの一方を支持体の結合部分に結び付けることができる。好ましい態様において、5'ビオチンを第一ユニバーサルアダプターに付加して、その後の鋳型ssDNAの単離およびビオチン結合タンパク質(例えば、ストレプトアビジン、NeutrAvidin(商標)またはアビジン)で飽和した固相支持体の表面とのユニバーサルアダプターの非共有結合を可能とする。適当な支持体には、以下に限定されることはないが、磁気ビーズ、アフィニティーカラム、膜(例えば、PDVF膜、ニトロセルロースなど)が含まれ、これらはストレプトアビジンまたは結合対のもう一方のメンバーで被覆することができる。他の結合が当技術分野において公知であり、ビオチン-ストレプトアビジンの代わりに使用されてもよい。例えば、抗体/抗原-抗原決定基、受容体/リガンドおよびオリゴヌクレオチド対合または相補結合を使用することができる。ある態様において、固相支持体はビーズ、好ましくはポリスチレンビーズである。一つの好ましい態様において、ビーズは約2.8 μmの直径を有するが、任意の適当なサイズを使用することができる。別の好ましい態様において、ビーズは常磁性ビーズ(例えば、Dynal Biotech, Inc., Lake Success, NY)である。本明細書で用いられる場合のこのビーズを「試料調製用ビーズ」という。
【0045】
各ユニバーサルアダプターは、一方がセンス配列を含み、もう一方がアンチセンス(相補)配列を含む、二つのssDNAオリゴヌクレオチドの組み合わせとアニーリングにより調製することができる。ユニバーサルアダプター設計の略図が、図2に描かれている。
【0046】
連結産物の単離
ユニバーサルアダプターの連結により、アダプターを各端に有する断片化したDNA、未結合の単一アダプター、およびアダプター二量体の形成が起こる。好ましい態様において、アダプターが連結されたDNAライブラリー集団を未連結の単一アダプターおよびアダプター二量体の集団から分離かつ単離する方法として、アガロースゲル電気泳動が使用される。他の態様において、断片は、サイズ排除クロマトグラフィー、ろ過、ショ糖沈降法、または当業者に公知の他の核酸分離技術により分離することができる。DNAのDNase I消化手順により通常、50〜700 bpに及ぶライブラリー集団が得られる。好ましい態様において、DNAマーカーの存在下でアガロースゲル電気泳動を行う際に、88 bpのユニバーサルアダプターセットの付加は、DNAライブラリー集団をより大きいサイズに変えて、約130〜800 bpのサイズ範囲の移動プロファイルをもたらすことになり; アダプター二量体は88 bpに移動することになり; および未連結のアダプターは44 bpに移動することになる。従って、サイズが200〜800 bpに及ぶ多数の二本鎖DNAライブラリーをアガロースゲルから物理的に単離して、標準的なゲル抽出技術により精製することができる。ある態様において、アダプターが連結されたDNAライブラリーのゲル分離により、サイズが200〜500 bpに及ぶライブラリー集団が回収されることになる。アダプターが連結された断片を識別する他の方法は、当業者に公知である。
【0047】
ニック修復
ユニバーサルアダプターに使用されるDNAオリゴヌクレオチドは5'リン酸化されていないので、リガーゼ処理の後、ギャップが断片化したDNAの3'連結部に存在することになる(図3Aを参照されたい)。これらの「ギャップ」または「ニック」は、ニックの入ったDNA断片に結合し、これを鎖置換、および伸長できるDNAポリメラーゼ酵素を使用することにより埋めることができる。3'から5'方向のエキソヌクレアーゼ活性はないが5'から3'方向のエキソヌクレアーゼ活性を示すDNAポリメラーゼは、ニックの修復およびニックの入っていない二本鎖DNAの形成をもたらすように、ニックを認識し、ニックの入った鎖を置換して、鎖を伸長する能力を有する(図3Bおよび3Cを参照されたい)( Hamilton, S.C., J.W. Farchaus and M.C. Davis. 2001. DNA polymerases as engines for biotechnology. BioTechniques 31:370)。
【0048】
ポリメラーゼ、リガーゼ、およびキナーゼを含むが、これらに限定されることはない、さまざまな修飾酵素がニック修復段階に使用される。本発明の方法で使用できるDNAポリメラーゼには、例えば、大腸菌(E. coli) DNAポリメラーゼI、サーモアンアエロバクター・サーモハイドロサルファリカス(Thermoanaerobacter thermohydrosulfuricus)ポリメラーゼI、およびバクテリオファージφ29が含まれる。好ましい態様において、鎖置換酵素のバチルス・ステアロサーモフィルス(Bacillus stearothermophilus)ポリメラーゼI(Bst DNAポリメラーゼI)を使用して、ニックの入ったdsDNAを修復し、ニックの入っていないdsDNAを作製する(図3Dを参照されたい)。別の好ましい態様において、リガーゼはT4 DNAリガーゼであり、キナーゼはT4ポリヌクレオチドキナーゼである。
【0049】
一本鎖DNAの単離
ニックの入っていないdsDNAの作製に続いて、第一および第二双方のアダプター分子を含むssDNAを単離することができる。二本鎖DNAライブラリーには、アダプターが以下の配置で結合される。
ユニバーサルアダプターA - DNA断片 - ユニバーサルアダプターA
ユニバーサルアダプターB - DNA断片 - ユニバーサルアダプターA*
ユニバーサルアダプターA - DNA断片 - ユニバーサルアダプターB*
ユニバーサルアダプターB - DNA断片 - ユニバーサルアダプターB
【0050】
「A」および「B」は、第一および第二アダプターに相当する。所望の集団は、星印で指定されている。
【0051】
ユニバーサルアダプターは、一方のユニバーサルアダプターのみが5'ビオチン部分を有するように設計されることが好ましい。例えば、ユニバーサルアダプターBが5'ビオチン部分を有する場合、ストレプトアビジンで被覆された試料調製用ビーズを使用して、ユニバーサルアダプターBを有する二本鎖DNAライブラリー種を全て結合することができる。二つのユニバーサルアダプターAを含むゲノムライブラリー集団種は、5'ビオチン部分を含んでおらず、ストレプトアビジン含有試料調製用ビーズに結合しないことになり、従って、洗い流すことができる。ビーズに付着したままであると思われる唯一の種は、ユニバーサルアダプターAとBとを有するものおよび二つのユニバーサルアダプターB配列を有するものである。
【0052】
二つのユニバーサルアダプターB配列(すなわち、各5'末端にビオチン部分)を有するDNA種は、二本鎖に含まれる各鎖が結合するので、ストレプトアビジン被覆試料調製用ビーズに各末端で結合することになる。ユニバーサルアダプターAとユニバーサルアダプターBとを有する二本鎖DNA種は、単一の5'ビオチン部分を含むことになり、従って、ストレプトアビジン被覆ビーズに一端のみで結合することになる。試料調製用ビーズが磁性である場合、ビーズは、磁化時には固相支持体に結合したままである。従って、低塩(「融解」または変性)溶液の存在下では、単一のユニバーサルアダプターAと単一のユニバーサルアダプターB配列とを含んだそのDNA断片だけが相補的な非結合鎖を遊離することになる。ビーズに付着したこの一本鎖DNAの集団を回収して、例えば、ピロリン酸配列決定法、リアルタイム定量PCR法、アガロースゲル電気泳動法、蛍光色素結合アッセイ法(PicoGreen(登録商標); Molecular Probes, Inc., Eugene, OR)、またはキャピラリーゲル電気泳動法により定量することができる。
【0053】
ある態様において、本発明の方法により作製されるssDNAライブラリーを定量して、単位容量あたりの分子数を算出する。例えば、分子を、ssDNA種のユニバーサルアダプター末端のPCRプライミング領域に相補的であるオリゴヌクレオチド捕捉プライマーを含んだ固相支持体にアニーリングさせることができる。
【0054】
ある種の態様において、ssDNAライブラリーの分子にアニーリングした捕捉プライマーを含むビーズをサーモサイクラーに移し、PCR増幅を可能とすることができる。DNAビーズに捕捉された一本鎖DNAの単一種のクローン集団を次に、配列決定することができる。ある態様において、固相支持体はビーズ、好ましくはセファロースビーズである。本明細書で用いられる場合のこのビーズを「DNA捕捉ビーズ」という。
【0055】
本明細書で使用されるビーズは、任意の手ごろなサイズのものとすることができ、さまざまな周知の材料から製造することができる。そのような材料の例としては、無機材料、天然高分子、および合成高分子が挙げられる。これらの材料の具体例としては、セルロース、セルロース誘導体、アクリル樹脂、ガラス; シリカゲル、ポリスチレン、ゼラチン、ポリビニル・ピロリドン、ビニルとアクリルアミドとの共重合体、ジビニルベンゼンまたは同様のもので架橋したポリスチレン(Merrifield Biochemistry 1964, 3, 1385-1390を参照されたい)、ポリアクリルアミド、ラテックスゲル、ポリスチレン、デキストラン、ゴム、シリコン、プラスチック、ニトロセルロース、セルロース、天然スポンジ、シリカゲル、ガラス、金属プラスチック、セルロース、架橋デキストラン(例えば、セファデックス(商標))およびアガロースゲル(セファロース(商標))ならびに当業者に周知の固相支持体が挙げられる。ある態様において、DNA捕捉ビーズの直径は20〜70 μmである。好ましい態様において、DNA捕捉ビーズの直径は20〜50 μmである。より好ましい態様において、DNA捕捉ビーズの直径は約30 μmである。
【0056】
ある局面において、本発明には、以下の段階を含む、固相支持体のライブラリーを作製するための方法が含まれる: (a) 本明細書に開示される方法により鋳型ssDNAの集団を調製する段階; (b) 固相支持体あたりDNA 1分子が存在するように各鋳型DNAを固相支持体に付着させる段階; (c) 増幅により各固相支持体上に各DNA断片のクローン集団が生じるように一本鎖の鋳型の集団を増幅する段階; (d) 鋳型ssDNAのクローン集団を配列決定する段階。
【0057】
ある態様において、固相支持体はDNA捕捉ビーズである。別の態様において、DNAはゲノムDNA、cDNA、またはRNA(例えば、ウイルスRNA)の逆転写産物である。DNAは、例えば、ビオチン-ストレプトアビジン結合、共有結合を介して、または相補オリゴヌクレオチドのハイブリダイゼーションにより固相支持体に付着することができる。ある態様において、各鋳型DNAはユニバーサルアダプターのセットに連結される。別の態様において、ユニバーサルアダプター対には、PCRプライマー配列、配列決定用プライマー配列、および識別可能なキー配列が含まれる。特有の末端を有する一本鎖DNAを単離し、その後、固相支持体に付着させて、クローン増幅のための増幅技術にさらす。DNAはPCRにより増幅することができる。ある局面において、本発明により、本明細書に記載の方法により作製された固相支持体に付着されたライブラリーが提供される。
【0058】
この方法により調製されるDNAは、鎖状伸長、ローリングサークル増幅、PCR、および配列決定のような、多くの分子生物学的手法に使用することができる。結合反応は、例えば、DNAに対するビーズのモル比を高くすることによって進められることができる。一本鎖DNA分子の捕捉は、ポアソン分布に従い、DNAが付着していないビーズ、DNA 1分子が付着したビーズ、またはDNA 2分子またはそれ以上の分子が付着したビーズの部分集合が生ずる。好ましい態様において、DNA 1分子が各ビーズに付着している。さらに、単離したライブラリーのさらなる操作に有用とできるアダプターによるさらなる修飾を含むことが可能である。
【0059】
捕捉ビーズとの鋳型核酸の結合
本発明のある種の態様において、増幅される一本鎖核酸の鋳型は、捕捉ビーズに付着される。核酸の鋳型は、当技術分野において公知の任意の方法で固相支持体の捕捉ビーズに付着することができる。好適な微細ビーズのような固相支持体にDNAを付着させるための多数の方法が当技術分野において存在する。本発明によって、ビーズとのDNAの共有結合性の化学結合は、ホスホアミデート結合を介してDNAの5'-リン酸をアミンで被覆した捕捉ビーズに結合させる、水溶性カルボジイミドのような、標準的なカップリング剤を用いることにより達成することができる。別の代替手段は、同様の化学反応を使用して、最初に特定のオリゴヌクレオチドリンカーをビーズに結合させて、次に、DNAリガーゼを用いてDNAをビーズ上のリンカーに結合させることである。オリゴヌクレオチドをビーズに結合させる他の結合化学反応には、N-ヒドロキシコハク酸アミド(NHS)およびその誘導体の使用が含まれる。そのような方法の場合、オリゴヌクレオチドの一端には、固相支持体と共有結合を形成する反応基(アミン基のような)が含まれてもよく、その一方、リンカーの他端には、固定化されるオリゴヌクレオチドと結合できる第二の反応基が含まれる。好ましい態様において、オリゴヌクレオチドは、共有結合によりDNA捕捉ビーズに結合される。しかしながら、キレート化または抗原-抗体複合体のような、非共有結合を使用して、オリゴヌクレオチドをビーズに結合させてもよい。
【0060】
制限酵素部位由来の重複末端またはバクテリオファージλを用いるクローニングベクターの「付着末端」のような、DNA断片の末端の特異配列に特異的にハイブリダイズするオリゴヌクレオチドリンカーを採用することができるが、平滑末端連結を有利に使用することもできる。これらの方法は、米国特許第5,674,743号に詳細に記載されている。ビーズを固定化するのに使用されるどの方法も、本発明の方法の各段階にわたって、固定化されたオリゴヌクレオチドを結合し続けることが好ましい。
【0061】
ある態様において、各捕捉ビーズは鋳型核酸の一部分を認識する(すなわち、その一部分に相補的である)核酸プライマーを複数有するように設計され、従って、鋳型核酸は捕捉ビーズにハイブリダイズされる。本明細書に記載される方法の場合、鋳型種のクローン増幅が好ましく、従って、ただ一つの特有の鋳型核酸がいずれか一つの捕捉ビーズに付着されることが好ましい。
【0062】
本明細書で使用されるビーズは、任意の手ごろなサイズのものとすることができ、さまざまな周知の材料から製造することができる。そのような材料の例としては、無機材料、天然高分子、および合成高分子が挙げられる。これらの材料の具体例としては、セルロース、セルロース誘導体、アクリル樹脂、ガラス、シリカゲル、ポリスチレン、ゼラチン、ポリビニル・ピロリドン、ビニルとアクリルアミドとの共重合体、ジビニルベンゼンまたは同様のもので架橋したポリスチレン(例えば、Merrifield Biochemistry 1964, 3, 1385-1390に記載されているような)、ポリアクリルアミド、ラテックスゲル、ポリスチレン、デキストラン、ゴム、シリコン、プラスチック、ニトロセルロース、天然スポンジ、シリカゲル、細孔性ガラス(control pore glass)、金属、架橋デキストラン(例えば、セファデックス(商標))、アガロースゲル(セファロース(商標))、および当業者に周知の固相支持体が挙げられる。好ましい態様において、捕捉ビーズは、直径が約25〜40 μmのセファロースビーズである。
【0063】
乳化
本発明で使用する場合、付着した鋳型核酸の有無にかかわらず捕捉ビーズは、熱安定性の油中水型乳濁液に懸濁させることができる。複数のマイクロリアクターには、ただ一つの鋳型および一つのビーズが含まれると考えられる。鋳型を含んでいないかまたはビーズを含んでいない多くの液滴が存在する可能性がある。同様に、二コピーまたはそれ以上の鋳型を含んだ液滴が存在する可能性がある。乳濁液は当技術分野において公知の任意の適当な方法により形成することができる。乳濁液を作製する一つの方法が以下に記載されるが、乳濁液を作製するための任意の方法を使用することができる。これらの方法は、当技術分野において公知であり、アジュバント法、向流法、逆流法、回転ドラム法、および膜法を含む。さらに、マイクロカプセルのサイズは、成分の流量および流速を変化させることにより調整することができる。例えば、液滴添加の場合、液滴のサイズおよび到達の総時間を変化させることができる。乳濁液は、封入ビーズ約3000個/マイクロリットルの密度を含むことが好ましい。
【0064】
生体反応に適した種々の乳濁液は、Griffiths and Tawfik, EMBO, 22, pp. 24-35 (2003); Ghadessy et al., Proc. Natl. Acad. Sci. USA 98, pp. 4552-4557(2001); 米国特許第6,489,103号および国際公開公報第02/22869号(それぞれが完全に参照により本明細書に組み入れられる)のなかで言及されている。Griffiths et al.,(米国特許第6,489,103号および国際公開公報第99/02671号)は、所望の活性を有する遺伝子産物をコードする一つまたは複数の遺伝要素のインビトロ選別のための方法について言及していることに留意されたい。この方法には、遺伝子の区画化、遺伝子の発現、および発現産物に基づく区画化遺伝子の選別が含まれる。本発明とは対照的に、Griffithのマイクロカプセル化による選別方法は、その核酸産物は固定されないおよび固定されることができないので、複数のマイクロカプセルの平行分析には適していない。Griffithsの核酸は固定されないので、それらは解乳化の間に混合されると思われる。
【0065】
乳濁液は、ビーズを増幅溶液に添加することにより作製されることが好ましい。本明細書で用いられる場合の「増幅溶液」という用語は、鋳型DNAの増幅を行うのに必要な試薬の十分な混合液を指す。増幅溶液の一つの例であるPCR増幅溶液が以下の実施例に示される。当然のことながら、実施される増幅のタイプや鋳型DNAがビーズに付着されるか溶液状態で供与されるかに基づいて、増幅溶液に種々の変更を行うことができる。ある態様において、ビーズと増幅溶液との混合液を生体適合性油(例えば、軽油, Sigma)の撹拌混合液に滴下して、乳化させる。別の態様において、ビーズおよび増幅溶液を十字流の生体適合性油に滴下する。使用される油には、一つまたは複数の生体適合性乳化安定剤が添加されてもよい。これらの乳化安定剤には、Atlox 4912、Span 80、および他の認識されかつ市販されている適当な安定剤が挙げられる。好ましい局面において、乳濁液は温度サイクリングを可能とするため、例えば、少なくとも94℃まで、少なくとも95℃まで、または少なくとも96℃まで熱安定性である。好ましくは、形成される液滴は、サイズが約5μm〜約500μmに、より好ましくは約10μm〜約350μmに、さらにより好ましくは約50〜250μmに、および最も好ましくは約100μm〜約200μmに及ぶ。十字流型の流体混合により、液滴形成の制御、および液滴サイズの均一性を可能とすることが好都合である。本発明者らは、ビーズを含有していない小さな水滴が乳濁液に存在し得ることに注目している。
【0066】
マイクロリアクターは、必要とされる増幅の程度に合う十分な増幅試薬を含むために十分に大きくするべきである。しかしながら、マイクロリアクターは、それぞれがDNAライブラリーのメンバーを含有するマイクロリアクターの集団を、従来の実験装置、例えば、PCRサーモサイクリング装置、試験管、インキュベーターおよび同様のものにより増幅できるように十分に小さくするべきである。とりわけ、マイクロリアクターの使用により、配列を混合させることなく鋳型の複合混合物(例えば、ゲノムDNA試料または全細胞RNA)の増幅、または一つもしくは複数の鋳型による優位性(例えば、PCR選択の偏り; Wagner et al., 1994, Suzuki and Giovannoni, 1996; Chandler et al., 1997, Polz and Cavanaugh, 1998を参照されたい)が可能となる。
【0067】
上記の制限により、マイクロリアクターの最適サイズは、直径を平均して100〜200μmとすることができる。このサイズのマイクロリアクターにより、容量が10 ml未満のマイクロリアクターの懸濁液中で約600,000個のメンバーからなるDNAライブラリーの増幅が可能になる。例えば、PCRが選択された増幅方法である場合、マイクロリアクター液10 mlをチューブ96 本収容の通常のサーモサイクラーのチューブ96本分に適合させることができる。好ましい態様において、600,000個のマイクロリアクターの懸濁液は、1 ml未満の容量を有する。1 ml未満の懸濁液は、従来のPCRサーモサイクラーのチューブ約10本中で増幅させることができる。最も好ましい態様において、600,000個のマイクロリアクターの懸濁液は、0.5 ml未満の容量を有する。
【0068】
本発明の別の態様は、鋳型とビーズとを用いるが、しかし鋳型をビーズに付着させずに核酸増幅を行う方法に向けられる。ある局面において、ビーズには、増幅後に増幅された核酸を結合できるリンカー分子が含まれてもよい。例えば、リンカーは、活性化できるリンカーであってもよい。そのようなリンカーは、公知であり、ストレプトアビジン/ビオチンおよび抗体/抗原のような温度感受性または塩感受性の結合対を含む。鋳型核酸は、ビーズで封入されて、増幅されてもよい。増幅後、増幅された核酸は、例えば、温度または塩濃度の調整によりビーズに結合されてもよい。
【0069】
増幅
鋳型核酸は、ビーズに付着していてもまたは付着していなくとも、転写に基づく増幅系(Kwoh D. et al., Proc. Natl. Acad Sci. (U.S.A.) 86: 1173 (1989); Gingeras T. R. et al., 国際公開公報88/10315号; Davey, C. et al., 欧州特許第329,822号; Miller, H.I. et al., 国際公開公報第89/06700号)、「RACE法」(Frohman, M. A., In: PCR Protocols: A Guide to Methods and Applications, Academic Press, NY (1990))および片側(one-sided)PCR法(Ohara, O. et al., Proc. Natl. Acad. Sci. (U.S.A.) 86. 5673-5677 (1989))を含む任意の適当な増幅方法により増幅することができる。ジオリゴヌクレオチド増幅法、等温増幅法(Walker, G. T. et al., Proc. Natl. Acad. Sci. (U.S.A.) 89: 392-396 (1992))、核酸配列に基づく増幅法(NASBA; 例えば、Deiman B et al., 2002, Mol Biotechnol. 20(2): 163-79を参照されたい)、全ゲノム増幅法(例えば、Hawkins TL et al., 2002, Curr Opin Biotechnol. 13(1): 65-7を参照されたい)、鎖置換増幅法(例えば、Andras SC, 2001, Mol Biotechnol. 19(1): 29-44を参照されたい)、ローリングサークル増幅法(米国特許第5,714,320号に概説されている)のようなさらに他の方法、および他の公知の技術を本発明により使用することができる。ある種の局面において、鋳型核酸は、マイクロリアクター中のビーズで封入後に増幅される。または、鋳型核酸は、マルチウェルの表面、例えば、PicoTiterプレートに分配後に増幅される。
【0070】
好ましい態様において、DNA増幅はPCRにより行われる。本発明によるPCRは、PCRに必要な試薬全てを含んだPCR溶液で標的核酸を封入することにより行われることができる。次に、PCRは、その乳濁液を当技術分野において公知の任意の適当なサーモサイクリング法にかけることにより達成することができる。好ましい態様において、30〜50サイクル、好ましくは約40サイクルの増幅が行われる。増幅手順が終わったら、増幅サイクルの後に1回または複数回のハイブリダイゼーションおよび伸長サイクルがあることが望ましいが、必ず必要とされるわけではない。好ましい態様において、10〜30サイクル、好ましくは約25サイクルのハイブリダイゼーションおよび伸長が行われる(例えば、実施例に記載のとおり)。日常的には、鋳型DNAは、通常、少なくとも10,000〜50,000,000コピーが各ビーズに固定化されるまで増幅される。核酸検出用途の場合、より少ない鋳型コピーが必要とされるものと認識される。核酸配列決定用途の場合、本発明者らは、少なくとも2百万〜5千万コピー、好ましくは約1千万〜3千万の鋳型DNAのコピーが各ビーズに固定化されることがよいと考えている。当業者は、ビーズのサイズ(およびその上の捕捉部位)により、捕捉プライマーをどのくらい結合することができるか(および従って増幅された鋳型をどのくらい各ビーズに捕捉することができるか)が決まることを認識すると思われる。
【0071】
ある局面において、本発明には、以下の段階を含む、複数の一本鎖DNA分子を含んだライブラリーをクローニングにより単離するための方法が含まれる: a) 大きい鋳型DNA分子を断片化して複数の断片化したDNA分子を作製する段階; b) 第一または第二の二本鎖ユニバーサルアダプターを断片化した各DNA分子の第一の末端におよび第一または第二のユニバーサルアダプターを断片化した各DNA分子の第二の末端に付着させて、アダプター連結DNA分子の混合物を形成させる段階; c) それぞれが第一の一本鎖ユニバーサルアダプターと第二の一本鎖ユニバーサルアダプターとを含む複数の一本鎖DNA分子を単離して、ライブラリーを得る段階; およびd) 複数のリアクターに一つのDNA分子が含まれるように一本鎖DNA分子をリアクターに送達し、それによってライブラリーをクローニングにより単離する段階。
【0072】
別の局面において、本発明には、以下の段階を含む、複数の一本鎖DNA分子を含んだライブラリーを作製するための方法が含まれる: a) 大きいまたは全ゲノムの鋳型DNA分子を断片化して複数の断片化したDNA分子を作製する段階; b) 第一の二本鎖ユニバーサルアダプターまたは第二のユニバーサルアダプターを断片化した各DNA分子の第一の末端におよび第一のユニバーサルアダプターまたは第二のユニバーサルアダプターを断片化した各DNA分子の第二の末端に連結させて、アダプター連結DNA分子の混合物を作製させる段階であって、その際に第一のユニバーサルアダプターには固相支持体に結合する部分が含まれる段階; c) 固相支持体に第一の二本鎖ユニバーサルアダプターを含むDNA分子を付着させる段階; d) 固相支持体に付着されていないアダプター連結DNA分子を洗い流す段階; e) 固相支持体に付着されたアダプター連結DNA分子を鎖分離させて、第一の一本鎖ユニバーサルアダプターを一端におよび第二の一本鎖アダプターを他端に含む複数の一本鎖DNA分子を遊離させる段階; およびf) 一本鎖DNA分子を単離し、それによってライブラリーを作製する段階。特定の局面において、(f)はi) 一本鎖DNA分子をリアクターアレイ上のある位置に送達する段階; またはii) 一本鎖DNA分子を油中水型乳濁液中の液滴内に送達する段階により達成することができる。
【0073】
これらの方法により、第一の二本鎖ユニバーサルアダプターまたは第二のユニバーサルアダプターを連結により断片化したDNA分子に付着させることができる。例えば、DNAリガーゼを使用することができる。これらの方法は、ポリメラーゼ、リガーゼ、キナーゼ、またはその組み合わせのような、DNA修復酵素およびDNA修飾酵素を用いて、アダプター連結DNA分子の混合物における一本鎖ニックを修復する段階をさらに含むことができる。特定の例として、この酵素には、バチルス・ステアロサーモフィルスのポリメラーゼI、T4リガーゼ、およびT4ポリヌクレオチドキナーゼが挙げられる。これらの方法のための鋳型DNAには、ゲノムDNA、cDNA、プラスミドDNA、コスミドDNA、人工染色体DNA、合成DNA、ファセミドDNA、ファージミドDNA、または逆転写産物を含むことができる。断片化は酵素的手段、化学的手段、または機械的手段により行われることができる。例えば、DNase I酵素は、10〜37℃の温度で1〜2分間行われる消化で使用することができる。または、制限酵素を使用することができる。機械的手段は、噴霧装置、フレンチプレス、超音波処理器、またはHydroShearとすることができる。
【0074】
これらの方法で使用する場合、断片化したDNA分子は、長さを50 bp〜700 bpとすることができる。適合末端は平滑末端とすることができる、または適合末端はAもしくはT突出部を含むことができる。平滑末端は、Pfuポリメラーゼ、T4 DNAポリメラーゼ、およびクレノーフラグメントのような酵素で生成することができる。第一または第二の二本鎖ユニバーサルアダプターは、一つまたは複数のホスホロチオエート結合を含んでもよく、ビオチン部分に付着されてもよい。さらに、第一の二本鎖ユニバーサルアダプターにもしくは第二の二本鎖ユニバーサルアダプターにまたはどちらの二本鎖ユニバーサルアダプターにも、識別可能なキー配列が含まれてもよい。例えば、識別可能なキー配列は、長さが3〜12ヌクレオチドであり、A、G、C、U、およびTからなる群より選択される少なくとも一つのヌクレオチドを含む。第一および第二の二本鎖ユニバーサルアダプターには、PCRプライミング配列および配列決定用プライマー配列が含まれてもよい。さまざまな局面において、PCRプライミング配列は長さが10〜20塩基対であり、配列決定用プライマー配列は長さが10〜20塩基対である。さらに、PCRプライミング配列および配列決定用プライマー配列は重複してもよい。
【0075】
これらの方法に合わせて、アダプター連結DNA分子の混合物をゲル電気泳動、ろ過、サイズ排除クロマトグラフィー、およびショ糖沈降法からなる群より選択される方法により分離する。複数の一本鎖DNA分子は、低塩処理、高pH処理、および化学的変性処理からなる群より選択される処理により得ることができる。さらなる局面において、複数の一本鎖DNA分子をDNA捕捉ビーズに付着することができる。さらに、DNA捕捉ビーズは、ビオチン/アビジン、リガンド/受容体、抗原/抗体または相補ヌクレオチドのような、結合対の成分を含むことができる。DNA捕捉ビーズは常磁性ビーズであることが好ましい。
【0076】
本発明には同様に、以下の段階を含む、固相支持体に付着された一本鎖DNAライブラリーを作製するための方法が含まれる: a) 複数の鋳型一本鎖DNAを作製する段階; b) 複数の鋳型ssDNAのそれぞれを固相支持体に付着させる段階; およびc) 鋳型一本鎖DNAが付着された固相支持体を単離する段階。
【0077】
本発明にはさらに、以下の段階を含む、固相支持体に付着された一本鎖DNAライブラリーを作製するための方法が含まれる: a) 大きい鋳型DNA分子を断片化して複数の断片化したDNA分子を作製する段階; b) 第一または第二の二本鎖ユニバーサルアダプターを断片化した各DNA分子の第一の末端におよび第一または第二のユニバーサルアダプターを断片化した各DNA分子の第二の末端に付着させて、アダプター連結DNA分子の混合物を作製する段階; c) 第一の一本鎖ユニバーサルアダプターと第二の一本鎖ユニバーサルアダプターとを含む一本鎖DNA分子を単離する段階; およびd) (c)より単離された一本鎖分子を固相支持体に付着させる段階。
【0078】
