
一酸化炭素同位体を用いる鈴木カップリング反応によるケトン及びアミンの炭素同位体の標識合成方法
炭素同位体で標識した一酸化炭素を用いる鈴木カップリング反応を介したカルボニル化により炭素同位体で標識したケトン及びアミンを合成する方法及び試薬が提供される。得られた炭素同位体で標識したケトン及びアミンは、特に陽電子放出断層撮影(PET)に使用される放射線医薬品として有用である。PET研究用の関連キットも提供される。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、陽電子放出核種で標識した生物活性化合物を含む、診断剤及び放射線診断剤に関する。本発明は更に、標識合成における一酸化炭素同位体の使用方法に関する。より具体的には、本発明は、最初の[11C]二酸化炭素混合気体から[11C]一酸化炭素の濃度が高い混合気体を製造し、鈴木カップリング反応を介したカルボニル化によるケトン及びアミンの標識合成に製造された混合気体を使用する方法に関する。本発明の放射標識されたケトン及びアミンは、具体的には陽電子放出断層撮影(PET)に使用される放射線医薬品として有用である。
【背景技術】
【0002】
短寿命の陽電子放出核種(例えば11C、t1/2=20.3分)で標識したトレーサーは、各種の非侵襲的in vivo研究に陽電子放出断層撮影(PET)との組合せで頻繁に使用される。標識物質の放射能、短い半減期、及びμM以下である量が理由で、これらのトレーサーの製造には特別な合成手順が必要である。これらの手順の考案に重要な部分を占めるのが、新規の11C標識前駆体の開発及び取扱いである。これは新種の化合物を標識するのに重要であるばかりでなく、異なる位置で所与の化合物を標識する可能性を高めるためにも重要である。
【0003】
過去20年間で、一酸化炭素を用いるカルボニル化反応は著しい発展を遂げた。パラジウム触媒を用いるカルボニル化カップリング反応などの方法の近年の発展は、一酸化炭素を異なるカルボニル化合物に変換する穏健で効率的な手段を提供してきた。
【0004】
生物活性物質がカルボニル基、又はカルボニル基から誘導可能な官能基をしばしば含むため、[11C]一酸化炭素を用いるカルボニル化反応はPETトレーサー合成において最も有益である。この合成は大部分の官能基に対して許容的であるため、カルボニル化段階で錯体構築ブロックを集積して目標化合物を得ることができる。これは、反応順序において非標識基質を標識前駆体とできるだけ遅く組み合わせて合成時間を減少させ、それにより未補正の放射化学収量を最適化しなければならないPETトレーサー合成で特に有益である。
【0005】
化合物を11Cで標識する場合、比放射能を最大化することが通常は重要である。これを実現するには、同位体希釈及び合成時間を最小限に抑える必要がある。[11C]二酸化炭素を標識反応に使用する場合、空気中の二酸化炭素からの同位体希釈が重要なことがある。一酸化炭素の反応性が低く、空気中濃度が低い(CO2の3.4×104ppmに対して0.1ppm)ため、[11C]一酸化炭素を使用することでこの問題は減少する。
【0006】
亜鉛、木炭又はモリブデンなどの還元剤を含む加熱カラムを用いた、[11C]二酸化炭素からの[11C]一酸化炭素の合成が、いくつかの刊行物に既に記載されている。[11C]一酸化炭素はヒトにおけるトレーサー実験に適用された最初の11C標識化合物の1つであったが、最近までPETトレーサーの製造には実際に使用されていなかった。この1つの理由は、[11C]一酸化炭素の溶解度が低く、反応速度が比較的遅いことにより、反応媒体中での捕捉効率が低くなるためである。[11C]ヨウ化メチル、[11C]シアン化水素又は[11C]二酸化炭素などの前駆体を用いる一般的手順は、気相の放射能を移動し、気体流を反応媒体に通すことにより放射能を捕捉することである。最近まで、標識合成で[11C]一酸化炭素を取扱う上でこれが唯一実施可能な手段であった。このアプローチでは、[11C]一酸化炭素を用いる標識合成の主要部分は、非常に低い収量を与えるか、完全に失敗することが予測され得る。
【0007】
高圧技術(300バール超)を用いる実用的に有益な11C標識合成の例はわずかしかない。原則として、高圧は、反応速度を高め、試薬の量を最小限に抑えるために利用することができる。このアプローチの1つの問題は、どのようにして標識前駆体を小さな高圧反応器に閉じ込めるかである。もう1つの問題は、反応器の構成である。一般的なカラム型の反応器を用いる場合(即ち各端部にチューブを取り付けた円筒)、気相は実際に、加圧により液相から効率的に排除される。その理由は、気相が圧縮された形態で、バルク量の液体試薬から取り付けられたチューブに逃れるためである。
【0008】
コールドトラップ技術は、特に[11C]一酸化炭素の場合、11C標識前駆体の取扱いに広く使用されている。しかし、この手順は単一の段階でしか行われず、標識化合物は常に、コールドトラップの加熱と同時に連続気体流中に放出されていた。更に、標識化合物を捕捉するのに使用される材料の体積は、標識化合物が移動された系に比較して大きかった。したがって、標識化合物のラジカル濃縮及び合成システムの小型化にこの技術を用いるというオプションは検討されなかった。このことは、11C標識化合物の量が通常、20〜60nmolの範囲であるという事実に鑑みて特に注目に値する。
【0009】
[11C]一酸化炭素の製造及び使用に関する近年の技術的発展により、この化合物が標識合成で有用になった。国際公開第02/102711号には、最初の二酸化炭素同位体混合気体から一酸化炭素同位体の濃度が高い混合気体を製造し、それを使用するためのシステム及び方法が記載されている。[11C]一酸化炭素は、サイクロトロンで製造された[11C]二酸化炭素から高い放射化学収量で得ることができ、高い比放射能を有する目標化合物を得るのに使用することができる。この反応器は、上記の問題点を克服しており、パラジウム又はセレンを媒介した反応における[11C]一酸化炭素を用いた11C標識化合物の合成に有用である。このような方法で、様々なカルボニル化合物を標識することができる(Kilhlberg,T.;Langstrom,B.J.,Org.Chem.64,1999,9201−9205;Kihlberg,T.,Karimi,F.,Langstrom,B.,J.Org.Chem.67,2002,3687−3692)。
【0010】
このような標識カルボニル化合物は、PETで使用される多くの薬学的に重要なトレーサーの合成に道を開いた。ケトンが生物学的に重要な分子の構築ブロックとして広く使用され、炭素同位体を用いた標識により反応前駆体又はPETトレーサーとして使用されることは注目に値する。多くの医薬品の生物活性はアミン官能基に依存するため、アミンは重要なクラスの化合物である。したがって、炭素同位体で標識したアミンは、重要で有用なPETトレーサーとして使用することができる。
【0011】
求電子剤、一酸化炭素及び有機水素化スズ(スティレカップリング)又は有機ホウ素化合物(鈴木カップリング)を用いた遷移金属を触媒とするカルボニル化は、ケトンの合成に多く使用されるアプローチである。11C標識化学反応では、スティレカルボニル化は多くの研究が行われてきたが、鈴木カルボニル化が用いられるのは稀であった。例えば、スティレカップリングは一連の11C標識ケトンの合成に使用されてきた(Lindstrom,P.,Kihlberg,T.,Langstrom,B.,J.Chem.Soc.Perkin Trans.1,1997,2701−2706)。しかし、このようなアプローチを使用する上で1つの欠点は、毒性の高い有機水素化スズ化合物の使用である。
【0012】
これとは別に、有機ホウ素及びヨードベンゼンを用いた11C標識ベンゾフェノンの合成(鈴木カップリング)が報告された(Zeisler,S.,Nader,M.Theobald,A.and Oberdorfer,F.,Appl,Radiat.Isot.1997,48,1091−1095)。しかし、この報告で使用された[11C]一酸化炭素の製造方法は、上記の低い比放射能という欠点がある。したがって、鈴木カップリング反応で使用される比放射能を増大させることが必要である。
【0013】
11C標識アミンの効率的な合成方法も未検討である。以前、カルボン酸及び[11C]ハロゲン化マグネシウムカルボキシレートの還元アミン化による11C標識アミンの合成に関する1つの報告が刊行された(Perrio−Huard,C.et al,J.Che.Soc.Perkin Trans.1,2000,311−316)。但し、このような方法では[11C]二酸化炭素が標識前駆体として使用される。そのため、このような方法は最適ではない。
【0014】
したがって、[11C]一酸化炭素を用いて、反応前駆体又はPETトレーサーとしての炭素同位体で標識したケトン及びアミンを合成する、新規の改善された方法が必要である。この方法は、有用なPETトレーサーの調製において[11C]一酸化炭素の有用性を更に高めることになろう。
【0015】
本明細書での参考文献の議論又は引用は、そのような参考文献が本発明の先行技術であることを承認するものではない。
【特許文献1】国際公開第02/102711号
【非特許文献1】Kilhlberg,T.;Langstrom,B.J.,Org.Chem.64,1999,9201−9205
【非特許文献2】Kihlberg,T.,Karimi,F.,Langstrom,B.,J.Org.Chem.67,2002,3687−3692
【非特許文献3】Lindstrom,P.,Kihlberg,T.,Langstrom,B.,J.Chem.Soc.Perkin Trans.1,1997,2701−2706
【非特許文献4】Zeisler,S.,Nader,M.Theobald,A.and Oberdorfer,F.,Appl,Radiat.Isot.1997,48,1091−1095
【非特許文献5】Perrio−Huard,C.et al,J.Che.Soc.Perkin Trans.1,2000,311−316
【発明の開示】
【課題を解決するための手段】
【0016】
本発明は、
(a)その底面に液体入口及び気体入口を有する高圧反応室を用意する段階と、
(b)遷移金属錯体と混合された、トリフレート、ボロン酸を含む標識すべき溶液を用意する段階と、
(c)気体入口を通じて反応室に一酸化炭素同位体の濃度が高い混合気体を導入する段階と、
(d)液体入口を通じて反応室に、遷移金属錯体と混合された前記溶液を高圧で導入する段階と、
(e)標識合成が行われている所定時間待機する段階と、
(f)反応室から標識ケトンを除去する段階とを含む、鈴木カップリング反応を介したケトンの標識合成方法を提供する。
【0017】
本発明はまた、本発明の方法により合成される炭素同位体で標識したケトンを提供する。
【0018】
本発明は更に、アミン、TiCl4及びNaBH3CNを用いて本発明の方法により得られる炭素同位体で標識したケトンを還元アミン化する段階を含む、アミンの標識合成方法を提供する。
【0019】
更に他の実施形態では、本発明はまた、本発明の方法により合成される炭素同位体で標識したアミンを提供する。更に他の実施形態では、本発明は、このような[11C]標識ケトン及び/又はアミンを含む、PETトレーサーとして使用されるキットを提供する。
【発明を実施するための最良の形態】
【0020】
本発明の1つの目的は、先行技術の装置の欠点を克服する標識合成における、一酸化炭素同位体を製造するための方法及びシステム、並びに一酸化炭素同位体の使用を提供することにある。これは本発明に記載の方法及びシステムにより実現される。
【0021】
このような方法及びシステムの1つの利点は、一酸化炭素同位体を標識生成物に、ほぼ定量的に転換できることである。
【0022】
本方法及びシステムには他にもいくつかの利点がある。高圧技術により、ジエチルエーテルなどの低沸点溶媒を高温(例えば200℃)で使用することが可能になる。気体の分散を妨げる材料からなる閉鎖系の使用により、感受性の高い化合物の安定性が増大し、適正製造規範(GMP)の点でも有利になることがある。
【0023】
得られる標識化合物が高濃度であり、合成システムの小型化により自動化、迅速な合成及び精製、並びに同位体希釈を最小限に抑えることによる比放射能の最適化が促進されるという、更なる他の利点も得られる。
【0024】
本発明で例示するように、最も重要なのは、完全に新規の合成の可能性が開かれることである。
【0025】
以下、図面を参照しながら本発明の実施形態を説明する。
【0026】
本出願を通じて使用される炭素同位体という用語は、11Cを意味することが好ましいが、所望であれば11Cを13C及び14Cなど他の炭素同位体に置き換えることができると理解されたい。
【0027】
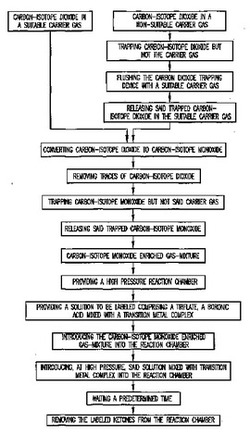
