
三次元集積回路用の層間充填材組成物、塗布液及び三次元集積回路の製造方法
【課題】半導体デバイスチップの3D積層化において、半導体デバイスチップ間のはんだバンプ等とランドの接合と同時に、熱伝導性の高い層間充填層を形成する層間充填材組成物、塗布液及び三次元集積回路の製造方法を提供する。
【解決手段】三次元集積回路用の層間充填材組成物が、120℃における溶融粘度が100Pa・s以下である樹脂(A)及びフラックス(B)を含有し、フラックス(B)の含有量が樹脂(A)100重量部当たり0.1重量部以上10重量部以下であるか、又は、120℃における溶融粘度が100Pa・s以下であって、熱伝導率が0.2W/mK以上である樹脂(A)と、熱伝導率が2W/mK以上、体積平均粒径が0.1μm以上5μm以下、かつ、最大体積粒径が10μm以下である無機フィラー(C)と、硬化剤(D)及び/またはフラックス(B)とを含有することを特徴とする。
【解決手段】三次元集積回路用の層間充填材組成物が、120℃における溶融粘度が100Pa・s以下である樹脂(A)及びフラックス(B)を含有し、フラックス(B)の含有量が樹脂(A)100重量部当たり0.1重量部以上10重量部以下であるか、又は、120℃における溶融粘度が100Pa・s以下であって、熱伝導率が0.2W/mK以上である樹脂(A)と、熱伝導率が2W/mK以上、体積平均粒径が0.1μm以上5μm以下、かつ、最大体積粒径が10μm以下である無機フィラー(C)と、硬化剤(D)及び/またはフラックス(B)とを含有することを特徴とする。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、三次元集積回路用の層間充填材組成物、該層間充填材組成物を含有する塗布液、及び該層間充填材組成物を含む三次元集積回路の製造方法に関する。
【背景技術】
【0002】
近年、半導体デバイスの更なる高速化・高容量化などの性能向上のために、トランジスタや配線の微細化に加えて、半導体デバイスチップを2層以上積み重ねた三次元(3D)積層化した三次元集積回路による性能向上に向けた研究開発が進められている。
三次元集積回路では、半導体デバイスチップ同士がそのチップ間において、はんだバンプ等の電気信号端子等で接続されていると同時に、層間充填材組成物を充填して形成された層間充填層により接着された構造を有している。
【0003】
具体的には、ウェハー上に塗布により層間充填材組成物の薄膜を形成した後に、Bステージ化を行い、次いでダイシングによりチップを切り出し、このチップを用いて加圧加熱による仮接合を繰り返し、最終的に加圧加熱条件下で本接合(半田接合)を行うプロセスが提案されている(非特許文献1参照)。
【0004】
このような三次元集積回路デバイスの実用化に向けて、種々の課題が指摘されているが、その内の一つにトランジスタや配線等のデバイスから発する熱の放熱問題がある。この問題は、半導体デバイスチップの積層の際に用いられる層間充填材組成物の熱伝導率が、金属やセラミックなどに比べ一般的に非常に低いことに起因し、積層デバイスチップ内での蓄熱によるパフォーマンスの低下が懸念されている。
【0005】
この課題を解決する一つの手法として、層間充填材組成物の高熱伝導化が挙げられる。例えば、層間充填材組成物を構成する樹脂単体として高熱伝導性のエポキシ樹脂を使用したり、そのような樹脂と高熱伝導性無機フィラーとを複合化させることで、層間充填材組成物を高熱伝導化することが行われている。例えば、特許文献1には、球状窒化ホウ素凝集体をフィラーとして配合した層間充填材組成物が記載されている。窒化ホウ素は通常板状粒子であり、その長径方向と短径方向とで熱伝導率が異なる性質を有するが、窒化ホウ素粒子をバインダーで結合して球状の凝集体とすることで、熱伝導率が各方向で均一化するため、該窒化ホウ素凝集体をフィラーとして樹脂に配合することで熱伝導率が向上することが開示されている。
【0006】
さらに、エポキシ樹脂自体の熱伝導性を向上させる発明として、エポキシ樹脂にメソゲン骨格を導入する方法が開示されている。例えば、非特許文献2には、種々のメソゲン骨格の導入によるエポキシ樹脂の熱伝導性向上についての記載があるが、熱伝導性の向上は見られるものの、コスト面、プロセス適合性、耐加水分解性や熱安定性などのバランスを考慮すると実用的とは言えない。
また、特許文献2には、ビフェニル骨格のみを用いた熱伝導性のよいエポキシ樹脂が開示されているが、合成されているのはごく低分子量のエポキシ樹脂のみであり、製膜性に欠けるため、薄膜として用いることが困難である。
【0007】
さらに、無機フィラーを含むエポキシ樹脂組成物においては、無機フィラー表面で樹脂の剥離が起こることがあり、所望の熱伝導率が達成されない場合があった。また、エポキシ当量が大きくないメソゲン骨格を有するエポキシ樹脂は、硬化後に樹脂が結晶性が高く固い構造をとることが多く、熱伝導性と低応力化とのバランスが求められている。
【0008】
一方、従来の半導体デバイスチップのインターポーザ等への搭載プロセスにおいては、初めに半導体デバイスチップ側のはんだバンプ等の電気信号端子をフラックスにより活性化処理を行い、次いでランド(電気接合電極)を有する基板に接合した後、基板間に液状樹脂又は液状樹脂に無機フィラーを添加したアンダーフィル材により充填・硬化により接合完了としている。この際、フラックスには、はんだバンプ等の金属電気信号端子及びランドの表面酸化膜除去や濡れ広がり性の向上、更には金属端子表面の再酸化防止などの活性化処理機能が求められている。
フラックスとしては、一般に、電気信号端子の金属酸化膜溶解能に優れたハロゲンを含む無機金属塩の他に、有機酸や有機酸塩、有機ハロゲン化合物やアミン類、ロジンやその構成成分の単独又は複数の組合せにより用いられている(例えば、非特許文献3参照)。
【0009】
また、半導体デバイスチップの3D積層プロセスにおいては、初めにフラックスを用いたはんだバンプ等の電気信号端子の活性化処理を行うと、端子表面に熱伝導性の低いフラックス層が形成され、層間充填材組成物による積層基板間の熱伝導性の阻害や、フラックス成分の残留による接合端子の腐食劣化等の要因となることが懸念されている。このため、高い熱伝導性を有する層間充填材組成物へ直接混合可能であり、且つ金属端子への腐食性の低いフラックスが求められている。
【0010】
上記のように、高熱伝導性の層間充填材組成物には、3D積層プロセスへの適合性や薄膜化に加えて、半導体デバイスチップ間における電気信号端子の接合性などが求められており、更なる技術開発が必要とされている。
【先行技術文献】
【特許文献】
【0011】
【特許文献1】特表2008−510878号公報
【特許文献2】特開2010−001427号公報
【非特許文献】
【0012】
【非特許文献1】エレクトロニクス実装学会講演大会講演論文集、61,23,2009)
【非特許文献2】電子部品用エポキシ樹脂の最新技術(シーエムシー出版、2006年、第1章P24〜31、第5章P114〜121)
【非特許文献3】はんだ付けの基礎と応用(工業調査会)
【発明の概要】
【発明が解決しようとする課題】
【0013】
三次元集積回路デバイスの実用化に向けての課題の一つに、トランジスタや配線等のデバイスから発する熱の放熱問題がある。この問題は、半導体デバイスチップの積層の際に用いられる層間充填材組成物の熱伝導率が、金属やセラミックなどに比べ一般的に非常に低いことに起因し、積層デバイスチップ内での蓄熱によるパフォーマンスの低下が懸念されており、より熱伝導率が高い層間充填材組成物が望まれている。
また、上記三次元集積回路は、更なる高速化・高容量化などの性能向上のために各チップ間の距離がチップ間距離10〜50μm程度にまで小さくなっている。
チップ間の層間充填層において、熱伝導率をより高めるために、場合によって配合されるフィラーの最大体積粒径は、層間充填層の厚みの1/3以下程度にする必要があるが、熱伝導率を高めるにはより多量のフィラーを配合することが好ましい。特に、微細なフィラーを使用する場合、多量のフィラーを配合する必要があるが、フィラーの配合量が多くなりすぎると、層間充填材組成物の接着力が低下したり、溶融時の柔軟性が低下してしまう場合がある。
上記特許文献1記載の球状窒化ホウ素凝集体は高熱伝導率を有するが、粒径が大きすぎるため、上記層間充填材組成物へのフィラーとして使用することができない。
そのため、より微細なフィラーを使用する必要があるが、粒径が小さなフィラーを使用すると、層間充填材組成物を構成する樹脂に配合したときに均一に混合することが困難であると共に、必要な熱伝導パス数が増加してチップ間の厚み方向に上から下まで繋がる確率が小さくなり、層間充填層の厚み方向への熱伝導率が不十分になる可能性がある。
【0014】
また、三次元集積回路用の層間充填材組成物としては、熱硬化性樹脂の反応の中間的な段階であって、材料は加熱により軟化して膨張するが、液体と接触しても完全には溶融又は溶解しない段階であるB−ステージ化膜後に、加温により軟化して溶融粘度が大きく低減することにより、3D積層プロセスにおける基板間の加圧によりはんだバンプ等の圧着接合を行う。この為、層間充填材組成物の硬化プロファイルとしては、B−ステージ化やはんだバンプの接合温度では完全硬化せず、短時間の流動性を有した後にゲル化して、その後に完全硬化することが重要である。そのため、層間充填材組成物を構成する樹脂には、チップ間を接合するための接着力や三次元集積回路の製造プロセスに適するように、該樹脂は加熱温度によって溶融粘度がコントロールできることが求められている。
【0015】
また、三次元集積回路では半導体デバイスチップ同士がそのチップ間において、はんだバンプ等の電気信号端子等で電気的に接続されていると同時に、層間充填材組成物を充填して形成された層間充填層により接着されている必要があるが、層間充填材組成物中で電気信号端子が電気的に確実に接続することが困難な場合があった。
本発明では、三次元集積回路用の層間充填材組成物中に、フラックスを含有することにより、層間充填材組成物中で電気信号端子が電気的に確実に接続できるようにする。ただし、この場合でも、一部のフラックス成分については、エポキシ樹脂成分のモノマー、オリゴマー及びポリマーや有機溶媒に対する溶解性が低く、層間充填材組成物との混合により均一に溶解することが困難になる場合があった。
【0016】
また、フラックスが有する酸性又は塩基性の官能基は、エポキシ樹脂成分に対して、フラックス作用と同時に硬化剤としての作用を呈し、B−ステージ化やはんだバンプ接合前の温度において、エポキシ樹脂の硬化を引き起こして、はんだバンプ等とランドの接合を阻害するという可能性も予想される。
【0017】
本発明は、上記課題に鑑みてなされたものであり、半導体デバイスチップの3D積層プロセスにおいて、半導体デバイスチップ間のはんだバンプ等とランドの接合性に優れると同時に、熱伝導性の高い層間充填層を形成する層間充填材組成物、該層間充填材組成物を含有する塗布液、及び該層間充填材組成物を含む三次元集積回路の製造方法を提供することを目的とするものである。
【課題を解決するための手段】
【0018】
本発明者等は、鋭意研究の結果、下記する発明が上記課題を解決出来ることを見出し、本発明を完成するに至った。
すなわち、本発明は、以下の要旨を有するものである。
1.120℃における溶融粘度が100Pa・s以下である樹脂(A)及びフラックス(B)を含有し、フラックス(B)の含有量が、樹脂(A)100重量部当たり0.1重量部以上10重量部以下であることを特徴とする三次元集積回路用の層間充填材組成物。
2.更に硬化剤(C)を含有する、上記1に記載の三次元集積回路用の層間充填材組成物。
3.熱伝導率が1W/mK以上の無機フィラー(D)を樹脂(A)100重量部当たり、50重量部以上400重量部以下更に含有する上記1又は2に記載の三次元集積回路用の層間充填材組成物。
4.120℃における溶融粘度が100Pa・s以下であって、熱伝導率が0.2W/mK以上である樹脂(A)と、熱伝導率が2W/mK以上、体積平均粒径が0.1μm以上5μm以下、かつ、最大体積粒径が10μm以下である無機フィラー(D)と、硬化剤(C)及び/またはフラックス(B)とを含有することを特徴とする三次元集積回路用の層間充填材組成物。
【0019】
5.前記樹脂(A)の50℃における溶融粘度が2000Pa・s以上である、上記1から4のいずれか1項に記載の三次元集積回路用の層間充填材組成物。
6.前記樹脂(A)が熱硬化性樹脂である、上記1から5のいずれか1項に記載の三次元集積回路用の層間充填材組成物。
7.前記樹脂(A)がエポキシ樹脂である、上記1から6のいずれか1項に記載の三次元集積回路用の層間充填材組成物。
8.前記エポキシ樹脂が、フェノキシ樹脂であるエポキシ樹脂(A1)、又はエポキシ樹脂(A1)と、分子内に2個以上のエポキシ基を有するエポキシ樹脂であるエポキシ樹脂(A2)との混合物である上記7に記載の三次元集積回路用の層間充填材組成物。
【0020】
9.前記エポキシ樹脂が、下記式(1)で表され、かつエポキシ当量が2,500g/当量以上30,000g/当量以下を有するエポキシ樹脂(B)である上記7に記載の三次元集積回路用の層間充填材組成物。
【化1】

(式(1)中、Aは下記式(2)で表されるビフェニル骨格であり、Bは水素原子または下記式(3)で表される基であり、nは繰り返し数であり、平均値は1<n<100である。)
【化2】
(式(2)中、R1は、水素原子、炭素数1〜10の炭化水素基、またはハロゲン元素であり、互いに同一であっても異なっていてもよい。)
【化3】
10.前記式(2)におけるR1が、水素原子または炭素数が1〜4のアルキル基であり、式(2)で表されるビフェニル骨格は少なくとも一つの水素原子と少なくとも一つの炭素数1〜4のアルキル基を有する、上記9に記載の三次元集積回路用の層間充填材組成物。
【0021】
11.更にエポキシ当量が2500g/当量未満であるエポキシ樹脂(C)を含有する上記9又は10に記載の三次元集積回路用の層間充填材組成物。
12.エポキシ樹脂(B)とエポキシ樹脂(C)を含む全エポキシ樹脂中のエポキシ樹脂(C)の割合が、10重量%以上80重量%以下である上記11に記載の三次元集積回路用の層間充填材組成物。
13.前記フラックス(B)が、有機カルボン酸である上記1から12のいずれか1項に記載の三次元集積回路用の層間充填材組成物。
14.前記有機カルボン酸の分解温度が、130℃以上である上記13に記載の三次元集積回路用の層間充填材組成物。
15.前記樹脂(A)と前期無機フィラー(D)との総体積に対し、無機フィラー(D)が5体積%以上60体積%以下である、上記3から14のいずれか1項に記載の層間充填材組成物。
16.前期無機フィラー(D)が、窒化ホウ素フィラーである、上記3から15のいずれか1項に記載三次元集積回路用の層間充填材組成物。
17.前記硬化剤(C)が、イミダゾール又はその誘導体である、上記2から16のいずれか1項に記載の三次元集積回路用の層間充填材組成物。
18.上記1から17のいずれか1項に記載の層間充填材組成物に、更に有機溶媒(E)を含有してなる、三次元集積回路用の層間充填材組成物の塗布液。
19.複数の半導体基板表面に、上記1から17のいずれか1項に記載の層間充填材組成物を成膜した後に、これらの半導体基板を加圧接着して積層する工程を含む、三次元集積回路の製造方法。
【発明の効果】
【0022】
本発明によると、半導体デバイス基板間のはんだバンプ等とランドの接合と同時に、熱伝導性の高い層間充填層を形成する層間充填材組成物、該層間充填材組成物を含有する塗布液、及び該層間充填材組成物を含む三次元集積回路の製造方法を提供することができる。
本発明の三次元集積回路用の層間充填材組成物によれば、半導体デバイスチップの3D積層プロセスにおいて、半導体デバイスチップ間のはんだバンプ等とランドとの接合性に優れると共に、特に、熱伝導率が高く微細な無機フィラーを熱伝導率が高いエポキシ樹脂によって接合するため、放熱性が高く、無機フィラーを使用した場合には更に、熱伝導性の高い層間充填層を形成することができる。
【図面の簡単な説明】
【0023】
【図1】接合評価に用いるはんだバンプ基板の模式図であり、(a)は平面図、(b)は断面図である。
【図2】実施例3の層間充填剤ペーストの粒度分布の評価結果である。
【図3】実施例3の層間充填剤ペーストから得られたB−ステージ化膜の表面写真である。
【図4】比較例3の層間充填剤ペーストの粒度分布の評価結果である。
【図5】比較例3の層間充填剤ペーストから得られたB−ステージ化膜の表面写真である。
【発明を実施するための形態】
【0024】
以下、本発明の実施の形態について説明するが、本発明は以下の実施の形態に限定されるものではなく、その要旨の範囲内で種々変形して実施することができる。
尚、本明細書において「〜」という表現を用いる場合、その前後の数値を含む表現として用いる。
【0025】
[三次元集積回路用の層間充填材組成物]
本発明の三次元集積回路用の層間充填材組成物(本発明では、単に層間充填材組成物ということがある。)は、三次元集積回路の各層を構成する半導体基板同士を接着し、半導体基板同士の間隙を充填する充填層を形成することができる組成物である。
この三次元集積回路用の層間充填材組成物においては、樹脂(A)の溶融粘度が120℃において100Pa・s以下であることにより、はんだバンプが融解する前に樹脂を溶融させて粘度を大きく低減することにより、加熱プレスによりはんだバンプとランド端子の接合を可能とし、更に半導体基板上に成膜された層間充填材組成物を200℃以上で加圧接着することにより、はんだバンプを融解させてランド端子との電気接続を実現することが出来る。この際、所定の硬化剤を添加することにより、B−ステージ化や、はんだバンプの接合温度では硬化せず、はんだバンプの接合後に短時間の流動性を有した後にゲル化して、その後に完全硬化することにより、安定な層間充填膜を形成することが出来る。
また、樹脂(A)の50℃における溶融粘度が2000Pa・s以上であると、B−ステージ化膜後の室温におけるタック性を低減し、基板の積み重ね時の位置合わせを実施することにより、三次元集積回路の積層基板同士の仮接着が可能になり、好ましい。
以下、各成分について説明する。
【0026】
[樹脂(A)]
本発明における樹脂(A)は、仮接着後に本接合を実施する際に、加温により層間充填材組成物を溶融して電気接合端子を接続させるために、120℃における溶融粘度が100Pa・s以下であることを必須とし、20Pa・s以下であることが好ましい。また、樹脂(A)は、層間充填材組成物として基板上に薄膜を形成後、仮接着前に接合対象の基板と位置合わせを行うために、50℃における溶融粘度が2000Pa・s以上であることが好ましく、10000Pa・s以上であることがより好ましい。
後述する無機フィラー(D)と組み合わせた際に十分な熱伝導性を得るために、熱伝導率が好ましくは0.2W/mK以上、より好ましくは0.22W/mK以上である。
【0027】
上記樹脂(A)として、上記溶融粘度条件を満たす樹脂が使用できるが、熱硬化性樹脂であるのが好ましい。かかる熱硬化性樹脂の好ましい例としては、アクリル樹脂、エポキシ樹脂、熱硬化性ポリイミド樹脂、熱硬化性フェノール樹脂、などが挙げられる。なかでも、エポキシ樹脂、熱硬化性ポリイミド樹脂が好ましい。
本発明における樹脂(A)は、なかでも、エポキシ樹脂であるのが好ましい。エポキシ樹脂は1種類の構造単位を有するエポキシ樹脂のみでもよいが、上記溶融粘度条件を満たすならば、構造単位の異なる複数のエポキシ樹脂を組み合わせてもよい。
エポキシ樹脂は、塗膜性ないしは成膜性や接着性と併せて、接合時のボイドを低減して高熱伝導の硬化物を得るために、後述するフェノキシ樹脂(以下、エポキシ樹脂(A1)と称す。)を少なくとも含むことが好ましく、特にエポキシ樹脂全量に対するエポキシ樹脂(A1)の重量比率が、5〜95重量%。より好ましくは10〜90重量%、さらに好ましくは20〜80重量%の範囲で含有されることが好ましい。
【0028】
[エポキシ樹脂(A1)及びエポキシ樹脂(A2)]
フェノキシ樹脂とは、通常、エピハロヒドリンと2価フェノール化合物とを反応させて得られる樹脂、または2価のエポキシ化合物と2価のフェノール化合物とを反応させて得られる樹脂を指す。本発明においてはこれらのうち、特に重量平均分子量10000以上の高分子量エポキシ樹脂であるフェノキシ樹脂をエポキシ樹脂(A1)と言う。
【0029】
エポキシ樹脂(A1)としては、ナフタレン骨格、フルオレン骨格、ビフェニル骨格、アントラセン骨格、ピレン骨格、キサンテン骨格、アダマンタン骨格およびジシクロペンタジエン骨格からなる群から選択された少なくとも1つの骨格を有するフェノキシ樹脂が好ましい。中でも、耐熱性がより一層高められるので、フルオレン骨格および/またはビフェニル骨格を有するフェノキシ樹脂が特に好ましい。
【0030】
上述のようにエポキシ樹脂(A)は、構造単位の異なる複数のエポキシ樹脂を含むものであってもよい。
上記エポキシ樹脂(A1)以外のエポキシ樹脂としては、分子内に2個以上のエポキシ基を有するエポキシ樹脂(以下、エポキシ樹脂(A2)と称す。)であることが好ましい。エポキシ樹脂(A2)としては、例えば、ビスフェノールA型エポキシ樹脂、ビスフェノールF型エポキシ樹脂、ナフタレン型エポキシ樹脂、フェノールノボラック型エポキシ樹脂、クレゾールノボラック型エポキシ樹脂、フェノールアラルキル型エポキシ樹脂、ビフェニル型エポキシ樹脂、トリフェニルメタン型エポキシ樹脂、ジシクロペンタジエン型エポキシ樹脂、グリシジルエステル型エポキシ樹脂、グリシジルアミン型エポキシ樹脂、多官能フェノール型エポキシ樹脂等が挙げられる。これらは1種を単独でまたは2種以上の混合体として使用することができる。
【0031】
エポキシ樹脂(A2)は、溶融粘度制御の観点から、その平均分子量が、好ましくは、100〜5000であり、より好ましくは、200〜2000である。平均分子量が100より低いものでは、耐熱性が劣る傾向にあり、5000より高いと、エポキシ樹脂の融点が高くなり、作業性が低下する傾向がある。
【0032】
また、本発明において、エポキシ樹脂としては、その目的を損なわない範囲において、エポキシ樹脂(A1)とエポキシ樹脂(A2)以外のエポキシ樹脂(以下、他のエポキシ樹脂)を使用してもよい。他のエポキシ樹脂の含有量は、エポキシ樹脂(A1)とエポキシ樹脂(A2)の合計に対して、通常、50重量%以下、好ましくは、30重量%以下である。
【0033】
本発明でエポキシ樹脂(A2)を使用する場合、エポキシ樹脂(A1)とエポキシ樹脂(A2)を含む全エポキシ樹脂中のエポキシ樹脂(A1)の割合は、その合計を100重量%として、10〜90重量%、好ましくは20〜80重量%である。なお、「エポキシ樹脂(A1)とエポキシ樹脂(A2)を含む全エポキシ樹脂」とは、本発明のエポキシ樹脂組成物に含まれるエポキシ樹脂が、エポキシ樹脂(A1)及びエポキシ樹脂(A2)のみの場合には、エポキシ樹脂(A1)とエポキシ樹脂(A2)の合計を意味し、さらに他のエポキシ樹脂を含む場合には、エポキシ樹脂(A1)、エポキシ樹脂(A2)及び他のエポキシ樹脂の合計を意味する。
エポキシ樹脂(A1)の割合が10重量%以上であることにより、エポキシ樹脂(A1)を配合することによる熱伝導性の向上効果を十分に得ることができ、所望の高熱伝導性を得ることができる。エポキシ樹脂(A1)の割合が90重量%未満でエポキシ樹脂(A2)が10重量%以上であることにより、エポキシ樹脂(A2)の配合効果が発揮され、硬化性、硬化物の物性が十分なものとなる。
【0034】
[エポキシ樹脂(B)]
本発明において、エポキシ樹脂は、なかでも、下記式(1)で表される構造を有し、かつ、エポキシ当量が2500g/当量以上であるエポキシ樹脂(B)が好ましい。
【0035】
【化4】
(式(1)中、Aは下記式(2)で表わされるビフェニル骨格であり、Bは水素原子又は下記式(3)であり、nは繰り返し数であり、平均値は10<n<50である。)
【化5】
(式(2)中、R1は、水素原子、炭素数1〜10の炭化水素基、又はハロゲン原子であり、互いに同一であっても異なっていてもよい。)
【化6】
【0036】
エポキシ樹脂(B)は、十分な伸び性を有し、熱伝導性、耐熱性とのバランスに優れたエポキシ樹脂である。
従来の高熱伝導性エポキシ樹脂はほぼすべてが熱伝導性を高めるために設計されたエポキシ樹脂であり、硬化条件を含めた硬化プロセス等において制限があることが多く、その選択の自由度は低かった。このため、部材や封止剤、接着剤などの製品に従来の高熱伝導性エポキシ樹脂を適用しようとした場合、コストを含めた製品の要求物性と高熱伝導性を両立させることが困難であった。
これに対し、エポキシ樹脂(B)は、それ自体熱伝導性に優れ、エポキシ樹脂成分として所望の量添加することで硬化物の熱伝導性を高めることが出来、また、エポキシ樹脂(B)の伸び性に由来して、材料に低応力性を付与しうる。
【0037】
エポキシ樹脂(B)が伸び性に優れる理由の詳細は明らかではないが、引っ張りの応力がかかった際の延伸に耐えうる分子鎖長を有し、さらにその応力を緩和するために、重なり合ったビフェニル骨格同士が「滑る」ことができるためであると推測される。また、この時、結晶性が高すぎると、脆く、伸びずに破断してしまうため、適度にアモルファス部分を有していることが重要であるが、エポキシ樹脂(B)では、ビフェニル骨格が置換基を有することにより、結晶性を適度に低下させることができ、このことが伸び性の発現に繋がっていると考えられる。従って、伸び性の観点からは、前記式(2)におけるR1がすべて水素原子ではなく、1つ以上のR1が炭化水素基、又はハロゲン原子であることが好ましい。
【0038】
熱伝導はフォノンと伝導電子に支配され、金属のように自由電子を有する場合は伝導電子による寄与が大きいが、エポキシ樹脂は一般的に絶縁体であり、絶縁体においてはフォノンが熱伝導の主因子である。フォノンによる熱伝導は振動エネルギーの伝播であるので、振動が減衰しにくく、硬い材料であるほど熱伝導性に優れる。
エポキシ樹脂(B)が熱伝導性に優れる理由の詳細は明確ではないが、全ての骨格がビフェニル骨格であることから構造の自由度が少なく、振動エネルギーが減衰しにくいこと、またビフェニル骨格は平面性が高いため、分子間の重なりが良く、より分子運動を拘束できることによるものであると推定される。
【0039】
エポキシ樹脂は、一般に、結晶性がよい方が耐熱性に優れる傾向があり、同一構造のエポキシ樹脂であれば、樹脂の分子量、あるいはエポキシ当量が高い方が耐熱性に優れる傾向にある。エポキシ樹脂(B)は適度な結晶性とエポキシ当量の高さを有することにより耐熱性にも優れる。
【0040】
前記エポキシ樹脂(B)を表す式(1)中、nは繰り返し数であり、平均値である。その値の範囲は10<n<50であるが、伸び性と樹脂の取り扱いの両面のバランスから、nの範囲は、15<n<50であることが好ましく、とりわけ20<n<50であることが好ましい。nが10以下であると、エポキシ樹脂(B)の伸び性が不十分となり、50以上であるとエポキシ樹脂(B)を含む組成物の粘度が高くなり、取り扱いが困難となる傾向がある。
【0041】
また、前記式(1)中、Aは前記式(2)で表されるビフェニル骨格であり、前記式(2)において、R1は、互いに同一であっても異なっていてもよく、水素原子、炭素数1〜10の炭化水素基、又はハロゲン原子を表すが、1分子のエポキシ樹脂において、R1としては水素原子と炭素数1〜10の炭化水素基との両方を含んでいるものがエポキシ樹脂(B)の結晶性とハンドリングの観点から好ましい。R1が同一であると結晶性が高くなり、熱伝導性を高めることが可能であるが、結晶性が高すぎるとエポキシ樹脂組成物をフィルム成形したときの伸びが小さくなる傾向にある。
【0042】
前記式(2)におけるR1が、炭素数1〜10の炭化水素基である場合には、R1は好ましくは1〜4のアルキル基、特に好ましくはメチル基である。
尚、R1の炭化水素基は置換基を有していてもよく、その置換基は特に限定されるものではないが、分子量で200以下のものである。
また、R1のハロゲン原子とは、フッ素原子、塩素原子、臭素原子を指し、これらは1種のみでも複数種を含んでいてもよい。
【0043】