この方法で使用する場合、固相支持体はDNA捕捉ビーズとすることができ、DNAはゲノムDNA、cDNA、プラスミドDNA、コスミドDNA、人工染色体DNA、合成DNA、ファセミドDNA、またはファージミドDNAとすることができる。ある種の局面において、DNAをアビジン/ビオチン、リガンド/受容体、抗原/抗体および相補ヌクレオチドのような結合対により固相支持体に付着させる。この方法により作製される可動性の固相支持体(mobile solid support)のライブラリーも包含される。
【0079】
さらに、本発明には、第一のアダプターと、鋳型DNAの断片と、第二のアダプターとを含む核酸分子が含まれ、その際に第一のアダプターと第二のアダプターそれぞれが、配列決定用プライマーと、PCRプラマーと識別可能なキー配列とを含み、およびその際に第一のアダプターと第二のアダプターは、解離した場合に、ストリンジェントなハイブリダイゼーション条件の下で相互に交差ハイブリダイズしない。この方法では、PCRプライマーは長さを10〜20塩基対とすることができ、配列決定用プライマーは長さを10〜20塩基対とすることができ、および識別可能なキー配列は長さを3〜12塩基対とすることができる。例として、鋳型DNAはゲノムDNA、cDNA、プラスミドDNA、コスミドDNA、人工染色体DNA、合成DNA、ファセミドDNA、またはファージミドDNAとすることができる。核酸分子は、解離した場合に、解離した鋳型DNAに対する交差ハイブリダイゼーションが最小であることが好ましい。
【0080】
本発明により同様に包含されるのは、以下の段階を含む、一本鎖DNA分子を調製するための方法である: a) 大きいまたは全ゲノムの鋳型DNA分子を断片化して複数の断片化したDNA分子を作製する段階; b) 第一の二本鎖ユニバーサルアダプターまたは第二のユニバーサルアダプターを断片化した各DNA分子の第一の末端におよび第一のユニバーサルアダプターまたは第二のユニバーサルアダプターを断片化した各DNA分子の第二の末端に連結させて、アダプター連結DNA分子の混合物を作製させる段階; c) 第一の二本鎖ユニバーサルアダプターの片鎖を介して固相支持体に第一の二本鎖ユニバーサルアダプターと第二の二本鎖アダプターとを含むアダプター連結DNA分子を付着させる段階; d) 固相支持体に付着されていないアダプター連結DNA分子を洗い流す段階; e) 固相支持体に付着されたアダプター連結DNA分子を鎖分離させて、第一の一本鎖ユニバーサルアダプターを一端におよび第二の一本鎖アダプターを他端に含む複数の一本鎖DNA分子を遊離させる段階; およびf) 一本鎖DNA分子を単離する段階。
【0081】
本発明によりさらに包含されるのは、以下の段階を含む、鋳型核酸を複数の反応中心に送達するための方法である: a) 鋳型核酸の集団を供与する段階; b) 各鋳型核酸を集団から隔離剤にまで単離して、隔離された鋳型核酸の集団を形成させる段階; およびc) 隔離された鋳型核酸の集団を複数の反応中心に送達する段階であって、その際に各反応中心は隔離された核酸を一つ受け取る段階。この方法の場合、単離する段階は、鋳型核酸をビーズに付着させる段階、または鋳型核酸を油中水型乳濁液の乳濁液中に封入する段階を含むことができる。鋳型核酸は、核酸を結合できるビーズで封入されてもよい。この方法の場合、送達する段階は、隔離された核酸を複数の反応中心に送達する段階を含むことができ、その際に各反応中心はpicotiterプレートのウェルである。この方法は、単離された一本鎖分子をそれぞれ個別的に固相支持体に付着させる段階をさらに含むことができる。
【0082】
本発明の他の特徴は、本発明の説明のために示されるのであって、その限定であるとは意図されない典型的な態様に関する以下の記載のなかで明らかになると思われる。本明細書を通じて、当技術分野の状況および内容を記載するため、さまざまな特許、公開特許公報および科学文献が引用される。それらの開示は、その全体が、参照により本明細書に組み入れられる。
【0083】
実施例
実施例1: 試料調製
DNA試料:
DNAは良質のものであるべきであり、タンパク質、ヌクレアーゼ、脂質、および他の化学物質(調製液からの残存EDTAのような)および塩のような混入物をなくすべきである。好ましくは、ゲノムDNAは1.8またはそれ以上の260/280比率を有するべきである。ただ一種類の生物のゲノムを配列決定することが望まれる場合には、DNAは、混入DNAが存在しないことを保証する検査が行われた品質とするべきである。例えば、ヒトDNAの調製は、細菌DNA分子による混入がないことを保証するPCRにより検査することができる。混入がないか検査する別の方法は、制限消化パターンおよび特に制限消化に続いて、生物(例えば、ヒトまたはマウス)に特異的であることが知られている適当なプローブや考えられる混入生物(例えば、大腸菌)に特異的であることが知られている第二のプローブを用いたサザンブロットによるものである。所望であれば、DNAは、生物の単一クローン(例えば、細菌の場合にはコロニー)を起源とすべきである。
【0084】
段階1: DNase I消化
DNase I消化段階の目的は、全ゲノムまたは大きいゲノム部分のようなDNAの大きいストレッチを小さな種に断片化することである。単一の鋳型DNAから作製された小サイズのDNA種のこの集団を「ライブラリー」という。デオキシリボヌクレアーゼI(DNaseI)は、二本鎖の鋳型DNAを切断するエンドヌクレアーゼである。DNaseIの切断特性により、鋳型DNAの無作為消化(すなわち、最小の配列の偏り)を可能にし、マンガンに基づく緩衝液の存在下で使用する場合には平滑末端の、二本鎖DNA断片が優勢になるあると考えられる(Melgar and Goldthwait 1968)。DNase Iによる鋳型ゲノムの消化は、次の三つの要因に依存する: i) 使用する酵素の量(単位数); ii) 消化の温度(℃); およびiii) インキュベーション時間(分)。以下に概略を説明するDNase I消化条件は、50〜700塩基対(bp)に及ぶサイズのDNAライブラリーを得るために最適化された。
1. DNAを得て、Tris-HCl (10 mM, pH 7〜8)で0.3 mg/mlの濃度に調製した。この調製には、全部でDNA(15 μg) 134 μlが必要とされた。EDTAを含有する緩衝液(すなわち、TE, Tris/EDTA)で希釈したDNA調製物を使わないことを推奨する。EDTAの存在は、DNaseIによる酵素消化に阻害的である。DNA調製物にEDTAが含まれる場合、DNAを溶液から「塩析」して、適当なTris-HCl緩衝液(10 mM, pH 7〜8)またはナノピュアH2O(pH 7〜8)で再構成することが重要である。
2. 0.2 mlチューブに、Tris pH 7.5(1M) 50 μl、MnCl2(1M) 10μl、BSA(100 mg/ml) 1 μl、および水39 μlを含有する、DNase I緩衝液を調製した。
3. 別の0.2 mlチューブに、DNase I緩衝液15 μlおよびDNase I(1U/ml) 1.5 μlを添加した。反応チューブを15℃設定のサーマルサイクラーにセットした。
4. DNA(0.3 mg/ml) 134 μlを15℃設定のサーマルサイクラーにセットしたDNase I反応チューブに添加した。蓋を閉じて、試料を正確に1分間インキュベートした。インキュベーション後、50 mM EDTA 50 μlを添加して、酵素消化を停止させた。
5. 消化したDNAは、QiaQuick PCR精製キットを用いて精製した。消化反応液を次に、4分割量に分けて、4本のスピンカラムを使用して各分割量を精製した(スピンカラムあたり37.5 μl)。製造元の手順書に従い、各カラムを溶出用緩衝液(EB) 30 μlで溶出した。次いで、溶出液を合わせて、最終反応容量120 μlを得た。
6. 消化反応液の一定分割量3 μl 1つをBioAnalzyer DNA 1000 LabChipによる分析のために確保しておいた。
【0085】
段階2: Pfuポリッシング
DNase Iによる鋳型DNAの消化により、主に平滑末端であるDNAの断片が得られるが、しかし、一部の断片は、1塩基または2塩基の長さの突出末端を含んだ末端を有することになる。Pfuポリッシングを使用して、5'突出部の埋め込み(すなわち、「平滑末端化」)により平滑末端種の量を増加させる。さらに、Pfu DNAポリメラーゼは、一つのおよび二つのヌクレオチド伸長部分の除去をもたらす3'から5'方向のエキソヌクレアーゼ活性を有する。Pfuポリッシングにより、アダプターの連結に使用可能な平滑末端DNA断片の量を増加させる(Costa 1994a, 1994b, 1994c)。以下のPfuポリッシング手順を使用した。
1. 0.2 mlチューブに、精製済みの、DNase I消化DNA断片115 μl、10×クローン化Pfu緩衝液15 μl、dNTP(10mM) 5 μl、およびクローン化Pfu DNAポリメラーゼ(2.5 U/μl)を順に添加した。
2. ポリッシング反応の各成分を十分に混合して、72℃で30分間インキュベートした。
3. インキュベーション後、反応チューブを取り出して、氷上に2分間置いた。
4. ポリッシング反応混合液を次に、4分割量に分けて、QiaQuick PCR精製カラムを使用して精製した(各カラムに37.5 μl)。製造元の手順書に従い、各カラムを緩衝液EB 30 μlで溶出した。次いで、溶出液を合わせて、最終反応容量120 μlを得た。
5. 最終的なポリッシング反応液の一定分割量3 μl 1つをBioAnalzyer DNA 1000 LabChipによる分析のために確保しておいた。
【0086】
段階3: 断片化したDNAライブラリーへのユニバーサルアダプターの連結
ゲノムDNAライブラリーの断片化およびポリッシング後、プライマー配列を各DNA断片の末端に付加する。これらのプライマー配列は「ユニバーサルアダプター」といわれ、PCR増幅およびヌクレオチド配列決定の双方を可能とする特異的なプライミング領域を含む二本鎖オリゴヌクレオチドからなる。ユニバーサルアダプターは、各デオキシリボヌクレオチド(すなわち、A、C、G、T)の1つからなる特有の4塩基の「キー」に先行する、長さが20 塩基対である特有の配列決定プライミング領域のセットに隣接して位置する長さが20 塩基対である特有のPCRプライミング領域のセットを含むように設計される。それぞれの特有のユニバーサルアダプター(「ユニバーサルアダプターA」および「ユニバーサルアダプターB」といわれる)は、長さが44塩基対(44 bp)である。ユニバーサルアダプターは、T4 DNAリガーゼにより、DNA断片の各末端に連結されて、各DNA断片に対し全体で88 bpのヌクレオチド付加をもたらす。異なるユニバーサルアダプターを各ゲノムDNAライブラリーの調製専用に設計して、その結果、各生物に特有の識別子を与えることができる。
【0087】
ユニバーサルアダプターの対を調製するため、一本鎖オリゴヌクレオチドを自家設計して、商業者を通じて製造する。ユニバーサルアダプターのDNAオリゴヌクレオチドは、ヌクレアーゼ活性を防ぐ働きをする、各オリゴヌクレオチド末端の二つのホスホロチオエート結合を考慮して設計する(Samini, T.D., B. Jolles, and A. Laigle. 2001. Best minimally modified antisense oligonucleotides according to cell nuclease activity. Antisense Nucleic Acid Drug Dev. 11(3):129., これらの開示は、その全体が、参照により本明細書に組み入れられる)。各オリゴヌクレオチドをHPLC精製して、最終調製物に混入または擬似DNAオリゴヌクレオチド配列が存在しないことを確実にする。
【0088】
ユニバーサルアダプターは、平滑末端にした、断片化ゲノムDNAに対する定方向連結を可能とするように設計される。各ユニバーサルアダプター対に対し、PCRプライミング領域には5'の4塩基突出部および平滑末端にした3'キー領域が含まれる。ユニバーサルアダプターの平滑末端側は平滑末端DNA断片に連結するが、アダプターの5'突出部は平滑末端DNA断片に連結することができないので、指向性が達成される。さらに、5'ビオチンをユニバーサルアダプターBに付加して、その後の鋳型ssDNAの単離(段階8)を可能とする。各ユニバーサルアダプターは、単一チューブ内で、二つの一本鎖の相補DNAオリゴヌクレオチド(すなわち、センス配列を含む一方のオリゴおよびアンチセンス配列を含むもう一方のオリゴ)をアニーリングさせることにより調製する。以下の連結手順を使用した。
1. 0.2 mlチューブに、nH2O(分子生物等級純水) 39 μl、消化済みの、ポリッシングしたDNAライブラリー 25 μl、2×クイックリガーゼ反応用緩衝液100μl、MMP1 (10 pm/μl)アダプターセット 20 μl(100倍比)、およびクイックリガーゼ 16 μlを順に添加した。この連結反応液を十分に混合して、室温で20分間インキュベートした。
2. その後、連結反応液を取り除いて、連結反応液の一定分割量10 μl 1つをBioAnalyzerで用いるために精製した。Qiagen Min-Eluteキットのシングルスピンカラムを使用した。製造元の手順書による手順に従い、カラムをEB 10 μlで溶出した。精製した連結反応液の一定分割量1 μl 1つをBioAnalyzer DNA 1000 LabChipにより添加した。未精製の連結反応液には、試料がBioAnalyzerで正しく泳動するのを阻害することになる多量の塩およびPEGが含まれるので、この精製段階を推奨する。
3. 連結反応液の残り(190 μL)を段階4のゲル分離に使用した。
【0089】
段階3a: マイクロコンろ過(Microcon Filtration)およびアダプター構築
全調製時間は約25分であった。
ユニバーサルアダプターの連結反応には、100倍過剰のアダプターが必要とされる。これらの過剰なアダプターの除去を補助するため、二本鎖gDNAライブラリーをMicrocon YM-100ろ過器に通す。Microcon YM-100膜を使用して、125 bpよりも小さな二本鎖DNAを除去することができる。従って、未結合のアダプター(44 bp)、ならびにアダプター二量体(88 bp)を連結化gDNAライブラリー集団から除去することができる。以下のろ過手順を使用した。
1. 段階4の連結反応液190 μLを組立型のMicrocon YM-100ろ過器に添加した。
2. ろ過器を遠心機にセットして、5000×gで約6分間、または膜がほぼ乾燥するまで回転させた。
3. 洗浄するため、1×TE 200 μlを添加した。
4. 試料を5000×gでさらに9分間、または膜がほぼ乾燥するまで回転させた。
5. 回収するため、ろ過容器を新たなバイアルに挿入して、3000×gで3分間回転させた。ろ過容器は廃棄した。回収された容量は、約10 μlであった。次に、TE 80 μlを添加した。
【0090】
アダプター(AおよびB)は、使用の前に、HPLC精製して、ホスホロチオエート結合で修飾した。アダプター「A」(10 μM)の場合、100 μMアダプターA(44 bp、センス) 10 μlを100 μMアダプターA(40 bp、アンチセンス) 10 μlと混合して、1×アニーリング用緩衝液30 μlを混合した(Vf = 50μl)。プライマーは、Sample Prep LabサーマルサイクラーのANNEALプログラムを用いてアニーリングさせた(下記参照)。アダプター「B」(10 μM)の場合、100 μMアダプターB(40 bp、センス) 10 μlを100 μMアダプターB(44 bp、アンチセンス) 10 μlと混合して、1×アニーリング用緩衝液30 μlを混合した(Vf = 50μl)。プライマーは、Sample Prep LabサーマルサイクラーのANNEALプログラムを用いてアニーリングさせた。アダプターの対は、使用するまで-20℃で保存することができる。
【0091】
ANNEAL-プライマーアニーリング用のプログラム
1. 95℃で1分インキュベートする。
2. 15℃まで0.1℃/秒で温度を下げる。ならびに
3. 15℃で保持する。
【0092】
ゲノムDNA挿入断片およびアダプターに必要な方向はなかった。断片は両端で連結可能とされた。ユニバーサルアダプターのセットには、4本の一本鎖DNAオリゴヌクレオチドが含まれた。各一本鎖オリゴヌクレオチドは、1マイクロモルのスケールで合成されて、HPLC精製された。各一本鎖オリゴヌクレオチドには、各末端に四つのホスホロチオエート結合が含まれた。
【0093】
段階4: 連結されたDNAライブラリーのゲル電気泳動および抽出
ユニバーサルアダプターの連結手順により以下のものが生ずる: 1) 両端にアダプターを有する断片化したDNA; 2) 未結合の単一アダプター; またはアダプター二量体の形成。アダプターが連結されたDNAライブラリー集団を未連結の、単一アダプターおよびアダプター二量体の集団から分離かつ単離する方法として、アガロースゲル電気泳動が使用される。ゲノムDNAのDNase I消化手順により、50〜700 bpに及ぶライブラリー集団が得られる(段階1)。88 bpのユニバーサルアダプターセットの付加は、その集団をより大きいサイズに変えて、約130〜800 bpのサイズ範囲の移動プロファイルをもたらすことになる。アダプター二量体は88 bpに移動することになり、未連結のアダプターは44 bpに移動することになる。従って、サイズ範囲が200 bpを超えるゲノムDNAライブラリーをアガロースゲルから物理的に単離して、標準的なゲル抽出技術により精製することができる。アダプターが連結されたDNAライブラリーのゲル分離により、サイズ範囲が200 bpまたはそれ以上であるライブラリー集団が回収されることになる(ライブラリーのサイズ範囲は、用途に応じて変化させることができる)。以下の電気泳動および抽出手順を使用した。
1. 2%アガロースゲルを調製した。
2. 10×Ready-Load Dye 10 μlをDNA連結混合液の残り90 μlに添加した。
3. 色素/連結反応混合液を4本の隣接レーンを使ってゲルに添加した(25 μl/レーン)。
4. 100 bp ラダー(0.1 μg/μl) 10 μlを連結反応液のレーンから2レーン離して添加した。
5. ゲルを100 Vで3時間泳動させた。
6. ゲルの泳動が完了した場合には、ゲルをゲル槽から取り出して、プラスチックラップで覆った平面に移した。DNAのバンドを手持ち式の長波長UVライトにより可視化した。無菌の、使い捨ての外科用メスを用いて、200〜400 bpの断片サイズをアガロースゲルから切り出した。この手法を用いて、任意のサイズ範囲のライブラリーを単離することができる。二つまたはそれ以上のサイズ範囲を単離することも可能である。ライブラリーのサイズ範囲が200〜900 bpである場合、単一ウェルからいくつかのサイズ範囲を単離することが可能である(すなわち、200〜400 bpおよび500〜700 bp)。
7. アガロースゲル内に埋まったDNAは、製造元の使用説明書に従い、Qiagen MinElute Gel Extractionキットを用いて単離した。手短に言えば、緩衝液QGをチューブ内のアガロースを覆うように添加した。アガロースを完全に溶解させた。緩衝液QGの色をQiagenの使用説明書に従ってpHを調整することにより維持し、試料の損失を最小限にした。2本のMinEluteスピンカラム(Qiagen)を精製のために使用した。溶解したアガロースの容量が多く、各カラムに何回も添加する必要があった。カラムを予め55℃に温めておいた緩衝液EB 10 μlで溶出した。溶出液をプールして、gDNAライブラリー 20 μlを得た。
8. それぞれ単離したDNAライブラリーの一定分割量1 μl 1つをBioAnalyzer DNA 1000 LabChipにより分析して、DNAライブラリー集団の正確な分布を評価した。
【0094】
段階5: ニックの入った二本鎖DNAライブラリーの鎖置換および鎖伸長
ユニバーサルアダプターに使用されるDNAオリゴヌクレオチドはリン酸化されないので、ギャップが断片化gDNAの3'連結部に存在する。これらの二つの「ギャップ」または「ニック」は、鎖置換DNAポリメラーゼを使用することにより埋めることができる。このポリメラーゼは、ニックの修復およびニックの入っていない二本鎖DNAの形成をもたらすように、ニックを認識し、ニックの入った鎖を置換して、鎖を伸長する。使用される鎖置換酵素は、Bst DNAポリメラーゼのラージフラグメントである。
1. 0.2 mlチューブに、ゲル抽出したDNAライブラリー 19 μl、nH2O 40 μl、10×ThermoPol反応用緩衝液(Reaction Buffer) 8 μl、BSA(1 mg/ml) 8 μl、dNTP(10 mM) 2 μl、およびBst Iポリメラーゼ(8 U/μl) 3 μlを順に添加した。
2. 試料を十分に混合してサーマルサイクラーにセットし、鎖置換インキュベーションプログラム:「BST」を用いてインキュベートした。ニックの入った二本鎖DNAの鎖置換および鎖伸長用のBSTプログラム
1. 65℃で30分インキュベートする。
2. 80℃で10分インキュベートする。
3. 58℃で10分インキュベートする。 ならびに
4. 14℃で保持する。
3. Bst処理DNAライブラリーの一定分割量1 μL 1つをBioAnalyzer DNA 1000 LabChipにより泳動した。
【0095】
段階6: ストレプトアビジンビーズの調製
ニックの入っていない二本鎖ゲノムDNAの作製に続いて、隣接のユニバーサルアダプター配列を含んだ一本鎖ゲノムDNAを単離することが必要である。この段階は、ストレプトアビジンビーズとのビオチン標識二本鎖DNAの結合について概説する。ストレプトアビジンビーズを調製するのに、以下の手順を使用した。
1. Dynal M-270ストレプトアビジンビーズ 100 μlは、この磁気ビーズをMPCに適用することにより1×結合用緩衝液(1 M NaCl, 0.5 mM EDTA, 5 mM Tris, pH 7.5) 200 μlで2回洗浄した。
2. ビーズを2×結合用緩衝液 100 μlに再懸濁させて、次にBst処理DNA試料(段階5からの)の残り79 μlおよび水20 μlを添加した。
3. ビーズ溶液を十分に混合して、チューブ回転装置に室温で20分間載せておいた。ビーズ混合液を、MPCを用いて、1×結合用緩衝液100 μlで2回、次いでnH2Oで2回洗浄した。結合および洗浄用(B & W)緩衝液 (2×および1×): 2×B & W緩衝液は、10 mM Tris・HCl (pH 7.5)、1 mM EDTA、および2 M NaClを混合することにより調製した。試薬を上記のように混ぜ合わせて、完全に混合した。この溶液は室温で6ヶ月間保存することができる。1×B & W緩衝液は、2×B & W緩衝液をnH2Oと1:1で混合することにより調製した。終濃度は上記の半分、すなわち、5 mM Tris・HCl (pH 7.5)、0.5 mM EDTA、および1 M NaClとなった。
【0096】
段階7: ストレプトアビジンビーズを用いた一本鎖DNAライブラリーの単離
ストレプトアビジンビーズとの二本鎖gDNAライブラリーの結合に続いて、連結されたプールからユニバーサルアダプターAとユニバーサルアダプターBとを含んだ一本鎖gDNAだけを単離することが好ましい(所望の集団は、星印で下記に指定されている)。二本鎖ゲノムDNA断片のプールには、以下の予想配置でアダプターが結合されることになる。
ユニバーサルアダプターA - gDNA断片 - ユニバーサルアダプターA
ユニバーサルアダプターB - gDNA断片 - ユニバーサルアダプターA*
ユニバーサルアダプターA - gDNA断片 - ユニバーサルアダプターB*
ユニバーサルアダプターB - gDNA断片 - ユニバーサルアダプターB
【0097】
ユニバーサルアダプターBだけが5'ビオチン部分を有するので、磁性のストレプトアビジン含有ビーズを使用して、ユニバーサルアダプターBを保有する全てのgDNAライブラリー種を結合することができる。二つのユニバーサルアダプターAを含むゲノムライブラリー集団種(または非連結種)は、ストレプトアビジン含有ビーズに結合せず、洗浄手順の間に除去される。洗浄後にビーズに結合したままの種には、ユニバーサルアダプターAとBとを有するものまたは二つのユニバーサルアダプターB末端を有するものが含まれる。
【0098】
二つのユニバーサルアダプターB配列を有し、二つのビオチン分子を有するゲノムDNA種は、ストレプトアビジン含有ビーズに両端で結合することができる。アダプターAとアダプターBとを有する種は一つのビオチン分子を有するのみで、ビーズに「B」末端でのみ結合することができる。一本鎖の集団を単離するため、ビーズに結合した二本鎖DNAを、相補DNA鎖間の水素結合を切断する働きをする水酸化ナトリウム溶液で処理する。DNA断片がビオチンをそれぞれの末端(ユニバーサルアダプターBの末端)に有する場合、生ずる一本鎖はどちらもビーズに結合したままである。断片が一つのビオチンのみ(ユニバーサルアダプターAおよびB)を有する場合には、相補鎖はDNA-ビーズ複合体から分離する。
【0099】
生ずる一本鎖ゲノムDNAライブラリーを液相から回収して、例えば、ピロリン酸配列決定(PyroSequence)法を使用して、またはRNA Pico 6000 LabChip (Agilent, Palo Alto, CA)を使用することにより定量化する。一本鎖ゲノムDNAライブラリーを、単位容量あたりの分子数を計算することにより定量化する。次いで、一本鎖gDNA分子を、DNA捕捉プライマー(PCRプライマーB)を含有する25〜30 μmのセファロースビーズにアニーリングさせる(1ビーズにつき1個の有効なコピーを得るためにビーズあたり半分のコピー数で)。次に、鋳型を乳濁液ポリメラーゼ連鎖反応手順により増幅させる。その後の配列決定は、周知技術を使用して行うことができる。一本鎖ライブラリーの単離のために、以下の手順を使用した。
1. 融解溶液(0.125 M NaOH, 0.1 M NaCl) 250 μlを上記段階6の洗浄済みのビーズに添加した。
2. ビーズ溶液を十分に混合して、ビーズ混合液をチューブ回転装置上で室温にて10分間インキュベートした。
3. Dynal MPC (磁性粒子濃縮器)を使用し、沈殿ビーズを注意深く取り除いて、上清を取っておいた。250 μlの上清には、一本鎖DNAライブラリーが含まれていた。
4. 別のチューブに、PB(QiaQuick精製キットからの) 1250 μlを添加して、この溶液を20%酢酸9 μlの添加により中和した。
5. Dynal MPCを用いて、一本鎖gDNAライブラリーを含有する250 μlの上清からビーズを沈殿させて、この上清を注意深く取り出し、新たに調製したPB/酢酸溶液に移した。
6. この溶液1500 μlを1個のQiaQuick精製スピンカラムにより精製した(試料を1回の添加につき750 μlとして2回同じカラムに添加して通す)。一本鎖DNAライブラリーをEB 50 μlで溶出した。
【0100】
段階8a: ピロリン酸配列決定による一本鎖gDNAの定量化
全調製時間は約1時間であった。
1. 0.2 mlチューブに、以下の試薬を順に添加した。
2. DNAをANNEAL-Sプログラム(以下の補遺を参照されたい)によりアニーリングさせた。
3. 試料をPSQ(ピロリン酸配列決定装置(jig))で泳動して、各試料中の鋳型のピコモル数を決定した(下記を参照されたい)。配列決定の方法は、米国特許第6,274,320号; 米国特許第4,863,849号; 米国特許第6,210,891号; および米国特許第6,258,568号のなかで見出すことができる(これらの開示は、その全体が、参照により本明細書に組み入れられる)。計算を行い、1マイクロリットルあたりの一本鎖gDNA鋳型分子の数を決定した。調製した一本鎖gDNAライブラリーの残り25 μlは、増幅およびその後の配列決定(反応数およそ1×106)に使用した。
【0101】
段階8b: RNA Pico 6000 LabChipによる一本鎖gDNAの定量化。全調製時間は約30分であった。
1. BioAnalyzer(ソフトウェアバージョン2.12)で、選択肢mRNA Picoアッセイを選択した。
2. BioAnalyzer上で、製造元のガイドラインに従い、RNA Pico 6000 LabChipを調製した。
3. RNA LabChipラダー(RNA 6000ラダー)を製造元(Ambion)の指示に従って調製した。手短に言えば、溶液状態のRNA LabChipラダーを70℃に2分間加熱した。ラダーを急冷するため、溶液を氷上で5分間冷却した。管壁から凝縮物を取り除くため、溶液を手短に遠心した。このRNA LabChipラダーは、氷上で保存し、1日のうちに使用した。
4. 分析されるssDNAライブラリーは、一定分割量1 μl 3つを用い、隣接レーンに入れて、3回通り泳動した。
5. BioAnalyzerのソフトウェアを使用して、各ssDNAライブラリーのレーンの濃度を計算した(下記表および図8を参照されたい)。以下に概略を説明した手順を用い、3つのレーン全ての平均を使って、ライブラリーのDNA濃度を計算した。
a. ピーク積分の下限線(図8の長い点線)をライブラリーのピークの直前に動かした(下記を参照されたい)。
b. ピーク積分の上限線(図8の長い点線)をライブラリーのピークの直後に動かした。このような方法で、下限および上限の積分線を結び付けるピーク積分線は、バックグラウンドの傾きに従った。
c. マウス矢印を使用してベースとなるピークの平均サイズ(通常、ピークの最高点付近)を決定した、またはソフトウェアにより選択されるような確定ピークを使用した。
d. ピーク中の物質量を求めるのに積分値を使用した。ピコグラムで得られた回収値を回収分子に変換した(下記表を参照されたい)。次いで、ライブラリーの濃度(1マイクロリットルあたりの分子数)を決定した。
【0102】
表
【0103】
上記の表に示されるように、ライブラリー1の濃度は1639 pg/μl(列5)と算出され、その平均断片サイズは434ヌクレオチド(列9)となった。これらの値は、上記の段階(a)〜(d)に記載されているようにAgilent 2100ソフトウェアから得られた。リボヌクレオチドの平均分子量(MW)は328.2 g/モル(列10)である。ライブラリー平均断片のMW (1.42×105 g/モル、列11)は、平均断片長(434)に平均リボヌクレオチド(328.2)を掛けることにより算出した。定量化したライブラリー(1639 pg/μl)を1マイクロリットルあたりのグラム数(1.64×10-9 g/μl、列12)に変換した。1マイクロリットルあたりのモル数(1.15×10-14モル/μl、列14)は、1マイクロリットルあたりのグラム数(1.64×10-9 g/μl、列12)をライブラリー断片の平均分子量(1.42×105、列11)で割ることにより算出した。最後に、1マイクロリットルあたりの分子数(6.93×109分子/μl、列15)は、1マイクロリットルあたりのモル数(1.15×10-14モル/μl、列14)にアボガドロ数(6.02×1023分子/モル)を掛けることにより得られた。
【0104】