図1は、第1に一酸化炭素同位体の濃度が高い混合気体の製造、第2に標識合成手順を含む本発明の方法に関するフローチャートを示す図である。より詳しくは、本方法の製造部分は以下の段階を含む。
【0028】
・以下に詳述する種類の好適なキャリアガス中の二酸化炭素同位体を用意する段階。
【0029】
・以下に詳述する反応装置内に前記混合気体を導入することで二酸化炭素同位体を一酸化炭素同位体に転換する段階。
【0030】
・一酸化炭素同位体でもキャリアガスでもなく二酸化炭素同位体が捕捉される二酸化炭素除去装置内に転換された混合気体を注ぎ込むことで微量の二酸化炭素同位体を除去する段階。二酸化炭素除去装置は以下に詳述する。
【0031】
・前記キャリアガスではなく一酸化炭素同位体が捕捉される一酸化炭素捕捉装置内に一酸化炭素同位体を捕捉する段階。一酸化炭素捕捉装置は以下に詳述する。
【0032】
・前記捕捉装置から前記捕捉された一酸化炭素同位体を放出することにより、ある体積の一酸化炭素同位体の濃度が高い混合気体を得る段階。
【0033】
最初の二酸化炭素同位体の混合気体が、二酸化炭素同位体、及び窒素などの、同様な分子特性等が理由で一酸化炭素のキャリアガスとして適さない第1のキャリアガスで構成される場合、製造段階は、最初の二酸化炭素同位体の混合気体のキャリアガスを変更する段階を更に含んでいてもよい。より詳しくは、He、Arなどの好適な第2のキャリアガス中の二酸化炭素同位体を用意する段階は、以下の段階を含む。
【0034】
・前記第1のキャリアガスではなく二酸化炭素同位体が捕捉される二酸化炭素捕捉装置内に最初の二酸化炭素同位体の混合気体を流し込む段階。二酸化炭素捕捉装置は以下に詳述する。
【0035】
・前記二酸化炭素捕捉装置を前記第2のキャリアガスで洗い流して前記第1のキャリアガスの残りを除去する段階。
【0036】
・前記好適な第2のキャリアガス中の前記捕捉された二酸化炭素同位体を放出する段階。
【0037】
製造段階に続くことができる標識合成段階では、製造された一酸化炭素同位体の濃度が高い混合気体を標識反応物として利用する。より詳しくは、標識合成段階は以下の段階を含む。
【0038】
・その底面に液体試薬入口及び標識反応物入口を有する高圧反応室を用意する段階。反応室は以下に詳述する。
【0039】
・遷移金属錯体と混合された、トリフレート、ボロン酸を含む標識すべき溶液を用意する段階。
【0040】
・標識反応物入口を通じて反応室に一酸化炭素同位体の濃度が高い混合気体を導入する段階。
【0041】
・液体試薬入口を通じて反応室に、遷移金属錯体と混合された前記溶液を高圧で導入する段階。
【0042】
・標識合成が行われている所定時間待機する段階。
【0043】
・反応室から標識化合物溶液を除去する段階。
【0044】
反応を促進するために、標識すべき物質は臭化リチウムを更に含んでいてもよい。遷移金属錯体はパラジウム錯体であることが好ましい。
【0045】
所定時間待機する段階は、標識合成が促進されるように反応室の温度を調整する段階を更に含んでいてもよい。
【0046】
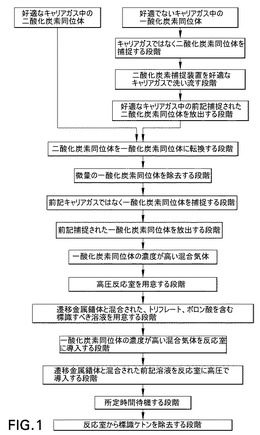
図2は、本発明の[11C]二酸化炭素製造及び標識システムを模式的に示す図である。このシステムは3つの主要ブロックで構成され、各ブロックは製造及び標識方法の3つの主要段階の1つを取扱う。
【0047】
ブロックAは、最初の二酸化炭素同位体の混合気体が、二酸化炭素同位体、及び一酸化炭素のキャリアガスとして適さない第1のキャリアガスで構成される場合、最初の二酸化炭素同位体の混合気体のキャリアガスを変更するために使用される。
【0048】
ブロックBは、二酸化炭素同位体を一酸化炭素同位体に転換し、転換された一酸化炭素同位体の混合気体を精製及び濃縮するために使用される。
【0049】
ブロックCは、一酸化炭素同位体の標識合成を行うために使用される。
【0050】
二酸化炭素同位体が通常、17MeVプロトン照射された窒素及び0.1%酸素を含む目標ガス中で14N(p,α)11C反応を用いて生成されることにより、最初の二酸化炭素同位体の混合気体が窒素をキャリアガスとして含むという事実があるため、ブロックAは通常必要である。しかし、窒素は分子特性において一酸化炭素に対してある種の類似性を示すため、例えば捕捉装置等において互いに分離することが困難であり、それによりこのような混合気体中での一酸化炭素同位体の濃度を上昇させることが困難である。代わりの好適なキャリアガスとしてはヘリウム、アルゴン等が挙げられる。外部の系がブロックB及びCに必要な気体圧力に耐容性を示さない場合、キャリアガスの圧力(例えば1〜4バール)を変更するために使用することもできる。一代替実施形態では、最初の二酸化炭素同位体の混合気体は、二酸化炭素同位体、及び一酸化炭素のキャリアガスとして好適な第1のキャリアガスで構成され、それによりブロックAを簡略化するか、排除さえすることができる。
【0051】
好ましい一実施形態(図2)によれば、ブロックAは、第1のバルブV1、二酸化炭素捕捉装置8及び第2のバルブV2で構成される。
【0052】
第1のバルブV1は、最初の二酸化炭素同位体の混合気体源12に接続される二酸化炭素入口10、ヘリウム、アルゴン等の好適なキャリアガス源16に接続されるキャリアガス入口14を有する。第1のバルブV1は、第2のバルブV2の第1の入口20に接続される第1の出口18、及び二酸化炭素捕捉装置8に接続される第2の出口22を更に有する。バルブV1は2つのモードA、Bで運転することができる。モードAでは、二酸化炭素入口10は第1の出口18に接続され、キャリアガス入口14は第2の出口22に接続される。モードBでは、二酸化炭素入口10は第2の出口22に接続され、キャリアガス入口14は第1の出口18に接続される。
【0053】
第2のバルブV2は、第1の入口20以外に、二酸化炭素捕捉装置8に接続される第2の入口24を有する。第2のバルブV2は、廃棄物出口26、及びブロックBの生成物入口30に接続される生成物出口28を更に有する。バルブV2は2つのモードA、Bで運転することができる。モードAでは、第1の入口20は廃棄物出口26に接続され、第2の入口24は生成物出口28に接続される。モードBでは、第1の入口20は生成物出口28に接続され、第2の入口24は廃棄物出口26に接続される。
【0054】
二酸化炭素捕捉装置8は、前記第1のキャリアガスではなく二酸化炭素を捕捉する装置であり、捕捉された二酸化炭素はその後、制御下で放出することができる。これは、冷却状態(例えば液体窒素中−196℃、又は液体アルゴン中−186℃)で二酸化炭素を選択的に捕捉し、加熱状態(例えば+50℃)で捕捉された二酸化炭素を放出する材料を含むカラムなどのコールドトラップを使用することで実現することが好ましい(本明細書では、「コールドトラップ」という表現は低温工学の使用に限定されない。したがって、室温で対象化合物を捕捉し、より高温でそれを放出する材料が含まれる)。好適な材料としては例えば、シリカ及びPorapacQ(登録商標)が挙げられる。シリカカラム又はポラパックカラムの捕捉挙動は、双極子−双極子相互作用、又は場合によってはファンデルワールス相互作用に関連している。前記カラム8は、捕捉材料の体積が二酸化炭素同位体を効率的に捕捉(95%超)する程度に大きく、捕捉された二酸化炭素のブロックBへの移動を遅延させない程度に小さくなるように形成されていることが好ましい。PorapacQ(登録商標)、窒素流量100ml/分の場合、体積は50〜150μlでなければならない。更に、二酸化炭素捕捉装置8の冷却及び加熱は、それが自動化プロセスとして、例えば自動的にカラムを液体窒素の位置まで引き下げ、そこから加熱装置に移動させることで行われるように設定できる。
【0055】
図2の好ましい実施形態によれば、ブロックBは、二酸化炭素同位体が一酸化炭素同位体に転換される反応装置32、二酸化炭素除去装置34、チェックバルブ36、及び一酸化炭素捕捉装置38で構成され、これらは直列に接続される。
【0056】
好ましい実施形態では、反応装置32は、正しい温度間隔に加熱された際に、二酸化炭素同位体を一酸化炭素同位体に転換する材料を含む反応炉である。二酸化炭素を一酸化炭素に変換する能力を有する様々な異なる材料、例えば鉛若しくはモリブデン又は同様の還元特性を有する任意の他の元素若しくは化合物を使用することができる。反応装置32が亜鉛炉の場合、350〜400℃に加熱しなければならず、温度を高精度で制御することが重要である。亜鉛の融点は420℃であり、亜鉛炉は、温度が410℃を超えた際に、おそらくは表面特性の変化により、二酸化炭素を一酸化炭素に変換する能力を急速に失う。この材料は、放射能を二酸化炭素捕捉装置8から後続の一酸化炭素捕捉装置38に移動するのに必要な時間を最小限に抑えるような少量を使用できるように、その量に対して効率的でなければならない。炉中の材料の量は、炉中での実用的な寿命(少なくとも数日)を確保する程度に多くなければならない。亜鉛粒質物の場合、体積は100〜1000μlでなければならない。
【0057】
二酸化炭素除去装置34は、反応装置32を出る混合気体から微量の二酸化炭素同位体を除去するために使用される。二酸化炭素除去装置34では、一酸化炭素同位体でもキャリアガスでもなく、二酸化炭素同位体が捕捉される。二酸化炭素除去装置34は、ascarite(登録商標)(即ちシリカで担持された水酸化ナトリウム)を含むカラムで構成することができる。一酸化炭素同位体が通過する際に、反応装置32で反応しなかった二酸化炭素同位体がこのカラム内に捕捉される(水酸化ナトリウムと反応して炭酸ナトリウムになる)。反応装置32が適切に機能していないことを高い値が示すと、二酸化炭素除去装置34の放射能が監視される。
【0058】
二酸化炭素捕捉装置8と同様に、一酸化炭素捕捉装置38は捕捉及び放出状態を有する。捕捉状態では、前記キャリアガスではなく一酸化炭素同位体が選択的に捕捉され、放出状態では、前記捕捉された一酸化炭素同位体が制御下で放出される。これは、モレキュラシーブなどの、シリカ又は同様の特性を有する材料を含むカラムなどのコールドトラップを用いて実現することが好ましい。このようなコールドトラップは、−100℃未満の冷却状態、例えば液体窒素中−196℃又は液体アルゴン中−186℃で一酸化炭素を選択的に捕捉し、加熱状態(例えば+50℃)で捕捉された一酸化炭素を放出する。シリカカラムの捕捉挙動は、双極子−双極子相互作用、又は場合によってはファンデルワール相互作用に関連している。放射能を運ぶヘリウムが窒素を含む場合、シリカカラムの一酸化炭素同位体を捕捉する能力が減少する。これは、窒素の物性が一酸化炭素と同様であるため、シリカ上の捕捉部位をめぐって窒素が一酸化炭素と競合するためである。
【0059】
図2の好ましい実施形態によれば、ブロックCは、第1及び第2の反応室バルブV3及びV4、前記反応室50、試薬バルブV5、注入ループ70、並びに溶媒バルブV6で構成される。
【0060】
第1の反応室バルブV3は、一酸化炭素捕捉装置38に接続される混合気体入口40、停止位置42、回収出口44、廃棄物出口46、及び反応室50の気体入口52に接続される反応室接続口48を有する。第1の反応室バルブV3は、4つの運転モードA〜Dを有する。反応室接続口48は、モードAでは混合気体入口40に接続され、モードBでは停止位置42に接続され、モードCでは回収出口44に接続され、モードDでは廃棄物出口46に接続される。
【0061】
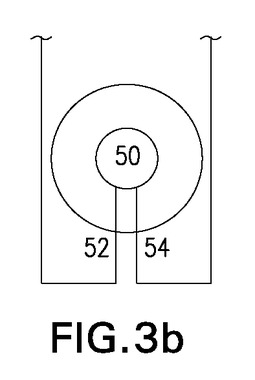
反応室50(マイクロオートクレーブ)は、気体入口52及び液体入口54を有し、これらは反応室の底面に端部を置くように配置される。気体入口52は、標識が終了した後に生成物出口として使用してもよい。運転中、一酸化炭素同位体の濃度が高い混合気体が気体入口52を通じて反応室50に導入された後、遷移金属錯体により高圧で標識すべき溶液が液体入口54を通じて反応室50に入る。図3a及び3bは、2つの好ましい反応室50の断面の模式図を示す。図3aはかなり製造が容易な円筒状反応室である一方、図3bの球状反応室は、反応室の表面積と体積の比が更に抑制されるため、最も好ましい実施形態である。表面積と体積の比を最小限に抑えることで、標識生成物の回収が最適化され、表面材料との反応の可能性が最小限に抑えられる。反応室50の「ダイビングベル構造」のため、気体入口52と液体入口54の両方が液体で満たされ、反応室50が下部から上方に向けて満たされる。一酸化炭素同位体を含む気体体積はこうして捕捉され、反応混合物と十分に接触する。液体の最終圧力が元々の気体圧力の約80倍であるため、気体の一般法則によれば、最終気体体積は液体体積の2%未満となる。こうして擬似一相系が得られる。本出願では、「擬似一相系」という用語は、200バールを超える圧力で96%を超える液体及び4%未満の気体を含む、表面積と体積の比が小さい閉じた体積を意味する。大部分の合成では、気相から液相への一酸化炭素の移動は、おそらく律速段階ではない。標識が終了した後、標識された体積はほぼ定量的に、反応室から内部圧力により気体入口/生成物出口52及び位置Cの第1の反応室バルブV3を通じて移動する。