Aのビフェニル骨格は、2,2’−ビフェニル骨格、2,3’−ビフェニル骨格、2,4’−ビフェニル骨格、3,3−ビフェニル骨格、3,4’−ビフェニル骨格、4,4’−ビフェニル骨格のいずれでも良いが、好ましくは4,4’−ビフェニル骨格である。 また、R1としては、2位及び/又は6位に水素原子があることが好ましく、3位及び/又は5位に炭化水素基があることが好ましい。
【0044】
エポキシ樹脂(B)を使用する場合のエポキシ当量は、2,500g/当量以上であることが好ましい。エポキシ樹脂(B)のエポキシ当量が2,500g/当量未満の場合には、十分な伸び性が得られず、フィルム成形・塗布などのプロセスに適用する際に取扱いが困難になる場合がある。
伸び性の観点からは、エポキシ樹脂(B)のエポキシ当量は、好ましくは3,000g/当量以上、より好ましくは4,000g/当量以上である。
一方、エポキシ当量の上限値は特に限定はないが、取り扱い性・作業性という点で、好ましくは30,000g/当量以下が好ましく、より好ましくは15,000g/当量以下、更に好ましくは10,000g/当量以下である。エポキシ樹脂(B)のエポキシ当量は、後述の実施例の項に記載される方法で求められる。
【0045】
エポキシ樹脂(B)の重量平均分子量Mwは、10,000以上200,000以下であることが好ましい。重量平均分子量が10,000より低いものでは伸び性が低くなる傾向にあり、200,000より高いと樹脂の取り扱いが困難となる傾向にある。エポキシ樹脂(B)の重量平均分子量は、後述の実施例の項に記載される方法で求められる。
エポキシ樹脂(B)の熱伝導率(硬化前の熱伝導率)は、通常0.18W/mK以上、好ましくは0.19W/mK以上、さらに好ましくは0.20W/mK以上である。尚、一般的にエポキシ樹脂の熱伝導率はエポキシ樹脂の硬化物として評価されることが多く、一般的な硬化していないビスフェノールA型エポキシ樹脂の熱伝導率は通常この値よりも低く、液状であるため伸び性を測定するサンプル作製も不可能である場合が多い。
【0046】
本発明に係るエポキシ樹脂(B)は、硬化前の樹脂そのものの状態でも十分な製膜性と熱伝導率を有し、かつ伸び性とのバランスにも優れるものである。なお、エポキシ樹脂(A)の熱伝導率は、後述の実施例の項に記載される方法で測定される。
エポキシ樹脂(B)は耐熱性に優れるものであり、後掲の実施例の項で示すガラス転移温度Tgで評価した場合、90℃以上、220℃以下を達成することができる。エポキシ樹脂のTgは、本発明の用途では高い方が好ましく、好ましくは95℃以上、より好ましくは100℃以上、更に好ましくは105℃以上であるが、Tgが高過ぎると、加工プロセスで使用する加熱温度で硬化反応が十分に進行せず、品質が安定しなかったり、要求される物性が発現しない、といった問題が生じうるため、その上限は通常200℃であることが好ましい。
【0047】
以下、エポキシ樹脂(B)の製造方法について説明する。
エポキシ樹脂(B)は、例えば、ビフェニル骨格を有する2官能エポキシ樹脂(X)とビフェノール化合物(Y)を反応させる、二段法によって得ることができる。また、1種類又は2種類以上のビフェノール化合物(Y)とエピクロロヒドリンを直接反応させる、一段法によっても得られる。しかし、ビフェノール化合物(Y)は溶剤溶解性が良くないため、一般的に一段法に用いられる溶剤がそのまま適用できない場合があるので、二段法を用いることが好ましい。
【0048】
エポキシ樹脂(B)の製造に用いられる2官能エポキシ樹脂(X)は、ビフェニル骨格を有し、分子内に2個のエポキシ基を持つ化合物であり、下記式(4)で表されるビフェノール化合物をエピハロヒドリンと縮合させて得られるエポキシ樹脂等が挙げられる。
【化7】
(式(4)中、R2は式(2)におけるR1と同義である。)
【0049】
前記式(4)で表されるビフェノール化合物としては、例えば、2,2’−ビフェノール、2,3’−ビフェノール、2,4’−ビフェノール、3,3’−ビフェノール、3,4’−ビフェノール、4,4’−ビフェノール、2−メチル−4,4’−ビフェノール、3−メチル−4,4’−ビフェノール、2,2’−ジメチル−4,4’−ビフェノール、3,3’−ジメチル−4,4’−ビフェノール、3,3’,5,5’−テトラメチル−4,4’−ビフェノール、2,2’,3,3’,5,5’−ヘキサメチル−4,4’−ビフェノール、2,2’,3,3’,5,5’,6,6’−オクタメチル−4,4’−ビフェノール等が挙げられる。これらの中で好ましいものは、4,4’−ビフェノール、3,3’−ジメチル−4,4’−ビフェノール、3,3’,5,5’−テトラメチル−4,4’−ビフェノールである。エピハロヒドリンとの縮合反応を行う際には、これらのビフェノール化合物は単独で用いてもよく、また複数種を併用してもよい。また、このようなビフェノール化合物とエピハロヒドリンとを縮合させて得られた2官能エポキシ樹脂(X)を複数種併用することもできる。
【0050】
2官能エポキシ樹脂(X)中としては、その末端基不純物である加水分解性塩素濃度が200ppm以下であり、αグリコール基濃度が100meq/kg以下である2官能エポキシ樹脂(X)を原料として使用することが好ましい。加水分解性塩素濃度が200ppmより大きい場合や、αグリコール基濃度が100meq/kgより大きい場合には、十分に高分子量化しなくなり、好ましくない。
【0051】
一方、ビフェノール化合物(Y)は、2個の水酸基がビフェニル骨格に結合した化合物であり、前記式(4)で表される。ビフェノール化合物(Y)としては、上記と同様、例えば、2,2’−ビフェノール、2,3’−ビフェノール、2,4’−ビフェノール、3,3’−ビフェノール、3,4’−ビフェノール、4,4’−ビフェノール、2−メチル−4,4’−ビフェノール、3−メチル−4,4’−ビフェノール、2,2’−ジメチル−4,4’−ビフェノール、3,3’−ジメチル−4,4’−ビフェノール、3,3’,5,5’−テトラメチル−4,4’−ビフェノール、2,2’,3,3’,5,5’−ヘキサメチル−4,4’−ビフェノール、2,2’,3,3’,5,5’,6,6’−オクタメチル−4,4’−ビフェノール等が挙げられる。これらの中で好ましいものは、4,4’−ビフェノール、3,3’−ジメチル−4,4’−ビフェノール、3,3’,5,5’−テトラメチル−4,4’−ビフェノールである。これらのビフェノール化合物は複数種を併用することもできる。
【0052】
なお、上記2官能エポキシ樹脂(X)とビフェノール化合物(Y)に含まれるビフェニル骨格が同時に無置換でないことが好ましく、一分子中に1つ以上の置換基を有することが好ましい。全てが無置換のビフェニル骨格であると、得られるエポキシ樹脂(B)の結晶性が高くなり、伸び性が悪くなる傾向にある。
【0053】
エポキシ樹脂(B)の製造において、上記の2官能エポキシ樹脂(X)とビフェノール化合物(Y)の使用量は、その配合当量比で、エポキシ基:フェノール性水酸基=1:0.90〜1.10となるようにするのが好ましい。この当量比が上記範囲であることにより十分な高分子量化が進行する。
エポキシ樹脂(B)の合成には触媒を用いてもよく、その触媒としては、エポキシ基とフェノール性水酸基、アルコール性水酸基やカルボキシル基との反応を進めるような触媒能を持つ化合物であればどのようなものでもよい。例えば、アルカリ金属化合物、有機リン化合物、第3級アミン、第4級アンモニウム塩、環状アミン類、イミダゾール類等が挙げられる。これらの触媒は1種のみを使用することも、2種以上組み合わせて使用することもできる。
【0054】
アルカリ金属化合物の具体例としては、水酸化ナトリウム、水酸化リチウム、水酸化カリウム等のアルカリ金属水酸化物、炭酸ナトリウム、重炭酸ナトリウム、塩化ナトリウム、塩化リチウム、塩化カリウム等のアルカリ金属塩、ナトリウムメトキシド、ナトリウムエトキシド等のアルカリ金属アルコキシド、アルカリ金属フェノキシド、水素化ナトリウム、水素化リチウム等のアルカリ金属の水素化物、酢酸ナトリウム、ステアリン酸ナトリウム等の有機酸のアルカリ金属塩が挙げられる。
【0055】
有機リン化合物の具体例としては、トリ−n−プロピルホスフィン、トリ−n−ブチルホスフィン、トリフェニルホスフィン、テトラメチルホスフォニウムブロマイド、テトラメチルホスフォニウムアイオダイド、テトラメチルホスフォニウムハイドロオキサイド、トリメチルシクロヘキシルホスホニウムクロライド、トリメチルシクロヘキシルホスホニウムブロマイド、トリメチルベンジルホスホニウムクロライド、トリメチルベンジルホスホニウムブロマイド、テトラフェニルホスホニウムブロマイド、トリフェニルメチルホスホニウムブロマイド、トリフェニルメチルホスホニウムアイオダイド、トリフェニルエチルホスホニウムクロライド、トリフェニルエチルホスホニウムブロマイド、トリフェニルエチルホスホニウムアイオダイド、トリフェニルベンジルホスホニウムクロライド、トリフェニルベンジルホスホニウムブロマイドなどが挙げられる。
【0056】
第3級アミンの具体例としては、トリエチルアミン、トリ−n−プロピルアミン、トリ−n−ブチルアミン、トリエタノールアミン、ベンジルジメチルアミンなどが挙げられる。
第4級アンモニウム塩の具体例としては、テトラメチルアンモニウムクロライド、テトラメチルアンモニウムブロマイド、テトラメチルアンモニウムハイドロオキサイド、トリエチルメチルアンモニウムクロライド、テトラエチルアンモニウムクロライド、テトラエチルアンモニウムブロマイド、テトラエチルアンモニウムアイオダイド、テトラプロピルアンモニウムブロマイド、テトラプロピルアンモニウムハイドロオキサイド、テトラブチルアンモニウムクロライド、テトラブチルアンモニウムブロマイド、テトラブチルアンモニウムアイオダイド、ベンジルトリメチルアンモニウムクロライド、ベンジルトリメチルアンモニウムブロマイド、ベンジルトリメチルアンモニウムハイドロオキサイド、ベンジルトリブチルアンモニウムクロライド、フェニルトリメチルアンモニウムクロライドなどが挙げられる。
【0057】
環状アミン類の具体例としては、1,8−ジアザビシクロ(5,4,0)ウンデセン−7,1,5−ジアザビシクロ(4,3,0)ノネン−5等が挙げられる。
イミダゾール類の具体例としては、2−メチルイミダゾール、2−エチル−4−メチルイミダゾール、2−フェニルイミダゾールなどが挙げられる。
【0058】
触媒の使用量は反応固形分中、通常0.001〜1重量%である。なお、触媒として、アルカリ金属化合物を使用すると得られるエポキシ樹脂(B)中にアルカリ金属分が残留し、エポキシ樹脂(B)を成分として含む、本発明のエポキシ樹脂組成物をプリント配線板に使用した場合、使用したプリント配線板の絶縁特性を悪化させる傾向があるため、エポキシ樹脂中のLi,NaおよびKの含有量の合計が60ppm以下、好ましくは50ppm以下とする必要がある。
【0059】
また、有機リン化合物、第3級アミン、第4級アンモニウム塩、環状アミン類、イミダゾール類等を触媒として使用した場合も、得られるエポキシ樹脂(B)中にこれらが触媒残渣として残留し、アルカリ金属分の残留と同様にプリント配線板の絶縁特性を悪化させるので、エポキシ樹脂(B)中の窒素の含有量が300ppm以下であり、エポキシ樹脂(B)中のリンの含有量が300ppm以下である必要がある。さらに好ましくは、エポキシ樹脂(B)中の窒素の含有量が200ppm以下であり、エポキシ樹脂(B)中のリンの含有量が200ppm以下である。
【0060】
エポキシ樹脂(B)は、その製造時の合成反応の工程において、溶剤として有機溶媒を用いてもよく、その有機溶媒としては、エポキシ樹脂(B)を溶解するものであればどのようなものでもよい。例えば、芳香族系溶剤、ケトン系溶剤、アミド系溶剤、グリコールエーテル系溶剤などが挙げられる。
芳香族系溶剤の具体例としては、ベンゼン、トルエン、キシレンなどが挙げられる。
ケトン系溶剤の具体例としては、アセトン、メチルエチルケトン、メチルイソブチルケトン、2−ヘプタノン、4−ヘプタノン、2−オクタノン、シクロヘキサノン、アセチルアセトン、ジオキサンなどが挙げられる。
【0061】
アミド系溶剤の具体例としては、ホルムアミド、N−メチルホルムアミド、N,N−ジメチルホルムアミド、アセトアミド、N−メチルアセトアミド、N,N−ジメチルアセトアミド、2−ピロリドン、N−メチルピロリドンなどが挙げられる。
グリコールエーテル系溶剤の具体例としては、エチレングリコールモノメチルエーテル、エチレングリコールモノエチルエーテル、エチレングリコールモノ−n−ブチルエーテル、エチレングリコールジメチルエーテル、エチレングリコールモノエチルエーテルアセテート、ジエチレングリコールモノメチルエーテル、ジエチレングリコールモノエチルエーテル、ジエチレングリコールモノ−n−ブチルエーテル、ジエチレングリコールジメチルエーテル、ジエチレングリコールモノエチルエーテルアセテート、プロピレングリコールモノメチルエーテル、プロピレングリコールモノ−n−ブチルエーテル、プロピレングリコールモノメチルエーテルアセテートなどが挙げられる。
これらの有機溶媒は単独で用いてもよく、2種以上を併用することもできる。
【0062】
エポキシ樹脂(B)の製造時の合成反応における固形分濃度は35〜95重量%が好ましい。また、反応途中で高粘性生成物が生じたときは溶剤(有機溶媒)を追加添加して反応を続けることもできる。反応終了後、溶剤(有機溶媒)は必要に応じて、除去することもできるし、更に追加することもできる。
【0063】
エポキシ樹脂(B)の製造において、2官能エポキシ樹脂(X)とビフェノール化合物(Y)との重合反応は使用する触媒が分解しない程度の反応温度で実施される。反応温度が高すぎると生成するエポキシ樹脂が劣化するおそれがある。逆に温度が低すぎると十分に反応が進まないことがある。これらの理由から反応温度は、好ましくは50〜230℃、より好ましくは120〜200℃である。また、反応時間は通常1〜12時間、好ましくは3〜10時間である。アセトンやメチルエチルケトンのような低沸点溶剤を使用する場合には、オートクレーブを使用して高圧下で反応を行うことで反応温度を確保することができる。
【0064】
[エポキシ樹脂(C)]
本発明において、エポキシ樹脂(B)は、単独でも使用できるが、エポキシ当量が2500g/当量未満であるエポキシ樹脂(以下、エポキシ樹脂(C)と称する。)と併用することができる。エポキシ樹脂(B)と共にエポキシ樹脂(C)を含むことにより、エポキシ樹脂成分の重合性およびフィラー表面への樹脂硬化物の密着性が向上するため、無機フィラー(D)を含有しても、十分な硬化物物性と熱伝導性を有する硬化物を得ることができる。
【0065】
また、上述のエポキシ樹脂(B)は、それ自体は硬化条件を含めた硬化プロセス等において制限が少なく、十分な伸び性を有し、熱伝導性、耐熱性とのバランスに優れたエポキシ樹脂であるが、エポキシ樹脂(C)を併用することにより、流動性と反応性を有するエポキシ樹脂成分が導入され、このエポキシ樹脂(B)とエポキシ樹脂(C)との混合物がフィラー表面での樹脂の剥離を抑制するため、十分な硬化物物性と熱伝導性を兼ね備える硬化物を得ることができる。
エポキシ樹脂(C)のエポキシ当量は、2500g/当量未満であることを必須するが、硬化物の反応性の観点からは、好ましくは、1000g/当量未満、より好ましくは、500g/当量未満である。
エポキシ樹脂(C)のエポキシ当量が、2500g/当量以上であると、十分な硬化物物性を有する硬化物が得られない。
【0066】
エポキシ樹脂(C)としては、上記エポキシ当量を満足するエポキシ樹脂であればよい。なお、上記式(1)で表される構造を有するエポキシ樹脂であっても、上記エポキシ当量を満足すれば、エポキシ樹脂(C)に該当する。このようなエポキシ樹脂の中でも、分子内に2個以上のエポキシ基を有するものであることが好ましく、例えば、ビスフェノールA型エポキシ樹脂、ビスフェノールF型エポキシ樹脂、ナフタレン型エポキシ樹脂、フェノールノボラック型エポキシ樹脂、クレゾールノボラック型エポキシ樹脂、フェノールアラルキル型エポキシ樹脂、ビフェニル型エポキシ樹脂、トリフェニルメタン型エポキシ樹脂、ジシクロペンタジエン型エポキシ樹脂、グリシジルエステル型エポキシ樹脂、グリシジルアミン型エポキシ樹脂、多官能フェノール型エポキシ樹脂等の、各種エポキシ樹脂を使用することができる。
これらは1種を単独で又は2種以上の混合体として使用することができる。
【0067】
エポキシ樹脂(C)は、上記エポキシ当量を満足するエポキシ樹脂であればよく、その他の物性は任意のものを使用することができる。
一方で、フィラー表面での樹脂剥離抑制の観点からは、エポキシ樹脂(C)の平均分子量は、好ましくは、100〜5000であり、より好ましくは、200〜2000である。平均分子量が100より低いものでは、耐熱性が劣る傾向にあり、5000より高いと、エポキシ樹脂の融点が高くなり、作業性が低下する傾向がある。
【0068】
また、本発明の目的を損なわない範囲において、エポキシ樹脂(B)とエポキシ樹脂(C)以外のエポキシ樹脂(以下、その他のエポキシ樹脂と称する。)を含んでいてもよい。その他のエポキシ樹脂の含有量は、エポキシ樹脂(B)とエポキシ樹脂(C)の合計に対して、通常、50重量%以下、好ましくは、30重量%以下である。
【0069】
エポキシ樹脂を含む組成物において、エポキシ樹脂(B)とエポキシ樹脂(C)を含む全エポキシ樹脂中のエポキシ樹脂(B)の割合は、その合計を100重量%として、10〜80重量%、好ましくは10〜70重量%である。なお、「エポキシ樹脂(B)とエポキシ樹脂(C)を含む全エポキシ樹脂」とは、本発明のエポキシ樹脂組成物に含まれるエポキシ樹脂が、エポキシ樹脂(B)及びエポキシ樹脂(C)のみの場合には、エポキシ樹脂(B)とエポキシ樹脂(C)の合計を意味し、さらにその他のエポキシ樹脂を含む場合には、エポキシ樹脂(B)、エポキシ樹脂(C)及びその他のエポキシ樹脂の合計を意味する。
エポキシ樹脂(B)の割合が10重量%以上であることにより、エポキシ樹脂(B)を配合することによる熱伝導性の向上効果を十分に得ることができ、所望の高熱伝導性を得ることができる。エポキシ樹脂(B)の割合が80重量%未満でエポキシ樹脂(C)が20重量%以上であることにより、エポキシ樹脂(C)の配合効果が発揮され、硬化性、硬化物の物性が十分なものとなる。
【0070】
[フラックス(B)]
本発明の層間充填材組成物はフラックス(B)を含有するが、本発明におけるフラックス(B)としては、はんだバンプ等の金属電気信号端子やランドなどの、金属表面の酸化膜を除去し、活性化させて溶融はんだの濡れ広がり性の向上に寄与するような化合物であれば、どのような化合物も使用することが可能である。より具体的には、金属端子のはんだ接合時において、はんだバンプ等の金属電気信号端子及びランドの表面酸化膜の溶解除去や、はんだバンプのランド表面における濡れ広がり性の向上、更にははんだバンプの金属端子表面の再酸化防止などの機能を有する化合物を言う。
【0071】
本発明で用いるフラックス(B)としては、蓚酸、マロン酸、コハク酸、グルタル酸、アジピン酸、リンゴ酸、酒石酸、クエン酸、乳酸、酢酸、プロピオン酸、酪酸、オレイン酸、ステアリン酸、安息香酸、アビエチン酸、ロジンなどの有機カルボン酸;有機カルボン酸をアルキルビニルエーテル類と反応して変換したヘミアセタールエステルである有機カルボン酸エステル;グルタミン酸塩酸塩、アニリン塩酸塩、ヒドラジン塩酸塩、臭化セチルピリジン、フェニルヒドラジン塩酸塩、テトラクロルナフタレン、メチルヒドラジン塩酸塩、メチルアミン塩酸塩、エチルアミン塩酸塩、ジエチルアミン塩酸塩、ブチルアミン塩酸塩などの有機ハロゲン化合物;尿素、ジエチレントリアミンヒドラジンなどのアミン類、エチレングリコール、ジエチレングリコール、トリエチレングリコール、テトラエチレングリコール、グリセリンなどの多価アルコール類、塩酸、フッ酸、燐酸、ホウフッ化水素酸などの無機酸;フッ化カリウム、フッ化ナトリウム、フッ化アンモニウム、フッ化銅、フッ化ニッケル、フッ化亜鉛などのフッ化物、塩化カリウム、塩化ナトリウム、塩化第一銅、塩化ニッケル、塩化アンモニウム、塩化亜鉛、塩化第一錫などの塩化物;臭化カリウム、臭化ナトリウム、臭化アンモニウム、臭化錫、臭化亜鉛などの臭化物などが挙げられる。
【0072】
これらの化合物は、そのまま用いても、また有機ポリマーや無機化合物等による被覆剤を用いてマイクロカプセル化したものを用いても良い。これらの化合物は1種を単独で用いてもよく、2種以上を任意の組み合わせおよび比率で混合して用いてもよい。
このうち、エポキシ樹脂や各種溶媒への溶解性より、カルボン酸若しくはカルボン酸エステルが好ましい。また、エポキシ樹脂に対して室温における硬化促進能が低いこと、及び層間充填材組成物の保存安定性より特に有機カルボン酸若しくは有機カルボン酸エステルが好ましい。
有機カルボン酸としては既に上記で例示した、蓚酸、マロン酸、コハク酸、グルタル酸、アジピン酸、リンゴ酸、酒石酸、クエン酸、乳酸、酢酸、プロピオン酸、酪酸、オレイン酸、ステアリン酸、安息香酸、アビエチン酸などがあげられるが、より好ましくは炭素数12以下の有機カルボン酸が好ましく、更に好ましくは炭素数8以下の有機カルボン酸が好ましい。
【0073】
有機カルボン酸エステルとしては、下記の反応式に従って、有機カルボン酸とアルキルビニルエーテル類を常温、常圧又は必要に応じて加温することにより得ることが出来る。尚、下記の反応式の反応は平衡反応でもあるので、有機カルボン酸エステルに転化する有機カルボン酸の割合を高めるにはアルキルビニルエーテル類を有機カルボン酸中のカルボキシル基に対して等量以上添加して反応させることが好ましい。
R1COOH+H2C=CHOR2⇔R1CO−O−CH(CH3)OR2
上記反応式において、R1はカルボン酸中の1つのカルボキシル基を除いた残りの分子鎖を示す。R2は炭素数1〜6のアルキル基を示す。
【0074】
有機カルボン酸エステルは、層間充填材組成物中において加熱により分解し、有機カルボン酸及びビニルエーテルを生成する。分解により生じる有機カルボン酸は、はんだボールに対するフラックス性を示す。
また、分解により生じる有機カルボン酸の中にはエポキシ樹脂に対する硬化作用を呈する可能性がある。これは、カルボキシル基はその解離により放出される水素イオンがエポキシ樹脂に対して硬化作用を呈する可能性があるためである。このカルボキシル基の解離による水素イオンの発生を抑制するために、有機カルボン酸をアルキルビニルエーテルにて保護した有機カルボン酸エステルが好ましく用いられる。
一方で、有機カルボン酸エステルを使用した場合においても、その分解温度が低すぎると、製造時における加圧加熱による仮接合時に、エポキシ樹脂が硬化してしまうおそれがある。
そのため、フラックス(B)における有機カルボン酸エステルの分解温度は、仮接合時での分解を回避または抑制するために、130℃以上であることが好ましく、より好ましくは140℃、更に好ましくは160℃以上、最も好ましくは180℃以上である。
【0075】
有機カルボン酸エステルの原料となる有機カルボン酸は、乳酸、酢酸、プロピオン酸、酪酸、オレイン酸、ステアリン酸、安息香酸、アビエチン酸、ロジンなどのモノカルボン酸;蓚酸、マロン酸、コハク酸、グルタル酸、アジピン酸、リンゴ酸、酒石酸、イソフタル酸、ピロメリット酸、マレイン酸、フマル酸、イタコン酸、などのジカルボン酸;クエン酸、1,2,4−トリメリット酸、トリス(2−カルボキシエチル)イソシアヌレートなどのトリカルボン酸;ピロメリット酸やブタンテトラカルボン酸などのテトラカルボン酸等を用いることが出来る。この中でもフラックスとしての反応性より、二個以上のカルボキシル基を有するポリカルボン酸類が好ましい。
【0076】
また、有機カルボン酸エステルの原料となるアルキルビニルエーテル類として、R2は炭素数1〜6のアルキル基であることが好ましく、この中でも、R2がメチル基、エチル基、プロピル基、ブチル基であることが特に好ましい。これらアルキル基の中でも、電子供与性の低いアルキル基ほど高温解離性を示すことから、アルキル基としては2級及び1級であることが好ましい。
【0077】
このうち有機カルボン酸エステルとしては、サンタシッドG(ジアルキルビニルエーテルブロック2官能ポリマー型カルボン酸)、サンタシッドH(モノアルキルビニルエーテルブロック2官能低分子量型カルボン酸)、サンタシッドI(モノアルキルビニルエーテルブロック2官能カルボン酸、いずれも、日油社製)などを好ましく用いることが出来る。
【0078】
本発明において、フラックス(B)の含有量は、樹脂(A)100重量部当たり、0.1重量部以上10重量部以下、好ましくは0.5重量部以上5重量部以下である。含有量が、0.1重量部未満では、酸化膜除去性低下によるはんだ接続不良のおそれがあり、また10重量部を超えると組成物の粘度上昇による接続不良のおそれがでてくる。
【0079】
[硬化剤(C)]
本発明において硬化剤(C)は、必要に応じて使用されるものであり、樹脂(A)が熱硬化性樹脂、特に、エポキシ樹脂である場合、樹脂を形成する際の架橋反応に寄与する物質を示す。
硬化剤(C)は、特に制限はなく既知のものが使用できる。例えば、エポキシ樹脂の場合、エポキシ樹脂硬化剤として知られているものはすべて使用できる。例えば、フェノール系硬化剤、脂肪族アミン、ポリエーテルアミン、脂環式アミン、芳香族アミンなどのアミン系硬化剤、酸無水物系硬化剤、アミド系硬化剤、第3級アミン、イミダゾール若しくはその誘導体、有機ホスフィン類、ホスホニウム塩、テトラフェニルボロン塩、有機酸ジヒドラジド、ハロゲン化ホウ素アミン錯体、ポリメルカプタン系硬化剤、イソシアネート系硬化剤、ブロックイソシアネート系硬化剤等が挙げられる。
【0080】
フェノール系硬化剤の具体例としては、ビスフェノールA、ビスフェノールF、4,4’−ジヒドロキシジフェニルメタン、4,4’−ジヒドロキシジフェニルエーテル、1,4−ビス(4−ヒドロキシフェノキシ)ベンゼン、1,3−ビス(4−ヒドロキシフェノキシ)ベンゼン、4,4’−ジヒドロキシジフェニルスルフィド、4,4’−ジヒドロキシジフェニルケトン、4,4’−ジヒドロキシジフェニルスルホン、4,4’−ジヒドロキシビフェニル、2,2’−ジヒドロキシビフェニル、10−(2,5−ジヒドロキシフェニル)−10H−9−オキサ−10−ホスファフェナンスレン−10−オキサイド、フェノールノボラック、ビスフェノールAノボラック、o−クレゾールノボラック、m−クレゾールノボラック、p−クレゾールノボラック、キシレノールノボラック、ポリ−p−ヒドロキシスチレン、ハイドロキノン、レゾルシン、カテコール、t−ブチルカテコール、t−ブチルハイドロキノン、フルオログリシノール、ピロガロール、t−ブチルピロガロール、アリル化ピロガロール、ポリアリル化ピロガロール、1,2,4−ベンゼントリオール、2,3,4−トリヒドロキシベンゾフェノン、1,2−ジヒドロキシナフタレン、1,3−ジヒドロキシナフタレン、1,4−ジヒドロキシナフタレン、1,5−ジヒドロキシナフタレン、1,6−ジヒドロキシナフタレン、1,7−ジヒドロキシナフタレン、1,8−ジヒドロキシナフタレン、2,3−ジヒドロキシナフタレン、2,4−ジヒドロキシナフタレン、2,5−ジヒドロキシナフタレン、2,6−ジヒドロキシナフタレン、2,7−ジヒドロキシナフタレン、2,8−ジヒドロキシナフタレン、上記ジヒドロキシナフタレンのアリル化物またはポリアリル化物、アリル化ビスフェノールA、アリル化ビスフェノールF、アリル化フェノールノボラック、アリル化ピロガロール等が例示される。
【0081】