最終のライブラリー濃度は、1×108分子/μlよりも大きくなると予想された。ライブラリーの質にとってさらに重要な要因は、アダプター二量体の濃度であった。図8で、ライブラリーのピークの高さは、アダプター二量体のピーク(マーカー後の最初のピーク)よりも約10倍高いと測定された。高品質のライブラリーは、二量体のピークよりも少なくとも2倍高いピーク高さを有すると予想される。提供のRNA Pico 6000 LabChipにより一本鎖gDNA濃度が500%の精度内で推定されることに留意されたい。従って、投入gDNAのビーズあたりのコピー数(cpb)を決定する鋳型の滴定を使用して、最初の配列決定の泳動を行うことが重要であった。推奨される投入DNAは、2.5 cpb、1 cpb、0.5 cpb、および0.1 cpbである。この滴定は、14×43 PTPの4スロットのビーズ充填室を使用して簡単に調べられた。
【0105】
段階9: 一本鎖gDNAライブラリーの希釈および保存
一本鎖gDNAライブラリーは、緩衝液EBで溶出かつ定量化した。分解を防ぐため、一本鎖gDNAライブラリーは、EDTAの存在下で-20℃にて凍結保存した。定量化後、等量の10 mM TEをライブラリー貯蔵液に添加した。以降の全ての希釈はTE中とした。収量は次の通りであった。
PSQ分析後のssDNAライブラリーの残りの最終容量 = 25 μl。
LabChip分析後のssDNAライブラリーの残りの最終容量 = 47 μl。
【0106】
最初の貯蔵液の希釈では、一本鎖gDNAライブラリーを100×106分子/μl 1×ライブラリー用溶出緩衝液に希釈した。共用の一本鎖gDNAライブラリーの一定分割量を調製した。このため、200,000分子/μlを1×ライブラリー用溶出緩衝液に希釈して、一定分割量20 μlを計量した。使い捨てライブラリーの一定分割量は、-20℃に保存した。
【0107】
段階10: 乳濁液ポリメラーゼ連鎖反応(Emulsion Polymerase Chain Reaction)
cpb数の増加が好ましい場合、ビーズ乳濁液のPCRを2003年6月6日付で出願された米国特許出願第06/476,504号(その全体が参照により本明細書に組み入れられる)に記載されているように行った。
【0108】
試薬調製
停止溶液(50 mM EDTA)には、50 mM EDTA溶液1.0 mlを得るためにnH2O 900 μlと混合した0.5 M EDTA 100 μlが含まれた。10 mM dNTPの場合、dCTP(100 mM) 10 μl、dATP(100 mM) 10 μl、dGTP(100 mM) 10 μl、およびdTTP(100 mM) 10 μlを分子生物等級純水60 μlと混合した。4種の100 mMヌクレオチド貯蔵液は全て氷上で融解させた。次いで、各ヌクレオチド10 μlをnH2O 60 μlと混合して最終容量を100 μlとし、完全に混合した。次に、一定分割量1 mlを1.5 ml微量遠心管に分注した。この貯蔵液は、-20℃で1年間保存することができる。
【0109】
10×アニーリング用緩衝液には、200 mM Tris (pH 7.5)および50 mM酢酸マグネシウムが含まれた。この溶液の場合、Tris 24.23 gをnH2O 800 mlに添加して、この混合液をpH 7.5に調整した。この溶液に、酢酸マグネシウム10.72 gを添加して、完全に溶解させた。この溶液は最終容量を1000 mlに合わせて、4℃で1ヶ月間保存することができる。10×TEには、100 mM Tris・HCl (pH 7.5)および50 mM EDTAが含まれた。これらの試薬を同時に添加して、完全に混合した。この溶液は室温で6ヶ月間保存することができる。
【0110】
実施例2: プライマー設計
上記のように、ユニバーサルアダプターは、以下を含むように設計される: 1) 長さが通常20 bpである特有のPCRプライミング領域のセット((2)に隣接して位置する); 2) 長さが通常20 bpである特有の配列決定プライミング領域のセット; および3) 任意で、続けて4種のデオキシリボヌクレオチド(すなわち、A、C、G、T)のそれぞれの少なくとも1つからなる特有の識別可能なキー配列。プライマーと関心対象のゲノムの意図しない領域との間の交差ハイブリダイゼーションの確率は、ゲノムサイズが増加するにつれおよびプライマーとの完全な適合性が低下するにつれて増加する。しかしながら、交差ハイブリダイズする領域(CHR)とのこの潜在的相互作用は、下記の理由から問題をもたらすとは予想されない。
【0111】
本発明の好ましい態様において、一本鎖DNAライブラリーは、PCR増幅およびその後の配列決定に使用される。配列決定法には、150〜500塩基対の断片への所定のゲノムの無作為消化が必要となり、その後、二つの特有の両方向性プライマー(PCRおよび配列決定領域の双方からなる)を断片の5'および3'末端に連結する(図5)。融解温度(Tm)、ゲノム内のプライミング配列の特異性および特定領域または関心対象の遺伝子との近接性に基づいて、ゲノムの既存部分をプライミング部位として選択する典型的なPCR増幅とは異なり、開示の方法では、合成によるプライミング部位を用いており、慎重な新規のプライマー設計が必要になる。
【0112】
四量体の選択:
デノボプライマー設計の戦略は、ハイブリダイゼーション実験のための分子タグ(Hensel, M. and D.W. Holden, Molecular genetic approaches for the study of virulence in both pathogenic bacteria and fungi. Microbiology, 1996. 142(Pt 5): p. 1049-58; Shoemaker, D.D., et al., Quantitative phenotypic analysis of yeast deletion mutants using a highly parallel molecular bar-coding strategy. Nat Genet, 1996. 14(4): p. 450-6を参照されたい)およびPCR/LDR(ポリメラーゼ連鎖反応/連結検出反応)のハイブリダイゼーションプライマー(Gerry, N.P., et al., Universal DNA microarray method for multiplex detection of low abundance point mutations. Journal of Molecular Biology, 1999. 292: p. 251-262; Witowski, N.E., et al., Microarray-based detection of select cardiovascular disease markers. BioTechniques, 2000. 29(5): p. 936-944.を参照されたい)に対して行われた研究に関する既刊文献に見出される。
【0113】
PCR/LDRの研究は、特に関連性があり、オリゴヌクレオチド「ジップコード」、つまり類似の最終Tmを有する6つの特異的に設計された四量体からなる24塩基のプライマーの設計に重点的に取り組んでいた(Gerry, N.P., et al., Universal DNA microarray method for multiplex detection of low abundance point mutations. Journal of Molecular Biology, 1999. 292: p. 251-262; 米国特許第6,506,594号を参照されたい)。四量体成分は、以下の判定基準に基づいて選択された: 各四量体は少なくとも2塩基だけ他のものと異なっていた、自己対合またはヘアピン形成を起こした四量体は除外した、および回文構造の四量体(AGCT)または反復性の四量体(TATA)は同様に取り除いた。考えられる256(44)通りの順列のうち36通りが必要条件を満たし、次に、許容可能なPCRプライマーの設計に必要なさらなる制約にかけられた(表1)。
【0114】
表1
表1には、Gerry et al. 1999. J. Mol. Bio. 292: 251-262により概説される判定基準に基づく四量体プライマー成分の選択を明示する行列が示されている。各四量体は少なくとも2塩基だけ他の全てのものと異なることが必要とされた。これらの四量体は他のどの四量体とも回文構造または相補的になることはない。36通りの四量体(太字、下線)を選択した。斜体の配列は、考慮から除外された回文構造の四量体を示す。
【0115】
プライマー設計:
PCRプライマーは、一般的なプライマー設計に共通の規格を満たすように設計し(Rubin, E. and A.A. Levy, A mathematical model and a computerized simulation of PCR using complex templates. Nucleic Acids Res, 1996. 24(18): p. 3538-45; Buck, G.A., et al., Design strategies and performance of custom DNA sequencing primers. Biotechniques, 1999. 27(3): p. 528-36を参照されたい)、実際の選択はコンピュータプログラムMMPにより行った。プライマーは、すべての両方向性PCR/配列決定用プライマーの効率的な合成のため、20塩基(5つの四量体)の長さに制限した。各プライマーは5'末端に2塩基のGCクランプ、および3'末端に1塩基のGCクランプを含み(表2)、全てのプライマーが類似のTm(+/-2℃)を共有していた(図10)。プライマー内でヘアピン形成(内部ヘアピンステム ΔG > -1.9 kcal/モル)させなかった。二量体化も同様に制御した。3塩基の最大許容二量体を許容したが、これは最後の6つの3'塩基で起こり、3'二量体に対する最大許容ΔGは-2.0 kcal/モルであった。さらに、3'末端がグループ内の他のものに過度に類似するプライマーに、ペナルティーを適用して、これにより、あるプライマーと他の逆相補体との間の交差ハイブリダイゼーションを阻止した。
【0116】
(表2)
【0117】
表2には、2つの5'および1つの3'G/Cクランプを与える、36通りの選択された四分子の考えられる順列が示されている。内部の位置は、残る四分子からなる。この結果、8×19×19×19×9通りの順列、または493,848通りの可能な組み合わせが生まれる。図10は初回通過、つまり493,848通りのプライマーの範囲を64〜66℃のTmを有する56,246通りの候補に減らす、Tmに基づく許容可能なプライマーの選択を示す。
【0118】
(表3)プライマーに対する完全な配列適合性の確率は、適合する長さの要件を減少させるにつれておよび関心対象のゲノムのサイズを増加させるにつれて増加する。
【0119】
複雑な試料集団でのミスマッチに対するPCRの許容度が報告されている(例えば、Rubin, E. and A.A. Levy, A mathematical model and a computerized simulation of PCR using complex templates. Nucleic Acids Res, 1996. 24(18): p. 3538-45を参照されたい)にもかかわらず、相補領域が関心対象のゲノム内に発生する確率は、プライマー設計過程において主要な懸案事項ではなかった。20塩基のプライマーに対して完全適合性を見出す確率は極めて低い(420)(表3)が、それほど連続的ではない適合性を見出す確率は、関心対象のゲノムのサイズとともに顕著に増加する。結果として、20塩基のうち少なくとも10塩基の完全適合性を見出す確率は、アデノウイルスのゲノムでは99.35%になる。16塩基の完全適合性を見出す確率は、NCBIデータベースの配列(アデノウイルスのゲノムよりも約100倍大きい)では97%になる。20塩基のプライマーに対して17塩基の完全適合性を見出す確率は、ヒトゲノムの配列(30億塩基)では99%になる。
【0120】
ゲノムの領域に対するプライマーの交差ハイブリダイゼーションの確率の高さは、鋳型断片を作製させるために使用される無作為DNA消化により、予想されるほどの問題にはならない。従って、交差ハイブリダイズする領域(CHR)の影響はほとんど害がない。CHRが溶液中のPCRプライマーとその鋳型との間の完全適合性と十分に競合できる可能性は低い。さらに、3'末端にミスマッチを含むプライマーはいずれも、著しく競合上不利な状態にあるものと思われる。たとえCHRが意図するPCRプライマーを打ち負かした(outcompete)としても、それにより、配列決定用プライマーに対する下流部位のない、切断型のPCR産物が作製されることになる。切断産物が捕捉ビーズに追いやられて、固定化される場合、二つの状況のうちの一つが生じることになる。CHRが液相のプライマーを打ち負かした場合には、固定化産物は配列決定用プライマー結合部位がないことになり、空のPicoTiterプレート(PTP)ウェルが生ずることになる。CHRがビーズ結合プライマーを打ち負かした場合には、配列決定用プライマーは依然として存在することになり、その唯一の影響は短い挿入断片ということになる。どちらの結果も配列決定の質を過度に落とすことにはならない。試料調製工程で使用されるゲノム材料の量が多い(今回、25 μg、35Kbのアデノウイルスゲノムを5.29×1016コピーを含有する)ことを考慮して、過剰なサンプリングを用い、完全なCHRのない断片を供与して、問題の領域の標準的PCR増幅を可能とすることができる。
【0121】
実施例3: 噴霧による試料調製
噴霧によるDNAの調製
噴霧段階の目的は、全ゲノムまたは大きいゲノム部分のようなDNAの大きいストレッチをDNA配列決定に適した小さな分子種に断片化することである。単一の鋳型DNAから作製された小サイズのDNA種のこの集団をライブラリーという。噴霧により二本鎖鋳型DNAが50〜900塩基対に及ぶ断片に剪断される。剪断されたライブラリーには、T4 DNAポリメラーゼ、大腸菌DNAポリメラーゼI(クレノーフラグメント)、およびT4ポリヌクレオチドキナーゼの組み合わせにより末端修復される一本鎖の末端が含まれる。T4 DNAポリメラーゼもクレノーDNAポリメラーゼも、その5'から3'方向のポリメラーゼ活性によってDNAの3'陥凹末端(5'突出部)を「埋める」ために使用される。T4およびクレノーポリメラーゼの一本鎖の3'から5'方向のエキソヌクレアーゼ活性により3'突出末端が除去されることとなり、T4ポリヌクレオチドキナーゼのキナーゼ活性により5'ヒドロキシル末端にリン酸が付加されることとなる。
【0122】
試料を以下のように調製した。
1. gDNA(ゲノムDNA) 15 μgを得て、10 mM TE (10 mM Tris, 0.1 mM EDTA, pH 7.6; 項の末尾の試薬リストを参照されたい)で最終容量100 μlに調整した。このDNAを吸光度260/280比の測定により混入がないか分析した。この比は1.8またはそれより高かった。gDNA最終濃度は約300 μg/mlになると予想された。
2. 氷冷の噴霧緩衝液(項の末尾を参照されたい) 1600 μlをgDNAに添加した。
3. この反応混合液を氷冷の噴霧装置(CIS-US, Bedford, MA)にセットした。
4. 15 mlスナップキャップ・ファルコンチューブのキャップを噴霧装置の先端に取り付けた(図7A)。
5. キャップを汚れのない噴霧装置のクランプ部品(ぴったりのカバー(ファルコンチューブの蓋に対し)と2つのゴム製O-リングとからなる)で固定した(図7B)。
6. 噴霧装置の底面を窒素供給器に取り付けて、装置全体をパラフィルムで包んだ(図7Cおよび7D)。
7. 噴霧装置を垂直に維持したまま(図7Dに示されるように)、50 psi(平方インチ当たりのポンド)の窒素を5分間加えた。噴霧装置の底面を数秒毎に硬質表面に叩きつけて、濃縮液を底面に押し進めた。
8. 窒素を5分後に止めた。圧力が平常値に戻った後(30秒)、窒素供給源を噴霧装置から取り外した。
9. パラフィルムを除去して、噴霧装置の先端の蓋を回して外した。試料を取り出して、1.5 ml微量遠心管に移した。
10. 噴霧装置の先端の蓋を再び取り付けて、噴霧装置を500 rpmで5分間遠心した。
11. 噴霧装置中の試料の残りを回収した。総回収量は約700 μlであった。
12. 回収された試料は、製造元の使用説明書に従い、QIAquickカラム(Qiagen社、Valencia, CA)を用いて精製した。量が多く、カラムに数回添加することが必要になった。試料は、55℃に予め暖めておいた緩衝液EB(10 mM Tris HC1, pH 8.5; Qiagenキットで供給される) 30 μlで溶出させた。
13. 試料はUV分光法により定量化した(100倍希釈の場合には水198 μlに2 μl)。
【0123】
酵素ポリッシング
鋳型DNAの噴霧により非相補的末端を有する多くのDNA断片が得られる。これらの末端は、3種の酵素、つまりT4 DNAポリメラーゼ、大腸菌DNAポリメラーゼ(クレノーフラグメント)およびT4ポリヌクレオチドキナーゼを用いることにより、平滑およびアダプター断片に連結される状態にされる。
【0124】
試料を以下のように調製した。
1. 0.2 mlチューブに、以下の試薬を順に添加した。
2. 段階1の溶液を十分に混合して、MJサーモサイクラー(任意の的確なインキュベーターを使用することができる)中で25℃にて10分間インキュベートした。
3. 大腸菌DNAポリメラーゼ(クレノーフラグメント)(5単位/ml) 1.25 μlを添加した。
4. 反応液を十分に混合して、MJサーモサイクラー中で25℃にて10分間および16℃にてさらに2時間インキュベートした。
5. 処理したDNAは、QiaQuickカラムを用いて精製し、55℃に予め暖めておいた緩衝液EB(10 mM Tris HC1, pH 8.5) 30 μlで溶出させた。
6. 以下の試薬を0.2 mlチューブに混合した。
7. 溶液を混合して、37℃で30分間、65℃で20分間のインキュベーション、その後、14℃で保存のT4 PNKプログラムを使用するMJサーマルサイクラーにセットした。
8. 試料は、QiaQuickカラムを用いて精製し、55℃に予め暖めておいた緩衝液EB 30 μlで溶出させた。
10. 最終ポリッシング反応液の一定分割量2 μlをBioAnalyzer DNA 1000 LabChipによる分析のためにとっておいた(以下を参照されたい)。
【0125】
アダプターの連結
アダプターを連結するための手順は、次のように行った。
1. 0.2 mlチューブに、以下の試薬を順に添加した。
上記の反応液は、5 μg用に設計されており、使用するgDNAの量に応じてスケールを変えた。
2. 試薬を十分に混合して、25℃で20分間インキュベートした。チューブは、ゲルがアガロースゲル電気泳動用に調製されるまで氷上とした。
【0126】
連結されたgDNAライブラリーのゲル電気泳動および抽出
ゲノムDNAの噴霧により、50〜900 bpに及ぶライブラリー集団が得られる。88 bpのユニバーサルアダプターセットの付加は、その集団をより大きいサイズに変えて、より大きいサイズ範囲(約130〜980 bp)の移動プロファイルをもたらすことになる。アダプター二量体は88 bpに移動することになり、連結されていないアダプターは44 bpに移動することになる。従って、サイズ範囲が250 bpまたはそれ以上に分離されるゲノムDNAライブラリーをアガロースゲルから物理的に単離して、標準的なゲル抽出技術により精製することができる。アダプターが連結されたgDNAライブラリーのゲル分離により、サイズ範囲が250 bpまたはそれ以上であるライブラリー集団が回収されることになる(ライブラリーのサイズ範囲は、用途に応じて変化させることができる)。アダプターの連結後のライブラリーのサイズ範囲は130〜980 bpである。この手順は、ゲルの異なる領域を切り出すことにより、例えば、130〜200 bp、200〜400 bp、250〜500 bp、300〜600 bp、500〜700 bpなどのような任意のバンドサイズ範囲の単離のために適合できることに留意されたい。下記の手順は、250 bp〜500 bpの単離断片に使用した。
【0127】
2%アガロース、1×TBE、およびエチジウムブロマイド(10 mg/ml貯蔵液) 4.5 μlを含むアガロースゲル150 mlを調製した。連結DNAを10×Ready Load Dyeと混合して、ゲルに添加した。さらに、100 bp ラダー(0.1 μg/μl) 10 μlを試料隣りの連結反応液から2レーン離して添加した。ゲルを100 Vで3時間電気泳動させた。ゲルの泳動が完了した場合には、ゲルをゲル槽から取り出し、GelDocに移して、プラスチックラップで覆った。DNAのバンドをPrep UVライトにより可視化した。無菌の、使い捨ての外科用メスを用いて、アガロースゲルから250〜500 bpの断片サイズを有するライブラリー集団を切り出した。この工程は、DNAのニッキングを防ぐために可能な限り素早く行った。このゲルスライスを15 mlファルコンチューブに入れた。アガロース内に埋まったgDNAライブラリーは、Qiagen MinElute Gel Extractionキットを用いて単離した。それぞれ単離したgDNAライブラリーの一定分割量をBioAnalyzer DNA 1000 LabChipにより分析して、gDNAライブラリー集団の正確な分布を評価した。
【0128】
gDNAライブラリーの鎖置換および鎖伸長ならびにストレプトアビジンビーズによる一本鎖gDNAライブラリーの単離
ニックの入った二本鎖gDNAライブラリーの鎖置換および鎖伸長は、Bst処理試料を65℃のサーマルサイクラー中で30分間インキュベートして、必要とされるまで氷上に置いたことを除いて、実施例1に記載されているように行った。ストレプトアビジンビーズは、最後の洗浄を1×結合用緩衝液200 μlで2回洗浄およびnH2O 200 μlで2回洗浄により行ったことを除いて、実施例1に記載されているように調製した。一本鎖gDNAライブラリーは、以下のようにストレプトアビジンビーズを用いて単離した。洗浄したビーズから水を除去して、融解溶液(以下を参照されたい) 250 μlを添加した。このビーズ懸濁液を十分に混合して、チューブ回転装置に載せて室温で10分間インキュベートした。別のチューブのなかで、PB(QiaQuick精製キットからの) 1250 μlおよび20%酢酸9 μlを混合した。融解溶液 250 μl中のビーズをDynal MPCにより沈殿させて、この上清を注意深く取り出し、新たに調製したPB/酢酸溶液に移した。この溶液1500 μlからDNAを1個のMinElute精製スピンカラムにより精製した。これは、試料を1回の添加につき750 μlとして2回同じカラムに添加して通すことで行った。一本鎖gDNAライブラリーを予め55℃に温めておいた緩衝液EB 15 μlで溶出した。
【0129】
一本鎖gDNAの定量化および保存
一本鎖gDNAは、実施例1に記載されているようにRNA Pico 6000 LabChipを用いて定量化した。場合によっては、一本鎖ライブラリーは、最初のAgilent 2100による定量化が正確に行われることを確実にするため、第二のアッセイにより定量化した。このために、RiboGreenによる定量化を記載のように(蛍光光度法によるssDNA定量化)行い、Agilent 2100による定量化を確認した。この二通りの推定値が3倍を超えるまでに異なっていた場合、各分析を繰り返した。定量化により二通りの手順の間で3倍を超える相違が明らかになった場合、ビーズに対してより広範囲の鋳型を使用した。
【0130】
一本鎖gDNAライブラリーの希釈および保存は、実施例1に記載されているように行った。収量は次の通りであった。
LabChip分析後のssDNAライブラリーの残りの最終容量 = 12 μl。
RiboGreen分析後のssDNAライブラリーの残りの最終容量 = 9 μl。
TEの添加後のssDNAライブラリーの最終容量 = 18 μl。
【0131】
等量のTEを一本鎖gDNAライブラリーの貯蔵液に添加した。一本鎖gDNAライブラリーを1×108分子/μl TE緩衝液に。貯蔵液を200,000分子/μl TEに希釈(500倍)して、一定分割量20 μlを調製した。
【0132】
噴霧後のライブラリー断片サイズの分布
噴霧およびポリッシング後の材料1 μlのAgilent 2100 DNA 1000 LabChip分析から得られた典型的な結果が図9Aに示されている。大部分の産物のサイズ範囲分布は、50〜900塩基対前後に収まると予想された。平均サイズ(ピークの最高点)は、約450 bpになると予想された。アダプターが連結されたライブラリー断片のゲル精製後に得られた典型的な結果が図9Bに示されている。
【0133】
試薬
特別の定めのない限り、実施例に記載の試薬は、市販されている標準的な試薬に相当する。例えば、クレノー、T4 DNAポリメラーゼ、T4 DNAポリメラーゼ緩衝液、T4 PNK、T4 PNK緩衝液、クイック T4 DNAリガーゼ、クイックライゲーション緩衝液、Bst DNAポリメラーゼ(ラージフラグメント)およびThermoPol反応用緩衝液は、New England Biolabs (Beverly, MA)から入手できる。dNTPミックスはPierce (Rockford, IL)から入手できる。アガロース、超高純度(UltraPure)TBE、ブルージュース・ゲルローディング緩衝液およびReady-Load 100 bp DNAラダーは、Invitrogen (Carlsbad, CA)から購入することができる。エチジウムブロマイドおよび2-プロパノールはFisher (Hampton, NH)から購入することができる。RNAラダーはAmbion (Austin, TX)から購入することができる。その他の試薬は、一般に公知であるおよび/または下記に記載されている。
【0134】
融解溶液:
融解溶液には、100 mM NaCl、および125 mM NaOHが含まれた。記載の試薬を混ぜ合わせて、完全に混合した。この溶液は室温で6ヶ月間保存することができる。
【0135】
結合および洗浄用(B & W)緩衝液 (2×および1×):
2×B & W緩衝液には、終濃度で10 mM Tris-HCl (pH 7.5)、1 mM EDTA、および2 M NaClが含まれた。記載の試薬を混ぜ合わせて、完全に混合した。この溶液は室温で6ヶ月間保存することができる。1×B & W緩衝液は、2×B & W緩衝液をピコピュアH2Oと1:1で混合することにより調製した。終濃度は上記のものの半分、すなわち、5 mM Tris-HCl (pH 7.5)、0.5 mM EDTA、および1 M NaClとなった。
【0136】
その他の緩衝液には、以下が含まれた。1×T4 DNAポリメラーゼ緩衝液: 50 mM NaCl、10 mM Tris-HCl、10 mM MgCl2、1 mMジチオスレイトール(25℃でpH 7.9)。TE: 10 mM Tris、1 mM EDTA。
【0137】
特殊試薬の調製:
TE (10 mM):
各試薬を混合した。この溶液は室温で6ヶ月間保存することができる。
【0138】
噴霧緩衝液:
全ての試薬をStericupに添加(グリセロールは最後に添加)して、十分に混合した。この溶液は、ラベル付けして、室温で6ヶ月間保存することができる。
【0139】
ATP (10 mM):
各試薬を混合した。この溶液は-20℃で6ヶ月間保存することができる。
【0140】
BSA (1 mg/ml):
試薬を混合した。この溶液は4℃で6ヶ月間保存することができる。
【0141】
ライブラリーアニーリング用緩衝液、10×:
10×アニーリング用緩衝液には、200 mM Tris (pH 7.5)および50 mM酢酸マグネシウムが含まれた。この緩衝液の場合、Tris 200 mlをピコピュアH2O 500 mlに添加した。次に、酢酸マグネシウム10.72 gを溶液に添加して、完全に溶解させた。この溶液を最終容量1000 mlに調整した。この溶液は4℃で6ヶ月間保存することができる。ライブラリーの混入の可能性を回避するため、この緩衝液を単回使用または短期使用のために分注した。
【0142】
アダプター:
アダプター「A」(400 μM):
この溶液の場合、1000 pmol/μl アダプターA(44 bp、センス) 10 μlを1000 pmol/μl アダプターA(40 bp、アンチセンス) 10 μl、10×ライブラリーアニーリング用緩衝液2.5 μl、および水2.5 μlと混合した(Vf = 25 μl)。アダプターは、Sample Prep LabサーマルサイクラーのANNEAL-Aプログラム(以下の補遺を参照されたい)を用いてアニーリングさせた。アダプター設計に関する詳細は、補遺に示されている。
【0143】
アダプター「B」(400 μM):
この溶液の場合、1000 pmol/μl アダプターB(40 bp、センス) 10 μlを1000 pmol/μl アダプターB(44 bp、アンチ) 10 μl、10×ライブラリーアニーリング用緩衝液2.5 μl、および水2.5 μlと混合した(Vf = 25 μl)。アダプターは、Sample Prep LabサーマルサイクラーのANNEAL-Aプログラム(補遺を参照されたい)を用いてアニーリングさせた。アニーリング後、アダプター「A」およびアダプター「B」を混合した(Vf = 50 μl)。アダプターセットは、使用前まで-20℃で保存することができる。
【0144】
20%酢酸:
この溶液の場合、氷酢酸を水に添加した。この溶液は室温で6ヶ月間保存することができる。
【0145】
補遺
アダプターアニーリングプログラム
ANNEAL-プライマーアニーリング用のプログラム
1. 95℃で1分インキュベートする。
2. 15℃まで0.1℃/秒で温度を下げる。ならびに
3. 14℃で保持する。
【0146】
末端修復用のT4ポリメラーゼ/クレノーPOLISHプログラム
1. 25℃で10分インキュベートする。
2. 16℃で2時間インキュベートする。ならびに
3. 4℃で保持する。
【0147】
末端修復用のT4 PNKプログラム
1. 37℃で30分インキュベートする。
2. 65℃で20分インキュベートする。ならびに
3. 14℃で保持する。
【0148】
ニックの入った二本鎖gDNAの鎖置換および鎖伸長用のBSTプログラム
1. 65℃で30分インキュベートする。ならびに
2. 14℃で保持する。
【0149】
参照文献
【0150】
本明細書を通じて、当技術分野の状況および内容を記載するため、さまざまな特許、公開された特許出願および科学文献が引用される。それらの開示は、その全体が、参照により本明細書に組み入れられる。
【0151】
他の態様
特定の態様を本明細書で詳細に開示してきたが、これは例示の目的のためにのみ一例として行われたものであり、先の、添付の特許請求の範囲に対して限定することを意図するものではない。特に、本発明者らは、特許請求の範囲にある本発明の精神および範囲から逸脱することなく、さまざまな置換、変更、および変形を本発明に対して行うことができるものと考える。
【図面の簡単な説明】
【0152】
【図1】鋳型DNAの断片化(図1A)、末端ポリッシング(図1B)、アダプター連結(図1C)、ニック修復、鎖伸長およびゲル分離(図1D)の段階を含むライブラリー調製の全過程の略図である。本発明の方法による180〜350塩基対のアデノウイルスDNAライブラリーの試料調製物を含有する典型的なアガロースゲルを同様に描写する。