【0062】
第2の反応室バルブV4は、反応室接続口56、廃棄物出口58、及び試薬入口60を有する。第2の反応室バルブV4は、2つの運転モードA及びBを有する。反応室接続口56は、モードAでは廃棄物入口58に接続され、モードBでは試薬入口60に接続される。
【0063】
試薬バルブV5は、第2の反応室バルブV4の試薬入口60に接続される試薬出口62、それらの間に注入ループ70が接続される注入ループ入口64及び出口66、廃棄物出口68、試薬源に接続される試薬入口71、並びに溶媒入口72を有する。試薬バルブV5は、2つの運転モードA及びBを有する。モードAでは、試薬入口71が注入ループ入口64に接続され、注入ループ出口66が廃棄物出口68に接続され、それにより試薬を注入ループ70に供給することができる。モードBでは、溶媒入口72が注入ループ入口64に接続され、注入ループ出口66が試薬出口62に接続され、それにより、高圧が溶媒入口72に加えられた際に、注入ループ70内に保存されている試薬を第2の反応室バルブV4を通じて反応室50に押し出すことができる。
【0064】
溶媒バルブV6は、試薬バルブV5の溶媒入口72に接続される溶媒出口74、停止位置76、廃棄物出口78、及び、溶媒供給HPLCポンプ(高速液体クロマトグラフィー)又は0〜10ml/分、最大400バールの圧力で有機溶媒を供給可能な任意の液体ポンプ(図示せず)に接続される溶媒入口80を有する。溶媒バルブV6は、2つの運転モードA及びBを有する。モードAでは、溶媒出口74が停止位置76に接続され、溶媒入口80が廃棄物出口78に接続される。モードBでは、溶媒出口74が溶媒入口80に接続され、それにより溶媒をHPLCポンプを用いて高圧でシステムに供給することができる。
【0065】
一酸化炭素捕捉装置38のシリカの体積が小さいことを除けば、二酸化炭素捕捉装置8及びすべての関連する先行技術との重大な相違は、二酸化炭素の放出に用いる手順にある。一酸化炭素を一酸化炭素捕捉装置8に捕捉した後、バルブV3を位置AからBに変えることで一酸化炭素捕捉装置38からの流れが停止し、設定された供給気体圧力(3〜5バール)まで一酸化炭素捕捉装置38の気体圧力が上昇する。次いで、一酸化炭素捕捉装置38を加熱して、キャリアガス中の一酸化炭素の体積を著しく膨張させることなくシリカ表面から一酸化炭素を放出する。バルブV4を位置AからBに変えた後、バルブV3を位置BからAに変える。この場合、一酸化炭素は急速且つほぼ定量的に、明確に定義されたマイクロプラグで反応室50に移動する。マイクロプラグは、対象物質(例えば1〜20μL)を含む、反応室50の体積の10%未満の気体体積として定義される。閉じられた出口を有する小さな反応室50へ効率的に物質移動を行うこの独自の方法は、以下の条件が必要である。
【0066】
・以下に定義するマイクロカラム38を使用しなければならない。捕捉材料(例えばシリカ)の体積は、一酸化炭素同位体を効率的に(95%超)捕捉できる程度に大きくなければならず、一酸化炭素同位体の濃度を最大限にできる程度に小さくなければならない(後続の反応器50の体積の1%未満)。シリカ及び反応室50体積200μlの場合、シリカ体積は0.1〜2μlでなければならない。
【0067】
・シリカカラムと反応室50を接続するチューブ及びバルブのデッドボリュームを最小限に抑えなければならない(マイクロオートクレーブ体積の10%未満)。
【0068】
・キャリアガスの圧力を移動前の反応室50中圧力(1気圧)の3〜5倍にしなければならない。
【0069】
具体的な好ましい一実施形態の仕様では、材料及び成分を以下のように選択する。Valco(登録商標)、Reodyne(登録商標)又はCheminert(登録商標)の高圧バルブを使用する。外径1/16インチのステンレスチューブを使用する。但し、ポラパックカラム8、シリカカラム38、及び反応室50への接続部には、並進運動を促進するため外径1/32インチのステンレスチューブを使用する。V1、V2及びV3の接続部の内径は0.2〜1mmでなければならない。この要件は、内径が、システムを通じてHeの最適流量(2〜50ml/分)が得られる可能性を妨げない程度に大きく、ポラパックカラム8からシリカカラム38への放射能の移動に必要な時間を長くしない程度に小さくなければならないということである。V3とオートクレーブの接続部のデッドボリュームは最小限に抑えなければならない(オートクレーブ体積の10%未満)。接続部の内径(0.05〜1mm)は最適なHe流量(2〜50ml/分)を可能にする程度に大きくなければならない。V4とV5の接続部のデッドボリュームはオートクレーブ体積の10%未満でなければならない。
【0070】
カラム8がポラパックカラムの場合、PorapacQ(登録商標)で充填され、ステンレススクリーンを取り付けたステンレスチューブ(外径=1/8インチ、内径=2mm、長さ=20mm)で構成されることが好ましい。シリカカラム38は、端部に空洞(深さ=1mm、高さ=1mm、体積=0.8μl)を有するステンレスチューブ(外径=1/16インチ、内径=0.1mm)で構成されることが好ましい。空洞は、GC固定相型のシリカ粉末(100/80メッシュ)で充填されている。カラムの端部はステンレススクリーンに取り付けられる。
【0071】
様々な異なる材料が捕捉装置に使用できることに留意されたい。GC材料を選ぶ場合、基準は、良好な遅延、並びに二酸化炭素及び一酸化炭素の各々に対する良好なピーク形状でなければならない。後者は、放射能の最適な回収を確実にする。
【0072】
上記に例示したシステムを用いる炭素同位体の製造方法を以下に詳述する。
【0073】
システムの準備は段階1〜5により行う。
【0074】
1.V1を位置Aに、V2を位置Aに、V3を位置Aに、V4を位置Aにし、ヘリウム流の最大圧力を5バールにする。この設定により、ヘリウム流は、[11C]二酸化炭素捕捉カラム、亜鉛炉、[11C]一酸化炭素捕捉カラム、反応室50を通ってV4から出ていく。システムが調節され、反応室50から溶媒が除去されて、ヘリウムがシステムを10ml/分以上で流れることができることが確認可能になる。
【0075】
2.亜鉛炉を作動させ、400℃に設定する。
【0076】
3.[11C]二酸化炭素及び[11C]一酸化炭素捕捉カラムを液体窒素で冷却する。−196℃で、ポラパック及びシリカカラムは、二酸化炭素同位体及び一酸化炭素同位体を各々効率的に捕捉する。
【0077】
4.V5を位置A(充填)にする。V5に取り付けた注入ループ(250μl)に反応混合物を充填する。
【0078】
5.HPLCポンプを、新たに蒸留したTHF(又は他の高品質溶媒)入りのフラスコに取り付け、呼び水を差す。V6を位置Aにする。
【0079】
二酸化炭素同位体の製造は段階6〜7で行うことができる。
【0080】
6.二酸化炭素同位体を、17MeVプロトン照射した窒素(AGA、窒素6.0)及び0.1%酸素(AGA、酸素4.8)を含む目標ガス中で14N(p,α)11C反応を用いて生成する。
【0081】
7.二酸化炭素同位体を流量100ml/分の窒素を用いる装置に移動する。
【0082】
炭素同位体の合成は以後、段階8〜16で行うことができる。
【0083】
8.V1を位置Bに、V2を位置Bにする。そこで、二酸化炭素同位体を含む窒素流をポラパックカラム(−196℃に冷却)の方向に流し、廃棄物ラインを通じて外に出す。ポラパックカラム内に捕捉された放射能を監視する。
【0084】
9.放射能がピークに達した時、V1を位置Aに変える。そこで、ヘリウム流をポラパックカラムの方向に流し、廃棄物ラインを通じて外に出す。この運転により、チューブ及びポラパックカラムから窒素が除去される。
【0085】
10.V2を位置Aにし、ポラパックカラムを約50℃に加熱する。そこで、放射能をポラパックカラムから放出し、ヘリウム流量10ml/分で亜鉛炉に移動し、そこで一酸化炭素同位体に変換する。
【0086】
11.シリカカラム(−196℃に冷却)に到達する前に、気体流はアスカライトカラムを通過する。そこで、一酸化炭素同位体をシリカカラム上に捕捉する。シリカカラムの放射能を監視し、値がピークに達した時、V3を位置Bに設定し、その後V4を位置Bに設定する。
【0087】
12.シリカカラムを約50℃に加熱し、一酸化炭素同位体を放出する。V3を位置Aに設定し、一酸化炭素同位体を反応室50に15秒以内に移動する。
【0088】
13.V3を位置Bに設定し、V5を位置Bに設定し、HPLCポンプを作動させ(流量7ml/分)、V6を位置Bに設定する。加圧THF(又は他の溶媒)を用いて、反応混合物を反応室50に移動させる。HPLCポンプがその設定された限界圧力(例えば40Mpa)に達して自動的に動作を停止した後、V6を位置Aに設定する。
【0089】
14.高沸点液体(例えばポリエチレングリコール又は鉱油)を含む加熱ブロックの空洞に反応室50を動かす。加熱ブロックの温度は通常100〜200℃の範囲である。
【0090】
15.十分な反応時間(通常5分)の後、V3を位置Cに設定し、反応室50の内容物を回収バイアルに移動する。
【0091】
16.反応室50を以下の手順でリンスすることができる。V3を位置Bに設定し、HPLCポンプを作動し、V6を位置Bに設定し、最大圧力に達したら、V6を位置Aに設定し、V3を位置3に設定し、それによりリンス体積を回収バイアルに移動する。
【0092】
本発明の反応室50システムの最近開発された完全自動バージョンでは、11C標識トレーサーの前駆体としての[11C]一酸化炭素の値は、[11C]ヨウ化メチルと同等である。製造及び取扱いが容易であり、[11C]ヨウ化メチルでの標識に適した基(例えばヘテロ原子が結合したメチル基)が一般的な生物活性物質であるため、現在、[11C]ヨウ化メチルが最も頻繁に使用される11C前駆体である。[11C]一酸化炭素で簡便に標識することができるカルボニル基も一般的な生物活性物質である。多くの場合、インビボでの代謝事象のため、カルボニル基が標識位置としてメチル基よりも一層有利であると思われる。したがって、PETトレーサーを製造するための[11C]一酸化炭素の使用は、[11C]ヨウ化メチルに対する興味深い補完となる。更に、同様の技術の使用により、この方法が13C及び14C置換化合物の合成に適用できる可能性が高まる。
【0093】
本発明の主な利点は、現行のカルボニル化反応標識合成の限界を克服し、
【0094】
【化1】

【0095】
[11C]一酸化炭素、並びにトリフレート及びボロン酸を含む、遷移金属錯体と混合された溶液を用いる鈴木カップリング反応により[11C]ケトンを合成して所望の標識化合物を得る新規の効率的な方法を提供する点にある。本発明は更に、本発明でアミン、TiCl4及びNaBH3CNを用いて得られた[11C]ケトンを還元アミン化することにより[11C]アミンを合成する新規の方法を提供する。比放射能レベルは、本発明の方法を用いると高くなる。本発明で前駆体として用いるトリフレートは式R1−OTfを有し、R1は直鎖若しくは環状アルキル若しくは置換アルキル、アリール又は置換アリールである。好ましくは、R1はC6H5、4−CH3O−C6H4、4−CH3−C6H4、4−NO2−C6H4、C10H7又はから選択される。
【0096】
本発明で用いるボロン酸は式RB(OH)2を有し、Rは直鎖若しくは環状アルキル若しくは置換アルキル、アリール又は置換アリールである。好ましくは、Rはフェニル、メチル、
【0097】
【化2】
【0098】
又はチエニルから選択される。得られた標識ケトンは式R1−C*O−Rを有し、*は標識炭素位置であり、R1及びRは上記定義の通りである。
【0099】
標識ケトンの合成の好ましい反応スキームを下記に例示する。
【0100】
【化3】
【0101】
この反応では、好ましい遷移金属錯体はパラジウム錯体である。反応を促進するために臭化リチウムを加えてもよい。放射化学収量を高めるために異なる塩基を使用してもよい。
【0102】
本発明の他の実施形態は、本発明の方法で得られた11C標識ケトンから11C標識アミンを合成する新規の方法である。11C標識ケトンは、本発明の方法に従って回収バイアルに回収された後で、更にTiCl4及びNaBH3CNの存在下で異なるアミンと還元アミン化することができる。標識アミンの合成の好ましい反応スキームの全体を下記に例示する。
【0103】
【化4】
【0104】
式中、R′はH、直鎖若しくは環状アルキル若しくは置換アルキル、アリール又は置換アリールであり、R″は直鎖若しくは環状アルキル若しくは置換アルキル、アリール又は置換アリールであり、R及びR1は上記定義の通りである。