アミン系硬化剤の具体例として、脂肪族アミン類としては、エチレンジアミン、1,3−ジアミノプロパン、1,4−ジアミノプロパン、ヘキサメチレンジアミン、2,5−ジメチルヘキサメチレンジアミン、トリメチルヘキサメチレンジアミン、ジエチレントリアミン、イミノビスプロピルアミン、ビス(ヘキサメチレン)トリアミン、トリエチレンテトラミン、テトラエチレンペンタミン、ペンタエチレンヘキサミン、N−ヒドロキシエチルエチレンジアミン、テトラ(ヒドロキシエチル)エチレンジアミン等が例示される。ポリエーテルアミン類としては、トリエチレングリコールジアミン、テトラエチレングリコールジアミン、ジエチレングリコールビス(プロピルアミン)、ポリオキシプロピレンジアミン、ポリオキシプロピレントリアミン類等が例示される。脂環式アミン類としては、イソホロンジアミン、メタセンジアミン、N−アミノエチルピペラジン、ビス(4−アミノ−3−メチルジシクロヘキシル)メタン、ビス(アミノメチル)シクロヘキサン、3,9−ビス(3−アミノプロピル)−2,4,8,10−テトラオキサスピロ(5,5)ウンデカン、ノルボルネンジアミン等が例示される。芳香族アミン類としては、テトラクロロ−p−キシレンジアミン、m−キシレンジアミン、p−キシレンジアミン、m−フェニレンジアミン、o−フェニレンジアミン、p−フェニレンジアミン、2,4−ジアミノアニソール、2,4−トルエンジアミン、2,4−ジアミノジフェニルメタン、4,4’−ジアミノジフェニルメタン、4,4’−ジアミノ−1,2−ジフェニルエタン、2,4−ジアミノジフェニルスルホン、4,4’−ジアミノジフェニルスルホン、m−アミノフェノール、m−アミノベンジルアミン、ベンジルジメチルアミン、2−ジメチルアミノメチル)フェノール、トリエタノールアミン、メチルベンジルアミン、α−(m−アミノフェ
ニル)エチルアミン、α−(p−アミノフェニル)エチルアミン、ジアミノジエチルジメチルジフェニルメタン、α,α’−ビス(4−アミノフェニル)−p−ジイソプロピルベンゼン等が例示される。
【0082】
酸無水物系硬化剤の具体例としては、ドデセニル無水コハク酸、ポリアジピン酸無水物、ポリアゼライン酸無水物、ポリセバシン酸無水物、ポリ(エチルオクタデカン二酸)無水物、ポリ(フェニルヘキサデカン二酸)無水物、メチルテトラヒドロ無水フタル酸、メチルヘキサヒドロ無水フタル酸、ヘキサヒドロ無水フタル酸、無水メチルハイミック酸、テトラヒドロ無水フタル酸、トリアルキルテトラヒドロ無水フタル酸、メチルシクロヘキセンジカルボン酸無水物、メチルシクロヘキセンテトラカルボン酸無水物、無水フタル酸、無水トリメリット酸、無水ピロメリット酸、ベンゾフェノンテトラカルボン酸無水物、エチレングリコールビストリメリテート二無水物、無水ヘット酸、無水ナジック酸、無水メチルナジック酸、5−(2,5−ジオキソテトラヒドロ−3−フラニル)−3−メチル−3−シクロヘキサン−1,2−ジカルボン酸無水物、3,4−ジカルボキシ−1,2,3,4−テトラヒドロ−1−ナフタレンコハク酸二無水物、1−メチル−ジカルボキシ−1,2,3,4−テトラヒドロ−1−ナフタレンコハク酸二無水物等が例示される。
【0083】
アミド系硬化剤としては、ジシアンジアミド、ポリアミド樹脂等が例示される。
第3級アミンとしては、1,8−ジアザビシクロ(5,4,0)ウンデセン−7、トリエチレンジアミン、ベンジルジメチルアミン、トリエタノールアミン、ジメチルアミノエタノール、トリス(ジメチルアミノメチル)フェノール等が例示される。
イミダゾール若しくはその誘導体としては、1−シアノエチル−2−フェニルイミダゾール、2−フェニルイミダゾール、2−エチル−4(5)−メチルイミダゾール、2−フェニル−4−メチルイミダゾール、1−ベンジル−2−メチルイミダゾール、1−ベンジル−2−フェニルイミダゾール、1−シアノエチル−2−ウンデシルイミダゾール、1−シアノ−2−フェニルイミダゾール、1−シアノエチル−2−ウンデシルイミダゾールトリメリテイト、1−シアノエチル−2−フェニルイミダゾリウムトリメリテイト、2,4−ジアミノ−6−[2’−メチルイミダゾリル−(1’)]−エチル−s−トリアジン、2,4−ジアミノ−6−[2’−エチル−4’−メチルイミダゾリル−(1’)]−エチル−s−トリアジン、2,4−ジアミノ−6−[2’−メチルイミダゾリル−(1’)]−エチル−s−トリアジンイソシアヌル酸付加体、2−フェニルイミダゾールイソシアヌル酸付加体、2−フェニル−4,5−ジヒドロキシメチルイミダゾール、2−フェニル−4−メチル−5−ヒドロキシメチルイミダゾール、又はエポキシ樹脂と上記イミダゾール類との付加体等が例示される。
【0084】
有機ホスフィン類としては、トリブチルホスフィン、メチルジフェニルホスフイン、トリフェニルホスフィン、ジフェニルホスフィン、フェニルホスフィン等が例示され、ホスホニウム塩としては、テトラフェニルホスホニウム・テトラフェニルボレート、テトラフェニルホスホニウム・エチルトリフェニルボレート、テトラブチルホスホニウム・テトラブチルボレート等が例示され、テトラフェニルボロン塩としては、2−エチル−4−メチルイミダゾール・テトラフェニルボレート、N−メチルモルホリン・テトラフェニルボレート等が例示される。
【0085】
また、上記した硬化剤は、1種を単独で用いてもよく、2種以上を任意の組み合わせおよび比率で混合して用いてもよい。
上記硬化剤の中でも、この中でも、イミダゾール又はその誘導体が好適に用いられる。
なお、フラックス(B)として、その分解生成物の有機カルボン酸がエポキシ樹脂の硬化作用を有す有機カルボン酸エステルを使用した場合には、該有機カルボン酸エステルを硬化剤(C)として用いてもよい。
【0086】
本発明の層間充填材組成物中の硬化剤(C)の含有量は、通常、樹脂(A)、特に、エポキシ樹脂100重量部に対して、0.1〜60重量部である。 ここで、硬化剤がフェノール系硬化剤、アミン系硬化剤、酸無水物系硬化剤の場合は、エポキシ樹脂中のエポキシ基と硬化剤中の官能基との当量比で0.8〜1.5の範囲となるように用いることが好ましい。この範囲外であると未反応のエポキシ基や硬化剤の官能基が残留し、所望の物性が得られないことがある。
また、硬化剤がアミド系硬化剤、第3級アミン、イミダゾール若しくはその誘導体、有機ホスフィン類、ホスホニウム塩、テトラフェニルボロン塩、有機酸ジヒドラジド、ハロゲン化ホウ素アミン錯体、ポリメルカプタン系硬化剤、イソシアネート系硬化剤、ブロックイソシアネート系硬化剤等の場合は、エポキシ樹脂100重量部に対して0.1〜20重量部の範囲で用いることが好ましい。
【0087】
[無機フィラー(D)]
本発明では、高い熱伝導率を有する無機フィラーを添加することにより、層間充填材組成物に更に高い熱伝導性を付与することが可能となる。これにより、半導体基板間の熱伝導を促進させて半導体デバイス基板の温度を低下させることにより、半導体デバイスを安定的に動作させることが可能となりより好ましい。
本発明で用いる無機フィラー(D)は高い熱伝導性を有するものが好ましく、特に熱伝導率が、好ましくは1W/mK以上が、より好ましく2W/mK以上の無機材料である。
【0088】
無機フィラー(D)は、体積平均粒径が0.1〜5μm、かつ、最大体積粒径が10μm以下であることが好ましく、より好ましくは体積平均粒径が0.1〜3μm、かつ、最大体積粒径が6μm以下であり、更に好ましくは、体積平均粒径が0.2〜1μm、かつ、最大体積粒径が3μm以下である。
上述のように高度に集積化された三次元集積回路において、層間充填材組成物からなるチップ間の層間充填層の厚みは、10〜50μm程度にまで小さくなっている。そのため、層間充填材組成物に配合するフィラーの体積粒径が10μmを超えると硬化した後の層間充填層の表面にフィラーが突出して、層間充填層の表面形状が悪化する傾向にある。
【0089】
一方、フィラーの粒径が小さ過ぎると、必要な熱伝導パス数が増加してチップ間の厚み方向に上から下まで繋がる確率が小さくなり、熱伝導性の高樹脂(A)と組み合わせても、層間充填層の厚み方向への熱伝導率が不十分になる。また、フィラーの粒径が小さ過ぎると、フィラーが凝集しやすくなり層間充填材組成物中での分散性が悪くなる。
本発明において、無機フィラー(D)の体積平均粒径を、上記範囲とすることにより、フィラー同士の過度の凝集が抑制され、厚み方向へ充分な熱伝導率を有する層間充填層を得ることができる。
【0090】
また、無機フィラー(D)として、体積平均粒径が異なる2種以上のフィラーを使用してもよい。例えば、体積平均粒径が比較的小さい、例えば0.1〜2μm、好ましくは0.2〜1.5μmの無機フィラーと、体積平均粒径が比較的大きい、例えば1〜5μm、好ましくは1〜3μmのフィラーとを併用することにより、体積平均粒径の大きい無機フィラー同士の熱伝導パスを体積平均粒径の小さい無機フィラーで繋ぐことにより、同一体積平均粒径のもののみを用いた場合に比べて高充填が可能となりより高い熱伝導性を得ることができる。
この場合、体積平均粒径の小さい無機フィラーと体積平均粒径の大きい無機フィラーとは重量比で10:1〜1:10の割合で用いることが、熱伝導パスの形成の上で好ましい。
【0091】
本発明において、無機フィラー(D)として使用される無機材料は、市販品や合成直後では、粉末が凝集して、上記粒径範囲を満たさない場合がある。そのため、無機フィラー(D)として使用される無機材料を上記粒径範囲を満たすように粉砕して用いることが好ましい。
無機材料の粉砕の方法は特に限定されず、ジルコニアビーズ等の粉砕用メディアと共に攪拌混合する方法や、ジェット噴射等の従来公知の粉砕方法を適用できる。 また、無機フィラー(D)は、樹脂(A)や塗布液中での分散性を高めるため、適宜表面処理をおこなってもよい。
【0092】
無機フィラー(D)としては、アルミナ(Al2O3:熱伝導率30W/mK)、窒化アルミニウム(AlN:熱伝導率260W/mK)、窒化ホウ素(BN:熱伝導率3W/mK(厚み方向)、275W/mK(面内方向))、窒化ケイ素(Si3N4:熱伝導率23W/mK)、シリカ(SiO2:熱伝導率1.4W/mK)などが挙げられるが、無機フィラーは更に酸素、水や高温暴露に対する安定性と低誘電性をも併せ持つことが接着したデバイスの信頼性の点で好ましく、このような無機フィラーとしては、Al2O3、AlN、BN、又はSiO2が好ましく、とりわけBNが好ましい。
これらの無機フィラー(D)は、1種を単独で用いてもよく、2種以上を任意の組み合わせおよび比率で混合して用いてもよい。
【0093】
本発明において、無機フィラー(D)の含有量は、樹脂(A)100重量部当たり、50重量部以上400重量部以下、好ましくは75重量部以上300重量部以下である。このような含有量とすることにより、本発明の層間充填材組成物は、充分な熱伝導性が得られ、かつ、均一な塗膜が形成できる程度の粘度を保つことができる。
無機フィラー(D)の含有量が、50重量部未満では、十分な熱伝導性が得られない場合があり、また、400重量部を超えると組成物の粘度が高くなり、均一な塗膜を形成出来ないとの問題が出てくる可能性がある。
【0094】
さらに、本発明の層間充填材組成物は、粘度調節等の目的で、本発明の効果を損なわない範囲で、無機フィラー(D)以外のフィラー(以下、その他のフィラーと称す。)を含有してもよい。例えば、フィラーを熱伝導性向上ではなく粘度調節を目的として添加する場合には、熱伝導率がそれほど高くない、汎用フィラーであるシリカ(SiO2:熱伝導率1.4W/mK)を使用することができる。
一方で、その他のフィラーの体積平均粒径及び最大体積粒径は、無機フィラー(D)と同様の範囲であることが好ましい。
【0095】
〔その他の添加剤〕
本発明の層間充填材組成物には、その機能性の更なる向上を目的として、本発明の効果を損なわない範囲において、各種の添加剤を含んでいてもよい。
このようなその他の添加剤としては、基材との接着性やマトリックス樹脂と無機フィラーとの接着性を向上させるための添加成分として、シランカップリング剤やチタネートカップリング剤等のカップリング剤、保存安定性向上のための紫外線防止剤、酸化防止剤、可塑剤、難燃剤、着色剤、分散剤、流動性改良剤、基材との密着性向上剤等が挙げられる。
これらは、いずれも1種を単独で用いてもよく、2種以上を任意の組み合わせおよび比率で混合して用いてもよい。
【0096】
上記添加剤の中でも、樹脂(A)と無機フィラー(D)との密着性を向上させる観点からは、シランカップリング剤、チタネートカップリング剤などのカップリング剤を含むことが好ましい。
シランカップリング剤としては、γ−グリシドキシプロピルトリメトキシシラン、γ−グリシドキシプロピルトリエトキシシラン、β−(3,4−エポキシシクロヘキシル)エチルトリメトキシシラン等のエポキシシラン;γ−アミノプロピルトリエトキシシラン、N−β(アミノエチル)γ−アミノプロピルトリメトキシシラン、N−β(アミノエチル)γ−アミノプロピルメチルジメトキシシラン、γ−アミノプロピルトリメトキシシラン、γ−ウレイドプロピルトリエトキシシラン等のアミノシラン;3−メルカプトプロピルトリメトキシシラン等のメルカプトシラン;p−スチリルトリメトキシシラン、ビニルトリクロルシラン、ビニルトリス(β−メトキシエトキシ)シラン、ビニルトリメトキシシラン、ビニルトリエトキシシラン、γ−メタクリロキシプロピルトリメトキシシラン等のビニルシラン、さらに、エポキシ系、アミノ系、ビニル系の高分子タイプのシラン等が挙げられる。
【0097】
チタネートカップリング剤としては、イソプロピルトリイソステアロイルチタネート、イソプロピルトリ(N−アミノエチル・アミノエチル)チタネート、ジイソプロピルビス(ジオクチルホスフェート)チタネート、テトライソプロピルビス(ジオクチルホスファイト)チタネート、テトラオクチルビス(ジトリデシルホスファイト)チタネート、テトラ(2,2−ジアリルオキシメチル−1−ブチル)ビス(ジトリデシル)ホスファイトチタネート、ビス(ジオクチルパイロホスフェート)オキシアセテートチタネート、ビス(ジオクチルパイロホスフェート)エチレンチタネート等が挙げられる。
【0098】
その他の添加剤の配合量には特に制限はなく、必要な機能性が得られる程度に、通常の樹脂組成物の配合量で用いられる。
なお、その他の添加剤のうち、カップリング剤の添加量は、層間充填材組成物中の全固形分に対して0.1〜2.0重量%程度とするのが好ましい。カップリング剤の配合量が少ないと、カップリング剤を配合したことによるマトリックス樹脂と無機フィラー(D)との密着性の向上効果を十分に得ることができず、多過ぎると得られる硬化物からカップリング剤がブリードアウトする問題がある。
【0099】
また、本発明の層間充填材組成物には、成形時の流動性改良や基材との密着性向上の観点より、熱可塑性のオリゴマー類を添加することができる。熱可塑性のオリゴマー類としては、C5系及びC9系の石油樹脂、スチレン樹脂、インデン樹脂、インデン・スチレン共重合樹脂、インデン・スチレン・フェノール共重合樹脂、インデン・クマロン共重合樹脂、インデン・ベンゾチオフェン共重合樹脂等が例示される。添加量としては、通常、樹脂(A)100重量部に対して、2〜30重量部の範囲である。
【0100】
〔塗布液〕
本発明の三次元集積回路用の層間充填材組成物塗布液(以下、単に、本発明の塗布液と称す。)は、上記層間充填材組成物、即ち、樹脂(A)、フラックス(B)及び、必要に応じて硬化剤(C)、無機フィラー(D)に、更に有機溶媒(E)を含有する。
【0101】
[有機溶媒(E)]
本発明の塗布液で用いる有機溶媒(E)としては、例えばアセトン、メチルエチルケトン(MEK)、メチルイソブチルケトン、メチルアミルケトン、シクロヘキサノン等のケトン類、酢酸エチル等のエステル類;エチレングリコールモノメチルエーテル等のエーテル類;N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド等のアミド類;メタノール、エタノール等のアルコール類;ヘキサン、シクロヘキサン等のアルカン類;トルエン、キシレン等の芳香族類などが挙げられる。
このうち、樹脂(A)の溶解性及び溶媒の沸点等を勘案すると、メチルエチルケトンやシクロヘキサノン等ケトン類、エステル類、又はエーテル類が好ましく、特にメチルエチルケトンやシクロヘキサノンのケトン類を用いることが特に好ましい。
これらの有機溶媒(E)は、1種を単独で用いてもよく、2種以上を任意の組み合わせおよび比率で混合して用いてもよい。
【0102】
本発明の塗布液において、有機溶媒(E)の他の成分に対する混合割合は、特に制限はないが、他の成分に対して好ましくは20重量%以上70重量%以下、特に好ましくは30重量%以上60重量%以下である。このような混合割合とすることにより、本発明の塗布液を使用することにより、任意の塗布法によって良好な塗布膜を形成することができる。
有機溶媒(E)の混合割合が、20重量%未満では塗布液の粘度が上昇し良好な塗布膜が得られない場合があり、または70重量%を超えると所定の膜厚が得られない等の問題が出てくる可能性がある。
【0103】
本発明の塗布液には、各種の添加剤を含んでいてもよい。
このような添加剤としては、上述の添加剤の他、塗布液中での各成分の分散性を向上させる界面活性剤、乳化剤、低弾性化剤、希釈剤、消泡剤、イオントラップ剤等が挙げられる。
ここで、界面活性剤としては、従来公知のアニオン系界面活性剤、ノニオン系界面活性剤、カチオン系界面活性剤のいずれも使用できる。
例えば、ポリオキシエチレンアルキルエーテル類、ポリオキシエチレンアルキルアリールエーテル類、ポリオキシエチレンアルキルエステル類、ソルビタンアルキルエステル類、モノグリセリドアルキルエステル類、アルキルベンゼンスルホン酸塩類、アルキルナフタレンスルホン酸塩類、アルキル硫酸塩類、アルキルスルホン酸塩類、スルホコハク酸エステル塩類、アルキルベタイン類、アミノ酸類などが挙げられる。
また、これら界面活性剤においてCH結合の一部又は全てがCF結合となったフッ素界面活性剤も好ましく用いることが出来る。
【0104】
界面活性剤の添加量として、層間充填材組成物中の全固形分に対して、0.001〜5重量%程度とするのが好ましい。0.001重量%未満では、所定の膜厚均一性が得られない場合があり、また5重量%を超えるとエポキシ樹脂成分との相分離等を引き起こす場合があり好ましくない。
【0105】
本発明の塗布液の製造方法は、特に限定されず従来公知の方法によればよく、塗布液の構成成分を混合することで製造することができる。なお、その際、組成物の均一性の向上、脱泡等を目的として、ペイントシェーカーやビーズミル、プラネタリミキサ、撹拌型分散機、自公転攪拌混合機、三本ロールなどを用いて混合することが好ましい。
各成分の混合順序も反応や沈殿物が発生するなど特段の問題がない限り任意であり、塗布液の構成成分のうち、何れか2成分又は3成分以上を予め配合し、その後に残りの成分を混合してもよいし、一度に全部を混合してもよい。
なお、上述のように無機フィラー(D)は、粒径の大きな凝集体とならないことが好ましいので、塗布液の製造前に粉砕してもよいし、他の成分と混合してから粉砕してもよい。無機材料の粉砕の方法は特に限定されず、従来公知の粉砕方法を適用できる。
【0106】
〔三次元集積回路の製造方法〕
以下、本発明の層間充填材組成物、又は層間充填材組成物塗布液を使用する三次元集積回路の製造方法について説明する。かかる製造方法は、複数の半導体基板に、上述の層間充填材組成物を成膜後に、これらの半導体基板を加圧接着して積層する工程を含んでいる。
【0107】
本発明では、始めに半導体基板上に層間充填材組成物の薄膜を形成する。本発明の層間充填材組成物の塗布液を用いる場合にはその塗布液を用いて、ディップ法、スピンコート法、スプレーコート、ブレード法やその他の任意の方法で塗布膜を形成することが出来る。得られた塗布膜から溶媒や低分子成分除去のために、50〜150℃の任意の温度でベーキング処理を行いB−ステージ化膜を形成する。この際、一定の温度においてベーキング処理を行ってもよいが、組成物中の揮発成分除去を円滑に進めるために、減圧条件下にてベーキング処理を行ってもよい。また、エポキシ樹脂の硬化が進行しない範囲で、段階的な昇温によるベーキング処理を行っても良い。例えば、初めに60℃、次に80℃、更に120℃で各5〜30分程度のべーキング処理を実施することが出来る。
また、本発明の塗布液を使用せずに、本発明の層間充填材組成物をそのまま用いてもよい。例えば、樹脂の硬化が始まらない温度範囲において加温して溶融させたものを用いて、任意の方法で半導体基板上に層間充填材組成物からなる膜を成膜としてもよい。
また、本発明の層間充填材組成物は、フィルム成形に適した十分な伸び性を有するため、本発明の層間充填材組成物をフィルム成形し、該フィルムを半導体基板上に設置することで成膜してもよい。
【0108】
次に上記方法にて成膜した層間充填材組成物からなる膜を加熱してタック性を発現させた後に、接合対象の半導体基板と仮接着を行う。仮接着の温度としては、樹脂(A)の組成にもよるが、80〜150℃の温度で行うことが好ましい。半導体基板の接合が複数層の場合には、前記仮接着を基板の層数分繰り返しても良いし、B−ステージ化膜を形成した基板を複数層重ね合わせた後に、加熱してまとめて仮接着させても良い。仮接着の際には必要に応じて基板間に1gf/cm2〜1Kgf/cm2の加重をかけて実施することが好ましい。
【0109】
仮接着の後には半導体基板の本接合を行う。仮接着させた半導体基板を200℃以上、好ましくは220℃以上に加圧接着することにより、層間充填材組成物の樹脂の溶融粘度を低下させて半導体基板間の電気端子の接続を促進すると同時に、組成物中のフラックスを活性化させて半導体基板間のはんだ接合を実現することが出来る。なお、加熱温度の上限は、使用するエポキシ樹脂が分解、変質しない温度であり、樹脂の種類、グレードにより適宜決定されるが、通常300℃以下で行われる。
また、加圧接着の際には必要に応じて基板間に10gf/cm2〜10Kgf/cm2の加重をかけて実施することが好ましい。
【実施例】
【0110】
以下、本発明について、実施例を用いてさらに詳細に説明するが、本発明はその趣旨を逸脱しない限り、以下の実施例に限定されるものではない。
【0111】
以下の実施例1〜3及び比較例1〜3において用いた層間充填材組成物の配合成分は次の通りである。
・エポキシ樹脂
エポキシ樹脂(A1):フェノキシ樹脂
重量平均分子量:26,000
エポキシ当量:4,600g/当量
30重量%メチルエチルケトン/シクロヘキサノンの1:1(重量比)の混合溶液
エポキシ樹脂(A2):三菱化学社製 品名「157S65」
80.6重量%MEK溶液
軟化点:65℃
150℃における溶融粘度:0.3Pa・s
・フラックス(B):日油社製 品名「サンタシッドG」ジアルキルビニルエーテル
ブロック2官能ポリマー型カルボン酸
・硬化剤(C):四国化成工業社製 品名「C11Z−CN」
1−シアノエチル−2−ウンデシルイミダゾール
・無機フィラー(D):日新リフラテック社製 窒化ホウ素 BN(熱伝導率3W/mK(厚み方向)、275W/mK(面内方向))
・有機溶媒(E)
有機溶媒(E1):和光純薬工業社製 試薬特級 メチルエチルケトン(以下、
MEKと略記することがある)
有機溶媒(E2):和光純薬工業社製 試薬特級 シクロヘキサノン(以下、
CHNと略記することがある)
【0112】
なお、前記エポキシ樹脂(A1)としてのフェノキシ樹脂は次のようにして作製した。
YL6121H(エポキシ当量171g/当量、4,4’-ビフェノール型エポキシ樹脂と3,3’,5,5’-テトラメチル-4,4’-ビフェノール型エポキシ樹脂の1:1混合物(三菱化学社製)215重量部、3,3’−ジメチル−4,4’−ビフェノール(OH当量107g/当量、本州化学社製)127重量部、27重量%テトラメチルアンモニウムヒドロキシド水溶液0.32重量部、及び、反応用溶媒としてシクロヘキサノン228重量部を撹拌機付き耐圧反応容器に入れ、窒素ガス雰囲気下180℃で5時間、反応を行った。次いで、希釈用溶剤としてシクロヘキサノン171重量部及びメチルエチルケトン399重量部を加えて固形分濃度を調整した。反応生成物から定法により溶剤を除去して30重量%の樹脂溶液を得た。
【0113】
なお、エポキシ樹脂の物性及び接合評価時のはんだバンプの電気抵抗、無機フィラーの粒径は次のようにして測定した。
(1)溶融粘度
アントンパール・ジャパン社製 粘弾性測定装置Physica MCR301を用いて溶融粘度(パラレルプレート動的粘度)を測定した。
まず、測定対象であるエポキシ樹脂から溶媒を留去して固形物を得、その後、この固形物に対してプレス成型を行い、厚さ約1mmの板状サンプルを得た。このサンプルを、パラレルプレートディッシュとパラレルプレート(φ25mm)の間に載置しパラレルプレート動的粘度測定を行った。
測定条件は、上記サンプルに正弦波歪みを20%与え、その歪みの角周波数は10rad/secとし、1分間に3℃の割合で昇温させる過程での粘度を40℃〜200℃まで測定した。
【0114】
(2)熱伝導率
以下の装置にて、熱拡散率、比重、比熱を測定し、この3つの測定値を乗じることで熱伝導率を求めた。
1)熱拡散率:アイフェイズ社「アイフェイズ・モバイル 1u」
2)比重:メトラー・トレド社 天秤 XS−204(「固体比重測定キット」使用)
3)比熱:セイコーインスツル社 DSC320/6200
【0115】
(3)電気抵抗
FLUKE77マルチメーター(JOHN FLUKE MFG. Co. INC) を用いて端子間の抵抗値を測定した。
(4)粒径測定
攪拌混合後の試料(実施例1参照)をシクロヘキサノンで分散させ、堀場製作所レーザ回折/散乱式粒度分布測定装置LA−920にて測定した。得られた粒度分布から粉砕後の無機フィラー(D)の体積平均粒径及び最大体積粒径を求めた。
【0116】
[実施例1、2、比較例1,2]
エポキシ樹脂として、エポキシ樹脂(A1)とエポキシ樹脂(A2)を所定の配合重量比(溶媒を除いた樹脂として20:80)とし、このエポキシ樹脂(A)の合計を100重量部とした。このときエポキシ樹脂の溶融粘度は、3000Pa・s(50℃)及び16Pa・s(120℃)であった。このエポキシ樹脂の100重量部に対して、フラックス(B)、有機溶媒(E)(メチルエチルケトン(E1)とシクロヘキサノン(E2)を配合重量比(35:65)で混合したもの)、及び硬化剤(C)を表1に記載の重量比にて配合した。次に自公転攪拌機を用いて1000rpmにて5分間撹拌、混練することにより、層間充填材の原料ペーストを得た。
【0117】
この原料ペーストを、図1(a),(b)に記載した、はんだバンプ基板上の10mm×10mmエリア(約8500バンプ)に膜厚50μmとなるように塗布を行い、120℃で1分間加熱により溶媒を留去してB−ステージ化膜とした。この基板を再度120℃に加温した後、前記塗布エリアに、銅基板の銅面を塗布エリア面に向けて0.1Kgf/cm2の加重で押し当てて仮接着を行った。室温まで冷却後、仮接着基板を小型のプレス機を用いて、10Kgf/cm2の加重で10分間、120℃にて加熱プレスを実施した。
次に、この基板を250℃のホットプレート上において、0.1Kgf/cm2の加重で1分間加熱プレスを行った。120℃プレス後、又は250℃プレス後に各々はんだバンプ基板−銅基板間の接合性評価のために電気抵抗測定を実施した。結果を表1に併せて示す。
【0118】
表1より、仮接着後及び120℃におけるプレス後では、はんだバンプ基板−銅基板間の電気抵抗は数Ω以上であったのに対して、200℃以上の温度にて加圧接着することにより電気抵抗はいずれも1Ω以下となった。更に、組成物にフラックスを含有させると何れも基板間の電気抵抗が0.1Ω未満となり、はんだバンプ基板及び銅基板が層間充填材組成物を介した接合でありながら低抵抗化しており、良好な接合状態を実現出来ていることが分かる。
【0119】
【表1】
【0120】
[参考例1〜16]
各種のフラックス(B)と有機溶媒(E)(メチルエチルケトン(E1)とシクロヘキサノン(E2)を配合重量比(35:65)で混合したもの)を表2に記載の重量濃度にて配合した後、撹拌混合し、フラックス溶液を得た。
なお、使用した薬品は以下のとおりである。
ロジン(アビエチン酸混合物;和光純薬工業社製)、サンタシッドG〜I(有機カルボン酸誘導体;日油社製)、2E4MZ(2−エチル−4−メチルイミダゾール;四国化成工業社製)、C11Z−CN(1−シアノエチル−2−ウンデシルイミダゾール;四国化成工業社製)
【0121】
このフラックス溶液を10mm×10mmの銅基板上に50μL滴下した後、このフラックス液滴中にはんだボール(Sn3.0Ag0.5Cu、直径300μm)を添加した。この基板をホットプレート上にて120℃で1分間加熱により溶媒を留去した。次に、この基板を250℃のホットプレート上において、250℃で10秒間加熱を行い銅基板に対するはんだボールの融解性を評価した。結果を表2に示す。