【図2】図2Aは、本発明のある態様によるユニバーサルアダプター設計の略図である。各ユニバーサルアダプターは、20 bpのPCRプライミング用のヌクレオチド配列、20 bpの配列決定プライミング用のヌクレオチド配列および非反復ヌクレオチド配列からなる特有の4 bpの識別可能な配列(すなわち、ACGT、CAGTなど)を含むように設計される二本の相補ssDNAオリゴヌクレオチドから作製される。図2Bは、本発明で用いる典型的なユニバーサルアダプターの配列対を描く。アダプターAセンス鎖: SEQ ID NO:1; アダプターAアンチセンス鎖: SEQ ID NO:2; アダプターBセンス鎖: SEQ ID NO:3; アダプターBアンチセンス鎖: SEQ ID NO:4。図2Cは、本発明で用いるユニバーサルアダプター設計の略図である。
【図3】本発明による、ニックの入った二本鎖DNA断片の鎖置換および鎖伸長を示す。合成オリゴヌクレオチドから作製されたユニバーサルアダプターの連結反応に従って、T4 DNAリガーゼ処理の後に2箇所のニック領域を含んだ二本鎖DNA断片が作製されることになる(図3A)。鎖置換酵素(すなわち、Bst DNAポリメラーゼI)の添加により、ニックに結合し(図3B)、ニックの入った鎖を鎖置換し、鎖のヌクレオチド伸長を完結して(図3C)、ニックの入っていない二本鎖DNA断片を作製することになる(図3D)。
【図4】ストレプトアビジン被覆磁気ビーズを用いた本発明による定方向連結一本鎖DNAの単離を示す。ユニバーサルアダプターAおよびB(この二つの異なるアダプターは「第一」および「第二」のユニバーサルアダプターといわれることもある)との連結反応後、二本鎖DNAには、4通りの可能な組み合わせ、つまりAA、BB、AB、およびBAのアダプターが含まれることになる。ユニバーサルアダプターBに5'-ビオチンが含まれる場合、磁性のストレプトアビジン被覆固相支持体を使用して、AB、BA、およびBBの集団を捕捉かつ単離する(AAの集団は洗い流される)。BBの集団は、二本鎖DNAの各末端がビーズに付着して、遊離されないので、ビーズ上に保持される。しかしながら、低塩緩衝液の存在下で洗浄すると、ABおよびBAの集団だけが、結合した鎖に相補的である一本鎖DNA断片を遊離することになる。一本鎖DNA断片を上清から単離して、その後の用途のための鋳型として使用する。この方法は、先に詳細に記載されている。
【図5】PCRプライマーおよび配列決定用プライマーが隣接した挿入断片を示す。
【図6】交差ハイブリダイゼーション反応(CHR)でミスマッチなPCRプライマーにより作製される切断型産物を示す。
【図7】本発明の方法に使用される噴霧装置の組立部品を描く。チューブキャップを噴霧装置の先端に取り付け(図7A)、キャップを噴霧装置のクランプ部品で固定した(図7B)。噴霧装置の底面を窒素供給器に取り付けて(図7C)、装置全体をパラフィルムで包んだ(図7D)。
【図8】一本鎖DNAライブラリーの分析からの典型的なBioAnalyzer出力結果を描く。
【図9】図9Aは、噴霧およびポリッシング後の一本鎖DNAライブラリーに対するLabChip分析の典型的な結果を描く。図9Bは、噴霧、ポリッシング、およびゲル精製後のアダプター連結一本鎖DNAライブラリーに対する典型的なサイズ分布結果を描く。
【図10】プライマー候補に対する融解温度に基づく推測を描く。
【技術分野】
【0001】
発明の分野
本発明は、タンパク質化学、分子生物学、および配列解析用の一本鎖ライブラリーの調製方法に関する。より具体的には、本発明は、増幅反応および配列決定反応に用いるDNAの処理方法を含む。
【0002】
関連出願
本出願は、以下の出願に対する優先権の恩典を主張するものである: 2003年1月29日付で出願した米国特許出願第60/443,471号、2003年4月23日付で出願した米国特許出願第60/465,071号; 2003年6月6日付で出願した米国特許出願第60/476,504号、2003年6月6日付で出願した米国特許出願第60/476,313号、2003年6月6日付で出願した米国特許出願第60/476,592号、2003年6月6日付で出願した米国特許出願第60/476,602号、2003年6月6日付で出願した米国特許出願第60/476,592号、および2003年8月25日付で出願した米国特許出願第60/497,985号。本段落の全ての特許および特許出願は、その全体が参照により本明細書に組み入れられる。
【0003】
本出願には同様に、以下の同時係属中の米国特許出願の2004年1月29日付で出願した「Bead Emulsion Nucleic Acid Amplification」、2004年1月29日付で出願した「Bead Emulsion Nucleic Acid Amplification with Continuous Flow」、2004年1月29日付で出願した「Double Ended Sequencing」、および2004年1月29日付で出願した「Methods Of Amplifying And Sequencing Nucleic Acids」が参照により組み入れられる。
【背景技術】
【0004】
発明の背景
ポリメラーゼ連鎖反応(PCR)による増幅では、二つのプライマーが、ある数のヌクレオチドによりDNA鋳型分子上で分離されるそれぞれのプライマーに相補的な位置で、鋳型DNAにハイブリダイズするように設計される。プライマー間のおよびプライマーを含む鋳型DNAの塩基配列が反復的な相補鎖伸長反応により増幅され、それにより、多数の標的DNA断片のコピーが数百倍分だけ増幅される。増幅は2nのように指数関数的である(式中nは増幅サイクルの数に等しい)。PCR後、増幅されたDNAは、従来の配列決定法により配列決定することができる(米国特許第6,274,320号を参照されたい)。
【0005】
大きい鋳型DNAを含む試料または長いヌクレオチド配列を含む全ゲノムDNAは、PCRによる効率的な増幅に寄与しない。これらの長い分子にはもともと、プライマーのハイブリダイゼーションに有用な配列がない。さらに、プライマーのハイブリダイゼーション配列を二本鎖DNA分子に付加する場合、増幅されたDNA分子の方向性を確定するのは困難であり、これが配列決定の取り組みの妨げになる。
【0006】
これらの欠点のいくつかを克服するため、さまざまな方法が設計されている。例えば、米国特許第5,508,169号は、核酸断片のサブセットを同一でない5'突出付着末端または3'突出付着末端に含まれる情報に基づいて指標付け(すなわち、選択または標的化)できることについて記載している。これには、II型制限酵素および断続性の回文構造を認識するII型制限酵素によるDNAの切断により露呈されるもののような、3塩基、4塩基または5塩基の付着末端を有する断片が含まれる。その特許は、(認識配列ではなく)制限酵素による切断部位の付着末端に相補的な突出一本鎖を含むアダプターに似た核酸分子(指標付けリンカー(indexing linker)といわれる)について記載している。さまざまな官能基または特定の用途のために設計した特異的な核酸配列を上記の断片のサブセットに選択的に付着させてもよい。周知の塩基配列を付着末端に有する指標付けリンカーの、その相補的な付着末端を持つ断片のサブセットとの選択的付着は、断片のサブセットの検出、同定、単離、増幅、および操作に使用することができる。
【0007】
米国特許第6,468,748号は、いくつかの段階を含む遺伝子および/または遺伝子断片の選別方法について記載している。第一に、ds cDNA分子を逆転写により、任意でポリ-T配列の上流に一般的プライマー-鋳型配列を有するポリ-Tプライマーを用いてmRNA分子から調製し、任意で一般的プライマー-鋳型配列を有する、ポリ-T配列を持つds cDNA分子を得る。第二に、ds cDNA分子を、一定数の任意のヌクレオチドのssDNA突出配列が入った付着末端を有する消化cDNA分子を生成する制限酵素で消化する。第三に、消化cDNA分子をdsDNAオリゴヌクレオチドアダプターのセットであって、そのアダプターのそれぞれが、消化cDNAの考えられるssDNA突出配列の一つに相補的な付着末端性ssDNAアダプター配列をその末端の一端に、ssDNAアダプター相補配列に特有の特異的プライマー-鋳型配列を他端に、およびそのセットの異なるアダプターの全てで同じである一定配列をその両端の間に有するdsDNAオリゴヌクレオチドアダプターのセットに連結させる。第四に、連結cDNA分子を、各個別のポリメラーゼ連鎖反応を目的に、任意でcDNAの一般的プライマー-鋳型(配列)を有するcDNAのポリ-T配列にアニーリングするプライマーと、cDNAの特異的プライマー-鋳型配列にアニーリングする異なる特異的プライマーのセットのあるプライマーとを使用して、個別のポリメラーゼ連鎖反応により増幅させる。第五に、増幅cDNA分子を各個別のポリメラーゼ連鎖反応後の増幅産物を回収することにより重複のない群(特異的プライマー-鋳型配列にアニーリングして、ポリメラーゼ連鎖反応を開始する特異的プライマーにより決定される増幅cDNA分子の各群)に選別する。
【0008】
米国特許第5,863,722号は、オリゴヌクレオチドタグを有するポリヌクレオチドを選別するための方法および材料について記載している。オリゴヌクレオチドタグは、天然型オリゴヌクレオチドと比べて結合強度および特異性の増加したサブユニットからなる相補的なオリゴマー化合物にハイブリダイズすることができる。そのような相補的なオリゴマー化合物は「タグ相補体」といわれる。タグ相補体のサブユニットは、「アンチセンス単量体」といわれる、非天然型ヌクレオチド類似体の単量体からなってもよく、またはそれらには、アンチセンス単量体を含む、3〜6個の範囲内の長さのヌクレオチドを有するオリゴマーもしくはその類似体であって、最小限に交差ハイブリダイズするセットから選択されるオリゴマーが含まれてもよい。そのようなセットの場合、セットのオリゴマーとセットの他のオリゴマーの相補体とからなる二重鎖には、少なくとも2つのミスマッチが含まれる。言い換えれば、最小限に交差ハイブリダイズするセットのオリゴマーは、せいぜい、同じセットの他のオリゴマーの相補体と少なくとも2つのミスマッチを有する二重鎖を形成するだけである。固相支持体に付着されるタグ相補体を使用して、ポリヌクレオチドの混合物からそれぞれタグを含んだポリヌクレオチドを選別する。各支持体の表面は、特定の配列を有するただ一種類のタグ相補体により誘導体化される。同様に、選別されるポリヌクレオチドはそれぞれ、同じポリヌクレオチドが同じタグを有し、異なるポリヌクレオチドが異なるタグを有するように、オリゴヌクレオチドタグをレパートリーとして含む。従って、支持体およびポリヌクレオチドの集団が、オリゴヌクレオチドタグのその各相補体との特異的ハイブリダイゼーションを可能とする条件の下で混合される場合、同じポリヌクレオチドの亜集団が特定のビーズまたは領域に選別される。その後、ポリヌクレオチドの亜集団をマイクロ生化学的技術により固相支持体上で操作することができる。
【0009】
米国特許第5,728,524号は、核酸配列の分類の方法であって、それぞれが所定のヌクレオチド塩基を含む配列に対する結合特異性を示すアダプター分子の集団に、その核酸配列を結合させる方法について記載している。得られる連結配列をその後、特定塩基に対する選択に基づいて分類する。
【0010】
しかしながら、当技術分野では、それぞれが他方とは異なる二つの既知配列を、それぞれが(未知断片配列の)各末端に隣接するようにしてさらに含んだ未知断片配列のライブラリーを作製するための方法については記載されていない。このように、先行技術の欠点を克服する方法に対する必要性が存在する。従って、本発明は、試料中の複数のDNA配列の操作を容易とするのに必要な方法、材料、およびキットを説明するものである。
【発明の開示】
【0011】
発明の簡単な概要
本発明は、ライブラリーがさらなる定量分析および比較分析に適するような、特に複数の核酸配列が未知であって、大きい鋳型DNAまたは全ての(もしくは一部の)ゲノムDNAに由来するような、試料から複数の核酸配列のライブラリーを調製するための新規な方法について記載する。本発明のある種の態様において、一本鎖DNA(ssDNA)の配列は、大きい鋳型DNAまたは全てのもしくは一部のゲノムDNAの試料から断片化、ポリッシング(polishing)、アダプター連結、ニック修復、およびssDNAの単離を経て調製される。
【0012】
従って、ある局面において、本発明により、複数のssDNAを含んだライブラリーをクローニングにより単離するための方法であって、その際に各ssDNAは第一の一本鎖ユニバーサルアダプターと第二の一本鎖ユニバーサルアダプターとを含み、以下の段階を含む方法が提供される:
(a) 大きい鋳型DNA分子を断片化して複数の断片化したDNA分子を作製する段階;
(b)第一または第二の二本鎖ユニバーサルアダプターを断片化した各DNA分子の第一の末端におよび第一または第二のユニバーサルアダプターを断片化した各DNA分子の第二の末端に付着させて、アダプター連結DNA分子の混合物を形成させる段階;
(c) それぞれが第一の一本鎖ユニバーサルアダプターと第二の一本鎖ユニバーサルアダプターとを含む複数の一本鎖DNA分子を単離する段階; および
(d) 複数のリアクターに一つのDNA分子が含まれるように一本鎖DNA分子をリアクターに送達し、それによってライブラリーをクローニングにより単離する段階。
【0013】
ある種の局面において、一本鎖DNA分子は、油中水型乳濁液中の液滴(すなわち、マイクロリアクター)内に、またはマルチウェル表面(例えば、PicoTiterプレート)上に送達される。一本鎖DNA分子は、固相支持体(例えば、ビーズ)への付着により送達されてもよい。
【0014】
他の局面において、第一の二本鎖ユニバーサルアダプターと第二の二本鎖ユニバーサルアダプターとを含むアダプター連結DNA分子は、二本鎖ユニバーサルアダプターの片鎖を介して(第一または第二のユニバーサルアダプターを介して)固相支持体に付着される。固相支持体に付着されていないアダプター連結DNA分子は洗い流されて、アダプター連結DNA分子の片鎖が遊離される。これにより、第一および第二のユニバーサルアダプター対を有する一本鎖分子の集団を含んだ複数のssDNAを含有する混合物が得られ、それによってライブラリーが得られる。
【0015】
断片化したDNAの配列は、既知であっても未知であってもよい。好ましい態様において、断片化したDNAの配列、特に断片化したDNAの末端の配列は未知である。
【0016】
別の局面において、本発明には、以下の段階を含む、固相支持体に結合されたssDNAライブラリーを作製するための方法が含まれる: (a) 鋳型ssDNAのライブラリーを作製する段階; (b) 鋳型ssDNAを固相支持体に付着させる段階; および(c) 一つの鋳型ssDNAが付着された固相支持体を単離する段階。別の局面において、本発明には、本明細書に記載の方法により作製される可動性の固相支持体のライブラリーが含まれる。
【0017】
発明の詳細な説明
本発明は、増幅反応および配列決定反応のための試料DNAの調製に関する。本発明には、以下の段階からなる試料DNAの調製方法が含まれる: (a) 大きい鋳型DNAまたは全ゲノムDNAの試料を断片化して複数のDNA消化断片を作製させる段階; (b) 複数のDNA消化試料に適合末端を作製する段階; (c) 断片化したDNA分子の末端にユニバーサルアダプター配列のセットを連結して複数のアダプター連結DNA分子を作製する段階であって、その際に各ユニバーサルアダプター配列はPCR用プライマー配列と、配列決定用プライマー配列と識別可能なキー配列とを含み、およびその際に一方のアダプターにはビオチンが付着している段階; (d) 複数の連結DNA断片を分離かつ単離する段階; (e) 複数の連結DNA断片の任意部分を取り出す段階; (f) 複数の連結DNA断片をニック修復および鎖伸長する段階; (g) それぞれの連結DNA断片を固相支持体に付着させる段階; および(h) 各末端に特有のアダプターが存在する(すなわち、方向性を与える)一本鎖のアダプター連結DNA断片を含んだ集団を単離する段階。
【0018】
特別の定めのない限り、本明細書で使用される全ての技術用語および科学用語は、当業者が通常理解しているのと同じ意味を有する。本発明の実施に際して、本明細書に記載のものに類似であるかまたは等価である方法および材料を使用することができるが、例示として適当な方法および材料を以下に記載する。例えば、三つまたはそれ以上の段階を含む方法が記載される場合がある。そのような方法では、全ての段階が規定の目標を達成するのに必要とされるとは限らない可能性があり、本発明はこれら(段階)の個別の目標を達成するのに、孤立的段階の使用を想定する。全ての刊行物、特許出願、特許、および他の参照文献の開示は、その全体が参照により本明細書に組み入れられる。さらに、材料、方法、および例は、単なる例示に過ぎず限定を意図するものではない。
【0019】
本明細書で用いられる場合の「ユニバーサルアダプター」という用語は、PCRのプライミングのためのヌクレオチド配列と配列決定のプライミングのためのヌクレオチド配列とを含むように設計される二本の相補的な且つアニーリングするオリゴヌクレオチドを指す。任意で、ユニバーサルアダプターは、非反復ヌクレオチド配列からなる特有の識別可能なキー配列(すなわち、ACGT、CAGTなど)をさらに含むことができる。ユニバーサルアダプターのセットには、二本鎖DNAの末端に連結され得る二本の特有であり且つ異なる二本鎖配列が含まれる。従って、同じユニバーサルアダプターまたは異なるユニバーサルアダプターをDNA分子の両端に連結することができる。一本鎖である大きいDNA分子に含まれる場合またはオリゴヌクレオチドとして存在する場合、ユニバーサルアダプターは、一本鎖のユニバーサルアダプターということができる。
【0020】
本明細書で用いられる場合の「識別可能なキー配列」という用語は、4種類のデオキシリボヌクレオチド(すなわち、A、C、G、T)の組み合わせを含む配列を指す。DNA断片の丸ごとのライブラリーには、識別可能な同一配列を使用することができる。または、異なる生物に由来するDNA断片のライブラリーを追跡するには、識別可能な異なるキー配列を使用することができる。二つまたはそれ以上のライブラリーの混合には、識別可能なより長いキー配列を使用することができる。
【0021】
本明細書で用いられる場合の「複数の分子」という用語は、同じ供給源から単離されたDNAを指し、したがって、異なる生物は同じ方法により別々に調製することができる。ある態様において、複数のDNA試料は、大きいDNA断片、例えば、ゲノムDNA、cDNA、ウイルスDNA、プラスミドDNA、コスミドDNA、人工染色体DNA(例えば、BAC、YAC、MAC、PAC)、合成DNA、ファージミドDNA、ファセミド(phasemid)DNAから、またはウイルスRNAの逆転写産物から得られる。このDNAは、任意の哺乳類(すなわち、ヒト、ヒト以外の霊長類、齧歯類、またはイヌ)、植物、鳥類、爬虫類、魚類、真菌、細菌、またはウイルスを含む、任意の供給源から得ることができる。
【0022】
本明細書で用いられる場合の「ライブラリー」という用語は、大きい鋳型DNA、例えば、断片化したゲノムまたは全ゲノムから作製された小さなサイズのDNA種のサブセットを指す。
【0023】
本明細書で用いられる場合の「特有のPCRプライミング領域」などで、「特有の」という用語は、増幅されるまたは配列決定されるDNA分子内に存在しないまたは極端に低いコピーレベルで存在する配列を指す。
【0024】
本明細書で用いられる場合の「適合する」という用語は、アダプター分子が付着できる二本鎖DNAの末端(すなわち、平滑末端または付着末端)を指す。
【0025】
本明細書で用いられる場合の「断片化する」という用語は、大きいDNA分子を小さなDNA断片に変換する工程を指す。
【0026】
本明細書で用いられる場合の「大きい鋳型DNA」とは、5 kb、10 kb、または25 kbを超える、好ましくは500 kbを超える、より好ましくは1 MBを超える、および最も好ましくは5 MBまたはそれ以上のDNAとすることができる。
【0027】
本明細書で用いられる場合の「ストリンジェントなハイブリダイゼーション条件」という用語は、完全に相補的な配列のみが互いにハイブリダイズできる条件を指す。
【0028】
以下の説明は、本発明の方法に含まれる基本的段階を要約している。各段階は特定の順序で列挙されているが、しかしながら、当業者に公知のように、各工程の順序は同じ結果を達成するように操作されてもよい。そのような操作は、本発明者らによって予想されている。さらに、一部の段階は、当業者に同様にして公知であるように最小化されてもよい。
【0029】
断片化
本発明の方法の実施に際して、DNA試料の断片化は、当業者に周知の任意の手段により行われることができる。断片化は酵素的手段、化学的手段、または機械的手段により行われることが好ましい。機械的手段には、超音波処理、フレンチプレス、HPLC、HydroShear(GeneMachines, San Carlos, CA)および噴霧を含むことができる。酵素的手段は、デオキシリボヌクレアーゼI(DNaseI)、非特異的ヌクレアーゼ、または単一のもしくは複合の制限エンドヌクレアーゼを用いた消化により行われることができる。好ましい態様において、断片化により、末端に隣接する配列が未知の末端が生ずる。末端に隣接する配列は、少なくとも5塩基、10塩基、20塩基、30塩基、または50塩基とすることができる。
【0030】
酵素による断片化
好ましい態様において、酵素的手段はDNaseIである。DNaseIは、二本鎖DNA(dsDNA)を非特異的に切断して5'-リン酸化オリゴヌクレオチド産物を放出する汎用酵素である。DNaseIは、Mn2+、Mg2+およびCa2+を含有する緩衝液中で至適活性を有する。DNaseIによる消化段階の目的は、大きいゲノムDNAを、ライブラリーを構成するより小さな種に断片化することである。DNaseIの切断特性により、鋳型DNAの無作為消化(すなわち、最小配列の偏り)をもたらし、マンガンに基づく緩衝液の存在下で使用する場合には平滑末端dsDNA断片が圧倒的に多い量になる(Melgar, E. and D.A. Goldthwait.1968. Deoxyribonucleic acid nucleases. II. The effects of metal on the mechanism of action of Deoxyribonuclease I. J. Biol. Chem. 243: 4409)。鋳型ゲノムのDNase I処理後に生ずる消化産物の範囲は、次の三つの要因に依存する: i) 使用する酵素の量(単位); ii) 消化の温度(℃); およびiii) インキュベーション時間(分)。以下に概略を説明するDNase I消化条件は、50〜700塩基対(bp)に及ぶサイズのゲノムライブラリーを得るために最適化された。
【0031】
好ましい態様において、DNase Iを使用して大きい鋳型DNAまたは全ゲノムDNAを1〜2分間消化し、50〜500 bp、または50〜700 bpに及ぶオリゴヌクレオチドの集団を作製する。別の好ましい態様において、DNase I消化を10℃〜37℃の温度で行う。別の好ましい態様において、消化されたDNA断片は長さが50 bp〜700 bpである。
【0032】
機械的断片化
核酸の断片化のための別の好ましい方法は、機械的な断片化である。機械的な断片化方法には、超音波処理および噴霧、ならびにHydroShear、HPLC、およびフレンチプレスの使用が含まれる。超音波処理は、適当な緩衝液(すなわち、10 mM Tris、0.1 mM EDTA)に入れたDNAを含有しており、さまざまな回数の10秒バーストの間、最大出力と連続出力を使用して超音波処理する管により行われることができる。超音波破砕機は、例えば、Misonix社(Farmingdale, NY)から市販されており、本質的にはBankier and Barrell (Bankier, A.T., Weston, K.M., and Barrell, B.G.,「Random cloning and sequencing by the M13/dideoxynucleotide chain termination method」, Meth. Enzymol. 155, 51-93 (1987)により記載されているように使用することができる。超音波処理の場合、試料を氷上に保持することにより、核酸を均一な温度に維持することが好ましい。例えば、0℃の一定温度条件は、均一の断片分布を維持するのに好ましい。超音波処理に最適な条件は、調製用の超音波処理を行う前に、所定のDNA試料に対して実験的に決定することができる。例えば、DNAの一定分割量を超音波処理の下で異なる時間、処理することができ、DNAのサイズと質をPAGEにより分析することができる。最適な超音波処理条件が決定されると、残りのDNAをその予め決められた条件に従って超音波処理することができる。
【0033】
核酸の断片化のための別の好ましい方法は、噴霧装置による処理である(例えば、GeneMachines, San Carlos, Californiaから得られる手順、およびハードウェア。同様に米国特許第5,506,100号および米国特許第5,610,010号を参照されたい)。噴霧の場合、流体力学的剪断力を使用して、DNA鎖を断片化する。例えば、急縮小流路(abrupt contraction)を備えた管にDNA水溶液を通すことができる。溶液が縮小流路に接近するにつれ、流体は縮小流路の狭い領域を通過する体積流量を維持するように加速する。この加速の間、流体抵抗により、DNAは切れるまで引き延ばされる。任意で、DNA溶液は、断片をさらに剪断するにはあまりにも短いところまで、縮小流路に数回(例えば、15〜20サイクル)通すことができる。縮小流路および流体の流量を調整することにより、最終的なDNA断片のサイズを決定することができる。反応条件を制御するおよび監視するためのソフトウェアは、噴霧過程の自動化を可能とするのに使用することができる。別の利点として、噴霧化のための特別な緩衝液の必要性がない。例えば、DNAは、以下に限定されることはないが、水、Tris緩衝液、Tris-EDTA緩衝液、および0.5 MまでのNaClを有するTris-EDTAを含む、さまざまな溶液に懸濁させることができる。
【0034】
ポリッシング
Mn2+の存在下でDNase Iを用いて鋳型ゲノムDNA(gDNA)をポリッシング消化することで、平滑末端であるかまたは1塩基もしくは2塩基の長さの突出末端を有するDNAの断片が生成される。同様に、機械的手段によるDNAの断片化により、平滑末端または突出末端を有する断片の組み合わせが得られる。これらのDNA断片は、酵素的にまたは機械的に作製されたかにかかわらず、下記の手順を使用して「ポリッシングされる」ことができる。
【0035】
ポリッシング(末端修復ともいわれる)とは、平滑末端DNAへの非平滑末端DNAの変換を指す。一つの方法として、ポリッシングは、BAL32ヌクレアーゼまたはマングビーン・ヌクレアーゼのような、一本鎖特異的エキソヌクレアーゼを用いた処理により行われることができる。一般に、ヌクレアーゼは使用前に正確に測定されるべきである。
【0036】
ある態様において、平滑末端は、Pfu DNAポリメラーゼによりもたらされる。他の態様において、平滑末端は、T4 DNAポリメラーゼまたはクレノーDNAポリメラーゼのような他のDNAポリメラーゼによりもたらされる。Pfu「ポリッシング」または平滑末端化を使用して、DNaseIによる鋳型ゲノムの消化後に作製される平滑末端種の量を増加させることができる。Pfu DNAポリメラーゼは、5'突出部を埋める。さらに、Pfu DNAポリメラーゼは、3'から5'方向のエキソヌクレアーゼ活性を示す。従って、この酵素を使用して一つのおよび二つのヌクレオチド伸長部分を除去し、アダプターの連結に使用可能な平滑末端DNA断片の量をさらに増加させることができる。
【0037】
アダプターの連結
DNAライブラリーの断片化および平滑末端化に続いて、ユニバーサルアダプター配列を各DNA断片に付加することができる。本発明の種々の態様において、ユニバーサルアダプターは、以下を含むように設計される: 1) 次の2)に隣接して位置する長さが通常10〜20 bpである(任意の適当なサイズを使用することができる)特有のPCRプライミング領域のセット; 2) 任意で次の3)に先行する、長さが通常10〜20 bpである(任意の適当なサイズを使用することができる)特有の配列決定プライミング領域のセット; 3) 4種のデオキシリボヌクレオチド(すなわち、A、C、G、T)のそれぞれの少なくとも1つの組み合わせを含む特有の識別可能なキー配列(例えば、長さが1〜12 bp)。好ましい態様において、識別可能なキー配列は4塩基長である。別の態様において、識別可能なキー配列は1〜4塩基の組み合わせとすることができる。別の態様において、キー配列は4種のヌクレオチドのそれぞれを1つ含む。ある種の態様において、キー配列は1つまたは複数のリボヌクレオチド、例えば、Uを含む。
【0038】
本発明のある態様において、それぞれの特有のユニバーサルアダプターは長さが44 bpであるが、任意の適当なサイズを使用することができる。好ましい態様において、ユニバーサルアダプターは、T4 DNAリガーゼによりDNA断片の各末端に連結されて、各DNA断片に対し全体で88 bpのヌクレオチド付加をもたらすが、任意の適当なサイズを使用することができる。異なるユニバーサルアダプターを各DNAライブラリーの調製専用に設計することができ、その結果、各生物に特有の識別子(identifier)を与えることができる。例えば、異なるライブラリーの調製に、異なるキー配列を採用することができる。当業者には明らかであるように、ユニバーサルアダプターのサイズおよび配列は変化させることができるものと理解される。従って、本発明で用いるアダプターは、本明細書に記載のサイズおよび配列に限定されることはない。
【0039】
例えば、二つの異なる(すなわち、「第一」および「第二」)ユニバーサルアダプターを調製するのに、一本鎖オリゴヌクレオチドを商業者(例えば、Integrated DNA Technologies, IAまたはOperon Technologies, CA)に注文することができる。本発明のある種の態様において、ライブラリーの第一アダプターの全てがPCRプライミング配列、配列決定用プライマー配列、および識別可能なキー配列を含む、あるヌクレオチド配列を共有し、その一方、第二アダプターの全てが別のヌクレオチド配列を共有する。別の態様において、ユニバーサルアダプターのオリゴヌクレオチド配列は、ホスホジエステル結合に代えて一つまたは複数のホスホロチオエート結合を用いた合成の間に修飾される。例えば、アダプターのオリゴヌクレオチドは、二つまたは三つのホスホロチオエート結合を5'と3'双方の末端に、または一端に含むことができる。未修飾のオリゴヌクレオチドは通常、ヌクレオチド塩基間のホスホジエステル結合の加水分解を触媒する混入ヌクレアーゼによる急速な分解を受けやすい。オリゴヌクレオチドの用途で用いるのに使用可能な一つの単純かつ広く使用されるヌクレアーゼ抵抗性の化学的性質がホスホロチオエート修飾である。ホスホロチオエートでは、硫黄原子がオリゴヌクレオチド骨格中の非架橋酸素に取って代わることで、これを全ての形態のヌクレアーゼ消化に抵抗性(すなわち、エンドヌクレアーゼ消化およびエキソヌクレアーゼ消化の双方に抵抗性)とする。各オリゴヌクレオチドをHPLC精製して、合成オリゴヌクレオチド調製物に混入しないことまたは擬似オリゴヌクレオチド配列が存在しないことを確実にする。
【0040】