【0105】
[11C]標識ケトン及び[11C]標識アミンは、各種PET研究で有益なPETトレーサーを提供する。本発明の一実施形態では、[11C]標識ケトン及び/又は[11C]標識アミンを含む、PETトレーサーとして使用されるキットを提供する。
【0106】
このようなキットは、ヒトへの投与、例えば血流に対する直接注射に適した無菌の製品が得られるよう設計されている。好適なキットは、アドレナリン作動性阻害剤及びアドレナリン作動性イメージング剤の前駆体を含む容器(例えばセプタムで密閉したバイアル)を含む。
【0107】
キットは、放射線防護剤、抗菌防腐剤、pH調整剤又は充填剤などの追加の成分を更に含んでいてもよい。
【0108】
「放射能防護剤」という用語は、水の放射線分解で生じる含酸素フリーラジカルなどの高反応性フリーラジカルを捕捉することにより、酸化還元プロセスなどの分解反応を阻害する化合物を意味する。本発明の放射能防護剤は、アスコルビン酸、p−アミノ安息香酸(即ち4−アミノ安息香酸)、ゲンチジン酸(即ち2,5−ジヒドロキシ安息香酸)、及びその生体適合性物質との塩から好適に選択される。
【0109】
「抗菌防腐剤」という用語は、細菌、酵母又はカビなどの有害の可能性がある微生物の増殖を阻害する薬剤を意味する。抗菌防腐剤は、用量に応じていくつかの殺菌特性をも発揮し得る。本発明の抗菌防腐剤の主な役割は、再構成後の医薬組成物、即ち、放射性診断用製品自体において、任意のこのような微生物の増殖を抑制することにある。しかしながら、抗菌防腐剤を、再構成前の本発明のキットの1種以上の成分中で、有害の可能性がある微生物の増殖を抑制するために用いてもよい。好適な抗菌防腐剤としては、パラベン、即ちエチル、プロピル若しくはブチルパラベン又はその混合物、ベンジルアルコール、フェノール、クレゾール、セトリミド及びチオマーサルが挙げられる。好ましい抗菌防腐剤はパラベンである。
【0110】
「pH調整剤」という用語は、再構成されたキットのpHがヒトへの投与に許容される範囲(約pH4.0〜10.5)内にあることを確実にするのに有用な化合物又は化合物の混合物を意味する。適切なこのようなpH調整剤としては、トリシン、リン酸塩又はTRIS[即ちトリス(ヒドロキシメチル)アミノメタン]などの薬学的に許容できる緩衝液、及び炭酸ナトリウム、重炭酸ナトリウム又はその混合物などの薬学的に許容できる塩基が挙げられる。リガンド結合体を酸性塩の形態で用いる場合、pH調整剤を別のバイアル又は容器に入れて、多段階手順の一部としてキットの使用者がpHを調整できるようにしてもよい。
【0111】
「充填剤」という用語は、製造及び凍結乾燥時の原料の取扱いを容易にできる、薬学的に許容できる増量剤を意味する。好適な充填剤としては、塩化ナトリウムなどの無機塩、及びスクロース、マルトース、マンニトール又はトレハロースなどの水溶性糖又は糖アルコールが挙げられる。
【実施例】
【0112】
本発明を以下の実施例で更に説明するが、実施例は本発明の範囲を決して限定するものではない。
【0113】
実施例1
実験セットアップ
[11C]二酸化炭素を、Uppsala Research Imaging Solution ABのScanditronix MC−17サイクロトロンにより、窒素(AGA、窒素6.0)及び0.1%酸素(AGA、酸素4.8)を含む目標ガス中で17Mevプロトンを用いる14N(p,α)11C反応で製造した。[11C]一酸化炭素を、400℃の亜鉛炉で遠隔操作ワークステーションを使用して[11C]二酸化炭素を還元することで製造した。
【0114】
液体クロマトグラフィー分析(LC)を、β+流量検出器と直列にしたBeckman 126グラジエントポンプ及びBeckman 166可変波長UV検出器を用いて行った。以下の移動相を使用した。0.05Mギ酸アンモニウム、pH3.5(A1)及びアセトニトリル(B1)、アセトニトリル/H2O:50/7(B2)、メタノール(B3)及びH2O中0.01Mギ酸(A2)。分析LCでは、Jones Chromatography Genesis C18、4μm、250×4.6mm(内径)カラムを流量1.5ml/分で使用した。セミ分取LCでは、Jones Chromatography Genesis C18、4μm、250×10mm(内径)カラムを流量4ml/分で使用した。自動合成システムSynthiaをLCでの注入及び画分回収に使用した。データ収集及びLC制御はBeckman System Goldクロマトグラフィーソフトウェアパッケージを用いて行った。
【0115】
放射能はVeenstra Instrumenten bvのVDC−202電離箱で測定した。製造時の放射能の概算ではLangenas Eltekniska ABの携帯線量率計を使用した。
【0116】
実施例2
[カルボニル−11C]ケトンの合成
テトラキス(トリフェニルホスフィン)パラジウム(0)(5.0mg、4.3μmol)をバイアル(1mL)に入れた。バイアルを窒素で洗い流し、内容物をTHF(200μL)に溶解した。アリールトリフレート(30.8μmol)及びLiBr(THF中0.46M溶液10μL、4.6μmol)を加えた。混合物を振盪して溶液を均一にした。アリール又はアルキルボロン酸(49.2μmol)をTHF(100μL)に溶解し、使用直前にパラジウム錯体、トリフレート及び臭化リチウムの溶液に加えた。得られた混合物を装置の注入ループに注入し、その装置から適量(200μL)を加圧下で、ヘリウム中の[11C]一酸化炭素で予め充填したマイクロオートクレーブに移した。マイクロオートクレーブを5分間加熱した(150℃)。粗生成物を、予め排気した、セプタムを取り付けたバイアル(5mL)に移した。マイクロオートクレーブをTHF(200μL)で充填し、回収バイアルに移し替えて空にした。バイアルを窒素で浄化した前後の放射能を測定した。75℃で加熱し窒素で浄化することで、溶媒体積を0.2mL未満に減少させた。アセトニトリル/水混合物(1:1、2mL)を加え、得られた溶液をセミ分取LC上に注入した。回収された画分の特性及び放射化学純度を分析LCで評価した。
【0117】
実施例3
11C−アミンの合成
テトラキス(トリフェニルホスフィン)パラジウム(0)(5.0mg、4.3μmol)、アリールトリフレート(30.8μmol)及びアリール又はメチルボロン酸(49.2μmol)を用いて上記のように[11C]カルボニル化を行った。粗生成物を、予め排気した、セプタムを取り付けたバイアル(5mL)に移し、放射能を測定した。バイアルを窒素で浄化して未反応[11C]一酸化炭素を除去し、放射能を再度測定した。四塩化チタン(CH2Cl2中1M溶液500μL、0.5mmol)及びアミン(0.13mmol)を加え、混合物を65℃で3分間加熱した。シアノ水素化ホウ素ナトリウム(MeOH中1M溶液500μL、0.5mmol)を加え、同温度で更に3分間加熱した。水酸化ナトリウム水溶液(2M水溶液500μL)をゆっくり加えることで反応液をクエンチし、濾過した。60℃で加熱し窒素で浄化することで濾液の体積を1mL未満に減少させた。粗生成物を、生成物同定用の少量の非放射性参照化合物の存在下、HPLCで分析した。放射化学収量を、生成物純度に捕捉効率を掛けることで求めた。
特定の実施形態、参考文献の引用
本発明の範囲は、本明細書に記載の特定の実施形態に限定されるものではない。実際、本明細書に記載の実施形態以外に、本発明の各種の変形形態が、上記の記載及び添付の図面より当業者には明らかであろう。このような変形形態は添付の特許請求の範囲内にあるものとする。
【0118】
各種刊行物及び特許出願が本明細書で引用され、その開示が全体として参照により組み込まれる。
【図面の簡単な説明】
【0119】
【図1】本発明の方法に関するフローチャートを示す図である。
【図2】本発明の一酸化炭素同位体の製造及び標識システムの模式図である。
【図3a】本発明の反応室の代替実施形態を示す図である。
【図3b】本発明の反応室の代替実施形態を示す図である。
【技術分野】
【0001】
本発明は、陽電子放出核種で標識した生物活性化合物を含む、診断剤及び放射線診断剤に関する。本発明は更に、標識合成における一酸化炭素同位体の使用方法に関する。より具体的には、本発明は、最初の[11C]二酸化炭素混合気体から[11C]一酸化炭素の濃度が高い混合気体を製造し、鈴木カップリング反応を介したカルボニル化によるケトン及びアミンの標識合成に製造された混合気体を使用する方法に関する。本発明の放射標識されたケトン及びアミンは、具体的には陽電子放出断層撮影(PET)に使用される放射線医薬品として有用である。
【背景技術】
【0002】
短寿命の陽電子放出核種(例えば11C、t1/2=20.3分)で標識したトレーサーは、各種の非侵襲的in vivo研究に陽電子放出断層撮影(PET)との組合せで頻繁に使用される。標識物質の放射能、短い半減期、及びμM以下である量が理由で、これらのトレーサーの製造には特別な合成手順が必要である。これらの手順の考案に重要な部分を占めるのが、新規の11C標識前駆体の開発及び取扱いである。これは新種の化合物を標識するのに重要であるばかりでなく、異なる位置で所与の化合物を標識する可能性を高めるためにも重要である。
【0003】
過去20年間で、一酸化炭素を用いるカルボニル化反応は著しい発展を遂げた。パラジウム触媒を用いるカルボニル化カップリング反応などの方法の近年の発展は、一酸化炭素を異なるカルボニル化合物に変換する穏健で効率的な手段を提供してきた。
【0004】
生物活性物質がカルボニル基、又はカルボニル基から誘導可能な官能基をしばしば含むため、[11C]一酸化炭素を用いるカルボニル化反応はPETトレーサー合成において最も有益である。この合成は大部分の官能基に対して許容的であるため、カルボニル化段階で錯体構築ブロックを集積して目標化合物を得ることができる。これは、反応順序において非標識基質を標識前駆体とできるだけ遅く組み合わせて合成時間を減少させ、それにより未補正の放射化学収量を最適化しなければならないPETトレーサー合成で特に有益である。
【0005】
化合物を11Cで標識する場合、比放射能を最大化することが通常は重要である。これを実現するには、同位体希釈及び合成時間を最小限に抑える必要がある。[11C]二酸化炭素を標識反応に使用する場合、空気中の二酸化炭素からの同位体希釈が重要なことがある。一酸化炭素の反応性が低く、空気中濃度が低い(CO2の3.4×104ppmに対して0.1ppm)ため、[11C]一酸化炭素を使用することでこの問題は減少する。
【0006】
亜鉛、木炭又はモリブデンなどの還元剤を含む加熱カラムを用いた、[11C]二酸化炭素からの[11C]一酸化炭素の合成が、いくつかの刊行物に既に記載されている。[11C]一酸化炭素はヒトにおけるトレーサー実験に適用された最初の11C標識化合物の1つであったが、最近までPETトレーサーの製造には実際に使用されていなかった。この1つの理由は、[11C]一酸化炭素の溶解度が低く、反応速度が比較的遅いことにより、反応媒体中での捕捉効率が低くなるためである。[11C]ヨウ化メチル、[11C]シアン化水素又は[11C]二酸化炭素などの前駆体を用いる一般的手順は、気相の放射能を移動し、気体流を反応媒体に通すことにより放射能を捕捉することである。最近まで、標識合成で[11C]一酸化炭素を取扱う上でこれが唯一実施可能な手段であった。このアプローチでは、[11C]一酸化炭素を用いる標識合成の主要部分は、非常に低い収量を与えるか、完全に失敗することが予測され得る。
【0007】
高圧技術(300バール超)を用いる実用的に有益な11C標識合成の例はわずかしかない。原則として、高圧は、反応速度を高め、試薬の量を最小限に抑えるために利用することができる。このアプローチの1つの問題は、どのようにして標識前駆体を小さな高圧反応器に閉じ込めるかである。もう1つの問題は、反応器の構成である。一般的なカラム型の反応器を用いる場合(即ち各端部にチューブを取り付けた円筒)、気相は実際に、加圧により液相から効率的に排除される。その理由は、気相が圧縮された形態で、バルク量の液体試薬から取り付けられたチューブに逃れるためである。
【0008】
コールドトラップ技術は、特に[11C]一酸化炭素の場合、11C標識前駆体の取扱いに広く使用されている。しかし、この手順は単一の段階でしか行われず、標識化合物は常に、コールドトラップの加熱と同時に連続気体流中に放出されていた。更に、標識化合物を捕捉するのに使用される材料の体積は、標識化合物が移動された系に比較して大きかった。したがって、標識化合物のラジカル濃縮及び合成システムの小型化にこの技術を用いるというオプションは検討されなかった。このことは、11C標識化合物の量が通常、20〜60nmolの範囲であるという事実に鑑みて特に注目に値する。