【0122】
表2より、フラックスとして有機カルボン酸又は有機カルボン酸エステルを用いた場合、ハンダボールが所定の温度において融解して、銅基板と良好な接合を形成した。一方、アミノ酸類は溶媒に対する溶解性が低く、またイミダゾール類はフラックスとしての作用が低くはんだボールと銅基板が接合出来なかった。
【0123】
【表2】
【0124】
[実施例3]
上記エポキシ樹脂(A1)溶液4.7gとエポキシ樹脂(A2)溶液7g(溶媒を除いた樹脂として配合重量比20:80、このときエポキシ樹脂の溶融粘度は、3000Pa・s(50℃)及び16Pa・s(120℃)であった)及び、硬化剤(C)の5%シクロヘキサノン溶液を0.7g加えて混合し、エポキシ樹脂溶液を得た。
【0125】
この樹脂溶液をセパレータ(シリコーン処理したポリエチレンテレフタレートフィルム、厚み:100μm)にドクターブレードで塗布し、60℃で15分、その後80℃で15分、その後、減圧下(圧力<4Torr)にて80℃で15分加熱して溶媒を除去してBステージ膜とした。その後、もう一枚のセパレータを得られたBステージ膜上に載せた後、まず100℃15分、次いで150℃15分、その後200℃で30分減圧プレス(圧力1MPa)することにより成型・硬化させて、膜厚約50μmの硬化膜を得た。得られたエポキシ樹脂の熱伝導率は、0.22W/mKであった。
【0126】
(層間充填材組成物の合成及び評価)
エポキシ樹脂として、上記エポキシ樹脂(A1)溶液4.7gとエポキシ樹脂(A2)溶液7g(溶媒を除いた樹脂として配合重量比20:80、このときエポキシ樹脂の溶融粘度は、3000Pa・s(50℃)及び16Pa・s(120℃)であった)、及び無機フィラー(D)19.8g、有機溶媒(E)20.5gをSUS製の容器(ガラス製蓋付き)に仕込む。SUS製の攪拌翼で550rpmで回転しながら、直径2mmのジルコニアボール(YTZ−2)を300g加え、5時間攪拌し、無機フィラー(D)を粉砕した。攪拌終了後、硬化剤(C)の5%シクロヘキサノン溶液を0.7g加え、更に5分間攪拌した。攪拌後、ろ過によりジルコニアボールを除き、層間充填剤ペースト(塗布液)を得た。
【0127】
得られた粉砕後の層間充填剤ペースト中の無機フィラー(D)の粒度分布を図2に示す。粒度分布から求めた無機フィラー(D)の体積平均粒径0.5μm、最大体積粒径1.5μmであった。
このペーストをスピンコーター(ミカサ社製)を用いて基板に塗布し、8℃×15分、120℃×15分で加熱乾燥しB−ステージ化膜とした。得られたB−ステージ化膜は、図3に示すように目視において均一であった。
【0128】
[比較例3]
エポキシ樹脂として、エポキシ樹脂(A1)溶液4.7gとエポキシ樹脂(A2)溶液7g(溶媒を除いた樹脂として配合重量比20:80、このときエポキシ樹脂の溶融粘度は、3000Pa・s(50℃)及び16Pa・s(120℃)であった)、及び無機フィラー(D)19.8g、硬化剤(C)(5%シクロヘキサノン溶液)0.7g、有機溶媒(E)20.5gをSUS製の容器(ガラス製蓋付き)に仕込んだ。SUS製の攪拌翼で550rpm×5分攪拌し、層間充填剤ペーストを得た。
実施例3と同じ装置にて層間充填剤ペーストの粒径分布を測定した結果を図4に示す。粒度分布から求めた無機フィラー(D)の体積平均粒径4.0μm、最大体積粒径20μmであった。
この層間充填剤ペーストを用いて、実施例3と同様の方法で、B−ステージ化膜を得た。得られたB−ステージ化膜には、図5に示すように目視で確認できるフィラーが多数存在し、不均一であった。
【0129】
〔合成例1〜3〕
これらの合成例におけるエポキシ樹脂(B)の評価は、下記の方法で行った。
<分子量>
高速GPC装置(東ソー(株)製「HLC−8320GPC EcoSEC(登録商標)」)を使用し、以下の測定条件で、標準ポリスチレンとして、TSK Standard Polystyrene:F−128(Mw1,090,000、Mn1,030,000)、F−10(Mw106,000、Mn103,000)、F−4(Mw43,000、Mn42,700)、F−2(Mw17,200、Mn16,900)、A−5000(Mw6,400、Mn6,100)、A−2500(Mw2,800、Mn2,700)、A−300(Mw453、Mn387)を使用した検量線を作成し、重量平均分子量および数平均分子量をポリスチレン換算値として測定した。
カラム:東ソー社製「TSKGEL SuperHM−H+H5000+H4000+H3000+H2000」
溶離液:テトラヒドロフラン
流速:0.5ml/min
検出:UV(波長254nm)
温度:40℃
試料濃度:0.1重量%
インジェクション量:10μL
【0130】
<n数>
前記エポキシ樹脂(B)の式(1)におけるnの値及びその平均値は、上記で求められた数平均分子量より算出した。
<エポキシ当量>
JIS K 7236に準じて測定し、固形分換算値として表記した。
<ガラス転移温度Tg>
溶剤を乾燥除去したエポキシ樹脂で、SIIナノテクノロジー社製「DSC7020」を使用し、30〜200℃まで10℃/minで昇温して測定した。
【0131】
<伸び>
エポキシ樹脂の溶液をセパレータ(シリコーン処理したポリエチレンテレフタレートフィルム、厚み:100μm)にアプリケーターで塗布し、60℃で1時間、その後150℃で1時間、更に200℃で1時間乾燥させ、厚さ約50μmのエポキシ樹脂フィルムを得た。これを幅1cmに切り出し、精密万能試験機(インストロン社製 INSTRON 5582型)を使用して5mm/minで3回測定した平均値を示した。
【0132】
また、下記する実施例で使用したエポキシ樹脂(B1)膜、実施例及び比較例で得られた硬化膜の熱伝導率は、下記の方法で評価した。なお、エポキシ樹脂(B1)膜は、上記の伸び測定用サンプルと同様の手法により得た。
<熱伝導率>
次の装置にて、熱拡散率、比重、比熱を測定し、この3つの測定値を乗じることで熱伝導率を求めた。
熱拡散率:アイフェイズ社「アイフェイズ・モバイル 1u」
比重:メトラー・トレド社「天秤 XS−204」(「固体比重測定キット」使用)
比熱:セイコーインスツル社「DSC320/6200」
【0133】
[エポキシ樹脂(B)の製造と評価]
<合成例1〜3>
表3に示した配合で化合物(X)、化合物(Y)、触媒および反応用溶剤を撹拌機付き耐圧反応容器に入れ、窒素ガス雰囲気下、180℃で5時間反応を行った後、希釈用溶剤を加えて固形分濃度を調整した。反応生成物から定法により溶剤を除去した後、得られた樹脂について分析を行った。結果を表3に示す。
【0134】
なお、反応に用いた化合物、触媒および溶剤は以下の通りである。
<化合物(X)>
(X−A):三菱化学社製 商品名「YL6121H」(4,4’−ビフェノール型エポキシ樹脂と3,3’,5,5’−テトラメチル−4,4’−ビフェノール型エポキシ樹脂の1:1混合物、エポキシ当量171g/当量)
(X−B):三菱化学社製 商品名「YX4000」(3,3’,5,5’−テトラメチル−4,4’−ビフェノールジグリシジルエーテル、エポキシ当量186g/当量)
【0135】
<化合物(Y)>
(Y−A):3,3’−ジメチル−4,4’−ビフェノール(OH当量107g/当量、本州化学社製)
(Y−B):3,3’,5,5’−テトラメチル−4,4’−ビフェノール(OH当量121g/当量)
<触媒>
(C−1):27重量%テトラメチルアンモニウムハイドロオキサイド水溶液
<溶剤>
(S−1):シクロヘキサノン
(S−2):メチルエチルケトン
(S−3):N,N’−ジメチルアセトアミド
【0136】
【表3】
【0137】
[エポキシ樹脂組成物の製造と評価]
<参考例4>
合成例1で得られたエポキシ樹脂(Mw59,741)2.5g(樹脂分0.75g;60重量部)と、ビスフェノールAノボラック型多官能エポキシ樹脂溶液(三菱化学社製 商品名「157S65(B80)」)0.625g(樹脂分0.5g;40重量部)と、硬化剤として2−エチル−4(5)−メチルイミダゾール(三菱化学社製 商品名「EMI24」)の20重量%溶液(溶剤MEK)0.032g(硬化剤重量0.00625g;0.5重量部)をはかり取り、自転公転ミキサーにて撹拌混合・脱泡を行った。このエポキシ樹脂組成物について硬化フィルムの熱伝導率を求めた。結果を表4に示す。
【0138】
<参考例5>
多官能エポキシ樹脂をビフェニル型多官能エポキシ樹脂(日本化薬社製 商品名「NC−3000−H」)の80重量%MEK溶液に変えた以外は参考例4と同様に、合成例1のエポキシ樹脂:NC−3000−H:EMI24=60:40:0.5(重量部)としてエポキシ樹脂組成物を製造し、得られたエポキシ樹脂組成物について、硬化フィルムの熱伝導率を求めた。結果を表4に示す。
【0139】
<参考例6>
合成例1のエポキシ樹脂:NC−3000−H:EMI24=30:70:0.5(重量部)としたこと以外は参考例5と同様にエポキシ樹脂組成物を製造し、得られたエポキシ樹脂組成物について、硬化フィルムの熱伝導率を求めた。結果を表4に示す。
【0140】
<参考例7>
多官能エポキシ樹脂をビフェニル型多官能エポキシ樹脂(日本化薬社製 商品名「NC−3000」)の70重量%シクロヘキサノン溶液に変えた以外は参考例4と同様に、合成例1のエポキシ樹脂:NC−3000:EMI24=60:40:0.5(重量部)として、エポキシ樹脂組成物を製造し、得られたエポキシ樹脂組成物について、硬化フィルムの熱伝導率を求めた。結果を表4に示す。
【0141】
<参考例8>
合成例1で得られたエポキシ樹脂:157S65(B80):EMI24=90:10:0.5(重量部)とした以外は参考例4と同様にエポキシ樹脂組成物を製造し、得られたエポキシ樹脂組成物について、硬化フィルムの熱伝導率を求めた。結果を表4に示す。
【0142】
【表4】
【0143】
以上の結果より、エポキシ樹脂(B)およびエポキシ樹脂(B)を含む組成物は、フィルム成形・塗布等のプロセスに適用するのに十分な伸び性を有し、かつ熱伝導性、耐熱性のバランスにも優れることが分かる。
【0144】
実施例4、5(比較例4、5)
これらの実施例及び比較例で使用したエポキシ樹脂(B)の1種であるエポキシ樹脂(B1)は、下記の方法で製造した。 使用した原料、触媒、溶剤を以下に示す。
・化合物(X):三菱化学社製 商品名「YL6121H」(4,4’−ビフェノール型 エポキシ樹脂と3,3’,5,5’−テトラメチル−4,4’−ビフェノール型エポキシ樹脂の1:1混合物、エポキシ当量171g/当量)
・化合物(Y):3,3’−ジメチル−4,4’−ビフェノール(OH当量107g/当量、本州化学社製)
・触媒:27重量%テトラメチルアンモニウムハイドロオキサイド水溶液
・溶剤
溶剤1:シクロヘキサノン
溶剤2:メチルエチルケトン
【0145】
化合物(X)210重量部、化合物(Y)127.6重量部、触媒0.78重量部、および溶剤1(シクロヘキサノン)181.8重量部を撹拌機付き耐圧反応容器に入れ、窒素ガス雰囲気下、180℃で5時間反応を行った。
その後、該耐圧反応容器に溶剤1(シクロヘキサノン)212.1重量部及び溶剤2(メチルエチルケトン)393.8重量部を加えて固形分濃度を30重量%に調整した。
反応生成物から定法により溶剤を除去した後、得られたエポキシ樹脂(B1)について分析を行った結果を以下に示す。
エポキシ樹脂(B1)
重量平均分子量(Mw):59000
数平均分子量(Mn) :14000
式(1)におけるn数 :49
エポキシ当量 :8150(g/当量)
Tg :110(℃)
伸び :72(%)
熱伝導率 :0.21(W/mK)
【0146】
下記の実施例及び比較例のエポキシ樹脂組成物の製造に使用した、エポキシ樹脂(B2)、エポキシ樹脂(C)、硬化剤(C)、無機フィラー(D)、及び溶媒(E)を以下に示す。
エポキシ樹脂(B2)
特殊骨格エポキシ樹脂(三菱化学社製、商品名YX6954BH30)
重量平均分子量(Mw):39000)
溶媒 :MEK:CHN=1:1
樹脂濃度 :30重量%溶液
・エポキシ樹脂(C)
エポキシ樹脂(C1):日本化薬社商品名「NC−3000−H」(80重量%M
EK溶液を調整)
エポキシ樹脂(C2): 三菱化学社製 商品名「157S65(B80)」(M
EK溶液;80重量%)
・硬化剤(C):三菱化学社製 商品名「EMI24」
2−エチル−4(5)−メチルイミダゾール
【0147】
・無機フィラー(D)
無機フィラー(D1):住友化学社製Al2O3 商品名「AA−3」
(体積平均粒径3μm)
無機フィラー(D2):住友化学社製Al2O3 商品名「AA−04」
(体積平均粒径0.4μm)
無機フィラー(D3):モメンティブ・パフォーマンス・マテリアルズ・ジャパ
ン合同会社製BN 商品名「PTX−25」(凝集粒子の体積平均粒径:25μm,一次粒子の体積平均粒径:3μm)
MEKとCHNの1:1(重量比)の混合溶媒
【0148】
[実施例4]
エポキシ樹脂(B1)とエポキシ樹脂(C1)を表5に示す配合重量比とし、エポキシ樹脂(B1)とエポキシ樹脂(C1)との合計100重量部に対して、硬化剤(C)を0.5重量部と、無機フィラー(D)として無機フィラー(D1)と無機フィラー(D2)を重量比8:2で混合したものを、硬化物中の無機フィラーの含有量が60体積%(エポキシ樹脂(B1)とエポキシ樹脂(C1)の合計100重量部として510重量部)となるように配合し、更に溶媒(E)を組成物中の固形分濃度が60重量%となるように添加して、ペースト状の塗布液を得た。
【0149】
このペースト状の塗布液をセパレータ(シリコーン処理したポリエチレンテレフタレートフィルム、厚み:100μm)にドクターブレードで塗布し、60℃で15分、その後80℃で15分、その後減圧(圧力<4Torr)にて80℃で15分加熱して溶媒を除去してBステージ膜とした。その後、もう一枚のセパレータを得られたBステージ膜上に載せた後、まず100℃15分、次いで150℃15分、その後200℃で30分減圧プレス(圧力1MPa)することにより成型・硬化させて、膜厚約50μmの硬化膜を得た。得られた硬化膜について、熱伝導率の評価を行った。結果を表5に示す。
【0150】
[実施例5]
実施例4において、無機フィラー(D)として無機フィラー(D3)を用い、エポキシ樹脂(C1)の代わりにエポキシ樹脂(C2)を用い、無機フィラー(D)の配合量を硬化物に対して40体積%(エポキシ樹脂(B1)とエポキシ樹脂(C2)の合計100重量部として129重量部)としたこと以外は実施例4と同様にして、硬化膜を得、その熱伝導率を評価した。結果を表5に示す。
【0151】
[比較例4]
エポキシ樹脂として、エポキシ樹脂(B1)の代わりにエポキシ樹脂(B2)を使用した以外は、実施例4と同様にしてエポキシ樹脂組成物を作製し、この組成物を硬化させて硬化物を得、その熱伝導率を評価した。結果を表5に示す。
【0152】
[比較例5]
エポキシ樹脂として、エポキシ樹脂(B1)の代わりにエポキシ樹脂(B2)を使用した以外は、は実施例5と同様にしてエポキシ樹脂組成物を作製し、この組成物を硬化させて硬化物を得、その熱伝導率を評価した。結果を表5に示す。
なお、表1中の各配合物の数字は重量部(溶液中の樹脂の重量)を示す。
【0153】
【表5】
【0154】
実施例6〜12
[配合成分]
実施例6〜12において用いた層間充填層形成用塗布液の配合成分は、次の通りである。
<エポキシ樹脂>
エポキシ樹脂(A1):実施例1で用いたのと同じエポキシ樹脂(A1)
エポキシ樹脂(A3):三菱化学株式会社製 品名「YL6800」
エポキシ樹脂(A4):三菱化学株式会社製 品名「1032H60」
エポキシ樹脂(A5):三菱化学株式会社製 品名「1001」
エポキシ樹脂(A6):三菱化学株式会社製 品名「4004」
エポキシ樹脂(A7):三菱化学株式会社製 品名「YX4000」
【0155】
<フラックス(B)>
和光純薬工業株式会社製 アジピン酸 試薬特級
<硬化剤(C)>
四国化成工業株式会社製 2−フェニル−4,5−ジヒドロキシメチルイミダゾール 品名「2PHZ−PW」
<無機フィラー(D)>
実施例1で用いたのと同じ、日新リフラテック社製 窒化ホウ素 BN
【0156】
<有機溶媒(E)>
有機溶媒(E)としては、前記有機溶媒(E1)(和光純薬工業社製 試薬特級 メチルエチルケトン)および有機溶媒(E2)(和光純薬工業社製 試薬特級 シクロヘキサノン)を用いた。
【0157】
[無機フィラーの粒径の測定]
攪拌混合後の層間充填層形成用塗布液をシクロヘキサノンで分散させ、島津製作所製粒度分布測定装置「SALD−2200」にて測定した。得られた粒度分布から粉砕後の無機フィラーの体積平均粒径及び最大体積粒径を求めた。
【0158】
[接合評価]
(1)チップへの塗布
攪拌混合後の層間充填層形成用塗布液について、マイクロピペットを用いて、Walts社製のSiチップ「CC80−0101JY ModelI」上に約10μLを塗布して塗り拡げた。
(2)B−Stage化
前記の層間充填層の塗布されたSiチップについて、ホットプレートを用いてはじめに80℃で15分間、続いて120℃で30分間それぞれ加熱を行いB−Stage化処理を行った。
(3)接合評価
B−Stage化された層間充填層の塗布された前記Siチップ及びWalts社製の有機基板「CC80−0102JY ModelI」について、東レエンジニアリング社製のフリップチップボンダ装置「FC3000S」を用いて、250℃まで昇温させて加熱圧着接合を行った。
(4)接合性(導通)評価
Siチップが接合された有機基板上の電極に、プローバー端子を接地させて、Siチップ及び有機基板の電気端子間に形成されたデイジーチェインについて、Keithley製マルチメータ「2100型」を用いて導通評価を行い、導通が確認出来たものを○とした。
【0159】
[層間充填層形成用塗布液の調製]
・実施例6の塗布液の調製
前記エポキシ樹脂(A1)のMEKとCHNの1:1(重量比)の混合溶液1.67g、エポキシ樹脂(A3)1.75g、およびエポキシ樹脂(A4)の80重量%シクロヘキサノン溶液0.31gを混合し、溶媒を除いた樹脂成分として、エポキシ樹脂(A1)とエポキシ樹脂(A3)とエポキシ樹脂(A4)との配合重量比が、20:70:10になるようにしてエポキシ樹脂組成物(I)を得た。このエポキシ樹脂組成物(I)の溶融粘度及び熱伝導率を表6に示す。
このエポキシ樹脂組成物(I)3.73g、無機フィラー(D)2.5g、および有機溶媒(E2)3.57gをポリエチレン製の容器に仕込み、更に直径0.5mmのジルコニアボールを20g加え、自公転攪拌機を用いて2000rpmで20分間攪拌した。攪拌終了後、ろ過によりジルコニアボールを取り除き、フラックス(B)を0.05gおよび硬化剤(C)を0.15g加え、更に自公転攪拌機にて6分間攪拌し、三次元集積回路用層間充填材組成物の塗布液を得た。この塗布液の固形分濃度は、50重量%で、無機フィラー(D)の含有量は、エポキシ樹脂組成物(I)及び無機フィラー(D)の合計量に対して50重量%であった。三次元集積回路用層間充填材組成物の塗布液中の無機フィラー(D)の体積平均粒径及び最大体積粒径を表7に示す。
【0160】
・実施例7の塗布液の調製
前記エポキシ樹脂(A1)のMEKとCHNの1:1(重量比)の混合溶液1.67g、エポキシ樹脂(A3)1.25g、エポキシ樹脂(A4)の80重量%シクロヘキサノン溶液0.31g、およびエポキシ樹脂(A5)の70重量%シクロヘキサノン溶液0.71gを混合し、溶媒を除いた樹脂成分として、エポキシ樹脂(A1)とエポキシ樹脂(A3)とエポキシ樹脂(A4)とエポキシ樹脂(A5)との配合重量比が、20:50:10:20になるようにしてエポキシ樹脂組成物(II)を得た。このエポキシ樹脂組成物(II)の溶融粘度及び熱伝導率を表6に示す。
エポキシ樹脂組成物(I)の代わりにエポキシ樹脂組成物(II)を3.94g、有機溶媒(E2)の代わりに有機溶媒(E1)を3.36g用いた以外は、実施例6の塗布液の調製と同様にして実施例7の三次元集積回路用層間充填材組成物の塗布液を調整した。三次元集積回路用層間充填材組成物の塗布液中の無機フィラー(D)の体積平均粒径及び最大体積粒径を表7に示す。
【0161】
・実施例8の塗布液の調製
エポキシ樹脂(A4)の80重量%シクロヘキサノン溶液の量を0.63g、エポキシ樹脂(A5)の70重量%シクロヘキサノン溶液の量を0.36gに変更し、溶媒を除いた樹脂として、エポキシ樹脂(A1)とエポキシ樹脂(A3)とエポキシ樹脂(A4)とエポキシ樹脂(A5)との配合重量比を20:50:20:10としたエポキシ樹脂組成物(III)を得た。このエポキシ樹脂組成物(III)の溶融粘度及び熱伝導率を表6に示す。
エポキシ樹脂組成物(III)を用いた以外は実施例7の塗布液の調製と同様にして、実施例8の三次元集積回路用層間充填材組成物の塗布液を調整した。この三次元集積回路用層間充填材組成物の塗布液中の無機フィラー(D)の体積平均粒径及び最大体積粒径を表7に示す。
【0162】
・実施例9の塗布液の調整
エポキシ樹脂(A6)を1.75g、エポキシ樹脂(A3)を0.7g、エポキシ樹脂(A4)の80重量%シクロヘキサノン溶液を0.35g、およびエポキシ樹脂(A5)の70重量%シクロヘキサノン溶液を0.70g混合し、溶媒を除いた樹脂成分として、エポキシ樹脂(A6)とエポキシ樹脂(A3)とエポキシ樹脂(A4)とエポキシ樹脂(A5)との配合重量比が、50:20:10:20になるようにしてエポキシ樹脂組成物(IV)を得た。このエポキシ樹脂組成物(IV)の溶融粘度及び熱伝導率を表6に示す。
エポキシ樹脂組成物(I)の代わりにエポキシ樹脂組成物(IV)3.50gを用い、フラックス(B)の使用量を0.07g、硬化剤(C)の使用量を0.21g、無機フィラー(D)の使用量を1.5gとして、有機溶媒(E2)の代わりに有機溶媒(E1)4.25gおよび有機溶媒(E2)0.47gを用いた以外は、実施例6の塗布液の調製と同様にして、実施例9の三次元集積回路用層間充填材組成物の塗布液を調整した。この塗布液の固形分濃度は、50重量%で、無機フィラー(D)の含有量は、エポキシ樹脂組成物(I)に対して30重量%であった。三次元集積回路用層間充填材組成物の塗布液中の無機フィラー(D)の体積平均粒径及び最大体積粒径を表7に示す。
【0163】
・実施例10〜13の塗布液の調整
エポキシ樹脂(A3)、エポキシ樹脂(A4)、エポキシ樹脂(A5)、エポキシ樹脂(A6)、およびエポキシ樹脂(A7)を表8に記載の比率にて混合した以外は、実施例9の三次元集積回路用層間充填材組成物の塗布液の調製と同様にして、樹脂組成物(V)〜(VIII)を調製した。このエポキシ樹脂組成物(V)〜(VIII)の溶融粘度及び熱伝導率を表6に示す。
当該樹脂組成物(V)〜(VIII)を使用した以外は、実施例9の三次元集積回路用層間充填材組成物の塗布液の調製と同様にして、実施例10〜13の三次元集積回路用層間充填材組成物の塗布液を調整した。三次元集積回路用層間充填材組成物の塗布液中の無機フィラー(D)の体積平均粒径及び最大体積粒径を表7に示す。
【0164】
[実施例6〜13]
前記三次元集積回路用層間充填材組成物の塗布液を用いて、Siチップ「CC80−0101JY ModelI」上に約10μLを塗布して塗り拡げた後、80℃で15分間、続いて120℃で30分間それぞれ加熱を行いB−Stage化処理を行った。このSiチップと有機基板「CC80−0102JY ModelI」を、フリップチップボンダ装置を用いて、250℃まで昇温させて加熱圧着接合を行った。接合後、有機基板上の電極に、プローバー端子を接地させて、Siチップ及び有機基板の電気端子間に形成されたデイジーチェインについて導通評価を行った。導通評価結果を、表7に示す。導通が確認出来たものを○、導通が確認されなかったものを×とした。
【0165】
【表6】
【表7】
【産業上の利用可能性】
【0166】
本発明によれば、半導体デバイス基板間のはんだバンプ等とランドの接合と同時に、熱伝導性の高い層間充填層を形成する層間充填材組成物、塗布液及び三次元集積回路の製造方法が提供される。
【技術分野】
【0001】
本発明は、三次元集積回路用の層間充填材組成物、該層間充填材組成物を含有する塗布液、及び該層間充填材組成物を含む三次元集積回路の製造方法に関する。
【背景技術】
【0002】
近年、半導体デバイスの更なる高速化・高容量化などの性能向上のために、トランジスタや配線の微細化に加えて、半導体デバイスチップを2層以上積み重ねた三次元(3D)積層化した三次元集積回路による性能向上に向けた研究開発が進められている。
三次元集積回路では、半導体デバイスチップ同士がそのチップ間において、はんだバンプ等の電気信号端子等で接続されていると同時に、層間充填材組成物を充填して形成された層間充填層により接着された構造を有している。
【0003】
具体的には、ウェハー上に塗布により層間充填材組成物の薄膜を形成した後に、Bステージ化を行い、次いでダイシングによりチップを切り出し、このチップを用いて加圧加熱による仮接合を繰り返し、最終的に加圧加熱条件下で本接合(半田接合)を行うプロセスが提案されている(非特許文献1参照)。
【0004】
このような三次元集積回路デバイスの実用化に向けて、種々の課題が指摘されているが、その内の一つにトランジスタや配線等のデバイスから発する熱の放熱問題がある。この問題は、半導体デバイスチップの積層の際に用いられる層間充填材組成物の熱伝導率が、金属やセラミックなどに比べ一般的に非常に低いことに起因し、積層デバイスチップ内での蓄熱によるパフォーマンスの低下が懸念されている。
【0005】
この課題を解決する一つの手法として、層間充填材組成物の高熱伝導化が挙げられる。例えば、層間充填材組成物を構成する樹脂単体として高熱伝導性のエポキシ樹脂を使用したり、そのような樹脂と高熱伝導性無機フィラーとを複合化させることで、層間充填材組成物を高熱伝導化することが行われている。例えば、特許文献1には、球状窒化ホウ素凝集体をフィラーとして配合した層間充填材組成物が記載されている。窒化ホウ素は通常板状粒子であり、その長径方向と短径方向とで熱伝導率が異なる性質を有するが、窒化ホウ素粒子をバインダーで結合して球状の凝集体とすることで、熱伝導率が各方向で均一化するため、該窒化ホウ素凝集体をフィラーとして樹脂に配合することで熱伝導率が向上することが開示されている。
【0006】
さらに、エポキシ樹脂自体の熱伝導性を向上させる発明として、エポキシ樹脂にメソゲン骨格を導入する方法が開示されている。例えば、非特許文献2には、種々のメソゲン骨格の導入によるエポキシ樹脂の熱伝導性向上についての記載があるが、熱伝導性の向上は見られるものの、コスト面、プロセス適合性、耐加水分解性や熱安定性などのバランスを考慮すると実用的とは言えない。
また、特許文献2には、ビフェニル骨格のみを用いた熱伝導性のよいエポキシ樹脂が開示されているが、合成されているのはごく低分子量のエポキシ樹脂のみであり、製膜性に欠けるため、薄膜として用いることが困難である。
【0007】
さらに、無機フィラーを含むエポキシ樹脂組成物においては、無機フィラー表面で樹脂の剥離が起こることがあり、所望の熱伝導率が達成されない場合があった。また、エポキシ当量が大きくないメソゲン骨格を有するエポキシ樹脂は、硬化後に樹脂が結晶性が高く固い構造をとることが多く、熱伝導性と低応力化とのバランスが求められている。
【0008】
一方、従来の半導体デバイスチップのインターポーザ等への搭載プロセスにおいては、初めに半導体デバイスチップ側のはんだバンプ等の電気信号端子をフラックスにより活性化処理を行い、次いでランド(電気接合電極)を有する基板に接合した後、基板間に液状樹脂又は液状樹脂に無機フィラーを添加したアンダーフィル材により充填・硬化により接合完了としている。この際、フラックスには、はんだバンプ等の金属電気信号端子及びランドの表面酸化膜除去や濡れ広がり性の向上、更には金属端子表面の再酸化防止などの活性化処理機能が求められている。
フラックスとしては、一般に、電気信号端子の金属酸化膜溶解能に優れたハロゲンを含む無機金属塩の他に、有機酸や有機酸塩、有機ハロゲン化合物やアミン類、ロジンやその構成成分の単独又は複数の組合せにより用いられている(例えば、非特許文献3参照)。