ユニバーサルアダプターは、断片化したDNAに対する定方向連結を可能とするように設計される。二本鎖ユニバーサルアダプターの各セットは、相互にまたは平滑末端DNA断片に連結できない非相補的な5'の4塩基突出部を含むPCRプライミング領域を考慮して設計される。従って、結合は、アダプターの3'末端とDNA断片の5'末端との間でまたはDNA断片の3'末端とアダプターの5'末端との間で起こる。二本鎖ユニバーサルアダプターの配列は、主に相補オリゴヌクレオチドをアニーリング可能として、二つの非相補オリゴヌクレオチド間の交差ハイブリダイゼーションを阻止可能とする配列を考慮して設計される一本鎖オリゴヌクレオチドを用いて作製される。
【0041】
ある態様において、ユニバーサルアダプターの95%が相補オリゴヌクレオチドのアニーリングから形成される。好ましい態様において、ユニバーサルアダプターの97%が相補オリゴヌクレオチドのアニーリングから形成される。より好ましい態様において、ユニバーサルアダプターの99%が相補オリゴヌクレオチドのアニーリングから形成される。最も好ましい態様において、ユニバーサルアダプターの100%が相補オリゴヌクレオチドのアニーリングから形成される。プライマーと擬似標的配列との間の交差ハイブリダイゼーションを最少化するためのプタイマー設計の典型的な方法が実施例2に示されている。
【0042】
本発明のある種の局面において、突出するヌクレオチド(例えば、T)を第一および第二アダプターの3'平滑末端に付加する。並行して、ポリメラーゼを使用して、鋳型DNAの5'平滑末端に突出するヌクレオチド(例えば、A)を付加する。アダプターおよび鋳型の突出ヌクレオチドは相補的であり、一層効率的なアダプターの連結を可能とする。
【0043】
他の局面において、プラスミド捕捉系を開示の方法に従って使用する。例えば、二本鎖ユニバーサルアダプターをプラスミドに挿入することができる。アダプター領域は、以下の配列を順番に含むことができる: 制限部位、PCRプライマー配列1、配列決定用プライマー配列1、キー配列1、制限部位、キー配列2、配列決定用プライマー配列2、PCRプライミング配列2、および制限部位。一つのアプローチでは、プラスミドをキー配列1とキー配列2との間を切断する一つまたは複数の制限酵素で消化する。断片化された鋳型DNAをキー配列1とキー配列2との間に連結する。連結されたコンストラクトを次に消化して、PCRプライミング配列2の後で切断する。PCRプライミング配列2に隣接する消化末端をビオチン化ヌクレオチドで埋める。ビオチン化コンストラクトを消化して、PCRプライマー配列1の前で切断する。アダプター-DNA断片-アダプター-ビオチン断片を切り出して、例えば、ストレプトアビジン磁気ビーズに結合させることで単離する。プラスミド捕捉系の他の態様が同様に、周知のクローニング技術の適用により可能である。これらの態様は同様に本発明に包含される。
【0044】
二つのアダプターのうちの一方を支持体の結合部分に結び付けることができる。好ましい態様において、5'ビオチンを第一ユニバーサルアダプターに付加して、その後の鋳型ssDNAの単離およびビオチン結合タンパク質(例えば、ストレプトアビジン、NeutrAvidin(商標)またはアビジン)で飽和した固相支持体の表面とのユニバーサルアダプターの非共有結合を可能とする。適当な支持体には、以下に限定されることはないが、磁気ビーズ、アフィニティーカラム、膜(例えば、PDVF膜、ニトロセルロースなど)が含まれ、これらはストレプトアビジンまたは結合対のもう一方のメンバーで被覆することができる。他の結合が当技術分野において公知であり、ビオチン-ストレプトアビジンの代わりに使用されてもよい。例えば、抗体/抗原-抗原決定基、受容体/リガンドおよびオリゴヌクレオチド対合または相補結合を使用することができる。ある態様において、固相支持体はビーズ、好ましくはポリスチレンビーズである。一つの好ましい態様において、ビーズは約2.8 μmの直径を有するが、任意の適当なサイズを使用することができる。別の好ましい態様において、ビーズは常磁性ビーズ(例えば、Dynal Biotech, Inc., Lake Success, NY)である。本明細書で用いられる場合のこのビーズを「試料調製用ビーズ」という。
【0045】
各ユニバーサルアダプターは、一方がセンス配列を含み、もう一方がアンチセンス(相補)配列を含む、二つのssDNAオリゴヌクレオチドの組み合わせとアニーリングにより調製することができる。ユニバーサルアダプター設計の略図が、図2に描かれている。
【0046】
連結産物の単離
ユニバーサルアダプターの連結により、アダプターを各端に有する断片化したDNA、未結合の単一アダプター、およびアダプター二量体の形成が起こる。好ましい態様において、アダプターが連結されたDNAライブラリー集団を未連結の単一アダプターおよびアダプター二量体の集団から分離かつ単離する方法として、アガロースゲル電気泳動が使用される。他の態様において、断片は、サイズ排除クロマトグラフィー、ろ過、ショ糖沈降法、または当業者に公知の他の核酸分離技術により分離することができる。DNAのDNase I消化手順により通常、50〜700 bpに及ぶライブラリー集団が得られる。好ましい態様において、DNAマーカーの存在下でアガロースゲル電気泳動を行う際に、88 bpのユニバーサルアダプターセットの付加は、DNAライブラリー集団をより大きいサイズに変えて、約130〜800 bpのサイズ範囲の移動プロファイルをもたらすことになり; アダプター二量体は88 bpに移動することになり; および未連結のアダプターは44 bpに移動することになる。従って、サイズが200〜800 bpに及ぶ多数の二本鎖DNAライブラリーをアガロースゲルから物理的に単離して、標準的なゲル抽出技術により精製することができる。ある態様において、アダプターが連結されたDNAライブラリーのゲル分離により、サイズが200〜500 bpに及ぶライブラリー集団が回収されることになる。アダプターが連結された断片を識別する他の方法は、当業者に公知である。
【0047】
ニック修復
ユニバーサルアダプターに使用されるDNAオリゴヌクレオチドは5'リン酸化されていないので、リガーゼ処理の後、ギャップが断片化したDNAの3'連結部に存在することになる(図3Aを参照されたい)。これらの「ギャップ」または「ニック」は、ニックの入ったDNA断片に結合し、これを鎖置換、および伸長できるDNAポリメラーゼ酵素を使用することにより埋めることができる。3'から5'方向のエキソヌクレアーゼ活性はないが5'から3'方向のエキソヌクレアーゼ活性を示すDNAポリメラーゼは、ニックの修復およびニックの入っていない二本鎖DNAの形成をもたらすように、ニックを認識し、ニックの入った鎖を置換して、鎖を伸長する能力を有する(図3Bおよび3Cを参照されたい)( Hamilton, S.C., J.W. Farchaus and M.C. Davis. 2001. DNA polymerases as engines for biotechnology. BioTechniques 31:370)。
【0048】
ポリメラーゼ、リガーゼ、およびキナーゼを含むが、これらに限定されることはない、さまざまな修飾酵素がニック修復段階に使用される。本発明の方法で使用できるDNAポリメラーゼには、例えば、大腸菌(E. coli) DNAポリメラーゼI、サーモアンアエロバクター・サーモハイドロサルファリカス(Thermoanaerobacter thermohydrosulfuricus)ポリメラーゼI、およびバクテリオファージφ29が含まれる。好ましい態様において、鎖置換酵素のバチルス・ステアロサーモフィルス(Bacillus stearothermophilus)ポリメラーゼI(Bst DNAポリメラーゼI)を使用して、ニックの入ったdsDNAを修復し、ニックの入っていないdsDNAを作製する(図3Dを参照されたい)。別の好ましい態様において、リガーゼはT4 DNAリガーゼであり、キナーゼはT4ポリヌクレオチドキナーゼである。
【0049】
一本鎖DNAの単離
ニックの入っていないdsDNAの作製に続いて、第一および第二双方のアダプター分子を含むssDNAを単離することができる。二本鎖DNAライブラリーには、アダプターが以下の配置で結合される。
ユニバーサルアダプターA - DNA断片 - ユニバーサルアダプターA
ユニバーサルアダプターB - DNA断片 - ユニバーサルアダプターA*
ユニバーサルアダプターA - DNA断片 - ユニバーサルアダプターB*
ユニバーサルアダプターB - DNA断片 - ユニバーサルアダプターB
【0050】
「A」および「B」は、第一および第二アダプターに相当する。所望の集団は、星印で指定されている。
【0051】
ユニバーサルアダプターは、一方のユニバーサルアダプターのみが5'ビオチン部分を有するように設計されることが好ましい。例えば、ユニバーサルアダプターBが5'ビオチン部分を有する場合、ストレプトアビジンで被覆された試料調製用ビーズを使用して、ユニバーサルアダプターBを有する二本鎖DNAライブラリー種を全て結合することができる。二つのユニバーサルアダプターAを含むゲノムライブラリー集団種は、5'ビオチン部分を含んでおらず、ストレプトアビジン含有試料調製用ビーズに結合しないことになり、従って、洗い流すことができる。ビーズに付着したままであると思われる唯一の種は、ユニバーサルアダプターAとBとを有するものおよび二つのユニバーサルアダプターB配列を有するものである。
【0052】
二つのユニバーサルアダプターB配列(すなわち、各5'末端にビオチン部分)を有するDNA種は、二本鎖に含まれる各鎖が結合するので、ストレプトアビジン被覆試料調製用ビーズに各末端で結合することになる。ユニバーサルアダプターAとユニバーサルアダプターBとを有する二本鎖DNA種は、単一の5'ビオチン部分を含むことになり、従って、ストレプトアビジン被覆ビーズに一端のみで結合することになる。試料調製用ビーズが磁性である場合、ビーズは、磁化時には固相支持体に結合したままである。従って、低塩(「融解」または変性)溶液の存在下では、単一のユニバーサルアダプターAと単一のユニバーサルアダプターB配列とを含んだそのDNA断片だけが相補的な非結合鎖を遊離することになる。ビーズに付着したこの一本鎖DNAの集団を回収して、例えば、ピロリン酸配列決定法、リアルタイム定量PCR法、アガロースゲル電気泳動法、蛍光色素結合アッセイ法(PicoGreen(登録商標); Molecular Probes, Inc., Eugene, OR)、またはキャピラリーゲル電気泳動法により定量することができる。
【0053】
ある態様において、本発明の方法により作製されるssDNAライブラリーを定量して、単位容量あたりの分子数を算出する。例えば、分子を、ssDNA種のユニバーサルアダプター末端のPCRプライミング領域に相補的であるオリゴヌクレオチド捕捉プライマーを含んだ固相支持体にアニーリングさせることができる。
【0054】
ある種の態様において、ssDNAライブラリーの分子にアニーリングした捕捉プライマーを含むビーズをサーモサイクラーに移し、PCR増幅を可能とすることができる。DNAビーズに捕捉された一本鎖DNAの単一種のクローン集団を次に、配列決定することができる。ある態様において、固相支持体はビーズ、好ましくはセファロースビーズである。本明細書で用いられる場合のこのビーズを「DNA捕捉ビーズ」という。
【0055】
本明細書で使用されるビーズは、任意の手ごろなサイズのものとすることができ、さまざまな周知の材料から製造することができる。そのような材料の例としては、無機材料、天然高分子、および合成高分子が挙げられる。これらの材料の具体例としては、セルロース、セルロース誘導体、アクリル樹脂、ガラス; シリカゲル、ポリスチレン、ゼラチン、ポリビニル・ピロリドン、ビニルとアクリルアミドとの共重合体、ジビニルベンゼンまたは同様のもので架橋したポリスチレン(Merrifield Biochemistry 1964, 3, 1385-1390を参照されたい)、ポリアクリルアミド、ラテックスゲル、ポリスチレン、デキストラン、ゴム、シリコン、プラスチック、ニトロセルロース、セルロース、天然スポンジ、シリカゲル、ガラス、金属プラスチック、セルロース、架橋デキストラン(例えば、セファデックス(商標))およびアガロースゲル(セファロース(商標))ならびに当業者に周知の固相支持体が挙げられる。ある態様において、DNA捕捉ビーズの直径は20〜70 μmである。好ましい態様において、DNA捕捉ビーズの直径は20〜50 μmである。より好ましい態様において、DNA捕捉ビーズの直径は約30 μmである。
【0056】
ある局面において、本発明には、以下の段階を含む、固相支持体のライブラリーを作製するための方法が含まれる: (a) 本明細書に開示される方法により鋳型ssDNAの集団を調製する段階; (b) 固相支持体あたりDNA 1分子が存在するように各鋳型DNAを固相支持体に付着させる段階; (c) 増幅により各固相支持体上に各DNA断片のクローン集団が生じるように一本鎖の鋳型の集団を増幅する段階; (d) 鋳型ssDNAのクローン集団を配列決定する段階。
【0057】
ある態様において、固相支持体はDNA捕捉ビーズである。別の態様において、DNAはゲノムDNA、cDNA、またはRNA(例えば、ウイルスRNA)の逆転写産物である。DNAは、例えば、ビオチン-ストレプトアビジン結合、共有結合を介して、または相補オリゴヌクレオチドのハイブリダイゼーションにより固相支持体に付着することができる。ある態様において、各鋳型DNAはユニバーサルアダプターのセットに連結される。別の態様において、ユニバーサルアダプター対には、PCRプライマー配列、配列決定用プライマー配列、および識別可能なキー配列が含まれる。特有の末端を有する一本鎖DNAを単離し、その後、固相支持体に付着させて、クローン増幅のための増幅技術にさらす。DNAはPCRにより増幅することができる。ある局面において、本発明により、本明細書に記載の方法により作製された固相支持体に付着されたライブラリーが提供される。
【0058】
この方法により調製されるDNAは、鎖状伸長、ローリングサークル増幅、PCR、および配列決定のような、多くの分子生物学的手法に使用することができる。結合反応は、例えば、DNAに対するビーズのモル比を高くすることによって進められることができる。一本鎖DNA分子の捕捉は、ポアソン分布に従い、DNAが付着していないビーズ、DNA 1分子が付着したビーズ、またはDNA 2分子またはそれ以上の分子が付着したビーズの部分集合が生ずる。好ましい態様において、DNA 1分子が各ビーズに付着している。さらに、単離したライブラリーのさらなる操作に有用とできるアダプターによるさらなる修飾を含むことが可能である。
【0059】
捕捉ビーズとの鋳型核酸の結合
本発明のある種の態様において、増幅される一本鎖核酸の鋳型は、捕捉ビーズに付着される。核酸の鋳型は、当技術分野において公知の任意の方法で固相支持体の捕捉ビーズに付着することができる。好適な微細ビーズのような固相支持体にDNAを付着させるための多数の方法が当技術分野において存在する。本発明によって、ビーズとのDNAの共有結合性の化学結合は、ホスホアミデート結合を介してDNAの5'-リン酸をアミンで被覆した捕捉ビーズに結合させる、水溶性カルボジイミドのような、標準的なカップリング剤を用いることにより達成することができる。別の代替手段は、同様の化学反応を使用して、最初に特定のオリゴヌクレオチドリンカーをビーズに結合させて、次に、DNAリガーゼを用いてDNAをビーズ上のリンカーに結合させることである。オリゴヌクレオチドをビーズに結合させる他の結合化学反応には、N-ヒドロキシコハク酸アミド(NHS)およびその誘導体の使用が含まれる。そのような方法の場合、オリゴヌクレオチドの一端には、固相支持体と共有結合を形成する反応基(アミン基のような)が含まれてもよく、その一方、リンカーの他端には、固定化されるオリゴヌクレオチドと結合できる第二の反応基が含まれる。好ましい態様において、オリゴヌクレオチドは、共有結合によりDNA捕捉ビーズに結合される。しかしながら、キレート化または抗原-抗体複合体のような、非共有結合を使用して、オリゴヌクレオチドをビーズに結合させてもよい。
【0060】
制限酵素部位由来の重複末端またはバクテリオファージλを用いるクローニングベクターの「付着末端」のような、DNA断片の末端の特異配列に特異的にハイブリダイズするオリゴヌクレオチドリンカーを採用することができるが、平滑末端連結を有利に使用することもできる。これらの方法は、米国特許第5,674,743号に詳細に記載されている。ビーズを固定化するのに使用されるどの方法も、本発明の方法の各段階にわたって、固定化されたオリゴヌクレオチドを結合し続けることが好ましい。
【0061】
ある態様において、各捕捉ビーズは鋳型核酸の一部分を認識する(すなわち、その一部分に相補的である)核酸プライマーを複数有するように設計され、従って、鋳型核酸は捕捉ビーズにハイブリダイズされる。本明細書に記載される方法の場合、鋳型種のクローン増幅が好ましく、従って、ただ一つの特有の鋳型核酸がいずれか一つの捕捉ビーズに付着されることが好ましい。
【0062】
本明細書で使用されるビーズは、任意の手ごろなサイズのものとすることができ、さまざまな周知の材料から製造することができる。そのような材料の例としては、無機材料、天然高分子、および合成高分子が挙げられる。これらの材料の具体例としては、セルロース、セルロース誘導体、アクリル樹脂、ガラス、シリカゲル、ポリスチレン、ゼラチン、ポリビニル・ピロリドン、ビニルとアクリルアミドとの共重合体、ジビニルベンゼンまたは同様のもので架橋したポリスチレン(例えば、Merrifield Biochemistry 1964, 3, 1385-1390に記載されているような)、ポリアクリルアミド、ラテックスゲル、ポリスチレン、デキストラン、ゴム、シリコン、プラスチック、ニトロセルロース、天然スポンジ、シリカゲル、細孔性ガラス(control pore glass)、金属、架橋デキストラン(例えば、セファデックス(商標))、アガロースゲル(セファロース(商標))、および当業者に周知の固相支持体が挙げられる。好ましい態様において、捕捉ビーズは、直径が約25〜40 μmのセファロースビーズである。
【0063】
乳化
本発明で使用する場合、付着した鋳型核酸の有無にかかわらず捕捉ビーズは、熱安定性の油中水型乳濁液に懸濁させることができる。複数のマイクロリアクターには、ただ一つの鋳型および一つのビーズが含まれると考えられる。鋳型を含んでいないかまたはビーズを含んでいない多くの液滴が存在する可能性がある。同様に、二コピーまたはそれ以上の鋳型を含んだ液滴が存在する可能性がある。乳濁液は当技術分野において公知の任意の適当な方法により形成することができる。乳濁液を作製する一つの方法が以下に記載されるが、乳濁液を作製するための任意の方法を使用することができる。これらの方法は、当技術分野において公知であり、アジュバント法、向流法、逆流法、回転ドラム法、および膜法を含む。さらに、マイクロカプセルのサイズは、成分の流量および流速を変化させることにより調整することができる。例えば、液滴添加の場合、液滴のサイズおよび到達の総時間を変化させることができる。乳濁液は、封入ビーズ約3000個/マイクロリットルの密度を含むことが好ましい。
【0064】
生体反応に適した種々の乳濁液は、Griffiths and Tawfik, EMBO, 22, pp. 24-35 (2003); Ghadessy et al., Proc. Natl. Acad. Sci. USA 98, pp. 4552-4557(2001); 米国特許第6,489,103号および国際公開公報第02/22869号(それぞれが完全に参照により本明細書に組み入れられる)のなかで言及されている。Griffiths et al.,(米国特許第6,489,103号および国際公開公報第99/02671号)は、所望の活性を有する遺伝子産物をコードする一つまたは複数の遺伝要素のインビトロ選別のための方法について言及していることに留意されたい。この方法には、遺伝子の区画化、遺伝子の発現、および発現産物に基づく区画化遺伝子の選別が含まれる。本発明とは対照的に、Griffithのマイクロカプセル化による選別方法は、その核酸産物は固定されないおよび固定されることができないので、複数のマイクロカプセルの平行分析には適していない。Griffithsの核酸は固定されないので、それらは解乳化の間に混合されると思われる。
【0065】
乳濁液は、ビーズを増幅溶液に添加することにより作製されることが好ましい。本明細書で用いられる場合の「増幅溶液」という用語は、鋳型DNAの増幅を行うのに必要な試薬の十分な混合液を指す。増幅溶液の一つの例であるPCR増幅溶液が以下の実施例に示される。当然のことながら、実施される増幅のタイプや鋳型DNAがビーズに付着されるか溶液状態で供与されるかに基づいて、増幅溶液に種々の変更を行うことができる。ある態様において、ビーズと増幅溶液との混合液を生体適合性油(例えば、軽油, Sigma)の撹拌混合液に滴下して、乳化させる。別の態様において、ビーズおよび増幅溶液を十字流の生体適合性油に滴下する。使用される油には、一つまたは複数の生体適合性乳化安定剤が添加されてもよい。これらの乳化安定剤には、Atlox 4912、Span 80、および他の認識されかつ市販されている適当な安定剤が挙げられる。好ましい局面において、乳濁液は温度サイクリングを可能とするため、例えば、少なくとも94℃まで、少なくとも95℃まで、または少なくとも96℃まで熱安定性である。好ましくは、形成される液滴は、サイズが約5μm〜約500μmに、より好ましくは約10μm〜約350μmに、さらにより好ましくは約50〜250μmに、および最も好ましくは約100μm〜約200μmに及ぶ。十字流型の流体混合により、液滴形成の制御、および液滴サイズの均一性を可能とすることが好都合である。本発明者らは、ビーズを含有していない小さな水滴が乳濁液に存在し得ることに注目している。
【0066】
マイクロリアクターは、必要とされる増幅の程度に合う十分な増幅試薬を含むために十分に大きくするべきである。しかしながら、マイクロリアクターは、それぞれがDNAライブラリーのメンバーを含有するマイクロリアクターの集団を、従来の実験装置、例えば、PCRサーモサイクリング装置、試験管、インキュベーターおよび同様のものにより増幅できるように十分に小さくするべきである。とりわけ、マイクロリアクターの使用により、配列を混合させることなく鋳型の複合混合物(例えば、ゲノムDNA試料または全細胞RNA)の増幅、または一つもしくは複数の鋳型による優位性(例えば、PCR選択の偏り; Wagner et al., 1994, Suzuki and Giovannoni, 1996; Chandler et al., 1997, Polz and Cavanaugh, 1998を参照されたい)が可能となる。
【0067】
上記の制限により、マイクロリアクターの最適サイズは、直径を平均して100〜200μmとすることができる。このサイズのマイクロリアクターにより、容量が10 ml未満のマイクロリアクターの懸濁液中で約600,000個のメンバーからなるDNAライブラリーの増幅が可能になる。例えば、PCRが選択された増幅方法である場合、マイクロリアクター液10 mlをチューブ96 本収容の通常のサーモサイクラーのチューブ96本分に適合させることができる。好ましい態様において、600,000個のマイクロリアクターの懸濁液は、1 ml未満の容量を有する。1 ml未満の懸濁液は、従来のPCRサーモサイクラーのチューブ約10本中で増幅させることができる。最も好ましい態様において、600,000個のマイクロリアクターの懸濁液は、0.5 ml未満の容量を有する。
【0068】
本発明の別の態様は、鋳型とビーズとを用いるが、しかし鋳型をビーズに付着させずに核酸増幅を行う方法に向けられる。ある局面において、ビーズには、増幅後に増幅された核酸を結合できるリンカー分子が含まれてもよい。例えば、リンカーは、活性化できるリンカーであってもよい。そのようなリンカーは、公知であり、ストレプトアビジン/ビオチンおよび抗体/抗原のような温度感受性または塩感受性の結合対を含む。鋳型核酸は、ビーズで封入されて、増幅されてもよい。増幅後、増幅された核酸は、例えば、温度または塩濃度の調整によりビーズに結合されてもよい。
【0069】
増幅
鋳型核酸は、ビーズに付着していてもまたは付着していなくとも、転写に基づく増幅系(Kwoh D. et al., Proc. Natl. Acad Sci. (U.S.A.) 86: 1173 (1989); Gingeras T. R. et al., 国際公開公報88/10315号; Davey, C. et al., 欧州特許第329,822号; Miller, H.I. et al., 国際公開公報第89/06700号)、「RACE法」(Frohman, M. A., In: PCR Protocols: A Guide to Methods and Applications, Academic Press, NY (1990))および片側(one-sided)PCR法(Ohara, O. et al., Proc. Natl. Acad. Sci. (U.S.A.) 86. 5673-5677 (1989))を含む任意の適当な増幅方法により増幅することができる。ジオリゴヌクレオチド増幅法、等温増幅法(Walker, G. T. et al., Proc. Natl. Acad. Sci. (U.S.A.) 89: 392-396 (1992))、核酸配列に基づく増幅法(NASBA; 例えば、Deiman B et al., 2002, Mol Biotechnol. 20(2): 163-79を参照されたい)、全ゲノム増幅法(例えば、Hawkins TL et al., 2002, Curr Opin Biotechnol. 13(1): 65-7を参照されたい)、鎖置換増幅法(例えば、Andras SC, 2001, Mol Biotechnol. 19(1): 29-44を参照されたい)、ローリングサークル増幅法(米国特許第5,714,320号に概説されている)のようなさらに他の方法、および他の公知の技術を本発明により使用することができる。ある種の局面において、鋳型核酸は、マイクロリアクター中のビーズで封入後に増幅される。または、鋳型核酸は、マルチウェルの表面、例えば、PicoTiterプレートに分配後に増幅される。
【0070】
好ましい態様において、DNA増幅はPCRにより行われる。本発明によるPCRは、PCRに必要な試薬全てを含んだPCR溶液で標的核酸を封入することにより行われることができる。次に、PCRは、その乳濁液を当技術分野において公知の任意の適当なサーモサイクリング法にかけることにより達成することができる。好ましい態様において、30〜50サイクル、好ましくは約40サイクルの増幅が行われる。増幅手順が終わったら、増幅サイクルの後に1回または複数回のハイブリダイゼーションおよび伸長サイクルがあることが望ましいが、必ず必要とされるわけではない。好ましい態様において、10〜30サイクル、好ましくは約25サイクルのハイブリダイゼーションおよび伸長が行われる(例えば、実施例に記載のとおり)。日常的には、鋳型DNAは、通常、少なくとも10,000〜50,000,000コピーが各ビーズに固定化されるまで増幅される。核酸検出用途の場合、より少ない鋳型コピーが必要とされるものと認識される。核酸配列決定用途の場合、本発明者らは、少なくとも2百万〜5千万コピー、好ましくは約1千万〜3千万の鋳型DNAのコピーが各ビーズに固定化されることがよいと考えている。当業者は、ビーズのサイズ(およびその上の捕捉部位)により、捕捉プライマーをどのくらい結合することができるか(および従って増幅された鋳型をどのくらい各ビーズに捕捉することができるか)が決まることを認識すると思われる。
【0071】
ある局面において、本発明には、以下の段階を含む、複数の一本鎖DNA分子を含んだライブラリーをクローニングにより単離するための方法が含まれる: a) 大きい鋳型DNA分子を断片化して複数の断片化したDNA分子を作製する段階; b) 第一または第二の二本鎖ユニバーサルアダプターを断片化した各DNA分子の第一の末端におよび第一または第二のユニバーサルアダプターを断片化した各DNA分子の第二の末端に付着させて、アダプター連結DNA分子の混合物を形成させる段階; c) それぞれが第一の一本鎖ユニバーサルアダプターと第二の一本鎖ユニバーサルアダプターとを含む複数の一本鎖DNA分子を単離して、ライブラリーを得る段階; およびd) 複数のリアクターに一つのDNA分子が含まれるように一本鎖DNA分子をリアクターに送達し、それによってライブラリーをクローニングにより単離する段階。
【0072】
別の局面において、本発明には、以下の段階を含む、複数の一本鎖DNA分子を含んだライブラリーを作製するための方法が含まれる: a) 大きいまたは全ゲノムの鋳型DNA分子を断片化して複数の断片化したDNA分子を作製する段階; b) 第一の二本鎖ユニバーサルアダプターまたは第二のユニバーサルアダプターを断片化した各DNA分子の第一の末端におよび第一のユニバーサルアダプターまたは第二のユニバーサルアダプターを断片化した各DNA分子の第二の末端に連結させて、アダプター連結DNA分子の混合物を作製させる段階であって、その際に第一のユニバーサルアダプターには固相支持体に結合する部分が含まれる段階; c) 固相支持体に第一の二本鎖ユニバーサルアダプターを含むDNA分子を付着させる段階; d) 固相支持体に付着されていないアダプター連結DNA分子を洗い流す段階; e) 固相支持体に付着されたアダプター連結DNA分子を鎖分離させて、第一の一本鎖ユニバーサルアダプターを一端におよび第二の一本鎖アダプターを他端に含む複数の一本鎖DNA分子を遊離させる段階; およびf) 一本鎖DNA分子を単離し、それによってライブラリーを作製する段階。特定の局面において、(f)はi) 一本鎖DNA分子をリアクターアレイ上のある位置に送達する段階; またはii) 一本鎖DNA分子を油中水型乳濁液中の液滴内に送達する段階により達成することができる。
【0073】
これらの方法により、第一の二本鎖ユニバーサルアダプターまたは第二のユニバーサルアダプターを連結により断片化したDNA分子に付着させることができる。例えば、DNAリガーゼを使用することができる。これらの方法は、ポリメラーゼ、リガーゼ、キナーゼ、またはその組み合わせのような、DNA修復酵素およびDNA修飾酵素を用いて、アダプター連結DNA分子の混合物における一本鎖ニックを修復する段階をさらに含むことができる。特定の例として、この酵素には、バチルス・ステアロサーモフィルスのポリメラーゼI、T4リガーゼ、およびT4ポリヌクレオチドキナーゼが挙げられる。これらの方法のための鋳型DNAには、ゲノムDNA、cDNA、プラスミドDNA、コスミドDNA、人工染色体DNA、合成DNA、ファセミドDNA、ファージミドDNA、または逆転写産物を含むことができる。断片化は酵素的手段、化学的手段、または機械的手段により行われることができる。例えば、DNase I酵素は、10〜37℃の温度で1〜2分間行われる消化で使用することができる。または、制限酵素を使用することができる。機械的手段は、噴霧装置、フレンチプレス、超音波処理器、またはHydroShearとすることができる。
【0074】