【0009】
[11C]一酸化炭素の製造及び使用に関する近年の技術的発展により、この化合物が標識合成で有用になった。国際公開第02/102711号には、最初の二酸化炭素同位体混合気体から一酸化炭素同位体の濃度が高い混合気体を製造し、それを使用するためのシステム及び方法が記載されている。[11C]一酸化炭素は、サイクロトロンで製造された[11C]二酸化炭素から高い放射化学収量で得ることができ、高い比放射能を有する目標化合物を得るのに使用することができる。この反応器は、上記の問題点を克服しており、パラジウム又はセレンを媒介した反応における[11C]一酸化炭素を用いた11C標識化合物の合成に有用である。このような方法で、様々なカルボニル化合物を標識することができる(Kilhlberg,T.;Langstrom,B.J.,Org.Chem.64,1999,9201−9205;Kihlberg,T.,Karimi,F.,Langstrom,B.,J.Org.Chem.67,2002,3687−3692)。
【0010】
このような標識カルボニル化合物は、PETで使用される多くの薬学的に重要なトレーサーの合成に道を開いた。ケトンが生物学的に重要な分子の構築ブロックとして広く使用され、炭素同位体を用いた標識により反応前駆体又はPETトレーサーとして使用されることは注目に値する。多くの医薬品の生物活性はアミン官能基に依存するため、アミンは重要なクラスの化合物である。したがって、炭素同位体で標識したアミンは、重要で有用なPETトレーサーとして使用することができる。
【0011】
求電子剤、一酸化炭素及び有機水素化スズ(スティレカップリング)又は有機ホウ素化合物(鈴木カップリング)を用いた遷移金属を触媒とするカルボニル化は、ケトンの合成に多く使用されるアプローチである。11C標識化学反応では、スティレカルボニル化は多くの研究が行われてきたが、鈴木カルボニル化が用いられるのは稀であった。例えば、スティレカップリングは一連の11C標識ケトンの合成に使用されてきた(Lindstrom,P.,Kihlberg,T.,Langstrom,B.,J.Chem.Soc.Perkin Trans.1,1997,2701−2706)。しかし、このようなアプローチを使用する上で1つの欠点は、毒性の高い有機水素化スズ化合物の使用である。
【0012】
これとは別に、有機ホウ素及びヨードベンゼンを用いた11C標識ベンゾフェノンの合成(鈴木カップリング)が報告された(Zeisler,S.,Nader,M.Theobald,A.and Oberdorfer,F.,Appl,Radiat.Isot.1997,48,1091−1095)。しかし、この報告で使用された[11C]一酸化炭素の製造方法は、上記の低い比放射能という欠点がある。したがって、鈴木カップリング反応で使用される比放射能を増大させることが必要である。
【0013】
11C標識アミンの効率的な合成方法も未検討である。以前、カルボン酸及び[11C]ハロゲン化マグネシウムカルボキシレートの還元アミン化による11C標識アミンの合成に関する1つの報告が刊行された(Perrio−Huard,C.et al,J.Che.Soc.Perkin Trans.1,2000,311−316)。但し、このような方法では[11C]二酸化炭素が標識前駆体として使用される。そのため、このような方法は最適ではない。
【0014】
したがって、[11C]一酸化炭素を用いて、反応前駆体又はPETトレーサーとしての炭素同位体で標識したケトン及びアミンを合成する、新規の改善された方法が必要である。この方法は、有用なPETトレーサーの調製において[11C]一酸化炭素の有用性を更に高めることになろう。
【0015】
本明細書での参考文献の議論又は引用は、そのような参考文献が本発明の先行技術であることを承認するものではない。
【特許文献1】国際公開第02/102711号
【非特許文献1】Kilhlberg,T.;Langstrom,B.J.,Org.Chem.64,1999,9201−9205
【非特許文献2】Kihlberg,T.,Karimi,F.,Langstrom,B.,J.Org.Chem.67,2002,3687−3692
【非特許文献3】Lindstrom,P.,Kihlberg,T.,Langstrom,B.,J.Chem.Soc.Perkin Trans.1,1997,2701−2706
【非特許文献4】Zeisler,S.,Nader,M.Theobald,A.and Oberdorfer,F.,Appl,Radiat.Isot.1997,48,1091−1095
【非特許文献5】Perrio−Huard,C.et al,J.Che.Soc.Perkin Trans.1,2000,311−316
【発明の開示】
【課題を解決するための手段】
【0016】
本発明は、
(a)その底面に液体入口及び気体入口を有する高圧反応室を用意する段階と、
(b)遷移金属錯体と混合された、トリフレート、ボロン酸を含む標識すべき溶液を用意する段階と、
(c)気体入口を通じて反応室に一酸化炭素同位体の濃度が高い混合気体を導入する段階と、
(d)液体入口を通じて反応室に、遷移金属錯体と混合された前記溶液を高圧で導入する段階と、
(e)標識合成が行われている所定時間待機する段階と、
(f)反応室から標識ケトンを除去する段階とを含む、鈴木カップリング反応を介したケトンの標識合成方法を提供する。
【0017】
本発明はまた、本発明の方法により合成される炭素同位体で標識したケトンを提供する。
【0018】
本発明は更に、アミン、TiCl4及びNaBH3CNを用いて本発明の方法により得られる炭素同位体で標識したケトンを還元アミン化する段階を含む、アミンの標識合成方法を提供する。
【0019】
更に他の実施形態では、本発明はまた、本発明の方法により合成される炭素同位体で標識したアミンを提供する。更に他の実施形態では、本発明は、このような[11C]標識ケトン及び/又はアミンを含む、PETトレーサーとして使用されるキットを提供する。
【発明を実施するための最良の形態】
【0020】
本発明の1つの目的は、先行技術の装置の欠点を克服する標識合成における、一酸化炭素同位体を製造するための方法及びシステム、並びに一酸化炭素同位体の使用を提供することにある。これは本発明に記載の方法及びシステムにより実現される。
【0021】
このような方法及びシステムの1つの利点は、一酸化炭素同位体を標識生成物に、ほぼ定量的に転換できることである。
【0022】
本方法及びシステムには他にもいくつかの利点がある。高圧技術により、ジエチルエーテルなどの低沸点溶媒を高温(例えば200℃)で使用することが可能になる。気体の分散を妨げる材料からなる閉鎖系の使用により、感受性の高い化合物の安定性が増大し、適正製造規範(GMP)の点でも有利になることがある。
【0023】
得られる標識化合物が高濃度であり、合成システムの小型化により自動化、迅速な合成及び精製、並びに同位体希釈を最小限に抑えることによる比放射能の最適化が促進されるという、更なる他の利点も得られる。
【0024】
本発明で例示するように、最も重要なのは、完全に新規の合成の可能性が開かれることである。
【0025】
以下、図面を参照しながら本発明の実施形態を説明する。
【0026】
本出願を通じて使用される炭素同位体という用語は、11Cを意味することが好ましいが、所望であれば11Cを13C及び14Cなど他の炭素同位体に置き換えることができると理解されたい。
【0027】
図1は、第1に一酸化炭素同位体の濃度が高い混合気体の製造、第2に標識合成手順を含む本発明の方法に関するフローチャートを示す図である。より詳しくは、本方法の製造部分は以下の段階を含む。
【0028】
・以下に詳述する種類の好適なキャリアガス中の二酸化炭素同位体を用意する段階。
【0029】
・以下に詳述する反応装置内に前記混合気体を導入することで二酸化炭素同位体を一酸化炭素同位体に転換する段階。
【0030】
・一酸化炭素同位体でもキャリアガスでもなく二酸化炭素同位体が捕捉される二酸化炭素除去装置内に転換された混合気体を注ぎ込むことで微量の二酸化炭素同位体を除去する段階。二酸化炭素除去装置は以下に詳述する。
【0031】
・前記キャリアガスではなく一酸化炭素同位体が捕捉される一酸化炭素捕捉装置内に一酸化炭素同位体を捕捉する段階。一酸化炭素捕捉装置は以下に詳述する。
【0032】
・前記捕捉装置から前記捕捉された一酸化炭素同位体を放出することにより、ある体積の一酸化炭素同位体の濃度が高い混合気体を得る段階。
【0033】
最初の二酸化炭素同位体の混合気体が、二酸化炭素同位体、及び窒素などの、同様な分子特性等が理由で一酸化炭素のキャリアガスとして適さない第1のキャリアガスで構成される場合、製造段階は、最初の二酸化炭素同位体の混合気体のキャリアガスを変更する段階を更に含んでいてもよい。より詳しくは、He、Arなどの好適な第2のキャリアガス中の二酸化炭素同位体を用意する段階は、以下の段階を含む。
【0034】
・前記第1のキャリアガスではなく二酸化炭素同位体が捕捉される二酸化炭素捕捉装置内に最初の二酸化炭素同位体の混合気体を流し込む段階。二酸化炭素捕捉装置は以下に詳述する。
【0035】
・前記二酸化炭素捕捉装置を前記第2のキャリアガスで洗い流して前記第1のキャリアガスの残りを除去する段階。
【0036】
・前記好適な第2のキャリアガス中の前記捕捉された二酸化炭素同位体を放出する段階。
【0037】
製造段階に続くことができる標識合成段階では、製造された一酸化炭素同位体の濃度が高い混合気体を標識反応物として利用する。より詳しくは、標識合成段階は以下の段階を含む。
【0038】
・その底面に液体試薬入口及び標識反応物入口を有する高圧反応室を用意する段階。反応室は以下に詳述する。
【0039】
・遷移金属錯体と混合された、トリフレート、ボロン酸を含む標識すべき溶液を用意する段階。
【0040】
・標識反応物入口を通じて反応室に一酸化炭素同位体の濃度が高い混合気体を導入する段階。
【0041】
・液体試薬入口を通じて反応室に、遷移金属錯体と混合された前記溶液を高圧で導入する段階。
【0042】
・標識合成が行われている所定時間待機する段階。
【0043】
・反応室から標識化合物溶液を除去する段階。
【0044】
反応を促進するために、標識すべき物質は臭化リチウムを更に含んでいてもよい。遷移金属錯体はパラジウム錯体であることが好ましい。
【0045】
所定時間待機する段階は、標識合成が促進されるように反応室の温度を調整する段階を更に含んでいてもよい。
【0046】
図2は、本発明の[11C]二酸化炭素製造及び標識システムを模式的に示す図である。このシステムは3つの主要ブロックで構成され、各ブロックは製造及び標識方法の3つの主要段階の1つを取扱う。
【0047】
ブロックAは、最初の二酸化炭素同位体の混合気体が、二酸化炭素同位体、及び一酸化炭素のキャリアガスとして適さない第1のキャリアガスで構成される場合、最初の二酸化炭素同位体の混合気体のキャリアガスを変更するために使用される。
【0048】
ブロックBは、二酸化炭素同位体を一酸化炭素同位体に転換し、転換された一酸化炭素同位体の混合気体を精製及び濃縮するために使用される。
【0049】
ブロックCは、一酸化炭素同位体の標識合成を行うために使用される。
【0050】
二酸化炭素同位体が通常、17MeVプロトン照射された窒素及び0.1%酸素を含む目標ガス中で14N(p,α)11C反応を用いて生成されることにより、最初の二酸化炭素同位体の混合気体が窒素をキャリアガスとして含むという事実があるため、ブロックAは通常必要である。しかし、窒素は分子特性において一酸化炭素に対してある種の類似性を示すため、例えば捕捉装置等において互いに分離することが困難であり、それによりこのような混合気体中での一酸化炭素同位体の濃度を上昇させることが困難である。代わりの好適なキャリアガスとしてはヘリウム、アルゴン等が挙げられる。外部の系がブロックB及びCに必要な気体圧力に耐容性を示さない場合、キャリアガスの圧力(例えば1〜4バール)を変更するために使用することもできる。一代替実施形態では、最初の二酸化炭素同位体の混合気体は、二酸化炭素同位体、及び一酸化炭素のキャリアガスとして好適な第1のキャリアガスで構成され、それによりブロックAを簡略化するか、排除さえすることができる。
【0051】
好ましい一実施形態(図2)によれば、ブロックAは、第1のバルブV1、二酸化炭素捕捉装置8及び第2のバルブV2で構成される。
【0052】
第1のバルブV1は、最初の二酸化炭素同位体の混合気体源12に接続される二酸化炭素入口10、ヘリウム、アルゴン等の好適なキャリアガス源16に接続されるキャリアガス入口14を有する。第1のバルブV1は、第2のバルブV2の第1の入口20に接続される第1の出口18、及び二酸化炭素捕捉装置8に接続される第2の出口22を更に有する。バルブV1は2つのモードA、Bで運転することができる。モードAでは、二酸化炭素入口10は第1の出口18に接続され、キャリアガス入口14は第2の出口22に接続される。モードBでは、二酸化炭素入口10は第2の出口22に接続され、キャリアガス入口14は第1の出口18に接続される。