【0009】
また、半導体デバイスチップの3D積層プロセスにおいては、初めにフラックスを用いたはんだバンプ等の電気信号端子の活性化処理を行うと、端子表面に熱伝導性の低いフラックス層が形成され、層間充填材組成物による積層基板間の熱伝導性の阻害や、フラックス成分の残留による接合端子の腐食劣化等の要因となることが懸念されている。このため、高い熱伝導性を有する層間充填材組成物へ直接混合可能であり、且つ金属端子への腐食性の低いフラックスが求められている。
【0010】
上記のように、高熱伝導性の層間充填材組成物には、3D積層プロセスへの適合性や薄膜化に加えて、半導体デバイスチップ間における電気信号端子の接合性などが求められており、更なる技術開発が必要とされている。
【先行技術文献】
【特許文献】
【0011】
【特許文献1】特表2008−510878号公報
【特許文献2】特開2010−001427号公報
【非特許文献】
【0012】
【非特許文献1】エレクトロニクス実装学会講演大会講演論文集、61,23,2009)
【非特許文献2】電子部品用エポキシ樹脂の最新技術(シーエムシー出版、2006年、第1章P24〜31、第5章P114〜121)
【非特許文献3】はんだ付けの基礎と応用(工業調査会)
【発明の概要】
【発明が解決しようとする課題】
【0013】
三次元集積回路デバイスの実用化に向けての課題の一つに、トランジスタや配線等のデバイスから発する熱の放熱問題がある。この問題は、半導体デバイスチップの積層の際に用いられる層間充填材組成物の熱伝導率が、金属やセラミックなどに比べ一般的に非常に低いことに起因し、積層デバイスチップ内での蓄熱によるパフォーマンスの低下が懸念されており、より熱伝導率が高い層間充填材組成物が望まれている。
また、上記三次元集積回路は、更なる高速化・高容量化などの性能向上のために各チップ間の距離がチップ間距離10〜50μm程度にまで小さくなっている。
チップ間の層間充填層において、熱伝導率をより高めるために、場合によって配合されるフィラーの最大体積粒径は、層間充填層の厚みの1/3以下程度にする必要があるが、熱伝導率を高めるにはより多量のフィラーを配合することが好ましい。特に、微細なフィラーを使用する場合、多量のフィラーを配合する必要があるが、フィラーの配合量が多くなりすぎると、層間充填材組成物の接着力が低下したり、溶融時の柔軟性が低下してしまう場合がある。
上記特許文献1記載の球状窒化ホウ素凝集体は高熱伝導率を有するが、粒径が大きすぎるため、上記層間充填材組成物へのフィラーとして使用することができない。
そのため、より微細なフィラーを使用する必要があるが、粒径が小さなフィラーを使用すると、層間充填材組成物を構成する樹脂に配合したときに均一に混合することが困難であると共に、必要な熱伝導パス数が増加してチップ間の厚み方向に上から下まで繋がる確率が小さくなり、層間充填層の厚み方向への熱伝導率が不十分になる可能性がある。
【0014】
また、三次元集積回路用の層間充填材組成物としては、熱硬化性樹脂の反応の中間的な段階であって、材料は加熱により軟化して膨張するが、液体と接触しても完全には溶融又は溶解しない段階であるB−ステージ化膜後に、加温により軟化して溶融粘度が大きく低減することにより、3D積層プロセスにおける基板間の加圧によりはんだバンプ等の圧着接合を行う。この為、層間充填材組成物の硬化プロファイルとしては、B−ステージ化やはんだバンプの接合温度では完全硬化せず、短時間の流動性を有した後にゲル化して、その後に完全硬化することが重要である。そのため、層間充填材組成物を構成する樹脂には、チップ間を接合するための接着力や三次元集積回路の製造プロセスに適するように、該樹脂は加熱温度によって溶融粘度がコントロールできることが求められている。
【0015】
また、三次元集積回路では半導体デバイスチップ同士がそのチップ間において、はんだバンプ等の電気信号端子等で電気的に接続されていると同時に、層間充填材組成物を充填して形成された層間充填層により接着されている必要があるが、層間充填材組成物中で電気信号端子が電気的に確実に接続することが困難な場合があった。
本発明では、三次元集積回路用の層間充填材組成物中に、フラックスを含有することにより、層間充填材組成物中で電気信号端子が電気的に確実に接続できるようにする。ただし、この場合でも、一部のフラックス成分については、エポキシ樹脂成分のモノマー、オリゴマー及びポリマーや有機溶媒に対する溶解性が低く、層間充填材組成物との混合により均一に溶解することが困難になる場合があった。
【0016】
また、フラックスが有する酸性又は塩基性の官能基は、エポキシ樹脂成分に対して、フラックス作用と同時に硬化剤としての作用を呈し、B−ステージ化やはんだバンプ接合前の温度において、エポキシ樹脂の硬化を引き起こして、はんだバンプ等とランドの接合を阻害するという可能性も予想される。
【0017】
本発明は、上記課題に鑑みてなされたものであり、半導体デバイスチップの3D積層プロセスにおいて、半導体デバイスチップ間のはんだバンプ等とランドの接合性に優れると同時に、熱伝導性の高い層間充填層を形成する層間充填材組成物、該層間充填材組成物を含有する塗布液、及び該層間充填材組成物を含む三次元集積回路の製造方法を提供することを目的とするものである。
【課題を解決するための手段】
【0018】
本発明者等は、鋭意研究の結果、下記する発明が上記課題を解決出来ることを見出し、本発明を完成するに至った。
すなわち、本発明は、以下の要旨を有するものである。
1.120℃における溶融粘度が100Pa・s以下である樹脂(A)及びフラックス(B)を含有し、フラックス(B)の含有量が、樹脂(A)100重量部当たり0.1重量部以上10重量部以下であることを特徴とする三次元集積回路用の層間充填材組成物。
2.更に硬化剤(C)を含有する、上記1に記載の三次元集積回路用の層間充填材組成物。
3.熱伝導率が1W/mK以上の無機フィラー(D)を樹脂(A)100重量部当たり、50重量部以上400重量部以下更に含有する上記1又は2に記載の三次元集積回路用の層間充填材組成物。
4.120℃における溶融粘度が100Pa・s以下であって、熱伝導率が0.2W/mK以上である樹脂(A)と、熱伝導率が2W/mK以上、体積平均粒径が0.1μm以上5μm以下、かつ、最大体積粒径が10μm以下である無機フィラー(D)と、硬化剤(C)及び/またはフラックス(B)とを含有することを特徴とする三次元集積回路用の層間充填材組成物。
【0019】
5.前記樹脂(A)の50℃における溶融粘度が2000Pa・s以上である、上記1から4のいずれか1項に記載の三次元集積回路用の層間充填材組成物。
6.前記樹脂(A)が熱硬化性樹脂である、上記1から5のいずれか1項に記載の三次元集積回路用の層間充填材組成物。
7.前記樹脂(A)がエポキシ樹脂である、上記1から6のいずれか1項に記載の三次元集積回路用の層間充填材組成物。
8.前記エポキシ樹脂が、フェノキシ樹脂であるエポキシ樹脂(A1)、又はエポキシ樹脂(A1)と、分子内に2個以上のエポキシ基を有するエポキシ樹脂であるエポキシ樹脂(A2)との混合物である上記7に記載の三次元集積回路用の層間充填材組成物。
【0020】
9.前記エポキシ樹脂が、下記式(1)で表され、かつエポキシ当量が2,500g/当量以上30,000g/当量以下を有するエポキシ樹脂(B)である上記7に記載の三次元集積回路用の層間充填材組成物。
【化1】
(式(1)中、Aは下記式(2)で表されるビフェニル骨格であり、Bは水素原子または下記式(3)で表される基であり、nは繰り返し数であり、平均値は1<n<100である。)
【化2】
(式(2)中、R1は、水素原子、炭素数1〜10の炭化水素基、またはハロゲン元素であり、互いに同一であっても異なっていてもよい。)
【化3】
10.前記式(2)におけるR1が、水素原子または炭素数が1〜4のアルキル基であり、式(2)で表されるビフェニル骨格は少なくとも一つの水素原子と少なくとも一つの炭素数1〜4のアルキル基を有する、上記9に記載の三次元集積回路用の層間充填材組成物。
【0021】
11.更にエポキシ当量が2500g/当量未満であるエポキシ樹脂(C)を含有する上記9又は10に記載の三次元集積回路用の層間充填材組成物。
12.エポキシ樹脂(B)とエポキシ樹脂(C)を含む全エポキシ樹脂中のエポキシ樹脂(C)の割合が、10重量%以上80重量%以下である上記11に記載の三次元集積回路用の層間充填材組成物。
13.前記フラックス(B)が、有機カルボン酸である上記1から12のいずれか1項に記載の三次元集積回路用の層間充填材組成物。
14.前記有機カルボン酸の分解温度が、130℃以上である上記13に記載の三次元集積回路用の層間充填材組成物。
15.前記樹脂(A)と前期無機フィラー(D)との総体積に対し、無機フィラー(D)が5体積%以上60体積%以下である、上記3から14のいずれか1項に記載の層間充填材組成物。
16.前期無機フィラー(D)が、窒化ホウ素フィラーである、上記3から15のいずれか1項に記載三次元集積回路用の層間充填材組成物。
17.前記硬化剤(C)が、イミダゾール又はその誘導体である、上記2から16のいずれか1項に記載の三次元集積回路用の層間充填材組成物。
18.上記1から17のいずれか1項に記載の層間充填材組成物に、更に有機溶媒(E)を含有してなる、三次元集積回路用の層間充填材組成物の塗布液。
19.複数の半導体基板表面に、上記1から17のいずれか1項に記載の層間充填材組成物を成膜した後に、これらの半導体基板を加圧接着して積層する工程を含む、三次元集積回路の製造方法。
【発明の効果】
【0022】
本発明によると、半導体デバイス基板間のはんだバンプ等とランドの接合と同時に、熱伝導性の高い層間充填層を形成する層間充填材組成物、該層間充填材組成物を含有する塗布液、及び該層間充填材組成物を含む三次元集積回路の製造方法を提供することができる。
本発明の三次元集積回路用の層間充填材組成物によれば、半導体デバイスチップの3D積層プロセスにおいて、半導体デバイスチップ間のはんだバンプ等とランドとの接合性に優れると共に、特に、熱伝導率が高く微細な無機フィラーを熱伝導率が高いエポキシ樹脂によって接合するため、放熱性が高く、無機フィラーを使用した場合には更に、熱伝導性の高い層間充填層を形成することができる。
【図面の簡単な説明】
【0023】
【図1】接合評価に用いるはんだバンプ基板の模式図であり、(a)は平面図、(b)は断面図である。
【図2】実施例3の層間充填剤ペーストの粒度分布の評価結果である。
【図3】実施例3の層間充填剤ペーストから得られたB−ステージ化膜の表面写真である。
【図4】比較例3の層間充填剤ペーストの粒度分布の評価結果である。
【図5】比較例3の層間充填剤ペーストから得られたB−ステージ化膜の表面写真である。
【発明を実施するための形態】
【0024】
以下、本発明の実施の形態について説明するが、本発明は以下の実施の形態に限定されるものではなく、その要旨の範囲内で種々変形して実施することができる。
尚、本明細書において「〜」という表現を用いる場合、その前後の数値を含む表現として用いる。
【0025】
[三次元集積回路用の層間充填材組成物]
本発明の三次元集積回路用の層間充填材組成物(本発明では、単に層間充填材組成物ということがある。)は、三次元集積回路の各層を構成する半導体基板同士を接着し、半導体基板同士の間隙を充填する充填層を形成することができる組成物である。
この三次元集積回路用の層間充填材組成物においては、樹脂(A)の溶融粘度が120℃において100Pa・s以下であることにより、はんだバンプが融解する前に樹脂を溶融させて粘度を大きく低減することにより、加熱プレスによりはんだバンプとランド端子の接合を可能とし、更に半導体基板上に成膜された層間充填材組成物を200℃以上で加圧接着することにより、はんだバンプを融解させてランド端子との電気接続を実現することが出来る。この際、所定の硬化剤を添加することにより、B−ステージ化や、はんだバンプの接合温度では硬化せず、はんだバンプの接合後に短時間の流動性を有した後にゲル化して、その後に完全硬化することにより、安定な層間充填膜を形成することが出来る。
また、樹脂(A)の50℃における溶融粘度が2000Pa・s以上であると、B−ステージ化膜後の室温におけるタック性を低減し、基板の積み重ね時の位置合わせを実施することにより、三次元集積回路の積層基板同士の仮接着が可能になり、好ましい。
以下、各成分について説明する。
【0026】
[樹脂(A)]
本発明における樹脂(A)は、仮接着後に本接合を実施する際に、加温により層間充填材組成物を溶融して電気接合端子を接続させるために、120℃における溶融粘度が100Pa・s以下であることを必須とし、20Pa・s以下であることが好ましい。また、樹脂(A)は、層間充填材組成物として基板上に薄膜を形成後、仮接着前に接合対象の基板と位置合わせを行うために、50℃における溶融粘度が2000Pa・s以上であることが好ましく、10000Pa・s以上であることがより好ましい。
後述する無機フィラー(D)と組み合わせた際に十分な熱伝導性を得るために、熱伝導率が好ましくは0.2W/mK以上、より好ましくは0.22W/mK以上である。
【0027】
上記樹脂(A)として、上記溶融粘度条件を満たす樹脂が使用できるが、熱硬化性樹脂であるのが好ましい。かかる熱硬化性樹脂の好ましい例としては、アクリル樹脂、エポキシ樹脂、熱硬化性ポリイミド樹脂、熱硬化性フェノール樹脂、などが挙げられる。なかでも、エポキシ樹脂、熱硬化性ポリイミド樹脂が好ましい。
本発明における樹脂(A)は、なかでも、エポキシ樹脂であるのが好ましい。エポキシ樹脂は1種類の構造単位を有するエポキシ樹脂のみでもよいが、上記溶融粘度条件を満たすならば、構造単位の異なる複数のエポキシ樹脂を組み合わせてもよい。
エポキシ樹脂は、塗膜性ないしは成膜性や接着性と併せて、接合時のボイドを低減して高熱伝導の硬化物を得るために、後述するフェノキシ樹脂(以下、エポキシ樹脂(A1)と称す。)を少なくとも含むことが好ましく、特にエポキシ樹脂全量に対するエポキシ樹脂(A1)の重量比率が、5〜95重量%。より好ましくは10〜90重量%、さらに好ましくは20〜80重量%の範囲で含有されることが好ましい。
【0028】
[エポキシ樹脂(A1)及びエポキシ樹脂(A2)]
フェノキシ樹脂とは、通常、エピハロヒドリンと2価フェノール化合物とを反応させて得られる樹脂、または2価のエポキシ化合物と2価のフェノール化合物とを反応させて得られる樹脂を指す。本発明においてはこれらのうち、特に重量平均分子量10000以上の高分子量エポキシ樹脂であるフェノキシ樹脂をエポキシ樹脂(A1)と言う。
【0029】
エポキシ樹脂(A1)としては、ナフタレン骨格、フルオレン骨格、ビフェニル骨格、アントラセン骨格、ピレン骨格、キサンテン骨格、アダマンタン骨格およびジシクロペンタジエン骨格からなる群から選択された少なくとも1つの骨格を有するフェノキシ樹脂が好ましい。中でも、耐熱性がより一層高められるので、フルオレン骨格および/またはビフェニル骨格を有するフェノキシ樹脂が特に好ましい。
【0030】
上述のようにエポキシ樹脂(A)は、構造単位の異なる複数のエポキシ樹脂を含むものであってもよい。
上記エポキシ樹脂(A1)以外のエポキシ樹脂としては、分子内に2個以上のエポキシ基を有するエポキシ樹脂(以下、エポキシ樹脂(A2)と称す。)であることが好ましい。エポキシ樹脂(A2)としては、例えば、ビスフェノールA型エポキシ樹脂、ビスフェノールF型エポキシ樹脂、ナフタレン型エポキシ樹脂、フェノールノボラック型エポキシ樹脂、クレゾールノボラック型エポキシ樹脂、フェノールアラルキル型エポキシ樹脂、ビフェニル型エポキシ樹脂、トリフェニルメタン型エポキシ樹脂、ジシクロペンタジエン型エポキシ樹脂、グリシジルエステル型エポキシ樹脂、グリシジルアミン型エポキシ樹脂、多官能フェノール型エポキシ樹脂等が挙げられる。これらは1種を単独でまたは2種以上の混合体として使用することができる。
【0031】
エポキシ樹脂(A2)は、溶融粘度制御の観点から、その平均分子量が、好ましくは、100〜5000であり、より好ましくは、200〜2000である。平均分子量が100より低いものでは、耐熱性が劣る傾向にあり、5000より高いと、エポキシ樹脂の融点が高くなり、作業性が低下する傾向がある。
【0032】
また、本発明において、エポキシ樹脂としては、その目的を損なわない範囲において、エポキシ樹脂(A1)とエポキシ樹脂(A2)以外のエポキシ樹脂(以下、他のエポキシ樹脂)を使用してもよい。他のエポキシ樹脂の含有量は、エポキシ樹脂(A1)とエポキシ樹脂(A2)の合計に対して、通常、50重量%以下、好ましくは、30重量%以下である。
【0033】
本発明でエポキシ樹脂(A2)を使用する場合、エポキシ樹脂(A1)とエポキシ樹脂(A2)を含む全エポキシ樹脂中のエポキシ樹脂(A1)の割合は、その合計を100重量%として、10〜90重量%、好ましくは20〜80重量%である。なお、「エポキシ樹脂(A1)とエポキシ樹脂(A2)を含む全エポキシ樹脂」とは、本発明のエポキシ樹脂組成物に含まれるエポキシ樹脂が、エポキシ樹脂(A1)及びエポキシ樹脂(A2)のみの場合には、エポキシ樹脂(A1)とエポキシ樹脂(A2)の合計を意味し、さらに他のエポキシ樹脂を含む場合には、エポキシ樹脂(A1)、エポキシ樹脂(A2)及び他のエポキシ樹脂の合計を意味する。
エポキシ樹脂(A1)の割合が10重量%以上であることにより、エポキシ樹脂(A1)を配合することによる熱伝導性の向上効果を十分に得ることができ、所望の高熱伝導性を得ることができる。エポキシ樹脂(A1)の割合が90重量%未満でエポキシ樹脂(A2)が10重量%以上であることにより、エポキシ樹脂(A2)の配合効果が発揮され、硬化性、硬化物の物性が十分なものとなる。
【0034】
[エポキシ樹脂(B)]
本発明において、エポキシ樹脂は、なかでも、下記式(1)で表される構造を有し、かつ、エポキシ当量が2500g/当量以上であるエポキシ樹脂(B)が好ましい。
【0035】
【化4】
(式(1)中、Aは下記式(2)で表わされるビフェニル骨格であり、Bは水素原子又は下記式(3)であり、nは繰り返し数であり、平均値は10<n<50である。)
【化5】
(式(2)中、R1は、水素原子、炭素数1〜10の炭化水素基、又はハロゲン原子であり、互いに同一であっても異なっていてもよい。)
【化6】
【0036】
エポキシ樹脂(B)は、十分な伸び性を有し、熱伝導性、耐熱性とのバランスに優れたエポキシ樹脂である。
従来の高熱伝導性エポキシ樹脂はほぼすべてが熱伝導性を高めるために設計されたエポキシ樹脂であり、硬化条件を含めた硬化プロセス等において制限があることが多く、その選択の自由度は低かった。このため、部材や封止剤、接着剤などの製品に従来の高熱伝導性エポキシ樹脂を適用しようとした場合、コストを含めた製品の要求物性と高熱伝導性を両立させることが困難であった。
これに対し、エポキシ樹脂(B)は、それ自体熱伝導性に優れ、エポキシ樹脂成分として所望の量添加することで硬化物の熱伝導性を高めることが出来、また、エポキシ樹脂(B)の伸び性に由来して、材料に低応力性を付与しうる。
【0037】
エポキシ樹脂(B)が伸び性に優れる理由の詳細は明らかではないが、引っ張りの応力がかかった際の延伸に耐えうる分子鎖長を有し、さらにその応力を緩和するために、重なり合ったビフェニル骨格同士が「滑る」ことができるためであると推測される。また、この時、結晶性が高すぎると、脆く、伸びずに破断してしまうため、適度にアモルファス部分を有していることが重要であるが、エポキシ樹脂(B)では、ビフェニル骨格が置換基を有することにより、結晶性を適度に低下させることができ、このことが伸び性の発現に繋がっていると考えられる。従って、伸び性の観点からは、前記式(2)におけるR1がすべて水素原子ではなく、1つ以上のR1が炭化水素基、又はハロゲン原子であることが好ましい。
【0038】
熱伝導はフォノンと伝導電子に支配され、金属のように自由電子を有する場合は伝導電子による寄与が大きいが、エポキシ樹脂は一般的に絶縁体であり、絶縁体においてはフォノンが熱伝導の主因子である。フォノンによる熱伝導は振動エネルギーの伝播であるので、振動が減衰しにくく、硬い材料であるほど熱伝導性に優れる。
エポキシ樹脂(B)が熱伝導性に優れる理由の詳細は明確ではないが、全ての骨格がビフェニル骨格であることから構造の自由度が少なく、振動エネルギーが減衰しにくいこと、またビフェニル骨格は平面性が高いため、分子間の重なりが良く、より分子運動を拘束できることによるものであると推定される。
【0039】
エポキシ樹脂は、一般に、結晶性がよい方が耐熱性に優れる傾向があり、同一構造のエポキシ樹脂であれば、樹脂の分子量、あるいはエポキシ当量が高い方が耐熱性に優れる傾向にある。エポキシ樹脂(B)は適度な結晶性とエポキシ当量の高さを有することにより耐熱性にも優れる。
【0040】
前記エポキシ樹脂(B)を表す式(1)中、nは繰り返し数であり、平均値である。その値の範囲は10<n<50であるが、伸び性と樹脂の取り扱いの両面のバランスから、nの範囲は、15<n<50であることが好ましく、とりわけ20<n<50であることが好ましい。nが10以下であると、エポキシ樹脂(B)の伸び性が不十分となり、50以上であるとエポキシ樹脂(B)を含む組成物の粘度が高くなり、取り扱いが困難となる傾向がある。
【0041】
また、前記式(1)中、Aは前記式(2)で表されるビフェニル骨格であり、前記式(2)において、R1は、互いに同一であっても異なっていてもよく、水素原子、炭素数1〜10の炭化水素基、又はハロゲン原子を表すが、1分子のエポキシ樹脂において、R1としては水素原子と炭素数1〜10の炭化水素基との両方を含んでいるものがエポキシ樹脂(B)の結晶性とハンドリングの観点から好ましい。R1が同一であると結晶性が高くなり、熱伝導性を高めることが可能であるが、結晶性が高すぎるとエポキシ樹脂組成物をフィルム成形したときの伸びが小さくなる傾向にある。
【0042】
前記式(2)におけるR1が、炭素数1〜10の炭化水素基である場合には、R1は好ましくは1〜4のアルキル基、特に好ましくはメチル基である。
尚、R1の炭化水素基は置換基を有していてもよく、その置換基は特に限定されるものではないが、分子量で200以下のものである。
また、R1のハロゲン原子とは、フッ素原子、塩素原子、臭素原子を指し、これらは1種のみでも複数種を含んでいてもよい。
【0043】
Aのビフェニル骨格は、2,2’−ビフェニル骨格、2,3’−ビフェニル骨格、2,4’−ビフェニル骨格、3,3−ビフェニル骨格、3,4’−ビフェニル骨格、4,4’−ビフェニル骨格のいずれでも良いが、好ましくは4,4’−ビフェニル骨格である。 また、R1としては、2位及び/又は6位に水素原子があることが好ましく、3位及び/又は5位に炭化水素基があることが好ましい。
【0044】
エポキシ樹脂(B)を使用する場合のエポキシ当量は、2,500g/当量以上であることが好ましい。エポキシ樹脂(B)のエポキシ当量が2,500g/当量未満の場合には、十分な伸び性が得られず、フィルム成形・塗布などのプロセスに適用する際に取扱いが困難になる場合がある。
伸び性の観点からは、エポキシ樹脂(B)のエポキシ当量は、好ましくは3,000g/当量以上、より好ましくは4,000g/当量以上である。
一方、エポキシ当量の上限値は特に限定はないが、取り扱い性・作業性という点で、好ましくは30,000g/当量以下が好ましく、より好ましくは15,000g/当量以下、更に好ましくは10,000g/当量以下である。エポキシ樹脂(B)のエポキシ当量は、後述の実施例の項に記載される方法で求められる。
【0045】
エポキシ樹脂(B)の重量平均分子量Mwは、10,000以上200,000以下であることが好ましい。重量平均分子量が10,000より低いものでは伸び性が低くなる傾向にあり、200,000より高いと樹脂の取り扱いが困難となる傾向にある。エポキシ樹脂(B)の重量平均分子量は、後述の実施例の項に記載される方法で求められる。
エポキシ樹脂(B)の熱伝導率(硬化前の熱伝導率)は、通常0.18W/mK以上、好ましくは0.19W/mK以上、さらに好ましくは0.20W/mK以上である。尚、一般的にエポキシ樹脂の熱伝導率はエポキシ樹脂の硬化物として評価されることが多く、一般的な硬化していないビスフェノールA型エポキシ樹脂の熱伝導率は通常この値よりも低く、液状であるため伸び性を測定するサンプル作製も不可能である場合が多い。
【0046】
本発明に係るエポキシ樹脂(B)は、硬化前の樹脂そのものの状態でも十分な製膜性と熱伝導率を有し、かつ伸び性とのバランスにも優れるものである。なお、エポキシ樹脂(A)の熱伝導率は、後述の実施例の項に記載される方法で測定される。
エポキシ樹脂(B)は耐熱性に優れるものであり、後掲の実施例の項で示すガラス転移温度Tgで評価した場合、90℃以上、220℃以下を達成することができる。エポキシ樹脂のTgは、本発明の用途では高い方が好ましく、好ましくは95℃以上、より好ましくは100℃以上、更に好ましくは105℃以上であるが、Tgが高過ぎると、加工プロセスで使用する加熱温度で硬化反応が十分に進行せず、品質が安定しなかったり、要求される物性が発現しない、といった問題が生じうるため、その上限は通常200℃であることが好ましい。
【0047】
以下、エポキシ樹脂(B)の製造方法について説明する。
エポキシ樹脂(B)は、例えば、ビフェニル骨格を有する2官能エポキシ樹脂(X)とビフェノール化合物(Y)を反応させる、二段法によって得ることができる。また、1種類又は2種類以上のビフェノール化合物(Y)とエピクロロヒドリンを直接反応させる、一段法によっても得られる。しかし、ビフェノール化合物(Y)は溶剤溶解性が良くないため、一般的に一段法に用いられる溶剤がそのまま適用できない場合があるので、二段法を用いることが好ましい。
【0048】
エポキシ樹脂(B)の製造に用いられる2官能エポキシ樹脂(X)は、ビフェニル骨格を有し、分子内に2個のエポキシ基を持つ化合物であり、下記式(4)で表されるビフェノール化合物をエピハロヒドリンと縮合させて得られるエポキシ樹脂等が挙げられる。
【化7】
(式(4)中、R2は式(2)におけるR1と同義である。)
【0049】
前記式(4)で表されるビフェノール化合物としては、例えば、2,2’−ビフェノール、2,3’−ビフェノール、2,4’−ビフェノール、3,3’−ビフェノール、3,4’−ビフェノール、4,4’−ビフェノール、2−メチル−4,4’−ビフェノール、3−メチル−4,4’−ビフェノール、2,2’−ジメチル−4,4’−ビフェノール、3,3’−ジメチル−4,4’−ビフェノール、3,3’,5,5’−テトラメチル−4,4’−ビフェノール、2,2’,3,3’,5,5’−ヘキサメチル−4,4’−ビフェノール、2,2’,3,3’,5,5’,6,6’−オクタメチル−4,4’−ビフェノール等が挙げられる。これらの中で好ましいものは、4,4’−ビフェノール、3,3’−ジメチル−4,4’−ビフェノール、3,3’,5,5’−テトラメチル−4,4’−ビフェノールである。エピハロヒドリンとの縮合反応を行う際には、これらのビフェノール化合物は単独で用いてもよく、また複数種を併用してもよい。また、このようなビフェノール化合物とエピハロヒドリンとを縮合させて得られた2官能エポキシ樹脂(X)を複数種併用することもできる。
【0050】
2官能エポキシ樹脂(X)中としては、その末端基不純物である加水分解性塩素濃度が200ppm以下であり、αグリコール基濃度が100meq/kg以下である2官能エポキシ樹脂(X)を原料として使用することが好ましい。加水分解性塩素濃度が200ppmより大きい場合や、αグリコール基濃度が100meq/kgより大きい場合には、十分に高分子量化しなくなり、好ましくない。
【0051】