これらの方法で使用する場合、断片化したDNA分子は、長さを50 bp〜700 bpとすることができる。適合末端は平滑末端とすることができる、または適合末端はAもしくはT突出部を含むことができる。平滑末端は、Pfuポリメラーゼ、T4 DNAポリメラーゼ、およびクレノーフラグメントのような酵素で生成することができる。第一または第二の二本鎖ユニバーサルアダプターは、一つまたは複数のホスホロチオエート結合を含んでもよく、ビオチン部分に付着されてもよい。さらに、第一の二本鎖ユニバーサルアダプターにもしくは第二の二本鎖ユニバーサルアダプターにまたはどちらの二本鎖ユニバーサルアダプターにも、識別可能なキー配列が含まれてもよい。例えば、識別可能なキー配列は、長さが3〜12ヌクレオチドであり、A、G、C、U、およびTからなる群より選択される少なくとも一つのヌクレオチドを含む。第一および第二の二本鎖ユニバーサルアダプターには、PCRプライミング配列および配列決定用プライマー配列が含まれてもよい。さまざまな局面において、PCRプライミング配列は長さが10〜20塩基対であり、配列決定用プライマー配列は長さが10〜20塩基対である。さらに、PCRプライミング配列および配列決定用プライマー配列は重複してもよい。
【0075】
これらの方法に合わせて、アダプター連結DNA分子の混合物をゲル電気泳動、ろ過、サイズ排除クロマトグラフィー、およびショ糖沈降法からなる群より選択される方法により分離する。複数の一本鎖DNA分子は、低塩処理、高pH処理、および化学的変性処理からなる群より選択される処理により得ることができる。さらなる局面において、複数の一本鎖DNA分子をDNA捕捉ビーズに付着することができる。さらに、DNA捕捉ビーズは、ビオチン/アビジン、リガンド/受容体、抗原/抗体または相補ヌクレオチドのような、結合対の成分を含むことができる。DNA捕捉ビーズは常磁性ビーズであることが好ましい。
【0076】
本発明には同様に、以下の段階を含む、固相支持体に付着された一本鎖DNAライブラリーを作製するための方法が含まれる: a) 複数の鋳型一本鎖DNAを作製する段階; b) 複数の鋳型ssDNAのそれぞれを固相支持体に付着させる段階; およびc) 鋳型一本鎖DNAが付着された固相支持体を単離する段階。
【0077】
本発明にはさらに、以下の段階を含む、固相支持体に付着された一本鎖DNAライブラリーを作製するための方法が含まれる: a) 大きい鋳型DNA分子を断片化して複数の断片化したDNA分子を作製する段階; b) 第一または第二の二本鎖ユニバーサルアダプターを断片化した各DNA分子の第一の末端におよび第一または第二のユニバーサルアダプターを断片化した各DNA分子の第二の末端に付着させて、アダプター連結DNA分子の混合物を作製する段階; c) 第一の一本鎖ユニバーサルアダプターと第二の一本鎖ユニバーサルアダプターとを含む一本鎖DNA分子を単離する段階; およびd) (c)より単離された一本鎖分子を固相支持体に付着させる段階。
【0078】
この方法で使用する場合、固相支持体はDNA捕捉ビーズとすることができ、DNAはゲノムDNA、cDNA、プラスミドDNA、コスミドDNA、人工染色体DNA、合成DNA、ファセミドDNA、またはファージミドDNAとすることができる。ある種の局面において、DNAをアビジン/ビオチン、リガンド/受容体、抗原/抗体および相補ヌクレオチドのような結合対により固相支持体に付着させる。この方法により作製される可動性の固相支持体(mobile solid support)のライブラリーも包含される。
【0079】
さらに、本発明には、第一のアダプターと、鋳型DNAの断片と、第二のアダプターとを含む核酸分子が含まれ、その際に第一のアダプターと第二のアダプターそれぞれが、配列決定用プライマーと、PCRプラマーと識別可能なキー配列とを含み、およびその際に第一のアダプターと第二のアダプターは、解離した場合に、ストリンジェントなハイブリダイゼーション条件の下で相互に交差ハイブリダイズしない。この方法では、PCRプライマーは長さを10〜20塩基対とすることができ、配列決定用プライマーは長さを10〜20塩基対とすることができ、および識別可能なキー配列は長さを3〜12塩基対とすることができる。例として、鋳型DNAはゲノムDNA、cDNA、プラスミドDNA、コスミドDNA、人工染色体DNA、合成DNA、ファセミドDNA、またはファージミドDNAとすることができる。核酸分子は、解離した場合に、解離した鋳型DNAに対する交差ハイブリダイゼーションが最小であることが好ましい。
【0080】
本発明により同様に包含されるのは、以下の段階を含む、一本鎖DNA分子を調製するための方法である: a) 大きいまたは全ゲノムの鋳型DNA分子を断片化して複数の断片化したDNA分子を作製する段階; b) 第一の二本鎖ユニバーサルアダプターまたは第二のユニバーサルアダプターを断片化した各DNA分子の第一の末端におよび第一のユニバーサルアダプターまたは第二のユニバーサルアダプターを断片化した各DNA分子の第二の末端に連結させて、アダプター連結DNA分子の混合物を作製させる段階; c) 第一の二本鎖ユニバーサルアダプターの片鎖を介して固相支持体に第一の二本鎖ユニバーサルアダプターと第二の二本鎖アダプターとを含むアダプター連結DNA分子を付着させる段階; d) 固相支持体に付着されていないアダプター連結DNA分子を洗い流す段階; e) 固相支持体に付着されたアダプター連結DNA分子を鎖分離させて、第一の一本鎖ユニバーサルアダプターを一端におよび第二の一本鎖アダプターを他端に含む複数の一本鎖DNA分子を遊離させる段階; およびf) 一本鎖DNA分子を単離する段階。
【0081】
本発明によりさらに包含されるのは、以下の段階を含む、鋳型核酸を複数の反応中心に送達するための方法である: a) 鋳型核酸の集団を供与する段階; b) 各鋳型核酸を集団から隔離剤にまで単離して、隔離された鋳型核酸の集団を形成させる段階; およびc) 隔離された鋳型核酸の集団を複数の反応中心に送達する段階であって、その際に各反応中心は隔離された核酸を一つ受け取る段階。この方法の場合、単離する段階は、鋳型核酸をビーズに付着させる段階、または鋳型核酸を油中水型乳濁液の乳濁液中に封入する段階を含むことができる。鋳型核酸は、核酸を結合できるビーズで封入されてもよい。この方法の場合、送達する段階は、隔離された核酸を複数の反応中心に送達する段階を含むことができ、その際に各反応中心はpicotiterプレートのウェルである。この方法は、単離された一本鎖分子をそれぞれ個別的に固相支持体に付着させる段階をさらに含むことができる。
【0082】
本発明の他の特徴は、本発明の説明のために示されるのであって、その限定であるとは意図されない典型的な態様に関する以下の記載のなかで明らかになると思われる。本明細書を通じて、当技術分野の状況および内容を記載するため、さまざまな特許、公開特許公報および科学文献が引用される。それらの開示は、その全体が、参照により本明細書に組み入れられる。
【0083】
実施例
実施例1: 試料調製
DNA試料:
DNAは良質のものであるべきであり、タンパク質、ヌクレアーゼ、脂質、および他の化学物質(調製液からの残存EDTAのような)および塩のような混入物をなくすべきである。好ましくは、ゲノムDNAは1.8またはそれ以上の260/280比率を有するべきである。ただ一種類の生物のゲノムを配列決定することが望まれる場合には、DNAは、混入DNAが存在しないことを保証する検査が行われた品質とするべきである。例えば、ヒトDNAの調製は、細菌DNA分子による混入がないことを保証するPCRにより検査することができる。混入がないか検査する別の方法は、制限消化パターンおよび特に制限消化に続いて、生物(例えば、ヒトまたはマウス)に特異的であることが知られている適当なプローブや考えられる混入生物(例えば、大腸菌)に特異的であることが知られている第二のプローブを用いたサザンブロットによるものである。所望であれば、DNAは、生物の単一クローン(例えば、細菌の場合にはコロニー)を起源とすべきである。
【0084】
段階1: DNase I消化
DNase I消化段階の目的は、全ゲノムまたは大きいゲノム部分のようなDNAの大きいストレッチを小さな種に断片化することである。単一の鋳型DNAから作製された小サイズのDNA種のこの集団を「ライブラリー」という。デオキシリボヌクレアーゼI(DNaseI)は、二本鎖の鋳型DNAを切断するエンドヌクレアーゼである。DNaseIの切断特性により、鋳型DNAの無作為消化(すなわち、最小の配列の偏り)を可能にし、マンガンに基づく緩衝液の存在下で使用する場合には平滑末端の、二本鎖DNA断片が優勢になるあると考えられる(Melgar and Goldthwait 1968)。DNase Iによる鋳型ゲノムの消化は、次の三つの要因に依存する: i) 使用する酵素の量(単位数); ii) 消化の温度(℃); およびiii) インキュベーション時間(分)。以下に概略を説明するDNase I消化条件は、50〜700塩基対(bp)に及ぶサイズのDNAライブラリーを得るために最適化された。
1. DNAを得て、Tris-HCl (10 mM, pH 7〜8)で0.3 mg/mlの濃度に調製した。この調製には、全部でDNA(15 μg) 134 μlが必要とされた。EDTAを含有する緩衝液(すなわち、TE, Tris/EDTA)で希釈したDNA調製物を使わないことを推奨する。EDTAの存在は、DNaseIによる酵素消化に阻害的である。DNA調製物にEDTAが含まれる場合、DNAを溶液から「塩析」して、適当なTris-HCl緩衝液(10 mM, pH 7〜8)またはナノピュアH2O(pH 7〜8)で再構成することが重要である。
2. 0.2 mlチューブに、Tris pH 7.5(1M) 50 μl、MnCl2(1M) 10μl、BSA(100 mg/ml) 1 μl、および水39 μlを含有する、DNase I緩衝液を調製した。
3. 別の0.2 mlチューブに、DNase I緩衝液15 μlおよびDNase I(1U/ml) 1.5 μlを添加した。反応チューブを15℃設定のサーマルサイクラーにセットした。
4. DNA(0.3 mg/ml) 134 μlを15℃設定のサーマルサイクラーにセットしたDNase I反応チューブに添加した。蓋を閉じて、試料を正確に1分間インキュベートした。インキュベーション後、50 mM EDTA 50 μlを添加して、酵素消化を停止させた。
5. 消化したDNAは、QiaQuick PCR精製キットを用いて精製した。消化反応液を次に、4分割量に分けて、4本のスピンカラムを使用して各分割量を精製した(スピンカラムあたり37.5 μl)。製造元の手順書に従い、各カラムを溶出用緩衝液(EB) 30 μlで溶出した。次いで、溶出液を合わせて、最終反応容量120 μlを得た。
6. 消化反応液の一定分割量3 μl 1つをBioAnalzyer DNA 1000 LabChipによる分析のために確保しておいた。
【0085】
段階2: Pfuポリッシング
DNase Iによる鋳型DNAの消化により、主に平滑末端であるDNAの断片が得られるが、しかし、一部の断片は、1塩基または2塩基の長さの突出末端を含んだ末端を有することになる。Pfuポリッシングを使用して、5'突出部の埋め込み(すなわち、「平滑末端化」)により平滑末端種の量を増加させる。さらに、Pfu DNAポリメラーゼは、一つのおよび二つのヌクレオチド伸長部分の除去をもたらす3'から5'方向のエキソヌクレアーゼ活性を有する。Pfuポリッシングにより、アダプターの連結に使用可能な平滑末端DNA断片の量を増加させる(Costa 1994a, 1994b, 1994c)。以下のPfuポリッシング手順を使用した。
1. 0.2 mlチューブに、精製済みの、DNase I消化DNA断片115 μl、10×クローン化Pfu緩衝液15 μl、dNTP(10mM) 5 μl、およびクローン化Pfu DNAポリメラーゼ(2.5 U/μl)を順に添加した。
2. ポリッシング反応の各成分を十分に混合して、72℃で30分間インキュベートした。
3. インキュベーション後、反応チューブを取り出して、氷上に2分間置いた。
4. ポリッシング反応混合液を次に、4分割量に分けて、QiaQuick PCR精製カラムを使用して精製した(各カラムに37.5 μl)。製造元の手順書に従い、各カラムを緩衝液EB 30 μlで溶出した。次いで、溶出液を合わせて、最終反応容量120 μlを得た。
5. 最終的なポリッシング反応液の一定分割量3 μl 1つをBioAnalzyer DNA 1000 LabChipによる分析のために確保しておいた。
【0086】
段階3: 断片化したDNAライブラリーへのユニバーサルアダプターの連結
ゲノムDNAライブラリーの断片化およびポリッシング後、プライマー配列を各DNA断片の末端に付加する。これらのプライマー配列は「ユニバーサルアダプター」といわれ、PCR増幅およびヌクレオチド配列決定の双方を可能とする特異的なプライミング領域を含む二本鎖オリゴヌクレオチドからなる。ユニバーサルアダプターは、各デオキシリボヌクレオチド(すなわち、A、C、G、T)の1つからなる特有の4塩基の「キー」に先行する、長さが20 塩基対である特有の配列決定プライミング領域のセットに隣接して位置する長さが20 塩基対である特有のPCRプライミング領域のセットを含むように設計される。それぞれの特有のユニバーサルアダプター(「ユニバーサルアダプターA」および「ユニバーサルアダプターB」といわれる)は、長さが44塩基対(44 bp)である。ユニバーサルアダプターは、T4 DNAリガーゼにより、DNA断片の各末端に連結されて、各DNA断片に対し全体で88 bpのヌクレオチド付加をもたらす。異なるユニバーサルアダプターを各ゲノムDNAライブラリーの調製専用に設計して、その結果、各生物に特有の識別子を与えることができる。
【0087】
ユニバーサルアダプターの対を調製するため、一本鎖オリゴヌクレオチドを自家設計して、商業者を通じて製造する。ユニバーサルアダプターのDNAオリゴヌクレオチドは、ヌクレアーゼ活性を防ぐ働きをする、各オリゴヌクレオチド末端の二つのホスホロチオエート結合を考慮して設計する(Samini, T.D., B. Jolles, and A. Laigle. 2001. Best minimally modified antisense oligonucleotides according to cell nuclease activity. Antisense Nucleic Acid Drug Dev. 11(3):129., これらの開示は、その全体が、参照により本明細書に組み入れられる)。各オリゴヌクレオチドをHPLC精製して、最終調製物に混入または擬似DNAオリゴヌクレオチド配列が存在しないことを確実にする。
【0088】
ユニバーサルアダプターは、平滑末端にした、断片化ゲノムDNAに対する定方向連結を可能とするように設計される。各ユニバーサルアダプター対に対し、PCRプライミング領域には5'の4塩基突出部および平滑末端にした3'キー領域が含まれる。ユニバーサルアダプターの平滑末端側は平滑末端DNA断片に連結するが、アダプターの5'突出部は平滑末端DNA断片に連結することができないので、指向性が達成される。さらに、5'ビオチンをユニバーサルアダプターBに付加して、その後の鋳型ssDNAの単離(段階8)を可能とする。各ユニバーサルアダプターは、単一チューブ内で、二つの一本鎖の相補DNAオリゴヌクレオチド(すなわち、センス配列を含む一方のオリゴおよびアンチセンス配列を含むもう一方のオリゴ)をアニーリングさせることにより調製する。以下の連結手順を使用した。
1. 0.2 mlチューブに、nH2O(分子生物等級純水) 39 μl、消化済みの、ポリッシングしたDNAライブラリー 25 μl、2×クイックリガーゼ反応用緩衝液100μl、MMP1 (10 pm/μl)アダプターセット 20 μl(100倍比)、およびクイックリガーゼ 16 μlを順に添加した。この連結反応液を十分に混合して、室温で20分間インキュベートした。
2. その後、連結反応液を取り除いて、連結反応液の一定分割量10 μl 1つをBioAnalyzerで用いるために精製した。Qiagen Min-Eluteキットのシングルスピンカラムを使用した。製造元の手順書による手順に従い、カラムをEB 10 μlで溶出した。精製した連結反応液の一定分割量1 μl 1つをBioAnalyzer DNA 1000 LabChipにより添加した。未精製の連結反応液には、試料がBioAnalyzerで正しく泳動するのを阻害することになる多量の塩およびPEGが含まれるので、この精製段階を推奨する。
3. 連結反応液の残り(190 μL)を段階4のゲル分離に使用した。
【0089】
段階3a: マイクロコンろ過(Microcon Filtration)およびアダプター構築
全調製時間は約25分であった。
ユニバーサルアダプターの連結反応には、100倍過剰のアダプターが必要とされる。これらの過剰なアダプターの除去を補助するため、二本鎖gDNAライブラリーをMicrocon YM-100ろ過器に通す。Microcon YM-100膜を使用して、125 bpよりも小さな二本鎖DNAを除去することができる。従って、未結合のアダプター(44 bp)、ならびにアダプター二量体(88 bp)を連結化gDNAライブラリー集団から除去することができる。以下のろ過手順を使用した。
1. 段階4の連結反応液190 μLを組立型のMicrocon YM-100ろ過器に添加した。
2. ろ過器を遠心機にセットして、5000×gで約6分間、または膜がほぼ乾燥するまで回転させた。
3. 洗浄するため、1×TE 200 μlを添加した。
4. 試料を5000×gでさらに9分間、または膜がほぼ乾燥するまで回転させた。
5. 回収するため、ろ過容器を新たなバイアルに挿入して、3000×gで3分間回転させた。ろ過容器は廃棄した。回収された容量は、約10 μlであった。次に、TE 80 μlを添加した。
【0090】
アダプター(AおよびB)は、使用の前に、HPLC精製して、ホスホロチオエート結合で修飾した。アダプター「A」(10 μM)の場合、100 μMアダプターA(44 bp、センス) 10 μlを100 μMアダプターA(40 bp、アンチセンス) 10 μlと混合して、1×アニーリング用緩衝液30 μlを混合した(Vf = 50μl)。プライマーは、Sample Prep LabサーマルサイクラーのANNEALプログラムを用いてアニーリングさせた(下記参照)。アダプター「B」(10 μM)の場合、100 μMアダプターB(40 bp、センス) 10 μlを100 μMアダプターB(44 bp、アンチセンス) 10 μlと混合して、1×アニーリング用緩衝液30 μlを混合した(Vf = 50μl)。プライマーは、Sample Prep LabサーマルサイクラーのANNEALプログラムを用いてアニーリングさせた。アダプターの対は、使用するまで-20℃で保存することができる。
【0091】
ANNEAL-プライマーアニーリング用のプログラム
1. 95℃で1分インキュベートする。
2. 15℃まで0.1℃/秒で温度を下げる。ならびに
3. 15℃で保持する。
【0092】
ゲノムDNA挿入断片およびアダプターに必要な方向はなかった。断片は両端で連結可能とされた。ユニバーサルアダプターのセットには、4本の一本鎖DNAオリゴヌクレオチドが含まれた。各一本鎖オリゴヌクレオチドは、1マイクロモルのスケールで合成されて、HPLC精製された。各一本鎖オリゴヌクレオチドには、各末端に四つのホスホロチオエート結合が含まれた。
【0093】
段階4: 連結されたDNAライブラリーのゲル電気泳動および抽出
ユニバーサルアダプターの連結手順により以下のものが生ずる: 1) 両端にアダプターを有する断片化したDNA; 2) 未結合の単一アダプター; またはアダプター二量体の形成。アダプターが連結されたDNAライブラリー集団を未連結の、単一アダプターおよびアダプター二量体の集団から分離かつ単離する方法として、アガロースゲル電気泳動が使用される。ゲノムDNAのDNase I消化手順により、50〜700 bpに及ぶライブラリー集団が得られる(段階1)。88 bpのユニバーサルアダプターセットの付加は、その集団をより大きいサイズに変えて、約130〜800 bpのサイズ範囲の移動プロファイルをもたらすことになる。アダプター二量体は88 bpに移動することになり、未連結のアダプターは44 bpに移動することになる。従って、サイズ範囲が200 bpを超えるゲノムDNAライブラリーをアガロースゲルから物理的に単離して、標準的なゲル抽出技術により精製することができる。アダプターが連結されたDNAライブラリーのゲル分離により、サイズ範囲が200 bpまたはそれ以上であるライブラリー集団が回収されることになる(ライブラリーのサイズ範囲は、用途に応じて変化させることができる)。以下の電気泳動および抽出手順を使用した。
1. 2%アガロースゲルを調製した。
2. 10×Ready-Load Dye 10 μlをDNA連結混合液の残り90 μlに添加した。
3. 色素/連結反応混合液を4本の隣接レーンを使ってゲルに添加した(25 μl/レーン)。
4. 100 bp ラダー(0.1 μg/μl) 10 μlを連結反応液のレーンから2レーン離して添加した。
5. ゲルを100 Vで3時間泳動させた。
6. ゲルの泳動が完了した場合には、ゲルをゲル槽から取り出して、プラスチックラップで覆った平面に移した。DNAのバンドを手持ち式の長波長UVライトにより可視化した。無菌の、使い捨ての外科用メスを用いて、200〜400 bpの断片サイズをアガロースゲルから切り出した。この手法を用いて、任意のサイズ範囲のライブラリーを単離することができる。二つまたはそれ以上のサイズ範囲を単離することも可能である。ライブラリーのサイズ範囲が200〜900 bpである場合、単一ウェルからいくつかのサイズ範囲を単離することが可能である(すなわち、200〜400 bpおよび500〜700 bp)。
7. アガロースゲル内に埋まったDNAは、製造元の使用説明書に従い、Qiagen MinElute Gel Extractionキットを用いて単離した。手短に言えば、緩衝液QGをチューブ内のアガロースを覆うように添加した。アガロースを完全に溶解させた。緩衝液QGの色をQiagenの使用説明書に従ってpHを調整することにより維持し、試料の損失を最小限にした。2本のMinEluteスピンカラム(Qiagen)を精製のために使用した。溶解したアガロースの容量が多く、各カラムに何回も添加する必要があった。カラムを予め55℃に温めておいた緩衝液EB 10 μlで溶出した。溶出液をプールして、gDNAライブラリー 20 μlを得た。
8. それぞれ単離したDNAライブラリーの一定分割量1 μl 1つをBioAnalyzer DNA 1000 LabChipにより分析して、DNAライブラリー集団の正確な分布を評価した。
【0094】
段階5: ニックの入った二本鎖DNAライブラリーの鎖置換および鎖伸長
ユニバーサルアダプターに使用されるDNAオリゴヌクレオチドはリン酸化されないので、ギャップが断片化gDNAの3'連結部に存在する。これらの二つの「ギャップ」または「ニック」は、鎖置換DNAポリメラーゼを使用することにより埋めることができる。このポリメラーゼは、ニックの修復およびニックの入っていない二本鎖DNAの形成をもたらすように、ニックを認識し、ニックの入った鎖を置換して、鎖を伸長する。使用される鎖置換酵素は、Bst DNAポリメラーゼのラージフラグメントである。
1. 0.2 mlチューブに、ゲル抽出したDNAライブラリー 19 μl、nH2O 40 μl、10×ThermoPol反応用緩衝液(Reaction Buffer) 8 μl、BSA(1 mg/ml) 8 μl、dNTP(10 mM) 2 μl、およびBst Iポリメラーゼ(8 U/μl) 3 μlを順に添加した。
2. 試料を十分に混合してサーマルサイクラーにセットし、鎖置換インキュベーションプログラム:「BST」を用いてインキュベートした。ニックの入った二本鎖DNAの鎖置換および鎖伸長用のBSTプログラム
1. 65℃で30分インキュベートする。
2. 80℃で10分インキュベートする。
3. 58℃で10分インキュベートする。 ならびに
4. 14℃で保持する。
3. Bst処理DNAライブラリーの一定分割量1 μL 1つをBioAnalyzer DNA 1000 LabChipにより泳動した。
【0095】
段階6: ストレプトアビジンビーズの調製
ニックの入っていない二本鎖ゲノムDNAの作製に続いて、隣接のユニバーサルアダプター配列を含んだ一本鎖ゲノムDNAを単離することが必要である。この段階は、ストレプトアビジンビーズとのビオチン標識二本鎖DNAの結合について概説する。ストレプトアビジンビーズを調製するのに、以下の手順を使用した。
1. Dynal M-270ストレプトアビジンビーズ 100 μlは、この磁気ビーズをMPCに適用することにより1×結合用緩衝液(1 M NaCl, 0.5 mM EDTA, 5 mM Tris, pH 7.5) 200 μlで2回洗浄した。
2. ビーズを2×結合用緩衝液 100 μlに再懸濁させて、次にBst処理DNA試料(段階5からの)の残り79 μlおよび水20 μlを添加した。
3. ビーズ溶液を十分に混合して、チューブ回転装置に室温で20分間載せておいた。ビーズ混合液を、MPCを用いて、1×結合用緩衝液100 μlで2回、次いでnH2Oで2回洗浄した。結合および洗浄用(B & W)緩衝液 (2×および1×): 2×B & W緩衝液は、10 mM Tris・HCl (pH 7.5)、1 mM EDTA、および2 M NaClを混合することにより調製した。試薬を上記のように混ぜ合わせて、完全に混合した。この溶液は室温で6ヶ月間保存することができる。1×B & W緩衝液は、2×B & W緩衝液をnH2Oと1:1で混合することにより調製した。終濃度は上記の半分、すなわち、5 mM Tris・HCl (pH 7.5)、0.5 mM EDTA、および1 M NaClとなった。
【0096】
段階7: ストレプトアビジンビーズを用いた一本鎖DNAライブラリーの単離
ストレプトアビジンビーズとの二本鎖gDNAライブラリーの結合に続いて、連結されたプールからユニバーサルアダプターAとユニバーサルアダプターBとを含んだ一本鎖gDNAだけを単離することが好ましい(所望の集団は、星印で下記に指定されている)。二本鎖ゲノムDNA断片のプールには、以下の予想配置でアダプターが結合されることになる。
ユニバーサルアダプターA - gDNA断片 - ユニバーサルアダプターA
ユニバーサルアダプターB - gDNA断片 - ユニバーサルアダプターA*
ユニバーサルアダプターA - gDNA断片 - ユニバーサルアダプターB*
ユニバーサルアダプターB - gDNA断片 - ユニバーサルアダプターB
【0097】
ユニバーサルアダプターBだけが5'ビオチン部分を有するので、磁性のストレプトアビジン含有ビーズを使用して、ユニバーサルアダプターBを保有する全てのgDNAライブラリー種を結合することができる。二つのユニバーサルアダプターAを含むゲノムライブラリー集団種(または非連結種)は、ストレプトアビジン含有ビーズに結合せず、洗浄手順の間に除去される。洗浄後にビーズに結合したままの種には、ユニバーサルアダプターAとBとを有するものまたは二つのユニバーサルアダプターB末端を有するものが含まれる。
【0098】
二つのユニバーサルアダプターB配列を有し、二つのビオチン分子を有するゲノムDNA種は、ストレプトアビジン含有ビーズに両端で結合することができる。アダプターAとアダプターBとを有する種は一つのビオチン分子を有するのみで、ビーズに「B」末端でのみ結合することができる。一本鎖の集団を単離するため、ビーズに結合した二本鎖DNAを、相補DNA鎖間の水素結合を切断する働きをする水酸化ナトリウム溶液で処理する。DNA断片がビオチンをそれぞれの末端(ユニバーサルアダプターBの末端)に有する場合、生ずる一本鎖はどちらもビーズに結合したままである。断片が一つのビオチンのみ(ユニバーサルアダプターAおよびB)を有する場合には、相補鎖はDNA-ビーズ複合体から分離する。
【0099】
生ずる一本鎖ゲノムDNAライブラリーを液相から回収して、例えば、ピロリン酸配列決定(PyroSequence)法を使用して、またはRNA Pico 6000 LabChip (Agilent, Palo Alto, CA)を使用することにより定量化する。一本鎖ゲノムDNAライブラリーを、単位容量あたりの分子数を計算することにより定量化する。次いで、一本鎖gDNA分子を、DNA捕捉プライマー(PCRプライマーB)を含有する25〜30 μmのセファロースビーズにアニーリングさせる(1ビーズにつき1個の有効なコピーを得るためにビーズあたり半分のコピー数で)。次に、鋳型を乳濁液ポリメラーゼ連鎖反応手順により増幅させる。その後の配列決定は、周知技術を使用して行うことができる。一本鎖ライブラリーの単離のために、以下の手順を使用した。
1. 融解溶液(0.125 M NaOH, 0.1 M NaCl) 250 μlを上記段階6の洗浄済みのビーズに添加した。
2. ビーズ溶液を十分に混合して、ビーズ混合液をチューブ回転装置上で室温にて10分間インキュベートした。
3. Dynal MPC (磁性粒子濃縮器)を使用し、沈殿ビーズを注意深く取り除いて、上清を取っておいた。250 μlの上清には、一本鎖DNAライブラリーが含まれていた。
4. 別のチューブに、PB(QiaQuick精製キットからの) 1250 μlを添加して、この溶液を20%酢酸9 μlの添加により中和した。
5. Dynal MPCを用いて、一本鎖gDNAライブラリーを含有する250 μlの上清からビーズを沈殿させて、この上清を注意深く取り出し、新たに調製したPB/酢酸溶液に移した。
6. この溶液1500 μlを1個のQiaQuick精製スピンカラムにより精製した(試料を1回の添加につき750 μlとして2回同じカラムに添加して通す)。一本鎖DNAライブラリーをEB 50 μlで溶出した。
【0100】
段階8a: ピロリン酸配列決定による一本鎖gDNAの定量化
全調製時間は約1時間であった。
1. 0.2 mlチューブに、以下の試薬を順に添加した。
2. DNAをANNEAL-Sプログラム(以下の補遺を参照されたい)によりアニーリングさせた。
3. 試料をPSQ(ピロリン酸配列決定装置(jig))で泳動して、各試料中の鋳型のピコモル数を決定した(下記を参照されたい)。配列決定の方法は、米国特許第6,274,320号; 米国特許第4,863,849号; 米国特許第6,210,891号; および米国特許第6,258,568号のなかで見出すことができる(これらの開示は、その全体が、参照により本明細書に組み入れられる)。計算を行い、1マイクロリットルあたりの一本鎖gDNA鋳型分子の数を決定した。調製した一本鎖gDNAライブラリーの残り25 μlは、増幅およびその後の配列決定(反応数およそ1×106)に使用した。
【0101】
段階8b: RNA Pico 6000 LabChipによる一本鎖gDNAの定量化。全調製時間は約30分であった。
1. BioAnalyzer(ソフトウェアバージョン2.12)で、選択肢mRNA Picoアッセイを選択した。
2. BioAnalyzer上で、製造元のガイドラインに従い、RNA Pico 6000 LabChipを調製した。
3. RNA LabChipラダー(RNA 6000ラダー)を製造元(Ambion)の指示に従って調製した。手短に言えば、溶液状態のRNA LabChipラダーを70℃に2分間加熱した。ラダーを急冷するため、溶液を氷上で5分間冷却した。管壁から凝縮物を取り除くため、溶液を手短に遠心した。このRNA LabChipラダーは、氷上で保存し、1日のうちに使用した。