【0053】
第2のバルブV2は、第1の入口20以外に、二酸化炭素捕捉装置8に接続される第2の入口24を有する。第2のバルブV2は、廃棄物出口26、及びブロックBの生成物入口30に接続される生成物出口28を更に有する。バルブV2は2つのモードA、Bで運転することができる。モードAでは、第1の入口20は廃棄物出口26に接続され、第2の入口24は生成物出口28に接続される。モードBでは、第1の入口20は生成物出口28に接続され、第2の入口24は廃棄物出口26に接続される。
【0054】
二酸化炭素捕捉装置8は、前記第1のキャリアガスではなく二酸化炭素を捕捉する装置であり、捕捉された二酸化炭素はその後、制御下で放出することができる。これは、冷却状態(例えば液体窒素中−196℃、又は液体アルゴン中−186℃)で二酸化炭素を選択的に捕捉し、加熱状態(例えば+50℃)で捕捉された二酸化炭素を放出する材料を含むカラムなどのコールドトラップを使用することで実現することが好ましい(本明細書では、「コールドトラップ」という表現は低温工学の使用に限定されない。したがって、室温で対象化合物を捕捉し、より高温でそれを放出する材料が含まれる)。好適な材料としては例えば、シリカ及びPorapacQ(登録商標)が挙げられる。シリカカラム又はポラパックカラムの捕捉挙動は、双極子−双極子相互作用、又は場合によってはファンデルワールス相互作用に関連している。前記カラム8は、捕捉材料の体積が二酸化炭素同位体を効率的に捕捉(95%超)する程度に大きく、捕捉された二酸化炭素のブロックBへの移動を遅延させない程度に小さくなるように形成されていることが好ましい。PorapacQ(登録商標)、窒素流量100ml/分の場合、体積は50〜150μlでなければならない。更に、二酸化炭素捕捉装置8の冷却及び加熱は、それが自動化プロセスとして、例えば自動的にカラムを液体窒素の位置まで引き下げ、そこから加熱装置に移動させることで行われるように設定できる。
【0055】
図2の好ましい実施形態によれば、ブロックBは、二酸化炭素同位体が一酸化炭素同位体に転換される反応装置32、二酸化炭素除去装置34、チェックバルブ36、及び一酸化炭素捕捉装置38で構成され、これらは直列に接続される。
【0056】
好ましい実施形態では、反応装置32は、正しい温度間隔に加熱された際に、二酸化炭素同位体を一酸化炭素同位体に転換する材料を含む反応炉である。二酸化炭素を一酸化炭素に変換する能力を有する様々な異なる材料、例えば鉛若しくはモリブデン又は同様の還元特性を有する任意の他の元素若しくは化合物を使用することができる。反応装置32が亜鉛炉の場合、350〜400℃に加熱しなければならず、温度を高精度で制御することが重要である。亜鉛の融点は420℃であり、亜鉛炉は、温度が410℃を超えた際に、おそらくは表面特性の変化により、二酸化炭素を一酸化炭素に変換する能力を急速に失う。この材料は、放射能を二酸化炭素捕捉装置8から後続の一酸化炭素捕捉装置38に移動するのに必要な時間を最小限に抑えるような少量を使用できるように、その量に対して効率的でなければならない。炉中の材料の量は、炉中での実用的な寿命(少なくとも数日)を確保する程度に多くなければならない。亜鉛粒質物の場合、体積は100〜1000μlでなければならない。
【0057】
二酸化炭素除去装置34は、反応装置32を出る混合気体から微量の二酸化炭素同位体を除去するために使用される。二酸化炭素除去装置34では、一酸化炭素同位体でもキャリアガスでもなく、二酸化炭素同位体が捕捉される。二酸化炭素除去装置34は、ascarite(登録商標)(即ちシリカで担持された水酸化ナトリウム)を含むカラムで構成することができる。一酸化炭素同位体が通過する際に、反応装置32で反応しなかった二酸化炭素同位体がこのカラム内に捕捉される(水酸化ナトリウムと反応して炭酸ナトリウムになる)。反応装置32が適切に機能していないことを高い値が示すと、二酸化炭素除去装置34の放射能が監視される。
【0058】
二酸化炭素捕捉装置8と同様に、一酸化炭素捕捉装置38は捕捉及び放出状態を有する。捕捉状態では、前記キャリアガスではなく一酸化炭素同位体が選択的に捕捉され、放出状態では、前記捕捉された一酸化炭素同位体が制御下で放出される。これは、モレキュラシーブなどの、シリカ又は同様の特性を有する材料を含むカラムなどのコールドトラップを用いて実現することが好ましい。このようなコールドトラップは、−100℃未満の冷却状態、例えば液体窒素中−196℃又は液体アルゴン中−186℃で一酸化炭素を選択的に捕捉し、加熱状態(例えば+50℃)で捕捉された一酸化炭素を放出する。シリカカラムの捕捉挙動は、双極子−双極子相互作用、又は場合によってはファンデルワール相互作用に関連している。放射能を運ぶヘリウムが窒素を含む場合、シリカカラムの一酸化炭素同位体を捕捉する能力が減少する。これは、窒素の物性が一酸化炭素と同様であるため、シリカ上の捕捉部位をめぐって窒素が一酸化炭素と競合するためである。
【0059】
図2の好ましい実施形態によれば、ブロックCは、第1及び第2の反応室バルブV3及びV4、前記反応室50、試薬バルブV5、注入ループ70、並びに溶媒バルブV6で構成される。
【0060】
第1の反応室バルブV3は、一酸化炭素捕捉装置38に接続される混合気体入口40、停止位置42、回収出口44、廃棄物出口46、及び反応室50の気体入口52に接続される反応室接続口48を有する。第1の反応室バルブV3は、4つの運転モードA〜Dを有する。反応室接続口48は、モードAでは混合気体入口40に接続され、モードBでは停止位置42に接続され、モードCでは回収出口44に接続され、モードDでは廃棄物出口46に接続される。
【0061】
反応室50(マイクロオートクレーブ)は、気体入口52及び液体入口54を有し、これらは反応室の底面に端部を置くように配置される。気体入口52は、標識が終了した後に生成物出口として使用してもよい。運転中、一酸化炭素同位体の濃度が高い混合気体が気体入口52を通じて反応室50に導入された後、遷移金属錯体により高圧で標識すべき溶液が液体入口54を通じて反応室50に入る。図3a及び3bは、2つの好ましい反応室50の断面の模式図を示す。図3aはかなり製造が容易な円筒状反応室である一方、図3bの球状反応室は、反応室の表面積と体積の比が更に抑制されるため、最も好ましい実施形態である。表面積と体積の比を最小限に抑えることで、標識生成物の回収が最適化され、表面材料との反応の可能性が最小限に抑えられる。反応室50の「ダイビングベル構造」のため、気体入口52と液体入口54の両方が液体で満たされ、反応室50が下部から上方に向けて満たされる。一酸化炭素同位体を含む気体体積はこうして捕捉され、反応混合物と十分に接触する。液体の最終圧力が元々の気体圧力の約80倍であるため、気体の一般法則によれば、最終気体体積は液体体積の2%未満となる。こうして擬似一相系が得られる。本出願では、「擬似一相系」という用語は、200バールを超える圧力で96%を超える液体及び4%未満の気体を含む、表面積と体積の比が小さい閉じた体積を意味する。大部分の合成では、気相から液相への一酸化炭素の移動は、おそらく律速段階ではない。標識が終了した後、標識された体積はほぼ定量的に、反応室から内部圧力により気体入口/生成物出口52及び位置Cの第1の反応室バルブV3を通じて移動する。
【0062】
第2の反応室バルブV4は、反応室接続口56、廃棄物出口58、及び試薬入口60を有する。第2の反応室バルブV4は、2つの運転モードA及びBを有する。反応室接続口56は、モードAでは廃棄物入口58に接続され、モードBでは試薬入口60に接続される。
【0063】
試薬バルブV5は、第2の反応室バルブV4の試薬入口60に接続される試薬出口62、それらの間に注入ループ70が接続される注入ループ入口64及び出口66、廃棄物出口68、試薬源に接続される試薬入口71、並びに溶媒入口72を有する。試薬バルブV5は、2つの運転モードA及びBを有する。モードAでは、試薬入口71が注入ループ入口64に接続され、注入ループ出口66が廃棄物出口68に接続され、それにより試薬を注入ループ70に供給することができる。モードBでは、溶媒入口72が注入ループ入口64に接続され、注入ループ出口66が試薬出口62に接続され、それにより、高圧が溶媒入口72に加えられた際に、注入ループ70内に保存されている試薬を第2の反応室バルブV4を通じて反応室50に押し出すことができる。
【0064】
溶媒バルブV6は、試薬バルブV5の溶媒入口72に接続される溶媒出口74、停止位置76、廃棄物出口78、及び、溶媒供給HPLCポンプ(高速液体クロマトグラフィー)又は0〜10ml/分、最大400バールの圧力で有機溶媒を供給可能な任意の液体ポンプ(図示せず)に接続される溶媒入口80を有する。溶媒バルブV6は、2つの運転モードA及びBを有する。モードAでは、溶媒出口74が停止位置76に接続され、溶媒入口80が廃棄物出口78に接続される。モードBでは、溶媒出口74が溶媒入口80に接続され、それにより溶媒をHPLCポンプを用いて高圧でシステムに供給することができる。
【0065】
一酸化炭素捕捉装置38のシリカの体積が小さいことを除けば、二酸化炭素捕捉装置8及びすべての関連する先行技術との重大な相違は、二酸化炭素の放出に用いる手順にある。一酸化炭素を一酸化炭素捕捉装置8に捕捉した後、バルブV3を位置AからBに変えることで一酸化炭素捕捉装置38からの流れが停止し、設定された供給気体圧力(3〜5バール)まで一酸化炭素捕捉装置38の気体圧力が上昇する。次いで、一酸化炭素捕捉装置38を加熱して、キャリアガス中の一酸化炭素の体積を著しく膨張させることなくシリカ表面から一酸化炭素を放出する。バルブV4を位置AからBに変えた後、バルブV3を位置BからAに変える。この場合、一酸化炭素は急速且つほぼ定量的に、明確に定義されたマイクロプラグで反応室50に移動する。マイクロプラグは、対象物質(例えば1〜20μL)を含む、反応室50の体積の10%未満の気体体積として定義される。閉じられた出口を有する小さな反応室50へ効率的に物質移動を行うこの独自の方法は、以下の条件が必要である。
【0066】
・以下に定義するマイクロカラム38を使用しなければならない。捕捉材料(例えばシリカ)の体積は、一酸化炭素同位体を効率的に(95%超)捕捉できる程度に大きくなければならず、一酸化炭素同位体の濃度を最大限にできる程度に小さくなければならない(後続の反応器50の体積の1%未満)。シリカ及び反応室50体積200μlの場合、シリカ体積は0.1〜2μlでなければならない。
【0067】
・シリカカラムと反応室50を接続するチューブ及びバルブのデッドボリュームを最小限に抑えなければならない(マイクロオートクレーブ体積の10%未満)。
【0068】
・キャリアガスの圧力を移動前の反応室50中圧力(1気圧)の3〜5倍にしなければならない。
【0069】
具体的な好ましい一実施形態の仕様では、材料及び成分を以下のように選択する。Valco(登録商標)、Reodyne(登録商標)又はCheminert(登録商標)の高圧バルブを使用する。外径1/16インチのステンレスチューブを使用する。但し、ポラパックカラム8、シリカカラム38、及び反応室50への接続部には、並進運動を促進するため外径1/32インチのステンレスチューブを使用する。V1、V2及びV3の接続部の内径は0.2〜1mmでなければならない。この要件は、内径が、システムを通じてHeの最適流量(2〜50ml/分)が得られる可能性を妨げない程度に大きく、ポラパックカラム8からシリカカラム38への放射能の移動に必要な時間を長くしない程度に小さくなければならないということである。V3とオートクレーブの接続部のデッドボリュームは最小限に抑えなければならない(オートクレーブ体積の10%未満)。接続部の内径(0.05〜1mm)は最適なHe流量(2〜50ml/分)を可能にする程度に大きくなければならない。V4とV5の接続部のデッドボリュームはオートクレーブ体積の10%未満でなければならない。
【0070】
カラム8がポラパックカラムの場合、PorapacQ(登録商標)で充填され、ステンレススクリーンを取り付けたステンレスチューブ(外径=1/8インチ、内径=2mm、長さ=20mm)で構成されることが好ましい。シリカカラム38は、端部に空洞(深さ=1mm、高さ=1mm、体積=0.8μl)を有するステンレスチューブ(外径=1/16インチ、内径=0.1mm)で構成されることが好ましい。空洞は、GC固定相型のシリカ粉末(100/80メッシュ)で充填されている。カラムの端部はステンレススクリーンに取り付けられる。