一方、ビフェノール化合物(Y)は、2個の水酸基がビフェニル骨格に結合した化合物であり、前記式(4)で表される。ビフェノール化合物(Y)としては、上記と同様、例えば、2,2’−ビフェノール、2,3’−ビフェノール、2,4’−ビフェノール、3,3’−ビフェノール、3,4’−ビフェノール、4,4’−ビフェノール、2−メチル−4,4’−ビフェノール、3−メチル−4,4’−ビフェノール、2,2’−ジメチル−4,4’−ビフェノール、3,3’−ジメチル−4,4’−ビフェノール、3,3’,5,5’−テトラメチル−4,4’−ビフェノール、2,2’,3,3’,5,5’−ヘキサメチル−4,4’−ビフェノール、2,2’,3,3’,5,5’,6,6’−オクタメチル−4,4’−ビフェノール等が挙げられる。これらの中で好ましいものは、4,4’−ビフェノール、3,3’−ジメチル−4,4’−ビフェノール、3,3’,5,5’−テトラメチル−4,4’−ビフェノールである。これらのビフェノール化合物は複数種を併用することもできる。
【0052】
なお、上記2官能エポキシ樹脂(X)とビフェノール化合物(Y)に含まれるビフェニル骨格が同時に無置換でないことが好ましく、一分子中に1つ以上の置換基を有することが好ましい。全てが無置換のビフェニル骨格であると、得られるエポキシ樹脂(B)の結晶性が高くなり、伸び性が悪くなる傾向にある。
【0053】
エポキシ樹脂(B)の製造において、上記の2官能エポキシ樹脂(X)とビフェノール化合物(Y)の使用量は、その配合当量比で、エポキシ基:フェノール性水酸基=1:0.90〜1.10となるようにするのが好ましい。この当量比が上記範囲であることにより十分な高分子量化が進行する。
エポキシ樹脂(B)の合成には触媒を用いてもよく、その触媒としては、エポキシ基とフェノール性水酸基、アルコール性水酸基やカルボキシル基との反応を進めるような触媒能を持つ化合物であればどのようなものでもよい。例えば、アルカリ金属化合物、有機リン化合物、第3級アミン、第4級アンモニウム塩、環状アミン類、イミダゾール類等が挙げられる。これらの触媒は1種のみを使用することも、2種以上組み合わせて使用することもできる。
【0054】
アルカリ金属化合物の具体例としては、水酸化ナトリウム、水酸化リチウム、水酸化カリウム等のアルカリ金属水酸化物、炭酸ナトリウム、重炭酸ナトリウム、塩化ナトリウム、塩化リチウム、塩化カリウム等のアルカリ金属塩、ナトリウムメトキシド、ナトリウムエトキシド等のアルカリ金属アルコキシド、アルカリ金属フェノキシド、水素化ナトリウム、水素化リチウム等のアルカリ金属の水素化物、酢酸ナトリウム、ステアリン酸ナトリウム等の有機酸のアルカリ金属塩が挙げられる。
【0055】
有機リン化合物の具体例としては、トリ−n−プロピルホスフィン、トリ−n−ブチルホスフィン、トリフェニルホスフィン、テトラメチルホスフォニウムブロマイド、テトラメチルホスフォニウムアイオダイド、テトラメチルホスフォニウムハイドロオキサイド、トリメチルシクロヘキシルホスホニウムクロライド、トリメチルシクロヘキシルホスホニウムブロマイド、トリメチルベンジルホスホニウムクロライド、トリメチルベンジルホスホニウムブロマイド、テトラフェニルホスホニウムブロマイド、トリフェニルメチルホスホニウムブロマイド、トリフェニルメチルホスホニウムアイオダイド、トリフェニルエチルホスホニウムクロライド、トリフェニルエチルホスホニウムブロマイド、トリフェニルエチルホスホニウムアイオダイド、トリフェニルベンジルホスホニウムクロライド、トリフェニルベンジルホスホニウムブロマイドなどが挙げられる。
【0056】
第3級アミンの具体例としては、トリエチルアミン、トリ−n−プロピルアミン、トリ−n−ブチルアミン、トリエタノールアミン、ベンジルジメチルアミンなどが挙げられる。
第4級アンモニウム塩の具体例としては、テトラメチルアンモニウムクロライド、テトラメチルアンモニウムブロマイド、テトラメチルアンモニウムハイドロオキサイド、トリエチルメチルアンモニウムクロライド、テトラエチルアンモニウムクロライド、テトラエチルアンモニウムブロマイド、テトラエチルアンモニウムアイオダイド、テトラプロピルアンモニウムブロマイド、テトラプロピルアンモニウムハイドロオキサイド、テトラブチルアンモニウムクロライド、テトラブチルアンモニウムブロマイド、テトラブチルアンモニウムアイオダイド、ベンジルトリメチルアンモニウムクロライド、ベンジルトリメチルアンモニウムブロマイド、ベンジルトリメチルアンモニウムハイドロオキサイド、ベンジルトリブチルアンモニウムクロライド、フェニルトリメチルアンモニウムクロライドなどが挙げられる。
【0057】
環状アミン類の具体例としては、1,8−ジアザビシクロ(5,4,0)ウンデセン−7,1,5−ジアザビシクロ(4,3,0)ノネン−5等が挙げられる。
イミダゾール類の具体例としては、2−メチルイミダゾール、2−エチル−4−メチルイミダゾール、2−フェニルイミダゾールなどが挙げられる。
【0058】
触媒の使用量は反応固形分中、通常0.001〜1重量%である。なお、触媒として、アルカリ金属化合物を使用すると得られるエポキシ樹脂(B)中にアルカリ金属分が残留し、エポキシ樹脂(B)を成分として含む、本発明のエポキシ樹脂組成物をプリント配線板に使用した場合、使用したプリント配線板の絶縁特性を悪化させる傾向があるため、エポキシ樹脂中のLi,NaおよびKの含有量の合計が60ppm以下、好ましくは50ppm以下とする必要がある。
【0059】
また、有機リン化合物、第3級アミン、第4級アンモニウム塩、環状アミン類、イミダゾール類等を触媒として使用した場合も、得られるエポキシ樹脂(B)中にこれらが触媒残渣として残留し、アルカリ金属分の残留と同様にプリント配線板の絶縁特性を悪化させるので、エポキシ樹脂(B)中の窒素の含有量が300ppm以下であり、エポキシ樹脂(B)中のリンの含有量が300ppm以下である必要がある。さらに好ましくは、エポキシ樹脂(B)中の窒素の含有量が200ppm以下であり、エポキシ樹脂(B)中のリンの含有量が200ppm以下である。
【0060】
エポキシ樹脂(B)は、その製造時の合成反応の工程において、溶剤として有機溶媒を用いてもよく、その有機溶媒としては、エポキシ樹脂(B)を溶解するものであればどのようなものでもよい。例えば、芳香族系溶剤、ケトン系溶剤、アミド系溶剤、グリコールエーテル系溶剤などが挙げられる。
芳香族系溶剤の具体例としては、ベンゼン、トルエン、キシレンなどが挙げられる。
ケトン系溶剤の具体例としては、アセトン、メチルエチルケトン、メチルイソブチルケトン、2−ヘプタノン、4−ヘプタノン、2−オクタノン、シクロヘキサノン、アセチルアセトン、ジオキサンなどが挙げられる。
【0061】
アミド系溶剤の具体例としては、ホルムアミド、N−メチルホルムアミド、N,N−ジメチルホルムアミド、アセトアミド、N−メチルアセトアミド、N,N−ジメチルアセトアミド、2−ピロリドン、N−メチルピロリドンなどが挙げられる。
グリコールエーテル系溶剤の具体例としては、エチレングリコールモノメチルエーテル、エチレングリコールモノエチルエーテル、エチレングリコールモノ−n−ブチルエーテル、エチレングリコールジメチルエーテル、エチレングリコールモノエチルエーテルアセテート、ジエチレングリコールモノメチルエーテル、ジエチレングリコールモノエチルエーテル、ジエチレングリコールモノ−n−ブチルエーテル、ジエチレングリコールジメチルエーテル、ジエチレングリコールモノエチルエーテルアセテート、プロピレングリコールモノメチルエーテル、プロピレングリコールモノ−n−ブチルエーテル、プロピレングリコールモノメチルエーテルアセテートなどが挙げられる。
これらの有機溶媒は単独で用いてもよく、2種以上を併用することもできる。
【0062】
エポキシ樹脂(B)の製造時の合成反応における固形分濃度は35〜95重量%が好ましい。また、反応途中で高粘性生成物が生じたときは溶剤(有機溶媒)を追加添加して反応を続けることもできる。反応終了後、溶剤(有機溶媒)は必要に応じて、除去することもできるし、更に追加することもできる。
【0063】
エポキシ樹脂(B)の製造において、2官能エポキシ樹脂(X)とビフェノール化合物(Y)との重合反応は使用する触媒が分解しない程度の反応温度で実施される。反応温度が高すぎると生成するエポキシ樹脂が劣化するおそれがある。逆に温度が低すぎると十分に反応が進まないことがある。これらの理由から反応温度は、好ましくは50〜230℃、より好ましくは120〜200℃である。また、反応時間は通常1〜12時間、好ましくは3〜10時間である。アセトンやメチルエチルケトンのような低沸点溶剤を使用する場合には、オートクレーブを使用して高圧下で反応を行うことで反応温度を確保することができる。
【0064】
[エポキシ樹脂(C)]
本発明において、エポキシ樹脂(B)は、単独でも使用できるが、エポキシ当量が2500g/当量未満であるエポキシ樹脂(以下、エポキシ樹脂(C)と称する。)と併用することができる。エポキシ樹脂(B)と共にエポキシ樹脂(C)を含むことにより、エポキシ樹脂成分の重合性およびフィラー表面への樹脂硬化物の密着性が向上するため、無機フィラー(D)を含有しても、十分な硬化物物性と熱伝導性を有する硬化物を得ることができる。
【0065】
また、上述のエポキシ樹脂(B)は、それ自体は硬化条件を含めた硬化プロセス等において制限が少なく、十分な伸び性を有し、熱伝導性、耐熱性とのバランスに優れたエポキシ樹脂であるが、エポキシ樹脂(C)を併用することにより、流動性と反応性を有するエポキシ樹脂成分が導入され、このエポキシ樹脂(B)とエポキシ樹脂(C)との混合物がフィラー表面での樹脂の剥離を抑制するため、十分な硬化物物性と熱伝導性を兼ね備える硬化物を得ることができる。
エポキシ樹脂(C)のエポキシ当量は、2500g/当量未満であることを必須するが、硬化物の反応性の観点からは、好ましくは、1000g/当量未満、より好ましくは、500g/当量未満である。
エポキシ樹脂(C)のエポキシ当量が、2500g/当量以上であると、十分な硬化物物性を有する硬化物が得られない。
【0066】
エポキシ樹脂(C)としては、上記エポキシ当量を満足するエポキシ樹脂であればよい。なお、上記式(1)で表される構造を有するエポキシ樹脂であっても、上記エポキシ当量を満足すれば、エポキシ樹脂(C)に該当する。このようなエポキシ樹脂の中でも、分子内に2個以上のエポキシ基を有するものであることが好ましく、例えば、ビスフェノールA型エポキシ樹脂、ビスフェノールF型エポキシ樹脂、ナフタレン型エポキシ樹脂、フェノールノボラック型エポキシ樹脂、クレゾールノボラック型エポキシ樹脂、フェノールアラルキル型エポキシ樹脂、ビフェニル型エポキシ樹脂、トリフェニルメタン型エポキシ樹脂、ジシクロペンタジエン型エポキシ樹脂、グリシジルエステル型エポキシ樹脂、グリシジルアミン型エポキシ樹脂、多官能フェノール型エポキシ樹脂等の、各種エポキシ樹脂を使用することができる。
これらは1種を単独で又は2種以上の混合体として使用することができる。
【0067】
エポキシ樹脂(C)は、上記エポキシ当量を満足するエポキシ樹脂であればよく、その他の物性は任意のものを使用することができる。
一方で、フィラー表面での樹脂剥離抑制の観点からは、エポキシ樹脂(C)の平均分子量は、好ましくは、100〜5000であり、より好ましくは、200〜2000である。平均分子量が100より低いものでは、耐熱性が劣る傾向にあり、5000より高いと、エポキシ樹脂の融点が高くなり、作業性が低下する傾向がある。
【0068】
また、本発明の目的を損なわない範囲において、エポキシ樹脂(B)とエポキシ樹脂(C)以外のエポキシ樹脂(以下、その他のエポキシ樹脂と称する。)を含んでいてもよい。その他のエポキシ樹脂の含有量は、エポキシ樹脂(B)とエポキシ樹脂(C)の合計に対して、通常、50重量%以下、好ましくは、30重量%以下である。
【0069】
エポキシ樹脂を含む組成物において、エポキシ樹脂(B)とエポキシ樹脂(C)を含む全エポキシ樹脂中のエポキシ樹脂(B)の割合は、その合計を100重量%として、10〜80重量%、好ましくは10〜70重量%である。なお、「エポキシ樹脂(B)とエポキシ樹脂(C)を含む全エポキシ樹脂」とは、本発明のエポキシ樹脂組成物に含まれるエポキシ樹脂が、エポキシ樹脂(B)及びエポキシ樹脂(C)のみの場合には、エポキシ樹脂(B)とエポキシ樹脂(C)の合計を意味し、さらにその他のエポキシ樹脂を含む場合には、エポキシ樹脂(B)、エポキシ樹脂(C)及びその他のエポキシ樹脂の合計を意味する。
エポキシ樹脂(B)の割合が10重量%以上であることにより、エポキシ樹脂(B)を配合することによる熱伝導性の向上効果を十分に得ることができ、所望の高熱伝導性を得ることができる。エポキシ樹脂(B)の割合が80重量%未満でエポキシ樹脂(C)が20重量%以上であることにより、エポキシ樹脂(C)の配合効果が発揮され、硬化性、硬化物の物性が十分なものとなる。
【0070】
[フラックス(B)]
本発明の層間充填材組成物はフラックス(B)を含有するが、本発明におけるフラックス(B)としては、はんだバンプ等の金属電気信号端子やランドなどの、金属表面の酸化膜を除去し、活性化させて溶融はんだの濡れ広がり性の向上に寄与するような化合物であれば、どのような化合物も使用することが可能である。より具体的には、金属端子のはんだ接合時において、はんだバンプ等の金属電気信号端子及びランドの表面酸化膜の溶解除去や、はんだバンプのランド表面における濡れ広がり性の向上、更にははんだバンプの金属端子表面の再酸化防止などの機能を有する化合物を言う。
【0071】
本発明で用いるフラックス(B)としては、蓚酸、マロン酸、コハク酸、グルタル酸、アジピン酸、リンゴ酸、酒石酸、クエン酸、乳酸、酢酸、プロピオン酸、酪酸、オレイン酸、ステアリン酸、安息香酸、アビエチン酸、ロジンなどの有機カルボン酸;有機カルボン酸をアルキルビニルエーテル類と反応して変換したヘミアセタールエステルである有機カルボン酸エステル;グルタミン酸塩酸塩、アニリン塩酸塩、ヒドラジン塩酸塩、臭化セチルピリジン、フェニルヒドラジン塩酸塩、テトラクロルナフタレン、メチルヒドラジン塩酸塩、メチルアミン塩酸塩、エチルアミン塩酸塩、ジエチルアミン塩酸塩、ブチルアミン塩酸塩などの有機ハロゲン化合物;尿素、ジエチレントリアミンヒドラジンなどのアミン類、エチレングリコール、ジエチレングリコール、トリエチレングリコール、テトラエチレングリコール、グリセリンなどの多価アルコール類、塩酸、フッ酸、燐酸、ホウフッ化水素酸などの無機酸;フッ化カリウム、フッ化ナトリウム、フッ化アンモニウム、フッ化銅、フッ化ニッケル、フッ化亜鉛などのフッ化物、塩化カリウム、塩化ナトリウム、塩化第一銅、塩化ニッケル、塩化アンモニウム、塩化亜鉛、塩化第一錫などの塩化物;臭化カリウム、臭化ナトリウム、臭化アンモニウム、臭化錫、臭化亜鉛などの臭化物などが挙げられる。
【0072】
これらの化合物は、そのまま用いても、また有機ポリマーや無機化合物等による被覆剤を用いてマイクロカプセル化したものを用いても良い。これらの化合物は1種を単独で用いてもよく、2種以上を任意の組み合わせおよび比率で混合して用いてもよい。
このうち、エポキシ樹脂や各種溶媒への溶解性より、カルボン酸若しくはカルボン酸エステルが好ましい。また、エポキシ樹脂に対して室温における硬化促進能が低いこと、及び層間充填材組成物の保存安定性より特に有機カルボン酸若しくは有機カルボン酸エステルが好ましい。
有機カルボン酸としては既に上記で例示した、蓚酸、マロン酸、コハク酸、グルタル酸、アジピン酸、リンゴ酸、酒石酸、クエン酸、乳酸、酢酸、プロピオン酸、酪酸、オレイン酸、ステアリン酸、安息香酸、アビエチン酸などがあげられるが、より好ましくは炭素数12以下の有機カルボン酸が好ましく、更に好ましくは炭素数8以下の有機カルボン酸が好ましい。
【0073】
有機カルボン酸エステルとしては、下記の反応式に従って、有機カルボン酸とアルキルビニルエーテル類を常温、常圧又は必要に応じて加温することにより得ることが出来る。尚、下記の反応式の反応は平衡反応でもあるので、有機カルボン酸エステルに転化する有機カルボン酸の割合を高めるにはアルキルビニルエーテル類を有機カルボン酸中のカルボキシル基に対して等量以上添加して反応させることが好ましい。
R1COOH+H2C=CHOR2⇔R1CO−O−CH(CH3)OR2
上記反応式において、R1はカルボン酸中の1つのカルボキシル基を除いた残りの分子鎖を示す。R2は炭素数1〜6のアルキル基を示す。
【0074】
有機カルボン酸エステルは、層間充填材組成物中において加熱により分解し、有機カルボン酸及びビニルエーテルを生成する。分解により生じる有機カルボン酸は、はんだボールに対するフラックス性を示す。
また、分解により生じる有機カルボン酸の中にはエポキシ樹脂に対する硬化作用を呈する可能性がある。これは、カルボキシル基はその解離により放出される水素イオンがエポキシ樹脂に対して硬化作用を呈する可能性があるためである。このカルボキシル基の解離による水素イオンの発生を抑制するために、有機カルボン酸をアルキルビニルエーテルにて保護した有機カルボン酸エステルが好ましく用いられる。
一方で、有機カルボン酸エステルを使用した場合においても、その分解温度が低すぎると、製造時における加圧加熱による仮接合時に、エポキシ樹脂が硬化してしまうおそれがある。
そのため、フラックス(B)における有機カルボン酸エステルの分解温度は、仮接合時での分解を回避または抑制するために、130℃以上であることが好ましく、より好ましくは140℃、更に好ましくは160℃以上、最も好ましくは180℃以上である。
【0075】
有機カルボン酸エステルの原料となる有機カルボン酸は、乳酸、酢酸、プロピオン酸、酪酸、オレイン酸、ステアリン酸、安息香酸、アビエチン酸、ロジンなどのモノカルボン酸;蓚酸、マロン酸、コハク酸、グルタル酸、アジピン酸、リンゴ酸、酒石酸、イソフタル酸、ピロメリット酸、マレイン酸、フマル酸、イタコン酸、などのジカルボン酸;クエン酸、1,2,4−トリメリット酸、トリス(2−カルボキシエチル)イソシアヌレートなどのトリカルボン酸;ピロメリット酸やブタンテトラカルボン酸などのテトラカルボン酸等を用いることが出来る。この中でもフラックスとしての反応性より、二個以上のカルボキシル基を有するポリカルボン酸類が好ましい。
【0076】
また、有機カルボン酸エステルの原料となるアルキルビニルエーテル類として、R2は炭素数1〜6のアルキル基であることが好ましく、この中でも、R2がメチル基、エチル基、プロピル基、ブチル基であることが特に好ましい。これらアルキル基の中でも、電子供与性の低いアルキル基ほど高温解離性を示すことから、アルキル基としては2級及び1級であることが好ましい。
【0077】
このうち有機カルボン酸エステルとしては、サンタシッドG(ジアルキルビニルエーテルブロック2官能ポリマー型カルボン酸)、サンタシッドH(モノアルキルビニルエーテルブロック2官能低分子量型カルボン酸)、サンタシッドI(モノアルキルビニルエーテルブロック2官能カルボン酸、いずれも、日油社製)などを好ましく用いることが出来る。
【0078】
本発明において、フラックス(B)の含有量は、樹脂(A)100重量部当たり、0.1重量部以上10重量部以下、好ましくは0.5重量部以上5重量部以下である。含有量が、0.1重量部未満では、酸化膜除去性低下によるはんだ接続不良のおそれがあり、また10重量部を超えると組成物の粘度上昇による接続不良のおそれがでてくる。
【0079】
[硬化剤(C)]
本発明において硬化剤(C)は、必要に応じて使用されるものであり、樹脂(A)が熱硬化性樹脂、特に、エポキシ樹脂である場合、樹脂を形成する際の架橋反応に寄与する物質を示す。
硬化剤(C)は、特に制限はなく既知のものが使用できる。例えば、エポキシ樹脂の場合、エポキシ樹脂硬化剤として知られているものはすべて使用できる。例えば、フェノール系硬化剤、脂肪族アミン、ポリエーテルアミン、脂環式アミン、芳香族アミンなどのアミン系硬化剤、酸無水物系硬化剤、アミド系硬化剤、第3級アミン、イミダゾール若しくはその誘導体、有機ホスフィン類、ホスホニウム塩、テトラフェニルボロン塩、有機酸ジヒドラジド、ハロゲン化ホウ素アミン錯体、ポリメルカプタン系硬化剤、イソシアネート系硬化剤、ブロックイソシアネート系硬化剤等が挙げられる。
【0080】
フェノール系硬化剤の具体例としては、ビスフェノールA、ビスフェノールF、4,4’−ジヒドロキシジフェニルメタン、4,4’−ジヒドロキシジフェニルエーテル、1,4−ビス(4−ヒドロキシフェノキシ)ベンゼン、1,3−ビス(4−ヒドロキシフェノキシ)ベンゼン、4,4’−ジヒドロキシジフェニルスルフィド、4,4’−ジヒドロキシジフェニルケトン、4,4’−ジヒドロキシジフェニルスルホン、4,4’−ジヒドロキシビフェニル、2,2’−ジヒドロキシビフェニル、10−(2,5−ジヒドロキシフェニル)−10H−9−オキサ−10−ホスファフェナンスレン−10−オキサイド、フェノールノボラック、ビスフェノールAノボラック、o−クレゾールノボラック、m−クレゾールノボラック、p−クレゾールノボラック、キシレノールノボラック、ポリ−p−ヒドロキシスチレン、ハイドロキノン、レゾルシン、カテコール、t−ブチルカテコール、t−ブチルハイドロキノン、フルオログリシノール、ピロガロール、t−ブチルピロガロール、アリル化ピロガロール、ポリアリル化ピロガロール、1,2,4−ベンゼントリオール、2,3,4−トリヒドロキシベンゾフェノン、1,2−ジヒドロキシナフタレン、1,3−ジヒドロキシナフタレン、1,4−ジヒドロキシナフタレン、1,5−ジヒドロキシナフタレン、1,6−ジヒドロキシナフタレン、1,7−ジヒドロキシナフタレン、1,8−ジヒドロキシナフタレン、2,3−ジヒドロキシナフタレン、2,4−ジヒドロキシナフタレン、2,5−ジヒドロキシナフタレン、2,6−ジヒドロキシナフタレン、2,7−ジヒドロキシナフタレン、2,8−ジヒドロキシナフタレン、上記ジヒドロキシナフタレンのアリル化物またはポリアリル化物、アリル化ビスフェノールA、アリル化ビスフェノールF、アリル化フェノールノボラック、アリル化ピロガロール等が例示される。
【0081】
アミン系硬化剤の具体例として、脂肪族アミン類としては、エチレンジアミン、1,3−ジアミノプロパン、1,4−ジアミノプロパン、ヘキサメチレンジアミン、2,5−ジメチルヘキサメチレンジアミン、トリメチルヘキサメチレンジアミン、ジエチレントリアミン、イミノビスプロピルアミン、ビス(ヘキサメチレン)トリアミン、トリエチレンテトラミン、テトラエチレンペンタミン、ペンタエチレンヘキサミン、N−ヒドロキシエチルエチレンジアミン、テトラ(ヒドロキシエチル)エチレンジアミン等が例示される。ポリエーテルアミン類としては、トリエチレングリコールジアミン、テトラエチレングリコールジアミン、ジエチレングリコールビス(プロピルアミン)、ポリオキシプロピレンジアミン、ポリオキシプロピレントリアミン類等が例示される。脂環式アミン類としては、イソホロンジアミン、メタセンジアミン、N−アミノエチルピペラジン、ビス(4−アミノ−3−メチルジシクロヘキシル)メタン、ビス(アミノメチル)シクロヘキサン、3,9−ビス(3−アミノプロピル)−2,4,8,10−テトラオキサスピロ(5,5)ウンデカン、ノルボルネンジアミン等が例示される。芳香族アミン類としては、テトラクロロ−p−キシレンジアミン、m−キシレンジアミン、p−キシレンジアミン、m−フェニレンジアミン、o−フェニレンジアミン、p−フェニレンジアミン、2,4−ジアミノアニソール、2,4−トルエンジアミン、2,4−ジアミノジフェニルメタン、4,4’−ジアミノジフェニルメタン、4,4’−ジアミノ−1,2−ジフェニルエタン、2,4−ジアミノジフェニルスルホン、4,4’−ジアミノジフェニルスルホン、m−アミノフェノール、m−アミノベンジルアミン、ベンジルジメチルアミン、2−ジメチルアミノメチル)フェノール、トリエタノールアミン、メチルベンジルアミン、α−(m−アミノフェ
ニル)エチルアミン、α−(p−アミノフェニル)エチルアミン、ジアミノジエチルジメチルジフェニルメタン、α,α’−ビス(4−アミノフェニル)−p−ジイソプロピルベンゼン等が例示される。
【0082】
酸無水物系硬化剤の具体例としては、ドデセニル無水コハク酸、ポリアジピン酸無水物、ポリアゼライン酸無水物、ポリセバシン酸無水物、ポリ(エチルオクタデカン二酸)無水物、ポリ(フェニルヘキサデカン二酸)無水物、メチルテトラヒドロ無水フタル酸、メチルヘキサヒドロ無水フタル酸、ヘキサヒドロ無水フタル酸、無水メチルハイミック酸、テトラヒドロ無水フタル酸、トリアルキルテトラヒドロ無水フタル酸、メチルシクロヘキセンジカルボン酸無水物、メチルシクロヘキセンテトラカルボン酸無水物、無水フタル酸、無水トリメリット酸、無水ピロメリット酸、ベンゾフェノンテトラカルボン酸無水物、エチレングリコールビストリメリテート二無水物、無水ヘット酸、無水ナジック酸、無水メチルナジック酸、5−(2,5−ジオキソテトラヒドロ−3−フラニル)−3−メチル−3−シクロヘキサン−1,2−ジカルボン酸無水物、3,4−ジカルボキシ−1,2,3,4−テトラヒドロ−1−ナフタレンコハク酸二無水物、1−メチル−ジカルボキシ−1,2,3,4−テトラヒドロ−1−ナフタレンコハク酸二無水物等が例示される。
【0083】
アミド系硬化剤としては、ジシアンジアミド、ポリアミド樹脂等が例示される。
第3級アミンとしては、1,8−ジアザビシクロ(5,4,0)ウンデセン−7、トリエチレンジアミン、ベンジルジメチルアミン、トリエタノールアミン、ジメチルアミノエタノール、トリス(ジメチルアミノメチル)フェノール等が例示される。
イミダゾール若しくはその誘導体としては、1−シアノエチル−2−フェニルイミダゾール、2−フェニルイミダゾール、2−エチル−4(5)−メチルイミダゾール、2−フェニル−4−メチルイミダゾール、1−ベンジル−2−メチルイミダゾール、1−ベンジル−2−フェニルイミダゾール、1−シアノエチル−2−ウンデシルイミダゾール、1−シアノ−2−フェニルイミダゾール、1−シアノエチル−2−ウンデシルイミダゾールトリメリテイト、1−シアノエチル−2−フェニルイミダゾリウムトリメリテイト、2,4−ジアミノ−6−[2’−メチルイミダゾリル−(1’)]−エチル−s−トリアジン、2,4−ジアミノ−6−[2’−エチル−4’−メチルイミダゾリル−(1’)]−エチル−s−トリアジン、2,4−ジアミノ−6−[2’−メチルイミダゾリル−(1’)]−エチル−s−トリアジンイソシアヌル酸付加体、2−フェニルイミダゾールイソシアヌル酸付加体、2−フェニル−4,5−ジヒドロキシメチルイミダゾール、2−フェニル−4−メチル−5−ヒドロキシメチルイミダゾール、又はエポキシ樹脂と上記イミダゾール類との付加体等が例示される。
【0084】
有機ホスフィン類としては、トリブチルホスフィン、メチルジフェニルホスフイン、トリフェニルホスフィン、ジフェニルホスフィン、フェニルホスフィン等が例示され、ホスホニウム塩としては、テトラフェニルホスホニウム・テトラフェニルボレート、テトラフェニルホスホニウム・エチルトリフェニルボレート、テトラブチルホスホニウム・テトラブチルボレート等が例示され、テトラフェニルボロン塩としては、2−エチル−4−メチルイミダゾール・テトラフェニルボレート、N−メチルモルホリン・テトラフェニルボレート等が例示される。
【0085】
また、上記した硬化剤は、1種を単独で用いてもよく、2種以上を任意の組み合わせおよび比率で混合して用いてもよい。
上記硬化剤の中でも、この中でも、イミダゾール又はその誘導体が好適に用いられる。
なお、フラックス(B)として、その分解生成物の有機カルボン酸がエポキシ樹脂の硬化作用を有す有機カルボン酸エステルを使用した場合には、該有機カルボン酸エステルを硬化剤(C)として用いてもよい。
【0086】
本発明の層間充填材組成物中の硬化剤(C)の含有量は、通常、樹脂(A)、特に、エポキシ樹脂100重量部に対して、0.1〜60重量部である。 ここで、硬化剤がフェノール系硬化剤、アミン系硬化剤、酸無水物系硬化剤の場合は、エポキシ樹脂中のエポキシ基と硬化剤中の官能基との当量比で0.8〜1.5の範囲となるように用いることが好ましい。この範囲外であると未反応のエポキシ基や硬化剤の官能基が残留し、所望の物性が得られないことがある。