4. 分析されるssDNAライブラリーは、一定分割量1 μl 3つを用い、隣接レーンに入れて、3回通り泳動した。
5. BioAnalyzerのソフトウェアを使用して、各ssDNAライブラリーのレーンの濃度を計算した(下記表および図8を参照されたい)。以下に概略を説明した手順を用い、3つのレーン全ての平均を使って、ライブラリーのDNA濃度を計算した。
a. ピーク積分の下限線(図8の長い点線)をライブラリーのピークの直前に動かした(下記を参照されたい)。
b. ピーク積分の上限線(図8の長い点線)をライブラリーのピークの直後に動かした。このような方法で、下限および上限の積分線を結び付けるピーク積分線は、バックグラウンドの傾きに従った。
c. マウス矢印を使用してベースとなるピークの平均サイズ(通常、ピークの最高点付近)を決定した、またはソフトウェアにより選択されるような確定ピークを使用した。
d. ピーク中の物質量を求めるのに積分値を使用した。ピコグラムで得られた回収値を回収分子に変換した(下記表を参照されたい)。次いで、ライブラリーの濃度(1マイクロリットルあたりの分子数)を決定した。
【0102】
表
【0103】
上記の表に示されるように、ライブラリー1の濃度は1639 pg/μl(列5)と算出され、その平均断片サイズは434ヌクレオチド(列9)となった。これらの値は、上記の段階(a)〜(d)に記載されているようにAgilent 2100ソフトウェアから得られた。リボヌクレオチドの平均分子量(MW)は328.2 g/モル(列10)である。ライブラリー平均断片のMW (1.42×105 g/モル、列11)は、平均断片長(434)に平均リボヌクレオチド(328.2)を掛けることにより算出した。定量化したライブラリー(1639 pg/μl)を1マイクロリットルあたりのグラム数(1.64×10-9 g/μl、列12)に変換した。1マイクロリットルあたりのモル数(1.15×10-14モル/μl、列14)は、1マイクロリットルあたりのグラム数(1.64×10-9 g/μl、列12)をライブラリー断片の平均分子量(1.42×105、列11)で割ることにより算出した。最後に、1マイクロリットルあたりの分子数(6.93×109分子/μl、列15)は、1マイクロリットルあたりのモル数(1.15×10-14モル/μl、列14)にアボガドロ数(6.02×1023分子/モル)を掛けることにより得られた。
【0104】
最終のライブラリー濃度は、1×108分子/μlよりも大きくなると予想された。ライブラリーの質にとってさらに重要な要因は、アダプター二量体の濃度であった。図8で、ライブラリーのピークの高さは、アダプター二量体のピーク(マーカー後の最初のピーク)よりも約10倍高いと測定された。高品質のライブラリーは、二量体のピークよりも少なくとも2倍高いピーク高さを有すると予想される。提供のRNA Pico 6000 LabChipにより一本鎖gDNA濃度が500%の精度内で推定されることに留意されたい。従って、投入gDNAのビーズあたりのコピー数(cpb)を決定する鋳型の滴定を使用して、最初の配列決定の泳動を行うことが重要であった。推奨される投入DNAは、2.5 cpb、1 cpb、0.5 cpb、および0.1 cpbである。この滴定は、14×43 PTPの4スロットのビーズ充填室を使用して簡単に調べられた。
【0105】
段階9: 一本鎖gDNAライブラリーの希釈および保存
一本鎖gDNAライブラリーは、緩衝液EBで溶出かつ定量化した。分解を防ぐため、一本鎖gDNAライブラリーは、EDTAの存在下で-20℃にて凍結保存した。定量化後、等量の10 mM TEをライブラリー貯蔵液に添加した。以降の全ての希釈はTE中とした。収量は次の通りであった。
PSQ分析後のssDNAライブラリーの残りの最終容量 = 25 μl。
LabChip分析後のssDNAライブラリーの残りの最終容量 = 47 μl。
【0106】
最初の貯蔵液の希釈では、一本鎖gDNAライブラリーを100×106分子/μl 1×ライブラリー用溶出緩衝液に希釈した。共用の一本鎖gDNAライブラリーの一定分割量を調製した。このため、200,000分子/μlを1×ライブラリー用溶出緩衝液に希釈して、一定分割量20 μlを計量した。使い捨てライブラリーの一定分割量は、-20℃に保存した。
【0107】
段階10: 乳濁液ポリメラーゼ連鎖反応(Emulsion Polymerase Chain Reaction)
cpb数の増加が好ましい場合、ビーズ乳濁液のPCRを2003年6月6日付で出願された米国特許出願第06/476,504号(その全体が参照により本明細書に組み入れられる)に記載されているように行った。
【0108】
試薬調製
停止溶液(50 mM EDTA)には、50 mM EDTA溶液1.0 mlを得るためにnH2O 900 μlと混合した0.5 M EDTA 100 μlが含まれた。10 mM dNTPの場合、dCTP(100 mM) 10 μl、dATP(100 mM) 10 μl、dGTP(100 mM) 10 μl、およびdTTP(100 mM) 10 μlを分子生物等級純水60 μlと混合した。4種の100 mMヌクレオチド貯蔵液は全て氷上で融解させた。次いで、各ヌクレオチド10 μlをnH2O 60 μlと混合して最終容量を100 μlとし、完全に混合した。次に、一定分割量1 mlを1.5 ml微量遠心管に分注した。この貯蔵液は、-20℃で1年間保存することができる。
【0109】
10×アニーリング用緩衝液には、200 mM Tris (pH 7.5)および50 mM酢酸マグネシウムが含まれた。この溶液の場合、Tris 24.23 gをnH2O 800 mlに添加して、この混合液をpH 7.5に調整した。この溶液に、酢酸マグネシウム10.72 gを添加して、完全に溶解させた。この溶液は最終容量を1000 mlに合わせて、4℃で1ヶ月間保存することができる。10×TEには、100 mM Tris・HCl (pH 7.5)および50 mM EDTAが含まれた。これらの試薬を同時に添加して、完全に混合した。この溶液は室温で6ヶ月間保存することができる。
【0110】
実施例2: プライマー設計
上記のように、ユニバーサルアダプターは、以下を含むように設計される: 1) 長さが通常20 bpである特有のPCRプライミング領域のセット((2)に隣接して位置する); 2) 長さが通常20 bpである特有の配列決定プライミング領域のセット; および3) 任意で、続けて4種のデオキシリボヌクレオチド(すなわち、A、C、G、T)のそれぞれの少なくとも1つからなる特有の識別可能なキー配列。プライマーと関心対象のゲノムの意図しない領域との間の交差ハイブリダイゼーションの確率は、ゲノムサイズが増加するにつれおよびプライマーとの完全な適合性が低下するにつれて増加する。しかしながら、交差ハイブリダイズする領域(CHR)とのこの潜在的相互作用は、下記の理由から問題をもたらすとは予想されない。
【0111】
本発明の好ましい態様において、一本鎖DNAライブラリーは、PCR増幅およびその後の配列決定に使用される。配列決定法には、150〜500塩基対の断片への所定のゲノムの無作為消化が必要となり、その後、二つの特有の両方向性プライマー(PCRおよび配列決定領域の双方からなる)を断片の5'および3'末端に連結する(図5)。融解温度(Tm)、ゲノム内のプライミング配列の特異性および特定領域または関心対象の遺伝子との近接性に基づいて、ゲノムの既存部分をプライミング部位として選択する典型的なPCR増幅とは異なり、開示の方法では、合成によるプライミング部位を用いており、慎重な新規のプライマー設計が必要になる。
【0112】
四量体の選択:
デノボプライマー設計の戦略は、ハイブリダイゼーション実験のための分子タグ(Hensel, M. and D.W. Holden, Molecular genetic approaches for the study of virulence in both pathogenic bacteria and fungi. Microbiology, 1996. 142(Pt 5): p. 1049-58; Shoemaker, D.D., et al., Quantitative phenotypic analysis of yeast deletion mutants using a highly parallel molecular bar-coding strategy. Nat Genet, 1996. 14(4): p. 450-6を参照されたい)およびPCR/LDR(ポリメラーゼ連鎖反応/連結検出反応)のハイブリダイゼーションプライマー(Gerry, N.P., et al., Universal DNA microarray method for multiplex detection of low abundance point mutations. Journal of Molecular Biology, 1999. 292: p. 251-262; Witowski, N.E., et al., Microarray-based detection of select cardiovascular disease markers. BioTechniques, 2000. 29(5): p. 936-944.を参照されたい)に対して行われた研究に関する既刊文献に見出される。
【0113】
PCR/LDRの研究は、特に関連性があり、オリゴヌクレオチド「ジップコード」、つまり類似の最終Tmを有する6つの特異的に設計された四量体からなる24塩基のプライマーの設計に重点的に取り組んでいた(Gerry, N.P., et al., Universal DNA microarray method for multiplex detection of low abundance point mutations. Journal of Molecular Biology, 1999. 292: p. 251-262; 米国特許第6,506,594号を参照されたい)。四量体成分は、以下の判定基準に基づいて選択された: 各四量体は少なくとも2塩基だけ他のものと異なっていた、自己対合またはヘアピン形成を起こした四量体は除外した、および回文構造の四量体(AGCT)または反復性の四量体(TATA)は同様に取り除いた。考えられる256(44)通りの順列のうち36通りが必要条件を満たし、次に、許容可能なPCRプライマーの設計に必要なさらなる制約にかけられた(表1)。
【0114】
表1
表1には、Gerry et al. 1999. J. Mol. Bio. 292: 251-262により概説される判定基準に基づく四量体プライマー成分の選択を明示する行列が示されている。各四量体は少なくとも2塩基だけ他の全てのものと異なることが必要とされた。これらの四量体は他のどの四量体とも回文構造または相補的になることはない。36通りの四量体(太字、下線)を選択した。斜体の配列は、考慮から除外された回文構造の四量体を示す。
【0115】
プライマー設計:
PCRプライマーは、一般的なプライマー設計に共通の規格を満たすように設計し(Rubin, E. and A.A. Levy, A mathematical model and a computerized simulation of PCR using complex templates. Nucleic Acids Res, 1996. 24(18): p. 3538-45; Buck, G.A., et al., Design strategies and performance of custom DNA sequencing primers. Biotechniques, 1999. 27(3): p. 528-36を参照されたい)、実際の選択はコンピュータプログラムMMPにより行った。プライマーは、すべての両方向性PCR/配列決定用プライマーの効率的な合成のため、20塩基(5つの四量体)の長さに制限した。各プライマーは5'末端に2塩基のGCクランプ、および3'末端に1塩基のGCクランプを含み(表2)、全てのプライマーが類似のTm(+/-2℃)を共有していた(図10)。プライマー内でヘアピン形成(内部ヘアピンステム ΔG > -1.9 kcal/モル)させなかった。二量体化も同様に制御した。3塩基の最大許容二量体を許容したが、これは最後の6つの3'塩基で起こり、3'二量体に対する最大許容ΔGは-2.0 kcal/モルであった。さらに、3'末端がグループ内の他のものに過度に類似するプライマーに、ペナルティーを適用して、これにより、あるプライマーと他の逆相補体との間の交差ハイブリダイゼーションを阻止した。
【0116】
(表2)
【0117】
表2には、2つの5'および1つの3'G/Cクランプを与える、36通りの選択された四分子の考えられる順列が示されている。内部の位置は、残る四分子からなる。この結果、8×19×19×19×9通りの順列、または493,848通りの可能な組み合わせが生まれる。図10は初回通過、つまり493,848通りのプライマーの範囲を64〜66℃のTmを有する56,246通りの候補に減らす、Tmに基づく許容可能なプライマーの選択を示す。
【0118】
(表3)プライマーに対する完全な配列適合性の確率は、適合する長さの要件を減少させるにつれておよび関心対象のゲノムのサイズを増加させるにつれて増加する。
【0119】
複雑な試料集団でのミスマッチに対するPCRの許容度が報告されている(例えば、Rubin, E. and A.A. Levy, A mathematical model and a computerized simulation of PCR using complex templates. Nucleic Acids Res, 1996. 24(18): p. 3538-45を参照されたい)にもかかわらず、相補領域が関心対象のゲノム内に発生する確率は、プライマー設計過程において主要な懸案事項ではなかった。20塩基のプライマーに対して完全適合性を見出す確率は極めて低い(420)(表3)が、それほど連続的ではない適合性を見出す確率は、関心対象のゲノムのサイズとともに顕著に増加する。結果として、20塩基のうち少なくとも10塩基の完全適合性を見出す確率は、アデノウイルスのゲノムでは99.35%になる。16塩基の完全適合性を見出す確率は、NCBIデータベースの配列(アデノウイルスのゲノムよりも約100倍大きい)では97%になる。20塩基のプライマーに対して17塩基の完全適合性を見出す確率は、ヒトゲノムの配列(30億塩基)では99%になる。
【0120】
ゲノムの領域に対するプライマーの交差ハイブリダイゼーションの確率の高さは、鋳型断片を作製させるために使用される無作為DNA消化により、予想されるほどの問題にはならない。従って、交差ハイブリダイズする領域(CHR)の影響はほとんど害がない。CHRが溶液中のPCRプライマーとその鋳型との間の完全適合性と十分に競合できる可能性は低い。さらに、3'末端にミスマッチを含むプライマーはいずれも、著しく競合上不利な状態にあるものと思われる。たとえCHRが意図するPCRプライマーを打ち負かした(outcompete)としても、それにより、配列決定用プライマーに対する下流部位のない、切断型のPCR産物が作製されることになる。切断産物が捕捉ビーズに追いやられて、固定化される場合、二つの状況のうちの一つが生じることになる。CHRが液相のプライマーを打ち負かした場合には、固定化産物は配列決定用プライマー結合部位がないことになり、空のPicoTiterプレート(PTP)ウェルが生ずることになる。CHRがビーズ結合プライマーを打ち負かした場合には、配列決定用プライマーは依然として存在することになり、その唯一の影響は短い挿入断片ということになる。どちらの結果も配列決定の質を過度に落とすことにはならない。試料調製工程で使用されるゲノム材料の量が多い(今回、25 μg、35Kbのアデノウイルスゲノムを5.29×1016コピーを含有する)ことを考慮して、過剰なサンプリングを用い、完全なCHRのない断片を供与して、問題の領域の標準的PCR増幅を可能とすることができる。
【0121】
実施例3: 噴霧による試料調製
噴霧によるDNAの調製
噴霧段階の目的は、全ゲノムまたは大きいゲノム部分のようなDNAの大きいストレッチをDNA配列決定に適した小さな分子種に断片化することである。単一の鋳型DNAから作製された小サイズのDNA種のこの集団をライブラリーという。噴霧により二本鎖鋳型DNAが50〜900塩基対に及ぶ断片に剪断される。剪断されたライブラリーには、T4 DNAポリメラーゼ、大腸菌DNAポリメラーゼI(クレノーフラグメント)、およびT4ポリヌクレオチドキナーゼの組み合わせにより末端修復される一本鎖の末端が含まれる。T4 DNAポリメラーゼもクレノーDNAポリメラーゼも、その5'から3'方向のポリメラーゼ活性によってDNAの3'陥凹末端(5'突出部)を「埋める」ために使用される。T4およびクレノーポリメラーゼの一本鎖の3'から5'方向のエキソヌクレアーゼ活性により3'突出末端が除去されることとなり、T4ポリヌクレオチドキナーゼのキナーゼ活性により5'ヒドロキシル末端にリン酸が付加されることとなる。
【0122】
試料を以下のように調製した。
1. gDNA(ゲノムDNA) 15 μgを得て、10 mM TE (10 mM Tris, 0.1 mM EDTA, pH 7.6; 項の末尾の試薬リストを参照されたい)で最終容量100 μlに調整した。このDNAを吸光度260/280比の測定により混入がないか分析した。この比は1.8またはそれより高かった。gDNA最終濃度は約300 μg/mlになると予想された。
2. 氷冷の噴霧緩衝液(項の末尾を参照されたい) 1600 μlをgDNAに添加した。
3. この反応混合液を氷冷の噴霧装置(CIS-US, Bedford, MA)にセットした。
4. 15 mlスナップキャップ・ファルコンチューブのキャップを噴霧装置の先端に取り付けた(図7A)。
5. キャップを汚れのない噴霧装置のクランプ部品(ぴったりのカバー(ファルコンチューブの蓋に対し)と2つのゴム製O-リングとからなる)で固定した(図7B)。
6. 噴霧装置の底面を窒素供給器に取り付けて、装置全体をパラフィルムで包んだ(図7Cおよび7D)。
7. 噴霧装置を垂直に維持したまま(図7Dに示されるように)、50 psi(平方インチ当たりのポンド)の窒素を5分間加えた。噴霧装置の底面を数秒毎に硬質表面に叩きつけて、濃縮液を底面に押し進めた。
8. 窒素を5分後に止めた。圧力が平常値に戻った後(30秒)、窒素供給源を噴霧装置から取り外した。
9. パラフィルムを除去して、噴霧装置の先端の蓋を回して外した。試料を取り出して、1.5 ml微量遠心管に移した。
10. 噴霧装置の先端の蓋を再び取り付けて、噴霧装置を500 rpmで5分間遠心した。
11. 噴霧装置中の試料の残りを回収した。総回収量は約700 μlであった。
12. 回収された試料は、製造元の使用説明書に従い、QIAquickカラム(Qiagen社、Valencia, CA)を用いて精製した。量が多く、カラムに数回添加することが必要になった。試料は、55℃に予め暖めておいた緩衝液EB(10 mM Tris HC1, pH 8.5; Qiagenキットで供給される) 30 μlで溶出させた。
13. 試料はUV分光法により定量化した(100倍希釈の場合には水198 μlに2 μl)。
【0123】
酵素ポリッシング
鋳型DNAの噴霧により非相補的末端を有する多くのDNA断片が得られる。これらの末端は、3種の酵素、つまりT4 DNAポリメラーゼ、大腸菌DNAポリメラーゼ(クレノーフラグメント)およびT4ポリヌクレオチドキナーゼを用いることにより、平滑およびアダプター断片に連結される状態にされる。
【0124】
試料を以下のように調製した。
1. 0.2 mlチューブに、以下の試薬を順に添加した。
2. 段階1の溶液を十分に混合して、MJサーモサイクラー(任意の的確なインキュベーターを使用することができる)中で25℃にて10分間インキュベートした。
3. 大腸菌DNAポリメラーゼ(クレノーフラグメント)(5単位/ml) 1.25 μlを添加した。
4. 反応液を十分に混合して、MJサーモサイクラー中で25℃にて10分間および16℃にてさらに2時間インキュベートした。
5. 処理したDNAは、QiaQuickカラムを用いて精製し、55℃に予め暖めておいた緩衝液EB(10 mM Tris HC1, pH 8.5) 30 μlで溶出させた。
6. 以下の試薬を0.2 mlチューブに混合した。
7. 溶液を混合して、37℃で30分間、65℃で20分間のインキュベーション、その後、14℃で保存のT4 PNKプログラムを使用するMJサーマルサイクラーにセットした。
8. 試料は、QiaQuickカラムを用いて精製し、55℃に予め暖めておいた緩衝液EB 30 μlで溶出させた。
10. 最終ポリッシング反応液の一定分割量2 μlをBioAnalyzer DNA 1000 LabChipによる分析のためにとっておいた(以下を参照されたい)。
【0125】
アダプターの連結
アダプターを連結するための手順は、次のように行った。
1. 0.2 mlチューブに、以下の試薬を順に添加した。
上記の反応液は、5 μg用に設計されており、使用するgDNAの量に応じてスケールを変えた。
2. 試薬を十分に混合して、25℃で20分間インキュベートした。チューブは、ゲルがアガロースゲル電気泳動用に調製されるまで氷上とした。
【0126】
連結されたgDNAライブラリーのゲル電気泳動および抽出
ゲノムDNAの噴霧により、50〜900 bpに及ぶライブラリー集団が得られる。88 bpのユニバーサルアダプターセットの付加は、その集団をより大きいサイズに変えて、より大きいサイズ範囲(約130〜980 bp)の移動プロファイルをもたらすことになる。アダプター二量体は88 bpに移動することになり、連結されていないアダプターは44 bpに移動することになる。従って、サイズ範囲が250 bpまたはそれ以上に分離されるゲノムDNAライブラリーをアガロースゲルから物理的に単離して、標準的なゲル抽出技術により精製することができる。アダプターが連結されたgDNAライブラリーのゲル分離により、サイズ範囲が250 bpまたはそれ以上であるライブラリー集団が回収されることになる(ライブラリーのサイズ範囲は、用途に応じて変化させることができる)。アダプターの連結後のライブラリーのサイズ範囲は130〜980 bpである。この手順は、ゲルの異なる領域を切り出すことにより、例えば、130〜200 bp、200〜400 bp、250〜500 bp、300〜600 bp、500〜700 bpなどのような任意のバンドサイズ範囲の単離のために適合できることに留意されたい。下記の手順は、250 bp〜500 bpの単離断片に使用した。
【0127】
2%アガロース、1×TBE、およびエチジウムブロマイド(10 mg/ml貯蔵液) 4.5 μlを含むアガロースゲル150 mlを調製した。連結DNAを10×Ready Load Dyeと混合して、ゲルに添加した。さらに、100 bp ラダー(0.1 μg/μl) 10 μlを試料隣りの連結反応液から2レーン離して添加した。ゲルを100 Vで3時間電気泳動させた。ゲルの泳動が完了した場合には、ゲルをゲル槽から取り出し、GelDocに移して、プラスチックラップで覆った。DNAのバンドをPrep UVライトにより可視化した。無菌の、使い捨ての外科用メスを用いて、アガロースゲルから250〜500 bpの断片サイズを有するライブラリー集団を切り出した。この工程は、DNAのニッキングを防ぐために可能な限り素早く行った。このゲルスライスを15 mlファルコンチューブに入れた。アガロース内に埋まったgDNAライブラリーは、Qiagen MinElute Gel Extractionキットを用いて単離した。それぞれ単離したgDNAライブラリーの一定分割量をBioAnalyzer DNA 1000 LabChipにより分析して、gDNAライブラリー集団の正確な分布を評価した。
【0128】
gDNAライブラリーの鎖置換および鎖伸長ならびにストレプトアビジンビーズによる一本鎖gDNAライブラリーの単離
ニックの入った二本鎖gDNAライブラリーの鎖置換および鎖伸長は、Bst処理試料を65℃のサーマルサイクラー中で30分間インキュベートして、必要とされるまで氷上に置いたことを除いて、実施例1に記載されているように行った。ストレプトアビジンビーズは、最後の洗浄を1×結合用緩衝液200 μlで2回洗浄およびnH2O 200 μlで2回洗浄により行ったことを除いて、実施例1に記載されているように調製した。一本鎖gDNAライブラリーは、以下のようにストレプトアビジンビーズを用いて単離した。洗浄したビーズから水を除去して、融解溶液(以下を参照されたい) 250 μlを添加した。このビーズ懸濁液を十分に混合して、チューブ回転装置に載せて室温で10分間インキュベートした。別のチューブのなかで、PB(QiaQuick精製キットからの) 1250 μlおよび20%酢酸9 μlを混合した。融解溶液 250 μl中のビーズをDynal MPCにより沈殿させて、この上清を注意深く取り出し、新たに調製したPB/酢酸溶液に移した。この溶液1500 μlからDNAを1個のMinElute精製スピンカラムにより精製した。これは、試料を1回の添加につき750 μlとして2回同じカラムに添加して通すことで行った。一本鎖gDNAライブラリーを予め55℃に温めておいた緩衝液EB 15 μlで溶出した。
【0129】
一本鎖gDNAの定量化および保存
一本鎖gDNAは、実施例1に記載されているようにRNA Pico 6000 LabChipを用いて定量化した。場合によっては、一本鎖ライブラリーは、最初のAgilent 2100による定量化が正確に行われることを確実にするため、第二のアッセイにより定量化した。このために、RiboGreenによる定量化を記載のように(蛍光光度法によるssDNA定量化)行い、Agilent 2100による定量化を確認した。この二通りの推定値が3倍を超えるまでに異なっていた場合、各分析を繰り返した。定量化により二通りの手順の間で3倍を超える相違が明らかになった場合、ビーズに対してより広範囲の鋳型を使用した。
【0130】
一本鎖gDNAライブラリーの希釈および保存は、実施例1に記載されているように行った。収量は次の通りであった。
LabChip分析後のssDNAライブラリーの残りの最終容量 = 12 μl。
RiboGreen分析後のssDNAライブラリーの残りの最終容量 = 9 μl。
TEの添加後のssDNAライブラリーの最終容量 = 18 μl。
【0131】
等量のTEを一本鎖gDNAライブラリーの貯蔵液に添加した。一本鎖gDNAライブラリーを1×108分子/μl TE緩衝液に。貯蔵液を200,000分子/μl TEに希釈(500倍)して、一定分割量20 μlを調製した。
【0132】
噴霧後のライブラリー断片サイズの分布
噴霧およびポリッシング後の材料1 μlのAgilent 2100 DNA 1000 LabChip分析から得られた典型的な結果が図9Aに示されている。大部分の産物のサイズ範囲分布は、50〜900塩基対前後に収まると予想された。平均サイズ(ピークの最高点)は、約450 bpになると予想された。アダプターが連結されたライブラリー断片のゲル精製後に得られた典型的な結果が図9Bに示されている。
【0133】
試薬
特別の定めのない限り、実施例に記載の試薬は、市販されている標準的な試薬に相当する。例えば、クレノー、T4 DNAポリメラーゼ、T4 DNAポリメラーゼ緩衝液、T4 PNK、T4 PNK緩衝液、クイック T4 DNAリガーゼ、クイックライゲーション緩衝液、Bst DNAポリメラーゼ(ラージフラグメント)およびThermoPol反応用緩衝液は、New England Biolabs (Beverly, MA)から入手できる。dNTPミックスはPierce (Rockford, IL)から入手できる。アガロース、超高純度(UltraPure)TBE、ブルージュース・ゲルローディング緩衝液およびReady-Load 100 bp DNAラダーは、Invitrogen (Carlsbad, CA)から購入することができる。エチジウムブロマイドおよび2-プロパノールはFisher (Hampton, NH)から購入することができる。RNAラダーはAmbion (Austin, TX)から購入することができる。その他の試薬は、一般に公知であるおよび/または下記に記載されている。
【0134】
融解溶液:
融解溶液には、100 mM NaCl、および125 mM NaOHが含まれた。記載の試薬を混ぜ合わせて、完全に混合した。この溶液は室温で6ヶ月間保存することができる。
【0135】
結合および洗浄用(B & W)緩衝液 (2×および1×):
2×B & W緩衝液には、終濃度で10 mM Tris-HCl (pH 7.5)、1 mM EDTA、および2 M NaClが含まれた。記載の試薬を混ぜ合わせて、完全に混合した。この溶液は室温で6ヶ月間保存することができる。1×B & W緩衝液は、2×B & W緩衝液をピコピュアH2Oと1:1で混合することにより調製した。終濃度は上記のものの半分、すなわち、5 mM Tris-HCl (pH 7.5)、0.5 mM EDTA、および1 M NaClとなった。
【0136】
その他の緩衝液には、以下が含まれた。1×T4 DNAポリメラーゼ緩衝液: 50 mM NaCl、10 mM Tris-HCl、10 mM MgCl2、1 mMジチオスレイトール(25℃でpH 7.9)。TE: 10 mM Tris、1 mM EDTA。
【0137】
特殊試薬の調製:
TE (10 mM):
各試薬を混合した。この溶液は室温で6ヶ月間保存することができる。
【0138】
噴霧緩衝液:
全ての試薬をStericupに添加(グリセロールは最後に添加)して、十分に混合した。この溶液は、ラベル付けして、室温で6ヶ月間保存することができる。
【0139】
ATP (10 mM):
各試薬を混合した。この溶液は-20℃で6ヶ月間保存することができる。
【0140】
BSA (1 mg/ml):
試薬を混合した。この溶液は4℃で6ヶ月間保存することができる。
【0141】
ライブラリーアニーリング用緩衝液、10×:
10×アニーリング用緩衝液には、200 mM Tris (pH 7.5)および50 mM酢酸マグネシウムが含まれた。この緩衝液の場合、Tris 200 mlをピコピュアH2O 500 mlに添加した。次に、酢酸マグネシウム10.72 gを溶液に添加して、完全に溶解させた。この溶液を最終容量1000 mlに調整した。この溶液は4℃で6ヶ月間保存することができる。ライブラリーの混入の可能性を回避するため、この緩衝液を単回使用または短期使用のために分注した。
【0142】
アダプター:
アダプター「A」(400 μM):
この溶液の場合、1000 pmol/μl アダプターA(44 bp、センス) 10 μlを1000 pmol/μl アダプターA(40 bp、アンチセンス) 10 μl、10×ライブラリーアニーリング用緩衝液2.5 μl、および水2.5 μlと混合した(Vf = 25 μl)。アダプターは、Sample Prep LabサーマルサイクラーのANNEAL-Aプログラム(以下の補遺を参照されたい)を用いてアニーリングさせた。アダプター設計に関する詳細は、補遺に示されている。
【0143】
アダプター「B」(400 μM):