【0071】
様々な異なる材料が捕捉装置に使用できることに留意されたい。GC材料を選ぶ場合、基準は、良好な遅延、並びに二酸化炭素及び一酸化炭素の各々に対する良好なピーク形状でなければならない。後者は、放射能の最適な回収を確実にする。
【0072】
上記に例示したシステムを用いる炭素同位体の製造方法を以下に詳述する。
【0073】
システムの準備は段階1〜5により行う。
【0074】
1.V1を位置Aに、V2を位置Aに、V3を位置Aに、V4を位置Aにし、ヘリウム流の最大圧力を5バールにする。この設定により、ヘリウム流は、[11C]二酸化炭素捕捉カラム、亜鉛炉、[11C]一酸化炭素捕捉カラム、反応室50を通ってV4から出ていく。システムが調節され、反応室50から溶媒が除去されて、ヘリウムがシステムを10ml/分以上で流れることができることが確認可能になる。
【0075】
2.亜鉛炉を作動させ、400℃に設定する。
【0076】
3.[11C]二酸化炭素及び[11C]一酸化炭素捕捉カラムを液体窒素で冷却する。−196℃で、ポラパック及びシリカカラムは、二酸化炭素同位体及び一酸化炭素同位体を各々効率的に捕捉する。
【0077】
4.V5を位置A(充填)にする。V5に取り付けた注入ループ(250μl)に反応混合物を充填する。
【0078】
5.HPLCポンプを、新たに蒸留したTHF(又は他の高品質溶媒)入りのフラスコに取り付け、呼び水を差す。V6を位置Aにする。
【0079】
二酸化炭素同位体の製造は段階6〜7で行うことができる。
【0080】
6.二酸化炭素同位体を、17MeVプロトン照射した窒素(AGA、窒素6.0)及び0.1%酸素(AGA、酸素4.8)を含む目標ガス中で14N(p,α)11C反応を用いて生成する。
【0081】
7.二酸化炭素同位体を流量100ml/分の窒素を用いる装置に移動する。
【0082】
炭素同位体の合成は以後、段階8〜16で行うことができる。
【0083】
8.V1を位置Bに、V2を位置Bにする。そこで、二酸化炭素同位体を含む窒素流をポラパックカラム(−196℃に冷却)の方向に流し、廃棄物ラインを通じて外に出す。ポラパックカラム内に捕捉された放射能を監視する。
【0084】
9.放射能がピークに達した時、V1を位置Aに変える。そこで、ヘリウム流をポラパックカラムの方向に流し、廃棄物ラインを通じて外に出す。この運転により、チューブ及びポラパックカラムから窒素が除去される。
【0085】
10.V2を位置Aにし、ポラパックカラムを約50℃に加熱する。そこで、放射能をポラパックカラムから放出し、ヘリウム流量10ml/分で亜鉛炉に移動し、そこで一酸化炭素同位体に変換する。
【0086】
11.シリカカラム(−196℃に冷却)に到達する前に、気体流はアスカライトカラムを通過する。そこで、一酸化炭素同位体をシリカカラム上に捕捉する。シリカカラムの放射能を監視し、値がピークに達した時、V3を位置Bに設定し、その後V4を位置Bに設定する。
【0087】
12.シリカカラムを約50℃に加熱し、一酸化炭素同位体を放出する。V3を位置Aに設定し、一酸化炭素同位体を反応室50に15秒以内に移動する。
【0088】
13.V3を位置Bに設定し、V5を位置Bに設定し、HPLCポンプを作動させ(流量7ml/分)、V6を位置Bに設定する。加圧THF(又は他の溶媒)を用いて、反応混合物を反応室50に移動させる。HPLCポンプがその設定された限界圧力(例えば40Mpa)に達して自動的に動作を停止した後、V6を位置Aに設定する。
【0089】
14.高沸点液体(例えばポリエチレングリコール又は鉱油)を含む加熱ブロックの空洞に反応室50を動かす。加熱ブロックの温度は通常100〜200℃の範囲である。
【0090】
15.十分な反応時間(通常5分)の後、V3を位置Cに設定し、反応室50の内容物を回収バイアルに移動する。
【0091】
16.反応室50を以下の手順でリンスすることができる。V3を位置Bに設定し、HPLCポンプを作動し、V6を位置Bに設定し、最大圧力に達したら、V6を位置Aに設定し、V3を位置3に設定し、それによりリンス体積を回収バイアルに移動する。
【0092】
本発明の反応室50システムの最近開発された完全自動バージョンでは、11C標識トレーサーの前駆体としての[11C]一酸化炭素の値は、[11C]ヨウ化メチルと同等である。製造及び取扱いが容易であり、[11C]ヨウ化メチルでの標識に適した基(例えばヘテロ原子が結合したメチル基)が一般的な生物活性物質であるため、現在、[11C]ヨウ化メチルが最も頻繁に使用される11C前駆体である。[11C]一酸化炭素で簡便に標識することができるカルボニル基も一般的な生物活性物質である。多くの場合、インビボでの代謝事象のため、カルボニル基が標識位置としてメチル基よりも一層有利であると思われる。したがって、PETトレーサーを製造するための[11C]一酸化炭素の使用は、[11C]ヨウ化メチルに対する興味深い補完となる。更に、同様の技術の使用により、この方法が13C及び14C置換化合物の合成に適用できる可能性が高まる。
【0093】
本発明の主な利点は、現行のカルボニル化反応標識合成の限界を克服し、
【0094】
【化1】
【0095】
[11C]一酸化炭素、並びにトリフレート及びボロン酸を含む、遷移金属錯体と混合された溶液を用いる鈴木カップリング反応により[11C]ケトンを合成して所望の標識化合物を得る新規の効率的な方法を提供する点にある。本発明は更に、本発明でアミン、TiCl4及びNaBH3CNを用いて得られた[11C]ケトンを還元アミン化することにより[11C]アミンを合成する新規の方法を提供する。比放射能レベルは、本発明の方法を用いると高くなる。本発明で前駆体として用いるトリフレートは式R1−OTfを有し、R1は直鎖若しくは環状アルキル若しくは置換アルキル、アリール又は置換アリールである。好ましくは、R1はC6H5、4−CH3O−C6H4、4−CH3−C6H4、4−NO2−C6H4、C10H7又はから選択される。
【0096】
本発明で用いるボロン酸は式RB(OH)2を有し、Rは直鎖若しくは環状アルキル若しくは置換アルキル、アリール又は置換アリールである。好ましくは、Rはフェニル、メチル、
【0097】
【化2】
【0098】
又はチエニルから選択される。得られた標識ケトンは式R1−C*O−Rを有し、*は標識炭素位置であり、R1及びRは上記定義の通りである。
【0099】
標識ケトンの合成の好ましい反応スキームを下記に例示する。
【0100】
【化3】
【0101】
この反応では、好ましい遷移金属錯体はパラジウム錯体である。反応を促進するために臭化リチウムを加えてもよい。放射化学収量を高めるために異なる塩基を使用してもよい。
【0102】
本発明の他の実施形態は、本発明の方法で得られた11C標識ケトンから11C標識アミンを合成する新規の方法である。11C標識ケトンは、本発明の方法に従って回収バイアルに回収された後で、更にTiCl4及びNaBH3CNの存在下で異なるアミンと還元アミン化することができる。標識アミンの合成の好ましい反応スキームの全体を下記に例示する。
【0103】
【化4】
【0104】
式中、R′はH、直鎖若しくは環状アルキル若しくは置換アルキル、アリール又は置換アリールであり、R″は直鎖若しくは環状アルキル若しくは置換アルキル、アリール又は置換アリールであり、R及びR1は上記定義の通りである。
【0105】
[11C]標識ケトン及び[11C]標識アミンは、各種PET研究で有益なPETトレーサーを提供する。本発明の一実施形態では、[11C]標識ケトン及び/又は[11C]標識アミンを含む、PETトレーサーとして使用されるキットを提供する。
【0106】
このようなキットは、ヒトへの投与、例えば血流に対する直接注射に適した無菌の製品が得られるよう設計されている。好適なキットは、アドレナリン作動性阻害剤及びアドレナリン作動性イメージング剤の前駆体を含む容器(例えばセプタムで密閉したバイアル)を含む。
【0107】
キットは、放射線防護剤、抗菌防腐剤、pH調整剤又は充填剤などの追加の成分を更に含んでいてもよい。
【0108】
「放射能防護剤」という用語は、水の放射線分解で生じる含酸素フリーラジカルなどの高反応性フリーラジカルを捕捉することにより、酸化還元プロセスなどの分解反応を阻害する化合物を意味する。本発明の放射能防護剤は、アスコルビン酸、p−アミノ安息香酸(即ち4−アミノ安息香酸)、ゲンチジン酸(即ち2,5−ジヒドロキシ安息香酸)、及びその生体適合性物質との塩から好適に選択される。
【0109】
「抗菌防腐剤」という用語は、細菌、酵母又はカビなどの有害の可能性がある微生物の増殖を阻害する薬剤を意味する。抗菌防腐剤は、用量に応じていくつかの殺菌特性をも発揮し得る。本発明の抗菌防腐剤の主な役割は、再構成後の医薬組成物、即ち、放射性診断用製品自体において、任意のこのような微生物の増殖を抑制することにある。しかしながら、抗菌防腐剤を、再構成前の本発明のキットの1種以上の成分中で、有害の可能性がある微生物の増殖を抑制するために用いてもよい。好適な抗菌防腐剤としては、パラベン、即ちエチル、プロピル若しくはブチルパラベン又はその混合物、ベンジルアルコール、フェノール、クレゾール、セトリミド及びチオマーサルが挙げられる。好ましい抗菌防腐剤はパラベンである。
【0110】
「pH調整剤」という用語は、再構成されたキットのpHがヒトへの投与に許容される範囲(約pH4.0〜10.5)内にあることを確実にするのに有用な化合物又は化合物の混合物を意味する。適切なこのようなpH調整剤としては、トリシン、リン酸塩又はTRIS[即ちトリス(ヒドロキシメチル)アミノメタン]などの薬学的に許容できる緩衝液、及び炭酸ナトリウム、重炭酸ナトリウム又はその混合物などの薬学的に許容できる塩基が挙げられる。リガンド結合体を酸性塩の形態で用いる場合、pH調整剤を別のバイアル又は容器に入れて、多段階手順の一部としてキットの使用者がpHを調整できるようにしてもよい。
【0111】
「充填剤」という用語は、製造及び凍結乾燥時の原料の取扱いを容易にできる、薬学的に許容できる増量剤を意味する。好適な充填剤としては、塩化ナトリウムなどの無機塩、及びスクロース、マルトース、マンニトール又はトレハロースなどの水溶性糖又は糖アルコールが挙げられる。
【実施例】
【0112】
本発明を以下の実施例で更に説明するが、実施例は本発明の範囲を決して限定するものではない。
【0113】
実施例1
実験セットアップ
[11C]二酸化炭素を、Uppsala Research Imaging Solution ABのScanditronix MC−17サイクロトロンにより、窒素(AGA、窒素6.0)及び0.1%酸素(AGA、酸素4.8)を含む目標ガス中で17Mevプロトンを用いる14N(p,α)11C反応で製造した。[11C]一酸化炭素を、400℃の亜鉛炉で遠隔操作ワークステーションを使用して[11C]二酸化炭素を還元することで製造した。
【0114】
液体クロマトグラフィー分析(LC)を、β+流量検出器と直列にしたBeckman 126グラジエントポンプ及びBeckman 166可変波長UV検出器を用いて行った。以下の移動相を使用した。0.05Mギ酸アンモニウム、pH3.5(A1)及びアセトニトリル(B1)、アセトニトリル/H2O:50/7(B2)、メタノール(B3)及びH2O中0.01Mギ酸(A2)。分析LCでは、Jones Chromatography Genesis C18、4μm、250×4.6mm(内径)カラムを流量1.5ml/分で使用した。セミ分取LCでは、Jones Chromatography Genesis C18、4μm、250×10mm(内径)カラムを流量4ml/分で使用した。自動合成システムSynthiaをLCでの注入及び画分回収に使用した。データ収集及びLC制御はBeckman System Goldクロマトグラフィーソフトウェアパッケージを用いて行った。
【0115】
放射能はVeenstra Instrumenten bvのVDC−202電離箱で測定した。製造時の放射能の概算ではLangenas Eltekniska ABの携帯線量率計を使用した。
【0116】
実施例2
[カルボニル−11C]ケトンの合成
テトラキス(トリフェニルホスフィン)パラジウム(0)(5.0mg、4.3μmol)をバイアル(1mL)に入れた。バイアルを窒素で洗い流し、内容物をTHF(200μL)に溶解した。アリールトリフレート(30.8μmol)及びLiBr(THF中0.46M溶液10μL、4.6μmol)を加えた。混合物を振盪して溶液を均一にした。アリール又はアルキルボロン酸(49.2μmol)をTHF(100μL)に溶解し、使用直前にパラジウム錯体、トリフレート及び臭化リチウムの溶液に加えた。得られた混合物を装置の注入ループに注入し、その装置から適量(200μL)を加圧下で、ヘリウム中の[11C]一酸化炭素で予め充填したマイクロオートクレーブに移した。マイクロオートクレーブを5分間加熱した(150℃)。粗生成物を、予め排気した、セプタムを取り付けたバイアル(5mL)に移した。マイクロオートクレーブをTHF(200μL)で充填し、回収バイアルに移し替えて空にした。バイアルを窒素で浄化した前後の放射能を測定した。75℃で加熱し窒素で浄化することで、溶媒体積を0.2mL未満に減少させた。アセトニトリル/水混合物(1:1、2mL)を加え、得られた溶液をセミ分取LC上に注入した。回収された画分の特性及び放射化学純度を分析LCで評価した。