また、硬化剤がアミド系硬化剤、第3級アミン、イミダゾール若しくはその誘導体、有機ホスフィン類、ホスホニウム塩、テトラフェニルボロン塩、有機酸ジヒドラジド、ハロゲン化ホウ素アミン錯体、ポリメルカプタン系硬化剤、イソシアネート系硬化剤、ブロックイソシアネート系硬化剤等の場合は、エポキシ樹脂100重量部に対して0.1〜20重量部の範囲で用いることが好ましい。
【0087】
[無機フィラー(D)]
本発明では、高い熱伝導率を有する無機フィラーを添加することにより、層間充填材組成物に更に高い熱伝導性を付与することが可能となる。これにより、半導体基板間の熱伝導を促進させて半導体デバイス基板の温度を低下させることにより、半導体デバイスを安定的に動作させることが可能となりより好ましい。
本発明で用いる無機フィラー(D)は高い熱伝導性を有するものが好ましく、特に熱伝導率が、好ましくは1W/mK以上が、より好ましく2W/mK以上の無機材料である。
【0088】
無機フィラー(D)は、体積平均粒径が0.1〜5μm、かつ、最大体積粒径が10μm以下であることが好ましく、より好ましくは体積平均粒径が0.1〜3μm、かつ、最大体積粒径が6μm以下であり、更に好ましくは、体積平均粒径が0.2〜1μm、かつ、最大体積粒径が3μm以下である。
上述のように高度に集積化された三次元集積回路において、層間充填材組成物からなるチップ間の層間充填層の厚みは、10〜50μm程度にまで小さくなっている。そのため、層間充填材組成物に配合するフィラーの体積粒径が10μmを超えると硬化した後の層間充填層の表面にフィラーが突出して、層間充填層の表面形状が悪化する傾向にある。
【0089】
一方、フィラーの粒径が小さ過ぎると、必要な熱伝導パス数が増加してチップ間の厚み方向に上から下まで繋がる確率が小さくなり、熱伝導性の高樹脂(A)と組み合わせても、層間充填層の厚み方向への熱伝導率が不十分になる。また、フィラーの粒径が小さ過ぎると、フィラーが凝集しやすくなり層間充填材組成物中での分散性が悪くなる。
本発明において、無機フィラー(D)の体積平均粒径を、上記範囲とすることにより、フィラー同士の過度の凝集が抑制され、厚み方向へ充分な熱伝導率を有する層間充填層を得ることができる。
【0090】
また、無機フィラー(D)として、体積平均粒径が異なる2種以上のフィラーを使用してもよい。例えば、体積平均粒径が比較的小さい、例えば0.1〜2μm、好ましくは0.2〜1.5μmの無機フィラーと、体積平均粒径が比較的大きい、例えば1〜5μm、好ましくは1〜3μmのフィラーとを併用することにより、体積平均粒径の大きい無機フィラー同士の熱伝導パスを体積平均粒径の小さい無機フィラーで繋ぐことにより、同一体積平均粒径のもののみを用いた場合に比べて高充填が可能となりより高い熱伝導性を得ることができる。
この場合、体積平均粒径の小さい無機フィラーと体積平均粒径の大きい無機フィラーとは重量比で10:1〜1:10の割合で用いることが、熱伝導パスの形成の上で好ましい。
【0091】
本発明において、無機フィラー(D)として使用される無機材料は、市販品や合成直後では、粉末が凝集して、上記粒径範囲を満たさない場合がある。そのため、無機フィラー(D)として使用される無機材料を上記粒径範囲を満たすように粉砕して用いることが好ましい。
無機材料の粉砕の方法は特に限定されず、ジルコニアビーズ等の粉砕用メディアと共に攪拌混合する方法や、ジェット噴射等の従来公知の粉砕方法を適用できる。 また、無機フィラー(D)は、樹脂(A)や塗布液中での分散性を高めるため、適宜表面処理をおこなってもよい。
【0092】
無機フィラー(D)としては、アルミナ(Al2O3:熱伝導率30W/mK)、窒化アルミニウム(AlN:熱伝導率260W/mK)、窒化ホウ素(BN:熱伝導率3W/mK(厚み方向)、275W/mK(面内方向))、窒化ケイ素(Si3N4:熱伝導率23W/mK)、シリカ(SiO2:熱伝導率1.4W/mK)などが挙げられるが、無機フィラーは更に酸素、水や高温暴露に対する安定性と低誘電性をも併せ持つことが接着したデバイスの信頼性の点で好ましく、このような無機フィラーとしては、Al2O3、AlN、BN、又はSiO2が好ましく、とりわけBNが好ましい。
これらの無機フィラー(D)は、1種を単独で用いてもよく、2種以上を任意の組み合わせおよび比率で混合して用いてもよい。
【0093】
本発明において、無機フィラー(D)の含有量は、樹脂(A)100重量部当たり、50重量部以上400重量部以下、好ましくは75重量部以上300重量部以下である。このような含有量とすることにより、本発明の層間充填材組成物は、充分な熱伝導性が得られ、かつ、均一な塗膜が形成できる程度の粘度を保つことができる。
無機フィラー(D)の含有量が、50重量部未満では、十分な熱伝導性が得られない場合があり、また、400重量部を超えると組成物の粘度が高くなり、均一な塗膜を形成出来ないとの問題が出てくる可能性がある。
【0094】
さらに、本発明の層間充填材組成物は、粘度調節等の目的で、本発明の効果を損なわない範囲で、無機フィラー(D)以外のフィラー(以下、その他のフィラーと称す。)を含有してもよい。例えば、フィラーを熱伝導性向上ではなく粘度調節を目的として添加する場合には、熱伝導率がそれほど高くない、汎用フィラーであるシリカ(SiO2:熱伝導率1.4W/mK)を使用することができる。
一方で、その他のフィラーの体積平均粒径及び最大体積粒径は、無機フィラー(D)と同様の範囲であることが好ましい。
【0095】
〔その他の添加剤〕
本発明の層間充填材組成物には、その機能性の更なる向上を目的として、本発明の効果を損なわない範囲において、各種の添加剤を含んでいてもよい。
このようなその他の添加剤としては、基材との接着性やマトリックス樹脂と無機フィラーとの接着性を向上させるための添加成分として、シランカップリング剤やチタネートカップリング剤等のカップリング剤、保存安定性向上のための紫外線防止剤、酸化防止剤、可塑剤、難燃剤、着色剤、分散剤、流動性改良剤、基材との密着性向上剤等が挙げられる。
これらは、いずれも1種を単独で用いてもよく、2種以上を任意の組み合わせおよび比率で混合して用いてもよい。
【0096】
上記添加剤の中でも、樹脂(A)と無機フィラー(D)との密着性を向上させる観点からは、シランカップリング剤、チタネートカップリング剤などのカップリング剤を含むことが好ましい。
シランカップリング剤としては、γ−グリシドキシプロピルトリメトキシシラン、γ−グリシドキシプロピルトリエトキシシラン、β−(3,4−エポキシシクロヘキシル)エチルトリメトキシシラン等のエポキシシラン;γ−アミノプロピルトリエトキシシラン、N−β(アミノエチル)γ−アミノプロピルトリメトキシシラン、N−β(アミノエチル)γ−アミノプロピルメチルジメトキシシラン、γ−アミノプロピルトリメトキシシラン、γ−ウレイドプロピルトリエトキシシラン等のアミノシラン;3−メルカプトプロピルトリメトキシシラン等のメルカプトシラン;p−スチリルトリメトキシシラン、ビニルトリクロルシラン、ビニルトリス(β−メトキシエトキシ)シラン、ビニルトリメトキシシラン、ビニルトリエトキシシラン、γ−メタクリロキシプロピルトリメトキシシラン等のビニルシラン、さらに、エポキシ系、アミノ系、ビニル系の高分子タイプのシラン等が挙げられる。
【0097】
チタネートカップリング剤としては、イソプロピルトリイソステアロイルチタネート、イソプロピルトリ(N−アミノエチル・アミノエチル)チタネート、ジイソプロピルビス(ジオクチルホスフェート)チタネート、テトライソプロピルビス(ジオクチルホスファイト)チタネート、テトラオクチルビス(ジトリデシルホスファイト)チタネート、テトラ(2,2−ジアリルオキシメチル−1−ブチル)ビス(ジトリデシル)ホスファイトチタネート、ビス(ジオクチルパイロホスフェート)オキシアセテートチタネート、ビス(ジオクチルパイロホスフェート)エチレンチタネート等が挙げられる。
【0098】
その他の添加剤の配合量には特に制限はなく、必要な機能性が得られる程度に、通常の樹脂組成物の配合量で用いられる。
なお、その他の添加剤のうち、カップリング剤の添加量は、層間充填材組成物中の全固形分に対して0.1〜2.0重量%程度とするのが好ましい。カップリング剤の配合量が少ないと、カップリング剤を配合したことによるマトリックス樹脂と無機フィラー(D)との密着性の向上効果を十分に得ることができず、多過ぎると得られる硬化物からカップリング剤がブリードアウトする問題がある。
【0099】
また、本発明の層間充填材組成物には、成形時の流動性改良や基材との密着性向上の観点より、熱可塑性のオリゴマー類を添加することができる。熱可塑性のオリゴマー類としては、C5系及びC9系の石油樹脂、スチレン樹脂、インデン樹脂、インデン・スチレン共重合樹脂、インデン・スチレン・フェノール共重合樹脂、インデン・クマロン共重合樹脂、インデン・ベンゾチオフェン共重合樹脂等が例示される。添加量としては、通常、樹脂(A)100重量部に対して、2〜30重量部の範囲である。
【0100】
〔塗布液〕
本発明の三次元集積回路用の層間充填材組成物塗布液(以下、単に、本発明の塗布液と称す。)は、上記層間充填材組成物、即ち、樹脂(A)、フラックス(B)及び、必要に応じて硬化剤(C)、無機フィラー(D)に、更に有機溶媒(E)を含有する。
【0101】
[有機溶媒(E)]
本発明の塗布液で用いる有機溶媒(E)としては、例えばアセトン、メチルエチルケトン(MEK)、メチルイソブチルケトン、メチルアミルケトン、シクロヘキサノン等のケトン類、酢酸エチル等のエステル類;エチレングリコールモノメチルエーテル等のエーテル類;N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド等のアミド類;メタノール、エタノール等のアルコール類;ヘキサン、シクロヘキサン等のアルカン類;トルエン、キシレン等の芳香族類などが挙げられる。
このうち、樹脂(A)の溶解性及び溶媒の沸点等を勘案すると、メチルエチルケトンやシクロヘキサノン等ケトン類、エステル類、又はエーテル類が好ましく、特にメチルエチルケトンやシクロヘキサノンのケトン類を用いることが特に好ましい。
これらの有機溶媒(E)は、1種を単独で用いてもよく、2種以上を任意の組み合わせおよび比率で混合して用いてもよい。
【0102】
本発明の塗布液において、有機溶媒(E)の他の成分に対する混合割合は、特に制限はないが、他の成分に対して好ましくは20重量%以上70重量%以下、特に好ましくは30重量%以上60重量%以下である。このような混合割合とすることにより、本発明の塗布液を使用することにより、任意の塗布法によって良好な塗布膜を形成することができる。
有機溶媒(E)の混合割合が、20重量%未満では塗布液の粘度が上昇し良好な塗布膜が得られない場合があり、または70重量%を超えると所定の膜厚が得られない等の問題が出てくる可能性がある。
【0103】
本発明の塗布液には、各種の添加剤を含んでいてもよい。
このような添加剤としては、上述の添加剤の他、塗布液中での各成分の分散性を向上させる界面活性剤、乳化剤、低弾性化剤、希釈剤、消泡剤、イオントラップ剤等が挙げられる。
ここで、界面活性剤としては、従来公知のアニオン系界面活性剤、ノニオン系界面活性剤、カチオン系界面活性剤のいずれも使用できる。
例えば、ポリオキシエチレンアルキルエーテル類、ポリオキシエチレンアルキルアリールエーテル類、ポリオキシエチレンアルキルエステル類、ソルビタンアルキルエステル類、モノグリセリドアルキルエステル類、アルキルベンゼンスルホン酸塩類、アルキルナフタレンスルホン酸塩類、アルキル硫酸塩類、アルキルスルホン酸塩類、スルホコハク酸エステル塩類、アルキルベタイン類、アミノ酸類などが挙げられる。
また、これら界面活性剤においてCH結合の一部又は全てがCF結合となったフッ素界面活性剤も好ましく用いることが出来る。
【0104】
界面活性剤の添加量として、層間充填材組成物中の全固形分に対して、0.001〜5重量%程度とするのが好ましい。0.001重量%未満では、所定の膜厚均一性が得られない場合があり、また5重量%を超えるとエポキシ樹脂成分との相分離等を引き起こす場合があり好ましくない。
【0105】
本発明の塗布液の製造方法は、特に限定されず従来公知の方法によればよく、塗布液の構成成分を混合することで製造することができる。なお、その際、組成物の均一性の向上、脱泡等を目的として、ペイントシェーカーやビーズミル、プラネタリミキサ、撹拌型分散機、自公転攪拌混合機、三本ロールなどを用いて混合することが好ましい。
各成分の混合順序も反応や沈殿物が発生するなど特段の問題がない限り任意であり、塗布液の構成成分のうち、何れか2成分又は3成分以上を予め配合し、その後に残りの成分を混合してもよいし、一度に全部を混合してもよい。
なお、上述のように無機フィラー(D)は、粒径の大きな凝集体とならないことが好ましいので、塗布液の製造前に粉砕してもよいし、他の成分と混合してから粉砕してもよい。無機材料の粉砕の方法は特に限定されず、従来公知の粉砕方法を適用できる。
【0106】
〔三次元集積回路の製造方法〕
以下、本発明の層間充填材組成物、又は層間充填材組成物塗布液を使用する三次元集積回路の製造方法について説明する。かかる製造方法は、複数の半導体基板に、上述の層間充填材組成物を成膜後に、これらの半導体基板を加圧接着して積層する工程を含んでいる。
【0107】
本発明では、始めに半導体基板上に層間充填材組成物の薄膜を形成する。本発明の層間充填材組成物の塗布液を用いる場合にはその塗布液を用いて、ディップ法、スピンコート法、スプレーコート、ブレード法やその他の任意の方法で塗布膜を形成することが出来る。得られた塗布膜から溶媒や低分子成分除去のために、50〜150℃の任意の温度でベーキング処理を行いB−ステージ化膜を形成する。この際、一定の温度においてベーキング処理を行ってもよいが、組成物中の揮発成分除去を円滑に進めるために、減圧条件下にてベーキング処理を行ってもよい。また、エポキシ樹脂の硬化が進行しない範囲で、段階的な昇温によるベーキング処理を行っても良い。例えば、初めに60℃、次に80℃、更に120℃で各5〜30分程度のべーキング処理を実施することが出来る。
また、本発明の塗布液を使用せずに、本発明の層間充填材組成物をそのまま用いてもよい。例えば、樹脂の硬化が始まらない温度範囲において加温して溶融させたものを用いて、任意の方法で半導体基板上に層間充填材組成物からなる膜を成膜としてもよい。
また、本発明の層間充填材組成物は、フィルム成形に適した十分な伸び性を有するため、本発明の層間充填材組成物をフィルム成形し、該フィルムを半導体基板上に設置することで成膜してもよい。
【0108】
次に上記方法にて成膜した層間充填材組成物からなる膜を加熱してタック性を発現させた後に、接合対象の半導体基板と仮接着を行う。仮接着の温度としては、樹脂(A)の組成にもよるが、80〜150℃の温度で行うことが好ましい。半導体基板の接合が複数層の場合には、前記仮接着を基板の層数分繰り返しても良いし、B−ステージ化膜を形成した基板を複数層重ね合わせた後に、加熱してまとめて仮接着させても良い。仮接着の際には必要に応じて基板間に1gf/cm2〜1Kgf/cm2の加重をかけて実施することが好ましい。
【0109】
仮接着の後には半導体基板の本接合を行う。仮接着させた半導体基板を200℃以上、好ましくは220℃以上に加圧接着することにより、層間充填材組成物の樹脂の溶融粘度を低下させて半導体基板間の電気端子の接続を促進すると同時に、組成物中のフラックスを活性化させて半導体基板間のはんだ接合を実現することが出来る。なお、加熱温度の上限は、使用するエポキシ樹脂が分解、変質しない温度であり、樹脂の種類、グレードにより適宜決定されるが、通常300℃以下で行われる。
また、加圧接着の際には必要に応じて基板間に10gf/cm2〜10Kgf/cm2の加重をかけて実施することが好ましい。
【実施例】
【0110】
以下、本発明について、実施例を用いてさらに詳細に説明するが、本発明はその趣旨を逸脱しない限り、以下の実施例に限定されるものではない。
【0111】
以下の実施例1〜3及び比較例1〜3において用いた層間充填材組成物の配合成分は次の通りである。
・エポキシ樹脂
エポキシ樹脂(A1):フェノキシ樹脂
重量平均分子量:26,000
エポキシ当量:4,600g/当量
30重量%メチルエチルケトン/シクロヘキサノンの1:1(重量比)の混合溶液
エポキシ樹脂(A2):三菱化学社製 品名「157S65」
80.6重量%MEK溶液
軟化点:65℃
150℃における溶融粘度:0.3Pa・s
・フラックス(B):日油社製 品名「サンタシッドG」ジアルキルビニルエーテル
ブロック2官能ポリマー型カルボン酸
・硬化剤(C):四国化成工業社製 品名「C11Z−CN」
1−シアノエチル−2−ウンデシルイミダゾール
・無機フィラー(D):日新リフラテック社製 窒化ホウ素 BN(熱伝導率3W/mK(厚み方向)、275W/mK(面内方向))
・有機溶媒(E)
有機溶媒(E1):和光純薬工業社製 試薬特級 メチルエチルケトン(以下、
MEKと略記することがある)
有機溶媒(E2):和光純薬工業社製 試薬特級 シクロヘキサノン(以下、
CHNと略記することがある)
【0112】
なお、前記エポキシ樹脂(A1)としてのフェノキシ樹脂は次のようにして作製した。
YL6121H(エポキシ当量171g/当量、4,4’-ビフェノール型エポキシ樹脂と3,3’,5,5’-テトラメチル-4,4’-ビフェノール型エポキシ樹脂の1:1混合物(三菱化学社製)215重量部、3,3’−ジメチル−4,4’−ビフェノール(OH当量107g/当量、本州化学社製)127重量部、27重量%テトラメチルアンモニウムヒドロキシド水溶液0.32重量部、及び、反応用溶媒としてシクロヘキサノン228重量部を撹拌機付き耐圧反応容器に入れ、窒素ガス雰囲気下180℃で5時間、反応を行った。次いで、希釈用溶剤としてシクロヘキサノン171重量部及びメチルエチルケトン399重量部を加えて固形分濃度を調整した。反応生成物から定法により溶剤を除去して30重量%の樹脂溶液を得た。
【0113】
なお、エポキシ樹脂の物性及び接合評価時のはんだバンプの電気抵抗、無機フィラーの粒径は次のようにして測定した。
(1)溶融粘度
アントンパール・ジャパン社製 粘弾性測定装置Physica MCR301を用いて溶融粘度(パラレルプレート動的粘度)を測定した。
まず、測定対象であるエポキシ樹脂から溶媒を留去して固形物を得、その後、この固形物に対してプレス成型を行い、厚さ約1mmの板状サンプルを得た。このサンプルを、パラレルプレートディッシュとパラレルプレート(φ25mm)の間に載置しパラレルプレート動的粘度測定を行った。
測定条件は、上記サンプルに正弦波歪みを20%与え、その歪みの角周波数は10rad/secとし、1分間に3℃の割合で昇温させる過程での粘度を40℃〜200℃まで測定した。
【0114】
(2)熱伝導率
以下の装置にて、熱拡散率、比重、比熱を測定し、この3つの測定値を乗じることで熱伝導率を求めた。
1)熱拡散率:アイフェイズ社「アイフェイズ・モバイル 1u」
2)比重:メトラー・トレド社 天秤 XS−204(「固体比重測定キット」使用)
3)比熱:セイコーインスツル社 DSC320/6200
【0115】
(3)電気抵抗
FLUKE77マルチメーター(JOHN FLUKE MFG. Co. INC) を用いて端子間の抵抗値を測定した。
(4)粒径測定
攪拌混合後の試料(実施例1参照)をシクロヘキサノンで分散させ、堀場製作所レーザ回折/散乱式粒度分布測定装置LA−920にて測定した。得られた粒度分布から粉砕後の無機フィラー(D)の体積平均粒径及び最大体積粒径を求めた。
【0116】
[実施例1、2、比較例1,2]
エポキシ樹脂として、エポキシ樹脂(A1)とエポキシ樹脂(A2)を所定の配合重量比(溶媒を除いた樹脂として20:80)とし、このエポキシ樹脂(A)の合計を100重量部とした。このときエポキシ樹脂の溶融粘度は、3000Pa・s(50℃)及び16Pa・s(120℃)であった。このエポキシ樹脂の100重量部に対して、フラックス(B)、有機溶媒(E)(メチルエチルケトン(E1)とシクロヘキサノン(E2)を配合重量比(35:65)で混合したもの)、及び硬化剤(C)を表1に記載の重量比にて配合した。次に自公転攪拌機を用いて1000rpmにて5分間撹拌、混練することにより、層間充填材の原料ペーストを得た。
【0117】
この原料ペーストを、図1(a),(b)に記載した、はんだバンプ基板上の10mm×10mmエリア(約8500バンプ)に膜厚50μmとなるように塗布を行い、120℃で1分間加熱により溶媒を留去してB−ステージ化膜とした。この基板を再度120℃に加温した後、前記塗布エリアに、銅基板の銅面を塗布エリア面に向けて0.1Kgf/cm2の加重で押し当てて仮接着を行った。室温まで冷却後、仮接着基板を小型のプレス機を用いて、10Kgf/cm2の加重で10分間、120℃にて加熱プレスを実施した。
次に、この基板を250℃のホットプレート上において、0.1Kgf/cm2の加重で1分間加熱プレスを行った。120℃プレス後、又は250℃プレス後に各々はんだバンプ基板−銅基板間の接合性評価のために電気抵抗測定を実施した。結果を表1に併せて示す。
【0118】
表1より、仮接着後及び120℃におけるプレス後では、はんだバンプ基板−銅基板間の電気抵抗は数Ω以上であったのに対して、200℃以上の温度にて加圧接着することにより電気抵抗はいずれも1Ω以下となった。更に、組成物にフラックスを含有させると何れも基板間の電気抵抗が0.1Ω未満となり、はんだバンプ基板及び銅基板が層間充填材組成物を介した接合でありながら低抵抗化しており、良好な接合状態を実現出来ていることが分かる。
【0119】
【表1】
【0120】
[参考例1〜16]
各種のフラックス(B)と有機溶媒(E)(メチルエチルケトン(E1)とシクロヘキサノン(E2)を配合重量比(35:65)で混合したもの)を表2に記載の重量濃度にて配合した後、撹拌混合し、フラックス溶液を得た。
なお、使用した薬品は以下のとおりである。
ロジン(アビエチン酸混合物;和光純薬工業社製)、サンタシッドG〜I(有機カルボン酸誘導体;日油社製)、2E4MZ(2−エチル−4−メチルイミダゾール;四国化成工業社製)、C11Z−CN(1−シアノエチル−2−ウンデシルイミダゾール;四国化成工業社製)
【0121】
このフラックス溶液を10mm×10mmの銅基板上に50μL滴下した後、このフラックス液滴中にはんだボール(Sn3.0Ag0.5Cu、直径300μm)を添加した。この基板をホットプレート上にて120℃で1分間加熱により溶媒を留去した。次に、この基板を250℃のホットプレート上において、250℃で10秒間加熱を行い銅基板に対するはんだボールの融解性を評価した。結果を表2に示す。
【0122】
表2より、フラックスとして有機カルボン酸又は有機カルボン酸エステルを用いた場合、ハンダボールが所定の温度において融解して、銅基板と良好な接合を形成した。一方、アミノ酸類は溶媒に対する溶解性が低く、またイミダゾール類はフラックスとしての作用が低くはんだボールと銅基板が接合出来なかった。
【0123】
【表2】
【0124】
[実施例3]
上記エポキシ樹脂(A1)溶液4.7gとエポキシ樹脂(A2)溶液7g(溶媒を除いた樹脂として配合重量比20:80、このときエポキシ樹脂の溶融粘度は、3000Pa・s(50℃)及び16Pa・s(120℃)であった)及び、硬化剤(C)の5%シクロヘキサノン溶液を0.7g加えて混合し、エポキシ樹脂溶液を得た。
【0125】
この樹脂溶液をセパレータ(シリコーン処理したポリエチレンテレフタレートフィルム、厚み:100μm)にドクターブレードで塗布し、60℃で15分、その後80℃で15分、その後、減圧下(圧力<4Torr)にて80℃で15分加熱して溶媒を除去してBステージ膜とした。その後、もう一枚のセパレータを得られたBステージ膜上に載せた後、まず100℃15分、次いで150℃15分、その後200℃で30分減圧プレス(圧力1MPa)することにより成型・硬化させて、膜厚約50μmの硬化膜を得た。得られたエポキシ樹脂の熱伝導率は、0.22W/mKであった。
【0126】
(層間充填材組成物の合成及び評価)
エポキシ樹脂として、上記エポキシ樹脂(A1)溶液4.7gとエポキシ樹脂(A2)溶液7g(溶媒を除いた樹脂として配合重量比20:80、このときエポキシ樹脂の溶融粘度は、3000Pa・s(50℃)及び16Pa・s(120℃)であった)、及び無機フィラー(D)19.8g、有機溶媒(E)20.5gをSUS製の容器(ガラス製蓋付き)に仕込む。SUS製の攪拌翼で550rpmで回転しながら、直径2mmのジルコニアボール(YTZ−2)を300g加え、5時間攪拌し、無機フィラー(D)を粉砕した。攪拌終了後、硬化剤(C)の5%シクロヘキサノン溶液を0.7g加え、更に5分間攪拌した。攪拌後、ろ過によりジルコニアボールを除き、層間充填剤ペースト(塗布液)を得た。
【0127】
得られた粉砕後の層間充填剤ペースト中の無機フィラー(D)の粒度分布を図2に示す。粒度分布から求めた無機フィラー(D)の体積平均粒径0.5μm、最大体積粒径1.5μmであった。
このペーストをスピンコーター(ミカサ社製)を用いて基板に塗布し、8℃×15分、120℃×15分で加熱乾燥しB−ステージ化膜とした。得られたB−ステージ化膜は、図3に示すように目視において均一であった。
【0128】
[比較例3]
エポキシ樹脂として、エポキシ樹脂(A1)溶液4.7gとエポキシ樹脂(A2)溶液7g(溶媒を除いた樹脂として配合重量比20:80、このときエポキシ樹脂の溶融粘度は、3000Pa・s(50℃)及び16Pa・s(120℃)であった)、及び無機フィラー(D)19.8g、硬化剤(C)(5%シクロヘキサノン溶液)0.7g、有機溶媒(E)20.5gをSUS製の容器(ガラス製蓋付き)に仕込んだ。SUS製の攪拌翼で550rpm×5分攪拌し、層間充填剤ペーストを得た。
実施例3と同じ装置にて層間充填剤ペーストの粒径分布を測定した結果を図4に示す。粒度分布から求めた無機フィラー(D)の体積平均粒径4.0μm、最大体積粒径20μmであった。
この層間充填剤ペーストを用いて、実施例3と同様の方法で、B−ステージ化膜を得た。得られたB−ステージ化膜には、図5に示すように目視で確認できるフィラーが多数存在し、不均一であった。
【0129】
〔合成例1〜3〕
これらの合成例におけるエポキシ樹脂(B)の評価は、下記の方法で行った。
<分子量>
高速GPC装置(東ソー(株)製「HLC−8320GPC EcoSEC(登録商標)」)を使用し、以下の測定条件で、標準ポリスチレンとして、TSK Standard Polystyrene:F−128(Mw1,090,000、Mn1,030,000)、F−10(Mw106,000、Mn103,000)、F−4(Mw43,000、Mn42,700)、F−2(Mw17,200、Mn16,900)、A−5000(Mw6,400、Mn6,100)、A−2500(Mw2,800、Mn2,700)、A−300(Mw453、Mn387)を使用した検量線を作成し、重量平均分子量および数平均分子量をポリスチレン換算値として測定した。
カラム:東ソー社製「TSKGEL SuperHM−H+H5000+H4000+H3000+H2000」
溶離液:テトラヒドロフラン
流速:0.5ml/min
検出:UV(波長254nm)
温度:40℃
試料濃度:0.1重量%
インジェクション量:10μL
【0130】
<n数>
前記エポキシ樹脂(B)の式(1)におけるnの値及びその平均値は、上記で求められた数平均分子量より算出した。
<エポキシ当量>
JIS K 7236に準じて測定し、固形分換算値として表記した。
<ガラス転移温度Tg>
溶剤を乾燥除去したエポキシ樹脂で、SIIナノテクノロジー社製「DSC7020」を使用し、30〜200℃まで10℃/minで昇温して測定した。
【0131】
<伸び>
エポキシ樹脂の溶液をセパレータ(シリコーン処理したポリエチレンテレフタレートフィルム、厚み:100μm)にアプリケーターで塗布し、60℃で1時間、その後150℃で1時間、更に200℃で1時間乾燥させ、厚さ約50μmのエポキシ樹脂フィルムを得た。これを幅1cmに切り出し、精密万能試験機(インストロン社製 INSTRON 5582型)を使用して5mm/minで3回測定した平均値を示した。