この溶液の場合、1000 pmol/μl アダプターB(40 bp、センス) 10 μlを1000 pmol/μl アダプターB(44 bp、アンチ) 10 μl、10×ライブラリーアニーリング用緩衝液2.5 μl、および水2.5 μlと混合した(Vf = 25 μl)。アダプターは、Sample Prep LabサーマルサイクラーのANNEAL-Aプログラム(補遺を参照されたい)を用いてアニーリングさせた。アニーリング後、アダプター「A」およびアダプター「B」を混合した(Vf = 50 μl)。アダプターセットは、使用前まで-20℃で保存することができる。
【0144】
20%酢酸:
この溶液の場合、氷酢酸を水に添加した。この溶液は室温で6ヶ月間保存することができる。
【0145】
補遺
アダプターアニーリングプログラム
ANNEAL-プライマーアニーリング用のプログラム
1. 95℃で1分インキュベートする。
2. 15℃まで0.1℃/秒で温度を下げる。ならびに
3. 14℃で保持する。
【0146】
末端修復用のT4ポリメラーゼ/クレノーPOLISHプログラム
1. 25℃で10分インキュベートする。
2. 16℃で2時間インキュベートする。ならびに
3. 4℃で保持する。
【0147】
末端修復用のT4 PNKプログラム
1. 37℃で30分インキュベートする。
2. 65℃で20分インキュベートする。ならびに
3. 14℃で保持する。
【0148】
ニックの入った二本鎖gDNAの鎖置換および鎖伸長用のBSTプログラム
1. 65℃で30分インキュベートする。ならびに
2. 14℃で保持する。
【0149】
参照文献
【0150】
本明細書を通じて、当技術分野の状況および内容を記載するため、さまざまな特許、公開された特許出願および科学文献が引用される。それらの開示は、その全体が、参照により本明細書に組み入れられる。
【0151】
他の態様
特定の態様を本明細書で詳細に開示してきたが、これは例示の目的のためにのみ一例として行われたものであり、先の、添付の特許請求の範囲に対して限定することを意図するものではない。特に、本発明者らは、特許請求の範囲にある本発明の精神および範囲から逸脱することなく、さまざまな置換、変更、および変形を本発明に対して行うことができるものと考える。
【図面の簡単な説明】
【0152】
【図1】鋳型DNAの断片化(図1A)、末端ポリッシング(図1B)、アダプター連結(図1C)、ニック修復、鎖伸長およびゲル分離(図1D)の段階を含むライブラリー調製の全過程の略図である。本発明の方法による180〜350塩基対のアデノウイルスDNAライブラリーの試料調製物を含有する典型的なアガロースゲルを同様に描写する。
【図2】図2Aは、本発明のある態様によるユニバーサルアダプター設計の略図である。各ユニバーサルアダプターは、20 bpのPCRプライミング用のヌクレオチド配列、20 bpの配列決定プライミング用のヌクレオチド配列および非反復ヌクレオチド配列からなる特有の4 bpの識別可能な配列(すなわち、ACGT、CAGTなど)を含むように設計される二本の相補ssDNAオリゴヌクレオチドから作製される。図2Bは、本発明で用いる典型的なユニバーサルアダプターの配列対を描く。アダプターAセンス鎖: SEQ ID NO:1; アダプターAアンチセンス鎖: SEQ ID NO:2; アダプターBセンス鎖: SEQ ID NO:3; アダプターBアンチセンス鎖: SEQ ID NO:4。図2Cは、本発明で用いるユニバーサルアダプター設計の略図である。
【図3】本発明による、ニックの入った二本鎖DNA断片の鎖置換および鎖伸長を示す。合成オリゴヌクレオチドから作製されたユニバーサルアダプターの連結反応に従って、T4 DNAリガーゼ処理の後に2箇所のニック領域を含んだ二本鎖DNA断片が作製されることになる(図3A)。鎖置換酵素(すなわち、Bst DNAポリメラーゼI)の添加により、ニックに結合し(図3B)、ニックの入った鎖を鎖置換し、鎖のヌクレオチド伸長を完結して(図3C)、ニックの入っていない二本鎖DNA断片を作製することになる(図3D)。
【図4】ストレプトアビジン被覆磁気ビーズを用いた本発明による定方向連結一本鎖DNAの単離を示す。ユニバーサルアダプターAおよびB(この二つの異なるアダプターは「第一」および「第二」のユニバーサルアダプターといわれることもある)との連結反応後、二本鎖DNAには、4通りの可能な組み合わせ、つまりAA、BB、AB、およびBAのアダプターが含まれることになる。ユニバーサルアダプターBに5'-ビオチンが含まれる場合、磁性のストレプトアビジン被覆固相支持体を使用して、AB、BA、およびBBの集団を捕捉かつ単離する(AAの集団は洗い流される)。BBの集団は、二本鎖DNAの各末端がビーズに付着して、遊離されないので、ビーズ上に保持される。しかしながら、低塩緩衝液の存在下で洗浄すると、ABおよびBAの集団だけが、結合した鎖に相補的である一本鎖DNA断片を遊離することになる。一本鎖DNA断片を上清から単離して、その後の用途のための鋳型として使用する。この方法は、先に詳細に記載されている。
【図5】PCRプライマーおよび配列決定用プライマーが隣接した挿入断片を示す。
【図6】交差ハイブリダイゼーション反応(CHR)でミスマッチなPCRプライマーにより作製される切断型産物を示す。
【図7】本発明の方法に使用される噴霧装置の組立部品を描く。チューブキャップを噴霧装置の先端に取り付け(図7A)、キャップを噴霧装置のクランプ部品で固定した(図7B)。噴霧装置の底面を窒素供給器に取り付けて(図7C)、装置全体をパラフィルムで包んだ(図7D)。
【図8】一本鎖DNAライブラリーの分析からの典型的なBioAnalyzer出力結果を描く。
【図9】図9Aは、噴霧およびポリッシング後の一本鎖DNAライブラリーに対するLabChip分析の典型的な結果を描く。図9Bは、噴霧、ポリッシング、およびゲル精製後のアダプター連結一本鎖DNAライブラリーに対する典型的なサイズ分布結果を描く。
【図10】プライマー候補に対する融解温度に基づく推測を描く。
【特許請求の範囲】
【請求項1】
以下の段階を含む、複数の一本鎖DNA分子を含んだライブラリーをクローニングにより単離するための方法:
(a) 大きい鋳型DNA分子を断片化して複数の断片化したDNA分子を作製する段階;
(b) 第一または第二の二本鎖ユニバーサルアダプターを断片化した各DNA分子の第一の末端におよび第一または第二のユニバーサルアダプターを断片化した各DNA分子の第二の末端に付着させて、アダプター連結DNA分子の混合物を形成させる段階;
(c) それぞれが第一の一本鎖ユニバーサルアダプターと第二の一本鎖ユニバーサルアダプターとを含む複数の一本鎖DNA分子を単離して、ライブラリーを得る段階; および
(d) 複数のリアクターに一つのDNA分子が含まれるように一本鎖DNA分子をリアクターに送達し、それによってライブラリーをクローニングにより単離する段階。
【請求項2】
以下の段階を含む、複数の一本鎖DNA分子を含んだライブラリーを作製するための方法:
(a) 大きいまたは全ゲノムの鋳型DNA分子を断片化して複数の断片化したDNA分子を作製する段階;
(b) 第一の二本鎖ユニバーサルアダプターまたは第二のユニバーサルアダプターを断片化した各DNA分子の第一の末端におよび第一のユニバーサルアダプターまたは第二のユニバーサルアダプターを断片化した各DNA分子の第二の末端に連結させて、アダプター連結DNA分子の混合物を作製させる段階であって、その際に第一のユニバーサルアダプターには固相支持体に結合する部分が含まれる段階;
(c) 固相支持体に第一の二本鎖ユニバーサルアダプターを含むDNA分子を付着させる段階;
(d) 固相支持体に付着されていないアダプター連結DNA分子を除去する段階;
(e) 一端のみで固相支持体に付着されたアダプター連結DNA分子を鎖分離させて、第一の一本鎖ユニバーサルアダプターを一端におよび第二の一本鎖アダプターを他端に有する複数の一本鎖DNA分子を遊離させる段階; および
(f) 段階(e)の遊離された一本鎖DNA分子のライブラリーを固相支持体に付着したままのそれらのDNA分子から単離する段階。
【請求項3】
第一の二本鎖ユニバーサルアダプターまたは第二のユニバーサルアダプターが、連結により付着される、請求項1または2記載の方法。
【請求項4】
(f)がi) 一本鎖DNA分子をリアクターアレイ上のある位置に送達する段階; またはii) 一本鎖DNA分子を油中水型乳濁液中の液滴内に送達する段階により達成される、請求項1記載の方法。
【請求項5】
DNA修復酵素およびDNA修飾酵素を用いて、アダプター連結DNA分子の混合物における一本鎖ニックを修復する段階をさらに含む、請求項1または2記載の方法。
【請求項6】
DNA修復酵素およびDNA修飾酵素が、ポリメラーゼ、リガーゼ、キナーゼ、およびその組み合わせからなる群より選択される、請求項5記載の方法。
【請求項7】
ポリメラーゼがバチルス・ステアロサーモフィルス(Bacillus stearothermophilus)ポリメラーゼIであり、リガーゼがT4リガーゼであり、およびキナーゼがT4ポリヌクレオチドキナーゼである、請求項6記載の方法。
【請求項8】
鋳型DNAが、ゲノムDNA、cDNA、プラスミドDNA、コスミドDNA、人工染色体DNA、合成DNA、ファセミド(phasemid)DNA、およびファージミドDNAからなる群より選択される、請求項1または2記載の方法。
【請求項9】
鋳型DNAには逆転写産物が含まれる、請求項1または2記載の方法。
【請求項10】
断片化が酵素的手段、化学的手段、および機械的手段からなる群より選択される手段により行われる、請求項1または2記載の方法。
【請求項11】
酵素的手段がDNaseIである、請求項10記載の方法。
【請求項12】
機械的手段が噴霧である、請求項10記載の方法。
【請求項13】
DNase I消化が10〜37℃の温度で1〜2分間行われる、請求項11記載の方法。
【請求項14】
酵素的手段が制限酵素である、請求項10記載の方法。
【請求項15】
機械的手段がフレンチプレス、超音波処理器、HydroShear、および噴霧装置からなる群より選択される、請求項10記載の方法。
【請求項16】
断片化したDNA分子の長さが50 bp〜700 bpである、請求項1または2記載の方法。
【請求項17】
適合末端が平滑末端である、請求項1または2記載の方法。
【請求項18】
平滑末端がPfuポリメラーゼ、T4 DNAポリメラーゼおよびクレノーフラグメントからなる群より選択される酵素で生成される、請求項17記載の方法。
【請求項19】
適合末端にはAまたはT突出部が含まれる、請求項1または2記載の方法。
【請求項20】
第一または第二の二本鎖ユニバーサルアダプターには、ホスホロチオエート結合が含まれる、請求項1または2記載の方法。
【請求項21】
ビオチン部分が第一または第二の二本鎖ユニバーサルアダプターに付着される、請求項1または2記載の方法。
【請求項22】
第一または第二の二本鎖ユニバーサルアダプターが、T4 DNAリガーゼで連結される、請求項1または2記載の方法。
【請求項23】
第一の二本鎖ユニバーサルアダプターにもしくは第二の二本鎖ユニバーサルアダプターにまたはどちらの二本鎖ユニバーサルアダプターにも、識別可能なキー配列が含まれる、請求項1または2記載の方法。
【請求項24】
識別可能なキー配列の長さが3〜12ヌクレオチドである、請求項23記載の方法。
【請求項25】
識別可能なキー配列が、A、G、C、U、およびTからなる群より選択される少なくとも一つのヌクレオチドを含む、請求項23記載の方法。
【請求項26】
第一および第二の二本鎖ユニバーサルアダプターには、PCRプライミング配列および配列決定用プライマー配列が含まれる、請求項1または2記載の方法。
【請求項27】
PCRプライミング配列の長さが10〜20塩基対である、請求項26記載の方法。
【請求項28】
配列決定用プライマー配列の長さが10〜20塩基対である、請求項26記載の方法。
【請求項29】
PCRプライミング配列および配列決定用プライマー配列が重複する、請求項26記載の方法。
【請求項30】
アダプター連結DNA分子の混合物がゲル電気泳動、ろ過、サイズ排除クロマトグラフィー、およびショ糖沈降法からなる群より選択される方法により分離される、請求項1または2記載の方法。
【請求項31】
複数の一本鎖DNA分子がDNA捕捉ビーズに付着される、請求項1または2記載の方法。
【請求項32】
DNA捕捉ビーズが結合対の成分を含む、請求項31記載の方法。
【請求項33】
結合対が、アビジン/ビオチン、リガンド/受容体、抗原/抗体および相補ヌクレオチドからなる群より選択される、請求項31記載の方法。
【請求項34】
DNA捕捉ビーズが常磁性ビーズである、請求項31記載の方法。
【請求項35】
複数の一本鎖DNA分子が、低塩処理、高pH処理、および化学的変性処理からなる群より選択される処理により得られる、請求項1または2記載の方法。
【請求項36】
以下の段階を含む、固相支持体に付着された一本鎖DNAライブラリーを作製するための方法:
(a) 複数の鋳型一本鎖DNAを作製する段階;
(b) 複数の鋳型ssDNAのそれぞれを固相支持体に付着させる段階; および
(c) 鋳型一本鎖DNAが付着された固相支持体を単離する段階。
【請求項37】
以下の段階を含む、固相支持体に付着された一本鎖DNAライブラリーを作製するための方法:
(a) 大きい鋳型DNA分子を断片化して複数の断片化したDNA分子を作製する段階;
(b) 第一または第二の二本鎖ユニバーサルアダプターを断片化した各DNA分子の第一の末端におよび第一または第二のユニバーサルアダプターを断片化した各DNA分子の第二の末端に付着させて、アダプター連結DNA分子の混合物を作製する段階;
(c) 第一の一本鎖ユニバーサルアダプターと第二の一本鎖ユニバーサルアダプターとを含む一本鎖DNA分子を単離する段階; および
(d) (c)からの単離された一本鎖分子を固相支持体に付着させる段階。
【請求項38】
固相支持体がDNA捕捉ビーズである、請求項37記載の方法。
【請求項39】
DNAが、ゲノムDNA、cDNA、プラスミドDNA、コスミドDNA、人工染色体DNA、合成DNA、ファセミドDNA、およびファージミドDNAからなる群より選択される、請求項37記載の方法。
【請求項40】
DNAが結合対により固相支持体に付着される、請求項37記載の方法。
【請求項41】
結合対が、アビジン/ビオチン、リガンド/受容体、抗原/抗体および相補ヌクレオチドからなる群より選択される、請求項40記載の方法。
【請求項42】
請求項37の方法により作製される可動性の固相支持体のライブラリー。
【請求項43】
第一のアダプターと、鋳型DNAの断片と、第二のアダプターとを含む核酸分子であって、その際に第一のアダプターと第二のアダプターそれぞれが、配列決定用プライマーと、PCRプラマーと、識別可能なキー配列とを含み、およびその際に第一のアダプターと第二のアダプターが、解離した場合に、ストリンジェントなハイブリダイゼーション条件の下で相互に交差ハイブリダイズしない核酸分子。
【請求項44】
PCRプライマーの長さが10〜20塩基対である、請求項43記載の核酸分子。
【請求項45】
配列決定用プライマーの長さが10〜20塩基対である、請求項43記載の核酸分子。
【請求項46】
識別可能なキー配列の長さが3〜12塩基対である、請求項43記載の核酸分子。
【請求項47】
鋳型DNAがゲノムDNA、cDNA、プラスミドDNA、コスミドDNA、人工染色体DNA、合成DNA、ファセミドDNA、およびファージミドDNAからなる群より選択される、請求項43記載の核酸分子。
【請求項48】
核酸分子が、解離した場合に、解離した鋳型DNAに対する交差ハイブリダイゼーションが最小である、請求項43記載の核酸分子。
【請求項49】
以下の段階を含む、一本鎖DNA分子を調製するための方法:
(a) 大きいまたは全ゲノムの鋳型DNA分子を断片化して複数の断片化したDNA分子を作製する段階;
(b) 第一の二本鎖ユニバーサルアダプターまたは第二のユニバーサルアダプターを断片化した各DNA分子の第一の末端におよび第一のユニバーサルアダプターまたは第二のユニバーサルアダプターを断片化した各DNA分子の第二の末端に連結させて、アダプター連結DNA分子の混合物を作製させる段階;
(c) 第一の二本鎖ユニバーサルアダプターの片鎖を介して固相支持体に第一の二本鎖ユニバーサルアダプターと第二の二本鎖アダプターとを含むアダプター連結DNA分子を付着させる段階;
(d) 固相支持体に付着されていないアダプター連結DNA分子を洗い流す段階;
(e) 一端のみで固相支持体に付着されたそれらのアダプター連結DNA分子を鎖分離させて、第一の一本鎖ユニバーサルアダプターを一端におよび第二の一本鎖アダプターを他端に含む複数の一本鎖DNA分子を遊離させる段階; および
(f) 一本鎖DNA分子を単離する段階。
【請求項50】
以下の段階を含む、鋳型核酸を複数の反応中心に送達するための方法:
(a) 鋳型核酸の集団を供与する段階;
(b) 各鋳型核酸を前記の集団から隔離剤までに単離して、隔離された鋳型核酸の集団を形成させる段階;
(c) 前記の隔離された鋳型核酸の集団を前記の複数の反応中心に送達する段階であって、その際に各反応中心が隔離された核酸を一つ受け取る段階。
【請求項51】
単離する段階が、鋳型核酸をビーズに付着させる段階を含む、請求項50記載の方法。
【請求項52】
単離する段階が、鋳型核酸を油中水型乳濁液の乳濁液中に封入する段階を含む、請求項50記載の方法。
【請求項53】
鋳型核酸が、核酸を結合できるビーズで封入される、請求項52記載の方法。
【請求項54】
送達する段階が、隔離された核酸を複数の反応中心に送達する段階を含み、その際に各反応中心がpicotiterプレートのウェルである、請求項50記載の方法。
【請求項55】
単離された一本鎖分子をそれぞれ個別的に固相支持体に付着させる段階をさらに含む、請求項1、2または49記載の方法。
【請求項1】
以下の段階を含む、複数の一本鎖DNA分子を含んだライブラリーをクローニングにより単離するための方法:
(a) 大きい鋳型DNA分子を断片化して複数の断片化したDNA分子を作製する段階;
(b) 第一または第二の二本鎖ユニバーサルアダプターを断片化した各DNA分子の第一の末端におよび第一または第二のユニバーサルアダプターを断片化した各DNA分子の第二の末端に付着させて、アダプター連結DNA分子の混合物を形成させる段階;
(c) それぞれが第一の一本鎖ユニバーサルアダプターと第二の一本鎖ユニバーサルアダプターとを含む複数の一本鎖DNA分子を単離して、ライブラリーを得る段階; および
(d) 複数のリアクターに一つのDNA分子が含まれるように一本鎖DNA分子をリアクターに送達し、それによってライブラリーをクローニングにより単離する段階。
【請求項2】
以下の段階を含む、複数の一本鎖DNA分子を含んだライブラリーを作製するための方法:
(a) 大きいまたは全ゲノムの鋳型DNA分子を断片化して複数の断片化したDNA分子を作製する段階;
(b) 第一の二本鎖ユニバーサルアダプターまたは第二のユニバーサルアダプターを断片化した各DNA分子の第一の末端におよび第一のユニバーサルアダプターまたは第二のユニバーサルアダプターを断片化した各DNA分子の第二の末端に連結させて、アダプター連結DNA分子の混合物を作製させる段階であって、その際に第一のユニバーサルアダプターには固相支持体に結合する部分が含まれる段階;
(c) 固相支持体に第一の二本鎖ユニバーサルアダプターを含むDNA分子を付着させる段階;
(d) 固相支持体に付着されていないアダプター連結DNA分子を除去する段階;
(e) 一端のみで固相支持体に付着されたアダプター連結DNA分子を鎖分離させて、第一の一本鎖ユニバーサルアダプターを一端におよび第二の一本鎖アダプターを他端に有する複数の一本鎖DNA分子を遊離させる段階; および
(f) 段階(e)の遊離された一本鎖DNA分子のライブラリーを固相支持体に付着したままのそれらのDNA分子から単離する段階。
【請求項3】
第一の二本鎖ユニバーサルアダプターまたは第二のユニバーサルアダプターが、連結により付着される、請求項1または2記載の方法。
【請求項4】
(f)がi) 一本鎖DNA分子をリアクターアレイ上のある位置に送達する段階; またはii) 一本鎖DNA分子を油中水型乳濁液中の液滴内に送達する段階により達成される、請求項1記載の方法。
【請求項5】
DNA修復酵素およびDNA修飾酵素を用いて、アダプター連結DNA分子の混合物における一本鎖ニックを修復する段階をさらに含む、請求項1または2記載の方法。
【請求項6】
DNA修復酵素およびDNA修飾酵素が、ポリメラーゼ、リガーゼ、キナーゼ、およびその組み合わせからなる群より選択される、請求項5記載の方法。
【請求項7】
ポリメラーゼがバチルス・ステアロサーモフィルス(Bacillus stearothermophilus)ポリメラーゼIであり、リガーゼがT4リガーゼであり、およびキナーゼがT4ポリヌクレオチドキナーゼである、請求項6記載の方法。
【請求項8】
鋳型DNAが、ゲノムDNA、cDNA、プラスミドDNA、コスミドDNA、人工染色体DNA、合成DNA、ファセミド(phasemid)DNA、およびファージミドDNAからなる群より選択される、請求項1または2記載の方法。
【請求項9】
鋳型DNAには逆転写産物が含まれる、請求項1または2記載の方法。
【請求項10】
断片化が酵素的手段、化学的手段、および機械的手段からなる群より選択される手段により行われる、請求項1または2記載の方法。
【請求項11】
酵素的手段がDNaseIである、請求項10記載の方法。
【請求項12】
機械的手段が噴霧である、請求項10記載の方法。
【請求項13】
DNase I消化が10〜37℃の温度で1〜2分間行われる、請求項11記載の方法。
【請求項14】
酵素的手段が制限酵素である、請求項10記載の方法。
【請求項15】
機械的手段がフレンチプレス、超音波処理器、HydroShear、および噴霧装置からなる群より選択される、請求項10記載の方法。
【請求項16】
断片化したDNA分子の長さが50 bp〜700 bpである、請求項1または2記載の方法。
【請求項17】
適合末端が平滑末端である、請求項1または2記載の方法。
【請求項18】
平滑末端がPfuポリメラーゼ、T4 DNAポリメラーゼおよびクレノーフラグメントからなる群より選択される酵素で生成される、請求項17記載の方法。
【請求項19】
適合末端にはAまたはT突出部が含まれる、請求項1または2記載の方法。
【請求項20】
第一または第二の二本鎖ユニバーサルアダプターには、ホスホロチオエート結合が含まれる、請求項1または2記載の方法。
【請求項21】
ビオチン部分が第一または第二の二本鎖ユニバーサルアダプターに付着される、請求項1または2記載の方法。
【請求項22】
第一または第二の二本鎖ユニバーサルアダプターが、T4 DNAリガーゼで連結される、請求項1または2記載の方法。
【請求項23】
第一の二本鎖ユニバーサルアダプターにもしくは第二の二本鎖ユニバーサルアダプターにまたはどちらの二本鎖ユニバーサルアダプターにも、識別可能なキー配列が含まれる、請求項1または2記載の方法。
【請求項24】
識別可能なキー配列の長さが3〜12ヌクレオチドである、請求項23記載の方法。
【請求項25】
識別可能なキー配列が、A、G、C、U、およびTからなる群より選択される少なくとも一つのヌクレオチドを含む、請求項23記載の方法。
【請求項26】
第一および第二の二本鎖ユニバーサルアダプターには、PCRプライミング配列および配列決定用プライマー配列が含まれる、請求項1または2記載の方法。
【請求項27】
PCRプライミング配列の長さが10〜20塩基対である、請求項26記載の方法。
【請求項28】
配列決定用プライマー配列の長さが10〜20塩基対である、請求項26記載の方法。
【請求項29】
PCRプライミング配列および配列決定用プライマー配列が重複する、請求項26記載の方法。
【請求項30】
アダプター連結DNA分子の混合物がゲル電気泳動、ろ過、サイズ排除クロマトグラフィー、およびショ糖沈降法からなる群より選択される方法により分離される、請求項1または2記載の方法。
【請求項31】
複数の一本鎖DNA分子がDNA捕捉ビーズに付着される、請求項1または2記載の方法。
【請求項32】
DNA捕捉ビーズが結合対の成分を含む、請求項31記載の方法。
【請求項33】
結合対が、アビジン/ビオチン、リガンド/受容体、抗原/抗体および相補ヌクレオチドからなる群より選択される、請求項31記載の方法。
【請求項34】
DNA捕捉ビーズが常磁性ビーズである、請求項31記載の方法。
【請求項35】
複数の一本鎖DNA分子が、低塩処理、高pH処理、および化学的変性処理からなる群より選択される処理により得られる、請求項1または2記載の方法。
【請求項36】
以下の段階を含む、固相支持体に付着された一本鎖DNAライブラリーを作製するための方法:
(a) 複数の鋳型一本鎖DNAを作製する段階;
(b) 複数の鋳型ssDNAのそれぞれを固相支持体に付着させる段階; および
(c) 鋳型一本鎖DNAが付着された固相支持体を単離する段階。
【請求項37】
以下の段階を含む、固相支持体に付着された一本鎖DNAライブラリーを作製するための方法:
(a) 大きい鋳型DNA分子を断片化して複数の断片化したDNA分子を作製する段階;
(b) 第一または第二の二本鎖ユニバーサルアダプターを断片化した各DNA分子の第一の末端におよび第一または第二のユニバーサルアダプターを断片化した各DNA分子の第二の末端に付着させて、アダプター連結DNA分子の混合物を作製する段階;
(c) 第一の一本鎖ユニバーサルアダプターと第二の一本鎖ユニバーサルアダプターとを含む一本鎖DNA分子を単離する段階; および
(d) (c)からの単離された一本鎖分子を固相支持体に付着させる段階。
【請求項38】
固相支持体がDNA捕捉ビーズである、請求項37記載の方法。
【請求項39】
DNAが、ゲノムDNA、cDNA、プラスミドDNA、コスミドDNA、人工染色体DNA、合成DNA、ファセミドDNA、およびファージミドDNAからなる群より選択される、請求項37記載の方法。
【請求項40】
DNAが結合対により固相支持体に付着される、請求項37記載の方法。
【請求項41】
結合対が、アビジン/ビオチン、リガンド/受容体、抗原/抗体および相補ヌクレオチドからなる群より選択される、請求項40記載の方法。
【請求項42】
請求項37の方法により作製される可動性の固相支持体のライブラリー。
【請求項43】
第一のアダプターと、鋳型DNAの断片と、第二のアダプターとを含む核酸分子であって、その際に第一のアダプターと第二のアダプターそれぞれが、配列決定用プライマーと、PCRプラマーと、識別可能なキー配列とを含み、およびその際に第一のアダプターと第二のアダプターが、解離した場合に、ストリンジェントなハイブリダイゼーション条件の下で相互に交差ハイブリダイズしない核酸分子。
【請求項44】
PCRプライマーの長さが10〜20塩基対である、請求項43記載の核酸分子。
【請求項45】
配列決定用プライマーの長さが10〜20塩基対である、請求項43記載の核酸分子。
【請求項46】
識別可能なキー配列の長さが3〜12塩基対である、請求項43記載の核酸分子。
【請求項47】
鋳型DNAがゲノムDNA、cDNA、プラスミドDNA、コスミドDNA、人工染色体DNA、合成DNA、ファセミドDNA、およびファージミドDNAからなる群より選択される、請求項43記載の核酸分子。
【請求項48】
核酸分子が、解離した場合に、解離した鋳型DNAに対する交差ハイブリダイゼーションが最小である、請求項43記載の核酸分子。
【請求項49】
以下の段階を含む、一本鎖DNA分子を調製するための方法:
(a) 大きいまたは全ゲノムの鋳型DNA分子を断片化して複数の断片化したDNA分子を作製する段階;
(b) 第一の二本鎖ユニバーサルアダプターまたは第二のユニバーサルアダプターを断片化した各DNA分子の第一の末端におよび第一のユニバーサルアダプターまたは第二のユニバーサルアダプターを断片化した各DNA分子の第二の末端に連結させて、アダプター連結DNA分子の混合物を作製させる段階;
(c) 第一の二本鎖ユニバーサルアダプターの片鎖を介して固相支持体に第一の二本鎖ユニバーサルアダプターと第二の二本鎖アダプターとを含むアダプター連結DNA分子を付着させる段階;
(d) 固相支持体に付着されていないアダプター連結DNA分子を洗い流す段階;
(e) 一端のみで固相支持体に付着されたそれらのアダプター連結DNA分子を鎖分離させて、第一の一本鎖ユニバーサルアダプターを一端におよび第二の一本鎖アダプターを他端に含む複数の一本鎖DNA分子を遊離させる段階; および
(f) 一本鎖DNA分子を単離する段階。
【請求項50】
以下の段階を含む、鋳型核酸を複数の反応中心に送達するための方法:
(a) 鋳型核酸の集団を供与する段階;
(b) 各鋳型核酸を前記の集団から隔離剤までに単離して、隔離された鋳型核酸の集団を形成させる段階;
(c) 前記の隔離された鋳型核酸の集団を前記の複数の反応中心に送達する段階であって、その際に各反応中心が隔離された核酸を一つ受け取る段階。
【請求項51】
単離する段階が、鋳型核酸をビーズに付着させる段階を含む、請求項50記載の方法。
【請求項52】
単離する段階が、鋳型核酸を油中水型乳濁液の乳濁液中に封入する段階を含む、請求項50記載の方法。
【請求項53】
鋳型核酸が、核酸を結合できるビーズで封入される、請求項52記載の方法。
【請求項54】
送達する段階が、隔離された核酸を複数の反応中心に送達する段階を含み、その際に各反応中心がpicotiterプレートのウェルである、請求項50記載の方法。
【請求項55】
単離された一本鎖分子をそれぞれ個別的に固相支持体に付着させる段階をさらに含む、請求項1、2または49記載の方法。
【図1】

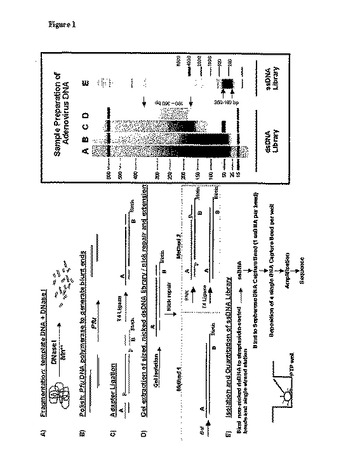

【図3】
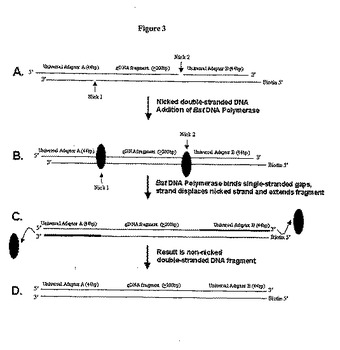
【図4】
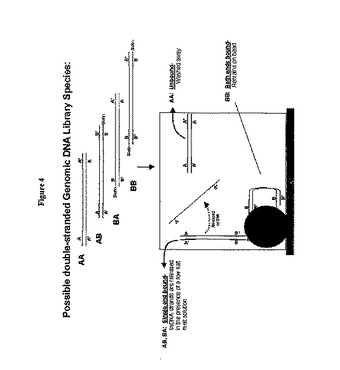

【図6】
【図7】

【図8】
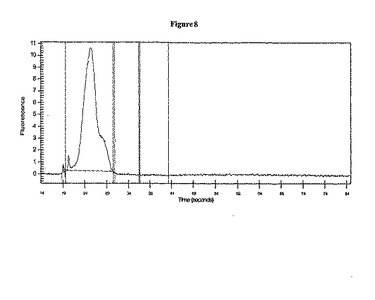
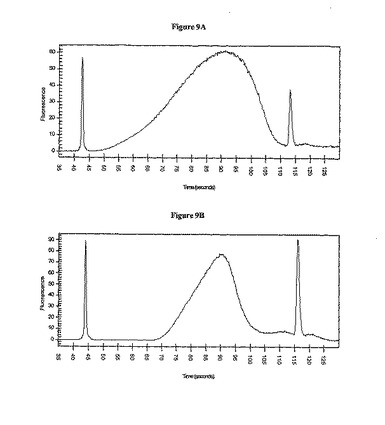
【図10】
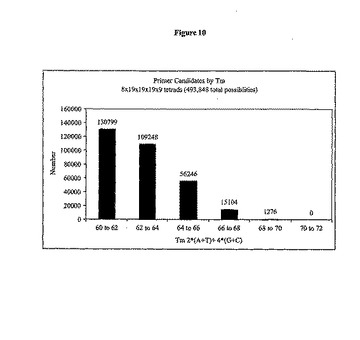
【図3】
【図4】
【図6】
【図7】
【図8】
【図10】
【公表番号】特表2007−525151(P2007−525151A)
【公表日】平成19年9月6日(2007.9.6)
【国際特許分類】
【出願番号】特願2006−503163(P2006−503163)
【出願日】平成16年1月28日(2004.1.28)
【国際出願番号】PCT/US2004/002571
【国際公開番号】WO2004/070007
【国際公開日】平成16年8月19日(2004.8.19)
【出願人】(505286730)454 コーポレーション (9)
【Fターム(参考)】
【公表日】平成19年9月6日(2007.9.6)
【国際特許分類】
【出願日】平成16年1月28日(2004.1.28)
【国際出願番号】PCT/US2004/002571
【国際公開番号】WO2004/070007
【国際公開日】平成16年8月19日(2004.8.19)
【出願人】(505286730)454 コーポレーション (9)
【Fターム(参考)】
[ Back to top ]