【0117】
実施例3
11C−アミンの合成
テトラキス(トリフェニルホスフィン)パラジウム(0)(5.0mg、4.3μmol)、アリールトリフレート(30.8μmol)及びアリール又はメチルボロン酸(49.2μmol)を用いて上記のように[11C]カルボニル化を行った。粗生成物を、予め排気した、セプタムを取り付けたバイアル(5mL)に移し、放射能を測定した。バイアルを窒素で浄化して未反応[11C]一酸化炭素を除去し、放射能を再度測定した。四塩化チタン(CH2Cl2中1M溶液500μL、0.5mmol)及びアミン(0.13mmol)を加え、混合物を65℃で3分間加熱した。シアノ水素化ホウ素ナトリウム(MeOH中1M溶液500μL、0.5mmol)を加え、同温度で更に3分間加熱した。水酸化ナトリウム水溶液(2M水溶液500μL)をゆっくり加えることで反応液をクエンチし、濾過した。60℃で加熱し窒素で浄化することで濾液の体積を1mL未満に減少させた。粗生成物を、生成物同定用の少量の非放射性参照化合物の存在下、HPLCで分析した。放射化学収量を、生成物純度に捕捉効率を掛けることで求めた。
特定の実施形態、参考文献の引用
本発明の範囲は、本明細書に記載の特定の実施形態に限定されるものではない。実際、本明細書に記載の実施形態以外に、本発明の各種の変形形態が、上記の記載及び添付の図面より当業者には明らかであろう。このような変形形態は添付の特許請求の範囲内にあるものとする。
【0118】
各種刊行物及び特許出願が本明細書で引用され、その開示が全体として参照により組み込まれる。
【図面の簡単な説明】
【0119】
【図1】本発明の方法に関するフローチャートを示す図である。
【図2】本発明の一酸化炭素同位体の製造及び標識システムの模式図である。
【図3a】本発明の反応室の代替実施形態を示す図である。
【図3b】本発明の反応室の代替実施形態を示す図である。
【特許請求の範囲】
【請求項1】
(a)その底面に液体入口及び気体入口を有する高圧反応室を用意する段階と、
(b)遷移金属錯体と混合された、トリフレート、ボロン酸を含む標識すべき溶液を用意する段階と、
(c)気体入口を通じてUV反応器アセンブリーの反応室に一酸化炭素同位体の濃度が高い混合気体を導入する段階と、
(d)液体入口を通じて反応室に、遷移金属錯体と混合された前記溶液を高圧で導入する段階と、
(e)標識合成が行われている所定時間待機する段階と、
(f)反応室から標識ケトンを除去する段階とを含むケトンの標識合成方法。
【請求項2】
一酸化炭素同位体の濃度が高い混合気体が、
(a)好適なキャリアガス中の二酸化炭素同位体を用意する段階と、
(b)反応装置内に前記混合気体を導入することで二酸化炭素同位体を一酸化炭素同位体に転換する段階と、
(c)前記キャリアガスではなく一酸化炭素同位体が捕捉される一酸化炭素捕捉装置内に一酸化炭素同位体を捕捉する段階と、
(d)前記捕捉装置から前記捕捉された一酸化炭素同位体を明確に定義されたマイクロプラグで放出することにより、ある体積の一酸化炭素同位体の濃度が高い混合気体を得る段階とを含む方法により製造される、請求項1記載の方法。
【請求項3】
炭素同位体が11C、13C又は14Cである、請求項1記載の方法。
【請求項4】
炭素同位体が11Cである、請求項1記載の方法。
【請求項5】
擬似一相系を維持するために、遷移金属錯体と混合された標識すべき溶液を導入する段階が、導入前の圧力の約80倍の圧力で行われる、請求項1記載の方法。
【請求項6】
所定時間待機する段階が、反応室の温度を調整して標識合成を促進する段階を含む、請求項1記載の方法。
【請求項7】
遷移金属錯体がパラジウム金属錯体である、請求項1記載の方法。
【請求項8】
トリフレートが式R1−OTfを有し、R1が直鎖若しくは環状アルキル若しくは置換アルキル、アリール又は置換アリールである、請求項1記載の方法。
【請求項9】
R1がC6H5、4−CH3O−C6H4、4−CH3−C6H4、4−NO2−C6H4、C10H7又は
【化1】
から選択される、請求項8記載の方法。
【請求項10】
ボロン酸が式RB(OH)2を有し、Rが直鎖若しくは環状アルキル若しくは置換アルキル、アリール又は置換アリールである、請求項1記載の方法。
【請求項11】
Rがフェニル、メチル、
【化2】
又はチエニルから選択される、請求項10記載の方法。
【請求項12】
反応を促進するために、標識すべき溶液が臭化リチウムと更に混合されている、請求項1記載の方法。
【請求項13】
式R1−C*O−Rを有し、*が標識炭素位置であり、R1及びRが独立に直鎖若しくは環状アルキル若しくは置換アルキル、アリール又は置換アリールである、請求項1記載の方法に従って合成された標識ケトン。
【請求項14】
(a)請求項13記載の標識ケトンを合成する段階と、
(b)TiCl4及びNaBH3CNの存在下、標識ケトンを異なるアミンで還元アミン化する段階とを含むアミンの標識合成方法。
【請求項15】
段階(b)のアミンが式R′R″NHを有し、R′がH、直鎖若しくは環状アルキル若しくは置換アルキル、アリール又は置換アリールであり、R″が直鎖若しくは環状アルキル若しくは置換アルキル、アリール又は置換アリールである、請求項14記載の方法。
【請求項16】
下記式を有する、請求項14記載の方法に従って合成された標識アミン。
式中、*は標識炭素位置であり、R1及びRは独立に直鎖若しくは環状アルキル若しくは置換アルキル、アリール又は置換アリールであり、R′はH、直鎖若しくは環状アルキル若しくは置換アルキル、アリール又は置換アリールであり、R″は直鎖若しくは環状アルキル若しくは置換アルキル、アリール又は置換アリールである。
【請求項17】
請求項13記載の炭素同位体で標識したケトンを含む、PET研究用キット。
【請求項18】
請求項16記載の炭素同位体で標識したケトンを含む、PET研究用キット。
【請求項19】
放射線防護剤、抗菌防腐剤、pH調整剤又は充填剤を更に含む、請求項18記載のキット。
【請求項20】
放射線防護剤がアスコルビン酸、p−アミノ安息香酸、ゲンチジン酸及びそれらの塩から選択される、請求項19記載のキット。
【請求項21】
抗菌防腐剤がパラベン、ベンジルアルコール、フェノール、クレゾール、セトリミド及びチオマーサルから選択される、請求項19記載のキット。
【請求項22】
pH調整剤が薬学的に許容できる緩衝液若しくは薬学的に許容できる塩基又はその混合物である、請求項19記載のキット。
【請求項23】
充填剤が無機塩、水溶性糖又は糖アルコールである、請求項19記載のキット。
【請求項1】
(a)その底面に液体入口及び気体入口を有する高圧反応室を用意する段階と、
(b)遷移金属錯体と混合された、トリフレート、ボロン酸を含む標識すべき溶液を用意する段階と、
(c)気体入口を通じてUV反応器アセンブリーの反応室に一酸化炭素同位体の濃度が高い混合気体を導入する段階と、
(d)液体入口を通じて反応室に、遷移金属錯体と混合された前記溶液を高圧で導入する段階と、
(e)標識合成が行われている所定時間待機する段階と、
(f)反応室から標識ケトンを除去する段階とを含むケトンの標識合成方法。
【請求項2】
一酸化炭素同位体の濃度が高い混合気体が、
(a)好適なキャリアガス中の二酸化炭素同位体を用意する段階と、
(b)反応装置内に前記混合気体を導入することで二酸化炭素同位体を一酸化炭素同位体に転換する段階と、
(c)前記キャリアガスではなく一酸化炭素同位体が捕捉される一酸化炭素捕捉装置内に一酸化炭素同位体を捕捉する段階と、
(d)前記捕捉装置から前記捕捉された一酸化炭素同位体を明確に定義されたマイクロプラグで放出することにより、ある体積の一酸化炭素同位体の濃度が高い混合気体を得る段階とを含む方法により製造される、請求項1記載の方法。
【請求項3】
炭素同位体が11C、13C又は14Cである、請求項1記載の方法。
【請求項4】
炭素同位体が11Cである、請求項1記載の方法。
【請求項5】
擬似一相系を維持するために、遷移金属錯体と混合された標識すべき溶液を導入する段階が、導入前の圧力の約80倍の圧力で行われる、請求項1記載の方法。
【請求項6】
所定時間待機する段階が、反応室の温度を調整して標識合成を促進する段階を含む、請求項1記載の方法。
【請求項7】
遷移金属錯体がパラジウム金属錯体である、請求項1記載の方法。
【請求項8】
トリフレートが式R1−OTfを有し、R1が直鎖若しくは環状アルキル若しくは置換アルキル、アリール又は置換アリールである、請求項1記載の方法。
【請求項9】
R1がC6H5、4−CH3O−C6H4、4−CH3−C6H4、4−NO2−C6H4、C10H7又は
【化1】
から選択される、請求項8記載の方法。
【請求項10】
ボロン酸が式RB(OH)2を有し、Rが直鎖若しくは環状アルキル若しくは置換アルキル、アリール又は置換アリールである、請求項1記載の方法。
【請求項11】
Rがフェニル、メチル、
【化2】
又はチエニルから選択される、請求項10記載の方法。
【請求項12】
反応を促進するために、標識すべき溶液が臭化リチウムと更に混合されている、請求項1記載の方法。
【請求項13】
式R1−C*O−Rを有し、*が標識炭素位置であり、R1及びRが独立に直鎖若しくは環状アルキル若しくは置換アルキル、アリール又は置換アリールである、請求項1記載の方法に従って合成された標識ケトン。
【請求項14】
(a)請求項13記載の標識ケトンを合成する段階と、
(b)TiCl4及びNaBH3CNの存在下、標識ケトンを異なるアミンで還元アミン化する段階とを含むアミンの標識合成方法。
【請求項15】
段階(b)のアミンが式R′R″NHを有し、R′がH、直鎖若しくは環状アルキル若しくは置換アルキル、アリール又は置換アリールであり、R″が直鎖若しくは環状アルキル若しくは置換アルキル、アリール又は置換アリールである、請求項14記載の方法。
【請求項16】
下記式を有する、請求項14記載の方法に従って合成された標識アミン。
式中、*は標識炭素位置であり、R1及びRは独立に直鎖若しくは環状アルキル若しくは置換アルキル、アリール又は置換アリールであり、R′はH、直鎖若しくは環状アルキル若しくは置換アルキル、アリール又は置換アリールであり、R″は直鎖若しくは環状アルキル若しくは置換アルキル、アリール又は置換アリールである。
【請求項17】
請求項13記載の炭素同位体で標識したケトンを含む、PET研究用キット。
【請求項18】
請求項16記載の炭素同位体で標識したケトンを含む、PET研究用キット。
【請求項19】
放射線防護剤、抗菌防腐剤、pH調整剤又は充填剤を更に含む、請求項18記載のキット。
【請求項20】
放射線防護剤がアスコルビン酸、p−アミノ安息香酸、ゲンチジン酸及びそれらの塩から選択される、請求項19記載のキット。
【請求項21】
抗菌防腐剤がパラベン、ベンジルアルコール、フェノール、クレゾール、セトリミド及びチオマーサルから選択される、請求項19記載のキット。
【請求項22】
pH調整剤が薬学的に許容できる緩衝液若しくは薬学的に許容できる塩基又はその混合物である、請求項19記載のキット。
【請求項23】
充填剤が無機塩、水溶性糖又は糖アルコールである、請求項19記載のキット。
【図1】

【図2】
【図3a】
【図3b】
【図2】
【図3a】
【図3b】
【公表番号】特表2007−522106(P2007−522106A)
【公表日】平成19年8月9日(2007.8.9)
【国際特許分類】
【出願番号】特願2006−546374(P2006−546374)
【出願日】平成16年12月20日(2004.12.20)
【国際出願番号】PCT/IB2004/004199
【国際公開番号】WO2005/066100
【国際公開日】平成17年7月21日(2005.7.21)
【出願人】(305040710)ジーイー・ヘルスケア・リミテッド (99)
【Fターム(参考)】
【公表日】平成19年8月9日(2007.8.9)
【国際特許分類】
【出願日】平成16年12月20日(2004.12.20)
【国際出願番号】PCT/IB2004/004199
【国際公開番号】WO2005/066100
【国際公開日】平成17年7月21日(2005.7.21)
【出願人】(305040710)ジーイー・ヘルスケア・リミテッド (99)
【Fターム(参考)】
[ Back to top ]