【0132】
また、下記する実施例で使用したエポキシ樹脂(B1)膜、実施例及び比較例で得られた硬化膜の熱伝導率は、下記の方法で評価した。なお、エポキシ樹脂(B1)膜は、上記の伸び測定用サンプルと同様の手法により得た。
<熱伝導率>
次の装置にて、熱拡散率、比重、比熱を測定し、この3つの測定値を乗じることで熱伝導率を求めた。
熱拡散率:アイフェイズ社「アイフェイズ・モバイル 1u」
比重:メトラー・トレド社「天秤 XS−204」(「固体比重測定キット」使用)
比熱:セイコーインスツル社「DSC320/6200」
【0133】
[エポキシ樹脂(B)の製造と評価]
<合成例1〜3>
表3に示した配合で化合物(X)、化合物(Y)、触媒および反応用溶剤を撹拌機付き耐圧反応容器に入れ、窒素ガス雰囲気下、180℃で5時間反応を行った後、希釈用溶剤を加えて固形分濃度を調整した。反応生成物から定法により溶剤を除去した後、得られた樹脂について分析を行った。結果を表3に示す。
【0134】
なお、反応に用いた化合物、触媒および溶剤は以下の通りである。
<化合物(X)>
(X−A):三菱化学社製 商品名「YL6121H」(4,4’−ビフェノール型エポキシ樹脂と3,3’,5,5’−テトラメチル−4,4’−ビフェノール型エポキシ樹脂の1:1混合物、エポキシ当量171g/当量)
(X−B):三菱化学社製 商品名「YX4000」(3,3’,5,5’−テトラメチル−4,4’−ビフェノールジグリシジルエーテル、エポキシ当量186g/当量)
【0135】
<化合物(Y)>
(Y−A):3,3’−ジメチル−4,4’−ビフェノール(OH当量107g/当量、本州化学社製)
(Y−B):3,3’,5,5’−テトラメチル−4,4’−ビフェノール(OH当量121g/当量)
<触媒>
(C−1):27重量%テトラメチルアンモニウムハイドロオキサイド水溶液
<溶剤>
(S−1):シクロヘキサノン
(S−2):メチルエチルケトン
(S−3):N,N’−ジメチルアセトアミド
【0136】
【表3】
【0137】
[エポキシ樹脂組成物の製造と評価]
<参考例4>
合成例1で得られたエポキシ樹脂(Mw59,741)2.5g(樹脂分0.75g;60重量部)と、ビスフェノールAノボラック型多官能エポキシ樹脂溶液(三菱化学社製 商品名「157S65(B80)」)0.625g(樹脂分0.5g;40重量部)と、硬化剤として2−エチル−4(5)−メチルイミダゾール(三菱化学社製 商品名「EMI24」)の20重量%溶液(溶剤MEK)0.032g(硬化剤重量0.00625g;0.5重量部)をはかり取り、自転公転ミキサーにて撹拌混合・脱泡を行った。このエポキシ樹脂組成物について硬化フィルムの熱伝導率を求めた。結果を表4に示す。
【0138】
<参考例5>
多官能エポキシ樹脂をビフェニル型多官能エポキシ樹脂(日本化薬社製 商品名「NC−3000−H」)の80重量%MEK溶液に変えた以外は参考例4と同様に、合成例1のエポキシ樹脂:NC−3000−H:EMI24=60:40:0.5(重量部)としてエポキシ樹脂組成物を製造し、得られたエポキシ樹脂組成物について、硬化フィルムの熱伝導率を求めた。結果を表4に示す。
【0139】
<参考例6>
合成例1のエポキシ樹脂:NC−3000−H:EMI24=30:70:0.5(重量部)としたこと以外は参考例5と同様にエポキシ樹脂組成物を製造し、得られたエポキシ樹脂組成物について、硬化フィルムの熱伝導率を求めた。結果を表4に示す。
【0140】
<参考例7>
多官能エポキシ樹脂をビフェニル型多官能エポキシ樹脂(日本化薬社製 商品名「NC−3000」)の70重量%シクロヘキサノン溶液に変えた以外は参考例4と同様に、合成例1のエポキシ樹脂:NC−3000:EMI24=60:40:0.5(重量部)として、エポキシ樹脂組成物を製造し、得られたエポキシ樹脂組成物について、硬化フィルムの熱伝導率を求めた。結果を表4に示す。
【0141】
<参考例8>
合成例1で得られたエポキシ樹脂:157S65(B80):EMI24=90:10:0.5(重量部)とした以外は参考例4と同様にエポキシ樹脂組成物を製造し、得られたエポキシ樹脂組成物について、硬化フィルムの熱伝導率を求めた。結果を表4に示す。
【0142】
【表4】
【0143】
以上の結果より、エポキシ樹脂(B)およびエポキシ樹脂(B)を含む組成物は、フィルム成形・塗布等のプロセスに適用するのに十分な伸び性を有し、かつ熱伝導性、耐熱性のバランスにも優れることが分かる。
【0144】
実施例4、5(比較例4、5)
これらの実施例及び比較例で使用したエポキシ樹脂(B)の1種であるエポキシ樹脂(B1)は、下記の方法で製造した。 使用した原料、触媒、溶剤を以下に示す。
・化合物(X):三菱化学社製 商品名「YL6121H」(4,4’−ビフェノール型 エポキシ樹脂と3,3’,5,5’−テトラメチル−4,4’−ビフェノール型エポキシ樹脂の1:1混合物、エポキシ当量171g/当量)
・化合物(Y):3,3’−ジメチル−4,4’−ビフェノール(OH当量107g/当量、本州化学社製)
・触媒:27重量%テトラメチルアンモニウムハイドロオキサイド水溶液
・溶剤
溶剤1:シクロヘキサノン
溶剤2:メチルエチルケトン
【0145】
化合物(X)210重量部、化合物(Y)127.6重量部、触媒0.78重量部、および溶剤1(シクロヘキサノン)181.8重量部を撹拌機付き耐圧反応容器に入れ、窒素ガス雰囲気下、180℃で5時間反応を行った。
その後、該耐圧反応容器に溶剤1(シクロヘキサノン)212.1重量部及び溶剤2(メチルエチルケトン)393.8重量部を加えて固形分濃度を30重量%に調整した。
反応生成物から定法により溶剤を除去した後、得られたエポキシ樹脂(B1)について分析を行った結果を以下に示す。
エポキシ樹脂(B1)
重量平均分子量(Mw):59000
数平均分子量(Mn) :14000
式(1)におけるn数 :49
エポキシ当量 :8150(g/当量)
Tg :110(℃)
伸び :72(%)
熱伝導率 :0.21(W/mK)
【0146】
下記の実施例及び比較例のエポキシ樹脂組成物の製造に使用した、エポキシ樹脂(B2)、エポキシ樹脂(C)、硬化剤(C)、無機フィラー(D)、及び溶媒(E)を以下に示す。
エポキシ樹脂(B2)
特殊骨格エポキシ樹脂(三菱化学社製、商品名YX6954BH30)
重量平均分子量(Mw):39000)
溶媒 :MEK:CHN=1:1
樹脂濃度 :30重量%溶液
・エポキシ樹脂(C)
エポキシ樹脂(C1):日本化薬社商品名「NC−3000−H」(80重量%M
EK溶液を調整)
エポキシ樹脂(C2): 三菱化学社製 商品名「157S65(B80)」(M
EK溶液;80重量%)
・硬化剤(C):三菱化学社製 商品名「EMI24」
2−エチル−4(5)−メチルイミダゾール
【0147】
・無機フィラー(D)
無機フィラー(D1):住友化学社製Al2O3 商品名「AA−3」
(体積平均粒径3μm)
無機フィラー(D2):住友化学社製Al2O3 商品名「AA−04」
(体積平均粒径0.4μm)
無機フィラー(D3):モメンティブ・パフォーマンス・マテリアルズ・ジャパ
ン合同会社製BN 商品名「PTX−25」(凝集粒子の体積平均粒径:25μm,一次粒子の体積平均粒径:3μm)
MEKとCHNの1:1(重量比)の混合溶媒
【0148】
[実施例4]
エポキシ樹脂(B1)とエポキシ樹脂(C1)を表5に示す配合重量比とし、エポキシ樹脂(B1)とエポキシ樹脂(C1)との合計100重量部に対して、硬化剤(C)を0.5重量部と、無機フィラー(D)として無機フィラー(D1)と無機フィラー(D2)を重量比8:2で混合したものを、硬化物中の無機フィラーの含有量が60体積%(エポキシ樹脂(B1)とエポキシ樹脂(C1)の合計100重量部として510重量部)となるように配合し、更に溶媒(E)を組成物中の固形分濃度が60重量%となるように添加して、ペースト状の塗布液を得た。
【0149】
このペースト状の塗布液をセパレータ(シリコーン処理したポリエチレンテレフタレートフィルム、厚み:100μm)にドクターブレードで塗布し、60℃で15分、その後80℃で15分、その後減圧(圧力<4Torr)にて80℃で15分加熱して溶媒を除去してBステージ膜とした。その後、もう一枚のセパレータを得られたBステージ膜上に載せた後、まず100℃15分、次いで150℃15分、その後200℃で30分減圧プレス(圧力1MPa)することにより成型・硬化させて、膜厚約50μmの硬化膜を得た。得られた硬化膜について、熱伝導率の評価を行った。結果を表5に示す。
【0150】
[実施例5]
実施例4において、無機フィラー(D)として無機フィラー(D3)を用い、エポキシ樹脂(C1)の代わりにエポキシ樹脂(C2)を用い、無機フィラー(D)の配合量を硬化物に対して40体積%(エポキシ樹脂(B1)とエポキシ樹脂(C2)の合計100重量部として129重量部)としたこと以外は実施例4と同様にして、硬化膜を得、その熱伝導率を評価した。結果を表5に示す。
【0151】
[比較例4]
エポキシ樹脂として、エポキシ樹脂(B1)の代わりにエポキシ樹脂(B2)を使用した以外は、実施例4と同様にしてエポキシ樹脂組成物を作製し、この組成物を硬化させて硬化物を得、その熱伝導率を評価した。結果を表5に示す。
【0152】
[比較例5]
エポキシ樹脂として、エポキシ樹脂(B1)の代わりにエポキシ樹脂(B2)を使用した以外は、は実施例5と同様にしてエポキシ樹脂組成物を作製し、この組成物を硬化させて硬化物を得、その熱伝導率を評価した。結果を表5に示す。
なお、表1中の各配合物の数字は重量部(溶液中の樹脂の重量)を示す。
【0153】
【表5】
【0154】
実施例6〜12
[配合成分]
実施例6〜12において用いた層間充填層形成用塗布液の配合成分は、次の通りである。
<エポキシ樹脂>
エポキシ樹脂(A1):実施例1で用いたのと同じエポキシ樹脂(A1)
エポキシ樹脂(A3):三菱化学株式会社製 品名「YL6800」
エポキシ樹脂(A4):三菱化学株式会社製 品名「1032H60」
エポキシ樹脂(A5):三菱化学株式会社製 品名「1001」
エポキシ樹脂(A6):三菱化学株式会社製 品名「4004」
エポキシ樹脂(A7):三菱化学株式会社製 品名「YX4000」
【0155】
<フラックス(B)>
和光純薬工業株式会社製 アジピン酸 試薬特級
<硬化剤(C)>
四国化成工業株式会社製 2−フェニル−4,5−ジヒドロキシメチルイミダゾール 品名「2PHZ−PW」
<無機フィラー(D)>
実施例1で用いたのと同じ、日新リフラテック社製 窒化ホウ素 BN
【0156】
<有機溶媒(E)>
有機溶媒(E)としては、前記有機溶媒(E1)(和光純薬工業社製 試薬特級 メチルエチルケトン)および有機溶媒(E2)(和光純薬工業社製 試薬特級 シクロヘキサノン)を用いた。
【0157】
[無機フィラーの粒径の測定]
攪拌混合後の層間充填層形成用塗布液をシクロヘキサノンで分散させ、島津製作所製粒度分布測定装置「SALD−2200」にて測定した。得られた粒度分布から粉砕後の無機フィラーの体積平均粒径及び最大体積粒径を求めた。
【0158】
[接合評価]
(1)チップへの塗布
攪拌混合後の層間充填層形成用塗布液について、マイクロピペットを用いて、Walts社製のSiチップ「CC80−0101JY ModelI」上に約10μLを塗布して塗り拡げた。
(2)B−Stage化
前記の層間充填層の塗布されたSiチップについて、ホットプレートを用いてはじめに80℃で15分間、続いて120℃で30分間それぞれ加熱を行いB−Stage化処理を行った。
(3)接合評価
B−Stage化された層間充填層の塗布された前記Siチップ及びWalts社製の有機基板「CC80−0102JY ModelI」について、東レエンジニアリング社製のフリップチップボンダ装置「FC3000S」を用いて、250℃まで昇温させて加熱圧着接合を行った。
(4)接合性(導通)評価
Siチップが接合された有機基板上の電極に、プローバー端子を接地させて、Siチップ及び有機基板の電気端子間に形成されたデイジーチェインについて、Keithley製マルチメータ「2100型」を用いて導通評価を行い、導通が確認出来たものを○とした。
【0159】
[層間充填層形成用塗布液の調製]
・実施例6の塗布液の調製
前記エポキシ樹脂(A1)のMEKとCHNの1:1(重量比)の混合溶液1.67g、エポキシ樹脂(A3)1.75g、およびエポキシ樹脂(A4)の80重量%シクロヘキサノン溶液0.31gを混合し、溶媒を除いた樹脂成分として、エポキシ樹脂(A1)とエポキシ樹脂(A3)とエポキシ樹脂(A4)との配合重量比が、20:70:10になるようにしてエポキシ樹脂組成物(I)を得た。このエポキシ樹脂組成物(I)の溶融粘度及び熱伝導率を表6に示す。
このエポキシ樹脂組成物(I)3.73g、無機フィラー(D)2.5g、および有機溶媒(E2)3.57gをポリエチレン製の容器に仕込み、更に直径0.5mmのジルコニアボールを20g加え、自公転攪拌機を用いて2000rpmで20分間攪拌した。攪拌終了後、ろ過によりジルコニアボールを取り除き、フラックス(B)を0.05gおよび硬化剤(C)を0.15g加え、更に自公転攪拌機にて6分間攪拌し、三次元集積回路用層間充填材組成物の塗布液を得た。この塗布液の固形分濃度は、50重量%で、無機フィラー(D)の含有量は、エポキシ樹脂組成物(I)及び無機フィラー(D)の合計量に対して50重量%であった。三次元集積回路用層間充填材組成物の塗布液中の無機フィラー(D)の体積平均粒径及び最大体積粒径を表7に示す。
【0160】
・実施例7の塗布液の調製
前記エポキシ樹脂(A1)のMEKとCHNの1:1(重量比)の混合溶液1.67g、エポキシ樹脂(A3)1.25g、エポキシ樹脂(A4)の80重量%シクロヘキサノン溶液0.31g、およびエポキシ樹脂(A5)の70重量%シクロヘキサノン溶液0.71gを混合し、溶媒を除いた樹脂成分として、エポキシ樹脂(A1)とエポキシ樹脂(A3)とエポキシ樹脂(A4)とエポキシ樹脂(A5)との配合重量比が、20:50:10:20になるようにしてエポキシ樹脂組成物(II)を得た。このエポキシ樹脂組成物(II)の溶融粘度及び熱伝導率を表6に示す。
エポキシ樹脂組成物(I)の代わりにエポキシ樹脂組成物(II)を3.94g、有機溶媒(E2)の代わりに有機溶媒(E1)を3.36g用いた以外は、実施例6の塗布液の調製と同様にして実施例7の三次元集積回路用層間充填材組成物の塗布液を調整した。三次元集積回路用層間充填材組成物の塗布液中の無機フィラー(D)の体積平均粒径及び最大体積粒径を表7に示す。
【0161】
・実施例8の塗布液の調製
エポキシ樹脂(A4)の80重量%シクロヘキサノン溶液の量を0.63g、エポキシ樹脂(A5)の70重量%シクロヘキサノン溶液の量を0.36gに変更し、溶媒を除いた樹脂として、エポキシ樹脂(A1)とエポキシ樹脂(A3)とエポキシ樹脂(A4)とエポキシ樹脂(A5)との配合重量比を20:50:20:10としたエポキシ樹脂組成物(III)を得た。このエポキシ樹脂組成物(III)の溶融粘度及び熱伝導率を表6に示す。
エポキシ樹脂組成物(III)を用いた以外は実施例7の塗布液の調製と同様にして、実施例8の三次元集積回路用層間充填材組成物の塗布液を調整した。この三次元集積回路用層間充填材組成物の塗布液中の無機フィラー(D)の体積平均粒径及び最大体積粒径を表7に示す。
【0162】
・実施例9の塗布液の調整
エポキシ樹脂(A6)を1.75g、エポキシ樹脂(A3)を0.7g、エポキシ樹脂(A4)の80重量%シクロヘキサノン溶液を0.35g、およびエポキシ樹脂(A5)の70重量%シクロヘキサノン溶液を0.70g混合し、溶媒を除いた樹脂成分として、エポキシ樹脂(A6)とエポキシ樹脂(A3)とエポキシ樹脂(A4)とエポキシ樹脂(A5)との配合重量比が、50:20:10:20になるようにしてエポキシ樹脂組成物(IV)を得た。このエポキシ樹脂組成物(IV)の溶融粘度及び熱伝導率を表6に示す。
エポキシ樹脂組成物(I)の代わりにエポキシ樹脂組成物(IV)3.50gを用い、フラックス(B)の使用量を0.07g、硬化剤(C)の使用量を0.21g、無機フィラー(D)の使用量を1.5gとして、有機溶媒(E2)の代わりに有機溶媒(E1)4.25gおよび有機溶媒(E2)0.47gを用いた以外は、実施例6の塗布液の調製と同様にして、実施例9の三次元集積回路用層間充填材組成物の塗布液を調整した。この塗布液の固形分濃度は、50重量%で、無機フィラー(D)の含有量は、エポキシ樹脂組成物(I)に対して30重量%であった。三次元集積回路用層間充填材組成物の塗布液中の無機フィラー(D)の体積平均粒径及び最大体積粒径を表7に示す。
【0163】
・実施例10〜13の塗布液の調整
エポキシ樹脂(A3)、エポキシ樹脂(A4)、エポキシ樹脂(A5)、エポキシ樹脂(A6)、およびエポキシ樹脂(A7)を表8に記載の比率にて混合した以外は、実施例9の三次元集積回路用層間充填材組成物の塗布液の調製と同様にして、樹脂組成物(V)〜(VIII)を調製した。このエポキシ樹脂組成物(V)〜(VIII)の溶融粘度及び熱伝導率を表6に示す。
当該樹脂組成物(V)〜(VIII)を使用した以外は、実施例9の三次元集積回路用層間充填材組成物の塗布液の調製と同様にして、実施例10〜13の三次元集積回路用層間充填材組成物の塗布液を調整した。三次元集積回路用層間充填材組成物の塗布液中の無機フィラー(D)の体積平均粒径及び最大体積粒径を表7に示す。
【0164】
[実施例6〜13]
前記三次元集積回路用層間充填材組成物の塗布液を用いて、Siチップ「CC80−0101JY ModelI」上に約10μLを塗布して塗り拡げた後、80℃で15分間、続いて120℃で30分間それぞれ加熱を行いB−Stage化処理を行った。このSiチップと有機基板「CC80−0102JY ModelI」を、フリップチップボンダ装置を用いて、250℃まで昇温させて加熱圧着接合を行った。接合後、有機基板上の電極に、プローバー端子を接地させて、Siチップ及び有機基板の電気端子間に形成されたデイジーチェインについて導通評価を行った。導通評価結果を、表7に示す。導通が確認出来たものを○、導通が確認されなかったものを×とした。
【0165】
【表6】
【表7】
【産業上の利用可能性】
【0166】
本発明によれば、半導体デバイス基板間のはんだバンプ等とランドの接合と同時に、熱伝導性の高い層間充填層を形成する層間充填材組成物、塗布液及び三次元集積回路の製造方法が提供される。
【特許請求の範囲】
【請求項1】
120℃における溶融粘度が100Pa・s以下である樹脂(A)及びフラックス(B)を含有し、フラックス(B)の含有量が、樹脂(A)100重量部当たり0.1重量部以上10重量部以下であることを特徴とする三次元集積回路用の層間充填材組成物。
【請求項2】
更に硬化剤(C)を含有する、請求項1に記載の三次元集積回路用の層間充填材組成物。
【請求項3】
熱伝導率が1W/mK以上の無機フィラー(D)を樹脂(A)100重量部当たり、50重量部以上400重量部以下更に含有する請求項1又は2に記載の三次元集積回路用の層間充填材組成物。
【請求項4】
120℃における溶融粘度が100Pa・s以下であって、熱伝導率が0.2W/mK以上である樹脂(A)と、熱伝導率が2W/mK以上、体積平均粒径が0.1μm以上5μm以下、かつ、最大体積粒径が10μm以下である無機フィラー(D)と、硬化剤(C)及び/またはフラックス(B)とを含有することを特徴とする三次元集積回路用の層間充填材組成物。
【請求項5】
前記樹脂(A)の50℃における溶融粘度が2000Pa・s以上である、請求項1から4のいずれか1項に記載の三次元集積回路用の層間充填材組成物。
【請求項6】
前記樹脂(A)が熱硬化性樹脂である、請求項1から5のいずれか1項に記載の三次元集積回路用の層間充填材組成物。
【請求項7】
前記樹脂(A)がエポキシ樹脂である、請求項1から6のいずれか1項に記載の三次元集積回路用の層間充填材組成物。
【請求項8】
前記エポキシ樹脂が、フェノキシ樹脂であるエポキシ樹脂(A1)、又はエポキシ樹脂(A1)と、分子内に2個以上のエポキシ基を有するエポキシ樹脂であるエポキシ樹脂(A2)との混合物である請求項7に記載の三次元集積回路用の層間充填材組成物。
【請求項9】
前記エポキシ樹脂が、下記式(1)で表され、かつエポキシ当量が2,500g/当量以上30,000g/当量以下を有するエポキシ樹脂(B)である請求項7に記載の三次元集積回路用の層間充填材組成物。
【化1】
(式(1)中、Aは下記式(2)で表されるビフェニル骨格であり、Bは水素原子または下記式(3)で表される基であり、nは繰り返し数であり、平均値は1<n<100である。)
【化2】
(式(2)中、R1は、水素原子、炭素数1〜10の炭化水素基、またはハロゲン元素であり、互いに同一であっても異なっていてもよい。)
【化3】
【請求項10】
前記式(2)におけるR1が、水素原子または炭素数が1〜4のアルキル基であり、式(2)で表されるビフェニル骨格は少なくとも一つの水素原子と少なくとも一つの炭素数1〜4のアルキル基を有する、請求項9に記載の三次元集積回路用の層間充填材組成物。
【請求項11】
更にエポキシ当量が2500g/当量未満であるエポキシ樹脂(C)を含有する
請求項9又は10に記載の三次元集積回路用の層間充填材組成物。
【請求項12】
エポキシ樹脂(B)とエポキシ樹脂(C)を含む全エポキシ樹脂中のエポキシ樹脂(C)の割合が、10重量%以上80重量%以下である請求項11に記載の三次元集積回路用の層間充填材組成物。
【請求項13】
前記フラックス(B)が、有機カルボン酸である請求項1から12のいずれか1項に記載の三次元集積回路用の層間充填材組成物。
【請求項14】
前記有機カルボン酸の分解温度が、130℃以上である請求項13に記載の三次元集積回路用の層間充填材組成物。
【請求項15】
前記樹脂(A)と前記無機フィラー(D)との総体積に対し、無機フィラー(D)が5体積%以上60体積%以下である、請求項3から14のいずれか1項に記載の三次元集積回路用の層間充填材組成物。
【請求項16】
前期無機フィラー(D)が、窒化ホウ素フィラーである、請求項3から15のいずれか1項に記載の三次元集積回路用の層間充填材組成物。
【請求項17】
前記硬化剤(C)が、イミダゾール又はその誘導体である、請求項2から16のいずれか1項に記載の三次元集積回路用の層間充填材組成物。
【請求項18】
請求項1から17のいずれか1項に記載の層間充填材組成物に、更に有機溶媒(E)を含有してなる、三次元集積回路用の層間充填材組成物の塗布液。
【請求項19】
複数の半導体基板表面に、請求項1から17のいずれか1項に記載の層間充填材組成物を成膜した後に、これらの半導体基板を加圧接着して積層する工程を含む、三次元集積回路の製造方法。
【請求項1】
120℃における溶融粘度が100Pa・s以下である樹脂(A)及びフラックス(B)を含有し、フラックス(B)の含有量が、樹脂(A)100重量部当たり0.1重量部以上10重量部以下であることを特徴とする三次元集積回路用の層間充填材組成物。
【請求項2】
更に硬化剤(C)を含有する、請求項1に記載の三次元集積回路用の層間充填材組成物。
【請求項3】
熱伝導率が1W/mK以上の無機フィラー(D)を樹脂(A)100重量部当たり、50重量部以上400重量部以下更に含有する請求項1又は2に記載の三次元集積回路用の層間充填材組成物。
【請求項4】
120℃における溶融粘度が100Pa・s以下であって、熱伝導率が0.2W/mK以上である樹脂(A)と、熱伝導率が2W/mK以上、体積平均粒径が0.1μm以上5μm以下、かつ、最大体積粒径が10μm以下である無機フィラー(D)と、硬化剤(C)及び/またはフラックス(B)とを含有することを特徴とする三次元集積回路用の層間充填材組成物。
【請求項5】
前記樹脂(A)の50℃における溶融粘度が2000Pa・s以上である、請求項1から4のいずれか1項に記載の三次元集積回路用の層間充填材組成物。
【請求項6】
前記樹脂(A)が熱硬化性樹脂である、請求項1から5のいずれか1項に記載の三次元集積回路用の層間充填材組成物。
【請求項7】
前記樹脂(A)がエポキシ樹脂である、請求項1から6のいずれか1項に記載の三次元集積回路用の層間充填材組成物。
【請求項8】
前記エポキシ樹脂が、フェノキシ樹脂であるエポキシ樹脂(A1)、又はエポキシ樹脂(A1)と、分子内に2個以上のエポキシ基を有するエポキシ樹脂であるエポキシ樹脂(A2)との混合物である請求項7に記載の三次元集積回路用の層間充填材組成物。
【請求項9】
前記エポキシ樹脂が、下記式(1)で表され、かつエポキシ当量が2,500g/当量以上30,000g/当量以下を有するエポキシ樹脂(B)である請求項7に記載の三次元集積回路用の層間充填材組成物。
【化1】
(式(1)中、Aは下記式(2)で表されるビフェニル骨格であり、Bは水素原子または下記式(3)で表される基であり、nは繰り返し数であり、平均値は1<n<100である。)
【化2】
(式(2)中、R1は、水素原子、炭素数1〜10の炭化水素基、またはハロゲン元素であり、互いに同一であっても異なっていてもよい。)
【化3】
【請求項10】
前記式(2)におけるR1が、水素原子または炭素数が1〜4のアルキル基であり、式(2)で表されるビフェニル骨格は少なくとも一つの水素原子と少なくとも一つの炭素数1〜4のアルキル基を有する、請求項9に記載の三次元集積回路用の層間充填材組成物。
【請求項11】
更にエポキシ当量が2500g/当量未満であるエポキシ樹脂(C)を含有する
請求項9又は10に記載の三次元集積回路用の層間充填材組成物。
【請求項12】
エポキシ樹脂(B)とエポキシ樹脂(C)を含む全エポキシ樹脂中のエポキシ樹脂(C)の割合が、10重量%以上80重量%以下である請求項11に記載の三次元集積回路用の層間充填材組成物。
【請求項13】
前記フラックス(B)が、有機カルボン酸である請求項1から12のいずれか1項に記載の三次元集積回路用の層間充填材組成物。
【請求項14】
前記有機カルボン酸の分解温度が、130℃以上である請求項13に記載の三次元集積回路用の層間充填材組成物。
【請求項15】
前記樹脂(A)と前記無機フィラー(D)との総体積に対し、無機フィラー(D)が5体積%以上60体積%以下である、請求項3から14のいずれか1項に記載の三次元集積回路用の層間充填材組成物。
【請求項16】
前期無機フィラー(D)が、窒化ホウ素フィラーである、請求項3から15のいずれか1項に記載の三次元集積回路用の層間充填材組成物。
【請求項17】
前記硬化剤(C)が、イミダゾール又はその誘導体である、請求項2から16のいずれか1項に記載の三次元集積回路用の層間充填材組成物。
【請求項18】
請求項1から17のいずれか1項に記載の層間充填材組成物に、更に有機溶媒(E)を含有してなる、三次元集積回路用の層間充填材組成物の塗布液。
【請求項19】
複数の半導体基板表面に、請求項1から17のいずれか1項に記載の層間充填材組成物を成膜した後に、これらの半導体基板を加圧接着して積層する工程を含む、三次元集積回路の製造方法。
【図1】

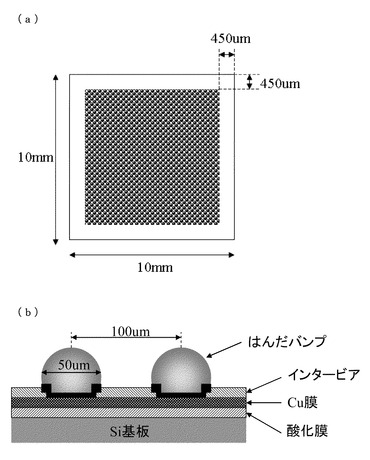
【図2】
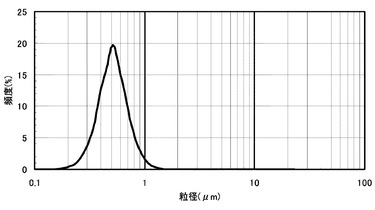
【図3】

【図4】
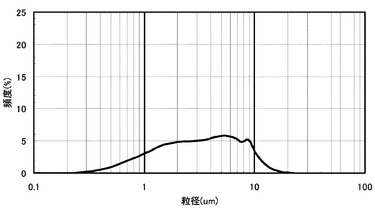
【図5】

【図2】
【図3】
【図4】
【図5】
【公開番号】特開2012−136689(P2012−136689A)
【公開日】平成24年7月19日(2012.7.19)
【国際特許分類】
【出願番号】特願2011−228311(P2011−228311)
【出願日】平成23年10月17日(2011.10.17)
【出願人】(000005968)三菱化学株式会社 (4,356)
【Fターム(参考)】
【公開日】平成24年7月19日(2012.7.19)
【国際特許分類】
【出願日】平成23年10月17日(2011.10.17)
【出願人】(000005968)三菱化学株式会社 (4,356)
【Fターム(参考)】
[ Back to top ]